User login
Metabolic surgery for treating type 2 diabetes mellitus: Now supported by the world's leading diabetes organizations

LIMITATIONS OF LIFESTYLE MANAGEMENT AND MEDICATIONS
First-line therapy with lifestyle management and second-line therapy with medications, including oral agents and insulin, are the mainstays of type 2 DM therapy. Although these approaches have reduced hyperglycemia and cardiovascular mortality, many patients have poor glycemic control and develop severe diabetes-related complications. A study using data from the National Health and Nutrition Examination Survey (N = 4,926) to evaluate success rates of lifestyle management plus drug therapy found that just 53% of patients with type 2 DM maintained a hemoglobin A1c (HbA1c) below 7%.6 Similarly, only 51% of those patients achieved a systolic and diastolic blood pressure less than 130/80 mm Hg, and only 56% achieved a low-density lipoprotein cholesterol level less than 100 mg/dL. Altogether, only 19% of the study cohort achieved all 3 therapy targets. Documented limitations of lifestyle counseling and drug therapy include behavior maladaptation, limitations in drug potency, nonadherence to medications, adverse effects, and economic deterrents.7
METABOLIC SURGERY FOR TYPE 2 DM
For patients with obesity and type 2 DM in whom lifestyle management and medications do not achieve desired treatment goals, bariatric surgery has emerged as the most effective treatment for attaining significant and durable weight loss. These gastrointestinal (GI) procedures, which reduce gastric volume with or without rerouting nutrient flow through the small intestine, were developed to yield long-term weight loss in patients with severe obesity. It is now known that they also cause dramatic improvement or remission of obesity-related comorbidities, especially type 2 DM. Research has shown that these effects are not only secondary to weight loss but also depend on neuroendocrine mechanisms secondary to changes in GI physiology. For these reasons, bariatric surgery is increasingly used with the primary intent to treat type 2 DM or metabolic disease, a practice referred to as metabolic surgery.

For more than 2 decades, indications for metabolic surgery reflected guidelines from a 1991 National Institutes of Health (NIH) consensus conference, which suggested considering surgery only in patients with a BMI of 40 kg/m2 or greater or a BMI of 35 kg/m2 or greater and significant obesity-related comorbidities.11 Guidelines published in 2013 expanded the recommendations to include adults with a BMI of at least 35 kg/m2 and an obesity-related comorbidity, such as diabetes, who are motivated to lose weight.4 These recommendations were primarily designed to guide the use of surgery as a weight-loss intervention for severe obesity. However, guidelines published in 2016 support use of metabolic surgery as a specific treatment for type 2 DM.5
Potential mechanisms resolving type 2 DM: More than weight loss
Bariatric surgery has been shown to have profound glucoregulatory effects. These include rapid improvement in hyperglycemia and reduction in exogenous insulin requirements that occur early after surgery and before the patient has any significant weight loss.12,13 Additionally, experiments in rodents showed that changes to GI anatomy can directly influence glucose homeostasis, independently of weight loss and caloric restriction.14
Although the exact molecular mechanisms underlying the effects of metabolic surgery on diabetes are not fully understood, many factors appear to play a role, including changes in bile acid metabolism, GI tract nutrient sensing, glucose utilization, insulin resistance, and intestinal microbiomes.15 These changes, acting through peripheral or central pathways, or perhaps both, lead to reduced hepatic glucose production, increased tissue glucose uptake, improved insulin sensitivity, and enhanced beta-cell function. A constellation of gut-derived neuroendocrine changes, rather than a single overarching mechanism, is the likely mediator of postoperative glycemic improvement, with the contributing factors varying according to the surgical procedure.
METABOLIC SURGERY OUTCOMES
Weight loss
Long-term reduction of excess body fat is a major goal of metabolic and bariatric surgery. Weight loss is usually expressed as either the percent of weight loss or the percent of excess weight loss (ie, weight loss above ideal weight). A meta-analysis of mostly short-term weight-loss outcomes (ie, < 5 years) from more than 22,000 procedures found an overall mean excess weight loss of 47.5% for patients who underwent LAGB, 61.6% for RYGB, 68.2% for vertical-banded gastroplasty, and 70.1% for BPD-DS.16 Vertical-banded gastroplasty differs from LAGB in that both a band and staples are used to create a small stomach pouch. Excess weight loss for SG generally averages 50% to 55%, which is intermediate between LAGB and RYGB.17,18
The Swedish Obese Subjects study (N = 4,047), a prospective study of bariatric surgery vs nonsurgical weight management of severely obese patients (BMI > 34), is the largest weight-loss study with the longest follow-up.19 At 20 years, the mean weight loss was 26% for gastric bypass, 18% for vertical-banded gastroplasty, 13% for gastric banding, and 1% for controls. A 10-year study in 1,787 severely obese patients (BMI ≥ 35) who underwent RYGB had 21% more weight loss from their baseline weight than the nonsurgical match.20 At 4-year follow-up in 2,410 patients, there were significant variations in weight loss depending on the procedure: 27.5% for RYGB, 17.8% for SG, and 10.6% in LAGB. Between 2% and 31% regained weight back to baseline: 30.5% for LAGB, 14.6% for SG, and 2.5% for RYGB.20 In contrast, long-term medical (nonsurgical) weight loss rarely exceeds 5%, even with intensive lifestyle intervention.21
Diabetes remission, cardiovascular risk factors, glycemic control
A meta-analysis of 19 mostly observational studies (N = 4,070 patients) reported an overall type 2 DM remission rate of 78% after bariatric surgery with 1 to 3 years of follow-up.22 Resolution or remission was typically defined as becoming “nondiabetic” with normal HbA1c without medications. In the Swedish Obese Subjects study, the remission rate was 72% at 2 years and 36% at 10 years compared with 21% and 13%, respectively, for the nonsurgical controls (P < .001).23 Bariatric surgery was also markedly more effective than nonsurgical treatment in preventing type 2 DM, with a relative risk reduction of 78%.
A systematic review published in 2012 evaluated long-term cardiovascular risk reduction after bariatric surgery in 73 studies and 19,543 patients.24 At a mean follow-up of 57.8 months, the average excess weight loss for all procedures was 54% and rates of remission or improvement were 63% for hypertension, 73% for type 2 DM, and 65% for hyperlipidemia. Results from 12 cohort-matched, nonrandomized studies comparing bariatric surgery vs nonsurgical controls suggest that improvements in surrogate disease markers such as HbA1c, blood pressure, lipids, and body weight after surgery translate to reduced macrovascular and microvascular events and death.25 One of these studies involving male veterans who were mostly at high cardiovascular risk reported a 42% reduction in mortality at 10 years compared with medical therapy.26
In the Swedish Obese Subjects study, the mortality rate from cardiovascular disease in the bariatric surgical group was lower than for control patients (adjusted hazard ratio, 0.47; P = .002) despite a greater prevalence of smoking and higher baseline weights and blood pressures in the surgical cohort.19 For patients with type 2 DM in this study, surgery was associated with a 50% reduction in microvascular complications.27 After 15 years of follow-up, the cumulative incidence of microvascular complications was 41.8 per 1,000 person-years for control patients and 20.6 per 1,000 person-years in the surgery group (hazard ratio, 0.44; P < .001).
These observational, nonrandomized study data suggest that in patients with type 2 DM, bariatric surgery is significantly better than medical management alone in improving glycemic control, reducing cardiovascular risk factors, and lowering long-term morbidity and mortality associated with type 2 DM.
METABOLIC SURGERY: CLINICAL TRIALS

Collectively, these RCTs showed that surgery was significantly superior to medical treatment in reaching the designated glycemic target (P < .05 for all). The one exception showed that diabetes remission for LAGB vs medical treatment was 33% and 23%, respectively.41 This result might be due to patients in this study having advanced type 2 DM (HbA1c 8.2% ± 1.2%, with 40% on insulin), and they likely had reduced beta-cell function. Overall, surgery decreased HbA1c by 2% to 3.5%, whereas medical treatment lowered it by only 1% to 1.5%. Most of these studies also showed superiority of surgery over medical treatment in achieving secondary end points such as weight loss, remission of metabolic syndrome, reduction in diabetes and cardiovascular medications, and improvement in triglycerides, lipids, and quality of life. Results were mixed in terms of improvements in systolic and diastolic blood pressure or low-density lipoproteins after surgery vs medical treatment, but many studies did show a corresponding reduction in medication usage.
Durability of the effects of surgery was demonstrated in a 5-year study that showed superior and durable weight loss and glycemic control (remission) with both RYGB and BPD in severely obese patients (BMI ≥ 35) vs medical therapy.32 Similarly, Schauer et al43 showed that RYGB and SG were more effective than intensive medical therapy in improving or, in some cases, resolving hyperglycemia for 5 years. In the RCTs, patients who preoperatively had shorter duration of diabetes, lower HbA1c levels, no insulin requirement, and more postoperative weight loss were more likely to achieve diabetes remission.
Although previous guidelines and payer coverage policies had limited metabolic surgery to severely obese patients (BMI ≥ 35 kg/m2), nearly all RCTs showed that the surgical procedures, especially RYGB and SG, were equally effective in patients with BMI 30 to 35 kg/m2. This is particularly important given that most patients with type 2 DM have a BMI less than 35 kg/m2. The effect of surgery in these patients with mild obesity is also durable out to at least 5 years.43
No RCT was sufficiently powered to detect differences in macrovascular or microvascular complications or death, especially at the relatively short follow-up, and no such differences have been detected thus far. The STAMPEDE (Surgical Therapy and Medications Potentially Eradicate Diabetes Efficiently) trial43 showed that bariatric surgery (RYGB or SG) did not appear to worsen or improve retinopathy outcomes at 5 years compared with intensive medical management.
METABOLIC SURGERY: ADVERSE EVENTS
Surgical complications

.")

Nutritional deficiencies
Postoperative nutritional deficiencies are typically associated with diminished nutrient intake or the malabsorptive effect of bariatric procedures. They are more common after RYGB and BPD-DS and less common after SG and LAGB. In addition, there is a high prevalence of nutritional deficiencies (35%–80%) in patients seeking bariatric surgery; thus, poor preoperative nutrition may be a factor in the development of postoperative deficiencies. Common preoperative nutrient deficiencies are vitamin A (11%), vitamin B12 (13%), vitamin D (40%), zinc (30%), iron (16%), ferritin (9%), selenium (58%), and folate (6%).51 Recommendations are to assess for these deficiencies and correct any identified before surgery.
Mild anemia after bariatric procedures is common, occurring in 15% to 20% of cases, and it is believed to result from reduced absorption of iron and B12, as well from pre-existing iron deficiency anemia in premenopausal patients.52 Deficiencies in trace minerals (selenium, zinc, and copper) and vitamins (B12, B1, A, E, D, and K) can occur after bariatric procedures, especially after BPD-DS.53 Nutrient deficiencies can be prevented or corrected with appropriate vitamin, iron, and calcium supplementation.54
Bone mineral density may decrease after bariatric surgery (14% in the proximal femur).55 Reduced mechanical loading after weight loss, reduced consumption and malabsorption of micronutrients (calcium, vitamin D), and neurohormonal alterations are potential underlying mechanisms of bone mineral density reduction after bariatric surgery. Rates of bone fracture and osteoporosis are not well delineated, raising questions about whether bone loss after bariatric surgery is clinically relevant or a functional adaptation to skeletal unloading. However, the extreme malabsorptive procedures of BPD-DS have been associated with severe calcium and vitamin D deficiencies, leading to decreased bone mineral density and osteoporosis.
Protein malnutrition also can occur after these extreme malabsorptive procedures. Patients require postoperative oral protein supplementation (80–100 g/day) and lifelong monitoring for nutritional complications after these procedures.56
Additional complications
Other late complications of bariatric surgery that are less clear in incidence and cause include kidney stones, alcohol abuse, depression, and suicide. One study of patients after RYGB (N = 4,690) reported a significantly higher prevalence of kidney stones than in obese controls: 7.5% vs 4.6%, respectively.57 Proposed causes of kidney stone formation following bariatric surgery include hyperoxaluria, hypocitraturia, and elevated urine acidity.58
The prevalence of alcohol-use disorder after bariatric surgery ranges from 7.6% to 11.8% and appears to be higher in patients with a history of alcohol use.59 Paradoxically, while bariatric surgery has been shown to significantly decrease depression,60 some studies suggest that a slight increase in the risk of suicide may occur,61 while others do not.62 A recent review concluded that accurate rates of suicide after bariatric surgery are not known, but practitioners should be aware of this concern and appropriately screen and counsel their patients.63
Although the 12 RCTs reported in Table 1 were not powered to detect differences in treatment-related complications, the overall rates of complications were consistent with those in observational studies.9 The most common surgical complications were anemia (15%), need for reoperation (8%), and GI (5%–10%). The 30-day surgical mortality rate was 0.2% (1 death) among the 465 surgical patients. Complications were not limited to the surgical patients. In the medical-treatment control group of the STAMPEDE trial,30 anemia (16%) and weight gain (16%) were common. Investigators reported challenges with medication compliance, including adverse effects leading to discontinuation of medications. Mild hypoglycemia was common, with no significant differences between the surgical and medical treatment groups.
METABOLIC SURGERY: COST EFFECTIVENESS
The cost of bariatric procedures varies considerably but, in general, ranges from $20,000 to $30,000, similar to the cost of cholecystectomy, hysterectomy, and colectomy. Retrospective analyses and modeling studies indicate that metabolic surgery is cost-effective and may present a cost savings in patients with type 2 DM, with a break-even time between 5 and 10 years.64,65 The cost savings, largely based on assumptions of long-term effectiveness and safety, result from reductions in medication use, outpatient care costs, and long-term complications of type 2 DM.
WHO SHOULD HAVE METABOLIC SURGERY?
Until recently, there was no clear national or international consensus on the role of metabolic surgery in treating type 2 DM. In 2015, the 2nd Diabetes Surgery Summit (DSS-II) Consensus Conference published guidelines that were endorsed by more than 50 diabetes and medical organizations.5 The recommendations cover many clinically relevant issues, including patient selection, preoperative evaluation, choice of procedure, and postoperative follow-up. The consensus conference delegates concluded that there is sufficient evidence demonstrating that metabolic surgery achieves excellent glycemic control and reduces cardiovascular risk factors.

The treatment algorithm from DSS-II incorporates appropriate use of all 3 treatment modalities: lifestyle intervention, drug therapy, and surgery (Figure 5).5 The 2017 Standards of Care for Diabetes from the American Diabetes Association include those key indications in the recommendations for metabolic surgery (Table 3).2
SUMMARY

