User login
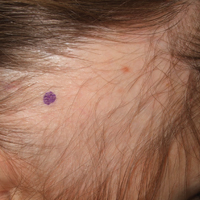
Brown patch on infant
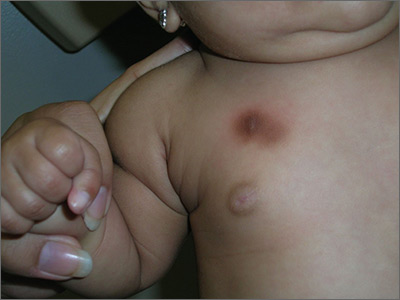
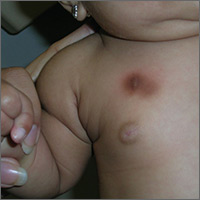
The FP diagnosed urticaria pigmentosa. If urticaria pigmentosa is suspected, it is helpful to stroke the lesion with a fingernail or the wooden end of a cotton-tipped applicator. This induces erythema and a wheal that is confined to the stroked site within the lesion—a positive Darier sign.
Urticaria pigmentosa is a form of mast cell activation syndrome (MCAS), in which there are too many mast cells in the skin. More serious forms of MCAS in adults include systemic mastocytosis and cutaneous mastocytosis. Urticaria pigmentosa is a self-limiting problem in young children that rarely causes sufficient symptoms to warrant treatment.
In this case, the FP assured the mother that this was not dangerous and would go away over time on its own. The FP wrote down the name of the condition for the mother and explained to her that if she noticed any changes or new symptoms, she should return for further evaluation and treatment. The mother was satisfied with this explanation.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Usatine R. Urticaria and angioedema. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013: 863-870.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
The FP diagnosed urticaria pigmentosa. If urticaria pigmentosa is suspected, it is helpful to stroke the lesion with a fingernail or the wooden end of a cotton-tipped applicator. This induces erythema and a wheal that is confined to the stroked site within the lesion—a positive Darier sign.
Urticaria pigmentosa is a form of mast cell activation syndrome (MCAS), in which there are too many mast cells in the skin. More serious forms of MCAS in adults include systemic mastocytosis and cutaneous mastocytosis. Urticaria pigmentosa is a self-limiting problem in young children that rarely causes sufficient symptoms to warrant treatment.
In this case, the FP assured the mother that this was not dangerous and would go away over time on its own. The FP wrote down the name of the condition for the mother and explained to her that if she noticed any changes or new symptoms, she should return for further evaluation and treatment. The mother was satisfied with this explanation.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Usatine R. Urticaria and angioedema. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013: 863-870.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
The FP diagnosed urticaria pigmentosa. If urticaria pigmentosa is suspected, it is helpful to stroke the lesion with a fingernail or the wooden end of a cotton-tipped applicator. This induces erythema and a wheal that is confined to the stroked site within the lesion—a positive Darier sign.
Urticaria pigmentosa is a form of mast cell activation syndrome (MCAS), in which there are too many mast cells in the skin. More serious forms of MCAS in adults include systemic mastocytosis and cutaneous mastocytosis. Urticaria pigmentosa is a self-limiting problem in young children that rarely causes sufficient symptoms to warrant treatment.
In this case, the FP assured the mother that this was not dangerous and would go away over time on its own. The FP wrote down the name of the condition for the mother and explained to her that if she noticed any changes or new symptoms, she should return for further evaluation and treatment. The mother was satisfied with this explanation.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Usatine R. Urticaria and angioedema. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013: 863-870.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
All FDA panel members go thumbs up for CTL019 in relapsed/refractory childhood ALL
The answer to the billion dollar question – Does the chimeric antigen receptor T-cell (CAR T) construct CTL019 (tisagenlecleucel-T) have a favorable risk-benefit profile for the treatment of children and young adults with relapsed/refractory B-cell precursor acute lymphoblastic leukemia? – was a unanimous “yes” at a July 12 meeting of the Food and Drug Administration’s Oncologic Drugs Advisory Committee.
“This is the most exciting thing I have seen in my lifetime, and probably since the introduction of ‘multiagent total cancer care,’ as it was called then, for treatment of childhood leukemia,” remarked Timothy P. Cripe, MD, PhD, from Nationwide Children’s Hospital in Columbus, Ohio, and a temporary voting member of the ODAC.
Catherine M. Bollard, MD, MBChB, from the Children’s National Medical Center in Washington, also a temporary ODAC member, said that she voted “yes” because “this is a very poor-risk patient population, this is an unmet need in the pediatric population, and as you saw in the data [presented to ODAC] today, the clinical responses are remarkable. I think Novartis [the maker of CTL019] has done a great job putting together a plan for mitigating risk going forward.”
CTL019 was shown in a pivotal phase 2 clinical trial to induce an overall remission rate of 83% in children and young adults with relapsed/refractory ALL for whom at least two prior lines of therapy had failed. Based on these results, the FDA accepted a biologics license application for the agent from Novartis.
At the meeting, panel members initially seemed favorably disposed toward recommending approval but heard concerns from FDA scientists about the potential for severe or fatal adverse events such as the cytokine release syndrome (CRS); the possible generation of replication competent retrovirus (RCR); and the potential for secondary malignancies from insertional mutagenesis, the incorporation of portions of the lentiviral vector into the patient’s genome.
In his opening remarks, Wilson W. Bryan, MD, from the FDA’s Office of Tissue and Advanced Therapies and Center for Biologics Evaluation and Drug Research, commented that “the clinical development of tisagenlecleucel suggests that this is a life-saving product.”
He went on, however, to frame the FDA’s concerns: “Clinical trials are not always a good predictor of the effectiveness and safety of a marketed product,” he said. “In particular, we are concerned that the same benefit and safety seen in clinical trials may not carry over to routine clinical use.”
The purpose of the hearing was to focus on manufacturing issues related to product quality, including replicability of the product for commercial use and safety issues such as prevention of CRS and neurotoxicities.
“We are also concerned about the hypothetical risk of secondary malignancies. Therefore, we are asking for the committee’s recommendations regarding the nature and duration of follow-up of patients who would receive this product,” Dr. Bryan said.
“CTL019 is a living drug, which demonstrates activity after a single infusion,” said Samit Hirawat, MD, head of oncology global development for Novartis.
But the nature of CTL019 as a living drug also means that it is subject to variations in the ability of autologous T cells harvested via leukapheresis to be infected with the lentiviral vector and expanded into a population of CAR T cells large enough to have therapeutic value, said Xiaobin Victor Lu, PhD, a chemistry, manufacturing, and controls reviewer for the FDA.
Mitigation plan
Novartis’ proposed plan includes specific, long-term steps for mitigating the risk of CRS and neurologic events, such as cerebral edema, the latter of which caused the FDA to call for a clinical hold of the phase 2 ROCKET trial for a different CAR T-cell construct.
Among the proposed elements of the mitigation plan are a 15-year minimum pharmacovigilance program and long-term safety follow-up for adverse events related to the therapy, efficacy, immunogenicity, transgene persistence of CD19 CAR, and the incidence of second malignancies possibly related to insertional mutagenesis.
Novartis also will train treatment center staff on processes for cell collection, cryopreservation, transport, chain of identity, safety management, and logistics for handling the CAR T-cell product. The company proposes to provide on-site training of personnel on CRS and neurotoxicity risk and management, as well as to offer information to patients and caregivers about the signs and symptoms of adverse events of concern.
Dr. Cripe expressed his concerns that Novartis’ proposal to initially limit the mitigation plan to 30 or 35 treatment sites would create problems of access and economic disparities among patients, and could cause inequities among treatment centers even with the same city.
David Lebwohl, MD, head of the CAR T global program for Novartis, said that the planned number of sites for the mitigation program would be expanded after 6 to 12 months if the CAR T construct receives final approval and clinical implementation goes well.
There was nearly unanimous agreement among the panel members that the planned 15-year follow-up and other mitigation measures would be adequate for detecting serious short- and long-term consequences of CAR T-cell therapy.
Patient/advocate perspective
In the public comment section of the proceedings, panel members were urged to vote in favor of CTL019 by parents of children with ALL, including Don McMahon, whose son Connor received the therapy after multiple relapses, and Tom Whitehead, father of Emily Whitehead, the first patient to receive CAR T cells for ALL.
Both children are alive and doing well.
CTL019 is produced by Novartis.
The unanimous recommendation by the Food and Drug Administration's Oncologic Drugs Advisory Committee means that the FDA is likely to approve CTL019 (tisagenlecleucel-T), and that approval may come quickly, possibly before the end of 2017. This approval was based on compelling data showing that 83% of children and young adults with refractory or relapsed acute lymphocytic leukemia (ALL) achieved remission with this therapy. This is exciting news for ALL patients as well as for the cell and gene therapy community. What remains to be determined are the labelling for CTL019, the cost of the therapy, and whether all patients who might benefit from this therapy will have the coverage to be able to access it.
While the response rates in patients treated in the trials presented to the FDA are very encouraging there are also concerns with the risks for cytokine release syndrome and neurotoxicity which can affect up to half of treated patients. As a result, Novartis, the manufacturer of CTL019, has proposed an extensive mitigation strategy and education process for the cell therapy centers that will offer the therapy. Initially, this is likely to be limited to around 30 centers that will be geographically distributed throughout the United States with gradual roll out to more centers as there is more experience with the use of CTL019.
Another issue for the centers is going to be operationalizing a new paradigm where CTL019 will be offered as a standard of care rather than in the context of a research study. Most of the initial pediatric centers will likely provide CTL019, within their transplant infrastructure since procurement, initial processing and infusion of cells will utilize their cell processing and collection facilities. The Foundation for Accreditation of Cell Therapy (FACT) has also anticipated this approval by publishing new standards for Immune Effectors earlier this year to promote quality practice in immune effector cell administration.
One other question is whether CTL019 will be transplant enabling or transplant replacing. While the initial response rates are very high and there are some well publicized patients who remain in remission over 5 years after CTL019 without other therapy, other responders proceeded to transplant and there is also a significant relapse rate. It is therefore an open question whether treating physicians will be happy to watch patients who attain remission after this therapy or whether they will still recommend transplant because there is not yet enough follow up on this product to know what the long-term cure rate is going to be.
Another CAR T-cell product is scheduled to come before an FDA advisory committee in October. The indication for KTE-C19 (axicabtagene ciloleucel) from Kite is for relapsed/refractory diffuse large B-cell lymphoma, a much bigger indication with a potentially much larger number of patients. The response rates for KTE-C19 in DLBCL (and indeed for CTL019 in DLBCL) are not as high as those for CTL019 in ALL and follow-up time is shorter, so it is not yet clear how many patients will have sustained long term responses. Nevertheless the response rate in patients who have failed all other therapies is high enough that this product will also likely be approved.
Helen Heslop, MD, is the Dan L. Duncan Chair and Professor of Medicine and Pediatrics at Baylor College of Medicine, Houston. She also is the Director of the Center for Cell and Gene Therapy at Baylor College of Medicine, Houston Methodist Hospital and Texas Children's Hospital. Dr. Heslop is a member of the editorial advisory board of Hematology News.
The unanimous recommendation by the Food and Drug Administration's Oncologic Drugs Advisory Committee means that the FDA is likely to approve CTL019 (tisagenlecleucel-T), and that approval may come quickly, possibly before the end of 2017. This approval was based on compelling data showing that 83% of children and young adults with refractory or relapsed acute lymphocytic leukemia (ALL) achieved remission with this therapy. This is exciting news for ALL patients as well as for the cell and gene therapy community. What remains to be determined are the labelling for CTL019, the cost of the therapy, and whether all patients who might benefit from this therapy will have the coverage to be able to access it.
While the response rates in patients treated in the trials presented to the FDA are very encouraging there are also concerns with the risks for cytokine release syndrome and neurotoxicity which can affect up to half of treated patients. As a result, Novartis, the manufacturer of CTL019, has proposed an extensive mitigation strategy and education process for the cell therapy centers that will offer the therapy. Initially, this is likely to be limited to around 30 centers that will be geographically distributed throughout the United States with gradual roll out to more centers as there is more experience with the use of CTL019.
Another issue for the centers is going to be operationalizing a new paradigm where CTL019 will be offered as a standard of care rather than in the context of a research study. Most of the initial pediatric centers will likely provide CTL019, within their transplant infrastructure since procurement, initial processing and infusion of cells will utilize their cell processing and collection facilities. The Foundation for Accreditation of Cell Therapy (FACT) has also anticipated this approval by publishing new standards for Immune Effectors earlier this year to promote quality practice in immune effector cell administration.
One other question is whether CTL019 will be transplant enabling or transplant replacing. While the initial response rates are very high and there are some well publicized patients who remain in remission over 5 years after CTL019 without other therapy, other responders proceeded to transplant and there is also a significant relapse rate. It is therefore an open question whether treating physicians will be happy to watch patients who attain remission after this therapy or whether they will still recommend transplant because there is not yet enough follow up on this product to know what the long-term cure rate is going to be.
Another CAR T-cell product is scheduled to come before an FDA advisory committee in October. The indication for KTE-C19 (axicabtagene ciloleucel) from Kite is for relapsed/refractory diffuse large B-cell lymphoma, a much bigger indication with a potentially much larger number of patients. The response rates for KTE-C19 in DLBCL (and indeed for CTL019 in DLBCL) are not as high as those for CTL019 in ALL and follow-up time is shorter, so it is not yet clear how many patients will have sustained long term responses. Nevertheless the response rate in patients who have failed all other therapies is high enough that this product will also likely be approved.
