User login
‘Flakka’: A low-cost, dangerous high
Use of α-pyrrolidinovalerophenone (α-PVP), a psychostimulant related to cathinone derivatives (“bath salts”), has been reported in the United States, especially in Florida.1 Known by the street names “flakka” or “gravel,” α-PVP is inexpensive, with a single dose (typically 100 mg) costing as little as $5.2 Alpha-PVP can be consumed via ingestion, injection, insufflation, or inhalation in vaporized forms, such as E-cigarettes, which deliver the drug quickly into the bloodstream and can make it easy to overdose.1 The low cost of this drug makes it likely to be abused. Here we review the mechanism of action and effects of α-PVP and summarize treatment options.
Mechanism of action
Alpha-PVP is a structural parent of 3,4-methylenedioxypyrovalerone (MDPV)—the first widely abused synthetic cathinone.3 Much like cocaine, α-PVP stimulates the CNS by acting as a potent dopamine and norepinephrine reuptake inhibitor. However, unlike cocaine, it lacks any action on serotonin transporters. The pyrrolidine ring in MDPV and α-PVP is responsible for the highly potent dopamine reuptake inhibitor action of these agents.3
A wide range of adverse effects
Use of α-PVP results in a state of “excited delirium,” with symptoms such as hyperthermia, hallucinations, paranoia, violent aggression, and self-harm.1 Alpha-PVP is known to cause rhabdomyolysis.4 Some studies have reported cardiovascular effects, such as arterial hypertension, palpitations, dyspnea, vasoconstriction, arrhythmia, myocardial infarction (MI), and myocarditis.5 Alpha-PVP also may result in neurologic symptoms, including headache, mydriasis, lightheadedness, paresthesia, seizures, dystonic movements, tremor, amnesia, dysgeusia, cerebral edema, motor automatisms, muscle spasm, nystagmus, parkinsonism, and stroke.5 Death may occur by cardiac arrest, renal damage, or suicide.
Case reports. The effects of α-PVP have been documented in the literature:
- A 17-year-old girl was brought to an emergency department in Florida with acute onset of bizarre behavior, agitation, and altered mental status. It took 6 days and repeated administrations of olanzapine and lorazepam for the patient to become calm, alert, and oriented.2
- ST-elevated MI with several intracardiac thrombi was reported in a 41-year-old woman who used α-PVP.4
- In 2015, 18 deaths related to α-PVP use were reported in South Florida.5
- Deaths related to α-PVP use also have been reported in Japan and Australia.5
Treatment options
There are no treatment guidelines for α-PVP-related psychiatric symptoms. Case reports describe remission of symptoms following aggressive treatment with antipsychotics and benzodiazepines.2 Guidelines for treatment of stimulant-induced behavioral and psychotic symptoms6 may be considered for patients who have used α-PVP.
Reassurance and supportive care are the basic principles of such interventions. A quiet environment and benzodiazepines may provide relief of agitation. Antipsychotics may be helpful if a patient exhibits psychotic symptoms.
Similar drugs may emerge
In 2014, the DEA classified α-PVP as a Schedule I substance. Laws against the import of such substances via the Internet or other means also may help control the spread of this drug. However, chemically similar drugs that may elude drug screens are continually emerging. The lack of evidence-based guidelines on recognizing and managing intoxication, withdrawal, and long-term effects of α-PVP and other “designer drugs” calls for greater research in this emerging area of substance use disorders.
1. National Institute on Drug Abuse. “Flakka” (alpha-PVP). https://www.drugabuse.gov/emerging-trends/flakka-alpha-pvp. Accessed July 26, 2017.
2. Crespi C. Flakka-induced prolonged psychosis. Case Rep Psychiatry. 2016;2016:3460849. doi: 10.1155/2016/3460849.
3. Glennon RA, Young R. Neurobiology of 3,4-methylenedioxypyrovalerone (MDPV) and α-pyrrolidinovalerophenone (α-PVP). Brain Res Bull. 2016;126(pt 1):111-126.
4. Cherry SV, Rodriguez YF. Synthetic stimulant reaching epidemic proportions: flakka-induced ST-elevation myocardial infarction with intracardiac thrombi. J Cardiothorac Vasc Anesth. 2017;31(1):e13-e14.
5. Katselou M, Papoutsis I, Nikolaou P, et al. α-PVP (“flakka”): a new synthetic cathinone invades the drug arena. Forensic Toxicol. 2016;34(1):41-50.
6. Sadock BJ, Sadock VA, Ruiz P. Hallucinogen-related disorders. In: Sadock BJ, Sadock VA, Ruiz P. Kaplan and Sadock’s synopsis of psychiatry: behavioral sciences/clinical psychiatry. 11th ed. Philadelphia, PA: Wolters Kluwer; 2015:648-655.
Use of α-pyrrolidinovalerophenone (α-PVP), a psychostimulant related to cathinone derivatives (“bath salts”), has been reported in the United States, especially in Florida.1 Known by the street names “flakka” or “gravel,” α-PVP is inexpensive, with a single dose (typically 100 mg) costing as little as $5.2 Alpha-PVP can be consumed via ingestion, injection, insufflation, or inhalation in vaporized forms, such as E-cigarettes, which deliver the drug quickly into the bloodstream and can make it easy to overdose.1 The low cost of this drug makes it likely to be abused. Here we review the mechanism of action and effects of α-PVP and summarize treatment options.
Mechanism of action
Alpha-PVP is a structural parent of 3,4-methylenedioxypyrovalerone (MDPV)—the first widely abused synthetic cathinone.3 Much like cocaine, α-PVP stimulates the CNS by acting as a potent dopamine and norepinephrine reuptake inhibitor. However, unlike cocaine, it lacks any action on serotonin transporters. The pyrrolidine ring in MDPV and α-PVP is responsible for the highly potent dopamine reuptake inhibitor action of these agents.3
A wide range of adverse effects
Use of α-PVP results in a state of “excited delirium,” with symptoms such as hyperthermia, hallucinations, paranoia, violent aggression, and self-harm.1 Alpha-PVP is known to cause rhabdomyolysis.4 Some studies have reported cardiovascular effects, such as arterial hypertension, palpitations, dyspnea, vasoconstriction, arrhythmia, myocardial infarction (MI), and myocarditis.5 Alpha-PVP also may result in neurologic symptoms, including headache, mydriasis, lightheadedness, paresthesia, seizures, dystonic movements, tremor, amnesia, dysgeusia, cerebral edema, motor automatisms, muscle spasm, nystagmus, parkinsonism, and stroke.5 Death may occur by cardiac arrest, renal damage, or suicide.
Case reports. The effects of α-PVP have been documented in the literature:
- A 17-year-old girl was brought to an emergency department in Florida with acute onset of bizarre behavior, agitation, and altered mental status. It took 6 days and repeated administrations of olanzapine and lorazepam for the patient to become calm, alert, and oriented.2
- ST-elevated MI with several intracardiac thrombi was reported in a 41-year-old woman who used α-PVP.4
- In 2015, 18 deaths related to α-PVP use were reported in South Florida.5
- Deaths related to α-PVP use also have been reported in Japan and Australia.5
Treatment options
There are no treatment guidelines for α-PVP-related psychiatric symptoms. Case reports describe remission of symptoms following aggressive treatment with antipsychotics and benzodiazepines.2 Guidelines for treatment of stimulant-induced behavioral and psychotic symptoms6 may be considered for patients who have used α-PVP.
Reassurance and supportive care are the basic principles of such interventions. A quiet environment and benzodiazepines may provide relief of agitation. Antipsychotics may be helpful if a patient exhibits psychotic symptoms.
Similar drugs may emerge
In 2014, the DEA classified α-PVP as a Schedule I substance. Laws against the import of such substances via the Internet or other means also may help control the spread of this drug. However, chemically similar drugs that may elude drug screens are continually emerging. The lack of evidence-based guidelines on recognizing and managing intoxication, withdrawal, and long-term effects of α-PVP and other “designer drugs” calls for greater research in this emerging area of substance use disorders.
Use of α-pyrrolidinovalerophenone (α-PVP), a psychostimulant related to cathinone derivatives (“bath salts”), has been reported in the United States, especially in Florida.1 Known by the street names “flakka” or “gravel,” α-PVP is inexpensive, with a single dose (typically 100 mg) costing as little as $5.2 Alpha-PVP can be consumed via ingestion, injection, insufflation, or inhalation in vaporized forms, such as E-cigarettes, which deliver the drug quickly into the bloodstream and can make it easy to overdose.1 The low cost of this drug makes it likely to be abused. Here we review the mechanism of action and effects of α-PVP and summarize treatment options.
Mechanism of action
Alpha-PVP is a structural parent of 3,4-methylenedioxypyrovalerone (MDPV)—the first widely abused synthetic cathinone.3 Much like cocaine, α-PVP stimulates the CNS by acting as a potent dopamine and norepinephrine reuptake inhibitor. However, unlike cocaine, it lacks any action on serotonin transporters. The pyrrolidine ring in MDPV and α-PVP is responsible for the highly potent dopamine reuptake inhibitor action of these agents.3
A wide range of adverse effects
Use of α-PVP results in a state of “excited delirium,” with symptoms such as hyperthermia, hallucinations, paranoia, violent aggression, and self-harm.1 Alpha-PVP is known to cause rhabdomyolysis.4 Some studies have reported cardiovascular effects, such as arterial hypertension, palpitations, dyspnea, vasoconstriction, arrhythmia, myocardial infarction (MI), and myocarditis.5 Alpha-PVP also may result in neurologic symptoms, including headache, mydriasis, lightheadedness, paresthesia, seizures, dystonic movements, tremor, amnesia, dysgeusia, cerebral edema, motor automatisms, muscle spasm, nystagmus, parkinsonism, and stroke.5 Death may occur by cardiac arrest, renal damage, or suicide.
Case reports. The effects of α-PVP have been documented in the literature:
- A 17-year-old girl was brought to an emergency department in Florida with acute onset of bizarre behavior, agitation, and altered mental status. It took 6 days and repeated administrations of olanzapine and lorazepam for the patient to become calm, alert, and oriented.2
- ST-elevated MI with several intracardiac thrombi was reported in a 41-year-old woman who used α-PVP.4
- In 2015, 18 deaths related to α-PVP use were reported in South Florida.5
- Deaths related to α-PVP use also have been reported in Japan and Australia.5
Treatment options
There are no treatment guidelines for α-PVP-related psychiatric symptoms. Case reports describe remission of symptoms following aggressive treatment with antipsychotics and benzodiazepines.2 Guidelines for treatment of stimulant-induced behavioral and psychotic symptoms6 may be considered for patients who have used α-PVP.
Reassurance and supportive care are the basic principles of such interventions. A quiet environment and benzodiazepines may provide relief of agitation. Antipsychotics may be helpful if a patient exhibits psychotic symptoms.
Similar drugs may emerge
In 2014, the DEA classified α-PVP as a Schedule I substance. Laws against the import of such substances via the Internet or other means also may help control the spread of this drug. However, chemically similar drugs that may elude drug screens are continually emerging. The lack of evidence-based guidelines on recognizing and managing intoxication, withdrawal, and long-term effects of α-PVP and other “designer drugs” calls for greater research in this emerging area of substance use disorders.
1. National Institute on Drug Abuse. “Flakka” (alpha-PVP). https://www.drugabuse.gov/emerging-trends/flakka-alpha-pvp. Accessed July 26, 2017.
2. Crespi C. Flakka-induced prolonged psychosis. Case Rep Psychiatry. 2016;2016:3460849. doi: 10.1155/2016/3460849.
3. Glennon RA, Young R. Neurobiology of 3,4-methylenedioxypyrovalerone (MDPV) and α-pyrrolidinovalerophenone (α-PVP). Brain Res Bull. 2016;126(pt 1):111-126.
4. Cherry SV, Rodriguez YF. Synthetic stimulant reaching epidemic proportions: flakka-induced ST-elevation myocardial infarction with intracardiac thrombi. J Cardiothorac Vasc Anesth. 2017;31(1):e13-e14.
5. Katselou M, Papoutsis I, Nikolaou P, et al. α-PVP (“flakka”): a new synthetic cathinone invades the drug arena. Forensic Toxicol. 2016;34(1):41-50.
6. Sadock BJ, Sadock VA, Ruiz P. Hallucinogen-related disorders. In: Sadock BJ, Sadock VA, Ruiz P. Kaplan and Sadock’s synopsis of psychiatry: behavioral sciences/clinical psychiatry. 11th ed. Philadelphia, PA: Wolters Kluwer; 2015:648-655.
1. National Institute on Drug Abuse. “Flakka” (alpha-PVP). https://www.drugabuse.gov/emerging-trends/flakka-alpha-pvp. Accessed July 26, 2017.
2. Crespi C. Flakka-induced prolonged psychosis. Case Rep Psychiatry. 2016;2016:3460849. doi: 10.1155/2016/3460849.
3. Glennon RA, Young R. Neurobiology of 3,4-methylenedioxypyrovalerone (MDPV) and α-pyrrolidinovalerophenone (α-PVP). Brain Res Bull. 2016;126(pt 1):111-126.
4. Cherry SV, Rodriguez YF. Synthetic stimulant reaching epidemic proportions: flakka-induced ST-elevation myocardial infarction with intracardiac thrombi. J Cardiothorac Vasc Anesth. 2017;31(1):e13-e14.
5. Katselou M, Papoutsis I, Nikolaou P, et al. α-PVP (“flakka”): a new synthetic cathinone invades the drug arena. Forensic Toxicol. 2016;34(1):41-50.
