User login
Acne severity shows negative impact on women’s self esteem
The severity of acne has a substantial negative impact on patients’ self-esteem in different age groups and cultures, with the greatest impact on women and in those with more severe acne, according to the authors of a review of studies that evaluated self-esteem in patients with acne.
Dermatologists Stephanie M. Gallitano, MD, of Columbia University, and Diane S. Berson, MD, of Cornell University, both in New York City, conducted a literature search of studies using the terms “acne vulgaris” and “self-esteem.” They identified 13 studies in 11 countries, including India, Singapore, Brazil, Greece, the United Kingdom, Egypt, South Korea, and Australia.
In the review, published in the International Journal of Women’s Dermatology, they wrote that most of the study authors determined that women with acne were more likely to have a greater degree of feelings that involved self-consciousness, lower self-esteem, and self-worth. In four studies, though, “men and women were equally affected by their disease,” and in one study in Egypt, self-esteem was significantly lower in males with acne than in women with acne, they wrote.
In all the studies, “self-esteem became lower as severity of acne increased,” they observed. There was evidence that subjective perceptions of acne severity affected self-esteem, with a few studies associating subjective evaluations of acne severity, but not objective evaluations, with lower self-esteem. They included a study of 550 students in Turkey, which found “a significant relationship between subjective acne severity and self-esteem but no relationship between objective acne severity and self-esteem.”
Most – between 70% and 80% – of patients used self-prescribed treatments, and 5% to almost 30% had seen a dermatologist. But patients with more severe acne were more likely to have seen a dermatologist, with 61% of those with moderate to severe acne having seen a dermatologist in a Greek study, for example. In addition, in an adult study and in an adolescent study, patients who felt that they had benefited from treatment also had improved self-esteem, improved quality of life, and less anxiety compared with those who did not feel they had benefited from treatment, the authors wrote.
Their review shows that acne “has a substantial negative impact on patients’ self-esteem” and that more severe acne and acne in women “tend to have the greatest impact across cultures,” they concluded.
Read the full study in the International Journal of Women’s Dermatology.
SOURCE: Gallitano S.M, et al. Int J Womens Dermatol 2017 Oct. doi: 10.1016/j.ijwd.2017.10.004
The severity of acne has a substantial negative impact on patients’ self-esteem in different age groups and cultures, with the greatest impact on women and in those with more severe acne, according to the authors of a review of studies that evaluated self-esteem in patients with acne.
Dermatologists Stephanie M. Gallitano, MD, of Columbia University, and Diane S. Berson, MD, of Cornell University, both in New York City, conducted a literature search of studies using the terms “acne vulgaris” and “self-esteem.” They identified 13 studies in 11 countries, including India, Singapore, Brazil, Greece, the United Kingdom, Egypt, South Korea, and Australia.
In the review, published in the International Journal of Women’s Dermatology, they wrote that most of the study authors determined that women with acne were more likely to have a greater degree of feelings that involved self-consciousness, lower self-esteem, and self-worth. In four studies, though, “men and women were equally affected by their disease,” and in one study in Egypt, self-esteem was significantly lower in males with acne than in women with acne, they wrote.
In all the studies, “self-esteem became lower as severity of acne increased,” they observed. There was evidence that subjective perceptions of acne severity affected self-esteem, with a few studies associating subjective evaluations of acne severity, but not objective evaluations, with lower self-esteem. They included a study of 550 students in Turkey, which found “a significant relationship between subjective acne severity and self-esteem but no relationship between objective acne severity and self-esteem.”
Most – between 70% and 80% – of patients used self-prescribed treatments, and 5% to almost 30% had seen a dermatologist. But patients with more severe acne were more likely to have seen a dermatologist, with 61% of those with moderate to severe acne having seen a dermatologist in a Greek study, for example. In addition, in an adult study and in an adolescent study, patients who felt that they had benefited from treatment also had improved self-esteem, improved quality of life, and less anxiety compared with those who did not feel they had benefited from treatment, the authors wrote.
Their review shows that acne “has a substantial negative impact on patients’ self-esteem” and that more severe acne and acne in women “tend to have the greatest impact across cultures,” they concluded.
Read the full study in the International Journal of Women’s Dermatology.
SOURCE: Gallitano S.M, et al. Int J Womens Dermatol 2017 Oct. doi: 10.1016/j.ijwd.2017.10.004
The severity of acne has a substantial negative impact on patients’ self-esteem in different age groups and cultures, with the greatest impact on women and in those with more severe acne, according to the authors of a review of studies that evaluated self-esteem in patients with acne.
Dermatologists Stephanie M. Gallitano, MD, of Columbia University, and Diane S. Berson, MD, of Cornell University, both in New York City, conducted a literature search of studies using the terms “acne vulgaris” and “self-esteem.” They identified 13 studies in 11 countries, including India, Singapore, Brazil, Greece, the United Kingdom, Egypt, South Korea, and Australia.
In the review, published in the International Journal of Women’s Dermatology, they wrote that most of the study authors determined that women with acne were more likely to have a greater degree of feelings that involved self-consciousness, lower self-esteem, and self-worth. In four studies, though, “men and women were equally affected by their disease,” and in one study in Egypt, self-esteem was significantly lower in males with acne than in women with acne, they wrote.
In all the studies, “self-esteem became lower as severity of acne increased,” they observed. There was evidence that subjective perceptions of acne severity affected self-esteem, with a few studies associating subjective evaluations of acne severity, but not objective evaluations, with lower self-esteem. They included a study of 550 students in Turkey, which found “a significant relationship between subjective acne severity and self-esteem but no relationship between objective acne severity and self-esteem.”
Most – between 70% and 80% – of patients used self-prescribed treatments, and 5% to almost 30% had seen a dermatologist. But patients with more severe acne were more likely to have seen a dermatologist, with 61% of those with moderate to severe acne having seen a dermatologist in a Greek study, for example. In addition, in an adult study and in an adolescent study, patients who felt that they had benefited from treatment also had improved self-esteem, improved quality of life, and less anxiety compared with those who did not feel they had benefited from treatment, the authors wrote.
Their review shows that acne “has a substantial negative impact on patients’ self-esteem” and that more severe acne and acne in women “tend to have the greatest impact across cultures,” they concluded.
Read the full study in the International Journal of Women’s Dermatology.
SOURCE: Gallitano S.M, et al. Int J Womens Dermatol 2017 Oct. doi: 10.1016/j.ijwd.2017.10.004
FROM THE INTERNATIONAL JOURNAL OF WOMEN’S DERMATOLOGY
CRB-410 update: Multiple myeloma response rates remain high with bb2121 CAR T-cell therapy
ATLANTA – A novel chimeric antigen receptor (CAR) T-cell therapy that targets B-cell maturation antigen showed promising efficacy with a manageable adverse event profile in heavily pretreated patients with relapsed/refractory multiple myeloma in the CRB-410 multicenter phase 1 dose escalation trial.
The product, known as bb2121, received breakthrough therapy designation from the Food and Drug Administration in November 2017 based on preliminary data from the ongoing trial. Those data showed that as of May 2017, the overall response rate at 1 month in 18 evaluable patients was 89%, whereas the response in those who received active dosing (150 x 106 CAR+ T cells or higher) was 100%.

Multiple myeloma currently is “essentially incurable,” and new treatments are desperately needed; B-cell maturation antigen (BCMA) – which is a member of the tumor necrosis factor superfamily that is expressed primarily by malignant myeloma cells, plasma cells, and some mature B cells – is a promising target, said Dr. Kochenderfer of the National Cancer Institute, Bethesda, Md.
The bb2121 product is a second-generation CAR construct targeting BCMA to redirect T cells to multiple myeloma cells. It was tested at doses of 50, 150, 450, and 800 x 106 CAR+ T cells in patients who first underwent chemotherapy as a conditioning regimen to enhance the activity of the CAR T cells.
A total of 24 patients were enrolled, but three had clinical deterioration and were not dosed. The remaining 21 patients had a median age of 58 years, performance scores of 0 or 1, and a median of 5 years since multiple myeloma diagnosis. A high percentage (43%) had high-risk cytogenetics. The median number of prior lines of therapy was seven, and all patients had undergone prior autologous stem cell transplant.
“Generally, this was a very well tolerated CAR T-cell product, especially in comparison to other protocols that I’ve participated in,” he said, noting that the incidence of adverse events, including dose-limiting toxicities, was the primary outcome measure of this phase of the study.
Cytokine release syndrome occurred in 71% of the 21 patients evaluable for response with a median follow-up of 35 weeks at the Oct. 2, 2017, data cutoff, but was grade 3 or greater in just 10% of those patients. Neurological toxicity occurred in 24% of patients, as well, but no cases were grade 3 or above, he said.
“The neurotoxicity was generally much milder and less prevalent than what I’ve seen in previous anti-CD19 CAR studies,” he said.
Neutropenia, thrombocytopenia, and anemia also occurred, but there were no dose-limiting toxicities observed during dose escalation.
Five deaths occurred. Three were due to disease progression and occurred in patients on the lowest dose (50 x 106 CAR+ T cells), which was deemed inactive. The other deaths occurred in patients receiving higher (active) doses; one was a result of myelodysplastic syndrome, and one from cardiac arrest, he said.
One or more serious adverse events occurred in 14 patients, and in some cases were characterized as such due to strict study protocols, Dr. Kochenderfer said.
Of note, one patient out of 12 in an ongoing dose expansion phase of the study, for which data have not yet been fully reported, experienced a delayed onset reversible grade 4 neurological toxicity associated with tumor lysis syndrome and cytokine release syndrome. The patient, who had the highest disease burden in the trial, completely recovered and has obtained a very good partial response despite low BCMA expression on the myeloma cells, Dr. Kochenderfer said.
In terms of response rates, 17 of 18 patients who received doses above 50 x 106 CAR+ T cells had overall responses, and 10 of the 18 achieved complete remission.
The median time to first response was 1 month, and the times to best response and complete response were 3.74 and 3.84 months, respectively. The rates of progression-free survival were 81% at 6 months, and 71% at 9 months, and responses deepened over time: as of May, the complete response rate was 27%, and as of October, it was 56%.
“Five of these patients so far have met the 1-year progression-free survival standard,” Dr. Kochenderfer said, adding that responses have endured for more than a year in several patients. The longest was 68 weeks at the time of the data presentation, and responses continued to improve as late as 15 months, with very good partial remission to complete remission transitions.
The median progression-free survival had not been reached in the active dose cohorts.
“So, in general, very impressive responses compared to my previous experience treating multiple myeloma,” he said.
The findings support the potential of CAR T therapy with bb2121 as a new treatment paradigm in relapsed/refractory multiple myeloma, he concluded, noting that a global pivotal trial of bb2121 (the phase 2 KarMMa trial) is now enrolling and will dose patients at between 150 and 350 x 106 CAR+ T cells. Under the breakthrough therapy designation granted for bb2121, the product will receive expedited review by the FDA.The CRB-410 trial is sponsored by bluebird bio and Celgene. Dr. Kochenderfer reported receiving research funding from bluebird bio and Kite Pharma, and having multiple patents in the CAR field.
ATLANTA – A novel chimeric antigen receptor (CAR) T-cell therapy that targets B-cell maturation antigen showed promising efficacy with a manageable adverse event profile in heavily pretreated patients with relapsed/refractory multiple myeloma in the CRB-410 multicenter phase 1 dose escalation trial.
The product, known as bb2121, received breakthrough therapy designation from the Food and Drug Administration in November 2017 based on preliminary data from the ongoing trial. Those data showed that as of May 2017, the overall response rate at 1 month in 18 evaluable patients was 89%, whereas the response in those who received active dosing (150 x 106 CAR+ T cells or higher) was 100%.
Multiple myeloma currently is “essentially incurable,” and new treatments are desperately needed; B-cell maturation antigen (BCMA) – which is a member of the tumor necrosis factor superfamily that is expressed primarily by malignant myeloma cells, plasma cells, and some mature B cells – is a promising target, said Dr. Kochenderfer of the National Cancer Institute, Bethesda, Md.
The bb2121 product is a second-generation CAR construct targeting BCMA to redirect T cells to multiple myeloma cells. It was tested at doses of 50, 150, 450, and 800 x 106 CAR+ T cells in patients who first underwent chemotherapy as a conditioning regimen to enhance the activity of the CAR T cells.
A total of 24 patients were enrolled, but three had clinical deterioration and were not dosed. The remaining 21 patients had a median age of 58 years, performance scores of 0 or 1, and a median of 5 years since multiple myeloma diagnosis. A high percentage (43%) had high-risk cytogenetics. The median number of prior lines of therapy was seven, and all patients had undergone prior autologous stem cell transplant.
“Generally, this was a very well tolerated CAR T-cell product, especially in comparison to other protocols that I’ve participated in,” he said, noting that the incidence of adverse events, including dose-limiting toxicities, was the primary outcome measure of this phase of the study.
Cytokine release syndrome occurred in 71% of the 21 patients evaluable for response with a median follow-up of 35 weeks at the Oct. 2, 2017, data cutoff, but was grade 3 or greater in just 10% of those patients. Neurological toxicity occurred in 24% of patients, as well, but no cases were grade 3 or above, he said.
“The neurotoxicity was generally much milder and less prevalent than what I’ve seen in previous anti-CD19 CAR studies,” he said.
Neutropenia, thrombocytopenia, and anemia also occurred, but there were no dose-limiting toxicities observed during dose escalation.
Five deaths occurred. Three were due to disease progression and occurred in patients on the lowest dose (50 x 106 CAR+ T cells), which was deemed inactive. The other deaths occurred in patients receiving higher (active) doses; one was a result of myelodysplastic syndrome, and one from cardiac arrest, he said.
One or more serious adverse events occurred in 14 patients, and in some cases were characterized as such due to strict study protocols, Dr. Kochenderfer said.
Of note, one patient out of 12 in an ongoing dose expansion phase of the study, for which data have not yet been fully reported, experienced a delayed onset reversible grade 4 neurological toxicity associated with tumor lysis syndrome and cytokine release syndrome. The patient, who had the highest disease burden in the trial, completely recovered and has obtained a very good partial response despite low BCMA expression on the myeloma cells, Dr. Kochenderfer said.
In terms of response rates, 17 of 18 patients who received doses above 50 x 106 CAR+ T cells had overall responses, and 10 of the 18 achieved complete remission.
