User login
IPV boost after initial OPV offers sustained protection to at least 11 months
Protection against the poliovirus is lower at 1 month but remains sustained at 6 and 11 months after an inactivated poliovirus vaccine (IPV) boost following initial oral poliovirus vaccination (OPV), according to Jacob John, MD, of Christian Medical College, Vellore, Tamil Nadu, India, and his associates.
In a randomized controlled trial from Nov. 4 and Dec. 17, 2014, 900 healthy children from ages 1 to 4 years were randomly assigned between three study groups. The groups had the children receive IPV boost at 5 months (arm A), at enrollment (arm B), or no vaccine (arm C). Poliovirus shedding in stool 7 days after challenge, determined by Fisher’s exact test, was significantly lower in arms A and B, compared with C (risk ratio, 0.68; P = .003, RR, 0.70; P = .006 for arm A vs. C and B vs. C, respectively). The reduction in shedding was more marked for serotype 3 (RR, 0.60; P = .004, RR, 0.54; P = .001 respectively) than for serotype 1 (RR, 0.72; P = .057, RR, 0.80; P = .215, respectively).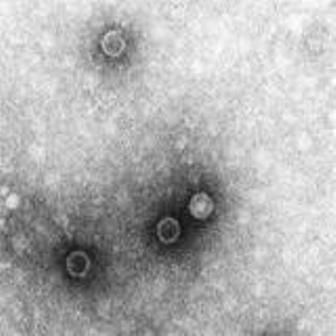
It was noted that 41 serious adverse events (11 in arm A, 17 in arm B, and 13 in arm C), including 2 deaths in arm A, were reported during the trial. However, the reported adverse events were classified as unrelated, and the deaths were from leukemia and from viral hemorrhagic fever.
“The boost to intestinal immunity against poliovirus that results from administration of IPV to OPV-vaccinated children is sustained at 6 and 11 months. It is clear that IPV is playing an increasingly important role in the polio endgame as the world transitions away from the use of OPV,” the researchers concluded. “Every effort needs to be made to ensure supply of this vaccine to meet this expanding role.”
Find the full study in the Journal of Infectious Diseases 2016. doi: 10.1093/infdis/jiw595.
Protection against the poliovirus is lower at 1 month but remains sustained at 6 and 11 months after an inactivated poliovirus vaccine (IPV) boost following initial oral poliovirus vaccination (OPV), according to Jacob John, MD, of Christian Medical College, Vellore, Tamil Nadu, India, and his associates.
In a randomized controlled trial from Nov. 4 and Dec. 17, 2014, 900 healthy children from ages 1 to 4 years were randomly assigned between three study groups. The groups had the children receive IPV boost at 5 months (arm A), at enrollment (arm B), or no vaccine (arm C). Poliovirus shedding in stool 7 days after challenge, determined by Fisher’s exact test, was significantly lower in arms A and B, compared with C (risk ratio, 0.68; P = .003, RR, 0.70; P = .006 for arm A vs. C and B vs. C, respectively). The reduction in shedding was more marked for serotype 3 (RR, 0.60; P = .004, RR, 0.54; P = .001 respectively) than for serotype 1 (RR, 0.72; P = .057, RR, 0.80; P = .215, respectively).
It was noted that 41 serious adverse events (11 in arm A, 17 in arm B, and 13 in arm C), including 2 deaths in arm A, were reported during the trial. However, the reported adverse events were classified as unrelated, and the deaths were from leukemia and from viral hemorrhagic fever.
“The boost to intestinal immunity against poliovirus that results from administration of IPV to OPV-vaccinated children is sustained at 6 and 11 months. It is clear that IPV is playing an increasingly important role in the polio endgame as the world transitions away from the use of OPV,” the researchers concluded. “Every effort needs to be made to ensure supply of this vaccine to meet this expanding role.”
Find the full study in the Journal of Infectious Diseases 2016. doi: 10.1093/infdis/jiw595.
Protection against the poliovirus is lower at 1 month but remains sustained at 6 and 11 months after an inactivated poliovirus vaccine (IPV) boost following initial oral poliovirus vaccination (OPV), according to Jacob John, MD, of Christian Medical College, Vellore, Tamil Nadu, India, and his associates.
In a randomized controlled trial from Nov. 4 and Dec. 17, 2014, 900 healthy children from ages 1 to 4 years were randomly assigned between three study groups. The groups had the children receive IPV boost at 5 months (arm A), at enrollment (arm B), or no vaccine (arm C). Poliovirus shedding in stool 7 days after challenge, determined by Fisher’s exact test, was significantly lower in arms A and B, compared with C (risk ratio, 0.68; P = .003, RR, 0.70; P = .006 for arm A vs. C and B vs. C, respectively). The reduction in shedding was more marked for serotype 3 (RR, 0.60; P = .004, RR, 0.54; P = .001 respectively) than for serotype 1 (RR, 0.72; P = .057, RR, 0.80; P = .215, respectively).
It was noted that 41 serious adverse events (11 in arm A, 17 in arm B, and 13 in arm C), including 2 deaths in arm A, were reported during the trial. However, the reported adverse events were classified as unrelated, and the deaths were from leukemia and from viral hemorrhagic fever.
“The boost to intestinal immunity against poliovirus that results from administration of IPV to OPV-vaccinated children is sustained at 6 and 11 months. It is clear that IPV is playing an increasingly important role in the polio endgame as the world transitions away from the use of OPV,” the researchers concluded. “Every effort needs to be made to ensure supply of this vaccine to meet this expanding role.”
Find the full study in the Journal of Infectious Diseases 2016. doi: 10.1093/infdis/jiw595.
FROM THE JOURNAL OF INFECTIOUS DISEASES
Leadership Initiatives in Patient-Centered Transgender Care
Patient-centered care is of fundamental importance when caring for the transgender population due to the well-established history of social stigma and systemic discrimination. Therefore, nursing education is mandated to equip graduates with culturally competent patient-centered care skills.1 In 2009, the Institute of Medicine (IOM) in partnership with the Robert Wood Johnson Foundation (RWJF) launched The Future of Nursing initiative, which outlined the major role nursing should play in transforming the health care system to meet the health care needs of diverse U.S. populations.
The initiative produced a blueprint of action-focused institutional recommendations at the local, state, and national levels that would facilitate the reforms necessary to transform the U.S. health care system. One of the recommendations of the IOM report was to increase opportunities for nurses to manage and lead collaborative efforts with physicians and other health care team members in the areas of systems redesign and research, to improve practice environments and health systems.2
The VHA is the largest integrated health care system in the U.S., serving more than 8.76 million veterans at more than 1,700 facilities. The VHA has an organizational structure that uses centralized control in Washington, DC, and branches out to 18 regional networks that are divided into local facilities in 50 states, the District of Columbia, Puerto Rico, the U.S. Virgin Islands, Guam, American Samoa, and the Philippines. This type of structure is known for promoting efficient standardization of processes and procedures across an organization.3
The VHA Blueprint for Excellence envisions the promotion of a positive culture of service and the advancement of health care innovations necessary to create an environment that all veterans deserve.4 To that end, the VHA can be a promising health care institution through which patient-centered initiatives can be standardized, promulgated nationally, and replicated as a model for the country and international health systems. However, it is important to note that the bureaucratic organizational structure of the VHA's national integrated system of care is based on a systemwide standardization effort.5 Therefore, more time may be required to implement organizational changes.
Transgender populations face significant social stigmatization, discrimination, and marginalization that contribute to negative patient outcomes. Consequently, this population experiences high rates of suicide, HIV/AIDS, substance use disorder, poverty, and homelessness.6 Due to the growing evidence of health disparities and negative health outcomes affecting transgender populations, the federal government has identified transgender patient care and outcomes as a major health concern and priority in the Healthy People Initiative 2020.2,7,8
In 2012, the VHA issued a directive mandating services for transgender veterans.9 Nevertheless, health care staff significantly lack the knowledge, skills, and cultural competencies that are vital in transgender care.
This article reviews the prevalence and demographics of the transgender population, social challenges, global health concerns, and public health policies. The article also examines how the doctor of nursing practice (DNP)-prepared nurse leader can provide transformational nursing leadership to facilitate culturally competent, patient-centered initiatives to improve access and services for transgender individuals in the VHA and provide a model for change in transgender population health.
Definitions
Gender is a behavioral, cultural, or psychological trait assigned by society that is associated with male or female sex. Sex denotes the biologic differences between males and females. Transgender is an umbrella term used to describe people whose gender identity or gender expression is different from that of their sex assigned at birth. Transsexualism is a subset of transgender persons who have taken steps to self-identify or transition to look like their preferred gender.
Demographics
Estimates of the prevalence of transgenderism are roughly drawn from less rigorous methods, such as the combination of parents who report transgenderism in children, the number of adults reportedly seeking clinical care (such as cross-sex or gender-affirming hormone therapy), and the number of surgical interventions reported in different countries.10 A meta-analysis of 21 studies concluded that the ratio of transsexuals (individuals who are altering or have already altered their birth sex) was predominantly 1:14,705 adult males and 1:38,461 adult females.11 Since all transgender persons do not identify as transsexual, these figures do not provide a precise estimation of the number of transgender persons worldwide.
About 700,000, or 0.3%, of the adult population in the U.S. identify themselves as transgender, and an estimated 134,300 identify as transgender veterans.6,12 The transgender population in the U.S. is estimated to be 55% white, 16% African American, 21% Hispanic, and 8% other races.13 The U.S. census data noted that the transgender population was geographically located across the nation. Transgender persons are more likely to be single, never married, divorced, and more educated but with significantly less household income.2 Data to provide an accurate reflection of the number of transgender people in the U.S. are lacking. Some transgender individuals also may identify as lesbian, gay, or bisexual, making population-based estimation even more challenging and difficult.
Transgender persons who have transitioned may not have changed their names or changed their identified sex on official Social Security records, which the Social Security Administration allows only if there is evidence that genital sexual reassignment surgery was performed.14 The number of transgender adults requesting treatment continues to rise.10
Social and Health Challenges
Transgender people face many challenges because of their gender identity. Surveys assessing the living conditions of transgender people have found that 43% to 60% report high levels of physical violence.15 By comparison, the National Intimate Partner and Sexual Violence Survey found that interpersonal violence and sexual violence were reported by lesbian and gay individuals at equal or higher levels than that reported by heterosexuals. Forty-four percent of lesbian women, 35% of heterosexual women, 29% of heterosexual men, and 26% of gay men reported experiencing rape or physical violence.16 A study in Spain reported 59% of transgender people experienced patterns of harassment, and in Canada, 34% of transgender people lived below the poverty level.17,18
In the U.S., the National Transgender Discrimination Survey of 6,450 transgender and nonconforming participants provided extensive data on challenges experienced by transgender people.6 Discrimination was frequently experienced in accessing health care. Due to transgender status, 19% were denied care, and 28% postponed care due to perceived harassment and violence within a health care setting.6 The same study also reported that as many as 41% live in extreme poverty with incomes of less than $10,000 per year reported. Twenty-six percent were physically assaulted, and 10% experienced sexual violence. More than 25% of the transgender population misused drugs or alcohol to cope with mistreatment.6
In the U.S., HIV infection rates for transgender individuals were more than 4 times (2.64%) the rate of the general population (0.6%).6 Internationally, there is a high prevalence of HIV in transgender women. The prevalence rate of HIV in U.S. transgender women was 21.74% of the estimated U.S. adult transgender population of about 700,000.19 One in 4 people living with HIV in the U.S. are women.20
Suicide attempt rates are extremely high among transgender people. A suicide rate of 22% to 43% has been reported across Europe, Canada, and the U.S.21 Depression and anxiety were commonly noted as a result of discrimination and social stigma. In the U.S., transgender persons reported high rates of depression, with 41% reporting attempted suicide compared with 1.6% of the general population.6 Access to health care services, such as mental health, psychosocial support, and stress management are critical for this vulnerable population.22
Health Policies
Since 1994, the UK has instituted legal employment protections for the transgender population. In the UK, transgender persons, including military and prisoners, have health care coverage that includes sexual reassignment surgery as part of the UK's National Health Service.23
In the U.S., the federal policy of "Don't Ask, Don't Tell" barring transgender persons from serving openly in the military was repealed in June 2016. This policy historically has had a silencing effect on perpetuating institutionalized biases.24 This remains problematic even after veterans have transitioned from military service to the VA for civilian care.
Between 2006 and 2013, the reported prevalence and incidence of transgender-related diagnoses in the VA have steadily increased with 40% of new diagnoses occurring since 2011.25 In fiscal year 2013, there were 32.9 per 100,000 veterans with transgender-related diagnoses.25 Health care staff, in particular health care providers (HCPs), can play a critical role in reducing health disparities and unequal treatment.26
With the passage of the U.S. Affordable Care Act (ACA), health insurance coverage for transgender persons is now guaranteed by law, and health disparities within the transgender population can begin to be properly addressed. The ACA offers the ability to purchase health insurance, possibly qualify for Medicaid, or obtain subsidies to purchase health insurance. Insurance coverage is accessible without regard to discrimination or preexisting conditions.27 As of May 2014, the Medicare program covered medically necessary hormone therapy and sex reassignment surgery.13 While VA benefits cover hormone therapy for transgender veterans, sex reassignment surgery is not currently a covered benefit.28 The ACA now increases access to primary care, preventative care, mental health services, and community health programs not previously available in the transgender community.
Healthy People 2020 Goals
One of the Healthy People 2020 stated goals is to improve the health and wellness of transgender people.29 The objective is to increase the number of population-based data collection systems used to monitor transgender people from the baseline of 2 to a total of 4 by 2020. The data systems would be assigned to collect relevant data, such as mental health; HIV status; illicit drug, alcohol, and tobacco use; cervical and breast cancer screening; health insurance coverage; and access to health care.
Health Care Staff Readiness
Transgender persons face health care challenges with major health disparities due to their gender identity. Transgender persons as a defined population are not well understood by HCPs. In a survey, 50% of transgender respondents reported that they had to teach their medical provider about transgender care.6 Negative perceptions of transgender persons are well established and have contribute to the poor health care access and services that transgender persons receive. Transgender persons are often denied access to care, denied visitation rights, and are hesitant to share information for fear of bureaucratic exclusion or isolation.
There is a lack of evidence-based studies to guide care and help HCPs gain greater understanding of this population's unique needs.30 Additionally, a significant lack of knowledge, skills, cultural competence, and awareness exist in providing transgender care. Research on nursing attitudes concerning transgender care consistently found negative attitudes, and physicians also frequently reported witnessing derogatory comments and discriminatory care from colleagues.31,32 The study by Carabez and colleagues found that practicing nurses rarely received the proper education or training in transgender health issues, and many were unaware of the needs of this population.33 In addition, many HCPs were uncomfortable working with transgender patients. Physicians also expressed knowledge deficits on gender identity disorders due to a lack of training and ethical concerns about their roles in providing gender-transitioning treatment.26
Although the VHA directive states that transgender services and treatment should be standardized, the VHA has not approved, defined, or endorsed specific standards of care or clinical guidelines within the organization for transgender care, further heightening HCP concerns.9 The clinical practice guidelines available for addressing preventive care for transgender patients are primarily based on consensus of expert opinion.34 Expert opinion has produced the Standards of Care (SOC) for the Health of Transsexual, Transgender, and Gender Nonconforming People, published by the World Professional Association for Transgender Health (WPATH) and cited by the IOM as the major clinical practice guidelines for providing care to transgender individuals.2 Transgender care at the VHA is guided by the WPATH standards of care.35
The VHA has created national educational programs and policies with targeted goals to provide uniform, culturally competent, patient-centered care. Online transgender health presentations are available, and at least 15 VHA facilities have transgender support groups.30 While the VHA supports a patient-centered philosophy for transgender patient care, many facilities do not currently have organizational initiatives that enhance clinical preparation of HCPs or have sufficiently modified the environment to better accommodate the health care needs of transgender veterans.
DNP Preparation
The DNP terminal degree provides nurses with doctoral-level training in organizational and systems leadership, leading quality improvement, and implementing systemwide initiatives by using scientific findings to drive processes that improve quality of care for a changing patient population.36 Preparation in research analysis of evidence-based interventions also is essential to evaluating practice patterns, patient outcomes, and systems of care that can identify gaps in practice. Training in health care policy and advocacy, information systems, patient care technology, and population health also is provided so that DNPs are competent to develop system strategies to transform health care through clinical prevention and health promotion.
QSEN Framework
In keeping with the IOM's Future of Nursing initiative recommendations that graduate nurses be prepared as leaders in education, practice, administration, and research, there is an increasing focus on providing graduate-level nursing education and training to ensure quality and efficiency of health outcomes.37 The Quality and Safety Education in Nursing (QSEN) project, initiated at the RWJF by Linda Cronenwett, PhD, RN, identifies a framework for knowledge, skills, and attitudes that defines the competencies that nurses need to deliver effective care to improve quality and safety within health care systems.38 These core competencies include quality improvement, safety, teamwork and collaboration, patient-centered care, evidence-based practice, and informatics. The RWJF and the American Association of Colleges of Nursing later expanded the project initiative to prepare nursing faculty to teach the QSEN competencies in graduate nursing programs.36
The DNP nurse leader is ideally suited to manage this project by applying competencies from the QSEN framework. Using open communication and mutual respect, the nurse leader is poised to effectively develop interprofessional teams to collaborate and initiate transformational changes that improve quality and patient-centered care delivered within the health care organization.
Public Health Resources
Public health resources addressing transgender patient care advocacy, public policy, community education, standards of care, cultural competency, mental health, hormone therapy, surgical interventions, reproductive health, primary care, preventative care, and research are available. For example, WPATH is an international multidisciplinary organization that has published comprehensive SOC for transgender, transsexual, and gender-nonconforming people. The seventh version of the SOC contains evidence-based guidelines for treatment.39 Additional online resources for transgender health are available from the CDC, the Center of Excellence for Transgender Health at the University of California, San Francisco; Department of Family and Community Medicine; and the National Center for Transgender Equality.13,40,41
Patient-Centered Transgender Care
The QSEN framework outlines competencies that provide applicable solutions that help prepare organizations to deliver culturally competent, patient-centered transgender care. The first step to creating patient-centered transgender care is to "analyze factors that create barriers to patient-centered care."42 The magnitude of the barriers to providing patient-centered transgender care also must be identified and understood. An assessment of individual values, beliefs, and attitudes can help to identify cultural characteristics and eliminate stereotypes that impact health practices.43
The nurse leader should solicit support from stakeholders to assess barriers to providing patient-centered transgender care at the system level. Stakeholders would include staff directly involved in patient care, such as physicians, nurse practitioners, physician assistants, registered nurses, nurse managers, nurse educators, licensed practical nurses, medical support assistants, psychologists, dieticians, and social workers. Other ancillary stakeholders with an interest in creating a patient-centered environment with positive patient outcomes include the executive leadership team of the organization, which consists of the chief of staff, director, administrative officers, and nurse executive.
The nurse leader should consult with experts in transgender care and present evidence-based research showing how deficits in staff knowledge, skills, and cultural competence negatively impact the quality of care provided to transgender persons. National data on the consequential health disparities and negative impacts on patient outcomes also should be discussed and presented to all stakeholders. The nurse leader in collaboration with the VA Office of Research and Development is ideally suited to obtain institutional review board approval of a proposal to conduct a needs assessment survey of health care staff barriers to providing patient-centered transgender care. Thereafter, the nurse leader would analyze, extract, and synthesize the data and evaluate the resources and technology available to translate this research knowledge into a clinical practice setting at the system level.44
The second solution uses the results of the survey to develop staff competency training within the organization. The nurse leader can facilitate collaboration and team building to develop practice guidelines and SOC. Competency training will prepare the staff to assist in developing strategies to improve the quality of care for transgender persons. Educationconcerning existing evidence-based clinical guidelines and SOC as well as anecdotal evidence of the needs of transgender patients should be included in competency training.45 One approach to competency training would be to trainintegrated multidisciplinary teams with expertise in transgender care to promote wellness and disease prevention.9 The nurse leader should collaborate with multiple disciplines to facilitate the development of interdisciplinary teams from nursing, medicine, social work, pharmacy, primary care, mental health, women's health, and endocrinology to participate in the Specialty Care Access Network Extension of Community Healthcare outcomes (SCAN-ECHO) training. Training can be offered by videoconferencing over several months and provides cost-effective, efficient training of providers in patient-centered transgender care.46,47 After the SCAN-ECHO program is completed, trained nursing experts could then develop a cultural sensitivity training program for nursing organizations to be offered to educate health care staff on an annual basis.
The third solution addresses the QSEN competency to "Analyze institutional features of the facilities that support or pose barriers to patient-centered care."42 Many veterans do not perceive VA environments as welcoming. In a study by Sherman and colleagues, less than one-third of veterans believed the VA environment was welcoming to sexual or gender minorities, and sexual orientation or gender identity was disclosed by only about 25% of veterans.48 Many veterans in this study felt uncomfortable disclosing their gender or sexual orientation. The majority felt that providers should not routinely ask about sexual orientation or gender identity, and 24% said they were very or somewhat uncomfortable discussing the issue. In another study, 202 VA providers were asked if they viewed the VA as welcoming, and 32% said the VA was somewhat or very unwelcoming.48
The nurse leader is trained in the essentials of health care policy advocacy, which is central to nursing practice.49 Nursing as a profession values social justice and equality, which are linked to fewer health disparities and more stable health indicators.50 Therefore, nursing can ideally provide organizational leaders by developing a culture wherein stable, patient-centered relationships can develop and thrive.
Organizational Culture
Strategies must be deployed to create an organizational culture that is welcoming, respectful, and supportive of transgender patients and family preferences. VA should develop support groups for transgender veterans in VA facilities. Support groups are helpful in diminishing stress, improving self-esteem, building confidence, and improving social relationships.51 Additionally, VA should develop community-based partnerships with other organizations that already provide institutional care and support from HCPs who support transgender persons' right to self-determination.52 These partnerships can foster environmental influences over time and lead to the development of trusting relationships between transgender veterans and the VA organization.
Another community partnership of importance for the nurse leader to develop is an alliance with local universities to train nursing students in cultural competencies in transgender care at VA facilities. The U.S. population continues to diversify in race and ethnicity and cultural influences; therefore, nurses must be prepared in cultural competencies in order to provide quality care that reduces health disparities.53
Under federal law, the VHA has a data sharing agreement with the DoD. Despite the repeal of the "Don't Ask, Don't Tell" federal law, which cleared the way for transgender persons to openly serve in the military, many transgender persons may remain fearful of reprisals, such as judgment, denial of care, or loss of benefits if gender identity is disclosed.54 Given the bureaucratic structure of the VHA, the implementation of cultural changes at the system level will require a collaborative effort between multidisciplinary teams and community partnerships to transform the VA environment over time. The authors believe that on this issue, external forces must guide and lead changes within the VA system in order to develop sustainable and trusting relationships with transgender veterans.
The fourth solution is implementation of policies that "empower patients or families in all aspects of the health care process."42 Again, the nurse leader is trained and prepared to advocate for a policy that implements a Patient Bill of Rights that explicitly guarantees health care and prohibits discrimination of gender-minority veterans. This change would foster trust and confidence from transgender individuals. A study found that 83% of providers and 83% of lesbian, gay, bisexual, and transgender veterans believe that this policy change would make the VHA environment more welcoming.48 Providing transgender-affirming materials and language on standard forms also would eliminate barriers, promote patient-centered care, and empower transgender patients by creating an environment that is more inclusive of everyone.48
Conclusion
The nurse leader is well positioned to implement the QSEN framework to integrate research, practice, and policy to create a more inclusive, patient-centered health care system for transgender veterans. By using the essential principles of doctoral education for advanced nursing practice, the nurse leader is prepared to advocate for changing the organization at the systems level. The nurse leader also is equipped to direct the implementation of patient-centered transgender care initiatives by ensuring the integration of the nursing organization as a partner in strategic planning as well as the development of solutions.
The VHA Blueprint of Excellence envisions organization and collaboration to promote new relationships that serve and benefit veterans. The DNP preparation allows the nurse leader to demonstrate the ability to collaborate with VHA stakeholders and develop alliances within and outside the organization by advocating for policy changes that will be transformational in improving health care delivery and patient outcomes to vulnerable transgender veteran populations. The IOM has tasked nurse executives with creating a health care infrastructure of doctorally prepared nurses to provide patient care that is increasingly growing more complex. With an increasing number of veterans using services, VHA has prioritized an expansion in the number of doctorally prepared nurses.55
As the largest integrated health care system in the U.S., the VHA provides an ideal setting for initiating these organizational changes as a result of having developed an integrated infrastructure to collect evidence-based data at the regional (network) and state facilities and make comparisons with national benchmarks. Therefore, changes are less difficult to disseminate throughout the hierarchy of the VHA. Consequently, the VHA has been a leader in the U.S. for equity in the health care arena and provides a model for international health care systems. Finally, these changes address an urgent need to reduce health disparities, morbidity, and mortality by improving quality care and health care delivery to a vulnerable transgender population.
1. Greiner AC, Knebel E, eds. Health Professions Education: A Bridge to Quality. Washington, DC: National Academies Press; 2003.
2. Institute of Medicine. Bisexual, and Transgender People: Building a Foundation for Better Understanding. Washington, DC: National Academy of Sciences; 2011.
3. Mintzberg H. The structuring of organizations: a synthesis of the research. https://papers.ssrn.com/sol3/papers.cfm?abstract_id=1496182 1979. Posted November 4, 2009. Accessed November 30, 2016.
4. U.S. Department of Veterans Affairs. VHA blue print for excellence. https://www.va.gov/health/docs/VHA_Blueprint_for_Excellence.pdf. Published September 21, 2014. Accessed November 30, 2016.
5. Morgan RO, Teal CR, Reddy SG, Ford ME, Ashton CM. Measurement in Veterans Affairs Health Services Research: veterans as a special population. Health Serv Res. 2005;40(5, part 2):1573-1583.
6. Grant JM, Mottet L, Tanis JE, Harrison J, Herman J, Keisling M. Injustice at every turn: a report of the National Transgender Discrimination Survey. http://www.thetaskforce.org/static_html/downloads /reports/reports/ntds_full.pdf. Published 2011. Accessed November 30, 2016.
7. Office of Disease Prevention and Health Promotion. Lesbian, gay, bisexual, and transgender health. http://www.healthypeople.gov/2020/topics-objec tives/topic/lesbian-gay-bisexual-and-transgender -health. Updated November 16, 2016. Accessed November 16, 2016.
8. Institute of Medicine Committee on Lesbian Gay, Bisexual, and Transgender Health Issues and Research Gaps and Opportunities. The Health of Lesbian, Gay, Bisexual, and Transgender People: Building a Foundation for Better Understanding. Washington, DC: National Academy of Sciences; 2011.
9. U.S. Department of Veterans Affairs. VHA Directive 2013-003: Providing Health Care for Transgender and Intersex Veterans. Washington, DC: U.S. Department of Veterans Affairs; 2013.
10. Zucker KJ, Lawrence AA. Epidemiology of gender identity disorder: recommendations for the Standards of Care of the World Professional Association for Transgender Health. Int J Transgenderism. 2009;11(1):8-18.
11. Arcelus J, Bouman WP, Van Den Noortgate W, Claes L, Witcomb G, Fernandez-Aranda F. Systematic review and meta-analysis of prevalence studies in transsexualism. Eur Psychiatry. 2015;30(6):807-815.
12. Gates GJ, Herman JL. Transgender military service in the United States. http://williamsinstitute.law.ucla.edu/wp-content/uploads/Transgender-Military -Service-May-2014.pdf. Published May 2014. Accessed November 30, 2016.
13. Flores AR, Brown TNT, and Herman JL. Race and ethnicity of adults who identify as transgender in the United States. http://williamsinstitute.law.ucla .edu/wp-content/uploads/Race-and-Ethnicity-of -Transgender-Identified-Adults-in-the-US.pdf. Published October 2016. Accessed December 13, 2016.
14. Harris BC. Likely Transgender individuals in US federal administrative records and the 2010 census. https://www.census.gov/srd/carra/15_03_Likely_Transgender_Individuals_in_ARs_and_2010Census.pdf. Published May 4, 2015. Accessed November 30, 2016.
15. Kenagy GP, Bostwick WB. Health and social service needs of transgender people in Chicago. Int J Transgenderism. 2005;8(2-3):57-66.
16. Centers for Disease Control and Prevention. National intimate partner and sexual violence survey, 2010 summary report. https://www.cdc.gov/viole nceprevention/pdf/nisvs_report2010-a.pdf. Published November 2011. Accessed December 12, 2016.
17. Bauer GR, Travers R, Scanlon K, Coleman TA. High heterogeneity of HIV-related sexual risk among transgender people in Ontario, Canada: a province-wide respondent-driven sampling survey. BMC Public Health. 2012;12(1):292-291.
18. Devis-Devis J, Pereira-Garcia S, Valencia-Peris A, Fuentes-Miguel J, López-Cañada E, Pérez-Samaniego V. Harassment patterns and risk profile in Spanish trans persons. J Homosex. 2016. [Epub ahead of print.]
19. Gates GJ. How many people are lesbian, gay, bisexual, and transgender? http://williamsinstitute.law.ucla.edu/wp-content/uploads/Gates-How-Many -People-LGBT-Apr-2011.pdf. Published April 2011. Accessed December 1, 2016.
20. Center for Disease Control and Prevention. HIV Among Women. http://www.cdc.gov/hiv/group/gender/women/index.html. Accessed December 10, 2016.
21. Bauer GR, Scheim AI, Pyne J, Travers R, Hammond R. Intervenable factors associated with suicide risk in transgender persons: a respondent driven sampling study in Ontario, Canada. BMC Public Health. 2015;15(1):525.
22. McCann E. People who are transgender: mental health concerns. J Psychiatr Ment Health Nurs. 2015;22(1):76-81.
23. Green R. Transsexual legal rights in the United States and United Kingdom: employment, medical treatment, and civil status. Arch Sex Behav. 2010;39(1):153-160.
24. Sharpe VA, Uchendu US. Ensuring appropriate care for LGBT veterans in the Veterans Health Administration. Hastings Cent Rep. 2014;44(suppl 4):S53-S55.
25. Kauth MR, Shipherd JC, Lindsay J, Blosnich JR, Brown GR, Jones KT. Access to care for transgender veterans in the Veterans Health Administration: 2006-2013. Am J Public Health. 2014;104(suppl 4):S532-S534.
26. Snelgrove JW, Jasudavisius AM, Rowe BW, Head EM, Bauer GR. "Completely out-at-sea" with "two-gender medicine": a qualitative analysis of physician-side barriers to providing healthcare for transgender patients. BMC Health Serv Res. 2012;12(1):110.
27. U.S. Department of Health and Human Services. Key features of the affordable care act. http://www .hhs.gov/healthcare/facts-and-features/key-features -of-aca/index.html. Last reviewed November 18, 2014. Accessed December 1, 2016.
28. U.S. Department of Veterans Affairs. Federal benefits for veterans, dependents, and survivors. https://www.va.gov/opa/publications/benefits_book/Chapter_1_Health_Care_Benefits.asp. Accessed December 1, 2016.