6. Sadock BJ, Sadock VA, Ruiz P. Hallucinogen-related disorders. In: Sadock BJ, Sadock VA, Ruiz P. Kaplan and Sadock’s synopsis of psychiatry: behavioral sciences/clinical psychiatry. 11th ed. Philadelphia, PA: Wolters Kluwer; 2015:648-655.
Confronting hate and violence against the LGBT community
It may be unusual for an LGBT health columnist to mention the horrendous events that occurred in Charlottesville, Va., in August 2017. It clearly was a demonstration of hate and violence against racial and ethnic minorities. Unfortunately, the LGBT community – especially LGBT communities of color – are often a target of that kind of hate and violence. This has a detrimental effect on the health of the LGBT community, and I believe that health care providers have a responsibility to address this hate and violence to promote the well-being of this marginalized community.
It cannot be overstated that LGBT individuals frequently experience anti-gay and anti-trans violence. According to the 2015 Federal Bureau of Investigation Hate Crime Statistics, about a fifth of hate crimes reported were based on sexual orientation or gender identity.1 In addition, LGBT youth are eight times as likely to experience bullying at school because of their sexual orientation or gender identity.2 Furthermore, on many surveys on anti-LGBT violence, people of color comprise more than half of the victims.3 There is a strong association between exposure to this violence and the health outcomes of LGBT youth. A study by Russell et al. showed that LGBT youth who were victims of physical violence at school are more likely to be depressed and suicidal and more likely to be diagnosed with an STD,4 and another study showed that LGBT youth who experienced anti-LGBT violence are more likely to engage in substance use.5 The health outcomes from anti-LGBT violence are not limited to the adolescent period – adolescents who experienced this kind of violence are more likely to report higher levels of depression as adults.6 Although researchers still are trying to determine the exact mechanism for these relationships, the most cited (and sensible) explanation is that exposure to anti-LGBT stigma, discrimination, and violence leads to a toxic environment, which in turn increases the risk for mental health problems and maladaptive coping mechanisms (such as substance use) as a response to such an environment.7

What can we do to stand up to the hate and violence against marginalized groups, such as the LGBT community? First, make your office a safe space. With the recent brazen display of hate and violence going around, members the LGBT community are desperate to feel protected. A good place to start is a guide by Advocates for Youth. Second, educate yourself and others. The title physician means “teacher,” and I feel it is your responsibility to teach your peers, colleagues, and the public about how anti-LGBT violence affects the health of LGBT individuals. To be an effective teacher, you need to be up to date on the research on how hatred and intolerance affects the health of the LGBT community. A good place to start is the Human Rights Campaign, which has accurate statistics on anti-LGBT violence and resources to address this problem. Finally, be an advocate. You don’t need to be in the streets with picket signs, nor do you necessarily need to lead the charge against anti-LGBT hate and violence – others will be at the front lines. What you can do is to call for your local, state, and federal government to institute policies that address anti-LGBT violence. Many medical organizations have resources that help health care providers engage with policy makers (check out the American Academy of Pediatrics advocacy page for these resources). Many of our elected officials take our professional opinions seriously.
Anti-gay and anti-trans violence is all too common in the LGBT community, especially violence against LGBT people of color, and this violence can adversely affect their health. Health care providers have a responsibility and the influence to confront these nexuses of hate and intolerance. You don’t need to do something heroic to accomplish this. You are members of a privileged and respected group of professionals, so small actions can coalesce into something that has a large impact on the health and well-being of the communities you serve.

Resources
• Advocates for Youth. Creating Safe Space for GLBTQ Youth: A Toolkit
• Human Rights Campaign. www.hrc.org/resources/
• American Academy of Pediatrics advocacy page: www.aap.org/en-us/advocacy-and-policy/
References
1. U.S. Department of Justice Federal Bureau of Investigation. Uniform Crime Report Hate Crime Statistics, 2015.
2. J Interpers Violence. 2017. doi: 10.1177/0886260517718830.
3. National Coalition of Anti-Violence Programs (NCAVP). Lesbian, Gay, Bisexual, Transgender, Queer and HIV-Affected Hate Violence in 2016.
4. J Sch Health. 2011 May;81(5):223-30.
5. Prev Sci. 2015 Jul;16(5):734-43.
6. Dev Psychol. 2010 Nov;46(6):1580-9.
7. Psychol Bull. 2003 Sep;129(5):674-97.
8. Gallup. Americans Rate Healthcare Providers High on Honesty, Ethics. 2016.
9. The Hippocratic Oath Today. 2001 or Do. No. Harm.
It may be unusual for an LGBT health columnist to mention the horrendous events that occurred in Charlottesville, Va., in August 2017. It clearly was a demonstration of hate and violence against racial and ethnic minorities. Unfortunately, the LGBT community – especially LGBT communities of color – are often a target of that kind of hate and violence. This has a detrimental effect on the health of the LGBT community, and I believe that health care providers have a responsibility to address this hate and violence to promote the well-being of this marginalized community.
It cannot be overstated that LGBT individuals frequently experience anti-gay and anti-trans violence. According to the 2015 Federal Bureau of Investigation Hate Crime Statistics, about a fifth of hate crimes reported were based on sexual orientation or gender identity.1 In addition, LGBT youth are eight times as likely to experience bullying at school because of their sexual orientation or gender identity.2 Furthermore, on many surveys on anti-LGBT violence, people of color comprise more than half of the victims.3 There is a strong association between exposure to this violence and the health outcomes of LGBT youth. A study by Russell et al. showed that LGBT youth who were victims of physical violence at school are more likely to be depressed and suicidal and more likely to be diagnosed with an STD,4 and another study showed that LGBT youth who experienced anti-LGBT violence are more likely to engage in substance use.5 The health outcomes from anti-LGBT violence are not limited to the adolescent period – adolescents who experienced this kind of violence are more likely to report higher levels of depression as adults.6 Although researchers still are trying to determine the exact mechanism for these relationships, the most cited (and sensible) explanation is that exposure to anti-LGBT stigma, discrimination, and violence leads to a toxic environment, which in turn increases the risk for mental health problems and maladaptive coping mechanisms (such as substance use) as a response to such an environment.7
What can we do to stand up to the hate and violence against marginalized groups, such as the LGBT community? First, make your office a safe space. With the recent brazen display of hate and violence going around, members the LGBT community are desperate to feel protected. A good place to start is a guide by Advocates for Youth. Second, educate yourself and others. The title physician means “teacher,” and I feel it is your responsibility to teach your peers, colleagues, and the public about how anti-LGBT violence affects the health of LGBT individuals. To be an effective teacher, you need to be up to date on the research on how hatred and intolerance affects the health of the LGBT community. A good place to start is the Human Rights Campaign, which has accurate statistics on anti-LGBT violence and resources to address this problem. Finally, be an advocate. You don’t need to be in the streets with picket signs, nor do you necessarily need to lead the charge against anti-LGBT hate and violence – others will be at the front lines. What you can do is to call for your local, state, and federal government to institute policies that address anti-LGBT violence. Many medical organizations have resources that help health care providers engage with policy makers (check out the American Academy of Pediatrics advocacy page for these resources). Many of our elected officials take our professional opinions seriously.
Anti-gay and anti-trans violence is all too common in the LGBT community, especially violence against LGBT people of color, and this violence can adversely affect their health. Health care providers have a responsibility and the influence to confront these nexuses of hate and intolerance. You don’t need to do something heroic to accomplish this. You are members of a privileged and respected group of professionals, so small actions can coalesce into something that has a large impact on the health and well-being of the communities you serve.
Resources
• Advocates for Youth. Creating Safe Space for GLBTQ Youth: A Toolkit
• Human Rights Campaign. www.hrc.org/resources/
• American Academy of Pediatrics advocacy page: www.aap.org/en-us/advocacy-and-policy/
References
1. U.S. Department of Justice Federal Bureau of Investigation. Uniform Crime Report Hate Crime Statistics, 2015.
2. J Interpers Violence. 2017. doi: 10.1177/0886260517718830.
3. National Coalition of Anti-Violence Programs (NCAVP). Lesbian, Gay, Bisexual, Transgender, Queer and HIV-Affected Hate Violence in 2016.
4. J Sch Health. 2011 May;81(5):223-30.
5. Prev Sci. 2015 Jul;16(5):734-43.
6. Dev Psychol. 2010 Nov;46(6):1580-9.
7. Psychol Bull. 2003 Sep;129(5):674-97.
8. Gallup. Americans Rate Healthcare Providers High on Honesty, Ethics. 2016.
9. The Hippocratic Oath Today. 2001 or Do. No. Harm.
It may be unusual for an LGBT health columnist to mention the horrendous events that occurred in Charlottesville, Va., in August 2017. It clearly was a demonstration of hate and violence against racial and ethnic minorities. Unfortunately, the LGBT community – especially LGBT communities of color – are often a target of that kind of hate and violence. This has a detrimental effect on the health of the LGBT community, and I believe that health care providers have a responsibility to address this hate and violence to promote the well-being of this marginalized community.
It cannot be overstated that LGBT individuals frequently experience anti-gay and anti-trans violence. According to the 2015 Federal Bureau of Investigation Hate Crime Statistics, about a fifth of hate crimes reported were based on sexual orientation or gender identity.1 In addition, LGBT youth are eight times as likely to experience bullying at school because of their sexual orientation or gender identity.2 Furthermore, on many surveys on anti-LGBT violence, people of color comprise more than half of the victims.3 There is a strong association between exposure to this violence and the health outcomes of LGBT youth. A study by Russell et al. showed that LGBT youth who were victims of physical violence at school are more likely to be depressed and suicidal and more likely to be diagnosed with an STD,4 and another study showed that LGBT youth who experienced anti-LGBT violence are more likely to engage in substance use.5 The health outcomes from anti-LGBT violence are not limited to the adolescent period – adolescents who experienced this kind of violence are more likely to report higher levels of depression as adults.6 Although researchers still are trying to determine the exact mechanism for these relationships, the most cited (and sensible) explanation is that exposure to anti-LGBT stigma, discrimination, and violence leads to a toxic environment, which in turn increases the risk for mental health problems and maladaptive coping mechanisms (such as substance use) as a response to such an environment.7
What can we do to stand up to the hate and violence against marginalized groups, such as the LGBT community? First, make your office a safe space. With the recent brazen display of hate and violence going around, members the LGBT community are desperate to feel protected. A good place to start is a guide by Advocates for Youth. Second, educate yourself and others. The title physician means “teacher,” and I feel it is your responsibility to teach your peers, colleagues, and the public about how anti-LGBT violence affects the health of LGBT individuals. To be an effective teacher, you need to be up to date on the research on how hatred and intolerance affects the health of the LGBT community. A good place to start is the Human Rights Campaign, which has accurate statistics on anti-LGBT violence and resources to address this problem. Finally, be an advocate. You don’t need to be in the streets with picket signs, nor do you necessarily need to lead the charge against anti-LGBT hate and violence – others will be at the front lines. What you can do is to call for your local, state, and federal government to institute policies that address anti-LGBT violence. Many medical organizations have resources that help health care providers engage with policy makers (check out the American Academy of Pediatrics advocacy page for these resources). Many of our elected officials take our professional opinions seriously.
Anti-gay and anti-trans violence is all too common in the LGBT community, especially violence against LGBT people of color, and this violence can adversely affect their health. Health care providers have a responsibility and the influence to confront these nexuses of hate and intolerance. You don’t need to do something heroic to accomplish this. You are members of a privileged and respected group of professionals, so small actions can coalesce into something that has a large impact on the health and well-being of the communities you serve.
Resources
• Advocates for Youth. Creating Safe Space for GLBTQ Youth: A Toolkit
• Human Rights Campaign. www.hrc.org/resources/
• American Academy of Pediatrics advocacy page: www.aap.org/en-us/advocacy-and-policy/
References
1. U.S. Department of Justice Federal Bureau of Investigation. Uniform Crime Report Hate Crime Statistics, 2015.
2. J Interpers Violence. 2017. doi: 10.1177/0886260517718830.
3. National Coalition of Anti-Violence Programs (NCAVP). Lesbian, Gay, Bisexual, Transgender, Queer and HIV-Affected Hate Violence in 2016.
4. J Sch Health. 2011 May;81(5):223-30.
5. Prev Sci. 2015 Jul;16(5):734-43.
6. Dev Psychol. 2010 Nov;46(6):1580-9.
7. Psychol Bull. 2003 Sep;129(5):674-97.
8. Gallup. Americans Rate Healthcare Providers High on Honesty, Ethics. 2016.
9. The Hippocratic Oath Today. 2001 or Do. No. Harm.

Cerebral palsy rate down in children born very or moderately preterm
, even in children who were born moderately preterm.
The researchers gathered data for cerebral palsy from the medical questionnaire, including information on head control, sitting, standing, walking, and quality of gait; trunk and limb tone; and any abnormal neurologic signs. Development was assessed using the second version of the 24-month Ages and Stages Questionnaire (ASQ), which is completed by parents.
Of 4,199 neonates born between 22 and 34 weeks’ gestation in 2011 enrolled in the EPIPAGE-2 study who lived until a median of 24.2 months corrected age, the rate of cerebral palsy dropped from 7% to 4% in those born between 24-26 and 27-31 weeks’ gestation, reported Véronique Pierrat, MD, PhD, of the Epidemiology and Biostatistics Sorbonne Paris Cité Research Center, INSERM, in Paris, and her associates. At 32-34 weeks’ gestation, the cerebral palsy rate was 1%. Only one child born at 22-23 weeks’ gestation lived beyond the neonatal period. Fewer than 1% of the children overall had severe auditory or visual impairment.