Helen Heslop, MD, is the Dan L. Duncan Chair and Professor of Medicine and Pediatrics at Baylor College of Medicine, Houston. She also is the Director of the Center for Cell and Gene Therapy at Baylor College of Medicine, Houston Methodist Hospital and Texas Children's Hospital. Dr. Heslop is a member of the editorial advisory board of Hematology News.
The unanimous recommendation by the Food and Drug Administration's Oncologic Drugs Advisory Committee means that the FDA is likely to approve CTL019 (tisagenlecleucel-T), and that approval may come quickly, possibly before the end of 2017. This approval was based on compelling data showing that 83% of children and young adults with refractory or relapsed acute lymphocytic leukemia (ALL) achieved remission with this therapy. This is exciting news for ALL patients as well as for the cell and gene therapy community. What remains to be determined are the labelling for CTL019, the cost of the therapy, and whether all patients who might benefit from this therapy will have the coverage to be able to access it.
While the response rates in patients treated in the trials presented to the FDA are very encouraging there are also concerns with the risks for cytokine release syndrome and neurotoxicity which can affect up to half of treated patients. As a result, Novartis, the manufacturer of CTL019, has proposed an extensive mitigation strategy and education process for the cell therapy centers that will offer the therapy. Initially, this is likely to be limited to around 30 centers that will be geographically distributed throughout the United States with gradual roll out to more centers as there is more experience with the use of CTL019.
Another issue for the centers is going to be operationalizing a new paradigm where CTL019 will be offered as a standard of care rather than in the context of a research study. Most of the initial pediatric centers will likely provide CTL019, within their transplant infrastructure since procurement, initial processing and infusion of cells will utilize their cell processing and collection facilities. The Foundation for Accreditation of Cell Therapy (FACT) has also anticipated this approval by publishing new standards for Immune Effectors earlier this year to promote quality practice in immune effector cell administration.
One other question is whether CTL019 will be transplant enabling or transplant replacing. While the initial response rates are very high and there are some well publicized patients who remain in remission over 5 years after CTL019 without other therapy, other responders proceeded to transplant and there is also a significant relapse rate. It is therefore an open question whether treating physicians will be happy to watch patients who attain remission after this therapy or whether they will still recommend transplant because there is not yet enough follow up on this product to know what the long-term cure rate is going to be.
Another CAR T-cell product is scheduled to come before an FDA advisory committee in October. The indication for KTE-C19 (axicabtagene ciloleucel) from Kite is for relapsed/refractory diffuse large B-cell lymphoma, a much bigger indication with a potentially much larger number of patients. The response rates for KTE-C19 in DLBCL (and indeed for CTL019 in DLBCL) are not as high as those for CTL019 in ALL and follow-up time is shorter, so it is not yet clear how many patients will have sustained long term responses. Nevertheless the response rate in patients who have failed all other therapies is high enough that this product will also likely be approved.
Helen Heslop, MD, is the Dan L. Duncan Chair and Professor of Medicine and Pediatrics at Baylor College of Medicine, Houston. She also is the Director of the Center for Cell and Gene Therapy at Baylor College of Medicine, Houston Methodist Hospital and Texas Children's Hospital. Dr. Heslop is a member of the editorial advisory board of Hematology News.
The answer to the billion dollar question – Does the chimeric antigen receptor T-cell (CAR T) construct CTL019 (tisagenlecleucel-T) have a favorable risk-benefit profile for the treatment of children and young adults with relapsed/refractory B-cell precursor acute lymphoblastic leukemia? – was a unanimous “yes” at a July 12 meeting of the Food and Drug Administration’s Oncologic Drugs Advisory Committee.
“This is the most exciting thing I have seen in my lifetime, and probably since the introduction of ‘multiagent total cancer care,’ as it was called then, for treatment of childhood leukemia,” remarked Timothy P. Cripe, MD, PhD, from Nationwide Children’s Hospital in Columbus, Ohio, and a temporary voting member of the ODAC.
Catherine M. Bollard, MD, MBChB, from the Children’s National Medical Center in Washington, also a temporary ODAC member, said that she voted “yes” because “this is a very poor-risk patient population, this is an unmet need in the pediatric population, and as you saw in the data [presented to ODAC] today, the clinical responses are remarkable. I think Novartis [the maker of CTL019] has done a great job putting together a plan for mitigating risk going forward.”
CTL019 was shown in a pivotal phase 2 clinical trial to induce an overall remission rate of 83% in children and young adults with relapsed/refractory ALL for whom at least two prior lines of therapy had failed. Based on these results, the FDA accepted a biologics license application for the agent from Novartis.
At the meeting, panel members initially seemed favorably disposed toward recommending approval but heard concerns from FDA scientists about the potential for severe or fatal adverse events such as the cytokine release syndrome (CRS); the possible generation of replication competent retrovirus (RCR); and the potential for secondary malignancies from insertional mutagenesis, the incorporation of portions of the lentiviral vector into the patient’s genome.
In his opening remarks, Wilson W. Bryan, MD, from the FDA’s Office of Tissue and Advanced Therapies and Center for Biologics Evaluation and Drug Research, commented that “the clinical development of tisagenlecleucel suggests that this is a life-saving product.”
He went on, however, to frame the FDA’s concerns: “Clinical trials are not always a good predictor of the effectiveness and safety of a marketed product,” he said. “In particular, we are concerned that the same benefit and safety seen in clinical trials may not carry over to routine clinical use.”
The purpose of the hearing was to focus on manufacturing issues related to product quality, including replicability of the product for commercial use and safety issues such as prevention of CRS and neurotoxicities.
“We are also concerned about the hypothetical risk of secondary malignancies. Therefore, we are asking for the committee’s recommendations regarding the nature and duration of follow-up of patients who would receive this product,” Dr. Bryan said.
“CTL019 is a living drug, which demonstrates activity after a single infusion,” said Samit Hirawat, MD, head of oncology global development for Novartis.
But the nature of CTL019 as a living drug also means that it is subject to variations in the ability of autologous T cells harvested via leukapheresis to be infected with the lentiviral vector and expanded into a population of CAR T cells large enough to have therapeutic value, said Xiaobin Victor Lu, PhD, a chemistry, manufacturing, and controls reviewer for the FDA.
Mitigation plan
Novartis’ proposed plan includes specific, long-term steps for mitigating the risk of CRS and neurologic events, such as cerebral edema, the latter of which caused the FDA to call for a clinical hold of the phase 2 ROCKET trial for a different CAR T-cell construct.
Among the proposed elements of the mitigation plan are a 15-year minimum pharmacovigilance program and long-term safety follow-up for adverse events related to the therapy, efficacy, immunogenicity, transgene persistence of CD19 CAR, and the incidence of second malignancies possibly related to insertional mutagenesis.
Novartis also will train treatment center staff on processes for cell collection, cryopreservation, transport, chain of identity, safety management, and logistics for handling the CAR T-cell product. The company proposes to provide on-site training of personnel on CRS and neurotoxicity risk and management, as well as to offer information to patients and caregivers about the signs and symptoms of adverse events of concern.
Dr. Cripe expressed his concerns that Novartis’ proposal to initially limit the mitigation plan to 30 or 35 treatment sites would create problems of access and economic disparities among patients, and could cause inequities among treatment centers even with the same city.
David Lebwohl, MD, head of the CAR T global program for Novartis, said that the planned number of sites for the mitigation program would be expanded after 6 to 12 months if the CAR T construct receives final approval and clinical implementation goes well.
There was nearly unanimous agreement among the panel members that the planned 15-year follow-up and other mitigation measures would be adequate for detecting serious short- and long-term consequences of CAR T-cell therapy.
Patient/advocate perspective
In the public comment section of the proceedings, panel members were urged to vote in favor of CTL019 by parents of children with ALL, including Don McMahon, whose son Connor received the therapy after multiple relapses, and Tom Whitehead, father of Emily Whitehead, the first patient to receive CAR T cells for ALL.
Both children are alive and doing well.
CTL019 is produced by Novartis.
The answer to the billion dollar question – Does the chimeric antigen receptor T-cell (CAR T) construct CTL019 (tisagenlecleucel-T) have a favorable risk-benefit profile for the treatment of children and young adults with relapsed/refractory B-cell precursor acute lymphoblastic leukemia? – was a unanimous “yes” at a July 12 meeting of the Food and Drug Administration’s Oncologic Drugs Advisory Committee.
“This is the most exciting thing I have seen in my lifetime, and probably since the introduction of ‘multiagent total cancer care,’ as it was called then, for treatment of childhood leukemia,” remarked Timothy P. Cripe, MD, PhD, from Nationwide Children’s Hospital in Columbus, Ohio, and a temporary voting member of the ODAC.
Catherine M. Bollard, MD, MBChB, from the Children’s National Medical Center in Washington, also a temporary ODAC member, said that she voted “yes” because “this is a very poor-risk patient population, this is an unmet need in the pediatric population, and as you saw in the data [presented to ODAC] today, the clinical responses are remarkable. I think Novartis [the maker of CTL019] has done a great job putting together a plan for mitigating risk going forward.”
CTL019 was shown in a pivotal phase 2 clinical trial to induce an overall remission rate of 83% in children and young adults with relapsed/refractory ALL for whom at least two prior lines of therapy had failed. Based on these results, the FDA accepted a biologics license application for the agent from Novartis.
At the meeting, panel members initially seemed favorably disposed toward recommending approval but heard concerns from FDA scientists about the potential for severe or fatal adverse events such as the cytokine release syndrome (CRS); the possible generation of replication competent retrovirus (RCR); and the potential for secondary malignancies from insertional mutagenesis, the incorporation of portions of the lentiviral vector into the patient’s genome.
In his opening remarks, Wilson W. Bryan, MD, from the FDA’s Office of Tissue and Advanced Therapies and Center for Biologics Evaluation and Drug Research, commented that “the clinical development of tisagenlecleucel suggests that this is a life-saving product.”
He went on, however, to frame the FDA’s concerns: “Clinical trials are not always a good predictor of the effectiveness and safety of a marketed product,” he said. “In particular, we are concerned that the same benefit and safety seen in clinical trials may not carry over to routine clinical use.”
The purpose of the hearing was to focus on manufacturing issues related to product quality, including replicability of the product for commercial use and safety issues such as prevention of CRS and neurotoxicities.
“We are also concerned about the hypothetical risk of secondary malignancies. Therefore, we are asking for the committee’s recommendations regarding the nature and duration of follow-up of patients who would receive this product,” Dr. Bryan said.
“CTL019 is a living drug, which demonstrates activity after a single infusion,” said Samit Hirawat, MD, head of oncology global development for Novartis.
But the nature of CTL019 as a living drug also means that it is subject to variations in the ability of autologous T cells harvested via leukapheresis to be infected with the lentiviral vector and expanded into a population of CAR T cells large enough to have therapeutic value, said Xiaobin Victor Lu, PhD, a chemistry, manufacturing, and controls reviewer for the FDA.
Mitigation plan
Novartis’ proposed plan includes specific, long-term steps for mitigating the risk of CRS and neurologic events, such as cerebral edema, the latter of which caused the FDA to call for a clinical hold of the phase 2 ROCKET trial for a different CAR T-cell construct.
Among the proposed elements of the mitigation plan are a 15-year minimum pharmacovigilance program and long-term safety follow-up for adverse events related to the therapy, efficacy, immunogenicity, transgene persistence of CD19 CAR, and the incidence of second malignancies possibly related to insertional mutagenesis.
Novartis also will train treatment center staff on processes for cell collection, cryopreservation, transport, chain of identity, safety management, and logistics for handling the CAR T-cell product. The company proposes to provide on-site training of personnel on CRS and neurotoxicity risk and management, as well as to offer information to patients and caregivers about the signs and symptoms of adverse events of concern.
Dr. Cripe expressed his concerns that Novartis’ proposal to initially limit the mitigation plan to 30 or 35 treatment sites would create problems of access and economic disparities among patients, and could cause inequities among treatment centers even with the same city.
David Lebwohl, MD, head of the CAR T global program for Novartis, said that the planned number of sites for the mitigation program would be expanded after 6 to 12 months if the CAR T construct receives final approval and clinical implementation goes well.
There was nearly unanimous agreement among the panel members that the planned 15-year follow-up and other mitigation measures would be adequate for detecting serious short- and long-term consequences of CAR T-cell therapy.
Patient/advocate perspective
In the public comment section of the proceedings, panel members were urged to vote in favor of CTL019 by parents of children with ALL, including Don McMahon, whose son Connor received the therapy after multiple relapses, and Tom Whitehead, father of Emily Whitehead, the first patient to receive CAR T cells for ALL.
Both children are alive and doing well.
CTL019 is produced by Novartis.
What works and what doesn’t in treating binge eating
SAN FRANCISCO – For patients who meet criteria for binge eating and who express a desire to lose weight, topiramate or lisdexamfetamine are effective treatment options, according to Judith Walsh, MD.
“Binge eating is the most common eating disorder in the United States,” Dr. Walsh said at the UCSF Annual Advances in Internal Medicine meeting. “The lifetime prevalence is 3.5% in women, compared with 2% in men, and 5%-30% of affected women are obese.”
