User login
Product News: 04 2017
Cutanea Life Sciences, Inc, launches Aktipak (erythromycin 3% and benzoyl peroxide 5%) Gel, a prescription combination therapy indicated for acne vulgaris. Aktipak is packaged in a pocket-sized, dual-chamber pouch that contains erythromycin and benzoyl peroxide in separate chambers to enable convenient on-the-go use. Immediately prior to use, the patient cuts or twists open the pouch, squeezes the 2 gels into the palm of the hand, mixes the gels together, and applies the mix to the area affected by acne. Aktipak has an 18-month shelf life and does not require refrigeration. Results can be seen within 8 weeks. For more information, visit www.aktipak.com.
Glytone Acne BPO Clearing Cleanser
Pierre Fabre Group introduces the Glytone Acne BPO Clearing Cleanser (4.5% encapsulated benzoyl peroxide [BPO]) with time-released technology to control the delivery of BPO and enhance penetration. The targeted delivery system adheres to the skin and penetrates the lipid layer while releasing the encapsulated BPO once warmed by the skin, providing optimal efficacy to inhibit the growth of acne-causing bacteria with minimal irritation. Glytone Acne BPO Clearing Cleanser is dispensed by physicians and can be used with other products in the Glytone acne product line for optimal results. For more information, visit www.glytone-usa.com.
Juvéderm Vollure XC
Allergan announces US Food and Drug Administration approval of Juvéderm Vollure XC for correction of moderate to severe facial wrinkles and folds such as the nasolabial folds in adults older than 21 years. It utilizes VYCROSS technology, which blends different weights of hyaluronic acid, contributing to the gel’s duration. Long-lasting results have been demonstrated up to 18 months. For more information, visit www.juvederm.com.
Neutrogena Light Therapy Acne Mask
Johnson & Johnson Consumer Inc presents the Neutrogena Light Therapy Acne Mask, an LED device utilizing red and blue light to treat acne at home. The mask contains 12 blue LED bulbs that kill Propionibacterium acnes bacteria and 9 red LED bulbs to penetrate deep into the skin to calm inflammation. The mask can be used for 10 minutes each night and shuts off automatically. Results have been seen in 1 week for mild to moderate acne. For more information, visit www.neutrogena.com.
3% Retinol Peel ProSystem
NeoStrata Company, Inc, introduces the 3% Retinol Peel ProSystem featuring Retinol Boosting Complex to exfoliate and improve the appearance of fine lines and winkles, help reduce acne, and improve skin laxity, while promoting a bright, even, and clear complexion. This physician-strength peel is applied in the office but is removed at home after 8 hours or overnight. This peel has demonstrated improvement in acne and skin texture as well as diminished pigmentation. For more information, visit www.neostrata.com.
Siliq
Valeant Pharmaceuticals International, Inc, announces US Food and Drug Administration approval of the Biologics License Application for Siliq (brodalumab) injection. Siliq, an IL-17 inhibitor, is indicated for the treatment of moderate to severe plaque psoriasis in adult patients who are candidates for systemic therapy or phototherapy and have failed to respond or have lost response to other systemic therapies. Siliq has a black box warning for patients with a history of suicidal thoughts or behavior and was approved with a Risk Evaluation and Mitigation Strategy involving a one-time enrollment for physicians and one-time informed consent for patients. Sales and marketing in the United States will begin in the second half of 2017. For more information, visit www.valeant.com.
Thermi
Thermi, an Almirall company, announces “The Art of Thermi” campaign focusing on 2 Thermi devices: ThermiRF and Thermi250. ThermiRF is temperature-controlled radiofrequency technology that uses heat to produce aesthetic outcomes for soft tissue applications. Thermi250 is a high-powered, temperature-controlled radiofrequency system emitting at 470 kHz designed with a user-friendly interface to offer versatility for targeting cellulite. For more information, visit www.thermi.com.
If you would like your product included in Product News, please email a press release to the Editorial Office at [email protected].
Cutanea Life Sciences, Inc, launches Aktipak (erythromycin 3% and benzoyl peroxide 5%) Gel, a prescription combination therapy indicated for acne vulgaris. Aktipak is packaged in a pocket-sized, dual-chamber pouch that contains erythromycin and benzoyl peroxide in separate chambers to enable convenient on-the-go use. Immediately prior to use, the patient cuts or twists open the pouch, squeezes the 2 gels into the palm of the hand, mixes the gels together, and applies the mix to the area affected by acne. Aktipak has an 18-month shelf life and does not require refrigeration. Results can be seen within 8 weeks. For more information, visit www.aktipak.com.
Glytone Acne BPO Clearing Cleanser
Pierre Fabre Group introduces the Glytone Acne BPO Clearing Cleanser (4.5% encapsulated benzoyl peroxide [BPO]) with time-released technology to control the delivery of BPO and enhance penetration. The targeted delivery system adheres to the skin and penetrates the lipid layer while releasing the encapsulated BPO once warmed by the skin, providing optimal efficacy to inhibit the growth of acne-causing bacteria with minimal irritation. Glytone Acne BPO Clearing Cleanser is dispensed by physicians and can be used with other products in the Glytone acne product line for optimal results. For more information, visit www.glytone-usa.com.
Juvéderm Vollure XC
Allergan announces US Food and Drug Administration approval of Juvéderm Vollure XC for correction of moderate to severe facial wrinkles and folds such as the nasolabial folds in adults older than 21 years. It utilizes VYCROSS technology, which blends different weights of hyaluronic acid, contributing to the gel’s duration. Long-lasting results have been demonstrated up to 18 months. For more information, visit www.juvederm.com.
Neutrogena Light Therapy Acne Mask
Johnson & Johnson Consumer Inc presents the Neutrogena Light Therapy Acne Mask, an LED device utilizing red and blue light to treat acne at home. The mask contains 12 blue LED bulbs that kill Propionibacterium acnes bacteria and 9 red LED bulbs to penetrate deep into the skin to calm inflammation. The mask can be used for 10 minutes each night and shuts off automatically. Results have been seen in 1 week for mild to moderate acne. For more information, visit www.neutrogena.com.
3% Retinol Peel ProSystem
NeoStrata Company, Inc, introduces the 3% Retinol Peel ProSystem featuring Retinol Boosting Complex to exfoliate and improve the appearance of fine lines and winkles, help reduce acne, and improve skin laxity, while promoting a bright, even, and clear complexion. This physician-strength peel is applied in the office but is removed at home after 8 hours or overnight. This peel has demonstrated improvement in acne and skin texture as well as diminished pigmentation. For more information, visit www.neostrata.com.
Siliq
Valeant Pharmaceuticals International, Inc, announces US Food and Drug Administration approval of the Biologics License Application for Siliq (brodalumab) injection. Siliq, an IL-17 inhibitor, is indicated for the treatment of moderate to severe plaque psoriasis in adult patients who are candidates for systemic therapy or phototherapy and have failed to respond or have lost response to other systemic therapies. Siliq has a black box warning for patients with a history of suicidal thoughts or behavior and was approved with a Risk Evaluation and Mitigation Strategy involving a one-time enrollment for physicians and one-time informed consent for patients. Sales and marketing in the United States will begin in the second half of 2017. For more information, visit www.valeant.com.
Thermi
Thermi, an Almirall company, announces “The Art of Thermi” campaign focusing on 2 Thermi devices: ThermiRF and Thermi250. ThermiRF is temperature-controlled radiofrequency technology that uses heat to produce aesthetic outcomes for soft tissue applications. Thermi250 is a high-powered, temperature-controlled radiofrequency system emitting at 470 kHz designed with a user-friendly interface to offer versatility for targeting cellulite. For more information, visit www.thermi.com.
If you would like your product included in Product News, please email a press release to the Editorial Office at [email protected].
Cutanea Life Sciences, Inc, launches Aktipak (erythromycin 3% and benzoyl peroxide 5%) Gel, a prescription combination therapy indicated for acne vulgaris. Aktipak is packaged in a pocket-sized, dual-chamber pouch that contains erythromycin and benzoyl peroxide in separate chambers to enable convenient on-the-go use. Immediately prior to use, the patient cuts or twists open the pouch, squeezes the 2 gels into the palm of the hand, mixes the gels together, and applies the mix to the area affected by acne. Aktipak has an 18-month shelf life and does not require refrigeration. Results can be seen within 8 weeks. For more information, visit www.aktipak.com.
Glytone Acne BPO Clearing Cleanser
Pierre Fabre Group introduces the Glytone Acne BPO Clearing Cleanser (4.5% encapsulated benzoyl peroxide [BPO]) with time-released technology to control the delivery of BPO and enhance penetration. The targeted delivery system adheres to the skin and penetrates the lipid layer while releasing the encapsulated BPO once warmed by the skin, providing optimal efficacy to inhibit the growth of acne-causing bacteria with minimal irritation. Glytone Acne BPO Clearing Cleanser is dispensed by physicians and can be used with other products in the Glytone acne product line for optimal results. For more information, visit www.glytone-usa.com.
Juvéderm Vollure XC
Allergan announces US Food and Drug Administration approval of Juvéderm Vollure XC for correction of moderate to severe facial wrinkles and folds such as the nasolabial folds in adults older than 21 years. It utilizes VYCROSS technology, which blends different weights of hyaluronic acid, contributing to the gel’s duration. Long-lasting results have been demonstrated up to 18 months. For more information, visit www.juvederm.com.
Neutrogena Light Therapy Acne Mask
Johnson & Johnson Consumer Inc presents the Neutrogena Light Therapy Acne Mask, an LED device utilizing red and blue light to treat acne at home. The mask contains 12 blue LED bulbs that kill Propionibacterium acnes bacteria and 9 red LED bulbs to penetrate deep into the skin to calm inflammation. The mask can be used for 10 minutes each night and shuts off automatically. Results have been seen in 1 week for mild to moderate acne. For more information, visit www.neutrogena.com.
3% Retinol Peel ProSystem
NeoStrata Company, Inc, introduces the 3% Retinol Peel ProSystem featuring Retinol Boosting Complex to exfoliate and improve the appearance of fine lines and winkles, help reduce acne, and improve skin laxity, while promoting a bright, even, and clear complexion. This physician-strength peel is applied in the office but is removed at home after 8 hours or overnight. This peel has demonstrated improvement in acne and skin texture as well as diminished pigmentation. For more information, visit www.neostrata.com.
Siliq
Valeant Pharmaceuticals International, Inc, announces US Food and Drug Administration approval of the Biologics License Application for Siliq (brodalumab) injection. Siliq, an IL-17 inhibitor, is indicated for the treatment of moderate to severe plaque psoriasis in adult patients who are candidates for systemic therapy or phototherapy and have failed to respond or have lost response to other systemic therapies. Siliq has a black box warning for patients with a history of suicidal thoughts or behavior and was approved with a Risk Evaluation and Mitigation Strategy involving a one-time enrollment for physicians and one-time informed consent for patients. Sales and marketing in the United States will begin in the second half of 2017. For more information, visit www.valeant.com.
Thermi
Thermi, an Almirall company, announces “The Art of Thermi” campaign focusing on 2 Thermi devices: ThermiRF and Thermi250. ThermiRF is temperature-controlled radiofrequency technology that uses heat to produce aesthetic outcomes for soft tissue applications. Thermi250 is a high-powered, temperature-controlled radiofrequency system emitting at 470 kHz designed with a user-friendly interface to offer versatility for targeting cellulite. For more information, visit www.thermi.com.
If you would like your product included in Product News, please email a press release to the Editorial Office at [email protected].

Eliminating hepatitis in the United States: A road map
An ambitious new report by the National Academies of Sciences, Engineering, and Medicine lays out a detailed path by which some 90,000 deaths from hepatitis B and C infection could be prevented by 2030.
The National Academies, a group of nongovernmental advisory bodies that includes the former Institute of Medicine, said that “the tools to prevent these deaths” exist – namely vaccination to prevent new hepatitis B infections and antiviral drugs, including new oral medications that can cure chronic hepatitis C infections within months.
The authors of the 200-plus-page report, led by Brian Strom, MD, MPH, of Rutgers University in Newark, NJ, calculate that deaths from hepatitis B infection could be halved by 2030 if 90% of patients are diagnosed, if 90% of those diagnosed are connected to care, and if 80% of those for whom treatment is indicated receive it. Treating everyone with chronic hepatitis C would reduce new infections by 90% by 2030, while reducing related deaths by 65%, Dr. Strom and his colleagues estimate.
But the authors also concede that drastic changes to current health policy would be required to reach these goals. These include the adoption of “aggressive testing, diagnosis, treatment, and prevention methods, such as needle exchange.”
They propose that the federal government seek a unique licensing arrangement with one or more manufacturers to bring down the notoriously high cost of direct-acting drugs used in hepatitis C, as a way of raising treatment rates. Currently, fewer than half the patients on Medicaid who are eligible for hepatitis C treatment receive it, and fewer than 1% of prisoners, who have high rates of infection.
Dr. Joseph Lim, director of the viral hepatitis program at Yale University in New Haven. Conn., who was not involved in the National Academies report, called it helpful in the sense that “it casts a spotlight on something that those of us involved in the care of people with viral hepatitis have long known – which is that this is a national and global public health burden that has been under the radar and in the shadow of other important health priorities.”
Both hepatitis B and C increase the risk of liver cancer and are associated with significant morbidity and mortality. Though approximately 4 million people in the United States are estimated to be infected with chronic hepatitis B (1.3 million) or C (2.7 million), these diseases account for less than 1% of the research budget at the National Institutes of Health, the report said. This compares unfavorably to funding for HIV, which affects about 1 million Americans.
As the report states, the tools to radically reduce hepatitis B and C deaths already exist. However, Dr. Lim cautioned in an interview, “the public health infrastructure to address viral hepatitis has been woefully inadequate.” In the United States, he noted, most states receive federal funding for at most a single person in charge of viral hepatitis epidemiology. “The resources currently available are in no way adequate to achieve the very aggressive goals described in the report,” he said.
Even among people with a known diagnosis of hepatitis B or C, only some receive confirmatory testing, Dr. Lim said. And of those with confirmed infections, “only a fraction are linked to care from the diagnosing clinician to a provider with the capacity to assess the state of liver disease and determine whether antiviral therapy is warranted.” Finally, he said, “many patients continue to face barriers to curative therapy due to cost and restrictions by public and private payers.”
Among the recommendations contained in the report is that unrestricted, mass treatment of hepatitis C infections be undertaken – regardless of disease stage. Currently, direct-acting antiviral agents remain costly and are poorly covered, notably by Medicaid. The National Academies advise that the government rectify this by purchasing “a license or assignment to the patent on a direct-acting antiviral drug, and use it only in those market segments where the government pays for treatment and access is now limited, such as Medicaid and prisons.”
Dr. Lim called the licensing proposal “very novel and bold,” but noted that there is no precedent in the United States for diseases such as hepatitis C. “If it could be done it would be an incredible model of government-pharma partnership for the public health good, and have a very significant impact.”
Steven Flamm, MD, chief of the liver transplantation program at Northwestern University in Chicago, who like Dr. Lim was not involved in the creation of the report, said in an interview that it contained innovative ideas and helped underscore the fact that “hepatitis has been given short shrift. The NIH and other agencies do not devote time and energy to this particular medical issue for reasons that are not completely clear.”
But “the problem with these kinds of analyses,” he said, “is that carrying them out is harder than making the recommendations.”
Dr. Flamm echoed Dr. Lim’s concerns about the practicability of implementing some of the recommendations in what he considers a resource-deprived health care environment for viral hepatitis.
“Is elimination possible or can you take a big bite out of it? The answer to that question is yes. We now have agents that can treat chronic viral hepatitis well, which we didn’t have a few years ago.”
Still, he emphasized, having the tools is only one part of the picture. Hepatitis C diagnostic tests have been available since the early 1990s. Yet, Dr. Flamm pointed out, fewer than half of patients have been diagnosed. “If the new CDC screening guidelines gain traction, we will do better than that.”
Dr. Flamm said that he considered the report’s call for a unique government licensing agreement for hepatitis C drugs a tall order. The drugs are already heavily discounted by manufacturers in many cases, he said, yet remain unavailable to those in need of them. In Illinois, Dr. Flamm said, few Medicaid patients with confirmed hepatitis C are given the short-acting antivirals that have revolutionized treatment. “The vast majority have no access to the therapy at all,” he said.
One of the report’s strengths, he said, is in detailing innovative prevention strategies such as delivering and promoting hepatitis B vaccinations to adults through local pharmacies, after the model of influenza vaccinations, and also conducting needle exchanges through pharmacies for intravenous drug users, who are at high risk of contracting both hepatitis B and C.
“Many of these strategies are not very costly,” he said. “The problem is you run into moral platitudes – to eliminate hepatitis, we will have to overcome that,” Dr. Flamm said, something that cannot be taken for granted in the current political environment.
But even if the goals outlined in the report seem ambitious, its authors have done an important service in underscoring the burden of viral hepatitis and laying out how some barriers to prevention, diagnosis, and treatment might be broken, he said.
Viral hepatitis “is a big deal, and it does cost a tremendous amount of money,” he added. “Everybody focuses on the therapeutic cost, but nobody focuses on the costs, direct and indirect, of all the sick people that are out there.”
An ambitious new report by the National Academies of Sciences, Engineering, and Medicine lays out a detailed path by which some 90,000 deaths from hepatitis B and C infection could be prevented by 2030.
The National Academies, a group of nongovernmental advisory bodies that includes the former Institute of Medicine, said that “the tools to prevent these deaths” exist – namely vaccination to prevent new hepatitis B infections and antiviral drugs, including new oral medications that can cure chronic hepatitis C infections within months.
The authors of the 200-plus-page report, led by Brian Strom, MD, MPH, of Rutgers University in Newark, NJ, calculate that deaths from hepatitis B infection could be halved by 2030 if 90% of patients are diagnosed, if 90% of those diagnosed are connected to care, and if 80% of those for whom treatment is indicated receive it. Treating everyone with chronic hepatitis C would reduce new infections by 90% by 2030, while reducing related deaths by 65%, Dr. Strom and his colleagues estimate.
But the authors also concede that drastic changes to current health policy would be required to reach these goals. These include the adoption of “aggressive testing, diagnosis, treatment, and prevention methods, such as needle exchange.”
They propose that the federal government seek a unique licensing arrangement with one or more manufacturers to bring down the notoriously high cost of direct-acting drugs used in hepatitis C, as a way of raising treatment rates. Currently, fewer than half the patients on Medicaid who are eligible for hepatitis C treatment receive it, and fewer than 1% of prisoners, who have high rates of infection.
Dr. Joseph Lim, director of the viral hepatitis program at Yale University in New Haven. Conn., who was not involved in the National Academies report, called it helpful in the sense that “it casts a spotlight on something that those of us involved in the care of people with viral hepatitis have long known – which is that this is a national and global public health burden that has been under the radar and in the shadow of other important health priorities.”
Both hepatitis B and C increase the risk of liver cancer and are associated with significant morbidity and mortality. Though approximately 4 million people in the United States are estimated to be infected with chronic hepatitis B (1.3 million) or C (2.7 million), these diseases account for less than 1% of the research budget at the National Institutes of Health, the report said. This compares unfavorably to funding for HIV, which affects about 1 million Americans.
