User login
Inaugural Innovation Summit Addressed CTS Creativity and Development
The first AATS Innovation Summit was launched on Saturday morning. The Summit was designed to assist cardiothoracic surgeons in developing new clinically applicable technology by giving them information on how to protect intellectual property, obtain funding, and conduct clinical trials. Attendees also got the latest updates on regulatory pathways and advice on building industry relationships needed to forge a novel product.
Pedro J. del Nido, MD, who was instrumental in coming up with the original idea for the Summit, introduced former AATS President W. Randolph Chitwood Jr., MD, who served as Program Director.

“The most fun part of this meeting,” Dr. Chitwood promised, would be the Commercialization Workshop at the end of the day “where we develop a fictitious product and carry it all the way to commercialization.”
Michael J. Mack, MD, a famous innovator in his own right, opened the didactic portion of the program by outlining the Summit Challenges.

“The whole field of cardiac surgery is moving to a catheter-based approach,” he declared.
He pointed out the tremendous growth in the invention of catheter-based products in the mitral regurgitation space. “The field is fertile with innovators,” he added.
In order to be innovative, he said, quoting what he called the Bavaria Rules, after Joseph E. Bavaria, MD, “You’ve got to have a vision; you’ve got to have a strategy; you need the tactics to implement that strategy, and at the end of the day, you have to execute that strategy.”
He concluded his talk saying, “Our field has a legacy of innovation. Our challenge is to adopt the tools to move that innovation field forward that our specialty has always been known for.”

He listed some of the major innovations in the cardiac space over the past 60+ years, from the beginning of aortic surgery, to mitral valve surgery, the Maze procedure, and even TAVR, pointing out that historically at least 10-20 years has been the lag time between the development of the innovation and its more-or-less widespread adoption, following the so-called S-curve.
The next stage of innovation is not starting from scratch, he said, but rather the recognition that the previous innovation can be improved upon, and the beginning of a new innovation S-curve at the peak of the old, a process of continual development and refinement.
Dr. Cox stressed that for innovation to be adopted, it couldn’t be overly complex, citing the Maze procedure for atrial fibrillation, which was highly superior to the alternatives for treating AF, but has still lagged behind pulmonary vein isolation, a much less effective, but much easier technique used by interventional cardiologists.
He showed data demonstrating that when it came to innovative techniques, cure rate effectively did not matter to adoption, compared to the level of complexity of the new procedure.
He added that it was important for any new innovation to not “get too far away” from nature and its lessons, and that it should not be too complex to be routinely trained for and adopted.
Dr. Cox concluded with an admonition to new innovators to “listen to what your peers say. They have ideas, experiences, and imaginations of their own.”
He added that “they may or may not have anything to say that will change your course, but you have to listen to them.”
The first AATS Innovation Summit was launched on Saturday morning. The Summit was designed to assist cardiothoracic surgeons in developing new clinically applicable technology by giving them information on how to protect intellectual property, obtain funding, and conduct clinical trials. Attendees also got the latest updates on regulatory pathways and advice on building industry relationships needed to forge a novel product.
Pedro J. del Nido, MD, who was instrumental in coming up with the original idea for the Summit, introduced former AATS President W. Randolph Chitwood Jr., MD, who served as Program Director.
“The most fun part of this meeting,” Dr. Chitwood promised, would be the Commercialization Workshop at the end of the day “where we develop a fictitious product and carry it all the way to commercialization.”
Michael J. Mack, MD, a famous innovator in his own right, opened the didactic portion of the program by outlining the Summit Challenges.
“The whole field of cardiac surgery is moving to a catheter-based approach,” he declared.
He pointed out the tremendous growth in the invention of catheter-based products in the mitral regurgitation space. “The field is fertile with innovators,” he added.
In order to be innovative, he said, quoting what he called the Bavaria Rules, after Joseph E. Bavaria, MD, “You’ve got to have a vision; you’ve got to have a strategy; you need the tactics to implement that strategy, and at the end of the day, you have to execute that strategy.”
He concluded his talk saying, “Our field has a legacy of innovation. Our challenge is to adopt the tools to move that innovation field forward that our specialty has always been known for.”
He listed some of the major innovations in the cardiac space over the past 60+ years, from the beginning of aortic surgery, to mitral valve surgery, the Maze procedure, and even TAVR, pointing out that historically at least 10-20 years has been the lag time between the development of the innovation and its more-or-less widespread adoption, following the so-called S-curve.
The next stage of innovation is not starting from scratch, he said, but rather the recognition that the previous innovation can be improved upon, and the beginning of a new innovation S-curve at the peak of the old, a process of continual development and refinement.
Dr. Cox stressed that for innovation to be adopted, it couldn’t be overly complex, citing the Maze procedure for atrial fibrillation, which was highly superior to the alternatives for treating AF, but has still lagged behind pulmonary vein isolation, a much less effective, but much easier technique used by interventional cardiologists.
He showed data demonstrating that when it came to innovative techniques, cure rate effectively did not matter to adoption, compared to the level of complexity of the new procedure.
He added that it was important for any new innovation to not “get too far away” from nature and its lessons, and that it should not be too complex to be routinely trained for and adopted.
Dr. Cox concluded with an admonition to new innovators to “listen to what your peers say. They have ideas, experiences, and imaginations of their own.”
He added that “they may or may not have anything to say that will change your course, but you have to listen to them.”
The first AATS Innovation Summit was launched on Saturday morning. The Summit was designed to assist cardiothoracic surgeons in developing new clinically applicable technology by giving them information on how to protect intellectual property, obtain funding, and conduct clinical trials. Attendees also got the latest updates on regulatory pathways and advice on building industry relationships needed to forge a novel product.
Pedro J. del Nido, MD, who was instrumental in coming up with the original idea for the Summit, introduced former AATS President W. Randolph Chitwood Jr., MD, who served as Program Director.
“The most fun part of this meeting,” Dr. Chitwood promised, would be the Commercialization Workshop at the end of the day “where we develop a fictitious product and carry it all the way to commercialization.”
Michael J. Mack, MD, a famous innovator in his own right, opened the didactic portion of the program by outlining the Summit Challenges.
“The whole field of cardiac surgery is moving to a catheter-based approach,” he declared.
He pointed out the tremendous growth in the invention of catheter-based products in the mitral regurgitation space. “The field is fertile with innovators,” he added.
In order to be innovative, he said, quoting what he called the Bavaria Rules, after Joseph E. Bavaria, MD, “You’ve got to have a vision; you’ve got to have a strategy; you need the tactics to implement that strategy, and at the end of the day, you have to execute that strategy.”
He concluded his talk saying, “Our field has a legacy of innovation. Our challenge is to adopt the tools to move that innovation field forward that our specialty has always been known for.”
He listed some of the major innovations in the cardiac space over the past 60+ years, from the beginning of aortic surgery, to mitral valve surgery, the Maze procedure, and even TAVR, pointing out that historically at least 10-20 years has been the lag time between the development of the innovation and its more-or-less widespread adoption, following the so-called S-curve.
The next stage of innovation is not starting from scratch, he said, but rather the recognition that the previous innovation can be improved upon, and the beginning of a new innovation S-curve at the peak of the old, a process of continual development and refinement.
Dr. Cox stressed that for innovation to be adopted, it couldn’t be overly complex, citing the Maze procedure for atrial fibrillation, which was highly superior to the alternatives for treating AF, but has still lagged behind pulmonary vein isolation, a much less effective, but much easier technique used by interventional cardiologists.
He showed data demonstrating that when it came to innovative techniques, cure rate effectively did not matter to adoption, compared to the level of complexity of the new procedure.
He added that it was important for any new innovation to not “get too far away” from nature and its lessons, and that it should not be too complex to be routinely trained for and adopted.
Dr. Cox concluded with an admonition to new innovators to “listen to what your peers say. They have ideas, experiences, and imaginations of their own.”
He added that “they may or may not have anything to say that will change your course, but you have to listen to them.”
Editorial Board Biographies
Matthew J. Matava, MD
Associate Editor for Professional Sports

Dr. Matava is a professor of Orthopedic Surgery and Physical Therapy, Chief of the Sports Medicine Service, and the Head Team Physician for the varsity athletic program at Washington University in St. Louis. He is also a team physician for the National Hockey League’s St. Louis Blues. Formerly, he was the Head Team Physician for the St. Louis Rams, and was President of the National Football League Physicians Society (NFLPS) from 2013-2015. Dr. Matava earned his Medical Degree from the University of Missouri-Kansas City. He completed his internship and orthopedic surgery residency at Emory University in Atlanta, GA, followed by a fellowship in sports medicine and arthroscopic surgery at the Cincinnati Sports Medicine and Orthopedic Center. He is the recipient of several research awards from Emory University, is a member of the Alpha Omega Medical Honor Society, and received the Palma Chironis Award for Excellence in Teaching from the Washington University Department of Orthopedic Surgery in 2012. Dr. Matava has been listed as a “Best Doctor in America” since 2005, and was recently hailed by Orthopedics This Week as one of the top 28 sports knee surgeons in the nation.
Jeffrey Sawyer, MD
Associate Editor for Pediatrics

Dr. Sawyer is a professor of Orthopaedic Surgery and the Pediatric Orthopaedic Fellowship Director at the University of Tennessee-Campbell Clinic. He also serves as a reviewer/editor for the Journal of Pediatric Orthopaedics and Orthopedic Clinics of North America. He graduated from the University of Rochester School of Medicine and completed his residency at the University of Pennsylvania, prior to completing his Pediatric Orthopaedic Fellowship at the University of Tennessee-Campbell Clinic. Dr. Sawyer has held numerous leadership positions in the Pediatric Orthopaedic Society of North America (POSNA). He also was a POSNA Traveling Fellow and won the POSNA Special Achievement Award for his work on the Pediatric Orthopaedic Workforce. He is a national authority on pediatric orthopedic trauma, and is on the Executive Committee of the Children’s Spine Foundation.
Brian K. Vickaryous, MD
Associate Editor for Trauma

Dr. Vickaryous is a specialist in orthopedic traumatology at the Florida Hospital Orthopedic Institute in Orlando, Florida, and has an additional subspecialty board certification in sports medicine. He attended the University of Miami, Florida through the combined degree Medical
Honors Program and completed his residency at the William Beaumont Army Medical Center/Texas Tech University of the Health Sciences. Dr. Vickaryous has also deployed overseas as Commander of the Trauma Unit, the 8th Forward Surgical Team, in Iraq in support of Operation Iraqi Freedom. He currently is a member of the American Academy of Orthopaedic
Surgeons (AAOS) and the Orthopaedic Trauma Association (OTA).
Michael B. Gerhardt, MD
Associate Editor for Sports Medicine

Dr. Gerhardt is a sports medicine specialist at the Kerlan-Jobe Institute and Santa Monica Orthopaedic Group in Los Angeles, CA. He also serves as faculty in the Department of Orthopaedic Surgery at Cedars-Sinai Medical Center. Dr. Gerhardt earned his undergraduate degree from UC San Diego and graduated medical school with honors from the Medical College of Pennsylvania. He received the Leonard Marmur Award for excellence in research and education during his orthopedic residency at the University of Southern California, prior to completing a Sports Medicine Fellowship in 2003. He received further training in hip arthroscopy at the Nashville Orthopaedic Sports Medicine and Orthopaedic Clinic, and maintains a leadership role in the area of sports medicine and hip preservation on a national and international level. Currently, he serves as Team Physician for the US Soccer Men’s National Team, the Los Angeles Galaxy, and Pepperdine University.
Matthew J. Matava, MD
Associate Editor for Professional Sports
Dr. Matava is a professor of Orthopedic Surgery and Physical Therapy, Chief of the Sports Medicine Service, and the Head Team Physician for the varsity athletic program at Washington University in St. Louis. He is also a team physician for the National Hockey League’s St. Louis Blues. Formerly, he was the Head Team Physician for the St. Louis Rams, and was President of the National Football League Physicians Society (NFLPS) from 2013-2015. Dr. Matava earned his Medical Degree from the University of Missouri-Kansas City. He completed his internship and orthopedic surgery residency at Emory University in Atlanta, GA, followed by a fellowship in sports medicine and arthroscopic surgery at the Cincinnati Sports Medicine and Orthopedic Center. He is the recipient of several research awards from Emory University, is a member of the Alpha Omega Medical Honor Society, and received the Palma Chironis Award for Excellence in Teaching from the Washington University Department of Orthopedic Surgery in 2012. Dr. Matava has been listed as a “Best Doctor in America” since 2005, and was recently hailed by Orthopedics This Week as one of the top 28 sports knee surgeons in the nation.
Jeffrey Sawyer, MD
Associate Editor for Pediatrics
Dr. Sawyer is a professor of Orthopaedic Surgery and the Pediatric Orthopaedic Fellowship Director at the University of Tennessee-Campbell Clinic. He also serves as a reviewer/editor for the Journal of Pediatric Orthopaedics and Orthopedic Clinics of North America. He graduated from the University of Rochester School of Medicine and completed his residency at the University of Pennsylvania, prior to completing his Pediatric Orthopaedic Fellowship at the University of Tennessee-Campbell Clinic. Dr. Sawyer has held numerous leadership positions in the Pediatric Orthopaedic Society of North America (POSNA). He also was a POSNA Traveling Fellow and won the POSNA Special Achievement Award for his work on the Pediatric Orthopaedic Workforce. He is a national authority on pediatric orthopedic trauma, and is on the Executive Committee of the Children’s Spine Foundation.
Brian K. Vickaryous, MD
Associate Editor for Trauma
Dr. Vickaryous is a specialist in orthopedic traumatology at the Florida Hospital Orthopedic Institute in Orlando, Florida, and has an additional subspecialty board certification in sports medicine. He attended the University of Miami, Florida through the combined degree Medical
Honors Program and completed his residency at the William Beaumont Army Medical Center/Texas Tech University of the Health Sciences. Dr. Vickaryous has also deployed overseas as Commander of the Trauma Unit, the 8th Forward Surgical Team, in Iraq in support of Operation Iraqi Freedom. He currently is a member of the American Academy of Orthopaedic
Surgeons (AAOS) and the Orthopaedic Trauma Association (OTA).
Michael B. Gerhardt, MD
Associate Editor for Sports Medicine
Dr. Gerhardt is a sports medicine specialist at the Kerlan-Jobe Institute and Santa Monica Orthopaedic Group in Los Angeles, CA. He also serves as faculty in the Department of Orthopaedic Surgery at Cedars-Sinai Medical Center. Dr. Gerhardt earned his undergraduate degree from UC San Diego and graduated medical school with honors from the Medical College of Pennsylvania. He received the Leonard Marmur Award for excellence in research and education during his orthopedic residency at the University of Southern California, prior to completing a Sports Medicine Fellowship in 2003. He received further training in hip arthroscopy at the Nashville Orthopaedic Sports Medicine and Orthopaedic Clinic, and maintains a leadership role in the area of sports medicine and hip preservation on a national and international level. Currently, he serves as Team Physician for the US Soccer Men’s National Team, the Los Angeles Galaxy, and Pepperdine University.
Matthew J. Matava, MD
Associate Editor for Professional Sports
Dr. Matava is a professor of Orthopedic Surgery and Physical Therapy, Chief of the Sports Medicine Service, and the Head Team Physician for the varsity athletic program at Washington University in St. Louis. He is also a team physician for the National Hockey League’s St. Louis Blues. Formerly, he was the Head Team Physician for the St. Louis Rams, and was President of the National Football League Physicians Society (NFLPS) from 2013-2015. Dr. Matava earned his Medical Degree from the University of Missouri-Kansas City. He completed his internship and orthopedic surgery residency at Emory University in Atlanta, GA, followed by a fellowship in sports medicine and arthroscopic surgery at the Cincinnati Sports Medicine and Orthopedic Center. He is the recipient of several research awards from Emory University, is a member of the Alpha Omega Medical Honor Society, and received the Palma Chironis Award for Excellence in Teaching from the Washington University Department of Orthopedic Surgery in 2012. Dr. Matava has been listed as a “Best Doctor in America” since 2005, and was recently hailed by Orthopedics This Week as one of the top 28 sports knee surgeons in the nation.
Jeffrey Sawyer, MD
Associate Editor for Pediatrics
Dr. Sawyer is a professor of Orthopaedic Surgery and the Pediatric Orthopaedic Fellowship Director at the University of Tennessee-Campbell Clinic. He also serves as a reviewer/editor for the Journal of Pediatric Orthopaedics and Orthopedic Clinics of North America. He graduated from the University of Rochester School of Medicine and completed his residency at the University of Pennsylvania, prior to completing his Pediatric Orthopaedic Fellowship at the University of Tennessee-Campbell Clinic. Dr. Sawyer has held numerous leadership positions in the Pediatric Orthopaedic Society of North America (POSNA). He also was a POSNA Traveling Fellow and won the POSNA Special Achievement Award for his work on the Pediatric Orthopaedic Workforce. He is a national authority on pediatric orthopedic trauma, and is on the Executive Committee of the Children’s Spine Foundation.
Brian K. Vickaryous, MD
Associate Editor for Trauma
Dr. Vickaryous is a specialist in orthopedic traumatology at the Florida Hospital Orthopedic Institute in Orlando, Florida, and has an additional subspecialty board certification in sports medicine. He attended the University of Miami, Florida through the combined degree Medical
Honors Program and completed his residency at the William Beaumont Army Medical Center/Texas Tech University of the Health Sciences. Dr. Vickaryous has also deployed overseas as Commander of the Trauma Unit, the 8th Forward Surgical Team, in Iraq in support of Operation Iraqi Freedom. He currently is a member of the American Academy of Orthopaedic
Surgeons (AAOS) and the Orthopaedic Trauma Association (OTA).
Michael B. Gerhardt, MD
Associate Editor for Sports Medicine
Dr. Gerhardt is a sports medicine specialist at the Kerlan-Jobe Institute and Santa Monica Orthopaedic Group in Los Angeles, CA. He also serves as faculty in the Department of Orthopaedic Surgery at Cedars-Sinai Medical Center. Dr. Gerhardt earned his undergraduate degree from UC San Diego and graduated medical school with honors from the Medical College of Pennsylvania. He received the Leonard Marmur Award for excellence in research and education during his orthopedic residency at the University of Southern California, prior to completing a Sports Medicine Fellowship in 2003. He received further training in hip arthroscopy at the Nashville Orthopaedic Sports Medicine and Orthopaedic Clinic, and maintains a leadership role in the area of sports medicine and hip preservation on a national and international level. Currently, he serves as Team Physician for the US Soccer Men’s National Team, the Los Angeles Galaxy, and Pepperdine University.
Presentation of the 2016 Resident Writer’s Award

Darla Conrad (left), Senior Director, North America Education Solutions, Johnson & Johnson Medical Devices, presents Kalpit N. Shah, MD (right) with his plaque for the second-place Resident Writer’s Award, and Christopher Rice, MD (center) with his plaque for the third-place Resident Writer’s Award at the 2017 Annual Meeting of the American Academy of Orthopaedic Surgeons (AAOS) in San Diego.
Winners of the 2016 Resident Writer’s Award
First-Place Award
An Original Study
Clinical Outcomes of Anatomical Total Shoulder Arthroplasty in a Young, Active Population

Dr. Kusnezov is a senior resident, completing his orthopedic surgery residency training, at the Texas Tech University Health Sciences Center/William Beaumont Army Medical Center joint military-civilian program in El Paso, Texas. Prior to residency, he completed both his undergraduate education and medical school at the University of California, Los Angeles, graduating Summa Cum Laude and AOA, respectively. Dr. Kusnezov is currently engaged in a multitude of ongoing projects with over 50 peer-reviewed publications to date. His research interests include trauma and limb salvage, complex total joint reconstruction, and interdisciplinary system improvement.
Second-Place Award
An Original Study
Patient-Reported Outcome Measures: How Do Digital Tablets Stack Up to Paper Forms? A Randomized, Controlled Study

Dr. Shah is currently in his third year of orthopedic surgery residency training at Brown University in Providence, Rhode Island. Prior to residency, he completed undergraduate education at the University of California, Berkeley, and medical school at the University of California, Irvine. He hopes to pursue a hand and upper extremity fellowship after residency. His research interests include upper extremity trauma and surgical complications, as well as technology and its implications on orthopedic surgery.
Third-Place Award
An Original Study
Treating Tibia Fractures With Far Cortical Locking Implants

Dr. Rice is an orthopedic surgery resident at the University of Wisconsin, Madison. He received his medical degree from the University of Utah and attended Brigham Young University for his undergraduate studies. He has a special interest in disorders of the hand and upper extremity trauma.
Darla Conrad (left), Senior Director, North America Education Solutions, Johnson & Johnson Medical Devices, presents Kalpit N. Shah, MD (right) with his plaque for the second-place Resident Writer’s Award, and Christopher Rice, MD (center) with his plaque for the third-place Resident Writer’s Award at the 2017 Annual Meeting of the American Academy of Orthopaedic Surgeons (AAOS) in San Diego.
Winners of the 2016 Resident Writer’s Award
First-Place Award
An Original Study
Clinical Outcomes of Anatomical Total Shoulder Arthroplasty in a Young, Active Population
Dr. Kusnezov is a senior resident, completing his orthopedic surgery residency training, at the Texas Tech University Health Sciences Center/William Beaumont Army Medical Center joint military-civilian program in El Paso, Texas. Prior to residency, he completed both his undergraduate education and medical school at the University of California, Los Angeles, graduating Summa Cum Laude and AOA, respectively. Dr. Kusnezov is currently engaged in a multitude of ongoing projects with over 50 peer-reviewed publications to date. His research interests include trauma and limb salvage, complex total joint reconstruction, and interdisciplinary system improvement.
Second-Place Award
An Original Study
Patient-Reported Outcome Measures: How Do Digital Tablets Stack Up to Paper Forms? A Randomized, Controlled Study
Dr. Shah is currently in his third year of orthopedic surgery residency training at Brown University in Providence, Rhode Island. Prior to residency, he completed undergraduate education at the University of California, Berkeley, and medical school at the University of California, Irvine. He hopes to pursue a hand and upper extremity fellowship after residency. His research interests include upper extremity trauma and surgical complications, as well as technology and its implications on orthopedic surgery.
Third-Place Award
An Original Study
Treating Tibia Fractures With Far Cortical Locking Implants
Dr. Rice is an orthopedic surgery resident at the University of Wisconsin, Madison. He received his medical degree from the University of Utah and attended Brigham Young University for his undergraduate studies. He has a special interest in disorders of the hand and upper extremity trauma.
Darla Conrad (left), Senior Director, North America Education Solutions, Johnson & Johnson Medical Devices, presents Kalpit N. Shah, MD (right) with his plaque for the second-place Resident Writer’s Award, and Christopher Rice, MD (center) with his plaque for the third-place Resident Writer’s Award at the 2017 Annual Meeting of the American Academy of Orthopaedic Surgeons (AAOS) in San Diego.
Winners of the 2016 Resident Writer’s Award
First-Place Award
An Original Study
Clinical Outcomes of Anatomical Total Shoulder Arthroplasty in a Young, Active Population
Dr. Kusnezov is a senior resident, completing his orthopedic surgery residency training, at the Texas Tech University Health Sciences Center/William Beaumont Army Medical Center joint military-civilian program in El Paso, Texas. Prior to residency, he completed both his undergraduate education and medical school at the University of California, Los Angeles, graduating Summa Cum Laude and AOA, respectively. Dr. Kusnezov is currently engaged in a multitude of ongoing projects with over 50 peer-reviewed publications to date. His research interests include trauma and limb salvage, complex total joint reconstruction, and interdisciplinary system improvement.
Second-Place Award
An Original Study
Patient-Reported Outcome Measures: How Do Digital Tablets Stack Up to Paper Forms? A Randomized, Controlled Study
Dr. Shah is currently in his third year of orthopedic surgery residency training at Brown University in Providence, Rhode Island. Prior to residency, he completed undergraduate education at the University of California, Berkeley, and medical school at the University of California, Irvine. He hopes to pursue a hand and upper extremity fellowship after residency. His research interests include upper extremity trauma and surgical complications, as well as technology and its implications on orthopedic surgery.
Third-Place Award
An Original Study
Treating Tibia Fractures With Far Cortical Locking Implants
Dr. Rice is an orthopedic surgery resident at the University of Wisconsin, Madison. He received his medical degree from the University of Utah and attended Brigham Young University for his undergraduate studies. He has a special interest in disorders of the hand and upper extremity trauma.
E-cigarettes and vapes: Do they work for smoking cessation and should we be recommending their use?
The popularity of electronic cigarettes (E-cigs) and “vapes” has grown dramatically, spawning a new industry of electronic nicotine delivery systems (ENDS). With the increasing use of E-cigs not only for smoking cessation, but also as a primary nicotine source, it is important for mental health professionals to be prepared to discuss use of these devices with patients. In this article, we will describe:
- the composition of E-cigs and their current use
- evidence for their use for smoking cessation
- adverse health effects
- recommendations of major regulatory agencies.
Finally, we will provide recommendations for E-cig use in clinical populations.
What is an electronic nicotine delivery system?
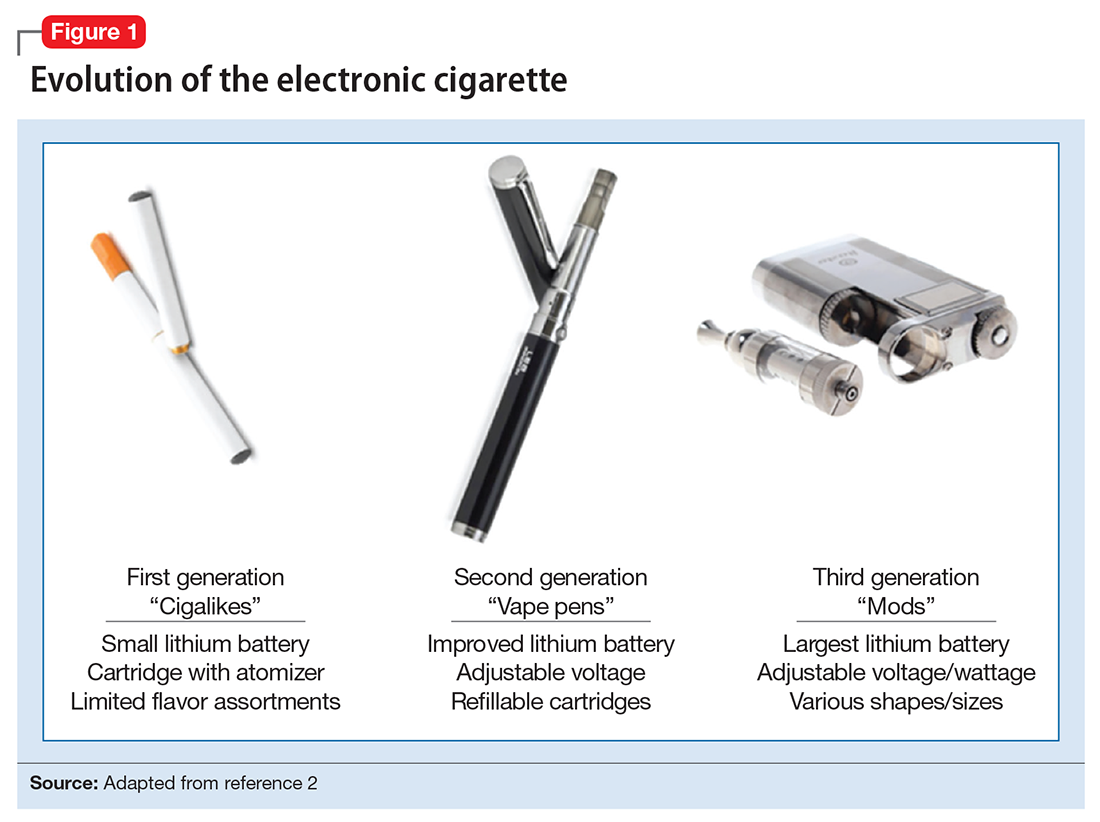
ENDS produce an aerosol with or without nicotine that is inhaled and is thought to mimic the use of combustible cigarettes. ENDS evolved from basic E-cigs into a less “cigarette-like” and more customizable product (Figure 1). ENDS include a range of designs and go by various names, including “personal vaporizers,” “e-cigars,” and “e-hookahs” (in this article, we will use the term “ENDS” to refer to these devices).
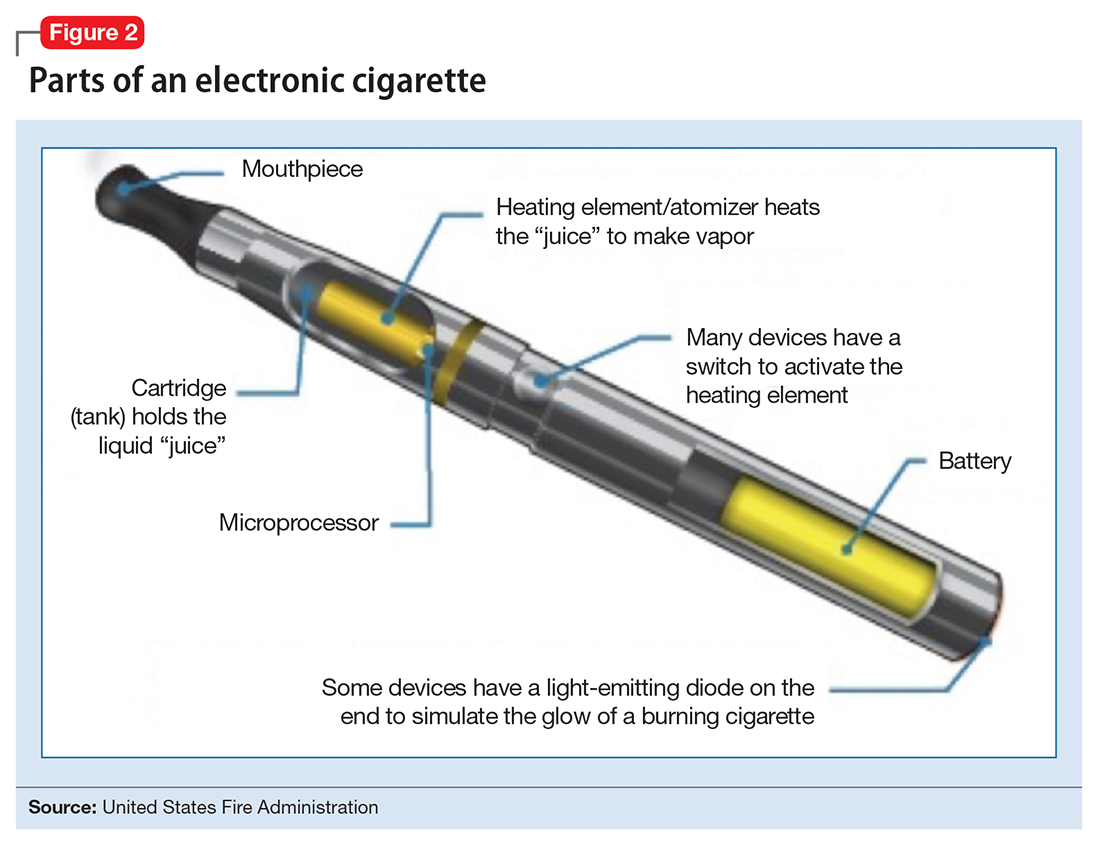
The general design of ENDS is a plastic tubing system that contains a mouthpiece, battery, electronic heating element (“vaporizer”), and a cartridge with liquid solvent with or without nicotine or flavoring (Figure 2). One draw on the mouthpiece or press of a button activates the device, heats the solution, and delivers a vapor in a similar manner to taking a puff of a cigarette. Although studies have shown that ENDS result in significant increases in plasma nicotine concentrations in 5 minutes,1 the plasma nicotine levels obtained with the first-generation “cigarette-like” ENDS are much lower than those caused by inhaling tobacco smoke.2 Over time nicotine delivery capability has improved as ENDS have evolved such that the rate of nicotine delivery and peak concentration obtained with newer models more closely mirror tobacco cigarettes.3 Whether the rapid delivery of larger amounts of nicotine helps or hinders one’s efforts to break nicotine addiction remains to be determined because of the reinforcing properties of the drug.
The liquid in the E-cig cartridge typically contains not only nicotine but a number of chemical compounds with potentially deleterious or unknown health risks. The 3 main ingredients include:
- a solvent of glycerin and/or propylene glycol
- nicotine in various concentrations
- flavorings.
The glycerin or propylene glycol forms the basis for the aerosol. Nicotine concentrations vary from 0 (denicotinized) to 35 mcg per puff.4 A study reported 7,700 unique flavors available for vaping liquid.5 The liquid also contains impurities, such as anabasine, which has effects on the α-7 nicotinic acetylcholine receptor and its principal use is as an insecticide and β-nicotyrine, which inhibits cytochrome P450 2A.
Epidemiology and end-user perspectives
In 2014, 12.4% of U.S. adults classified themselves as “ever users” of ENDS (used at least once) and 3.7% of adults classified themselves as current users, according to the National Health Interview Study.6 Importantly, among E-cig users who had not used combustible cigarettes, young adults (age 18 to 24) were more likely to have tried ENDS than older adults. ENDS are becoming more popular across the globe. A study in the European Union found that ever users of ENDS most commonly were current cigarette smokers (31%) followed by former (10.8%) and never smokers (2.3%).7
ENDS use is relevant for mental health professionals because of the high rate of comorbid tobacco use disorder in individuals with psychiatric conditions. For example, 2 U.S. population surveys8,9 revealed those with mental health conditions were 1.5 to 2 times more likely to have tried ENDS and 2 to 3 times more likely to be current users. Those with psychiatric illness reported similar reasons for ENDS use as other individuals, including “just because,” use as a smoking cessation aid, ease of use, and perceived safety vs combustible cigarettes.
A recent review that included 9 studies focusing on ENDS use in those with mental illness reported mixed findings on the utility of these devices to reduce or stop use of combustible cigarettes.10 Additionally, it is important to monitor the use of cigarettes and ENDS in patients with psychiatric illness because the byproducts of tobacco smoke can affect the metabolism of some psychotropic medications.11 Although reduced use of combustible cigarettes could lead to lower dosing of some psychotropics, an unreported decrease in combustible cigarette use could lead to supratherapeutic drug levels. There are no data on the effect of ENDS on the metabolism of psychotropics.
ENDS are increasingly popular among adolescents. In 2015, there were an estimated 4.6 million current tobacco users among middle/high school youths in the United States and 3 million current ENDS users, according to the National Youth Tobacco Surveys.12 The shift from combustible cigarettes to ENDS is notable, with an increase in the percentage of current E-cig users and a decrease in the percentage of exclusive combustible cigarette users. In addition, there has been no change in the prevalence of lifetime tobacco users.12 This is a global issue, as reports of ever use of ENDS by adolescents range from 6.5% to 31% in the United States, 14.6% in Canada, and 4.7% to 38.5% in Europe.13 Based on these trends, the U.S. Surgeon General released a statement warning against the use of ENDS in youth because of the lack of safety data and strong association with use of tobacco products.14
There are a number of possible reasons for the increasing popularity of ENDS, including the product’s novelty, lack of regulations regarding their sale, availability of flavorings, and the perception that ENDS are safe alternatives to cigarettes. E-cig–using youths have described ENDS as “not at all harmful” and “not at all addictive” and believe that ENDS with flavoring are less harmful than those without.15 Although studies in adults show some users reporting that ENDS are less satisfying, they are seen as useful in decreasing craving and a safer alternative to cigarettes.16,17
Are ENDS effective for smoking cessation?
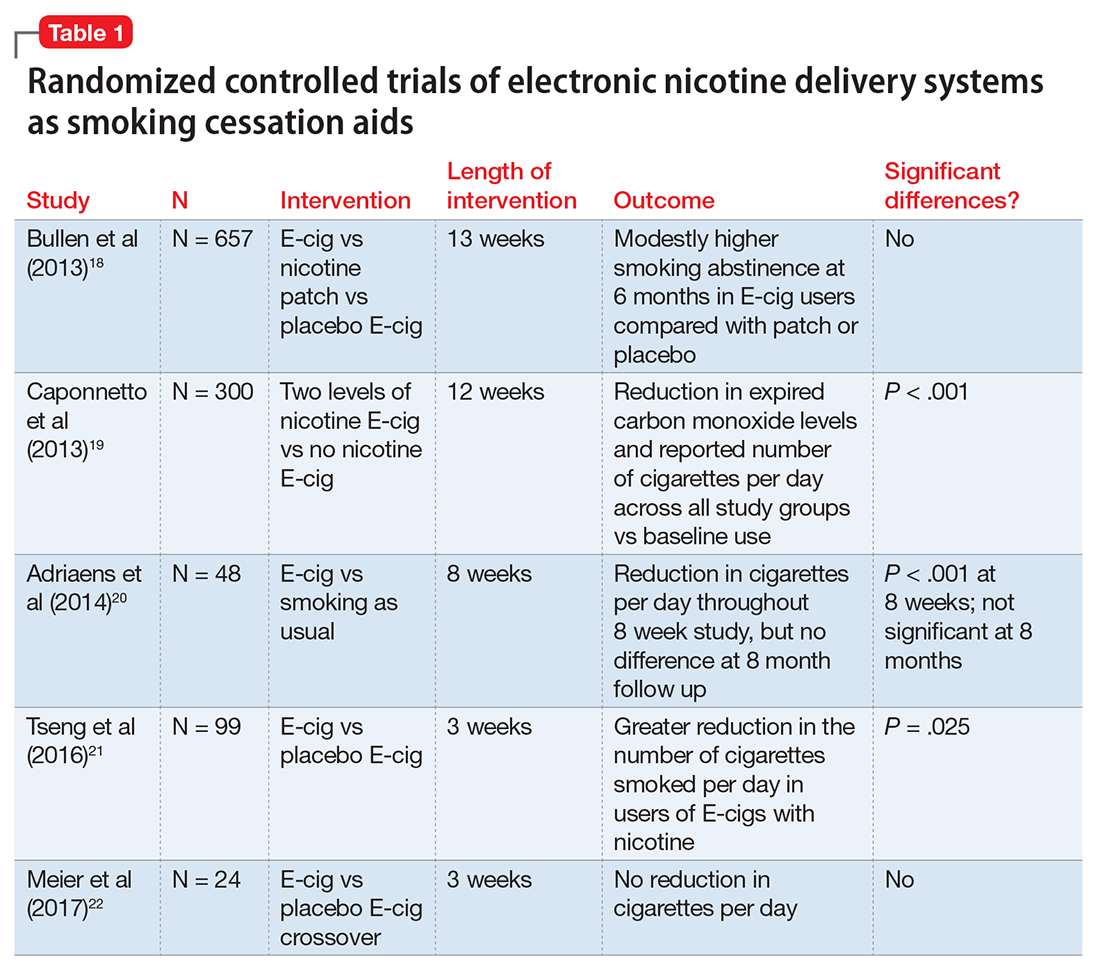
The evidence for ENDS as aids to smoking cessation remains murky (Table 118-22). There is a paucity of randomized controlled clinical trials (RCTs) investigating ENDS for smoking cessation or reduction, and it is difficult to quantify the amount of nicotine used in ENDS because of the variety of delivery systems and cartridges. In a recent Cochrane review, those using ENDS to quit smoking were more likely to be abstinent from combustible cigarettes at 6 months vs those using nicotine-free ENDS (relative risk = 2.29; 95% CI, 1.05 to 4.96), but there was no significant difference in quit rates compared with nicotine patches.23 However, the confidence in this finding was rated as low because of the limited number of RCTs. Of note, the authors found 15 ongoing RCTs at the time of publication that might be eligible for later evaluation.
Non-RCTs reveal mixed data. Positive results include 1 study with an odds ratio of 6.07 to quit for intensive ENDS users vs non-users,24 and another with dual users of combustible and electronic cigarettes having a 46% quit rate at 1 year.25 Additionally, in a pilot study providing ENDS to 14 patients with schizophrenia who had no previous desire to quit smoking, authors noted a reduction in the number of cigarettes smoked per day by 50% in one-half of participants and abstinence in 14% of participants at 52 weeks.26 Studies with neutral or negative results include those showing ENDS users to be current combustible tobacco smokers, and use of ENDS not predicting smoking cessation.4,27 Data also are mixed regarding the use of ENDS as a harm reduction strategy. One study found that ENDS decreased cigarette consumption, but did not increase the likelihood of quitting,28 while another reported that daily use of ENDS increased the odds of reducing smoking by as much as 2.5 times compared with non-use of such aids.29 In a 24-month prospective cohort study following tobacco users, there was no difference in the number of cigarettes smoked per day in those who started the trial as users of combustible cigarettes alone vs combustible cigarettes plus ENDS users.30 Interestingly, those who started the study as combustible cigarette users and switched to ENDS and those who had continued dual use throughout the 24 months smoked fewer combustible cigarettes per day than those who never tried ENDS or quit during the study period.
Health effects

