User login
Understanding a rare hemoglobin mutation

Smoking can prevent anemia in individuals with a rare hemoglobin mutation, according to research published in the Journal of Biological Chemistry.
The so-called Kirklareli mutation was found to be the cause of mild anemia in a young woman in Germany.
But a smoking habit protected the young woman’s father, who also carried the mutation, from developing anemia.
The Kirklareli mutation is one of more than 1000 discovered so far in adult human hemoglobin.
Most of these mutations appear to have no effect on people, but when medical problems occur, the disease is called a hemoglobinopathy and often named after the city or hospital where it was discovered. In this case, the family was living in Mannheim, Germany, but the father was born in the Turkish city of Kirklareli.
The Kirklareli mutation did not affect the iron content of the father’s blood, but it did appear to be the root cause of the young woman’s chronic anemia, according to researchers.
Further investigation revealed that absorbing carbon monoxide from cigarette smoke is therapeutic for individuals with this rare genetic disorder.
The Kirklareli mutation is in the alpha subunit of human hemoglobin (H58L) and causes it to rapidly auto-oxidize, which causes the protein to fall apart, lose heme, and precipitate. As a result, the protein loses its ability to carry oxygen. Eventually, red cells become deformed and are destroyed.
This mutation also gives the protein an 80,000-fold higher affinity for carbon monoxide than for oxygen. Carbon monoxide from a cigarette will be selectively taken up by the mutant hemoglobin and prevent it from oxidizing and denaturing.
This high affinity for carbon monoxide explained why the father showed no signs of anemia, the researchers said.
“He may never be an athlete because his blood can’t carry as much oxygen, but smoking has prevented him from being anemic,” said study author John Olson, PhD, of Rice University in Houston, Texas.
“And there’s a side benefit. People with this trait are more resistant to carbon monoxide poisoning.”
Dr Olson said he doesn’t know how or if doctors treated the young woman, but he suspects her iron-deficiency anemia was more an annoyance than a threat to her life and would not recommend she start smoking to relieve it.
“She shouldn’t smoke,” Dr Olson said. “But she could take antioxidants, such as a lot of vitamin C, which would help prevent oxidation of her mutant hemoglobin. Her anemia is not that severe. At the same time, she shouldn’t worry too much about secondhand smoke, which might have a positive effect.”
After ruling out common causes of anemia—such as blood loss, gastritis, or congenital defects—the woman’s doctors were curious enough about her ailment to call upon Emmanuel Bissé, MD, PhD, a researcher at Universitätsklinikum Freiburg in Freiburg, Germany, who discovered the Kirklareli mutation after sequencing the woman’s DNA.
Dr Bissé, in turn, recruited Dr Olson and his team to help determine why the histidine-to-leucine change caused anemia in the daughter but not the father.
Coincidentally, Ivan Birukou, a graduate student in Dr Olson’s lab, had already generated the Kirklareli mutation in human hemoglobin to study how the protein rapidly and selectively binds oxygen.
“Emmanuel wrote to me and said, ‘I know you’ve been making all these mutants in hemoglobin, and you’ve probably done the H58L mutation in [alpha] chains. Does this phenotype make sense?’” Dr Olson recalled.
“I said, ‘We can do a really neat study here, because we’ve already made the mutant hemoglobin in a recombinant system.’ We actually had a crystal structure [matching Kirklareli] that Ivan and [staff scientist] Jayashree Soman never published but had deposited in the Protein Data Bank. We had made this mutation to try to understand what the distal histidine was doing in alpha subunits.”
The researchers found in a 2010 study that replacing the histidine, which forms a strong hydrogen bond to oxygen, with leucine caused a dramatic decrease in oxygen affinity and an increase in carbon monoxide binding.
Dr Olson and Birukou realized back then that histidine played a key role in discriminating between oxygen and carbon monoxide in hemoglobin.
“When Emmanuel wrote to me about his discovery, I already ‘knew’ what was happening with respect to carbon monoxide binding,” Dr Olson said.
He said the normal hydrogen bond causes bound oxygen to stick more tightly to hemoglobin in the same way hydrogen bonds cause spilled soda to feel sticky.
“When you touch it, the sugar oxygens and hydrogens make hydrogen bonds with the polysaccharides on your finger,” Dr Olson said. “That stickiness helps hold onto oxygen. But leucine is more like an oil, like butane or hexane, and oxygen does not stick well inside hemoglobin. In contrast, bound carbon monoxide is more like methane or ethane and can’t form hydrogen bonds.”
Andres Benitez Cardenas, PhD, a researcher in Dr Olson’s lab, did the experiment in which he put carbon monoxide on the mutant alpha subunit of hemoglobin Kirklareli. The bound carbon monoxide slowed down oxidation of the protein and prevented loss of heme and precipitation.
“In effect, Andres did the ‘smoking experiment’ to show why the father’s hemoglobin didn’t denature and cause anemia,” Dr Olson said.
He noted that the effect caused by Kirklareli, though unusual, is not unique. Patients with hemoglobin Zurich also have an abnormal form of hemoglobin that more readily binds to carbon monoxide. ![]()
Smoking can prevent anemia in individuals with a rare hemoglobin mutation, according to research published in the Journal of Biological Chemistry.
The so-called Kirklareli mutation was found to be the cause of mild anemia in a young woman in Germany.
But a smoking habit protected the young woman’s father, who also carried the mutation, from developing anemia.
The Kirklareli mutation is one of more than 1000 discovered so far in adult human hemoglobin.
Most of these mutations appear to have no effect on people, but when medical problems occur, the disease is called a hemoglobinopathy and often named after the city or hospital where it was discovered. In this case, the family was living in Mannheim, Germany, but the father was born in the Turkish city of Kirklareli.
The Kirklareli mutation did not affect the iron content of the father’s blood, but it did appear to be the root cause of the young woman’s chronic anemia, according to researchers.
Further investigation revealed that absorbing carbon monoxide from cigarette smoke is therapeutic for individuals with this rare genetic disorder.
The Kirklareli mutation is in the alpha subunit of human hemoglobin (H58L) and causes it to rapidly auto-oxidize, which causes the protein to fall apart, lose heme, and precipitate. As a result, the protein loses its ability to carry oxygen. Eventually, red cells become deformed and are destroyed.
This mutation also gives the protein an 80,000-fold higher affinity for carbon monoxide than for oxygen. Carbon monoxide from a cigarette will be selectively taken up by the mutant hemoglobin and prevent it from oxidizing and denaturing.
This high affinity for carbon monoxide explained why the father showed no signs of anemia, the researchers said.
“He may never be an athlete because his blood can’t carry as much oxygen, but smoking has prevented him from being anemic,” said study author John Olson, PhD, of Rice University in Houston, Texas.
“And there’s a side benefit. People with this trait are more resistant to carbon monoxide poisoning.”
Dr Olson said he doesn’t know how or if doctors treated the young woman, but he suspects her iron-deficiency anemia was more an annoyance than a threat to her life and would not recommend she start smoking to relieve it.
“She shouldn’t smoke,” Dr Olson said. “But she could take antioxidants, such as a lot of vitamin C, which would help prevent oxidation of her mutant hemoglobin. Her anemia is not that severe. At the same time, she shouldn’t worry too much about secondhand smoke, which might have a positive effect.”
After ruling out common causes of anemia—such as blood loss, gastritis, or congenital defects—the woman’s doctors were curious enough about her ailment to call upon Emmanuel Bissé, MD, PhD, a researcher at Universitätsklinikum Freiburg in Freiburg, Germany, who discovered the Kirklareli mutation after sequencing the woman’s DNA.
Dr Bissé, in turn, recruited Dr Olson and his team to help determine why the histidine-to-leucine change caused anemia in the daughter but not the father.
Coincidentally, Ivan Birukou, a graduate student in Dr Olson’s lab, had already generated the Kirklareli mutation in human hemoglobin to study how the protein rapidly and selectively binds oxygen.
“Emmanuel wrote to me and said, ‘I know you’ve been making all these mutants in hemoglobin, and you’ve probably done the H58L mutation in [alpha] chains. Does this phenotype make sense?’” Dr Olson recalled.
“I said, ‘We can do a really neat study here, because we’ve already made the mutant hemoglobin in a recombinant system.’ We actually had a crystal structure [matching Kirklareli] that Ivan and [staff scientist] Jayashree Soman never published but had deposited in the Protein Data Bank. We had made this mutation to try to understand what the distal histidine was doing in alpha subunits.”
The researchers found in a 2010 study that replacing the histidine, which forms a strong hydrogen bond to oxygen, with leucine caused a dramatic decrease in oxygen affinity and an increase in carbon monoxide binding.
Dr Olson and Birukou realized back then that histidine played a key role in discriminating between oxygen and carbon monoxide in hemoglobin.
“When Emmanuel wrote to me about his discovery, I already ‘knew’ what was happening with respect to carbon monoxide binding,” Dr Olson said.
He said the normal hydrogen bond causes bound oxygen to stick more tightly to hemoglobin in the same way hydrogen bonds cause spilled soda to feel sticky.
“When you touch it, the sugar oxygens and hydrogens make hydrogen bonds with the polysaccharides on your finger,” Dr Olson said. “That stickiness helps hold onto oxygen. But leucine is more like an oil, like butane or hexane, and oxygen does not stick well inside hemoglobin. In contrast, bound carbon monoxide is more like methane or ethane and can’t form hydrogen bonds.”
Andres Benitez Cardenas, PhD, a researcher in Dr Olson’s lab, did the experiment in which he put carbon monoxide on the mutant alpha subunit of hemoglobin Kirklareli. The bound carbon monoxide slowed down oxidation of the protein and prevented loss of heme and precipitation.
“In effect, Andres did the ‘smoking experiment’ to show why the father’s hemoglobin didn’t denature and cause anemia,” Dr Olson said.
He noted that the effect caused by Kirklareli, though unusual, is not unique. Patients with hemoglobin Zurich also have an abnormal form of hemoglobin that more readily binds to carbon monoxide. ![]()
Smoking can prevent anemia in individuals with a rare hemoglobin mutation, according to research published in the Journal of Biological Chemistry.
The so-called Kirklareli mutation was found to be the cause of mild anemia in a young woman in Germany.
But a smoking habit protected the young woman’s father, who also carried the mutation, from developing anemia.
The Kirklareli mutation is one of more than 1000 discovered so far in adult human hemoglobin.
Most of these mutations appear to have no effect on people, but when medical problems occur, the disease is called a hemoglobinopathy and often named after the city or hospital where it was discovered. In this case, the family was living in Mannheim, Germany, but the father was born in the Turkish city of Kirklareli.
The Kirklareli mutation did not affect the iron content of the father’s blood, but it did appear to be the root cause of the young woman’s chronic anemia, according to researchers.
Further investigation revealed that absorbing carbon monoxide from cigarette smoke is therapeutic for individuals with this rare genetic disorder.
The Kirklareli mutation is in the alpha subunit of human hemoglobin (H58L) and causes it to rapidly auto-oxidize, which causes the protein to fall apart, lose heme, and precipitate. As a result, the protein loses its ability to carry oxygen. Eventually, red cells become deformed and are destroyed.
This mutation also gives the protein an 80,000-fold higher affinity for carbon monoxide than for oxygen. Carbon monoxide from a cigarette will be selectively taken up by the mutant hemoglobin and prevent it from oxidizing and denaturing.
This high affinity for carbon monoxide explained why the father showed no signs of anemia, the researchers said.
“He may never be an athlete because his blood can’t carry as much oxygen, but smoking has prevented him from being anemic,” said study author John Olson, PhD, of Rice University in Houston, Texas.
“And there’s a side benefit. People with this trait are more resistant to carbon monoxide poisoning.”
Dr Olson said he doesn’t know how or if doctors treated the young woman, but he suspects her iron-deficiency anemia was more an annoyance than a threat to her life and would not recommend she start smoking to relieve it.
“She shouldn’t smoke,” Dr Olson said. “But she could take antioxidants, such as a lot of vitamin C, which would help prevent oxidation of her mutant hemoglobin. Her anemia is not that severe. At the same time, she shouldn’t worry too much about secondhand smoke, which might have a positive effect.”
After ruling out common causes of anemia—such as blood loss, gastritis, or congenital defects—the woman’s doctors were curious enough about her ailment to call upon Emmanuel Bissé, MD, PhD, a researcher at Universitätsklinikum Freiburg in Freiburg, Germany, who discovered the Kirklareli mutation after sequencing the woman’s DNA.
Dr Bissé, in turn, recruited Dr Olson and his team to help determine why the histidine-to-leucine change caused anemia in the daughter but not the father.
Coincidentally, Ivan Birukou, a graduate student in Dr Olson’s lab, had already generated the Kirklareli mutation in human hemoglobin to study how the protein rapidly and selectively binds oxygen.
“Emmanuel wrote to me and said, ‘I know you’ve been making all these mutants in hemoglobin, and you’ve probably done the H58L mutation in [alpha] chains. Does this phenotype make sense?’” Dr Olson recalled.