As the report states, the tools to radically reduce hepatitis B and C deaths already exist. However, Dr. Lim cautioned in an interview, “the public health infrastructure to address viral hepatitis has been woefully inadequate.” In the United States, he noted, most states receive federal funding for at most a single person in charge of viral hepatitis epidemiology. “The resources currently available are in no way adequate to achieve the very aggressive goals described in the report,” he said.
Even among people with a known diagnosis of hepatitis B or C, only some receive confirmatory testing, Dr. Lim said. And of those with confirmed infections, “only a fraction are linked to care from the diagnosing clinician to a provider with the capacity to assess the state of liver disease and determine whether antiviral therapy is warranted.” Finally, he said, “many patients continue to face barriers to curative therapy due to cost and restrictions by public and private payers.”
Among the recommendations contained in the report is that unrestricted, mass treatment of hepatitis C infections be undertaken – regardless of disease stage. Currently, direct-acting antiviral agents remain costly and are poorly covered, notably by Medicaid. The National Academies advise that the government rectify this by purchasing “a license or assignment to the patent on a direct-acting antiviral drug, and use it only in those market segments where the government pays for treatment and access is now limited, such as Medicaid and prisons.”
Dr. Lim called the licensing proposal “very novel and bold,” but noted that there is no precedent in the United States for diseases such as hepatitis C. “If it could be done it would be an incredible model of government-pharma partnership for the public health good, and have a very significant impact.”
Steven Flamm, MD, chief of the liver transplantation program at Northwestern University in Chicago, who like Dr. Lim was not involved in the creation of the report, said in an interview that it contained innovative ideas and helped underscore the fact that “hepatitis has been given short shrift. The NIH and other agencies do not devote time and energy to this particular medical issue for reasons that are not completely clear.”
But “the problem with these kinds of analyses,” he said, “is that carrying them out is harder than making the recommendations.”
Dr. Flamm echoed Dr. Lim’s concerns about the practicability of implementing some of the recommendations in what he considers a resource-deprived health care environment for viral hepatitis.
“Is elimination possible or can you take a big bite out of it? The answer to that question is yes. We now have agents that can treat chronic viral hepatitis well, which we didn’t have a few years ago.”
Still, he emphasized, having the tools is only one part of the picture. Hepatitis C diagnostic tests have been available since the early 1990s. Yet, Dr. Flamm pointed out, fewer than half of patients have been diagnosed. “If the new CDC screening guidelines gain traction, we will do better than that.”
Dr. Flamm said that he considered the report’s call for a unique government licensing agreement for hepatitis C drugs a tall order. The drugs are already heavily discounted by manufacturers in many cases, he said, yet remain unavailable to those in need of them. In Illinois, Dr. Flamm said, few Medicaid patients with confirmed hepatitis C are given the short-acting antivirals that have revolutionized treatment. “The vast majority have no access to the therapy at all,” he said.
One of the report’s strengths, he said, is in detailing innovative prevention strategies such as delivering and promoting hepatitis B vaccinations to adults through local pharmacies, after the model of influenza vaccinations, and also conducting needle exchanges through pharmacies for intravenous drug users, who are at high risk of contracting both hepatitis B and C.
“Many of these strategies are not very costly,” he said. “The problem is you run into moral platitudes – to eliminate hepatitis, we will have to overcome that,” Dr. Flamm said, something that cannot be taken for granted in the current political environment.
But even if the goals outlined in the report seem ambitious, its authors have done an important service in underscoring the burden of viral hepatitis and laying out how some barriers to prevention, diagnosis, and treatment might be broken, he said.
Viral hepatitis “is a big deal, and it does cost a tremendous amount of money,” he added. “Everybody focuses on the therapeutic cost, but nobody focuses on the costs, direct and indirect, of all the sick people that are out there.”
An ambitious new report by the National Academies of Sciences, Engineering, and Medicine lays out a detailed path by which some 90,000 deaths from hepatitis B and C infection could be prevented by 2030.
The National Academies, a group of nongovernmental advisory bodies that includes the former Institute of Medicine, said that “the tools to prevent these deaths” exist – namely vaccination to prevent new hepatitis B infections and antiviral drugs, including new oral medications that can cure chronic hepatitis C infections within months.
The authors of the 200-plus-page report, led by Brian Strom, MD, MPH, of Rutgers University in Newark, NJ, calculate that deaths from hepatitis B infection could be halved by 2030 if 90% of patients are diagnosed, if 90% of those diagnosed are connected to care, and if 80% of those for whom treatment is indicated receive it. Treating everyone with chronic hepatitis C would reduce new infections by 90% by 2030, while reducing related deaths by 65%, Dr. Strom and his colleagues estimate.
But the authors also concede that drastic changes to current health policy would be required to reach these goals. These include the adoption of “aggressive testing, diagnosis, treatment, and prevention methods, such as needle exchange.”
They propose that the federal government seek a unique licensing arrangement with one or more manufacturers to bring down the notoriously high cost of direct-acting drugs used in hepatitis C, as a way of raising treatment rates. Currently, fewer than half the patients on Medicaid who are eligible for hepatitis C treatment receive it, and fewer than 1% of prisoners, who have high rates of infection.
Dr. Joseph Lim, director of the viral hepatitis program at Yale University in New Haven. Conn., who was not involved in the National Academies report, called it helpful in the sense that “it casts a spotlight on something that those of us involved in the care of people with viral hepatitis have long known – which is that this is a national and global public health burden that has been under the radar and in the shadow of other important health priorities.”
Both hepatitis B and C increase the risk of liver cancer and are associated with significant morbidity and mortality. Though approximately 4 million people in the United States are estimated to be infected with chronic hepatitis B (1.3 million) or C (2.7 million), these diseases account for less than 1% of the research budget at the National Institutes of Health, the report said. This compares unfavorably to funding for HIV, which affects about 1 million Americans.
As the report states, the tools to radically reduce hepatitis B and C deaths already exist. However, Dr. Lim cautioned in an interview, “the public health infrastructure to address viral hepatitis has been woefully inadequate.” In the United States, he noted, most states receive federal funding for at most a single person in charge of viral hepatitis epidemiology. “The resources currently available are in no way adequate to achieve the very aggressive goals described in the report,” he said.
Even among people with a known diagnosis of hepatitis B or C, only some receive confirmatory testing, Dr. Lim said. And of those with confirmed infections, “only a fraction are linked to care from the diagnosing clinician to a provider with the capacity to assess the state of liver disease and determine whether antiviral therapy is warranted.” Finally, he said, “many patients continue to face barriers to curative therapy due to cost and restrictions by public and private payers.”
Among the recommendations contained in the report is that unrestricted, mass treatment of hepatitis C infections be undertaken – regardless of disease stage. Currently, direct-acting antiviral agents remain costly and are poorly covered, notably by Medicaid. The National Academies advise that the government rectify this by purchasing “a license or assignment to the patent on a direct-acting antiviral drug, and use it only in those market segments where the government pays for treatment and access is now limited, such as Medicaid and prisons.”
Dr. Lim called the licensing proposal “very novel and bold,” but noted that there is no precedent in the United States for diseases such as hepatitis C. “If it could be done it would be an incredible model of government-pharma partnership for the public health good, and have a very significant impact.”
Steven Flamm, MD, chief of the liver transplantation program at Northwestern University in Chicago, who like Dr. Lim was not involved in the creation of the report, said in an interview that it contained innovative ideas and helped underscore the fact that “hepatitis has been given short shrift. The NIH and other agencies do not devote time and energy to this particular medical issue for reasons that are not completely clear.”
But “the problem with these kinds of analyses,” he said, “is that carrying them out is harder than making the recommendations.”
Dr. Flamm echoed Dr. Lim’s concerns about the practicability of implementing some of the recommendations in what he considers a resource-deprived health care environment for viral hepatitis.
“Is elimination possible or can you take a big bite out of it? The answer to that question is yes. We now have agents that can treat chronic viral hepatitis well, which we didn’t have a few years ago.”
Still, he emphasized, having the tools is only one part of the picture. Hepatitis C diagnostic tests have been available since the early 1990s. Yet, Dr. Flamm pointed out, fewer than half of patients have been diagnosed. “If the new CDC screening guidelines gain traction, we will do better than that.”
Dr. Flamm said that he considered the report’s call for a unique government licensing agreement for hepatitis C drugs a tall order. The drugs are already heavily discounted by manufacturers in many cases, he said, yet remain unavailable to those in need of them. In Illinois, Dr. Flamm said, few Medicaid patients with confirmed hepatitis C are given the short-acting antivirals that have revolutionized treatment. “The vast majority have no access to the therapy at all,” he said.
One of the report’s strengths, he said, is in detailing innovative prevention strategies such as delivering and promoting hepatitis B vaccinations to adults through local pharmacies, after the model of influenza vaccinations, and also conducting needle exchanges through pharmacies for intravenous drug users, who are at high risk of contracting both hepatitis B and C.
“Many of these strategies are not very costly,” he said. “The problem is you run into moral platitudes – to eliminate hepatitis, we will have to overcome that,” Dr. Flamm said, something that cannot be taken for granted in the current political environment.
But even if the goals outlined in the report seem ambitious, its authors have done an important service in underscoring the burden of viral hepatitis and laying out how some barriers to prevention, diagnosis, and treatment might be broken, he said.
Viral hepatitis “is a big deal, and it does cost a tremendous amount of money,” he added. “Everybody focuses on the therapeutic cost, but nobody focuses on the costs, direct and indirect, of all the sick people that are out there.”
FROM THE NATIONAL ACADEMIES OF SCIENCES, ENGINEERING, AND MEDICINE
Psoriasis on the Hands and Feet: How Patients Should Care for These Areas
What does your patient need to know at the first visit?
Patients with this condition need to avoid friction and excessive moisture. They should be counseled to use gloves for excessive wet work. I recommend they use cotton gloves on the hands, and then cover those with rubber gloves. Patients should use a hand emollient regularly, including after each time they wash their hands or have exposure to water. If the patient lifts weights, I recommend he/she use weight-lifting gloves to reduce friction.
What are your go to treatments? What are the side effects?
The first line of therapy for hand and foot psoriasis is a topical agent. I most often use a combination of topical steroids and a topical vitamin D analogue. If insurance is amenable, I may use a fixed combination of topical steroid and vitamin D analogue.
If topical therapies are not successful, I often consider using excimer laser therapy, which requires the patient to come to the office twice weekly, so it is important to determine if this therapy is compatible with the patient's schedule. Other options include oral and biological therapies. Apremilast is a reasonable first-line systemic therapy given that it is an oral therapy, requires no laboratory monitoring, and has a favorable safety profile. Alternatively, biologic agents can be utilized. There are several analyses available looking at the efficacy of different biologics in hand and foot psoriasis, but at this point there is no consensus first choice for a biologic in this condition. Many available biologics may have a notable impact though.
The side effects of therapies for psoriasis are well established. Topical therapies and excimer laser are relatively safe choices. Apremilast has been associated with early gastrointestinal tract side effects that tend to resolve over time. Each biologic has a unique safety profile, with a rare incidence of side effects that should be reviewed carefully with any prospective patients before starting therapy.
How do you keep patients compliant with treatment?
It is important to reinforce gentle hand care and foot care. Patients need to understand that lack of compliance with treatment will lead to recurrence of disease.
What do you do if patients refuse treatment?
I try to educate them as best as possible, and ask them to return and reconsider therapy if they find that this condition affects their quality of life.
What does your patient need to know at the first visit?
Patients with this condition need to avoid friction and excessive moisture. They should be counseled to use gloves for excessive wet work. I recommend they use cotton gloves on the hands, and then cover those with rubber gloves. Patients should use a hand emollient regularly, including after each time they wash their hands or have exposure to water. If the patient lifts weights, I recommend he/she use weight-lifting gloves to reduce friction.
What are your go to treatments? What are the side effects?
The first line of therapy for hand and foot psoriasis is a topical agent. I most often use a combination of topical steroids and a topical vitamin D analogue. If insurance is amenable, I may use a fixed combination of topical steroid and vitamin D analogue.
If topical therapies are not successful, I often consider using excimer laser therapy, which requires the patient to come to the office twice weekly, so it is important to determine if this therapy is compatible with the patient's schedule. Other options include oral and biological therapies. Apremilast is a reasonable first-line systemic therapy given that it is an oral therapy, requires no laboratory monitoring, and has a favorable safety profile. Alternatively, biologic agents can be utilized. There are several analyses available looking at the efficacy of different biologics in hand and foot psoriasis, but at this point there is no consensus first choice for a biologic in this condition. Many available biologics may have a notable impact though.
The side effects of therapies for psoriasis are well established. Topical therapies and excimer laser are relatively safe choices. Apremilast has been associated with early gastrointestinal tract side effects that tend to resolve over time. Each biologic has a unique safety profile, with a rare incidence of side effects that should be reviewed carefully with any prospective patients before starting therapy.
How do you keep patients compliant with treatment?
It is important to reinforce gentle hand care and foot care. Patients need to understand that lack of compliance with treatment will lead to recurrence of disease.
What do you do if patients refuse treatment?
I try to educate them as best as possible, and ask them to return and reconsider therapy if they find that this condition affects their quality of life.
What does your patient need to know at the first visit?
Patients with this condition need to avoid friction and excessive moisture. They should be counseled to use gloves for excessive wet work. I recommend they use cotton gloves on the hands, and then cover those with rubber gloves. Patients should use a hand emollient regularly, including after each time they wash their hands or have exposure to water. If the patient lifts weights, I recommend he/she use weight-lifting gloves to reduce friction.
What are your go to treatments? What are the side effects?
The first line of therapy for hand and foot psoriasis is a topical agent. I most often use a combination of topical steroids and a topical vitamin D analogue. If insurance is amenable, I may use a fixed combination of topical steroid and vitamin D analogue.
If topical therapies are not successful, I often consider using excimer laser therapy, which requires the patient to come to the office twice weekly, so it is important to determine if this therapy is compatible with the patient's schedule. Other options include oral and biological therapies. Apremilast is a reasonable first-line systemic therapy given that it is an oral therapy, requires no laboratory monitoring, and has a favorable safety profile. Alternatively, biologic agents can be utilized. There are several analyses available looking at the efficacy of different biologics in hand and foot psoriasis, but at this point there is no consensus first choice for a biologic in this condition. Many available biologics may have a notable impact though.
The side effects of therapies for psoriasis are well established. Topical therapies and excimer laser are relatively safe choices. Apremilast has been associated with early gastrointestinal tract side effects that tend to resolve over time. Each biologic has a unique safety profile, with a rare incidence of side effects that should be reviewed carefully with any prospective patients before starting therapy.
How do you keep patients compliant with treatment?
It is important to reinforce gentle hand care and foot care. Patients need to understand that lack of compliance with treatment will lead to recurrence of disease.
What do you do if patients refuse treatment?
I try to educate them as best as possible, and ask them to return and reconsider therapy if they find that this condition affects their quality of life.

Microneedling Therapy With and Without Platelet-Rich Plasma
Microneedling therapy, also known as collagen induction therapy or percutaneous collagen induction, is an increasingly popular treatment modality for skin rejuvenation. The approach employs small needles to puncture the skin and stimulate local collagen production in a minimally invasive manner. Recently, clinicians have incorporated the use of platelet-rich plasma (PRP) with the aim of augmenting cosmetic outcomes. In this article, we examine the utility of this approach by reviewing comparison studies of microneedling therapy with and without the application of PRP.
Dr. Gary Goldenberg demonstrates microneedling with platelet-rich plasma in a procedural video available here.
Microneedling Therapy
The use of microneedling first gained attention in the 1990s. Initially, Camirand and Doucet1 described tattooing without pigment for the treatment of achromatic and hypertrophic scars. Fernandes2 evolved this concept and developed a drum-shaped device with fine protruding needles to puncture the skin. Microneedling devices have expanded in recent years and now include both cord- and battery-powered pens and rollers, with needles ranging in length from 0.25 to 3.0 mm.
