User login
Product News: 07 2017
Glytone Sunscreen Lotion Broad Spectrum SPF 40
Pierre Fabre Dermo-Cosmetique USA introduces Glytone Sunscreen Lotion Broad Spectrum SPF 40, a mineral-based formula for face and body with micronized zinc oxide, octinoxate, and octisalate. The lightweight formula is water resistant for up to 40 minutes and contains hyaluronic acid to nourish the skin and help boost natural moisture levels to visibly reduce the appearance of fine lines and wrinkles. For more information, visit www.glytone-usa.com.
proactivMD
The Proactiv Company launches the proactivMD Essentials System, a 3-step acne regimen that has been reformulated to include
If you would like your product included in Product News, please email a press release to the Editorial Office at [email protected].
Glytone Sunscreen Lotion Broad Spectrum SPF 40
Pierre Fabre Dermo-Cosmetique USA introduces Glytone Sunscreen Lotion Broad Spectrum SPF 40, a mineral-based formula for face and body with micronized zinc oxide, octinoxate, and octisalate. The lightweight formula is water resistant for up to 40 minutes and contains hyaluronic acid to nourish the skin and help boost natural moisture levels to visibly reduce the appearance of fine lines and wrinkles. For more information, visit www.glytone-usa.com.
proactivMD
The Proactiv Company launches the proactivMD Essentials System, a 3-step acne regimen that has been reformulated to include
If you would like your product included in Product News, please email a press release to the Editorial Office at [email protected].
Glytone Sunscreen Lotion Broad Spectrum SPF 40
Pierre Fabre Dermo-Cosmetique USA introduces Glytone Sunscreen Lotion Broad Spectrum SPF 40, a mineral-based formula for face and body with micronized zinc oxide, octinoxate, and octisalate. The lightweight formula is water resistant for up to 40 minutes and contains hyaluronic acid to nourish the skin and help boost natural moisture levels to visibly reduce the appearance of fine lines and wrinkles. For more information, visit www.glytone-usa.com.
proactivMD
The Proactiv Company launches the proactivMD Essentials System, a 3-step acne regimen that has been reformulated to include
If you would like your product included in Product News, please email a press release to the Editorial Office at [email protected].

Hair and Scalp Disorders in Adult and Pediatric Patients With Skin of Color
One of the most common concerns among black patients is hair- and scalp-related disease. As increasing numbers of black patients opt to see dermatologists, it is imperative that all dermatologists be adequately trained to address the concerns of this patient population. When patients ask for help with common skin diseases of the hair and scalp, there are details that must be included in diagnosis, treatment, and hair care recommendations to reach goals for excellence in patient care. Herein, we provide must-know information to effectively approach this patient population.
Seborrheic Dermatitis
A study utilizing data from the National Ambulatory Medical Care Survey from 1993 to 2009 revealed seborrheic dermatitis (SD) as the second most common diagnosis for black patients who visit a dermatologist.1 Prevalence data from a population of 1408 white, black, and Chinese patients from the United States and China revealed scalp flaking in 81% to 95% of black patients, 66% to 82% in white patients, and 30% to 42% in Chinese patients.2 Seborrheic dermatitis has a notable prevalence in black women and often is considered normal by patients. It can be exacerbated by infrequent shampooing (ranging from once per month or longer in between shampoos) and the inappropriate use of hair oils and pomades; it also has been associated with hair breakage, lichen simplex chronicus, and folliculitis. Seborrheic dermatitis must be distinguished from other disorders including sarcoidosis, psoriasis, discoid lupus erythematosus, tinea capitis, and lichen simplex chronicus.
Although there is a paucity of literature on the treatment of SD in black patients, components of treatment are similar to those recommended for other populations. Black women are advised to carefully utilize antidandruff shampoos containing zinc pyrithione, selenium sulfide, or tar to avoid hair shaft damage and dryness. Ketoconazole shampoo rarely is recommended and may be more appropriately used in men and boys, as hair fragility is less of a concern for them. The shampoo should be applied directly to the scalp rather than the hair shafts to minimize dryness, with no particular elongated contact time needed for these medicated shampoos to be effective. Because conditioners can wash off the active ingredients in therapeutic shampoos, antidandruff conditioners are recommended. Potent or ultrapotent topical corticosteroids applied to the scalp 3 to 4 times weekly initially will control the symptoms of itching as well as scaling, and mid-potency topical corticosteroid oil may be used at weekly intervals.
Hairline and facial involvement of SD often co-occurs, and low-potency topical steroids may be applied to the affected areas twice daily for 3 to 4 weeks, which may be repeated for flares. Topical calcineurin inhibitors or antifungal creams such as ketoconazole or econazole may then provide effective control. Encouraging patients to increase shampooing to once weekly or every 2 weeks and discontinue use of scalp pomades and oils also is recommended. Patients must know that an itchy scaly scalp represents a treatable disorder.
Acquired Trichorrhexis Nodosa
Hair fragility and breakage is common and multifactorial in black patients. Hair shaft breakage can occur on the vertex scalp in central centrifugal cicatricial alopecia (CCCA), with random localized breakage due to scratching in SD. Heat, hair colorants, and chemical relaxers may result in diffuse damage and breakage.3 Sodium-, potassium-, and guanine hydroxide–containing chemical relaxers change the physical properties of the hair by rearranging disulfide bonds. They remove the monomolecular layer of fatty acids covalently bound to the cuticle that help prevent penetration of water into the hair shaft. Additionally, chemical relaxers weaken the hair shaft and decrease tensile strength.
Unlike hair relaxers, colorants are less likely to lead to catastrophic hair breakage after a single use and require frequent use, which leads to cumulative damage. Thermal straightening is another cause of hair-shaft weakening in black patients.4,5 Flat irons and curling irons can cause substantially more damage than blow-dryers due to the amount of heat generated. Flat irons may reach a high temperature of 230ºC (450ºF) as compared to 100°C (210°F) for a blow-dryer. Even the simple act of combing the hair can cause hair breakage, as demonstrated in African volunteers whose hair remained short in contrast to white and Asian volunteers, despite the fact that they had not cut their hair for 1 or more years.6,7 These volunteers had many hair strand knots that led to breakage during combing and hair grooming.6
There is no known prevalence data for acquired trichorrhexis nodosa, though a study of 30 white and black women demonstrated that broken hairs were significantly increased in black women (P=.0001).8 Another study by Hall et al9 of 103 black women showed that 55% of the women reported breakage of hair shafts with normal styling. Khumalo et al6 investigated hair shaft fragility and reported no trichothiodystrophy; the authors concluded that the cause of the hair fragility likely was physical trauma or an undiscovered structural abnormality. Franbourg et al10 examined the structure of hair fibers in white, Asian, and black patients and found no differences, but microfractures were only present in black patients and were determined to be the cause of hair breakage. These studies underscore the need for specific questioning of the patient on hair care including combing, washing, drying, and using products and chemicals.
The approach to the treatment of hair breakage involves correcting underlying abnormalities (eg, iron deficiency, hypothyroidism, nutritional deficiencies). Patients should “give their hair a rest” by discontinuing use of heat, colorants, and chemical relaxers. For patients who are unable to comply, advising them to stop these processes for 6 to 12 months will allow for repair of the hair shaft. To minimize damage from colorants, recommend semipermanent, demipermanent, or temporary dyes. Patients should be counseled to stop bleaching their hair or using permanent colorants. The use of heat protectant products on the hair before styling as well as layering moisturizing regimens starting with a moisturizing shampoo followed by a leave-in, dimethicone-containing conditioner marketed for dry damaged hair is suggested. Dimethicone thinly coats the hair shaft to restore hydrophobicity, smoothes cuticular scales, decreases frizz, and protects the hair from damage. Use of a 2-in-1 shampoo and conditioner containing anionic surfactants and wide-toothed, smooth (no jagged edges in the grooves) combs along with rare brushing are recommended. The hair may be worn in its natural state, but straightening with heat should be avoided. Air drying the hair can minimize breakage, but if thermal styling is necessary, patients should turn the temperature setting of the flat or curling iron down. Protective hair care practices may include placing a loosely sewn-in hair weave that will allow for good hair care, wearing loose braids, or using a wig. Serial trimming of the hair every 6 to 8 weeks is recommended. Improvement may take time, and patients should be advised of this timeline to prevent frustration.
Acne Keloidalis Nuchae
Acne keloidalis nuchae (AKN) is characterized by papules and pustules located on the occipital scalp and/or the nape of the neck, which may result in keloidal papules and plaques. The etiology is unknown, but ingrown hairs, genetics, trauma, infection, inflammation, and androgen hormones have been proposed to play a role.11 Although AKN may occur in black women, it is primarily a disorder in black men. The diagnosis is made based primarily on clinical findings, and a history of short haircuts may support the diagnosis. Treatment is tailored to the severity of the disease (Table 1). Avoidance of short haircuts and irritation from shirt collars may be helpful. Patients should be advised that the condition is controllable but not curable.

Pseudofolliculitis Barbae
Pseudofolliculitis barbae (PFB) is characterized by papules and pustules in the beard region that may result in postinflammatory hyperpigmentation, keloidal scar formation, and/or linear scarring. The coarse curled hairs characteristic of black men penetrate the follicle before exiting the skin and penetrate the skin after exiting the follicle, resulting in inflammation. Shaving methods and genetics also may contribute to the development of PFB. As with AKN, diagnosis is made clinically and does not require a skin biopsy. Important components of the patient’s history that should be obtained are hair removal practices and the use of over-the-counter products (eg, shave [pre and post] moisturizers, exfoliants, shaving creams or gels, keratin-softening agents containing α- or β-hydroxy acids). A bacterial culture may be appropriate if a notable pustular component is present. The patient should be advised to discontinue shaving if possible, which may require a physician’s letter explaining the necessity to the patient’s employer. Pseudofolliculitis barbae often can be prevented or lessened with the right hair removal strategy. Because there is not one optimal hair removal strategy that suits every patient, encourage the patient to experiment with different hair removal techniques, from depilatories to electric shavers, foil-guard razors, and multiple-blade razors. Preshave hydration and postshave moisturiza-tion also should be encouraged.12 Benzoyl peroxide–containing shave gels and cleansers, as well as moisturizers containing glycolic, salicylic, and phytic acids, may minimize ingrown hairs, papules, and inflammation.
Other useful topical agents include eflornithine hydrochloride to decrease hair growth, retinoids to soften hair fibers, mild topical steroids to reduce inflammation, and/or topical erythromycin or clindamycin if pustules are present.13 Oral antibiotics such as doxycycline, minocycline, or erythromycin can be added for more severe cases of inflammation or infection. Procedural interventions include laser hair removal to prevent PFB and intralesional triamcinolone 10 to 40 mg/cc every 4 to 6 weeks, with the total volume depending on the size and number of lesions.
Alopecia
Alopecia is the sixth most common diagnosis seen in black patients visiting a dermatologist.14 The physician’s response to the patient’s chief concern of hair loss is key to building a relationship of confidence and trust. Trivializing the concern or dismissing it will undermine the physician-patient relationship. A survey by Gathers and Mahan15 revealed that 68% of patients thought that physicians did not understand their hair.
Hair loss negatively impacts quality of life, and a study of 50 black South African women with alopecia demonstrated a notable disease burden. Factors with the highest impact were those related to self-image, relationships, and interactions with others.16
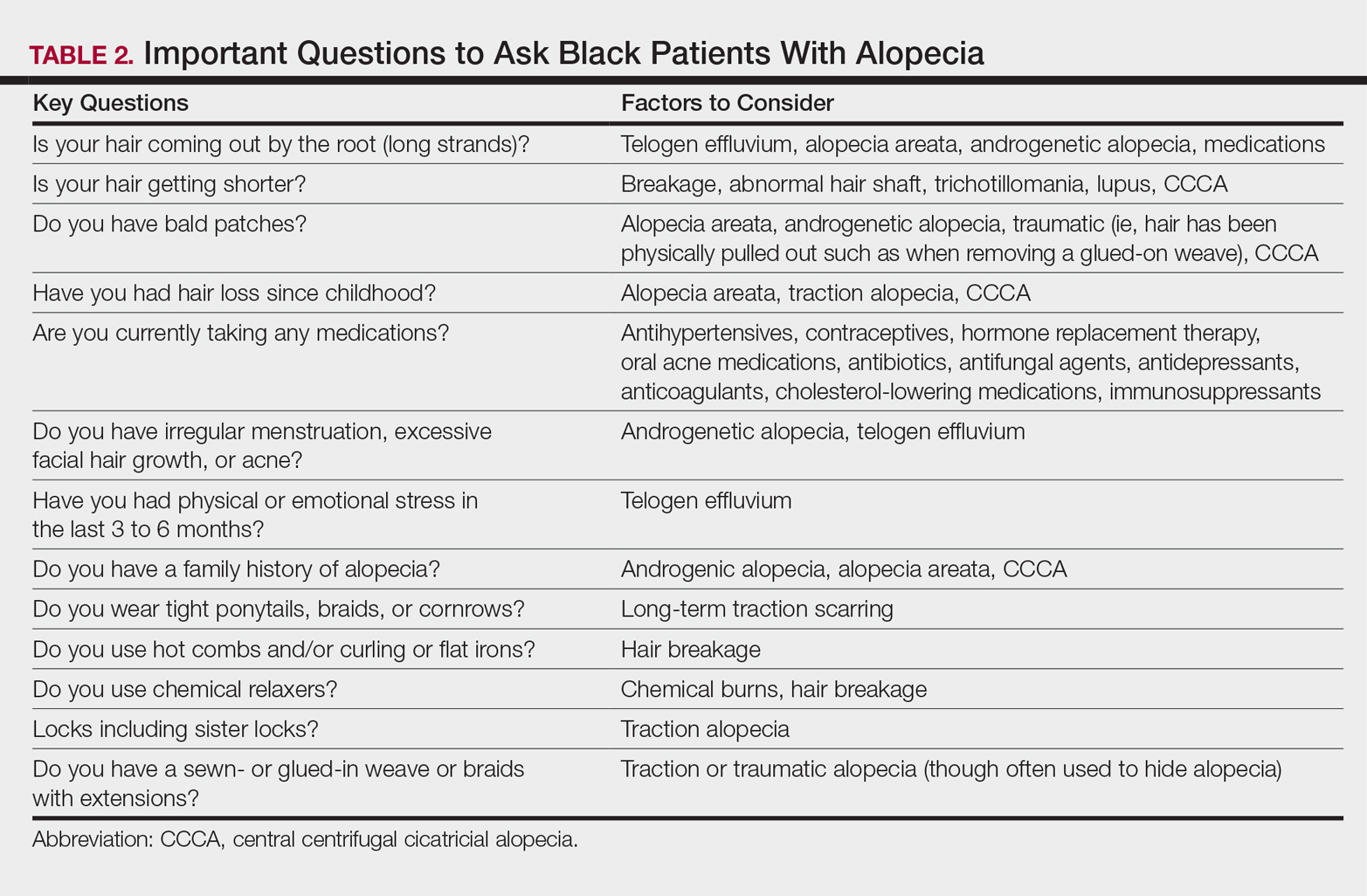
It is not unusual for black women to have multiple types of alopecia identified in one biopsy specimen. Wohltmann and Sperling17 demonstrated 2 or more different types of alopecia in more than 10% of biopsy specimens of alopecia, including CCCA, androgenetic alopecia, end-stage traction alopecia, telogen effluvium, and tinea capitis. A complete history, physical examination, and appropriate procedures (eg, hair pull test, dermatoscopic examination and scalp biopsy) likely will yield an accurate diagnosis. Table 2 highlights important questions that should be asked about the patient’s history.
Physical examination of the scalp including dermatoscopic examination and a hair pull test as well as an evaluation of other hair-bearing areas may suggest a diagnosis that can be confirmed with a scalp biopsy.18,19 Selection of a biopsy site at the periphery of the alopecic area that includes hair and consultation with a dermatopathologist familiar with features of CCCA, traction, and traumatic alopecia are important for making an accurate diagnosis.
Tinea Capitis in Black Pediatric Patients
Tinea capitis, a fungal infection of the scalp and hair, is one of the most common issues in children with skin of color. Clinical presentation may include widely distributed scaling, annular scaly plaques, annular patches of alopecia studded with black dots (broken hairs), and/or annular inflammatory plaques. Although scalp hyperkeratosis often is a hallmark of pediatric tinea capitis, it is not diagnostic. The differential diagnosis of pediatric scalp hyperkeratosis/scaling includes tinea capitis, SD, atopic dermatitis, psoriasis, and sebopsoriasis.20,21 Clues to accurate diagnosis of tinea capitis may be found by examination of the adult who combs the child’s hair, as erythematous annular scaly plaques representing tinea corporis may be observed on the forearms or thighs. Although the thighs are a seemingly unusual location, the frequent practice of the child sitting on the floor between the legs of the adult during hairstyling provides a point of contact for the transmission of tinea from the child’s scalp to the thighs or forearms of the adult. Once tinea capitis is clinically suspected, the diagnosis is confirmed by a fungal culture. Adequate sampling is obtained by clipping hairs in an area of scaling for submission and vigorously rubbing the area of black dots or hyperkeratosis with a cotton swab.
Hubbard22 shed light on the decision to treat tinea capitis empirically or await the culture results. One hundred consecutive children (98 were black) presented with the constellation of scalp alopecia, scaling, pruritus, and occipital lymphadenopathy. Sixty-eight of those children had positive fungal cultures, and of them, 60 had both occipital lymphadenopathy and scaling and 55 had both occipital lymphadenopathy and alopecia.22 Thus, occipital lymphadenopathy in conjunction with alopecia and/or scaling is predictive of tinea capitis in this population and suggests that the initiation of treatment prior to confirmative culture results is appropriate.
The mainstay of treatment for tinea capitis is griseofulvin, but it is often underdosed and not continued for an adequate period of time to ensure clearance of the infection. Griseofulvin microsize (125 mg/5 mL) at the dosage of 20 to 25 mg/kg once daily for 8 to 12 weeks is recommended instead of a lower-dosed 4- to 6-week course.23,24
Options for treating a child with residual disease include increasing and/or extending the griseofulvin dosage, encouraging ingestion of fatty foods to enhance absorption, dividing the dosage of griseofulvin from once daily to twice daily, changing therapy to oral terbinafine due to resistance to griseofulvin, examining siblings as a source of reinfection, and reviewing the positive fungal culture report to distinguish Trichophyton tonsurans versus Microsporum canis as the causative agent and adjust treatment accordingly. Although griseofulvin is the first-line treatment for M canis, terbinafine, which is approved for children 4 years and older for tineacapitis, is most efficacious for T tonsurans.25 Treatment with terbinafine is weight based and should extend for 2 to 4 weeksfor T tonsurans and 8 to 12 weeks for M canis.
Antifungal shampoos may help reduce household spread of tinea and decrease transmissible fungal spores, but they may cause hair dryness and breakage.26,27 Antifungal shampoos can be applied directly onto the scalp for a 5- to 10-minute contact time and rinsed, and then the hair should be shampooed with a moisturizing shampoo followed by a moisturizing conditioner. Hair conditioners may decrease household spread of tinea capitis and should be used by the patient and other members of the household.28 Infection control may be enhanced by advising parents to dispose of hair pomades and washing hair accessories, combs, and brushes in hot soapy water, preferably in the dishwasher.
Hair Growth
The inability of the hair of black children to grow long is a common concern for parents of toddlers and preschool-aged children. Although the hair does grow, it grows more slowly than hair in white children (0.259 vs 0.330 mm per day), and it is likely to break faster than it is growing in black versus white children (146.6 vs 13.13 total broken hairs).8 Reassurance that the hair is indeed growing and that the length will increase as the child matures is important. Avoidance of hairstyles that promote traction and use of hair extensions, as well as use of moisturizing shampoos and conditioners, may minimize breakage and support the growth of healthy hair.
Conclusion
Hair- and scalp-related disease in black adults and children is commonly encountered in dermatology practice. It is important to understand the intrinsic characteristics of facial and scalp hair as well as hair care practices in this patient population that differ from those of white and Asian populations, such as frequency of shampooing, products, and styling. Familiarity with these differences may aid in effective diagnosis, treatment, and hair care recommendations in patients with these conditions.
- Davis SA, Naarahari S, Feldman SR, et al. Top dermatologic conditions in patients of color: an analysis of nationally representative data. J Drugs Dermatol. 2012;11:466-473.
- Hickman JG, Cardin C, Dawson TL, et al. Dandruff, part I: scalp disease prevalence in Caucasians, African Americans, and Chinese and the effects of shampoo frequency on scalp health. Poster presented at: 60th Annual Meeting of the American Academy of Dermatology; February 22-27, 2002; New Orleans, LA.
- Swee W, Klontz KC, Lambert LA. A nationwide outbreak of alopecia associated with the use of a hair-relaxing formulation. Arch Dermatol. 2000;136:1104-1108.
- Nicholson AG, Harland CC, Bull RH, et al. Chemically induced cosmetic alopecia. Br J Dermatol. 1993;128:537-541.
- Detwiler SP, Carson JL, Woosley JT, et al. Bubble hair. case caused by an overheating hair dryer and reproducibility in normal hair with heat. J Am Acad Dermatol. 1994;30:54-60.
- Khumalo NP, Dawber RP, Ferguson DJ. Apparent fragility of African hair is unrelated to the cystine-rich protein distribution: a cytochemical electron microscopic study. Exp Dermatol. 2005;14:311-314.
- Robbins C. Hair breakage during combing. I. pathways of breakage. J Cosmet Sci. 2006;57:233-243.
- Lewallen R, Francis S, Fisher B, et al. Hair care practices and structural evaluation of scalp and hair shaft parameter in African American and Caucasian women. J Cosmet Dermatol. 2015;14:216-223.
- Hall RR, Francis S, Whitt-Glover M, et al. Hair care practices as a barrier to physical activity in African American women. JAMA Dermatol. 2013;149:310-314.
- Franbourg A, Hallegot P, Baltenneck F, et al. Current research on ethnic hair. J Am Acad Dermatol. 2003;48(6 suppl):S115-S119.
- Ogunbiyi A. Acne keloidalis nuchae: prevalence, impact, and management challenges. Clin Cosmet Investig Dermatol. 2016;9:483-489.
- Gray J, McMichael AJ. Pseudofolliculitis barbae: understanding the condition and the role of facial grooming. Int J Cosmet Sci. 2016;38(suppl 1):24-27.
- Kundu RV, Patterson S. Dermatologic conditions in skin of color: part II. disorders occurring predominately in skin of color. Am Fam Physician. 2013;87:859-865.
- Davis SA, Naarahari S, Feldman SR, et al. Top dermatologic conditions in patients of color: an analysis of nationally representative data. J Drugs Dermatol. 2012;11:466-473.
- Gathers RC, Mahan MG. African American women, hair care and health barriers. J Clin Aesthet Dermatol. 2014;7:26-29.
- Dlova NC, Fabbrocini G, Lauro C, et al. Quality of life in South African black women with alopecia: a pilot study. Int J Dermatol. 2016;55:875-881.
- Wohltmann WE, Sperling L. Histopathologic diagnosis of multifactorial alopecia. J Cutan Pathol. 2016;43:483-491.
- McDonald KA, Shelley AJ, Colantonio S, et al. Hair pull test: evidence-based update and revision of guidelines. J Am Acad Dermatol. 2017;76:472-477.
- Miteva M, Tosti A. Dermatoscopic features of central centrifugal cicatricial alopecia. J Am Acad Dermatol. 2014;71:443-444.
- Coley MK, Bhanusali DG, Silverberg JI, et al. Scalp hyperkeratosis and alopecia in children of color. J Drugs Dermatol. 2011;10:511-516.
- Silverberg NB. Scalp hyperkeratosis in children with skin of color: diagnostic and therapeutic considerations. Cutis. 2015;95:199-204, 207.
- Hubbard TW. The predictive value of symptoms in diagnosing childhood tinea capitis. Arch Pediatr Adolesc Med. 1999;153:1150-1153.
- Kakourou T, Uksal U; European Society for Pediatric Dermatology. Guidelines for the management of tinea capitis in children. Pediatr Dermatol. 2010;27:226-228.
- Sethi A, Antanya R. Systemic antifungal therapy for cutaneous infections in children. Pediatr Infect Dis J. 2006;25:643-644.
- Gupta AK. Drummond-Main C. Meta-analysis of randomized, controlled trials comparing particular doses of griseofulvin and terbinafine for the treatment of tinea capitis. Pediatr Dermatol. 2013;30:1-6.
- Greer DL. Successful treatment of tinea capitis with 2% ketoconazole shampoo. Int J Dermatol 2000;39:302-304.
- Sharma V, Silverberg NB, Howard R, et al. Do hair care practices affect the acquisition of tinea capitis? a case-control study. Arch Pediatr Adolesc Med. 2001;155:818-821.
- Greer DL. Successful treatment of tinea capitis with 2% ketoconazole shampoo. Int J Dermatol. 2000;39:302-304.
One of the most common concerns among black patients is hair- and scalp-related disease. As increasing numbers of black patients opt to see dermatologists, it is imperative that all dermatologists be adequately trained to address the concerns of this patient population. When patients ask for help with common skin diseases of the hair and scalp, there are details that must be included in diagnosis, treatment, and hair care recommendations to reach goals for excellence in patient care. Herein, we provide must-know information to effectively approach this patient population.
Seborrheic Dermatitis
A study utilizing data from the National Ambulatory Medical Care Survey from 1993 to 2009 revealed seborrheic dermatitis (SD) as the second most common diagnosis for black patients who visit a dermatologist.1 Prevalence data from a population of 1408 white, black, and Chinese patients from the United States and China revealed scalp flaking in 81% to 95% of black patients, 66% to 82% in white patients, and 30% to 42% in Chinese patients.2 Seborrheic dermatitis has a notable prevalence in black women and often is considered normal by patients. It can be exacerbated by infrequent shampooing (ranging from once per month or longer in between shampoos) and the inappropriate use of hair oils and pomades; it also has been associated with hair breakage, lichen simplex chronicus, and folliculitis. Seborrheic dermatitis must be distinguished from other disorders including sarcoidosis, psoriasis, discoid lupus erythematosus, tinea capitis, and lichen simplex chronicus.
Although there is a paucity of literature on the treatment of SD in black patients, components of treatment are similar to those recommended for other populations. Black women are advised to carefully utilize antidandruff shampoos containing zinc pyrithione, selenium sulfide, or tar to avoid hair shaft damage and dryness. Ketoconazole shampoo rarely is recommended and may be more appropriately used in men and boys, as hair fragility is less of a concern for them. The shampoo should be applied directly to the scalp rather than the hair shafts to minimize dryness, with no particular elongated contact time needed for these medicated shampoos to be effective. Because conditioners can wash off the active ingredients in therapeutic shampoos, antidandruff conditioners are recommended. Potent or ultrapotent topical corticosteroids applied to the scalp 3 to 4 times weekly initially will control the symptoms of itching as well as scaling, and mid-potency topical corticosteroid oil may be used at weekly intervals.
Hairline and facial involvement of SD often co-occurs, and low-potency topical steroids may be applied to the affected areas twice daily for 3 to 4 weeks, which may be repeated for flares. Topical calcineurin inhibitors or antifungal creams such as ketoconazole or econazole may then provide effective control. Encouraging patients to increase shampooing to once weekly or every 2 weeks and discontinue use of scalp pomades and oils also is recommended. Patients must know that an itchy scaly scalp represents a treatable disorder.
Acquired Trichorrhexis Nodosa
Hair fragility and breakage is common and multifactorial in black patients. Hair shaft breakage can occur on the vertex scalp in central centrifugal cicatricial alopecia (CCCA), with random localized breakage due to scratching in SD. Heat, hair colorants, and chemical relaxers may result in diffuse damage and breakage.3 Sodium-, potassium-, and guanine hydroxide–containing chemical relaxers change the physical properties of the hair by rearranging disulfide bonds. They remove the monomolecular layer of fatty acids covalently bound to the cuticle that help prevent penetration of water into the hair shaft. Additionally, chemical relaxers weaken the hair shaft and decrease tensile strength.
Unlike hair relaxers, colorants are less likely to lead to catastrophic hair breakage after a single use and require frequent use, which leads to cumulative damage. Thermal straightening is another cause of hair-shaft weakening in black patients.4,5 Flat irons and curling irons can cause substantially more damage than blow-dryers due to the amount of heat generated. Flat irons may reach a high temperature of 230ºC (450ºF) as compared to 100°C (210°F) for a blow-dryer. Even the simple act of combing the hair can cause hair breakage, as demonstrated in African volunteers whose hair remained short in contrast to white and Asian volunteers, despite the fact that they had not cut their hair for 1 or more years.6,7 These volunteers had many hair strand knots that led to breakage during combing and hair grooming.6
There is no known prevalence data for acquired trichorrhexis nodosa, though a study of 30 white and black women demonstrated that broken hairs were significantly increased in black women (P=.0001).8 Another study by Hall et al9 of 103 black women showed that 55% of the women reported breakage of hair shafts with normal styling. Khumalo et al6 investigated hair shaft fragility and reported no trichothiodystrophy; the authors concluded that the cause of the hair fragility likely was physical trauma or an undiscovered structural abnormality. Franbourg et al10 examined the structure of hair fibers in white, Asian, and black patients and found no differences, but microfractures were only present in black patients and were determined to be the cause of hair breakage. These studies underscore the need for specific questioning of the patient on hair care including combing, washing, drying, and using products and chemicals.
The approach to the treatment of hair breakage involves correcting underlying abnormalities (eg, iron deficiency, hypothyroidism, nutritional deficiencies). Patients should “give their hair a rest” by discontinuing use of heat, colorants, and chemical relaxers. For patients who are unable to comply, advising them to stop these processes for 6 to 12 months will allow for repair of the hair shaft. To minimize damage from colorants, recommend semipermanent, demipermanent, or temporary dyes. Patients should be counseled to stop bleaching their hair or using permanent colorants. The use of heat protectant products on the hair before styling as well as layering moisturizing regimens starting with a moisturizing shampoo followed by a leave-in, dimethicone-containing conditioner marketed for dry damaged hair is suggested. Dimethicone thinly coats the hair shaft to restore hydrophobicity, smoothes cuticular scales, decreases frizz, and protects the hair from damage. Use of a 2-in-1 shampoo and conditioner containing anionic surfactants and wide-toothed, smooth (no jagged edges in the grooves) combs along with rare brushing are recommended. The hair may be worn in its natural state, but straightening with heat should be avoided. Air drying the hair can minimize breakage, but if thermal styling is necessary, patients should turn the temperature setting of the flat or curling iron down. Protective hair care practices may include placing a loosely sewn-in hair weave that will allow for good hair care, wearing loose braids, or using a wig. Serial trimming of the hair every 6 to 8 weeks is recommended. Improvement may take time, and patients should be advised of this timeline to prevent frustration.
Acne Keloidalis Nuchae
Acne keloidalis nuchae (AKN) is characterized by papules and pustules located on the occipital scalp and/or the nape of the neck, which may result in keloidal papules and plaques. The etiology is unknown, but ingrown hairs, genetics, trauma, infection, inflammation, and androgen hormones have been proposed to play a role.11 Although AKN may occur in black women, it is primarily a disorder in black men. The diagnosis is made based primarily on clinical findings, and a history of short haircuts may support the diagnosis. Treatment is tailored to the severity of the disease (Table 1). Avoidance of short haircuts and irritation from shirt collars may be helpful. Patients should be advised that the condition is controllable but not curable.
Pseudofolliculitis Barbae
Pseudofolliculitis barbae (PFB) is characterized by papules and pustules in the beard region that may result in postinflammatory hyperpigmentation, keloidal scar formation, and/or linear scarring. The coarse curled hairs characteristic of black men penetrate the follicle before exiting the skin and penetrate the skin after exiting the follicle, resulting in inflammation. Shaving methods and genetics also may contribute to the development of PFB. As with AKN, diagnosis is made clinically and does not require a skin biopsy. Important components of the patient’s history that should be obtained are hair removal practices and the use of over-the-counter products (eg, shave [pre and post] moisturizers, exfoliants, shaving creams or gels, keratin-softening agents containing α- or β-hydroxy acids). A bacterial culture may be appropriate if a notable pustular component is present. The patient should be advised to discontinue shaving if possible, which may require a physician’s letter explaining the necessity to the patient’s employer. Pseudofolliculitis barbae often can be prevented or lessened with the right hair removal strategy. Because there is not one optimal hair removal strategy that suits every patient, encourage the patient to experiment with different hair removal techniques, from depilatories to electric shavers, foil-guard razors, and multiple-blade razors. Preshave hydration and postshave moisturiza-tion also should be encouraged.12 Benzoyl peroxide–containing shave gels and cleansers, as well as moisturizers containing glycolic, salicylic, and phytic acids, may minimize ingrown hairs, papules, and inflammation.
Other useful topical agents include eflornithine hydrochloride to decrease hair growth, retinoids to soften hair fibers, mild topical steroids to reduce inflammation, and/or topical erythromycin or clindamycin if pustules are present.13 Oral antibiotics such as doxycycline, minocycline, or erythromycin can be added for more severe cases of inflammation or infection. Procedural interventions include laser hair removal to prevent PFB and intralesional triamcinolone 10 to 40 mg/cc every 4 to 6 weeks, with the total volume depending on the size and number of lesions.
Alopecia
Alopecia is the sixth most common diagnosis seen in black patients visiting a dermatologist.14 The physician’s response to the patient’s chief concern of hair loss is key to building a relationship of confidence and trust. Trivializing the concern or dismissing it will undermine the physician-patient relationship. A survey by Gathers and Mahan15 revealed that 68% of patients thought that physicians did not understand their hair.
Hair loss negatively impacts quality of life, and a study of 50 black South African women with alopecia demonstrated a notable disease burden. Factors with the highest impact were those related to self-image, relationships, and interactions with others.16
It is not unusual for black women to have multiple types of alopecia identified in one biopsy specimen. Wohltmann and Sperling17 demonstrated 2 or more different types of alopecia in more than 10% of biopsy specimens of alopecia, including CCCA, androgenetic alopecia, end-stage traction alopecia, telogen effluvium, and tinea capitis. A complete history, physical examination, and appropriate procedures (eg, hair pull test, dermatoscopic examination and scalp biopsy) likely will yield an accurate diagnosis. Table 2 highlights important questions that should be asked about the patient’s history.
Physical examination of the scalp including dermatoscopic examination and a hair pull test as well as an evaluation of other hair-bearing areas may suggest a diagnosis that can be confirmed with a scalp biopsy.18,19 Selection of a biopsy site at the periphery of the alopecic area that includes hair and consultation with a dermatopathologist familiar with features of CCCA, traction, and traumatic alopecia are important for making an accurate diagnosis.
Tinea Capitis in Black Pediatric Patients
Tinea capitis, a fungal infection of the scalp and hair, is one of the most common issues in children with skin of color. Clinical presentation may include widely distributed scaling, annular scaly plaques, annular patches of alopecia studded with black dots (broken hairs), and/or annular inflammatory plaques. Although scalp hyperkeratosis often is a hallmark of pediatric tinea capitis, it is not diagnostic. The differential diagnosis of pediatric scalp hyperkeratosis/scaling includes tinea capitis, SD, atopic dermatitis, psoriasis, and sebopsoriasis.20,21 Clues to accurate diagnosis of tinea capitis may be found by examination of the adult who combs the child’s hair, as erythematous annular scaly plaques representing tinea corporis may be observed on the forearms or thighs. Although the thighs are a seemingly unusual location, the frequent practice of the child sitting on the floor between the legs of the adult during hairstyling provides a point of contact for the transmission of tinea from the child’s scalp to the thighs or forearms of the adult. Once tinea capitis is clinically suspected, the diagnosis is confirmed by a fungal culture. Adequate sampling is obtained by clipping hairs in an area of scaling for submission and vigorously rubbing the area of black dots or hyperkeratosis with a cotton swab.
Hubbard22 shed light on the decision to treat tinea capitis empirically or await the culture results. One hundred consecutive children (98 were black) presented with the constellation of scalp alopecia, scaling, pruritus, and occipital lymphadenopathy. Sixty-eight of those children had positive fungal cultures, and of them, 60 had both occipital lymphadenopathy and scaling and 55 had both occipital lymphadenopathy and alopecia.22 Thus, occipital lymphadenopathy in conjunction with alopecia and/or scaling is predictive of tinea capitis in this population and suggests that the initiation of treatment prior to confirmative culture results is appropriate.
The mainstay of treatment for tinea capitis is griseofulvin, but it is often underdosed and not continued for an adequate period of time to ensure clearance of the infection. Griseofulvin microsize (125 mg/5 mL) at the dosage of 20 to 25 mg/kg once daily for 8 to 12 weeks is recommended instead of a lower-dosed 4- to 6-week course.23,24
Options for treating a child with residual disease include increasing and/or extending the griseofulvin dosage, encouraging ingestion of fatty foods to enhance absorption, dividing the dosage of griseofulvin from once daily to twice daily, changing therapy to oral terbinafine due to resistance to griseofulvin, examining siblings as a source of reinfection, and reviewing the positive fungal culture report to distinguish Trichophyton tonsurans versus Microsporum canis as the causative agent and adjust treatment accordingly. Although griseofulvin is the first-line treatment for M canis, terbinafine, which is approved for children 4 years and older for tineacapitis, is most efficacious for T tonsurans.25 Treatment with terbinafine is weight based and should extend for 2 to 4 weeksfor T tonsurans and 8 to 12 weeks for M canis.
Antifungal shampoos may help reduce household spread of tinea and decrease transmissible fungal spores, but they may cause hair dryness and breakage.26,27 Antifungal shampoos can be applied directly onto the scalp for a 5- to 10-minute contact time and rinsed, and then the hair should be shampooed with a moisturizing shampoo followed by a moisturizing conditioner. Hair conditioners may decrease household spread of tinea capitis and should be used by the patient and other members of the household.28 Infection control may be enhanced by advising parents to dispose of hair pomades and washing hair accessories, combs, and brushes in hot soapy water, preferably in the dishwasher.
Hair Growth
The inability of the hair of black children to grow long is a common concern for parents of toddlers and preschool-aged children. Although the hair does grow, it grows more slowly than hair in white children (0.259 vs 0.330 mm per day), and it is likely to break faster than it is growing in black versus white children (146.6 vs 13.13 total broken hairs).8 Reassurance that the hair is indeed growing and that the length will increase as the child matures is important. Avoidance of hairstyles that promote traction and use of hair extensions, as well as use of moisturizing shampoos and conditioners, may minimize breakage and support the growth of healthy hair.
Conclusion
Hair- and scalp-related disease in black adults and children is commonly encountered in dermatology practice. It is important to understand the intrinsic characteristics of facial and scalp hair as well as hair care practices in this patient population that differ from those of white and Asian populations, such as frequency of shampooing, products, and styling. Familiarity with these differences may aid in effective diagnosis, treatment, and hair care recommendations in patients with these conditions.
One of the most common concerns among black patients is hair- and scalp-related disease. As increasing numbers of black patients opt to see dermatologists, it is imperative that all dermatologists be adequately trained to address the concerns of this patient population. When patients ask for help with common skin diseases of the hair and scalp, there are details that must be included in diagnosis, treatment, and hair care recommendations to reach goals for excellence in patient care. Herein, we provide must-know information to effectively approach this patient population.