29. HealthyPeople.gov. Lesbian, gay, bisexual, and transgender health. https://www.healthypeople.gov/2020/topics-objectives/topic/lesbian-gay-bi sexual-and-transgender-health. Updated December 1, 2016. Accessed December 1, 2016.
30. Lutwak N, Byne W, Erickson-Schroth L, et al. Transgender veterans are inadequately understood by health care providers. Mil Med. 2014;179(5):483-485.
31. Dorsen C. An integrative review of nurse attitudes towards lesbian, gay, bisexual, and transgender patients. Can J Nurs Res. 2012;44(3):18-43.
32. Eliason MJ, Dibble SL, Robertson PA. Lesbian, gay, bisexual, and transgender (LGBT) physicians' experiences in the workplace. J Homosex. 2011;58(10):1355-1371.
33. Carabez R, Pellegrini M, Mankovitz A, Eliason M, Ciano M, Scott M. "Never in All My Years...": Nurses' education about LGBT health. J Prof Nurs. 2015;31(4):323-329
34. Buchholz L. Transgender care moves into the mainstream. JAMA. 2015;314(17):1785-1787.
35. VA Boston Healthcare System. Patient Care Memorandum-11-046-LM. Management of transgender veteran patients. http://www.boston.va.gov/services/images/lgbt_patient_care_memo_transgender_care.pdf. Published May 2011. Accessed December 1, 2016.
36. Cronenwett L, Sherwood G, Pohl J, et al. Quality and safety education for advanced nursing practice. Nurs Outlook. 2009;57(6):338-348.
37. Institute of Medicine. Committee on the Robert Wood Johnson Foundation Initiative on the Future of Nursing. The Future of Nursing: Leading Change, Advancing Health. Washington, DC: National Academies Press; 2011.
38. Smith EL, Cronenwett L, Sherwood G. Current assessments of quality and safety education in nursing. Nurs Outlook. 2007;55(3):132-137.
39. World Professional Association for Transgender Health (WPATH).The standards of care. http://www.wpath.org/site_page.cfm?pk_association _webpage_menu=1351&pk_association_web page=4655. Accessed December 1, 2016.
40. University of California San Francisco Department of Family and Community Medicine. Center of Excellence for Transgender Health. http://www.tran shealth.ucsf.edu/trans?page=home-00-00 Accessed December 1, 2016.
41. Center for Disease Control and Prevention. Lesbian, gay, bisexual and transgender health. http://www.cdc.gov/lgbthealth/transgender.htm. Accessed December 1, 2016.
42. American Association of Colleges of Nursing. QSEN education consortium: graduate-level QSEN competencies, knowledge, skills and attitudes. http://www.aacn.nche.edu/faculty/qsen/competen cies.pdf. Accessed December 1, 2016.
43. Andrews MM, Boyle JS. Transcultural Concepts in Nursing Care. Philadelphia, PA: Lippincott Williams & Wilkins; 2008.
44. Moran KJ, Burson R, Conrad D. The Doctor of Nursing Practice Scholarly Project: A Framework for Success. Burlington, MA: Jones & Bartlett; 2013.
45. Hanssmann C, Morrison D, Russian E, Shiu-Thornton S, Bowen D. A community-based program evaluation of community competency trainings. J Assoc Nurses AIDS Care.
46. Knapp H, Fletcher M, Taylor A, Chan K, Goetz MB. No clinic left behind: providing cost-effective in-services via distance learning. J Healthc Qual. 2011;33(5):17-24.
47. Kauth MR, Shipherd JC, Lindsay JA, Kirsh S, Knapp H, Matza L. Teleconsultation and training of VHA providers on transgender care: implementation of a multisite hub system. Telemed J E Health. 2015;21(12):1012-1018.
48. Sherman MD, Kauth MR, Ridener L, Shipherd JC, Bratkovich K, Beaulieu G. An empirical investigation of challenges and recommendations for welcoming sexual and gender minority veterans into VA care. Prof Psychol: Res Pract. 2014;45(6):433-442.
49. American Association of Colleges of Nursing. The essentials of doctoral education for advanced nursing practice. http://www.aacn.nche.edu/pub lications/position/DNPEssentials.pdf. Published October 2006. Accessed December 1, 2016.
50. Boutain DM. Social justice as a framework for professional nursing. J Nurs Educ. 2005;44(9):404-408.
51. Poteat T, German D, Kerrigan D. Managing uncertainty: a grounded theory of stigma in transgender health care encounters. Soc Sci Med. 2013;84:22-29.
52. Thornhill L, Klein P. Creating environments of care with transgender communities. J Assoc Nurs AIDS Care. 2010;21(3):230-239.
53. Collins J. Nursing cultural competencies: Improving patient care quality and satisfaction. Ohio Nurses Rev. 2015;90(1):10-11.
54. Sherman MD, Kauth MR, Shipherd JC, Street RL Jr. Communication between VA providers and sexual and gender minority veterans: a pilot study. Psychol Serv. 2014;11(2):235-242.
55. Cowan L, Fasoli DR, Hagle ME, et al. Creating an infrastructure to advance nursing practice and care for veterans. Nurse Leader. 2013;11(5):33-36.
Patient-centered care is of fundamental importance when caring for the transgender population due to the well-established history of social stigma and systemic discrimination. Therefore, nursing education is mandated to equip graduates with culturally competent patient-centered care skills.1 In 2009, the Institute of Medicine (IOM) in partnership with the Robert Wood Johnson Foundation (RWJF) launched The Future of Nursing initiative, which outlined the major role nursing should play in transforming the health care system to meet the health care needs of diverse U.S. populations.
The initiative produced a blueprint of action-focused institutional recommendations at the local, state, and national levels that would facilitate the reforms necessary to transform the U.S. health care system. One of the recommendations of the IOM report was to increase opportunities for nurses to manage and lead collaborative efforts with physicians and other health care team members in the areas of systems redesign and research, to improve practice environments and health systems.2
The VHA is the largest integrated health care system in the U.S., serving more than 8.76 million veterans at more than 1,700 facilities. The VHA has an organizational structure that uses centralized control in Washington, DC, and branches out to 18 regional networks that are divided into local facilities in 50 states, the District of Columbia, Puerto Rico, the U.S. Virgin Islands, Guam, American Samoa, and the Philippines. This type of structure is known for promoting efficient standardization of processes and procedures across an organization.3
The VHA Blueprint for Excellence envisions the promotion of a positive culture of service and the advancement of health care innovations necessary to create an environment that all veterans deserve.4 To that end, the VHA can be a promising health care institution through which patient-centered initiatives can be standardized, promulgated nationally, and replicated as a model for the country and international health systems. However, it is important to note that the bureaucratic organizational structure of the VHA's national integrated system of care is based on a systemwide standardization effort.5 Therefore, more time may be required to implement organizational changes.
Transgender populations face significant social stigmatization, discrimination, and marginalization that contribute to negative patient outcomes. Consequently, this population experiences high rates of suicide, HIV/AIDS, substance use disorder, poverty, and homelessness.6 Due to the growing evidence of health disparities and negative health outcomes affecting transgender populations, the federal government has identified transgender patient care and outcomes as a major health concern and priority in the Healthy People Initiative 2020.2,7,8
In 2012, the VHA issued a directive mandating services for transgender veterans.9 Nevertheless, health care staff significantly lack the knowledge, skills, and cultural competencies that are vital in transgender care.
This article reviews the prevalence and demographics of the transgender population, social challenges, global health concerns, and public health policies. The article also examines how the doctor of nursing practice (DNP)-prepared nurse leader can provide transformational nursing leadership to facilitate culturally competent, patient-centered initiatives to improve access and services for transgender individuals in the VHA and provide a model for change in transgender population health.
Definitions
Gender is a behavioral, cultural, or psychological trait assigned by society that is associated with male or female sex. Sex denotes the biologic differences between males and females. Transgender is an umbrella term used to describe people whose gender identity or gender expression is different from that of their sex assigned at birth. Transsexualism is a subset of transgender persons who have taken steps to self-identify or transition to look like their preferred gender.
Demographics
Estimates of the prevalence of transgenderism are roughly drawn from less rigorous methods, such as the combination of parents who report transgenderism in children, the number of adults reportedly seeking clinical care (such as cross-sex or gender-affirming hormone therapy), and the number of surgical interventions reported in different countries.10 A meta-analysis of 21 studies concluded that the ratio of transsexuals (individuals who are altering or have already altered their birth sex) was predominantly 1:14,705 adult males and 1:38,461 adult females.11 Since all transgender persons do not identify as transsexual, these figures do not provide a precise estimation of the number of transgender persons worldwide.
About 700,000, or 0.3%, of the adult population in the U.S. identify themselves as transgender, and an estimated 134,300 identify as transgender veterans.6,12 The transgender population in the U.S. is estimated to be 55% white, 16% African American, 21% Hispanic, and 8% other races.13 The U.S. census data noted that the transgender population was geographically located across the nation. Transgender persons are more likely to be single, never married, divorced, and more educated but with significantly less household income.2 Data to provide an accurate reflection of the number of transgender people in the U.S. are lacking. Some transgender individuals also may identify as lesbian, gay, or bisexual, making population-based estimation even more challenging and difficult.
Transgender persons who have transitioned may not have changed their names or changed their identified sex on official Social Security records, which the Social Security Administration allows only if there is evidence that genital sexual reassignment surgery was performed.14 The number of transgender adults requesting treatment continues to rise.10
Social and Health Challenges
Transgender people face many challenges because of their gender identity. Surveys assessing the living conditions of transgender people have found that 43% to 60% report high levels of physical violence.15 By comparison, the National Intimate Partner and Sexual Violence Survey found that interpersonal violence and sexual violence were reported by lesbian and gay individuals at equal or higher levels than that reported by heterosexuals. Forty-four percent of lesbian women, 35% of heterosexual women, 29% of heterosexual men, and 26% of gay men reported experiencing rape or physical violence.16 A study in Spain reported 59% of transgender people experienced patterns of harassment, and in Canada, 34% of transgender people lived below the poverty level.17,18
In the U.S., the National Transgender Discrimination Survey of 6,450 transgender and nonconforming participants provided extensive data on challenges experienced by transgender people.6 Discrimination was frequently experienced in accessing health care. Due to transgender status, 19% were denied care, and 28% postponed care due to perceived harassment and violence within a health care setting.6 The same study also reported that as many as 41% live in extreme poverty with incomes of less than $10,000 per year reported. Twenty-six percent were physically assaulted, and 10% experienced sexual violence. More than 25% of the transgender population misused drugs or alcohol to cope with mistreatment.6
In the U.S., HIV infection rates for transgender individuals were more than 4 times (2.64%) the rate of the general population (0.6%).6 Internationally, there is a high prevalence of HIV in transgender women. The prevalence rate of HIV in U.S. transgender women was 21.74% of the estimated U.S. adult transgender population of about 700,000.19 One in 4 people living with HIV in the U.S. are women.20
Suicide attempt rates are extremely high among transgender people. A suicide rate of 22% to 43% has been reported across Europe, Canada, and the U.S.21 Depression and anxiety were commonly noted as a result of discrimination and social stigma. In the U.S., transgender persons reported high rates of depression, with 41% reporting attempted suicide compared with 1.6% of the general population.6 Access to health care services, such as mental health, psychosocial support, and stress management are critical for this vulnerable population.22
Health Policies
Since 1994, the UK has instituted legal employment protections for the transgender population. In the UK, transgender persons, including military and prisoners, have health care coverage that includes sexual reassignment surgery as part of the UK's National Health Service.23
In the U.S., the federal policy of "Don't Ask, Don't Tell" barring transgender persons from serving openly in the military was repealed in June 2016. This policy historically has had a silencing effect on perpetuating institutionalized biases.24 This remains problematic even after veterans have transitioned from military service to the VA for civilian care.
Between 2006 and 2013, the reported prevalence and incidence of transgender-related diagnoses in the VA have steadily increased with 40% of new diagnoses occurring since 2011.25 In fiscal year 2013, there were 32.9 per 100,000 veterans with transgender-related diagnoses.25 Health care staff, in particular health care providers (HCPs), can play a critical role in reducing health disparities and unequal treatment.26
With the passage of the U.S. Affordable Care Act (ACA), health insurance coverage for transgender persons is now guaranteed by law, and health disparities within the transgender population can begin to be properly addressed. The ACA offers the ability to purchase health insurance, possibly qualify for Medicaid, or obtain subsidies to purchase health insurance. Insurance coverage is accessible without regard to discrimination or preexisting conditions.27 As of May 2014, the Medicare program covered medically necessary hormone therapy and sex reassignment surgery.13 While VA benefits cover hormone therapy for transgender veterans, sex reassignment surgery is not currently a covered benefit.28 The ACA now increases access to primary care, preventative care, mental health services, and community health programs not previously available in the transgender community.
Healthy People 2020 Goals
One of the Healthy People 2020 stated goals is to improve the health and wellness of transgender people.29 The objective is to increase the number of population-based data collection systems used to monitor transgender people from the baseline of 2 to a total of 4 by 2020. The data systems would be assigned to collect relevant data, such as mental health; HIV status; illicit drug, alcohol, and tobacco use; cervical and breast cancer screening; health insurance coverage; and access to health care.
Health Care Staff Readiness
Transgender persons face health care challenges with major health disparities due to their gender identity. Transgender persons as a defined population are not well understood by HCPs. In a survey, 50% of transgender respondents reported that they had to teach their medical provider about transgender care.6 Negative perceptions of transgender persons are well established and have contribute to the poor health care access and services that transgender persons receive. Transgender persons are often denied access to care, denied visitation rights, and are hesitant to share information for fear of bureaucratic exclusion or isolation.
There is a lack of evidence-based studies to guide care and help HCPs gain greater understanding of this population's unique needs.30 Additionally, a significant lack of knowledge, skills, cultural competence, and awareness exist in providing transgender care. Research on nursing attitudes concerning transgender care consistently found negative attitudes, and physicians also frequently reported witnessing derogatory comments and discriminatory care from colleagues.31,32 The study by Carabez and colleagues found that practicing nurses rarely received the proper education or training in transgender health issues, and many were unaware of the needs of this population.33 In addition, many HCPs were uncomfortable working with transgender patients. Physicians also expressed knowledge deficits on gender identity disorders due to a lack of training and ethical concerns about their roles in providing gender-transitioning treatment.26
Although the VHA directive states that transgender services and treatment should be standardized, the VHA has not approved, defined, or endorsed specific standards of care or clinical guidelines within the organization for transgender care, further heightening HCP concerns.9 The clinical practice guidelines available for addressing preventive care for transgender patients are primarily based on consensus of expert opinion.34 Expert opinion has produced the Standards of Care (SOC) for the Health of Transsexual, Transgender, and Gender Nonconforming People, published by the World Professional Association for Transgender Health (WPATH) and cited by the IOM as the major clinical practice guidelines for providing care to transgender individuals.2 Transgender care at the VHA is guided by the WPATH standards of care.35
The VHA has created national educational programs and policies with targeted goals to provide uniform, culturally competent, patient-centered care. Online transgender health presentations are available, and at least 15 VHA facilities have transgender support groups.30 While the VHA supports a patient-centered philosophy for transgender patient care, many facilities do not currently have organizational initiatives that enhance clinical preparation of HCPs or have sufficiently modified the environment to better accommodate the health care needs of transgender veterans.
DNP Preparation
The DNP terminal degree provides nurses with doctoral-level training in organizational and systems leadership, leading quality improvement, and implementing systemwide initiatives by using scientific findings to drive processes that improve quality of care for a changing patient population.36 Preparation in research analysis of evidence-based interventions also is essential to evaluating practice patterns, patient outcomes, and systems of care that can identify gaps in practice. Training in health care policy and advocacy, information systems, patient care technology, and population health also is provided so that DNPs are competent to develop system strategies to transform health care through clinical prevention and health promotion.
QSEN Framework
In keeping with the IOM's Future of Nursing initiative recommendations that graduate nurses be prepared as leaders in education, practice, administration, and research, there is an increasing focus on providing graduate-level nursing education and training to ensure quality and efficiency of health outcomes.37 The Quality and Safety Education in Nursing (QSEN) project, initiated at the RWJF by Linda Cronenwett, PhD, RN, identifies a framework for knowledge, skills, and attitudes that defines the competencies that nurses need to deliver effective care to improve quality and safety within health care systems.38 These core competencies include quality improvement, safety, teamwork and collaboration, patient-centered care, evidence-based practice, and informatics. The RWJF and the American Association of Colleges of Nursing later expanded the project initiative to prepare nursing faculty to teach the QSEN competencies in graduate nursing programs.36
The DNP nurse leader is ideally suited to manage this project by applying competencies from the QSEN framework. Using open communication and mutual respect, the nurse leader is poised to effectively develop interprofessional teams to collaborate and initiate transformational changes that improve quality and patient-centered care delivered within the health care organization.
Public Health Resources
Public health resources addressing transgender patient care advocacy, public policy, community education, standards of care, cultural competency, mental health, hormone therapy, surgical interventions, reproductive health, primary care, preventative care, and research are available. For example, WPATH is an international multidisciplinary organization that has published comprehensive SOC for transgender, transsexual, and gender-nonconforming people. The seventh version of the SOC contains evidence-based guidelines for treatment.39 Additional online resources for transgender health are available from the CDC, the Center of Excellence for Transgender Health at the University of California, San Francisco; Department of Family and Community Medicine; and the National Center for Transgender Equality.13,40,41
Patient-Centered Transgender Care
The QSEN framework outlines competencies that provide applicable solutions that help prepare organizations to deliver culturally competent, patient-centered transgender care. The first step to creating patient-centered transgender care is to "analyze factors that create barriers to patient-centered care."42 The magnitude of the barriers to providing patient-centered transgender care also must be identified and understood. An assessment of individual values, beliefs, and attitudes can help to identify cultural characteristics and eliminate stereotypes that impact health practices.43
The nurse leader should solicit support from stakeholders to assess barriers to providing patient-centered transgender care at the system level. Stakeholders would include staff directly involved in patient care, such as physicians, nurse practitioners, physician assistants, registered nurses, nurse managers, nurse educators, licensed practical nurses, medical support assistants, psychologists, dieticians, and social workers. Other ancillary stakeholders with an interest in creating a patient-centered environment with positive patient outcomes include the executive leadership team of the organization, which consists of the chief of staff, director, administrative officers, and nurse executive.
The nurse leader should consult with experts in transgender care and present evidence-based research showing how deficits in staff knowledge, skills, and cultural competence negatively impact the quality of care provided to transgender persons. National data on the consequential health disparities and negative impacts on patient outcomes also should be discussed and presented to all stakeholders. The nurse leader in collaboration with the VA Office of Research and Development is ideally suited to obtain institutional review board approval of a proposal to conduct a needs assessment survey of health care staff barriers to providing patient-centered transgender care. Thereafter, the nurse leader would analyze, extract, and synthesize the data and evaluate the resources and technology available to translate this research knowledge into a clinical practice setting at the system level.44
The second solution uses the results of the survey to develop staff competency training within the organization. The nurse leader can facilitate collaboration and team building to develop practice guidelines and SOC. Competency training will prepare the staff to assist in developing strategies to improve the quality of care for transgender persons. Educationconcerning existing evidence-based clinical guidelines and SOC as well as anecdotal evidence of the needs of transgender patients should be included in competency training.45 One approach to competency training would be to trainintegrated multidisciplinary teams with expertise in transgender care to promote wellness and disease prevention.9 The nurse leader should collaborate with multiple disciplines to facilitate the development of interdisciplinary teams from nursing, medicine, social work, pharmacy, primary care, mental health, women's health, and endocrinology to participate in the Specialty Care Access Network Extension of Community Healthcare outcomes (SCAN-ECHO) training. Training can be offered by videoconferencing over several months and provides cost-effective, efficient training of providers in patient-centered transgender care.46,47 After the SCAN-ECHO program is completed, trained nursing experts could then develop a cultural sensitivity training program for nursing organizations to be offered to educate health care staff on an annual basis.
The third solution addresses the QSEN competency to "Analyze institutional features of the facilities that support or pose barriers to patient-centered care."42 Many veterans do not perceive VA environments as welcoming. In a study by Sherman and colleagues, less than one-third of veterans believed the VA environment was welcoming to sexual or gender minorities, and sexual orientation or gender identity was disclosed by only about 25% of veterans.48 Many veterans in this study felt uncomfortable disclosing their gender or sexual orientation. The majority felt that providers should not routinely ask about sexual orientation or gender identity, and 24% said they were very or somewhat uncomfortable discussing the issue. In another study, 202 VA providers were asked if they viewed the VA as welcoming, and 32% said the VA was somewhat or very unwelcoming.48
The nurse leader is trained in the essentials of health care policy advocacy, which is central to nursing practice.49 Nursing as a profession values social justice and equality, which are linked to fewer health disparities and more stable health indicators.50 Therefore, nursing can ideally provide organizational leaders by developing a culture wherein stable, patient-centered relationships can develop and thrive.
Organizational Culture
Strategies must be deployed to create an organizational culture that is welcoming, respectful, and supportive of transgender patients and family preferences. VA should develop support groups for transgender veterans in VA facilities. Support groups are helpful in diminishing stress, improving self-esteem, building confidence, and improving social relationships.51 Additionally, VA should develop community-based partnerships with other organizations that already provide institutional care and support from HCPs who support transgender persons' right to self-determination.52 These partnerships can foster environmental influences over time and lead to the development of trusting relationships between transgender veterans and the VA organization.
Another community partnership of importance for the nurse leader to develop is an alliance with local universities to train nursing students in cultural competencies in transgender care at VA facilities. The U.S. population continues to diversify in race and ethnicity and cultural influences; therefore, nurses must be prepared in cultural competencies in order to provide quality care that reduces health disparities.53
Under federal law, the VHA has a data sharing agreement with the DoD. Despite the repeal of the "Don't Ask, Don't Tell" federal law, which cleared the way for transgender persons to openly serve in the military, many transgender persons may remain fearful of reprisals, such as judgment, denial of care, or loss of benefits if gender identity is disclosed.54 Given the bureaucratic structure of the VHA, the implementation of cultural changes at the system level will require a collaborative effort between multidisciplinary teams and community partnerships to transform the VA environment over time. The authors believe that on this issue, external forces must guide and lead changes within the VA system in order to develop sustainable and trusting relationships with transgender veterans.
The fourth solution is implementation of policies that "empower patients or families in all aspects of the health care process."42 Again, the nurse leader is trained and prepared to advocate for a policy that implements a Patient Bill of Rights that explicitly guarantees health care and prohibits discrimination of gender-minority veterans. This change would foster trust and confidence from transgender individuals. A study found that 83% of providers and 83% of lesbian, gay, bisexual, and transgender veterans believe that this policy change would make the VHA environment more welcoming.48 Providing transgender-affirming materials and language on standard forms also would eliminate barriers, promote patient-centered care, and empower transgender patients by creating an environment that is more inclusive of everyone.48
Conclusion
The nurse leader is well positioned to implement the QSEN framework to integrate research, practice, and policy to create a more inclusive, patient-centered health care system for transgender veterans. By using the essential principles of doctoral education for advanced nursing practice, the nurse leader is prepared to advocate for changing the organization at the systems level. The nurse leader also is equipped to direct the implementation of patient-centered transgender care initiatives by ensuring the integration of the nursing organization as a partner in strategic planning as well as the development of solutions.
The VHA Blueprint of Excellence envisions organization and collaboration to promote new relationships that serve and benefit veterans. The DNP preparation allows the nurse leader to demonstrate the ability to collaborate with VHA stakeholders and develop alliances within and outside the organization by advocating for policy changes that will be transformational in improving health care delivery and patient outcomes to vulnerable transgender veteran populations. The IOM has tasked nurse executives with creating a health care infrastructure of doctorally prepared nurses to provide patient care that is increasingly growing more complex. With an increasing number of veterans using services, VHA has prioritized an expansion in the number of doctorally prepared nurses.55
As the largest integrated health care system in the U.S., the VHA provides an ideal setting for initiating these organizational changes as a result of having developed an integrated infrastructure to collect evidence-based data at the regional (network) and state facilities and make comparisons with national benchmarks. Therefore, changes are less difficult to disseminate throughout the hierarchy of the VHA. Consequently, the VHA has been a leader in the U.S. for equity in the health care arena and provides a model for international health care systems. Finally, these changes address an urgent need to reduce health disparities, morbidity, and mortality by improving quality care and health care delivery to a vulnerable transgender population.
Patient-centered care is of fundamental importance when caring for the transgender population due to the well-established history of social stigma and systemic discrimination. Therefore, nursing education is mandated to equip graduates with culturally competent patient-centered care skills.1 In 2009, the Institute of Medicine (IOM) in partnership with the Robert Wood Johnson Foundation (RWJF) launched The Future of Nursing initiative, which outlined the major role nursing should play in transforming the health care system to meet the health care needs of diverse U.S. populations.
The initiative produced a blueprint of action-focused institutional recommendations at the local, state, and national levels that would facilitate the reforms necessary to transform the U.S. health care system. One of the recommendations of the IOM report was to increase opportunities for nurses to manage and lead collaborative efforts with physicians and other health care team members in the areas of systems redesign and research, to improve practice environments and health systems.2
The VHA is the largest integrated health care system in the U.S., serving more than 8.76 million veterans at more than 1,700 facilities. The VHA has an organizational structure that uses centralized control in Washington, DC, and branches out to 18 regional networks that are divided into local facilities in 50 states, the District of Columbia, Puerto Rico, the U.S. Virgin Islands, Guam, American Samoa, and the Philippines. This type of structure is known for promoting efficient standardization of processes and procedures across an organization.3
The VHA Blueprint for Excellence envisions the promotion of a positive culture of service and the advancement of health care innovations necessary to create an environment that all veterans deserve.4 To that end, the VHA can be a promising health care institution through which patient-centered initiatives can be standardized, promulgated nationally, and replicated as a model for the country and international health systems. However, it is important to note that the bureaucratic organizational structure of the VHA's national integrated system of care is based on a systemwide standardization effort.5 Therefore, more time may be required to implement organizational changes.
Transgender populations face significant social stigmatization, discrimination, and marginalization that contribute to negative patient outcomes. Consequently, this population experiences high rates of suicide, HIV/AIDS, substance use disorder, poverty, and homelessness.6 Due to the growing evidence of health disparities and negative health outcomes affecting transgender populations, the federal government has identified transgender patient care and outcomes as a major health concern and priority in the Healthy People Initiative 2020.2,7,8
In 2012, the VHA issued a directive mandating services for transgender veterans.9 Nevertheless, health care staff significantly lack the knowledge, skills, and cultural competencies that are vital in transgender care.
This article reviews the prevalence and demographics of the transgender population, social challenges, global health concerns, and public health policies. The article also examines how the doctor of nursing practice (DNP)-prepared nurse leader can provide transformational nursing leadership to facilitate culturally competent, patient-centered initiatives to improve access and services for transgender individuals in the VHA and provide a model for change in transgender population health.
Definitions
Gender is a behavioral, cultural, or psychological trait assigned by society that is associated with male or female sex. Sex denotes the biologic differences between males and females. Transgender is an umbrella term used to describe people whose gender identity or gender expression is different from that of their sex assigned at birth. Transsexualism is a subset of transgender persons who have taken steps to self-identify or transition to look like their preferred gender.
Demographics
Estimates of the prevalence of transgenderism are roughly drawn from less rigorous methods, such as the combination of parents who report transgenderism in children, the number of adults reportedly seeking clinical care (such as cross-sex or gender-affirming hormone therapy), and the number of surgical interventions reported in different countries.10 A meta-analysis of 21 studies concluded that the ratio of transsexuals (individuals who are altering or have already altered their birth sex) was predominantly 1:14,705 adult males and 1:38,461 adult females.11 Since all transgender persons do not identify as transsexual, these figures do not provide a precise estimation of the number of transgender persons worldwide.
About 700,000, or 0.3%, of the adult population in the U.S. identify themselves as transgender, and an estimated 134,300 identify as transgender veterans.6,12 The transgender population in the U.S. is estimated to be 55% white, 16% African American, 21% Hispanic, and 8% other races.13 The U.S. census data noted that the transgender population was geographically located across the nation. Transgender persons are more likely to be single, never married, divorced, and more educated but with significantly less household income.2 Data to provide an accurate reflection of the number of transgender people in the U.S. are lacking. Some transgender individuals also may identify as lesbian, gay, or bisexual, making population-based estimation even more challenging and difficult.
Transgender persons who have transitioned may not have changed their names or changed their identified sex on official Social Security records, which the Social Security Administration allows only if there is evidence that genital sexual reassignment surgery was performed.14 The number of transgender adults requesting treatment continues to rise.10
Social and Health Challenges
Transgender people face many challenges because of their gender identity. Surveys assessing the living conditions of transgender people have found that 43% to 60% report high levels of physical violence.15 By comparison, the National Intimate Partner and Sexual Violence Survey found that interpersonal violence and sexual violence were reported by lesbian and gay individuals at equal or higher levels than that reported by heterosexuals. Forty-four percent of lesbian women, 35% of heterosexual women, 29% of heterosexual men, and 26% of gay men reported experiencing rape or physical violence.16 A study in Spain reported 59% of transgender people experienced patterns of harassment, and in Canada, 34% of transgender people lived below the poverty level.17,18
In the U.S., the National Transgender Discrimination Survey of 6,450 transgender and nonconforming participants provided extensive data on challenges experienced by transgender people.6 Discrimination was frequently experienced in accessing health care. Due to transgender status, 19% were denied care, and 28% postponed care due to perceived harassment and violence within a health care setting.6 The same study also reported that as many as 41% live in extreme poverty with incomes of less than $10,000 per year reported. Twenty-six percent were physically assaulted, and 10% experienced sexual violence. More than 25% of the transgender population misused drugs or alcohol to cope with mistreatment.6
In the U.S., HIV infection rates for transgender individuals were more than 4 times (2.64%) the rate of the general population (0.6%).6 Internationally, there is a high prevalence of HIV in transgender women. The prevalence rate of HIV in U.S. transgender women was 21.74% of the estimated U.S. adult transgender population of about 700,000.19 One in 4 people living with HIV in the U.S. are women.20
Suicide attempt rates are extremely high among transgender people. A suicide rate of 22% to 43% has been reported across Europe, Canada, and the U.S.21 Depression and anxiety were commonly noted as a result of discrimination and social stigma. In the U.S., transgender persons reported high rates of depression, with 41% reporting attempted suicide compared with 1.6% of the general population.6 Access to health care services, such as mental health, psychosocial support, and stress management are critical for this vulnerable population.22
Health Policies
Since 1994, the UK has instituted legal employment protections for the transgender population. In the UK, transgender persons, including military and prisoners, have health care coverage that includes sexual reassignment surgery as part of the UK's National Health Service.23
In the U.S., the federal policy of "Don't Ask, Don't Tell" barring transgender persons from serving openly in the military was repealed in June 2016. This policy historically has had a silencing effect on perpetuating institutionalized biases.24 This remains problematic even after veterans have transitioned from military service to the VA for civilian care.