To better understand the adverse health effects of ENDS, one must consider potential short- and long-term consequences (Table 2). In the short-term, ENDS have been found to increase markers of inflammation and oxidative stress acutely as evidenced by in vivo laboratory studies.31,32 ENDS also have been linked to upper respiratory irritation, in part, because of the transformation of glycerin in the nicotine cartridge to acrolein upon combustion.33 Even 5 minutes of ad lib E-cig use has been found to significantly increase airflow resistance during pulmonary function tests34—changes that have been shown to precede more persistent alterations in peak expiratory flow, such as those seen in chronic obstructive pulmonary disease. The more common patient-reported side effects include:
- daytime cough (27%)
- phlegm production (25%)
- headache (21%)
- dry mouth/throat (20%)
- vertigo, headache, or nausea (9%).35,36
A RCT investigating efficacy of E-cigs vs nicotine patches vs denicotinized E-cigs found no difference among the groups in the number of reported adverse events.18 Interestingly, another RCT found a decrease in adverse events, such as dry cough, mouth irritation, throat irritation, shortness of breath, and headache, compared with baseline in combustible cigarette smokers who used regular or denicotinized E-cigs.19
Although no studies have directly investigated long-term health consequences of ENDS because of their relative novelty, one can extrapolate potential harmful long-term effects based on knowledge of the products’ chemical constituents. For example, propylene glycol can degrade into propylene oxide, a class 2B carcinogen.37 Other potential carcinogens in the aerosol include formaldehyde and acetaldehyde. On a broader scale, many of the particulates have been shown to cause systemic inflammation, which is thought to increase cardiovascular and respiratory disease and death.38 Flavorings in ENDS include a variety of components including, but not limited to, aldehydes, which are irritants, and other additives that have been associated with respiratory disease.39
Second-hand exposure. There are no long-term studies of second-hand vapor exposure, but similar to long-term health on primary users, one can glean some observations from the literature. It is promising that compared with cigarettes, ENDS lack sidestream smoke and the vapor has not been found to contain carbon monoxide.40 Some research has demonstrated that the size and spray of fine particles in the aerosol is as large or larger than combustible cigarettes.41 Formaldehyde, acetaldehyde, isoprene, and acetic acid have been found in ENDS vapor.40 Interestingly, a simulated café study found elevated nicotine, glycerine, hydrocarbon, and other materials classified as carcinogens in the air.42
Although it is popularly thought that ENDS are less toxic than tobacco cigarettes, there is not enough evidence to estimate precisely as to how much less toxic or the consequences of use. ENDS are increasingly popular and are being used by never smokers who should be educated on the potential harm that ENDS pose.
Recommendations from agencies and medical organizations
The World Health Organization (WHO) recommended prohibiting the use of ENDS in indoor spaces to minimize potential health risks to users and non-users. The WHO also aims to prevent dissemination of unproven health claims, including claims that ENDS are effective—or not—or that the devices are innocuous.36 In the United States, the FDA has stated that ENDS are not recommended for safe quitting (2009). In August 2016, the FDA introduced regulations banning the sale of ENDS to individuals age <18 and required manufacturers to submit documents detailing all ingredients for review and possible approval.
The American Lung Association has stated its concerns about the use of ENDS but has not made any direct recommendations. The American Heart Association reports a potential negative public health impact and provides clinical guideline recommendations.43 Prominent psychiatric organizations such as the American Psychiatric Association, American Academy of Addiction Psychiatry (AAAP), the Substance Abuse and Mental Health Services Administration (SAMHSA), and the National Institute of Drug Abuse do not have official statements supporting or rejecting the use of ENDS. However, they do note the potential harm and lack of substantial evidence for efficacy of ENDS as a smoking cessation tool, and the AAAP and SAMHSA state that they will work with regulatory agencies to reduce the use of toxic products with addictive potential including ENDS.44-46
Clinical recommendations
We do not recommend ENDS as a first-line treatment for smoking cessation because there is no evidence they are superior to the FDA-approved nicotine replacement therapies (NRTs), the paucity of research into the potential short- and long-term health risks of ENDS, and the fact that these products are not regulated for use as smoking cessation aids. It is, however, advisable to discuss ENDS use with patients by:
- asking if they are using the products
- assessing whether the user also is a smoker
- advising the patient to quit.
It also is important to assess the patient’s knowledge and attitudes regarding ENDS use and provide education about the products. Some patients firmly believe that ENDS are the lesser of 2 evils, and they are decreasing the harms of smoking by using these devices. While the debate over a potential harm reduction strategy unfolds,47 we think that because of the state of the evidence it is prudent to adopt a more precautionary stance and recommend that patients work toward abstinence from nicotine in any form.
For dual tobacco/ENDS users and for patients using ENDS who want to quit smoking, we recommend treatment with an approved pharmacotherapy (ie, NRTs, bupropion, and varenicline) combined with counseling. A 2013 Cochrane Review found that all pharamacotherapy options are more effective than placebo, and combination NRT and varenicline are superior to single NRT or bupropion (Box).23,48

1. Hajek P, Goniewicz ML, Phillips A, et al. Nicotine intake from electronic cigarettes on initial use and after 4 weeks of regular use. Nicotine Tob Res. 2015;17(2):175-179.
2. Farsalinos KE, Polosa R. Safety evaluation and risk assessment of electronic cigarettes as tobacco cigarette substitutes: a systematic review. Ther Adv Drug Saf. 2014;5(2):67-86.
3. St Helen G, Havel C, Dempsey DA, et al. Nicotine delivery, retention and pharmacokinetics from various electronic cigarettes. Addiction. 2016;111(3):535-544.
4. Grana R, Benowitz N, Glantz SA. E-cigarettes: a scientific review. Circulation. 2014;129(19):1972-1986.
5. Zhu SH, Sun JY, Bonnevie E, et al. Four hundred and sixty brands of e-cigarettes and counting: implications for product regulation. Tob Control. 2014;23(suppl 3):iii3-iii9. doi: 10.1136/tobaccocontrol-2014-051670.
6. Schoenborn CA, Gindi RM. Electronic cigarette use among adults: United States, 2014. NCHS Data Brief. 2015;(217):1-8.
7. Farsalinos KE, Poulas K, Voudris V, et al. Electronic cigarette use in the European Union: analysis of a representative sample of 27 460 Europeans from 28 countries. Addiction. 2016;111(11):2032-2040.
8. Cummins SE, Zhu SH, Tedeschi GJ, et al. Use of e-cigarettes by individuals with mental health conditions. Tob Control. 2015;23(suppl 3):iii48-iii53. doi: 10.1136/tobaccocontrol-2013-051511.
9. Spears CA, Jones DM, Weaver SR, et al. Use of electronic nicotine delivery systems among adults with mental health conditions, 2015. Int J Environ Res Public Heal. 2017;14(1):10.
10. Hefner K, Valentine G, Sofuoglu M. Electronic cigarettes and mental illness: reviewing the evidence for help and harm among those with psychiatric and substance use disorders [published online February 2, 2017]. Am J Addict. doi: 10.1111/ajad.12504.
11. Anthenelli R. How—and why—to help psychiatric patients stop smoking. Current Psychiatry. 2005;4(1):77-87.
12. Singh T, Arrazola RA, Corey CG, et al. Tobacco use among middle and high school students—United States, 2011-2015. MMWR Morb Mortal Wkly Rep. 2016;65(14):361-367.
13. Greenhill R, Dawkins L, Notley C, et al. Adolescent awareness and use of electronic cigarettes: a review of emerging trends and findings. J Adolesc Heal. 2016;59(6):612-619.
14. U.S. Department of Health and Human Services. E-cigarette use among youth and young adults: a report of the Surgeon General. Atlanta, GA: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention; 2016.
15. Cooper M, Harrell MB, Pérez A, et al. Flavorings and perceived harm and addictiveness of e-cigarettes among youth. Tob Regul Sci. 2016;2(3):278-289.
16. Kim H, Davis AH, Dohack JL, et al. E-cigarettes use behavior and experience of adults: qualitative research findings to inform e-cigarette use measure development. Nicotine Tob Res. 2017;19(2):190-196.
17. Czoli CD, Fong GT, Mays D, et al. How do consumers perceive differences in risk across nicotine products? A review of relative risk perceptions across smokeless tobacco, e-cigarettes, nicotine replacement therapy and combustible cigarettes. Tob Control. 2017;26(e1):e49-e58.
18. Bullen C, Howe C, Laugesen M, et al. Electronic cigarettes for smoking cessation: a randomised controlled trial. Lancet. 2013;382(9905):1629-1637.
19. Caponnetto P, Campagna D, Cibella F, et al. EffiCiency and safety of an eLectronic cigAreTte (ECLAT) as tobacco cigarettes substitute: a prospective 12-month randomized control design study. PLoS One. 2013;8(6):e66317. doi: 10.1371/journal.pone.0066317.
20. Adriaens K, Van Gucht D, Declerck P, et al. Effectiveness of the electronic cigarette: an eight-week Flemish study with six-month follow-up on smoking reduction, craving and experienced benefits and complaints. Int J Environ Res Public Health. 2014;11(11):11220-11248.
21. Tseng TY, Ostroff JS, Campo A, et al. A randomized trial comparing the effect of nicotine versus placebo electronic cigarettes on smoking reduction among young adult smokers. Nicotine Tob Res. 2016;18(10):1937-1943.
22. Meier E, Wahlquist AE, Heckman BW, et al. A pilot randomized crossover trial of electronic cigarette sampling among smokers. Nicotine Tob Res. 2017;19(2):176-182.
23. Hartmann-Boyce J, McRobbie H, Bullen C, et al. Electronic cigarettes for smoking cessation [published online September 14, 2016]. Cochrane Database Syst Rev. 2016;9:CD010216.
24. Biener L, Hargraves JL. A longitudinal study of electronic cigarette use among a population-based sample of adult smokers: association with smoking cessation and motivation to quit. Nicotine Tob Res. 2014;17(2):127-133.
25. Etter JF, Bullen C. A longitudinal study of electronic cigarette users. Addict Behav. 2014;39(2):491-494.
26. Caponnetto P, Auditore R, Russo C, et al. Impact of an electronic cigarette on smoking reduction and cessation in schizophrenic smokers: a prospective 12-month pilot study. Int J Environ Res Public Health. 2013;10(2):446-461.
27. Popova L, Ling PM. Alternative tobacco product use and smoking cessation: a national study. Am J Public Health. 2013;103(5):923-930.
28. Adkison SE, O’Connor RJ, Bansal-Travers M, et al. Electronic nicotine delivery systems: International Tobacco Control Four-Country Survey. Am J Prev Med. 2013;44(3):207-215.
29. Brose LS, Hitchman SC, Brown J, et al. Is the use of electronic cigarettes while smoking associated with smoking cessation attempts, cessation and reduced cigarette consumption? A survey with a 1-year follow-up. Addiction. 2015;110(7):1160-1168.
30. Manzoli L, Flacco ME, Ferrante M, et al; ISLESE Working Group. Cohort study of electronic cigarette use: effectiveness and safety at 24 months [published online June 6, 2016]. Tob Control. doi: 10.1136/tobaccocontrol-2015-052822.
31. Lerner CA, Sundar IK, Yao H, et al. Vapors produced by electronic cigarettes and E-juices with flavorings induce toxicity, oxidative stress, and inflammatory response in lung epithelial cells and in mouse lung. PLoS One. 2015;10(2):e0116732. doi: 10.1371/journal.pone.0116732.
32. Sussan TE, Gajghate S, Thimmulappa RK, et al. Exposure to electronic cigarettes impairs pulmonary anti-bacterial and anti-viral defenses in a mouse model. PLoS One. 2015;10(2):e0116861. doi: 10.1371/journal.pone.0116861.
33. US Environmental Protection Agency. Acrolein. https://www.epa.gov/sites/production/files/2016-08/documents/acrolein.pdf. Updated September 2009. Accessed April 7, 2017.
34. Vardavas CI, Anagnostopoulos N, Kougias M, et al. Short-term pulmonary effects of using an electronic cigarette: impact on respiratory flow resistance, impedance, and exhaled nitric oxide. Chest. 2012;141(6):1400-1406.
35. Etter JF. Electronic cigarettes: a survey of users. BMC Public Health. 2010;10:231.
36. Goniewicz ML, Lingas EO, Hajek P. Patterns of electronic cigarette use and user beliefs about their safety and benefits: an internet survey. Drug Alcohol Rev. 2013;32(2):133-140.
37. Laino T, Tuma C, Moor P, et al. Mechanisms of propylene glycol and triacetin pyrolysis. J Phys Chem A. 2012;116(18):4602-4609.
38. Brook RD, Rajagopalan S, Pope CA 3rd, et al; American Heart Association Council on Epidemiology and Prevention; Council on the Kidney in Cardiovascular Disease; Council on Nutrition, Physical Activity and Metabolism. Particulate matter air pollution and cardiovascular disease: an update to the scientific statement from the American Heart Association. Circulation. 2010;121(21):2331-2378.
39. Barrington-Trimis JL, Samet JM, McConnell R. Flavorings in electronic cigarettes: an unrecognized respiratory health hazard? JAMA. 2014;312(23):2493-2494.
40. Schripp T, Markewitz D, Uhde E, et al. Does e-cigarette consumption cause passive vaping? Indoor Air. 2013;23(1):25-31.
41. Fuoco FC, Buonanno G, Stabile L, et al. Influential parameters on particle concentration and size distribution in the mainstream of e-cigarettes. Environ Pollut. 2014;184:523-529.
42. Schober W, Szendrei K, Matzen W, et al. Use of electronic cigarettes (e-cigarettes) impairs indoor air quality and increases FeNO levels of e-cigarette consumers. Int J Hyg Environ Health. 2014;217(6):628-637.
43. Bhatnagar A, Whitsel L, Ribisl K, et al; American Heart Association Advocacy Coordinating Committee; Council on Cardiovascular and Stroke Nursing; Council on Clinical Cardiology; Council on Quality of Care and Outcomes Research. Electronic cigarettes: a policy statement from the American Heart Association. Circulation. 2014;130(16):1418-1436.
44. E-cigarettes pose risks. SAMHSA News. https://www.samhsa.gov/samhsaNewsLetter/Volume_22_Number_3/e_cigarettes. Published 2014. Accessed April 7, 2017.
45. National Institute on Drug Abuse. Electronic cigarettes (e-cigarettes). https://www.drugabuse.gov/publications/drugfacts/electronic-cigarettes-e-cigarettes. Revised May 2016. Accessed April 7, 2017.
46. American Academy of Addiction Psychiatry. Nicotine dependence. East Providence, RI: American Academy of Addition Psychiatry; 2015.
47. Green SH, Bayer R, Fairchild AL. Evidence, policy, and e-cigarettes — will England reframe the debate. N Engl J Med. 2016;374(14):1301-1303.
48. Cahill K, Stevens S, Lancaster T. Pharmacological treatments for smoking cessation. JAMA. 2014;311(2):193-194.
The popularity of electronic cigarettes (E-cigs) and “vapes” has grown dramatically, spawning a new industry of electronic nicotine delivery systems (ENDS). With the increasing use of E-cigs not only for smoking cessation, but also as a primary nicotine source, it is important for mental health professionals to be prepared to discuss use of these devices with patients. In this article, we will describe:
- the composition of E-cigs and their current use
- evidence for their use for smoking cessation
- adverse health effects
- recommendations of major regulatory agencies.
Finally, we will provide recommendations for E-cig use in clinical populations.
What is an electronic nicotine delivery system?
ENDS produce an aerosol with or without nicotine that is inhaled and is thought to mimic the use of combustible cigarettes. ENDS evolved from basic E-cigs into a less “cigarette-like” and more customizable product (Figure 1). ENDS include a range of designs and go by various names, including “personal vaporizers,” “e-cigars,” and “e-hookahs” (in this article, we will use the term “ENDS” to refer to these devices).
The general design of ENDS is a plastic tubing system that contains a mouthpiece, battery, electronic heating element (“vaporizer”), and a cartridge with liquid solvent with or without nicotine or flavoring (Figure 2). One draw on the mouthpiece or press of a button activates the device, heats the solution, and delivers a vapor in a similar manner to taking a puff of a cigarette. Although studies have shown that ENDS result in significant increases in plasma nicotine concentrations in 5 minutes,1 the plasma nicotine levels obtained with the first-generation “cigarette-like” ENDS are much lower than those caused by inhaling tobacco smoke.2 Over time nicotine delivery capability has improved as ENDS have evolved such that the rate of nicotine delivery and peak concentration obtained with newer models more closely mirror tobacco cigarettes.3 Whether the rapid delivery of larger amounts of nicotine helps or hinders one’s efforts to break nicotine addiction remains to be determined because of the reinforcing properties of the drug.
The liquid in the E-cig cartridge typically contains not only nicotine but a number of chemical compounds with potentially deleterious or unknown health risks. The 3 main ingredients include:
- a solvent of glycerin and/or propylene glycol
- nicotine in various concentrations
- flavorings.
The glycerin or propylene glycol forms the basis for the aerosol. Nicotine concentrations vary from 0 (denicotinized) to 35 mcg per puff.4 A study reported 7,700 unique flavors available for vaping liquid.5 The liquid also contains impurities, such as anabasine, which has effects on the α-7 nicotinic acetylcholine receptor and its principal use is as an insecticide and β-nicotyrine, which inhibits cytochrome P450 2A.
Epidemiology and end-user perspectives
In 2014, 12.4% of U.S. adults classified themselves as “ever users” of ENDS (used at least once) and 3.7% of adults classified themselves as current users, according to the National Health Interview Study.6 Importantly, among E-cig users who had not used combustible cigarettes, young adults (age 18 to 24) were more likely to have tried ENDS than older adults. ENDS are becoming more popular across the globe. A study in the European Union found that ever users of ENDS most commonly were current cigarette smokers (31%) followed by former (10.8%) and never smokers (2.3%).7
ENDS use is relevant for mental health professionals because of the high rate of comorbid tobacco use disorder in individuals with psychiatric conditions. For example, 2 U.S. population surveys8,9 revealed those with mental health conditions were 1.5 to 2 times more likely to have tried ENDS and 2 to 3 times more likely to be current users. Those with psychiatric illness reported similar reasons for ENDS use as other individuals, including “just because,” use as a smoking cessation aid, ease of use, and perceived safety vs combustible cigarettes.
A recent review that included 9 studies focusing on ENDS use in those with mental illness reported mixed findings on the utility of these devices to reduce or stop use of combustible cigarettes.10 Additionally, it is important to monitor the use of cigarettes and ENDS in patients with psychiatric illness because the byproducts of tobacco smoke can affect the metabolism of some psychotropic medications.11 Although reduced use of combustible cigarettes could lead to lower dosing of some psychotropics, an unreported decrease in combustible cigarette use could lead to supratherapeutic drug levels. There are no data on the effect of ENDS on the metabolism of psychotropics.
ENDS are increasingly popular among adolescents. In 2015, there were an estimated 4.6 million current tobacco users among middle/high school youths in the United States and 3 million current ENDS users, according to the National Youth Tobacco Surveys.12 The shift from combustible cigarettes to ENDS is notable, with an increase in the percentage of current E-cig users and a decrease in the percentage of exclusive combustible cigarette users. In addition, there has been no change in the prevalence of lifetime tobacco users.12 This is a global issue, as reports of ever use of ENDS by adolescents range from 6.5% to 31% in the United States, 14.6% in Canada, and 4.7% to 38.5% in Europe.13 Based on these trends, the U.S. Surgeon General released a statement warning against the use of ENDS in youth because of the lack of safety data and strong association with use of tobacco products.14
There are a number of possible reasons for the increasing popularity of ENDS, including the product’s novelty, lack of regulations regarding their sale, availability of flavorings, and the perception that ENDS are safe alternatives to cigarettes. E-cig–using youths have described ENDS as “not at all harmful” and “not at all addictive” and believe that ENDS with flavoring are less harmful than those without.15 Although studies in adults show some users reporting that ENDS are less satisfying, they are seen as useful in decreasing craving and a safer alternative to cigarettes.16,17
Are ENDS effective for smoking cessation?
The evidence for ENDS as aids to smoking cessation remains murky (Table 118-22). There is a paucity of randomized controlled clinical trials (RCTs) investigating ENDS for smoking cessation or reduction, and it is difficult to quantify the amount of nicotine used in ENDS because of the variety of delivery systems and cartridges. In a recent Cochrane review, those using ENDS to quit smoking were more likely to be abstinent from combustible cigarettes at 6 months vs those using nicotine-free ENDS (relative risk = 2.29; 95% CI, 1.05 to 4.96), but there was no significant difference in quit rates compared with nicotine patches.23 However, the confidence in this finding was rated as low because of the limited number of RCTs. Of note, the authors found 15 ongoing RCTs at the time of publication that might be eligible for later evaluation.
Non-RCTs reveal mixed data. Positive results include 1 study with an odds ratio of 6.07 to quit for intensive ENDS users vs non-users,24 and another with dual users of combustible and electronic cigarettes having a 46% quit rate at 1 year.25 Additionally, in a pilot study providing ENDS to 14 patients with schizophrenia who had no previous desire to quit smoking, authors noted a reduction in the number of cigarettes smoked per day by 50% in one-half of participants and abstinence in 14% of participants at 52 weeks.26 Studies with neutral or negative results include those showing ENDS users to be current combustible tobacco smokers, and use of ENDS not predicting smoking cessation.4,27 Data also are mixed regarding the use of ENDS as a harm reduction strategy. One study found that ENDS decreased cigarette consumption, but did not increase the likelihood of quitting,28 while another reported that daily use of ENDS increased the odds of reducing smoking by as much as 2.5 times compared with non-use of such aids.29 In a 24-month prospective cohort study following tobacco users, there was no difference in the number of cigarettes smoked per day in those who started the trial as users of combustible cigarettes alone vs combustible cigarettes plus ENDS users.30 Interestingly, those who started the study as combustible cigarette users and switched to ENDS and those who had continued dual use throughout the 24 months smoked fewer combustible cigarettes per day than those who never tried ENDS or quit during the study period.
Health effects
To better understand the adverse health effects of ENDS, one must consider potential short- and long-term consequences (Table 2). In the short-term, ENDS have been found to increase markers of inflammation and oxidative stress acutely as evidenced by in vivo laboratory studies.31,32 ENDS also have been linked to upper respiratory irritation, in part, because of the transformation of glycerin in the nicotine cartridge to acrolein upon combustion.33 Even 5 minutes of ad lib E-cig use has been found to significantly increase airflow resistance during pulmonary function tests34—changes that have been shown to precede more persistent alterations in peak expiratory flow, such as those seen in chronic obstructive pulmonary disease. The more common patient-reported side effects include:
- daytime cough (27%)
- phlegm production (25%)
- headache (21%)
- dry mouth/throat (20%)
- vertigo, headache, or nausea (9%).35,36
A RCT investigating efficacy of E-cigs vs nicotine patches vs denicotinized E-cigs found no difference among the groups in the number of reported adverse events.18 Interestingly, another RCT found a decrease in adverse events, such as dry cough, mouth irritation, throat irritation, shortness of breath, and headache, compared with baseline in combustible cigarette smokers who used regular or denicotinized E-cigs.19
Although no studies have directly investigated long-term health consequences of ENDS because of their relative novelty, one can extrapolate potential harmful long-term effects based on knowledge of the products’ chemical constituents. For example, propylene glycol can degrade into propylene oxide, a class 2B carcinogen.37 Other potential carcinogens in the aerosol include formaldehyde and acetaldehyde. On a broader scale, many of the particulates have been shown to cause systemic inflammation, which is thought to increase cardiovascular and respiratory disease and death.38 Flavorings in ENDS include a variety of components including, but not limited to, aldehydes, which are irritants, and other additives that have been associated with respiratory disease.39
Second-hand exposure. There are no long-term studies of second-hand vapor exposure, but similar to long-term health on primary users, one can glean some observations from the literature. It is promising that compared with cigarettes, ENDS lack sidestream smoke and the vapor has not been found to contain carbon monoxide.40 Some research has demonstrated that the size and spray of fine particles in the aerosol is as large or larger than combustible cigarettes.41 Formaldehyde, acetaldehyde, isoprene, and acetic acid have been found in ENDS vapor.40 Interestingly, a simulated café study found elevated nicotine, glycerine, hydrocarbon, and other materials classified as carcinogens in the air.42
Although it is popularly thought that ENDS are less toxic than tobacco cigarettes, there is not enough evidence to estimate precisely as to how much less toxic or the consequences of use. ENDS are increasingly popular and are being used by never smokers who should be educated on the potential harm that ENDS pose.
Recommendations from agencies and medical organizations
The World Health Organization (WHO) recommended prohibiting the use of ENDS in indoor spaces to minimize potential health risks to users and non-users. The WHO also aims to prevent dissemination of unproven health claims, including claims that ENDS are effective—or not—or that the devices are innocuous.36 In the United States, the FDA has stated that ENDS are not recommended for safe quitting (2009). In August 2016, the FDA introduced regulations banning the sale of ENDS to individuals age <18 and required manufacturers to submit documents detailing all ingredients for review and possible approval.
The American Lung Association has stated its concerns about the use of ENDS but has not made any direct recommendations. The American Heart Association reports a potential negative public health impact and provides clinical guideline recommendations.43 Prominent psychiatric organizations such as the American Psychiatric Association, American Academy of Addiction Psychiatry (AAAP), the Substance Abuse and Mental Health Services Administration (SAMHSA), and the National Institute of Drug Abuse do not have official statements supporting or rejecting the use of ENDS. However, they do note the potential harm and lack of substantial evidence for efficacy of ENDS as a smoking cessation tool, and the AAAP and SAMHSA state that they will work with regulatory agencies to reduce the use of toxic products with addictive potential including ENDS.44-46
Clinical recommendations
We do not recommend ENDS as a first-line treatment for smoking cessation because there is no evidence they are superior to the FDA-approved nicotine replacement therapies (NRTs), the paucity of research into the potential short- and long-term health risks of ENDS, and the fact that these products are not regulated for use as smoking cessation aids. It is, however, advisable to discuss ENDS use with patients by:
- asking if they are using the products
- assessing whether the user also is a smoker
- advising the patient to quit.
It also is important to assess the patient’s knowledge and attitudes regarding ENDS use and provide education about the products. Some patients firmly believe that ENDS are the lesser of 2 evils, and they are decreasing the harms of smoking by using these devices. While the debate over a potential harm reduction strategy unfolds,47 we think that because of the state of the evidence it is prudent to adopt a more precautionary stance and recommend that patients work toward abstinence from nicotine in any form.
For dual tobacco/ENDS users and for patients using ENDS who want to quit smoking, we recommend treatment with an approved pharmacotherapy (ie, NRTs, bupropion, and varenicline) combined with counseling. A 2013 Cochrane Review found that all pharamacotherapy options are more effective than placebo, and combination NRT and varenicline are superior to single NRT or bupropion (Box).23,48
The popularity of electronic cigarettes (E-cigs) and “vapes” has grown dramatically, spawning a new industry of electronic nicotine delivery systems (ENDS). With the increasing use of E-cigs not only for smoking cessation, but also as a primary nicotine source, it is important for mental health professionals to be prepared to discuss use of these devices with patients. In this article, we will describe:
- the composition of E-cigs and their current use
- evidence for their use for smoking cessation
- adverse health effects
- recommendations of major regulatory agencies.
Finally, we will provide recommendations for E-cig use in clinical populations.
What is an electronic nicotine delivery system?
ENDS produce an aerosol with or without nicotine that is inhaled and is thought to mimic the use of combustible cigarettes. ENDS evolved from basic E-cigs into a less “cigarette-like” and more customizable product (Figure 1). ENDS include a range of designs and go by various names, including “personal vaporizers,” “e-cigars,” and “e-hookahs” (in this article, we will use the term “ENDS” to refer to these devices).
The general design of ENDS is a plastic tubing system that contains a mouthpiece, battery, electronic heating element (“vaporizer”), and a cartridge with liquid solvent with or without nicotine or flavoring (Figure 2). One draw on the mouthpiece or press of a button activates the device, heats the solution, and delivers a vapor in a similar manner to taking a puff of a cigarette. Although studies have shown that ENDS result in significant increases in plasma nicotine concentrations in 5 minutes,1 the plasma nicotine levels obtained with the first-generation “cigarette-like” ENDS are much lower than those caused by inhaling tobacco smoke.2 Over time nicotine delivery capability has improved as ENDS have evolved such that the rate of nicotine delivery and peak concentration obtained with newer models more closely mirror tobacco cigarettes.3 Whether the rapid delivery of larger amounts of nicotine helps or hinders one’s efforts to break nicotine addiction remains to be determined because of the reinforcing properties of the drug.
The liquid in the E-cig cartridge typically contains not only nicotine but a number of chemical compounds with potentially deleterious or unknown health risks. The 3 main ingredients include:
- a solvent of glycerin and/or propylene glycol
- nicotine in various concentrations
- flavorings.
The glycerin or propylene glycol forms the basis for the aerosol. Nicotine concentrations vary from 0 (denicotinized) to 35 mcg per puff.4 A study reported 7,700 unique flavors available for vaping liquid.5 The liquid also contains impurities, such as anabasine, which has effects on the α-7 nicotinic acetylcholine receptor and its principal use is as an insecticide and β-nicotyrine, which inhibits cytochrome P450 2A.
Epidemiology and end-user perspectives
In 2014, 12.4% of U.S. adults classified themselves as “ever users” of ENDS (used at least once) and 3.7% of adults classified themselves as current users, according to the National Health Interview Study.6 Importantly, among E-cig users who had not used combustible cigarettes, young adults (age 18 to 24) were more likely to have tried ENDS than older adults. ENDS are becoming more popular across the globe. A study in the European Union found that ever users of ENDS most commonly were current cigarette smokers (31%) followed by former (10.8%) and never smokers (2.3%).7
ENDS use is relevant for mental health professionals because of the high rate of comorbid tobacco use disorder in individuals with psychiatric conditions. For example, 2 U.S. population surveys8,9 revealed those with mental health conditions were 1.5 to 2 times more likely to have tried ENDS and 2 to 3 times more likely to be current users. Those with psychiatric illness reported similar reasons for ENDS use as other individuals, including “just because,” use as a smoking cessation aid, ease of use, and perceived safety vs combustible cigarettes.
A recent review that included 9 studies focusing on ENDS use in those with mental illness reported mixed findings on the utility of these devices to reduce or stop use of combustible cigarettes.10 Additionally, it is important to monitor the use of cigarettes and ENDS in patients with psychiatric illness because the byproducts of tobacco smoke can affect the metabolism of some psychotropic medications.11 Although reduced use of combustible cigarettes could lead to lower dosing of some psychotropics, an unreported decrease in combustible cigarette use could lead to supratherapeutic drug levels. There are no data on the effect of ENDS on the metabolism of psychotropics.
ENDS are increasingly popular among adolescents. In 2015, there were an estimated 4.6 million current tobacco users among middle/high school youths in the United States and 3 million current ENDS users, according to the National Youth Tobacco Surveys.12 The shift from combustible cigarettes to ENDS is notable, with an increase in the percentage of current E-cig users and a decrease in the percentage of exclusive combustible cigarette users. In addition, there has been no change in the prevalence of lifetime tobacco users.12 This is a global issue, as reports of ever use of ENDS by adolescents range from 6.5% to 31% in the United States, 14.6% in Canada, and 4.7% to 38.5% in Europe.13 Based on these trends, the U.S. Surgeon General released a statement warning against the use of ENDS in youth because of the lack of safety data and strong association with use of tobacco products.14
There are a number of possible reasons for the increasing popularity of ENDS, including the product’s novelty, lack of regulations regarding their sale, availability of flavorings, and the perception that ENDS are safe alternatives to cigarettes. E-cig–using youths have described ENDS as “not at all harmful” and “not at all addictive” and believe that ENDS with flavoring are less harmful than those without.15 Although studies in adults show some users reporting that ENDS are less satisfying, they are seen as useful in decreasing craving and a safer alternative to cigarettes.16,17
Are ENDS effective for smoking cessation?
The evidence for ENDS as aids to smoking cessation remains murky (Table 118-22). There is a paucity of randomized controlled clinical trials (RCTs) investigating ENDS for smoking cessation or reduction, and it is difficult to quantify the amount of nicotine used in ENDS because of the variety of delivery systems and cartridges. In a recent Cochrane review, those using ENDS to quit smoking were more likely to be abstinent from combustible cigarettes at 6 months vs those using nicotine-free ENDS (relative risk = 2.29; 95% CI, 1.05 to 4.96), but there was no significant difference in quit rates compared with nicotine patches.23 However, the confidence in this finding was rated as low because of the limited number of RCTs. Of note, the authors found 15 ongoing RCTs at the time of publication that might be eligible for later evaluation.
Non-RCTs reveal mixed data. Positive results include 1 study with an odds ratio of 6.07 to quit for intensive ENDS users vs non-users,24 and another with dual users of combustible and electronic cigarettes having a 46% quit rate at 1 year.25 Additionally, in a pilot study providing ENDS to 14 patients with schizophrenia who had no previous desire to quit smoking, authors noted a reduction in the number of cigarettes smoked per day by 50% in one-half of participants and abstinence in 14% of participants at 52 weeks.26 Studies with neutral or negative results include those showing ENDS users to be current combustible tobacco smokers, and use of ENDS not predicting smoking cessation.4,27 Data also are mixed regarding the use of ENDS as a harm reduction strategy. One study found that ENDS decreased cigarette consumption, but did not increase the likelihood of quitting,28 while another reported that daily use of ENDS increased the odds of reducing smoking by as much as 2.5 times compared with non-use of such aids.29 In a 24-month prospective cohort study following tobacco users, there was no difference in the number of cigarettes smoked per day in those who started the trial as users of combustible cigarettes alone vs combustible cigarettes plus ENDS users.30 Interestingly, those who started the study as combustible cigarette users and switched to ENDS and those who had continued dual use throughout the 24 months smoked fewer combustible cigarettes per day than those who never tried ENDS or quit during the study period.
Health effects
To better understand the adverse health effects of ENDS, one must consider potential short- and long-term consequences (Table 2). In the short-term, ENDS have been found to increase markers of inflammation and oxidative stress acutely as evidenced by in vivo laboratory studies.31,32 ENDS also have been linked to upper respiratory irritation, in part, because of the transformation of glycerin in the nicotine cartridge to acrolein upon combustion.33 Even 5 minutes of ad lib E-cig use has been found to significantly increase airflow resistance during pulmonary function tests34—changes that have been shown to precede more persistent alterations in peak expiratory flow, such as those seen in chronic obstructive pulmonary disease. The more common patient-reported side effects include:
- daytime cough (27%)
- phlegm production (25%)
- headache (21%)
- dry mouth/throat (20%)
- vertigo, headache, or nausea (9%).35,36
A RCT investigating efficacy of E-cigs vs nicotine patches vs denicotinized E-cigs found no difference among the groups in the number of reported adverse events.18 Interestingly, another RCT found a decrease in adverse events, such as dry cough, mouth irritation, throat irritation, shortness of breath, and headache, compared with baseline in combustible cigarette smokers who used regular or denicotinized E-cigs.19
Although no studies have directly investigated long-term health consequences of ENDS because of their relative novelty, one can extrapolate potential harmful long-term effects based on knowledge of the products’ chemical constituents. For example, propylene glycol can degrade into propylene oxide, a class 2B carcinogen.37 Other potential carcinogens in the aerosol include formaldehyde and acetaldehyde. On a broader scale, many of the particulates have been shown to cause systemic inflammation, which is thought to increase cardiovascular and respiratory disease and death.38 Flavorings in ENDS include a variety of components including, but not limited to, aldehydes, which are irritants, and other additives that have been associated with respiratory disease.39
Second-hand exposure. There are no long-term studies of second-hand vapor exposure, but similar to long-term health on primary users, one can glean some observations from the literature. It is promising that compared with cigarettes, ENDS lack sidestream smoke and the vapor has not been found to contain carbon monoxide.40 Some research has demonstrated that the size and spray of fine particles in the aerosol is as large or larger than combustible cigarettes.41 Formaldehyde, acetaldehyde, isoprene, and acetic acid have been found in ENDS vapor.40 Interestingly, a simulated café study found elevated nicotine, glycerine, hydrocarbon, and other materials classified as carcinogens in the air.42
Although it is popularly thought that ENDS are less toxic than tobacco cigarettes, there is not enough evidence to estimate precisely as to how much less toxic or the consequences of use. ENDS are increasingly popular and are being used by never smokers who should be educated on the potential harm that ENDS pose.
Recommendations from agencies and medical organizations
The World Health Organization (WHO) recommended prohibiting the use of ENDS in indoor spaces to minimize potential health risks to users and non-users. The WHO also aims to prevent dissemination of unproven health claims, including claims that ENDS are effective—or not—or that the devices are innocuous.36 In the United States, the FDA has stated that ENDS are not recommended for safe quitting (2009). In August 2016, the FDA introduced regulations banning the sale of ENDS to individuals age <18 and required manufacturers to submit documents detailing all ingredients for review and possible approval.
The American Lung Association has stated its concerns about the use of ENDS but has not made any direct recommendations. The American Heart Association reports a potential negative public health impact and provides clinical guideline recommendations.43 Prominent psychiatric organizations such as the American Psychiatric Association, American Academy of Addiction Psychiatry (AAAP), the Substance Abuse and Mental Health Services Administration (SAMHSA), and the National Institute of Drug Abuse do not have official statements supporting or rejecting the use of ENDS. However, they do note the potential harm and lack of substantial evidence for efficacy of ENDS as a smoking cessation tool, and the AAAP and SAMHSA state that they will work with regulatory agencies to reduce the use of toxic products with addictive potential including ENDS.44-46
Clinical recommendations
We do not recommend ENDS as a first-line treatment for smoking cessation because there is no evidence they are superior to the FDA-approved nicotine replacement therapies (NRTs), the paucity of research into the potential short- and long-term health risks of ENDS, and the fact that these products are not regulated for use as smoking cessation aids. It is, however, advisable to discuss ENDS use with patients by:
- asking if they are using the products
- assessing whether the user also is a smoker
- advising the patient to quit.
It also is important to assess the patient’s knowledge and attitudes regarding ENDS use and provide education about the products. Some patients firmly believe that ENDS are the lesser of 2 evils, and they are decreasing the harms of smoking by using these devices. While the debate over a potential harm reduction strategy unfolds,47 we think that because of the state of the evidence it is prudent to adopt a more precautionary stance and recommend that patients work toward abstinence from nicotine in any form.
For dual tobacco/ENDS users and for patients using ENDS who want to quit smoking, we recommend treatment with an approved pharmacotherapy (ie, NRTs, bupropion, and varenicline) combined with counseling. A 2013 Cochrane Review found that all pharamacotherapy options are more effective than placebo, and combination NRT and varenicline are superior to single NRT or bupropion (Box).23,48
1. Hajek P, Goniewicz ML, Phillips A, et al. Nicotine intake from electronic cigarettes on initial use and after 4 weeks of regular use. Nicotine Tob Res. 2015;17(2):175-179.
2. Farsalinos KE, Polosa R. Safety evaluation and risk assessment of electronic cigarettes as tobacco cigarette substitutes: a systematic review. Ther Adv Drug Saf. 2014;5(2):67-86.
3. St Helen G, Havel C, Dempsey DA, et al. Nicotine delivery, retention and pharmacokinetics from various electronic cigarettes. Addiction. 2016;111(3):535-544.
4. Grana R, Benowitz N, Glantz SA. E-cigarettes: a scientific review. Circulation. 2014;129(19):1972-1986.
5. Zhu SH, Sun JY, Bonnevie E, et al. Four hundred and sixty brands of e-cigarettes and counting: implications for product regulation. Tob Control. 2014;23(suppl 3):iii3-iii9. doi: 10.1136/tobaccocontrol-2014-051670.
6. Schoenborn CA, Gindi RM. Electronic cigarette use among adults: United States, 2014. NCHS Data Brief. 2015;(217):1-8.
7. Farsalinos KE, Poulas K, Voudris V, et al. Electronic cigarette use in the European Union: analysis of a representative sample of 27 460 Europeans from 28 countries. Addiction. 2016;111(11):2032-2040.
8. Cummins SE, Zhu SH, Tedeschi GJ, et al. Use of e-cigarettes by individuals with mental health conditions. Tob Control. 2015;23(suppl 3):iii48-iii53. doi: 10.1136/tobaccocontrol-2013-051511.
9. Spears CA, Jones DM, Weaver SR, et al. Use of electronic nicotine delivery systems among adults with mental health conditions, 2015. Int J Environ Res Public Heal. 2017;14(1):10.
10. Hefner K, Valentine G, Sofuoglu M. Electronic cigarettes and mental illness: reviewing the evidence for help and harm among those with psychiatric and substance use disorders [published online February 2, 2017]. Am J Addict. doi: 10.1111/ajad.12504.
11. Anthenelli R. How—and why—to help psychiatric patients stop smoking. Current Psychiatry. 2005;4(1):77-87.
12. Singh T, Arrazola RA, Corey CG, et al. Tobacco use among middle and high school students—United States, 2011-2015. MMWR Morb Mortal Wkly Rep. 2016;65(14):361-367.
13. Greenhill R, Dawkins L, Notley C, et al. Adolescent awareness and use of electronic cigarettes: a review of emerging trends and findings. J Adolesc Heal. 2016;59(6):612-619.
14. U.S. Department of Health and Human Services. E-cigarette use among youth and young adults: a report of the Surgeon General. Atlanta, GA: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention; 2016.
15. Cooper M, Harrell MB, Pérez A, et al. Flavorings and perceived harm and addictiveness of e-cigarettes among youth. Tob Regul Sci. 2016;2(3):278-289.
16. Kim H, Davis AH, Dohack JL, et al. E-cigarettes use behavior and experience of adults: qualitative research findings to inform e-cigarette use measure development. Nicotine Tob Res. 2017;19(2):190-196.
17. Czoli CD, Fong GT, Mays D, et al. How do consumers perceive differences in risk across nicotine products? A review of relative risk perceptions across smokeless tobacco, e-cigarettes, nicotine replacement therapy and combustible cigarettes. Tob Control. 2017;26(e1):e49-e58.
18. Bullen C, Howe C, Laugesen M, et al. Electronic cigarettes for smoking cessation: a randomised controlled trial. Lancet. 2013;382(9905):1629-1637.
19. Caponnetto P, Campagna D, Cibella F, et al. EffiCiency and safety of an eLectronic cigAreTte (ECLAT) as tobacco cigarettes substitute: a prospective 12-month randomized control design study. PLoS One. 2013;8(6):e66317. doi: 10.1371/journal.pone.0066317.
20. Adriaens K, Van Gucht D, Declerck P, et al. Effectiveness of the electronic cigarette: an eight-week Flemish study with six-month follow-up on smoking reduction, craving and experienced benefits and complaints. Int J Environ Res Public Health. 2014;11(11):11220-11248.
21. Tseng TY, Ostroff JS, Campo A, et al. A randomized trial comparing the effect of nicotine versus placebo electronic cigarettes on smoking reduction among young adult smokers. Nicotine Tob Res. 2016;18(10):1937-1943.
22. Meier E, Wahlquist AE, Heckman BW, et al. A pilot randomized crossover trial of electronic cigarette sampling among smokers. Nicotine Tob Res. 2017;19(2):176-182.
23. Hartmann-Boyce J, McRobbie H, Bullen C, et al. Electronic cigarettes for smoking cessation [published online September 14, 2016]. Cochrane Database Syst Rev. 2016;9:CD010216.
24. Biener L, Hargraves JL. A longitudinal study of electronic cigarette use among a population-based sample of adult smokers: association with smoking cessation and motivation to quit. Nicotine Tob Res. 2014;17(2):127-133.
25. Etter JF, Bullen C. A longitudinal study of electronic cigarette users. Addict Behav. 2014;39(2):491-494.
26. Caponnetto P, Auditore R, Russo C, et al. Impact of an electronic cigarette on smoking reduction and cessation in schizophrenic smokers: a prospective 12-month pilot study. Int J Environ Res Public Health. 2013;10(2):446-461.
27. Popova L, Ling PM. Alternative tobacco product use and smoking cessation: a national study. Am J Public Health. 2013;103(5):923-930.
28. Adkison SE, O’Connor RJ, Bansal-Travers M, et al. Electronic nicotine delivery systems: International Tobacco Control Four-Country Survey. Am J Prev Med. 2013;44(3):207-215.
29. Brose LS, Hitchman SC, Brown J, et al. Is the use of electronic cigarettes while smoking associated with smoking cessation attempts, cessation and reduced cigarette consumption? A survey with a 1-year follow-up. Addiction. 2015;110(7):1160-1168.
30. Manzoli L, Flacco ME, Ferrante M, et al; ISLESE Working Group. Cohort study of electronic cigarette use: effectiveness and safety at 24 months [published online June 6, 2016]. Tob Control. doi: 10.1136/tobaccocontrol-2015-052822.
31. Lerner CA, Sundar IK, Yao H, et al. Vapors produced by electronic cigarettes and E-juices with flavorings induce toxicity, oxidative stress, and inflammatory response in lung epithelial cells and in mouse lung. PLoS One. 2015;10(2):e0116732. doi: 10.1371/journal.pone.0116732.
32. Sussan TE, Gajghate S, Thimmulappa RK, et al. Exposure to electronic cigarettes impairs pulmonary anti-bacterial and anti-viral defenses in a mouse model. PLoS One. 2015;10(2):e0116861. doi: 10.1371/journal.pone.0116861.
33. US Environmental Protection Agency. Acrolein. https://www.epa.gov/sites/production/files/2016-08/documents/acrolein.pdf. Updated September 2009. Accessed April 7, 2017.
34. Vardavas CI, Anagnostopoulos N, Kougias M, et al. Short-term pulmonary effects of using an electronic cigarette: impact on respiratory flow resistance, impedance, and exhaled nitric oxide. Chest. 2012;141(6):1400-1406.
35. Etter JF. Electronic cigarettes: a survey of users. BMC Public Health. 2010;10:231.
36. Goniewicz ML, Lingas EO, Hajek P. Patterns of electronic cigarette use and user beliefs about their safety and benefits: an internet survey. Drug Alcohol Rev. 2013;32(2):133-140.
37. Laino T, Tuma C, Moor P, et al. Mechanisms of propylene glycol and triacetin pyrolysis. J Phys Chem A. 2012;116(18):4602-4609.
38. Brook RD, Rajagopalan S, Pope CA 3rd, et al; American Heart Association Council on Epidemiology and Prevention; Council on the Kidney in Cardiovascular Disease; Council on Nutrition, Physical Activity and Metabolism. Particulate matter air pollution and cardiovascular disease: an update to the scientific statement from the American Heart Association. Circulation. 2010;121(21):2331-2378.
39. Barrington-Trimis JL, Samet JM, McConnell R. Flavorings in electronic cigarettes: an unrecognized respiratory health hazard? JAMA. 2014;312(23):2493-2494.
40. Schripp T, Markewitz D, Uhde E, et al. Does e-cigarette consumption cause passive vaping? Indoor Air. 2013;23(1):25-31.
41. Fuoco FC, Buonanno G, Stabile L, et al. Influential parameters on particle concentration and size distribution in the mainstream of e-cigarettes. Environ Pollut. 2014;184:523-529.
42. Schober W, Szendrei K, Matzen W, et al. Use of electronic cigarettes (e-cigarettes) impairs indoor air quality and increases FeNO levels of e-cigarette consumers. Int J Hyg Environ Health. 2014;217(6):628-637.
43. Bhatnagar A, Whitsel L, Ribisl K, et al; American Heart Association Advocacy Coordinating Committee; Council on Cardiovascular and Stroke Nursing; Council on Clinical Cardiology; Council on Quality of Care and Outcomes Research. Electronic cigarettes: a policy statement from the American Heart Association. Circulation. 2014;130(16):1418-1436.
44. E-cigarettes pose risks. SAMHSA News. https://www.samhsa.gov/samhsaNewsLetter/Volume_22_Number_3/e_cigarettes. Published 2014. Accessed April 7, 2017.
45. National Institute on Drug Abuse. Electronic cigarettes (e-cigarettes). https://www.drugabuse.gov/publications/drugfacts/electronic-cigarettes-e-cigarettes. Revised May 2016. Accessed April 7, 2017.
46. American Academy of Addiction Psychiatry. Nicotine dependence. East Providence, RI: American Academy of Addition Psychiatry; 2015.
47. Green SH, Bayer R, Fairchild AL. Evidence, policy, and e-cigarettes — will England reframe the debate. N Engl J Med. 2016;374(14):1301-1303.
48. Cahill K, Stevens S, Lancaster T. Pharmacological treatments for smoking cessation. JAMA. 2014;311(2):193-194.
1. Hajek P, Goniewicz ML, Phillips A, et al. Nicotine intake from electronic cigarettes on initial use and after 4 weeks of regular use. Nicotine Tob Res. 2015;17(2):175-179.
2. Farsalinos KE, Polosa R. Safety evaluation and risk assessment of electronic cigarettes as tobacco cigarette substitutes: a systematic review. Ther Adv Drug Saf. 2014;5(2):67-86.
3. St Helen G, Havel C, Dempsey DA, et al. Nicotine delivery, retention and pharmacokinetics from various electronic cigarettes. Addiction. 2016;111(3):535-544.
4. Grana R, Benowitz N, Glantz SA. E-cigarettes: a scientific review. Circulation. 2014;129(19):1972-1986.
5. Zhu SH, Sun JY, Bonnevie E, et al. Four hundred and sixty brands of e-cigarettes and counting: implications for product regulation. Tob Control. 2014;23(suppl 3):iii3-iii9. doi: 10.1136/tobaccocontrol-2014-051670.
6. Schoenborn CA, Gindi RM. Electronic cigarette use among adults: United States, 2014. NCHS Data Brief. 2015;(217):1-8.
7. Farsalinos KE, Poulas K, Voudris V, et al. Electronic cigarette use in the European Union: analysis of a representative sample of 27 460 Europeans from 28 countries. Addiction. 2016;111(11):2032-2040.
8. Cummins SE, Zhu SH, Tedeschi GJ, et al. Use of e-cigarettes by individuals with mental health conditions. Tob Control. 2015;23(suppl 3):iii48-iii53. doi: 10.1136/tobaccocontrol-2013-051511.
9. Spears CA, Jones DM, Weaver SR, et al. Use of electronic nicotine delivery systems among adults with mental health conditions, 2015. Int J Environ Res Public Heal. 2017;14(1):10.
10. Hefner K, Valentine G, Sofuoglu M. Electronic cigarettes and mental illness: reviewing the evidence for help and harm among those with psychiatric and substance use disorders [published online February 2, 2017]. Am J Addict. doi: 10.1111/ajad.12504.
11. Anthenelli R. How—and why—to help psychiatric patients stop smoking. Current Psychiatry. 2005;4(1):77-87.
12. Singh T, Arrazola RA, Corey CG, et al. Tobacco use among middle and high school students—United States, 2011-2015. MMWR Morb Mortal Wkly Rep. 2016;65(14):361-367.
13. Greenhill R, Dawkins L, Notley C, et al. Adolescent awareness and use of electronic cigarettes: a review of emerging trends and findings. J Adolesc Heal. 2016;59(6):612-619.
14. U.S. Department of Health and Human Services. E-cigarette use among youth and young adults: a report of the Surgeon General. Atlanta, GA: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention; 2016.
15. Cooper M, Harrell MB, Pérez A, et al. Flavorings and perceived harm and addictiveness of e-cigarettes among youth. Tob Regul Sci. 2016;2(3):278-289.
16. Kim H, Davis AH, Dohack JL, et al. E-cigarettes use behavior and experience of adults: qualitative research findings to inform e-cigarette use measure development. Nicotine Tob Res. 2017;19(2):190-196.
17. Czoli CD, Fong GT, Mays D, et al. How do consumers perceive differences in risk across nicotine products? A review of relative risk perceptions across smokeless tobacco, e-cigarettes, nicotine replacement therapy and combustible cigarettes. Tob Control. 2017;26(e1):e49-e58.
18. Bullen C, Howe C, Laugesen M, et al. Electronic cigarettes for smoking cessation: a randomised controlled trial. Lancet. 2013;382(9905):1629-1637.
19. Caponnetto P, Campagna D, Cibella F, et al. EffiCiency and safety of an eLectronic cigAreTte (ECLAT) as tobacco cigarettes substitute: a prospective 12-month randomized control design study. PLoS One. 2013;8(6):e66317. doi: 10.1371/journal.pone.0066317.
20. Adriaens K, Van Gucht D, Declerck P, et al. Effectiveness of the electronic cigarette: an eight-week Flemish study with six-month follow-up on smoking reduction, craving and experienced benefits and complaints. Int J Environ Res Public Health. 2014;11(11):11220-11248.
21. Tseng TY, Ostroff JS, Campo A, et al. A randomized trial comparing the effect of nicotine versus placebo electronic cigarettes on smoking reduction among young adult smokers. Nicotine Tob Res. 2016;18(10):1937-1943.
22. Meier E, Wahlquist AE, Heckman BW, et al. A pilot randomized crossover trial of electronic cigarette sampling among smokers. Nicotine Tob Res. 2017;19(2):176-182.
23. Hartmann-Boyce J, McRobbie H, Bullen C, et al. Electronic cigarettes for smoking cessation [published online September 14, 2016]. Cochrane Database Syst Rev. 2016;9:CD010216.
24. Biener L, Hargraves JL. A longitudinal study of electronic cigarette use among a population-based sample of adult smokers: association with smoking cessation and motivation to quit. Nicotine Tob Res. 2014;17(2):127-133.
25. Etter JF, Bullen C. A longitudinal study of electronic cigarette users. Addict Behav. 2014;39(2):491-494.
26. Caponnetto P, Auditore R, Russo C, et al. Impact of an electronic cigarette on smoking reduction and cessation in schizophrenic smokers: a prospective 12-month pilot study. Int J Environ Res Public Health. 2013;10(2):446-461.
27. Popova L, Ling PM. Alternative tobacco product use and smoking cessation: a national study. Am J Public Health. 2013;103(5):923-930.
28. Adkison SE, O’Connor RJ, Bansal-Travers M, et al. Electronic nicotine delivery systems: International Tobacco Control Four-Country Survey. Am J Prev Med. 2013;44(3):207-215.
29. Brose LS, Hitchman SC, Brown J, et al. Is the use of electronic cigarettes while smoking associated with smoking cessation attempts, cessation and reduced cigarette consumption? A survey with a 1-year follow-up. Addiction. 2015;110(7):1160-1168.
30. Manzoli L, Flacco ME, Ferrante M, et al; ISLESE Working Group. Cohort study of electronic cigarette use: effectiveness and safety at 24 months [published online June 6, 2016]. Tob Control. doi: 10.1136/tobaccocontrol-2015-052822.
31. Lerner CA, Sundar IK, Yao H, et al. Vapors produced by electronic cigarettes and E-juices with flavorings induce toxicity, oxidative stress, and inflammatory response in lung epithelial cells and in mouse lung. PLoS One. 2015;10(2):e0116732. doi: 10.1371/journal.pone.0116732.
32. Sussan TE, Gajghate S, Thimmulappa RK, et al. Exposure to electronic cigarettes impairs pulmonary anti-bacterial and anti-viral defenses in a mouse model. PLoS One. 2015;10(2):e0116861. doi: 10.1371/journal.pone.0116861.
33. US Environmental Protection Agency. Acrolein. https://www.epa.gov/sites/production/files/2016-08/documents/acrolein.pdf. Updated September 2009. Accessed April 7, 2017.
34. Vardavas CI, Anagnostopoulos N, Kougias M, et al. Short-term pulmonary effects of using an electronic cigarette: impact on respiratory flow resistance, impedance, and exhaled nitric oxide. Chest. 2012;141(6):1400-1406.
35. Etter JF. Electronic cigarettes: a survey of users. BMC Public Health. 2010;10:231.
36. Goniewicz ML, Lingas EO, Hajek P. Patterns of electronic cigarette use and user beliefs about their safety and benefits: an internet survey. Drug Alcohol Rev. 2013;32(2):133-140.
37. Laino T, Tuma C, Moor P, et al. Mechanisms of propylene glycol and triacetin pyrolysis. J Phys Chem A. 2012;116(18):4602-4609.
38. Brook RD, Rajagopalan S, Pope CA 3rd, et al; American Heart Association Council on Epidemiology and Prevention; Council on the Kidney in Cardiovascular Disease; Council on Nutrition, Physical Activity and Metabolism. Particulate matter air pollution and cardiovascular disease: an update to the scientific statement from the American Heart Association. Circulation. 2010;121(21):2331-2378.
39. Barrington-Trimis JL, Samet JM, McConnell R. Flavorings in electronic cigarettes: an unrecognized respiratory health hazard? JAMA. 2014;312(23):2493-2494.
40. Schripp T, Markewitz D, Uhde E, et al. Does e-cigarette consumption cause passive vaping? Indoor Air. 2013;23(1):25-31.
41. Fuoco FC, Buonanno G, Stabile L, et al. Influential parameters on particle concentration and size distribution in the mainstream of e-cigarettes. Environ Pollut. 2014;184:523-529.
42. Schober W, Szendrei K, Matzen W, et al. Use of electronic cigarettes (e-cigarettes) impairs indoor air quality and increases FeNO levels of e-cigarette consumers. Int J Hyg Environ Health. 2014;217(6):628-637.
43. Bhatnagar A, Whitsel L, Ribisl K, et al; American Heart Association Advocacy Coordinating Committee; Council on Cardiovascular and Stroke Nursing; Council on Clinical Cardiology; Council on Quality of Care and Outcomes Research. Electronic cigarettes: a policy statement from the American Heart Association. Circulation. 2014;130(16):1418-1436.
44. E-cigarettes pose risks. SAMHSA News. https://www.samhsa.gov/samhsaNewsLetter/Volume_22_Number_3/e_cigarettes. Published 2014. Accessed April 7, 2017.
45. National Institute on Drug Abuse. Electronic cigarettes (e-cigarettes). https://www.drugabuse.gov/publications/drugfacts/electronic-cigarettes-e-cigarettes. Revised May 2016. Accessed April 7, 2017.
46. American Academy of Addiction Psychiatry. Nicotine dependence. East Providence, RI: American Academy of Addition Psychiatry; 2015.
47. Green SH, Bayer R, Fairchild AL. Evidence, policy, and e-cigarettes — will England reframe the debate. N Engl J Med. 2016;374(14):1301-1303.
48. Cahill K, Stevens S, Lancaster T. Pharmacological treatments for smoking cessation. JAMA. 2014;311(2):193-194.