Seborrheic Dermatitis
A study utilizing data from the National Ambulatory Medical Care Survey from 1993 to 2009 revealed seborrheic dermatitis (SD) as the second most common diagnosis for black patients who visit a dermatologist.1 Prevalence data from a population of 1408 white, black, and Chinese patients from the United States and China revealed scalp flaking in 81% to 95% of black patients, 66% to 82% in white patients, and 30% to 42% in Chinese patients.2 Seborrheic dermatitis has a notable prevalence in black women and often is considered normal by patients. It can be exacerbated by infrequent shampooing (ranging from once per month or longer in between shampoos) and the inappropriate use of hair oils and pomades; it also has been associated with hair breakage, lichen simplex chronicus, and folliculitis. Seborrheic dermatitis must be distinguished from other disorders including sarcoidosis, psoriasis, discoid lupus erythematosus, tinea capitis, and lichen simplex chronicus.
Although there is a paucity of literature on the treatment of SD in black patients, components of treatment are similar to those recommended for other populations. Black women are advised to carefully utilize antidandruff shampoos containing zinc pyrithione, selenium sulfide, or tar to avoid hair shaft damage and dryness. Ketoconazole shampoo rarely is recommended and may be more appropriately used in men and boys, as hair fragility is less of a concern for them. The shampoo should be applied directly to the scalp rather than the hair shafts to minimize dryness, with no particular elongated contact time needed for these medicated shampoos to be effective. Because conditioners can wash off the active ingredients in therapeutic shampoos, antidandruff conditioners are recommended. Potent or ultrapotent topical corticosteroids applied to the scalp 3 to 4 times weekly initially will control the symptoms of itching as well as scaling, and mid-potency topical corticosteroid oil may be used at weekly intervals.
Hairline and facial involvement of SD often co-occurs, and low-potency topical steroids may be applied to the affected areas twice daily for 3 to 4 weeks, which may be repeated for flares. Topical calcineurin inhibitors or antifungal creams such as ketoconazole or econazole may then provide effective control. Encouraging patients to increase shampooing to once weekly or every 2 weeks and discontinue use of scalp pomades and oils also is recommended. Patients must know that an itchy scaly scalp represents a treatable disorder.
Acquired Trichorrhexis Nodosa
Hair fragility and breakage is common and multifactorial in black patients. Hair shaft breakage can occur on the vertex scalp in central centrifugal cicatricial alopecia (CCCA), with random localized breakage due to scratching in SD. Heat, hair colorants, and chemical relaxers may result in diffuse damage and breakage.3 Sodium-, potassium-, and guanine hydroxide–containing chemical relaxers change the physical properties of the hair by rearranging disulfide bonds. They remove the monomolecular layer of fatty acids covalently bound to the cuticle that help prevent penetration of water into the hair shaft. Additionally, chemical relaxers weaken the hair shaft and decrease tensile strength.
Unlike hair relaxers, colorants are less likely to lead to catastrophic hair breakage after a single use and require frequent use, which leads to cumulative damage. Thermal straightening is another cause of hair-shaft weakening in black patients.4,5 Flat irons and curling irons can cause substantially more damage than blow-dryers due to the amount of heat generated. Flat irons may reach a high temperature of 230ºC (450ºF) as compared to 100°C (210°F) for a blow-dryer. Even the simple act of combing the hair can cause hair breakage, as demonstrated in African volunteers whose hair remained short in contrast to white and Asian volunteers, despite the fact that they had not cut their hair for 1 or more years.6,7 These volunteers had many hair strand knots that led to breakage during combing and hair grooming.6
There is no known prevalence data for acquired trichorrhexis nodosa, though a study of 30 white and black women demonstrated that broken hairs were significantly increased in black women (P=.0001).8 Another study by Hall et al9 of 103 black women showed that 55% of the women reported breakage of hair shafts with normal styling. Khumalo et al6 investigated hair shaft fragility and reported no trichothiodystrophy; the authors concluded that the cause of the hair fragility likely was physical trauma or an undiscovered structural abnormality. Franbourg et al10 examined the structure of hair fibers in white, Asian, and black patients and found no differences, but microfractures were only present in black patients and were determined to be the cause of hair breakage. These studies underscore the need for specific questioning of the patient on hair care including combing, washing, drying, and using products and chemicals.
The approach to the treatment of hair breakage involves correcting underlying abnormalities (eg, iron deficiency, hypothyroidism, nutritional deficiencies). Patients should “give their hair a rest” by discontinuing use of heat, colorants, and chemical relaxers. For patients who are unable to comply, advising them to stop these processes for 6 to 12 months will allow for repair of the hair shaft. To minimize damage from colorants, recommend semipermanent, demipermanent, or temporary dyes. Patients should be counseled to stop bleaching their hair or using permanent colorants. The use of heat protectant products on the hair before styling as well as layering moisturizing regimens starting with a moisturizing shampoo followed by a leave-in, dimethicone-containing conditioner marketed for dry damaged hair is suggested. Dimethicone thinly coats the hair shaft to restore hydrophobicity, smoothes cuticular scales, decreases frizz, and protects the hair from damage. Use of a 2-in-1 shampoo and conditioner containing anionic surfactants and wide-toothed, smooth (no jagged edges in the grooves) combs along with rare brushing are recommended. The hair may be worn in its natural state, but straightening with heat should be avoided. Air drying the hair can minimize breakage, but if thermal styling is necessary, patients should turn the temperature setting of the flat or curling iron down. Protective hair care practices may include placing a loosely sewn-in hair weave that will allow for good hair care, wearing loose braids, or using a wig. Serial trimming of the hair every 6 to 8 weeks is recommended. Improvement may take time, and patients should be advised of this timeline to prevent frustration.
Acne Keloidalis Nuchae
Acne keloidalis nuchae (AKN) is characterized by papules and pustules located on the occipital scalp and/or the nape of the neck, which may result in keloidal papules and plaques. The etiology is unknown, but ingrown hairs, genetics, trauma, infection, inflammation, and androgen hormones have been proposed to play a role.11 Although AKN may occur in black women, it is primarily a disorder in black men. The diagnosis is made based primarily on clinical findings, and a history of short haircuts may support the diagnosis. Treatment is tailored to the severity of the disease (Table 1). Avoidance of short haircuts and irritation from shirt collars may be helpful. Patients should be advised that the condition is controllable but not curable.
Pseudofolliculitis Barbae
Pseudofolliculitis barbae (PFB) is characterized by papules and pustules in the beard region that may result in postinflammatory hyperpigmentation, keloidal scar formation, and/or linear scarring. The coarse curled hairs characteristic of black men penetrate the follicle before exiting the skin and penetrate the skin after exiting the follicle, resulting in inflammation. Shaving methods and genetics also may contribute to the development of PFB. As with AKN, diagnosis is made clinically and does not require a skin biopsy. Important components of the patient’s history that should be obtained are hair removal practices and the use of over-the-counter products (eg, shave [pre and post] moisturizers, exfoliants, shaving creams or gels, keratin-softening agents containing α- or β-hydroxy acids). A bacterial culture may be appropriate if a notable pustular component is present. The patient should be advised to discontinue shaving if possible, which may require a physician’s letter explaining the necessity to the patient’s employer. Pseudofolliculitis barbae often can be prevented or lessened with the right hair removal strategy. Because there is not one optimal hair removal strategy that suits every patient, encourage the patient to experiment with different hair removal techniques, from depilatories to electric shavers, foil-guard razors, and multiple-blade razors. Preshave hydration and postshave moisturiza-tion also should be encouraged.12 Benzoyl peroxide–containing shave gels and cleansers, as well as moisturizers containing glycolic, salicylic, and phytic acids, may minimize ingrown hairs, papules, and inflammation.
Other useful topical agents include eflornithine hydrochloride to decrease hair growth, retinoids to soften hair fibers, mild topical steroids to reduce inflammation, and/or topical erythromycin or clindamycin if pustules are present.13 Oral antibiotics such as doxycycline, minocycline, or erythromycin can be added for more severe cases of inflammation or infection. Procedural interventions include laser hair removal to prevent PFB and intralesional triamcinolone 10 to 40 mg/cc every 4 to 6 weeks, with the total volume depending on the size and number of lesions.
Alopecia
Alopecia is the sixth most common diagnosis seen in black patients visiting a dermatologist.14 The physician’s response to the patient’s chief concern of hair loss is key to building a relationship of confidence and trust. Trivializing the concern or dismissing it will undermine the physician-patient relationship. A survey by Gathers and Mahan15 revealed that 68% of patients thought that physicians did not understand their hair.
Hair loss negatively impacts quality of life, and a study of 50 black South African women with alopecia demonstrated a notable disease burden. Factors with the highest impact were those related to self-image, relationships, and interactions with others.16
It is not unusual for black women to have multiple types of alopecia identified in one biopsy specimen. Wohltmann and Sperling17 demonstrated 2 or more different types of alopecia in more than 10% of biopsy specimens of alopecia, including CCCA, androgenetic alopecia, end-stage traction alopecia, telogen effluvium, and tinea capitis. A complete history, physical examination, and appropriate procedures (eg, hair pull test, dermatoscopic examination and scalp biopsy) likely will yield an accurate diagnosis. Table 2 highlights important questions that should be asked about the patient’s history.
Physical examination of the scalp including dermatoscopic examination and a hair pull test as well as an evaluation of other hair-bearing areas may suggest a diagnosis that can be confirmed with a scalp biopsy.18,19 Selection of a biopsy site at the periphery of the alopecic area that includes hair and consultation with a dermatopathologist familiar with features of CCCA, traction, and traumatic alopecia are important for making an accurate diagnosis.
Tinea Capitis in Black Pediatric Patients
Tinea capitis, a fungal infection of the scalp and hair, is one of the most common issues in children with skin of color. Clinical presentation may include widely distributed scaling, annular scaly plaques, annular patches of alopecia studded with black dots (broken hairs), and/or annular inflammatory plaques. Although scalp hyperkeratosis often is a hallmark of pediatric tinea capitis, it is not diagnostic. The differential diagnosis of pediatric scalp hyperkeratosis/scaling includes tinea capitis, SD, atopic dermatitis, psoriasis, and sebopsoriasis.20,21 Clues to accurate diagnosis of tinea capitis may be found by examination of the adult who combs the child’s hair, as erythematous annular scaly plaques representing tinea corporis may be observed on the forearms or thighs. Although the thighs are a seemingly unusual location, the frequent practice of the child sitting on the floor between the legs of the adult during hairstyling provides a point of contact for the transmission of tinea from the child’s scalp to the thighs or forearms of the adult. Once tinea capitis is clinically suspected, the diagnosis is confirmed by a fungal culture. Adequate sampling is obtained by clipping hairs in an area of scaling for submission and vigorously rubbing the area of black dots or hyperkeratosis with a cotton swab.
Hubbard22 shed light on the decision to treat tinea capitis empirically or await the culture results. One hundred consecutive children (98 were black) presented with the constellation of scalp alopecia, scaling, pruritus, and occipital lymphadenopathy. Sixty-eight of those children had positive fungal cultures, and of them, 60 had both occipital lymphadenopathy and scaling and 55 had both occipital lymphadenopathy and alopecia.22 Thus, occipital lymphadenopathy in conjunction with alopecia and/or scaling is predictive of tinea capitis in this population and suggests that the initiation of treatment prior to confirmative culture results is appropriate.
The mainstay of treatment for tinea capitis is griseofulvin, but it is often underdosed and not continued for an adequate period of time to ensure clearance of the infection. Griseofulvin microsize (125 mg/5 mL) at the dosage of 20 to 25 mg/kg once daily for 8 to 12 weeks is recommended instead of a lower-dosed 4- to 6-week course.23,24
Options for treating a child with residual disease include increasing and/or extending the griseofulvin dosage, encouraging ingestion of fatty foods to enhance absorption, dividing the dosage of griseofulvin from once daily to twice daily, changing therapy to oral terbinafine due to resistance to griseofulvin, examining siblings as a source of reinfection, and reviewing the positive fungal culture report to distinguish Trichophyton tonsurans versus Microsporum canis as the causative agent and adjust treatment accordingly. Although griseofulvin is the first-line treatment for M canis, terbinafine, which is approved for children 4 years and older for tineacapitis, is most efficacious for T tonsurans.25 Treatment with terbinafine is weight based and should extend for 2 to 4 weeksfor T tonsurans and 8 to 12 weeks for M canis.
Antifungal shampoos may help reduce household spread of tinea and decrease transmissible fungal spores, but they may cause hair dryness and breakage.26,27 Antifungal shampoos can be applied directly onto the scalp for a 5- to 10-minute contact time and rinsed, and then the hair should be shampooed with a moisturizing shampoo followed by a moisturizing conditioner. Hair conditioners may decrease household spread of tinea capitis and should be used by the patient and other members of the household.28 Infection control may be enhanced by advising parents to dispose of hair pomades and washing hair accessories, combs, and brushes in hot soapy water, preferably in the dishwasher.
Hair Growth
The inability of the hair of black children to grow long is a common concern for parents of toddlers and preschool-aged children. Although the hair does grow, it grows more slowly than hair in white children (0.259 vs 0.330 mm per day), and it is likely to break faster than it is growing in black versus white children (146.6 vs 13.13 total broken hairs).8 Reassurance that the hair is indeed growing and that the length will increase as the child matures is important. Avoidance of hairstyles that promote traction and use of hair extensions, as well as use of moisturizing shampoos and conditioners, may minimize breakage and support the growth of healthy hair.
Conclusion
Hair- and scalp-related disease in black adults and children is commonly encountered in dermatology practice. It is important to understand the intrinsic characteristics of facial and scalp hair as well as hair care practices in this patient population that differ from those of white and Asian populations, such as frequency of shampooing, products, and styling. Familiarity with these differences may aid in effective diagnosis, treatment, and hair care recommendations in patients with these conditions.
- Davis SA, Naarahari S, Feldman SR, et al. Top dermatologic conditions in patients of color: an analysis of nationally representative data. J Drugs Dermatol. 2012;11:466-473.
- Hickman JG, Cardin C, Dawson TL, et al. Dandruff, part I: scalp disease prevalence in Caucasians, African Americans, and Chinese and the effects of shampoo frequency on scalp health. Poster presented at: 60th Annual Meeting of the American Academy of Dermatology; February 22-27, 2002; New Orleans, LA.
- Swee W, Klontz KC, Lambert LA. A nationwide outbreak of alopecia associated with the use of a hair-relaxing formulation. Arch Dermatol. 2000;136:1104-1108.
- Nicholson AG, Harland CC, Bull RH, et al. Chemically induced cosmetic alopecia. Br J Dermatol. 1993;128:537-541.
- Detwiler SP, Carson JL, Woosley JT, et al. Bubble hair. case caused by an overheating hair dryer and reproducibility in normal hair with heat. J Am Acad Dermatol. 1994;30:54-60.
- Khumalo NP, Dawber RP, Ferguson DJ. Apparent fragility of African hair is unrelated to the cystine-rich protein distribution: a cytochemical electron microscopic study. Exp Dermatol. 2005;14:311-314.
- Robbins C. Hair breakage during combing. I. pathways of breakage. J Cosmet Sci. 2006;57:233-243.
- Lewallen R, Francis S, Fisher B, et al. Hair care practices and structural evaluation of scalp and hair shaft parameter in African American and Caucasian women. J Cosmet Dermatol. 2015;14:216-223.
- Hall RR, Francis S, Whitt-Glover M, et al. Hair care practices as a barrier to physical activity in African American women. JAMA Dermatol. 2013;149:310-314.
- Franbourg A, Hallegot P, Baltenneck F, et al. Current research on ethnic hair. J Am Acad Dermatol. 2003;48(6 suppl):S115-S119.
- Ogunbiyi A. Acne keloidalis nuchae: prevalence, impact, and management challenges. Clin Cosmet Investig Dermatol. 2016;9:483-489.
- Gray J, McMichael AJ. Pseudofolliculitis barbae: understanding the condition and the role of facial grooming. Int J Cosmet Sci. 2016;38(suppl 1):24-27.
- Kundu RV, Patterson S. Dermatologic conditions in skin of color: part II. disorders occurring predominately in skin of color. Am Fam Physician. 2013;87:859-865.
- Davis SA, Naarahari S, Feldman SR, et al. Top dermatologic conditions in patients of color: an analysis of nationally representative data. J Drugs Dermatol. 2012;11:466-473.
- Gathers RC, Mahan MG. African American women, hair care and health barriers. J Clin Aesthet Dermatol. 2014;7:26-29.
- Dlova NC, Fabbrocini G, Lauro C, et al. Quality of life in South African black women with alopecia: a pilot study. Int J Dermatol. 2016;55:875-881.
- Wohltmann WE, Sperling L. Histopathologic diagnosis of multifactorial alopecia. J Cutan Pathol. 2016;43:483-491.
- McDonald KA, Shelley AJ, Colantonio S, et al. Hair pull test: evidence-based update and revision of guidelines. J Am Acad Dermatol. 2017;76:472-477.
- Miteva M, Tosti A. Dermatoscopic features of central centrifugal cicatricial alopecia. J Am Acad Dermatol. 2014;71:443-444.
- Coley MK, Bhanusali DG, Silverberg JI, et al. Scalp hyperkeratosis and alopecia in children of color. J Drugs Dermatol. 2011;10:511-516.
- Silverberg NB. Scalp hyperkeratosis in children with skin of color: diagnostic and therapeutic considerations. Cutis. 2015;95:199-204, 207.
- Hubbard TW. The predictive value of symptoms in diagnosing childhood tinea capitis. Arch Pediatr Adolesc Med. 1999;153:1150-1153.
- Kakourou T, Uksal U; European Society for Pediatric Dermatology. Guidelines for the management of tinea capitis in children. Pediatr Dermatol. 2010;27:226-228.
- Sethi A, Antanya R. Systemic antifungal therapy for cutaneous infections in children. Pediatr Infect Dis J. 2006;25:643-644.
- Gupta AK. Drummond-Main C. Meta-analysis of randomized, controlled trials comparing particular doses of griseofulvin and terbinafine for the treatment of tinea capitis. Pediatr Dermatol. 2013;30:1-6.
- Greer DL. Successful treatment of tinea capitis with 2% ketoconazole shampoo. Int J Dermatol 2000;39:302-304.
- Sharma V, Silverberg NB, Howard R, et al. Do hair care practices affect the acquisition of tinea capitis? a case-control study. Arch Pediatr Adolesc Med. 2001;155:818-821.
- Greer DL. Successful treatment of tinea capitis with 2% ketoconazole shampoo. Int J Dermatol. 2000;39:302-304.
- Davis SA, Naarahari S, Feldman SR, et al. Top dermatologic conditions in patients of color: an analysis of nationally representative data. J Drugs Dermatol. 2012;11:466-473.
- Hickman JG, Cardin C, Dawson TL, et al. Dandruff, part I: scalp disease prevalence in Caucasians, African Americans, and Chinese and the effects of shampoo frequency on scalp health. Poster presented at: 60th Annual Meeting of the American Academy of Dermatology; February 22-27, 2002; New Orleans, LA.
- Swee W, Klontz KC, Lambert LA. A nationwide outbreak of alopecia associated with the use of a hair-relaxing formulation. Arch Dermatol. 2000;136:1104-1108.
- Nicholson AG, Harland CC, Bull RH, et al. Chemically induced cosmetic alopecia. Br J Dermatol. 1993;128:537-541.
- Detwiler SP, Carson JL, Woosley JT, et al. Bubble hair. case caused by an overheating hair dryer and reproducibility in normal hair with heat. J Am Acad Dermatol. 1994;30:54-60.
- Khumalo NP, Dawber RP, Ferguson DJ. Apparent fragility of African hair is unrelated to the cystine-rich protein distribution: a cytochemical electron microscopic study. Exp Dermatol. 2005;14:311-314.
- Robbins C. Hair breakage during combing. I. pathways of breakage. J Cosmet Sci. 2006;57:233-243.
- Lewallen R, Francis S, Fisher B, et al. Hair care practices and structural evaluation of scalp and hair shaft parameter in African American and Caucasian women. J Cosmet Dermatol. 2015;14:216-223.
- Hall RR, Francis S, Whitt-Glover M, et al. Hair care practices as a barrier to physical activity in African American women. JAMA Dermatol. 2013;149:310-314.
- Franbourg A, Hallegot P, Baltenneck F, et al. Current research on ethnic hair. J Am Acad Dermatol. 2003;48(6 suppl):S115-S119.
- Ogunbiyi A. Acne keloidalis nuchae: prevalence, impact, and management challenges. Clin Cosmet Investig Dermatol. 2016;9:483-489.
- Gray J, McMichael AJ. Pseudofolliculitis barbae: understanding the condition and the role of facial grooming. Int J Cosmet Sci. 2016;38(suppl 1):24-27.
- Kundu RV, Patterson S. Dermatologic conditions in skin of color: part II. disorders occurring predominately in skin of color. Am Fam Physician. 2013;87:859-865.
- Davis SA, Naarahari S, Feldman SR, et al. Top dermatologic conditions in patients of color: an analysis of nationally representative data. J Drugs Dermatol. 2012;11:466-473.
- Gathers RC, Mahan MG. African American women, hair care and health barriers. J Clin Aesthet Dermatol. 2014;7:26-29.
- Dlova NC, Fabbrocini G, Lauro C, et al. Quality of life in South African black women with alopecia: a pilot study. Int J Dermatol. 2016;55:875-881.
- Wohltmann WE, Sperling L. Histopathologic diagnosis of multifactorial alopecia. J Cutan Pathol. 2016;43:483-491.
- McDonald KA, Shelley AJ, Colantonio S, et al. Hair pull test: evidence-based update and revision of guidelines. J Am Acad Dermatol. 2017;76:472-477.
- Miteva M, Tosti A. Dermatoscopic features of central centrifugal cicatricial alopecia. J Am Acad Dermatol. 2014;71:443-444.
- Coley MK, Bhanusali DG, Silverberg JI, et al. Scalp hyperkeratosis and alopecia in children of color. J Drugs Dermatol. 2011;10:511-516.
- Silverberg NB. Scalp hyperkeratosis in children with skin of color: diagnostic and therapeutic considerations. Cutis. 2015;95:199-204, 207.
- Hubbard TW. The predictive value of symptoms in diagnosing childhood tinea capitis. Arch Pediatr Adolesc Med. 1999;153:1150-1153.
- Kakourou T, Uksal U; European Society for Pediatric Dermatology. Guidelines for the management of tinea capitis in children. Pediatr Dermatol. 2010;27:226-228.
- Sethi A, Antanya R. Systemic antifungal therapy for cutaneous infections in children. Pediatr Infect Dis J. 2006;25:643-644.
- Gupta AK. Drummond-Main C. Meta-analysis of randomized, controlled trials comparing particular doses of griseofulvin and terbinafine for the treatment of tinea capitis. Pediatr Dermatol. 2013;30:1-6.
- Greer DL. Successful treatment of tinea capitis with 2% ketoconazole shampoo. Int J Dermatol 2000;39:302-304.
- Sharma V, Silverberg NB, Howard R, et al. Do hair care practices affect the acquisition of tinea capitis? a case-control study. Arch Pediatr Adolesc Med. 2001;155:818-821.
- Greer DL. Successful treatment of tinea capitis with 2% ketoconazole shampoo. Int J Dermatol. 2000;39:302-304.
Practice Points
- Instruct patients with acquired trichorrhexis nodosa to discontinue use of heat, colorants, and chemical relaxers on their hair.
- Create a contract with your seborrheic dermatitis patients to have them shampoo at least weekly or every 2 weeks.
- For children with treated tinea capitis that has not completely resolved, increase or extend the griseofulvin dosage, encourage ingestion of fatty foods to enhance absorption, and divide dosage of griseofulvin from once to twice daily.
- Selection of a biopsy site at the periphery of an alopecic area that includes hair and hair follicles and evaluation by a dermatopathologist familiar with the features of central centrifugal cicatricial, traction, and traumatic alopecias will ensure an accurate diagnosis of alopecia.

ED-to-ED Transfers: Optimizing Patient Safety
Emergency Department (ED)-to-ED transfers are a reality of practice in emergency medicine, and they can certainly present a tall order for ensuring patient safety. Challenges abound in getting the right patient to the right place at the right time by the right transportation method.1 A critically ill patient becomes a metaphorical baton to be passed on, requiring the best care along the way--even when the patient is not completely aware of the reasons for the transfer of care. For some EDs, ED-to-ED transfers have become a common daily occurrence. The realities of freestanding EDs, hospital overcrowding, and subspecialty coverage gaps create challenges in direct hospital admission, necessitating a second ED stop before the patient reaches an appropriate destination and provider for definitive care.
The transfer of a patient is much more complex than arranging and carrying out an ambulance ride across town. If thought of as a process, with pre-transfer planning on the sending end, the transfer itself, and post-transfer assurance of care continuity on the receiving end, the quality of care and patient safety can be elevated. Emergency department-to-ED transfers require careful attention to communication, with important hand-offs between the sending facility, the ambulance personnel, and the receiving facility. To lead the discussion around the perils of interhospital ED-to-ED transfers, the following case reports illustrate some of the challenges encountered.
Case Scenarios
Case 1
A 58-year-old man presented to a freestanding ED at 10:30
The nursing staff obtained intravenous (IV) access, and blood samples were drawn. Parenteral pain control and antiemetics were administered while a computed tomography (CT) scan of the abdomen and pelvis with contrast was emergently in progress. Meanwhile, the laboratory test results included the following: lactate, 3.8 mmol/L; lipase 42 U/L; carbon monoxide, 14 mmol/L; white blood cell count (WBC), 12 x 109/L without bandemia; serum creatinine, 1.0 mg/dL; liver function tests with a mild elevation of transaminases; and normal coagulation studies.
After reviewing the CT scan, the radiologist called to report a hyperdensity in the lumen of the superior mesenteric artery, which might represent a subsegmental dissection or a partial occlusion or plaque, with no radiographic evidence of bowel ischemia. Vascular surgery service was consulted, and the decision to start IV heparin was agreed upon. The vascular surgeon requested that a mesenteric peripheral vascular laboratory examination (PVL) be arranged on arrival at the hospital ED, and an ED-to-ED transfer to the hospital was arranged. The case was discussed with the receiving day shift emergency physician (EP), who planned to order the mesenteric PVL immediately upon the patient’s arrival.
Emergent transportation via an advanced life support (ALS) ambulance was arranged. The nursing report was called in to the hospital ED at noon, and the ALS unit had arrived and was ready to transfer the patient. Repeat vital signs were obtained, revealing an HR of 45 beats/min and a BP of 200/100 mm Hg.
After an uneventful transport, the patient arrived at the hospital ED at 12:45
Case Commentary
In this case, when the nursing assessment preceding transfer revealed sustained abnormal vital signs particularly the significant hypertension, reassessment and blood pressure management by the sending EP prior to transport may have diminished the poor outcome resulting from the intracranial hemorrhage. Ideally, BP control should have been implemented prior to transport—especially in the context of possible arterial dissection/occlusion with ongoing anticoagulation therapy. If such attempts to control BP prior to transport prove inadequate, a hand-off communication with the receiving EP is indicated, emphasizing the need for immediate evaluation and critical intervention upon patient arrival. On the receiving end of a patient transfer, it is good practice that all critically ill or injured patients be immediately assessed upon arrival at the ED, regardless of planned interventions by any other department.

Transport from another ED cannot mislead to a false sense of security that ED care is completed. Patients geographically located in the ED (especially those who are newly arrived) are the responsibility of the ED providers until that point where the next specialist provider clearly assumes care of the patient (see “ED-to-ED Transfers: Summary of Responsibilities”). This point in handoff time can be murky and unclear; yet, as illustrated by this case, it is best to re-evaluate and ensure appropriate emergency care of the patient upon arrival. In addition, as with any other ED patient with a change in condition, timely re-evaluation of the transferred patient is indicated upon receiving the report from nursing that the patient’s condition had changed.
Transfers between EDs should be viewed as a process, and that each phase in the process is important—from the pre-transfer preparation at the sending facility, the physical transfer itself by transport personnel, and the post-transfer arrival that requires the receiving facility to ensure care continues seamlessly and appropriately. Even in situations of high acuity and/or high volume, anticipation and timely attention is required by the receiving staff to ensure continuity of safe patient care. The metaphorical baton was dropped in this transfer.
Opportunities for Patient Safety Improvement. The sending facility should always address any abnormal vital signs prior to patient transfer. The receiving facility should evaluate all transferred patients at the bedside as soon as possible upon arrival. Both facilities should take timely advantage of the information the nurses provide, especially when there is a change in the patient’s condition. All involved physicians from the sending facility should communicate to the receiving ED staff critical and potentially critical patient care information and concerns that pose a risk of deterioration of the patient’s condition.
Case 2
A 23-year-old woman presented to a community hospital ED with a sore throat, fever, and difficulty swallowing. The PA on duty saw the patient in the fast track section of the ED. The patient reported the sore throat had been persistent for the past 3 days, and that she began having difficulty swallowing the day of presentation. Her reported temperature at home was 102°F, but the patient said she had been unable to take acetaminophen or ibuprofen because it was too painful to swallow. The patient had no significant medical history and reported no known recent streptococcal exposure. She denied alcohol use, but admitted to smoking an average of 10 cigarettes per day.
On physical examination, the patient’s vital signs were: HR, 92 beats/min; RR, 11 breaths/min; BP, 122/65 mm Hg; and T, 99.5°F. Oxygen saturation was 98% on room air. She was not drooling or tripoding. Throat examination revealed posterior oropharyngeal erythema, edema, and exudate, with a uvula displaced to the left with a right-sided asymmetric tonsillar swelling consistent with a significant peritonsillar abscess. The remainder of the physical examination was unremarkable.
Rapid strep and monospot testing were negative; the patient’s WBC was 9.1 x 109/L with a normal differential. After discussion with the attending EP, an IV line was started, and clindamycin 900 mg and dexamethasone 10 mg were administered. Arrangements were made with the university hospital ED for ALS ambulance transfer, as there were no otolaryngologist services available at the community hospital.
Upon arrival, the patient was examined by the university attending EP and was found to have mild asymmetry of the tonsils, but no midline disruption or uvula shift. The patient was given advice on symptomatic management and was discharged home.
Case Commentary
It is likely that transfer of this patient and its inherent risks could have been averted had the community EP personally assessed the patient prior to transfer arrangements. Supervision of physician extenders and residents in the ED may present challenges to patient safety, diagnostic accuracy, and appropriate treatment, especially in this era of volume and time-driven throughput metrics.
Emergency department transfers are costly and place patients and transport staff at a certain degree of risk. Both ground and air transfer include the possibility of collision, and ED-to-ED transfers should be reserved only for patients who need it. Furthermore, inappropriate transfers remove a transport vehicle and team from use by another patient in true need, resulting in added cost for no value to the patient, and negatively impact the receiving EP, who is left to answer the patient’s questions regarding why they had to be transferred.
An additional point to consider is the management of patient’s expectations when they are being transferred to a facility for more specialized care. At times, patients are led to believe they are being transferred for a certain test or procedure, yet when they arrive at the receiving facility, it is determined that intervention is no longer needed. Better patient communication on the part of the sending facility could help lessen the burden to the staff of the receiving facility when they need to explain why a certain test or procedure was actually not needed, despite the patient’s transfer. This is especially important in rare circumstances when the sending facility is staffed only by a PA or NP and not an ED attending.
Opportunities for Patient Safety Improvement. Active involvement of supervising attending physicians can mitigate the risk of inappropriate ED-to-ED transfers. The active supervisory role of attending EPs in patient care administered by physician extenders and residents is a serious responsibility that deserves priority. Communication with patients regarding their expectations should be initiated by the sending ED provider prior to transport.
Case 3
A primigravid 19-year-old woman at 24 weeks gestation with no prior prenatal care presented to a community hospital ED at 1:50
Since this community hospital had closed its obstetrical unit and moved all obstetrical and pediatric services to a sister hospital approximately 9 miles (13 minutes) away, the EP on duty immediately started IV fluids, ordered fetal heart tones (there was no fetal monitoring capability in the hospital), paged the obstetrician (OB) at the sister hospital, and activated an ALS ground transfer unit, all in parallel sequence. The OB on duty returned the page at 2:20
The discussion between the OB and EP included the risks and benefits of immediate transfer in the antenatal period versus the postpartum period; from the perspective of the EP, who had no access to safe fetal monitoring, labor and delivery support, or neonatal intensive care unit (NICU)/pediatric services, such transport was indicated. The EP felt strongly that the benefit of antenatal transfer outweighed the risk of delivering a late second-trimester fetus in an unsupported environment. However, the OB remained firm in his stance, and stated the patient was unstable and therefore could not be transferred under the law.
Hospital administration at the receiving hospital was paged to assist. The hospital administrator on duty returned the EP’s call at 2:57
The specialized pediatric transport team, with medical control from the pediatric hospital, arrived to transport the premature neonate in critical condition. Care was transferred to the transport team, but while preparing to load the patient into the transport incubator, the team questioned the position of the ET tube; they decided to extubate and reintubate the patient using their specialized equipment. The EP was not made aware of this decision. Unfortunately, after extubation, the transport team was unable to reintubate the neonate, who went into cardiopulmonary arrest and expired in the ED.
Case Commentary
Obstetrical emergencies are challenging even in a fully supported ED, and these challenges are heightened significantly in EDs that lack obstetric and pediatric support. In retrospect, it is truly difficult to determine if any action could or would have altered the outcome of this case.
In some circumstances, determining that a patient is “stable for transfer” or that the benefits of a transfer outweigh the risks is complicated and difficult. In this case, the patient was never “stable,” as she was in active labor. The EMTALA statute and its provisions govern when and how a patient may be transferred from one hospital to another when an unstable medical condition exists, but does not prohibit transferring an unstable patient. The OB’s understanding of the law was mistaken by the assumption that the patient was unstable and therefore could not be transferred at all.2 The essential provisions of the statute state that any patient who comes to the ED requesting examination or treatment for a medical condition must be provided with an appropriate medical screening examination to determine if (s)he is suffering from an emergency medical condition.3 If (s)he is, then the hospital is obligated to either provide him/her with treatment until (s)he is stable or transfer him/her to another hospital that has the capability to provide definitive care for the patient, and the benefit of transfer for this stabilizing care outweighs the risk of the transfer.3
Under the circumstances of this case scenario, it seems reasonable to transfer a pregnant patient in labor if the transferring physician felt that the safety of both mother and baby would be best served at the receiving hospital with specialized services and that the timing of the transfer was appropriate, considering the clinical findings and distance to the receiving hospital—with anticipation that delivery is most likely to occur after arrival at the receiving hospital.4 Again, this is a very complex situation, and the possibility exists that if the transfer proceeds, delivery could occur in the ambulance, which may introduce an additional potentially adverse event.
There is no time to delay in this decision-making process, and the risks and benefits of transfer are not clearly defined. The additional circumstance of an extremely preterm infant who will require specialized NICU care augments the need for expeditious transport to the sister hospital, as contrasted with active labor in a full-term gestation.
Part of EMTALA states “hospitals with specialized capabilities are obligated to accept transfers from hospitals who lack the capability to treat unstable emergency medical conditions.” In this case, the risk of delivering such a preterm infant at a hospital not equipped with even basic obstetrical and pediatric services may outweigh any potential risks of transport to a sister hospital 13 minutes away by ground transport. To mitigate the risk of an in-transit delivery, supporting the transport team with a physician or registered nurse to ride along may have been an option.
Finally, delivery of the premature newborn created a second unstable patient in even greater danger than the mother. Interhospital transfers of critically ill and injured pediatric patients to pediatric hospitals often involve specialized transfer units staffed by expertly trained paramedic and/or nurse teams under the medical control of the pediatric hospital. The unfortunate outcome of this premature infant may have been the ultimate outcome at 24 weeks, despite the extubation and inability of the team to re-intubate. However, communication with the EP in the department in the decision to change the ET tube may have been helpful to the team in the face of a difficult re-intubation.
Opportunities for Patient Safety Improvement. A solid understanding of the EMTALA statute and its provisions is essential not only for providers in the ED, but also for consultants who must understand their responsibilities under the law. Timely transfer arrangements cannot be underestimated, and hospital policy should support expeditious positive responses in emergent situations. Active communication between the sending EP and transport team while still in the ED is prudent.
Conclusion
Interhospital ED-to-ED transfers are frequent occurrences in many EDs. An ED-to-ED transfer of a patient is a process that often involves complex decision-making and a rapid but thorough assessment of the potential risks and benefits. At each stage of the transfer process, each party involved must anticipate, to best degree possible, patient risks and communicate these risks clearly from the pretransfer phase to the transfer team and to the receiving facility. Assurance of the six aims of the Institute of Medicine5 are central to good decision-making that leads to an appropriate disposition of patient transfer to another ED. These aims demand that care delivered is safe, timely, effective, patient-centered, efficient, and equitable.5 When interhospital ED-to-ED transfer is deemed necessary, the sending provider generally is responsible for making certain the right care at the right time is safeguarded from the time the patient enters the ED until he arrives at the receiving ED. The receiving ED then completely assumes the responsibility to evaluate and manage the patient until the definitive caregiver takes over.
1. Appropriate Interfacility Patient Transfer. ACEP Clinical Policy. https://www.acep.org/clinical---practice-management/appropriate-interfacility-patient-transfer/. Accessed December 14, 2016.
2. Frequently Asked Questions About The Emergency Medical Treatment and Active Labor Act. http://www.emtala.com/faq.htm Accessed January 14, 2017.
3. The Emergency Medical Treatment and Active Labor Act, as established under the Consolidated Omnibus Budget Reconciliation Act (COBRA) of 1985 (42 USC 1395 dd) and 42 CFR 489.24; 42 CFR489.20 (EMTALA regulations).
4. The American College of Obstetricians and Gynecologists. Committee Opinion. Hospital Based Triage of Obstetric Patients. http://www.acog.org/Resources-And-Publications/Committee-Opinions/Committee-on-Obstetric-Practice/Hospital-Based-Triage-of-Obstetric-Patients. Accessed January 19, 2017.
5. Institute of Medicine Committee on Quality in Healthcare in America. Crossing the Quality Chasm: A New Health System for the 21st Century. Washington, DC: National Academies Press; 2001.
Emergency Department (ED)-to-ED transfers are a reality of practice in emergency medicine, and they can certainly present a tall order for ensuring patient safety. Challenges abound in getting the right patient to the right place at the right time by the right transportation method.1 A critically ill patient becomes a metaphorical baton to be passed on, requiring the best care along the way--even when the patient is not completely aware of the reasons for the transfer of care. For some EDs, ED-to-ED transfers have become a common daily occurrence. The realities of freestanding EDs, hospital overcrowding, and subspecialty coverage gaps create challenges in direct hospital admission, necessitating a second ED stop before the patient reaches an appropriate destination and provider for definitive care.
The transfer of a patient is much more complex than arranging and carrying out an ambulance ride across town. If thought of as a process, with pre-transfer planning on the sending end, the transfer itself, and post-transfer assurance of care continuity on the receiving end, the quality of care and patient safety can be elevated. Emergency department-to-ED transfers require careful attention to communication, with important hand-offs between the sending facility, the ambulance personnel, and the receiving facility. To lead the discussion around the perils of interhospital ED-to-ED transfers, the following case reports illustrate some of the challenges encountered.
Case Scenarios
Case 1
A 58-year-old man presented to a freestanding ED at 10:30
The nursing staff obtained intravenous (IV) access, and blood samples were drawn. Parenteral pain control and antiemetics were administered while a computed tomography (CT) scan of the abdomen and pelvis with contrast was emergently in progress. Meanwhile, the laboratory test results included the following: lactate, 3.8 mmol/L; lipase 42 U/L; carbon monoxide, 14 mmol/L; white blood cell count (WBC), 12 x 109/L without bandemia; serum creatinine, 1.0 mg/dL; liver function tests with a mild elevation of transaminases; and normal coagulation studies.
