User login
Back to Basics: An Uncommon, Unrelated Presentation of a Common Disease
The early initial ulcerative lesion (chancre) caused by Treponema pallidum infection, has a median incubation period of 21 days (primary syphilis). When untreated, secondary syphilis will develop within weeks to months and is characterized by generalized symptoms such as malaise, fevers, headaches, sore throat, and myalgia. However, the most characteristic finding in secondary syphilis remains a rash that is classically identified as symmetric, macular, or papular, and involving the entire trunk and extremities, including the palms and soles.
When secondary syphilis is left untreated, late syphilis or tertiary syphilis can develop, which is characterized by cardiovascular involvement, including aortitis, gummatous syphilis (granulomatous nodules in a variety of organs but typically the skin and bones), or central nervous system involvement.1-3 The following case describes a patient with nondescript symptoms, including malaise and cough, who had a characteristic rash of secondary syphilis that was diagnosed and treated in our Houston-area community hospital.
Case
In late autumn, a 30-year-old man presented to our community ED for evaluation of a cough productive of green sputum along with mild chest discomfort, malaise, and generalized myalgia, which were intermittent over the course of the past month. The patient denied rhinorrhea, fevers, chills, dyspnea, or any other systemic complaints. He also denied any sick contacts, but noted that his influenza vaccine was not up to date.
The patient denied any remote or recent medical or surgical history. He further denied taking any medications, and noted that his only medical allergy was to penicillin. His family history was noncontributory. Regarding his social history, the patient admitted to smoking one pack of cigarettes per day and to a daily alcohol intake of approximately one 6-pack of beer. He also admitted to frequently smoking crystal methamphetamine, which he stated he had last used 2 days prior to presentation. The patient said his current chest pain was similar to prior episodes, noting that when the pain occurred, he would temporarily stop smoking crystal methamphetamine.
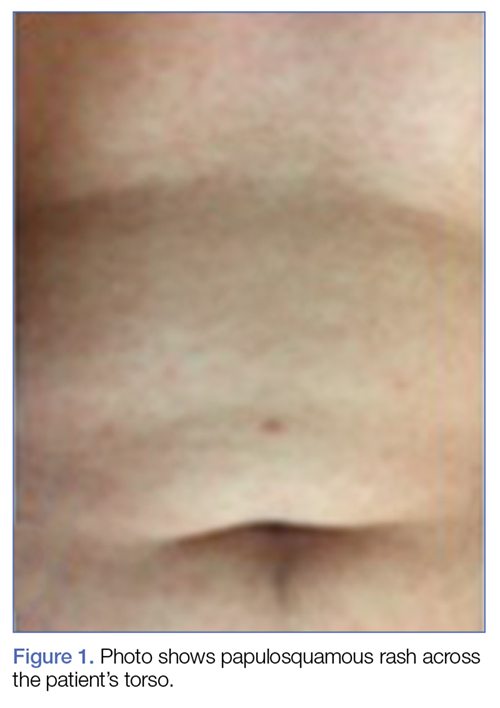
Plain chest radiography, electrocardiogram, complete metabolic panel, complete blood count, B-natriuretic peptide, and troponin levels were all unremarkable. Due to the presence and nature of the patient’s rash, a rapid plasma reagin (RPR) screen was also taken, the results of which were reactive.
On further questioning, the patient admitted to having multiple female sexual partners with whom he used barrier protection sporadically. A more detailed physical examination revealed multiple painless ulcerations/chancres over the penile shaft and scrotum, without urethral drainage or inguinal lymphadenopathy. The patient denied dysuria or hematuria.
Since the patient was allergic to penicillin, he was given a single oral dose of azithromycin 2 g, and started on a 2-week course of oral doxycycline 100 mg. Further laboratory studies included gonorrhea and chlamydia cultures, both of which were negative. He was instructed to follow-up with his primary care physician for extended sexually transmitted infection (STI) panel-testing, including HIV, hepatitis, and confirmatory syphilis testing. Unfortunately, it is not known whether the patient complied with discharge instructions as he was lost to follow-up.
Discussion
Diagnostic algorithms for syphilis, one of the best studied STIs, have changed with technological advancement, but diagnosis and treatment for the most part has remained mostly the same. The uniqueness of this case is really focused around the patient’s chief complaint. While it is classic to present with malaise, headache, and rash, our patient complained of cough productive of sputum with chest pain—a rare presentation of secondary syphilis. The fortuitous key finding of the truncal rash directed the emergency physician toward the appropriate diagnosis.
Diagnosis
In the ED, where patients such as the one in our case are often lost to follow-up, and consistent infectious disease and primary care follow-up is unavailable, prompt treatment based on history and physical examination alone is recommended. Patients should be tested for syphilis, as well as other STIs including chlamydia, gonorrhea, hepatitis, and HIV as an outpatient. In addition, any partners with whom the patient has had sexual contact within the last 90 days should also undergo STI testing; sexual partners from over 90 days should be notified of the patient’s status and evaluated with testing as indicated.4 All positive test results should be reported to the Centers for Disease Control and Prevention (CDC).5
Nontreponemal and Treponemal Testing
For patients with clinical signs and symptoms of syphilis, recommended laboratory evaluation includes both nontreponemal and treponemal testing. Nontreponemal tests include RPR, venereal disease research laboratory test, and toluidine red unheated serum test. Treponemal tests include fluorescent treponemal antibody absorption, microhemagglutination test for antibodies to T pallidum, T pallidum particle agglutination assay, T pallidum enzyme immunoassay, and chemiluminescence immunoassay. Patients who test positive for treponemal antibody will typically remain reactive for life.5,6
In the setting of discordant test results, patients with a nonreactive treponemal result are generally considered to be negative for syphilis. Discordant results with a negative nontreponemal test are more complicated, and recommendations are based on symptomatology and repeat testing.5
Treatment
When a patient has a positive nontreponemal and treponemal test, treatment is usually indicated. As with the patient in this case, treatment is always indicated for patients who have no prior history of syphilis. For patients who have a history of treated syphilis, attention must be given to titer levels on previous testing and to patient symptomatology.
The treatment for early (primary and secondary) syphilis in patients with no penicillin allergy is a single dose of penicillin G benzathine intramuscularly, at a dose of 2.4 million U. Alternative regimens include doxycycline 100 mg orally twice daily for 14 days, and azithromycin 2 g orally as a single dose; however, there is an association of treatment failure with azithromycin due to macrolide resistance.5 The patient in this case received empiric treatment targeting syphilis, gonorrhea, and chlamydia.
Conclusion
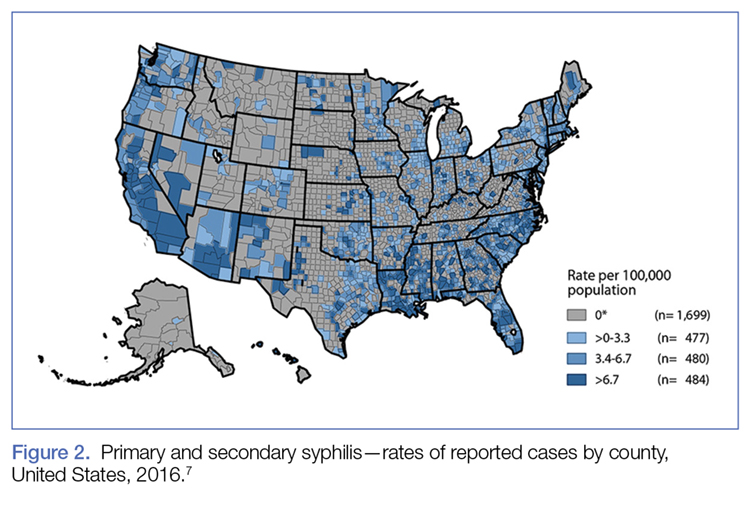
Ten years ago, the rates of primary and secondary syphilis were low, leading the infectious disease community to believe that preventive efforts had been effective. According to the CDC, however, “[current] rates…are the highest they have been in more than 20 years.”5Figure 2 demonstrates the geographic distribution of syphilis cases in the United States in 2016.7
Heightened concern has prompted the CDC to promote the theme “Syphilis Strikes Back” in April 2017, which was STI Awareness Month.8 Identification of disease is critical in the ED, especially when a previously common disease has become uncommon, like the resurgence of syphilis we are now seeing.
1. Clark EG, Danbolt N. The Oslo study of the natural course of untreated syphilis: An epidemiologic investigation based on a re-study of the Boeck-Bruusgaard material. Med Clin North Am. 1964;48:613.
2. Rockwell DH, Yobs AR, Moore MB Jr. The Tuskegee study of untreated syphilis; the 30th year of observation. Arch Intern Med. 1964;114:792-798.
3. Sparling PF, Swartz MN, Musher DM, Healy BP. Clinical manifestations of syphilis. In: Holmes KK, Sparling PF, Stamm WE, et al, eds. Sexually Transmitted Diseases. 4th ed. New York, NY: McGraw-Hill; 1999:661-684.
4. Birnbaumer DM. Sexually transmitted diseases. In: Marx JA, Hockberger RS, Walls RM, eds. Rosen’s Emergency Medicine: Concepts and Clinical Practice. Vol 2. 8th ed. Philadelphia, PA: Saunders; 2014:1312-1325.
5. Workowski KA, Bolan GA; Centers for Disease Control and Prevention. Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep. 2015;64(RR-03):1-137.
6. Larsen SA. Syphilis. Clin Lab Med. 1989;9(3):545-557.
7. Centers for Disease Control Prevention. Primary and secondary syphilis—rates of reported cases by county, United States, 2016. https://www.cdc.gov/std/stats16/figures/33.htm. Updated September 26, 2017. Accessed October 31 2017.]
8. Centers for Disease Control and Prevention. STD Awareness Month. Syphilis Strikes Back. https://www.cdc.gov/std/sam/index.htm?s_cid=tw_SAM_17001. Updated April 6, 2017. Accessed October 31, 2017.
The early initial ulcerative lesion (chancre) caused by Treponema pallidum infection, has a median incubation period of 21 days (primary syphilis). When untreated, secondary syphilis will develop within weeks to months and is characterized by generalized symptoms such as malaise, fevers, headaches, sore throat, and myalgia. However, the most characteristic finding in secondary syphilis remains a rash that is classically identified as symmetric, macular, or papular, and involving the entire trunk and extremities, including the palms and soles.
When secondary syphilis is left untreated, late syphilis or tertiary syphilis can develop, which is characterized by cardiovascular involvement, including aortitis, gummatous syphilis (granulomatous nodules in a variety of organs but typically the skin and bones), or central nervous system involvement.1-3 The following case describes a patient with nondescript symptoms, including malaise and cough, who had a characteristic rash of secondary syphilis that was diagnosed and treated in our Houston-area community hospital.
Case
In late autumn, a 30-year-old man presented to our community ED for evaluation of a cough productive of green sputum along with mild chest discomfort, malaise, and generalized myalgia, which were intermittent over the course of the past month. The patient denied rhinorrhea, fevers, chills, dyspnea, or any other systemic complaints. He also denied any sick contacts, but noted that his influenza vaccine was not up to date.
The patient denied any remote or recent medical or surgical history. He further denied taking any medications, and noted that his only medical allergy was to penicillin. His family history was noncontributory. Regarding his social history, the patient admitted to smoking one pack of cigarettes per day and to a daily alcohol intake of approximately one 6-pack of beer. He also admitted to frequently smoking crystal methamphetamine, which he stated he had last used 2 days prior to presentation. The patient said his current chest pain was similar to prior episodes, noting that when the pain occurred, he would temporarily stop smoking crystal methamphetamine.
Plain chest radiography, electrocardiogram, complete metabolic panel, complete blood count, B-natriuretic peptide, and troponin levels were all unremarkable. Due to the presence and nature of the patient’s rash, a rapid plasma reagin (RPR) screen was also taken, the results of which were reactive.
On further questioning, the patient admitted to having multiple female sexual partners with whom he used barrier protection sporadically. A more detailed physical examination revealed multiple painless ulcerations/chancres over the penile shaft and scrotum, without urethral drainage or inguinal lymphadenopathy. The patient denied dysuria or hematuria.
Since the patient was allergic to penicillin, he was given a single oral dose of azithromycin 2 g, and started on a 2-week course of oral doxycycline 100 mg. Further laboratory studies included gonorrhea and chlamydia cultures, both of which were negative. He was instructed to follow-up with his primary care physician for extended sexually transmitted infection (STI) panel-testing, including HIV, hepatitis, and confirmatory syphilis testing. Unfortunately, it is not known whether the patient complied with discharge instructions as he was lost to follow-up.
Discussion
Diagnostic algorithms for syphilis, one of the best studied STIs, have changed with technological advancement, but diagnosis and treatment for the most part has remained mostly the same. The uniqueness of this case is really focused around the patient’s chief complaint. While it is classic to present with malaise, headache, and rash, our patient complained of cough productive of sputum with chest pain—a rare presentation of secondary syphilis. The fortuitous key finding of the truncal rash directed the emergency physician toward the appropriate diagnosis.
Diagnosis
In the ED, where patients such as the one in our case are often lost to follow-up, and consistent infectious disease and primary care follow-up is unavailable, prompt treatment based on history and physical examination alone is recommended. Patients should be tested for syphilis, as well as other STIs including chlamydia, gonorrhea, hepatitis, and HIV as an outpatient. In addition, any partners with whom the patient has had sexual contact within the last 90 days should also undergo STI testing; sexual partners from over 90 days should be notified of the patient’s status and evaluated with testing as indicated.4 All positive test results should be reported to the Centers for Disease Control and Prevention (CDC).5
Nontreponemal and Treponemal Testing
For patients with clinical signs and symptoms of syphilis, recommended laboratory evaluation includes both nontreponemal and treponemal testing. Nontreponemal tests include RPR, venereal disease research laboratory test, and toluidine red unheated serum test. Treponemal tests include fluorescent treponemal antibody absorption, microhemagglutination test for antibodies to T pallidum, T pallidum particle agglutination assay, T pallidum enzyme immunoassay, and chemiluminescence immunoassay. Patients who test positive for treponemal antibody will typically remain reactive for life.5,6
In the setting of discordant test results, patients with a nonreactive treponemal result are generally considered to be negative for syphilis. Discordant results with a negative nontreponemal test are more complicated, and recommendations are based on symptomatology and repeat testing.5
Treatment
When a patient has a positive nontreponemal and treponemal test, treatment is usually indicated. As with the patient in this case, treatment is always indicated for patients who have no prior history of syphilis. For patients who have a history of treated syphilis, attention must be given to titer levels on previous testing and to patient symptomatology.
The treatment for early (primary and secondary) syphilis in patients with no penicillin allergy is a single dose of penicillin G benzathine intramuscularly, at a dose of 2.4 million U. Alternative regimens include doxycycline 100 mg orally twice daily for 14 days, and azithromycin 2 g orally as a single dose; however, there is an association of treatment failure with azithromycin due to macrolide resistance.5 The patient in this case received empiric treatment targeting syphilis, gonorrhea, and chlamydia.
Conclusion
Ten years ago, the rates of primary and secondary syphilis were low, leading the infectious disease community to believe that preventive efforts had been effective. According to the CDC, however, “[current] rates…are the highest they have been in more than 20 years.”5Figure 2 demonstrates the geographic distribution of syphilis cases in the United States in 2016.7
Heightened concern has prompted the CDC to promote the theme “Syphilis Strikes Back” in April 2017, which was STI Awareness Month.8 Identification of disease is critical in the ED, especially when a previously common disease has become uncommon, like the resurgence of syphilis we are now seeing.
The early initial ulcerative lesion (chancre) caused by Treponema pallidum infection, has a median incubation period of 21 days (primary syphilis). When untreated, secondary syphilis will develop within weeks to months and is characterized by generalized symptoms such as malaise, fevers, headaches, sore throat, and myalgia. However, the most characteristic finding in secondary syphilis remains a rash that is classically identified as symmetric, macular, or papular, and involving the entire trunk and extremities, including the palms and soles.