“I said, ‘We can do a really neat study here, because we’ve already made the mutant hemoglobin in a recombinant system.’ We actually had a crystal structure [matching Kirklareli] that Ivan and [staff scientist] Jayashree Soman never published but had deposited in the Protein Data Bank. We had made this mutation to try to understand what the distal histidine was doing in alpha subunits.”
The researchers found in a 2010 study that replacing the histidine, which forms a strong hydrogen bond to oxygen, with leucine caused a dramatic decrease in oxygen affinity and an increase in carbon monoxide binding.
Dr Olson and Birukou realized back then that histidine played a key role in discriminating between oxygen and carbon monoxide in hemoglobin.
“When Emmanuel wrote to me about his discovery, I already ‘knew’ what was happening with respect to carbon monoxide binding,” Dr Olson said.
He said the normal hydrogen bond causes bound oxygen to stick more tightly to hemoglobin in the same way hydrogen bonds cause spilled soda to feel sticky.
“When you touch it, the sugar oxygens and hydrogens make hydrogen bonds with the polysaccharides on your finger,” Dr Olson said. “That stickiness helps hold onto oxygen. But leucine is more like an oil, like butane or hexane, and oxygen does not stick well inside hemoglobin. In contrast, bound carbon monoxide is more like methane or ethane and can’t form hydrogen bonds.”
Andres Benitez Cardenas, PhD, a researcher in Dr Olson’s lab, did the experiment in which he put carbon monoxide on the mutant alpha subunit of hemoglobin Kirklareli. The bound carbon monoxide slowed down oxidation of the protein and prevented loss of heme and precipitation.
“In effect, Andres did the ‘smoking experiment’ to show why the father’s hemoglobin didn’t denature and cause anemia,” Dr Olson said.
He noted that the effect caused by Kirklareli, though unusual, is not unique. Patients with hemoglobin Zurich also have an abnormal form of hemoglobin that more readily binds to carbon monoxide. ![]()
Mitigating Stress Levels May Impact Seizures
Clinicians would be wise to recommend stress reduction techniques to patients with epilepsy, despite the fact that randomized controlled trials have yet to demonstrate that stress management reduces the frequency of seizures. One survey has suggested that most patients who report stress-triggered seizures use some sort of stress reduction methods and most say they are effective. McKee et al also point out that studies have found that stress management does improve quality of life in this patient population. The investigators also recommended that stressed patients with epilepsy should be screened for depression, anxiety, and other treatable mood disorders since they are more common in these patients.
McKee HR, Privitera MD. Stress as a seizure precipitant: Identification, associated factors, and treatment options. Seizure. 2017; 44:21-26.
Clinicians would be wise to recommend stress reduction techniques to patients with epilepsy, despite the fact that randomized controlled trials have yet to demonstrate that stress management reduces the frequency of seizures. One survey has suggested that most patients who report stress-triggered seizures use some sort of stress reduction methods and most say they are effective. McKee et al also point out that studies have found that stress management does improve quality of life in this patient population. The investigators also recommended that stressed patients with epilepsy should be screened for depression, anxiety, and other treatable mood disorders since they are more common in these patients.
McKee HR, Privitera MD. Stress as a seizure precipitant: Identification, associated factors, and treatment options. Seizure. 2017; 44:21-26.
Clinicians would be wise to recommend stress reduction techniques to patients with epilepsy, despite the fact that randomized controlled trials have yet to demonstrate that stress management reduces the frequency of seizures. One survey has suggested that most patients who report stress-triggered seizures use some sort of stress reduction methods and most say they are effective. McKee et al also point out that studies have found that stress management does improve quality of life in this patient population. The investigators also recommended that stressed patients with epilepsy should be screened for depression, anxiety, and other treatable mood disorders since they are more common in these patients.
McKee HR, Privitera MD. Stress as a seizure precipitant: Identification, associated factors, and treatment options. Seizure. 2017; 44:21-26.
Restless Leg Syndrome More Common in Temporal Lobe Epilepsy
Patients with temporal lobe epilepsy (TLE) are more likely to experience restless leg syndrome (RLS) than the general public, according to a recent outpatient clinic analysis, which compared 98 TLE patients to 50 controls who did not have a history of epilepsy or any family members with the disorder. The investigators also found that the odds of developing RLS were 4.6 times greater in patients with right-sided TLE, when compared to left-sided TLE. They also suggested that worsening RLS may serve as an early warning of an impending seizure in some patients.
Geyer JD, Geyer EE, Fetterman Z, Carney PR. Epilepsy and restless legs syndrome. Epilepsy Behav. 2017;68:41-44.
Patients with temporal lobe epilepsy (TLE) are more likely to experience restless leg syndrome (RLS) than the general public, according to a recent outpatient clinic analysis, which compared 98 TLE patients to 50 controls who did not have a history of epilepsy or any family members with the disorder. The investigators also found that the odds of developing RLS were 4.6 times greater in patients with right-sided TLE, when compared to left-sided TLE. They also suggested that worsening RLS may serve as an early warning of an impending seizure in some patients.
Geyer JD, Geyer EE, Fetterman Z, Carney PR. Epilepsy and restless legs syndrome. Epilepsy Behav. 2017;68:41-44.
Patients with temporal lobe epilepsy (TLE) are more likely to experience restless leg syndrome (RLS) than the general public, according to a recent outpatient clinic analysis, which compared 98 TLE patients to 50 controls who did not have a history of epilepsy or any family members with the disorder. The investigators also found that the odds of developing RLS were 4.6 times greater in patients with right-sided TLE, when compared to left-sided TLE. They also suggested that worsening RLS may serve as an early warning of an impending seizure in some patients.
Geyer JD, Geyer EE, Fetterman Z, Carney PR. Epilepsy and restless legs syndrome. Epilepsy Behav. 2017;68:41-44.
Half of Patients With Epilepsy Do Not Receive Medication Soon Enough
A recent analysis of Medicare records has found that among 3706 new cases of epilepsy, 79.6% had received 1 antiepilepsy drug within 1 year of follow-up. However, only 50% of patients had received prompt therapy, which was defined as receiving the first medication within 30 days of diagnosis. The delay in initiating monotherapy was detected when researchers performed retrospective analyses of 2008–2010 Medicare administrative claims that were obtained from a 5% random sample of patients. The investigators have called for additional research to determine the reasons for the delays and have urged the development of new paradigms to improve patient care.
Martin RC, Faught E, Szaflarski JP, et al. What does the U.S. Medicare administrative claims database tell us about initial antiepileptic drug treatment for older adults with new-onset epilepsy? Epilepsia. 2017[Epub ahead of print]
A recent analysis of Medicare records has found that among 3706 new cases of epilepsy, 79.6% had received 1 antiepilepsy drug within 1 year of follow-up. However, only 50% of patients had received prompt therapy, which was defined as receiving the first medication within 30 days of diagnosis. The delay in initiating monotherapy was detected when researchers performed retrospective analyses of 2008–2010 Medicare administrative claims that were obtained from a 5% random sample of patients. The investigators have called for additional research to determine the reasons for the delays and have urged the development of new paradigms to improve patient care.
Martin RC, Faught E, Szaflarski JP, et al. What does the U.S. Medicare administrative claims database tell us about initial antiepileptic drug treatment for older adults with new-onset epilepsy? Epilepsia. 2017[Epub ahead of print]
A recent analysis of Medicare records has found that among 3706 new cases of epilepsy, 79.6% had received 1 antiepilepsy drug within 1 year of follow-up. However, only 50% of patients had received prompt therapy, which was defined as receiving the first medication within 30 days of diagnosis. The delay in initiating monotherapy was detected when researchers performed retrospective analyses of 2008–2010 Medicare administrative claims that were obtained from a 5% random sample of patients. The investigators have called for additional research to determine the reasons for the delays and have urged the development of new paradigms to improve patient care.
Martin RC, Faught E, Szaflarski JP, et al. What does the U.S. Medicare administrative claims database tell us about initial antiepileptic drug treatment for older adults with new-onset epilepsy? Epilepsia. 2017[Epub ahead of print]
Urgent colonoscopy for LGIB: Consider case by case
Colonoscopy performed within 24 hours of lower gastrointestinal bleeding appears safe and well tolerated but does not appear to improve a number of important clinical outcomes when compared with elective colonoscopy, according to the findings of a systematic review and meta-analysis.
Such “urgent colonoscopy” may, however, reduce hospital length of stay and cost, Abdul M. Kouanda, MD, of the University of California, San Francisco, and his colleagues reported online in Gastrointestinal Endoscopy.
In a pooled analysis of data from 12 studies with a total of 10,172 patients who underwent urgent colonoscopy, and 14,224 patients who underwent elective colonoscopy, the former was associated with increased use of endoscopic therapeutic interventions, compared with elective colonoscopy (relative risk, 1.70), but not with improved bleeding source localization (RR, 1.08), adverse event rates (RR, 1.05), rebleeding rates (RR, 1.14), transfusion requirements (RR, 1.02), or mortality (RR, 1.17), the investigators found (Gastrointest Endosc. 2017 Feb 4. doi: 10.1016/j.gie.2017.01.035).
The findings are based on nine studies from the United States, two from Japan, and one from Spain. Nine were retrospective cohort studies, two were randomized controlled trials, and one was a prospective cohort study.
With respect to the 70% greater use of therapeutic interventions with urgent colonoscopy, a subanalysis showed that the difference between urgent and elective colonoscopy was evident only in the randomized trials; no difference was seen in the prospective trials. With further stratification of urgent colonoscopy into procedures performed within 12 hours, the observation of increased therapeutic interventions was no longer statistically significant (RR, 3.46), they said.
As for bleeding source localization, the outcomes remained similar when retrospective studies were analyzed separately, and with colonoscopy performed within 12 hours. Blood transfusions decreased with urgent colonoscopy when only retrospective studies were analyzed (RR, 0.84).
The investigators noted that there was a trend toward decreased length of hospital stay among those undergoing urgent colonoscopy (mean of 4.8 days vs. 6.4 days with elective colonoscopy). Only two studies looked at cost: One showed a decrease in hospital costs with urgent vs. elective colonoscopy, while one showed no difference.
“In our pooled analysis, the mean hospital costs in the urgent colonoscopy group were $24,866, compared with $27,691 in the elective group; however, the difference between the two was not statistically significant,” they wrote.
The annual incidence of lower gastrointestinal bleeding (LGIB) in the United States is 20.5-35.7 out of 100,000 patients, and the incidence increases with age; there is a 200-fold increase in incidence from the 3rd to 9th decade of life, the investigators said, adding that the incidence is rising as the population ages.
“Such a trend has important implications for both the quality of care for treating LGIB and the associated costs to the overall U.S. health care system,” they wrote, noting that while colonoscopy is appropriate for evaluating LGIB in most cases, no clear consensus exists with respect to timing of colonoscopy.
Even a recent American Society for Gastrointestinal Endoscopy guideline recommending that initial colonoscopy for severe and hemodynamically stable hematochezia be performed within 8-24 hours of admission is based only on moderate-quality level evidence that is “fraught with a number of limitations,” they wrote.
The current study was designed to “further clarify the utility of urgent versus elective colonoscopy in evaluating patients hospitalized with a lower GI bleed,” they added.
The lack of clinical benefit seen in this study “may be secondary to the benign, often self-resolving natural history in the majority of LGIB cases. However, there may be a subset of patients who could benefit from early intervention (such as severe blood loss, hemodynamically unstable patients), and thus the decision to pursue urgent colonoscopy should be made on a case-by-case basis,” they said.
Further, although several critical patient outcomes did not appear to be impacted by urgent vs. elective colonoscopy in this study, the trends toward a decrease in length of stay suggest that earlier performance of colonoscopy may lead to earlier and better identification of low-risk and high-risk stigmata, allowing those with low-risk lesions to be discharged much earlier.
“Additionally, earlier discharge of patients could also reduce their risk of health care–associated infections and adverse events,” the investigators noted.
The findings with respect to length of stay and cost “align perfectly with the new focus in health care on providing high quality and safe care to patients while at the same time containing medical costs,” they wrote, adding that clinicians should carefully consider all factors when deciding to pursue urgent colonoscopy.”
The study is limited by heterogeneity and publications bias, and by factors inherent in meta-analyses, but it also has several strengths, including a large number of studies and patients. Also, it is the first of its kind to examine “all of the available literature to elucidate the time frame for performing colonoscopy in patients with hematochezia,” the investigators said, concluding that further research is needed to identify subsets of patients who will benefit from early intervention, to evaluate the cost effectiveness of urgent colonoscopy, and to look at – in larger randomized controlled trials – the overall benefit of urgent colonoscopy.
The authors reported having no disclosures.
Colonoscopy performed within 24 hours of lower gastrointestinal bleeding appears safe and well tolerated but does not appear to improve a number of important clinical outcomes when compared with elective colonoscopy, according to the findings of a systematic review and meta-analysis.
Such “urgent colonoscopy” may, however, reduce hospital length of stay and cost, Abdul M. Kouanda, MD, of the University of California, San Francisco, and his colleagues reported online in Gastrointestinal Endoscopy.