The safety of metabolic surgery has significantly improved with the advent of laparoscopic surgery and recent national quality improvement initiatives that have made gastric bypass and SG as safe as cholecystectomy and appendectomy. Although observational studies suggest that metabolic surgery is associated with a reduction in cardiovascular and diabetes complications and mortality, these observations have not been confirmed in long-term RCTs.
Based on the published evidence, metabolic surgery is now endorsed as a standard treatment option, which provides patients and practitioners with a powerful tool to help combat the life-impairing effects of type 2 DM.
- Bays HE, Chapman RH, Grandy S; for the SHIELD Investigators Group. The relationship of body mass index to diabetes mellitus, hypertension and dyslipidaemia: comparison of data from two national surveys. Int J Clin Pract May 2007; 61:737–747.
- Marathe PH, Gao HX, Close KL. American Diabetes Association standards of medical care in diabetes—2017. Diabetes Care 2017; 40(suppl 1):S1–S135.
- Fox CS, Golden SH, Anderson C, et al; American Heart Association; American Diabetes Association. Update on prevention of cardiovascular disease in adults with type 2 diabetes mellitus in light of recent evidence: a scientific statement from the American Heart Association and the American Diabetes Association. Circulation 2015; 132:691–718.
- Jensen MD, Ryan DH, Apovian CM, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines; Obesity Society. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and The Obesity Society. J Am Coll Cardiol 2014; 63:2985–3023.
- Rubino F, Nathan DM, Eckel RH, et al; Delegates of the 2nd Diabetes Surgery Summit. Metabolic surgery in the treatment algorithm for type 2 diabetes: a joint statement by international diabetes organizations. Diabetes Care 2016; 39:861–877.
- Stark Casagrande S, Fradkin JE, Saydah SH, Rust KF, Cowie CC. The prevalence of meeting A1C, blood pressure, and LDL goals among people with diabetes, 1988–2010. Diabetes Care 2013; 36:2271–2279.
- Kolandaivelu K, Leiden BB, O’Gara PT, Bhatt DL. Non-adherence to cardiovascular medications. Eur Heart J 2014; 35:3267–3276.
- Angrisani L, Santonicola A, Iovino P, Formisano G, Buchwald H, Scopinaro N. Bariatric surgery worldwide 2013. Obes Surg 2015; 25:1822–1832.
- Schauer PR, Mingrone G, Ikramuddin S, Wolfe B. Clinical outcomes of metabolic surgery: efficacy of glycemic control, weight loss, and remission of diabetes. Diabetes Care 2016; 39:902–911.
- Khorgami Z, Andalib A, Corcelles R, Aminian A, Brethauer S, Schauer P. Recent national trends in the surgical treatment of obesity: sleeve gastrectomy dominates. Surg Obes Relat Dis 2015; 11(suppl):S1–S34 [Abstract A111].
- Consensus Development Conference Panel. NIH conference. Gastrointestinal surgery for severe obesity. Ann Intern Med 1991; 115:956–961.
- Pories WJ, MacDonald KG Jr, Flickinger EG, et al. Is type II diabetes mellitus (NIDDM) a surgical disease? Ann Surg 1992; 215:633–642.
- Schauer PR, Burguera B, Ikramuddin S, et al. Effect of laparoscopic Roux-en Y gastric bypass on type 2 diabetes mellitus. Ann Surg 2003; 238:467–484.
- Rubino F, Marescaux J. Effect of duodenal-jejunal exclusion in a non-obese animal model of type 2 diabetes: a new perspective for an old disease. Ann Surg 2004; 239:1–11.
- Batterham RL, Cummings DE. Mechanisms of diabetes improvement following bariatric/metabolic surgery. Diabetes Care 2016; 39:893–901.
- Buchwald H, Avidor Y, Braunwald E, et al. Bariatric surgery: a systematic review and meta-analysis. JAMA 2004; 292:1724–1737.
- Brethauer SA, Hammel JP, Schauer PR. Systematic review of sleeve gastrectomy as staging and primary bariatric procedure. Surg Obes Relat Dis 2009; 5:469–475.
- Eid GM, Brethauer S, Mattar SG, Titchner RL, Gourash W, Schauer PR. Laparoscopic sleeve gastrectomy for super obese patients: forty-eight percent excess weight loss after 6 to 8 years with 93% follow-up. Ann Surg 2012; 256:262–265.
- Sjöström L, Peltonen M, Jacobson P, et al. Bariatric surgery and long-term cardiovascular events. JAMA 2012; 307:56–65.
- Maciejewski ML, Arterburn DE, Van Scoyoc L, et al. Bariatric surgery and long-term durability of weight loss. JAMA Surg 2016; 151:1046–1055.
- Wing RR, Bolin P, Brancati FL, et al; for the Look AHEAD Research Group. Cardiovascular effects of intensive lifestyle intervention in type 2 diabetes. N Engl J Med 2013; 369:145–154.
- Buchwald H, Estok R, Fahrbach K, et al. Weight and type 2 diabetes after bariatric surgery: systematic review and meta-analysis. Am J Med 2009; 122:248–256.
- Sjöström L, Lindroos AK, Peltonen M, et al; Swedish Obese Subjects Study Scientific Group. Lifestyle, diabetes, and cardiovascular risk factors 10 years after bariatric surgery. N Engl J Med 2004; 351:2683–2693.
- Vest AR, Heneghan HM, Agarwal S, Schauer PR, Young JB. Bariatric surgery and cardiovascular outcomes: a systematic review. Heart 2012; 98:1763–1777.
- Vest AR, Heneghan HM, Schauer PR, Young JB. Surgical management of obesity and the relationship to cardiovascular disease. Circulation 2013; 127:945–959.
- Arterburn DE, Olsen MK, Smith VA, et al. Association between bariatric surgery and long-term survival. JAMA 2015; 313:62–70.
- Sjöström L, Peltonen M, Jacobson P, et al. Association of bariatric surgery with long-term remission of type 2 diabetes and with microvascular and macrovascular complications. JAMA 2014; 311:2297–2304.
- Dixon JB, O’Brien PE, Playfair J, et al. Adjustable gastric banding and conventional therapy for type 2 diabetes: a randomized controlled trial. JAMA 2008; 299:316–323.
- Schauer PR, Kashyap SR, Wolski K, et al. Bariatric surgery versus intensive medical therapy in obese patients with diabetes. N Engl J Med 2012; 366:1567–1576.
- Schauer PR, Bhatt DL, Kirwan JP, et al; STAMPEDE Investigators. Bariatric surgery versus intensive medical therapy for diabetes—3-year outcomes. N Engl J Med 2014; 370:2002–2013.
- Mingrone G, Panunzi S, De Gaetano A, et al. Bariatric surgery versus conventional medical therapy for type 2 diabetes. N Engl J Med 2012; 366:1577–1585.
- Mingrone G, Panunzi S, De Gaetano A, et al. Bariatric-metabolic surgery versus conventional medical treatment in obese patients with type 2 diabetes: 5 year follow-up of an open-label, single-centre, randomized controlled trial. Lancet 2015; 386:964–973.
- Ikramuddin S, Korner J, Lee WJ, et al. Roux-en-Y gastric bypass vs intensive medical management for the control of type 2 diabetes, hypertension, and hyperlipidemia: the Diabetes Surgery Study randomized clinical trial. JAMA 2013; 309:2240–2249.
- Ikramuddin S, Billington CJ, Lee WJ, et al. Roux-en-Y gastric bypass for diabetes (the Diabetes Surgery Study): 2-year outcomes of a 5-year, randomized, controlled trial. Lancet Diabetes Endocrinol 2015; 3:413–422.
- Liang Z, Wu Q, Chen B, Yu P, Zhao H, Ouyang X. Effect of laparoscopic Roux-en-Y gastric bypass surgery on type 2 diabetes mellitus with hypertension: a randomized controlled trial. Diabetes Res Clin Pract 2013; 101:50–56.
- Halperin F, Ding SA, Simonson DC, et al. Roux-en-Y gastric bypass surgery or lifestyle with intensive medical management in patients with type 2 diabetes: feasibility and 1-year results of a randomized clinical trial. JAMA Surg 2014; 149:716–726.
- Courcoulas AP, Goodpaster BH, Eagleton JK, et al. Surgical vs medical treatments for type 2 diabetes mellitus: a randomized clinical trial. JAMA Surg 2014; 149:707–715.
- Courcoulas AP, Belle SH, Neiberg RH, et al. Three-year outcomes of bariatric surgery vs. lifestyle intervention for type 2 diabetes mellitus treatment: a randomized clinical trial. JAMA Surg 2015; 150:931–940.
- Wentworth JM, Playfair J, Laurie C, et al. Multidisciplinary diabetes care with and without bariatric surgery in overweight people: a randomised controlled trial. Lancet Diabetes Endocrinol 2014; 2:545–552.
- Parikh M, Chung M, Sheth S, et al. Randomized pilot trial of bariatric surgery versus intensive medical weight management on diabetes remission in type 2 diabetic patients who do not meet NIH criteria for surgery and the role of soluble RAGE as a novel biomarker of success. Ann Surg 2014; 260:617–622.
- Ding SA, Simonson DC, Wewalka M, et al. Adjustable gastric band surgery or medical management in patients with type 2 diabetes: a randomized clinical trial. J Clin Endocrinol Metab 2015; 100:2546–2556.
- Cummings DE, Arterburn DE, Westbrook EO, et al. Gastric bypass surgery vs. intensive lifestyle and medical intervention for type 2 diabetes: the CROSSROADS randomized controlled trial. Diabetologia 2016; 59:945–953.
- Schauer PR, Bhatt DL, Kirwan JP, et al; STAMPEDE Investigators. Metabolic surgery vs. intensive medical therapy for diabetes: 5-year outcomes. N Engl J Med 2017; 376:641–651.
- Shah SS, Todkar J, Phadake U, et al. Gastric bypass vs. medical/lifestyle care for type 2 diabetes in South Asians with BMI 25-40 kg/m2: the COSMID randomized trial [261-OR]. Presented at the American Diabetes Association’s 76th Scientific Session; June 10–14, 2016; New Orleans, LA.
- Flum DR, Belle SH, King WC, et al; Longitudinal Assessment of Bariatric Surgery (LABS) Consortium. Perioperative safety in the longitudinal assessment of bariatric surgery. N Engl J Med 2009; 361:445–454.
- Aminian A, Brethauer SA, Kirwan JP, Kashyap SR, Burguera B, Schauer PR. How safe is metabolic/diabetes surgery? Diabetes Obes Metab 2015; 17:198–201.
- Thodiyil PA, Yenumula P, Rogula T, et al. Selective non operative management of leaks after gastric bypass: lessons learned from 2675 consecutive patients. Ann Surg 2008; 248:782–792.
- Rogula T, Yenumula PR, Schauer PR. A complication of Roux-en-Y gastric bypass: intestinal obstruction. Surg Endosc 2007; 21:1914–1918.
- Thornton CM, Rozen WM, So D, Kaplan ED, Wilkinson S. Reducing band slippage in laparoscopic adjustable gastric banding: the mesh plication pars flaccida technique. Obes Surg 2009; 19:1702–1706.
- Himpens J, Cadière G-B, Bazi M, Vouche M, Cadière B, Dapri G. Long-term outcomes of laparoscopic adjustable gastric banding. Arch Surg 2011; 146:802–807.
- Madan AK, Orth WS, Tichansky DS, Ternovits CA. Vitamin and trace mineral levels after laparoscopic gastric bypass. Obes Surg 2006; 16:603–606.
- Love AL, Billett HH. Obesity, bariatric surgery, and iron deficiency: true, true, true and related. Am J Hematol 2008; 83:403–409.
- Shankar P, Boylan M, Sriram K. Micronutrient deficiencies after bariatric surgery. Nutrition 2010; 26:1031–1037.
- Gong K, Gagner M, Pomp A, Almahmeed T, Bardaro SJ. Micronutrient deficiencies after laparoscopic gastric bypass: recommendations. Obes Surg 2008; 18:1062–1066.
- Scibora LM. Skeletal effects of bariatric surgery: examining bone loss, potential mechanisms and clinical relevance. Diabetes Obes Metab 2014; 16:1204–1213.
- Baptista V, Wassef W. Bariatric procedures: an update on techniques, outcomes and complications. Curr Opin Gastroenterol 2013; 29:684–693.
- Matlaga BR, Shore AD, Magnuson T, Clark JM, Johns R, Makary MA. Effect of gastric bypass surgery on kidney stone disease. J Urol 2009; 181:2573–2577.
- Sakhaee K, Poindexter J, Aguirre C. The effects of bariatric surgery on bone and nephrolithiasis. Bone 2016; 84:1–8.
- Li L, Wu LT. Substance use after bariatric surgery: a review. J Psychiatr Res 2016; 76:16–29.
- Ayloo S, Thompson K, Choudhury N, Sheriffdeen R. Correlation between the Beck Depression Inventory and bariatric surgical procedures. Surg Obes Relat Dis 2015; 11:637–342.
- Adams TD, Gress RE, Smith SC, et al. Long-term mortality after gastric bypass surgery. N Engl J Med 2007; 357:753–761.
- Sjöström L, Narbro K, Sjöström CD, et al; Swedish Obese Subjects Study. Effects of bariatric surgery on mortality in Swedish obese subjects. N Engl J Med 2007; 357:741–752.
- Mitchell JE, Crosby R, de Zwaan M, et al. Possible risk factors for increased suicide following bariatric surgery. Obesity (Silver Spring) 2013; 21:665–672.
- Fouse T, Schauer P. The socioeconomic impact of morbid obesity and factors affecting access to obesity surgery. Surg Clin North Am 2016; 96:669–679.
- Rubin JK, Hinrichs-Krapels S, Hesketh R, Martin A, Herman WH, Rubino F. Identifying barriers to appropriate use of metabolic/bariatric surgery for type 2 diabetes treatment: policy lab results. Diabetes Care 2016; 39:954–963.
LIMITATIONS OF LIFESTYLE MANAGEMENT AND MEDICATIONS
First-line therapy with lifestyle management and second-line therapy with medications, including oral agents and insulin, are the mainstays of type 2 DM therapy. Although these approaches have reduced hyperglycemia and cardiovascular mortality, many patients have poor glycemic control and develop severe diabetes-related complications. A study using data from the National Health and Nutrition Examination Survey (N = 4,926) to evaluate success rates of lifestyle management plus drug therapy found that just 53% of patients with type 2 DM maintained a hemoglobin A1c (HbA1c) below 7%.6 Similarly, only 51% of those patients achieved a systolic and diastolic blood pressure less than 130/80 mm Hg, and only 56% achieved a low-density lipoprotein cholesterol level less than 100 mg/dL. Altogether, only 19% of the study cohort achieved all 3 therapy targets. Documented limitations of lifestyle counseling and drug therapy include behavior maladaptation, limitations in drug potency, nonadherence to medications, adverse effects, and economic deterrents.7
METABOLIC SURGERY FOR TYPE 2 DM
For patients with obesity and type 2 DM in whom lifestyle management and medications do not achieve desired treatment goals, bariatric surgery has emerged as the most effective treatment for attaining significant and durable weight loss. These gastrointestinal (GI) procedures, which reduce gastric volume with or without rerouting nutrient flow through the small intestine, were developed to yield long-term weight loss in patients with severe obesity. It is now known that they also cause dramatic improvement or remission of obesity-related comorbidities, especially type 2 DM. Research has shown that these effects are not only secondary to weight loss but also depend on neuroendocrine mechanisms secondary to changes in GI physiology. For these reasons, bariatric surgery is increasingly used with the primary intent to treat type 2 DM or metabolic disease, a practice referred to as metabolic surgery.
For more than 2 decades, indications for metabolic surgery reflected guidelines from a 1991 National Institutes of Health (NIH) consensus conference, which suggested considering surgery only in patients with a BMI of 40 kg/m2 or greater or a BMI of 35 kg/m2 or greater and significant obesity-related comorbidities.11 Guidelines published in 2013 expanded the recommendations to include adults with a BMI of at least 35 kg/m2 and an obesity-related comorbidity, such as diabetes, who are motivated to lose weight.4 These recommendations were primarily designed to guide the use of surgery as a weight-loss intervention for severe obesity. However, guidelines published in 2016 support use of metabolic surgery as a specific treatment for type 2 DM.5
Potential mechanisms resolving type 2 DM: More than weight loss
Bariatric surgery has been shown to have profound glucoregulatory effects. These include rapid improvement in hyperglycemia and reduction in exogenous insulin requirements that occur early after surgery and before the patient has any significant weight loss.12,13 Additionally, experiments in rodents showed that changes to GI anatomy can directly influence glucose homeostasis, independently of weight loss and caloric restriction.14
Although the exact molecular mechanisms underlying the effects of metabolic surgery on diabetes are not fully understood, many factors appear to play a role, including changes in bile acid metabolism, GI tract nutrient sensing, glucose utilization, insulin resistance, and intestinal microbiomes.15 These changes, acting through peripheral or central pathways, or perhaps both, lead to reduced hepatic glucose production, increased tissue glucose uptake, improved insulin sensitivity, and enhanced beta-cell function. A constellation of gut-derived neuroendocrine changes, rather than a single overarching mechanism, is the likely mediator of postoperative glycemic improvement, with the contributing factors varying according to the surgical procedure.
METABOLIC SURGERY OUTCOMES
Weight loss
Long-term reduction of excess body fat is a major goal of metabolic and bariatric surgery. Weight loss is usually expressed as either the percent of weight loss or the percent of excess weight loss (ie, weight loss above ideal weight). A meta-analysis of mostly short-term weight-loss outcomes (ie, < 5 years) from more than 22,000 procedures found an overall mean excess weight loss of 47.5% for patients who underwent LAGB, 61.6% for RYGB, 68.2% for vertical-banded gastroplasty, and 70.1% for BPD-DS.16 Vertical-banded gastroplasty differs from LAGB in that both a band and staples are used to create a small stomach pouch. Excess weight loss for SG generally averages 50% to 55%, which is intermediate between LAGB and RYGB.17,18
The Swedish Obese Subjects study (N = 4,047), a prospective study of bariatric surgery vs nonsurgical weight management of severely obese patients (BMI > 34), is the largest weight-loss study with the longest follow-up.19 At 20 years, the mean weight loss was 26% for gastric bypass, 18% for vertical-banded gastroplasty, 13% for gastric banding, and 1% for controls. A 10-year study in 1,787 severely obese patients (BMI ≥ 35) who underwent RYGB had 21% more weight loss from their baseline weight than the nonsurgical match.20 At 4-year follow-up in 2,410 patients, there were significant variations in weight loss depending on the procedure: 27.5% for RYGB, 17.8% for SG, and 10.6% in LAGB. Between 2% and 31% regained weight back to baseline: 30.5% for LAGB, 14.6% for SG, and 2.5% for RYGB.20 In contrast, long-term medical (nonsurgical) weight loss rarely exceeds 5%, even with intensive lifestyle intervention.21
Diabetes remission, cardiovascular risk factors, glycemic control
A meta-analysis of 19 mostly observational studies (N = 4,070 patients) reported an overall type 2 DM remission rate of 78% after bariatric surgery with 1 to 3 years of follow-up.22 Resolution or remission was typically defined as becoming “nondiabetic” with normal HbA1c without medications. In the Swedish Obese Subjects study, the remission rate was 72% at 2 years and 36% at 10 years compared with 21% and 13%, respectively, for the nonsurgical controls (P < .001).23 Bariatric surgery was also markedly more effective than nonsurgical treatment in preventing type 2 DM, with a relative risk reduction of 78%.
A systematic review published in 2012 evaluated long-term cardiovascular risk reduction after bariatric surgery in 73 studies and 19,543 patients.24 At a mean follow-up of 57.8 months, the average excess weight loss for all procedures was 54% and rates of remission or improvement were 63% for hypertension, 73% for type 2 DM, and 65% for hyperlipidemia. Results from 12 cohort-matched, nonrandomized studies comparing bariatric surgery vs nonsurgical controls suggest that improvements in surrogate disease markers such as HbA1c, blood pressure, lipids, and body weight after surgery translate to reduced macrovascular and microvascular events and death.25 One of these studies involving male veterans who were mostly at high cardiovascular risk reported a 42% reduction in mortality at 10 years compared with medical therapy.26
In the Swedish Obese Subjects study, the mortality rate from cardiovascular disease in the bariatric surgical group was lower than for control patients (adjusted hazard ratio, 0.47; P = .002) despite a greater prevalence of smoking and higher baseline weights and blood pressures in the surgical cohort.19 For patients with type 2 DM in this study, surgery was associated with a 50% reduction in microvascular complications.27 After 15 years of follow-up, the cumulative incidence of microvascular complications was 41.8 per 1,000 person-years for control patients and 20.6 per 1,000 person-years in the surgery group (hazard ratio, 0.44; P < .001).
These observational, nonrandomized study data suggest that in patients with type 2 DM, bariatric surgery is significantly better than medical management alone in improving glycemic control, reducing cardiovascular risk factors, and lowering long-term morbidity and mortality associated with type 2 DM.
METABOLIC SURGERY: CLINICAL TRIALS
Collectively, these RCTs showed that surgery was significantly superior to medical treatment in reaching the designated glycemic target (P < .05 for all). The one exception showed that diabetes remission for LAGB vs medical treatment was 33% and 23%, respectively.41 This result might be due to patients in this study having advanced type 2 DM (HbA1c 8.2% ± 1.2%, with 40% on insulin), and they likely had reduced beta-cell function. Overall, surgery decreased HbA1c by 2% to 3.5%, whereas medical treatment lowered it by only 1% to 1.5%. Most of these studies also showed superiority of surgery over medical treatment in achieving secondary end points such as weight loss, remission of metabolic syndrome, reduction in diabetes and cardiovascular medications, and improvement in triglycerides, lipids, and quality of life. Results were mixed in terms of improvements in systolic and diastolic blood pressure or low-density lipoproteins after surgery vs medical treatment, but many studies did show a corresponding reduction in medication usage.
Durability of the effects of surgery was demonstrated in a 5-year study that showed superior and durable weight loss and glycemic control (remission) with both RYGB and BPD in severely obese patients (BMI ≥ 35) vs medical therapy.32 Similarly, Schauer et al43 showed that RYGB and SG were more effective than intensive medical therapy in improving or, in some cases, resolving hyperglycemia for 5 years. In the RCTs, patients who preoperatively had shorter duration of diabetes, lower HbA1c levels, no insulin requirement, and more postoperative weight loss were more likely to achieve diabetes remission.
Although previous guidelines and payer coverage policies had limited metabolic surgery to severely obese patients (BMI ≥ 35 kg/m2), nearly all RCTs showed that the surgical procedures, especially RYGB and SG, were equally effective in patients with BMI 30 to 35 kg/m2. This is particularly important given that most patients with type 2 DM have a BMI less than 35 kg/m2. The effect of surgery in these patients with mild obesity is also durable out to at least 5 years.43
No RCT was sufficiently powered to detect differences in macrovascular or microvascular complications or death, especially at the relatively short follow-up, and no such differences have been detected thus far. The STAMPEDE (Surgical Therapy and Medications Potentially Eradicate Diabetes Efficiently) trial43 showed that bariatric surgery (RYGB or SG) did not appear to worsen or improve retinopathy outcomes at 5 years compared with intensive medical management.
METABOLIC SURGERY: ADVERSE EVENTS
Surgical complications
Nutritional deficiencies
Postoperative nutritional deficiencies are typically associated with diminished nutrient intake or the malabsorptive effect of bariatric procedures. They are more common after RYGB and BPD-DS and less common after SG and LAGB. In addition, there is a high prevalence of nutritional deficiencies (35%–80%) in patients seeking bariatric surgery; thus, poor preoperative nutrition may be a factor in the development of postoperative deficiencies. Common preoperative nutrient deficiencies are vitamin A (11%), vitamin B12 (13%), vitamin D (40%), zinc (30%), iron (16%), ferritin (9%), selenium (58%), and folate (6%).51 Recommendations are to assess for these deficiencies and correct any identified before surgery.
Mild anemia after bariatric procedures is common, occurring in 15% to 20% of cases, and it is believed to result from reduced absorption of iron and B12, as well from pre-existing iron deficiency anemia in premenopausal patients.52 Deficiencies in trace minerals (selenium, zinc, and copper) and vitamins (B12, B1, A, E, D, and K) can occur after bariatric procedures, especially after BPD-DS.53 Nutrient deficiencies can be prevented or corrected with appropriate vitamin, iron, and calcium supplementation.54
Bone mineral density may decrease after bariatric surgery (14% in the proximal femur).55 Reduced mechanical loading after weight loss, reduced consumption and malabsorption of micronutrients (calcium, vitamin D), and neurohormonal alterations are potential underlying mechanisms of bone mineral density reduction after bariatric surgery. Rates of bone fracture and osteoporosis are not well delineated, raising questions about whether bone loss after bariatric surgery is clinically relevant or a functional adaptation to skeletal unloading. However, the extreme malabsorptive procedures of BPD-DS have been associated with severe calcium and vitamin D deficiencies, leading to decreased bone mineral density and osteoporosis.
Protein malnutrition also can occur after these extreme malabsorptive procedures. Patients require postoperative oral protein supplementation (80–100 g/day) and lifelong monitoring for nutritional complications after these procedures.56
Additional complications
Other late complications of bariatric surgery that are less clear in incidence and cause include kidney stones, alcohol abuse, depression, and suicide. One study of patients after RYGB (N = 4,690) reported a significantly higher prevalence of kidney stones than in obese controls: 7.5% vs 4.6%, respectively.57 Proposed causes of kidney stone formation following bariatric surgery include hyperoxaluria, hypocitraturia, and elevated urine acidity.58
The prevalence of alcohol-use disorder after bariatric surgery ranges from 7.6% to 11.8% and appears to be higher in patients with a history of alcohol use.59 Paradoxically, while bariatric surgery has been shown to significantly decrease depression,60 some studies suggest that a slight increase in the risk of suicide may occur,61 while others do not.62 A recent review concluded that accurate rates of suicide after bariatric surgery are not known, but practitioners should be aware of this concern and appropriately screen and counsel their patients.63
Although the 12 RCTs reported in Table 1 were not powered to detect differences in treatment-related complications, the overall rates of complications were consistent with those in observational studies.9 The most common surgical complications were anemia (15%), need for reoperation (8%), and GI (5%–10%). The 30-day surgical mortality rate was 0.2% (1 death) among the 465 surgical patients. Complications were not limited to the surgical patients. In the medical-treatment control group of the STAMPEDE trial,30 anemia (16%) and weight gain (16%) were common. Investigators reported challenges with medication compliance, including adverse effects leading to discontinuation of medications. Mild hypoglycemia was common, with no significant differences between the surgical and medical treatment groups.
METABOLIC SURGERY: COST EFFECTIVENESS
The cost of bariatric procedures varies considerably but, in general, ranges from $20,000 to $30,000, similar to the cost of cholecystectomy, hysterectomy, and colectomy. Retrospective analyses and modeling studies indicate that metabolic surgery is cost-effective and may present a cost savings in patients with type 2 DM, with a break-even time between 5 and 10 years.64,65 The cost savings, largely based on assumptions of long-term effectiveness and safety, result from reductions in medication use, outpatient care costs, and long-term complications of type 2 DM.
WHO SHOULD HAVE METABOLIC SURGERY?
Until recently, there was no clear national or international consensus on the role of metabolic surgery in treating type 2 DM. In 2015, the 2nd Diabetes Surgery Summit (DSS-II) Consensus Conference published guidelines that were endorsed by more than 50 diabetes and medical organizations.5 The recommendations cover many clinically relevant issues, including patient selection, preoperative evaluation, choice of procedure, and postoperative follow-up. The consensus conference delegates concluded that there is sufficient evidence demonstrating that metabolic surgery achieves excellent glycemic control and reduces cardiovascular risk factors.
The treatment algorithm from DSS-II incorporates appropriate use of all 3 treatment modalities: lifestyle intervention, drug therapy, and surgery (Figure 5).5 The 2017 Standards of Care for Diabetes from the American Diabetes Association include those key indications in the recommendations for metabolic surgery (Table 3).2
SUMMARY
The safety of metabolic surgery has significantly improved with the advent of laparoscopic surgery and recent national quality improvement initiatives that have made gastric bypass and SG as safe as cholecystectomy and appendectomy. Although observational studies suggest that metabolic surgery is associated with a reduction in cardiovascular and diabetes complications and mortality, these observations have not been confirmed in long-term RCTs.
Based on the published evidence, metabolic surgery is now endorsed as a standard treatment option, which provides patients and practitioners with a powerful tool to help combat the life-impairing effects of type 2 DM.
LIMITATIONS OF LIFESTYLE MANAGEMENT AND MEDICATIONS
First-line therapy with lifestyle management and second-line therapy with medications, including oral agents and insulin, are the mainstays of type 2 DM therapy. Although these approaches have reduced hyperglycemia and cardiovascular mortality, many patients have poor glycemic control and develop severe diabetes-related complications. A study using data from the National Health and Nutrition Examination Survey (N = 4,926) to evaluate success rates of lifestyle management plus drug therapy found that just 53% of patients with type 2 DM maintained a hemoglobin A1c (HbA1c) below 7%.6 Similarly, only 51% of those patients achieved a systolic and diastolic blood pressure less than 130/80 mm Hg, and only 56% achieved a low-density lipoprotein cholesterol level less than 100 mg/dL. Altogether, only 19% of the study cohort achieved all 3 therapy targets. Documented limitations of lifestyle counseling and drug therapy include behavior maladaptation, limitations in drug potency, nonadherence to medications, adverse effects, and economic deterrents.7
METABOLIC SURGERY FOR TYPE 2 DM
For patients with obesity and type 2 DM in whom lifestyle management and medications do not achieve desired treatment goals, bariatric surgery has emerged as the most effective treatment for attaining significant and durable weight loss. These gastrointestinal (GI) procedures, which reduce gastric volume with or without rerouting nutrient flow through the small intestine, were developed to yield long-term weight loss in patients with severe obesity. It is now known that they also cause dramatic improvement or remission of obesity-related comorbidities, especially type 2 DM. Research has shown that these effects are not only secondary to weight loss but also depend on neuroendocrine mechanisms secondary to changes in GI physiology. For these reasons, bariatric surgery is increasingly used with the primary intent to treat type 2 DM or metabolic disease, a practice referred to as metabolic surgery.
For more than 2 decades, indications for metabolic surgery reflected guidelines from a 1991 National Institutes of Health (NIH) consensus conference, which suggested considering surgery only in patients with a BMI of 40 kg/m2 or greater or a BMI of 35 kg/m2 or greater and significant obesity-related comorbidities.11 Guidelines published in 2013 expanded the recommendations to include adults with a BMI of at least 35 kg/m2 and an obesity-related comorbidity, such as diabetes, who are motivated to lose weight.4 These recommendations were primarily designed to guide the use of surgery as a weight-loss intervention for severe obesity. However, guidelines published in 2016 support use of metabolic surgery as a specific treatment for type 2 DM.5
Potential mechanisms resolving type 2 DM: More than weight loss
Bariatric surgery has been shown to have profound glucoregulatory effects. These include rapid improvement in hyperglycemia and reduction in exogenous insulin requirements that occur early after surgery and before the patient has any significant weight loss.12,13 Additionally, experiments in rodents showed that changes to GI anatomy can directly influence glucose homeostasis, independently of weight loss and caloric restriction.14
Although the exact molecular mechanisms underlying the effects of metabolic surgery on diabetes are not fully understood, many factors appear to play a role, including changes in bile acid metabolism, GI tract nutrient sensing, glucose utilization, insulin resistance, and intestinal microbiomes.15 These changes, acting through peripheral or central pathways, or perhaps both, lead to reduced hepatic glucose production, increased tissue glucose uptake, improved insulin sensitivity, and enhanced beta-cell function. A constellation of gut-derived neuroendocrine changes, rather than a single overarching mechanism, is the likely mediator of postoperative glycemic improvement, with the contributing factors varying according to the surgical procedure.
METABOLIC SURGERY OUTCOMES
Weight loss
Long-term reduction of excess body fat is a major goal of metabolic and bariatric surgery. Weight loss is usually expressed as either the percent of weight loss or the percent of excess weight loss (ie, weight loss above ideal weight). A meta-analysis of mostly short-term weight-loss outcomes (ie, < 5 years) from more than 22,000 procedures found an overall mean excess weight loss of 47.5% for patients who underwent LAGB, 61.6% for RYGB, 68.2% for vertical-banded gastroplasty, and 70.1% for BPD-DS.16 Vertical-banded gastroplasty differs from LAGB in that both a band and staples are used to create a small stomach pouch. Excess weight loss for SG generally averages 50% to 55%, which is intermediate between LAGB and RYGB.17,18
The Swedish Obese Subjects study (N = 4,047), a prospective study of bariatric surgery vs nonsurgical weight management of severely obese patients (BMI > 34), is the largest weight-loss study with the longest follow-up.19 At 20 years, the mean weight loss was 26% for gastric bypass, 18% for vertical-banded gastroplasty, 13% for gastric banding, and 1% for controls. A 10-year study in 1,787 severely obese patients (BMI ≥ 35) who underwent RYGB had 21% more weight loss from their baseline weight than the nonsurgical match.20 At 4-year follow-up in 2,410 patients, there were significant variations in weight loss depending on the procedure: 27.5% for RYGB, 17.8% for SG, and 10.6% in LAGB. Between 2% and 31% regained weight back to baseline: 30.5% for LAGB, 14.6% for SG, and 2.5% for RYGB.20 In contrast, long-term medical (nonsurgical) weight loss rarely exceeds 5%, even with intensive lifestyle intervention.21
Diabetes remission, cardiovascular risk factors, glycemic control
A meta-analysis of 19 mostly observational studies (N = 4,070 patients) reported an overall type 2 DM remission rate of 78% after bariatric surgery with 1 to 3 years of follow-up.22 Resolution or remission was typically defined as becoming “nondiabetic” with normal HbA1c without medications. In the Swedish Obese Subjects study, the remission rate was 72% at 2 years and 36% at 10 years compared with 21% and 13%, respectively, for the nonsurgical controls (P < .001).23 Bariatric surgery was also markedly more effective than nonsurgical treatment in preventing type 2 DM, with a relative risk reduction of 78%.
A systematic review published in 2012 evaluated long-term cardiovascular risk reduction after bariatric surgery in 73 studies and 19,543 patients.24 At a mean follow-up of 57.8 months, the average excess weight loss for all procedures was 54% and rates of remission or improvement were 63% for hypertension, 73% for type 2 DM, and 65% for hyperlipidemia. Results from 12 cohort-matched, nonrandomized studies comparing bariatric surgery vs nonsurgical controls suggest that improvements in surrogate disease markers such as HbA1c, blood pressure, lipids, and body weight after surgery translate to reduced macrovascular and microvascular events and death.25 One of these studies involving male veterans who were mostly at high cardiovascular risk reported a 42% reduction in mortality at 10 years compared with medical therapy.26
In the Swedish Obese Subjects study, the mortality rate from cardiovascular disease in the bariatric surgical group was lower than for control patients (adjusted hazard ratio, 0.47; P = .002) despite a greater prevalence of smoking and higher baseline weights and blood pressures in the surgical cohort.19 For patients with type 2 DM in this study, surgery was associated with a 50% reduction in microvascular complications.27 After 15 years of follow-up, the cumulative incidence of microvascular complications was 41.8 per 1,000 person-years for control patients and 20.6 per 1,000 person-years in the surgery group (hazard ratio, 0.44; P < .001).
These observational, nonrandomized study data suggest that in patients with type 2 DM, bariatric surgery is significantly better than medical management alone in improving glycemic control, reducing cardiovascular risk factors, and lowering long-term morbidity and mortality associated with type 2 DM.
METABOLIC SURGERY: CLINICAL TRIALS
Collectively, these RCTs showed that surgery was significantly superior to medical treatment in reaching the designated glycemic target (P < .05 for all). The one exception showed that diabetes remission for LAGB vs medical treatment was 33% and 23%, respectively.41 This result might be due to patients in this study having advanced type 2 DM (HbA1c 8.2% ± 1.2%, with 40% on insulin), and they likely had reduced beta-cell function. Overall, surgery decreased HbA1c by 2% to 3.5%, whereas medical treatment lowered it by only 1% to 1.5%. Most of these studies also showed superiority of surgery over medical treatment in achieving secondary end points such as weight loss, remission of metabolic syndrome, reduction in diabetes and cardiovascular medications, and improvement in triglycerides, lipids, and quality of life. Results were mixed in terms of improvements in systolic and diastolic blood pressure or low-density lipoproteins after surgery vs medical treatment, but many studies did show a corresponding reduction in medication usage.
Durability of the effects of surgery was demonstrated in a 5-year study that showed superior and durable weight loss and glycemic control (remission) with both RYGB and BPD in severely obese patients (BMI ≥ 35) vs medical therapy.32 Similarly, Schauer et al43 showed that RYGB and SG were more effective than intensive medical therapy in improving or, in some cases, resolving hyperglycemia for 5 years. In the RCTs, patients who preoperatively had shorter duration of diabetes, lower HbA1c levels, no insulin requirement, and more postoperative weight loss were more likely to achieve diabetes remission.
Although previous guidelines and payer coverage policies had limited metabolic surgery to severely obese patients (BMI ≥ 35 kg/m2), nearly all RCTs showed that the surgical procedures, especially RYGB and SG, were equally effective in patients with BMI 30 to 35 kg/m2. This is particularly important given that most patients with type 2 DM have a BMI less than 35 kg/m2. The effect of surgery in these patients with mild obesity is also durable out to at least 5 years.43
No RCT was sufficiently powered to detect differences in macrovascular or microvascular complications or death, especially at the relatively short follow-up, and no such differences have been detected thus far. The STAMPEDE (Surgical Therapy and Medications Potentially Eradicate Diabetes Efficiently) trial43 showed that bariatric surgery (RYGB or SG) did not appear to worsen or improve retinopathy outcomes at 5 years compared with intensive medical management.
METABOLIC SURGERY: ADVERSE EVENTS
Surgical complications
Nutritional deficiencies
Postoperative nutritional deficiencies are typically associated with diminished nutrient intake or the malabsorptive effect of bariatric procedures. They are more common after RYGB and BPD-DS and less common after SG and LAGB. In addition, there is a high prevalence of nutritional deficiencies (35%–80%) in patients seeking bariatric surgery; thus, poor preoperative nutrition may be a factor in the development of postoperative deficiencies. Common preoperative nutrient deficiencies are vitamin A (11%), vitamin B12 (13%), vitamin D (40%), zinc (30%), iron (16%), ferritin (9%), selenium (58%), and folate (6%).51 Recommendations are to assess for these deficiencies and correct any identified before surgery.
Mild anemia after bariatric procedures is common, occurring in 15% to 20% of cases, and it is believed to result from reduced absorption of iron and B12, as well from pre-existing iron deficiency anemia in premenopausal patients.52 Deficiencies in trace minerals (selenium, zinc, and copper) and vitamins (B12, B1, A, E, D, and K) can occur after bariatric procedures, especially after BPD-DS.53 Nutrient deficiencies can be prevented or corrected with appropriate vitamin, iron, and calcium supplementation.54
Bone mineral density may decrease after bariatric surgery (14% in the proximal femur).55 Reduced mechanical loading after weight loss, reduced consumption and malabsorption of micronutrients (calcium, vitamin D), and neurohormonal alterations are potential underlying mechanisms of bone mineral density reduction after bariatric surgery. Rates of bone fracture and osteoporosis are not well delineated, raising questions about whether bone loss after bariatric surgery is clinically relevant or a functional adaptation to skeletal unloading. However, the extreme malabsorptive procedures of BPD-DS have been associated with severe calcium and vitamin D deficiencies, leading to decreased bone mineral density and osteoporosis.
Protein malnutrition also can occur after these extreme malabsorptive procedures. Patients require postoperative oral protein supplementation (80–100 g/day) and lifelong monitoring for nutritional complications after these procedures.56
Additional complications
Other late complications of bariatric surgery that are less clear in incidence and cause include kidney stones, alcohol abuse, depression, and suicide. One study of patients after RYGB (N = 4,690) reported a significantly higher prevalence of kidney stones than in obese controls: 7.5% vs 4.6%, respectively.57 Proposed causes of kidney stone formation following bariatric surgery include hyperoxaluria, hypocitraturia, and elevated urine acidity.58
The prevalence of alcohol-use disorder after bariatric surgery ranges from 7.6% to 11.8% and appears to be higher in patients with a history of alcohol use.59 Paradoxically, while bariatric surgery has been shown to significantly decrease depression,60 some studies suggest that a slight increase in the risk of suicide may occur,61 while others do not.62 A recent review concluded that accurate rates of suicide after bariatric surgery are not known, but practitioners should be aware of this concern and appropriately screen and counsel their patients.63
Although the 12 RCTs reported in Table 1 were not powered to detect differences in treatment-related complications, the overall rates of complications were consistent with those in observational studies.9 The most common surgical complications were anemia (15%), need for reoperation (8%), and GI (5%–10%). The 30-day surgical mortality rate was 0.2% (1 death) among the 465 surgical patients. Complications were not limited to the surgical patients. In the medical-treatment control group of the STAMPEDE trial,30 anemia (16%) and weight gain (16%) were common. Investigators reported challenges with medication compliance, including adverse effects leading to discontinuation of medications. Mild hypoglycemia was common, with no significant differences between the surgical and medical treatment groups.
METABOLIC SURGERY: COST EFFECTIVENESS
The cost of bariatric procedures varies considerably but, in general, ranges from $20,000 to $30,000, similar to the cost of cholecystectomy, hysterectomy, and colectomy. Retrospective analyses and modeling studies indicate that metabolic surgery is cost-effective and may present a cost savings in patients with type 2 DM, with a break-even time between 5 and 10 years.64,65 The cost savings, largely based on assumptions of long-term effectiveness and safety, result from reductions in medication use, outpatient care costs, and long-term complications of type 2 DM.
WHO SHOULD HAVE METABOLIC SURGERY?
Until recently, there was no clear national or international consensus on the role of metabolic surgery in treating type 2 DM. In 2015, the 2nd Diabetes Surgery Summit (DSS-II) Consensus Conference published guidelines that were endorsed by more than 50 diabetes and medical organizations.5 The recommendations cover many clinically relevant issues, including patient selection, preoperative evaluation, choice of procedure, and postoperative follow-up. The consensus conference delegates concluded that there is sufficient evidence demonstrating that metabolic surgery achieves excellent glycemic control and reduces cardiovascular risk factors.
The treatment algorithm from DSS-II incorporates appropriate use of all 3 treatment modalities: lifestyle intervention, drug therapy, and surgery (Figure 5).5 The 2017 Standards of Care for Diabetes from the American Diabetes Association include those key indications in the recommendations for metabolic surgery (Table 3).2
SUMMARY
The safety of metabolic surgery has significantly improved with the advent of laparoscopic surgery and recent national quality improvement initiatives that have made gastric bypass and SG as safe as cholecystectomy and appendectomy. Although observational studies suggest that metabolic surgery is associated with a reduction in cardiovascular and diabetes complications and mortality, these observations have not been confirmed in long-term RCTs.
Based on the published evidence, metabolic surgery is now endorsed as a standard treatment option, which provides patients and practitioners with a powerful tool to help combat the life-impairing effects of type 2 DM.
- Bays HE, Chapman RH, Grandy S; for the SHIELD Investigators Group. The relationship of body mass index to diabetes mellitus, hypertension and dyslipidaemia: comparison of data from two national surveys. Int J Clin Pract May 2007; 61:737–747.
- Marathe PH, Gao HX, Close KL. American Diabetes Association standards of medical care in diabetes—2017. Diabetes Care 2017; 40(suppl 1):S1–S135.
- Fox CS, Golden SH, Anderson C, et al; American Heart Association; American Diabetes Association. Update on prevention of cardiovascular disease in adults with type 2 diabetes mellitus in light of recent evidence: a scientific statement from the American Heart Association and the American Diabetes Association. Circulation 2015; 132:691–718.
- Jensen MD, Ryan DH, Apovian CM, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines; Obesity Society. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and The Obesity Society. J Am Coll Cardiol 2014; 63:2985–3023.
- Rubino F, Nathan DM, Eckel RH, et al; Delegates of the 2nd Diabetes Surgery Summit. Metabolic surgery in the treatment algorithm for type 2 diabetes: a joint statement by international diabetes organizations. Diabetes Care 2016; 39:861–877.
- Stark Casagrande S, Fradkin JE, Saydah SH, Rust KF, Cowie CC. The prevalence of meeting A1C, blood pressure, and LDL goals among people with diabetes, 1988–2010. Diabetes Care 2013; 36:2271–2279.
- Kolandaivelu K, Leiden BB, O’Gara PT, Bhatt DL. Non-adherence to cardiovascular medications. Eur Heart J 2014; 35:3267–3276.
- Angrisani L, Santonicola A, Iovino P, Formisano G, Buchwald H, Scopinaro N. Bariatric surgery worldwide 2013. Obes Surg 2015; 25:1822–1832.
- Schauer PR, Mingrone G, Ikramuddin S, Wolfe B. Clinical outcomes of metabolic surgery: efficacy of glycemic control, weight loss, and remission of diabetes. Diabetes Care 2016; 39:902–911.
- Khorgami Z, Andalib A, Corcelles R, Aminian A, Brethauer S, Schauer P. Recent national trends in the surgical treatment of obesity: sleeve gastrectomy dominates. Surg Obes Relat Dis 2015; 11(suppl):S1–S34 [Abstract A111].
- Consensus Development Conference Panel. NIH conference. Gastrointestinal surgery for severe obesity. Ann Intern Med 1991; 115:956–961.
- Pories WJ, MacDonald KG Jr, Flickinger EG, et al. Is type II diabetes mellitus (NIDDM) a surgical disease? Ann Surg 1992; 215:633–642.
- Schauer PR, Burguera B, Ikramuddin S, et al. Effect of laparoscopic Roux-en Y gastric bypass on type 2 diabetes mellitus. Ann Surg 2003; 238:467–484.
- Rubino F, Marescaux J. Effect of duodenal-jejunal exclusion in a non-obese animal model of type 2 diabetes: a new perspective for an old disease. Ann Surg 2004; 239:1–11.
- Batterham RL, Cummings DE. Mechanisms of diabetes improvement following bariatric/metabolic surgery. Diabetes Care 2016; 39:893–901.
- Buchwald H, Avidor Y, Braunwald E, et al. Bariatric surgery: a systematic review and meta-analysis. JAMA 2004; 292:1724–1737.
- Brethauer SA, Hammel JP, Schauer PR. Systematic review of sleeve gastrectomy as staging and primary bariatric procedure. Surg Obes Relat Dis 2009; 5:469–475.
- Eid GM, Brethauer S, Mattar SG, Titchner RL, Gourash W, Schauer PR. Laparoscopic sleeve gastrectomy for super obese patients: forty-eight percent excess weight loss after 6 to 8 years with 93% follow-up. Ann Surg 2012; 256:262–265.
- Sjöström L, Peltonen M, Jacobson P, et al. Bariatric surgery and long-term cardiovascular events. JAMA 2012; 307:56–65.
- Maciejewski ML, Arterburn DE, Van Scoyoc L, et al. Bariatric surgery and long-term durability of weight loss. JAMA Surg 2016; 151:1046–1055.
- Wing RR, Bolin P, Brancati FL, et al; for the Look AHEAD Research Group. Cardiovascular effects of intensive lifestyle intervention in type 2 diabetes. N Engl J Med 2013; 369:145–154.
- Buchwald H, Estok R, Fahrbach K, et al. Weight and type 2 diabetes after bariatric surgery: systematic review and meta-analysis. Am J Med 2009; 122:248–256.
- Sjöström L, Lindroos AK, Peltonen M, et al; Swedish Obese Subjects Study Scientific Group. Lifestyle, diabetes, and cardiovascular risk factors 10 years after bariatric surgery. N Engl J Med 2004; 351:2683–2693.
- Vest AR, Heneghan HM, Agarwal S, Schauer PR, Young JB. Bariatric surgery and cardiovascular outcomes: a systematic review. Heart 2012; 98:1763–1777.
- Vest AR, Heneghan HM, Schauer PR, Young JB. Surgical management of obesity and the relationship to cardiovascular disease. Circulation 2013; 127:945–959.
- Arterburn DE, Olsen MK, Smith VA, et al. Association between bariatric surgery and long-term survival. JAMA 2015; 313:62–70.
- Sjöström L, Peltonen M, Jacobson P, et al. Association of bariatric surgery with long-term remission of type 2 diabetes and with microvascular and macrovascular complications. JAMA 2014; 311:2297–2304.
- Dixon JB, O’Brien PE, Playfair J, et al. Adjustable gastric banding and conventional therapy for type 2 diabetes: a randomized controlled trial. JAMA 2008; 299:316–323.
- Schauer PR, Kashyap SR, Wolski K, et al. Bariatric surgery versus intensive medical therapy in obese patients with diabetes. N Engl J Med 2012; 366:1567–1576.
- Schauer PR, Bhatt DL, Kirwan JP, et al; STAMPEDE Investigators. Bariatric surgery versus intensive medical therapy for diabetes—3-year outcomes. N Engl J Med 2014; 370:2002–2013.
- Mingrone G, Panunzi S, De Gaetano A, et al. Bariatric surgery versus conventional medical therapy for type 2 diabetes. N Engl J Med 2012; 366:1577–1585.
- Mingrone G, Panunzi S, De Gaetano A, et al. Bariatric-metabolic surgery versus conventional medical treatment in obese patients with type 2 diabetes: 5 year follow-up of an open-label, single-centre, randomized controlled trial. Lancet 2015; 386:964–973.
- Ikramuddin S, Korner J, Lee WJ, et al. Roux-en-Y gastric bypass vs intensive medical management for the control of type 2 diabetes, hypertension, and hyperlipidemia: the Diabetes Surgery Study randomized clinical trial. JAMA 2013; 309:2240–2249.
- Ikramuddin S, Billington CJ, Lee WJ, et al. Roux-en-Y gastric bypass for diabetes (the Diabetes Surgery Study): 2-year outcomes of a 5-year, randomized, controlled trial. Lancet Diabetes Endocrinol 2015; 3:413–422.
- Liang Z, Wu Q, Chen B, Yu P, Zhao H, Ouyang X. Effect of laparoscopic Roux-en-Y gastric bypass surgery on type 2 diabetes mellitus with hypertension: a randomized controlled trial. Diabetes Res Clin Pract 2013; 101:50–56.
- Halperin F, Ding SA, Simonson DC, et al. Roux-en-Y gastric bypass surgery or lifestyle with intensive medical management in patients with type 2 diabetes: feasibility and 1-year results of a randomized clinical trial. JAMA Surg 2014; 149:716–726.
- Courcoulas AP, Goodpaster BH, Eagleton JK, et al. Surgical vs medical treatments for type 2 diabetes mellitus: a randomized clinical trial. JAMA Surg 2014; 149:707–715.
- Courcoulas AP, Belle SH, Neiberg RH, et al. Three-year outcomes of bariatric surgery vs. lifestyle intervention for type 2 diabetes mellitus treatment: a randomized clinical trial. JAMA Surg 2015; 150:931–940.
- Wentworth JM, Playfair J, Laurie C, et al. Multidisciplinary diabetes care with and without bariatric surgery in overweight people: a randomised controlled trial. Lancet Diabetes Endocrinol 2014; 2:545–552.
- Parikh M, Chung M, Sheth S, et al. Randomized pilot trial of bariatric surgery versus intensive medical weight management on diabetes remission in type 2 diabetic patients who do not meet NIH criteria for surgery and the role of soluble RAGE as a novel biomarker of success. Ann Surg 2014; 260:617–622.
- Ding SA, Simonson DC, Wewalka M, et al. Adjustable gastric band surgery or medical management in patients with type 2 diabetes: a randomized clinical trial. J Clin Endocrinol Metab 2015; 100:2546–2556.
- Cummings DE, Arterburn DE, Westbrook EO, et al. Gastric bypass surgery vs. intensive lifestyle and medical intervention for type 2 diabetes: the CROSSROADS randomized controlled trial. Diabetologia 2016; 59:945–953.
- Schauer PR, Bhatt DL, Kirwan JP, et al; STAMPEDE Investigators. Metabolic surgery vs. intensive medical therapy for diabetes: 5-year outcomes. N Engl J Med 2017; 376:641–651.
- Shah SS, Todkar J, Phadake U, et al. Gastric bypass vs. medical/lifestyle care for type 2 diabetes in South Asians with BMI 25-40 kg/m2: the COSMID randomized trial [261-OR]. Presented at the American Diabetes Association’s 76th Scientific Session; June 10–14, 2016; New Orleans, LA.
- Flum DR, Belle SH, King WC, et al; Longitudinal Assessment of Bariatric Surgery (LABS) Consortium. Perioperative safety in the longitudinal assessment of bariatric surgery. N Engl J Med 2009; 361:445–454.
- Aminian A, Brethauer SA, Kirwan JP, Kashyap SR, Burguera B, Schauer PR. How safe is metabolic/diabetes surgery? Diabetes Obes Metab 2015; 17:198–201.
- Thodiyil PA, Yenumula P, Rogula T, et al. Selective non operative management of leaks after gastric bypass: lessons learned from 2675 consecutive patients. Ann Surg 2008; 248:782–792.
- Rogula T, Yenumula PR, Schauer PR. A complication of Roux-en-Y gastric bypass: intestinal obstruction. Surg Endosc 2007; 21:1914–1918.
- Thornton CM, Rozen WM, So D, Kaplan ED, Wilkinson S. Reducing band slippage in laparoscopic adjustable gastric banding: the mesh plication pars flaccida technique. Obes Surg 2009; 19:1702–1706.
- Himpens J, Cadière G-B, Bazi M, Vouche M, Cadière B, Dapri G. Long-term outcomes of laparoscopic adjustable gastric banding. Arch Surg 2011; 146:802–807.
- Madan AK, Orth WS, Tichansky DS, Ternovits CA. Vitamin and trace mineral levels after laparoscopic gastric bypass. Obes Surg 2006; 16:603–606.
- Love AL, Billett HH. Obesity, bariatric surgery, and iron deficiency: true, true, true and related. Am J Hematol 2008; 83:403–409.
- Shankar P, Boylan M, Sriram K. Micronutrient deficiencies after bariatric surgery. Nutrition 2010; 26:1031–1037.
- Gong K, Gagner M, Pomp A, Almahmeed T, Bardaro SJ. Micronutrient deficiencies after laparoscopic gastric bypass: recommendations. Obes Surg 2008; 18:1062–1066.
- Scibora LM. Skeletal effects of bariatric surgery: examining bone loss, potential mechanisms and clinical relevance. Diabetes Obes Metab 2014; 16:1204–1213.
- Baptista V, Wassef W. Bariatric procedures: an update on techniques, outcomes and complications. Curr Opin Gastroenterol 2013; 29:684–693.
- Matlaga BR, Shore AD, Magnuson T, Clark JM, Johns R, Makary MA. Effect of gastric bypass surgery on kidney stone disease. J Urol 2009; 181:2573–2577.
- Sakhaee K, Poindexter J, Aguirre C. The effects of bariatric surgery on bone and nephrolithiasis. Bone 2016; 84:1–8.
- Li L, Wu LT. Substance use after bariatric surgery: a review. J Psychiatr Res 2016; 76:16–29.
- Ayloo S, Thompson K, Choudhury N, Sheriffdeen R. Correlation between the Beck Depression Inventory and bariatric surgical procedures. Surg Obes Relat Dis 2015; 11:637–342.
- Adams TD, Gress RE, Smith SC, et al. Long-term mortality after gastric bypass surgery. N Engl J Med 2007; 357:753–761.
- Sjöström L, Narbro K, Sjöström CD, et al; Swedish Obese Subjects Study. Effects of bariatric surgery on mortality in Swedish obese subjects. N Engl J Med 2007; 357:741–752.
- Mitchell JE, Crosby R, de Zwaan M, et al. Possible risk factors for increased suicide following bariatric surgery. Obesity (Silver Spring) 2013; 21:665–672.
- Fouse T, Schauer P. The socioeconomic impact of morbid obesity and factors affecting access to obesity surgery. Surg Clin North Am 2016; 96:669–679.
- Rubin JK, Hinrichs-Krapels S, Hesketh R, Martin A, Herman WH, Rubino F. Identifying barriers to appropriate use of metabolic/bariatric surgery for type 2 diabetes treatment: policy lab results. Diabetes Care 2016; 39:954–963.
- Bays HE, Chapman RH, Grandy S; for the SHIELD Investigators Group. The relationship of body mass index to diabetes mellitus, hypertension and dyslipidaemia: comparison of data from two national surveys. Int J Clin Pract May 2007; 61:737–747.
- Marathe PH, Gao HX, Close KL. American Diabetes Association standards of medical care in diabetes—2017. Diabetes Care 2017; 40(suppl 1):S1–S135.
- Fox CS, Golden SH, Anderson C, et al; American Heart Association; American Diabetes Association. Update on prevention of cardiovascular disease in adults with type 2 diabetes mellitus in light of recent evidence: a scientific statement from the American Heart Association and the American Diabetes Association. Circulation 2015; 132:691–718.
- Jensen MD, Ryan DH, Apovian CM, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines; Obesity Society. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and The Obesity Society. J Am Coll Cardiol 2014; 63:2985–3023.
- Rubino F, Nathan DM, Eckel RH, et al; Delegates of the 2nd Diabetes Surgery Summit. Metabolic surgery in the treatment algorithm for type 2 diabetes: a joint statement by international diabetes organizations. Diabetes Care 2016; 39:861–877.
- Stark Casagrande S, Fradkin JE, Saydah SH, Rust KF, Cowie CC. The prevalence of meeting A1C, blood pressure, and LDL goals among people with diabetes, 1988–2010. Diabetes Care 2013; 36:2271–2279.
- Kolandaivelu K, Leiden BB, O’Gara PT, Bhatt DL. Non-adherence to cardiovascular medications. Eur Heart J 2014; 35:3267–3276.
- Angrisani L, Santonicola A, Iovino P, Formisano G, Buchwald H, Scopinaro N. Bariatric surgery worldwide 2013. Obes Surg 2015; 25:1822–1832.
- Schauer PR, Mingrone G, Ikramuddin S, Wolfe B. Clinical outcomes of metabolic surgery: efficacy of glycemic control, weight loss, and remission of diabetes. Diabetes Care 2016; 39:902–911.
- Khorgami Z, Andalib A, Corcelles R, Aminian A, Brethauer S, Schauer P. Recent national trends in the surgical treatment of obesity: sleeve gastrectomy dominates. Surg Obes Relat Dis 2015; 11(suppl):S1–S34 [Abstract A111].
- Consensus Development Conference Panel. NIH conference. Gastrointestinal surgery for severe obesity. Ann Intern Med 1991; 115:956–961.
- Pories WJ, MacDonald KG Jr, Flickinger EG, et al. Is type II diabetes mellitus (NIDDM) a surgical disease? Ann Surg 1992; 215:633–642.
- Schauer PR, Burguera B, Ikramuddin S, et al. Effect of laparoscopic Roux-en Y gastric bypass on type 2 diabetes mellitus. Ann Surg 2003; 238:467–484.
- Rubino F, Marescaux J. Effect of duodenal-jejunal exclusion in a non-obese animal model of type 2 diabetes: a new perspective for an old disease. Ann Surg 2004; 239:1–11.
- Batterham RL, Cummings DE. Mechanisms of diabetes improvement following bariatric/metabolic surgery. Diabetes Care 2016; 39:893–901.
- Buchwald H, Avidor Y, Braunwald E, et al. Bariatric surgery: a systematic review and meta-analysis. JAMA 2004; 292:1724–1737.
- Brethauer SA, Hammel JP, Schauer PR. Systematic review of sleeve gastrectomy as staging and primary bariatric procedure. Surg Obes Relat Dis 2009; 5:469–475.
- Eid GM, Brethauer S, Mattar SG, Titchner RL, Gourash W, Schauer PR. Laparoscopic sleeve gastrectomy for super obese patients: forty-eight percent excess weight loss after 6 to 8 years with 93% follow-up. Ann Surg 2012; 256:262–265.
- Sjöström L, Peltonen M, Jacobson P, et al. Bariatric surgery and long-term cardiovascular events. JAMA 2012; 307:56–65.
- Maciejewski ML, Arterburn DE, Van Scoyoc L, et al. Bariatric surgery and long-term durability of weight loss. JAMA Surg 2016; 151:1046–1055.
- Wing RR, Bolin P, Brancati FL, et al; for the Look AHEAD Research Group. Cardiovascular effects of intensive lifestyle intervention in type 2 diabetes. N Engl J Med 2013; 369:145–154.
- Buchwald H, Estok R, Fahrbach K, et al. Weight and type 2 diabetes after bariatric surgery: systematic review and meta-analysis. Am J Med 2009; 122:248–256.
- Sjöström L, Lindroos AK, Peltonen M, et al; Swedish Obese Subjects Study Scientific Group. Lifestyle, diabetes, and cardiovascular risk factors 10 years after bariatric surgery. N Engl J Med 2004; 351:2683–2693.
- Vest AR, Heneghan HM, Agarwal S, Schauer PR, Young JB. Bariatric surgery and cardiovascular outcomes: a systematic review. Heart 2012; 98:1763–1777.
- Vest AR, Heneghan HM, Schauer PR, Young JB. Surgical management of obesity and the relationship to cardiovascular disease. Circulation 2013; 127:945–959.
- Arterburn DE, Olsen MK, Smith VA, et al. Association between bariatric surgery and long-term survival. JAMA 2015; 313:62–70.
- Sjöström L, Peltonen M, Jacobson P, et al. Association of bariatric surgery with long-term remission of type 2 diabetes and with microvascular and macrovascular complications. JAMA 2014; 311:2297–2304.
- Dixon JB, O’Brien PE, Playfair J, et al. Adjustable gastric banding and conventional therapy for type 2 diabetes: a randomized controlled trial. JAMA 2008; 299:316–323.
- Schauer PR, Kashyap SR, Wolski K, et al. Bariatric surgery versus intensive medical therapy in obese patients with diabetes. N Engl J Med 2012; 366:1567–1576.
- Schauer PR, Bhatt DL, Kirwan JP, et al; STAMPEDE Investigators. Bariatric surgery versus intensive medical therapy for diabetes—3-year outcomes. N Engl J Med 2014; 370:2002–2013.
- Mingrone G, Panunzi S, De Gaetano A, et al. Bariatric surgery versus conventional medical therapy for type 2 diabetes. N Engl J Med 2012; 366:1577–1585.
- Mingrone G, Panunzi S, De Gaetano A, et al. Bariatric-metabolic surgery versus conventional medical treatment in obese patients with type 2 diabetes: 5 year follow-up of an open-label, single-centre, randomized controlled trial. Lancet 2015; 386:964–973.
- Ikramuddin S, Korner J, Lee WJ, et al. Roux-en-Y gastric bypass vs intensive medical management for the control of type 2 diabetes, hypertension, and hyperlipidemia: the Diabetes Surgery Study randomized clinical trial. JAMA 2013; 309:2240–2249.
- Ikramuddin S, Billington CJ, Lee WJ, et al. Roux-en-Y gastric bypass for diabetes (the Diabetes Surgery Study): 2-year outcomes of a 5-year, randomized, controlled trial. Lancet Diabetes Endocrinol 2015; 3:413–422.
- Liang Z, Wu Q, Chen B, Yu P, Zhao H, Ouyang X. Effect of laparoscopic Roux-en-Y gastric bypass surgery on type 2 diabetes mellitus with hypertension: a randomized controlled trial. Diabetes Res Clin Pract 2013; 101:50–56.
- Halperin F, Ding SA, Simonson DC, et al. Roux-en-Y gastric bypass surgery or lifestyle with intensive medical management in patients with type 2 diabetes: feasibility and 1-year results of a randomized clinical trial. JAMA Surg 2014; 149:716–726.
- Courcoulas AP, Goodpaster BH, Eagleton JK, et al. Surgical vs medical treatments for type 2 diabetes mellitus: a randomized clinical trial. JAMA Surg 2014; 149:707–715.
- Courcoulas AP, Belle SH, Neiberg RH, et al. Three-year outcomes of bariatric surgery vs. lifestyle intervention for type 2 diabetes mellitus treatment: a randomized clinical trial. JAMA Surg 2015; 150:931–940.
- Wentworth JM, Playfair J, Laurie C, et al. Multidisciplinary diabetes care with and without bariatric surgery in overweight people: a randomised controlled trial. Lancet Diabetes Endocrinol 2014; 2:545–552.
- Parikh M, Chung M, Sheth S, et al. Randomized pilot trial of bariatric surgery versus intensive medical weight management on diabetes remission in type 2 diabetic patients who do not meet NIH criteria for surgery and the role of soluble RAGE as a novel biomarker of success. Ann Surg 2014; 260:617–622.
- Ding SA, Simonson DC, Wewalka M, et al. Adjustable gastric band surgery or medical management in patients with type 2 diabetes: a randomized clinical trial. J Clin Endocrinol Metab 2015; 100:2546–2556.
- Cummings DE, Arterburn DE, Westbrook EO, et al. Gastric bypass surgery vs. intensive lifestyle and medical intervention for type 2 diabetes: the CROSSROADS randomized controlled trial. Diabetologia 2016; 59:945–953.
- Schauer PR, Bhatt DL, Kirwan JP, et al; STAMPEDE Investigators. Metabolic surgery vs. intensive medical therapy for diabetes: 5-year outcomes. N Engl J Med 2017; 376:641–651.
- Shah SS, Todkar J, Phadake U, et al. Gastric bypass vs. medical/lifestyle care for type 2 diabetes in South Asians with BMI 25-40 kg/m2: the COSMID randomized trial [261-OR]. Presented at the American Diabetes Association’s 76th Scientific Session; June 10–14, 2016; New Orleans, LA.
- Flum DR, Belle SH, King WC, et al; Longitudinal Assessment of Bariatric Surgery (LABS) Consortium. Perioperative safety in the longitudinal assessment of bariatric surgery. N Engl J Med 2009; 361:445–454.
- Aminian A, Brethauer SA, Kirwan JP, Kashyap SR, Burguera B, Schauer PR. How safe is metabolic/diabetes surgery? Diabetes Obes Metab 2015; 17:198–201.
- Thodiyil PA, Yenumula P, Rogula T, et al. Selective non operative management of leaks after gastric bypass: lessons learned from 2675 consecutive patients. Ann Surg 2008; 248:782–792.
- Rogula T, Yenumula PR, Schauer PR. A complication of Roux-en-Y gastric bypass: intestinal obstruction. Surg Endosc 2007; 21:1914–1918.
- Thornton CM, Rozen WM, So D, Kaplan ED, Wilkinson S. Reducing band slippage in laparoscopic adjustable gastric banding: the mesh plication pars flaccida technique. Obes Surg 2009; 19:1702–1706.
- Himpens J, Cadière G-B, Bazi M, Vouche M, Cadière B, Dapri G. Long-term outcomes of laparoscopic adjustable gastric banding. Arch Surg 2011; 146:802–807.
- Madan AK, Orth WS, Tichansky DS, Ternovits CA. Vitamin and trace mineral levels after laparoscopic gastric bypass. Obes Surg 2006; 16:603–606.
- Love AL, Billett HH. Obesity, bariatric surgery, and iron deficiency: true, true, true and related. Am J Hematol 2008; 83:403–409.
- Shankar P, Boylan M, Sriram K. Micronutrient deficiencies after bariatric surgery. Nutrition 2010; 26:1031–1037.
- Gong K, Gagner M, Pomp A, Almahmeed T, Bardaro SJ. Micronutrient deficiencies after laparoscopic gastric bypass: recommendations. Obes Surg 2008; 18:1062–1066.
- Scibora LM. Skeletal effects of bariatric surgery: examining bone loss, potential mechanisms and clinical relevance. Diabetes Obes Metab 2014; 16:1204–1213.
- Baptista V, Wassef W. Bariatric procedures: an update on techniques, outcomes and complications. Curr Opin Gastroenterol 2013; 29:684–693.
- Matlaga BR, Shore AD, Magnuson T, Clark JM, Johns R, Makary MA. Effect of gastric bypass surgery on kidney stone disease. J Urol 2009; 181:2573–2577.
- Sakhaee K, Poindexter J, Aguirre C. The effects of bariatric surgery on bone and nephrolithiasis. Bone 2016; 84:1–8.
- Li L, Wu LT. Substance use after bariatric surgery: a review. J Psychiatr Res 2016; 76:16–29.
- Ayloo S, Thompson K, Choudhury N, Sheriffdeen R. Correlation between the Beck Depression Inventory and bariatric surgical procedures. Surg Obes Relat Dis 2015; 11:637–342.
- Adams TD, Gress RE, Smith SC, et al. Long-term mortality after gastric bypass surgery. N Engl J Med 2007; 357:753–761.
- Sjöström L, Narbro K, Sjöström CD, et al; Swedish Obese Subjects Study. Effects of bariatric surgery on mortality in Swedish obese subjects. N Engl J Med 2007; 357:741–752.
- Mitchell JE, Crosby R, de Zwaan M, et al. Possible risk factors for increased suicide following bariatric surgery. Obesity (Silver Spring) 2013; 21:665–672.
- Fouse T, Schauer P. The socioeconomic impact of morbid obesity and factors affecting access to obesity surgery. Surg Clin North Am 2016; 96:669–679.
- Rubin JK, Hinrichs-Krapels S, Hesketh R, Martin A, Herman WH, Rubino F. Identifying barriers to appropriate use of metabolic/bariatric surgery for type 2 diabetes treatment: policy lab results. Diabetes Care 2016; 39:954–963.
KEY POINTS
- Randomized clinical trials have shown that metabolic surgery is statistically superior to medical treatment in achieving targeted glycemic levels along with improvements in weight loss, remission of metabolic syndrome, reduction in medications, and improvements in lipid levels.
- The safety of metabolic and bariatric surgery has significantly improved with the advent of laparoscopic surgery, resulting in complication profiles similar to those of cholecystectomy and appendectomy.
- Metabolic surgery is now recommended as standard treatment option for type 2 diabetes in patients with body mass index levels as low as 30 kg/m2.
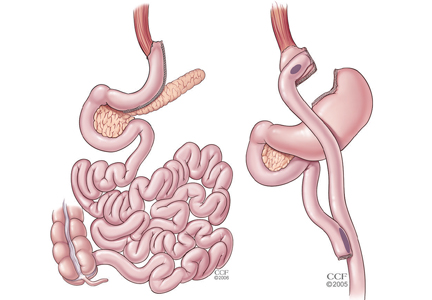
— Bonus Article — Medical Treatment of Diabetes Mellitus
Medical Treatment of Diabetes Mellitus
In the United States, 57.9% of patients with diabetes mellitus (DM) have at least 1 diabetes-related complication and 14.3% of patients with diabetes have 3 or more diabetes-related complications.1 Achieving glycemic control in patients with DM reduces the development and progression of retinopathy, nephropathy, and neuropathy. Aggressive treatment of dyslipidemia and hypertension decreases macrovascular complications.2–4 The techniques for monitoring blood glucose and the various treatment options available to manage glycemic control in patients with diabetes are reviewed below.
Measuring Glycemic Control
The primary techniques available to assess the quality of a patient’s glycemic control are self-monitoring of blood glucose and interval measurement of hemoglobin A1c (HbA1c). Continuous glucose monitoring is also available and may be appropriate for select patients, such as patients with brittle diabetes and those using insulin pumps.
Self-monitoring of blood glucose
For patients with type 1 DM and patients with insulin-dependent type 2 DM, self-monitoring of blood glucose allows patients to adjust insulin dosing to prevent hypoglycemia and hyperglycemia.2,5–7 The American Diabetes Association (ADA) guidelines recommend that patients with type 1 DM self-monitor their glucose:
- Before eating
- At bedtime
- Before exercise
- If hypoglycemia is suspected
- Until hypoglycemia is corrected
- Postprandially upon occasion
- And before critical tasks (ie, driving).8
Patients should be educated about how to use real-time blood glucose values to adjust their food intake and medical therapy.
It is commonly recommended that patients with type 2 DM self-monitor their blood glucose levels, but the evidence to support the effectiveness of this practice is inconclusive. Initial studies showed reductions in HbA1c with self-monitoring; however, the inclusion of beneficial health behaviors such as diet and exercise in the analyses makes it difficult to assess the effectiveness of self-monitor blood glucose alone.2,9
The ADA recommends that nonpregnant adults maintain blood glucose levels of 80 mg/dL to 130 mg/dL preprandial and less than 180 mg/dL postprandial.8 The blood glucose goals for patients with gestational diabetes are 95 mg/dL or less preprandial and either 140 mg/dL or less 1-hour postprandial or 120 mg/dL or less 2-hours postprandial.
HbA1c
HbA1c tests reflect the mean blood glucose values over a 3-month period and can predict patients’ risk of microvascular complications.10,11 The ADA recommends that patients with stable glycemic control have an HbA1c test at least twice a year. Quarterly HbA1c testing is suggested for patients with a recent change in therapy or for patients not meeting their glycemic goals.8
Measurement of HbA1c is influenced by the red blood cell turnover rate; therefore, anemia, transfusions, and hemoglobinopathies can cause inaccurate test values. The ADA recommends that nonpregnant adults maintain HbA1c levels near 7%. For patients with diabetes who become pregnant, the goal is HbA1c levels less than 6.0%.8 The ADA also recommends that select patients, especially those with a long life expectancy and little comorbidity, adopt glycemic targets near normal levels (HbA1c < 6.5%), providing the target can be achieved without significant hypoglycemia.8
Glycemic Treatment