When secondary syphilis is left untreated, late syphilis or tertiary syphilis can develop, which is characterized by cardiovascular involvement, including aortitis, gummatous syphilis (granulomatous nodules in a variety of organs but typically the skin and bones), or central nervous system involvement.1-3 The following case describes a patient with nondescript symptoms, including malaise and cough, who had a characteristic rash of secondary syphilis that was diagnosed and treated in our Houston-area community hospital.
Case
In late autumn, a 30-year-old man presented to our community ED for evaluation of a cough productive of green sputum along with mild chest discomfort, malaise, and generalized myalgia, which were intermittent over the course of the past month. The patient denied rhinorrhea, fevers, chills, dyspnea, or any other systemic complaints. He also denied any sick contacts, but noted that his influenza vaccine was not up to date.
The patient denied any remote or recent medical or surgical history. He further denied taking any medications, and noted that his only medical allergy was to penicillin. His family history was noncontributory. Regarding his social history, the patient admitted to smoking one pack of cigarettes per day and to a daily alcohol intake of approximately one 6-pack of beer. He also admitted to frequently smoking crystal methamphetamine, which he stated he had last used 2 days prior to presentation. The patient said his current chest pain was similar to prior episodes, noting that when the pain occurred, he would temporarily stop smoking crystal methamphetamine.
Plain chest radiography, electrocardiogram, complete metabolic panel, complete blood count, B-natriuretic peptide, and troponin levels were all unremarkable. Due to the presence and nature of the patient’s rash, a rapid plasma reagin (RPR) screen was also taken, the results of which were reactive.
On further questioning, the patient admitted to having multiple female sexual partners with whom he used barrier protection sporadically. A more detailed physical examination revealed multiple painless ulcerations/chancres over the penile shaft and scrotum, without urethral drainage or inguinal lymphadenopathy. The patient denied dysuria or hematuria.
Since the patient was allergic to penicillin, he was given a single oral dose of azithromycin 2 g, and started on a 2-week course of oral doxycycline 100 mg. Further laboratory studies included gonorrhea and chlamydia cultures, both of which were negative. He was instructed to follow-up with his primary care physician for extended sexually transmitted infection (STI) panel-testing, including HIV, hepatitis, and confirmatory syphilis testing. Unfortunately, it is not known whether the patient complied with discharge instructions as he was lost to follow-up.
Discussion
Diagnostic algorithms for syphilis, one of the best studied STIs, have changed with technological advancement, but diagnosis and treatment for the most part has remained mostly the same. The uniqueness of this case is really focused around the patient’s chief complaint. While it is classic to present with malaise, headache, and rash, our patient complained of cough productive of sputum with chest pain—a rare presentation of secondary syphilis. The fortuitous key finding of the truncal rash directed the emergency physician toward the appropriate diagnosis.
Diagnosis
In the ED, where patients such as the one in our case are often lost to follow-up, and consistent infectious disease and primary care follow-up is unavailable, prompt treatment based on history and physical examination alone is recommended. Patients should be tested for syphilis, as well as other STIs including chlamydia, gonorrhea, hepatitis, and HIV as an outpatient. In addition, any partners with whom the patient has had sexual contact within the last 90 days should also undergo STI testing; sexual partners from over 90 days should be notified of the patient’s status and evaluated with testing as indicated.4 All positive test results should be reported to the Centers for Disease Control and Prevention (CDC).5
Nontreponemal and Treponemal Testing
For patients with clinical signs and symptoms of syphilis, recommended laboratory evaluation includes both nontreponemal and treponemal testing. Nontreponemal tests include RPR, venereal disease research laboratory test, and toluidine red unheated serum test. Treponemal tests include fluorescent treponemal antibody absorption, microhemagglutination test for antibodies to T pallidum, T pallidum particle agglutination assay, T pallidum enzyme immunoassay, and chemiluminescence immunoassay. Patients who test positive for treponemal antibody will typically remain reactive for life.5,6
In the setting of discordant test results, patients with a nonreactive treponemal result are generally considered to be negative for syphilis. Discordant results with a negative nontreponemal test are more complicated, and recommendations are based on symptomatology and repeat testing.5
Treatment
When a patient has a positive nontreponemal and treponemal test, treatment is usually indicated. As with the patient in this case, treatment is always indicated for patients who have no prior history of syphilis. For patients who have a history of treated syphilis, attention must be given to titer levels on previous testing and to patient symptomatology.
The treatment for early (primary and secondary) syphilis in patients with no penicillin allergy is a single dose of penicillin G benzathine intramuscularly, at a dose of 2.4 million U. Alternative regimens include doxycycline 100 mg orally twice daily for 14 days, and azithromycin 2 g orally as a single dose; however, there is an association of treatment failure with azithromycin due to macrolide resistance.5 The patient in this case received empiric treatment targeting syphilis, gonorrhea, and chlamydia.
Conclusion
Ten years ago, the rates of primary and secondary syphilis were low, leading the infectious disease community to believe that preventive efforts had been effective. According to the CDC, however, “[current] rates…are the highest they have been in more than 20 years.”5Figure 2 demonstrates the geographic distribution of syphilis cases in the United States in 2016.7
Heightened concern has prompted the CDC to promote the theme “Syphilis Strikes Back” in April 2017, which was STI Awareness Month.8 Identification of disease is critical in the ED, especially when a previously common disease has become uncommon, like the resurgence of syphilis we are now seeing.
1. Clark EG, Danbolt N. The Oslo study of the natural course of untreated syphilis: An epidemiologic investigation based on a re-study of the Boeck-Bruusgaard material. Med Clin North Am. 1964;48:613.
2. Rockwell DH, Yobs AR, Moore MB Jr. The Tuskegee study of untreated syphilis; the 30th year of observation. Arch Intern Med. 1964;114:792-798.
3. Sparling PF, Swartz MN, Musher DM, Healy BP. Clinical manifestations of syphilis. In: Holmes KK, Sparling PF, Stamm WE, et al, eds. Sexually Transmitted Diseases. 4th ed. New York, NY: McGraw-Hill; 1999:661-684.
4. Birnbaumer DM. Sexually transmitted diseases. In: Marx JA, Hockberger RS, Walls RM, eds. Rosen’s Emergency Medicine: Concepts and Clinical Practice. Vol 2. 8th ed. Philadelphia, PA: Saunders; 2014:1312-1325.
5. Workowski KA, Bolan GA; Centers for Disease Control and Prevention. Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep. 2015;64(RR-03):1-137.
6. Larsen SA. Syphilis. Clin Lab Med. 1989;9(3):545-557.
7. Centers for Disease Control Prevention. Primary and secondary syphilis—rates of reported cases by county, United States, 2016. https://www.cdc.gov/std/stats16/figures/33.htm. Updated September 26, 2017. Accessed October 31 2017.]
8. Centers for Disease Control and Prevention. STD Awareness Month. Syphilis Strikes Back. https://www.cdc.gov/std/sam/index.htm?s_cid=tw_SAM_17001. Updated April 6, 2017. Accessed October 31, 2017.
1. Clark EG, Danbolt N. The Oslo study of the natural course of untreated syphilis: An epidemiologic investigation based on a re-study of the Boeck-Bruusgaard material. Med Clin North Am. 1964;48:613.
2. Rockwell DH, Yobs AR, Moore MB Jr. The Tuskegee study of untreated syphilis; the 30th year of observation. Arch Intern Med. 1964;114:792-798.
3. Sparling PF, Swartz MN, Musher DM, Healy BP. Clinical manifestations of syphilis. In: Holmes KK, Sparling PF, Stamm WE, et al, eds. Sexually Transmitted Diseases. 4th ed. New York, NY: McGraw-Hill; 1999:661-684.
4. Birnbaumer DM. Sexually transmitted diseases. In: Marx JA, Hockberger RS, Walls RM, eds. Rosen’s Emergency Medicine: Concepts and Clinical Practice. Vol 2. 8th ed. Philadelphia, PA: Saunders; 2014:1312-1325.
5. Workowski KA, Bolan GA; Centers for Disease Control and Prevention. Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep. 2015;64(RR-03):1-137.
6. Larsen SA. Syphilis. Clin Lab Med. 1989;9(3):545-557.
7. Centers for Disease Control Prevention. Primary and secondary syphilis—rates of reported cases by county, United States, 2016. https://www.cdc.gov/std/stats16/figures/33.htm. Updated September 26, 2017. Accessed October 31 2017.]
8. Centers for Disease Control and Prevention. STD Awareness Month. Syphilis Strikes Back. https://www.cdc.gov/std/sam/index.htm?s_cid=tw_SAM_17001. Updated April 6, 2017. Accessed October 31, 2017.
Home noninvasive ventilation reduces COPD readmissions
Clinical question: Is there a benefit to home noninvasive ventilation (NIV) following a hospital admission for chronic obstructive pulmonary disease (COPD) exacerbation?
Background: Preventing hospital readmission following a COPD exacerbation is a priority; however, the role of NIV in this situation remains uncertain.

Setting: 13 medical centers in the United Kingdom.
Synopsis: Investigators randomized 116 patients with COPD and persistent hypercapnia (paCO2 less than 53) 2-4 weeks following a COPD exacerbation to either home oxygen therapy with NIV or to home oxygen therapy alone. The study’s primary endpoint was a composite of time to readmission or death within 12 months. They found that the median time to this endpoint was significantly longer in the intervention group (1.4 vs. 4.3 months; 95% CI, 0.31-0.77; P = .002) and that the absolute risk reduction was 17.0% (80.4% vs. 63.4%; 95% CI, 0.1%-34.0%). The differences were driven by readmissions, as the mortality rate did not differ significantly between groups, although the study was not powered to evaluate this. Of note, the median NIV settings were 24/4, which constitutes a “high-pressure strategy” which may account for the benefits seen in this study that have been absent in some other trials.
Bottom line: NIV reduced readmissions in patients with COPD and persistent hypercapnia several weeks following an acute exacerbation.
Citation: Murphy PB, Rehal S, Arbane G, et al. Effect of home noninvasive ventilation with oxygen therapy vs. oxygen therapy alone on hospital readmission or death after an acute COPD exacerbation, a randomized clinical trial. JAMA. 2017;317(21):2177-86.
Dr. Herscher is assistant professor, division of hospital medicine, Icahn School of Medicine of the Mount Sinai Health System.
Clinical question: Is there a benefit to home noninvasive ventilation (NIV) following a hospital admission for chronic obstructive pulmonary disease (COPD) exacerbation?
Background: Preventing hospital readmission following a COPD exacerbation is a priority; however, the role of NIV in this situation remains uncertain.
Setting: 13 medical centers in the United Kingdom.
Synopsis: Investigators randomized 116 patients with COPD and persistent hypercapnia (paCO2 less than 53) 2-4 weeks following a COPD exacerbation to either home oxygen therapy with NIV or to home oxygen therapy alone. The study’s primary endpoint was a composite of time to readmission or death within 12 months. They found that the median time to this endpoint was significantly longer in the intervention group (1.4 vs. 4.3 months; 95% CI, 0.31-0.77; P = .002) and that the absolute risk reduction was 17.0% (80.4% vs. 63.4%; 95% CI, 0.1%-34.0%). The differences were driven by readmissions, as the mortality rate did not differ significantly between groups, although the study was not powered to evaluate this. Of note, the median NIV settings were 24/4, which constitutes a “high-pressure strategy” which may account for the benefits seen in this study that have been absent in some other trials.
Bottom line: NIV reduced readmissions in patients with COPD and persistent hypercapnia several weeks following an acute exacerbation.
Citation: Murphy PB, Rehal S, Arbane G, et al. Effect of home noninvasive ventilation with oxygen therapy vs. oxygen therapy alone on hospital readmission or death after an acute COPD exacerbation, a randomized clinical trial. JAMA. 2017;317(21):2177-86.
Dr. Herscher is assistant professor, division of hospital medicine, Icahn School of Medicine of the Mount Sinai Health System.
Clinical question: Is there a benefit to home noninvasive ventilation (NIV) following a hospital admission for chronic obstructive pulmonary disease (COPD) exacerbation?
Background: Preventing hospital readmission following a COPD exacerbation is a priority; however, the role of NIV in this situation remains uncertain.
Setting: 13 medical centers in the United Kingdom.
Synopsis: Investigators randomized 116 patients with COPD and persistent hypercapnia (paCO2 less than 53) 2-4 weeks following a COPD exacerbation to either home oxygen therapy with NIV or to home oxygen therapy alone. The study’s primary endpoint was a composite of time to readmission or death within 12 months. They found that the median time to this endpoint was significantly longer in the intervention group (1.4 vs. 4.3 months; 95% CI, 0.31-0.77; P = .002) and that the absolute risk reduction was 17.0% (80.4% vs. 63.4%; 95% CI, 0.1%-34.0%). The differences were driven by readmissions, as the mortality rate did not differ significantly between groups, although the study was not powered to evaluate this. Of note, the median NIV settings were 24/4, which constitutes a “high-pressure strategy” which may account for the benefits seen in this study that have been absent in some other trials.
Bottom line: NIV reduced readmissions in patients with COPD and persistent hypercapnia several weeks following an acute exacerbation.
Citation: Murphy PB, Rehal S, Arbane G, et al. Effect of home noninvasive ventilation with oxygen therapy vs. oxygen therapy alone on hospital readmission or death after an acute COPD exacerbation, a randomized clinical trial. JAMA. 2017;317(21):2177-86.
Dr. Herscher is assistant professor, division of hospital medicine, Icahn School of Medicine of the Mount Sinai Health System.
Duodenal Perforation After Endoscopic Procedure
Tension pneumoperitoneum (TPP), also known as hyperacute abdominal compartment syndrome or abdominal tamponade, is a rare condition most commonly associated with gastrointestinal (GI) perforation during endoscopy and iatrogenic insufflation of gas into the peritoneal cavity.1 Other reported causes of TPP include gastric rupture after cardiopulmonary resuscitation, barotrauma during scuba diving, positive pressure ventilation through pleural-peritoneal channels, and spontaneous TPP of uncertain mechanism.1-4
Case Presentation
A 76-year-old male with a history of ischemic cardiomyopathy, hypertension, and diabetes mellitus presented to the VA Puget Sound Health Care System in Seattle, Washington emergency department with painless jaundice, hematemesis, melena, and acute renal failure. On esophagogastroduodenoscopy (EGD), he was found to have an ulcer on the posterior wall of the duodenal bulb. The ulcer was coagulated and injected with epinephrine. The patient’s subsequent hospital course was complicated by worsening liver function, the need for renal replacement therapy, and recurrence of upper GI bleeding that required a transcatheter embolization of 2 separate superior pancreaticoduodenal arteries (SPDA) and the inferior pancreaticoduodenal artery (IPDA).
Once clinically stable, an endoscopic retrograde cholangiopancreatography (ERCP) was performed to evaluate for cholangiocarcinoma. A stricture was discovered in the common hepatic duct, brushings were taken, and a 15 cm, 7 Fr stent was placed in the common hepatic duct. The procedure was performed with an Olympus TJF Type Q180V duodenovideoscope (Tokyo, Japan) with an external diameter of 13.7 mm. The patient became hypotensive during the procedure and was treated with phenylephrine and ephedrine boluses. There was no endoscopic evidence of bleeding or bowel trauma.