ID ‘boot camp’ emphasizes practice pearls
An intensive, day-long precourse on infectious diseases will return to the SHM annual meeting program for the first time in several years.
“Bugs, Drugs, and You: Infectious Diseases ‘Boot Camp’ for Hospitalists” will focus on a range of important clinical issues, including infectious diseases that appear with frequency and the high-stakes nature of which require correct diagnosis and treatment.

Cochair Jennifer Hanrahan, DO, also of MetroHealth Medical Center, agreed, noting that hospitalists must be able to recognize infectious disease emergencies and have some familiarity with HIV as it presents in hospitalized patients.
“We want [attendees] to think about which tests to order and when to stop antibiotic therapy,” she said.
The faculty includes infectious diseases specialists with extensive teaching backgrounds. Several are practicing hospitalists. John Sanders, MD, MPH, of Wake Forest University, Winston-Salem, N.C., and Glenn Wortmann, MD, of MedStar Washington Hospital Center will join Dr. Hanrahan and Dr. Pile as faculty members.

The precourse has five objectives:
- Describe best practices in skin and soft tissue infection treatment based on current guidelines and recently approved antimicrobial agents.
- Outline optimal strategies for prevention and treatment of Clostridium difficile, including disease recurrences.
- Identify key strategies for prevention of selected infections and discuss resistance issues impacting antimicrobial use.
- Focus on prompt recognition and treatment of certain infectious disease emergencies.
- Cover significant trends in endocarditis and other endovascular infections and their impact on treatment approaches.
When the infectious disease precourse was last presented, it received highly positive evaluations. “We believe that this new, improved iteration will be better than ever,” Dr. Pile said. “It will be well worth attendees’ time and money, providing them with renewed confidence in their ability to manage common and less common infectious diseases. They will be able to return to their practices armed with new strategies that they can immediately put to use.”
Bugs, Drugs, and You: Infectious Diseases “Boot Camp” for Hospitalists
Monday, May 1, 8:15 a.m.–4:30 p.m.
An intensive, day-long precourse on infectious diseases will return to the SHM annual meeting program for the first time in several years.
“Bugs, Drugs, and You: Infectious Diseases ‘Boot Camp’ for Hospitalists” will focus on a range of important clinical issues, including infectious diseases that appear with frequency and the high-stakes nature of which require correct diagnosis and treatment.
Cochair Jennifer Hanrahan, DO, also of MetroHealth Medical Center, agreed, noting that hospitalists must be able to recognize infectious disease emergencies and have some familiarity with HIV as it presents in hospitalized patients.
“We want [attendees] to think about which tests to order and when to stop antibiotic therapy,” she said.
The faculty includes infectious diseases specialists with extensive teaching backgrounds. Several are practicing hospitalists. John Sanders, MD, MPH, of Wake Forest University, Winston-Salem, N.C., and Glenn Wortmann, MD, of MedStar Washington Hospital Center will join Dr. Hanrahan and Dr. Pile as faculty members.
The precourse has five objectives:
- Describe best practices in skin and soft tissue infection treatment based on current guidelines and recently approved antimicrobial agents.
- Outline optimal strategies for prevention and treatment of Clostridium difficile, including disease recurrences.
- Identify key strategies for prevention of selected infections and discuss resistance issues impacting antimicrobial use.
- Focus on prompt recognition and treatment of certain infectious disease emergencies.
- Cover significant trends in endocarditis and other endovascular infections and their impact on treatment approaches.
When the infectious disease precourse was last presented, it received highly positive evaluations. “We believe that this new, improved iteration will be better than ever,” Dr. Pile said. “It will be well worth attendees’ time and money, providing them with renewed confidence in their ability to manage common and less common infectious diseases. They will be able to return to their practices armed with new strategies that they can immediately put to use.”
Bugs, Drugs, and You: Infectious Diseases “Boot Camp” for Hospitalists
Monday, May 1, 8:15 a.m.–4:30 p.m.
An intensive, day-long precourse on infectious diseases will return to the SHM annual meeting program for the first time in several years.
“Bugs, Drugs, and You: Infectious Diseases ‘Boot Camp’ for Hospitalists” will focus on a range of important clinical issues, including infectious diseases that appear with frequency and the high-stakes nature of which require correct diagnosis and treatment.
Cochair Jennifer Hanrahan, DO, also of MetroHealth Medical Center, agreed, noting that hospitalists must be able to recognize infectious disease emergencies and have some familiarity with HIV as it presents in hospitalized patients.
“We want [attendees] to think about which tests to order and when to stop antibiotic therapy,” she said.
The faculty includes infectious diseases specialists with extensive teaching backgrounds. Several are practicing hospitalists. John Sanders, MD, MPH, of Wake Forest University, Winston-Salem, N.C., and Glenn Wortmann, MD, of MedStar Washington Hospital Center will join Dr. Hanrahan and Dr. Pile as faculty members.
The precourse has five objectives:
- Describe best practices in skin and soft tissue infection treatment based on current guidelines and recently approved antimicrobial agents.
- Outline optimal strategies for prevention and treatment of Clostridium difficile, including disease recurrences.
- Identify key strategies for prevention of selected infections and discuss resistance issues impacting antimicrobial use.
- Focus on prompt recognition and treatment of certain infectious disease emergencies.
- Cover significant trends in endocarditis and other endovascular infections and their impact on treatment approaches.
When the infectious disease precourse was last presented, it received highly positive evaluations. “We believe that this new, improved iteration will be better than ever,” Dr. Pile said. “It will be well worth attendees’ time and money, providing them with renewed confidence in their ability to manage common and less common infectious diseases. They will be able to return to their practices armed with new strategies that they can immediately put to use.”
Bugs, Drugs, and You: Infectious Diseases “Boot Camp” for Hospitalists
Monday, May 1, 8:15 a.m.–4:30 p.m.
Use and misuse of opioid agonists in opioid addiction
For a patient struggling with opioid addiction, opioid agonist therapy with methadone or buprenorphine can reduce craving and opioid use and may even save his or her life. But many clinicians are unfamiliar with this evidence-based treatment,1,2 which is best started early in the course of addiction.3
This article outlines the pharmacology of these drugs, their clinical uses, and the challenges of using them to treat opioid addiction.
DIAGNOSTIC CRITERIA
Opioid addiction, formally known as opioid use disorder, is a pattern of opioid misuse leading to clinically significant impairment in multiple areas of life. The Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition, lists 11 diagnostic criteria, but only 2 need to be present within the past year to make the diagnosis4:
- Taking opioids longer or in higher doses than was intended
- A persistent desire or unsuccessful efforts to cut down or control opioid use
- Spending a great deal of time obtaining, using, or recovering from using opioids
- Craving opioids
- Repeatedly failing to fulfill obligations at work, school, or home due to opioid use
- Continuing to use opioids even though it causes or exacerbates social or interpersonal problems
- Giving up or curtailing important social, occupational, or recreational activities because of opioid use
- Repeatedly using opioids in situations in which it is physically hazardous
- Continuing to use opioids despite knowledge of having a persistent or recurrent physical or psychological problem that is likely to have been caused or exacerbated by the substance
- Tolerance
- Withdrawal.
Recent estimates indicate that 2.23 million people in the United States have opioid use disorder (426,000 with heroin and 1.8 million with prescription opioids).5
Progression from prescription opioids to heroin
We have observed that many patients with opioid use disorder start by misusing prescription opioids. Over time, tolerance can develop, which drives patients to use higher and higher doses.6
As the addiction progresses, a subset of prescription opioid users advances to using heroin, which is typically less expensive and easier to obtain.7 Most patients start with the intranasal route but eventually inject it intravenously.6,7
For many addicts, heroin use has medical consequences such as hepatitis C virus (HCV) and human immunodeficiency virus (HIV) infection, psychiatric problems such as depression and anxiety, and illegal activities such as theft and sex work.8 People who use heroin appear to have more severe addiction and a lower socioeconomic status than prescription opioid users.9–11 But recently, a growing number of middle class individuals are becoming addicted to heroin.12
METHADONE
Methadone is a long-acting synthetic opioid that functions as a full agonist on the mu-opioid receptor. The drug binds, occupies, and stimulates the receptor, preventing withdrawal symptoms and reducing opioid cravings for at least 24 hours.13
Adverse effects of methadone
The most common adverse effects include lightheadedness, dizziness, sedation, nausea, vomiting, and sweating.14 Other adverse effects:
Unintentional overdose. The risk is serious, as a single 30-mg dose can be fatal in people who are opioid-naïve.13
QTc prolongation, which can lead to torsade de pointes. This risk, which is dose-related, must be taken into consideration in patients who have any cardiac symptoms (eg, syncope, arrhythmia), pathology (familial QT prolongation), or other risk factors for QTc prolongation (eg, hypokalemia, QTc-prolonging medications).15
Respiratory depression, which can be fatal. This dose-related risk is heightened during the first 4 weeks of treatment if titration is too rapid or if methadone is used in combination with other drugs that cause central nervous system or respiratory depression.13,14
Starting methadone
To prevent respiratory depression and death related to rapid induction, the general rule is to start methadone at a low daily dose (20–30 mg) depending on the patient’s withdrawal symptoms.14 During this period, patients need to be closely monitored and educated on the perils of concomitant use of central nervous system depressants.14
In most patients, the dose is titrated up until their withdrawal symptoms and cravings are eliminated, which generally requires 60 to 120 mg daily.14 Hepatic and renal impairment, pregnancy, and advanced age can alter methadone pharmacokinetics and may therefore necessitate dose adjustment.
BUPRENORPHINE
Buprenorphine is an alkaloid thebaine opioid derivative that acts as a partial mu-opioid agonist and a kappa antagonist.16 Like methadone, buprenorphine is used to manage cravings and withdrawal symptoms.16 Dosages of 4 to 16 mg (up to 32 mg) per day of buprenorphine are usually required to adequately control opioid cravings.16
Sublingual and subdermal products
Buprenorphine is currently available in the United States in sublingual and subdermal formulations.16,17
Sublingual buprenorphine is usually combined with naloxone in a 4:1 ratio to deter intravenous use. Intravenous injection of the combination product can precipitate withdrawal due to the antagonist action of naloxone. (Taken orally or sublingually, naloxone is poorly absorbed and has little or no clinical effect.) Buprenorphine-naloxone is available in tablets, a sublingual film strip, and a buccal film strip. Buprenorphine is also available by itself in a sublingual formulation.
The US Food and Drug Administration has approved a buprenorphine subdermal implant, Probuphine. Four rods, about 1 inch long, are placed under the skin in the inner aspect of the upper arm and provide the equivalent of 8 mg of buprenorphine daily for 6 months.17 However, this method is formulated only for maintenance treatment and cannot be used for induction. Additionally, it is recommended that the implants be surgically removed at the end of 6 months, after which another set of implants can be inserted in the other arm or the patient can switch to sublingual therapy, depending on the clinical situation and patient preference.17
Generally safer than methadone
Buprenorphine works on the same receptor as methadone and therefore has a similar side effect profile. However, buprenorphine has a ceiling effect, which greatly reduces the risk of fatal respiratory depression.18 It also does not cause clinically significant QTc prolongation and is preferable in patients who have cardiac risk factors.18
Another advantage is that buprenorphine has fewer identified medication interactions than methadone.18 Further, induction of buprenorphine in patients with opioid use disorder has been shown to be safer than methadone.19
Although buprenorphine has been found to be 6 times safer than methadone with regard to overdose among the general population,20 it can still cause fatal intoxication if used in combination with central nervous system depressants.21
Buprenorphine has been also associated with hepatotoxicity, though the risk of new-onset liver disease appears to be low.22
NALTREXONE IS LESS EFFECTIVE THAN METHADONE, BUPRENORPHINE
Besides methadone and buprenorphine, the only other approved option for treating opioid use disorder is the opioid antagonist naltrexone.
Naltrexone has significantly less abuse potential, as it provides no euphoria, but patients do not like it. Even with the long-acting formulation (Vivitrol), naltrexone treatment is significantly less effective than methadone or buprenorphine.23–25 Further, although naltrexone is not a controlled substance and so does not face the same scrutiny as the agonist therapies, there are other significant barriers. Additional information on naltrexone is presented in reviews by Modesto-Lowe and Van Kirk24 and Woody.25
OBSTACLES TO TREATMENT
People hold conflicting views about opioid agonist therapy. Some believe that “trading one drug for another” is not a legitimate therapeutic strategy, and they may feel ashamed of being on maintenance therapy.26 Similarly, some argue that the answer to establishing stable abstinence does not lie simply in prescribing medications.
The contrary argument is that these medications, if used appropriately, confer many benefits such as reducing the medical and psychosocial sequelae of opioid addiction.18 In fact, properly treated patients no longer meet the diagnostic criteria of opioid use disorder, and both methadone and buprenorphine are on the World Health Organization’s (WHO) list of essential medicines.27
Despite endorsement by the WHO, the stigma attached to the opioid agonists has been difficult to overcome. Patients with opioid use disorder may be viewed with distrust by healthcare providers and often do not feel welcome in healthcare settings or in self-help recovery groups.28
Barriers to methadone therapy
Federal regulations on methadone prescribing and use were established to promote patient safety and decrease diversion, but they may also complicate access to care.29 They stipulate that to qualify for methadone maintenance, patients need to demonstrate opioid addiction for 1 year, except for pregnant women and those who have been incarcerated in the past 6 months. Patients under the age of 18 must have 2 documented failed treatment episodes as well as approval by a guardian to receive treatment.
Inconvenience. Methadone can be prescribed for opioid dependence only by an accredited treatment program. Patients must therefore travel to the clinic and wait to be evaluated on a daily basis for a minimum of 90 days. Only after they demonstrate consistent responsible behavior and negative results on urine testing do they become eligible to take methadone home.29 If a patient is to travel out of the area during the initial 90 days of treatment, he or she must make arrangements in advance to find a clinic that will provide a “guest dose.”
The inconvenience arising from the regulations may deter some patients from seeking methadone therapy. In spite of this, once patients are started on methadone, more of them continue treatment than with buprenorphine.18 A proposed reason is that methadone is a potent full opioid agonist and therefore relieves withdrawal symptoms and craving more effectively than buprenorphine, which is a partial agonist.30 Another possible reason is the higher level of supervision afforded by methadone clinics, which require daily contact for at least 90 days.
Safety concerns arise from methadone diversion, as illicit use may have lethal consequences. In the past decade, deaths from methadone overdose have risen significantly, most of them due to respiratory depression or torsade de pointes.13 However, most cases of diversion and overdose involve methadone that is prescribed for pain by individual practitioners and not from maintenance programs.13
Advantages of buprenorphine
Together, methadone’s lethality, stigma, and inconvenience may contribute to patients preferring buprenorphine.31
The regulations governing buprenorphine’s use are less restrictive than those with methadone. For example, patients must have a diagnosis of opioid addiction to be prescribed buprenorphine, but they are not required to carry the diagnosis for a year before treatment.31 Additionally, they do not need to travel to a federally approved opioid treatment center daily and can receive buprenorphine directly from a physician in an outpatient setting.
Under the Drug Abuse Treatment Act (DATA) of 2000, any physician can apply for a waiver to prescribe and dispense buprenorphine in his or her office. To qualify for an initial waiver, physicians must either obtain certification in the fields of addiction medicine or addiction psychiatry or complete an approved 8-hour training session.32 Each physician starts with a maximum of 30 patients, but can apply to treat up to 100 patients after 1 year and eventually up to 275 patients. Physicians must document every buprenorphine prescription they write and be able to refer patients for counseling.31
As of February 2017, nurse practitioners and physician assistants can also apply for a DATA 2000 waiver. All waivered providers are subject to unannounced visits from the Drug Enforcement Administration once every 5 years.32
While there are no federal restrictions on the amount of buprenorphine that can be dispensed, some states and some insurance companies have placed restrictions on dose or length of treatment.33 Buprenorphine patients can fill their prescriptions at any pharmacy and are permitted to bring their medication home, which improves access to care. However, office-based outpatient treatment is not without risk, and preventing buprenorphine diversion remains a challenge.34
‘Lending’ buprenorphine is a felony
Addicts have illegally used buprenorphine to self-treat opioid withdrawal, craving, and dependence.35 Its misuse has also been coupled with self-treatment of conditions that include depression and pain.36
A survey found that 83.7% of patients deem buprenorphine diversion to be appropriate; further, most patients said they consider it unethical to withhold prescribed buprenorphine from individuals showing symptoms of withdrawal.34 Physicians who prescribe buprenorphine must inform their patients that even “lending” or giving away their medication is a felony.
Prescribing physicians must also be diligent about monitoring for signs of diversion such as inconsistent urine toxicology screens, “lost” medication, and requests for early refills or escalating doses.37
EVALUATING PATIENTS FOR OPIOID REPLACEMENT THERAPY
In addition to federal regulations, we propose a 4-step approach to evaluate eligibility for opioid replacement therapy based on existing guidelines.37–39
Step 1: History and physical examination
The history should give particular attention to the patient’s cardiac, pulmonary, and hepatic status, with consideration of the risks of any medical comorbidities (eg, bacterial endocarditis, HIV and HCV infection) that might influence treatment.37
It is also essential to evaluate for any contraindications or drug interactions before prescribing methadone or buprenorphine.38
Contraindications to methadone maintenance include40:
- Cor pulmonale
- Methadone hypersensitivity
- Pseudomembranous colitis
- Selegiline use (due to risk of serotonin syndrome)
- Ileum paralyticus.
Contraindications to buprenorphine use include:
- Hypersensitivity to naloxone or buprenorphine
- Impaired liver function (due to the risk of inadvertent overdose associated with slowed metabolism).
Concurrent use of alcohol or illicit benzodiazepines is a relative contraindication to both methadone and buprenorphine due to the risk of respiratory depression and overdose.37 Likewise, avoid coprescribing opioid agonists and benzodiazepines whenever possible. Obtain a complete list of current medications and query a prescription-monitoring database to determine whether any controlled substances are currently prescribed.37
During the physical examination, look for stigmata of intravenous drug use such as track marks or abscesses37 and document any physical findings consistent with intoxication or withdrawal. Patients must be completely detoxed or in withdrawal before beginning buprenorphine induction; premature induction can precipitate withdrawal.38
A discussion of pregnant patients with opioid use disorder is beyond the scope of this paper. However, it is incumbent on the prescriber to inquire whether the client is pregnant or intends to become pregnant and what birth control methods are in place.
Step 2: Assess psychiatric status
Assessment of the patient’s psychiatric status, including an assessment of alcohol and other drug use, will help determine his or her eligibility for opioid agonists.37 To prepare for the need to manage patients with psychiatrically complex issues, it is helpful to develop relationships with addiction specialists and psychiatrists who are familiar with opioid replacement therapy in your area. This will make it easier to collaborate on patients’ care.
Ask all patients directly about suicidal or homicidal ideation. Any patient with active suicidal or homicidal ideation should be assessed for need of immediate hospitalization by a psychiatrist or another qualified mental health professional. Patients with a history of suicidal ideation should be monitored closely by a mental health professional throughout treatment.37
Many if not most patients with opioid use disorder have concurrent psychiatric disorders, and the interplay between these disorders is complex.40,41 Depression, for example, can precede and even precipitate drug use (an observation supporting the “self-medication theory”).42 If the underlying depressive disorder is not addressed, relapse is nearly inevitable.
It has also been shown that both chronic opioid use and withdrawal can exacerbate aversive emotional states. This escalation of symptoms may result from the pharmacologic effects of opioids or from psychosocial sequelae that can arise from chronic opioid use.41 In this situation, maintaining abstinence can lead to resolution of depressive symptoms. As depression and opioid use can occur together, successful treatment requires equal attention to both illnesses.
Other common comorbidities in patients with opioid use disorder include posttraumatic stress disorder, attention deficit hyperactivity disorder, antisocial personality disorder, and concurrent substance abuse disorders.43 The confluence of antisocial personality disorder is particularly important, as patients with antisocial personality disorder display disruptive and maladaptive behaviors.
Identify any psychotropic medication that is prescribed and check carefully for drug interactions. This applies especially to methadone, as many psychiatric medications also prolong the QT interval. Moreover, patients may not be forthcoming about the use of psychiatric medication.
Find out whether the patient is using any other addictive substances, particularly those that affect the central nervous system, as those who use fentanyl, benzodiazepines, or alcohol are at the highest risk of overdose.31 Often the best option for those with concurrent substance use disorders is inpatient detoxification followed by residential rehabilitation care. Either buprenorphine or methadone can then be initiated upon return to an outpatient setting.
Step 3: Assess psychosocial status
To what extent do the patient’s home environment and support systems promote a drug-free lifestyle? Unfortunately, the psychosocial status of many of these patients is fragile, and they may live in areas where illicit drugs are readily available (which can be urban, suburban, or rural), making it difficult to stay substance-free.38
Generally, lifestyle modifications are needed to transform maladaptive behaviors and promote an environment conducive to long-term recovery. Referrals to social services to address housing, vocational needs, and entitlements may be helpful.39
Step 4: Assess readiness to change
According to one model, people go through 5 stages when changing a behavior: precontemplation, contemplation, preparation for action, action, and maintenance.43 In general, the further along the stages a patient is, the more appropriate he or she is for office-based treatment with buprenorphine.39
The level of change can be assessed with tools such as Stages of Change Readiness and Treatment Eagerness Scale (SOCRATES). Use of stage-specific strategies may enhance a patient’s readiness to cease opioid use.43
Precontemplation. Those in the precontemplation stage are not ready to think about changing their behavior.43 They may be unaware of or unwilling to consider the risks associated with their opioid use and resistant to the idea of quitting. Engagement with opioid agonists for individuals in this stage is low and dropout rates are likely high.
Thus, the proper approach for “precontemplators” is to help them develop some ambivalence about their opioid use. One tactic is to involve the patient in a discussion of the personal benefits and risks of opioid use.
Contemplation. Individuals in the contemplation stage have begun to weigh the costs and benefits of opioid use and express ambivalence about it.44 Because the patient is willing to explore the risks of ongoing use and consider the benefits of treatment, the goal in this stage is to elicit a commitment from the individual to seek treatment.
Preparation. The person in this stage moves from thinking about treatment to planning what action to take.45 As the individual prepares to enter treatment, indecision tends to resurface, as well as self-doubt about his or her ability to change. During this stage, it is important for the provider to spell out goals (abstinence) and strategies (eg, counseling, medication) and enhance a sense of self-efficacy.
Action and maintenance. Patients in these stages engage in treatment and employ new strategies to abstain from opioid use. Maintaining these behaviors can be a daily struggle. Expressing confidence in the patient’s ability to abstain from use will support his or her progress. Behavioral interventions such as strategic avoidance of triggers and engagement in alternative activities (eg, support groups, exercise, faith-based practices) will help to maintain abstinence.
A CHRONIC CONDITION
Opioid use disorder, like many chronic illnesses, requires long-term attention to attain successful patient outcomes. The opioid agonists methadone and buprenorphine are the mainstay of treatment for it, conferring benefits such as reducing opioid use and preventing relapse.
Candidates for opioid agonist therapy should undergo a multidisciplinary assessment, including an evaluation on the patient’s readiness to change his or her opioid use.
Patient education should include a discussion of the risks of methadone (eg, respiratory depression, fatal overdose, and QTc prolongation) and buprenorphine (eg hepatotoxicity) and their benefits (eg, controlling craving, decreasing the risk of relapse). Patients should also be educated about overdose and diversion.
Despite the difficulties inherent in treating patients with opioid use disorder, when used appropriately, opioid agonist therapy can be lifesaving for patients struggling with long-term opioid addiction.
Acknowledgment: We thank Katelyn Colosi, BS, and Drs. Susan Wolfe, Dennis Bouffard, and Sinha Shirshendu for their helpful comments.
- Wakeman SE, Pham-Kanter G, Donelan K. Attitudes, practices, and preparedness to care for patients with substance use disorder: results from a survey of general internists. Subst Abus 2016; 37:635–641.
- Samuels EA, Dwyer K, Mello MJ, Baird J, Kellogg AR, Bernstein E. Emergency department-based opioid harm reduction: moving physicians from willing to doing. Acad Emerg Med 2016; 23:455–465.
- Mohlman MK, Tanzman B, Finison K, Pinette M, Jones C. Impact of medication-assisted treatment for opioid addiction on Medicaid expenditures and health services utilization rates in Vermont. J Subst Abuse Treat 2016; 67:9–14.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition. Arlington, VA, American Psychiatric Association, 2013.
- Center for Behavioral Health Statistics and Quality. Behavioral health trends in the United States: results from the 2014 National Survey on Drug Use and Health. www.samhsa.gov/data. Accessed April 6, 2017.
- Compton WM, Jones CM, Baldwin GT. Relationship between nonmedical prescription-opioid use and heroin use. N Engl J Med 2016; 374:154–163.
- Ruan X, Wyche MQ, Kaye AD. Analyzing the relationship between nonmedical prescription-opioid use and heroin use. J Opioid Manage 2016; 12:11–14.
- Hser YI, Evans E, Grella C, Ling W, Anglin D. Long-term course of opioid addiction. Harv Rev Psychiatry 2015; 23:76–89.
- Nielsen S, Hillhouse M, Mooney L, Ang A, Ling W. Buprenorphine pharmacotherapy and behavioral treatment: comparison of outcomes among prescription opioid users, heroin users and combination users. J Subst Abuse Treat 2015; 48:70–76.
- Moore BA, Fiellin DA, Barry DT, et al. Primary care office-based buprenorphine treatment: comparison of heroin and prescription opioid dependent patients. J Gen Intern Med 2007; 22:527–530.
- Fischer B, Patra J, Cruz MF, Gittins J, Rehm J. Comparing heroin users and prescription opioid users in a Canadian multi-site population of illicit opioid users. Drug Alcohol Rev 2008; 27:625–632.
- Compton WM, Jones CM, Baldwin GT. Relationship between nonmedical prescription-opioid use and heroin use. N Engl J Med 2016; 374:154–163.
- Jones CM, Baldwin GT, Manocchio T, White JO, Mack KA. Trends in methadone distribution for pain treatment, methadone diversion, and overdose deaths—United States, 2002–2014. MMWR Morb Mortal Wkly Rep 2016; 65:667–671.
- Baxter LE Sr, Campbell A, Deshields M, et al. Safe methadone induction and stabilization: report of an expert panel. J Addict Med 2013; 7:377–386.
- Alinejad S, Kazemi T, Zamani N, Hoffman RS, Mehrpour O. A systematic review of the cardiotoxicity of methadone. EXCLI J 2015; 14:577–600.
- Johnson RE, Strain EC, Amass L. Buprenorphine: how to use it right. Drug Alcohol Depend 2003; 70(suppl 2):S59–S77.
- Ling W. Buprenorphine implant for opioid addiction. Pain Manage 2012; 2:345–350.
- Saxon AJ, Hser YI, Woody G, Ling W. Medication-assisted treatment for opioid addiction: methadone and buprenorphine. J Food Drug Anal 2013; 21:S69–S72.
- Kimber J, Larney S, Hickman M, Randall D, Degenhardt L. Mortality risk of opioid substitution therapy with methadone versus buprenorphine: a retrospective cohort study. Lancet Psychiatry 2015; 2:901–908.
- Marteau D, McDonald R, Patel K. The relative risk of fatal poisoning by methadone or buprenorphine within the wider population of England and Wales. BMJ Open 2015; 5: e007629.
- Kintz P. Deaths involving buprenorphine: a compendium of French cases. Forensic Sci Int 2001; 121:65–69.
- Zuin M, Giorgini A, Selmi C, et al. Acute liver and renal failure during treatment with buprenorphine at therapeutic dose. Dig Liver Dis 2009; 41:e8–e10.
- Klein JW. Pharmacotherapy for substance use disorders. Med Clin North Am 2016; 100:891–910.
- Modesto-Lowe V, Van Kirk J. Clinical uses of naltrexone: a review of the evidence. Exp Clin Psychopharmocol 2002; 10:213–227.
- Woody GE. Agonist models for treating persons with substance use disorders. Curr Psychiatry Rep 2014; 16:489.
- Sanders JJ, Roose RJ, Lubrano MC, Lucan SC. Meaning and methadone: patient perceptions of methadone dose and a model to promote adherence to maintenance treatment. J Addict Med 2013; 7:307–313.
- Herget G. Methadone and buprenorphine added to the WHO list of essential medicines. HIV/AIDS Policy Law Rev 2005; 10:23–24.
- Suzuki J, Dodds T. Clinical recommendation of 12-step meeting attendance and discussion regarding disclosure of buprenorphine use among patients in office-based opioid treatment. Subst Abus 2016; 37:31–34.
- Rettig RA, Yarmolinsky A. Federal Regulation of Methadone Treatment. Washington, DC: National Academies Press; 1995.
- Srivastava A, Kahan M, Nader M. Primary care management of opioid use disorders: abstinence, methadone, or buprenorphine-naloxone? Can Fam Physician 2017; 63:200–205.
- Substance Abuse and Mental Health Services Administration. Federal Guidelines for Opioid Treatment Programs. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2015.
- Substance Abuse and Mental Health Services Administration SAMSHA. Buprenorphine waiver management. www.samhsa.gov/medication-assisted-treatment/buprenorphine-waiver-management. Accessed April 6, 2017.
- Mark TL, Lubran R, McCance-Kats EF, Chalk M, Richardson J. Medicaid coverage of medications to treat alcohol and opioid dependence. J Subst Abuse Treat 2015; 55:1–5.
- Johnson B, Richert T. Diversion of methadone and buprenorphine from opioid substitution treatment: the importance of patients’ attitudes and norms. J Subst Abuse Treat 2015; 54:50–55.
- Yokell MA, Zaller ND, Green TC, Rich JD. Buprenorphine and buprenorphine/naloxone diversion, misuse, and illicit use: an international review. Curr Drug Abuse Rev 2011; 4:28–41.
- Schuman-Olivier Z, Albanese M, Nelson SE, et al. Self-treatment: illicit buprenorphine use by opioid-dependent treatment seekers. J Subst Abuse Treat 2010; 39:41–50.
- American Society of Addiction Medicine. National practice guidelines for the use of medications in the treatment of addiction involving opioid use. www.asam.org/docs/default-source/practice-support/guidelines-and-consensus-docs/asam-national-practice-guideline-supplement.pdf. Accessed April 6, 2017.
- McNicholas L. Clinical guidelines for the use of buprenorphine in the treatment of opioid addiction. Rockville, MD: US Department of Health and Human Services, Substance Abuse and Mental Health Service Administration; 2004.
- Center for Substance Abuse Treatment. Clinical guidelines for the use of buprenorphine in the treatment of opioid addiction. Rockville (MD): Substance Abuse and Mental Health Services Administration (US); 2004. (Treatment Improvement Protocol (TIP) Series, No. 40.) www.ncbi.nlm.nih.gov/books/NBK64245. Accessed April 6, 2017.
- Zippel-Schultz B, Specka M, Cimander K, et al. Outcomes of patients in long-term opioid maintenance treatment. Subst Use Misuse 2016; 51:1493–1503.
- Martins SS, Keyes KM, Storr CL, Zhu H, Chilcoat HD. Pathways between nonmedical opioid use/dependence and psychiatric disorders: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Drug Alcohol Depend 2009; 103:16–24.
- Khantzian EJ. The self-medication hypothesis of addictive disorders: focus on heroin and cocaine dependence. Am J Psychiatry 1985; 142:1259–1264.
- Belding MA, Iguchi MY, Lamb RJ, Lakin M, Terry R. Stages and processes of change among polydrug users in methadone maintenance treatment. Drug Alcohol Depend 1995; 39:45–53.
- Peteet JR, Brenner S, Curtiss D, Ferrigno M, Kauffman J. A stage of change approach to addiction in the medical setting. Gen Hosp Psychiatry 1998; 20:267–273.
- Vijay A, Bazazi AR, Yee I, Kamarulzaman A, Altice FL. Treatment readiness, attitudes toward, and experiences with methadone and buprenorphine maintenance therapy among people who inject drugs in Malaysia. J Subst Abuse Treat 2015; 54:29–36.
For a patient struggling with opioid addiction, opioid agonist therapy with methadone or buprenorphine can reduce craving and opioid use and may even save his or her life. But many clinicians are unfamiliar with this evidence-based treatment,1,2 which is best started early in the course of addiction.3
This article outlines the pharmacology of these drugs, their clinical uses, and the challenges of using them to treat opioid addiction.
DIAGNOSTIC CRITERIA
Opioid addiction, formally known as opioid use disorder, is a pattern of opioid misuse leading to clinically significant impairment in multiple areas of life. The Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition, lists 11 diagnostic criteria, but only 2 need to be present within the past year to make the diagnosis4:
- Taking opioids longer or in higher doses than was intended
- A persistent desire or unsuccessful efforts to cut down or control opioid use
- Spending a great deal of time obtaining, using, or recovering from using opioids
- Craving opioids
- Repeatedly failing to fulfill obligations at work, school, or home due to opioid use
- Continuing to use opioids even though it causes or exacerbates social or interpersonal problems
- Giving up or curtailing important social, occupational, or recreational activities because of opioid use
- Repeatedly using opioids in situations in which it is physically hazardous
- Continuing to use opioids despite knowledge of having a persistent or recurrent physical or psychological problem that is likely to have been caused or exacerbated by the substance
- Tolerance
- Withdrawal.
Recent estimates indicate that 2.23 million people in the United States have opioid use disorder (426,000 with heroin and 1.8 million with prescription opioids).5
Progression from prescription opioids to heroin
We have observed that many patients with opioid use disorder start by misusing prescription opioids. Over time, tolerance can develop, which drives patients to use higher and higher doses.6
As the addiction progresses, a subset of prescription opioid users advances to using heroin, which is typically less expensive and easier to obtain.7 Most patients start with the intranasal route but eventually inject it intravenously.6,7
For many addicts, heroin use has medical consequences such as hepatitis C virus (HCV) and human immunodeficiency virus (HIV) infection, psychiatric problems such as depression and anxiety, and illegal activities such as theft and sex work.8 People who use heroin appear to have more severe addiction and a lower socioeconomic status than prescription opioid users.9–11 But recently, a growing number of middle class individuals are becoming addicted to heroin.12
METHADONE
Methadone is a long-acting synthetic opioid that functions as a full agonist on the mu-opioid receptor. The drug binds, occupies, and stimulates the receptor, preventing withdrawal symptoms and reducing opioid cravings for at least 24 hours.13
Adverse effects of methadone
The most common adverse effects include lightheadedness, dizziness, sedation, nausea, vomiting, and sweating.14 Other adverse effects:
Unintentional overdose. The risk is serious, as a single 30-mg dose can be fatal in people who are opioid-naïve.13
QTc prolongation, which can lead to torsade de pointes. This risk, which is dose-related, must be taken into consideration in patients who have any cardiac symptoms (eg, syncope, arrhythmia), pathology (familial QT prolongation), or other risk factors for QTc prolongation (eg, hypokalemia, QTc-prolonging medications).15
Respiratory depression, which can be fatal. This dose-related risk is heightened during the first 4 weeks of treatment if titration is too rapid or if methadone is used in combination with other drugs that cause central nervous system or respiratory depression.13,14
Starting methadone
To prevent respiratory depression and death related to rapid induction, the general rule is to start methadone at a low daily dose (20–30 mg) depending on the patient’s withdrawal symptoms.14 During this period, patients need to be closely monitored and educated on the perils of concomitant use of central nervous system depressants.14
In most patients, the dose is titrated up until their withdrawal symptoms and cravings are eliminated, which generally requires 60 to 120 mg daily.14 Hepatic and renal impairment, pregnancy, and advanced age can alter methadone pharmacokinetics and may therefore necessitate dose adjustment.
BUPRENORPHINE
Buprenorphine is an alkaloid thebaine opioid derivative that acts as a partial mu-opioid agonist and a kappa antagonist.16 Like methadone, buprenorphine is used to manage cravings and withdrawal symptoms.16 Dosages of 4 to 16 mg (up to 32 mg) per day of buprenorphine are usually required to adequately control opioid cravings.16
Sublingual and subdermal products
Buprenorphine is currently available in the United States in sublingual and subdermal formulations.16,17
Sublingual buprenorphine is usually combined with naloxone in a 4:1 ratio to deter intravenous use. Intravenous injection of the combination product can precipitate withdrawal due to the antagonist action of naloxone. (Taken orally or sublingually, naloxone is poorly absorbed and has little or no clinical effect.) Buprenorphine-naloxone is available in tablets, a sublingual film strip, and a buccal film strip. Buprenorphine is also available by itself in a sublingual formulation.
The US Food and Drug Administration has approved a buprenorphine subdermal implant, Probuphine. Four rods, about 1 inch long, are placed under the skin in the inner aspect of the upper arm and provide the equivalent of 8 mg of buprenorphine daily for 6 months.17 However, this method is formulated only for maintenance treatment and cannot be used for induction. Additionally, it is recommended that the implants be surgically removed at the end of 6 months, after which another set of implants can be inserted in the other arm or the patient can switch to sublingual therapy, depending on the clinical situation and patient preference.17
Generally safer than methadone
Buprenorphine works on the same receptor as methadone and therefore has a similar side effect profile. However, buprenorphine has a ceiling effect, which greatly reduces the risk of fatal respiratory depression.18 It also does not cause clinically significant QTc prolongation and is preferable in patients who have cardiac risk factors.18
Another advantage is that buprenorphine has fewer identified medication interactions than methadone.18 Further, induction of buprenorphine in patients with opioid use disorder has been shown to be safer than methadone.19
Although buprenorphine has been found to be 6 times safer than methadone with regard to overdose among the general population,20 it can still cause fatal intoxication if used in combination with central nervous system depressants.21
Buprenorphine has been also associated with hepatotoxicity, though the risk of new-onset liver disease appears to be low.22
NALTREXONE IS LESS EFFECTIVE THAN METHADONE, BUPRENORPHINE
Besides methadone and buprenorphine, the only other approved option for treating opioid use disorder is the opioid antagonist naltrexone.
Naltrexone has significantly less abuse potential, as it provides no euphoria, but patients do not like it. Even with the long-acting formulation (Vivitrol), naltrexone treatment is significantly less effective than methadone or buprenorphine.23–25 Further, although naltrexone is not a controlled substance and so does not face the same scrutiny as the agonist therapies, there are other significant barriers. Additional information on naltrexone is presented in reviews by Modesto-Lowe and Van Kirk24 and Woody.25
OBSTACLES TO TREATMENT
People hold conflicting views about opioid agonist therapy. Some believe that “trading one drug for another” is not a legitimate therapeutic strategy, and they may feel ashamed of being on maintenance therapy.26 Similarly, some argue that the answer to establishing stable abstinence does not lie simply in prescribing medications.
The contrary argument is that these medications, if used appropriately, confer many benefits such as reducing the medical and psychosocial sequelae of opioid addiction.18 In fact, properly treated patients no longer meet the diagnostic criteria of opioid use disorder, and both methadone and buprenorphine are on the World Health Organization’s (WHO) list of essential medicines.27
Despite endorsement by the WHO, the stigma attached to the opioid agonists has been difficult to overcome. Patients with opioid use disorder may be viewed with distrust by healthcare providers and often do not feel welcome in healthcare settings or in self-help recovery groups.28
Barriers to methadone therapy
Federal regulations on methadone prescribing and use were established to promote patient safety and decrease diversion, but they may also complicate access to care.29 They stipulate that to qualify for methadone maintenance, patients need to demonstrate opioid addiction for 1 year, except for pregnant women and those who have been incarcerated in the past 6 months. Patients under the age of 18 must have 2 documented failed treatment episodes as well as approval by a guardian to receive treatment.
Inconvenience. Methadone can be prescribed for opioid dependence only by an accredited treatment program. Patients must therefore travel to the clinic and wait to be evaluated on a daily basis for a minimum of 90 days. Only after they demonstrate consistent responsible behavior and negative results on urine testing do they become eligible to take methadone home.29 If a patient is to travel out of the area during the initial 90 days of treatment, he or she must make arrangements in advance to find a clinic that will provide a “guest dose.”
The inconvenience arising from the regulations may deter some patients from seeking methadone therapy. In spite of this, once patients are started on methadone, more of them continue treatment than with buprenorphine.18 A proposed reason is that methadone is a potent full opioid agonist and therefore relieves withdrawal symptoms and craving more effectively than buprenorphine, which is a partial agonist.30 Another possible reason is the higher level of supervision afforded by methadone clinics, which require daily contact for at least 90 days.
Safety concerns arise from methadone diversion, as illicit use may have lethal consequences. In the past decade, deaths from methadone overdose have risen significantly, most of them due to respiratory depression or torsade de pointes.13 However, most cases of diversion and overdose involve methadone that is prescribed for pain by individual practitioners and not from maintenance programs.13
Advantages of buprenorphine
Together, methadone’s lethality, stigma, and inconvenience may contribute to patients preferring buprenorphine.31
The regulations governing buprenorphine’s use are less restrictive than those with methadone. For example, patients must have a diagnosis of opioid addiction to be prescribed buprenorphine, but they are not required to carry the diagnosis for a year before treatment.31 Additionally, they do not need to travel to a federally approved opioid treatment center daily and can receive buprenorphine directly from a physician in an outpatient setting.
Under the Drug Abuse Treatment Act (DATA) of 2000, any physician can apply for a waiver to prescribe and dispense buprenorphine in his or her office. To qualify for an initial waiver, physicians must either obtain certification in the fields of addiction medicine or addiction psychiatry or complete an approved 8-hour training session.32 Each physician starts with a maximum of 30 patients, but can apply to treat up to 100 patients after 1 year and eventually up to 275 patients. Physicians must document every buprenorphine prescription they write and be able to refer patients for counseling.31
As of February 2017, nurse practitioners and physician assistants can also apply for a DATA 2000 waiver. All waivered providers are subject to unannounced visits from the Drug Enforcement Administration once every 5 years.32
While there are no federal restrictions on the amount of buprenorphine that can be dispensed, some states and some insurance companies have placed restrictions on dose or length of treatment.33 Buprenorphine patients can fill their prescriptions at any pharmacy and are permitted to bring their medication home, which improves access to care. However, office-based outpatient treatment is not without risk, and preventing buprenorphine diversion remains a challenge.34
‘Lending’ buprenorphine is a felony
Addicts have illegally used buprenorphine to self-treat opioid withdrawal, craving, and dependence.35 Its misuse has also been coupled with self-treatment of conditions that include depression and pain.36
A survey found that 83.7% of patients deem buprenorphine diversion to be appropriate; further, most patients said they consider it unethical to withhold prescribed buprenorphine from individuals showing symptoms of withdrawal.34 Physicians who prescribe buprenorphine must inform their patients that even “lending” or giving away their medication is a felony.
Prescribing physicians must also be diligent about monitoring for signs of diversion such as inconsistent urine toxicology screens, “lost” medication, and requests for early refills or escalating doses.37
EVALUATING PATIENTS FOR OPIOID REPLACEMENT THERAPY
In addition to federal regulations, we propose a 4-step approach to evaluate eligibility for opioid replacement therapy based on existing guidelines.37–39
Step 1: History and physical examination
The history should give particular attention to the patient’s cardiac, pulmonary, and hepatic status, with consideration of the risks of any medical comorbidities (eg, bacterial endocarditis, HIV and HCV infection) that might influence treatment.37
It is also essential to evaluate for any contraindications or drug interactions before prescribing methadone or buprenorphine.38
Contraindications to methadone maintenance include40:
- Cor pulmonale
- Methadone hypersensitivity
- Pseudomembranous colitis
- Selegiline use (due to risk of serotonin syndrome)
- Ileum paralyticus.
Contraindications to buprenorphine use include:
- Hypersensitivity to naloxone or buprenorphine
- Impaired liver function (due to the risk of inadvertent overdose associated with slowed metabolism).
Concurrent use of alcohol or illicit benzodiazepines is a relative contraindication to both methadone and buprenorphine due to the risk of respiratory depression and overdose.37 Likewise, avoid coprescribing opioid agonists and benzodiazepines whenever possible. Obtain a complete list of current medications and query a prescription-monitoring database to determine whether any controlled substances are currently prescribed.37
During the physical examination, look for stigmata of intravenous drug use such as track marks or abscesses37 and document any physical findings consistent with intoxication or withdrawal. Patients must be completely detoxed or in withdrawal before beginning buprenorphine induction; premature induction can precipitate withdrawal.38
A discussion of pregnant patients with opioid use disorder is beyond the scope of this paper. However, it is incumbent on the prescriber to inquire whether the client is pregnant or intends to become pregnant and what birth control methods are in place.
Step 2: Assess psychiatric status
Assessment of the patient’s psychiatric status, including an assessment of alcohol and other drug use, will help determine his or her eligibility for opioid agonists.37 To prepare for the need to manage patients with psychiatrically complex issues, it is helpful to develop relationships with addiction specialists and psychiatrists who are familiar with opioid replacement therapy in your area. This will make it easier to collaborate on patients’ care.
Ask all patients directly about suicidal or homicidal ideation. Any patient with active suicidal or homicidal ideation should be assessed for need of immediate hospitalization by a psychiatrist or another qualified mental health professional. Patients with a history of suicidal ideation should be monitored closely by a mental health professional throughout treatment.37
Many if not most patients with opioid use disorder have concurrent psychiatric disorders, and the interplay between these disorders is complex.40,41 Depression, for example, can precede and even precipitate drug use (an observation supporting the “self-medication theory”).42 If the underlying depressive disorder is not addressed, relapse is nearly inevitable.
It has also been shown that both chronic opioid use and withdrawal can exacerbate aversive emotional states. This escalation of symptoms may result from the pharmacologic effects of opioids or from psychosocial sequelae that can arise from chronic opioid use.41 In this situation, maintaining abstinence can lead to resolution of depressive symptoms. As depression and opioid use can occur together, successful treatment requires equal attention to both illnesses.
Other common comorbidities in patients with opioid use disorder include posttraumatic stress disorder, attention deficit hyperactivity disorder, antisocial personality disorder, and concurrent substance abuse disorders.43 The confluence of antisocial personality disorder is particularly important, as patients with antisocial personality disorder display disruptive and maladaptive behaviors.
Identify any psychotropic medication that is prescribed and check carefully for drug interactions. This applies especially to methadone, as many psychiatric medications also prolong the QT interval. Moreover, patients may not be forthcoming about the use of psychiatric medication.
Find out whether the patient is using any other addictive substances, particularly those that affect the central nervous system, as those who use fentanyl, benzodiazepines, or alcohol are at the highest risk of overdose.31 Often the best option for those with concurrent substance use disorders is inpatient detoxification followed by residential rehabilitation care. Either buprenorphine or methadone can then be initiated upon return to an outpatient setting.
Step 3: Assess psychosocial status
To what extent do the patient’s home environment and support systems promote a drug-free lifestyle? Unfortunately, the psychosocial status of many of these patients is fragile, and they may live in areas where illicit drugs are readily available (which can be urban, suburban, or rural), making it difficult to stay substance-free.38
Generally, lifestyle modifications are needed to transform maladaptive behaviors and promote an environment conducive to long-term recovery. Referrals to social services to address housing, vocational needs, and entitlements may be helpful.39
Step 4: Assess readiness to change
According to one model, people go through 5 stages when changing a behavior: precontemplation, contemplation, preparation for action, action, and maintenance.43 In general, the further along the stages a patient is, the more appropriate he or she is for office-based treatment with buprenorphine.39
The level of change can be assessed with tools such as Stages of Change Readiness and Treatment Eagerness Scale (SOCRATES). Use of stage-specific strategies may enhance a patient’s readiness to cease opioid use.43
Precontemplation. Those in the precontemplation stage are not ready to think about changing their behavior.43 They may be unaware of or unwilling to consider the risks associated with their opioid use and resistant to the idea of quitting. Engagement with opioid agonists for individuals in this stage is low and dropout rates are likely high.
Thus, the proper approach for “precontemplators” is to help them develop some ambivalence about their opioid use. One tactic is to involve the patient in a discussion of the personal benefits and risks of opioid use.
Contemplation. Individuals in the contemplation stage have begun to weigh the costs and benefits of opioid use and express ambivalence about it.44 Because the patient is willing to explore the risks of ongoing use and consider the benefits of treatment, the goal in this stage is to elicit a commitment from the individual to seek treatment.
Preparation. The person in this stage moves from thinking about treatment to planning what action to take.45 As the individual prepares to enter treatment, indecision tends to resurface, as well as self-doubt about his or her ability to change. During this stage, it is important for the provider to spell out goals (abstinence) and strategies (eg, counseling, medication) and enhance a sense of self-efficacy.
Action and maintenance. Patients in these stages engage in treatment and employ new strategies to abstain from opioid use. Maintaining these behaviors can be a daily struggle. Expressing confidence in the patient’s ability to abstain from use will support his or her progress. Behavioral interventions such as strategic avoidance of triggers and engagement in alternative activities (eg, support groups, exercise, faith-based practices) will help to maintain abstinence.
A CHRONIC CONDITION
Opioid use disorder, like many chronic illnesses, requires long-term attention to attain successful patient outcomes. The opioid agonists methadone and buprenorphine are the mainstay of treatment for it, conferring benefits such as reducing opioid use and preventing relapse.
Candidates for opioid agonist therapy should undergo a multidisciplinary assessment, including an evaluation on the patient’s readiness to change his or her opioid use.
Patient education should include a discussion of the risks of methadone (eg, respiratory depression, fatal overdose, and QTc prolongation) and buprenorphine (eg hepatotoxicity) and their benefits (eg, controlling craving, decreasing the risk of relapse). Patients should also be educated about overdose and diversion.
Despite the difficulties inherent in treating patients with opioid use disorder, when used appropriately, opioid agonist therapy can be lifesaving for patients struggling with long-term opioid addiction.
Acknowledgment: We thank Katelyn Colosi, BS, and Drs. Susan Wolfe, Dennis Bouffard, and Sinha Shirshendu for their helpful comments.
For a patient struggling with opioid addiction, opioid agonist therapy with methadone or buprenorphine can reduce craving and opioid use and may even save his or her life. But many clinicians are unfamiliar with this evidence-based treatment,1,2 which is best started early in the course of addiction.3
This article outlines the pharmacology of these drugs, their clinical uses, and the challenges of using them to treat opioid addiction.
DIAGNOSTIC CRITERIA
Opioid addiction, formally known as opioid use disorder, is a pattern of opioid misuse leading to clinically significant impairment in multiple areas of life. The Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition, lists 11 diagnostic criteria, but only 2 need to be present within the past year to make the diagnosis4:
- Taking opioids longer or in higher doses than was intended
- A persistent desire or unsuccessful efforts to cut down or control opioid use
- Spending a great deal of time obtaining, using, or recovering from using opioids
- Craving opioids
- Repeatedly failing to fulfill obligations at work, school, or home due to opioid use
- Continuing to use opioids even though it causes or exacerbates social or interpersonal problems
- Giving up or curtailing important social, occupational, or recreational activities because of opioid use
- Repeatedly using opioids in situations in which it is physically hazardous
- Continuing to use opioids despite knowledge of having a persistent or recurrent physical or psychological problem that is likely to have been caused or exacerbated by the substance
- Tolerance
- Withdrawal.
Recent estimates indicate that 2.23 million people in the United States have opioid use disorder (426,000 with heroin and 1.8 million with prescription opioids).5
Progression from prescription opioids to heroin
We have observed that many patients with opioid use disorder start by misusing prescription opioids. Over time, tolerance can develop, which drives patients to use higher and higher doses.6
As the addiction progresses, a subset of prescription opioid users advances to using heroin, which is typically less expensive and easier to obtain.7 Most patients start with the intranasal route but eventually inject it intravenously.6,7
For many addicts, heroin use has medical consequences such as hepatitis C virus (HCV) and human immunodeficiency virus (HIV) infection, psychiatric problems such as depression and anxiety, and illegal activities such as theft and sex work.8 People who use heroin appear to have more severe addiction and a lower socioeconomic status than prescription opioid users.9–11 But recently, a growing number of middle class individuals are becoming addicted to heroin.12
METHADONE
Methadone is a long-acting synthetic opioid that functions as a full agonist on the mu-opioid receptor. The drug binds, occupies, and stimulates the receptor, preventing withdrawal symptoms and reducing opioid cravings for at least 24 hours.13
Adverse effects of methadone
The most common adverse effects include lightheadedness, dizziness, sedation, nausea, vomiting, and sweating.14 Other adverse effects:
Unintentional overdose. The risk is serious, as a single 30-mg dose can be fatal in people who are opioid-naïve.13
QTc prolongation, which can lead to torsade de pointes. This risk, which is dose-related, must be taken into consideration in patients who have any cardiac symptoms (eg, syncope, arrhythmia), pathology (familial QT prolongation), or other risk factors for QTc prolongation (eg, hypokalemia, QTc-prolonging medications).15
Respiratory depression, which can be fatal. This dose-related risk is heightened during the first 4 weeks of treatment if titration is too rapid or if methadone is used in combination with other drugs that cause central nervous system or respiratory depression.13,14
Starting methadone
To prevent respiratory depression and death related to rapid induction, the general rule is to start methadone at a low daily dose (20–30 mg) depending on the patient’s withdrawal symptoms.14 During this period, patients need to be closely monitored and educated on the perils of concomitant use of central nervous system depressants.14
In most patients, the dose is titrated up until their withdrawal symptoms and cravings are eliminated, which generally requires 60 to 120 mg daily.14 Hepatic and renal impairment, pregnancy, and advanced age can alter methadone pharmacokinetics and may therefore necessitate dose adjustment.
BUPRENORPHINE
Buprenorphine is an alkaloid thebaine opioid derivative that acts as a partial mu-opioid agonist and a kappa antagonist.16 Like methadone, buprenorphine is used to manage cravings and withdrawal symptoms.16 Dosages of 4 to 16 mg (up to 32 mg) per day of buprenorphine are usually required to adequately control opioid cravings.16
Sublingual and subdermal products
Buprenorphine is currently available in the United States in sublingual and subdermal formulations.16,17
Sublingual buprenorphine is usually combined with naloxone in a 4:1 ratio to deter intravenous use. Intravenous injection of the combination product can precipitate withdrawal due to the antagonist action of naloxone. (Taken orally or sublingually, naloxone is poorly absorbed and has little or no clinical effect.) Buprenorphine-naloxone is available in tablets, a sublingual film strip, and a buccal film strip. Buprenorphine is also available by itself in a sublingual formulation.
The US Food and Drug Administration has approved a buprenorphine subdermal implant, Probuphine. Four rods, about 1 inch long, are placed under the skin in the inner aspect of the upper arm and provide the equivalent of 8 mg of buprenorphine daily for 6 months.17 However, this method is formulated only for maintenance treatment and cannot be used for induction. Additionally, it is recommended that the implants be surgically removed at the end of 6 months, after which another set of implants can be inserted in the other arm or the patient can switch to sublingual therapy, depending on the clinical situation and patient preference.17