In a pooled analysis of data from 12 studies with a total of 10,172 patients who underwent urgent colonoscopy, and 14,224 patients who underwent elective colonoscopy, the former was associated with increased use of endoscopic therapeutic interventions, compared with elective colonoscopy (relative risk, 1.70), but not with improved bleeding source localization (RR, 1.08), adverse event rates (RR, 1.05), rebleeding rates (RR, 1.14), transfusion requirements (RR, 1.02), or mortality (RR, 1.17), the investigators found (Gastrointest Endosc. 2017 Feb 4. doi: 10.1016/j.gie.2017.01.035).
The findings are based on nine studies from the United States, two from Japan, and one from Spain. Nine were retrospective cohort studies, two were randomized controlled trials, and one was a prospective cohort study.
With respect to the 70% greater use of therapeutic interventions with urgent colonoscopy, a subanalysis showed that the difference between urgent and elective colonoscopy was evident only in the randomized trials; no difference was seen in the prospective trials. With further stratification of urgent colonoscopy into procedures performed within 12 hours, the observation of increased therapeutic interventions was no longer statistically significant (RR, 3.46), they said.
As for bleeding source localization, the outcomes remained similar when retrospective studies were analyzed separately, and with colonoscopy performed within 12 hours. Blood transfusions decreased with urgent colonoscopy when only retrospective studies were analyzed (RR, 0.84).
The investigators noted that there was a trend toward decreased length of hospital stay among those undergoing urgent colonoscopy (mean of 4.8 days vs. 6.4 days with elective colonoscopy). Only two studies looked at cost: One showed a decrease in hospital costs with urgent vs. elective colonoscopy, while one showed no difference.
“In our pooled analysis, the mean hospital costs in the urgent colonoscopy group were $24,866, compared with $27,691 in the elective group; however, the difference between the two was not statistically significant,” they wrote.
The annual incidence of lower gastrointestinal bleeding (LGIB) in the United States is 20.5-35.7 out of 100,000 patients, and the incidence increases with age; there is a 200-fold increase in incidence from the 3rd to 9th decade of life, the investigators said, adding that the incidence is rising as the population ages.
“Such a trend has important implications for both the quality of care for treating LGIB and the associated costs to the overall U.S. health care system,” they wrote, noting that while colonoscopy is appropriate for evaluating LGIB in most cases, no clear consensus exists with respect to timing of colonoscopy.
Even a recent American Society for Gastrointestinal Endoscopy guideline recommending that initial colonoscopy for severe and hemodynamically stable hematochezia be performed within 8-24 hours of admission is based only on moderate-quality level evidence that is “fraught with a number of limitations,” they wrote.
The current study was designed to “further clarify the utility of urgent versus elective colonoscopy in evaluating patients hospitalized with a lower GI bleed,” they added.
The lack of clinical benefit seen in this study “may be secondary to the benign, often self-resolving natural history in the majority of LGIB cases. However, there may be a subset of patients who could benefit from early intervention (such as severe blood loss, hemodynamically unstable patients), and thus the decision to pursue urgent colonoscopy should be made on a case-by-case basis,” they said.
Further, although several critical patient outcomes did not appear to be impacted by urgent vs. elective colonoscopy in this study, the trends toward a decrease in length of stay suggest that earlier performance of colonoscopy may lead to earlier and better identification of low-risk and high-risk stigmata, allowing those with low-risk lesions to be discharged much earlier.
“Additionally, earlier discharge of patients could also reduce their risk of health care–associated infections and adverse events,” the investigators noted.
The findings with respect to length of stay and cost “align perfectly with the new focus in health care on providing high quality and safe care to patients while at the same time containing medical costs,” they wrote, adding that clinicians should carefully consider all factors when deciding to pursue urgent colonoscopy.”
The study is limited by heterogeneity and publications bias, and by factors inherent in meta-analyses, but it also has several strengths, including a large number of studies and patients. Also, it is the first of its kind to examine “all of the available literature to elucidate the time frame for performing colonoscopy in patients with hematochezia,” the investigators said, concluding that further research is needed to identify subsets of patients who will benefit from early intervention, to evaluate the cost effectiveness of urgent colonoscopy, and to look at – in larger randomized controlled trials – the overall benefit of urgent colonoscopy.
The authors reported having no disclosures.
Colonoscopy performed within 24 hours of lower gastrointestinal bleeding appears safe and well tolerated but does not appear to improve a number of important clinical outcomes when compared with elective colonoscopy, according to the findings of a systematic review and meta-analysis.
Such “urgent colonoscopy” may, however, reduce hospital length of stay and cost, Abdul M. Kouanda, MD, of the University of California, San Francisco, and his colleagues reported online in Gastrointestinal Endoscopy.
In a pooled analysis of data from 12 studies with a total of 10,172 patients who underwent urgent colonoscopy, and 14,224 patients who underwent elective colonoscopy, the former was associated with increased use of endoscopic therapeutic interventions, compared with elective colonoscopy (relative risk, 1.70), but not with improved bleeding source localization (RR, 1.08), adverse event rates (RR, 1.05), rebleeding rates (RR, 1.14), transfusion requirements (RR, 1.02), or mortality (RR, 1.17), the investigators found (Gastrointest Endosc. 2017 Feb 4. doi: 10.1016/j.gie.2017.01.035).
The findings are based on nine studies from the United States, two from Japan, and one from Spain. Nine were retrospective cohort studies, two were randomized controlled trials, and one was a prospective cohort study.
With respect to the 70% greater use of therapeutic interventions with urgent colonoscopy, a subanalysis showed that the difference between urgent and elective colonoscopy was evident only in the randomized trials; no difference was seen in the prospective trials. With further stratification of urgent colonoscopy into procedures performed within 12 hours, the observation of increased therapeutic interventions was no longer statistically significant (RR, 3.46), they said.
As for bleeding source localization, the outcomes remained similar when retrospective studies were analyzed separately, and with colonoscopy performed within 12 hours. Blood transfusions decreased with urgent colonoscopy when only retrospective studies were analyzed (RR, 0.84).
The investigators noted that there was a trend toward decreased length of hospital stay among those undergoing urgent colonoscopy (mean of 4.8 days vs. 6.4 days with elective colonoscopy). Only two studies looked at cost: One showed a decrease in hospital costs with urgent vs. elective colonoscopy, while one showed no difference.
“In our pooled analysis, the mean hospital costs in the urgent colonoscopy group were $24,866, compared with $27,691 in the elective group; however, the difference between the two was not statistically significant,” they wrote.
The annual incidence of lower gastrointestinal bleeding (LGIB) in the United States is 20.5-35.7 out of 100,000 patients, and the incidence increases with age; there is a 200-fold increase in incidence from the 3rd to 9th decade of life, the investigators said, adding that the incidence is rising as the population ages.
“Such a trend has important implications for both the quality of care for treating LGIB and the associated costs to the overall U.S. health care system,” they wrote, noting that while colonoscopy is appropriate for evaluating LGIB in most cases, no clear consensus exists with respect to timing of colonoscopy.
Even a recent American Society for Gastrointestinal Endoscopy guideline recommending that initial colonoscopy for severe and hemodynamically stable hematochezia be performed within 8-24 hours of admission is based only on moderate-quality level evidence that is “fraught with a number of limitations,” they wrote.
The current study was designed to “further clarify the utility of urgent versus elective colonoscopy in evaluating patients hospitalized with a lower GI bleed,” they added.
The lack of clinical benefit seen in this study “may be secondary to the benign, often self-resolving natural history in the majority of LGIB cases. However, there may be a subset of patients who could benefit from early intervention (such as severe blood loss, hemodynamically unstable patients), and thus the decision to pursue urgent colonoscopy should be made on a case-by-case basis,” they said.
Further, although several critical patient outcomes did not appear to be impacted by urgent vs. elective colonoscopy in this study, the trends toward a decrease in length of stay suggest that earlier performance of colonoscopy may lead to earlier and better identification of low-risk and high-risk stigmata, allowing those with low-risk lesions to be discharged much earlier.
“Additionally, earlier discharge of patients could also reduce their risk of health care–associated infections and adverse events,” the investigators noted.
The findings with respect to length of stay and cost “align perfectly with the new focus in health care on providing high quality and safe care to patients while at the same time containing medical costs,” they wrote, adding that clinicians should carefully consider all factors when deciding to pursue urgent colonoscopy.”
The study is limited by heterogeneity and publications bias, and by factors inherent in meta-analyses, but it also has several strengths, including a large number of studies and patients. Also, it is the first of its kind to examine “all of the available literature to elucidate the time frame for performing colonoscopy in patients with hematochezia,” the investigators said, concluding that further research is needed to identify subsets of patients who will benefit from early intervention, to evaluate the cost effectiveness of urgent colonoscopy, and to look at – in larger randomized controlled trials – the overall benefit of urgent colonoscopy.
The authors reported having no disclosures.
FROM GASTROINTESTINAL ENDOSCOPY
Key clinical point:
Major finding: Urgent vs. elective colonoscopy was not associated with improved bleeding source localization (RR, 1.08), adverse event rates (RR, 1.05), rebleeding rates (RR, 1.14), transfusion requirements (RR, 1.02) or mortality (RR, 1.17).
Data source: A systematic review and meta-analysis of 12 studies including more than 24,000 patients.
Disclosures: The authors reported having no disclosures.
No AR in CTCs linked with better survival in advanced prostate cancer
The presence and amount of full-length androgen receptor biomarker detected in the circulating tumor cells of people with metastatic castration-resistant prostate cancer can inform prognosis, a prospective study reveals.
Investigators report significant differences in prostate-specific antigen 50 (PSA50) values, PSA progression-free survival, clinical and/or radiologic progression-free survival, as well as overall survival, based on baseline levels of the amplified androgen receptor full-length (AR-FL) marker. The findings suggest quantification of AR-FL could serve as a clinically useful molecular biomarker in addition to AR-V7 status.
Prognosis differed among the 48% of patients with no detectable AR-FL marker, the 26% with amplification values below a median, and the remaining 26% with values above the median. The study included 202 men tested before starting hormonal treatment with either abiraterone or enzalutamide.
“Despite androgen deprivation, the androgen receptor continues to play a crucial role in prostate cancer,” Emmanuel S. Antonarakis, MBBCh, of Johns Hopkins University in Baltimore, said at in a press briefing held at the 2017 genitourinary cancers symposium sponsored by the American Society of Clinical Oncology, ASTRO, and the Society of Urologic Oncology. Dr. Antonarakis presented the findings on behalf of lead author John Silberstein, MD, and their coinvestigators.
Researchers found an inverse association with higher level of AR-FL and PSA50 responses. Also, men who did not achieve a PSA50 response had a mean of 55.4 transcripts, compared with 6.7 transcripts for those who did. Analyzed another way, the AR-FL–negative patients had a 62% PSA response rate, compared with 54% among the AR-FL–positive patients with amplification below the median and 28% for AR-FL–positive patients with values above the median.
In a multivariate analysis, controlling for AR-V7 and clinical variables, AR-FL remained prognostic for inferior PSA progression-free survival (hazard ratio, 1.06, P = .04). “A similar picture was seen with radiographic progression-free survival,” Dr. Antonarakis said. The best prognosis was for patients with undetectable AR-FL and the worst was for patients with detectable values above the median (HR, 1.04). However, AR-FL only trended toward significance (P = .13).
Similarly, for overall survival, AR-FL–negative patients had the best prognosis and patients with AR-FL above median had the worst in the multivariate analysis (HR, 1.07). “AR-FL reached borderline clinical significance,” he said (P = .06).
The presence of AR-V7 was independently prognostic in the multivariate analysis as well. “In conjunction with AR-V7, AR-FL quantification could serve as an additional biomarker to detect abiraterone or enzalutamide sensitivity or resistance,” Dr. Antonarakis said.
The current research builds on previous findings in this patient population. For example, genetic aberrations in circulating tumor DNA were associated with treatment resistance and inferior outcomes, including a worse progression-free survival, Dr. Antonarakis said (Clin. Cancer Res. 2015;21:2315-24). Other researchers demonstrated similar outcomes, both worse progression-free survival and overall survival among patients who had amplification or mutation of AR, compared with wild type, Dr. Antonarakis said.
These investigators used cell-free DNA to quantify AR, and the current study assessed circulating tumor cell–derived AR.
“Our vision is, very shortly in the future, we will have a liquid biopsy in patients to fully characterize their full complement of AR – patients with copy number gains, mutations in their genes, and splicing variance in the clinic,” Dr. Antonarakis said. It’s important to consider all three factors, he added.
Did you see any patients who were AR-V7 positive but AR-FL negative? study discussant Angelo Demarzo, MD, PhD, of Johns Hopkins University in Baltimore asked. “We have yet to find a patient like this. AR full length so far is always present when AR-V7 is positive,” Dr. Antonarakis replied. He added, however, “There is a subset of patients who are AR-V7 negative who have a high burden of AR full length, and they will still have a high risk.”