Between 2006 and 2013, the reported prevalence and incidence of transgender-related diagnoses in the VA have steadily increased with 40% of new diagnoses occurring since 2011.25 In fiscal year 2013, there were 32.9 per 100,000 veterans with transgender-related diagnoses.25 Health care staff, in particular health care providers (HCPs), can play a critical role in reducing health disparities and unequal treatment.26
With the passage of the U.S. Affordable Care Act (ACA), health insurance coverage for transgender persons is now guaranteed by law, and health disparities within the transgender population can begin to be properly addressed. The ACA offers the ability to purchase health insurance, possibly qualify for Medicaid, or obtain subsidies to purchase health insurance. Insurance coverage is accessible without regard to discrimination or preexisting conditions.27 As of May 2014, the Medicare program covered medically necessary hormone therapy and sex reassignment surgery.13 While VA benefits cover hormone therapy for transgender veterans, sex reassignment surgery is not currently a covered benefit.28 The ACA now increases access to primary care, preventative care, mental health services, and community health programs not previously available in the transgender community.
Healthy People 2020 Goals
One of the Healthy People 2020 stated goals is to improve the health and wellness of transgender people.29 The objective is to increase the number of population-based data collection systems used to monitor transgender people from the baseline of 2 to a total of 4 by 2020. The data systems would be assigned to collect relevant data, such as mental health; HIV status; illicit drug, alcohol, and tobacco use; cervical and breast cancer screening; health insurance coverage; and access to health care.
Health Care Staff Readiness
Transgender persons face health care challenges with major health disparities due to their gender identity. Transgender persons as a defined population are not well understood by HCPs. In a survey, 50% of transgender respondents reported that they had to teach their medical provider about transgender care.6 Negative perceptions of transgender persons are well established and have contribute to the poor health care access and services that transgender persons receive. Transgender persons are often denied access to care, denied visitation rights, and are hesitant to share information for fear of bureaucratic exclusion or isolation.
There is a lack of evidence-based studies to guide care and help HCPs gain greater understanding of this population's unique needs.30 Additionally, a significant lack of knowledge, skills, cultural competence, and awareness exist in providing transgender care. Research on nursing attitudes concerning transgender care consistently found negative attitudes, and physicians also frequently reported witnessing derogatory comments and discriminatory care from colleagues.31,32 The study by Carabez and colleagues found that practicing nurses rarely received the proper education or training in transgender health issues, and many were unaware of the needs of this population.33 In addition, many HCPs were uncomfortable working with transgender patients. Physicians also expressed knowledge deficits on gender identity disorders due to a lack of training and ethical concerns about their roles in providing gender-transitioning treatment.26
Although the VHA directive states that transgender services and treatment should be standardized, the VHA has not approved, defined, or endorsed specific standards of care or clinical guidelines within the organization for transgender care, further heightening HCP concerns.9 The clinical practice guidelines available for addressing preventive care for transgender patients are primarily based on consensus of expert opinion.34 Expert opinion has produced the Standards of Care (SOC) for the Health of Transsexual, Transgender, and Gender Nonconforming People, published by the World Professional Association for Transgender Health (WPATH) and cited by the IOM as the major clinical practice guidelines for providing care to transgender individuals.2 Transgender care at the VHA is guided by the WPATH standards of care.35
The VHA has created national educational programs and policies with targeted goals to provide uniform, culturally competent, patient-centered care. Online transgender health presentations are available, and at least 15 VHA facilities have transgender support groups.30 While the VHA supports a patient-centered philosophy for transgender patient care, many facilities do not currently have organizational initiatives that enhance clinical preparation of HCPs or have sufficiently modified the environment to better accommodate the health care needs of transgender veterans.
DNP Preparation
The DNP terminal degree provides nurses with doctoral-level training in organizational and systems leadership, leading quality improvement, and implementing systemwide initiatives by using scientific findings to drive processes that improve quality of care for a changing patient population.36 Preparation in research analysis of evidence-based interventions also is essential to evaluating practice patterns, patient outcomes, and systems of care that can identify gaps in practice. Training in health care policy and advocacy, information systems, patient care technology, and population health also is provided so that DNPs are competent to develop system strategies to transform health care through clinical prevention and health promotion.
QSEN Framework
In keeping with the IOM's Future of Nursing initiative recommendations that graduate nurses be prepared as leaders in education, practice, administration, and research, there is an increasing focus on providing graduate-level nursing education and training to ensure quality and efficiency of health outcomes.37 The Quality and Safety Education in Nursing (QSEN) project, initiated at the RWJF by Linda Cronenwett, PhD, RN, identifies a framework for knowledge, skills, and attitudes that defines the competencies that nurses need to deliver effective care to improve quality and safety within health care systems.38 These core competencies include quality improvement, safety, teamwork and collaboration, patient-centered care, evidence-based practice, and informatics. The RWJF and the American Association of Colleges of Nursing later expanded the project initiative to prepare nursing faculty to teach the QSEN competencies in graduate nursing programs.36
The DNP nurse leader is ideally suited to manage this project by applying competencies from the QSEN framework. Using open communication and mutual respect, the nurse leader is poised to effectively develop interprofessional teams to collaborate and initiate transformational changes that improve quality and patient-centered care delivered within the health care organization.
Public Health Resources
Public health resources addressing transgender patient care advocacy, public policy, community education, standards of care, cultural competency, mental health, hormone therapy, surgical interventions, reproductive health, primary care, preventative care, and research are available. For example, WPATH is an international multidisciplinary organization that has published comprehensive SOC for transgender, transsexual, and gender-nonconforming people. The seventh version of the SOC contains evidence-based guidelines for treatment.39 Additional online resources for transgender health are available from the CDC, the Center of Excellence for Transgender Health at the University of California, San Francisco; Department of Family and Community Medicine; and the National Center for Transgender Equality.13,40,41
Patient-Centered Transgender Care
The QSEN framework outlines competencies that provide applicable solutions that help prepare organizations to deliver culturally competent, patient-centered transgender care. The first step to creating patient-centered transgender care is to "analyze factors that create barriers to patient-centered care."42 The magnitude of the barriers to providing patient-centered transgender care also must be identified and understood. An assessment of individual values, beliefs, and attitudes can help to identify cultural characteristics and eliminate stereotypes that impact health practices.43
The nurse leader should solicit support from stakeholders to assess barriers to providing patient-centered transgender care at the system level. Stakeholders would include staff directly involved in patient care, such as physicians, nurse practitioners, physician assistants, registered nurses, nurse managers, nurse educators, licensed practical nurses, medical support assistants, psychologists, dieticians, and social workers. Other ancillary stakeholders with an interest in creating a patient-centered environment with positive patient outcomes include the executive leadership team of the organization, which consists of the chief of staff, director, administrative officers, and nurse executive.
The nurse leader should consult with experts in transgender care and present evidence-based research showing how deficits in staff knowledge, skills, and cultural competence negatively impact the quality of care provided to transgender persons. National data on the consequential health disparities and negative impacts on patient outcomes also should be discussed and presented to all stakeholders. The nurse leader in collaboration with the VA Office of Research and Development is ideally suited to obtain institutional review board approval of a proposal to conduct a needs assessment survey of health care staff barriers to providing patient-centered transgender care. Thereafter, the nurse leader would analyze, extract, and synthesize the data and evaluate the resources and technology available to translate this research knowledge into a clinical practice setting at the system level.44
The second solution uses the results of the survey to develop staff competency training within the organization. The nurse leader can facilitate collaboration and team building to develop practice guidelines and SOC. Competency training will prepare the staff to assist in developing strategies to improve the quality of care for transgender persons. Educationconcerning existing evidence-based clinical guidelines and SOC as well as anecdotal evidence of the needs of transgender patients should be included in competency training.45 One approach to competency training would be to trainintegrated multidisciplinary teams with expertise in transgender care to promote wellness and disease prevention.9 The nurse leader should collaborate with multiple disciplines to facilitate the development of interdisciplinary teams from nursing, medicine, social work, pharmacy, primary care, mental health, women's health, and endocrinology to participate in the Specialty Care Access Network Extension of Community Healthcare outcomes (SCAN-ECHO) training. Training can be offered by videoconferencing over several months and provides cost-effective, efficient training of providers in patient-centered transgender care.46,47 After the SCAN-ECHO program is completed, trained nursing experts could then develop a cultural sensitivity training program for nursing organizations to be offered to educate health care staff on an annual basis.
The third solution addresses the QSEN competency to "Analyze institutional features of the facilities that support or pose barriers to patient-centered care."42 Many veterans do not perceive VA environments as welcoming. In a study by Sherman and colleagues, less than one-third of veterans believed the VA environment was welcoming to sexual or gender minorities, and sexual orientation or gender identity was disclosed by only about 25% of veterans.48 Many veterans in this study felt uncomfortable disclosing their gender or sexual orientation. The majority felt that providers should not routinely ask about sexual orientation or gender identity, and 24% said they were very or somewhat uncomfortable discussing the issue. In another study, 202 VA providers were asked if they viewed the VA as welcoming, and 32% said the VA was somewhat or very unwelcoming.48
The nurse leader is trained in the essentials of health care policy advocacy, which is central to nursing practice.49 Nursing as a profession values social justice and equality, which are linked to fewer health disparities and more stable health indicators.50 Therefore, nursing can ideally provide organizational leaders by developing a culture wherein stable, patient-centered relationships can develop and thrive.
Organizational Culture
Strategies must be deployed to create an organizational culture that is welcoming, respectful, and supportive of transgender patients and family preferences. VA should develop support groups for transgender veterans in VA facilities. Support groups are helpful in diminishing stress, improving self-esteem, building confidence, and improving social relationships.51 Additionally, VA should develop community-based partnerships with other organizations that already provide institutional care and support from HCPs who support transgender persons' right to self-determination.52 These partnerships can foster environmental influences over time and lead to the development of trusting relationships between transgender veterans and the VA organization.
Another community partnership of importance for the nurse leader to develop is an alliance with local universities to train nursing students in cultural competencies in transgender care at VA facilities. The U.S. population continues to diversify in race and ethnicity and cultural influences; therefore, nurses must be prepared in cultural competencies in order to provide quality care that reduces health disparities.53
Under federal law, the VHA has a data sharing agreement with the DoD. Despite the repeal of the "Don't Ask, Don't Tell" federal law, which cleared the way for transgender persons to openly serve in the military, many transgender persons may remain fearful of reprisals, such as judgment, denial of care, or loss of benefits if gender identity is disclosed.54 Given the bureaucratic structure of the VHA, the implementation of cultural changes at the system level will require a collaborative effort between multidisciplinary teams and community partnerships to transform the VA environment over time. The authors believe that on this issue, external forces must guide and lead changes within the VA system in order to develop sustainable and trusting relationships with transgender veterans.
The fourth solution is implementation of policies that "empower patients or families in all aspects of the health care process."42 Again, the nurse leader is trained and prepared to advocate for a policy that implements a Patient Bill of Rights that explicitly guarantees health care and prohibits discrimination of gender-minority veterans. This change would foster trust and confidence from transgender individuals. A study found that 83% of providers and 83% of lesbian, gay, bisexual, and transgender veterans believe that this policy change would make the VHA environment more welcoming.48 Providing transgender-affirming materials and language on standard forms also would eliminate barriers, promote patient-centered care, and empower transgender patients by creating an environment that is more inclusive of everyone.48
Conclusion
The nurse leader is well positioned to implement the QSEN framework to integrate research, practice, and policy to create a more inclusive, patient-centered health care system for transgender veterans. By using the essential principles of doctoral education for advanced nursing practice, the nurse leader is prepared to advocate for changing the organization at the systems level. The nurse leader also is equipped to direct the implementation of patient-centered transgender care initiatives by ensuring the integration of the nursing organization as a partner in strategic planning as well as the development of solutions.
The VHA Blueprint of Excellence envisions organization and collaboration to promote new relationships that serve and benefit veterans. The DNP preparation allows the nurse leader to demonstrate the ability to collaborate with VHA stakeholders and develop alliances within and outside the organization by advocating for policy changes that will be transformational in improving health care delivery and patient outcomes to vulnerable transgender veteran populations. The IOM has tasked nurse executives with creating a health care infrastructure of doctorally prepared nurses to provide patient care that is increasingly growing more complex. With an increasing number of veterans using services, VHA has prioritized an expansion in the number of doctorally prepared nurses.55
As the largest integrated health care system in the U.S., the VHA provides an ideal setting for initiating these organizational changes as a result of having developed an integrated infrastructure to collect evidence-based data at the regional (network) and state facilities and make comparisons with national benchmarks. Therefore, changes are less difficult to disseminate throughout the hierarchy of the VHA. Consequently, the VHA has been a leader in the U.S. for equity in the health care arena and provides a model for international health care systems. Finally, these changes address an urgent need to reduce health disparities, morbidity, and mortality by improving quality care and health care delivery to a vulnerable transgender population.
1. Greiner AC, Knebel E, eds. Health Professions Education: A Bridge to Quality. Washington, DC: National Academies Press; 2003.
2. Institute of Medicine. Bisexual, and Transgender People: Building a Foundation for Better Understanding. Washington, DC: National Academy of Sciences; 2011.
3. Mintzberg H. The structuring of organizations: a synthesis of the research. https://papers.ssrn.com/sol3/papers.cfm?abstract_id=1496182 1979. Posted November 4, 2009. Accessed November 30, 2016.
4. U.S. Department of Veterans Affairs. VHA blue print for excellence. https://www.va.gov/health/docs/VHA_Blueprint_for_Excellence.pdf. Published September 21, 2014. Accessed November 30, 2016.
5. Morgan RO, Teal CR, Reddy SG, Ford ME, Ashton CM. Measurement in Veterans Affairs Health Services Research: veterans as a special population. Health Serv Res. 2005;40(5, part 2):1573-1583.
6. Grant JM, Mottet L, Tanis JE, Harrison J, Herman J, Keisling M. Injustice at every turn: a report of the National Transgender Discrimination Survey. http://www.thetaskforce.org/static_html/downloads /reports/reports/ntds_full.pdf. Published 2011. Accessed November 30, 2016.
7. Office of Disease Prevention and Health Promotion. Lesbian, gay, bisexual, and transgender health. http://www.healthypeople.gov/2020/topics-objec tives/topic/lesbian-gay-bisexual-and-transgender -health. Updated November 16, 2016. Accessed November 16, 2016.
8. Institute of Medicine Committee on Lesbian Gay, Bisexual, and Transgender Health Issues and Research Gaps and Opportunities. The Health of Lesbian, Gay, Bisexual, and Transgender People: Building a Foundation for Better Understanding. Washington, DC: National Academy of Sciences; 2011.
9. U.S. Department of Veterans Affairs. VHA Directive 2013-003: Providing Health Care for Transgender and Intersex Veterans. Washington, DC: U.S. Department of Veterans Affairs; 2013.
10. Zucker KJ, Lawrence AA. Epidemiology of gender identity disorder: recommendations for the Standards of Care of the World Professional Association for Transgender Health. Int J Transgenderism. 2009;11(1):8-18.
11. Arcelus J, Bouman WP, Van Den Noortgate W, Claes L, Witcomb G, Fernandez-Aranda F. Systematic review and meta-analysis of prevalence studies in transsexualism. Eur Psychiatry. 2015;30(6):807-815.
12. Gates GJ, Herman JL. Transgender military service in the United States. http://williamsinstitute.law.ucla.edu/wp-content/uploads/Transgender-Military -Service-May-2014.pdf. Published May 2014. Accessed November 30, 2016.
13. Flores AR, Brown TNT, and Herman JL. Race and ethnicity of adults who identify as transgender in the United States. http://williamsinstitute.law.ucla .edu/wp-content/uploads/Race-and-Ethnicity-of -Transgender-Identified-Adults-in-the-US.pdf. Published October 2016. Accessed December 13, 2016.
14. Harris BC. Likely Transgender individuals in US federal administrative records and the 2010 census. https://www.census.gov/srd/carra/15_03_Likely_Transgender_Individuals_in_ARs_and_2010Census.pdf. Published May 4, 2015. Accessed November 30, 2016.
15. Kenagy GP, Bostwick WB. Health and social service needs of transgender people in Chicago. Int J Transgenderism. 2005;8(2-3):57-66.
16. Centers for Disease Control and Prevention. National intimate partner and sexual violence survey, 2010 summary report. https://www.cdc.gov/viole nceprevention/pdf/nisvs_report2010-a.pdf. Published November 2011. Accessed December 12, 2016.
17. Bauer GR, Travers R, Scanlon K, Coleman TA. High heterogeneity of HIV-related sexual risk among transgender people in Ontario, Canada: a province-wide respondent-driven sampling survey. BMC Public Health. 2012;12(1):292-291.
18. Devis-Devis J, Pereira-Garcia S, Valencia-Peris A, Fuentes-Miguel J, López-Cañada E, Pérez-Samaniego V. Harassment patterns and risk profile in Spanish trans persons. J Homosex. 2016. [Epub ahead of print.]
19. Gates GJ. How many people are lesbian, gay, bisexual, and transgender? http://williamsinstitute.law.ucla.edu/wp-content/uploads/Gates-How-Many -People-LGBT-Apr-2011.pdf. Published April 2011. Accessed December 1, 2016.
20. Center for Disease Control and Prevention. HIV Among Women. http://www.cdc.gov/hiv/group/gender/women/index.html. Accessed December 10, 2016.
21. Bauer GR, Scheim AI, Pyne J, Travers R, Hammond R. Intervenable factors associated with suicide risk in transgender persons: a respondent driven sampling study in Ontario, Canada. BMC Public Health. 2015;15(1):525.
22. McCann E. People who are transgender: mental health concerns. J Psychiatr Ment Health Nurs. 2015;22(1):76-81.
23. Green R. Transsexual legal rights in the United States and United Kingdom: employment, medical treatment, and civil status. Arch Sex Behav. 2010;39(1):153-160.
24. Sharpe VA, Uchendu US. Ensuring appropriate care for LGBT veterans in the Veterans Health Administration. Hastings Cent Rep. 2014;44(suppl 4):S53-S55.
25. Kauth MR, Shipherd JC, Lindsay J, Blosnich JR, Brown GR, Jones KT. Access to care for transgender veterans in the Veterans Health Administration: 2006-2013. Am J Public Health. 2014;104(suppl 4):S532-S534.
26. Snelgrove JW, Jasudavisius AM, Rowe BW, Head EM, Bauer GR. "Completely out-at-sea" with "two-gender medicine": a qualitative analysis of physician-side barriers to providing healthcare for transgender patients. BMC Health Serv Res. 2012;12(1):110.
27. U.S. Department of Health and Human Services. Key features of the affordable care act. http://www .hhs.gov/healthcare/facts-and-features/key-features -of-aca/index.html. Last reviewed November 18, 2014. Accessed December 1, 2016.
28. U.S. Department of Veterans Affairs. Federal benefits for veterans, dependents, and survivors. https://www.va.gov/opa/publications/benefits_book/Chapter_1_Health_Care_Benefits.asp. Accessed December 1, 2016.
29. HealthyPeople.gov. Lesbian, gay, bisexual, and transgender health. https://www.healthypeople.gov/2020/topics-objectives/topic/lesbian-gay-bi sexual-and-transgender-health. Updated December 1, 2016. Accessed December 1, 2016.
30. Lutwak N, Byne W, Erickson-Schroth L, et al. Transgender veterans are inadequately understood by health care providers. Mil Med. 2014;179(5):483-485.
31. Dorsen C. An integrative review of nurse attitudes towards lesbian, gay, bisexual, and transgender patients. Can J Nurs Res. 2012;44(3):18-43.
32. Eliason MJ, Dibble SL, Robertson PA. Lesbian, gay, bisexual, and transgender (LGBT) physicians' experiences in the workplace. J Homosex. 2011;58(10):1355-1371.
33. Carabez R, Pellegrini M, Mankovitz A, Eliason M, Ciano M, Scott M. "Never in All My Years...": Nurses' education about LGBT health. J Prof Nurs. 2015;31(4):323-329
34. Buchholz L. Transgender care moves into the mainstream. JAMA. 2015;314(17):1785-1787.
35. VA Boston Healthcare System. Patient Care Memorandum-11-046-LM. Management of transgender veteran patients. http://www.boston.va.gov/services/images/lgbt_patient_care_memo_transgender_care.pdf. Published May 2011. Accessed December 1, 2016.
36. Cronenwett L, Sherwood G, Pohl J, et al. Quality and safety education for advanced nursing practice. Nurs Outlook. 2009;57(6):338-348.
37. Institute of Medicine. Committee on the Robert Wood Johnson Foundation Initiative on the Future of Nursing. The Future of Nursing: Leading Change, Advancing Health. Washington, DC: National Academies Press; 2011.
38. Smith EL, Cronenwett L, Sherwood G. Current assessments of quality and safety education in nursing. Nurs Outlook. 2007;55(3):132-137.
39. World Professional Association for Transgender Health (WPATH).The standards of care. http://www.wpath.org/site_page.cfm?pk_association _webpage_menu=1351&pk_association_web page=4655. Accessed December 1, 2016.
40. University of California San Francisco Department of Family and Community Medicine. Center of Excellence for Transgender Health. http://www.tran shealth.ucsf.edu/trans?page=home-00-00 Accessed December 1, 2016.
41. Center for Disease Control and Prevention. Lesbian, gay, bisexual and transgender health. http://www.cdc.gov/lgbthealth/transgender.htm. Accessed December 1, 2016.
42. American Association of Colleges of Nursing. QSEN education consortium: graduate-level QSEN competencies, knowledge, skills and attitudes. http://www.aacn.nche.edu/faculty/qsen/competen cies.pdf. Accessed December 1, 2016.
43. Andrews MM, Boyle JS. Transcultural Concepts in Nursing Care. Philadelphia, PA: Lippincott Williams & Wilkins; 2008.
44. Moran KJ, Burson R, Conrad D. The Doctor of Nursing Practice Scholarly Project: A Framework for Success. Burlington, MA: Jones & Bartlett; 2013.
45. Hanssmann C, Morrison D, Russian E, Shiu-Thornton S, Bowen D. A community-based program evaluation of community competency trainings. J Assoc Nurses AIDS Care.
46. Knapp H, Fletcher M, Taylor A, Chan K, Goetz MB. No clinic left behind: providing cost-effective in-services via distance learning. J Healthc Qual. 2011;33(5):17-24.
47. Kauth MR, Shipherd JC, Lindsay JA, Kirsh S, Knapp H, Matza L. Teleconsultation and training of VHA providers on transgender care: implementation of a multisite hub system. Telemed J E Health. 2015;21(12):1012-1018.
48. Sherman MD, Kauth MR, Ridener L, Shipherd JC, Bratkovich K, Beaulieu G. An empirical investigation of challenges and recommendations for welcoming sexual and gender minority veterans into VA care. Prof Psychol: Res Pract. 2014;45(6):433-442.
49. American Association of Colleges of Nursing. The essentials of doctoral education for advanced nursing practice. http://www.aacn.nche.edu/pub lications/position/DNPEssentials.pdf. Published October 2006. Accessed December 1, 2016.
50. Boutain DM. Social justice as a framework for professional nursing. J Nurs Educ. 2005;44(9):404-408.
51. Poteat T, German D, Kerrigan D. Managing uncertainty: a grounded theory of stigma in transgender health care encounters. Soc Sci Med. 2013;84:22-29.
52. Thornhill L, Klein P. Creating environments of care with transgender communities. J Assoc Nurs AIDS Care. 2010;21(3):230-239.
53. Collins J. Nursing cultural competencies: Improving patient care quality and satisfaction. Ohio Nurses Rev. 2015;90(1):10-11.
54. Sherman MD, Kauth MR, Shipherd JC, Street RL Jr. Communication between VA providers and sexual and gender minority veterans: a pilot study. Psychol Serv. 2014;11(2):235-242.
55. Cowan L, Fasoli DR, Hagle ME, et al. Creating an infrastructure to advance nursing practice and care for veterans. Nurse Leader. 2013;11(5):33-36.
1. Greiner AC, Knebel E, eds. Health Professions Education: A Bridge to Quality. Washington, DC: National Academies Press; 2003.
2. Institute of Medicine. Bisexual, and Transgender People: Building a Foundation for Better Understanding. Washington, DC: National Academy of Sciences; 2011.
3. Mintzberg H. The structuring of organizations: a synthesis of the research. https://papers.ssrn.com/sol3/papers.cfm?abstract_id=1496182 1979. Posted November 4, 2009. Accessed November 30, 2016.
4. U.S. Department of Veterans Affairs. VHA blue print for excellence. https://www.va.gov/health/docs/VHA_Blueprint_for_Excellence.pdf. Published September 21, 2014. Accessed November 30, 2016.
5. Morgan RO, Teal CR, Reddy SG, Ford ME, Ashton CM. Measurement in Veterans Affairs Health Services Research: veterans as a special population. Health Serv Res. 2005;40(5, part 2):1573-1583.
6. Grant JM, Mottet L, Tanis JE, Harrison J, Herman J, Keisling M. Injustice at every turn: a report of the National Transgender Discrimination Survey. http://www.thetaskforce.org/static_html/downloads /reports/reports/ntds_full.pdf. Published 2011. Accessed November 30, 2016.
7. Office of Disease Prevention and Health Promotion. Lesbian, gay, bisexual, and transgender health. http://www.healthypeople.gov/2020/topics-objec tives/topic/lesbian-gay-bisexual-and-transgender -health. Updated November 16, 2016. Accessed November 16, 2016.
8. Institute of Medicine Committee on Lesbian Gay, Bisexual, and Transgender Health Issues and Research Gaps and Opportunities. The Health of Lesbian, Gay, Bisexual, and Transgender People: Building a Foundation for Better Understanding. Washington, DC: National Academy of Sciences; 2011.
9. U.S. Department of Veterans Affairs. VHA Directive 2013-003: Providing Health Care for Transgender and Intersex Veterans. Washington, DC: U.S. Department of Veterans Affairs; 2013.
10. Zucker KJ, Lawrence AA. Epidemiology of gender identity disorder: recommendations for the Standards of Care of the World Professional Association for Transgender Health. Int J Transgenderism. 2009;11(1):8-18.
11. Arcelus J, Bouman WP, Van Den Noortgate W, Claes L, Witcomb G, Fernandez-Aranda F. Systematic review and meta-analysis of prevalence studies in transsexualism. Eur Psychiatry. 2015;30(6):807-815.
12. Gates GJ, Herman JL. Transgender military service in the United States. http://williamsinstitute.law.ucla.edu/wp-content/uploads/Transgender-Military -Service-May-2014.pdf. Published May 2014. Accessed November 30, 2016.
13. Flores AR, Brown TNT, and Herman JL. Race and ethnicity of adults who identify as transgender in the United States. http://williamsinstitute.law.ucla .edu/wp-content/uploads/Race-and-Ethnicity-of -Transgender-Identified-Adults-in-the-US.pdf. Published October 2016. Accessed December 13, 2016.
14. Harris BC. Likely Transgender individuals in US federal administrative records and the 2010 census. https://www.census.gov/srd/carra/15_03_Likely_Transgender_Individuals_in_ARs_and_2010Census.pdf. Published May 4, 2015. Accessed November 30, 2016.
15. Kenagy GP, Bostwick WB. Health and social service needs of transgender people in Chicago. Int J Transgenderism. 2005;8(2-3):57-66.
16. Centers for Disease Control and Prevention. National intimate partner and sexual violence survey, 2010 summary report. https://www.cdc.gov/viole nceprevention/pdf/nisvs_report2010-a.pdf. Published November 2011. Accessed December 12, 2016.
17. Bauer GR, Travers R, Scanlon K, Coleman TA. High heterogeneity of HIV-related sexual risk among transgender people in Ontario, Canada: a province-wide respondent-driven sampling survey. BMC Public Health. 2012;12(1):292-291.
18. Devis-Devis J, Pereira-Garcia S, Valencia-Peris A, Fuentes-Miguel J, López-Cañada E, Pérez-Samaniego V. Harassment patterns and risk profile in Spanish trans persons. J Homosex. 2016. [Epub ahead of print.]
19. Gates GJ. How many people are lesbian, gay, bisexual, and transgender? http://williamsinstitute.law.ucla.edu/wp-content/uploads/Gates-How-Many -People-LGBT-Apr-2011.pdf. Published April 2011. Accessed December 1, 2016.
20. Center for Disease Control and Prevention. HIV Among Women. http://www.cdc.gov/hiv/group/gender/women/index.html. Accessed December 10, 2016.
21. Bauer GR, Scheim AI, Pyne J, Travers R, Hammond R. Intervenable factors associated with suicide risk in transgender persons: a respondent driven sampling study in Ontario, Canada. BMC Public Health. 2015;15(1):525.
22. McCann E. People who are transgender: mental health concerns. J Psychiatr Ment Health Nurs. 2015;22(1):76-81.
23. Green R. Transsexual legal rights in the United States and United Kingdom: employment, medical treatment, and civil status. Arch Sex Behav. 2010;39(1):153-160.
24. Sharpe VA, Uchendu US. Ensuring appropriate care for LGBT veterans in the Veterans Health Administration. Hastings Cent Rep. 2014;44(suppl 4):S53-S55.
25. Kauth MR, Shipherd JC, Lindsay J, Blosnich JR, Brown GR, Jones KT. Access to care for transgender veterans in the Veterans Health Administration: 2006-2013. Am J Public Health. 2014;104(suppl 4):S532-S534.
26. Snelgrove JW, Jasudavisius AM, Rowe BW, Head EM, Bauer GR. "Completely out-at-sea" with "two-gender medicine": a qualitative analysis of physician-side barriers to providing healthcare for transgender patients. BMC Health Serv Res. 2012;12(1):110.
27. U.S. Department of Health and Human Services. Key features of the affordable care act. http://www .hhs.gov/healthcare/facts-and-features/key-features -of-aca/index.html. Last reviewed November 18, 2014. Accessed December 1, 2016.
28. U.S. Department of Veterans Affairs. Federal benefits for veterans, dependents, and survivors. https://www.va.gov/opa/publications/benefits_book/Chapter_1_Health_Care_Benefits.asp. Accessed December 1, 2016.
29. HealthyPeople.gov. Lesbian, gay, bisexual, and transgender health. https://www.healthypeople.gov/2020/topics-objectives/topic/lesbian-gay-bi sexual-and-transgender-health. Updated December 1, 2016. Accessed December 1, 2016.
30. Lutwak N, Byne W, Erickson-Schroth L, et al. Transgender veterans are inadequately understood by health care providers. Mil Med. 2014;179(5):483-485.
31. Dorsen C. An integrative review of nurse attitudes towards lesbian, gay, bisexual, and transgender patients. Can J Nurs Res. 2012;44(3):18-43.
32. Eliason MJ, Dibble SL, Robertson PA. Lesbian, gay, bisexual, and transgender (LGBT) physicians' experiences in the workplace. J Homosex. 2011;58(10):1355-1371.
33. Carabez R, Pellegrini M, Mankovitz A, Eliason M, Ciano M, Scott M. "Never in All My Years...": Nurses' education about LGBT health. J Prof Nurs. 2015;31(4):323-329
34. Buchholz L. Transgender care moves into the mainstream. JAMA. 2015;314(17):1785-1787.
35. VA Boston Healthcare System. Patient Care Memorandum-11-046-LM. Management of transgender veteran patients. http://www.boston.va.gov/services/images/lgbt_patient_care_memo_transgender_care.pdf. Published May 2011. Accessed December 1, 2016.