ASQ analysis was considered for 2,506 children, after excluding children with cerebral palsy, deafness or blindness, or severe congenital brain malformations. ASQ scores were below threshold in 50%, 41%, and 36% of children born at 24-26, 27-31, and 32-34 weeks’ gestation, respectively. “The domains most frequently scoring below threshold were communication and personal-social in all gestational age groups. Proportions of children scoring below the threshold in either of these domains decreased with increasing gestational age but still were 18% and 13%, respectively, at 32-34 weeks’ gestation,” the investigators said.
Only 1% or fewer of the children in this cohort had severe gastrointestinal or respiratory disabilities.
In a comparison of 1997 data and this 2011 data after adjustment for baseline characteristics in children born at 22-31 weeks’ gestation, survival increased by a mean 6% , and survival without neuromotor or sensory impairment by 7%; in children born at 24-31 weeks’ gestation, cerebral palsy decreased by a mean 3%. No statistically significant changes were found between the two periods for survival, survival without neuromotor or sensory disabilities, and rates of cerebral palsy in children born at 24 weeks’ gestation, but “noticeable improvements were seen at 25-26 weeks and, to a lesser extent, at 27-31 weeks,” Dr. Pierrat and her associates said. At 32-34 weeks’ gestation, the cerebral palsy rate dropped by 3%, but survival and survival without severe neuromotor or sensory impairment were similar between the two time periods.
In regard to the 2011 data, “the proportion of infants at risk of developmental delay was high, even for those born at 32-34 weeks’ gestation. Our results invite questioning perinatal strategies in France, and in countries with similar recommendations. However, improving outcomes at extremely low gestational age requires a complex change in philosophy of care and close cooperation not only between obstetricians and neonatologists, but also developmental specialists, parent associations, and policy makers,” Dr. Pierrat and her associates concluded.
The investigators said they had no relevant financial disclosures. The study was funded by the French Institute of Public Health Research/Institute of Public Health and several of its partners, the PREMUP Foundation, Fondation de France, and Fondation pour la Recherche Médicale.
, even in children who were born moderately preterm.
The researchers gathered data for cerebral palsy from the medical questionnaire, including information on head control, sitting, standing, walking, and quality of gait; trunk and limb tone; and any abnormal neurologic signs. Development was assessed using the second version of the 24-month Ages and Stages Questionnaire (ASQ), which is completed by parents.
Of 4,199 neonates born between 22 and 34 weeks’ gestation in 2011 enrolled in the EPIPAGE-2 study who lived until a median of 24.2 months corrected age, the rate of cerebral palsy dropped from 7% to 4% in those born between 24-26 and 27-31 weeks’ gestation, reported Véronique Pierrat, MD, PhD, of the Epidemiology and Biostatistics Sorbonne Paris Cité Research Center, INSERM, in Paris, and her associates. At 32-34 weeks’ gestation, the cerebral palsy rate was 1%. Only one child born at 22-23 weeks’ gestation lived beyond the neonatal period. Fewer than 1% of the children overall had severe auditory or visual impairment.
ASQ analysis was considered for 2,506 children, after excluding children with cerebral palsy, deafness or blindness, or severe congenital brain malformations. ASQ scores were below threshold in 50%, 41%, and 36% of children born at 24-26, 27-31, and 32-34 weeks’ gestation, respectively. “The domains most frequently scoring below threshold were communication and personal-social in all gestational age groups. Proportions of children scoring below the threshold in either of these domains decreased with increasing gestational age but still were 18% and 13%, respectively, at 32-34 weeks’ gestation,” the investigators said.
Only 1% or fewer of the children in this cohort had severe gastrointestinal or respiratory disabilities.
In a comparison of 1997 data and this 2011 data after adjustment for baseline characteristics in children born at 22-31 weeks’ gestation, survival increased by a mean 6% , and survival without neuromotor or sensory impairment by 7%; in children born at 24-31 weeks’ gestation, cerebral palsy decreased by a mean 3%. No statistically significant changes were found between the two periods for survival, survival without neuromotor or sensory disabilities, and rates of cerebral palsy in children born at 24 weeks’ gestation, but “noticeable improvements were seen at 25-26 weeks and, to a lesser extent, at 27-31 weeks,” Dr. Pierrat and her associates said. At 32-34 weeks’ gestation, the cerebral palsy rate dropped by 3%, but survival and survival without severe neuromotor or sensory impairment were similar between the two time periods.
In regard to the 2011 data, “the proportion of infants at risk of developmental delay was high, even for those born at 32-34 weeks’ gestation. Our results invite questioning perinatal strategies in France, and in countries with similar recommendations. However, improving outcomes at extremely low gestational age requires a complex change in philosophy of care and close cooperation not only between obstetricians and neonatologists, but also developmental specialists, parent associations, and policy makers,” Dr. Pierrat and her associates concluded.
The investigators said they had no relevant financial disclosures. The study was funded by the French Institute of Public Health Research/Institute of Public Health and several of its partners, the PREMUP Foundation, Fondation de France, and Fondation pour la Recherche Médicale.
, even in children who were born moderately preterm.
The researchers gathered data for cerebral palsy from the medical questionnaire, including information on head control, sitting, standing, walking, and quality of gait; trunk and limb tone; and any abnormal neurologic signs. Development was assessed using the second version of the 24-month Ages and Stages Questionnaire (ASQ), which is completed by parents.
Of 4,199 neonates born between 22 and 34 weeks’ gestation in 2011 enrolled in the EPIPAGE-2 study who lived until a median of 24.2 months corrected age, the rate of cerebral palsy dropped from 7% to 4% in those born between 24-26 and 27-31 weeks’ gestation, reported Véronique Pierrat, MD, PhD, of the Epidemiology and Biostatistics Sorbonne Paris Cité Research Center, INSERM, in Paris, and her associates. At 32-34 weeks’ gestation, the cerebral palsy rate was 1%. Only one child born at 22-23 weeks’ gestation lived beyond the neonatal period. Fewer than 1% of the children overall had severe auditory or visual impairment.
ASQ analysis was considered for 2,506 children, after excluding children with cerebral palsy, deafness or blindness, or severe congenital brain malformations. ASQ scores were below threshold in 50%, 41%, and 36% of children born at 24-26, 27-31, and 32-34 weeks’ gestation, respectively. “The domains most frequently scoring below threshold were communication and personal-social in all gestational age groups. Proportions of children scoring below the threshold in either of these domains decreased with increasing gestational age but still were 18% and 13%, respectively, at 32-34 weeks’ gestation,” the investigators said.
Only 1% or fewer of the children in this cohort had severe gastrointestinal or respiratory disabilities.
In a comparison of 1997 data and this 2011 data after adjustment for baseline characteristics in children born at 22-31 weeks’ gestation, survival increased by a mean 6% , and survival without neuromotor or sensory impairment by 7%; in children born at 24-31 weeks’ gestation, cerebral palsy decreased by a mean 3%. No statistically significant changes were found between the two periods for survival, survival without neuromotor or sensory disabilities, and rates of cerebral palsy in children born at 24 weeks’ gestation, but “noticeable improvements were seen at 25-26 weeks and, to a lesser extent, at 27-31 weeks,” Dr. Pierrat and her associates said. At 32-34 weeks’ gestation, the cerebral palsy rate dropped by 3%, but survival and survival without severe neuromotor or sensory impairment were similar between the two time periods.
In regard to the 2011 data, “the proportion of infants at risk of developmental delay was high, even for those born at 32-34 weeks’ gestation. Our results invite questioning perinatal strategies in France, and in countries with similar recommendations. However, improving outcomes at extremely low gestational age requires a complex change in philosophy of care and close cooperation not only between obstetricians and neonatologists, but also developmental specialists, parent associations, and policy makers,” Dr. Pierrat and her associates concluded.
The investigators said they had no relevant financial disclosures. The study was funded by the French Institute of Public Health Research/Institute of Public Health and several of its partners, the PREMUP Foundation, Fondation de France, and Fondation pour la Recherche Médicale.
FROM BMJ
Key clinical point: Cerebral palsy rates are down in children born very and moderately preterm, but the risk of developmental delay remains high.
Major finding: ASQ scores were below threshold in 50%, 41%, and 36% of children born at 24-26, 27-31, and 32-34 weeks’ gestation, respectively.
Data source: A national population study of 4,199 French neonates born between 22 and 34 weeks’ gestation in 2011.
Disclosures: The investigators said they had no relevant financial disclosures. The study was funded by the French Institute of Public Health Research/Institute of Public Health and several of its partners, the PREMUP Foundation, Fondation de France, and Fondation pour la Recherche Médicale.
Standardized infection ratio for CLABSI almost halved since 2009
The standardized infection ratio (SIR) for central line–associated bloodstream infections dropped 42% from 2009 to 2014, according to the Agency for Healthcare Research and Quality.
For acute care hospitalizations, the SIR for central line–associated bloodstream infections (CLABSIs) fell from 0.854 in 2009 to 0.495 in 2014. Over that same time period, the SIR for surgical site infections involving Surgical Care Improvement Project procedures decreased from 0.981 to 0.827 – almost 16%, the AHRQ said in its annual National Healthcare Quality and Disparities Report.
From 2010 to 2014, the SIR for catheter-associated urinary tract infections increased 6.7% from 0.937 to 1.000, but that change was not significant. For laboratory-identified hospital-onset Clostridium difficile infection, the SIR dropped from 0.963 to 0.924 – about 4% – from 2012 to 2014, the AHRQ reported using data from the National Center for Emerging and Zoonotic Infectious Diseases and the National Healthcare Safety Network.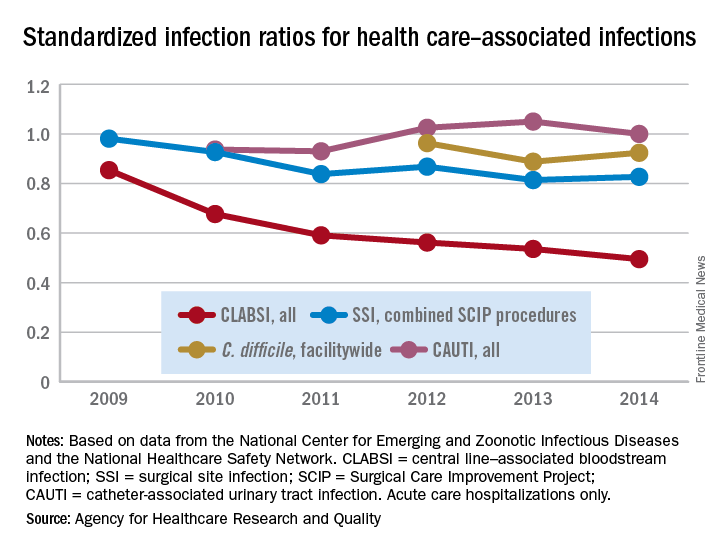
The standardized infection ratio (SIR) for central line–associated bloodstream infections dropped 42% from 2009 to 2014, according to the Agency for Healthcare Research and Quality.
For acute care hospitalizations, the SIR for central line–associated bloodstream infections (CLABSIs) fell from 0.854 in 2009 to 0.495 in 2014. Over that same time period, the SIR for surgical site infections involving Surgical Care Improvement Project procedures decreased from 0.981 to 0.827 – almost 16%, the AHRQ said in its annual National Healthcare Quality and Disparities Report.
From 2010 to 2014, the SIR for catheter-associated urinary tract infections increased 6.7% from 0.937 to 1.000, but that change was not significant. For laboratory-identified hospital-onset Clostridium difficile infection, the SIR dropped from 0.963 to 0.924 – about 4% – from 2012 to 2014, the AHRQ reported using data from the National Center for Emerging and Zoonotic Infectious Diseases and the National Healthcare Safety Network.
The standardized infection ratio (SIR) for central line–associated bloodstream infections dropped 42% from 2009 to 2014, according to the Agency for Healthcare Research and Quality.
For acute care hospitalizations, the SIR for central line–associated bloodstream infections (CLABSIs) fell from 0.854 in 2009 to 0.495 in 2014. Over that same time period, the SIR for surgical site infections involving Surgical Care Improvement Project procedures decreased from 0.981 to 0.827 – almost 16%, the AHRQ said in its annual National Healthcare Quality and Disparities Report.
From 2010 to 2014, the SIR for catheter-associated urinary tract infections increased 6.7% from 0.937 to 1.000, but that change was not significant. For laboratory-identified hospital-onset Clostridium difficile infection, the SIR dropped from 0.963 to 0.924 – about 4% – from 2012 to 2014, the AHRQ reported using data from the National Center for Emerging and Zoonotic Infectious Diseases and the National Healthcare Safety Network.
Rare Case of Orbital Involvement from Multiple Myeloma
An orbital mass is often the “tip of the iceberg”—it may be secondary to systemic malignancy, warn clinicians from University Sains Malaysia-Health Campus and Hospital Sultanah Bahiyah, both in Malaysia. Orbital metastases usually originate from lung and breast cancers, but these authors report on an unusual case of a patient whose orbital involvement stemmed from multiple myeloma (MM).
Related: A Mysterious Massive Hemorrhage
The 85-year-old woman presented with right-eye proptosis, reduced visual acuity and diplopia. She had been bedridden with chronic back pain but had no symptoms of thyroid disorder or malignancy. Cardiovascular, breast, abdominal, and neurologic examinations were normal. She had no palpable lymph nodes. Blood investigations for infective and inflammatory causes were unremarkable.