According to the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5), the condition is characterized by binge eating episodes during which patients consume larger amounts of food than most people would under similar circumstances. “They feel a lack control over eating, [the notion that] ‘I’m eating and I can’t stop,’” said Dr. Walsh of the UCSF division of internal medicine and the Women’s Center for Excellence. Affected patients engage in the behavior of binge eating more than once per week over a period of at least 3 months, and the episodes typically last fewer than 2 hours.
While therapist-led cognitive-behavioral therapy (CBT) is generally considered to be the mainstay of treatment for binge eating disorder, guidelines from the American Psychiatric Association and the National Institute for Health and Care Excellence are conflicting, she said.
A recent meta-analysis funded by the Agency for Healthcare Research and Quality set out to summarize the benefits and harms of psychological and pharmacologic therapies for adults with binge eating disorder (Ann Intern Med. 2016 Sep 20;165[6]:409-20). The researchers evaluated 34 published trials with a low to medium risk of bias, including 25 placebo-controlled trials and 9 waitlist-controlled psychological trials. The majority of trial participants (77%) were women, their mean body mass index was 28.8 kg/m2, and their treatment duration ranged from 6 weeks to 6 months.
The review found that second-generation antidepressants such as citalopram, escitalopram, fluoxetine, fluvoxamine, paroxetine, and sertraline improved abstinence from binge eating (relative risk, 1.67) and depression symptoms (RR, 1.97), but resulted in no weight change. Therapist-led CBT was found to strongly improve abstinence from binge eating (RR, 4.95), but it resulted in no change in depressive symptoms or in weight.
Use of lisdexamfetamine was associated with improved abstinence from binge eating (RR, 2.61) and weight loss of 5.2%-6.3%, while use of topiramate was found in two trials to have a moderate benefit on abstinence from binge eating (by 58%-64% in one trial and by 29%-30% in the other), and weight loss of about 4.9 kg.
“Harms of treatment are generally not reported in the psychological studies, nor in 20 of the 25 pharmacologic studies included in the analysis,” said Dr. Walsh, who was not involved with review. The three trials of lisdexamfetamine found an increased risk of sympathetic nervous system arousal (RR, 4.28), insomnia (RR, 2.8), and GI upset (RR, 2.71).
“So, in adults with binge eating disorder, the primary therapy to increase abstinence is cognitive-behavioral therapy, topiramate, lisdexamfetamine, and second-generation antidepressants,” Dr. Walsh concluded. If reducing weight is a priority, the best available treatment options are topiramate and lisdexamfetamine, she said.
Dr. Walsh reported having no financial disclosures.
SAN FRANCISCO – For patients who meet criteria for binge eating and who express a desire to lose weight, topiramate or lisdexamfetamine are effective treatment options, according to Judith Walsh, MD.
“Binge eating is the most common eating disorder in the United States,” Dr. Walsh said at the UCSF Annual Advances in Internal Medicine meeting. “The lifetime prevalence is 3.5% in women, compared with 2% in men, and 5%-30% of affected women are obese.”
According to the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5), the condition is characterized by binge eating episodes during which patients consume larger amounts of food than most people would under similar circumstances. “They feel a lack control over eating, [the notion that] ‘I’m eating and I can’t stop,’” said Dr. Walsh of the UCSF division of internal medicine and the Women’s Center for Excellence. Affected patients engage in the behavior of binge eating more than once per week over a period of at least 3 months, and the episodes typically last fewer than 2 hours.
While therapist-led cognitive-behavioral therapy (CBT) is generally considered to be the mainstay of treatment for binge eating disorder, guidelines from the American Psychiatric Association and the National Institute for Health and Care Excellence are conflicting, she said.
A recent meta-analysis funded by the Agency for Healthcare Research and Quality set out to summarize the benefits and harms of psychological and pharmacologic therapies for adults with binge eating disorder (Ann Intern Med. 2016 Sep 20;165[6]:409-20). The researchers evaluated 34 published trials with a low to medium risk of bias, including 25 placebo-controlled trials and 9 waitlist-controlled psychological trials. The majority of trial participants (77%) were women, their mean body mass index was 28.8 kg/m2, and their treatment duration ranged from 6 weeks to 6 months.
The review found that second-generation antidepressants such as citalopram, escitalopram, fluoxetine, fluvoxamine, paroxetine, and sertraline improved abstinence from binge eating (relative risk, 1.67) and depression symptoms (RR, 1.97), but resulted in no weight change. Therapist-led CBT was found to strongly improve abstinence from binge eating (RR, 4.95), but it resulted in no change in depressive symptoms or in weight.
Use of lisdexamfetamine was associated with improved abstinence from binge eating (RR, 2.61) and weight loss of 5.2%-6.3%, while use of topiramate was found in two trials to have a moderate benefit on abstinence from binge eating (by 58%-64% in one trial and by 29%-30% in the other), and weight loss of about 4.9 kg.
“Harms of treatment are generally not reported in the psychological studies, nor in 20 of the 25 pharmacologic studies included in the analysis,” said Dr. Walsh, who was not involved with review. The three trials of lisdexamfetamine found an increased risk of sympathetic nervous system arousal (RR, 4.28), insomnia (RR, 2.8), and GI upset (RR, 2.71).
“So, in adults with binge eating disorder, the primary therapy to increase abstinence is cognitive-behavioral therapy, topiramate, lisdexamfetamine, and second-generation antidepressants,” Dr. Walsh concluded. If reducing weight is a priority, the best available treatment options are topiramate and lisdexamfetamine, she said.
Dr. Walsh reported having no financial disclosures.
SAN FRANCISCO – For patients who meet criteria for binge eating and who express a desire to lose weight, topiramate or lisdexamfetamine are effective treatment options, according to Judith Walsh, MD.
“Binge eating is the most common eating disorder in the United States,” Dr. Walsh said at the UCSF Annual Advances in Internal Medicine meeting. “The lifetime prevalence is 3.5% in women, compared with 2% in men, and 5%-30% of affected women are obese.”
According to the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5), the condition is characterized by binge eating episodes during which patients consume larger amounts of food than most people would under similar circumstances. “They feel a lack control over eating, [the notion that] ‘I’m eating and I can’t stop,’” said Dr. Walsh of the UCSF division of internal medicine and the Women’s Center for Excellence. Affected patients engage in the behavior of binge eating more than once per week over a period of at least 3 months, and the episodes typically last fewer than 2 hours.
While therapist-led cognitive-behavioral therapy (CBT) is generally considered to be the mainstay of treatment for binge eating disorder, guidelines from the American Psychiatric Association and the National Institute for Health and Care Excellence are conflicting, she said.
A recent meta-analysis funded by the Agency for Healthcare Research and Quality set out to summarize the benefits and harms of psychological and pharmacologic therapies for adults with binge eating disorder (Ann Intern Med. 2016 Sep 20;165[6]:409-20). The researchers evaluated 34 published trials with a low to medium risk of bias, including 25 placebo-controlled trials and 9 waitlist-controlled psychological trials. The majority of trial participants (77%) were women, their mean body mass index was 28.8 kg/m2, and their treatment duration ranged from 6 weeks to 6 months.
The review found that second-generation antidepressants such as citalopram, escitalopram, fluoxetine, fluvoxamine, paroxetine, and sertraline improved abstinence from binge eating (relative risk, 1.67) and depression symptoms (RR, 1.97), but resulted in no weight change. Therapist-led CBT was found to strongly improve abstinence from binge eating (RR, 4.95), but it resulted in no change in depressive symptoms or in weight.
Use of lisdexamfetamine was associated with improved abstinence from binge eating (RR, 2.61) and weight loss of 5.2%-6.3%, while use of topiramate was found in two trials to have a moderate benefit on abstinence from binge eating (by 58%-64% in one trial and by 29%-30% in the other), and weight loss of about 4.9 kg.
“Harms of treatment are generally not reported in the psychological studies, nor in 20 of the 25 pharmacologic studies included in the analysis,” said Dr. Walsh, who was not involved with review. The three trials of lisdexamfetamine found an increased risk of sympathetic nervous system arousal (RR, 4.28), insomnia (RR, 2.8), and GI upset (RR, 2.71).
“So, in adults with binge eating disorder, the primary therapy to increase abstinence is cognitive-behavioral therapy, topiramate, lisdexamfetamine, and second-generation antidepressants,” Dr. Walsh concluded. If reducing weight is a priority, the best available treatment options are topiramate and lisdexamfetamine, she said.
Dr. Walsh reported having no financial disclosures.
EXPERT ANALYSIS AT THE ANNUAL ADVANCES IN INTERNAL MEDICINE
Improving diet over time reduces mortality
Improving diet over time consistently reduces all-cause and cardiovascular mortality, according to an analysis of two large U.S. databases.
Researchers examined the association between dietary change over a 12-year period (1986-1998) and subsequent mortality during a further 12 years of follow-up (1998-2010), using data for 47,994 women participating in the Nurses’ Health Study and 25,745 men participating in the Health Professionals Follow-Up Study. They calculated dietary quality using three different methods: the Alternate Healthy Eating Index-2010 score, the Alternate Mediterranean Diet score, and the Dietary Approaches to Stop Hypertension (DASH) diet score.
Through numerous analyses of the data, they found a consistent inverse relationship between dietary quality and mortality. Overall, a 20-percentile increase in dietary quality over time was associated with an 8%-17% reduction in all-cause mortality, regardless of which scoring method was used. In contrast, a similar decline in dietary quality was associated with a 6%-12% increase in mortality.
“The pooled hazard ratios among participants with the greatest improvement in diet quality (13%-33% improvement), as compared with those whose diet quality remained relatively stable (0%-3% improvement) in the 12-year period, were the following: 0.91 according to changes in the Alternate Healthy Eating Index score, 0.84 according to changes in the Alternate Mediterranean Diet score, and 0.89 according to changes in the DASH score,” the investigators said (N Engl J Med. 2017 Jul 13. doi: 10.1056/NEJMoa1613502).
In addition, participants who maintained a high-quality diet throughout the study period also reduced their risk of death from any cause by 9%-14%, compared with those who maintained a poor-quality diet over time.
“Our findings provide support for the recommendation of the 2015 Dietary Guidelines Advisory Committee that it is not necessary to conform to a single diet plan to achieve healthy eating patterns. These three dietary patterns, although different in description and composition, capture the essential elements of a healthy diet. Common food groups in each score that contributed the most to improvements were whole grains, vegetables, fruits, and fish or n-3 fatty acids,” Dr. Sotos-Prieto and her associates noted.
Improving diet over time consistently reduces all-cause and cardiovascular mortality, according to an analysis of two large U.S. databases.
Researchers examined the association between dietary change over a 12-year period (1986-1998) and subsequent mortality during a further 12 years of follow-up (1998-2010), using data for 47,994 women participating in the Nurses’ Health Study and 25,745 men participating in the Health Professionals Follow-Up Study. They calculated dietary quality using three different methods: the Alternate Healthy Eating Index-2010 score, the Alternate Mediterranean Diet score, and the Dietary Approaches to Stop Hypertension (DASH) diet score.
Through numerous analyses of the data, they found a consistent inverse relationship between dietary quality and mortality. Overall, a 20-percentile increase in dietary quality over time was associated with an 8%-17% reduction in all-cause mortality, regardless of which scoring method was used. In contrast, a similar decline in dietary quality was associated with a 6%-12% increase in mortality.
“The pooled hazard ratios among participants with the greatest improvement in diet quality (13%-33% improvement), as compared with those whose diet quality remained relatively stable (0%-3% improvement) in the 12-year period, were the following: 0.91 according to changes in the Alternate Healthy Eating Index score, 0.84 according to changes in the Alternate Mediterranean Diet score, and 0.89 according to changes in the DASH score,” the investigators said (N Engl J Med. 2017 Jul 13. doi: 10.1056/NEJMoa1613502).
In addition, participants who maintained a high-quality diet throughout the study period also reduced their risk of death from any cause by 9%-14%, compared with those who maintained a poor-quality diet over time.
“Our findings provide support for the recommendation of the 2015 Dietary Guidelines Advisory Committee that it is not necessary to conform to a single diet plan to achieve healthy eating patterns. These three dietary patterns, although different in description and composition, capture the essential elements of a healthy diet. Common food groups in each score that contributed the most to improvements were whole grains, vegetables, fruits, and fish or n-3 fatty acids,” Dr. Sotos-Prieto and her associates noted.
Improving diet over time consistently reduces all-cause and cardiovascular mortality, according to an analysis of two large U.S. databases.
Researchers examined the association between dietary change over a 12-year period (1986-1998) and subsequent mortality during a further 12 years of follow-up (1998-2010), using data for 47,994 women participating in the Nurses’ Health Study and 25,745 men participating in the Health Professionals Follow-Up Study. They calculated dietary quality using three different methods: the Alternate Healthy Eating Index-2010 score, the Alternate Mediterranean Diet score, and the Dietary Approaches to Stop Hypertension (DASH) diet score.
Through numerous analyses of the data, they found a consistent inverse relationship between dietary quality and mortality. Overall, a 20-percentile increase in dietary quality over time was associated with an 8%-17% reduction in all-cause mortality, regardless of which scoring method was used. In contrast, a similar decline in dietary quality was associated with a 6%-12% increase in mortality.
“The pooled hazard ratios among participants with the greatest improvement in diet quality (13%-33% improvement), as compared with those whose diet quality remained relatively stable (0%-3% improvement) in the 12-year period, were the following: 0.91 according to changes in the Alternate Healthy Eating Index score, 0.84 according to changes in the Alternate Mediterranean Diet score, and 0.89 according to changes in the DASH score,” the investigators said (N Engl J Med. 2017 Jul 13. doi: 10.1056/NEJMoa1613502).