36. Cronenwett L, Sherwood G, Pohl J, et al. Quality and safety education for advanced nursing practice. Nurs Outlook. 2009;57(6):338-348.
37. Institute of Medicine. Committee on the Robert Wood Johnson Foundation Initiative on the Future of Nursing. The Future of Nursing: Leading Change, Advancing Health. Washington, DC: National Academies Press; 2011.
38. Smith EL, Cronenwett L, Sherwood G. Current assessments of quality and safety education in nursing. Nurs Outlook. 2007;55(3):132-137.
39. World Professional Association for Transgender Health (WPATH).The standards of care. http://www.wpath.org/site_page.cfm?pk_association _webpage_menu=1351&pk_association_web page=4655. Accessed December 1, 2016.
40. University of California San Francisco Department of Family and Community Medicine. Center of Excellence for Transgender Health. http://www.tran shealth.ucsf.edu/trans?page=home-00-00 Accessed December 1, 2016.
41. Center for Disease Control and Prevention. Lesbian, gay, bisexual and transgender health. http://www.cdc.gov/lgbthealth/transgender.htm. Accessed December 1, 2016.
42. American Association of Colleges of Nursing. QSEN education consortium: graduate-level QSEN competencies, knowledge, skills and attitudes. http://www.aacn.nche.edu/faculty/qsen/competen cies.pdf. Accessed December 1, 2016.
43. Andrews MM, Boyle JS. Transcultural Concepts in Nursing Care. Philadelphia, PA: Lippincott Williams & Wilkins; 2008.
44. Moran KJ, Burson R, Conrad D. The Doctor of Nursing Practice Scholarly Project: A Framework for Success. Burlington, MA: Jones & Bartlett; 2013.
45. Hanssmann C, Morrison D, Russian E, Shiu-Thornton S, Bowen D. A community-based program evaluation of community competency trainings. J Assoc Nurses AIDS Care.
46. Knapp H, Fletcher M, Taylor A, Chan K, Goetz MB. No clinic left behind: providing cost-effective in-services via distance learning. J Healthc Qual. 2011;33(5):17-24.
47. Kauth MR, Shipherd JC, Lindsay JA, Kirsh S, Knapp H, Matza L. Teleconsultation and training of VHA providers on transgender care: implementation of a multisite hub system. Telemed J E Health. 2015;21(12):1012-1018.
48. Sherman MD, Kauth MR, Ridener L, Shipherd JC, Bratkovich K, Beaulieu G. An empirical investigation of challenges and recommendations for welcoming sexual and gender minority veterans into VA care. Prof Psychol: Res Pract. 2014;45(6):433-442.
49. American Association of Colleges of Nursing. The essentials of doctoral education for advanced nursing practice. http://www.aacn.nche.edu/pub lications/position/DNPEssentials.pdf. Published October 2006. Accessed December 1, 2016.
50. Boutain DM. Social justice as a framework for professional nursing. J Nurs Educ. 2005;44(9):404-408.
51. Poteat T, German D, Kerrigan D. Managing uncertainty: a grounded theory of stigma in transgender health care encounters. Soc Sci Med. 2013;84:22-29.
52. Thornhill L, Klein P. Creating environments of care with transgender communities. J Assoc Nurs AIDS Care. 2010;21(3):230-239.
53. Collins J. Nursing cultural competencies: Improving patient care quality and satisfaction. Ohio Nurses Rev. 2015;90(1):10-11.
54. Sherman MD, Kauth MR, Shipherd JC, Street RL Jr. Communication between VA providers and sexual and gender minority veterans: a pilot study. Psychol Serv. 2014;11(2):235-242.
55. Cowan L, Fasoli DR, Hagle ME, et al. Creating an infrastructure to advance nursing practice and care for veterans. Nurse Leader. 2013;11(5):33-36.
Oral Rehydration Therapy for KidsA More Palatable Alternative

A 3-year-old boy is brought in by his mother for vomiting and diarrhea that started in the middle of the night. On examination, he is slightly dehydrated but does not have an acute abdomen or other source of infection. He is drinking from a sippy cup. What fluids should you recommend?
Acute gastroenteritis is a common cause of vomiting and/or diarrhea in children, resulting in 1.5 million outpatient visits and 200,000 hospital admissions annually in the United States.2 Children with gastroenteritis are at risk for dehydration, and the recommended treatment for anything less than severe dehydration is oral rehydration therapy (ORT) and early resumption of feeding upon rehydration.2
In 2002, the World Health Organization recommended an ORT with an osmolarity of 245 mOsm/L.3 However, cultural preferences, cost, taste, availability, and caregiver and professional preference for IV hydration have all been barriers to the use of ORT.2,4-8 In fact, a study of ORT preferences in 66 children ages 5 to 10 years found that less than half of the children would voluntarily drink the ORT again.5
This study evaluated the use of diluted apple juice as a more palatable alternative to ORT in children with vomiting and/or diarrhea.
STUDY SUMMARY
In kids older than 2, apple juice will do
This study was a single-center, single-blind, noninferiority RCT conducted in the emergency department (ED) of a tertiary care pediatric hospital in Canada. The researchers compared the use of half-strength apple juice to a standard ORT for rehydration in simple gastroenteritis.1 Participants were 6 months to 5 years of age, weighed more than 8 kg (17.7 lb), and had vomiting and/or diarrhea for less than 96 hours (with ≥ 3 episodes over the past 24 hours). They also had a Clinical Dehydration Scale (CDS) score < 5 and a capillary refill of < 2 seconds (see Table).9 Of the total, 68% of the children had a CDS score of 0; 25.5%, of 1 to 2; and 6.4%, of 3 to 4. Exclusion criteria included chronic gastrointestinal disease or other significant comorbidities (eg, diabetes) that could affect the clinical state and potential acute abdominal pathology.

Children were randomly assigned to receive half-strength apple juice (intervention group, n = 323) or an apple-flavored sucralose-sweetened electrolyte maintenance solution (EMS; control group, n = 324). Immediately on triage, each child received 2 L of their assigned fluid, to be used while in the ED and then at home. The children received 5 mL of fluid every two to five minutes. If a child vomited after starting the fluid, he or she was given oral ondansetron.
At discharge, caregivers were encouraged to replace 2 mL/kg of fluid for a vomiting episode and 10 mL/kg of fluid for a diarrhea episode. At home, children in the juice group could also drink any other preferred fluid, including sports beverages. The EMS group was instructed to drink only the solution provided or a comparable ORT. Caregivers were contacted daily by phone until the child had no symptoms for 24 hours. They were also asked to keep a daily log of vomiting and diarrhea frequency, as well as any subsequent health care visits. At least one follow-up contact occurred with 99.5% of the children.
The primary outcome was treatment failure, defined as the occurrence of any of the following within seven days of the ED visit: hospitalization, IV rehydration, further health care visits for diarrhea/vomiting in any setting, protracted symptoms (ie, ≥ 3 episodes of vomiting or diarrhea within a 24-hour period occurring > 7 days after enrollment), 3% or greater weight loss, or CDS score ≥ 5 at follow-up.
Treatment failure occurred in 16.7% of the juice group, compared to 25% of the EMS group (difference, 8.3 percentage points; number needed to treat [NNT], 12), consistent with noninferior effectiveness. The benefit was seen primarily in children ≥ 24 months of age. In children < 24 months, the treatment failure for juice was 23.9% and for EMS, 24.1%. In older children (those ≥ 24 months to 5 years), the treatment failure with juice was 9.8% and with EMS, 25.9% (difference, 16.2 percentage points; NNT, 6.2).
IV rehydration in the ED or within seven days of the initial visit was needed in 2.5% of the juice group and in 9% of the EMS group (difference, 6.5 percentage points; NNT, 15.4). There were no differences in hospitalization rate or in diarrhea or vomiting frequency between groups.
WHAT’S NEW
Kids drink more of what they like
This study, in a developed country, found rehydration with diluted apple juice worked just as well as ORT. In children ≥ 24 months of age, there were fewer treatment failures.
CAVEATS
Infants may not benefit; ondansetron played a role
Children in this study were only mildly dehydrated. The study did not include infants younger than 6 months of age, and the greatest benefit was seen in children ≥ 24 months of age.
Also noteworthy was that most of the children (67.4%) received an oral dose of ondansetron (0.1 mg/kg). Although ondansetron is expensive, it would be considered cost-effective if one dose prevents a hospitalization. Previous studies of oral ondansetron show it reduces vomiting (NNT, 5); lowers the rate of IV hydration in the ED (NNT, 5); and reduces the hospitalization rate from the ED (NNT, 17).10
Lastly, there are a variety of fluid replacement guidelines. In this study, fluid replacement was 2 mL/kg for a vomiting episode and 10 mL/kg for a diarrhea episode.
CHALLENGES TO IMPLEMENTATION
Given the ease of swapping diluted apple juice for ORT, there are no foreseen barriers to implementation.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2016. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice. 2016;65(12): 924-926.
1. Freedman SB, Willan AR, Boutis K, et al. Effect of dilute apple juice and preferred fluids vs electrolyte maintenance solution on treatment failure among children with mild gastroenteritis: a randomized clinical trial. JAMA. 2016;315:1966-1974.
2. King CK, Glass R, Bresee JS, et al. Managing acute gastroenteritis among children: oral rehydration, maintenance, and nutritional therapy. MMWR Recomm Rep. 2003;52:1-16.
3. World Health Organization. New formula oral rehydration salts. WHO Drug Information. 2002;16(2). http://apps.who.int/medicinedocs/en/d/Js4950e/2.4.html. Accessed December 5, 2016.
4. Cohen MB, Hardin J. Medicaid coverage of oral rehydration solutions. N Engl J Med. 1993;329:211.
5. Freedman SB, Cho D, Boutis K, et al. Assessing the palatability of oral rehydration solutions in school-aged children: a randomized crossover trial. Arch Pediatr Adolesc Med. 2010;164:696-702.
6. Reis EC, Goepp JG, Katz S, et al. Barriers to use of oral rehydration therapy. Pediatrics. 1994;93:708-711.
7. Karpas A, Finkelstein M, Reid S. Parental preference for rehydration method for children in the emergency department. Pediatr Emerg Care. 2009;25:301-306.
8. Ozuah PO, Avner JR, Stein RE. Oral rehydration, emergency physicians, and practice parameters: a national survey. Pediatrics. 2002;109:259-261.
9. Goldman RD, Friedman JN, Parkin PC. Validation of the clinical dehydration scale for children with acute gastroenteritis. Pediatrics. 2008;122:545-549.
10. Fedorowicz Z, Jagannath VA, Carter B. Antiemetics for reducing vomiting related to acute gastroenteritis in children and adolescents. Cochrane Database Syst Rev. 2011; CD005506.
A 3-year-old boy is brought in by his mother for vomiting and diarrhea that started in the middle of the night. On examination, he is slightly dehydrated but does not have an acute abdomen or other source of infection. He is drinking from a sippy cup. What fluids should you recommend?
Acute gastroenteritis is a common cause of vomiting and/or diarrhea in children, resulting in 1.5 million outpatient visits and 200,000 hospital admissions annually in the United States.2 Children with gastroenteritis are at risk for dehydration, and the recommended treatment for anything less than severe dehydration is oral rehydration therapy (ORT) and early resumption of feeding upon rehydration.2
In 2002, the World Health Organization recommended an ORT with an osmolarity of 245 mOsm/L.3 However, cultural preferences, cost, taste, availability, and caregiver and professional preference for IV hydration have all been barriers to the use of ORT.2,4-8 In fact, a study of ORT preferences in 66 children ages 5 to 10 years found that less than half of the children would voluntarily drink the ORT again.5
This study evaluated the use of diluted apple juice as a more palatable alternative to ORT in children with vomiting and/or diarrhea.
STUDY SUMMARY
In kids older than 2, apple juice will do
This study was a single-center, single-blind, noninferiority RCT conducted in the emergency department (ED) of a tertiary care pediatric hospital in Canada. The researchers compared the use of half-strength apple juice to a standard ORT for rehydration in simple gastroenteritis.1 Participants were 6 months to 5 years of age, weighed more than 8 kg (17.7 lb), and had vomiting and/or diarrhea for less than 96 hours (with ≥ 3 episodes over the past 24 hours). They also had a Clinical Dehydration Scale (CDS) score < 5 and a capillary refill of < 2 seconds (see Table).9 Of the total, 68% of the children had a CDS score of 0; 25.5%, of 1 to 2; and 6.4%, of 3 to 4. Exclusion criteria included chronic gastrointestinal disease or other significant comorbidities (eg, diabetes) that could affect the clinical state and potential acute abdominal pathology.
Children were randomly assigned to receive half-strength apple juice (intervention group, n = 323) or an apple-flavored sucralose-sweetened electrolyte maintenance solution (EMS; control group, n = 324). Immediately on triage, each child received 2 L of their assigned fluid, to be used while in the ED and then at home. The children received 5 mL of fluid every two to five minutes. If a child vomited after starting the fluid, he or she was given oral ondansetron.
At discharge, caregivers were encouraged to replace 2 mL/kg of fluid for a vomiting episode and 10 mL/kg of fluid for a diarrhea episode. At home, children in the juice group could also drink any other preferred fluid, including sports beverages. The EMS group was instructed to drink only the solution provided or a comparable ORT. Caregivers were contacted daily by phone until the child had no symptoms for 24 hours. They were also asked to keep a daily log of vomiting and diarrhea frequency, as well as any subsequent health care visits. At least one follow-up contact occurred with 99.5% of the children.
The primary outcome was treatment failure, defined as the occurrence of any of the following within seven days of the ED visit: hospitalization, IV rehydration, further health care visits for diarrhea/vomiting in any setting, protracted symptoms (ie, ≥ 3 episodes of vomiting or diarrhea within a 24-hour period occurring > 7 days after enrollment), 3% or greater weight loss, or CDS score ≥ 5 at follow-up.
Treatment failure occurred in 16.7% of the juice group, compared to 25% of the EMS group (difference, 8.3 percentage points; number needed to treat [NNT], 12), consistent with noninferior effectiveness. The benefit was seen primarily in children ≥ 24 months of age. In children < 24 months, the treatment failure for juice was 23.9% and for EMS, 24.1%. In older children (those ≥ 24 months to 5 years), the treatment failure with juice was 9.8% and with EMS, 25.9% (difference, 16.2 percentage points; NNT, 6.2).
IV rehydration in the ED or within seven days of the initial visit was needed in 2.5% of the juice group and in 9% of the EMS group (difference, 6.5 percentage points; NNT, 15.4). There were no differences in hospitalization rate or in diarrhea or vomiting frequency between groups.
WHAT’S NEW
Kids drink more of what they like
This study, in a developed country, found rehydration with diluted apple juice worked just as well as ORT. In children ≥ 24 months of age, there were fewer treatment failures.
CAVEATS
Infants may not benefit; ondansetron played a role
Children in this study were only mildly dehydrated. The study did not include infants younger than 6 months of age, and the greatest benefit was seen in children ≥ 24 months of age.
Also noteworthy was that most of the children (67.4%) received an oral dose of ondansetron (0.1 mg/kg). Although ondansetron is expensive, it would be considered cost-effective if one dose prevents a hospitalization. Previous studies of oral ondansetron show it reduces vomiting (NNT, 5); lowers the rate of IV hydration in the ED (NNT, 5); and reduces the hospitalization rate from the ED (NNT, 17).10
Lastly, there are a variety of fluid replacement guidelines. In this study, fluid replacement was 2 mL/kg for a vomiting episode and 10 mL/kg for a diarrhea episode.
CHALLENGES TO IMPLEMENTATION
Given the ease of swapping diluted apple juice for ORT, there are no foreseen barriers to implementation.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2016. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice. 2016;65(12): 924-926.
A 3-year-old boy is brought in by his mother for vomiting and diarrhea that started in the middle of the night. On examination, he is slightly dehydrated but does not have an acute abdomen or other source of infection. He is drinking from a sippy cup. What fluids should you recommend?
Acute gastroenteritis is a common cause of vomiting and/or diarrhea in children, resulting in 1.5 million outpatient visits and 200,000 hospital admissions annually in the United States.2 Children with gastroenteritis are at risk for dehydration, and the recommended treatment for anything less than severe dehydration is oral rehydration therapy (ORT) and early resumption of feeding upon rehydration.2
In 2002, the World Health Organization recommended an ORT with an osmolarity of 245 mOsm/L.3 However, cultural preferences, cost, taste, availability, and caregiver and professional preference for IV hydration have all been barriers to the use of ORT.2,4-8 In fact, a study of ORT preferences in 66 children ages 5 to 10 years found that less than half of the children would voluntarily drink the ORT again.5
This study evaluated the use of diluted apple juice as a more palatable alternative to ORT in children with vomiting and/or diarrhea.
STUDY SUMMARY
In kids older than 2, apple juice will do
This study was a single-center, single-blind, noninferiority RCT conducted in the emergency department (ED) of a tertiary care pediatric hospital in Canada. The researchers compared the use of half-strength apple juice to a standard ORT for rehydration in simple gastroenteritis.1 Participants were 6 months to 5 years of age, weighed more than 8 kg (17.7 lb), and had vomiting and/or diarrhea for less than 96 hours (with ≥ 3 episodes over the past 24 hours). They also had a Clinical Dehydration Scale (CDS) score < 5 and a capillary refill of < 2 seconds (see Table).9 Of the total, 68% of the children had a CDS score of 0; 25.5%, of 1 to 2; and 6.4%, of 3 to 4. Exclusion criteria included chronic gastrointestinal disease or other significant comorbidities (eg, diabetes) that could affect the clinical state and potential acute abdominal pathology.
Children were randomly assigned to receive half-strength apple juice (intervention group, n = 323) or an apple-flavored sucralose-sweetened electrolyte maintenance solution (EMS; control group, n = 324). Immediately on triage, each child received 2 L of their assigned fluid, to be used while in the ED and then at home. The children received 5 mL of fluid every two to five minutes. If a child vomited after starting the fluid, he or she was given oral ondansetron.
At discharge, caregivers were encouraged to replace 2 mL/kg of fluid for a vomiting episode and 10 mL/kg of fluid for a diarrhea episode. At home, children in the juice group could also drink any other preferred fluid, including sports beverages. The EMS group was instructed to drink only the solution provided or a comparable ORT. Caregivers were contacted daily by phone until the child had no symptoms for 24 hours. They were also asked to keep a daily log of vomiting and diarrhea frequency, as well as any subsequent health care visits. At least one follow-up contact occurred with 99.5% of the children.
The primary outcome was treatment failure, defined as the occurrence of any of the following within seven days of the ED visit: hospitalization, IV rehydration, further health care visits for diarrhea/vomiting in any setting, protracted symptoms (ie, ≥ 3 episodes of vomiting or diarrhea within a 24-hour period occurring > 7 days after enrollment), 3% or greater weight loss, or CDS score ≥ 5 at follow-up.
Treatment failure occurred in 16.7% of the juice group, compared to 25% of the EMS group (difference, 8.3 percentage points; number needed to treat [NNT], 12), consistent with noninferior effectiveness. The benefit was seen primarily in children ≥ 24 months of age. In children < 24 months, the treatment failure for juice was 23.9% and for EMS, 24.1%. In older children (those ≥ 24 months to 5 years), the treatment failure with juice was 9.8% and with EMS, 25.9% (difference, 16.2 percentage points; NNT, 6.2).
IV rehydration in the ED or within seven days of the initial visit was needed in 2.5% of the juice group and in 9% of the EMS group (difference, 6.5 percentage points; NNT, 15.4). There were no differences in hospitalization rate or in diarrhea or vomiting frequency between groups.
WHAT’S NEW
Kids drink more of what they like
This study, in a developed country, found rehydration with diluted apple juice worked just as well as ORT. In children ≥ 24 months of age, there were fewer treatment failures.
CAVEATS
Infants may not benefit; ondansetron played a role
Children in this study were only mildly dehydrated. The study did not include infants younger than 6 months of age, and the greatest benefit was seen in children ≥ 24 months of age.
Also noteworthy was that most of the children (67.4%) received an oral dose of ondansetron (0.1 mg/kg). Although ondansetron is expensive, it would be considered cost-effective if one dose prevents a hospitalization. Previous studies of oral ondansetron show it reduces vomiting (NNT, 5); lowers the rate of IV hydration in the ED (NNT, 5); and reduces the hospitalization rate from the ED (NNT, 17).10
Lastly, there are a variety of fluid replacement guidelines. In this study, fluid replacement was 2 mL/kg for a vomiting episode and 10 mL/kg for a diarrhea episode.
CHALLENGES TO IMPLEMENTATION
Given the ease of swapping diluted apple juice for ORT, there are no foreseen barriers to implementation.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2016. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice. 2016;65(12): 924-926.
1. Freedman SB, Willan AR, Boutis K, et al. Effect of dilute apple juice and preferred fluids vs electrolyte maintenance solution on treatment failure among children with mild gastroenteritis: a randomized clinical trial. JAMA. 2016;315:1966-1974.
2. King CK, Glass R, Bresee JS, et al. Managing acute gastroenteritis among children: oral rehydration, maintenance, and nutritional therapy. MMWR Recomm Rep. 2003;52:1-16.
3. World Health Organization. New formula oral rehydration salts. WHO Drug Information. 2002;16(2). http://apps.who.int/medicinedocs/en/d/Js4950e/2.4.html. Accessed December 5, 2016.
4. Cohen MB, Hardin J. Medicaid coverage of oral rehydration solutions. N Engl J Med. 1993;329:211.
5. Freedman SB, Cho D, Boutis K, et al. Assessing the palatability of oral rehydration solutions in school-aged children: a randomized crossover trial. Arch Pediatr Adolesc Med. 2010;164:696-702.
6. Reis EC, Goepp JG, Katz S, et al. Barriers to use of oral rehydration therapy. Pediatrics. 1994;93:708-711.
7. Karpas A, Finkelstein M, Reid S. Parental preference for rehydration method for children in the emergency department. Pediatr Emerg Care. 2009;25:301-306.
8. Ozuah PO, Avner JR, Stein RE. Oral rehydration, emergency physicians, and practice parameters: a national survey. Pediatrics. 2002;109:259-261.
9. Goldman RD, Friedman JN, Parkin PC. Validation of the clinical dehydration scale for children with acute gastroenteritis. Pediatrics. 2008;122:545-549.
10. Fedorowicz Z, Jagannath VA, Carter B. Antiemetics for reducing vomiting related to acute gastroenteritis in children and adolescents. Cochrane Database Syst Rev. 2011; CD005506.
1. Freedman SB, Willan AR, Boutis K, et al. Effect of dilute apple juice and preferred fluids vs electrolyte maintenance solution on treatment failure among children with mild gastroenteritis: a randomized clinical trial. JAMA. 2016;315:1966-1974.
2. King CK, Glass R, Bresee JS, et al. Managing acute gastroenteritis among children: oral rehydration, maintenance, and nutritional therapy. MMWR Recomm Rep. 2003;52:1-16.
3. World Health Organization. New formula oral rehydration salts. WHO Drug Information. 2002;16(2). http://apps.who.int/medicinedocs/en/d/Js4950e/2.4.html. Accessed December 5, 2016.
4. Cohen MB, Hardin J. Medicaid coverage of oral rehydration solutions. N Engl J Med. 1993;329:211.
5. Freedman SB, Cho D, Boutis K, et al. Assessing the palatability of oral rehydration solutions in school-aged children: a randomized crossover trial. Arch Pediatr Adolesc Med. 2010;164:696-702.
6. Reis EC, Goepp JG, Katz S, et al. Barriers to use of oral rehydration therapy. Pediatrics. 1994;93:708-711.
7. Karpas A, Finkelstein M, Reid S. Parental preference for rehydration method for children in the emergency department. Pediatr Emerg Care. 2009;25:301-306.
8. Ozuah PO, Avner JR, Stein RE. Oral rehydration, emergency physicians, and practice parameters: a national survey. Pediatrics. 2002;109:259-261.
9. Goldman RD, Friedman JN, Parkin PC. Validation of the clinical dehydration scale for children with acute gastroenteritis. Pediatrics. 2008;122:545-549.
10. Fedorowicz Z, Jagannath VA, Carter B. Antiemetics for reducing vomiting related to acute gastroenteritis in children and adolescents. Cochrane Database Syst Rev. 2011; CD005506.
Silver
Silver is a naturally occurring chemical element (number 47 on the periodic table) with the chemical symbol Ag, which is derived from the Latin word argentum (árgyros in Greek), from the Indo-European root “arg-,” meaning “shining,” “white,” or “gray.” It has been used for medical purposes since ancient times, with Hippocrates (circa 460-370 BCE) noting the beneficial healing and disease-altering activity of the element.1
In modern times, silver compounds – in metallic, nanocrystalline, and ionic formulations – have exhibited broad antibacterial activity and attracted interest for topical antiseptic use in wound dressings.2 Nanocrystalline silver dressings were introduced commercially as antimicrobial dressings in 1998.3 Silver is now used in dressings, catheters, cleansers, ophthalmic ointments, and myriad other medical products. In 2010 alone, an estimated 15 metric tons of silver were incorporated into medical products worldwide.4 It also is included in personal care products, textiles, and water purification devices. In topical skin preparations, the noble metal is included as colloidal silver (suspension of silver particles in an aqueous base) or nanosilver (as nanoparticles ranging from 1 nm to 100 nm in at least one dimension).5 Silver has been used to treat burns and wounds, but this discussion will be limited to acne, atopic dermatitis, and the anti-inflammatory response.

Anti-inflammatory uses
For several decades, noble metals including silver have been known to exert anti-inflammatory activity.7-11 In the case of silver, its anti-inflammatory properties appear to be mediated by its influence on the cytokine system. Silver nanoparticles inhibit the activity of interleukin-6 (IL-6), IL-12, IL-1beta, and tumor necrosis factor–alpha (TNF-alpha). This impact on the cytokine system is responsible for the impact of silver in demonstrably alleviating symptoms of rheumatoid arthritis.3
In 2004, Bhol et al. used dinitrochlorobenzene (DNCB) to induce allergic contact dermatitis in a guinea pig model, finding that topical nanocrystalline silver cream dose-dependently decreased erythema and as effectively as topical steroids and immunosuppressants.12 The next year, Bhol and Schechter showed that nanocrystalline silver suppressed allergic contact dermatitis in mice, inhibited TNF-alpha and IL-12 expression, and induced inflammatory cell apoptosis.13
In 2008, Nadworny et al. used a porcine contact dermatitis model to investigate the anti-inflammatory activity of nanocrystalline silver. They found that nanocrystalline silver treatments reduced DNCB-induced erythema and edema, promoted apoptosis in dermal cells, and diminished matrix metalloproteinase (MMP) and proinflammatory cytokine expression.3 The investigators speculated that the lower TNF-alpha observed in the silver-treated animals occurred due to apoptosis of the inflammatory cells.
Acne
Silver acts as a bactericidal and anti-inflammatory agent, without generating free radicals, as seen with benzoyl peroxide. Therefore, it is a compelling option for responding to the presence of Propionibacterium acnes. However, silver has not been approved by the Food and Drug Administration for this use. Even though formal acne studies have not been performed with silver sulfadiazine, it has long been used “off-label” for this purpose. As suggested above, the use of silver sulfadiazine for acne is limited by the risk of sulfa allergy. Cosmetic appearance and ease of use also are limiting factors, as silver sulfadiazine preparations are characterized by a thick, white pasty consistency. Other options for use of silver to treat acne include silver-containing cleansers and textiles.
Atopic dermatitis
A 2006 study in patients with atopic dermatitis demonstrated that silver-coated textiles could significantly diminish Staphylococcus aureus density after 2 days of wearing, with the effect enduring through the end of 7 days of treatment and then 1 week after removal of the textiles.14 Within 2 weeks, objective and subjective symptoms of atopic dermatitis were significantly enhanced in association with the silver-coated textiles, compared with cotton, without measurable adverse effects. A technology called Padycare incorporates silver into micromesh material (82% polyamide, 18% Lycra) used in clothing and bedding.15 As compared with topical formulations applied directly to the skin, textiles confer certain advantages such as preventing scratching and protecting against irritating substances and allergens. Washing of silver-infused textiles is a possible disadvantage, though, as the amount of silver lost from textiles can range from a 100% loss after four washings to less than a 1% loss.16 It also is important to note that there are concerns regarding the potential of silver to leak from textiles into the water supply, and eradicating the beneficial bacteria used to treat the water.
Conclusion
Despite centuries of medical use, silver has not been approved by the FDA for any medical applications. Further study, particularly in terms of safety and efficacy, is necessary. Nevertheless, it is used off-label before and after minimally invasive dermatologic procedures (for example, dermal filling, botulinum toxin injections, chemical peeling) because of its antimicrobial and anti-inflammatory activities as well as soothing qualities for facial skin and the skin barrier. Silver appears to be particularly suitable for use as an acne therapy option due to the low risk of bacterial resistance, lack of irritation, and its preservation of the skin barrier unlike harsher options such as retinoids, antibiotics, and benzoyl peroxide.
References
1. Adv Skin Wound Care. 2006 Nov-Dec;19(9):472-4.
2. Clin Infect Dis. 2009 Nov 15;49(10):1541-9.
3. Nanomedicine. 2008 Sep;4(3):241-51.
4. J Antimicrob Chemother. 2013 Jan;68(1):131-8.
5. Nanocrystalline Silver: Use in wound care, in Current Advances in the Medical Application of Nanotechnology (Manchester, England: Bentham Books, 2012, pp. 25-31).
6. Nanomedicine. 2013 Jan;9(1):39-54.
7. Jpn J Pharmacol. 1965 Jun;15(2):131-4.
8. J Allergy Clin Immunol. 1995 Aug;96(2):251-6.
9. Inflamm Res. 2003 Dec;52(12):487-501.
10. J Nutr Environ Med. 1997;7(4):295-305.
11. Clin Exp Pharmacol Physiol. 2000 Mar;27(3):139-44.
12. Clin Exp Dermatol. 2004 May;29(3):282-7.
13. Br J Dermatol. 2005 Jun;152(6):1235-42.
14. Curr Probl Dermatol. 2006;33:152-64.
15. J Eur Acad Dermatol Venereol. 2006 May;20(5):534-41.
16. Environ Sci Technol. 2008 Jun 1;42(11):4133-9.
Dr. Baumann is chief executive officer of the Baumann Cosmetic & Research Institute in the Design District in Miami. She founded the Cosmetic Dermatology Center at the University of Miami in 1997. Dr. Baumann wrote the textbook “Cosmetic Dermatology: Principles and Practice” (New York: McGraw-Hill, 2002), and a book for consumers, “The Skin Type Solution” (New York: Bantam Dell, 2006). Her latest book, “Cosmeceuticals and Cosmetic Ingredients,” was published in November 2014. Dr. Baumann has received funding for clinical grants from Allergan, Aveeno, Avon, Evolus, Galderma, GlaxoSmithKline, Kythera Biopharmaceuticals, Mary Kay, Medicis, Neutrogena, Philosophy, Topix, and Unilever. Dr. Baumann also developed and owns the Baumann Skin Type Solution skin typing systems and related products.