After completion of the procedure, in the recovery area the patient became severely hypotensive and unresponsive. The physical examination was noteworthy for gross abdominal distention. Arterial blood gas analysis revealed severe metabolic and respiratory acidosis. Chest radiography demonstrated massive pneumoperitoneum, low lung volumes, and diaphragmatic compression (Figure). 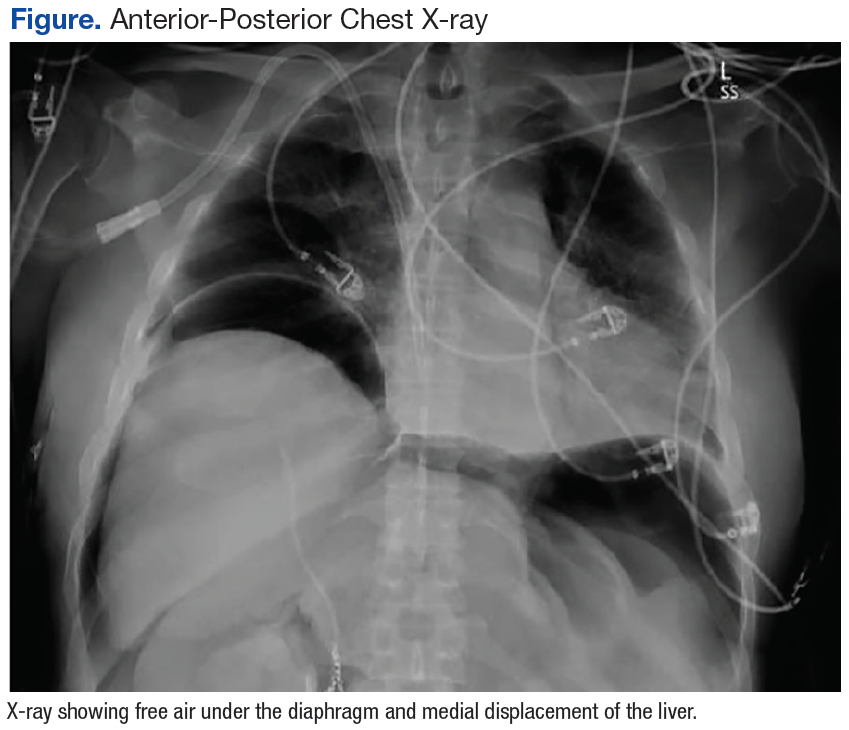
A diagnosis of tension pneumoperitoneum was made, and as the patient was transported to the operating room he became bradycardic without a pulse, requiring initiation of cardiopulmonary resuscitation. The abdomen was decompressed with a 14-gauge needle, followed by insertion of a laparoscopic trocar as a decompressive maneuver. This procedure resulted in return of spontaneous circulation.
An exploratory laparotomy was performed, and a massive rush of air was noted on opening the peritoneum. A pinhole perforation of the anterior wall of the second portion of the duodenum was found along with large-volume bilious ascites. This perforation was repaired with a Graham patch, and the patient was taken to the intensive care unit. Postoperatively, the patient developed disseminated intravascular coagulation, shock liver, and acute respiratory distress syndrome, expiring 10 days later from sequelae of multiorgan failure.
Discussion
In relation to upper GI endoscopic procedures, TPP has been reported after diagnostic EGD, endoscopic sphincterotomy, and submucosal tumor dissection.5-7 During these interventions, clinically apparent or overt iatrogenic perforations can occur either in the stomach or duodenum. These perforations may function as one-way valves that cause massive air accumulation and marked elevation of the diaphragm, which severely decreases lung volumes, pulmonary compliance, and limits gas exchange. Hemodynamically, compression of the inferior vena cava restricts venous return to the heart, resulting in decreased cardiac output.8
Patients with TPP present in acute distress with dyspnea, abdominal pain, and shock. On physical examination the abdomen is markedly distended, tympanic, and rigid. Rectal prolapse and subcutaneous emphysema also may be present.9 Roentographic features of TPP include findings of intraperitoneal air with elevation of the diaphragm, medial displacement of the liver (saddlebag sign), and juxtaposition of air in visceral interfaces, making intra-abdominal structures (spleen and gallbladder) appear more discrete.10 Abdominal computer tomography may show massive pneumoperitoneum with bowel loop compression and centralization of abdominal organs.4
Treatment strategies include emergent decompression either with percutaneous catheter insertion or abdominal drain placement followed by a definitive surgical repair. As with management of tension pneumothorax, treatment should not be delayed while awaiting confirmatory radiologic studies.9 When percutaneous needle decompression is undertaken, it is preferable to use a large bore (14-gauge venous catheter) and to advance a catheter over a needle to minimize the risk of visceral injury with egress of air and return of abdominal organs to their normal anatomical positions. The needle should be inserted directly above or below the umbilicus or in the left or right lower quadrants to avoid solid organ (ie, liver or spleen) injury.
Etiologic possibilities for the duodenal perforation in this case include mechanical trauma from the endoscope and duodenal tissue infarction after embolization of a bleeding duodenal ulcer. The duodenum and pancreatic head have a dual blood supply from the SPDA, a branch of the gastroduodenal artery, and the IPDA, a branch of the superior mesenteric artery.11 After failed endoscopic management of persistent duodenal hemorrhage, the patient underwent synchronous embolization of 2 separate SPDAs and the IPDA. This might have put the first 2 segments of the duodenum at risk for ischemic damage and caused it to perforate at some point during the patient’s hospitalization (as evidenced by the bilious ascitis) or rendered them vulnerable to perforation from intraluminal insufflation during endoscopy.12
During the laparotomy, a pinhole-sized perforation was noted in the anterior wall of the second part of the duodenum, distinct form the duodenal ulcer present on the posterior wall. This perforation likely provided a pathway for the intraluminal gas to escape into the peritoneal cavity, culminating in abdominal tamponade, cardiopulmonary deterioration, and arrest. Needle decompression of the abdominal cavity provided an effective, though temporizing relief of this pressure, enabling return of spontaneous circulation.
This case highlights the need for a high index of suspicion for TPP in a patient with cardiopulmonary compromise and abdominal distension after upper GI endoscopic procedures even in the absence of identifiable perforations. Close coordination among gastroenterologists, anesthesiologists, and surgeons is key in prevention, recognition, and management of this rare but catastrophic complication.
1. Bunni J, Bryson PJ, Higgs SM. Abdominal compartment syndrome caused by tension pneumoperitoneum in a scuba diver. Ann R Coll Surg Engl. 2012;94(8):e237-e239.
2. Cameron PA, Rosengarten PL, Johnson WR, Dziukas L. Tension pneumoperitoneum after cardiopulmonary resuscitation. Med J Aust. 1991;155(1):44-47.
3. Burdett-Smith P, Jaffey L. Tension pneumoperitoneum. J Accid Emerg Med. 1996;13(3):220-221.
4. Joshi D, Ganai B. Radiological features of tension pneumoperitoneum. BMJ Case Rep. 2015;2015.
5. Rai A, Iftikhar S. Tension pneumothorax complicating diagnostic upper endoscopy: a case report. Am J Gastroenterol. 1999;94(3):845-847.
6. Iyilikci L, Akarsu M, Duran E, et al. Duodenal perforation and bilateral tension pneumothorax following endoscopic sphincterotomy. J Anesth. 2009;23(1):164-165.
7. Siboni S, Bona D, Abate E, Bonavina L. Tension pneumoperitoneum following endoscopic submucosal dissection of leiomyoma of the cardia. Endoscopy. 2010;42(suppl 2):E152.
8. Deenichin GP. Abdominal compartment syndrome. Surg Today. 2008;38(1):5-19.
9. Chiapponi C, Stocker U, Korner M, et al. Emergency percutaneous needle decompression for tension pneumoperitoneum. BMC Gastroenterol. 2011;11:48.
10. Lin BW, Thanassi W. Tension pneumoperitoneum. J Emerg Med. 2010;38(1):57-59.
11. Bell SD, Lau KY, Sniderman KW. Synchronous embolization of the gastroduodenal artery and the inferior pancreaticoduodenal artery in patients with massive duodenal hemorrhage. J Vasc Interv Radiol. 1995;6(4):531-536.
12. Wang YL, Cheng YS, Liu LZ, He ZH, Ding KH. Emergency transcatheter arterial embolization for patients with acute massive duodenal ulcer hemorrhage. World J Gastroenterol. 2012;18(34):4765-4770.
Tension pneumoperitoneum (TPP), also known as hyperacute abdominal compartment syndrome or abdominal tamponade, is a rare condition most commonly associated with gastrointestinal (GI) perforation during endoscopy and iatrogenic insufflation of gas into the peritoneal cavity.1 Other reported causes of TPP include gastric rupture after cardiopulmonary resuscitation, barotrauma during scuba diving, positive pressure ventilation through pleural-peritoneal channels, and spontaneous TPP of uncertain mechanism.1-4
Case Presentation
A 76-year-old male with a history of ischemic cardiomyopathy, hypertension, and diabetes mellitus presented to the VA Puget Sound Health Care System in Seattle, Washington emergency department with painless jaundice, hematemesis, melena, and acute renal failure. On esophagogastroduodenoscopy (EGD), he was found to have an ulcer on the posterior wall of the duodenal bulb. The ulcer was coagulated and injected with epinephrine. The patient’s subsequent hospital course was complicated by worsening liver function, the need for renal replacement therapy, and recurrence of upper GI bleeding that required a transcatheter embolization of 2 separate superior pancreaticoduodenal arteries (SPDA) and the inferior pancreaticoduodenal artery (IPDA).
Once clinically stable, an endoscopic retrograde cholangiopancreatography (ERCP) was performed to evaluate for cholangiocarcinoma. A stricture was discovered in the common hepatic duct, brushings were taken, and a 15 cm, 7 Fr stent was placed in the common hepatic duct. The procedure was performed with an Olympus TJF Type Q180V duodenovideoscope (Tokyo, Japan) with an external diameter of 13.7 mm. The patient became hypotensive during the procedure and was treated with phenylephrine and ephedrine boluses. There was no endoscopic evidence of bleeding or bowel trauma.
After completion of the procedure, in the recovery area the patient became severely hypotensive and unresponsive. The physical examination was noteworthy for gross abdominal distention. Arterial blood gas analysis revealed severe metabolic and respiratory acidosis. Chest radiography demonstrated massive pneumoperitoneum, low lung volumes, and diaphragmatic compression (Figure).
A diagnosis of tension pneumoperitoneum was made, and as the patient was transported to the operating room he became bradycardic without a pulse, requiring initiation of cardiopulmonary resuscitation. The abdomen was decompressed with a 14-gauge needle, followed by insertion of a laparoscopic trocar as a decompressive maneuver. This procedure resulted in return of spontaneous circulation.
An exploratory laparotomy was performed, and a massive rush of air was noted on opening the peritoneum. A pinhole perforation of the anterior wall of the second portion of the duodenum was found along with large-volume bilious ascites. This perforation was repaired with a Graham patch, and the patient was taken to the intensive care unit. Postoperatively, the patient developed disseminated intravascular coagulation, shock liver, and acute respiratory distress syndrome, expiring 10 days later from sequelae of multiorgan failure.
Discussion
In relation to upper GI endoscopic procedures, TPP has been reported after diagnostic EGD, endoscopic sphincterotomy, and submucosal tumor dissection.5-7 During these interventions, clinically apparent or overt iatrogenic perforations can occur either in the stomach or duodenum. These perforations may function as one-way valves that cause massive air accumulation and marked elevation of the diaphragm, which severely decreases lung volumes, pulmonary compliance, and limits gas exchange. Hemodynamically, compression of the inferior vena cava restricts venous return to the heart, resulting in decreased cardiac output.8
Patients with TPP present in acute distress with dyspnea, abdominal pain, and shock. On physical examination the abdomen is markedly distended, tympanic, and rigid. Rectal prolapse and subcutaneous emphysema also may be present.9 Roentographic features of TPP include findings of intraperitoneal air with elevation of the diaphragm, medial displacement of the liver (saddlebag sign), and juxtaposition of air in visceral interfaces, making intra-abdominal structures (spleen and gallbladder) appear more discrete.10 Abdominal computer tomography may show massive pneumoperitoneum with bowel loop compression and centralization of abdominal organs.4
Treatment strategies include emergent decompression either with percutaneous catheter insertion or abdominal drain placement followed by a definitive surgical repair. As with management of tension pneumothorax, treatment should not be delayed while awaiting confirmatory radiologic studies.9 When percutaneous needle decompression is undertaken, it is preferable to use a large bore (14-gauge venous catheter) and to advance a catheter over a needle to minimize the risk of visceral injury with egress of air and return of abdominal organs to their normal anatomical positions. The needle should be inserted directly above or below the umbilicus or in the left or right lower quadrants to avoid solid organ (ie, liver or spleen) injury.
Etiologic possibilities for the duodenal perforation in this case include mechanical trauma from the endoscope and duodenal tissue infarction after embolization of a bleeding duodenal ulcer. The duodenum and pancreatic head have a dual blood supply from the SPDA, a branch of the gastroduodenal artery, and the IPDA, a branch of the superior mesenteric artery.11 After failed endoscopic management of persistent duodenal hemorrhage, the patient underwent synchronous embolization of 2 separate SPDAs and the IPDA. This might have put the first 2 segments of the duodenum at risk for ischemic damage and caused it to perforate at some point during the patient’s hospitalization (as evidenced by the bilious ascitis) or rendered them vulnerable to perforation from intraluminal insufflation during endoscopy.12
During the laparotomy, a pinhole-sized perforation was noted in the anterior wall of the second part of the duodenum, distinct form the duodenal ulcer present on the posterior wall. This perforation likely provided a pathway for the intraluminal gas to escape into the peritoneal cavity, culminating in abdominal tamponade, cardiopulmonary deterioration, and arrest. Needle decompression of the abdominal cavity provided an effective, though temporizing relief of this pressure, enabling return of spontaneous circulation.
This case highlights the need for a high index of suspicion for TPP in a patient with cardiopulmonary compromise and abdominal distension after upper GI endoscopic procedures even in the absence of identifiable perforations. Close coordination among gastroenterologists, anesthesiologists, and surgeons is key in prevention, recognition, and management of this rare but catastrophic complication.
Tension pneumoperitoneum (TPP), also known as hyperacute abdominal compartment syndrome or abdominal tamponade, is a rare condition most commonly associated with gastrointestinal (GI) perforation during endoscopy and iatrogenic insufflation of gas into the peritoneal cavity.1 Other reported causes of TPP include gastric rupture after cardiopulmonary resuscitation, barotrauma during scuba diving, positive pressure ventilation through pleural-peritoneal channels, and spontaneous TPP of uncertain mechanism.1-4
Case Presentation
A 76-year-old male with a history of ischemic cardiomyopathy, hypertension, and diabetes mellitus presented to the VA Puget Sound Health Care System in Seattle, Washington emergency department with painless jaundice, hematemesis, melena, and acute renal failure. On esophagogastroduodenoscopy (EGD), he was found to have an ulcer on the posterior wall of the duodenal bulb. The ulcer was coagulated and injected with epinephrine. The patient’s subsequent hospital course was complicated by worsening liver function, the need for renal replacement therapy, and recurrence of upper GI bleeding that required a transcatheter embolization of 2 separate superior pancreaticoduodenal arteries (SPDA) and the inferior pancreaticoduodenal artery (IPDA).
Once clinically stable, an endoscopic retrograde cholangiopancreatography (ERCP) was performed to evaluate for cholangiocarcinoma. A stricture was discovered in the common hepatic duct, brushings were taken, and a 15 cm, 7 Fr stent was placed in the common hepatic duct. The procedure was performed with an Olympus TJF Type Q180V duodenovideoscope (Tokyo, Japan) with an external diameter of 13.7 mm. The patient became hypotensive during the procedure and was treated with phenylephrine and ephedrine boluses. There was no endoscopic evidence of bleeding or bowel trauma.
After completion of the procedure, in the recovery area the patient became severely hypotensive and unresponsive. The physical examination was noteworthy for gross abdominal distention. Arterial blood gas analysis revealed severe metabolic and respiratory acidosis. Chest radiography demonstrated massive pneumoperitoneum, low lung volumes, and diaphragmatic compression (Figure).