After reviewing the CT scan, the radiologist called to report a hyperdensity in the lumen of the superior mesenteric artery, which might represent a subsegmental dissection or a partial occlusion or plaque, with no radiographic evidence of bowel ischemia. Vascular surgery service was consulted, and the decision to start IV heparin was agreed upon. The vascular surgeon requested that a mesenteric peripheral vascular laboratory examination (PVL) be arranged on arrival at the hospital ED, and an ED-to-ED transfer to the hospital was arranged. The case was discussed with the receiving day shift emergency physician (EP), who planned to order the mesenteric PVL immediately upon the patient’s arrival.
Emergent transportation via an advanced life support (ALS) ambulance was arranged. The nursing report was called in to the hospital ED at noon, and the ALS unit had arrived and was ready to transfer the patient. Repeat vital signs were obtained, revealing an HR of 45 beats/min and a BP of 200/100 mm Hg.
After an uneventful transport, the patient arrived at the hospital ED at 12:45
Case Commentary
In this case, when the nursing assessment preceding transfer revealed sustained abnormal vital signs particularly the significant hypertension, reassessment and blood pressure management by the sending EP prior to transport may have diminished the poor outcome resulting from the intracranial hemorrhage. Ideally, BP control should have been implemented prior to transport—especially in the context of possible arterial dissection/occlusion with ongoing anticoagulation therapy. If such attempts to control BP prior to transport prove inadequate, a hand-off communication with the receiving EP is indicated, emphasizing the need for immediate evaluation and critical intervention upon patient arrival. On the receiving end of a patient transfer, it is good practice that all critically ill or injured patients be immediately assessed upon arrival at the ED, regardless of planned interventions by any other department.
Transport from another ED cannot mislead to a false sense of security that ED care is completed. Patients geographically located in the ED (especially those who are newly arrived) are the responsibility of the ED providers until that point where the next specialist provider clearly assumes care of the patient (see “ED-to-ED Transfers: Summary of Responsibilities”). This point in handoff time can be murky and unclear; yet, as illustrated by this case, it is best to re-evaluate and ensure appropriate emergency care of the patient upon arrival. In addition, as with any other ED patient with a change in condition, timely re-evaluation of the transferred patient is indicated upon receiving the report from nursing that the patient’s condition had changed.
Transfers between EDs should be viewed as a process, and that each phase in the process is important—from the pre-transfer preparation at the sending facility, the physical transfer itself by transport personnel, and the post-transfer arrival that requires the receiving facility to ensure care continues seamlessly and appropriately. Even in situations of high acuity and/or high volume, anticipation and timely attention is required by the receiving staff to ensure continuity of safe patient care. The metaphorical baton was dropped in this transfer.
Opportunities for Patient Safety Improvement. The sending facility should always address any abnormal vital signs prior to patient transfer. The receiving facility should evaluate all transferred patients at the bedside as soon as possible upon arrival. Both facilities should take timely advantage of the information the nurses provide, especially when there is a change in the patient’s condition. All involved physicians from the sending facility should communicate to the receiving ED staff critical and potentially critical patient care information and concerns that pose a risk of deterioration of the patient’s condition.
Case 2
A 23-year-old woman presented to a community hospital ED with a sore throat, fever, and difficulty swallowing. The PA on duty saw the patient in the fast track section of the ED. The patient reported the sore throat had been persistent for the past 3 days, and that she began having difficulty swallowing the day of presentation. Her reported temperature at home was 102°F, but the patient said she had been unable to take acetaminophen or ibuprofen because it was too painful to swallow. The patient had no significant medical history and reported no known recent streptococcal exposure. She denied alcohol use, but admitted to smoking an average of 10 cigarettes per day.
On physical examination, the patient’s vital signs were: HR, 92 beats/min; RR, 11 breaths/min; BP, 122/65 mm Hg; and T, 99.5°F. Oxygen saturation was 98% on room air. She was not drooling or tripoding. Throat examination revealed posterior oropharyngeal erythema, edema, and exudate, with a uvula displaced to the left with a right-sided asymmetric tonsillar swelling consistent with a significant peritonsillar abscess. The remainder of the physical examination was unremarkable.
Rapid strep and monospot testing were negative; the patient’s WBC was 9.1 x 109/L with a normal differential. After discussion with the attending EP, an IV line was started, and clindamycin 900 mg and dexamethasone 10 mg were administered. Arrangements were made with the university hospital ED for ALS ambulance transfer, as there were no otolaryngologist services available at the community hospital.
Upon arrival, the patient was examined by the university attending EP and was found to have mild asymmetry of the tonsils, but no midline disruption or uvula shift. The patient was given advice on symptomatic management and was discharged home.
Case Commentary
It is likely that transfer of this patient and its inherent risks could have been averted had the community EP personally assessed the patient prior to transfer arrangements. Supervision of physician extenders and residents in the ED may present challenges to patient safety, diagnostic accuracy, and appropriate treatment, especially in this era of volume and time-driven throughput metrics.
Emergency department transfers are costly and place patients and transport staff at a certain degree of risk. Both ground and air transfer include the possibility of collision, and ED-to-ED transfers should be reserved only for patients who need it. Furthermore, inappropriate transfers remove a transport vehicle and team from use by another patient in true need, resulting in added cost for no value to the patient, and negatively impact the receiving EP, who is left to answer the patient’s questions regarding why they had to be transferred.
An additional point to consider is the management of patient’s expectations when they are being transferred to a facility for more specialized care. At times, patients are led to believe they are being transferred for a certain test or procedure, yet when they arrive at the receiving facility, it is determined that intervention is no longer needed. Better patient communication on the part of the sending facility could help lessen the burden to the staff of the receiving facility when they need to explain why a certain test or procedure was actually not needed, despite the patient’s transfer. This is especially important in rare circumstances when the sending facility is staffed only by a PA or NP and not an ED attending.
Opportunities for Patient Safety Improvement. Active involvement of supervising attending physicians can mitigate the risk of inappropriate ED-to-ED transfers. The active supervisory role of attending EPs in patient care administered by physician extenders and residents is a serious responsibility that deserves priority. Communication with patients regarding their expectations should be initiated by the sending ED provider prior to transport.
Case 3
A primigravid 19-year-old woman at 24 weeks gestation with no prior prenatal care presented to a community hospital ED at 1:50
Since this community hospital had closed its obstetrical unit and moved all obstetrical and pediatric services to a sister hospital approximately 9 miles (13 minutes) away, the EP on duty immediately started IV fluids, ordered fetal heart tones (there was no fetal monitoring capability in the hospital), paged the obstetrician (OB) at the sister hospital, and activated an ALS ground transfer unit, all in parallel sequence. The OB on duty returned the page at 2:20
The discussion between the OB and EP included the risks and benefits of immediate transfer in the antenatal period versus the postpartum period; from the perspective of the EP, who had no access to safe fetal monitoring, labor and delivery support, or neonatal intensive care unit (NICU)/pediatric services, such transport was indicated. The EP felt strongly that the benefit of antenatal transfer outweighed the risk of delivering a late second-trimester fetus in an unsupported environment. However, the OB remained firm in his stance, and stated the patient was unstable and therefore could not be transferred under the law.
Hospital administration at the receiving hospital was paged to assist. The hospital administrator on duty returned the EP’s call at 2:57
The specialized pediatric transport team, with medical control from the pediatric hospital, arrived to transport the premature neonate in critical condition. Care was transferred to the transport team, but while preparing to load the patient into the transport incubator, the team questioned the position of the ET tube; they decided to extubate and reintubate the patient using their specialized equipment. The EP was not made aware of this decision. Unfortunately, after extubation, the transport team was unable to reintubate the neonate, who went into cardiopulmonary arrest and expired in the ED.
Case Commentary
Obstetrical emergencies are challenging even in a fully supported ED, and these challenges are heightened significantly in EDs that lack obstetric and pediatric support. In retrospect, it is truly difficult to determine if any action could or would have altered the outcome of this case.
In some circumstances, determining that a patient is “stable for transfer” or that the benefits of a transfer outweigh the risks is complicated and difficult. In this case, the patient was never “stable,” as she was in active labor. The EMTALA statute and its provisions govern when and how a patient may be transferred from one hospital to another when an unstable medical condition exists, but does not prohibit transferring an unstable patient. The OB’s understanding of the law was mistaken by the assumption that the patient was unstable and therefore could not be transferred at all.2 The essential provisions of the statute state that any patient who comes to the ED requesting examination or treatment for a medical condition must be provided with an appropriate medical screening examination to determine if (s)he is suffering from an emergency medical condition.3 If (s)he is, then the hospital is obligated to either provide him/her with treatment until (s)he is stable or transfer him/her to another hospital that has the capability to provide definitive care for the patient, and the benefit of transfer for this stabilizing care outweighs the risk of the transfer.3
Under the circumstances of this case scenario, it seems reasonable to transfer a pregnant patient in labor if the transferring physician felt that the safety of both mother and baby would be best served at the receiving hospital with specialized services and that the timing of the transfer was appropriate, considering the clinical findings and distance to the receiving hospital—with anticipation that delivery is most likely to occur after arrival at the receiving hospital.4 Again, this is a very complex situation, and the possibility exists that if the transfer proceeds, delivery could occur in the ambulance, which may introduce an additional potentially adverse event.
There is no time to delay in this decision-making process, and the risks and benefits of transfer are not clearly defined. The additional circumstance of an extremely preterm infant who will require specialized NICU care augments the need for expeditious transport to the sister hospital, as contrasted with active labor in a full-term gestation.
Part of EMTALA states “hospitals with specialized capabilities are obligated to accept transfers from hospitals who lack the capability to treat unstable emergency medical conditions.” In this case, the risk of delivering such a preterm infant at a hospital not equipped with even basic obstetrical and pediatric services may outweigh any potential risks of transport to a sister hospital 13 minutes away by ground transport. To mitigate the risk of an in-transit delivery, supporting the transport team with a physician or registered nurse to ride along may have been an option.
Finally, delivery of the premature newborn created a second unstable patient in even greater danger than the mother. Interhospital transfers of critically ill and injured pediatric patients to pediatric hospitals often involve specialized transfer units staffed by expertly trained paramedic and/or nurse teams under the medical control of the pediatric hospital. The unfortunate outcome of this premature infant may have been the ultimate outcome at 24 weeks, despite the extubation and inability of the team to re-intubate. However, communication with the EP in the department in the decision to change the ET tube may have been helpful to the team in the face of a difficult re-intubation.
Opportunities for Patient Safety Improvement. A solid understanding of the EMTALA statute and its provisions is essential not only for providers in the ED, but also for consultants who must understand their responsibilities under the law. Timely transfer arrangements cannot be underestimated, and hospital policy should support expeditious positive responses in emergent situations. Active communication between the sending EP and transport team while still in the ED is prudent.
Conclusion
Interhospital ED-to-ED transfers are frequent occurrences in many EDs. An ED-to-ED transfer of a patient is a process that often involves complex decision-making and a rapid but thorough assessment of the potential risks and benefits. At each stage of the transfer process, each party involved must anticipate, to best degree possible, patient risks and communicate these risks clearly from the pretransfer phase to the transfer team and to the receiving facility. Assurance of the six aims of the Institute of Medicine5 are central to good decision-making that leads to an appropriate disposition of patient transfer to another ED. These aims demand that care delivered is safe, timely, effective, patient-centered, efficient, and equitable.5 When interhospital ED-to-ED transfer is deemed necessary, the sending provider generally is responsible for making certain the right care at the right time is safeguarded from the time the patient enters the ED until he arrives at the receiving ED. The receiving ED then completely assumes the responsibility to evaluate and manage the patient until the definitive caregiver takes over.
Emergency Department (ED)-to-ED transfers are a reality of practice in emergency medicine, and they can certainly present a tall order for ensuring patient safety. Challenges abound in getting the right patient to the right place at the right time by the right transportation method.1 A critically ill patient becomes a metaphorical baton to be passed on, requiring the best care along the way--even when the patient is not completely aware of the reasons for the transfer of care. For some EDs, ED-to-ED transfers have become a common daily occurrence. The realities of freestanding EDs, hospital overcrowding, and subspecialty coverage gaps create challenges in direct hospital admission, necessitating a second ED stop before the patient reaches an appropriate destination and provider for definitive care.
The transfer of a patient is much more complex than arranging and carrying out an ambulance ride across town. If thought of as a process, with pre-transfer planning on the sending end, the transfer itself, and post-transfer assurance of care continuity on the receiving end, the quality of care and patient safety can be elevated. Emergency department-to-ED transfers require careful attention to communication, with important hand-offs between the sending facility, the ambulance personnel, and the receiving facility. To lead the discussion around the perils of interhospital ED-to-ED transfers, the following case reports illustrate some of the challenges encountered.
Case Scenarios
Case 1
A 58-year-old man presented to a freestanding ED at 10:30
The nursing staff obtained intravenous (IV) access, and blood samples were drawn. Parenteral pain control and antiemetics were administered while a computed tomography (CT) scan of the abdomen and pelvis with contrast was emergently in progress. Meanwhile, the laboratory test results included the following: lactate, 3.8 mmol/L; lipase 42 U/L; carbon monoxide, 14 mmol/L; white blood cell count (WBC), 12 x 109/L without bandemia; serum creatinine, 1.0 mg/dL; liver function tests with a mild elevation of transaminases; and normal coagulation studies.
After reviewing the CT scan, the radiologist called to report a hyperdensity in the lumen of the superior mesenteric artery, which might represent a subsegmental dissection or a partial occlusion or plaque, with no radiographic evidence of bowel ischemia. Vascular surgery service was consulted, and the decision to start IV heparin was agreed upon. The vascular surgeon requested that a mesenteric peripheral vascular laboratory examination (PVL) be arranged on arrival at the hospital ED, and an ED-to-ED transfer to the hospital was arranged. The case was discussed with the receiving day shift emergency physician (EP), who planned to order the mesenteric PVL immediately upon the patient’s arrival.
Emergent transportation via an advanced life support (ALS) ambulance was arranged. The nursing report was called in to the hospital ED at noon, and the ALS unit had arrived and was ready to transfer the patient. Repeat vital signs were obtained, revealing an HR of 45 beats/min and a BP of 200/100 mm Hg.
After an uneventful transport, the patient arrived at the hospital ED at 12:45
Case Commentary
In this case, when the nursing assessment preceding transfer revealed sustained abnormal vital signs particularly the significant hypertension, reassessment and blood pressure management by the sending EP prior to transport may have diminished the poor outcome resulting from the intracranial hemorrhage. Ideally, BP control should have been implemented prior to transport—especially in the context of possible arterial dissection/occlusion with ongoing anticoagulation therapy. If such attempts to control BP prior to transport prove inadequate, a hand-off communication with the receiving EP is indicated, emphasizing the need for immediate evaluation and critical intervention upon patient arrival. On the receiving end of a patient transfer, it is good practice that all critically ill or injured patients be immediately assessed upon arrival at the ED, regardless of planned interventions by any other department.
Transport from another ED cannot mislead to a false sense of security that ED care is completed. Patients geographically located in the ED (especially those who are newly arrived) are the responsibility of the ED providers until that point where the next specialist provider clearly assumes care of the patient (see “ED-to-ED Transfers: Summary of Responsibilities”). This point in handoff time can be murky and unclear; yet, as illustrated by this case, it is best to re-evaluate and ensure appropriate emergency care of the patient upon arrival. In addition, as with any other ED patient with a change in condition, timely re-evaluation of the transferred patient is indicated upon receiving the report from nursing that the patient’s condition had changed.
Transfers between EDs should be viewed as a process, and that each phase in the process is important—from the pre-transfer preparation at the sending facility, the physical transfer itself by transport personnel, and the post-transfer arrival that requires the receiving facility to ensure care continues seamlessly and appropriately. Even in situations of high acuity and/or high volume, anticipation and timely attention is required by the receiving staff to ensure continuity of safe patient care. The metaphorical baton was dropped in this transfer.
Opportunities for Patient Safety Improvement. The sending facility should always address any abnormal vital signs prior to patient transfer. The receiving facility should evaluate all transferred patients at the bedside as soon as possible upon arrival. Both facilities should take timely advantage of the information the nurses provide, especially when there is a change in the patient’s condition. All involved physicians from the sending facility should communicate to the receiving ED staff critical and potentially critical patient care information and concerns that pose a risk of deterioration of the patient’s condition.
Case 2
A 23-year-old woman presented to a community hospital ED with a sore throat, fever, and difficulty swallowing. The PA on duty saw the patient in the fast track section of the ED. The patient reported the sore throat had been persistent for the past 3 days, and that she began having difficulty swallowing the day of presentation. Her reported temperature at home was 102°F, but the patient said she had been unable to take acetaminophen or ibuprofen because it was too painful to swallow. The patient had no significant medical history and reported no known recent streptococcal exposure. She denied alcohol use, but admitted to smoking an average of 10 cigarettes per day.
On physical examination, the patient’s vital signs were: HR, 92 beats/min; RR, 11 breaths/min; BP, 122/65 mm Hg; and T, 99.5°F. Oxygen saturation was 98% on room air. She was not drooling or tripoding. Throat examination revealed posterior oropharyngeal erythema, edema, and exudate, with a uvula displaced to the left with a right-sided asymmetric tonsillar swelling consistent with a significant peritonsillar abscess. The remainder of the physical examination was unremarkable.
Rapid strep and monospot testing were negative; the patient’s WBC was 9.1 x 109/L with a normal differential. After discussion with the attending EP, an IV line was started, and clindamycin 900 mg and dexamethasone 10 mg were administered. Arrangements were made with the university hospital ED for ALS ambulance transfer, as there were no otolaryngologist services available at the community hospital.
Upon arrival, the patient was examined by the university attending EP and was found to have mild asymmetry of the tonsils, but no midline disruption or uvula shift. The patient was given advice on symptomatic management and was discharged home.
Case Commentary
It is likely that transfer of this patient and its inherent risks could have been averted had the community EP personally assessed the patient prior to transfer arrangements. Supervision of physician extenders and residents in the ED may present challenges to patient safety, diagnostic accuracy, and appropriate treatment, especially in this era of volume and time-driven throughput metrics.
Emergency department transfers are costly and place patients and transport staff at a certain degree of risk. Both ground and air transfer include the possibility of collision, and ED-to-ED transfers should be reserved only for patients who need it. Furthermore, inappropriate transfers remove a transport vehicle and team from use by another patient in true need, resulting in added cost for no value to the patient, and negatively impact the receiving EP, who is left to answer the patient’s questions regarding why they had to be transferred.
An additional point to consider is the management of patient’s expectations when they are being transferred to a facility for more specialized care. At times, patients are led to believe they are being transferred for a certain test or procedure, yet when they arrive at the receiving facility, it is determined that intervention is no longer needed. Better patient communication on the part of the sending facility could help lessen the burden to the staff of the receiving facility when they need to explain why a certain test or procedure was actually not needed, despite the patient’s transfer. This is especially important in rare circumstances when the sending facility is staffed only by a PA or NP and not an ED attending.
Opportunities for Patient Safety Improvement. Active involvement of supervising attending physicians can mitigate the risk of inappropriate ED-to-ED transfers. The active supervisory role of attending EPs in patient care administered by physician extenders and residents is a serious responsibility that deserves priority. Communication with patients regarding their expectations should be initiated by the sending ED provider prior to transport.
Case 3
A primigravid 19-year-old woman at 24 weeks gestation with no prior prenatal care presented to a community hospital ED at 1:50
Since this community hospital had closed its obstetrical unit and moved all obstetrical and pediatric services to a sister hospital approximately 9 miles (13 minutes) away, the EP on duty immediately started IV fluids, ordered fetal heart tones (there was no fetal monitoring capability in the hospital), paged the obstetrician (OB) at the sister hospital, and activated an ALS ground transfer unit, all in parallel sequence. The OB on duty returned the page at 2:20
The discussion between the OB and EP included the risks and benefits of immediate transfer in the antenatal period versus the postpartum period; from the perspective of the EP, who had no access to safe fetal monitoring, labor and delivery support, or neonatal intensive care unit (NICU)/pediatric services, such transport was indicated. The EP felt strongly that the benefit of antenatal transfer outweighed the risk of delivering a late second-trimester fetus in an unsupported environment. However, the OB remained firm in his stance, and stated the patient was unstable and therefore could not be transferred under the law.
Hospital administration at the receiving hospital was paged to assist. The hospital administrator on duty returned the EP’s call at 2:57
The specialized pediatric transport team, with medical control from the pediatric hospital, arrived to transport the premature neonate in critical condition. Care was transferred to the transport team, but while preparing to load the patient into the transport incubator, the team questioned the position of the ET tube; they decided to extubate and reintubate the patient using their specialized equipment. The EP was not made aware of this decision. Unfortunately, after extubation, the transport team was unable to reintubate the neonate, who went into cardiopulmonary arrest and expired in the ED.
Case Commentary
Obstetrical emergencies are challenging even in a fully supported ED, and these challenges are heightened significantly in EDs that lack obstetric and pediatric support. In retrospect, it is truly difficult to determine if any action could or would have altered the outcome of this case.
In some circumstances, determining that a patient is “stable for transfer” or that the benefits of a transfer outweigh the risks is complicated and difficult. In this case, the patient was never “stable,” as she was in active labor. The EMTALA statute and its provisions govern when and how a patient may be transferred from one hospital to another when an unstable medical condition exists, but does not prohibit transferring an unstable patient. The OB’s understanding of the law was mistaken by the assumption that the patient was unstable and therefore could not be transferred at all.2 The essential provisions of the statute state that any patient who comes to the ED requesting examination or treatment for a medical condition must be provided with an appropriate medical screening examination to determine if (s)he is suffering from an emergency medical condition.3 If (s)he is, then the hospital is obligated to either provide him/her with treatment until (s)he is stable or transfer him/her to another hospital that has the capability to provide definitive care for the patient, and the benefit of transfer for this stabilizing care outweighs the risk of the transfer.3
Under the circumstances of this case scenario, it seems reasonable to transfer a pregnant patient in labor if the transferring physician felt that the safety of both mother and baby would be best served at the receiving hospital with specialized services and that the timing of the transfer was appropriate, considering the clinical findings and distance to the receiving hospital—with anticipation that delivery is most likely to occur after arrival at the receiving hospital.4 Again, this is a very complex situation, and the possibility exists that if the transfer proceeds, delivery could occur in the ambulance, which may introduce an additional potentially adverse event.
There is no time to delay in this decision-making process, and the risks and benefits of transfer are not clearly defined. The additional circumstance of an extremely preterm infant who will require specialized NICU care augments the need for expeditious transport to the sister hospital, as contrasted with active labor in a full-term gestation.
Part of EMTALA states “hospitals with specialized capabilities are obligated to accept transfers from hospitals who lack the capability to treat unstable emergency medical conditions.” In this case, the risk of delivering such a preterm infant at a hospital not equipped with even basic obstetrical and pediatric services may outweigh any potential risks of transport to a sister hospital 13 minutes away by ground transport. To mitigate the risk of an in-transit delivery, supporting the transport team with a physician or registered nurse to ride along may have been an option.
Finally, delivery of the premature newborn created a second unstable patient in even greater danger than the mother. Interhospital transfers of critically ill and injured pediatric patients to pediatric hospitals often involve specialized transfer units staffed by expertly trained paramedic and/or nurse teams under the medical control of the pediatric hospital. The unfortunate outcome of this premature infant may have been the ultimate outcome at 24 weeks, despite the extubation and inability of the team to re-intubate. However, communication with the EP in the department in the decision to change the ET tube may have been helpful to the team in the face of a difficult re-intubation.
Opportunities for Patient Safety Improvement. A solid understanding of the EMTALA statute and its provisions is essential not only for providers in the ED, but also for consultants who must understand their responsibilities under the law. Timely transfer arrangements cannot be underestimated, and hospital policy should support expeditious positive responses in emergent situations. Active communication between the sending EP and transport team while still in the ED is prudent.
Conclusion
Interhospital ED-to-ED transfers are frequent occurrences in many EDs. An ED-to-ED transfer of a patient is a process that often involves complex decision-making and a rapid but thorough assessment of the potential risks and benefits. At each stage of the transfer process, each party involved must anticipate, to best degree possible, patient risks and communicate these risks clearly from the pretransfer phase to the transfer team and to the receiving facility. Assurance of the six aims of the Institute of Medicine5 are central to good decision-making that leads to an appropriate disposition of patient transfer to another ED. These aims demand that care delivered is safe, timely, effective, patient-centered, efficient, and equitable.5 When interhospital ED-to-ED transfer is deemed necessary, the sending provider generally is responsible for making certain the right care at the right time is safeguarded from the time the patient enters the ED until he arrives at the receiving ED. The receiving ED then completely assumes the responsibility to evaluate and manage the patient until the definitive caregiver takes over.
1. Appropriate Interfacility Patient Transfer. ACEP Clinical Policy. https://www.acep.org/clinical---practice-management/appropriate-interfacility-patient-transfer/. Accessed December 14, 2016.
2. Frequently Asked Questions About The Emergency Medical Treatment and Active Labor Act. http://www.emtala.com/faq.htm Accessed January 14, 2017.
3. The Emergency Medical Treatment and Active Labor Act, as established under the Consolidated Omnibus Budget Reconciliation Act (COBRA) of 1985 (42 USC 1395 dd) and 42 CFR 489.24; 42 CFR489.20 (EMTALA regulations).
4. The American College of Obstetricians and Gynecologists. Committee Opinion. Hospital Based Triage of Obstetric Patients. http://www.acog.org/Resources-And-Publications/Committee-Opinions/Committee-on-Obstetric-Practice/Hospital-Based-Triage-of-Obstetric-Patients. Accessed January 19, 2017.
5. Institute of Medicine Committee on Quality in Healthcare in America. Crossing the Quality Chasm: A New Health System for the 21st Century. Washington, DC: National Academies Press; 2001.
1. Appropriate Interfacility Patient Transfer. ACEP Clinical Policy. https://www.acep.org/clinical---practice-management/appropriate-interfacility-patient-transfer/. Accessed December 14, 2016.
2. Frequently Asked Questions About The Emergency Medical Treatment and Active Labor Act. http://www.emtala.com/faq.htm Accessed January 14, 2017.
3. The Emergency Medical Treatment and Active Labor Act, as established under the Consolidated Omnibus Budget Reconciliation Act (COBRA) of 1985 (42 USC 1395 dd) and 42 CFR 489.24; 42 CFR489.20 (EMTALA regulations).
4. The American College of Obstetricians and Gynecologists. Committee Opinion. Hospital Based Triage of Obstetric Patients. http://www.acog.org/Resources-And-Publications/Committee-Opinions/Committee-on-Obstetric-Practice/Hospital-Based-Triage-of-Obstetric-Patients. Accessed January 19, 2017.
5. Institute of Medicine Committee on Quality in Healthcare in America. Crossing the Quality Chasm: A New Health System for the 21st Century. Washington, DC: National Academies Press; 2001.

Management of Trauma and Burn Scars: The Dermatologist’s Role in Expanding Patient Access to Care
Hypertrophic scarring secondary to trauma, burns, and surgical interventions is a major source of morbidity worldwide and often is mechanically, aesthetically, and symptomatically debilitating. Modern advances in acute trauma care protocols have resulted in survival rates greater than 90% in both civilian and military populations.1,2 Patients with wounds that have historically proven fatal are now surviving and are confronted with the long-term sequelae of their injuries. With more than 52,000 service members injured in military engagements from 2001 to 2015 and 8.5 million civilians presenting annually with injury patterns at risk for hypertrophic scarring, it is paramount that we ensure access to safe and effective long-term scar care.2,3
At its simplest level, hypertrophic scarring is believed to result from a disequilibrium between collagen production and degradation. This failure to properly transition through the stages of wound healing results in bothersome symptoms, a disfigured appearance, and mechanical dysfunction of the skin (Figure, A). Decreased elasticity and extensibility, increased dermal thickness, and scar contractures impair patient range of motion and functional mobility. Those affected commonly experience varying degrees of pruritus and dysesthesia along the scar. Combined with aesthetic variations in pigmentation, erythema, texture, and thickness, hypertrophic scarring often leads to long-term psychosocial impairment and decreased health-related quality of life.4
Treatment Approach
Treatment of hypertrophic scars requires a multimodal approach due to the spectrum of associated concerns and the natural recalcitrance of the scar to therapy. Protocols should be tailored to the individual but generally begin with tissue-conserving surgical interventions followed by selective photothermolysis of the scar vasculature. Subsequently, deep and superficial ablative fractional laser (AFL) treatment and local pharmacotherapy also are employed. Treatment can be accomplished in the outpatient setting under local anesthesia in a serial fashion. In the authors’ experience, these therapies behave in a synergistic fashion, achieving outcomes that far exceed the sum of their parts, often obviating the need for scar excision in the majority of cases (Figure, B).
Tissue-Conserving Surgical Intervention
Z-plasty is an indispensable surgical tool due to its ability to lengthen scars and reduce wound tension. Treatment is easily customizable to the patient and can be performed using the individual or multiple Z-plasty techniques. Undermining and step-off correction while suturing allow the physician to lower raised scars, elevate depressed scars, and obscure scar presence by minimizing the straight lines that draw the eye to the scar. Z-plasties rely on the creation and transposition of 2 triangular flaps and permit a 75% increase in length along the desired tension vector. As such, Z-plasties decrease wound tension and facilitate scar maturation.
Selective Photothermolysis of the Vasculature
Although there are several devices available to treat vascular and immature hypertrophic scars, the majority of studies have been conducted with the 595-nm pulsed dye laser. By preferentially heating oxyhemoglobin within the dermal microvasculature, the pulsed dye laser irreparably injures the vascular endothelium. The subsequent tissue hypoxia and collagen fiber heating results in collagen fiber realignment, normalization of collagen subtypes, and neocollagenesis.5 Pulsed dye laser therapy most effectively reduces erythema and pruritus; however, improvements in scar volume, pliability, and elasticity also have been reported.5 When targeting the fine vasculature of the scar, thermal confinement is critical to prevent injury to the surrounding dermis. As such, pulse widths of 0.45 to 1.5 milliseconds are routinely utilized with a fluence just sufficient to elicit transient purpura lasting 3 to 5 seconds. Employing a spot size of 7 to 10 mm, typical fluences range from 4.5 to 6.5 J/cm2. Engagement of the dynamic cooling device reduces the risk for complications, allowing the patient to proceed to the next step in their therapy regimen: the AFL.
Ablative Fractional Laser
The AFL creates a pixilated pattern of injury throughout the epidermis and dermis of the treatment area. Ablative fractional laser platforms include the 10,600-nm CO2 and 2940-nm erbium-doped YAG lasers, both targeting intracellular water. The AFL vaporizes columns of tissue, leaving minute vertical channels with narrow rims of protein coagulation referred to as microscopic treatment zones (MTZs).6 Scar collagen analysis after AFL treatment has shown a profile resembling unaffected skin.7 Consistently, patients report improvements in stiffness, range of motion, pain, pruritus, pigmentation, and erythema.Physician observers also have reported similar improvements in these end points.8,9 Recently, interim data from a prospective controlled trial were presented showing objective improvements in dermal thickness, elasticity, and extensibility after 3 treatments with the CO2 AFL.6 The UltraPulse CO2 laser (Lumenis) is the most well-studied and widely available AFL for scar therapy and as such we will outline common treatment parameters with this device. Of note, treatment end points may be generalized to any AFL.
The DeepFX UltraPulse configuration is utilized to achieve deep AFL therapy and has a fixed pulse width of 0.8 milliseconds, slightly less than the thermal relaxation time of the skin. The diameter of the MTZs is 120 µm, and MTZ density for scar treatment ranges from 1% to 10% with a goal depth of at least 80% of scar thickness. Maximal penetration of the AFL is 4 mm, which is directly proportional to fluence. The goal of deep AFL is the removal of scar tissue to facilitate remodeling and neocollagenesis. Superficial fractional ablation can then be achieved utilizing the ActiveFX UltraPulse configuration generating a 1.3-mm MTZ spot size. We commonly use a treatment level of 3 (82% density). Typical treatment energy ranges from 80 to 125 mJ, which correlates with depths of approximately 50 to 115 µm. With both configurations, the size and shape of the treatment area can be customized to the scar. In addition, frequency may be adjusted to control the speed of treatment while balancing the risk of bulk heating. The goal of superficial AFL is to minimize scar surface irregularities and ensure blending of deep AFL treatment. Once AFL treatment is complete, local pharmacotherapy can then be employed.
Pharmacotherapy
Intralesional corticosteroids have long represented the standard of care for hypertrophic scars, with concentrations between 2.5 and 40 mg/mL that are titrated to scar thickness and location to avoid unwanted atrophy. Visual blanching of the scar represents the clinical end point for treatment. Corticosteroids act by inhibiting fibroblast proliferation and enhancing collagen degradation.10 5-Fluorouracil (5-FU) also is used in scar management. In addition to inhibiting fibroblast proliferation and inducing fibroblast apoptosis, 5-FU inhibits myofibroblast proliferation, which is helpful in the prevention and treatment of scar contracture.11 As monotherapy, weekly injections with 1 to 3 mL of 50 mg/mL 5-FU has been safe and effective. Combination intralesional corticosteroid and 5-FU therapy has been reported and is associated with improved scar regression, reduced reoccurrence, and fewer side effects.11 In our experience, a 1:1 suspension is effective with appropriate titration of the corticosteroid component. Although less well defined, topical application of pharmacotherapy and massage to the newly created MTZs appears beneficial and offers another option for delivery of corticosteroids and 5-FU, in addition to a number of promising medications such as bimatoprost, poly-L-lactic acid, timolol, and rapamycin.12
Conclusion
Advances in laser surgery and our understanding of wound healing have created a paradigm shift in the treatment approach to trauma and burn scars. In lieu of extensive scar excisions, the summarized multimodal regimen emphasizing tissue conservation and autologous remodeling is gaining favor in the military, academic medical centers, and scar centers of excellence, but patients are finding local access to care difficult. Dermatologists are uniquely positioned to cost-effectively deliver this care in the outpatient setting utilizing devices and techniques they already possess. With the end goal of optimization of functional, symptomatic, and aesthetic state of the patient, it is critical that dermatologists seize this opportunity to truly make a difference for the military and civilian patients that need it most.
- American Burn Association, National Burn Repository. 2015 National burn repository report of data from 2005-2014. http://www.ameriburn.org/2015NBRAnnualReport.pdf. Accessed May 10, 2017.
- Centers for Disease Control and Prevention. 2013 National hospital ambulatory medical care survey emergency department summary tables. https://www.cdc.gov/nchs/data/ahcd/nhamcs_emergency/2013_ed_web_tables.pdf. Accessed May 10, 2017.
- Fischer H. A guide to U.S. Military casualty statistics: Operation Freedom’s Sentinel, Operation Inherent Resolve, Operation New Dawn, Operation Iraqi Freedom, and Operation Enduring Freedom. Congressional Research Service website. https://fas.org/sgp/crs/natsec/RS22452.pdf. Published August 7, 2015. Accessed May 10, 2017.
- Van Loey NE, Van Son MJ. Psychopathology and psychological problems in patients with burn scars: epidemiology and management. Am J Clin Dermatol. 2003;4:245-272.
- Vrijman C, van Drooge AM, Limpens J, et al. Laser and intense pulsed light therapy for the treatment of hypertrophic scars: a systematic review. Br J Dermatol. 2011;165:934-942.
- Miletta N, Lee K, Siwy K, et al. Objective improvement in burn scars after treatment with fractionated CO2 laser. Paper presented at: American Society for Laser Medicine and Surgery 36th Annual Conference; April 1-3, 2016; Boston, MA.
- Ozog DM, Liu A, Chaffins ML, et al. Evaluation of clinical results, histological architecture, and collagen expression following treatment of mature burn scars with a fraction carbon dioxide laser. JAMA Dermatol. 2013;149:50-57.
- Levi B, Ibrahim A, Mathews K, et al. The use of CO2 fractional photothermolysis for the treatment of burn scars. J Burn Care Res. 2016;37:106-114.
- van Drooge AM, Vrijman C, van der Veen W, et al. A randomized controlled pilot study on ablative fractional CO2 laser for consecutive patients presenting with various scar types. Dermatol Surg. 2015;41:371-377.
- Wang XQ, Lui YK, Wang ZY, et al. Antimitotic drug injections and radiotherapy: a review of the effectiveness of treatment for hypertrophic scars and keloids. Int J Low Extrem Wounds. 2008;7:151-159.
- Gupta S, Kalra A. Efficacy and safety of intralesional 5-fluorouracil in the treatment of keloids. Dermatology. 2002;204:130-132.
- Haedersdal M, Erlendsson AM, Paasch U, et al. Translational medicine in the field of AFL (AFXL)-assisted drug delivery: a critical review from basics to current clinical status. J Am Acad Dermatol. 2016;74:981-1004.
Hypertrophic scarring secondary to trauma, burns, and surgical interventions is a major source of morbidity worldwide and often is mechanically, aesthetically, and symptomatically debilitating. Modern advances in acute trauma care protocols have resulted in survival rates greater than 90% in both civilian and military populations.1,2 Patients with wounds that have historically proven fatal are now surviving and are confronted with the long-term sequelae of their injuries. With more than 52,000 service members injured in military engagements from 2001 to 2015 and 8.5 million civilians presenting annually with injury patterns at risk for hypertrophic scarring, it is paramount that we ensure access to safe and effective long-term scar care.2,3
At its simplest level, hypertrophic scarring is believed to result from a disequilibrium between collagen production and degradation. This failure to properly transition through the stages of wound healing results in bothersome symptoms, a disfigured appearance, and mechanical dysfunction of the skin (Figure, A). Decreased elasticity and extensibility, increased dermal thickness, and scar contractures impair patient range of motion and functional mobility. Those affected commonly experience varying degrees of pruritus and dysesthesia along the scar. Combined with aesthetic variations in pigmentation, erythema, texture, and thickness, hypertrophic scarring often leads to long-term psychosocial impairment and decreased health-related quality of life.4
Treatment Approach
Treatment of hypertrophic scars requires a multimodal approach due to the spectrum of associated concerns and the natural recalcitrance of the scar to therapy. Protocols should be tailored to the individual but generally begin with tissue-conserving surgical interventions followed by selective photothermolysis of the scar vasculature. Subsequently, deep and superficial ablative fractional laser (AFL) treatment and local pharmacotherapy also are employed. Treatment can be accomplished in the outpatient setting under local anesthesia in a serial fashion. In the authors’ experience, these therapies behave in a synergistic fashion, achieving outcomes that far exceed the sum of their parts, often obviating the need for scar excision in the majority of cases (Figure, B).
Tissue-Conserving Surgical Intervention
Z-plasty is an indispensable surgical tool due to its ability to lengthen scars and reduce wound tension. Treatment is easily customizable to the patient and can be performed using the individual or multiple Z-plasty techniques. Undermining and step-off correction while suturing allow the physician to lower raised scars, elevate depressed scars, and obscure scar presence by minimizing the straight lines that draw the eye to the scar. Z-plasties rely on the creation and transposition of 2 triangular flaps and permit a 75% increase in length along the desired tension vector. As such, Z-plasties decrease wound tension and facilitate scar maturation.