Silver is a naturally occurring chemical element (number 47 on the periodic table) with the chemical symbol Ag, which is derived from the Latin word argentum (árgyros in Greek), from the Indo-European root “arg-,” meaning “shining,” “white,” or “gray.” It has been used for medical purposes since ancient times, with Hippocrates (circa 460-370 BCE) noting the beneficial healing and disease-altering activity of the element.1
In modern times, silver compounds – in metallic, nanocrystalline, and ionic formulations – have exhibited broad antibacterial activity and attracted interest for topical antiseptic use in wound dressings.2 Nanocrystalline silver dressings were introduced commercially as antimicrobial dressings in 1998.3 Silver is now used in dressings, catheters, cleansers, ophthalmic ointments, and myriad other medical products. In 2010 alone, an estimated 15 metric tons of silver were incorporated into medical products worldwide.4 It also is included in personal care products, textiles, and water purification devices. In topical skin preparations, the noble metal is included as colloidal silver (suspension of silver particles in an aqueous base) or nanosilver (as nanoparticles ranging from 1 nm to 100 nm in at least one dimension).5 Silver has been used to treat burns and wounds, but this discussion will be limited to acne, atopic dermatitis, and the anti-inflammatory response.
Anti-inflammatory uses
For several decades, noble metals including silver have been known to exert anti-inflammatory activity.7-11 In the case of silver, its anti-inflammatory properties appear to be mediated by its influence on the cytokine system. Silver nanoparticles inhibit the activity of interleukin-6 (IL-6), IL-12, IL-1beta, and tumor necrosis factor–alpha (TNF-alpha). This impact on the cytokine system is responsible for the impact of silver in demonstrably alleviating symptoms of rheumatoid arthritis.3
In 2004, Bhol et al. used dinitrochlorobenzene (DNCB) to induce allergic contact dermatitis in a guinea pig model, finding that topical nanocrystalline silver cream dose-dependently decreased erythema and as effectively as topical steroids and immunosuppressants.12 The next year, Bhol and Schechter showed that nanocrystalline silver suppressed allergic contact dermatitis in mice, inhibited TNF-alpha and IL-12 expression, and induced inflammatory cell apoptosis.13
In 2008, Nadworny et al. used a porcine contact dermatitis model to investigate the anti-inflammatory activity of nanocrystalline silver. They found that nanocrystalline silver treatments reduced DNCB-induced erythema and edema, promoted apoptosis in dermal cells, and diminished matrix metalloproteinase (MMP) and proinflammatory cytokine expression.3 The investigators speculated that the lower TNF-alpha observed in the silver-treated animals occurred due to apoptosis of the inflammatory cells.
Acne
Silver acts as a bactericidal and anti-inflammatory agent, without generating free radicals, as seen with benzoyl peroxide. Therefore, it is a compelling option for responding to the presence of Propionibacterium acnes. However, silver has not been approved by the Food and Drug Administration for this use. Even though formal acne studies have not been performed with silver sulfadiazine, it has long been used “off-label” for this purpose. As suggested above, the use of silver sulfadiazine for acne is limited by the risk of sulfa allergy. Cosmetic appearance and ease of use also are limiting factors, as silver sulfadiazine preparations are characterized by a thick, white pasty consistency. Other options for use of silver to treat acne include silver-containing cleansers and textiles.
Atopic dermatitis
A 2006 study in patients with atopic dermatitis demonstrated that silver-coated textiles could significantly diminish Staphylococcus aureus density after 2 days of wearing, with the effect enduring through the end of 7 days of treatment and then 1 week after removal of the textiles.14 Within 2 weeks, objective and subjective symptoms of atopic dermatitis were significantly enhanced in association with the silver-coated textiles, compared with cotton, without measurable adverse effects. A technology called Padycare incorporates silver into micromesh material (82% polyamide, 18% Lycra) used in clothing and bedding.15 As compared with topical formulations applied directly to the skin, textiles confer certain advantages such as preventing scratching and protecting against irritating substances and allergens. Washing of silver-infused textiles is a possible disadvantage, though, as the amount of silver lost from textiles can range from a 100% loss after four washings to less than a 1% loss.16 It also is important to note that there are concerns regarding the potential of silver to leak from textiles into the water supply, and eradicating the beneficial bacteria used to treat the water.
Conclusion
Despite centuries of medical use, silver has not been approved by the FDA for any medical applications. Further study, particularly in terms of safety and efficacy, is necessary. Nevertheless, it is used off-label before and after minimally invasive dermatologic procedures (for example, dermal filling, botulinum toxin injections, chemical peeling) because of its antimicrobial and anti-inflammatory activities as well as soothing qualities for facial skin and the skin barrier. Silver appears to be particularly suitable for use as an acne therapy option due to the low risk of bacterial resistance, lack of irritation, and its preservation of the skin barrier unlike harsher options such as retinoids, antibiotics, and benzoyl peroxide.
References
1. Adv Skin Wound Care. 2006 Nov-Dec;19(9):472-4.
2. Clin Infect Dis. 2009 Nov 15;49(10):1541-9.
3. Nanomedicine. 2008 Sep;4(3):241-51.
4. J Antimicrob Chemother. 2013 Jan;68(1):131-8.
5. Nanocrystalline Silver: Use in wound care, in Current Advances in the Medical Application of Nanotechnology (Manchester, England: Bentham Books, 2012, pp. 25-31).
6. Nanomedicine. 2013 Jan;9(1):39-54.
7. Jpn J Pharmacol. 1965 Jun;15(2):131-4.
8. J Allergy Clin Immunol. 1995 Aug;96(2):251-6.
9. Inflamm Res. 2003 Dec;52(12):487-501.
10. J Nutr Environ Med. 1997;7(4):295-305.
11. Clin Exp Pharmacol Physiol. 2000 Mar;27(3):139-44.
12. Clin Exp Dermatol. 2004 May;29(3):282-7.
13. Br J Dermatol. 2005 Jun;152(6):1235-42.
14. Curr Probl Dermatol. 2006;33:152-64.
15. J Eur Acad Dermatol Venereol. 2006 May;20(5):534-41.
16. Environ Sci Technol. 2008 Jun 1;42(11):4133-9.
Dr. Baumann is chief executive officer of the Baumann Cosmetic & Research Institute in the Design District in Miami. She founded the Cosmetic Dermatology Center at the University of Miami in 1997. Dr. Baumann wrote the textbook “Cosmetic Dermatology: Principles and Practice” (New York: McGraw-Hill, 2002), and a book for consumers, “The Skin Type Solution” (New York: Bantam Dell, 2006). Her latest book, “Cosmeceuticals and Cosmetic Ingredients,” was published in November 2014. Dr. Baumann has received funding for clinical grants from Allergan, Aveeno, Avon, Evolus, Galderma, GlaxoSmithKline, Kythera Biopharmaceuticals, Mary Kay, Medicis, Neutrogena, Philosophy, Topix, and Unilever. Dr. Baumann also developed and owns the Baumann Skin Type Solution skin typing systems and related products.
Silver is a naturally occurring chemical element (number 47 on the periodic table) with the chemical symbol Ag, which is derived from the Latin word argentum (árgyros in Greek), from the Indo-European root “arg-,” meaning “shining,” “white,” or “gray.” It has been used for medical purposes since ancient times, with Hippocrates (circa 460-370 BCE) noting the beneficial healing and disease-altering activity of the element.1
In modern times, silver compounds – in metallic, nanocrystalline, and ionic formulations – have exhibited broad antibacterial activity and attracted interest for topical antiseptic use in wound dressings.2 Nanocrystalline silver dressings were introduced commercially as antimicrobial dressings in 1998.3 Silver is now used in dressings, catheters, cleansers, ophthalmic ointments, and myriad other medical products. In 2010 alone, an estimated 15 metric tons of silver were incorporated into medical products worldwide.4 It also is included in personal care products, textiles, and water purification devices. In topical skin preparations, the noble metal is included as colloidal silver (suspension of silver particles in an aqueous base) or nanosilver (as nanoparticles ranging from 1 nm to 100 nm in at least one dimension).5 Silver has been used to treat burns and wounds, but this discussion will be limited to acne, atopic dermatitis, and the anti-inflammatory response.
Anti-inflammatory uses
For several decades, noble metals including silver have been known to exert anti-inflammatory activity.7-11 In the case of silver, its anti-inflammatory properties appear to be mediated by its influence on the cytokine system. Silver nanoparticles inhibit the activity of interleukin-6 (IL-6), IL-12, IL-1beta, and tumor necrosis factor–alpha (TNF-alpha). This impact on the cytokine system is responsible for the impact of silver in demonstrably alleviating symptoms of rheumatoid arthritis.3
In 2004, Bhol et al. used dinitrochlorobenzene (DNCB) to induce allergic contact dermatitis in a guinea pig model, finding that topical nanocrystalline silver cream dose-dependently decreased erythema and as effectively as topical steroids and immunosuppressants.12 The next year, Bhol and Schechter showed that nanocrystalline silver suppressed allergic contact dermatitis in mice, inhibited TNF-alpha and IL-12 expression, and induced inflammatory cell apoptosis.13
In 2008, Nadworny et al. used a porcine contact dermatitis model to investigate the anti-inflammatory activity of nanocrystalline silver. They found that nanocrystalline silver treatments reduced DNCB-induced erythema and edema, promoted apoptosis in dermal cells, and diminished matrix metalloproteinase (MMP) and proinflammatory cytokine expression.3 The investigators speculated that the lower TNF-alpha observed in the silver-treated animals occurred due to apoptosis of the inflammatory cells.
Acne
Silver acts as a bactericidal and anti-inflammatory agent, without generating free radicals, as seen with benzoyl peroxide. Therefore, it is a compelling option for responding to the presence of Propionibacterium acnes. However, silver has not been approved by the Food and Drug Administration for this use. Even though formal acne studies have not been performed with silver sulfadiazine, it has long been used “off-label” for this purpose. As suggested above, the use of silver sulfadiazine for acne is limited by the risk of sulfa allergy. Cosmetic appearance and ease of use also are limiting factors, as silver sulfadiazine preparations are characterized by a thick, white pasty consistency. Other options for use of silver to treat acne include silver-containing cleansers and textiles.
Atopic dermatitis
A 2006 study in patients with atopic dermatitis demonstrated that silver-coated textiles could significantly diminish Staphylococcus aureus density after 2 days of wearing, with the effect enduring through the end of 7 days of treatment and then 1 week after removal of the textiles.14 Within 2 weeks, objective and subjective symptoms of atopic dermatitis were significantly enhanced in association with the silver-coated textiles, compared with cotton, without measurable adverse effects. A technology called Padycare incorporates silver into micromesh material (82% polyamide, 18% Lycra) used in clothing and bedding.15 As compared with topical formulations applied directly to the skin, textiles confer certain advantages such as preventing scratching and protecting against irritating substances and allergens. Washing of silver-infused textiles is a possible disadvantage, though, as the amount of silver lost from textiles can range from a 100% loss after four washings to less than a 1% loss.16 It also is important to note that there are concerns regarding the potential of silver to leak from textiles into the water supply, and eradicating the beneficial bacteria used to treat the water.
Conclusion
Despite centuries of medical use, silver has not been approved by the FDA for any medical applications. Further study, particularly in terms of safety and efficacy, is necessary. Nevertheless, it is used off-label before and after minimally invasive dermatologic procedures (for example, dermal filling, botulinum toxin injections, chemical peeling) because of its antimicrobial and anti-inflammatory activities as well as soothing qualities for facial skin and the skin barrier. Silver appears to be particularly suitable for use as an acne therapy option due to the low risk of bacterial resistance, lack of irritation, and its preservation of the skin barrier unlike harsher options such as retinoids, antibiotics, and benzoyl peroxide.
References
1. Adv Skin Wound Care. 2006 Nov-Dec;19(9):472-4.
2. Clin Infect Dis. 2009 Nov 15;49(10):1541-9.
3. Nanomedicine. 2008 Sep;4(3):241-51.
4. J Antimicrob Chemother. 2013 Jan;68(1):131-8.
5. Nanocrystalline Silver: Use in wound care, in Current Advances in the Medical Application of Nanotechnology (Manchester, England: Bentham Books, 2012, pp. 25-31).
6. Nanomedicine. 2013 Jan;9(1):39-54.
7. Jpn J Pharmacol. 1965 Jun;15(2):131-4.
8. J Allergy Clin Immunol. 1995 Aug;96(2):251-6.
9. Inflamm Res. 2003 Dec;52(12):487-501.
10. J Nutr Environ Med. 1997;7(4):295-305.
11. Clin Exp Pharmacol Physiol. 2000 Mar;27(3):139-44.
12. Clin Exp Dermatol. 2004 May;29(3):282-7.
13. Br J Dermatol. 2005 Jun;152(6):1235-42.
14. Curr Probl Dermatol. 2006;33:152-64.
15. J Eur Acad Dermatol Venereol. 2006 May;20(5):534-41.
16. Environ Sci Technol. 2008 Jun 1;42(11):4133-9.
Dr. Baumann is chief executive officer of the Baumann Cosmetic & Research Institute in the Design District in Miami. She founded the Cosmetic Dermatology Center at the University of Miami in 1997. Dr. Baumann wrote the textbook “Cosmetic Dermatology: Principles and Practice” (New York: McGraw-Hill, 2002), and a book for consumers, “The Skin Type Solution” (New York: Bantam Dell, 2006). Her latest book, “Cosmeceuticals and Cosmetic Ingredients,” was published in November 2014. Dr. Baumann has received funding for clinical grants from Allergan, Aveeno, Avon, Evolus, Galderma, GlaxoSmithKline, Kythera Biopharmaceuticals, Mary Kay, Medicis, Neutrogena, Philosophy, Topix, and Unilever. Dr. Baumann also developed and owns the Baumann Skin Type Solution skin typing systems and related products.

Emergency Ultrasound: Ultrasound-Guided Hip Arthrocentesis
Hip ultrasound has long been considered an effective diagnostic and interventional tool to identify hip effusions and perform guided arthrocentesis in patients with suspected septic arthritis. Although imaging and interventional techniques are typically performed by interventional radiologists, several case reports support the use of these techniques by the emergency physician (EP) in both pediatric and adult patients presenting with hip pain.1,2
Hip ultrasound permits rapid visualization of the joint space to assess the presence of a hip effusion, and provides the opportunity for the clinician to quickly perform hip arthrocentesis and to obtain synovial fluid for analysis—the current gold standard of diagnosis. The current literature shows treatment of effusion in the adult hip via ultrasound-guided interventional methods to be more convenient and less painful than traditional fluoroscopic-guided techniques, and to have the same procedural success rate.3 With the increasing utilization of point-of-care (POC) ultrasound in the ED, ultrasound-guided hip arthrocentesis has become a powerful tool in the EP’s armamentarium to aid in evaluating and treating patients in the ED presenting with hip pain.
Imaging Technique
To perform an ultrasound-guided arthrocentesis, the patient should be placed in the supine position, with both knees bent and the hips externally rotated in the frog leg position (Figure 1). 

Arthrocentesis
When an effusion is present, arthrocentesis is warranted. To perform this procedure, the femoral vessels should be identified inferior to the inguinal ligament and avoided laterally. The hip should be prepared in a sterile fashion and a lubricated probe should be placed in a sterile dressing with a cord cover. The effusion should be visualized again, and the area should be anesthetized superficially and deeply with local anesthetic, aspirating prior to infusing at the deeper levels. An 18-gauge spinal needle affixed to a 20-mL syringe should be introduced and advanced while aspirating under direct visualization through the capsule of the hip into the effusion. The fluid is then aspirated and sent for laboratory analysis.
Summary
A delayed diagnosis of hip effusion and failure to initiate prompt treatment are the most common causes of late complications of septic arthritis.4 Point-of-care diagnostic and interventional ultrasound of the hip permit instant visualization and implementation of immediate diagnostic and therapeutic measures, which decrease morbidity in adult patients with septic arthritis. Hip arthrocentesis with subsequent synovial fluid analysis, the gold standard of diagnosis, has traditionally been performed by radiology services. Recent literature, however, has shown performance of these ultrasound-guided techniques by EPs to be safe and efficient, facilitating time to treatment.
1. Freeman K, Dewitz A, Baker WE. Ultrasound-guided hip arthrocentesis in the ED. Am J Emerg Med. 2007;25(1):80-86. doi:10.1016/j.ajem.2006.08.002.
2. Minardi JJ, Lander OM. Septic hip arthritis: diagnosis and arthrocentesis using bedside ultrasound. J Emerg Med. 2012;43(2):316-318. doi:10.1016/j.jemermed.2011.09.029.
3. Byrd JW, Potts EA, Allison RK, Jones KS. Ultrasound-guided hip injections: a comparative study with fluoroscopy-guided injections. Arthroscopy. 2014;30(1):42-46. doi:10.1016/j.arthro.2013.09.083.
4. Mascioli AA, Park AL. Infectious arthritis. In: Canale ST, Beaty JH eds. Campbell’s Operative Orthopaedics. Vol 1. 13th ed. Philadelphia, PA: Elsevier Mosby; 2013:749-772.
Hip ultrasound has long been considered an effective diagnostic and interventional tool to identify hip effusions and perform guided arthrocentesis in patients with suspected septic arthritis. Although imaging and interventional techniques are typically performed by interventional radiologists, several case reports support the use of these techniques by the emergency physician (EP) in both pediatric and adult patients presenting with hip pain.1,2
Hip ultrasound permits rapid visualization of the joint space to assess the presence of a hip effusion, and provides the opportunity for the clinician to quickly perform hip arthrocentesis and to obtain synovial fluid for analysis—the current gold standard of diagnosis. The current literature shows treatment of effusion in the adult hip via ultrasound-guided interventional methods to be more convenient and less painful than traditional fluoroscopic-guided techniques, and to have the same procedural success rate.3 With the increasing utilization of point-of-care (POC) ultrasound in the ED, ultrasound-guided hip arthrocentesis has become a powerful tool in the EP’s armamentarium to aid in evaluating and treating patients in the ED presenting with hip pain.
Imaging Technique
To perform an ultrasound-guided arthrocentesis, the patient should be placed in the supine position, with both knees bent and the hips externally rotated in the frog leg position (Figure 1).
Arthrocentesis
When an effusion is present, arthrocentesis is warranted. To perform this procedure, the femoral vessels should be identified inferior to the inguinal ligament and avoided laterally. The hip should be prepared in a sterile fashion and a lubricated probe should be placed in a sterile dressing with a cord cover. The effusion should be visualized again, and the area should be anesthetized superficially and deeply with local anesthetic, aspirating prior to infusing at the deeper levels. An 18-gauge spinal needle affixed to a 20-mL syringe should be introduced and advanced while aspirating under direct visualization through the capsule of the hip into the effusion. The fluid is then aspirated and sent for laboratory analysis.
Summary
A delayed diagnosis of hip effusion and failure to initiate prompt treatment are the most common causes of late complications of septic arthritis.4 Point-of-care diagnostic and interventional ultrasound of the hip permit instant visualization and implementation of immediate diagnostic and therapeutic measures, which decrease morbidity in adult patients with septic arthritis. Hip arthrocentesis with subsequent synovial fluid analysis, the gold standard of diagnosis, has traditionally been performed by radiology services. Recent literature, however, has shown performance of these ultrasound-guided techniques by EPs to be safe and efficient, facilitating time to treatment.
Hip ultrasound has long been considered an effective diagnostic and interventional tool to identify hip effusions and perform guided arthrocentesis in patients with suspected septic arthritis. Although imaging and interventional techniques are typically performed by interventional radiologists, several case reports support the use of these techniques by the emergency physician (EP) in both pediatric and adult patients presenting with hip pain.1,2
Hip ultrasound permits rapid visualization of the joint space to assess the presence of a hip effusion, and provides the opportunity for the clinician to quickly perform hip arthrocentesis and to obtain synovial fluid for analysis—the current gold standard of diagnosis. The current literature shows treatment of effusion in the adult hip via ultrasound-guided interventional methods to be more convenient and less painful than traditional fluoroscopic-guided techniques, and to have the same procedural success rate.3 With the increasing utilization of point-of-care (POC) ultrasound in the ED, ultrasound-guided hip arthrocentesis has become a powerful tool in the EP’s armamentarium to aid in evaluating and treating patients in the ED presenting with hip pain.
Imaging Technique
To perform an ultrasound-guided arthrocentesis, the patient should be placed in the supine position, with both knees bent and the hips externally rotated in the frog leg position (Figure 1).
Arthrocentesis
When an effusion is present, arthrocentesis is warranted. To perform this procedure, the femoral vessels should be identified inferior to the inguinal ligament and avoided laterally. The hip should be prepared in a sterile fashion and a lubricated probe should be placed in a sterile dressing with a cord cover. The effusion should be visualized again, and the area should be anesthetized superficially and deeply with local anesthetic, aspirating prior to infusing at the deeper levels. An 18-gauge spinal needle affixed to a 20-mL syringe should be introduced and advanced while aspirating under direct visualization through the capsule of the hip into the effusion. The fluid is then aspirated and sent for laboratory analysis.
Summary
A delayed diagnosis of hip effusion and failure to initiate prompt treatment are the most common causes of late complications of septic arthritis.4 Point-of-care diagnostic and interventional ultrasound of the hip permit instant visualization and implementation of immediate diagnostic and therapeutic measures, which decrease morbidity in adult patients with septic arthritis. Hip arthrocentesis with subsequent synovial fluid analysis, the gold standard of diagnosis, has traditionally been performed by radiology services. Recent literature, however, has shown performance of these ultrasound-guided techniques by EPs to be safe and efficient, facilitating time to treatment.
1. Freeman K, Dewitz A, Baker WE. Ultrasound-guided hip arthrocentesis in the ED. Am J Emerg Med. 2007;25(1):80-86. doi:10.1016/j.ajem.2006.08.002.
2. Minardi JJ, Lander OM. Septic hip arthritis: diagnosis and arthrocentesis using bedside ultrasound. J Emerg Med. 2012;43(2):316-318. doi:10.1016/j.jemermed.2011.09.029.
3. Byrd JW, Potts EA, Allison RK, Jones KS. Ultrasound-guided hip injections: a comparative study with fluoroscopy-guided injections. Arthroscopy. 2014;30(1):42-46. doi:10.1016/j.arthro.2013.09.083.
4. Mascioli AA, Park AL. Infectious arthritis. In: Canale ST, Beaty JH eds. Campbell’s Operative Orthopaedics. Vol 1. 13th ed. Philadelphia, PA: Elsevier Mosby; 2013:749-772.
1. Freeman K, Dewitz A, Baker WE. Ultrasound-guided hip arthrocentesis in the ED. Am J Emerg Med. 2007;25(1):80-86. doi:10.1016/j.ajem.2006.08.002.
2. Minardi JJ, Lander OM. Septic hip arthritis: diagnosis and arthrocentesis using bedside ultrasound. J Emerg Med. 2012;43(2):316-318. doi:10.1016/j.jemermed.2011.09.029.
3. Byrd JW, Potts EA, Allison RK, Jones KS. Ultrasound-guided hip injections: a comparative study with fluoroscopy-guided injections. Arthroscopy. 2014;30(1):42-46. doi:10.1016/j.arthro.2013.09.083.
4. Mascioli AA, Park AL. Infectious arthritis. In: Canale ST, Beaty JH eds. Campbell’s Operative Orthopaedics. Vol 1. 13th ed. Philadelphia, PA: Elsevier Mosby; 2013:749-772.
Pediatric ENT Complaints: An Update
Among all of the causes of ear, nose, and throat (ENT) complaints, acute otitis media (AOM), bacterial sinusitis, and streptococcal pharyngitis (SP) are the most common infections prompting pediatric presentation to the ED. Through a series of case scenarios, along with key questions to help guide the clinician’s work-up, this review covers the proper evaluation and management of pediatric ENT complaints.
Case Scenario 1
A 13-month-old girl presented to the ED with a 1-day history of fever and runny nose. According to her parents, the child had been continually pulling on her ears in apparent discomfort. During history-taking, the parents further informed the emergency physician (EP) that the patient started daycare 4 months earlier and had two elementary school-aged siblings. The patient’s medical history was significant for otitis media, but the parents stated she had not been on antibiotics for over 4 months.
On physical examination, the patient’s vital signs were: blood pressure (BP), 75/50 mm Hg; temperature (T), 101.3°F; slight tachycardia; and normal age-adjusted respiratory rate (RR). Oxygen saturation was 100% on room air. The lungs were clear to auscultation and heart sounds were normal and without murmur. The otolaryngologic examination revealed copious yellow discharge from both nostrils, non-erythematous posterior oropharynx, and erythema to the right tympanic membrane (TM). Questions to Guide the Work-Up: (1) What physical examination findings should be present for accurate diagnosis of otitis media? (2) Will this patient require antibiotics immediately, or is a “wait-and-see” approach indicated? (3) If treatment with antibiotic therapy is warranted, what are the appropriate therapeutic regimen and duration of therapy?
Otitis Media
Acute otitis media is one of the most common presentations in young children. Defined as the rapid onset of signs and symptoms of middle ear inflammation, in conjunction with middle ear effusion (MEE), AOM can develop secondary to a viral or bacterial infection. It is estimated that more than 80% of the pediatric population will experience at least one episode of AOM by age 3 years.1-3
Risk factors for AOM include upper respiratory infection (URI), daycare attendance, siblings, parental smoking, and formula-feeding versus breastfeeding. The patient’s history may include rapid-onset otalgia, fever, irritability, anorexia, and concurrent URI symptoms, as well as other nonspecific symptoms (eg, ear rubbing and/or pulling, crying, changes in behavior and sleep patterns).2-4 In general, otalgia and ear-rubbing in the nonverbal patient seem to have the best predictive value for AOM.3
Signs and Symptoms
A normal TM should be translucent and pearly gray, with visible landmarks of the manubrium of malleus and pars flaccida. A TM that is bulging, cloudy, and immobile is the most consistent finding in AOM, with bulging having a specificity of 97%. Redness of the tympanic membrane is not a useful predictor of AOM as this finding is noted in upward of 30% of pediatric patients on general examination but in <1% of AOM diagnoses in the absence of a bulging TM.
Diagnosis
Pneumatic otoscopy is the gold standard for diagnosing for MEE; however, this examination can be difficult in younger, often uncooperative, patients. A TM that does not perceptibly move with either positive or negative insufflation pressure greatly enhances the diagnostic accuracy for MEE over the use of visible eardrum characteristics alone.2-5
Acute otitis media is a clinical diagnosis and does not require imaging studies or laboratory evaluation unless more serious processes, such as skull fracture, mastoiditis, or intracranial abscess, are being considered.2,3
Treatment and Management
Analgesia. The first step in managing patients with AOM is to provide analgesia. In most cases, acetaminophen in patients over 2 months of age, or ibuprofen in patients over 6 months of age, are adequate choices for managing pain. When either of these analgesics is administered in the clinic/ED setting, patients should be monitored to assure adequate pain relief prior to discharge.
While topical agents such as combination antipyrine-benzocaine suspensions were commonly given in the past to alleviate the pain associated with AOM, there are limited data to support their effectiveness. As such, in July 2015, the US Food and Drug Administration ordered manufacturers to halt production on these unapproved prescription products.3,4,6 There are also no randomized controlled trials (RCTs) to support the use of decongestants or antihistamines for resolution of AOM or otalgia.3,7
Antibiotic Therapy. The most common bacteria associated with AOM are Streptococcus pneumonia, nontypeable Hemophilus influenza, and Moraxella catarrhalis. In 30% of patients, the causative etiology is viral. When the decision is made to treat AOM, high-dose amoxicillin is still considered the first-line treatment, despite ever evolving susceptibilities of bacteria. 
When a child is noted to have been treated with amoxicillin within a 30-day period or who has concurrent conjunctivitis, amoxicillin-clavulanate is considered the first-line treatment.2-4,7,8 The current American Academy of Pediatrics (AAP) guidelines recommend 10 days of antibiotic therapy for children younger than age 2 years, and 5 to 7 days for children older than age 2 years who have uncomplicated AOM. Intramuscular (IM) ceftriaxone is an acceptable first-line agent in a child who is unable to tolerate oral medications or who is suffering persistent emesis. Intramuscular ceftriaxone can be given as a single dose of 50 mg/kg, though the patient should be followed closely as studies show that a second dose may be necessary 5 to 7 days later to prevent infection recurrence. The IM dose of ceftriaxone 50 mg/kg can also be given if treatment with other antibiotics fails to resolve the AOM (failure is defined as no improvement in the patient’s condition 48 to 72 hours from treatment). In such cases, ceftriaxone is given in three consecutive doses.3,4,7
Wait-and-See Approach. Studies of patients whose AOM was confirmed via culture (19% were positive for S pneumoniae, 48% for H influenza, and 78% for M catarrhalis) showed bacterial clearance without antibiotic intervention.4 Based on these findings, the 2013 revised AAP evidence-based clinical practice guidelines indicate an initial watching-and-waiting period combined with pain management for patients older than 6 months of age who are diagnosed with unilateral AOM in the absence of severe symptoms (ie, fever is lower than 102.2˚F or patient has severe otalgia).4 A period of observation prior to treatment is also endorsed for children older than age 2 years who exhibit nonsevere symptoms—even if they have bilateral disease.4
Conversely, all patients younger than age 6 months and all children with severe symptoms should be treated with antibiotics at diagnosis.3,4 The wait-and-see approach, recommends an observation period of 24 to 48 hours for children in the lower risk group prior to antibiotic administration. Delayed antibiotic administration can be performed by a physician in an office/ED follow-up or as a safety-net antibiotic prescription (SNAP) sent home with the family on the initial ED encounter.2-4,8,9
Case 1 Resolution
Given this patient’s unilateral and nonsevere symptoms (minor otalgia, fever <102.2°F), age older than 6 months, and no recent antibiotic use), she was treated with oral ibuprofen. At discharge, the parents were given a 10-day SNAP prescription of high-dose amoxicillin (90 mg/kg/d, divided into two daily doses) and instructed to fill the prescription only if the patient’s otalgia did not improve in 1 or 2 days.
Case Scenario 2
A 5-year-old boy was presented for evaluation by his parents, who stated that their son had been sick since he had started kindergarten in the fall. The patient had a 10-day history of cough, thick runny nose, and facial pain, and a 1-day history of new-onset fever and headache. His parents further noted that the patient had been seen by his pediatrician several times over the past week. At each of these visits, the pediatrician had informed them that their son had a virus.
Vital signs on examination were: BP, 100/60 mm Hg; heart rate (HR), 112 beats/min; normal age-adjusted RR; and T, 102.6oF. Oxygen saturation was 100% on room air. The patient did not appear toxic, his lungs were clear on auscultation, and there were no other clinical signs suggestive of meningitis. The otolaryngologic examination revealed bilateral thick mucoid drainage and visible edema and erythema of the nasal turbinates. The patient was noted to have some facial pain in the maxillary area bilaterally.
Questions to Guide the Work-Up: (1) Does the patient have a prolonged URI or pediatric sinusitis, and what differentiates the two conditions? (2) What sinuses are present in a 5-year-old patient? (3) What treatment modalities are available for sinusitis? (4) Is imaging of the sinuses helpful in confirming the diagnosis?