A diagnosis of tension pneumoperitoneum was made, and as the patient was transported to the operating room he became bradycardic without a pulse, requiring initiation of cardiopulmonary resuscitation. The abdomen was decompressed with a 14-gauge needle, followed by insertion of a laparoscopic trocar as a decompressive maneuver. This procedure resulted in return of spontaneous circulation.
An exploratory laparotomy was performed, and a massive rush of air was noted on opening the peritoneum. A pinhole perforation of the anterior wall of the second portion of the duodenum was found along with large-volume bilious ascites. This perforation was repaired with a Graham patch, and the patient was taken to the intensive care unit. Postoperatively, the patient developed disseminated intravascular coagulation, shock liver, and acute respiratory distress syndrome, expiring 10 days later from sequelae of multiorgan failure.
Discussion
In relation to upper GI endoscopic procedures, TPP has been reported after diagnostic EGD, endoscopic sphincterotomy, and submucosal tumor dissection.5-7 During these interventions, clinically apparent or overt iatrogenic perforations can occur either in the stomach or duodenum. These perforations may function as one-way valves that cause massive air accumulation and marked elevation of the diaphragm, which severely decreases lung volumes, pulmonary compliance, and limits gas exchange. Hemodynamically, compression of the inferior vena cava restricts venous return to the heart, resulting in decreased cardiac output.8
Patients with TPP present in acute distress with dyspnea, abdominal pain, and shock. On physical examination the abdomen is markedly distended, tympanic, and rigid. Rectal prolapse and subcutaneous emphysema also may be present.9 Roentographic features of TPP include findings of intraperitoneal air with elevation of the diaphragm, medial displacement of the liver (saddlebag sign), and juxtaposition of air in visceral interfaces, making intra-abdominal structures (spleen and gallbladder) appear more discrete.10 Abdominal computer tomography may show massive pneumoperitoneum with bowel loop compression and centralization of abdominal organs.4
Treatment strategies include emergent decompression either with percutaneous catheter insertion or abdominal drain placement followed by a definitive surgical repair. As with management of tension pneumothorax, treatment should not be delayed while awaiting confirmatory radiologic studies.9 When percutaneous needle decompression is undertaken, it is preferable to use a large bore (14-gauge venous catheter) and to advance a catheter over a needle to minimize the risk of visceral injury with egress of air and return of abdominal organs to their normal anatomical positions. The needle should be inserted directly above or below the umbilicus or in the left or right lower quadrants to avoid solid organ (ie, liver or spleen) injury.
Etiologic possibilities for the duodenal perforation in this case include mechanical trauma from the endoscope and duodenal tissue infarction after embolization of a bleeding duodenal ulcer. The duodenum and pancreatic head have a dual blood supply from the SPDA, a branch of the gastroduodenal artery, and the IPDA, a branch of the superior mesenteric artery.11 After failed endoscopic management of persistent duodenal hemorrhage, the patient underwent synchronous embolization of 2 separate SPDAs and the IPDA. This might have put the first 2 segments of the duodenum at risk for ischemic damage and caused it to perforate at some point during the patient’s hospitalization (as evidenced by the bilious ascitis) or rendered them vulnerable to perforation from intraluminal insufflation during endoscopy.12
During the laparotomy, a pinhole-sized perforation was noted in the anterior wall of the second part of the duodenum, distinct form the duodenal ulcer present on the posterior wall. This perforation likely provided a pathway for the intraluminal gas to escape into the peritoneal cavity, culminating in abdominal tamponade, cardiopulmonary deterioration, and arrest. Needle decompression of the abdominal cavity provided an effective, though temporizing relief of this pressure, enabling return of spontaneous circulation.
This case highlights the need for a high index of suspicion for TPP in a patient with cardiopulmonary compromise and abdominal distension after upper GI endoscopic procedures even in the absence of identifiable perforations. Close coordination among gastroenterologists, anesthesiologists, and surgeons is key in prevention, recognition, and management of this rare but catastrophic complication.
1. Bunni J, Bryson PJ, Higgs SM. Abdominal compartment syndrome caused by tension pneumoperitoneum in a scuba diver. Ann R Coll Surg Engl. 2012;94(8):e237-e239.
2. Cameron PA, Rosengarten PL, Johnson WR, Dziukas L. Tension pneumoperitoneum after cardiopulmonary resuscitation. Med J Aust. 1991;155(1):44-47.
3. Burdett-Smith P, Jaffey L. Tension pneumoperitoneum. J Accid Emerg Med. 1996;13(3):220-221.
4. Joshi D, Ganai B. Radiological features of tension pneumoperitoneum. BMJ Case Rep. 2015;2015.
5. Rai A, Iftikhar S. Tension pneumothorax complicating diagnostic upper endoscopy: a case report. Am J Gastroenterol. 1999;94(3):845-847.
6. Iyilikci L, Akarsu M, Duran E, et al. Duodenal perforation and bilateral tension pneumothorax following endoscopic sphincterotomy. J Anesth. 2009;23(1):164-165.
7. Siboni S, Bona D, Abate E, Bonavina L. Tension pneumoperitoneum following endoscopic submucosal dissection of leiomyoma of the cardia. Endoscopy. 2010;42(suppl 2):E152.
8. Deenichin GP. Abdominal compartment syndrome. Surg Today. 2008;38(1):5-19.
9. Chiapponi C, Stocker U, Korner M, et al. Emergency percutaneous needle decompression for tension pneumoperitoneum. BMC Gastroenterol. 2011;11:48.
10. Lin BW, Thanassi W. Tension pneumoperitoneum. J Emerg Med. 2010;38(1):57-59.
11. Bell SD, Lau KY, Sniderman KW. Synchronous embolization of the gastroduodenal artery and the inferior pancreaticoduodenal artery in patients with massive duodenal hemorrhage. J Vasc Interv Radiol. 1995;6(4):531-536.
12. Wang YL, Cheng YS, Liu LZ, He ZH, Ding KH. Emergency transcatheter arterial embolization for patients with acute massive duodenal ulcer hemorrhage. World J Gastroenterol. 2012;18(34):4765-4770.
1. Bunni J, Bryson PJ, Higgs SM. Abdominal compartment syndrome caused by tension pneumoperitoneum in a scuba diver. Ann R Coll Surg Engl. 2012;94(8):e237-e239.
2. Cameron PA, Rosengarten PL, Johnson WR, Dziukas L. Tension pneumoperitoneum after cardiopulmonary resuscitation. Med J Aust. 1991;155(1):44-47.
3. Burdett-Smith P, Jaffey L. Tension pneumoperitoneum. J Accid Emerg Med. 1996;13(3):220-221.
4. Joshi D, Ganai B. Radiological features of tension pneumoperitoneum. BMJ Case Rep. 2015;2015.
5. Rai A, Iftikhar S. Tension pneumothorax complicating diagnostic upper endoscopy: a case report. Am J Gastroenterol. 1999;94(3):845-847.
6. Iyilikci L, Akarsu M, Duran E, et al. Duodenal perforation and bilateral tension pneumothorax following endoscopic sphincterotomy. J Anesth. 2009;23(1):164-165.
7. Siboni S, Bona D, Abate E, Bonavina L. Tension pneumoperitoneum following endoscopic submucosal dissection of leiomyoma of the cardia. Endoscopy. 2010;42(suppl 2):E152.
8. Deenichin GP. Abdominal compartment syndrome. Surg Today. 2008;38(1):5-19.
9. Chiapponi C, Stocker U, Korner M, et al. Emergency percutaneous needle decompression for tension pneumoperitoneum. BMC Gastroenterol. 2011;11:48.
10. Lin BW, Thanassi W. Tension pneumoperitoneum. J Emerg Med. 2010;38(1):57-59.
11. Bell SD, Lau KY, Sniderman KW. Synchronous embolization of the gastroduodenal artery and the inferior pancreaticoduodenal artery in patients with massive duodenal hemorrhage. J Vasc Interv Radiol. 1995;6(4):531-536.
12. Wang YL, Cheng YS, Liu LZ, He ZH, Ding KH. Emergency transcatheter arterial embolization for patients with acute massive duodenal ulcer hemorrhage. World J Gastroenterol. 2012;18(34):4765-4770.
FDA approves letermovir as CMV prophylaxis

The US Food and Drug Administration (FDA) has approved the oral and intravenous formulations of letermovir (PREVYMIS™).
Letermovir is a member of a new class of non-nucleoside CMV inhibitors known as 3,4 dihydro-quinazolines.
The FDA approved letermovir as prophylaxis for cytomegalovirus (CMV) infection and disease in adult recipients of allogeneic hematopoietic stem cell transplants (HSCTs) who are CMV-seropositive.
“PREVYMIS is the first new medicine for CMV infection approved in the US in 15 years,” said Roy Baynes, senior vice president, head of clinical development, and chief medical officer of Merck Research Laboratories, the company marketing letermovir.
Letermovir is expected to be available in December. The list price (wholesaler acquisition cost) per day is $195.00 for letermovir tablets and $270.00 for letermovir injections. (Wholesaler acquisition costs do not include discounts that may be paid on the product.)
The recommended dosage of letermovir is 480 mg once daily, initiated as early as day 0 and up to day 28 post-transplant (before or after engraftment) and continued through day 100. If letermovir is co-administered with cyclosporine, the dosage of letermovir should be decreased to 240 mg once daily.
Letermovir is available as 240 mg and 480 mg tablets, which may be administered with or without food. Letermovir is also available as a 240 mg and 480 mg injection for intravenous infusion via a peripheral catheter or central venous line at a constant rate over 1 hour.
For more details on letermovir, see the full prescribing information.
Trial results
The FDA’s approval of letermovir was supported by results of a phase 3 trial of adult recipients of allogeneic HSCTs who were CMV-seropositive. Patients were randomized (2:1) to receive either letermovir (at a dose of 480 mg once-daily, adjusted to 240 mg when co-administered with cyclosporine) or placebo.
Study drug was initiated after HSCT (at any time from day 0 to 28 post-transplant) and continued through week 14 post-transplant. Patients were monitored through week 24 post-HSCT for the primary efficacy endpoint, with continued follow-up through week 48.
Among the 565 treated patients, 34% were engrafted at baseline, and 30% had one or more factors associated with additional risk for CMV reactivation. The most common primary reasons for transplant were acute myeloid leukemia (38%), myelodysplastic syndromes (16%), and lymphoma (12%).
Thirty eight percent of patients in the letermovir arm and 61% in the placebo arm failed prophylaxis.
Reasons for failure (in the letermovir and placebo arms, respectively) included:
- Clinically significant CMV infection—18% vs 42%
- Initiation of PET based on documented CMV viremia—16% vs 40%
- CMV end-organ disease—2% for both
- Study discontinuation before week 24—17% vs 16%
- Missing outcome in week 24 visit window—3% for both.
The stratum-adjusted treatment difference for letermovir vs placebo was -23.5 (95% CI, -32.5, -14.6, P<0.0001).
The Kaplan-Meier event rate for all-cause mortality in the letermovir and placebo arms, respectively, was 12% and 17% at week 24 and 24% and 28% at week 48.
Common adverse events (in the letermovir and placebo arms, respectively) were nausea (27% vs 23%), diarrhea (26% vs 24%), vomiting (19% vs 14%), peripheral edema (14% vs 9%), cough (14% vs 10%), headache (14% vs 9%), fatigue (13% vs 11%), and abdominal pain (12% vs 9%).
The cardiac adverse event rate (regardless of investigator-assessed causality) was 13% in the letermovir arm and 6% in the placebo arm. The most common cardiac adverse events (in the letermovir and placebo arms, respectively) were tachycardia (4% vs 2%) and atrial fibrillation (3% vs 1%).
Results from this trial were presented at the 2017 BMT Tandem Meetings. ![]()
The US Food and Drug Administration (FDA) has approved the oral and intravenous formulations of letermovir (PREVYMIS™).
Letermovir is a member of a new class of non-nucleoside CMV inhibitors known as 3,4 dihydro-quinazolines.
The FDA approved letermovir as prophylaxis for cytomegalovirus (CMV) infection and disease in adult recipients of allogeneic hematopoietic stem cell transplants (HSCTs) who are CMV-seropositive.
“PREVYMIS is the first new medicine for CMV infection approved in the US in 15 years,” said Roy Baynes, senior vice president, head of clinical development, and chief medical officer of Merck Research Laboratories, the company marketing letermovir.
Letermovir is expected to be available in December. The list price (wholesaler acquisition cost) per day is $195.00 for letermovir tablets and $270.00 for letermovir injections. (Wholesaler acquisition costs do not include discounts that may be paid on the product.)
The recommended dosage of letermovir is 480 mg once daily, initiated as early as day 0 and up to day 28 post-transplant (before or after engraftment) and continued through day 100. If letermovir is co-administered with cyclosporine, the dosage of letermovir should be decreased to 240 mg once daily.
Letermovir is available as 240 mg and 480 mg tablets, which may be administered with or without food. Letermovir is also available as a 240 mg and 480 mg injection for intravenous infusion via a peripheral catheter or central venous line at a constant rate over 1 hour.
For more details on letermovir, see the full prescribing information.
Trial results
The FDA’s approval of letermovir was supported by results of a phase 3 trial of adult recipients of allogeneic HSCTs who were CMV-seropositive. Patients were randomized (2:1) to receive either letermovir (at a dose of 480 mg once-daily, adjusted to 240 mg when co-administered with cyclosporine) or placebo.
Study drug was initiated after HSCT (at any time from day 0 to 28 post-transplant) and continued through week 14 post-transplant. Patients were monitored through week 24 post-HSCT for the primary efficacy endpoint, with continued follow-up through week 48.
Among the 565 treated patients, 34% were engrafted at baseline, and 30% had one or more factors associated with additional risk for CMV reactivation. The most common primary reasons for transplant were acute myeloid leukemia (38%), myelodysplastic syndromes (16%), and lymphoma (12%).
Thirty eight percent of patients in the letermovir arm and 61% in the placebo arm failed prophylaxis.
Reasons for failure (in the letermovir and placebo arms, respectively) included:
- Clinically significant CMV infection—18% vs 42%
- Initiation of PET based on documented CMV viremia—16% vs 40%
- CMV end-organ disease—2% for both
- Study discontinuation before week 24—17% vs 16%
- Missing outcome in week 24 visit window—3% for both.
The stratum-adjusted treatment difference for letermovir vs placebo was -23.5 (95% CI, -32.5, -14.6, P<0.0001).
The Kaplan-Meier event rate for all-cause mortality in the letermovir and placebo arms, respectively, was 12% and 17% at week 24 and 24% and 28% at week 48.
Common adverse events (in the letermovir and placebo arms, respectively) were nausea (27% vs 23%), diarrhea (26% vs 24%), vomiting (19% vs 14%), peripheral edema (14% vs 9%), cough (14% vs 10%), headache (14% vs 9%), fatigue (13% vs 11%), and abdominal pain (12% vs 9%).
The cardiac adverse event rate (regardless of investigator-assessed causality) was 13% in the letermovir arm and 6% in the placebo arm. The most common cardiac adverse events (in the letermovir and placebo arms, respectively) were tachycardia (4% vs 2%) and atrial fibrillation (3% vs 1%).
Results from this trial were presented at the 2017 BMT Tandem Meetings. ![]()
The US Food and Drug Administration (FDA) has approved the oral and intravenous formulations of letermovir (PREVYMIS™).
Letermovir is a member of a new class of non-nucleoside CMV inhibitors known as 3,4 dihydro-quinazolines.
The FDA approved letermovir as prophylaxis for cytomegalovirus (CMV) infection and disease in adult recipients of allogeneic hematopoietic stem cell transplants (HSCTs) who are CMV-seropositive.