Selective Photothermolysis of the Vasculature
Although there are several devices available to treat vascular and immature hypertrophic scars, the majority of studies have been conducted with the 595-nm pulsed dye laser. By preferentially heating oxyhemoglobin within the dermal microvasculature, the pulsed dye laser irreparably injures the vascular endothelium. The subsequent tissue hypoxia and collagen fiber heating results in collagen fiber realignment, normalization of collagen subtypes, and neocollagenesis.5 Pulsed dye laser therapy most effectively reduces erythema and pruritus; however, improvements in scar volume, pliability, and elasticity also have been reported.5 When targeting the fine vasculature of the scar, thermal confinement is critical to prevent injury to the surrounding dermis. As such, pulse widths of 0.45 to 1.5 milliseconds are routinely utilized with a fluence just sufficient to elicit transient purpura lasting 3 to 5 seconds. Employing a spot size of 7 to 10 mm, typical fluences range from 4.5 to 6.5 J/cm2. Engagement of the dynamic cooling device reduces the risk for complications, allowing the patient to proceed to the next step in their therapy regimen: the AFL.
Ablative Fractional Laser
The AFL creates a pixilated pattern of injury throughout the epidermis and dermis of the treatment area. Ablative fractional laser platforms include the 10,600-nm CO2 and 2940-nm erbium-doped YAG lasers, both targeting intracellular water. The AFL vaporizes columns of tissue, leaving minute vertical channels with narrow rims of protein coagulation referred to as microscopic treatment zones (MTZs).6 Scar collagen analysis after AFL treatment has shown a profile resembling unaffected skin.7 Consistently, patients report improvements in stiffness, range of motion, pain, pruritus, pigmentation, and erythema.Physician observers also have reported similar improvements in these end points.8,9 Recently, interim data from a prospective controlled trial were presented showing objective improvements in dermal thickness, elasticity, and extensibility after 3 treatments with the CO2 AFL.6 The UltraPulse CO2 laser (Lumenis) is the most well-studied and widely available AFL for scar therapy and as such we will outline common treatment parameters with this device. Of note, treatment end points may be generalized to any AFL.
The DeepFX UltraPulse configuration is utilized to achieve deep AFL therapy and has a fixed pulse width of 0.8 milliseconds, slightly less than the thermal relaxation time of the skin. The diameter of the MTZs is 120 µm, and MTZ density for scar treatment ranges from 1% to 10% with a goal depth of at least 80% of scar thickness. Maximal penetration of the AFL is 4 mm, which is directly proportional to fluence. The goal of deep AFL is the removal of scar tissue to facilitate remodeling and neocollagenesis. Superficial fractional ablation can then be achieved utilizing the ActiveFX UltraPulse configuration generating a 1.3-mm MTZ spot size. We commonly use a treatment level of 3 (82% density). Typical treatment energy ranges from 80 to 125 mJ, which correlates with depths of approximately 50 to 115 µm. With both configurations, the size and shape of the treatment area can be customized to the scar. In addition, frequency may be adjusted to control the speed of treatment while balancing the risk of bulk heating. The goal of superficial AFL is to minimize scar surface irregularities and ensure blending of deep AFL treatment. Once AFL treatment is complete, local pharmacotherapy can then be employed.
Pharmacotherapy
Intralesional corticosteroids have long represented the standard of care for hypertrophic scars, with concentrations between 2.5 and 40 mg/mL that are titrated to scar thickness and location to avoid unwanted atrophy. Visual blanching of the scar represents the clinical end point for treatment. Corticosteroids act by inhibiting fibroblast proliferation and enhancing collagen degradation.10 5-Fluorouracil (5-FU) also is used in scar management. In addition to inhibiting fibroblast proliferation and inducing fibroblast apoptosis, 5-FU inhibits myofibroblast proliferation, which is helpful in the prevention and treatment of scar contracture.11 As monotherapy, weekly injections with 1 to 3 mL of 50 mg/mL 5-FU has been safe and effective. Combination intralesional corticosteroid and 5-FU therapy has been reported and is associated with improved scar regression, reduced reoccurrence, and fewer side effects.11 In our experience, a 1:1 suspension is effective with appropriate titration of the corticosteroid component. Although less well defined, topical application of pharmacotherapy and massage to the newly created MTZs appears beneficial and offers another option for delivery of corticosteroids and 5-FU, in addition to a number of promising medications such as bimatoprost, poly-L-lactic acid, timolol, and rapamycin.12
Conclusion
Advances in laser surgery and our understanding of wound healing have created a paradigm shift in the treatment approach to trauma and burn scars. In lieu of extensive scar excisions, the summarized multimodal regimen emphasizing tissue conservation and autologous remodeling is gaining favor in the military, academic medical centers, and scar centers of excellence, but patients are finding local access to care difficult. Dermatologists are uniquely positioned to cost-effectively deliver this care in the outpatient setting utilizing devices and techniques they already possess. With the end goal of optimization of functional, symptomatic, and aesthetic state of the patient, it is critical that dermatologists seize this opportunity to truly make a difference for the military and civilian patients that need it most.
Hypertrophic scarring secondary to trauma, burns, and surgical interventions is a major source of morbidity worldwide and often is mechanically, aesthetically, and symptomatically debilitating. Modern advances in acute trauma care protocols have resulted in survival rates greater than 90% in both civilian and military populations.1,2 Patients with wounds that have historically proven fatal are now surviving and are confronted with the long-term sequelae of their injuries. With more than 52,000 service members injured in military engagements from 2001 to 2015 and 8.5 million civilians presenting annually with injury patterns at risk for hypertrophic scarring, it is paramount that we ensure access to safe and effective long-term scar care.2,3
At its simplest level, hypertrophic scarring is believed to result from a disequilibrium between collagen production and degradation. This failure to properly transition through the stages of wound healing results in bothersome symptoms, a disfigured appearance, and mechanical dysfunction of the skin (Figure, A). Decreased elasticity and extensibility, increased dermal thickness, and scar contractures impair patient range of motion and functional mobility. Those affected commonly experience varying degrees of pruritus and dysesthesia along the scar. Combined with aesthetic variations in pigmentation, erythema, texture, and thickness, hypertrophic scarring often leads to long-term psychosocial impairment and decreased health-related quality of life.4
Treatment Approach
Treatment of hypertrophic scars requires a multimodal approach due to the spectrum of associated concerns and the natural recalcitrance of the scar to therapy. Protocols should be tailored to the individual but generally begin with tissue-conserving surgical interventions followed by selective photothermolysis of the scar vasculature. Subsequently, deep and superficial ablative fractional laser (AFL) treatment and local pharmacotherapy also are employed. Treatment can be accomplished in the outpatient setting under local anesthesia in a serial fashion. In the authors’ experience, these therapies behave in a synergistic fashion, achieving outcomes that far exceed the sum of their parts, often obviating the need for scar excision in the majority of cases (Figure, B).
Tissue-Conserving Surgical Intervention
Z-plasty is an indispensable surgical tool due to its ability to lengthen scars and reduce wound tension. Treatment is easily customizable to the patient and can be performed using the individual or multiple Z-plasty techniques. Undermining and step-off correction while suturing allow the physician to lower raised scars, elevate depressed scars, and obscure scar presence by minimizing the straight lines that draw the eye to the scar. Z-plasties rely on the creation and transposition of 2 triangular flaps and permit a 75% increase in length along the desired tension vector. As such, Z-plasties decrease wound tension and facilitate scar maturation.
Selective Photothermolysis of the Vasculature
Although there are several devices available to treat vascular and immature hypertrophic scars, the majority of studies have been conducted with the 595-nm pulsed dye laser. By preferentially heating oxyhemoglobin within the dermal microvasculature, the pulsed dye laser irreparably injures the vascular endothelium. The subsequent tissue hypoxia and collagen fiber heating results in collagen fiber realignment, normalization of collagen subtypes, and neocollagenesis.5 Pulsed dye laser therapy most effectively reduces erythema and pruritus; however, improvements in scar volume, pliability, and elasticity also have been reported.5 When targeting the fine vasculature of the scar, thermal confinement is critical to prevent injury to the surrounding dermis. As such, pulse widths of 0.45 to 1.5 milliseconds are routinely utilized with a fluence just sufficient to elicit transient purpura lasting 3 to 5 seconds. Employing a spot size of 7 to 10 mm, typical fluences range from 4.5 to 6.5 J/cm2. Engagement of the dynamic cooling device reduces the risk for complications, allowing the patient to proceed to the next step in their therapy regimen: the AFL.
Ablative Fractional Laser
The AFL creates a pixilated pattern of injury throughout the epidermis and dermis of the treatment area. Ablative fractional laser platforms include the 10,600-nm CO2 and 2940-nm erbium-doped YAG lasers, both targeting intracellular water. The AFL vaporizes columns of tissue, leaving minute vertical channels with narrow rims of protein coagulation referred to as microscopic treatment zones (MTZs).6 Scar collagen analysis after AFL treatment has shown a profile resembling unaffected skin.7 Consistently, patients report improvements in stiffness, range of motion, pain, pruritus, pigmentation, and erythema.Physician observers also have reported similar improvements in these end points.8,9 Recently, interim data from a prospective controlled trial were presented showing objective improvements in dermal thickness, elasticity, and extensibility after 3 treatments with the CO2 AFL.6 The UltraPulse CO2 laser (Lumenis) is the most well-studied and widely available AFL for scar therapy and as such we will outline common treatment parameters with this device. Of note, treatment end points may be generalized to any AFL.
The DeepFX UltraPulse configuration is utilized to achieve deep AFL therapy and has a fixed pulse width of 0.8 milliseconds, slightly less than the thermal relaxation time of the skin. The diameter of the MTZs is 120 µm, and MTZ density for scar treatment ranges from 1% to 10% with a goal depth of at least 80% of scar thickness. Maximal penetration of the AFL is 4 mm, which is directly proportional to fluence. The goal of deep AFL is the removal of scar tissue to facilitate remodeling and neocollagenesis. Superficial fractional ablation can then be achieved utilizing the ActiveFX UltraPulse configuration generating a 1.3-mm MTZ spot size. We commonly use a treatment level of 3 (82% density). Typical treatment energy ranges from 80 to 125 mJ, which correlates with depths of approximately 50 to 115 µm. With both configurations, the size and shape of the treatment area can be customized to the scar. In addition, frequency may be adjusted to control the speed of treatment while balancing the risk of bulk heating. The goal of superficial AFL is to minimize scar surface irregularities and ensure blending of deep AFL treatment. Once AFL treatment is complete, local pharmacotherapy can then be employed.
Pharmacotherapy
Intralesional corticosteroids have long represented the standard of care for hypertrophic scars, with concentrations between 2.5 and 40 mg/mL that are titrated to scar thickness and location to avoid unwanted atrophy. Visual blanching of the scar represents the clinical end point for treatment. Corticosteroids act by inhibiting fibroblast proliferation and enhancing collagen degradation.10 5-Fluorouracil (5-FU) also is used in scar management. In addition to inhibiting fibroblast proliferation and inducing fibroblast apoptosis, 5-FU inhibits myofibroblast proliferation, which is helpful in the prevention and treatment of scar contracture.11 As monotherapy, weekly injections with 1 to 3 mL of 50 mg/mL 5-FU has been safe and effective. Combination intralesional corticosteroid and 5-FU therapy has been reported and is associated with improved scar regression, reduced reoccurrence, and fewer side effects.11 In our experience, a 1:1 suspension is effective with appropriate titration of the corticosteroid component. Although less well defined, topical application of pharmacotherapy and massage to the newly created MTZs appears beneficial and offers another option for delivery of corticosteroids and 5-FU, in addition to a number of promising medications such as bimatoprost, poly-L-lactic acid, timolol, and rapamycin.12
Conclusion
Advances in laser surgery and our understanding of wound healing have created a paradigm shift in the treatment approach to trauma and burn scars. In lieu of extensive scar excisions, the summarized multimodal regimen emphasizing tissue conservation and autologous remodeling is gaining favor in the military, academic medical centers, and scar centers of excellence, but patients are finding local access to care difficult. Dermatologists are uniquely positioned to cost-effectively deliver this care in the outpatient setting utilizing devices and techniques they already possess. With the end goal of optimization of functional, symptomatic, and aesthetic state of the patient, it is critical that dermatologists seize this opportunity to truly make a difference for the military and civilian patients that need it most.
- American Burn Association, National Burn Repository. 2015 National burn repository report of data from 2005-2014. http://www.ameriburn.org/2015NBRAnnualReport.pdf. Accessed May 10, 2017.
- Centers for Disease Control and Prevention. 2013 National hospital ambulatory medical care survey emergency department summary tables. https://www.cdc.gov/nchs/data/ahcd/nhamcs_emergency/2013_ed_web_tables.pdf. Accessed May 10, 2017.
- Fischer H. A guide to U.S. Military casualty statistics: Operation Freedom’s Sentinel, Operation Inherent Resolve, Operation New Dawn, Operation Iraqi Freedom, and Operation Enduring Freedom. Congressional Research Service website. https://fas.org/sgp/crs/natsec/RS22452.pdf. Published August 7, 2015. Accessed May 10, 2017.
- Van Loey NE, Van Son MJ. Psychopathology and psychological problems in patients with burn scars: epidemiology and management. Am J Clin Dermatol. 2003;4:245-272.
- Vrijman C, van Drooge AM, Limpens J, et al. Laser and intense pulsed light therapy for the treatment of hypertrophic scars: a systematic review. Br J Dermatol. 2011;165:934-942.
- Miletta N, Lee K, Siwy K, et al. Objective improvement in burn scars after treatment with fractionated CO2 laser. Paper presented at: American Society for Laser Medicine and Surgery 36th Annual Conference; April 1-3, 2016; Boston, MA.
- Ozog DM, Liu A, Chaffins ML, et al. Evaluation of clinical results, histological architecture, and collagen expression following treatment of mature burn scars with a fraction carbon dioxide laser. JAMA Dermatol. 2013;149:50-57.
- Levi B, Ibrahim A, Mathews K, et al. The use of CO2 fractional photothermolysis for the treatment of burn scars. J Burn Care Res. 2016;37:106-114.
- van Drooge AM, Vrijman C, van der Veen W, et al. A randomized controlled pilot study on ablative fractional CO2 laser for consecutive patients presenting with various scar types. Dermatol Surg. 2015;41:371-377.
- Wang XQ, Lui YK, Wang ZY, et al. Antimitotic drug injections and radiotherapy: a review of the effectiveness of treatment for hypertrophic scars and keloids. Int J Low Extrem Wounds. 2008;7:151-159.
- Gupta S, Kalra A. Efficacy and safety of intralesional 5-fluorouracil in the treatment of keloids. Dermatology. 2002;204:130-132.
- Haedersdal M, Erlendsson AM, Paasch U, et al. Translational medicine in the field of AFL (AFXL)-assisted drug delivery: a critical review from basics to current clinical status. J Am Acad Dermatol. 2016;74:981-1004.
- American Burn Association, National Burn Repository. 2015 National burn repository report of data from 2005-2014. http://www.ameriburn.org/2015NBRAnnualReport.pdf. Accessed May 10, 2017.
- Centers for Disease Control and Prevention. 2013 National hospital ambulatory medical care survey emergency department summary tables. https://www.cdc.gov/nchs/data/ahcd/nhamcs_emergency/2013_ed_web_tables.pdf. Accessed May 10, 2017.
- Fischer H. A guide to U.S. Military casualty statistics: Operation Freedom’s Sentinel, Operation Inherent Resolve, Operation New Dawn, Operation Iraqi Freedom, and Operation Enduring Freedom. Congressional Research Service website. https://fas.org/sgp/crs/natsec/RS22452.pdf. Published August 7, 2015. Accessed May 10, 2017.
- Van Loey NE, Van Son MJ. Psychopathology and psychological problems in patients with burn scars: epidemiology and management. Am J Clin Dermatol. 2003;4:245-272.
- Vrijman C, van Drooge AM, Limpens J, et al. Laser and intense pulsed light therapy for the treatment of hypertrophic scars: a systematic review. Br J Dermatol. 2011;165:934-942.
- Miletta N, Lee K, Siwy K, et al. Objective improvement in burn scars after treatment with fractionated CO2 laser. Paper presented at: American Society for Laser Medicine and Surgery 36th Annual Conference; April 1-3, 2016; Boston, MA.
- Ozog DM, Liu A, Chaffins ML, et al. Evaluation of clinical results, histological architecture, and collagen expression following treatment of mature burn scars with a fraction carbon dioxide laser. JAMA Dermatol. 2013;149:50-57.
- Levi B, Ibrahim A, Mathews K, et al. The use of CO2 fractional photothermolysis for the treatment of burn scars. J Burn Care Res. 2016;37:106-114.
- van Drooge AM, Vrijman C, van der Veen W, et al. A randomized controlled pilot study on ablative fractional CO2 laser for consecutive patients presenting with various scar types. Dermatol Surg. 2015;41:371-377.
- Wang XQ, Lui YK, Wang ZY, et al. Antimitotic drug injections and radiotherapy: a review of the effectiveness of treatment for hypertrophic scars and keloids. Int J Low Extrem Wounds. 2008;7:151-159.
- Gupta S, Kalra A. Efficacy and safety of intralesional 5-fluorouracil in the treatment of keloids. Dermatology. 2002;204:130-132.
- Haedersdal M, Erlendsson AM, Paasch U, et al. Translational medicine in the field of AFL (AFXL)-assisted drug delivery: a critical review from basics to current clinical status. J Am Acad Dermatol. 2016;74:981-1004.
Practice Points
- Burn and trauma scarring represents a major source of morbidity in both the civilian and military populations worldwide and often is mechanically, aesthetically, and symptomatically debilitating.
- Advances in laser surgery and our understanding of wound healing have resulted in a scar therapy paradigm shift from large scar excisions and repair to a multimodal, tissue-conserving approach that relies on remodeling of the existing tissue.
- Dermatologists are uniquely positioned to increase patient access to cost-effective, outpatient-based burn and trauma scar care utilizing devices and techniques that they currently possess.
MS News From the AAN & CMSC Annual Meetings

The Changing Landscape of Trauma Care, Part 1
Introduction
There has been a fundamental change in the face of injury in the United States. Traditionally, injury was thought to be a disease of the young male population, with motor vehicle collision (MVC) being the most common mechanism of injury. Depending on the trauma center, blunt trauma would comprise up to 99% of patients admitted. This profile has fundamentally changed over the last 15 years. Trauma center performance is often benchmarked against local, regional, or national norms, and as all medical centers now measure quality as the primary endpoint, these changes in demographics can be very important.
Certainly, the most important change has been the “graying” of trauma patients. When I (TS) started working in Baltimore 20 years ago, patients over age 65 years comprised approximately 5% of our total trauma admissions. Last year, over 30% of our 7,000 primary admissions were patients over age 65 years who had sustained ground-level falls.
Injury patterns in the elderly differ compared to standard blunt trauma in which traumatic brain injury (TBI) is common. Extremity fractures, particularly hip fractures, are common, whereas torso injuries other than rib fractures are relatively uncommon. As this article points out, elderly trauma patients almost universally have significant medical problems. Cognitive deficits and balance issues may explain ground-level falls in this population. Syncope from a myriad of underlying medical conditions and/or medications may have contributed to their falls as well.
The evaluation process for elderly trauma patients must be directed not only at diagnosing injury but also at attempting to identify the reason for the injury. This may involve a number of diagnostic tests in the ED, in the outpatient setting, or even on an inpatient floor.
Unfortunately, elderly patients can succumb to relatively minor injuries, and those who survive such afflictions often have difficulty making a full recovery. Many elderly patients who were able to function preinjury were marginally compensated at home. Operative therapy, often needed to treat injuries such as a hip or extremity fracture, by itself represents physiological burden to an elderly patient. Likewise, full recovery after even a mild TBI can be quite difficult.
Admitting an elderly patient to the hospital can present several challenges. For example, elderly patients are often on a number of prescription and nonprescription medications, including over-the-counter nutritional and herbal supplements, many of which interact with the newly prescribed medications given to treat trauma (eg, analgesics, sedatives, antiseizure drugs). Moreover, elderly patients often become disoriented and agitated when they are out of their home environment. All too often, the therapy for these and other problems is another medication, and thus the cycle continues. Therefore, elderly patients are ultimately at increased risk for death from seemingly trivial injury, which in turn may create significant perceived quality issues for a medical center.
The use of systemic anticoagulation has become almost ubiquitous in older patients. Some days it seems like every patient I (TS) admit is taking an anticoagulant—at least aspirin. While primary care providers (PCPs) correctly realize the important role these anticoagulants have in treating chronic medical conditions, they often do not recognize the dangers associated with increased traumatic bleeding following an injury.
Frequently, we knowingly take patients with conditions such as rate-controlled atrial fibrillation (AF) off their prescribed anticoagulant, believing they are simply not candidates for anticoagulation because of their propensity to fall. Even though we attempt to communicate our concerns to the PCP, when these patients are readmitted, it is common to find that they have been placed back on an anticoagulant.
The advent of novel oral anticoagulants (NOACs) has made routine laboratory testing obsolete. One need only to turn on the television to see the many advertisements explaining why this agent or that agent is preferable to warfarin. While, fresh frozen plasma (FFP) and/or prothrombin complex concentrates (PCC) are quite effective at reversing the anticoagulant effect of warfarin, reversal of these newer agents is either extremely difficult or impossible.
Anticoagulant reversal can be more or less important, depending on the situation. For instance, while subcutaneous bleeding is concerning, it can be temporized by operative exploration and/or packing. When necessary, blood can be transfused to replace the blood lost. However, the same is not true for a patient with significant TBI, because even a small volume of ongoing hemorrhage can prove lethal. Cavitary hemorrhage in the chest and/or abdomen is also extremely difficult to treat if the anticoagulant effect cannot be reversed. Given the popularity of the new anticoagulants, I (TS) am afraid that this problem will be with us for years to come.
There has been a significant spike in interpersonal violence in the United States over the past few years. While the cause is often difficult to identify, its existence is impossible to ignore. The violence seems to be concentrated in a number of municipal areas, but violence can occur in any community. Certainly, even mass casualties have become part of our everyday life.
In 2016, homicides and nonfatal shootings increased dramatically relative to 2015. In 201 7 , we are tracking a 40% increase in homicides and a 30% increase in nonfatal shootings—particularly concerning when one considers that these numbers are being compared to the previously increased 2016 statistics.
Many community EDs are not accustomed to dealing with a significant volume of penetrating trauma, and thus they may not be as familiar with the newest means of resuscitation, evaluation, and treatment of these injuries. It will be important for every medical center to do what is necessary to be able to effectively triage and provide initial treatment for patients with penetrating trauma.
The victims of penetrating trauma are often young, and unfortunately, despite our best efforts, these patients often die in the ED. This creates a huge emotional burden on people who work in the ED, particularly those who are not used to seeing large volumes of gunshot wounds (GSWs) or stab wounds. Even those of us working in busy urban trauma centers feel the emotional burden of this new epidemic. Each of us will need to cope with these issues and help each other deal with them.
It is important to recognize the dramatic change in trauma demographics over the last few years, and make plans to care for the changing face of trauma to optimize results and save as many lives as possible. In part 1 of our 2-part, “The Changing Landscape of Trauma Care,” we focus on the specific issues and concerns encountered in elderly trauma patients, as well as victims of all ages presenting with penetrating trauma from stab and GSWs.
Trauma in the Elderly Population
There has been and continues to be an increase in the elderly population in the United States. In 2014, 46 million Americans representing 15% of the total population were older than age 65 years.1 Of all age groups in the United States, the elderly population is one of the fastest growing and, according to the 2010 Census, grew at a faster rate than in previous years.2 This growth is expected to continue as many of the post-World War II baby boomer generation age. By the year 2030, an estimated 1 in 5 Americans will be older than 65 years of age—representing a 7% absolute increase from 2010 to 2030.1
Furthermore, men and women in this population are maintaining an active lifestyle well into their seventh and eighth decade, which has led to an increased incidence of trauma in this age group, primarily from falls and low-velocity MVCs. According to data from the National Trauma Data Bank in 2016, nearly 43% of all traumatic incidents occurred in patients older than age 55 years, as compared to only 32% in 2010.3,4 Today, injury is the seventh leading cause of death among the elderly population.5
Pre-existing Conditions and Comorbidities
The elderly population tends to have more complex medical histories, with pre-existing conditions and comorbidities—both of which result in intolerance to alterations from normal physiology after acute trauma and may place them at risk for complications and death. This point was highlighted in an invited commentary by one of us (TS) over 20 years ago, in which he stated, “Resting organ function often is preserved, but the ability to augment performance in response to stress is greatly compromised.”6
Studies in the early 1990s established a link between trauma outcomes and comorbidities.7-9 Morris et al7 found that ischemic heart disease, diabetes, chronic obstructive pulmonary disease, congenital coagulopathy, and cirrhosis highly influence trauma outcomes. They also noted that 25% of trauma patients over age 65 years had at least one of these five comorbidities and were nearly two times more likely to die. These findings were confirmed in 2002 by Grossman et al,8 who demonstrated that each year over age 65 years held a mortality increase of 6.8%.8 Additionally, they found that congestive heart failure, cancer, renal disease, and hepatic disease were the comorbidities with the highest impact on mortality.8
The presence of pre-existing conditions or comorbidities has also been associated with increased risk for complications, and subsequent increased mortality. In 2010, Aitken et al9 found that 6.2% of elderly trauma patients developed pneumonia postinjury, which was associated with increased intensive care unit (ICU) and hospital length of stay.Pre-existing pulmonary disease and higher Injury Severity Scores (ISS) were also found to be risk factors, demonstrating a 5.9% incidence of acute kidney injury in this group, conferring a 10-fold increased risk of mortality.
In efforts to improve outcomes in elderly trauma patients, many centers have integrated geriatric consults in the ED for all patients over a certain age, following injury. Though Olufajo et al10 were unable to demonstrate an in-house or 30-day mortality benefit after implementing a mandatory geriatrics consult for patients over age 70 years, they did show a nonstatistically significant trend toward fewer ICU readmissions with the consults.
In 2001, Demetriades et al11 reported a 50% mortality rate among patients aged 70 years and older who met criteria for full trauma team activation. Interestingly, the mortality rate for patients over age 70 years was 24%, compared to 7.6% for younger patients admitted during the same period. Those in the 70 years and older age group who did not meet criteria for full trauma team activation still had a 16% mortality rate, and 24% required ICU admission.
Demetriades et al11 also demonstrated that prehospital/admission vital signs in patients 70 years and older were often normal but misleading. In this group, 63% of patients with an ISS greater than 15, and 25% with an ISS greater than 30 did not have tachycardia or hypotension criteria for full trauma activation.11 These findings have led to recommendations for a lower threshold for trauma activations in geriatric patients.12
Recent studies have suggested that adding an age threshold to the trauma activation criteria may improve outcomes without leading to an unacceptable overtriage rate. In 2016, Hammer, et al13 reported improved outcomes, with only 2% of patients being overtriaged, when they added to their trauma activation criteria an age threshold of 70 years, regardless of physiology or mechanism of injury. They ultimately concluded that it was appropriate and cost-effective. In 2017, Cooper et al14 published a position paper on the Geriatric Trauma Coalition (GeriTraC) covering the convergence of aging and injury. The mission of GeriTraC is to improve geriatric trauma care from prevention to transition of care.14
Fall-Related Trauma
Falls are the most common cause of fatal and nonfatal injury in patients over age 65 years.15 Most fall injuries occur at home and during the winter months, and tend to be from ground level.16 Although most result in only minor trauma, many cause significant injuries requiring hospitalization. In 2006, Stevens et al17 estimated that both fatal and nonfatal falls in the elderly accounted for almost $20 billion in direct medical costs.
Motor Vehicle Collisions
Motor vehicle collisions/pedestrian struck are the second most significant causes of fatal and nonfatal injury in elderly patients. Older drivers who are hospitalized following an MVC have significantly longer hospital lengths of stay and an overall higher mortality rate.16 Elderly patients are more likely to be victims of “pedestrian-struck-by-vehicle” due to their decreased visual and auditory acuity, reduced reaction time, slower movement, and confusion.
Suicide
Suicide is the third leading cause of injury-related death for those aged 65 years and older.15 Risk factors for suicide in the elderly population include psychiatric disorders, particularly depression; medical conditions, especially cancer or chronic lung disease; moderate-to-large alcohol use; and social isolation. Changes in behavior, such as altering a will, new preoccupation with religion, or giving away life possessions, may be warning signs of impending suicide.
Novel Oral Anticoagulants
Many people, both old and young, are taking oral anticoagulants for various conditions. Warfarin has traditionally been the medication of choice, with readily available reversal agents, if needed. However, the development of NOACs, which antagonize activity of a single step in the coagulation cascade, has presented trauma care providers with a new challenge in achieving hemostasis. The NOACs include a direct thrombin inhibitor (dabigatran), and the Factor Xa (FXa) inhibitors (apixaban, edoxaban, and rivaroxaban). These NOACs have been shown to be as effective as traditional vitamin K antagonists (warfarin) with a comparable or lower spontaneous risk of bleeding. Along with an acceptable safety profile, these drugs cause significantly less drug and food interactions and are easier to dose, with no need for monitoring levels.18 Since the arrival of the first NOAC dabigatran in 2010, use of these drugs has continued to increase, and are becoming more popular in the treatment of venous thromboembolism in younger patients as well. A study by Desai et al19 examining newly initiated anticoagulation for AF between 2010 and 2013 found that 62% of all new anticoagulant prescriptions were for NOACs.
Hemostasis Challenges
Because of the lack of reversal agents or antidotes available, the NOACs present a unique challenge and major concern when anticoagulation properties must be reversed quickly. Among the NOACs, dabigatran is the only NOAC that is 35% protein bound and can be effectively cleared by hemodialysis (HD). Rivaroxaban and apixaban, in contrast, are highly protein bound (95% and 87%, respectively), which renders HD ineffective for clearance. Even for dabigatran, though HD may be a treatment option in the presence of potentially life-threatening bleeding associated with dabigatran alone, this is only a possibility if the patient’s hemodynamics can tolerate HD.
Extrapolating from experience with warfarin-associated bleeding, the use of FFP, PCC, and recombinant activated factor VII for NOAC-associated bleeding has been proposed and attempted.20 Though FFP may be necessary to restore circulating blood volume as part of a massive transfusion protocol in a patient with NOAC-associated hemorrhage, it is generally not a reasonable sole strategy for reversal of NOACs because the coagulation factors in FFP are not present in high enough concentrations to be effective.18
Prothrombin Complex Concentrates
Three- and Four-Factor PCCs. Four-factor PCC (4F-PCC), which became available for use in the United States in April 2013, contains concentrated amounts of all four of the vitamin K dependent factors (II, VII, IX, and X), as well as proteins C and S. Three-factor PCC (3F-PCC) does not contain significant levels of factor VII,20 and preclinical studies on its efficacy in reversing NOACs have not been consistent.
Early studies using animal models showed promising results for both 3F-PCC and 4F-PCC in correcting derangements in laboratory coagulation markers as well as observed bleeding time.21-23 However, other animal studies failed to demonstrate an improvement in observed bleeding time or volume despite full or partial correction of coagulation studies after PCC.24,25 In human studies, PCC has been observed to correct some laboratory parameters of coagulation, but not others.26,27 Thus far, these studies have been limited to healthy volunteers without active bleeding and have been largely ex vivo and in vitro studies, so it is difficult to determine if the demonstrated correction of coagulation studies translates into clinical benefit. Both 3F-PCC and 4F-PCC have shown promise, though studies with 4F-PCC have yielded more consistent results.26,27Activated PCC. Activated PCC (aPCC), which contains the same vitamin K dependent factors (factors II, VII, IX, X) with some in their activated form, has shown similar results. In fact, ex vivo and in vitro studies thus far seem to suggest that aPCC is more effective than PCC in correcting coagulation test parameters, as well as thrombin generation indices.28-31 However, an aPCC has also been demonstrated to be more procoagulant and, thus, may increase the risk of thrombotic complications.32
Recombinant Activated Factor VII
Recombinant activated factor VII has shown less promise than PCC or aPCC in the reversal of NOAC-associated bleeding. Additionally, similar to aPCC, it may increase the risk of thrombosis.20,33
Monoclonal Antibody Agent
In October 2015, the US Food and Drug Administration approved idarucizumab, a monoclonal antibody agent for the reversal of dabigatran. Idarucizumab has a binding affinity approximately 350 times higher than the binding affinity of dabigatran for thrombin with no demonstrated procoagulant effects.20 To date, there are no commercially available antidotes or reversal agents for the FXa inhibitors, though two promising agents are in various phases of clinical trials. The first, andexanet alfa, is a modified, recombinant factor X which binds FXa inhibitors with high affinity. This agent has shown promising results in the reversal of apixaban and rivaroxaban.20 The second is called aripazine (PER977) and has the potential to reverse unfractionated heparin, low molecular weight heparins, fondaparinux, FXa inhibitors, and thrombin inhibitors. Early in vivo human studies have been promising.18
Currently, there are no well-designed clinical studies examining the use of PCC for NOAC reversal in trauma. There are only a few published case reports, showing both successful and unsuccessful results, and a small retrospective series of only 18 patients specifically looking at both traumatic and spontaneous intracranial hemorrhage.34-37 There are also no universally agreed upon published guidelines for the management of NOAC-associated bleeding in the absence of drug-specific reversal agents.
Penetrating Trauma
The United States leads all high-income nations in GSW mortality,38 and its rate of firearm homicide is almost 20 times that of other high-income countries. In 2014, there were more than 33,000 firearm-related deaths in the United States, almost two-thirds of which were suicide-related.38 These numbers represent 16.8% of all deaths from injury. For each fatal firearm injury, there were nearly two nonfatal firearm injuries (65,106) the same year.39 Since 2001, the leading cause of death among black males aged 15 to 44 years has been firearm-related homicide. In 2015, that age demographic was lowered to include 10- to 14-year-old black males. In 2015, suicide by firearm was the second leading cause of death among white males over the age of 55 years and the third leading cause of death among white males aged 10 to 54 years.40
Incidents of gun violence are on the rise. These incidents are becoming more frequent and more often fatal. In a retrospective review of their trauma registry, as well as county records, Sauaia et al41 examined trends of GSW severity and mortality in Denver, Colorado from 2000 to 2013. They noted the proportion of GSW admissions remained stable over time, but injury severity and mortality from GSWs increased significantly, contrary to mortality and survival trends for all other injury mechanisms.41
The increasing GSW severity and mortality trend is not unique to Denver. Many media sources in cities across the country have reported similar statistics obtained from their local police departments in the past year. Though gun violence is a subject that is in desperate need of prevention research, current legislation makes these studies challenging to undertake. In 1996, Congress passed the Dickey Amendment to the Omnibus Consolidated Appropriations Act for the 1997 fiscal year, which states that “none of the funds made available for injury prevention and control at the Centers for Disease Control and Prevention may be used to advocate or promote gun control.”42,43 In the 2011 Consolidated Appropriations Act for the fiscal year 2012, this restriction was expanded to include the National Institutes of Health (NIH).44,45 These measures largely explain the paucity of primary research in gun violence in the last two decades—despite the increasing role and costs gun violence contributes to the US health care system. Gun violence is an epidemic, and like all other epidemics in the United States, it requires government-funded research to help protect the people.44
Conclusion
The last decade has seen some significant changes in trauma demographics in the United States. As the population of US men and women older than age 65 years continues to grow, trauma can no longer be considered a disease of young people. In addition to elderly men and women being more active than ever before, comorbid diseases place them at higher risk for complications and death following injury. For these reasons, many trauma triage algorithms now include age as an independent factor in activating a trauma alert. In addition to age, medications, and especially polypharmacy, can place patients at greater risk of injury and complications following trauma.
The last 10 years also has seen an increase in the number of patients on anticoagulants. The development of the NOACs further complicates the care of trauma patients taking these medications. Although designed to simplify care for patients and providers by minimizing bleeding risks and eliminating blood monitoring, there are only limited, and sometimes no reliable reversal agents available for NOACs, creating challenges when treating trauma patients who are on these medications. Finally, despite efforts by many individuals and groups, gun violence still remains a large and growing problem in the United States. Hopefully, continued efforts of national, state and local programs will begin to improve the current situation.
Editor’s Note: Part 2 of “The Changing Landscape of Trauma Care” will appear in the August 2017 issue of Emergency Medicine and will cover the changes in strategies and techniques to care for injured patients.
1. Federal Interagency Forum on Aging-Related Statistics. Older Americans 2016: key indicators of well-being. US Gov Print Off; Washington, DC. 2016; August. https://agingstats.gov/docs/LatestReport/Older-Americans-2016-Key-Indicators-of-WellBeing.pdf. Accessed June 8, 2017.
2. Howden L, Meyer J. Age and sex composition: 2010. 2010 Census Briefs. 2011;(May):1-16. http://www.census.gov/library/publications/2011/dec/c2010br-03.html. Accessed June 8, 2017.
3. American College of Surgeons Committee on Trauma. National Trauma Data Bank 2010 Annual Report. 2010:1-93. https://www.facs.org/~/media/files/quality%20programs/trauma/ntdb/ntdbannualreport2010.ashx. Accesssed June 8, 2017.
4. American College of Surgeons Committee on Trauma. National Trauma Data Bank 2016 Annual Report. 2016:1-147. https://www.facs.org/~/media/files/quality%20programs/trauma/ntdb/ntdb%20annual%20report%202016.ashx. Accessed June 8, 2017.
5. Health, United States 2015. With Special Feature on Racial and Ethnic Health Disparities. US Department of Health and Human Services. Centers for Disease Control and Prevention. 2016:126. https://www.cdc.gov/nchs/data/hus/hus15.pdf. Accessed June 8, 2017.
6. Scalea TM. Invited commentary (for McMahon DJ, William S, Kauder D. Comorbidity and trauma in the elderly. World J Surg. 1996;20(8):116. doi:10.1007/s002689900170.
7. Morris JA Jr, MacKenzie EJ, Edelstein SL. The effect of preexisting conditions
8. Grossman MD, Miller D, Scaff DW, Arcona S. When is an elder old? Effect of preexisting conditions on mortality in geriatric trauma. J Trauma. 2002;52(2):242-246.
9. Aitken LM, Burmeister E, Lang J, Chaboyer W, Richmond TS. Characteristics and outcomes of injured older adults after hospital admission. J Am Geriatr Soc. 2010;58(3):442-449. doi:10.1111/j.1532-5415.2010.02728.x.
10. Olufajo OA, Tulebaev S, Javedan H, et al. Integrating geriatric consults into routine care of older trauma patients: one-year experience of a level I trauma center. J Am Coll Surg. 2016;222(6):1029-1035. doi:10.1016/j.jamcollsurg.2015.12.058.
11. Demetriades D, Sava J, Alo K, et al. Old age as a criterion for trauma team activation. J Trauma. 2001;51(4):754-756; discussion 756-757.
12. Calland JF, Ingraham AM, Martin N, et al; Eastern Association for the Surgery of Trauma. Evaluation and management of geriatric trauma: an Eastern Association for the Surgery of Trauma practice management guideline. J Trauma Acute Care Surg. 2012;73(5 Suppl 4):S345-S350. doi:10.1097/TA.0b013e318270191f.
13. Hammer PM, Storey AC, Bell T, et al. Improving geriatric trauma outcomes: A small step toward a big problem. J Trauma Acute Care Surg. 2016;81(1):162-167. doi:10.1097/TA.0000000000001063.
14. Cooper Z, Maxwell CA, Fakhry SM, et al. A position paper: The convergence of aging and injury and the need for a Geriatric Trauma Coalition (GeriTraC). J Trauma Acute Care Surg. 2017;82(2):419-422. doi:10.1097/TA.0000000000001317.