Generally safer than methadone
Buprenorphine works on the same receptor as methadone and therefore has a similar side effect profile. However, buprenorphine has a ceiling effect, which greatly reduces the risk of fatal respiratory depression.18 It also does not cause clinically significant QTc prolongation and is preferable in patients who have cardiac risk factors.18
Another advantage is that buprenorphine has fewer identified medication interactions than methadone.18 Further, induction of buprenorphine in patients with opioid use disorder has been shown to be safer than methadone.19
Although buprenorphine has been found to be 6 times safer than methadone with regard to overdose among the general population,20 it can still cause fatal intoxication if used in combination with central nervous system depressants.21
Buprenorphine has been also associated with hepatotoxicity, though the risk of new-onset liver disease appears to be low.22
NALTREXONE IS LESS EFFECTIVE THAN METHADONE, BUPRENORPHINE
Besides methadone and buprenorphine, the only other approved option for treating opioid use disorder is the opioid antagonist naltrexone.
Naltrexone has significantly less abuse potential, as it provides no euphoria, but patients do not like it. Even with the long-acting formulation (Vivitrol), naltrexone treatment is significantly less effective than methadone or buprenorphine.23–25 Further, although naltrexone is not a controlled substance and so does not face the same scrutiny as the agonist therapies, there are other significant barriers. Additional information on naltrexone is presented in reviews by Modesto-Lowe and Van Kirk24 and Woody.25
OBSTACLES TO TREATMENT
People hold conflicting views about opioid agonist therapy. Some believe that “trading one drug for another” is not a legitimate therapeutic strategy, and they may feel ashamed of being on maintenance therapy.26 Similarly, some argue that the answer to establishing stable abstinence does not lie simply in prescribing medications.
The contrary argument is that these medications, if used appropriately, confer many benefits such as reducing the medical and psychosocial sequelae of opioid addiction.18 In fact, properly treated patients no longer meet the diagnostic criteria of opioid use disorder, and both methadone and buprenorphine are on the World Health Organization’s (WHO) list of essential medicines.27
Despite endorsement by the WHO, the stigma attached to the opioid agonists has been difficult to overcome. Patients with opioid use disorder may be viewed with distrust by healthcare providers and often do not feel welcome in healthcare settings or in self-help recovery groups.28
Barriers to methadone therapy
Federal regulations on methadone prescribing and use were established to promote patient safety and decrease diversion, but they may also complicate access to care.29 They stipulate that to qualify for methadone maintenance, patients need to demonstrate opioid addiction for 1 year, except for pregnant women and those who have been incarcerated in the past 6 months. Patients under the age of 18 must have 2 documented failed treatment episodes as well as approval by a guardian to receive treatment.
Inconvenience. Methadone can be prescribed for opioid dependence only by an accredited treatment program. Patients must therefore travel to the clinic and wait to be evaluated on a daily basis for a minimum of 90 days. Only after they demonstrate consistent responsible behavior and negative results on urine testing do they become eligible to take methadone home.29 If a patient is to travel out of the area during the initial 90 days of treatment, he or she must make arrangements in advance to find a clinic that will provide a “guest dose.”
The inconvenience arising from the regulations may deter some patients from seeking methadone therapy. In spite of this, once patients are started on methadone, more of them continue treatment than with buprenorphine.18 A proposed reason is that methadone is a potent full opioid agonist and therefore relieves withdrawal symptoms and craving more effectively than buprenorphine, which is a partial agonist.30 Another possible reason is the higher level of supervision afforded by methadone clinics, which require daily contact for at least 90 days.
Safety concerns arise from methadone diversion, as illicit use may have lethal consequences. In the past decade, deaths from methadone overdose have risen significantly, most of them due to respiratory depression or torsade de pointes.13 However, most cases of diversion and overdose involve methadone that is prescribed for pain by individual practitioners and not from maintenance programs.13
Advantages of buprenorphine
Together, methadone’s lethality, stigma, and inconvenience may contribute to patients preferring buprenorphine.31
The regulations governing buprenorphine’s use are less restrictive than those with methadone. For example, patients must have a diagnosis of opioid addiction to be prescribed buprenorphine, but they are not required to carry the diagnosis for a year before treatment.31 Additionally, they do not need to travel to a federally approved opioid treatment center daily and can receive buprenorphine directly from a physician in an outpatient setting.
Under the Drug Abuse Treatment Act (DATA) of 2000, any physician can apply for a waiver to prescribe and dispense buprenorphine in his or her office. To qualify for an initial waiver, physicians must either obtain certification in the fields of addiction medicine or addiction psychiatry or complete an approved 8-hour training session.32 Each physician starts with a maximum of 30 patients, but can apply to treat up to 100 patients after 1 year and eventually up to 275 patients. Physicians must document every buprenorphine prescription they write and be able to refer patients for counseling.31
As of February 2017, nurse practitioners and physician assistants can also apply for a DATA 2000 waiver. All waivered providers are subject to unannounced visits from the Drug Enforcement Administration once every 5 years.32
While there are no federal restrictions on the amount of buprenorphine that can be dispensed, some states and some insurance companies have placed restrictions on dose or length of treatment.33 Buprenorphine patients can fill their prescriptions at any pharmacy and are permitted to bring their medication home, which improves access to care. However, office-based outpatient treatment is not without risk, and preventing buprenorphine diversion remains a challenge.34
‘Lending’ buprenorphine is a felony
Addicts have illegally used buprenorphine to self-treat opioid withdrawal, craving, and dependence.35 Its misuse has also been coupled with self-treatment of conditions that include depression and pain.36
A survey found that 83.7% of patients deem buprenorphine diversion to be appropriate; further, most patients said they consider it unethical to withhold prescribed buprenorphine from individuals showing symptoms of withdrawal.34 Physicians who prescribe buprenorphine must inform their patients that even “lending” or giving away their medication is a felony.
Prescribing physicians must also be diligent about monitoring for signs of diversion such as inconsistent urine toxicology screens, “lost” medication, and requests for early refills or escalating doses.37
EVALUATING PATIENTS FOR OPIOID REPLACEMENT THERAPY
In addition to federal regulations, we propose a 4-step approach to evaluate eligibility for opioid replacement therapy based on existing guidelines.37–39
Step 1: History and physical examination
The history should give particular attention to the patient’s cardiac, pulmonary, and hepatic status, with consideration of the risks of any medical comorbidities (eg, bacterial endocarditis, HIV and HCV infection) that might influence treatment.37
It is also essential to evaluate for any contraindications or drug interactions before prescribing methadone or buprenorphine.38
Contraindications to methadone maintenance include40:
- Cor pulmonale
- Methadone hypersensitivity
- Pseudomembranous colitis
- Selegiline use (due to risk of serotonin syndrome)
- Ileum paralyticus.
Contraindications to buprenorphine use include:
- Hypersensitivity to naloxone or buprenorphine
- Impaired liver function (due to the risk of inadvertent overdose associated with slowed metabolism).
Concurrent use of alcohol or illicit benzodiazepines is a relative contraindication to both methadone and buprenorphine due to the risk of respiratory depression and overdose.37 Likewise, avoid coprescribing opioid agonists and benzodiazepines whenever possible. Obtain a complete list of current medications and query a prescription-monitoring database to determine whether any controlled substances are currently prescribed.37
During the physical examination, look for stigmata of intravenous drug use such as track marks or abscesses37 and document any physical findings consistent with intoxication or withdrawal. Patients must be completely detoxed or in withdrawal before beginning buprenorphine induction; premature induction can precipitate withdrawal.38
A discussion of pregnant patients with opioid use disorder is beyond the scope of this paper. However, it is incumbent on the prescriber to inquire whether the client is pregnant or intends to become pregnant and what birth control methods are in place.
Step 2: Assess psychiatric status
Assessment of the patient’s psychiatric status, including an assessment of alcohol and other drug use, will help determine his or her eligibility for opioid agonists.37 To prepare for the need to manage patients with psychiatrically complex issues, it is helpful to develop relationships with addiction specialists and psychiatrists who are familiar with opioid replacement therapy in your area. This will make it easier to collaborate on patients’ care.
Ask all patients directly about suicidal or homicidal ideation. Any patient with active suicidal or homicidal ideation should be assessed for need of immediate hospitalization by a psychiatrist or another qualified mental health professional. Patients with a history of suicidal ideation should be monitored closely by a mental health professional throughout treatment.37
Many if not most patients with opioid use disorder have concurrent psychiatric disorders, and the interplay between these disorders is complex.40,41 Depression, for example, can precede and even precipitate drug use (an observation supporting the “self-medication theory”).42 If the underlying depressive disorder is not addressed, relapse is nearly inevitable.
It has also been shown that both chronic opioid use and withdrawal can exacerbate aversive emotional states. This escalation of symptoms may result from the pharmacologic effects of opioids or from psychosocial sequelae that can arise from chronic opioid use.41 In this situation, maintaining abstinence can lead to resolution of depressive symptoms. As depression and opioid use can occur together, successful treatment requires equal attention to both illnesses.
Other common comorbidities in patients with opioid use disorder include posttraumatic stress disorder, attention deficit hyperactivity disorder, antisocial personality disorder, and concurrent substance abuse disorders.43 The confluence of antisocial personality disorder is particularly important, as patients with antisocial personality disorder display disruptive and maladaptive behaviors.
Identify any psychotropic medication that is prescribed and check carefully for drug interactions. This applies especially to methadone, as many psychiatric medications also prolong the QT interval. Moreover, patients may not be forthcoming about the use of psychiatric medication.
Find out whether the patient is using any other addictive substances, particularly those that affect the central nervous system, as those who use fentanyl, benzodiazepines, or alcohol are at the highest risk of overdose.31 Often the best option for those with concurrent substance use disorders is inpatient detoxification followed by residential rehabilitation care. Either buprenorphine or methadone can then be initiated upon return to an outpatient setting.
Step 3: Assess psychosocial status
To what extent do the patient’s home environment and support systems promote a drug-free lifestyle? Unfortunately, the psychosocial status of many of these patients is fragile, and they may live in areas where illicit drugs are readily available (which can be urban, suburban, or rural), making it difficult to stay substance-free.38
Generally, lifestyle modifications are needed to transform maladaptive behaviors and promote an environment conducive to long-term recovery. Referrals to social services to address housing, vocational needs, and entitlements may be helpful.39
Step 4: Assess readiness to change
According to one model, people go through 5 stages when changing a behavior: precontemplation, contemplation, preparation for action, action, and maintenance.43 In general, the further along the stages a patient is, the more appropriate he or she is for office-based treatment with buprenorphine.39
The level of change can be assessed with tools such as Stages of Change Readiness and Treatment Eagerness Scale (SOCRATES). Use of stage-specific strategies may enhance a patient’s readiness to cease opioid use.43
Precontemplation. Those in the precontemplation stage are not ready to think about changing their behavior.43 They may be unaware of or unwilling to consider the risks associated with their opioid use and resistant to the idea of quitting. Engagement with opioid agonists for individuals in this stage is low and dropout rates are likely high.
Thus, the proper approach for “precontemplators” is to help them develop some ambivalence about their opioid use. One tactic is to involve the patient in a discussion of the personal benefits and risks of opioid use.
Contemplation. Individuals in the contemplation stage have begun to weigh the costs and benefits of opioid use and express ambivalence about it.44 Because the patient is willing to explore the risks of ongoing use and consider the benefits of treatment, the goal in this stage is to elicit a commitment from the individual to seek treatment.
Preparation. The person in this stage moves from thinking about treatment to planning what action to take.45 As the individual prepares to enter treatment, indecision tends to resurface, as well as self-doubt about his or her ability to change. During this stage, it is important for the provider to spell out goals (abstinence) and strategies (eg, counseling, medication) and enhance a sense of self-efficacy.
Action and maintenance. Patients in these stages engage in treatment and employ new strategies to abstain from opioid use. Maintaining these behaviors can be a daily struggle. Expressing confidence in the patient’s ability to abstain from use will support his or her progress. Behavioral interventions such as strategic avoidance of triggers and engagement in alternative activities (eg, support groups, exercise, faith-based practices) will help to maintain abstinence.
A CHRONIC CONDITION
Opioid use disorder, like many chronic illnesses, requires long-term attention to attain successful patient outcomes. The opioid agonists methadone and buprenorphine are the mainstay of treatment for it, conferring benefits such as reducing opioid use and preventing relapse.
Candidates for opioid agonist therapy should undergo a multidisciplinary assessment, including an evaluation on the patient’s readiness to change his or her opioid use.
Patient education should include a discussion of the risks of methadone (eg, respiratory depression, fatal overdose, and QTc prolongation) and buprenorphine (eg hepatotoxicity) and their benefits (eg, controlling craving, decreasing the risk of relapse). Patients should also be educated about overdose and diversion.
Despite the difficulties inherent in treating patients with opioid use disorder, when used appropriately, opioid agonist therapy can be lifesaving for patients struggling with long-term opioid addiction.
Acknowledgment: We thank Katelyn Colosi, BS, and Drs. Susan Wolfe, Dennis Bouffard, and Sinha Shirshendu for their helpful comments.
- Wakeman SE, Pham-Kanter G, Donelan K. Attitudes, practices, and preparedness to care for patients with substance use disorder: results from a survey of general internists. Subst Abus 2016; 37:635–641.
- Samuels EA, Dwyer K, Mello MJ, Baird J, Kellogg AR, Bernstein E. Emergency department-based opioid harm reduction: moving physicians from willing to doing. Acad Emerg Med 2016; 23:455–465.
- Mohlman MK, Tanzman B, Finison K, Pinette M, Jones C. Impact of medication-assisted treatment for opioid addiction on Medicaid expenditures and health services utilization rates in Vermont. J Subst Abuse Treat 2016; 67:9–14.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition. Arlington, VA, American Psychiatric Association, 2013.
- Center for Behavioral Health Statistics and Quality. Behavioral health trends in the United States: results from the 2014 National Survey on Drug Use and Health. www.samhsa.gov/data. Accessed April 6, 2017.
- Compton WM, Jones CM, Baldwin GT. Relationship between nonmedical prescription-opioid use and heroin use. N Engl J Med 2016; 374:154–163.
- Ruan X, Wyche MQ, Kaye AD. Analyzing the relationship between nonmedical prescription-opioid use and heroin use. J Opioid Manage 2016; 12:11–14.
- Hser YI, Evans E, Grella C, Ling W, Anglin D. Long-term course of opioid addiction. Harv Rev Psychiatry 2015; 23:76–89.
- Nielsen S, Hillhouse M, Mooney L, Ang A, Ling W. Buprenorphine pharmacotherapy and behavioral treatment: comparison of outcomes among prescription opioid users, heroin users and combination users. J Subst Abuse Treat 2015; 48:70–76.
- Moore BA, Fiellin DA, Barry DT, et al. Primary care office-based buprenorphine treatment: comparison of heroin and prescription opioid dependent patients. J Gen Intern Med 2007; 22:527–530.
- Fischer B, Patra J, Cruz MF, Gittins J, Rehm J. Comparing heroin users and prescription opioid users in a Canadian multi-site population of illicit opioid users. Drug Alcohol Rev 2008; 27:625–632.
- Compton WM, Jones CM, Baldwin GT. Relationship between nonmedical prescription-opioid use and heroin use. N Engl J Med 2016; 374:154–163.
- Jones CM, Baldwin GT, Manocchio T, White JO, Mack KA. Trends in methadone distribution for pain treatment, methadone diversion, and overdose deaths—United States, 2002–2014. MMWR Morb Mortal Wkly Rep 2016; 65:667–671.
- Baxter LE Sr, Campbell A, Deshields M, et al. Safe methadone induction and stabilization: report of an expert panel. J Addict Med 2013; 7:377–386.
- Alinejad S, Kazemi T, Zamani N, Hoffman RS, Mehrpour O. A systematic review of the cardiotoxicity of methadone. EXCLI J 2015; 14:577–600.
- Johnson RE, Strain EC, Amass L. Buprenorphine: how to use it right. Drug Alcohol Depend 2003; 70(suppl 2):S59–S77.
- Ling W. Buprenorphine implant for opioid addiction. Pain Manage 2012; 2:345–350.
- Saxon AJ, Hser YI, Woody G, Ling W. Medication-assisted treatment for opioid addiction: methadone and buprenorphine. J Food Drug Anal 2013; 21:S69–S72.
- Kimber J, Larney S, Hickman M, Randall D, Degenhardt L. Mortality risk of opioid substitution therapy with methadone versus buprenorphine: a retrospective cohort study. Lancet Psychiatry 2015; 2:901–908.
- Marteau D, McDonald R, Patel K. The relative risk of fatal poisoning by methadone or buprenorphine within the wider population of England and Wales. BMJ Open 2015; 5: e007629.
- Kintz P. Deaths involving buprenorphine: a compendium of French cases. Forensic Sci Int 2001; 121:65–69.
- Zuin M, Giorgini A, Selmi C, et al. Acute liver and renal failure during treatment with buprenorphine at therapeutic dose. Dig Liver Dis 2009; 41:e8–e10.
- Klein JW. Pharmacotherapy for substance use disorders. Med Clin North Am 2016; 100:891–910.
- Modesto-Lowe V, Van Kirk J. Clinical uses of naltrexone: a review of the evidence. Exp Clin Psychopharmocol 2002; 10:213–227.
- Woody GE. Agonist models for treating persons with substance use disorders. Curr Psychiatry Rep 2014; 16:489.
- Sanders JJ, Roose RJ, Lubrano MC, Lucan SC. Meaning and methadone: patient perceptions of methadone dose and a model to promote adherence to maintenance treatment. J Addict Med 2013; 7:307–313.
- Herget G. Methadone and buprenorphine added to the WHO list of essential medicines. HIV/AIDS Policy Law Rev 2005; 10:23–24.
- Suzuki J, Dodds T. Clinical recommendation of 12-step meeting attendance and discussion regarding disclosure of buprenorphine use among patients in office-based opioid treatment. Subst Abus 2016; 37:31–34.
- Rettig RA, Yarmolinsky A. Federal Regulation of Methadone Treatment. Washington, DC: National Academies Press; 1995.
- Srivastava A, Kahan M, Nader M. Primary care management of opioid use disorders: abstinence, methadone, or buprenorphine-naloxone? Can Fam Physician 2017; 63:200–205.
- Substance Abuse and Mental Health Services Administration. Federal Guidelines for Opioid Treatment Programs. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2015.
- Substance Abuse and Mental Health Services Administration SAMSHA. Buprenorphine waiver management. www.samhsa.gov/medication-assisted-treatment/buprenorphine-waiver-management. Accessed April 6, 2017.
- Mark TL, Lubran R, McCance-Kats EF, Chalk M, Richardson J. Medicaid coverage of medications to treat alcohol and opioid dependence. J Subst Abuse Treat 2015; 55:1–5.
- Johnson B, Richert T. Diversion of methadone and buprenorphine from opioid substitution treatment: the importance of patients’ attitudes and norms. J Subst Abuse Treat 2015; 54:50–55.
- Yokell MA, Zaller ND, Green TC, Rich JD. Buprenorphine and buprenorphine/naloxone diversion, misuse, and illicit use: an international review. Curr Drug Abuse Rev 2011; 4:28–41.
- Schuman-Olivier Z, Albanese M, Nelson SE, et al. Self-treatment: illicit buprenorphine use by opioid-dependent treatment seekers. J Subst Abuse Treat 2010; 39:41–50.
- American Society of Addiction Medicine. National practice guidelines for the use of medications in the treatment of addiction involving opioid use. www.asam.org/docs/default-source/practice-support/guidelines-and-consensus-docs/asam-national-practice-guideline-supplement.pdf. Accessed April 6, 2017.
- McNicholas L. Clinical guidelines for the use of buprenorphine in the treatment of opioid addiction. Rockville, MD: US Department of Health and Human Services, Substance Abuse and Mental Health Service Administration; 2004.
- Center for Substance Abuse Treatment. Clinical guidelines for the use of buprenorphine in the treatment of opioid addiction. Rockville (MD): Substance Abuse and Mental Health Services Administration (US); 2004. (Treatment Improvement Protocol (TIP) Series, No. 40.) www.ncbi.nlm.nih.gov/books/NBK64245. Accessed April 6, 2017.
- Zippel-Schultz B, Specka M, Cimander K, et al. Outcomes of patients in long-term opioid maintenance treatment. Subst Use Misuse 2016; 51:1493–1503.
- Martins SS, Keyes KM, Storr CL, Zhu H, Chilcoat HD. Pathways between nonmedical opioid use/dependence and psychiatric disorders: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Drug Alcohol Depend 2009; 103:16–24.
- Khantzian EJ. The self-medication hypothesis of addictive disorders: focus on heroin and cocaine dependence. Am J Psychiatry 1985; 142:1259–1264.
- Belding MA, Iguchi MY, Lamb RJ, Lakin M, Terry R. Stages and processes of change among polydrug users in methadone maintenance treatment. Drug Alcohol Depend 1995; 39:45–53.
- Peteet JR, Brenner S, Curtiss D, Ferrigno M, Kauffman J. A stage of change approach to addiction in the medical setting. Gen Hosp Psychiatry 1998; 20:267–273.
- Vijay A, Bazazi AR, Yee I, Kamarulzaman A, Altice FL. Treatment readiness, attitudes toward, and experiences with methadone and buprenorphine maintenance therapy among people who inject drugs in Malaysia. J Subst Abuse Treat 2015; 54:29–36.
- Wakeman SE, Pham-Kanter G, Donelan K. Attitudes, practices, and preparedness to care for patients with substance use disorder: results from a survey of general internists. Subst Abus 2016; 37:635–641.
- Samuels EA, Dwyer K, Mello MJ, Baird J, Kellogg AR, Bernstein E. Emergency department-based opioid harm reduction: moving physicians from willing to doing. Acad Emerg Med 2016; 23:455–465.
- Mohlman MK, Tanzman B, Finison K, Pinette M, Jones C. Impact of medication-assisted treatment for opioid addiction on Medicaid expenditures and health services utilization rates in Vermont. J Subst Abuse Treat 2016; 67:9–14.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition. Arlington, VA, American Psychiatric Association, 2013.
- Center for Behavioral Health Statistics and Quality. Behavioral health trends in the United States: results from the 2014 National Survey on Drug Use and Health. www.samhsa.gov/data. Accessed April 6, 2017.
- Compton WM, Jones CM, Baldwin GT. Relationship between nonmedical prescription-opioid use and heroin use. N Engl J Med 2016; 374:154–163.
- Ruan X, Wyche MQ, Kaye AD. Analyzing the relationship between nonmedical prescription-opioid use and heroin use. J Opioid Manage 2016; 12:11–14.
- Hser YI, Evans E, Grella C, Ling W, Anglin D. Long-term course of opioid addiction. Harv Rev Psychiatry 2015; 23:76–89.
- Nielsen S, Hillhouse M, Mooney L, Ang A, Ling W. Buprenorphine pharmacotherapy and behavioral treatment: comparison of outcomes among prescription opioid users, heroin users and combination users. J Subst Abuse Treat 2015; 48:70–76.
- Moore BA, Fiellin DA, Barry DT, et al. Primary care office-based buprenorphine treatment: comparison of heroin and prescription opioid dependent patients. J Gen Intern Med 2007; 22:527–530.
- Fischer B, Patra J, Cruz MF, Gittins J, Rehm J. Comparing heroin users and prescription opioid users in a Canadian multi-site population of illicit opioid users. Drug Alcohol Rev 2008; 27:625–632.
- Compton WM, Jones CM, Baldwin GT. Relationship between nonmedical prescription-opioid use and heroin use. N Engl J Med 2016; 374:154–163.
- Jones CM, Baldwin GT, Manocchio T, White JO, Mack KA. Trends in methadone distribution for pain treatment, methadone diversion, and overdose deaths—United States, 2002–2014. MMWR Morb Mortal Wkly Rep 2016; 65:667–671.
- Baxter LE Sr, Campbell A, Deshields M, et al. Safe methadone induction and stabilization: report of an expert panel. J Addict Med 2013; 7:377–386.
- Alinejad S, Kazemi T, Zamani N, Hoffman RS, Mehrpour O. A systematic review of the cardiotoxicity of methadone. EXCLI J 2015; 14:577–600.
- Johnson RE, Strain EC, Amass L. Buprenorphine: how to use it right. Drug Alcohol Depend 2003; 70(suppl 2):S59–S77.
- Ling W. Buprenorphine implant for opioid addiction. Pain Manage 2012; 2:345–350.
- Saxon AJ, Hser YI, Woody G, Ling W. Medication-assisted treatment for opioid addiction: methadone and buprenorphine. J Food Drug Anal 2013; 21:S69–S72.
- Kimber J, Larney S, Hickman M, Randall D, Degenhardt L. Mortality risk of opioid substitution therapy with methadone versus buprenorphine: a retrospective cohort study. Lancet Psychiatry 2015; 2:901–908.
- Marteau D, McDonald R, Patel K. The relative risk of fatal poisoning by methadone or buprenorphine within the wider population of England and Wales. BMJ Open 2015; 5: e007629.
- Kintz P. Deaths involving buprenorphine: a compendium of French cases. Forensic Sci Int 2001; 121:65–69.
- Zuin M, Giorgini A, Selmi C, et al. Acute liver and renal failure during treatment with buprenorphine at therapeutic dose. Dig Liver Dis 2009; 41:e8–e10.
- Klein JW. Pharmacotherapy for substance use disorders. Med Clin North Am 2016; 100:891–910.
- Modesto-Lowe V, Van Kirk J. Clinical uses of naltrexone: a review of the evidence. Exp Clin Psychopharmocol 2002; 10:213–227.
- Woody GE. Agonist models for treating persons with substance use disorders. Curr Psychiatry Rep 2014; 16:489.
- Sanders JJ, Roose RJ, Lubrano MC, Lucan SC. Meaning and methadone: patient perceptions of methadone dose and a model to promote adherence to maintenance treatment. J Addict Med 2013; 7:307–313.
- Herget G. Methadone and buprenorphine added to the WHO list of essential medicines. HIV/AIDS Policy Law Rev 2005; 10:23–24.
- Suzuki J, Dodds T. Clinical recommendation of 12-step meeting attendance and discussion regarding disclosure of buprenorphine use among patients in office-based opioid treatment. Subst Abus 2016; 37:31–34.
- Rettig RA, Yarmolinsky A. Federal Regulation of Methadone Treatment. Washington, DC: National Academies Press; 1995.
- Srivastava A, Kahan M, Nader M. Primary care management of opioid use disorders: abstinence, methadone, or buprenorphine-naloxone? Can Fam Physician 2017; 63:200–205.
- Substance Abuse and Mental Health Services Administration. Federal Guidelines for Opioid Treatment Programs. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2015.
- Substance Abuse and Mental Health Services Administration SAMSHA. Buprenorphine waiver management. www.samhsa.gov/medication-assisted-treatment/buprenorphine-waiver-management. Accessed April 6, 2017.
- Mark TL, Lubran R, McCance-Kats EF, Chalk M, Richardson J. Medicaid coverage of medications to treat alcohol and opioid dependence. J Subst Abuse Treat 2015; 55:1–5.
- Johnson B, Richert T. Diversion of methadone and buprenorphine from opioid substitution treatment: the importance of patients’ attitudes and norms. J Subst Abuse Treat 2015; 54:50–55.
- Yokell MA, Zaller ND, Green TC, Rich JD. Buprenorphine and buprenorphine/naloxone diversion, misuse, and illicit use: an international review. Curr Drug Abuse Rev 2011; 4:28–41.
- Schuman-Olivier Z, Albanese M, Nelson SE, et al. Self-treatment: illicit buprenorphine use by opioid-dependent treatment seekers. J Subst Abuse Treat 2010; 39:41–50.
- American Society of Addiction Medicine. National practice guidelines for the use of medications in the treatment of addiction involving opioid use. www.asam.org/docs/default-source/practice-support/guidelines-and-consensus-docs/asam-national-practice-guideline-supplement.pdf. Accessed April 6, 2017.
- McNicholas L. Clinical guidelines for the use of buprenorphine in the treatment of opioid addiction. Rockville, MD: US Department of Health and Human Services, Substance Abuse and Mental Health Service Administration; 2004.
- Center for Substance Abuse Treatment. Clinical guidelines for the use of buprenorphine in the treatment of opioid addiction. Rockville (MD): Substance Abuse and Mental Health Services Administration (US); 2004. (Treatment Improvement Protocol (TIP) Series, No. 40.) www.ncbi.nlm.nih.gov/books/NBK64245. Accessed April 6, 2017.
- Zippel-Schultz B, Specka M, Cimander K, et al. Outcomes of patients in long-term opioid maintenance treatment. Subst Use Misuse 2016; 51:1493–1503.
- Martins SS, Keyes KM, Storr CL, Zhu H, Chilcoat HD. Pathways between nonmedical opioid use/dependence and psychiatric disorders: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Drug Alcohol Depend 2009; 103:16–24.
- Khantzian EJ. The self-medication hypothesis of addictive disorders: focus on heroin and cocaine dependence. Am J Psychiatry 1985; 142:1259–1264.
- Belding MA, Iguchi MY, Lamb RJ, Lakin M, Terry R. Stages and processes of change among polydrug users in methadone maintenance treatment. Drug Alcohol Depend 1995; 39:45–53.
- Peteet JR, Brenner S, Curtiss D, Ferrigno M, Kauffman J. A stage of change approach to addiction in the medical setting. Gen Hosp Psychiatry 1998; 20:267–273.
- Vijay A, Bazazi AR, Yee I, Kamarulzaman A, Altice FL. Treatment readiness, attitudes toward, and experiences with methadone and buprenorphine maintenance therapy among people who inject drugs in Malaysia. J Subst Abuse Treat 2015; 54:29–36.
KEY POINTS
- Opioid use disorder is potentially lethal and has become more prevalent in the United States over the past few decades.
- The opioid agonist methadone and the partial agonist buprenorphine are the currently recommended treatments for patients who need opioid maintenance therapy. However, they carry the risk of adverse effects (eg, respiratory depression, QTc interval prolongation, hepatotoxicity), diversion, and overdose.
- Patients being considered for opioid agonist therapy need a comprehensive assessment including a thorough medical history and physical examination, psychiatric evaluation, psychosocial appraisal, and determination of readiness to change.
- When methadone and buprenorphine are properly prescribed they confer significant benefits, including reduction or elimination of opioid use, reductions in overdose risk, and positive changes in behavior and lifestyle.
A rational approach to opioid use disorder in primary care
As a medical student, I understood that dealing with death was part of the practice of medicine. I was prepared to help my patients face the end of life from disease and old age and had steeled myself against the inevitable losses I would see from trauma and infection. However, I had no sense of the incredible burden that opioid addiction and death from unintentional overdose would one day cause.
MORE DEATHS FROM OVERDOSE THAN FROM MOTOR VEHICLE ACCIDENTS
To highlight the point, unintentional overdose deaths in 2008 exceeded motor vehicle accidents as the leading cause of accidental death in the United States.1 Since then, the problem has only worsened; by 2014 the US Centers for Disease Control and Prevention reported that 78 Americans were dying each day from unintentional opioid overdose.2
Yet the scourge of deaths from opioid overdose is only the most obvious way that opioid use disorder destroys the lives of patients suffering from addiction, as well as their friends and family. Among many other heartaches, opioid use disorder is associated with severely impaired social function, increased rates of hepatitis C and human immunodeficiency virus (HIV) infection, and serious legal consequences and incarceration.3 Sadly, opioid use disorder has torn apart countless families. Addiction may be a brain disease, but its scope of morbidity extends far beyond the individual with the affliction.
PLENTY OF BLAME TO GO AROUND
To some extent, physicians are culpable in propagating this epidemic, and not just in their obvious role as opioid suppliers. To be certain, opioid overprescribing is a tremendous problem; in 2014, more than 240 million prescriptions for opioids were issued, enough for every American adult to have his or her own bottle of pills.4
However, there is plenty of blame to go around in the medical system for the problems of overprescribing and inappropriate opioid use. Among other factors, medical schools have historically failed to teach young physicians how to treat pain or prescribe opioids safely,5 and pain specialists are often inaccessible to primary care providers.6 Additionally, pharmaceutical companies have been found guilty of marketing opioids to prescribers in misleading ways,7 and well-intentioned but misguided campaigns such as the “pain as a fifth vital sign” movement may have inadvertently contributed to opioid overprescribing as well.8
TACKLING THE CHALLENGE
Prescribers need to tackle these challenges by educating themselves about when and how to prescribe opioids for chronic pain. Breaking the cycle of overprescribing can be achieved by learning to prescribe opioids rationally, cautiously, and as part of a comprehensive multimodal pain management plan with a commitment to risk assessment and harm reduction. It also means having an exit strategy at the start of opioid therapy. This must include recognizing problematic opioid use when it occurs and having options to offer patients when opioid use disorder becomes the primary problem.
Recognizing the problem
Physicians are notoriously poor at predicting and detecting the presence of aberrant drug use behaviors and opioid use disorder. For example, in a study of patients clinicians thought were not at risk for misuse of medications, 60% had urine drug tests showing either the presence of illicit drugs or no evidence of the prescribed drug.9
The prevalence of problematic opioid use in patients on chronic opioid therapy for pain has been variably reported in the literature, but one systematic review found that misuse rates ranged from 21% to 29% (95% confidence interval 13%–38%) and addiction rates averaged 8% to 12% (3%–17%).10 These numbers are alarming, and prescribers need to know how to screen for and diagnose opioid addiction when they see it.
Importantly, there is a wide spectrum of opioid misuse behaviors, and the wise prescriber will thoughtfully consider each circumstance before assuming a patient has a substance use disorder. For example, one patient may skip doses and “hoard” unused pills for fear that he or she will run out of medication during a pain flare, while another may use opioids for nonmedical reasons such as to get high. Both examples represent aberrant drug use, but in the first case patient education may sufficiently address the problem, while the second may herald a more dangerous and less correctable problem.
Responding with empathy
Simply recognizing that a problem exists is not enough. Once we identify problematic opioid use, we also need to know how to address it.
Managing opioid misuse behaviors requires empathy, and prescribers should consider a patient’s motivation and emotive response to counsel. For instance, the patient who skips doses and hoards pills may fear that their well-controlled pain will suddenly worsen if their doctor’s opioid prescribing becomes more restrictive as new guidelines are released.
The lesson is that safe opioid prescribing may require a more restrictive approach than was understood in prior years, but rational prescribing also means careful consideration before arbitrarily tapering or discontinuing opioids in a patient who has demonstrated benefit without evidence of harm, even if new guidelines now recommend against starting opioid therapy for similar pain syndromes. For example, the American College of Physicians released a guideline earlier this year that recommended against opioids to treat low back pain, but it did not recommend stopping opioids if patients were already taking them and benefiting from their use.11
Sometimes the best course of action is to discontinue opioid therapy. This decision may trigger a grief-like reaction in some patients and there can be distinct communication challenges during each coping phase.12 The prescriber should frame opioid prescribing discussions on the changing balance of perceived benefits, risks, and harms; in some cases, the treatment may have “failed” or no longer be appropriate, but the patient may still be suffering from pain. Further, the patient may now need help with a newly recognized substance use disorder and may be particularly vulnerable during this time.
The wrong approach, in my opinion, is to discharge the patient from care because of addiction. This approach may seem justified to the provider who feels betrayed by a patient who has used a prescription differently than intended and has thus placed everyone at risk. However, providers should not take it personally; by definition, a patient with addiction has lost control over use of a drug and may have a stronger relationship with the drug than with you. Instead, we should attempt to intervene to protect a patient’s health and chances of survival. It is critical that physicians learn to leverage treatment resources to provide the support patients need to start the long process of recovery. This may involve detoxification and rehabilitation programs, but in many cases opioid agonist therapy also has a role.
Medication-assisted therapy
Medication-assisted therapy with methadone or buprenorphine can be an extremely important part of this process and is a strategy that Modesto-Lowe et al explore in this issue of the Journal.13 As they point out, patients and providers often misunderstand the use of opioid agonists to treat opioid use disorder; many perceive this as merely substituting one form of addiction for another. However, compelling data support this approach. Studies have shown that opioid agonist therapy is associated with decreased illicit opioid use, better retention in substance use treatment programs, reduced hepatitis C and HIV seroconversion, reduced rates of criminal activity and incarceration, decreased overdose risk, and improved survival.14
Opioid agonists are not a cure-all and come with their own challenges, but for many patients they can “create the space” needed to do the real work of recovery—healing their damaged relationships with themselves, their family, and their society.
Providers need to educate themselves regarding the options available and when and how to use them. They should familiarize themselves with methadone and buprenorphine treatment programs in their community. Better yet, with only 8 hours of additional training, primary care physicians can become waivered to prescribe buprenorphine to treat opioid addiction right in the office. Treating addiction is quickly becoming part of primary care, and clinicians in practice can no longer turn a blind eye toward this problem.
- Miniño AM, Murphy SL, Xu J, Kochanek KD. Deaths: final data for 2008. Natl Vital Stat Rep 2011; 59:1–126.
- Rudd RA, Aleshire N, Zibbell JE, Gladden RM. Increases in drug and opioid overdose deaths—United States, 2000–2014. MMWR Morb Mortal Wkly Rep 2016; 64(50–51):1378–1382.
- Hser YI, Evans E, Grella C, Ling W, Anglin D. Long-term course of opioid addiction. Harv Rev Psychiatry 2015; 23:76–89.
- The opioid epidemic: by the numbers. Department of Health and Human Services; 2016 [updated June 2016.] www.hhs.gov/sites/default/files/Factsheet-opioids-061516.pdf. Accessed April 18, 2017.
- Roehr B. US needs new strategy to help 116 million patients in chronic pain. BMJ 2011; 343:d4206.
- Breuer B, Pappagallo M, Tai JY, Portenoy RK. U.S. board-certified pain physician practices: uniformity and census data of their locations. J Pain 2007; 8:244–250.
- Morreale M. Why is the pendulum swinging? The opiate epidemic in the USA. Acad Psychiatry 2016; 40:839–840.
- Hirsch R. The opioid epidemic: It’s time to place blame where it belongs. KevinMD.com. April 6, 2016. http://www.kevinmd.com/blog/2016/04/the-opioid-epidemic-its-time-to-place-blame-where-it-belongs.html. Accessed April 8, 2017.
- Bronstein K, Passik S, Munitz L, Leider H. Can clinicians accurately predict which patients are misusing their medications? J Pain 2011; 12(suppl):P3. Abstract 111.
- Vowles KE, McEntee ML, Julnes PS, Frohe T, Ney JP, van der Goes DN. Rates of opioid misuse, abuse, and addiction in chronic pain: a systematic review and data synthesis. Pain 2015; 156:569–576.
- Qaseem A, Wilt T, McClean R, Forciea MA. Noninvasive treatments for acute, subacute, and chronic low back pain: a clinical practice guideline from the American College of Physicians. Ann Intern Med 2017; 166:514–530.
- Tobin D, Andrews R, Becker W. Prescribing opioids in primary care: safely starting, monitoring, and stopping. Cleve Clin J Med 2016; 83:207–215.
- Modesto-Lowe V, Sweizbin B, Cheplin M, Hoefer G. Use and misuse of opioid agonists in opioid addiction. Cleve Clin J Med 2017; 84:377–384.
- Nielsen S, Larance B, Degenhardt L, Gowing L, Kehler C, Lintzeris N. Opioid agonist treatment for pharmaceutical opioid dependent people. Cochrane Database Syst Rev 2016(5):CD011117.
As a medical student, I understood that dealing with death was part of the practice of medicine. I was prepared to help my patients face the end of life from disease and old age and had steeled myself against the inevitable losses I would see from trauma and infection. However, I had no sense of the incredible burden that opioid addiction and death from unintentional overdose would one day cause.
MORE DEATHS FROM OVERDOSE THAN FROM MOTOR VEHICLE ACCIDENTS
To highlight the point, unintentional overdose deaths in 2008 exceeded motor vehicle accidents as the leading cause of accidental death in the United States.1 Since then, the problem has only worsened; by 2014 the US Centers for Disease Control and Prevention reported that 78 Americans were dying each day from unintentional opioid overdose.2
Yet the scourge of deaths from opioid overdose is only the most obvious way that opioid use disorder destroys the lives of patients suffering from addiction, as well as their friends and family. Among many other heartaches, opioid use disorder is associated with severely impaired social function, increased rates of hepatitis C and human immunodeficiency virus (HIV) infection, and serious legal consequences and incarceration.3 Sadly, opioid use disorder has torn apart countless families. Addiction may be a brain disease, but its scope of morbidity extends far beyond the individual with the affliction.
PLENTY OF BLAME TO GO AROUND
To some extent, physicians are culpable in propagating this epidemic, and not just in their obvious role as opioid suppliers. To be certain, opioid overprescribing is a tremendous problem; in 2014, more than 240 million prescriptions for opioids were issued, enough for every American adult to have his or her own bottle of pills.4
However, there is plenty of blame to go around in the medical system for the problems of overprescribing and inappropriate opioid use. Among other factors, medical schools have historically failed to teach young physicians how to treat pain or prescribe opioids safely,5 and pain specialists are often inaccessible to primary care providers.6 Additionally, pharmaceutical companies have been found guilty of marketing opioids to prescribers in misleading ways,7 and well-intentioned but misguided campaigns such as the “pain as a fifth vital sign” movement may have inadvertently contributed to opioid overprescribing as well.8
TACKLING THE CHALLENGE
Prescribers need to tackle these challenges by educating themselves about when and how to prescribe opioids for chronic pain. Breaking the cycle of overprescribing can be achieved by learning to prescribe opioids rationally, cautiously, and as part of a comprehensive multimodal pain management plan with a commitment to risk assessment and harm reduction. It also means having an exit strategy at the start of opioid therapy. This must include recognizing problematic opioid use when it occurs and having options to offer patients when opioid use disorder becomes the primary problem.
Recognizing the problem
Physicians are notoriously poor at predicting and detecting the presence of aberrant drug use behaviors and opioid use disorder. For example, in a study of patients clinicians thought were not at risk for misuse of medications, 60% had urine drug tests showing either the presence of illicit drugs or no evidence of the prescribed drug.9
The prevalence of problematic opioid use in patients on chronic opioid therapy for pain has been variably reported in the literature, but one systematic review found that misuse rates ranged from 21% to 29% (95% confidence interval 13%–38%) and addiction rates averaged 8% to 12% (3%–17%).10 These numbers are alarming, and prescribers need to know how to screen for and diagnose opioid addiction when they see it.
Importantly, there is a wide spectrum of opioid misuse behaviors, and the wise prescriber will thoughtfully consider each circumstance before assuming a patient has a substance use disorder. For example, one patient may skip doses and “hoard” unused pills for fear that he or she will run out of medication during a pain flare, while another may use opioids for nonmedical reasons such as to get high. Both examples represent aberrant drug use, but in the first case patient education may sufficiently address the problem, while the second may herald a more dangerous and less correctable problem.
Responding with empathy
Simply recognizing that a problem exists is not enough. Once we identify problematic opioid use, we also need to know how to address it.
Managing opioid misuse behaviors requires empathy, and prescribers should consider a patient’s motivation and emotive response to counsel. For instance, the patient who skips doses and hoards pills may fear that their well-controlled pain will suddenly worsen if their doctor’s opioid prescribing becomes more restrictive as new guidelines are released.
The lesson is that safe opioid prescribing may require a more restrictive approach than was understood in prior years, but rational prescribing also means careful consideration before arbitrarily tapering or discontinuing opioids in a patient who has demonstrated benefit without evidence of harm, even if new guidelines now recommend against starting opioid therapy for similar pain syndromes. For example, the American College of Physicians released a guideline earlier this year that recommended against opioids to treat low back pain, but it did not recommend stopping opioids if patients were already taking them and benefiting from their use.11
Sometimes the best course of action is to discontinue opioid therapy. This decision may trigger a grief-like reaction in some patients and there can be distinct communication challenges during each coping phase.12 The prescriber should frame opioid prescribing discussions on the changing balance of perceived benefits, risks, and harms; in some cases, the treatment may have “failed” or no longer be appropriate, but the patient may still be suffering from pain. Further, the patient may now need help with a newly recognized substance use disorder and may be particularly vulnerable during this time.
The wrong approach, in my opinion, is to discharge the patient from care because of addiction. This approach may seem justified to the provider who feels betrayed by a patient who has used a prescription differently than intended and has thus placed everyone at risk. However, providers should not take it personally; by definition, a patient with addiction has lost control over use of a drug and may have a stronger relationship with the drug than with you. Instead, we should attempt to intervene to protect a patient’s health and chances of survival. It is critical that physicians learn to leverage treatment resources to provide the support patients need to start the long process of recovery. This may involve detoxification and rehabilitation programs, but in many cases opioid agonist therapy also has a role.
Medication-assisted therapy
Medication-assisted therapy with methadone or buprenorphine can be an extremely important part of this process and is a strategy that Modesto-Lowe et al explore in this issue of the Journal.13 As they point out, patients and providers often misunderstand the use of opioid agonists to treat opioid use disorder; many perceive this as merely substituting one form of addiction for another. However, compelling data support this approach. Studies have shown that opioid agonist therapy is associated with decreased illicit opioid use, better retention in substance use treatment programs, reduced hepatitis C and HIV seroconversion, reduced rates of criminal activity and incarceration, decreased overdose risk, and improved survival.14
Opioid agonists are not a cure-all and come with their own challenges, but for many patients they can “create the space” needed to do the real work of recovery—healing their damaged relationships with themselves, their family, and their society.
Providers need to educate themselves regarding the options available and when and how to use them. They should familiarize themselves with methadone and buprenorphine treatment programs in their community. Better yet, with only 8 hours of additional training, primary care physicians can become waivered to prescribe buprenorphine to treat opioid addiction right in the office. Treating addiction is quickly becoming part of primary care, and clinicians in practice can no longer turn a blind eye toward this problem.
As a medical student, I understood that dealing with death was part of the practice of medicine. I was prepared to help my patients face the end of life from disease and old age and had steeled myself against the inevitable losses I would see from trauma and infection. However, I had no sense of the incredible burden that opioid addiction and death from unintentional overdose would one day cause.
MORE DEATHS FROM OVERDOSE THAN FROM MOTOR VEHICLE ACCIDENTS
To highlight the point, unintentional overdose deaths in 2008 exceeded motor vehicle accidents as the leading cause of accidental death in the United States.1 Since then, the problem has only worsened; by 2014 the US Centers for Disease Control and Prevention reported that 78 Americans were dying each day from unintentional opioid overdose.2
Yet the scourge of deaths from opioid overdose is only the most obvious way that opioid use disorder destroys the lives of patients suffering from addiction, as well as their friends and family. Among many other heartaches, opioid use disorder is associated with severely impaired social function, increased rates of hepatitis C and human immunodeficiency virus (HIV) infection, and serious legal consequences and incarceration.3 Sadly, opioid use disorder has torn apart countless families. Addiction may be a brain disease, but its scope of morbidity extends far beyond the individual with the affliction.
PLENTY OF BLAME TO GO AROUND
To some extent, physicians are culpable in propagating this epidemic, and not just in their obvious role as opioid suppliers. To be certain, opioid overprescribing is a tremendous problem; in 2014, more than 240 million prescriptions for opioids were issued, enough for every American adult to have his or her own bottle of pills.4
However, there is plenty of blame to go around in the medical system for the problems of overprescribing and inappropriate opioid use. Among other factors, medical schools have historically failed to teach young physicians how to treat pain or prescribe opioids safely,5 and pain specialists are often inaccessible to primary care providers.6 Additionally, pharmaceutical companies have been found guilty of marketing opioids to prescribers in misleading ways,7 and well-intentioned but misguided campaigns such as the “pain as a fifth vital sign” movement may have inadvertently contributed to opioid overprescribing as well.8
TACKLING THE CHALLENGE
Prescribers need to tackle these challenges by educating themselves about when and how to prescribe opioids for chronic pain. Breaking the cycle of overprescribing can be achieved by learning to prescribe opioids rationally, cautiously, and as part of a comprehensive multimodal pain management plan with a commitment to risk assessment and harm reduction. It also means having an exit strategy at the start of opioid therapy. This must include recognizing problematic opioid use when it occurs and having options to offer patients when opioid use disorder becomes the primary problem.
Recognizing the problem
Physicians are notoriously poor at predicting and detecting the presence of aberrant drug use behaviors and opioid use disorder. For example, in a study of patients clinicians thought were not at risk for misuse of medications, 60% had urine drug tests showing either the presence of illicit drugs or no evidence of the prescribed drug.9
The prevalence of problematic opioid use in patients on chronic opioid therapy for pain has been variably reported in the literature, but one systematic review found that misuse rates ranged from 21% to 29% (95% confidence interval 13%–38%) and addiction rates averaged 8% to 12% (3%–17%).10 These numbers are alarming, and prescribers need to know how to screen for and diagnose opioid addiction when they see it.
Importantly, there is a wide spectrum of opioid misuse behaviors, and the wise prescriber will thoughtfully consider each circumstance before assuming a patient has a substance use disorder. For example, one patient may skip doses and “hoard” unused pills for fear that he or she will run out of medication during a pain flare, while another may use opioids for nonmedical reasons such as to get high. Both examples represent aberrant drug use, but in the first case patient education may sufficiently address the problem, while the second may herald a more dangerous and less correctable problem.
Responding with empathy
Simply recognizing that a problem exists is not enough. Once we identify problematic opioid use, we also need to know how to address it.
Managing opioid misuse behaviors requires empathy, and prescribers should consider a patient’s motivation and emotive response to counsel. For instance, the patient who skips doses and hoards pills may fear that their well-controlled pain will suddenly worsen if their doctor’s opioid prescribing becomes more restrictive as new guidelines are released.
The lesson is that safe opioid prescribing may require a more restrictive approach than was understood in prior years, but rational prescribing also means careful consideration before arbitrarily tapering or discontinuing opioids in a patient who has demonstrated benefit without evidence of harm, even if new guidelines now recommend against starting opioid therapy for similar pain syndromes. For example, the American College of Physicians released a guideline earlier this year that recommended against opioids to treat low back pain, but it did not recommend stopping opioids if patients were already taking them and benefiting from their use.11
Sometimes the best course of action is to discontinue opioid therapy. This decision may trigger a grief-like reaction in some patients and there can be distinct communication challenges during each coping phase.12 The prescriber should frame opioid prescribing discussions on the changing balance of perceived benefits, risks, and harms; in some cases, the treatment may have “failed” or no longer be appropriate, but the patient may still be suffering from pain. Further, the patient may now need help with a newly recognized substance use disorder and may be particularly vulnerable during this time.
The wrong approach, in my opinion, is to discharge the patient from care because of addiction. This approach may seem justified to the provider who feels betrayed by a patient who has used a prescription differently than intended and has thus placed everyone at risk. However, providers should not take it personally; by definition, a patient with addiction has lost control over use of a drug and may have a stronger relationship with the drug than with you. Instead, we should attempt to intervene to protect a patient’s health and chances of survival. It is critical that physicians learn to leverage treatment resources to provide the support patients need to start the long process of recovery. This may involve detoxification and rehabilitation programs, but in many cases opioid agonist therapy also has a role.
Medication-assisted therapy
Medication-assisted therapy with methadone or buprenorphine can be an extremely important part of this process and is a strategy that Modesto-Lowe et al explore in this issue of the Journal.13 As they point out, patients and providers often misunderstand the use of opioid agonists to treat opioid use disorder; many perceive this as merely substituting one form of addiction for another. However, compelling data support this approach. Studies have shown that opioid agonist therapy is associated with decreased illicit opioid use, better retention in substance use treatment programs, reduced hepatitis C and HIV seroconversion, reduced rates of criminal activity and incarceration, decreased overdose risk, and improved survival.14
Opioid agonists are not a cure-all and come with their own challenges, but for many patients they can “create the space” needed to do the real work of recovery—healing their damaged relationships with themselves, their family, and their society.
Providers need to educate themselves regarding the options available and when and how to use them. They should familiarize themselves with methadone and buprenorphine treatment programs in their community. Better yet, with only 8 hours of additional training, primary care physicians can become waivered to prescribe buprenorphine to treat opioid addiction right in the office. Treating addiction is quickly becoming part of primary care, and clinicians in practice can no longer turn a blind eye toward this problem.
- Miniño AM, Murphy SL, Xu J, Kochanek KD. Deaths: final data for 2008. Natl Vital Stat Rep 2011; 59:1–126.
- Rudd RA, Aleshire N, Zibbell JE, Gladden RM. Increases in drug and opioid overdose deaths—United States, 2000–2014. MMWR Morb Mortal Wkly Rep 2016; 64(50–51):1378–1382.
- Hser YI, Evans E, Grella C, Ling W, Anglin D. Long-term course of opioid addiction. Harv Rev Psychiatry 2015; 23:76–89.
- The opioid epidemic: by the numbers. Department of Health and Human Services; 2016 [updated June 2016.] www.hhs.gov/sites/default/files/Factsheet-opioids-061516.pdf. Accessed April 18, 2017.
- Roehr B. US needs new strategy to help 116 million patients in chronic pain. BMJ 2011; 343:d4206.
- Breuer B, Pappagallo M, Tai JY, Portenoy RK. U.S. board-certified pain physician practices: uniformity and census data of their locations. J Pain 2007; 8:244–250.
- Morreale M. Why is the pendulum swinging? The opiate epidemic in the USA. Acad Psychiatry 2016; 40:839–840.
- Hirsch R. The opioid epidemic: It’s time to place blame where it belongs. KevinMD.com. April 6, 2016. http://www.kevinmd.com/blog/2016/04/the-opioid-epidemic-its-time-to-place-blame-where-it-belongs.html. Accessed April 8, 2017.
- Bronstein K, Passik S, Munitz L, Leider H. Can clinicians accurately predict which patients are misusing their medications? J Pain 2011; 12(suppl):P3. Abstract 111.
- Vowles KE, McEntee ML, Julnes PS, Frohe T, Ney JP, van der Goes DN. Rates of opioid misuse, abuse, and addiction in chronic pain: a systematic review and data synthesis. Pain 2015; 156:569–576.
- Qaseem A, Wilt T, McClean R, Forciea MA. Noninvasive treatments for acute, subacute, and chronic low back pain: a clinical practice guideline from the American College of Physicians. Ann Intern Med 2017; 166:514–530.
- Tobin D, Andrews R, Becker W. Prescribing opioids in primary care: safely starting, monitoring, and stopping. Cleve Clin J Med 2016; 83:207–215.
- Modesto-Lowe V, Sweizbin B, Cheplin M, Hoefer G. Use and misuse of opioid agonists in opioid addiction. Cleve Clin J Med 2017; 84:377–384.
- Nielsen S, Larance B, Degenhardt L, Gowing L, Kehler C, Lintzeris N. Opioid agonist treatment for pharmaceutical opioid dependent people. Cochrane Database Syst Rev 2016(5):CD011117.
- Miniño AM, Murphy SL, Xu J, Kochanek KD. Deaths: final data for 2008. Natl Vital Stat Rep 2011; 59:1–126.
- Rudd RA, Aleshire N, Zibbell JE, Gladden RM. Increases in drug and opioid overdose deaths—United States, 2000–2014. MMWR Morb Mortal Wkly Rep 2016; 64(50–51):1378–1382.
- Hser YI, Evans E, Grella C, Ling W, Anglin D. Long-term course of opioid addiction. Harv Rev Psychiatry 2015; 23:76–89.
- The opioid epidemic: by the numbers. Department of Health and Human Services; 2016 [updated June 2016.] www.hhs.gov/sites/default/files/Factsheet-opioids-061516.pdf. Accessed April 18, 2017.
- Roehr B. US needs new strategy to help 116 million patients in chronic pain. BMJ 2011; 343:d4206.
- Breuer B, Pappagallo M, Tai JY, Portenoy RK. U.S. board-certified pain physician practices: uniformity and census data of their locations. J Pain 2007; 8:244–250.
- Morreale M. Why is the pendulum swinging? The opiate epidemic in the USA. Acad Psychiatry 2016; 40:839–840.
- Hirsch R. The opioid epidemic: It’s time to place blame where it belongs. KevinMD.com. April 6, 2016. http://www.kevinmd.com/blog/2016/04/the-opioid-epidemic-its-time-to-place-blame-where-it-belongs.html. Accessed April 8, 2017.
- Bronstein K, Passik S, Munitz L, Leider H. Can clinicians accurately predict which patients are misusing their medications? J Pain 2011; 12(suppl):P3. Abstract 111.
- Vowles KE, McEntee ML, Julnes PS, Frohe T, Ney JP, van der Goes DN. Rates of opioid misuse, abuse, and addiction in chronic pain: a systematic review and data synthesis. Pain 2015; 156:569–576.
- Qaseem A, Wilt T, McClean R, Forciea MA. Noninvasive treatments for acute, subacute, and chronic low back pain: a clinical practice guideline from the American College of Physicians. Ann Intern Med 2017; 166:514–530.
- Tobin D, Andrews R, Becker W. Prescribing opioids in primary care: safely starting, monitoring, and stopping. Cleve Clin J Med 2016; 83:207–215.
- Modesto-Lowe V, Sweizbin B, Cheplin M, Hoefer G. Use and misuse of opioid agonists in opioid addiction. Cleve Clin J Med 2017; 84:377–384.
- Nielsen S, Larance B, Degenhardt L, Gowing L, Kehler C, Lintzeris N. Opioid agonist treatment for pharmaceutical opioid dependent people. Cochrane Database Syst Rev 2016(5):CD011117.

