User login
Improving Hospital Telemetry Usage
Hospitalists often rely on inpatient telemetry monitoring to identify arrhythmias, ischemia, and QT prolongation, but research has shown that its inappropriate usage increases costs to the healthcare system. An abstract presented at the 2016 meeting of the Society of Hospital Medicine looked at one hospital’s telemetry usage and how it might be improved.

The study revolved around a progress note template the authors developed, which incorporated documentation for telemetry use indications and need for telemetry continuation on non-ICU internal medicine services. The authors also provided an educational session describing American College of Cardiology and American Heart Association (ACC/AHA) telemetry use guidelines for internal medicine residents with a pretest and posttest.
Application of ACA/AHA guidelines was assessed with five scenarios before and after instruction on the guidelines. On pretest, only 29% of trainees answered all five questions correctly; on posttest, 63% did. A comparison between charts of admitted patients with telemetry orders from 2015 with charts from 2013 indicated that the appropriate initiation of telemetry improved significantly as did telemetry documentation. Inappropriate continuation rates were cut in half.
The success of the study suggests further work.
“We plan expansion of telemetry utilization education to internal medicine faculty and nursing to encourage daily review of telemetry usage,” the authors write. “We are also working to develop telemetry orders that end during standard work hours to prevent inadvertent continuation by overnight providers.”
Reference
1. Kuehn C, Steyers CM III, Glenn K, Fang M. Resident-based telemetry utilization innovations lead to improved outcomes [abstract]. J Hosp Med. 2016;11(suppl 1). Accessed October 17, 2016.
Hospitalists often rely on inpatient telemetry monitoring to identify arrhythmias, ischemia, and QT prolongation, but research has shown that its inappropriate usage increases costs to the healthcare system. An abstract presented at the 2016 meeting of the Society of Hospital Medicine looked at one hospital’s telemetry usage and how it might be improved.
The study revolved around a progress note template the authors developed, which incorporated documentation for telemetry use indications and need for telemetry continuation on non-ICU internal medicine services. The authors also provided an educational session describing American College of Cardiology and American Heart Association (ACC/AHA) telemetry use guidelines for internal medicine residents with a pretest and posttest.
Application of ACA/AHA guidelines was assessed with five scenarios before and after instruction on the guidelines. On pretest, only 29% of trainees answered all five questions correctly; on posttest, 63% did. A comparison between charts of admitted patients with telemetry orders from 2015 with charts from 2013 indicated that the appropriate initiation of telemetry improved significantly as did telemetry documentation. Inappropriate continuation rates were cut in half.
The success of the study suggests further work.
“We plan expansion of telemetry utilization education to internal medicine faculty and nursing to encourage daily review of telemetry usage,” the authors write. “We are also working to develop telemetry orders that end during standard work hours to prevent inadvertent continuation by overnight providers.”
Reference
1. Kuehn C, Steyers CM III, Glenn K, Fang M. Resident-based telemetry utilization innovations lead to improved outcomes [abstract]. J Hosp Med. 2016;11(suppl 1). Accessed October 17, 2016.
Hospitalists often rely on inpatient telemetry monitoring to identify arrhythmias, ischemia, and QT prolongation, but research has shown that its inappropriate usage increases costs to the healthcare system. An abstract presented at the 2016 meeting of the Society of Hospital Medicine looked at one hospital’s telemetry usage and how it might be improved.
The study revolved around a progress note template the authors developed, which incorporated documentation for telemetry use indications and need for telemetry continuation on non-ICU internal medicine services. The authors also provided an educational session describing American College of Cardiology and American Heart Association (ACC/AHA) telemetry use guidelines for internal medicine residents with a pretest and posttest.
Application of ACA/AHA guidelines was assessed with five scenarios before and after instruction on the guidelines. On pretest, only 29% of trainees answered all five questions correctly; on posttest, 63% did. A comparison between charts of admitted patients with telemetry orders from 2015 with charts from 2013 indicated that the appropriate initiation of telemetry improved significantly as did telemetry documentation. Inappropriate continuation rates were cut in half.
The success of the study suggests further work.
“We plan expansion of telemetry utilization education to internal medicine faculty and nursing to encourage daily review of telemetry usage,” the authors write. “We are also working to develop telemetry orders that end during standard work hours to prevent inadvertent continuation by overnight providers.”
Reference
1. Kuehn C, Steyers CM III, Glenn K, Fang M. Resident-based telemetry utilization innovations lead to improved outcomes [abstract]. J Hosp Med. 2016;11(suppl 1). Accessed October 17, 2016.
Measuring Excellent Comportment among Hospitalists
The hospitalist’s performance is among the major determinants of a patient’s hospital experience. But what are the elements of a successful interaction? The authors of an article published in the Journal of Hospital Medicine set out to establish metrics to answer—and measure the answer—to that question, to assess hospitalists’ behaviors, and to establish norms and expectations.
“This study represents a first step to specifically characterize comportment and communication in hospital medicine,” the authors write.
Patient satisfaction surveys, they state, have some shortcomings in providing useful answers to that question.
“First, the attribution to specific providers is questionable,” the authors write. “Second, recall about the provider by the patients may be poor because surveys are sent to patients days after they return home. Third, the patients’ recovery and health outcomes are likely to influence their assessment of the doctor. Finally, feedback is known to be most valuable and transformative when it is specific and given in real time.”
Researchers asked the chiefs of hospital medicine divisions at five hospitals to identify their “most clinically excellent” hospitalists. Each hospitalist was observed during a routine clinical shift, and behaviors were recorded that were believed to be associated with excellent comportment and communication using the hospital medicine comportment and communication tool (HMCCOT), the final version of which has 23 variables. The physicians’ HMCCOT scores were associated with their patient satisfaction survey scores, suggesting that improved comportment might translate into enhanced patient satisfaction.
The results showed extensive variability in comportment and communication at the bedside. One variable that stood out to the researchers was that teach-back was employed in only 13% of the encounters.
“Previous studies have shown that teach-back corroborates patient comprehension and can be used to engage patients (and caregivers) in realistic goal setting and optimal health service utilization,” the researchers write. “Further, patients who clearly understand their post-discharge plan are 30% less likely to be readmitted or visit the emergency department. The data for our group have helped us to see areas of strengths, such as hand washing, where we are above compliance rates across hospitals in the United States, as well as those matters that represent opportunities for improvement such as connecting more deeply with our patients.”
The researchers call for future studies to determine whether hospitalists can improve feedback from this tool and whether enhancing comportment and communication can improve both patient satisfaction and clinical outcomes.
Reference
- Kotwal S, Khaliq W, Landis R, Wright S. Developing a comportment and communication tool for use in hospital medicine [published online ahead of print August 13, 2016]. J Hosp Med. doi:10.1002/jhm.2647.
The hospitalist’s performance is among the major determinants of a patient’s hospital experience. But what are the elements of a successful interaction? The authors of an article published in the Journal of Hospital Medicine set out to establish metrics to answer—and measure the answer—to that question, to assess hospitalists’ behaviors, and to establish norms and expectations.
“This study represents a first step to specifically characterize comportment and communication in hospital medicine,” the authors write.
Patient satisfaction surveys, they state, have some shortcomings in providing useful answers to that question.
“First, the attribution to specific providers is questionable,” the authors write. “Second, recall about the provider by the patients may be poor because surveys are sent to patients days after they return home. Third, the patients’ recovery and health outcomes are likely to influence their assessment of the doctor. Finally, feedback is known to be most valuable and transformative when it is specific and given in real time.”
Researchers asked the chiefs of hospital medicine divisions at five hospitals to identify their “most clinically excellent” hospitalists. Each hospitalist was observed during a routine clinical shift, and behaviors were recorded that were believed to be associated with excellent comportment and communication using the hospital medicine comportment and communication tool (HMCCOT), the final version of which has 23 variables. The physicians’ HMCCOT scores were associated with their patient satisfaction survey scores, suggesting that improved comportment might translate into enhanced patient satisfaction.
The results showed extensive variability in comportment and communication at the bedside. One variable that stood out to the researchers was that teach-back was employed in only 13% of the encounters.
“Previous studies have shown that teach-back corroborates patient comprehension and can be used to engage patients (and caregivers) in realistic goal setting and optimal health service utilization,” the researchers write. “Further, patients who clearly understand their post-discharge plan are 30% less likely to be readmitted or visit the emergency department. The data for our group have helped us to see areas of strengths, such as hand washing, where we are above compliance rates across hospitals in the United States, as well as those matters that represent opportunities for improvement such as connecting more deeply with our patients.”
The researchers call for future studies to determine whether hospitalists can improve feedback from this tool and whether enhancing comportment and communication can improve both patient satisfaction and clinical outcomes.
Reference
- Kotwal S, Khaliq W, Landis R, Wright S. Developing a comportment and communication tool for use in hospital medicine [published online ahead of print August 13, 2016]. J Hosp Med. doi:10.1002/jhm.2647.