The median time to first response was 1 month, and the times to best response and complete response were 3.74 and 3.84 months, respectively. The rates of progression-free survival were 81% at 6 months, and 71% at 9 months, and responses deepened over time: as of May, the complete response rate was 27%, and as of October, it was 56%.
“Five of these patients so far have met the 1-year progression-free survival standard,” Dr. Kochenderfer said, adding that responses have endured for more than a year in several patients. The longest was 68 weeks at the time of the data presentation, and responses continued to improve as late as 15 months, with very good partial remission to complete remission transitions.
The median progression-free survival had not been reached in the active dose cohorts.
“So, in general, very impressive responses compared to my previous experience treating multiple myeloma,” he said.
The findings support the potential of CAR T therapy with bb2121 as a new treatment paradigm in relapsed/refractory multiple myeloma, he concluded, noting that a global pivotal trial of bb2121 (the phase 2 KarMMa trial) is now enrolling and will dose patients at between 150 and 350 x 106 CAR+ T cells. Under the breakthrough therapy designation granted for bb2121, the product will receive expedited review by the FDA.The CRB-410 trial is sponsored by bluebird bio and Celgene. Dr. Kochenderfer reported receiving research funding from bluebird bio and Kite Pharma, and having multiple patents in the CAR field.
ATLANTA – A novel chimeric antigen receptor (CAR) T-cell therapy that targets B-cell maturation antigen showed promising efficacy with a manageable adverse event profile in heavily pretreated patients with relapsed/refractory multiple myeloma in the CRB-410 multicenter phase 1 dose escalation trial.
The product, known as bb2121, received breakthrough therapy designation from the Food and Drug Administration in November 2017 based on preliminary data from the ongoing trial. Those data showed that as of May 2017, the overall response rate at 1 month in 18 evaluable patients was 89%, whereas the response in those who received active dosing (150 x 106 CAR+ T cells or higher) was 100%.
Multiple myeloma currently is “essentially incurable,” and new treatments are desperately needed; B-cell maturation antigen (BCMA) – which is a member of the tumor necrosis factor superfamily that is expressed primarily by malignant myeloma cells, plasma cells, and some mature B cells – is a promising target, said Dr. Kochenderfer of the National Cancer Institute, Bethesda, Md.
The bb2121 product is a second-generation CAR construct targeting BCMA to redirect T cells to multiple myeloma cells. It was tested at doses of 50, 150, 450, and 800 x 106 CAR+ T cells in patients who first underwent chemotherapy as a conditioning regimen to enhance the activity of the CAR T cells.
A total of 24 patients were enrolled, but three had clinical deterioration and were not dosed. The remaining 21 patients had a median age of 58 years, performance scores of 0 or 1, and a median of 5 years since multiple myeloma diagnosis. A high percentage (43%) had high-risk cytogenetics. The median number of prior lines of therapy was seven, and all patients had undergone prior autologous stem cell transplant.
“Generally, this was a very well tolerated CAR T-cell product, especially in comparison to other protocols that I’ve participated in,” he said, noting that the incidence of adverse events, including dose-limiting toxicities, was the primary outcome measure of this phase of the study.
Cytokine release syndrome occurred in 71% of the 21 patients evaluable for response with a median follow-up of 35 weeks at the Oct. 2, 2017, data cutoff, but was grade 3 or greater in just 10% of those patients. Neurological toxicity occurred in 24% of patients, as well, but no cases were grade 3 or above, he said.
“The neurotoxicity was generally much milder and less prevalent than what I’ve seen in previous anti-CD19 CAR studies,” he said.
Neutropenia, thrombocytopenia, and anemia also occurred, but there were no dose-limiting toxicities observed during dose escalation.
Five deaths occurred. Three were due to disease progression and occurred in patients on the lowest dose (50 x 106 CAR+ T cells), which was deemed inactive. The other deaths occurred in patients receiving higher (active) doses; one was a result of myelodysplastic syndrome, and one from cardiac arrest, he said.
One or more serious adverse events occurred in 14 patients, and in some cases were characterized as such due to strict study protocols, Dr. Kochenderfer said.
Of note, one patient out of 12 in an ongoing dose expansion phase of the study, for which data have not yet been fully reported, experienced a delayed onset reversible grade 4 neurological toxicity associated with tumor lysis syndrome and cytokine release syndrome. The patient, who had the highest disease burden in the trial, completely recovered and has obtained a very good partial response despite low BCMA expression on the myeloma cells, Dr. Kochenderfer said.
In terms of response rates, 17 of 18 patients who received doses above 50 x 106 CAR+ T cells had overall responses, and 10 of the 18 achieved complete remission.
The median time to first response was 1 month, and the times to best response and complete response were 3.74 and 3.84 months, respectively. The rates of progression-free survival were 81% at 6 months, and 71% at 9 months, and responses deepened over time: as of May, the complete response rate was 27%, and as of October, it was 56%.
“Five of these patients so far have met the 1-year progression-free survival standard,” Dr. Kochenderfer said, adding that responses have endured for more than a year in several patients. The longest was 68 weeks at the time of the data presentation, and responses continued to improve as late as 15 months, with very good partial remission to complete remission transitions.
The median progression-free survival had not been reached in the active dose cohorts.
“So, in general, very impressive responses compared to my previous experience treating multiple myeloma,” he said.
The findings support the potential of CAR T therapy with bb2121 as a new treatment paradigm in relapsed/refractory multiple myeloma, he concluded, noting that a global pivotal trial of bb2121 (the phase 2 KarMMa trial) is now enrolling and will dose patients at between 150 and 350 x 106 CAR+ T cells. Under the breakthrough therapy designation granted for bb2121, the product will receive expedited review by the FDA.The CRB-410 trial is sponsored by bluebird bio and Celgene. Dr. Kochenderfer reported receiving research funding from bluebird bio and Kite Pharma, and having multiple patents in the CAR field.
REPORTING FROM ASH 2017
Key clinical point:
Major finding: The overall response rate was 94%.
Study details: An update from the phase 1 CRB-410 dose trial of 21 patients.
Disclosures: The CRB-410 trial is sponsored by bluebird bio and Celgene. Dr. Kochenderfer reported receiving research funding from bluebird bio and Kite Pharma, and having multiple patents in the CAR field.
Source: Berdeja J et al. ASH 2017 Abstract 740.

Medicaid’s share of state budgets continues to grow
State spending on Medicaid in fiscal 2017 was up 6.1% over 2016, and the program’s share of state budgets increased for the fifth year in a row, according to the National Association of State Budget Officers.
Total spending by the states on Medicaid benefits for more than 74 million individuals was an estimated $574 billion in 2017, which represented 29% of all expenditures. That compares with 28.7% in 2016 and 23.6% in 2012 – the last year that Medicaid’s share of state spending decreased, NASBO said in its annual State Expenditure Report.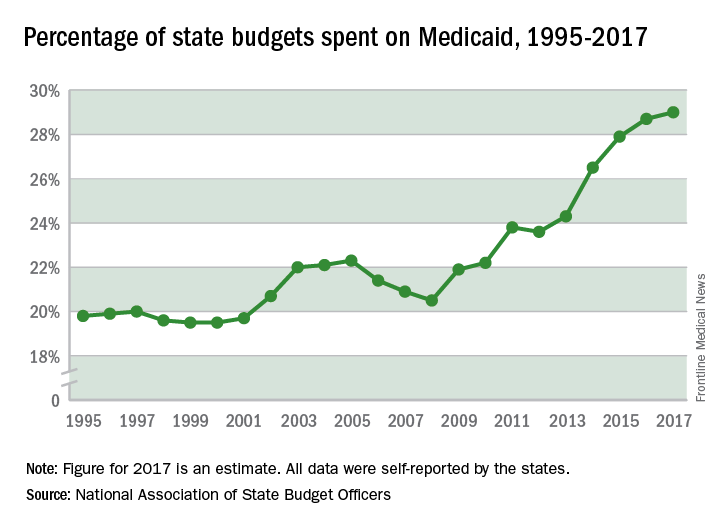
Enrollment rose by 2.9% from 2016 to 2017, which was down from the 3.9% increase seen from 2015 to 2016. Since October 2013, Medicaid enrollment is up 38% in expansion states and 12% in nonexpansion states, the report noted.
Enrollment increases are “the major driver” of spending growth, NASBO said, and “Medicaid’s annual spending growth from all fund sources has averaged 8.1% over the past 5 years, while the rest of total spending growth has averaged 2.2% annually.”
State spending on Medicaid in fiscal 2017 was up 6.1% over 2016, and the program’s share of state budgets increased for the fifth year in a row, according to the National Association of State Budget Officers.
Total spending by the states on Medicaid benefits for more than 74 million individuals was an estimated $574 billion in 2017, which represented 29% of all expenditures. That compares with 28.7% in 2016 and 23.6% in 2012 – the last year that Medicaid’s share of state spending decreased, NASBO said in its annual State Expenditure Report.
Enrollment rose by 2.9% from 2016 to 2017, which was down from the 3.9% increase seen from 2015 to 2016. Since October 2013, Medicaid enrollment is up 38% in expansion states and 12% in nonexpansion states, the report noted.
Enrollment increases are “the major driver” of spending growth, NASBO said, and “Medicaid’s annual spending growth from all fund sources has averaged 8.1% over the past 5 years, while the rest of total spending growth has averaged 2.2% annually.”
State spending on Medicaid in fiscal 2017 was up 6.1% over 2016, and the program’s share of state budgets increased for the fifth year in a row, according to the National Association of State Budget Officers.
Total spending by the states on Medicaid benefits for more than 74 million individuals was an estimated $574 billion in 2017, which represented 29% of all expenditures. That compares with 28.7% in 2016 and 23.6% in 2012 – the last year that Medicaid’s share of state spending decreased, NASBO said in its annual State Expenditure Report.
Enrollment rose by 2.9% from 2016 to 2017, which was down from the 3.9% increase seen from 2015 to 2016. Since October 2013, Medicaid enrollment is up 38% in expansion states and 12% in nonexpansion states, the report noted.
Enrollment increases are “the major driver” of spending growth, NASBO said, and “Medicaid’s annual spending growth from all fund sources has averaged 8.1% over the past 5 years, while the rest of total spending growth has averaged 2.2% annually.”
Guselkumab crushes skin disease in psoriatic arthritis patients
GENEVA – The interleukin-23 inhibitor guselkumab generates the same impressive improvement in skin disease in psoriatic arthritis patients as has been seen in psoriasis without joint disease, Alexa B. Kimball, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
However, psoriatic arthritis patients’ improvement in Dermatology Life Quality Index (DLQI) scores is less robust than in patients with psoriasis only, added Dr. Kimball, professor of dermatology at Harvard Medical School, Boston, and CEO of Harvard Medical Faculty Physicians at Beth Israel Deaconess Medical Center.

The psoriatic arthritis group as a whole had more severe psoriasis, with a baseline mean PASI score of 24.3 and involvement of 32.7% of their body surface area as compared with a PASI score of 21.2 and 27.2% BSA in psoriasis patients without arthritis. A total of 28% of the psoriatic arthritis patients had previously been on other biologics and 77% had been on nonbiologic systemic agents, compared with 19% and 60% of the psoriasis patients, respectively. The psoriatic arthritis group had a mean 19.2-year history of psoriasis, 1.9 years longer than the psoriasis-only group.
Participants were randomized to 100 mg of guselkumab administered subcutaneously at weeks 0, 4, 12, and 20; placebo through week 12, followed by a switch to adalimumab (Humira); or adalimumab at 80 mg at week 0, then 40 mg at week 2 and 40 mg again every 2 weeks until week 23.
The key findings:
The PASI 90 response rate – that is, at least a 90% improvement in Psoriasis Area and Severity Index – in guselkumab-treated patients at week 16 was 72% in patients with psoriatic arthritis and 71% in those without. At week 24, the PASI 90 rate was 74% in guselkumab-treated patients with psoriatic arthritis and similar at 78% in those without. In contrast, the PASI 90 rate at week 24 in patients on adalimumab was significantly lower: 48% in the psoriatic arthritis group and 55% in those with psoriasis only. The PASI 90 rate in placebo-treated controls was single digit.
At week 24, 82% of psoriatic arthritis patients on guselkumab had clear or almost clear skin as reflected in an Investigator’s Global Assessment score of 0 or 1, as did 84% of psoriasis-only patients.
A DLQI score of 0 or 1, meaning the dermatologic disease had no impact on patient quality of life, was documented at week 16 in 46% of psoriatic arthritis patients and 55% of psoriasis-only patients, a trend that didn’t achieve statistical significance. However, by week 24 the difference became significant, with a DLQI of 0 or 1 in 48% of the psoriatic arthritis patients, compared with 62% of psoriasis-only patients.
VOYAGE 1 and 2 were dermatologic studies that didn’t measure changes in joint symptom scores or other psoriatic arthritis outcomes. Guselkumab as a potential treatment for psoriatic arthritis is under investigation in other studies.
The VOYAGE trials and this analysis were sponsored by Janssen. Dr. Kimball reported receiving research funding from and serving as a consultant to Janssen and numerous other pharmaceutical companies.
SOURCE: Kimball A et al. https://eadvgeneva2017.org/
GENEVA – The interleukin-23 inhibitor guselkumab generates the same impressive improvement in skin disease in psoriatic arthritis patients as has been seen in psoriasis without joint disease, Alexa B. Kimball, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
However, psoriatic arthritis patients’ improvement in Dermatology Life Quality Index (DLQI) scores is less robust than in patients with psoriasis only, added Dr. Kimball, professor of dermatology at Harvard Medical School, Boston, and CEO of Harvard Medical Faculty Physicians at Beth Israel Deaconess Medical Center.
The psoriatic arthritis group as a whole had more severe psoriasis, with a baseline mean PASI score of 24.3 and involvement of 32.7% of their body surface area as compared with a PASI score of 21.2 and 27.2% BSA in psoriasis patients without arthritis. A total of 28% of the psoriatic arthritis patients had previously been on other biologics and 77% had been on nonbiologic systemic agents, compared with 19% and 60% of the psoriasis patients, respectively. The psoriatic arthritis group had a mean 19.2-year history of psoriasis, 1.9 years longer than the psoriasis-only group.
Participants were randomized to 100 mg of guselkumab administered subcutaneously at weeks 0, 4, 12, and 20; placebo through week 12, followed by a switch to adalimumab (Humira); or adalimumab at 80 mg at week 0, then 40 mg at week 2 and 40 mg again every 2 weeks until week 23.