A broken pacemaker lead in a 69-year-old woman
A 69-year-old woman presented with fatigue, cough, and lightheadedness. She had a history of atrial fibrillation and complete heart block, for which she had a pacemaker (dual-pacing, dual-sensing, dual-response, and rate-adaptive mode) inserted in 2005. Her heart rate was 30 beats per minute.
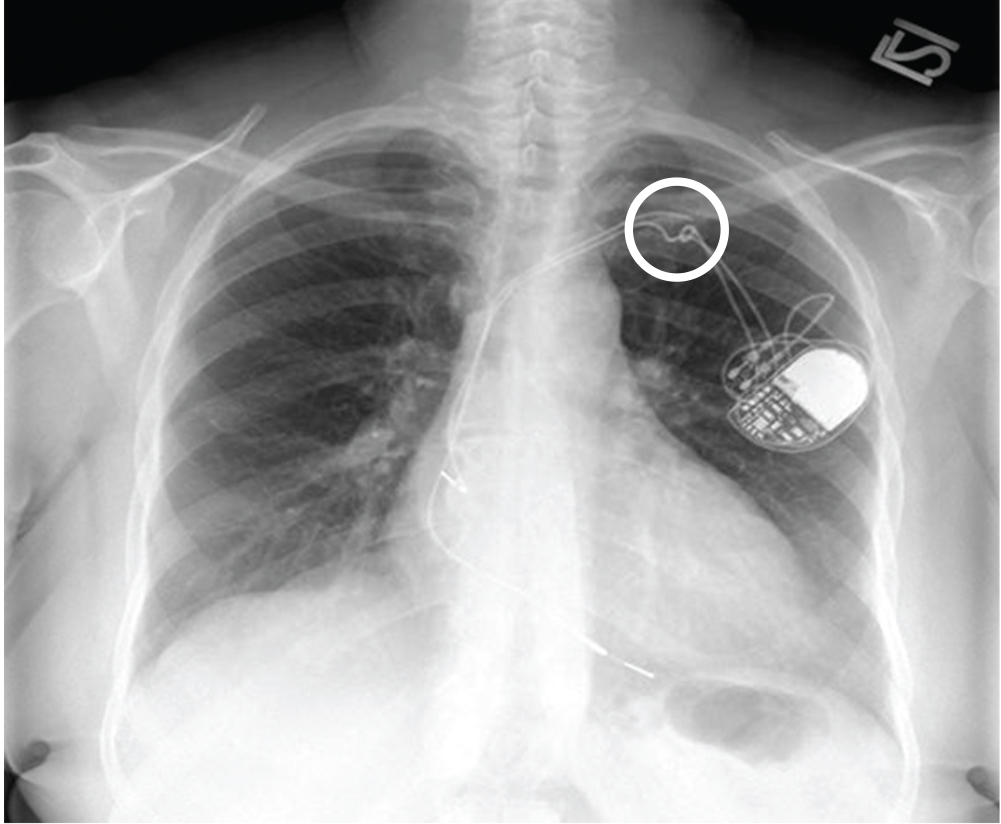
A chest radiograph showed a fractured right ventricular pacemaker lead (Figure 1). Electrocardiography showed sinus rhythm with a high-grade atrioventricular block (Figure 2). Pacemaker interrogation confirmed the diagnosis of lead fracture. A new lead was placed, and the old lead was abandoned.
HOW LEADS BREAK
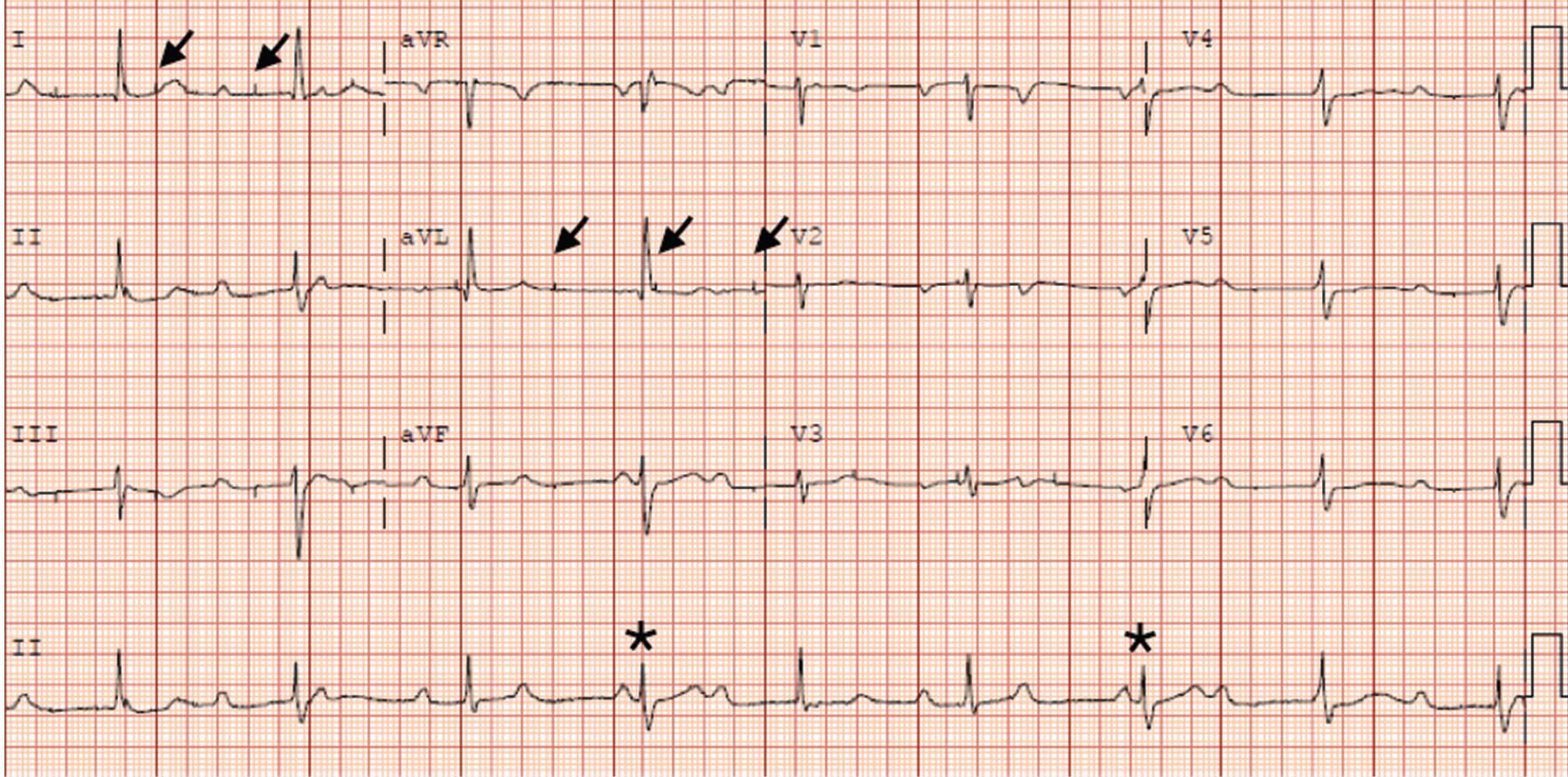
The rate of lead fracture ranges from 0.1% to 4.2% per patient-year, and the annual failure rate increases progressively with time after implantation.1,2
Extrinsic pressure on the lead can eventually break it. This can happen between the first rib and clavicle, in “subclavian crush” injury, or with any anatomical abnormality that narrows the thoracic outlet. Typically, classic subclavian crush results from entrapment of the pacemaker leads by the subclavius muscle or the costoclavicular ligament as the lead follows the needle course of the antecedent access puncture of the subclavian vein. This results in intermittent flexing of the lead and potential lead fracture3 and was likely the cause of lead fracture in our patient.
The risk of fracture is higher in patients under the age of 50, those who perform intense physical activity, women, and patients with greater left ventricular ejection fraction.4,5 Certain leads are prone to fracture due to design flaws. One of these was the Medtronic Sprint Fidelis cardioverter defibrillator lead, which was recalled in 2007.5
DETECTING LEAD FRACTURE
Symptoms of lead fracture vary, depending on the patient’s pacemaker-dependency and on the degree of loss of capture (ie, the degree to which the heart fails to respond to the pacemaker’s signals), and may include lightheadedness, syncope, and extracardiac stimulation.
The electrical integrity of a lead can be tested by measuring the circuit impedance, which normally ranges from 300 to 1,000 ohms.6 An insulation failure results in very low impedance, while a disrupted circuit due to lead fracture commonly causes a sudden rather than gradual increase in impedance.6
Simple imaging studies such as chest radiography or fluoroscopy may establish the diagnosis of lead fracture. One should carefully trace every lead along its entire course and look for any conductor discontinuity, kinks, or sharp bends.6
REMOVE THE OLD LEAD, OR LEAVE IT IN PLACE?
The treatment for lead fracture is usually to put in a new lead, with or without extracting the old one.
In view of the potential complications of lead removal such as cardiac perforation or vascular tear, lead abandonment with placement of a new lead may be performed.7 There are no controlled clinical studies comparing lead abandonment vs lead extraction.8 However, extraction is currently recommended only in patients in whom the old lead causes life-threatening arrhythmias, interferes with the operation of implanted cardiac devices, interferes with radiation therapy or needed surgery, or, due to its design or failure, poses an immediate threat to the patient if left in place.7 Lead removal is reasonable in patients who require specific imaging studies such as magnetic resonance imaging with no available imaging alternative for the diagnosis.7
In our patient, a new lead was placed without removing the fractured lead, with no complications. Afterward, the patient’s heart rhythm was observed to be appropriately paced, and she was discharged home the following day.
- Alt E, Völker R, Blömer H. Lead fracture in pacemaker patients. Thorac Cardiovasc Surg 1987; 35:101–104.
- Kleemann T, Becker T, Doenges K, et al. Annual rate of transvenous defibrillation lead defects in implantable cardioverter-defibrillators over a period of > 10 years. Circulation 2007; 115:2474–2480.
- Magney JE, Flynn DM, Parsons JA, et al. Anatomical mechanisms explaining damage to pacemaker leads, defibrillator leads, and failure of central venous catheters adjacent to the sternoclavicular joint. Pacing Clin Electrophysiol 1993; 16:445–457.
- Farwell D, Green MS, Lemery R, Gollob MH, Birnie DH. Accelerating risk of Fidelis lead fracture. Heart Rhythm 2008; 5:1375–1379.
- Morrison TB, Rea RF, Hodge DO, et al. Risk factors for implantable defibrillator lead fracture in a recalled and a nonrecalled lead. J Cardiovasc Electrophysiol 2010; 21:671–677.
- Swerdlow CD, Ellenbogen KA. Implantable cardioverter-defibrillator leads: design, diagnostics, and management. Circulation 2013; 128:2062–2071.
- Wilkoff BL, Love CJ, Byrd CL, et al; Heart Rhythm Society; American Heart Association. Transvenous lead extraction: Heart Rhythm Society expert consensus on facilities, training, indications, and patient management: this document was endorsed by the American Heart Association (AHA). Heart Rhythm 2009; 6:1085–1104.
- Maytin M, Epstein LM, Henrikson CA. Lead extraction is preferred for lead revisions and system upgrades: when less is more. Circ Arrhythm Electrophysiol 2010; 3:413–424.
A 69-year-old woman presented with fatigue, cough, and lightheadedness. She had a history of atrial fibrillation and complete heart block, for which she had a pacemaker (dual-pacing, dual-sensing, dual-response, and rate-adaptive mode) inserted in 2005. Her heart rate was 30 beats per minute.
A chest radiograph showed a fractured right ventricular pacemaker lead (Figure 1). Electrocardiography showed sinus rhythm with a high-grade atrioventricular block (Figure 2). Pacemaker interrogation confirmed the diagnosis of lead fracture. A new lead was placed, and the old lead was abandoned.
HOW LEADS BREAK
The rate of lead fracture ranges from 0.1% to 4.2% per patient-year, and the annual failure rate increases progressively with time after implantation.1,2
Extrinsic pressure on the lead can eventually break it. This can happen between the first rib and clavicle, in “subclavian crush” injury, or with any anatomical abnormality that narrows the thoracic outlet. Typically, classic subclavian crush results from entrapment of the pacemaker leads by the subclavius muscle or the costoclavicular ligament as the lead follows the needle course of the antecedent access puncture of the subclavian vein. This results in intermittent flexing of the lead and potential lead fracture3 and was likely the cause of lead fracture in our patient.
The risk of fracture is higher in patients under the age of 50, those who perform intense physical activity, women, and patients with greater left ventricular ejection fraction.4,5 Certain leads are prone to fracture due to design flaws. One of these was the Medtronic Sprint Fidelis cardioverter defibrillator lead, which was recalled in 2007.5
DETECTING LEAD FRACTURE
Symptoms of lead fracture vary, depending on the patient’s pacemaker-dependency and on the degree of loss of capture (ie, the degree to which the heart fails to respond to the pacemaker’s signals), and may include lightheadedness, syncope, and extracardiac stimulation.
The electrical integrity of a lead can be tested by measuring the circuit impedance, which normally ranges from 300 to 1,000 ohms.6 An insulation failure results in very low impedance, while a disrupted circuit due to lead fracture commonly causes a sudden rather than gradual increase in impedance.6
Simple imaging studies such as chest radiography or fluoroscopy may establish the diagnosis of lead fracture. One should carefully trace every lead along its entire course and look for any conductor discontinuity, kinks, or sharp bends.6
REMOVE THE OLD LEAD, OR LEAVE IT IN PLACE?
The treatment for lead fracture is usually to put in a new lead, with or without extracting the old one.
In view of the potential complications of lead removal such as cardiac perforation or vascular tear, lead abandonment with placement of a new lead may be performed.7 There are no controlled clinical studies comparing lead abandonment vs lead extraction.8 However, extraction is currently recommended only in patients in whom the old lead causes life-threatening arrhythmias, interferes with the operation of implanted cardiac devices, interferes with radiation therapy or needed surgery, or, due to its design or failure, poses an immediate threat to the patient if left in place.7 Lead removal is reasonable in patients who require specific imaging studies such as magnetic resonance imaging with no available imaging alternative for the diagnosis.7
In our patient, a new lead was placed without removing the fractured lead, with no complications. Afterward, the patient’s heart rhythm was observed to be appropriately paced, and she was discharged home the following day.
A 69-year-old woman presented with fatigue, cough, and lightheadedness. She had a history of atrial fibrillation and complete heart block, for which she had a pacemaker (dual-pacing, dual-sensing, dual-response, and rate-adaptive mode) inserted in 2005. Her heart rate was 30 beats per minute.
A chest radiograph showed a fractured right ventricular pacemaker lead (Figure 1). Electrocardiography showed sinus rhythm with a high-grade atrioventricular block (Figure 2). Pacemaker interrogation confirmed the diagnosis of lead fracture. A new lead was placed, and the old lead was abandoned.
HOW LEADS BREAK
The rate of lead fracture ranges from 0.1% to 4.2% per patient-year, and the annual failure rate increases progressively with time after implantation.1,2
Extrinsic pressure on the lead can eventually break it. This can happen between the first rib and clavicle, in “subclavian crush” injury, or with any anatomical abnormality that narrows the thoracic outlet. Typically, classic subclavian crush results from entrapment of the pacemaker leads by the subclavius muscle or the costoclavicular ligament as the lead follows the needle course of the antecedent access puncture of the subclavian vein. This results in intermittent flexing of the lead and potential lead fracture3 and was likely the cause of lead fracture in our patient.
The risk of fracture is higher in patients under the age of 50, those who perform intense physical activity, women, and patients with greater left ventricular ejection fraction.4,5 Certain leads are prone to fracture due to design flaws. One of these was the Medtronic Sprint Fidelis cardioverter defibrillator lead, which was recalled in 2007.5
DETECTING LEAD FRACTURE
Symptoms of lead fracture vary, depending on the patient’s pacemaker-dependency and on the degree of loss of capture (ie, the degree to which the heart fails to respond to the pacemaker’s signals), and may include lightheadedness, syncope, and extracardiac stimulation.
The electrical integrity of a lead can be tested by measuring the circuit impedance, which normally ranges from 300 to 1,000 ohms.6 An insulation failure results in very low impedance, while a disrupted circuit due to lead fracture commonly causes a sudden rather than gradual increase in impedance.6
Simple imaging studies such as chest radiography or fluoroscopy may establish the diagnosis of lead fracture. One should carefully trace every lead along its entire course and look for any conductor discontinuity, kinks, or sharp bends.6
REMOVE THE OLD LEAD, OR LEAVE IT IN PLACE?
The treatment for lead fracture is usually to put in a new lead, with or without extracting the old one.
In view of the potential complications of lead removal such as cardiac perforation or vascular tear, lead abandonment with placement of a new lead may be performed.7 There are no controlled clinical studies comparing lead abandonment vs lead extraction.8 However, extraction is currently recommended only in patients in whom the old lead causes life-threatening arrhythmias, interferes with the operation of implanted cardiac devices, interferes with radiation therapy or needed surgery, or, due to its design or failure, poses an immediate threat to the patient if left in place.7 Lead removal is reasonable in patients who require specific imaging studies such as magnetic resonance imaging with no available imaging alternative for the diagnosis.7
In our patient, a new lead was placed without removing the fractured lead, with no complications. Afterward, the patient’s heart rhythm was observed to be appropriately paced, and she was discharged home the following day.
- Alt E, Völker R, Blömer H. Lead fracture in pacemaker patients. Thorac Cardiovasc Surg 1987; 35:101–104.
- Kleemann T, Becker T, Doenges K, et al. Annual rate of transvenous defibrillation lead defects in implantable cardioverter-defibrillators over a period of > 10 years. Circulation 2007; 115:2474–2480.
- Magney JE, Flynn DM, Parsons JA, et al. Anatomical mechanisms explaining damage to pacemaker leads, defibrillator leads, and failure of central venous catheters adjacent to the sternoclavicular joint. Pacing Clin Electrophysiol 1993; 16:445–457.
- Farwell D, Green MS, Lemery R, Gollob MH, Birnie DH. Accelerating risk of Fidelis lead fracture. Heart Rhythm 2008; 5:1375–1379.
- Morrison TB, Rea RF, Hodge DO, et al. Risk factors for implantable defibrillator lead fracture in a recalled and a nonrecalled lead. J Cardiovasc Electrophysiol 2010; 21:671–677.
- Swerdlow CD, Ellenbogen KA. Implantable cardioverter-defibrillator leads: design, diagnostics, and management. Circulation 2013; 128:2062–2071.
- Wilkoff BL, Love CJ, Byrd CL, et al; Heart Rhythm Society; American Heart Association. Transvenous lead extraction: Heart Rhythm Society expert consensus on facilities, training, indications, and patient management: this document was endorsed by the American Heart Association (AHA). Heart Rhythm 2009; 6:1085–1104.
- Maytin M, Epstein LM, Henrikson CA. Lead extraction is preferred for lead revisions and system upgrades: when less is more. Circ Arrhythm Electrophysiol 2010; 3:413–424.
- Alt E, Völker R, Blömer H. Lead fracture in pacemaker patients. Thorac Cardiovasc Surg 1987; 35:101–104.
- Kleemann T, Becker T, Doenges K, et al. Annual rate of transvenous defibrillation lead defects in implantable cardioverter-defibrillators over a period of > 10 years. Circulation 2007; 115:2474–2480.
- Magney JE, Flynn DM, Parsons JA, et al. Anatomical mechanisms explaining damage to pacemaker leads, defibrillator leads, and failure of central venous catheters adjacent to the sternoclavicular joint. Pacing Clin Electrophysiol 1993; 16:445–457.
- Farwell D, Green MS, Lemery R, Gollob MH, Birnie DH. Accelerating risk of Fidelis lead fracture. Heart Rhythm 2008; 5:1375–1379.
- Morrison TB, Rea RF, Hodge DO, et al. Risk factors for implantable defibrillator lead fracture in a recalled and a nonrecalled lead. J Cardiovasc Electrophysiol 2010; 21:671–677.
- Swerdlow CD, Ellenbogen KA. Implantable cardioverter-defibrillator leads: design, diagnostics, and management. Circulation 2013; 128:2062–2071.
- Wilkoff BL, Love CJ, Byrd CL, et al; Heart Rhythm Society; American Heart Association. Transvenous lead extraction: Heart Rhythm Society expert consensus on facilities, training, indications, and patient management: this document was endorsed by the American Heart Association (AHA). Heart Rhythm 2009; 6:1085–1104.
- Maytin M, Epstein LM, Henrikson CA. Lead extraction is preferred for lead revisions and system upgrades: when less is more. Circ Arrhythm Electrophysiol 2010; 3:413–424.
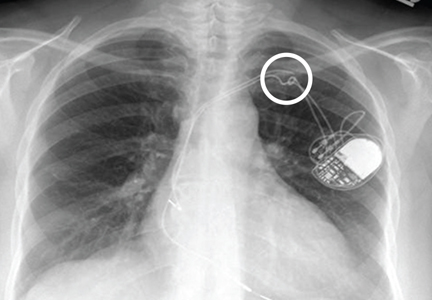
Preventing herpes zoster through vaccination: New developments
Herpes zoster (HZ), or shingles, represents a reactivation of the varicella-zoster virus (VZV). Following primary infection, usually in childhood, the virus typically lies dormant in the dorsal root and sensory nerve ganglia for decades. The precise mechanism of reactivation is not well understood, but it is associated with a decline in cell-mediated immunity that occurs with advancing age, immune-compromising conditions such as HIV infection and cancer, or immunosuppressive therapies, including corticosteroids.1 HZ is usually a self-limited disease characterized by unilateral dermatomal rash and pain, but can cause disseminated infection in immunocompromised individuals.2
Treatment with antiviral medications within 72 hours of rash onset can reduce acute HZ symptoms.1 However, antiviral agents are only minimally effective in preventing postherpetic neuralgia, the most common complication of HZ.3 Therefore, efforts to reduce the burden of HZ morbidity have focused on prevention through vaccination.
Currently, the only shingles vaccine approved by the US Food and Drug Administration (FDA) is Zostavax (Merck), which contains the live-attenuated Oka strain of VZV at a concentration 14 times greater than that of the varicella vaccine (Varivax, Merck). The live-attenuated vaccine boosts VZV-specific cell-mediated immunity, preventing reactivation of the latent virus.
In this article, we describe the burden of disease and review recent developments in the literature on HZ vaccine, including duration of efficacy, uptake and barriers to vaccination, cost-effectiveness, and the outlook for future vaccines.
INCIDENCE INCREASES WITH AGE
The incidence of herpes zoster in the general population is between 3 and 5 per 1,000 person-years4 and increases with age, especially after age 60 when the incidence can approach 13 to 15 per 1,000 person-years.5,6 An estimated 1 million new cases occur each year in the United States, and about 6% of patients experience a second episode of HZ within 8 years.7,8 In immunocompromised patients, the incidence of HZ is 2 to 10 times higher than in the general population.9
The incidence of HZ has been increasing for reasons that are unclear. After varicella vaccine was introduced into the routine childhood immunization schedule in 1995, it was hypothesized that the resultant decrease in primary varicella infections would remove a natural source of immune boosting and cause an increase in HZ incidence for up to 20 years.10 However, recent studies demonstrate that the observed increase in HZ incidence actually predates the introduction of varicella vaccine,11–13 and the widespread use of varicella vaccine has not resulted in an increase in the incidence of HZ.14
Other potential explanations for the rise in reported incidence include increasing awareness among patients, who might previously not have sought care and among physicians, who may be more likely to make the diagnosis. Advertisement of new treatments for HZ, including gabapentin and capsaicin, probably began to increase awareness in the 1990s, as did promotion of the HZ vaccine after its licensure in 2006.
HZ can occur in people who have been vaccinated against varicella due to reactivation of the vaccine-strain virus, but the risk is lower than after infection with wild-type varicella.15 Given that the varicella vaccine has been routinely used in children for only 20 years, the long-term effect of varicella vaccination on the incidence of HZ in elderly people is unknown.
Serious complications
HZ can cause rare but serious complications including encephalitis, herpes ophthalmicus, herpes oticus, myelitis, and retinitis.1 These can lead to long-term disability including unilateral blindness and deafness.
The most common and debilitating complication is postherpetic neuralgia, a persistent pain lasting at least 3 months, with a mean duration of 3.3 years and sometimes as long as 10 years.16 Postherpetic neuralgia occurs in 8% to 32% of patients after acute HZ,4 and the incidence increases with age, being most common after age 70. The chronic pain of postherpetic neuralgia has a significant adverse impact on patients’ quality of life, including physical disability and emotional distress.17 Some pain is intense, and anecdotal reports of patients committing suicide were included in the Advisory Committee on Immunization Practices (ACIP) recommendations regarding herpes zoster vaccine.18
HZ and its complications also impose a substantial economic burden on society.19 In a population-based study, the mean direct medical costs of HZ ranged from $620 to $1,160 (2015 dollars) depending on age,20 and the mean costs of postherpetic neuralgia were 2 to 5 times higher than that.20–22 Immunocompromised patients had costs 2 to 3 times higher than those of immunocompetent adults.23 In addition, for employed patients, HZ resulted in an average loss of 32 hours of work due to absenteeism and 84 hours due to presenteeism (ie, working while sick and therefore suboptimally).24
Assuming there are 1 million cases of HZ each year, if 8% to 32% of patients go on to develop postherpetic neuralgia, that would translate into approximately $1 to $2 billion in direct medical costs. With 60% of adult patients working,25 at an average wage of $23.23 per hour,26 HZ illness could be responsible for another $1.6 billion in lost productivity.
EFFICACY AND SAFETY OF HZ VACCINE
In 2006, the FDA approved the live-attenuated Oka strain VZV vaccine for prevention of HZ and postherpetic neuralgia in adults age 60 and older based on findings from the Shingles Prevention Study (SPS).27
The Shingles Prevention Study
This multicenter randomized placebo-controlled trial27 enrolled 38,546 immunocompetent persons age 60 and older. Subjects in the intervention group received a single dose of live-attenuated vaccine, and all participants were followed for up to 4.9 years after vaccination.
 vaccines")
HZ occurred in 315 (1.636%) of the 19,254 participants in the vaccine group and in 642 (3.336%) of the 19,247 participants in the placebo group, an absolute risk reduction of 1.7%, number needed to treat 59, relative risk reduction 51%, P < .001. Similarly, postherpetic neuralgia occurred in 27 (0.140%) of the 19,254 vaccine recipients and in 80 (0.416%) of the placebo recipients (an absolute risk reduction of 0.276%, number needed to treat 362, relative risk reduction 66%, P < .001). The investigators calculated that vaccination reduced the overall burden of illness by 61% (Table 1).
The efficacy against HZ incidence decreased with age,28 but the efficacy against postherpetic neuralgia did not. In addition, vaccine recipients who developed HZ generally had less severe manifestations.
The safety of the vaccine was assessed for all participants in the SPS. In addition, one-sixth of SPS participants were enrolled in a safety substudy. These participants completed a detailed report card regarding all medically important events within the first 42 days. Forty-eight percent of the vaccine group and 17% of the placebo group (P < .05) experienced adverse events, primarily at the injection site. Less than 1% of all local reactions were severe.29 Serious adverse events were rare (< 2%), but occurred significantly more often in the vaccinated group.
Short-Term Persistence Substudy
Short-term efficacy of the live-attenuated vaccine (up to 7 years) was assessed in the Short-Term Persistence Substudy (STPS), which involved 14,270 of the initial participants and reported yearly and overall vaccine efficacy.30 After 5 years, the yearly efficacy against postherpetic neuralgia incidence declined to 32% and was no longer statistically significant. Efficacy against HZ incidence and burden of illness displayed the same pattern. After the end of the STPS, all subjects in the placebo group received vaccination.
Long-Term Persistence Substudy
Those in the intervention group were followed for an additional 4 years in the Long-Term Persistence Substudy (LTPS).31 Due to the lack of concurrent controls in the LTPS, the authors used regression models based on historical controls to estimate contemporary population incidence of HZ and postherpetic neuralgia for comparison.
Efficacy continued to decline over time, and by 10 years after vaccination there was no difference between vaccinated patients and historical controls in the rate of any end point (ie, efficacy declined to zero).
A trial of booster vaccination
Because many patients are vaccinated at age 60, waning immunity could leave them vulnerable to HZ and postherpetic neuralgia by age 70. A potential solution would be to give a booster dose after 10 years.
A recent phase 3 clinical trial of adults age 70 years and older found that a booster dose of live-attenuated vaccine was as safe and immunogenic as an initial dose.32 While antibody responses were similar in the boosted group and the newly vaccinated group, cell-mediated immunity was higher in the boosted group.
Because prevention of HZ is generally via cell-mediated immunity, the booster might be more effective than the initial vaccination, but clinical trials measuring actual cases prevented will be required to prove it. A booster dose is not currently recommended.
A trial of vaccination in adults 50 to 59
In 2011, the FDA extended its approval of HZ vaccine for use in adults ages 50 to 59.33
In a randomized, double-blind, placebo-controlled trial in this age group,33 the vaccine reduced HZ incidence by almost 70% (absolute risk reduction 0.614%, number needed to treat 156; Table 1), but the severity of HZ cases was not affected. There were too few cases of postherpetic neuralgia to assess the efficacy for this end point. The study followed patients for only 1.5 years after vaccination, so the duration of efficacy is unknown.
As in the older recipients, the vaccine was well tolerated; injection-site reactions and headache were the major adverse effects reported among vaccine recipients.33
INDICATIONS AND CONTRAINDICATIONS
Although HZ vaccine is licensed for use in adults age 50 and older, the ACIP recommends it only for immunocompetent adults age 60 and older. At this time, the ACIP does not recommend HZ vaccine in those younger than 60 because of the low risk of HZ in this age group.34
Any person age 60 or older should receive a single dose of the live-attenuated HZ vaccine subcutaneously, regardless of past history of HZ.
The vaccine is contraindicated in patients who have a history of allergic reaction to any vaccine component, immunosuppression or immunodeficiency conditions, and pregnancy. Specifically, people who will receive immunosuppressive therapies should have the vaccine at least 14 days before beginning treatment. Antiviral medications such as acyclovir, famciclovir, and valacyclovir should be discontinued at least 24 hours before vaccination and not resumed until 14 days later. Patients taking high-dose corticosteroids for more than 2 weeks should not be vaccinated until at least 1 month after therapy is completed.
In contrast, HZ vaccine is not contraindicated for leukemia patients who are in remission and who have not received chemotherapy or radiation for at least 3 months, or for patients receiving short-term, low-to-moderate dose, topical, intra-articular, bursal, or tendon injections of corticosteroids. Patients on low-dose methotrexate, azathioprine, or 6-mercaptopurine can also receive the vaccine.18
VACCINATION RATES ARE LOW