The presence and amount of full-length androgen receptor biomarker detected in the circulating tumor cells of people with metastatic castration-resistant prostate cancer can inform prognosis, a prospective study reveals.
Investigators report significant differences in prostate-specific antigen 50 (PSA50) values, PSA progression-free survival, clinical and/or radiologic progression-free survival, as well as overall survival, based on baseline levels of the amplified androgen receptor full-length (AR-FL) marker. The findings suggest quantification of AR-FL could serve as a clinically useful molecular biomarker in addition to AR-V7 status.
Prognosis differed among the 48% of patients with no detectable AR-FL marker, the 26% with amplification values below a median, and the remaining 26% with values above the median. The study included 202 men tested before starting hormonal treatment with either abiraterone or enzalutamide.
“Despite androgen deprivation, the androgen receptor continues to play a crucial role in prostate cancer,” Emmanuel S. Antonarakis, MBBCh, of Johns Hopkins University in Baltimore, said at in a press briefing held at the 2017 genitourinary cancers symposium sponsored by the American Society of Clinical Oncology, ASTRO, and the Society of Urologic Oncology. Dr. Antonarakis presented the findings on behalf of lead author John Silberstein, MD, and their coinvestigators.
Researchers found an inverse association with higher level of AR-FL and PSA50 responses. Also, men who did not achieve a PSA50 response had a mean of 55.4 transcripts, compared with 6.7 transcripts for those who did. Analyzed another way, the AR-FL–negative patients had a 62% PSA response rate, compared with 54% among the AR-FL–positive patients with amplification below the median and 28% for AR-FL–positive patients with values above the median.
In a multivariate analysis, controlling for AR-V7 and clinical variables, AR-FL remained prognostic for inferior PSA progression-free survival (hazard ratio, 1.06, P = .04). “A similar picture was seen with radiographic progression-free survival,” Dr. Antonarakis said. The best prognosis was for patients with undetectable AR-FL and the worst was for patients with detectable values above the median (HR, 1.04). However, AR-FL only trended toward significance (P = .13).
Similarly, for overall survival, AR-FL–negative patients had the best prognosis and patients with AR-FL above median had the worst in the multivariate analysis (HR, 1.07). “AR-FL reached borderline clinical significance,” he said (P = .06).
The presence of AR-V7 was independently prognostic in the multivariate analysis as well. “In conjunction with AR-V7, AR-FL quantification could serve as an additional biomarker to detect abiraterone or enzalutamide sensitivity or resistance,” Dr. Antonarakis said.
The current research builds on previous findings in this patient population. For example, genetic aberrations in circulating tumor DNA were associated with treatment resistance and inferior outcomes, including a worse progression-free survival, Dr. Antonarakis said (Clin. Cancer Res. 2015;21:2315-24). Other researchers demonstrated similar outcomes, both worse progression-free survival and overall survival among patients who had amplification or mutation of AR, compared with wild type, Dr. Antonarakis said.
These investigators used cell-free DNA to quantify AR, and the current study assessed circulating tumor cell–derived AR.
“Our vision is, very shortly in the future, we will have a liquid biopsy in patients to fully characterize their full complement of AR – patients with copy number gains, mutations in their genes, and splicing variance in the clinic,” Dr. Antonarakis said. It’s important to consider all three factors, he added.
Did you see any patients who were AR-V7 positive but AR-FL negative? study discussant Angelo Demarzo, MD, PhD, of Johns Hopkins University in Baltimore asked. “We have yet to find a patient like this. AR full length so far is always present when AR-V7 is positive,” Dr. Antonarakis replied. He added, however, “There is a subset of patients who are AR-V7 negative who have a high burden of AR full length, and they will still have a high risk.”
The presence and amount of full-length androgen receptor biomarker detected in the circulating tumor cells of people with metastatic castration-resistant prostate cancer can inform prognosis, a prospective study reveals.
Investigators report significant differences in prostate-specific antigen 50 (PSA50) values, PSA progression-free survival, clinical and/or radiologic progression-free survival, as well as overall survival, based on baseline levels of the amplified androgen receptor full-length (AR-FL) marker. The findings suggest quantification of AR-FL could serve as a clinically useful molecular biomarker in addition to AR-V7 status.
Prognosis differed among the 48% of patients with no detectable AR-FL marker, the 26% with amplification values below a median, and the remaining 26% with values above the median. The study included 202 men tested before starting hormonal treatment with either abiraterone or enzalutamide.
“Despite androgen deprivation, the androgen receptor continues to play a crucial role in prostate cancer,” Emmanuel S. Antonarakis, MBBCh, of Johns Hopkins University in Baltimore, said at in a press briefing held at the 2017 genitourinary cancers symposium sponsored by the American Society of Clinical Oncology, ASTRO, and the Society of Urologic Oncology. Dr. Antonarakis presented the findings on behalf of lead author John Silberstein, MD, and their coinvestigators.
Researchers found an inverse association with higher level of AR-FL and PSA50 responses. Also, men who did not achieve a PSA50 response had a mean of 55.4 transcripts, compared with 6.7 transcripts for those who did. Analyzed another way, the AR-FL–negative patients had a 62% PSA response rate, compared with 54% among the AR-FL–positive patients with amplification below the median and 28% for AR-FL–positive patients with values above the median.
In a multivariate analysis, controlling for AR-V7 and clinical variables, AR-FL remained prognostic for inferior PSA progression-free survival (hazard ratio, 1.06, P = .04). “A similar picture was seen with radiographic progression-free survival,” Dr. Antonarakis said. The best prognosis was for patients with undetectable AR-FL and the worst was for patients with detectable values above the median (HR, 1.04). However, AR-FL only trended toward significance (P = .13).
Similarly, for overall survival, AR-FL–negative patients had the best prognosis and patients with AR-FL above median had the worst in the multivariate analysis (HR, 1.07). “AR-FL reached borderline clinical significance,” he said (P = .06).
The presence of AR-V7 was independently prognostic in the multivariate analysis as well. “In conjunction with AR-V7, AR-FL quantification could serve as an additional biomarker to detect abiraterone or enzalutamide sensitivity or resistance,” Dr. Antonarakis said.
The current research builds on previous findings in this patient population. For example, genetic aberrations in circulating tumor DNA were associated with treatment resistance and inferior outcomes, including a worse progression-free survival, Dr. Antonarakis said (Clin. Cancer Res. 2015;21:2315-24). Other researchers demonstrated similar outcomes, both worse progression-free survival and overall survival among patients who had amplification or mutation of AR, compared with wild type, Dr. Antonarakis said.
These investigators used cell-free DNA to quantify AR, and the current study assessed circulating tumor cell–derived AR.
“Our vision is, very shortly in the future, we will have a liquid biopsy in patients to fully characterize their full complement of AR – patients with copy number gains, mutations in their genes, and splicing variance in the clinic,” Dr. Antonarakis said. It’s important to consider all three factors, he added.
Did you see any patients who were AR-V7 positive but AR-FL negative? study discussant Angelo Demarzo, MD, PhD, of Johns Hopkins University in Baltimore asked. “We have yet to find a patient like this. AR full length so far is always present when AR-V7 is positive,” Dr. Antonarakis replied. He added, however, “There is a subset of patients who are AR-V7 negative who have a high burden of AR full length, and they will still have a high risk.”
Key clinical point: A full-length androgen receptor biomarker can classify patients with metastatic castration-resistant prostate cancer and inform prognosis.
Major finding: Biomarker-negative patients had the best prognosis for overall survival, compared with those AR-FL levels above the median (HR, 1.07; P = .06).
Data source: Prospective study of 202 patients with advanced prostate cancer treated with abiraterone or enzalutamide.
Disclosures: The study was funded with support from the Prostate Cancer Foundation, the Department of Defense Prostate Cancer Research Program, and the Patrick C. Walsh Fund. Dr. Antonarakis is a consultant/advisor to Sanofi, Dendreon, Medivation, Janssen Biotech, ESSA, and Astellas Pharma; receives honoraria from Sanofi, Dendreon, Medivation, Janssen Biotech, ESSA, and Astellas Pharma; and receives travel and accommodation expense support from Sanofi, Dendreon, and Medivation.
Laser resurfacing can effectively minimize post surgery scars
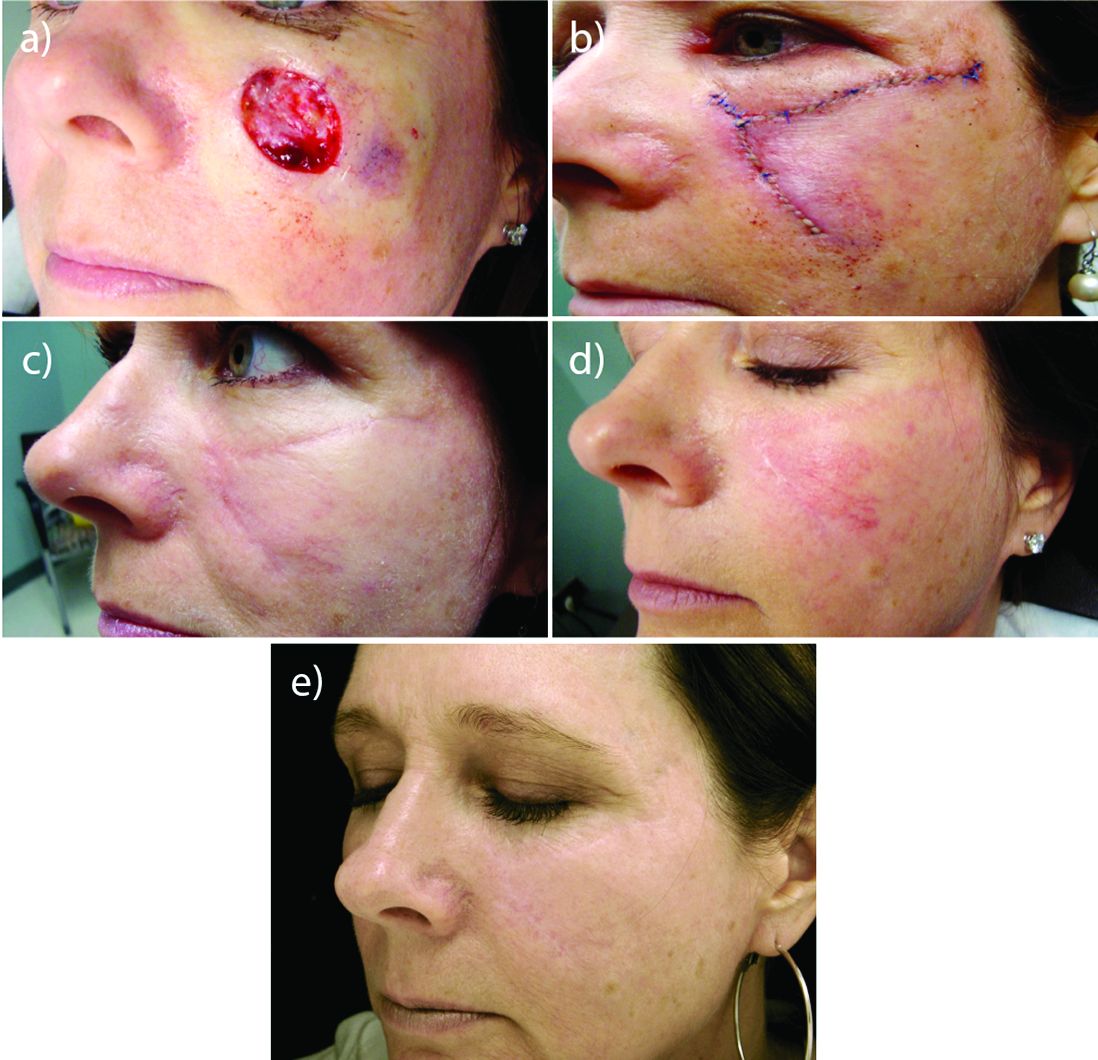
MIAMI – In his practice, Joel L. Cohen, MD, spends a good part of his day doing Mohs surgery, “with the goal of cancer removal, and after surgery, having the patient look good,” he said at the Orlando Dermatology Aesthetic and Clinical Conference.
“Having resurfacing in my practice has allowed me to treat not only wrinkles and etched lines, but also help skin cancer patients by blending and minimizing their skin cancer scars,” said Dr. Cohen, an aesthetic dermatologist and Mohs surgeon in private practice in Denver.

Resurfacing in his practice using a variety of lasers is very helpful, Dr. Cohen said. He published a study in November that compared pulse dye laser, CO2 ablative fractional lasers, or a combination of both for modification of scars following Mohs surgery (J Drugs Dermatol. 2016 Nov 1;15[11]:1315-9).
The prospective, multicenter study revealed that although both monotherapy approaches were safe and effective, the combination of pulse dye laser and fractional ablative laser offered some synergy that was preferred by patients.
Perioral resurfacing possible
Beyond the world of treating scars, a typical cosmetic patient in Dr. Cohen’s practice presents with numerous lines around the perioral area. “When people think about rejuvenation of the lips, they only think of fillers. But fillers are not the only way to rejuvenate this area, and it is really about choosing the right tool for the right job – where resurfacing lasers are needed.”
