User login
Introducing our new Editorial Board Members


Kidney Disease Progression: How to Calculate Risk
Q)When I diagnose patients with minor kidney disease, they often ask if they will require dialysis. I know it is unlikely, but I wish I could give them a better answer. Can you help me?
The diagnosis of chronic kidney disease (CKD) is understandably concerning for many patients. Being able to estimate CKD progression helps patients gain a better understanding of their condition while allowing clinicians to develop more personalized care plans. Tangri and colleagues developed a model that can be used to predict risk for kidney failure requiring dialysis or transplantation in patients with stage III to V CKD. This model has been validated in multiple diverse populations in North America and worldwide.1
The Kidney Failure Risk Equation (found at www.kidneyfailurerisk.com) uses four variables—age, gender, glomerular filtration rate (GFR), and urine albumin-to-creatinine ratio (ACR)—to assess two- and five-year risk for kidney failure.1,2 For example
- A 63-year-old woman with a GFR of 45 mL/min and an ACR of 30 mg/g has a 0.4% two-year risk and a 1.3% five-year risk for progression to kidney failure requiring dialysis or transplant.1
- Alternatively, a 55-year-old man with a GFR of 38 mL/min and an ACR of 150 mg/g has a 2.9% two-year risk and a 9% five-year risk for progression to end-stage renal disease (ESRD).1
Per proposed thresholds, patients with a score < 5% would be deemed “low risk”; with scores of 5% to 15%, “intermediate risk”; and with scores > 15%, “high risk.”1,2
The Kidney Failure Risk Equation can be incorporated into clinic visits to provide context for lab results. For patients with low risk for progression, optimal care and lifestyle measures can be reinforced. For those with intermediate or high risk, more intensive treatments and appropriate referrals can be initiated. (The National Kidney Foundation advises referral when a patient’s estimated GFR is 20 mL/min or the urine ACR is ≥ 300 mg/g.3) Providing a numeric risk for progression can help alleviate the patient’s uncertainty surrounding the diagnosis of CKD. —NDM
Nicole D. McCormick, MS, MBA, NP-C, CCTC
University of Colorado Renal Transplant Clinic, Aurora, Colorado
1. Tangri N, Grams ME, Levey AS, et al. Multinational assessment of accuracy of equations for predicting risk of kidney failure: a meta-analysis. JAMA. 2016;315(2):164-174.
2. Tangri N, Stevens LA, Griffith J, et al. A predictive model for progression of chronic kidney disease to kidney failure. JAMA. 2011;305(15):1553-1559.
3. National Kidney Foundation. Renal Replacement Therapy: What the PCP Needs to Know. www.kidney.org/sites/default/files/PCP%20in%20a%20Box%20-%20Module%203.pptx. Accessed December 5, 2016.
Clinician Reviews in partnership with

Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a semi-retired PA who works with the American Academy of Nephrology PAs and is a past chair of the NKF-CAP. This month’s responses were authored by Nicole D. McCormick, MS, MBA, NP-C, CCTC, who practices at the University of Colorado Renal Transplant Clinic in Aurora, Colorado, and Mary Rogers Sorey, MSN, who is an Assistant in Medicine in the Division of Nephrology and Hypertension at Vanderbilt University Medical Center, Nashville.
Clinician Reviews in partnership with
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a semi-retired PA who works with the American Academy of Nephrology PAs and is a past chair of the NKF-CAP. This month’s responses were authored by Nicole D. McCormick, MS, MBA, NP-C, CCTC, who practices at the University of Colorado Renal Transplant Clinic in Aurora, Colorado, and Mary Rogers Sorey, MSN, who is an Assistant in Medicine in the Division of Nephrology and Hypertension at Vanderbilt University Medical Center, Nashville.
Clinician Reviews in partnership with
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a semi-retired PA who works with the American Academy of Nephrology PAs and is a past chair of the NKF-CAP. This month’s responses were authored by Nicole D. McCormick, MS, MBA, NP-C, CCTC, who practices at the University of Colorado Renal Transplant Clinic in Aurora, Colorado, and Mary Rogers Sorey, MSN, who is an Assistant in Medicine in the Division of Nephrology and Hypertension at Vanderbilt University Medical Center, Nashville.
Q)When I diagnose patients with minor kidney disease, they often ask if they will require dialysis. I know it is unlikely, but I wish I could give them a better answer. Can you help me?
The diagnosis of chronic kidney disease (CKD) is understandably concerning for many patients. Being able to estimate CKD progression helps patients gain a better understanding of their condition while allowing clinicians to develop more personalized care plans. Tangri and colleagues developed a model that can be used to predict risk for kidney failure requiring dialysis or transplantation in patients with stage III to V CKD. This model has been validated in multiple diverse populations in North America and worldwide.1
The Kidney Failure Risk Equation (found at www.kidneyfailurerisk.com) uses four variables—age, gender, glomerular filtration rate (GFR), and urine albumin-to-creatinine ratio (ACR)—to assess two- and five-year risk for kidney failure.1,2 For example
- A 63-year-old woman with a GFR of 45 mL/min and an ACR of 30 mg/g has a 0.4% two-year risk and a 1.3% five-year risk for progression to kidney failure requiring dialysis or transplant.1
- Alternatively, a 55-year-old man with a GFR of 38 mL/min and an ACR of 150 mg/g has a 2.9% two-year risk and a 9% five-year risk for progression to end-stage renal disease (ESRD).1
Per proposed thresholds, patients with a score < 5% would be deemed “low risk”; with scores of 5% to 15%, “intermediate risk”; and with scores > 15%, “high risk.”1,2
The Kidney Failure Risk Equation can be incorporated into clinic visits to provide context for lab results. For patients with low risk for progression, optimal care and lifestyle measures can be reinforced. For those with intermediate or high risk, more intensive treatments and appropriate referrals can be initiated. (The National Kidney Foundation advises referral when a patient’s estimated GFR is 20 mL/min or the urine ACR is ≥ 300 mg/g.3) Providing a numeric risk for progression can help alleviate the patient’s uncertainty surrounding the diagnosis of CKD. —NDM
Nicole D. McCormick, MS, MBA, NP-C, CCTC
University of Colorado Renal Transplant Clinic, Aurora, Colorado
Q)When I diagnose patients with minor kidney disease, they often ask if they will require dialysis. I know it is unlikely, but I wish I could give them a better answer. Can you help me?
The diagnosis of chronic kidney disease (CKD) is understandably concerning for many patients. Being able to estimate CKD progression helps patients gain a better understanding of their condition while allowing clinicians to develop more personalized care plans. Tangri and colleagues developed a model that can be used to predict risk for kidney failure requiring dialysis or transplantation in patients with stage III to V CKD. This model has been validated in multiple diverse populations in North America and worldwide.1
The Kidney Failure Risk Equation (found at www.kidneyfailurerisk.com) uses four variables—age, gender, glomerular filtration rate (GFR), and urine albumin-to-creatinine ratio (ACR)—to assess two- and five-year risk for kidney failure.1,2 For example
- A 63-year-old woman with a GFR of 45 mL/min and an ACR of 30 mg/g has a 0.4% two-year risk and a 1.3% five-year risk for progression to kidney failure requiring dialysis or transplant.1
- Alternatively, a 55-year-old man with a GFR of 38 mL/min and an ACR of 150 mg/g has a 2.9% two-year risk and a 9% five-year risk for progression to end-stage renal disease (ESRD).1
Per proposed thresholds, patients with a score < 5% would be deemed “low risk”; with scores of 5% to 15%, “intermediate risk”; and with scores > 15%, “high risk.”1,2
The Kidney Failure Risk Equation can be incorporated into clinic visits to provide context for lab results. For patients with low risk for progression, optimal care and lifestyle measures can be reinforced. For those with intermediate or high risk, more intensive treatments and appropriate referrals can be initiated. (The National Kidney Foundation advises referral when a patient’s estimated GFR is 20 mL/min or the urine ACR is ≥ 300 mg/g.3) Providing a numeric risk for progression can help alleviate the patient’s uncertainty surrounding the diagnosis of CKD. —NDM
Nicole D. McCormick, MS, MBA, NP-C, CCTC
University of Colorado Renal Transplant Clinic, Aurora, Colorado
1. Tangri N, Grams ME, Levey AS, et al. Multinational assessment of accuracy of equations for predicting risk of kidney failure: a meta-analysis. JAMA. 2016;315(2):164-174.
2. Tangri N, Stevens LA, Griffith J, et al. A predictive model for progression of chronic kidney disease to kidney failure. JAMA. 2011;305(15):1553-1559.
3. National Kidney Foundation. Renal Replacement Therapy: What the PCP Needs to Know. www.kidney.org/sites/default/files/PCP%20in%20a%20Box%20-%20Module%203.pptx. Accessed December 5, 2016.
1. Tangri N, Grams ME, Levey AS, et al. Multinational assessment of accuracy of equations for predicting risk of kidney failure: a meta-analysis. JAMA. 2016;315(2):164-174.
2. Tangri N, Stevens LA, Griffith J, et al. A predictive model for progression of chronic kidney disease to kidney failure. JAMA. 2011;305(15):1553-1559.
3. National Kidney Foundation. Renal Replacement Therapy: What the PCP Needs to Know. www.kidney.org/sites/default/files/PCP%20in%20a%20Box%20-%20Module%203.pptx. Accessed December 5, 2016.
Pulmonary embolism in COPD exacerbations
Clinical question: How frequent is pulmonary embolism (PE) in patients with unexplained acute chronic obstructive pulmonary disease (COPD) exacerbation?
Study design: Systematic review.
Setting: U.S. hospitals and EDs.
Synopsis: PE prevalence was 16.1% (95% CI, 8.3%-25.8%) in patients with unexplained COPD exacerbations. Thirty-two percent were subsegmental, 35% affected one of the main pulmonary arteries, and 32% were located in the lobar and interlobar arteries. Heterogeneity between the included studies was high. In-hospital and 1-year mortality were increased in patients with PE and COPD exacerbations in one study but not in another.
Signs of cardiac failure, hypotension, and syncope were more frequently found in patients with COPD exacerbation and PE, compared with patients with COPD exacerbation without PE.
Bottom line: PE is a common occurrence in patients with unexplained COPD exacerbations; two-thirds of those emboli involved segmental circulation and therefore were clinically relevant.
Citation: Aleva FE, Voets LW, Simons SO, de Mast Q, van der Ven A, Heijdra YF. Prevalence and localization of pulmonary embolism in unexplained acute exacerbations of COPD: a systematic review and meta-analysis [published online ahead of print Aug. 11, 2016]. Chest. doi: 10.1016/j.chest.2016.07.034.
Clinical question: How frequent is pulmonary embolism (PE) in patients with unexplained acute chronic obstructive pulmonary disease (COPD) exacerbation?
Study design: Systematic review.
Setting: U.S. hospitals and EDs.
Synopsis: PE prevalence was 16.1% (95% CI, 8.3%-25.8%) in patients with unexplained COPD exacerbations. Thirty-two percent were subsegmental, 35% affected one of the main pulmonary arteries, and 32% were located in the lobar and interlobar arteries. Heterogeneity between the included studies was high. In-hospital and 1-year mortality were increased in patients with PE and COPD exacerbations in one study but not in another.
Signs of cardiac failure, hypotension, and syncope were more frequently found in patients with COPD exacerbation and PE, compared with patients with COPD exacerbation without PE.
Bottom line: PE is a common occurrence in patients with unexplained COPD exacerbations; two-thirds of those emboli involved segmental circulation and therefore were clinically relevant.
Citation: Aleva FE, Voets LW, Simons SO, de Mast Q, van der Ven A, Heijdra YF. Prevalence and localization of pulmonary embolism in unexplained acute exacerbations of COPD: a systematic review and meta-analysis [published online ahead of print Aug. 11, 2016]. Chest. doi: 10.1016/j.chest.2016.07.034.
Clinical question: How frequent is pulmonary embolism (PE) in patients with unexplained acute chronic obstructive pulmonary disease (COPD) exacerbation?
Study design: Systematic review.
Setting: U.S. hospitals and EDs.
Synopsis: PE prevalence was 16.1% (95% CI, 8.3%-25.8%) in patients with unexplained COPD exacerbations. Thirty-two percent were subsegmental, 35% affected one of the main pulmonary arteries, and 32% were located in the lobar and interlobar arteries. Heterogeneity between the included studies was high. In-hospital and 1-year mortality were increased in patients with PE and COPD exacerbations in one study but not in another.
Signs of cardiac failure, hypotension, and syncope were more frequently found in patients with COPD exacerbation and PE, compared with patients with COPD exacerbation without PE.
Bottom line: PE is a common occurrence in patients with unexplained COPD exacerbations; two-thirds of those emboli involved segmental circulation and therefore were clinically relevant.
Citation: Aleva FE, Voets LW, Simons SO, de Mast Q, van der Ven A, Heijdra YF. Prevalence and localization of pulmonary embolism in unexplained acute exacerbations of COPD: a systematic review and meta-analysis [published online ahead of print Aug. 11, 2016]. Chest. doi: 10.1016/j.chest.2016.07.034.
Have you Googled yourself lately?