The hospitalist’s performance is among the major determinants of a patient’s hospital experience. But what are the elements of a successful interaction? The authors of an article published in the Journal of Hospital Medicine set out to establish metrics to answer—and measure the answer—to that question, to assess hospitalists’ behaviors, and to establish norms and expectations.
“This study represents a first step to specifically characterize comportment and communication in hospital medicine,” the authors write.
Patient satisfaction surveys, they state, have some shortcomings in providing useful answers to that question.
“First, the attribution to specific providers is questionable,” the authors write. “Second, recall about the provider by the patients may be poor because surveys are sent to patients days after they return home. Third, the patients’ recovery and health outcomes are likely to influence their assessment of the doctor. Finally, feedback is known to be most valuable and transformative when it is specific and given in real time.”
Researchers asked the chiefs of hospital medicine divisions at five hospitals to identify their “most clinically excellent” hospitalists. Each hospitalist was observed during a routine clinical shift, and behaviors were recorded that were believed to be associated with excellent comportment and communication using the hospital medicine comportment and communication tool (HMCCOT), the final version of which has 23 variables. The physicians’ HMCCOT scores were associated with their patient satisfaction survey scores, suggesting that improved comportment might translate into enhanced patient satisfaction.
The results showed extensive variability in comportment and communication at the bedside. One variable that stood out to the researchers was that teach-back was employed in only 13% of the encounters.
“Previous studies have shown that teach-back corroborates patient comprehension and can be used to engage patients (and caregivers) in realistic goal setting and optimal health service utilization,” the researchers write. “Further, patients who clearly understand their post-discharge plan are 30% less likely to be readmitted or visit the emergency department. The data for our group have helped us to see areas of strengths, such as hand washing, where we are above compliance rates across hospitals in the United States, as well as those matters that represent opportunities for improvement such as connecting more deeply with our patients.”
The researchers call for future studies to determine whether hospitalists can improve feedback from this tool and whether enhancing comportment and communication can improve both patient satisfaction and clinical outcomes.
Reference
- Kotwal S, Khaliq W, Landis R, Wright S. Developing a comportment and communication tool for use in hospital medicine [published online ahead of print August 13, 2016]. J Hosp Med. doi:10.1002/jhm.2647.
Which Patients With Cancer Best Survive the ICU?
Because cancer is a complex disease, admitting a patient with cancer to the intensive care unit (ICU) can be challenging triage. Often the reason for the admission is acute complications related to the cancer or its treatment. Understanding how those complications might affect the patient’s outcome is critical to planning care, gauging use of ICU resources, and counseling patients and their families.
Related: Delirium in the Cardiac ICU
Although some studies have identified important determinants of mortality, the existing literature is “scarce,” say researchers who studied outcomes in ICU patients with cancer. They analyzed data from 2 cohort studies of 1,737 patients with solid tumors and 291 with hematologic malignancies. Of those, 456 (23%) had cancer-related complications at ICU admission, most frequently chemotherapy and radiation therapy toxicity, venous thromboembolism (VTE), and respiratory failure by tumor.
Patients with complications tended to have worse performance status scores and active disease. They also were more likely to have more severe organ dysfunctions, greater need for invasive support, and infection at ICU admission.
Complications occurred more often in patients with metastatic solid tumors, particularly patients with lung and breast cancer (although less common in patients with gastrointestinal [GI] tumors), and in patients with more aggressive hematologic malignancies, especially acute leukemia and aggressive non-Hodgkin lymphoma.
Related: Survival After Long-Term Residence in an Intensive Care Unit
Their study had several major findings, the researchers say. First, 1 in 4 patients with cancer admitted to the ICU has acute complications related to the underlying cancer or treatment adverse effects. However, although there were many cancer-related complications with varying degrees of prognostic impact and despite high mortality rates, outcomes in these patients were “better than perceived a priori,” the researchers say.
A high Sequential Organ Failure Assessment score on the first day of ICU stay, worse performance status, and need for mechanical ventilation were independent predictors of mortality, and all in accord with current literature. However, among the individual cancer-related complications studied, only vena cava syndrome, GI involvement, and respiratory failure were independently associated with in-hospital mortality. A “substantial” mortality rate (73%) among patients with GI involvement emphasizes the importance of discussing the appropriateness of ICU admission in these patients, the researchers caution. Although VTE was one of the most common complications, it was not a major determinant of outcome.
Related: Patients Benefit From ICU Telemedicine
Another important point, the researchers note, was the high frequency of chemotherapy/radiation therapy-induced toxicity. Treatment-related neutropenia is not a good predictor of outcome, they say, since research has found it is not a relevant predictor of mortality.
Source:
Torres VBL, Vassalo J, Silva UVA, et al. PLoS One. 2016;11(10): e0164537.
doi: 10.1371/journal.pone.0164537.
Because cancer is a complex disease, admitting a patient with cancer to the intensive care unit (ICU) can be challenging triage. Often the reason for the admission is acute complications related to the cancer or its treatment. Understanding how those complications might affect the patient’s outcome is critical to planning care, gauging use of ICU resources, and counseling patients and their families.
Related: Delirium in the Cardiac ICU
Although some studies have identified important determinants of mortality, the existing literature is “scarce,” say researchers who studied outcomes in ICU patients with cancer. They analyzed data from 2 cohort studies of 1,737 patients with solid tumors and 291 with hematologic malignancies. Of those, 456 (23%) had cancer-related complications at ICU admission, most frequently chemotherapy and radiation therapy toxicity, venous thromboembolism (VTE), and respiratory failure by tumor.
Patients with complications tended to have worse performance status scores and active disease. They also were more likely to have more severe organ dysfunctions, greater need for invasive support, and infection at ICU admission.
Complications occurred more often in patients with metastatic solid tumors, particularly patients with lung and breast cancer (although less common in patients with gastrointestinal [GI] tumors), and in patients with more aggressive hematologic malignancies, especially acute leukemia and aggressive non-Hodgkin lymphoma.
Related: Survival After Long-Term Residence in an Intensive Care Unit
Their study had several major findings, the researchers say. First, 1 in 4 patients with cancer admitted to the ICU has acute complications related to the underlying cancer or treatment adverse effects. However, although there were many cancer-related complications with varying degrees of prognostic impact and despite high mortality rates, outcomes in these patients were “better than perceived a priori,” the researchers say.
A high Sequential Organ Failure Assessment score on the first day of ICU stay, worse performance status, and need for mechanical ventilation were independent predictors of mortality, and all in accord with current literature. However, among the individual cancer-related complications studied, only vena cava syndrome, GI involvement, and respiratory failure were independently associated with in-hospital mortality. A “substantial” mortality rate (73%) among patients with GI involvement emphasizes the importance of discussing the appropriateness of ICU admission in these patients, the researchers caution. Although VTE was one of the most common complications, it was not a major determinant of outcome.
Related: Patients Benefit From ICU Telemedicine
Another important point, the researchers note, was the high frequency of chemotherapy/radiation therapy-induced toxicity. Treatment-related neutropenia is not a good predictor of outcome, they say, since research has found it is not a relevant predictor of mortality.
Source:
Torres VBL, Vassalo J, Silva UVA, et al. PLoS One. 2016;11(10): e0164537.
doi: 10.1371/journal.pone.0164537.
Because cancer is a complex disease, admitting a patient with cancer to the intensive care unit (ICU) can be challenging triage. Often the reason for the admission is acute complications related to the cancer or its treatment. Understanding how those complications might affect the patient’s outcome is critical to planning care, gauging use of ICU resources, and counseling patients and their families.
Related: Delirium in the Cardiac ICU
Although some studies have identified important determinants of mortality, the existing literature is “scarce,” say researchers who studied outcomes in ICU patients with cancer. They analyzed data from 2 cohort studies of 1,737 patients with solid tumors and 291 with hematologic malignancies. Of those, 456 (23%) had cancer-related complications at ICU admission, most frequently chemotherapy and radiation therapy toxicity, venous thromboembolism (VTE), and respiratory failure by tumor.
Patients with complications tended to have worse performance status scores and active disease. They also were more likely to have more severe organ dysfunctions, greater need for invasive support, and infection at ICU admission.
Complications occurred more often in patients with metastatic solid tumors, particularly patients with lung and breast cancer (although less common in patients with gastrointestinal [GI] tumors), and in patients with more aggressive hematologic malignancies, especially acute leukemia and aggressive non-Hodgkin lymphoma.
Related: Survival After Long-Term Residence in an Intensive Care Unit
Their study had several major findings, the researchers say. First, 1 in 4 patients with cancer admitted to the ICU has acute complications related to the underlying cancer or treatment adverse effects. However, although there were many cancer-related complications with varying degrees of prognostic impact and despite high mortality rates, outcomes in these patients were “better than perceived a priori,” the researchers say.