Insulin sensitizers
Biguanides (metformin)
Metformin is the only available biguanide. Metformin should be used as a first-line therapy in patients with type 2 DM whenever possible.13 Metformin suppresses hepatic glucose output and primarily affects fasting glycemia; however, reduced postprandial glucose concentrations also occur.
The most common side effects of metformin are diarrhea, nausea, and abdominal discomfort. Metformin has the potential to produce very rare but life-threatening lactic acidosis (< 1 in 100,000). The use of metformin is contraindicated in patients with a glomerular filtration rate less than 30 mL/min, with acidosis, hypoxia, or dehydration.8
Metformin usually does not lead to hypoglycemia when used as monotherapy. It can lead to weight loss (3%–5% of body weight), and it has been shown to decrease plasma triglyceride concentrations (10%–20%).8,14,15
Thiazolidinediones
Thiazolidinediones (TZDs) primarily enhance the insulin sensitivity of muscle and fat tissue and mildly enhance insulin sensitivity of the liver. TZDs lower fasting and postprandial blood glucose levels.
Major side effects of TZDs include weight gain, with an increase in subcutaneous adiposity, and fluid retention. Fluid retention typically manifests as peripheral edema, but heart failure can occur on occasion. These agents should be avoided in patients with functional class III or IV heart failure. The PROactive trial of the TZD pioglitazone found that pioglitazone did not increase cardiovascular risk compared with placebo.16 TZDs have been associated with an increased risk of fractures, particularly in women. When used as monotherapy, TZDs do not cause hypoglycemia. Pioglitazone lowers triglyceride levels, increases high-density lipoprotein cholesterol, and increases the low-density lipoprotein cholesterol particle size.8,16–18
Insulin secretagogues
Insulin secretagogues such as sulfonylureas and glinides stimulate secretion of insulin from the pancreas regardless of the ambient glucose concentration.
Sulfonylureas
Sulfonylureas lower fasting and postprandial glucose levels. The main side effects include weight gain (about 2 kg upon initiation) and hypoglycemia. The UK Prospective Diabetes Study (UKPDS) trial showed a decrease in microvascular complications with the use of sulfonylureas.19 Caution should be used in patients with liver or kidney dysfunction or patients who frequently skip meals. Newer, second-generation sulfonylureas (ie, glipizide and glimepiride) may have less risk of hypoglycemia because their action is somewhat glucose dependent.8,17,19
Glinides
Glinides, which include repaglinide and nateglenide, have a rapid onset of action and a short duration of action, so they are a good option for patients with erratically timed meals. Glinides have a lower risk of hypoglycemia than sulfonylureas. Caution must be used with glinides in patients with liver dysfunction. Dosing is immediately before meals.8,17
Alpha-glucosidase inhibitors
Alpha-glucosidase inhibitors such as acarbose, miglitol, and voglibose block the enzyme alpha-glucosidase in the cells of the brush border of the small intestine, which delays absorption of carbohydrates. Alpha-glucosidase inhibitors primarily affect postprandial hyperglycemia without causing hypoglycemia. Abdominal cramps, bloating, flatulence, and diarrhea are the most common side effects. Use of alpha-glucosidase inhibitors should be avoided in patients with severe hepatic or renal impairment. Dosing is prior to carbohydrate-containing meals.8,20
Incretin-based therapies
Therapies that target the incretin hormones to increase insulin production include glucagon-like peptide-1 (GLP-1) receptor agonists and dipeptidyl peptidase-4 (DPP-4) inhibitors.
GLP-1 agonists
Exenatide, liraglutide, albiglutide, and dulaglutide are synthetic analogs of the GLP-1 hormone. GLP-1 is produced in the small intestine; it stimulates insulin secretion and inhibits glucagon secretion in a glucose-dependent manner. It also delays gastric emptying and suppresses appetite through central pathways. GLP-1 agonists primarily decrease postprandial blood glucose levels; however, a moderate reduction in fasting blood glucose and some weight loss can also occur.
The major side effects are gastrointestinal complaints such as nausea, vomiting, and diarrhea. Hypoglycemia does not occur unless GLP-1 analogues are combined with a sulfonylurea or insulin. There is a slightly increased risk of acute pancreatitis in patients using GLP-1 agonist medications, and patients must be warned to discontinue use of these medications if abdominal pain occurs.
Dosing of GLP-1 agonist medications is either twice daily, daily, or weekly by subcutaneous injection.8,21
DPP-4 inhibitors
DPP-4 is an enzyme that rapidly degrades GLP-1. Suppression of DPP-4 leads to higher levels of insulin secretion and suppression of glucagon secretion in a glucose-dependent manner.
The DPP-4 inhibitors such as linagliptin, sitagliptin, saxagliptin, and alogliptin are given orally once daily. An increased risk of acute pancreatitis has been reported in some patients. Dose reduction is needed in patients with renal impairment for most of these medications.8,22
SLGT-2 inhibitors
SGLT-2 inhibitors include canagliflozin, dapagliflozin, and empagliflozin and are the newest group of antidiabetic medications. These medications inhibit glucose reabsorption in proximal tubule of the kidney leading to glycosuria, which lowers the blood glucose concentration, lowers blood pressure, and leads to some weight loss. Empagliflozin was shown to be cardioprotective in some patients.23
SGLT-2 inhibitors are given once a day in the morning and the primary side effects are polyuria and genital yeast infections. These medications are contraindicated in patients with severe end-stage renal disease and those who are on dialysis.8,24
Pramlintide (amylinomimetics)
Pramlintide, an amylinomimetic, is a synthetic drug that acts like amylin, a hormone secreted by beta cells that suppresses glucagon secretion, slows gastric emptying, and suppresses appetite through central pathways. Pramlintide acts primarily on postprandial blood glucose levels.
The side effects of pramlintide are gastrointestinal complaints, especially nausea. Currently, pramlintide is approved only as an adjunctive therapy with insulin, and it can be used in patients with type 1 DM or type 2 DM. The dose for type 1 DM is 15 µg before each meal subcutaneously, and for type 2 DM it is generally 60 µg before meals.25
Dopamine-receptor agonist (bromocriptine)
Bromocriptine is a central dopamine-receptor agonist, and when given in rapid-release form within 2 hours of awakening in the morning, it improves glycemic control for patients with type 2 DM. The mechanism of action resulting in improved glycemic control is unknown. Studies have demonstrated the cardiovascular safety of bromocriptine.26
Side effects of bromocriptine include hypotension, somnolence, and nausea. Individuals with psychiatric disorders may experience exacerbation while taking bromocriptine. Bromocriptine is taken with food to diminish nausea.27
Insulin
Insulin and insulin analogues remain the most direct method of reducing hyperglycemia. There is no upper limit in dosing for therapeutic effect, so it can be used to bring any HbA1c down to near-normal levels. Other benefits of insulin include reducing triglyceride levels and increasing high-density lipoprotein cholesterol.
Hypoglycemia is a concern with use of insulin, and studies have shown that episodes for which the patient required assistance due to the hypoglycemia occurred between 1 and 3 times per 100 patient-years.13 Weight gain can occur after initiation of insulin therapy, and patients typically gain 2 kg to 4 kg.8
Initiation and Titration of Therapy
All patients with type 1 DM require insulin therapy. There are 2 regimens available: basal-bolus and insulin-pump therapy. Patients with type 2 DM often require insulin, which can be combined with oral hypoglycemic agents. Regimens include basal insulin only, twice-daily premixed insulin, basal-bolus therapy, and insulin-pump therapy.28
Basal-bolus therapy
The basal-bolus regimen combines a long-acting agent for basal-insulin needs that is used once or twice daily and a rapid-acting agent for prandial coverage. Traditionally, 50% of the total daily dose is given as basal insulin (detemir, glargine, degludec) and the remaining dose as prandial insulin divided equally before meals (regular, lispro, glulisine, or aspart).
The meal dose of insulin can be fixed, but it is better to determine the dose based on the carbohydrate content of the meal. To do so, patients should be educated about carbohydrate counting and the dose of insulin required to cover the carbohydrate content of the meal. Consultation with a diabetes educator is needed for patients to effectively dose insulin based on the carbohydrate content of meals. Patients are also provided with a sliding scale of supplemental insulin to use as a third component of therapy when the blood glucose level is higher than desired.
The starting total daily insulin dose is typically 0.3 U/kg for patients with type 1 DM and 0.5 U/kg for patients with type 2 DM if no other medications are used. The ADA recommends adding basal insulin at 0.1 to 0.2 U/kg for patients with type 2 DM once they need it. The key to good glycemic control is self-monitoring of blood glucose by the patient and frequent adjustment of the regimen until control is achieved.8
Insulin-pump therapy
The insulin pump allows the use of different basal insulin rates at different periods of the day for greater flexibility with daily dosing. The insulin pump also allows administration of the meal bolus as a single discrete bolus or as an extended bolus (square bolus) over a certain period of time, which allows a better match between insulin delivery and glucose absorption from the meal in patients with abnormalities of gastric emptying. Use of an insulin pump should be considered in the following patients:
- Patients unable to achieve target goals with basal-bolus regimens
- Patients with frequent hypoglycemia, dawn phenomenon, or brittle diabetes
- Pregnant patients
- Patients with insulin sensitivity or those requiring more intense monitoring due to complications.
Recently, continuous glucose monitors have been developed that measure interstitial glucose levels. Continuous glucose monitoring has been shown to lower HbA1c in adult patients with type 1 DM.29
Gestational diabetes
In patients with gestational diabetes, insulin therapy is indicated when exercise and nutritional therapy are ineffective in controlling prandial and fasting blood glucose levels. Basal therapy alone may be sufficient, but a basal-bolus regimen is often required.8
Summary
- Glycemic control reduces the development and progression of complications of diabetes such as retinopathy, nephropathy, and neuropathy.
- The primary techniques available to assess the quality of a patient’s glycemic control are self-monitoring of blood glucose and interval measurement of HbA1c.
- Available treatment options to control blood glucose include insulin sensitizers, insulin secretagogues, alpha-glucosidase inhibitors, incretin-based therapies, SGLT-2 inhibitors, amylinomimetics (pramlintide), dopamine-receptor agonist (bromocriptine), and insulin.
- Mitka M. Report quantifies diabetes complications. JAMA 2007; 297:2337–2338.
- Welschen LM, Bloemendal E, Nijpels G, et al. Self-monitoring of blood glucose in patients with type 2 diabetes who are not using insulin: a systematic review. Diabetes Care 2005; 28:1510–1517.
- UK Prospective Diabetes Study (UKPDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34) [published erratum appears in Lancet 1998; 352:1558]. Lancet 1998; 352:854–865.
- Chase HP, Jackson WE, Hoops SL, Cockerham RS, Archer PG, O’Brien D. Glucose control and the renal and retinal complications of insulin-dependent diabetes. JAMA 1989; 261:1155–1160.
- The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 1993; 329:977–986.
- Evans JM, Newton RW, Ruta DA, MacDonald TM, Stevenson RJ, Morris AD. Frequency of blood glucose monitoring in relation to glycaemic control: observational study with diabetes database. BMJ 1999; 319:83–86.
- Bergenstal, RM, James GR III; Global Consensus Conference on Glucose Monitoring Panel. The role of self-monitoring of blood glucose in the care of people with diabetes: report of a global consensus conference. Am J Med 2005; 118(suppl 9A):1S–6S.
- American Diabetes Association. Standards of medical care in diabetes—2017: summary of revisions. Diabetes Care 2017; 40(suppl 1):S1–S135.
- Schwedes U, Siebolds M, Mertes G; for the SMBG Study Group. Meal-related structured self-monitoring of blood glucose: effect on diabetes control in non-insulin-treated type 2 diabetic patients. Diabetes Care 2002; 25:1928–1932.
- Saudek CD, Derr RL, Kalyani RR. Assessing glycemia in diabetes using self-monitoring blood glucose and hemoglobin A1c. JAMA 2006; 295:1688–1697.
- Delamater A. Clinical use of hemoglobin A1c to improve diabetes management. Clinical Diabetes 2006; 24:6–8.
- Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes, 2015: a patient-centered approach: update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2015; 38:140–149.
- Nathan DM, Buse JB, Davidson MB, et al; American Diabetes Association; European Association for Study of Diabetes. Medical management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy: a consensus statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2009; 32:193–203.
- Bailey CJ, Turner RC. Metformin. N Engl J Med 1996; 334:574–579.
- Bailey CJ. Biguanides and NIDDM. Diabetes Care 1992; 15:755–772.
- Dormandy JA, Charbonnel C, Eckland DJ, et al; PROactive investigators. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial. Lancet 2005; 366:1279–1289.
- Fonseca VA, Kulkarni KD. Management of type 2 diabetes: oral agents, insulin, and injectables. J Am Diet Assoc 2008; 108(4 suppl 1):S29–S33.
- Nathan DM, Buse JB, Davidson MB, et al. Management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy—update regarding thiazolidinediones: a consensus statement from the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2008; 31:173–175.
- UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33) [erratum published in Lancet 1999; 354:602]. Lancet 1998; 352:837–853.
- Chiasson J-L, Josse RG, Gomis R, Hanefeld M, Karsik A, Laakso M; for the STOP-NIDDM Trial Research Group. Acarbose treatment and the risk of cardiovascular disease and hypertension in patients with impaired glucose tolerance: The STOP-NIDDM Trial. JAMA 2003; 290:486–494.
- Victoza [package insert]. Bagsvaerd, Denmark: Novo Nordisk; 2010. https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/022341lbl.pdf. Accessed June 26, 2017.
- Nauck MA, Vilsbøll T, Gallwitz B, Garber A, Madsbad S. Incretin-based therapies viewpoints on the way to consensus. Diabetes Care 2009; 32(suppl 2):S223–S231.
- ZinmanB, Wanner C, Larchin JM; EMPA-REG OUTCOME Investigators. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med 2015; 373:2117–2128.
- Abdul-Ghani MA, Norton L, DeFronzo RA. Role of sodium-glucose cotransporter 2 (SGLT 2) inhibitors in the treatment of type 2 diabetes. Endocr Rev 2011; 32:515–531.
- Symlin (Pramlintide acetate) [package insert]. Wilmington DE: AstraZeneca; 2015. Pharmaceuticals LP. http://www.azpicentral.com/symlin/pi_symlin.pdf#page=1. Accessed June 26, 2017.
- Gaziano JM, Cincotta AH, O’Connor CM, et al. Randomized clinical trial of quick-release bromocriptine among patients with type 2 diabetes on overall safety and cardiovascular outcomes. Diabetes Care 2010; 33:1503–1508.
- Cycloset [package insert]. Tiverton, RI: VeroScience LLC; 2016. http://www.veroscience.com/documents/CyclosetPackageInsertFeb062017.pdf. Accessed June 26, 2017.
- Hirsch IB, Bergenstal RM, Parkin CG, Wright Jr, E, Buse JB. A real-world approach to insulin therapy in primary care practice. Clinical Diabetes 2005; 23:78–86.
- Juvenile Diabetes Research Foundation Continuous Glucose Monitoring Study Group; Tamborlane WV, Beck RW, Bode BW, et al. Continuous glucose monitoring and intensive treatment of type 1 diabetes. N Engl J Med 2008; 359:1464–1476.
In the United States, 57.9% of patients with diabetes mellitus (DM) have at least 1 diabetes-related complication and 14.3% of patients with diabetes have 3 or more diabetes-related complications.1 Achieving glycemic control in patients with DM reduces the development and progression of retinopathy, nephropathy, and neuropathy. Aggressive treatment of dyslipidemia and hypertension decreases macrovascular complications.2–4 The techniques for monitoring blood glucose and the various treatment options available to manage glycemic control in patients with diabetes are reviewed below.
Measuring Glycemic Control
The primary techniques available to assess the quality of a patient’s glycemic control are self-monitoring of blood glucose and interval measurement of hemoglobin A1c (HbA1c). Continuous glucose monitoring is also available and may be appropriate for select patients, such as patients with brittle diabetes and those using insulin pumps.
Self-monitoring of blood glucose
For patients with type 1 DM and patients with insulin-dependent type 2 DM, self-monitoring of blood glucose allows patients to adjust insulin dosing to prevent hypoglycemia and hyperglycemia.2,5–7 The American Diabetes Association (ADA) guidelines recommend that patients with type 1 DM self-monitor their glucose:
- Before eating
- At bedtime
- Before exercise
- If hypoglycemia is suspected
- Until hypoglycemia is corrected
- Postprandially upon occasion
- And before critical tasks (ie, driving).8
Patients should be educated about how to use real-time blood glucose values to adjust their food intake and medical therapy.
It is commonly recommended that patients with type 2 DM self-monitor their blood glucose levels, but the evidence to support the effectiveness of this practice is inconclusive. Initial studies showed reductions in HbA1c with self-monitoring; however, the inclusion of beneficial health behaviors such as diet and exercise in the analyses makes it difficult to assess the effectiveness of self-monitor blood glucose alone.2,9
The ADA recommends that nonpregnant adults maintain blood glucose levels of 80 mg/dL to 130 mg/dL preprandial and less than 180 mg/dL postprandial.8 The blood glucose goals for patients with gestational diabetes are 95 mg/dL or less preprandial and either 140 mg/dL or less 1-hour postprandial or 120 mg/dL or less 2-hours postprandial.
HbA1c
HbA1c tests reflect the mean blood glucose values over a 3-month period and can predict patients’ risk of microvascular complications.10,11 The ADA recommends that patients with stable glycemic control have an HbA1c test at least twice a year. Quarterly HbA1c testing is suggested for patients with a recent change in therapy or for patients not meeting their glycemic goals.8
Measurement of HbA1c is influenced by the red blood cell turnover rate; therefore, anemia, transfusions, and hemoglobinopathies can cause inaccurate test values. The ADA recommends that nonpregnant adults maintain HbA1c levels near 7%. For patients with diabetes who become pregnant, the goal is HbA1c levels less than 6.0%.8 The ADA also recommends that select patients, especially those with a long life expectancy and little comorbidity, adopt glycemic targets near normal levels (HbA1c < 6.5%), providing the target can be achieved without significant hypoglycemia.8
Glycemic Treatment
Insulin sensitizers
Biguanides (metformin)
Metformin is the only available biguanide. Metformin should be used as a first-line therapy in patients with type 2 DM whenever possible.13 Metformin suppresses hepatic glucose output and primarily affects fasting glycemia; however, reduced postprandial glucose concentrations also occur.
The most common side effects of metformin are diarrhea, nausea, and abdominal discomfort. Metformin has the potential to produce very rare but life-threatening lactic acidosis (< 1 in 100,000). The use of metformin is contraindicated in patients with a glomerular filtration rate less than 30 mL/min, with acidosis, hypoxia, or dehydration.8
Metformin usually does not lead to hypoglycemia when used as monotherapy. It can lead to weight loss (3%–5% of body weight), and it has been shown to decrease plasma triglyceride concentrations (10%–20%).8,14,15
Thiazolidinediones
Thiazolidinediones (TZDs) primarily enhance the insulin sensitivity of muscle and fat tissue and mildly enhance insulin sensitivity of the liver. TZDs lower fasting and postprandial blood glucose levels.
Major side effects of TZDs include weight gain, with an increase in subcutaneous adiposity, and fluid retention. Fluid retention typically manifests as peripheral edema, but heart failure can occur on occasion. These agents should be avoided in patients with functional class III or IV heart failure. The PROactive trial of the TZD pioglitazone found that pioglitazone did not increase cardiovascular risk compared with placebo.16 TZDs have been associated with an increased risk of fractures, particularly in women. When used as monotherapy, TZDs do not cause hypoglycemia. Pioglitazone lowers triglyceride levels, increases high-density lipoprotein cholesterol, and increases the low-density lipoprotein cholesterol particle size.8,16–18
Insulin secretagogues
Insulin secretagogues such as sulfonylureas and glinides stimulate secretion of insulin from the pancreas regardless of the ambient glucose concentration.
Sulfonylureas
Sulfonylureas lower fasting and postprandial glucose levels. The main side effects include weight gain (about 2 kg upon initiation) and hypoglycemia. The UK Prospective Diabetes Study (UKPDS) trial showed a decrease in microvascular complications with the use of sulfonylureas.19 Caution should be used in patients with liver or kidney dysfunction or patients who frequently skip meals. Newer, second-generation sulfonylureas (ie, glipizide and glimepiride) may have less risk of hypoglycemia because their action is somewhat glucose dependent.8,17,19
Glinides
Glinides, which include repaglinide and nateglenide, have a rapid onset of action and a short duration of action, so they are a good option for patients with erratically timed meals. Glinides have a lower risk of hypoglycemia than sulfonylureas. Caution must be used with glinides in patients with liver dysfunction. Dosing is immediately before meals.8,17
Alpha-glucosidase inhibitors
Alpha-glucosidase inhibitors such as acarbose, miglitol, and voglibose block the enzyme alpha-glucosidase in the cells of the brush border of the small intestine, which delays absorption of carbohydrates. Alpha-glucosidase inhibitors primarily affect postprandial hyperglycemia without causing hypoglycemia. Abdominal cramps, bloating, flatulence, and diarrhea are the most common side effects. Use of alpha-glucosidase inhibitors should be avoided in patients with severe hepatic or renal impairment. Dosing is prior to carbohydrate-containing meals.8,20
Incretin-based therapies
Therapies that target the incretin hormones to increase insulin production include glucagon-like peptide-1 (GLP-1) receptor agonists and dipeptidyl peptidase-4 (DPP-4) inhibitors.
GLP-1 agonists
Exenatide, liraglutide, albiglutide, and dulaglutide are synthetic analogs of the GLP-1 hormone. GLP-1 is produced in the small intestine; it stimulates insulin secretion and inhibits glucagon secretion in a glucose-dependent manner. It also delays gastric emptying and suppresses appetite through central pathways. GLP-1 agonists primarily decrease postprandial blood glucose levels; however, a moderate reduction in fasting blood glucose and some weight loss can also occur.
The major side effects are gastrointestinal complaints such as nausea, vomiting, and diarrhea. Hypoglycemia does not occur unless GLP-1 analogues are combined with a sulfonylurea or insulin. There is a slightly increased risk of acute pancreatitis in patients using GLP-1 agonist medications, and patients must be warned to discontinue use of these medications if abdominal pain occurs.
Dosing of GLP-1 agonist medications is either twice daily, daily, or weekly by subcutaneous injection.8,21
DPP-4 inhibitors
DPP-4 is an enzyme that rapidly degrades GLP-1. Suppression of DPP-4 leads to higher levels of insulin secretion and suppression of glucagon secretion in a glucose-dependent manner.
The DPP-4 inhibitors such as linagliptin, sitagliptin, saxagliptin, and alogliptin are given orally once daily. An increased risk of acute pancreatitis has been reported in some patients. Dose reduction is needed in patients with renal impairment for most of these medications.8,22
SLGT-2 inhibitors
SGLT-2 inhibitors include canagliflozin, dapagliflozin, and empagliflozin and are the newest group of antidiabetic medications. These medications inhibit glucose reabsorption in proximal tubule of the kidney leading to glycosuria, which lowers the blood glucose concentration, lowers blood pressure, and leads to some weight loss. Empagliflozin was shown to be cardioprotective in some patients.23
SGLT-2 inhibitors are given once a day in the morning and the primary side effects are polyuria and genital yeast infections. These medications are contraindicated in patients with severe end-stage renal disease and those who are on dialysis.8,24
Pramlintide (amylinomimetics)
Pramlintide, an amylinomimetic, is a synthetic drug that acts like amylin, a hormone secreted by beta cells that suppresses glucagon secretion, slows gastric emptying, and suppresses appetite through central pathways. Pramlintide acts primarily on postprandial blood glucose levels.
The side effects of pramlintide are gastrointestinal complaints, especially nausea. Currently, pramlintide is approved only as an adjunctive therapy with insulin, and it can be used in patients with type 1 DM or type 2 DM. The dose for type 1 DM is 15 µg before each meal subcutaneously, and for type 2 DM it is generally 60 µg before meals.25
Dopamine-receptor agonist (bromocriptine)
Bromocriptine is a central dopamine-receptor agonist, and when given in rapid-release form within 2 hours of awakening in the morning, it improves glycemic control for patients with type 2 DM. The mechanism of action resulting in improved glycemic control is unknown. Studies have demonstrated the cardiovascular safety of bromocriptine.26
Side effects of bromocriptine include hypotension, somnolence, and nausea. Individuals with psychiatric disorders may experience exacerbation while taking bromocriptine. Bromocriptine is taken with food to diminish nausea.27
Insulin
Insulin and insulin analogues remain the most direct method of reducing hyperglycemia. There is no upper limit in dosing for therapeutic effect, so it can be used to bring any HbA1c down to near-normal levels. Other benefits of insulin include reducing triglyceride levels and increasing high-density lipoprotein cholesterol.
Hypoglycemia is a concern with use of insulin, and studies have shown that episodes for which the patient required assistance due to the hypoglycemia occurred between 1 and 3 times per 100 patient-years.13 Weight gain can occur after initiation of insulin therapy, and patients typically gain 2 kg to 4 kg.8
Initiation and Titration of Therapy
All patients with type 1 DM require insulin therapy. There are 2 regimens available: basal-bolus and insulin-pump therapy. Patients with type 2 DM often require insulin, which can be combined with oral hypoglycemic agents. Regimens include basal insulin only, twice-daily premixed insulin, basal-bolus therapy, and insulin-pump therapy.28
Basal-bolus therapy
The basal-bolus regimen combines a long-acting agent for basal-insulin needs that is used once or twice daily and a rapid-acting agent for prandial coverage. Traditionally, 50% of the total daily dose is given as basal insulin (detemir, glargine, degludec) and the remaining dose as prandial insulin divided equally before meals (regular, lispro, glulisine, or aspart).
The meal dose of insulin can be fixed, but it is better to determine the dose based on the carbohydrate content of the meal. To do so, patients should be educated about carbohydrate counting and the dose of insulin required to cover the carbohydrate content of the meal. Consultation with a diabetes educator is needed for patients to effectively dose insulin based on the carbohydrate content of meals. Patients are also provided with a sliding scale of supplemental insulin to use as a third component of therapy when the blood glucose level is higher than desired.
The starting total daily insulin dose is typically 0.3 U/kg for patients with type 1 DM and 0.5 U/kg for patients with type 2 DM if no other medications are used. The ADA recommends adding basal insulin at 0.1 to 0.2 U/kg for patients with type 2 DM once they need it. The key to good glycemic control is self-monitoring of blood glucose by the patient and frequent adjustment of the regimen until control is achieved.8
Insulin-pump therapy
The insulin pump allows the use of different basal insulin rates at different periods of the day for greater flexibility with daily dosing. The insulin pump also allows administration of the meal bolus as a single discrete bolus or as an extended bolus (square bolus) over a certain period of time, which allows a better match between insulin delivery and glucose absorption from the meal in patients with abnormalities of gastric emptying. Use of an insulin pump should be considered in the following patients:
- Patients unable to achieve target goals with basal-bolus regimens
- Patients with frequent hypoglycemia, dawn phenomenon, or brittle diabetes
- Pregnant patients
- Patients with insulin sensitivity or those requiring more intense monitoring due to complications.
Recently, continuous glucose monitors have been developed that measure interstitial glucose levels. Continuous glucose monitoring has been shown to lower HbA1c in adult patients with type 1 DM.29
Gestational diabetes
In patients with gestational diabetes, insulin therapy is indicated when exercise and nutritional therapy are ineffective in controlling prandial and fasting blood glucose levels. Basal therapy alone may be sufficient, but a basal-bolus regimen is often required.8
Summary
- Glycemic control reduces the development and progression of complications of diabetes such as retinopathy, nephropathy, and neuropathy.
- The primary techniques available to assess the quality of a patient’s glycemic control are self-monitoring of blood glucose and interval measurement of HbA1c.
- Available treatment options to control blood glucose include insulin sensitizers, insulin secretagogues, alpha-glucosidase inhibitors, incretin-based therapies, SGLT-2 inhibitors, amylinomimetics (pramlintide), dopamine-receptor agonist (bromocriptine), and insulin.
In the United States, 57.9% of patients with diabetes mellitus (DM) have at least 1 diabetes-related complication and 14.3% of patients with diabetes have 3 or more diabetes-related complications.1 Achieving glycemic control in patients with DM reduces the development and progression of retinopathy, nephropathy, and neuropathy. Aggressive treatment of dyslipidemia and hypertension decreases macrovascular complications.2–4 The techniques for monitoring blood glucose and the various treatment options available to manage glycemic control in patients with diabetes are reviewed below.
Measuring Glycemic Control
The primary techniques available to assess the quality of a patient’s glycemic control are self-monitoring of blood glucose and interval measurement of hemoglobin A1c (HbA1c). Continuous glucose monitoring is also available and may be appropriate for select patients, such as patients with brittle diabetes and those using insulin pumps.
Self-monitoring of blood glucose
For patients with type 1 DM and patients with insulin-dependent type 2 DM, self-monitoring of blood glucose allows patients to adjust insulin dosing to prevent hypoglycemia and hyperglycemia.2,5–7 The American Diabetes Association (ADA) guidelines recommend that patients with type 1 DM self-monitor their glucose:
- Before eating
- At bedtime
- Before exercise
- If hypoglycemia is suspected
- Until hypoglycemia is corrected
- Postprandially upon occasion
- And before critical tasks (ie, driving).8
Patients should be educated about how to use real-time blood glucose values to adjust their food intake and medical therapy.
It is commonly recommended that patients with type 2 DM self-monitor their blood glucose levels, but the evidence to support the effectiveness of this practice is inconclusive. Initial studies showed reductions in HbA1c with self-monitoring; however, the inclusion of beneficial health behaviors such as diet and exercise in the analyses makes it difficult to assess the effectiveness of self-monitor blood glucose alone.2,9
The ADA recommends that nonpregnant adults maintain blood glucose levels of 80 mg/dL to 130 mg/dL preprandial and less than 180 mg/dL postprandial.8 The blood glucose goals for patients with gestational diabetes are 95 mg/dL or less preprandial and either 140 mg/dL or less 1-hour postprandial or 120 mg/dL or less 2-hours postprandial.
HbA1c
HbA1c tests reflect the mean blood glucose values over a 3-month period and can predict patients’ risk of microvascular complications.10,11 The ADA recommends that patients with stable glycemic control have an HbA1c test at least twice a year. Quarterly HbA1c testing is suggested for patients with a recent change in therapy or for patients not meeting their glycemic goals.8
Measurement of HbA1c is influenced by the red blood cell turnover rate; therefore, anemia, transfusions, and hemoglobinopathies can cause inaccurate test values. The ADA recommends that nonpregnant adults maintain HbA1c levels near 7%. For patients with diabetes who become pregnant, the goal is HbA1c levels less than 6.0%.8 The ADA also recommends that select patients, especially those with a long life expectancy and little comorbidity, adopt glycemic targets near normal levels (HbA1c < 6.5%), providing the target can be achieved without significant hypoglycemia.8
Glycemic Treatment
Insulin sensitizers
Biguanides (metformin)
Metformin is the only available biguanide. Metformin should be used as a first-line therapy in patients with type 2 DM whenever possible.13 Metformin suppresses hepatic glucose output and primarily affects fasting glycemia; however, reduced postprandial glucose concentrations also occur.
The most common side effects of metformin are diarrhea, nausea, and abdominal discomfort. Metformin has the potential to produce very rare but life-threatening lactic acidosis (< 1 in 100,000). The use of metformin is contraindicated in patients with a glomerular filtration rate less than 30 mL/min, with acidosis, hypoxia, or dehydration.8
Metformin usually does not lead to hypoglycemia when used as monotherapy. It can lead to weight loss (3%–5% of body weight), and it has been shown to decrease plasma triglyceride concentrations (10%–20%).8,14,15
Thiazolidinediones
Thiazolidinediones (TZDs) primarily enhance the insulin sensitivity of muscle and fat tissue and mildly enhance insulin sensitivity of the liver. TZDs lower fasting and postprandial blood glucose levels.
Major side effects of TZDs include weight gain, with an increase in subcutaneous adiposity, and fluid retention. Fluid retention typically manifests as peripheral edema, but heart failure can occur on occasion. These agents should be avoided in patients with functional class III or IV heart failure. The PROactive trial of the TZD pioglitazone found that pioglitazone did not increase cardiovascular risk compared with placebo.16 TZDs have been associated with an increased risk of fractures, particularly in women. When used as monotherapy, TZDs do not cause hypoglycemia. Pioglitazone lowers triglyceride levels, increases high-density lipoprotein cholesterol, and increases the low-density lipoprotein cholesterol particle size.8,16–18
Insulin secretagogues
Insulin secretagogues such as sulfonylureas and glinides stimulate secretion of insulin from the pancreas regardless of the ambient glucose concentration.
Sulfonylureas
Sulfonylureas lower fasting and postprandial glucose levels. The main side effects include weight gain (about 2 kg upon initiation) and hypoglycemia. The UK Prospective Diabetes Study (UKPDS) trial showed a decrease in microvascular complications with the use of sulfonylureas.19 Caution should be used in patients with liver or kidney dysfunction or patients who frequently skip meals. Newer, second-generation sulfonylureas (ie, glipizide and glimepiride) may have less risk of hypoglycemia because their action is somewhat glucose dependent.8,17,19
Glinides
Glinides, which include repaglinide and nateglenide, have a rapid onset of action and a short duration of action, so they are a good option for patients with erratically timed meals. Glinides have a lower risk of hypoglycemia than sulfonylureas. Caution must be used with glinides in patients with liver dysfunction. Dosing is immediately before meals.8,17
Alpha-glucosidase inhibitors
Alpha-glucosidase inhibitors such as acarbose, miglitol, and voglibose block the enzyme alpha-glucosidase in the cells of the brush border of the small intestine, which delays absorption of carbohydrates. Alpha-glucosidase inhibitors primarily affect postprandial hyperglycemia without causing hypoglycemia. Abdominal cramps, bloating, flatulence, and diarrhea are the most common side effects. Use of alpha-glucosidase inhibitors should be avoided in patients with severe hepatic or renal impairment. Dosing is prior to carbohydrate-containing meals.8,20
Incretin-based therapies
Therapies that target the incretin hormones to increase insulin production include glucagon-like peptide-1 (GLP-1) receptor agonists and dipeptidyl peptidase-4 (DPP-4) inhibitors.
GLP-1 agonists
Exenatide, liraglutide, albiglutide, and dulaglutide are synthetic analogs of the GLP-1 hormone. GLP-1 is produced in the small intestine; it stimulates insulin secretion and inhibits glucagon secretion in a glucose-dependent manner. It also delays gastric emptying and suppresses appetite through central pathways. GLP-1 agonists primarily decrease postprandial blood glucose levels; however, a moderate reduction in fasting blood glucose and some weight loss can also occur.
The major side effects are gastrointestinal complaints such as nausea, vomiting, and diarrhea. Hypoglycemia does not occur unless GLP-1 analogues are combined with a sulfonylurea or insulin. There is a slightly increased risk of acute pancreatitis in patients using GLP-1 agonist medications, and patients must be warned to discontinue use of these medications if abdominal pain occurs.
Dosing of GLP-1 agonist medications is either twice daily, daily, or weekly by subcutaneous injection.8,21
DPP-4 inhibitors
DPP-4 is an enzyme that rapidly degrades GLP-1. Suppression of DPP-4 leads to higher levels of insulin secretion and suppression of glucagon secretion in a glucose-dependent manner.
The DPP-4 inhibitors such as linagliptin, sitagliptin, saxagliptin, and alogliptin are given orally once daily. An increased risk of acute pancreatitis has been reported in some patients. Dose reduction is needed in patients with renal impairment for most of these medications.8,22
SLGT-2 inhibitors
SGLT-2 inhibitors include canagliflozin, dapagliflozin, and empagliflozin and are the newest group of antidiabetic medications. These medications inhibit glucose reabsorption in proximal tubule of the kidney leading to glycosuria, which lowers the blood glucose concentration, lowers blood pressure, and leads to some weight loss. Empagliflozin was shown to be cardioprotective in some patients.23
SGLT-2 inhibitors are given once a day in the morning and the primary side effects are polyuria and genital yeast infections. These medications are contraindicated in patients with severe end-stage renal disease and those who are on dialysis.8,24
Pramlintide (amylinomimetics)
Pramlintide, an amylinomimetic, is a synthetic drug that acts like amylin, a hormone secreted by beta cells that suppresses glucagon secretion, slows gastric emptying, and suppresses appetite through central pathways. Pramlintide acts primarily on postprandial blood glucose levels.
The side effects of pramlintide are gastrointestinal complaints, especially nausea. Currently, pramlintide is approved only as an adjunctive therapy with insulin, and it can be used in patients with type 1 DM or type 2 DM. The dose for type 1 DM is 15 µg before each meal subcutaneously, and for type 2 DM it is generally 60 µg before meals.25
Dopamine-receptor agonist (bromocriptine)
Bromocriptine is a central dopamine-receptor agonist, and when given in rapid-release form within 2 hours of awakening in the morning, it improves glycemic control for patients with type 2 DM. The mechanism of action resulting in improved glycemic control is unknown. Studies have demonstrated the cardiovascular safety of bromocriptine.26
Side effects of bromocriptine include hypotension, somnolence, and nausea. Individuals with psychiatric disorders may experience exacerbation while taking bromocriptine. Bromocriptine is taken with food to diminish nausea.27
Insulin
Insulin and insulin analogues remain the most direct method of reducing hyperglycemia. There is no upper limit in dosing for therapeutic effect, so it can be used to bring any HbA1c down to near-normal levels. Other benefits of insulin include reducing triglyceride levels and increasing high-density lipoprotein cholesterol.
Hypoglycemia is a concern with use of insulin, and studies have shown that episodes for which the patient required assistance due to the hypoglycemia occurred between 1 and 3 times per 100 patient-years.13 Weight gain can occur after initiation of insulin therapy, and patients typically gain 2 kg to 4 kg.8
Initiation and Titration of Therapy
All patients with type 1 DM require insulin therapy. There are 2 regimens available: basal-bolus and insulin-pump therapy. Patients with type 2 DM often require insulin, which can be combined with oral hypoglycemic agents. Regimens include basal insulin only, twice-daily premixed insulin, basal-bolus therapy, and insulin-pump therapy.28
Basal-bolus therapy
The basal-bolus regimen combines a long-acting agent for basal-insulin needs that is used once or twice daily and a rapid-acting agent for prandial coverage. Traditionally, 50% of the total daily dose is given as basal insulin (detemir, glargine, degludec) and the remaining dose as prandial insulin divided equally before meals (regular, lispro, glulisine, or aspart).
The meal dose of insulin can be fixed, but it is better to determine the dose based on the carbohydrate content of the meal. To do so, patients should be educated about carbohydrate counting and the dose of insulin required to cover the carbohydrate content of the meal. Consultation with a diabetes educator is needed for patients to effectively dose insulin based on the carbohydrate content of meals. Patients are also provided with a sliding scale of supplemental insulin to use as a third component of therapy when the blood glucose level is higher than desired.
The starting total daily insulin dose is typically 0.3 U/kg for patients with type 1 DM and 0.5 U/kg for patients with type 2 DM if no other medications are used. The ADA recommends adding basal insulin at 0.1 to 0.2 U/kg for patients with type 2 DM once they need it. The key to good glycemic control is self-monitoring of blood glucose by the patient and frequent adjustment of the regimen until control is achieved.8
Insulin-pump therapy
The insulin pump allows the use of different basal insulin rates at different periods of the day for greater flexibility with daily dosing. The insulin pump also allows administration of the meal bolus as a single discrete bolus or as an extended bolus (square bolus) over a certain period of time, which allows a better match between insulin delivery and glucose absorption from the meal in patients with abnormalities of gastric emptying. Use of an insulin pump should be considered in the following patients:
- Patients unable to achieve target goals with basal-bolus regimens
- Patients with frequent hypoglycemia, dawn phenomenon, or brittle diabetes
- Pregnant patients
- Patients with insulin sensitivity or those requiring more intense monitoring due to complications.
Recently, continuous glucose monitors have been developed that measure interstitial glucose levels. Continuous glucose monitoring has been shown to lower HbA1c in adult patients with type 1 DM.29
Gestational diabetes
In patients with gestational diabetes, insulin therapy is indicated when exercise and nutritional therapy are ineffective in controlling prandial and fasting blood glucose levels. Basal therapy alone may be sufficient, but a basal-bolus regimen is often required.8
Summary
- Glycemic control reduces the development and progression of complications of diabetes such as retinopathy, nephropathy, and neuropathy.
- The primary techniques available to assess the quality of a patient’s glycemic control are self-monitoring of blood glucose and interval measurement of HbA1c.
- Available treatment options to control blood glucose include insulin sensitizers, insulin secretagogues, alpha-glucosidase inhibitors, incretin-based therapies, SGLT-2 inhibitors, amylinomimetics (pramlintide), dopamine-receptor agonist (bromocriptine), and insulin.
- Mitka M. Report quantifies diabetes complications. JAMA 2007; 297:2337–2338.
- Welschen LM, Bloemendal E, Nijpels G, et al. Self-monitoring of blood glucose in patients with type 2 diabetes who are not using insulin: a systematic review. Diabetes Care 2005; 28:1510–1517.
- UK Prospective Diabetes Study (UKPDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34) [published erratum appears in Lancet 1998; 352:1558]. Lancet 1998; 352:854–865.
- Chase HP, Jackson WE, Hoops SL, Cockerham RS, Archer PG, O’Brien D. Glucose control and the renal and retinal complications of insulin-dependent diabetes. JAMA 1989; 261:1155–1160.
- The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 1993; 329:977–986.
- Evans JM, Newton RW, Ruta DA, MacDonald TM, Stevenson RJ, Morris AD. Frequency of blood glucose monitoring in relation to glycaemic control: observational study with diabetes database. BMJ 1999; 319:83–86.
- Bergenstal, RM, James GR III; Global Consensus Conference on Glucose Monitoring Panel. The role of self-monitoring of blood glucose in the care of people with diabetes: report of a global consensus conference. Am J Med 2005; 118(suppl 9A):1S–6S.
- American Diabetes Association. Standards of medical care in diabetes—2017: summary of revisions. Diabetes Care 2017; 40(suppl 1):S1–S135.
- Schwedes U, Siebolds M, Mertes G; for the SMBG Study Group. Meal-related structured self-monitoring of blood glucose: effect on diabetes control in non-insulin-treated type 2 diabetic patients. Diabetes Care 2002; 25:1928–1932.
- Saudek CD, Derr RL, Kalyani RR. Assessing glycemia in diabetes using self-monitoring blood glucose and hemoglobin A1c. JAMA 2006; 295:1688–1697.
- Delamater A. Clinical use of hemoglobin A1c to improve diabetes management. Clinical Diabetes 2006; 24:6–8.
- Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes, 2015: a patient-centered approach: update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2015; 38:140–149.
- Nathan DM, Buse JB, Davidson MB, et al; American Diabetes Association; European Association for Study of Diabetes. Medical management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy: a consensus statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2009; 32:193–203.
- Bailey CJ, Turner RC. Metformin. N Engl J Med 1996; 334:574–579.
- Bailey CJ. Biguanides and NIDDM. Diabetes Care 1992; 15:755–772.
- Dormandy JA, Charbonnel C, Eckland DJ, et al; PROactive investigators. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial. Lancet 2005; 366:1279–1289.
- Fonseca VA, Kulkarni KD. Management of type 2 diabetes: oral agents, insulin, and injectables. J Am Diet Assoc 2008; 108(4 suppl 1):S29–S33.
- Nathan DM, Buse JB, Davidson MB, et al. Management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy—update regarding thiazolidinediones: a consensus statement from the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2008; 31:173–175.
- UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33) [erratum published in Lancet 1999; 354:602]. Lancet 1998; 352:837–853.
- Chiasson J-L, Josse RG, Gomis R, Hanefeld M, Karsik A, Laakso M; for the STOP-NIDDM Trial Research Group. Acarbose treatment and the risk of cardiovascular disease and hypertension in patients with impaired glucose tolerance: The STOP-NIDDM Trial. JAMA 2003; 290:486–494.
- Victoza [package insert]. Bagsvaerd, Denmark: Novo Nordisk; 2010. https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/022341lbl.pdf. Accessed June 26, 2017.
- Nauck MA, Vilsbøll T, Gallwitz B, Garber A, Madsbad S. Incretin-based therapies viewpoints on the way to consensus. Diabetes Care 2009; 32(suppl 2):S223–S231.
- ZinmanB, Wanner C, Larchin JM; EMPA-REG OUTCOME Investigators. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med 2015; 373:2117–2128.
- Abdul-Ghani MA, Norton L, DeFronzo RA. Role of sodium-glucose cotransporter 2 (SGLT 2) inhibitors in the treatment of type 2 diabetes. Endocr Rev 2011; 32:515–531.
- Symlin (Pramlintide acetate) [package insert]. Wilmington DE: AstraZeneca; 2015. Pharmaceuticals LP. http://www.azpicentral.com/symlin/pi_symlin.pdf#page=1. Accessed June 26, 2017.
- Gaziano JM, Cincotta AH, O’Connor CM, et al. Randomized clinical trial of quick-release bromocriptine among patients with type 2 diabetes on overall safety and cardiovascular outcomes. Diabetes Care 2010; 33:1503–1508.
- Cycloset [package insert]. Tiverton, RI: VeroScience LLC; 2016. http://www.veroscience.com/documents/CyclosetPackageInsertFeb062017.pdf. Accessed June 26, 2017.
- Hirsch IB, Bergenstal RM, Parkin CG, Wright Jr, E, Buse JB. A real-world approach to insulin therapy in primary care practice. Clinical Diabetes 2005; 23:78–86.
- Juvenile Diabetes Research Foundation Continuous Glucose Monitoring Study Group; Tamborlane WV, Beck RW, Bode BW, et al. Continuous glucose monitoring and intensive treatment of type 1 diabetes. N Engl J Med 2008; 359:1464–1476.
- Mitka M. Report quantifies diabetes complications. JAMA 2007; 297:2337–2338.
- Welschen LM, Bloemendal E, Nijpels G, et al. Self-monitoring of blood glucose in patients with type 2 diabetes who are not using insulin: a systematic review. Diabetes Care 2005; 28:1510–1517.
- UK Prospective Diabetes Study (UKPDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34) [published erratum appears in Lancet 1998; 352:1558]. Lancet 1998; 352:854–865.
- Chase HP, Jackson WE, Hoops SL, Cockerham RS, Archer PG, O’Brien D. Glucose control and the renal and retinal complications of insulin-dependent diabetes. JAMA 1989; 261:1155–1160.
- The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 1993; 329:977–986.
- Evans JM, Newton RW, Ruta DA, MacDonald TM, Stevenson RJ, Morris AD. Frequency of blood glucose monitoring in relation to glycaemic control: observational study with diabetes database. BMJ 1999; 319:83–86.
- Bergenstal, RM, James GR III; Global Consensus Conference on Glucose Monitoring Panel. The role of self-monitoring of blood glucose in the care of people with diabetes: report of a global consensus conference. Am J Med 2005; 118(suppl 9A):1S–6S.
- American Diabetes Association. Standards of medical care in diabetes—2017: summary of revisions. Diabetes Care 2017; 40(suppl 1):S1–S135.
- Schwedes U, Siebolds M, Mertes G; for the SMBG Study Group. Meal-related structured self-monitoring of blood glucose: effect on diabetes control in non-insulin-treated type 2 diabetic patients. Diabetes Care 2002; 25:1928–1932.
- Saudek CD, Derr RL, Kalyani RR. Assessing glycemia in diabetes using self-monitoring blood glucose and hemoglobin A1c. JAMA 2006; 295:1688–1697.
- Delamater A. Clinical use of hemoglobin A1c to improve diabetes management. Clinical Diabetes 2006; 24:6–8.
- Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes, 2015: a patient-centered approach: update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2015; 38:140–149.
- Nathan DM, Buse JB, Davidson MB, et al; American Diabetes Association; European Association for Study of Diabetes. Medical management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy: a consensus statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2009; 32:193–203.
- Bailey CJ, Turner RC. Metformin. N Engl J Med 1996; 334:574–579.
- Bailey CJ. Biguanides and NIDDM. Diabetes Care 1992; 15:755–772.
- Dormandy JA, Charbonnel C, Eckland DJ, et al; PROactive investigators. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial. Lancet 2005; 366:1279–1289.
- Fonseca VA, Kulkarni KD. Management of type 2 diabetes: oral agents, insulin, and injectables. J Am Diet Assoc 2008; 108(4 suppl 1):S29–S33.
- Nathan DM, Buse JB, Davidson MB, et al. Management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy—update regarding thiazolidinediones: a consensus statement from the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2008; 31:173–175.
- UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33) [erratum published in Lancet 1999; 354:602]. Lancet 1998; 352:837–853.
- Chiasson J-L, Josse RG, Gomis R, Hanefeld M, Karsik A, Laakso M; for the STOP-NIDDM Trial Research Group. Acarbose treatment and the risk of cardiovascular disease and hypertension in patients with impaired glucose tolerance: The STOP-NIDDM Trial. JAMA 2003; 290:486–494.
- Victoza [package insert]. Bagsvaerd, Denmark: Novo Nordisk; 2010. https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/022341lbl.pdf. Accessed June 26, 2017.
- Nauck MA, Vilsbøll T, Gallwitz B, Garber A, Madsbad S. Incretin-based therapies viewpoints on the way to consensus. Diabetes Care 2009; 32(suppl 2):S223–S231.
- ZinmanB, Wanner C, Larchin JM; EMPA-REG OUTCOME Investigators. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med 2015; 373:2117–2128.
- Abdul-Ghani MA, Norton L, DeFronzo RA. Role of sodium-glucose cotransporter 2 (SGLT 2) inhibitors in the treatment of type 2 diabetes. Endocr Rev 2011; 32:515–531.
- Symlin (Pramlintide acetate) [package insert]. Wilmington DE: AstraZeneca; 2015. Pharmaceuticals LP. http://www.azpicentral.com/symlin/pi_symlin.pdf#page=1. Accessed June 26, 2017.
- Gaziano JM, Cincotta AH, O’Connor CM, et al. Randomized clinical trial of quick-release bromocriptine among patients with type 2 diabetes on overall safety and cardiovascular outcomes. Diabetes Care 2010; 33:1503–1508.
- Cycloset [package insert]. Tiverton, RI: VeroScience LLC; 2016. http://www.veroscience.com/documents/CyclosetPackageInsertFeb062017.pdf. Accessed June 26, 2017.
- Hirsch IB, Bergenstal RM, Parkin CG, Wright Jr, E, Buse JB. A real-world approach to insulin therapy in primary care practice. Clinical Diabetes 2005; 23:78–86.
- Juvenile Diabetes Research Foundation Continuous Glucose Monitoring Study Group; Tamborlane WV, Beck RW, Bode BW, et al. Continuous glucose monitoring and intensive treatment of type 1 diabetes. N Engl J Med 2008; 359:1464–1476.
Medical Treatment of Diabetes Mellitus
Medical Treatment of Diabetes Mellitus
Dosing accuracy of direct oral anticoagulants in an academic medical center
Direct-acting oral anticoagulants (DOACs) have been introduced into clinical use for stroke prevention in patients with nonvalvular atrial fibrillation (NVAF), prevention of venous thrombosis after hip or knee surgery, and treatment of deep vein thrombosis (DVT) and pulmonary embolism (PE).1-7 Advantages of DOACs over warfarin are often stated as fixed dosing, minor drug and food interactions, wider therapeutic index, and no need for laboratory test monitoring.1,8 Yet, recommended DOAC dosages vary by renal function and therapeutic indications. Dosing recommendations for prevention of stroke in patients with NVAF are based on estimated creatinine clearance (dabigatran, rivaroxaban, edoxaban), age (apixaban), weight (apixaban, edoxaban), serum creatinine level (apixaban, edoxaban), and presence of cirrhosis by Child-Pugh class9,10 (apixaban, edoxaban).4-6,11,12 Dosing recommendations based on coadministration of strong CYP34A and P-glycoprotein inhibitors or inducers vary by DOAC. In addition, dabigatran cannot be crushed and must be stored in its original packaging, and rivaroxaban should be taken with food when the dose is over 10 mg.
We studied DOAC prescribing in adults admitted to a large academic medical center by comparing initial prescribed dosing with FDA-approved prescribing information. We hypothesized that the complexity of DOAC dosing may not be recognized by prescribers.
METHODS
Our study protocol was approved by the Committee on Human Research (Institutional Review Board) of the University of California San Francisco.
Data Collection
We used electronic medical records (EMRs) to identify adult inpatients who were prescribed a DOAC (apixaban, dabigatran, edoxaban, or rivaroxaban) at the University of California San Francisco Medical Center, a large academic hospital, between July 1, 2014 and June 30, 2015. Demographic and medical information related to therapeutic indications, contraindications, and indications for dose adjustments were collected and included diagnoses classified by International Classification of Diseases, Ninth Revision (ICD-9) and Tenth Revision (ICD-10) for venous thromboses; phlebitis or thrombophlebitis; PE or venous embolism; atrial arrhythmias; surgical procedures; cirrhosis and/or ascites or liver disease; coagulopathies; artificial heart valves or implanted devices; prior use of medications including parenteral anticoagulants; and laboratory data obtained before the first DOAC order (serum creatinine level, estimated glomerular filtration rate [eGFR] determined by Chronic Kidney Disease Epidemiology Collaboration,13 international normalized ratio, or, if available, activated partial thromboplastin time and bilirubin level). Creatinine clearance was calculated with the Cockcroft-Gault method14 using total body weight, per drug label recommendation. Child-Pugh class was calculated if cirrhosis was diagnosed.10 DOAC dose, frequency, dosing directions, and prescriber medical specialty were determined.
Accuracy of search results was confirmed by review of the first 200 patients’ records. Records were manually reviewed for encounters lacking ICD-9/10 codes and approved DOAC indications (30%) and encounters having multiple coded diagnostic indications (to identify the indication). ICD-9 codes for venous thrombosis were reviewed to differentiate acute from chronic events.
Data Analysis
The main outcome was concordance or discordance between the first DOAC prescribing order and the FDA-approved prescribing information at the time. Initial classification, performed by 2 independent reviewers (a pharmacist and a physician, or 2 pharmacists), was followed by adjudication and individual record review (by 2 independent reviewers) of all initial prescribing orders classified as discordant. A third reviewer adjudicated any disagreement. Records and notes were reviewed to identify stated or potential reasons for dosing variation and pre-admission prescriptions. Data are presented as means and standard deviations (SDs) and as raw numbers and percentages. Differences in patient characteristics by DOAC or therapeutic indication were determined by analysis of variance (ANOVA) with Bonferroni correction for post hoc comparisons. Dosing information was categorized as the same as recommended, lower than recommended, higher than recommended, or avoid drug use (drug–drug or drug–disease interaction), per FDA-approved prescribing information, and χ2 tests were used to determine whether variation in dosing occurred by individual DOAC, therapeutic indication, or prescriber specialty. Relationships between dosing variation and age or renal function were tested by ANOVA with Bonferroni correction for post hoc comparisons.
RESULTS


Patients with AF were older with lower creatinine clearance compared to patients with other diagnoses. Mean (SD) patient age was 72.1 (12.7) years for AF, 53.1 (10.9) years for chronic PE, 55.5 (14) years for acute PE, 56.4 (15.9) years for chronic DVT, 57.9 (18.4) years for acute DVT, and 61.4 (11.6) years for DVT prevention after hip or knee surgery (P < 0.0001 for all comparisons). Mean (SD) estimated creatinine clearance was 76.8 (43.5) mL/min for AF, 92.4 (44.4) mL/min for DVT prevention after hip or knee surgery, 111 (53) mL/min for chronic DVT, 118 (55) mL/min for acute DVT, 126 (60) mL/min for chronic PE, and 127 (54) mL/min for acute PE (P < 0.0001 for all comparisons). Differences between patient groups by therapeutic indication were not detected for weight, body mass index, or serum creatinine level.
The most frequent deviation from prescribing recommendations was omission of directions to administer rivaroxaban with food—93% (248/268) of orders—but not for DVT prevention after hip or knee surgery, for which the 10-mg dose is appropriately administered without food. Doses were the same as recommended for 82% of apixaban orders, 84% of rivaroxaban orders, and 93% of initial dabigatran orders (P < 0.05 for differences; Table 3). Dosages not concordant with FDA recommendations were prescribed in 44 (18.1%) of 243 apixaban orders, 41 (14.3%) of 286 rivaroxaban orders, and 7 (7.2%) of 89 initial dabigatran orders. Lower than recommended doses were more common than higher than recommended doses (Table 3, Figure 1): 15.2% versus 2.1% of apixaban orders, 9.4% versus 3.5% of rivaroxaban orders, and 4.2% versus 1.0% of initial dabigatran orders (P < 0.05). Failure to avoid drug use (for potential drug–drug or drug–disease interactions) was uncommon (1%-2%). There were more deviations from recommended doses for patients with AF or DVT prevention after hip or knee surgery than for patients with acute or chronic PE or acute DVT (Table 3). No significant differences were detected between prescribed and recommended doses by prescriber specialty.


