User login
Federal Health Care Data Trends 2017 Introduction
Military service comes with many health care costs, both immediate and long term. These costs are incurred by active-duty and veteran patients, as well as the VA and DoD systems that have struggled to adequately meet their health care needs. The VA and DoD health care systems face a myriad of challenges in treating their diverse populations.
Identifying and responding to health care challenges requires reliable and detailed data. The 2017 Federal Health Care Data Trends was developed not only to report important data on the most significant health care challenges in federal medicine, but also to simplify and identify emergent trends.
The men and women who serve in the U.S. military are more likely to be diagnosed with posttraumatic stress disorder, diabetes mellitus, and chronic obstructive pulmonary disease. Agent Orange, burn pits, and other toxic exposures also increase their risk of developing multiple types of cancer, multiple sclerosis, asthma, and many other conditions. Veterans are likely to be older than nonveterans (the median age of male veterans is 64 years, compared with 41 years for nonveterans), and therefore at risk for age-related conditions.
Of the nearly 22 million veterans in the U.S., 8.9 million are enrolled in the VA, and just short of 6 million access health care services annually. Active-duty service members make up just 15% of the military health system, while the family members of active-duty service members, National Guard members, reservists, and retirees constitute more than half of the TRICARE population.
Click here to continue reading.
Military service comes with many health care costs, both immediate and long term. These costs are incurred by active-duty and veteran patients, as well as the VA and DoD systems that have struggled to adequately meet their health care needs. The VA and DoD health care systems face a myriad of challenges in treating their diverse populations.
Identifying and responding to health care challenges requires reliable and detailed data. The 2017 Federal Health Care Data Trends was developed not only to report important data on the most significant health care challenges in federal medicine, but also to simplify and identify emergent trends.
The men and women who serve in the U.S. military are more likely to be diagnosed with posttraumatic stress disorder, diabetes mellitus, and chronic obstructive pulmonary disease. Agent Orange, burn pits, and other toxic exposures also increase their risk of developing multiple types of cancer, multiple sclerosis, asthma, and many other conditions. Veterans are likely to be older than nonveterans (the median age of male veterans is 64 years, compared with 41 years for nonveterans), and therefore at risk for age-related conditions.
Of the nearly 22 million veterans in the U.S., 8.9 million are enrolled in the VA, and just short of 6 million access health care services annually. Active-duty service members make up just 15% of the military health system, while the family members of active-duty service members, National Guard members, reservists, and retirees constitute more than half of the TRICARE population.
Click here to continue reading.
Military service comes with many health care costs, both immediate and long term. These costs are incurred by active-duty and veteran patients, as well as the VA and DoD systems that have struggled to adequately meet their health care needs. The VA and DoD health care systems face a myriad of challenges in treating their diverse populations.
Identifying and responding to health care challenges requires reliable and detailed data. The 2017 Federal Health Care Data Trends was developed not only to report important data on the most significant health care challenges in federal medicine, but also to simplify and identify emergent trends.
The men and women who serve in the U.S. military are more likely to be diagnosed with posttraumatic stress disorder, diabetes mellitus, and chronic obstructive pulmonary disease. Agent Orange, burn pits, and other toxic exposures also increase their risk of developing multiple types of cancer, multiple sclerosis, asthma, and many other conditions. Veterans are likely to be older than nonveterans (the median age of male veterans is 64 years, compared with 41 years for nonveterans), and therefore at risk for age-related conditions.
Of the nearly 22 million veterans in the U.S., 8.9 million are enrolled in the VA, and just short of 6 million access health care services annually. Active-duty service members make up just 15% of the military health system, while the family members of active-duty service members, National Guard members, reservists, and retirees constitute more than half of the TRICARE population.
Click here to continue reading.
Studies support testing for iron deficiency in young women
A pair of studies suggest physicians should consider testing female adolescents for iron deficiency within a few years of starting menses.
Women are typically tested for anemia in their teens, with a quick and affordable hemoglobin test.
However, iron deficiency can develop years before anemia and can be missed by hemoglobin testing alone.
Blood tests for iron deficiency without anemia are more costly and more difficult to obtain than hemoglobin testing for anemia.
Deepa Sekhar, MD, of Penn State College of Medicine in Hershey, Pennsylvania, and her colleagues set out to determine risk factors for iron deficiency without anemia in order to pinpoint which women could benefit most from the more costly testing.
The results of the researchers’ 2 studies were published in PLOS ONE and The Journal of Pediatrics.
PLOS ONE study
The researchers evaluated data from 6216 females, ages 12 to 49, who took part in the National Health and Nutrition Examination Survey (NHANES) between 2003 and 2010. As part of the survey, participants were tested for both iron deficiency and anemia.
Eight percent of all subjects (n=494) had iron deficiency.
Nine percent (n=250) of non-anemic younger women (ages 12-21) had iron deficiency, as did 7% (n=244) of older women (ages 22-49) who were not anemic.
The researchers looked at potential risk factors for iron deficiency, including the age when women started menstruating, as well as their race/ethnicity, poverty status, food insecurity, tobacco or nicotine use, dietary information, body mass index, and physical activity.
All of these factors have been associated with iron-deficiency anemia in women in prior studies.
In this study, there was only 1 risk factor significantly associated with iron deficiency without anemia.
Young women (ages 12-21) who had been menstruating for more than 3 years had a significantly higher risk of iron deficiency without anemia (risk ratio=3.18).
The Journal of Pediatrics study
In this study, the researchers looked at whether a questionnaire could better predict iron status.
The questionnaire included questions on depression, poor attention, and daytime sleepiness, all of which have been associated with iron deficiency or iron-deficiency anemia, but were not captured in the prior NHANES analyses.
This questionnaire was compared to the 4 questions assessing iron-deficiency anemia risk in the Bright Futures Adolescent Previsit Questionnaire, a survey recommended for physician use by the American Academy of Pediatrics.
Ninety-six female adolescents participated in this study. Eighteen percent of them (n=17) had iron deficiency, and 5% (n=5) had iron-deficiency anemia.
Both the Bright Futures questions and the researchers’ risk assessment questionnaire poorly predicted ferritin and hemoglobin values in these subjects.
Mean differences in depression, poor attention, food insecurity, daytime sleepiness, and body mass index percentile were not significantly associated with ferritin or hemoglobin.
Conclusions
The results of these 2 studies suggest that risk factors and assessments cannot accurately determine which young women should receive testing for iron deficiency, although results from the first study might be used to determine when testing should occur.
“I think we need to establish the optimal timing for an objective assessment of adolescent iron deficiency and anemia,” Dr Sekhar said.
She believes the appropriate age may be 16 years old, when most females will have been menstruating for at least 3 years.
Further research will be needed to determine which blood test for iron deficiency without anemia is accurate, cost-efficient, and practical for routine doctor’s office use.
This test should be given with hemoglobin testing to catch all young women on the spectrum of iron deficiency, Dr Sekhar said. ![]()
A pair of studies suggest physicians should consider testing female adolescents for iron deficiency within a few years of starting menses.
Women are typically tested for anemia in their teens, with a quick and affordable hemoglobin test.
However, iron deficiency can develop years before anemia and can be missed by hemoglobin testing alone.
Blood tests for iron deficiency without anemia are more costly and more difficult to obtain than hemoglobin testing for anemia.
Deepa Sekhar, MD, of Penn State College of Medicine in Hershey, Pennsylvania, and her colleagues set out to determine risk factors for iron deficiency without anemia in order to pinpoint which women could benefit most from the more costly testing.
The results of the researchers’ 2 studies were published in PLOS ONE and The Journal of Pediatrics.
PLOS ONE study
The researchers evaluated data from 6216 females, ages 12 to 49, who took part in the National Health and Nutrition Examination Survey (NHANES) between 2003 and 2010. As part of the survey, participants were tested for both iron deficiency and anemia.
Eight percent of all subjects (n=494) had iron deficiency.
Nine percent (n=250) of non-anemic younger women (ages 12-21) had iron deficiency, as did 7% (n=244) of older women (ages 22-49) who were not anemic.
The researchers looked at potential risk factors for iron deficiency, including the age when women started menstruating, as well as their race/ethnicity, poverty status, food insecurity, tobacco or nicotine use, dietary information, body mass index, and physical activity.
All of these factors have been associated with iron-deficiency anemia in women in prior studies.
In this study, there was only 1 risk factor significantly associated with iron deficiency without anemia.
Young women (ages 12-21) who had been menstruating for more than 3 years had a significantly higher risk of iron deficiency without anemia (risk ratio=3.18).
The Journal of Pediatrics study
In this study, the researchers looked at whether a questionnaire could better predict iron status.
The questionnaire included questions on depression, poor attention, and daytime sleepiness, all of which have been associated with iron deficiency or iron-deficiency anemia, but were not captured in the prior NHANES analyses.
This questionnaire was compared to the 4 questions assessing iron-deficiency anemia risk in the Bright Futures Adolescent Previsit Questionnaire, a survey recommended for physician use by the American Academy of Pediatrics.
Ninety-six female adolescents participated in this study. Eighteen percent of them (n=17) had iron deficiency, and 5% (n=5) had iron-deficiency anemia.
Both the Bright Futures questions and the researchers’ risk assessment questionnaire poorly predicted ferritin and hemoglobin values in these subjects.
Mean differences in depression, poor attention, food insecurity, daytime sleepiness, and body mass index percentile were not significantly associated with ferritin or hemoglobin.
Conclusions
The results of these 2 studies suggest that risk factors and assessments cannot accurately determine which young women should receive testing for iron deficiency, although results from the first study might be used to determine when testing should occur.
“I think we need to establish the optimal timing for an objective assessment of adolescent iron deficiency and anemia,” Dr Sekhar said.
She believes the appropriate age may be 16 years old, when most females will have been menstruating for at least 3 years.
Further research will be needed to determine which blood test for iron deficiency without anemia is accurate, cost-efficient, and practical for routine doctor’s office use.
This test should be given with hemoglobin testing to catch all young women on the spectrum of iron deficiency, Dr Sekhar said. ![]()
A pair of studies suggest physicians should consider testing female adolescents for iron deficiency within a few years of starting menses.
Women are typically tested for anemia in their teens, with a quick and affordable hemoglobin test.
However, iron deficiency can develop years before anemia and can be missed by hemoglobin testing alone.
Blood tests for iron deficiency without anemia are more costly and more difficult to obtain than hemoglobin testing for anemia.
Deepa Sekhar, MD, of Penn State College of Medicine in Hershey, Pennsylvania, and her colleagues set out to determine risk factors for iron deficiency without anemia in order to pinpoint which women could benefit most from the more costly testing.
The results of the researchers’ 2 studies were published in PLOS ONE and The Journal of Pediatrics.
PLOS ONE study
The researchers evaluated data from 6216 females, ages 12 to 49, who took part in the National Health and Nutrition Examination Survey (NHANES) between 2003 and 2010. As part of the survey, participants were tested for both iron deficiency and anemia.
Eight percent of all subjects (n=494) had iron deficiency.
Nine percent (n=250) of non-anemic younger women (ages 12-21) had iron deficiency, as did 7% (n=244) of older women (ages 22-49) who were not anemic.
The researchers looked at potential risk factors for iron deficiency, including the age when women started menstruating, as well as their race/ethnicity, poverty status, food insecurity, tobacco or nicotine use, dietary information, body mass index, and physical activity.
All of these factors have been associated with iron-deficiency anemia in women in prior studies.
In this study, there was only 1 risk factor significantly associated with iron deficiency without anemia.
Young women (ages 12-21) who had been menstruating for more than 3 years had a significantly higher risk of iron deficiency without anemia (risk ratio=3.18).
The Journal of Pediatrics study
In this study, the researchers looked at whether a questionnaire could better predict iron status.
The questionnaire included questions on depression, poor attention, and daytime sleepiness, all of which have been associated with iron deficiency or iron-deficiency anemia, but were not captured in the prior NHANES analyses.
This questionnaire was compared to the 4 questions assessing iron-deficiency anemia risk in the Bright Futures Adolescent Previsit Questionnaire, a survey recommended for physician use by the American Academy of Pediatrics.
Ninety-six female adolescents participated in this study. Eighteen percent of them (n=17) had iron deficiency, and 5% (n=5) had iron-deficiency anemia.
Both the Bright Futures questions and the researchers’ risk assessment questionnaire poorly predicted ferritin and hemoglobin values in these subjects.
Mean differences in depression, poor attention, food insecurity, daytime sleepiness, and body mass index percentile were not significantly associated with ferritin or hemoglobin.
Conclusions
The results of these 2 studies suggest that risk factors and assessments cannot accurately determine which young women should receive testing for iron deficiency, although results from the first study might be used to determine when testing should occur.
“I think we need to establish the optimal timing for an objective assessment of adolescent iron deficiency and anemia,” Dr Sekhar said.