A high Sequential Organ Failure Assessment score on the first day of ICU stay, worse performance status, and need for mechanical ventilation were independent predictors of mortality, and all in accord with current literature. However, among the individual cancer-related complications studied, only vena cava syndrome, GI involvement, and respiratory failure were independently associated with in-hospital mortality. A “substantial” mortality rate (73%) among patients with GI involvement emphasizes the importance of discussing the appropriateness of ICU admission in these patients, the researchers caution. Although VTE was one of the most common complications, it was not a major determinant of outcome.
Related: Patients Benefit From ICU Telemedicine
Another important point, the researchers note, was the high frequency of chemotherapy/radiation therapy-induced toxicity. Treatment-related neutropenia is not a good predictor of outcome, they say, since research has found it is not a relevant predictor of mortality.
Source:
Torres VBL, Vassalo J, Silva UVA, et al. PLoS One. 2016;11(10): e0164537.
doi: 10.1371/journal.pone.0164537.
Targeting CD98 to treat AML
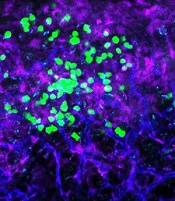
with blood vessels (blue).
Image courtesy of
UC San Diego Health
Preclinical research suggests the cell surface molecule CD98 promotes acute myeloid leukemia (AML), and the anti-CD98 antibody IGN523 can inhibit AML growth.
In AML patient cells and mouse models of the disease, IGN523 disrupted the interactions between leukemia cells and the surrounding blood vessels, thereby inhibiting the growth of AML.
Tannishtha Reya, PhD, of the University of California San Diego School of Medicine, and her colleagues reported these findings in Cancer Cell.
The team believes their results suggest IGN523 or other anti-CD98 antibodies might be useful for treating AML, particularly in children.
However, in a phase 1 study presented at the 2015 ASH Annual Meeting, IGN523 demonstrated only modest anti-leukemic activity in adults with AML.
Still, the researchers involved in the phase 1 study said IGN523 may prove effective in combination with other drugs used to treat AML.
Cancer Cell study
“To improve therapeutic strategies for [AML], we need to look not just at the cancer cells themselves but also at their interactions with surrounding cells, tissues, molecules, and blood vessels in the body,” Dr Reya said.
“In this study, we identified CD98 as a critical molecule driving AML growth. We showed that blocking CD98 can effectively reduce leukemia burden and improve survival by preventing cancer cells from receiving support from the surrounding environment.”
Dr Reya’s team engineered mouse models that lacked CD98 and found that loss of this molecule blocked AML growth and improved survival. Furthermore, CD98 loss largely spared normal blood cells, which the researchers said indicates a potential therapeutic window.
Additional experiments revealed that leukemia cells lacking CD98 had fewer stable interactions with the lining of blood vessels—interactions that were needed to fuel AML growth.
So the researchers decided to test the effects of blocking CD98 with a therapeutic inhibitor—IGN523. The team found that IGN523 blocks CD98’s AML-promoting activity in mouse models and human AML cells.
The researchers also transplanted patient-derived AML cells into mice and treated the recipients with either IGN523 or a control antibody. Anti-CD98 treatment effectively eliminated AML cells, while AML in the control mice expanded more than 100-fold.
“This study suggests that human AML can’t get established without CD98 and that blocking the molecule with anti-CD98 antibodies could be beneficial for the treatment of AML in both adults and children,” Dr Reya said.
Moving forward, Dr Reya and her colleagues are working to further define whether CD98 could be used to treat pediatric AML.
“Many of the models we used in this work were based on mutations found in childhood AML,” Dr Reya said. “While many childhood cancers have become very treatable, childhood AML continues to have a high rate of relapse and death.”
“We plan to work with pediatric oncologists to test if anti-CD98 agents can be effective against pediatric AML and whether it can improve responses to current treatments. I think this is particularly important to pursue since the anti-CD98 antibody has already been through phase 1 trials and could be more easily positioned to test in drug-resistant pediatric AML.”
Igenica Biotherapeutics Inc., the company developing IGN523, provided the drug for this study, and one of the study’s authors is an employee of the company. ![]()
with blood vessels (blue).
Image courtesy of
UC San Diego Health
Preclinical research suggests the cell surface molecule CD98 promotes acute myeloid leukemia (AML), and the anti-CD98 antibody IGN523 can inhibit AML growth.
In AML patient cells and mouse models of the disease, IGN523 disrupted the interactions between leukemia cells and the surrounding blood vessels, thereby inhibiting the growth of AML.
Tannishtha Reya, PhD, of the University of California San Diego School of Medicine, and her colleagues reported these findings in Cancer Cell.
The team believes their results suggest IGN523 or other anti-CD98 antibodies might be useful for treating AML, particularly in children.
However, in a phase 1 study presented at the 2015 ASH Annual Meeting, IGN523 demonstrated only modest anti-leukemic activity in adults with AML.
Still, the researchers involved in the phase 1 study said IGN523 may prove effective in combination with other drugs used to treat AML.
Cancer Cell study
“To improve therapeutic strategies for [AML], we need to look not just at the cancer cells themselves but also at their interactions with surrounding cells, tissues, molecules, and blood vessels in the body,” Dr Reya said.
“In this study, we identified CD98 as a critical molecule driving AML growth. We showed that blocking CD98 can effectively reduce leukemia burden and improve survival by preventing cancer cells from receiving support from the surrounding environment.”
Dr Reya’s team engineered mouse models that lacked CD98 and found that loss of this molecule blocked AML growth and improved survival. Furthermore, CD98 loss largely spared normal blood cells, which the researchers said indicates a potential therapeutic window.
Additional experiments revealed that leukemia cells lacking CD98 had fewer stable interactions with the lining of blood vessels—interactions that were needed to fuel AML growth.
So the researchers decided to test the effects of blocking CD98 with a therapeutic inhibitor—IGN523. The team found that IGN523 blocks CD98’s AML-promoting activity in mouse models and human AML cells.
The researchers also transplanted patient-derived AML cells into mice and treated the recipients with either IGN523 or a control antibody. Anti-CD98 treatment effectively eliminated AML cells, while AML in the control mice expanded more than 100-fold.
“This study suggests that human AML can’t get established without CD98 and that blocking the molecule with anti-CD98 antibodies could be beneficial for the treatment of AML in both adults and children,” Dr Reya said.
Moving forward, Dr Reya and her colleagues are working to further define whether CD98 could be used to treat pediatric AML.
“Many of the models we used in this work were based on mutations found in childhood AML,” Dr Reya said. “While many childhood cancers have become very treatable, childhood AML continues to have a high rate of relapse and death.”
“We plan to work with pediatric oncologists to test if anti-CD98 agents can be effective against pediatric AML and whether it can improve responses to current treatments. I think this is particularly important to pursue since the anti-CD98 antibody has already been through phase 1 trials and could be more easily positioned to test in drug-resistant pediatric AML.”
Igenica Biotherapeutics Inc., the company developing IGN523, provided the drug for this study, and one of the study’s authors is an employee of the company. ![]()
with blood vessels (blue).
Image courtesy of
UC San Diego Health
Preclinical research suggests the cell surface molecule CD98 promotes acute myeloid leukemia (AML), and the anti-CD98 antibody IGN523 can inhibit AML growth.
In AML patient cells and mouse models of the disease, IGN523 disrupted the interactions between leukemia cells and the surrounding blood vessels, thereby inhibiting the growth of AML.
Tannishtha Reya, PhD, of the University of California San Diego School of Medicine, and her colleagues reported these findings in Cancer Cell.
The team believes their results suggest IGN523 or other anti-CD98 antibodies might be useful for treating AML, particularly in children.
However, in a phase 1 study presented at the 2015 ASH Annual Meeting, IGN523 demonstrated only modest anti-leukemic activity in adults with AML.
Still, the researchers involved in the phase 1 study said IGN523 may prove effective in combination with other drugs used to treat AML.
Cancer Cell study
“To improve therapeutic strategies for [AML], we need to look not just at the cancer cells themselves but also at their interactions with surrounding cells, tissues, molecules, and blood vessels in the body,” Dr Reya said.
“In this study, we identified CD98 as a critical molecule driving AML growth. We showed that blocking CD98 can effectively reduce leukemia burden and improve survival by preventing cancer cells from receiving support from the surrounding environment.”
Dr Reya’s team engineered mouse models that lacked CD98 and found that loss of this molecule blocked AML growth and improved survival. Furthermore, CD98 loss largely spared normal blood cells, which the researchers said indicates a potential therapeutic window.
Additional experiments revealed that leukemia cells lacking CD98 had fewer stable interactions with the lining of blood vessels—interactions that were needed to fuel AML growth.
So the researchers decided to test the effects of blocking CD98 with a therapeutic inhibitor—IGN523. The team found that IGN523 blocks CD98’s AML-promoting activity in mouse models and human AML cells.
The researchers also transplanted patient-derived AML cells into mice and treated the recipients with either IGN523 or a control antibody. Anti-CD98 treatment effectively eliminated AML cells, while AML in the control mice expanded more than 100-fold.