However, a chest radiograph showed osteopenic bones, a pathologic fracture of the right clavicle, and an opacity obscuring the left retrocardiac region, suggesting a mass in the lower lobe of the left lung. The patient declined further imaging but underwent biopsy for the right orbital mass. Histopathologic examination revealed cells suggestive of MM. She was diagnosed with osseous plasmacytoma.
Orbital involvement in MM may be the first manifestation of systemic disease, the clinicians say. The diagnosis is usually based on clinical suspicion. Patients tend to present with nonspecific symptoms like back pain and fatigue. Computed tomography scanning is the imaging modality of choice, the authors say, but in older patients the findings may be hard to interpret. Thinning of the bone, for instance, may mimic metastases. Biopsy provides a definitive diagnosis and guides further management.
Related: Less Lenalidomide May Be More in Frail Elderly Multiple Myeloma Patients
Orbital involvement in MM is rare but treatable. Discovery of a plasmacytoma should always prompt investigation for systemic involvement, the authors advise, because the treatment and prognosis differ between the two. In their patient, proptosis secondary to the orbital plasmacytoma led them to discover end-organ damage in the form of multiple bone lesions. Solitary plasmacytoma would be treated with radiotherapy and resection; active MM with end-organ damage requires systemic chemotherapy.
Getting to the root of the problem can be difficult when the presentation is “insidious” and clinical features are nonspecific, the authors say. Patient and thorough investigation can make the difference in resolving the diagnostic imaging challenges.
Source:
Tai E, Sim SK, Haron J, Wan Hitam WH. BMJ Case Rep. 2017;2017: pii: bcr-2017-220895.
doi: 10.1136/bcr-2017-220895.
An orbital mass is often the “tip of the iceberg”—it may be secondary to systemic malignancy, warn clinicians from University Sains Malaysia-Health Campus and Hospital Sultanah Bahiyah, both in Malaysia. Orbital metastases usually originate from lung and breast cancers, but these authors report on an unusual case of a patient whose orbital involvement stemmed from multiple myeloma (MM).
Related: A Mysterious Massive Hemorrhage
The 85-year-old woman presented with right-eye proptosis, reduced visual acuity and diplopia. She had been bedridden with chronic back pain but had no symptoms of thyroid disorder or malignancy. Cardiovascular, breast, abdominal, and neurologic examinations were normal. She had no palpable lymph nodes. Blood investigations for infective and inflammatory causes were unremarkable.
However, a chest radiograph showed osteopenic bones, a pathologic fracture of the right clavicle, and an opacity obscuring the left retrocardiac region, suggesting a mass in the lower lobe of the left lung. The patient declined further imaging but underwent biopsy for the right orbital mass. Histopathologic examination revealed cells suggestive of MM. She was diagnosed with osseous plasmacytoma.
Orbital involvement in MM may be the first manifestation of systemic disease, the clinicians say. The diagnosis is usually based on clinical suspicion. Patients tend to present with nonspecific symptoms like back pain and fatigue. Computed tomography scanning is the imaging modality of choice, the authors say, but in older patients the findings may be hard to interpret. Thinning of the bone, for instance, may mimic metastases. Biopsy provides a definitive diagnosis and guides further management.
Related: Less Lenalidomide May Be More in Frail Elderly Multiple Myeloma Patients
Orbital involvement in MM is rare but treatable. Discovery of a plasmacytoma should always prompt investigation for systemic involvement, the authors advise, because the treatment and prognosis differ between the two. In their patient, proptosis secondary to the orbital plasmacytoma led them to discover end-organ damage in the form of multiple bone lesions. Solitary plasmacytoma would be treated with radiotherapy and resection; active MM with end-organ damage requires systemic chemotherapy.
Getting to the root of the problem can be difficult when the presentation is “insidious” and clinical features are nonspecific, the authors say. Patient and thorough investigation can make the difference in resolving the diagnostic imaging challenges.
Source:
Tai E, Sim SK, Haron J, Wan Hitam WH. BMJ Case Rep. 2017;2017: pii: bcr-2017-220895.
doi: 10.1136/bcr-2017-220895.
An orbital mass is often the “tip of the iceberg”—it may be secondary to systemic malignancy, warn clinicians from University Sains Malaysia-Health Campus and Hospital Sultanah Bahiyah, both in Malaysia. Orbital metastases usually originate from lung and breast cancers, but these authors report on an unusual case of a patient whose orbital involvement stemmed from multiple myeloma (MM).
Related: A Mysterious Massive Hemorrhage
The 85-year-old woman presented with right-eye proptosis, reduced visual acuity and diplopia. She had been bedridden with chronic back pain but had no symptoms of thyroid disorder or malignancy. Cardiovascular, breast, abdominal, and neurologic examinations were normal. She had no palpable lymph nodes. Blood investigations for infective and inflammatory causes were unremarkable.
However, a chest radiograph showed osteopenic bones, a pathologic fracture of the right clavicle, and an opacity obscuring the left retrocardiac region, suggesting a mass in the lower lobe of the left lung. The patient declined further imaging but underwent biopsy for the right orbital mass. Histopathologic examination revealed cells suggestive of MM. She was diagnosed with osseous plasmacytoma.
Orbital involvement in MM may be the first manifestation of systemic disease, the clinicians say. The diagnosis is usually based on clinical suspicion. Patients tend to present with nonspecific symptoms like back pain and fatigue. Computed tomography scanning is the imaging modality of choice, the authors say, but in older patients the findings may be hard to interpret. Thinning of the bone, for instance, may mimic metastases. Biopsy provides a definitive diagnosis and guides further management.
Related: Less Lenalidomide May Be More in Frail Elderly Multiple Myeloma Patients
Orbital involvement in MM is rare but treatable. Discovery of a plasmacytoma should always prompt investigation for systemic involvement, the authors advise, because the treatment and prognosis differ between the two. In their patient, proptosis secondary to the orbital plasmacytoma led them to discover end-organ damage in the form of multiple bone lesions. Solitary plasmacytoma would be treated with radiotherapy and resection; active MM with end-organ damage requires systemic chemotherapy.
Getting to the root of the problem can be difficult when the presentation is “insidious” and clinical features are nonspecific, the authors say. Patient and thorough investigation can make the difference in resolving the diagnostic imaging challenges.
Source:
Tai E, Sim SK, Haron J, Wan Hitam WH. BMJ Case Rep. 2017;2017: pii: bcr-2017-220895.
doi: 10.1136/bcr-2017-220895.
Vitamin C could help treat TET2-mutant leukemias

Preclinical research suggests high-dose vitamin C may be effective against TET2-mutant leukemias.
Investigators found that vitamin C mimics TET2 restoration, thereby suppressing leukemic colony formation, inhibiting leukemia progression in mice, and enhancing leukemia cells’ sensitivity to treatment with a PARP inhibitor.
“We’re excited by the prospect that high-dose vitamin C might become a safe treatment for blood diseases caused by TET2-deficient leukemia stem cells, most likely in combination with other targeted therapies,” said study author Benjamin G. Neel, MD, PhD, of NYU School of Medicine in New York, New York.
Dr Neel and his colleagues reported their findings in Cell.
Previous research had shown that vitamin C could stimulate the activity of TET2 as well as TET1 and TET3.
Because only 1 copy of the TET2 gene in each stem cell is usually affected in TET2-mutant blood diseases, the investigators hypothesized that high doses of vitamin C might reverse the effects of TET2 deficiency by turning up the action of the remaining functional gene.
Indeed, the team found that vitamin C had the same effect as restoring TET2 function genetically. By promoting DNA demethylation, high-dose vitamin C induced stem cells to mature and suppressed the growth of leukemic stem cells (LSCs) implanted in mice.
“Interestingly, we also found that vitamin C treatment had an effect on leukemic stem cells that resembled damage to their DNA,” said study author Luisa Cimmino, PhD, of NYU School of Medicine.
“For this reason, we decided to combine vitamin C with a PARP inhibitor, a drug type known to cause cancer cell death by blocking the repair of DNA damage, and already approved for treating certain patients with ovarian cancer.”
The combination had an enhanced effect on LSCs, further shifting them from self-renewal back toward maturity and cell death.
Dr Cimmino said these results suggest vitamin C might also be effective against leukemias without TET2 mutations. As vitamin C turns up any TET2 activity normally in place, it might drive LSCs without TET2 mutations toward death as well. ![]()
Preclinical research suggests high-dose vitamin C may be effective against TET2-mutant leukemias.
Investigators found that vitamin C mimics TET2 restoration, thereby suppressing leukemic colony formation, inhibiting leukemia progression in mice, and enhancing leukemia cells’ sensitivity to treatment with a PARP inhibitor.
“We’re excited by the prospect that high-dose vitamin C might become a safe treatment for blood diseases caused by TET2-deficient leukemia stem cells, most likely in combination with other targeted therapies,” said study author Benjamin G. Neel, MD, PhD, of NYU School of Medicine in New York, New York.
Dr Neel and his colleagues reported their findings in Cell.
Previous research had shown that vitamin C could stimulate the activity of TET2 as well as TET1 and TET3.
Because only 1 copy of the TET2 gene in each stem cell is usually affected in TET2-mutant blood diseases, the investigators hypothesized that high doses of vitamin C might reverse the effects of TET2 deficiency by turning up the action of the remaining functional gene.
Indeed, the team found that vitamin C had the same effect as restoring TET2 function genetically. By promoting DNA demethylation, high-dose vitamin C induced stem cells to mature and suppressed the growth of leukemic stem cells (LSCs) implanted in mice.
“Interestingly, we also found that vitamin C treatment had an effect on leukemic stem cells that resembled damage to their DNA,” said study author Luisa Cimmino, PhD, of NYU School of Medicine.
“For this reason, we decided to combine vitamin C with a PARP inhibitor, a drug type known to cause cancer cell death by blocking the repair of DNA damage, and already approved for treating certain patients with ovarian cancer.”
The combination had an enhanced effect on LSCs, further shifting them from self-renewal back toward maturity and cell death.
Dr Cimmino said these results suggest vitamin C might also be effective against leukemias without TET2 mutations. As vitamin C turns up any TET2 activity normally in place, it might drive LSCs without TET2 mutations toward death as well. ![]()
Preclinical research suggests high-dose vitamin C may be effective against TET2-mutant leukemias.
Investigators found that vitamin C mimics TET2 restoration, thereby suppressing leukemic colony formation, inhibiting leukemia progression in mice, and enhancing leukemia cells’ sensitivity to treatment with a PARP inhibitor.
“We’re excited by the prospect that high-dose vitamin C might become a safe treatment for blood diseases caused by TET2-deficient leukemia stem cells, most likely in combination with other targeted therapies,” said study author Benjamin G. Neel, MD, PhD, of NYU School of Medicine in New York, New York.
Dr Neel and his colleagues reported their findings in Cell.
Previous research had shown that vitamin C could stimulate the activity of TET2 as well as TET1 and TET3.
Because only 1 copy of the TET2 gene in each stem cell is usually affected in TET2-mutant blood diseases, the investigators hypothesized that high doses of vitamin C might reverse the effects of TET2 deficiency by turning up the action of the remaining functional gene.
Indeed, the team found that vitamin C had the same effect as restoring TET2 function genetically. By promoting DNA demethylation, high-dose vitamin C induced stem cells to mature and suppressed the growth of leukemic stem cells (LSCs) implanted in mice.
“Interestingly, we also found that vitamin C treatment had an effect on leukemic stem cells that resembled damage to their DNA,” said study author Luisa Cimmino, PhD, of NYU School of Medicine.
“For this reason, we decided to combine vitamin C with a PARP inhibitor, a drug type known to cause cancer cell death by blocking the repair of DNA damage, and already approved for treating certain patients with ovarian cancer.”
The combination had an enhanced effect on LSCs, further shifting them from self-renewal back toward maturity and cell death.
Dr Cimmino said these results suggest vitamin C might also be effective against leukemias without TET2 mutations. As vitamin C turns up any TET2 activity normally in place, it might drive LSCs without TET2 mutations toward death as well. ![]()
AAP releases revised guidelines on screening, treatment of hypertension
which includes revised BP tables based on normal-weight children only.
The document, published Aug. 21 in Pediatrics, is the first update since 2004, and recommends significant changes in both screening and treatment of hypertension (HTN).
The guidelines also include a simplified screening table for initial screening, which lists the 90th percentile BP for age and sex, for children at the fifth percentile of height. These values give the table a negative predictive value of greater than 99%, although the committee stressed that the table should only be used for screening, and not for diagnosis.
“To diagnose elevated BP or HTN, it is important to locate the actual cutoffs in the complete BP tables because the [systolic] BP and [diastolic] BP cutoffs may be as much as 9 mm Hg higher depending on a child’s age and length or height,” wrote Joseph T. Flynn, MD, and his colleagues on the AAP subcommittee on screening and management of high blood pressure in children.
To ensure consistency between these guidelines and the 2017 adult guidelines from the American Heart Association and American College of Cardiology, the committee also decided to replace the term “prehypertension” with “elevated blood pressure.”
Similarly, the committee recommended adopting revised stage 1 and stage 2 HTN criteria, to enable the staging scheme for children aged 13 years and over to “seamlessly interface” with the 2017 AHA and ACC adult guidelines.
“There are still no data to identify a specific level of BP in childhood that leads to adverse [cardiovascular] outcomes in adulthood,” the committee wrote. “Therefore, the subcommittee decided to maintain a statistical definition for childhood HTN.”