She believes the appropriate age may be 16 years old, when most females will have been menstruating for at least 3 years.
Further research will be needed to determine which blood test for iron deficiency without anemia is accurate, cost-efficient, and practical for routine doctor’s office use.
This test should be given with hemoglobin testing to catch all young women on the spectrum of iron deficiency, Dr Sekhar said. ![]()
ATV use by children still leads to significant trauma center admittance
The incidence of children admitted to Pennsylvania trauma centers because of accidents while riding all-terrain vehicles (ATV) fell 13% from the first 5 years to the last 6 years of a 2004-2014 study – but that decrease was not statistically or clinically significant, reported Mariano Garay, MD, of Pennsylvania State University, Hershey, and his associates.
In the American Academy of Pediatrics’ most recent policy statement in 2000, the academy recommended that use of ATVs be restricted to people older than 16 years and to off-road use only, with no passengers.
The median age of patients was 14 years, with a range of 1-17 years. Boys accounted for three-quarters of the patients. Being a passenger or being pulled behind the ATVs accounted for 24% of the injured patients. Of the crashes, 15% occurred on a street or roadway, and 49% of the riders reportedly wore a helmet. The majority of children who died were age 12-15 years, 25% were passengers, and 32% were injured while the ATV was being used on a street or roadway.
“Researchers in numerous studies, as well as professional organizations and ATV manufacturers, have concluded that children less than 16 years of age do not have the capacity to safely operate ATVs,” Dr. Garay and associates cautioned. “We advise primary care providers to be the forefront of the prevention effort and to continue to provide families with safety information and recommendations of age restrictions for ATV use by children.”
The incidence of children admitted to Pennsylvania trauma centers because of accidents while riding all-terrain vehicles (ATV) fell 13% from the first 5 years to the last 6 years of a 2004-2014 study – but that decrease was not statistically or clinically significant, reported Mariano Garay, MD, of Pennsylvania State University, Hershey, and his associates.
In the American Academy of Pediatrics’ most recent policy statement in 2000, the academy recommended that use of ATVs be restricted to people older than 16 years and to off-road use only, with no passengers.
The median age of patients was 14 years, with a range of 1-17 years. Boys accounted for three-quarters of the patients. Being a passenger or being pulled behind the ATVs accounted for 24% of the injured patients. Of the crashes, 15% occurred on a street or roadway, and 49% of the riders reportedly wore a helmet. The majority of children who died were age 12-15 years, 25% were passengers, and 32% were injured while the ATV was being used on a street or roadway.
“Researchers in numerous studies, as well as professional organizations and ATV manufacturers, have concluded that children less than 16 years of age do not have the capacity to safely operate ATVs,” Dr. Garay and associates cautioned. “We advise primary care providers to be the forefront of the prevention effort and to continue to provide families with safety information and recommendations of age restrictions for ATV use by children.”
The incidence of children admitted to Pennsylvania trauma centers because of accidents while riding all-terrain vehicles (ATV) fell 13% from the first 5 years to the last 6 years of a 2004-2014 study – but that decrease was not statistically or clinically significant, reported Mariano Garay, MD, of Pennsylvania State University, Hershey, and his associates.
In the American Academy of Pediatrics’ most recent policy statement in 2000, the academy recommended that use of ATVs be restricted to people older than 16 years and to off-road use only, with no passengers.
The median age of patients was 14 years, with a range of 1-17 years. Boys accounted for three-quarters of the patients. Being a passenger or being pulled behind the ATVs accounted for 24% of the injured patients. Of the crashes, 15% occurred on a street or roadway, and 49% of the riders reportedly wore a helmet. The majority of children who died were age 12-15 years, 25% were passengers, and 32% were injured while the ATV was being used on a street or roadway.
“Researchers in numerous studies, as well as professional organizations and ATV manufacturers, have concluded that children less than 16 years of age do not have the capacity to safely operate ATVs,” Dr. Garay and associates cautioned. “We advise primary care providers to be the forefront of the prevention effort and to continue to provide families with safety information and recommendations of age restrictions for ATV use by children.”
FROM PEDIATRICS
Something is Afoot
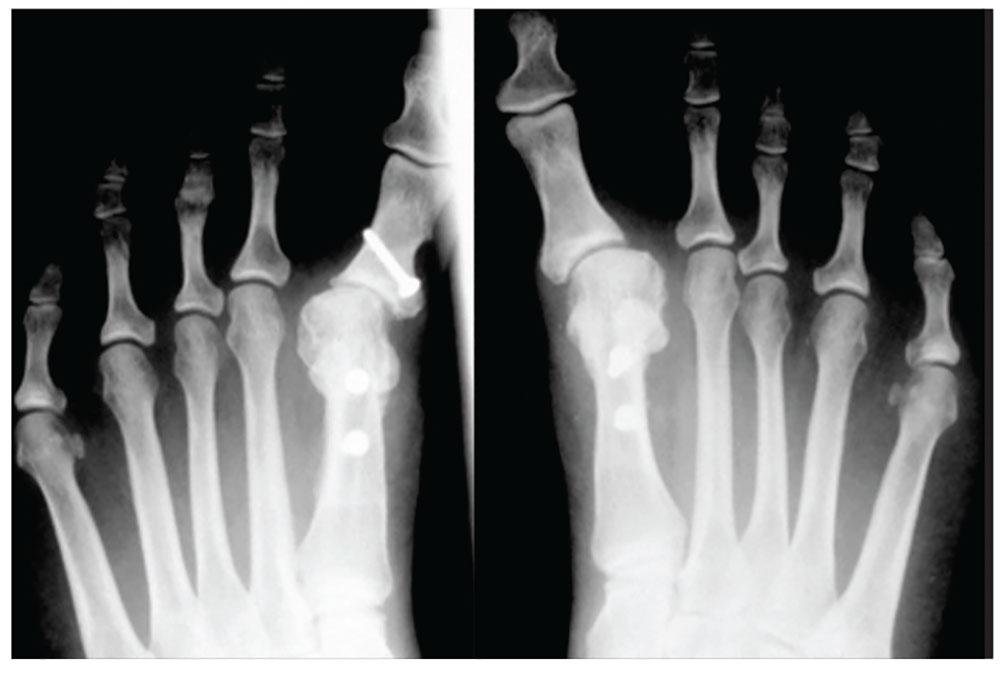
1. A 43-year-old woman presents with progressively worsening bilateral great toe pain that began during pregnancy and increased following the birth of her daughter three years ago. Both of her feet have developed a crescent moon shape, making it painful and difficult to wear normal shoes. This patient has
a) Degenerative arthritis
b) Hallux varus
c) Gout
d) Traumatic sesamoiditis
Diagnosis: Physical exam revealed bilateral hallux varus deformity of the great toe, which was greater on the left foot than on the right (23° and 16°, respectively). The deformities were easily, passively, correctable. Standing radiographs showed evidence of previous proximal osteotomies and well-healed distal first metatarsal osteotomies. Due to unsuccessful nonoperative management, surgical reconstruction was offered.
For further information, see “Bilateral Hallux Varus Deformity Correction With a Suture Button Construct.” Am J Orthop. 2013;42(3):121-124.

2. Following treatment for onychomycosis, this 49-year-old man’s toenails demonstrate inflammation of the medial and lateral nail borders of the hallux and second toes of the left foot. This patient’s diagnosis is
a) Subungual exostosis
b) Primary osteomyelitis of the phalanx
c) Tumors of the nail bed
d) Onychocryptosis
Diagnosis: Onychocryptosis, also known as ingrown toenail, is a rare complication of oral antifungal therapy. As the healthy nail plate advances, it may adhere to the nail bed and cut into the lateral nail folds. In this case, the site of the onychocryptosis corresponded to the proximal clearing of the nail plate. The patient required excision of the nail borders, after which the secondary inflammation resolved. The condition was treated with chemical matrixectomy.
For more information, see “Multiple Onychocryptosis Following Treatment of Onychomycosis With Oral Terbinafine.” Cutis. 2000;66(3):211-212.
3. A 39-year-old man was playing a game of pick-up basketball when he felt a pop, immediately followed by a sharp pain in the back of his ankle and lower leg. He now walks with a limp. The cause is
a) Achilles tendon rupture
b) Medial gastrocnemius tear
c) Calf muscle strain
d) Posterior tibial stress syndrome
Diagnosis: Often diagnosed as an ankle sprain, an Achilles tendon rupture most commonly occurs in middle-aged men from overexertion in sports—usually tennis, racquetball, basketball, or badminton, which involve bursts of jumping, pivoting, and running. Rupture may also occur from a sudden stumble, fall from a significant height, or abrupt step into a hole or off a curb, which causes the tendon to overstretch forcefully.
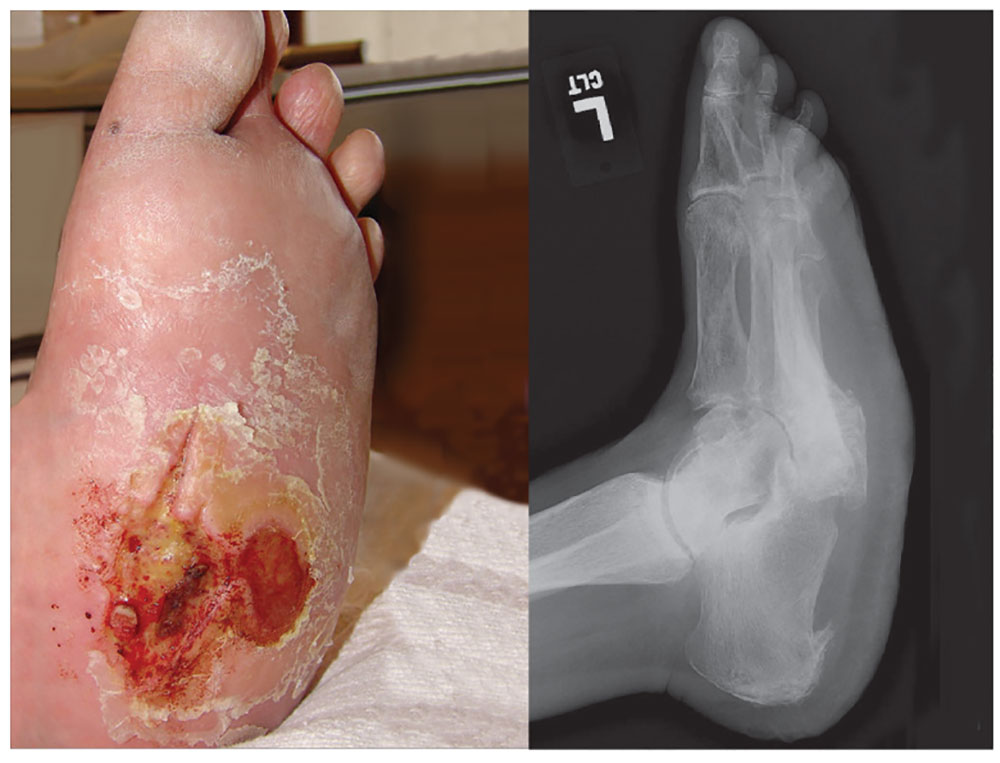
4. A 60-year-old woman seeks relief for a foot wound of several months’ duration that persists despite use of antibiotics and proper care. The skin over the plantar surface has a full-thickness ulcer, with partial necrosis of the subcutaneous tissue. Her history is significant for diabetes with neuropathy, nephropathy, and retinopathy. The diagnosis is
a) Osteomyelitis
b) Deep venous thrombosis
c) Charcot joint
d) Septic joint
Diagnosis: Charcot joint changes, and an associated stage III pressure ulcer, account for the extensive collapse of the inner arch and “rocker bottom foot” seen on the radiograph. Also known as neurogenic arthropathy, Charcot joint is commonly seen with diabetic neuropathy. In affected patients, secondary degenerative changes to the joints occur with loss of normal muscle tone, proprioception, temperature perception, and pain perception. The joints become loose, enlarged, and boggy. There can be extensive cartilage erosion or osteophyte formation. The normal plantar and heel forces are increased, producing eccentric loading of the foot and leading to microfractures, ligament laxity, and bone destruction.
For more information, see “A disfigured foot with ulcer.” J Fam Pract. 2008;57(5):321-323.
1. A 43-year-old woman presents with progressively worsening bilateral great toe pain that began during pregnancy and increased following the birth of her daughter three years ago. Both of her feet have developed a crescent moon shape, making it painful and difficult to wear normal shoes. This patient has
a) Degenerative arthritis
b) Hallux varus
c) Gout
d) Traumatic sesamoiditis
Diagnosis: Physical exam revealed bilateral hallux varus deformity of the great toe, which was greater on the left foot than on the right (23° and 16°, respectively). The deformities were easily, passively, correctable. Standing radiographs showed evidence of previous proximal osteotomies and well-healed distal first metatarsal osteotomies. Due to unsuccessful nonoperative management, surgical reconstruction was offered.