The key findings:
The PASI 90 response rate – that is, at least a 90% improvement in Psoriasis Area and Severity Index – in guselkumab-treated patients at week 16 was 72% in patients with psoriatic arthritis and 71% in those without. At week 24, the PASI 90 rate was 74% in guselkumab-treated patients with psoriatic arthritis and similar at 78% in those without. In contrast, the PASI 90 rate at week 24 in patients on adalimumab was significantly lower: 48% in the psoriatic arthritis group and 55% in those with psoriasis only. The PASI 90 rate in placebo-treated controls was single digit.
At week 24, 82% of psoriatic arthritis patients on guselkumab had clear or almost clear skin as reflected in an Investigator’s Global Assessment score of 0 or 1, as did 84% of psoriasis-only patients.
A DLQI score of 0 or 1, meaning the dermatologic disease had no impact on patient quality of life, was documented at week 16 in 46% of psoriatic arthritis patients and 55% of psoriasis-only patients, a trend that didn’t achieve statistical significance. However, by week 24 the difference became significant, with a DLQI of 0 or 1 in 48% of the psoriatic arthritis patients, compared with 62% of psoriasis-only patients.
VOYAGE 1 and 2 were dermatologic studies that didn’t measure changes in joint symptom scores or other psoriatic arthritis outcomes. Guselkumab as a potential treatment for psoriatic arthritis is under investigation in other studies.
The VOYAGE trials and this analysis were sponsored by Janssen. Dr. Kimball reported receiving research funding from and serving as a consultant to Janssen and numerous other pharmaceutical companies.
SOURCE: Kimball A et al. https://eadvgeneva2017.org/
GENEVA – The interleukin-23 inhibitor guselkumab generates the same impressive improvement in skin disease in psoriatic arthritis patients as has been seen in psoriasis without joint disease, Alexa B. Kimball, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
However, psoriatic arthritis patients’ improvement in Dermatology Life Quality Index (DLQI) scores is less robust than in patients with psoriasis only, added Dr. Kimball, professor of dermatology at Harvard Medical School, Boston, and CEO of Harvard Medical Faculty Physicians at Beth Israel Deaconess Medical Center.
The psoriatic arthritis group as a whole had more severe psoriasis, with a baseline mean PASI score of 24.3 and involvement of 32.7% of their body surface area as compared with a PASI score of 21.2 and 27.2% BSA in psoriasis patients without arthritis. A total of 28% of the psoriatic arthritis patients had previously been on other biologics and 77% had been on nonbiologic systemic agents, compared with 19% and 60% of the psoriasis patients, respectively. The psoriatic arthritis group had a mean 19.2-year history of psoriasis, 1.9 years longer than the psoriasis-only group.
Participants were randomized to 100 mg of guselkumab administered subcutaneously at weeks 0, 4, 12, and 20; placebo through week 12, followed by a switch to adalimumab (Humira); or adalimumab at 80 mg at week 0, then 40 mg at week 2 and 40 mg again every 2 weeks until week 23.
The key findings:
The PASI 90 response rate – that is, at least a 90% improvement in Psoriasis Area and Severity Index – in guselkumab-treated patients at week 16 was 72% in patients with psoriatic arthritis and 71% in those without. At week 24, the PASI 90 rate was 74% in guselkumab-treated patients with psoriatic arthritis and similar at 78% in those without. In contrast, the PASI 90 rate at week 24 in patients on adalimumab was significantly lower: 48% in the psoriatic arthritis group and 55% in those with psoriasis only. The PASI 90 rate in placebo-treated controls was single digit.
At week 24, 82% of psoriatic arthritis patients on guselkumab had clear or almost clear skin as reflected in an Investigator’s Global Assessment score of 0 or 1, as did 84% of psoriasis-only patients.
A DLQI score of 0 or 1, meaning the dermatologic disease had no impact on patient quality of life, was documented at week 16 in 46% of psoriatic arthritis patients and 55% of psoriasis-only patients, a trend that didn’t achieve statistical significance. However, by week 24 the difference became significant, with a DLQI of 0 or 1 in 48% of the psoriatic arthritis patients, compared with 62% of psoriasis-only patients.
VOYAGE 1 and 2 were dermatologic studies that didn’t measure changes in joint symptom scores or other psoriatic arthritis outcomes. Guselkumab as a potential treatment for psoriatic arthritis is under investigation in other studies.
The VOYAGE trials and this analysis were sponsored by Janssen. Dr. Kimball reported receiving research funding from and serving as a consultant to Janssen and numerous other pharmaceutical companies.
SOURCE: Kimball A et al. https://eadvgeneva2017.org/
REPORTING FROM THE EADV CONGRESS
Key clinical point:
Major finding: After 16 weeks on guselkumab, 72% of psoriatic arthritis patients and 71% with psoriasis-only had a PASI 90 response.
Study details: This was a comparison of skin and DLQI outcomes in 335 patients with psoriatic arthritis and 1,494 with psoriasis only who participated in two randomized, double-blind, phase 3 clinical trials.
Disclosures: Janssen sponsored the study. The presenter reported receiving research grants from and serving as a consultant to Janssen and numerous other pharmaceutical companies.
Source: Kimball A et al. https://eadvgeneva2017.org/.
Transition in care from the MICU to the ward
Editor’s Note: The Society of Hospital Medicine’s (SHM’s) Physician in Training Committee launched a scholarship program in 2015 for medical students to help transform healthcare and revolutionize patient care. The program has been expanded for the 2017-18 year, offering two options for students to receive funding and engage in scholarly work during their first, second and third years of medical school. As a part of the program, recipients are required to write about their experience on a biweekly basis.
This summer, my research project focused on the highly vulnerable patients who are transferred from the medical intensive care unit to the general floor. Patients who are readmitted tend to have worse health outcomes, longer stays, higher mortality rates, and higher health care costs. Previous research shows that higher quality handoffs, where receiving and transferring providers share the same shared mental model, result in better outcomes. We were interested in learning whether these shared mental models are being formed as a result of handoffs between the ward and the MICU.
The current results reveal that 18% of MICU teams shared a complete mental model, 25% shared a strong shared mental model, 9% shared a weak mental model, 30% shared no mental model, and 18% of patient encounters did not have a sufficient number of MICU respondents. Regarding inter-team communication, 7% shared a full shared mental model, 49% shared a partial mental model, 30% shared no shared mental model, and 14% of unique patient encounters did not have enough respondents.
With complex patient cases, it can be difficult to identify the most important factor of care for a particular patient. However, I think this information would be very useful in identifying whether these exchanges result in individuals prioritizing the same factor of care for their respective patient. I think this information would be very useful in future quality improvement, and seeing whether this communication results in the formation of shared mental models.
Anton Garazha is a medical student at Chicago Medical School at Rosalind Franklin University in North Chicago. He received his bachelor of science degree in biology from Loyola University in Chicago in 2015 and his master of biomedical science degree from Rosalind Franklin University in 2016. Anton is very interested in community outreach and quality improvement, and in his spare time tutors students in science-based subjects.
Editor’s Note: The Society of Hospital Medicine’s (SHM’s) Physician in Training Committee launched a scholarship program in 2015 for medical students to help transform healthcare and revolutionize patient care. The program has been expanded for the 2017-18 year, offering two options for students to receive funding and engage in scholarly work during their first, second and third years of medical school. As a part of the program, recipients are required to write about their experience on a biweekly basis.
This summer, my research project focused on the highly vulnerable patients who are transferred from the medical intensive care unit to the general floor. Patients who are readmitted tend to have worse health outcomes, longer stays, higher mortality rates, and higher health care costs. Previous research shows that higher quality handoffs, where receiving and transferring providers share the same shared mental model, result in better outcomes. We were interested in learning whether these shared mental models are being formed as a result of handoffs between the ward and the MICU.
The current results reveal that 18% of MICU teams shared a complete mental model, 25% shared a strong shared mental model, 9% shared a weak mental model, 30% shared no mental model, and 18% of patient encounters did not have a sufficient number of MICU respondents. Regarding inter-team communication, 7% shared a full shared mental model, 49% shared a partial mental model, 30% shared no shared mental model, and 14% of unique patient encounters did not have enough respondents.
With complex patient cases, it can be difficult to identify the most important factor of care for a particular patient. However, I think this information would be very useful in identifying whether these exchanges result in individuals prioritizing the same factor of care for their respective patient. I think this information would be very useful in future quality improvement, and seeing whether this communication results in the formation of shared mental models.
Anton Garazha is a medical student at Chicago Medical School at Rosalind Franklin University in North Chicago. He received his bachelor of science degree in biology from Loyola University in Chicago in 2015 and his master of biomedical science degree from Rosalind Franklin University in 2016. Anton is very interested in community outreach and quality improvement, and in his spare time tutors students in science-based subjects.
Editor’s Note: The Society of Hospital Medicine’s (SHM’s) Physician in Training Committee launched a scholarship program in 2015 for medical students to help transform healthcare and revolutionize patient care. The program has been expanded for the 2017-18 year, offering two options for students to receive funding and engage in scholarly work during their first, second and third years of medical school. As a part of the program, recipients are required to write about their experience on a biweekly basis.
This summer, my research project focused on the highly vulnerable patients who are transferred from the medical intensive care unit to the general floor. Patients who are readmitted tend to have worse health outcomes, longer stays, higher mortality rates, and higher health care costs. Previous research shows that higher quality handoffs, where receiving and transferring providers share the same shared mental model, result in better outcomes. We were interested in learning whether these shared mental models are being formed as a result of handoffs between the ward and the MICU.
The current results reveal that 18% of MICU teams shared a complete mental model, 25% shared a strong shared mental model, 9% shared a weak mental model, 30% shared no mental model, and 18% of patient encounters did not have a sufficient number of MICU respondents. Regarding inter-team communication, 7% shared a full shared mental model, 49% shared a partial mental model, 30% shared no shared mental model, and 14% of unique patient encounters did not have enough respondents.
With complex patient cases, it can be difficult to identify the most important factor of care for a particular patient. However, I think this information would be very useful in identifying whether these exchanges result in individuals prioritizing the same factor of care for their respective patient. I think this information would be very useful in future quality improvement, and seeing whether this communication results in the formation of shared mental models.
Anton Garazha is a medical student at Chicago Medical School at Rosalind Franklin University in North Chicago. He received his bachelor of science degree in biology from Loyola University in Chicago in 2015 and his master of biomedical science degree from Rosalind Franklin University in 2016. Anton is very interested in community outreach and quality improvement, and in his spare time tutors students in science-based subjects.
Cancer care in 2017: the promise of more cures with the challenges of an unstable health care system
This past year will likely be remembered as one of breakthrough advances in reducing the burden of cancer, with some landmark “firsts” coming out of the US Food and Drug Admini
Our excitement has also been tempered by the rapid rise in the cost of effective biologic, immunologic, and targeted therapies. With the approval of trastuzumab-dkst (Ogivri), the first targeted biosimilar for HER2-positive breast and gastrointestinal cancers, we can look forward to price decreases possibly in the 20%-30% range over time from a targeted therapy with remarkable clinical efficacy. We know that approved biosimilars have demonstrated clinical efficacy along with similar minor biologic diversity that is also seen in the reference biologic.1 We can also hope that increasing competition among biosimilar and reference compounds will lead to improvements in production methodologies that can allow further price reductions so that even more patients can gain access to these highly effective therapies.
In addition, the first FDA approval for the next-generation sequencing (NGS) FoundationOne profiling test and the rapid announcement by the Centers for Medicare & Medicaid Services (CMS) that it will cover the cost of that testing brings us a step closer to knowing which patients most likely will or won’t benefit from costly and toxic targeted therapies. Along with the many clinical trials studying which mutations predict which efficacies of individual or combinations of targeted agents, the approval and CMS coverage policy will help us improve value to our patients; when we can recommend the most beneficial therapies and avoid futile ones.
Finally, the approval for the DigniCap Scalp Cooling System for patients on chemotherapy for all solid tumors is of great importance. Pending coverage availability, it may influence some patients to get chemotherapy they might otherwise have forgone to avoid hair loss (see related article).
More consolidation: the best of all worlds?
In my 27 years in private practice, during which practice revenues grew with the favorable profit margins on novel therapies, forward-thinking physician leaders piloted innovations in oncology electronic medical records (EMRs), the delivery of team-based care, clinical research partnerships, and more comprehensive care services to better serve diverse communities, including those in rural areas. At my previous practice, that included adding clinicians to our group to serve patients at hospital clinics in 2 counties in southern California, each county with populations larger than 15 states. Our private practice worked with these public entities to bring state-of-the art care and private practice efficiencies to the uninsured and underserved in our region.
Unfortunately, revenues plummeted with changes in reimbursement after passage of the Medicare Modernization Act in 2003 and they continue to destabilize and reduce the number of community practices across the country. Many oncologists and oncology practices, including mine, chose to join larger academic or hospital systems or larger oncology networks at a time they are also facing growing pressures to contain costs, focus on out-patient care, complex clinical trials, and expanded access to care.
Although we may lament the shrinking landscape of private oncology practices, we can also be inspired by the physicians who have joined ranks with the better-funded, better-resourced, more traditional hospital and academic systems. These larger systems have more resources, more clincial trial offerings, staffing, technology, and analytics to expand value-based care initiatives to larger numbers of patients.
The hub-and-spoke models of oncology care with integrated networks linked by technology, and networked into larger analytic and decision support systems such as CancerLinQ, the health information technology program of the American Society of Clinical Oncology (ASCO),2 could facilitate documentable delivery of comprehensive, evidence-based care, moving us closer to meeting the Quadruple Aim of optimal health care: improving the patient experience of care (including quality and satisfaction); improving the health of populations; reducing the per capita cost of health care; and improving the work life of those who deliver care.3,4
Payment reform: working to align incentives
Everyone seems to agree that the fee-for-service payment models do not align incentives for improving total health outcomes at the lowest costs, but at the moment, there seems to be no best way of aligning them. Robinson has reported on the oncology payment initiatives at four major health insurance plans – Medicare (public) and Anthem, Aetna, and UnitedHealthcare (all private), noting that:5
- Medicare is testing its Oncology Care Model at more than 200 sites in the United States, and early data are expected to be released in 2018.
- Anthem continues with its Cancer Care Quality Program that includes adherence to 2 key requirements: that participants are compliant with Anthem-approved drug pathways, and that they register their patients at the insurer’s oncology website and enter their clinical data. Anthem is also considering expanding the management fee for certain high priority clinical trials.