“This study suggests that human AML can’t get established without CD98 and that blocking the molecule with anti-CD98 antibodies could be beneficial for the treatment of AML in both adults and children,” Dr Reya said.
Moving forward, Dr Reya and her colleagues are working to further define whether CD98 could be used to treat pediatric AML.
“Many of the models we used in this work were based on mutations found in childhood AML,” Dr Reya said. “While many childhood cancers have become very treatable, childhood AML continues to have a high rate of relapse and death.”
“We plan to work with pediatric oncologists to test if anti-CD98 agents can be effective against pediatric AML and whether it can improve responses to current treatments. I think this is particularly important to pursue since the anti-CD98 antibody has already been through phase 1 trials and could be more easily positioned to test in drug-resistant pediatric AML.”
Igenica Biotherapeutics Inc., the company developing IGN523, provided the drug for this study, and one of the study’s authors is an employee of the company. ![]()
Study reveals ‘high-traffic’ routes of malaria importation
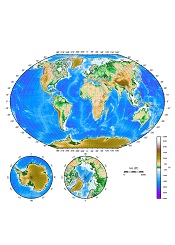
Results of an international study suggest France and the UK experience the highest number of malaria cases imported from other countries.
Researchers mapped the movement of malaria from endemic countries to 40 countries defined as being malaria-free.
The countries with the highest average number of imported infections per year over the past decade were France (2169), the UK (1898), the US (1511), Italy (637), and Germany (401).
Infection movement was strongly skewed to a small number of “high-traffic” routes, with malaria cases originating from West Africa accounting for 56% (13,947) of all cases detected in non-endemic countries.
These results were published in The Lancet Infectious Diseases.
“This is the first world-wide assessment of imported malaria cases in 20 years, and mapping this data is hugely valuable in helping us understand how we can mitigate against the effects of the global movements of the disease,” said study author Andrew Tatem, PhD, of the University of Southampton in the UK.
“Imported malaria can be expensive to treat, contribute to drug resistance, sometimes cause secondary local transmission, and threaten the long-term goal of eradication. This study forms part of wider efforts to understand patterns of human and malaria parasite movement to help guide elimination strategies.”
For this study, researchers analyzed a database of nationally reported statistics on imported malaria covering more than 50,000 individual cases over 10 years.
Although most incidents of malaria in non-endemic countries originated in West Africa, the study showed that 20% were from India (4988), 13% were from East Africa (3242), and 3% were from Papua New Guinea (748).
And although the routes from West Africa to France and the UK showed the strongest imported malaria link, there were other high-traffic routes. These included India to the US (149 cases on average per year), Papua New Guinea to Australia (97), Pakistan to the UK (69), and Haiti to the US (52).
By mapping this network of malaria movements across continents, the researchers showed that a number of factors, beyond geographic ones, may influence the strength of importation levels.
For example, the researchers believe that historical, economic, language, and cultural ties all play a part. They said population movements with former colonies had particular influence; such as Nigeria, Ghana, and Kenya with the UK, and Mali, Niger, and Chad with France.
The researchers hope to conduct further studies to examine which factors are the drivers behind the patterns of malaria spread between endemic and non-endemic countries. ![]()
Results of an international study suggest France and the UK experience the highest number of malaria cases imported from other countries.
Researchers mapped the movement of malaria from endemic countries to 40 countries defined as being malaria-free.
The countries with the highest average number of imported infections per year over the past decade were France (2169), the UK (1898), the US (1511), Italy (637), and Germany (401).
Infection movement was strongly skewed to a small number of “high-traffic” routes, with malaria cases originating from West Africa accounting for 56% (13,947) of all cases detected in non-endemic countries.
These results were published in The Lancet Infectious Diseases.
“This is the first world-wide assessment of imported malaria cases in 20 years, and mapping this data is hugely valuable in helping us understand how we can mitigate against the effects of the global movements of the disease,” said study author Andrew Tatem, PhD, of the University of Southampton in the UK.
“Imported malaria can be expensive to treat, contribute to drug resistance, sometimes cause secondary local transmission, and threaten the long-term goal of eradication. This study forms part of wider efforts to understand patterns of human and malaria parasite movement to help guide elimination strategies.”
For this study, researchers analyzed a database of nationally reported statistics on imported malaria covering more than 50,000 individual cases over 10 years.
Although most incidents of malaria in non-endemic countries originated in West Africa, the study showed that 20% were from India (4988), 13% were from East Africa (3242), and 3% were from Papua New Guinea (748).
And although the routes from West Africa to France and the UK showed the strongest imported malaria link, there were other high-traffic routes. These included India to the US (149 cases on average per year), Papua New Guinea to Australia (97), Pakistan to the UK (69), and Haiti to the US (52).
By mapping this network of malaria movements across continents, the researchers showed that a number of factors, beyond geographic ones, may influence the strength of importation levels.
For example, the researchers believe that historical, economic, language, and cultural ties all play a part. They said population movements with former colonies had particular influence; such as Nigeria, Ghana, and Kenya with the UK, and Mali, Niger, and Chad with France.
The researchers hope to conduct further studies to examine which factors are the drivers behind the patterns of malaria spread between endemic and non-endemic countries. ![]()
Results of an international study suggest France and the UK experience the highest number of malaria cases imported from other countries.
Researchers mapped the movement of malaria from endemic countries to 40 countries defined as being malaria-free.
The countries with the highest average number of imported infections per year over the past decade were France (2169), the UK (1898), the US (1511), Italy (637), and Germany (401).
Infection movement was strongly skewed to a small number of “high-traffic” routes, with malaria cases originating from West Africa accounting for 56% (13,947) of all cases detected in non-endemic countries.
These results were published in The Lancet Infectious Diseases.
“This is the first world-wide assessment of imported malaria cases in 20 years, and mapping this data is hugely valuable in helping us understand how we can mitigate against the effects of the global movements of the disease,” said study author Andrew Tatem, PhD, of the University of Southampton in the UK.
“Imported malaria can be expensive to treat, contribute to drug resistance, sometimes cause secondary local transmission, and threaten the long-term goal of eradication. This study forms part of wider efforts to understand patterns of human and malaria parasite movement to help guide elimination strategies.”
For this study, researchers analyzed a database of nationally reported statistics on imported malaria covering more than 50,000 individual cases over 10 years.
Although most incidents of malaria in non-endemic countries originated in West Africa, the study showed that 20% were from India (4988), 13% were from East Africa (3242), and 3% were from Papua New Guinea (748).
And although the routes from West Africa to France and the UK showed the strongest imported malaria link, there were other high-traffic routes. These included India to the US (149 cases on average per year), Papua New Guinea to Australia (97), Pakistan to the UK (69), and Haiti to the US (52).
By mapping this network of malaria movements across continents, the researchers showed that a number of factors, beyond geographic ones, may influence the strength of importation levels.
For example, the researchers believe that historical, economic, language, and cultural ties all play a part. They said population movements with former colonies had particular influence; such as Nigeria, Ghana, and Kenya with the UK, and Mali, Niger, and Chad with France.
The researchers hope to conduct further studies to examine which factors are the drivers behind the patterns of malaria spread between endemic and non-endemic countries. ![]()
Testing could ID cancer patients at high risk of VTE

Image by Andre E.X. Brown
Genetic testing could help identify breast cancer patients with a high risk of developing venous thromboembolism (VTE), according to a study published in Clinical Cancer Research.
The study showed that patients who received chemotherapy, had a higher genetic susceptibility for VTE according to a polygenic risk score (PRS), or had both of these risk factors were more likely to develop VTE than patients without these risk factors.
In addition, researchers found the impact of genetic susceptibility was most pronounced in older patients.
“The risk for [VTE] is increased in cancer patients, particularly in those receiving chemotherapy,” said study author Judith S. Brand, PhD, of Karolinska Institutet in Stockholm, Sweden.
“As one of the most common cancers, breast cancer accounts for a large number of cancer-associated VTE cases.”
Dr Brand and her colleagues sought to identify the individual and joint effects of chemotherapy and genetic susceptibility on VTE risk. They studied 4261 women in the Stockholm region diagnosed with primary invasive breast cancer between 2001 and 2008, and followed until 2012.
Risks were stratified based on chemotherapy status and genetic susceptibility, as determined by a PRS based on 9 established VTE loci, with the top 5% classified as having high genetic susceptibility.
The median follow-up was 7.6 years, and 276 patients experienced a VTE during that time.
The researchers found that receiving chemotherapy and having high genetic susceptibility independently increased the risk of VTE. The hazard ratio was 1.98 for patients receiving chemotherapy and 1.90 for patients in the highest 5% of the PRS.
The 1-year cumulative incidence of VTE was 9.5% among patients who both received chemotherapy and had high genetic susceptibility, compared with 1.3% in the patients who did not have either of these risk factors (P<0.001).
The researchers also found that patient age played a role in VTE risk. The team said the risk-increasing effect of the PRS was stronger in older patients (P interaction = 0.04).