Treatment with microneedling promotes skin rejuvenation by creating small puncture wounds in the epidermis and dermis. This injury triggers the wound healing cascade and alters the modulation of growth factors to promote regenerative effects.3,4 Following microneedling therapy, increases occur in elastic fiber formation, collagen deposition, and dermal thickness (Figure).5 Of interesting histologic note, collagen is deposited in the normal lattice pattern following this treatment rather than in the parallel bundles typical of scars.6 Microneedling preserves the overall integrity of the epidermal layers and basement membrane, allowing the epidermis to heal without abnormality, verified on histology by a normal stratum corneum, enhanced stratum granulosum, and normal rete ridges.7
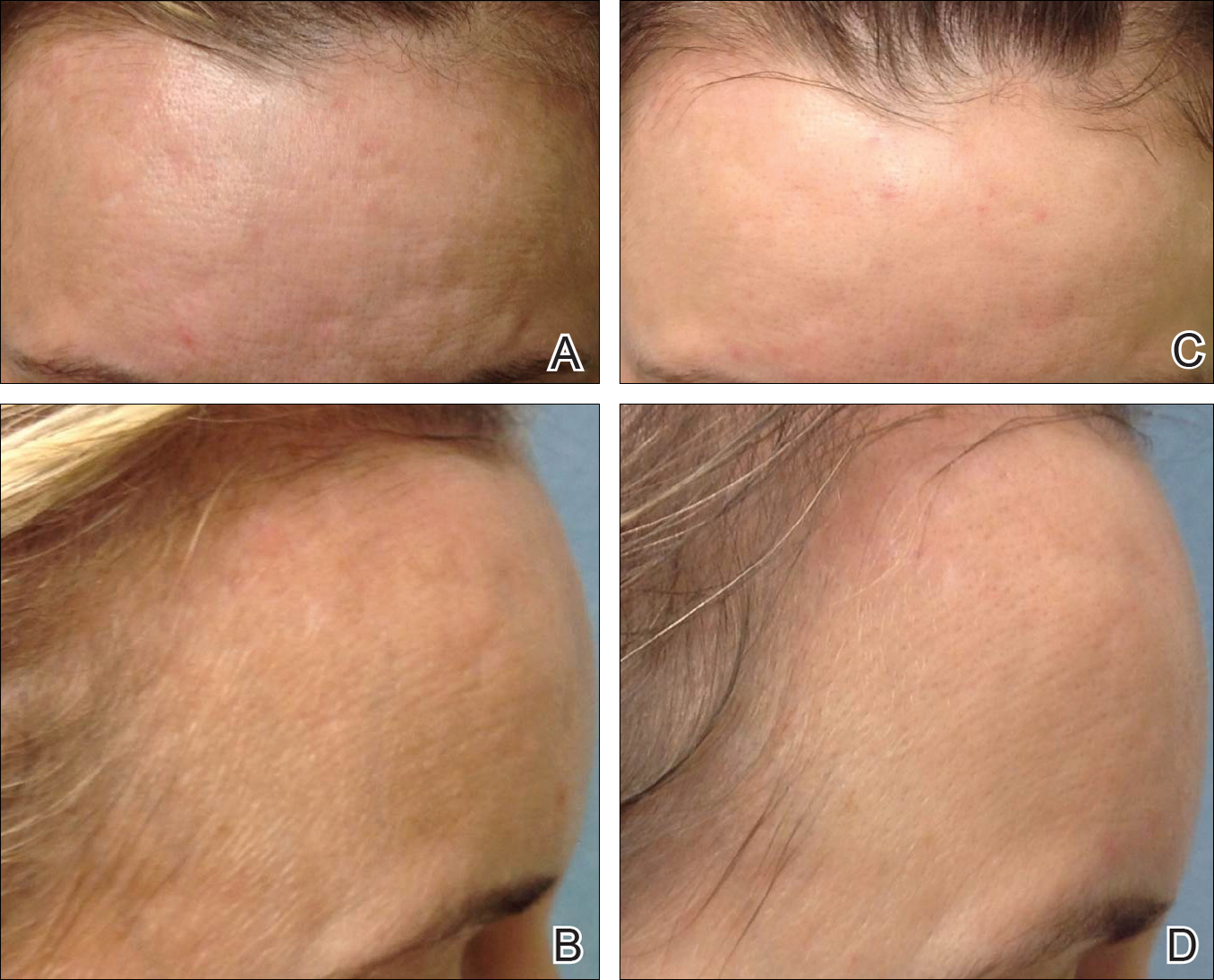
Microneedling has demonstrated several uses beyond general skin rejuvenation. In patients with atrophic acne scars, therapy can lead to improved scar appearance, skin texture, and patient satisfaction.8,9 Hypertrophic and dyspigmented burn scars on the body, face, arms, and legs have shown to be receptive to repeated treatments.10 Microneedling also has shown promise in treating androgenic alopecia, increasing hair regrowth in patients who previously showed poor response to conventional therapy with minoxidil and finasteride.11,12
Platelet-Rich Plasma
Platelet-rich plasma is developed by enriching blood with an autologous concentration of platelets. The preparation of PRP begins with whole blood, commonly obtained peripherally by venipuncture. Samples undergo centrifugation to allow separation of the blood into 3 layers: platelet-poor plasma, PRP, and erythrocytes.13 The typical platelet count of whole blood is approximately 200,000/µL; PRP aims to prepare a platelet count of at least 1,000,000/µL in a 5-mL volume.14
An attractive component of PRP is its high concentration of growth factors, including platelet-derived growth factor, transforming growth factor, vascular endothelial growth factor, and epithelial growth factor.15 Because of the regenerative effects of these proteins, PRP has been investigated as a modality to augment wound healing in a variety of clinical areas, such as maxillofacial surgery, orthopedics, cardiovascular surgery, and treatment of soft tissue ulcers.16
Combination Use of Microneedling and PRP
Several studies have compared the effects of microneedling with and without the application of PRP (Table).17-20 In an animal model, Akcal et al17 examined the effects of microneedling and PRP on skin flap survival. Eight rats were randomly divided into 5 groups: sham, control, microneedling alone, microneedling plus PRP, and microneedling plus platelet-poor plasma. Treatments were applied to skin flaps after 4 hours of induced ischemia. The surviving flap area was measured, with results demonstrating significantly higher viable areas in the microneedling plus PRP group relative to all other groups (P<.01). On histologic examination, the microneedling plus PRP group showed well-organized epidermal layers and a dermal integrity that matched the dermis of the sham group.17
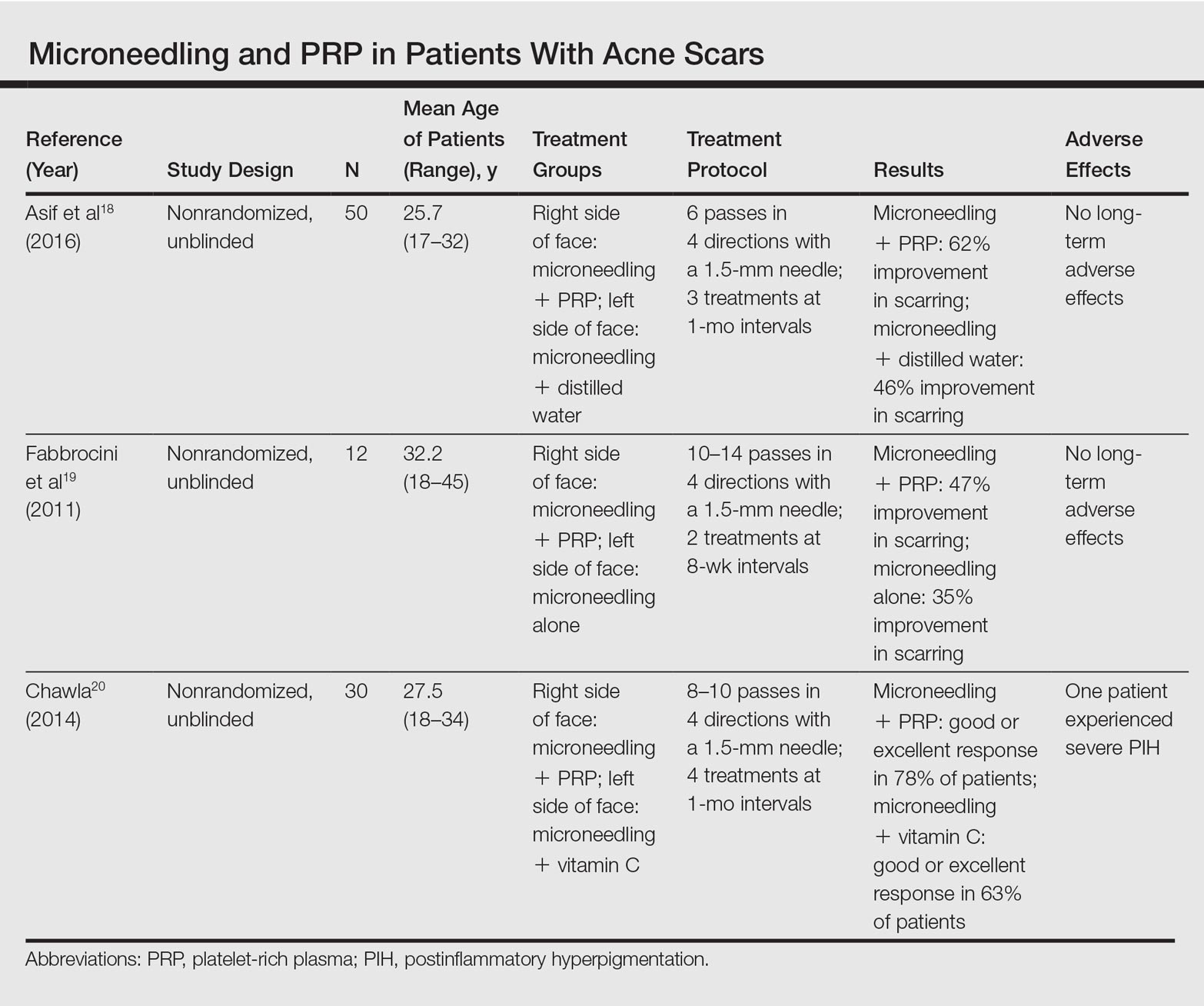
Asif et al18 performed a split-face comparison study of 50 patients with atrophic acne scars. On the right side, microneedling was performed followed by intradermal injections and topical application of PRP. On the left side, microneedling was performed followed by intradermal injections of distilled water. The study included 3 treatment sessions with 1 month between each session. Scars were assessed using the Goodman and Baron scale,21 which is designed to grade the morphology of postacne scarring. Scars on the right side improved by 62.2% and scars on the left side improved by 45.8%; prior to treatment, both sides demonstrated similar severity scores, but final severity scores were significantly reduced in the microneedling plus PRP group relative to the microneedling plus distilled water group (P<.00001). No residual side effects from treatment were reported.18
Examining the degree of improvement more carefully, microneedling plus PRP yielded excellent improvement in 40% (20/50) of patients and good improvement in 60% (30/50).18 Microneedling plus distilled water led to excellent improvement in 10% (5/50) and good improvement in 84% (42/50). Given that microneedling plus distilled water still provided good to excellent results in 94% of patients, the addition of PRP was helpful though not necessary in achieving meaningful benefit.18
In another split-face study, Fabbrocini et al19 evaluated 12 adult patients with acne scars. The right side of the face received microneedling plus PRP, while the left side received microneedling alone. Two treatments were performed 8 weeks apart. Severity scores (0=no lesions; 10=maximum severity) were used to assess patient outcomes throughout the study. Acne scars improved on both sides of the face following the treatment period, but the reduction in scar severity with microneedling plus PRP (3.5 points) was significantly greater than with microneedling alone (2.6 points)(P<.05). Patients tended to experience2 to 3 days of mild swelling and erythema after treatment regardless of PRP addition. With only 12 patients, the study was limited by a small sample size. The 10-point grading system differed from the Goodman and Baron scale in that it lacked corresponding qualitative markers, likely decreasing reproducibility.19
Chawla20 compared the effectiveness of combination therapy with microneedling plus PRP versus microneedling and vitamin C application. In a split-face study of 30 patients with atrophic acne scars, the right side of the face was treated with microneedling plus PRP and the left side was treated with microneedling plus vitamin C. Four sessions were performed with an interval of 1 month in between treatments. The Goodman and Baron Scale was used to assess treatment efficacy. Overall, both treatments led to improved outcomes, but in categorizing patients who demonstrated poor responses, a significantly larger percentage existed in the microneedling plus vitamin C group (37% [10/27]) versus the microneedling plus PRP group (22% [6/27])(P=.021). Additionally, aggregate patient satisfaction scores were higher with microneedling plus PRP relative to microneedling plus vitamin C (P=.01). Of note, assessments of improvement were performed by the treating physician and patient satisfaction reports were completed with knowledge of the therapies and cost factor, which may have influenced results.20
Conclusion
Microneedling therapy continues to evolve with a range of applications now emerging in dermatology. As PRP has gained popularity, there has been increased interest in its utilization to amplify the regenerative effects of microneedling. Although the number of direct comparisons examining microneedling with and without PRP is limited, the available evidence indicates that the addition of PRP may improve cosmetic outcomes. These results have been demonstrated primarily in the management of acne scars, but favorable effects may extend to other indications. Continued study is warranted to further quantify the degree of these benefits and to elucidate optimal treatment schedules.
In addition, it is important to consider a cost-benefit analysis of PRP. The price of PRP varies depending on the clinical site but in certain cases may double the cost of a microneedling treatment session. Although studies have demonstrated a statistically significant benefit to PRP, the clinical significance of this supplementary treatment must be weighed against the increased expense. A discussion should take place with the consideration that microneedling alone can provide a satisfactory result for some patients.
- Camirand A, Doucet J. Needle dermabrasion. Aesthetic Plast Surg. 1997;21:48-51.
- Fernandes D. Percutaneous collagen induction: an alternative to laser resurfacing. Aesthet Surg J. 2002;22:307-309.
- Fabbrocini G, Fardella N, Monfrecola A, et al. Acne scarring treatment using skin needling. Clin Exp Dermatol. 2009;34:874-879.
- Zeitter S, Sikora Z, Jahn S, et al. Microneedling: matching the results of medical needling and repetitive treatments to maximize potential for skin regeneration [published online February 7, 2014]. Burns. 2014;40:966-973.
- Schwarz M, Laaff H. A prospective controlled assessment of microneedling with the Dermaroller device. Plast Reconstr Surg. 2011;127:E146-E148.
- Fernandes D, Signorini M. Combating photoaging with percutaneous collagen induction. Clin Dermatol. 2008;26:192-199.
- Aust MC, Fernandes D, Kolokythas P, et al. Percutaneous collagen induction therapy: an alternative treatment for scars, wrinkles, and skin laxity. Plast Reconstr Surg. 2008;121:1421-1429.
- El-Domyati M, Barakat M, Awad S, et al. Microneedling therapy for atrophic acne scars: an objective evaluation. J Clin Aesthet Dermatol. 2015;8:36-42.
- Leheta T, El Tawdy A, Abdel Hay R, et al. Percutaneous collagen induction versus full-concentration trichloroacetic acid in the treatment of atrophic acne scars. Dermatol Surg. 2011;37:207-216.
- Aust MC, Knobloch K, Reimers K, et al. Percutaneous collagen induction therapy: an alternative treatment for burn scars. Burns. 2010;36:836-843.
- Dhurat R, Mathapati S. Response to microneedling treatment in men with androgenetic alopecia who failed to respond to conventional therapy. Indian J Dermatol. 2015;60:260-263.
- Dhurat R, Sukesh M, Avhad G, et al. A randomized evaluator blinded study of effect of microneedling in androgenetic alopecia: a pilot study. Int J Trichology. 2013;5:6-11.
- Wang HL, Avila G. Platelet rich plasma: myth or reality? Eur J Dent. 2007;1:192-194.
- Marx RE. Platelet-rich plasma (PRP): what is PRP and what is not PRP? Implant Dent. 2001;10:225-228.
- Lubkowska A, Dolegowska B, Banfi G. Growth factor content in PRP and their applicability in medicine. J Biol Regul Homeost Agents. 2012;26(2 suppl 1):3S-22S.
- Pietrzak WS, Eppley BL. Platelet rich plasma: biology and new technology. J Craniofac Surg. 2005;16:1043-1054.
- Akcal A, Savas SA, Gorgulu T, et al. The effect of platelete rich plasma combined with microneedling on full venous outflow compromise in a rat skin flap model. Plast Reconstr Surg. 2015;136(4 suppl):71-72.
- Asif M, Kanodia S, Singh K. Combined autologous platelet-rich plasma with microneedling verses microneedling with distilled water in the treatment of atrophic acne scars: a concurrent split-face study [published online January 8, 2016]. J Cosmet Dermatol. 2016;15:434-443.
- Fabbrocini G, De Vita V, Pastore F, et al. Combined use of skin needling and platelet-rich plasma in acne scarring treatment. Cosmet Dermatol. 2011;24:177-183.
- Chawla S. Split face comparative study of microneedling with PRP versus microneedling with vitamin C in treating atrophic post acne scars. J Cutan Aesthet Surg. 2014;7:209-212.
- Goodman GJ, Baron JA. Postacne scarring: a qualitative global scarring grading system. Dermatol Surg. 2006;32:1458-1466.
Microneedling therapy, also known as collagen induction therapy or percutaneous collagen induction, is an increasingly popular treatment modality for skin rejuvenation. The approach employs small needles to puncture the skin and stimulate local collagen production in a minimally invasive manner. Recently, clinicians have incorporated the use of platelet-rich plasma (PRP) with the aim of augmenting cosmetic outcomes. In this article, we examine the utility of this approach by reviewing comparison studies of microneedling therapy with and without the application of PRP.
Dr. Gary Goldenberg demonstrates microneedling with platelet-rich plasma in a procedural video available here.
Microneedling Therapy
The use of microneedling first gained attention in the 1990s. Initially, Camirand and Doucet1 described tattooing without pigment for the treatment of achromatic and hypertrophic scars. Fernandes2 evolved this concept and developed a drum-shaped device with fine protruding needles to puncture the skin. Microneedling devices have expanded in recent years and now include both cord- and battery-powered pens and rollers, with needles ranging in length from 0.25 to 3.0 mm.
Treatment with microneedling promotes skin rejuvenation by creating small puncture wounds in the epidermis and dermis. This injury triggers the wound healing cascade and alters the modulation of growth factors to promote regenerative effects.3,4 Following microneedling therapy, increases occur in elastic fiber formation, collagen deposition, and dermal thickness (Figure).5 Of interesting histologic note, collagen is deposited in the normal lattice pattern following this treatment rather than in the parallel bundles typical of scars.6 Microneedling preserves the overall integrity of the epidermal layers and basement membrane, allowing the epidermis to heal without abnormality, verified on histology by a normal stratum corneum, enhanced stratum granulosum, and normal rete ridges.7
Microneedling has demonstrated several uses beyond general skin rejuvenation. In patients with atrophic acne scars, therapy can lead to improved scar appearance, skin texture, and patient satisfaction.8,9 Hypertrophic and dyspigmented burn scars on the body, face, arms, and legs have shown to be receptive to repeated treatments.10 Microneedling also has shown promise in treating androgenic alopecia, increasing hair regrowth in patients who previously showed poor response to conventional therapy with minoxidil and finasteride.11,12
Platelet-Rich Plasma
Platelet-rich plasma is developed by enriching blood with an autologous concentration of platelets. The preparation of PRP begins with whole blood, commonly obtained peripherally by venipuncture. Samples undergo centrifugation to allow separation of the blood into 3 layers: platelet-poor plasma, PRP, and erythrocytes.13 The typical platelet count of whole blood is approximately 200,000/µL; PRP aims to prepare a platelet count of at least 1,000,000/µL in a 5-mL volume.14
An attractive component of PRP is its high concentration of growth factors, including platelet-derived growth factor, transforming growth factor, vascular endothelial growth factor, and epithelial growth factor.15 Because of the regenerative effects of these proteins, PRP has been investigated as a modality to augment wound healing in a variety of clinical areas, such as maxillofacial surgery, orthopedics, cardiovascular surgery, and treatment of soft tissue ulcers.16
Combination Use of Microneedling and PRP
Several studies have compared the effects of microneedling with and without the application of PRP (Table).17-20 In an animal model, Akcal et al17 examined the effects of microneedling and PRP on skin flap survival. Eight rats were randomly divided into 5 groups: sham, control, microneedling alone, microneedling plus PRP, and microneedling plus platelet-poor plasma. Treatments were applied to skin flaps after 4 hours of induced ischemia. The surviving flap area was measured, with results demonstrating significantly higher viable areas in the microneedling plus PRP group relative to all other groups (P<.01). On histologic examination, the microneedling plus PRP group showed well-organized epidermal layers and a dermal integrity that matched the dermis of the sham group.17
Asif et al18 performed a split-face comparison study of 50 patients with atrophic acne scars. On the right side, microneedling was performed followed by intradermal injections and topical application of PRP. On the left side, microneedling was performed followed by intradermal injections of distilled water. The study included 3 treatment sessions with 1 month between each session. Scars were assessed using the Goodman and Baron scale,21 which is designed to grade the morphology of postacne scarring. Scars on the right side improved by 62.2% and scars on the left side improved by 45.8%; prior to treatment, both sides demonstrated similar severity scores, but final severity scores were significantly reduced in the microneedling plus PRP group relative to the microneedling plus distilled water group (P<.00001). No residual side effects from treatment were reported.18
Examining the degree of improvement more carefully, microneedling plus PRP yielded excellent improvement in 40% (20/50) of patients and good improvement in 60% (30/50).18 Microneedling plus distilled water led to excellent improvement in 10% (5/50) and good improvement in 84% (42/50). Given that microneedling plus distilled water still provided good to excellent results in 94% of patients, the addition of PRP was helpful though not necessary in achieving meaningful benefit.18
In another split-face study, Fabbrocini et al19 evaluated 12 adult patients with acne scars. The right side of the face received microneedling plus PRP, while the left side received microneedling alone. Two treatments were performed 8 weeks apart. Severity scores (0=no lesions; 10=maximum severity) were used to assess patient outcomes throughout the study. Acne scars improved on both sides of the face following the treatment period, but the reduction in scar severity with microneedling plus PRP (3.5 points) was significantly greater than with microneedling alone (2.6 points)(P<.05). Patients tended to experience2 to 3 days of mild swelling and erythema after treatment regardless of PRP addition. With only 12 patients, the study was limited by a small sample size. The 10-point grading system differed from the Goodman and Baron scale in that it lacked corresponding qualitative markers, likely decreasing reproducibility.19
Chawla20 compared the effectiveness of combination therapy with microneedling plus PRP versus microneedling and vitamin C application. In a split-face study of 30 patients with atrophic acne scars, the right side of the face was treated with microneedling plus PRP and the left side was treated with microneedling plus vitamin C. Four sessions were performed with an interval of 1 month in between treatments. The Goodman and Baron Scale was used to assess treatment efficacy. Overall, both treatments led to improved outcomes, but in categorizing patients who demonstrated poor responses, a significantly larger percentage existed in the microneedling plus vitamin C group (37% [10/27]) versus the microneedling plus PRP group (22% [6/27])(P=.021). Additionally, aggregate patient satisfaction scores were higher with microneedling plus PRP relative to microneedling plus vitamin C (P=.01). Of note, assessments of improvement were performed by the treating physician and patient satisfaction reports were completed with knowledge of the therapies and cost factor, which may have influenced results.20
Conclusion
Microneedling therapy continues to evolve with a range of applications now emerging in dermatology. As PRP has gained popularity, there has been increased interest in its utilization to amplify the regenerative effects of microneedling. Although the number of direct comparisons examining microneedling with and without PRP is limited, the available evidence indicates that the addition of PRP may improve cosmetic outcomes. These results have been demonstrated primarily in the management of acne scars, but favorable effects may extend to other indications. Continued study is warranted to further quantify the degree of these benefits and to elucidate optimal treatment schedules.