15. Centers for Disease Control and Prevention. Web-based injury statistics query and reporting system (WISQARS). National Center for Injury Prevention and Control, Centers for Disease Control and Prevention. http://www.cdc.gov/injury/wisqars/index.html. Updated June 1, 2017. Accessed June 8, 2017.
16. Menaker J, Scalea TM. Care of the injured elderly. In Rosenthal RA, Zenilman K, Katlic MR, eds. Principles and Practice of Geriatric Surgery. 2nd ed. Springer: New York, NY: Springer; 2011:391-410.
17. Stevens JA, Corso PS, Finkelstein EA, Miller TR. The costs of fatal and non-fatal falls among older adults. Inj Prev. 2006;12(5):290-295. doi:10.1136/ip.2005.011015.
18. von Heymann C, Rosenthal C, Kaufner L, Sander M. Management of direct oral anticoagulants-associated bleeding in the trauma patient. Curr Opin Anaesthesiol. 2016;29(2):220-228. doi:10.1097/ACO.0000000000000294.
19. Desai NR, Krumme AA, Schneeweiss S, et al. Patterns of initiation of oral anticoagulants in patients with atrial fibrillation- quality and cost implications. Am J Med. 2014;127(11):1075-1082.e1. doi:10.1016/j.amjmed.2014.05.013.
20. Marano G, Vaglio S, Pupella S, Liumbruno GM, Franchini M. How we treat bleeding associated with direct oral anticoagulants. Blood Transfus. 2016;14(5):465-473. doi:10.2450/2016.0180-15.
21. Zhou W, Schwarting S, Illanes S, et al. Hemostatic therapy in experimental intracerebral hemorrhage associated with the direct thrombin inhibitor dabigatran. Stroke. 2011;42(12):3594-3599. doi:10.1161/STROKEAHA.111.624650.
22. Pragst I, Zeitler SH, Doerr B, et al. Reversal of dabigatran anticoagulation by prothrombin complex concentrate (Beriplex P/N) in a rabbit model. J Thromb Haemost. 2012;10(9):1841-1848. doi:10.1111/j.1538-7836.2012.04859.x.
23. van Ryn J, Schurer J, Kink-Eiband M, Clemens A. The successful reversal of dabigatran-induced bleeding by coagulation factor concentrates in a rat tail bleeding model do not correlate with ex vivo markers of anticoagulation. Blood. 2011;118(2316).
24. Herzog E, Kaspereit F, Krege W, Joanne R Van, Dickneite G, Pragst I. Non-clinical safety and efficacy of prothrombin complex concentrates (pcc) for the reversal of dabigatran mediated anticoagulation. J Thromb Haemost. 2013;11:693.
25. Godier A, Miclot A, Le Bonniec B, et al. Evaluation of prothrombin complex concentrate and recombinant activated factor VII to reverse rivaroxaban in a rabbit model. Anesthesiology. 2012;116(1):94-102. doi:10.1097/ALN.0b013e318238c036.
26. Eerenberg ES, Kamphuisen PW, Sijpkens MK, Meijers JC, Buller HR, Levi M. Reversal of rivaroxaban and dabigatran by prothrombin complex concentrate: a randomized, placebo-controlled, crossover study in healthy subjects. Circulation. 2011;124(14):1573-1579. doi:10.1161/CIRCULATIONAHA.111.029017.
27. Escolar G, Fernandez-Gallego V, Arellano-Rodrigo E, et al. Reversal of apixaban induced alterations in hemostasis by different coagulation factor concentrates: significance of studies in vitro with circulating human blood. PLoS One. 2013;8(11):e78696. doi:10.1371/journal.pone.0078696.
28. Galan AM, Arellano-Rodrigo E, Sanz V, et al. Effects of rivaroxaban and dabigatran on hemostasis and reversion of their antithrombotic effects by different coagulation factors: evidence raised from a clinical study in healthy volunteers. J Thromb Haemost. 2013;11:418-419.
29. Escolar G, Arellano-Rodrigo E, Lopez-Vilchez I, et al. Reversal of rivaroxaban-induced alterations on hemostasis by different coagulation factor concentrates—in vitro studies with steady and circulating human blood. Circ J. 2015;79(2):331-338. doi:0.1253/circj.CJ-14-0909.
30. Perzborn E, Gruber A, Tinel H, et al. Reversal of rivaroxaban anticoagulation by haemostatic agents in rats and primates. Thromb Haemost. 2013;110(1):162-172. doi:10.1160/TH12-12-0907.
31. Chan HHW, Atkinson HM, Goncharenko M, Berry LR, Chan AKC. Reversal of dabigatran using recombinant activated factor VII and activated prothrombin complex concentrates in thromboelastography assay. J Thromb Haemost. 2011;9:576-577.
32. Hoffman M, Monroe DM. Reversing targeted oral anticoagulants. Hematology Am Soc Hematol Educ Program. 2014;2014(1):518-523. doi:10.1182/asheducation-2014.1.518.
33. Marlu R, Hodaj E, Paris A, Albaladejo P, Cracowski JL, Pernod G. Effect of non-specific reversal agents on anticoagulant activity of dabigatran and rivaroxaban: a randomised crossover ex vivo study in healthy volunteers. Thromb Haemost. 2012;108(2):217-224. doi:10.1160/TH12-03-0179.
34. Grandhi R, Newman WC, Zhang X, et al. Administration of 4-factor prothrombin complex concentrate as an antidote for intracranial bleeding in patients taking direct factor xa inhibitors. World Neurosurg. 2015;84(6):1956-61. doi:10.1016/j.wneu.2015.08.042.
35. Durie R, Kohute M, Fernandez C, Knight M. Prothrombin complex concentrate for the management of severe traumatic bleeding in a patient anticoagulated with apixaban. J Clin Pharm Ther. 2016;41(1):92-93. doi:10.1111/jcpt.12339.
36. Kauffmann S, Chabanne R, Coste A, et al. Favorable outcome of rivaroxaban-associated intracerebral hemorrhage reversed by 4-factor prothrombin complex concentrate: impact on thrombin generation. A&A Case Rep. 2015;4(11):151-154. doi:10.1213/XAA.0000000000000143.
37. Maurice-Szamburski A, Graillon T, Bruder N. Favorable outcome after a subdural hematoma treated with feiba in a 77-year-old patient treated by rivaroxaban. J Neurosurg Anesthesiol. 2014;26(2):183. doi:10.1097/ANA.0000000000000030.
38. Richardson EG, Hemenway D. Homicide, suicide, and unintentional firearm fatality: comparing the United States with other high-income countries, 2003. J Trauma. 2011;70(1):238-243. doi:10.1097/TA.0b013e3181dbaddf.
39. Kochanek KD, Murphy SL, Xu J, Tejada-Vera B. Deaths: final data for 2014. Natl Vital Stat Rep. 2016;65(4):1-122. https://www.cdc.gov/nchs/data/nvsr/nvsr65/nvsr65_04.pdf. Accessed June 8, 2017.
40. Centers for Disease Control and Prevention. Leading causes of death reports, national and regional, 1999-2015. https://webappa.cdc.gov/sasweb/ncipc/leadcaus10_us.html. Accessed June 8, 2017.
41. Sauaia A, Gonzalez E, Moore HB, Bol K, Moore EE. Fatality and severity of firearm injuries in a denver trauma center, 2000-2013. JAMA. 2016;315(22):2465-2467. doi:10.1001/jama.2016.5978.
42. Omnibus Consolidated Appropriations Bill. HR 3610, Pub L No. 104-208. http://www.gpo.gov/fdsys/pkg/PLAW-104publ208/pdf/PLAW-104publ208.pdf. September 1996. Accessed June 13, 2017.
43. Kellermann AL, Rivara FP. Silencing the science on gun research. JAMA. 2013;309(6):549-550. doi:10.1001/jama.2012.208207.
44. Consolidated Appropriations Act of 2012. HR 2055: Pub L No. 112-174. https://www.gpo.gov/fdsys/pkg/PLAW-112publ74/pdf/PLAW-112publ74.pdf. December 2011. Accessed June 13, 2017.
45. Rubin R. Tale of 2 agencies: CDC avoids gun violence research but NIH funds it. JAMA. 2016;315(16):1689-1691. doi:10.1001/jama.2016.1707.
46. Cook A, Osler T, Hosmer D, et al. Gunshot wounds resulting in hospitalization in the United States: 2004-2013. Injury. 2017;48(3):621-627. doi:10.1016/j.injury.2017.01.044.
Introduction
There has been a fundamental change in the face of injury in the United States. Traditionally, injury was thought to be a disease of the young male population, with motor vehicle collision (MVC) being the most common mechanism of injury. Depending on the trauma center, blunt trauma would comprise up to 99% of patients admitted. This profile has fundamentally changed over the last 15 years. Trauma center performance is often benchmarked against local, regional, or national norms, and as all medical centers now measure quality as the primary endpoint, these changes in demographics can be very important.
Certainly, the most important change has been the “graying” of trauma patients. When I (TS) started working in Baltimore 20 years ago, patients over age 65 years comprised approximately 5% of our total trauma admissions. Last year, over 30% of our 7,000 primary admissions were patients over age 65 years who had sustained ground-level falls.
Injury patterns in the elderly differ compared to standard blunt trauma in which traumatic brain injury (TBI) is common. Extremity fractures, particularly hip fractures, are common, whereas torso injuries other than rib fractures are relatively uncommon. As this article points out, elderly trauma patients almost universally have significant medical problems. Cognitive deficits and balance issues may explain ground-level falls in this population. Syncope from a myriad of underlying medical conditions and/or medications may have contributed to their falls as well.
The evaluation process for elderly trauma patients must be directed not only at diagnosing injury but also at attempting to identify the reason for the injury. This may involve a number of diagnostic tests in the ED, in the outpatient setting, or even on an inpatient floor.
Unfortunately, elderly patients can succumb to relatively minor injuries, and those who survive such afflictions often have difficulty making a full recovery. Many elderly patients who were able to function preinjury were marginally compensated at home. Operative therapy, often needed to treat injuries such as a hip or extremity fracture, by itself represents physiological burden to an elderly patient. Likewise, full recovery after even a mild TBI can be quite difficult.
Admitting an elderly patient to the hospital can present several challenges. For example, elderly patients are often on a number of prescription and nonprescription medications, including over-the-counter nutritional and herbal supplements, many of which interact with the newly prescribed medications given to treat trauma (eg, analgesics, sedatives, antiseizure drugs). Moreover, elderly patients often become disoriented and agitated when they are out of their home environment. All too often, the therapy for these and other problems is another medication, and thus the cycle continues. Therefore, elderly patients are ultimately at increased risk for death from seemingly trivial injury, which in turn may create significant perceived quality issues for a medical center.
The use of systemic anticoagulation has become almost ubiquitous in older patients. Some days it seems like every patient I (TS) admit is taking an anticoagulant—at least aspirin. While primary care providers (PCPs) correctly realize the important role these anticoagulants have in treating chronic medical conditions, they often do not recognize the dangers associated with increased traumatic bleeding following an injury.
Frequently, we knowingly take patients with conditions such as rate-controlled atrial fibrillation (AF) off their prescribed anticoagulant, believing they are simply not candidates for anticoagulation because of their propensity to fall. Even though we attempt to communicate our concerns to the PCP, when these patients are readmitted, it is common to find that they have been placed back on an anticoagulant.
The advent of novel oral anticoagulants (NOACs) has made routine laboratory testing obsolete. One need only to turn on the television to see the many advertisements explaining why this agent or that agent is preferable to warfarin. While, fresh frozen plasma (FFP) and/or prothrombin complex concentrates (PCC) are quite effective at reversing the anticoagulant effect of warfarin, reversal of these newer agents is either extremely difficult or impossible.
Anticoagulant reversal can be more or less important, depending on the situation. For instance, while subcutaneous bleeding is concerning, it can be temporized by operative exploration and/or packing. When necessary, blood can be transfused to replace the blood lost. However, the same is not true for a patient with significant TBI, because even a small volume of ongoing hemorrhage can prove lethal. Cavitary hemorrhage in the chest and/or abdomen is also extremely difficult to treat if the anticoagulant effect cannot be reversed. Given the popularity of the new anticoagulants, I (TS) am afraid that this problem will be with us for years to come.
There has been a significant spike in interpersonal violence in the United States over the past few years. While the cause is often difficult to identify, its existence is impossible to ignore. The violence seems to be concentrated in a number of municipal areas, but violence can occur in any community. Certainly, even mass casualties have become part of our everyday life.
In 2016, homicides and nonfatal shootings increased dramatically relative to 2015. In 201 7 , we are tracking a 40% increase in homicides and a 30% increase in nonfatal shootings—particularly concerning when one considers that these numbers are being compared to the previously increased 2016 statistics.
Many community EDs are not accustomed to dealing with a significant volume of penetrating trauma, and thus they may not be as familiar with the newest means of resuscitation, evaluation, and treatment of these injuries. It will be important for every medical center to do what is necessary to be able to effectively triage and provide initial treatment for patients with penetrating trauma.
The victims of penetrating trauma are often young, and unfortunately, despite our best efforts, these patients often die in the ED. This creates a huge emotional burden on people who work in the ED, particularly those who are not used to seeing large volumes of gunshot wounds (GSWs) or stab wounds. Even those of us working in busy urban trauma centers feel the emotional burden of this new epidemic. Each of us will need to cope with these issues and help each other deal with them.
It is important to recognize the dramatic change in trauma demographics over the last few years, and make plans to care for the changing face of trauma to optimize results and save as many lives as possible. In part 1 of our 2-part, “The Changing Landscape of Trauma Care,” we focus on the specific issues and concerns encountered in elderly trauma patients, as well as victims of all ages presenting with penetrating trauma from stab and GSWs.
Trauma in the Elderly Population
There has been and continues to be an increase in the elderly population in the United States. In 2014, 46 million Americans representing 15% of the total population were older than age 65 years.1 Of all age groups in the United States, the elderly population is one of the fastest growing and, according to the 2010 Census, grew at a faster rate than in previous years.2 This growth is expected to continue as many of the post-World War II baby boomer generation age. By the year 2030, an estimated 1 in 5 Americans will be older than 65 years of age—representing a 7% absolute increase from 2010 to 2030.1
Furthermore, men and women in this population are maintaining an active lifestyle well into their seventh and eighth decade, which has led to an increased incidence of trauma in this age group, primarily from falls and low-velocity MVCs. According to data from the National Trauma Data Bank in 2016, nearly 43% of all traumatic incidents occurred in patients older than age 55 years, as compared to only 32% in 2010.3,4 Today, injury is the seventh leading cause of death among the elderly population.5
Pre-existing Conditions and Comorbidities
The elderly population tends to have more complex medical histories, with pre-existing conditions and comorbidities—both of which result in intolerance to alterations from normal physiology after acute trauma and may place them at risk for complications and death. This point was highlighted in an invited commentary by one of us (TS) over 20 years ago, in which he stated, “Resting organ function often is preserved, but the ability to augment performance in response to stress is greatly compromised.”6
Studies in the early 1990s established a link between trauma outcomes and comorbidities.7-9 Morris et al7 found that ischemic heart disease, diabetes, chronic obstructive pulmonary disease, congenital coagulopathy, and cirrhosis highly influence trauma outcomes. They also noted that 25% of trauma patients over age 65 years had at least one of these five comorbidities and were nearly two times more likely to die. These findings were confirmed in 2002 by Grossman et al,8 who demonstrated that each year over age 65 years held a mortality increase of 6.8%.8 Additionally, they found that congestive heart failure, cancer, renal disease, and hepatic disease were the comorbidities with the highest impact on mortality.8
The presence of pre-existing conditions or comorbidities has also been associated with increased risk for complications, and subsequent increased mortality. In 2010, Aitken et al9 found that 6.2% of elderly trauma patients developed pneumonia postinjury, which was associated with increased intensive care unit (ICU) and hospital length of stay.Pre-existing pulmonary disease and higher Injury Severity Scores (ISS) were also found to be risk factors, demonstrating a 5.9% incidence of acute kidney injury in this group, conferring a 10-fold increased risk of mortality.
In efforts to improve outcomes in elderly trauma patients, many centers have integrated geriatric consults in the ED for all patients over a certain age, following injury. Though Olufajo et al10 were unable to demonstrate an in-house or 30-day mortality benefit after implementing a mandatory geriatrics consult for patients over age 70 years, they did show a nonstatistically significant trend toward fewer ICU readmissions with the consults.
In 2001, Demetriades et al11 reported a 50% mortality rate among patients aged 70 years and older who met criteria for full trauma team activation. Interestingly, the mortality rate for patients over age 70 years was 24%, compared to 7.6% for younger patients admitted during the same period. Those in the 70 years and older age group who did not meet criteria for full trauma team activation still had a 16% mortality rate, and 24% required ICU admission.
Demetriades et al11 also demonstrated that prehospital/admission vital signs in patients 70 years and older were often normal but misleading. In this group, 63% of patients with an ISS greater than 15, and 25% with an ISS greater than 30 did not have tachycardia or hypotension criteria for full trauma activation.11 These findings have led to recommendations for a lower threshold for trauma activations in geriatric patients.12
Recent studies have suggested that adding an age threshold to the trauma activation criteria may improve outcomes without leading to an unacceptable overtriage rate. In 2016, Hammer, et al13 reported improved outcomes, with only 2% of patients being overtriaged, when they added to their trauma activation criteria an age threshold of 70 years, regardless of physiology or mechanism of injury. They ultimately concluded that it was appropriate and cost-effective. In 2017, Cooper et al14 published a position paper on the Geriatric Trauma Coalition (GeriTraC) covering the convergence of aging and injury. The mission of GeriTraC is to improve geriatric trauma care from prevention to transition of care.14
Fall-Related Trauma
Falls are the most common cause of fatal and nonfatal injury in patients over age 65 years.15 Most fall injuries occur at home and during the winter months, and tend to be from ground level.16 Although most result in only minor trauma, many cause significant injuries requiring hospitalization. In 2006, Stevens et al17 estimated that both fatal and nonfatal falls in the elderly accounted for almost $20 billion in direct medical costs.
Motor Vehicle Collisions
Motor vehicle collisions/pedestrian struck are the second most significant causes of fatal and nonfatal injury in elderly patients. Older drivers who are hospitalized following an MVC have significantly longer hospital lengths of stay and an overall higher mortality rate.16 Elderly patients are more likely to be victims of “pedestrian-struck-by-vehicle” due to their decreased visual and auditory acuity, reduced reaction time, slower movement, and confusion.
Suicide
Suicide is the third leading cause of injury-related death for those aged 65 years and older.15 Risk factors for suicide in the elderly population include psychiatric disorders, particularly depression; medical conditions, especially cancer or chronic lung disease; moderate-to-large alcohol use; and social isolation. Changes in behavior, such as altering a will, new preoccupation with religion, or giving away life possessions, may be warning signs of impending suicide.
Novel Oral Anticoagulants
Many people, both old and young, are taking oral anticoagulants for various conditions. Warfarin has traditionally been the medication of choice, with readily available reversal agents, if needed. However, the development of NOACs, which antagonize activity of a single step in the coagulation cascade, has presented trauma care providers with a new challenge in achieving hemostasis. The NOACs include a direct thrombin inhibitor (dabigatran), and the Factor Xa (FXa) inhibitors (apixaban, edoxaban, and rivaroxaban). These NOACs have been shown to be as effective as traditional vitamin K antagonists (warfarin) with a comparable or lower spontaneous risk of bleeding. Along with an acceptable safety profile, these drugs cause significantly less drug and food interactions and are easier to dose, with no need for monitoring levels.18 Since the arrival of the first NOAC dabigatran in 2010, use of these drugs has continued to increase, and are becoming more popular in the treatment of venous thromboembolism in younger patients as well. A study by Desai et al19 examining newly initiated anticoagulation for AF between 2010 and 2013 found that 62% of all new anticoagulant prescriptions were for NOACs.
Hemostasis Challenges
Because of the lack of reversal agents or antidotes available, the NOACs present a unique challenge and major concern when anticoagulation properties must be reversed quickly. Among the NOACs, dabigatran is the only NOAC that is 35% protein bound and can be effectively cleared by hemodialysis (HD). Rivaroxaban and apixaban, in contrast, are highly protein bound (95% and 87%, respectively), which renders HD ineffective for clearance. Even for dabigatran, though HD may be a treatment option in the presence of potentially life-threatening bleeding associated with dabigatran alone, this is only a possibility if the patient’s hemodynamics can tolerate HD.
Extrapolating from experience with warfarin-associated bleeding, the use of FFP, PCC, and recombinant activated factor VII for NOAC-associated bleeding has been proposed and attempted.20 Though FFP may be necessary to restore circulating blood volume as part of a massive transfusion protocol in a patient with NOAC-associated hemorrhage, it is generally not a reasonable sole strategy for reversal of NOACs because the coagulation factors in FFP are not present in high enough concentrations to be effective.18
Prothrombin Complex Concentrates
Three- and Four-Factor PCCs. Four-factor PCC (4F-PCC), which became available for use in the United States in April 2013, contains concentrated amounts of all four of the vitamin K dependent factors (II, VII, IX, and X), as well as proteins C and S. Three-factor PCC (3F-PCC) does not contain significant levels of factor VII,20 and preclinical studies on its efficacy in reversing NOACs have not been consistent.
Early studies using animal models showed promising results for both 3F-PCC and 4F-PCC in correcting derangements in laboratory coagulation markers as well as observed bleeding time.21-23 However, other animal studies failed to demonstrate an improvement in observed bleeding time or volume despite full or partial correction of coagulation studies after PCC.24,25 In human studies, PCC has been observed to correct some laboratory parameters of coagulation, but not others.26,27 Thus far, these studies have been limited to healthy volunteers without active bleeding and have been largely ex vivo and in vitro studies, so it is difficult to determine if the demonstrated correction of coagulation studies translates into clinical benefit. Both 3F-PCC and 4F-PCC have shown promise, though studies with 4F-PCC have yielded more consistent results.26,27Activated PCC. Activated PCC (aPCC), which contains the same vitamin K dependent factors (factors II, VII, IX, X) with some in their activated form, has shown similar results. In fact, ex vivo and in vitro studies thus far seem to suggest that aPCC is more effective than PCC in correcting coagulation test parameters, as well as thrombin generation indices.28-31 However, an aPCC has also been demonstrated to be more procoagulant and, thus, may increase the risk of thrombotic complications.32
Recombinant Activated Factor VII
Recombinant activated factor VII has shown less promise than PCC or aPCC in the reversal of NOAC-associated bleeding. Additionally, similar to aPCC, it may increase the risk of thrombosis.20,33
Monoclonal Antibody Agent
In October 2015, the US Food and Drug Administration approved idarucizumab, a monoclonal antibody agent for the reversal of dabigatran. Idarucizumab has a binding affinity approximately 350 times higher than the binding affinity of dabigatran for thrombin with no demonstrated procoagulant effects.20 To date, there are no commercially available antidotes or reversal agents for the FXa inhibitors, though two promising agents are in various phases of clinical trials. The first, andexanet alfa, is a modified, recombinant factor X which binds FXa inhibitors with high affinity. This agent has shown promising results in the reversal of apixaban and rivaroxaban.20 The second is called aripazine (PER977) and has the potential to reverse unfractionated heparin, low molecular weight heparins, fondaparinux, FXa inhibitors, and thrombin inhibitors. Early in vivo human studies have been promising.18
Currently, there are no well-designed clinical studies examining the use of PCC for NOAC reversal in trauma. There are only a few published case reports, showing both successful and unsuccessful results, and a small retrospective series of only 18 patients specifically looking at both traumatic and spontaneous intracranial hemorrhage.34-37 There are also no universally agreed upon published guidelines for the management of NOAC-associated bleeding in the absence of drug-specific reversal agents.
Penetrating Trauma
The United States leads all high-income nations in GSW mortality,38 and its rate of firearm homicide is almost 20 times that of other high-income countries. In 2014, there were more than 33,000 firearm-related deaths in the United States, almost two-thirds of which were suicide-related.38 These numbers represent 16.8% of all deaths from injury. For each fatal firearm injury, there were nearly two nonfatal firearm injuries (65,106) the same year.39 Since 2001, the leading cause of death among black males aged 15 to 44 years has been firearm-related homicide. In 2015, that age demographic was lowered to include 10- to 14-year-old black males. In 2015, suicide by firearm was the second leading cause of death among white males over the age of 55 years and the third leading cause of death among white males aged 10 to 54 years.40
Incidents of gun violence are on the rise. These incidents are becoming more frequent and more often fatal. In a retrospective review of their trauma registry, as well as county records, Sauaia et al41 examined trends of GSW severity and mortality in Denver, Colorado from 2000 to 2013. They noted the proportion of GSW admissions remained stable over time, but injury severity and mortality from GSWs increased significantly, contrary to mortality and survival trends for all other injury mechanisms.41
The increasing GSW severity and mortality trend is not unique to Denver. Many media sources in cities across the country have reported similar statistics obtained from their local police departments in the past year. Though gun violence is a subject that is in desperate need of prevention research, current legislation makes these studies challenging to undertake. In 1996, Congress passed the Dickey Amendment to the Omnibus Consolidated Appropriations Act for the 1997 fiscal year, which states that “none of the funds made available for injury prevention and control at the Centers for Disease Control and Prevention may be used to advocate or promote gun control.”42,43 In the 2011 Consolidated Appropriations Act for the fiscal year 2012, this restriction was expanded to include the National Institutes of Health (NIH).44,45 These measures largely explain the paucity of primary research in gun violence in the last two decades—despite the increasing role and costs gun violence contributes to the US health care system. Gun violence is an epidemic, and like all other epidemics in the United States, it requires government-funded research to help protect the people.44
Conclusion
The last decade has seen some significant changes in trauma demographics in the United States. As the population of US men and women older than age 65 years continues to grow, trauma can no longer be considered a disease of young people. In addition to elderly men and women being more active than ever before, comorbid diseases place them at higher risk for complications and death following injury. For these reasons, many trauma triage algorithms now include age as an independent factor in activating a trauma alert. In addition to age, medications, and especially polypharmacy, can place patients at greater risk of injury and complications following trauma.
The last 10 years also has seen an increase in the number of patients on anticoagulants. The development of the NOACs further complicates the care of trauma patients taking these medications. Although designed to simplify care for patients and providers by minimizing bleeding risks and eliminating blood monitoring, there are only limited, and sometimes no reliable reversal agents available for NOACs, creating challenges when treating trauma patients who are on these medications. Finally, despite efforts by many individuals and groups, gun violence still remains a large and growing problem in the United States. Hopefully, continued efforts of national, state and local programs will begin to improve the current situation.
Editor’s Note: Part 2 of “The Changing Landscape of Trauma Care” will appear in the August 2017 issue of Emergency Medicine and will cover the changes in strategies and techniques to care for injured patients.
Introduction
There has been a fundamental change in the face of injury in the United States. Traditionally, injury was thought to be a disease of the young male population, with motor vehicle collision (MVC) being the most common mechanism of injury. Depending on the trauma center, blunt trauma would comprise up to 99% of patients admitted. This profile has fundamentally changed over the last 15 years. Trauma center performance is often benchmarked against local, regional, or national norms, and as all medical centers now measure quality as the primary endpoint, these changes in demographics can be very important.
Certainly, the most important change has been the “graying” of trauma patients. When I (TS) started working in Baltimore 20 years ago, patients over age 65 years comprised approximately 5% of our total trauma admissions. Last year, over 30% of our 7,000 primary admissions were patients over age 65 years who had sustained ground-level falls.
Injury patterns in the elderly differ compared to standard blunt trauma in which traumatic brain injury (TBI) is common. Extremity fractures, particularly hip fractures, are common, whereas torso injuries other than rib fractures are relatively uncommon. As this article points out, elderly trauma patients almost universally have significant medical problems. Cognitive deficits and balance issues may explain ground-level falls in this population. Syncope from a myriad of underlying medical conditions and/or medications may have contributed to their falls as well.
The evaluation process for elderly trauma patients must be directed not only at diagnosing injury but also at attempting to identify the reason for the injury. This may involve a number of diagnostic tests in the ED, in the outpatient setting, or even on an inpatient floor.
Unfortunately, elderly patients can succumb to relatively minor injuries, and those who survive such afflictions often have difficulty making a full recovery. Many elderly patients who were able to function preinjury were marginally compensated at home. Operative therapy, often needed to treat injuries such as a hip or extremity fracture, by itself represents physiological burden to an elderly patient. Likewise, full recovery after even a mild TBI can be quite difficult.
Admitting an elderly patient to the hospital can present several challenges. For example, elderly patients are often on a number of prescription and nonprescription medications, including over-the-counter nutritional and herbal supplements, many of which interact with the newly prescribed medications given to treat trauma (eg, analgesics, sedatives, antiseizure drugs). Moreover, elderly patients often become disoriented and agitated when they are out of their home environment. All too often, the therapy for these and other problems is another medication, and thus the cycle continues. Therefore, elderly patients are ultimately at increased risk for death from seemingly trivial injury, which in turn may create significant perceived quality issues for a medical center.
The use of systemic anticoagulation has become almost ubiquitous in older patients. Some days it seems like every patient I (TS) admit is taking an anticoagulant—at least aspirin. While primary care providers (PCPs) correctly realize the important role these anticoagulants have in treating chronic medical conditions, they often do not recognize the dangers associated with increased traumatic bleeding following an injury.
Frequently, we knowingly take patients with conditions such as rate-controlled atrial fibrillation (AF) off their prescribed anticoagulant, believing they are simply not candidates for anticoagulation because of their propensity to fall. Even though we attempt to communicate our concerns to the PCP, when these patients are readmitted, it is common to find that they have been placed back on an anticoagulant.
The advent of novel oral anticoagulants (NOACs) has made routine laboratory testing obsolete. One need only to turn on the television to see the many advertisements explaining why this agent or that agent is preferable to warfarin. While, fresh frozen plasma (FFP) and/or prothrombin complex concentrates (PCC) are quite effective at reversing the anticoagulant effect of warfarin, reversal of these newer agents is either extremely difficult or impossible.
Anticoagulant reversal can be more or less important, depending on the situation. For instance, while subcutaneous bleeding is concerning, it can be temporized by operative exploration and/or packing. When necessary, blood can be transfused to replace the blood lost. However, the same is not true for a patient with significant TBI, because even a small volume of ongoing hemorrhage can prove lethal. Cavitary hemorrhage in the chest and/or abdomen is also extremely difficult to treat if the anticoagulant effect cannot be reversed. Given the popularity of the new anticoagulants, I (TS) am afraid that this problem will be with us for years to come.
There has been a significant spike in interpersonal violence in the United States over the past few years. While the cause is often difficult to identify, its existence is impossible to ignore. The violence seems to be concentrated in a number of municipal areas, but violence can occur in any community. Certainly, even mass casualties have become part of our everyday life.
In 2016, homicides and nonfatal shootings increased dramatically relative to 2015. In 201 7 , we are tracking a 40% increase in homicides and a 30% increase in nonfatal shootings—particularly concerning when one considers that these numbers are being compared to the previously increased 2016 statistics.
Many community EDs are not accustomed to dealing with a significant volume of penetrating trauma, and thus they may not be as familiar with the newest means of resuscitation, evaluation, and treatment of these injuries. It will be important for every medical center to do what is necessary to be able to effectively triage and provide initial treatment for patients with penetrating trauma.
The victims of penetrating trauma are often young, and unfortunately, despite our best efforts, these patients often die in the ED. This creates a huge emotional burden on people who work in the ED, particularly those who are not used to seeing large volumes of gunshot wounds (GSWs) or stab wounds. Even those of us working in busy urban trauma centers feel the emotional burden of this new epidemic. Each of us will need to cope with these issues and help each other deal with them.
It is important to recognize the dramatic change in trauma demographics over the last few years, and make plans to care for the changing face of trauma to optimize results and save as many lives as possible. In part 1 of our 2-part, “The Changing Landscape of Trauma Care,” we focus on the specific issues and concerns encountered in elderly trauma patients, as well as victims of all ages presenting with penetrating trauma from stab and GSWs.
Trauma in the Elderly Population
There has been and continues to be an increase in the elderly population in the United States. In 2014, 46 million Americans representing 15% of the total population were older than age 65 years.1 Of all age groups in the United States, the elderly population is one of the fastest growing and, according to the 2010 Census, grew at a faster rate than in previous years.2 This growth is expected to continue as many of the post-World War II baby boomer generation age. By the year 2030, an estimated 1 in 5 Americans will be older than 65 years of age—representing a 7% absolute increase from 2010 to 2030.1
Furthermore, men and women in this population are maintaining an active lifestyle well into their seventh and eighth decade, which has led to an increased incidence of trauma in this age group, primarily from falls and low-velocity MVCs. According to data from the National Trauma Data Bank in 2016, nearly 43% of all traumatic incidents occurred in patients older than age 55 years, as compared to only 32% in 2010.3,4 Today, injury is the seventh leading cause of death among the elderly population.5
Pre-existing Conditions and Comorbidities
The elderly population tends to have more complex medical histories, with pre-existing conditions and comorbidities—both of which result in intolerance to alterations from normal physiology after acute trauma and may place them at risk for complications and death. This point was highlighted in an invited commentary by one of us (TS) over 20 years ago, in which he stated, “Resting organ function often is preserved, but the ability to augment performance in response to stress is greatly compromised.”6
Studies in the early 1990s established a link between trauma outcomes and comorbidities.7-9 Morris et al7 found that ischemic heart disease, diabetes, chronic obstructive pulmonary disease, congenital coagulopathy, and cirrhosis highly influence trauma outcomes. They also noted that 25% of trauma patients over age 65 years had at least one of these five comorbidities and were nearly two times more likely to die. These findings were confirmed in 2002 by Grossman et al,8 who demonstrated that each year over age 65 years held a mortality increase of 6.8%.8 Additionally, they found that congestive heart failure, cancer, renal disease, and hepatic disease were the comorbidities with the highest impact on mortality.8
The presence of pre-existing conditions or comorbidities has also been associated with increased risk for complications, and subsequent increased mortality. In 2010, Aitken et al9 found that 6.2% of elderly trauma patients developed pneumonia postinjury, which was associated with increased intensive care unit (ICU) and hospital length of stay.Pre-existing pulmonary disease and higher Injury Severity Scores (ISS) were also found to be risk factors, demonstrating a 5.9% incidence of acute kidney injury in this group, conferring a 10-fold increased risk of mortality.
In efforts to improve outcomes in elderly trauma patients, many centers have integrated geriatric consults in the ED for all patients over a certain age, following injury. Though Olufajo et al10 were unable to demonstrate an in-house or 30-day mortality benefit after implementing a mandatory geriatrics consult for patients over age 70 years, they did show a nonstatistically significant trend toward fewer ICU readmissions with the consults.
In 2001, Demetriades et al11 reported a 50% mortality rate among patients aged 70 years and older who met criteria for full trauma team activation. Interestingly, the mortality rate for patients over age 70 years was 24%, compared to 7.6% for younger patients admitted during the same period. Those in the 70 years and older age group who did not meet criteria for full trauma team activation still had a 16% mortality rate, and 24% required ICU admission.
Demetriades et al11 also demonstrated that prehospital/admission vital signs in patients 70 years and older were often normal but misleading. In this group, 63% of patients with an ISS greater than 15, and 25% with an ISS greater than 30 did not have tachycardia or hypotension criteria for full trauma activation.11 These findings have led to recommendations for a lower threshold for trauma activations in geriatric patients.12
Recent studies have suggested that adding an age threshold to the trauma activation criteria may improve outcomes without leading to an unacceptable overtriage rate. In 2016, Hammer, et al13 reported improved outcomes, with only 2% of patients being overtriaged, when they added to their trauma activation criteria an age threshold of 70 years, regardless of physiology or mechanism of injury. They ultimately concluded that it was appropriate and cost-effective. In 2017, Cooper et al14 published a position paper on the Geriatric Trauma Coalition (GeriTraC) covering the convergence of aging and injury. The mission of GeriTraC is to improve geriatric trauma care from prevention to transition of care.14
Fall-Related Trauma
Falls are the most common cause of fatal and nonfatal injury in patients over age 65 years.15 Most fall injuries occur at home and during the winter months, and tend to be from ground level.16 Although most result in only minor trauma, many cause significant injuries requiring hospitalization. In 2006, Stevens et al17 estimated that both fatal and nonfatal falls in the elderly accounted for almost $20 billion in direct medical costs.
Motor Vehicle Collisions
Motor vehicle collisions/pedestrian struck are the second most significant causes of fatal and nonfatal injury in elderly patients. Older drivers who are hospitalized following an MVC have significantly longer hospital lengths of stay and an overall higher mortality rate.16 Elderly patients are more likely to be victims of “pedestrian-struck-by-vehicle” due to their decreased visual and auditory acuity, reduced reaction time, slower movement, and confusion.
Suicide
Suicide is the third leading cause of injury-related death for those aged 65 years and older.15 Risk factors for suicide in the elderly population include psychiatric disorders, particularly depression; medical conditions, especially cancer or chronic lung disease; moderate-to-large alcohol use; and social isolation. Changes in behavior, such as altering a will, new preoccupation with religion, or giving away life possessions, may be warning signs of impending suicide.
Novel Oral Anticoagulants
Many people, both old and young, are taking oral anticoagulants for various conditions. Warfarin has traditionally been the medication of choice, with readily available reversal agents, if needed. However, the development of NOACs, which antagonize activity of a single step in the coagulation cascade, has presented trauma care providers with a new challenge in achieving hemostasis. The NOACs include a direct thrombin inhibitor (dabigatran), and the Factor Xa (FXa) inhibitors (apixaban, edoxaban, and rivaroxaban). These NOACs have been shown to be as effective as traditional vitamin K antagonists (warfarin) with a comparable or lower spontaneous risk of bleeding. Along with an acceptable safety profile, these drugs cause significantly less drug and food interactions and are easier to dose, with no need for monitoring levels.18 Since the arrival of the first NOAC dabigatran in 2010, use of these drugs has continued to increase, and are becoming more popular in the treatment of venous thromboembolism in younger patients as well. A study by Desai et al19 examining newly initiated anticoagulation for AF between 2010 and 2013 found that 62% of all new anticoagulant prescriptions were for NOACs.
Hemostasis Challenges
Because of the lack of reversal agents or antidotes available, the NOACs present a unique challenge and major concern when anticoagulation properties must be reversed quickly. Among the NOACs, dabigatran is the only NOAC that is 35% protein bound and can be effectively cleared by hemodialysis (HD). Rivaroxaban and apixaban, in contrast, are highly protein bound (95% and 87%, respectively), which renders HD ineffective for clearance. Even for dabigatran, though HD may be a treatment option in the presence of potentially life-threatening bleeding associated with dabigatran alone, this is only a possibility if the patient’s hemodynamics can tolerate HD.
Extrapolating from experience with warfarin-associated bleeding, the use of FFP, PCC, and recombinant activated factor VII for NOAC-associated bleeding has been proposed and attempted.20 Though FFP may be necessary to restore circulating blood volume as part of a massive transfusion protocol in a patient with NOAC-associated hemorrhage, it is generally not a reasonable sole strategy for reversal of NOACs because the coagulation factors in FFP are not present in high enough concentrations to be effective.18
Prothrombin Complex Concentrates
Three- and Four-Factor PCCs. Four-factor PCC (4F-PCC), which became available for use in the United States in April 2013, contains concentrated amounts of all four of the vitamin K dependent factors (II, VII, IX, and X), as well as proteins C and S. Three-factor PCC (3F-PCC) does not contain significant levels of factor VII,20 and preclinical studies on its efficacy in reversing NOACs have not been consistent.
