User login
Letters to the Editor: Risk-reducing surgery for BRCA mutation carriers

“SHOULD RISK-REDUCING GYNECOLOGIC SURGERY FOR BRCA MUTATION CARRIERS INCLUDE HYSTERECTOMY?”
ANDREW M. KAUNITZ, MD (WEB EXCLUSIVE, AUGUST 28, 2016)
Hysterectomy warranted?
I am wondering if Dr. Kaunitz really is recommending performing 270 hysterectomies to prevent one endometrial cancer? Is this justified given the risks from the hysterectomy itself, the economics of the disease, or any significant reductions in endometrial cancer mortality?
David O. Holtz, MD
Paoli, Pennsylvania
Dr. Kaunitz responds
I appreciate Dr. Holtz’s interest in my commentary on the role of hysterectomy as part of risk-reducing surgery in BRCA mutation carriers. Women who are mutation carriers are at increased risk for serous or serous-like endometrial cancers. Further, hysterectomy offers specific advantages for young mutation carriers for whom menopausal hormone therapy is often indicated after risk-reducing salpingo-oophorectomy. Accordingly, I would indeed encourage such women to consider hysterectomy as part of risk-reducing gynecologic surgery if such surgery can be accomplished via minimally invasive techniques.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
“SHOULD RISK-REDUCING GYNECOLOGIC SURGERY FOR BRCA MUTATION CARRIERS INCLUDE HYSTERECTOMY?”
ANDREW M. KAUNITZ, MD (WEB EXCLUSIVE, AUGUST 28, 2016)
Hysterectomy warranted?
I am wondering if Dr. Kaunitz really is recommending performing 270 hysterectomies to prevent one endometrial cancer? Is this justified given the risks from the hysterectomy itself, the economics of the disease, or any significant reductions in endometrial cancer mortality?
David O. Holtz, MD
Paoli, Pennsylvania
Dr. Kaunitz responds
I appreciate Dr. Holtz’s interest in my commentary on the role of hysterectomy as part of risk-reducing surgery in BRCA mutation carriers. Women who are mutation carriers are at increased risk for serous or serous-like endometrial cancers. Further, hysterectomy offers specific advantages for young mutation carriers for whom menopausal hormone therapy is often indicated after risk-reducing salpingo-oophorectomy. Accordingly, I would indeed encourage such women to consider hysterectomy as part of risk-reducing gynecologic surgery if such surgery can be accomplished via minimally invasive techniques.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
“SHOULD RISK-REDUCING GYNECOLOGIC SURGERY FOR BRCA MUTATION CARRIERS INCLUDE HYSTERECTOMY?”
ANDREW M. KAUNITZ, MD (WEB EXCLUSIVE, AUGUST 28, 2016)
Hysterectomy warranted?
I am wondering if Dr. Kaunitz really is recommending performing 270 hysterectomies to prevent one endometrial cancer? Is this justified given the risks from the hysterectomy itself, the economics of the disease, or any significant reductions in endometrial cancer mortality?
David O. Holtz, MD
Paoli, Pennsylvania
Dr. Kaunitz responds
I appreciate Dr. Holtz’s interest in my commentary on the role of hysterectomy as part of risk-reducing surgery in BRCA mutation carriers. Women who are mutation carriers are at increased risk for serous or serous-like endometrial cancers. Further, hysterectomy offers specific advantages for young mutation carriers for whom menopausal hormone therapy is often indicated after risk-reducing salpingo-oophorectomy. Accordingly, I would indeed encourage such women to consider hysterectomy as part of risk-reducing gynecologic surgery if such surgery can be accomplished via minimally invasive techniques.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.

VIDEO: Biologic therapy for multidrug-resistant HIV offers new hope
NEW ORLEANS – Ibalizumab, the first monoclonal antibody to reach a phase III trial for multidrug resistant HIV therapy, has demonstrated efficacy, safety, and a novel mechanism of action, offering promise to many patients with few remaining options.
“The drug has now shown very significant antiretroviral activity, with 83% of patients demonstrating a half log decrease after 7 days, and a mean and median decrease of 1.1 log,” Jacob Lalezari, MD, lead author of the study and medical director of Quest Clinical Research in San Francisco, said at IDWeek 2016, the combined annual meetings of the Infectious Diseases Society of America, the Society for Healthcare Epidemiology of America, the HIV Medicine Association, and the Pediatric Infectious Diseases Society. “The big message here is the novel mechanism of action provides new treatment to patients with limited options, really offering hope to those left behind by an otherwise amazing evolution of HIV treatment.”

Dr. Lalezari said there is currently no evidence of cross-resistance with existing antiretrovirals. “I think we got lucky with that,” he noted. “For the primary care HIV doc, who is increasingly overwhelmed by drug-drug interactions, the good news is ibalizumab does not have obvious drug-drug interactions with antiretrovirals or other [HIV] drugs in other classes.”
Daniel Kuritzkes, MD, chief of infectious diseases at Brigham and Women’s Hospital and professor at Harvard Medical School, both in Boston, announced the findings with Dr. Lalezari at an IDWeek 2016 press conference. “This study is very important, because although we have terrific therapies for initial and second line treatment, we’ve really come to need treatment for the core group of patients who have developed resistance to all the drugs in the armamentarium,” he said.
Multidrug resistance emerges in less than 5% of people with HIV, affecting up to about 10,000 people in the United States. “It’s not a huge population, but it’s the most vulnerable and in need,” Dr. Lalezari said.
The 40 treatment-experienced patients in the study had their viral load and CD4+ counts measured at day 0. At day 7, they received a 2,000 mg IV loading dose of ibalizumab. At day 14, the response to ibalizumab monotherapy was measured and participants began an optimized background regimen with at least one other agent to which HIV showed sensitivity. Unfortunately, for about 50% of the patients, there was no such agent remaining, so researchers added BMS-663068, an investigational oral attachment inhibitor.
The cohort was 85% men, 45% nonwhite, and had a mean duration of HIV infection of 21 years. At study entry, patients’ mean viral load was approximately 100,000 copies/mL and mean CD4+ T-cell count was 160/mcL. However, half of the patients had T-cell counts below 100, and one third had counts below 10, “meaning they are at the very edge of sustainability,” Dr. Lalezari said.
Efficacy and safety
“The story is pretty simple – the drug worked,” Dr. Lalezari said. In addition to the 83% who met the primary endpoint of a half-life log decrease in HIV-1 RNA, 60% had a full log decrease or more at day 14. The mean and median HIV-1 RNA decrease for the entire cohort was 1.1 log10, a significant difference, compared with the day 0 to 7 control period (P less than .0001).
Putting the findings in perspective, Dr. Lalezari said, “So 1 log is not the most potent drug we see for HIV, but it’s pretty good. And in the setting of multidrug-resistant virus, it’s very good.” Although the agent is not potent enough for monotherapy, he said it is a strong candidate for combination therapy. The goal is to “give somebody a chance, potentially their last chance, to get control of the virus and prevent the progression of disease, and importantly, prevent them from spreading it to somebody else.”
“It’s a bit of a mystery” why 7 of the 40 patients did not meet the primary endpoint, Dr. Lalezari added. None of the factors the investigators compared between responders and nonresponders were significantly different.
Ibalizumab appears safe with no discontinuations and no treatment-related serious adverse events, Dr. Lalezari said. “We have not seen anything that strikes me as concerning at all in terms of safety, and it’s in a patient population that is quite ill.”
When asked during the press conference to address cost concerns, an issue in other specialties when biologics are introduced, Dr. Lalezari responded, “The one comment I will make is that whatever this drug is, it’s not a ‘me too drug.’ It’s unique and offers a unique mechanism of action. For those patients, the patients we saw whose T cells were under 10, whose health was failing and they were getting ready to die, their viral loads got suppressed and now they’re living, so it brings great value.”
Ibalizumab’s antiretroviral activity stems from blocking post-attachment conformational changes that are required to enable the HIV virus to bind with its co-receptors and ultimately gain entry into the target T cell. “Importantly the drug is away from the binding site for MHC class II molecules, and therefore not thought to cause T-cell depletion or be immunosuppressive,” Dr. Lalezari said.
In terms of the bigger picture, unlike most HIV drugs taken daily, ibalizumab is the first of the long-acting antiretrovirals, he noted. “I do think a paradigm shift is looming for at least some patients for whom long-acting therapy might be advantageous. There is also a movement toward IM and hopefully subcutaneous therapy as well, which would be very advantageous for patient self-administration.”
Although Dr. Lalezari and Dr. Kuritzkes presented the 14-day findings, the study is ongoing and participants continue to receive 800 mg ibalizumab intravenously every two weeks.
TaiMed Biologics, the sponsor of the study, has an expanded access program. Dr. Lalezari said, “So if you [have] a patient who is in trouble, in need of rescue therapy, there is an option for you now to consider.”
Dr. Lalezari receives research funding from TaiMed Biologics. Dr. Kuritzkes had no relevant financial disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
NEW ORLEANS – Ibalizumab, the first monoclonal antibody to reach a phase III trial for multidrug resistant HIV therapy, has demonstrated efficacy, safety, and a novel mechanism of action, offering promise to many patients with few remaining options.
“The drug has now shown very significant antiretroviral activity, with 83% of patients demonstrating a half log decrease after 7 days, and a mean and median decrease of 1.1 log,” Jacob Lalezari, MD, lead author of the study and medical director of Quest Clinical Research in San Francisco, said at IDWeek 2016, the combined annual meetings of the Infectious Diseases Society of America, the Society for Healthcare Epidemiology of America, the HIV Medicine Association, and the Pediatric Infectious Diseases Society. “The big message here is the novel mechanism of action provides new treatment to patients with limited options, really offering hope to those left behind by an otherwise amazing evolution of HIV treatment.”
Dr. Lalezari said there is currently no evidence of cross-resistance with existing antiretrovirals. “I think we got lucky with that,” he noted. “For the primary care HIV doc, who is increasingly overwhelmed by drug-drug interactions, the good news is ibalizumab does not have obvious drug-drug interactions with antiretrovirals or other [HIV] drugs in other classes.”
Daniel Kuritzkes, MD, chief of infectious diseases at Brigham and Women’s Hospital and professor at Harvard Medical School, both in Boston, announced the findings with Dr. Lalezari at an IDWeek 2016 press conference. “This study is very important, because although we have terrific therapies for initial and second line treatment, we’ve really come to need treatment for the core group of patients who have developed resistance to all the drugs in the armamentarium,” he said.
Multidrug resistance emerges in less than 5% of people with HIV, affecting up to about 10,000 people in the United States. “It’s not a huge population, but it’s the most vulnerable and in need,” Dr. Lalezari said.
The 40 treatment-experienced patients in the study had their viral load and CD4+ counts measured at day 0. At day 7, they received a 2,000 mg IV loading dose of ibalizumab. At day 14, the response to ibalizumab monotherapy was measured and participants began an optimized background regimen with at least one other agent to which HIV showed sensitivity. Unfortunately, for about 50% of the patients, there was no such agent remaining, so researchers added BMS-663068, an investigational oral attachment inhibitor.
The cohort was 85% men, 45% nonwhite, and had a mean duration of HIV infection of 21 years. At study entry, patients’ mean viral load was approximately 100,000 copies/mL and mean CD4+ T-cell count was 160/mcL. However, half of the patients had T-cell counts below 100, and one third had counts below 10, “meaning they are at the very edge of sustainability,” Dr. Lalezari said.
Efficacy and safety
“The story is pretty simple – the drug worked,” Dr. Lalezari said. In addition to the 83% who met the primary endpoint of a half-life log decrease in HIV-1 RNA, 60% had a full log decrease or more at day 14. The mean and median HIV-1 RNA decrease for the entire cohort was 1.1 log10, a significant difference, compared with the day 0 to 7 control period (P less than .0001).
Putting the findings in perspective, Dr. Lalezari said, “So 1 log is not the most potent drug we see for HIV, but it’s pretty good. And in the setting of multidrug-resistant virus, it’s very good.” Although the agent is not potent enough for monotherapy, he said it is a strong candidate for combination therapy. The goal is to “give somebody a chance, potentially their last chance, to get control of the virus and prevent the progression of disease, and importantly, prevent them from spreading it to somebody else.”
“It’s a bit of a mystery” why 7 of the 40 patients did not meet the primary endpoint, Dr. Lalezari added. None of the factors the investigators compared between responders and nonresponders were significantly different.
Ibalizumab appears safe with no discontinuations and no treatment-related serious adverse events, Dr. Lalezari said. “We have not seen anything that strikes me as concerning at all in terms of safety, and it’s in a patient population that is quite ill.”