Set realistic expectations
Setting the right expectations for people is extremely important, Dr. Cohen said. “You can educate the patient that if you’re putting the needle into the lines, you’re only treating the larger lines that you can get a 30-g needle into, but there are often a host of other lines in that area – many of which are too small to get a needle into.”
As a starting point, neuromodulators can have a role in trying to prevent or delay etched-in lines from forming around the mouth in the first place. “These are the lines between the musculature, the ones you see when you ask the patient to purse their lips,” Dr. Cohen said. He typically injects a medium dose of one of three neuromodulators – such as 6-10 U of onabotulinumtoxinA (Botox), 6-10 U of incobotulinumtoxinA (Xeomin) or 14-18 U of abobotulinumtoxinA (Dysport). “Then somewhere between week 8 and 10, there is an attenuation of the effect, and I often will see patients back then for additional treatment with a neuromodulator,” he added.
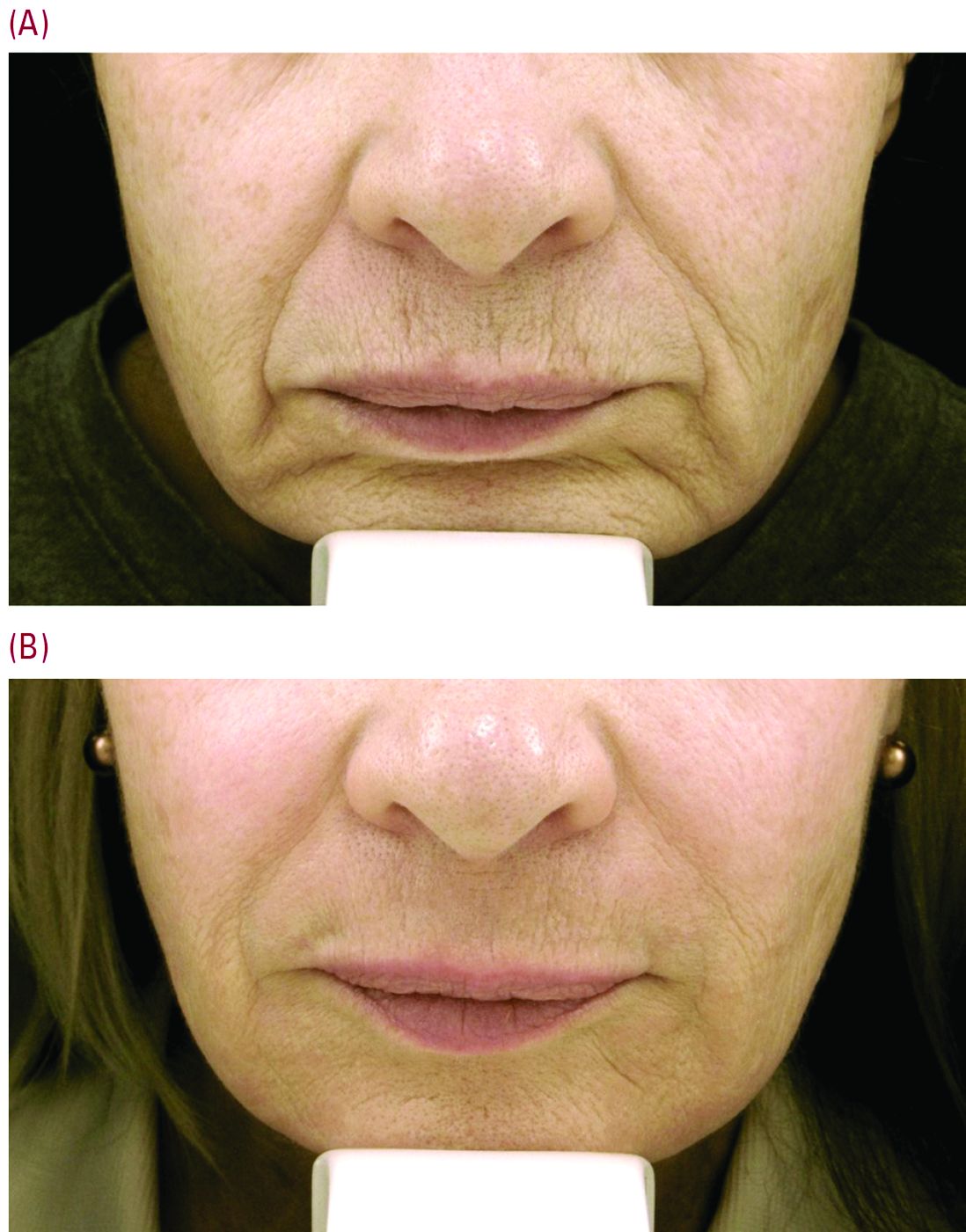
“For our every day patient complaining of lots of etched perioral lines, we have laser resurfacing,” Dr. Cohen noted. He is a bigger proponent of full-field erbium treatment versus fractional ablative laser resurfacing for these prominent upper cutaneous lip lines because the results are much more impressive with a single treatment. He added that dermatologists could do fractional treatment around the rest of the face, and reserve the erbium resurfacing to improve the appearance of lines around the mouth and prominent creping skin around the eyes.
Realistic postprocedure expectations are especially essential in the days after erbium laser resurfacing – as it is a tough downtime procedure for patients, often taking 7-9 days to re-epithelialize. “Having photos to show patients what they will look like is really helpful,” Dr. Cohen said. He suggested showing patients a chronologic set of photos of the downtime period as well as the results – so they realize improvement occurs slowly over time. “Getting people to understand they are gong to look terrible for 1.5-2 weeks is superimportant.”
“I like to have them back in the office for a postprocedure check a few days after the bigger laser resurfacing procedures are done, just to check on them,” Dr. Cohen said. “A lot of hand holding is often needed, as there is significantly more healing time with the full-field ablative resurfacing than there is with fractional. Full-field resurfacing patients will experience postprocedure erythema for a few weeks or even months,” Dr. Cohen said. A prescription of topical steroids, and sometimes some brimonidine topical gel (Mirvaso) as well can help reduce the redness.
Toxin injection then laser resurfacing
For some patients, injection of a neuromodulator a week or 2 before laser resurfacing treatment can decrease some of the movement and contraction of the muscle, “and hopefully give them better results,” Dr. Cohen said.
Timing is important. “You don’t want to use neuromodulators on the same day of treatment,” he advised. “The thinking is swelling could potentially cause the neuromodulators to spread to unwanted adjacent muscles.”
Safety first
Another tip for the postprocedure period is to supply patients with very specific written instructions. “I wish they would follow them. Patients don’t always listen to what we advise, demonstrate, and also have written down for them,” he commented. For example, one patient had resurfacing several weeks before leaving on an undisclosed kayaking trip. Despite instructions to use sunscreen, she said she wore a hat for sun protection and developed postinflammatory hyperpigmentation around the mouth that lasted for several months, Dr. Cohen said.*
With heavy resurfacing and ablative resurfacing in general, it is advised to always give patients an antiviral prophylaxis course such as valacyclovir, but it is unfortunate that not all patients will adhere to the recommended regimen, he added.
Another patient had an adverse reaction after resurfacing because she did not follow instructions to apply white petrolatum to her chest following laser resurfacing, Dr. Cohen said. She used Neosporin, “even though in all our paperwork we say never use Neosporin and just use the petrolatum. She had a big contact dermatitis reaction to the Neosporin.”
“So you really need to caution people about the importance of following instructions very carefully,” he emphasized.
Dr. Cohen is a consultant for Sciton and for companies that manufacture injectables, including Allergan, Galderma, and Merz.
Correction 2/24/17: An earlier version of this article mischaracterized the type of pigmentation disorder that the patient developed.
MIAMI – In his practice, Joel L. Cohen, MD, spends a good part of his day doing Mohs surgery, “with the goal of cancer removal, and after surgery, having the patient look good,” he said at the Orlando Dermatology Aesthetic and Clinical Conference.
“Having resurfacing in my practice has allowed me to treat not only wrinkles and etched lines, but also help skin cancer patients by blending and minimizing their skin cancer scars,” said Dr. Cohen, an aesthetic dermatologist and Mohs surgeon in private practice in Denver.
Resurfacing in his practice using a variety of lasers is very helpful, Dr. Cohen said. He published a study in November that compared pulse dye laser, CO2 ablative fractional lasers, or a combination of both for modification of scars following Mohs surgery (J Drugs Dermatol. 2016 Nov 1;15[11]:1315-9).
The prospective, multicenter study revealed that although both monotherapy approaches were safe and effective, the combination of pulse dye laser and fractional ablative laser offered some synergy that was preferred by patients.
Perioral resurfacing possible
Beyond the world of treating scars, a typical cosmetic patient in Dr. Cohen’s practice presents with numerous lines around the perioral area. “When people think about rejuvenation of the lips, they only think of fillers. But fillers are not the only way to rejuvenate this area, and it is really about choosing the right tool for the right job – where resurfacing lasers are needed.”
Set realistic expectations
Setting the right expectations for people is extremely important, Dr. Cohen said. “You can educate the patient that if you’re putting the needle into the lines, you’re only treating the larger lines that you can get a 30-g needle into, but there are often a host of other lines in that area – many of which are too small to get a needle into.”
As a starting point, neuromodulators can have a role in trying to prevent or delay etched-in lines from forming around the mouth in the first place. “These are the lines between the musculature, the ones you see when you ask the patient to purse their lips,” Dr. Cohen said. He typically injects a medium dose of one of three neuromodulators – such as 6-10 U of onabotulinumtoxinA (Botox), 6-10 U of incobotulinumtoxinA (Xeomin) or 14-18 U of abobotulinumtoxinA (Dysport). “Then somewhere between week 8 and 10, there is an attenuation of the effect, and I often will see patients back then for additional treatment with a neuromodulator,” he added.
“For our every day patient complaining of lots of etched perioral lines, we have laser resurfacing,” Dr. Cohen noted. He is a bigger proponent of full-field erbium treatment versus fractional ablative laser resurfacing for these prominent upper cutaneous lip lines because the results are much more impressive with a single treatment. He added that dermatologists could do fractional treatment around the rest of the face, and reserve the erbium resurfacing to improve the appearance of lines around the mouth and prominent creping skin around the eyes.
Realistic postprocedure expectations are especially essential in the days after erbium laser resurfacing – as it is a tough downtime procedure for patients, often taking 7-9 days to re-epithelialize. “Having photos to show patients what they will look like is really helpful,” Dr. Cohen said. He suggested showing patients a chronologic set of photos of the downtime period as well as the results – so they realize improvement occurs slowly over time. “Getting people to understand they are gong to look terrible for 1.5-2 weeks is superimportant.”
“I like to have them back in the office for a postprocedure check a few days after the bigger laser resurfacing procedures are done, just to check on them,” Dr. Cohen said. “A lot of hand holding is often needed, as there is significantly more healing time with the full-field ablative resurfacing than there is with fractional. Full-field resurfacing patients will experience postprocedure erythema for a few weeks or even months,” Dr. Cohen said. A prescription of topical steroids, and sometimes some brimonidine topical gel (Mirvaso) as well can help reduce the redness.
Toxin injection then laser resurfacing
For some patients, injection of a neuromodulator a week or 2 before laser resurfacing treatment can decrease some of the movement and contraction of the muscle, “and hopefully give them better results,” Dr. Cohen said.
Timing is important. “You don’t want to use neuromodulators on the same day of treatment,” he advised. “The thinking is swelling could potentially cause the neuromodulators to spread to unwanted adjacent muscles.”
Safety first
Another tip for the postprocedure period is to supply patients with very specific written instructions. “I wish they would follow them. Patients don’t always listen to what we advise, demonstrate, and also have written down for them,” he commented. For example, one patient had resurfacing several weeks before leaving on an undisclosed kayaking trip. Despite instructions to use sunscreen, she said she wore a hat for sun protection and developed postinflammatory hyperpigmentation around the mouth that lasted for several months, Dr. Cohen said.*
With heavy resurfacing and ablative resurfacing in general, it is advised to always give patients an antiviral prophylaxis course such as valacyclovir, but it is unfortunate that not all patients will adhere to the recommended regimen, he added.
Another patient had an adverse reaction after resurfacing because she did not follow instructions to apply white petrolatum to her chest following laser resurfacing, Dr. Cohen said. She used Neosporin, “even though in all our paperwork we say never use Neosporin and just use the petrolatum. She had a big contact dermatitis reaction to the Neosporin.”
“So you really need to caution people about the importance of following instructions very carefully,” he emphasized.
Dr. Cohen is a consultant for Sciton and for companies that manufacture injectables, including Allergan, Galderma, and Merz.
Correction 2/24/17: An earlier version of this article mischaracterized the type of pigmentation disorder that the patient developed.
MIAMI – In his practice, Joel L. Cohen, MD, spends a good part of his day doing Mohs surgery, “with the goal of cancer removal, and after surgery, having the patient look good,” he said at the Orlando Dermatology Aesthetic and Clinical Conference.