The online rating business is proliferating in the medical industry. This should really come as no surprise as health care is a service industry and online ratings have long been a staple in most other service industries. It has become routine practice for most of us to search such online reviews when seeking a pair of shoes, a toaster, or a restaurant; we almost can’t help but scour these sites to help us make the best decision possible.

Not dissimilarly, patients these days seek care and make decisions by using a variety of inputs, including:
- Anticipated cost (is the physician or practice in or out of network?).
- Availability or access to the service (location of the practice and how long it will take to be seen).
- How good the services and care will be when they get there.
That same article found that for those who used online physician ratings, about one-third had selected a physician based on good ratings, and about one-third had avoided a physician based on poor ratings. So patients do seem to be paying attention to these sites and seeking or avoiding care based on what information they find.
Based on that evidence, it is not surprising that so many physician rating sites have sprung up; not only is there a market demand for the availability of this information, the rating sites are also profitable for the host companies. Vitals.com, for example, makes most of its revenue from advertisements and turns a sizable profit every year. Other profitable health care rating sites include Healthgrades, Yelp, Zocdoc, and WebMD.
When I Google my own name, for example, Vitals.com is the first ratings website that appears in the search results. The first pop-up asks you to rate me and then it takes you to a site with all sorts of facts about me (most of which are notably inaccurate). If I had any online ratings (which I do not currently), you would then see my star ratings and any comments.
The second rating site that comes up for me via Google search is PhysicianWiki.com.There is a whole host of information on me (most of which is accurate), along with a set of personal ratings, including my office, my staff, and my waiting times (which, of course, do not make any sense given I am a hospitalist!). It is unclear how those ratings were generated or what volume of responses they represent.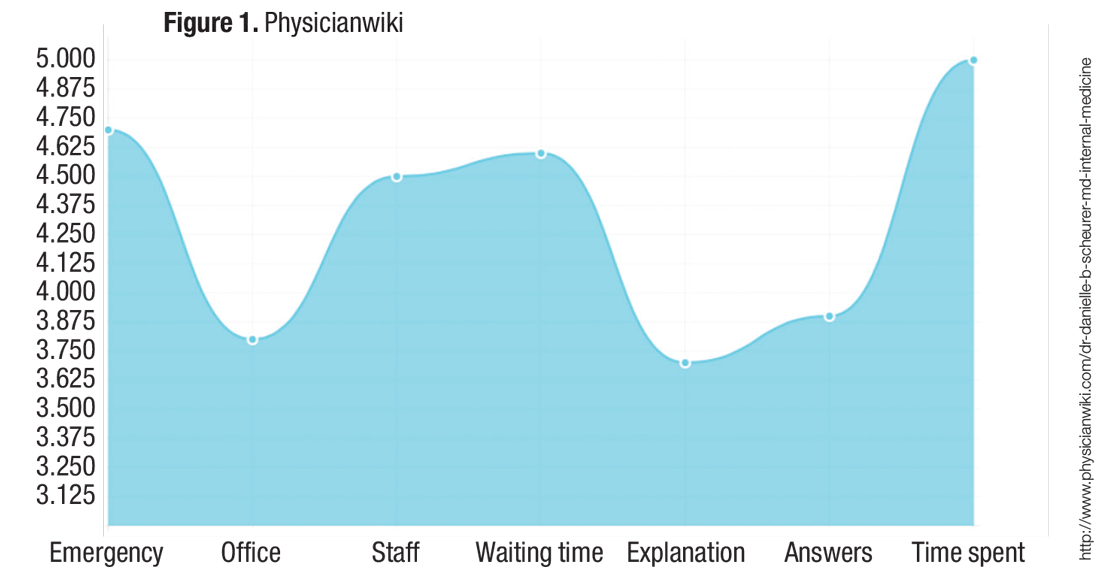
My health care system proposed rolling out a similar online rating system, and it was met with great skepticism from many physicians. There were two primary concerns:
- They felt it was “tacky” and that the profession of medicine should not be relegated to oversimplified service ratings. They worried that they would feel pressured to please the patient rather than “do the right thing” for the patient. For example, they would be less likely to give difficult advice (such as lose weight or stop smoking) or to resist prescribing medications that they deemed unnecessary or frankly dangerous (for example, antibiotics or narcotics).
Although these are valid concerns, it is hard to ignore the proliferation and traffic of these online websites. For you and your team, I would recommend taking a look at what is online about the members of your group and thinking about online strategies to take control of the conversation.
I don’t think the controversy over online physician ratings will wane anytime soon, but there is no doubt that they are profitable for companies and are therefore highly likely to continue to multiply.
References
1.Hanauer DA, Zheng K, Singer DC, Gebremariam A, Davis MM. Public awareness, perception, and use of online physician rating sites. JAMA. 2014;311(7):734-735. 2. A to Z provider listing: find a U of U Health Care physician by last name. University of Utah website. Available at http://healthcare.utah.edu/fad. Accessed Nov. 16, 2016.
Danielle Scheurer, MD, MSc, SFHM, is a hospitalist and chief quality officer at the Medical University of South Carolina in Charleston. She is physician editor of The Hospitalist. Email her at [email protected].
The online rating business is proliferating in the medical industry. This should really come as no surprise as health care is a service industry and online ratings have long been a staple in most other service industries. It has become routine practice for most of us to search such online reviews when seeking a pair of shoes, a toaster, or a restaurant; we almost can’t help but scour these sites to help us make the best decision possible.
Not dissimilarly, patients these days seek care and make decisions by using a variety of inputs, including:
- Anticipated cost (is the physician or practice in or out of network?).
- Availability or access to the service (location of the practice and how long it will take to be seen).
- How good the services and care will be when they get there.
That same article found that for those who used online physician ratings, about one-third had selected a physician based on good ratings, and about one-third had avoided a physician based on poor ratings. So patients do seem to be paying attention to these sites and seeking or avoiding care based on what information they find.
Based on that evidence, it is not surprising that so many physician rating sites have sprung up; not only is there a market demand for the availability of this information, the rating sites are also profitable for the host companies. Vitals.com, for example, makes most of its revenue from advertisements and turns a sizable profit every year. Other profitable health care rating sites include Healthgrades, Yelp, Zocdoc, and WebMD.
When I Google my own name, for example, Vitals.com is the first ratings website that appears in the search results. The first pop-up asks you to rate me and then it takes you to a site with all sorts of facts about me (most of which are notably inaccurate). If I had any online ratings (which I do not currently), you would then see my star ratings and any comments.
The second rating site that comes up for me via Google search is PhysicianWiki.com.There is a whole host of information on me (most of which is accurate), along with a set of personal ratings, including my office, my staff, and my waiting times (which, of course, do not make any sense given I am a hospitalist!). It is unclear how those ratings were generated or what volume of responses they represent.
My health care system proposed rolling out a similar online rating system, and it was met with great skepticism from many physicians. There were two primary concerns:
- They felt it was “tacky” and that the profession of medicine should not be relegated to oversimplified service ratings. They worried that they would feel pressured to please the patient rather than “do the right thing” for the patient. For example, they would be less likely to give difficult advice (such as lose weight or stop smoking) or to resist prescribing medications that they deemed unnecessary or frankly dangerous (for example, antibiotics or narcotics).
Although these are valid concerns, it is hard to ignore the proliferation and traffic of these online websites. For you and your team, I would recommend taking a look at what is online about the members of your group and thinking about online strategies to take control of the conversation.
I don’t think the controversy over online physician ratings will wane anytime soon, but there is no doubt that they are profitable for companies and are therefore highly likely to continue to multiply.
References
1.Hanauer DA, Zheng K, Singer DC, Gebremariam A, Davis MM. Public awareness, perception, and use of online physician rating sites. JAMA. 2014;311(7):734-735. 2. A to Z provider listing: find a U of U Health Care physician by last name. University of Utah website. Available at http://healthcare.utah.edu/fad. Accessed Nov. 16, 2016.
Danielle Scheurer, MD, MSc, SFHM, is a hospitalist and chief quality officer at the Medical University of South Carolina in Charleston. She is physician editor of The Hospitalist. Email her at [email protected].
The online rating business is proliferating in the medical industry. This should really come as no surprise as health care is a service industry and online ratings have long been a staple in most other service industries. It has become routine practice for most of us to search such online reviews when seeking a pair of shoes, a toaster, or a restaurant; we almost can’t help but scour these sites to help us make the best decision possible.
Not dissimilarly, patients these days seek care and make decisions by using a variety of inputs, including:
- Anticipated cost (is the physician or practice in or out of network?).
- Availability or access to the service (location of the practice and how long it will take to be seen).
- How good the services and care will be when they get there.
That same article found that for those who used online physician ratings, about one-third had selected a physician based on good ratings, and about one-third had avoided a physician based on poor ratings. So patients do seem to be paying attention to these sites and seeking or avoiding care based on what information they find.
Based on that evidence, it is not surprising that so many physician rating sites have sprung up; not only is there a market demand for the availability of this information, the rating sites are also profitable for the host companies. Vitals.com, for example, makes most of its revenue from advertisements and turns a sizable profit every year. Other profitable health care rating sites include Healthgrades, Yelp, Zocdoc, and WebMD.
When I Google my own name, for example, Vitals.com is the first ratings website that appears in the search results. The first pop-up asks you to rate me and then it takes you to a site with all sorts of facts about me (most of which are notably inaccurate). If I had any online ratings (which I do not currently), you would then see my star ratings and any comments.
The second rating site that comes up for me via Google search is PhysicianWiki.com.There is a whole host of information on me (most of which is accurate), along with a set of personal ratings, including my office, my staff, and my waiting times (which, of course, do not make any sense given I am a hospitalist!). It is unclear how those ratings were generated or what volume of responses they represent.
My health care system proposed rolling out a similar online rating system, and it was met with great skepticism from many physicians. There were two primary concerns:
- They felt it was “tacky” and that the profession of medicine should not be relegated to oversimplified service ratings. They worried that they would feel pressured to please the patient rather than “do the right thing” for the patient. For example, they would be less likely to give difficult advice (such as lose weight or stop smoking) or to resist prescribing medications that they deemed unnecessary or frankly dangerous (for example, antibiotics or narcotics).
Although these are valid concerns, it is hard to ignore the proliferation and traffic of these online websites. For you and your team, I would recommend taking a look at what is online about the members of your group and thinking about online strategies to take control of the conversation.
I don’t think the controversy over online physician ratings will wane anytime soon, but there is no doubt that they are profitable for companies and are therefore highly likely to continue to multiply.
References
1.Hanauer DA, Zheng K, Singer DC, Gebremariam A, Davis MM. Public awareness, perception, and use of online physician rating sites. JAMA. 2014;311(7):734-735. 2. A to Z provider listing: find a U of U Health Care physician by last name. University of Utah website. Available at http://healthcare.utah.edu/fad. Accessed Nov. 16, 2016.
Danielle Scheurer, MD, MSc, SFHM, is a hospitalist and chief quality officer at the Medical University of South Carolina in Charleston. She is physician editor of The Hospitalist. Email her at [email protected].
In Memoriam
CHEST has been informed of the following members’ deaths. We extend our sincere condolences.
Anthony Cosentino, MD, FCCP (January 2016)
Ben Branscomb, MD (July 2016)
Steven Sahn, MD, FCCP (Aug 2016)
Thomas Aldrich, MD (September 2016)
John C. Baldwin, MD, FCCP (September 2016)
David Cugell, MD, FCCP (December 2016)
CHEST has been informed of the following members’ deaths. We extend our sincere condolences.
Anthony Cosentino, MD, FCCP (January 2016)
Ben Branscomb, MD (July 2016)
Steven Sahn, MD, FCCP (Aug 2016)
Thomas Aldrich, MD (September 2016)
John C. Baldwin, MD, FCCP (September 2016)
David Cugell, MD, FCCP (December 2016)
CHEST has been informed of the following members’ deaths. We extend our sincere condolences.
Anthony Cosentino, MD, FCCP (January 2016)
Ben Branscomb, MD (July 2016)
Steven Sahn, MD, FCCP (Aug 2016)
Thomas Aldrich, MD (September 2016)
John C. Baldwin, MD, FCCP (September 2016)
David Cugell, MD, FCCP (December 2016)
Winners-All at CHEST 2016
We all know that, with the great success of CHEST 2016, everyone who shared that event is a winner. But, we would especially like to call out some of the special winners who were recognized during our annual meeting.
CHEST Awards
College Medalist Award
Lewis J. Rubin, MD, FCCP
Distinguished Service Award
Kim D. French, MHSA, CAPPM, FCCP
Alfred Soffer Award for Editorial Excellence
Seth J. Koenig, MD, FCCP
Master Clinician Educator Award
Jack D. Buckley, MD, MPH, FCCP
Distinguished Scientist Honor Lecture
Jay Nadel, MD
Edward C. Rosenow III, MD, Master FCCP/Master Teacher Honor Lecture
Suhail Raoof, MBBS, FCCP
Murray Kornfeld Memorial Founders Lecture
Michael Niederman, MD, FCCP Pasquale Ciaglia Memorial Lecture Kevin L. Kovitz, MD, FCCP
Roger C. Bone Memorial Lecture
Robert A. Berg, MD Thomas L. Petty, MD, Master FCCP Memorial Lecture Nicola A. Hanania, MD, MS, FCCP Margaret Pfrommer Memorial Lecture in Long-term Mechanical Ventilation Thomas G. Keens, MD Om P. Sharma, MD, Master FCCP Memorial Lecture Robert P. Baughman, MD, FCCP
Early Career Educator
Gabriel Bosslet, MD, FCCP
CHEST Challenge Championship 2016
1st Place
The University of Arizona
Huthayfa Ateeli, MBBS Naser Mahmoud, MD
Muna Omar, MD, MBBS
PD: James L. Knepler Jr.