In addition, it is important to consider a cost-benefit analysis of PRP. The price of PRP varies depending on the clinical site but in certain cases may double the cost of a microneedling treatment session. Although studies have demonstrated a statistically significant benefit to PRP, the clinical significance of this supplementary treatment must be weighed against the increased expense. A discussion should take place with the consideration that microneedling alone can provide a satisfactory result for some patients.
Microneedling therapy, also known as collagen induction therapy or percutaneous collagen induction, is an increasingly popular treatment modality for skin rejuvenation. The approach employs small needles to puncture the skin and stimulate local collagen production in a minimally invasive manner. Recently, clinicians have incorporated the use of platelet-rich plasma (PRP) with the aim of augmenting cosmetic outcomes. In this article, we examine the utility of this approach by reviewing comparison studies of microneedling therapy with and without the application of PRP.
Dr. Gary Goldenberg demonstrates microneedling with platelet-rich plasma in a procedural video available here.
Microneedling Therapy
The use of microneedling first gained attention in the 1990s. Initially, Camirand and Doucet1 described tattooing without pigment for the treatment of achromatic and hypertrophic scars. Fernandes2 evolved this concept and developed a drum-shaped device with fine protruding needles to puncture the skin. Microneedling devices have expanded in recent years and now include both cord- and battery-powered pens and rollers, with needles ranging in length from 0.25 to 3.0 mm.
Treatment with microneedling promotes skin rejuvenation by creating small puncture wounds in the epidermis and dermis. This injury triggers the wound healing cascade and alters the modulation of growth factors to promote regenerative effects.3,4 Following microneedling therapy, increases occur in elastic fiber formation, collagen deposition, and dermal thickness (Figure).5 Of interesting histologic note, collagen is deposited in the normal lattice pattern following this treatment rather than in the parallel bundles typical of scars.6 Microneedling preserves the overall integrity of the epidermal layers and basement membrane, allowing the epidermis to heal without abnormality, verified on histology by a normal stratum corneum, enhanced stratum granulosum, and normal rete ridges.7
Microneedling has demonstrated several uses beyond general skin rejuvenation. In patients with atrophic acne scars, therapy can lead to improved scar appearance, skin texture, and patient satisfaction.8,9 Hypertrophic and dyspigmented burn scars on the body, face, arms, and legs have shown to be receptive to repeated treatments.10 Microneedling also has shown promise in treating androgenic alopecia, increasing hair regrowth in patients who previously showed poor response to conventional therapy with minoxidil and finasteride.11,12
Platelet-Rich Plasma
Platelet-rich plasma is developed by enriching blood with an autologous concentration of platelets. The preparation of PRP begins with whole blood, commonly obtained peripherally by venipuncture. Samples undergo centrifugation to allow separation of the blood into 3 layers: platelet-poor plasma, PRP, and erythrocytes.13 The typical platelet count of whole blood is approximately 200,000/µL; PRP aims to prepare a platelet count of at least 1,000,000/µL in a 5-mL volume.14
An attractive component of PRP is its high concentration of growth factors, including platelet-derived growth factor, transforming growth factor, vascular endothelial growth factor, and epithelial growth factor.15 Because of the regenerative effects of these proteins, PRP has been investigated as a modality to augment wound healing in a variety of clinical areas, such as maxillofacial surgery, orthopedics, cardiovascular surgery, and treatment of soft tissue ulcers.16
Combination Use of Microneedling and PRP
Several studies have compared the effects of microneedling with and without the application of PRP (Table).17-20 In an animal model, Akcal et al17 examined the effects of microneedling and PRP on skin flap survival. Eight rats were randomly divided into 5 groups: sham, control, microneedling alone, microneedling plus PRP, and microneedling plus platelet-poor plasma. Treatments were applied to skin flaps after 4 hours of induced ischemia. The surviving flap area was measured, with results demonstrating significantly higher viable areas in the microneedling plus PRP group relative to all other groups (P<.01). On histologic examination, the microneedling plus PRP group showed well-organized epidermal layers and a dermal integrity that matched the dermis of the sham group.17
Asif et al18 performed a split-face comparison study of 50 patients with atrophic acne scars. On the right side, microneedling was performed followed by intradermal injections and topical application of PRP. On the left side, microneedling was performed followed by intradermal injections of distilled water. The study included 3 treatment sessions with 1 month between each session. Scars were assessed using the Goodman and Baron scale,21 which is designed to grade the morphology of postacne scarring. Scars on the right side improved by 62.2% and scars on the left side improved by 45.8%; prior to treatment, both sides demonstrated similar severity scores, but final severity scores were significantly reduced in the microneedling plus PRP group relative to the microneedling plus distilled water group (P<.00001). No residual side effects from treatment were reported.18
Examining the degree of improvement more carefully, microneedling plus PRP yielded excellent improvement in 40% (20/50) of patients and good improvement in 60% (30/50).18 Microneedling plus distilled water led to excellent improvement in 10% (5/50) and good improvement in 84% (42/50). Given that microneedling plus distilled water still provided good to excellent results in 94% of patients, the addition of PRP was helpful though not necessary in achieving meaningful benefit.18
In another split-face study, Fabbrocini et al19 evaluated 12 adult patients with acne scars. The right side of the face received microneedling plus PRP, while the left side received microneedling alone. Two treatments were performed 8 weeks apart. Severity scores (0=no lesions; 10=maximum severity) were used to assess patient outcomes throughout the study. Acne scars improved on both sides of the face following the treatment period, but the reduction in scar severity with microneedling plus PRP (3.5 points) was significantly greater than with microneedling alone (2.6 points)(P<.05). Patients tended to experience2 to 3 days of mild swelling and erythema after treatment regardless of PRP addition. With only 12 patients, the study was limited by a small sample size. The 10-point grading system differed from the Goodman and Baron scale in that it lacked corresponding qualitative markers, likely decreasing reproducibility.19
Chawla20 compared the effectiveness of combination therapy with microneedling plus PRP versus microneedling and vitamin C application. In a split-face study of 30 patients with atrophic acne scars, the right side of the face was treated with microneedling plus PRP and the left side was treated with microneedling plus vitamin C. Four sessions were performed with an interval of 1 month in between treatments. The Goodman and Baron Scale was used to assess treatment efficacy. Overall, both treatments led to improved outcomes, but in categorizing patients who demonstrated poor responses, a significantly larger percentage existed in the microneedling plus vitamin C group (37% [10/27]) versus the microneedling plus PRP group (22% [6/27])(P=.021). Additionally, aggregate patient satisfaction scores were higher with microneedling plus PRP relative to microneedling plus vitamin C (P=.01). Of note, assessments of improvement were performed by the treating physician and patient satisfaction reports were completed with knowledge of the therapies and cost factor, which may have influenced results.20
Conclusion
Microneedling therapy continues to evolve with a range of applications now emerging in dermatology. As PRP has gained popularity, there has been increased interest in its utilization to amplify the regenerative effects of microneedling. Although the number of direct comparisons examining microneedling with and without PRP is limited, the available evidence indicates that the addition of PRP may improve cosmetic outcomes. These results have been demonstrated primarily in the management of acne scars, but favorable effects may extend to other indications. Continued study is warranted to further quantify the degree of these benefits and to elucidate optimal treatment schedules.
In addition, it is important to consider a cost-benefit analysis of PRP. The price of PRP varies depending on the clinical site but in certain cases may double the cost of a microneedling treatment session. Although studies have demonstrated a statistically significant benefit to PRP, the clinical significance of this supplementary treatment must be weighed against the increased expense. A discussion should take place with the consideration that microneedling alone can provide a satisfactory result for some patients.
- Camirand A, Doucet J. Needle dermabrasion. Aesthetic Plast Surg. 1997;21:48-51.
- Fernandes D. Percutaneous collagen induction: an alternative to laser resurfacing. Aesthet Surg J. 2002;22:307-309.
- Fabbrocini G, Fardella N, Monfrecola A, et al. Acne scarring treatment using skin needling. Clin Exp Dermatol. 2009;34:874-879.
- Zeitter S, Sikora Z, Jahn S, et al. Microneedling: matching the results of medical needling and repetitive treatments to maximize potential for skin regeneration [published online February 7, 2014]. Burns. 2014;40:966-973.
- Schwarz M, Laaff H. A prospective controlled assessment of microneedling with the Dermaroller device. Plast Reconstr Surg. 2011;127:E146-E148.
- Fernandes D, Signorini M. Combating photoaging with percutaneous collagen induction. Clin Dermatol. 2008;26:192-199.
- Aust MC, Fernandes D, Kolokythas P, et al. Percutaneous collagen induction therapy: an alternative treatment for scars, wrinkles, and skin laxity. Plast Reconstr Surg. 2008;121:1421-1429.
- El-Domyati M, Barakat M, Awad S, et al. Microneedling therapy for atrophic acne scars: an objective evaluation. J Clin Aesthet Dermatol. 2015;8:36-42.
- Leheta T, El Tawdy A, Abdel Hay R, et al. Percutaneous collagen induction versus full-concentration trichloroacetic acid in the treatment of atrophic acne scars. Dermatol Surg. 2011;37:207-216.
- Aust MC, Knobloch K, Reimers K, et al. Percutaneous collagen induction therapy: an alternative treatment for burn scars. Burns. 2010;36:836-843.
- Dhurat R, Mathapati S. Response to microneedling treatment in men with androgenetic alopecia who failed to respond to conventional therapy. Indian J Dermatol. 2015;60:260-263.
- Dhurat R, Sukesh M, Avhad G, et al. A randomized evaluator blinded study of effect of microneedling in androgenetic alopecia: a pilot study. Int J Trichology. 2013;5:6-11.
- Wang HL, Avila G. Platelet rich plasma: myth or reality? Eur J Dent. 2007;1:192-194.
- Marx RE. Platelet-rich plasma (PRP): what is PRP and what is not PRP? Implant Dent. 2001;10:225-228.
- Lubkowska A, Dolegowska B, Banfi G. Growth factor content in PRP and their applicability in medicine. J Biol Regul Homeost Agents. 2012;26(2 suppl 1):3S-22S.
- Pietrzak WS, Eppley BL. Platelet rich plasma: biology and new technology. J Craniofac Surg. 2005;16:1043-1054.
- Akcal A, Savas SA, Gorgulu T, et al. The effect of platelete rich plasma combined with microneedling on full venous outflow compromise in a rat skin flap model. Plast Reconstr Surg. 2015;136(4 suppl):71-72.
- Asif M, Kanodia S, Singh K. Combined autologous platelet-rich plasma with microneedling verses microneedling with distilled water in the treatment of atrophic acne scars: a concurrent split-face study [published online January 8, 2016]. J Cosmet Dermatol. 2016;15:434-443.
- Fabbrocini G, De Vita V, Pastore F, et al. Combined use of skin needling and platelet-rich plasma in acne scarring treatment. Cosmet Dermatol. 2011;24:177-183.
- Chawla S. Split face comparative study of microneedling with PRP versus microneedling with vitamin C in treating atrophic post acne scars. J Cutan Aesthet Surg. 2014;7:209-212.
- Goodman GJ, Baron JA. Postacne scarring: a qualitative global scarring grading system. Dermatol Surg. 2006;32:1458-1466.
- Camirand A, Doucet J. Needle dermabrasion. Aesthetic Plast Surg. 1997;21:48-51.
- Fernandes D. Percutaneous collagen induction: an alternative to laser resurfacing. Aesthet Surg J. 2002;22:307-309.
- Fabbrocini G, Fardella N, Monfrecola A, et al. Acne scarring treatment using skin needling. Clin Exp Dermatol. 2009;34:874-879.
- Zeitter S, Sikora Z, Jahn S, et al. Microneedling: matching the results of medical needling and repetitive treatments to maximize potential for skin regeneration [published online February 7, 2014]. Burns. 2014;40:966-973.
- Schwarz M, Laaff H. A prospective controlled assessment of microneedling with the Dermaroller device. Plast Reconstr Surg. 2011;127:E146-E148.
- Fernandes D, Signorini M. Combating photoaging with percutaneous collagen induction. Clin Dermatol. 2008;26:192-199.
- Aust MC, Fernandes D, Kolokythas P, et al. Percutaneous collagen induction therapy: an alternative treatment for scars, wrinkles, and skin laxity. Plast Reconstr Surg. 2008;121:1421-1429.
- El-Domyati M, Barakat M, Awad S, et al. Microneedling therapy for atrophic acne scars: an objective evaluation. J Clin Aesthet Dermatol. 2015;8:36-42.
- Leheta T, El Tawdy A, Abdel Hay R, et al. Percutaneous collagen induction versus full-concentration trichloroacetic acid in the treatment of atrophic acne scars. Dermatol Surg. 2011;37:207-216.
- Aust MC, Knobloch K, Reimers K, et al. Percutaneous collagen induction therapy: an alternative treatment for burn scars. Burns. 2010;36:836-843.
- Dhurat R, Mathapati S. Response to microneedling treatment in men with androgenetic alopecia who failed to respond to conventional therapy. Indian J Dermatol. 2015;60:260-263.
- Dhurat R, Sukesh M, Avhad G, et al. A randomized evaluator blinded study of effect of microneedling in androgenetic alopecia: a pilot study. Int J Trichology. 2013;5:6-11.
- Wang HL, Avila G. Platelet rich plasma: myth or reality? Eur J Dent. 2007;1:192-194.
- Marx RE. Platelet-rich plasma (PRP): what is PRP and what is not PRP? Implant Dent. 2001;10:225-228.
- Lubkowska A, Dolegowska B, Banfi G. Growth factor content in PRP and their applicability in medicine. J Biol Regul Homeost Agents. 2012;26(2 suppl 1):3S-22S.
- Pietrzak WS, Eppley BL. Platelet rich plasma: biology and new technology. J Craniofac Surg. 2005;16:1043-1054.
- Akcal A, Savas SA, Gorgulu T, et al. The effect of platelete rich plasma combined with microneedling on full venous outflow compromise in a rat skin flap model. Plast Reconstr Surg. 2015;136(4 suppl):71-72.
- Asif M, Kanodia S, Singh K. Combined autologous platelet-rich plasma with microneedling verses microneedling with distilled water in the treatment of atrophic acne scars: a concurrent split-face study [published online January 8, 2016]. J Cosmet Dermatol. 2016;15:434-443.
- Fabbrocini G, De Vita V, Pastore F, et al. Combined use of skin needling and platelet-rich plasma in acne scarring treatment. Cosmet Dermatol. 2011;24:177-183.
- Chawla S. Split face comparative study of microneedling with PRP versus microneedling with vitamin C in treating atrophic post acne scars. J Cutan Aesthet Surg. 2014;7:209-212.
- Goodman GJ, Baron JA. Postacne scarring: a qualitative global scarring grading system. Dermatol Surg. 2006;32:1458-1466.
Practice Points
- Microneedling is an effective therapy for skin rejuvenation.
- Preliminary evidence indicates that the addition of platelet-rich plasma to microneedling improves cosmetic outcomes.
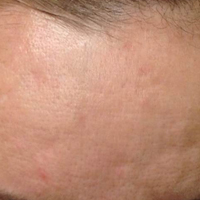
Elbasvir, grazoprevir beat HCV despite compensated cirrhosis
Twelve weeks of combination therapy with elbasvir and grazoprevir (EBR/GZR) achieved sustained virologic response in 98% of treatment-naive patients with compensated cirrhosis and chronic hepatitis C (HCV) genotype 1, 4, or 6 infections, and in 89% of treatment-experienced patients, according to a pooled analysis of six industry-sponsored trials.
Concomitant ribavirin offered “no incremental benefit” for treatment-naive patients, while 16 or 18 weeks of EBR and GZR with ribavirin achieved SVR12 in 100% of treatment-experienced patients, wrote Ira M. Jacobson, MD, of Mount Sinai Beth Israel and Icahn School of Medicine at Mount Sinai, New York, and his associates. The report was published in the May issue of Gastroenterology (doi: 10.1053/j.gastro.2017.01.050).
Regardless of treatment history, genotype 1a patients with resistance-associated variants (RAV) in HCV nonstructural protein 5A (NS5A) needed ribavirin to achieve sustained virologic response (SVR) rates above 90%, the researchers emphasized. “Both patients with HCV genotype 1a infection with baseline RAVs who received 16 or 18 weeks of EBR/GZR and ribavirin achieved SVR12,” the researchers noted. Elbasvir is an HCV NS5A inhibitor, and GZR is an HCV NS3/4A protease inhibitor. In 2016, the Food and Drug Administration approved them in combination (Zepatier) for chronic genotype 1 and genotype 4 HCV. Studies have confirmed the benefits of treating HCV even when patients have cirrhosis, but they can be challenging to treat, especially if they have already failed a regimen that included a direct-acting antiviral agent, the investigators noted.
To explore the efficacy of EBR/GZR in compensated, Child-Pugh A cirrhosis, they studied 402 such patients with HCV genotype 1, 4, or 6 infections whose baseline HCV RNA level exceeded 10,000 IU. Patients had participated in one of six phase II/III clinical trials and had received EBR/GZR 50 mg/100 mg once daily for 12-18 weeks, with or without ribavirin (800-1400 mg/day based on body weight). Treatment-naive patients received 12 weeks of treatment, while treatment-experienced patients received 12, 16, or 18 weeks of treatment.
Twelve weeks of ribavirin did not boost SVR for either treatment-naive (90%) or treatment-experienced patients (91%), the researchers said. However, all 49 treatment-experienced patients who received EBR/GZR plus ribavirin for 16 or 18 weeks achieved SVR12, compared to 94% of patients who received EBR/GZR without ribavirin for 16 or 18 weeks.
Virologic failure was more common with HCV genotype 1a infections than with genotype 1b or 4 infection, especially if patients previously had not responded to interferon, the researchers noted. Eight of 11 (73%) patients with HCV genotype 1a infection and baseline NS5A RAVs achieved SVR12, compared with 98% of GT1a-infected patients without RAVs at baseline. But EBR/GZR was effective in various other subgroups, including patients with less than 100,000 platelets per microL, serum albumin below 3.5 g/dL, and FibroScan scores below 25 kPa. These findings suggest that EBR/GZR remains effective in patients with advanced compensated cirrhosis, the investigators said.