In patients age 60 or older who underwent chemotherapy and had a high genetic susceptibility, the 1-year cumulative incidence of VTE was 25%.
“Breast cancer patients receiving chemotherapy are not routinely being examined for VTE prevention in today’s clinical practice,” Dr Brand said. “Our study demonstrates that information on genetic susceptibility can be used to identify patients at high risk of developing VTE.”
“Combined with other clinical risk factors and biomarkers, these findings will guide future studies evaluating routine VTE risk assessment in chemotherapy outpatients, and prophylaxis for those at highest risk. Because older patients demonstrated a stronger genetic effect and higher VTE incidence, this group requires special attention in future risk stratification efforts.”
Dr Brand added that a limitation of this study is the small number of older patients who had chemotherapy and a high genetic susceptibility. She said larger-scale studies would be necessary to provide more precise risk estimates. And further research is needed to assess the safety and potential benefit of thromboprophylaxis in high-risk cancer patients. ![]()
Image by Andre E.X. Brown
Genetic testing could help identify breast cancer patients with a high risk of developing venous thromboembolism (VTE), according to a study published in Clinical Cancer Research.
The study showed that patients who received chemotherapy, had a higher genetic susceptibility for VTE according to a polygenic risk score (PRS), or had both of these risk factors were more likely to develop VTE than patients without these risk factors.
In addition, researchers found the impact of genetic susceptibility was most pronounced in older patients.
“The risk for [VTE] is increased in cancer patients, particularly in those receiving chemotherapy,” said study author Judith S. Brand, PhD, of Karolinska Institutet in Stockholm, Sweden.
“As one of the most common cancers, breast cancer accounts for a large number of cancer-associated VTE cases.”
Dr Brand and her colleagues sought to identify the individual and joint effects of chemotherapy and genetic susceptibility on VTE risk. They studied 4261 women in the Stockholm region diagnosed with primary invasive breast cancer between 2001 and 2008, and followed until 2012.
Risks were stratified based on chemotherapy status and genetic susceptibility, as determined by a PRS based on 9 established VTE loci, with the top 5% classified as having high genetic susceptibility.
The median follow-up was 7.6 years, and 276 patients experienced a VTE during that time.
The researchers found that receiving chemotherapy and having high genetic susceptibility independently increased the risk of VTE. The hazard ratio was 1.98 for patients receiving chemotherapy and 1.90 for patients in the highest 5% of the PRS.
The 1-year cumulative incidence of VTE was 9.5% among patients who both received chemotherapy and had high genetic susceptibility, compared with 1.3% in the patients who did not have either of these risk factors (P<0.001).
The researchers also found that patient age played a role in VTE risk. The team said the risk-increasing effect of the PRS was stronger in older patients (P interaction = 0.04).
In patients age 60 or older who underwent chemotherapy and had a high genetic susceptibility, the 1-year cumulative incidence of VTE was 25%.
“Breast cancer patients receiving chemotherapy are not routinely being examined for VTE prevention in today’s clinical practice,” Dr Brand said. “Our study demonstrates that information on genetic susceptibility can be used to identify patients at high risk of developing VTE.”
“Combined with other clinical risk factors and biomarkers, these findings will guide future studies evaluating routine VTE risk assessment in chemotherapy outpatients, and prophylaxis for those at highest risk. Because older patients demonstrated a stronger genetic effect and higher VTE incidence, this group requires special attention in future risk stratification efforts.”
Dr Brand added that a limitation of this study is the small number of older patients who had chemotherapy and a high genetic susceptibility. She said larger-scale studies would be necessary to provide more precise risk estimates. And further research is needed to assess the safety and potential benefit of thromboprophylaxis in high-risk cancer patients. ![]()
Image by Andre E.X. Brown
Genetic testing could help identify breast cancer patients with a high risk of developing venous thromboembolism (VTE), according to a study published in Clinical Cancer Research.
The study showed that patients who received chemotherapy, had a higher genetic susceptibility for VTE according to a polygenic risk score (PRS), or had both of these risk factors were more likely to develop VTE than patients without these risk factors.
In addition, researchers found the impact of genetic susceptibility was most pronounced in older patients.
“The risk for [VTE] is increased in cancer patients, particularly in those receiving chemotherapy,” said study author Judith S. Brand, PhD, of Karolinska Institutet in Stockholm, Sweden.
“As one of the most common cancers, breast cancer accounts for a large number of cancer-associated VTE cases.”
Dr Brand and her colleagues sought to identify the individual and joint effects of chemotherapy and genetic susceptibility on VTE risk. They studied 4261 women in the Stockholm region diagnosed with primary invasive breast cancer between 2001 and 2008, and followed until 2012.
Risks were stratified based on chemotherapy status and genetic susceptibility, as determined by a PRS based on 9 established VTE loci, with the top 5% classified as having high genetic susceptibility.
The median follow-up was 7.6 years, and 276 patients experienced a VTE during that time.
The researchers found that receiving chemotherapy and having high genetic susceptibility independently increased the risk of VTE. The hazard ratio was 1.98 for patients receiving chemotherapy and 1.90 for patients in the highest 5% of the PRS.
The 1-year cumulative incidence of VTE was 9.5% among patients who both received chemotherapy and had high genetic susceptibility, compared with 1.3% in the patients who did not have either of these risk factors (P<0.001).
The researchers also found that patient age played a role in VTE risk. The team said the risk-increasing effect of the PRS was stronger in older patients (P interaction = 0.04).
In patients age 60 or older who underwent chemotherapy and had a high genetic susceptibility, the 1-year cumulative incidence of VTE was 25%.
“Breast cancer patients receiving chemotherapy are not routinely being examined for VTE prevention in today’s clinical practice,” Dr Brand said. “Our study demonstrates that information on genetic susceptibility can be used to identify patients at high risk of developing VTE.”
“Combined with other clinical risk factors and biomarkers, these findings will guide future studies evaluating routine VTE risk assessment in chemotherapy outpatients, and prophylaxis for those at highest risk. Because older patients demonstrated a stronger genetic effect and higher VTE incidence, this group requires special attention in future risk stratification efforts.”
Dr Brand added that a limitation of this study is the small number of older patients who had chemotherapy and a high genetic susceptibility. She said larger-scale studies would be necessary to provide more precise risk estimates. And further research is needed to assess the safety and potential benefit of thromboprophylaxis in high-risk cancer patients. ![]()
CTC analysis as good as BM biopsy in MM, team says

Photo by Juan D. Alfonso
Analysis of circulating tumor cells (CTCs) can provide at least as much genetic information about multiple myeloma (MM) as a bone marrow (BM) biopsy, according to a new study.
Researchers found evidence to suggest that analyzing CTCs isolated from the peripheral blood of MM patients could help physicians monitor disease progression over time, track the emergence of drug resistance, and tailor therapies to individual patients.
Jens Lohr, MD, PhD, of the Broad Institute of MIT and Harvard in Cambridge, Massachusetts, and colleagues conducted this research and detailed their findings in Science Translational Medicine.
The researchers said they devised a method that was “highly sensitive” in detecting and sequencing single CTCs, even from patients with low tumor burden.
Specifically, the team found they could isolate CTCs from, and detect mutations in, blood samples from an MM patient who had achieved a very good partial response to treatment and a patient with monoclonal gammopathy of undetermined significance.
Overall, the researchers found that CTCs could be used to identify mutations relevant for prognosis, and some of these mutations were more abundant in CTCs than in BM samples.
For example, the team detected somatic mutations in the KRAS, BRAF, IRF4, and TP53 genes in single CTCs from 3 MM patients. And the same mutations were present in single BM-derived MM cells.
In another 3 patients, the proportion of CTCs harboring TP53 R273C, BRAF G469A, and NRAS G13D mutations was higher than that observed in BM-derived cells. In 2 of the patients, the mutations weren’t detectable in BM cells because of insufficient sample material.
The researchers also found that CTCs could reveal patients with an overabundance of molecules expressed by MM cells—such as CD38 and SLAMF7—that can be targeted by therapies currently approved to treat MM—such as daratumumab and elotuzumab.
The team therefore believes that, with further development, CTC analysis could be a valuable tool for advancing precision medicine in MM. ![]()
Photo by Juan D. Alfonso
Analysis of circulating tumor cells (CTCs) can provide at least as much genetic information about multiple myeloma (MM) as a bone marrow (BM) biopsy, according to a new study.
Researchers found evidence to suggest that analyzing CTCs isolated from the peripheral blood of MM patients could help physicians monitor disease progression over time, track the emergence of drug resistance, and tailor therapies to individual patients.
Jens Lohr, MD, PhD, of the Broad Institute of MIT and Harvard in Cambridge, Massachusetts, and colleagues conducted this research and detailed their findings in Science Translational Medicine.
The researchers said they devised a method that was “highly sensitive” in detecting and sequencing single CTCs, even from patients with low tumor burden.
Specifically, the team found they could isolate CTCs from, and detect mutations in, blood samples from an MM patient who had achieved a very good partial response to treatment and a patient with monoclonal gammopathy of undetermined significance.