In addition, participants who maintained a high-quality diet throughout the study period also reduced their risk of death from any cause by 9%-14%, compared with those who maintained a poor-quality diet over time.
“Our findings provide support for the recommendation of the 2015 Dietary Guidelines Advisory Committee that it is not necessary to conform to a single diet plan to achieve healthy eating patterns. These three dietary patterns, although different in description and composition, capture the essential elements of a healthy diet. Common food groups in each score that contributed the most to improvements were whole grains, vegetables, fruits, and fish or n-3 fatty acids,” Dr. Sotos-Prieto and her associates noted.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: Improving one’s diet over time consistently reduces all-cause and cardiovascular mortality.
Major finding: A 20-percentile increase in dietary quality over time was associated with an 8%-17% reduction in all-cause mortality, while a similar decline in dietary quality was associated with a 6%-12% increase in mortality.
Data source: A secondary analysis of data for 47,994 women participating in the Nurses’ Health Study and 25,745 men in the Health Professionals Follow-up Study.
Disclosures: This study was funded by the National Institutes of Health. Dr. Sotos-Prieto reported having no relevant financial disclosures. One of her associates reported ties to Metagenics and the California Walnut Commission.
Temporal Triangular Alopecia Acquired in Adulthood
To the Editor:
Temporal triangular alopecia (TTA), a condition first described by Sabouraud1 in 1905, is a circumscribed nonscarring form of alopecia. Also referred to as congenital triangular alopecia, TTA presents as a triangular or lancet-shaped area of hair loss involving the frontotemporal hairline. Temporal triangular alopecia is characterized histologically by a normal number of miniaturized hair follicles without notable inflammation.2 Although the majority of cases arise between birth and 9 years of age,3,4 rare cases of adult-onset TTA also have been reported.5,6 Adult-onset cases can cause notable diagnostic confusion and inappropriate treatment, as reported in our patient.
A 25-year-old woman with a history of Hashimoto thyroiditis presented with hair loss affecting the right temporal scalp of 3 years' duration that was first noticed by her husband. The lesion was an asymptomatic, 6×8-cm, roughly lancet-shaped patch of alopecia located on the right temporal scalp, bordering on the frontal hairline (Figure 1). Centrally, the patch appeared almost hairless with a few retained terminal hairs. The frontal hairline was thinned but still present. There was no scaling or erythema, and fine vellus hairs and a few isolated terminal hairs covered the area. The corresponding skin on the contralateral temporal scalp showed normal hair density. The patient insisted that she had normal hair at the affected area until 22 years of age, and she denied a history of trauma or tight hairstyles. Initially diagnosed with alopecia areata by her primary care provider, the patient was treated with topical corticosteroids for 6 months without benefit. She was subsequently referred to a dermatologist who again offered a diagnosis of alopecia areata and treated the lesions with 2 intralesional corticosteroid injections without benefit. No biopsies of the affected area were performed, and the patient was given a trial of topical minoxidil.
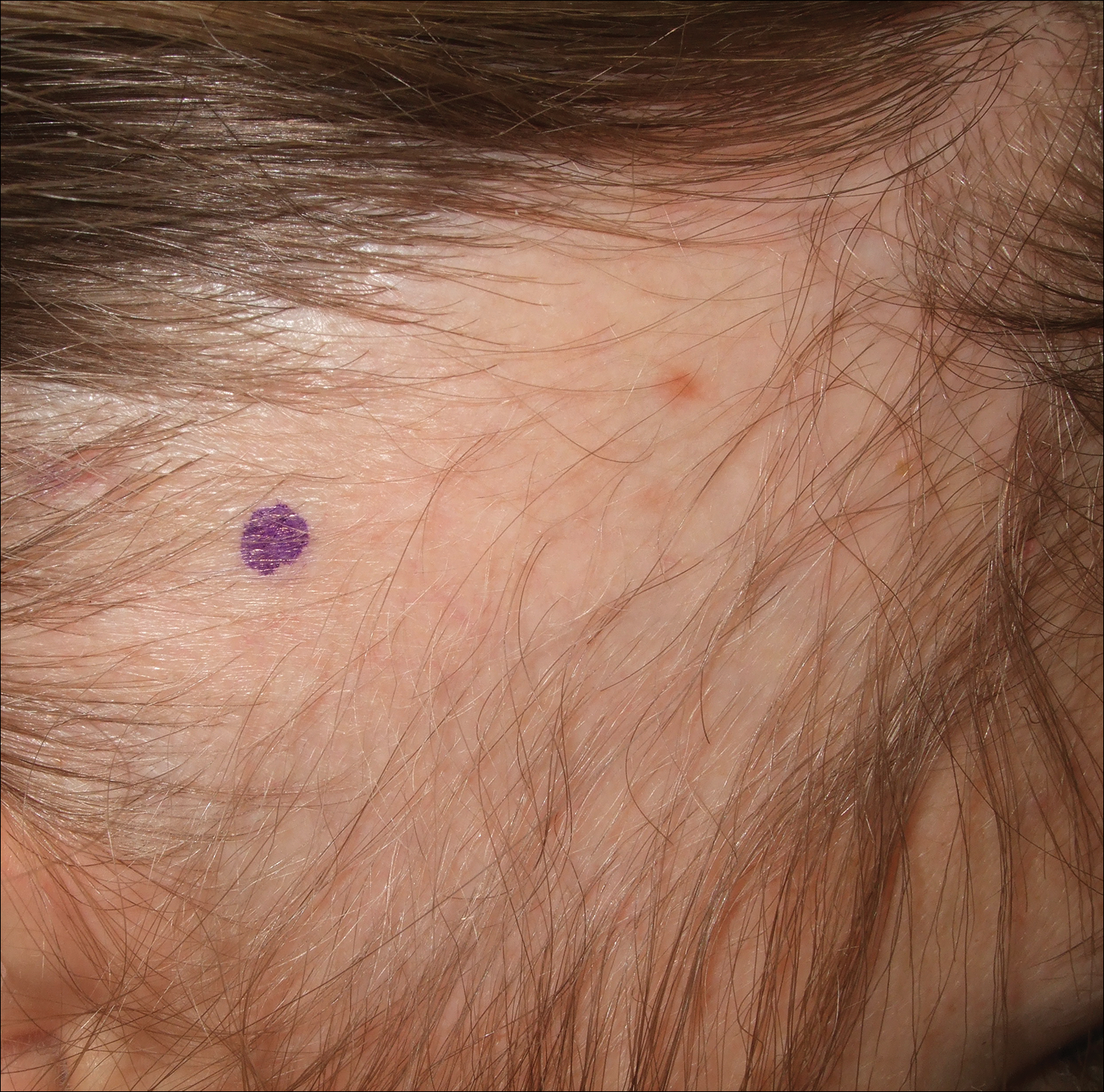
The patient consulted a new primary care provider and was diagnosed with scarring alopecia. She was referred to our dermatology department for further treatment. An initial biopsy at the edge of the affected area was interpreted as normal, but after failing additional intralesional corticosteroid injections, she was referred to our hair clinic where another biopsy was performed in the central portion of the lesion. A 4-mm diameter punch biopsy specimen revealed a normal epidermis and dermis; however, in the lower dermis only a single terminal follicle was seen (Figure 2). Sections through the upper dermis (Figure 3) showed that the total number of hairs was normal or nearly normal with at least 22 follicles, but most were vellus and indeterminate hairs with only a single terminal hair. The dermal architecture was otherwise normal. Given the clinical and histologic findings, a diagnosis of TTA was made. Subsequent to the diagnosis, the patient did not pursue any additional treatment options and preferred to style her hair so that the area of TTA remained covered.
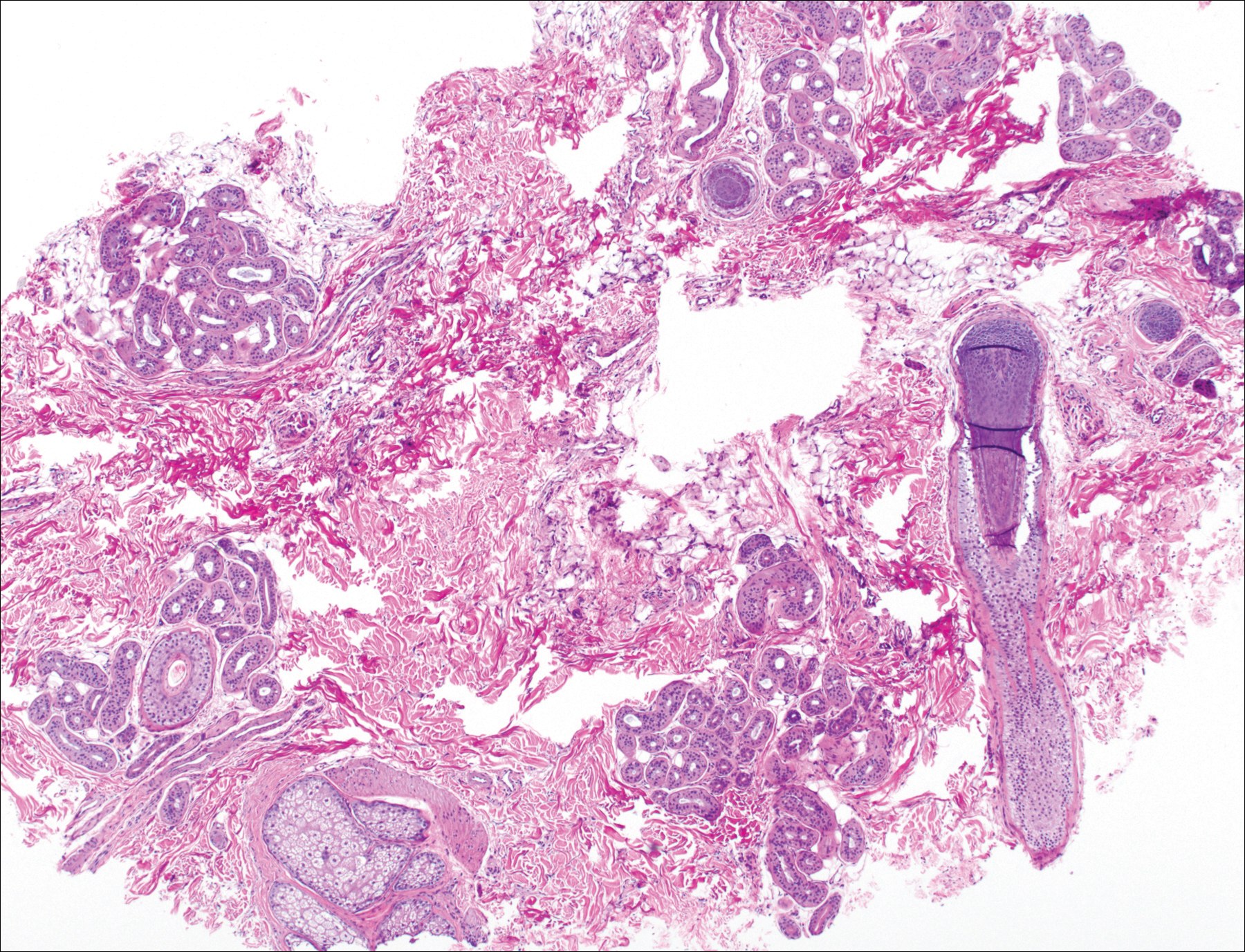
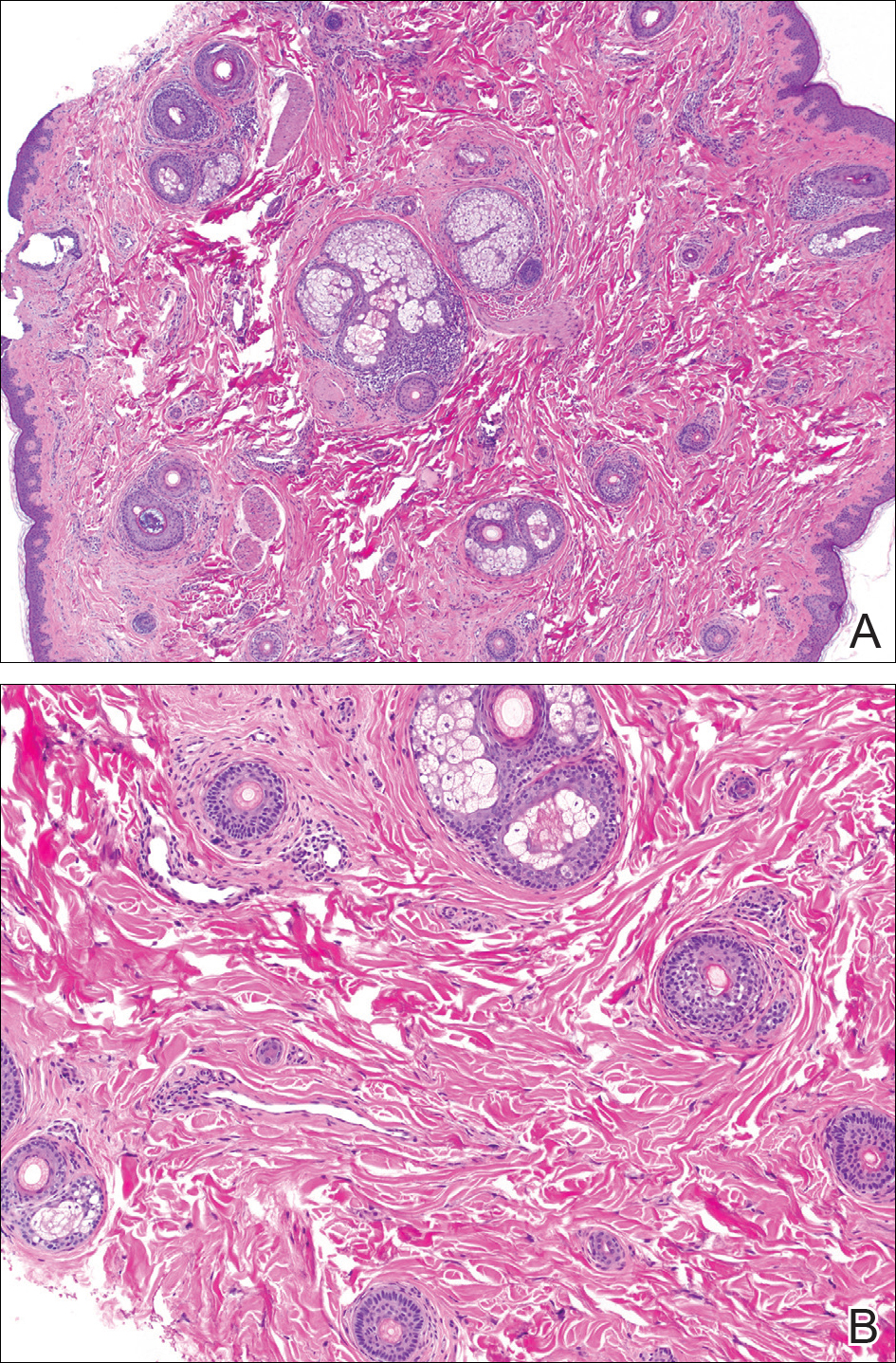
The differential diagnosis in adults presenting with a patch of localized alopecia includes alopecia areata, trichotillomania, pressure-induced alopecia, traction alopecia, lichen planopilaris, discoid lupus erythematosus, and rarely TTA. Temporal triangular alopecia is a fairly common, if underreported, nonscarring form of alopecia that mainly affects young children. A PubMed search of articles indexed for MEDLINE using the terms temporal triangular alopecia or congenital triangular alopecia or triangular alopecia documented only 76 cases of TTA including our own, with the majority of patients diagnosed before 9 years of age. Only 2 cases of adult-onset TTA have been reported,5,6 possibly leading to misdiagnosis of adult patients who present with similar areas of hair loss. As with some prior cases of TTA,5,7 our patient was misdiagnosed with alopecia areata and scarring alopecia, both treated unsuccessfully before a diagnosis of TTA was considered. Clues to the diagnosis included the location, the lack of change in size and shape, the lack of response to intralesional corticosteroids, and the presence of numerous vellus hairs on the surface. A biopsy of the visibly hairless zone was confirmatory. The normal or nearly normal number of miniaturized hairs in specimens of TTA suggest that topical minoxidil therapy (eg, 5% solution twice daily for at least 6 months) might be useful, but the authors have tried it on a few other patients with clinically typical TTA without discernible benefit. When lesions are small, excision provides a fast and permanent solution to the problem, albeit with the usual risks of minor surgery.