Early studies using animal models showed promising results for both 3F-PCC and 4F-PCC in correcting derangements in laboratory coagulation markers as well as observed bleeding time.21-23 However, other animal studies failed to demonstrate an improvement in observed bleeding time or volume despite full or partial correction of coagulation studies after PCC.24,25 In human studies, PCC has been observed to correct some laboratory parameters of coagulation, but not others.26,27 Thus far, these studies have been limited to healthy volunteers without active bleeding and have been largely ex vivo and in vitro studies, so it is difficult to determine if the demonstrated correction of coagulation studies translates into clinical benefit. Both 3F-PCC and 4F-PCC have shown promise, though studies with 4F-PCC have yielded more consistent results.26,27Activated PCC. Activated PCC (aPCC), which contains the same vitamin K dependent factors (factors II, VII, IX, X) with some in their activated form, has shown similar results. In fact, ex vivo and in vitro studies thus far seem to suggest that aPCC is more effective than PCC in correcting coagulation test parameters, as well as thrombin generation indices.28-31 However, an aPCC has also been demonstrated to be more procoagulant and, thus, may increase the risk of thrombotic complications.32
Recombinant Activated Factor VII
Recombinant activated factor VII has shown less promise than PCC or aPCC in the reversal of NOAC-associated bleeding. Additionally, similar to aPCC, it may increase the risk of thrombosis.20,33
Monoclonal Antibody Agent
In October 2015, the US Food and Drug Administration approved idarucizumab, a monoclonal antibody agent for the reversal of dabigatran. Idarucizumab has a binding affinity approximately 350 times higher than the binding affinity of dabigatran for thrombin with no demonstrated procoagulant effects.20 To date, there are no commercially available antidotes or reversal agents for the FXa inhibitors, though two promising agents are in various phases of clinical trials. The first, andexanet alfa, is a modified, recombinant factor X which binds FXa inhibitors with high affinity. This agent has shown promising results in the reversal of apixaban and rivaroxaban.20 The second is called aripazine (PER977) and has the potential to reverse unfractionated heparin, low molecular weight heparins, fondaparinux, FXa inhibitors, and thrombin inhibitors. Early in vivo human studies have been promising.18
Currently, there are no well-designed clinical studies examining the use of PCC for NOAC reversal in trauma. There are only a few published case reports, showing both successful and unsuccessful results, and a small retrospective series of only 18 patients specifically looking at both traumatic and spontaneous intracranial hemorrhage.34-37 There are also no universally agreed upon published guidelines for the management of NOAC-associated bleeding in the absence of drug-specific reversal agents.
Penetrating Trauma
The United States leads all high-income nations in GSW mortality,38 and its rate of firearm homicide is almost 20 times that of other high-income countries. In 2014, there were more than 33,000 firearm-related deaths in the United States, almost two-thirds of which were suicide-related.38 These numbers represent 16.8% of all deaths from injury. For each fatal firearm injury, there were nearly two nonfatal firearm injuries (65,106) the same year.39 Since 2001, the leading cause of death among black males aged 15 to 44 years has been firearm-related homicide. In 2015, that age demographic was lowered to include 10- to 14-year-old black males. In 2015, suicide by firearm was the second leading cause of death among white males over the age of 55 years and the third leading cause of death among white males aged 10 to 54 years.40
Incidents of gun violence are on the rise. These incidents are becoming more frequent and more often fatal. In a retrospective review of their trauma registry, as well as county records, Sauaia et al41 examined trends of GSW severity and mortality in Denver, Colorado from 2000 to 2013. They noted the proportion of GSW admissions remained stable over time, but injury severity and mortality from GSWs increased significantly, contrary to mortality and survival trends for all other injury mechanisms.41
The increasing GSW severity and mortality trend is not unique to Denver. Many media sources in cities across the country have reported similar statistics obtained from their local police departments in the past year. Though gun violence is a subject that is in desperate need of prevention research, current legislation makes these studies challenging to undertake. In 1996, Congress passed the Dickey Amendment to the Omnibus Consolidated Appropriations Act for the 1997 fiscal year, which states that “none of the funds made available for injury prevention and control at the Centers for Disease Control and Prevention may be used to advocate or promote gun control.”42,43 In the 2011 Consolidated Appropriations Act for the fiscal year 2012, this restriction was expanded to include the National Institutes of Health (NIH).44,45 These measures largely explain the paucity of primary research in gun violence in the last two decades—despite the increasing role and costs gun violence contributes to the US health care system. Gun violence is an epidemic, and like all other epidemics in the United States, it requires government-funded research to help protect the people.44
Conclusion
The last decade has seen some significant changes in trauma demographics in the United States. As the population of US men and women older than age 65 years continues to grow, trauma can no longer be considered a disease of young people. In addition to elderly men and women being more active than ever before, comorbid diseases place them at higher risk for complications and death following injury. For these reasons, many trauma triage algorithms now include age as an independent factor in activating a trauma alert. In addition to age, medications, and especially polypharmacy, can place patients at greater risk of injury and complications following trauma.
The last 10 years also has seen an increase in the number of patients on anticoagulants. The development of the NOACs further complicates the care of trauma patients taking these medications. Although designed to simplify care for patients and providers by minimizing bleeding risks and eliminating blood monitoring, there are only limited, and sometimes no reliable reversal agents available for NOACs, creating challenges when treating trauma patients who are on these medications. Finally, despite efforts by many individuals and groups, gun violence still remains a large and growing problem in the United States. Hopefully, continued efforts of national, state and local programs will begin to improve the current situation.
Editor’s Note: Part 2 of “The Changing Landscape of Trauma Care” will appear in the August 2017 issue of Emergency Medicine and will cover the changes in strategies and techniques to care for injured patients.
1. Federal Interagency Forum on Aging-Related Statistics. Older Americans 2016: key indicators of well-being. US Gov Print Off; Washington, DC. 2016; August. https://agingstats.gov/docs/LatestReport/Older-Americans-2016-Key-Indicators-of-WellBeing.pdf. Accessed June 8, 2017.
2. Howden L, Meyer J. Age and sex composition: 2010. 2010 Census Briefs. 2011;(May):1-16. http://www.census.gov/library/publications/2011/dec/c2010br-03.html. Accessed June 8, 2017.
3. American College of Surgeons Committee on Trauma. National Trauma Data Bank 2010 Annual Report. 2010:1-93. https://www.facs.org/~/media/files/quality%20programs/trauma/ntdb/ntdbannualreport2010.ashx. Accesssed June 8, 2017.
4. American College of Surgeons Committee on Trauma. National Trauma Data Bank 2016 Annual Report. 2016:1-147. https://www.facs.org/~/media/files/quality%20programs/trauma/ntdb/ntdb%20annual%20report%202016.ashx. Accessed June 8, 2017.
5. Health, United States 2015. With Special Feature on Racial and Ethnic Health Disparities. US Department of Health and Human Services. Centers for Disease Control and Prevention. 2016:126. https://www.cdc.gov/nchs/data/hus/hus15.pdf. Accessed June 8, 2017.
6. Scalea TM. Invited commentary (for McMahon DJ, William S, Kauder D. Comorbidity and trauma in the elderly. World J Surg. 1996;20(8):116. doi:10.1007/s002689900170.
7. Morris JA Jr, MacKenzie EJ, Edelstein SL. The effect of preexisting conditions
8. Grossman MD, Miller D, Scaff DW, Arcona S. When is an elder old? Effect of preexisting conditions on mortality in geriatric trauma. J Trauma. 2002;52(2):242-246.
9. Aitken LM, Burmeister E, Lang J, Chaboyer W, Richmond TS. Characteristics and outcomes of injured older adults after hospital admission. J Am Geriatr Soc. 2010;58(3):442-449. doi:10.1111/j.1532-5415.2010.02728.x.
10. Olufajo OA, Tulebaev S, Javedan H, et al. Integrating geriatric consults into routine care of older trauma patients: one-year experience of a level I trauma center. J Am Coll Surg. 2016;222(6):1029-1035. doi:10.1016/j.jamcollsurg.2015.12.058.
11. Demetriades D, Sava J, Alo K, et al. Old age as a criterion for trauma team activation. J Trauma. 2001;51(4):754-756; discussion 756-757.
12. Calland JF, Ingraham AM, Martin N, et al; Eastern Association for the Surgery of Trauma. Evaluation and management of geriatric trauma: an Eastern Association for the Surgery of Trauma practice management guideline. J Trauma Acute Care Surg. 2012;73(5 Suppl 4):S345-S350. doi:10.1097/TA.0b013e318270191f.
13. Hammer PM, Storey AC, Bell T, et al. Improving geriatric trauma outcomes: A small step toward a big problem. J Trauma Acute Care Surg. 2016;81(1):162-167. doi:10.1097/TA.0000000000001063.
14. Cooper Z, Maxwell CA, Fakhry SM, et al. A position paper: The convergence of aging and injury and the need for a Geriatric Trauma Coalition (GeriTraC). J Trauma Acute Care Surg. 2017;82(2):419-422. doi:10.1097/TA.0000000000001317.
15. Centers for Disease Control and Prevention. Web-based injury statistics query and reporting system (WISQARS). National Center for Injury Prevention and Control, Centers for Disease Control and Prevention. http://www.cdc.gov/injury/wisqars/index.html. Updated June 1, 2017. Accessed June 8, 2017.
16. Menaker J, Scalea TM. Care of the injured elderly. In Rosenthal RA, Zenilman K, Katlic MR, eds. Principles and Practice of Geriatric Surgery. 2nd ed. Springer: New York, NY: Springer; 2011:391-410.
17. Stevens JA, Corso PS, Finkelstein EA, Miller TR. The costs of fatal and non-fatal falls among older adults. Inj Prev. 2006;12(5):290-295. doi:10.1136/ip.2005.011015.
18. von Heymann C, Rosenthal C, Kaufner L, Sander M. Management of direct oral anticoagulants-associated bleeding in the trauma patient. Curr Opin Anaesthesiol. 2016;29(2):220-228. doi:10.1097/ACO.0000000000000294.
19. Desai NR, Krumme AA, Schneeweiss S, et al. Patterns of initiation of oral anticoagulants in patients with atrial fibrillation- quality and cost implications. Am J Med. 2014;127(11):1075-1082.e1. doi:10.1016/j.amjmed.2014.05.013.
20. Marano G, Vaglio S, Pupella S, Liumbruno GM, Franchini M. How we treat bleeding associated with direct oral anticoagulants. Blood Transfus. 2016;14(5):465-473. doi:10.2450/2016.0180-15.
21. Zhou W, Schwarting S, Illanes S, et al. Hemostatic therapy in experimental intracerebral hemorrhage associated with the direct thrombin inhibitor dabigatran. Stroke. 2011;42(12):3594-3599. doi:10.1161/STROKEAHA.111.624650.
22. Pragst I, Zeitler SH, Doerr B, et al. Reversal of dabigatran anticoagulation by prothrombin complex concentrate (Beriplex P/N) in a rabbit model. J Thromb Haemost. 2012;10(9):1841-1848. doi:10.1111/j.1538-7836.2012.04859.x.
23. van Ryn J, Schurer J, Kink-Eiband M, Clemens A. The successful reversal of dabigatran-induced bleeding by coagulation factor concentrates in a rat tail bleeding model do not correlate with ex vivo markers of anticoagulation. Blood. 2011;118(2316).
24. Herzog E, Kaspereit F, Krege W, Joanne R Van, Dickneite G, Pragst I. Non-clinical safety and efficacy of prothrombin complex concentrates (pcc) for the reversal of dabigatran mediated anticoagulation. J Thromb Haemost. 2013;11:693.
25. Godier A, Miclot A, Le Bonniec B, et al. Evaluation of prothrombin complex concentrate and recombinant activated factor VII to reverse rivaroxaban in a rabbit model. Anesthesiology. 2012;116(1):94-102. doi:10.1097/ALN.0b013e318238c036.
26. Eerenberg ES, Kamphuisen PW, Sijpkens MK, Meijers JC, Buller HR, Levi M. Reversal of rivaroxaban and dabigatran by prothrombin complex concentrate: a randomized, placebo-controlled, crossover study in healthy subjects. Circulation. 2011;124(14):1573-1579. doi:10.1161/CIRCULATIONAHA.111.029017.
27. Escolar G, Fernandez-Gallego V, Arellano-Rodrigo E, et al. Reversal of apixaban induced alterations in hemostasis by different coagulation factor concentrates: significance of studies in vitro with circulating human blood. PLoS One. 2013;8(11):e78696. doi:10.1371/journal.pone.0078696.
28. Galan AM, Arellano-Rodrigo E, Sanz V, et al. Effects of rivaroxaban and dabigatran on hemostasis and reversion of their antithrombotic effects by different coagulation factors: evidence raised from a clinical study in healthy volunteers. J Thromb Haemost. 2013;11:418-419.
29. Escolar G, Arellano-Rodrigo E, Lopez-Vilchez I, et al. Reversal of rivaroxaban-induced alterations on hemostasis by different coagulation factor concentrates—in vitro studies with steady and circulating human blood. Circ J. 2015;79(2):331-338. doi:0.1253/circj.CJ-14-0909.
30. Perzborn E, Gruber A, Tinel H, et al. Reversal of rivaroxaban anticoagulation by haemostatic agents in rats and primates. Thromb Haemost. 2013;110(1):162-172. doi:10.1160/TH12-12-0907.
31. Chan HHW, Atkinson HM, Goncharenko M, Berry LR, Chan AKC. Reversal of dabigatran using recombinant activated factor VII and activated prothrombin complex concentrates in thromboelastography assay. J Thromb Haemost. 2011;9:576-577.
32. Hoffman M, Monroe DM. Reversing targeted oral anticoagulants. Hematology Am Soc Hematol Educ Program. 2014;2014(1):518-523. doi:10.1182/asheducation-2014.1.518.
33. Marlu R, Hodaj E, Paris A, Albaladejo P, Cracowski JL, Pernod G. Effect of non-specific reversal agents on anticoagulant activity of dabigatran and rivaroxaban: a randomised crossover ex vivo study in healthy volunteers. Thromb Haemost. 2012;108(2):217-224. doi:10.1160/TH12-03-0179.
34. Grandhi R, Newman WC, Zhang X, et al. Administration of 4-factor prothrombin complex concentrate as an antidote for intracranial bleeding in patients taking direct factor xa inhibitors. World Neurosurg. 2015;84(6):1956-61. doi:10.1016/j.wneu.2015.08.042.
35. Durie R, Kohute M, Fernandez C, Knight M. Prothrombin complex concentrate for the management of severe traumatic bleeding in a patient anticoagulated with apixaban. J Clin Pharm Ther. 2016;41(1):92-93. doi:10.1111/jcpt.12339.
36. Kauffmann S, Chabanne R, Coste A, et al. Favorable outcome of rivaroxaban-associated intracerebral hemorrhage reversed by 4-factor prothrombin complex concentrate: impact on thrombin generation. A&A Case Rep. 2015;4(11):151-154. doi:10.1213/XAA.0000000000000143.
37. Maurice-Szamburski A, Graillon T, Bruder N. Favorable outcome after a subdural hematoma treated with feiba in a 77-year-old patient treated by rivaroxaban. J Neurosurg Anesthesiol. 2014;26(2):183. doi:10.1097/ANA.0000000000000030.
38. Richardson EG, Hemenway D. Homicide, suicide, and unintentional firearm fatality: comparing the United States with other high-income countries, 2003. J Trauma. 2011;70(1):238-243. doi:10.1097/TA.0b013e3181dbaddf.
39. Kochanek KD, Murphy SL, Xu J, Tejada-Vera B. Deaths: final data for 2014. Natl Vital Stat Rep. 2016;65(4):1-122. https://www.cdc.gov/nchs/data/nvsr/nvsr65/nvsr65_04.pdf. Accessed June 8, 2017.
40. Centers for Disease Control and Prevention. Leading causes of death reports, national and regional, 1999-2015. https://webappa.cdc.gov/sasweb/ncipc/leadcaus10_us.html. Accessed June 8, 2017.
41. Sauaia A, Gonzalez E, Moore HB, Bol K, Moore EE. Fatality and severity of firearm injuries in a denver trauma center, 2000-2013. JAMA. 2016;315(22):2465-2467. doi:10.1001/jama.2016.5978.
42. Omnibus Consolidated Appropriations Bill. HR 3610, Pub L No. 104-208. http://www.gpo.gov/fdsys/pkg/PLAW-104publ208/pdf/PLAW-104publ208.pdf. September 1996. Accessed June 13, 2017.
43. Kellermann AL, Rivara FP. Silencing the science on gun research. JAMA. 2013;309(6):549-550. doi:10.1001/jama.2012.208207.
44. Consolidated Appropriations Act of 2012. HR 2055: Pub L No. 112-174. https://www.gpo.gov/fdsys/pkg/PLAW-112publ74/pdf/PLAW-112publ74.pdf. December 2011. Accessed June 13, 2017.
45. Rubin R. Tale of 2 agencies: CDC avoids gun violence research but NIH funds it. JAMA. 2016;315(16):1689-1691. doi:10.1001/jama.2016.1707.
46. Cook A, Osler T, Hosmer D, et al. Gunshot wounds resulting in hospitalization in the United States: 2004-2013. Injury. 2017;48(3):621-627. doi:10.1016/j.injury.2017.01.044.
1. Federal Interagency Forum on Aging-Related Statistics. Older Americans 2016: key indicators of well-being. US Gov Print Off; Washington, DC. 2016; August. https://agingstats.gov/docs/LatestReport/Older-Americans-2016-Key-Indicators-of-WellBeing.pdf. Accessed June 8, 2017.
2. Howden L, Meyer J. Age and sex composition: 2010. 2010 Census Briefs. 2011;(May):1-16. http://www.census.gov/library/publications/2011/dec/c2010br-03.html. Accessed June 8, 2017.
3. American College of Surgeons Committee on Trauma. National Trauma Data Bank 2010 Annual Report. 2010:1-93. https://www.facs.org/~/media/files/quality%20programs/trauma/ntdb/ntdbannualreport2010.ashx. Accesssed June 8, 2017.
4. American College of Surgeons Committee on Trauma. National Trauma Data Bank 2016 Annual Report. 2016:1-147. https://www.facs.org/~/media/files/quality%20programs/trauma/ntdb/ntdb%20annual%20report%202016.ashx. Accessed June 8, 2017.
5. Health, United States 2015. With Special Feature on Racial and Ethnic Health Disparities. US Department of Health and Human Services. Centers for Disease Control and Prevention. 2016:126. https://www.cdc.gov/nchs/data/hus/hus15.pdf. Accessed June 8, 2017.
6. Scalea TM. Invited commentary (for McMahon DJ, William S, Kauder D. Comorbidity and trauma in the elderly. World J Surg. 1996;20(8):116. doi:10.1007/s002689900170.
7. Morris JA Jr, MacKenzie EJ, Edelstein SL. The effect of preexisting conditions
8. Grossman MD, Miller D, Scaff DW, Arcona S. When is an elder old? Effect of preexisting conditions on mortality in geriatric trauma. J Trauma. 2002;52(2):242-246.
9. Aitken LM, Burmeister E, Lang J, Chaboyer W, Richmond TS. Characteristics and outcomes of injured older adults after hospital admission. J Am Geriatr Soc. 2010;58(3):442-449. doi:10.1111/j.1532-5415.2010.02728.x.
10. Olufajo OA, Tulebaev S, Javedan H, et al. Integrating geriatric consults into routine care of older trauma patients: one-year experience of a level I trauma center. J Am Coll Surg. 2016;222(6):1029-1035. doi:10.1016/j.jamcollsurg.2015.12.058.
11. Demetriades D, Sava J, Alo K, et al. Old age as a criterion for trauma team activation. J Trauma. 2001;51(4):754-756; discussion 756-757.
12. Calland JF, Ingraham AM, Martin N, et al; Eastern Association for the Surgery of Trauma. Evaluation and management of geriatric trauma: an Eastern Association for the Surgery of Trauma practice management guideline. J Trauma Acute Care Surg. 2012;73(5 Suppl 4):S345-S350. doi:10.1097/TA.0b013e318270191f.
13. Hammer PM, Storey AC, Bell T, et al. Improving geriatric trauma outcomes: A small step toward a big problem. J Trauma Acute Care Surg. 2016;81(1):162-167. doi:10.1097/TA.0000000000001063.
14. Cooper Z, Maxwell CA, Fakhry SM, et al. A position paper: The convergence of aging and injury and the need for a Geriatric Trauma Coalition (GeriTraC). J Trauma Acute Care Surg. 2017;82(2):419-422. doi:10.1097/TA.0000000000001317.
15. Centers for Disease Control and Prevention. Web-based injury statistics query and reporting system (WISQARS). National Center for Injury Prevention and Control, Centers for Disease Control and Prevention. http://www.cdc.gov/injury/wisqars/index.html. Updated June 1, 2017. Accessed June 8, 2017.
16. Menaker J, Scalea TM. Care of the injured elderly. In Rosenthal RA, Zenilman K, Katlic MR, eds. Principles and Practice of Geriatric Surgery. 2nd ed. Springer: New York, NY: Springer; 2011:391-410.
17. Stevens JA, Corso PS, Finkelstein EA, Miller TR. The costs of fatal and non-fatal falls among older adults. Inj Prev. 2006;12(5):290-295. doi:10.1136/ip.2005.011015.
18. von Heymann C, Rosenthal C, Kaufner L, Sander M. Management of direct oral anticoagulants-associated bleeding in the trauma patient. Curr Opin Anaesthesiol. 2016;29(2):220-228. doi:10.1097/ACO.0000000000000294.
19. Desai NR, Krumme AA, Schneeweiss S, et al. Patterns of initiation of oral anticoagulants in patients with atrial fibrillation- quality and cost implications. Am J Med. 2014;127(11):1075-1082.e1. doi:10.1016/j.amjmed.2014.05.013.
20. Marano G, Vaglio S, Pupella S, Liumbruno GM, Franchini M. How we treat bleeding associated with direct oral anticoagulants. Blood Transfus. 2016;14(5):465-473. doi:10.2450/2016.0180-15.
21. Zhou W, Schwarting S, Illanes S, et al. Hemostatic therapy in experimental intracerebral hemorrhage associated with the direct thrombin inhibitor dabigatran. Stroke. 2011;42(12):3594-3599. doi:10.1161/STROKEAHA.111.624650.
22. Pragst I, Zeitler SH, Doerr B, et al. Reversal of dabigatran anticoagulation by prothrombin complex concentrate (Beriplex P/N) in a rabbit model. J Thromb Haemost. 2012;10(9):1841-1848. doi:10.1111/j.1538-7836.2012.04859.x.
23. van Ryn J, Schurer J, Kink-Eiband M, Clemens A. The successful reversal of dabigatran-induced bleeding by coagulation factor concentrates in a rat tail bleeding model do not correlate with ex vivo markers of anticoagulation. Blood. 2011;118(2316).
24. Herzog E, Kaspereit F, Krege W, Joanne R Van, Dickneite G, Pragst I. Non-clinical safety and efficacy of prothrombin complex concentrates (pcc) for the reversal of dabigatran mediated anticoagulation. J Thromb Haemost. 2013;11:693.
25. Godier A, Miclot A, Le Bonniec B, et al. Evaluation of prothrombin complex concentrate and recombinant activated factor VII to reverse rivaroxaban in a rabbit model. Anesthesiology. 2012;116(1):94-102. doi:10.1097/ALN.0b013e318238c036.
26. Eerenberg ES, Kamphuisen PW, Sijpkens MK, Meijers JC, Buller HR, Levi M. Reversal of rivaroxaban and dabigatran by prothrombin complex concentrate: a randomized, placebo-controlled, crossover study in healthy subjects. Circulation. 2011;124(14):1573-1579. doi:10.1161/CIRCULATIONAHA.111.029017.
27. Escolar G, Fernandez-Gallego V, Arellano-Rodrigo E, et al. Reversal of apixaban induced alterations in hemostasis by different coagulation factor concentrates: significance of studies in vitro with circulating human blood. PLoS One. 2013;8(11):e78696. doi:10.1371/journal.pone.0078696.
28. Galan AM, Arellano-Rodrigo E, Sanz V, et al. Effects of rivaroxaban and dabigatran on hemostasis and reversion of their antithrombotic effects by different coagulation factors: evidence raised from a clinical study in healthy volunteers. J Thromb Haemost. 2013;11:418-419.
29. Escolar G, Arellano-Rodrigo E, Lopez-Vilchez I, et al. Reversal of rivaroxaban-induced alterations on hemostasis by different coagulation factor concentrates—in vitro studies with steady and circulating human blood. Circ J. 2015;79(2):331-338. doi:0.1253/circj.CJ-14-0909.
30. Perzborn E, Gruber A, Tinel H, et al. Reversal of rivaroxaban anticoagulation by haemostatic agents in rats and primates. Thromb Haemost. 2013;110(1):162-172. doi:10.1160/TH12-12-0907.
31. Chan HHW, Atkinson HM, Goncharenko M, Berry LR, Chan AKC. Reversal of dabigatran using recombinant activated factor VII and activated prothrombin complex concentrates in thromboelastography assay. J Thromb Haemost. 2011;9:576-577.
32. Hoffman M, Monroe DM. Reversing targeted oral anticoagulants. Hematology Am Soc Hematol Educ Program. 2014;2014(1):518-523. doi:10.1182/asheducation-2014.1.518.
33. Marlu R, Hodaj E, Paris A, Albaladejo P, Cracowski JL, Pernod G. Effect of non-specific reversal agents on anticoagulant activity of dabigatran and rivaroxaban: a randomised crossover ex vivo study in healthy volunteers. Thromb Haemost. 2012;108(2):217-224. doi:10.1160/TH12-03-0179.
34. Grandhi R, Newman WC, Zhang X, et al. Administration of 4-factor prothrombin complex concentrate as an antidote for intracranial bleeding in patients taking direct factor xa inhibitors. World Neurosurg. 2015;84(6):1956-61. doi:10.1016/j.wneu.2015.08.042.
35. Durie R, Kohute M, Fernandez C, Knight M. Prothrombin complex concentrate for the management of severe traumatic bleeding in a patient anticoagulated with apixaban. J Clin Pharm Ther. 2016;41(1):92-93. doi:10.1111/jcpt.12339.
36. Kauffmann S, Chabanne R, Coste A, et al. Favorable outcome of rivaroxaban-associated intracerebral hemorrhage reversed by 4-factor prothrombin complex concentrate: impact on thrombin generation. A&A Case Rep. 2015;4(11):151-154. doi:10.1213/XAA.0000000000000143.
37. Maurice-Szamburski A, Graillon T, Bruder N. Favorable outcome after a subdural hematoma treated with feiba in a 77-year-old patient treated by rivaroxaban. J Neurosurg Anesthesiol. 2014;26(2):183. doi:10.1097/ANA.0000000000000030.
38. Richardson EG, Hemenway D. Homicide, suicide, and unintentional firearm fatality: comparing the United States with other high-income countries, 2003. J Trauma. 2011;70(1):238-243. doi:10.1097/TA.0b013e3181dbaddf.
39. Kochanek KD, Murphy SL, Xu J, Tejada-Vera B. Deaths: final data for 2014. Natl Vital Stat Rep. 2016;65(4):1-122. https://www.cdc.gov/nchs/data/nvsr/nvsr65/nvsr65_04.pdf. Accessed June 8, 2017.
40. Centers for Disease Control and Prevention. Leading causes of death reports, national and regional, 1999-2015. https://webappa.cdc.gov/sasweb/ncipc/leadcaus10_us.html. Accessed June 8, 2017.
41. Sauaia A, Gonzalez E, Moore HB, Bol K, Moore EE. Fatality and severity of firearm injuries in a denver trauma center, 2000-2013. JAMA. 2016;315(22):2465-2467. doi:10.1001/jama.2016.5978.
42. Omnibus Consolidated Appropriations Bill. HR 3610, Pub L No. 104-208. http://www.gpo.gov/fdsys/pkg/PLAW-104publ208/pdf/PLAW-104publ208.pdf. September 1996. Accessed June 13, 2017.
43. Kellermann AL, Rivara FP. Silencing the science on gun research. JAMA. 2013;309(6):549-550. doi:10.1001/jama.2012.208207.
44. Consolidated Appropriations Act of 2012. HR 2055: Pub L No. 112-174. https://www.gpo.gov/fdsys/pkg/PLAW-112publ74/pdf/PLAW-112publ74.pdf. December 2011. Accessed June 13, 2017.
45. Rubin R. Tale of 2 agencies: CDC avoids gun violence research but NIH funds it. JAMA. 2016;315(16):1689-1691. doi:10.1001/jama.2016.1707.
46. Cook A, Osler T, Hosmer D, et al. Gunshot wounds resulting in hospitalization in the United States: 2004-2013. Injury. 2017;48(3):621-627. doi:10.1016/j.injury.2017.01.044.

Rosacea Treatment Schema: An Update
When tasked with outlining updated therapy regimens for rosacea, specific patient vignettes come to mind.
A 53-year-old male golfer presents with years of central facial flushing, prominent telangiectases, erythema, and scattered pink papules. He attempted various over-the-counter topical products indicated for acne, such as salicylic acid scrub and benzoyl peroxide cream, with no improvement and much irritation. Recently, his wife has been helping him apply redness-concealing makeup in the morning and over-the-counter hydrocortisone cream in the evening, which has been slightly helpful.
This patient’s rosacea could conceivably be labeled under the papulopustular rosacea subtype; however, the conventional categories are fluid with subtype overlap and imprecise diagnostic criteria. He also seemed to display features of the erythematotelangiectatic subtype, perhaps with underlying photodamage as well as steroid rebound erythema and/or atrophy.1 Nevertheless, it is a common presentation, and certain baseline tenets should be applied. First, all steroid products and irritants (eg, benzoyl peroxide and salicylic acid ingredients, any scrub vehicle) should be discontinued. Education about avoidance of triggers (ie, sun, heat, spicy food, alcohol, stress is paramount. Because barrier inadequacy is a recent insight into rosacea pathogenesis, mild syndet- or lipid-free cleansers, daily sunscreen, and evening emollients dictate baseline skin care, as does meticulous situation-specific sun protection.2,3 The papular component and immediate erythema in and around the papules can be managed topically (prior to sunscreen or emollient application) with metronidazole gel or cream up to twice daily, ivermectin cream once daily, or azelaic acid gel or foam up to twice daily. Oral doxycycline 40 mg (delayed release) on an empty stomach or 50 mg (immediate release) with food to avert antimicrobial dosing and antibiotic resistance also could be considered if topical therapy is inadequate or irritating, though gastrointestinal comorbidities with rosacea also should be delineated before initiating oral antibiotics.4-6 (Management of this patient’s nonlesional fixed erythema, telangiectases, and flushing is discussed after the next vignette.)
What if a woman presented in a similar fashion as above, only without papules? Her family physician prescribed metronidazole gel twice daily for years with no improvement in flushing, redness, or telangiectases.
Background erythema in rosacea often is persistent with trigger-specific intensification, with or without episodic facial flushing; undoubtedly, these symptoms can be difficult to compartmentalize depending on the clarity of the patient’s history and frequency of clinic visits. The aforementioned baseline skin care and sun-protection regimen applies, and newer topical agents such as α-adrenergics (daily oxymetazoline cream or brimonidine gel) may be considered for persistent erythema; however, irritant potential and rebound erythema are common.7-9 Topical therapies such as metronidazole gel, as in this case, are inadequate for persistent background erythema or flushing. Persistent erythema and telangiectases can be reduced with pulsed dye laser or intense pulsed light modalities, particularly following conservative management of acute inflammation.5 Episodic flushing is poorly controlled with the above tactics, but anecdotally, topical or oral α-adrenergics or oral nonselective beta-blockers could be considered; the latter is also applicable to migraine therapy, which is perhaps comorbid with rosacea.5,10
A 35-year-old Hispanic woman states that the scalp, forehead, and cheeks have been flaky, pink, and pruritic for years. She saw several aestheticians for it and the admixed “acne” on the face, receiving salicylic acid chemical peels with no improvement and much dyspigmentation.
Although underreported, the commingling of rosacea with seborrheic dermatitis is common, perhaps with mutual Demodex mite overpopulation, assigning topical therapies to its management such as daily ivermectin cream or steroid-sparing pimecrolimus cream for inflammatory papules and scaly regions of the face and scalp.11-13 Further, this case exemplifies the increasing incidence and awareness of rosacea in darker skin types, along with its postinflammatory pigmentary perturbations, which necessitate repeated education about barrier control and sun protection.14
A 72-year-old male farmer presents with his wife whoinsists that his nose has been increasing in size for years; she procures a prior driver’s license photograph as proof. She also notes that he has been snoring at night and having more trouble breathing while working outdoors. The patient had not noticed.
Phymatous rosacea may exist as an additional feature of any rosacea subtype or as a singular finding, presenting as actively inflamed, fibrotic/noninflamed, or both. Management, particularly if inflamed, involves baseline gentle skin care and sun protection, avoidance of rosacea triggers, and implementation of oral therapy such as doxycycline or isotretinoin. Many cases, particularly those with a fibrotic component, warrant surgical methods such as fractionated CO2 laser or Shaw scalpel surgical sculpting. These cases frequently demonstrate varying degrees of airway compromise, validating surgery as a legitimate medical, not merely cosmetic, presentation.5,15
Final Thoughts
The Table, constructed as a concise therapy compendium by the ROSacea COnsensus (ROSCO) international panel of dermatologists and ophthalmologists, outlines data-driven and expert experience-based therapies for rosacea.5 This panel asserts that phenotypical features, not rigid subtypes, oblige patient-specific treatment schema. Also, as these cases outline, an evolving understanding of rosacea’s multifaceted pathogenesis, assorted presentations, and frequent pitfalls in daily skin care and initial management require individualized care.
- Tan J, Steinhoff M, Berg M, et al; Rosacea International Study Group. Shortcomings in rosacea diagnosis and classification. Br J Dermatol. 2017;176:197-199.
- Levin J, Miller R. A guide to the ingredients and potential benefits of over-the-counter cleansers and moisturizers for rosacea patients. J Clin Aesthet Dermatol. 2011;4:31-49.
- Del Rosso JQ. Adjunctive skin care in the management of rosacea: cleansers, moisturizers, and photoprotectants. Cutis. 2005;75(suppl 3):17-21;discussion 33-36.
- van Zuuren EJ, Fedorowicz Z. Interventions for rosacea: abridged updated Cochrane systematic review including GRADE assessments [published online August 30, 2015]. Br J Dermatol. 2015;173:651-662.
- Schaller M, Almeida LM, Bewley A, et al. Rosacea treatment update: recommendations from the global ROSacea COnsensus (ROSCO) panel. Br J Dermatol. 2017;176:465-471.
- Egeberg A, Weinstock LB, Thvssen EP, et al. Rosacea and gastrointestinal disorders: a population-based cohort study. Br J Dermatol. 2017;176:100-106.
- Layton AM, Schaller M, Homey B, et al. Brimonidine gel 0.33% rapidly improves patient-reported outcomes by controlling facial erythema of rosacea: a randomized, double-blind, vehicle-controlled study. J Eur Acad Dermatol Venereol. 2015;29:2405-2410.
- Docherty JR, Steinhoff M, Lorton D, et al. Multidisciplinary consideration of potential pathophysiologic mechanisms of paradoxical erythema with topical brimonidine therapy [published online August 25, 2016]. Adv Ther. 2016;33:1885-1895.
- Shanler SD, Ondo AL. Successful treatment of the erythema and flushing of rosacea using a topically applied selective alpha1-adrenergic receptor agonist, oxymetazoline. Arch Dermatol. 2007;143:1369-1371.
- Egeberg A, Ashina M, Gaist D, et al. Prevalence and risk of migraine in patients with rosacea: a population-based cohort study. J Am Acad Dermatol. 2017;76:454-458.
- Zhao YE, Peng Y, Wang XL, et al. Facial dermatosis associated with Demodex: a case-control study. J Zhejiang Univ Sci B. 2011;12:1008-1015.
- Siddiqui K, Stein Gold L, Gill J. The efficacy, safety, and tolerability of ivermectin compared with current topical treatments for the inflammatory lesions of rosacea: a network meta-analysis. Springerplus. 2016;5:1151. doi: 10.1186/s40064-016-2819-8.
- Kim MB, Kim GW, Park HJ, et al. Pimecrolimus 1% cream for the treatment of rosacea. J Dermatol. 2011;38:1135-1139.
- Al-Dabagh A, Davis SA, McMichael AJ, et al. Rosacea in skin of color: not a rare diagnosis. Dermatol Online J. 2014;20. pii:13030/qt1mv9r0ss.
- Little SC, Stucker FJ, Compton A, et al. Nuances in the management of rhinophyma. Facial Plast Surg. 2012;28:231-237.
When tasked with outlining updated therapy regimens for rosacea, specific patient vignettes come to mind.
A 53-year-old male golfer presents with years of central facial flushing, prominent telangiectases, erythema, and scattered pink papules. He attempted various over-the-counter topical products indicated for acne, such as salicylic acid scrub and benzoyl peroxide cream, with no improvement and much irritation. Recently, his wife has been helping him apply redness-concealing makeup in the morning and over-the-counter hydrocortisone cream in the evening, which has been slightly helpful.
This patient’s rosacea could conceivably be labeled under the papulopustular rosacea subtype; however, the conventional categories are fluid with subtype overlap and imprecise diagnostic criteria. He also seemed to display features of the erythematotelangiectatic subtype, perhaps with underlying photodamage as well as steroid rebound erythema and/or atrophy.1 Nevertheless, it is a common presentation, and certain baseline tenets should be applied. First, all steroid products and irritants (eg, benzoyl peroxide and salicylic acid ingredients, any scrub vehicle) should be discontinued. Education about avoidance of triggers (ie, sun, heat, spicy food, alcohol, stress is paramount. Because barrier inadequacy is a recent insight into rosacea pathogenesis, mild syndet- or lipid-free cleansers, daily sunscreen, and evening emollients dictate baseline skin care, as does meticulous situation-specific sun protection.2,3 The papular component and immediate erythema in and around the papules can be managed topically (prior to sunscreen or emollient application) with metronidazole gel or cream up to twice daily, ivermectin cream once daily, or azelaic acid gel or foam up to twice daily. Oral doxycycline 40 mg (delayed release) on an empty stomach or 50 mg (immediate release) with food to avert antimicrobial dosing and antibiotic resistance also could be considered if topical therapy is inadequate or irritating, though gastrointestinal comorbidities with rosacea also should be delineated before initiating oral antibiotics.4-6 (Management of this patient’s nonlesional fixed erythema, telangiectases, and flushing is discussed after the next vignette.)
What if a woman presented in a similar fashion as above, only without papules? Her family physician prescribed metronidazole gel twice daily for years with no improvement in flushing, redness, or telangiectases.
Background erythema in rosacea often is persistent with trigger-specific intensification, with or without episodic facial flushing; undoubtedly, these symptoms can be difficult to compartmentalize depending on the clarity of the patient’s history and frequency of clinic visits. The aforementioned baseline skin care and sun-protection regimen applies, and newer topical agents such as α-adrenergics (daily oxymetazoline cream or brimonidine gel) may be considered for persistent erythema; however, irritant potential and rebound erythema are common.7-9 Topical therapies such as metronidazole gel, as in this case, are inadequate for persistent background erythema or flushing. Persistent erythema and telangiectases can be reduced with pulsed dye laser or intense pulsed light modalities, particularly following conservative management of acute inflammation.5 Episodic flushing is poorly controlled with the above tactics, but anecdotally, topical or oral α-adrenergics or oral nonselective beta-blockers could be considered; the latter is also applicable to migraine therapy, which is perhaps comorbid with rosacea.5,10
A 35-year-old Hispanic woman states that the scalp, forehead, and cheeks have been flaky, pink, and pruritic for years. She saw several aestheticians for it and the admixed “acne” on the face, receiving salicylic acid chemical peels with no improvement and much dyspigmentation.