For apixaban, patients who were prescribed lower than recommended doses were older than those prescribed recommended doses: mean (SD), 78.1 (12.2) years versus 71 (13.6) years (P = 0.003). Seventy-six percent of those prescribed lower than recommended doses were older than 75. Prescriptions for apixaban at lower than recommended doses were continuations of prior outpatient prescriptions in 20 of 37 cases (almost half), and in 12 cases (one-fourth) antiplatelet drugs were coprescribed (aspirin in 10 cases, clopidogrel in 1, prasugrel in 1). For rivaroxaban, older age was associated with both lower than recommended dosing (P = 0.003) and higher than recommended dosing (P < 0.001). Variations from prescribing recommendations were continuations of outpatient rivaroxaban doses in about two-thirds (26 of 41; 63.4 %) with 13 receiving antiplatelet drugs. For dabigatran, 6 of 7 orders not in agreement with recommendations were continuations of outpatient dosing.
The specific equation used to estimate renal function also had the potential to lead to dosing errors. Among the 41 rivaroxaban patients categorized as receiving doses discordant with recommendations, 8 would have had an inappropriate DOAC dose if eGFR were used instead of eCrCL as recommended. No relationships were detected for other patient variables/measures and dosing deviations from recommendations.
DISCUSSION
We examined initial hospital orders for DOACs in adults admitted to a single academic medical center during 2014-2015. Dabigatran, apixaban and rivaroxaban were prescribed for prevention of stroke in patients with atrial fibrillation/flutter (AF) in three quarters of the encounters similar to national patterns. (15) Prescribing departures from FDA-approved recommendations ranged from failure to prescribe rivaroxaban with food to failure to recognize drug-drug interactions in 1% to 2%. Unexpectedly, lower than recommended dosing was more common than higher than recommended dosing of the three DOACs.
Rivaroxaban bioavailability is dose dependent with the presence of food required to enhance absorption for doses over 10 mg that are used for prevention of stroke in patients with non-valvular AF or treatment of DVT or PE.5,16 Peak rivaroxaban concentrations are 75% higher and the total area under the concentration vs. time curve after dosing is 40% higher when rivaroxaban is administered with high fat high calorie meals compared to the fasting state.16 If rivaroxaban is not administered with food, drug concentrations and pharmacologic effects may be less than in clinical trials that specified co-administration with food.17-19 A small survey of outpatients receiving rivaroxaban found that 23% reported taking it without food.20 With electronic pharmacy systems in almost all hospitals and electronic prescriber order entry in most, automated addition of directions for rivaroxaban administration with food for doses over 10 mg to labels or dispensing instructions could easily correct this deviation from recommended practice.
Lower than recommended doses were prescribed in 9.4% of orders for rivaroxaban and 15.2% of orders for apixaban, with dose-deviations often appearing to be a continuation of outpatient doses. Patients 75 years or older were more likely to receive lower than recommended dosing of apixaban. Reductions in apixaban doses from 5 mg twice daily to 2.5 mg twice daily are recommended in patients with non-valvular AF with two of the following criteria: age ≥80 y, weight ≤60 kg, serum creatinine ≥1.5 mg/dL or co-administration of a strong PgP inhibitor to a patient without 2 of the 3 dose reduction criteria. Our study was not designed to determine reasons for under-dosing, but we speculate that clinicians may have considered patients aged 75-79 years to be similar to those 80 years of age or older, or, older and not as healthy as those enrolled in randomized trials.21-25 The median age of our patients with AF receiving apixaban was 75y (interquartile range of 16) vs 70y ( interquartile range 63-76) in the pivotal trial comparing warfarin to apixaban.21 Renal function was also lower with 37% having eCrCL below 50 mL/min compared to 17% in ARISTOTLE. (21). Twenty-six percent of our apixaban-treated AF patients qualified for the lower 2.5 mg twice daily compared to only 5% of ARISTOTLE participants,21 further suggesting differences between patients in our sample compared to randomized trial participants.
Concerns regarding bleeding or falls in older patients, may also have contributed to lower than recommended doses. Recent analyses of patients at risk for falls confirmed that increased risk of falling was associated with more bone fractures, bleeding and all-cause death but not stroke or systemic emboli, and with less severe bleeding with the DOAC edoxaban compared to warfarin.26 While a rationale for personalized or lower than recommended dosing of apixaban may exist in very old patients and those at risk of falls and bleeding, more data are needed to determine outcomes of lower than recommended doses of DOACs before such an approach can be endorsed. Monitoring of anticoagulant effect in patients who receive doses lower than those investigated in clinical trials could provide important information. The assays that measure DOAC effects are likely to be more available because of the use of reversal agents in the setting of bleeding with DOACs.27
We had anticipated higher than recommended dosing for rivaroxaban as recommendations are based on creatinine clearance while laboratories routinely report estimated glomerular filtration rate (eGFR) that can provide higher estimates of renal clearance and estimated DOAC doses in older and smaller individuals.28 Higher than recommended dosing was found in only 3.5% of our sample. In half, eGFR estimates were higher than creatinine clearance estimates. An international postmarketing registry of rivaroxaban use for the prevention of stroke in patients with NVAF, which included outpatients, found that 36% of those with creatinine clearances below 50 mL/min received a dose higher than recommended, and 15% received a dose lower than expected.29 A more recent outpatient registry report on patients with NVAF, in which apixaban, dabigatran, or rivaroxaban was administered, found that overall 9.4% received a dose lower than recommended, and 3.4% were overdosed, with a similar percentage (34%) of rivaroxaban patients with creatinine clearance of 15 to 50 mL/min receiving higher than recommended dosing.30 The lower rate of higher-than-recommended doses that we observed may have been related to the routine measurement of serum creatinine and attention to dosing adjustments for renal function in the inpatient setting compared to the outpatient setting. In addition, renal function data may not be available to outpatient pharmacies, limiting potential input on dosing recommendations. At least one cardiac society recommends monitoring of renal function in patients treated with DOACs, annually in patients with normal estimated creatinine clearance and more frequently (at intervals in months equal to the creatinine clearance divided by 10) in patients with abnormal creatinine clearance.11 A hospital encounter provides an opportunity to assess or reassess renal status to optimize DOAC dosing.
Dabigatran was the first DOAC introduced into use in the United States with the same dose recommended for prevention of stroke in patients with AF or venous thromboembolic disease with reductions for creatinine clearance below 30 mL/min or creatinine clearance between 30 and 50 mL/min and concomitant use of the potent P-glycoprotein inhibitor dronedarone or systemic ketoconazole. The relative simplicity of dosing may have been responsible for the lowest rate of prescribing outside of recommendations observed in this study, but the low dabigatran use limits analyses of contributing factors.
Failure to avoid drug use in combination with use of strong P-glycoprotein inducers or inhibitors was infrequent but should be preventable. Current prescribing recommendations refer to “strong” P-glycoprotein inhibitors and list different specific agents that interact with each DOAC without a standardized definition or classification. Standardized classifications or reference sources would be helpful.
Our primary goal in this study was to compare initial prescribed dosing of DOACs with FDA-approved prescribing directions. However, therapeutic indication data warrant discussion. In our sample, 7.5% of patients with AF had bioprosthetic valves or recent mitral valve repair or replacement. Using the NVAF definition found in the 2014 AHA/ACC/HRS (American Heart Association, American College of Cardiology, Heart Rhythm Society) AF guidelines1—“absence of rheumatic mitral valve disease, a prosthetic heart valve, or mitral valve repair”—these patients would not appear to be candidates for DOACs. However, arguments have been made that a bioprosthetic heart valve or native valve after valve repair does not have a risk profile for thromboembolism that differs from other forms of NVAF and would be equally responsive to DOAC therapy.31 Data are sparse, but retrospective subanalyses of limited numbers of patients with valvular disease (including bioprosthesis and mitral repair patients but excluding mechanical valve patients) enrolled in the pivotal DOAC studies support this conclusion.32 For the first months after biological valve replacement (including catheter-based valve replacement), recent European guidelines recommend vitamin K antagonists but also state, “NOACs probably deliver the same protection.”8 DOACs were also used for management of venous thromboembolic disease (both acute and chronic) in patients with active cancer. Our data predate the most recent American College of Chest Physician guidelines on treatment of venous thromboembolism in patients with cancer, which provide grade 2B recommendations for use of low-molecular-weight heparin (LMWH) over vitamin K antagonists and grade 2C recommendations for use of LMWH over dabigatran, rivaroxaban, apixaban, or edoxaban.33
Our study had several limitations. First, data were from a single US academic medical center, though similar rates of prescribing deviation from recommendations have been reported for rivaroxaban and dabigatran in NVAF patients in other countries.29,34 Second, therapeutic indications may have been misclassified because of errors, incomplete EMR data, or multiple indications. Third, we analyzed the first DOAC order and not dispensing information or subsequent corrections. Therefore, deviations from recommendations should not be interpreted as errors that reached patients. We evaluated dosing based on the measures used at the time of hospital admission, noting that, in a significant fraction of deviations from recommended doses, they represented continuations of outpatient doses when renal function or weight may have differed, and it is unknown whether patients were counseled to take rivaroxaban with food in the outpatient setting. Fourth, the number of patients with acute DVT was small, so firm conclusions cannot be drawn for this specific population. Fifth, our estimates of off-label dosing may have been underestimates, as data on cancer and cancer activity or cardiac valvular disease may not have been complete.
CONCLUSION
Healthcare professionals are prescribing DOACs in ways that differ from recommendations. These differences may reflect the older ages and reduced renal function of clinical populations relative to randomized clinical trial groups, but they could also potentially alter clinical efficacy. Our findings support the need to evaluate the appropriateness and dosing of DOACs at each encounter and to determine the outcomes of patients treated with lower than recommended doses of DOACs and the outcomes of DOAC-treated patients with bioprostheses or active malignancies.
Acknowledgment
The authors thank Tobias Schmelzinger for electronic data extraction and compilation and University of California San Francisco students Eduardo De La Torre Cruz (School of Pharmacy) and Carlos Mikell (School of Medicine) for assistance with data review.
Disclosure
Dr. Schwartz reports receiving personal fees from Bristol-Myers Squibb and Amgen and grants from Bristol-Myers Squibb and Pfizer, outside the submitted work. The other authors have nothing to report.
1. January CT, Wann LS, Alpert JS, et al. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: executive summary. A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol. 2014;64(21):2246-2280. PubMed
2. Saraf K, Morris PD, Garg P, Sheridan P, Storey R. Non–vitamin K antagonist oral anticoagulants (NOACs): clinical evidence and therapeutic considerations. Postgrad Med J. 2014;90(1067):520-528. PubMed
3. Yeh CH, Gross PL, Weitz JI. Evolving use of new oral anticoagulants for treatment of venous thromboembolism. Blood. 2014;124(7):1020-1028. PubMed
4. Pradaxa website. https://www.pradaxa.com. Accessed June 1, 2017.
5. Xarelto website. https://www.xarelto-us.com. Accessed June 1, 2017.
6. Eliquis website. http://www.eliquis.com. Accessed June 1, 2017.
7. Savaysa [prescribing information]. Tokyo, Japan: Daiichi Sankyo; 2015.
8. Kirchhof P, Benussi S, Kotecha D, et al. 2016 ESC guidelines for the management of atrial fibrillation developed in collaboration with EACTS. Eur Heart J. 2016;37(38):2893-2962. PubMed
9. Child C, Turcotte J. Surgery and portal hypertension. In: Child CG, ed. The Liver and Portal Hypertension. Philadelphia, PA: Saunders; 1964:50-64. PubMed
10. Pugh RN, Murray-Lyon IM, Dawson JL, Pietroni MC, Williams R. Transection of the oesophagus for bleeding oesophageal varices. Br J Surg. 1973;60(8):646-649. PubMed
11. Heidbuchel H, Verhamme P, Alings M, et al. Updated European Heart Rhythm Association practical guide on the use of non–vitamin K antagonist anticoagulants in patients with non-valvular atrial fibrillation. Europace. 2015;17(10):1467-1507. PubMed
12. Savaysa website. https://savaysahcp.com. Accessed June 1, 2017.
13. Levey AS, Stevens LA, Schmid CH, et al; CKD-EPI (Chronic Kidney Disease Epidemiology Collaboration). A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150(9):604-612. PubMed
14. Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16(1):31-41. PubMed
15. Rose AJ, Reisman JI, Allen AL, Miller DR. Potentially inappropriate prescribing of direct-acting oral anticoagulants in the Veterans Health Administration. Am J Pharm Benefits. 2016;4(4):e75-e80.
16. Stampfuss J, Kubitza D, Becka M, Mueck W. The effect of food on the absorption and pharmacokinetics of rivaroxaban. Int J Clin Pharmacol Ther. 2013;51(7):549-561. PubMed
17. Patel MR, Mahaffey KW, Garg J, et al; ROCKET AF Investigators. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med. 2011;365(10):883-891. PubMed
18. EINSTEIN Investigators, Bauersachs R, Berkowitz SD, et al. Oral rivaroxaban for symptomatic venous thromboembolism. N Engl J Med. 2010;363(26):2499-2510. PubMed
19. EINSTEIN-PE Investigators, Büller HR, Prins MH, et al. Oral rivaroxaban for the treatment of symptomatic pulmonary embolism. N Engl J Med. 2012;366(14):1287-1297. PubMed
20. Simon J, Hawes E, Deyo Z, Bryant-Shilliday B. Evaluation of prescribing and patient use of target-specific oral anticoagulants in the outpatient setting. J Clin Pharm Ther. 2015;40(5):525-530. PubMed
21. Granger CB, Alexander JH, McMurray JJ, et al; ARISTOTLE Committees and Investigators. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med. 2011;365(11):981-992. PubMed
22. Ruff CT, Giugliano RP, Braunwald E, et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: a meta-analysis of randomised trials. Lancet. 2014;383(9921):955-962. PubMed
23. van der Hulle T, Kooiman J, den Exter PL, Dekkers OM, Klok FA, Huisman MV. Effectiveness and safety of novel oral anticoagulants as compared with vitamin K antagonists in the treatment of acute symptomatic venous thromboembolism: a systematic review and meta-analysis. J Thromb Haemost. 2014;12(3):320-328. PubMed
24. Schuh T, Reichardt B, Finsterer J, Stöllberger C. Age-dependency of prescribing patterns of oral anticoagulant drugs in Austria during 2011–2014. J Thromb Thrombolysis. 2016;42(3):447-451. PubMed
25. Stöllberger C, Brooks R, Finsterer J, Pachofszky T. Use of direct-acting oral anticoagulants in nonagenarians: a call for more data. Drugs Aging. 2016;33(5):315-320. PubMed
26. Steffel J, Giugliano RP, Braunwald E, et al. Edoxaban versus warfarin in atrial fibrillation patients at risk of falling: ENGAGE AF-TIMI 48 analysis. J Am Coll Cardiol. 2016;68(11):1169-1178. PubMed
27. Ruff CT, Giugliano RP, Antman EM. Management of bleeding with non–vitamin K antagonist oral anticoagulants in the era of specific reversal agents. Circulation. 2016;134(3):248-261. PubMed
28. Schwartz JB. Potential impact of substituting estimated glomerular filtration rate for estimated creatinine clearance for dosing of direct oral anticoagulants. J Am Geriatr Soc. 2016;64(10):1996-2002. PubMed
29. Camm AJ, Amarenco P, Haas S, et al; XANTUS Investigators. XANTUS: a real-world, prospective, observational study of patients treated with rivaroxaban for stroke prevention in atrial fibrillation. Eur Heart J. 2016;37(14):1145-1153. PubMed
30. Steinberg BA, Shrader P, Thomas L, et al; ORBIT-AF Investigators and Patients. Off-label dosing of non–vitamin K antagonist oral anticoagulants and adverse outcomes: the ORBIT-AF II Registry. J Am Coll Cardiol. 2016;68(24):2597-2604. PubMed
31. Fauchier L, Philippart R, Clementy N, et al. How to define valvular atrial fibrillation? Arch Cardiovasc Dis. 2015;108(10):530-539. PubMed
32. Di Biase L. Use of direct oral anticoagulants in patients with atrial fibrillation and valvular heart lesions. J Am Heart Assoc. 2016;5(2). PubMed
33. Kearon C, Akl EA, Ornelas J, et al. Antithrombotic therapy for VTE disease:
CHEST guideline and expert panel report. Chest. 2016;149(2):315-352. PubMed
34. Larock AS, Mullier F, Sennesael AL, et al. Appropriateness of prescribing dabigatran
etexilate and rivaroxaban in patients with nonvalvular atrial fibrillation: a prospective
study. Ann Pharmacother. 2014;48(10):1258-1268. PubMed
Direct-acting oral anticoagulants (DOACs) have been introduced into clinical use for stroke prevention in patients with nonvalvular atrial fibrillation (NVAF), prevention of venous thrombosis after hip or knee surgery, and treatment of deep vein thrombosis (DVT) and pulmonary embolism (PE).1-7 Advantages of DOACs over warfarin are often stated as fixed dosing, minor drug and food interactions, wider therapeutic index, and no need for laboratory test monitoring.1,8 Yet, recommended DOAC dosages vary by renal function and therapeutic indications. Dosing recommendations for prevention of stroke in patients with NVAF are based on estimated creatinine clearance (dabigatran, rivaroxaban, edoxaban), age (apixaban), weight (apixaban, edoxaban), serum creatinine level (apixaban, edoxaban), and presence of cirrhosis by Child-Pugh class9,10 (apixaban, edoxaban).4-6,11,12 Dosing recommendations based on coadministration of strong CYP34A and P-glycoprotein inhibitors or inducers vary by DOAC. In addition, dabigatran cannot be crushed and must be stored in its original packaging, and rivaroxaban should be taken with food when the dose is over 10 mg.
We studied DOAC prescribing in adults admitted to a large academic medical center by comparing initial prescribed dosing with FDA-approved prescribing information. We hypothesized that the complexity of DOAC dosing may not be recognized by prescribers.
METHODS
Our study protocol was approved by the Committee on Human Research (Institutional Review Board) of the University of California San Francisco.
Data Collection
We used electronic medical records (EMRs) to identify adult inpatients who were prescribed a DOAC (apixaban, dabigatran, edoxaban, or rivaroxaban) at the University of California San Francisco Medical Center, a large academic hospital, between July 1, 2014 and June 30, 2015. Demographic and medical information related to therapeutic indications, contraindications, and indications for dose adjustments were collected and included diagnoses classified by International Classification of Diseases, Ninth Revision (ICD-9) and Tenth Revision (ICD-10) for venous thromboses; phlebitis or thrombophlebitis; PE or venous embolism; atrial arrhythmias; surgical procedures; cirrhosis and/or ascites or liver disease; coagulopathies; artificial heart valves or implanted devices; prior use of medications including parenteral anticoagulants; and laboratory data obtained before the first DOAC order (serum creatinine level, estimated glomerular filtration rate [eGFR] determined by Chronic Kidney Disease Epidemiology Collaboration,13 international normalized ratio, or, if available, activated partial thromboplastin time and bilirubin level). Creatinine clearance was calculated with the Cockcroft-Gault method14 using total body weight, per drug label recommendation. Child-Pugh class was calculated if cirrhosis was diagnosed.10 DOAC dose, frequency, dosing directions, and prescriber medical specialty were determined.
Accuracy of search results was confirmed by review of the first 200 patients’ records. Records were manually reviewed for encounters lacking ICD-9/10 codes and approved DOAC indications (30%) and encounters having multiple coded diagnostic indications (to identify the indication). ICD-9 codes for venous thrombosis were reviewed to differentiate acute from chronic events.
Data Analysis
The main outcome was concordance or discordance between the first DOAC prescribing order and the FDA-approved prescribing information at the time. Initial classification, performed by 2 independent reviewers (a pharmacist and a physician, or 2 pharmacists), was followed by adjudication and individual record review (by 2 independent reviewers) of all initial prescribing orders classified as discordant. A third reviewer adjudicated any disagreement. Records and notes were reviewed to identify stated or potential reasons for dosing variation and pre-admission prescriptions. Data are presented as means and standard deviations (SDs) and as raw numbers and percentages. Differences in patient characteristics by DOAC or therapeutic indication were determined by analysis of variance (ANOVA) with Bonferroni correction for post hoc comparisons. Dosing information was categorized as the same as recommended, lower than recommended, higher than recommended, or avoid drug use (drug–drug or drug–disease interaction), per FDA-approved prescribing information, and χ2 tests were used to determine whether variation in dosing occurred by individual DOAC, therapeutic indication, or prescriber specialty. Relationships between dosing variation and age or renal function were tested by ANOVA with Bonferroni correction for post hoc comparisons.
RESULTS
Patients with AF were older with lower creatinine clearance compared to patients with other diagnoses. Mean (SD) patient age was 72.1 (12.7) years for AF, 53.1 (10.9) years for chronic PE, 55.5 (14) years for acute PE, 56.4 (15.9) years for chronic DVT, 57.9 (18.4) years for acute DVT, and 61.4 (11.6) years for DVT prevention after hip or knee surgery (P < 0.0001 for all comparisons). Mean (SD) estimated creatinine clearance was 76.8 (43.5) mL/min for AF, 92.4 (44.4) mL/min for DVT prevention after hip or knee surgery, 111 (53) mL/min for chronic DVT, 118 (55) mL/min for acute DVT, 126 (60) mL/min for chronic PE, and 127 (54) mL/min for acute PE (P < 0.0001 for all comparisons). Differences between patient groups by therapeutic indication were not detected for weight, body mass index, or serum creatinine level.
The most frequent deviation from prescribing recommendations was omission of directions to administer rivaroxaban with food—93% (248/268) of orders—but not for DVT prevention after hip or knee surgery, for which the 10-mg dose is appropriately administered without food. Doses were the same as recommended for 82% of apixaban orders, 84% of rivaroxaban orders, and 93% of initial dabigatran orders (P < 0.05 for differences; Table 3). Dosages not concordant with FDA recommendations were prescribed in 44 (18.1%) of 243 apixaban orders, 41 (14.3%) of 286 rivaroxaban orders, and 7 (7.2%) of 89 initial dabigatran orders. Lower than recommended doses were more common than higher than recommended doses (Table 3, Figure 1): 15.2% versus 2.1% of apixaban orders, 9.4% versus 3.5% of rivaroxaban orders, and 4.2% versus 1.0% of initial dabigatran orders (P < 0.05). Failure to avoid drug use (for potential drug–drug or drug–disease interactions) was uncommon (1%-2%). There were more deviations from recommended doses for patients with AF or DVT prevention after hip or knee surgery than for patients with acute or chronic PE or acute DVT (Table 3). No significant differences were detected between prescribed and recommended doses by prescriber specialty.
For apixaban, patients who were prescribed lower than recommended doses were older than those prescribed recommended doses: mean (SD), 78.1 (12.2) years versus 71 (13.6) years (P = 0.003). Seventy-six percent of those prescribed lower than recommended doses were older than 75. Prescriptions for apixaban at lower than recommended doses were continuations of prior outpatient prescriptions in 20 of 37 cases (almost half), and in 12 cases (one-fourth) antiplatelet drugs were coprescribed (aspirin in 10 cases, clopidogrel in 1, prasugrel in 1). For rivaroxaban, older age was associated with both lower than recommended dosing (P = 0.003) and higher than recommended dosing (P < 0.001). Variations from prescribing recommendations were continuations of outpatient rivaroxaban doses in about two-thirds (26 of 41; 63.4 %) with 13 receiving antiplatelet drugs. For dabigatran, 6 of 7 orders not in agreement with recommendations were continuations of outpatient dosing.
The specific equation used to estimate renal function also had the potential to lead to dosing errors. Among the 41 rivaroxaban patients categorized as receiving doses discordant with recommendations, 8 would have had an inappropriate DOAC dose if eGFR were used instead of eCrCL as recommended. No relationships were detected for other patient variables/measures and dosing deviations from recommendations.
DISCUSSION
We examined initial hospital orders for DOACs in adults admitted to a single academic medical center during 2014-2015. Dabigatran, apixaban and rivaroxaban were prescribed for prevention of stroke in patients with atrial fibrillation/flutter (AF) in three quarters of the encounters similar to national patterns. (15) Prescribing departures from FDA-approved recommendations ranged from failure to prescribe rivaroxaban with food to failure to recognize drug-drug interactions in 1% to 2%. Unexpectedly, lower than recommended dosing was more common than higher than recommended dosing of the three DOACs.
Rivaroxaban bioavailability is dose dependent with the presence of food required to enhance absorption for doses over 10 mg that are used for prevention of stroke in patients with non-valvular AF or treatment of DVT or PE.5,16 Peak rivaroxaban concentrations are 75% higher and the total area under the concentration vs. time curve after dosing is 40% higher when rivaroxaban is administered with high fat high calorie meals compared to the fasting state.16 If rivaroxaban is not administered with food, drug concentrations and pharmacologic effects may be less than in clinical trials that specified co-administration with food.17-19 A small survey of outpatients receiving rivaroxaban found that 23% reported taking it without food.20 With electronic pharmacy systems in almost all hospitals and electronic prescriber order entry in most, automated addition of directions for rivaroxaban administration with food for doses over 10 mg to labels or dispensing instructions could easily correct this deviation from recommended practice.
Lower than recommended doses were prescribed in 9.4% of orders for rivaroxaban and 15.2% of orders for apixaban, with dose-deviations often appearing to be a continuation of outpatient doses. Patients 75 years or older were more likely to receive lower than recommended dosing of apixaban. Reductions in apixaban doses from 5 mg twice daily to 2.5 mg twice daily are recommended in patients with non-valvular AF with two of the following criteria: age ≥80 y, weight ≤60 kg, serum creatinine ≥1.5 mg/dL or co-administration of a strong PgP inhibitor to a patient without 2 of the 3 dose reduction criteria. Our study was not designed to determine reasons for under-dosing, but we speculate that clinicians may have considered patients aged 75-79 years to be similar to those 80 years of age or older, or, older and not as healthy as those enrolled in randomized trials.21-25 The median age of our patients with AF receiving apixaban was 75y (interquartile range of 16) vs 70y ( interquartile range 63-76) in the pivotal trial comparing warfarin to apixaban.21 Renal function was also lower with 37% having eCrCL below 50 mL/min compared to 17% in ARISTOTLE. (21). Twenty-six percent of our apixaban-treated AF patients qualified for the lower 2.5 mg twice daily compared to only 5% of ARISTOTLE participants,21 further suggesting differences between patients in our sample compared to randomized trial participants.
Concerns regarding bleeding or falls in older patients, may also have contributed to lower than recommended doses. Recent analyses of patients at risk for falls confirmed that increased risk of falling was associated with more bone fractures, bleeding and all-cause death but not stroke or systemic emboli, and with less severe bleeding with the DOAC edoxaban compared to warfarin.26 While a rationale for personalized or lower than recommended dosing of apixaban may exist in very old patients and those at risk of falls and bleeding, more data are needed to determine outcomes of lower than recommended doses of DOACs before such an approach can be endorsed. Monitoring of anticoagulant effect in patients who receive doses lower than those investigated in clinical trials could provide important information. The assays that measure DOAC effects are likely to be more available because of the use of reversal agents in the setting of bleeding with DOACs.27
We had anticipated higher than recommended dosing for rivaroxaban as recommendations are based on creatinine clearance while laboratories routinely report estimated glomerular filtration rate (eGFR) that can provide higher estimates of renal clearance and estimated DOAC doses in older and smaller individuals.28 Higher than recommended dosing was found in only 3.5% of our sample. In half, eGFR estimates were higher than creatinine clearance estimates. An international postmarketing registry of rivaroxaban use for the prevention of stroke in patients with NVAF, which included outpatients, found that 36% of those with creatinine clearances below 50 mL/min received a dose higher than recommended, and 15% received a dose lower than expected.29 A more recent outpatient registry report on patients with NVAF, in which apixaban, dabigatran, or rivaroxaban was administered, found that overall 9.4% received a dose lower than recommended, and 3.4% were overdosed, with a similar percentage (34%) of rivaroxaban patients with creatinine clearance of 15 to 50 mL/min receiving higher than recommended dosing.30 The lower rate of higher-than-recommended doses that we observed may have been related to the routine measurement of serum creatinine and attention to dosing adjustments for renal function in the inpatient setting compared to the outpatient setting. In addition, renal function data may not be available to outpatient pharmacies, limiting potential input on dosing recommendations. At least one cardiac society recommends monitoring of renal function in patients treated with DOACs, annually in patients with normal estimated creatinine clearance and more frequently (at intervals in months equal to the creatinine clearance divided by 10) in patients with abnormal creatinine clearance.11 A hospital encounter provides an opportunity to assess or reassess renal status to optimize DOAC dosing.
Dabigatran was the first DOAC introduced into use in the United States with the same dose recommended for prevention of stroke in patients with AF or venous thromboembolic disease with reductions for creatinine clearance below 30 mL/min or creatinine clearance between 30 and 50 mL/min and concomitant use of the potent P-glycoprotein inhibitor dronedarone or systemic ketoconazole. The relative simplicity of dosing may have been responsible for the lowest rate of prescribing outside of recommendations observed in this study, but the low dabigatran use limits analyses of contributing factors.
Failure to avoid drug use in combination with use of strong P-glycoprotein inducers or inhibitors was infrequent but should be preventable. Current prescribing recommendations refer to “strong” P-glycoprotein inhibitors and list different specific agents that interact with each DOAC without a standardized definition or classification. Standardized classifications or reference sources would be helpful.
Our primary goal in this study was to compare initial prescribed dosing of DOACs with FDA-approved prescribing directions. However, therapeutic indication data warrant discussion. In our sample, 7.5% of patients with AF had bioprosthetic valves or recent mitral valve repair or replacement. Using the NVAF definition found in the 2014 AHA/ACC/HRS (American Heart Association, American College of Cardiology, Heart Rhythm Society) AF guidelines1—“absence of rheumatic mitral valve disease, a prosthetic heart valve, or mitral valve repair”—these patients would not appear to be candidates for DOACs. However, arguments have been made that a bioprosthetic heart valve or native valve after valve repair does not have a risk profile for thromboembolism that differs from other forms of NVAF and would be equally responsive to DOAC therapy.31 Data are sparse, but retrospective subanalyses of limited numbers of patients with valvular disease (including bioprosthesis and mitral repair patients but excluding mechanical valve patients) enrolled in the pivotal DOAC studies support this conclusion.32 For the first months after biological valve replacement (including catheter-based valve replacement), recent European guidelines recommend vitamin K antagonists but also state, “NOACs probably deliver the same protection.”8 DOACs were also used for management of venous thromboembolic disease (both acute and chronic) in patients with active cancer. Our data predate the most recent American College of Chest Physician guidelines on treatment of venous thromboembolism in patients with cancer, which provide grade 2B recommendations for use of low-molecular-weight heparin (LMWH) over vitamin K antagonists and grade 2C recommendations for use of LMWH over dabigatran, rivaroxaban, apixaban, or edoxaban.33
Our study had several limitations. First, data were from a single US academic medical center, though similar rates of prescribing deviation from recommendations have been reported for rivaroxaban and dabigatran in NVAF patients in other countries.29,34 Second, therapeutic indications may have been misclassified because of errors, incomplete EMR data, or multiple indications. Third, we analyzed the first DOAC order and not dispensing information or subsequent corrections. Therefore, deviations from recommendations should not be interpreted as errors that reached patients. We evaluated dosing based on the measures used at the time of hospital admission, noting that, in a significant fraction of deviations from recommended doses, they represented continuations of outpatient doses when renal function or weight may have differed, and it is unknown whether patients were counseled to take rivaroxaban with food in the outpatient setting. Fourth, the number of patients with acute DVT was small, so firm conclusions cannot be drawn for this specific population. Fifth, our estimates of off-label dosing may have been underestimates, as data on cancer and cancer activity or cardiac valvular disease may not have been complete.
CONCLUSION
Healthcare professionals are prescribing DOACs in ways that differ from recommendations. These differences may reflect the older ages and reduced renal function of clinical populations relative to randomized clinical trial groups, but they could also potentially alter clinical efficacy. Our findings support the need to evaluate the appropriateness and dosing of DOACs at each encounter and to determine the outcomes of patients treated with lower than recommended doses of DOACs and the outcomes of DOAC-treated patients with bioprostheses or active malignancies.
Acknowledgment
The authors thank Tobias Schmelzinger for electronic data extraction and compilation and University of California San Francisco students Eduardo De La Torre Cruz (School of Pharmacy) and Carlos Mikell (School of Medicine) for assistance with data review.
Disclosure
Dr. Schwartz reports receiving personal fees from Bristol-Myers Squibb and Amgen and grants from Bristol-Myers Squibb and Pfizer, outside the submitted work. The other authors have nothing to report.
Direct-acting oral anticoagulants (DOACs) have been introduced into clinical use for stroke prevention in patients with nonvalvular atrial fibrillation (NVAF), prevention of venous thrombosis after hip or knee surgery, and treatment of deep vein thrombosis (DVT) and pulmonary embolism (PE).1-7 Advantages of DOACs over warfarin are often stated as fixed dosing, minor drug and food interactions, wider therapeutic index, and no need for laboratory test monitoring.1,8 Yet, recommended DOAC dosages vary by renal function and therapeutic indications. Dosing recommendations for prevention of stroke in patients with NVAF are based on estimated creatinine clearance (dabigatran, rivaroxaban, edoxaban), age (apixaban), weight (apixaban, edoxaban), serum creatinine level (apixaban, edoxaban), and presence of cirrhosis by Child-Pugh class9,10 (apixaban, edoxaban).4-6,11,12 Dosing recommendations based on coadministration of strong CYP34A and P-glycoprotein inhibitors or inducers vary by DOAC. In addition, dabigatran cannot be crushed and must be stored in its original packaging, and rivaroxaban should be taken with food when the dose is over 10 mg.
We studied DOAC prescribing in adults admitted to a large academic medical center by comparing initial prescribed dosing with FDA-approved prescribing information. We hypothesized that the complexity of DOAC dosing may not be recognized by prescribers.
METHODS
Our study protocol was approved by the Committee on Human Research (Institutional Review Board) of the University of California San Francisco.
Data Collection
We used electronic medical records (EMRs) to identify adult inpatients who were prescribed a DOAC (apixaban, dabigatran, edoxaban, or rivaroxaban) at the University of California San Francisco Medical Center, a large academic hospital, between July 1, 2014 and June 30, 2015. Demographic and medical information related to therapeutic indications, contraindications, and indications for dose adjustments were collected and included diagnoses classified by International Classification of Diseases, Ninth Revision (ICD-9) and Tenth Revision (ICD-10) for venous thromboses; phlebitis or thrombophlebitis; PE or venous embolism; atrial arrhythmias; surgical procedures; cirrhosis and/or ascites or liver disease; coagulopathies; artificial heart valves or implanted devices; prior use of medications including parenteral anticoagulants; and laboratory data obtained before the first DOAC order (serum creatinine level, estimated glomerular filtration rate [eGFR] determined by Chronic Kidney Disease Epidemiology Collaboration,13 international normalized ratio, or, if available, activated partial thromboplastin time and bilirubin level). Creatinine clearance was calculated with the Cockcroft-Gault method14 using total body weight, per drug label recommendation. Child-Pugh class was calculated if cirrhosis was diagnosed.10 DOAC dose, frequency, dosing directions, and prescriber medical specialty were determined.
Accuracy of search results was confirmed by review of the first 200 patients’ records. Records were manually reviewed for encounters lacking ICD-9/10 codes and approved DOAC indications (30%) and encounters having multiple coded diagnostic indications (to identify the indication). ICD-9 codes for venous thrombosis were reviewed to differentiate acute from chronic events.
Data Analysis
The main outcome was concordance or discordance between the first DOAC prescribing order and the FDA-approved prescribing information at the time. Initial classification, performed by 2 independent reviewers (a pharmacist and a physician, or 2 pharmacists), was followed by adjudication and individual record review (by 2 independent reviewers) of all initial prescribing orders classified as discordant. A third reviewer adjudicated any disagreement. Records and notes were reviewed to identify stated or potential reasons for dosing variation and pre-admission prescriptions. Data are presented as means and standard deviations (SDs) and as raw numbers and percentages. Differences in patient characteristics by DOAC or therapeutic indication were determined by analysis of variance (ANOVA) with Bonferroni correction for post hoc comparisons. Dosing information was categorized as the same as recommended, lower than recommended, higher than recommended, or avoid drug use (drug–drug or drug–disease interaction), per FDA-approved prescribing information, and χ2 tests were used to determine whether variation in dosing occurred by individual DOAC, therapeutic indication, or prescriber specialty. Relationships between dosing variation and age or renal function were tested by ANOVA with Bonferroni correction for post hoc comparisons.
RESULTS
Patients with AF were older with lower creatinine clearance compared to patients with other diagnoses. Mean (SD) patient age was 72.1 (12.7) years for AF, 53.1 (10.9) years for chronic PE, 55.5 (14) years for acute PE, 56.4 (15.9) years for chronic DVT, 57.9 (18.4) years for acute DVT, and 61.4 (11.6) years for DVT prevention after hip or knee surgery (P < 0.0001 for all comparisons). Mean (SD) estimated creatinine clearance was 76.8 (43.5) mL/min for AF, 92.4 (44.4) mL/min for DVT prevention after hip or knee surgery, 111 (53) mL/min for chronic DVT, 118 (55) mL/min for acute DVT, 126 (60) mL/min for chronic PE, and 127 (54) mL/min for acute PE (P < 0.0001 for all comparisons). Differences between patient groups by therapeutic indication were not detected for weight, body mass index, or serum creatinine level.
The most frequent deviation from prescribing recommendations was omission of directions to administer rivaroxaban with food—93% (248/268) of orders—but not for DVT prevention after hip or knee surgery, for which the 10-mg dose is appropriately administered without food. Doses were the same as recommended for 82% of apixaban orders, 84% of rivaroxaban orders, and 93% of initial dabigatran orders (P < 0.05 for differences; Table 3). Dosages not concordant with FDA recommendations were prescribed in 44 (18.1%) of 243 apixaban orders, 41 (14.3%) of 286 rivaroxaban orders, and 7 (7.2%) of 89 initial dabigatran orders. Lower than recommended doses were more common than higher than recommended doses (Table 3, Figure 1): 15.2% versus 2.1% of apixaban orders, 9.4% versus 3.5% of rivaroxaban orders, and 4.2% versus 1.0% of initial dabigatran orders (P < 0.05). Failure to avoid drug use (for potential drug–drug or drug–disease interactions) was uncommon (1%-2%). There were more deviations from recommended doses for patients with AF or DVT prevention after hip or knee surgery than for patients with acute or chronic PE or acute DVT (Table 3). No significant differences were detected between prescribed and recommended doses by prescriber specialty.
For apixaban, patients who were prescribed lower than recommended doses were older than those prescribed recommended doses: mean (SD), 78.1 (12.2) years versus 71 (13.6) years (P = 0.003). Seventy-six percent of those prescribed lower than recommended doses were older than 75. Prescriptions for apixaban at lower than recommended doses were continuations of prior outpatient prescriptions in 20 of 37 cases (almost half), and in 12 cases (one-fourth) antiplatelet drugs were coprescribed (aspirin in 10 cases, clopidogrel in 1, prasugrel in 1). For rivaroxaban, older age was associated with both lower than recommended dosing (P = 0.003) and higher than recommended dosing (P < 0.001). Variations from prescribing recommendations were continuations of outpatient rivaroxaban doses in about two-thirds (26 of 41; 63.4 %) with 13 receiving antiplatelet drugs. For dabigatran, 6 of 7 orders not in agreement with recommendations were continuations of outpatient dosing.
The specific equation used to estimate renal function also had the potential to lead to dosing errors. Among the 41 rivaroxaban patients categorized as receiving doses discordant with recommendations, 8 would have had an inappropriate DOAC dose if eGFR were used instead of eCrCL as recommended. No relationships were detected for other patient variables/measures and dosing deviations from recommendations.
DISCUSSION
We examined initial hospital orders for DOACs in adults admitted to a single academic medical center during 2014-2015. Dabigatran, apixaban and rivaroxaban were prescribed for prevention of stroke in patients with atrial fibrillation/flutter (AF) in three quarters of the encounters similar to national patterns. (15) Prescribing departures from FDA-approved recommendations ranged from failure to prescribe rivaroxaban with food to failure to recognize drug-drug interactions in 1% to 2%. Unexpectedly, lower than recommended dosing was more common than higher than recommended dosing of the three DOACs.
Rivaroxaban bioavailability is dose dependent with the presence of food required to enhance absorption for doses over 10 mg that are used for prevention of stroke in patients with non-valvular AF or treatment of DVT or PE.5,16 Peak rivaroxaban concentrations are 75% higher and the total area under the concentration vs. time curve after dosing is 40% higher when rivaroxaban is administered with high fat high calorie meals compared to the fasting state.16 If rivaroxaban is not administered with food, drug concentrations and pharmacologic effects may be less than in clinical trials that specified co-administration with food.17-19 A small survey of outpatients receiving rivaroxaban found that 23% reported taking it without food.20 With electronic pharmacy systems in almost all hospitals and electronic prescriber order entry in most, automated addition of directions for rivaroxaban administration with food for doses over 10 mg to labels or dispensing instructions could easily correct this deviation from recommended practice.
Lower than recommended doses were prescribed in 9.4% of orders for rivaroxaban and 15.2% of orders for apixaban, with dose-deviations often appearing to be a continuation of outpatient doses. Patients 75 years or older were more likely to receive lower than recommended dosing of apixaban. Reductions in apixaban doses from 5 mg twice daily to 2.5 mg twice daily are recommended in patients with non-valvular AF with two of the following criteria: age ≥80 y, weight ≤60 kg, serum creatinine ≥1.5 mg/dL or co-administration of a strong PgP inhibitor to a patient without 2 of the 3 dose reduction criteria. Our study was not designed to determine reasons for under-dosing, but we speculate that clinicians may have considered patients aged 75-79 years to be similar to those 80 years of age or older, or, older and not as healthy as those enrolled in randomized trials.21-25 The median age of our patients with AF receiving apixaban was 75y (interquartile range of 16) vs 70y ( interquartile range 63-76) in the pivotal trial comparing warfarin to apixaban.21 Renal function was also lower with 37% having eCrCL below 50 mL/min compared to 17% in ARISTOTLE. (21). Twenty-six percent of our apixaban-treated AF patients qualified for the lower 2.5 mg twice daily compared to only 5% of ARISTOTLE participants,21 further suggesting differences between patients in our sample compared to randomized trial participants.
Concerns regarding bleeding or falls in older patients, may also have contributed to lower than recommended doses. Recent analyses of patients at risk for falls confirmed that increased risk of falling was associated with more bone fractures, bleeding and all-cause death but not stroke or systemic emboli, and with less severe bleeding with the DOAC edoxaban compared to warfarin.26 While a rationale for personalized or lower than recommended dosing of apixaban may exist in very old patients and those at risk of falls and bleeding, more data are needed to determine outcomes of lower than recommended doses of DOACs before such an approach can be endorsed. Monitoring of anticoagulant effect in patients who receive doses lower than those investigated in clinical trials could provide important information. The assays that measure DOAC effects are likely to be more available because of the use of reversal agents in the setting of bleeding with DOACs.27
We had anticipated higher than recommended dosing for rivaroxaban as recommendations are based on creatinine clearance while laboratories routinely report estimated glomerular filtration rate (eGFR) that can provide higher estimates of renal clearance and estimated DOAC doses in older and smaller individuals.28 Higher than recommended dosing was found in only 3.5% of our sample. In half, eGFR estimates were higher than creatinine clearance estimates. An international postmarketing registry of rivaroxaban use for the prevention of stroke in patients with NVAF, which included outpatients, found that 36% of those with creatinine clearances below 50 mL/min received a dose higher than recommended, and 15% received a dose lower than expected.29 A more recent outpatient registry report on patients with NVAF, in which apixaban, dabigatran, or rivaroxaban was administered, found that overall 9.4% received a dose lower than recommended, and 3.4% were overdosed, with a similar percentage (34%) of rivaroxaban patients with creatinine clearance of 15 to 50 mL/min receiving higher than recommended dosing.30 The lower rate of higher-than-recommended doses that we observed may have been related to the routine measurement of serum creatinine and attention to dosing adjustments for renal function in the inpatient setting compared to the outpatient setting. In addition, renal function data may not be available to outpatient pharmacies, limiting potential input on dosing recommendations. At least one cardiac society recommends monitoring of renal function in patients treated with DOACs, annually in patients with normal estimated creatinine clearance and more frequently (at intervals in months equal to the creatinine clearance divided by 10) in patients with abnormal creatinine clearance.11 A hospital encounter provides an opportunity to assess or reassess renal status to optimize DOAC dosing.
Dabigatran was the first DOAC introduced into use in the United States with the same dose recommended for prevention of stroke in patients with AF or venous thromboembolic disease with reductions for creatinine clearance below 30 mL/min or creatinine clearance between 30 and 50 mL/min and concomitant use of the potent P-glycoprotein inhibitor dronedarone or systemic ketoconazole. The relative simplicity of dosing may have been responsible for the lowest rate of prescribing outside of recommendations observed in this study, but the low dabigatran use limits analyses of contributing factors.
Failure to avoid drug use in combination with use of strong P-glycoprotein inducers or inhibitors was infrequent but should be preventable. Current prescribing recommendations refer to “strong” P-glycoprotein inhibitors and list different specific agents that interact with each DOAC without a standardized definition or classification. Standardized classifications or reference sources would be helpful.
Our primary goal in this study was to compare initial prescribed dosing of DOACs with FDA-approved prescribing directions. However, therapeutic indication data warrant discussion. In our sample, 7.5% of patients with AF had bioprosthetic valves or recent mitral valve repair or replacement. Using the NVAF definition found in the 2014 AHA/ACC/HRS (American Heart Association, American College of Cardiology, Heart Rhythm Society) AF guidelines1—“absence of rheumatic mitral valve disease, a prosthetic heart valve, or mitral valve repair”—these patients would not appear to be candidates for DOACs. However, arguments have been made that a bioprosthetic heart valve or native valve after valve repair does not have a risk profile for thromboembolism that differs from other forms of NVAF and would be equally responsive to DOAC therapy.31 Data are sparse, but retrospective subanalyses of limited numbers of patients with valvular disease (including bioprosthesis and mitral repair patients but excluding mechanical valve patients) enrolled in the pivotal DOAC studies support this conclusion.32 For the first months after biological valve replacement (including catheter-based valve replacement), recent European guidelines recommend vitamin K antagonists but also state, “NOACs probably deliver the same protection.”8 DOACs were also used for management of venous thromboembolic disease (both acute and chronic) in patients with active cancer. Our data predate the most recent American College of Chest Physician guidelines on treatment of venous thromboembolism in patients with cancer, which provide grade 2B recommendations for use of low-molecular-weight heparin (LMWH) over vitamin K antagonists and grade 2C recommendations for use of LMWH over dabigatran, rivaroxaban, apixaban, or edoxaban.33
Our study had several limitations. First, data were from a single US academic medical center, though similar rates of prescribing deviation from recommendations have been reported for rivaroxaban and dabigatran in NVAF patients in other countries.29,34 Second, therapeutic indications may have been misclassified because of errors, incomplete EMR data, or multiple indications. Third, we analyzed the first DOAC order and not dispensing information or subsequent corrections. Therefore, deviations from recommendations should not be interpreted as errors that reached patients. We evaluated dosing based on the measures used at the time of hospital admission, noting that, in a significant fraction of deviations from recommended doses, they represented continuations of outpatient doses when renal function or weight may have differed, and it is unknown whether patients were counseled to take rivaroxaban with food in the outpatient setting. Fourth, the number of patients with acute DVT was small, so firm conclusions cannot be drawn for this specific population. Fifth, our estimates of off-label dosing may have been underestimates, as data on cancer and cancer activity or cardiac valvular disease may not have been complete.
CONCLUSION
Healthcare professionals are prescribing DOACs in ways that differ from recommendations. These differences may reflect the older ages and reduced renal function of clinical populations relative to randomized clinical trial groups, but they could also potentially alter clinical efficacy. Our findings support the need to evaluate the appropriateness and dosing of DOACs at each encounter and to determine the outcomes of patients treated with lower than recommended doses of DOACs and the outcomes of DOAC-treated patients with bioprostheses or active malignancies.
Acknowledgment
The authors thank Tobias Schmelzinger for electronic data extraction and compilation and University of California San Francisco students Eduardo De La Torre Cruz (School of Pharmacy) and Carlos Mikell (School of Medicine) for assistance with data review.
Disclosure
Dr. Schwartz reports receiving personal fees from Bristol-Myers Squibb and Amgen and grants from Bristol-Myers Squibb and Pfizer, outside the submitted work. The other authors have nothing to report.
1. January CT, Wann LS, Alpert JS, et al. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: executive summary. A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol. 2014;64(21):2246-2280. PubMed
2. Saraf K, Morris PD, Garg P, Sheridan P, Storey R. Non–vitamin K antagonist oral anticoagulants (NOACs): clinical evidence and therapeutic considerations. Postgrad Med J. 2014;90(1067):520-528. PubMed
3. Yeh CH, Gross PL, Weitz JI. Evolving use of new oral anticoagulants for treatment of venous thromboembolism. Blood. 2014;124(7):1020-1028. PubMed
4. Pradaxa website. https://www.pradaxa.com. Accessed June 1, 2017.
5. Xarelto website. https://www.xarelto-us.com. Accessed June 1, 2017.
6. Eliquis website. http://www.eliquis.com. Accessed June 1, 2017.
7. Savaysa [prescribing information]. Tokyo, Japan: Daiichi Sankyo; 2015.
8. Kirchhof P, Benussi S, Kotecha D, et al. 2016 ESC guidelines for the management of atrial fibrillation developed in collaboration with EACTS. Eur Heart J. 2016;37(38):2893-2962. PubMed
9. Child C, Turcotte J. Surgery and portal hypertension. In: Child CG, ed. The Liver and Portal Hypertension. Philadelphia, PA: Saunders; 1964:50-64. PubMed
10. Pugh RN, Murray-Lyon IM, Dawson JL, Pietroni MC, Williams R. Transection of the oesophagus for bleeding oesophageal varices. Br J Surg. 1973;60(8):646-649. PubMed
11. Heidbuchel H, Verhamme P, Alings M, et al. Updated European Heart Rhythm Association practical guide on the use of non–vitamin K antagonist anticoagulants in patients with non-valvular atrial fibrillation. Europace. 2015;17(10):1467-1507. PubMed
12. Savaysa website. https://savaysahcp.com. Accessed June 1, 2017.
13. Levey AS, Stevens LA, Schmid CH, et al; CKD-EPI (Chronic Kidney Disease Epidemiology Collaboration). A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150(9):604-612. PubMed
14. Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16(1):31-41. PubMed
15. Rose AJ, Reisman JI, Allen AL, Miller DR. Potentially inappropriate prescribing of direct-acting oral anticoagulants in the Veterans Health Administration. Am J Pharm Benefits. 2016;4(4):e75-e80.
16. Stampfuss J, Kubitza D, Becka M, Mueck W. The effect of food on the absorption and pharmacokinetics of rivaroxaban. Int J Clin Pharmacol Ther. 2013;51(7):549-561. PubMed
17. Patel MR, Mahaffey KW, Garg J, et al; ROCKET AF Investigators. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med. 2011;365(10):883-891. PubMed
18. EINSTEIN Investigators, Bauersachs R, Berkowitz SD, et al. Oral rivaroxaban for symptomatic venous thromboembolism. N Engl J Med. 2010;363(26):2499-2510. PubMed
19. EINSTEIN-PE Investigators, Büller HR, Prins MH, et al. Oral rivaroxaban for the treatment of symptomatic pulmonary embolism. N Engl J Med. 2012;366(14):1287-1297. PubMed
20. Simon J, Hawes E, Deyo Z, Bryant-Shilliday B. Evaluation of prescribing and patient use of target-specific oral anticoagulants in the outpatient setting. J Clin Pharm Ther. 2015;40(5):525-530. PubMed
21. Granger CB, Alexander JH, McMurray JJ, et al; ARISTOTLE Committees and Investigators. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med. 2011;365(11):981-992. PubMed
22. Ruff CT, Giugliano RP, Braunwald E, et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: a meta-analysis of randomised trials. Lancet. 2014;383(9921):955-962. PubMed
23. van der Hulle T, Kooiman J, den Exter PL, Dekkers OM, Klok FA, Huisman MV. Effectiveness and safety of novel oral anticoagulants as compared with vitamin K antagonists in the treatment of acute symptomatic venous thromboembolism: a systematic review and meta-analysis. J Thromb Haemost. 2014;12(3):320-328. PubMed
24. Schuh T, Reichardt B, Finsterer J, Stöllberger C. Age-dependency of prescribing patterns of oral anticoagulant drugs in Austria during 2011–2014. J Thromb Thrombolysis. 2016;42(3):447-451. PubMed
25. Stöllberger C, Brooks R, Finsterer J, Pachofszky T. Use of direct-acting oral anticoagulants in nonagenarians: a call for more data. Drugs Aging. 2016;33(5):315-320. PubMed
26. Steffel J, Giugliano RP, Braunwald E, et al. Edoxaban versus warfarin in atrial fibrillation patients at risk of falling: ENGAGE AF-TIMI 48 analysis. J Am Coll Cardiol. 2016;68(11):1169-1178. PubMed
27. Ruff CT, Giugliano RP, Antman EM. Management of bleeding with non–vitamin K antagonist oral anticoagulants in the era of specific reversal agents. Circulation. 2016;134(3):248-261. PubMed
28. Schwartz JB. Potential impact of substituting estimated glomerular filtration rate for estimated creatinine clearance for dosing of direct oral anticoagulants. J Am Geriatr Soc. 2016;64(10):1996-2002. PubMed
29. Camm AJ, Amarenco P, Haas S, et al; XANTUS Investigators. XANTUS: a real-world, prospective, observational study of patients treated with rivaroxaban for stroke prevention in atrial fibrillation. Eur Heart J. 2016;37(14):1145-1153. PubMed
30. Steinberg BA, Shrader P, Thomas L, et al; ORBIT-AF Investigators and Patients. Off-label dosing of non–vitamin K antagonist oral anticoagulants and adverse outcomes: the ORBIT-AF II Registry. J Am Coll Cardiol. 2016;68(24):2597-2604. PubMed
31. Fauchier L, Philippart R, Clementy N, et al. How to define valvular atrial fibrillation? Arch Cardiovasc Dis. 2015;108(10):530-539. PubMed
32. Di Biase L. Use of direct oral anticoagulants in patients with atrial fibrillation and valvular heart lesions. J Am Heart Assoc. 2016;5(2). PubMed
33. Kearon C, Akl EA, Ornelas J, et al. Antithrombotic therapy for VTE disease:
CHEST guideline and expert panel report. Chest. 2016;149(2):315-352. PubMed
34. Larock AS, Mullier F, Sennesael AL, et al. Appropriateness of prescribing dabigatran
etexilate and rivaroxaban in patients with nonvalvular atrial fibrillation: a prospective
study. Ann Pharmacother. 2014;48(10):1258-1268. PubMed
1. January CT, Wann LS, Alpert JS, et al. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: executive summary. A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol. 2014;64(21):2246-2280. PubMed
2. Saraf K, Morris PD, Garg P, Sheridan P, Storey R. Non–vitamin K antagonist oral anticoagulants (NOACs): clinical evidence and therapeutic considerations. Postgrad Med J. 2014;90(1067):520-528. PubMed
3. Yeh CH, Gross PL, Weitz JI. Evolving use of new oral anticoagulants for treatment of venous thromboembolism. Blood. 2014;124(7):1020-1028. PubMed
4. Pradaxa website. https://www.pradaxa.com. Accessed June 1, 2017.
5. Xarelto website. https://www.xarelto-us.com. Accessed June 1, 2017.
6. Eliquis website. http://www.eliquis.com. Accessed June 1, 2017.
7. Savaysa [prescribing information]. Tokyo, Japan: Daiichi Sankyo; 2015.
8. Kirchhof P, Benussi S, Kotecha D, et al. 2016 ESC guidelines for the management of atrial fibrillation developed in collaboration with EACTS. Eur Heart J. 2016;37(38):2893-2962. PubMed
9. Child C, Turcotte J. Surgery and portal hypertension. In: Child CG, ed. The Liver and Portal Hypertension. Philadelphia, PA: Saunders; 1964:50-64. PubMed
10. Pugh RN, Murray-Lyon IM, Dawson JL, Pietroni MC, Williams R. Transection of the oesophagus for bleeding oesophageal varices. Br J Surg. 1973;60(8):646-649. PubMed
11. Heidbuchel H, Verhamme P, Alings M, et al. Updated European Heart Rhythm Association practical guide on the use of non–vitamin K antagonist anticoagulants in patients with non-valvular atrial fibrillation. Europace. 2015;17(10):1467-1507. PubMed
12. Savaysa website. https://savaysahcp.com. Accessed June 1, 2017.
13. Levey AS, Stevens LA, Schmid CH, et al; CKD-EPI (Chronic Kidney Disease Epidemiology Collaboration). A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150(9):604-612. PubMed
14. Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16(1):31-41. PubMed
15. Rose AJ, Reisman JI, Allen AL, Miller DR. Potentially inappropriate prescribing of direct-acting oral anticoagulants in the Veterans Health Administration. Am J Pharm Benefits. 2016;4(4):e75-e80.
16. Stampfuss J, Kubitza D, Becka M, Mueck W. The effect of food on the absorption and pharmacokinetics of rivaroxaban. Int J Clin Pharmacol Ther. 2013;51(7):549-561. PubMed
17. Patel MR, Mahaffey KW, Garg J, et al; ROCKET AF Investigators. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med. 2011;365(10):883-891. PubMed
18. EINSTEIN Investigators, Bauersachs R, Berkowitz SD, et al. Oral rivaroxaban for symptomatic venous thromboembolism. N Engl J Med. 2010;363(26):2499-2510. PubMed
19. EINSTEIN-PE Investigators, Büller HR, Prins MH, et al. Oral rivaroxaban for the treatment of symptomatic pulmonary embolism. N Engl J Med. 2012;366(14):1287-1297. PubMed
20. Simon J, Hawes E, Deyo Z, Bryant-Shilliday B. Evaluation of prescribing and patient use of target-specific oral anticoagulants in the outpatient setting. J Clin Pharm Ther. 2015;40(5):525-530. PubMed
21. Granger CB, Alexander JH, McMurray JJ, et al; ARISTOTLE Committees and Investigators. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med. 2011;365(11):981-992. PubMed
22. Ruff CT, Giugliano RP, Braunwald E, et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: a meta-analysis of randomised trials. Lancet. 2014;383(9921):955-962. PubMed
23. van der Hulle T, Kooiman J, den Exter PL, Dekkers OM, Klok FA, Huisman MV. Effectiveness and safety of novel oral anticoagulants as compared with vitamin K antagonists in the treatment of acute symptomatic venous thromboembolism: a systematic review and meta-analysis. J Thromb Haemost. 2014;12(3):320-328. PubMed
24. Schuh T, Reichardt B, Finsterer J, Stöllberger C. Age-dependency of prescribing patterns of oral anticoagulant drugs in Austria during 2011–2014. J Thromb Thrombolysis. 2016;42(3):447-451. PubMed
25. Stöllberger C, Brooks R, Finsterer J, Pachofszky T. Use of direct-acting oral anticoagulants in nonagenarians: a call for more data. Drugs Aging. 2016;33(5):315-320. PubMed
26. Steffel J, Giugliano RP, Braunwald E, et al. Edoxaban versus warfarin in atrial fibrillation patients at risk of falling: ENGAGE AF-TIMI 48 analysis. J Am Coll Cardiol. 2016;68(11):1169-1178. PubMed
27. Ruff CT, Giugliano RP, Antman EM. Management of bleeding with non–vitamin K antagonist oral anticoagulants in the era of specific reversal agents. Circulation. 2016;134(3):248-261. PubMed
28. Schwartz JB. Potential impact of substituting estimated glomerular filtration rate for estimated creatinine clearance for dosing of direct oral anticoagulants. J Am Geriatr Soc. 2016;64(10):1996-2002. PubMed
29. Camm AJ, Amarenco P, Haas S, et al; XANTUS Investigators. XANTUS: a real-world, prospective, observational study of patients treated with rivaroxaban for stroke prevention in atrial fibrillation. Eur Heart J. 2016;37(14):1145-1153. PubMed
30. Steinberg BA, Shrader P, Thomas L, et al; ORBIT-AF Investigators and Patients. Off-label dosing of non–vitamin K antagonist oral anticoagulants and adverse outcomes: the ORBIT-AF II Registry. J Am Coll Cardiol. 2016;68(24):2597-2604. PubMed
31. Fauchier L, Philippart R, Clementy N, et al. How to define valvular atrial fibrillation? Arch Cardiovasc Dis. 2015;108(10):530-539. PubMed
32. Di Biase L. Use of direct oral anticoagulants in patients with atrial fibrillation and valvular heart lesions. J Am Heart Assoc. 2016;5(2). PubMed
33. Kearon C, Akl EA, Ornelas J, et al. Antithrombotic therapy for VTE disease:
CHEST guideline and expert panel report. Chest. 2016;149(2):315-352. PubMed
34. Larock AS, Mullier F, Sennesael AL, et al. Appropriateness of prescribing dabigatran
etexilate and rivaroxaban in patients with nonvalvular atrial fibrillation: a prospective
study. Ann Pharmacother. 2014;48(10):1258-1268. PubMed
© 2017 Society of Hospital Medicine
July 2017 Digital Edition
Click here to access the July 2017 Digital Edition.
Table of Contents
- Nonpathologic Postdeployment Transition Symptoms in Combat National Guard Members and Reservists
- Sarcopenia and the New ICD-10-CM Code: Screening, Staging, and Diagnosis Considerations
- Cerebral Venous Thrombosis
- Peer Support for Whistleblowers
- Symptoms Mimicking Those of Hypokalemic Periodic Paralysis Induced by Soluble Barium Poisoning
- Implementation of a Patient Medication Disposal Program at a VA Medical Center
- The VA Is in Critical Condition, but What Is the Prognosis?
- Leonard Wood: Advocate of Military Preparedness