- Aetna’s Oncology Solutions takes a different approach by providing increased payments for generic chemotherapies.
- United has eliminated the mark-up for new drugs and continues to mark up the prices of the older and generic therapies. Its episode-based pricing gives practices upfront payments based on expected drug margins so that practices can fund more comprehensive evidence-based care. In a presentation at a Washington State Medical Oncology Society meeting recently, United’s Lee Newcomer, reported that the insurer continues to see improved clinical and financial outcomes as well as encouraging early data showing that patients might do better in the real-world setting on some therapies that have not been fully compared in head-to-head randomized clinical trials.6,7
ASCO is pulling these ideas together at the national level with its Patient-Centered Oncology Payment (PCOP) model, which is similar to Medicare’s alternative payment model. The PCOP model focuses on high-value, quality care. Higher upfront payments would cover the additional diagnostic services, care planning, and management to improve compliance and adherence as well as clinical trial evaluations. The model was developed and vetted by the ASCO Clinical Practice Committee and practicing oncologists, and is supported by staff and consultants. It is currently in its second year of operation with a commercial payer and will be submitted for review to the Physician-Focused Payment Model Technical Advisory Committee of the Health and Human Services. The results of the review are expected in 2018. If the model is approved, it could provide a uniform approach for payers that would align incentives for high-quality cancer care and allow for better predictive modeling for practices, irrespective of size, to invest in infrastructure and staffing to meet the growing demand for high-quality, value-based cancer care.
Better science: the promise of more cures
The FDA approved a record number drugs and biologics in 2017 for various cancers,8 including the landmark approval of the first CART therapy for cancer, tisagenlecleucel, which targets CD19 on B cells in the treatment of acute leukemia. That approval was rapidly followed by a second anti-CD19 CART therapy, axicabtagene ciloluecel, for refractory, aggressive B-cell non-Hodgkin lymphoma.9,10 Although these therapies can achieve remarkable response and even complete response rates in otherwise refractory patients, only some achieve a long-term remission, and the costs are an order of magnitude above most other cancer therapies. That raises the question of what duration of benefit we should expect for treatments that cost in the range of $500,000 for the therapy alone, along with the additional costs for care, hospitalization, monitoring, expensive biologics (eg, tocilizumab, for the severe and potentially life-threatening cytokine-release syndrome associated with CART therapies), and significant neurologic and other therapy-related toxicities.
Novel arrangements between pharmaceutical companies and payers are currently being discussed so that only patients who meet specific response criteria would be charged for the therapy. In addition, we await findings from ongoing research to see if new approaches can find specific targetable sites on solid tumors that could spare the healthy organ tissues while eliminating highly resistant or heterogeneous populations of mutations in patients with advanced solid tumors. Such development of highly specific targets for CART therapies would improve their efficacy and safety, and with defined protocols in place to address toxicities and efforts to reduce the costs of the therapies, we can hopefully ensure broader access for patients to this potentially transformative therapeutic tool.
In addition to the excitement around the CART therapies, many of the years other new approvals will bring incremental but meaningful improvement in outcomes for patients with common cancers. The approval of neratinib, the first agent approved as extended adjuvant therapy for women with early-stage HER2/neu-positive breast cancer, is welcome, given the current 30% recurrence risk that extends past 10 years for women in that disease population who have completed standard adjuvant HER2-directed therapies. The 34% reduction in recurrence risk with a year of extended oral adjuvant therapy, as reported by Martin and colleagues,11 with benefits sustained out to 5 years and with controllable diarrhea as the major toxicity, are encouraging. This oral therapy may be especially beneficial for hormone-receptor–positive women in whom blocking the HER2/neu pathway may enhance cell signaling through the hormone pathways, which can be blocked with oral agents at the same time to provide significant reduction of recurrence risk.
Diagnostics
The concept of personalized medicine is based on identifying biomarkers that are predictive of a patient’s response to treatment. There has been much progress toward applying NGS of tumors for use in the clinic, but we are still awaiting evidence from randomized clinical trials that such approaches prolong overall or progression-free survival.12 Dr Julie Lange, an associate professor of clinical surgery and director of the Breast Cancer Program at the Keck School of Medicine at the University of Southern California, Los Angeles, provided me with the references to key studies in this field in which she is a leading researcher.13 However, she pointed out that in the absence of effective therapies, advanced biomarker testing may be less helpful, as is the case in heavily pretreated patients,14 unless a molecular test can pinpoint a potentially clinically actionable mutation. With the plethora of available assays and the high costs of molecular testing, clinicians are challenged in knowing what testing is best for which patients. Findings from a number of key ongoing national trials may eventually help us understand which tumor mutations in which tumor types can be most effectively targeted when multiple targetable mutations are found (TAPUR,15 MATCH,16 and QUILT17 and other basket trials18). The complexity of molecular testing has led to the development of institutional, trial-based, or co-operative group molecular tumor boards to provide guidance on specific targeted therapies for specific tumor mutations.
ASCO has launched a monthly series called Molecular Oncology Tumor Boards19 to expand the knowledge base in this field. It is presented as user-driven discussions designed to help providers integrate the use of the new genetic and genomic tests and their results into the day-to-day clinical care of patients with cancer.20
Liquid biopsies
As busy clinicians, we need to understand the differences in liquid biopsy tests and their correlation with actionable targets, especially given the rapid progress in this field. Again, Dr Lange offered clarity on those differences. Liquid biopsy, refers to using a blood draw to isolate circulating tumor cells (CTCs) or circulating tumor DNA (ctDNA) to assess tumor biomarkers.21 Both CTCs and ctDNA tests have been shown to be prognostic of worse survival.22-24 Liquid biopsies are currently supplemental to direct tumor biopsies, not replacements for them. The theoretical advantage of liquid biopsies is that they may reflect tumor heterogeneity by examining the repertoire of mutations contributed by diverse metastatic sites that shed CTCs or ctDNA into the circulation. The question is which type of testing can best inform therapy decisions.
Assays for ctDNA using droplet digital PCR [polymerase chain reaction], a digital PCR method based on water-oil emulsion droplet technology, require a priori knowledge of the specific mutation associated with response or lack of response to a specific therapy.25,26 Technical issues related to the detection of rare alleles present within a mixed population of leukocytes, and ctDNA remains a challenge for many ctDNA assays. However, there is evidence to suggest that whole-exome sequencing of ctDNA is concordant with mutations in metastases,27 however benchmarking ctDNA against tissue biopsies of metastases was not possible in all studies because tumor blocks were not available or because of the failure of tumor NGS assays. 28,29
Newer generations of CTC assays take advantage of the circulating tumor cell as a functional assay for mutational status, gene expression, proteomics, epigenetics, and/or chemosensitivity of cultured cells. The relationship between CTCs and ctDNA remains uncertain as to whether CTCs are the cell of origin for ctDNA or if ctDNA may reflect responding or resistant tumor populations. The use of NGS on tumor specimens, ctDNA, and CTCs as a discovery tool is advancing the field by improving the understanding of disease heterogeneity and potential treatment targets. These results require correlation with patterns of response to therapy, and ultimately require validation in randomized clinical trials to provide strong evidence justifying their use outside of clinical trials. We can look forward to a time in the not distant future when specific liquid biopsy assays will reflect the array of mutations in different metastatic sites with validation that they correlate with efficacy of targeting those mutations that have targetable therapies.
From the FDA
New approvals
- Trastuzumab-dkst (Ogivri, Mylan; Dec 1) was approved as a biosimilar to trastuzumab (Herceptin, Genentech) for the treatment of patients with HER2-overexpressing breast or metastatic stomach cancer (gastric or gastroesophageal junction adenocarcinoma).
- Sunitinib malate (Sutent, Pfizer; Nov 16) was approved for the adjuvant treatment of adult patients at high risk of recurrent renal cell carcinoma after nephrectomy.
- Obinutuzumab (Gazyva, Genentech; Nov 16) received regular approval in combination with chemotherapy, followed by obinutuzumab monotherapy in patients achieving partial remission, for adult patients with previously untreated stage II bulky, III, or IV follicular lymphoma.
- Emicizumab-kxwh (Hemlibra, Genentech; Nov 16) was approved for routine prophylaxis to prevent or reduce the frequency of bleeding episodes in adult and pediatric patients with hemophilia A with factor VIII inhibitors.
- Dasatinib (Sprycel, Bristol-Myers Squibb; Nov 9) was approved for the treatment of pediatric patients with Philadelphia chromosome-positive chronic myeloid leukemia (CML) in the chronic phase.
- Brentuximab vedotin (Adcetris, Seattle Genetics; Nov 9) for the treatment of previously treated adult patients with primary cutaneous anaplastic large cell lymphoma (pcALCL) or CD30-expressing mycosis fungoides.
- Alectinib (Alecensa, Hoffmann-La Roche/Genentech; Nov 6) was approved for treatment of patients with anaplastic lymphoma kinase–positive metastatic non-small cell lung cancer (NSCLC), as detected by an FDA-approved test.
- Vemurafenib (Zelboraf, Hoffmann-La Roche; Nov 6) received approval for the treatment of Acalabrutinib (Calquence, AstraZeneca/Acerta; Oct 31) was granted accelerated approval for treatment of adult patients with mantle cell lymphoma (MCL) who have received at least one previous therapy.
- Axicabtagene ciloleucel (Yescarta, Kite; Oct 18), a CART therapy, was approved for treatment of adult patients with relapsed or refractory large B-cell lymphoma after two or more lines of systemic therapy, including diffuse large B-cell lymphoma (DLBCL) not otherwise specified, primary mediastinal large B-cell lymphoma, high-grade B-cell lymphoma, and DLBCL arising from follicular lymphoma. The complete remission rate reviewed by the FDA for trial patients was 51%.30 It was the second CART therapy this year to receive approval (see tisagenlecleucel; Aug 30). The agency granted orphan drug designation and priority review to therapy for this indication.
- Abemaciclib (Verzenio, Eli Lilly; Sep 28) was approved in combination with fulvestrant for women with hormone receptor-positive, HER2-negative advanced or metastatic breast cancer with disease progression following endocrine therapy.
- Copanlisib (Aliqopa, Bayer; Sep 14) got accelerated approval for the treatment of adult patients with relapsed follicular lymphoma who have received at least two prior systemic therapies.
- Bevacizumab-awwb (Mvasi, Amgen; Sep 14) was approved as a biosimilar to bevacizumab (Avastin, Genentech) for treating multiple types of cancer. It was the first biosimilar approved in the US for the treatment of cancer.
- Gemtuzumab ozogamicin (Mylotarg, Pfizer; Sep 1) was approved for the treatment of newly diagnosed CD33-positive acute myeloid leukemia (AML) in adults and of relapsed/refractory CD33-positive AML in adults and pediatric patients aged 2 or older. It can be used in combination with daunorubicin and cytarabine for adults with newly diagnosed AML, or as a standalone treatment for certain adult and pediatric patients. The drug was originally approved in 2000 as a standalone treatment for CD33-positive AML in patients older than 60 years, but was withdrawn in 2010 because of safety concerns and postmarketing trials could not confirm benefit. The current approval is for a lower recommended dose and schedule.31
- Tisagenlecleucel (Kymriah, Novartis; Aug 30) was approved for the treatment of patients up to age 25 years with B-cell precursor acute lymphoblastic leukemia (ALL) that is refractory or in second or later relapse. It is the first CART immunotherapy approved by the agency.
- Inotuzumab ozogamicin (Besponsa, Wyeth; Aug 17) was approved for the treatment of adults with relapsed or refractory B-cell precursor ALL.
- A liposome-encapsulated combination of daunorubicin and cytarabine (Vyxeos, Jazz; Aug 3) was approved for the treatment of adults with newly diagnosed therapy-related AML (t-AML) or AML with myelodysplasia-related changes (AML-MRC), two types of AML having a poor prognosis.
- Enasidenib (Idhifa, Celgene; Aug 1) was approved for the treatment of adult patients with relapsed or refractory AML with an isocitrate dehydrogenase-2 mutation as detected by an FDA-approved test.
- Neratinib (Nerlynx, Puma; Jul 17) was approved as the first extended adjuvant therapy for adult patients with early stage HER2-overexpressed/amplified breast cancer, to follow adjuvant trastuzumab-based therapy.
- Blinatumomab (Blincyto, Amgen; Jul 11) was approved for the treatment of relapsed or refractory B-cell precursor acute lymphoblastic leukemia in adults and children.
- L-glutamine oral powder (Endari, Emmaus; Jul 7) was approved for oral administration to reduce the acute complications of sickle cell disease in adult and pediatric patients 5 years and older.
- Betrixaban (Bevyxxa, Portola; Jun 23) was approved for the prophylaxis of venous thromboembolism (VTE) in adult patients hospitalized for an acute medical illness who are at risk for thromboembolic complications because of moderate or severe restricted mobility and other risk factors for VTE.
- The combination of rituximab and hyaluronidase human (Rituxan Hycela, Genentech; Jun 22) was approved for adult patients with follicular lymphoma, DLBCL, and chronic lymphocytic leukemia. Hyaluronidase human is an enzyme that helps deliver the rituximab. This formulation allows subcutaneous administration of the combination, which will shorten patient visit times and potentially even allow at-home therapy delivery.
- Ceritinib (Zykadia, Novartis; May 26) was approved for patients with metastatic NSCLC whose tumors are anaplastic lymphoma kinase (ALK)-positive as detected by an FDA-approved test.
- Avelumab (Bavencio, EMD Serono; May 9) got accelerated approval for patients with locally advanced or metastatic urothelial carcinoma whose disease progressed during or following platinum-containing chemotherapy or within 12 months of neoadjuvant or adjuvant platinum-containing chemotherapy.
- Durvalumab (ImfinzI, AstraZeneca; May 1) got accelerated approval for the treatment of patients with locally advanced or metastatic urothelial carcinoma who have disease progression during or following platinum-containing chemotherapy or who have disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy.
- Brigatinib (Alunbrig tablets, Takeda through Ariad; Apr 28) got accelerated approval for the treatment of patients with metastatic anaplastic lymphoma kinase (ALK)-positive NSCLC who have progressed on or are intolerant to crizotinib.
- Midostaurin (Rydapt, Novartis; Apr 28) was approved for the treatment of adult patients with newly diagnosed AML who are FLT3 mutation-positive, as detected by an FDA-approved test, in combination with standard cytarabine and daunorubicin induction and cytarabine consolidation.