In terms of screening children for hypertension, the guidelines review committee made the recommendation that BP be measured annually in children and adolescents aged 3 years or above. However, if the child is at greater risk of hypertension because of obesity, medications known to increase BP – such as stimulants for ADHD – renal disease, a history of aortic arch obstruction or coarctation, or have diabetes, they should have their BP measured at every health care encounter.
They also stressed it was important to take more than one measurement over time before diagnosing HTN, and to use appropriately-sized cuffs to ensure an accurate measurement.
If a child or adolescent has confirmed auscultatory BP readings at or above the 95th percentile on three different visits, this justifies a diagnosis of HTN, they wrote.
The committee strongly recommended the routine performance of ambulatory BP monitoring in patients with high-risk conditions, such as diabetes, secondary hypertension, or renal disease, to look for abnormal circadian BP patterns that might point to an increased risk of target organ damage.
They also issued revised recommendations on when to perform echocardiography in children newly diagnosed with HTN.
“It is recommended that echocardiography be performed to assess for cardiac target organ damage (left ventricular mass, geometry, and function) at the time of consideration of pharmacologic treatment of HTN,” they wrote, suggesting repeat echocardiography could be used to monitor target organ damage at 6-12 month intervals.
They offered a revised definition of left ventricular HTN as a left ventricular mass greater than 51 g/m2.7 for children and adolescents older than 8 years (greater than 115 g/body surface area for boys and greater than 95 g/body surface area for girls).
While previous treatment recommendations used a treatment target of a systolic and diastolic BP below the 95th percentile in children without chronic kidney disease, new evidence prompted a revised recommendation of a target either below the 90th percentile or less than 130/80 mm Hg.
“Longitudinal studies on BP from childhood to adulthood that include indirect measures of [cardiovascular] injury indicate that the risk for subsequent [cardiovascular disease] in early adulthood increases as the BP level in adolescence exceeds 120/80 mm Hg,” they wrote. “In addition, there is some evidence that targeting a BP less than 90th percentile results in reductions in [left ventricular mass index] and prevalence of [left ventricular hypertrophy].”
In hypertensive children and adolescents who have failed lifestyle modifications, such as physical activity, weight loss, and stress reduction (particularly those who have left ventricular hypertrophy, symptomatic HTN, or stage 2 HTN without a clearly modifiable factor such as obesity), pharmacologic treatment with an angiotensin-converting enzyme inhibitor, angiotensin receptor blocker, long-acting calcium channel blocker, or thiazide diuretic is indicated, according to the revised guidelines.
The guidelines were supported by the American Academy of Pediatrics and endorsed by the American Heart Association. No conflicts of interest were declared.
which includes revised BP tables based on normal-weight children only.
The document, published Aug. 21 in Pediatrics, is the first update since 2004, and recommends significant changes in both screening and treatment of hypertension (HTN).
The guidelines also include a simplified screening table for initial screening, which lists the 90th percentile BP for age and sex, for children at the fifth percentile of height. These values give the table a negative predictive value of greater than 99%, although the committee stressed that the table should only be used for screening, and not for diagnosis.
“To diagnose elevated BP or HTN, it is important to locate the actual cutoffs in the complete BP tables because the [systolic] BP and [diastolic] BP cutoffs may be as much as 9 mm Hg higher depending on a child’s age and length or height,” wrote Joseph T. Flynn, MD, and his colleagues on the AAP subcommittee on screening and management of high blood pressure in children.
To ensure consistency between these guidelines and the 2017 adult guidelines from the American Heart Association and American College of Cardiology, the committee also decided to replace the term “prehypertension” with “elevated blood pressure.”
Similarly, the committee recommended adopting revised stage 1 and stage 2 HTN criteria, to enable the staging scheme for children aged 13 years and over to “seamlessly interface” with the 2017 AHA and ACC adult guidelines.
“There are still no data to identify a specific level of BP in childhood that leads to adverse [cardiovascular] outcomes in adulthood,” the committee wrote. “Therefore, the subcommittee decided to maintain a statistical definition for childhood HTN.”
In terms of screening children for hypertension, the guidelines review committee made the recommendation that BP be measured annually in children and adolescents aged 3 years or above. However, if the child is at greater risk of hypertension because of obesity, medications known to increase BP – such as stimulants for ADHD – renal disease, a history of aortic arch obstruction or coarctation, or have diabetes, they should have their BP measured at every health care encounter.
They also stressed it was important to take more than one measurement over time before diagnosing HTN, and to use appropriately-sized cuffs to ensure an accurate measurement.
If a child or adolescent has confirmed auscultatory BP readings at or above the 95th percentile on three different visits, this justifies a diagnosis of HTN, they wrote.
The committee strongly recommended the routine performance of ambulatory BP monitoring in patients with high-risk conditions, such as diabetes, secondary hypertension, or renal disease, to look for abnormal circadian BP patterns that might point to an increased risk of target organ damage.
They also issued revised recommendations on when to perform echocardiography in children newly diagnosed with HTN.
“It is recommended that echocardiography be performed to assess for cardiac target organ damage (left ventricular mass, geometry, and function) at the time of consideration of pharmacologic treatment of HTN,” they wrote, suggesting repeat echocardiography could be used to monitor target organ damage at 6-12 month intervals.
They offered a revised definition of left ventricular HTN as a left ventricular mass greater than 51 g/m2.7 for children and adolescents older than 8 years (greater than 115 g/body surface area for boys and greater than 95 g/body surface area for girls).
While previous treatment recommendations used a treatment target of a systolic and diastolic BP below the 95th percentile in children without chronic kidney disease, new evidence prompted a revised recommendation of a target either below the 90th percentile or less than 130/80 mm Hg.
“Longitudinal studies on BP from childhood to adulthood that include indirect measures of [cardiovascular] injury indicate that the risk for subsequent [cardiovascular disease] in early adulthood increases as the BP level in adolescence exceeds 120/80 mm Hg,” they wrote. “In addition, there is some evidence that targeting a BP less than 90th percentile results in reductions in [left ventricular mass index] and prevalence of [left ventricular hypertrophy].”
In hypertensive children and adolescents who have failed lifestyle modifications, such as physical activity, weight loss, and stress reduction (particularly those who have left ventricular hypertrophy, symptomatic HTN, or stage 2 HTN without a clearly modifiable factor such as obesity), pharmacologic treatment with an angiotensin-converting enzyme inhibitor, angiotensin receptor blocker, long-acting calcium channel blocker, or thiazide diuretic is indicated, according to the revised guidelines.
The guidelines were supported by the American Academy of Pediatrics and endorsed by the American Heart Association. No conflicts of interest were declared.
which includes revised BP tables based on normal-weight children only.
The document, published Aug. 21 in Pediatrics, is the first update since 2004, and recommends significant changes in both screening and treatment of hypertension (HTN).
The guidelines also include a simplified screening table for initial screening, which lists the 90th percentile BP for age and sex, for children at the fifth percentile of height. These values give the table a negative predictive value of greater than 99%, although the committee stressed that the table should only be used for screening, and not for diagnosis.
“To diagnose elevated BP or HTN, it is important to locate the actual cutoffs in the complete BP tables because the [systolic] BP and [diastolic] BP cutoffs may be as much as 9 mm Hg higher depending on a child’s age and length or height,” wrote Joseph T. Flynn, MD, and his colleagues on the AAP subcommittee on screening and management of high blood pressure in children.
To ensure consistency between these guidelines and the 2017 adult guidelines from the American Heart Association and American College of Cardiology, the committee also decided to replace the term “prehypertension” with “elevated blood pressure.”
Similarly, the committee recommended adopting revised stage 1 and stage 2 HTN criteria, to enable the staging scheme for children aged 13 years and over to “seamlessly interface” with the 2017 AHA and ACC adult guidelines.
“There are still no data to identify a specific level of BP in childhood that leads to adverse [cardiovascular] outcomes in adulthood,” the committee wrote. “Therefore, the subcommittee decided to maintain a statistical definition for childhood HTN.”
In terms of screening children for hypertension, the guidelines review committee made the recommendation that BP be measured annually in children and adolescents aged 3 years or above. However, if the child is at greater risk of hypertension because of obesity, medications known to increase BP – such as stimulants for ADHD – renal disease, a history of aortic arch obstruction or coarctation, or have diabetes, they should have their BP measured at every health care encounter.
They also stressed it was important to take more than one measurement over time before diagnosing HTN, and to use appropriately-sized cuffs to ensure an accurate measurement.
If a child or adolescent has confirmed auscultatory BP readings at or above the 95th percentile on three different visits, this justifies a diagnosis of HTN, they wrote.
The committee strongly recommended the routine performance of ambulatory BP monitoring in patients with high-risk conditions, such as diabetes, secondary hypertension, or renal disease, to look for abnormal circadian BP patterns that might point to an increased risk of target organ damage.
They also issued revised recommendations on when to perform echocardiography in children newly diagnosed with HTN.
“It is recommended that echocardiography be performed to assess for cardiac target organ damage (left ventricular mass, geometry, and function) at the time of consideration of pharmacologic treatment of HTN,” they wrote, suggesting repeat echocardiography could be used to monitor target organ damage at 6-12 month intervals.
They offered a revised definition of left ventricular HTN as a left ventricular mass greater than 51 g/m2.7 for children and adolescents older than 8 years (greater than 115 g/body surface area for boys and greater than 95 g/body surface area for girls).
While previous treatment recommendations used a treatment target of a systolic and diastolic BP below the 95th percentile in children without chronic kidney disease, new evidence prompted a revised recommendation of a target either below the 90th percentile or less than 130/80 mm Hg.
“Longitudinal studies on BP from childhood to adulthood that include indirect measures of [cardiovascular] injury indicate that the risk for subsequent [cardiovascular disease] in early adulthood increases as the BP level in adolescence exceeds 120/80 mm Hg,” they wrote. “In addition, there is some evidence that targeting a BP less than 90th percentile results in reductions in [left ventricular mass index] and prevalence of [left ventricular hypertrophy].”
In hypertensive children and adolescents who have failed lifestyle modifications, such as physical activity, weight loss, and stress reduction (particularly those who have left ventricular hypertrophy, symptomatic HTN, or stage 2 HTN without a clearly modifiable factor such as obesity), pharmacologic treatment with an angiotensin-converting enzyme inhibitor, angiotensin receptor blocker, long-acting calcium channel blocker, or thiazide diuretic is indicated, according to the revised guidelines.
The guidelines were supported by the American Academy of Pediatrics and endorsed by the American Heart Association. No conflicts of interest were declared.
FROM PEDIATRICS
Recommendations for infant sleep position are not being followed
Despite the American Academy of Pediatrics recommendation that parents place infants supine for sleeping, many mothers do not do so, according to the results of the Study of Attitudes and Factors Affecting Infant Care.
In 32 hospitals, 3,297 mothers were surveyed to determine their intentions, the actual infant sleeping position used, and the factors that affected their behaviors. Of those, 2,491 (77%) reported that they usually place their infants in supine position, but only 49% reported that they exclusively place their infants supine. In addition, 14% reported that they place infants on their sides and 8% prone, reported Eve R. Colson, MD, of the department of pediatrics at Yale University, New Haven, Conn., and her coauthors (Pediatrics. 2017. doi: 10.1542/peds.2017-0596).
Several other discrepancies in compliance with the AAP recommendation also were noted. African American mothers were more likely to intend to use prone position, compared with white mothers (adjusted odds ratio, 2.5; 95% confidence interval, 1.57-3.85). Those who did not complete high school were also more likely to intend to use the prone position (aOR, 2.1; 95% CI, 1.16-3.73). On the other hand, those who received recommendation-compliant advice from a doctor were less likely to place their infants in the prone (aOR, 0.6; 95% CI, 0.39-0.93) or side (aOR, 0.5; 95% CI, 0.36-0.67) positions.
“Of particular note, those who reported that their social norms supported placing the infant in the prone position were much more likely to do so, compared with those who felt that their social norms supported using only the supine position (aOR, 11.6; 95% CI, 7.24-18.7). And, most remarkably, those who had positive attitudes about the prone sleep position ... were more likely to choose the prone position (aOR, 130; 95% CI, 71.8-236),” the researchers wrote. These findings indicate that choices in infant sleeping position are directly influenced by attitudes toward the choice, subjective social norms, and perceptions about control.
“These beliefs persist and are potentially modifiable, so they should be considered an important part of any intervention to change practice,” Dr. Colson and her colleagues wrote.
The study was a nationally representative sample of mothers of infants aged 2-6 months. Although the data were taken from patient surveys, which could have been misreported, they are supported by the findings of other studies.
[polldaddy:{"method":"iframe","type":"survey","src":"//newspolls2017.polldaddy.com/s/recommendations-for-infant-sleep-position-are-not-being-followed?iframe=1"}]
“Maternal race and education continue to be factors associated with choice of infant sleeping position as does advice from a doctor. Factors that appear to be of equal or greater importance are those related to attitudes, subjective social norms, and perceived control, all of which can potentially be altered through educational interventions,” Dr. Colson and her colleagues concluded.
The Eunice Kennedy Shriver National Institute of Child Health and Human Development and the National Institutes of Health funded the study. The authors reported no financial disclosures.
Over the 10 years spanning 1994-2004, the sudden infant death syndrome rate in the United States fell by 53%, correlating with an increase in exclusive supine sleep from less than 10% to 78%. However, since then, rates of both supine sleep and SIDS death have remained stagnant.
To make progress in these areas, current data are needed on supine sleep to enhance understanding of how families make these decisions. Colson et al. provide exactly the kind of information we need to guide providers and public health officials in their efforts to help families maintain the safest sleep environments for their infants.