Although the vaccine has been recommended since 2008, uptake has been slow. Figure 1 shows the rate of HZ vaccination in adults age 60 and older surveyed in the National Health Interview Survey from 2007 to 2013.35 Eight years after the vaccine was licensed, only 28% of eligible patients had been vaccinated. Assuming the current rate of increase remains constant, it will take 7 more years to reach a 60% coverage rate—the same as for pneumococcal vaccine36—and 18 years to reach universal coverage.
Barriers to vaccination
Several barriers to HZ vaccination might account for the slow uptake.
For the first few years the vaccine was available, the requirement to store it frozen presented an obstacle for some physicians.37 Physicians may also have been discouraged by the cumbersome Medicare reimbursement process because while the administration fee is covered through Medicare Part B, the live-attenuated vaccine is reimbursed only through Medicare Part D, a benefit that varies by plans. Other barriers to physicians are supply shortages, high up-front costs, and uncertainties regarding the duration of vaccine protection, its safety, and side effects.38–40
Patient barriers include lack of physician recommendation, lack of familiarity with the vaccine, high out-of-pocket costs, the perception that they are at low risk for HZ, underestimation of the pain associated with HZ and postherpetic neuralgia, and fear of vaccine adverse effects.39,41,42
Interventions to increase vaccination rates
Certain interventions have been shown to increase vaccination adherence in general and HZ vaccination in particular. In randomized trials involving other vaccines, electronic medical record reminders supporting panel management or nurse-initiated protocols have been proven to increase vaccination rates, but these methods have not been tested for HZ vaccine specifically.43,44
In an observational study, Chaudhry et al found that the number of HZ vaccinations administered at the Mayo Clinic increased 43% in one practice and 54% in another after the implementation of an electronic alert.45 A randomized controlled trial showed that an informational package discussing HZ and the vaccine sent to patients via either their electronic personal health record or traditional mail increased HZ vaccination by almost 3 times.46
Pharmacists can also influence vaccination rates. States that provide full immunization privileges to pharmacists have vaccination rates significantly higher than states with restricted or no authorization.47
COST-EFFECTIVENESS CONSIDERATIONS
Unlike the Centers for Medicare and Medicaid Services, the ACIP does consider cost-effectiveness in their vaccine recommendations. Because of the morbidity associated with HZ and postherpetic neuralgia as well as the economic impact, vaccination is generally considered cost-effective for adults age 60 and older.48,49
Analyses have demonstrated that cost-effectiveness hinges on 4 factors: initial vaccine efficacy, the duration of efficacy, the age-specific incidence of HZ, and the cost of the vaccine.
For patients ages 50 to 59, the incidence of HZ is low, and because the duration of vaccine efficacy is short even though initial vaccine efficacy is high, vaccination in this age group offers poor value.50 At older ages, the incidence of HZ and postherpetic neuralgia rises, making vaccination more cost-effective. After age 60, the vaccine is cost-effective at all ages, although age 70 appears to offer the optimal trade-off between increasing incidence and declining vaccine efficacy.48,49
For patients who plan to be vaccinated only once, waiting until age 70 would appear to offer the best value.51 For those who are willing to receive a booster dose, the optimal age for vaccination is unknown, but will likely depend on the effectiveness, cost, and duration of the booster.
A NEW HZ VACCINE
In 2015, GlaxoSmithKline tested a new HZ vaccine containing a single VZV glycoprotein in an AS01B adjuvant system (HZ/su vaccine).52 In a phase 3 randomized trial involving 15,411 immunocompetent persons age 50 and older, a 2-dose schedule of HZ/su vaccine was 97% effective in preventing HZ (Table 1).53 Importantly, the vaccine was equally effective in older patients.
This vaccine also had a high rate of adverse reactions, with 17% of vaccine recipients vs 3% of placebo recipients reporting events that prevented normal everyday activities for at least 1 day. However, the rate of serious adverse reactions was the same in both groups (9%). The company announced that they intended to submit a regulatory application for HZ/su vaccine in the second half of 2016.54
Because of its high efficacy, HZ/su vaccine has the potential to change practice, but several issues must be resolved before it can supplant the current vaccine.
First, the AS01B adjuvant is not currently licensed in the United States, so it is unclear if the HZ/su vaccine can get FDA approval.52,55
Second, there are several questions about the efficacy of the vaccine, including long-term efficacy, efficacy in the elderly, and efficacy in the case of a patient receiving only 1 of the 2 required doses.
Third, the impact of HZ/su vaccine on complications such as postherpetic neuralgia has not been established. The clinical trial (NCT01165229) examining vaccine efficacy against postherpetic neuralgia incidence and other complications in adults age 70 and older has recently been completed and data should be available soon. Given the extremely high efficacy against HZ, it is likely that it will be close to 100% effective against this complication.
Fourth, there is uncertainty as to how the HZ/su vaccine should be used in patients who have already received the live-attenuated vaccine, if it is determined that a booster is necessary.
Finally, the vaccine is not yet priced. Given its superior effectiveness, particularly in older individuals, competitive pricing could dramatically affect the market. How Medicare or other insurers cover the new vaccine will likely influence its acceptance.
HZ VACCINATION OF IMMUNOCOMPROMISED PATIENTS
Immunocompromised patients are at highest risk for developing HZ. Unfortunately, there are currently no HZ vaccines approved for use in this population. The current live-attenuated vaccine has been demonstrated to be safe, well tolerated, and immunogenic in patients age 60 and older who are receiving chronic or maintenance low to moderate doses of corticosteroids.56
A clinical trial is being conducted to assess the immunogenicity, clinical effectiveness, and safety of the vaccine in rheumatoid arthritis patients receiving antitumor necrosis factor therapy (NCT01967316). Other trials are examining vaccine efficacy and safety in patients with solid organ tumors prior to chemotherapy (NCT02444936) and in patients who will be undergoing living donor kidney transplantation (NCT00940940). Researchers are also investigating the possibility of vaccinating allogeneic stem cell donors before donation in order to protect transplant recipients against HZ (NCT01573182).
ZVHT and HZ/su vaccination in immunocompromised patients
Heat-treated varicella-zoster vaccine (ZVHT) is a potential alternative for immunocompromised patients. A 4-dose regimen has been proven to reduce the risk of HZ in patients receiving autologous hematopoietic-cell transplants for non-Hodgkin or Hodgkin lymphoma.57
In another trial, the 4-dose ZVHT was safe and elicited significant VZV-specific T-cell response through 28 days in immunosuppressed patients with solid tumor malignancy, hematologic malignancy, human immunodeficiency virus infection with CD4 counts of 200 cells/mm3 or less, and autologous hematopoietic-cell transplants. The T-cell response was poor in allogeneic hematopoietic-cell transplant recipients, however.58
Because the HZ/su vaccine does not contain live virus, it seems particularly promising for immunocompromised patients. In phase 1 and 2 studies, a 3-dose regimen has been shown to be safe and immunogenic in hematopoietic-cell transplant recipients and HIV-infected adults with CD4 count higher than 200 cells/mm3.59,60 A phase 3 trial assessing the efficacy of HZ/su vaccine in autologous hematopoietic-cell transplant recipients is under way (NCT01610414). Changes in recommendations for HZ vaccine in these most vulnerable populations await the results of these studies.
- Dworkin RH, Johnson RW, Breuer J, et al. Recommendations for the management of herpes zoster. Clin Infect Dis 2007; 44(suppl 1):S1–S26.
- Johnson RW. Herpes zoster and postherpetic neuralgia. Expert Rev Vaccines 2010; 9(suppl):21–26.
- Chen N, Li Q, Yang J, Zhou M, Zhou D, He L. Antiviral treatment for preventing postherpetic neuralgia. Cochrane Database Syst Rev 2014; 2:CD006866.
- Kawai K, Gebremeskel BG, Acosta CJ. Systematic review of incidence and complications of herpes zoster: towards a global perspective. BMJ Open 2014; 4:e004833.
- Tseng HF, Smith N, Harpaz R, Bialek SR, Sy LS, Jacobsen SJ. Herpes zoster vaccine in older adults and the risk of subsequent herpes zoster disease. JAMA 2011; 305:160–166.
- Langan SM, Smeeth L, Margolis DJ, Thomas SL. Herpes zoster vaccine effectiveness against incident herpes zoster and post-herpetic neuralgia in an older US population: a cohort study. PLoS Med 2013; 10:e1001420.
- Yawn BP, Saddier P, Wollan PC, St. Sauver JL, Kurland MJ, Sy LS. A population-based study of the incidence and complication rates of herpes zoster before zoster vaccine introduction. Mayo Clin Proc 2007; 82:1341–1349.
- Yawn BP, Wollan PC, Kurland MJ, St. Sauver JL, Saddier P. Herpes zoster recurrences more frequent than previously reported. Mayo Clin Proc 2011; 86:88–93.
- Chen SY, Suaya JA, Li Q, et al. Incidence of herpes zoster in patients with altered immune function. Infection 2014; 42:325–334.
- Edmunds WJ, Brisson M. The effect of vaccination on the epidemiology of varicella zoster virus. J Infect 2002; 44:211–219.
- Hales CM, Harpaz R, Joesoef MR, Bialek SR. Examination of links between herpes zoster incidence and childhood varicella vaccination. Ann Intern Med 2013; 159:739–745.
- Leung J, Harpaz R, Molinari NA, Jumaan A, Zhou F. Herpes zoster incidence among insured persons in the United States, 1993-2006: evaluation of impact of varicella vaccination. Clin Infect Dis 2011; 52:332–340.
- Rimland D, Moanna A. Increasing incidence of herpes zoster among veterans. Clin Infect Dis 2010; 50:1000–1005.
- Jumaan AO, Yu O, Jackson LA, Bohlke K, Galil K, Seward JF. Incidence of herpes zoster, before and after varicella-vaccination-associated decreases in the incidence of varicella, 1992-2002. J Infect Dis 2005; 191:2002–2007.
- Plotkin SA, Starr SE, Connor K, Morton D. Zoster in normal children after varicella vaccine. J Infect Dis 1989; 159:1000–1001.
- Oster G, Harding G, Dukes E, Edelsberg J, Cleary PD. Pain, medication use, and health-related quality of life in older persons with postherpetic neuralgia: results from a population-based survey. J Pain 2005; 6:356–363.
- Johnson RW, Bouhassira D, Kassianos G, Leplege A, Schmader KE, Weinke T. The impact of herpes zoster and post-herpetic neuralgia on quality-of-life. BMC Med 2010; 8:37.
- Harpaz R, Ortega-Sanchez IR, Seward JF; Advisory Committee on Immunization Practices (ACIP) Centers for Disease Control and Prevention (CDC). Prevention of herpes zoster: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2008; 57:1–30.
- Panatto D, Bragazzi NL, Rizzitelli E, et al. Evaluation of the economic burden of herpes zoster (HZ) infection. Hum Vaccin Immunother 2015; 11:245–262.
- Yawn BP, Itzler RF, Wollan PC, Pellissier JM, Sy LS, Saddier P. Health care utilization and cost burden of herpes zoster in a community population. Mayo Clin Proc 2009; 84:787–794.
- Dworkin RH, White R, O’Connor AB, Hawkins K. Health care expenditure burden of persisting herpes zoster pain. Pain Med 2008; 9:348–353.
- White RR, Lenhart G, Singhal PK, et al. Incremental 1-year medical resource utilization and costs for patients with herpes zoster from a set of US health plans. Pharmacoeconomics 2009; 27:781–792.
- Insinga RP, Itzler RF, Pellissier JM. Acute/subacute herpes zoster: healthcare resource utilisation and costs in a group of US health plans. Pharmacoeconomics 2007; 25:155–169.
- Singhal PK, Makin C, Pellissier J, Sy L, White R, Saddier P. Work and productivity loss related to herpes zoster. J Med Econ 2011; 14:639–645.
- US Bureau of Labor Statistics. Labor force statistics from the current population survey. www.bls.gov/web/empsit/cpseea13.htm. Accessed April 6, 2017.
- US Bureau of Labor Statistic. Occupational employment statistics. www.bls.gov/oes/current/oes_nat.htm. Accessed April 6, 2017.
- Oxman MN, Levin MJ, Johnson GR, et al; Shingles Prevention Study Group. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N Engl J Med 2005; 352:2271–2284.
- Food and Drug Administration (FDA). FDA clinical briefing document for Merck & Co., Inc. Zoster vaccine live (Oka/Merck) Zostavax. www.fda.gov/ohrms/dockets/ac/05/briefing/5-4198b2_1.pdf. Accessed April 6, 2017.
- Simberkoff MS, Arbeit RD, Johnson GR, et al; Shingles Prevention Study Group. Safety of herpes zoster vaccine in the shingles prevention study: a randomized trial. Ann Intern Med 2010; 152:545–554.
- Schmader KE, Oxman MN, Levin MJ, et al; Shingles Prevention Study Group. Persistence of the efficacy of zoster vaccine in the shingles prevention study and the short-term persistence substudy. Clin Infect Dis 2012; 55:1320–1328.
- Morrison VA, Johnson GR, Schmader KE, et al; Shingles Prevention Study Group. Long-term persistence of zoster vaccine efficacy. Clin Infect Dis 2015; 60:900–909.
- Levin MJ, Schmader KE, Pang L, et al. Cellular and humoral responses to a second dose of herpes zoster vaccine administered 10 years after the first dose among older adults. J Infect Dis 2016; 213:14–22.
- Schmader KE, Levin MJ, Gnann JW Jr, et al. Efficacy, safety, and tolerability of herpes zoster vaccine in persons aged 50-59 years. Clin Infect Dis 2012; 54:922–928.
- Hales CM, Harpaz R, Ortega-Sanchez I, Bialek SR; Centers for Disease Control and Prevention (CDC). Update on recommendations for use of herpes zoster vaccine. MMWR Morb Mortal Wkly Rep 2014; 63:729–731.
- Centers for Disease Control and Prevention (CDC). Surveillance of vaccination coverage among adult populations—United States, 2014. MMWR Morb Mortal Wkly Rep 2016; 65(1):1–36. Accessed April 12, 2017.
- Williams WW, Lu PJ, O’Halloran A, et al; Centers for Disease Control and Prevention (CDC). Vaccination coverage among adults, excluding influenza vaccination—United States, 2013. MMWR Morb Mortal Wkly Rep 2015; 64:95–102.
- Oxman MN. Zoster vaccine: current status and future prospects. Clin Infect Dis 2010; 51:197–213.
- Hurley LP, Lindley MC, Harpaz R, et al. Barriers to the use of herpes zoster vaccine. Ann Intern Med 2010; 152:555–560.
- Lu PJ, Euler GL, Jumaan AO, Harpaz R. Herpes zoster vaccination among adults aged 60 years or older in the United States, 2007: uptake of the first new vaccine to target seniors. Vaccine 2009; 27:882–887.
- Hurley LP, Harpaz R, Daley MF, et al. National survey of primary care physicians regarding herpes zoster and the herpes zoster vaccine. J Infect Dis 2008; 197(suppl 2):S216–S223.
- Joon Lee T, Hayes S, Cummings DM, et al. Herpes zoster knowledge, prevalence, and vaccination rate by race. J Am Board Fam Med 2013; 26:45–51.
- Opstelten W, van Essen GA, Hak E. Determinants of non-compliance with herpes zoster vaccination in the community-dwelling elderly. Vaccine 2009; 27:192–196.
- Loo TS, Davis RB, Lipsitz LA, et al. Electronic medical record reminders and panel management to improve primary care of elderly patients. Arch Intern Med 2011; 171:1552–1558.
- Rhew DC, Glassman PA, Goetz MB. Improving pneumococcal vaccine rates. Nurse protocols versus clinical reminders. J Gen Intern Med 1999; 14:351–356.
- Chaudhry R, Schietel SM, North F, Dejesus R, Kesman RL, Stroebel RJ. Improving rates of herpes zoster vaccination with a clinical decision support system in a primary care practice. J Eval Clin Pract 2013; 19:263–266.
- Otsuka SH, Tayal NH, Porter K, Embi PJ, Beatty SJ. Improving herpes zoster vaccination rates through use of a clinical pharmacist and a personal health record. Am J Med 2013; 126:832.e1–832.e6.
- Taitel MS, Fensterheim LE, Cannon AE, Cohen ES. Improving pneumococcal and herpes zoster vaccination uptake: expanding pharmacist privileges. Am J Manag Care 2013; 19:e309–e313.
- Kawai K, Preaud E, Baron-Papillon F, Largeron N, Acosta CJ. Cost-effectiveness of vaccination against herpes zoster and postherpetic neuralgia: a critical review. Vaccine 2014; 32:1645–1653.
- Szucs TD, Pfeil AM. A systematic review of the cost effectiveness of herpes zoster vaccination. Pharmacoeconomics 2013; 31:125–136.
- Le P, Rothberg MB. Cost-effectiveness of herpes zoster vaccine for persons aged 50 years. Ann Intern Med 2015; 163:489–497.
- Le P, Rothberg MB. Determining the optimal age to vaccinate against herpes zoster: a cost-effectiveness analysis. Society for Medical Decision Making 37th Annual North American Meeting. St. Louis, MO; October 18-21, 2015.
- Cohen JI. Clinical practice: herpes zoster. N Engl J Med 2013; 369:255–263.
- Lal H, Cunningham AL, Godeaux O, et al; ZOE-50 Study Group. Efficacy of an adjuvanted herpes zoster subunit vaccine in older adults. N Engl J Med 2015; 372:2087–2096.
- GlaxoSmithKline plc. GSK’s candidate shingles vaccine demonstrates 90% efficacy against shingles in people 70 years of age and over. www.gsk.com/en-gb/media/press-releases/gsk-s-candidate-shingles-vaccine-demonstrates-90-efficacy-against-shingles-in-people-70-years-of-age-and-over/. Accessed April 6, 2017.
- Reed SG, Orr MT, Fox CB. Key roles of adjuvants in modern vaccines. Nat Med 2013; 19:1597–1608.
- Russell AF, Parrino J, Fisher CL Jr, et al. Safety, tolerability, and immunogenicity of zoster vaccine in subjects on chronic/maintenance corticosteroids. Vaccine 2015; 33:3129–3134.
- Hata A, Asanuma H, Rinki M, et al. Use of an inactivated varicella vaccine in recipients of hematopoietic-cell transplants. N Engl J Med 2002; 347:26–34.
- Mullane KM, Winston DJ, Wertheim MS, et al. Safety and immunogenicity of heat-treated zoster vaccine (ZVHT) in immunocompromised adults. J Infect Dis 2013; 208:1375–1385.
- Stadtmauer EA, Sullivan KM, Marty FM, et al. A phase 1/2 study of an adjuvanted varicella-zoster virus subunit vaccine in autologous hematopoietic cell transplant recipients. Blood 2014; 124:2921–2929.
- Berkowitz EM, Moyle G, Stellbrink HJ, et al. Safety and immunogenicity of an adjuvanted herpes zoster subunit candidate vaccine in HIV-infected adults: a phase 1/2a randomized, placebo-controlled study. J Infect Dis 2015; 211:1279–1287.
Herpes zoster (HZ), or shingles, represents a reactivation of the varicella-zoster virus (VZV). Following primary infection, usually in childhood, the virus typically lies dormant in the dorsal root and sensory nerve ganglia for decades. The precise mechanism of reactivation is not well understood, but it is associated with a decline in cell-mediated immunity that occurs with advancing age, immune-compromising conditions such as HIV infection and cancer, or immunosuppressive therapies, including corticosteroids.1 HZ is usually a self-limited disease characterized by unilateral dermatomal rash and pain, but can cause disseminated infection in immunocompromised individuals.2
Treatment with antiviral medications within 72 hours of rash onset can reduce acute HZ symptoms.1 However, antiviral agents are only minimally effective in preventing postherpetic neuralgia, the most common complication of HZ.3 Therefore, efforts to reduce the burden of HZ morbidity have focused on prevention through vaccination.
Currently, the only shingles vaccine approved by the US Food and Drug Administration (FDA) is Zostavax (Merck), which contains the live-attenuated Oka strain of VZV at a concentration 14 times greater than that of the varicella vaccine (Varivax, Merck). The live-attenuated vaccine boosts VZV-specific cell-mediated immunity, preventing reactivation of the latent virus.
In this article, we describe the burden of disease and review recent developments in the literature on HZ vaccine, including duration of efficacy, uptake and barriers to vaccination, cost-effectiveness, and the outlook for future vaccines.
INCIDENCE INCREASES WITH AGE
The incidence of herpes zoster in the general population is between 3 and 5 per 1,000 person-years4 and increases with age, especially after age 60 when the incidence can approach 13 to 15 per 1,000 person-years.5,6 An estimated 1 million new cases occur each year in the United States, and about 6% of patients experience a second episode of HZ within 8 years.7,8 In immunocompromised patients, the incidence of HZ is 2 to 10 times higher than in the general population.9
The incidence of HZ has been increasing for reasons that are unclear. After varicella vaccine was introduced into the routine childhood immunization schedule in 1995, it was hypothesized that the resultant decrease in primary varicella infections would remove a natural source of immune boosting and cause an increase in HZ incidence for up to 20 years.10 However, recent studies demonstrate that the observed increase in HZ incidence actually predates the introduction of varicella vaccine,11–13 and the widespread use of varicella vaccine has not resulted in an increase in the incidence of HZ.14
Other potential explanations for the rise in reported incidence include increasing awareness among patients, who might previously not have sought care and among physicians, who may be more likely to make the diagnosis. Advertisement of new treatments for HZ, including gabapentin and capsaicin, probably began to increase awareness in the 1990s, as did promotion of the HZ vaccine after its licensure in 2006.
HZ can occur in people who have been vaccinated against varicella due to reactivation of the vaccine-strain virus, but the risk is lower than after infection with wild-type varicella.15 Given that the varicella vaccine has been routinely used in children for only 20 years, the long-term effect of varicella vaccination on the incidence of HZ in elderly people is unknown.
Serious complications
HZ can cause rare but serious complications including encephalitis, herpes ophthalmicus, herpes oticus, myelitis, and retinitis.1 These can lead to long-term disability including unilateral blindness and deafness.
The most common and debilitating complication is postherpetic neuralgia, a persistent pain lasting at least 3 months, with a mean duration of 3.3 years and sometimes as long as 10 years.16 Postherpetic neuralgia occurs in 8% to 32% of patients after acute HZ,4 and the incidence increases with age, being most common after age 70. The chronic pain of postherpetic neuralgia has a significant adverse impact on patients’ quality of life, including physical disability and emotional distress.17 Some pain is intense, and anecdotal reports of patients committing suicide were included in the Advisory Committee on Immunization Practices (ACIP) recommendations regarding herpes zoster vaccine.18
HZ and its complications also impose a substantial economic burden on society.19 In a population-based study, the mean direct medical costs of HZ ranged from $620 to $1,160 (2015 dollars) depending on age,20 and the mean costs of postherpetic neuralgia were 2 to 5 times higher than that.20–22 Immunocompromised patients had costs 2 to 3 times higher than those of immunocompetent adults.23 In addition, for employed patients, HZ resulted in an average loss of 32 hours of work due to absenteeism and 84 hours due to presenteeism (ie, working while sick and therefore suboptimally).24
Assuming there are 1 million cases of HZ each year, if 8% to 32% of patients go on to develop postherpetic neuralgia, that would translate into approximately $1 to $2 billion in direct medical costs. With 60% of adult patients working,25 at an average wage of $23.23 per hour,26 HZ illness could be responsible for another $1.6 billion in lost productivity.
EFFICACY AND SAFETY OF HZ VACCINE
In 2006, the FDA approved the live-attenuated Oka strain VZV vaccine for prevention of HZ and postherpetic neuralgia in adults age 60 and older based on findings from the Shingles Prevention Study (SPS).27
The Shingles Prevention Study
This multicenter randomized placebo-controlled trial27 enrolled 38,546 immunocompetent persons age 60 and older. Subjects in the intervention group received a single dose of live-attenuated vaccine, and all participants were followed for up to 4.9 years after vaccination.
HZ occurred in 315 (1.636%) of the 19,254 participants in the vaccine group and in 642 (3.336%) of the 19,247 participants in the placebo group, an absolute risk reduction of 1.7%, number needed to treat 59, relative risk reduction 51%, P < .001. Similarly, postherpetic neuralgia occurred in 27 (0.140%) of the 19,254 vaccine recipients and in 80 (0.416%) of the placebo recipients (an absolute risk reduction of 0.276%, number needed to treat 362, relative risk reduction 66%, P < .001). The investigators calculated that vaccination reduced the overall burden of illness by 61% (Table 1).
The efficacy against HZ incidence decreased with age,28 but the efficacy against postherpetic neuralgia did not. In addition, vaccine recipients who developed HZ generally had less severe manifestations.
The safety of the vaccine was assessed for all participants in the SPS. In addition, one-sixth of SPS participants were enrolled in a safety substudy. These participants completed a detailed report card regarding all medically important events within the first 42 days. Forty-eight percent of the vaccine group and 17% of the placebo group (P < .05) experienced adverse events, primarily at the injection site. Less than 1% of all local reactions were severe.29 Serious adverse events were rare (< 2%), but occurred significantly more often in the vaccinated group.
Short-Term Persistence Substudy
Short-term efficacy of the live-attenuated vaccine (up to 7 years) was assessed in the Short-Term Persistence Substudy (STPS), which involved 14,270 of the initial participants and reported yearly and overall vaccine efficacy.30 After 5 years, the yearly efficacy against postherpetic neuralgia incidence declined to 32% and was no longer statistically significant. Efficacy against HZ incidence and burden of illness displayed the same pattern. After the end of the STPS, all subjects in the placebo group received vaccination.
Long-Term Persistence Substudy
Those in the intervention group were followed for an additional 4 years in the Long-Term Persistence Substudy (LTPS).31 Due to the lack of concurrent controls in the LTPS, the authors used regression models based on historical controls to estimate contemporary population incidence of HZ and postherpetic neuralgia for comparison.
Efficacy continued to decline over time, and by 10 years after vaccination there was no difference between vaccinated patients and historical controls in the rate of any end point (ie, efficacy declined to zero).
A trial of booster vaccination
Because many patients are vaccinated at age 60, waning immunity could leave them vulnerable to HZ and postherpetic neuralgia by age 70. A potential solution would be to give a booster dose after 10 years.
A recent phase 3 clinical trial of adults age 70 years and older found that a booster dose of live-attenuated vaccine was as safe and immunogenic as an initial dose.32 While antibody responses were similar in the boosted group and the newly vaccinated group, cell-mediated immunity was higher in the boosted group.
Because prevention of HZ is generally via cell-mediated immunity, the booster might be more effective than the initial vaccination, but clinical trials measuring actual cases prevented will be required to prove it. A booster dose is not currently recommended.
A trial of vaccination in adults 50 to 59
In 2011, the FDA extended its approval of HZ vaccine for use in adults ages 50 to 59.33
In a randomized, double-blind, placebo-controlled trial in this age group,33 the vaccine reduced HZ incidence by almost 70% (absolute risk reduction 0.614%, number needed to treat 156; Table 1), but the severity of HZ cases was not affected. There were too few cases of postherpetic neuralgia to assess the efficacy for this end point. The study followed patients for only 1.5 years after vaccination, so the duration of efficacy is unknown.
As in the older recipients, the vaccine was well tolerated; injection-site reactions and headache were the major adverse effects reported among vaccine recipients.33
INDICATIONS AND CONTRAINDICATIONS
Although HZ vaccine is licensed for use in adults age 50 and older, the ACIP recommends it only for immunocompetent adults age 60 and older. At this time, the ACIP does not recommend HZ vaccine in those younger than 60 because of the low risk of HZ in this age group.34
Any person age 60 or older should receive a single dose of the live-attenuated HZ vaccine subcutaneously, regardless of past history of HZ.
The vaccine is contraindicated in patients who have a history of allergic reaction to any vaccine component, immunosuppression or immunodeficiency conditions, and pregnancy. Specifically, people who will receive immunosuppressive therapies should have the vaccine at least 14 days before beginning treatment. Antiviral medications such as acyclovir, famciclovir, and valacyclovir should be discontinued at least 24 hours before vaccination and not resumed until 14 days later. Patients taking high-dose corticosteroids for more than 2 weeks should not be vaccinated until at least 1 month after therapy is completed.
In contrast, HZ vaccine is not contraindicated for leukemia patients who are in remission and who have not received chemotherapy or radiation for at least 3 months, or for patients receiving short-term, low-to-moderate dose, topical, intra-articular, bursal, or tendon injections of corticosteroids. Patients on low-dose methotrexate, azathioprine, or 6-mercaptopurine can also receive the vaccine.18
VACCINATION RATES ARE LOW
Although the vaccine has been recommended since 2008, uptake has been slow. Figure 1 shows the rate of HZ vaccination in adults age 60 and older surveyed in the National Health Interview Survey from 2007 to 2013.35 Eight years after the vaccine was licensed, only 28% of eligible patients had been vaccinated. Assuming the current rate of increase remains constant, it will take 7 more years to reach a 60% coverage rate—the same as for pneumococcal vaccine36—and 18 years to reach universal coverage.
Barriers to vaccination
Several barriers to HZ vaccination might account for the slow uptake.
For the first few years the vaccine was available, the requirement to store it frozen presented an obstacle for some physicians.37 Physicians may also have been discouraged by the cumbersome Medicare reimbursement process because while the administration fee is covered through Medicare Part B, the live-attenuated vaccine is reimbursed only through Medicare Part D, a benefit that varies by plans. Other barriers to physicians are supply shortages, high up-front costs, and uncertainties regarding the duration of vaccine protection, its safety, and side effects.38–40
Patient barriers include lack of physician recommendation, lack of familiarity with the vaccine, high out-of-pocket costs, the perception that they are at low risk for HZ, underestimation of the pain associated with HZ and postherpetic neuralgia, and fear of vaccine adverse effects.39,41,42
Interventions to increase vaccination rates
Certain interventions have been shown to increase vaccination adherence in general and HZ vaccination in particular. In randomized trials involving other vaccines, electronic medical record reminders supporting panel management or nurse-initiated protocols have been proven to increase vaccination rates, but these methods have not been tested for HZ vaccine specifically.43,44
In an observational study, Chaudhry et al found that the number of HZ vaccinations administered at the Mayo Clinic increased 43% in one practice and 54% in another after the implementation of an electronic alert.45 A randomized controlled trial showed that an informational package discussing HZ and the vaccine sent to patients via either their electronic personal health record or traditional mail increased HZ vaccination by almost 3 times.46
Pharmacists can also influence vaccination rates. States that provide full immunization privileges to pharmacists have vaccination rates significantly higher than states with restricted or no authorization.47
COST-EFFECTIVENESS CONSIDERATIONS
Unlike the Centers for Medicare and Medicaid Services, the ACIP does consider cost-effectiveness in their vaccine recommendations. Because of the morbidity associated with HZ and postherpetic neuralgia as well as the economic impact, vaccination is generally considered cost-effective for adults age 60 and older.48,49
Analyses have demonstrated that cost-effectiveness hinges on 4 factors: initial vaccine efficacy, the duration of efficacy, the age-specific incidence of HZ, and the cost of the vaccine.
For patients ages 50 to 59, the incidence of HZ is low, and because the duration of vaccine efficacy is short even though initial vaccine efficacy is high, vaccination in this age group offers poor value.50 At older ages, the incidence of HZ and postherpetic neuralgia rises, making vaccination more cost-effective. After age 60, the vaccine is cost-effective at all ages, although age 70 appears to offer the optimal trade-off between increasing incidence and declining vaccine efficacy.48,49
For patients who plan to be vaccinated only once, waiting until age 70 would appear to offer the best value.51 For those who are willing to receive a booster dose, the optimal age for vaccination is unknown, but will likely depend on the effectiveness, cost, and duration of the booster.
A NEW HZ VACCINE
In 2015, GlaxoSmithKline tested a new HZ vaccine containing a single VZV glycoprotein in an AS01B adjuvant system (HZ/su vaccine).52 In a phase 3 randomized trial involving 15,411 immunocompetent persons age 50 and older, a 2-dose schedule of HZ/su vaccine was 97% effective in preventing HZ (Table 1).53 Importantly, the vaccine was equally effective in older patients.
This vaccine also had a high rate of adverse reactions, with 17% of vaccine recipients vs 3% of placebo recipients reporting events that prevented normal everyday activities for at least 1 day. However, the rate of serious adverse reactions was the same in both groups (9%). The company announced that they intended to submit a regulatory application for HZ/su vaccine in the second half of 2016.54
Because of its high efficacy, HZ/su vaccine has the potential to change practice, but several issues must be resolved before it can supplant the current vaccine.
First, the AS01B adjuvant is not currently licensed in the United States, so it is unclear if the HZ/su vaccine can get FDA approval.52,55
Second, there are several questions about the efficacy of the vaccine, including long-term efficacy, efficacy in the elderly, and efficacy in the case of a patient receiving only 1 of the 2 required doses.
Third, the impact of HZ/su vaccine on complications such as postherpetic neuralgia has not been established. The clinical trial (NCT01165229) examining vaccine efficacy against postherpetic neuralgia incidence and other complications in adults age 70 and older has recently been completed and data should be available soon. Given the extremely high efficacy against HZ, it is likely that it will be close to 100% effective against this complication.
Fourth, there is uncertainty as to how the HZ/su vaccine should be used in patients who have already received the live-attenuated vaccine, if it is determined that a booster is necessary.
Finally, the vaccine is not yet priced. Given its superior effectiveness, particularly in older individuals, competitive pricing could dramatically affect the market. How Medicare or other insurers cover the new vaccine will likely influence its acceptance.
HZ VACCINATION OF IMMUNOCOMPROMISED PATIENTS
Immunocompromised patients are at highest risk for developing HZ. Unfortunately, there are currently no HZ vaccines approved for use in this population. The current live-attenuated vaccine has been demonstrated to be safe, well tolerated, and immunogenic in patients age 60 and older who are receiving chronic or maintenance low to moderate doses of corticosteroids.56
A clinical trial is being conducted to assess the immunogenicity, clinical effectiveness, and safety of the vaccine in rheumatoid arthritis patients receiving antitumor necrosis factor therapy (NCT01967316). Other trials are examining vaccine efficacy and safety in patients with solid organ tumors prior to chemotherapy (NCT02444936) and in patients who will be undergoing living donor kidney transplantation (NCT00940940). Researchers are also investigating the possibility of vaccinating allogeneic stem cell donors before donation in order to protect transplant recipients against HZ (NCT01573182).
ZVHT and HZ/su vaccination in immunocompromised patients
Heat-treated varicella-zoster vaccine (ZVHT) is a potential alternative for immunocompromised patients. A 4-dose regimen has been proven to reduce the risk of HZ in patients receiving autologous hematopoietic-cell transplants for non-Hodgkin or Hodgkin lymphoma.57
In another trial, the 4-dose ZVHT was safe and elicited significant VZV-specific T-cell response through 28 days in immunosuppressed patients with solid tumor malignancy, hematologic malignancy, human immunodeficiency virus infection with CD4 counts of 200 cells/mm3 or less, and autologous hematopoietic-cell transplants. The T-cell response was poor in allogeneic hematopoietic-cell transplant recipients, however.58
Because the HZ/su vaccine does not contain live virus, it seems particularly promising for immunocompromised patients. In phase 1 and 2 studies, a 3-dose regimen has been shown to be safe and immunogenic in hematopoietic-cell transplant recipients and HIV-infected adults with CD4 count higher than 200 cells/mm3.59,60 A phase 3 trial assessing the efficacy of HZ/su vaccine in autologous hematopoietic-cell transplant recipients is under way (NCT01610414). Changes in recommendations for HZ vaccine in these most vulnerable populations await the results of these studies.
Herpes zoster (HZ), or shingles, represents a reactivation of the varicella-zoster virus (VZV). Following primary infection, usually in childhood, the virus typically lies dormant in the dorsal root and sensory nerve ganglia for decades. The precise mechanism of reactivation is not well understood, but it is associated with a decline in cell-mediated immunity that occurs with advancing age, immune-compromising conditions such as HIV infection and cancer, or immunosuppressive therapies, including corticosteroids.1 HZ is usually a self-limited disease characterized by unilateral dermatomal rash and pain, but can cause disseminated infection in immunocompromised individuals.2
Treatment with antiviral medications within 72 hours of rash onset can reduce acute HZ symptoms.1 However, antiviral agents are only minimally effective in preventing postherpetic neuralgia, the most common complication of HZ.3 Therefore, efforts to reduce the burden of HZ morbidity have focused on prevention through vaccination.
Currently, the only shingles vaccine approved by the US Food and Drug Administration (FDA) is Zostavax (Merck), which contains the live-attenuated Oka strain of VZV at a concentration 14 times greater than that of the varicella vaccine (Varivax, Merck). The live-attenuated vaccine boosts VZV-specific cell-mediated immunity, preventing reactivation of the latent virus.