2nd Place
New York Methodist Hospital
Anu R. Jacob, MD
Stephen D. Milan, MD
Jordan Taillon, MD
PD: Anthony G. Saleh, MD, FCCP
3rd Place
Interfaith Medical Center
Chidozie C. Agu, MD
Saroj P. Kandel, MBBS Divya Salhan, MD, MBBS
PD: Marie Frances J. Schmidt, MD, FCCP
CHEST Foundation Grant Winners
GlaxoSmithKline Distinguished Scholar in Respiratory Health
Don Hayes Jr., MD, FCCP The Research Institute at Nationwide Children’s Hospital
Implications of the Lung Allocation Score in Prioritizing Critically Ill Patients for Lung Transplantation
Supported by GlaxoSmithKline.
2016 Research Grantees
Alice Turner, MBChB, MRCP, PhD
University of Birmingham, United Kingdom
CHEST Foundation and the Alpha-1 Foundation Research Grant in Alpha-1 Antitrypsin Deficiency
Improving Access to Augmentation: A Propensity-Matching Study Between the UK AATD Registry and AlphaNet
This grant is jointly supported by the CHEST Foundation and the Alpha-1 Foundation.
Robert Busch, MD
Brigham and Women’s Hospital, Channing Division of Network Medicine
CHEST Foundation Research Grant in Chronic Obstructive Pulmonary Disease
Methylation Quantitative Trait Loci: Markers of Race-Specific Disparities in African Americans With COPD
This grant is supported by AstraZeneca.
Clemens Grassberger, PhD
Massachusetts General Hospital – Harvard University
CHEST Foundation Research Grant in Lung Cancer
Dynamic FLT-PET as Biomarker for Early Response in Locally Advanced Lung Cancer Patients
This grant is supported by Genentech Inc.
Cristina Russo, MD, PhD
Bambino Gesù Children’s Hospital, Rome, Italy
CHEST Foundation Research Grant in Nontuberculous Mycobacteria
A Proteomic-Metaproteomic Analysis Approach Allows Identification of Drug Target Candidates for the Future Design of Preventive, Diagnostic, and Therapeutic Strategies Against Nontuberculous Mycobacteria Diseases
This grant is supported by Insmed.
Peter Leary, MD, MS
University of Washington
CHEST Foundation Research Grant in Pulmonary Arterial Hypertension
Expression Profiling in Pulmonary Arterial Hypertension
This grant is supported by Actelion Pharmaceuticals, US, Inc.
Brett Ley, MD
University of California, San Francisco
CHEST Foundation Research Grant in Pulmonary Fibrosis
Extracellular Circulation RNAs as Predictors of Disease Progression in Idiopathic Pulmonary Fibrosis
This grant is supported by Boehringer Ingelheim Pharmaceuticals & Genentech Inc.
Sydney Montesi, MD
Massachusetts General Hospital
CHEST Foundation Research Grant in Pulmonary Fibrosis
Gadofosveset-Enhanced Lung MRI to Detect Idiopathic Pulmonary Fibrosis Disease Activity
This grant is supported by Boehringer Ingelheim Pharmaceuticals & Genentech Inc.
Farbod Rahaghi, MD, PhD
Brigham and Women’s Hospital
CHEST Foundation Research Grant in Venous Thromboembolism
CT Scan-Based Markers for Prediction of Outcomes in Acute Pulmonary Embolism
This grant is supported by Daiichi Sankyo.
Catherine Oberg, MD
Icahn School of Medicine at Mount Sinai
CHEST Foundation Research Grant in Women’s Lung Health
Effects of Household Air Pollution on Airway Inflammation, Lung Function, and Respiratory Symptoms
This grant is supported in full by the CHEST Foundation.
2016 Community Service Grantee
Ethel Jane Carter, MD, FCCP
Warren Alpert School of Medicine at Brown University
CHEST Foundation Community Service Grant Honoring D. Robert McCaffree, MD, Master FCCP
East African Training Initiative ( EATI) in Pulmonary Medicine
2016 NetWorks Challenge Travel Grantees
Debarsee Banerjee, MS, MD
Women’s Health NetWork
Drew Harris, MD
Occupational and Environmental Health NetWork
Kerry Hena, MD
Occupational and Environmental Health NetWork
Amanpreet Kaur, MD
Women’s Health NetWork
2016 Diversity Travel Grant Winners
John B. Bishara, DO
Renato F. Blanco Jr., MD
Angel Coz-Yataco, MD Sherie A. Gause, MD
Anthony Nebor, MD
James T. Williams, MD
Alfred Soffer Research Award Winners
Kerry Hena, MD
Deepak Pradhan, MD, FCCP
Young Investigator Award Winners
Elizabeth Becker: Clinical Characteristics of Sarcoidosis in World Trade Center (WTC) Exposed Fire Department of the City of New York (FDNY) Firefighters
Daniel Altman, MD : Cost-Effectiveness of Universally Funding Smoking Cessation Pharmacotherapy
Top 3 Poster Winners
Epaminondas Kosmas, MD, PhD, FCCP : Bronchiectasis in Patients With COPD: An Irrelevant Imaging Finding or a Clinically Important Phenotype?
Mark Regala, MD, BS : Evaluation of Outcomes of Post-Extubation Dysphagia in Elderly Patients
Massa Zantah, MD : Correlation of Esophageal Dilatation and Pulmonary Fibrosis in Scleroderma
Runner-up: Alev Gurgun, MD : Pulmonary Rehabilitation Response in Elderly and Younger Patients With COPD
Case Report Slide Winners
John Egan, MD, BA : An Unusual Cause of Tracheal Stenosis Due to a Vascular Anomaly Successfully Managed With Silicone Airway Stenting Prior to Definitive Vascular Repair
Harprett Grewal, MD : Bladder PTLD: First Reported Case of Post-Transplant Lymphoproliferative Disorder (PTLD) in the Bladder in a Lung Transplant Recipient
Michael Fingerhood, MD, MPH : Pulmonary Overlap Histiocytosis: A Rare Case of Interstitial Lung Disease Due to Erdheim Chester Disease in a Patient With Langerhans Cell Histiocytosis and Myelodisplastic Syndrome
Yihenew Negatu, MD : Acute ST Elevation Myocardial Infarction Related to Carbon Monoxide Poisoning in a Young Patient Without Coronary Artery Disease
Stephanie Wappel, MD : False-Negative Pet Imaging in Early Stage Malignant Pleural Mesothelioma
Lina Miyakawa, MD : Restrictive EGFR Mutation
Jeffrey Bonenfant, DO : A Unique Case of Follicular Bronchiolitis
Melissa Myers, MD : Seeing the Forest and Not Just the Trees: A Case of Recurrent Fever, Cough, and Respiratory Failure
Carly Fabrizio, DO : An Unusual Case of Submassive Hemoptysis
Meilinh Thi, DO : A Case to Make Your Skin Crawl
Garrett Harp, MD : Lambertosis: A Lung Cancer Mimic
Malik Khan, MD : Pleural Epithelioid Hemangioendothelioma: A Case Report
Priya Patel, MD : A Troubling Trifecta: Pulmonary Alveolar Proteinosis and Pneumocystis Pneumonia in Acute Myeloid Leukemia
Atul Palkar, MD : SGLT2 Inhibitors: Mind the Gap
Ji Yeon Lee, MD : Making Unusual Connections: Fibrosing Mediastinitis Leading to Bronchoesophageal Fistula
Sailm Daouk, MD : A Rare Form of Invasive Aspergillus Infection in a Severely Immunocompromised Host
Venkata Ravi Kumar Angirekula, MD : Vanishing Lung
Stephen Milan, MD : An Unexpected Mass
Lelia Logue, MD : A Rare Cause of Dysphagia
Daniel Hershberger, MD : Rapidly Progressive Hypoxic Respiratory Failure After a Rash: A Case of Clinically Amyopathic Dermatomyositis (CADM)-Associated ILD
Fellow Case Report Poster Winners
Krishna Siva Sai Kakkera
An Unusual Case of Crypotococcal Pleural Effusion
George Cheng
Use of Laparoscopic Suction Irrigator With Rigid Pleuroscope in Medical Thoracoscopy
Matt Koroscil
Wong Type Dermatomyositis Complicated by Interstitial Lung Disease
Derek Hansen
Acute Fibrinous and Organizing Pneumonia Following Hematopoietic Stem Cell Transplantation Responsive to Corticosteroid Therapy
Ala Eddin Sagar
Pulmonary Embolism Caused by Thrombin-Based Hemostatic Matrix After Discectomy
Sandeep Chennadi
Systemic Lupus Erythematosus (SLE) With Refractory Bilateral Chylothorax and Chylous Ascites
Medical Student/Resident Case Report Poster Winners
Justin Fiala
Pulmonary Presentation Without Concurrent Bone Involvement in Erdheim-Chester Disease: A Report of Two Cases
Navitha Ramesh
A Fatal Migration: A Case of Intra-Cardiac Embolization of a Peripheral Stent
Humna Abid Memon
Use of Extracorporeal Membrane Oxygenation in Postpartum Management of a Patient With PAH
Vanessa Ohleyer
A Case of Unusual Anatomy for an Uncommon Mediastinal Tumor
Tanushree Gahlot
Three Unusual Presentations of Job’s Syndrome (Hyper Immunoglobulin E Syndrome)
NetWorks Challenge Winners
Round 1
Women’s Lung Health NetWork
Round 2
Practice and Operations NetWork-1st place
Home-Based Mechanical Ventilation and Neuromuscular Disease NetWorks – 2nd place
Round 3
Home-Based Mechanical Ventilation, Neuromuscular Disease, and the Women’s Lung Health NetWorks
CHEST Bingo Winners
Youseff Anid, MD, FCCP
Karen Cochran, ACNP
Molly Howsware, DO
Katie Jeans, MD
Genovena Medina, RN
Gregory Eisinger, MD
Saurabh Mittal, MD, MBBS
Navitha Ramesh, MD
Dalvinder Dhillon, MD
Teresita Saylor, MD, FCCP
Carl Kaplan, MD, FCCP
Vishal Patel, MBBS, FCCP
Erin Peterson, CNP
Lilian Pereira, DO
We all know that, with the great success of CHEST 2016, everyone who shared that event is a winner. But, we would especially like to call out some of the special winners who were recognized during our annual meeting.
CHEST Awards
College Medalist Award
Lewis J. Rubin, MD, FCCP
Distinguished Service Award
Kim D. French, MHSA, CAPPM, FCCP
Alfred Soffer Award for Editorial Excellence
Seth J. Koenig, MD, FCCP
Master Clinician Educator Award
Jack D. Buckley, MD, MPH, FCCP
Distinguished Scientist Honor Lecture
Jay Nadel, MD
Edward C. Rosenow III, MD, Master FCCP/Master Teacher Honor Lecture
Suhail Raoof, MBBS, FCCP
Murray Kornfeld Memorial Founders Lecture
Michael Niederman, MD, FCCP Pasquale Ciaglia Memorial Lecture Kevin L. Kovitz, MD, FCCP
Roger C. Bone Memorial Lecture
Robert A. Berg, MD Thomas L. Petty, MD, Master FCCP Memorial Lecture Nicola A. Hanania, MD, MS, FCCP Margaret Pfrommer Memorial Lecture in Long-term Mechanical Ventilation Thomas G. Keens, MD Om P. Sharma, MD, Master FCCP Memorial Lecture Robert P. Baughman, MD, FCCP
Early Career Educator
Gabriel Bosslet, MD, FCCP
CHEST Challenge Championship 2016
1st Place
The University of Arizona
Huthayfa Ateeli, MBBS Naser Mahmoud, MD
Muna Omar, MD, MBBS
PD: James L. Knepler Jr.
2nd Place
New York Methodist Hospital
Anu R. Jacob, MD
Stephen D. Milan, MD
Jordan Taillon, MD
PD: Anthony G. Saleh, MD, FCCP
3rd Place
Interfaith Medical Center
Chidozie C. Agu, MD
Saroj P. Kandel, MBBS Divya Salhan, MD, MBBS
PD: Marie Frances J. Schmidt, MD, FCCP
CHEST Foundation Grant Winners
GlaxoSmithKline Distinguished Scholar in Respiratory Health
Don Hayes Jr., MD, FCCP The Research Institute at Nationwide Children’s Hospital
Implications of the Lung Allocation Score in Prioritizing Critically Ill Patients for Lung Transplantation
Supported by GlaxoSmithKline.
2016 Research Grantees
Alice Turner, MBChB, MRCP, PhD
University of Birmingham, United Kingdom
CHEST Foundation and the Alpha-1 Foundation Research Grant in Alpha-1 Antitrypsin Deficiency
Improving Access to Augmentation: A Propensity-Matching Study Between the UK AATD Registry and AlphaNet
This grant is jointly supported by the CHEST Foundation and the Alpha-1 Foundation.