As a start, mothers who want to practice safe sleep need to be empowered to insist that other caregivers in their lives support their parenting decisions, because the study shows that mothers who feel that they have more control are more likely to use the recommended position. We also must look at how we can help change personal attitudes and societal norms in favor of supine sleep because these issues were found to be some of the strongest predictors of prone sleep position.
We, as health care providers, need to provide clear and consistent messaging in both word and behavior to help mothers make safe decisions for their infants.
Michael H. Goodstein, MD, is a neonatologist at WellSpan York (Pa.) Hospital. Barbara M. Ostfeld, PhD, is the program director of the SIDS Center of New Jersey at Rutgers University, New Brunswick. Their remarks accompanied the article by Colson et al. (Pediatrics 2017 Aug 21. doi: 10.1542/peds.2017-2068). Neither author reported any financial disclosures.
Over the 10 years spanning 1994-2004, the sudden infant death syndrome rate in the United States fell by 53%, correlating with an increase in exclusive supine sleep from less than 10% to 78%. However, since then, rates of both supine sleep and SIDS death have remained stagnant.
To make progress in these areas, current data are needed on supine sleep to enhance understanding of how families make these decisions. Colson et al. provide exactly the kind of information we need to guide providers and public health officials in their efforts to help families maintain the safest sleep environments for their infants.
As a start, mothers who want to practice safe sleep need to be empowered to insist that other caregivers in their lives support their parenting decisions, because the study shows that mothers who feel that they have more control are more likely to use the recommended position. We also must look at how we can help change personal attitudes and societal norms in favor of supine sleep because these issues were found to be some of the strongest predictors of prone sleep position.
We, as health care providers, need to provide clear and consistent messaging in both word and behavior to help mothers make safe decisions for their infants.
Michael H. Goodstein, MD, is a neonatologist at WellSpan York (Pa.) Hospital. Barbara M. Ostfeld, PhD, is the program director of the SIDS Center of New Jersey at Rutgers University, New Brunswick. Their remarks accompanied the article by Colson et al. (Pediatrics 2017 Aug 21. doi: 10.1542/peds.2017-2068). Neither author reported any financial disclosures.
Over the 10 years spanning 1994-2004, the sudden infant death syndrome rate in the United States fell by 53%, correlating with an increase in exclusive supine sleep from less than 10% to 78%. However, since then, rates of both supine sleep and SIDS death have remained stagnant.
To make progress in these areas, current data are needed on supine sleep to enhance understanding of how families make these decisions. Colson et al. provide exactly the kind of information we need to guide providers and public health officials in their efforts to help families maintain the safest sleep environments for their infants.
As a start, mothers who want to practice safe sleep need to be empowered to insist that other caregivers in their lives support their parenting decisions, because the study shows that mothers who feel that they have more control are more likely to use the recommended position. We also must look at how we can help change personal attitudes and societal norms in favor of supine sleep because these issues were found to be some of the strongest predictors of prone sleep position.
We, as health care providers, need to provide clear and consistent messaging in both word and behavior to help mothers make safe decisions for their infants.
Michael H. Goodstein, MD, is a neonatologist at WellSpan York (Pa.) Hospital. Barbara M. Ostfeld, PhD, is the program director of the SIDS Center of New Jersey at Rutgers University, New Brunswick. Their remarks accompanied the article by Colson et al. (Pediatrics 2017 Aug 21. doi: 10.1542/peds.2017-2068). Neither author reported any financial disclosures.
Despite the American Academy of Pediatrics recommendation that parents place infants supine for sleeping, many mothers do not do so, according to the results of the Study of Attitudes and Factors Affecting Infant Care.
In 32 hospitals, 3,297 mothers were surveyed to determine their intentions, the actual infant sleeping position used, and the factors that affected their behaviors. Of those, 2,491 (77%) reported that they usually place their infants in supine position, but only 49% reported that they exclusively place their infants supine. In addition, 14% reported that they place infants on their sides and 8% prone, reported Eve R. Colson, MD, of the department of pediatrics at Yale University, New Haven, Conn., and her coauthors (Pediatrics. 2017. doi: 10.1542/peds.2017-0596).
Several other discrepancies in compliance with the AAP recommendation also were noted. African American mothers were more likely to intend to use prone position, compared with white mothers (adjusted odds ratio, 2.5; 95% confidence interval, 1.57-3.85). Those who did not complete high school were also more likely to intend to use the prone position (aOR, 2.1; 95% CI, 1.16-3.73). On the other hand, those who received recommendation-compliant advice from a doctor were less likely to place their infants in the prone (aOR, 0.6; 95% CI, 0.39-0.93) or side (aOR, 0.5; 95% CI, 0.36-0.67) positions.
“Of particular note, those who reported that their social norms supported placing the infant in the prone position were much more likely to do so, compared with those who felt that their social norms supported using only the supine position (aOR, 11.6; 95% CI, 7.24-18.7). And, most remarkably, those who had positive attitudes about the prone sleep position ... were more likely to choose the prone position (aOR, 130; 95% CI, 71.8-236),” the researchers wrote. These findings indicate that choices in infant sleeping position are directly influenced by attitudes toward the choice, subjective social norms, and perceptions about control.
“These beliefs persist and are potentially modifiable, so they should be considered an important part of any intervention to change practice,” Dr. Colson and her colleagues wrote.
The study was a nationally representative sample of mothers of infants aged 2-6 months. Although the data were taken from patient surveys, which could have been misreported, they are supported by the findings of other studies.
[polldaddy:{"method":"iframe","type":"survey","src":"//newspolls2017.polldaddy.com/s/recommendations-for-infant-sleep-position-are-not-being-followed?iframe=1"}]
“Maternal race and education continue to be factors associated with choice of infant sleeping position as does advice from a doctor. Factors that appear to be of equal or greater importance are those related to attitudes, subjective social norms, and perceived control, all of which can potentially be altered through educational interventions,” Dr. Colson and her colleagues concluded.
The Eunice Kennedy Shriver National Institute of Child Health and Human Development and the National Institutes of Health funded the study. The authors reported no financial disclosures.
Despite the American Academy of Pediatrics recommendation that parents place infants supine for sleeping, many mothers do not do so, according to the results of the Study of Attitudes and Factors Affecting Infant Care.
In 32 hospitals, 3,297 mothers were surveyed to determine their intentions, the actual infant sleeping position used, and the factors that affected their behaviors. Of those, 2,491 (77%) reported that they usually place their infants in supine position, but only 49% reported that they exclusively place their infants supine. In addition, 14% reported that they place infants on their sides and 8% prone, reported Eve R. Colson, MD, of the department of pediatrics at Yale University, New Haven, Conn., and her coauthors (Pediatrics. 2017. doi: 10.1542/peds.2017-0596).
Several other discrepancies in compliance with the AAP recommendation also were noted. African American mothers were more likely to intend to use prone position, compared with white mothers (adjusted odds ratio, 2.5; 95% confidence interval, 1.57-3.85). Those who did not complete high school were also more likely to intend to use the prone position (aOR, 2.1; 95% CI, 1.16-3.73). On the other hand, those who received recommendation-compliant advice from a doctor were less likely to place their infants in the prone (aOR, 0.6; 95% CI, 0.39-0.93) or side (aOR, 0.5; 95% CI, 0.36-0.67) positions.
“Of particular note, those who reported that their social norms supported placing the infant in the prone position were much more likely to do so, compared with those who felt that their social norms supported using only the supine position (aOR, 11.6; 95% CI, 7.24-18.7). And, most remarkably, those who had positive attitudes about the prone sleep position ... were more likely to choose the prone position (aOR, 130; 95% CI, 71.8-236),” the researchers wrote. These findings indicate that choices in infant sleeping position are directly influenced by attitudes toward the choice, subjective social norms, and perceptions about control.
“These beliefs persist and are potentially modifiable, so they should be considered an important part of any intervention to change practice,” Dr. Colson and her colleagues wrote.
The study was a nationally representative sample of mothers of infants aged 2-6 months. Although the data were taken from patient surveys, which could have been misreported, they are supported by the findings of other studies.
[polldaddy:{"method":"iframe","type":"survey","src":"//newspolls2017.polldaddy.com/s/recommendations-for-infant-sleep-position-are-not-being-followed?iframe=1"}]
“Maternal race and education continue to be factors associated with choice of infant sleeping position as does advice from a doctor. Factors that appear to be of equal or greater importance are those related to attitudes, subjective social norms, and perceived control, all of which can potentially be altered through educational interventions,” Dr. Colson and her colleagues concluded.
The Eunice Kennedy Shriver National Institute of Child Health and Human Development and the National Institutes of Health funded the study. The authors reported no financial disclosures.
FROM PEDIATRICS
Key clinical point:
Major finding: Of the 3,297 mothers surveyed, 2,491 (77%) reported that they usually place their infants in supine position, but only 49% reported that they exclusively place their infants supine.
Data source: The Study of Attitudes and Factors Affecting Infant Care, involving 3,297 mothers.
Disclosures: The Eunice Kennedy Shriver National Institute of Child Health and Human Development and the National Institutes of Health funded the study. The authors reported no financial disclosures.

Ribociclib: another CDK inhibitor hits the mark in breast cancer
This spring, the US Food and Drug Administration approved a second cyclin-dependent kinase (CDK) inhibitor for the treatment of postmenopausal women with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced/metastatic breast cancer in combination with aromatase inhibitors (AIs).1 The drug, ribociclib, joins palbociclib as the second drug in this class, which targets key regulators of the mammalian cell cycle and can help to overcome resistance to endocrine therapy–like AIs, a standard front-line treatment option in this group of patients. Palbociclib (Ibrance) was approved last year in combination with the AI letrozole, which was recently expanded to include its use in combination with all AIs, the same indication for which ribociclib received approval.
The ribociclib approval was based on the results of a phase 3, randomized, double-blind, placebo-controlled, international clinical trial called MONALEESA-2.2 The trial, conducted in 29 countries, compared the effects of ribociclib plus letrozole with letrozole plus placebo in 668 postmenopausal women with locally confirmed, HR-positive, HER2-negative, recurrent or metastatic breast cancer.
Patients had not received previous systemic therapy for advanced disease, had measurable disease according to Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1), had an Eastern Cooperative Oncology Group performance status of 0 or 1 (range, 1-5; 0, fully active and 5, dead), and had adequate bone marrow and organ function.
Patients were excluded if they had received previous CDK4/6 therapy, any previous systemic chemotherapy, endocrine therapy for advanced disease, previous neoadjuvant or adjuvant therapy with any nonsteroidal AI (unless they had been disease free for more than 12 months), and had inflammatory breast cancer, central nervous system metastases, history of cardiac disease or dysfunction, or impaired gastrointestinal function that alters drug absorption.
Patients were treated with ribociclib at a dose of 600 mg daily on a 3-weeks-on, 1-week-off schedule in 28-day cycles or placebo, which were combined with letrozole at a dose of 2.5 mg a day on a continuous schedule. Randomization was stratified according to the presence or absence of liver or lung metastases and treatment was continued until disease progression, unacceptable toxicity, death or discontinuation of treatment. Dose reductions of ribociclib were allowed, to manage adverse events (AEs), but treatment crossover was not permitted.
Tumor assessments were performed at screening, every 8 weeks during the first 18 months, every 12 weeks thereafter until disease progression, and at the end of treatment, and were assessed by an independent review committee. The baseline characteristics of the patient population were well balanced; patients had a median age of 62 years, all were HR positive except 1 patient who was HER2 positive.
The trial was ended prematurely after an initial interim analysis demonstrated a significant benefit in favor of ribociclib in the primary endpoint, progression-free survival (PFS). Over a median duration of follow-up of 15.3 months, the median PFS was not yet reached in the ribociclib arm, compared with 14.7 months in the placebo arm (hazard ratio, 0.556; P < .0001). In a subsequent analysis with 11 months of additional follow-up, the median PFS was 25.3 months in the combination arm, compared with 16 months in the placebo arm, which translated into a 44% reduction in the risk of disease progression or death. The PFS benefit with ribociclib was observed across all preplanned subgroup analyses. The objective response rates were 52.7% in the ribociclib arm, compared with 37.1% in the placebo arm, but overall survival data were immature.
The frequency and severity of AEs were increased in the combination arm; most common were neutropenia, nausea, fatigue, diarrhea, leukopenia, alopecia, vomiting, constipation, headache, and back pain. The most common grade 3 or 4 AEs experienced with ribociclib were neutropenia, leukopenia, abnormal liver function tests, lymphopenia, and vomiting.
Ribociclib is accompanied by warnings and precautions about QT interval prolongation, hepatobiliary toxicity, and neutropenia. Clinicians are advised to monitor electrocardiograms and electrolytes before the start of ribociclib therapy and to begin treatment only in patients with QTcF values <450 ms and in whom electrolyte abnormalities have been corrected. ECG should be repeated at around day 14 of the first cycle, the beginning of the second cycle, and as deemed clinically necessary.
Liver function tests should be performed before starting treatment, every 2 weeks for the first 2 cycles, at the beginning of each of the subsequent 4 cycles, and as clinically indicated. For aspartate aminotransferase (AST) and/or alanine aminotransferase (ALT) levels greater than 3-5 times the upper limit of normal (ULN, grade 2), ribociclib should be interrupted until recovery to baseline or lower. For levels >5-20 times the ULN (grade 3) or recurring grade 2 increases, treatment should be interrupted until recovery to baseline or lower and then resumed at the next lowest dose level. Treatment with ribociclib should be discontinued in the event of recurring grade 3 elevations or for AST/ALT elevations >3 times ULN in combination with total bilirubin >2 times ULN.