Overall, the researchers found that CTCs could be used to identify mutations relevant for prognosis, and some of these mutations were more abundant in CTCs than in BM samples.
For example, the team detected somatic mutations in the KRAS, BRAF, IRF4, and TP53 genes in single CTCs from 3 MM patients. And the same mutations were present in single BM-derived MM cells.
In another 3 patients, the proportion of CTCs harboring TP53 R273C, BRAF G469A, and NRAS G13D mutations was higher than that observed in BM-derived cells. In 2 of the patients, the mutations weren’t detectable in BM cells because of insufficient sample material.
The researchers also found that CTCs could reveal patients with an overabundance of molecules expressed by MM cells—such as CD38 and SLAMF7—that can be targeted by therapies currently approved to treat MM—such as daratumumab and elotuzumab.
The team therefore believes that, with further development, CTC analysis could be a valuable tool for advancing precision medicine in MM. ![]()
Photo by Juan D. Alfonso
Analysis of circulating tumor cells (CTCs) can provide at least as much genetic information about multiple myeloma (MM) as a bone marrow (BM) biopsy, according to a new study.
Researchers found evidence to suggest that analyzing CTCs isolated from the peripheral blood of MM patients could help physicians monitor disease progression over time, track the emergence of drug resistance, and tailor therapies to individual patients.
Jens Lohr, MD, PhD, of the Broad Institute of MIT and Harvard in Cambridge, Massachusetts, and colleagues conducted this research and detailed their findings in Science Translational Medicine.
The researchers said they devised a method that was “highly sensitive” in detecting and sequencing single CTCs, even from patients with low tumor burden.
Specifically, the team found they could isolate CTCs from, and detect mutations in, blood samples from an MM patient who had achieved a very good partial response to treatment and a patient with monoclonal gammopathy of undetermined significance.
Overall, the researchers found that CTCs could be used to identify mutations relevant for prognosis, and some of these mutations were more abundant in CTCs than in BM samples.
For example, the team detected somatic mutations in the KRAS, BRAF, IRF4, and TP53 genes in single CTCs from 3 MM patients. And the same mutations were present in single BM-derived MM cells.
In another 3 patients, the proportion of CTCs harboring TP53 R273C, BRAF G469A, and NRAS G13D mutations was higher than that observed in BM-derived cells. In 2 of the patients, the mutations weren’t detectable in BM cells because of insufficient sample material.
The researchers also found that CTCs could reveal patients with an overabundance of molecules expressed by MM cells—such as CD38 and SLAMF7—that can be targeted by therapies currently approved to treat MM—such as daratumumab and elotuzumab.
The team therefore believes that, with further development, CTC analysis could be a valuable tool for advancing precision medicine in MM. ![]()
Making Fall Prevention “Routine”
Every second of every day in the U.S. an older adult falls, according to CDC data. Falls are the number one cause of injuries and deaths from injury among older Americans. The report cites 29 million falls in 2014 alone.
“Older adult falls are increasing and, sadly, often herald the end of independence,” said CDC Director Tom Frieden, MD, MPH. But he adds: “Health care providers can make fall prevention a routine part of care in their practice, and older adults can take steps to protect themselves.”
Related: Preventing Falls and Saving Costs
The CDC created Stopping Elderly Accidents, Deaths, and Injuries (STEADI), an initiative to make fall prevention routine in health care. The program provides information on how to screen for falls, online training for providers, videos on conducting functional assessments, and brochures for health care providers (HCPs), patients, and caregivers.
Among the suggestions for HCPs:
- Ask patients whether they have fallen in the past year, feel unsteady, or worry about falling
- Review medications and stop, switch, or reduce medicines that could increase the risk of falls
- Recommend vitamin D supplements
For older adults, the CDC recommends:
- Talk to a HCP about falls and fall prevention
- Tell a HCP if you have fallen—fewer than half of Americans who fall tell their doctor
- Have your eyes checked, and update eye prescriptions
- Participate in evidence-based programs like tai chi to improve balance and strengthen legs
- Get rid of fall hazards in your home
Every second of every day in the U.S. an older adult falls, according to CDC data. Falls are the number one cause of injuries and deaths from injury among older Americans. The report cites 29 million falls in 2014 alone.
“Older adult falls are increasing and, sadly, often herald the end of independence,” said CDC Director Tom Frieden, MD, MPH. But he adds: “Health care providers can make fall prevention a routine part of care in their practice, and older adults can take steps to protect themselves.”
Related: Preventing Falls and Saving Costs
The CDC created Stopping Elderly Accidents, Deaths, and Injuries (STEADI), an initiative to make fall prevention routine in health care. The program provides information on how to screen for falls, online training for providers, videos on conducting functional assessments, and brochures for health care providers (HCPs), patients, and caregivers.
Among the suggestions for HCPs:
- Ask patients whether they have fallen in the past year, feel unsteady, or worry about falling
- Review medications and stop, switch, or reduce medicines that could increase the risk of falls
- Recommend vitamin D supplements
For older adults, the CDC recommends:
- Talk to a HCP about falls and fall prevention
- Tell a HCP if you have fallen—fewer than half of Americans who fall tell their doctor
- Have your eyes checked, and update eye prescriptions
- Participate in evidence-based programs like tai chi to improve balance and strengthen legs
- Get rid of fall hazards in your home
Every second of every day in the U.S. an older adult falls, according to CDC data. Falls are the number one cause of injuries and deaths from injury among older Americans. The report cites 29 million falls in 2014 alone.
“Older adult falls are increasing and, sadly, often herald the end of independence,” said CDC Director Tom Frieden, MD, MPH. But he adds: “Health care providers can make fall prevention a routine part of care in their practice, and older adults can take steps to protect themselves.”
Related: Preventing Falls and Saving Costs
The CDC created Stopping Elderly Accidents, Deaths, and Injuries (STEADI), an initiative to make fall prevention routine in health care. The program provides information on how to screen for falls, online training for providers, videos on conducting functional assessments, and brochures for health care providers (HCPs), patients, and caregivers.
Among the suggestions for HCPs:
- Ask patients whether they have fallen in the past year, feel unsteady, or worry about falling
- Review medications and stop, switch, or reduce medicines that could increase the risk of falls
- Recommend vitamin D supplements
For older adults, the CDC recommends:
- Talk to a HCP about falls and fall prevention
- Tell a HCP if you have fallen—fewer than half of Americans who fall tell their doctor
- Have your eyes checked, and update eye prescriptions
- Participate in evidence-based programs like tai chi to improve balance and strengthen legs
- Get rid of fall hazards in your home
A Severe Sitchuation
This 49-year-old man was first diagnosed with Darier disease at age 8. It has been contained, for the most part, by topical medications and acitretin. But the itching on his legs is especially severe, making it difficult for him to leave them alone.
EXAMINATION
Both legs have heavy scaling and lichenification from the lateral aspects of the knees down to the ankles. There is modest erythema but no suppurative signs. Other areas of scaling are seen on his chest and back, but these are less coarse and rough than the leg rash.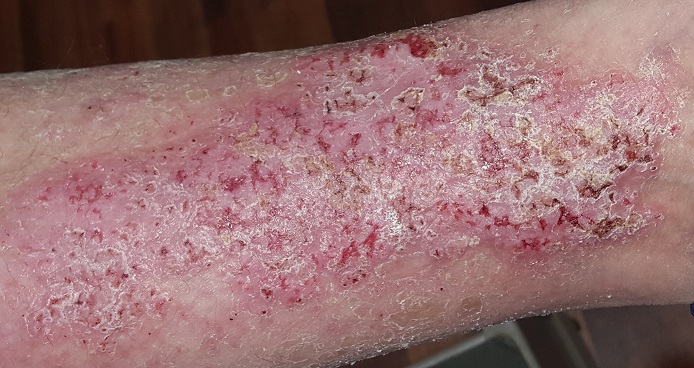
What is the diagnosis?
DISCUSSION
Lichen simplex chronicus (LSC) is the term given to this phenomenon. LSC is always secondary to a primary trigger, which can be dry skin, eczema, psoriasis, or even a bug bite. Excessive scratching induces more itching, and before the patient realizes it, he has jumped aboard the itch-scratch-itch train—a vicious cycle that can be hard to break.
The skin reacts to all this attention by thickening and becoming rough; on a microscopic level, the nerves become more sensitive. The urge to scratch becomes so irresistible that some patients fantasize about giving in.
The first goal of treating LSC is to stop the itching by helping the patient avoid contact with the site. High-potency topical steroids can be effective for this, especially if used under occlusion, which not only potentiates the steroid but also provides a barrier to protect the area from the patient’s fingernails (or hairbrush). Once the condition improves, steroids can be slowly withdrawn.
A biopsy, if it had been necessary, would have shown hypertrophic epidermis and acanthotic changes.
TAKE-HOME LEARNING POINTS
• Lichen simplex chronicus (LSC) is secondary to an inciting condition such as eczema, xerosis, psoriasis, or Darier disease.