- Sabouraud RJA. Manuel Élémentaire de Dermatologie Topographique Régionale. Paris, France: Masson & Cie; 1905:197.
- Trakimas C, Sperling LC, Skelton HG 3rd, et al. Clinical and histologic findings in temporal triangular alopecia. J Am Acad Dermatol. 1994;31:205-209.
- Yamazaki M, Irisawa R, Tsuboi R. Temporal triangular alopecia and a review of 52 past cases. J Dermatol. 2010;37:360-362.
- Sarifakioglu E, Yilmaz AE, Gorpelioglu C, et al. Prevalence of scalp disorders and hair loss in children. Cutis. 2012;90:225-229.
- Trakimas CA, Sperling LC. Temporal triangular alopecia acquired in adulthood. J Am Acad Dermatol. 1999;40:842-844.
- Akan IM, Yildirim S, Avci G, et al. Bilateral temporal triangular alopecia acquired in adulthood. Plast Reconstr Surg. 2001;107:1616-1617.
- Gupta LK, Khare AK, Garg A, et al. Congenital triangular alopecia--a close mimicker of alopecia areata. Int J Trichology. 2011;3:40-41.
To the Editor:
Temporal triangular alopecia (TTA), a condition first described by Sabouraud1 in 1905, is a circumscribed nonscarring form of alopecia. Also referred to as congenital triangular alopecia, TTA presents as a triangular or lancet-shaped area of hair loss involving the frontotemporal hairline. Temporal triangular alopecia is characterized histologically by a normal number of miniaturized hair follicles without notable inflammation.2 Although the majority of cases arise between birth and 9 years of age,3,4 rare cases of adult-onset TTA also have been reported.5,6 Adult-onset cases can cause notable diagnostic confusion and inappropriate treatment, as reported in our patient.
A 25-year-old woman with a history of Hashimoto thyroiditis presented with hair loss affecting the right temporal scalp of 3 years' duration that was first noticed by her husband. The lesion was an asymptomatic, 6×8-cm, roughly lancet-shaped patch of alopecia located on the right temporal scalp, bordering on the frontal hairline (Figure 1). Centrally, the patch appeared almost hairless with a few retained terminal hairs. The frontal hairline was thinned but still present. There was no scaling or erythema, and fine vellus hairs and a few isolated terminal hairs covered the area. The corresponding skin on the contralateral temporal scalp showed normal hair density. The patient insisted that she had normal hair at the affected area until 22 years of age, and she denied a history of trauma or tight hairstyles. Initially diagnosed with alopecia areata by her primary care provider, the patient was treated with topical corticosteroids for 6 months without benefit. She was subsequently referred to a dermatologist who again offered a diagnosis of alopecia areata and treated the lesions with 2 intralesional corticosteroid injections without benefit. No biopsies of the affected area were performed, and the patient was given a trial of topical minoxidil.
The patient consulted a new primary care provider and was diagnosed with scarring alopecia. She was referred to our dermatology department for further treatment. An initial biopsy at the edge of the affected area was interpreted as normal, but after failing additional intralesional corticosteroid injections, she was referred to our hair clinic where another biopsy was performed in the central portion of the lesion. A 4-mm diameter punch biopsy specimen revealed a normal epidermis and dermis; however, in the lower dermis only a single terminal follicle was seen (Figure 2). Sections through the upper dermis (Figure 3) showed that the total number of hairs was normal or nearly normal with at least 22 follicles, but most were vellus and indeterminate hairs with only a single terminal hair. The dermal architecture was otherwise normal. Given the clinical and histologic findings, a diagnosis of TTA was made. Subsequent to the diagnosis, the patient did not pursue any additional treatment options and preferred to style her hair so that the area of TTA remained covered.
The differential diagnosis in adults presenting with a patch of localized alopecia includes alopecia areata, trichotillomania, pressure-induced alopecia, traction alopecia, lichen planopilaris, discoid lupus erythematosus, and rarely TTA. Temporal triangular alopecia is a fairly common, if underreported, nonscarring form of alopecia that mainly affects young children. A PubMed search of articles indexed for MEDLINE using the terms temporal triangular alopecia or congenital triangular alopecia or triangular alopecia documented only 76 cases of TTA including our own, with the majority of patients diagnosed before 9 years of age. Only 2 cases of adult-onset TTA have been reported,5,6 possibly leading to misdiagnosis of adult patients who present with similar areas of hair loss. As with some prior cases of TTA,5,7 our patient was misdiagnosed with alopecia areata and scarring alopecia, both treated unsuccessfully before a diagnosis of TTA was considered. Clues to the diagnosis included the location, the lack of change in size and shape, the lack of response to intralesional corticosteroids, and the presence of numerous vellus hairs on the surface. A biopsy of the visibly hairless zone was confirmatory. The normal or nearly normal number of miniaturized hairs in specimens of TTA suggest that topical minoxidil therapy (eg, 5% solution twice daily for at least 6 months) might be useful, but the authors have tried it on a few other patients with clinically typical TTA without discernible benefit. When lesions are small, excision provides a fast and permanent solution to the problem, albeit with the usual risks of minor surgery.
To the Editor:
Temporal triangular alopecia (TTA), a condition first described by Sabouraud1 in 1905, is a circumscribed nonscarring form of alopecia. Also referred to as congenital triangular alopecia, TTA presents as a triangular or lancet-shaped area of hair loss involving the frontotemporal hairline. Temporal triangular alopecia is characterized histologically by a normal number of miniaturized hair follicles without notable inflammation.2 Although the majority of cases arise between birth and 9 years of age,3,4 rare cases of adult-onset TTA also have been reported.5,6 Adult-onset cases can cause notable diagnostic confusion and inappropriate treatment, as reported in our patient.
A 25-year-old woman with a history of Hashimoto thyroiditis presented with hair loss affecting the right temporal scalp of 3 years' duration that was first noticed by her husband. The lesion was an asymptomatic, 6×8-cm, roughly lancet-shaped patch of alopecia located on the right temporal scalp, bordering on the frontal hairline (Figure 1). Centrally, the patch appeared almost hairless with a few retained terminal hairs. The frontal hairline was thinned but still present. There was no scaling or erythema, and fine vellus hairs and a few isolated terminal hairs covered the area. The corresponding skin on the contralateral temporal scalp showed normal hair density. The patient insisted that she had normal hair at the affected area until 22 years of age, and she denied a history of trauma or tight hairstyles. Initially diagnosed with alopecia areata by her primary care provider, the patient was treated with topical corticosteroids for 6 months without benefit. She was subsequently referred to a dermatologist who again offered a diagnosis of alopecia areata and treated the lesions with 2 intralesional corticosteroid injections without benefit. No biopsies of the affected area were performed, and the patient was given a trial of topical minoxidil.
The patient consulted a new primary care provider and was diagnosed with scarring alopecia. She was referred to our dermatology department for further treatment. An initial biopsy at the edge of the affected area was interpreted as normal, but after failing additional intralesional corticosteroid injections, she was referred to our hair clinic where another biopsy was performed in the central portion of the lesion. A 4-mm diameter punch biopsy specimen revealed a normal epidermis and dermis; however, in the lower dermis only a single terminal follicle was seen (Figure 2). Sections through the upper dermis (Figure 3) showed that the total number of hairs was normal or nearly normal with at least 22 follicles, but most were vellus and indeterminate hairs with only a single terminal hair. The dermal architecture was otherwise normal. Given the clinical and histologic findings, a diagnosis of TTA was made. Subsequent to the diagnosis, the patient did not pursue any additional treatment options and preferred to style her hair so that the area of TTA remained covered.
The differential diagnosis in adults presenting with a patch of localized alopecia includes alopecia areata, trichotillomania, pressure-induced alopecia, traction alopecia, lichen planopilaris, discoid lupus erythematosus, and rarely TTA. Temporal triangular alopecia is a fairly common, if underreported, nonscarring form of alopecia that mainly affects young children. A PubMed search of articles indexed for MEDLINE using the terms temporal triangular alopecia or congenital triangular alopecia or triangular alopecia documented only 76 cases of TTA including our own, with the majority of patients diagnosed before 9 years of age. Only 2 cases of adult-onset TTA have been reported,5,6 possibly leading to misdiagnosis of adult patients who present with similar areas of hair loss. As with some prior cases of TTA,5,7 our patient was misdiagnosed with alopecia areata and scarring alopecia, both treated unsuccessfully before a diagnosis of TTA was considered. Clues to the diagnosis included the location, the lack of change in size and shape, the lack of response to intralesional corticosteroids, and the presence of numerous vellus hairs on the surface. A biopsy of the visibly hairless zone was confirmatory. The normal or nearly normal number of miniaturized hairs in specimens of TTA suggest that topical minoxidil therapy (eg, 5% solution twice daily for at least 6 months) might be useful, but the authors have tried it on a few other patients with clinically typical TTA without discernible benefit. When lesions are small, excision provides a fast and permanent solution to the problem, albeit with the usual risks of minor surgery.
- Sabouraud RJA. Manuel Élémentaire de Dermatologie Topographique Régionale. Paris, France: Masson & Cie; 1905:197.
- Trakimas C, Sperling LC, Skelton HG 3rd, et al. Clinical and histologic findings in temporal triangular alopecia. J Am Acad Dermatol. 1994;31:205-209.
- Yamazaki M, Irisawa R, Tsuboi R. Temporal triangular alopecia and a review of 52 past cases. J Dermatol. 2010;37:360-362.
- Sarifakioglu E, Yilmaz AE, Gorpelioglu C, et al. Prevalence of scalp disorders and hair loss in children. Cutis. 2012;90:225-229.
- Trakimas CA, Sperling LC. Temporal triangular alopecia acquired in adulthood. J Am Acad Dermatol. 1999;40:842-844.
- Akan IM, Yildirim S, Avci G, et al. Bilateral temporal triangular alopecia acquired in adulthood. Plast Reconstr Surg. 2001;107:1616-1617.
- Gupta LK, Khare AK, Garg A, et al. Congenital triangular alopecia--a close mimicker of alopecia areata. Int J Trichology. 2011;3:40-41.