Although underreported, the commingling of rosacea with seborrheic dermatitis is common, perhaps with mutual Demodex mite overpopulation, assigning topical therapies to its management such as daily ivermectin cream or steroid-sparing pimecrolimus cream for inflammatory papules and scaly regions of the face and scalp.11-13 Further, this case exemplifies the increasing incidence and awareness of rosacea in darker skin types, along with its postinflammatory pigmentary perturbations, which necessitate repeated education about barrier control and sun protection.14
A 72-year-old male farmer presents with his wife whoinsists that his nose has been increasing in size for years; she procures a prior driver’s license photograph as proof. She also notes that he has been snoring at night and having more trouble breathing while working outdoors. The patient had not noticed.
Phymatous rosacea may exist as an additional feature of any rosacea subtype or as a singular finding, presenting as actively inflamed, fibrotic/noninflamed, or both. Management, particularly if inflamed, involves baseline gentle skin care and sun protection, avoidance of rosacea triggers, and implementation of oral therapy such as doxycycline or isotretinoin. Many cases, particularly those with a fibrotic component, warrant surgical methods such as fractionated CO2 laser or Shaw scalpel surgical sculpting. These cases frequently demonstrate varying degrees of airway compromise, validating surgery as a legitimate medical, not merely cosmetic, presentation.5,15
Final Thoughts
The Table, constructed as a concise therapy compendium by the ROSacea COnsensus (ROSCO) international panel of dermatologists and ophthalmologists, outlines data-driven and expert experience-based therapies for rosacea.5 This panel asserts that phenotypical features, not rigid subtypes, oblige patient-specific treatment schema. Also, as these cases outline, an evolving understanding of rosacea’s multifaceted pathogenesis, assorted presentations, and frequent pitfalls in daily skin care and initial management require individualized care.
When tasked with outlining updated therapy regimens for rosacea, specific patient vignettes come to mind.
A 53-year-old male golfer presents with years of central facial flushing, prominent telangiectases, erythema, and scattered pink papules. He attempted various over-the-counter topical products indicated for acne, such as salicylic acid scrub and benzoyl peroxide cream, with no improvement and much irritation. Recently, his wife has been helping him apply redness-concealing makeup in the morning and over-the-counter hydrocortisone cream in the evening, which has been slightly helpful.
This patient’s rosacea could conceivably be labeled under the papulopustular rosacea subtype; however, the conventional categories are fluid with subtype overlap and imprecise diagnostic criteria. He also seemed to display features of the erythematotelangiectatic subtype, perhaps with underlying photodamage as well as steroid rebound erythema and/or atrophy.1 Nevertheless, it is a common presentation, and certain baseline tenets should be applied. First, all steroid products and irritants (eg, benzoyl peroxide and salicylic acid ingredients, any scrub vehicle) should be discontinued. Education about avoidance of triggers (ie, sun, heat, spicy food, alcohol, stress is paramount. Because barrier inadequacy is a recent insight into rosacea pathogenesis, mild syndet- or lipid-free cleansers, daily sunscreen, and evening emollients dictate baseline skin care, as does meticulous situation-specific sun protection.2,3 The papular component and immediate erythema in and around the papules can be managed topically (prior to sunscreen or emollient application) with metronidazole gel or cream up to twice daily, ivermectin cream once daily, or azelaic acid gel or foam up to twice daily. Oral doxycycline 40 mg (delayed release) on an empty stomach or 50 mg (immediate release) with food to avert antimicrobial dosing and antibiotic resistance also could be considered if topical therapy is inadequate or irritating, though gastrointestinal comorbidities with rosacea also should be delineated before initiating oral antibiotics.4-6 (Management of this patient’s nonlesional fixed erythema, telangiectases, and flushing is discussed after the next vignette.)
What if a woman presented in a similar fashion as above, only without papules? Her family physician prescribed metronidazole gel twice daily for years with no improvement in flushing, redness, or telangiectases.
Background erythema in rosacea often is persistent with trigger-specific intensification, with or without episodic facial flushing; undoubtedly, these symptoms can be difficult to compartmentalize depending on the clarity of the patient’s history and frequency of clinic visits. The aforementioned baseline skin care and sun-protection regimen applies, and newer topical agents such as α-adrenergics (daily oxymetazoline cream or brimonidine gel) may be considered for persistent erythema; however, irritant potential and rebound erythema are common.7-9 Topical therapies such as metronidazole gel, as in this case, are inadequate for persistent background erythema or flushing. Persistent erythema and telangiectases can be reduced with pulsed dye laser or intense pulsed light modalities, particularly following conservative management of acute inflammation.5 Episodic flushing is poorly controlled with the above tactics, but anecdotally, topical or oral α-adrenergics or oral nonselective beta-blockers could be considered; the latter is also applicable to migraine therapy, which is perhaps comorbid with rosacea.5,10
A 35-year-old Hispanic woman states that the scalp, forehead, and cheeks have been flaky, pink, and pruritic for years. She saw several aestheticians for it and the admixed “acne” on the face, receiving salicylic acid chemical peels with no improvement and much dyspigmentation.
Although underreported, the commingling of rosacea with seborrheic dermatitis is common, perhaps with mutual Demodex mite overpopulation, assigning topical therapies to its management such as daily ivermectin cream or steroid-sparing pimecrolimus cream for inflammatory papules and scaly regions of the face and scalp.11-13 Further, this case exemplifies the increasing incidence and awareness of rosacea in darker skin types, along with its postinflammatory pigmentary perturbations, which necessitate repeated education about barrier control and sun protection.14
A 72-year-old male farmer presents with his wife whoinsists that his nose has been increasing in size for years; she procures a prior driver’s license photograph as proof. She also notes that he has been snoring at night and having more trouble breathing while working outdoors. The patient had not noticed.
Phymatous rosacea may exist as an additional feature of any rosacea subtype or as a singular finding, presenting as actively inflamed, fibrotic/noninflamed, or both. Management, particularly if inflamed, involves baseline gentle skin care and sun protection, avoidance of rosacea triggers, and implementation of oral therapy such as doxycycline or isotretinoin. Many cases, particularly those with a fibrotic component, warrant surgical methods such as fractionated CO2 laser or Shaw scalpel surgical sculpting. These cases frequently demonstrate varying degrees of airway compromise, validating surgery as a legitimate medical, not merely cosmetic, presentation.5,15
Final Thoughts
The Table, constructed as a concise therapy compendium by the ROSacea COnsensus (ROSCO) international panel of dermatologists and ophthalmologists, outlines data-driven and expert experience-based therapies for rosacea.5 This panel asserts that phenotypical features, not rigid subtypes, oblige patient-specific treatment schema. Also, as these cases outline, an evolving understanding of rosacea’s multifaceted pathogenesis, assorted presentations, and frequent pitfalls in daily skin care and initial management require individualized care.
- Tan J, Steinhoff M, Berg M, et al; Rosacea International Study Group. Shortcomings in rosacea diagnosis and classification. Br J Dermatol. 2017;176:197-199.
- Levin J, Miller R. A guide to the ingredients and potential benefits of over-the-counter cleansers and moisturizers for rosacea patients. J Clin Aesthet Dermatol. 2011;4:31-49.
- Del Rosso JQ. Adjunctive skin care in the management of rosacea: cleansers, moisturizers, and photoprotectants. Cutis. 2005;75(suppl 3):17-21;discussion 33-36.
- van Zuuren EJ, Fedorowicz Z. Interventions for rosacea: abridged updated Cochrane systematic review including GRADE assessments [published online August 30, 2015]. Br J Dermatol. 2015;173:651-662.
- Schaller M, Almeida LM, Bewley A, et al. Rosacea treatment update: recommendations from the global ROSacea COnsensus (ROSCO) panel. Br J Dermatol. 2017;176:465-471.
- Egeberg A, Weinstock LB, Thvssen EP, et al. Rosacea and gastrointestinal disorders: a population-based cohort study. Br J Dermatol. 2017;176:100-106.
- Layton AM, Schaller M, Homey B, et al. Brimonidine gel 0.33% rapidly improves patient-reported outcomes by controlling facial erythema of rosacea: a randomized, double-blind, vehicle-controlled study. J Eur Acad Dermatol Venereol. 2015;29:2405-2410.
- Docherty JR, Steinhoff M, Lorton D, et al. Multidisciplinary consideration of potential pathophysiologic mechanisms of paradoxical erythema with topical brimonidine therapy [published online August 25, 2016]. Adv Ther. 2016;33:1885-1895.
- Shanler SD, Ondo AL. Successful treatment of the erythema and flushing of rosacea using a topically applied selective alpha1-adrenergic receptor agonist, oxymetazoline. Arch Dermatol. 2007;143:1369-1371.
- Egeberg A, Ashina M, Gaist D, et al. Prevalence and risk of migraine in patients with rosacea: a population-based cohort study. J Am Acad Dermatol. 2017;76:454-458.
- Zhao YE, Peng Y, Wang XL, et al. Facial dermatosis associated with Demodex: a case-control study. J Zhejiang Univ Sci B. 2011;12:1008-1015.
- Siddiqui K, Stein Gold L, Gill J. The efficacy, safety, and tolerability of ivermectin compared with current topical treatments for the inflammatory lesions of rosacea: a network meta-analysis. Springerplus. 2016;5:1151. doi: 10.1186/s40064-016-2819-8.
- Kim MB, Kim GW, Park HJ, et al. Pimecrolimus 1% cream for the treatment of rosacea. J Dermatol. 2011;38:1135-1139.
- Al-Dabagh A, Davis SA, McMichael AJ, et al. Rosacea in skin of color: not a rare diagnosis. Dermatol Online J. 2014;20. pii:13030/qt1mv9r0ss.
- Little SC, Stucker FJ, Compton A, et al. Nuances in the management of rhinophyma. Facial Plast Surg. 2012;28:231-237.
- Tan J, Steinhoff M, Berg M, et al; Rosacea International Study Group. Shortcomings in rosacea diagnosis and classification. Br J Dermatol. 2017;176:197-199.
- Levin J, Miller R. A guide to the ingredients and potential benefits of over-the-counter cleansers and moisturizers for rosacea patients. J Clin Aesthet Dermatol. 2011;4:31-49.
- Del Rosso JQ. Adjunctive skin care in the management of rosacea: cleansers, moisturizers, and photoprotectants. Cutis. 2005;75(suppl 3):17-21;discussion 33-36.
- van Zuuren EJ, Fedorowicz Z. Interventions for rosacea: abridged updated Cochrane systematic review including GRADE assessments [published online August 30, 2015]. Br J Dermatol. 2015;173:651-662.
- Schaller M, Almeida LM, Bewley A, et al. Rosacea treatment update: recommendations from the global ROSacea COnsensus (ROSCO) panel. Br J Dermatol. 2017;176:465-471.
- Egeberg A, Weinstock LB, Thvssen EP, et al. Rosacea and gastrointestinal disorders: a population-based cohort study. Br J Dermatol. 2017;176:100-106.
- Layton AM, Schaller M, Homey B, et al. Brimonidine gel 0.33% rapidly improves patient-reported outcomes by controlling facial erythema of rosacea: a randomized, double-blind, vehicle-controlled study. J Eur Acad Dermatol Venereol. 2015;29:2405-2410.
- Docherty JR, Steinhoff M, Lorton D, et al. Multidisciplinary consideration of potential pathophysiologic mechanisms of paradoxical erythema with topical brimonidine therapy [published online August 25, 2016]. Adv Ther. 2016;33:1885-1895.
- Shanler SD, Ondo AL. Successful treatment of the erythema and flushing of rosacea using a topically applied selective alpha1-adrenergic receptor agonist, oxymetazoline. Arch Dermatol. 2007;143:1369-1371.
- Egeberg A, Ashina M, Gaist D, et al. Prevalence and risk of migraine in patients with rosacea: a population-based cohort study. J Am Acad Dermatol. 2017;76:454-458.
- Zhao YE, Peng Y, Wang XL, et al. Facial dermatosis associated with Demodex: a case-control study. J Zhejiang Univ Sci B. 2011;12:1008-1015.
- Siddiqui K, Stein Gold L, Gill J. The efficacy, safety, and tolerability of ivermectin compared with current topical treatments for the inflammatory lesions of rosacea: a network meta-analysis. Springerplus. 2016;5:1151. doi: 10.1186/s40064-016-2819-8.
- Kim MB, Kim GW, Park HJ, et al. Pimecrolimus 1% cream for the treatment of rosacea. J Dermatol. 2011;38:1135-1139.
- Al-Dabagh A, Davis SA, McMichael AJ, et al. Rosacea in skin of color: not a rare diagnosis. Dermatol Online J. 2014;20. pii:13030/qt1mv9r0ss.
- Little SC, Stucker FJ, Compton A, et al. Nuances in the management of rhinophyma. Facial Plast Surg. 2012;28:231-237.

First EDition: ED Visits Increased in States That Expanded Medicaid, more
BY JEFF BAUER
There was a substantial increase in the number of ED visits in states that expanded Medicaid coverage in 2014, after the Affordable Care Act was implemented, and a decrease in the number of ED visits by uninsured patients, according to a study published in Annals of Emergency Medicine.
Researchers analyzed quarterly data on ED visits from the Agency for Healthcare Research and Quality’s Fast Stats database, which is an early-release, aggregated version of the State Emergency Department Databases and State Inpatient Databases. They compared changes in ED visits per capita and changes in share of ED visits by payer (Medicaid, uninsured, and private insurance) in states that did and did not expand Medicaid coverage in 2014.
The analysis included 25 states: 14 Medicaid expansion states (Arizona, California, Hawaii, Iowa, Illinois, Kentucky, Maryland, Minnesota, North Dakota, New Jersey, Nevada, New York, Rhode Island, and Vermont) and 11 nonexpansion states (Florida, Georgia, Indiana, Kansas, Missouri, North Carolina, Nebraska, South Carolina, South Dakota, Tennessee, and Wisconsin). Researchers defined visits that occurred during all 4 quarters of 2012 and the first 3 quarters of 2013 as the pre-expansion period, and visits from the first through fourth quarters of 2014 as the postexpansion period. Visits that occurred during the fourth quarter of 2013 were not included in the analysis because Medicaid coverage began to increase in the final quarter of 2013 for most states.
Overall, researchers found that after 2014, ED use per 1,000 people per quarter increased by 2.5 visits more in expansion states compared to nonexpansion states. Researchers estimated that 1.13 million ED visits in 2014 could be attributed to Medicaid expansion in these states. In expansion states, the share of ED visits by Medicaid patients increased by 8.8 percentage points and the share of visits by insured patients decreased by 5.3 percentage points, compared to nonexpansion states. The share of visits by insured patients did not change for expansion states but increased slightly for nonexpansion states.
An American College of Emergency Physicians press release about this study included editorial comments by Ari Friedman, MD, of Beth Israel Deaconess Medical Center in Boston, who said, “More emergency department visits by Medicaid beneficiaries is neither clearly bad nor clearly good. Insurance increases access to care, including emergency department care. We need to move beyond the value judgments that have dominated so much study of emergency department utilization towards a more rational basis for how we structure unscheduled visits in the health system. If we want to meet patients’ care needs as patients themselves define them, the emergency department has a key role to play in a flexible system.”
Nikpay S, Freedman S, Levy H, Buchmueller T. Effect of the Affordable Care Act Medicaid expansion on emergency department visits: evidence from state-level emergency department databases. Ann Emerg Med. 2017 June 26. [Epub ahead of print]. doi:http://dx.doi.org/10.1016/j.annemergmed.2017.03.023.
Child Firearm Suicide at Highest Rate in More Than a Decade
MOLLIE KALAYCIO
FRONTLINE MEDICAL NEWS
Boys, older children, and minorities are disproportionately affected when it comes to firearm injuries and deaths in US children and adolescents, and child firearm suicide rates are at the highest they have been in more than a decade, new study results revealed.
Approximately 19 children are either medically treated for a gunshot wound or killed by one every day in the United States. “The majority of these children are boys 13-17 years old, African American in the case of firearm homicide, and white and American Indian in the case of firearm suicide. Pediatric firearm injuries and deaths are an important public health problem in the United States, contributing substantially each year to premature death, illness, and disability of children,” said Katherine A. Fowler, PhD, of the National Center for Injury Prevention and Control, Atlanta, and her associates. “Finding ways to prevent such injuries and ensure that all children have safe, stable, nurturing relationships and environments remains one of our most important priorities.”
National data on fatal firearm injuries in 2011-2014 for this study were derived from death certificate data from the Centers for Disease Control and Prevention’s (CDC’s) National Vital Statistics System, obtained via the CDC’s Web-based Injury Statistics Query and Reporting System. Data on nonfatal firearm injuries for 2011-2014 were obtained from the National Electronic Injury Surveillance System.
“From 2012 to 2014, the average annual case fatality rate was 74% for firearm-related self-harm, 14% for firearm-related assaults, and 6% for unintentional firearm injuries,” the investigators reported.
Boys accounted for 82% of all child firearm deaths from 2012 to 2014. In this time period, the annual rate of firearm death for boys was 4.5 times higher than the annual rate for girls (2.8 vs. 0.6 per 100,000). This difference was even more pronounced by age, with the rate for 13- to 17-year-old boys being six times higher than the rate for same-aged girls. Similarly, boys suffer the majority of nonfatal firearm injuries treated in US EDs, accounting for 84% of all nonfatal firearm injuries medically treated each year from 2012 to 2014. The average annual rate of nonfatal firearm injuries for boys was five times the rate for girls at 13 vs. 3 per 100,000.
The annual rate of firearm homicide was 10 times higher among 13- to 17-year-olds vs. 0- to 12-year-olds (3 vs. 0.3 per 100,000). Unintentional firearm death rates were approximately twice as high when comparing these two groups (0.2 vs. 0.1 per 100,000).
Dr Fowler and her associates wrote, “Our findings indicate that most children who died of unintentional firearm injuries were shot by another child in their own age range and most often in the context of playing with a gun or showing it to others. More than one-third of the deaths of older children occurred in incidents in which the shooter thought that the gun was unloaded or thought that the safety was engaged.”
“Child firearm suicide rates showed a significant upward trend between 2007 and 2014, increasing 60% from 1.0 to 1.6 (P < .05) to the highest rate seen over the period examined,” Dr Fowler and her associates said.
Firearm suicide rates were 11 times higher among 13- to 17-year-olds vs. 10- to 12-year-olds (2 vs. 0.2 per 100,000). Older children also accounted for 88% of all nonfatal firearm injuries treated in an ED. The overall average annual rate of nonfatal firearm injuries for older children was 19 times that of younger children (24 vs. 1 per 100,000).
The annual firearm homicide rate for African American children was nearly 10 times higher than the rate for white children (4 vs. 0.4 per 100,000). However, the annual rate of firearm suicide among white children was nearly four times higher than the rate for African American children (2. vs. 0.6 per 100,000).
Awareness of the availability of firearms during times of crisis is crucial because suicides are often impulsive in young people, Dr Fowler and her associates said, “with previous findings indicating that many who attempt suicide spend 10 minutes or less deliberating. Safe storage practices (ie, unloading and locking all firearms and ammunition) can potentially be lifesaving in these instances,” as the results of previous studies in this age group attest.
Firearm deaths are the third leading cause of
Fowler KA, Dahlberg LL, Haileyesus T, Gutierrez C, Bacon S. Childhood firearm injuries in the united states. Pediatrics. 2017;140(1):e20163486.
BY JEFF BAUER
There was a substantial increase in the number of ED visits in states that expanded Medicaid coverage in 2014, after the Affordable Care Act was implemented, and a decrease in the number of ED visits by uninsured patients, according to a study published in Annals of Emergency Medicine.
Researchers analyzed quarterly data on ED visits from the Agency for Healthcare Research and Quality’s Fast Stats database, which is an early-release, aggregated version of the State Emergency Department Databases and State Inpatient Databases. They compared changes in ED visits per capita and changes in share of ED visits by payer (Medicaid, uninsured, and private insurance) in states that did and did not expand Medicaid coverage in 2014.
The analysis included 25 states: 14 Medicaid expansion states (Arizona, California, Hawaii, Iowa, Illinois, Kentucky, Maryland, Minnesota, North Dakota, New Jersey, Nevada, New York, Rhode Island, and Vermont) and 11 nonexpansion states (Florida, Georgia, Indiana, Kansas, Missouri, North Carolina, Nebraska, South Carolina, South Dakota, Tennessee, and Wisconsin). Researchers defined visits that occurred during all 4 quarters of 2012 and the first 3 quarters of 2013 as the pre-expansion period, and visits from the first through fourth quarters of 2014 as the postexpansion period. Visits that occurred during the fourth quarter of 2013 were not included in the analysis because Medicaid coverage began to increase in the final quarter of 2013 for most states.
Overall, researchers found that after 2014, ED use per 1,000 people per quarter increased by 2.5 visits more in expansion states compared to nonexpansion states. Researchers estimated that 1.13 million ED visits in 2014 could be attributed to Medicaid expansion in these states. In expansion states, the share of ED visits by Medicaid patients increased by 8.8 percentage points and the share of visits by insured patients decreased by 5.3 percentage points, compared to nonexpansion states. The share of visits by insured patients did not change for expansion states but increased slightly for nonexpansion states.
An American College of Emergency Physicians press release about this study included editorial comments by Ari Friedman, MD, of Beth Israel Deaconess Medical Center in Boston, who said, “More emergency department visits by Medicaid beneficiaries is neither clearly bad nor clearly good. Insurance increases access to care, including emergency department care. We need to move beyond the value judgments that have dominated so much study of emergency department utilization towards a more rational basis for how we structure unscheduled visits in the health system. If we want to meet patients’ care needs as patients themselves define them, the emergency department has a key role to play in a flexible system.”
Nikpay S, Freedman S, Levy H, Buchmueller T. Effect of the Affordable Care Act Medicaid expansion on emergency department visits: evidence from state-level emergency department databases. Ann Emerg Med. 2017 June 26. [Epub ahead of print]. doi:http://dx.doi.org/10.1016/j.annemergmed.2017.03.023.
Child Firearm Suicide at Highest Rate in More Than a Decade
MOLLIE KALAYCIO
FRONTLINE MEDICAL NEWS
Boys, older children, and minorities are disproportionately affected when it comes to firearm injuries and deaths in US children and adolescents, and child firearm suicide rates are at the highest they have been in more than a decade, new study results revealed.
Approximately 19 children are either medically treated for a gunshot wound or killed by one every day in the United States. “The majority of these children are boys 13-17 years old, African American in the case of firearm homicide, and white and American Indian in the case of firearm suicide. Pediatric firearm injuries and deaths are an important public health problem in the United States, contributing substantially each year to premature death, illness, and disability of children,” said Katherine A. Fowler, PhD, of the National Center for Injury Prevention and Control, Atlanta, and her associates. “Finding ways to prevent such injuries and ensure that all children have safe, stable, nurturing relationships and environments remains one of our most important priorities.”
National data on fatal firearm injuries in 2011-2014 for this study were derived from death certificate data from the Centers for Disease Control and Prevention’s (CDC’s) National Vital Statistics System, obtained via the CDC’s Web-based Injury Statistics Query and Reporting System. Data on nonfatal firearm injuries for 2011-2014 were obtained from the National Electronic Injury Surveillance System.
“From 2012 to 2014, the average annual case fatality rate was 74% for firearm-related self-harm, 14% for firearm-related assaults, and 6% for unintentional firearm injuries,” the investigators reported.
Boys accounted for 82% of all child firearm deaths from 2012 to 2014. In this time period, the annual rate of firearm death for boys was 4.5 times higher than the annual rate for girls (2.8 vs. 0.6 per 100,000). This difference was even more pronounced by age, with the rate for 13- to 17-year-old boys being six times higher than the rate for same-aged girls. Similarly, boys suffer the majority of nonfatal firearm injuries treated in US EDs, accounting for 84% of all nonfatal firearm injuries medically treated each year from 2012 to 2014. The average annual rate of nonfatal firearm injuries for boys was five times the rate for girls at 13 vs. 3 per 100,000.
The annual rate of firearm homicide was 10 times higher among 13- to 17-year-olds vs. 0- to 12-year-olds (3 vs. 0.3 per 100,000). Unintentional firearm death rates were approximately twice as high when comparing these two groups (0.2 vs. 0.1 per 100,000).
Dr Fowler and her associates wrote, “Our findings indicate that most children who died of unintentional firearm injuries were shot by another child in their own age range and most often in the context of playing with a gun or showing it to others. More than one-third of the deaths of older children occurred in incidents in which the shooter thought that the gun was unloaded or thought that the safety was engaged.”
“Child firearm suicide rates showed a significant upward trend between 2007 and 2014, increasing 60% from 1.0 to 1.6 (P < .05) to the highest rate seen over the period examined,” Dr Fowler and her associates said.
Firearm suicide rates were 11 times higher among 13- to 17-year-olds vs. 10- to 12-year-olds (2 vs. 0.2 per 100,000). Older children also accounted for 88% of all nonfatal firearm injuries treated in an ED. The overall average annual rate of nonfatal firearm injuries for older children was 19 times that of younger children (24 vs. 1 per 100,000).
The annual firearm homicide rate for African American children was nearly 10 times higher than the rate for white children (4 vs. 0.4 per 100,000). However, the annual rate of firearm suicide among white children was nearly four times higher than the rate for African American children (2. vs. 0.6 per 100,000).
Awareness of the availability of firearms during times of crisis is crucial because suicides are often impulsive in young people, Dr Fowler and her associates said, “with previous findings indicating that many who attempt suicide spend 10 minutes or less deliberating. Safe storage practices (ie, unloading and locking all firearms and ammunition) can potentially be lifesaving in these instances,” as the results of previous studies in this age group attest.
Firearm deaths are the third leading cause of
Fowler KA, Dahlberg LL, Haileyesus T, Gutierrez C, Bacon S. Childhood firearm injuries in the united states. Pediatrics. 2017;140(1):e20163486.
BY JEFF BAUER
There was a substantial increase in the number of ED visits in states that expanded Medicaid coverage in 2014, after the Affordable Care Act was implemented, and a decrease in the number of ED visits by uninsured patients, according to a study published in Annals of Emergency Medicine.
Researchers analyzed quarterly data on ED visits from the Agency for Healthcare Research and Quality’s Fast Stats database, which is an early-release, aggregated version of the State Emergency Department Databases and State Inpatient Databases. They compared changes in ED visits per capita and changes in share of ED visits by payer (Medicaid, uninsured, and private insurance) in states that did and did not expand Medicaid coverage in 2014.
The analysis included 25 states: 14 Medicaid expansion states (Arizona, California, Hawaii, Iowa, Illinois, Kentucky, Maryland, Minnesota, North Dakota, New Jersey, Nevada, New York, Rhode Island, and Vermont) and 11 nonexpansion states (Florida, Georgia, Indiana, Kansas, Missouri, North Carolina, Nebraska, South Carolina, South Dakota, Tennessee, and Wisconsin). Researchers defined visits that occurred during all 4 quarters of 2012 and the first 3 quarters of 2013 as the pre-expansion period, and visits from the first through fourth quarters of 2014 as the postexpansion period. Visits that occurred during the fourth quarter of 2013 were not included in the analysis because Medicaid coverage began to increase in the final quarter of 2013 for most states.
Overall, researchers found that after 2014, ED use per 1,000 people per quarter increased by 2.5 visits more in expansion states compared to nonexpansion states. Researchers estimated that 1.13 million ED visits in 2014 could be attributed to Medicaid expansion in these states. In expansion states, the share of ED visits by Medicaid patients increased by 8.8 percentage points and the share of visits by insured patients decreased by 5.3 percentage points, compared to nonexpansion states. The share of visits by insured patients did not change for expansion states but increased slightly for nonexpansion states.
An American College of Emergency Physicians press release about this study included editorial comments by Ari Friedman, MD, of Beth Israel Deaconess Medical Center in Boston, who said, “More emergency department visits by Medicaid beneficiaries is neither clearly bad nor clearly good. Insurance increases access to care, including emergency department care. We need to move beyond the value judgments that have dominated so much study of emergency department utilization towards a more rational basis for how we structure unscheduled visits in the health system. If we want to meet patients’ care needs as patients themselves define them, the emergency department has a key role to play in a flexible system.”
Nikpay S, Freedman S, Levy H, Buchmueller T. Effect of the Affordable Care Act Medicaid expansion on emergency department visits: evidence from state-level emergency department databases. Ann Emerg Med. 2017 June 26. [Epub ahead of print]. doi:http://dx.doi.org/10.1016/j.annemergmed.2017.03.023.
Child Firearm Suicide at Highest Rate in More Than a Decade
MOLLIE KALAYCIO
FRONTLINE MEDICAL NEWS
Boys, older children, and minorities are disproportionately affected when it comes to firearm injuries and deaths in US children and adolescents, and child firearm suicide rates are at the highest they have been in more than a decade, new study results revealed.
Approximately 19 children are either medically treated for a gunshot wound or killed by one every day in the United States. “The majority of these children are boys 13-17 years old, African American in the case of firearm homicide, and white and American Indian in the case of firearm suicide. Pediatric firearm injuries and deaths are an important public health problem in the United States, contributing substantially each year to premature death, illness, and disability of children,” said Katherine A. Fowler, PhD, of the National Center for Injury Prevention and Control, Atlanta, and her associates. “Finding ways to prevent such injuries and ensure that all children have safe, stable, nurturing relationships and environments remains one of our most important priorities.”
National data on fatal firearm injuries in 2011-2014 for this study were derived from death certificate data from the Centers for Disease Control and Prevention’s (CDC’s) National Vital Statistics System, obtained via the CDC’s Web-based Injury Statistics Query and Reporting System. Data on nonfatal firearm injuries for 2011-2014 were obtained from the National Electronic Injury Surveillance System.
“From 2012 to 2014, the average annual case fatality rate was 74% for firearm-related self-harm, 14% for firearm-related assaults, and 6% for unintentional firearm injuries,” the investigators reported.
Boys accounted for 82% of all child firearm deaths from 2012 to 2014. In this time period, the annual rate of firearm death for boys was 4.5 times higher than the annual rate for girls (2.8 vs. 0.6 per 100,000). This difference was even more pronounced by age, with the rate for 13- to 17-year-old boys being six times higher than the rate for same-aged girls. Similarly, boys suffer the majority of nonfatal firearm injuries treated in US EDs, accounting for 84% of all nonfatal firearm injuries medically treated each year from 2012 to 2014. The average annual rate of nonfatal firearm injuries for boys was five times the rate for girls at 13 vs. 3 per 100,000.
The annual rate of firearm homicide was 10 times higher among 13- to 17-year-olds vs. 0- to 12-year-olds (3 vs. 0.3 per 100,000). Unintentional firearm death rates were approximately twice as high when comparing these two groups (0.2 vs. 0.1 per 100,000).
Dr Fowler and her associates wrote, “Our findings indicate that most children who died of unintentional firearm injuries were shot by another child in their own age range and most often in the context of playing with a gun or showing it to others. More than one-third of the deaths of older children occurred in incidents in which the shooter thought that the gun was unloaded or thought that the safety was engaged.”
“Child firearm suicide rates showed a significant upward trend between 2007 and 2014, increasing 60% from 1.0 to 1.6 (P < .05) to the highest rate seen over the period examined,” Dr Fowler and her associates said.
Firearm suicide rates were 11 times higher among 13- to 17-year-olds vs. 10- to 12-year-olds (2 vs. 0.2 per 100,000). Older children also accounted for 88% of all nonfatal firearm injuries treated in an ED. The overall average annual rate of nonfatal firearm injuries for older children was 19 times that of younger children (24 vs. 1 per 100,000).
The annual firearm homicide rate for African American children was nearly 10 times higher than the rate for white children (4 vs. 0.4 per 100,000). However, the annual rate of firearm suicide among white children was nearly four times higher than the rate for African American children (2. vs. 0.6 per 100,000).
Awareness of the availability of firearms during times of crisis is crucial because suicides are often impulsive in young people, Dr Fowler and her associates said, “with previous findings indicating that many who attempt suicide spend 10 minutes or less deliberating. Safe storage practices (ie, unloading and locking all firearms and ammunition) can potentially be lifesaving in these instances,” as the results of previous studies in this age group attest.
Firearm deaths are the third leading cause of
Fowler KA, Dahlberg LL, Haileyesus T, Gutierrez C, Bacon S. Childhood firearm injuries in the united states. Pediatrics. 2017;140(1):e20163486.
Topical Cannabinoids in Dermatology
The prevalence of topical cannabinoids has risen sharply in recent years. Commercial advertisers promote their usage as a safe means to treat a multitude of skin disorders, including atopic dermatitis (AD), psoriasis, and acne. Topical compounds have garnered interest in laboratory studies, but the purchase of commercial formulations is limited to over-the-counter products from unregulated suppliers. In this article, we review the scientific evidence behind topical cannabinoids and evaluate their role in clinical dermatology.
Background
Cannabis is designated as a Schedule I drug, according to the Controlled Substances Act of 1970. This listing is given to substances with no therapeutic value and a high potential for abuse. However, as of 2017, 29 states and the District of Columbia have laws legalizing cannabis in some capacity. These regulations typically apply to medicinal use, though several states have now legalized recreational use.
Cannabinoids represent a broad class of chemical compounds derived from the cannabis plant. Originally, this class only comprised phytocannabinoids, cannabinoids produced by the cannabis plant. Tetrahydrocannabinol (THC) is the most well-known phytocannabinoid and leads to the psychoactive effects typically associated with cannabis use. Later investigation led to the discovery of endocannabinoids, cannabinoids that are naturally produced by human and animal bodies, as well as synthetic cannabinoids.1 Cannabidiol is a phytocannabinoid that has been investigated in neurologic and anti-inflammatory conditions.2-4
Cannabinoids act as agonists on 2 principal receptors— cannabinoid receptor type 1 (CB1) and cannabinoid receptor type 2 (CB2)—which are both G protein–coupled receptors (Figure).5 Both have distinct distributions throughout different organ systems, to which cannabinoids (eg, THC, cannabidiol, endocannabinoids) show differential binding.6,7 Importantly, the expression of CB1 and CB2 has been identified on sensory nerve fibers, inflammatory cells, and adnexal structures of human skin.8 Based on these associations, topical application of cannabinoids has become a modality of interest for dermatological disorders. These formulations aim to influence cutaneous morphology without producing psychoactive effects.