“PREVYMIS is the first new medicine for CMV infection approved in the US in 15 years,” said Roy Baynes, senior vice president, head of clinical development, and chief medical officer of Merck Research Laboratories, the company marketing letermovir.
Letermovir is expected to be available in December. The list price (wholesaler acquisition cost) per day is $195.00 for letermovir tablets and $270.00 for letermovir injections. (Wholesaler acquisition costs do not include discounts that may be paid on the product.)
The recommended dosage of letermovir is 480 mg once daily, initiated as early as day 0 and up to day 28 post-transplant (before or after engraftment) and continued through day 100. If letermovir is co-administered with cyclosporine, the dosage of letermovir should be decreased to 240 mg once daily.
Letermovir is available as 240 mg and 480 mg tablets, which may be administered with or without food. Letermovir is also available as a 240 mg and 480 mg injection for intravenous infusion via a peripheral catheter or central venous line at a constant rate over 1 hour.
For more details on letermovir, see the full prescribing information.
Trial results
The FDA’s approval of letermovir was supported by results of a phase 3 trial of adult recipients of allogeneic HSCTs who were CMV-seropositive. Patients were randomized (2:1) to receive either letermovir (at a dose of 480 mg once-daily, adjusted to 240 mg when co-administered with cyclosporine) or placebo.
Study drug was initiated after HSCT (at any time from day 0 to 28 post-transplant) and continued through week 14 post-transplant. Patients were monitored through week 24 post-HSCT for the primary efficacy endpoint, with continued follow-up through week 48.
Among the 565 treated patients, 34% were engrafted at baseline, and 30% had one or more factors associated with additional risk for CMV reactivation. The most common primary reasons for transplant were acute myeloid leukemia (38%), myelodysplastic syndromes (16%), and lymphoma (12%).
Thirty eight percent of patients in the letermovir arm and 61% in the placebo arm failed prophylaxis.
Reasons for failure (in the letermovir and placebo arms, respectively) included:
- Clinically significant CMV infection—18% vs 42%
- Initiation of PET based on documented CMV viremia—16% vs 40%
- CMV end-organ disease—2% for both
- Study discontinuation before week 24—17% vs 16%
- Missing outcome in week 24 visit window—3% for both.
The stratum-adjusted treatment difference for letermovir vs placebo was -23.5 (95% CI, -32.5, -14.6, P<0.0001).
The Kaplan-Meier event rate for all-cause mortality in the letermovir and placebo arms, respectively, was 12% and 17% at week 24 and 24% and 28% at week 48.
Common adverse events (in the letermovir and placebo arms, respectively) were nausea (27% vs 23%), diarrhea (26% vs 24%), vomiting (19% vs 14%), peripheral edema (14% vs 9%), cough (14% vs 10%), headache (14% vs 9%), fatigue (13% vs 11%), and abdominal pain (12% vs 9%).
The cardiac adverse event rate (regardless of investigator-assessed causality) was 13% in the letermovir arm and 6% in the placebo arm. The most common cardiac adverse events (in the letermovir and placebo arms, respectively) were tachycardia (4% vs 2%) and atrial fibrillation (3% vs 1%).
Results from this trial were presented at the 2017 BMT Tandem Meetings. ![]()
FDA approves brentuximab vedotin for pcALCL, MF

The US Food and Drug Administration (FDA) has expanded the approved use of brentuximab vedotin (BV, ADCETRIS).
BV is now approved for adults with primary cutaneous anaplastic large-cell lymphoma (pcALCL) and CD30-expressing mycosis fungoides (MF) who have received prior systemic therapy.
This is the fourth FDA-approved indication for BV. The drug has regular approval for 2 indications in classical Hodgkin lymphoma and accelerated approval for the treatment of systemic ALCL.
In November 2016, the FDA granted BV breakthrough therapy designation for the treatment of patients with pcALCL and CD30-expressing MF who require systemic therapy and have received one prior systemic therapy. The agency also granted the supplemental biologics license application priority review.
The approval for BV in pcALCL and CD30-expressing MF is based on data from the phase 3 ALCANZA trial and a pair of phase 2 investigator-sponsored trials.
Phase 3 trial
Results from ALCANZA were presented at the 9th Annual T-cell Lymphoma Forum in January and published in The Lancet in June.
There were 128 patients in the intent-to-treat and safety populations. Sixty-four patients (48 with MF and 16 with pcALCL) were randomized to receive BV at 1.8 mg/kg every 3 weeks for up to 48 weeks.
The other 64 patients (49 with MF and 15 with pcALCL) were randomized to receive standard of care (SOC)—methotrexate at 5 mg to 50 mg weekly or bexarotene at a target dose of 300 mg/m² daily for up to 48 weeks.
The study’s primary endpoint was the rate of objective response lasting at least 4 months (ORR4). The ORR4 rate was significantly higher with BV than with SOC—56.3% and 12.5%, respectively (P<0.0001).
For patients with MF, the ORR4 was 50% with BV and 10% with SOC. For patients with pcALCL, the ORR4 was 75% with BV and 20% with SOC.
Overall, the complete response (CR) rates were 15.6% in the BV arm and 1.6% in the SOC arm (P=0.0046).
For patients with MF, the CR rate was 10% with BV and 0% with SOC. For patients with pcALCL, the CR rate was 31% with BV and 7% with SOC.
Progression-free survival (PFS) was significantly longer in the BV arm than the SOC arm. The median PFS was 16.7 months and 3.5 months, respectively. The hazard ratio was 0.270 (P<0.0001).
For patients with MF, the median PFS was 15.9 months with BV and 3.5 months with SOC. For patients with pcALCL, the median PFS was 27.5 months with BV and 5.3 months with SOC.
The most common adverse events (AEs) of any grade (occurring in 15% or more of patients in the BV and SOC arms, respectively) were peripheral neuropathy (67% and 6%), nausea (36% and 13%), diarrhea (29% and 6%), fatigue (29% and 27%), vomiting (17% and 5%), alopecia (15% and 3%), pruritus (17% and 13%), pyrexia (17% and 18%), decreased appetite (15% and 5%), and hypertriglyceridemia (2% and 18%).
Phase 2 trials
Data from the investigator-sponsored trials were published in the Journal of Clinical Oncology in 2015.
The first study was published in July of that year. The trial enrolled 32 patients with MF or Sézary syndrome. Thirty patients were evaluable for efficacy, and more than half had received 3 or more prior systemic therapies.
Patients received BV (1.8 mg/kg) every 3 weeks for a maximum of 16 doses. The primary endpoint was objective clinical response rate.
Seventy percent of patients (21/30) achieved an objective response across all stages of disease. One patient had a CR, 20 had a partial response, 4 had stable disease, 5 had progressive disease, and 2 were not evaluable for response.
The most common related AEs of any grade were peripheral neuropathy (66%), fatigue (47%), nausea (28%), hair loss (22%), and neutropenia (19%). Grade 3/4 related AEs included neutropenia (n=4), rash (n=3), and peripheral neuropathy (n=1).
The second phase 2 trial was published in August 2015. This trial enrolled CD30-positive patients with lymphomatoid papulosis (LyP), pcALCL, and MF.
Fifty-four patients were enrolled, and 48 were evaluable at the time of analysis. Patients had received an infusion of BV (1.8 mg/kg) every 21 days.
Seventy-three percent of patients (35/48) achieved an objective response, including 100% (20/20) with LyP and/or pcALCL and 54% (15/28) with MF. The CR rate was 35% (n=17).
The most common AEs were peripheral neuropathy (67%), fatigue (35%), skin rash (24%), diarrhea (15%), muscle pain (17%), localized skin infection (15%), neutropenia (15%), and hair loss (11%).
Grade 3/4 AEs included neutropenia (n=3), nausea (n=2), unstable angina or myocardial infarction (n=2), infection (n=2), joint pain (n=2), fatigue (n=1), deep vein thrombosis (n=1), pulmonary embolism (n=1), aminotransferase elevation (n=1), and dehydration (n=1). ![]()
The US Food and Drug Administration (FDA) has expanded the approved use of brentuximab vedotin (BV, ADCETRIS).
BV is now approved for adults with primary cutaneous anaplastic large-cell lymphoma (pcALCL) and CD30-expressing mycosis fungoides (MF) who have received prior systemic therapy.
This is the fourth FDA-approved indication for BV. The drug has regular approval for 2 indications in classical Hodgkin lymphoma and accelerated approval for the treatment of systemic ALCL.
In November 2016, the FDA granted BV breakthrough therapy designation for the treatment of patients with pcALCL and CD30-expressing MF who require systemic therapy and have received one prior systemic therapy. The agency also granted the supplemental biologics license application priority review.
The approval for BV in pcALCL and CD30-expressing MF is based on data from the phase 3 ALCANZA trial and a pair of phase 2 investigator-sponsored trials.
Phase 3 trial
Results from ALCANZA were presented at the 9th Annual T-cell Lymphoma Forum in January and published in The Lancet in June.
There were 128 patients in the intent-to-treat and safety populations. Sixty-four patients (48 with MF and 16 with pcALCL) were randomized to receive BV at 1.8 mg/kg every 3 weeks for up to 48 weeks.
The other 64 patients (49 with MF and 15 with pcALCL) were randomized to receive standard of care (SOC)—methotrexate at 5 mg to 50 mg weekly or bexarotene at a target dose of 300 mg/m² daily for up to 48 weeks.
The study’s primary endpoint was the rate of objective response lasting at least 4 months (ORR4). The ORR4 rate was significantly higher with BV than with SOC—56.3% and 12.5%, respectively (P<0.0001).
For patients with MF, the ORR4 was 50% with BV and 10% with SOC. For patients with pcALCL, the ORR4 was 75% with BV and 20% with SOC.
Overall, the complete response (CR) rates were 15.6% in the BV arm and 1.6% in the SOC arm (P=0.0046).
For patients with MF, the CR rate was 10% with BV and 0% with SOC. For patients with pcALCL, the CR rate was 31% with BV and 7% with SOC.
Progression-free survival (PFS) was significantly longer in the BV arm than the SOC arm. The median PFS was 16.7 months and 3.5 months, respectively. The hazard ratio was 0.270 (P<0.0001).
For patients with MF, the median PFS was 15.9 months with BV and 3.5 months with SOC. For patients with pcALCL, the median PFS was 27.5 months with BV and 5.3 months with SOC.
The most common adverse events (AEs) of any grade (occurring in 15% or more of patients in the BV and SOC arms, respectively) were peripheral neuropathy (67% and 6%), nausea (36% and 13%), diarrhea (29% and 6%), fatigue (29% and 27%), vomiting (17% and 5%), alopecia (15% and 3%), pruritus (17% and 13%), pyrexia (17% and 18%), decreased appetite (15% and 5%), and hypertriglyceridemia (2% and 18%).
Phase 2 trials
Data from the investigator-sponsored trials were published in the Journal of Clinical Oncology in 2015.
The first study was published in July of that year. The trial enrolled 32 patients with MF or Sézary syndrome. Thirty patients were evaluable for efficacy, and more than half had received 3 or more prior systemic therapies.
Patients received BV (1.8 mg/kg) every 3 weeks for a maximum of 16 doses. The primary endpoint was objective clinical response rate.
Seventy percent of patients (21/30) achieved an objective response across all stages of disease. One patient had a CR, 20 had a partial response, 4 had stable disease, 5 had progressive disease, and 2 were not evaluable for response.
The most common related AEs of any grade were peripheral neuropathy (66%), fatigue (47%), nausea (28%), hair loss (22%), and neutropenia (19%). Grade 3/4 related AEs included neutropenia (n=4), rash (n=3), and peripheral neuropathy (n=1).
The second phase 2 trial was published in August 2015. This trial enrolled CD30-positive patients with lymphomatoid papulosis (LyP), pcALCL, and MF.
Fifty-four patients were enrolled, and 48 were evaluable at the time of analysis. Patients had received an infusion of BV (1.8 mg/kg) every 21 days.
Seventy-three percent of patients (35/48) achieved an objective response, including 100% (20/20) with LyP and/or pcALCL and 54% (15/28) with MF. The CR rate was 35% (n=17).
The most common AEs were peripheral neuropathy (67%), fatigue (35%), skin rash (24%), diarrhea (15%), muscle pain (17%), localized skin infection (15%), neutropenia (15%), and hair loss (11%).
Grade 3/4 AEs included neutropenia (n=3), nausea (n=2), unstable angina or myocardial infarction (n=2), infection (n=2), joint pain (n=2), fatigue (n=1), deep vein thrombosis (n=1), pulmonary embolism (n=1), aminotransferase elevation (n=1), and dehydration (n=1). ![]()
The US Food and Drug Administration (FDA) has expanded the approved use of brentuximab vedotin (BV, ADCETRIS).
BV is now approved for adults with primary cutaneous anaplastic large-cell lymphoma (pcALCL) and CD30-expressing mycosis fungoides (MF) who have received prior systemic therapy.
This is the fourth FDA-approved indication for BV. The drug has regular approval for 2 indications in classical Hodgkin lymphoma and accelerated approval for the treatment of systemic ALCL.
In November 2016, the FDA granted BV breakthrough therapy designation for the treatment of patients with pcALCL and CD30-expressing MF who require systemic therapy and have received one prior systemic therapy. The agency also granted the supplemental biologics license application priority review.
The approval for BV in pcALCL and CD30-expressing MF is based on data from the phase 3 ALCANZA trial and a pair of phase 2 investigator-sponsored trials.
Phase 3 trial
Results from ALCANZA were presented at the 9th Annual T-cell Lymphoma Forum in January and published in The Lancet in June.
There were 128 patients in the intent-to-treat and safety populations. Sixty-four patients (48 with MF and 16 with pcALCL) were randomized to receive BV at 1.8 mg/kg every 3 weeks for up to 48 weeks.
The other 64 patients (49 with MF and 15 with pcALCL) were randomized to receive standard of care (SOC)—methotrexate at 5 mg to 50 mg weekly or bexarotene at a target dose of 300 mg/m² daily for up to 48 weeks.
The study’s primary endpoint was the rate of objective response lasting at least 4 months (ORR4). The ORR4 rate was significantly higher with BV than with SOC—56.3% and 12.5%, respectively (P<0.0001).
For patients with MF, the ORR4 was 50% with BV and 10% with SOC. For patients with pcALCL, the ORR4 was 75% with BV and 20% with SOC.
Overall, the complete response (CR) rates were 15.6% in the BV arm and 1.6% in the SOC arm (P=0.0046).
For patients with MF, the CR rate was 10% with BV and 0% with SOC. For patients with pcALCL, the CR rate was 31% with BV and 7% with SOC.
Progression-free survival (PFS) was significantly longer in the BV arm than the SOC arm. The median PFS was 16.7 months and 3.5 months, respectively. The hazard ratio was 0.270 (P<0.0001).
For patients with MF, the median PFS was 15.9 months with BV and 3.5 months with SOC. For patients with pcALCL, the median PFS was 27.5 months with BV and 5.3 months with SOC.
The most common adverse events (AEs) of any grade (occurring in 15% or more of patients in the BV and SOC arms, respectively) were peripheral neuropathy (67% and 6%), nausea (36% and 13%), diarrhea (29% and 6%), fatigue (29% and 27%), vomiting (17% and 5%), alopecia (15% and 3%), pruritus (17% and 13%), pyrexia (17% and 18%), decreased appetite (15% and 5%), and hypertriglyceridemia (2% and 18%).
Phase 2 trials
Data from the investigator-sponsored trials were published in the Journal of Clinical Oncology in 2015.
The first study was published in July of that year. The trial enrolled 32 patients with MF or Sézary syndrome. Thirty patients were evaluable for efficacy, and more than half had received 3 or more prior systemic therapies.
Patients received BV (1.8 mg/kg) every 3 weeks for a maximum of 16 doses. The primary endpoint was objective clinical response rate.