Click here to access the July 2017 Digital Edition.
Table of Contents
- Nonpathologic Postdeployment Transition Symptoms in Combat National Guard Members and Reservists
- Sarcopenia and the New ICD-10-CM Code: Screening, Staging, and Diagnosis Considerations
- Cerebral Venous Thrombosis
- Peer Support for Whistleblowers
- Symptoms Mimicking Those of Hypokalemic Periodic Paralysis Induced by Soluble Barium Poisoning
- Implementation of a Patient Medication Disposal Program at a VA Medical Center
- The VA Is in Critical Condition, but What Is the Prognosis?
- Leonard Wood: Advocate of Military Preparedness
Click here to access the July 2017 Digital Edition.
Table of Contents
- Nonpathologic Postdeployment Transition Symptoms in Combat National Guard Members and Reservists
- Sarcopenia and the New ICD-10-CM Code: Screening, Staging, and Diagnosis Considerations
- Cerebral Venous Thrombosis
- Peer Support for Whistleblowers
- Symptoms Mimicking Those of Hypokalemic Periodic Paralysis Induced by Soluble Barium Poisoning
- Implementation of a Patient Medication Disposal Program at a VA Medical Center
- The VA Is in Critical Condition, but What Is the Prognosis?
- Leonard Wood: Advocate of Military Preparedness