Robert Busch, MD
Brigham and Women’s Hospital, Channing Division of Network Medicine
CHEST Foundation Research Grant in Chronic Obstructive Pulmonary Disease
Methylation Quantitative Trait Loci: Markers of Race-Specific Disparities in African Americans With COPD
This grant is supported by AstraZeneca.
Clemens Grassberger, PhD
Massachusetts General Hospital – Harvard University
CHEST Foundation Research Grant in Lung Cancer
Dynamic FLT-PET as Biomarker for Early Response in Locally Advanced Lung Cancer Patients
This grant is supported by Genentech Inc.
Cristina Russo, MD, PhD
Bambino Gesù Children’s Hospital, Rome, Italy
CHEST Foundation Research Grant in Nontuberculous Mycobacteria
A Proteomic-Metaproteomic Analysis Approach Allows Identification of Drug Target Candidates for the Future Design of Preventive, Diagnostic, and Therapeutic Strategies Against Nontuberculous Mycobacteria Diseases
This grant is supported by Insmed.
Peter Leary, MD, MS
University of Washington
CHEST Foundation Research Grant in Pulmonary Arterial Hypertension
Expression Profiling in Pulmonary Arterial Hypertension
This grant is supported by Actelion Pharmaceuticals, US, Inc.
Brett Ley, MD
University of California, San Francisco
CHEST Foundation Research Grant in Pulmonary Fibrosis
Extracellular Circulation RNAs as Predictors of Disease Progression in Idiopathic Pulmonary Fibrosis
This grant is supported by Boehringer Ingelheim Pharmaceuticals & Genentech Inc.
Sydney Montesi, MD
Massachusetts General Hospital
CHEST Foundation Research Grant in Pulmonary Fibrosis
Gadofosveset-Enhanced Lung MRI to Detect Idiopathic Pulmonary Fibrosis Disease Activity
This grant is supported by Boehringer Ingelheim Pharmaceuticals & Genentech Inc.
Farbod Rahaghi, MD, PhD
Brigham and Women’s Hospital
CHEST Foundation Research Grant in Venous Thromboembolism
CT Scan-Based Markers for Prediction of Outcomes in Acute Pulmonary Embolism
This grant is supported by Daiichi Sankyo.
Catherine Oberg, MD
Icahn School of Medicine at Mount Sinai
CHEST Foundation Research Grant in Women’s Lung Health
Effects of Household Air Pollution on Airway Inflammation, Lung Function, and Respiratory Symptoms
This grant is supported in full by the CHEST Foundation.
2016 Community Service Grantee
Ethel Jane Carter, MD, FCCP
Warren Alpert School of Medicine at Brown University
CHEST Foundation Community Service Grant Honoring D. Robert McCaffree, MD, Master FCCP
East African Training Initiative ( EATI) in Pulmonary Medicine
2016 NetWorks Challenge Travel Grantees
Debarsee Banerjee, MS, MD
Women’s Health NetWork
Drew Harris, MD
Occupational and Environmental Health NetWork
Kerry Hena, MD
Occupational and Environmental Health NetWork
Amanpreet Kaur, MD
Women’s Health NetWork
2016 Diversity Travel Grant Winners
John B. Bishara, DO
Renato F. Blanco Jr., MD
Angel Coz-Yataco, MD Sherie A. Gause, MD
Anthony Nebor, MD
James T. Williams, MD
Alfred Soffer Research Award Winners
Kerry Hena, MD
Deepak Pradhan, MD, FCCP
Young Investigator Award Winners
Elizabeth Becker: Clinical Characteristics of Sarcoidosis in World Trade Center (WTC) Exposed Fire Department of the City of New York (FDNY) Firefighters
Daniel Altman, MD : Cost-Effectiveness of Universally Funding Smoking Cessation Pharmacotherapy
Top 3 Poster Winners
Epaminondas Kosmas, MD, PhD, FCCP : Bronchiectasis in Patients With COPD: An Irrelevant Imaging Finding or a Clinically Important Phenotype?
Mark Regala, MD, BS : Evaluation of Outcomes of Post-Extubation Dysphagia in Elderly Patients
Massa Zantah, MD : Correlation of Esophageal Dilatation and Pulmonary Fibrosis in Scleroderma
Runner-up: Alev Gurgun, MD : Pulmonary Rehabilitation Response in Elderly and Younger Patients With COPD
Case Report Slide Winners
John Egan, MD, BA : An Unusual Cause of Tracheal Stenosis Due to a Vascular Anomaly Successfully Managed With Silicone Airway Stenting Prior to Definitive Vascular Repair
Harprett Grewal, MD : Bladder PTLD: First Reported Case of Post-Transplant Lymphoproliferative Disorder (PTLD) in the Bladder in a Lung Transplant Recipient
Michael Fingerhood, MD, MPH : Pulmonary Overlap Histiocytosis: A Rare Case of Interstitial Lung Disease Due to Erdheim Chester Disease in a Patient With Langerhans Cell Histiocytosis and Myelodisplastic Syndrome
Yihenew Negatu, MD : Acute ST Elevation Myocardial Infarction Related to Carbon Monoxide Poisoning in a Young Patient Without Coronary Artery Disease
Stephanie Wappel, MD : False-Negative Pet Imaging in Early Stage Malignant Pleural Mesothelioma
Lina Miyakawa, MD : Restrictive EGFR Mutation
Jeffrey Bonenfant, DO : A Unique Case of Follicular Bronchiolitis
Melissa Myers, MD : Seeing the Forest and Not Just the Trees: A Case of Recurrent Fever, Cough, and Respiratory Failure
Carly Fabrizio, DO : An Unusual Case of Submassive Hemoptysis
Meilinh Thi, DO : A Case to Make Your Skin Crawl
Garrett Harp, MD : Lambertosis: A Lung Cancer Mimic
Malik Khan, MD : Pleural Epithelioid Hemangioendothelioma: A Case Report
Priya Patel, MD : A Troubling Trifecta: Pulmonary Alveolar Proteinosis and Pneumocystis Pneumonia in Acute Myeloid Leukemia
Atul Palkar, MD : SGLT2 Inhibitors: Mind the Gap
Ji Yeon Lee, MD : Making Unusual Connections: Fibrosing Mediastinitis Leading to Bronchoesophageal Fistula
Sailm Daouk, MD : A Rare Form of Invasive Aspergillus Infection in a Severely Immunocompromised Host
Venkata Ravi Kumar Angirekula, MD : Vanishing Lung
Stephen Milan, MD : An Unexpected Mass
Lelia Logue, MD : A Rare Cause of Dysphagia
Daniel Hershberger, MD : Rapidly Progressive Hypoxic Respiratory Failure After a Rash: A Case of Clinically Amyopathic Dermatomyositis (CADM)-Associated ILD
Fellow Case Report Poster Winners
Krishna Siva Sai Kakkera
An Unusual Case of Crypotococcal Pleural Effusion
George Cheng
Use of Laparoscopic Suction Irrigator With Rigid Pleuroscope in Medical Thoracoscopy
Matt Koroscil
Wong Type Dermatomyositis Complicated by Interstitial Lung Disease
Derek Hansen
Acute Fibrinous and Organizing Pneumonia Following Hematopoietic Stem Cell Transplantation Responsive to Corticosteroid Therapy
Ala Eddin Sagar
Pulmonary Embolism Caused by Thrombin-Based Hemostatic Matrix After Discectomy
Sandeep Chennadi
Systemic Lupus Erythematosus (SLE) With Refractory Bilateral Chylothorax and Chylous Ascites
Medical Student/Resident Case Report Poster Winners
Justin Fiala
Pulmonary Presentation Without Concurrent Bone Involvement in Erdheim-Chester Disease: A Report of Two Cases
Navitha Ramesh
A Fatal Migration: A Case of Intra-Cardiac Embolization of a Peripheral Stent
Humna Abid Memon
Use of Extracorporeal Membrane Oxygenation in Postpartum Management of a Patient With PAH
Vanessa Ohleyer
A Case of Unusual Anatomy for an Uncommon Mediastinal Tumor
Tanushree Gahlot
Three Unusual Presentations of Job’s Syndrome (Hyper Immunoglobulin E Syndrome)
NetWorks Challenge Winners
Round 1
Women’s Lung Health NetWork
Round 2
Practice and Operations NetWork-1st place
Home-Based Mechanical Ventilation and Neuromuscular Disease NetWorks – 2nd place
Round 3
Home-Based Mechanical Ventilation, Neuromuscular Disease, and the Women’s Lung Health NetWorks
CHEST Bingo Winners
Youseff Anid, MD, FCCP
Karen Cochran, ACNP
Molly Howsware, DO
Katie Jeans, MD
Genovena Medina, RN
Gregory Eisinger, MD
Saurabh Mittal, MD, MBBS
Navitha Ramesh, MD
Dalvinder Dhillon, MD
Teresita Saylor, MD, FCCP
Carl Kaplan, MD, FCCP
Vishal Patel, MBBS, FCCP
Erin Peterson, CNP
Lilian Pereira, DO
We all know that, with the great success of CHEST 2016, everyone who shared that event is a winner. But, we would especially like to call out some of the special winners who were recognized during our annual meeting.
CHEST Awards
College Medalist Award
Lewis J. Rubin, MD, FCCP
Distinguished Service Award
Kim D. French, MHSA, CAPPM, FCCP
Alfred Soffer Award for Editorial Excellence
Seth J. Koenig, MD, FCCP
Master Clinician Educator Award
Jack D. Buckley, MD, MPH, FCCP
Distinguished Scientist Honor Lecture
Jay Nadel, MD
Edward C. Rosenow III, MD, Master FCCP/Master Teacher Honor Lecture
Suhail Raoof, MBBS, FCCP
Murray Kornfeld Memorial Founders Lecture
Michael Niederman, MD, FCCP Pasquale Ciaglia Memorial Lecture Kevin L. Kovitz, MD, FCCP
Roger C. Bone Memorial Lecture
Robert A. Berg, MD Thomas L. Petty, MD, Master FCCP Memorial Lecture Nicola A. Hanania, MD, MS, FCCP Margaret Pfrommer Memorial Lecture in Long-term Mechanical Ventilation Thomas G. Keens, MD Om P. Sharma, MD, Master FCCP Memorial Lecture Robert P. Baughman, MD, FCCP
Early Career Educator
Gabriel Bosslet, MD, FCCP
CHEST Challenge Championship 2016
1st Place
The University of Arizona
Huthayfa Ateeli, MBBS Naser Mahmoud, MD
Muna Omar, MD, MBBS
PD: James L. Knepler Jr.
2nd Place
New York Methodist Hospital
Anu R. Jacob, MD
Stephen D. Milan, MD
Jordan Taillon, MD
PD: Anthony G. Saleh, MD, FCCP
3rd Place
Interfaith Medical Center
Chidozie C. Agu, MD
Saroj P. Kandel, MBBS Divya Salhan, MD, MBBS
PD: Marie Frances J. Schmidt, MD, FCCP
CHEST Foundation Grant Winners
GlaxoSmithKline Distinguished Scholar in Respiratory Health
Don Hayes Jr., MD, FCCP The Research Institute at Nationwide Children’s Hospital
Implications of the Lung Allocation Score in Prioritizing Critically Ill Patients for Lung Transplantation
Supported by GlaxoSmithKline.
2016 Research Grantees
Alice Turner, MBChB, MRCP, PhD
University of Birmingham, United Kingdom
CHEST Foundation and the Alpha-1 Foundation Research Grant in Alpha-1 Antitrypsin Deficiency
Improving Access to Augmentation: A Propensity-Matching Study Between the UK AATD Registry and AlphaNet
This grant is jointly supported by the CHEST Foundation and the Alpha-1 Foundation.
Robert Busch, MD
Brigham and Women’s Hospital, Channing Division of Network Medicine
CHEST Foundation Research Grant in Chronic Obstructive Pulmonary Disease
Methylation Quantitative Trait Loci: Markers of Race-Specific Disparities in African Americans With COPD
This grant is supported by AstraZeneca.
Clemens Grassberger, PhD
Massachusetts General Hospital – Harvard University
CHEST Foundation Research Grant in Lung Cancer
Dynamic FLT-PET as Biomarker for Early Response in Locally Advanced Lung Cancer Patients
This grant is supported by Genentech Inc.
Cristina Russo, MD, PhD
Bambino Gesù Children’s Hospital, Rome, Italy
CHEST Foundation Research Grant in Nontuberculous Mycobacteria
A Proteomic-Metaproteomic Analysis Approach Allows Identification of Drug Target Candidates for the Future Design of Preventive, Diagnostic, and Therapeutic Strategies Against Nontuberculous Mycobacteria Diseases
This grant is supported by Insmed.
Peter Leary, MD, MS
University of Washington
CHEST Foundation Research Grant in Pulmonary Arterial Hypertension
Expression Profiling in Pulmonary Arterial Hypertension
This grant is supported by Actelion Pharmaceuticals, US, Inc.
Brett Ley, MD
University of California, San Francisco
CHEST Foundation Research Grant in Pulmonary Fibrosis
Extracellular Circulation RNAs as Predictors of Disease Progression in Idiopathic Pulmonary Fibrosis
This grant is supported by Boehringer Ingelheim Pharmaceuticals & Genentech Inc.