When asked during the press conference to address cost concerns, an issue in other specialties when biologics are introduced, Dr. Lalezari responded, “The one comment I will make is that whatever this drug is, it’s not a ‘me too drug.’ It’s unique and offers a unique mechanism of action. For those patients, the patients we saw whose T cells were under 10, whose health was failing and they were getting ready to die, their viral loads got suppressed and now they’re living, so it brings great value.”
Ibalizumab’s antiretroviral activity stems from blocking post-attachment conformational changes that are required to enable the HIV virus to bind with its co-receptors and ultimately gain entry into the target T cell. “Importantly the drug is away from the binding site for MHC class II molecules, and therefore not thought to cause T-cell depletion or be immunosuppressive,” Dr. Lalezari said.
In terms of the bigger picture, unlike most HIV drugs taken daily, ibalizumab is the first of the long-acting antiretrovirals, he noted. “I do think a paradigm shift is looming for at least some patients for whom long-acting therapy might be advantageous. There is also a movement toward IM and hopefully subcutaneous therapy as well, which would be very advantageous for patient self-administration.”
Although Dr. Lalezari and Dr. Kuritzkes presented the 14-day findings, the study is ongoing and participants continue to receive 800 mg ibalizumab intravenously every two weeks.
TaiMed Biologics, the sponsor of the study, has an expanded access program. Dr. Lalezari said, “So if you [have] a patient who is in trouble, in need of rescue therapy, there is an option for you now to consider.”
Dr. Lalezari receives research funding from TaiMed Biologics. Dr. Kuritzkes had no relevant financial disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
NEW ORLEANS – Ibalizumab, the first monoclonal antibody to reach a phase III trial for multidrug resistant HIV therapy, has demonstrated efficacy, safety, and a novel mechanism of action, offering promise to many patients with few remaining options.
“The drug has now shown very significant antiretroviral activity, with 83% of patients demonstrating a half log decrease after 7 days, and a mean and median decrease of 1.1 log,” Jacob Lalezari, MD, lead author of the study and medical director of Quest Clinical Research in San Francisco, said at IDWeek 2016, the combined annual meetings of the Infectious Diseases Society of America, the Society for Healthcare Epidemiology of America, the HIV Medicine Association, and the Pediatric Infectious Diseases Society. “The big message here is the novel mechanism of action provides new treatment to patients with limited options, really offering hope to those left behind by an otherwise amazing evolution of HIV treatment.”
Dr. Lalezari said there is currently no evidence of cross-resistance with existing antiretrovirals. “I think we got lucky with that,” he noted. “For the primary care HIV doc, who is increasingly overwhelmed by drug-drug interactions, the good news is ibalizumab does not have obvious drug-drug interactions with antiretrovirals or other [HIV] drugs in other classes.”
Daniel Kuritzkes, MD, chief of infectious diseases at Brigham and Women’s Hospital and professor at Harvard Medical School, both in Boston, announced the findings with Dr. Lalezari at an IDWeek 2016 press conference. “This study is very important, because although we have terrific therapies for initial and second line treatment, we’ve really come to need treatment for the core group of patients who have developed resistance to all the drugs in the armamentarium,” he said.
Multidrug resistance emerges in less than 5% of people with HIV, affecting up to about 10,000 people in the United States. “It’s not a huge population, but it’s the most vulnerable and in need,” Dr. Lalezari said.
The 40 treatment-experienced patients in the study had their viral load and CD4+ counts measured at day 0. At day 7, they received a 2,000 mg IV loading dose of ibalizumab. At day 14, the response to ibalizumab monotherapy was measured and participants began an optimized background regimen with at least one other agent to which HIV showed sensitivity. Unfortunately, for about 50% of the patients, there was no such agent remaining, so researchers added BMS-663068, an investigational oral attachment inhibitor.
The cohort was 85% men, 45% nonwhite, and had a mean duration of HIV infection of 21 years. At study entry, patients’ mean viral load was approximately 100,000 copies/mL and mean CD4+ T-cell count was 160/mcL. However, half of the patients had T-cell counts below 100, and one third had counts below 10, “meaning they are at the very edge of sustainability,” Dr. Lalezari said.
Efficacy and safety
“The story is pretty simple – the drug worked,” Dr. Lalezari said. In addition to the 83% who met the primary endpoint of a half-life log decrease in HIV-1 RNA, 60% had a full log decrease or more at day 14. The mean and median HIV-1 RNA decrease for the entire cohort was 1.1 log10, a significant difference, compared with the day 0 to 7 control period (P less than .0001).
Putting the findings in perspective, Dr. Lalezari said, “So 1 log is not the most potent drug we see for HIV, but it’s pretty good. And in the setting of multidrug-resistant virus, it’s very good.” Although the agent is not potent enough for monotherapy, he said it is a strong candidate for combination therapy. The goal is to “give somebody a chance, potentially their last chance, to get control of the virus and prevent the progression of disease, and importantly, prevent them from spreading it to somebody else.”
“It’s a bit of a mystery” why 7 of the 40 patients did not meet the primary endpoint, Dr. Lalezari added. None of the factors the investigators compared between responders and nonresponders were significantly different.
Ibalizumab appears safe with no discontinuations and no treatment-related serious adverse events, Dr. Lalezari said. “We have not seen anything that strikes me as concerning at all in terms of safety, and it’s in a patient population that is quite ill.”
When asked during the press conference to address cost concerns, an issue in other specialties when biologics are introduced, Dr. Lalezari responded, “The one comment I will make is that whatever this drug is, it’s not a ‘me too drug.’ It’s unique and offers a unique mechanism of action. For those patients, the patients we saw whose T cells were under 10, whose health was failing and they were getting ready to die, their viral loads got suppressed and now they’re living, so it brings great value.”
Ibalizumab’s antiretroviral activity stems from blocking post-attachment conformational changes that are required to enable the HIV virus to bind with its co-receptors and ultimately gain entry into the target T cell. “Importantly the drug is away from the binding site for MHC class II molecules, and therefore not thought to cause T-cell depletion or be immunosuppressive,” Dr. Lalezari said.
In terms of the bigger picture, unlike most HIV drugs taken daily, ibalizumab is the first of the long-acting antiretrovirals, he noted. “I do think a paradigm shift is looming for at least some patients for whom long-acting therapy might be advantageous. There is also a movement toward IM and hopefully subcutaneous therapy as well, which would be very advantageous for patient self-administration.”
Although Dr. Lalezari and Dr. Kuritzkes presented the 14-day findings, the study is ongoing and participants continue to receive 800 mg ibalizumab intravenously every two weeks.
TaiMed Biologics, the sponsor of the study, has an expanded access program. Dr. Lalezari said, “So if you [have] a patient who is in trouble, in need of rescue therapy, there is an option for you now to consider.”
Dr. Lalezari receives research funding from TaiMed Biologics. Dr. Kuritzkes had no relevant financial disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT IDWEEK 2016
Key clinical point: Ibalizumab, a new biologic in a phase III trial, significantly reduces viral loads in patients with multidrug-resistant HIV infection.
Major finding: At 14 days, 83% of 40 trial participants experienced at least a one-half log reduction in HIV viral load.
Data source: Single arm, 24-week study of ibalizumab plus optimized background regimen in treatment experienced patients with multidrug-resistant HIV-1.
Disclosures: TaiMed Biologics, the manufacturer of ibalizumab, sponsored the study. Dr. Lalezari receives research funding from TaiMed Biologics. Dr. Kuritzkes had no relevant financial disclosures.

Staple line reinforcement linked to increased leak risk in bariatric surgery
Laparoscopic sleeve gastrectomy is safe and effective overall, but staple line reinforcement appears to increase the rate of postsurgical leaks – which were associated with readmissions and, in some cases, reoperations.
A large review of quality improvement data found that staple line reinforcement – an extremely common technique – was associated with a 60% increased risk of leak, compared with closures without staple line reinforcement, Elizabeth R. Berger, MD, and her colleagues reported in the October issue of the Annals of Surgery (2016;264:464-73).
“This study also demonstrates that leaks were significantly more morbid than bleeding with higher readmission and reoperation rates in patients with a leak vs. a bleed,” wrote Dr. Berger of Loyola University, Chicago, and her coauthors. “Therefore, a surgeon should consider the benefits, risks, and costs of each surgical technique in performing a laparoscopic sleeve gastrectomy and selectively utilize those that, in their hands, minimize morbidity while maximizing clinical effectiveness.”
The team examined outcomes in 189,477 laparoscopic sleeve gastrectomies performed by 1,634 surgeons at 720 centers from 2012 to 2014. All of the data were extracted from the Metabolic and Bariatric Surgery Accreditation and Quality Improvement Program, created in 2012 by the American College of Surgeons and the American Society for Metabolic and Bariatric Surgery.
They examined the impact of staple line reinforcement, oversewing the staple line, bougie size, and distance of the staple line from the pylorus on 30-day outcomes, and their effect on weight loss and weight-related comorbidities at 1 year. Outcomes included morbidity, leak rates, and bleeding, which were examined at both the patient and surgeon levels.
Most patients (126,904; 67%) patients had some type of staple line reinforcement (SLR); the rest had only oversewn staple lines (OSL) or no reinforcement. Leaks occurred in 1,703 patients and bleeds in 1,436 patients. Leaks were more serious than bleeds: Patients with a leak were almost 28% more likely to readmitted and 11% more likely to need a reoperation than were patients who had only a bleed.
At the patient level, those with SLR with or without OSL were 20%-46% more likely to experience a leak than were those who had neither. Bleeding rates were about 70% lower in the SLR groups.
Most surgeons in the analysis (80%) used some type of SLR; almost 20% routinely used only OSL, and 30% routinely used only SLR. At the surgeon level, SLR was associated with a 60% increased risk of a postoperative leak, compared with no reinforcement. There was no association between SLR and bleeding risk, however.
Oversewing had an effect on 1-year weight loss. Patients with oversewn staple lines lost an additional 1.3 points on the body mass index (BMI) scale, compared with patients with no type of reinforcement.
“The reason for increased leaks from SLR is relatively unclear,” the authors wrote. “The two layers of material that are placed within the staple line could increase ischemia or decrease the relative staple heights. At the notches, where one staple firing ends and the next one begins, there is sandwiching of the two layers of staples and a combined four layers of SLR. This bulk may predispose to leaks.”
Larger bougie sizes (BS) seemed more beneficial than did smaller ones, in both the surgeon- and patient-level analyses. A BS of at least 38 French was associated with a 28% decreased risk of a leak (odds ratio 0.72) at the patient level and a 10% decreased risk at the surgeon level (OR 0.90). There were no associations with bleeding.
“Our findings support literature that describes narrower BSs leads to increased ischemia secondary to increased intraluminal pressure, causing more leaks,” the authors wrote.
A BS of at least 40 French had a significant impact on weight loss. At 1 year, patients with the larger BS had lost 2.45 points more on the BMI scale than did those with smaller sizes.
This finding is in accord with other studies, including one that found the best weight-loss outcomes associated with a BS of more than 60 French. “Perhaps the sleeve works because of more rapid emptying, which is favored by a relatively larger BS, rather than because of restriction,” they said.
The distance to the pylorus (DP) from the staple line initiation point was divided into four sections: less than 4 cm; 4-5 cm; 5-6 cm; and 6 cm or more.
On a patient level, there was no association between DP and leak rates. There was, however, an association with bleeding. A DP of 4-4.99 cm had the highest rate, 90%, while a DP of 5-5.99 cm had the lowest (71%). DP was also associated with weight loss on this level, with a distance of more than 6 cm being associated with the biggest BMI decrease (3.7 points).
“Our data show significantly increased excess weight loss in a stepwise fashion as the DP increases,” the authors said. “Our data suggest that as DP increased, there was an increased excess weight loss, possibly explained by preserving the ‘antral mill.’ Stapling further from the pylorus perhaps keeps the antrum’s functional component intact and allows food to enter the distal gut more quickly, leading to earlier satiety and increased weight loss.”
Only 114 surgeons (8%) used a DP of less than 4 cm. There were no significant associations with any 30-day outcomes and DP after adjustment.
The authors had no financial disclosures.
Before drawing overarching conclusions and implementing recommendations based on this study, there are several limitations that must be borne in mind when considering data-mining exercises such as this one:
• It should be taken into account that there was significant intraoperative variation in technique and experience among the surgeons that was not captured through the data acquisition.