- Sabouraud RJA. Manuel Élémentaire de Dermatologie Topographique Régionale. Paris, France: Masson & Cie; 1905:197.
- Trakimas C, Sperling LC, Skelton HG 3rd, et al. Clinical and histologic findings in temporal triangular alopecia. J Am Acad Dermatol. 1994;31:205-209.
- Yamazaki M, Irisawa R, Tsuboi R. Temporal triangular alopecia and a review of 52 past cases. J Dermatol. 2010;37:360-362.
- Sarifakioglu E, Yilmaz AE, Gorpelioglu C, et al. Prevalence of scalp disorders and hair loss in children. Cutis. 2012;90:225-229.
- Trakimas CA, Sperling LC. Temporal triangular alopecia acquired in adulthood. J Am Acad Dermatol. 1999;40:842-844.
- Akan IM, Yildirim S, Avci G, et al. Bilateral temporal triangular alopecia acquired in adulthood. Plast Reconstr Surg. 2001;107:1616-1617.
- Gupta LK, Khare AK, Garg A, et al. Congenital triangular alopecia--a close mimicker of alopecia areata. Int J Trichology. 2011;3:40-41.
Practice Points
- Temporal triangular alopecia (TTA) in adults often is confused with alopecia areata.
- An acquired, persistent, unchanging, circumscribed hairless spot in an adult that does not respond to intralesional corticosteroids may represent TTA.
- Hair miniaturization without peribulbar inflammation is consistent with a diagnosis of TTA.
The burden of health care–associated C. difficile infection in a nonmetropolitan setting
Clostridium difficile infection (CDI) remains a major cause of health care–associated diarrhea in industrialized countries and is a common target of antimicrobial stewardship programs (ASPs).
While the burden of CDI has been well described in tertiary metropolitan hospitals, there is a lack of published evidence from regional and rural hospitals. Our recent study published in the Journal of Hospital Infection explores the effect of an ASP on health care–associated CDI rates and the impact of CDI on length of stay and hospital costs.

The ASP functioned alongside infection control practices, including isolation of patients with antimicrobial-resistant organisms (including C. difficile), hand hygiene, personal protective equipment, and terminal cleaning. Timely feedback emails to medical officers contained information on patient-specific risk factors for CDI, current and prior antimicrobials, and suggestions for CDI treatment. The effect of health care–associated CDI on length of stay and hospital costs was investigated using a group of matched controls, identified retrospectively using hospital performance data. Prior antimicrobial and proton pump inhibitor use were also measured and compared with background use.
The results of our study demonstrated a stable health care–associated CDI rate of around four cases per 10,000 occupied bed days over a 5-year period, similar to the average Australian rate. The length of time over which CDI rates could be effectively examined prior to the intervention was limited by changes to C. difficile stool testing methods. Median length of stay was 11 days greater, and median hospital costs were AU$11,361 higher for patients with health care–associated CDI (n = 91) than for their matched controls (n = 172). It is likely that the increase in costs was associated with additional length of stay but also with increased investigation and treatment costs. Among the group of patients with severe disease (n = 8), only four received oral vancomycin according to Australian guidelines, possibly because of under-recognition of severity criteria. The response rate to emails was low at 19%, showing that other methods are additionally necessary to communicate CDI case feedback.
Third generation cephalosporins and beta-lactamase inhibitor combinations were over-represented in the health care–associated CDI group, where narrower spectrum antimicrobials such as beta-lactamase sensitive penicillins were under represented. Rates of prior antimicrobial use and proton pump inhibitor use were broadly in agreement with the literature.
Our study demonstrated that, in the Australian nonmetropolitan setting, there was a high burden of health care–associated CDI in terms of hospital costs and length of stay, even though our health district experienced CDI rates that were similar to the Australian average. Challenges associated with the study included maintenance of consistent data collection across multiple hospital sites without comprehensive electronic medical records, provision of timely email feedback, and dissemination of study results.
Analysis of prior antimicrobial use has allowed us to identify targets for ongoing antimicrobial stewardship activities, and we also intend to provide further education on recognition of severity criteria. These activities can be supported through daily antimicrobial stewardship ward rounds. Future research could involve application of appropriateness criteria to prior and current antimicrobial use in CDI patients in order to identify avoidable cases and use of more advanced statistical techniques such as multistate modeling to determine differences in outcomes between cases and controls.
Stuart Bond, BPharm, DipPharmPrac, is an antimicrobial stewardship pharmacist based at Wollongong Hospital in New South Wales, Australia.
Clostridium difficile infection (CDI) remains a major cause of health care–associated diarrhea in industrialized countries and is a common target of antimicrobial stewardship programs (ASPs).
While the burden of CDI has been well described in tertiary metropolitan hospitals, there is a lack of published evidence from regional and rural hospitals. Our recent study published in the Journal of Hospital Infection explores the effect of an ASP on health care–associated CDI rates and the impact of CDI on length of stay and hospital costs.
The ASP functioned alongside infection control practices, including isolation of patients with antimicrobial-resistant organisms (including C. difficile), hand hygiene, personal protective equipment, and terminal cleaning. Timely feedback emails to medical officers contained information on patient-specific risk factors for CDI, current and prior antimicrobials, and suggestions for CDI treatment. The effect of health care–associated CDI on length of stay and hospital costs was investigated using a group of matched controls, identified retrospectively using hospital performance data. Prior antimicrobial and proton pump inhibitor use were also measured and compared with background use.
The results of our study demonstrated a stable health care–associated CDI rate of around four cases per 10,000 occupied bed days over a 5-year period, similar to the average Australian rate. The length of time over which CDI rates could be effectively examined prior to the intervention was limited by changes to C. difficile stool testing methods. Median length of stay was 11 days greater, and median hospital costs were AU$11,361 higher for patients with health care–associated CDI (n = 91) than for their matched controls (n = 172). It is likely that the increase in costs was associated with additional length of stay but also with increased investigation and treatment costs. Among the group of patients with severe disease (n = 8), only four received oral vancomycin according to Australian guidelines, possibly because of under-recognition of severity criteria. The response rate to emails was low at 19%, showing that other methods are additionally necessary to communicate CDI case feedback.
Third generation cephalosporins and beta-lactamase inhibitor combinations were over-represented in the health care–associated CDI group, where narrower spectrum antimicrobials such as beta-lactamase sensitive penicillins were under represented. Rates of prior antimicrobial use and proton pump inhibitor use were broadly in agreement with the literature.
Our study demonstrated that, in the Australian nonmetropolitan setting, there was a high burden of health care–associated CDI in terms of hospital costs and length of stay, even though our health district experienced CDI rates that were similar to the Australian average. Challenges associated with the study included maintenance of consistent data collection across multiple hospital sites without comprehensive electronic medical records, provision of timely email feedback, and dissemination of study results.
Analysis of prior antimicrobial use has allowed us to identify targets for ongoing antimicrobial stewardship activities, and we also intend to provide further education on recognition of severity criteria. These activities can be supported through daily antimicrobial stewardship ward rounds. Future research could involve application of appropriateness criteria to prior and current antimicrobial use in CDI patients in order to identify avoidable cases and use of more advanced statistical techniques such as multistate modeling to determine differences in outcomes between cases and controls.
Stuart Bond, BPharm, DipPharmPrac, is an antimicrobial stewardship pharmacist based at Wollongong Hospital in New South Wales, Australia.
Clostridium difficile infection (CDI) remains a major cause of health care–associated diarrhea in industrialized countries and is a common target of antimicrobial stewardship programs (ASPs).
While the burden of CDI has been well described in tertiary metropolitan hospitals, there is a lack of published evidence from regional and rural hospitals. Our recent study published in the Journal of Hospital Infection explores the effect of an ASP on health care–associated CDI rates and the impact of CDI on length of stay and hospital costs.
The ASP functioned alongside infection control practices, including isolation of patients with antimicrobial-resistant organisms (including C. difficile), hand hygiene, personal protective equipment, and terminal cleaning. Timely feedback emails to medical officers contained information on patient-specific risk factors for CDI, current and prior antimicrobials, and suggestions for CDI treatment. The effect of health care–associated CDI on length of stay and hospital costs was investigated using a group of matched controls, identified retrospectively using hospital performance data. Prior antimicrobial and proton pump inhibitor use were also measured and compared with background use.
The results of our study demonstrated a stable health care–associated CDI rate of around four cases per 10,000 occupied bed days over a 5-year period, similar to the average Australian rate. The length of time over which CDI rates could be effectively examined prior to the intervention was limited by changes to C. difficile stool testing methods. Median length of stay was 11 days greater, and median hospital costs were AU$11,361 higher for patients with health care–associated CDI (n = 91) than for their matched controls (n = 172). It is likely that the increase in costs was associated with additional length of stay but also with increased investigation and treatment costs. Among the group of patients with severe disease (n = 8), only four received oral vancomycin according to Australian guidelines, possibly because of under-recognition of severity criteria. The response rate to emails was low at 19%, showing that other methods are additionally necessary to communicate CDI case feedback.
Third generation cephalosporins and beta-lactamase inhibitor combinations were over-represented in the health care–associated CDI group, where narrower spectrum antimicrobials such as beta-lactamase sensitive penicillins were under represented. Rates of prior antimicrobial use and proton pump inhibitor use were broadly in agreement with the literature.
Our study demonstrated that, in the Australian nonmetropolitan setting, there was a high burden of health care–associated CDI in terms of hospital costs and length of stay, even though our health district experienced CDI rates that were similar to the Australian average. Challenges associated with the study included maintenance of consistent data collection across multiple hospital sites without comprehensive electronic medical records, provision of timely email feedback, and dissemination of study results.
Analysis of prior antimicrobial use has allowed us to identify targets for ongoing antimicrobial stewardship activities, and we also intend to provide further education on recognition of severity criteria. These activities can be supported through daily antimicrobial stewardship ward rounds. Future research could involve application of appropriateness criteria to prior and current antimicrobial use in CDI patients in order to identify avoidable cases and use of more advanced statistical techniques such as multistate modeling to determine differences in outcomes between cases and controls.
Stuart Bond, BPharm, DipPharmPrac, is an antimicrobial stewardship pharmacist based at Wollongong Hospital in New South Wales, Australia.
ABS wants feedback on 10-year exam alternatives
Many surgeons let out a collective cheer recently when the American Board of Surgery announced that, as of 2018, they will no longer have to take a high-stakes, pass-fail exam every 10 years to maintain certification.
The Board also announced, effective immediately, that it’s extending its continuing medical education (CME) reporting cycle from 3 to 5 years and reducing the number of required self-assessment CMEs – lectures and articles with a short quiz at the end – by a third. Surgeons now have to report 150 credits over 5 years, 50 from self-assessment CMEs. Under the old system, it was 90 credits over 3 years, 60 of which had to include self-assessment.

Alternative evaluation options
What’s on the minds of many, though, is what the alternatives to the 10-year exam will be. “That’s the $64,000 question,” said Emery Chen, MD, FACS, a general surgeon in private practice in Lancaster, Calif.
ABS is considering models that have worked well for other medical boards and hopes to roll out the alternative pathways at its winter meeting in January 2018, according to ABS Executive Director Frank Lewis, MD, FACS.

“I think it will probably be less stressful. People worry about the high-stakes exam. They don’t know what’s going to be on it, and they worry that failing could result in losing their staff privileges. We don’t think that’s particularly useful. The purpose is not to either pass or fail you but for you to learn the material,” Dr. Lewis said.
Meanwhile, surgeons who want to take the 10-year exam will still have the option.
Among the many options, surgeons could be given the two dozen journal articles deemed by experts to be the most cutting-edge for a given period, and quizzed on the material, with a chance to be re-quizzed as needed. There could be CME mini-courses or open-book exams on the areas most relevant to a surgeon’s practice and opportunities to relearn what might have been forgotten. Questions could be pushed out by smart phone to assess surgeons’ knowledge, with follow-up review material for incorrect answers and additional questions until the material is aced.
“Everything’s on the table,” said ABS at-large Board Member Tyler G. Hughes, MD, FACS, clinical faculty member at Kansas University, Salina.
A changing world
Like the country’s many other medical boards, ABS has been under pressure to make its MOC process less burdensome and more useful. A major problem is that general surgery isn’t very general anymore; it covers everything from transplants and bariatrics to trauma, endocrine, and vascular operations and more. Surgeons who have specialized have chafed at being examined every 10 years on areas that are no longer part of their practice and at questions about rare diseases such as multiple endocrine neoplasia I and II.
“They remember that one question that kind of stuck in their craw,” said Carol Scott-Conner, MD, FACS, emeritus professor and former head of surgery at the University of Iowa, Iowa City. At one point in her career, Dr. Scott-Conner helped write questions for the exam. “It wasn’t designed to be tricky, but, if you were not practicing in all these fields,” it was tough. “What you’re tested on should be relevant to your practice and help you get better at what you are doing. I think it’s a good change,” she said.

ABS has taken note. “There are a whole variety of things targeted at ways of maintaining longitudinal learning and providing some demonstration of it. We haven’t settled on any one of those. In fact, we may have more than one. We aim to get a good deal of feedback and see what our diplomates feel would be most helpful for them. It’s going to require a lot more detail, but hopefully it’s going to be more effective and more useful.” Dr. Lewis said.
The group will be at national and regional medical meetings this summer and fall to ask surgeons, in person, what they want their MOC system to be. There might be surveys as well, and ABS plans to be at the ACS Clinical Congress in October to solicit input.