Sydney Montesi, MD
Massachusetts General Hospital
CHEST Foundation Research Grant in Pulmonary Fibrosis
Gadofosveset-Enhanced Lung MRI to Detect Idiopathic Pulmonary Fibrosis Disease Activity
This grant is supported by Boehringer Ingelheim Pharmaceuticals & Genentech Inc.
Farbod Rahaghi, MD, PhD
Brigham and Women’s Hospital
CHEST Foundation Research Grant in Venous Thromboembolism
CT Scan-Based Markers for Prediction of Outcomes in Acute Pulmonary Embolism
This grant is supported by Daiichi Sankyo.
Catherine Oberg, MD
Icahn School of Medicine at Mount Sinai
CHEST Foundation Research Grant in Women’s Lung Health
Effects of Household Air Pollution on Airway Inflammation, Lung Function, and Respiratory Symptoms
This grant is supported in full by the CHEST Foundation.
2016 Community Service Grantee
Ethel Jane Carter, MD, FCCP
Warren Alpert School of Medicine at Brown University
CHEST Foundation Community Service Grant Honoring D. Robert McCaffree, MD, Master FCCP
East African Training Initiative ( EATI) in Pulmonary Medicine
2016 NetWorks Challenge Travel Grantees
Debarsee Banerjee, MS, MD
Women’s Health NetWork
Drew Harris, MD
Occupational and Environmental Health NetWork
Kerry Hena, MD
Occupational and Environmental Health NetWork
Amanpreet Kaur, MD
Women’s Health NetWork
2016 Diversity Travel Grant Winners
John B. Bishara, DO
Renato F. Blanco Jr., MD
Angel Coz-Yataco, MD Sherie A. Gause, MD
Anthony Nebor, MD
James T. Williams, MD
Alfred Soffer Research Award Winners
Kerry Hena, MD
Deepak Pradhan, MD, FCCP
Young Investigator Award Winners
Elizabeth Becker: Clinical Characteristics of Sarcoidosis in World Trade Center (WTC) Exposed Fire Department of the City of New York (FDNY) Firefighters
Daniel Altman, MD : Cost-Effectiveness of Universally Funding Smoking Cessation Pharmacotherapy
Top 3 Poster Winners
Epaminondas Kosmas, MD, PhD, FCCP : Bronchiectasis in Patients With COPD: An Irrelevant Imaging Finding or a Clinically Important Phenotype?
Mark Regala, MD, BS : Evaluation of Outcomes of Post-Extubation Dysphagia in Elderly Patients
Massa Zantah, MD : Correlation of Esophageal Dilatation and Pulmonary Fibrosis in Scleroderma
Runner-up: Alev Gurgun, MD : Pulmonary Rehabilitation Response in Elderly and Younger Patients With COPD
Case Report Slide Winners
John Egan, MD, BA : An Unusual Cause of Tracheal Stenosis Due to a Vascular Anomaly Successfully Managed With Silicone Airway Stenting Prior to Definitive Vascular Repair
Harprett Grewal, MD : Bladder PTLD: First Reported Case of Post-Transplant Lymphoproliferative Disorder (PTLD) in the Bladder in a Lung Transplant Recipient
Michael Fingerhood, MD, MPH : Pulmonary Overlap Histiocytosis: A Rare Case of Interstitial Lung Disease Due to Erdheim Chester Disease in a Patient With Langerhans Cell Histiocytosis and Myelodisplastic Syndrome
Yihenew Negatu, MD : Acute ST Elevation Myocardial Infarction Related to Carbon Monoxide Poisoning in a Young Patient Without Coronary Artery Disease
Stephanie Wappel, MD : False-Negative Pet Imaging in Early Stage Malignant Pleural Mesothelioma
Lina Miyakawa, MD : Restrictive EGFR Mutation
Jeffrey Bonenfant, DO : A Unique Case of Follicular Bronchiolitis
Melissa Myers, MD : Seeing the Forest and Not Just the Trees: A Case of Recurrent Fever, Cough, and Respiratory Failure
Carly Fabrizio, DO : An Unusual Case of Submassive Hemoptysis
Meilinh Thi, DO : A Case to Make Your Skin Crawl
Garrett Harp, MD : Lambertosis: A Lung Cancer Mimic
Malik Khan, MD : Pleural Epithelioid Hemangioendothelioma: A Case Report
Priya Patel, MD : A Troubling Trifecta: Pulmonary Alveolar Proteinosis and Pneumocystis Pneumonia in Acute Myeloid Leukemia
Atul Palkar, MD : SGLT2 Inhibitors: Mind the Gap
Ji Yeon Lee, MD : Making Unusual Connections: Fibrosing Mediastinitis Leading to Bronchoesophageal Fistula
Sailm Daouk, MD : A Rare Form of Invasive Aspergillus Infection in a Severely Immunocompromised Host
Venkata Ravi Kumar Angirekula, MD : Vanishing Lung
Stephen Milan, MD : An Unexpected Mass
Lelia Logue, MD : A Rare Cause of Dysphagia
Daniel Hershberger, MD : Rapidly Progressive Hypoxic Respiratory Failure After a Rash: A Case of Clinically Amyopathic Dermatomyositis (CADM)-Associated ILD
Fellow Case Report Poster Winners
Krishna Siva Sai Kakkera
An Unusual Case of Crypotococcal Pleural Effusion
George Cheng
Use of Laparoscopic Suction Irrigator With Rigid Pleuroscope in Medical Thoracoscopy
Matt Koroscil
Wong Type Dermatomyositis Complicated by Interstitial Lung Disease
Derek Hansen
Acute Fibrinous and Organizing Pneumonia Following Hematopoietic Stem Cell Transplantation Responsive to Corticosteroid Therapy
Ala Eddin Sagar
Pulmonary Embolism Caused by Thrombin-Based Hemostatic Matrix After Discectomy
Sandeep Chennadi
Systemic Lupus Erythematosus (SLE) With Refractory Bilateral Chylothorax and Chylous Ascites
Medical Student/Resident Case Report Poster Winners
Justin Fiala
Pulmonary Presentation Without Concurrent Bone Involvement in Erdheim-Chester Disease: A Report of Two Cases
Navitha Ramesh
A Fatal Migration: A Case of Intra-Cardiac Embolization of a Peripheral Stent
Humna Abid Memon
Use of Extracorporeal Membrane Oxygenation in Postpartum Management of a Patient With PAH
Vanessa Ohleyer
A Case of Unusual Anatomy for an Uncommon Mediastinal Tumor
Tanushree Gahlot
Three Unusual Presentations of Job’s Syndrome (Hyper Immunoglobulin E Syndrome)
NetWorks Challenge Winners
Round 1
Women’s Lung Health NetWork
Round 2
Practice and Operations NetWork-1st place
Home-Based Mechanical Ventilation and Neuromuscular Disease NetWorks – 2nd place
Round 3
Home-Based Mechanical Ventilation, Neuromuscular Disease, and the Women’s Lung Health NetWorks
CHEST Bingo Winners
Youseff Anid, MD, FCCP
Karen Cochran, ACNP
Molly Howsware, DO
Katie Jeans, MD
Genovena Medina, RN
Gregory Eisinger, MD
Saurabh Mittal, MD, MBBS
Navitha Ramesh, MD
Dalvinder Dhillon, MD
Teresita Saylor, MD, FCCP
Carl Kaplan, MD, FCCP
Vishal Patel, MBBS, FCCP
Erin Peterson, CNP
Lilian Pereira, DO
Joint CHEST-SGP Congress 2017
Basel, Switzerland June 7-9
Join leaders in CHEST medicine for a program designed by clinicians for clinicians.
The Joint Congress organized by CHEST and the Swiss Society of Pneumology will be held from June 7-9 in Basel, Switzerland. The program has been designed by more than 140 faculty members from both the United States and Europe, and it aims to provide a robust overview of all aspects of respiratory medicine through interactive sessions, plenary discussions, critical appraisals on controversial topics, and a review of the last year of published works.
The Joint Congress also provides the opportunity to take part in hands-on simulation in areas such as lung function techniques including body plethysmography, N2 washout techniques, and respiratory physiotherapy. Another hands-on opportunity is the interventional pneumology CHEST experience course, which will be held from 8:00 AM-11:00 AM on June 7 and 8 on site. This course will provide an overview of conventional and EBUS-guided TBNA, an anatomy identification of airway nodes, management of airway bleeding, and management of pneumothorax. This course is ideal for clinicians and health-care professionals with specialties in pulmonary, critical care, and intensive care medicine, as well as thoracic surgery.
The program at the Joint CHEST-SGP Congress aims to improve the patient care abilities of every attendee, as well as provide an ideal environment for networking with leaders in your field.
The call for abstracts remains open until January 24, 2017. The abstract topic areas are:
- Airway disease
- Interstitial lung disease
- Sleep/Breathing
- Lung cancer
- Epidemiology/Rehabilitation
- Interventional pneumology
- Pulmonary hypertension
- Basic science
- Thoracic surgery
- Pediatrics
All abstracts must be submitted via the Joint Congress abstracts web portal www.chest-sgp-switzerland2017.org.
CHEST recognizes the value of international outreach, and this Joint Congress advances that initiative. CHEST aims to standardize the patient care across borders and to encourage international collaboration to build the future of chest medicine. To further this mission, an application has been made to the European Accreditation Council for Continuing Medical Education (EACCME®) for CME accreditation of this event. Additionally, an application has been made to the European Board for Accreditation in Pneumology (EBAP) to provide quality assurance and CME for the event.
For more information or to register, visit the CHEST Joint Congress website www.chest-sgp-switzerland2017.org. Early registration ends on March 16, 2017.
Basel, Switzerland June 7-9
Join leaders in CHEST medicine for a program designed by clinicians for clinicians.
The Joint Congress organized by CHEST and the Swiss Society of Pneumology will be held from June 7-9 in Basel, Switzerland. The program has been designed by more than 140 faculty members from both the United States and Europe, and it aims to provide a robust overview of all aspects of respiratory medicine through interactive sessions, plenary discussions, critical appraisals on controversial topics, and a review of the last year of published works.
The Joint Congress also provides the opportunity to take part in hands-on simulation in areas such as lung function techniques including body plethysmography, N2 washout techniques, and respiratory physiotherapy. Another hands-on opportunity is the interventional pneumology CHEST experience course, which will be held from 8:00 AM-11:00 AM on June 7 and 8 on site. This course will provide an overview of conventional and EBUS-guided TBNA, an anatomy identification of airway nodes, management of airway bleeding, and management of pneumothorax. This course is ideal for clinicians and health-care professionals with specialties in pulmonary, critical care, and intensive care medicine, as well as thoracic surgery.
The program at the Joint CHEST-SGP Congress aims to improve the patient care abilities of every attendee, as well as provide an ideal environment for networking with leaders in your field.
The call for abstracts remains open until January 24, 2017. The abstract topic areas are:
- Airway disease
- Interstitial lung disease
- Sleep/Breathing
- Lung cancer
- Epidemiology/Rehabilitation
- Interventional pneumology
- Pulmonary hypertension
- Basic science
- Thoracic surgery
- Pediatrics
All abstracts must be submitted via the Joint Congress abstracts web portal www.chest-sgp-switzerland2017.org.
CHEST recognizes the value of international outreach, and this Joint Congress advances that initiative. CHEST aims to standardize the patient care across borders and to encourage international collaboration to build the future of chest medicine. To further this mission, an application has been made to the European Accreditation Council for Continuing Medical Education (EACCME®) for CME accreditation of this event. Additionally, an application has been made to the European Board for Accreditation in Pneumology (EBAP) to provide quality assurance and CME for the event.
For more information or to register, visit the CHEST Joint Congress website www.chest-sgp-switzerland2017.org. Early registration ends on March 16, 2017.
Basel, Switzerland June 7-9
Join leaders in CHEST medicine for a program designed by clinicians for clinicians.
The Joint Congress organized by CHEST and the Swiss Society of Pneumology will be held from June 7-9 in Basel, Switzerland. The program has been designed by more than 140 faculty members from both the United States and Europe, and it aims to provide a robust overview of all aspects of respiratory medicine through interactive sessions, plenary discussions, critical appraisals on controversial topics, and a review of the last year of published works.
The Joint Congress also provides the opportunity to take part in hands-on simulation in areas such as lung function techniques including body plethysmography, N2 washout techniques, and respiratory physiotherapy. Another hands-on opportunity is the interventional pneumology CHEST experience course, which will be held from 8:00 AM-11:00 AM on June 7 and 8 on site. This course will provide an overview of conventional and EBUS-guided TBNA, an anatomy identification of airway nodes, management of airway bleeding, and management of pneumothorax. This course is ideal for clinicians and health-care professionals with specialties in pulmonary, critical care, and intensive care medicine, as well as thoracic surgery.
The program at the Joint CHEST-SGP Congress aims to improve the patient care abilities of every attendee, as well as provide an ideal environment for networking with leaders in your field.
The call for abstracts remains open until January 24, 2017. The abstract topic areas are:
- Airway disease
- Interstitial lung disease
- Sleep/Breathing
- Lung cancer
- Epidemiology/Rehabilitation
- Interventional pneumology
- Pulmonary hypertension
- Basic science
- Thoracic surgery
- Pediatrics
All abstracts must be submitted via the Joint Congress abstracts web portal www.chest-sgp-switzerland2017.org.