• The manifestation of the itch-scratch-itch cycle leads to thickening and lichenification of the skin. The itch of LSC is so compelling that scratching is hard to resist.
• Despite treatment and patient education, recurrences are quite common.
This 49-year-old man was first diagnosed with Darier disease at age 8. It has been contained, for the most part, by topical medications and acitretin. But the itching on his legs is especially severe, making it difficult for him to leave them alone.
EXAMINATION
Both legs have heavy scaling and lichenification from the lateral aspects of the knees down to the ankles. There is modest erythema but no suppurative signs. Other areas of scaling are seen on his chest and back, but these are less coarse and rough than the leg rash.
What is the diagnosis?
DISCUSSION
Lichen simplex chronicus (LSC) is the term given to this phenomenon. LSC is always secondary to a primary trigger, which can be dry skin, eczema, psoriasis, or even a bug bite. Excessive scratching induces more itching, and before the patient realizes it, he has jumped aboard the itch-scratch-itch train—a vicious cycle that can be hard to break.
The skin reacts to all this attention by thickening and becoming rough; on a microscopic level, the nerves become more sensitive. The urge to scratch becomes so irresistible that some patients fantasize about giving in.
The first goal of treating LSC is to stop the itching by helping the patient avoid contact with the site. High-potency topical steroids can be effective for this, especially if used under occlusion, which not only potentiates the steroid but also provides a barrier to protect the area from the patient’s fingernails (or hairbrush). Once the condition improves, steroids can be slowly withdrawn.
A biopsy, if it had been necessary, would have shown hypertrophic epidermis and acanthotic changes.
TAKE-HOME LEARNING POINTS
• Lichen simplex chronicus (LSC) is secondary to an inciting condition such as eczema, xerosis, psoriasis, or Darier disease.
• The manifestation of the itch-scratch-itch cycle leads to thickening and lichenification of the skin. The itch of LSC is so compelling that scratching is hard to resist.
• Despite treatment and patient education, recurrences are quite common.
This 49-year-old man was first diagnosed with Darier disease at age 8. It has been contained, for the most part, by topical medications and acitretin. But the itching on his legs is especially severe, making it difficult for him to leave them alone.
EXAMINATION
Both legs have heavy scaling and lichenification from the lateral aspects of the knees down to the ankles. There is modest erythema but no suppurative signs. Other areas of scaling are seen on his chest and back, but these are less coarse and rough than the leg rash.
What is the diagnosis?
DISCUSSION
Lichen simplex chronicus (LSC) is the term given to this phenomenon. LSC is always secondary to a primary trigger, which can be dry skin, eczema, psoriasis, or even a bug bite. Excessive scratching induces more itching, and before the patient realizes it, he has jumped aboard the itch-scratch-itch train—a vicious cycle that can be hard to break.
The skin reacts to all this attention by thickening and becoming rough; on a microscopic level, the nerves become more sensitive. The urge to scratch becomes so irresistible that some patients fantasize about giving in.
The first goal of treating LSC is to stop the itching by helping the patient avoid contact with the site. High-potency topical steroids can be effective for this, especially if used under occlusion, which not only potentiates the steroid but also provides a barrier to protect the area from the patient’s fingernails (or hairbrush). Once the condition improves, steroids can be slowly withdrawn.
A biopsy, if it had been necessary, would have shown hypertrophic epidermis and acanthotic changes.
TAKE-HOME LEARNING POINTS
• Lichen simplex chronicus (LSC) is secondary to an inciting condition such as eczema, xerosis, psoriasis, or Darier disease.
• The manifestation of the itch-scratch-itch cycle leads to thickening and lichenification of the skin. The itch of LSC is so compelling that scratching is hard to resist.
• Despite treatment and patient education, recurrences are quite common.
The Paradox of Pain Management
Pain was introduced as the “fifth vital sign” in the 1990s, ranking it as important a measure as blood pressure, heart and respiratory rate, and temperature.1 The American Pain Society promoted this notion to increase awareness of pain treatment among health care professionals. Emphasizing its importance, the Veterans Health Administration in 1999 launched the “Pain as the 5th Vital Sign” initiative, which mandated a pain intensity rating at all clinical encounters.2
Interestingly, the Joint Commission standards never stated that pain needed to be treated as a vital sign. But many organizations started to require documentation of routine pain screening for all patients. Health care providers were instructed to inquire about pain and to treat it as an essential element of health history.
These changes were quite controversial. The additional measure, while important, competed with other priority screening needs, including diabetes, cancer, and hypertension. There was—and continues to be—quite the debate on whether pain actually can be measured and what impact that information has on the quality of care.
I do not intend to enter that debate here. Instead, I want to discuss what continues to be a conundrum for me: the paradox of pain management.
For many patients, especially those in acute or emergency care settings, the presenting complaint is pain. I would submit that for many the expectation is for pain to be immediately and permanently relieved. But is this a realistic goal?
I recall a lecture on pain management I attended years ago; at that time, the approach involved early identification and prompt, aggressive treatment. When asked “How much medication and for how long?” the lecturer used diabetes as a treatment model, stating, “You would increase insulin until the blood glucose was controlled—don’t be afraid to increase pain medication until the pain is controlled.” In the early days of pain management, that was the accepted norm. The possibility that a “zero” on the pain scale was unattainable for some patients was not considered.
Yet seemingly overnight, once pain was decreed a vital sign, health care providers were mandated to measure it and faced with the responsibility to treat it. This resulted in a vague 0-1
Faced with growing concern for undertreated pain in the US, however, many of us strove to achieve a balance of sufficient yet appropriate treatment. We struggled to determine how to relieve the pain our patients experienced without creating other problems, such as undesirable side effects, misuse, or addiction. That predicament, paired with the ever-increasing direct-to-consumer advertisements about pain relief and the insistence by (some, not all) patients that nonnarcotic pain medication is ineffective, bred the crisis of opioid overuse and addiction we now face.
But just as I chose not to debate the impact of pain measurement on quality of care, I also choose not to debate the existence of the opioid crisis. What I want to emphasize is that all policy changes have consequences. I reach out to you, my colleagues, for innovative ideas to strike the delicate balance of appropriate use of narcotics. How do we address the needs of patients whose pain is more than just an inconvenience and for whom daily use of a narcotic allows them to function—while also avoiding the pitfalls that we are now regularly warned about?
I have no doubt that each of us knows at least one person—a patient, a family member, a neighbor—for whom pain is a daily occurrence. But we must put that in perspective; not all pain is a barrier to physical and emotional functioning. Data suggest that a “33% to 50% decrease in pain intensity is meaningful from a patient’s perspective and represents a reasonable standard of intervention efficacy.”3 For those who deal with chronic pain, even a slight improvement is progress.
So, while the American Medical Association and the American Pain Society bicker about whether pain is the “fifth vital sign,” we must find a better means to resolve the discord in our society.4 Banning all opioid use is not the answer, but neither is considering narcotics the default treatment for pain.
We must remind our patients, our policymakers, and ourselves that identifying and assessing pain is not equated with writing an opioid or narcotic prescription. Nor will removing those medications from our formulary mitigate the crisis. We need to communicate a clear, consistent message that pain is real, that some pain is a fact of life, and that we will help our patients.
However, it is incumbent upon us to adopt a systematic yet personalized plan of care that is effective, cost conscious, culturally and developmentally appropriate, and safe—and that plan may or may not include prescribing narcotics. We have much work ahead of us in order to minimize the potential for misuse of these medications without impeding patients’ access to necessary health care.
Please share your thoughts on this conundrum by writing to [email protected].
1. Veterans Health Administration. Pain as the 5th vital sign toolkit. www.va.gov/PAINMAN AGEMENT/docs/Pain_As_the_5th_Vital_Sign_Toolkit.pdf. Accessed October 5, 2016.
2. Mularski RA, White-Chu F, Overbay D, et al. Measuring pain as the 5th vital sign does not improve quality of pain management. J Gen Intern Med . 2006;21(6):607-612.
3. Gordon DB, Dahl JL, Miaskowski C, et al. American Pain Society recommendations for improving the quality of acute and cancer pain management. Arch Intern Med . 2005; 165(14):1574-1580.
4. Anson P. AMA drops pain as a vital sign . Pain News Network. June 16, 2016. www.painnewsnetwork.org/stories/2016/6/16/ama-drops-pain-as-vital-sign. Accessed October 5, 2016.
Marie-Eileen Onieal, PhD, CPNP, FAANP

Marie-Eileen Onieal, PhD, CPNP, FAANP
Marie-Eileen Onieal, PhD, CPNP, FAANP
Pain was introduced as the “fifth vital sign” in the 1990s, ranking it as important a measure as blood pressure, heart and respiratory rate, and temperature.1 The American Pain Society promoted this notion to increase awareness of pain treatment among health care professionals. Emphasizing its importance, the Veterans Health Administration in 1999 launched the “Pain as the 5th Vital Sign” initiative, which mandated a pain intensity rating at all clinical encounters.2
Interestingly, the Joint Commission standards never stated that pain needed to be treated as a vital sign. But many organizations started to require documentation of routine pain screening for all patients. Health care providers were instructed to inquire about pain and to treat it as an essential element of health history.