The Board is looking to help surgeons with multiple certificates, whether from ABS or other boards, as well. “If we can create a more flexible assessment program that helps them maintain all their certifications, that’s what we want to do. We are looking at options to make it easier for them,” said ABS Director of Communications and Public Affairs Christine Shiffer, who noted that ABS will still likely require a 12-month case log every 10 years, even if surgeons opt out of the 10-year exam.
Will the new system have a negative impact on patient outcomes? After all, even specialized surgeons take night call sometimes. “ABS is responsible to surgeons but also the American public. If we say somebody is certified, it has to mean something. People would argue that there’s no real evidence that the recertification process improves surgical competence. Time will tell,” Dr. Scott-Conner said.
Dr. Lewis said he doesn’t know if the changes will save any money on MOC but noted that, instead of one $1,600 fee every 10 years, there’ll be an option to spread payments out.
Dr. Hughes is on the editorial advisory board of this publication.
Many surgeons let out a collective cheer recently when the American Board of Surgery announced that, as of 2018, they will no longer have to take a high-stakes, pass-fail exam every 10 years to maintain certification.
The Board also announced, effective immediately, that it’s extending its continuing medical education (CME) reporting cycle from 3 to 5 years and reducing the number of required self-assessment CMEs – lectures and articles with a short quiz at the end – by a third. Surgeons now have to report 150 credits over 5 years, 50 from self-assessment CMEs. Under the old system, it was 90 credits over 3 years, 60 of which had to include self-assessment.
Alternative evaluation options
What’s on the minds of many, though, is what the alternatives to the 10-year exam will be. “That’s the $64,000 question,” said Emery Chen, MD, FACS, a general surgeon in private practice in Lancaster, Calif.
ABS is considering models that have worked well for other medical boards and hopes to roll out the alternative pathways at its winter meeting in January 2018, according to ABS Executive Director Frank Lewis, MD, FACS.
“I think it will probably be less stressful. People worry about the high-stakes exam. They don’t know what’s going to be on it, and they worry that failing could result in losing their staff privileges. We don’t think that’s particularly useful. The purpose is not to either pass or fail you but for you to learn the material,” Dr. Lewis said.
Meanwhile, surgeons who want to take the 10-year exam will still have the option.
Among the many options, surgeons could be given the two dozen journal articles deemed by experts to be the most cutting-edge for a given period, and quizzed on the material, with a chance to be re-quizzed as needed. There could be CME mini-courses or open-book exams on the areas most relevant to a surgeon’s practice and opportunities to relearn what might have been forgotten. Questions could be pushed out by smart phone to assess surgeons’ knowledge, with follow-up review material for incorrect answers and additional questions until the material is aced.
“Everything’s on the table,” said ABS at-large Board Member Tyler G. Hughes, MD, FACS, clinical faculty member at Kansas University, Salina.
A changing world
Like the country’s many other medical boards, ABS has been under pressure to make its MOC process less burdensome and more useful. A major problem is that general surgery isn’t very general anymore; it covers everything from transplants and bariatrics to trauma, endocrine, and vascular operations and more. Surgeons who have specialized have chafed at being examined every 10 years on areas that are no longer part of their practice and at questions about rare diseases such as multiple endocrine neoplasia I and II.
“They remember that one question that kind of stuck in their craw,” said Carol Scott-Conner, MD, FACS, emeritus professor and former head of surgery at the University of Iowa, Iowa City. At one point in her career, Dr. Scott-Conner helped write questions for the exam. “It wasn’t designed to be tricky, but, if you were not practicing in all these fields,” it was tough. “What you’re tested on should be relevant to your practice and help you get better at what you are doing. I think it’s a good change,” she said.
ABS has taken note. “There are a whole variety of things targeted at ways of maintaining longitudinal learning and providing some demonstration of it. We haven’t settled on any one of those. In fact, we may have more than one. We aim to get a good deal of feedback and see what our diplomates feel would be most helpful for them. It’s going to require a lot more detail, but hopefully it’s going to be more effective and more useful.” Dr. Lewis said.
The group will be at national and regional medical meetings this summer and fall to ask surgeons, in person, what they want their MOC system to be. There might be surveys as well, and ABS plans to be at the ACS Clinical Congress in October to solicit input.
The Board is looking to help surgeons with multiple certificates, whether from ABS or other boards, as well. “If we can create a more flexible assessment program that helps them maintain all their certifications, that’s what we want to do. We are looking at options to make it easier for them,” said ABS Director of Communications and Public Affairs Christine Shiffer, who noted that ABS will still likely require a 12-month case log every 10 years, even if surgeons opt out of the 10-year exam.
Will the new system have a negative impact on patient outcomes? After all, even specialized surgeons take night call sometimes. “ABS is responsible to surgeons but also the American public. If we say somebody is certified, it has to mean something. People would argue that there’s no real evidence that the recertification process improves surgical competence. Time will tell,” Dr. Scott-Conner said.
Dr. Lewis said he doesn’t know if the changes will save any money on MOC but noted that, instead of one $1,600 fee every 10 years, there’ll be an option to spread payments out.
Dr. Hughes is on the editorial advisory board of this publication.
Many surgeons let out a collective cheer recently when the American Board of Surgery announced that, as of 2018, they will no longer have to take a high-stakes, pass-fail exam every 10 years to maintain certification.
The Board also announced, effective immediately, that it’s extending its continuing medical education (CME) reporting cycle from 3 to 5 years and reducing the number of required self-assessment CMEs – lectures and articles with a short quiz at the end – by a third. Surgeons now have to report 150 credits over 5 years, 50 from self-assessment CMEs. Under the old system, it was 90 credits over 3 years, 60 of which had to include self-assessment.
Alternative evaluation options
What’s on the minds of many, though, is what the alternatives to the 10-year exam will be. “That’s the $64,000 question,” said Emery Chen, MD, FACS, a general surgeon in private practice in Lancaster, Calif.
ABS is considering models that have worked well for other medical boards and hopes to roll out the alternative pathways at its winter meeting in January 2018, according to ABS Executive Director Frank Lewis, MD, FACS.
“I think it will probably be less stressful. People worry about the high-stakes exam. They don’t know what’s going to be on it, and they worry that failing could result in losing their staff privileges. We don’t think that’s particularly useful. The purpose is not to either pass or fail you but for you to learn the material,” Dr. Lewis said.
Meanwhile, surgeons who want to take the 10-year exam will still have the option.
Among the many options, surgeons could be given the two dozen journal articles deemed by experts to be the most cutting-edge for a given period, and quizzed on the material, with a chance to be re-quizzed as needed. There could be CME mini-courses or open-book exams on the areas most relevant to a surgeon’s practice and opportunities to relearn what might have been forgotten. Questions could be pushed out by smart phone to assess surgeons’ knowledge, with follow-up review material for incorrect answers and additional questions until the material is aced.
“Everything’s on the table,” said ABS at-large Board Member Tyler G. Hughes, MD, FACS, clinical faculty member at Kansas University, Salina.
A changing world
Like the country’s many other medical boards, ABS has been under pressure to make its MOC process less burdensome and more useful. A major problem is that general surgery isn’t very general anymore; it covers everything from transplants and bariatrics to trauma, endocrine, and vascular operations and more. Surgeons who have specialized have chafed at being examined every 10 years on areas that are no longer part of their practice and at questions about rare diseases such as multiple endocrine neoplasia I and II.
“They remember that one question that kind of stuck in their craw,” said Carol Scott-Conner, MD, FACS, emeritus professor and former head of surgery at the University of Iowa, Iowa City. At one point in her career, Dr. Scott-Conner helped write questions for the exam. “It wasn’t designed to be tricky, but, if you were not practicing in all these fields,” it was tough. “What you’re tested on should be relevant to your practice and help you get better at what you are doing. I think it’s a good change,” she said.
ABS has taken note. “There are a whole variety of things targeted at ways of maintaining longitudinal learning and providing some demonstration of it. We haven’t settled on any one of those. In fact, we may have more than one. We aim to get a good deal of feedback and see what our diplomates feel would be most helpful for them. It’s going to require a lot more detail, but hopefully it’s going to be more effective and more useful.” Dr. Lewis said.
The group will be at national and regional medical meetings this summer and fall to ask surgeons, in person, what they want their MOC system to be. There might be surveys as well, and ABS plans to be at the ACS Clinical Congress in October to solicit input.
The Board is looking to help surgeons with multiple certificates, whether from ABS or other boards, as well. “If we can create a more flexible assessment program that helps them maintain all their certifications, that’s what we want to do. We are looking at options to make it easier for them,” said ABS Director of Communications and Public Affairs Christine Shiffer, who noted that ABS will still likely require a 12-month case log every 10 years, even if surgeons opt out of the 10-year exam.
Will the new system have a negative impact on patient outcomes? After all, even specialized surgeons take night call sometimes. “ABS is responsible to surgeons but also the American public. If we say somebody is certified, it has to mean something. People would argue that there’s no real evidence that the recertification process improves surgical competence. Time will tell,” Dr. Scott-Conner said.
Dr. Lewis said he doesn’t know if the changes will save any money on MOC but noted that, instead of one $1,600 fee every 10 years, there’ll be an option to spread payments out.
Dr. Hughes is on the editorial advisory board of this publication.
Pneumococcal conjugate vaccines modestly reduce complex acute otitis media
Incidence rate ratios of children hospitalized with acute otitis media (hAOM) or more advanced stages, such as mastoidismus and acute mastoiditis (M+AM) declined by 10% and 20%, respectively, after introduction of pneumococcal conjugate vaccines (PCV) in the central region of Denmark, reported Bjarke B. Laursen of Aarhus University in Denmark, and associates.
The use of antibiotics for AOM prior to hospitalization increased markedly during the study period, and that may have impacted the modest reduction in hAOM and M+AM, the researchers said.
Incidence of Streptococcus pneumoniae cases decreased from 38% in the prevaccine era to 31% in the PCV-7 era and decreased even more to 16% in the PCV-13 era. The incidence of the second most frequently isolated bacteria, group A streptococcus (GAS), fell from 17% in the prevaccine era to 16% in the PCV-13 era, becoming equal to S. pneumoniae. The “no growth” results rose from 12% in the prevaccine era to 38% and 44% in the PCV-7 and PCV-13 eras, respectively (Int J Pediatr Otorhinolaryngol. 2017 Jul 4. doi: 10.1016/j.ijporl.2017.07.002) .
In the M+AM group, S. pneumoniae–positive cases fell to 10% in the PCV-13 era, compared with 44% in the prevaccine era and 41% in the PCV-7 era. A rise in GAS-positive cultures was seen in the M+AM group, from 15% in the prevaccine era to 30% in the PCV-13 era. The “no growth” group rose in incidence from 13% of cases in the prevaccine era to 26% in the PCV-7 era and 33% in the PCV-13 era.
“This microbiological shift is worrisome, because GAS may cause very serious infections due to its invasive nature,” Dr. Laursen and associates wrote. “An increase in GAS as described in this study has not been demonstrated elsewhere so far. In contrast to our findings, the most common species to ‘take over’ in the United States was Haemophilus influenzae.
“As [S. pneumoniae] and GAS are potentially invasive pathogens causing more severe clinical signs and symptoms, such cases are more likely to be admitted compared to cases caused by non-typable Haemophilus influenzae that gives rise to more subtle middle ear infections and clinical pictures,” the researchers noted.
Considering how many patients received antibiotics prior to admission in the hAOM group, there was a slight increase from 45% in the prevaccine era to 54% in the PCV-13 era, but a “more pronounced increase was found in the M+AM group, from 27% in the prevaccine era to 46% in the PCV-13 era,” they said.
The investigators called for continued surveillance of the microbiology associated with complex AOM.
No study funding or investigator disclosure information was provided.
Incidence rate ratios of children hospitalized with acute otitis media (hAOM) or more advanced stages, such as mastoidismus and acute mastoiditis (M+AM) declined by 10% and 20%, respectively, after introduction of pneumococcal conjugate vaccines (PCV) in the central region of Denmark, reported Bjarke B. Laursen of Aarhus University in Denmark, and associates.
The use of antibiotics for AOM prior to hospitalization increased markedly during the study period, and that may have impacted the modest reduction in hAOM and M+AM, the researchers said.
Incidence of Streptococcus pneumoniae cases decreased from 38% in the prevaccine era to 31% in the PCV-7 era and decreased even more to 16% in the PCV-13 era. The incidence of the second most frequently isolated bacteria, group A streptococcus (GAS), fell from 17% in the prevaccine era to 16% in the PCV-13 era, becoming equal to S. pneumoniae. The “no growth” results rose from 12% in the prevaccine era to 38% and 44% in the PCV-7 and PCV-13 eras, respectively (Int J Pediatr Otorhinolaryngol. 2017 Jul 4. doi: 10.1016/j.ijporl.2017.07.002) .
In the M+AM group, S. pneumoniae–positive cases fell to 10% in the PCV-13 era, compared with 44% in the prevaccine era and 41% in the PCV-7 era. A rise in GAS-positive cultures was seen in the M+AM group, from 15% in the prevaccine era to 30% in the PCV-13 era. The “no growth” group rose in incidence from 13% of cases in the prevaccine era to 26% in the PCV-7 era and 33% in the PCV-13 era.