- Osimertinib (Tagrisso, AstraZeneca; Mar 30) got regular approval for the treatment of patients with metastatic epidermal growth factor receptor (EGFR) T790M mutation-positive NSCLC, as detected by an FDA-approved test, whose disease has progressed on or after EGFR tyrosine-kinase inhibitor therapy.
- Niraparib (Zejula, Tesaro; Mar 27), a poly ADP-ribose polymerase (PARP) inhibitor, was approved for the maintenance treatment of adult patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in complete or partial response to platinum-based chemotherapy.
- Avelumab (Mar 23), a PD-L1–blocking human IgG1 lambda monoclonal antibody, got accelerated approval for the treatment of patients 12 years and older with metastatic Merkel cell carcinoma. It is the first FDA-approved product to treat this type of cancer.
- Ribociclib (Kisqali, Novartis; Mar 13), a CDK4/6 inhibitor, was approved as a breakthrough therapy after priority review for use in combination with an aromatase inhibitor as initial endocrine-based therapy for the treatment of postmenopausal women with HR-positive, HER2-negative advanced or metastatic breast cancer.
Expanded/additional indications
- Nivolumab (Opdivo, Bristol-Myers Squibb; Sep 22) got accelerated expanded indication approval for treatment of hepatocellular carcinoma (HCC) in patients previously treated with sorafenib.
- Pembrolizumab (Keytruda, Merck; Sep 22) got accelerated expanded indication approval for recurrent locally advanced or metastatic gastric or gastroesophageal junction adenocarcinoma in patients whose tumors express PD-L1 as determined by an FDA-approved test.
- DigniCap Scalp Cooling System (Dignitana Inc; Jul 3) was cleared for expanded use for reducing hair loss during chemotherapy for all solid tumors. Marketing authorization for the cooling cap had been granted in 2015 for patients with breast cancer.
- Olaparib tablets (Lynparza, AstraZeneca; Aug 17) got approval for an expanded indication as maintenance treatment of adult patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer, who are in a complete or partial response to platinum-based chemotherapy.
- Ibrutinib (Imbruvica, Pharmacyclics; Aug 2) got expanded indication approval for the treatment of adult patients with chronic graft-versus-host disease (cGVHD) after failure of one or more lines of systemic therapy. It was the first FDA-approved therapy for the treatment of cGVHD. (Ibrutinib was previously approved for chronic lymphocytic leukemia/small lymphocytic lymphoma, chronic lymphocytic leukemia/small lymphocytic lymphoma with 17p deletion, Waldenström’s macroglobulinemia, marginal zone lymphoma, and mantle cell lymphoma).
- Nivolumab (Aug 2) got an accelerated expanded indication for the treatment of patients 12 years and older with mismatch repair deficient (dMMR) and microsatellite instability-high (MSI-H) metastatic colorectal cancer that has progressed after treatment with a fluoropyrimidine, oxaliplatin, and irinotecan.
- Dabrafenib and trametinib (Tafinlar and Mekinist, Novartis; Jun 22) were approved for the expanded indication in combination for patients with metastatic NSCLC with BRAF V600E mutation as detected by an FDA-approved test. The combination demonstrated superior efficacy compared with dabrafenib alone (overall response rate: 61% and 27%, respectively).32
- Pembrolizumab (May 23) got approved for expanded indication for adult and pediatric patients with unresectable or metastatic, MSI-H or dMMR solid tumors that have progressed after treatment and who have no satisfactory alternative treatment options or with MSI-H or dMMR colorectal cancer that has progressed after treatment with a fluoropyrimidine, oxaliplatin, and irinotecan.
- Pembrolizumab (May 18) got approval for expanded indication for patients with locally advanced or metastatic urothelial carcinoma who have disease progression during or following platinum-containing chemotherapy or within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy.
- Pembrolizumab (May 10) got accelerated expanded indication for use combination with pemetrexed and carboplatin for the treatment of patients with previously untreated metastatic NSCLC.
- Regorafenib (Stivarga, Bayer; Apr 27) got an additional indication for the treatment of patients with HCC who have been previously treated with sorafenib.
- Palbociclib (Ibrance, Pfizer; Mar 31) got an expanded indication that includes first-line therapy for the treatment of hormone receptor–positive, HER2-negative advanced or metastatic breast cancer in combination with an aromatase inhibitor as initial endocrine based therapy in postmenopausal women.
- Pembrolizumab (Mar 15) got an accelerated additional indication approval for treatment of adult and pediatric patients with refractory classical Hodgkin lymphoma, or those who have relapsed after three or more previous lines of therapy.
- Lenalidomide (Revlimid, Celgene; Feb 22) got an additional indication as maintenance therapy for patients with multiple myeloma following autologous stem cell transplant.
- Nivolumab (Feb 2) got an accelerated expanded indication for treatment of patients with locally advanced or metastatic urothelial carcinoma who have disease progression during or following platinum-containing chemotherapy or have disease progression within 12 months of neoadjuvant or adjuvant treatment with a platinum-containing chemotherapy.
Modified use
- Cabazitaxel (Jevtana, Sanofi-Aventis; Sep 14) in combination with prednisone was approved at a lower dose of 20 mg/m2 every 3 weeks for the treatment of patients with metastatic castration-resistant prostate cancer previously treated with a docetaxel-containing treatment regimen. It had been approved at 25 mg/m2 every 3 weeks for this indication in 2010.
Tests/diagnostics
- Marketing approval was given to the FoundationOne CDx (Foundation Medicine; Nov 30), an NGS-based in vitro diagnostic to detect genetic mutations in 324 genes and 2 genomic signatures in any solid tumor type.
- Marketing approval was given to the Praxis Extended RAS Panel (Illumina; Jun 29), a next generation sequencing test to detect certain genetic mutations in RAS genes in tumor samples of patients with metastatic colorectal cancer. The test is used to aid in the identification of patients who may be eligible for treatment with panitumumab (Vectibix, Amgen).
- Marketing was approved for ipsogen JAK2 RGQ PCR Kit (Qiagen ; Mar 27) to detect mutations affecting the Janus tyrosine kinase 2 gene. This is the first FDA-authorized test intended to help physicians in evaluating patients for suspected polycythemia vera.
Imaging and pathology aids
- Aminolevulinic acid hydrochloride, known as ALA HCl (Gleolan, NX; Jun 6) was approved as an optical imaging agent indicated in patients with gliomas (suspected World Health Organization grades III or IV on preoperative imaging) as an adjunct for the visualization of malignant tissue during surgery.
- Marketing was approved for the Philips IntelliSite Pathology Solution (PIPS, Philips Medical Systems Nederland; Apr 17), as an aid to the pathologist to review and interpret digital images of surgical pathology slides prepared from formalin-fixed paraffin embedded tissue.
Challenges and uncertainties
The current administration’s initiatives to reduce administrative burdens is underway with the Patients Over Paperwork initiative. Eliminating and streamlining regulations to increase efficiency and improve beneficiary experience could be helpful to both oncologists and patients. For now, the Medicare Access and CHIP Reauthorization Act (MACRA) program, allows you to “pick your pace” in the 2017 performance year and report on at least one measure to avoid a payment reduction penalty on your Medicare payments in 2019. In the final rule for 2018, the CMS finalized a proposal to apply the MIPS [Merit-based Incentive Payment System] adjustment to all Part B items and services, which will include Part B drugs. This would be unfair to oncologists who treat on the basis of evidence-based guidelines and pathways and have no control over the costs of the drugs they prescribe.
In addition, more requirements will be imposed in 2018 in a move toward full MACRA implementation. All four composite categories (Quality – 60% for 2017; Advancing Care Information (ACI, renamed from Meaningful Use) – 25% for 2017; Improvement Activities (IA) – 15% for 2017; and Cost – 0% for 2017, but weighted in the future) will be scored, including resource use (cost) at 10%. CMS will collect data to assess the total cost of care and the Medicare Spend per Beneficiary to assess use. Full program implementation, with cost being assessed at 30% of your score is expected in the 2019 performance year. ASCO’s clinical affairs and policy experts have studied the implications of Part B chemotherapy drugs being included in the cost component of the MIPS scoring and will continue advocating for policies that hold clinicians responsible only for the aspects of care they can control, such as providing high-quality care based on the patient’s disease, biomarkers, comorbidities, and preferences, and not the costs of the evidence-based therapies needed by patients.
Toward a better 2018 for ourselves and our patients
As an eternal optimist, I remain enthusiastic that despite the many challenges, we will find effective ways to bring standard as well as newer, cell-based and targeted therapies to our patients and cover the costs of highly effective therapies. I also remain hopeful that improving technological capabilities and payment reforms will be used by innovative clinical and administrative care teams to give clinicians more time to improve the care and health of patients while validating the methodologies so that real world data can help us further craft therapies to improve the health of each individual who needs our care. As we close this 15th year of our journal, we hope our presentations of practical science and implementation content has helped support your work while freeing some time for you to enjoy the journey. Our best wishes for a joyful holiday season celebrated with friends and family and the patients who entrust us to help them face and live beyond their cancer diagnoses.
1. US Food & Drug Administration. Biosimilar and interchangeable
products. https://www.fda.gov/Drugs/DevelopmentApprovalProcess/
HowDrugsareDevelopedandApproved/ApprovalApplications/
¬erapeuticBiologicApplications/Biosimilars/ucm580419.htm. Last
updated October 23, 2017. Accessed December 11, 2017.
2. ASCO CancerLinQ website. https://cancerlinq.org/. Publishing/
update information not available. Accessed November 3, 2017.
3. Bodenheimer T, Sinsky C. From triple to quadruple aim: care of the
patient requires care of the provider. https://www.ncbi.nlm.nih.gov/
pmc/articles/PMC4226781/. Published November 2014. Accessed
November 7.
4. Institute for Healthcare Improvement. http://www.ihi.org/Engage/
Initiatives/TripleAim/Pages/default.aspx. No update information
given. Accessed November 3, 2017.
5. Robinson JC. Value-based physician payment in oncology: public and
private insurer initiatives. Milbank Q. 2017;95(1);184-203.
6. Newcomer L. Oral communication: Washington State Medical
Oncology Society meeting, August 19, 2017.
7. Newcomer LN, Gould B, Page RD, Donelan SA, Perkins M.
Changing physician incentives for aordable, quality cancer
care: results of an episode payment model. J Oncol Pract.
2014;10(5):322-326.
8. US Department of Health and Human Services website. Hematology/
oncology (cancer) approvals & safety notications. https://www.fda.
gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm279174.htm.
Last updated December 1, 2017. Accessed December 3, 2017.
9. Hem-Onc Today website. CAR T-cell therapy approval huge
step for oncology, but only ‘beginning of story’. https://www.
healio.com/hematology-oncology/leukemia/news/print/hemonctoday/%
7B33119631-5996-45cf-9be6-8e36466ded9e%7D/car-tcell-
therapy-approval-huge-step-for-oncology-but-only-beginningof-
story. Published September 25, 2017. Accessed November 9, 2017.
10. Gauthier J, Yakoub-Agha I. Chimeric antigen-receptor T-cell therapy
for hematological malignancies and solid tumors: Clinical data
to date, current limitations and perspectives. Curr Res in Transl Med.
2017;65(3):93-102.
11. Martin M, Holmes FA, Ejlertsen B, et al. Neratinib after trastuzumab-
based adjuvant therapy in early-stage HER2+ breast cancer:
5-year analysis of the phase III ExteNET trial [ESMO oral presentation].
Ann Oncol. 2017;28(suppl 5):v43-v67.
12. Le Tourneau C, Delord JP, Goncalves A, et al. Molecularly targeted
therapy based on tumour molecular proling versus conventional
therapy for advanced cancer SHIVA): a multicentre, openlabel,
proof-of-concept, randomised, controlled phase 2 trial. Lancet
Oncol. 2015;16(13):1324-1334.
13. Forte V, Barrak DK, Elhodaky M, Tung L, Snow A, Lang JE. ¬e
potential for liquid biopsies in the precision medical treatment of
breast cancer. Cancer Biol Med. 2016;13(1):19-40.
14. Smerage JB, Barlow WE, Hortobagyi GN, et al. Circulating tumor
cells and response to chemotherapy in metastatic breast cancer:
SWOG S0500. J Clin Oncol. 2014;32(31):3483-3489.
15. US National Library of Medicine. TAPUR: testing the use of food
and drug administration (FDA) approved drugs that target a specific
abnormality in a tumor gene in people with advanced stage cancer
(TAPUR). https://clinicaltrials.gov/ct2/show/NCT02693535.
First posted February 26, 2016; last updated September 18, 2017.
Accessed November 10, 2017.
16. US National Library of Medicine. NCI-MATCH: Targeted therapy
directed by genetic testing in treating patients with advanced refractory
solid tumors, lymphomas, or multiple myeloma. https://clinicaltrials.
gov/ct2/show/NCT02465060. First posted June 8, 2015; last
updated November 9, 2017. Accessed November 10, 2017.
17. US National Library of Medicine. QUILT-3.039: NANT Pancreatic
cancer vaccine: combination immunotherapy in subjects with pancreatic
cancer who have progressed on or after standard-of-care therapy.
https://clinicaltrials.gov/ct2/show/NCT03136406. First posted May
2, 2017; last updated October 30, 2017. Accessed November 10,
2017.
18. Cunanan KM, Gonen M, Shen R, et al. Basket trials in oncology:
A trade-o between complexity and eciency. J Clin Oncol.
2017;35(3):271-273.
19. ASCO website. https://university.asco.org/motb. Last update
November 2017. Accessed November 10, 2017.
20. ASCO website. Molecular oncology tumor boards invite discussion
of growing eld in cancer care. http://www.ascopost.com/issues/
february-25-2015/molecular-oncology-tumor-boards-invite-discussion-
of-growing-eld-in-cancer-care/. Published February 25, 2017.
Accessed November 10, 2017.
21. de Lartigue J. Liquid gold: blood-based biopsies make headway.
JCSO 2017;15(1):49-54.
22. Cristofanilli M, Budd GT, Ellis MJ, et al. Circulating tumor cells,
disease progression, and survival in metastatic breast cancer. N Engl J
Med. 2004;351(8):781-791.
23. Lucci A, Hall CS, Lodhi AK, et al. Circulating tumour cells in
non-metastatic breast cancer: a prospective study. Lancet Oncol.
2012;13(7):688-695.