Acute Bacterial Sinusitis
Rhinosinusitis is an inflammation of the mucosal lining of the nasal passages and paranasal sinuses. Most cases occur secondary to a viral URI and resolve spontaneously in 99% of the pediatric population.10,11
Acute bacterial sinusitis (ABS) is an inflammation of the same mucosal lining of the nasal passages secondary to bacterial overgrowth that lasts more than 10 days, with complete resolution by 30 days.12,13 When evaluating a pediatric patient for ABS, it is important to consider the sinus growth and development: If the sinus is not yet formed, it therefore cannot be the location of an ABS.13 The ethmoid and maxillary sinuses are present at birth, aerated within 4 months of life, and are fully developed by age 12 years. The sphenoid sinuses begin development around age 3 years, are aerated by age 7 or 8 years, and are fully developed by age 18 to 20 years. The frontal sinuses begin development around age 8 years and are aerated and fully developed by age 12 to 15 years.10,13,14 While most guidelines focus on children older than age 1 year (due to very small infantile sinuses), ABS does occur in children younger than age 1 year.12,14
Signs and Symptoms
Differentiation between a viral URI/rhinosinusitis and ABS is a challenge and can be based upon severity of symptoms as well as length of illness. Symptoms of ABS are typically present and persistent for more than 10 days, without improvement. Continuing illness and worsening of symptoms are identifying features of ABS given most viral URIs gradually resolve within a 10-day timeframe. Other common symptoms include milky/thick nasal discharge, fever, predominantly nocturnal cough, and headache. Other less common symptoms include facial pain, toothache, malodorous breath, and periorbital edema. On physical examination, erythema and edema of the turbinates, as well as reproducible pain over aerated sinuses, are suggestive of ABS.10-14
Diagnosis
In the acute care setting, diagnosis of ABS should be clinical in nature. Neither imaging nor laboratory work-up is generally required secondary to their poor diagnostic specificity for ABS. The bacteria involved in ABS are similar to those associated with AOM, with S pneumonia, nontypeable H influenza, and M catarrhalis being the predominant organisms.10-15
Treatment and Management
Treatment of ABS is generally recommended once the diagnosis is made, though this is based largely on expert opinion as there are limited RCTs available.13 However, available studies do show a more rapid improvement in children on antibiotic therapy than those on placebo.15,16
Antibiotic Therapy. Amoxicillin remains the antimicrobial agent of choice for first-line treatment of uncomplicated ABS forsituations in which antimicrobial resistance is not suspected. In communities with a high prevalence of nonsusceptible S pneumoniae (>10%, including intermediate- and high-level resistance), treatment may be initiated at 80 to 90 mg/kg/d in two divided doses, with a maximum of 2 g per dose.
Patients presenting with moderate to severe illness, as well as those who are younger than 2 years, attend childcare, or have recently been treated with an antimicrobial, may receive high-dose amoxicillin-clavulanate as initial therapy given the elevated beta-lactamase production of the common bacteria that cause ABS.
Second-line alternatives include azithromycin, cefdinir, and sulfamethoxizole-trimethoprim (Table 1). There are data to suggest higher rates of decreased susceptibility of S pneumonia and H influenza to third-generation cephalosporins, and the addition of clindamycin may be warranted when utilizing those medications. Treatment is recommended for 10 to 14 days, though improvement should be noted within 1 to 3 days.10-12,14-17
Adjuvant Therapy. Additional therapies include nasal irrigation, decongestants, antihistamines, and intranasal steroids; however, there are only anecdotal reports of their efficacy in providing symptom relief. Therefore, there are insufficient evidence-based data to support or refute the role of these adjuvant therapies in treating pediatric patients with ABS.9,13
Case 2 Resolution
The prolonged duration and severity of symptoms (high fever and headache) and the gradual worsening of the clinical course (ie, late-onset fever) in this patient all suggest ABS rather than a simple prolonged URI. The physical examination findings of inflamed turbinates and facial pain further increase the specificity for ABS. The patient was started on oral amoxicillin-clavulanate with planned treatment for 14 days. At discharge, his parents were instructed to follow-up with the patient’s pediatrician in 3 days to ensure a degree of clinical resolution.
Case Scenario 3
A 4-year-old boy was presented by his parents for evaluation of a 2-day history of a persistent and unimproved sore throat. The patient’s mother indicated that the child’s oral T upon returning home earlier from preschool was 101.2oF. She further noted that her 17-month-old daughter and 8-year-old son also experienced similar symptoms which had self-resolved. Triage vital signs were: T, 100.8oF, orally; BP, HR, and RR were all within normal limits. Oxygen saturation was 100% on room air.
On physical examination, the child was noted to have anterior cervical lymph nodes bilaterally and an erythematous oropharynx with exudate noted on both tonsils. There were no cutaneous abnormalities, nasal edema, erythema, or drainage. Based on the clinical examination, the EP was suspicious for SP.
Questions to Guide the Work-Up: (1) Is SP diagnosed based on clinical findings alone in this patient’s age group? (2) At what age in the pediatric population is it appropriate to perform a rapid streptococcal antigen test? (3) Are there medications other than antibiotics that are beneficial in treating symptomatic SP?
Streptococcal Pharyngitis
Streptococcal pharyngitis is a clinical condition caused by group A beta-hemolytic S pyogens. This bacterium is responsible for multiple conditions, including pharyngitis, skin infections, poststreptococcal glomerulonephritis, and rheumatic fever, as well as invasive syndromes. (This case focuses solely on SP).
Pharyngitis can occur secondary to a viral or bacterial infection, and SP is the most common cause of pediatric bacterial pharyngitis. It is estimated that children aged 5 to 15 years are more commonly diagnosed with SP, although approximately 24% of children younger than age 5 years with pharyngitis symptoms will be ultimately diagnosed with SP.
Signs and Symptoms
Typical symptoms include fever, pharyngitis, generalized abdominal pain, nausea, vomiting, headache, and absence of viral URI symptoms (eg, cough, nasal discharge). However, younger patients with SP may have clinical findings of prolonged nasal drainage and excoriated nares. Examination findings may include swollen and tender anterior cervical lymph nodes; generalized edema and erythema of the posterior pharynx; tonsillar exudates; and palatal petechiae.
Diagnosis
Centor Criteria. The Centor criteria were developed to assist practitioners in identifying patients with potential SP. Criteria for patients older than age 15 years include fever, absence of cough, tonsillar exudates, and tender anterior cervical lymphadenopathy. A modified Centor criteria was later established to include children older than age 5 years, with children between ages 5 and 15 years being the fifth variable in the modified score. In general, patients with a score of 4 or 5 (presence of each variable = 1 point) are most likely to test positive for SP on rapid antigen testing (RAT) or culture.18-20
Swab, Rapid Antigen Testing, and Culture. Swabbing the throat and RAT and/or culture should be performed in most children with suspected SP because the clinical features alone do not reliably discriminate SP from viral pharyngitis. Rapid antigen testing is only specific for group A beta-hemolytic streptococcal species, which is the only streptococcal species that is routinely treated with antibiotics in the setting of acute pharyngitis. It is unlikely for a patient with a score of 0 or 1 to have SP, and several sources suggest neither testing nor treating this cohort, but rather to consider an alternative diagnosis.18-20
Within the population of children and young adolescents, due to a RAT sensitivity of 70% to 90%, a negative result should always be backed-up by a throat culture, and treatment initiated if results of the culture are later found to be positive. As the current generation of RAT tests have a high specificity, a positive RAT does not necessitate a back-up culture, and treatment is indicated without further investigation.19,20
Routine RAT is not recommended in children younger than age 3 years as patients in this age group are at low-risk of developing rheumatic fever. One notable exception for these very young children would be if there are siblings in the home with confirmed SP, in which case, RAT should be considered in the clinical context of SP.21 Adolescents over age 15 years are another cohort with a low likelihood of developing rheumatic fever, though they can develop other poststreptococcal complications, such as glomerulonephritis.
The US Centers for Disease Control and Prevention/American Academy of Family Practitioners (AAFP) guidelines suggest that pharyngitis in older adolescents can be approached in a similar fashion to adults, with empiric therapy for a Centor score of 3 or 4, RAT (without the need for follow-up culture) for Centor score of 2, and neither testing nor treating patients with a score of 0 or 1.19
Treatment and Management
Streptococcal pharyngitis is treated mainly to prevent the poststreptococcal complications of rheumatic fever, though it will not prevent poststreptococcal glomerulonephritis. Treatment of SP also facilitates resolution of symptoms and return to baseline activities.
Antibiotic Therapy. Patients who have a positive RAT or a follow-up throat culture positive for group A streptococcus should be given antibiotics. The gold standard treatment is penicillin V orally for 10 days. 
Corticosteroid Therapy. The use of corticosteroids for symptom control of SP in pediatric patients is controversial. Although the Infectious Disease Society of America does not recommend corticosteroid therapy in the treatment of SP, several studies show such therapy (namely dexamethasone), improves pain in children and adolescents diagnosed with SP, but without significant change to the overall disease course.21,23-26
Case 3 Resolution
The patient had a modified Centor criteria score of 4, as well as siblings with similar symptoms. In following current guidelines, the EP performed a RAT and back-up culture. The RAT was negative in the ED, but the back-up culture was subsequently positive, and the child was started on a 10-day course of oral amoxicillin.
Conclusion
When evaluating pediatric patients presenting with ENT signs and symptoms such as ear pain and erythema, fever, sore throat, nasal congestion and discharge, a thorough physical examination and history-taking—including recent illness of any siblings—along with testing when indicated, is essential to guide the diagnosis and determine appropriate treatment and management. In addition to administering antibiotic therapy when such is warranted, the EP should provide appropriate analgesia to manage the patient’s pain and assure relief prior to discharge.
1. Rosenfeld RM, Shin JJ, Schwartz SR, et al. Clinical practice guideline: otitis media with effusion (Update). Otolaryngol Head Neck Surg. 2016;154(1 Suppl):S1-S41. doi:10.1177/0194599815623467.
2. Acute Otitis Media Guideline Team, Cincinnati Children’s Hospital Medical Center. Evidence-based care guideline for medical management of acute otitis media in children 2 months to 13 years of age. http://f.i-md.com/medinfo/material/4f4/4eb132ba44ae4ffe12a814f4/4eb132d744ae4ffe12a814f7.pdf. August 2006. Accessed December 29, 2016.
3. Nesbit CE, Powers MC. An evidence-based approach to managing acute otitis media. Pediatr Emerg Med Pract. 2013;10(4):1-26; quiz 26-27.
4. Lieberthal AS, Carroll AE, Chonmaitree T, et al. The diagnosis and management of acute otitis media. Pediatrics. 2013;131(3):e964-e999. doi:10.1542/peds.2012-3488.
5. American Academy of Family Physicians; American Academy of Otolaryngology-Head and Neck Surgery; American Academy of Pediatrics Subcommittee on Otitis Media With Effusion. Otitis media with effusion. Pediatrics. 2004;113(5):1412-1429.
6. US Food and Drug Administration Web site. FDA: Use only approved prescription ear drops. http://www.fda.gov/ForConsumers/ConsumerUpdates/-ucm453087.htm. Updated July 10, 2015. Accessed December 15, 2016.
7. Sack F. An evidence based approach to the management of uncomplicated acute otitis media in children. Int Pediatrics. 2005;20(1):44-46.
8. Johnson NC, Holger JS. Pediatric acute otitis media: the case for delayed antibiotic treatment. J Emerg Med. 2007;32(3):279-284. doi:10.1016/j.jemermed.2006.07.029.
9. Spiro DM, Tay KY, Arnold DH, Dziura JD, Baker MD, Shapiro ED. Wait-and-see prescription for the treatment of acute otitis media: a randomized controlled trial. JAMA. 2006;296(10):1235-1241. doi:10.1001/jama.296.10.1235.
10. Brook I. Management of acute rhinosinusitis in pediatric patients. Pediatr Emerg Med Pract. 2012;9(5):1-24.
11. Ferdman RM, Linzer JF Jr. The runny nose in the emergency department: rhinitis and sinusitis. Clin Pediatr Emerg Med. 2007;8(2):123-130.
12. Acute Bacterial Sinusitis Guideline Team, Cincinnati Children’s Hospital Medical Center: Evidence-based care guideline for medical management of acute bacterial sinusitis in children 1 through 18 years of age. http://www.antibioticos.msssi.gob.es/PDF/sinusitisguideline.pdf. July 7, 2006. Accessed December 29, 2016
13. Holt KR, Murdoch Cuenca M, Cuenca PJ, Johnston GM. acute pediatric sinusitis and “the 10-day rule.” Pediatr Emerg Med Pract. 2006;3(2):1-16.
14. American Academy of Pediatrics. Subcommittee on Management of Sinusitis and Committee on Quality Improvement. Clinical practice guideline: management of sinusitis. Pediatrics. 2001;108(3):798-808.
15. Wald ER, Nash D, Eickhoff J. Effectiveness of amoxicillin/clavulanate potassium in the treatment of acute bacterial sinusitis in children. Pediatrics. 2009;124(1):9-15. doi:10.1542/peds.2008-2902.
16. Arroll B, Kenealy T. Are antibiotics effective for acute purulent rhinitis? Systematic review and meta-analysis of placebo controlled randomised trials. BMJ. 2006;333(7562):279. doi:10.1136/bmj.38891.681215.AE.
17. McQuillan L, Crane LA, Kempe A. Diagnosis and management of acute sinusitis by pediatricians. Pediatrics. 2009;123(2):e193-e198.
18. Singer JI, Fontanette R. Recognizable and suspected group A beta-hemolytic streptococcal syndromes. Pediatr Emerg Med Rep. 2010;15(11):129-144.
19. Weglowski J. An evidence-based approach to the evaluation and treatment of pharyngitis in children. Pediatr Emerg Med Pract. 2011;8(12):1-28.
20. Gerber MA, Baltimore RS, Eaton CB, et al. Prevention of rheumatic fever and diagnosis and treatment of acute Streptococcal pharyngitis: a scientific statement from the American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee of the Council on Cardiovascular Disease in the Young, the Interdisciplinary Council on Functional Genomics and Translational Biology, and the Interdisciplinary Council on Quality of Care and Outcomes Research: endorsed by the American Academy of Pediatrics. Circulation. 2009;119(11):1541-1551. doi:10.1161/CIRCULATIONAHA.109.191959.
21. Shulman ST, Bisno AL, Clegg HW, et al. Clinical practice guideline for the diagnosis and management of group A streptococcal pharyngitis: 2012 update by the Infectious Diseases Society of America. Clin Infect Dis. 2012;55(10):1279-1282. doi:10.1093/cid/cis847.
22. Clegg HW, Ryan AG, Dallas SD, et al. Treatment of streptococcal pharyngitis with once-daily compared with twice-daily amoxicillin: a noninferiority trial. Pediatr Infect Dis J. 2006;25(9):761-767. doi:10.1097/01.inf.0000235678.46805.92.
23. Bulloch B, Kabani A, Tenenbein M. Oral dexamethasone for the treatment of pain in children with acute pharyngitis: a randomized, double-blind, placebo-controlled trial. Ann Emerg Med. 2003;41(5):601-608. doi:10.1067/mem.2003.136.
24. Niland ML, Bonsu BK, Nuss KE, Goodman DG. A pilot study of 1 versus 3 days of dexamethasone as add-on therapy in children with streptococcal pharyngitis. Pediatr Infect Dis J. 2006;25(6):477-481. doi:10.1097/01.inf.0000219469.95772.3f.
25. Wei JL, Kasperbauer JL, Weaver AL, Boggust AJ. Efficacy of single-dose dexamethasone as adjuvant therapy for acute pharyngitis. Laryngoscope. 2002;112(1):87-93. doi:10.1097/00005537-200201000-00016.
26. Hayward G, Thompson M, Heneghan C, Perera R, Del Mar C, Glasziou P. Corticosteroids for pain relief in sore throat: systematic review and meta-analysis. BMJ. 2009;339:b2976. doi:10.1136/bmj.b2976.
Among all of the causes of ear, nose, and throat (ENT) complaints, acute otitis media (AOM), bacterial sinusitis, and streptococcal pharyngitis (SP) are the most common infections prompting pediatric presentation to the ED. Through a series of case scenarios, along with key questions to help guide the clinician’s work-up, this review covers the proper evaluation and management of pediatric ENT complaints.
Case Scenario 1
A 13-month-old girl presented to the ED with a 1-day history of fever and runny nose. According to her parents, the child had been continually pulling on her ears in apparent discomfort. During history-taking, the parents further informed the emergency physician (EP) that the patient started daycare 4 months earlier and had two elementary school-aged siblings. The patient’s medical history was significant for otitis media, but the parents stated she had not been on antibiotics for over 4 months.
On physical examination, the patient’s vital signs were: blood pressure (BP), 75/50 mm Hg; temperature (T), 101.3°F; slight tachycardia; and normal age-adjusted respiratory rate (RR). Oxygen saturation was 100% on room air. The lungs were clear to auscultation and heart sounds were normal and without murmur. The otolaryngologic examination revealed copious yellow discharge from both nostrils, non-erythematous posterior oropharynx, and erythema to the right tympanic membrane (TM). Questions to Guide the Work-Up: (1) What physical examination findings should be present for accurate diagnosis of otitis media? (2) Will this patient require antibiotics immediately, or is a “wait-and-see” approach indicated? (3) If treatment with antibiotic therapy is warranted, what are the appropriate therapeutic regimen and duration of therapy?
Otitis Media
Acute otitis media is one of the most common presentations in young children. Defined as the rapid onset of signs and symptoms of middle ear inflammation, in conjunction with middle ear effusion (MEE), AOM can develop secondary to a viral or bacterial infection. It is estimated that more than 80% of the pediatric population will experience at least one episode of AOM by age 3 years.1-3
Risk factors for AOM include upper respiratory infection (URI), daycare attendance, siblings, parental smoking, and formula-feeding versus breastfeeding. The patient’s history may include rapid-onset otalgia, fever, irritability, anorexia, and concurrent URI symptoms, as well as other nonspecific symptoms (eg, ear rubbing and/or pulling, crying, changes in behavior and sleep patterns).2-4 In general, otalgia and ear-rubbing in the nonverbal patient seem to have the best predictive value for AOM.3
Signs and Symptoms
A normal TM should be translucent and pearly gray, with visible landmarks of the manubrium of malleus and pars flaccida. A TM that is bulging, cloudy, and immobile is the most consistent finding in AOM, with bulging having a specificity of 97%. Redness of the tympanic membrane is not a useful predictor of AOM as this finding is noted in upward of 30% of pediatric patients on general examination but in <1% of AOM diagnoses in the absence of a bulging TM.
Diagnosis
Pneumatic otoscopy is the gold standard for diagnosing for MEE; however, this examination can be difficult in younger, often uncooperative, patients. A TM that does not perceptibly move with either positive or negative insufflation pressure greatly enhances the diagnostic accuracy for MEE over the use of visible eardrum characteristics alone.2-5
Acute otitis media is a clinical diagnosis and does not require imaging studies or laboratory evaluation unless more serious processes, such as skull fracture, mastoiditis, or intracranial abscess, are being considered.2,3
Treatment and Management
Analgesia. The first step in managing patients with AOM is to provide analgesia. In most cases, acetaminophen in patients over 2 months of age, or ibuprofen in patients over 6 months of age, are adequate choices for managing pain. When either of these analgesics is administered in the clinic/ED setting, patients should be monitored to assure adequate pain relief prior to discharge.
While topical agents such as combination antipyrine-benzocaine suspensions were commonly given in the past to alleviate the pain associated with AOM, there are limited data to support their effectiveness. As such, in July 2015, the US Food and Drug Administration ordered manufacturers to halt production on these unapproved prescription products.3,4,6 There are also no randomized controlled trials (RCTs) to support the use of decongestants or antihistamines for resolution of AOM or otalgia.3,7
Antibiotic Therapy. The most common bacteria associated with AOM are Streptococcus pneumonia, nontypeable Hemophilus influenza, and Moraxella catarrhalis. In 30% of patients, the causative etiology is viral. When the decision is made to treat AOM, high-dose amoxicillin is still considered the first-line treatment, despite ever evolving susceptibilities of bacteria.
When a child is noted to have been treated with amoxicillin within a 30-day period or who has concurrent conjunctivitis, amoxicillin-clavulanate is considered the first-line treatment.2-4,7,8 The current American Academy of Pediatrics (AAP) guidelines recommend 10 days of antibiotic therapy for children younger than age 2 years, and 5 to 7 days for children older than age 2 years who have uncomplicated AOM. Intramuscular (IM) ceftriaxone is an acceptable first-line agent in a child who is unable to tolerate oral medications or who is suffering persistent emesis. Intramuscular ceftriaxone can be given as a single dose of 50 mg/kg, though the patient should be followed closely as studies show that a second dose may be necessary 5 to 7 days later to prevent infection recurrence. The IM dose of ceftriaxone 50 mg/kg can also be given if treatment with other antibiotics fails to resolve the AOM (failure is defined as no improvement in the patient’s condition 48 to 72 hours from treatment). In such cases, ceftriaxone is given in three consecutive doses.3,4,7
Wait-and-See Approach. Studies of patients whose AOM was confirmed via culture (19% were positive for S pneumoniae, 48% for H influenza, and 78% for M catarrhalis) showed bacterial clearance without antibiotic intervention.4 Based on these findings, the 2013 revised AAP evidence-based clinical practice guidelines indicate an initial watching-and-waiting period combined with pain management for patients older than 6 months of age who are diagnosed with unilateral AOM in the absence of severe symptoms (ie, fever is lower than 102.2˚F or patient has severe otalgia).4 A period of observation prior to treatment is also endorsed for children older than age 2 years who exhibit nonsevere symptoms—even if they have bilateral disease.4
Conversely, all patients younger than age 6 months and all children with severe symptoms should be treated with antibiotics at diagnosis.3,4 The wait-and-see approach, recommends an observation period of 24 to 48 hours for children in the lower risk group prior to antibiotic administration. Delayed antibiotic administration can be performed by a physician in an office/ED follow-up or as a safety-net antibiotic prescription (SNAP) sent home with the family on the initial ED encounter.2-4,8,9
Case 1 Resolution
Given this patient’s unilateral and nonsevere symptoms (minor otalgia, fever <102.2°F), age older than 6 months, and no recent antibiotic use), she was treated with oral ibuprofen. At discharge, the parents were given a 10-day SNAP prescription of high-dose amoxicillin (90 mg/kg/d, divided into two daily doses) and instructed to fill the prescription only if the patient’s otalgia did not improve in 1 or 2 days.
Case Scenario 2
A 5-year-old boy was presented for evaluation by his parents, who stated that their son had been sick since he had started kindergarten in the fall. The patient had a 10-day history of cough, thick runny nose, and facial pain, and a 1-day history of new-onset fever and headache. His parents further noted that the patient had been seen by his pediatrician several times over the past week. At each of these visits, the pediatrician had informed them that their son had a virus.
Vital signs on examination were: BP, 100/60 mm Hg; heart rate (HR), 112 beats/min; normal age-adjusted RR; and T, 102.6oF. Oxygen saturation was 100% on room air. The patient did not appear toxic, his lungs were clear on auscultation, and there were no other clinical signs suggestive of meningitis. The otolaryngologic examination revealed bilateral thick mucoid drainage and visible edema and erythema of the nasal turbinates. The patient was noted to have some facial pain in the maxillary area bilaterally.
Questions to Guide the Work-Up: (1) Does the patient have a prolonged URI or pediatric sinusitis, and what differentiates the two conditions? (2) What sinuses are present in a 5-year-old patient? (3) What treatment modalities are available for sinusitis? (4) Is imaging of the sinuses helpful in confirming the diagnosis?
Acute Bacterial Sinusitis
Rhinosinusitis is an inflammation of the mucosal lining of the nasal passages and paranasal sinuses. Most cases occur secondary to a viral URI and resolve spontaneously in 99% of the pediatric population.10,11
Acute bacterial sinusitis (ABS) is an inflammation of the same mucosal lining of the nasal passages secondary to bacterial overgrowth that lasts more than 10 days, with complete resolution by 30 days.12,13 When evaluating a pediatric patient for ABS, it is important to consider the sinus growth and development: If the sinus is not yet formed, it therefore cannot be the location of an ABS.13 The ethmoid and maxillary sinuses are present at birth, aerated within 4 months of life, and are fully developed by age 12 years. The sphenoid sinuses begin development around age 3 years, are aerated by age 7 or 8 years, and are fully developed by age 18 to 20 years. The frontal sinuses begin development around age 8 years and are aerated and fully developed by age 12 to 15 years.10,13,14 While most guidelines focus on children older than age 1 year (due to very small infantile sinuses), ABS does occur in children younger than age 1 year.12,14
Signs and Symptoms
Differentiation between a viral URI/rhinosinusitis and ABS is a challenge and can be based upon severity of symptoms as well as length of illness. Symptoms of ABS are typically present and persistent for more than 10 days, without improvement. Continuing illness and worsening of symptoms are identifying features of ABS given most viral URIs gradually resolve within a 10-day timeframe. Other common symptoms include milky/thick nasal discharge, fever, predominantly nocturnal cough, and headache. Other less common symptoms include facial pain, toothache, malodorous breath, and periorbital edema. On physical examination, erythema and edema of the turbinates, as well as reproducible pain over aerated sinuses, are suggestive of ABS.10-14
Diagnosis
In the acute care setting, diagnosis of ABS should be clinical in nature. Neither imaging nor laboratory work-up is generally required secondary to their poor diagnostic specificity for ABS. The bacteria involved in ABS are similar to those associated with AOM, with S pneumonia, nontypeable H influenza, and M catarrhalis being the predominant organisms.10-15
Treatment and Management
Treatment of ABS is generally recommended once the diagnosis is made, though this is based largely on expert opinion as there are limited RCTs available.13 However, available studies do show a more rapid improvement in children on antibiotic therapy than those on placebo.15,16
Antibiotic Therapy. Amoxicillin remains the antimicrobial agent of choice for first-line treatment of uncomplicated ABS forsituations in which antimicrobial resistance is not suspected. In communities with a high prevalence of nonsusceptible S pneumoniae (>10%, including intermediate- and high-level resistance), treatment may be initiated at 80 to 90 mg/kg/d in two divided doses, with a maximum of 2 g per dose.
Patients presenting with moderate to severe illness, as well as those who are younger than 2 years, attend childcare, or have recently been treated with an antimicrobial, may receive high-dose amoxicillin-clavulanate as initial therapy given the elevated beta-lactamase production of the common bacteria that cause ABS.
Second-line alternatives include azithromycin, cefdinir, and sulfamethoxizole-trimethoprim (Table 1). There are data to suggest higher rates of decreased susceptibility of S pneumonia and H influenza to third-generation cephalosporins, and the addition of clindamycin may be warranted when utilizing those medications. Treatment is recommended for 10 to 14 days, though improvement should be noted within 1 to 3 days.10-12,14-17
Adjuvant Therapy. Additional therapies include nasal irrigation, decongestants, antihistamines, and intranasal steroids; however, there are only anecdotal reports of their efficacy in providing symptom relief. Therefore, there are insufficient evidence-based data to support or refute the role of these adjuvant therapies in treating pediatric patients with ABS.9,13
Case 2 Resolution
The prolonged duration and severity of symptoms (high fever and headache) and the gradual worsening of the clinical course (ie, late-onset fever) in this patient all suggest ABS rather than a simple prolonged URI. The physical examination findings of inflamed turbinates and facial pain further increase the specificity for ABS. The patient was started on oral amoxicillin-clavulanate with planned treatment for 14 days. At discharge, his parents were instructed to follow-up with the patient’s pediatrician in 3 days to ensure a degree of clinical resolution.
Case Scenario 3
A 4-year-old boy was presented by his parents for evaluation of a 2-day history of a persistent and unimproved sore throat. The patient’s mother indicated that the child’s oral T upon returning home earlier from preschool was 101.2oF. She further noted that her 17-month-old daughter and 8-year-old son also experienced similar symptoms which had self-resolved. Triage vital signs were: T, 100.8oF, orally; BP, HR, and RR were all within normal limits. Oxygen saturation was 100% on room air.
On physical examination, the child was noted to have anterior cervical lymph nodes bilaterally and an erythematous oropharynx with exudate noted on both tonsils. There were no cutaneous abnormalities, nasal edema, erythema, or drainage. Based on the clinical examination, the EP was suspicious for SP.
Questions to Guide the Work-Up: (1) Is SP diagnosed based on clinical findings alone in this patient’s age group? (2) At what age in the pediatric population is it appropriate to perform a rapid streptococcal antigen test? (3) Are there medications other than antibiotics that are beneficial in treating symptomatic SP?
Streptococcal Pharyngitis
Streptococcal pharyngitis is a clinical condition caused by group A beta-hemolytic S pyogens. This bacterium is responsible for multiple conditions, including pharyngitis, skin infections, poststreptococcal glomerulonephritis, and rheumatic fever, as well as invasive syndromes. (This case focuses solely on SP).
Pharyngitis can occur secondary to a viral or bacterial infection, and SP is the most common cause of pediatric bacterial pharyngitis. It is estimated that children aged 5 to 15 years are more commonly diagnosed with SP, although approximately 24% of children younger than age 5 years with pharyngitis symptoms will be ultimately diagnosed with SP.
Signs and Symptoms
Typical symptoms include fever, pharyngitis, generalized abdominal pain, nausea, vomiting, headache, and absence of viral URI symptoms (eg, cough, nasal discharge). However, younger patients with SP may have clinical findings of prolonged nasal drainage and excoriated nares. Examination findings may include swollen and tender anterior cervical lymph nodes; generalized edema and erythema of the posterior pharynx; tonsillar exudates; and palatal petechiae.
Diagnosis
Centor Criteria. The Centor criteria were developed to assist practitioners in identifying patients with potential SP. Criteria for patients older than age 15 years include fever, absence of cough, tonsillar exudates, and tender anterior cervical lymphadenopathy. A modified Centor criteria was later established to include children older than age 5 years, with children between ages 5 and 15 years being the fifth variable in the modified score. In general, patients with a score of 4 or 5 (presence of each variable = 1 point) are most likely to test positive for SP on rapid antigen testing (RAT) or culture.18-20
Swab, Rapid Antigen Testing, and Culture. Swabbing the throat and RAT and/or culture should be performed in most children with suspected SP because the clinical features alone do not reliably discriminate SP from viral pharyngitis. Rapid antigen testing is only specific for group A beta-hemolytic streptococcal species, which is the only streptococcal species that is routinely treated with antibiotics in the setting of acute pharyngitis. It is unlikely for a patient with a score of 0 or 1 to have SP, and several sources suggest neither testing nor treating this cohort, but rather to consider an alternative diagnosis.18-20
Within the population of children and young adolescents, due to a RAT sensitivity of 70% to 90%, a negative result should always be backed-up by a throat culture, and treatment initiated if results of the culture are later found to be positive. As the current generation of RAT tests have a high specificity, a positive RAT does not necessitate a back-up culture, and treatment is indicated without further investigation.19,20
Routine RAT is not recommended in children younger than age 3 years as patients in this age group are at low-risk of developing rheumatic fever. One notable exception for these very young children would be if there are siblings in the home with confirmed SP, in which case, RAT should be considered in the clinical context of SP.21 Adolescents over age 15 years are another cohort with a low likelihood of developing rheumatic fever, though they can develop other poststreptococcal complications, such as glomerulonephritis.