CHEST recognizes the value of international outreach, and this Joint Congress advances that initiative. CHEST aims to standardize the patient care across borders and to encourage international collaboration to build the future of chest medicine. To further this mission, an application has been made to the European Accreditation Council for Continuing Medical Education (EACCME®) for CME accreditation of this event. Additionally, an application has been made to the European Board for Accreditation in Pneumology (EBAP) to provide quality assurance and CME for the event.
For more information or to register, visit the CHEST Joint Congress website www.chest-sgp-switzerland2017.org. Early registration ends on March 16, 2017.
Adding epoetin alfa to lenalidomide boosted myelodysplastic syndrome responses
SAN DIEGO – Dual therapy with lenalidomide and epoetin alfa was safe and led to freedom from transfusion significantly more often than lenalidomide alone in patients with erythropoietin-refractory, lower-risk, non-del(5q) myelodysplastic syndromes, according to a randomized phase III head-to-head trial.
After 16 weeks of treatment, 33% of patients who received both lenalidomide and epoetin alfa met International Working Group 2000 criteria for major erythroid response, compared with only 14% of patients receiving lenalidomide monotherapy (P = .03), Alan F. List, MD, reported at the annual meeting of the American Society of Hematology.

Recombinant human erythropoietin improves anemia in some cases of MDS, but salvage options are limited. “Cytokine therapy is generally ineffective in patients with high transfusion burden or elevated serum erythropoietin level,” Dr. List said.
Lenalidomide (Revlimid) promotes the in vitro expansion of primitive erythroid precursors, and in a recent phase III, placebo-controlled trial, the immunomodulator improved erythropoiesis in about 25% of lower-risk, non-del(5q) MDS patients who were azanucleoside-naïve and transfusion-dependent, with effects lasting about 8 months. In another pilot study, adding epoetin alfa to lenalidomide induced erythroid responses in 28% of MDS patients who were not responding to lenalidomide alone. “This suggests that lenalidomide overcomes resistance and augments response to recombinant human erythropoietin,” Dr. List explained.
For their phase III trial, he and his associates randomly assigned erythropoietin-refractory, lower-risk, non-del(5q) MDS patients with hemoglobin levels under 9.5 g/dL to receive lenalidomide (10 mg per day for 21 days every 28 days) with or without epoetin alfa (weekly dose, 60,000 units subcutaneously). A total of 14% of patients had previously received azanucleoside therapy, about 92% had received erythropoietic stimulating agents, and median serum erythropoietin levels were 167 and 143 mU per mL in the monotherapy and dual therapy arms, respectively.
In accordance with International Working Group 2000 criteria, the researchers defined major erythroid response as transfusion independence for least 8 consecutive weeks, with at least a 1 g/dL increase in hemoglobin levels if patients were transfusion-dependent at baseline, and at least a 2 g/dL rise in hemoglobin if they were transfusion-independent.
In an interim analysis of 163 patients, 26% of the dual therapy group and 11% of lenalidomide-only patients met this primary endpoint (P = .02). These results met predefined criteria for stopping the study, after which 34 lenalidomide nonresponders crossed over to dual therapy. In all, 21% of these patients also had a major erythroid response, Dr. List said.
A multivariable analysis that included disease duration, International Prognostic Scoring System low versus intermediate-1 risk status, baseline erythropoietin level, and prior azanucleoside exposure showed that only dual lenalidomide–epoetin alfa therapy predicted major erythroid response. Specifically, dual therapy increased the odds of this outcome by about 63% when compared with lenalidomide monotherapy (P = .03).
Secondary analyses linked major erythroid response to having more low than high molecular weight CD45 isoform. In fact, the median ratio of high to low molecular weight CD45 was 1.5 among responders and 4.2 among nonresponders (P = .04) This finding fits the hypothesis that larger CD45 isoforms keep lenalidomide from enhancing erythropoietin receptor signaling, Dr. List said. Indeed, rates of major erythroid response to lenalidomide–epoetin alfa therapy were 73% when patients had a low isoform ratio, but were only 18% when they had a high isoform ratio (P = .03). The CD45 isoform ratio distinguished responders from nonresponders with a sensitivity and specificity of 80% and 75%, respectively, Dr. List said.
Grade 3 or higher nonhematologic events affected about a quarter of patients in each arm, and rates of individual events were similar. The most common serious adverse event was fatigue (5% of patients), followed by elevated serum creatinine (3.7%). About 10% of patients in each arm died while on study.
The National Institutes of Health supported the study. Dr. List had no relevant financial disclosures.
SAN DIEGO – Dual therapy with lenalidomide and epoetin alfa was safe and led to freedom from transfusion significantly more often than lenalidomide alone in patients with erythropoietin-refractory, lower-risk, non-del(5q) myelodysplastic syndromes, according to a randomized phase III head-to-head trial.
After 16 weeks of treatment, 33% of patients who received both lenalidomide and epoetin alfa met International Working Group 2000 criteria for major erythroid response, compared with only 14% of patients receiving lenalidomide monotherapy (P = .03), Alan F. List, MD, reported at the annual meeting of the American Society of Hematology.
Recombinant human erythropoietin improves anemia in some cases of MDS, but salvage options are limited. “Cytokine therapy is generally ineffective in patients with high transfusion burden or elevated serum erythropoietin level,” Dr. List said.
Lenalidomide (Revlimid) promotes the in vitro expansion of primitive erythroid precursors, and in a recent phase III, placebo-controlled trial, the immunomodulator improved erythropoiesis in about 25% of lower-risk, non-del(5q) MDS patients who were azanucleoside-naïve and transfusion-dependent, with effects lasting about 8 months. In another pilot study, adding epoetin alfa to lenalidomide induced erythroid responses in 28% of MDS patients who were not responding to lenalidomide alone. “This suggests that lenalidomide overcomes resistance and augments response to recombinant human erythropoietin,” Dr. List explained.
For their phase III trial, he and his associates randomly assigned erythropoietin-refractory, lower-risk, non-del(5q) MDS patients with hemoglobin levels under 9.5 g/dL to receive lenalidomide (10 mg per day for 21 days every 28 days) with or without epoetin alfa (weekly dose, 60,000 units subcutaneously). A total of 14% of patients had previously received azanucleoside therapy, about 92% had received erythropoietic stimulating agents, and median serum erythropoietin levels were 167 and 143 mU per mL in the monotherapy and dual therapy arms, respectively.
In accordance with International Working Group 2000 criteria, the researchers defined major erythroid response as transfusion independence for least 8 consecutive weeks, with at least a 1 g/dL increase in hemoglobin levels if patients were transfusion-dependent at baseline, and at least a 2 g/dL rise in hemoglobin if they were transfusion-independent.
In an interim analysis of 163 patients, 26% of the dual therapy group and 11% of lenalidomide-only patients met this primary endpoint (P = .02). These results met predefined criteria for stopping the study, after which 34 lenalidomide nonresponders crossed over to dual therapy. In all, 21% of these patients also had a major erythroid response, Dr. List said.
A multivariable analysis that included disease duration, International Prognostic Scoring System low versus intermediate-1 risk status, baseline erythropoietin level, and prior azanucleoside exposure showed that only dual lenalidomide–epoetin alfa therapy predicted major erythroid response. Specifically, dual therapy increased the odds of this outcome by about 63% when compared with lenalidomide monotherapy (P = .03).
Secondary analyses linked major erythroid response to having more low than high molecular weight CD45 isoform. In fact, the median ratio of high to low molecular weight CD45 was 1.5 among responders and 4.2 among nonresponders (P = .04) This finding fits the hypothesis that larger CD45 isoforms keep lenalidomide from enhancing erythropoietin receptor signaling, Dr. List said. Indeed, rates of major erythroid response to lenalidomide–epoetin alfa therapy were 73% when patients had a low isoform ratio, but were only 18% when they had a high isoform ratio (P = .03). The CD45 isoform ratio distinguished responders from nonresponders with a sensitivity and specificity of 80% and 75%, respectively, Dr. List said.
Grade 3 or higher nonhematologic events affected about a quarter of patients in each arm, and rates of individual events were similar. The most common serious adverse event was fatigue (5% of patients), followed by elevated serum creatinine (3.7%). About 10% of patients in each arm died while on study.
The National Institutes of Health supported the study. Dr. List had no relevant financial disclosures.
SAN DIEGO – Dual therapy with lenalidomide and epoetin alfa was safe and led to freedom from transfusion significantly more often than lenalidomide alone in patients with erythropoietin-refractory, lower-risk, non-del(5q) myelodysplastic syndromes, according to a randomized phase III head-to-head trial.
After 16 weeks of treatment, 33% of patients who received both lenalidomide and epoetin alfa met International Working Group 2000 criteria for major erythroid response, compared with only 14% of patients receiving lenalidomide monotherapy (P = .03), Alan F. List, MD, reported at the annual meeting of the American Society of Hematology.
Recombinant human erythropoietin improves anemia in some cases of MDS, but salvage options are limited. “Cytokine therapy is generally ineffective in patients with high transfusion burden or elevated serum erythropoietin level,” Dr. List said.
Lenalidomide (Revlimid) promotes the in vitro expansion of primitive erythroid precursors, and in a recent phase III, placebo-controlled trial, the immunomodulator improved erythropoiesis in about 25% of lower-risk, non-del(5q) MDS patients who were azanucleoside-naïve and transfusion-dependent, with effects lasting about 8 months. In another pilot study, adding epoetin alfa to lenalidomide induced erythroid responses in 28% of MDS patients who were not responding to lenalidomide alone. “This suggests that lenalidomide overcomes resistance and augments response to recombinant human erythropoietin,” Dr. List explained.
For their phase III trial, he and his associates randomly assigned erythropoietin-refractory, lower-risk, non-del(5q) MDS patients with hemoglobin levels under 9.5 g/dL to receive lenalidomide (10 mg per day for 21 days every 28 days) with or without epoetin alfa (weekly dose, 60,000 units subcutaneously). A total of 14% of patients had previously received azanucleoside therapy, about 92% had received erythropoietic stimulating agents, and median serum erythropoietin levels were 167 and 143 mU per mL in the monotherapy and dual therapy arms, respectively.
In accordance with International Working Group 2000 criteria, the researchers defined major erythroid response as transfusion independence for least 8 consecutive weeks, with at least a 1 g/dL increase in hemoglobin levels if patients were transfusion-dependent at baseline, and at least a 2 g/dL rise in hemoglobin if they were transfusion-independent.
In an interim analysis of 163 patients, 26% of the dual therapy group and 11% of lenalidomide-only patients met this primary endpoint (P = .02). These results met predefined criteria for stopping the study, after which 34 lenalidomide nonresponders crossed over to dual therapy. In all, 21% of these patients also had a major erythroid response, Dr. List said.
A multivariable analysis that included disease duration, International Prognostic Scoring System low versus intermediate-1 risk status, baseline erythropoietin level, and prior azanucleoside exposure showed that only dual lenalidomide–epoetin alfa therapy predicted major erythroid response. Specifically, dual therapy increased the odds of this outcome by about 63% when compared with lenalidomide monotherapy (P = .03).
Secondary analyses linked major erythroid response to having more low than high molecular weight CD45 isoform. In fact, the median ratio of high to low molecular weight CD45 was 1.5 among responders and 4.2 among nonresponders (P = .04) This finding fits the hypothesis that larger CD45 isoforms keep lenalidomide from enhancing erythropoietin receptor signaling, Dr. List said. Indeed, rates of major erythroid response to lenalidomide–epoetin alfa therapy were 73% when patients had a low isoform ratio, but were only 18% when they had a high isoform ratio (P = .03). The CD45 isoform ratio distinguished responders from nonresponders with a sensitivity and specificity of 80% and 75%, respectively, Dr. List said.
Grade 3 or higher nonhematologic events affected about a quarter of patients in each arm, and rates of individual events were similar. The most common serious adverse event was fatigue (5% of patients), followed by elevated serum creatinine (3.7%). About 10% of patients in each arm died while on study.
The National Institutes of Health supported the study. Dr. List had no relevant financial disclosures.
Key clinical point: Dual therapy with lenalidomide and epoetin alfa was more effective than lenalidomide monotherapy in patients with erythropoietin-refractory, lower-risk, non-del(5q) myelodysplastic syndrome.
Major finding: After 16 weeks of treatment, 33% of patients who received both agents met International Working Group 2000 criteria for major erythroid response, compared with 14% of patients receiving lenalidomide monotherapy (P = .03).
Data source: An interim analysis of 163 patients in the phase III ECOG-ACRIN E2905 Intergroup Study.
Disclosures: The National Institutes of Health supported the study. Dr. List had no relevant financial disclosures.
ACGME seeks to return trainees’ maximum shifts to 24 hours
First-year residents may be permitted to work up to 24 consecutive hours – 8 hours longer than they can now – under a proposal from a task force of the Accreditation Council for Graduate Medical Education (ACGME).
The ACGME Duty Hour Task Force proposes to raise first-year trainees’ work hour limit from 16 hours, reverting to the 24-hour maximum that remained in effect until 2011 – and the existing limit in place for all other residents.