24. Dawson SJ, Tsui DW, Murtaza M, et al. Analysis of circulating
tumor DNA to monitor metastatic breast cancer. N Engl J Med.
2013;368(13):1199-1209.
25. Chandarlapaty S, Chen D, He W, et al. Prevalence of ESR1 mutations
in cell-free DNA and outcomes in metastatic breast cancer: a
secondary analysis of the BOLERO-2 clinical trial. JAMA Oncol.
2016;2(10):1310-1315.
26. Kim SB, Dent R, Wongchenko WJ, et al. Concordance between
plasma-based and tissue-based next-generation sequencing in
LOTUS [Correspondence]. http://www.thelancet.com/journals/
lanonc/article/PIIS1470-2045(17)30785-4/fulltext. Published
November 2017. Accessed December 3, 2017.
27. Adalsteinsson VA, Ha G, Freeman SS, et al. Scalable whole-exome
sequencing of cell-free DNA reveals high concordance with metastatic
tumors. https://www.nature.com/articles/s41467-017-00965-y.
Published online November 6, 2017. Accessed November 19, 2017.
28. Parsons DW, Roy A, Yang Y, et al. Clinical genomics for children
with solid tumors: current realities and future opportunities
[Abstract]. Clin Cancer Res. 2016;22(1 Suppl):abstract IA16.
29. ¬ompson JC, Yee SS, Troxel AB, et al. Detection of therapeutically
targetable driver and resistance mutations in lung cancer patients by
next-generation sequencing of cell-free circulating tumor DNA. Clin
Cancer Res. 2016;22(23):5772-5782.
30. Press release, FDA. FDA approves axicabtagene ciloleucel
for large B-cell lymphoma. https://www.fda.gov/Drugs/
InformationOnDrugs/ApprovedDrugs/ucm581296.htm. Last
updated October 25, 2017. Accessed November 6, 2017.
31. Press release, FDA. FDA Approves gemtuzumab ozogamicin
for CD33-positive AML. https://www.fda.gov/Drugs/
InformationOnDrugs/ApprovedDrugs/ucm574518.htm. Last
updated September 1, 2017. Accessed November 6, 2017.
32. Press release, FDA. FDA grants regular approval to dabrafenib and
trametinib combination for metastatic NSCLC with BRAF V600E
mutation. https://www.fda.gov/Drugs/InformationOnDrugs/
ApprovedDrugs/ucm564331.htm. Last updated June 22, 2017.
Accessed November 6, 2017.
This past year will likely be remembered as one of breakthrough advances in reducing the burden of cancer, with some landmark “firsts” coming out of the US Food and Drug Admini
Our excitement has also been tempered by the rapid rise in the cost of effective biologic, immunologic, and targeted therapies. With the approval of trastuzumab-dkst (Ogivri), the first targeted biosimilar for HER2-positive breast and gastrointestinal cancers, we can look forward to price decreases possibly in the 20%-30% range over time from a targeted therapy with remarkable clinical efficacy. We know that approved biosimilars have demonstrated clinical efficacy along with similar minor biologic diversity that is also seen in the reference biologic.1 We can also hope that increasing competition among biosimilar and reference compounds will lead to improvements in production methodologies that can allow further price reductions so that even more patients can gain access to these highly effective therapies.
In addition, the first FDA approval for the next-generation sequencing (NGS) FoundationOne profiling test and the rapid announcement by the Centers for Medicare & Medicaid Services (CMS) that it will cover the cost of that testing brings us a step closer to knowing which patients most likely will or won’t benefit from costly and toxic targeted therapies. Along with the many clinical trials studying which mutations predict which efficacies of individual or combinations of targeted agents, the approval and CMS coverage policy will help us improve value to our patients; when we can recommend the most beneficial therapies and avoid futile ones.
Finally, the approval for the DigniCap Scalp Cooling System for patients on chemotherapy for all solid tumors is of great importance. Pending coverage availability, it may influence some patients to get chemotherapy they might otherwise have forgone to avoid hair loss (see related article).
More consolidation: the best of all worlds?
In my 27 years in private practice, during which practice revenues grew with the favorable profit margins on novel therapies, forward-thinking physician leaders piloted innovations in oncology electronic medical records (EMRs), the delivery of team-based care, clinical research partnerships, and more comprehensive care services to better serve diverse communities, including those in rural areas. At my previous practice, that included adding clinicians to our group to serve patients at hospital clinics in 2 counties in southern California, each county with populations larger than 15 states. Our private practice worked with these public entities to bring state-of-the art care and private practice efficiencies to the uninsured and underserved in our region.
Unfortunately, revenues plummeted with changes in reimbursement after passage of the Medicare Modernization Act in 2003 and they continue to destabilize and reduce the number of community practices across the country. Many oncologists and oncology practices, including mine, chose to join larger academic or hospital systems or larger oncology networks at a time they are also facing growing pressures to contain costs, focus on out-patient care, complex clinical trials, and expanded access to care.
Although we may lament the shrinking landscape of private oncology practices, we can also be inspired by the physicians who have joined ranks with the better-funded, better-resourced, more traditional hospital and academic systems. These larger systems have more resources, more clincial trial offerings, staffing, technology, and analytics to expand value-based care initiatives to larger numbers of patients.
The hub-and-spoke models of oncology care with integrated networks linked by technology, and networked into larger analytic and decision support systems such as CancerLinQ, the health information technology program of the American Society of Clinical Oncology (ASCO),2 could facilitate documentable delivery of comprehensive, evidence-based care, moving us closer to meeting the Quadruple Aim of optimal health care: improving the patient experience of care (including quality and satisfaction); improving the health of populations; reducing the per capita cost of health care; and improving the work life of those who deliver care.3,4
Payment reform: working to align incentives
Everyone seems to agree that the fee-for-service payment models do not align incentives for improving total health outcomes at the lowest costs, but at the moment, there seems to be no best way of aligning them. Robinson has reported on the oncology payment initiatives at four major health insurance plans – Medicare (public) and Anthem, Aetna, and UnitedHealthcare (all private), noting that:5
- Medicare is testing its Oncology Care Model at more than 200 sites in the United States, and early data are expected to be released in 2018.
- Anthem continues with its Cancer Care Quality Program that includes adherence to 2 key requirements: that participants are compliant with Anthem-approved drug pathways, and that they register their patients at the insurer’s oncology website and enter their clinical data. Anthem is also considering expanding the management fee for certain high priority clinical trials.
- Aetna’s Oncology Solutions takes a different approach by providing increased payments for generic chemotherapies.
- United has eliminated the mark-up for new drugs and continues to mark up the prices of the older and generic therapies. Its episode-based pricing gives practices upfront payments based on expected drug margins so that practices can fund more comprehensive evidence-based care. In a presentation at a Washington State Medical Oncology Society meeting recently, United’s Lee Newcomer, reported that the insurer continues to see improved clinical and financial outcomes as well as encouraging early data showing that patients might do better in the real-world setting on some therapies that have not been fully compared in head-to-head randomized clinical trials.6,7
ASCO is pulling these ideas together at the national level with its Patient-Centered Oncology Payment (PCOP) model, which is similar to Medicare’s alternative payment model. The PCOP model focuses on high-value, quality care. Higher upfront payments would cover the additional diagnostic services, care planning, and management to improve compliance and adherence as well as clinical trial evaluations. The model was developed and vetted by the ASCO Clinical Practice Committee and practicing oncologists, and is supported by staff and consultants. It is currently in its second year of operation with a commercial payer and will be submitted for review to the Physician-Focused Payment Model Technical Advisory Committee of the Health and Human Services. The results of the review are expected in 2018. If the model is approved, it could provide a uniform approach for payers that would align incentives for high-quality cancer care and allow for better predictive modeling for practices, irrespective of size, to invest in infrastructure and staffing to meet the growing demand for high-quality, value-based cancer care.
Better science: the promise of more cures
The FDA approved a record number drugs and biologics in 2017 for various cancers,8 including the landmark approval of the first CART therapy for cancer, tisagenlecleucel, which targets CD19 on B cells in the treatment of acute leukemia. That approval was rapidly followed by a second anti-CD19 CART therapy, axicabtagene ciloluecel, for refractory, aggressive B-cell non-Hodgkin lymphoma.9,10 Although these therapies can achieve remarkable response and even complete response rates in otherwise refractory patients, only some achieve a long-term remission, and the costs are an order of magnitude above most other cancer therapies. That raises the question of what duration of benefit we should expect for treatments that cost in the range of $500,000 for the therapy alone, along with the additional costs for care, hospitalization, monitoring, expensive biologics (eg, tocilizumab, for the severe and potentially life-threatening cytokine-release syndrome associated with CART therapies), and significant neurologic and other therapy-related toxicities.
Novel arrangements between pharmaceutical companies and payers are currently being discussed so that only patients who meet specific response criteria would be charged for the therapy. In addition, we await findings from ongoing research to see if new approaches can find specific targetable sites on solid tumors that could spare the healthy organ tissues while eliminating highly resistant or heterogeneous populations of mutations in patients with advanced solid tumors. Such development of highly specific targets for CART therapies would improve their efficacy and safety, and with defined protocols in place to address toxicities and efforts to reduce the costs of the therapies, we can hopefully ensure broader access for patients to this potentially transformative therapeutic tool.
In addition to the excitement around the CART therapies, many of the years other new approvals will bring incremental but meaningful improvement in outcomes for patients with common cancers. The approval of neratinib, the first agent approved as extended adjuvant therapy for women with early-stage HER2/neu-positive breast cancer, is welcome, given the current 30% recurrence risk that extends past 10 years for women in that disease population who have completed standard adjuvant HER2-directed therapies. The 34% reduction in recurrence risk with a year of extended oral adjuvant therapy, as reported by Martin and colleagues,11 with benefits sustained out to 5 years and with controllable diarrhea as the major toxicity, are encouraging. This oral therapy may be especially beneficial for hormone-receptor–positive women in whom blocking the HER2/neu pathway may enhance cell signaling through the hormone pathways, which can be blocked with oral agents at the same time to provide significant reduction of recurrence risk.
Diagnostics
The concept of personalized medicine is based on identifying biomarkers that are predictive of a patient’s response to treatment. There has been much progress toward applying NGS of tumors for use in the clinic, but we are still awaiting evidence from randomized clinical trials that such approaches prolong overall or progression-free survival.12 Dr Julie Lange, an associate professor of clinical surgery and director of the Breast Cancer Program at the Keck School of Medicine at the University of Southern California, Los Angeles, provided me with the references to key studies in this field in which she is a leading researcher.13 However, she pointed out that in the absence of effective therapies, advanced biomarker testing may be less helpful, as is the case in heavily pretreated patients,14 unless a molecular test can pinpoint a potentially clinically actionable mutation. With the plethora of available assays and the high costs of molecular testing, clinicians are challenged in knowing what testing is best for which patients. Findings from a number of key ongoing national trials may eventually help us understand which tumor mutations in which tumor types can be most effectively targeted when multiple targetable mutations are found (TAPUR,15 MATCH,16 and QUILT17 and other basket trials18). The complexity of molecular testing has led to the development of institutional, trial-based, or co-operative group molecular tumor boards to provide guidance on specific targeted therapies for specific tumor mutations.
ASCO has launched a monthly series called Molecular Oncology Tumor Boards19 to expand the knowledge base in this field. It is presented as user-driven discussions designed to help providers integrate the use of the new genetic and genomic tests and their results into the day-to-day clinical care of patients with cancer.20
Liquid biopsies
As busy clinicians, we need to understand the differences in liquid biopsy tests and their correlation with actionable targets, especially given the rapid progress in this field. Again, Dr Lange offered clarity on those differences. Liquid biopsy, refers to using a blood draw to isolate circulating tumor cells (CTCs) or circulating tumor DNA (ctDNA) to assess tumor biomarkers.21 Both CTCs and ctDNA tests have been shown to be prognostic of worse survival.22-24 Liquid biopsies are currently supplemental to direct tumor biopsies, not replacements for them. The theoretical advantage of liquid biopsies is that they may reflect tumor heterogeneity by examining the repertoire of mutations contributed by diverse metastatic sites that shed CTCs or ctDNA into the circulation. The question is which type of testing can best inform therapy decisions.
Assays for ctDNA using droplet digital PCR [polymerase chain reaction], a digital PCR method based on water-oil emulsion droplet technology, require a priori knowledge of the specific mutation associated with response or lack of response to a specific therapy.25,26 Technical issues related to the detection of rare alleles present within a mixed population of leukocytes, and ctDNA remains a challenge for many ctDNA assays. However, there is evidence to suggest that whole-exome sequencing of ctDNA is concordant with mutations in metastases,27 however benchmarking ctDNA against tissue biopsies of metastases was not possible in all studies because tumor blocks were not available or because of the failure of tumor NGS assays. 28,29
Newer generations of CTC assays take advantage of the circulating tumor cell as a functional assay for mutational status, gene expression, proteomics, epigenetics, and/or chemosensitivity of cultured cells. The relationship between CTCs and ctDNA remains uncertain as to whether CTCs are the cell of origin for ctDNA or if ctDNA may reflect responding or resistant tumor populations. The use of NGS on tumor specimens, ctDNA, and CTCs as a discovery tool is advancing the field by improving the understanding of disease heterogeneity and potential treatment targets. These results require correlation with patterns of response to therapy, and ultimately require validation in randomized clinical trials to provide strong evidence justifying their use outside of clinical trials. We can look forward to a time in the not distant future when specific liquid biopsy assays will reflect the array of mutations in different metastatic sites with validation that they correlate with efficacy of targeting those mutations that have targetable therapies.
From the FDA
New approvals
- Trastuzumab-dkst (Ogivri, Mylan; Dec 1) was approved as a biosimilar to trastuzumab (Herceptin, Genentech) for the treatment of patients with HER2-overexpressing breast or metastatic stomach cancer (gastric or gastroesophageal junction adenocarcinoma).
- Sunitinib malate (Sutent, Pfizer; Nov 16) was approved for the adjuvant treatment of adult patients at high risk of recurrent renal cell carcinoma after nephrectomy.
- Obinutuzumab (Gazyva, Genentech; Nov 16) received regular approval in combination with chemotherapy, followed by obinutuzumab monotherapy in patients achieving partial remission, for adult patients with previously untreated stage II bulky, III, or IV follicular lymphoma.
- Emicizumab-kxwh (Hemlibra, Genentech; Nov 16) was approved for routine prophylaxis to prevent or reduce the frequency of bleeding episodes in adult and pediatric patients with hemophilia A with factor VIII inhibitors.