These changes were quite controversial. The additional measure, while important, competed with other priority screening needs, including diabetes, cancer, and hypertension. There was—and continues to be—quite the debate on whether pain actually can be measured and what impact that information has on the quality of care.
I do not intend to enter that debate here. Instead, I want to discuss what continues to be a conundrum for me: the paradox of pain management.
For many patients, especially those in acute or emergency care settings, the presenting complaint is pain. I would submit that for many the expectation is for pain to be immediately and permanently relieved. But is this a realistic goal?
I recall a lecture on pain management I attended years ago; at that time, the approach involved early identification and prompt, aggressive treatment. When asked “How much medication and for how long?” the lecturer used diabetes as a treatment model, stating, “You would increase insulin until the blood glucose was controlled—don’t be afraid to increase pain medication until the pain is controlled.” In the early days of pain management, that was the accepted norm. The possibility that a “zero” on the pain scale was unattainable for some patients was not considered.
Yet seemingly overnight, once pain was decreed a vital sign, health care providers were mandated to measure it and faced with the responsibility to treat it. This resulted in a vague 0-1
Faced with growing concern for undertreated pain in the US, however, many of us strove to achieve a balance of sufficient yet appropriate treatment. We struggled to determine how to relieve the pain our patients experienced without creating other problems, such as undesirable side effects, misuse, or addiction. That predicament, paired with the ever-increasing direct-to-consumer advertisements about pain relief and the insistence by (some, not all) patients that nonnarcotic pain medication is ineffective, bred the crisis of opioid overuse and addiction we now face.
But just as I chose not to debate the impact of pain measurement on quality of care, I also choose not to debate the existence of the opioid crisis. What I want to emphasize is that all policy changes have consequences. I reach out to you, my colleagues, for innovative ideas to strike the delicate balance of appropriate use of narcotics. How do we address the needs of patients whose pain is more than just an inconvenience and for whom daily use of a narcotic allows them to function—while also avoiding the pitfalls that we are now regularly warned about?
I have no doubt that each of us knows at least one person—a patient, a family member, a neighbor—for whom pain is a daily occurrence. But we must put that in perspective; not all pain is a barrier to physical and emotional functioning. Data suggest that a “33% to 50% decrease in pain intensity is meaningful from a patient’s perspective and represents a reasonable standard of intervention efficacy.”3 For those who deal with chronic pain, even a slight improvement is progress.
So, while the American Medical Association and the American Pain Society bicker about whether pain is the “fifth vital sign,” we must find a better means to resolve the discord in our society.4 Banning all opioid use is not the answer, but neither is considering narcotics the default treatment for pain.
We must remind our patients, our policymakers, and ourselves that identifying and assessing pain is not equated with writing an opioid or narcotic prescription. Nor will removing those medications from our formulary mitigate the crisis. We need to communicate a clear, consistent message that pain is real, that some pain is a fact of life, and that we will help our patients.
However, it is incumbent upon us to adopt a systematic yet personalized plan of care that is effective, cost conscious, culturally and developmentally appropriate, and safe—and that plan may or may not include prescribing narcotics. We have much work ahead of us in order to minimize the potential for misuse of these medications without impeding patients’ access to necessary health care.
Please share your thoughts on this conundrum by writing to [email protected].
Pain was introduced as the “fifth vital sign” in the 1990s, ranking it as important a measure as blood pressure, heart and respiratory rate, and temperature.1 The American Pain Society promoted this notion to increase awareness of pain treatment among health care professionals. Emphasizing its importance, the Veterans Health Administration in 1999 launched the “Pain as the 5th Vital Sign” initiative, which mandated a pain intensity rating at all clinical encounters.2
Interestingly, the Joint Commission standards never stated that pain needed to be treated as a vital sign. But many organizations started to require documentation of routine pain screening for all patients. Health care providers were instructed to inquire about pain and to treat it as an essential element of health history.
These changes were quite controversial. The additional measure, while important, competed with other priority screening needs, including diabetes, cancer, and hypertension. There was—and continues to be—quite the debate on whether pain actually can be measured and what impact that information has on the quality of care.
I do not intend to enter that debate here. Instead, I want to discuss what continues to be a conundrum for me: the paradox of pain management.
For many patients, especially those in acute or emergency care settings, the presenting complaint is pain. I would submit that for many the expectation is for pain to be immediately and permanently relieved. But is this a realistic goal?
I recall a lecture on pain management I attended years ago; at that time, the approach involved early identification and prompt, aggressive treatment. When asked “How much medication and for how long?” the lecturer used diabetes as a treatment model, stating, “You would increase insulin until the blood glucose was controlled—don’t be afraid to increase pain medication until the pain is controlled.” In the early days of pain management, that was the accepted norm. The possibility that a “zero” on the pain scale was unattainable for some patients was not considered.
Yet seemingly overnight, once pain was decreed a vital sign, health care providers were mandated to measure it and faced with the responsibility to treat it. This resulted in a vague 0-1
Faced with growing concern for undertreated pain in the US, however, many of us strove to achieve a balance of sufficient yet appropriate treatment. We struggled to determine how to relieve the pain our patients experienced without creating other problems, such as undesirable side effects, misuse, or addiction. That predicament, paired with the ever-increasing direct-to-consumer advertisements about pain relief and the insistence by (some, not all) patients that nonnarcotic pain medication is ineffective, bred the crisis of opioid overuse and addiction we now face.
But just as I chose not to debate the impact of pain measurement on quality of care, I also choose not to debate the existence of the opioid crisis. What I want to emphasize is that all policy changes have consequences. I reach out to you, my colleagues, for innovative ideas to strike the delicate balance of appropriate use of narcotics. How do we address the needs of patients whose pain is more than just an inconvenience and for whom daily use of a narcotic allows them to function—while also avoiding the pitfalls that we are now regularly warned about?
I have no doubt that each of us knows at least one person—a patient, a family member, a neighbor—for whom pain is a daily occurrence. But we must put that in perspective; not all pain is a barrier to physical and emotional functioning. Data suggest that a “33% to 50% decrease in pain intensity is meaningful from a patient’s perspective and represents a reasonable standard of intervention efficacy.”3 For those who deal with chronic pain, even a slight improvement is progress.
So, while the American Medical Association and the American Pain Society bicker about whether pain is the “fifth vital sign,” we must find a better means to resolve the discord in our society.4 Banning all opioid use is not the answer, but neither is considering narcotics the default treatment for pain.
We must remind our patients, our policymakers, and ourselves that identifying and assessing pain is not equated with writing an opioid or narcotic prescription. Nor will removing those medications from our formulary mitigate the crisis. We need to communicate a clear, consistent message that pain is real, that some pain is a fact of life, and that we will help our patients.
However, it is incumbent upon us to adopt a systematic yet personalized plan of care that is effective, cost conscious, culturally and developmentally appropriate, and safe—and that plan may or may not include prescribing narcotics. We have much work ahead of us in order to minimize the potential for misuse of these medications without impeding patients’ access to necessary health care.
Please share your thoughts on this conundrum by writing to [email protected].
1. Veterans Health Administration. Pain as the 5th vital sign toolkit. www.va.gov/PAINMAN AGEMENT/docs/Pain_As_the_5th_Vital_Sign_Toolkit.pdf. Accessed October 5, 2016.
2. Mularski RA, White-Chu F, Overbay D, et al. Measuring pain as the 5th vital sign does not improve quality of pain management. J Gen Intern Med . 2006;21(6):607-612.
3. Gordon DB, Dahl JL, Miaskowski C, et al. American Pain Society recommendations for improving the quality of acute and cancer pain management. Arch Intern Med . 2005; 165(14):1574-1580.
4. Anson P. AMA drops pain as a vital sign . Pain News Network. June 16, 2016. www.painnewsnetwork.org/stories/2016/6/16/ama-drops-pain-as-vital-sign. Accessed October 5, 2016.
1. Veterans Health Administration. Pain as the 5th vital sign toolkit. www.va.gov/PAINMAN AGEMENT/docs/Pain_As_the_5th_Vital_Sign_Toolkit.pdf. Accessed October 5, 2016.
2. Mularski RA, White-Chu F, Overbay D, et al. Measuring pain as the 5th vital sign does not improve quality of pain management. J Gen Intern Med . 2006;21(6):607-612.
3. Gordon DB, Dahl JL, Miaskowski C, et al. American Pain Society recommendations for improving the quality of acute and cancer pain management. Arch Intern Med . 2005; 165(14):1574-1580.
4. Anson P. AMA drops pain as a vital sign . Pain News Network. June 16, 2016. www.painnewsnetwork.org/stories/2016/6/16/ama-drops-pain-as-vital-sign. Accessed October 5, 2016.