Seventy percent of patients (21/30) achieved an objective response across all stages of disease. One patient had a CR, 20 had a partial response, 4 had stable disease, 5 had progressive disease, and 2 were not evaluable for response.
The most common related AEs of any grade were peripheral neuropathy (66%), fatigue (47%), nausea (28%), hair loss (22%), and neutropenia (19%). Grade 3/4 related AEs included neutropenia (n=4), rash (n=3), and peripheral neuropathy (n=1).
The second phase 2 trial was published in August 2015. This trial enrolled CD30-positive patients with lymphomatoid papulosis (LyP), pcALCL, and MF.
Fifty-four patients were enrolled, and 48 were evaluable at the time of analysis. Patients had received an infusion of BV (1.8 mg/kg) every 21 days.
Seventy-three percent of patients (35/48) achieved an objective response, including 100% (20/20) with LyP and/or pcALCL and 54% (15/28) with MF. The CR rate was 35% (n=17).
The most common AEs were peripheral neuropathy (67%), fatigue (35%), skin rash (24%), diarrhea (15%), muscle pain (17%), localized skin infection (15%), neutropenia (15%), and hair loss (11%).
Grade 3/4 AEs included neutropenia (n=3), nausea (n=2), unstable angina or myocardial infarction (n=2), infection (n=2), joint pain (n=2), fatigue (n=1), deep vein thrombosis (n=1), pulmonary embolism (n=1), aminotransferase elevation (n=1), and dehydration (n=1). ![]()
FDA approves IV formulation of aprepitant for CINV

The US Food and Drug Administration (FDA) has approved use of an intravenous (IV) formulation of aprepitant (CINVANTI™) to prevent chemotherapy-induced nausea and vomiting (CINV).
CINVANTI is intended to be used in combination with other antiemetic agents to prevent acute and delayed nausea and vomiting associated with initial and repeat courses of highly emetogenic chemotherapy (HEC) and moderately emetogenic chemotherapy (MEC).
CINVANTI is to be used in combination with a 5-HT3 receptor antagonist and dexamethasone.
The full prescribing information is available at www.cinvanti.com.
The US commercial launch of CINVANTI is planned for January 2018.
CINVANTI is the first IV formulation to directly deliver aprepitant, a substance P/neurokinin-1 (NK1) receptor antagonist.
Aprepitant is also the active ingredient in EMEND® capsules, which were approved by the FDA in 2003. EMEND IV®, which was approved in 2008, contains aprepitant’s prodrug, fosaprepitant.
Heron Therapeutics, Inc., developed CINVANTI in an attempt to provide an IV formulation of aprepitant that has the same efficacy as IV fosaprepitant but does not pose the risk of adverse events (AEs) related to polysorbate 80.
“Aprepitant has long been the standard in the NK1 class, and it remains the only single-agent NK1 with proven efficacy in preventing CINV in both the acute and delayed phases in HEC and MEC,” said Rudolph M. Navari, MD, PhD, of the University of Alabama Birmingham School of Medicine.
“Because CINVANTI is a novel, polysorbate 80-free, IV formulation of aprepitant, it enables physicians to provide patients with standard-of-care efficacy without the potential risk of polysorbate 80-related adverse events, such as infusion-site reactions.”
The FDA approved CINVANTI based on data demonstrating the bioequivalence of CINVANTI to EMEND IV.
A phase 1, randomized, 2-way cross-over study comparing the drugs enrolled 100 healthy subjects. The subjects received CINVANTI at 130 mg or EMEND IV at 150 mg, given over 30 minutes on day 1 of periods 1 and 2.
The researchers said 90% confidence intervals for CINVANTI AUC0-t (area under the time-concentration curve from time 0 to the last measurable concentration), AUC0-inf (area under the time-concentration curve from time 0 extrapolated to infinity), and C12h (plasma concentration at 12 hours) “were well within bioequivalence bounds,” which was 80% to 125%.
The team also found the incidence of treatment-emergent AEs was lower with CINVANTI than EMEND IV—21% and 28%, respectively. The same was true for related treatment-emergent AEs—15% and 28%, respectively.
These data were presented at the Hematology/Oncology Pharmacy Association Annual Conference in March/April and the Multinational Association of Supportive Care in Cancer (MASCC)/International Society of Oral Oncology (ISOO) Annual Meeting in June. ![]()
The US Food and Drug Administration (FDA) has approved use of an intravenous (IV) formulation of aprepitant (CINVANTI™) to prevent chemotherapy-induced nausea and vomiting (CINV).
CINVANTI is intended to be used in combination with other antiemetic agents to prevent acute and delayed nausea and vomiting associated with initial and repeat courses of highly emetogenic chemotherapy (HEC) and moderately emetogenic chemotherapy (MEC).
CINVANTI is to be used in combination with a 5-HT3 receptor antagonist and dexamethasone.
The full prescribing information is available at www.cinvanti.com.
The US commercial launch of CINVANTI is planned for January 2018.
CINVANTI is the first IV formulation to directly deliver aprepitant, a substance P/neurokinin-1 (NK1) receptor antagonist.
Aprepitant is also the active ingredient in EMEND® capsules, which were approved by the FDA in 2003. EMEND IV®, which was approved in 2008, contains aprepitant’s prodrug, fosaprepitant.
Heron Therapeutics, Inc., developed CINVANTI in an attempt to provide an IV formulation of aprepitant that has the same efficacy as IV fosaprepitant but does not pose the risk of adverse events (AEs) related to polysorbate 80.
“Aprepitant has long been the standard in the NK1 class, and it remains the only single-agent NK1 with proven efficacy in preventing CINV in both the acute and delayed phases in HEC and MEC,” said Rudolph M. Navari, MD, PhD, of the University of Alabama Birmingham School of Medicine.
“Because CINVANTI is a novel, polysorbate 80-free, IV formulation of aprepitant, it enables physicians to provide patients with standard-of-care efficacy without the potential risk of polysorbate 80-related adverse events, such as infusion-site reactions.”
The FDA approved CINVANTI based on data demonstrating the bioequivalence of CINVANTI to EMEND IV.
A phase 1, randomized, 2-way cross-over study comparing the drugs enrolled 100 healthy subjects. The subjects received CINVANTI at 130 mg or EMEND IV at 150 mg, given over 30 minutes on day 1 of periods 1 and 2.
The researchers said 90% confidence intervals for CINVANTI AUC0-t (area under the time-concentration curve from time 0 to the last measurable concentration), AUC0-inf (area under the time-concentration curve from time 0 extrapolated to infinity), and C12h (plasma concentration at 12 hours) “were well within bioequivalence bounds,” which was 80% to 125%.
The team also found the incidence of treatment-emergent AEs was lower with CINVANTI than EMEND IV—21% and 28%, respectively. The same was true for related treatment-emergent AEs—15% and 28%, respectively.
These data were presented at the Hematology/Oncology Pharmacy Association Annual Conference in March/April and the Multinational Association of Supportive Care in Cancer (MASCC)/International Society of Oral Oncology (ISOO) Annual Meeting in June. ![]()
The US Food and Drug Administration (FDA) has approved use of an intravenous (IV) formulation of aprepitant (CINVANTI™) to prevent chemotherapy-induced nausea and vomiting (CINV).
CINVANTI is intended to be used in combination with other antiemetic agents to prevent acute and delayed nausea and vomiting associated with initial and repeat courses of highly emetogenic chemotherapy (HEC) and moderately emetogenic chemotherapy (MEC).
CINVANTI is to be used in combination with a 5-HT3 receptor antagonist and dexamethasone.
The full prescribing information is available at www.cinvanti.com.
The US commercial launch of CINVANTI is planned for January 2018.
CINVANTI is the first IV formulation to directly deliver aprepitant, a substance P/neurokinin-1 (NK1) receptor antagonist.
Aprepitant is also the active ingredient in EMEND® capsules, which were approved by the FDA in 2003. EMEND IV®, which was approved in 2008, contains aprepitant’s prodrug, fosaprepitant.
Heron Therapeutics, Inc., developed CINVANTI in an attempt to provide an IV formulation of aprepitant that has the same efficacy as IV fosaprepitant but does not pose the risk of adverse events (AEs) related to polysorbate 80.
“Aprepitant has long been the standard in the NK1 class, and it remains the only single-agent NK1 with proven efficacy in preventing CINV in both the acute and delayed phases in HEC and MEC,” said Rudolph M. Navari, MD, PhD, of the University of Alabama Birmingham School of Medicine.
“Because CINVANTI is a novel, polysorbate 80-free, IV formulation of aprepitant, it enables physicians to provide patients with standard-of-care efficacy without the potential risk of polysorbate 80-related adverse events, such as infusion-site reactions.”
The FDA approved CINVANTI based on data demonstrating the bioequivalence of CINVANTI to EMEND IV.
A phase 1, randomized, 2-way cross-over study comparing the drugs enrolled 100 healthy subjects. The subjects received CINVANTI at 130 mg or EMEND IV at 150 mg, given over 30 minutes on day 1 of periods 1 and 2.
The researchers said 90% confidence intervals for CINVANTI AUC0-t (area under the time-concentration curve from time 0 to the last measurable concentration), AUC0-inf (area under the time-concentration curve from time 0 extrapolated to infinity), and C12h (plasma concentration at 12 hours) “were well within bioequivalence bounds,” which was 80% to 125%.
The team also found the incidence of treatment-emergent AEs was lower with CINVANTI than EMEND IV—21% and 28%, respectively. The same was true for related treatment-emergent AEs—15% and 28%, respectively.
These data were presented at the Hematology/Oncology Pharmacy Association Annual Conference in March/April and the Multinational Association of Supportive Care in Cancer (MASCC)/International Society of Oral Oncology (ISOO) Annual Meeting in June. ![]()
When to “CAP” Off Pneumonia Treatment

A 65-year-old woman is admitted to your inpatient service from the family health center. She is diagnosed with community-acquired pneumonia (CAP) based on a five-day history of cough and fever and a positive chest x-ray. She now requires oxygen at rest. She has a history of hypertension and diabetes, both of which have been controlled by oral medications. Antibiotic therapy is initiated—but what treatment duration is ideal?
The World Health Organization estimates that pneumonia is the third most common cause of mortality worldwide, causing 3.2 million deaths per year.2 Appropriate prescribing of antibiotics is critical for successful treatment of CAP.
In 2007, the Infectious Diseases Society of America (IDSA) and the American Thoracic Society (ATS) created consensus guidelines for the treatment of CAP.3 These guidelines recommend a minimum five-day course of antibiotics if the patient is clinically stable (defined as afebrile for 48 h; heart rate ≤ 100 beats/min; respiratory rate ≤ 24 breaths/min-1
However, these recommendations are not routinely followed. Practitioners often make it their custom to prescribe longer courses of antibiotics.4 Yet, we know that there are several reasons to consider shorter courses of antibiotics, including lower health care costs, fewer adverse effects, and lower rates of bacterial resistance.5-7
Two meta-analyses were performed to compare the safety and efficacy of short- and long-course antibiotic therapy in CAP (≤ 7 d vs > 7 d, respectively).8,9 Both meta-analyses found no difference in efficacy or safety between shorter and longer courses of antibiotics for CAP. Secondary outcomes noted a trend toward decreased antibiotic-associated adverse events with shorter courses of therapy.8,9
However, there are limitations to broad implementation. Studies included in these analyses utilized a variety of antibiotic treatment regimens and longer courses (7 d vs 5 d) that are not recommended by the IDSA/ATS guidelines. Additionally, studies included both inpatient and outpatient treatment groups, so findings may not apply to an exclusively inpatient CAP population.8,9
This study sought to validate the IDSA/ATS guidelines recommending a five-day course of antibiotics for hospitalized patients with CAP.1
STUDY SUMMARY
No differences in clinical outcomes
This multicenter, double-blind, noninferiority randomized trial compared short-term antibiotic treatment duration (5 d) to physician-discretion antibiotic treatment duration among 312 patients (ages 18 and older) admitted for CAP to one of four teaching hospitals in Spain.1 Pneumonia was diagnosed on chest radiograph with at least one symptom: cough, fever, dyspnea, or chest pain. Patients were excluded if, among other things, they had an immunocompromising condition, lived in a nursing home, had a recent hospital stay, used antibiotics within the previous 30 days, or had an uncommon pathogen, such as Pseudomonas aeruginosa or Staphylococcus aureus.1
After receiving a minimum of five days of antibiotics, patients were randomly assigned to an intervention group (where, if clinically stable, no further antibiotics were given) or a control group (where physicians determined antibiotic duration).1
Primary outcomes were clinical success rate at days 10 and 30 from admission (defined as resolution of signs and symptoms of CAP without further antibiotics) and improvement of CAP-related symptoms (as determined by an 18-item questionnaire scored 0-90, with higher scores indicative of greater severity). Secondary outcomes included duration of antibiotic use, time to clinical improvement, mortality, hospital readmission, hospital length of stay, and CAP recurrence.1
Of the 312 participants, 162 were randomized to the intervention group and 150 to the control group. Mean age of patients in the intervention and control groups was 66.2 and 64.7, respectively. Other baseline demographics were similar between the groups. Nearly 80% of patients received quinolone treatment; < 10% received a ß-lactam plus a macrolide.1
Clinical success rates were similar for the control and intervention groups at day 10 (49% vs 56%, respectively) and day 30 (89% vs 92%). Median antibiotic treatment duration was shorter in the intervention group than in the control group (5 d vs 10 d); the intervention group also had a lower rate of 30-day hospital readmissions (1.4% vs 6.6%). There were no differences for other secondary outcomes.1
WHAT’S NEW
Clinical support for 2007 guidelines
This is the first study to clinically support the IDSA/ATS guidelines, which state that a five-day course of antibiotic therapy for hospitalized adults with CAP is effective and without increased risk for adverse events.
CAVEATS
Generalizability is unclear
This study focused on antibiotic duration for the treatment of CAP in hospitalized patients and mainly used quinolone antibiotics. It remains unclear if duration of therapy is as effective in the outpatient setting or when using alternative antibiotic regimens.
If patients continued to have symptoms (eg, fever or low oxygen saturation on room air) after five days of antibiotics, treatment was continued in the study. Thus, patients in real life who continue to have symptoms may need individualized therapy and may require more than five days of antibiotics.
CHALLENGES TO IMPLEMENTATION
Antibiotics end before clinical improvement
In this study, it took an average of 12 days in both groups for patients to achieve clinical improvement, and upwards of 15 to 18 days for patients to return to normal activity. Patients and providers may be dissatisfied if the treatment course ends days before clinical improvement of symptoms. This may cause prescribers to lengthen the duration of antibiotic therapy inappropriately.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2017. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice (2017; 66[10]:629-631).
1. Uranga A, España PP, Bilbao A, et al. Duration of antibiotic treatment in community-acquired pneumonia: a multicenter randomized clinical trial. JAMA Intern Med. 2016;176:1257-1265.
2. World Health Organization. The top 10 causes of death. www.who.int/mediacentre/factsheets/fs310/en/index.html. Accessed October 18, 2017.
3. Mandell LA, Wunderink RG, Anzueto A, et al. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis. 2007;44(suppl 2):S27-S72.
4. Aliberti S, Blasi F, Zanaboni AM, et al. Duration of antibiotic therapy in hospitalised patients with community-acquired pneumonia. Eur Respir J. 2010;36:128-134.
5. Guillemot D, Carbon C, Balkau B, et al. Low dosage and long treatment duration of ß-lactam: risk factors for carriage of penicillin-resistant Streptococcus pneumoniae. JAMA. 1998; 279:365-370.
6. Opmeer BC, el Moussaoui R, Bossuyt PM, et al. Costs associated with shorter duration of antibiotic therapy in hospitalized patients with mild-to-moderate severe community-acquired pneumonia. J Antimicrob Chemother. 2007;60: 1131-1136.