The US Centers for Disease Control and Prevention/American Academy of Family Practitioners (AAFP) guidelines suggest that pharyngitis in older adolescents can be approached in a similar fashion to adults, with empiric therapy for a Centor score of 3 or 4, RAT (without the need for follow-up culture) for Centor score of 2, and neither testing nor treating patients with a score of 0 or 1.19
Treatment and Management
Streptococcal pharyngitis is treated mainly to prevent the poststreptococcal complications of rheumatic fever, though it will not prevent poststreptococcal glomerulonephritis. Treatment of SP also facilitates resolution of symptoms and return to baseline activities.
Antibiotic Therapy. Patients who have a positive RAT or a follow-up throat culture positive for group A streptococcus should be given antibiotics. The gold standard treatment is penicillin V orally for 10 days.
Corticosteroid Therapy. The use of corticosteroids for symptom control of SP in pediatric patients is controversial. Although the Infectious Disease Society of America does not recommend corticosteroid therapy in the treatment of SP, several studies show such therapy (namely dexamethasone), improves pain in children and adolescents diagnosed with SP, but without significant change to the overall disease course.21,23-26
Case 3 Resolution
The patient had a modified Centor criteria score of 4, as well as siblings with similar symptoms. In following current guidelines, the EP performed a RAT and back-up culture. The RAT was negative in the ED, but the back-up culture was subsequently positive, and the child was started on a 10-day course of oral amoxicillin.
Conclusion
When evaluating pediatric patients presenting with ENT signs and symptoms such as ear pain and erythema, fever, sore throat, nasal congestion and discharge, a thorough physical examination and history-taking—including recent illness of any siblings—along with testing when indicated, is essential to guide the diagnosis and determine appropriate treatment and management. In addition to administering antibiotic therapy when such is warranted, the EP should provide appropriate analgesia to manage the patient’s pain and assure relief prior to discharge.
Among all of the causes of ear, nose, and throat (ENT) complaints, acute otitis media (AOM), bacterial sinusitis, and streptococcal pharyngitis (SP) are the most common infections prompting pediatric presentation to the ED. Through a series of case scenarios, along with key questions to help guide the clinician’s work-up, this review covers the proper evaluation and management of pediatric ENT complaints.
Case Scenario 1
A 13-month-old girl presented to the ED with a 1-day history of fever and runny nose. According to her parents, the child had been continually pulling on her ears in apparent discomfort. During history-taking, the parents further informed the emergency physician (EP) that the patient started daycare 4 months earlier and had two elementary school-aged siblings. The patient’s medical history was significant for otitis media, but the parents stated she had not been on antibiotics for over 4 months.
On physical examination, the patient’s vital signs were: blood pressure (BP), 75/50 mm Hg; temperature (T), 101.3°F; slight tachycardia; and normal age-adjusted respiratory rate (RR). Oxygen saturation was 100% on room air. The lungs were clear to auscultation and heart sounds were normal and without murmur. The otolaryngologic examination revealed copious yellow discharge from both nostrils, non-erythematous posterior oropharynx, and erythema to the right tympanic membrane (TM). Questions to Guide the Work-Up: (1) What physical examination findings should be present for accurate diagnosis of otitis media? (2) Will this patient require antibiotics immediately, or is a “wait-and-see” approach indicated? (3) If treatment with antibiotic therapy is warranted, what are the appropriate therapeutic regimen and duration of therapy?
Otitis Media
Acute otitis media is one of the most common presentations in young children. Defined as the rapid onset of signs and symptoms of middle ear inflammation, in conjunction with middle ear effusion (MEE), AOM can develop secondary to a viral or bacterial infection. It is estimated that more than 80% of the pediatric population will experience at least one episode of AOM by age 3 years.1-3
Risk factors for AOM include upper respiratory infection (URI), daycare attendance, siblings, parental smoking, and formula-feeding versus breastfeeding. The patient’s history may include rapid-onset otalgia, fever, irritability, anorexia, and concurrent URI symptoms, as well as other nonspecific symptoms (eg, ear rubbing and/or pulling, crying, changes in behavior and sleep patterns).2-4 In general, otalgia and ear-rubbing in the nonverbal patient seem to have the best predictive value for AOM.3
Signs and Symptoms
A normal TM should be translucent and pearly gray, with visible landmarks of the manubrium of malleus and pars flaccida. A TM that is bulging, cloudy, and immobile is the most consistent finding in AOM, with bulging having a specificity of 97%. Redness of the tympanic membrane is not a useful predictor of AOM as this finding is noted in upward of 30% of pediatric patients on general examination but in <1% of AOM diagnoses in the absence of a bulging TM.
Diagnosis
Pneumatic otoscopy is the gold standard for diagnosing for MEE; however, this examination can be difficult in younger, often uncooperative, patients. A TM that does not perceptibly move with either positive or negative insufflation pressure greatly enhances the diagnostic accuracy for MEE over the use of visible eardrum characteristics alone.2-5
Acute otitis media is a clinical diagnosis and does not require imaging studies or laboratory evaluation unless more serious processes, such as skull fracture, mastoiditis, or intracranial abscess, are being considered.2,3
Treatment and Management
Analgesia. The first step in managing patients with AOM is to provide analgesia. In most cases, acetaminophen in patients over 2 months of age, or ibuprofen in patients over 6 months of age, are adequate choices for managing pain. When either of these analgesics is administered in the clinic/ED setting, patients should be monitored to assure adequate pain relief prior to discharge.
While topical agents such as combination antipyrine-benzocaine suspensions were commonly given in the past to alleviate the pain associated with AOM, there are limited data to support their effectiveness. As such, in July 2015, the US Food and Drug Administration ordered manufacturers to halt production on these unapproved prescription products.3,4,6 There are also no randomized controlled trials (RCTs) to support the use of decongestants or antihistamines for resolution of AOM or otalgia.3,7
Antibiotic Therapy. The most common bacteria associated with AOM are Streptococcus pneumonia, nontypeable Hemophilus influenza, and Moraxella catarrhalis. In 30% of patients, the causative etiology is viral. When the decision is made to treat AOM, high-dose amoxicillin is still considered the first-line treatment, despite ever evolving susceptibilities of bacteria.
When a child is noted to have been treated with amoxicillin within a 30-day period or who has concurrent conjunctivitis, amoxicillin-clavulanate is considered the first-line treatment.2-4,7,8 The current American Academy of Pediatrics (AAP) guidelines recommend 10 days of antibiotic therapy for children younger than age 2 years, and 5 to 7 days for children older than age 2 years who have uncomplicated AOM. Intramuscular (IM) ceftriaxone is an acceptable first-line agent in a child who is unable to tolerate oral medications or who is suffering persistent emesis. Intramuscular ceftriaxone can be given as a single dose of 50 mg/kg, though the patient should be followed closely as studies show that a second dose may be necessary 5 to 7 days later to prevent infection recurrence. The IM dose of ceftriaxone 50 mg/kg can also be given if treatment with other antibiotics fails to resolve the AOM (failure is defined as no improvement in the patient’s condition 48 to 72 hours from treatment). In such cases, ceftriaxone is given in three consecutive doses.3,4,7
Wait-and-See Approach. Studies of patients whose AOM was confirmed via culture (19% were positive for S pneumoniae, 48% for H influenza, and 78% for M catarrhalis) showed bacterial clearance without antibiotic intervention.4 Based on these findings, the 2013 revised AAP evidence-based clinical practice guidelines indicate an initial watching-and-waiting period combined with pain management for patients older than 6 months of age who are diagnosed with unilateral AOM in the absence of severe symptoms (ie, fever is lower than 102.2˚F or patient has severe otalgia).4 A period of observation prior to treatment is also endorsed for children older than age 2 years who exhibit nonsevere symptoms—even if they have bilateral disease.4
Conversely, all patients younger than age 6 months and all children with severe symptoms should be treated with antibiotics at diagnosis.3,4 The wait-and-see approach, recommends an observation period of 24 to 48 hours for children in the lower risk group prior to antibiotic administration. Delayed antibiotic administration can be performed by a physician in an office/ED follow-up or as a safety-net antibiotic prescription (SNAP) sent home with the family on the initial ED encounter.2-4,8,9
Case 1 Resolution
Given this patient’s unilateral and nonsevere symptoms (minor otalgia, fever <102.2°F), age older than 6 months, and no recent antibiotic use), she was treated with oral ibuprofen. At discharge, the parents were given a 10-day SNAP prescription of high-dose amoxicillin (90 mg/kg/d, divided into two daily doses) and instructed to fill the prescription only if the patient’s otalgia did not improve in 1 or 2 days.
Case Scenario 2
A 5-year-old boy was presented for evaluation by his parents, who stated that their son had been sick since he had started kindergarten in the fall. The patient had a 10-day history of cough, thick runny nose, and facial pain, and a 1-day history of new-onset fever and headache. His parents further noted that the patient had been seen by his pediatrician several times over the past week. At each of these visits, the pediatrician had informed them that their son had a virus.
Vital signs on examination were: BP, 100/60 mm Hg; heart rate (HR), 112 beats/min; normal age-adjusted RR; and T, 102.6oF. Oxygen saturation was 100% on room air. The patient did not appear toxic, his lungs were clear on auscultation, and there were no other clinical signs suggestive of meningitis. The otolaryngologic examination revealed bilateral thick mucoid drainage and visible edema and erythema of the nasal turbinates. The patient was noted to have some facial pain in the maxillary area bilaterally.
Questions to Guide the Work-Up: (1) Does the patient have a prolonged URI or pediatric sinusitis, and what differentiates the two conditions? (2) What sinuses are present in a 5-year-old patient? (3) What treatment modalities are available for sinusitis? (4) Is imaging of the sinuses helpful in confirming the diagnosis?
Acute Bacterial Sinusitis
Rhinosinusitis is an inflammation of the mucosal lining of the nasal passages and paranasal sinuses. Most cases occur secondary to a viral URI and resolve spontaneously in 99% of the pediatric population.10,11
Acute bacterial sinusitis (ABS) is an inflammation of the same mucosal lining of the nasal passages secondary to bacterial overgrowth that lasts more than 10 days, with complete resolution by 30 days.12,13 When evaluating a pediatric patient for ABS, it is important to consider the sinus growth and development: If the sinus is not yet formed, it therefore cannot be the location of an ABS.13 The ethmoid and maxillary sinuses are present at birth, aerated within 4 months of life, and are fully developed by age 12 years. The sphenoid sinuses begin development around age 3 years, are aerated by age 7 or 8 years, and are fully developed by age 18 to 20 years. The frontal sinuses begin development around age 8 years and are aerated and fully developed by age 12 to 15 years.10,13,14 While most guidelines focus on children older than age 1 year (due to very small infantile sinuses), ABS does occur in children younger than age 1 year.12,14
Signs and Symptoms
Differentiation between a viral URI/rhinosinusitis and ABS is a challenge and can be based upon severity of symptoms as well as length of illness. Symptoms of ABS are typically present and persistent for more than 10 days, without improvement. Continuing illness and worsening of symptoms are identifying features of ABS given most viral URIs gradually resolve within a 10-day timeframe. Other common symptoms include milky/thick nasal discharge, fever, predominantly nocturnal cough, and headache. Other less common symptoms include facial pain, toothache, malodorous breath, and periorbital edema. On physical examination, erythema and edema of the turbinates, as well as reproducible pain over aerated sinuses, are suggestive of ABS.10-14
Diagnosis
In the acute care setting, diagnosis of ABS should be clinical in nature. Neither imaging nor laboratory work-up is generally required secondary to their poor diagnostic specificity for ABS. The bacteria involved in ABS are similar to those associated with AOM, with S pneumonia, nontypeable H influenza, and M catarrhalis being the predominant organisms.10-15
Treatment and Management
Treatment of ABS is generally recommended once the diagnosis is made, though this is based largely on expert opinion as there are limited RCTs available.13 However, available studies do show a more rapid improvement in children on antibiotic therapy than those on placebo.15,16
Antibiotic Therapy. Amoxicillin remains the antimicrobial agent of choice for first-line treatment of uncomplicated ABS forsituations in which antimicrobial resistance is not suspected. In communities with a high prevalence of nonsusceptible S pneumoniae (>10%, including intermediate- and high-level resistance), treatment may be initiated at 80 to 90 mg/kg/d in two divided doses, with a maximum of 2 g per dose.
Patients presenting with moderate to severe illness, as well as those who are younger than 2 years, attend childcare, or have recently been treated with an antimicrobial, may receive high-dose amoxicillin-clavulanate as initial therapy given the elevated beta-lactamase production of the common bacteria that cause ABS.
Second-line alternatives include azithromycin, cefdinir, and sulfamethoxizole-trimethoprim (Table 1). There are data to suggest higher rates of decreased susceptibility of S pneumonia and H influenza to third-generation cephalosporins, and the addition of clindamycin may be warranted when utilizing those medications. Treatment is recommended for 10 to 14 days, though improvement should be noted within 1 to 3 days.10-12,14-17
Adjuvant Therapy. Additional therapies include nasal irrigation, decongestants, antihistamines, and intranasal steroids; however, there are only anecdotal reports of their efficacy in providing symptom relief. Therefore, there are insufficient evidence-based data to support or refute the role of these adjuvant therapies in treating pediatric patients with ABS.9,13
Case 2 Resolution
The prolonged duration and severity of symptoms (high fever and headache) and the gradual worsening of the clinical course (ie, late-onset fever) in this patient all suggest ABS rather than a simple prolonged URI. The physical examination findings of inflamed turbinates and facial pain further increase the specificity for ABS. The patient was started on oral amoxicillin-clavulanate with planned treatment for 14 days. At discharge, his parents were instructed to follow-up with the patient’s pediatrician in 3 days to ensure a degree of clinical resolution.
Case Scenario 3
A 4-year-old boy was presented by his parents for evaluation of a 2-day history of a persistent and unimproved sore throat. The patient’s mother indicated that the child’s oral T upon returning home earlier from preschool was 101.2oF. She further noted that her 17-month-old daughter and 8-year-old son also experienced similar symptoms which had self-resolved. Triage vital signs were: T, 100.8oF, orally; BP, HR, and RR were all within normal limits. Oxygen saturation was 100% on room air.
On physical examination, the child was noted to have anterior cervical lymph nodes bilaterally and an erythematous oropharynx with exudate noted on both tonsils. There were no cutaneous abnormalities, nasal edema, erythema, or drainage. Based on the clinical examination, the EP was suspicious for SP.
Questions to Guide the Work-Up: (1) Is SP diagnosed based on clinical findings alone in this patient’s age group? (2) At what age in the pediatric population is it appropriate to perform a rapid streptococcal antigen test? (3) Are there medications other than antibiotics that are beneficial in treating symptomatic SP?
Streptococcal Pharyngitis
Streptococcal pharyngitis is a clinical condition caused by group A beta-hemolytic S pyogens. This bacterium is responsible for multiple conditions, including pharyngitis, skin infections, poststreptococcal glomerulonephritis, and rheumatic fever, as well as invasive syndromes. (This case focuses solely on SP).
Pharyngitis can occur secondary to a viral or bacterial infection, and SP is the most common cause of pediatric bacterial pharyngitis. It is estimated that children aged 5 to 15 years are more commonly diagnosed with SP, although approximately 24% of children younger than age 5 years with pharyngitis symptoms will be ultimately diagnosed with SP.
Signs and Symptoms
Typical symptoms include fever, pharyngitis, generalized abdominal pain, nausea, vomiting, headache, and absence of viral URI symptoms (eg, cough, nasal discharge). However, younger patients with SP may have clinical findings of prolonged nasal drainage and excoriated nares. Examination findings may include swollen and tender anterior cervical lymph nodes; generalized edema and erythema of the posterior pharynx; tonsillar exudates; and palatal petechiae.
Diagnosis
Centor Criteria. The Centor criteria were developed to assist practitioners in identifying patients with potential SP. Criteria for patients older than age 15 years include fever, absence of cough, tonsillar exudates, and tender anterior cervical lymphadenopathy. A modified Centor criteria was later established to include children older than age 5 years, with children between ages 5 and 15 years being the fifth variable in the modified score. In general, patients with a score of 4 or 5 (presence of each variable = 1 point) are most likely to test positive for SP on rapid antigen testing (RAT) or culture.18-20
Swab, Rapid Antigen Testing, and Culture. Swabbing the throat and RAT and/or culture should be performed in most children with suspected SP because the clinical features alone do not reliably discriminate SP from viral pharyngitis. Rapid antigen testing is only specific for group A beta-hemolytic streptococcal species, which is the only streptococcal species that is routinely treated with antibiotics in the setting of acute pharyngitis. It is unlikely for a patient with a score of 0 or 1 to have SP, and several sources suggest neither testing nor treating this cohort, but rather to consider an alternative diagnosis.18-20
Within the population of children and young adolescents, due to a RAT sensitivity of 70% to 90%, a negative result should always be backed-up by a throat culture, and treatment initiated if results of the culture are later found to be positive. As the current generation of RAT tests have a high specificity, a positive RAT does not necessitate a back-up culture, and treatment is indicated without further investigation.19,20
Routine RAT is not recommended in children younger than age 3 years as patients in this age group are at low-risk of developing rheumatic fever. One notable exception for these very young children would be if there are siblings in the home with confirmed SP, in which case, RAT should be considered in the clinical context of SP.21 Adolescents over age 15 years are another cohort with a low likelihood of developing rheumatic fever, though they can develop other poststreptococcal complications, such as glomerulonephritis.
The US Centers for Disease Control and Prevention/American Academy of Family Practitioners (AAFP) guidelines suggest that pharyngitis in older adolescents can be approached in a similar fashion to adults, with empiric therapy for a Centor score of 3 or 4, RAT (without the need for follow-up culture) for Centor score of 2, and neither testing nor treating patients with a score of 0 or 1.19
Treatment and Management
Streptococcal pharyngitis is treated mainly to prevent the poststreptococcal complications of rheumatic fever, though it will not prevent poststreptococcal glomerulonephritis. Treatment of SP also facilitates resolution of symptoms and return to baseline activities.
Antibiotic Therapy. Patients who have a positive RAT or a follow-up throat culture positive for group A streptococcus should be given antibiotics. The gold standard treatment is penicillin V orally for 10 days.
Corticosteroid Therapy. The use of corticosteroids for symptom control of SP in pediatric patients is controversial. Although the Infectious Disease Society of America does not recommend corticosteroid therapy in the treatment of SP, several studies show such therapy (namely dexamethasone), improves pain in children and adolescents diagnosed with SP, but without significant change to the overall disease course.21,23-26
Case 3 Resolution
The patient had a modified Centor criteria score of 4, as well as siblings with similar symptoms. In following current guidelines, the EP performed a RAT and back-up culture. The RAT was negative in the ED, but the back-up culture was subsequently positive, and the child was started on a 10-day course of oral amoxicillin.
Conclusion
When evaluating pediatric patients presenting with ENT signs and symptoms such as ear pain and erythema, fever, sore throat, nasal congestion and discharge, a thorough physical examination and history-taking—including recent illness of any siblings—along with testing when indicated, is essential to guide the diagnosis and determine appropriate treatment and management. In addition to administering antibiotic therapy when such is warranted, the EP should provide appropriate analgesia to manage the patient’s pain and assure relief prior to discharge.
1. Rosenfeld RM, Shin JJ, Schwartz SR, et al. Clinical practice guideline: otitis media with effusion (Update). Otolaryngol Head Neck Surg. 2016;154(1 Suppl):S1-S41. doi:10.1177/0194599815623467.
2. Acute Otitis Media Guideline Team, Cincinnati Children’s Hospital Medical Center. Evidence-based care guideline for medical management of acute otitis media in children 2 months to 13 years of age. http://f.i-md.com/medinfo/material/4f4/4eb132ba44ae4ffe12a814f4/4eb132d744ae4ffe12a814f7.pdf. August 2006. Accessed December 29, 2016.
3. Nesbit CE, Powers MC. An evidence-based approach to managing acute otitis media. Pediatr Emerg Med Pract. 2013;10(4):1-26; quiz 26-27.
4. Lieberthal AS, Carroll AE, Chonmaitree T, et al. The diagnosis and management of acute otitis media. Pediatrics. 2013;131(3):e964-e999. doi:10.1542/peds.2012-3488.
5. American Academy of Family Physicians; American Academy of Otolaryngology-Head and Neck Surgery; American Academy of Pediatrics Subcommittee on Otitis Media With Effusion. Otitis media with effusion. Pediatrics. 2004;113(5):1412-1429.
6. US Food and Drug Administration Web site. FDA: Use only approved prescription ear drops. http://www.fda.gov/ForConsumers/ConsumerUpdates/-ucm453087.htm. Updated July 10, 2015. Accessed December 15, 2016.
7. Sack F. An evidence based approach to the management of uncomplicated acute otitis media in children. Int Pediatrics. 2005;20(1):44-46.
8. Johnson NC, Holger JS. Pediatric acute otitis media: the case for delayed antibiotic treatment. J Emerg Med. 2007;32(3):279-284. doi:10.1016/j.jemermed.2006.07.029.
9. Spiro DM, Tay KY, Arnold DH, Dziura JD, Baker MD, Shapiro ED. Wait-and-see prescription for the treatment of acute otitis media: a randomized controlled trial. JAMA. 2006;296(10):1235-1241. doi:10.1001/jama.296.10.1235.
10. Brook I. Management of acute rhinosinusitis in pediatric patients. Pediatr Emerg Med Pract. 2012;9(5):1-24.
11. Ferdman RM, Linzer JF Jr. The runny nose in the emergency department: rhinitis and sinusitis. Clin Pediatr Emerg Med. 2007;8(2):123-130.
12. Acute Bacterial Sinusitis Guideline Team, Cincinnati Children’s Hospital Medical Center: Evidence-based care guideline for medical management of acute bacterial sinusitis in children 1 through 18 years of age. http://www.antibioticos.msssi.gob.es/PDF/sinusitisguideline.pdf. July 7, 2006. Accessed December 29, 2016
13. Holt KR, Murdoch Cuenca M, Cuenca PJ, Johnston GM. acute pediatric sinusitis and “the 10-day rule.” Pediatr Emerg Med Pract. 2006;3(2):1-16.
14. American Academy of Pediatrics. Subcommittee on Management of Sinusitis and Committee on Quality Improvement. Clinical practice guideline: management of sinusitis. Pediatrics. 2001;108(3):798-808.
15. Wald ER, Nash D, Eickhoff J. Effectiveness of amoxicillin/clavulanate potassium in the treatment of acute bacterial sinusitis in children. Pediatrics. 2009;124(1):9-15. doi:10.1542/peds.2008-2902.
16. Arroll B, Kenealy T. Are antibiotics effective for acute purulent rhinitis? Systematic review and meta-analysis of placebo controlled randomised trials. BMJ. 2006;333(7562):279. doi:10.1136/bmj.38891.681215.AE.
17. McQuillan L, Crane LA, Kempe A. Diagnosis and management of acute sinusitis by pediatricians. Pediatrics. 2009;123(2):e193-e198.
18. Singer JI, Fontanette R. Recognizable and suspected group A beta-hemolytic streptococcal syndromes. Pediatr Emerg Med Rep. 2010;15(11):129-144.
19. Weglowski J. An evidence-based approach to the evaluation and treatment of pharyngitis in children. Pediatr Emerg Med Pract. 2011;8(12):1-28.
20. Gerber MA, Baltimore RS, Eaton CB, et al. Prevention of rheumatic fever and diagnosis and treatment of acute Streptococcal pharyngitis: a scientific statement from the American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee of the Council on Cardiovascular Disease in the Young, the Interdisciplinary Council on Functional Genomics and Translational Biology, and the Interdisciplinary Council on Quality of Care and Outcomes Research: endorsed by the American Academy of Pediatrics. Circulation. 2009;119(11):1541-1551. doi:10.1161/CIRCULATIONAHA.109.191959.
21. Shulman ST, Bisno AL, Clegg HW, et al. Clinical practice guideline for the diagnosis and management of group A streptococcal pharyngitis: 2012 update by the Infectious Diseases Society of America. Clin Infect Dis. 2012;55(10):1279-1282. doi:10.1093/cid/cis847.
22. Clegg HW, Ryan AG, Dallas SD, et al. Treatment of streptococcal pharyngitis with once-daily compared with twice-daily amoxicillin: a noninferiority trial. Pediatr Infect Dis J. 2006;25(9):761-767. doi:10.1097/01.inf.0000235678.46805.92.
23. Bulloch B, Kabani A, Tenenbein M. Oral dexamethasone for the treatment of pain in children with acute pharyngitis: a randomized, double-blind, placebo-controlled trial. Ann Emerg Med. 2003;41(5):601-608. doi:10.1067/mem.2003.136.
24. Niland ML, Bonsu BK, Nuss KE, Goodman DG. A pilot study of 1 versus 3 days of dexamethasone as add-on therapy in children with streptococcal pharyngitis. Pediatr Infect Dis J. 2006;25(6):477-481. doi:10.1097/01.inf.0000219469.95772.3f.
25. Wei JL, Kasperbauer JL, Weaver AL, Boggust AJ. Efficacy of single-dose dexamethasone as adjuvant therapy for acute pharyngitis. Laryngoscope. 2002;112(1):87-93. doi:10.1097/00005537-200201000-00016.
26. Hayward G, Thompson M, Heneghan C, Perera R, Del Mar C, Glasziou P. Corticosteroids for pain relief in sore throat: systematic review and meta-analysis. BMJ. 2009;339:b2976. doi:10.1136/bmj.b2976.
1. Rosenfeld RM, Shin JJ, Schwartz SR, et al. Clinical practice guideline: otitis media with effusion (Update). Otolaryngol Head Neck Surg. 2016;154(1 Suppl):S1-S41. doi:10.1177/0194599815623467.
2. Acute Otitis Media Guideline Team, Cincinnati Children’s Hospital Medical Center. Evidence-based care guideline for medical management of acute otitis media in children 2 months to 13 years of age. http://f.i-md.com/medinfo/material/4f4/4eb132ba44ae4ffe12a814f4/4eb132d744ae4ffe12a814f7.pdf. August 2006. Accessed December 29, 2016.
3. Nesbit CE, Powers MC. An evidence-based approach to managing acute otitis media. Pediatr Emerg Med Pract. 2013;10(4):1-26; quiz 26-27.
4. Lieberthal AS, Carroll AE, Chonmaitree T, et al. The diagnosis and management of acute otitis media. Pediatrics. 2013;131(3):e964-e999. doi:10.1542/peds.2012-3488.
5. American Academy of Family Physicians; American Academy of Otolaryngology-Head and Neck Surgery; American Academy of Pediatrics Subcommittee on Otitis Media With Effusion. Otitis media with effusion. Pediatrics. 2004;113(5):1412-1429.
6. US Food and Drug Administration Web site. FDA: Use only approved prescription ear drops. http://www.fda.gov/ForConsumers/ConsumerUpdates/-ucm453087.htm. Updated July 10, 2015. Accessed December 15, 2016.
7. Sack F. An evidence based approach to the management of uncomplicated acute otitis media in children. Int Pediatrics. 2005;20(1):44-46.
8. Johnson NC, Holger JS. Pediatric acute otitis media: the case for delayed antibiotic treatment. J Emerg Med. 2007;32(3):279-284. doi:10.1016/j.jemermed.2006.07.029.
9. Spiro DM, Tay KY, Arnold DH, Dziura JD, Baker MD, Shapiro ED. Wait-and-see prescription for the treatment of acute otitis media: a randomized controlled trial. JAMA. 2006;296(10):1235-1241. doi:10.1001/jama.296.10.1235.
10. Brook I. Management of acute rhinosinusitis in pediatric patients. Pediatr Emerg Med Pract. 2012;9(5):1-24.
11. Ferdman RM, Linzer JF Jr. The runny nose in the emergency department: rhinitis and sinusitis. Clin Pediatr Emerg Med. 2007;8(2):123-130.
12. Acute Bacterial Sinusitis Guideline Team, Cincinnati Children’s Hospital Medical Center: Evidence-based care guideline for medical management of acute bacterial sinusitis in children 1 through 18 years of age. http://www.antibioticos.msssi.gob.es/PDF/sinusitisguideline.pdf. July 7, 2006. Accessed December 29, 2016
13. Holt KR, Murdoch Cuenca M, Cuenca PJ, Johnston GM. acute pediatric sinusitis and “the 10-day rule.” Pediatr Emerg Med Pract. 2006;3(2):1-16.
14. American Academy of Pediatrics. Subcommittee on Management of Sinusitis and Committee on Quality Improvement. Clinical practice guideline: management of sinusitis. Pediatrics. 2001;108(3):798-808.
15. Wald ER, Nash D, Eickhoff J. Effectiveness of amoxicillin/clavulanate potassium in the treatment of acute bacterial sinusitis in children. Pediatrics. 2009;124(1):9-15. doi:10.1542/peds.2008-2902.
16. Arroll B, Kenealy T. Are antibiotics effective for acute purulent rhinitis? Systematic review and meta-analysis of placebo controlled randomised trials. BMJ. 2006;333(7562):279. doi:10.1136/bmj.38891.681215.AE.
17. McQuillan L, Crane LA, Kempe A. Diagnosis and management of acute sinusitis by pediatricians. Pediatrics. 2009;123(2):e193-e198.
18. Singer JI, Fontanette R. Recognizable and suspected group A beta-hemolytic streptococcal syndromes. Pediatr Emerg Med Rep. 2010;15(11):129-144.
19. Weglowski J. An evidence-based approach to the evaluation and treatment of pharyngitis in children. Pediatr Emerg Med Pract. 2011;8(12):1-28.
20. Gerber MA, Baltimore RS, Eaton CB, et al. Prevention of rheumatic fever and diagnosis and treatment of acute Streptococcal pharyngitis: a scientific statement from the American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee of the Council on Cardiovascular Disease in the Young, the Interdisciplinary Council on Functional Genomics and Translational Biology, and the Interdisciplinary Council on Quality of Care and Outcomes Research: endorsed by the American Academy of Pediatrics. Circulation. 2009;119(11):1541-1551. doi:10.1161/CIRCULATIONAHA.109.191959.
21. Shulman ST, Bisno AL, Clegg HW, et al. Clinical practice guideline for the diagnosis and management of group A streptococcal pharyngitis: 2012 update by the Infectious Diseases Society of America. Clin Infect Dis. 2012;55(10):1279-1282. doi:10.1093/cid/cis847.
22. Clegg HW, Ryan AG, Dallas SD, et al. Treatment of streptococcal pharyngitis with once-daily compared with twice-daily amoxicillin: a noninferiority trial. Pediatr Infect Dis J. 2006;25(9):761-767. doi:10.1097/01.inf.0000235678.46805.92.
23. Bulloch B, Kabani A, Tenenbein M. Oral dexamethasone for the treatment of pain in children with acute pharyngitis: a randomized, double-blind, placebo-controlled trial. Ann Emerg Med. 2003;41(5):601-608. doi:10.1067/mem.2003.136.
24. Niland ML, Bonsu BK, Nuss KE, Goodman DG. A pilot study of 1 versus 3 days of dexamethasone as add-on therapy in children with streptococcal pharyngitis. Pediatr Infect Dis J. 2006;25(6):477-481. doi:10.1097/01.inf.0000219469.95772.3f.