Federal Health Care Data Trends 2017


Dozing off: Examining excessive daytime sleepiness in psychiatric patients
Excessive daytime sleepiness (EDS) is “the inability to maintain wakefulness and alertness during the major waking periods of the day, with sleep occurring unintentionally or at inappropriate times, almost daily for at least 3 months,” according to the American Academy of Sleep Medicine.1 EDS is common, with a prevalence up to 25% to 30% in the general population.1-4 The prevalence rate varies in different studies, primarily because of inconsistent definitions of EDS, and therefore differences in diagnosis and assessment.1,2,4 In a study of 300 psychiatric outpatients, 34% had EDS.3 However, studies and evidence reviewing EDS in psychiatric patients are limited.
The causes of EDS are many and varied,1,8 including medical and psychiatric etiologies. A thorough history, screening at-risk patients, and timely sleep center referral are vital to detect and appropriately manage the cause of EDS.5
This article reviews the literature on EDS, with a focus on the risks of untreated EDS, common etiologies of the condition, as well as a brief description of screening and treatment strategies.
EDS vs fatigue
Many patients describe EDS as “fatigue”1; however, a patient’s report of fatigue could be mistaken for EDS.4 Although there is overlap, it is important for physicians to distinguish between these 2 entities for accurate identification and treatment.1,4
Risk of inadequate screening
A study of 117 patients with symptomatic coronary artery disease showed that EDS is associated with significantly greater incidence of cardiovascular adverse events at 16-month follow up.2 This study had limitations such as small sample size; therefore, more studies are needed. Because of these risks, timely and accurate diagnosis not only improves the patient’s quality of life and reduces polypharmacy but also can be life-saving.
Common causes of EDS in psychiatric patients
Because of the high prevalence and severity of impairments caused by EDS, it is essential for psychiatrists to be informed about causes of EDS and thoroughly assess for the potential underlying etiology before concluding that the sleep problem is a manifestation of the psychiatric disorder and prescribing psychotropic medication for it.
Some common causes of EDS in psychiatric patients include:
Sleep-disordered breathing.8 Obstructive sleep apnea (OSA) is often underdiagnosed,6,7 and considering how common it is,6 psychiatrists likely will see many patients with OSA in their practice.5 OSA has a higher prevalence among patients with psychiatric disorders such as depression6,9 and schizophrenia. Additionally, there is evidence suggesting that patients with OSA are more likely to suffer from depression and EDS than healthy controls6,9,10; some of the proposed mechanisms are sleep fragmentation and hypoxemia.6,9-11 OSA is the most common form of sleep-disordered breathing and is a common cause of EDS.1,2,12 Also, undiagnosed and untreated OSA in patients with depression could cause refractoriness to pharmacological treatment of depression.6,9,10
When unrecognized and untreated, OSA can be life-threatening. Despite this, OSA is not regularly screened for in clinical psychiatric practice.6,10 Therefore, it is imperative that psychiatrists be well-acquainted with measures to identify at-risk patients and refer to a sleep specialist when appropriate.
OSA is accompanied by irritability, cognitive difficulties, and poor sleep, creating an overlap with symptoms of depressive disorders.6,10 Use of sedative hypnotic medications, such as benzodiazepines, which further reduces muscle tone in the airway and suppresses respiratory effort, can worsen OSA symptoms5,6,10 and pose cerebrovascular, cardiovascular, and potentially life-threatening risks, and therefore is not indicated in this population.9,13
Obesity is a risk factor for OSA.6 Patients with mood disorders or schizophrenia or other psychotic disorders are at higher risk of obesity because of psychotropic-induced weight gain, stress-induced mechanisms, and/or lower levels of self-care. When these patients have unrecognized or untreated OSA and are prescribed sedative medications at night or stimulant medications during the day, they could be at increased cardiac or respiratory risks without resolving their underlying condition. A diligent psychiatrist can dramatically reduce the risks by referring a patient for nocturnal polysomnography,1 helping the patient implement lifestyle modifications (eg, exercise, weight loss, and healthy nutrition), prescribing judiciously, and monitoring closely for such risks. An accurate diagnosis of and treatment for OSA can improve sleep6 dramatically and help depressive symptoms through better sleep, more daytime energy and concentration, and adequate oxygenation of the brain while sleeping.
Psychiatrists can screen for OSA using the STOP-Bang (Snoring, Tired, Observed apnea, Pressure, Body mass index, Age, Neck circumference, Gender) Questionnaire, which is a quick, 8-item screening scale that helps to categorize OSA risk as mild, moderate, or severe.12 Hypertension, snoring, and/or gasping for breath (“observed apnea”)—a history which often is provided by spouses or significant others—daytime dozing and/or tiredness, having a large neck circumference or volume, body mass index, male sex, and age are items on the STOP-Bang Questionnaire and also are features that should raise high clinical suspicion of OSA.12 Referral for nocturnal polysomnography in at-risk patients should be the next step1,5 in any sleep-related breathing disorder.
Treatment for OSA involves continuous positive airway pressure (CPAP) therapy, which has been shown to relieve OSA and decrease related EDS.5,6 Other treatment modalities, such as oral appliances and surgery, may be used5 in some cases, but more studies are needed for conclusive results.
Several studies have shown improved depression, mood, and cognition after administering treatment such as CPAP6,9,14 in patients with OSA and depression. Considering the significant risks of cardiovascular,8 cerebrovascular,8 and overall morbidity and mortality associated with untreated OSA,12 it is important to routinely screen for sleep-disordered breathing in patients with depression9 or other psychiatric disorders and refer for specialized sleep evaluation and treatment, when indicated.
Medications. EDS can result from some prescription and over-the-counter medications.1,2,5,7 Sedating antidepressants, antihistamines, antipsychotics, anticonvulsants,1,8 and beta blockers2 could cause sedation, which can persist during daytime, although a few studies did not find an association between antipsychotic use and EDS.3 Benzodiazepines and other sedative-hypnotics,1,7 especially long-acting agents or higher dosages,5 can lead to EDS and decreased alertness. Non-psychotropics, such as opioid pain medications,1,7 antitussives, and skeletal muscle relaxants, also can contribute to or cause daytime sedation.7 When using these agents, psychiatrists should monitor and routinely assess patients while aiming for the lowest effective dosage when feasible.
This strategy creates a framework for psychiatrists to routinely educate patients about these commonly encountered side effects, reduce polypharmacy when possible, and help patients effectively manage or prevent these adverse effects.
Depression.1 Some studies found >45% patients with depression had EDS.3,13,15 Besides an association between depression and EDS,13,16 Chellappa and Araújo13 also found a significant association between EDS and suicidal ideation. The causes of EDS in patients with depression may be varied, ranging from restless legs syndrome, residual depressive symptoms,15 to OSA. Depression is often comorbid with OSA,6 with up to 20% of patients with depression suffering from OSA,10 creating higher risk for EDS. Depressive disorders are routinely assessed during an evaluation of OSA at sleep centers, but OSA often is not screened in psychiatric practice.10
There is a strong need for regular screening for OSA in patients with depression, particularly because most studies show a link between the 2 conditions.10 Both depression and OSA have some common risk factors, such as obesity, hypertension, and metabolic syndrome.10 Patients with these conditions are at greater risk for OSA, and therefore a psychiatrist should proactively screen and refer such patients for nocturnal polysomnography when they suspect OSA. Patients with OSA and depression often present to the psychiatrist with depressive symptoms that appear to be resistant to pharmacological treatment,10 therefore underscoring the importance of screening and ruling out OSA in patients with depression.
Circadian rhythm disorders, restless legs syndrome, alcohol and other substance use, and use of prescription sedative-hypnotics are more common in patients with depression; therefore, this population is at high risk for EDS.
Circadian rhythm disorders and insufficient sleep syndrome. Insufficient sleep syndrome1,2,8 frequently causes EDS and occurs more commonly in busy people who try to get by with less sleep.8 Over time, the effect of sleep loss is cumulative and can be accompanied by mood symptoms, such as irritability, fatigue, and problems with concentration.8 Shift workers1,8 commonly experience insufficient sleep as well as circadian rhythm disorders and EDS. Modafinil is FDA-approved for EDS in shift work sleep disorder.
Geriatric patients may experience advanced sleep phase syndrome involving early awakenings.8 Adolescents, on the other hand, often suffer from delayed sleep phase syndrome, which is a type of circadian rhythm disorder, related to increasing academic and social pressures, natural pubertal shift to later sleep onset, pervading technology use, and often nebulous bedtime routines. This can be a cause of sleep persisting into daytime.8 Taking a careful history and a sleep diary may be useful because this disorder might be confused for insomnia. Treatment involves gradual shifting of the time of sleep onset through bright light exposure and other modalities.8
Adolescents might not be forthcoming about the severity of their sleep problems; therefore, psychiatrists should screen proactively through clinical interviews of patients and parents and consider this possibility when encountering an adolescent with recent-onset attention or cognitive difficulties.
Treatment for circadian rhythm disorders usually includes planned or prescribed sleep scheduling, timed light exposure,8 and occasional use of melatonin or other sedative agents.17
Hypersomnia of central origin, which includes narcolepsy, idiopathic hypersomnia, and recurrent hypersomnia, can present with EDS.1,18,19 Narcolepsy is a rare, debilitating sleep disorder that manifests as EDS or sleep attacks, with or without cataplexy, and sleep paralysis.5,8,18,19 The Multiple Sleep Latency Test and polysomnography are used for diagnosis.1,5 Shortened REM latency is a classic finding often noted on polysomnography. Treatment involves pharmacologic and behavioral strategies and education.5,8 Modafinil is FDA-approved for EDS associated with narcolepsy. Stimulant medications have been used for narcolepsy in the past; further studies are needed to establish benefit–risk ratio of use in this population.18
Kleine-Levin syndrome is a form of recurrent hypersomnia, a less common sleep disorder, characterized by episodes of excessive sleepiness accompanied by hyperphagia and hypersexuality.5,18,19
Other medical conditions,1 such as the rare familial fatal insomnia, neurological conditions1 such as encephalitis,8 epilepsy,8 Alzheimer’s disease or other types of dementia,8 Parkinson’s disease,1 or multiple sclerosis,1,18 can cause excessive daytime fatigue by causing secondary insomnia or hypersomnia.
Treating the underlying disorder is an important first step in these cases. In addition, coordinating with neurologists or other specialists involved in caring for patients with these conditions is important. Regularly reviewing and simplifying the often complex medication regimen, when possible, can go a long way in mitigating EDS in this population.
Other disorders affecting sleep. Restless legs syndrome and periodic limb movement disorder are other causes of EDS.3 Treatment involves lifestyle modifications, iron supplementation in certain patients, and use of dopaminergic agents such as ropinirole, pramipexole, and other medications, depending on severity of the condition, comorbidities, and other factors.20
Alcohol or substance use. Substance use or withdrawal can be associated with sleep disorders, such as hypersomnia,19 insomnia,19 and related EDS.5 For example, alcohol use disorder affects REM sleep, and can cause EDS. Secondary central apnea can be the result of long-standing opioid use19 and can present like EDS.
Insomnia. Primary insomnia rarely causes EDS.5 Insomnia due to a medical or psychiatric condition may be an indirect cause of EDS by causing sleep deprivation.
Steps for timely and accurate diagnosis
Utilize the following steps for facilitating timely diagnosis and treatment of EDS:
Thorough history. Patients often describe “tiredness” instead of sleepiness.8 Therefore, the astute psychiatrist should explore further when patients are presenting with this concern, especially by asking more specific questions such as the tendency to doze off during daytime.8
Family members can be vital sources for obtaining a complete history,5 especially because patients might deny,8 minimize, or not be fully aware1 of the extent of their symptoms. Asking family members about patient’s snoring, irregular breathing, or gasping at night can be particularly valuable.5 Obtaining a family history of sleep disorders can be particularly important, especially in conditions such as OSA and narcolepsy.
Asking about any history of safety issues,8 including sleepiness during driving, cooking, or other activities, is also important.
Use of scales and other screening measures. Psychiatrists can use initial screening measures in the office setting. Epworth Sleepiness Scale15,21 is a validated,2 short, self-administered measure to assess the level of daytime sleepiness; however, it has some limitations such as not being able to measure changes in sleepiness from hour to hour or day to day. Because of its limitations, the Epworth Sleepiness Scale should not be used by itself as a diagnostic tool.3 It has been commonly used for detecting OSA2 and narcolepsy. The Stanford Sleepiness Scale is a self-rating scale that measures the subjective degree of sleepiness and alertness; it has limitations as well, such as having little correlation with chronic sleep loss.8 Other tools such as visual analogue scales also could be helpful.8 For more specialized testing, such as Multiple Sleep Latency Test or polysomnography, referral to a sleep specialist is ideal.8
Education. The assessment is an opportunity for the psychiatrist to educate patients about sleep hygiene, the importance of regular bedtimes, and getting adequate sleep to avoid accumulating a sleep deficit.
Urgent referral of at-risk populations. Prompt or urgent referral of at-risk populations, such as geriatric patients or those with a history of dozing off during driving, is invaluable in preventing morbidity and mortality from untreated sleep disorders.
Patients with severe daytime sleepiness should be advised to not drive or operate heavy machinery until this condition is adequately controlled.18
Bottom Line
1. Chervin RD. Approach to the patient with excessive daytime sleepiness. http://www.uptodate.com/contents/approach-to-the-patient-with-excessive-daytime-sleepiness. Updated January 2016. Accessed June 5, 2017.
2. Lee CH, Ng WY, Hau W, et al. Excessive daytime sleepiness is associated with longer culprit lesion and adverse outcomes in patients with coronary artery disease. J Clin Sleep Med. 2013;9(12):1267-1272.
3. Hawley CJ, Gale TM, Sivakumaran T, et al. Excessive daytime sleepiness in psychiatric disorders: prevalence, correlates and clinical significance. Psychiatry Res. 2010;175(1-2):138-141.
4. Pigeon WR, Sateia MJ, Ferguson RJ. Distinguishing between excessive daytime sleepiness and fatigue: toward improved detection and treatment. J Psychosom Res. 2003;54(1):61-69.
5. Krahn LE. Excessive daytime sleepiness: diagnosing the causes. Current Psychiatry. 2002;1(1):49-57.
6. Ejaz SM, Khawaja IS, Bhatia S, et al. Obstructive sleep apnea and depression: a review. Innov Clin Neurosci. 2011;8(8):17-25.
7. Pagel JF. Excessive daytime sleepiness. Am Fam Physician. 2009;79(5):391-396.
8. Guilleminault C, Brooks SN. Excessive daytime sleepiness: a challenge for the practising neurologist. Brain. 2001;124(pt 8):1482-1491.
9. Cheng P, Casement M, Chen CF, et al. Sleep disordered breathing in major depressive disorder. J Sleep Res. 2013;22(4):459-462.
10. Schröder CM, O’Hara R. Depression and obstructive sleep apnea (OSA). Ann Gen Psychiatry. 2005;4:13.
11. Bardwell WA, Berry CC, Ancoli-Israel S, et al. Psychological correlates of sleep apnea. J Psychosom Res. 1999;47(6):583-596.
12. Chung F, Abdullah HR, Liao P. STOP-Bang Questionnaire: a practical approach to screen for obstructive sleep apnea. Chest. 2016;149(3):631-638.
13. Chellappa SL, Araújo JF. Excessive daytime sleepiness in patients with depressive disorder. Rev Bras Psiquiatr. 2006;28(2):126-129.
14. Habukawa M, Uchimura N, Kakuma T, et al. Effect of CPAP treatment on residual depressive symptoms in patients with major depression and coexisting sleep apnea: contribution of daytime sleepiness to residual depressive symptoms. Sleep Med. 2010;11(6):552-557.
15. Lundt L. Use of the Epworth Sleepiness Scale to evaluate the symptom of excessive sleepiness in major depressive disorder. Gen Hosp Psychiatry. 2005;27(2):146-148.
16. Hawley CJ. Excessive daytime sleepiness in psychiatry: a relevant focus for clinical attention and treatment? Int J Psychiatry Clin Pract. 2006;10(2):117-123.
17. Dodson ER, Zee PC. Therapeutics for circadian rhythm sleep disorders. Sleep Med Clin. 2010;5(4):701-715.
18. Morgenthaler TI, Kapur VK, Brown TM, et al; Standards of Practice Committee of the American Academy of Sleep Medicine. Practice parameters for the treatment of narcolepsy and other hypersomnias of central origin. Sleep. 2007;30(12):1705-1711.
19. Thorpy MJ. Classification of sleep disorders. Neurotherapeutics. 2012;9(4):687-701.
20. National Institute of Neurological Disorders and Stroke. Restless legs syndrome information page. https://www.ninds.nih.gov/Disorders/All-Disorders/Restless-Legs-Syndrome-Information-Page. Accessed June 2, 2017.
21. Johns MW. Reliability and factor analysis of the Epworth Sleepiness Scale. Sleep. 1992;15(4):376-381.
Excessive daytime sleepiness (EDS) is “the inability to maintain wakefulness and alertness during the major waking periods of the day, with sleep occurring unintentionally or at inappropriate times, almost daily for at least 3 months,” according to the American Academy of Sleep Medicine.1 EDS is common, with a prevalence up to 25% to 30% in the general population.1-4 The prevalence rate varies in different studies, primarily because of inconsistent definitions of EDS, and therefore differences in diagnosis and assessment.1,2,4 In a study of 300 psychiatric outpatients, 34% had EDS.3 However, studies and evidence reviewing EDS in psychiatric patients are limited.
The causes of EDS are many and varied,1,8 including medical and psychiatric etiologies. A thorough history, screening at-risk patients, and timely sleep center referral are vital to detect and appropriately manage the cause of EDS.5
This article reviews the literature on EDS, with a focus on the risks of untreated EDS, common etiologies of the condition, as well as a brief description of screening and treatment strategies.
EDS vs fatigue
Many patients describe EDS as “fatigue”1; however, a patient’s report of fatigue could be mistaken for EDS.4 Although there is overlap, it is important for physicians to distinguish between these 2 entities for accurate identification and treatment.1,4
Risk of inadequate screening
A study of 117 patients with symptomatic coronary artery disease showed that EDS is associated with significantly greater incidence of cardiovascular adverse events at 16-month follow up.2 This study had limitations such as small sample size; therefore, more studies are needed. Because of these risks, timely and accurate diagnosis not only improves the patient’s quality of life and reduces polypharmacy but also can be life-saving.
Common causes of EDS in psychiatric patients
Because of the high prevalence and severity of impairments caused by EDS, it is essential for psychiatrists to be informed about causes of EDS and thoroughly assess for the potential underlying etiology before concluding that the sleep problem is a manifestation of the psychiatric disorder and prescribing psychotropic medication for it.
Some common causes of EDS in psychiatric patients include:
Sleep-disordered breathing.8 Obstructive sleep apnea (OSA) is often underdiagnosed,6,7 and considering how common it is,6 psychiatrists likely will see many patients with OSA in their practice.5 OSA has a higher prevalence among patients with psychiatric disorders such as depression6,9 and schizophrenia. Additionally, there is evidence suggesting that patients with OSA are more likely to suffer from depression and EDS than healthy controls6,9,10; some of the proposed mechanisms are sleep fragmentation and hypoxemia.6,9-11 OSA is the most common form of sleep-disordered breathing and is a common cause of EDS.1,2,12 Also, undiagnosed and untreated OSA in patients with depression could cause refractoriness to pharmacological treatment of depression.6,9,10
When unrecognized and untreated, OSA can be life-threatening. Despite this, OSA is not regularly screened for in clinical psychiatric practice.6,10 Therefore, it is imperative that psychiatrists be well-acquainted with measures to identify at-risk patients and refer to a sleep specialist when appropriate.
OSA is accompanied by irritability, cognitive difficulties, and poor sleep, creating an overlap with symptoms of depressive disorders.6,10 Use of sedative hypnotic medications, such as benzodiazepines, which further reduces muscle tone in the airway and suppresses respiratory effort, can worsen OSA symptoms5,6,10 and pose cerebrovascular, cardiovascular, and potentially life-threatening risks, and therefore is not indicated in this population.9,13
Obesity is a risk factor for OSA.6 Patients with mood disorders or schizophrenia or other psychotic disorders are at higher risk of obesity because of psychotropic-induced weight gain, stress-induced mechanisms, and/or lower levels of self-care. When these patients have unrecognized or untreated OSA and are prescribed sedative medications at night or stimulant medications during the day, they could be at increased cardiac or respiratory risks without resolving their underlying condition. A diligent psychiatrist can dramatically reduce the risks by referring a patient for nocturnal polysomnography,1 helping the patient implement lifestyle modifications (eg, exercise, weight loss, and healthy nutrition), prescribing judiciously, and monitoring closely for such risks. An accurate diagnosis of and treatment for OSA can improve sleep6 dramatically and help depressive symptoms through better sleep, more daytime energy and concentration, and adequate oxygenation of the brain while sleeping.
Psychiatrists can screen for OSA using the STOP-Bang (Snoring, Tired, Observed apnea, Pressure, Body mass index, Age, Neck circumference, Gender) Questionnaire, which is a quick, 8-item screening scale that helps to categorize OSA risk as mild, moderate, or severe.12 Hypertension, snoring, and/or gasping for breath (“observed apnea”)—a history which often is provided by spouses or significant others—daytime dozing and/or tiredness, having a large neck circumference or volume, body mass index, male sex, and age are items on the STOP-Bang Questionnaire and also are features that should raise high clinical suspicion of OSA.12 Referral for nocturnal polysomnography in at-risk patients should be the next step1,5 in any sleep-related breathing disorder.
Treatment for OSA involves continuous positive airway pressure (CPAP) therapy, which has been shown to relieve OSA and decrease related EDS.5,6 Other treatment modalities, such as oral appliances and surgery, may be used5 in some cases, but more studies are needed for conclusive results.
Several studies have shown improved depression, mood, and cognition after administering treatment such as CPAP6,9,14 in patients with OSA and depression. Considering the significant risks of cardiovascular,8 cerebrovascular,8 and overall morbidity and mortality associated with untreated OSA,12 it is important to routinely screen for sleep-disordered breathing in patients with depression9 or other psychiatric disorders and refer for specialized sleep evaluation and treatment, when indicated.
Medications. EDS can result from some prescription and over-the-counter medications.1,2,5,7 Sedating antidepressants, antihistamines, antipsychotics, anticonvulsants,1,8 and beta blockers2 could cause sedation, which can persist during daytime, although a few studies did not find an association between antipsychotic use and EDS.3 Benzodiazepines and other sedative-hypnotics,1,7 especially long-acting agents or higher dosages,5 can lead to EDS and decreased alertness. Non-psychotropics, such as opioid pain medications,1,7 antitussives, and skeletal muscle relaxants, also can contribute to or cause daytime sedation.7 When using these agents, psychiatrists should monitor and routinely assess patients while aiming for the lowest effective dosage when feasible.
This strategy creates a framework for psychiatrists to routinely educate patients about these commonly encountered side effects, reduce polypharmacy when possible, and help patients effectively manage or prevent these adverse effects.
Depression.1 Some studies found >45% patients with depression had EDS.3,13,15 Besides an association between depression and EDS,13,16 Chellappa and Araújo13 also found a significant association between EDS and suicidal ideation. The causes of EDS in patients with depression may be varied, ranging from restless legs syndrome, residual depressive symptoms,15 to OSA. Depression is often comorbid with OSA,6 with up to 20% of patients with depression suffering from OSA,10 creating higher risk for EDS. Depressive disorders are routinely assessed during an evaluation of OSA at sleep centers, but OSA often is not screened in psychiatric practice.10
There is a strong need for regular screening for OSA in patients with depression, particularly because most studies show a link between the 2 conditions.10 Both depression and OSA have some common risk factors, such as obesity, hypertension, and metabolic syndrome.10 Patients with these conditions are at greater risk for OSA, and therefore a psychiatrist should proactively screen and refer such patients for nocturnal polysomnography when they suspect OSA. Patients with OSA and depression often present to the psychiatrist with depressive symptoms that appear to be resistant to pharmacological treatment,10 therefore underscoring the importance of screening and ruling out OSA in patients with depression.
Circadian rhythm disorders, restless legs syndrome, alcohol and other substance use, and use of prescription sedative-hypnotics are more common in patients with depression; therefore, this population is at high risk for EDS.
Circadian rhythm disorders and insufficient sleep syndrome. Insufficient sleep syndrome1,2,8 frequently causes EDS and occurs more commonly in busy people who try to get by with less sleep.8 Over time, the effect of sleep loss is cumulative and can be accompanied by mood symptoms, such as irritability, fatigue, and problems with concentration.8 Shift workers1,8 commonly experience insufficient sleep as well as circadian rhythm disorders and EDS. Modafinil is FDA-approved for EDS in shift work sleep disorder.
Geriatric patients may experience advanced sleep phase syndrome involving early awakenings.8 Adolescents, on the other hand, often suffer from delayed sleep phase syndrome, which is a type of circadian rhythm disorder, related to increasing academic and social pressures, natural pubertal shift to later sleep onset, pervading technology use, and often nebulous bedtime routines. This can be a cause of sleep persisting into daytime.8 Taking a careful history and a sleep diary may be useful because this disorder might be confused for insomnia. Treatment involves gradual shifting of the time of sleep onset through bright light exposure and other modalities.8
Adolescents might not be forthcoming about the severity of their sleep problems; therefore, psychiatrists should screen proactively through clinical interviews of patients and parents and consider this possibility when encountering an adolescent with recent-onset attention or cognitive difficulties.
Treatment for circadian rhythm disorders usually includes planned or prescribed sleep scheduling, timed light exposure,8 and occasional use of melatonin or other sedative agents.17
Hypersomnia of central origin, which includes narcolepsy, idiopathic hypersomnia, and recurrent hypersomnia, can present with EDS.1,18,19 Narcolepsy is a rare, debilitating sleep disorder that manifests as EDS or sleep attacks, with or without cataplexy, and sleep paralysis.5,8,18,19 The Multiple Sleep Latency Test and polysomnography are used for diagnosis.1,5 Shortened REM latency is a classic finding often noted on polysomnography. Treatment involves pharmacologic and behavioral strategies and education.5,8 Modafinil is FDA-approved for EDS associated with narcolepsy. Stimulant medications have been used for narcolepsy in the past; further studies are needed to establish benefit–risk ratio of use in this population.18
Kleine-Levin syndrome is a form of recurrent hypersomnia, a less common sleep disorder, characterized by episodes of excessive sleepiness accompanied by hyperphagia and hypersexuality.5,18,19
Other medical conditions,1 such as the rare familial fatal insomnia, neurological conditions1 such as encephalitis,8 epilepsy,8 Alzheimer’s disease or other types of dementia,8 Parkinson’s disease,1 or multiple sclerosis,1,18 can cause excessive daytime fatigue by causing secondary insomnia or hypersomnia.
Treating the underlying disorder is an important first step in these cases. In addition, coordinating with neurologists or other specialists involved in caring for patients with these conditions is important. Regularly reviewing and simplifying the often complex medication regimen, when possible, can go a long way in mitigating EDS in this population.
Other disorders affecting sleep. Restless legs syndrome and periodic limb movement disorder are other causes of EDS.3 Treatment involves lifestyle modifications, iron supplementation in certain patients, and use of dopaminergic agents such as ropinirole, pramipexole, and other medications, depending on severity of the condition, comorbidities, and other factors.20
Alcohol or substance use. Substance use or withdrawal can be associated with sleep disorders, such as hypersomnia,19 insomnia,19 and related EDS.5 For example, alcohol use disorder affects REM sleep, and can cause EDS. Secondary central apnea can be the result of long-standing opioid use19 and can present like EDS.
Insomnia. Primary insomnia rarely causes EDS.5 Insomnia due to a medical or psychiatric condition may be an indirect cause of EDS by causing sleep deprivation.
Steps for timely and accurate diagnosis
Utilize the following steps for facilitating timely diagnosis and treatment of EDS:
Thorough history. Patients often describe “tiredness” instead of sleepiness.8 Therefore, the astute psychiatrist should explore further when patients are presenting with this concern, especially by asking more specific questions such as the tendency to doze off during daytime.8
Family members can be vital sources for obtaining a complete history,5 especially because patients might deny,8 minimize, or not be fully aware1 of the extent of their symptoms. Asking family members about patient’s snoring, irregular breathing, or gasping at night can be particularly valuable.5 Obtaining a family history of sleep disorders can be particularly important, especially in conditions such as OSA and narcolepsy.
Asking about any history of safety issues,8 including sleepiness during driving, cooking, or other activities, is also important.
Use of scales and other screening measures. Psychiatrists can use initial screening measures in the office setting. Epworth Sleepiness Scale15,21 is a validated,2 short, self-administered measure to assess the level of daytime sleepiness; however, it has some limitations such as not being able to measure changes in sleepiness from hour to hour or day to day. Because of its limitations, the Epworth Sleepiness Scale should not be used by itself as a diagnostic tool.3 It has been commonly used for detecting OSA2 and narcolepsy. The Stanford Sleepiness Scale is a self-rating scale that measures the subjective degree of sleepiness and alertness; it has limitations as well, such as having little correlation with chronic sleep loss.8 Other tools such as visual analogue scales also could be helpful.8 For more specialized testing, such as Multiple Sleep Latency Test or polysomnography, referral to a sleep specialist is ideal.8
Education. The assessment is an opportunity for the psychiatrist to educate patients about sleep hygiene, the importance of regular bedtimes, and getting adequate sleep to avoid accumulating a sleep deficit.
Urgent referral of at-risk populations. Prompt or urgent referral of at-risk populations, such as geriatric patients or those with a history of dozing off during driving, is invaluable in preventing morbidity and mortality from untreated sleep disorders.
Patients with severe daytime sleepiness should be advised to not drive or operate heavy machinery until this condition is adequately controlled.18
Bottom Line
Excessive daytime sleepiness (EDS) is “the inability to maintain wakefulness and alertness during the major waking periods of the day, with sleep occurring unintentionally or at inappropriate times, almost daily for at least 3 months,” according to the American Academy of Sleep Medicine.1 EDS is common, with a prevalence up to 25% to 30% in the general population.1-4 The prevalence rate varies in different studies, primarily because of inconsistent definitions of EDS, and therefore differences in diagnosis and assessment.1,2,4 In a study of 300 psychiatric outpatients, 34% had EDS.3 However, studies and evidence reviewing EDS in psychiatric patients are limited.
The causes of EDS are many and varied,1,8 including medical and psychiatric etiologies. A thorough history, screening at-risk patients, and timely sleep center referral are vital to detect and appropriately manage the cause of EDS.5
This article reviews the literature on EDS, with a focus on the risks of untreated EDS, common etiologies of the condition, as well as a brief description of screening and treatment strategies.
EDS vs fatigue
Many patients describe EDS as “fatigue”1; however, a patient’s report of fatigue could be mistaken for EDS.4 Although there is overlap, it is important for physicians to distinguish between these 2 entities for accurate identification and treatment.1,4
Risk of inadequate screening
A study of 117 patients with symptomatic coronary artery disease showed that EDS is associated with significantly greater incidence of cardiovascular adverse events at 16-month follow up.2 This study had limitations such as small sample size; therefore, more studies are needed. Because of these risks, timely and accurate diagnosis not only improves the patient’s quality of life and reduces polypharmacy but also can be life-saving.
Common causes of EDS in psychiatric patients
Because of the high prevalence and severity of impairments caused by EDS, it is essential for psychiatrists to be informed about causes of EDS and thoroughly assess for the potential underlying etiology before concluding that the sleep problem is a manifestation of the psychiatric disorder and prescribing psychotropic medication for it.
Some common causes of EDS in psychiatric patients include:
Sleep-disordered breathing.8 Obstructive sleep apnea (OSA) is often underdiagnosed,6,7 and considering how common it is,6 psychiatrists likely will see many patients with OSA in their practice.5 OSA has a higher prevalence among patients with psychiatric disorders such as depression6,9 and schizophrenia. Additionally, there is evidence suggesting that patients with OSA are more likely to suffer from depression and EDS than healthy controls6,9,10; some of the proposed mechanisms are sleep fragmentation and hypoxemia.6,9-11 OSA is the most common form of sleep-disordered breathing and is a common cause of EDS.1,2,12 Also, undiagnosed and untreated OSA in patients with depression could cause refractoriness to pharmacological treatment of depression.6,9,10
When unrecognized and untreated, OSA can be life-threatening. Despite this, OSA is not regularly screened for in clinical psychiatric practice.6,10 Therefore, it is imperative that psychiatrists be well-acquainted with measures to identify at-risk patients and refer to a sleep specialist when appropriate.
OSA is accompanied by irritability, cognitive difficulties, and poor sleep, creating an overlap with symptoms of depressive disorders.6,10 Use of sedative hypnotic medications, such as benzodiazepines, which further reduces muscle tone in the airway and suppresses respiratory effort, can worsen OSA symptoms5,6,10 and pose cerebrovascular, cardiovascular, and potentially life-threatening risks, and therefore is not indicated in this population.9,13
Obesity is a risk factor for OSA.6 Patients with mood disorders or schizophrenia or other psychotic disorders are at higher risk of obesity because of psychotropic-induced weight gain, stress-induced mechanisms, and/or lower levels of self-care. When these patients have unrecognized or untreated OSA and are prescribed sedative medications at night or stimulant medications during the day, they could be at increased cardiac or respiratory risks without resolving their underlying condition. A diligent psychiatrist can dramatically reduce the risks by referring a patient for nocturnal polysomnography,1 helping the patient implement lifestyle modifications (eg, exercise, weight loss, and healthy nutrition), prescribing judiciously, and monitoring closely for such risks. An accurate diagnosis of and treatment for OSA can improve sleep6 dramatically and help depressive symptoms through better sleep, more daytime energy and concentration, and adequate oxygenation of the brain while sleeping.
Psychiatrists can screen for OSA using the STOP-Bang (Snoring, Tired, Observed apnea, Pressure, Body mass index, Age, Neck circumference, Gender) Questionnaire, which is a quick, 8-item screening scale that helps to categorize OSA risk as mild, moderate, or severe.12 Hypertension, snoring, and/or gasping for breath (“observed apnea”)—a history which often is provided by spouses or significant others—daytime dozing and/or tiredness, having a large neck circumference or volume, body mass index, male sex, and age are items on the STOP-Bang Questionnaire and also are features that should raise high clinical suspicion of OSA.12 Referral for nocturnal polysomnography in at-risk patients should be the next step1,5 in any sleep-related breathing disorder.
Treatment for OSA involves continuous positive airway pressure (CPAP) therapy, which has been shown to relieve OSA and decrease related EDS.5,6 Other treatment modalities, such as oral appliances and surgery, may be used5 in some cases, but more studies are needed for conclusive results.
Several studies have shown improved depression, mood, and cognition after administering treatment such as CPAP6,9,14 in patients with OSA and depression. Considering the significant risks of cardiovascular,8 cerebrovascular,8 and overall morbidity and mortality associated with untreated OSA,12 it is important to routinely screen for sleep-disordered breathing in patients with depression9 or other psychiatric disorders and refer for specialized sleep evaluation and treatment, when indicated.
Medications. EDS can result from some prescription and over-the-counter medications.1,2,5,7 Sedating antidepressants, antihistamines, antipsychotics, anticonvulsants,1,8 and beta blockers2 could cause sedation, which can persist during daytime, although a few studies did not find an association between antipsychotic use and EDS.3 Benzodiazepines and other sedative-hypnotics,1,7 especially long-acting agents or higher dosages,5 can lead to EDS and decreased alertness. Non-psychotropics, such as opioid pain medications,1,7 antitussives, and skeletal muscle relaxants, also can contribute to or cause daytime sedation.7 When using these agents, psychiatrists should monitor and routinely assess patients while aiming for the lowest effective dosage when feasible.
This strategy creates a framework for psychiatrists to routinely educate patients about these commonly encountered side effects, reduce polypharmacy when possible, and help patients effectively manage or prevent these adverse effects.
Depression.1 Some studies found >45% patients with depression had EDS.3,13,15 Besides an association between depression and EDS,13,16 Chellappa and Araújo13 also found a significant association between EDS and suicidal ideation. The causes of EDS in patients with depression may be varied, ranging from restless legs syndrome, residual depressive symptoms,15 to OSA. Depression is often comorbid with OSA,6 with up to 20% of patients with depression suffering from OSA,10 creating higher risk for EDS. Depressive disorders are routinely assessed during an evaluation of OSA at sleep centers, but OSA often is not screened in psychiatric practice.10
There is a strong need for regular screening for OSA in patients with depression, particularly because most studies show a link between the 2 conditions.10 Both depression and OSA have some common risk factors, such as obesity, hypertension, and metabolic syndrome.10 Patients with these conditions are at greater risk for OSA, and therefore a psychiatrist should proactively screen and refer such patients for nocturnal polysomnography when they suspect OSA. Patients with OSA and depression often present to the psychiatrist with depressive symptoms that appear to be resistant to pharmacological treatment,10 therefore underscoring the importance of screening and ruling out OSA in patients with depression.
Circadian rhythm disorders, restless legs syndrome, alcohol and other substance use, and use of prescription sedative-hypnotics are more common in patients with depression; therefore, this population is at high risk for EDS.
Circadian rhythm disorders and insufficient sleep syndrome. Insufficient sleep syndrome1,2,8 frequently causes EDS and occurs more commonly in busy people who try to get by with less sleep.8 Over time, the effect of sleep loss is cumulative and can be accompanied by mood symptoms, such as irritability, fatigue, and problems with concentration.8 Shift workers1,8 commonly experience insufficient sleep as well as circadian rhythm disorders and EDS. Modafinil is FDA-approved for EDS in shift work sleep disorder.
Geriatric patients may experience advanced sleep phase syndrome involving early awakenings.8 Adolescents, on the other hand, often suffer from delayed sleep phase syndrome, which is a type of circadian rhythm disorder, related to increasing academic and social pressures, natural pubertal shift to later sleep onset, pervading technology use, and often nebulous bedtime routines. This can be a cause of sleep persisting into daytime.8 Taking a careful history and a sleep diary may be useful because this disorder might be confused for insomnia. Treatment involves gradual shifting of the time of sleep onset through bright light exposure and other modalities.8
Adolescents might not be forthcoming about the severity of their sleep problems; therefore, psychiatrists should screen proactively through clinical interviews of patients and parents and consider this possibility when encountering an adolescent with recent-onset attention or cognitive difficulties.
Treatment for circadian rhythm disorders usually includes planned or prescribed sleep scheduling, timed light exposure,8 and occasional use of melatonin or other sedative agents.17
Hypersomnia of central origin, which includes narcolepsy, idiopathic hypersomnia, and recurrent hypersomnia, can present with EDS.1,18,19 Narcolepsy is a rare, debilitating sleep disorder that manifests as EDS or sleep attacks, with or without cataplexy, and sleep paralysis.5,8,18,19 The Multiple Sleep Latency Test and polysomnography are used for diagnosis.1,5 Shortened REM latency is a classic finding often noted on polysomnography. Treatment involves pharmacologic and behavioral strategies and education.5,8 Modafinil is FDA-approved for EDS associated with narcolepsy. Stimulant medications have been used for narcolepsy in the past; further studies are needed to establish benefit–risk ratio of use in this population.18
Kleine-Levin syndrome is a form of recurrent hypersomnia, a less common sleep disorder, characterized by episodes of excessive sleepiness accompanied by hyperphagia and hypersexuality.5,18,19
Other medical conditions,1 such as the rare familial fatal insomnia, neurological conditions1 such as encephalitis,8 epilepsy,8 Alzheimer’s disease or other types of dementia,8 Parkinson’s disease,1 or multiple sclerosis,1,18 can cause excessive daytime fatigue by causing secondary insomnia or hypersomnia.
Treating the underlying disorder is an important first step in these cases. In addition, coordinating with neurologists or other specialists involved in caring for patients with these conditions is important. Regularly reviewing and simplifying the often complex medication regimen, when possible, can go a long way in mitigating EDS in this population.
Other disorders affecting sleep. Restless legs syndrome and periodic limb movement disorder are other causes of EDS.3 Treatment involves lifestyle modifications, iron supplementation in certain patients, and use of dopaminergic agents such as ropinirole, pramipexole, and other medications, depending on severity of the condition, comorbidities, and other factors.20
Alcohol or substance use. Substance use or withdrawal can be associated with sleep disorders, such as hypersomnia,19 insomnia,19 and related EDS.5 For example, alcohol use disorder affects REM sleep, and can cause EDS. Secondary central apnea can be the result of long-standing opioid use19 and can present like EDS.
Insomnia. Primary insomnia rarely causes EDS.5 Insomnia due to a medical or psychiatric condition may be an indirect cause of EDS by causing sleep deprivation.
Steps for timely and accurate diagnosis
Utilize the following steps for facilitating timely diagnosis and treatment of EDS:
Thorough history. Patients often describe “tiredness” instead of sleepiness.8 Therefore, the astute psychiatrist should explore further when patients are presenting with this concern, especially by asking more specific questions such as the tendency to doze off during daytime.8
Family members can be vital sources for obtaining a complete history,5 especially because patients might deny,8 minimize, or not be fully aware1 of the extent of their symptoms. Asking family members about patient’s snoring, irregular breathing, or gasping at night can be particularly valuable.5 Obtaining a family history of sleep disorders can be particularly important, especially in conditions such as OSA and narcolepsy.
Asking about any history of safety issues,8 including sleepiness during driving, cooking, or other activities, is also important.
Use of scales and other screening measures. Psychiatrists can use initial screening measures in the office setting. Epworth Sleepiness Scale15,21 is a validated,2 short, self-administered measure to assess the level of daytime sleepiness; however, it has some limitations such as not being able to measure changes in sleepiness from hour to hour or day to day. Because of its limitations, the Epworth Sleepiness Scale should not be used by itself as a diagnostic tool.3 It has been commonly used for detecting OSA2 and narcolepsy. The Stanford Sleepiness Scale is a self-rating scale that measures the subjective degree of sleepiness and alertness; it has limitations as well, such as having little correlation with chronic sleep loss.8 Other tools such as visual analogue scales also could be helpful.8 For more specialized testing, such as Multiple Sleep Latency Test or polysomnography, referral to a sleep specialist is ideal.8
Education. The assessment is an opportunity for the psychiatrist to educate patients about sleep hygiene, the importance of regular bedtimes, and getting adequate sleep to avoid accumulating a sleep deficit.
Urgent referral of at-risk populations. Prompt or urgent referral of at-risk populations, such as geriatric patients or those with a history of dozing off during driving, is invaluable in preventing morbidity and mortality from untreated sleep disorders.
Patients with severe daytime sleepiness should be advised to not drive or operate heavy machinery until this condition is adequately controlled.18
Bottom Line
1. Chervin RD. Approach to the patient with excessive daytime sleepiness. http://www.uptodate.com/contents/approach-to-the-patient-with-excessive-daytime-sleepiness. Updated January 2016. Accessed June 5, 2017.
2. Lee CH, Ng WY, Hau W, et al. Excessive daytime sleepiness is associated with longer culprit lesion and adverse outcomes in patients with coronary artery disease. J Clin Sleep Med. 2013;9(12):1267-1272.
3. Hawley CJ, Gale TM, Sivakumaran T, et al. Excessive daytime sleepiness in psychiatric disorders: prevalence, correlates and clinical significance. Psychiatry Res. 2010;175(1-2):138-141.
4. Pigeon WR, Sateia MJ, Ferguson RJ. Distinguishing between excessive daytime sleepiness and fatigue: toward improved detection and treatment. J Psychosom Res. 2003;54(1):61-69.
5. Krahn LE. Excessive daytime sleepiness: diagnosing the causes. Current Psychiatry. 2002;1(1):49-57.
6. Ejaz SM, Khawaja IS, Bhatia S, et al. Obstructive sleep apnea and depression: a review. Innov Clin Neurosci. 2011;8(8):17-25.
7. Pagel JF. Excessive daytime sleepiness. Am Fam Physician. 2009;79(5):391-396.
8. Guilleminault C, Brooks SN. Excessive daytime sleepiness: a challenge for the practising neurologist. Brain. 2001;124(pt 8):1482-1491.
9. Cheng P, Casement M, Chen CF, et al. Sleep disordered breathing in major depressive disorder. J Sleep Res. 2013;22(4):459-462.
10. Schröder CM, O’Hara R. Depression and obstructive sleep apnea (OSA). Ann Gen Psychiatry. 2005;4:13.
11. Bardwell WA, Berry CC, Ancoli-Israel S, et al. Psychological correlates of sleep apnea. J Psychosom Res. 1999;47(6):583-596.
12. Chung F, Abdullah HR, Liao P. STOP-Bang Questionnaire: a practical approach to screen for obstructive sleep apnea. Chest. 2016;149(3):631-638.
13. Chellappa SL, Araújo JF. Excessive daytime sleepiness in patients with depressive disorder. Rev Bras Psiquiatr. 2006;28(2):126-129.
14. Habukawa M, Uchimura N, Kakuma T, et al. Effect of CPAP treatment on residual depressive symptoms in patients with major depression and coexisting sleep apnea: contribution of daytime sleepiness to residual depressive symptoms. Sleep Med. 2010;11(6):552-557.
15. Lundt L. Use of the Epworth Sleepiness Scale to evaluate the symptom of excessive sleepiness in major depressive disorder. Gen Hosp Psychiatry. 2005;27(2):146-148.
16. Hawley CJ. Excessive daytime sleepiness in psychiatry: a relevant focus for clinical attention and treatment? Int J Psychiatry Clin Pract. 2006;10(2):117-123.
17. Dodson ER, Zee PC. Therapeutics for circadian rhythm sleep disorders. Sleep Med Clin. 2010;5(4):701-715.
18. Morgenthaler TI, Kapur VK, Brown TM, et al; Standards of Practice Committee of the American Academy of Sleep Medicine. Practice parameters for the treatment of narcolepsy and other hypersomnias of central origin. Sleep. 2007;30(12):1705-1711.
19. Thorpy MJ. Classification of sleep disorders. Neurotherapeutics. 2012;9(4):687-701.
20. National Institute of Neurological Disorders and Stroke. Restless legs syndrome information page. https://www.ninds.nih.gov/Disorders/All-Disorders/Restless-Legs-Syndrome-Information-Page. Accessed June 2, 2017.
21. Johns MW. Reliability and factor analysis of the Epworth Sleepiness Scale. Sleep. 1992;15(4):376-381.
1. Chervin RD. Approach to the patient with excessive daytime sleepiness. http://www.uptodate.com/contents/approach-to-the-patient-with-excessive-daytime-sleepiness. Updated January 2016. Accessed June 5, 2017.
2. Lee CH, Ng WY, Hau W, et al. Excessive daytime sleepiness is associated with longer culprit lesion and adverse outcomes in patients with coronary artery disease. J Clin Sleep Med. 2013;9(12):1267-1272.
3. Hawley CJ, Gale TM, Sivakumaran T, et al. Excessive daytime sleepiness in psychiatric disorders: prevalence, correlates and clinical significance. Psychiatry Res. 2010;175(1-2):138-141.
4. Pigeon WR, Sateia MJ, Ferguson RJ. Distinguishing between excessive daytime sleepiness and fatigue: toward improved detection and treatment. J Psychosom Res. 2003;54(1):61-69.
5. Krahn LE. Excessive daytime sleepiness: diagnosing the causes. Current Psychiatry. 2002;1(1):49-57.
6. Ejaz SM, Khawaja IS, Bhatia S, et al. Obstructive sleep apnea and depression: a review. Innov Clin Neurosci. 2011;8(8):17-25.
7. Pagel JF. Excessive daytime sleepiness. Am Fam Physician. 2009;79(5):391-396.
8. Guilleminault C, Brooks SN. Excessive daytime sleepiness: a challenge for the practising neurologist. Brain. 2001;124(pt 8):1482-1491.
9. Cheng P, Casement M, Chen CF, et al. Sleep disordered breathing in major depressive disorder. J Sleep Res. 2013;22(4):459-462.
10. Schröder CM, O’Hara R. Depression and obstructive sleep apnea (OSA). Ann Gen Psychiatry. 2005;4:13.
11. Bardwell WA, Berry CC, Ancoli-Israel S, et al. Psychological correlates of sleep apnea. J Psychosom Res. 1999;47(6):583-596.
12. Chung F, Abdullah HR, Liao P. STOP-Bang Questionnaire: a practical approach to screen for obstructive sleep apnea. Chest. 2016;149(3):631-638.
13. Chellappa SL, Araújo JF. Excessive daytime sleepiness in patients with depressive disorder. Rev Bras Psiquiatr. 2006;28(2):126-129.
14. Habukawa M, Uchimura N, Kakuma T, et al. Effect of CPAP treatment on residual depressive symptoms in patients with major depression and coexisting sleep apnea: contribution of daytime sleepiness to residual depressive symptoms. Sleep Med. 2010;11(6):552-557.
15. Lundt L. Use of the Epworth Sleepiness Scale to evaluate the symptom of excessive sleepiness in major depressive disorder. Gen Hosp Psychiatry. 2005;27(2):146-148.
16. Hawley CJ. Excessive daytime sleepiness in psychiatry: a relevant focus for clinical attention and treatment? Int J Psychiatry Clin Pract. 2006;10(2):117-123.
17. Dodson ER, Zee PC. Therapeutics for circadian rhythm sleep disorders. Sleep Med Clin. 2010;5(4):701-715.
18. Morgenthaler TI, Kapur VK, Brown TM, et al; Standards of Practice Committee of the American Academy of Sleep Medicine. Practice parameters for the treatment of narcolepsy and other hypersomnias of central origin. Sleep. 2007;30(12):1705-1711.
19. Thorpy MJ. Classification of sleep disorders. Neurotherapeutics. 2012;9(4):687-701.
20. National Institute of Neurological Disorders and Stroke. Restless legs syndrome information page. https://www.ninds.nih.gov/Disorders/All-Disorders/Restless-Legs-Syndrome-Information-Page. Accessed June 2, 2017.
21. Johns MW. Reliability and factor analysis of the Epworth Sleepiness Scale. Sleep. 1992;15(4):376-381.