• Similarly, the true distance between the stapler and the selected bougie is also variable, adding an inherent lack of accuracy of the true real diameter of the completed gastric tube.
• There is a lack of granular information, including the type of SLR or staplers used, thereby also limiting any reliable conclusions that could be drawn.
• There are additional techniques, such as omental buttressing, and the use of clips, sutures, or hemostatic agents that are not reported, yet may have an impact on leak and bleeding rates.
• The reported follow-up rate of 39.4% at 1 year is typically considered to be suboptimal.
• SLR techniques may also include oversewing, and these are also subject to wide variation, including the type of suture material used, and the actual suturing technique that was implemented.
• Only those patients whose bleeding was severe enough to warrant transfusions were included, such that lower level bleeding would have not been represented in this report.
• There were also deficiencies in correlating leaks or bleeding rates with staple height selection, or the experience and learning curve of the surgeon.

Samer Mattar, MD, is a bariatric surgeon and professor of surgery at Oregon Health and Science University, Portland. Dr. Mattar has no disclosures.
Before drawing overarching conclusions and implementing recommendations based on this study, there are several limitations that must be borne in mind when considering data-mining exercises such as this one:
• It should be taken into account that there was significant intraoperative variation in technique and experience among the surgeons that was not captured through the data acquisition.
• Similarly, the true distance between the stapler and the selected bougie is also variable, adding an inherent lack of accuracy of the true real diameter of the completed gastric tube.
• There is a lack of granular information, including the type of SLR or staplers used, thereby also limiting any reliable conclusions that could be drawn.
• There are additional techniques, such as omental buttressing, and the use of clips, sutures, or hemostatic agents that are not reported, yet may have an impact on leak and bleeding rates.
• The reported follow-up rate of 39.4% at 1 year is typically considered to be suboptimal.
• SLR techniques may also include oversewing, and these are also subject to wide variation, including the type of suture material used, and the actual suturing technique that was implemented.
• Only those patients whose bleeding was severe enough to warrant transfusions were included, such that lower level bleeding would have not been represented in this report.
• There were also deficiencies in correlating leaks or bleeding rates with staple height selection, or the experience and learning curve of the surgeon.
Samer Mattar, MD, is a bariatric surgeon and professor of surgery at Oregon Health and Science University, Portland. Dr. Mattar has no disclosures.
Before drawing overarching conclusions and implementing recommendations based on this study, there are several limitations that must be borne in mind when considering data-mining exercises such as this one:
• It should be taken into account that there was significant intraoperative variation in technique and experience among the surgeons that was not captured through the data acquisition.
• Similarly, the true distance between the stapler and the selected bougie is also variable, adding an inherent lack of accuracy of the true real diameter of the completed gastric tube.
• There is a lack of granular information, including the type of SLR or staplers used, thereby also limiting any reliable conclusions that could be drawn.
• There are additional techniques, such as omental buttressing, and the use of clips, sutures, or hemostatic agents that are not reported, yet may have an impact on leak and bleeding rates.
• The reported follow-up rate of 39.4% at 1 year is typically considered to be suboptimal.
• SLR techniques may also include oversewing, and these are also subject to wide variation, including the type of suture material used, and the actual suturing technique that was implemented.
• Only those patients whose bleeding was severe enough to warrant transfusions were included, such that lower level bleeding would have not been represented in this report.
• There were also deficiencies in correlating leaks or bleeding rates with staple height selection, or the experience and learning curve of the surgeon.
Samer Mattar, MD, is a bariatric surgeon and professor of surgery at Oregon Health and Science University, Portland. Dr. Mattar has no disclosures.
Laparoscopic sleeve gastrectomy is safe and effective overall, but staple line reinforcement appears to increase the rate of postsurgical leaks – which were associated with readmissions and, in some cases, reoperations.
A large review of quality improvement data found that staple line reinforcement – an extremely common technique – was associated with a 60% increased risk of leak, compared with closures without staple line reinforcement, Elizabeth R. Berger, MD, and her colleagues reported in the October issue of the Annals of Surgery (2016;264:464-73).
“This study also demonstrates that leaks were significantly more morbid than bleeding with higher readmission and reoperation rates in patients with a leak vs. a bleed,” wrote Dr. Berger of Loyola University, Chicago, and her coauthors. “Therefore, a surgeon should consider the benefits, risks, and costs of each surgical technique in performing a laparoscopic sleeve gastrectomy and selectively utilize those that, in their hands, minimize morbidity while maximizing clinical effectiveness.”
The team examined outcomes in 189,477 laparoscopic sleeve gastrectomies performed by 1,634 surgeons at 720 centers from 2012 to 2014. All of the data were extracted from the Metabolic and Bariatric Surgery Accreditation and Quality Improvement Program, created in 2012 by the American College of Surgeons and the American Society for Metabolic and Bariatric Surgery.
They examined the impact of staple line reinforcement, oversewing the staple line, bougie size, and distance of the staple line from the pylorus on 30-day outcomes, and their effect on weight loss and weight-related comorbidities at 1 year. Outcomes included morbidity, leak rates, and bleeding, which were examined at both the patient and surgeon levels.
Most patients (126,904; 67%) patients had some type of staple line reinforcement (SLR); the rest had only oversewn staple lines (OSL) or no reinforcement. Leaks occurred in 1,703 patients and bleeds in 1,436 patients. Leaks were more serious than bleeds: Patients with a leak were almost 28% more likely to readmitted and 11% more likely to need a reoperation than were patients who had only a bleed.
At the patient level, those with SLR with or without OSL were 20%-46% more likely to experience a leak than were those who had neither. Bleeding rates were about 70% lower in the SLR groups.
Most surgeons in the analysis (80%) used some type of SLR; almost 20% routinely used only OSL, and 30% routinely used only SLR. At the surgeon level, SLR was associated with a 60% increased risk of a postoperative leak, compared with no reinforcement. There was no association between SLR and bleeding risk, however.
Oversewing had an effect on 1-year weight loss. Patients with oversewn staple lines lost an additional 1.3 points on the body mass index (BMI) scale, compared with patients with no type of reinforcement.
“The reason for increased leaks from SLR is relatively unclear,” the authors wrote. “The two layers of material that are placed within the staple line could increase ischemia or decrease the relative staple heights. At the notches, where one staple firing ends and the next one begins, there is sandwiching of the two layers of staples and a combined four layers of SLR. This bulk may predispose to leaks.”
Larger bougie sizes (BS) seemed more beneficial than did smaller ones, in both the surgeon- and patient-level analyses. A BS of at least 38 French was associated with a 28% decreased risk of a leak (odds ratio 0.72) at the patient level and a 10% decreased risk at the surgeon level (OR 0.90). There were no associations with bleeding.
“Our findings support literature that describes narrower BSs leads to increased ischemia secondary to increased intraluminal pressure, causing more leaks,” the authors wrote.
A BS of at least 40 French had a significant impact on weight loss. At 1 year, patients with the larger BS had lost 2.45 points more on the BMI scale than did those with smaller sizes.
This finding is in accord with other studies, including one that found the best weight-loss outcomes associated with a BS of more than 60 French. “Perhaps the sleeve works because of more rapid emptying, which is favored by a relatively larger BS, rather than because of restriction,” they said.
The distance to the pylorus (DP) from the staple line initiation point was divided into four sections: less than 4 cm; 4-5 cm; 5-6 cm; and 6 cm or more.
On a patient level, there was no association between DP and leak rates. There was, however, an association with bleeding. A DP of 4-4.99 cm had the highest rate, 90%, while a DP of 5-5.99 cm had the lowest (71%). DP was also associated with weight loss on this level, with a distance of more than 6 cm being associated with the biggest BMI decrease (3.7 points).
“Our data show significantly increased excess weight loss in a stepwise fashion as the DP increases,” the authors said. “Our data suggest that as DP increased, there was an increased excess weight loss, possibly explained by preserving the ‘antral mill.’ Stapling further from the pylorus perhaps keeps the antrum’s functional component intact and allows food to enter the distal gut more quickly, leading to earlier satiety and increased weight loss.”
Only 114 surgeons (8%) used a DP of less than 4 cm. There were no significant associations with any 30-day outcomes and DP after adjustment.
The authors had no financial disclosures.
Laparoscopic sleeve gastrectomy is safe and effective overall, but staple line reinforcement appears to increase the rate of postsurgical leaks – which were associated with readmissions and, in some cases, reoperations.
A large review of quality improvement data found that staple line reinforcement – an extremely common technique – was associated with a 60% increased risk of leak, compared with closures without staple line reinforcement, Elizabeth R. Berger, MD, and her colleagues reported in the October issue of the Annals of Surgery (2016;264:464-73).
“This study also demonstrates that leaks were significantly more morbid than bleeding with higher readmission and reoperation rates in patients with a leak vs. a bleed,” wrote Dr. Berger of Loyola University, Chicago, and her coauthors. “Therefore, a surgeon should consider the benefits, risks, and costs of each surgical technique in performing a laparoscopic sleeve gastrectomy and selectively utilize those that, in their hands, minimize morbidity while maximizing clinical effectiveness.”
The team examined outcomes in 189,477 laparoscopic sleeve gastrectomies performed by 1,634 surgeons at 720 centers from 2012 to 2014. All of the data were extracted from the Metabolic and Bariatric Surgery Accreditation and Quality Improvement Program, created in 2012 by the American College of Surgeons and the American Society for Metabolic and Bariatric Surgery.
They examined the impact of staple line reinforcement, oversewing the staple line, bougie size, and distance of the staple line from the pylorus on 30-day outcomes, and their effect on weight loss and weight-related comorbidities at 1 year. Outcomes included morbidity, leak rates, and bleeding, which were examined at both the patient and surgeon levels.
Most patients (126,904; 67%) patients had some type of staple line reinforcement (SLR); the rest had only oversewn staple lines (OSL) or no reinforcement. Leaks occurred in 1,703 patients and bleeds in 1,436 patients. Leaks were more serious than bleeds: Patients with a leak were almost 28% more likely to readmitted and 11% more likely to need a reoperation than were patients who had only a bleed.
At the patient level, those with SLR with or without OSL were 20%-46% more likely to experience a leak than were those who had neither. Bleeding rates were about 70% lower in the SLR groups.
Most surgeons in the analysis (80%) used some type of SLR; almost 20% routinely used only OSL, and 30% routinely used only SLR. At the surgeon level, SLR was associated with a 60% increased risk of a postoperative leak, compared with no reinforcement. There was no association between SLR and bleeding risk, however.
Oversewing had an effect on 1-year weight loss. Patients with oversewn staple lines lost an additional 1.3 points on the body mass index (BMI) scale, compared with patients with no type of reinforcement.
“The reason for increased leaks from SLR is relatively unclear,” the authors wrote. “The two layers of material that are placed within the staple line could increase ischemia or decrease the relative staple heights. At the notches, where one staple firing ends and the next one begins, there is sandwiching of the two layers of staples and a combined four layers of SLR. This bulk may predispose to leaks.”
Larger bougie sizes (BS) seemed more beneficial than did smaller ones, in both the surgeon- and patient-level analyses. A BS of at least 38 French was associated with a 28% decreased risk of a leak (odds ratio 0.72) at the patient level and a 10% decreased risk at the surgeon level (OR 0.90). There were no associations with bleeding.
“Our findings support literature that describes narrower BSs leads to increased ischemia secondary to increased intraluminal pressure, causing more leaks,” the authors wrote.
A BS of at least 40 French had a significant impact on weight loss. At 1 year, patients with the larger BS had lost 2.45 points more on the BMI scale than did those with smaller sizes.
This finding is in accord with other studies, including one that found the best weight-loss outcomes associated with a BS of more than 60 French. “Perhaps the sleeve works because of more rapid emptying, which is favored by a relatively larger BS, rather than because of restriction,” they said.
The distance to the pylorus (DP) from the staple line initiation point was divided into four sections: less than 4 cm; 4-5 cm; 5-6 cm; and 6 cm or more.
On a patient level, there was no association between DP and leak rates. There was, however, an association with bleeding. A DP of 4-4.99 cm had the highest rate, 90%, while a DP of 5-5.99 cm had the lowest (71%). DP was also associated with weight loss on this level, with a distance of more than 6 cm being associated with the biggest BMI decrease (3.7 points).
“Our data show significantly increased excess weight loss in a stepwise fashion as the DP increases,” the authors said. “Our data suggest that as DP increased, there was an increased excess weight loss, possibly explained by preserving the ‘antral mill.’ Stapling further from the pylorus perhaps keeps the antrum’s functional component intact and allows food to enter the distal gut more quickly, leading to earlier satiety and increased weight loss.”