Most patients tolerated therapy, but six stopped treatment because of adverse events, including one episode of severe, possibly treatment-related abdominal pain. Also, four patients had late, asymptomatic rises in alanine aminotransferase (ALT) after first normalizing on treatment, and one patient stopped treatment because of grade 4 ALT elevation with eosinophilia. “There were no decompensation events in this generally healthy cirrhotic population, and no other evidence of declining liver function while on treatment,” the researchers added.
The integrated analysis was not prespecified, nor was it powered to compare outcomes between treatment arms. Only three patients had genotype 6 infection, and all were treatment-experienced. Only 23 patients had genotype 4 infection. Also, most patients had well-compensated cirrhosis. Finally, the trials varied in terms of how they defined cirrhosis, the investigators noted.
Merck, which funded the study, makes elbasvir and grazoprevir. The investigators acknowledged medical writing and editorial assistance from ApotheCom, which Merck funded. Dr. Jacobson disclosed consulting relationships and grant funding from Merck, AbbVie, Bristol-Myers Squibb, Gilead, Intercept, Janssen, and Trek.
Twelve weeks of combination therapy with elbasvir and grazoprevir (EBR/GZR) achieved sustained virologic response in 98% of treatment-naive patients with compensated cirrhosis and chronic hepatitis C (HCV) genotype 1, 4, or 6 infections, and in 89% of treatment-experienced patients, according to a pooled analysis of six industry-sponsored trials.
Concomitant ribavirin offered “no incremental benefit” for treatment-naive patients, while 16 or 18 weeks of EBR and GZR with ribavirin achieved SVR12 in 100% of treatment-experienced patients, wrote Ira M. Jacobson, MD, of Mount Sinai Beth Israel and Icahn School of Medicine at Mount Sinai, New York, and his associates. The report was published in the May issue of Gastroenterology (doi: 10.1053/j.gastro.2017.01.050).
Regardless of treatment history, genotype 1a patients with resistance-associated variants (RAV) in HCV nonstructural protein 5A (NS5A) needed ribavirin to achieve sustained virologic response (SVR) rates above 90%, the researchers emphasized. “Both patients with HCV genotype 1a infection with baseline RAVs who received 16 or 18 weeks of EBR/GZR and ribavirin achieved SVR12,” the researchers noted. Elbasvir is an HCV NS5A inhibitor, and GZR is an HCV NS3/4A protease inhibitor. In 2016, the Food and Drug Administration approved them in combination (Zepatier) for chronic genotype 1 and genotype 4 HCV. Studies have confirmed the benefits of treating HCV even when patients have cirrhosis, but they can be challenging to treat, especially if they have already failed a regimen that included a direct-acting antiviral agent, the investigators noted.
To explore the efficacy of EBR/GZR in compensated, Child-Pugh A cirrhosis, they studied 402 such patients with HCV genotype 1, 4, or 6 infections whose baseline HCV RNA level exceeded 10,000 IU. Patients had participated in one of six phase II/III clinical trials and had received EBR/GZR 50 mg/100 mg once daily for 12-18 weeks, with or without ribavirin (800-1400 mg/day based on body weight). Treatment-naive patients received 12 weeks of treatment, while treatment-experienced patients received 12, 16, or 18 weeks of treatment.
Twelve weeks of ribavirin did not boost SVR for either treatment-naive (90%) or treatment-experienced patients (91%), the researchers said. However, all 49 treatment-experienced patients who received EBR/GZR plus ribavirin for 16 or 18 weeks achieved SVR12, compared to 94% of patients who received EBR/GZR without ribavirin for 16 or 18 weeks.
Virologic failure was more common with HCV genotype 1a infections than with genotype 1b or 4 infection, especially if patients previously had not responded to interferon, the researchers noted. Eight of 11 (73%) patients with HCV genotype 1a infection and baseline NS5A RAVs achieved SVR12, compared with 98% of GT1a-infected patients without RAVs at baseline. But EBR/GZR was effective in various other subgroups, including patients with less than 100,000 platelets per microL, serum albumin below 3.5 g/dL, and FibroScan scores below 25 kPa. These findings suggest that EBR/GZR remains effective in patients with advanced compensated cirrhosis, the investigators said.
Most patients tolerated therapy, but six stopped treatment because of adverse events, including one episode of severe, possibly treatment-related abdominal pain. Also, four patients had late, asymptomatic rises in alanine aminotransferase (ALT) after first normalizing on treatment, and one patient stopped treatment because of grade 4 ALT elevation with eosinophilia. “There were no decompensation events in this generally healthy cirrhotic population, and no other evidence of declining liver function while on treatment,” the researchers added.
The integrated analysis was not prespecified, nor was it powered to compare outcomes between treatment arms. Only three patients had genotype 6 infection, and all were treatment-experienced. Only 23 patients had genotype 4 infection. Also, most patients had well-compensated cirrhosis. Finally, the trials varied in terms of how they defined cirrhosis, the investigators noted.
Merck, which funded the study, makes elbasvir and grazoprevir. The investigators acknowledged medical writing and editorial assistance from ApotheCom, which Merck funded. Dr. Jacobson disclosed consulting relationships and grant funding from Merck, AbbVie, Bristol-Myers Squibb, Gilead, Intercept, Janssen, and Trek.
Twelve weeks of combination therapy with elbasvir and grazoprevir (EBR/GZR) achieved sustained virologic response in 98% of treatment-naive patients with compensated cirrhosis and chronic hepatitis C (HCV) genotype 1, 4, or 6 infections, and in 89% of treatment-experienced patients, according to a pooled analysis of six industry-sponsored trials.
Concomitant ribavirin offered “no incremental benefit” for treatment-naive patients, while 16 or 18 weeks of EBR and GZR with ribavirin achieved SVR12 in 100% of treatment-experienced patients, wrote Ira M. Jacobson, MD, of Mount Sinai Beth Israel and Icahn School of Medicine at Mount Sinai, New York, and his associates. The report was published in the May issue of Gastroenterology (doi: 10.1053/j.gastro.2017.01.050).
Regardless of treatment history, genotype 1a patients with resistance-associated variants (RAV) in HCV nonstructural protein 5A (NS5A) needed ribavirin to achieve sustained virologic response (SVR) rates above 90%, the researchers emphasized. “Both patients with HCV genotype 1a infection with baseline RAVs who received 16 or 18 weeks of EBR/GZR and ribavirin achieved SVR12,” the researchers noted. Elbasvir is an HCV NS5A inhibitor, and GZR is an HCV NS3/4A protease inhibitor. In 2016, the Food and Drug Administration approved them in combination (Zepatier) for chronic genotype 1 and genotype 4 HCV. Studies have confirmed the benefits of treating HCV even when patients have cirrhosis, but they can be challenging to treat, especially if they have already failed a regimen that included a direct-acting antiviral agent, the investigators noted.
To explore the efficacy of EBR/GZR in compensated, Child-Pugh A cirrhosis, they studied 402 such patients with HCV genotype 1, 4, or 6 infections whose baseline HCV RNA level exceeded 10,000 IU. Patients had participated in one of six phase II/III clinical trials and had received EBR/GZR 50 mg/100 mg once daily for 12-18 weeks, with or without ribavirin (800-1400 mg/day based on body weight). Treatment-naive patients received 12 weeks of treatment, while treatment-experienced patients received 12, 16, or 18 weeks of treatment.
Twelve weeks of ribavirin did not boost SVR for either treatment-naive (90%) or treatment-experienced patients (91%), the researchers said. However, all 49 treatment-experienced patients who received EBR/GZR plus ribavirin for 16 or 18 weeks achieved SVR12, compared to 94% of patients who received EBR/GZR without ribavirin for 16 or 18 weeks.
Virologic failure was more common with HCV genotype 1a infections than with genotype 1b or 4 infection, especially if patients previously had not responded to interferon, the researchers noted. Eight of 11 (73%) patients with HCV genotype 1a infection and baseline NS5A RAVs achieved SVR12, compared with 98% of GT1a-infected patients without RAVs at baseline. But EBR/GZR was effective in various other subgroups, including patients with less than 100,000 platelets per microL, serum albumin below 3.5 g/dL, and FibroScan scores below 25 kPa. These findings suggest that EBR/GZR remains effective in patients with advanced compensated cirrhosis, the investigators said.
Most patients tolerated therapy, but six stopped treatment because of adverse events, including one episode of severe, possibly treatment-related abdominal pain. Also, four patients had late, asymptomatic rises in alanine aminotransferase (ALT) after first normalizing on treatment, and one patient stopped treatment because of grade 4 ALT elevation with eosinophilia. “There were no decompensation events in this generally healthy cirrhotic population, and no other evidence of declining liver function while on treatment,” the researchers added.
The integrated analysis was not prespecified, nor was it powered to compare outcomes between treatment arms. Only three patients had genotype 6 infection, and all were treatment-experienced. Only 23 patients had genotype 4 infection. Also, most patients had well-compensated cirrhosis. Finally, the trials varied in terms of how they defined cirrhosis, the investigators noted.
Merck, which funded the study, makes elbasvir and grazoprevir. The investigators acknowledged medical writing and editorial assistance from ApotheCom, which Merck funded. Dr. Jacobson disclosed consulting relationships and grant funding from Merck, AbbVie, Bristol-Myers Squibb, Gilead, Intercept, Janssen, and Trek.
FROM GASTROENTEROLOGY
Key clinical point: Twelve weeks of combination therapy with elbasvir and grazoprevir was relatively well tolerated and achieved sustained viral response for most patients with chronic genotype 1, 4, or 6 hepatitis C virus infections and compensated cirrhosis.
Major finding: Rates of sustained virologic response were 98% for treatment-naive patients and 89% for treatment-experienced patients.
Data source: An integrated analysis of 402 patients with treatment-naive or treatment-experienced HCV GT1, 4, or 6 infection and compensated, Child-Pugh A cirrhosis.
Disclosures: Merck, which funded the study, makes elbasvir and grazoprevir. The investigators acknowledged medical writing and editorial assistance from ApotheCom, which Merck funded. Dr. Jacobson disclosed consulting relationships and grant funding from Merck, AbbVie, Bristol-Myers Squibb, Gilead, Intercept, Janssen, and Trek.
LGBT Access to Health Care: A Dermatologist’s Role in Building a Therapeutic Relationship
The last decade has been a period of advancement for the lesbian, gay, bisexual, and transgender (LGBT) community for legal protections and visibility. Although the journey to acceptance and equality is far from over, this progress has appropriately extended to medical academia as physicians search for ways to become more inclusive and effective care providers for their LGBT patients.1 In a recent cross-sectional study, Ginsberg et al2 examined the role for dermatologists in the care of transgender patients. The investigators concluded that dermatologists should play a larger role in a transgender patient’s physical transformation.2 It is our opinion that dermatologists need to be comfortable building rapport with LGBT patients and to become attuned to their specific needs to provide effective care.
When forging a relationship with an LGBT patient, assumptions can damage rapport. Two assumptions that should be avoided include presuming heterosexuality or, on the other hand, assuming risk for disease based on known LGBT status. A dermatologist who takes a cursory sexual history, or none at all, assuming his/her patient is heterosexual creates an environment in which a nonheterosexual patient feels uncomfortable being honest and open. Although there is enough literature to support the claim that some sexual minority groups have increased risk for sexually transmitted infections (STIs),3 it is dangerous to assume a patient’s risk based solely on sexual orientation. An abstinent patient or a patient in a long-term, monogamous, same-sex relationship, for instance, may feel stereotyped by a dermatologist who wants to screen him/her for an STI. The best step in building a therapeutic relationship is to cast out these assumptions and allow LGBT patients to be open about themselves and their sexual practices. Sexual histories should be asked in nonjudgmental ways that are related to the health of the patient, leading to relevant and useful information for their care. For example, ask patients, “Do you have sex with men, women, or both?” This question should be delivered in a matter-of-fact tone, which conveys to the patient that the provider merely wants an answer to guide patient care.
Dermatologists can tailor their encounters to the specific needs of sexual minority patients. The medical literature is rich with examples of conditions that occur at greater frequency in specific sexual minority groups. Sexually transmitted infections, particularly human immunodeficiency virus, are important causes of morbidity and mortality among sexual minorities, especially men who have sex with men (MSM).3,4 Anal and penile human papillomavirus (HPV) infection and HPV-associated anal carcinoma risk are increased in MSM.5,6 The literature has remained inconclusive on the use of anal Papanicolaou tests for diagnosis; however, dermatologists have a duty to at least examine the perianal and genital area of any patient at risk for HPV-related disease or STIs.7,8 For younger patients, the HPV vaccine can help prevent certain types of HPV infection and likely reduce a patient’s risk for condyloma acuminatum and other sequelae of the virus. Guidelines have been expanded to include men aged 13 to 21 years and up to 26 years.9 More research is needed to determine if detection and prevention of these types of HPV infection using the vaccine in MSM actually leads to a decreased incidence of anal carcinoma.
Certain LGBT groups may benefit from a dermatologist’s care outside the realm of infectious diseases. One study found that increased indoor tanning use in MSM correlated with increased risk for nonmelanoma skin cancer.10 Lesbians have been found to be less likely to pursue preventative health examinations in general, including skin checks.11 Finally, transgender patients can utilize dermatologists for help with transformative procedures and side effects of hormonal treatment such as androgenic acne.1,4
Cutaneous and beyond, the future of LGBT health care in the United States is affected by the institutions that train future physicians. There is a trend toward incorporating formal LGBT curricula into medical schools and academic centers.12 The Penn Medicine Program for LGBT Health (Philadelphia, Pennsylvania) is a pilot program geared toward both educating future clinicians and providing equal and unbiased care to LGBT patients.12 Programs such as this one give rise to a new generation of physicians who feel comfortable and aware of the needs of their LGBT patients.
In a time when LGBT patients are becoming more comfortable claiming their sexual and gender identities openly, there is a need for dermatologists to provide individualized unbiased care, which can best be achieved by building rapport through assumption-free history taking, performing thorough physical examinations that include the genital and perianal area, and passing these good practices on to trainees.
- Snyder JE. Trend analysis of medical publications about LGBT persons: 1950-2007. J Homosex. 2011;58:164-188.
- Ginsberg BA, Calderon M, Seminara NM, et al. A potential role for the dermatologist in the physical transformation of transgender people: a survey of attitudes and practices within the transgender community. J Am Acad Dermatol. 2016;74:303-308.
- Gee R. Primary care health issues among men who have sex with men. J Am Acad Nurse Pract. 2006;18:144-153.
- Katz KA, Furnish TJ. Dermatology-related epidemiologic and clinical concerns of men who have sex with men, women who have sex with women, and transgender individuals. Arch Dermatol. 2005;141:1303-1310.
- Fenkl EA, Jones SG, Schochet E, et al. HPV and anal cancer knowledge among HIV-infected and non-infected men who have sex with men [published online December 11, 2015]. LGBT Health. 2016;3:42-48. doi:10.1089/lgbt.2015.0086.
- Chin-Hong PV, Vittinghoff E, Cranston RD, et al. Age-related prevalence of anal cancer precursors in homosexual men: the EXPLORE Study. J Natl Cancer Inst. 2005;97:896-905.
- Schofield AM, Sadler L, Nelson L, et al. A prospective study of anal cancer screening in HIV-positive and negative MSM. AIDS. 2016;30:1375-1383.
- Katz MH, Katz KA, Bernestein KT, et al. We need data on anal screening effectiveness before focusing on increasing it [published online September 23, 2010]. Am J Public Health. 2010;100:2016.
- Petrosky E, Bocchini JA, Hariri S, et al. Use of 9-Valent human papillomavirus (HPV) vaccine: updated HPV vaccination recommendations of the advisory committee on immunization practices. MMWR Morb Mortal Wkly Rep. 2015;64:300-304.
- Mansh M, Katz KA, Linos E, et al. Association of skin cancer and indoor tanning in sexual minority men and women. JAMA Dermatol. 2015;151:1308-1316.
- Conron KJ, Mimiaga MJ, Landers SJ. A population-based study of sexual orientation identity and gender differences in adult health. Am J Public Health. 2010;100:1953-1960.
- Yehia BR, Calder D, Flesch JD, et al. Advancing LGBT health at an academic medical center: a case study. LGBT Health. 2015;2:362-366.
The last decade has been a period of advancement for the lesbian, gay, bisexual, and transgender (LGBT) community for legal protections and visibility. Although the journey to acceptance and equality is far from over, this progress has appropriately extended to medical academia as physicians search for ways to become more inclusive and effective care providers for their LGBT patients.1 In a recent cross-sectional study, Ginsberg et al2 examined the role for dermatologists in the care of transgender patients. The investigators concluded that dermatologists should play a larger role in a transgender patient’s physical transformation.2 It is our opinion that dermatologists need to be comfortable building rapport with LGBT patients and to become attuned to their specific needs to provide effective care.
When forging a relationship with an LGBT patient, assumptions can damage rapport. Two assumptions that should be avoided include presuming heterosexuality or, on the other hand, assuming risk for disease based on known LGBT status. A dermatologist who takes a cursory sexual history, or none at all, assuming his/her patient is heterosexual creates an environment in which a nonheterosexual patient feels uncomfortable being honest and open. Although there is enough literature to support the claim that some sexual minority groups have increased risk for sexually transmitted infections (STIs),3 it is dangerous to assume a patient’s risk based solely on sexual orientation. An abstinent patient or a patient in a long-term, monogamous, same-sex relationship, for instance, may feel stereotyped by a dermatologist who wants to screen him/her for an STI. The best step in building a therapeutic relationship is to cast out these assumptions and allow LGBT patients to be open about themselves and their sexual practices. Sexual histories should be asked in nonjudgmental ways that are related to the health of the patient, leading to relevant and useful information for their care. For example, ask patients, “Do you have sex with men, women, or both?” This question should be delivered in a matter-of-fact tone, which conveys to the patient that the provider merely wants an answer to guide patient care.
Dermatologists can tailor their encounters to the specific needs of sexual minority patients. The medical literature is rich with examples of conditions that occur at greater frequency in specific sexual minority groups. Sexually transmitted infections, particularly human immunodeficiency virus, are important causes of morbidity and mortality among sexual minorities, especially men who have sex with men (MSM).3,4 Anal and penile human papillomavirus (HPV) infection and HPV-associated anal carcinoma risk are increased in MSM.5,6 The literature has remained inconclusive on the use of anal Papanicolaou tests for diagnosis; however, dermatologists have a duty to at least examine the perianal and genital area of any patient at risk for HPV-related disease or STIs.7,8 For younger patients, the HPV vaccine can help prevent certain types of HPV infection and likely reduce a patient’s risk for condyloma acuminatum and other sequelae of the virus. Guidelines have been expanded to include men aged 13 to 21 years and up to 26 years.9 More research is needed to determine if detection and prevention of these types of HPV infection using the vaccine in MSM actually leads to a decreased incidence of anal carcinoma.
Certain LGBT groups may benefit from a dermatologist’s care outside the realm of infectious diseases. One study found that increased indoor tanning use in MSM correlated with increased risk for nonmelanoma skin cancer.10 Lesbians have been found to be less likely to pursue preventative health examinations in general, including skin checks.11 Finally, transgender patients can utilize dermatologists for help with transformative procedures and side effects of hormonal treatment such as androgenic acne.1,4
Cutaneous and beyond, the future of LGBT health care in the United States is affected by the institutions that train future physicians. There is a trend toward incorporating formal LGBT curricula into medical schools and academic centers.12 The Penn Medicine Program for LGBT Health (Philadelphia, Pennsylvania) is a pilot program geared toward both educating future clinicians and providing equal and unbiased care to LGBT patients.12 Programs such as this one give rise to a new generation of physicians who feel comfortable and aware of the needs of their LGBT patients.