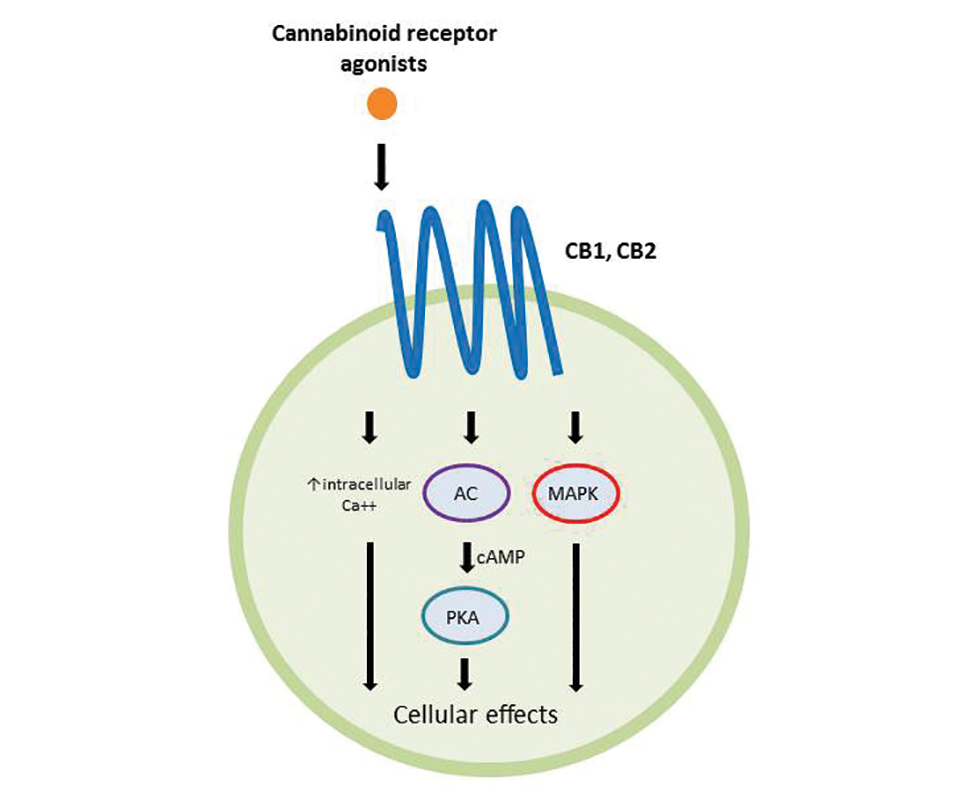
Topical Cannabinoids in Inflammatory Disorders
Atopic dermatitis has emerged as an active area of investigation for cannabinoid receptors and topical agonists (Table 1). In an animal model, Kim et al9 examined the effects of CB1 agonism on skin inflammation. Mice treated with topical CB1 agonists showed greater recovery of epidermal barrier function in acutely abrogated skin relative to those treated with a vehicle preparation. In addition, agonism of CB1 led to significant (P<.001) decreases in skin fold thickness among models of acute and chronic skin inflammation.9
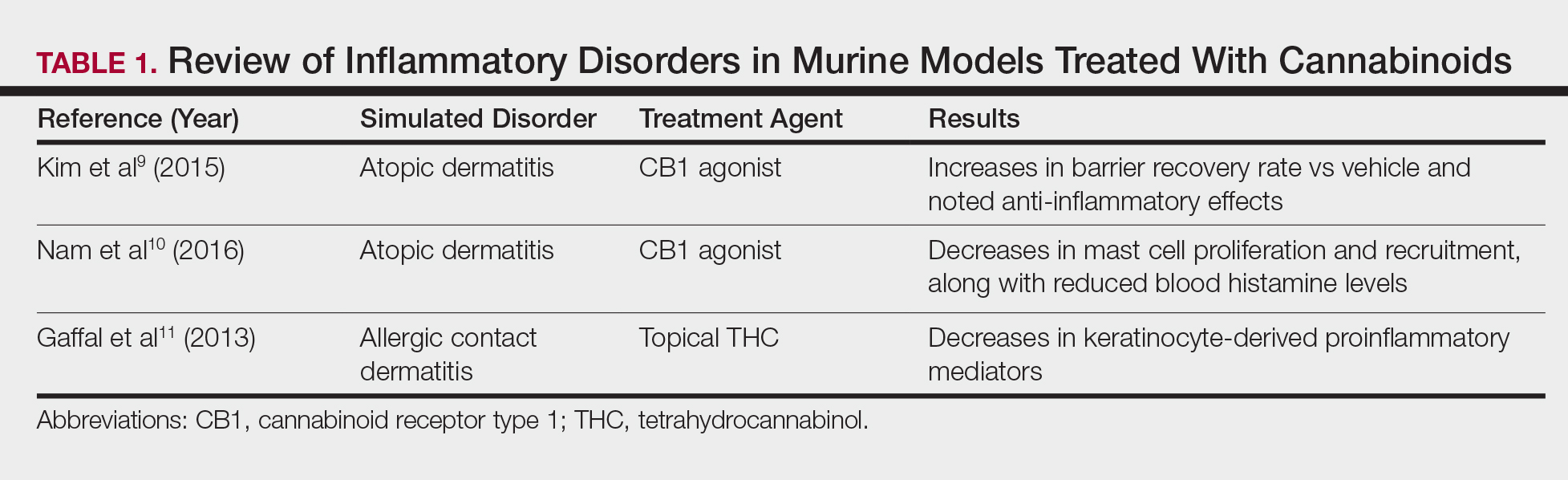
Nam et al10 also examined the role of topical CB1 agonists in mice with induced AD-like symptoms. Relative to treatment with vehicle, CB1 agonists significantly reduced the recruitment of mast cells (P<.01) and lowered the blood concentration of histamine (P<.05). Given the noted decrease in the release of inflammatory mediators, the authors speculated that topical agonsim of CB1 may prove useful in several conditions related to mast cell activation, such as AD, contact dermatitis, and psoriasis.10
The anti-inflammatory properties of topical THC were evaluated by Gaffal et al.11 In a mouse model of allergic contact dermatitis, mice treated with topical THC showed decreases in myeloid immune cell infiltration, with these beneficial effects existing even in mice with deficient CB1 and CB2 receptors. These results support a potentially wide anti-inflammatory activity of topical THC.11
Topical Cannabinoids in Pain Management
The effects of smoked cannabis in treating pain have undergone thorough investigation over recent years. Benefits have been noted in treating neuropathic pain, particularly in human immunodeficiency virus–associated sensory neuropathy.12-15 Smoked cannabis also may provide value as a synergistic therapy with opioids, thereby allowing for lower opioid doses.16
In contrast, research into the relationship between topical application of cannabinoids and nociception remains in preliminary stages (Table 2). In a mouse model, Dogrul et al17 assessed the topical antinociceptive potential of a mixed CB1-CB2 agonist. Results showed significant (P<.01) and dose-dependent antinociceptive effects relative to treatment with a vehicle.17 In a related study, Yesilyurt et al18 evaluated whether a mixed CB1-CB2 agonist could enhance the antinociceptive effects of topical opioids. Among mice treated with the combination of a cannabinoid agonist and topical morphine, a significantly (P<.05) greater analgesic effect was demonstrated relative to topical morphine alone.18
Studies in humans have been far more limited. Phan et al19 conducted a small, nonrandomized, open-label trial of a topical cannabinoid cream in patients with facial postherpetic neuralgia. Of 8 patients treated, 5 noted a mean pain reduction of 87.8%. No comparison vehicle was used. Based on this narrow study design, it is difficult to extrapolate these positive results to a broader patient population.19
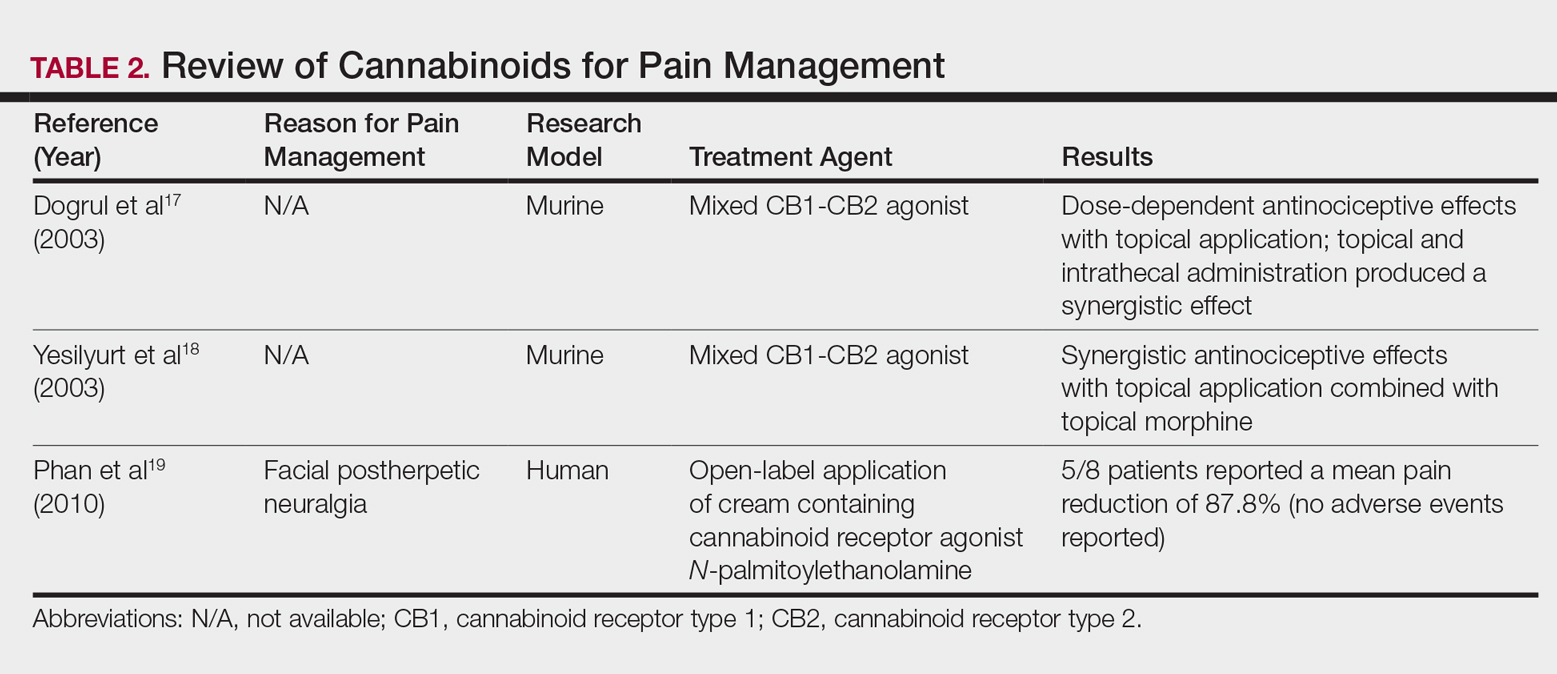
Commercial Products
Although preliminary models with topical cannabinoids have shown potential, large-scale clinical trials in humans have yet to be performed. Despite this lack of investigation, commercial formulations of topical cannabinoids are available to dermatology patients. These formulations are nonstandardized, and no safety data exists regarding their use. Topical cannabinoids on the market may contain various amounts of active ingredient and may be combined with a range of other compounds.
In dermatology offices, it is not uncommon for patients to express an intention to use topical cannabinoid products following their planned treatment or procedure. Patients also have been known to use topical cannabinoid products prior to dermatologic procedures, sometimes in place of an approved topical anesthetic, without consulting the physician performing the procedure. With interventions that lead to active areas of wound healing, the application of such products may increase the risk for contamination and infection. Therefore, patients should be counseled that the use of commercial topical cannabinoids could jeopardize the success of their planned procedure, put them at risk for infection, and possibly lead to systemic absorption and/or changes in wound-healing capacities.
Conclusion
Based on the results from recent animal models, cannabinoids may have a role in future treatment algorithms for several inflammatory conditions. However, current efficacy and safety data are almost entirely limited to preliminary animal studies in rodents. In addition, the formulation of topical cannabinoid products is nonstandardized and poorly regulated. As such, the present evidence does not support the use of topical cannabinoids in dermatology practices. Dermatologists should ask patients about the use of any cannabinoid products as part of a treatment program, especially given the unsubstantiated claims often made by unscrupulous advertisers. This issue highlights the need for further research and regulation.
- Pacher P, Batkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev. 2006;58:389-462.
- Giacoppo S, Galuppo M, Pollastro F, et al. A new formulation of cannabidiol in cream shows therapeutic effects in a mouse model of experimental autoimmune encephalomyelitis. Daru. 2015;23:48.
- Hammell DC, Zhang LP, Ma F, et al. Transdermal cannabidiol reduces inflammation and pain-related behaviours in a rat model of arthritis. Eur J Pain. 2016;20:936-948.
- Schicho R, Storr M. Topical and systemic cannabidiol improves trinitrobenzene sulfonic acid colitis in mice. Pharmacology. 2012;89:149-155.
- Howlett AC, Barth F, Bonner TI, et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev. 2002;54:161-202.
- Pertwee RG. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: delta9-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin. Br J Pharmacol. 2008;153:199-215.
- Svizenska I, Dubovy P, Sulcova A. Cannabinoid receptors 1 and 2 (CB1 and CB2), their distribution, ligands and functional involvement in nervous system structures—a short review. Pharmacol Biochem Behav. 2008;90:501-511.
- Stander S, Schmelz M, Metze D, et al. Distribution of cannabinoid receptor 1 (CB1) and 2 (CB2) on sensory nerve fibers and adnexal structures in human skin. J Dermatol Sci. 2005;38:177-188.
- Kim HJ, Kim B, Park BM, et al. Topical cannabinoid receptor 1 agonist attenuates the cutaneous inflammatory responses in oxazolone-induced atopic dermatitis model. Int J Dermatol. 2015;54:E401-E408.
- Nam G, Jeong SK, Park BM, et al. Selective cannabinoid receptor-1 agonists regulate mast cell activation in an oxazolone-induced atopic dermatitis model. Ann Dermatol. 2016;28:22-29.
- Gaffal E, Cron M, Glodde N, et al. Anti-inflammatory activity of topical THC in DNFB-mediated mouse allergic contact dermatitis independent of CB1 and CB2 receptors. Allergy. 2013;68:994-1000.
- Abrams DI, Jay CA, Shade SB, et al. Cannabis in painful HIV-associated sensory neuropathy: a randomized placebo-controlled trial. Neurology. 2007;68:515-521.
- Ellis RJ, Toperoff W, Vaida F, et al. Smoked medicinal cannabis for neuropathic pain in HIV: a randomized, crossover clinical trial. Neuropsychopharmacology. 2009;34:672-680.
- Wilsey B, Marcotte T, Deutsch R, et al. Low-dose vaporized cannabis significantly improves neuropathic pain. J Pain. 2013;14:136-148.
- Wilsey B, Marcotte T, Tsodikov A, et al. A randomized, placebo-controlled, crossover trial of cannabis cigarettes in neuropathic pain. J Pain. 2008;9:506-521.
- Abrams DI, Couey P, Shade SB, et al. Cannabinoid-opioid interaction in chronic pain. Clin Pharmacol Ther. 2011;90:844-851.
- Dogrul A, Gul H, Akar A, et al. Topical cannabinoid antinociception: synergy with spinal sites. Pain. 2003;105:11-16.
- Yesilyurt O, Dogrul A, Gul H, et al. Topical cannabinoid enhances topical morphine antinociception. Pain. 2003;105:303-308.
- Phan NQ, Siepmann D, Gralow I, et al. Adjuvant topical therapy with a cannabinoid receptor agonist in facial postherpetic neuralgia. J Dtsch Dermatol Ges. 2010;8:88-91.
The prevalence of topical cannabinoids has risen sharply in recent years. Commercial advertisers promote their usage as a safe means to treat a multitude of skin disorders, including atopic dermatitis (AD), psoriasis, and acne. Topical compounds have garnered interest in laboratory studies, but the purchase of commercial formulations is limited to over-the-counter products from unregulated suppliers. In this article, we review the scientific evidence behind topical cannabinoids and evaluate their role in clinical dermatology.
Background
Cannabis is designated as a Schedule I drug, according to the Controlled Substances Act of 1970. This listing is given to substances with no therapeutic value and a high potential for abuse. However, as of 2017, 29 states and the District of Columbia have laws legalizing cannabis in some capacity. These regulations typically apply to medicinal use, though several states have now legalized recreational use.
Cannabinoids represent a broad class of chemical compounds derived from the cannabis plant. Originally, this class only comprised phytocannabinoids, cannabinoids produced by the cannabis plant. Tetrahydrocannabinol (THC) is the most well-known phytocannabinoid and leads to the psychoactive effects typically associated with cannabis use. Later investigation led to the discovery of endocannabinoids, cannabinoids that are naturally produced by human and animal bodies, as well as synthetic cannabinoids.1 Cannabidiol is a phytocannabinoid that has been investigated in neurologic and anti-inflammatory conditions.2-4
Cannabinoids act as agonists on 2 principal receptors— cannabinoid receptor type 1 (CB1) and cannabinoid receptor type 2 (CB2)—which are both G protein–coupled receptors (Figure).5 Both have distinct distributions throughout different organ systems, to which cannabinoids (eg, THC, cannabidiol, endocannabinoids) show differential binding.6,7 Importantly, the expression of CB1 and CB2 has been identified on sensory nerve fibers, inflammatory cells, and adnexal structures of human skin.8 Based on these associations, topical application of cannabinoids has become a modality of interest for dermatological disorders. These formulations aim to influence cutaneous morphology without producing psychoactive effects.
Topical Cannabinoids in Inflammatory Disorders
Atopic dermatitis has emerged as an active area of investigation for cannabinoid receptors and topical agonists (Table 1). In an animal model, Kim et al9 examined the effects of CB1 agonism on skin inflammation. Mice treated with topical CB1 agonists showed greater recovery of epidermal barrier function in acutely abrogated skin relative to those treated with a vehicle preparation. In addition, agonism of CB1 led to significant (P<.001) decreases in skin fold thickness among models of acute and chronic skin inflammation.9
Nam et al10 also examined the role of topical CB1 agonists in mice with induced AD-like symptoms. Relative to treatment with vehicle, CB1 agonists significantly reduced the recruitment of mast cells (P<.01) and lowered the blood concentration of histamine (P<.05). Given the noted decrease in the release of inflammatory mediators, the authors speculated that topical agonsim of CB1 may prove useful in several conditions related to mast cell activation, such as AD, contact dermatitis, and psoriasis.10
The anti-inflammatory properties of topical THC were evaluated by Gaffal et al.11 In a mouse model of allergic contact dermatitis, mice treated with topical THC showed decreases in myeloid immune cell infiltration, with these beneficial effects existing even in mice with deficient CB1 and CB2 receptors. These results support a potentially wide anti-inflammatory activity of topical THC.11
Topical Cannabinoids in Pain Management
The effects of smoked cannabis in treating pain have undergone thorough investigation over recent years. Benefits have been noted in treating neuropathic pain, particularly in human immunodeficiency virus–associated sensory neuropathy.12-15 Smoked cannabis also may provide value as a synergistic therapy with opioids, thereby allowing for lower opioid doses.16
In contrast, research into the relationship between topical application of cannabinoids and nociception remains in preliminary stages (Table 2). In a mouse model, Dogrul et al17 assessed the topical antinociceptive potential of a mixed CB1-CB2 agonist. Results showed significant (P<.01) and dose-dependent antinociceptive effects relative to treatment with a vehicle.17 In a related study, Yesilyurt et al18 evaluated whether a mixed CB1-CB2 agonist could enhance the antinociceptive effects of topical opioids. Among mice treated with the combination of a cannabinoid agonist and topical morphine, a significantly (P<.05) greater analgesic effect was demonstrated relative to topical morphine alone.18
Studies in humans have been far more limited. Phan et al19 conducted a small, nonrandomized, open-label trial of a topical cannabinoid cream in patients with facial postherpetic neuralgia. Of 8 patients treated, 5 noted a mean pain reduction of 87.8%. No comparison vehicle was used. Based on this narrow study design, it is difficult to extrapolate these positive results to a broader patient population.19
Commercial Products
Although preliminary models with topical cannabinoids have shown potential, large-scale clinical trials in humans have yet to be performed. Despite this lack of investigation, commercial formulations of topical cannabinoids are available to dermatology patients. These formulations are nonstandardized, and no safety data exists regarding their use. Topical cannabinoids on the market may contain various amounts of active ingredient and may be combined with a range of other compounds.
In dermatology offices, it is not uncommon for patients to express an intention to use topical cannabinoid products following their planned treatment or procedure. Patients also have been known to use topical cannabinoid products prior to dermatologic procedures, sometimes in place of an approved topical anesthetic, without consulting the physician performing the procedure. With interventions that lead to active areas of wound healing, the application of such products may increase the risk for contamination and infection. Therefore, patients should be counseled that the use of commercial topical cannabinoids could jeopardize the success of their planned procedure, put them at risk for infection, and possibly lead to systemic absorption and/or changes in wound-healing capacities.
Conclusion
Based on the results from recent animal models, cannabinoids may have a role in future treatment algorithms for several inflammatory conditions. However, current efficacy and safety data are almost entirely limited to preliminary animal studies in rodents. In addition, the formulation of topical cannabinoid products is nonstandardized and poorly regulated. As such, the present evidence does not support the use of topical cannabinoids in dermatology practices. Dermatologists should ask patients about the use of any cannabinoid products as part of a treatment program, especially given the unsubstantiated claims often made by unscrupulous advertisers. This issue highlights the need for further research and regulation.
The prevalence of topical cannabinoids has risen sharply in recent years. Commercial advertisers promote their usage as a safe means to treat a multitude of skin disorders, including atopic dermatitis (AD), psoriasis, and acne. Topical compounds have garnered interest in laboratory studies, but the purchase of commercial formulations is limited to over-the-counter products from unregulated suppliers. In this article, we review the scientific evidence behind topical cannabinoids and evaluate their role in clinical dermatology.
Background
Cannabis is designated as a Schedule I drug, according to the Controlled Substances Act of 1970. This listing is given to substances with no therapeutic value and a high potential for abuse. However, as of 2017, 29 states and the District of Columbia have laws legalizing cannabis in some capacity. These regulations typically apply to medicinal use, though several states have now legalized recreational use.
Cannabinoids represent a broad class of chemical compounds derived from the cannabis plant. Originally, this class only comprised phytocannabinoids, cannabinoids produced by the cannabis plant. Tetrahydrocannabinol (THC) is the most well-known phytocannabinoid and leads to the psychoactive effects typically associated with cannabis use. Later investigation led to the discovery of endocannabinoids, cannabinoids that are naturally produced by human and animal bodies, as well as synthetic cannabinoids.1 Cannabidiol is a phytocannabinoid that has been investigated in neurologic and anti-inflammatory conditions.2-4
Cannabinoids act as agonists on 2 principal receptors— cannabinoid receptor type 1 (CB1) and cannabinoid receptor type 2 (CB2)—which are both G protein–coupled receptors (Figure).5 Both have distinct distributions throughout different organ systems, to which cannabinoids (eg, THC, cannabidiol, endocannabinoids) show differential binding.6,7 Importantly, the expression of CB1 and CB2 has been identified on sensory nerve fibers, inflammatory cells, and adnexal structures of human skin.8 Based on these associations, topical application of cannabinoids has become a modality of interest for dermatological disorders. These formulations aim to influence cutaneous morphology without producing psychoactive effects.
Topical Cannabinoids in Inflammatory Disorders
Atopic dermatitis has emerged as an active area of investigation for cannabinoid receptors and topical agonists (Table 1). In an animal model, Kim et al9 examined the effects of CB1 agonism on skin inflammation. Mice treated with topical CB1 agonists showed greater recovery of epidermal barrier function in acutely abrogated skin relative to those treated with a vehicle preparation. In addition, agonism of CB1 led to significant (P<.001) decreases in skin fold thickness among models of acute and chronic skin inflammation.9
Nam et al10 also examined the role of topical CB1 agonists in mice with induced AD-like symptoms. Relative to treatment with vehicle, CB1 agonists significantly reduced the recruitment of mast cells (P<.01) and lowered the blood concentration of histamine (P<.05). Given the noted decrease in the release of inflammatory mediators, the authors speculated that topical agonsim of CB1 may prove useful in several conditions related to mast cell activation, such as AD, contact dermatitis, and psoriasis.10
The anti-inflammatory properties of topical THC were evaluated by Gaffal et al.11 In a mouse model of allergic contact dermatitis, mice treated with topical THC showed decreases in myeloid immune cell infiltration, with these beneficial effects existing even in mice with deficient CB1 and CB2 receptors. These results support a potentially wide anti-inflammatory activity of topical THC.11
Topical Cannabinoids in Pain Management
The effects of smoked cannabis in treating pain have undergone thorough investigation over recent years. Benefits have been noted in treating neuropathic pain, particularly in human immunodeficiency virus–associated sensory neuropathy.12-15 Smoked cannabis also may provide value as a synergistic therapy with opioids, thereby allowing for lower opioid doses.16
In contrast, research into the relationship between topical application of cannabinoids and nociception remains in preliminary stages (Table 2). In a mouse model, Dogrul et al17 assessed the topical antinociceptive potential of a mixed CB1-CB2 agonist. Results showed significant (P<.01) and dose-dependent antinociceptive effects relative to treatment with a vehicle.17 In a related study, Yesilyurt et al18 evaluated whether a mixed CB1-CB2 agonist could enhance the antinociceptive effects of topical opioids. Among mice treated with the combination of a cannabinoid agonist and topical morphine, a significantly (P<.05) greater analgesic effect was demonstrated relative to topical morphine alone.18
Studies in humans have been far more limited. Phan et al19 conducted a small, nonrandomized, open-label trial of a topical cannabinoid cream in patients with facial postherpetic neuralgia. Of 8 patients treated, 5 noted a mean pain reduction of 87.8%. No comparison vehicle was used. Based on this narrow study design, it is difficult to extrapolate these positive results to a broader patient population.19
Commercial Products
Although preliminary models with topical cannabinoids have shown potential, large-scale clinical trials in humans have yet to be performed. Despite this lack of investigation, commercial formulations of topical cannabinoids are available to dermatology patients. These formulations are nonstandardized, and no safety data exists regarding their use. Topical cannabinoids on the market may contain various amounts of active ingredient and may be combined with a range of other compounds.
In dermatology offices, it is not uncommon for patients to express an intention to use topical cannabinoid products following their planned treatment or procedure. Patients also have been known to use topical cannabinoid products prior to dermatologic procedures, sometimes in place of an approved topical anesthetic, without consulting the physician performing the procedure. With interventions that lead to active areas of wound healing, the application of such products may increase the risk for contamination and infection. Therefore, patients should be counseled that the use of commercial topical cannabinoids could jeopardize the success of their planned procedure, put them at risk for infection, and possibly lead to systemic absorption and/or changes in wound-healing capacities.
Conclusion
Based on the results from recent animal models, cannabinoids may have a role in future treatment algorithms for several inflammatory conditions. However, current efficacy and safety data are almost entirely limited to preliminary animal studies in rodents. In addition, the formulation of topical cannabinoid products is nonstandardized and poorly regulated. As such, the present evidence does not support the use of topical cannabinoids in dermatology practices. Dermatologists should ask patients about the use of any cannabinoid products as part of a treatment program, especially given the unsubstantiated claims often made by unscrupulous advertisers. This issue highlights the need for further research and regulation.
- Pacher P, Batkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev. 2006;58:389-462.
- Giacoppo S, Galuppo M, Pollastro F, et al. A new formulation of cannabidiol in cream shows therapeutic effects in a mouse model of experimental autoimmune encephalomyelitis. Daru. 2015;23:48.
- Hammell DC, Zhang LP, Ma F, et al. Transdermal cannabidiol reduces inflammation and pain-related behaviours in a rat model of arthritis. Eur J Pain. 2016;20:936-948.
- Schicho R, Storr M. Topical and systemic cannabidiol improves trinitrobenzene sulfonic acid colitis in mice. Pharmacology. 2012;89:149-155.
- Howlett AC, Barth F, Bonner TI, et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev. 2002;54:161-202.
- Pertwee RG. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: delta9-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin. Br J Pharmacol. 2008;153:199-215.
- Svizenska I, Dubovy P, Sulcova A. Cannabinoid receptors 1 and 2 (CB1 and CB2), their distribution, ligands and functional involvement in nervous system structures—a short review. Pharmacol Biochem Behav. 2008;90:501-511.
- Stander S, Schmelz M, Metze D, et al. Distribution of cannabinoid receptor 1 (CB1) and 2 (CB2) on sensory nerve fibers and adnexal structures in human skin. J Dermatol Sci. 2005;38:177-188.
- Kim HJ, Kim B, Park BM, et al. Topical cannabinoid receptor 1 agonist attenuates the cutaneous inflammatory responses in oxazolone-induced atopic dermatitis model. Int J Dermatol. 2015;54:E401-E408.
- Nam G, Jeong SK, Park BM, et al. Selective cannabinoid receptor-1 agonists regulate mast cell activation in an oxazolone-induced atopic dermatitis model. Ann Dermatol. 2016;28:22-29.
- Gaffal E, Cron M, Glodde N, et al. Anti-inflammatory activity of topical THC in DNFB-mediated mouse allergic contact dermatitis independent of CB1 and CB2 receptors. Allergy. 2013;68:994-1000.
- Abrams DI, Jay CA, Shade SB, et al. Cannabis in painful HIV-associated sensory neuropathy: a randomized placebo-controlled trial. Neurology. 2007;68:515-521.
- Ellis RJ, Toperoff W, Vaida F, et al. Smoked medicinal cannabis for neuropathic pain in HIV: a randomized, crossover clinical trial. Neuropsychopharmacology. 2009;34:672-680.
- Wilsey B, Marcotte T, Deutsch R, et al. Low-dose vaporized cannabis significantly improves neuropathic pain. J Pain. 2013;14:136-148.
- Wilsey B, Marcotte T, Tsodikov A, et al. A randomized, placebo-controlled, crossover trial of cannabis cigarettes in neuropathic pain. J Pain. 2008;9:506-521.
- Abrams DI, Couey P, Shade SB, et al. Cannabinoid-opioid interaction in chronic pain. Clin Pharmacol Ther. 2011;90:844-851.
- Dogrul A, Gul H, Akar A, et al. Topical cannabinoid antinociception: synergy with spinal sites. Pain. 2003;105:11-16.
- Yesilyurt O, Dogrul A, Gul H, et al. Topical cannabinoid enhances topical morphine antinociception. Pain. 2003;105:303-308.
- Phan NQ, Siepmann D, Gralow I, et al. Adjuvant topical therapy with a cannabinoid receptor agonist in facial postherpetic neuralgia. J Dtsch Dermatol Ges. 2010;8:88-91.
- Pacher P, Batkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev. 2006;58:389-462.
- Giacoppo S, Galuppo M, Pollastro F, et al. A new formulation of cannabidiol in cream shows therapeutic effects in a mouse model of experimental autoimmune encephalomyelitis. Daru. 2015;23:48.
- Hammell DC, Zhang LP, Ma F, et al. Transdermal cannabidiol reduces inflammation and pain-related behaviours in a rat model of arthritis. Eur J Pain. 2016;20:936-948.
- Schicho R, Storr M. Topical and systemic cannabidiol improves trinitrobenzene sulfonic acid colitis in mice. Pharmacology. 2012;89:149-155.
- Howlett AC, Barth F, Bonner TI, et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev. 2002;54:161-202.
- Pertwee RG. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: delta9-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin. Br J Pharmacol. 2008;153:199-215.
- Svizenska I, Dubovy P, Sulcova A. Cannabinoid receptors 1 and 2 (CB1 and CB2), their distribution, ligands and functional involvement in nervous system structures—a short review. Pharmacol Biochem Behav. 2008;90:501-511.
- Stander S, Schmelz M, Metze D, et al. Distribution of cannabinoid receptor 1 (CB1) and 2 (CB2) on sensory nerve fibers and adnexal structures in human skin. J Dermatol Sci. 2005;38:177-188.
- Kim HJ, Kim B, Park BM, et al. Topical cannabinoid receptor 1 agonist attenuates the cutaneous inflammatory responses in oxazolone-induced atopic dermatitis model. Int J Dermatol. 2015;54:E401-E408.
- Nam G, Jeong SK, Park BM, et al. Selective cannabinoid receptor-1 agonists regulate mast cell activation in an oxazolone-induced atopic dermatitis model. Ann Dermatol. 2016;28:22-29.
- Gaffal E, Cron M, Glodde N, et al. Anti-inflammatory activity of topical THC in DNFB-mediated mouse allergic contact dermatitis independent of CB1 and CB2 receptors. Allergy. 2013;68:994-1000.
- Abrams DI, Jay CA, Shade SB, et al. Cannabis in painful HIV-associated sensory neuropathy: a randomized placebo-controlled trial. Neurology. 2007;68:515-521.
- Ellis RJ, Toperoff W, Vaida F, et al. Smoked medicinal cannabis for neuropathic pain in HIV: a randomized, crossover clinical trial. Neuropsychopharmacology. 2009;34:672-680.
- Wilsey B, Marcotte T, Deutsch R, et al. Low-dose vaporized cannabis significantly improves neuropathic pain. J Pain. 2013;14:136-148.
- Wilsey B, Marcotte T, Tsodikov A, et al. A randomized, placebo-controlled, crossover trial of cannabis cigarettes in neuropathic pain. J Pain. 2008;9:506-521.
- Abrams DI, Couey P, Shade SB, et al. Cannabinoid-opioid interaction in chronic pain. Clin Pharmacol Ther. 2011;90:844-851.
- Dogrul A, Gul H, Akar A, et al. Topical cannabinoid antinociception: synergy with spinal sites. Pain. 2003;105:11-16.
- Yesilyurt O, Dogrul A, Gul H, et al. Topical cannabinoid enhances topical morphine antinociception. Pain. 2003;105:303-308.
- Phan NQ, Siepmann D, Gralow I, et al. Adjuvant topical therapy with a cannabinoid receptor agonist in facial postherpetic neuralgia. J Dtsch Dermatol Ges. 2010;8:88-91.
Practice Points
- Topical cannabinoids are advertised by companies as treatment options for numerous dermatologic conditions.
- Despite promising data in rodent models, there have been no rigorous studies to date confirming efficacy or safety in humans.
- Dermatologists should therefore inquire with patients about the use of any topical cannabinoid products, especially around the time of planned procedures, as they may affect treatment outcomes.

Trauma Care: The “Golden Hour” Meets the “Golden Years”
In Emergency Medicine this month and next, Drs. Tom Scalea (See “The Golden Hourglass,” EM, April 2007), Ashley Menne, Daniel Haase, and Jay Menaker of the University of Maryland’s R Adams Cowley Shock Trauma Center paint a detailed picture of the changing landscape of trauma care over the past two decades.
In his introduction, Dr. Scalea writes “Certainly, the most important change has been the ‘graying’ of trauma patients…[whose evaluation and care] may involve a number of diagnostic tests in the ED…”, and whose care must include dealing with comorbidities, and a large number of medications that might interact with the analgesics, sedatives, and anti-seizure meds needed to treat trauma. These considerations have led many Level I trauma centers to add advanced patient age as an independent determinant for both trauma activations and subsequent ICU admissions, and to include “geriatric” consultants in the initial management.
The aging trauma patient, however, is not the only factor responsible for major changes in the management of serious trauma, as the Shock Trauma group describes the current difficulties in attempting to rapidly reverse the anticoagulation effects of the novel oral anticoagulants (NOACs) that are increasingly being prescribed instead of warfarin to manage the thromboembolic complications of atrial fibrillation, valve replacement, venous thrombosis, and pulmonary embolism in both younger and older patients. They also explain a major change in thinking regarding the optimal degree of blood pressure control in favor of “permissive hypotension” as part of “damage control resuscitation,” and in the amount and types of volume replacement, optimal blood component ratios for transfusion, monitoring, and faster and less invasive endovascular repair techniques for hemostasis. The authors also note the persistent and rising incidence of penetrating trauma from gunshot and knife wounds.
But the increasing percentages of elderly trauma victims requiring care for devastating falls and low-speed vehicular injuries in even the busiest “knife and gun club” trauma centers mandate the attention of all health care providers. In recent months, much space in this and other journals has been devoted to the health care issues of the elderly (see “Recognizing and Managing Elder Abuse in the Emergency Department,” and “Elder Abuse: A New Old Problem,” EM, May 2017) that necessitate significantly increased resources and provider time and effort now, and for at least the first half of the 21st century.
The main reason for this seismic demographic shift, dubbed by some “the silver tsunami”, is the aging post World War II “baby boomer” generation that has commanded center stage in western society throughout their development since the late 1940s. As a member of that generation, I often wonder how subsequent generations such as “Gen X” and “Millennials” view this phenomenon. Do they resent the attention, resources, and expenditures now demanded by baby boomers? If so, there is an important lesson to be learned from the changes in trauma care described in the following pages: virtually every measure now employed to enhance recovery of an elderly trauma victim will benefit younger trauma victims, as well. At most, some of the measures may not be absolutely necessary because younger adults have greater functional reserve and are more likely to survive less precise management, even if their posttraumatic courses are longer and more difficult. But younger trauma victims with comorbidities can also benefit from a more inclusive team approach from the start, as well as measures such as permissive hypotension, vascular stents and less invasive endovascular approaches, more precise blood component replacement, more accurate monitoring, and a better approach to anticoagulation and its reversal.
Faster and better quality survival of all trauma victims—including, but not limited to, the elderly—will free up needed and expensive resources for other patients and trauma victims, including those who continue to butt heads, drive two and four wheel vehicles at excessive speeds, and engage in trauma of an “interpersonal nature.”
In Emergency Medicine this month and next, Drs. Tom Scalea (See “The Golden Hourglass,” EM, April 2007), Ashley Menne, Daniel Haase, and Jay Menaker of the University of Maryland’s R Adams Cowley Shock Trauma Center paint a detailed picture of the changing landscape of trauma care over the past two decades.
In his introduction, Dr. Scalea writes “Certainly, the most important change has been the ‘graying’ of trauma patients…[whose evaluation and care] may involve a number of diagnostic tests in the ED…”, and whose care must include dealing with comorbidities, and a large number of medications that might interact with the analgesics, sedatives, and anti-seizure meds needed to treat trauma. These considerations have led many Level I trauma centers to add advanced patient age as an independent determinant for both trauma activations and subsequent ICU admissions, and to include “geriatric” consultants in the initial management.
The aging trauma patient, however, is not the only factor responsible for major changes in the management of serious trauma, as the Shock Trauma group describes the current difficulties in attempting to rapidly reverse the anticoagulation effects of the novel oral anticoagulants (NOACs) that are increasingly being prescribed instead of warfarin to manage the thromboembolic complications of atrial fibrillation, valve replacement, venous thrombosis, and pulmonary embolism in both younger and older patients. They also explain a major change in thinking regarding the optimal degree of blood pressure control in favor of “permissive hypotension” as part of “damage control resuscitation,” and in the amount and types of volume replacement, optimal blood component ratios for transfusion, monitoring, and faster and less invasive endovascular repair techniques for hemostasis. The authors also note the persistent and rising incidence of penetrating trauma from gunshot and knife wounds.
But the increasing percentages of elderly trauma victims requiring care for devastating falls and low-speed vehicular injuries in even the busiest “knife and gun club” trauma centers mandate the attention of all health care providers. In recent months, much space in this and other journals has been devoted to the health care issues of the elderly (see “Recognizing and Managing Elder Abuse in the Emergency Department,” and “Elder Abuse: A New Old Problem,” EM, May 2017) that necessitate significantly increased resources and provider time and effort now, and for at least the first half of the 21st century.
The main reason for this seismic demographic shift, dubbed by some “the silver tsunami”, is the aging post World War II “baby boomer” generation that has commanded center stage in western society throughout their development since the late 1940s. As a member of that generation, I often wonder how subsequent generations such as “Gen X” and “Millennials” view this phenomenon. Do they resent the attention, resources, and expenditures now demanded by baby boomers? If so, there is an important lesson to be learned from the changes in trauma care described in the following pages: virtually every measure now employed to enhance recovery of an elderly trauma victim will benefit younger trauma victims, as well. At most, some of the measures may not be absolutely necessary because younger adults have greater functional reserve and are more likely to survive less precise management, even if their posttraumatic courses are longer and more difficult. But younger trauma victims with comorbidities can also benefit from a more inclusive team approach from the start, as well as measures such as permissive hypotension, vascular stents and less invasive endovascular approaches, more precise blood component replacement, more accurate monitoring, and a better approach to anticoagulation and its reversal.
Faster and better quality survival of all trauma victims—including, but not limited to, the elderly—will free up needed and expensive resources for other patients and trauma victims, including those who continue to butt heads, drive two and four wheel vehicles at excessive speeds, and engage in trauma of an “interpersonal nature.”
In Emergency Medicine this month and next, Drs. Tom Scalea (See “The Golden Hourglass,” EM, April 2007), Ashley Menne, Daniel Haase, and Jay Menaker of the University of Maryland’s R Adams Cowley Shock Trauma Center paint a detailed picture of the changing landscape of trauma care over the past two decades.
In his introduction, Dr. Scalea writes “Certainly, the most important change has been the ‘graying’ of trauma patients…[whose evaluation and care] may involve a number of diagnostic tests in the ED…”, and whose care must include dealing with comorbidities, and a large number of medications that might interact with the analgesics, sedatives, and anti-seizure meds needed to treat trauma. These considerations have led many Level I trauma centers to add advanced patient age as an independent determinant for both trauma activations and subsequent ICU admissions, and to include “geriatric” consultants in the initial management.
The aging trauma patient, however, is not the only factor responsible for major changes in the management of serious trauma, as the Shock Trauma group describes the current difficulties in attempting to rapidly reverse the anticoagulation effects of the novel oral anticoagulants (NOACs) that are increasingly being prescribed instead of warfarin to manage the thromboembolic complications of atrial fibrillation, valve replacement, venous thrombosis, and pulmonary embolism in both younger and older patients. They also explain a major change in thinking regarding the optimal degree of blood pressure control in favor of “permissive hypotension” as part of “damage control resuscitation,” and in the amount and types of volume replacement, optimal blood component ratios for transfusion, monitoring, and faster and less invasive endovascular repair techniques for hemostasis. The authors also note the persistent and rising incidence of penetrating trauma from gunshot and knife wounds.
But the increasing percentages of elderly trauma victims requiring care for devastating falls and low-speed vehicular injuries in even the busiest “knife and gun club” trauma centers mandate the attention of all health care providers. In recent months, much space in this and other journals has been devoted to the health care issues of the elderly (see “Recognizing and Managing Elder Abuse in the Emergency Department,” and “Elder Abuse: A New Old Problem,” EM, May 2017) that necessitate significantly increased resources and provider time and effort now, and for at least the first half of the 21st century.
The main reason for this seismic demographic shift, dubbed by some “the silver tsunami”, is the aging post World War II “baby boomer” generation that has commanded center stage in western society throughout their development since the late 1940s. As a member of that generation, I often wonder how subsequent generations such as “Gen X” and “Millennials” view this phenomenon. Do they resent the attention, resources, and expenditures now demanded by baby boomers? If so, there is an important lesson to be learned from the changes in trauma care described in the following pages: virtually every measure now employed to enhance recovery of an elderly trauma victim will benefit younger trauma victims, as well. At most, some of the measures may not be absolutely necessary because younger adults have greater functional reserve and are more likely to survive less precise management, even if their posttraumatic courses are longer and more difficult. But younger trauma victims with comorbidities can also benefit from a more inclusive team approach from the start, as well as measures such as permissive hypotension, vascular stents and less invasive endovascular approaches, more precise blood component replacement, more accurate monitoring, and a better approach to anticoagulation and its reversal.
Faster and better quality survival of all trauma victims—including, but not limited to, the elderly—will free up needed and expensive resources for other patients and trauma victims, including those who continue to butt heads, drive two and four wheel vehicles at excessive speeds, and engage in trauma of an “interpersonal nature.”