- Dasatinib (Sprycel, Bristol-Myers Squibb; Nov 9) was approved for the treatment of pediatric patients with Philadelphia chromosome-positive chronic myeloid leukemia (CML) in the chronic phase.
- Brentuximab vedotin (Adcetris, Seattle Genetics; Nov 9) for the treatment of previously treated adult patients with primary cutaneous anaplastic large cell lymphoma (pcALCL) or CD30-expressing mycosis fungoides.
- Alectinib (Alecensa, Hoffmann-La Roche/Genentech; Nov 6) was approved for treatment of patients with anaplastic lymphoma kinase–positive metastatic non-small cell lung cancer (NSCLC), as detected by an FDA-approved test.
- Vemurafenib (Zelboraf, Hoffmann-La Roche; Nov 6) received approval for the treatment of Acalabrutinib (Calquence, AstraZeneca/Acerta; Oct 31) was granted accelerated approval for treatment of adult patients with mantle cell lymphoma (MCL) who have received at least one previous therapy.
- Axicabtagene ciloleucel (Yescarta, Kite; Oct 18), a CART therapy, was approved for treatment of adult patients with relapsed or refractory large B-cell lymphoma after two or more lines of systemic therapy, including diffuse large B-cell lymphoma (DLBCL) not otherwise specified, primary mediastinal large B-cell lymphoma, high-grade B-cell lymphoma, and DLBCL arising from follicular lymphoma. The complete remission rate reviewed by the FDA for trial patients was 51%.30 It was the second CART therapy this year to receive approval (see tisagenlecleucel; Aug 30). The agency granted orphan drug designation and priority review to therapy for this indication.
- Abemaciclib (Verzenio, Eli Lilly; Sep 28) was approved in combination with fulvestrant for women with hormone receptor-positive, HER2-negative advanced or metastatic breast cancer with disease progression following endocrine therapy.
- Copanlisib (Aliqopa, Bayer; Sep 14) got accelerated approval for the treatment of adult patients with relapsed follicular lymphoma who have received at least two prior systemic therapies.
- Bevacizumab-awwb (Mvasi, Amgen; Sep 14) was approved as a biosimilar to bevacizumab (Avastin, Genentech) for treating multiple types of cancer. It was the first biosimilar approved in the US for the treatment of cancer.
- Gemtuzumab ozogamicin (Mylotarg, Pfizer; Sep 1) was approved for the treatment of newly diagnosed CD33-positive acute myeloid leukemia (AML) in adults and of relapsed/refractory CD33-positive AML in adults and pediatric patients aged 2 or older. It can be used in combination with daunorubicin and cytarabine for adults with newly diagnosed AML, or as a standalone treatment for certain adult and pediatric patients. The drug was originally approved in 2000 as a standalone treatment for CD33-positive AML in patients older than 60 years, but was withdrawn in 2010 because of safety concerns and postmarketing trials could not confirm benefit. The current approval is for a lower recommended dose and schedule.31
- Tisagenlecleucel (Kymriah, Novartis; Aug 30) was approved for the treatment of patients up to age 25 years with B-cell precursor acute lymphoblastic leukemia (ALL) that is refractory or in second or later relapse. It is the first CART immunotherapy approved by the agency.
- Inotuzumab ozogamicin (Besponsa, Wyeth; Aug 17) was approved for the treatment of adults with relapsed or refractory B-cell precursor ALL.
- A liposome-encapsulated combination of daunorubicin and cytarabine (Vyxeos, Jazz; Aug 3) was approved for the treatment of adults with newly diagnosed therapy-related AML (t-AML) or AML with myelodysplasia-related changes (AML-MRC), two types of AML having a poor prognosis.
- Enasidenib (Idhifa, Celgene; Aug 1) was approved for the treatment of adult patients with relapsed or refractory AML with an isocitrate dehydrogenase-2 mutation as detected by an FDA-approved test.
- Neratinib (Nerlynx, Puma; Jul 17) was approved as the first extended adjuvant therapy for adult patients with early stage HER2-overexpressed/amplified breast cancer, to follow adjuvant trastuzumab-based therapy.
- Blinatumomab (Blincyto, Amgen; Jul 11) was approved for the treatment of relapsed or refractory B-cell precursor acute lymphoblastic leukemia in adults and children.
- L-glutamine oral powder (Endari, Emmaus; Jul 7) was approved for oral administration to reduce the acute complications of sickle cell disease in adult and pediatric patients 5 years and older.
- Betrixaban (Bevyxxa, Portola; Jun 23) was approved for the prophylaxis of venous thromboembolism (VTE) in adult patients hospitalized for an acute medical illness who are at risk for thromboembolic complications because of moderate or severe restricted mobility and other risk factors for VTE.
- The combination of rituximab and hyaluronidase human (Rituxan Hycela, Genentech; Jun 22) was approved for adult patients with follicular lymphoma, DLBCL, and chronic lymphocytic leukemia. Hyaluronidase human is an enzyme that helps deliver the rituximab. This formulation allows subcutaneous administration of the combination, which will shorten patient visit times and potentially even allow at-home therapy delivery.
- Ceritinib (Zykadia, Novartis; May 26) was approved for patients with metastatic NSCLC whose tumors are anaplastic lymphoma kinase (ALK)-positive as detected by an FDA-approved test.
- Avelumab (Bavencio, EMD Serono; May 9) got accelerated approval for patients with locally advanced or metastatic urothelial carcinoma whose disease progressed during or following platinum-containing chemotherapy or within 12 months of neoadjuvant or adjuvant platinum-containing chemotherapy.
- Durvalumab (ImfinzI, AstraZeneca; May 1) got accelerated approval for the treatment of patients with locally advanced or metastatic urothelial carcinoma who have disease progression during or following platinum-containing chemotherapy or who have disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy.
- Brigatinib (Alunbrig tablets, Takeda through Ariad; Apr 28) got accelerated approval for the treatment of patients with metastatic anaplastic lymphoma kinase (ALK)-positive NSCLC who have progressed on or are intolerant to crizotinib.
- Midostaurin (Rydapt, Novartis; Apr 28) was approved for the treatment of adult patients with newly diagnosed AML who are FLT3 mutation-positive, as detected by an FDA-approved test, in combination with standard cytarabine and daunorubicin induction and cytarabine consolidation.
- Osimertinib (Tagrisso, AstraZeneca; Mar 30) got regular approval for the treatment of patients with metastatic epidermal growth factor receptor (EGFR) T790M mutation-positive NSCLC, as detected by an FDA-approved test, whose disease has progressed on or after EGFR tyrosine-kinase inhibitor therapy.
- Niraparib (Zejula, Tesaro; Mar 27), a poly ADP-ribose polymerase (PARP) inhibitor, was approved for the maintenance treatment of adult patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in complete or partial response to platinum-based chemotherapy.
- Avelumab (Mar 23), a PD-L1–blocking human IgG1 lambda monoclonal antibody, got accelerated approval for the treatment of patients 12 years and older with metastatic Merkel cell carcinoma. It is the first FDA-approved product to treat this type of cancer.
- Ribociclib (Kisqali, Novartis; Mar 13), a CDK4/6 inhibitor, was approved as a breakthrough therapy after priority review for use in combination with an aromatase inhibitor as initial endocrine-based therapy for the treatment of postmenopausal women with HR-positive, HER2-negative advanced or metastatic breast cancer.
Expanded/additional indications
- Nivolumab (Opdivo, Bristol-Myers Squibb; Sep 22) got accelerated expanded indication approval for treatment of hepatocellular carcinoma (HCC) in patients previously treated with sorafenib.
- Pembrolizumab (Keytruda, Merck; Sep 22) got accelerated expanded indication approval for recurrent locally advanced or metastatic gastric or gastroesophageal junction adenocarcinoma in patients whose tumors express PD-L1 as determined by an FDA-approved test.
- DigniCap Scalp Cooling System (Dignitana Inc; Jul 3) was cleared for expanded use for reducing hair loss during chemotherapy for all solid tumors. Marketing authorization for the cooling cap had been granted in 2015 for patients with breast cancer.
- Olaparib tablets (Lynparza, AstraZeneca; Aug 17) got approval for an expanded indication as maintenance treatment of adult patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer, who are in a complete or partial response to platinum-based chemotherapy.
- Ibrutinib (Imbruvica, Pharmacyclics; Aug 2) got expanded indication approval for the treatment of adult patients with chronic graft-versus-host disease (cGVHD) after failure of one or more lines of systemic therapy. It was the first FDA-approved therapy for the treatment of cGVHD. (Ibrutinib was previously approved for chronic lymphocytic leukemia/small lymphocytic lymphoma, chronic lymphocytic leukemia/small lymphocytic lymphoma with 17p deletion, Waldenström’s macroglobulinemia, marginal zone lymphoma, and mantle cell lymphoma).
- Nivolumab (Aug 2) got an accelerated expanded indication for the treatment of patients 12 years and older with mismatch repair deficient (dMMR) and microsatellite instability-high (MSI-H) metastatic colorectal cancer that has progressed after treatment with a fluoropyrimidine, oxaliplatin, and irinotecan.
- Dabrafenib and trametinib (Tafinlar and Mekinist, Novartis; Jun 22) were approved for the expanded indication in combination for patients with metastatic NSCLC with BRAF V600E mutation as detected by an FDA-approved test. The combination demonstrated superior efficacy compared with dabrafenib alone (overall response rate: 61% and 27%, respectively).32
- Pembrolizumab (May 23) got approved for expanded indication for adult and pediatric patients with unresectable or metastatic, MSI-H or dMMR solid tumors that have progressed after treatment and who have no satisfactory alternative treatment options or with MSI-H or dMMR colorectal cancer that has progressed after treatment with a fluoropyrimidine, oxaliplatin, and irinotecan.
- Pembrolizumab (May 18) got approval for expanded indication for patients with locally advanced or metastatic urothelial carcinoma who have disease progression during or following platinum-containing chemotherapy or within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy.
- Pembrolizumab (May 10) got accelerated expanded indication for use combination with pemetrexed and carboplatin for the treatment of patients with previously untreated metastatic NSCLC.
- Regorafenib (Stivarga, Bayer; Apr 27) got an additional indication for the treatment of patients with HCC who have been previously treated with sorafenib.
- Palbociclib (Ibrance, Pfizer; Mar 31) got an expanded indication that includes first-line therapy for the treatment of hormone receptor–positive, HER2-negative advanced or metastatic breast cancer in combination with an aromatase inhibitor as initial endocrine based therapy in postmenopausal women.
- Pembrolizumab (Mar 15) got an accelerated additional indication approval for treatment of adult and pediatric patients with refractory classical Hodgkin lymphoma, or those who have relapsed after three or more previous lines of therapy.
- Lenalidomide (Revlimid, Celgene; Feb 22) got an additional indication as maintenance therapy for patients with multiple myeloma following autologous stem cell transplant.
- Nivolumab (Feb 2) got an accelerated expanded indication for treatment of patients with locally advanced or metastatic urothelial carcinoma who have disease progression during or following platinum-containing chemotherapy or have disease progression within 12 months of neoadjuvant or adjuvant treatment with a platinum-containing chemotherapy.
Modified use
- Cabazitaxel (Jevtana, Sanofi-Aventis; Sep 14) in combination with prednisone was approved at a lower dose of 20 mg/m2 every 3 weeks for the treatment of patients with metastatic castration-resistant prostate cancer previously treated with a docetaxel-containing treatment regimen. It had been approved at 25 mg/m2 every 3 weeks for this indication in 2010.
Tests/diagnostics
- Marketing approval was given to the FoundationOne CDx (Foundation Medicine; Nov 30), an NGS-based in vitro diagnostic to detect genetic mutations in 324 genes and 2 genomic signatures in any solid tumor type.
- Marketing approval was given to the Praxis Extended RAS Panel (Illumina; Jun 29), a next generation sequencing test to detect certain genetic mutations in RAS genes in tumor samples of patients with metastatic colorectal cancer. The test is used to aid in the identification of patients who may be eligible for treatment with panitumumab (Vectibix, Amgen).
- Marketing was approved for ipsogen JAK2 RGQ PCR Kit (Qiagen ; Mar 27) to detect mutations affecting the Janus tyrosine kinase 2 gene. This is the first FDA-authorized test intended to help physicians in evaluating patients for suspected polycythemia vera.
Imaging and pathology aids
- Aminolevulinic acid hydrochloride, known as ALA HCl (Gleolan, NX; Jun 6) was approved as an optical imaging agent indicated in patients with gliomas (suspected World Health Organization grades III or IV on preoperative imaging) as an adjunct for the visualization of malignant tissue during surgery.
- Marketing was approved for the Philips IntelliSite Pathology Solution (PIPS, Philips Medical Systems Nederland; Apr 17), as an aid to the pathologist to review and interpret digital images of surgical pathology slides prepared from formalin-fixed paraffin embedded tissue.
Challenges and uncertainties
The current administration’s initiatives to reduce administrative burdens is underway with the Patients Over Paperwork initiative. Eliminating and streamlining regulations to increase efficiency and improve beneficiary experience could be helpful to both oncologists and patients. For now, the Medicare Access and CHIP Reauthorization Act (MACRA) program, allows you to “pick your pace” in the 2017 performance year and report on at least one measure to avoid a payment reduction penalty on your Medicare payments in 2019. In the final rule for 2018, the CMS finalized a proposal to apply the MIPS [Merit-based Incentive Payment System] adjustment to all Part B items and services, which will include Part B drugs. This would be unfair to oncologists who treat on the basis of evidence-based guidelines and pathways and have no control over the costs of the drugs they prescribe.
In addition, more requirements will be imposed in 2018 in a move toward full MACRA implementation. All four composite categories (Quality – 60% for 2017; Advancing Care Information (ACI, renamed from Meaningful Use) – 25% for 2017; Improvement Activities (IA) – 15% for 2017; and Cost – 0% for 2017, but weighted in the future) will be scored, including resource use (cost) at 10%. CMS will collect data to assess the total cost of care and the Medicare Spend per Beneficiary to assess use. Full program implementation, with cost being assessed at 30% of your score is expected in the 2019 performance year. ASCO’s clinical affairs and policy experts have studied the implications of Part B chemotherapy drugs being included in the cost component of the MIPS scoring and will continue advocating for policies that hold clinicians responsible only for the aspects of care they can control, such as providing high-quality care based on the patient’s disease, biomarkers, comorbidities, and preferences, and not the costs of the evidence-based therapies needed by patients.