In a time when LGBT patients are becoming more comfortable claiming their sexual and gender identities openly, there is a need for dermatologists to provide individualized unbiased care, which can best be achieved by building rapport through assumption-free history taking, performing thorough physical examinations that include the genital and perianal area, and passing these good practices on to trainees.
The last decade has been a period of advancement for the lesbian, gay, bisexual, and transgender (LGBT) community for legal protections and visibility. Although the journey to acceptance and equality is far from over, this progress has appropriately extended to medical academia as physicians search for ways to become more inclusive and effective care providers for their LGBT patients.1 In a recent cross-sectional study, Ginsberg et al2 examined the role for dermatologists in the care of transgender patients. The investigators concluded that dermatologists should play a larger role in a transgender patient’s physical transformation.2 It is our opinion that dermatologists need to be comfortable building rapport with LGBT patients and to become attuned to their specific needs to provide effective care.
When forging a relationship with an LGBT patient, assumptions can damage rapport. Two assumptions that should be avoided include presuming heterosexuality or, on the other hand, assuming risk for disease based on known LGBT status. A dermatologist who takes a cursory sexual history, or none at all, assuming his/her patient is heterosexual creates an environment in which a nonheterosexual patient feels uncomfortable being honest and open. Although there is enough literature to support the claim that some sexual minority groups have increased risk for sexually transmitted infections (STIs),3 it is dangerous to assume a patient’s risk based solely on sexual orientation. An abstinent patient or a patient in a long-term, monogamous, same-sex relationship, for instance, may feel stereotyped by a dermatologist who wants to screen him/her for an STI. The best step in building a therapeutic relationship is to cast out these assumptions and allow LGBT patients to be open about themselves and their sexual practices. Sexual histories should be asked in nonjudgmental ways that are related to the health of the patient, leading to relevant and useful information for their care. For example, ask patients, “Do you have sex with men, women, or both?” This question should be delivered in a matter-of-fact tone, which conveys to the patient that the provider merely wants an answer to guide patient care.
Dermatologists can tailor their encounters to the specific needs of sexual minority patients. The medical literature is rich with examples of conditions that occur at greater frequency in specific sexual minority groups. Sexually transmitted infections, particularly human immunodeficiency virus, are important causes of morbidity and mortality among sexual minorities, especially men who have sex with men (MSM).3,4 Anal and penile human papillomavirus (HPV) infection and HPV-associated anal carcinoma risk are increased in MSM.5,6 The literature has remained inconclusive on the use of anal Papanicolaou tests for diagnosis; however, dermatologists have a duty to at least examine the perianal and genital area of any patient at risk for HPV-related disease or STIs.7,8 For younger patients, the HPV vaccine can help prevent certain types of HPV infection and likely reduce a patient’s risk for condyloma acuminatum and other sequelae of the virus. Guidelines have been expanded to include men aged 13 to 21 years and up to 26 years.9 More research is needed to determine if detection and prevention of these types of HPV infection using the vaccine in MSM actually leads to a decreased incidence of anal carcinoma.
Certain LGBT groups may benefit from a dermatologist’s care outside the realm of infectious diseases. One study found that increased indoor tanning use in MSM correlated with increased risk for nonmelanoma skin cancer.10 Lesbians have been found to be less likely to pursue preventative health examinations in general, including skin checks.11 Finally, transgender patients can utilize dermatologists for help with transformative procedures and side effects of hormonal treatment such as androgenic acne.1,4
Cutaneous and beyond, the future of LGBT health care in the United States is affected by the institutions that train future physicians. There is a trend toward incorporating formal LGBT curricula into medical schools and academic centers.12 The Penn Medicine Program for LGBT Health (Philadelphia, Pennsylvania) is a pilot program geared toward both educating future clinicians and providing equal and unbiased care to LGBT patients.12 Programs such as this one give rise to a new generation of physicians who feel comfortable and aware of the needs of their LGBT patients.
In a time when LGBT patients are becoming more comfortable claiming their sexual and gender identities openly, there is a need for dermatologists to provide individualized unbiased care, which can best be achieved by building rapport through assumption-free history taking, performing thorough physical examinations that include the genital and perianal area, and passing these good practices on to trainees.
- Snyder JE. Trend analysis of medical publications about LGBT persons: 1950-2007. J Homosex. 2011;58:164-188.
- Ginsberg BA, Calderon M, Seminara NM, et al. A potential role for the dermatologist in the physical transformation of transgender people: a survey of attitudes and practices within the transgender community. J Am Acad Dermatol. 2016;74:303-308.
- Gee R. Primary care health issues among men who have sex with men. J Am Acad Nurse Pract. 2006;18:144-153.
- Katz KA, Furnish TJ. Dermatology-related epidemiologic and clinical concerns of men who have sex with men, women who have sex with women, and transgender individuals. Arch Dermatol. 2005;141:1303-1310.
- Fenkl EA, Jones SG, Schochet E, et al. HPV and anal cancer knowledge among HIV-infected and non-infected men who have sex with men [published online December 11, 2015]. LGBT Health. 2016;3:42-48. doi:10.1089/lgbt.2015.0086.
- Chin-Hong PV, Vittinghoff E, Cranston RD, et al. Age-related prevalence of anal cancer precursors in homosexual men: the EXPLORE Study. J Natl Cancer Inst. 2005;97:896-905.
- Schofield AM, Sadler L, Nelson L, et al. A prospective study of anal cancer screening in HIV-positive and negative MSM. AIDS. 2016;30:1375-1383.
- Katz MH, Katz KA, Bernestein KT, et al. We need data on anal screening effectiveness before focusing on increasing it [published online September 23, 2010]. Am J Public Health. 2010;100:2016.
- Petrosky E, Bocchini JA, Hariri S, et al. Use of 9-Valent human papillomavirus (HPV) vaccine: updated HPV vaccination recommendations of the advisory committee on immunization practices. MMWR Morb Mortal Wkly Rep. 2015;64:300-304.
- Mansh M, Katz KA, Linos E, et al. Association of skin cancer and indoor tanning in sexual minority men and women. JAMA Dermatol. 2015;151:1308-1316.
- Conron KJ, Mimiaga MJ, Landers SJ. A population-based study of sexual orientation identity and gender differences in adult health. Am J Public Health. 2010;100:1953-1960.
- Yehia BR, Calder D, Flesch JD, et al. Advancing LGBT health at an academic medical center: a case study. LGBT Health. 2015;2:362-366.
- Snyder JE. Trend analysis of medical publications about LGBT persons: 1950-2007. J Homosex. 2011;58:164-188.
- Ginsberg BA, Calderon M, Seminara NM, et al. A potential role for the dermatologist in the physical transformation of transgender people: a survey of attitudes and practices within the transgender community. J Am Acad Dermatol. 2016;74:303-308.
- Gee R. Primary care health issues among men who have sex with men. J Am Acad Nurse Pract. 2006;18:144-153.
- Katz KA, Furnish TJ. Dermatology-related epidemiologic and clinical concerns of men who have sex with men, women who have sex with women, and transgender individuals. Arch Dermatol. 2005;141:1303-1310.
- Fenkl EA, Jones SG, Schochet E, et al. HPV and anal cancer knowledge among HIV-infected and non-infected men who have sex with men [published online December 11, 2015]. LGBT Health. 2016;3:42-48. doi:10.1089/lgbt.2015.0086.
- Chin-Hong PV, Vittinghoff E, Cranston RD, et al. Age-related prevalence of anal cancer precursors in homosexual men: the EXPLORE Study. J Natl Cancer Inst. 2005;97:896-905.
- Schofield AM, Sadler L, Nelson L, et al. A prospective study of anal cancer screening in HIV-positive and negative MSM. AIDS. 2016;30:1375-1383.
- Katz MH, Katz KA, Bernestein KT, et al. We need data on anal screening effectiveness before focusing on increasing it [published online September 23, 2010]. Am J Public Health. 2010;100:2016.
- Petrosky E, Bocchini JA, Hariri S, et al. Use of 9-Valent human papillomavirus (HPV) vaccine: updated HPV vaccination recommendations of the advisory committee on immunization practices. MMWR Morb Mortal Wkly Rep. 2015;64:300-304.
- Mansh M, Katz KA, Linos E, et al. Association of skin cancer and indoor tanning in sexual minority men and women. JAMA Dermatol. 2015;151:1308-1316.
- Conron KJ, Mimiaga MJ, Landers SJ. A population-based study of sexual orientation identity and gender differences in adult health. Am J Public Health. 2010;100:1953-1960.
- Yehia BR, Calder D, Flesch JD, et al. Advancing LGBT health at an academic medical center: a case study. LGBT Health. 2015;2:362-366.

Here’s what’s trending at SHM
Hospitalist specialty code becomes official designation
On April 3, 2017, “hospitalist” becomes an official specialty designation under Medicare – the code itself is C6. Starting on that date, hospitalists can change their specialty designation on the Medicare enrollment application. Specialty codes are self-designated and describe the kind of medicine that health care providers practice. Appropriate use of specialty codes helps distinguish differences among providers and improves the quality of utilization data. SHM applied for a specialty code for hospitalists nearly 3 years ago, and the Centers for Medicare & Medicaid Services approved the application in February 2016.
Stand with your fellow hospitalists and make sure to declare “I’m a C6.”
Assess your knowledge in hospital medicine with SPARK ONE
SHM recently launched SPARK ONE, a comprehensive online self-assessment tool created specifically for hospital medicine professionals. The activity contains 450-plus vignette-style multiple-choice questions covering 100% of the American Board of Internal Medicine’s Focused Practice in Hospital Medicine (FPHM) exam blueprint. This online tool is your complete resource for successfully preparing for the FPHM exam, or assessing your general knowledge in hospital medicine.
White paper now available: Hospitalist Attitudes Toward Electronic Medical Records
SHM’s Health Information Technology Committee diligently analyzed survey results that captured hospitalists’ attitudes towards electronic medical records, resulting in a white paper now available. The purpose of this paper is to effect change on EHR systems by informing conversations with decision makers and to provide hospital medicine a definitive voice in the landscape of the tumultuous world of electronic medical record systems.
SHM believes hospitalists are especially qualified to evaluate these systems, and the survey results paint a grim picture of the effectiveness and usability of the systems that hospitalists spend the majority of their time interacting with. These results should serve as a call to action to accelerate the pace of advancement and innovation in health care technology.
By sharing these results, SHM hopes to raise awareness of the unacceptable performance of existing systems that contributes to slower-than-desired improvement in quality and safety as well as increasing provider frustration. SHM strongly believes that health care needs a renewed focus on initial goals of technology adoption. View the white paper at hospitalmedicine.org/EHR.
Join the Early-Career Academic Hospitalist Speed Mentoring Session at Hospital Medicine 2017
The SHM “speed mentoring” session, held on Tuesday, May 2 from noon to 1:00 p.m. at Hospital Medicine 2017, is designed to assist early-career hospitalists in specific areas of career development by providing a fresh perspective and rapid advice.
Early-career hospitalists will be matched with three senior advisors by area of interest. The “mentee” will spend 10-15 minutes with each advisor and will then rotate to the next advisor. After the session, there will be time for additional informal discussion and networking among advisors and peers.
Pre-registration by March 31, 2017 is required; it will not be possible to register for this activity on-site at HM17. There is no additional fee to register. Registration will be limited to the first 20 participants. Visit hospitalmedicine2017.org/academic today.
Now available: 4 new antimicrobial stewardship modules
SHM has developed four new antimicrobial stewardship modules to help you demonstrate an understanding of best practices to optimize and improve antimicrobial prescribing within your hospital:
1. Optimizing Antibiotic Use for Hospitalized Patients
2. Best Practices in Treatment of UTIs: “Low-Hanging Fruit”
3. Best Practices in Acute Bacterial Skin Infection
4. Antibiotic Use for Inpatient Respiratory Infections
View these resources at hospitalmedicine.org/abx.
Network with the largest gathering of pediatric hospital medicine professionals
Pediatric Hospital Medicine 2017 (PHM 2017) will be held July 20-23 at the Omni Nashville located in Nashville, Tenn. PHM 2017 offers an all-inclusive arrangement of educational and networking opportunities planned specifically for the pediatric hospital medicine professional. More than 100 concurrent sessions to choose from over the 4 days of the conference allow participants to get the best out of their PHM 2017 experience.

Earn free CME in the enhanced SHM Learning Portal
You asked, and we listened: Introducing the enhanced SHM Learning Portal! The SHM Learning Portal, the online learning home for hospitalists with all eLearning initiatives in one place, just launched a brand-new responsive design in March 2016.
Launching this summer, SHM’s new Learning Portal will offer a better way to access and track online CME, with member discounts to a growing library of content. For more information, visit www.shmlearningportal.org.
Bringing SHM to you with local chapter meetings
Attend a chapter meeting and experience SHM locally. Chapters provide focused educational topics through key speakers and presentations and the opportunity to network with other hospitalists in your area. Find a chapter meeting close to you at hospitalmedicine.org/chapters.
A new look for SHM’s Center for Hospital Innovation & Improvement
Just as hospital medicine is evolving, so is SHM’s group dedicated to developing quality improvement safety tools and programs to meet health care’s changing needs. SHM is proud to unveil a new look for its Center for Hospital Innovation & Improvement this month. Stay tuned for more, and visit hospitalmedicine.org/QI for the latest offerings in a variety of quality improvement topics and clinical areas.
Stand out as a leader with the Fellow in Hospital Medicine designation
Applications for SHM’s Fellow in Hospital Medicine designation open on April 17, 2017. You may be eligible if you have been a member of SHM for at least 3 years and have been involved in key quality improvement programs and leadership roles in hospital medicine. Learn more and apply at hospitalmedicine.org/fellows.
Mr. Radler is Communications Specialist at the Society of Hospital Medicine.
Hospitalist specialty code becomes official designation
On April 3, 2017, “hospitalist” becomes an official specialty designation under Medicare – the code itself is C6. Starting on that date, hospitalists can change their specialty designation on the Medicare enrollment application. Specialty codes are self-designated and describe the kind of medicine that health care providers practice. Appropriate use of specialty codes helps distinguish differences among providers and improves the quality of utilization data. SHM applied for a specialty code for hospitalists nearly 3 years ago, and the Centers for Medicare & Medicaid Services approved the application in February 2016.
Stand with your fellow hospitalists and make sure to declare “I’m a C6.”
Assess your knowledge in hospital medicine with SPARK ONE
SHM recently launched SPARK ONE, a comprehensive online self-assessment tool created specifically for hospital medicine professionals. The activity contains 450-plus vignette-style multiple-choice questions covering 100% of the American Board of Internal Medicine’s Focused Practice in Hospital Medicine (FPHM) exam blueprint. This online tool is your complete resource for successfully preparing for the FPHM exam, or assessing your general knowledge in hospital medicine.
White paper now available: Hospitalist Attitudes Toward Electronic Medical Records
SHM’s Health Information Technology Committee diligently analyzed survey results that captured hospitalists’ attitudes towards electronic medical records, resulting in a white paper now available. The purpose of this paper is to effect change on EHR systems by informing conversations with decision makers and to provide hospital medicine a definitive voice in the landscape of the tumultuous world of electronic medical record systems.
SHM believes hospitalists are especially qualified to evaluate these systems, and the survey results paint a grim picture of the effectiveness and usability of the systems that hospitalists spend the majority of their time interacting with. These results should serve as a call to action to accelerate the pace of advancement and innovation in health care technology.
By sharing these results, SHM hopes to raise awareness of the unacceptable performance of existing systems that contributes to slower-than-desired improvement in quality and safety as well as increasing provider frustration. SHM strongly believes that health care needs a renewed focus on initial goals of technology adoption. View the white paper at hospitalmedicine.org/EHR.
Join the Early-Career Academic Hospitalist Speed Mentoring Session at Hospital Medicine 2017
The SHM “speed mentoring” session, held on Tuesday, May 2 from noon to 1:00 p.m. at Hospital Medicine 2017, is designed to assist early-career hospitalists in specific areas of career development by providing a fresh perspective and rapid advice.
Early-career hospitalists will be matched with three senior advisors by area of interest. The “mentee” will spend 10-15 minutes with each advisor and will then rotate to the next advisor. After the session, there will be time for additional informal discussion and networking among advisors and peers.
Pre-registration by March 31, 2017 is required; it will not be possible to register for this activity on-site at HM17. There is no additional fee to register. Registration will be limited to the first 20 participants. Visit hospitalmedicine2017.org/academic today.
Now available: 4 new antimicrobial stewardship modules
SHM has developed four new antimicrobial stewardship modules to help you demonstrate an understanding of best practices to optimize and improve antimicrobial prescribing within your hospital:
1. Optimizing Antibiotic Use for Hospitalized Patients
2. Best Practices in Treatment of UTIs: “Low-Hanging Fruit”
3. Best Practices in Acute Bacterial Skin Infection
4. Antibiotic Use for Inpatient Respiratory Infections
View these resources at hospitalmedicine.org/abx.
Network with the largest gathering of pediatric hospital medicine professionals
Pediatric Hospital Medicine 2017 (PHM 2017) will be held July 20-23 at the Omni Nashville located in Nashville, Tenn. PHM 2017 offers an all-inclusive arrangement of educational and networking opportunities planned specifically for the pediatric hospital medicine professional. More than 100 concurrent sessions to choose from over the 4 days of the conference allow participants to get the best out of their PHM 2017 experience.
Earn free CME in the enhanced SHM Learning Portal
You asked, and we listened: Introducing the enhanced SHM Learning Portal! The SHM Learning Portal, the online learning home for hospitalists with all eLearning initiatives in one place, just launched a brand-new responsive design in March 2016.
Launching this summer, SHM’s new Learning Portal will offer a better way to access and track online CME, with member discounts to a growing library of content. For more information, visit www.shmlearningportal.org.
Bringing SHM to you with local chapter meetings
Attend a chapter meeting and experience SHM locally. Chapters provide focused educational topics through key speakers and presentations and the opportunity to network with other hospitalists in your area. Find a chapter meeting close to you at hospitalmedicine.org/chapters.
A new look for SHM’s Center for Hospital Innovation & Improvement
Just as hospital medicine is evolving, so is SHM’s group dedicated to developing quality improvement safety tools and programs to meet health care’s changing needs. SHM is proud to unveil a new look for its Center for Hospital Innovation & Improvement this month. Stay tuned for more, and visit hospitalmedicine.org/QI for the latest offerings in a variety of quality improvement topics and clinical areas.
Stand out as a leader with the Fellow in Hospital Medicine designation
Applications for SHM’s Fellow in Hospital Medicine designation open on April 17, 2017. You may be eligible if you have been a member of SHM for at least 3 years and have been involved in key quality improvement programs and leadership roles in hospital medicine. Learn more and apply at hospitalmedicine.org/fellows.
Mr. Radler is Communications Specialist at the Society of Hospital Medicine.
Hospitalist specialty code becomes official designation
On April 3, 2017, “hospitalist” becomes an official specialty designation under Medicare – the code itself is C6. Starting on that date, hospitalists can change their specialty designation on the Medicare enrollment application. Specialty codes are self-designated and describe the kind of medicine that health care providers practice. Appropriate use of specialty codes helps distinguish differences among providers and improves the quality of utilization data. SHM applied for a specialty code for hospitalists nearly 3 years ago, and the Centers for Medicare & Medicaid Services approved the application in February 2016.