“Having resurfacing in my practice has allowed me to treat not only wrinkles and etched lines, but also help skin cancer patients by blending and minimizing their skin cancer scars,” said Dr. Cohen, an aesthetic dermatologist and Mohs surgeon in private practice in Denver.
Resurfacing in his practice using a variety of lasers is very helpful, Dr. Cohen said. He published a study in November that compared pulse dye laser, CO2 ablative fractional lasers, or a combination of both for modification of scars following Mohs surgery (J Drugs Dermatol. 2016 Nov 1;15[11]:1315-9).
The prospective, multicenter study revealed that although both monotherapy approaches were safe and effective, the combination of pulse dye laser and fractional ablative laser offered some synergy that was preferred by patients.
Perioral resurfacing possible
Beyond the world of treating scars, a typical cosmetic patient in Dr. Cohen’s practice presents with numerous lines around the perioral area. “When people think about rejuvenation of the lips, they only think of fillers. But fillers are not the only way to rejuvenate this area, and it is really about choosing the right tool for the right job – where resurfacing lasers are needed.”
Set realistic expectations
Setting the right expectations for people is extremely important, Dr. Cohen said. “You can educate the patient that if you’re putting the needle into the lines, you’re only treating the larger lines that you can get a 30-g needle into, but there are often a host of other lines in that area – many of which are too small to get a needle into.”
As a starting point, neuromodulators can have a role in trying to prevent or delay etched-in lines from forming around the mouth in the first place. “These are the lines between the musculature, the ones you see when you ask the patient to purse their lips,” Dr. Cohen said. He typically injects a medium dose of one of three neuromodulators – such as 6-10 U of onabotulinumtoxinA (Botox), 6-10 U of incobotulinumtoxinA (Xeomin) or 14-18 U of abobotulinumtoxinA (Dysport). “Then somewhere between week 8 and 10, there is an attenuation of the effect, and I often will see patients back then for additional treatment with a neuromodulator,” he added.
“For our every day patient complaining of lots of etched perioral lines, we have laser resurfacing,” Dr. Cohen noted. He is a bigger proponent of full-field erbium treatment versus fractional ablative laser resurfacing for these prominent upper cutaneous lip lines because the results are much more impressive with a single treatment. He added that dermatologists could do fractional treatment around the rest of the face, and reserve the erbium resurfacing to improve the appearance of lines around the mouth and prominent creping skin around the eyes.
Realistic postprocedure expectations are especially essential in the days after erbium laser resurfacing – as it is a tough downtime procedure for patients, often taking 7-9 days to re-epithelialize. “Having photos to show patients what they will look like is really helpful,” Dr. Cohen said. He suggested showing patients a chronologic set of photos of the downtime period as well as the results – so they realize improvement occurs slowly over time. “Getting people to understand they are gong to look terrible for 1.5-2 weeks is superimportant.”
“I like to have them back in the office for a postprocedure check a few days after the bigger laser resurfacing procedures are done, just to check on them,” Dr. Cohen said. “A lot of hand holding is often needed, as there is significantly more healing time with the full-field ablative resurfacing than there is with fractional. Full-field resurfacing patients will experience postprocedure erythema for a few weeks or even months,” Dr. Cohen said. A prescription of topical steroids, and sometimes some brimonidine topical gel (Mirvaso) as well can help reduce the redness.
Toxin injection then laser resurfacing
For some patients, injection of a neuromodulator a week or 2 before laser resurfacing treatment can decrease some of the movement and contraction of the muscle, “and hopefully give them better results,” Dr. Cohen said.
Timing is important. “You don’t want to use neuromodulators on the same day of treatment,” he advised. “The thinking is swelling could potentially cause the neuromodulators to spread to unwanted adjacent muscles.”
Safety first
Another tip for the postprocedure period is to supply patients with very specific written instructions. “I wish they would follow them. Patients don’t always listen to what we advise, demonstrate, and also have written down for them,” he commented. For example, one patient had resurfacing several weeks before leaving on an undisclosed kayaking trip. Despite instructions to use sunscreen, she said she wore a hat for sun protection and developed postinflammatory hyperpigmentation around the mouth that lasted for several months, Dr. Cohen said.*
With heavy resurfacing and ablative resurfacing in general, it is advised to always give patients an antiviral prophylaxis course such as valacyclovir, but it is unfortunate that not all patients will adhere to the recommended regimen, he added.
Another patient had an adverse reaction after resurfacing because she did not follow instructions to apply white petrolatum to her chest following laser resurfacing, Dr. Cohen said. She used Neosporin, “even though in all our paperwork we say never use Neosporin and just use the petrolatum. She had a big contact dermatitis reaction to the Neosporin.”
“So you really need to caution people about the importance of following instructions very carefully,” he emphasized.
Dr. Cohen is a consultant for Sciton and for companies that manufacture injectables, including Allergan, Galderma, and Merz.
Correction 2/24/17: An earlier version of this article mischaracterized the type of pigmentation disorder that the patient developed.
EXPERT ANALYSIS FROM THE ODAC CONFERENCE
Can a nomogram foretell invasive pulmonary adenocarcinoma?
The diagnosis of solitary peripheral subsolid nodule carries with it an undefined risk of invasive pulmonary carcinoma, but clinicians have not had a tool that can help guide their planning for surgery. However, researchers in China have developed a nomogram that they said may aid clinicians to predict the risk of invasive pulmonary adenocarcinoma in these patients.
“Validation by the use of bootstrap resampling revealed optimal discrimination and calibration, indicating that the nomogram may have clinical utility,” said Chenghua Jin, MD, and Jinlin Cao, MD, of Zhejiang University, Hangzhou, China, and coauthors. They reported their findings in the February issue of the Journal of Thoracic and Cardiovascular Surgery (2017;153:42-9).
The nomogram accounts for the following factors: computed tomography attenuation; nodule size; spiculation; signs of vascular convergence; pleural tags; and solid proportion. “The nomogram showed a robust discrimination with an area under the receiver operating characteristic curve of 0.894,” Dr. Jin and coauthors reported. An area under the curve of 1 is equivalent to 100%, so the area under the curve this study reported shows close to 90% accuracy.
The study involved a retrospective analysis of 273 consecutive patients who had resection of a solitary peripheral subsolid nodule at Zhejiang University School of Medicine from January 2013 to December 2014. Subsolid pulmonary nodules include pure ground-glass nodules and part-solid nodules that feature both solid and ground-glass components. “The optimal management of patients with a subsolid nodule is of growing clinical concern, because the most common diagnosis for resected subsolid nodules is lung adenocarcinoma,” Dr. Jin and colleagues indicated.
Of the study population, 58% were diagnosed with invasive pulmonary adenocarcinoma. Other diagnoses within the group were benign (13%), atypical adenomatous hyperplasia (1%), adenocarcinoma in situ (6.5%) and minimally invasive adenocarcinoma (21%).
Results of the multivariable analyses showed that invasive pulmonary adenocarcinoma correlated with the following characteristics: lesion size; spiculation; vascular convergence; and pleural tag. Factors that were not significant included age, family history of lung cancer, CT attenuation, and solid proportion. However, the researchers did include CT attenuation, along with solid proportion, in the final regression analysis based on their contributions to the statistical analysis.
For the model, CT attenuation of –500 to –200 Hounsfield units carried an odds ratio of 1.690 (P = .228) while CT attenuation greater than –200 HU had an OR of 1.791 (P = .645). Positive spiculation had an OR of 3.312 (no P value given) and negative vascular convergence an OR of 0.300 (no P value given).
While a number of prediction models have been devised and validated to evaluate the likelihood of malignancy in pulmonary nodules, they have not given subsolid nodules “specific or detailed consideration,” Dr. Jin and and coauthors said. “To our knowledge, this study was the first to construct a quantitative nomogram to predict the probability of invasive pulmonary adenocarcinoma in patients with subsolid nodules,” the researchers wrote.
One limitation of the study is its selection bias toward patients with a greater probability of having a malignancy. Also, validation of the nomogram requires external analysis with additional databases from other countries and with more diverse ethnic groups. Another shortcoming is the retrospective nature of the study and a small number of patients who had positron emission tomography. “Further data collection, wider geographic recruitment, and incorporation of positron emission tomography results and some molecular factors could improve this model for future use,” Dr. Jin and coauthors concluded.
Dr. Jin and Dr. Cao had no relevant financial disclosures. The study received funding from the Zhejiang Province Science and Technology Plan.
The nomogram Dr. Jin and coauthors present can be a valuable tool for determining the extent of resection of subsolid pulmonary nodules and to distinguish invasive from preinvasive disease where preoperative needle biopsy and intraopertiave frozen section typically cannot, Bryan Burt, MD, of Baylor College of Medicine, Houston, said in his invited commentary (J Thorac Cardiovasc Surg. 2017;153:460-1).
“However,” Dr. Burt added, “as the accuracy of frozen section for this disease improves, as it has in select centers, the clinical utility of such a nomogram will diminish.”

Use of the nomogram relies on experienced chest radiologists to aid in scoring variables and a validation methodology that a retrospective trial cannot meet, Dr. Burt said. “Of note, this nomogram was constructed from a dataset composed of only surgically resected lesions, and it will be imperative to validate these methods among a larger cohort of individuals with subsolid pulmonary nodules treated both surgically and nonsurgically, ideally in a prospective trial,” Dr. Burt concluded.
Dr. Burt had no relevant financial disclosures.
The nomogram Dr. Jin and coauthors present can be a valuable tool for determining the extent of resection of subsolid pulmonary nodules and to distinguish invasive from preinvasive disease where preoperative needle biopsy and intraopertiave frozen section typically cannot, Bryan Burt, MD, of Baylor College of Medicine, Houston, said in his invited commentary (J Thorac Cardiovasc Surg. 2017;153:460-1).
“However,” Dr. Burt added, “as the accuracy of frozen section for this disease improves, as it has in select centers, the clinical utility of such a nomogram will diminish.”
Use of the nomogram relies on experienced chest radiologists to aid in scoring variables and a validation methodology that a retrospective trial cannot meet, Dr. Burt said. “Of note, this nomogram was constructed from a dataset composed of only surgically resected lesions, and it will be imperative to validate these methods among a larger cohort of individuals with subsolid pulmonary nodules treated both surgically and nonsurgically, ideally in a prospective trial,” Dr. Burt concluded.
Dr. Burt had no relevant financial disclosures.
The nomogram Dr. Jin and coauthors present can be a valuable tool for determining the extent of resection of subsolid pulmonary nodules and to distinguish invasive from preinvasive disease where preoperative needle biopsy and intraopertiave frozen section typically cannot, Bryan Burt, MD, of Baylor College of Medicine, Houston, said in his invited commentary (J Thorac Cardiovasc Surg. 2017;153:460-1).
“However,” Dr. Burt added, “as the accuracy of frozen section for this disease improves, as it has in select centers, the clinical utility of such a nomogram will diminish.”
Use of the nomogram relies on experienced chest radiologists to aid in scoring variables and a validation methodology that a retrospective trial cannot meet, Dr. Burt said. “Of note, this nomogram was constructed from a dataset composed of only surgically resected lesions, and it will be imperative to validate these methods among a larger cohort of individuals with subsolid pulmonary nodules treated both surgically and nonsurgically, ideally in a prospective trial,” Dr. Burt concluded.
Dr. Burt had no relevant financial disclosures.
The diagnosis of solitary peripheral subsolid nodule carries with it an undefined risk of invasive pulmonary carcinoma, but clinicians have not had a tool that can help guide their planning for surgery. However, researchers in China have developed a nomogram that they said may aid clinicians to predict the risk of invasive pulmonary adenocarcinoma in these patients.
“Validation by the use of bootstrap resampling revealed optimal discrimination and calibration, indicating that the nomogram may have clinical utility,” said Chenghua Jin, MD, and Jinlin Cao, MD, of Zhejiang University, Hangzhou, China, and coauthors. They reported their findings in the February issue of the Journal of Thoracic and Cardiovascular Surgery (2017;153:42-9).
The nomogram accounts for the following factors: computed tomography attenuation; nodule size; spiculation; signs of vascular convergence; pleural tags; and solid proportion. “The nomogram showed a robust discrimination with an area under the receiver operating characteristic curve of 0.894,” Dr. Jin and coauthors reported. An area under the curve of 1 is equivalent to 100%, so the area under the curve this study reported shows close to 90% accuracy.
The study involved a retrospective analysis of 273 consecutive patients who had resection of a solitary peripheral subsolid nodule at Zhejiang University School of Medicine from January 2013 to December 2014. Subsolid pulmonary nodules include pure ground-glass nodules and part-solid nodules that feature both solid and ground-glass components. “The optimal management of patients with a subsolid nodule is of growing clinical concern, because the most common diagnosis for resected subsolid nodules is lung adenocarcinoma,” Dr. Jin and colleagues indicated.