Accelerated aging in schizophrenia

Excessive daytime sleepiness
Nutraceuticals for traumatic brain injury: Should you recommend their use?
Traumatic brain injury (TBI) affects more than 2 million people in the United States each year.1 TBI can trigger a cascade of secondary injury mechanisms, such as inflammation, hypoxic/ischemic injury, excitotoxicity, and oxidative stress,2 that could contribute to cognitive and behavioral changes. Although neuropsychiatric symptoms might not be obvious after a TBI, they have a high prevalence in these patients, can last long term, and may be difficult to treat.3 Despite research advances in understanding the biological basis of TBI and identifying potential therapeutic targets, treatment options for individuals with TBI remain limited.
As a result, clinicians have turned to alternative treatments for TBI, including nutraceuticals. In this article, we will:
- provide an overview of nutraceuticals used in treating TBI, first exploring outcomes soon after TBI, then concentrating on neuropsychiatric outcomes
- evaluate the existing evidence, including recommended dietary allowances (Table 1)4,5 and side effects (Table 2)
- review recommendations for their clinical use.
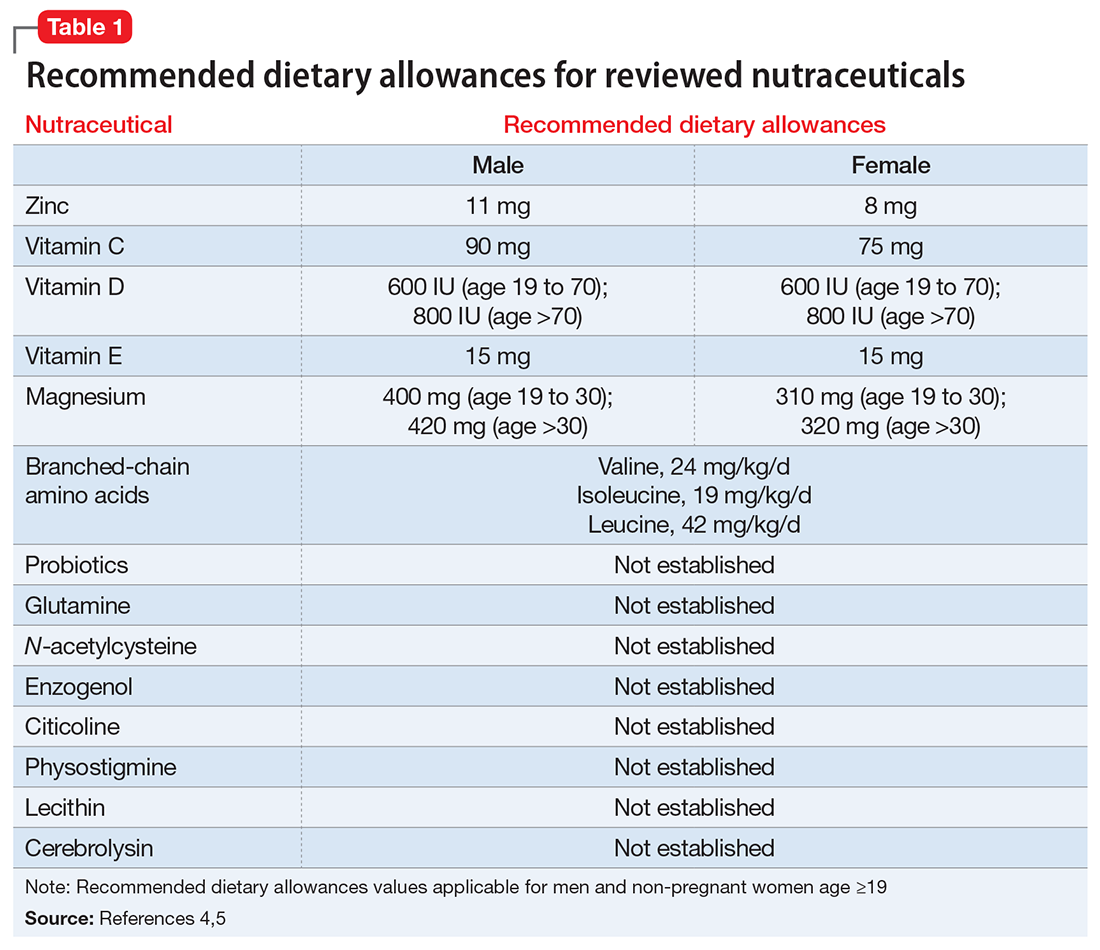
Pharmacologic approaches are limited
Nutraceuticals have gained attention for managing TBI-associated neuropsychiatric disorders because of the limited evidence supporting current approaches. Existing strategies encompass pharmacologic and non-pharmacologic interventions, psychoeducation, supportive and behavioral psychotherapies, and cognitive rehabilitation.6
Many pharmacologic options exist for specific neurobehavioral symptoms, but the evidence for their use is based on small studies, case reports, and knowledge extrapolated from their use in idiopathic psychiatric disorders.7,8 No FDA-approved drugs have been effective for treating neuropsychiatric disturbances after a TBI. Off-label use of antidepressants, anticonvulsants, dopaminergic agents, and cholinesterase inhibitors in TBI has been associated with inadequate clinical response and/or intolerable side effects.9,10
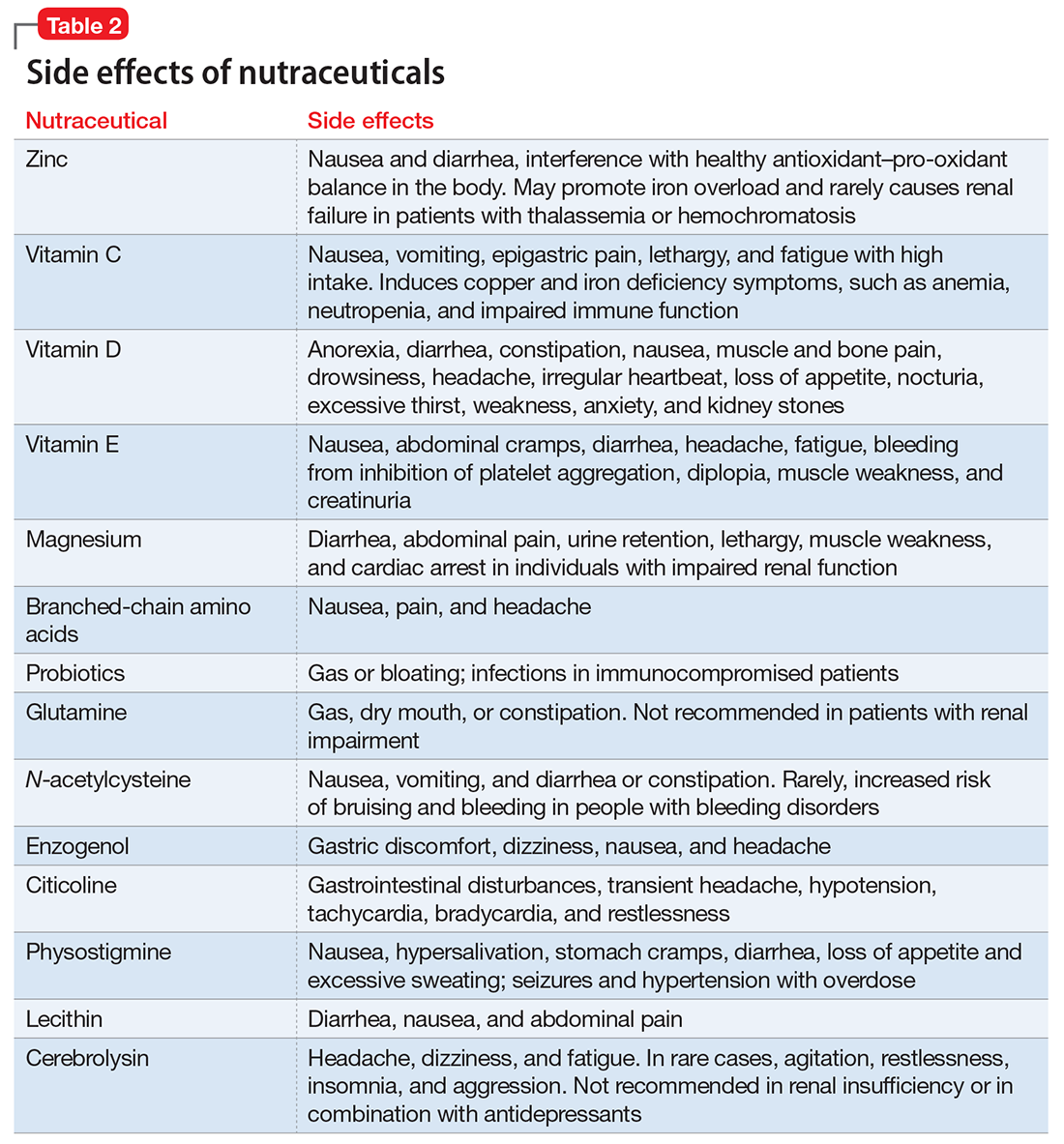
What are nutraceuticals?
DeFelice11 introduced the term “nutraceutical” to refer to “any substance that is a food or part of a food and provides medical or health benefits, including the prevention and treatment of disease.” The term has been expanded to include dietary supplements, such as vitamins, minerals, amino acids, herbal or other botanicals, and food products that provide health benefits beyond what they normally provide in food form. The FDA does not regulate the marketing or manufacturing of nutraceuticals; therefore, their bioavailability and metabolism can vary.
Despite their widespread use, the evidence supporting the efficacy of nutraceuticals for patients with TBI is limited. Their effects might vary by population and depend on dose, timing, TBI severity, and whether taken alone or in combination with other nutraceutical or pharmaceutical agents. Fourteen randomized controlled trials (RCTs) have addressed the use of nutraceuticals in TBI (Table 3), but further research is needed to clarify for which conditions they provide maximum benefit.
Nutraceuticals and their potential use in TBI
Zinc is considered essential for optimal CNS functioning. Patients with TBI might be at risk for zinc deficiency, which has been associated with increased cell death and behavioral deficits.12,13 A randomized, prospective, double-blinded controlled trial examined the effects of supplemental zinc administration (12 mg for 15 days) compared with standard zinc therapy (2.5 mg for 15 days) over 1 month in 68 adults with acute severe closed head injury.14 The supplemental zinc group showed improved visceral protein levels, lower mortality, and more favorable neurologic recovery based on higher adjusted mean Glasgow Coma Scale score on day 28 and mean motor score on days 15 and 21.
Rodent studies have shown that zinc supplementation could reduce deficits in spatial learning and memory and depression-like behaviors and help decrease stress and anxiety,12 although no human clinical trials have been conducted. Despite the potential neuroprotective effects of zinc supplementation, evidence exists that endogenous zinc release and accumulation following TBI can trigger cellular changes that result in neuronal death.13
Vitamins C and E. Oxidative damage is believed to play a significant role in secondary injury in TBI, so research has focused on the role of antioxidants, such as vitamins C and E, to promote post-TBI recovery.15 One RCT16 of 100 adults with acute severe head injury reported that vitamin E administration was associated with reduced mortality and lower Glasgow Outcome Scale (GOS) scores, and vitamin C was associated with stabilized or reduced perilesional edema/infarct on CT scan.
Vitamin D. An animal study reported that vitamin D supplementation can help reduce inflammation, oxidative stress, and cell death in TBI, and that vitamin D deficiency has been associated with increased inflammation and behavioral deficits.17 Further evidence suggests that vitamin D may have a synergistic effect when used in combination with the hormone progesterone. A RCT of 60 patients with severe TBI reported that 60% of those who received progesterone plus vitamin D had GOS scores of 4 (good recovery) or 5 (moderate disability) vs 45% receiving progesterone alone or 25% receiving placebo.18
Magnesium, one of the most widely used nutraceuticals, is considered essential for CNS functioning, including the regulation of N-methyl-
A RCT evaluated the safety and efficacy of magnesium supplementation in 60 patients with severe closed TBI, with one-half randomized to standard care and the other also receiving magnesium sulfate (MgSO4; initiation dose of 4 g IV and 10 g IM, continuation dose of 5 g IM every 4 hours for 24 hours).20 After 3 months, more patients in the MgSO4 group had higher GOS scores than controls (73.3% vs 40%), lower 1-month mortality rates (13.3% vs 43.3%), and lower rates of intraoperative brain swelling (29.4% vs 73.3%).
However, a larger RCT of 499 patients with moderate or severe TBI randomized to high-dose (1.25 to 2.5 mmol/L) or low-dose (1.0 to 1.85 mmol/L) IV MgSO4 or placebo provided conflicting results.21 Participants received MgSO4 8 hours after injury and continued for 5 days. After 6 months, patients in the high-dose MgSO4 and placebo groups had similar composite primary outcome measures (eg, seizures, neuropsychological measures, functional status measures), although the high-dose group had a higher mortality rate than the placebo group. Patients who received low-dose MgSO4 showed worse outcomes than those assigned to placebo.
Amino acids. Branched-chain amino acids (BCAAs), including valine, isoleucine, and leucine, are essential in protein and neurotransmitter synthesis. Reduced levels of endogenous BCAAs have been reported in patients with mild or severe TBI.22 Preclinical studies suggest that BCAAs can improve hippocampal-dependent cognitive functioning following TBI.23
Two RCTs of BCAAs have been conducted in humans. One study randomized 40 men with severe TBI to IV BCAAs or placebo.24 After 15 days, the BCAA group showed greater improvement in Disability Rating Scale scores. The study also found that supplementation increased total BCAA levels without negatively affecting plasma levels of neurotransmitter precursors tyrosine and tryptophan. A second study found that 41 patients in a vegetative or minimally conscious state who received BCAA supplementation for 15 days had higher Disability Rating Scale scores than those receiving placebo.25
Probiotics and glutamine. Probiotics are non-pathogenic microorganisms that have been shown to modulate the host’s immune system.26 TBI is associated with immunological changes, including a shift from T-helper type 1 (TH1) cells to T-helper type 2 (TH2) cells that increase susceptibility to infection.27
A RCT of 52 patients with severe TBI suggested a correlation between probiotic administration-modulated cytokine levels and TH1/TH2 balance.28 A 3-times daily probiotic mix of Bifidobacterium longum, Lactobacillus bulgaricus, and Streptococcus thermophilus for 21 days led to shorter average ICU stays (6.8 vs 10.7 days, P = .034) and a decrease in nosocomial infections (34.6% vs 57.7%, P = .095) vs placebo, although the latter difference was not statistically significant.28
A prospective RCT of 20 patients with brain injury29 found a similar impact of early enteral nutrition supplemented with Lactobacillus johnsonii and glutamine, 30 g, vs a standard enteral nutrition formula. The treatment group experienced fewer nosocomial infections (50% vs 100%, P = .03), shorter ICU stays (10 vs 22 days, P < .01), and fewer days on mechanical ventilation (7 vs 14, P = .04). Despite these studies, evidence for the use of glutamine in patients with TBI is scarce and inconclusive.
N-acetylcysteine (NAC) comes from the amino acid L-cysteine. NAC is an effective scavenger of free radicals and improves cerebral microcirculatory blood flow and tissue oxygenation.30 A randomized, double-blind, placebo-controlled study of oral NAC supplementation in 81 active duty service members with mild TBI found NAC had a significant effect on outcomes.31 Oral NAC supplementation led to improved neuropsychological test results, number of mild TBI symptoms, complete symptom resolution by day 7 of treatment compared with placebo, and NAC was well tolerated. Lack of replication studies and generalizability of findings to civilian, moderate, or chronic TBI populations are key limitations of this study.
Proposed mechanisms for the neuroprotective benefit of NAC include its antioxidant and inflammatory activation of cysteine/glutamate exchange, metabotropic glutamate receptor modulation, and glutathione synthesis.32 NAC has poor blood–brain permeability, but the vascular disruption seen in acute TBI might facilitate its delivery to affected neural sites.31 As such, the benefits of NAC in subacute or chronic TBI are questionable.
Neuropsychiatric outcomes of nutraceuticals
Enzogenol. This flavonoid-rich extract from the bark of the Monterey pine tree (Pinus radiata), known by the trade name Enzogenol, reportedly has antioxidant and anti-inflammatory properties that may counter oxidative damage and neuroinflammation following TBI. A phase II trial randomized participants to Enzogenol, 1,000 mg/d, or placebo for 6 weeks, then all participants received Enzogenol for 6 weeks followed by placebo for 4 weeks.33 Enzogenol was well tolerated with few side effects.
Compared with placebo, participants receiving Enzogenol showed no significant change in mood, as measured by the Hospital Anxiety and Depression Scale, and greater improvement in overall cognition as assessed by the Cognitive Failures Questionnaire. However, measures of working memory (digit span, arithmetic, and letter–number sequencing subtests of the Wechsler Adult Intelligence Scale) and episodic memory (California Verbal Learning Test) showed no benefit from Enzogenol.
Citicoline (CDP-choline) is an endogenous compound widely available as a nutraceutical that has been approved for TBI therapy in 59 countries.34 Animal studies indicate that it could possess neuroprotective properties. Proposed mechanisms for such effects have included stabilizing cell membranes, reducing inflammation, reducing the presence of free radicals, or stimulating production of acetylcholine.35,36 A study in rats found that CDP-choline was associated with increased levels of acetylcholine in the hippocampus and neocortex, which may help reduce neurobehavioral deficits.37
A study of 14 adults with mild to moderate closed head injury38 found that patients who received CDP-choline showed a greater reduction in post-concussion symptoms and improvement in recognition memory than controls who received placebo. However, the Citicoline Brain Injury Treatment Trial, a large randomized trial of 1,213 adults with complicated mild, moderate, or severe TBI, reported that CDP-choline did not improve functional and cognitive status.39
Physostigmine and lecithin. The cholinergic system is a key modulatory neurotransmitter system of the brain that mediates conscious awareness, attention, learning, and working memory.40 A double-blind, placebo-controlled study of 16 patients with moderate to severe closed head injury provided inconsistent evidence for the efficacy of physostigmine and lecithin in the treatment of memory and attention disturbances.41The results showed no differences between the physostigmine–lecithin combination vs lecithin alone, although sustained attention on the Continuous Performance Test was more efficient with physostigmine than placebo when the drug condition occurred first in the crossover design. The lack of encouraging data and concerns about its cardiovascular and proconvulsant properties in patients with TBI may explain the dearth of studies with physostigmine.
Cerebrolysin. A peptide preparation produced from purified pig brain proteins, known by the trade name Cerebrolysin, is popular in Asia and Europe for its nootropic properties. Cerebrolysin may activate cerebral mechanisms related to attention and memory processes,42 and some data have shown efficacy in improving cognitive symptoms and daily activities in patients with Alzheimer’s disease43 and TBI.44
A blinded 12-week study of 32 participants with acute mild TBI reported that those randomized to Cerebrolysin showed improvement in cognitive functioning vs the placebo group.45 The authors concluded that Cerebrolysin provides an advantage for patients with mild TBI and brain contusion if treatment starts within 24 hours of mild TBI onset. Cerebrolysin was well tolerated. Major limitations of this study were small sample size, lack of information regarding comorbid neuropsychiatric conditions and treatments, and short treatment duration.
A recent Cochrane review of 6 RCTs with 1,501 participants found no clinical benefit of Cerebrolysin for treating acute ischemic stroke, and found moderate-quality evidence of an increase with non-fatal serious adverse events but not in total serious adverse events.46 We do not recommend Cerebrolysin use in patients with TBI at this time until additional efficacy and safety data are available.
Nutraceuticals used in other populations
Other nutraceuticals with preclinical evidence of possible benefit in TBI but lacking evidence from human clinical trials include omega-3 fatty acids,47 curcumin,48 and resveratrol,49 providing further proof that results from experimental studies do not necessarily extend to clinical trials.50
Studies of nutraceuticals in other neurological and psychiatric populations have yielded some promising results. Significant interest has focused on the association between vitamin D deficiency, dementia, and neurodegenerative conditions such as Alzheimer’s disease, multiple sclerosis, and Parkinson’s disease.51 The role of vitamin D in regulation of calcium-mediated neuronal excitotoxicity and oxidative stress and in the induction of synaptic structural proteins, neurotrophic factors, and deficient neurotransmitters makes it an attractive candidate as a neuroprotective agent.52
RCTs of nutraceuticals also have reported positive findings for a variety of mood and anxiety disorders, such as St. John’s wort, S-adenosylmethionine, omega-3 fatty acids for major depression53 and bipolar depression,54 and kava for generalized anxiety disorder.55 More research, however, is needed in these areas.
The use of nonpharmacologic agents in TBI often relies on similar neuropsychiatric symptom profiles of idiopathic psychiatric disorders. Attention-deficit/hyperactivity disorder (ADHD) closely resembles TBI, but systemic reviews of studies of zinc, magnesium, and polyunsaturated fatty acids supplementation in ADHD provide no evidence of therapeutic benefit.56-58
Educate patients in role of nutraceuticals
Despite lack of FDA oversight and limited empirical support, nutraceuticals continue to be widely marketed and used for their putative health benefits59 and have gained increased attention among clinicians.60 Because nutritional deficiency may make the brain less able than other organs to recover from injury,61 supplementation is an option, especially in individuals who could be at greater risk of TBI (eg, athletes and military personnel).
Lacking robust scientific evidence to support the use of nutraceuticals either for enhancing TBI recovery or treating neuropsychiatric disturbances, clinicians must educate patients that these agents are not completely benign and can have significant side effects and drug interactions.62,63 Nutraceuticals may contain multiple ingredients, some of which can be toxic, particularly at higher doses. Many patients may not volunteer information about their nutraceutical use to their health care providers,64 so we must ask them about that and inform them of the potential for adverse events and drug interactions.
1. Centers for Disease Control and Prevention. Report to Congress on traumatic brain injury in the United States: epidemiology and rehabilitation. https://www.cdc.gov/traumaticbraininjury/pubs/congress_epi_rehab.html. Updated January 22, 2016. Accessed June 5, 2017.
2. Werner C, Engelhard K. Pathophysiology of traumatic brain injury. Br J Anaesth. 2007;99(1):4-9.
3. Vaishnavi S, Rao V, Fann JR. Neuropsychiatric problems after traumatic brain injury: unraveling the silent epidemic. Psychosomatics. 2009;50(3):198-205.
4. National Institutes of Health Office of Dietary Supplements. Dietary supplement fact sheets. https://ods.od.nih.gov/factsheets/list-all. Accessed June 5, 2017.
5. Institute of Medicine, Food and Nutrition Board. Dietary reference intakes for energy, carbohydrate, fiber, fat, fatty acids, cholesterol, protein, and amino acids. Washington, DC: National Academy of Sciences; 2002.
6. Rao V, Koliatsos V, Ahmed F, et al. Neuropsychiatric disturbances associated with traumatic brain injury: a practical approach to evaluation and management. Semin Neurol. 2015;35(1):64-82.
7. Chew E, Zafonte RD. Pharmacological management of neurobehavioral disorders following traumatic brain injury—a state-of-the-art review. J Rehabil Res Dev. 2009;46(6):851-879.
8. Petraglia AL, Maroon JC, Bailes JE. From the field of play to the field of combat: a review of the pharmacological management of concussion. Neurosurgery. 2012;70(6):1520-1533; discussion 1533.
9. Bengtsson M, Godbolt AK. Effects of acetylcholinesterase inhibitors on cognitive function in patients with chronic traumatic brain injury: a systematic review. J Rehabil Med. 2016;48(1):1-5.
10. Neurobehavioral Guidelines Working Group; Warden DL, Gordon B, McAllister TW, et al. Guidelines for the pharmacologic treatment of neurobehavioral sequelae of traumatic brain injury. J Neurotrauma. 2006;23(10):1468-1501.
11. DeFelice SL. The nutraceutical revolution: its impact on food industry R&D. Trends Food Sci Technol. 1995;6(2):59-61.
12. Cope EC, Morris DR, Levenson CW. Improving treatments and outcomes: an emerging role for zinc in traumatic brain injury. Nutr Rev. 2012;70(7):410-413.
13. Morris DR, Levenson CW. Zinc in traumatic brain injury: from neuroprotection to neurotoxicity. Curr Opin Clin Nutr Metab Care. 2013;16(6):708-711.
14. Young B, Ott L, Kasarskis E, et al. Zinc supplementation is associated with improved neurologic recovery rate and visceral protein levels of patients with severe closed head injury. J Neurotrauma. 1996;13(1):25-34.
15. Fernández-Gajardo R, Matamala JM, Carrasco R, et al. Novel therapeutic strategies for traumatic brain injury: acute antioxidant reinforcement. CNS Drugs. 2014;28(3):229-248.
16. Razmkon A, Sadidi A, Sherafat-Kazemzadeh E, et al. Administration of vitamin C and vitamin E in severe head injury: a randomized double-blind controlled trial. Clin Neurosurg. 2011;58:133-137.
17. Cekic M, Cutler SM, VanLandingham JW, et al. Vitamin D deficiency reduces the benefits of progesterone treatment after brain injury in aged rats. Neurobiol Aging. 2011;32(5):864-874.
18. Aminmansour B, Nikbakht H, Ghorbani A, et al. Comparison of the administration of progesterone versus progesterone and vitamin D in improvement of outcomes in patients with traumatic brain injury: a randomized clinical trial with placebo group. Adv Biomed Res. 2012;1:58.
19. Cernak I, Savic VJ, Kotur J, et al. Characterization of plasma magnesium concentration and oxidative stress following graded traumatic brain injury in humans. J Neurotrauma. 2000;17(1):53-68.
20. Dhandapani SS, Gupta A, Vivekanandhan S, et al. Randomized controlled trial of magnesium sulphate in severe closed traumatic brain injury. The Indian Journal of Neurotrauma. 2008;5(1):27-33.
21. Temkin NR, Anderson GD, Winn HR, et al. Magnesium sulfate for neuroprotection after traumatic brain injury: a randomised controlled trial. Lancet Neurol. 2007;6(1):29-38.
22. Jeter CB, Hergenroeder GW, Ward NH 3rd, et al. Human mild traumatic brain injury decreases circulating branched-chain amino acids and their metabolite levels. J Neurotrauma. 2013;30(8):671-679.
23. Cole JT, Mitala CM, Kundu S, et al. Dietary branched chain amino acids ameliorate injury-induced cognitive impairment. Proc Natl Acad Sci U S A. 2010;107(1):366-371.
24. Aquilani R, Iadarola P, Contardi A, et al. Branched-chain amino acids enhance the cognitive recovery of patients with severe traumatic brain injury. Arch Phys Med Rehabil. 2005;86(9):1729-1735.
25. Aquilani R, Boselli M, Boschi F, et al. Branched-chain amino acids may improve recovery from a vegetative or minimally conscious state in patients with traumatic brain injury: a pilot study. Arch Phys Med Rehabil. 2008;89(9):1642-1647.
26. Kang HJ, Im SH. Probiotics as an immune modulator. J Nutr Sci Vitaminol (Tokyo). 2015;61(suppl):S103-S105.
27. DiPiro JT, Howdieshell TR, Goddard JK, et al. Association of interleukin-4 plasma levels with traumatic injury and clinical course. Arch Surg. 1995;130(11):1159-1162; discussion 1162-1163.
28. Tan M, Zhu JC, Du J, et al. Effects of probiotics on serum levels of Th1/Th2 cytokine and clinical outcomes in severe traumatic brain-injured patients: a prospective randomized pilot study. Crit Care. 2011;15(6):R290.
29. Falcão de Arruda IS, de Aguilar-Nascimento JE. Benefits of early enteral nutrition with glutamine and probiotics in brain injury patients. Clin Sci (Lond). 2004;106(3):287-292.
30. Cuzzocrea S, Mazzon E, Costantino G, et al. Beneficial effects of n-acetylcysteine on ischaemic brain injury. Br J Pharmacol. 2000;130(6):1219-1226.
31. Hoffer ME, Balaban C, Slade MD, et al. Amelioration of acute sequelae of blast induced mild traumatic brain injury by N-acetyl cysteine: a double-blind, placebo controlled study. PLoS One. 2013;8(1):e54163.
32. Eakin K, Baratz-Goldstein R, Pick CG, et al. Efficacy of N-acetyl cysteine in traumatic brain injury. PLoS One. 2014;9(4):e90617.
33. Theadom A, Mahon S, Barker-Collo S, et al. Enzogenol for cognitive functioning in traumatic brain injury: a pilot placebo-controlled RCT. Eur J Neurol. 2013;20(8):1135-1144.
34. Arenth PM, Russell KC, Ricker JH, et al. CDP-choline as a biological supplement during neurorecovery: a focused review. PM R. 2011;3(6 suppl 1):S123-S131.
35. Clark WM. Efficacy of citicoline as an acute stroke treatment. Expert Opin Pharmacother. 2009;10(5):839-846.
36. Guseva MV, Hopkins DM, Scheff SW, et al. Dietary choline supplementation improves behavioral, histological, and neurochemical outcomes in a rat model of traumatic brain injury. J Neurotrauma. 2008;25(8):975-983.
37. Dixon CE, Ma X, Marion DW. Effects of CDP-choline treatment on neurobehavioral deficits after TBI and on hippocampal and neocortical acetylcholine release. J Neurotrauma. 1997;14(3):161-169.
38. Levin HS. Treatment of postconcussional symptoms with CDP-choline. J Neurol Sci. 1991;103(suppl):S39-S42.
39. Zafonte RD, Bagiella E, Ansel BM, et al. Effect of citicoline on functional and cognitive status among patients with traumatic brain injury: Citicoline Brain Injury Treatment Trial (COBRIT). JAMA. 2012;308(19):1993-2000.
40. Perry E, Walker M, Grace J, et al. Acetylcholine in mind: a neurotransmitter correlate of consciousness? Trends Neurosci. 1999;22(6):273-280.
41. Levin HS, Peters BH, Kalisky Z, et al. Effects of oral physostigmine and lecithin on memory and attention in closed head-injured patients. Cent Nerv Syst Trauma. 1986;3(4):333-342.
42. Alvarez XA, Lombardi VR, Corzo L, et al. Oral cerebrolysin enhances brain alpha activity and improves cognitive performance in elderly control subjects. J Neural Transm Suppl. 2000;59:315-328.
43. Ruether E, Husmann R, Kinzler E, et al. A 28-week, double-blind, placebo-controlled study with cerebrolysin in patients with mild to moderate Alzheimer’s disease. Int Clin Psychopharmacol. 2001;16(5):253-263.
44. Wong GK, Zhu XL, Poon WS. Beneficial effect of cerebrolysin on moderate and severe head injury patients: result of a cohort study. Acta Neurochir Suppl. 2005;95:59-60.
45. Chen CC, Wei ST, Tsaia SC, et al. Cerebrolysin enhances cognitive recovery of mild traumatic brain injury patients: double-blind, placebo-controlled, randomized study. Br J Neurosurg. 2013;27(6):803-807.
46. Ziganshina LE, Abakumova T, Vernay L. Cerebrolysin for acute ischaemic stroke. Cochrane Database Syst Rev. 2016;12:CD007026.
47. Barrett EC, McBurney MI, Ciappio ED. ω-3 fatty acid supplementation as a potential therapeutic aid for the recovery from mild traumatic brain injury/concussion. Adv Nutr. 2014;5(3):268-277.
48. Sharma S, Zhuang Y, Ying Z, et al. Dietary curcumin supplementation counteracts reduction in levels of molecules involved in energy homeostasis after brain trauma. Neuroscience. 2009;161(4):1037-1044.
49. Gatson JW, Liu MM, Abdelfattah K, et al. Resveratrol decreases inflammation in the brain of mice with mild traumatic brain injury. J Trauma Acute Care Surg. 2013;74(2):470-475; discussion 474-475.
50. Grey A, Bolland M. Clinical trial evidence and use of fish oil supplements. JAMA Intern Med. 2014;174(3):460-462.
51. Mpandzou G, Aït Ben Haddou E, Regragui W, et al. Vitamin D deficiency and its role in neurological conditions: a review. Rev Neurol (Paris). 2016;172(2):109-122.
52. Karakis I, Pase MP, Beiser A, et al. Association of serum vitamin D with the risk of incident dementia and subclinical indices of brain aging: The Framingham Heart Study. J Alzheimers Dis. 2016;51(2):451-461.
53. Sarris J, Papakostas GI, Vitolo O, et al. S-adenosyl methionine (SAMe) versus escitalopram and placebo in major depression RCT: efficacy and effects of histamine and carnitine as moderators of response. J Affect Disord. 2014;164:76-81.
54. Sarris J, Mischoulon D, Schweitzer I. Omega-3 for bipolar disorder: meta-analyses of use in mania and bipolar depression. J Clin Psychiatry. 2012;73(1):81-86.
55. Sarris J, Stough C, Bousman C, et al. Kava in the treatment of generalized anxiety disorder: a double-blind, randomized, placebo-controlled study. J Clin Psychopharmacol. 2013;33(5):643-648.
56. Hariri M, Azadbakht L. Magnesium, iron, and zinc supplementation for the treatment of attention deficit hyperactivity disorder: a systematic review on the recent literature. Int J Prev Med. 2015;6:83.
57. Gillies D, Sinn JKh, Lad SS, et al. Polyunsaturated fatty acids (PUFA) for attention deficit hyperactivity disorder (ADHD) in children and adolescents. Cochrane Database Syst Rev. 2012;7:CD007986.
58. Ghanizadeh A, Berk M. Zinc for treating of children and adolescents with attention-deficit hyperactivity disorder: a systematic review of randomized controlled clinical trials. Eur J Clin Nutr. 2013;67(1):122-124.
59. U.S. Food and Drug Administration. Can a dietary supplement treat a concussion? No! http://www.fda.gov/forconsumers/consumerupdates/ucm378845.htm. Updated February 13, 2015. Accessed June 5, 2017.
60. Sarris J, Logan AC, Akbaraly TN, et al; International Society for Nutritional Psychiatry Research. Nutritional medicine as mainstream in psychiatry. Lancet Psychiatry. 2015;2(3):271-274.
61. Desai A, Kevala K, Kim HY. Depletion of brain docosahexaenoic acid impairs recovery from traumatic brain injury. PLoS One. 2014;9(1):e86472.
62. Edie CF, Dewan N. Which psychotropics interact with four common supplements. Current Psychiatry. 2005;4(1):16-30.
63. Di Lorenzo C, Ceschi A, Kupferschmidt H, et al. Adverse effects of plant food supplements and botanical preparations: a systematic review with critical evaluation of causality. Br J Clin Pharmacol. 2015;79(4):578-592.
64. National Center for Complementary and Integrative Health. Complementary and alternative medicine: what people aged 50 and older discuss with their health care providers. https://nccih.nih.gov/research/statistics/2010. Published 2011. Accessed June 5, 2017.
Traumatic brain injury (TBI) affects more than 2 million people in the United States each year.1 TBI can trigger a cascade of secondary injury mechanisms, such as inflammation, hypoxic/ischemic injury, excitotoxicity, and oxidative stress,2 that could contribute to cognitive and behavioral changes. Although neuropsychiatric symptoms might not be obvious after a TBI, they have a high prevalence in these patients, can last long term, and may be difficult to treat.3 Despite research advances in understanding the biological basis of TBI and identifying potential therapeutic targets, treatment options for individuals with TBI remain limited.
As a result, clinicians have turned to alternative treatments for TBI, including nutraceuticals. In this article, we will:
- provide an overview of nutraceuticals used in treating TBI, first exploring outcomes soon after TBI, then concentrating on neuropsychiatric outcomes
- evaluate the existing evidence, including recommended dietary allowances (Table 1)4,5 and side effects (Table 2)
- review recommendations for their clinical use.
Pharmacologic approaches are limited
Nutraceuticals have gained attention for managing TBI-associated neuropsychiatric disorders because of the limited evidence supporting current approaches. Existing strategies encompass pharmacologic and non-pharmacologic interventions, psychoeducation, supportive and behavioral psychotherapies, and cognitive rehabilitation.6
Many pharmacologic options exist for specific neurobehavioral symptoms, but the evidence for their use is based on small studies, case reports, and knowledge extrapolated from their use in idiopathic psychiatric disorders.7,8 No FDA-approved drugs have been effective for treating neuropsychiatric disturbances after a TBI. Off-label use of antidepressants, anticonvulsants, dopaminergic agents, and cholinesterase inhibitors in TBI has been associated with inadequate clinical response and/or intolerable side effects.9,10
What are nutraceuticals?
DeFelice11 introduced the term “nutraceutical” to refer to “any substance that is a food or part of a food and provides medical or health benefits, including the prevention and treatment of disease.” The term has been expanded to include dietary supplements, such as vitamins, minerals, amino acids, herbal or other botanicals, and food products that provide health benefits beyond what they normally provide in food form. The FDA does not regulate the marketing or manufacturing of nutraceuticals; therefore, their bioavailability and metabolism can vary.
Despite their widespread use, the evidence supporting the efficacy of nutraceuticals for patients with TBI is limited. Their effects might vary by population and depend on dose, timing, TBI severity, and whether taken alone or in combination with other nutraceutical or pharmaceutical agents. Fourteen randomized controlled trials (RCTs) have addressed the use of nutraceuticals in TBI (Table 3), but further research is needed to clarify for which conditions they provide maximum benefit.
Nutraceuticals and their potential use in TBI
Zinc is considered essential for optimal CNS functioning. Patients with TBI might be at risk for zinc deficiency, which has been associated with increased cell death and behavioral deficits.12,13 A randomized, prospective, double-blinded controlled trial examined the effects of supplemental zinc administration (12 mg for 15 days) compared with standard zinc therapy (2.5 mg for 15 days) over 1 month in 68 adults with acute severe closed head injury.14 The supplemental zinc group showed improved visceral protein levels, lower mortality, and more favorable neurologic recovery based on higher adjusted mean Glasgow Coma Scale score on day 28 and mean motor score on days 15 and 21.
Rodent studies have shown that zinc supplementation could reduce deficits in spatial learning and memory and depression-like behaviors and help decrease stress and anxiety,12 although no human clinical trials have been conducted. Despite the potential neuroprotective effects of zinc supplementation, evidence exists that endogenous zinc release and accumulation following TBI can trigger cellular changes that result in neuronal death.13
Vitamins C and E. Oxidative damage is believed to play a significant role in secondary injury in TBI, so research has focused on the role of antioxidants, such as vitamins C and E, to promote post-TBI recovery.15 One RCT16 of 100 adults with acute severe head injury reported that vitamin E administration was associated with reduced mortality and lower Glasgow Outcome Scale (GOS) scores, and vitamin C was associated with stabilized or reduced perilesional edema/infarct on CT scan.
Vitamin D. An animal study reported that vitamin D supplementation can help reduce inflammation, oxidative stress, and cell death in TBI, and that vitamin D deficiency has been associated with increased inflammation and behavioral deficits.17 Further evidence suggests that vitamin D may have a synergistic effect when used in combination with the hormone progesterone. A RCT of 60 patients with severe TBI reported that 60% of those who received progesterone plus vitamin D had GOS scores of 4 (good recovery) or 5 (moderate disability) vs 45% receiving progesterone alone or 25% receiving placebo.18
Magnesium, one of the most widely used nutraceuticals, is considered essential for CNS functioning, including the regulation of N-methyl-
A RCT evaluated the safety and efficacy of magnesium supplementation in 60 patients with severe closed TBI, with one-half randomized to standard care and the other also receiving magnesium sulfate (MgSO4; initiation dose of 4 g IV and 10 g IM, continuation dose of 5 g IM every 4 hours for 24 hours).20 After 3 months, more patients in the MgSO4 group had higher GOS scores than controls (73.3% vs 40%), lower 1-month mortality rates (13.3% vs 43.3%), and lower rates of intraoperative brain swelling (29.4% vs 73.3%).
However, a larger RCT of 499 patients with moderate or severe TBI randomized to high-dose (1.25 to 2.5 mmol/L) or low-dose (1.0 to 1.85 mmol/L) IV MgSO4 or placebo provided conflicting results.21 Participants received MgSO4 8 hours after injury and continued for 5 days. After 6 months, patients in the high-dose MgSO4 and placebo groups had similar composite primary outcome measures (eg, seizures, neuropsychological measures, functional status measures), although the high-dose group had a higher mortality rate than the placebo group. Patients who received low-dose MgSO4 showed worse outcomes than those assigned to placebo.
Amino acids. Branched-chain amino acids (BCAAs), including valine, isoleucine, and leucine, are essential in protein and neurotransmitter synthesis. Reduced levels of endogenous BCAAs have been reported in patients with mild or severe TBI.22 Preclinical studies suggest that BCAAs can improve hippocampal-dependent cognitive functioning following TBI.23
Two RCTs of BCAAs have been conducted in humans. One study randomized 40 men with severe TBI to IV BCAAs or placebo.24 After 15 days, the BCAA group showed greater improvement in Disability Rating Scale scores. The study also found that supplementation increased total BCAA levels without negatively affecting plasma levels of neurotransmitter precursors tyrosine and tryptophan. A second study found that 41 patients in a vegetative or minimally conscious state who received BCAA supplementation for 15 days had higher Disability Rating Scale scores than those receiving placebo.25
Probiotics and glutamine. Probiotics are non-pathogenic microorganisms that have been shown to modulate the host’s immune system.26 TBI is associated with immunological changes, including a shift from T-helper type 1 (TH1) cells to T-helper type 2 (TH2) cells that increase susceptibility to infection.27
A RCT of 52 patients with severe TBI suggested a correlation between probiotic administration-modulated cytokine levels and TH1/TH2 balance.28 A 3-times daily probiotic mix of Bifidobacterium longum, Lactobacillus bulgaricus, and Streptococcus thermophilus for 21 days led to shorter average ICU stays (6.8 vs 10.7 days, P = .034) and a decrease in nosocomial infections (34.6% vs 57.7%, P = .095) vs placebo, although the latter difference was not statistically significant.28
A prospective RCT of 20 patients with brain injury29 found a similar impact of early enteral nutrition supplemented with Lactobacillus johnsonii and glutamine, 30 g, vs a standard enteral nutrition formula. The treatment group experienced fewer nosocomial infections (50% vs 100%, P = .03), shorter ICU stays (10 vs 22 days, P < .01), and fewer days on mechanical ventilation (7 vs 14, P = .04). Despite these studies, evidence for the use of glutamine in patients with TBI is scarce and inconclusive.
N-acetylcysteine (NAC) comes from the amino acid L-cysteine. NAC is an effective scavenger of free radicals and improves cerebral microcirculatory blood flow and tissue oxygenation.30 A randomized, double-blind, placebo-controlled study of oral NAC supplementation in 81 active duty service members with mild TBI found NAC had a significant effect on outcomes.31 Oral NAC supplementation led to improved neuropsychological test results, number of mild TBI symptoms, complete symptom resolution by day 7 of treatment compared with placebo, and NAC was well tolerated. Lack of replication studies and generalizability of findings to civilian, moderate, or chronic TBI populations are key limitations of this study.
Proposed mechanisms for the neuroprotective benefit of NAC include its antioxidant and inflammatory activation of cysteine/glutamate exchange, metabotropic glutamate receptor modulation, and glutathione synthesis.32 NAC has poor blood–brain permeability, but the vascular disruption seen in acute TBI might facilitate its delivery to affected neural sites.31 As such, the benefits of NAC in subacute or chronic TBI are questionable.
Neuropsychiatric outcomes of nutraceuticals
Enzogenol. This flavonoid-rich extract from the bark of the Monterey pine tree (Pinus radiata), known by the trade name Enzogenol, reportedly has antioxidant and anti-inflammatory properties that may counter oxidative damage and neuroinflammation following TBI. A phase II trial randomized participants to Enzogenol, 1,000 mg/d, or placebo for 6 weeks, then all participants received Enzogenol for 6 weeks followed by placebo for 4 weeks.33 Enzogenol was well tolerated with few side effects.
Compared with placebo, participants receiving Enzogenol showed no significant change in mood, as measured by the Hospital Anxiety and Depression Scale, and greater improvement in overall cognition as assessed by the Cognitive Failures Questionnaire. However, measures of working memory (digit span, arithmetic, and letter–number sequencing subtests of the Wechsler Adult Intelligence Scale) and episodic memory (California Verbal Learning Test) showed no benefit from Enzogenol.
Citicoline (CDP-choline) is an endogenous compound widely available as a nutraceutical that has been approved for TBI therapy in 59 countries.34 Animal studies indicate that it could possess neuroprotective properties. Proposed mechanisms for such effects have included stabilizing cell membranes, reducing inflammation, reducing the presence of free radicals, or stimulating production of acetylcholine.35,36 A study in rats found that CDP-choline was associated with increased levels of acetylcholine in the hippocampus and neocortex, which may help reduce neurobehavioral deficits.37
A study of 14 adults with mild to moderate closed head injury38 found that patients who received CDP-choline showed a greater reduction in post-concussion symptoms and improvement in recognition memory than controls who received placebo. However, the Citicoline Brain Injury Treatment Trial, a large randomized trial of 1,213 adults with complicated mild, moderate, or severe TBI, reported that CDP-choline did not improve functional and cognitive status.39
Physostigmine and lecithin. The cholinergic system is a key modulatory neurotransmitter system of the brain that mediates conscious awareness, attention, learning, and working memory.40 A double-blind, placebo-controlled study of 16 patients with moderate to severe closed head injury provided inconsistent evidence for the efficacy of physostigmine and lecithin in the treatment of memory and attention disturbances.41The results showed no differences between the physostigmine–lecithin combination vs lecithin alone, although sustained attention on the Continuous Performance Test was more efficient with physostigmine than placebo when the drug condition occurred first in the crossover design. The lack of encouraging data and concerns about its cardiovascular and proconvulsant properties in patients with TBI may explain the dearth of studies with physostigmine.
Cerebrolysin. A peptide preparation produced from purified pig brain proteins, known by the trade name Cerebrolysin, is popular in Asia and Europe for its nootropic properties. Cerebrolysin may activate cerebral mechanisms related to attention and memory processes,42 and some data have shown efficacy in improving cognitive symptoms and daily activities in patients with Alzheimer’s disease43 and TBI.44
A blinded 12-week study of 32 participants with acute mild TBI reported that those randomized to Cerebrolysin showed improvement in cognitive functioning vs the placebo group.45 The authors concluded that Cerebrolysin provides an advantage for patients with mild TBI and brain contusion if treatment starts within 24 hours of mild TBI onset. Cerebrolysin was well tolerated. Major limitations of this study were small sample size, lack of information regarding comorbid neuropsychiatric conditions and treatments, and short treatment duration.
A recent Cochrane review of 6 RCTs with 1,501 participants found no clinical benefit of Cerebrolysin for treating acute ischemic stroke, and found moderate-quality evidence of an increase with non-fatal serious adverse events but not in total serious adverse events.46 We do not recommend Cerebrolysin use in patients with TBI at this time until additional efficacy and safety data are available.
Nutraceuticals used in other populations
Other nutraceuticals with preclinical evidence of possible benefit in TBI but lacking evidence from human clinical trials include omega-3 fatty acids,47 curcumin,48 and resveratrol,49 providing further proof that results from experimental studies do not necessarily extend to clinical trials.50
Studies of nutraceuticals in other neurological and psychiatric populations have yielded some promising results. Significant interest has focused on the association between vitamin D deficiency, dementia, and neurodegenerative conditions such as Alzheimer’s disease, multiple sclerosis, and Parkinson’s disease.51 The role of vitamin D in regulation of calcium-mediated neuronal excitotoxicity and oxidative stress and in the induction of synaptic structural proteins, neurotrophic factors, and deficient neurotransmitters makes it an attractive candidate as a neuroprotective agent.52
RCTs of nutraceuticals also have reported positive findings for a variety of mood and anxiety disorders, such as St. John’s wort, S-adenosylmethionine, omega-3 fatty acids for major depression53 and bipolar depression,54 and kava for generalized anxiety disorder.55 More research, however, is needed in these areas.
The use of nonpharmacologic agents in TBI often relies on similar neuropsychiatric symptom profiles of idiopathic psychiatric disorders. Attention-deficit/hyperactivity disorder (ADHD) closely resembles TBI, but systemic reviews of studies of zinc, magnesium, and polyunsaturated fatty acids supplementation in ADHD provide no evidence of therapeutic benefit.56-58
Educate patients in role of nutraceuticals
Despite lack of FDA oversight and limited empirical support, nutraceuticals continue to be widely marketed and used for their putative health benefits59 and have gained increased attention among clinicians.60 Because nutritional deficiency may make the brain less able than other organs to recover from injury,61 supplementation is an option, especially in individuals who could be at greater risk of TBI (eg, athletes and military personnel).
Lacking robust scientific evidence to support the use of nutraceuticals either for enhancing TBI recovery or treating neuropsychiatric disturbances, clinicians must educate patients that these agents are not completely benign and can have significant side effects and drug interactions.62,63 Nutraceuticals may contain multiple ingredients, some of which can be toxic, particularly at higher doses. Many patients may not volunteer information about their nutraceutical use to their health care providers,64 so we must ask them about that and inform them of the potential for adverse events and drug interactions.
Traumatic brain injury (TBI) affects more than 2 million people in the United States each year.1 TBI can trigger a cascade of secondary injury mechanisms, such as inflammation, hypoxic/ischemic injury, excitotoxicity, and oxidative stress,2 that could contribute to cognitive and behavioral changes. Although neuropsychiatric symptoms might not be obvious after a TBI, they have a high prevalence in these patients, can last long term, and may be difficult to treat.3 Despite research advances in understanding the biological basis of TBI and identifying potential therapeutic targets, treatment options for individuals with TBI remain limited.
As a result, clinicians have turned to alternative treatments for TBI, including nutraceuticals. In this article, we will:
- provide an overview of nutraceuticals used in treating TBI, first exploring outcomes soon after TBI, then concentrating on neuropsychiatric outcomes
- evaluate the existing evidence, including recommended dietary allowances (Table 1)4,5 and side effects (Table 2)
- review recommendations for their clinical use.
Pharmacologic approaches are limited
Nutraceuticals have gained attention for managing TBI-associated neuropsychiatric disorders because of the limited evidence supporting current approaches. Existing strategies encompass pharmacologic and non-pharmacologic interventions, psychoeducation, supportive and behavioral psychotherapies, and cognitive rehabilitation.6
Many pharmacologic options exist for specific neurobehavioral symptoms, but the evidence for their use is based on small studies, case reports, and knowledge extrapolated from their use in idiopathic psychiatric disorders.7,8 No FDA-approved drugs have been effective for treating neuropsychiatric disturbances after a TBI. Off-label use of antidepressants, anticonvulsants, dopaminergic agents, and cholinesterase inhibitors in TBI has been associated with inadequate clinical response and/or intolerable side effects.9,10
What are nutraceuticals?
DeFelice11 introduced the term “nutraceutical” to refer to “any substance that is a food or part of a food and provides medical or health benefits, including the prevention and treatment of disease.” The term has been expanded to include dietary supplements, such as vitamins, minerals, amino acids, herbal or other botanicals, and food products that provide health benefits beyond what they normally provide in food form. The FDA does not regulate the marketing or manufacturing of nutraceuticals; therefore, their bioavailability and metabolism can vary.
Despite their widespread use, the evidence supporting the efficacy of nutraceuticals for patients with TBI is limited. Their effects might vary by population and depend on dose, timing, TBI severity, and whether taken alone or in combination with other nutraceutical or pharmaceutical agents. Fourteen randomized controlled trials (RCTs) have addressed the use of nutraceuticals in TBI (Table 3), but further research is needed to clarify for which conditions they provide maximum benefit.
Nutraceuticals and their potential use in TBI
Zinc is considered essential for optimal CNS functioning. Patients with TBI might be at risk for zinc deficiency, which has been associated with increased cell death and behavioral deficits.12,13 A randomized, prospective, double-blinded controlled trial examined the effects of supplemental zinc administration (12 mg for 15 days) compared with standard zinc therapy (2.5 mg for 15 days) over 1 month in 68 adults with acute severe closed head injury.14 The supplemental zinc group showed improved visceral protein levels, lower mortality, and more favorable neurologic recovery based on higher adjusted mean Glasgow Coma Scale score on day 28 and mean motor score on days 15 and 21.
Rodent studies have shown that zinc supplementation could reduce deficits in spatial learning and memory and depression-like behaviors and help decrease stress and anxiety,12 although no human clinical trials have been conducted. Despite the potential neuroprotective effects of zinc supplementation, evidence exists that endogenous zinc release and accumulation following TBI can trigger cellular changes that result in neuronal death.13
Vitamins C and E. Oxidative damage is believed to play a significant role in secondary injury in TBI, so research has focused on the role of antioxidants, such as vitamins C and E, to promote post-TBI recovery.15 One RCT16 of 100 adults with acute severe head injury reported that vitamin E administration was associated with reduced mortality and lower Glasgow Outcome Scale (GOS) scores, and vitamin C was associated with stabilized or reduced perilesional edema/infarct on CT scan.
Vitamin D. An animal study reported that vitamin D supplementation can help reduce inflammation, oxidative stress, and cell death in TBI, and that vitamin D deficiency has been associated with increased inflammation and behavioral deficits.17 Further evidence suggests that vitamin D may have a synergistic effect when used in combination with the hormone progesterone. A RCT of 60 patients with severe TBI reported that 60% of those who received progesterone plus vitamin D had GOS scores of 4 (good recovery) or 5 (moderate disability) vs 45% receiving progesterone alone or 25% receiving placebo.18
Magnesium, one of the most widely used nutraceuticals, is considered essential for CNS functioning, including the regulation of N-methyl-
A RCT evaluated the safety and efficacy of magnesium supplementation in 60 patients with severe closed TBI, with one-half randomized to standard care and the other also receiving magnesium sulfate (MgSO4; initiation dose of 4 g IV and 10 g IM, continuation dose of 5 g IM every 4 hours for 24 hours).20 After 3 months, more patients in the MgSO4 group had higher GOS scores than controls (73.3% vs 40%), lower 1-month mortality rates (13.3% vs 43.3%), and lower rates of intraoperative brain swelling (29.4% vs 73.3%).
However, a larger RCT of 499 patients with moderate or severe TBI randomized to high-dose (1.25 to 2.5 mmol/L) or low-dose (1.0 to 1.85 mmol/L) IV MgSO4 or placebo provided conflicting results.21 Participants received MgSO4 8 hours after injury and continued for 5 days. After 6 months, patients in the high-dose MgSO4 and placebo groups had similar composite primary outcome measures (eg, seizures, neuropsychological measures, functional status measures), although the high-dose group had a higher mortality rate than the placebo group. Patients who received low-dose MgSO4 showed worse outcomes than those assigned to placebo.
Amino acids. Branched-chain amino acids (BCAAs), including valine, isoleucine, and leucine, are essential in protein and neurotransmitter synthesis. Reduced levels of endogenous BCAAs have been reported in patients with mild or severe TBI.22 Preclinical studies suggest that BCAAs can improve hippocampal-dependent cognitive functioning following TBI.23
Two RCTs of BCAAs have been conducted in humans. One study randomized 40 men with severe TBI to IV BCAAs or placebo.24 After 15 days, the BCAA group showed greater improvement in Disability Rating Scale scores. The study also found that supplementation increased total BCAA levels without negatively affecting plasma levels of neurotransmitter precursors tyrosine and tryptophan. A second study found that 41 patients in a vegetative or minimally conscious state who received BCAA supplementation for 15 days had higher Disability Rating Scale scores than those receiving placebo.25
Probiotics and glutamine. Probiotics are non-pathogenic microorganisms that have been shown to modulate the host’s immune system.26 TBI is associated with immunological changes, including a shift from T-helper type 1 (TH1) cells to T-helper type 2 (TH2) cells that increase susceptibility to infection.27
A RCT of 52 patients with severe TBI suggested a correlation between probiotic administration-modulated cytokine levels and TH1/TH2 balance.28 A 3-times daily probiotic mix of Bifidobacterium longum, Lactobacillus bulgaricus, and Streptococcus thermophilus for 21 days led to shorter average ICU stays (6.8 vs 10.7 days, P = .034) and a decrease in nosocomial infections (34.6% vs 57.7%, P = .095) vs placebo, although the latter difference was not statistically significant.28
A prospective RCT of 20 patients with brain injury29 found a similar impact of early enteral nutrition supplemented with Lactobacillus johnsonii and glutamine, 30 g, vs a standard enteral nutrition formula. The treatment group experienced fewer nosocomial infections (50% vs 100%, P = .03), shorter ICU stays (10 vs 22 days, P < .01), and fewer days on mechanical ventilation (7 vs 14, P = .04). Despite these studies, evidence for the use of glutamine in patients with TBI is scarce and inconclusive.
N-acetylcysteine (NAC) comes from the amino acid L-cysteine. NAC is an effective scavenger of free radicals and improves cerebral microcirculatory blood flow and tissue oxygenation.30 A randomized, double-blind, placebo-controlled study of oral NAC supplementation in 81 active duty service members with mild TBI found NAC had a significant effect on outcomes.31 Oral NAC supplementation led to improved neuropsychological test results, number of mild TBI symptoms, complete symptom resolution by day 7 of treatment compared with placebo, and NAC was well tolerated. Lack of replication studies and generalizability of findings to civilian, moderate, or chronic TBI populations are key limitations of this study.
Proposed mechanisms for the neuroprotective benefit of NAC include its antioxidant and inflammatory activation of cysteine/glutamate exchange, metabotropic glutamate receptor modulation, and glutathione synthesis.32 NAC has poor blood–brain permeability, but the vascular disruption seen in acute TBI might facilitate its delivery to affected neural sites.31 As such, the benefits of NAC in subacute or chronic TBI are questionable.
Neuropsychiatric outcomes of nutraceuticals
Enzogenol. This flavonoid-rich extract from the bark of the Monterey pine tree (Pinus radiata), known by the trade name Enzogenol, reportedly has antioxidant and anti-inflammatory properties that may counter oxidative damage and neuroinflammation following TBI. A phase II trial randomized participants to Enzogenol, 1,000 mg/d, or placebo for 6 weeks, then all participants received Enzogenol for 6 weeks followed by placebo for 4 weeks.33 Enzogenol was well tolerated with few side effects.
Compared with placebo, participants receiving Enzogenol showed no significant change in mood, as measured by the Hospital Anxiety and Depression Scale, and greater improvement in overall cognition as assessed by the Cognitive Failures Questionnaire. However, measures of working memory (digit span, arithmetic, and letter–number sequencing subtests of the Wechsler Adult Intelligence Scale) and episodic memory (California Verbal Learning Test) showed no benefit from Enzogenol.
Citicoline (CDP-choline) is an endogenous compound widely available as a nutraceutical that has been approved for TBI therapy in 59 countries.34 Animal studies indicate that it could possess neuroprotective properties. Proposed mechanisms for such effects have included stabilizing cell membranes, reducing inflammation, reducing the presence of free radicals, or stimulating production of acetylcholine.35,36 A study in rats found that CDP-choline was associated with increased levels of acetylcholine in the hippocampus and neocortex, which may help reduce neurobehavioral deficits.37
A study of 14 adults with mild to moderate closed head injury38 found that patients who received CDP-choline showed a greater reduction in post-concussion symptoms and improvement in recognition memory than controls who received placebo. However, the Citicoline Brain Injury Treatment Trial, a large randomized trial of 1,213 adults with complicated mild, moderate, or severe TBI, reported that CDP-choline did not improve functional and cognitive status.39
Physostigmine and lecithin. The cholinergic system is a key modulatory neurotransmitter system of the brain that mediates conscious awareness, attention, learning, and working memory.40 A double-blind, placebo-controlled study of 16 patients with moderate to severe closed head injury provided inconsistent evidence for the efficacy of physostigmine and lecithin in the treatment of memory and attention disturbances.41The results showed no differences between the physostigmine–lecithin combination vs lecithin alone, although sustained attention on the Continuous Performance Test was more efficient with physostigmine than placebo when the drug condition occurred first in the crossover design. The lack of encouraging data and concerns about its cardiovascular and proconvulsant properties in patients with TBI may explain the dearth of studies with physostigmine.
Cerebrolysin. A peptide preparation produced from purified pig brain proteins, known by the trade name Cerebrolysin, is popular in Asia and Europe for its nootropic properties. Cerebrolysin may activate cerebral mechanisms related to attention and memory processes,42 and some data have shown efficacy in improving cognitive symptoms and daily activities in patients with Alzheimer’s disease43 and TBI.44
A blinded 12-week study of 32 participants with acute mild TBI reported that those randomized to Cerebrolysin showed improvement in cognitive functioning vs the placebo group.45 The authors concluded that Cerebrolysin provides an advantage for patients with mild TBI and brain contusion if treatment starts within 24 hours of mild TBI onset. Cerebrolysin was well tolerated. Major limitations of this study were small sample size, lack of information regarding comorbid neuropsychiatric conditions and treatments, and short treatment duration.
A recent Cochrane review of 6 RCTs with 1,501 participants found no clinical benefit of Cerebrolysin for treating acute ischemic stroke, and found moderate-quality evidence of an increase with non-fatal serious adverse events but not in total serious adverse events.46 We do not recommend Cerebrolysin use in patients with TBI at this time until additional efficacy and safety data are available.
Nutraceuticals used in other populations
Other nutraceuticals with preclinical evidence of possible benefit in TBI but lacking evidence from human clinical trials include omega-3 fatty acids,47 curcumin,48 and resveratrol,49 providing further proof that results from experimental studies do not necessarily extend to clinical trials.50
Studies of nutraceuticals in other neurological and psychiatric populations have yielded some promising results. Significant interest has focused on the association between vitamin D deficiency, dementia, and neurodegenerative conditions such as Alzheimer’s disease, multiple sclerosis, and Parkinson’s disease.51 The role of vitamin D in regulation of calcium-mediated neuronal excitotoxicity and oxidative stress and in the induction of synaptic structural proteins, neurotrophic factors, and deficient neurotransmitters makes it an attractive candidate as a neuroprotective agent.52
RCTs of nutraceuticals also have reported positive findings for a variety of mood and anxiety disorders, such as St. John’s wort, S-adenosylmethionine, omega-3 fatty acids for major depression53 and bipolar depression,54 and kava for generalized anxiety disorder.55 More research, however, is needed in these areas.
The use of nonpharmacologic agents in TBI often relies on similar neuropsychiatric symptom profiles of idiopathic psychiatric disorders. Attention-deficit/hyperactivity disorder (ADHD) closely resembles TBI, but systemic reviews of studies of zinc, magnesium, and polyunsaturated fatty acids supplementation in ADHD provide no evidence of therapeutic benefit.56-58
Educate patients in role of nutraceuticals
Despite lack of FDA oversight and limited empirical support, nutraceuticals continue to be widely marketed and used for their putative health benefits59 and have gained increased attention among clinicians.60 Because nutritional deficiency may make the brain less able than other organs to recover from injury,61 supplementation is an option, especially in individuals who could be at greater risk of TBI (eg, athletes and military personnel).
Lacking robust scientific evidence to support the use of nutraceuticals either for enhancing TBI recovery or treating neuropsychiatric disturbances, clinicians must educate patients that these agents are not completely benign and can have significant side effects and drug interactions.62,63 Nutraceuticals may contain multiple ingredients, some of which can be toxic, particularly at higher doses. Many patients may not volunteer information about their nutraceutical use to their health care providers,64 so we must ask them about that and inform them of the potential for adverse events and drug interactions.
1. Centers for Disease Control and Prevention. Report to Congress on traumatic brain injury in the United States: epidemiology and rehabilitation. https://www.cdc.gov/traumaticbraininjury/pubs/congress_epi_rehab.html. Updated January 22, 2016. Accessed June 5, 2017.
2. Werner C, Engelhard K. Pathophysiology of traumatic brain injury. Br J Anaesth. 2007;99(1):4-9.
3. Vaishnavi S, Rao V, Fann JR. Neuropsychiatric problems after traumatic brain injury: unraveling the silent epidemic. Psychosomatics. 2009;50(3):198-205.
4. National Institutes of Health Office of Dietary Supplements. Dietary supplement fact sheets. https://ods.od.nih.gov/factsheets/list-all. Accessed June 5, 2017.
5. Institute of Medicine, Food and Nutrition Board. Dietary reference intakes for energy, carbohydrate, fiber, fat, fatty acids, cholesterol, protein, and amino acids. Washington, DC: National Academy of Sciences; 2002.
6. Rao V, Koliatsos V, Ahmed F, et al. Neuropsychiatric disturbances associated with traumatic brain injury: a practical approach to evaluation and management. Semin Neurol. 2015;35(1):64-82.
7. Chew E, Zafonte RD. Pharmacological management of neurobehavioral disorders following traumatic brain injury—a state-of-the-art review. J Rehabil Res Dev. 2009;46(6):851-879.
8. Petraglia AL, Maroon JC, Bailes JE. From the field of play to the field of combat: a review of the pharmacological management of concussion. Neurosurgery. 2012;70(6):1520-1533; discussion 1533.
9. Bengtsson M, Godbolt AK. Effects of acetylcholinesterase inhibitors on cognitive function in patients with chronic traumatic brain injury: a systematic review. J Rehabil Med. 2016;48(1):1-5.
10. Neurobehavioral Guidelines Working Group; Warden DL, Gordon B, McAllister TW, et al. Guidelines for the pharmacologic treatment of neurobehavioral sequelae of traumatic brain injury. J Neurotrauma. 2006;23(10):1468-1501.
11. DeFelice SL. The nutraceutical revolution: its impact on food industry R&D. Trends Food Sci Technol. 1995;6(2):59-61.
12. Cope EC, Morris DR, Levenson CW. Improving treatments and outcomes: an emerging role for zinc in traumatic brain injury. Nutr Rev. 2012;70(7):410-413.
13. Morris DR, Levenson CW. Zinc in traumatic brain injury: from neuroprotection to neurotoxicity. Curr Opin Clin Nutr Metab Care. 2013;16(6):708-711.
14. Young B, Ott L, Kasarskis E, et al. Zinc supplementation is associated with improved neurologic recovery rate and visceral protein levels of patients with severe closed head injury. J Neurotrauma. 1996;13(1):25-34.
15. Fernández-Gajardo R, Matamala JM, Carrasco R, et al. Novel therapeutic strategies for traumatic brain injury: acute antioxidant reinforcement. CNS Drugs. 2014;28(3):229-248.
16. Razmkon A, Sadidi A, Sherafat-Kazemzadeh E, et al. Administration of vitamin C and vitamin E in severe head injury: a randomized double-blind controlled trial. Clin Neurosurg. 2011;58:133-137.
17. Cekic M, Cutler SM, VanLandingham JW, et al. Vitamin D deficiency reduces the benefits of progesterone treatment after brain injury in aged rats. Neurobiol Aging. 2011;32(5):864-874.
18. Aminmansour B, Nikbakht H, Ghorbani A, et al. Comparison of the administration of progesterone versus progesterone and vitamin D in improvement of outcomes in patients with traumatic brain injury: a randomized clinical trial with placebo group. Adv Biomed Res. 2012;1:58.
19. Cernak I, Savic VJ, Kotur J, et al. Characterization of plasma magnesium concentration and oxidative stress following graded traumatic brain injury in humans. J Neurotrauma. 2000;17(1):53-68.
20. Dhandapani SS, Gupta A, Vivekanandhan S, et al. Randomized controlled trial of magnesium sulphate in severe closed traumatic brain injury. The Indian Journal of Neurotrauma. 2008;5(1):27-33.
21. Temkin NR, Anderson GD, Winn HR, et al. Magnesium sulfate for neuroprotection after traumatic brain injury: a randomised controlled trial. Lancet Neurol. 2007;6(1):29-38.
22. Jeter CB, Hergenroeder GW, Ward NH 3rd, et al. Human mild traumatic brain injury decreases circulating branched-chain amino acids and their metabolite levels. J Neurotrauma. 2013;30(8):671-679.
23. Cole JT, Mitala CM, Kundu S, et al. Dietary branched chain amino acids ameliorate injury-induced cognitive impairment. Proc Natl Acad Sci U S A. 2010;107(1):366-371.
24. Aquilani R, Iadarola P, Contardi A, et al. Branched-chain amino acids enhance the cognitive recovery of patients with severe traumatic brain injury. Arch Phys Med Rehabil. 2005;86(9):1729-1735.
25. Aquilani R, Boselli M, Boschi F, et al. Branched-chain amino acids may improve recovery from a vegetative or minimally conscious state in patients with traumatic brain injury: a pilot study. Arch Phys Med Rehabil. 2008;89(9):1642-1647.
26. Kang HJ, Im SH. Probiotics as an immune modulator. J Nutr Sci Vitaminol (Tokyo). 2015;61(suppl):S103-S105.
27. DiPiro JT, Howdieshell TR, Goddard JK, et al. Association of interleukin-4 plasma levels with traumatic injury and clinical course. Arch Surg. 1995;130(11):1159-1162; discussion 1162-1163.
28. Tan M, Zhu JC, Du J, et al. Effects of probiotics on serum levels of Th1/Th2 cytokine and clinical outcomes in severe traumatic brain-injured patients: a prospective randomized pilot study. Crit Care. 2011;15(6):R290.
29. Falcão de Arruda IS, de Aguilar-Nascimento JE. Benefits of early enteral nutrition with glutamine and probiotics in brain injury patients. Clin Sci (Lond). 2004;106(3):287-292.
30. Cuzzocrea S, Mazzon E, Costantino G, et al. Beneficial effects of n-acetylcysteine on ischaemic brain injury. Br J Pharmacol. 2000;130(6):1219-1226.
31. Hoffer ME, Balaban C, Slade MD, et al. Amelioration of acute sequelae of blast induced mild traumatic brain injury by N-acetyl cysteine: a double-blind, placebo controlled study. PLoS One. 2013;8(1):e54163.
32. Eakin K, Baratz-Goldstein R, Pick CG, et al. Efficacy of N-acetyl cysteine in traumatic brain injury. PLoS One. 2014;9(4):e90617.
33. Theadom A, Mahon S, Barker-Collo S, et al. Enzogenol for cognitive functioning in traumatic brain injury: a pilot placebo-controlled RCT. Eur J Neurol. 2013;20(8):1135-1144.
34. Arenth PM, Russell KC, Ricker JH, et al. CDP-choline as a biological supplement during neurorecovery: a focused review. PM R. 2011;3(6 suppl 1):S123-S131.
35. Clark WM. Efficacy of citicoline as an acute stroke treatment. Expert Opin Pharmacother. 2009;10(5):839-846.
36. Guseva MV, Hopkins DM, Scheff SW, et al. Dietary choline supplementation improves behavioral, histological, and neurochemical outcomes in a rat model of traumatic brain injury. J Neurotrauma. 2008;25(8):975-983.
37. Dixon CE, Ma X, Marion DW. Effects of CDP-choline treatment on neurobehavioral deficits after TBI and on hippocampal and neocortical acetylcholine release. J Neurotrauma. 1997;14(3):161-169.
38. Levin HS. Treatment of postconcussional symptoms with CDP-choline. J Neurol Sci. 1991;103(suppl):S39-S42.
39. Zafonte RD, Bagiella E, Ansel BM, et al. Effect of citicoline on functional and cognitive status among patients with traumatic brain injury: Citicoline Brain Injury Treatment Trial (COBRIT). JAMA. 2012;308(19):1993-2000.
40. Perry E, Walker M, Grace J, et al. Acetylcholine in mind: a neurotransmitter correlate of consciousness? Trends Neurosci. 1999;22(6):273-280.
41. Levin HS, Peters BH, Kalisky Z, et al. Effects of oral physostigmine and lecithin on memory and attention in closed head-injured patients. Cent Nerv Syst Trauma. 1986;3(4):333-342.
42. Alvarez XA, Lombardi VR, Corzo L, et al. Oral cerebrolysin enhances brain alpha activity and improves cognitive performance in elderly control subjects. J Neural Transm Suppl. 2000;59:315-328.
43. Ruether E, Husmann R, Kinzler E, et al. A 28-week, double-blind, placebo-controlled study with cerebrolysin in patients with mild to moderate Alzheimer’s disease. Int Clin Psychopharmacol. 2001;16(5):253-263.
44. Wong GK, Zhu XL, Poon WS. Beneficial effect of cerebrolysin on moderate and severe head injury patients: result of a cohort study. Acta Neurochir Suppl. 2005;95:59-60.
45. Chen CC, Wei ST, Tsaia SC, et al. Cerebrolysin enhances cognitive recovery of mild traumatic brain injury patients: double-blind, placebo-controlled, randomized study. Br J Neurosurg. 2013;27(6):803-807.
46. Ziganshina LE, Abakumova T, Vernay L. Cerebrolysin for acute ischaemic stroke. Cochrane Database Syst Rev. 2016;12:CD007026.
47. Barrett EC, McBurney MI, Ciappio ED. ω-3 fatty acid supplementation as a potential therapeutic aid for the recovery from mild traumatic brain injury/concussion. Adv Nutr. 2014;5(3):268-277.
48. Sharma S, Zhuang Y, Ying Z, et al. Dietary curcumin supplementation counteracts reduction in levels of molecules involved in energy homeostasis after brain trauma. Neuroscience. 2009;161(4):1037-1044.
49. Gatson JW, Liu MM, Abdelfattah K, et al. Resveratrol decreases inflammation in the brain of mice with mild traumatic brain injury. J Trauma Acute Care Surg. 2013;74(2):470-475; discussion 474-475.
50. Grey A, Bolland M. Clinical trial evidence and use of fish oil supplements. JAMA Intern Med. 2014;174(3):460-462.
51. Mpandzou G, Aït Ben Haddou E, Regragui W, et al. Vitamin D deficiency and its role in neurological conditions: a review. Rev Neurol (Paris). 2016;172(2):109-122.
52. Karakis I, Pase MP, Beiser A, et al. Association of serum vitamin D with the risk of incident dementia and subclinical indices of brain aging: The Framingham Heart Study. J Alzheimers Dis. 2016;51(2):451-461.
53. Sarris J, Papakostas GI, Vitolo O, et al. S-adenosyl methionine (SAMe) versus escitalopram and placebo in major depression RCT: efficacy and effects of histamine and carnitine as moderators of response. J Affect Disord. 2014;164:76-81.
54. Sarris J, Mischoulon D, Schweitzer I. Omega-3 for bipolar disorder: meta-analyses of use in mania and bipolar depression. J Clin Psychiatry. 2012;73(1):81-86.
55. Sarris J, Stough C, Bousman C, et al. Kava in the treatment of generalized anxiety disorder: a double-blind, randomized, placebo-controlled study. J Clin Psychopharmacol. 2013;33(5):643-648.
56. Hariri M, Azadbakht L. Magnesium, iron, and zinc supplementation for the treatment of attention deficit hyperactivity disorder: a systematic review on the recent literature. Int J Prev Med. 2015;6:83.
57. Gillies D, Sinn JKh, Lad SS, et al. Polyunsaturated fatty acids (PUFA) for attention deficit hyperactivity disorder (ADHD) in children and adolescents. Cochrane Database Syst Rev. 2012;7:CD007986.
58. Ghanizadeh A, Berk M. Zinc for treating of children and adolescents with attention-deficit hyperactivity disorder: a systematic review of randomized controlled clinical trials. Eur J Clin Nutr. 2013;67(1):122-124.
59. U.S. Food and Drug Administration. Can a dietary supplement treat a concussion? No! http://www.fda.gov/forconsumers/consumerupdates/ucm378845.htm. Updated February 13, 2015. Accessed June 5, 2017.
60. Sarris J, Logan AC, Akbaraly TN, et al; International Society for Nutritional Psychiatry Research. Nutritional medicine as mainstream in psychiatry. Lancet Psychiatry. 2015;2(3):271-274.
61. Desai A, Kevala K, Kim HY. Depletion of brain docosahexaenoic acid impairs recovery from traumatic brain injury. PLoS One. 2014;9(1):e86472.
62. Edie CF, Dewan N. Which psychotropics interact with four common supplements. Current Psychiatry. 2005;4(1):16-30.
63. Di Lorenzo C, Ceschi A, Kupferschmidt H, et al. Adverse effects of plant food supplements and botanical preparations: a systematic review with critical evaluation of causality. Br J Clin Pharmacol. 2015;79(4):578-592.
64. National Center for Complementary and Integrative Health. Complementary and alternative medicine: what people aged 50 and older discuss with their health care providers. https://nccih.nih.gov/research/statistics/2010. Published 2011. Accessed June 5, 2017.
1. Centers for Disease Control and Prevention. Report to Congress on traumatic brain injury in the United States: epidemiology and rehabilitation. https://www.cdc.gov/traumaticbraininjury/pubs/congress_epi_rehab.html. Updated January 22, 2016. Accessed June 5, 2017.
2. Werner C, Engelhard K. Pathophysiology of traumatic brain injury. Br J Anaesth. 2007;99(1):4-9.
3. Vaishnavi S, Rao V, Fann JR. Neuropsychiatric problems after traumatic brain injury: unraveling the silent epidemic. Psychosomatics. 2009;50(3):198-205.
4. National Institutes of Health Office of Dietary Supplements. Dietary supplement fact sheets. https://ods.od.nih.gov/factsheets/list-all. Accessed June 5, 2017.
5. Institute of Medicine, Food and Nutrition Board. Dietary reference intakes for energy, carbohydrate, fiber, fat, fatty acids, cholesterol, protein, and amino acids. Washington, DC: National Academy of Sciences; 2002.
6. Rao V, Koliatsos V, Ahmed F, et al. Neuropsychiatric disturbances associated with traumatic brain injury: a practical approach to evaluation and management. Semin Neurol. 2015;35(1):64-82.
7. Chew E, Zafonte RD. Pharmacological management of neurobehavioral disorders following traumatic brain injury—a state-of-the-art review. J Rehabil Res Dev. 2009;46(6):851-879.
8. Petraglia AL, Maroon JC, Bailes JE. From the field of play to the field of combat: a review of the pharmacological management of concussion. Neurosurgery. 2012;70(6):1520-1533; discussion 1533.
9. Bengtsson M, Godbolt AK. Effects of acetylcholinesterase inhibitors on cognitive function in patients with chronic traumatic brain injury: a systematic review. J Rehabil Med. 2016;48(1):1-5.
10. Neurobehavioral Guidelines Working Group; Warden DL, Gordon B, McAllister TW, et al. Guidelines for the pharmacologic treatment of neurobehavioral sequelae of traumatic brain injury. J Neurotrauma. 2006;23(10):1468-1501.
11. DeFelice SL. The nutraceutical revolution: its impact on food industry R&D. Trends Food Sci Technol. 1995;6(2):59-61.
12. Cope EC, Morris DR, Levenson CW. Improving treatments and outcomes: an emerging role for zinc in traumatic brain injury. Nutr Rev. 2012;70(7):410-413.
13. Morris DR, Levenson CW. Zinc in traumatic brain injury: from neuroprotection to neurotoxicity. Curr Opin Clin Nutr Metab Care. 2013;16(6):708-711.
14. Young B, Ott L, Kasarskis E, et al. Zinc supplementation is associated with improved neurologic recovery rate and visceral protein levels of patients with severe closed head injury. J Neurotrauma. 1996;13(1):25-34.
15. Fernández-Gajardo R, Matamala JM, Carrasco R, et al. Novel therapeutic strategies for traumatic brain injury: acute antioxidant reinforcement. CNS Drugs. 2014;28(3):229-248.
16. Razmkon A, Sadidi A, Sherafat-Kazemzadeh E, et al. Administration of vitamin C and vitamin E in severe head injury: a randomized double-blind controlled trial. Clin Neurosurg. 2011;58:133-137.
17. Cekic M, Cutler SM, VanLandingham JW, et al. Vitamin D deficiency reduces the benefits of progesterone treatment after brain injury in aged rats. Neurobiol Aging. 2011;32(5):864-874.
18. Aminmansour B, Nikbakht H, Ghorbani A, et al. Comparison of the administration of progesterone versus progesterone and vitamin D in improvement of outcomes in patients with traumatic brain injury: a randomized clinical trial with placebo group. Adv Biomed Res. 2012;1:58.
19. Cernak I, Savic VJ, Kotur J, et al. Characterization of plasma magnesium concentration and oxidative stress following graded traumatic brain injury in humans. J Neurotrauma. 2000;17(1):53-68.
20. Dhandapani SS, Gupta A, Vivekanandhan S, et al. Randomized controlled trial of magnesium sulphate in severe closed traumatic brain injury. The Indian Journal of Neurotrauma. 2008;5(1):27-33.
21. Temkin NR, Anderson GD, Winn HR, et al. Magnesium sulfate for neuroprotection after traumatic brain injury: a randomised controlled trial. Lancet Neurol. 2007;6(1):29-38.
22. Jeter CB, Hergenroeder GW, Ward NH 3rd, et al. Human mild traumatic brain injury decreases circulating branched-chain amino acids and their metabolite levels. J Neurotrauma. 2013;30(8):671-679.
23. Cole JT, Mitala CM, Kundu S, et al. Dietary branched chain amino acids ameliorate injury-induced cognitive impairment. Proc Natl Acad Sci U S A. 2010;107(1):366-371.
24. Aquilani R, Iadarola P, Contardi A, et al. Branched-chain amino acids enhance the cognitive recovery of patients with severe traumatic brain injury. Arch Phys Med Rehabil. 2005;86(9):1729-1735.
25. Aquilani R, Boselli M, Boschi F, et al. Branched-chain amino acids may improve recovery from a vegetative or minimally conscious state in patients with traumatic brain injury: a pilot study. Arch Phys Med Rehabil. 2008;89(9):1642-1647.
26. Kang HJ, Im SH. Probiotics as an immune modulator. J Nutr Sci Vitaminol (Tokyo). 2015;61(suppl):S103-S105.
27. DiPiro JT, Howdieshell TR, Goddard JK, et al. Association of interleukin-4 plasma levels with traumatic injury and clinical course. Arch Surg. 1995;130(11):1159-1162; discussion 1162-1163.
28. Tan M, Zhu JC, Du J, et al. Effects of probiotics on serum levels of Th1/Th2 cytokine and clinical outcomes in severe traumatic brain-injured patients: a prospective randomized pilot study. Crit Care. 2011;15(6):R290.
29. Falcão de Arruda IS, de Aguilar-Nascimento JE. Benefits of early enteral nutrition with glutamine and probiotics in brain injury patients. Clin Sci (Lond). 2004;106(3):287-292.
30. Cuzzocrea S, Mazzon E, Costantino G, et al. Beneficial effects of n-acetylcysteine on ischaemic brain injury. Br J Pharmacol. 2000;130(6):1219-1226.
31. Hoffer ME, Balaban C, Slade MD, et al. Amelioration of acute sequelae of blast induced mild traumatic brain injury by N-acetyl cysteine: a double-blind, placebo controlled study. PLoS One. 2013;8(1):e54163.
32. Eakin K, Baratz-Goldstein R, Pick CG, et al. Efficacy of N-acetyl cysteine in traumatic brain injury. PLoS One. 2014;9(4):e90617.
33. Theadom A, Mahon S, Barker-Collo S, et al. Enzogenol for cognitive functioning in traumatic brain injury: a pilot placebo-controlled RCT. Eur J Neurol. 2013;20(8):1135-1144.
34. Arenth PM, Russell KC, Ricker JH, et al. CDP-choline as a biological supplement during neurorecovery: a focused review. PM R. 2011;3(6 suppl 1):S123-S131.
35. Clark WM. Efficacy of citicoline as an acute stroke treatment. Expert Opin Pharmacother. 2009;10(5):839-846.
36. Guseva MV, Hopkins DM, Scheff SW, et al. Dietary choline supplementation improves behavioral, histological, and neurochemical outcomes in a rat model of traumatic brain injury. J Neurotrauma. 2008;25(8):975-983.
37. Dixon CE, Ma X, Marion DW. Effects of CDP-choline treatment on neurobehavioral deficits after TBI and on hippocampal and neocortical acetylcholine release. J Neurotrauma. 1997;14(3):161-169.
38. Levin HS. Treatment of postconcussional symptoms with CDP-choline. J Neurol Sci. 1991;103(suppl):S39-S42.
39. Zafonte RD, Bagiella E, Ansel BM, et al. Effect of citicoline on functional and cognitive status among patients with traumatic brain injury: Citicoline Brain Injury Treatment Trial (COBRIT). JAMA. 2012;308(19):1993-2000.
40. Perry E, Walker M, Grace J, et al. Acetylcholine in mind: a neurotransmitter correlate of consciousness? Trends Neurosci. 1999;22(6):273-280.
41. Levin HS, Peters BH, Kalisky Z, et al. Effects of oral physostigmine and lecithin on memory and attention in closed head-injured patients. Cent Nerv Syst Trauma. 1986;3(4):333-342.
42. Alvarez XA, Lombardi VR, Corzo L, et al. Oral cerebrolysin enhances brain alpha activity and improves cognitive performance in elderly control subjects. J Neural Transm Suppl. 2000;59:315-328.
43. Ruether E, Husmann R, Kinzler E, et al. A 28-week, double-blind, placebo-controlled study with cerebrolysin in patients with mild to moderate Alzheimer’s disease. Int Clin Psychopharmacol. 2001;16(5):253-263.
44. Wong GK, Zhu XL, Poon WS. Beneficial effect of cerebrolysin on moderate and severe head injury patients: result of a cohort study. Acta Neurochir Suppl. 2005;95:59-60.
45. Chen CC, Wei ST, Tsaia SC, et al. Cerebrolysin enhances cognitive recovery of mild traumatic brain injury patients: double-blind, placebo-controlled, randomized study. Br J Neurosurg. 2013;27(6):803-807.
46. Ziganshina LE, Abakumova T, Vernay L. Cerebrolysin for acute ischaemic stroke. Cochrane Database Syst Rev. 2016;12:CD007026.
47. Barrett EC, McBurney MI, Ciappio ED. ω-3 fatty acid supplementation as a potential therapeutic aid for the recovery from mild traumatic brain injury/concussion. Adv Nutr. 2014;5(3):268-277.
48. Sharma S, Zhuang Y, Ying Z, et al. Dietary curcumin supplementation counteracts reduction in levels of molecules involved in energy homeostasis after brain trauma. Neuroscience. 2009;161(4):1037-1044.
49. Gatson JW, Liu MM, Abdelfattah K, et al. Resveratrol decreases inflammation in the brain of mice with mild traumatic brain injury. J Trauma Acute Care Surg. 2013;74(2):470-475; discussion 474-475.
50. Grey A, Bolland M. Clinical trial evidence and use of fish oil supplements. JAMA Intern Med. 2014;174(3):460-462.
51. Mpandzou G, Aït Ben Haddou E, Regragui W, et al. Vitamin D deficiency and its role in neurological conditions: a review. Rev Neurol (Paris). 2016;172(2):109-122.
52. Karakis I, Pase MP, Beiser A, et al. Association of serum vitamin D with the risk of incident dementia and subclinical indices of brain aging: The Framingham Heart Study. J Alzheimers Dis. 2016;51(2):451-461.
53. Sarris J, Papakostas GI, Vitolo O, et al. S-adenosyl methionine (SAMe) versus escitalopram and placebo in major depression RCT: efficacy and effects of histamine and carnitine as moderators of response. J Affect Disord. 2014;164:76-81.
54. Sarris J, Mischoulon D, Schweitzer I. Omega-3 for bipolar disorder: meta-analyses of use in mania and bipolar depression. J Clin Psychiatry. 2012;73(1):81-86.
55. Sarris J, Stough C, Bousman C, et al. Kava in the treatment of generalized anxiety disorder: a double-blind, randomized, placebo-controlled study. J Clin Psychopharmacol. 2013;33(5):643-648.
56. Hariri M, Azadbakht L. Magnesium, iron, and zinc supplementation for the treatment of attention deficit hyperactivity disorder: a systematic review on the recent literature. Int J Prev Med. 2015;6:83.
57. Gillies D, Sinn JKh, Lad SS, et al. Polyunsaturated fatty acids (PUFA) for attention deficit hyperactivity disorder (ADHD) in children and adolescents. Cochrane Database Syst Rev. 2012;7:CD007986.
58. Ghanizadeh A, Berk M. Zinc for treating of children and adolescents with attention-deficit hyperactivity disorder: a systematic review of randomized controlled clinical trials. Eur J Clin Nutr. 2013;67(1):122-124.
59. U.S. Food and Drug Administration. Can a dietary supplement treat a concussion? No! http://www.fda.gov/forconsumers/consumerupdates/ucm378845.htm. Updated February 13, 2015. Accessed June 5, 2017.
60. Sarris J, Logan AC, Akbaraly TN, et al; International Society for Nutritional Psychiatry Research. Nutritional medicine as mainstream in psychiatry. Lancet Psychiatry. 2015;2(3):271-274.
61. Desai A, Kevala K, Kim HY. Depletion of brain docosahexaenoic acid impairs recovery from traumatic brain injury. PLoS One. 2014;9(1):e86472.
62. Edie CF, Dewan N. Which psychotropics interact with four common supplements. Current Psychiatry. 2005;4(1):16-30.
63. Di Lorenzo C, Ceschi A, Kupferschmidt H, et al. Adverse effects of plant food supplements and botanical preparations: a systematic review with critical evaluation of causality. Br J Clin Pharmacol. 2015;79(4):578-592.
64. National Center for Complementary and Integrative Health. Complementary and alternative medicine: what people aged 50 and older discuss with their health care providers. https://nccih.nih.gov/research/statistics/2010. Published 2011. Accessed June 5, 2017.