Stand with your fellow hospitalists and make sure to declare “I’m a C6.”
Assess your knowledge in hospital medicine with SPARK ONE
SHM recently launched SPARK ONE, a comprehensive online self-assessment tool created specifically for hospital medicine professionals. The activity contains 450-plus vignette-style multiple-choice questions covering 100% of the American Board of Internal Medicine’s Focused Practice in Hospital Medicine (FPHM) exam blueprint. This online tool is your complete resource for successfully preparing for the FPHM exam, or assessing your general knowledge in hospital medicine.
White paper now available: Hospitalist Attitudes Toward Electronic Medical Records
SHM’s Health Information Technology Committee diligently analyzed survey results that captured hospitalists’ attitudes towards electronic medical records, resulting in a white paper now available. The purpose of this paper is to effect change on EHR systems by informing conversations with decision makers and to provide hospital medicine a definitive voice in the landscape of the tumultuous world of electronic medical record systems.
SHM believes hospitalists are especially qualified to evaluate these systems, and the survey results paint a grim picture of the effectiveness and usability of the systems that hospitalists spend the majority of their time interacting with. These results should serve as a call to action to accelerate the pace of advancement and innovation in health care technology.
By sharing these results, SHM hopes to raise awareness of the unacceptable performance of existing systems that contributes to slower-than-desired improvement in quality and safety as well as increasing provider frustration. SHM strongly believes that health care needs a renewed focus on initial goals of technology adoption. View the white paper at hospitalmedicine.org/EHR.
Join the Early-Career Academic Hospitalist Speed Mentoring Session at Hospital Medicine 2017
The SHM “speed mentoring” session, held on Tuesday, May 2 from noon to 1:00 p.m. at Hospital Medicine 2017, is designed to assist early-career hospitalists in specific areas of career development by providing a fresh perspective and rapid advice.
Early-career hospitalists will be matched with three senior advisors by area of interest. The “mentee” will spend 10-15 minutes with each advisor and will then rotate to the next advisor. After the session, there will be time for additional informal discussion and networking among advisors and peers.
Pre-registration by March 31, 2017 is required; it will not be possible to register for this activity on-site at HM17. There is no additional fee to register. Registration will be limited to the first 20 participants. Visit hospitalmedicine2017.org/academic today.
Now available: 4 new antimicrobial stewardship modules
SHM has developed four new antimicrobial stewardship modules to help you demonstrate an understanding of best practices to optimize and improve antimicrobial prescribing within your hospital:
1. Optimizing Antibiotic Use for Hospitalized Patients
2. Best Practices in Treatment of UTIs: “Low-Hanging Fruit”
3. Best Practices in Acute Bacterial Skin Infection
4. Antibiotic Use for Inpatient Respiratory Infections
View these resources at hospitalmedicine.org/abx.
Network with the largest gathering of pediatric hospital medicine professionals
Pediatric Hospital Medicine 2017 (PHM 2017) will be held July 20-23 at the Omni Nashville located in Nashville, Tenn. PHM 2017 offers an all-inclusive arrangement of educational and networking opportunities planned specifically for the pediatric hospital medicine professional. More than 100 concurrent sessions to choose from over the 4 days of the conference allow participants to get the best out of their PHM 2017 experience.
Earn free CME in the enhanced SHM Learning Portal
You asked, and we listened: Introducing the enhanced SHM Learning Portal! The SHM Learning Portal, the online learning home for hospitalists with all eLearning initiatives in one place, just launched a brand-new responsive design in March 2016.
Launching this summer, SHM’s new Learning Portal will offer a better way to access and track online CME, with member discounts to a growing library of content. For more information, visit www.shmlearningportal.org.
Bringing SHM to you with local chapter meetings
Attend a chapter meeting and experience SHM locally. Chapters provide focused educational topics through key speakers and presentations and the opportunity to network with other hospitalists in your area. Find a chapter meeting close to you at hospitalmedicine.org/chapters.
A new look for SHM’s Center for Hospital Innovation & Improvement
Just as hospital medicine is evolving, so is SHM’s group dedicated to developing quality improvement safety tools and programs to meet health care’s changing needs. SHM is proud to unveil a new look for its Center for Hospital Innovation & Improvement this month. Stay tuned for more, and visit hospitalmedicine.org/QI for the latest offerings in a variety of quality improvement topics and clinical areas.
Stand out as a leader with the Fellow in Hospital Medicine designation
Applications for SHM’s Fellow in Hospital Medicine designation open on April 17, 2017. You may be eligible if you have been a member of SHM for at least 3 years and have been involved in key quality improvement programs and leadership roles in hospital medicine. Learn more and apply at hospitalmedicine.org/fellows.
Mr. Radler is Communications Specialist at the Society of Hospital Medicine.

Data-driven HR, RR parameters might help reduce alarm fatigue
Clinical question: Can alarm fatigue in pediatric inpatient settings be safely mitigated by modifying alarm limits with data-driven vital sign reference ranges?
Background: The management of patient alarms in the hospital is a significant safety issue, with the large majority of alarms (85%-99%) either false or not clinically significant. This leads to provider desensitization or alarm fatigue, which has been shown to contribute to adverse events.
In 2014, the Joint Commission made the issue of alarm system safety and alarm fatigue a priority for hospitals.1 Multiple studies have been published addressing alarm fatigue in hospitalized adult patients, but this issue is less well studied in pediatrics, including little guidance on optimizing alarm parameters. Widely used reference ranges and guides are based on limited evidence, primarily based on observational data in healthy outpatients or consensus data.

A 2013 study used vital sign data from hospitalized children to develop percentile curves for heart rate (HR) and respiratory rate (RR) and estimated that 54% of vital sign measurements in hospitalized children are out of range using currently accepted reference ranges.2
To safely decrease the number of out-of-range vital sign measurements resulting from current reference ranges, this study used data from non–critically ill hospitalized children to develop HR and RR percentile charts, and then performed retrospective safety analysis by evaluating effects of modifying the alarm limits on identification of cardiorespiratory arrests (CRA) and rapid response team (RRT) activations.
Study Design: Retrospective, cross-sectional study.
Setting: Single-site, 311-bed quaternary-care academic hospital, both general medical and surgical units.
Synopsis: Vital signs were extracted from the institution’s electronic health record (EHR) for all general medical and surgical patients discharged between Jan. 1, 3013, and May 3, 2014, excluding critically ill children and physiologically implausible vital signs. Two different sets were used, a training set (patients discharged between Jan. 1, 2013, and Dec. 31, 2013) and a validation set (Jan. 1, 2014-May 3, 2014). One HR and RR pair was randomly selected for each 4-hour interval during hospitalization, with a maximum of 10 HR and RR pairs per patient. Age-stratified percentiles were calculated using this data. The 5th and 95th percentile limits using the study data were compared with the 5th and 95th percentile values in the 2013 study, and the reference ranges currently in use at the institution (2004 National Institutes of Health ranges).2
The training set used 62,508 vital sign measurements for 7,202 patients to calculate percentiles for HR and RR among 14 different age groups. The validation set consisted of 82,993 vital sign measurements for 2,287 patients. Using the 5th and 95th percentiles for HR and RR resulted in 24,045 (55.6%) fewer out-of-range measurements in the validation set compared to NIH reference ranges (45% fewer HR values, 61% fewer RR values). This finding, as well as the vital sign percentile ranges, was consistent with the data published in the 2013 study.2
Data for all 148 out-of-ICU RRT and CRA events during the same time period were reviewed using manual chart review. Evaluating vital signs within the 12 hours preceding the events, 144 patients had out-of-range HR or RR measurements using NIH ranges. One hundred thirty-six (94.4%) of these 144 patients also had out-of-range measurements using the study-derived 5th and 95th percentile values.
Manual chart review of the remaining eight patients who had normal HR or RR demonstrated that the RRT or CRA interventions occurred for clinical indications that did no rely on HR or RR measurement (for example, desaturations, difficulty breathing, hematemesis), so the data-driven parameters did not miss any of these events.
Bottom line: In this retrospective study, using data-driven HR and RR parameters was at least as safe as the NIH-published reference ranges currently in use in this hospital. In addition to maintaining safety related to RRT and CRA events, use of the data-driven parameters resulted in 55.6% fewer out-of-range vital sign measurements in the studied population. This may reduce the frequency of false alarms and improve alarm fatigue, and should be studied prospectively in the future.
Citation: Goel VV, Poole SF, Longhurst CA, et al. Safety analysis of proposed data-driven physiologic alarm parameters for hospitalized children. J Hosp Med. 2016;11(12):817-823.
Dr. Galloway is a pediatric hospitalist at Sanford Children’s Hospital in Sioux Falls, S.D., assistant professor of pediatrics at the University of South Dakota Sanford School of Medicine, and vice chief of the division of hospital pediatrics at USD SSOM and Sanford Children’s Hospital.
References
1. The Joint Commission. Alarm system safety. Available at: https://www.jointcommission.org/assets/1/18/R3_Report_Issue_5_12_2_13_Final.pdf. Published December 11, 2013.
2. Bonafide CP, Brady PW, Keren R, Conway PH, Marsolo K, Daymont C. Development of heart and respiratory rate percentile curves for hospitalized children. Pediatrics. 2013;131(4):e1150-1157.
Clinical question: Can alarm fatigue in pediatric inpatient settings be safely mitigated by modifying alarm limits with data-driven vital sign reference ranges?
Background: The management of patient alarms in the hospital is a significant safety issue, with the large majority of alarms (85%-99%) either false or not clinically significant. This leads to provider desensitization or alarm fatigue, which has been shown to contribute to adverse events.
In 2014, the Joint Commission made the issue of alarm system safety and alarm fatigue a priority for hospitals.1 Multiple studies have been published addressing alarm fatigue in hospitalized adult patients, but this issue is less well studied in pediatrics, including little guidance on optimizing alarm parameters. Widely used reference ranges and guides are based on limited evidence, primarily based on observational data in healthy outpatients or consensus data.
A 2013 study used vital sign data from hospitalized children to develop percentile curves for heart rate (HR) and respiratory rate (RR) and estimated that 54% of vital sign measurements in hospitalized children are out of range using currently accepted reference ranges.2
To safely decrease the number of out-of-range vital sign measurements resulting from current reference ranges, this study used data from non–critically ill hospitalized children to develop HR and RR percentile charts, and then performed retrospective safety analysis by evaluating effects of modifying the alarm limits on identification of cardiorespiratory arrests (CRA) and rapid response team (RRT) activations.
Study Design: Retrospective, cross-sectional study.
Setting: Single-site, 311-bed quaternary-care academic hospital, both general medical and surgical units.
Synopsis: Vital signs were extracted from the institution’s electronic health record (EHR) for all general medical and surgical patients discharged between Jan. 1, 3013, and May 3, 2014, excluding critically ill children and physiologically implausible vital signs. Two different sets were used, a training set (patients discharged between Jan. 1, 2013, and Dec. 31, 2013) and a validation set (Jan. 1, 2014-May 3, 2014). One HR and RR pair was randomly selected for each 4-hour interval during hospitalization, with a maximum of 10 HR and RR pairs per patient. Age-stratified percentiles were calculated using this data. The 5th and 95th percentile limits using the study data were compared with the 5th and 95th percentile values in the 2013 study, and the reference ranges currently in use at the institution (2004 National Institutes of Health ranges).2
The training set used 62,508 vital sign measurements for 7,202 patients to calculate percentiles for HR and RR among 14 different age groups. The validation set consisted of 82,993 vital sign measurements for 2,287 patients. Using the 5th and 95th percentiles for HR and RR resulted in 24,045 (55.6%) fewer out-of-range measurements in the validation set compared to NIH reference ranges (45% fewer HR values, 61% fewer RR values). This finding, as well as the vital sign percentile ranges, was consistent with the data published in the 2013 study.2
Data for all 148 out-of-ICU RRT and CRA events during the same time period were reviewed using manual chart review. Evaluating vital signs within the 12 hours preceding the events, 144 patients had out-of-range HR or RR measurements using NIH ranges. One hundred thirty-six (94.4%) of these 144 patients also had out-of-range measurements using the study-derived 5th and 95th percentile values.
Manual chart review of the remaining eight patients who had normal HR or RR demonstrated that the RRT or CRA interventions occurred for clinical indications that did no rely on HR or RR measurement (for example, desaturations, difficulty breathing, hematemesis), so the data-driven parameters did not miss any of these events.
Bottom line: In this retrospective study, using data-driven HR and RR parameters was at least as safe as the NIH-published reference ranges currently in use in this hospital. In addition to maintaining safety related to RRT and CRA events, use of the data-driven parameters resulted in 55.6% fewer out-of-range vital sign measurements in the studied population. This may reduce the frequency of false alarms and improve alarm fatigue, and should be studied prospectively in the future.
Citation: Goel VV, Poole SF, Longhurst CA, et al. Safety analysis of proposed data-driven physiologic alarm parameters for hospitalized children. J Hosp Med. 2016;11(12):817-823.
Dr. Galloway is a pediatric hospitalist at Sanford Children’s Hospital in Sioux Falls, S.D., assistant professor of pediatrics at the University of South Dakota Sanford School of Medicine, and vice chief of the division of hospital pediatrics at USD SSOM and Sanford Children’s Hospital.
References
1. The Joint Commission. Alarm system safety. Available at: https://www.jointcommission.org/assets/1/18/R3_Report_Issue_5_12_2_13_Final.pdf. Published December 11, 2013.
2. Bonafide CP, Brady PW, Keren R, Conway PH, Marsolo K, Daymont C. Development of heart and respiratory rate percentile curves for hospitalized children. Pediatrics. 2013;131(4):e1150-1157.
Clinical question: Can alarm fatigue in pediatric inpatient settings be safely mitigated by modifying alarm limits with data-driven vital sign reference ranges?
Background: The management of patient alarms in the hospital is a significant safety issue, with the large majority of alarms (85%-99%) either false or not clinically significant. This leads to provider desensitization or alarm fatigue, which has been shown to contribute to adverse events.
In 2014, the Joint Commission made the issue of alarm system safety and alarm fatigue a priority for hospitals.1 Multiple studies have been published addressing alarm fatigue in hospitalized adult patients, but this issue is less well studied in pediatrics, including little guidance on optimizing alarm parameters. Widely used reference ranges and guides are based on limited evidence, primarily based on observational data in healthy outpatients or consensus data.
A 2013 study used vital sign data from hospitalized children to develop percentile curves for heart rate (HR) and respiratory rate (RR) and estimated that 54% of vital sign measurements in hospitalized children are out of range using currently accepted reference ranges.2
To safely decrease the number of out-of-range vital sign measurements resulting from current reference ranges, this study used data from non–critically ill hospitalized children to develop HR and RR percentile charts, and then performed retrospective safety analysis by evaluating effects of modifying the alarm limits on identification of cardiorespiratory arrests (CRA) and rapid response team (RRT) activations.
Study Design: Retrospective, cross-sectional study.
Setting: Single-site, 311-bed quaternary-care academic hospital, both general medical and surgical units.
Synopsis: Vital signs were extracted from the institution’s electronic health record (EHR) for all general medical and surgical patients discharged between Jan. 1, 3013, and May 3, 2014, excluding critically ill children and physiologically implausible vital signs. Two different sets were used, a training set (patients discharged between Jan. 1, 2013, and Dec. 31, 2013) and a validation set (Jan. 1, 2014-May 3, 2014). One HR and RR pair was randomly selected for each 4-hour interval during hospitalization, with a maximum of 10 HR and RR pairs per patient. Age-stratified percentiles were calculated using this data. The 5th and 95th percentile limits using the study data were compared with the 5th and 95th percentile values in the 2013 study, and the reference ranges currently in use at the institution (2004 National Institutes of Health ranges).2
The training set used 62,508 vital sign measurements for 7,202 patients to calculate percentiles for HR and RR among 14 different age groups. The validation set consisted of 82,993 vital sign measurements for 2,287 patients. Using the 5th and 95th percentiles for HR and RR resulted in 24,045 (55.6%) fewer out-of-range measurements in the validation set compared to NIH reference ranges (45% fewer HR values, 61% fewer RR values). This finding, as well as the vital sign percentile ranges, was consistent with the data published in the 2013 study.2
Data for all 148 out-of-ICU RRT and CRA events during the same time period were reviewed using manual chart review. Evaluating vital signs within the 12 hours preceding the events, 144 patients had out-of-range HR or RR measurements using NIH ranges. One hundred thirty-six (94.4%) of these 144 patients also had out-of-range measurements using the study-derived 5th and 95th percentile values.
Manual chart review of the remaining eight patients who had normal HR or RR demonstrated that the RRT or CRA interventions occurred for clinical indications that did no rely on HR or RR measurement (for example, desaturations, difficulty breathing, hematemesis), so the data-driven parameters did not miss any of these events.
Bottom line: In this retrospective study, using data-driven HR and RR parameters was at least as safe as the NIH-published reference ranges currently in use in this hospital. In addition to maintaining safety related to RRT and CRA events, use of the data-driven parameters resulted in 55.6% fewer out-of-range vital sign measurements in the studied population. This may reduce the frequency of false alarms and improve alarm fatigue, and should be studied prospectively in the future.
Citation: Goel VV, Poole SF, Longhurst CA, et al. Safety analysis of proposed data-driven physiologic alarm parameters for hospitalized children. J Hosp Med. 2016;11(12):817-823.
Dr. Galloway is a pediatric hospitalist at Sanford Children’s Hospital in Sioux Falls, S.D., assistant professor of pediatrics at the University of South Dakota Sanford School of Medicine, and vice chief of the division of hospital pediatrics at USD SSOM and Sanford Children’s Hospital.
References
1. The Joint Commission. Alarm system safety. Available at: https://www.jointcommission.org/assets/1/18/R3_Report_Issue_5_12_2_13_Final.pdf. Published December 11, 2013.
2. Bonafide CP, Brady PW, Keren R, Conway PH, Marsolo K, Daymont C. Development of heart and respiratory rate percentile curves for hospitalized children. Pediatrics. 2013;131(4):e1150-1157.
Sneak Peak: The Hospital Leader Blog
FEATURED POST: “A Renewed Call to Overhaul Hospital Observation Care”
In response to concerns about Medicare beneficiary out-of-pocket financial risk, Congress unanimously passed the NOTICE Act, which President Obama signed into law August 5, 2015. This law states that all Medicare beneficiaries hospitalized for 24 hours or more as outpatients under observation must to be notified in writing that they are outpatients “not later than 36 hours after the time such individual begins receiving such services” as well as the associated “implications for cost-sharing.” Last month, the Centers for Medicare & Medicaid Services (CMS) released the final Medicare Outpatient Observation Notice (MOON) that hospitals will start delivering to patients no later than March 8, 2017 to comply with the law. Patients or their representatives must sign the form to acknowledge receipt.

First, Medicare beneficiaries are notified after they have been hospitalized, certainly after they could make an informed decision about accepting observation care. Second, patients or their representative must sign the form, yet it is unclear if this signature holds the patient financially liable, particularly if signed by a representative with no legal authority over the patient’s financial affairs. Third, the form does nothing for a patient’s right to appeal their status. And because observation is a billing distinction, the field at the top of the form requiring hospitals to specify why the patient is not an inpatient is circular reasoning, as patients are outpatients only when they fail to meet Medicare inpatient billing criteria.