For further information, see “Bilateral Hallux Varus Deformity Correction With a Suture Button Construct.” Am J Orthop. 2013;42(3):121-124.
2. Following treatment for onychomycosis, this 49-year-old man’s toenails demonstrate inflammation of the medial and lateral nail borders of the hallux and second toes of the left foot. This patient’s diagnosis is
a) Subungual exostosis
b) Primary osteomyelitis of the phalanx
c) Tumors of the nail bed
d) Onychocryptosis
Diagnosis: Onychocryptosis, also known as ingrown toenail, is a rare complication of oral antifungal therapy. As the healthy nail plate advances, it may adhere to the nail bed and cut into the lateral nail folds. In this case, the site of the onychocryptosis corresponded to the proximal clearing of the nail plate. The patient required excision of the nail borders, after which the secondary inflammation resolved. The condition was treated with chemical matrixectomy.
For more information, see “Multiple Onychocryptosis Following Treatment of Onychomycosis With Oral Terbinafine.” Cutis. 2000;66(3):211-212.
3. A 39-year-old man was playing a game of pick-up basketball when he felt a pop, immediately followed by a sharp pain in the back of his ankle and lower leg. He now walks with a limp. The cause is
a) Achilles tendon rupture
b) Medial gastrocnemius tear
c) Calf muscle strain
d) Posterior tibial stress syndrome
Diagnosis: Often diagnosed as an ankle sprain, an Achilles tendon rupture most commonly occurs in middle-aged men from overexertion in sports—usually tennis, racquetball, basketball, or badminton, which involve bursts of jumping, pivoting, and running. Rupture may also occur from a sudden stumble, fall from a significant height, or abrupt step into a hole or off a curb, which causes the tendon to overstretch forcefully.
4. A 60-year-old woman seeks relief for a foot wound of several months’ duration that persists despite use of antibiotics and proper care. The skin over the plantar surface has a full-thickness ulcer, with partial necrosis of the subcutaneous tissue. Her history is significant for diabetes with neuropathy, nephropathy, and retinopathy. The diagnosis is
a) Osteomyelitis
b) Deep venous thrombosis
c) Charcot joint
d) Septic joint
Diagnosis: Charcot joint changes, and an associated stage III pressure ulcer, account for the extensive collapse of the inner arch and “rocker bottom foot” seen on the radiograph. Also known as neurogenic arthropathy, Charcot joint is commonly seen with diabetic neuropathy. In affected patients, secondary degenerative changes to the joints occur with loss of normal muscle tone, proprioception, temperature perception, and pain perception. The joints become loose, enlarged, and boggy. There can be extensive cartilage erosion or osteophyte formation. The normal plantar and heel forces are increased, producing eccentric loading of the foot and leading to microfractures, ligament laxity, and bone destruction.
For more information, see “A disfigured foot with ulcer.” J Fam Pract. 2008;57(5):321-323.
1. A 43-year-old woman presents with progressively worsening bilateral great toe pain that began during pregnancy and increased following the birth of her daughter three years ago. Both of her feet have developed a crescent moon shape, making it painful and difficult to wear normal shoes. This patient has
a) Degenerative arthritis
b) Hallux varus
c) Gout
d) Traumatic sesamoiditis
Diagnosis: Physical exam revealed bilateral hallux varus deformity of the great toe, which was greater on the left foot than on the right (23° and 16°, respectively). The deformities were easily, passively, correctable. Standing radiographs showed evidence of previous proximal osteotomies and well-healed distal first metatarsal osteotomies. Due to unsuccessful nonoperative management, surgical reconstruction was offered.
For further information, see “Bilateral Hallux Varus Deformity Correction With a Suture Button Construct.” Am J Orthop. 2013;42(3):121-124.
2. Following treatment for onychomycosis, this 49-year-old man’s toenails demonstrate inflammation of the medial and lateral nail borders of the hallux and second toes of the left foot. This patient’s diagnosis is
a) Subungual exostosis
b) Primary osteomyelitis of the phalanx
c) Tumors of the nail bed
d) Onychocryptosis
Diagnosis: Onychocryptosis, also known as ingrown toenail, is a rare complication of oral antifungal therapy. As the healthy nail plate advances, it may adhere to the nail bed and cut into the lateral nail folds. In this case, the site of the onychocryptosis corresponded to the proximal clearing of the nail plate. The patient required excision of the nail borders, after which the secondary inflammation resolved. The condition was treated with chemical matrixectomy.
For more information, see “Multiple Onychocryptosis Following Treatment of Onychomycosis With Oral Terbinafine.” Cutis. 2000;66(3):211-212.
3. A 39-year-old man was playing a game of pick-up basketball when he felt a pop, immediately followed by a sharp pain in the back of his ankle and lower leg. He now walks with a limp. The cause is
a) Achilles tendon rupture
b) Medial gastrocnemius tear
c) Calf muscle strain
d) Posterior tibial stress syndrome
Diagnosis: Often diagnosed as an ankle sprain, an Achilles tendon rupture most commonly occurs in middle-aged men from overexertion in sports—usually tennis, racquetball, basketball, or badminton, which involve bursts of jumping, pivoting, and running. Rupture may also occur from a sudden stumble, fall from a significant height, or abrupt step into a hole or off a curb, which causes the tendon to overstretch forcefully.
4. A 60-year-old woman seeks relief for a foot wound of several months’ duration that persists despite use of antibiotics and proper care. The skin over the plantar surface has a full-thickness ulcer, with partial necrosis of the subcutaneous tissue. Her history is significant for diabetes with neuropathy, nephropathy, and retinopathy. The diagnosis is
a) Osteomyelitis
b) Deep venous thrombosis
c) Charcot joint
d) Septic joint
Diagnosis: Charcot joint changes, and an associated stage III pressure ulcer, account for the extensive collapse of the inner arch and “rocker bottom foot” seen on the radiograph. Also known as neurogenic arthropathy, Charcot joint is commonly seen with diabetic neuropathy. In affected patients, secondary degenerative changes to the joints occur with loss of normal muscle tone, proprioception, temperature perception, and pain perception. The joints become loose, enlarged, and boggy. There can be extensive cartilage erosion or osteophyte formation. The normal plantar and heel forces are increased, producing eccentric loading of the foot and leading to microfractures, ligament laxity, and bone destruction.
For more information, see “A disfigured foot with ulcer.” J Fam Pract. 2008;57(5):321-323.
Vaccines protect unborn mice from Zika infection
Researchers have reported that 2 different vaccines can protect mice from Zika virus-induced congenital disease.
Female mice that were vaccinated before pregnancy and infected with Zika virus while pregnant bore pups that showed no trace of the virus.
These results, published in Cell, offer the first evidence that a vaccine administered prior to pregnancy can protect fetuses from Zika infection and resulting injury.
“There are several vaccines in human trials right now, but, to date, none of them has been shown to protect during pregnancy,” said Michael S. Diamond, MD, PhD, of Washington University School of Medicine in St. Louis, Missouri.
“We tested 2 different vaccines, and they both provided substantial protection.”
Last year, Dr Diamond and his colleagues developed a mouse model of Zika infection that mimics the effects of the infection in pregnant women.
Using this model, the researchers evaluated the ability of 2 vaccines to protect fetuses whose mothers were infected during pregnancy.
One of the vaccines is a messenger RNA (mRNA) vaccine, developed by Moderna Therapeutics, that is currently in safety testing in men and women who are not pregnant.
The other is a live-attenuated vaccine, developed by the University of Texas Medical Branch, that is being tested in animals.
For this study, groups of 18 to 20 female mice were vaccinated with one of the vaccines or a placebo, and some animals received a second dose of the same vaccine or placebo a month later.
Three weeks later, the researchers measured antibody levels in the mice’s blood as a measure of the strength of their immune response.
Both vaccines elicited very high levels of neutralizing antibodies against Zika, while the placebos did not.
After the mice became pregnant, they were infected with Zika on the sixth day of pregnancy, to mimic the experience of a woman bitten by a Zika-carrying mosquito early in pregnancy.
One week after infection, the researchers measured the amount of virus in the mothers and fetuses.
With both vaccines, fetuses and placentas from vaccinated mice contained very low levels of Zika’s genetic material.
For the mRNA vaccine, more than half of the placentas and fetuses had no detectable viral genetic material at all.
The live-attenuated vaccine was even more effective. In 78% of the placentas and 83% of the fetuses, no viral genetic material was found.
“The amount of viral genetic material in the placentas and fetuses from the vaccinated females was just above the limit of detection,” Dr Diamond said. “It’s not totally clear whether it was infectious virus or just remnants of viruses that had already been killed.”
In contrast, the amount of detectable viral material in the placentas and fetuses of unvaccinated mice was hundreds to thousands of times higher.
The researchers repeated the experiment with different mice so they could evaluate the pups at birth.
None of the mothers in the placebo group made it to term. The mothers became seriously ill, many of the fetuses showed high levels of infection and died in utero, and the placentas showed severe damage.
In contrast, the vaccinated mothers remained healthy, all of their pups were born without obvious signs of injury, and the newborn pups had no measurable Zika virus in their heads.
“In general, most doctors don’t want to vaccinate during pregnancy on the outside chance that the immune response itself could harm the fetus,” Dr Diamond said. “But if you’re in an area where Zika is circulating, you might vaccinate during pregnancy because the risk of Zika infection is worse than some theoretical risk of immune-mediated damage.”
The researchers did not assess whether the vaccines are safe and effective for use during pregnancy.
Such studies couldn’t really be done in mice, Dr Diamond explained, because mouse pregnancies only last 19 days. That doesn’t allow enough time for a protective immune response to develop before the pups are born.
Additionally, this study did not address the question of whether the vaccines work when pregnant women are infected with Zika through sexual contact. It is possible that Zika virus in semen can travel to the uterus and then to the fetus without passing through the bloodstream.
“The question is, ‘Would the immunity still hold up if the virus does not pass through the bloodstream?’“ Dr Diamond said. “We think it will hold up, but that has to be tested.” ![]()
Researchers have reported that 2 different vaccines can protect mice from Zika virus-induced congenital disease.
Female mice that were vaccinated before pregnancy and infected with Zika virus while pregnant bore pups that showed no trace of the virus.
These results, published in Cell, offer the first evidence that a vaccine administered prior to pregnancy can protect fetuses from Zika infection and resulting injury.
“There are several vaccines in human trials right now, but, to date, none of them has been shown to protect during pregnancy,” said Michael S. Diamond, MD, PhD, of Washington University School of Medicine in St. Louis, Missouri.
“We tested 2 different vaccines, and they both provided substantial protection.”
Last year, Dr Diamond and his colleagues developed a mouse model of Zika infection that mimics the effects of the infection in pregnant women.
Using this model, the researchers evaluated the ability of 2 vaccines to protect fetuses whose mothers were infected during pregnancy.
One of the vaccines is a messenger RNA (mRNA) vaccine, developed by Moderna Therapeutics, that is currently in safety testing in men and women who are not pregnant.
The other is a live-attenuated vaccine, developed by the University of Texas Medical Branch, that is being tested in animals.
For this study, groups of 18 to 20 female mice were vaccinated with one of the vaccines or a placebo, and some animals received a second dose of the same vaccine or placebo a month later.
Three weeks later, the researchers measured antibody levels in the mice’s blood as a measure of the strength of their immune response.
Both vaccines elicited very high levels of neutralizing antibodies against Zika, while the placebos did not.
After the mice became pregnant, they were infected with Zika on the sixth day of pregnancy, to mimic the experience of a woman bitten by a Zika-carrying mosquito early in pregnancy.
One week after infection, the researchers measured the amount of virus in the mothers and fetuses.
With both vaccines, fetuses and placentas from vaccinated mice contained very low levels of Zika’s genetic material.
For the mRNA vaccine, more than half of the placentas and fetuses had no detectable viral genetic material at all.
The live-attenuated vaccine was even more effective. In 78% of the placentas and 83% of the fetuses, no viral genetic material was found.
“The amount of viral genetic material in the placentas and fetuses from the vaccinated females was just above the limit of detection,” Dr Diamond said. “It’s not totally clear whether it was infectious virus or just remnants of viruses that had already been killed.”
In contrast, the amount of detectable viral material in the placentas and fetuses of unvaccinated mice was hundreds to thousands of times higher.
The researchers repeated the experiment with different mice so they could evaluate the pups at birth.
None of the mothers in the placebo group made it to term. The mothers became seriously ill, many of the fetuses showed high levels of infection and died in utero, and the placentas showed severe damage.
In contrast, the vaccinated mothers remained healthy, all of their pups were born without obvious signs of injury, and the newborn pups had no measurable Zika virus in their heads.