Only 114 surgeons (8%) used a DP of less than 4 cm. There were no significant associations with any 30-day outcomes and DP after adjustment.
The authors had no financial disclosures.
FROM THE ANNALS OF SURGERY
Key clinical point:
Major finding: Compared to not reinforcing the staple line, doing sow as associated with up to a 60% increase in the risk of a postsurgical leak.
Data source: The database review contained outcomes on 189,477 laparoscopic sleeve gastrectomies.
Disclosures: None of the study authors had any financial disclosures.
Heart failure targets African Americans
ORLANDO – The disparity in U.S. heart failure incidence continued undiminished during 2002-2013, with African Americans maintaining a steady 2.3-fold increased rate of heart failure, compared with whites, based on national levels of heart failure hospitalizations, a reasonable surrogate for incidence rates, Boback Ziaeian, MD, reported at the annual scientific meeting of the Heart Failure Society of America.
The same period also showed a substantial relative improvement in the heart failure hospitalization rates among U.S. Hispanics, compared with whites, so that, by 2013, the ethnic disparity seen in 2002 between Hispanics and whites largely disappeared, reported Dr. Ziaeian, a cardiologist at the University of California, Los Angeles. The data he analyzed also showed that Asian Americans had the lowest heart failure hospitalization rates of any racial or ethnic group throughout the 11-year period, and that the incidence of heart failure fell more sharply in women than in men during the period, based on the hospitalization numbers.

Age-adjusted heart failure hospitalizations among whites dropped by 30%, and among African Americans by a nearly identical 29%. But this maintained a greater than twofold disparity in rates between the two groups. Among whites, the rate per 100,000 fell from 448 to 315; among African Americans, it dropped from 1,048 to 741. In 2013, the rate of heart failure hospitalizations was 2.4-fold higher in African Americans, compared with whites.
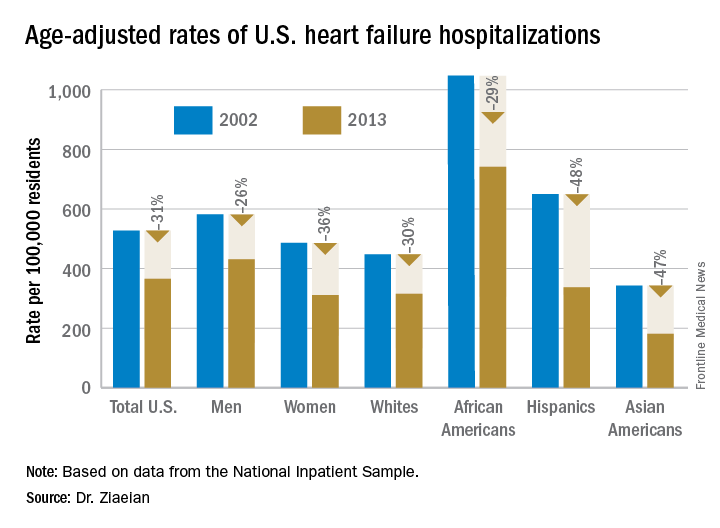
Heart failure hospitalizations fell among Hispanics from 650 per 100,000 to 337 per 100,000 in 2013, a 48% drop that brought the rate among Hispanics to nearly the same as among whites. Asian Americans remained the group with the least heart failure throughout the period, falling from 343 hospitalizations per 100,000 in 2002 to 181 per 100,000 in 2013, a 47% drop.
Among women, the age-adjusted rate per 100,000 fell from 486 to 311, a 36% drop, compared with a decrease from 582 to 431 per 100,000 in men, a 26% reduction. Lower incidence in women may reflect better risk factor control during the study period, compared with men, such as a higher rate of quiting smoking and better treatment compliance, Dr. Ziaeian suggested.
[email protected]
On Twitter @mitchelzoler
ORLANDO – The disparity in U.S. heart failure incidence continued undiminished during 2002-2013, with African Americans maintaining a steady 2.3-fold increased rate of heart failure, compared with whites, based on national levels of heart failure hospitalizations, a reasonable surrogate for incidence rates, Boback Ziaeian, MD, reported at the annual scientific meeting of the Heart Failure Society of America.
The same period also showed a substantial relative improvement in the heart failure hospitalization rates among U.S. Hispanics, compared with whites, so that, by 2013, the ethnic disparity seen in 2002 between Hispanics and whites largely disappeared, reported Dr. Ziaeian, a cardiologist at the University of California, Los Angeles. The data he analyzed also showed that Asian Americans had the lowest heart failure hospitalization rates of any racial or ethnic group throughout the 11-year period, and that the incidence of heart failure fell more sharply in women than in men during the period, based on the hospitalization numbers.
Age-adjusted heart failure hospitalizations among whites dropped by 30%, and among African Americans by a nearly identical 29%. But this maintained a greater than twofold disparity in rates between the two groups. Among whites, the rate per 100,000 fell from 448 to 315; among African Americans, it dropped from 1,048 to 741. In 2013, the rate of heart failure hospitalizations was 2.4-fold higher in African Americans, compared with whites.
Heart failure hospitalizations fell among Hispanics from 650 per 100,000 to 337 per 100,000 in 2013, a 48% drop that brought the rate among Hispanics to nearly the same as among whites. Asian Americans remained the group with the least heart failure throughout the period, falling from 343 hospitalizations per 100,000 in 2002 to 181 per 100,000 in 2013, a 47% drop.
Among women, the age-adjusted rate per 100,000 fell from 486 to 311, a 36% drop, compared with a decrease from 582 to 431 per 100,000 in men, a 26% reduction. Lower incidence in women may reflect better risk factor control during the study period, compared with men, such as a higher rate of quiting smoking and better treatment compliance, Dr. Ziaeian suggested.
[email protected]
On Twitter @mitchelzoler
ORLANDO – The disparity in U.S. heart failure incidence continued undiminished during 2002-2013, with African Americans maintaining a steady 2.3-fold increased rate of heart failure, compared with whites, based on national levels of heart failure hospitalizations, a reasonable surrogate for incidence rates, Boback Ziaeian, MD, reported at the annual scientific meeting of the Heart Failure Society of America.
The same period also showed a substantial relative improvement in the heart failure hospitalization rates among U.S. Hispanics, compared with whites, so that, by 2013, the ethnic disparity seen in 2002 between Hispanics and whites largely disappeared, reported Dr. Ziaeian, a cardiologist at the University of California, Los Angeles. The data he analyzed also showed that Asian Americans had the lowest heart failure hospitalization rates of any racial or ethnic group throughout the 11-year period, and that the incidence of heart failure fell more sharply in women than in men during the period, based on the hospitalization numbers.
Age-adjusted heart failure hospitalizations among whites dropped by 30%, and among African Americans by a nearly identical 29%. But this maintained a greater than twofold disparity in rates between the two groups. Among whites, the rate per 100,000 fell from 448 to 315; among African Americans, it dropped from 1,048 to 741. In 2013, the rate of heart failure hospitalizations was 2.4-fold higher in African Americans, compared with whites.
Heart failure hospitalizations fell among Hispanics from 650 per 100,000 to 337 per 100,000 in 2013, a 48% drop that brought the rate among Hispanics to nearly the same as among whites. Asian Americans remained the group with the least heart failure throughout the period, falling from 343 hospitalizations per 100,000 in 2002 to 181 per 100,000 in 2013, a 47% drop.
Among women, the age-adjusted rate per 100,000 fell from 486 to 311, a 36% drop, compared with a decrease from 582 to 431 per 100,000 in men, a 26% reduction. Lower incidence in women may reflect better risk factor control during the study period, compared with men, such as a higher rate of quiting smoking and better treatment compliance, Dr. Ziaeian suggested.
[email protected]
On Twitter @mitchelzoler
AT THE HFSA ANNUAL SCIENTIFIC MEETING
Key clinical point:
Major finding: In 2013, age-adjusted heart failure hospitalization was 741/100,000 in African Americans and 315/100,000 in whites.
Data source: The National Inpatient Sample and U.S. Census data.
Disclosures: Dr. Ziaeian had no disclosures.
Young adults and anxiety: Marriage may not be protective
A new study of anxiety disorders among young adults aged 18-24 shows that the illnesses are less prevalent among African American and Hispanic young adults, compared with whites. Furthermore, anxiety disorders are 1.5 times as prevalent among married people in this age group, compared with their unmarried peers.
For their research, presented at the annual meeting of the American Academy of Child and Adolescent Psychiatry, Cristiane S. Duarte, PhD, MPH, of Columbia University, New York, and her colleagues looked at data from the 2012/2013 National Epidemiologic Survey on Alcohol and Related Conditions (NESARC), a nationally representative sample of U.S. households.
“We were trying to look specifically at young adulthood, which there’s emerging consensus to regard as a key developmental period,” said Dr. Duarte, whose research focuses on anxiety disorders in young adults. “It’s a period where several psychiatric disorders tend to become much more prevalent. Having untreated anxiety disorders at this age can put young adults at risk for worse outcomes down the line. If anxiety disorders can be resolved, a young adult’s trajectory can be quite different; it’s a time in life in which the right intervention can have a really big impact,” she said.
The NESARC III survey data used structured diagnostic interviews and DSM-5 criteria to assess anxiety disorders occurring in the past year. These included specific phobia, generalized anxiety disorder, social anxiety, panic disorder, and agoraphobia.
For the most part, Dr. Duarte said, her group’s findings on anxiety disorders reflected earlier prevalence studies that had used DSM-IV criteria. Women were more likely than were men to report any past-year anxiety disorder (odds ratio, 2.26; 95% confidence interval, 1.80-2.84), as were people with lower personal and family incomes. Rates of anxiety disorders were highest in groups with the lowest personal and family incomes, and among people neither employed nor in an educational program.
Dr. Duarte said in an interview that the latter findings were generally anticipated. However, the finding that African Americans and Hispanics at this age had lower risk relative to whites (OR, 0.52; 95% CI, 0.40-0.67) and (OR, 0.63; 95% CI, 0.49-0.83) was interesting, because it appeared to mirror the lower relative prevalence seen among adults in those two groups, rather than the higher prevalence seen among children in the same groups. More research will be needed, she said, to verify and, if correct, understand this reversing trend in prevalence seen between childhood and adulthood.
The study’s most unexpected finding, Dr. Duarte said, was that married individuals aged 18-24 had higher prevalence of anxiety (OR, 1.54; 95% CI, 1.05-2.26). “Across the board, marriage is protective for many health and mental health conditions,” Dr. Duarte said, but she acknowledged that many factors could be in play. Marriage might not, in fact, be protective in this age group; the institution might be reflective of cultural factors promoting early marriage; or the findings could reflect a selection into marriage possibly related to existing anxiety disorders.
“To better understand this finding, we will need to consider several complexities which are part of young adulthood as a unique developmental period,” she said.
Dr. Duarte’s and her colleagues’ study was funded by the Youth Anxiety Center at New York–Presbyterian Hospital. Three coauthors reported research support from pharmaceutical manufacturers and royalties from commercial publishers.
A new study of anxiety disorders among young adults aged 18-24 shows that the illnesses are less prevalent among African American and Hispanic young adults, compared with whites. Furthermore, anxiety disorders are 1.5 times as prevalent among married people in this age group, compared with their unmarried peers.
For their research, presented at the annual meeting of the American Academy of Child and Adolescent Psychiatry, Cristiane S. Duarte, PhD, MPH, of Columbia University, New York, and her colleagues looked at data from the 2012/2013 National Epidemiologic Survey on Alcohol and Related Conditions (NESARC), a nationally representative sample of U.S. households.
“We were trying to look specifically at young adulthood, which there’s emerging consensus to regard as a key developmental period,” said Dr. Duarte, whose research focuses on anxiety disorders in young adults. “It’s a period where several psychiatric disorders tend to become much more prevalent. Having untreated anxiety disorders at this age can put young adults at risk for worse outcomes down the line. If anxiety disorders can be resolved, a young adult’s trajectory can be quite different; it’s a time in life in which the right intervention can have a really big impact,” she said.
The NESARC III survey data used structured diagnostic interviews and DSM-5 criteria to assess anxiety disorders occurring in the past year. These included specific phobia, generalized anxiety disorder, social anxiety, panic disorder, and agoraphobia.