“There is better team continuity of care provided with less micromanagement of resident duty hours,” said Thomas J. Nasca, MD, ACGME chief executive and vice chairman of the task force.
The ACGME instituted the 16-hour cap for first-year residents in the wake of a December 2008 report released by the Institute of Medicine (IOM), “Resident Duty Hours: Enhancing Sleep, Supervision, and Safety.” The ACGME also prohibited 30-hour shifts, which some trainees had been clocking.
“Every 5 years, the ACGME reviews its program requirements,” Dr. Nasca said. “That’s a promise we made to the community and to the public” in July 2003. The most recent changes occurred in July 2011.
Since then, some faculty have contended that shorter work hour limits for the first-year residents are accelerating the frequency of patient transitions and disrupting continuity of care. Many educators also noted that residents would gain more knowledge by monitoring a hospitalized patient during the initial 24 hours.
The American College of Physicians “approves and commends ACGME for explicitly addressing [resident] well being in the Common Program Requirements,” ACP President Nitin S. Damle, MD, said in a statement. “We support ACGME in removing the standard limiting duty hours for PGY-1 residents to no more than 16 hours. By mandating different hours for PGY-residents and more senior residents, the old standard disrupted the continuity of the resident team and resulted in considerable unintended consequences without commensurate improvement in patient safety or educational outcomes.”
The ACP does not support a proposed additional 4-hour extension to the current 4-hour extension to a 24-hour call shift. “We believe that the [current] 24+ rule adequately addresses all clinical and educational needs,” Dr. Damle said in the statement. “The new standard places residents at risk for 28 consecutive hours of duty.”
The proposed revisions to training requirements also include phasing out the term “duty” hours in favor of “clinical experience and education,” highlighting that residents’ responsibility to patients takes precedence over any adherence to a schedule or clock. Working a certain number of hospital hours is only one aspect of delivering safe and quality care, the ACGME Duty Hour Task Force noted in its proposal.
The proposal does not change the following, which all are averaged over a period of 4 weeks: a maximum of 80 hours per week, 1 day free from clinical experience or education in 7, and in-house call no more often than every third night.
“It is important to note that the absence of a common 16-hour limit does not imply that programs may no longer configure their clinical schedules in 16-hour increments if that is the preferred option for a given setting or clinical context,” Dr. Nasca wrote in a Nov. 4 letter to the graduate medical education community.
In its December 2008 report, the IOM noted that revamping residents’ work hours alone offered no guarantee of patient safety. It called for increased supervision by seasoned physicians, restrictions on patient caseloads based on residents’ levels of experience and specialty, overlap in schedules around shift changes to decrease the possibility of error in transitioning patients from one provider to another, and broad-based research to examine the outcomes from these changes.
“Surgical residents, when they’re operating, come under very direct supervision by attending physicians,” said Jay Bhattacharya, MD, professor of medicine at Stanford (Calif.) University, who served on the 2008 IOM committee. “That supervision is generally pretty good. Even for a tired resident, the supervision makes it so that the fatigue the trainee faces doesn’t endanger the patient.”
Acknowledging the emerging evidence that physicians are at heightened risk for burnout and perhaps depression, the proposal underscores the need for residency programs and institutions to prioritize the well-being of residents as well as nurses and other hospital personnel.
“Burnout and depression impair a physician’s ability to provide excellent care. Self-care is an important aspect of professionalism, and a skill that must be learned and nurtured under the guidance and role modeling of faculty members,” Dr. Nasca wrote in his Nov. 4 letter.
If the proposal is approved by the ACGME board of directors in February, the changes will be rolled out in July.
The ACGME accepted comments on the proposal through Dec. 19; comments will be reviewed and considered before a final set of proposed requirements are sent to the ACGME board for approval.
“We’re here to serve the public, and so the focus is on the comments that reflect the most recent and comprehensive evidence and educational outcomes rather than the individual opinions we receive,” Dr. Nasca said.
First-year residents may be permitted to work up to 24 consecutive hours – 8 hours longer than they can now – under a proposal from a task force of the Accreditation Council for Graduate Medical Education (ACGME).
The ACGME Duty Hour Task Force proposes to raise first-year trainees’ work hour limit from 16 hours, reverting to the 24-hour maximum that remained in effect until 2011 – and the existing limit in place for all other residents.
“There is better team continuity of care provided with less micromanagement of resident duty hours,” said Thomas J. Nasca, MD, ACGME chief executive and vice chairman of the task force.
The ACGME instituted the 16-hour cap for first-year residents in the wake of a December 2008 report released by the Institute of Medicine (IOM), “Resident Duty Hours: Enhancing Sleep, Supervision, and Safety.” The ACGME also prohibited 30-hour shifts, which some trainees had been clocking.
“Every 5 years, the ACGME reviews its program requirements,” Dr. Nasca said. “That’s a promise we made to the community and to the public” in July 2003. The most recent changes occurred in July 2011.
Since then, some faculty have contended that shorter work hour limits for the first-year residents are accelerating the frequency of patient transitions and disrupting continuity of care. Many educators also noted that residents would gain more knowledge by monitoring a hospitalized patient during the initial 24 hours.
The American College of Physicians “approves and commends ACGME for explicitly addressing [resident] well being in the Common Program Requirements,” ACP President Nitin S. Damle, MD, said in a statement. “We support ACGME in removing the standard limiting duty hours for PGY-1 residents to no more than 16 hours. By mandating different hours for PGY-residents and more senior residents, the old standard disrupted the continuity of the resident team and resulted in considerable unintended consequences without commensurate improvement in patient safety or educational outcomes.”
The ACP does not support a proposed additional 4-hour extension to the current 4-hour extension to a 24-hour call shift. “We believe that the [current] 24+ rule adequately addresses all clinical and educational needs,” Dr. Damle said in the statement. “The new standard places residents at risk for 28 consecutive hours of duty.”
The proposed revisions to training requirements also include phasing out the term “duty” hours in favor of “clinical experience and education,” highlighting that residents’ responsibility to patients takes precedence over any adherence to a schedule or clock. Working a certain number of hospital hours is only one aspect of delivering safe and quality care, the ACGME Duty Hour Task Force noted in its proposal.
The proposal does not change the following, which all are averaged over a period of 4 weeks: a maximum of 80 hours per week, 1 day free from clinical experience or education in 7, and in-house call no more often than every third night.
“It is important to note that the absence of a common 16-hour limit does not imply that programs may no longer configure their clinical schedules in 16-hour increments if that is the preferred option for a given setting or clinical context,” Dr. Nasca wrote in a Nov. 4 letter to the graduate medical education community.
In its December 2008 report, the IOM noted that revamping residents’ work hours alone offered no guarantee of patient safety. It called for increased supervision by seasoned physicians, restrictions on patient caseloads based on residents’ levels of experience and specialty, overlap in schedules around shift changes to decrease the possibility of error in transitioning patients from one provider to another, and broad-based research to examine the outcomes from these changes.
“Surgical residents, when they’re operating, come under very direct supervision by attending physicians,” said Jay Bhattacharya, MD, professor of medicine at Stanford (Calif.) University, who served on the 2008 IOM committee. “That supervision is generally pretty good. Even for a tired resident, the supervision makes it so that the fatigue the trainee faces doesn’t endanger the patient.”
Acknowledging the emerging evidence that physicians are at heightened risk for burnout and perhaps depression, the proposal underscores the need for residency programs and institutions to prioritize the well-being of residents as well as nurses and other hospital personnel.
“Burnout and depression impair a physician’s ability to provide excellent care. Self-care is an important aspect of professionalism, and a skill that must be learned and nurtured under the guidance and role modeling of faculty members,” Dr. Nasca wrote in his Nov. 4 letter.
If the proposal is approved by the ACGME board of directors in February, the changes will be rolled out in July.
The ACGME accepted comments on the proposal through Dec. 19; comments will be reviewed and considered before a final set of proposed requirements are sent to the ACGME board for approval.
“We’re here to serve the public, and so the focus is on the comments that reflect the most recent and comprehensive evidence and educational outcomes rather than the individual opinions we receive,” Dr. Nasca said.
First-year residents may be permitted to work up to 24 consecutive hours – 8 hours longer than they can now – under a proposal from a task force of the Accreditation Council for Graduate Medical Education (ACGME).
The ACGME Duty Hour Task Force proposes to raise first-year trainees’ work hour limit from 16 hours, reverting to the 24-hour maximum that remained in effect until 2011 – and the existing limit in place for all other residents.
“There is better team continuity of care provided with less micromanagement of resident duty hours,” said Thomas J. Nasca, MD, ACGME chief executive and vice chairman of the task force.
The ACGME instituted the 16-hour cap for first-year residents in the wake of a December 2008 report released by the Institute of Medicine (IOM), “Resident Duty Hours: Enhancing Sleep, Supervision, and Safety.” The ACGME also prohibited 30-hour shifts, which some trainees had been clocking.
“Every 5 years, the ACGME reviews its program requirements,” Dr. Nasca said. “That’s a promise we made to the community and to the public” in July 2003. The most recent changes occurred in July 2011.
Since then, some faculty have contended that shorter work hour limits for the first-year residents are accelerating the frequency of patient transitions and disrupting continuity of care. Many educators also noted that residents would gain more knowledge by monitoring a hospitalized patient during the initial 24 hours.
The American College of Physicians “approves and commends ACGME for explicitly addressing [resident] well being in the Common Program Requirements,” ACP President Nitin S. Damle, MD, said in a statement. “We support ACGME in removing the standard limiting duty hours for PGY-1 residents to no more than 16 hours. By mandating different hours for PGY-residents and more senior residents, the old standard disrupted the continuity of the resident team and resulted in considerable unintended consequences without commensurate improvement in patient safety or educational outcomes.”
The ACP does not support a proposed additional 4-hour extension to the current 4-hour extension to a 24-hour call shift. “We believe that the [current] 24+ rule adequately addresses all clinical and educational needs,” Dr. Damle said in the statement. “The new standard places residents at risk for 28 consecutive hours of duty.”
The proposed revisions to training requirements also include phasing out the term “duty” hours in favor of “clinical experience and education,” highlighting that residents’ responsibility to patients takes precedence over any adherence to a schedule or clock. Working a certain number of hospital hours is only one aspect of delivering safe and quality care, the ACGME Duty Hour Task Force noted in its proposal.
The proposal does not change the following, which all are averaged over a period of 4 weeks: a maximum of 80 hours per week, 1 day free from clinical experience or education in 7, and in-house call no more often than every third night.
“It is important to note that the absence of a common 16-hour limit does not imply that programs may no longer configure their clinical schedules in 16-hour increments if that is the preferred option for a given setting or clinical context,” Dr. Nasca wrote in a Nov. 4 letter to the graduate medical education community.
In its December 2008 report, the IOM noted that revamping residents’ work hours alone offered no guarantee of patient safety. It called for increased supervision by seasoned physicians, restrictions on patient caseloads based on residents’ levels of experience and specialty, overlap in schedules around shift changes to decrease the possibility of error in transitioning patients from one provider to another, and broad-based research to examine the outcomes from these changes.
“Surgical residents, when they’re operating, come under very direct supervision by attending physicians,” said Jay Bhattacharya, MD, professor of medicine at Stanford (Calif.) University, who served on the 2008 IOM committee. “That supervision is generally pretty good. Even for a tired resident, the supervision makes it so that the fatigue the trainee faces doesn’t endanger the patient.”
Acknowledging the emerging evidence that physicians are at heightened risk for burnout and perhaps depression, the proposal underscores the need for residency programs and institutions to prioritize the well-being of residents as well as nurses and other hospital personnel.
“Burnout and depression impair a physician’s ability to provide excellent care. Self-care is an important aspect of professionalism, and a skill that must be learned and nurtured under the guidance and role modeling of faculty members,” Dr. Nasca wrote in his Nov. 4 letter.
If the proposal is approved by the ACGME board of directors in February, the changes will be rolled out in July.
The ACGME accepted comments on the proposal through Dec. 19; comments will be reviewed and considered before a final set of proposed requirements are sent to the ACGME board for approval.
“We’re here to serve the public, and so the focus is on the comments that reflect the most recent and comprehensive evidence and educational outcomes rather than the individual opinions we receive,” Dr. Nasca said.
Transient Benign Neonatal Skin Findings
Review the PDF of the fact sheet on transient benign neonatal skin findings with board-relevant, easy-to-review material. This fact sheet lists benign findings that can be seen in neonates and infants.