“In general, most doctors don’t want to vaccinate during pregnancy on the outside chance that the immune response itself could harm the fetus,” Dr Diamond said. “But if you’re in an area where Zika is circulating, you might vaccinate during pregnancy because the risk of Zika infection is worse than some theoretical risk of immune-mediated damage.”
The researchers did not assess whether the vaccines are safe and effective for use during pregnancy.
Such studies couldn’t really be done in mice, Dr Diamond explained, because mouse pregnancies only last 19 days. That doesn’t allow enough time for a protective immune response to develop before the pups are born.
Additionally, this study did not address the question of whether the vaccines work when pregnant women are infected with Zika through sexual contact. It is possible that Zika virus in semen can travel to the uterus and then to the fetus without passing through the bloodstream.
“The question is, ‘Would the immunity still hold up if the virus does not pass through the bloodstream?’“ Dr Diamond said. “We think it will hold up, but that has to be tested.” ![]()
Researchers have reported that 2 different vaccines can protect mice from Zika virus-induced congenital disease.
Female mice that were vaccinated before pregnancy and infected with Zika virus while pregnant bore pups that showed no trace of the virus.
These results, published in Cell, offer the first evidence that a vaccine administered prior to pregnancy can protect fetuses from Zika infection and resulting injury.
“There are several vaccines in human trials right now, but, to date, none of them has been shown to protect during pregnancy,” said Michael S. Diamond, MD, PhD, of Washington University School of Medicine in St. Louis, Missouri.
“We tested 2 different vaccines, and they both provided substantial protection.”
Last year, Dr Diamond and his colleagues developed a mouse model of Zika infection that mimics the effects of the infection in pregnant women.
Using this model, the researchers evaluated the ability of 2 vaccines to protect fetuses whose mothers were infected during pregnancy.
One of the vaccines is a messenger RNA (mRNA) vaccine, developed by Moderna Therapeutics, that is currently in safety testing in men and women who are not pregnant.
The other is a live-attenuated vaccine, developed by the University of Texas Medical Branch, that is being tested in animals.
For this study, groups of 18 to 20 female mice were vaccinated with one of the vaccines or a placebo, and some animals received a second dose of the same vaccine or placebo a month later.
Three weeks later, the researchers measured antibody levels in the mice’s blood as a measure of the strength of their immune response.
Both vaccines elicited very high levels of neutralizing antibodies against Zika, while the placebos did not.
After the mice became pregnant, they were infected with Zika on the sixth day of pregnancy, to mimic the experience of a woman bitten by a Zika-carrying mosquito early in pregnancy.
One week after infection, the researchers measured the amount of virus in the mothers and fetuses.
With both vaccines, fetuses and placentas from vaccinated mice contained very low levels of Zika’s genetic material.
For the mRNA vaccine, more than half of the placentas and fetuses had no detectable viral genetic material at all.
The live-attenuated vaccine was even more effective. In 78% of the placentas and 83% of the fetuses, no viral genetic material was found.
“The amount of viral genetic material in the placentas and fetuses from the vaccinated females was just above the limit of detection,” Dr Diamond said. “It’s not totally clear whether it was infectious virus or just remnants of viruses that had already been killed.”
In contrast, the amount of detectable viral material in the placentas and fetuses of unvaccinated mice was hundreds to thousands of times higher.
The researchers repeated the experiment with different mice so they could evaluate the pups at birth.
None of the mothers in the placebo group made it to term. The mothers became seriously ill, many of the fetuses showed high levels of infection and died in utero, and the placentas showed severe damage.
In contrast, the vaccinated mothers remained healthy, all of their pups were born without obvious signs of injury, and the newborn pups had no measurable Zika virus in their heads.
“In general, most doctors don’t want to vaccinate during pregnancy on the outside chance that the immune response itself could harm the fetus,” Dr Diamond said. “But if you’re in an area where Zika is circulating, you might vaccinate during pregnancy because the risk of Zika infection is worse than some theoretical risk of immune-mediated damage.”
The researchers did not assess whether the vaccines are safe and effective for use during pregnancy.
Such studies couldn’t really be done in mice, Dr Diamond explained, because mouse pregnancies only last 19 days. That doesn’t allow enough time for a protective immune response to develop before the pups are born.
Additionally, this study did not address the question of whether the vaccines work when pregnant women are infected with Zika through sexual contact. It is possible that Zika virus in semen can travel to the uterus and then to the fetus without passing through the bloodstream.
“The question is, ‘Would the immunity still hold up if the virus does not pass through the bloodstream?’“ Dr Diamond said. “We think it will hold up, but that has to be tested.” ![]()
Children with psoriasis face multitude of comorbidities
CHICAGO – Children with psoriasis face a multitude of potential problems and comorbidities, ranging from anxiety and depression to obesity and metabolic disease, so early and proactive identification is key.
“These children are more likely to engage in high-risk behavior such as use of alcohol, tobacco, and drugs – a trend that continues into adult ages,” Kelly M. Cordoro, MD, said at the World Congress for Pediatric Dermatology. “They also have a higher association with inflammatory bowel disease, among other conditions. Those of us who care for pediatric psoriasis patients are on the front lines of recognition of these potential comorbidities, which allow for, ideally, prevention and certainly, early intervention.”

Obesity ranks as the most well understood comorbidity of psoriasis in children. Study after study has demonstrated this association. In addition, obese children with psoriasis may also harbor components of the metabolic syndrome – hypertension, dyslipidemia, and diabetes. “They’re not as much at risk for metabolic syndrome in the absence of obesity, but there’s still a small signal,” Dr. Cordoro said. “We ask ourselves this question as clinicians: Are these pediatric patients at risk for cardiovascular and cerebrovascular disease as they get older? In other words, what is the health of a 6-year-old, obese child with severe psoriasis, who may also have other components of the metabolic syndrome, going to be like when he is 35 or 40? Are these the children who go on to have cardiovascular events as documented in adult studies of psoriasis?”
To date, several studies have identified a clear link between psoriasis and obesity, and between psoriasis and hypertension, diabetes, and dyslipidemia in certain populations. “There is a dose-response effect,” Dr. Cordoro said. “The more severe the psoriasis, the more likely the patient is to be obese, and vice versa.” In one study, researchers analyzed 409 psoriasis patients up to age 17 years in nine countries (JAMA Dermatol. 2013;149:166-76). They concluded that globally, children with psoriasis have excess adiposity and increased central adiposity regardless of psoriasis severity. The researchers used multiple measures of adiposity, not just body mass index, but also waist circumference and waist-to-height ratio. “Waist circumference and waist-to-height ratio are surrogates for central and visceral adiposity,” said Dr. Cordoro, who was involved with the study. “And central adiposity may be a more sensitive indicator of metabolic disease and cardiovascular risk than BMI [body mass index] alone.”
Another study demonstrated that high adiposity preceded psoriasis by up to 2 years in 93% of overweight or obese psoriatic children (JAMA Dermatol. 2014;150:573-4).
In a more recent analysis, researchers evaluated lipid function in 44 psoriatic children (J Invest Dermatol. 2016;136[1]:67-73). Compared with age-matched controls, children with psoriasis were found to have higher waist-to-hip ratio, higher insulin resistance, and 27% were obese. “There was no difference in fasting lipid levels but the blood profiles had atherogenic markers that are worrisome for ongoing risk for atherosclerosis, cardiovascular disease, and cerebrovascular disease,” Dr. Cordoro said.
Research among adults has demonstrated that psoriasis confers an independent risk of atherosclerosis, MI, stroke, and early cardiovascular-related mortality, the so-called “psoriatic march.” Theoretically, Dr. Cordoro said, severe psoriasis sets up a state of chronic systemic inflammation, which leads to insulin resistance, which predisposes affected individuals to endothelial dysfunction, and eventually can lead to atherosclerosis. “When atherosclerosis becomes unstable, now you’ve gone from having severe psoriasis into a situation where the chronic inflammation may have predisposed you to having a thrombotic event such as a heart attack or stroke,” she said. “Obesity replicates that same pattern. What does this all mean? Is this real or is this just a theory? We don’t know, but it’s certainly biologically plausible. It’s not been proven with long-term prospective studies, which we need.”
Dr. Cordoro went on to discuss the importance of assessing young psoriasis patients for psychiatric and emotional comorbidities, including anxiety, depression, and eating disorders. “These kids can become socially isolated, which can lead to more downstream effects: more anxiety, more depression, sometimes overeating and obesity,” she said. “It’s not only that the patient has situational anxiety or depression, the notion that ‘My skin looks terrible. I’m really depressed about it;’ it’s more than that. It turns out that the same inflammatory milieu in psoriasis lesions can be replicated in the brain inflammatory milieu in patients with depression and other psychiatric disorders. That’s fascinating to recognize that these comorbidities can be intrinsic. There’s a biological basis and not just a downstream effect.”
She advises clinicians who care for children with psoriasis to keep potential comorbidities in mind, and to make sure families understand that there can be psychiatric, emotional, and physical consequences to undertreated disease. “We do not yet know how to risk stratify these patients. At the very least, you want to identify overweight or obese children with moderate to severe disease for early intervention,” Dr. Cordoro said. “Weight loss and lifestyle interventions are the hardest goals to accomplish but are really critical. Prevention is the best strategy. We can help ourselves and help our patients by referring to obesity and nutrition experts who can not only help the child but get the entire family involved.”
In a consensus statement published online in JAMA Dermatology, a multidisciplinary panel of experts including Dr. Cordoro offer an evidence- and consensus-based approach to screening children with psoriasis, based on a review of 153 manuscripts in the medical literature. The panel recommends that all psoriasis patients 2-21 years of age should undergo annual measurements of blood pressure and BMI, and screenings for arthritis and mood disorders. “These don’t have to be formal mood disorder screens,” Dr. Cordoro said. “They can be informal questioning about anxiety and depression, like ‘How is your psoriasis impacting you? How do you feel about your psoriasis? What do you say when people ask you about your psoriasis?’ It’s also important to ask overweight patients what they’re doing to keep their weight in check. Oftentimes when you ask a question about mood or impact of disease or stigma or bullying, the child will be completely silent and either stay silent or start crying or start telling you their stories. It’s really important to ask, because it validates that their concerns are more than just about vanity but about their overall health, and that is a critical difference.”
Dr. Cordoro disclosed that she is a consultant for Pfizer and Valeant.
CHICAGO – Children with psoriasis face a multitude of potential problems and comorbidities, ranging from anxiety and depression to obesity and metabolic disease, so early and proactive identification is key.
“These children are more likely to engage in high-risk behavior such as use of alcohol, tobacco, and drugs – a trend that continues into adult ages,” Kelly M. Cordoro, MD, said at the World Congress for Pediatric Dermatology. “They also have a higher association with inflammatory bowel disease, among other conditions. Those of us who care for pediatric psoriasis patients are on the front lines of recognition of these potential comorbidities, which allow for, ideally, prevention and certainly, early intervention.”
Obesity ranks as the most well understood comorbidity of psoriasis in children. Study after study has demonstrated this association. In addition, obese children with psoriasis may also harbor components of the metabolic syndrome – hypertension, dyslipidemia, and diabetes. “They’re not as much at risk for metabolic syndrome in the absence of obesity, but there’s still a small signal,” Dr. Cordoro said. “We ask ourselves this question as clinicians: Are these pediatric patients at risk for cardiovascular and cerebrovascular disease as they get older? In other words, what is the health of a 6-year-old, obese child with severe psoriasis, who may also have other components of the metabolic syndrome, going to be like when he is 35 or 40? Are these the children who go on to have cardiovascular events as documented in adult studies of psoriasis?”
To date, several studies have identified a clear link between psoriasis and obesity, and between psoriasis and hypertension, diabetes, and dyslipidemia in certain populations. “There is a dose-response effect,” Dr. Cordoro said. “The more severe the psoriasis, the more likely the patient is to be obese, and vice versa.” In one study, researchers analyzed 409 psoriasis patients up to age 17 years in nine countries (JAMA Dermatol. 2013;149:166-76). They concluded that globally, children with psoriasis have excess adiposity and increased central adiposity regardless of psoriasis severity. The researchers used multiple measures of adiposity, not just body mass index, but also waist circumference and waist-to-height ratio. “Waist circumference and waist-to-height ratio are surrogates for central and visceral adiposity,” said Dr. Cordoro, who was involved with the study. “And central adiposity may be a more sensitive indicator of metabolic disease and cardiovascular risk than BMI [body mass index] alone.”
Another study demonstrated that high adiposity preceded psoriasis by up to 2 years in 93% of overweight or obese psoriatic children (JAMA Dermatol. 2014;150:573-4).