For the most part, Dr. Duarte said, her group’s findings on anxiety disorders reflected earlier prevalence studies that had used DSM-IV criteria. Women were more likely than were men to report any past-year anxiety disorder (odds ratio, 2.26; 95% confidence interval, 1.80-2.84), as were people with lower personal and family incomes. Rates of anxiety disorders were highest in groups with the lowest personal and family incomes, and among people neither employed nor in an educational program.
Dr. Duarte said in an interview that the latter findings were generally anticipated. However, the finding that African Americans and Hispanics at this age had lower risk relative to whites (OR, 0.52; 95% CI, 0.40-0.67) and (OR, 0.63; 95% CI, 0.49-0.83) was interesting, because it appeared to mirror the lower relative prevalence seen among adults in those two groups, rather than the higher prevalence seen among children in the same groups. More research will be needed, she said, to verify and, if correct, understand this reversing trend in prevalence seen between childhood and adulthood.
The study’s most unexpected finding, Dr. Duarte said, was that married individuals aged 18-24 had higher prevalence of anxiety (OR, 1.54; 95% CI, 1.05-2.26). “Across the board, marriage is protective for many health and mental health conditions,” Dr. Duarte said, but she acknowledged that many factors could be in play. Marriage might not, in fact, be protective in this age group; the institution might be reflective of cultural factors promoting early marriage; or the findings could reflect a selection into marriage possibly related to existing anxiety disorders.
“To better understand this finding, we will need to consider several complexities which are part of young adulthood as a unique developmental period,” she said.
Dr. Duarte’s and her colleagues’ study was funded by the Youth Anxiety Center at New York–Presbyterian Hospital. Three coauthors reported research support from pharmaceutical manufacturers and royalties from commercial publishers.
A new study of anxiety disorders among young adults aged 18-24 shows that the illnesses are less prevalent among African American and Hispanic young adults, compared with whites. Furthermore, anxiety disorders are 1.5 times as prevalent among married people in this age group, compared with their unmarried peers.
For their research, presented at the annual meeting of the American Academy of Child and Adolescent Psychiatry, Cristiane S. Duarte, PhD, MPH, of Columbia University, New York, and her colleagues looked at data from the 2012/2013 National Epidemiologic Survey on Alcohol and Related Conditions (NESARC), a nationally representative sample of U.S. households.
“We were trying to look specifically at young adulthood, which there’s emerging consensus to regard as a key developmental period,” said Dr. Duarte, whose research focuses on anxiety disorders in young adults. “It’s a period where several psychiatric disorders tend to become much more prevalent. Having untreated anxiety disorders at this age can put young adults at risk for worse outcomes down the line. If anxiety disorders can be resolved, a young adult’s trajectory can be quite different; it’s a time in life in which the right intervention can have a really big impact,” she said.
The NESARC III survey data used structured diagnostic interviews and DSM-5 criteria to assess anxiety disorders occurring in the past year. These included specific phobia, generalized anxiety disorder, social anxiety, panic disorder, and agoraphobia.
For the most part, Dr. Duarte said, her group’s findings on anxiety disorders reflected earlier prevalence studies that had used DSM-IV criteria. Women were more likely than were men to report any past-year anxiety disorder (odds ratio, 2.26; 95% confidence interval, 1.80-2.84), as were people with lower personal and family incomes. Rates of anxiety disorders were highest in groups with the lowest personal and family incomes, and among people neither employed nor in an educational program.
Dr. Duarte said in an interview that the latter findings were generally anticipated. However, the finding that African Americans and Hispanics at this age had lower risk relative to whites (OR, 0.52; 95% CI, 0.40-0.67) and (OR, 0.63; 95% CI, 0.49-0.83) was interesting, because it appeared to mirror the lower relative prevalence seen among adults in those two groups, rather than the higher prevalence seen among children in the same groups. More research will be needed, she said, to verify and, if correct, understand this reversing trend in prevalence seen between childhood and adulthood.
The study’s most unexpected finding, Dr. Duarte said, was that married individuals aged 18-24 had higher prevalence of anxiety (OR, 1.54; 95% CI, 1.05-2.26). “Across the board, marriage is protective for many health and mental health conditions,” Dr. Duarte said, but she acknowledged that many factors could be in play. Marriage might not, in fact, be protective in this age group; the institution might be reflective of cultural factors promoting early marriage; or the findings could reflect a selection into marriage possibly related to existing anxiety disorders.
“To better understand this finding, we will need to consider several complexities which are part of young adulthood as a unique developmental period,” she said.
Dr. Duarte’s and her colleagues’ study was funded by the Youth Anxiety Center at New York–Presbyterian Hospital. Three coauthors reported research support from pharmaceutical manufacturers and royalties from commercial publishers.
FROM AACAP 2016
Key clinical point:
Major finding: African Americans and Hispanics who are young adults have a lower risk relative to their white peers (OR, 0.52; 95% confidence interval, 0.40-.067) and (OR, 0.63; 95% CI, 0.49-0.83). In addition, married individuals aged 18-24 had higher prevalence of anxiety (OR, 1.54; 95% CI, 1.05-2.26) than did their unmarried peers.
Data source: Data from the National Epidemiologic Survey on Alcohol and Related Conditions, a nationally representative sample of U.S. households.
Disclosures: The Youth Anxiety Center at New York–Presbyterian Hospital funded the study. Three coauthors reported research support from pharmaceutical manufacturers and royalties from commercial publishers.
Study identifies SSI risk factors after open LEB
COLUMBUS, OHIO – A study of vascular procedures at 35 Michigan hospitals has identified three risk factors for surgical site infection after lower-extremity bypass that hospitals and vascular surgery teams may be able to modify.
“Patients who had iodine-only skin antiseptic preparation, a high-peak intraoperative glucose, or long operative times were more likely to have substantially increased risk for surgical site infection (SSI),” Frank Davis, MD, of the University of Michigan said in reporting the study results at the annual meeting of the Midwestern Vascular Surgical Society. Those risk factors are modifiable, Dr. Davis said.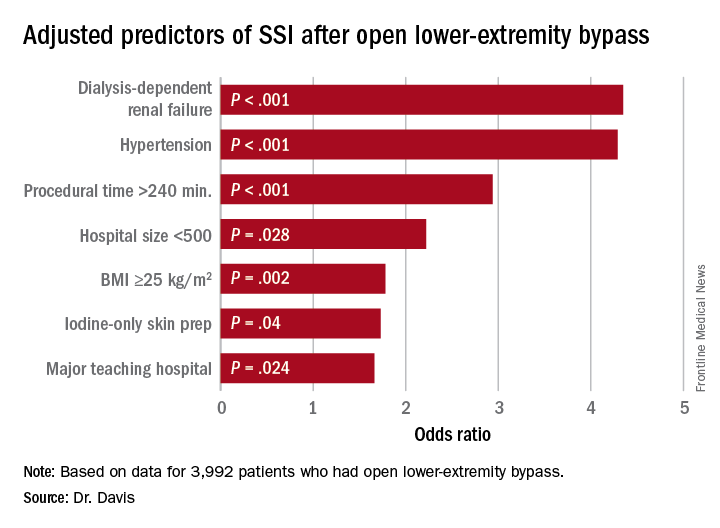
“Specific attention needs to be served moving forward in attempts to decrease the risk of SSI for lower-extremity bypass,” Dr. Davis said. “The incidence of SSI in our cohort across the state of Michigan was approximately 9.2%, and for those who did develop a SSI, there was a substantial increase in 30-day morbidity.”
Patients who had an SSI were more than three times more likely to have a major amputation (9% vs. 2.3%) than those without, and more than five times more likely to have a reoperation (3.9% vs. 0.7%), Dr. Davis said.
“With regard to preoperative symptomatology, those with lower peripheral artery questionnaire scores, resting pain, or acute ischemia were more likely to develop SSI postoperatively,” Dr. Davis said. “Patients who underwent an interim coronal bypass had a significant increase of SSI in comparison to all other bypass configurations.”
He also noted that major teaching hospitals or hospitals with 500 or fewer beds had higher rates of SSI.
“Targeted improvements in preoperative care may decrease complications and improve vascular patient outcomes,” Dr. Davis said.
Dr. Davis had no relationships to disclose.
COLUMBUS, OHIO – A study of vascular procedures at 35 Michigan hospitals has identified three risk factors for surgical site infection after lower-extremity bypass that hospitals and vascular surgery teams may be able to modify.
“Patients who had iodine-only skin antiseptic preparation, a high-peak intraoperative glucose, or long operative times were more likely to have substantially increased risk for surgical site infection (SSI),” Frank Davis, MD, of the University of Michigan said in reporting the study results at the annual meeting of the Midwestern Vascular Surgical Society. Those risk factors are modifiable, Dr. Davis said.
“Specific attention needs to be served moving forward in attempts to decrease the risk of SSI for lower-extremity bypass,” Dr. Davis said. “The incidence of SSI in our cohort across the state of Michigan was approximately 9.2%, and for those who did develop a SSI, there was a substantial increase in 30-day morbidity.”
Patients who had an SSI were more than three times more likely to have a major amputation (9% vs. 2.3%) than those without, and more than five times more likely to have a reoperation (3.9% vs. 0.7%), Dr. Davis said.
“With regard to preoperative symptomatology, those with lower peripheral artery questionnaire scores, resting pain, or acute ischemia were more likely to develop SSI postoperatively,” Dr. Davis said. “Patients who underwent an interim coronal bypass had a significant increase of SSI in comparison to all other bypass configurations.”
He also noted that major teaching hospitals or hospitals with 500 or fewer beds had higher rates of SSI.
“Targeted improvements in preoperative care may decrease complications and improve vascular patient outcomes,” Dr. Davis said.
Dr. Davis had no relationships to disclose.
COLUMBUS, OHIO – A study of vascular procedures at 35 Michigan hospitals has identified three risk factors for surgical site infection after lower-extremity bypass that hospitals and vascular surgery teams may be able to modify.
“Patients who had iodine-only skin antiseptic preparation, a high-peak intraoperative glucose, or long operative times were more likely to have substantially increased risk for surgical site infection (SSI),” Frank Davis, MD, of the University of Michigan said in reporting the study results at the annual meeting of the Midwestern Vascular Surgical Society. Those risk factors are modifiable, Dr. Davis said.
“Specific attention needs to be served moving forward in attempts to decrease the risk of SSI for lower-extremity bypass,” Dr. Davis said. “The incidence of SSI in our cohort across the state of Michigan was approximately 9.2%, and for those who did develop a SSI, there was a substantial increase in 30-day morbidity.”
Patients who had an SSI were more than three times more likely to have a major amputation (9% vs. 2.3%) than those without, and more than five times more likely to have a reoperation (3.9% vs. 0.7%), Dr. Davis said.
“With regard to preoperative symptomatology, those with lower peripheral artery questionnaire scores, resting pain, or acute ischemia were more likely to develop SSI postoperatively,” Dr. Davis said. “Patients who underwent an interim coronal bypass had a significant increase of SSI in comparison to all other bypass configurations.”
He also noted that major teaching hospitals or hospitals with 500 or fewer beds had higher rates of SSI.
“Targeted improvements in preoperative care may decrease complications and improve vascular patient outcomes,” Dr. Davis said.
Dr. Davis had no relationships to disclose.
AT MIDWESTERN VASCULAR 2016
Key clinical point: Study identified three key modifiable risk factors in surgical site infection (SSI) open after lower-extremity bypass (LEB).
Major finding: Incidence of SSI was 9.2% in the study cohort.
Data source: Blue Cross Blue Shield Michigan Vascular Intervention Collaborative database of 3,992 open LEB operations at 35 centers from January 2012 to June 2015.
Disclosures: Dr. Davis reported having no financial disclosures.
Syphilis testing before and after stillbirth is suboptimal
ATLANTA – Physicians are falling short on syphilis testing in both the prenatal period and at the time of delivery, suggest the findings of a study examining insurance claims from nearly 10,000 women who experienced stillbirths.
Overall, less than 10% of women in the study were tested for syphilis following a stillbirth delivery, while less than two-thirds of women who experienced a stillbirth had received prenatal syphilis testing.
Dr. Patel and his coinvestigators examined data from the Truven Health MarketScan Medicaid and commercial claims database to evaluate the proportion of women who had syphilis testing within at least 1 week before and 1 week after a stillbirth delivery.
The investigators identified women aged 15-44 years who had a stillbirth delivery in 2013. Stillbirths were identified via ICD-9 codes and these codes were also used to track prenatal syphilis testing, as well as syphilis testing, placental examination and complete blood count (CBC) performed at the time of delivery.