Complete blood counts should be performed before starting treatment and monitored every 2 weeks for the first 2 cycles, at the beginning of each of the 4 subsequent cycles, and as clinically needed. If absolute neutrophil counts are 500-1,000 mm3 (grade 3), treatment should be discontinued until recovery to grade 2 or lower. If grade 3 neutropenia recurs or for grade 3 febrile neutropenia or grade 4 neutropenia, treatment should resume at a lower dose level upon recovery to grade 2 or lower.
Pregnant women and those of reproductive age should be warned of the risk of fetal harm and the need for effective contraception during treatment and for at least 3 weeks after the last dose. Ribociclib is marketed as Kisqali by Novartis.
1. Ribociclib (Kisqali). US Food and Drug Administration website. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm546438.htm. Last updated March 14, 2017. Accessed April 3, 2017.
2. Kisqali (ribociclib) tables, for oral use. Prescribing information. Novartis Pharmaceuticals Corp. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/kisqali.pdf. March 2017. Accessed April 3, 2017.
3. Horobagyi GN, Stemmer SN, Burris HA, et al. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N Engl J Med. 2016;375:1738-1748.
This spring, the US Food and Drug Administration approved a second cyclin-dependent kinase (CDK) inhibitor for the treatment of postmenopausal women with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced/metastatic breast cancer in combination with aromatase inhibitors (AIs).1 The drug, ribociclib, joins palbociclib as the second drug in this class, which targets key regulators of the mammalian cell cycle and can help to overcome resistance to endocrine therapy–like AIs, a standard front-line treatment option in this group of patients. Palbociclib (Ibrance) was approved last year in combination with the AI letrozole, which was recently expanded to include its use in combination with all AIs, the same indication for which ribociclib received approval.
The ribociclib approval was based on the results of a phase 3, randomized, double-blind, placebo-controlled, international clinical trial called MONALEESA-2.2 The trial, conducted in 29 countries, compared the effects of ribociclib plus letrozole with letrozole plus placebo in 668 postmenopausal women with locally confirmed, HR-positive, HER2-negative, recurrent or metastatic breast cancer.
Patients had not received previous systemic therapy for advanced disease, had measurable disease according to Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1), had an Eastern Cooperative Oncology Group performance status of 0 or 1 (range, 1-5; 0, fully active and 5, dead), and had adequate bone marrow and organ function.
Patients were excluded if they had received previous CDK4/6 therapy, any previous systemic chemotherapy, endocrine therapy for advanced disease, previous neoadjuvant or adjuvant therapy with any nonsteroidal AI (unless they had been disease free for more than 12 months), and had inflammatory breast cancer, central nervous system metastases, history of cardiac disease or dysfunction, or impaired gastrointestinal function that alters drug absorption.
Patients were treated with ribociclib at a dose of 600 mg daily on a 3-weeks-on, 1-week-off schedule in 28-day cycles or placebo, which were combined with letrozole at a dose of 2.5 mg a day on a continuous schedule. Randomization was stratified according to the presence or absence of liver or lung metastases and treatment was continued until disease progression, unacceptable toxicity, death or discontinuation of treatment. Dose reductions of ribociclib were allowed, to manage adverse events (AEs), but treatment crossover was not permitted.
Tumor assessments were performed at screening, every 8 weeks during the first 18 months, every 12 weeks thereafter until disease progression, and at the end of treatment, and were assessed by an independent review committee. The baseline characteristics of the patient population were well balanced; patients had a median age of 62 years, all were HR positive except 1 patient who was HER2 positive.
The trial was ended prematurely after an initial interim analysis demonstrated a significant benefit in favor of ribociclib in the primary endpoint, progression-free survival (PFS). Over a median duration of follow-up of 15.3 months, the median PFS was not yet reached in the ribociclib arm, compared with 14.7 months in the placebo arm (hazard ratio, 0.556; P < .0001). In a subsequent analysis with 11 months of additional follow-up, the median PFS was 25.3 months in the combination arm, compared with 16 months in the placebo arm, which translated into a 44% reduction in the risk of disease progression or death. The PFS benefit with ribociclib was observed across all preplanned subgroup analyses. The objective response rates were 52.7% in the ribociclib arm, compared with 37.1% in the placebo arm, but overall survival data were immature.
The frequency and severity of AEs were increased in the combination arm; most common were neutropenia, nausea, fatigue, diarrhea, leukopenia, alopecia, vomiting, constipation, headache, and back pain. The most common grade 3 or 4 AEs experienced with ribociclib were neutropenia, leukopenia, abnormal liver function tests, lymphopenia, and vomiting.
Ribociclib is accompanied by warnings and precautions about QT interval prolongation, hepatobiliary toxicity, and neutropenia. Clinicians are advised to monitor electrocardiograms and electrolytes before the start of ribociclib therapy and to begin treatment only in patients with QTcF values <450 ms and in whom electrolyte abnormalities have been corrected. ECG should be repeated at around day 14 of the first cycle, the beginning of the second cycle, and as deemed clinically necessary.
Liver function tests should be performed before starting treatment, every 2 weeks for the first 2 cycles, at the beginning of each of the subsequent 4 cycles, and as clinically indicated. For aspartate aminotransferase (AST) and/or alanine aminotransferase (ALT) levels greater than 3-5 times the upper limit of normal (ULN, grade 2), ribociclib should be interrupted until recovery to baseline or lower. For levels >5-20 times the ULN (grade 3) or recurring grade 2 increases, treatment should be interrupted until recovery to baseline or lower and then resumed at the next lowest dose level. Treatment with ribociclib should be discontinued in the event of recurring grade 3 elevations or for AST/ALT elevations >3 times ULN in combination with total bilirubin >2 times ULN.
Complete blood counts should be performed before starting treatment and monitored every 2 weeks for the first 2 cycles, at the beginning of each of the 4 subsequent cycles, and as clinically needed. If absolute neutrophil counts are 500-1,000 mm3 (grade 3), treatment should be discontinued until recovery to grade 2 or lower. If grade 3 neutropenia recurs or for grade 3 febrile neutropenia or grade 4 neutropenia, treatment should resume at a lower dose level upon recovery to grade 2 or lower.
Pregnant women and those of reproductive age should be warned of the risk of fetal harm and the need for effective contraception during treatment and for at least 3 weeks after the last dose. Ribociclib is marketed as Kisqali by Novartis.
This spring, the US Food and Drug Administration approved a second cyclin-dependent kinase (CDK) inhibitor for the treatment of postmenopausal women with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced/metastatic breast cancer in combination with aromatase inhibitors (AIs).1 The drug, ribociclib, joins palbociclib as the second drug in this class, which targets key regulators of the mammalian cell cycle and can help to overcome resistance to endocrine therapy–like AIs, a standard front-line treatment option in this group of patients. Palbociclib (Ibrance) was approved last year in combination with the AI letrozole, which was recently expanded to include its use in combination with all AIs, the same indication for which ribociclib received approval.
The ribociclib approval was based on the results of a phase 3, randomized, double-blind, placebo-controlled, international clinical trial called MONALEESA-2.2 The trial, conducted in 29 countries, compared the effects of ribociclib plus letrozole with letrozole plus placebo in 668 postmenopausal women with locally confirmed, HR-positive, HER2-negative, recurrent or metastatic breast cancer.
Patients had not received previous systemic therapy for advanced disease, had measurable disease according to Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1), had an Eastern Cooperative Oncology Group performance status of 0 or 1 (range, 1-5; 0, fully active and 5, dead), and had adequate bone marrow and organ function.
Patients were excluded if they had received previous CDK4/6 therapy, any previous systemic chemotherapy, endocrine therapy for advanced disease, previous neoadjuvant or adjuvant therapy with any nonsteroidal AI (unless they had been disease free for more than 12 months), and had inflammatory breast cancer, central nervous system metastases, history of cardiac disease or dysfunction, or impaired gastrointestinal function that alters drug absorption.
Patients were treated with ribociclib at a dose of 600 mg daily on a 3-weeks-on, 1-week-off schedule in 28-day cycles or placebo, which were combined with letrozole at a dose of 2.5 mg a day on a continuous schedule. Randomization was stratified according to the presence or absence of liver or lung metastases and treatment was continued until disease progression, unacceptable toxicity, death or discontinuation of treatment. Dose reductions of ribociclib were allowed, to manage adverse events (AEs), but treatment crossover was not permitted.
Tumor assessments were performed at screening, every 8 weeks during the first 18 months, every 12 weeks thereafter until disease progression, and at the end of treatment, and were assessed by an independent review committee. The baseline characteristics of the patient population were well balanced; patients had a median age of 62 years, all were HR positive except 1 patient who was HER2 positive.
The trial was ended prematurely after an initial interim analysis demonstrated a significant benefit in favor of ribociclib in the primary endpoint, progression-free survival (PFS). Over a median duration of follow-up of 15.3 months, the median PFS was not yet reached in the ribociclib arm, compared with 14.7 months in the placebo arm (hazard ratio, 0.556; P < .0001). In a subsequent analysis with 11 months of additional follow-up, the median PFS was 25.3 months in the combination arm, compared with 16 months in the placebo arm, which translated into a 44% reduction in the risk of disease progression or death. The PFS benefit with ribociclib was observed across all preplanned subgroup analyses. The objective response rates were 52.7% in the ribociclib arm, compared with 37.1% in the placebo arm, but overall survival data were immature.
The frequency and severity of AEs were increased in the combination arm; most common were neutropenia, nausea, fatigue, diarrhea, leukopenia, alopecia, vomiting, constipation, headache, and back pain. The most common grade 3 or 4 AEs experienced with ribociclib were neutropenia, leukopenia, abnormal liver function tests, lymphopenia, and vomiting.
Ribociclib is accompanied by warnings and precautions about QT interval prolongation, hepatobiliary toxicity, and neutropenia. Clinicians are advised to monitor electrocardiograms and electrolytes before the start of ribociclib therapy and to begin treatment only in patients with QTcF values <450 ms and in whom electrolyte abnormalities have been corrected. ECG should be repeated at around day 14 of the first cycle, the beginning of the second cycle, and as deemed clinically necessary.
Liver function tests should be performed before starting treatment, every 2 weeks for the first 2 cycles, at the beginning of each of the subsequent 4 cycles, and as clinically indicated. For aspartate aminotransferase (AST) and/or alanine aminotransferase (ALT) levels greater than 3-5 times the upper limit of normal (ULN, grade 2), ribociclib should be interrupted until recovery to baseline or lower. For levels >5-20 times the ULN (grade 3) or recurring grade 2 increases, treatment should be interrupted until recovery to baseline or lower and then resumed at the next lowest dose level. Treatment with ribociclib should be discontinued in the event of recurring grade 3 elevations or for AST/ALT elevations >3 times ULN in combination with total bilirubin >2 times ULN.
Complete blood counts should be performed before starting treatment and monitored every 2 weeks for the first 2 cycles, at the beginning of each of the 4 subsequent cycles, and as clinically needed. If absolute neutrophil counts are 500-1,000 mm3 (grade 3), treatment should be discontinued until recovery to grade 2 or lower. If grade 3 neutropenia recurs or for grade 3 febrile neutropenia or grade 4 neutropenia, treatment should resume at a lower dose level upon recovery to grade 2 or lower.
Pregnant women and those of reproductive age should be warned of the risk of fetal harm and the need for effective contraception during treatment and for at least 3 weeks after the last dose. Ribociclib is marketed as Kisqali by Novartis.
1. Ribociclib (Kisqali). US Food and Drug Administration website. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm546438.htm. Last updated March 14, 2017. Accessed April 3, 2017.
2. Kisqali (ribociclib) tables, for oral use. Prescribing information. Novartis Pharmaceuticals Corp. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/kisqali.pdf. March 2017. Accessed April 3, 2017.
3. Horobagyi GN, Stemmer SN, Burris HA, et al. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N Engl J Med. 2016;375:1738-1748.
1. Ribociclib (Kisqali). US Food and Drug Administration website. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm546438.htm. Last updated March 14, 2017. Accessed April 3, 2017.
2. Kisqali (ribociclib) tables, for oral use. Prescribing information. Novartis Pharmaceuticals Corp. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/kisqali.pdf. March 2017. Accessed April 3, 2017.
3. Horobagyi GN, Stemmer SN, Burris HA, et al. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N Engl J Med. 2016;375:1738-1748.
Approval makes olaratumab the first first-line treatment option for soft tissue sarcoma in more than 40 years
When the US Food and Drug Administration approved olaratumab as a first-line treatment for patients with soft tissue sarcoma (STS) in the fall of 2016, it marked the first approval since the chemotherapy drug doxorubicin became standard of care more than 40 years ago.1 Though rare, STS, which comprises a host of different histologic subtypes, has proven difficult to treat. Like pazopanib, which was approved in 2012 for the treatment of STS in the second-line setting, olaratumab targets the platelet-derived growth factor receptor alpha (PDGFRα), a tyrosine kinase receptor involved in cell signaling pathways that promotes key hallmark abilities in both cancer cells and the cells of the tumor microenvironment. Olaratumab, however, is a much more specific inhibitor of PDGFRα compared with pazopanib.
Accelerated approval was granted for the treatment of patients with STS that is not amenable to curative treatment with radiotherapy or surgery and with a subtype that cannot be treated effectively with an anthracycline-containing regimen. The approval was based on the phase 2 JGDG study, a randomized, active-controlled clinical trial in which 133 patients were randomized 1:1 to receive olaratumab plus doxorubicin, or doxorubicin alone.2
Eligible patients included those aged 18 years and over, with histologically confirmed diagnosis of locally advanced or metastatic STS not previously treated with an anthracycline, with an Eastern Cooperative Oncology Group (ECOG) performance status of 0-2 (range, 1-5; 0, fully active and 5, dead), and with available tumor tissue for determination of PDGFRα expression by immunohistochemistry. Patients were enrolled at 16 clinical sites in 16 cities and 15 states in the United States from October 2010 to January 2013.