In a more recent analysis, researchers evaluated lipid function in 44 psoriatic children (J Invest Dermatol. 2016;136[1]:67-73). Compared with age-matched controls, children with psoriasis were found to have higher waist-to-hip ratio, higher insulin resistance, and 27% were obese. “There was no difference in fasting lipid levels but the blood profiles had atherogenic markers that are worrisome for ongoing risk for atherosclerosis, cardiovascular disease, and cerebrovascular disease,” Dr. Cordoro said.
Research among adults has demonstrated that psoriasis confers an independent risk of atherosclerosis, MI, stroke, and early cardiovascular-related mortality, the so-called “psoriatic march.” Theoretically, Dr. Cordoro said, severe psoriasis sets up a state of chronic systemic inflammation, which leads to insulin resistance, which predisposes affected individuals to endothelial dysfunction, and eventually can lead to atherosclerosis. “When atherosclerosis becomes unstable, now you’ve gone from having severe psoriasis into a situation where the chronic inflammation may have predisposed you to having a thrombotic event such as a heart attack or stroke,” she said. “Obesity replicates that same pattern. What does this all mean? Is this real or is this just a theory? We don’t know, but it’s certainly biologically plausible. It’s not been proven with long-term prospective studies, which we need.”
Dr. Cordoro went on to discuss the importance of assessing young psoriasis patients for psychiatric and emotional comorbidities, including anxiety, depression, and eating disorders. “These kids can become socially isolated, which can lead to more downstream effects: more anxiety, more depression, sometimes overeating and obesity,” she said. “It’s not only that the patient has situational anxiety or depression, the notion that ‘My skin looks terrible. I’m really depressed about it;’ it’s more than that. It turns out that the same inflammatory milieu in psoriasis lesions can be replicated in the brain inflammatory milieu in patients with depression and other psychiatric disorders. That’s fascinating to recognize that these comorbidities can be intrinsic. There’s a biological basis and not just a downstream effect.”
She advises clinicians who care for children with psoriasis to keep potential comorbidities in mind, and to make sure families understand that there can be psychiatric, emotional, and physical consequences to undertreated disease. “We do not yet know how to risk stratify these patients. At the very least, you want to identify overweight or obese children with moderate to severe disease for early intervention,” Dr. Cordoro said. “Weight loss and lifestyle interventions are the hardest goals to accomplish but are really critical. Prevention is the best strategy. We can help ourselves and help our patients by referring to obesity and nutrition experts who can not only help the child but get the entire family involved.”
In a consensus statement published online in JAMA Dermatology, a multidisciplinary panel of experts including Dr. Cordoro offer an evidence- and consensus-based approach to screening children with psoriasis, based on a review of 153 manuscripts in the medical literature. The panel recommends that all psoriasis patients 2-21 years of age should undergo annual measurements of blood pressure and BMI, and screenings for arthritis and mood disorders. “These don’t have to be formal mood disorder screens,” Dr. Cordoro said. “They can be informal questioning about anxiety and depression, like ‘How is your psoriasis impacting you? How do you feel about your psoriasis? What do you say when people ask you about your psoriasis?’ It’s also important to ask overweight patients what they’re doing to keep their weight in check. Oftentimes when you ask a question about mood or impact of disease or stigma or bullying, the child will be completely silent and either stay silent or start crying or start telling you their stories. It’s really important to ask, because it validates that their concerns are more than just about vanity but about their overall health, and that is a critical difference.”
Dr. Cordoro disclosed that she is a consultant for Pfizer and Valeant.
CHICAGO – Children with psoriasis face a multitude of potential problems and comorbidities, ranging from anxiety and depression to obesity and metabolic disease, so early and proactive identification is key.
“These children are more likely to engage in high-risk behavior such as use of alcohol, tobacco, and drugs – a trend that continues into adult ages,” Kelly M. Cordoro, MD, said at the World Congress for Pediatric Dermatology. “They also have a higher association with inflammatory bowel disease, among other conditions. Those of us who care for pediatric psoriasis patients are on the front lines of recognition of these potential comorbidities, which allow for, ideally, prevention and certainly, early intervention.”
Obesity ranks as the most well understood comorbidity of psoriasis in children. Study after study has demonstrated this association. In addition, obese children with psoriasis may also harbor components of the metabolic syndrome – hypertension, dyslipidemia, and diabetes. “They’re not as much at risk for metabolic syndrome in the absence of obesity, but there’s still a small signal,” Dr. Cordoro said. “We ask ourselves this question as clinicians: Are these pediatric patients at risk for cardiovascular and cerebrovascular disease as they get older? In other words, what is the health of a 6-year-old, obese child with severe psoriasis, who may also have other components of the metabolic syndrome, going to be like when he is 35 or 40? Are these the children who go on to have cardiovascular events as documented in adult studies of psoriasis?”
To date, several studies have identified a clear link between psoriasis and obesity, and between psoriasis and hypertension, diabetes, and dyslipidemia in certain populations. “There is a dose-response effect,” Dr. Cordoro said. “The more severe the psoriasis, the more likely the patient is to be obese, and vice versa.” In one study, researchers analyzed 409 psoriasis patients up to age 17 years in nine countries (JAMA Dermatol. 2013;149:166-76). They concluded that globally, children with psoriasis have excess adiposity and increased central adiposity regardless of psoriasis severity. The researchers used multiple measures of adiposity, not just body mass index, but also waist circumference and waist-to-height ratio. “Waist circumference and waist-to-height ratio are surrogates for central and visceral adiposity,” said Dr. Cordoro, who was involved with the study. “And central adiposity may be a more sensitive indicator of metabolic disease and cardiovascular risk than BMI [body mass index] alone.”
Another study demonstrated that high adiposity preceded psoriasis by up to 2 years in 93% of overweight or obese psoriatic children (JAMA Dermatol. 2014;150:573-4).
In a more recent analysis, researchers evaluated lipid function in 44 psoriatic children (J Invest Dermatol. 2016;136[1]:67-73). Compared with age-matched controls, children with psoriasis were found to have higher waist-to-hip ratio, higher insulin resistance, and 27% were obese. “There was no difference in fasting lipid levels but the blood profiles had atherogenic markers that are worrisome for ongoing risk for atherosclerosis, cardiovascular disease, and cerebrovascular disease,” Dr. Cordoro said.
Research among adults has demonstrated that psoriasis confers an independent risk of atherosclerosis, MI, stroke, and early cardiovascular-related mortality, the so-called “psoriatic march.” Theoretically, Dr. Cordoro said, severe psoriasis sets up a state of chronic systemic inflammation, which leads to insulin resistance, which predisposes affected individuals to endothelial dysfunction, and eventually can lead to atherosclerosis. “When atherosclerosis becomes unstable, now you’ve gone from having severe psoriasis into a situation where the chronic inflammation may have predisposed you to having a thrombotic event such as a heart attack or stroke,” she said. “Obesity replicates that same pattern. What does this all mean? Is this real or is this just a theory? We don’t know, but it’s certainly biologically plausible. It’s not been proven with long-term prospective studies, which we need.”
Dr. Cordoro went on to discuss the importance of assessing young psoriasis patients for psychiatric and emotional comorbidities, including anxiety, depression, and eating disorders. “These kids can become socially isolated, which can lead to more downstream effects: more anxiety, more depression, sometimes overeating and obesity,” she said. “It’s not only that the patient has situational anxiety or depression, the notion that ‘My skin looks terrible. I’m really depressed about it;’ it’s more than that. It turns out that the same inflammatory milieu in psoriasis lesions can be replicated in the brain inflammatory milieu in patients with depression and other psychiatric disorders. That’s fascinating to recognize that these comorbidities can be intrinsic. There’s a biological basis and not just a downstream effect.”
She advises clinicians who care for children with psoriasis to keep potential comorbidities in mind, and to make sure families understand that there can be psychiatric, emotional, and physical consequences to undertreated disease. “We do not yet know how to risk stratify these patients. At the very least, you want to identify overweight or obese children with moderate to severe disease for early intervention,” Dr. Cordoro said. “Weight loss and lifestyle interventions are the hardest goals to accomplish but are really critical. Prevention is the best strategy. We can help ourselves and help our patients by referring to obesity and nutrition experts who can not only help the child but get the entire family involved.”
In a consensus statement published online in JAMA Dermatology, a multidisciplinary panel of experts including Dr. Cordoro offer an evidence- and consensus-based approach to screening children with psoriasis, based on a review of 153 manuscripts in the medical literature. The panel recommends that all psoriasis patients 2-21 years of age should undergo annual measurements of blood pressure and BMI, and screenings for arthritis and mood disorders. “These don’t have to be formal mood disorder screens,” Dr. Cordoro said. “They can be informal questioning about anxiety and depression, like ‘How is your psoriasis impacting you? How do you feel about your psoriasis? What do you say when people ask you about your psoriasis?’ It’s also important to ask overweight patients what they’re doing to keep their weight in check. Oftentimes when you ask a question about mood or impact of disease or stigma or bullying, the child will be completely silent and either stay silent or start crying or start telling you their stories. It’s really important to ask, because it validates that their concerns are more than just about vanity but about their overall health, and that is a critical difference.”
Dr. Cordoro disclosed that she is a consultant for Pfizer and Valeant.
AT WCPD 2017
Surgeon, primary care collaboration needed to catch hyperparathyroidism
A report from the University of Alabama at Birmingham provides further evidence that hyperparathyroidism is often missed in the United States health care system.
Investigators reviewed the electronic health records for 682,704 patients at the university from 2011 to 2015 and identified hypercalcemia (serum calcium greater than 10.5 mg/dL) – usually the first indication of disease – in 10,432 patients. The next step should have been a parathyroid hormone (PTH) measurement, but PTH was measured in only 3,200 patients (31%), and it usually took multiple abnormal calcium levels before PTH was checked, reported Courtney Balentine, MD, and her colleagues at the University of Alabama at Birmingham.
In addition, 592 of 2,666 patients (22%) with both elevated calcium and PTH levels were referred to surgeons for a parathyroidectomy consult, although parathyroidectomy is a low-risk outpatient procedure that cures up to 95% of patients, and surgeons are best suited to discuss the risks and benefits of the procedure with patients, investigators said (Ann Surg. 2017 Jul 3. doi: 10.1097/SLA.0000000000002370).
Underdiagnosis and treatment of hyperparathyroidism can lead to fractures, kidney stones, depression, cognitive impairment, hypertension, stroke, and myocardial infarction.
The investigators plan to meet with primary care doctors to hear how they think the problem should be addressed. Alerts and automatic referrals are also being considered for the EHR. “The combination of systems changes and stakeholder engagement is more likely to succeed than focusing on one component to the exclusion of others,” Dr. Balentine and colleagues said.
The issue could be that primary care physicians are just too overwhelmed to notice or be concerned about an isolated abnormal calcium level in an otherwise routine assessment. Or perhaps they assume surgeons come into play only if there are kidney stones, bone changes, or other obvious signs of trouble, they said.
The team has started to look at charts to get a better understanding of what’s going wrong. “I have been a little bit flabbergasted by how many excuses there are for either not checking a PTH or not referring once the diagnosis is there. … ‘This patient probably would not benefit from the surgery; the risk is too high; he or she does not want X, Y, or Z.’ I think if they just refer [patients to surgeons] to have the conversation, we might very well agree with them, but at least we [could] have the conversation with the patient, and I think [that] would make more sense,” Dr. Balentine said in a transcript of a question and answer session that was published with the report.
“Indeed, the recent American Association of Endocrine Surgery guidelines emphasize the importance of referring patients to surgical experts for discussion of treatment options,” the investigators said in the report.
It’s possible that elevated calcium and PTH levels were evaluated and treated at other institutions and so were not captured by the analysis. “Our mean follow-up was 16 months, however, which suggests that most patients were seen in the UAB system long enough to undergo appropriate evaluation and referral.” Also, patients with “two or more abnormal calcium values had similarly low rates of surgical referral, which suggests that loss to follow-up is unlikely to explain our findings,” they said.
The mean age of the cohort was 54 years; 56% of the patients were white, and 61% were women.
The work was supported by the Agency for Healthcare Research and Quality. The authors reported no conflicts of interest.
A report from the University of Alabama at Birmingham provides further evidence that hyperparathyroidism is often missed in the United States health care system.
Investigators reviewed the electronic health records for 682,704 patients at the university from 2011 to 2015 and identified hypercalcemia (serum calcium greater than 10.5 mg/dL) – usually the first indication of disease – in 10,432 patients. The next step should have been a parathyroid hormone (PTH) measurement, but PTH was measured in only 3,200 patients (31%), and it usually took multiple abnormal calcium levels before PTH was checked, reported Courtney Balentine, MD, and her colleagues at the University of Alabama at Birmingham.
In addition, 592 of 2,666 patients (22%) with both elevated calcium and PTH levels were referred to surgeons for a parathyroidectomy consult, although parathyroidectomy is a low-risk outpatient procedure that cures up to 95% of patients, and surgeons are best suited to discuss the risks and benefits of the procedure with patients, investigators said (Ann Surg. 2017 Jul 3. doi: 10.1097/SLA.0000000000002370).