In total, there were 3,731 women enrolled in Medicaid and 6,096 commercially-insured women who experienced stillbirths and were included in the study. Of these women, 65.5% of Medicaid-covered women and 56.6% of commercially-insured women received prenatal syphilis testing. At delivery, 6.5% of Medicaid-insured women and 9.3% of commercially-insured women received syphilis testing.
Most women in the study were receiving prenatal care. In all, 73.2% of Medicaid-covered women and 76.5% of commercially-insured women received it. Placental examination at the time of delivery occurred for 61.5% of Medicaid-covered women and 58.0% of commercially-insured women, while CBC was performed in 31.2% and 35.8% of women, respectively.
“Overall, prenatal syphilis testing was significantly higher than syphilis testing at the time of delivery,” Dr. Patel said. “Women with prenatal syphilis testing were more likely to be tested for syphilis at delivery than those not tested, regardless of [their] insurance.”
Dr. Patel did not report information on financial disclosures.
ATLANTA – Physicians are falling short on syphilis testing in both the prenatal period and at the time of delivery, suggest the findings of a study examining insurance claims from nearly 10,000 women who experienced stillbirths.
Overall, less than 10% of women in the study were tested for syphilis following a stillbirth delivery, while less than two-thirds of women who experienced a stillbirth had received prenatal syphilis testing.
Dr. Patel and his coinvestigators examined data from the Truven Health MarketScan Medicaid and commercial claims database to evaluate the proportion of women who had syphilis testing within at least 1 week before and 1 week after a stillbirth delivery.
The investigators identified women aged 15-44 years who had a stillbirth delivery in 2013. Stillbirths were identified via ICD-9 codes and these codes were also used to track prenatal syphilis testing, as well as syphilis testing, placental examination and complete blood count (CBC) performed at the time of delivery.
In total, there were 3,731 women enrolled in Medicaid and 6,096 commercially-insured women who experienced stillbirths and were included in the study. Of these women, 65.5% of Medicaid-covered women and 56.6% of commercially-insured women received prenatal syphilis testing. At delivery, 6.5% of Medicaid-insured women and 9.3% of commercially-insured women received syphilis testing.
Most women in the study were receiving prenatal care. In all, 73.2% of Medicaid-covered women and 76.5% of commercially-insured women received it. Placental examination at the time of delivery occurred for 61.5% of Medicaid-covered women and 58.0% of commercially-insured women, while CBC was performed in 31.2% and 35.8% of women, respectively.
“Overall, prenatal syphilis testing was significantly higher than syphilis testing at the time of delivery,” Dr. Patel said. “Women with prenatal syphilis testing were more likely to be tested for syphilis at delivery than those not tested, regardless of [their] insurance.”
Dr. Patel did not report information on financial disclosures.
ATLANTA – Physicians are falling short on syphilis testing in both the prenatal period and at the time of delivery, suggest the findings of a study examining insurance claims from nearly 10,000 women who experienced stillbirths.
Overall, less than 10% of women in the study were tested for syphilis following a stillbirth delivery, while less than two-thirds of women who experienced a stillbirth had received prenatal syphilis testing.
Dr. Patel and his coinvestigators examined data from the Truven Health MarketScan Medicaid and commercial claims database to evaluate the proportion of women who had syphilis testing within at least 1 week before and 1 week after a stillbirth delivery.
The investigators identified women aged 15-44 years who had a stillbirth delivery in 2013. Stillbirths were identified via ICD-9 codes and these codes were also used to track prenatal syphilis testing, as well as syphilis testing, placental examination and complete blood count (CBC) performed at the time of delivery.
In total, there were 3,731 women enrolled in Medicaid and 6,096 commercially-insured women who experienced stillbirths and were included in the study. Of these women, 65.5% of Medicaid-covered women and 56.6% of commercially-insured women received prenatal syphilis testing. At delivery, 6.5% of Medicaid-insured women and 9.3% of commercially-insured women received syphilis testing.
Most women in the study were receiving prenatal care. In all, 73.2% of Medicaid-covered women and 76.5% of commercially-insured women received it. Placental examination at the time of delivery occurred for 61.5% of Medicaid-covered women and 58.0% of commercially-insured women, while CBC was performed in 31.2% and 35.8% of women, respectively.
“Overall, prenatal syphilis testing was significantly higher than syphilis testing at the time of delivery,” Dr. Patel said. “Women with prenatal syphilis testing were more likely to be tested for syphilis at delivery than those not tested, regardless of [their] insurance.”
Dr. Patel did not report information on financial disclosures.
AT THE 2016 STD PREVENTION CONFERENCE
Key clinical point:
Major finding: A total of 65.5% of Medicaid-covered women and 56.6% of commercially-insured women received prenatal syphilis testing. At delivery, 6.5% of Medicaid-covered women and 9.3% of commercially-insured women received syphilis testing.
Data source: Review of claims data from 3,731 women enrolled in Medicaid and 6,096 commercially-insured women who had stillbirth deliveries in 2013.
Disclosures: Dr. Patel did not report information on financial disclosures.
Close monitoring of psoriasis patients can delay PsA onset
NEWPORT BEACH, CALIF. – A patient with psoriasis can develop crippling psoriatic arthritis (PsA) within 5 to 10 years of diagnosis, but monitoring patients for signs of trouble can help prevent the onset of PsA, according to Alan Menter, MD.
Even a simple foot examination can make a huge difference, noted Dr. Menter, chief of the division of dermatology and director of the Psoriasis Research Institute at Baylor University Medical Center, Dallas. “At every visit, you and I should be looking for early signs of joint disease,” he said at the Skin Disease Education Foundation’s Women’s & Pediatric Dermatology Seminar. “We should not let these patients develop any joint disease because we have drugs that can prevent joint destruction.”

Dr. Menter pointed out that PsA is a disease that is distinct from psoriasis. “It’s linked to psoriasis, but genetically, there are differences,” he said, “and immunologically, what goes on in skin is not identical.”
He provided the following pearls regarding diagnosing PsA:
• Be on the lookout for “sausage fingers” and “sausage toes,” both signs of PsA. “You and I are very visual people, and we can see a swollen toe or finger very easily,” Dr. Menter said. “I take the shoes off every psoriasis patient at every visit and run my thumb and index finger down the Achilles. I look for a swollen Achilles – classic enthesitis.” In some cases, swollen big toes in psoriasis patients may be misdiagnosed as gout instead of PsA, he noted.
• Ask patients about how their joints feel when they wake up in the morning: Do they have swelling and tenderness? “That’s an early marker of psoriatic arthritis disease,” Dr. Menter said. In contrast, in a patient with osteoarthritis, “the more they use their joints, the worse it gets.”
• The severity of psoriasis has nothing to do with the severity of PsA. “You can have 50% of the body covered with psoriasis but no arthritis,” he said. “Or you can have someone with one patch of psoriasis on the scalp with devastating joint disease.”
• Be aware that there are five PsA subtypes that can occur in combination with each other:
1. Dactylitis. This is the form that causes the “sausage digit.”
2. Asymmetric oligoarthritis. This is the type most commonly seen on presentation, when there are few joints affected.
3. Symmetric arthritis. This form is more common in females and difficult to differentiate from rheumatoid arthritis.
4. Distal interphalangeal joint arthritis. This type is often linked to dactylitis and nail dystrophy.
5. Arthritis mutilans. This is more common in females, linked to long disease duration, and present in an estimated 5% of cases.
Dr. Menter suggested that dermatologists refer suspected cases of PsA to a rheumatologist. Since patients may have to wait 6-10 weeks for an appointment, he recommended that dermatologists consider NSAIDs, such as the over-the-counter naproxen and prescription meloxicam and celecoxib in the meantime. Dermatologists may also consider bringing up the use of methotrexate and biologics, he said.
Dr. Menter disclosed relationships with multiple pharmaceutical companies, including AbbVie, Allergan, Amgen, Boehringer Ingelheim, Eli Lilly, Merck, Novartis, and Pfizer.
SDEF and this news organization are owned by Frontline Medical Communications.
NEWPORT BEACH, CALIF. – A patient with psoriasis can develop crippling psoriatic arthritis (PsA) within 5 to 10 years of diagnosis, but monitoring patients for signs of trouble can help prevent the onset of PsA, according to Alan Menter, MD.
Even a simple foot examination can make a huge difference, noted Dr. Menter, chief of the division of dermatology and director of the Psoriasis Research Institute at Baylor University Medical Center, Dallas. “At every visit, you and I should be looking for early signs of joint disease,” he said at the Skin Disease Education Foundation’s Women’s & Pediatric Dermatology Seminar. “We should not let these patients develop any joint disease because we have drugs that can prevent joint destruction.”
Dr. Menter pointed out that PsA is a disease that is distinct from psoriasis. “It’s linked to psoriasis, but genetically, there are differences,” he said, “and immunologically, what goes on in skin is not identical.”
He provided the following pearls regarding diagnosing PsA:
• Be on the lookout for “sausage fingers” and “sausage toes,” both signs of PsA. “You and I are very visual people, and we can see a swollen toe or finger very easily,” Dr. Menter said. “I take the shoes off every psoriasis patient at every visit and run my thumb and index finger down the Achilles. I look for a swollen Achilles – classic enthesitis.” In some cases, swollen big toes in psoriasis patients may be misdiagnosed as gout instead of PsA, he noted.
• Ask patients about how their joints feel when they wake up in the morning: Do they have swelling and tenderness? “That’s an early marker of psoriatic arthritis disease,” Dr. Menter said. In contrast, in a patient with osteoarthritis, “the more they use their joints, the worse it gets.”
• The severity of psoriasis has nothing to do with the severity of PsA. “You can have 50% of the body covered with psoriasis but no arthritis,” he said. “Or you can have someone with one patch of psoriasis on the scalp with devastating joint disease.”
• Be aware that there are five PsA subtypes that can occur in combination with each other:
1. Dactylitis. This is the form that causes the “sausage digit.”
2. Asymmetric oligoarthritis. This is the type most commonly seen on presentation, when there are few joints affected.
3. Symmetric arthritis. This form is more common in females and difficult to differentiate from rheumatoid arthritis.
4. Distal interphalangeal joint arthritis. This type is often linked to dactylitis and nail dystrophy.
5. Arthritis mutilans. This is more common in females, linked to long disease duration, and present in an estimated 5% of cases.
Dr. Menter suggested that dermatologists refer suspected cases of PsA to a rheumatologist. Since patients may have to wait 6-10 weeks for an appointment, he recommended that dermatologists consider NSAIDs, such as the over-the-counter naproxen and prescription meloxicam and celecoxib in the meantime. Dermatologists may also consider bringing up the use of methotrexate and biologics, he said.
Dr. Menter disclosed relationships with multiple pharmaceutical companies, including AbbVie, Allergan, Amgen, Boehringer Ingelheim, Eli Lilly, Merck, Novartis, and Pfizer.
SDEF and this news organization are owned by Frontline Medical Communications.
NEWPORT BEACH, CALIF. – A patient with psoriasis can develop crippling psoriatic arthritis (PsA) within 5 to 10 years of diagnosis, but monitoring patients for signs of trouble can help prevent the onset of PsA, according to Alan Menter, MD.
Even a simple foot examination can make a huge difference, noted Dr. Menter, chief of the division of dermatology and director of the Psoriasis Research Institute at Baylor University Medical Center, Dallas. “At every visit, you and I should be looking for early signs of joint disease,” he said at the Skin Disease Education Foundation’s Women’s & Pediatric Dermatology Seminar. “We should not let these patients develop any joint disease because we have drugs that can prevent joint destruction.”
Dr. Menter pointed out that PsA is a disease that is distinct from psoriasis. “It’s linked to psoriasis, but genetically, there are differences,” he said, “and immunologically, what goes on in skin is not identical.”
He provided the following pearls regarding diagnosing PsA:
• Be on the lookout for “sausage fingers” and “sausage toes,” both signs of PsA. “You and I are very visual people, and we can see a swollen toe or finger very easily,” Dr. Menter said. “I take the shoes off every psoriasis patient at every visit and run my thumb and index finger down the Achilles. I look for a swollen Achilles – classic enthesitis.” In some cases, swollen big toes in psoriasis patients may be misdiagnosed as gout instead of PsA, he noted.
• Ask patients about how their joints feel when they wake up in the morning: Do they have swelling and tenderness? “That’s an early marker of psoriatic arthritis disease,” Dr. Menter said. In contrast, in a patient with osteoarthritis, “the more they use their joints, the worse it gets.”
• The severity of psoriasis has nothing to do with the severity of PsA. “You can have 50% of the body covered with psoriasis but no arthritis,” he said. “Or you can have someone with one patch of psoriasis on the scalp with devastating joint disease.”