Perhaps most importantly, the primary purpose of the NOTICE Act – to inform beneficiaries of the “implications for cost-sharing” when hospitalized under observation – cannot truly be accomplished.
On December 19, 2016, the Department of Health and Human Services Office of Inspector General (OIG) issued the best cost-sharing data available to date describing observation hospital care under the 2-midnight rule. In their report, the OIG used FY 2014 data to compare cost of short outpatient and inpatient stays with similar diagnoses. But because hospitalized outpatients under observation pay a copayment for each individual hospital service, financial risk is not directly correlated with a diagnosis but instead the result of the number, cost, and complexity of services rendered in the hospital, with no limit on the additive amount of per-service deductibles. In contrast, the inpatient deductible is finite per benefit period.
As the OIG report does not provide an accounting of services rendered nor comparison based on equivalent services, it isn’t clear how these cost estimates will help inform discussions when my observation patients receive their MOON.
Dr. Sheehy is a physician and associate professor at the University of Wisconsin School of Medicine and Public Health.
Read the full text of this blog post at http://blogs.hospitalmedicine.org/Blog/a-renewed-call-to-overhaul-hospital-observation-care/
Also on The Hospital Leader…
• New ABIM MOC Two-Year Plan for Internal Medicine Threatens the Focused Practice in Hospital Medicine By Burke Kealey, MD, SFHM
• The Nursing Home Get Out of Jail Card (“We Don’t Want Our Patient Back”). It’s Now Adios. By Brad Flansbaum, DO, MPH, MHM
• The Inmates Are Running the Asylum By Tracy Cardin, ACNP-BC, SFHM
• Do Clinicians Understand Quality Metric Data? By Danielle Scheurer, MD, MSCR, SFHM
• Fake News! Get Your Fake News Here! By Jordan Messler, MD, SFHM
FEATURED POST: “A Renewed Call to Overhaul Hospital Observation Care”
In response to concerns about Medicare beneficiary out-of-pocket financial risk, Congress unanimously passed the NOTICE Act, which President Obama signed into law August 5, 2015. This law states that all Medicare beneficiaries hospitalized for 24 hours or more as outpatients under observation must to be notified in writing that they are outpatients “not later than 36 hours after the time such individual begins receiving such services” as well as the associated “implications for cost-sharing.” Last month, the Centers for Medicare & Medicaid Services (CMS) released the final Medicare Outpatient Observation Notice (MOON) that hospitals will start delivering to patients no later than March 8, 2017 to comply with the law. Patients or their representatives must sign the form to acknowledge receipt.
First, Medicare beneficiaries are notified after they have been hospitalized, certainly after they could make an informed decision about accepting observation care. Second, patients or their representative must sign the form, yet it is unclear if this signature holds the patient financially liable, particularly if signed by a representative with no legal authority over the patient’s financial affairs. Third, the form does nothing for a patient’s right to appeal their status. And because observation is a billing distinction, the field at the top of the form requiring hospitals to specify why the patient is not an inpatient is circular reasoning, as patients are outpatients only when they fail to meet Medicare inpatient billing criteria.
Perhaps most importantly, the primary purpose of the NOTICE Act – to inform beneficiaries of the “implications for cost-sharing” when hospitalized under observation – cannot truly be accomplished.
On December 19, 2016, the Department of Health and Human Services Office of Inspector General (OIG) issued the best cost-sharing data available to date describing observation hospital care under the 2-midnight rule. In their report, the OIG used FY 2014 data to compare cost of short outpatient and inpatient stays with similar diagnoses. But because hospitalized outpatients under observation pay a copayment for each individual hospital service, financial risk is not directly correlated with a diagnosis but instead the result of the number, cost, and complexity of services rendered in the hospital, with no limit on the additive amount of per-service deductibles. In contrast, the inpatient deductible is finite per benefit period.
As the OIG report does not provide an accounting of services rendered nor comparison based on equivalent services, it isn’t clear how these cost estimates will help inform discussions when my observation patients receive their MOON.
Dr. Sheehy is a physician and associate professor at the University of Wisconsin School of Medicine and Public Health.
Read the full text of this blog post at http://blogs.hospitalmedicine.org/Blog/a-renewed-call-to-overhaul-hospital-observation-care/
Also on The Hospital Leader…
• New ABIM MOC Two-Year Plan for Internal Medicine Threatens the Focused Practice in Hospital Medicine By Burke Kealey, MD, SFHM
• The Nursing Home Get Out of Jail Card (“We Don’t Want Our Patient Back”). It’s Now Adios. By Brad Flansbaum, DO, MPH, MHM
• The Inmates Are Running the Asylum By Tracy Cardin, ACNP-BC, SFHM
• Do Clinicians Understand Quality Metric Data? By Danielle Scheurer, MD, MSCR, SFHM
• Fake News! Get Your Fake News Here! By Jordan Messler, MD, SFHM
FEATURED POST: “A Renewed Call to Overhaul Hospital Observation Care”
In response to concerns about Medicare beneficiary out-of-pocket financial risk, Congress unanimously passed the NOTICE Act, which President Obama signed into law August 5, 2015. This law states that all Medicare beneficiaries hospitalized for 24 hours or more as outpatients under observation must to be notified in writing that they are outpatients “not later than 36 hours after the time such individual begins receiving such services” as well as the associated “implications for cost-sharing.” Last month, the Centers for Medicare & Medicaid Services (CMS) released the final Medicare Outpatient Observation Notice (MOON) that hospitals will start delivering to patients no later than March 8, 2017 to comply with the law. Patients or their representatives must sign the form to acknowledge receipt.
First, Medicare beneficiaries are notified after they have been hospitalized, certainly after they could make an informed decision about accepting observation care. Second, patients or their representative must sign the form, yet it is unclear if this signature holds the patient financially liable, particularly if signed by a representative with no legal authority over the patient’s financial affairs. Third, the form does nothing for a patient’s right to appeal their status. And because observation is a billing distinction, the field at the top of the form requiring hospitals to specify why the patient is not an inpatient is circular reasoning, as patients are outpatients only when they fail to meet Medicare inpatient billing criteria.
Perhaps most importantly, the primary purpose of the NOTICE Act – to inform beneficiaries of the “implications for cost-sharing” when hospitalized under observation – cannot truly be accomplished.
On December 19, 2016, the Department of Health and Human Services Office of Inspector General (OIG) issued the best cost-sharing data available to date describing observation hospital care under the 2-midnight rule. In their report, the OIG used FY 2014 data to compare cost of short outpatient and inpatient stays with similar diagnoses. But because hospitalized outpatients under observation pay a copayment for each individual hospital service, financial risk is not directly correlated with a diagnosis but instead the result of the number, cost, and complexity of services rendered in the hospital, with no limit on the additive amount of per-service deductibles. In contrast, the inpatient deductible is finite per benefit period.
As the OIG report does not provide an accounting of services rendered nor comparison based on equivalent services, it isn’t clear how these cost estimates will help inform discussions when my observation patients receive their MOON.
Dr. Sheehy is a physician and associate professor at the University of Wisconsin School of Medicine and Public Health.
Read the full text of this blog post at http://blogs.hospitalmedicine.org/Blog/a-renewed-call-to-overhaul-hospital-observation-care/
Also on The Hospital Leader…
• New ABIM MOC Two-Year Plan for Internal Medicine Threatens the Focused Practice in Hospital Medicine By Burke Kealey, MD, SFHM
• The Nursing Home Get Out of Jail Card (“We Don’t Want Our Patient Back”). It’s Now Adios. By Brad Flansbaum, DO, MPH, MHM
• The Inmates Are Running the Asylum By Tracy Cardin, ACNP-BC, SFHM
• Do Clinicians Understand Quality Metric Data? By Danielle Scheurer, MD, MSCR, SFHM
• Fake News! Get Your Fake News Here! By Jordan Messler, MD, SFHM

VIDEO: Study estimates prevalence of pediatric celiac disease, autoimmunity
By age 15 years, 3.1% of adolescents in Denver developed celiac disease, and another 2% developed a lesser degree of celiac disease autoimmunity, according to a 20-year prospective longitudinal study.
“Although more than 5% of children may experience a period of celiac disease autoimmunity [CDA], not all develop celiac disease [CD] or require gluten-free diets,” Edwin Liu, MD, of University of Colorado School of Medicine and Children’s Hospital Colorado (Aurora, Colo.), wrote with his associates in the May issue of Gastroenterology (doi: 10.1053/j.gastro.2017.02.002). Most celiac autoimmunity probably develops before age 10, “which informs future efforts for universal screening,” they added.
Source: American Gastroenterological Association
About 40% of the general population has the HLA-DQ2 or DQ8 risk genotypes for celiac disease [CD], but little is known about rates of celiac disease among children in the United States, the researchers said. To help fill this gap, they analyzed celiac-risk HLA genotypes for 31,766 infants born between 1993 and 2004 from the Diabetes Autoimmunity Study in the Young. The 1,339 children with HLA risk genotypes were followed for up to 20 years.
By age 15 years, 66 of these children (4.9%) had developed tissue transglutaminase autoantibodies (tTGA) consistent with CDA, and also met criteria for CD, the researchers said. Another 46 (3.4%) children developed only CDA, of whom 46% experienced spontaneous resolution of tTGA seropositivity without treatment. By using genotype-specific risk weighting for population frequencies of HLA, the researchers estimated that 2.4% of the general population of Denver had CDA by age 5 years, 4.3% had CDA by age 10 years, and 5.1% had CDA by age 15 years. Estimated rates of CD were 1.6%, 2.8%, and 3.1%, respectively.
These findings suggest a significant rise in the incidence of CD compared with historical estimates in the United States, and reflect recent studies “using different approaches in North America,” the researchers said. Reasons for the “dramatic increase” are unknown, but environmental causes seem likely, especially given the absence of identified genetic differences and marked changes in the prevalence of CD during the past 2 decades, they added.
Several other reports have documented fluctuating and transient tTGA antibodies in children, the researchers noted. Awareness of transient CD autoantibodies might limit public acceptance of universal screening programs for CD, they said. “Continued long-term follow-up will identify whether the autoimmunity in these subjects truly abates and tolerance develops, or if CDA will recur in time, possibly in response to additional stimulating events,” they added. “At present, low positive tTGA results should be interpreted with caution, and do not necessarily indicate need for biopsy or for treatment.”
The study did not include the DR5/DR7 risk genotype, which accounts for less than 5% of CD cases. The study also did not account for the estimated 2.5% of the general population that has DR3/DR7, which can be considered high risk, the researchers said. Thus, the study is conservative and might underestimate the real incidence of CD or CDA, they added.
The National Institutes of Health provided funding. The investigators reported having no conflicts of interest.
This study calls into question the incidence of celiac disease in the modern pediatric population and, by extension, future prevalence in adults. This is a unique prospective cohort study that followed children over a decade and a half and estimated a cumulative incidence of celiac disease of 3.1% by age 15. In sharp contrast, previous retrospective population-based studies estimated a prevalence of approximately 0.75%-1% in adult and pediatric populations. A recent publication by the United States Preventive Services Task Force used the previously accepted prevalence estimates to recommend against routine screening for celiac disease in the asymptomatic general population as well as targeted screening in those at higher risk. Increases in disease incidence as reported by the current study may call these recommendations into question, particularly in young children where cumulative incidence was high and potential for treatment benefit is substantial.

Dawn Wiese Adams, MD, MS, is assistant professor, director of celiac clinic, in the department of gastroenterology, hepatology, and nutrition, Vanderbilt University Medical Center, Nashville, Tenn. She has no conflicts of interest.
This study calls into question the incidence of celiac disease in the modern pediatric population and, by extension, future prevalence in adults. This is a unique prospective cohort study that followed children over a decade and a half and estimated a cumulative incidence of celiac disease of 3.1% by age 15. In sharp contrast, previous retrospective population-based studies estimated a prevalence of approximately 0.75%-1% in adult and pediatric populations. A recent publication by the United States Preventive Services Task Force used the previously accepted prevalence estimates to recommend against routine screening for celiac disease in the asymptomatic general population as well as targeted screening in those at higher risk. Increases in disease incidence as reported by the current study may call these recommendations into question, particularly in young children where cumulative incidence was high and potential for treatment benefit is substantial.
Dawn Wiese Adams, MD, MS, is assistant professor, director of celiac clinic, in the department of gastroenterology, hepatology, and nutrition, Vanderbilt University Medical Center, Nashville, Tenn. She has no conflicts of interest.
This study calls into question the incidence of celiac disease in the modern pediatric population and, by extension, future prevalence in adults. This is a unique prospective cohort study that followed children over a decade and a half and estimated a cumulative incidence of celiac disease of 3.1% by age 15. In sharp contrast, previous retrospective population-based studies estimated a prevalence of approximately 0.75%-1% in adult and pediatric populations. A recent publication by the United States Preventive Services Task Force used the previously accepted prevalence estimates to recommend against routine screening for celiac disease in the asymptomatic general population as well as targeted screening in those at higher risk. Increases in disease incidence as reported by the current study may call these recommendations into question, particularly in young children where cumulative incidence was high and potential for treatment benefit is substantial.
Dawn Wiese Adams, MD, MS, is assistant professor, director of celiac clinic, in the department of gastroenterology, hepatology, and nutrition, Vanderbilt University Medical Center, Nashville, Tenn. She has no conflicts of interest.
By age 15 years, 3.1% of adolescents in Denver developed celiac disease, and another 2% developed a lesser degree of celiac disease autoimmunity, according to a 20-year prospective longitudinal study.
“Although more than 5% of children may experience a period of celiac disease autoimmunity [CDA], not all develop celiac disease [CD] or require gluten-free diets,” Edwin Liu, MD, of University of Colorado School of Medicine and Children’s Hospital Colorado (Aurora, Colo.), wrote with his associates in the May issue of Gastroenterology (doi: 10.1053/j.gastro.2017.02.002). Most celiac autoimmunity probably develops before age 10, “which informs future efforts for universal screening,” they added.
Source: American Gastroenterological Association
About 40% of the general population has the HLA-DQ2 or DQ8 risk genotypes for celiac disease [CD], but little is known about rates of celiac disease among children in the United States, the researchers said. To help fill this gap, they analyzed celiac-risk HLA genotypes for 31,766 infants born between 1993 and 2004 from the Diabetes Autoimmunity Study in the Young. The 1,339 children with HLA risk genotypes were followed for up to 20 years.
By age 15 years, 66 of these children (4.9%) had developed tissue transglutaminase autoantibodies (tTGA) consistent with CDA, and also met criteria for CD, the researchers said. Another 46 (3.4%) children developed only CDA, of whom 46% experienced spontaneous resolution of tTGA seropositivity without treatment. By using genotype-specific risk weighting for population frequencies of HLA, the researchers estimated that 2.4% of the general population of Denver had CDA by age 5 years, 4.3% had CDA by age 10 years, and 5.1% had CDA by age 15 years. Estimated rates of CD were 1.6%, 2.8%, and 3.1%, respectively.
These findings suggest a significant rise in the incidence of CD compared with historical estimates in the United States, and reflect recent studies “using different approaches in North America,” the researchers said. Reasons for the “dramatic increase” are unknown, but environmental causes seem likely, especially given the absence of identified genetic differences and marked changes in the prevalence of CD during the past 2 decades, they added.
Several other reports have documented fluctuating and transient tTGA antibodies in children, the researchers noted. Awareness of transient CD autoantibodies might limit public acceptance of universal screening programs for CD, they said. “Continued long-term follow-up will identify whether the autoimmunity in these subjects truly abates and tolerance develops, or if CDA will recur in time, possibly in response to additional stimulating events,” they added. “At present, low positive tTGA results should be interpreted with caution, and do not necessarily indicate need for biopsy or for treatment.”
The study did not include the DR5/DR7 risk genotype, which accounts for less than 5% of CD cases. The study also did not account for the estimated 2.5% of the general population that has DR3/DR7, which can be considered high risk, the researchers said. Thus, the study is conservative and might underestimate the real incidence of CD or CDA, they added.
The National Institutes of Health provided funding. The investigators reported having no conflicts of interest.
By age 15 years, 3.1% of adolescents in Denver developed celiac disease, and another 2% developed a lesser degree of celiac disease autoimmunity, according to a 20-year prospective longitudinal study.
“Although more than 5% of children may experience a period of celiac disease autoimmunity [CDA], not all develop celiac disease [CD] or require gluten-free diets,” Edwin Liu, MD, of University of Colorado School of Medicine and Children’s Hospital Colorado (Aurora, Colo.), wrote with his associates in the May issue of Gastroenterology (doi: 10.1053/j.gastro.2017.02.002). Most celiac autoimmunity probably develops before age 10, “which informs future efforts for universal screening,” they added.
Source: American Gastroenterological Association
About 40% of the general population has the HLA-DQ2 or DQ8 risk genotypes for celiac disease [CD], but little is known about rates of celiac disease among children in the United States, the researchers said. To help fill this gap, they analyzed celiac-risk HLA genotypes for 31,766 infants born between 1993 and 2004 from the Diabetes Autoimmunity Study in the Young. The 1,339 children with HLA risk genotypes were followed for up to 20 years.
By age 15 years, 66 of these children (4.9%) had developed tissue transglutaminase autoantibodies (tTGA) consistent with CDA, and also met criteria for CD, the researchers said. Another 46 (3.4%) children developed only CDA, of whom 46% experienced spontaneous resolution of tTGA seropositivity without treatment. By using genotype-specific risk weighting for population frequencies of HLA, the researchers estimated that 2.4% of the general population of Denver had CDA by age 5 years, 4.3% had CDA by age 10 years, and 5.1% had CDA by age 15 years. Estimated rates of CD were 1.6%, 2.8%, and 3.1%, respectively.
These findings suggest a significant rise in the incidence of CD compared with historical estimates in the United States, and reflect recent studies “using different approaches in North America,” the researchers said. Reasons for the “dramatic increase” are unknown, but environmental causes seem likely, especially given the absence of identified genetic differences and marked changes in the prevalence of CD during the past 2 decades, they added.
Several other reports have documented fluctuating and transient tTGA antibodies in children, the researchers noted. Awareness of transient CD autoantibodies might limit public acceptance of universal screening programs for CD, they said. “Continued long-term follow-up will identify whether the autoimmunity in these subjects truly abates and tolerance develops, or if CDA will recur in time, possibly in response to additional stimulating events,” they added. “At present, low positive tTGA results should be interpreted with caution, and do not necessarily indicate need for biopsy or for treatment.”
The study did not include the DR5/DR7 risk genotype, which accounts for less than 5% of CD cases. The study also did not account for the estimated 2.5% of the general population that has DR3/DR7, which can be considered high risk, the researchers said. Thus, the study is conservative and might underestimate the real incidence of CD or CDA, they added.
The National Institutes of Health provided funding. The investigators reported having no conflicts of interest.
Key clinical point: The presence of celiac disease autoimmunity does not predict universal progression to celiac disease.
Major finding: By age 15 years, an estimated 3.1% of children in Denver developed celiac disease, and another 2% developed a lesser degree of celiac disease autoimmunity that often resolved spontaneously without treatment.
Data source: A 20-year prospective study of 1,339 children with genetic risk factors for celiac disease, with extrapolation based on the prevalence of human leukocyte antigen genotypes in the general population.
Disclosures: The National Institutes of Health provided funding. The investigators reported having no conflicts of interest.