Underdiagnosis and treatment of hyperparathyroidism can lead to fractures, kidney stones, depression, cognitive impairment, hypertension, stroke, and myocardial infarction.
The investigators plan to meet with primary care doctors to hear how they think the problem should be addressed. Alerts and automatic referrals are also being considered for the EHR. “The combination of systems changes and stakeholder engagement is more likely to succeed than focusing on one component to the exclusion of others,” Dr. Balentine and colleagues said.
The issue could be that primary care physicians are just too overwhelmed to notice or be concerned about an isolated abnormal calcium level in an otherwise routine assessment. Or perhaps they assume surgeons come into play only if there are kidney stones, bone changes, or other obvious signs of trouble, they said.
The team has started to look at charts to get a better understanding of what’s going wrong. “I have been a little bit flabbergasted by how many excuses there are for either not checking a PTH or not referring once the diagnosis is there. … ‘This patient probably would not benefit from the surgery; the risk is too high; he or she does not want X, Y, or Z.’ I think if they just refer [patients to surgeons] to have the conversation, we might very well agree with them, but at least we [could] have the conversation with the patient, and I think [that] would make more sense,” Dr. Balentine said in a transcript of a question and answer session that was published with the report.
“Indeed, the recent American Association of Endocrine Surgery guidelines emphasize the importance of referring patients to surgical experts for discussion of treatment options,” the investigators said in the report.
It’s possible that elevated calcium and PTH levels were evaluated and treated at other institutions and so were not captured by the analysis. “Our mean follow-up was 16 months, however, which suggests that most patients were seen in the UAB system long enough to undergo appropriate evaluation and referral.” Also, patients with “two or more abnormal calcium values had similarly low rates of surgical referral, which suggests that loss to follow-up is unlikely to explain our findings,” they said.
The mean age of the cohort was 54 years; 56% of the patients were white, and 61% were women.
The work was supported by the Agency for Healthcare Research and Quality. The authors reported no conflicts of interest.
A report from the University of Alabama at Birmingham provides further evidence that hyperparathyroidism is often missed in the United States health care system.
Investigators reviewed the electronic health records for 682,704 patients at the university from 2011 to 2015 and identified hypercalcemia (serum calcium greater than 10.5 mg/dL) – usually the first indication of disease – in 10,432 patients. The next step should have been a parathyroid hormone (PTH) measurement, but PTH was measured in only 3,200 patients (31%), and it usually took multiple abnormal calcium levels before PTH was checked, reported Courtney Balentine, MD, and her colleagues at the University of Alabama at Birmingham.
In addition, 592 of 2,666 patients (22%) with both elevated calcium and PTH levels were referred to surgeons for a parathyroidectomy consult, although parathyroidectomy is a low-risk outpatient procedure that cures up to 95% of patients, and surgeons are best suited to discuss the risks and benefits of the procedure with patients, investigators said (Ann Surg. 2017 Jul 3. doi: 10.1097/SLA.0000000000002370).
Underdiagnosis and treatment of hyperparathyroidism can lead to fractures, kidney stones, depression, cognitive impairment, hypertension, stroke, and myocardial infarction.
The investigators plan to meet with primary care doctors to hear how they think the problem should be addressed. Alerts and automatic referrals are also being considered for the EHR. “The combination of systems changes and stakeholder engagement is more likely to succeed than focusing on one component to the exclusion of others,” Dr. Balentine and colleagues said.
The issue could be that primary care physicians are just too overwhelmed to notice or be concerned about an isolated abnormal calcium level in an otherwise routine assessment. Or perhaps they assume surgeons come into play only if there are kidney stones, bone changes, or other obvious signs of trouble, they said.
The team has started to look at charts to get a better understanding of what’s going wrong. “I have been a little bit flabbergasted by how many excuses there are for either not checking a PTH or not referring once the diagnosis is there. … ‘This patient probably would not benefit from the surgery; the risk is too high; he or she does not want X, Y, or Z.’ I think if they just refer [patients to surgeons] to have the conversation, we might very well agree with them, but at least we [could] have the conversation with the patient, and I think [that] would make more sense,” Dr. Balentine said in a transcript of a question and answer session that was published with the report.
“Indeed, the recent American Association of Endocrine Surgery guidelines emphasize the importance of referring patients to surgical experts for discussion of treatment options,” the investigators said in the report.
It’s possible that elevated calcium and PTH levels were evaluated and treated at other institutions and so were not captured by the analysis. “Our mean follow-up was 16 months, however, which suggests that most patients were seen in the UAB system long enough to undergo appropriate evaluation and referral.” Also, patients with “two or more abnormal calcium values had similarly low rates of surgical referral, which suggests that loss to follow-up is unlikely to explain our findings,” they said.
The mean age of the cohort was 54 years; 56% of the patients were white, and 61% were women.
The work was supported by the Agency for Healthcare Research and Quality. The authors reported no conflicts of interest.
FROM ANNALS OF SURGERY
Key clinical point:
Major finding: Just 31% of patients with a finding of hypercalcemia went on to have their parathyroid hormone levels checked.
Data source: Electronic health record review of 682,704 patients at a large American university.
Disclosures: The work was supported by the Agency for Healthcare Research and Quality. The authors reported no conflicts of interest.
Intense exercise reduces hypoglycemia awareness in type 1 diabetes
A single 15-minute session of high-intensity exercise masked symptoms of subsequent hypoglycemia in patients with type 1 diabetes and normal hypoglycemia awareness, according to Dutch investigators.
Twenty patients with type 1 diabetes – 10 with normal hypoglycemia awareness and 10 with impaired awareness – and 10 healthy subjects were asked to push themselves as hard as they could for 30-second bursts during 15 minutes of stationary bicycling and to go at an easier pace in between, reported Hanne Rooijackers, MD, of the Radboud University Medical Center, Nijmegen, the Netherlands, and colleagues.
The subjects had intravenous cannulae in both forearms, one for blood draws and the other for glucose and insulin administration. All three groups were kept euglycemic during exercise, but then were subjected to hyperinsulinemic-hypoglycemic clamps afterwards. Plasma glucose levels were allowed to fall to 2.6 mmol/L over about 35 min., and were kept there for another 60 min. Trembling, palpitations, anxiety, and other symptoms were serially assessed while patients were hypoglycemic, along with cognitive function and levels of hormones involved with hypoglycemic defense.
For comparison, the subjects all had clamps applied and hypoglycemia symptoms and physiologic responses assessed after a 15-minute session of rest at least 2 weeks apart from the exercise session.
The healthy subjects had a peak of about 20 points on a composite score of hypoglycemia symptoms during rest; exercise reduced the peak score only a small amount to about 18 points. Diabetic patients with normal hypoglycemia awareness hit a peak symptom score of 31 points during rest, which fell substantially after exercise to 22 points. Exercise, meanwhile, had no effect on diabetic patients with impaired awareness; after both rest and exercise, they had a peak composite symptom score of about 11 points, Dr. Rooijackers and colleagues reported (Diabetes. 2017 Jul;66[7]:1990-8).
High-intensity interval training (HIIT) did not affect hypoglycemic awareness in patients with impaired awareness probably because of “a ‘floor’ effect, in that symptom responses could not be further suppressed than they already were,” the investigators speculated.
Regular exercise is recommended for patients with type 1 diabetes, and, like others, they are turning to HIIT – short bursts of intense exercise broken up by brief periods of rest or lower intensity movement – because it appears to deliver the benefits for more moderate exercise in less time.
The findings suggest, however, that high-intensity exercise might increase the risk of severe hypoglycemia in type 1 patients by reducing awareness of its symptoms and blunting hormonal defenses.
The team suspects elevated lactate levels account for the findings. Exercise increases plasma lactate levels, and as blood glucose levels fall, the brain uses lactate as an alternative fuel, which likely blunts the effects of hypoglycemia. Plasma lactate spiked in the study subjects about 15 minutes after exercise.
Participants presented early in the morning after fasting overnight and abstaining from strenuous exercise for 2 days. They were in their mid-20s on average, normal weight, and fairly well balanced between men and women. Patients with type 1 diabetes were eligible for the study if they had hemoglobin A1c levels below 9% and no vascular complications beyond retinopathy. Their duration of diabetes was about 10 years.
The work was funded by the Dutch Diabetes Research Foundation and the European Foundation for the Study of Diabetes. The authors had no conflicts of interest.
A single 15-minute session of high-intensity exercise masked symptoms of subsequent hypoglycemia in patients with type 1 diabetes and normal hypoglycemia awareness, according to Dutch investigators.
Twenty patients with type 1 diabetes – 10 with normal hypoglycemia awareness and 10 with impaired awareness – and 10 healthy subjects were asked to push themselves as hard as they could for 30-second bursts during 15 minutes of stationary bicycling and to go at an easier pace in between, reported Hanne Rooijackers, MD, of the Radboud University Medical Center, Nijmegen, the Netherlands, and colleagues.
The subjects had intravenous cannulae in both forearms, one for blood draws and the other for glucose and insulin administration. All three groups were kept euglycemic during exercise, but then were subjected to hyperinsulinemic-hypoglycemic clamps afterwards. Plasma glucose levels were allowed to fall to 2.6 mmol/L over about 35 min., and were kept there for another 60 min. Trembling, palpitations, anxiety, and other symptoms were serially assessed while patients were hypoglycemic, along with cognitive function and levels of hormones involved with hypoglycemic defense.
For comparison, the subjects all had clamps applied and hypoglycemia symptoms and physiologic responses assessed after a 15-minute session of rest at least 2 weeks apart from the exercise session.
The healthy subjects had a peak of about 20 points on a composite score of hypoglycemia symptoms during rest; exercise reduced the peak score only a small amount to about 18 points. Diabetic patients with normal hypoglycemia awareness hit a peak symptom score of 31 points during rest, which fell substantially after exercise to 22 points. Exercise, meanwhile, had no effect on diabetic patients with impaired awareness; after both rest and exercise, they had a peak composite symptom score of about 11 points, Dr. Rooijackers and colleagues reported (Diabetes. 2017 Jul;66[7]:1990-8).
High-intensity interval training (HIIT) did not affect hypoglycemic awareness in patients with impaired awareness probably because of “a ‘floor’ effect, in that symptom responses could not be further suppressed than they already were,” the investigators speculated.
Regular exercise is recommended for patients with type 1 diabetes, and, like others, they are turning to HIIT – short bursts of intense exercise broken up by brief periods of rest or lower intensity movement – because it appears to deliver the benefits for more moderate exercise in less time.
The findings suggest, however, that high-intensity exercise might increase the risk of severe hypoglycemia in type 1 patients by reducing awareness of its symptoms and blunting hormonal defenses.
The team suspects elevated lactate levels account for the findings. Exercise increases plasma lactate levels, and as blood glucose levels fall, the brain uses lactate as an alternative fuel, which likely blunts the effects of hypoglycemia. Plasma lactate spiked in the study subjects about 15 minutes after exercise.
Participants presented early in the morning after fasting overnight and abstaining from strenuous exercise for 2 days. They were in their mid-20s on average, normal weight, and fairly well balanced between men and women. Patients with type 1 diabetes were eligible for the study if they had hemoglobin A1c levels below 9% and no vascular complications beyond retinopathy. Their duration of diabetes was about 10 years.
The work was funded by the Dutch Diabetes Research Foundation and the European Foundation for the Study of Diabetes. The authors had no conflicts of interest.
A single 15-minute session of high-intensity exercise masked symptoms of subsequent hypoglycemia in patients with type 1 diabetes and normal hypoglycemia awareness, according to Dutch investigators.
Twenty patients with type 1 diabetes – 10 with normal hypoglycemia awareness and 10 with impaired awareness – and 10 healthy subjects were asked to push themselves as hard as they could for 30-second bursts during 15 minutes of stationary bicycling and to go at an easier pace in between, reported Hanne Rooijackers, MD, of the Radboud University Medical Center, Nijmegen, the Netherlands, and colleagues.
The subjects had intravenous cannulae in both forearms, one for blood draws and the other for glucose and insulin administration. All three groups were kept euglycemic during exercise, but then were subjected to hyperinsulinemic-hypoglycemic clamps afterwards. Plasma glucose levels were allowed to fall to 2.6 mmol/L over about 35 min., and were kept there for another 60 min. Trembling, palpitations, anxiety, and other symptoms were serially assessed while patients were hypoglycemic, along with cognitive function and levels of hormones involved with hypoglycemic defense.
For comparison, the subjects all had clamps applied and hypoglycemia symptoms and physiologic responses assessed after a 15-minute session of rest at least 2 weeks apart from the exercise session.