In this article, we describe the burden of disease and review recent developments in the literature on HZ vaccine, including duration of efficacy, uptake and barriers to vaccination, cost-effectiveness, and the outlook for future vaccines.
INCIDENCE INCREASES WITH AGE
The incidence of herpes zoster in the general population is between 3 and 5 per 1,000 person-years4 and increases with age, especially after age 60 when the incidence can approach 13 to 15 per 1,000 person-years.5,6 An estimated 1 million new cases occur each year in the United States, and about 6% of patients experience a second episode of HZ within 8 years.7,8 In immunocompromised patients, the incidence of HZ is 2 to 10 times higher than in the general population.9
The incidence of HZ has been increasing for reasons that are unclear. After varicella vaccine was introduced into the routine childhood immunization schedule in 1995, it was hypothesized that the resultant decrease in primary varicella infections would remove a natural source of immune boosting and cause an increase in HZ incidence for up to 20 years.10 However, recent studies demonstrate that the observed increase in HZ incidence actually predates the introduction of varicella vaccine,11–13 and the widespread use of varicella vaccine has not resulted in an increase in the incidence of HZ.14
Other potential explanations for the rise in reported incidence include increasing awareness among patients, who might previously not have sought care and among physicians, who may be more likely to make the diagnosis. Advertisement of new treatments for HZ, including gabapentin and capsaicin, probably began to increase awareness in the 1990s, as did promotion of the HZ vaccine after its licensure in 2006.
HZ can occur in people who have been vaccinated against varicella due to reactivation of the vaccine-strain virus, but the risk is lower than after infection with wild-type varicella.15 Given that the varicella vaccine has been routinely used in children for only 20 years, the long-term effect of varicella vaccination on the incidence of HZ in elderly people is unknown.
Serious complications
HZ can cause rare but serious complications including encephalitis, herpes ophthalmicus, herpes oticus, myelitis, and retinitis.1 These can lead to long-term disability including unilateral blindness and deafness.
The most common and debilitating complication is postherpetic neuralgia, a persistent pain lasting at least 3 months, with a mean duration of 3.3 years and sometimes as long as 10 years.16 Postherpetic neuralgia occurs in 8% to 32% of patients after acute HZ,4 and the incidence increases with age, being most common after age 70. The chronic pain of postherpetic neuralgia has a significant adverse impact on patients’ quality of life, including physical disability and emotional distress.17 Some pain is intense, and anecdotal reports of patients committing suicide were included in the Advisory Committee on Immunization Practices (ACIP) recommendations regarding herpes zoster vaccine.18
HZ and its complications also impose a substantial economic burden on society.19 In a population-based study, the mean direct medical costs of HZ ranged from $620 to $1,160 (2015 dollars) depending on age,20 and the mean costs of postherpetic neuralgia were 2 to 5 times higher than that.20–22 Immunocompromised patients had costs 2 to 3 times higher than those of immunocompetent adults.23 In addition, for employed patients, HZ resulted in an average loss of 32 hours of work due to absenteeism and 84 hours due to presenteeism (ie, working while sick and therefore suboptimally).24
Assuming there are 1 million cases of HZ each year, if 8% to 32% of patients go on to develop postherpetic neuralgia, that would translate into approximately $1 to $2 billion in direct medical costs. With 60% of adult patients working,25 at an average wage of $23.23 per hour,26 HZ illness could be responsible for another $1.6 billion in lost productivity.
EFFICACY AND SAFETY OF HZ VACCINE
In 2006, the FDA approved the live-attenuated Oka strain VZV vaccine for prevention of HZ and postherpetic neuralgia in adults age 60 and older based on findings from the Shingles Prevention Study (SPS).27
The Shingles Prevention Study
This multicenter randomized placebo-controlled trial27 enrolled 38,546 immunocompetent persons age 60 and older. Subjects in the intervention group received a single dose of live-attenuated vaccine, and all participants were followed for up to 4.9 years after vaccination.
HZ occurred in 315 (1.636%) of the 19,254 participants in the vaccine group and in 642 (3.336%) of the 19,247 participants in the placebo group, an absolute risk reduction of 1.7%, number needed to treat 59, relative risk reduction 51%, P < .001. Similarly, postherpetic neuralgia occurred in 27 (0.140%) of the 19,254 vaccine recipients and in 80 (0.416%) of the placebo recipients (an absolute risk reduction of 0.276%, number needed to treat 362, relative risk reduction 66%, P < .001). The investigators calculated that vaccination reduced the overall burden of illness by 61% (Table 1).
The efficacy against HZ incidence decreased with age,28 but the efficacy against postherpetic neuralgia did not. In addition, vaccine recipients who developed HZ generally had less severe manifestations.
The safety of the vaccine was assessed for all participants in the SPS. In addition, one-sixth of SPS participants were enrolled in a safety substudy. These participants completed a detailed report card regarding all medically important events within the first 42 days. Forty-eight percent of the vaccine group and 17% of the placebo group (P < .05) experienced adverse events, primarily at the injection site. Less than 1% of all local reactions were severe.29 Serious adverse events were rare (< 2%), but occurred significantly more often in the vaccinated group.
Short-Term Persistence Substudy
Short-term efficacy of the live-attenuated vaccine (up to 7 years) was assessed in the Short-Term Persistence Substudy (STPS), which involved 14,270 of the initial participants and reported yearly and overall vaccine efficacy.30 After 5 years, the yearly efficacy against postherpetic neuralgia incidence declined to 32% and was no longer statistically significant. Efficacy against HZ incidence and burden of illness displayed the same pattern. After the end of the STPS, all subjects in the placebo group received vaccination.
Long-Term Persistence Substudy
Those in the intervention group were followed for an additional 4 years in the Long-Term Persistence Substudy (LTPS).31 Due to the lack of concurrent controls in the LTPS, the authors used regression models based on historical controls to estimate contemporary population incidence of HZ and postherpetic neuralgia for comparison.
Efficacy continued to decline over time, and by 10 years after vaccination there was no difference between vaccinated patients and historical controls in the rate of any end point (ie, efficacy declined to zero).
A trial of booster vaccination
Because many patients are vaccinated at age 60, waning immunity could leave them vulnerable to HZ and postherpetic neuralgia by age 70. A potential solution would be to give a booster dose after 10 years.
A recent phase 3 clinical trial of adults age 70 years and older found that a booster dose of live-attenuated vaccine was as safe and immunogenic as an initial dose.32 While antibody responses were similar in the boosted group and the newly vaccinated group, cell-mediated immunity was higher in the boosted group.
Because prevention of HZ is generally via cell-mediated immunity, the booster might be more effective than the initial vaccination, but clinical trials measuring actual cases prevented will be required to prove it. A booster dose is not currently recommended.
A trial of vaccination in adults 50 to 59
In 2011, the FDA extended its approval of HZ vaccine for use in adults ages 50 to 59.33
In a randomized, double-blind, placebo-controlled trial in this age group,33 the vaccine reduced HZ incidence by almost 70% (absolute risk reduction 0.614%, number needed to treat 156; Table 1), but the severity of HZ cases was not affected. There were too few cases of postherpetic neuralgia to assess the efficacy for this end point. The study followed patients for only 1.5 years after vaccination, so the duration of efficacy is unknown.
As in the older recipients, the vaccine was well tolerated; injection-site reactions and headache were the major adverse effects reported among vaccine recipients.33
INDICATIONS AND CONTRAINDICATIONS
Although HZ vaccine is licensed for use in adults age 50 and older, the ACIP recommends it only for immunocompetent adults age 60 and older. At this time, the ACIP does not recommend HZ vaccine in those younger than 60 because of the low risk of HZ in this age group.34
Any person age 60 or older should receive a single dose of the live-attenuated HZ vaccine subcutaneously, regardless of past history of HZ.
The vaccine is contraindicated in patients who have a history of allergic reaction to any vaccine component, immunosuppression or immunodeficiency conditions, and pregnancy. Specifically, people who will receive immunosuppressive therapies should have the vaccine at least 14 days before beginning treatment. Antiviral medications such as acyclovir, famciclovir, and valacyclovir should be discontinued at least 24 hours before vaccination and not resumed until 14 days later. Patients taking high-dose corticosteroids for more than 2 weeks should not be vaccinated until at least 1 month after therapy is completed.
In contrast, HZ vaccine is not contraindicated for leukemia patients who are in remission and who have not received chemotherapy or radiation for at least 3 months, or for patients receiving short-term, low-to-moderate dose, topical, intra-articular, bursal, or tendon injections of corticosteroids. Patients on low-dose methotrexate, azathioprine, or 6-mercaptopurine can also receive the vaccine.18
VACCINATION RATES ARE LOW
Although the vaccine has been recommended since 2008, uptake has been slow. Figure 1 shows the rate of HZ vaccination in adults age 60 and older surveyed in the National Health Interview Survey from 2007 to 2013.35 Eight years after the vaccine was licensed, only 28% of eligible patients had been vaccinated. Assuming the current rate of increase remains constant, it will take 7 more years to reach a 60% coverage rate—the same as for pneumococcal vaccine36—and 18 years to reach universal coverage.
Barriers to vaccination
Several barriers to HZ vaccination might account for the slow uptake.
For the first few years the vaccine was available, the requirement to store it frozen presented an obstacle for some physicians.37 Physicians may also have been discouraged by the cumbersome Medicare reimbursement process because while the administration fee is covered through Medicare Part B, the live-attenuated vaccine is reimbursed only through Medicare Part D, a benefit that varies by plans. Other barriers to physicians are supply shortages, high up-front costs, and uncertainties regarding the duration of vaccine protection, its safety, and side effects.38–40
Patient barriers include lack of physician recommendation, lack of familiarity with the vaccine, high out-of-pocket costs, the perception that they are at low risk for HZ, underestimation of the pain associated with HZ and postherpetic neuralgia, and fear of vaccine adverse effects.39,41,42
Interventions to increase vaccination rates
Certain interventions have been shown to increase vaccination adherence in general and HZ vaccination in particular. In randomized trials involving other vaccines, electronic medical record reminders supporting panel management or nurse-initiated protocols have been proven to increase vaccination rates, but these methods have not been tested for HZ vaccine specifically.43,44
In an observational study, Chaudhry et al found that the number of HZ vaccinations administered at the Mayo Clinic increased 43% in one practice and 54% in another after the implementation of an electronic alert.45 A randomized controlled trial showed that an informational package discussing HZ and the vaccine sent to patients via either their electronic personal health record or traditional mail increased HZ vaccination by almost 3 times.46
Pharmacists can also influence vaccination rates. States that provide full immunization privileges to pharmacists have vaccination rates significantly higher than states with restricted or no authorization.47
COST-EFFECTIVENESS CONSIDERATIONS
Unlike the Centers for Medicare and Medicaid Services, the ACIP does consider cost-effectiveness in their vaccine recommendations. Because of the morbidity associated with HZ and postherpetic neuralgia as well as the economic impact, vaccination is generally considered cost-effective for adults age 60 and older.48,49
Analyses have demonstrated that cost-effectiveness hinges on 4 factors: initial vaccine efficacy, the duration of efficacy, the age-specific incidence of HZ, and the cost of the vaccine.
For patients ages 50 to 59, the incidence of HZ is low, and because the duration of vaccine efficacy is short even though initial vaccine efficacy is high, vaccination in this age group offers poor value.50 At older ages, the incidence of HZ and postherpetic neuralgia rises, making vaccination more cost-effective. After age 60, the vaccine is cost-effective at all ages, although age 70 appears to offer the optimal trade-off between increasing incidence and declining vaccine efficacy.48,49
For patients who plan to be vaccinated only once, waiting until age 70 would appear to offer the best value.51 For those who are willing to receive a booster dose, the optimal age for vaccination is unknown, but will likely depend on the effectiveness, cost, and duration of the booster.
A NEW HZ VACCINE
In 2015, GlaxoSmithKline tested a new HZ vaccine containing a single VZV glycoprotein in an AS01B adjuvant system (HZ/su vaccine).52 In a phase 3 randomized trial involving 15,411 immunocompetent persons age 50 and older, a 2-dose schedule of HZ/su vaccine was 97% effective in preventing HZ (Table 1).53 Importantly, the vaccine was equally effective in older patients.
This vaccine also had a high rate of adverse reactions, with 17% of vaccine recipients vs 3% of placebo recipients reporting events that prevented normal everyday activities for at least 1 day. However, the rate of serious adverse reactions was the same in both groups (9%). The company announced that they intended to submit a regulatory application for HZ/su vaccine in the second half of 2016.54
Because of its high efficacy, HZ/su vaccine has the potential to change practice, but several issues must be resolved before it can supplant the current vaccine.
First, the AS01B adjuvant is not currently licensed in the United States, so it is unclear if the HZ/su vaccine can get FDA approval.52,55
Second, there are several questions about the efficacy of the vaccine, including long-term efficacy, efficacy in the elderly, and efficacy in the case of a patient receiving only 1 of the 2 required doses.
Third, the impact of HZ/su vaccine on complications such as postherpetic neuralgia has not been established. The clinical trial (NCT01165229) examining vaccine efficacy against postherpetic neuralgia incidence and other complications in adults age 70 and older has recently been completed and data should be available soon. Given the extremely high efficacy against HZ, it is likely that it will be close to 100% effective against this complication.
Fourth, there is uncertainty as to how the HZ/su vaccine should be used in patients who have already received the live-attenuated vaccine, if it is determined that a booster is necessary.
Finally, the vaccine is not yet priced. Given its superior effectiveness, particularly in older individuals, competitive pricing could dramatically affect the market. How Medicare or other insurers cover the new vaccine will likely influence its acceptance.
HZ VACCINATION OF IMMUNOCOMPROMISED PATIENTS
Immunocompromised patients are at highest risk for developing HZ. Unfortunately, there are currently no HZ vaccines approved for use in this population. The current live-attenuated vaccine has been demonstrated to be safe, well tolerated, and immunogenic in patients age 60 and older who are receiving chronic or maintenance low to moderate doses of corticosteroids.56
A clinical trial is being conducted to assess the immunogenicity, clinical effectiveness, and safety of the vaccine in rheumatoid arthritis patients receiving antitumor necrosis factor therapy (NCT01967316). Other trials are examining vaccine efficacy and safety in patients with solid organ tumors prior to chemotherapy (NCT02444936) and in patients who will be undergoing living donor kidney transplantation (NCT00940940). Researchers are also investigating the possibility of vaccinating allogeneic stem cell donors before donation in order to protect transplant recipients against HZ (NCT01573182).
ZVHT and HZ/su vaccination in immunocompromised patients
Heat-treated varicella-zoster vaccine (ZVHT) is a potential alternative for immunocompromised patients. A 4-dose regimen has been proven to reduce the risk of HZ in patients receiving autologous hematopoietic-cell transplants for non-Hodgkin or Hodgkin lymphoma.57
In another trial, the 4-dose ZVHT was safe and elicited significant VZV-specific T-cell response through 28 days in immunosuppressed patients with solid tumor malignancy, hematologic malignancy, human immunodeficiency virus infection with CD4 counts of 200 cells/mm3 or less, and autologous hematopoietic-cell transplants. The T-cell response was poor in allogeneic hematopoietic-cell transplant recipients, however.58
Because the HZ/su vaccine does not contain live virus, it seems particularly promising for immunocompromised patients. In phase 1 and 2 studies, a 3-dose regimen has been shown to be safe and immunogenic in hematopoietic-cell transplant recipients and HIV-infected adults with CD4 count higher than 200 cells/mm3.59,60 A phase 3 trial assessing the efficacy of HZ/su vaccine in autologous hematopoietic-cell transplant recipients is under way (NCT01610414). Changes in recommendations for HZ vaccine in these most vulnerable populations await the results of these studies.
- Dworkin RH, Johnson RW, Breuer J, et al. Recommendations for the management of herpes zoster. Clin Infect Dis 2007; 44(suppl 1):S1–S26.
- Johnson RW. Herpes zoster and postherpetic neuralgia. Expert Rev Vaccines 2010; 9(suppl):21–26.
- Chen N, Li Q, Yang J, Zhou M, Zhou D, He L. Antiviral treatment for preventing postherpetic neuralgia. Cochrane Database Syst Rev 2014; 2:CD006866.
- Kawai K, Gebremeskel BG, Acosta CJ. Systematic review of incidence and complications of herpes zoster: towards a global perspective. BMJ Open 2014; 4:e004833.
- Tseng HF, Smith N, Harpaz R, Bialek SR, Sy LS, Jacobsen SJ. Herpes zoster vaccine in older adults and the risk of subsequent herpes zoster disease. JAMA 2011; 305:160–166.
- Langan SM, Smeeth L, Margolis DJ, Thomas SL. Herpes zoster vaccine effectiveness against incident herpes zoster and post-herpetic neuralgia in an older US population: a cohort study. PLoS Med 2013; 10:e1001420.
- Yawn BP, Saddier P, Wollan PC, St. Sauver JL, Kurland MJ, Sy LS. A population-based study of the incidence and complication rates of herpes zoster before zoster vaccine introduction. Mayo Clin Proc 2007; 82:1341–1349.
- Yawn BP, Wollan PC, Kurland MJ, St. Sauver JL, Saddier P. Herpes zoster recurrences more frequent than previously reported. Mayo Clin Proc 2011; 86:88–93.
- Chen SY, Suaya JA, Li Q, et al. Incidence of herpes zoster in patients with altered immune function. Infection 2014; 42:325–334.
- Edmunds WJ, Brisson M. The effect of vaccination on the epidemiology of varicella zoster virus. J Infect 2002; 44:211–219.
- Hales CM, Harpaz R, Joesoef MR, Bialek SR. Examination of links between herpes zoster incidence and childhood varicella vaccination. Ann Intern Med 2013; 159:739–745.
- Leung J, Harpaz R, Molinari NA, Jumaan A, Zhou F. Herpes zoster incidence among insured persons in the United States, 1993-2006: evaluation of impact of varicella vaccination. Clin Infect Dis 2011; 52:332–340.
- Rimland D, Moanna A. Increasing incidence of herpes zoster among veterans. Clin Infect Dis 2010; 50:1000–1005.
- Jumaan AO, Yu O, Jackson LA, Bohlke K, Galil K, Seward JF. Incidence of herpes zoster, before and after varicella-vaccination-associated decreases in the incidence of varicella, 1992-2002. J Infect Dis 2005; 191:2002–2007.
- Plotkin SA, Starr SE, Connor K, Morton D. Zoster in normal children after varicella vaccine. J Infect Dis 1989; 159:1000–1001.
- Oster G, Harding G, Dukes E, Edelsberg J, Cleary PD. Pain, medication use, and health-related quality of life in older persons with postherpetic neuralgia: results from a population-based survey. J Pain 2005; 6:356–363.
- Johnson RW, Bouhassira D, Kassianos G, Leplege A, Schmader KE, Weinke T. The impact of herpes zoster and post-herpetic neuralgia on quality-of-life. BMC Med 2010; 8:37.
- Harpaz R, Ortega-Sanchez IR, Seward JF; Advisory Committee on Immunization Practices (ACIP) Centers for Disease Control and Prevention (CDC). Prevention of herpes zoster: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2008; 57:1–30.
- Panatto D, Bragazzi NL, Rizzitelli E, et al. Evaluation of the economic burden of herpes zoster (HZ) infection. Hum Vaccin Immunother 2015; 11:245–262.
- Yawn BP, Itzler RF, Wollan PC, Pellissier JM, Sy LS, Saddier P. Health care utilization and cost burden of herpes zoster in a community population. Mayo Clin Proc 2009; 84:787–794.
- Dworkin RH, White R, O’Connor AB, Hawkins K. Health care expenditure burden of persisting herpes zoster pain. Pain Med 2008; 9:348–353.
- White RR, Lenhart G, Singhal PK, et al. Incremental 1-year medical resource utilization and costs for patients with herpes zoster from a set of US health plans. Pharmacoeconomics 2009; 27:781–792.
- Insinga RP, Itzler RF, Pellissier JM. Acute/subacute herpes zoster: healthcare resource utilisation and costs in a group of US health plans. Pharmacoeconomics 2007; 25:155–169.
- Singhal PK, Makin C, Pellissier J, Sy L, White R, Saddier P. Work and productivity loss related to herpes zoster. J Med Econ 2011; 14:639–645.
- US Bureau of Labor Statistics. Labor force statistics from the current population survey. www.bls.gov/web/empsit/cpseea13.htm. Accessed April 6, 2017.
- US Bureau of Labor Statistic. Occupational employment statistics. www.bls.gov/oes/current/oes_nat.htm. Accessed April 6, 2017.
- Oxman MN, Levin MJ, Johnson GR, et al; Shingles Prevention Study Group. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N Engl J Med 2005; 352:2271–2284.
- Food and Drug Administration (FDA). FDA clinical briefing document for Merck & Co., Inc. Zoster vaccine live (Oka/Merck) Zostavax. www.fda.gov/ohrms/dockets/ac/05/briefing/5-4198b2_1.pdf. Accessed April 6, 2017.
- Simberkoff MS, Arbeit RD, Johnson GR, et al; Shingles Prevention Study Group. Safety of herpes zoster vaccine in the shingles prevention study: a randomized trial. Ann Intern Med 2010; 152:545–554.
- Schmader KE, Oxman MN, Levin MJ, et al; Shingles Prevention Study Group. Persistence of the efficacy of zoster vaccine in the shingles prevention study and the short-term persistence substudy. Clin Infect Dis 2012; 55:1320–1328.
- Morrison VA, Johnson GR, Schmader KE, et al; Shingles Prevention Study Group. Long-term persistence of zoster vaccine efficacy. Clin Infect Dis 2015; 60:900–909.
- Levin MJ, Schmader KE, Pang L, et al. Cellular and humoral responses to a second dose of herpes zoster vaccine administered 10 years after the first dose among older adults. J Infect Dis 2016; 213:14–22.
- Schmader KE, Levin MJ, Gnann JW Jr, et al. Efficacy, safety, and tolerability of herpes zoster vaccine in persons aged 50-59 years. Clin Infect Dis 2012; 54:922–928.
- Hales CM, Harpaz R, Ortega-Sanchez I, Bialek SR; Centers for Disease Control and Prevention (CDC). Update on recommendations for use of herpes zoster vaccine. MMWR Morb Mortal Wkly Rep 2014; 63:729–731.
- Centers for Disease Control and Prevention (CDC). Surveillance of vaccination coverage among adult populations—United States, 2014. MMWR Morb Mortal Wkly Rep 2016; 65(1):1–36. Accessed April 12, 2017.
- Williams WW, Lu PJ, O’Halloran A, et al; Centers for Disease Control and Prevention (CDC). Vaccination coverage among adults, excluding influenza vaccination—United States, 2013. MMWR Morb Mortal Wkly Rep 2015; 64:95–102.
- Oxman MN. Zoster vaccine: current status and future prospects. Clin Infect Dis 2010; 51:197–213.
- Hurley LP, Lindley MC, Harpaz R, et al. Barriers to the use of herpes zoster vaccine. Ann Intern Med 2010; 152:555–560.
- Lu PJ, Euler GL, Jumaan AO, Harpaz R. Herpes zoster vaccination among adults aged 60 years or older in the United States, 2007: uptake of the first new vaccine to target seniors. Vaccine 2009; 27:882–887.
- Hurley LP, Harpaz R, Daley MF, et al. National survey of primary care physicians regarding herpes zoster and the herpes zoster vaccine. J Infect Dis 2008; 197(suppl 2):S216–S223.
- Joon Lee T, Hayes S, Cummings DM, et al. Herpes zoster knowledge, prevalence, and vaccination rate by race. J Am Board Fam Med 2013; 26:45–51.
- Opstelten W, van Essen GA, Hak E. Determinants of non-compliance with herpes zoster vaccination in the community-dwelling elderly. Vaccine 2009; 27:192–196.
- Loo TS, Davis RB, Lipsitz LA, et al. Electronic medical record reminders and panel management to improve primary care of elderly patients. Arch Intern Med 2011; 171:1552–1558.
- Rhew DC, Glassman PA, Goetz MB. Improving pneumococcal vaccine rates. Nurse protocols versus clinical reminders. J Gen Intern Med 1999; 14:351–356.
- Chaudhry R, Schietel SM, North F, Dejesus R, Kesman RL, Stroebel RJ. Improving rates of herpes zoster vaccination with a clinical decision support system in a primary care practice. J Eval Clin Pract 2013; 19:263–266.
- Otsuka SH, Tayal NH, Porter K, Embi PJ, Beatty SJ. Improving herpes zoster vaccination rates through use of a clinical pharmacist and a personal health record. Am J Med 2013; 126:832.e1–832.e6.
- Taitel MS, Fensterheim LE, Cannon AE, Cohen ES. Improving pneumococcal and herpes zoster vaccination uptake: expanding pharmacist privileges. Am J Manag Care 2013; 19:e309–e313.
- Kawai K, Preaud E, Baron-Papillon F, Largeron N, Acosta CJ. Cost-effectiveness of vaccination against herpes zoster and postherpetic neuralgia: a critical review. Vaccine 2014; 32:1645–1653.
- Szucs TD, Pfeil AM. A systematic review of the cost effectiveness of herpes zoster vaccination. Pharmacoeconomics 2013; 31:125–136.
- Le P, Rothberg MB. Cost-effectiveness of herpes zoster vaccine for persons aged 50 years. Ann Intern Med 2015; 163:489–497.
- Le P, Rothberg MB. Determining the optimal age to vaccinate against herpes zoster: a cost-effectiveness analysis. Society for Medical Decision Making 37th Annual North American Meeting. St. Louis, MO; October 18-21, 2015.
- Cohen JI. Clinical practice: herpes zoster. N Engl J Med 2013; 369:255–263.
- Lal H, Cunningham AL, Godeaux O, et al; ZOE-50 Study Group. Efficacy of an adjuvanted herpes zoster subunit vaccine in older adults. N Engl J Med 2015; 372:2087–2096.
- GlaxoSmithKline plc. GSK’s candidate shingles vaccine demonstrates 90% efficacy against shingles in people 70 years of age and over. www.gsk.com/en-gb/media/press-releases/gsk-s-candidate-shingles-vaccine-demonstrates-90-efficacy-against-shingles-in-people-70-years-of-age-and-over/. Accessed April 6, 2017.
- Reed SG, Orr MT, Fox CB. Key roles of adjuvants in modern vaccines. Nat Med 2013; 19:1597–1608.
- Russell AF, Parrino J, Fisher CL Jr, et al. Safety, tolerability, and immunogenicity of zoster vaccine in subjects on chronic/maintenance corticosteroids. Vaccine 2015; 33:3129–3134.
- Hata A, Asanuma H, Rinki M, et al. Use of an inactivated varicella vaccine in recipients of hematopoietic-cell transplants. N Engl J Med 2002; 347:26–34.
- Mullane KM, Winston DJ, Wertheim MS, et al. Safety and immunogenicity of heat-treated zoster vaccine (ZVHT) in immunocompromised adults. J Infect Dis 2013; 208:1375–1385.
- Stadtmauer EA, Sullivan KM, Marty FM, et al. A phase 1/2 study of an adjuvanted varicella-zoster virus subunit vaccine in autologous hematopoietic cell transplant recipients. Blood 2014; 124:2921–2929.
- Berkowitz EM, Moyle G, Stellbrink HJ, et al. Safety and immunogenicity of an adjuvanted herpes zoster subunit candidate vaccine in HIV-infected adults: a phase 1/2a randomized, placebo-controlled study. J Infect Dis 2015; 211:1279–1287.
- Dworkin RH, Johnson RW, Breuer J, et al. Recommendations for the management of herpes zoster. Clin Infect Dis 2007; 44(suppl 1):S1–S26.
- Johnson RW. Herpes zoster and postherpetic neuralgia. Expert Rev Vaccines 2010; 9(suppl):21–26.
- Chen N, Li Q, Yang J, Zhou M, Zhou D, He L. Antiviral treatment for preventing postherpetic neuralgia. Cochrane Database Syst Rev 2014; 2:CD006866.
- Kawai K, Gebremeskel BG, Acosta CJ. Systematic review of incidence and complications of herpes zoster: towards a global perspective. BMJ Open 2014; 4:e004833.
- Tseng HF, Smith N, Harpaz R, Bialek SR, Sy LS, Jacobsen SJ. Herpes zoster vaccine in older adults and the risk of subsequent herpes zoster disease. JAMA 2011; 305:160–166.
- Langan SM, Smeeth L, Margolis DJ, Thomas SL. Herpes zoster vaccine effectiveness against incident herpes zoster and post-herpetic neuralgia in an older US population: a cohort study. PLoS Med 2013; 10:e1001420.
- Yawn BP, Saddier P, Wollan PC, St. Sauver JL, Kurland MJ, Sy LS. A population-based study of the incidence and complication rates of herpes zoster before zoster vaccine introduction. Mayo Clin Proc 2007; 82:1341–1349.
- Yawn BP, Wollan PC, Kurland MJ, St. Sauver JL, Saddier P. Herpes zoster recurrences more frequent than previously reported. Mayo Clin Proc 2011; 86:88–93.
- Chen SY, Suaya JA, Li Q, et al. Incidence of herpes zoster in patients with altered immune function. Infection 2014; 42:325–334.
- Edmunds WJ, Brisson M. The effect of vaccination on the epidemiology of varicella zoster virus. J Infect 2002; 44:211–219.
- Hales CM, Harpaz R, Joesoef MR, Bialek SR. Examination of links between herpes zoster incidence and childhood varicella vaccination. Ann Intern Med 2013; 159:739–745.
- Leung J, Harpaz R, Molinari NA, Jumaan A, Zhou F. Herpes zoster incidence among insured persons in the United States, 1993-2006: evaluation of impact of varicella vaccination. Clin Infect Dis 2011; 52:332–340.
- Rimland D, Moanna A. Increasing incidence of herpes zoster among veterans. Clin Infect Dis 2010; 50:1000–1005.
- Jumaan AO, Yu O, Jackson LA, Bohlke K, Galil K, Seward JF. Incidence of herpes zoster, before and after varicella-vaccination-associated decreases in the incidence of varicella, 1992-2002. J Infect Dis 2005; 191:2002–2007.
- Plotkin SA, Starr SE, Connor K, Morton D. Zoster in normal children after varicella vaccine. J Infect Dis 1989; 159:1000–1001.
- Oster G, Harding G, Dukes E, Edelsberg J, Cleary PD. Pain, medication use, and health-related quality of life in older persons with postherpetic neuralgia: results from a population-based survey. J Pain 2005; 6:356–363.
- Johnson RW, Bouhassira D, Kassianos G, Leplege A, Schmader KE, Weinke T. The impact of herpes zoster and post-herpetic neuralgia on quality-of-life. BMC Med 2010; 8:37.
- Harpaz R, Ortega-Sanchez IR, Seward JF; Advisory Committee on Immunization Practices (ACIP) Centers for Disease Control and Prevention (CDC). Prevention of herpes zoster: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2008; 57:1–30.
- Panatto D, Bragazzi NL, Rizzitelli E, et al. Evaluation of the economic burden of herpes zoster (HZ) infection. Hum Vaccin Immunother 2015; 11:245–262.
- Yawn BP, Itzler RF, Wollan PC, Pellissier JM, Sy LS, Saddier P. Health care utilization and cost burden of herpes zoster in a community population. Mayo Clin Proc 2009; 84:787–794.
- Dworkin RH, White R, O’Connor AB, Hawkins K. Health care expenditure burden of persisting herpes zoster pain. Pain Med 2008; 9:348–353.
- White RR, Lenhart G, Singhal PK, et al. Incremental 1-year medical resource utilization and costs for patients with herpes zoster from a set of US health plans. Pharmacoeconomics 2009; 27:781–792.
- Insinga RP, Itzler RF, Pellissier JM. Acute/subacute herpes zoster: healthcare resource utilisation and costs in a group of US health plans. Pharmacoeconomics 2007; 25:155–169.
- Singhal PK, Makin C, Pellissier J, Sy L, White R, Saddier P. Work and productivity loss related to herpes zoster. J Med Econ 2011; 14:639–645.
- US Bureau of Labor Statistics. Labor force statistics from the current population survey. www.bls.gov/web/empsit/cpseea13.htm. Accessed April 6, 2017.
- US Bureau of Labor Statistic. Occupational employment statistics. www.bls.gov/oes/current/oes_nat.htm. Accessed April 6, 2017.
- Oxman MN, Levin MJ, Johnson GR, et al; Shingles Prevention Study Group. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N Engl J Med 2005; 352:2271–2284.
- Food and Drug Administration (FDA). FDA clinical briefing document for Merck & Co., Inc. Zoster vaccine live (Oka/Merck) Zostavax. www.fda.gov/ohrms/dockets/ac/05/briefing/5-4198b2_1.pdf. Accessed April 6, 2017.
- Simberkoff MS, Arbeit RD, Johnson GR, et al; Shingles Prevention Study Group. Safety of herpes zoster vaccine in the shingles prevention study: a randomized trial. Ann Intern Med 2010; 152:545–554.
- Schmader KE, Oxman MN, Levin MJ, et al; Shingles Prevention Study Group. Persistence of the efficacy of zoster vaccine in the shingles prevention study and the short-term persistence substudy. Clin Infect Dis 2012; 55:1320–1328.
- Morrison VA, Johnson GR, Schmader KE, et al; Shingles Prevention Study Group. Long-term persistence of zoster vaccine efficacy. Clin Infect Dis 2015; 60:900–909.
- Levin MJ, Schmader KE, Pang L, et al. Cellular and humoral responses to a second dose of herpes zoster vaccine administered 10 years after the first dose among older adults. J Infect Dis 2016; 213:14–22.
- Schmader KE, Levin MJ, Gnann JW Jr, et al. Efficacy, safety, and tolerability of herpes zoster vaccine in persons aged 50-59 years. Clin Infect Dis 2012; 54:922–928.
- Hales CM, Harpaz R, Ortega-Sanchez I, Bialek SR; Centers for Disease Control and Prevention (CDC). Update on recommendations for use of herpes zoster vaccine. MMWR Morb Mortal Wkly Rep 2014; 63:729–731.
- Centers for Disease Control and Prevention (CDC). Surveillance of vaccination coverage among adult populations—United States, 2014. MMWR Morb Mortal Wkly Rep 2016; 65(1):1–36. Accessed April 12, 2017.
- Williams WW, Lu PJ, O’Halloran A, et al; Centers for Disease Control and Prevention (CDC). Vaccination coverage among adults, excluding influenza vaccination—United States, 2013. MMWR Morb Mortal Wkly Rep 2015; 64:95–102.
- Oxman MN. Zoster vaccine: current status and future prospects. Clin Infect Dis 2010; 51:197–213.
- Hurley LP, Lindley MC, Harpaz R, et al. Barriers to the use of herpes zoster vaccine. Ann Intern Med 2010; 152:555–560.
- Lu PJ, Euler GL, Jumaan AO, Harpaz R. Herpes zoster vaccination among adults aged 60 years or older in the United States, 2007: uptake of the first new vaccine to target seniors. Vaccine 2009; 27:882–887.
- Hurley LP, Harpaz R, Daley MF, et al. National survey of primary care physicians regarding herpes zoster and the herpes zoster vaccine. J Infect Dis 2008; 197(suppl 2):S216–S223.
- Joon Lee T, Hayes S, Cummings DM, et al. Herpes zoster knowledge, prevalence, and vaccination rate by race. J Am Board Fam Med 2013; 26:45–51.
- Opstelten W, van Essen GA, Hak E. Determinants of non-compliance with herpes zoster vaccination in the community-dwelling elderly. Vaccine 2009; 27:192–196.
- Loo TS, Davis RB, Lipsitz LA, et al. Electronic medical record reminders and panel management to improve primary care of elderly patients. Arch Intern Med 2011; 171:1552–1558.
- Rhew DC, Glassman PA, Goetz MB. Improving pneumococcal vaccine rates. Nurse protocols versus clinical reminders. J Gen Intern Med 1999; 14:351–356.
- Chaudhry R, Schietel SM, North F, Dejesus R, Kesman RL, Stroebel RJ. Improving rates of herpes zoster vaccination with a clinical decision support system in a primary care practice. J Eval Clin Pract 2013; 19:263–266.
- Otsuka SH, Tayal NH, Porter K, Embi PJ, Beatty SJ. Improving herpes zoster vaccination rates through use of a clinical pharmacist and a personal health record. Am J Med 2013; 126:832.e1–832.e6.
- Taitel MS, Fensterheim LE, Cannon AE, Cohen ES. Improving pneumococcal and herpes zoster vaccination uptake: expanding pharmacist privileges. Am J Manag Care 2013; 19:e309–e313.
- Kawai K, Preaud E, Baron-Papillon F, Largeron N, Acosta CJ. Cost-effectiveness of vaccination against herpes zoster and postherpetic neuralgia: a critical review. Vaccine 2014; 32:1645–1653.
- Szucs TD, Pfeil AM. A systematic review of the cost effectiveness of herpes zoster vaccination. Pharmacoeconomics 2013; 31:125–136.
- Le P, Rothberg MB. Cost-effectiveness of herpes zoster vaccine for persons aged 50 years. Ann Intern Med 2015; 163:489–497.
- Le P, Rothberg MB. Determining the optimal age to vaccinate against herpes zoster: a cost-effectiveness analysis. Society for Medical Decision Making 37th Annual North American Meeting. St. Louis, MO; October 18-21, 2015.
- Cohen JI. Clinical practice: herpes zoster. N Engl J Med 2013; 369:255–263.
- Lal H, Cunningham AL, Godeaux O, et al; ZOE-50 Study Group. Efficacy of an adjuvanted herpes zoster subunit vaccine in older adults. N Engl J Med 2015; 372:2087–2096.
- GlaxoSmithKline plc. GSK’s candidate shingles vaccine demonstrates 90% efficacy against shingles in people 70 years of age and over. www.gsk.com/en-gb/media/press-releases/gsk-s-candidate-shingles-vaccine-demonstrates-90-efficacy-against-shingles-in-people-70-years-of-age-and-over/. Accessed April 6, 2017.
- Reed SG, Orr MT, Fox CB. Key roles of adjuvants in modern vaccines. Nat Med 2013; 19:1597–1608.
- Russell AF, Parrino J, Fisher CL Jr, et al. Safety, tolerability, and immunogenicity of zoster vaccine in subjects on chronic/maintenance corticosteroids. Vaccine 2015; 33:3129–3134.
- Hata A, Asanuma H, Rinki M, et al. Use of an inactivated varicella vaccine in recipients of hematopoietic-cell transplants. N Engl J Med 2002; 347:26–34.
- Mullane KM, Winston DJ, Wertheim MS, et al. Safety and immunogenicity of heat-treated zoster vaccine (ZVHT) in immunocompromised adults. J Infect Dis 2013; 208:1375–1385.
- Stadtmauer EA, Sullivan KM, Marty FM, et al. A phase 1/2 study of an adjuvanted varicella-zoster virus subunit vaccine in autologous hematopoietic cell transplant recipients. Blood 2014; 124:2921–2929.
- Berkowitz EM, Moyle G, Stellbrink HJ, et al. Safety and immunogenicity of an adjuvanted herpes zoster subunit candidate vaccine in HIV-infected adults: a phase 1/2a randomized, placebo-controlled study. J Infect Dis 2015; 211:1279–1287.
KEY POINTS
- HZ continues to be an important public health problem, with substantial morbidity and economic impact. Because of the lack of effective treatment, vaccination provides the best strategy for disease mitigation.
- Physicians can reduce the impact of HZ by educating patients about its complications and recommending immunization for all patients age 60 and older. Patients can protect themselves by seeking vaccination.
- Vaccine protection wanes completely after 10 years, and physicians should be prepared to offer a booster dose should the Advisory Committee on Immunization Practices issue such recommendations.
- Newer vaccines offer promise for greater efficacy, especially for the elderly. For immunocompromised patients, a safe and effective vaccine may be available in the near future.