• Be aware that there are five PsA subtypes that can occur in combination with each other:
1. Dactylitis. This is the form that causes the “sausage digit.”
2. Asymmetric oligoarthritis. This is the type most commonly seen on presentation, when there are few joints affected.
3. Symmetric arthritis. This form is more common in females and difficult to differentiate from rheumatoid arthritis.
4. Distal interphalangeal joint arthritis. This type is often linked to dactylitis and nail dystrophy.
5. Arthritis mutilans. This is more common in females, linked to long disease duration, and present in an estimated 5% of cases.
Dr. Menter suggested that dermatologists refer suspected cases of PsA to a rheumatologist. Since patients may have to wait 6-10 weeks for an appointment, he recommended that dermatologists consider NSAIDs, such as the over-the-counter naproxen and prescription meloxicam and celecoxib in the meantime. Dermatologists may also consider bringing up the use of methotrexate and biologics, he said.
Dr. Menter disclosed relationships with multiple pharmaceutical companies, including AbbVie, Allergan, Amgen, Boehringer Ingelheim, Eli Lilly, Merck, Novartis, and Pfizer.
SDEF and this news organization are owned by Frontline Medical Communications.
EXPERT ANALYSIS FROM SDEF WOMEN'S & PEDIATRIC DERMATOLOGY SEMINAR
Follow-Up of Infants With Zika Virus Identifies Several Neurologic Impairments
A report on 11 infants in Brazil suggests the term “congenital Zika syndrome” be used to describe the abnormalities associated with Zika virus infection because microcephaly is only one clinical sign of this congenital malformation disorder. The report was published online ahead of print October 3 in JAMA Neurology.
“To our knowledge,” the researchers wrote, “most reports to date have focused on select aspects of the maternal or fetal infection and fetal effects.” To provide a fuller description, the researchers sought to characterize the prenatal evolution and perinatal outcomes of 11 neonates who had developmental abnormalities and neurologic damage associated with Zika infection.
Follow-Up of 11 Neonates
Amilcar Tanuri, MD, PhD, Professor of Genetics and Chief of the Laboratory of Molecular Virology at the Institute of Biology, Federal University of Rio de Janeiro, and coauthors observed 11 infants with congenital Zika infection from gestation to six months in the state of Paraíba, Brazil. Cases were referred between October 2015 and February 2016. Ten of 11 women included in the study presented with symptoms of Zika infection during the first half of pregnancy, and all 11 had laboratory evidence of infection in several tissues by serology or polymerase chain reaction. Brain damage was confirmed through intrauterine ultrasonography and was complemented by MRI. Histopathologic analysis was performed on the placenta and brain tissue from infants who died. The ZIKV genome was investigated in several tissues and sequenced for further phylogenetic analysis.
Of the 11 infants, seven (63.6%) were female, and the median maternal age at delivery was 25. Three of the neonates died, giving a perinatal mortality rate of 27.3%. Zika virus was identified in amniotic fluid, placenta, cord blood, and neonatal tissues collected post mortem in the three babies who died within 48 hours of delivery.
Brain damage and neurologic impairments were identified in all patients, including microcephaly, a reduction in cerebral volume, ventriculomegaly, cerebellar hypoplasia, lissencephaly with hydrocephalus, and fetal akinesia deformation sequence. Testing for other causes of microcephaly, such as genetic disorders and infections, was negative. The ZIKV virus genome was found in tissues of the mothers and their babies.
“Combined findings from clinical, laboratory, imaging, and pathologic examinations provided a more complete picture of the severe damage and developmental abnormalities caused by ZIKV infection than has been previously reported,” Dr. Tanuri and colleagues said.
Formulating a Plan of Action
“Although we have limited ways to stop emerging pathogens, we now have powerful techniques to quickly identify the culprit, such as polymerase chain reaction and whole genome sequencing,” said Raymond P. Roos, MD, Marjorie and Robert E. Straus Professor in Neurologic Science in the Department of Neurology at the University of Chicago, in an accompanying editorial. “We also have novel methods to control vectors and produce vaccines in an accelerated time frame.”
But many unanswered questions remain, said Dr. Roos. Among those questions is what neurologists can do about the Zika virus. “It would be valuable to have adult and pediatric neurologists network with the US Centers for Disease Control and Prevention to establish a surveillance system that could track Zika virus-induced Guillain-Barré syndrome (GBS) and CNS disease. This [cooperation] would facilitate the identification and characterization of disorders, the formation of a registry, and the mounting of comprehensive epidemiologic studies. This approach would also help to identify long-term sequelae of intrauterine infection and clarify effective treatments of the GBS syndrome.”
—Glenn S. Williams
Suggested Reading
Melo AS, Aguiar RS, Amorim MM, et al. Congenital Zika virus infection: beyond neonatal microcephaly. JAMA Neurol. 2016 Oct 3 [Epub ahead of print].
Roos RP. Zika virus-a public health emergency of international concern. JAMA Neurol. 2016 Oct 3 [Epub ahead of print].
A report on 11 infants in Brazil suggests the term “congenital Zika syndrome” be used to describe the abnormalities associated with Zika virus infection because microcephaly is only one clinical sign of this congenital malformation disorder. The report was published online ahead of print October 3 in JAMA Neurology.
“To our knowledge,” the researchers wrote, “most reports to date have focused on select aspects of the maternal or fetal infection and fetal effects.” To provide a fuller description, the researchers sought to characterize the prenatal evolution and perinatal outcomes of 11 neonates who had developmental abnormalities and neurologic damage associated with Zika infection.
Follow-Up of 11 Neonates
Amilcar Tanuri, MD, PhD, Professor of Genetics and Chief of the Laboratory of Molecular Virology at the Institute of Biology, Federal University of Rio de Janeiro, and coauthors observed 11 infants with congenital Zika infection from gestation to six months in the state of Paraíba, Brazil. Cases were referred between October 2015 and February 2016. Ten of 11 women included in the study presented with symptoms of Zika infection during the first half of pregnancy, and all 11 had laboratory evidence of infection in several tissues by serology or polymerase chain reaction. Brain damage was confirmed through intrauterine ultrasonography and was complemented by MRI. Histopathologic analysis was performed on the placenta and brain tissue from infants who died. The ZIKV genome was investigated in several tissues and sequenced for further phylogenetic analysis.
Of the 11 infants, seven (63.6%) were female, and the median maternal age at delivery was 25. Three of the neonates died, giving a perinatal mortality rate of 27.3%. Zika virus was identified in amniotic fluid, placenta, cord blood, and neonatal tissues collected post mortem in the three babies who died within 48 hours of delivery.
Brain damage and neurologic impairments were identified in all patients, including microcephaly, a reduction in cerebral volume, ventriculomegaly, cerebellar hypoplasia, lissencephaly with hydrocephalus, and fetal akinesia deformation sequence. Testing for other causes of microcephaly, such as genetic disorders and infections, was negative. The ZIKV virus genome was found in tissues of the mothers and their babies.
“Combined findings from clinical, laboratory, imaging, and pathologic examinations provided a more complete picture of the severe damage and developmental abnormalities caused by ZIKV infection than has been previously reported,” Dr. Tanuri and colleagues said.
Formulating a Plan of Action
“Although we have limited ways to stop emerging pathogens, we now have powerful techniques to quickly identify the culprit, such as polymerase chain reaction and whole genome sequencing,” said Raymond P. Roos, MD, Marjorie and Robert E. Straus Professor in Neurologic Science in the Department of Neurology at the University of Chicago, in an accompanying editorial. “We also have novel methods to control vectors and produce vaccines in an accelerated time frame.”
But many unanswered questions remain, said Dr. Roos. Among those questions is what neurologists can do about the Zika virus. “It would be valuable to have adult and pediatric neurologists network with the US Centers for Disease Control and Prevention to establish a surveillance system that could track Zika virus-induced Guillain-Barré syndrome (GBS) and CNS disease. This [cooperation] would facilitate the identification and characterization of disorders, the formation of a registry, and the mounting of comprehensive epidemiologic studies. This approach would also help to identify long-term sequelae of intrauterine infection and clarify effective treatments of the GBS syndrome.”
—Glenn S. Williams
Suggested Reading
Melo AS, Aguiar RS, Amorim MM, et al. Congenital Zika virus infection: beyond neonatal microcephaly. JAMA Neurol. 2016 Oct 3 [Epub ahead of print].
Roos RP. Zika virus-a public health emergency of international concern. JAMA Neurol. 2016 Oct 3 [Epub ahead of print].
A report on 11 infants in Brazil suggests the term “congenital Zika syndrome” be used to describe the abnormalities associated with Zika virus infection because microcephaly is only one clinical sign of this congenital malformation disorder. The report was published online ahead of print October 3 in JAMA Neurology.
“To our knowledge,” the researchers wrote, “most reports to date have focused on select aspects of the maternal or fetal infection and fetal effects.” To provide a fuller description, the researchers sought to characterize the prenatal evolution and perinatal outcomes of 11 neonates who had developmental abnormalities and neurologic damage associated with Zika infection.
Follow-Up of 11 Neonates
Amilcar Tanuri, MD, PhD, Professor of Genetics and Chief of the Laboratory of Molecular Virology at the Institute of Biology, Federal University of Rio de Janeiro, and coauthors observed 11 infants with congenital Zika infection from gestation to six months in the state of Paraíba, Brazil. Cases were referred between October 2015 and February 2016. Ten of 11 women included in the study presented with symptoms of Zika infection during the first half of pregnancy, and all 11 had laboratory evidence of infection in several tissues by serology or polymerase chain reaction. Brain damage was confirmed through intrauterine ultrasonography and was complemented by MRI. Histopathologic analysis was performed on the placenta and brain tissue from infants who died. The ZIKV genome was investigated in several tissues and sequenced for further phylogenetic analysis.
Of the 11 infants, seven (63.6%) were female, and the median maternal age at delivery was 25. Three of the neonates died, giving a perinatal mortality rate of 27.3%. Zika virus was identified in amniotic fluid, placenta, cord blood, and neonatal tissues collected post mortem in the three babies who died within 48 hours of delivery.
Brain damage and neurologic impairments were identified in all patients, including microcephaly, a reduction in cerebral volume, ventriculomegaly, cerebellar hypoplasia, lissencephaly with hydrocephalus, and fetal akinesia deformation sequence. Testing for other causes of microcephaly, such as genetic disorders and infections, was negative. The ZIKV virus genome was found in tissues of the mothers and their babies.
“Combined findings from clinical, laboratory, imaging, and pathologic examinations provided a more complete picture of the severe damage and developmental abnormalities caused by ZIKV infection than has been previously reported,” Dr. Tanuri and colleagues said.
Formulating a Plan of Action
“Although we have limited ways to stop emerging pathogens, we now have powerful techniques to quickly identify the culprit, such as polymerase chain reaction and whole genome sequencing,” said Raymond P. Roos, MD, Marjorie and Robert E. Straus Professor in Neurologic Science in the Department of Neurology at the University of Chicago, in an accompanying editorial. “We also have novel methods to control vectors and produce vaccines in an accelerated time frame.”
But many unanswered questions remain, said Dr. Roos. Among those questions is what neurologists can do about the Zika virus. “It would be valuable to have adult and pediatric neurologists network with the US Centers for Disease Control and Prevention to establish a surveillance system that could track Zika virus-induced Guillain-Barré syndrome (GBS) and CNS disease. This [cooperation] would facilitate the identification and characterization of disorders, the formation of a registry, and the mounting of comprehensive epidemiologic studies. This approach would also help to identify long-term sequelae of intrauterine infection and clarify effective treatments of the GBS syndrome.”
—Glenn S. Williams
Suggested Reading
Melo AS, Aguiar RS, Amorim MM, et al. Congenital Zika virus infection: beyond neonatal microcephaly. JAMA Neurol. 2016 Oct 3 [Epub ahead of print].
Roos RP. Zika virus-a public health emergency of international concern. JAMA Neurol. 2016 Oct 3 [Epub ahead of print].
‘Excellent’ real-world experience with LAA closure device
A device that closes the left atrial appendage to prevent stroke in patients with nonvalvular atrial fibrillation showed a 95.6% procedural success rate in a study of real-world experience since it was approved by the FDA in 2015, according to a report presented at the Transcatheter Cardiovascular Therapeutics annual meeting and published simultaneously in the Journal of the American College of Cardiology.