“This microbiological shift is worrisome, because GAS may cause very serious infections due to its invasive nature,” Dr. Laursen and associates wrote. “An increase in GAS as described in this study has not been demonstrated elsewhere so far. In contrast to our findings, the most common species to ‘take over’ in the United States was Haemophilus influenzae.
“As [S. pneumoniae] and GAS are potentially invasive pathogens causing more severe clinical signs and symptoms, such cases are more likely to be admitted compared to cases caused by non-typable Haemophilus influenzae that gives rise to more subtle middle ear infections and clinical pictures,” the researchers noted.
Considering how many patients received antibiotics prior to admission in the hAOM group, there was a slight increase from 45% in the prevaccine era to 54% in the PCV-13 era, but a “more pronounced increase was found in the M+AM group, from 27% in the prevaccine era to 46% in the PCV-13 era,” they said.
The investigators called for continued surveillance of the microbiology associated with complex AOM.
No study funding or investigator disclosure information was provided.
Incidence rate ratios of children hospitalized with acute otitis media (hAOM) or more advanced stages, such as mastoidismus and acute mastoiditis (M+AM) declined by 10% and 20%, respectively, after introduction of pneumococcal conjugate vaccines (PCV) in the central region of Denmark, reported Bjarke B. Laursen of Aarhus University in Denmark, and associates.
The use of antibiotics for AOM prior to hospitalization increased markedly during the study period, and that may have impacted the modest reduction in hAOM and M+AM, the researchers said.
Incidence of Streptococcus pneumoniae cases decreased from 38% in the prevaccine era to 31% in the PCV-7 era and decreased even more to 16% in the PCV-13 era. The incidence of the second most frequently isolated bacteria, group A streptococcus (GAS), fell from 17% in the prevaccine era to 16% in the PCV-13 era, becoming equal to S. pneumoniae. The “no growth” results rose from 12% in the prevaccine era to 38% and 44% in the PCV-7 and PCV-13 eras, respectively (Int J Pediatr Otorhinolaryngol. 2017 Jul 4. doi: 10.1016/j.ijporl.2017.07.002) .
In the M+AM group, S. pneumoniae–positive cases fell to 10% in the PCV-13 era, compared with 44% in the prevaccine era and 41% in the PCV-7 era. A rise in GAS-positive cultures was seen in the M+AM group, from 15% in the prevaccine era to 30% in the PCV-13 era. The “no growth” group rose in incidence from 13% of cases in the prevaccine era to 26% in the PCV-7 era and 33% in the PCV-13 era.
“This microbiological shift is worrisome, because GAS may cause very serious infections due to its invasive nature,” Dr. Laursen and associates wrote. “An increase in GAS as described in this study has not been demonstrated elsewhere so far. In contrast to our findings, the most common species to ‘take over’ in the United States was Haemophilus influenzae.
“As [S. pneumoniae] and GAS are potentially invasive pathogens causing more severe clinical signs and symptoms, such cases are more likely to be admitted compared to cases caused by non-typable Haemophilus influenzae that gives rise to more subtle middle ear infections and clinical pictures,” the researchers noted.
Considering how many patients received antibiotics prior to admission in the hAOM group, there was a slight increase from 45% in the prevaccine era to 54% in the PCV-13 era, but a “more pronounced increase was found in the M+AM group, from 27% in the prevaccine era to 46% in the PCV-13 era,” they said.
The investigators called for continued surveillance of the microbiology associated with complex AOM.
No study funding or investigator disclosure information was provided.
FROM THE INTERNATIONAL JOURNAL OF PEDIATRIC OTORHINOLARYNGOLOGY
Key clinical point:
Major finding: Overall incidence in hAOM decreased numerically from 6.45/100,000 annually in the prevaccine era to 2.43/100,000 in the PCV-7 era, and to 5.88/100,000 annually in the PCV-13 era.
Data source: A retrospective study of 246 cases of children hospitalized with acute otitis media.
Disclosures: No study funding or investigator disclosure information was provided.
Target childhood obesity now to prevent knee OA later
LAS VEGAS – Childhood overweight was associated with increased risk of patellar cartilage defects in young adulthood independent of adult weight status in what’s believed to be the first long-term prospective study to address the issue using informative MRI imaging.
“Our data indicate the importance of intervening in childhood obesity for adult joint health,” Benny E. Antony, MD, reported at the World Congress on Osteoarthritis, sponsored by the Osteoarthritis Research Society International.
He presented the 25-year prospective follow-up from the population-based Childhood Determinants of Adult Knee Cartilage Study, a substudy of the Australian Schools Health and Fitness Survey of 1985. The analysis included 322 nationally representative participants who were 7-15 years old at enrollment and 31-41 years old at follow-up, when they underwent screening MRI knee scans in which cartilage defects in the tibial, femoral, and patellar zones were rated by a modified Outerbridge scoring system.

The increased prevalence of patellar compared with tibiofemoral cartilage defects is consistent with growing evidence that knee OA typically starts in the patellar region and then spreads through the knee over time, according to Dr. Antony of the University of Tasmania in Hobart, Australia.
Among the other key findings:
• Women had a higher prevalence of cartilage defects: 43% in the whole knee and 30% at the patella, compared with rates of 34% and 20%, respectively, in men.
• The prevalence of patellar cartilage defects in young adulthood was 24.2% in those who had a normal weight both as children and young adults compared with 40% in participants who were overweight at both time points, for an adjusted 1.77-fold increased risk in subjects who were overweight across the decades, .
• Excess childhood weight per kilogram, fat mass per kilogram, and body mass per unit were each associated with 5%-12% increased risks of patellar cartilage defects 25 years later, independent of adult body weight status, in an analysis adjusted for childhood age, sex, height, duration of follow-up, and history of pediatric or adult knee injury.
• A dose-response relationship was evident between the degree of childhood overweight and the severity of cartilage defects as young adults on a 0-4 rating scale.
Dr. Antony reported having no financial conflicts of interest regarding the study, which was supported by the National Health and Medical Research Council of Australia.
LAS VEGAS – Childhood overweight was associated with increased risk of patellar cartilage defects in young adulthood independent of adult weight status in what’s believed to be the first long-term prospective study to address the issue using informative MRI imaging.
“Our data indicate the importance of intervening in childhood obesity for adult joint health,” Benny E. Antony, MD, reported at the World Congress on Osteoarthritis, sponsored by the Osteoarthritis Research Society International.
He presented the 25-year prospective follow-up from the population-based Childhood Determinants of Adult Knee Cartilage Study, a substudy of the Australian Schools Health and Fitness Survey of 1985. The analysis included 322 nationally representative participants who were 7-15 years old at enrollment and 31-41 years old at follow-up, when they underwent screening MRI knee scans in which cartilage defects in the tibial, femoral, and patellar zones were rated by a modified Outerbridge scoring system.
The increased prevalence of patellar compared with tibiofemoral cartilage defects is consistent with growing evidence that knee OA typically starts in the patellar region and then spreads through the knee over time, according to Dr. Antony of the University of Tasmania in Hobart, Australia.
Among the other key findings:
• Women had a higher prevalence of cartilage defects: 43% in the whole knee and 30% at the patella, compared with rates of 34% and 20%, respectively, in men.
• The prevalence of patellar cartilage defects in young adulthood was 24.2% in those who had a normal weight both as children and young adults compared with 40% in participants who were overweight at both time points, for an adjusted 1.77-fold increased risk in subjects who were overweight across the decades, .
• Excess childhood weight per kilogram, fat mass per kilogram, and body mass per unit were each associated with 5%-12% increased risks of patellar cartilage defects 25 years later, independent of adult body weight status, in an analysis adjusted for childhood age, sex, height, duration of follow-up, and history of pediatric or adult knee injury.
• A dose-response relationship was evident between the degree of childhood overweight and the severity of cartilage defects as young adults on a 0-4 rating scale.
Dr. Antony reported having no financial conflicts of interest regarding the study, which was supported by the National Health and Medical Research Council of Australia.
LAS VEGAS – Childhood overweight was associated with increased risk of patellar cartilage defects in young adulthood independent of adult weight status in what’s believed to be the first long-term prospective study to address the issue using informative MRI imaging.
“Our data indicate the importance of intervening in childhood obesity for adult joint health,” Benny E. Antony, MD, reported at the World Congress on Osteoarthritis, sponsored by the Osteoarthritis Research Society International.
He presented the 25-year prospective follow-up from the population-based Childhood Determinants of Adult Knee Cartilage Study, a substudy of the Australian Schools Health and Fitness Survey of 1985. The analysis included 322 nationally representative participants who were 7-15 years old at enrollment and 31-41 years old at follow-up, when they underwent screening MRI knee scans in which cartilage defects in the tibial, femoral, and patellar zones were rated by a modified Outerbridge scoring system.
The increased prevalence of patellar compared with tibiofemoral cartilage defects is consistent with growing evidence that knee OA typically starts in the patellar region and then spreads through the knee over time, according to Dr. Antony of the University of Tasmania in Hobart, Australia.
Among the other key findings:
• Women had a higher prevalence of cartilage defects: 43% in the whole knee and 30% at the patella, compared with rates of 34% and 20%, respectively, in men.
• The prevalence of patellar cartilage defects in young adulthood was 24.2% in those who had a normal weight both as children and young adults compared with 40% in participants who were overweight at both time points, for an adjusted 1.77-fold increased risk in subjects who were overweight across the decades, .
• Excess childhood weight per kilogram, fat mass per kilogram, and body mass per unit were each associated with 5%-12% increased risks of patellar cartilage defects 25 years later, independent of adult body weight status, in an analysis adjusted for childhood age, sex, height, duration of follow-up, and history of pediatric or adult knee injury.
• A dose-response relationship was evident between the degree of childhood overweight and the severity of cartilage defects as young adults on a 0-4 rating scale.
Dr. Antony reported having no financial conflicts of interest regarding the study, which was supported by the National Health and Medical Research Council of Australia.
AT OARSI 2017
Key clinical point:
Major finding: The prevalence of patellar cartilage defects in young adults was 24.2% in those who were normal weight both as children and young adults, compared with 40% in subjects who were overweight at both time points.
Data source: A prospective population-based cohort study of 322 subjects followed from childhood to age 31-41 years, when they were assessed via MRI for knee cartilage defects.
Disclosures: The National Health and Medical Research Council of Australia supported the study. The presenter reported having no financial conflicts.
Clinical trial: Mesh Type in Ventral Hernia Repair
The Mesh Type in Ventral Hernia Repair trial is an interventional study currently recruiting patients scheduled for open ventral hernia repair.
Half of ventral hernia repairs utilize synthetic mesh, while the other half use biologic mesh. There is currently little solid evidence that one mesh type is better than the other, although the study investigators hypothesize that biologic mesh is superior to synthetic.
Patients will be included in the trial if they are scheduled for open ventral hernia repair at LBJ General Hospital in Houston and are at least 18 years old. Patients will be excluded if they have an active infection, are unlikely to survive the next 2 years, are individuals in whom a prosthetic would not normally be placed, or are unlikely to follow up.
The primary endpoint goal is zero complications 1 year after the operation. Secondary endpoint goals include patient-centered outcomes, cost, Dindo-Clavien complications (grades I-IV), and to be complication free 3 years after the operation.
Recruitment began on March 27, 2017, and the study is expected to include 50 people. The primary study endpoint will be completed March 31, 2019, with the full study being completed March 31, 2022.
Find more information at the study page on Clinicaltrials.gov.
The Mesh Type in Ventral Hernia Repair trial is an interventional study currently recruiting patients scheduled for open ventral hernia repair.
Half of ventral hernia repairs utilize synthetic mesh, while the other half use biologic mesh. There is currently little solid evidence that one mesh type is better than the other, although the study investigators hypothesize that biologic mesh is superior to synthetic.
Patients will be included in the trial if they are scheduled for open ventral hernia repair at LBJ General Hospital in Houston and are at least 18 years old. Patients will be excluded if they have an active infection, are unlikely to survive the next 2 years, are individuals in whom a prosthetic would not normally be placed, or are unlikely to follow up.
The primary endpoint goal is zero complications 1 year after the operation. Secondary endpoint goals include patient-centered outcomes, cost, Dindo-Clavien complications (grades I-IV), and to be complication free 3 years after the operation.
Recruitment began on March 27, 2017, and the study is expected to include 50 people. The primary study endpoint will be completed March 31, 2019, with the full study being completed March 31, 2022.
Find more information at the study page on Clinicaltrials.gov.
The Mesh Type in Ventral Hernia Repair trial is an interventional study currently recruiting patients scheduled for open ventral hernia repair.
Half of ventral hernia repairs utilize synthetic mesh, while the other half use biologic mesh. There is currently little solid evidence that one mesh type is better than the other, although the study investigators hypothesize that biologic mesh is superior to synthetic.
Patients will be included in the trial if they are scheduled for open ventral hernia repair at LBJ General Hospital in Houston and are at least 18 years old. Patients will be excluded if they have an active infection, are unlikely to survive the next 2 years, are individuals in whom a prosthetic would not normally be placed, or are unlikely to follow up.
The primary endpoint goal is zero complications 1 year after the operation. Secondary endpoint goals include patient-centered outcomes, cost, Dindo-Clavien complications (grades I-IV), and to be complication free 3 years after the operation.
Recruitment began on March 27, 2017, and the study is expected to include 50 people. The primary study endpoint will be completed March 31, 2019, with the full study being completed March 31, 2022.
Find more information at the study page on Clinicaltrials.gov.
SUMMARY FROM CLINICALTRIALS.GOV