Thoughtful vaccination

Few in mainstream medicine doubt the efficacy of vaccination and the net positive value of a thoughtful vaccination policy. We have witnessed within a professional lifetime the virtual eradication, at least regionally, of a number of previously devastating infectious diseases including polio, smallpox, pertussis, and measles. With growing understanding of disease mechanisms, the potential value of vaccination has expanded to include preventing sequelae of certain infections and malignancy—a holy grail in oncology, the true prevention of cancer. Sporadic local resistance to uniform vaccination against measles has resulted in geographic reappearance of the disease, providing further support for a uniform approach to vaccination against communicable diseases, with some potential to opt out, perhaps with associated societal repercussions for those who do so.
For most clinicians, certainly those of us dealing with chronically ill or immunosuppressed patients, the decision to recommend annual influenza vaccination and pneumococcal vaccinations per guidelines is an easy one. Vaccination against certain infections provides some protection for the individual patient and for the population, contributing to “herd immunity” and helping to protect against the occurrence of pandemics. But this is not the case for all vaccines. For some vaccines the issues of immunity and vaccination are more individual and complicated and warrant more education, reflection, and conversation.
The vaccine to protect against human papillomavirus is effective at reducing the incidence of cervical and anal cancers triggered by infection with certain strains of human papillomavirus. The viral infection itself is not immediately life-threatening. Thus, patients (and their parents) are asked to consider vaccination against a sexually transmitted virus (before sexual transmission of infection) to prevent a possible malignancy later in life. This decision can create social angst. Perhaps less socially challenging, but medically more complicated, is vaccination against the hepatitis B virus (HBV). HBV is also sexually transmissible, but the source in many patients infected with this virus is unclear. The HBV vaccine helps protect against clinically meaningful hepatitis and chronic liver disease from HBV and also will reduce the occurrence of HBV-associated hepatocellular carcinoma and progression of cirrhosis in patients coinfected with hepatitis C virus.
More complex yet is vaccination against the varicella-zoster virus (VZV) to reduce the likelihood of shingles and its possible consequence, postherpetic neuralgia. Le, Sabella, and Rothberg, in this issue of the Journal, review clinical and public health challenges affecting the use of this vaccine.
We are almost all exposed to this virus as children, through natural infection (chickenpox) or vaccination; many infections are seemingly subclinical. My older son had the distinct misfortune of getting chickenpox in a quite memorable way, developing a concentrated collection of the pruritic vesicles underneath a newly placed arm cast. Whether we remember the initial infection or not, VZV sets up housekeeping and lies dormant for decades within sensory neurons. Decades later, it may erupt as shingles along the distribution of the infected nerves with characteristic painful vesicles, sometimes with persistent, extremely painful, and debilitating residua within the same dermatomal distribution: postherpetic neuralgia. The triggers for this fairly common scenario are only generically understood and include waning cellular immunity attributed to aging, malignancy, immunosuppressive medications, human immunodeficiency virus, and perhaps stress and depression.
The zoster vaccine is unique in that it is given to bolster already present cellular immunity to prevent clinical recurrence of the earlier infection, decades after the virus has been dormant—not, as for the other vaccines noted above, to prevent primary infection. It doesn’t matter if the patient has already experienced an episode of shingles. The currently available vaccine is a live-attenuated strain (Oka) of VZV. Another vaccine in clinical testing uses isolated viral components with adjuvant and thus eliminates current concerns of giving the live-attenuated vaccine to immunosuppressed and elderly patients, those who may benefit the most from it.
Fortunately, it seems that even significantly immunosuppressed and elderly patients tolerate the current vaccine, but at present it is suggested that these groups not receive the vaccine, and vaccination rates in these patients is low. Hopefully, additional data will accumulate regarding the safety of the vaccine and will permit its more widespread use within these patient groups, or a replacement “dead” vaccine will become available.
As Le et al nicely discuss, the current vaccine efficacy wanes over about 10 years, and then a booster vaccine should be considered, but there are few data to provide safety and efficacy outcome measures from patients who received a booster vaccination. The potential need to receive a booster injection after about a decade, the demonstrated greater efficacy against zoster when the initial vaccination is provided to patients ages 60 through 69 than in those over 70, and the lower absolute impact when given to patients ages 50 through 59 (baseline zoster incidence increases with age) should enter into the conversation with patients as to when they should receive the vaccine.
Additionally, there are the extremely provocative and as yet unanswered questions surrounding the implications of vaccination if VZV infection is a trigger of inflammatory vascular diseases such as giant cell arteritis in the elderly, as was proposed by the late Dr. Don Gilden.1
And yes, I did get the vaccine. I was 64 at the time.
- Gilden D, Nagel MA. Varicella zoster virus triggers the immunopathology of giant cell arteritis. Curr Opin Rheumatol 2016; 28:376–382.
Few in mainstream medicine doubt the efficacy of vaccination and the net positive value of a thoughtful vaccination policy. We have witnessed within a professional lifetime the virtual eradication, at least regionally, of a number of previously devastating infectious diseases including polio, smallpox, pertussis, and measles. With growing understanding of disease mechanisms, the potential value of vaccination has expanded to include preventing sequelae of certain infections and malignancy—a holy grail in oncology, the true prevention of cancer. Sporadic local resistance to uniform vaccination against measles has resulted in geographic reappearance of the disease, providing further support for a uniform approach to vaccination against communicable diseases, with some potential to opt out, perhaps with associated societal repercussions for those who do so.
For most clinicians, certainly those of us dealing with chronically ill or immunosuppressed patients, the decision to recommend annual influenza vaccination and pneumococcal vaccinations per guidelines is an easy one. Vaccination against certain infections provides some protection for the individual patient and for the population, contributing to “herd immunity” and helping to protect against the occurrence of pandemics. But this is not the case for all vaccines. For some vaccines the issues of immunity and vaccination are more individual and complicated and warrant more education, reflection, and conversation.
The vaccine to protect against human papillomavirus is effective at reducing the incidence of cervical and anal cancers triggered by infection with certain strains of human papillomavirus. The viral infection itself is not immediately life-threatening. Thus, patients (and their parents) are asked to consider vaccination against a sexually transmitted virus (before sexual transmission of infection) to prevent a possible malignancy later in life. This decision can create social angst. Perhaps less socially challenging, but medically more complicated, is vaccination against the hepatitis B virus (HBV). HBV is also sexually transmissible, but the source in many patients infected with this virus is unclear. The HBV vaccine helps protect against clinically meaningful hepatitis and chronic liver disease from HBV and also will reduce the occurrence of HBV-associated hepatocellular carcinoma and progression of cirrhosis in patients coinfected with hepatitis C virus.
More complex yet is vaccination against the varicella-zoster virus (VZV) to reduce the likelihood of shingles and its possible consequence, postherpetic neuralgia. Le, Sabella, and Rothberg, in this issue of the Journal, review clinical and public health challenges affecting the use of this vaccine.
We are almost all exposed to this virus as children, through natural infection (chickenpox) or vaccination; many infections are seemingly subclinical. My older son had the distinct misfortune of getting chickenpox in a quite memorable way, developing a concentrated collection of the pruritic vesicles underneath a newly placed arm cast. Whether we remember the initial infection or not, VZV sets up housekeeping and lies dormant for decades within sensory neurons. Decades later, it may erupt as shingles along the distribution of the infected nerves with characteristic painful vesicles, sometimes with persistent, extremely painful, and debilitating residua within the same dermatomal distribution: postherpetic neuralgia. The triggers for this fairly common scenario are only generically understood and include waning cellular immunity attributed to aging, malignancy, immunosuppressive medications, human immunodeficiency virus, and perhaps stress and depression.
The zoster vaccine is unique in that it is given to bolster already present cellular immunity to prevent clinical recurrence of the earlier infection, decades after the virus has been dormant—not, as for the other vaccines noted above, to prevent primary infection. It doesn’t matter if the patient has already experienced an episode of shingles. The currently available vaccine is a live-attenuated strain (Oka) of VZV. Another vaccine in clinical testing uses isolated viral components with adjuvant and thus eliminates current concerns of giving the live-attenuated vaccine to immunosuppressed and elderly patients, those who may benefit the most from it.
Fortunately, it seems that even significantly immunosuppressed and elderly patients tolerate the current vaccine, but at present it is suggested that these groups not receive the vaccine, and vaccination rates in these patients is low. Hopefully, additional data will accumulate regarding the safety of the vaccine and will permit its more widespread use within these patient groups, or a replacement “dead” vaccine will become available.
As Le et al nicely discuss, the current vaccine efficacy wanes over about 10 years, and then a booster vaccine should be considered, but there are few data to provide safety and efficacy outcome measures from patients who received a booster vaccination. The potential need to receive a booster injection after about a decade, the demonstrated greater efficacy against zoster when the initial vaccination is provided to patients ages 60 through 69 than in those over 70, and the lower absolute impact when given to patients ages 50 through 59 (baseline zoster incidence increases with age) should enter into the conversation with patients as to when they should receive the vaccine.
Additionally, there are the extremely provocative and as yet unanswered questions surrounding the implications of vaccination if VZV infection is a trigger of inflammatory vascular diseases such as giant cell arteritis in the elderly, as was proposed by the late Dr. Don Gilden.1
And yes, I did get the vaccine. I was 64 at the time.
Few in mainstream medicine doubt the efficacy of vaccination and the net positive value of a thoughtful vaccination policy. We have witnessed within a professional lifetime the virtual eradication, at least regionally, of a number of previously devastating infectious diseases including polio, smallpox, pertussis, and measles. With growing understanding of disease mechanisms, the potential value of vaccination has expanded to include preventing sequelae of certain infections and malignancy—a holy grail in oncology, the true prevention of cancer. Sporadic local resistance to uniform vaccination against measles has resulted in geographic reappearance of the disease, providing further support for a uniform approach to vaccination against communicable diseases, with some potential to opt out, perhaps with associated societal repercussions for those who do so.
For most clinicians, certainly those of us dealing with chronically ill or immunosuppressed patients, the decision to recommend annual influenza vaccination and pneumococcal vaccinations per guidelines is an easy one. Vaccination against certain infections provides some protection for the individual patient and for the population, contributing to “herd immunity” and helping to protect against the occurrence of pandemics. But this is not the case for all vaccines. For some vaccines the issues of immunity and vaccination are more individual and complicated and warrant more education, reflection, and conversation.
The vaccine to protect against human papillomavirus is effective at reducing the incidence of cervical and anal cancers triggered by infection with certain strains of human papillomavirus. The viral infection itself is not immediately life-threatening. Thus, patients (and their parents) are asked to consider vaccination against a sexually transmitted virus (before sexual transmission of infection) to prevent a possible malignancy later in life. This decision can create social angst. Perhaps less socially challenging, but medically more complicated, is vaccination against the hepatitis B virus (HBV). HBV is also sexually transmissible, but the source in many patients infected with this virus is unclear. The HBV vaccine helps protect against clinically meaningful hepatitis and chronic liver disease from HBV and also will reduce the occurrence of HBV-associated hepatocellular carcinoma and progression of cirrhosis in patients coinfected with hepatitis C virus.
More complex yet is vaccination against the varicella-zoster virus (VZV) to reduce the likelihood of shingles and its possible consequence, postherpetic neuralgia. Le, Sabella, and Rothberg, in this issue of the Journal, review clinical and public health challenges affecting the use of this vaccine.
We are almost all exposed to this virus as children, through natural infection (chickenpox) or vaccination; many infections are seemingly subclinical. My older son had the distinct misfortune of getting chickenpox in a quite memorable way, developing a concentrated collection of the pruritic vesicles underneath a newly placed arm cast. Whether we remember the initial infection or not, VZV sets up housekeeping and lies dormant for decades within sensory neurons. Decades later, it may erupt as shingles along the distribution of the infected nerves with characteristic painful vesicles, sometimes with persistent, extremely painful, and debilitating residua within the same dermatomal distribution: postherpetic neuralgia. The triggers for this fairly common scenario are only generically understood and include waning cellular immunity attributed to aging, malignancy, immunosuppressive medications, human immunodeficiency virus, and perhaps stress and depression.
The zoster vaccine is unique in that it is given to bolster already present cellular immunity to prevent clinical recurrence of the earlier infection, decades after the virus has been dormant—not, as for the other vaccines noted above, to prevent primary infection. It doesn’t matter if the patient has already experienced an episode of shingles. The currently available vaccine is a live-attenuated strain (Oka) of VZV. Another vaccine in clinical testing uses isolated viral components with adjuvant and thus eliminates current concerns of giving the live-attenuated vaccine to immunosuppressed and elderly patients, those who may benefit the most from it.
Fortunately, it seems that even significantly immunosuppressed and elderly patients tolerate the current vaccine, but at present it is suggested that these groups not receive the vaccine, and vaccination rates in these patients is low. Hopefully, additional data will accumulate regarding the safety of the vaccine and will permit its more widespread use within these patient groups, or a replacement “dead” vaccine will become available.
As Le et al nicely discuss, the current vaccine efficacy wanes over about 10 years, and then a booster vaccine should be considered, but there are few data to provide safety and efficacy outcome measures from patients who received a booster vaccination. The potential need to receive a booster injection after about a decade, the demonstrated greater efficacy against zoster when the initial vaccination is provided to patients ages 60 through 69 than in those over 70, and the lower absolute impact when given to patients ages 50 through 59 (baseline zoster incidence increases with age) should enter into the conversation with patients as to when they should receive the vaccine.
Additionally, there are the extremely provocative and as yet unanswered questions surrounding the implications of vaccination if VZV infection is a trigger of inflammatory vascular diseases such as giant cell arteritis in the elderly, as was proposed by the late Dr. Don Gilden.1
And yes, I did get the vaccine. I was 64 at the time.
- Gilden D, Nagel MA. Varicella zoster virus triggers the immunopathology of giant cell arteritis. Curr Opin Rheumatol 2016; 28:376–382.
- Gilden D, Nagel MA. Varicella zoster virus triggers the immunopathology of giant cell arteritis. Curr Opin Rheumatol 2016; 28:376–382.