Glutamate’s exciting roles in body, brain, and mind: A fertile future pharmacotherapy target
GLU is now recognized as the most abundant neurotransmitter in the brain, and its excitatory properties are vital for brain structure and function. Importantly, it also is the precursor of γ-aminobutyric acid, the ubiquitous inhibitory neurotransmitter in the brain. GLU is one of the first molecules produced during fetal life and plays a critical role in brain development and in organ development because it is a building block for protein synthesis and for manufacturing muscle and other body tissue. Therefore, aberrations in GLU activity can have a major impact on neurodevelopment—the underpinning of most psychiatric disorders due to genetic and environmental factors—and the general health of the brain and body.
GLU is derived from glutamic acid, which is not considered an essential amino acid because it is synthesized in the body via the citric acid cycle. It is readily available from many food items, including cheese, soy, and tomatoes. Monosodium GLU2 is used as a food additive to enhance flavor (Chinese food, anyone?). Incidentally, GLU represents >50% of all amino acids in breast milk, which underscores its importance for a baby’s brain and body development.
GLU’s many brain receptors
Amazingly, although it has been long known that GLU is present in all body tissues, the role of GLU in the CNS and brain was not recognized until the 1980s. This was several decades after the discovery of other neurotransmitters, such as acetylcholine, norepinephrine, and serotonin, which are less widely distributed in the CNS. Over the past 30 years, advances in psychiatric research have elucidated the numerous effects of GLU and its receptors on neuropsychiatric disorders. Multiple receptors of GLU have been discovered, including 16 ion channel receptors (7 for N-methyl-
GLU and neurodegeneration
An excess of GLU activity can be neurotoxic and can lead to brain damage.3 Therefore, it is not surprising that excess GLU activity has been found in many neurodegenerative disorders (Table). Similar to other neurologic disorders that are considered neurodegenerative, such as amyotrophic lateral sclerosis (ALS), multiple sclerosis, Alzheimer’s disease (AD), Huntington’s disease, and Parkinson’s disease, major psychiatric disorders, such as schizophrenia, depression, and bipolar disorder, also are neurodegenerative if left untreated or if multiple relapses recur because of treatment discontinuation (Table). Several neuroimaging studies have documented brain tissue loss in psychotic and mood disorders after repeated episodes. Therefore, targeting GLU in psychotic and mood disorders is legitimately a “hot” research area in psychiatry.
GLU models of psychiatric neurobiology
Advances in biological psychiatry have moved GLU to the forefront of the neurobiology and pathophysiology of the most serious psychiatric disorders. Overactivity or underactivity of the GLU NMDA receptor has emerged as scientifically plausible mechanisms underlying psychotic and mood disorders. The GLU hypothesis of schizophrenia4 grew out of the observation that phencyclidine, a drug of abuse that is a potent NMDA antagonist (50-fold stronger than ketamine), can trigger in healthy individuals a severe psychosis indistinguishable from schizophrenia, with positive and negative symptoms, cognitive impairment, thought disorder, catatonia, and agitation. Similarly, the recently discovered paraneoplastic encephalitis caused by an ovarian teratoma that secretes antibodies to the NMDA receptor produces acute psychosis, seizures, delirium, dyskinesia, headache, bizarre behavior, confusion, paranoia, auditory and visual hallucinations, and cognitive deficits.5 This demonstrates how the GLU NMDA receptor and its 7 subunits are intimately associated with various psychotic symptoms when genetic or non-genetic factors (antagonists or antibodies) drastically reduce its activity.
On the other hand, there is an impressive body of evidence that, unlike the hypofunction of NMDA receptors in schizophrenia, there appears to be increased activity of NMDA receptors in both unipolar and bipolar depression.6 Several NMDA antagonists have been shown in controlled clinical trials to be highly effective in rapidly reversing severe, chronic depression that did not respond to standard antidepressants.7 A number of NMDA antagonists have been reported to rapidly reverse—within a few hours—severe and chronic depression when administered intravenously (ketamine, rapastinel, scopolamine), intranasally (S-ketamine), or via inhalation (nitrous oxide). NMDA antagonists also show promise in other serious psychiatric disorders such as obsessive-compulsive disorder.8 Riluzole and memantine reduce GLU activity and both are FDA-approved for treating neurodegenerative disorders, such as ALS and AD, respectively.9,10 Therefore, antagonism of GLU is considered neuroprotective and can be therapeutically beneficial in managing neurodegenerative brain disorders.
GLU and the future of psychopharmacology
Based on the wealth of data generated over the past 2 decades regarding the central role of GLU receptors (NMDA, AMPA, kainate, and others) in brain health and disease, modulating GLU pathways is rapidly emerging as a key target for drug development for neuropsychiatric disorders. This approach could help with some medical comorbidities, such as diabetes11 and pain,12 that co-occur frequently with schizophrenia and depression. GLU has been implicated in diabetes via toxicity that destroys pancreatic beta cells.11 It is possible that novel drug development in the future could exploit GLU signaling and pathways to concurrently repair disorders of the brain and body, such as schizophrenia with comorbid diabetes or depression with comorbid pain. It is worth noting that glucose dysregulation has been shown to exist at the onset of schizophrenia before treatment is started.13 This might be related to GLU toxicity occurring simultaneously in the body and the brain. Also worth noting is that ketamine, an NMDA antagonist which has emerged as an ultra-rapid acting antidepressant, is an anesthetic, suggesting that perhaps it may help mitigate the pain symptoms that often accompany major depression.
It is logical to conclude that GLU pathways show exciting prospects for therapeutic advances for the brain, body, and mind. This merits intensive scientific effort for novel drug development in neuropsychiatric disorder that may parsimoniously rectify co-occurring GLU-related diseases of the brain, body, and mind.
1. Meldrum BS. Glutamate as a neurotransmitter in the brain: review of physiology and pathology. J Nutr. 2000;130(4S suppl):1007S-1015S.
2. Freeman M. Reconsidering the effects of monosodium glutamate: a literature review. J Am Acad Nurse Pract. 2005;18(10):482-486.
3. Novelli A, Pérez-Basterrechea M, Fernández-Sánchez MT. Glutamate and neurodegeneration. In: Schmidt WJ, Reith MEA, eds. Dopamine and glutamate in psychiatric disorders. Totowa, NJ: Humana Press; 2005:447-474.
4. Javitt DC. Glutamate and schizophrenia: phencyclidine, N-methyl-D-aspartate receptors, and dopamine-glutamate interactions. Int Rev Neurobiol. 2007;78:69-108.
5. Dalmau E, Tüzün E, Wu HY, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol. 2007;61(1):25-36.
6. Iadarola ND, Niciu MJ, Richards EM, et al. Ketamine and other N-methyl-D-aspartate receptor antagonists in the treatment of depression: a perspective review. Ther Adv Chronic Dis. 2015;6(3):97-114.
7. Wohleb ES, Gerhard D, Thomas A, et al. Molecular and cellular mechanisms of rapid-acting antidepressants ketamine and scopolamine. Curr Neuropharmacol. 2017;15(1):11-20.
8. Pittenger C. Glutamate modulators in the treatment of obsessive-compulsive disorder. Psychiatr Ann. 2015;45(6):308-315.
9. Farrimond LE, Roberts E, McShane R. Memantine and cholinesterase inhibitor combination therapy for Alzheimer’s disease: a systematic review. BMJ Open. 2012;2(3). doi: 10.1136/bmjopen-2012-000917.
10. Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med. 1994;330(9):585-591.
11. Davalli AM, Perego C, Folli FB. The potential role of glutamate in the current diabetes epidemic. Acta Diabetol. 2012;49(3):167-183.
12. Wozniak KM, Rojas C, Wu Y, et al. The role of glutamate signaling in pain processes and its regulation by GCP II inhibition. Curr Med Chem. 2012;19(9):1323-1334.
13. Pillinger T, Beck K, Gobjila C, et al. Impaired glucose homeostasis in first-episode schizophrenia: a systematic review and meta-analysis. JAMA Psychiatry. 2017;74(3):261-269.
GLU is now recognized as the most abundant neurotransmitter in the brain, and its excitatory properties are vital for brain structure and function. Importantly, it also is the precursor of γ-aminobutyric acid, the ubiquitous inhibitory neurotransmitter in the brain. GLU is one of the first molecules produced during fetal life and plays a critical role in brain development and in organ development because it is a building block for protein synthesis and for manufacturing muscle and other body tissue. Therefore, aberrations in GLU activity can have a major impact on neurodevelopment—the underpinning of most psychiatric disorders due to genetic and environmental factors—and the general health of the brain and body.
GLU is derived from glutamic acid, which is not considered an essential amino acid because it is synthesized in the body via the citric acid cycle. It is readily available from many food items, including cheese, soy, and tomatoes. Monosodium GLU2 is used as a food additive to enhance flavor (Chinese food, anyone?). Incidentally, GLU represents >50% of all amino acids in breast milk, which underscores its importance for a baby’s brain and body development.
GLU’s many brain receptors
Amazingly, although it has been long known that GLU is present in all body tissues, the role of GLU in the CNS and brain was not recognized until the 1980s. This was several decades after the discovery of other neurotransmitters, such as acetylcholine, norepinephrine, and serotonin, which are less widely distributed in the CNS. Over the past 30 years, advances in psychiatric research have elucidated the numerous effects of GLU and its receptors on neuropsychiatric disorders. Multiple receptors of GLU have been discovered, including 16 ion channel receptors (7 for N-methyl-
GLU and neurodegeneration
An excess of GLU activity can be neurotoxic and can lead to brain damage.3 Therefore, it is not surprising that excess GLU activity has been found in many neurodegenerative disorders (Table). Similar to other neurologic disorders that are considered neurodegenerative, such as amyotrophic lateral sclerosis (ALS), multiple sclerosis, Alzheimer’s disease (AD), Huntington’s disease, and Parkinson’s disease, major psychiatric disorders, such as schizophrenia, depression, and bipolar disorder, also are neurodegenerative if left untreated or if multiple relapses recur because of treatment discontinuation (Table). Several neuroimaging studies have documented brain tissue loss in psychotic and mood disorders after repeated episodes. Therefore, targeting GLU in psychotic and mood disorders is legitimately a “hot” research area in psychiatry.
GLU models of psychiatric neurobiology
Advances in biological psychiatry have moved GLU to the forefront of the neurobiology and pathophysiology of the most serious psychiatric disorders. Overactivity or underactivity of the GLU NMDA receptor has emerged as scientifically plausible mechanisms underlying psychotic and mood disorders. The GLU hypothesis of schizophrenia4 grew out of the observation that phencyclidine, a drug of abuse that is a potent NMDA antagonist (50-fold stronger than ketamine), can trigger in healthy individuals a severe psychosis indistinguishable from schizophrenia, with positive and negative symptoms, cognitive impairment, thought disorder, catatonia, and agitation. Similarly, the recently discovered paraneoplastic encephalitis caused by an ovarian teratoma that secretes antibodies to the NMDA receptor produces acute psychosis, seizures, delirium, dyskinesia, headache, bizarre behavior, confusion, paranoia, auditory and visual hallucinations, and cognitive deficits.5 This demonstrates how the GLU NMDA receptor and its 7 subunits are intimately associated with various psychotic symptoms when genetic or non-genetic factors (antagonists or antibodies) drastically reduce its activity.
On the other hand, there is an impressive body of evidence that, unlike the hypofunction of NMDA receptors in schizophrenia, there appears to be increased activity of NMDA receptors in both unipolar and bipolar depression.6 Several NMDA antagonists have been shown in controlled clinical trials to be highly effective in rapidly reversing severe, chronic depression that did not respond to standard antidepressants.7 A number of NMDA antagonists have been reported to rapidly reverse—within a few hours—severe and chronic depression when administered intravenously (ketamine, rapastinel, scopolamine), intranasally (S-ketamine), or via inhalation (nitrous oxide). NMDA antagonists also show promise in other serious psychiatric disorders such as obsessive-compulsive disorder.8 Riluzole and memantine reduce GLU activity and both are FDA-approved for treating neurodegenerative disorders, such as ALS and AD, respectively.9,10 Therefore, antagonism of GLU is considered neuroprotective and can be therapeutically beneficial in managing neurodegenerative brain disorders.
GLU and the future of psychopharmacology
Based on the wealth of data generated over the past 2 decades regarding the central role of GLU receptors (NMDA, AMPA, kainate, and others) in brain health and disease, modulating GLU pathways is rapidly emerging as a key target for drug development for neuropsychiatric disorders. This approach could help with some medical comorbidities, such as diabetes11 and pain,12 that co-occur frequently with schizophrenia and depression. GLU has been implicated in diabetes via toxicity that destroys pancreatic beta cells.11 It is possible that novel drug development in the future could exploit GLU signaling and pathways to concurrently repair disorders of the brain and body, such as schizophrenia with comorbid diabetes or depression with comorbid pain. It is worth noting that glucose dysregulation has been shown to exist at the onset of schizophrenia before treatment is started.13 This might be related to GLU toxicity occurring simultaneously in the body and the brain. Also worth noting is that ketamine, an NMDA antagonist which has emerged as an ultra-rapid acting antidepressant, is an anesthetic, suggesting that perhaps it may help mitigate the pain symptoms that often accompany major depression.
It is logical to conclude that GLU pathways show exciting prospects for therapeutic advances for the brain, body, and mind. This merits intensive scientific effort for novel drug development in neuropsychiatric disorder that may parsimoniously rectify co-occurring GLU-related diseases of the brain, body, and mind.
GLU is now recognized as the most abundant neurotransmitter in the brain, and its excitatory properties are vital for brain structure and function. Importantly, it also is the precursor of γ-aminobutyric acid, the ubiquitous inhibitory neurotransmitter in the brain. GLU is one of the first molecules produced during fetal life and plays a critical role in brain development and in organ development because it is a building block for protein synthesis and for manufacturing muscle and other body tissue. Therefore, aberrations in GLU activity can have a major impact on neurodevelopment—the underpinning of most psychiatric disorders due to genetic and environmental factors—and the general health of the brain and body.
GLU is derived from glutamic acid, which is not considered an essential amino acid because it is synthesized in the body via the citric acid cycle. It is readily available from many food items, including cheese, soy, and tomatoes. Monosodium GLU2 is used as a food additive to enhance flavor (Chinese food, anyone?). Incidentally, GLU represents >50% of all amino acids in breast milk, which underscores its importance for a baby’s brain and body development.
GLU’s many brain receptors
Amazingly, although it has been long known that GLU is present in all body tissues, the role of GLU in the CNS and brain was not recognized until the 1980s. This was several decades after the discovery of other neurotransmitters, such as acetylcholine, norepinephrine, and serotonin, which are less widely distributed in the CNS. Over the past 30 years, advances in psychiatric research have elucidated the numerous effects of GLU and its receptors on neuropsychiatric disorders. Multiple receptors of GLU have been discovered, including 16 ion channel receptors (7 for N-methyl-
GLU and neurodegeneration
An excess of GLU activity can be neurotoxic and can lead to brain damage.3 Therefore, it is not surprising that excess GLU activity has been found in many neurodegenerative disorders (Table). Similar to other neurologic disorders that are considered neurodegenerative, such as amyotrophic lateral sclerosis (ALS), multiple sclerosis, Alzheimer’s disease (AD), Huntington’s disease, and Parkinson’s disease, major psychiatric disorders, such as schizophrenia, depression, and bipolar disorder, also are neurodegenerative if left untreated or if multiple relapses recur because of treatment discontinuation (Table). Several neuroimaging studies have documented brain tissue loss in psychotic and mood disorders after repeated episodes. Therefore, targeting GLU in psychotic and mood disorders is legitimately a “hot” research area in psychiatry.
GLU models of psychiatric neurobiology
Advances in biological psychiatry have moved GLU to the forefront of the neurobiology and pathophysiology of the most serious psychiatric disorders. Overactivity or underactivity of the GLU NMDA receptor has emerged as scientifically plausible mechanisms underlying psychotic and mood disorders. The GLU hypothesis of schizophrenia4 grew out of the observation that phencyclidine, a drug of abuse that is a potent NMDA antagonist (50-fold stronger than ketamine), can trigger in healthy individuals a severe psychosis indistinguishable from schizophrenia, with positive and negative symptoms, cognitive impairment, thought disorder, catatonia, and agitation. Similarly, the recently discovered paraneoplastic encephalitis caused by an ovarian teratoma that secretes antibodies to the NMDA receptor produces acute psychosis, seizures, delirium, dyskinesia, headache, bizarre behavior, confusion, paranoia, auditory and visual hallucinations, and cognitive deficits.5 This demonstrates how the GLU NMDA receptor and its 7 subunits are intimately associated with various psychotic symptoms when genetic or non-genetic factors (antagonists or antibodies) drastically reduce its activity.
On the other hand, there is an impressive body of evidence that, unlike the hypofunction of NMDA receptors in schizophrenia, there appears to be increased activity of NMDA receptors in both unipolar and bipolar depression.6 Several NMDA antagonists have been shown in controlled clinical trials to be highly effective in rapidly reversing severe, chronic depression that did not respond to standard antidepressants.7 A number of NMDA antagonists have been reported to rapidly reverse—within a few hours—severe and chronic depression when administered intravenously (ketamine, rapastinel, scopolamine), intranasally (S-ketamine), or via inhalation (nitrous oxide). NMDA antagonists also show promise in other serious psychiatric disorders such as obsessive-compulsive disorder.8 Riluzole and memantine reduce GLU activity and both are FDA-approved for treating neurodegenerative disorders, such as ALS and AD, respectively.9,10 Therefore, antagonism of GLU is considered neuroprotective and can be therapeutically beneficial in managing neurodegenerative brain disorders.
GLU and the future of psychopharmacology
Based on the wealth of data generated over the past 2 decades regarding the central role of GLU receptors (NMDA, AMPA, kainate, and others) in brain health and disease, modulating GLU pathways is rapidly emerging as a key target for drug development for neuropsychiatric disorders. This approach could help with some medical comorbidities, such as diabetes11 and pain,12 that co-occur frequently with schizophrenia and depression. GLU has been implicated in diabetes via toxicity that destroys pancreatic beta cells.11 It is possible that novel drug development in the future could exploit GLU signaling and pathways to concurrently repair disorders of the brain and body, such as schizophrenia with comorbid diabetes or depression with comorbid pain. It is worth noting that glucose dysregulation has been shown to exist at the onset of schizophrenia before treatment is started.13 This might be related to GLU toxicity occurring simultaneously in the body and the brain. Also worth noting is that ketamine, an NMDA antagonist which has emerged as an ultra-rapid acting antidepressant, is an anesthetic, suggesting that perhaps it may help mitigate the pain symptoms that often accompany major depression.
It is logical to conclude that GLU pathways show exciting prospects for therapeutic advances for the brain, body, and mind. This merits intensive scientific effort for novel drug development in neuropsychiatric disorder that may parsimoniously rectify co-occurring GLU-related diseases of the brain, body, and mind.
1. Meldrum BS. Glutamate as a neurotransmitter in the brain: review of physiology and pathology. J Nutr. 2000;130(4S suppl):1007S-1015S.
2. Freeman M. Reconsidering the effects of monosodium glutamate: a literature review. J Am Acad Nurse Pract. 2005;18(10):482-486.
3. Novelli A, Pérez-Basterrechea M, Fernández-Sánchez MT. Glutamate and neurodegeneration. In: Schmidt WJ, Reith MEA, eds. Dopamine and glutamate in psychiatric disorders. Totowa, NJ: Humana Press; 2005:447-474.
4. Javitt DC. Glutamate and schizophrenia: phencyclidine, N-methyl-D-aspartate receptors, and dopamine-glutamate interactions. Int Rev Neurobiol. 2007;78:69-108.
5. Dalmau E, Tüzün E, Wu HY, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol. 2007;61(1):25-36.
6. Iadarola ND, Niciu MJ, Richards EM, et al. Ketamine and other N-methyl-D-aspartate receptor antagonists in the treatment of depression: a perspective review. Ther Adv Chronic Dis. 2015;6(3):97-114.
7. Wohleb ES, Gerhard D, Thomas A, et al. Molecular and cellular mechanisms of rapid-acting antidepressants ketamine and scopolamine. Curr Neuropharmacol. 2017;15(1):11-20.
8. Pittenger C. Glutamate modulators in the treatment of obsessive-compulsive disorder. Psychiatr Ann. 2015;45(6):308-315.
9. Farrimond LE, Roberts E, McShane R. Memantine and cholinesterase inhibitor combination therapy for Alzheimer’s disease: a systematic review. BMJ Open. 2012;2(3). doi: 10.1136/bmjopen-2012-000917.
10. Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med. 1994;330(9):585-591.
11. Davalli AM, Perego C, Folli FB. The potential role of glutamate in the current diabetes epidemic. Acta Diabetol. 2012;49(3):167-183.
12. Wozniak KM, Rojas C, Wu Y, et al. The role of glutamate signaling in pain processes and its regulation by GCP II inhibition. Curr Med Chem. 2012;19(9):1323-1334.
13. Pillinger T, Beck K, Gobjila C, et al. Impaired glucose homeostasis in first-episode schizophrenia: a systematic review and meta-analysis. JAMA Psychiatry. 2017;74(3):261-269.
1. Meldrum BS. Glutamate as a neurotransmitter in the brain: review of physiology and pathology. J Nutr. 2000;130(4S suppl):1007S-1015S.
2. Freeman M. Reconsidering the effects of monosodium glutamate: a literature review. J Am Acad Nurse Pract. 2005;18(10):482-486.
3. Novelli A, Pérez-Basterrechea M, Fernández-Sánchez MT. Glutamate and neurodegeneration. In: Schmidt WJ, Reith MEA, eds. Dopamine and glutamate in psychiatric disorders. Totowa, NJ: Humana Press; 2005:447-474.
4. Javitt DC. Glutamate and schizophrenia: phencyclidine, N-methyl-D-aspartate receptors, and dopamine-glutamate interactions. Int Rev Neurobiol. 2007;78:69-108.
5. Dalmau E, Tüzün E, Wu HY, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol. 2007;61(1):25-36.
6. Iadarola ND, Niciu MJ, Richards EM, et al. Ketamine and other N-methyl-D-aspartate receptor antagonists in the treatment of depression: a perspective review. Ther Adv Chronic Dis. 2015;6(3):97-114.
7. Wohleb ES, Gerhard D, Thomas A, et al. Molecular and cellular mechanisms of rapid-acting antidepressants ketamine and scopolamine. Curr Neuropharmacol. 2017;15(1):11-20.
8. Pittenger C. Glutamate modulators in the treatment of obsessive-compulsive disorder. Psychiatr Ann. 2015;45(6):308-315.
9. Farrimond LE, Roberts E, McShane R. Memantine and cholinesterase inhibitor combination therapy for Alzheimer’s disease: a systematic review. BMJ Open. 2012;2(3). doi: 10.1136/bmjopen-2012-000917.
10. Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med. 1994;330(9):585-591.
11. Davalli AM, Perego C, Folli FB. The potential role of glutamate in the current diabetes epidemic. Acta Diabetol. 2012;49(3):167-183.
12. Wozniak KM, Rojas C, Wu Y, et al. The role of glutamate signaling in pain processes and its regulation by GCP II inhibition. Curr Med Chem. 2012;19(9):1323-1334.
13. Pillinger T, Beck K, Gobjila C, et al. Impaired glucose homeostasis in first-episode schizophrenia: a systematic review and meta-analysis. JAMA Psychiatry. 2017;74(3):261-269.