7. File TM Jr. Clinical efficacy of newer agents in short-duration therapy for community-acquired pneumonia. Clin Infect Dis. 2004;39(suppl 3):S159-S164.
8. Li JZ, Winston LG, Moore DH, et al. Efficacy of short-course antibiotic regimens for community-acquired pneumonia: a meta-analysis. Am J Med. 2007;120:783-790.
9. Dimopoulos G, Matthaiou DK, Karageorgopoulos DE, et al. Short- versus long-course antibacterial therapy for community-acquired pneumonia: a meta-analysis. Drugs. 2008;68: 1841-1854.
A 65-year-old woman is admitted to your inpatient service from the family health center. She is diagnosed with community-acquired pneumonia (CAP) based on a five-day history of cough and fever and a positive chest x-ray. She now requires oxygen at rest. She has a history of hypertension and diabetes, both of which have been controlled by oral medications. Antibiotic therapy is initiated—but what treatment duration is ideal?
The World Health Organization estimates that pneumonia is the third most common cause of mortality worldwide, causing 3.2 million deaths per year.2 Appropriate prescribing of antibiotics is critical for successful treatment of CAP.
In 2007, the Infectious Diseases Society of America (IDSA) and the American Thoracic Society (ATS) created consensus guidelines for the treatment of CAP.3 These guidelines recommend a minimum five-day course of antibiotics if the patient is clinically stable (defined as afebrile for 48 h; heart rate ≤ 100 beats/min; respiratory rate ≤ 24 breaths/min-1
However, these recommendations are not routinely followed. Practitioners often make it their custom to prescribe longer courses of antibiotics.4 Yet, we know that there are several reasons to consider shorter courses of antibiotics, including lower health care costs, fewer adverse effects, and lower rates of bacterial resistance.5-7
Two meta-analyses were performed to compare the safety and efficacy of short- and long-course antibiotic therapy in CAP (≤ 7 d vs > 7 d, respectively).8,9 Both meta-analyses found no difference in efficacy or safety between shorter and longer courses of antibiotics for CAP. Secondary outcomes noted a trend toward decreased antibiotic-associated adverse events with shorter courses of therapy.8,9
However, there are limitations to broad implementation. Studies included in these analyses utilized a variety of antibiotic treatment regimens and longer courses (7 d vs 5 d) that are not recommended by the IDSA/ATS guidelines. Additionally, studies included both inpatient and outpatient treatment groups, so findings may not apply to an exclusively inpatient CAP population.8,9
This study sought to validate the IDSA/ATS guidelines recommending a five-day course of antibiotics for hospitalized patients with CAP.1
STUDY SUMMARY
No differences in clinical outcomes
This multicenter, double-blind, noninferiority randomized trial compared short-term antibiotic treatment duration (5 d) to physician-discretion antibiotic treatment duration among 312 patients (ages 18 and older) admitted for CAP to one of four teaching hospitals in Spain.1 Pneumonia was diagnosed on chest radiograph with at least one symptom: cough, fever, dyspnea, or chest pain. Patients were excluded if, among other things, they had an immunocompromising condition, lived in a nursing home, had a recent hospital stay, used antibiotics within the previous 30 days, or had an uncommon pathogen, such as Pseudomonas aeruginosa or Staphylococcus aureus.1
After receiving a minimum of five days of antibiotics, patients were randomly assigned to an intervention group (where, if clinically stable, no further antibiotics were given) or a control group (where physicians determined antibiotic duration).1
Primary outcomes were clinical success rate at days 10 and 30 from admission (defined as resolution of signs and symptoms of CAP without further antibiotics) and improvement of CAP-related symptoms (as determined by an 18-item questionnaire scored 0-90, with higher scores indicative of greater severity). Secondary outcomes included duration of antibiotic use, time to clinical improvement, mortality, hospital readmission, hospital length of stay, and CAP recurrence.1
Of the 312 participants, 162 were randomized to the intervention group and 150 to the control group. Mean age of patients in the intervention and control groups was 66.2 and 64.7, respectively. Other baseline demographics were similar between the groups. Nearly 80% of patients received quinolone treatment; < 10% received a ß-lactam plus a macrolide.1
Clinical success rates were similar for the control and intervention groups at day 10 (49% vs 56%, respectively) and day 30 (89% vs 92%). Median antibiotic treatment duration was shorter in the intervention group than in the control group (5 d vs 10 d); the intervention group also had a lower rate of 30-day hospital readmissions (1.4% vs 6.6%). There were no differences for other secondary outcomes.1
WHAT’S NEW
Clinical support for 2007 guidelines
This is the first study to clinically support the IDSA/ATS guidelines, which state that a five-day course of antibiotic therapy for hospitalized adults with CAP is effective and without increased risk for adverse events.
CAVEATS
Generalizability is unclear
This study focused on antibiotic duration for the treatment of CAP in hospitalized patients and mainly used quinolone antibiotics. It remains unclear if duration of therapy is as effective in the outpatient setting or when using alternative antibiotic regimens.
If patients continued to have symptoms (eg, fever or low oxygen saturation on room air) after five days of antibiotics, treatment was continued in the study. Thus, patients in real life who continue to have symptoms may need individualized therapy and may require more than five days of antibiotics.
CHALLENGES TO IMPLEMENTATION
Antibiotics end before clinical improvement
In this study, it took an average of 12 days in both groups for patients to achieve clinical improvement, and upwards of 15 to 18 days for patients to return to normal activity. Patients and providers may be dissatisfied if the treatment course ends days before clinical improvement of symptoms. This may cause prescribers to lengthen the duration of antibiotic therapy inappropriately.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2017. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice (2017; 66[10]:629-631).
A 65-year-old woman is admitted to your inpatient service from the family health center. She is diagnosed with community-acquired pneumonia (CAP) based on a five-day history of cough and fever and a positive chest x-ray. She now requires oxygen at rest. She has a history of hypertension and diabetes, both of which have been controlled by oral medications. Antibiotic therapy is initiated—but what treatment duration is ideal?
The World Health Organization estimates that pneumonia is the third most common cause of mortality worldwide, causing 3.2 million deaths per year.2 Appropriate prescribing of antibiotics is critical for successful treatment of CAP.
In 2007, the Infectious Diseases Society of America (IDSA) and the American Thoracic Society (ATS) created consensus guidelines for the treatment of CAP.3 These guidelines recommend a minimum five-day course of antibiotics if the patient is clinically stable (defined as afebrile for 48 h; heart rate ≤ 100 beats/min; respiratory rate ≤ 24 breaths/min-1
However, these recommendations are not routinely followed. Practitioners often make it their custom to prescribe longer courses of antibiotics.4 Yet, we know that there are several reasons to consider shorter courses of antibiotics, including lower health care costs, fewer adverse effects, and lower rates of bacterial resistance.5-7
Two meta-analyses were performed to compare the safety and efficacy of short- and long-course antibiotic therapy in CAP (≤ 7 d vs > 7 d, respectively).8,9 Both meta-analyses found no difference in efficacy or safety between shorter and longer courses of antibiotics for CAP. Secondary outcomes noted a trend toward decreased antibiotic-associated adverse events with shorter courses of therapy.8,9
However, there are limitations to broad implementation. Studies included in these analyses utilized a variety of antibiotic treatment regimens and longer courses (7 d vs 5 d) that are not recommended by the IDSA/ATS guidelines. Additionally, studies included both inpatient and outpatient treatment groups, so findings may not apply to an exclusively inpatient CAP population.8,9
This study sought to validate the IDSA/ATS guidelines recommending a five-day course of antibiotics for hospitalized patients with CAP.1
STUDY SUMMARY
No differences in clinical outcomes
This multicenter, double-blind, noninferiority randomized trial compared short-term antibiotic treatment duration (5 d) to physician-discretion antibiotic treatment duration among 312 patients (ages 18 and older) admitted for CAP to one of four teaching hospitals in Spain.1 Pneumonia was diagnosed on chest radiograph with at least one symptom: cough, fever, dyspnea, or chest pain. Patients were excluded if, among other things, they had an immunocompromising condition, lived in a nursing home, had a recent hospital stay, used antibiotics within the previous 30 days, or had an uncommon pathogen, such as Pseudomonas aeruginosa or Staphylococcus aureus.1
After receiving a minimum of five days of antibiotics, patients were randomly assigned to an intervention group (where, if clinically stable, no further antibiotics were given) or a control group (where physicians determined antibiotic duration).1
Primary outcomes were clinical success rate at days 10 and 30 from admission (defined as resolution of signs and symptoms of CAP without further antibiotics) and improvement of CAP-related symptoms (as determined by an 18-item questionnaire scored 0-90, with higher scores indicative of greater severity). Secondary outcomes included duration of antibiotic use, time to clinical improvement, mortality, hospital readmission, hospital length of stay, and CAP recurrence.1
Of the 312 participants, 162 were randomized to the intervention group and 150 to the control group. Mean age of patients in the intervention and control groups was 66.2 and 64.7, respectively. Other baseline demographics were similar between the groups. Nearly 80% of patients received quinolone treatment; < 10% received a ß-lactam plus a macrolide.1
Clinical success rates were similar for the control and intervention groups at day 10 (49% vs 56%, respectively) and day 30 (89% vs 92%). Median antibiotic treatment duration was shorter in the intervention group than in the control group (5 d vs 10 d); the intervention group also had a lower rate of 30-day hospital readmissions (1.4% vs 6.6%). There were no differences for other secondary outcomes.1
WHAT’S NEW
Clinical support for 2007 guidelines
This is the first study to clinically support the IDSA/ATS guidelines, which state that a five-day course of antibiotic therapy for hospitalized adults with CAP is effective and without increased risk for adverse events.
CAVEATS
Generalizability is unclear
This study focused on antibiotic duration for the treatment of CAP in hospitalized patients and mainly used quinolone antibiotics. It remains unclear if duration of therapy is as effective in the outpatient setting or when using alternative antibiotic regimens.
If patients continued to have symptoms (eg, fever or low oxygen saturation on room air) after five days of antibiotics, treatment was continued in the study. Thus, patients in real life who continue to have symptoms may need individualized therapy and may require more than five days of antibiotics.
CHALLENGES TO IMPLEMENTATION
Antibiotics end before clinical improvement
In this study, it took an average of 12 days in both groups for patients to achieve clinical improvement, and upwards of 15 to 18 days for patients to return to normal activity. Patients and providers may be dissatisfied if the treatment course ends days before clinical improvement of symptoms. This may cause prescribers to lengthen the duration of antibiotic therapy inappropriately.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2017. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice (2017; 66[10]:629-631).
1. Uranga A, España PP, Bilbao A, et al. Duration of antibiotic treatment in community-acquired pneumonia: a multicenter randomized clinical trial. JAMA Intern Med. 2016;176:1257-1265.
2. World Health Organization. The top 10 causes of death. www.who.int/mediacentre/factsheets/fs310/en/index.html. Accessed October 18, 2017.
3. Mandell LA, Wunderink RG, Anzueto A, et al. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis. 2007;44(suppl 2):S27-S72.
4. Aliberti S, Blasi F, Zanaboni AM, et al. Duration of antibiotic therapy in hospitalised patients with community-acquired pneumonia. Eur Respir J. 2010;36:128-134.
5. Guillemot D, Carbon C, Balkau B, et al. Low dosage and long treatment duration of ß-lactam: risk factors for carriage of penicillin-resistant Streptococcus pneumoniae. JAMA. 1998; 279:365-370.
6. Opmeer BC, el Moussaoui R, Bossuyt PM, et al. Costs associated with shorter duration of antibiotic therapy in hospitalized patients with mild-to-moderate severe community-acquired pneumonia. J Antimicrob Chemother. 2007;60: 1131-1136.
7. File TM Jr. Clinical efficacy of newer agents in short-duration therapy for community-acquired pneumonia. Clin Infect Dis. 2004;39(suppl 3):S159-S164.
8. Li JZ, Winston LG, Moore DH, et al. Efficacy of short-course antibiotic regimens for community-acquired pneumonia: a meta-analysis. Am J Med. 2007;120:783-790.
9. Dimopoulos G, Matthaiou DK, Karageorgopoulos DE, et al. Short- versus long-course antibacterial therapy for community-acquired pneumonia: a meta-analysis. Drugs. 2008;68: 1841-1854.
1. Uranga A, España PP, Bilbao A, et al. Duration of antibiotic treatment in community-acquired pneumonia: a multicenter randomized clinical trial. JAMA Intern Med. 2016;176:1257-1265.
2. World Health Organization. The top 10 causes of death. www.who.int/mediacentre/factsheets/fs310/en/index.html. Accessed October 18, 2017.
3. Mandell LA, Wunderink RG, Anzueto A, et al. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis. 2007;44(suppl 2):S27-S72.
4. Aliberti S, Blasi F, Zanaboni AM, et al. Duration of antibiotic therapy in hospitalised patients with community-acquired pneumonia. Eur Respir J. 2010;36:128-134.
5. Guillemot D, Carbon C, Balkau B, et al. Low dosage and long treatment duration of ß-lactam: risk factors for carriage of penicillin-resistant Streptococcus pneumoniae. JAMA. 1998; 279:365-370.
6. Opmeer BC, el Moussaoui R, Bossuyt PM, et al. Costs associated with shorter duration of antibiotic therapy in hospitalized patients with mild-to-moderate severe community-acquired pneumonia. J Antimicrob Chemother. 2007;60: 1131-1136.
7. File TM Jr. Clinical efficacy of newer agents in short-duration therapy for community-acquired pneumonia. Clin Infect Dis. 2004;39(suppl 3):S159-S164.
8. Li JZ, Winston LG, Moore DH, et al. Efficacy of short-course antibiotic regimens for community-acquired pneumonia: a meta-analysis. Am J Med. 2007;120:783-790.
9. Dimopoulos G, Matthaiou DK, Karageorgopoulos DE, et al. Short- versus long-course antibacterial therapy for community-acquired pneumonia: a meta-analysis. Drugs. 2008;68: 1841-1854.
FDA approves brentuximab vedotin for primary cutaneous anaplastic large cell lymphoma
Approval was based on a 56% objective response rate for brentuximab vedotin versus 12% for physician’s choice in a phase 3 trial (ALCANZA) of 131 patients with mycosis fungoides or pcALCL. All patients had received one prior systemic therapy and were randomized (1:1) to receive either brentuximab vedotin or the physician’s choice of methotrexate or bexarotene, the Food and Drug Administration said in a press statement.
The most common adverse reactions for patients in the brentuximab vedotin arm were anemia, peripheral sensory neuropathy, nausea, diarrhea, fatigue, and neutropenia. The most common adverse event leading to discontinuation of brentuximab vedotin was peripheral neuropathy.
The recommended dose of brentuximab vedotin is 1.8 mg/kg up to a maximum of 180 mg/kg as an IV infusion over 30 minutes every 3 weeks until a maximum of 16 cycles, disease progression, or unacceptable toxicity, the FDA wrote.
Brentuximab vedotin is marketed as Adcetris by Seattle Genetics.
ALCANZA results were presented at ASH 2016 and published in the Lancet in Aug. 5, 2017.
Approval was based on a 56% objective response rate for brentuximab vedotin versus 12% for physician’s choice in a phase 3 trial (ALCANZA) of 131 patients with mycosis fungoides or pcALCL. All patients had received one prior systemic therapy and were randomized (1:1) to receive either brentuximab vedotin or the physician’s choice of methotrexate or bexarotene, the Food and Drug Administration said in a press statement.
The most common adverse reactions for patients in the brentuximab vedotin arm were anemia, peripheral sensory neuropathy, nausea, diarrhea, fatigue, and neutropenia. The most common adverse event leading to discontinuation of brentuximab vedotin was peripheral neuropathy.