The healthy subjects had a peak of about 20 points on a composite score of hypoglycemia symptoms during rest; exercise reduced the peak score only a small amount to about 18 points. Diabetic patients with normal hypoglycemia awareness hit a peak symptom score of 31 points during rest, which fell substantially after exercise to 22 points. Exercise, meanwhile, had no effect on diabetic patients with impaired awareness; after both rest and exercise, they had a peak composite symptom score of about 11 points, Dr. Rooijackers and colleagues reported (Diabetes. 2017 Jul;66[7]:1990-8).
High-intensity interval training (HIIT) did not affect hypoglycemic awareness in patients with impaired awareness probably because of “a ‘floor’ effect, in that symptom responses could not be further suppressed than they already were,” the investigators speculated.
Regular exercise is recommended for patients with type 1 diabetes, and, like others, they are turning to HIIT – short bursts of intense exercise broken up by brief periods of rest or lower intensity movement – because it appears to deliver the benefits for more moderate exercise in less time.
The findings suggest, however, that high-intensity exercise might increase the risk of severe hypoglycemia in type 1 patients by reducing awareness of its symptoms and blunting hormonal defenses.
The team suspects elevated lactate levels account for the findings. Exercise increases plasma lactate levels, and as blood glucose levels fall, the brain uses lactate as an alternative fuel, which likely blunts the effects of hypoglycemia. Plasma lactate spiked in the study subjects about 15 minutes after exercise.
Participants presented early in the morning after fasting overnight and abstaining from strenuous exercise for 2 days. They were in their mid-20s on average, normal weight, and fairly well balanced between men and women. Patients with type 1 diabetes were eligible for the study if they had hemoglobin A1c levels below 9% and no vascular complications beyond retinopathy. Their duration of diabetes was about 10 years.
The work was funded by the Dutch Diabetes Research Foundation and the European Foundation for the Study of Diabetes. The authors had no conflicts of interest.
FROM DIABETES
Key clinical point:
Major finding: Subjects without diabetes hit a peak of about 20 points on a composite score of hypoglycemia symptoms during rest; exercise reduced the peak score only a small amount to about 18 points. Diabetic patients with normal hypoglycemia awareness hit a peak symptom score of 31 points during rest, which fell substantially after exercise to 22 points.
Data source: Hyperinsulinemic-hypoglycemic clamp experiments in 30 patients.
Disclosures: The work was funded by the Dutch Diabetes Research Foundation and the European Foundation for the Study of Diabetes. The authors had no conflicts of interest.
CABG with arterial grafts provides excellent outcomes for CTO
PARIS – Eighty-eight percent of chronic total occlusions (CTOs) in a large series of patients undergoing coronary artery bypass graft surgery were successfully bypassed using arterial conduits with durable patency.
“Bypass graft surgery using arterial grafts is an acceptable modality of treatment for patients with CTOs and perhaps can be a benchmark against which PCI [percutaneous coronary intervention] for CTOs should be measured,” Teresa May Kieser, MD, said at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.

“We have previously shown in another paper that arterial grafting mitigates the adverse effect of incomplete revascularization,” said Dr. Kieser, a cardiothoracic surgeon at the University of Calgary (Alt.).
In recent years, treatment of CTOs has increasingly drawn the attention of interventional cardiologists. Dr. Kieser presented what she thinks is the first study of coronary artery bypass graft (CABG) surgery for management of CTOs. It included 1,333 consecutive CABG patients with a total of 3,906 bypasses, a whopping 98% of which were arterial grafts, with a mean of 2.9 grafts per patient. Eleven percent of the CABGs were done emergently, 48% urgently, and 41% electively.
The key epidemiologic finding to emerge from the study is that CTOs are quite common in CABG patients. In this series, 47% of CABG patients had a mean of 1.35 chronically occluded coronary arteries.
Of 843 CTOs in three major territories, 88% were able to be bypassed. All of the 246 CTOs in the left anterior descending coronary artery were able to be bypassed, as were 84% of 415 CTOs in the right coronary artery and 85% in the circumflex system.
The CTO group as a whole had significantly greater impairment of left ventricular function. Thirty-seven percent of them had an ejection fraction of 30%-50%, compared with 22% of the non-CTO patients. The 10% prevalence of an left ventricular ejection fraction (LVEF) below 30% in the CTO group was twice that of the non-CTO group. The CTO group was also significantly more likely to undergo incomplete revascularization, by a margin of 21% versus 5.7%.
Operative mortality was 3.7% overall and just 0.55% in the elective CABG patients. In a multivariate logistic regression analysis controlled for surgical urgency, incomplete revascularization, and EuroSCORE risk, operative mortality didn’t differ significantly between the CTO and non-CTO groups.
However, in the presence of CTOs, incomplete revascularization was associated with an 11.6% operative mortality, compared with a 2.8% rate in fully revascularized CTO patients.
A total of 110 patients with bypassed CTOs underwent symptom-driven follow-up coronary angiography at a median of 3.6 years after CABG. Reassuringly, CTO graft patency was noted in 95% of the LAD grafts, 92% of the right coronary artery grafts, and 79% of the circumflex grafts.
Dr. Kieser’s audience of interventional cardiologists was clearly bowled over by her results, not only the high rate of successful surgical bypass of CTOs, but also by her use of arterial grafts 98% of the time.
“This is my personal practice,” she explained. “I just believe in arterial grafting so much. They perform best in CTO arteries because of their lack of competitive flow.”
Session chair Oliver Gämperli, MD, of University Hospital Zurich, commented, “We are very concerned about patency rates, and you showed us fantastic patency rates. This is much better than what we’re used to with saphenous vein grafts. I think we need to talk to our surgeons and try to get them to do more arterial grafts of CTOs.”
It’s worth noting that in the CTO subgroup from the landmark randomized SYNTAX trial, the complete revascularization rate was only about 50% in the PCI group, compared with nearly 65% in the CABG group, he added.
Asked why a cardiac surgeon wouldn’t bypass a CTO, Dr. Kieser rattled off several technical reasons, including a vessel size of less than 1 mm, diffuse disease, extensive scar, or an inaccessible location. But that’s not the whole story, she added. She has heard surgical colleagues say, “The patient doesn’t need that artery, he’s learned to live without it.” That burns her up.
“Patients need every artery in the heart, and the one with a CTO is the best one for an arterial graft because it almost cannot fail, especially to the left anterior descending artery. I think we have to change the mentality of the surgeons to ‘If it can be done, it should be done,’ ” Dr. Kieser said.
She reported having no financial conflicts of interest regarding her study.
PARIS – Eighty-eight percent of chronic total occlusions (CTOs) in a large series of patients undergoing coronary artery bypass graft surgery were successfully bypassed using arterial conduits with durable patency.
“Bypass graft surgery using arterial grafts is an acceptable modality of treatment for patients with CTOs and perhaps can be a benchmark against which PCI [percutaneous coronary intervention] for CTOs should be measured,” Teresa May Kieser, MD, said at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
“We have previously shown in another paper that arterial grafting mitigates the adverse effect of incomplete revascularization,” said Dr. Kieser, a cardiothoracic surgeon at the University of Calgary (Alt.).
In recent years, treatment of CTOs has increasingly drawn the attention of interventional cardiologists. Dr. Kieser presented what she thinks is the first study of coronary artery bypass graft (CABG) surgery for management of CTOs. It included 1,333 consecutive CABG patients with a total of 3,906 bypasses, a whopping 98% of which were arterial grafts, with a mean of 2.9 grafts per patient. Eleven percent of the CABGs were done emergently, 48% urgently, and 41% electively.
The key epidemiologic finding to emerge from the study is that CTOs are quite common in CABG patients. In this series, 47% of CABG patients had a mean of 1.35 chronically occluded coronary arteries.
Of 843 CTOs in three major territories, 88% were able to be bypassed. All of the 246 CTOs in the left anterior descending coronary artery were able to be bypassed, as were 84% of 415 CTOs in the right coronary artery and 85% in the circumflex system.
The CTO group as a whole had significantly greater impairment of left ventricular function. Thirty-seven percent of them had an ejection fraction of 30%-50%, compared with 22% of the non-CTO patients. The 10% prevalence of an left ventricular ejection fraction (LVEF) below 30% in the CTO group was twice that of the non-CTO group. The CTO group was also significantly more likely to undergo incomplete revascularization, by a margin of 21% versus 5.7%.
Operative mortality was 3.7% overall and just 0.55% in the elective CABG patients. In a multivariate logistic regression analysis controlled for surgical urgency, incomplete revascularization, and EuroSCORE risk, operative mortality didn’t differ significantly between the CTO and non-CTO groups.
However, in the presence of CTOs, incomplete revascularization was associated with an 11.6% operative mortality, compared with a 2.8% rate in fully revascularized CTO patients.
A total of 110 patients with bypassed CTOs underwent symptom-driven follow-up coronary angiography at a median of 3.6 years after CABG. Reassuringly, CTO graft patency was noted in 95% of the LAD grafts, 92% of the right coronary artery grafts, and 79% of the circumflex grafts.
Dr. Kieser’s audience of interventional cardiologists was clearly bowled over by her results, not only the high rate of successful surgical bypass of CTOs, but also by her use of arterial grafts 98% of the time.
“This is my personal practice,” she explained. “I just believe in arterial grafting so much. They perform best in CTO arteries because of their lack of competitive flow.”
Session chair Oliver Gämperli, MD, of University Hospital Zurich, commented, “We are very concerned about patency rates, and you showed us fantastic patency rates. This is much better than what we’re used to with saphenous vein grafts. I think we need to talk to our surgeons and try to get them to do more arterial grafts of CTOs.”
It’s worth noting that in the CTO subgroup from the landmark randomized SYNTAX trial, the complete revascularization rate was only about 50% in the PCI group, compared with nearly 65% in the CABG group, he added.
Asked why a cardiac surgeon wouldn’t bypass a CTO, Dr. Kieser rattled off several technical reasons, including a vessel size of less than 1 mm, diffuse disease, extensive scar, or an inaccessible location. But that’s not the whole story, she added. She has heard surgical colleagues say, “The patient doesn’t need that artery, he’s learned to live without it.” That burns her up.
“Patients need every artery in the heart, and the one with a CTO is the best one for an arterial graft because it almost cannot fail, especially to the left anterior descending artery. I think we have to change the mentality of the surgeons to ‘If it can be done, it should be done,’ ” Dr. Kieser said.
She reported having no financial conflicts of interest regarding her study.
PARIS – Eighty-eight percent of chronic total occlusions (CTOs) in a large series of patients undergoing coronary artery bypass graft surgery were successfully bypassed using arterial conduits with durable patency.
“Bypass graft surgery using arterial grafts is an acceptable modality of treatment for patients with CTOs and perhaps can be a benchmark against which PCI [percutaneous coronary intervention] for CTOs should be measured,” Teresa May Kieser, MD, said at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
“We have previously shown in another paper that arterial grafting mitigates the adverse effect of incomplete revascularization,” said Dr. Kieser, a cardiothoracic surgeon at the University of Calgary (Alt.).
In recent years, treatment of CTOs has increasingly drawn the attention of interventional cardiologists. Dr. Kieser presented what she thinks is the first study of coronary artery bypass graft (CABG) surgery for management of CTOs. It included 1,333 consecutive CABG patients with a total of 3,906 bypasses, a whopping 98% of which were arterial grafts, with a mean of 2.9 grafts per patient. Eleven percent of the CABGs were done emergently, 48% urgently, and 41% electively.
The key epidemiologic finding to emerge from the study is that CTOs are quite common in CABG patients. In this series, 47% of CABG patients had a mean of 1.35 chronically occluded coronary arteries.
Of 843 CTOs in three major territories, 88% were able to be bypassed. All of the 246 CTOs in the left anterior descending coronary artery were able to be bypassed, as were 84% of 415 CTOs in the right coronary artery and 85% in the circumflex system.
The CTO group as a whole had significantly greater impairment of left ventricular function. Thirty-seven percent of them had an ejection fraction of 30%-50%, compared with 22% of the non-CTO patients. The 10% prevalence of an left ventricular ejection fraction (LVEF) below 30% in the CTO group was twice that of the non-CTO group. The CTO group was also significantly more likely to undergo incomplete revascularization, by a margin of 21% versus 5.7%.
Operative mortality was 3.7% overall and just 0.55% in the elective CABG patients. In a multivariate logistic regression analysis controlled for surgical urgency, incomplete revascularization, and EuroSCORE risk, operative mortality didn’t differ significantly between the CTO and non-CTO groups.
However, in the presence of CTOs, incomplete revascularization was associated with an 11.6% operative mortality, compared with a 2.8% rate in fully revascularized CTO patients.
A total of 110 patients with bypassed CTOs underwent symptom-driven follow-up coronary angiography at a median of 3.6 years after CABG. Reassuringly, CTO graft patency was noted in 95% of the LAD grafts, 92% of the right coronary artery grafts, and 79% of the circumflex grafts.