Patients were excluded if they had histologically or cytologically confirmed Kaposi sarcoma; untreated central nervous system metastases; received prior treatment with doxorubicin or other anthracyclines and anthracenediones, or any drug targeting PDGF or the PDGFRs; received concurrent treatment with other anticancer therapy within 4 weeks before study entry; unstable angina pectoris, angioplasty, cardiac stenting, or myocardial infarction within 6 months before study entry; HIV infection; or if they were pregnant or lactating.
Olaratumab was administered at 15 mg/kg as an intravenous infusion on days 1 and 8 of each 21-day cycle, and doxorubicin at 75 mg/m2 as an intravenous infusion on day 1 of each cycle, for a maximum of 8 cycles. Patients were permitted to receive dexarozoxane on cycles 5-8 and crossover was permitted. Tumor response was assessed by Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1) every 6 weeks, and survival assessed every 2 months, until study completion. PDGFR expression was assessed by immunohistochemistry at a central academic laboratory before randomization.
The primary endpoint of the study was progression-free survival (PFS) and the combination of olaratumab–doxorubicin significantly extended PFS in this patient population: median PFS was 6.6 months in the combination arm, compared with 4.1 months in the doxorubicin-alone arm (hazard ratio [HR], 0.672; P = .0615). The objective response rate (ORR) and median overall survival (OS), which were secondary endpoints in the trial, were also significantly improved with combination therapy compared with doxorubicin alone (ORR, 18.2% vs 11.9%, respectively; median OS, 26.5 months vs 14.7 months). The benefits of combination therapy were observed across prespecified subgroups, including histological tumor type, number of previous treatments, and PDGFRα expression level.
The most common adverse events (AEs) in the patients taking olaratumab were nausea, fatigue, neutropenia, musculoskeletal pain, mucositis, alopecia, vomiting, diarrhea, decreased appetite, abdominal pain, neuropathy, and headache. Grade 3/4 AEs were also higher for the combination than for doxorubicin alone. The most common AE leading to discontinuation of olaratumab was infusion-related reactions, which occurred in 13% of patients.
According to the prescribing information, the recommended dose for olaratumab is 15 mg/kg as an intravenous infusion over 60 minutes on days 1 and 8 of each 21-day cycle until disease progression or unacceptable toxicity, in combination with doxorubicin for the first 8 cycles. Patients should be premedicated with dexamethasone and diphenhydramine, to help protect against infusion-related reactions.
Olaratumab, marketed as Lartruvo by Lilly Oncology, has warnings and precautions relating to infusion-related reactions and embryofetal toxicity. Patients should be monitored for signs and symptoms of the former during and after infusion and olaratumab should be administered in a setting with available resuscitation equipment. Olaratumab should be permanently discontinued in the event of grade 3/4 infusion-related reactions. Olaratumab can cause fetal harm and female patients should be advised of the potential risk to a fetus and the need for effective contraception during treatment and for 3 months after the last dose.
1. FDA grants accelerated approval to new treatment for advanced soft tissue sarcoma. FDA News Release. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm525878.htm. Last updated October 19, 2016. Accessed March 6, 2017.
2. Tap WD, Jones RL, Van Tine BA, et al. Olaratumumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet. 2016;388(10043):488-497.
3. Lartruvo (olaratumumab) injection, for intravenous use. Prescribing information. Eli Lilly and Co. http://pi.lilly.com/us/lartruvo-uspi.pdf. Last update October 2016. Accessed March 6, 2017.
When the US Food and Drug Administration approved olaratumab as a first-line treatment for patients with soft tissue sarcoma (STS) in the fall of 2016, it marked the first approval since the chemotherapy drug doxorubicin became standard of care more than 40 years ago.1 Though rare, STS, which comprises a host of different histologic subtypes, has proven difficult to treat. Like pazopanib, which was approved in 2012 for the treatment of STS in the second-line setting, olaratumab targets the platelet-derived growth factor receptor alpha (PDGFRα), a tyrosine kinase receptor involved in cell signaling pathways that promotes key hallmark abilities in both cancer cells and the cells of the tumor microenvironment. Olaratumab, however, is a much more specific inhibitor of PDGFRα compared with pazopanib.
Accelerated approval was granted for the treatment of patients with STS that is not amenable to curative treatment with radiotherapy or surgery and with a subtype that cannot be treated effectively with an anthracycline-containing regimen. The approval was based on the phase 2 JGDG study, a randomized, active-controlled clinical trial in which 133 patients were randomized 1:1 to receive olaratumab plus doxorubicin, or doxorubicin alone.2
Eligible patients included those aged 18 years and over, with histologically confirmed diagnosis of locally advanced or metastatic STS not previously treated with an anthracycline, with an Eastern Cooperative Oncology Group (ECOG) performance status of 0-2 (range, 1-5; 0, fully active and 5, dead), and with available tumor tissue for determination of PDGFRα expression by immunohistochemistry. Patients were enrolled at 16 clinical sites in 16 cities and 15 states in the United States from October 2010 to January 2013.
Patients were excluded if they had histologically or cytologically confirmed Kaposi sarcoma; untreated central nervous system metastases; received prior treatment with doxorubicin or other anthracyclines and anthracenediones, or any drug targeting PDGF or the PDGFRs; received concurrent treatment with other anticancer therapy within 4 weeks before study entry; unstable angina pectoris, angioplasty, cardiac stenting, or myocardial infarction within 6 months before study entry; HIV infection; or if they were pregnant or lactating.
Olaratumab was administered at 15 mg/kg as an intravenous infusion on days 1 and 8 of each 21-day cycle, and doxorubicin at 75 mg/m2 as an intravenous infusion on day 1 of each cycle, for a maximum of 8 cycles. Patients were permitted to receive dexarozoxane on cycles 5-8 and crossover was permitted. Tumor response was assessed by Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1) every 6 weeks, and survival assessed every 2 months, until study completion. PDGFR expression was assessed by immunohistochemistry at a central academic laboratory before randomization.
The primary endpoint of the study was progression-free survival (PFS) and the combination of olaratumab–doxorubicin significantly extended PFS in this patient population: median PFS was 6.6 months in the combination arm, compared with 4.1 months in the doxorubicin-alone arm (hazard ratio [HR], 0.672; P = .0615). The objective response rate (ORR) and median overall survival (OS), which were secondary endpoints in the trial, were also significantly improved with combination therapy compared with doxorubicin alone (ORR, 18.2% vs 11.9%, respectively; median OS, 26.5 months vs 14.7 months). The benefits of combination therapy were observed across prespecified subgroups, including histological tumor type, number of previous treatments, and PDGFRα expression level.
The most common adverse events (AEs) in the patients taking olaratumab were nausea, fatigue, neutropenia, musculoskeletal pain, mucositis, alopecia, vomiting, diarrhea, decreased appetite, abdominal pain, neuropathy, and headache. Grade 3/4 AEs were also higher for the combination than for doxorubicin alone. The most common AE leading to discontinuation of olaratumab was infusion-related reactions, which occurred in 13% of patients.
According to the prescribing information, the recommended dose for olaratumab is 15 mg/kg as an intravenous infusion over 60 minutes on days 1 and 8 of each 21-day cycle until disease progression or unacceptable toxicity, in combination with doxorubicin for the first 8 cycles. Patients should be premedicated with dexamethasone and diphenhydramine, to help protect against infusion-related reactions.
Olaratumab, marketed as Lartruvo by Lilly Oncology, has warnings and precautions relating to infusion-related reactions and embryofetal toxicity. Patients should be monitored for signs and symptoms of the former during and after infusion and olaratumab should be administered in a setting with available resuscitation equipment. Olaratumab should be permanently discontinued in the event of grade 3/4 infusion-related reactions. Olaratumab can cause fetal harm and female patients should be advised of the potential risk to a fetus and the need for effective contraception during treatment and for 3 months after the last dose.
When the US Food and Drug Administration approved olaratumab as a first-line treatment for patients with soft tissue sarcoma (STS) in the fall of 2016, it marked the first approval since the chemotherapy drug doxorubicin became standard of care more than 40 years ago.1 Though rare, STS, which comprises a host of different histologic subtypes, has proven difficult to treat. Like pazopanib, which was approved in 2012 for the treatment of STS in the second-line setting, olaratumab targets the platelet-derived growth factor receptor alpha (PDGFRα), a tyrosine kinase receptor involved in cell signaling pathways that promotes key hallmark abilities in both cancer cells and the cells of the tumor microenvironment. Olaratumab, however, is a much more specific inhibitor of PDGFRα compared with pazopanib.
Accelerated approval was granted for the treatment of patients with STS that is not amenable to curative treatment with radiotherapy or surgery and with a subtype that cannot be treated effectively with an anthracycline-containing regimen. The approval was based on the phase 2 JGDG study, a randomized, active-controlled clinical trial in which 133 patients were randomized 1:1 to receive olaratumab plus doxorubicin, or doxorubicin alone.2
Eligible patients included those aged 18 years and over, with histologically confirmed diagnosis of locally advanced or metastatic STS not previously treated with an anthracycline, with an Eastern Cooperative Oncology Group (ECOG) performance status of 0-2 (range, 1-5; 0, fully active and 5, dead), and with available tumor tissue for determination of PDGFRα expression by immunohistochemistry. Patients were enrolled at 16 clinical sites in 16 cities and 15 states in the United States from October 2010 to January 2013.
Patients were excluded if they had histologically or cytologically confirmed Kaposi sarcoma; untreated central nervous system metastases; received prior treatment with doxorubicin or other anthracyclines and anthracenediones, or any drug targeting PDGF or the PDGFRs; received concurrent treatment with other anticancer therapy within 4 weeks before study entry; unstable angina pectoris, angioplasty, cardiac stenting, or myocardial infarction within 6 months before study entry; HIV infection; or if they were pregnant or lactating.
Olaratumab was administered at 15 mg/kg as an intravenous infusion on days 1 and 8 of each 21-day cycle, and doxorubicin at 75 mg/m2 as an intravenous infusion on day 1 of each cycle, for a maximum of 8 cycles. Patients were permitted to receive dexarozoxane on cycles 5-8 and crossover was permitted. Tumor response was assessed by Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1) every 6 weeks, and survival assessed every 2 months, until study completion. PDGFR expression was assessed by immunohistochemistry at a central academic laboratory before randomization.
The primary endpoint of the study was progression-free survival (PFS) and the combination of olaratumab–doxorubicin significantly extended PFS in this patient population: median PFS was 6.6 months in the combination arm, compared with 4.1 months in the doxorubicin-alone arm (hazard ratio [HR], 0.672; P = .0615). The objective response rate (ORR) and median overall survival (OS), which were secondary endpoints in the trial, were also significantly improved with combination therapy compared with doxorubicin alone (ORR, 18.2% vs 11.9%, respectively; median OS, 26.5 months vs 14.7 months). The benefits of combination therapy were observed across prespecified subgroups, including histological tumor type, number of previous treatments, and PDGFRα expression level.
The most common adverse events (AEs) in the patients taking olaratumab were nausea, fatigue, neutropenia, musculoskeletal pain, mucositis, alopecia, vomiting, diarrhea, decreased appetite, abdominal pain, neuropathy, and headache. Grade 3/4 AEs were also higher for the combination than for doxorubicin alone. The most common AE leading to discontinuation of olaratumab was infusion-related reactions, which occurred in 13% of patients.
According to the prescribing information, the recommended dose for olaratumab is 15 mg/kg as an intravenous infusion over 60 minutes on days 1 and 8 of each 21-day cycle until disease progression or unacceptable toxicity, in combination with doxorubicin for the first 8 cycles. Patients should be premedicated with dexamethasone and diphenhydramine, to help protect against infusion-related reactions.
Olaratumab, marketed as Lartruvo by Lilly Oncology, has warnings and precautions relating to infusion-related reactions and embryofetal toxicity. Patients should be monitored for signs and symptoms of the former during and after infusion and olaratumab should be administered in a setting with available resuscitation equipment. Olaratumab should be permanently discontinued in the event of grade 3/4 infusion-related reactions. Olaratumab can cause fetal harm and female patients should be advised of the potential risk to a fetus and the need for effective contraception during treatment and for 3 months after the last dose.
1. FDA grants accelerated approval to new treatment for advanced soft tissue sarcoma. FDA News Release. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm525878.htm. Last updated October 19, 2016. Accessed March 6, 2017.
2. Tap WD, Jones RL, Van Tine BA, et al. Olaratumumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet. 2016;388(10043):488-497.
3. Lartruvo (olaratumumab) injection, for intravenous use. Prescribing information. Eli Lilly and Co. http://pi.lilly.com/us/lartruvo-uspi.pdf. Last update October 2016. Accessed March 6, 2017.
1. FDA grants accelerated approval to new treatment for advanced soft tissue sarcoma. FDA News Release. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm525878.htm. Last updated October 19, 2016. Accessed March 6, 2017.
2. Tap WD, Jones RL, Van Tine BA, et al. Olaratumumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet. 2016;388(10043):488-497.
3. Lartruvo (olaratumumab) injection, for intravenous use. Prescribing information. Eli Lilly and Co. http://pi.lilly.com/us/lartruvo-uspi.pdf. Last update October 2016. Accessed March 6, 2017.