The recommended dose of brentuximab vedotin is 1.8 mg/kg up to a maximum of 180 mg/kg as an IV infusion over 30 minutes every 3 weeks until a maximum of 16 cycles, disease progression, or unacceptable toxicity, the FDA wrote.
Brentuximab vedotin is marketed as Adcetris by Seattle Genetics.
ALCANZA results were presented at ASH 2016 and published in the Lancet in Aug. 5, 2017.
Approval was based on a 56% objective response rate for brentuximab vedotin versus 12% for physician’s choice in a phase 3 trial (ALCANZA) of 131 patients with mycosis fungoides or pcALCL. All patients had received one prior systemic therapy and were randomized (1:1) to receive either brentuximab vedotin or the physician’s choice of methotrexate or bexarotene, the Food and Drug Administration said in a press statement.
The most common adverse reactions for patients in the brentuximab vedotin arm were anemia, peripheral sensory neuropathy, nausea, diarrhea, fatigue, and neutropenia. The most common adverse event leading to discontinuation of brentuximab vedotin was peripheral neuropathy.
The recommended dose of brentuximab vedotin is 1.8 mg/kg up to a maximum of 180 mg/kg as an IV infusion over 30 minutes every 3 weeks until a maximum of 16 cycles, disease progression, or unacceptable toxicity, the FDA wrote.
Brentuximab vedotin is marketed as Adcetris by Seattle Genetics.
ALCANZA results were presented at ASH 2016 and published in the Lancet in Aug. 5, 2017.
Adjunctive minocycline for schizophrenia found beneficial
PARIS – A report that adjunctive minocycline was found safe and effective for treatment of schizophrenia must be considered one of the year’s highlights in the field of psychosis, Pascal Steullet, PhD, said at the annual congress of the European College of Neuropsychopharmacology.
The report came in the form of a meta-analysis conducted by investigators in China and Australia. This was the largest meta-analysis looking at the topic to date, and the only one to include a search of the Chinese language database, which provided three of the eight randomized, placebo-controlled clinical trials that were examined. Seven of the eight randomized trials were double-blind and deemed high quality by widely used criteria, including the Jadad scale and GRADE (Grading of Recommendations Assessment, Development and Evaluation) methodology, noted Dr. Steullet, a neuroscientist at the University of Lausanne (Switzerland).

The primary outcome was change in the PANSS (Positive and Negative Syndrome Scale) total psychopathology score, and the positive, negative, and general symptom subscale scores.
The biggest benefit was on PANSS negative symptoms. Minocycline brought significantly greater improvement in this domain than placebo, with a standard mean difference (SMD) of –0.69 and a P value of less than .00001 (Eur Psychopharmacol. 2017 Jan;27[1]:8-18).
“That’s quite a good effect size,” Dr. Steullet commented.
The benefit on PANSS positive symptoms, while statistically significant, was far less robust, with an SMD of –0.22 in favor of minocycline.
The PANSS total psychopathology score favored minocycline with an SMD of –0.64, which Dr. Steullet again deemed “a quite significant effect size.” The PANSS general symptom score also showed a significant benefit in favor of minocycline, with an SMD of –0.45.
Among various secondary endpoints that were evaluated: A significant benefit was found favoring minocycline on the Clinical Global Impressions scale (SMD of –0.53) and the Abnormal Involuntary Movement Scale (SMD of –0.56). However, no differences were found between the minocycline and control groups on the Global Assessment of Functioning Scale, the Extrapyramidal Symptom Rating Scale, or the Calgary Depression Scale for Schizophrenia.
Although the average dose of minocycline prescribed in the eight trials was 171.9 mg/day, dosing strategies varied widely from trial to trial, and the investigators concluded that future studies are needed in order to pin down the optimal dosing and duration.
There was no increase in adverse drug reactions in the minocycline-treated group.
Neurocognitive function as assessed by the MATRICS Consensus Cognitive Battery showed no differences between the minocycline and placebo groups in working memory, problem solving, attention/vigilance, or other elements.
Proposed mechanisms of minocycline’s benefit as adjunctive therapy alongside antipsychotics for schizophrenia include the antimicrobial’s good CNS penetration and its anti-inflammatory effects, which could reduce neuroinflammation, dampen activated microglia, and enhance glutamate neurotransmitters.
The meta-analysis was supported by the University of Macau. Its authors reported having no financial conflicts of interest. Dr. Steullet, who was not involved in the work, also reported having no financial conflicts of interest.
PARIS – A report that adjunctive minocycline was found safe and effective for treatment of schizophrenia must be considered one of the year’s highlights in the field of psychosis, Pascal Steullet, PhD, said at the annual congress of the European College of Neuropsychopharmacology.
The report came in the form of a meta-analysis conducted by investigators in China and Australia. This was the largest meta-analysis looking at the topic to date, and the only one to include a search of the Chinese language database, which provided three of the eight randomized, placebo-controlled clinical trials that were examined. Seven of the eight randomized trials were double-blind and deemed high quality by widely used criteria, including the Jadad scale and GRADE (Grading of Recommendations Assessment, Development and Evaluation) methodology, noted Dr. Steullet, a neuroscientist at the University of Lausanne (Switzerland).
The primary outcome was change in the PANSS (Positive and Negative Syndrome Scale) total psychopathology score, and the positive, negative, and general symptom subscale scores.
The biggest benefit was on PANSS negative symptoms. Minocycline brought significantly greater improvement in this domain than placebo, with a standard mean difference (SMD) of –0.69 and a P value of less than .00001 (Eur Psychopharmacol. 2017 Jan;27[1]:8-18).
“That’s quite a good effect size,” Dr. Steullet commented.
The benefit on PANSS positive symptoms, while statistically significant, was far less robust, with an SMD of –0.22 in favor of minocycline.
The PANSS total psychopathology score favored minocycline with an SMD of –0.64, which Dr. Steullet again deemed “a quite significant effect size.” The PANSS general symptom score also showed a significant benefit in favor of minocycline, with an SMD of –0.45.
Among various secondary endpoints that were evaluated: A significant benefit was found favoring minocycline on the Clinical Global Impressions scale (SMD of –0.53) and the Abnormal Involuntary Movement Scale (SMD of –0.56). However, no differences were found between the minocycline and control groups on the Global Assessment of Functioning Scale, the Extrapyramidal Symptom Rating Scale, or the Calgary Depression Scale for Schizophrenia.
Although the average dose of minocycline prescribed in the eight trials was 171.9 mg/day, dosing strategies varied widely from trial to trial, and the investigators concluded that future studies are needed in order to pin down the optimal dosing and duration.
There was no increase in adverse drug reactions in the minocycline-treated group.
Neurocognitive function as assessed by the MATRICS Consensus Cognitive Battery showed no differences between the minocycline and placebo groups in working memory, problem solving, attention/vigilance, or other elements.
Proposed mechanisms of minocycline’s benefit as adjunctive therapy alongside antipsychotics for schizophrenia include the antimicrobial’s good CNS penetration and its anti-inflammatory effects, which could reduce neuroinflammation, dampen activated microglia, and enhance glutamate neurotransmitters.
The meta-analysis was supported by the University of Macau. Its authors reported having no financial conflicts of interest. Dr. Steullet, who was not involved in the work, also reported having no financial conflicts of interest.
PARIS – A report that adjunctive minocycline was found safe and effective for treatment of schizophrenia must be considered one of the year’s highlights in the field of psychosis, Pascal Steullet, PhD, said at the annual congress of the European College of Neuropsychopharmacology.
The report came in the form of a meta-analysis conducted by investigators in China and Australia. This was the largest meta-analysis looking at the topic to date, and the only one to include a search of the Chinese language database, which provided three of the eight randomized, placebo-controlled clinical trials that were examined. Seven of the eight randomized trials were double-blind and deemed high quality by widely used criteria, including the Jadad scale and GRADE (Grading of Recommendations Assessment, Development and Evaluation) methodology, noted Dr. Steullet, a neuroscientist at the University of Lausanne (Switzerland).
The primary outcome was change in the PANSS (Positive and Negative Syndrome Scale) total psychopathology score, and the positive, negative, and general symptom subscale scores.
The biggest benefit was on PANSS negative symptoms. Minocycline brought significantly greater improvement in this domain than placebo, with a standard mean difference (SMD) of –0.69 and a P value of less than .00001 (Eur Psychopharmacol. 2017 Jan;27[1]:8-18).
“That’s quite a good effect size,” Dr. Steullet commented.
The benefit on PANSS positive symptoms, while statistically significant, was far less robust, with an SMD of –0.22 in favor of minocycline.
The PANSS total psychopathology score favored minocycline with an SMD of –0.64, which Dr. Steullet again deemed “a quite significant effect size.” The PANSS general symptom score also showed a significant benefit in favor of minocycline, with an SMD of –0.45.
Among various secondary endpoints that were evaluated: A significant benefit was found favoring minocycline on the Clinical Global Impressions scale (SMD of –0.53) and the Abnormal Involuntary Movement Scale (SMD of –0.56). However, no differences were found between the minocycline and control groups on the Global Assessment of Functioning Scale, the Extrapyramidal Symptom Rating Scale, or the Calgary Depression Scale for Schizophrenia.
Although the average dose of minocycline prescribed in the eight trials was 171.9 mg/day, dosing strategies varied widely from trial to trial, and the investigators concluded that future studies are needed in order to pin down the optimal dosing and duration.
There was no increase in adverse drug reactions in the minocycline-treated group.
Neurocognitive function as assessed by the MATRICS Consensus Cognitive Battery showed no differences between the minocycline and placebo groups in working memory, problem solving, attention/vigilance, or other elements.
Proposed mechanisms of minocycline’s benefit as adjunctive therapy alongside antipsychotics for schizophrenia include the antimicrobial’s good CNS penetration and its anti-inflammatory effects, which could reduce neuroinflammation, dampen activated microglia, and enhance glutamate neurotransmitters.
The meta-analysis was supported by the University of Macau. Its authors reported having no financial conflicts of interest. Dr. Steullet, who was not involved in the work, also reported having no financial conflicts of interest.
EXPERT ANALYSIS FROM THE ECNP CONGRESS
Late-Breaking Science preview: Wednesday, Nov. 15
The final Late-Breaking Science session delves into innovative therapies and novel applications, including two phase 1-2 stem cell trials, an early trial in toxin treatments to prevent atrial fibrillation, a phase 1 test of an interatrial shunt device for heart failure with preserved ejection fraction, and more:
- TNT-POAF: Nathan Waldron, MD, of Duke University, Durham, N.C., will present results of a trial aiming to prevent postoperative atrial fibrillation with the use of temporary toxin treatment.
- REDUCE LAP–HF 1: In what the investigators call the first randomized controlled trial of a device-based therapy to reduce left atrial pressure in HFpEF, an inter-atrial shunt device designed to provide continuous and dynamic decompression of the left atrium. Sanjiv J Shah, MD, of Northwestern University will present the study, which holds the hypothesis that the device may reduce symptoms and slow the progression of heart failure.
- PROPEL: This study tested the hypothesis that granulocyte-macrophage colony-stimulating factor (GM-CSF) combined with supervised treadmill exercise in patients with peripheral artery disease would significantly improve functional performance more than GM-CSF alone or supervised treadmill exercise alone. Mary McDermott, MD, of Northwestern University, Chicago, will present the primary endpoint of change in 6-minute walk performance at 12-weeks’ follow-up, as well as several secondary outcomes.
- ALLSTAR: Timothy Henry, MD, of the Cedars-Sinai Heart Institute, Los Angeles, will present the phase 1-2 ALLSTAR (Allogeneic Heart Stem Cells to Achieve Myocardial Regeneration) study, which compared allogeneic cardiosphere-derived cells (CAP-1002) to placebo in order to find whether it is safe and effective in decreasing infarct size in patients with an MI.
- HOPE-Duchenne: This phase 1-2 study randomized men with cardiomyopathy secondary to Duchenne muscular dystrophy to receive CAP-1002 cells or usual care; its primary outcome is safety. Ronald Victor, MD, will present the results.
[email protected]
On Twitter @cardionews
The final Late-Breaking Science session delves into innovative therapies and novel applications, including two phase 1-2 stem cell trials, an early trial in toxin treatments to prevent atrial fibrillation, a phase 1 test of an interatrial shunt device for heart failure with preserved ejection fraction, and more:
- TNT-POAF: Nathan Waldron, MD, of Duke University, Durham, N.C., will present results of a trial aiming to prevent postoperative atrial fibrillation with the use of temporary toxin treatment.
- REDUCE LAP–HF 1: In what the investigators call the first randomized controlled trial of a device-based therapy to reduce left atrial pressure in HFpEF, an inter-atrial shunt device designed to provide continuous and dynamic decompression of the left atrium. Sanjiv J Shah, MD, of Northwestern University will present the study, which holds the hypothesis that the device may reduce symptoms and slow the progression of heart failure.
- PROPEL: This study tested the hypothesis that granulocyte-macrophage colony-stimulating factor (GM-CSF) combined with supervised treadmill exercise in patients with peripheral artery disease would significantly improve functional performance more than GM-CSF alone or supervised treadmill exercise alone. Mary McDermott, MD, of Northwestern University, Chicago, will present the primary endpoint of change in 6-minute walk performance at 12-weeks’ follow-up, as well as several secondary outcomes.
- ALLSTAR: Timothy Henry, MD, of the Cedars-Sinai Heart Institute, Los Angeles, will present the phase 1-2 ALLSTAR (Allogeneic Heart Stem Cells to Achieve Myocardial Regeneration) study, which compared allogeneic cardiosphere-derived cells (CAP-1002) to placebo in order to find whether it is safe and effective in decreasing infarct size in patients with an MI.
- HOPE-Duchenne: This phase 1-2 study randomized men with cardiomyopathy secondary to Duchenne muscular dystrophy to receive CAP-1002 cells or usual care; its primary outcome is safety. Ronald Victor, MD, will present the results.
[email protected]
On Twitter @cardionews
The final Late-Breaking Science session delves into innovative therapies and novel applications, including two phase 1-2 stem cell trials, an early trial in toxin treatments to prevent atrial fibrillation, a phase 1 test of an interatrial shunt device for heart failure with preserved ejection fraction, and more:
- TNT-POAF: Nathan Waldron, MD, of Duke University, Durham, N.C., will present results of a trial aiming to prevent postoperative atrial fibrillation with the use of temporary toxin treatment.
- REDUCE LAP–HF 1: In what the investigators call the first randomized controlled trial of a device-based therapy to reduce left atrial pressure in HFpEF, an inter-atrial shunt device designed to provide continuous and dynamic decompression of the left atrium. Sanjiv J Shah, MD, of Northwestern University will present the study, which holds the hypothesis that the device may reduce symptoms and slow the progression of heart failure.
- PROPEL: This study tested the hypothesis that granulocyte-macrophage colony-stimulating factor (GM-CSF) combined with supervised treadmill exercise in patients with peripheral artery disease would significantly improve functional performance more than GM-CSF alone or supervised treadmill exercise alone. Mary McDermott, MD, of Northwestern University, Chicago, will present the primary endpoint of change in 6-minute walk performance at 12-weeks’ follow-up, as well as several secondary outcomes.
- ALLSTAR: Timothy Henry, MD, of the Cedars-Sinai Heart Institute, Los Angeles, will present the phase 1-2 ALLSTAR (Allogeneic Heart Stem Cells to Achieve Myocardial Regeneration) study, which compared allogeneic cardiosphere-derived cells (CAP-1002) to placebo in order to find whether it is safe and effective in decreasing infarct size in patients with an MI.
- HOPE-Duchenne: This phase 1-2 study randomized men with cardiomyopathy secondary to Duchenne muscular dystrophy to receive CAP-1002 cells or usual care; its primary outcome is safety. Ronald Victor, MD, will present the results.
[email protected]
On Twitter @cardionews