Of the study population, 58% were diagnosed with invasive pulmonary adenocarcinoma. Other diagnoses within the group were benign (13%), atypical adenomatous hyperplasia (1%), adenocarcinoma in situ (6.5%) and minimally invasive adenocarcinoma (21%).
Results of the multivariable analyses showed that invasive pulmonary adenocarcinoma correlated with the following characteristics: lesion size; spiculation; vascular convergence; and pleural tag. Factors that were not significant included age, family history of lung cancer, CT attenuation, and solid proportion. However, the researchers did include CT attenuation, along with solid proportion, in the final regression analysis based on their contributions to the statistical analysis.
For the model, CT attenuation of –500 to –200 Hounsfield units carried an odds ratio of 1.690 (P = .228) while CT attenuation greater than –200 HU had an OR of 1.791 (P = .645). Positive spiculation had an OR of 3.312 (no P value given) and negative vascular convergence an OR of 0.300 (no P value given).
While a number of prediction models have been devised and validated to evaluate the likelihood of malignancy in pulmonary nodules, they have not given subsolid nodules “specific or detailed consideration,” Dr. Jin and and coauthors said. “To our knowledge, this study was the first to construct a quantitative nomogram to predict the probability of invasive pulmonary adenocarcinoma in patients with subsolid nodules,” the researchers wrote.
One limitation of the study is its selection bias toward patients with a greater probability of having a malignancy. Also, validation of the nomogram requires external analysis with additional databases from other countries and with more diverse ethnic groups. Another shortcoming is the retrospective nature of the study and a small number of patients who had positron emission tomography. “Further data collection, wider geographic recruitment, and incorporation of positron emission tomography results and some molecular factors could improve this model for future use,” Dr. Jin and coauthors concluded.
Dr. Jin and Dr. Cao had no relevant financial disclosures. The study received funding from the Zhejiang Province Science and Technology Plan.
The diagnosis of solitary peripheral subsolid nodule carries with it an undefined risk of invasive pulmonary carcinoma, but clinicians have not had a tool that can help guide their planning for surgery. However, researchers in China have developed a nomogram that they said may aid clinicians to predict the risk of invasive pulmonary adenocarcinoma in these patients.
“Validation by the use of bootstrap resampling revealed optimal discrimination and calibration, indicating that the nomogram may have clinical utility,” said Chenghua Jin, MD, and Jinlin Cao, MD, of Zhejiang University, Hangzhou, China, and coauthors. They reported their findings in the February issue of the Journal of Thoracic and Cardiovascular Surgery (2017;153:42-9).
The nomogram accounts for the following factors: computed tomography attenuation; nodule size; spiculation; signs of vascular convergence; pleural tags; and solid proportion. “The nomogram showed a robust discrimination with an area under the receiver operating characteristic curve of 0.894,” Dr. Jin and coauthors reported. An area under the curve of 1 is equivalent to 100%, so the area under the curve this study reported shows close to 90% accuracy.
The study involved a retrospective analysis of 273 consecutive patients who had resection of a solitary peripheral subsolid nodule at Zhejiang University School of Medicine from January 2013 to December 2014. Subsolid pulmonary nodules include pure ground-glass nodules and part-solid nodules that feature both solid and ground-glass components. “The optimal management of patients with a subsolid nodule is of growing clinical concern, because the most common diagnosis for resected subsolid nodules is lung adenocarcinoma,” Dr. Jin and colleagues indicated.
Of the study population, 58% were diagnosed with invasive pulmonary adenocarcinoma. Other diagnoses within the group were benign (13%), atypical adenomatous hyperplasia (1%), adenocarcinoma in situ (6.5%) and minimally invasive adenocarcinoma (21%).
Results of the multivariable analyses showed that invasive pulmonary adenocarcinoma correlated with the following characteristics: lesion size; spiculation; vascular convergence; and pleural tag. Factors that were not significant included age, family history of lung cancer, CT attenuation, and solid proportion. However, the researchers did include CT attenuation, along with solid proportion, in the final regression analysis based on their contributions to the statistical analysis.
For the model, CT attenuation of –500 to –200 Hounsfield units carried an odds ratio of 1.690 (P = .228) while CT attenuation greater than –200 HU had an OR of 1.791 (P = .645). Positive spiculation had an OR of 3.312 (no P value given) and negative vascular convergence an OR of 0.300 (no P value given).
While a number of prediction models have been devised and validated to evaluate the likelihood of malignancy in pulmonary nodules, they have not given subsolid nodules “specific or detailed consideration,” Dr. Jin and and coauthors said. “To our knowledge, this study was the first to construct a quantitative nomogram to predict the probability of invasive pulmonary adenocarcinoma in patients with subsolid nodules,” the researchers wrote.
One limitation of the study is its selection bias toward patients with a greater probability of having a malignancy. Also, validation of the nomogram requires external analysis with additional databases from other countries and with more diverse ethnic groups. Another shortcoming is the retrospective nature of the study and a small number of patients who had positron emission tomography. “Further data collection, wider geographic recruitment, and incorporation of positron emission tomography results and some molecular factors could improve this model for future use,” Dr. Jin and coauthors concluded.
Dr. Jin and Dr. Cao had no relevant financial disclosures. The study received funding from the Zhejiang Province Science and Technology Plan.
EXPERT ANALYSIS FROM THE JOURNAL OF THORACIC AND CARDIOVASCULAR SURGERY
Key clinical point: Investigators developed a nomogram that may help predict the risk of invasive pulmonary adenocarcinoma for patients with a solitary peripheral subsolid nodule.
Major finding: This nomogram may help clinicians individualize each patient’s prognosis for invasive pulmonary adenocarcinoma and develop treatment plans accordingly.
Data source: Retrospective analysis of 273 consecutive patients who had surgery to remove a solitary peripheral subsolid nodule at a single center.
Disclosure: The investigators received support from the Zhejiang Province Science and Technology Plan. Dr. Jin and Dr. Cao reported having no relevant financial disclosures.
Site of care may impact survival for AYAs with ALL, AML

Receiving treatment at specialized cancer centers may improve survival for adolescents and young adults (AYAs) with acute leukemia, according to a study published in Cancer Epidemiology, Biomarkers & Prevention.
The study showed that, when patients were treated at specialized cancer centers, survival rates were similar for younger AYAs and children with acute lymphoblastic leukemia (ALL) or acute myeloid leukemia (AML).
However, older AYAs did not appear to reap the same survival benefit from receiving treatment at a specialized cancer center.
And AYAs of all ages had significantly worse survival than children if they were not treated at specialized cancer centers.
AYAs (patients ages 15 to 39) with ALL and AML have significantly worse survival outcomes than children ages 14 and under, according to study author Julie Wolfson, MD, of the University of Alabama at Birmingham.
“A much smaller percentage of AYAs with cancer are treated at specialized cancer centers than children with cancer,” she added. “We wanted to understand whether this difference in the site of cancer care is associated with the difference in survival outcomes.”
Dr Wolfson and her colleagues used data from the Los Angeles County Cancer Surveillance Program to identify patients diagnosed with ALL or AML from ages 1 to 39.
Patients were said to have been treated at a specialized cancer center if, at any age, they were cared for at any of the National Cancer Institute-designated Comprehensive Cancer Centers (CCC) in Los Angeles County or if, at the age of 21 or younger, they were cared for at any of the Children’s Oncology Group (COG) sites not designated a Comprehensive Cancer Center.
Included in the analysis were 978 patients diagnosed with ALL as a child (ages 1 to 14), 402 patients diagnosed with ALL as an AYA (ages 15 to 39), 131 patients diagnosed with AML as a child, and 359 patients diagnosed with AML as an AYA.
Seventy percent of the children with ALL and 30% of the AYAs with ALL were treated at CCC/COG sites. Seventy-four percent of the children with AML and 22% of the AYAs with AML were treated at CCC/COG sites.
Results in ALL
AYAs diagnosed with ALL at ages 15 to 21 and 22 to 29 who were treated at CCC/COG sites had comparable survival to children who were diagnosed with ALL from ages 10 to 14 and treated at CCC/COG sites. The hazard ratios (HRs) were 1.3 for the 15-21 age group (P=0.3) and 1.2 for the 22-29 age group (P=0.8).
The reference group for this analysis was 10- to 14-year-olds treated at CCC/COG sites because the researchers excluded data from children with ALL ages 1 to 9. These patients were excluded because they have significantly better survival than the other groups, potentially as a result of different disease biology.
The researchers also found that treatment at CCC/COG sites did not improve survival for 30- to 39-year-olds with ALL. The HR for them was 3.4 (P<0.001).
Likewise, all AYAs with ALL who were not treated at CCC/COG sites had worse survival than 10- to 14-year-olds treated at CCC/COG sites. The HRs were 1.9 for the 15-21 age group (P=0.005), 2.6 for the 22-29 age group (P<0.001), and 3.0 for the 30-39 age group (P<0.001).
Results in AML
For AML patients, the reference group was 1- to 14-year-olds treated at CCC/COG sites. Compared to these patients, 15- to 21-year-olds not treated at CCC/COG sites had an increased hazard of death (HR=1.7, P=0.02), whereas 15- to 21-year-olds treated at CCC/COG sites did not (HR=1.3; P=0.4).
All 22- to 39-year-olds, regardless of the site of care, had an increased hazard of death compared to the children treated at CCC/COG sites. The HR was 3.4 (P<0.001) for 22- to 39-year-olds treated at CCC/COG sites, and the HR was 3.0 (P<0.001) for 22- to 39-year-olds not treated at CCC/COG sites.
“The fact that the older AYAs did not appear to benefit from treatment at a specialized cancer center suggests to us that the biology of the disease in these patients differs from that in younger individuals,” Dr Wolfson said.
“The most important thing we can do to help these patients is to enroll them on clinical trials so that we can better understand the biology of the disease and its response to therapy. ![]()
Receiving treatment at specialized cancer centers may improve survival for adolescents and young adults (AYAs) with acute leukemia, according to a study published in Cancer Epidemiology, Biomarkers & Prevention.
The study showed that, when patients were treated at specialized cancer centers, survival rates were similar for younger AYAs and children with acute lymphoblastic leukemia (ALL) or acute myeloid leukemia (AML).
However, older AYAs did not appear to reap the same survival benefit from receiving treatment at a specialized cancer center.
And AYAs of all ages had significantly worse survival than children if they were not treated at specialized cancer centers.
AYAs (patients ages 15 to 39) with ALL and AML have significantly worse survival outcomes than children ages 14 and under, according to study author Julie Wolfson, MD, of the University of Alabama at Birmingham.
“A much smaller percentage of AYAs with cancer are treated at specialized cancer centers than children with cancer,” she added. “We wanted to understand whether this difference in the site of cancer care is associated with the difference in survival outcomes.”
Dr Wolfson and her colleagues used data from the Los Angeles County Cancer Surveillance Program to identify patients diagnosed with ALL or AML from ages 1 to 39.
Patients were said to have been treated at a specialized cancer center if, at any age, they were cared for at any of the National Cancer Institute-designated Comprehensive Cancer Centers (CCC) in Los Angeles County or if, at the age of 21 or younger, they were cared for at any of the Children’s Oncology Group (COG) sites not designated a Comprehensive Cancer Center.
Included in the analysis were 978 patients diagnosed with ALL as a child (ages 1 to 14), 402 patients diagnosed with ALL as an AYA (ages 15 to 39), 131 patients diagnosed with AML as a child, and 359 patients diagnosed with AML as an AYA.
Seventy percent of the children with ALL and 30% of the AYAs with ALL were treated at CCC/COG sites. Seventy-four percent of the children with AML and 22% of the AYAs with AML were treated at CCC/COG sites.
Results in ALL
AYAs diagnosed with ALL at ages 15 to 21 and 22 to 29 who were treated at CCC/COG sites had comparable survival to children who were diagnosed with ALL from ages 10 to 14 and treated at CCC/COG sites. The hazard ratios (HRs) were 1.3 for the 15-21 age group (P=0.3) and 1.2 for the 22-29 age group (P=0.8).
The reference group for this analysis was 10- to 14-year-olds treated at CCC/COG sites because the researchers excluded data from children with ALL ages 1 to 9. These patients were excluded because they have significantly better survival than the other groups, potentially as a result of different disease biology.
The researchers also found that treatment at CCC/COG sites did not improve survival for 30- to 39-year-olds with ALL. The HR for them was 3.4 (P<0.001).
Likewise, all AYAs with ALL who were not treated at CCC/COG sites had worse survival than 10- to 14-year-olds treated at CCC/COG sites. The HRs were 1.9 for the 15-21 age group (P=0.005), 2.6 for the 22-29 age group (P<0.001), and 3.0 for the 30-39 age group (P<0.001).
Results in AML
For AML patients, the reference group was 1- to 14-year-olds treated at CCC/COG sites. Compared to these patients, 15- to 21-year-olds not treated at CCC/COG sites had an increased hazard of death (HR=1.7, P=0.02), whereas 15- to 21-year-olds treated at CCC/COG sites did not (HR=1.3; P=0.4).