Dr. Kieser’s audience of interventional cardiologists was clearly bowled over by her results, not only the high rate of successful surgical bypass of CTOs, but also by her use of arterial grafts 98% of the time.
“This is my personal practice,” she explained. “I just believe in arterial grafting so much. They perform best in CTO arteries because of their lack of competitive flow.”
Session chair Oliver Gämperli, MD, of University Hospital Zurich, commented, “We are very concerned about patency rates, and you showed us fantastic patency rates. This is much better than what we’re used to with saphenous vein grafts. I think we need to talk to our surgeons and try to get them to do more arterial grafts of CTOs.”
It’s worth noting that in the CTO subgroup from the landmark randomized SYNTAX trial, the complete revascularization rate was only about 50% in the PCI group, compared with nearly 65% in the CABG group, he added.
Asked why a cardiac surgeon wouldn’t bypass a CTO, Dr. Kieser rattled off several technical reasons, including a vessel size of less than 1 mm, diffuse disease, extensive scar, or an inaccessible location. But that’s not the whole story, she added. She has heard surgical colleagues say, “The patient doesn’t need that artery, he’s learned to live without it.” That burns her up.
“Patients need every artery in the heart, and the one with a CTO is the best one for an arterial graft because it almost cannot fail, especially to the left anterior descending artery. I think we have to change the mentality of the surgeons to ‘If it can be done, it should be done,’ ” Dr. Kieser said.
She reported having no financial conflicts of interest regarding her study.
AT EUROPCR
Key clinical point:
Major finding: Chronic total occlusions were present in 47% of 1,333 consecutive CABG patients, and 88% of the CTOs were successfully bypassed using arterial conduits.
Data source: A retrospective observational study of 1,333 consecutive CABG patients, 47% of whom had one or more chronic total occlusions.
Disclosures: The study presenter reported having no financial conflicts.
Plasma biomarker distinguishes ARDS, acute heart failure
WASHINGTON – Plasma levels of an interleukin-33 receptor that’s involved in inflammation regulation appeared able to discriminate between acute respiratory distress syndrome and acute decompensated heart failure in an analysis with 72 patients.
In a second study, high plasma levels of the same interleukin-33 receptor, known as soluble suppressor of tumorgenicity 2 (sST2), identified acute respiratory distress syndrome (ARDS) patients who were sicker and more responsive to conservative fluid management, Sean D. Levy, MD, said at an international conference of the American Thoracic Society.

In order to assess the ability of sST2 to reliably distinguish patients with ARDS from those with acute decompensated heart failure, he and his associates selected 72 patients seen at the Massachusetts General Hospital in Boston with an initial diagnosis of acute decompensated heart failure accompanied by bilateral lung infiltrates and acute hypoxemia respiratory failure requiring endotracheal intubation and mechanical ventilation. The investigators measured the sST2 level in a plasma specimen from each patient. In addition, after each patient either left the hospital or died, their case underwent review by two critical care physicians who retrospectively rediagnosed the patients as either having ARDS or acute decompensated heart failure. This divided the cohort into 30 patients with ARDS and 42 with true acute heart failure. The two subgroups matched up fairly closely for most clinical measures and comorbidities, but APACHE III (Acute Physiology and Chronic Health Evaluation III) scores averaged significantly higher in the ARDS patients.
The plasma levels of sST2 showed a dramatic split between the two subgroups. The 30 patients retrospectively diagnosed with ARDS had an average level of 386 ng/mL with an interquartile range of 318-611 ng/mL. The 42 acute decompensated heart failure patients averaged a sST2 level of 148 ng/mL, with an interquartile range of 84-225 ng/mL. The area under the receiver operator curve for discriminating between ARDS and acute heart failure using a cutpoint of 271 mg/mL was 0.86, showing “good” discrimination, Dr. Levy said. This cutpoint had a sensitivity of 83% and specificity of 88% for correctly distinguishing between ARDS and acute heart failure.
In a second analysis, Dr. Levy and his associates looked at the ability of sST2 levels to separate out patients with acute lung injury who had a more robust response to either the conservative or liberal fluid-management strategies tested in the Fluid and Catheter Treatment Trial (FACTT), run by the National Heart, Lung, and Blood Institute’s ARDS Clinical Trials Network. The primary outcome of FACTT was death from any cause 60 days after entry, and this showed no significant difference between conservative (restricted fluids and increased urine output) and liberal (the reverse) fluid management strategies in acute lung injury patients (N Engl J Med. 2006 Jun 15;354[14]:2564-75). From among the 1,001 patients enrolled in FACTT, 826 had specimens available for measuring sST2 (Crit Care Med. 2013 Nov;41[11]:2521-31),
The researchers applied the sST2 cut point they derived in the first analysis to the FACTT cohort and identified 133 (16%) patients with a low sST2 level and 693 (84%) with a high level. The patients with high sST2 were sicker, with significantly higher APACHE III scores, worse acidemia, and worse renal function.
Patients with high sST2 levels had a significant increase in ventilator-free days on conservative fluid management, compared with liberal management, while the two management strategies produced virtually identical results in the patients with low levels of sST2. Patients with high sST2 also had a significantly quicker time to extubation on a conservative strategy compared with the liberal strategy, and again this correlation did not exist among patients with low sST2. However, as in the overall trial a conservative strategy had no discernible impact on 60-day mortality, compared with the liberal strategy, even in the subgroup with high sST2.
Dr. Levy had no disclosures.
[email protected]
On Twitter @mitchelzoler
WASHINGTON – Plasma levels of an interleukin-33 receptor that’s involved in inflammation regulation appeared able to discriminate between acute respiratory distress syndrome and acute decompensated heart failure in an analysis with 72 patients.
In a second study, high plasma levels of the same interleukin-33 receptor, known as soluble suppressor of tumorgenicity 2 (sST2), identified acute respiratory distress syndrome (ARDS) patients who were sicker and more responsive to conservative fluid management, Sean D. Levy, MD, said at an international conference of the American Thoracic Society.
In order to assess the ability of sST2 to reliably distinguish patients with ARDS from those with acute decompensated heart failure, he and his associates selected 72 patients seen at the Massachusetts General Hospital in Boston with an initial diagnosis of acute decompensated heart failure accompanied by bilateral lung infiltrates and acute hypoxemia respiratory failure requiring endotracheal intubation and mechanical ventilation. The investigators measured the sST2 level in a plasma specimen from each patient. In addition, after each patient either left the hospital or died, their case underwent review by two critical care physicians who retrospectively rediagnosed the patients as either having ARDS or acute decompensated heart failure. This divided the cohort into 30 patients with ARDS and 42 with true acute heart failure. The two subgroups matched up fairly closely for most clinical measures and comorbidities, but APACHE III (Acute Physiology and Chronic Health Evaluation III) scores averaged significantly higher in the ARDS patients.
The plasma levels of sST2 showed a dramatic split between the two subgroups. The 30 patients retrospectively diagnosed with ARDS had an average level of 386 ng/mL with an interquartile range of 318-611 ng/mL. The 42 acute decompensated heart failure patients averaged a sST2 level of 148 ng/mL, with an interquartile range of 84-225 ng/mL. The area under the receiver operator curve for discriminating between ARDS and acute heart failure using a cutpoint of 271 mg/mL was 0.86, showing “good” discrimination, Dr. Levy said. This cutpoint had a sensitivity of 83% and specificity of 88% for correctly distinguishing between ARDS and acute heart failure.
In a second analysis, Dr. Levy and his associates looked at the ability of sST2 levels to separate out patients with acute lung injury who had a more robust response to either the conservative or liberal fluid-management strategies tested in the Fluid and Catheter Treatment Trial (FACTT), run by the National Heart, Lung, and Blood Institute’s ARDS Clinical Trials Network. The primary outcome of FACTT was death from any cause 60 days after entry, and this showed no significant difference between conservative (restricted fluids and increased urine output) and liberal (the reverse) fluid management strategies in acute lung injury patients (N Engl J Med. 2006 Jun 15;354[14]:2564-75). From among the 1,001 patients enrolled in FACTT, 826 had specimens available for measuring sST2 (Crit Care Med. 2013 Nov;41[11]:2521-31),
The researchers applied the sST2 cut point they derived in the first analysis to the FACTT cohort and identified 133 (16%) patients with a low sST2 level and 693 (84%) with a high level. The patients with high sST2 were sicker, with significantly higher APACHE III scores, worse acidemia, and worse renal function.
Patients with high sST2 levels had a significant increase in ventilator-free days on conservative fluid management, compared with liberal management, while the two management strategies produced virtually identical results in the patients with low levels of sST2. Patients with high sST2 also had a significantly quicker time to extubation on a conservative strategy compared with the liberal strategy, and again this correlation did not exist among patients with low sST2. However, as in the overall trial a conservative strategy had no discernible impact on 60-day mortality, compared with the liberal strategy, even in the subgroup with high sST2.
Dr. Levy had no disclosures.
[email protected]
On Twitter @mitchelzoler
WASHINGTON – Plasma levels of an interleukin-33 receptor that’s involved in inflammation regulation appeared able to discriminate between acute respiratory distress syndrome and acute decompensated heart failure in an analysis with 72 patients.
In a second study, high plasma levels of the same interleukin-33 receptor, known as soluble suppressor of tumorgenicity 2 (sST2), identified acute respiratory distress syndrome (ARDS) patients who were sicker and more responsive to conservative fluid management, Sean D. Levy, MD, said at an international conference of the American Thoracic Society.
In order to assess the ability of sST2 to reliably distinguish patients with ARDS from those with acute decompensated heart failure, he and his associates selected 72 patients seen at the Massachusetts General Hospital in Boston with an initial diagnosis of acute decompensated heart failure accompanied by bilateral lung infiltrates and acute hypoxemia respiratory failure requiring endotracheal intubation and mechanical ventilation. The investigators measured the sST2 level in a plasma specimen from each patient. In addition, after each patient either left the hospital or died, their case underwent review by two critical care physicians who retrospectively rediagnosed the patients as either having ARDS or acute decompensated heart failure. This divided the cohort into 30 patients with ARDS and 42 with true acute heart failure. The two subgroups matched up fairly closely for most clinical measures and comorbidities, but APACHE III (Acute Physiology and Chronic Health Evaluation III) scores averaged significantly higher in the ARDS patients.
The plasma levels of sST2 showed a dramatic split between the two subgroups. The 30 patients retrospectively diagnosed with ARDS had an average level of 386 ng/mL with an interquartile range of 318-611 ng/mL. The 42 acute decompensated heart failure patients averaged a sST2 level of 148 ng/mL, with an interquartile range of 84-225 ng/mL. The area under the receiver operator curve for discriminating between ARDS and acute heart failure using a cutpoint of 271 mg/mL was 0.86, showing “good” discrimination, Dr. Levy said. This cutpoint had a sensitivity of 83% and specificity of 88% for correctly distinguishing between ARDS and acute heart failure.
In a second analysis, Dr. Levy and his associates looked at the ability of sST2 levels to separate out patients with acute lung injury who had a more robust response to either the conservative or liberal fluid-management strategies tested in the Fluid and Catheter Treatment Trial (FACTT), run by the National Heart, Lung, and Blood Institute’s ARDS Clinical Trials Network. The primary outcome of FACTT was death from any cause 60 days after entry, and this showed no significant difference between conservative (restricted fluids and increased urine output) and liberal (the reverse) fluid management strategies in acute lung injury patients (N Engl J Med. 2006 Jun 15;354[14]:2564-75). From among the 1,001 patients enrolled in FACTT, 826 had specimens available for measuring sST2 (Crit Care Med. 2013 Nov;41[11]:2521-31),
The researchers applied the sST2 cut point they derived in the first analysis to the FACTT cohort and identified 133 (16%) patients with a low sST2 level and 693 (84%) with a high level. The patients with high sST2 were sicker, with significantly higher APACHE III scores, worse acidemia, and worse renal function.
Patients with high sST2 levels had a significant increase in ventilator-free days on conservative fluid management, compared with liberal management, while the two management strategies produced virtually identical results in the patients with low levels of sST2. Patients with high sST2 also had a significantly quicker time to extubation on a conservative strategy compared with the liberal strategy, and again this correlation did not exist among patients with low sST2. However, as in the overall trial a conservative strategy had no discernible impact on 60-day mortality, compared with the liberal strategy, even in the subgroup with high sST2.
Dr. Levy had no disclosures.
[email protected]
On Twitter @mitchelzoler
AT ATS 2017
Key clinical point:
Major finding: An sST2 cutpoint of 271 ng/mL discriminated between ARDS and acute heart failure with 83% sensitivity and 88% specificity.
Data source: Review of 72 patients admitted for acute decompensated heart failure at one U.S. center.
Disclosures: Dr. Levy had no disclosures.