The study was based on data collected by Boston Scientific, the manufacturer of the Watchman device, regarding 3,822 consecutive patients who underwent the implantation during a 14-month period. The “excellent” procedural success rate, together with low short-term complication rates, are especially “remarkable” because 71% of the interventional cardiologists and electrophysiologists who performed these procedures had no experience with the device prior to FDA approval, said Vivek Y. Reddy, MD, of Mount Sinai Medical Center, New York.

For this study, the implantations were done by 382 physicians at 169 U.S. medical centers. A total of 3,653 procedures were successful. The median duration of the implantation was “an acceptable” 50 minutes (range, 10-210 minutes), and an average of 1.38 devices (range, 1-6) were required per patient. In 23% of cases, a “partial recapture” of a device was necessary to reposition it (J Am Coll Cardiol. 2016 Nov. doi: 10.1016/j.jacc.2016.10.010).
The rates of major complications within 1 week – pericardial tamponade (<1%), procedure-related stroke (0.08%), and mortality (0.08%) – were characterized as “favorable.”
The most common complication was pericardial effusion requiring intervention, which developed in 39 patients (1.02%). The effusions were drained percutaneously in most (24) of these patients. Another 11 patients (0.29%) developed mild pericardial effusions requiring only conservative management.
Three strokes, two ischemic and one hemorrhagic, were deemed related to the procedure, though the hemorrhagic bleed may have resulted chiefly from anticoagulation medications. Three deaths were judged to be related to the procedure: All were secondary to pericardial tamponade associated with perforation by the device.
“It is worth comparing [this] cardiac tamponade rate with [that of] another left atrial cardiovascular procedure, catheter ablation of atrial fibrillation,” Dr. Reddy noted.
A worldwide survey of more than 20,000 catheter ablations reported a pericardial tamponade rate of 1.31%, and another study of more than 93,000 ablation procedures performed during a 1-year period reported a rate of 1.52%, he said at the meeting, which was sponsored by the Cardiovascular Research Foundation.
Other short-term complications in this study included nine cases of device embolization (0.24%). Six of these required surgical removal of the device, while three were retrieved percutaneously.
No sponsor was cited for this study. Boston Scientific, maker of the Watchman left atrial appendage closure device, collected the data on all implantations of the device in the United States following FDA approval. Dr. Reddy and his associates reported ties to Boston Scientific, Coherex, SentreHeart, Abbott Vascular, and St. Jude Medical.
The results reported by Dr. Reddy and his colleagues are remarkably favorable for the earliest phase of widespread dissemination of this technology, especially in the context of the much higher rates of medication-related adverse events that occur with long-term oral anticoagulation. These findings should reassure us that left atrial appendage device closure has been safely introduced into clinical practice in the United States.
The study design, however, raises some concerns, and clinicians should be aware that complications may have been underreported. The manufacturer’s employees collected the data regarding complications in a somewhat informal manner, so this was not an objective study meeting the rigorous standards of clinical trials or postmarketing registries. These “clinical specialists” only included complications that developed within 1 week of the procedure, which fails to address possible delayed complications such as device-related thrombus. And they also didn’t track some important complications such as vascular events and bleeding events.
Jacqueline Saw, MD, of Vancouver General Hospital, and Matthew J. Price, MD, of Scripps Clinic in La Jolla, Calif., made these remarks in an editorial (J Am Coll Cardiol. 2016 Nov. doi: 10.1016/j.jacc.2016.10.019) accompanying Dr. Reddy’s report. Dr. Saw reported ties to Boston Scientific, AstraZeneca, Abbott Vascular, St. Jude Medical, Servier, Bayer, and Sunovion. Dr. Price reported ties to Boston Scientific, St. Jude Medical, W.L. Gore, Medtronic, AstraZeneca, Abbott Vascular, and Terumo.
The results reported by Dr. Reddy and his colleagues are remarkably favorable for the earliest phase of widespread dissemination of this technology, especially in the context of the much higher rates of medication-related adverse events that occur with long-term oral anticoagulation. These findings should reassure us that left atrial appendage device closure has been safely introduced into clinical practice in the United States.
The study design, however, raises some concerns, and clinicians should be aware that complications may have been underreported. The manufacturer’s employees collected the data regarding complications in a somewhat informal manner, so this was not an objective study meeting the rigorous standards of clinical trials or postmarketing registries. These “clinical specialists” only included complications that developed within 1 week of the procedure, which fails to address possible delayed complications such as device-related thrombus. And they also didn’t track some important complications such as vascular events and bleeding events.
Jacqueline Saw, MD, of Vancouver General Hospital, and Matthew J. Price, MD, of Scripps Clinic in La Jolla, Calif., made these remarks in an editorial (J Am Coll Cardiol. 2016 Nov. doi: 10.1016/j.jacc.2016.10.019) accompanying Dr. Reddy’s report. Dr. Saw reported ties to Boston Scientific, AstraZeneca, Abbott Vascular, St. Jude Medical, Servier, Bayer, and Sunovion. Dr. Price reported ties to Boston Scientific, St. Jude Medical, W.L. Gore, Medtronic, AstraZeneca, Abbott Vascular, and Terumo.
The results reported by Dr. Reddy and his colleagues are remarkably favorable for the earliest phase of widespread dissemination of this technology, especially in the context of the much higher rates of medication-related adverse events that occur with long-term oral anticoagulation. These findings should reassure us that left atrial appendage device closure has been safely introduced into clinical practice in the United States.
The study design, however, raises some concerns, and clinicians should be aware that complications may have been underreported. The manufacturer’s employees collected the data regarding complications in a somewhat informal manner, so this was not an objective study meeting the rigorous standards of clinical trials or postmarketing registries. These “clinical specialists” only included complications that developed within 1 week of the procedure, which fails to address possible delayed complications such as device-related thrombus. And they also didn’t track some important complications such as vascular events and bleeding events.
Jacqueline Saw, MD, of Vancouver General Hospital, and Matthew J. Price, MD, of Scripps Clinic in La Jolla, Calif., made these remarks in an editorial (J Am Coll Cardiol. 2016 Nov. doi: 10.1016/j.jacc.2016.10.019) accompanying Dr. Reddy’s report. Dr. Saw reported ties to Boston Scientific, AstraZeneca, Abbott Vascular, St. Jude Medical, Servier, Bayer, and Sunovion. Dr. Price reported ties to Boston Scientific, St. Jude Medical, W.L. Gore, Medtronic, AstraZeneca, Abbott Vascular, and Terumo.
A device that closes the left atrial appendage to prevent stroke in patients with nonvalvular atrial fibrillation showed a 95.6% procedural success rate in a study of real-world experience since it was approved by the FDA in 2015, according to a report presented at the Transcatheter Cardiovascular Therapeutics annual meeting and published simultaneously in the Journal of the American College of Cardiology.
The study was based on data collected by Boston Scientific, the manufacturer of the Watchman device, regarding 3,822 consecutive patients who underwent the implantation during a 14-month period. The “excellent” procedural success rate, together with low short-term complication rates, are especially “remarkable” because 71% of the interventional cardiologists and electrophysiologists who performed these procedures had no experience with the device prior to FDA approval, said Vivek Y. Reddy, MD, of Mount Sinai Medical Center, New York.
For this study, the implantations were done by 382 physicians at 169 U.S. medical centers. A total of 3,653 procedures were successful. The median duration of the implantation was “an acceptable” 50 minutes (range, 10-210 minutes), and an average of 1.38 devices (range, 1-6) were required per patient. In 23% of cases, a “partial recapture” of a device was necessary to reposition it (J Am Coll Cardiol. 2016 Nov. doi: 10.1016/j.jacc.2016.10.010).
The rates of major complications within 1 week – pericardial tamponade (<1%), procedure-related stroke (0.08%), and mortality (0.08%) – were characterized as “favorable.”
The most common complication was pericardial effusion requiring intervention, which developed in 39 patients (1.02%). The effusions were drained percutaneously in most (24) of these patients. Another 11 patients (0.29%) developed mild pericardial effusions requiring only conservative management.
Three strokes, two ischemic and one hemorrhagic, were deemed related to the procedure, though the hemorrhagic bleed may have resulted chiefly from anticoagulation medications. Three deaths were judged to be related to the procedure: All were secondary to pericardial tamponade associated with perforation by the device.
“It is worth comparing [this] cardiac tamponade rate with [that of] another left atrial cardiovascular procedure, catheter ablation of atrial fibrillation,” Dr. Reddy noted.
A worldwide survey of more than 20,000 catheter ablations reported a pericardial tamponade rate of 1.31%, and another study of more than 93,000 ablation procedures performed during a 1-year period reported a rate of 1.52%, he said at the meeting, which was sponsored by the Cardiovascular Research Foundation.
Other short-term complications in this study included nine cases of device embolization (0.24%). Six of these required surgical removal of the device, while three were retrieved percutaneously.
No sponsor was cited for this study. Boston Scientific, maker of the Watchman left atrial appendage closure device, collected the data on all implantations of the device in the United States following FDA approval. Dr. Reddy and his associates reported ties to Boston Scientific, Coherex, SentreHeart, Abbott Vascular, and St. Jude Medical.
A device that closes the left atrial appendage to prevent stroke in patients with nonvalvular atrial fibrillation showed a 95.6% procedural success rate in a study of real-world experience since it was approved by the FDA in 2015, according to a report presented at the Transcatheter Cardiovascular Therapeutics annual meeting and published simultaneously in the Journal of the American College of Cardiology.
The study was based on data collected by Boston Scientific, the manufacturer of the Watchman device, regarding 3,822 consecutive patients who underwent the implantation during a 14-month period. The “excellent” procedural success rate, together with low short-term complication rates, are especially “remarkable” because 71% of the interventional cardiologists and electrophysiologists who performed these procedures had no experience with the device prior to FDA approval, said Vivek Y. Reddy, MD, of Mount Sinai Medical Center, New York.
For this study, the implantations were done by 382 physicians at 169 U.S. medical centers. A total of 3,653 procedures were successful. The median duration of the implantation was “an acceptable” 50 minutes (range, 10-210 minutes), and an average of 1.38 devices (range, 1-6) were required per patient. In 23% of cases, a “partial recapture” of a device was necessary to reposition it (J Am Coll Cardiol. 2016 Nov. doi: 10.1016/j.jacc.2016.10.010).
The rates of major complications within 1 week – pericardial tamponade (<1%), procedure-related stroke (0.08%), and mortality (0.08%) – were characterized as “favorable.”
The most common complication was pericardial effusion requiring intervention, which developed in 39 patients (1.02%). The effusions were drained percutaneously in most (24) of these patients. Another 11 patients (0.29%) developed mild pericardial effusions requiring only conservative management.
Three strokes, two ischemic and one hemorrhagic, were deemed related to the procedure, though the hemorrhagic bleed may have resulted chiefly from anticoagulation medications. Three deaths were judged to be related to the procedure: All were secondary to pericardial tamponade associated with perforation by the device.
“It is worth comparing [this] cardiac tamponade rate with [that of] another left atrial cardiovascular procedure, catheter ablation of atrial fibrillation,” Dr. Reddy noted.
A worldwide survey of more than 20,000 catheter ablations reported a pericardial tamponade rate of 1.31%, and another study of more than 93,000 ablation procedures performed during a 1-year period reported a rate of 1.52%, he said at the meeting, which was sponsored by the Cardiovascular Research Foundation.
Other short-term complications in this study included nine cases of device embolization (0.24%). Six of these required surgical removal of the device, while three were retrieved percutaneously.
No sponsor was cited for this study. Boston Scientific, maker of the Watchman left atrial appendage closure device, collected the data on all implantations of the device in the United States following FDA approval. Dr. Reddy and his associates reported ties to Boston Scientific, Coherex, SentreHeart, Abbott Vascular, and St. Jude Medical.
FROM TCT 2016
Key clinical point: A left atrial appendage closure device to prevent stroke in patients with nonvalvular atrial fibrillation showed a 95.6% procedural success rate.
Major finding: Of 3,822 implantations, 3,653 (95.6%) were successful, and 1-week complication rates were low, with <1% pericardial tamponade, 0.08% procedure-related stroke, and 0.08% mortality.
Data source: An analysis of manufacturer-collected data on all 3,822 consecutive device implantations at 169 medical centers from March 2015 to May 2016.
Disclosures: No sponsor was cited for this study. Boston Scientific, maker of the Watchman left atrial appendage closure device, collected the data on all implantations of the device in the United States following FDA approval. Dr. Reddy and his associates reported ties to Boston Scientific, Coherex, SentreHeart, Abbott Vascular, and St. Jude Medical.