All 22- to 39-year-olds, regardless of the site of care, had an increased hazard of death compared to the children treated at CCC/COG sites. The HR was 3.4 (P<0.001) for 22- to 39-year-olds treated at CCC/COG sites, and the HR was 3.0 (P<0.001) for 22- to 39-year-olds not treated at CCC/COG sites.
“The fact that the older AYAs did not appear to benefit from treatment at a specialized cancer center suggests to us that the biology of the disease in these patients differs from that in younger individuals,” Dr Wolfson said.
“The most important thing we can do to help these patients is to enroll them on clinical trials so that we can better understand the biology of the disease and its response to therapy. ![]()
Receiving treatment at specialized cancer centers may improve survival for adolescents and young adults (AYAs) with acute leukemia, according to a study published in Cancer Epidemiology, Biomarkers & Prevention.
The study showed that, when patients were treated at specialized cancer centers, survival rates were similar for younger AYAs and children with acute lymphoblastic leukemia (ALL) or acute myeloid leukemia (AML).
However, older AYAs did not appear to reap the same survival benefit from receiving treatment at a specialized cancer center.
And AYAs of all ages had significantly worse survival than children if they were not treated at specialized cancer centers.
AYAs (patients ages 15 to 39) with ALL and AML have significantly worse survival outcomes than children ages 14 and under, according to study author Julie Wolfson, MD, of the University of Alabama at Birmingham.
“A much smaller percentage of AYAs with cancer are treated at specialized cancer centers than children with cancer,” she added. “We wanted to understand whether this difference in the site of cancer care is associated with the difference in survival outcomes.”
Dr Wolfson and her colleagues used data from the Los Angeles County Cancer Surveillance Program to identify patients diagnosed with ALL or AML from ages 1 to 39.
Patients were said to have been treated at a specialized cancer center if, at any age, they were cared for at any of the National Cancer Institute-designated Comprehensive Cancer Centers (CCC) in Los Angeles County or if, at the age of 21 or younger, they were cared for at any of the Children’s Oncology Group (COG) sites not designated a Comprehensive Cancer Center.
Included in the analysis were 978 patients diagnosed with ALL as a child (ages 1 to 14), 402 patients diagnosed with ALL as an AYA (ages 15 to 39), 131 patients diagnosed with AML as a child, and 359 patients diagnosed with AML as an AYA.
Seventy percent of the children with ALL and 30% of the AYAs with ALL were treated at CCC/COG sites. Seventy-four percent of the children with AML and 22% of the AYAs with AML were treated at CCC/COG sites.
Results in ALL
AYAs diagnosed with ALL at ages 15 to 21 and 22 to 29 who were treated at CCC/COG sites had comparable survival to children who were diagnosed with ALL from ages 10 to 14 and treated at CCC/COG sites. The hazard ratios (HRs) were 1.3 for the 15-21 age group (P=0.3) and 1.2 for the 22-29 age group (P=0.8).
The reference group for this analysis was 10- to 14-year-olds treated at CCC/COG sites because the researchers excluded data from children with ALL ages 1 to 9. These patients were excluded because they have significantly better survival than the other groups, potentially as a result of different disease biology.
The researchers also found that treatment at CCC/COG sites did not improve survival for 30- to 39-year-olds with ALL. The HR for them was 3.4 (P<0.001).
Likewise, all AYAs with ALL who were not treated at CCC/COG sites had worse survival than 10- to 14-year-olds treated at CCC/COG sites. The HRs were 1.9 for the 15-21 age group (P=0.005), 2.6 for the 22-29 age group (P<0.001), and 3.0 for the 30-39 age group (P<0.001).
Results in AML
For AML patients, the reference group was 1- to 14-year-olds treated at CCC/COG sites. Compared to these patients, 15- to 21-year-olds not treated at CCC/COG sites had an increased hazard of death (HR=1.7, P=0.02), whereas 15- to 21-year-olds treated at CCC/COG sites did not (HR=1.3; P=0.4).
All 22- to 39-year-olds, regardless of the site of care, had an increased hazard of death compared to the children treated at CCC/COG sites. The HR was 3.4 (P<0.001) for 22- to 39-year-olds treated at CCC/COG sites, and the HR was 3.0 (P<0.001) for 22- to 39-year-olds not treated at CCC/COG sites.
“The fact that the older AYAs did not appear to benefit from treatment at a specialized cancer center suggests to us that the biology of the disease in these patients differs from that in younger individuals,” Dr Wolfson said.
“The most important thing we can do to help these patients is to enroll them on clinical trials so that we can better understand the biology of the disease and its response to therapy. ![]()
Using mRNA to treat hemophilia B

Researchers have reported the successful treatment of hemophilia B in mice using messenger RNA (mRNA).
In experiments, protein-replacement therapy using mRNA was effective longer than recombinant factor IX (FIX) therapy.
The mRNA therapy also appeared to be safe and produced mild, transient immune responses.
In addition, the researchers noted that the mRNA therapy can be produced more cheaply than recombinant FIX therapy.
They said this research, published in PNAS, is proof-of-concept that mRNA therapy could be used to treat hemophilia B and other genetic diseases.
“We are really excited about this work because, short of correcting a faulty gene, protein-replacement therapy using mRNA is one of the most promising techniques we have at our disposal,” said study author Inder Verma, PhD, of the Salk Institute for Biological Studies in La Jolla, California.
“Now, we have proof that we can successfully treat a disease—with virtually no side effects—at a lower cost than manufacturing the needed protein.”
This work is a collaboration between the Verma lab and Arcturus Therapeutics, a biotech company that developed a system of encapsulating mRNA within lipid nanoparticles.
The researchers created an mRNA blueprint for human FIX nanoparticles and delivered them, via injection, to mice with a faulty FIX gene.
Once in the bloodstream, the nanoparticles traveled to the liver, where their fatty casing helped them ease into cells and deliver to the protein-making machinery the mRNA instructions to assemble FIX.
The hemophilic mice were given 3 injections over a 5-month period, during which their coagulation and immune responses were carefully monitored.
Normal clotting occurred in the mice within 4 hours of receiving the therapy, and the results lasted for up to 6 days.
The mice had a weak immune response to the treatment, which quickly returned to baseline.
“One of the issues with both nanoparticles and mRNA treatment is toxicity, and, in our study, we did not see much evidence of that,” said study author Suvasini Ramaswamy, PhD, of the Salk Institute for Biological Studies.
“We gave the treatment over a long span to give the immune system time to see it and react to it, but the immune response looked more like a mild allergic response and quickly returned to normal, so the technology seems pretty reliable and safe in our mouse model.”
The researchers also gave mice recombinant FIX, directly comparing it to the mRNA therapy. The mRNA therapy proved more effective, maintaining 20% more clotting activity 4 days after injection.
“Conceptually, in vivo mRNA delivery has been around for a long time, but its therapeutic use has been limited by poor stability, immune-reactivity, and problems with reproducible systemic delivery,” said Pad Chivukula, chief scientific officer at Arcturus Therapeutics.
“The results suggest that nanoparticle delivery technology overcomes these challenges and might allow for the development of novel, cost-effective mRNA therapeutics.”
This work was funded by the National Institutes of Health, the Ipsen Foundation, the H.N. and Frances C. Berger Foundation, the Glenn Center for Research on Aging, The Leona M. and Harry B. Helmsley Charitable Trust, and the California Institute for Regenerative Medicine. The lipid nanoparticles were developed by Arcturus Therapeutics. ![]()
Researchers have reported the successful treatment of hemophilia B in mice using messenger RNA (mRNA).
In experiments, protein-replacement therapy using mRNA was effective longer than recombinant factor IX (FIX) therapy.
The mRNA therapy also appeared to be safe and produced mild, transient immune responses.
In addition, the researchers noted that the mRNA therapy can be produced more cheaply than recombinant FIX therapy.
They said this research, published in PNAS, is proof-of-concept that mRNA therapy could be used to treat hemophilia B and other genetic diseases.
“We are really excited about this work because, short of correcting a faulty gene, protein-replacement therapy using mRNA is one of the most promising techniques we have at our disposal,” said study author Inder Verma, PhD, of the Salk Institute for Biological Studies in La Jolla, California.
“Now, we have proof that we can successfully treat a disease—with virtually no side effects—at a lower cost than manufacturing the needed protein.”
This work is a collaboration between the Verma lab and Arcturus Therapeutics, a biotech company that developed a system of encapsulating mRNA within lipid nanoparticles.
The researchers created an mRNA blueprint for human FIX nanoparticles and delivered them, via injection, to mice with a faulty FIX gene.
Once in the bloodstream, the nanoparticles traveled to the liver, where their fatty casing helped them ease into cells and deliver to the protein-making machinery the mRNA instructions to assemble FIX.
The hemophilic mice were given 3 injections over a 5-month period, during which their coagulation and immune responses were carefully monitored.
Normal clotting occurred in the mice within 4 hours of receiving the therapy, and the results lasted for up to 6 days.
The mice had a weak immune response to the treatment, which quickly returned to baseline.
“One of the issues with both nanoparticles and mRNA treatment is toxicity, and, in our study, we did not see much evidence of that,” said study author Suvasini Ramaswamy, PhD, of the Salk Institute for Biological Studies.
“We gave the treatment over a long span to give the immune system time to see it and react to it, but the immune response looked more like a mild allergic response and quickly returned to normal, so the technology seems pretty reliable and safe in our mouse model.”
The researchers also gave mice recombinant FIX, directly comparing it to the mRNA therapy. The mRNA therapy proved more effective, maintaining 20% more clotting activity 4 days after injection.
“Conceptually, in vivo mRNA delivery has been around for a long time, but its therapeutic use has been limited by poor stability, immune-reactivity, and problems with reproducible systemic delivery,” said Pad Chivukula, chief scientific officer at Arcturus Therapeutics.
“The results suggest that nanoparticle delivery technology overcomes these challenges and might allow for the development of novel, cost-effective mRNA therapeutics.”
This work was funded by the National Institutes of Health, the Ipsen Foundation, the H.N. and Frances C. Berger Foundation, the Glenn Center for Research on Aging, The Leona M. and Harry B. Helmsley Charitable Trust, and the California Institute for Regenerative Medicine. The lipid nanoparticles were developed by Arcturus Therapeutics. ![]()
Researchers have reported the successful treatment of hemophilia B in mice using messenger RNA (mRNA).
In experiments, protein-replacement therapy using mRNA was effective longer than recombinant factor IX (FIX) therapy.
The mRNA therapy also appeared to be safe and produced mild, transient immune responses.
In addition, the researchers noted that the mRNA therapy can be produced more cheaply than recombinant FIX therapy.
They said this research, published in PNAS, is proof-of-concept that mRNA therapy could be used to treat hemophilia B and other genetic diseases.
“We are really excited about this work because, short of correcting a faulty gene, protein-replacement therapy using mRNA is one of the most promising techniques we have at our disposal,” said study author Inder Verma, PhD, of the Salk Institute for Biological Studies in La Jolla, California.
“Now, we have proof that we can successfully treat a disease—with virtually no side effects—at a lower cost than manufacturing the needed protein.”
This work is a collaboration between the Verma lab and Arcturus Therapeutics, a biotech company that developed a system of encapsulating mRNA within lipid nanoparticles.
The researchers created an mRNA blueprint for human FIX nanoparticles and delivered them, via injection, to mice with a faulty FIX gene.
Once in the bloodstream, the nanoparticles traveled to the liver, where their fatty casing helped them ease into cells and deliver to the protein-making machinery the mRNA instructions to assemble FIX.
The hemophilic mice were given 3 injections over a 5-month period, during which their coagulation and immune responses were carefully monitored.
Normal clotting occurred in the mice within 4 hours of receiving the therapy, and the results lasted for up to 6 days.
The mice had a weak immune response to the treatment, which quickly returned to baseline.
“One of the issues with both nanoparticles and mRNA treatment is toxicity, and, in our study, we did not see much evidence of that,” said study author Suvasini Ramaswamy, PhD, of the Salk Institute for Biological Studies.
“We gave the treatment over a long span to give the immune system time to see it and react to it, but the immune response looked more like a mild allergic response and quickly returned to normal, so the technology seems pretty reliable and safe in our mouse model.”
The researchers also gave mice recombinant FIX, directly comparing it to the mRNA therapy. The mRNA therapy proved more effective, maintaining 20% more clotting activity 4 days after injection.
“Conceptually, in vivo mRNA delivery has been around for a long time, but its therapeutic use has been limited by poor stability, immune-reactivity, and problems with reproducible systemic delivery,” said Pad Chivukula, chief scientific officer at Arcturus Therapeutics.
“The results suggest that nanoparticle delivery technology overcomes these challenges and might allow for the development of novel, cost-effective mRNA therapeutics.”
This work was funded by the National Institutes of Health, the Ipsen Foundation, the H.N. and Frances C. Berger Foundation, the Glenn Center for Research on Aging, The Leona M. and Harry B. Helmsley Charitable Trust, and the California Institute for Regenerative Medicine. The lipid nanoparticles were developed by Arcturus Therapeutics. ![]()