25. Wei JL, Kasperbauer JL, Weaver AL, Boggust AJ. Efficacy of single-dose dexamethasone as adjuvant therapy for acute pharyngitis. Laryngoscope. 2002;112(1):87-93. doi:10.1097/00005537-200201000-00016.
26. Hayward G, Thompson M, Heneghan C, Perera R, Del Mar C, Glasziou P. Corticosteroids for pain relief in sore throat: systematic review and meta-analysis. BMJ. 2009;339:b2976. doi:10.1136/bmj.b2976.
First-trimester blood glucose predicts congenital heart disease risk
NEW ORLEANS – A single, random, first-trimester maternal plasma glucose measurement is superior to an oral glucose tolerance test later in pregnancy as a predictor of congenital heart disease in newborns, Emmi Helle, MD, reported at the American Heart Association scientific sessions.
This finding from a large retrospective study, if confirmed in a prospective data set, is likely to be practice changing. At present, a 1-hour oral glucose tolerance test in the second or third trimester is considered the best means of identifying pregnant women who ought to undergo fetal echocardiography for prenatal diagnosis of congenital heart disease, noted Dr. Helle of Stanford (Calif.) University.
An elevated random plasma glucose value in the first trimester was broadly predictive of increased risk for a variety of congenital heart anomalies, not just, for example, cyanotic conditions.
Fetal heart development is completed during the first trimester, Dr. Helle observed.
Her study received a warm reception. Michael A. Portman, MD, singled it out in his final-day wrap-up of the meeting’s highlights in the field of congenital heart disease.
Several studies have demonstrated that prenatal diagnosis of congenital heart disease results in improved surgical outcomes in newborns. The question is, how to get the right women – those at increased risk – to diagnostic fetal echocardiography. Guidelines suggest but don’t mandate on the basis of weak evidence that an oral glucose tolerance test performed in the second or early third trimester may be a useful means of screening mothers for fetal imaging. Dr. Helle’s study points to a better way.
“Hopefully we can change our guidelines and make them more scientific for identification of mothers who should undergo fetal echocardiography,” said Dr. Portman, professor of pediatrics at the University of Washington, Seattle, and director of pediatric cardiovascular research at Seattle Children’s Hospital.
Dr. Helle and Dr. Portman reported having no relevant financial interests.
NEW ORLEANS – A single, random, first-trimester maternal plasma glucose measurement is superior to an oral glucose tolerance test later in pregnancy as a predictor of congenital heart disease in newborns, Emmi Helle, MD, reported at the American Heart Association scientific sessions.
This finding from a large retrospective study, if confirmed in a prospective data set, is likely to be practice changing. At present, a 1-hour oral glucose tolerance test in the second or third trimester is considered the best means of identifying pregnant women who ought to undergo fetal echocardiography for prenatal diagnosis of congenital heart disease, noted Dr. Helle of Stanford (Calif.) University.
An elevated random plasma glucose value in the first trimester was broadly predictive of increased risk for a variety of congenital heart anomalies, not just, for example, cyanotic conditions.
Fetal heart development is completed during the first trimester, Dr. Helle observed.
Her study received a warm reception. Michael A. Portman, MD, singled it out in his final-day wrap-up of the meeting’s highlights in the field of congenital heart disease.
Several studies have demonstrated that prenatal diagnosis of congenital heart disease results in improved surgical outcomes in newborns. The question is, how to get the right women – those at increased risk – to diagnostic fetal echocardiography. Guidelines suggest but don’t mandate on the basis of weak evidence that an oral glucose tolerance test performed in the second or early third trimester may be a useful means of screening mothers for fetal imaging. Dr. Helle’s study points to a better way.
“Hopefully we can change our guidelines and make them more scientific for identification of mothers who should undergo fetal echocardiography,” said Dr. Portman, professor of pediatrics at the University of Washington, Seattle, and director of pediatric cardiovascular research at Seattle Children’s Hospital.
Dr. Helle and Dr. Portman reported having no relevant financial interests.
NEW ORLEANS – A single, random, first-trimester maternal plasma glucose measurement is superior to an oral glucose tolerance test later in pregnancy as a predictor of congenital heart disease in newborns, Emmi Helle, MD, reported at the American Heart Association scientific sessions.
This finding from a large retrospective study, if confirmed in a prospective data set, is likely to be practice changing. At present, a 1-hour oral glucose tolerance test in the second or third trimester is considered the best means of identifying pregnant women who ought to undergo fetal echocardiography for prenatal diagnosis of congenital heart disease, noted Dr. Helle of Stanford (Calif.) University.
An elevated random plasma glucose value in the first trimester was broadly predictive of increased risk for a variety of congenital heart anomalies, not just, for example, cyanotic conditions.
Fetal heart development is completed during the first trimester, Dr. Helle observed.
Her study received a warm reception. Michael A. Portman, MD, singled it out in his final-day wrap-up of the meeting’s highlights in the field of congenital heart disease.
Several studies have demonstrated that prenatal diagnosis of congenital heart disease results in improved surgical outcomes in newborns. The question is, how to get the right women – those at increased risk – to diagnostic fetal echocardiography. Guidelines suggest but don’t mandate on the basis of weak evidence that an oral glucose tolerance test performed in the second or early third trimester may be a useful means of screening mothers for fetal imaging. Dr. Helle’s study points to a better way.
“Hopefully we can change our guidelines and make them more scientific for identification of mothers who should undergo fetal echocardiography,” said Dr. Portman, professor of pediatrics at the University of Washington, Seattle, and director of pediatric cardiovascular research at Seattle Children’s Hospital.
Dr. Helle and Dr. Portman reported having no relevant financial interests.
AT THE AHA SCIENTIFIC SESSIONS
Key clinical point:
Major finding: For every 10-mg/dL increase in maternal plasma glucose on a random first-trimester measurement, the risk of giving birth to a baby with congenital heart disease rose by 8%.
Data source: A retrospective study of 19,197 pregnancies, 811 of which resulted in congenital heart disease in the offspring.
Disclosures: The presenter reported having no financial conflicts of interest regarding the study.
Fewer people having problems with medical bills
The number of people under age 65 years who were in families having trouble paying medical bills dropped by more than 22% from 2011 to 2016, according to the National Center for Health Statistics.
For the first 6 months of 2016, there were 43.8 million people, or 16.2% of the population under age 65 years, who were in families that had problems paying medical bills in the past year, which was down from 56.5 million (21.3 % of the population) in 2011, the NCHS reported.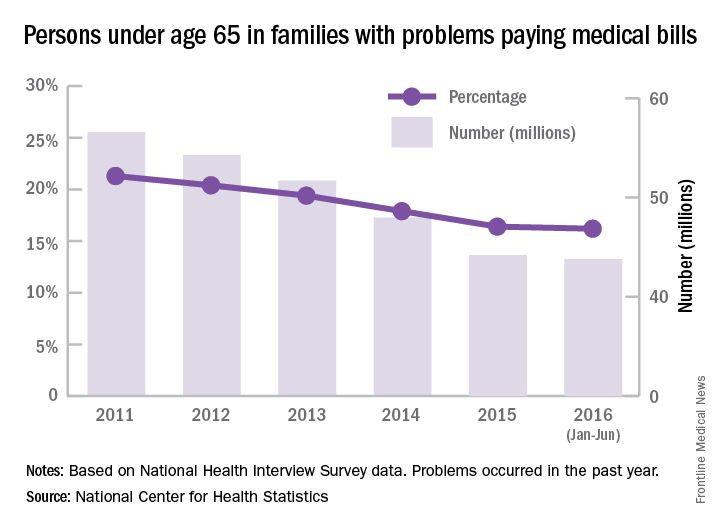
The drop was consistent across race/ethnicity lines, but not the start and endpoints. The percentage of non-Hispanic blacks in families having trouble paying their medical bills dropped from 27.3% in 2011 to 23% in 2016, although there was actually a small increase from 2015 to 2016. Hispanics dropped from 24.3% in 2011 to 17.4% in 2016, non-Hispanic whites dropped from 19.8% to 15.1%, and non-Hispanic Asians went from 11% to 6%, according to data collected from 579,379 people for the National Health Interview Survey.
The number of people under age 65 years who were in families having trouble paying medical bills dropped by more than 22% from 2011 to 2016, according to the National Center for Health Statistics.
For the first 6 months of 2016, there were 43.8 million people, or 16.2% of the population under age 65 years, who were in families that had problems paying medical bills in the past year, which was down from 56.5 million (21.3 % of the population) in 2011, the NCHS reported.
The drop was consistent across race/ethnicity lines, but not the start and endpoints. The percentage of non-Hispanic blacks in families having trouble paying their medical bills dropped from 27.3% in 2011 to 23% in 2016, although there was actually a small increase from 2015 to 2016. Hispanics dropped from 24.3% in 2011 to 17.4% in 2016, non-Hispanic whites dropped from 19.8% to 15.1%, and non-Hispanic Asians went from 11% to 6%, according to data collected from 579,379 people for the National Health Interview Survey.
The number of people under age 65 years who were in families having trouble paying medical bills dropped by more than 22% from 2011 to 2016, according to the National Center for Health Statistics.
For the first 6 months of 2016, there were 43.8 million people, or 16.2% of the population under age 65 years, who were in families that had problems paying medical bills in the past year, which was down from 56.5 million (21.3 % of the population) in 2011, the NCHS reported.
The drop was consistent across race/ethnicity lines, but not the start and endpoints. The percentage of non-Hispanic blacks in families having trouble paying their medical bills dropped from 27.3% in 2011 to 23% in 2016, although there was actually a small increase from 2015 to 2016. Hispanics dropped from 24.3% in 2011 to 17.4% in 2016, non-Hispanic whites dropped from 19.8% to 15.1%, and non-Hispanic Asians went from 11% to 6%, according to data collected from 579,379 people for the National Health Interview Survey.
Sorafenib survival benefit in HCC depends on etiology
The impact of sorafenib in treating patients with advanced hepatocellular cancer may be dependent on their hepatitis status, according to a meta-analysis of patient-level data from three large prospective clinical trials using sorafenib as the control treatment.
The analysis indicated that sorafenib had a favorable effect on overall survival for hepatocellular carcinoma patients who were positive for hepatitis C (HCV), but negative for hepatitis B (HBV) only.
For HCV-positive/HBV-negative patients with advanced unresectable hepatocellular carcinoma (aHCC) who received sorafenib, median unadjusted survival was 12.6 months, compared to 10.2 months for patients who received other treatments, yielding a log hazard ratio of –0.27 (95% confidence interval [CI] –0.46 to –0.06).
Though the study did not shed light on the reasons for this difference in overall survival (OS), the results were seen consistently in data from all trials, and sorafenib did not confer any significant survival benefit for individuals who were not HCV positive and HBV negative. “Irrespective of the mechanism, our data suggest that in future trials in aHCC, particularly where sorafenib is the control arm, there should be stratification according to etiology,” wrote Richard Jackson, MSc, and his coauthors (J Clin Oncol. 2017 Jan 3:JCO2016695197 [Epub ahead of print]).
Mr. Jackson, a medical statistician with the Institute of Translational Medicine at the University of Liverpool, England, and his collaborators examined data from 3,256 patients with aHCC. Of these, 1,643 (50%) received sorafenib. The remainder was aggregated into an “other treatment” group, pooling data from patients who received brivanib, sunitinib, and linifanib.
All patients were also divided into four etiologic subgroups: HBV-negative/HCV-negative; HBV-negative/HCV-positive; HBV-positive/HCV-negative; and HBV-positive/HCV-positive. Mr. Jackson and his colleagues then examined the pooled data to see what effect treatment type (sorafenib versus pooled comparator treatments) had on overall survival for each etiologic subgroup. They found no statistically significant survival benefit for sorafenib in the three etiologic subgroups that were not HCV positive/HBV negative, though the data showed a statistically insignificant trend favoring sorafenib. Race, when examined as a potential confounding factor, was not associated with a difference in OS benefit for sorafenib.
The sponsors of three large clinical trials of treatments for aHCC that used sorafenib as the control arm provided deidentified patient-level data to the study’s authors, who then undertook an individual patient data (IPD) meta-analysis. “IPD meta-analyses have a major advantage over aggregate meta-analyses in that they ensure consistent analytic techniques and allow for detailed inspection of interaction of subgroup effects that are not available in published evidence,” wrote Mr. Jackson and his coauthors.
In discussing their findings, the researchers called for more of the data-sharing that allowed their IPD meta-analysis, citing a proposal on the topic from the International Committee of Medical Journal Editors. “[O]ur study showed how the benefits of access to completed trial data are not necessarily confined to reanalysis of the original hypothesis tested by the trial. Here, by a meta-analysis, we arrive at an answer to a question that was not considered when the trials were conceived and could not have been answered by any of the trials individually,” they wrote.
The study data were provided by Bristol-Myers Squibb, Pfizer, and AbbVie from studies they sponsored. Mr. Jackson reported no conflicts of interest.
[email protected]
On Twitter @karioakes
The impact of sorafenib in treating patients with advanced hepatocellular cancer may be dependent on their hepatitis status, according to a meta-analysis of patient-level data from three large prospective clinical trials using sorafenib as the control treatment.
The analysis indicated that sorafenib had a favorable effect on overall survival for hepatocellular carcinoma patients who were positive for hepatitis C (HCV), but negative for hepatitis B (HBV) only.
For HCV-positive/HBV-negative patients with advanced unresectable hepatocellular carcinoma (aHCC) who received sorafenib, median unadjusted survival was 12.6 months, compared to 10.2 months for patients who received other treatments, yielding a log hazard ratio of –0.27 (95% confidence interval [CI] –0.46 to –0.06).
Though the study did not shed light on the reasons for this difference in overall survival (OS), the results were seen consistently in data from all trials, and sorafenib did not confer any significant survival benefit for individuals who were not HCV positive and HBV negative. “Irrespective of the mechanism, our data suggest that in future trials in aHCC, particularly where sorafenib is the control arm, there should be stratification according to etiology,” wrote Richard Jackson, MSc, and his coauthors (J Clin Oncol. 2017 Jan 3:JCO2016695197 [Epub ahead of print]).
Mr. Jackson, a medical statistician with the Institute of Translational Medicine at the University of Liverpool, England, and his collaborators examined data from 3,256 patients with aHCC. Of these, 1,643 (50%) received sorafenib. The remainder was aggregated into an “other treatment” group, pooling data from patients who received brivanib, sunitinib, and linifanib.
All patients were also divided into four etiologic subgroups: HBV-negative/HCV-negative; HBV-negative/HCV-positive; HBV-positive/HCV-negative; and HBV-positive/HCV-positive. Mr. Jackson and his colleagues then examined the pooled data to see what effect treatment type (sorafenib versus pooled comparator treatments) had on overall survival for each etiologic subgroup. They found no statistically significant survival benefit for sorafenib in the three etiologic subgroups that were not HCV positive/HBV negative, though the data showed a statistically insignificant trend favoring sorafenib. Race, when examined as a potential confounding factor, was not associated with a difference in OS benefit for sorafenib.
The sponsors of three large clinical trials of treatments for aHCC that used sorafenib as the control arm provided deidentified patient-level data to the study’s authors, who then undertook an individual patient data (IPD) meta-analysis. “IPD meta-analyses have a major advantage over aggregate meta-analyses in that they ensure consistent analytic techniques and allow for detailed inspection of interaction of subgroup effects that are not available in published evidence,” wrote Mr. Jackson and his coauthors.
In discussing their findings, the researchers called for more of the data-sharing that allowed their IPD meta-analysis, citing a proposal on the topic from the International Committee of Medical Journal Editors. “[O]ur study showed how the benefits of access to completed trial data are not necessarily confined to reanalysis of the original hypothesis tested by the trial. Here, by a meta-analysis, we arrive at an answer to a question that was not considered when the trials were conceived and could not have been answered by any of the trials individually,” they wrote.
The study data were provided by Bristol-Myers Squibb, Pfizer, and AbbVie from studies they sponsored. Mr. Jackson reported no conflicts of interest.
[email protected]
On Twitter @karioakes
The impact of sorafenib in treating patients with advanced hepatocellular cancer may be dependent on their hepatitis status, according to a meta-analysis of patient-level data from three large prospective clinical trials using sorafenib as the control treatment.
The analysis indicated that sorafenib had a favorable effect on overall survival for hepatocellular carcinoma patients who were positive for hepatitis C (HCV), but negative for hepatitis B (HBV) only.
For HCV-positive/HBV-negative patients with advanced unresectable hepatocellular carcinoma (aHCC) who received sorafenib, median unadjusted survival was 12.6 months, compared to 10.2 months for patients who received other treatments, yielding a log hazard ratio of –0.27 (95% confidence interval [CI] –0.46 to –0.06).
Though the study did not shed light on the reasons for this difference in overall survival (OS), the results were seen consistently in data from all trials, and sorafenib did not confer any significant survival benefit for individuals who were not HCV positive and HBV negative. “Irrespective of the mechanism, our data suggest that in future trials in aHCC, particularly where sorafenib is the control arm, there should be stratification according to etiology,” wrote Richard Jackson, MSc, and his coauthors (J Clin Oncol. 2017 Jan 3:JCO2016695197 [Epub ahead of print]).
Mr. Jackson, a medical statistician with the Institute of Translational Medicine at the University of Liverpool, England, and his collaborators examined data from 3,256 patients with aHCC. Of these, 1,643 (50%) received sorafenib. The remainder was aggregated into an “other treatment” group, pooling data from patients who received brivanib, sunitinib, and linifanib.
All patients were also divided into four etiologic subgroups: HBV-negative/HCV-negative; HBV-negative/HCV-positive; HBV-positive/HCV-negative; and HBV-positive/HCV-positive. Mr. Jackson and his colleagues then examined the pooled data to see what effect treatment type (sorafenib versus pooled comparator treatments) had on overall survival for each etiologic subgroup. They found no statistically significant survival benefit for sorafenib in the three etiologic subgroups that were not HCV positive/HBV negative, though the data showed a statistically insignificant trend favoring sorafenib. Race, when examined as a potential confounding factor, was not associated with a difference in OS benefit for sorafenib.
The sponsors of three large clinical trials of treatments for aHCC that used sorafenib as the control arm provided deidentified patient-level data to the study’s authors, who then undertook an individual patient data (IPD) meta-analysis. “IPD meta-analyses have a major advantage over aggregate meta-analyses in that they ensure consistent analytic techniques and allow for detailed inspection of interaction of subgroup effects that are not available in published evidence,” wrote Mr. Jackson and his coauthors.
In discussing their findings, the researchers called for more of the data-sharing that allowed their IPD meta-analysis, citing a proposal on the topic from the International Committee of Medical Journal Editors. “[O]ur study showed how the benefits of access to completed trial data are not necessarily confined to reanalysis of the original hypothesis tested by the trial. Here, by a meta-analysis, we arrive at an answer to a question that was not considered when the trials were conceived and could not have been answered by any of the trials individually,” they wrote.
The study data were provided by Bristol-Myers Squibb, Pfizer, and AbbVie from studies they sponsored. Mr. Jackson reported no conflicts of interest.
[email protected]
On Twitter @karioakes
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Key clinical point:
Major finding: Median unadjusted survival time for HCV-positive/HBV-negative patients was 12.6 months on sorafenib, compared to 10.2 months for these patients who received other treatments.
Data source: Individual patient data meta-analysis of three randomized phase III clinical trials, examining data from 3,256 patients.
Disclosures: The study data were provided by Bristol-Myers Squibb, Pfizer, and AbbVie. Mr. Jackson reported no conflicts of interest.
Gastric cancer yields to growth hormone antagonist in lab
It sounds counterintuitive, but targeting a neuropeptide hormone produced in the hypothalamus may be an effective strategy for treating gastric cancer, the second most common cause of cancer deaths worldwide, investigators from China and the United States contend.
Growth hormone–releasing hormone (GHRH) and its receptor (GHRH-R) are found primarily in the anterior pituitary gland, but are also present in gastric cancers, other solid tumors, and lymphomas. Increased levels of GHRH-R in tumor samples from patients with gastric cancer are associated with poor outcomes, noted Andrew V. Schally, PhD, MD, DSc, of the University of Miami, and his colleagues at the Shantou (China) University Medical College.
Furthermore, an experimental peptide drug labeled MIA-602 that targets GHRH-R inhibited the growth of gastric cancer cell lines and human tumor xenografts in mice, the investigators reported in the journal PNAS.
“The GHRH receptor is both a biomarker that can confirm prognosis and a therapeutic target,” Dr. Schally said in a statement.
Elevated GHRH-R expression in tumors
GHRH-R antagonists such as MIA-602 work through downregulation of the p21-activated kinase 1 (PAK1)–mediated signal transducer and activator of transcription 3 (STAT3)/nuclear factor–kappaB (NF-kappaB) inflammatory pathway. This pathway is involved in the interplay between inflammatory processes and intracellular signaling thought to be the cause of gastric cancer tumorigenesis and progression, the investigators explained.
They first looked for GHRH-R expression in gastric cancer samples from 106 patients, using immunohistochemistry staining of primary tumors and adjacent normal tissues. They found that gastric cancer tissues “exhibited robust expression of GHRH-R, compared with normal tissues.”
In 50 samples, GHRH-R was determined to be overexpressed, and this overexpression was significantly associated with both greater tumor size (P = .031) and high pathologic tumor stage (P = .001). Increasing expression of GHRH-R was also significantly associated with worse overall survival (P less than .001).
They confirmed these findings in samples from a multinational cohort of patients, which again showed that the highest levels of GHRH-R expression were associated with poor overall survival (P less than .001).
The authors also looked at messenger RNA expression and gene copy number in 65 gastric cancer samples and 19 adjacent normal tissue samples, and found that GHRH-R mRNA was significantly higher in tumor tissues than normal control tissues (P less than .001).
MAI-602 in vitro and in vivo
To see whether MAI-602 could inhibit the growth of gastric cancer cells, the investigators tried it at various doses in three human gastric cancer cell lines, and found that it inhibited cells in a dose-dependent fashion, compared with vehicle used as a control (P less than .001).
In addition, the experimental agent “exhibited remarkable inhibitory effects on tumor growth in vivo” in mice with human tumor xenografts (P less than .001).
Finally, they showed that the cancer suppression effects of MAI-602 work through inhibition of STAT3/NF-kappaB inflammatory signaling. In vitro and in vivo, MAI-602 decreased the expression of both GHRH and GHRH-R, whereas as a GHRH-R agonist increased levels of both the hormone and its receptor. They also demonstrated that PAK1 appears to be a critical mediator of STAT3/NF-kappaB activity, and that MAI-602 works primarily by blocking PAK1-mediated inflammatory signaling.
“MIA-602 remarkably inhibits the growth of human in vitro and in vivo through the suppression of PAK1–STAT3/NF-kappaB signaling. Our study strongly highlights the therapeutic potential of GHRH-R antagonists in the treatment of gastric cancer patients. Knowledge gained in our study will shed light on how to select the appropriate patients for personalized cancer therapy using GHRH-R antagonists,” Dr. Schally and his coauthors wrote.
The study was supported by the Li Ka Shing Foundation, Chinese foundation, and government grants to individual researchers, as well as support from the the Medical Research Service of the U.S. Department of Veterans Affairs, South Florida Veterans Affairs Foundation for Research and Education, and the University of Miami.
It sounds counterintuitive, but targeting a neuropeptide hormone produced in the hypothalamus may be an effective strategy for treating gastric cancer, the second most common cause of cancer deaths worldwide, investigators from China and the United States contend.
Growth hormone–releasing hormone (GHRH) and its receptor (GHRH-R) are found primarily in the anterior pituitary gland, but are also present in gastric cancers, other solid tumors, and lymphomas. Increased levels of GHRH-R in tumor samples from patients with gastric cancer are associated with poor outcomes, noted Andrew V. Schally, PhD, MD, DSc, of the University of Miami, and his colleagues at the Shantou (China) University Medical College.
Furthermore, an experimental peptide drug labeled MIA-602 that targets GHRH-R inhibited the growth of gastric cancer cell lines and human tumor xenografts in mice, the investigators reported in the journal PNAS.
“The GHRH receptor is both a biomarker that can confirm prognosis and a therapeutic target,” Dr. Schally said in a statement.
Elevated GHRH-R expression in tumors
GHRH-R antagonists such as MIA-602 work through downregulation of the p21-activated kinase 1 (PAK1)–mediated signal transducer and activator of transcription 3 (STAT3)/nuclear factor–kappaB (NF-kappaB) inflammatory pathway. This pathway is involved in the interplay between inflammatory processes and intracellular signaling thought to be the cause of gastric cancer tumorigenesis and progression, the investigators explained.
They first looked for GHRH-R expression in gastric cancer samples from 106 patients, using immunohistochemistry staining of primary tumors and adjacent normal tissues. They found that gastric cancer tissues “exhibited robust expression of GHRH-R, compared with normal tissues.”
In 50 samples, GHRH-R was determined to be overexpressed, and this overexpression was significantly associated with both greater tumor size (P = .031) and high pathologic tumor stage (P = .001). Increasing expression of GHRH-R was also significantly associated with worse overall survival (P less than .001).
They confirmed these findings in samples from a multinational cohort of patients, which again showed that the highest levels of GHRH-R expression were associated with poor overall survival (P less than .001).
The authors also looked at messenger RNA expression and gene copy number in 65 gastric cancer samples and 19 adjacent normal tissue samples, and found that GHRH-R mRNA was significantly higher in tumor tissues than normal control tissues (P less than .001).
MAI-602 in vitro and in vivo
To see whether MAI-602 could inhibit the growth of gastric cancer cells, the investigators tried it at various doses in three human gastric cancer cell lines, and found that it inhibited cells in a dose-dependent fashion, compared with vehicle used as a control (P less than .001).
In addition, the experimental agent “exhibited remarkable inhibitory effects on tumor growth in vivo” in mice with human tumor xenografts (P less than .001).
Finally, they showed that the cancer suppression effects of MAI-602 work through inhibition of STAT3/NF-kappaB inflammatory signaling. In vitro and in vivo, MAI-602 decreased the expression of both GHRH and GHRH-R, whereas as a GHRH-R agonist increased levels of both the hormone and its receptor. They also demonstrated that PAK1 appears to be a critical mediator of STAT3/NF-kappaB activity, and that MAI-602 works primarily by blocking PAK1-mediated inflammatory signaling.
“MIA-602 remarkably inhibits the growth of human in vitro and in vivo through the suppression of PAK1–STAT3/NF-kappaB signaling. Our study strongly highlights the therapeutic potential of GHRH-R antagonists in the treatment of gastric cancer patients. Knowledge gained in our study will shed light on how to select the appropriate patients for personalized cancer therapy using GHRH-R antagonists,” Dr. Schally and his coauthors wrote.
The study was supported by the Li Ka Shing Foundation, Chinese foundation, and government grants to individual researchers, as well as support from the the Medical Research Service of the U.S. Department of Veterans Affairs, South Florida Veterans Affairs Foundation for Research and Education, and the University of Miami.
It sounds counterintuitive, but targeting a neuropeptide hormone produced in the hypothalamus may be an effective strategy for treating gastric cancer, the second most common cause of cancer deaths worldwide, investigators from China and the United States contend.
Growth hormone–releasing hormone (GHRH) and its receptor (GHRH-R) are found primarily in the anterior pituitary gland, but are also present in gastric cancers, other solid tumors, and lymphomas. Increased levels of GHRH-R in tumor samples from patients with gastric cancer are associated with poor outcomes, noted Andrew V. Schally, PhD, MD, DSc, of the University of Miami, and his colleagues at the Shantou (China) University Medical College.
Furthermore, an experimental peptide drug labeled MIA-602 that targets GHRH-R inhibited the growth of gastric cancer cell lines and human tumor xenografts in mice, the investigators reported in the journal PNAS.
“The GHRH receptor is both a biomarker that can confirm prognosis and a therapeutic target,” Dr. Schally said in a statement.
Elevated GHRH-R expression in tumors
GHRH-R antagonists such as MIA-602 work through downregulation of the p21-activated kinase 1 (PAK1)–mediated signal transducer and activator of transcription 3 (STAT3)/nuclear factor–kappaB (NF-kappaB) inflammatory pathway. This pathway is involved in the interplay between inflammatory processes and intracellular signaling thought to be the cause of gastric cancer tumorigenesis and progression, the investigators explained.
They first looked for GHRH-R expression in gastric cancer samples from 106 patients, using immunohistochemistry staining of primary tumors and adjacent normal tissues. They found that gastric cancer tissues “exhibited robust expression of GHRH-R, compared with normal tissues.”
In 50 samples, GHRH-R was determined to be overexpressed, and this overexpression was significantly associated with both greater tumor size (P = .031) and high pathologic tumor stage (P = .001). Increasing expression of GHRH-R was also significantly associated with worse overall survival (P less than .001).
They confirmed these findings in samples from a multinational cohort of patients, which again showed that the highest levels of GHRH-R expression were associated with poor overall survival (P less than .001).
The authors also looked at messenger RNA expression and gene copy number in 65 gastric cancer samples and 19 adjacent normal tissue samples, and found that GHRH-R mRNA was significantly higher in tumor tissues than normal control tissues (P less than .001).
MAI-602 in vitro and in vivo
To see whether MAI-602 could inhibit the growth of gastric cancer cells, the investigators tried it at various doses in three human gastric cancer cell lines, and found that it inhibited cells in a dose-dependent fashion, compared with vehicle used as a control (P less than .001).
In addition, the experimental agent “exhibited remarkable inhibitory effects on tumor growth in vivo” in mice with human tumor xenografts (P less than .001).
Finally, they showed that the cancer suppression effects of MAI-602 work through inhibition of STAT3/NF-kappaB inflammatory signaling. In vitro and in vivo, MAI-602 decreased the expression of both GHRH and GHRH-R, whereas as a GHRH-R agonist increased levels of both the hormone and its receptor. They also demonstrated that PAK1 appears to be a critical mediator of STAT3/NF-kappaB activity, and that MAI-602 works primarily by blocking PAK1-mediated inflammatory signaling.
“MIA-602 remarkably inhibits the growth of human in vitro and in vivo through the suppression of PAK1–STAT3/NF-kappaB signaling. Our study strongly highlights the therapeutic potential of GHRH-R antagonists in the treatment of gastric cancer patients. Knowledge gained in our study will shed light on how to select the appropriate patients for personalized cancer therapy using GHRH-R antagonists,” Dr. Schally and his coauthors wrote.
The study was supported by the Li Ka Shing Foundation, Chinese foundation, and government grants to individual researchers, as well as support from the the Medical Research Service of the U.S. Department of Veterans Affairs, South Florida Veterans Affairs Foundation for Research and Education, and the University of Miami.
FROM PNAS
Key clinical point:
Major finding: The experimental GHRH-R antagonist MAI-602 inhibited gastric cancer growth in cell lines and human tumor xenograft models.
Data source: Proof of concept experiments showing the relationship between GHRH-R and gastric cancer, and elucidation of a method for targeting GHRH-R with an investigational peptide compound.
Disclosures: The study was supported by the Li Ka Shing Foundation, Chinese foundation, and government grants to individual researchers, as well as support from the the Medical Research Service of the U.S. Department of Veterans Affairs, South Florida Veterans Affairs Foundation for Research and Education, and the University of Miami.