Practice Questions
1. The parents of a 2-month-old infant present with their child. They are worried because the infant has “acne” that is not going away. Friends told them to try gentle cleansers and they have avoided using lotions or cream on her face. However, the bumps will not go away. On examination she has papules and pustules. Comedones cannot be identified. What are your next steps?
a. adapalene cream 0.1% every night at bedtime
b. benzoyl peroxide cream 4%
c. benzoyl peroxide wash 2.5%
d. erythromycin gel 2%
e. ketoconazole cream 2% twice daily
2. While in the newborn nursery prior to discharge, the attending pediatrician notices a rash on a 2-day-old neonate who is otherwise completely healthy. The pediatrician consults a dermatologist for his/her opinion. The dermatologist sees erythematous macules with central pustules located predominately on the trunk and proximal extremities. A pustule is unroofed with a blade, the contents smeared on a glass slide, and a Giemsa stain is performed. What is the predominant cell type you would expect to see on histological examination?
a. eosinophils
b. Langerhans cells
c. lymphocytes
d. neutrophils
e. no cells are visualized
3. Shortly after delivery, the pediatricians notice that the baby has numerous hyperpigmented macules on the back. No other primary lesions are seen. The neonate is otherwise normal in appearance and nontoxic appearing. A dermatologist is consulted for a recommendation for further workup or potential biopsy. The dermatologist examines the newborn. He is a well-appearing black boy with skin that is otherwise intact. A few pustules on the back are present that have a collarette of scale. The dermatologist reviews the mother’s prenatal history and the review shows that she was screened for syphilis and had a negative screening test with no other history of infectious diseases. What is the most appropriate next step to confirm your suspicions?
a. do a swab of a pustule and send it for viral culture
b. have his blood drawn and check for signs of neonatal herpes simplex virus infection
c. perform a biopsy of a pustule
d. perform a Giemsa stain on a smear of the pustule
e. start treatment with permethrin
4. Which intraoral cysts occur on the alveolar ridge of a neonate?
a. Bohn nodule
b. branchial cleft cyst
c. Epstein pearls
d. median raphe cyst
e. palatal cysts of the newborn
5. Miliaria rubra is associated with inflammation of the sweat glands in what portion of the skin?
a. basement membrane zone
b. dermis
c. dermoepidermal junction
d. intraepidermal
e. subcutis
Answers to practice questions provided on next page
Practice Question Answers
1. The parents of a 2-month-old infant present with their child. They are worried because the infant has “acne” that is not going away. Friends told them to try gentle cleansers and they have avoided using lotions or cream on her face. However, the bumps will not go away. On examination she has papules and pustules. Comedones cannot be identified. What are your next steps?
a. adapalene cream 0.1% every night at bedtime
b. benzoyl peroxide cream 4%
c. benzoyl peroxide wash 2.5%
d. erythromycin gel 2%
e. ketoconazole cream 2% twice daily
2. While in the newborn nursery prior to discharge, the attending pediatrician notices a rash on a 2-day-old neonate who is otherwise completely healthy. The pediatrician consults a dermatologist for his/her opinion. The dermatologist sees erythematous macules with central pustules located predominately on the trunk and proximal extremities. A pustule is unroofed with a blade, the contents smeared on a glass slide, and a Giemsa stain is performed. What is the predominant cell type you would expect to see on histological examination?
a. eosinophils
b. Langerhans cells
c. lymphocytes
d. neutrophils
e. no cells are visualized
3. Shortly after delivery, the pediatricians notice that the baby has numerous hyperpigmented macules on the back. No other primary lesions are seen. The neonate is otherwise normal in appearance and nontoxic appearing. A dermatologist is consulted for a recommendation for further workup or potential biopsy. The dermatologist examines the newborn. He is a well-appearing black boy with skin that is otherwise intact. A few pustules on the back are present that have a collarette of scale. The dermatologist reviews the mother’s prenatal history and the review shows that she was screened for syphilis and had a negative screening test with no other history of infectious diseases. What is the most appropriate next step to confirm your suspicions?
a. do a swab of a pustule and send it for viral culture
b. have his blood drawn and check for signs of neonatal herpes simplex virus infection
c. perform a biopsy of a pustule
d. perform a Giemsa stain on a smear of the pustule
e. start treatment with permethrin
4. Which intraoral cysts occur on the alveolar ridge of a neonate?
a. Bohn nodule
b. branchial cleft cyst
c. Epstein pearls
d. median raphe cyst
e. palatal cysts of the newborn
5. Miliaria rubra is associated with inflammation of the sweat glands in what portion of the skin?
a. basement membrane zone
b. dermis
c. dermoepidermal junction
d. intraepidermal
e. subcutis
Review the PDF of the fact sheet on transient benign neonatal skin findings with board-relevant, easy-to-review material. This fact sheet lists benign findings that can be seen in neonates and infants.
Practice Questions
1. The parents of a 2-month-old infant present with their child. They are worried because the infant has “acne” that is not going away. Friends told them to try gentle cleansers and they have avoided using lotions or cream on her face. However, the bumps will not go away. On examination she has papules and pustules. Comedones cannot be identified. What are your next steps?
a. adapalene cream 0.1% every night at bedtime
b. benzoyl peroxide cream 4%
c. benzoyl peroxide wash 2.5%
d. erythromycin gel 2%
e. ketoconazole cream 2% twice daily
2. While in the newborn nursery prior to discharge, the attending pediatrician notices a rash on a 2-day-old neonate who is otherwise completely healthy. The pediatrician consults a dermatologist for his/her opinion. The dermatologist sees erythematous macules with central pustules located predominately on the trunk and proximal extremities. A pustule is unroofed with a blade, the contents smeared on a glass slide, and a Giemsa stain is performed. What is the predominant cell type you would expect to see on histological examination?
a. eosinophils
b. Langerhans cells
c. lymphocytes
d. neutrophils
e. no cells are visualized
3. Shortly after delivery, the pediatricians notice that the baby has numerous hyperpigmented macules on the back. No other primary lesions are seen. The neonate is otherwise normal in appearance and nontoxic appearing. A dermatologist is consulted for a recommendation for further workup or potential biopsy. The dermatologist examines the newborn. He is a well-appearing black boy with skin that is otherwise intact. A few pustules on the back are present that have a collarette of scale. The dermatologist reviews the mother’s prenatal history and the review shows that she was screened for syphilis and had a negative screening test with no other history of infectious diseases. What is the most appropriate next step to confirm your suspicions?
a. do a swab of a pustule and send it for viral culture
b. have his blood drawn and check for signs of neonatal herpes simplex virus infection
c. perform a biopsy of a pustule
d. perform a Giemsa stain on a smear of the pustule
e. start treatment with permethrin
4. Which intraoral cysts occur on the alveolar ridge of a neonate?
a. Bohn nodule
b. branchial cleft cyst
c. Epstein pearls
d. median raphe cyst
e. palatal cysts of the newborn
5. Miliaria rubra is associated with inflammation of the sweat glands in what portion of the skin?
a. basement membrane zone
b. dermis
c. dermoepidermal junction
d. intraepidermal
e. subcutis
Answers to practice questions provided on next page
Practice Question Answers
1. The parents of a 2-month-old infant present with their child. They are worried because the infant has “acne” that is not going away. Friends told them to try gentle cleansers and they have avoided using lotions or cream on her face. However, the bumps will not go away. On examination she has papules and pustules. Comedones cannot be identified. What are your next steps?
a. adapalene cream 0.1% every night at bedtime
b. benzoyl peroxide cream 4%
c. benzoyl peroxide wash 2.5%
d. erythromycin gel 2%
e. ketoconazole cream 2% twice daily
2. While in the newborn nursery prior to discharge, the attending pediatrician notices a rash on a 2-day-old neonate who is otherwise completely healthy. The pediatrician consults a dermatologist for his/her opinion. The dermatologist sees erythematous macules with central pustules located predominately on the trunk and proximal extremities. A pustule is unroofed with a blade, the contents smeared on a glass slide, and a Giemsa stain is performed. What is the predominant cell type you would expect to see on histological examination?
a. eosinophils
b. Langerhans cells
c. lymphocytes
d. neutrophils
e. no cells are visualized
3. Shortly after delivery, the pediatricians notice that the baby has numerous hyperpigmented macules on the back. No other primary lesions are seen. The neonate is otherwise normal in appearance and nontoxic appearing. A dermatologist is consulted for a recommendation for further workup or potential biopsy. The dermatologist examines the newborn. He is a well-appearing black boy with skin that is otherwise intact. A few pustules on the back are present that have a collarette of scale. The dermatologist reviews the mother’s prenatal history and the review shows that she was screened for syphilis and had a negative screening test with no other history of infectious diseases. What is the most appropriate next step to confirm your suspicions?
a. do a swab of a pustule and send it for viral culture
b. have his blood drawn and check for signs of neonatal herpes simplex virus infection
c. perform a biopsy of a pustule
d. perform a Giemsa stain on a smear of the pustule
e. start treatment with permethrin
4. Which intraoral cysts occur on the alveolar ridge of a neonate?
a. Bohn nodule
b. branchial cleft cyst
c. Epstein pearls
d. median raphe cyst
e. palatal cysts of the newborn
5. Miliaria rubra is associated with inflammation of the sweat glands in what portion of the skin?
a. basement membrane zone
b. dermis
c. dermoepidermal junction
d. intraepidermal
e. subcutis
Review the PDF of the fact sheet on transient benign neonatal skin findings with board-relevant, easy-to-review material. This fact sheet lists benign findings that can be seen in neonates and infants.
Practice Questions
1. The parents of a 2-month-old infant present with their child. They are worried because the infant has “acne” that is not going away. Friends told them to try gentle cleansers and they have avoided using lotions or cream on her face. However, the bumps will not go away. On examination she has papules and pustules. Comedones cannot be identified. What are your next steps?
a. adapalene cream 0.1% every night at bedtime
b. benzoyl peroxide cream 4%
c. benzoyl peroxide wash 2.5%
d. erythromycin gel 2%
e. ketoconazole cream 2% twice daily
2. While in the newborn nursery prior to discharge, the attending pediatrician notices a rash on a 2-day-old neonate who is otherwise completely healthy. The pediatrician consults a dermatologist for his/her opinion. The dermatologist sees erythematous macules with central pustules located predominately on the trunk and proximal extremities. A pustule is unroofed with a blade, the contents smeared on a glass slide, and a Giemsa stain is performed. What is the predominant cell type you would expect to see on histological examination?
a. eosinophils
b. Langerhans cells
c. lymphocytes
d. neutrophils
e. no cells are visualized
3. Shortly after delivery, the pediatricians notice that the baby has numerous hyperpigmented macules on the back. No other primary lesions are seen. The neonate is otherwise normal in appearance and nontoxic appearing. A dermatologist is consulted for a recommendation for further workup or potential biopsy. The dermatologist examines the newborn. He is a well-appearing black boy with skin that is otherwise intact. A few pustules on the back are present that have a collarette of scale. The dermatologist reviews the mother’s prenatal history and the review shows that she was screened for syphilis and had a negative screening test with no other history of infectious diseases. What is the most appropriate next step to confirm your suspicions?
a. do a swab of a pustule and send it for viral culture
b. have his blood drawn and check for signs of neonatal herpes simplex virus infection
c. perform a biopsy of a pustule
d. perform a Giemsa stain on a smear of the pustule
e. start treatment with permethrin
4. Which intraoral cysts occur on the alveolar ridge of a neonate?
a. Bohn nodule
b. branchial cleft cyst
c. Epstein pearls
d. median raphe cyst
e. palatal cysts of the newborn
5. Miliaria rubra is associated with inflammation of the sweat glands in what portion of the skin?
a. basement membrane zone
b. dermis
c. dermoepidermal junction
d. intraepidermal
e. subcutis
Answers to practice questions provided on next page
Practice Question Answers
1. The parents of a 2-month-old infant present with their child. They are worried because the infant has “acne” that is not going away. Friends told them to try gentle cleansers and they have avoided using lotions or cream on her face. However, the bumps will not go away. On examination she has papules and pustules. Comedones cannot be identified. What are your next steps?
a. adapalene cream 0.1% every night at bedtime
b. benzoyl peroxide cream 4%
c. benzoyl peroxide wash 2.5%
d. erythromycin gel 2%
e. ketoconazole cream 2% twice daily
2. While in the newborn nursery prior to discharge, the attending pediatrician notices a rash on a 2-day-old neonate who is otherwise completely healthy. The pediatrician consults a dermatologist for his/her opinion. The dermatologist sees erythematous macules with central pustules located predominately on the trunk and proximal extremities. A pustule is unroofed with a blade, the contents smeared on a glass slide, and a Giemsa stain is performed. What is the predominant cell type you would expect to see on histological examination?
a. eosinophils
b. Langerhans cells
c. lymphocytes
d. neutrophils
e. no cells are visualized
3. Shortly after delivery, the pediatricians notice that the baby has numerous hyperpigmented macules on the back. No other primary lesions are seen. The neonate is otherwise normal in appearance and nontoxic appearing. A dermatologist is consulted for a recommendation for further workup or potential biopsy. The dermatologist examines the newborn. He is a well-appearing black boy with skin that is otherwise intact. A few pustules on the back are present that have a collarette of scale. The dermatologist reviews the mother’s prenatal history and the review shows that she was screened for syphilis and had a negative screening test with no other history of infectious diseases. What is the most appropriate next step to confirm your suspicions?
a. do a swab of a pustule and send it for viral culture
b. have his blood drawn and check for signs of neonatal herpes simplex virus infection
c. perform a biopsy of a pustule
d. perform a Giemsa stain on a smear of the pustule
e. start treatment with permethrin
4. Which intraoral cysts occur on the alveolar ridge of a neonate?
a. Bohn nodule
b. branchial cleft cyst
c. Epstein pearls
d. median raphe cyst
e. palatal cysts of the newborn
5. Miliaria rubra is associated with inflammation of the sweat glands in what portion of the skin?
a. basement membrane zone
b. dermis
c. dermoepidermal junction
d. intraepidermal
e. subcutis