Toward a better 2018 for ourselves and our patients
As an eternal optimist, I remain enthusiastic that despite the many challenges, we will find effective ways to bring standard as well as newer, cell-based and targeted therapies to our patients and cover the costs of highly effective therapies. I also remain hopeful that improving technological capabilities and payment reforms will be used by innovative clinical and administrative care teams to give clinicians more time to improve the care and health of patients while validating the methodologies so that real world data can help us further craft therapies to improve the health of each individual who needs our care. As we close this 15th year of our journal, we hope our presentations of practical science and implementation content has helped support your work while freeing some time for you to enjoy the journey. Our best wishes for a joyful holiday season celebrated with friends and family and the patients who entrust us to help them face and live beyond their cancer diagnoses.
This past year will likely be remembered as one of breakthrough advances in reducing the burden of cancer, with some landmark “firsts” coming out of the US Food and Drug Admini
Our excitement has also been tempered by the rapid rise in the cost of effective biologic, immunologic, and targeted therapies. With the approval of trastuzumab-dkst (Ogivri), the first targeted biosimilar for HER2-positive breast and gastrointestinal cancers, we can look forward to price decreases possibly in the 20%-30% range over time from a targeted therapy with remarkable clinical efficacy. We know that approved biosimilars have demonstrated clinical efficacy along with similar minor biologic diversity that is also seen in the reference biologic.1 We can also hope that increasing competition among biosimilar and reference compounds will lead to improvements in production methodologies that can allow further price reductions so that even more patients can gain access to these highly effective therapies.
In addition, the first FDA approval for the next-generation sequencing (NGS) FoundationOne profiling test and the rapid announcement by the Centers for Medicare & Medicaid Services (CMS) that it will cover the cost of that testing brings us a step closer to knowing which patients most likely will or won’t benefit from costly and toxic targeted therapies. Along with the many clinical trials studying which mutations predict which efficacies of individual or combinations of targeted agents, the approval and CMS coverage policy will help us improve value to our patients; when we can recommend the most beneficial therapies and avoid futile ones.
Finally, the approval for the DigniCap Scalp Cooling System for patients on chemotherapy for all solid tumors is of great importance. Pending coverage availability, it may influence some patients to get chemotherapy they might otherwise have forgone to avoid hair loss (see related article).
More consolidation: the best of all worlds?
In my 27 years in private practice, during which practice revenues grew with the favorable profit margins on novel therapies, forward-thinking physician leaders piloted innovations in oncology electronic medical records (EMRs), the delivery of team-based care, clinical research partnerships, and more comprehensive care services to better serve diverse communities, including those in rural areas. At my previous practice, that included adding clinicians to our group to serve patients at hospital clinics in 2 counties in southern California, each county with populations larger than 15 states. Our private practice worked with these public entities to bring state-of-the art care and private practice efficiencies to the uninsured and underserved in our region.
Unfortunately, revenues plummeted with changes in reimbursement after passage of the Medicare Modernization Act in 2003 and they continue to destabilize and reduce the number of community practices across the country. Many oncologists and oncology practices, including mine, chose to join larger academic or hospital systems or larger oncology networks at a time they are also facing growing pressures to contain costs, focus on out-patient care, complex clinical trials, and expanded access to care.
Although we may lament the shrinking landscape of private oncology practices, we can also be inspired by the physicians who have joined ranks with the better-funded, better-resourced, more traditional hospital and academic systems. These larger systems have more resources, more clincial trial offerings, staffing, technology, and analytics to expand value-based care initiatives to larger numbers of patients.
The hub-and-spoke models of oncology care with integrated networks linked by technology, and networked into larger analytic and decision support systems such as CancerLinQ, the health information technology program of the American Society of Clinical Oncology (ASCO),2 could facilitate documentable delivery of comprehensive, evidence-based care, moving us closer to meeting the Quadruple Aim of optimal health care: improving the patient experience of care (including quality and satisfaction); improving the health of populations; reducing the per capita cost of health care; and improving the work life of those who deliver care.3,4
Payment reform: working to align incentives
Everyone seems to agree that the fee-for-service payment models do not align incentives for improving total health outcomes at the lowest costs, but at the moment, there seems to be no best way of aligning them. Robinson has reported on the oncology payment initiatives at four major health insurance plans – Medicare (public) and Anthem, Aetna, and UnitedHealthcare (all private), noting that:5
- Medicare is testing its Oncology Care Model at more than 200 sites in the United States, and early data are expected to be released in 2018.
- Anthem continues with its Cancer Care Quality Program that includes adherence to 2 key requirements: that participants are compliant with Anthem-approved drug pathways, and that they register their patients at the insurer’s oncology website and enter their clinical data. Anthem is also considering expanding the management fee for certain high priority clinical trials.
- Aetna’s Oncology Solutions takes a different approach by providing increased payments for generic chemotherapies.
- United has eliminated the mark-up for new drugs and continues to mark up the prices of the older and generic therapies. Its episode-based pricing gives practices upfront payments based on expected drug margins so that practices can fund more comprehensive evidence-based care. In a presentation at a Washington State Medical Oncology Society meeting recently, United’s Lee Newcomer, reported that the insurer continues to see improved clinical and financial outcomes as well as encouraging early data showing that patients might do better in the real-world setting on some therapies that have not been fully compared in head-to-head randomized clinical trials.6,7
ASCO is pulling these ideas together at the national level with its Patient-Centered Oncology Payment (PCOP) model, which is similar to Medicare’s alternative payment model. The PCOP model focuses on high-value, quality care. Higher upfront payments would cover the additional diagnostic services, care planning, and management to improve compliance and adherence as well as clinical trial evaluations. The model was developed and vetted by the ASCO Clinical Practice Committee and practicing oncologists, and is supported by staff and consultants. It is currently in its second year of operation with a commercial payer and will be submitted for review to the Physician-Focused Payment Model Technical Advisory Committee of the Health and Human Services. The results of the review are expected in 2018. If the model is approved, it could provide a uniform approach for payers that would align incentives for high-quality cancer care and allow for better predictive modeling for practices, irrespective of size, to invest in infrastructure and staffing to meet the growing demand for high-quality, value-based cancer care.
Better science: the promise of more cures
The FDA approved a record number drugs and biologics in 2017 for various cancers,8 including the landmark approval of the first CART therapy for cancer, tisagenlecleucel, which targets CD19 on B cells in the treatment of acute leukemia. That approval was rapidly followed by a second anti-CD19 CART therapy, axicabtagene ciloluecel, for refractory, aggressive B-cell non-Hodgkin lymphoma.9,10 Although these therapies can achieve remarkable response and even complete response rates in otherwise refractory patients, only some achieve a long-term remission, and the costs are an order of magnitude above most other cancer therapies. That raises the question of what duration of benefit we should expect for treatments that cost in the range of $500,000 for the therapy alone, along with the additional costs for care, hospitalization, monitoring, expensive biologics (eg, tocilizumab, for the severe and potentially life-threatening cytokine-release syndrome associated with CART therapies), and significant neurologic and other therapy-related toxicities.
Novel arrangements between pharmaceutical companies and payers are currently being discussed so that only patients who meet specific response criteria would be charged for the therapy. In addition, we await findings from ongoing research to see if new approaches can find specific targetable sites on solid tumors that could spare the healthy organ tissues while eliminating highly resistant or heterogeneous populations of mutations in patients with advanced solid tumors. Such development of highly specific targets for CART therapies would improve their efficacy and safety, and with defined protocols in place to address toxicities and efforts to reduce the costs of the therapies, we can hopefully ensure broader access for patients to this potentially transformative therapeutic tool.
In addition to the excitement around the CART therapies, many of the years other new approvals will bring incremental but meaningful improvement in outcomes for patients with common cancers. The approval of neratinib, the first agent approved as extended adjuvant therapy for women with early-stage HER2/neu-positive breast cancer, is welcome, given the current 30% recurrence risk that extends past 10 years for women in that disease population who have completed standard adjuvant HER2-directed therapies. The 34% reduction in recurrence risk with a year of extended oral adjuvant therapy, as reported by Martin and colleagues,11 with benefits sustained out to 5 years and with controllable diarrhea as the major toxicity, are encouraging. This oral therapy may be especially beneficial for hormone-receptor–positive women in whom blocking the HER2/neu pathway may enhance cell signaling through the hormone pathways, which can be blocked with oral agents at the same time to provide significant reduction of recurrence risk.
Diagnostics
The concept of personalized medicine is based on identifying biomarkers that are predictive of a patient’s response to treatment. There has been much progress toward applying NGS of tumors for use in the clinic, but we are still awaiting evidence from randomized clinical trials that such approaches prolong overall or progression-free survival.12 Dr Julie Lange, an associate professor of clinical surgery and director of the Breast Cancer Program at the Keck School of Medicine at the University of Southern California, Los Angeles, provided me with the references to key studies in this field in which she is a leading researcher.13 However, she pointed out that in the absence of effective therapies, advanced biomarker testing may be less helpful, as is the case in heavily pretreated patients,14 unless a molecular test can pinpoint a potentially clinically actionable mutation. With the plethora of available assays and the high costs of molecular testing, clinicians are challenged in knowing what testing is best for which patients. Findings from a number of key ongoing national trials may eventually help us understand which tumor mutations in which tumor types can be most effectively targeted when multiple targetable mutations are found (TAPUR,15 MATCH,16 and QUILT17 and other basket trials18). The complexity of molecular testing has led to the development of institutional, trial-based, or co-operative group molecular tumor boards to provide guidance on specific targeted therapies for specific tumor mutations.
ASCO has launched a monthly series called Molecular Oncology Tumor Boards19 to expand the knowledge base in this field. It is presented as user-driven discussions designed to help providers integrate the use of the new genetic and genomic tests and their results into the day-to-day clinical care of patients with cancer.20
Liquid biopsies
As busy clinicians, we need to understand the differences in liquid biopsy tests and their correlation with actionable targets, especially given the rapid progress in this field. Again, Dr Lange offered clarity on those differences. Liquid biopsy, refers to using a blood draw to isolate circulating tumor cells (CTCs) or circulating tumor DNA (ctDNA) to assess tumor biomarkers.21 Both CTCs and ctDNA tests have been shown to be prognostic of worse survival.22-24 Liquid biopsies are currently supplemental to direct tumor biopsies, not replacements for them. The theoretical advantage of liquid biopsies is that they may reflect tumor heterogeneity by examining the repertoire of mutations contributed by diverse metastatic sites that shed CTCs or ctDNA into the circulation. The question is which type of testing can best inform therapy decisions.
Assays for ctDNA using droplet digital PCR [polymerase chain reaction], a digital PCR method based on water-oil emulsion droplet technology, require a priori knowledge of the specific mutation associated with response or lack of response to a specific therapy.25,26 Technical issues related to the detection of rare alleles present within a mixed population of leukocytes, and ctDNA remains a challenge for many ctDNA assays. However, there is evidence to suggest that whole-exome sequencing of ctDNA is concordant with mutations in metastases,27 however benchmarking ctDNA against tissue biopsies of metastases was not possible in all studies because tumor blocks were not available or because of the failure of tumor NGS assays. 28,29
Newer generations of CTC assays take advantage of the circulating tumor cell as a functional assay for mutational status, gene expression, proteomics, epigenetics, and/or chemosensitivity of cultured cells. The relationship between CTCs and ctDNA remains uncertain as to whether CTCs are the cell of origin for ctDNA or if ctDNA may reflect responding or resistant tumor populations. The use of NGS on tumor specimens, ctDNA, and CTCs as a discovery tool is advancing the field by improving the understanding of disease heterogeneity and potential treatment targets. These results require correlation with patterns of response to therapy, and ultimately require validation in randomized clinical trials to provide strong evidence justifying their use outside of clinical trials. We can look forward to a time in the not distant future when specific liquid biopsy assays will reflect the array of mutations in different metastatic sites with validation that they correlate with efficacy of targeting those mutations that have targetable therapies.
From the FDA
New approvals
- Trastuzumab-dkst (Ogivri, Mylan; Dec 1) was approved as a biosimilar to trastuzumab (Herceptin, Genentech) for the treatment of patients with HER2-overexpressing breast or metastatic stomach cancer (gastric or gastroesophageal junction adenocarcinoma).
- Sunitinib malate (Sutent, Pfizer; Nov 16) was approved for the adjuvant treatment of adult patients at high risk of recurrent renal cell carcinoma after nephrectomy.
- Obinutuzumab (Gazyva, Genentech; Nov 16) received regular approval in combination with chemotherapy, followed by obinutuzumab monotherapy in patients achieving partial remission, for adult patients with previously untreated stage II bulky, III, or IV follicular lymphoma.
- Emicizumab-kxwh (Hemlibra, Genentech; Nov 16) was approved for routine prophylaxis to prevent or reduce the frequency of bleeding episodes in adult and pediatric patients with hemophilia A with factor VIII inhibitors.
- Dasatinib (Sprycel, Bristol-Myers Squibb; Nov 9) was approved for the treatment of pediatric patients with Philadelphia chromosome-positive chronic myeloid leukemia (CML) in the chronic phase.
- Brentuximab vedotin (Adcetris, Seattle Genetics; Nov 9) for the treatment of previously treated adult patients with primary cutaneous anaplastic large cell lymphoma (pcALCL) or CD30-expressing mycosis fungoides.
- Alectinib (Alecensa, Hoffmann-La Roche/Genentech; Nov 6) was approved for treatment of patients with anaplastic lymphoma kinase–positive metastatic non-small cell lung cancer (NSCLC), as detected by an FDA-approved test.
- Vemurafenib (Zelboraf, Hoffmann-La Roche; Nov 6) received approval for the treatment of Acalabrutinib (Calquence, AstraZeneca/Acerta; Oct 31) was granted accelerated approval for treatment of adult patients with mantle cell lymphoma (MCL) who have received at least one previous therapy.
- Axicabtagene ciloleucel (Yescarta, Kite; Oct 18), a CART therapy, was approved for treatment of adult patients with relapsed or refractory large B-cell lymphoma after two or more lines of systemic therapy, including diffuse large B-cell lymphoma (DLBCL) not otherwise specified, primary mediastinal large B-cell lymphoma, high-grade B-cell lymphoma, and DLBCL arising from follicular lymphoma. The complete remission rate reviewed by the FDA for trial patients was 51%.30 It was the second CART therapy this year to receive approval (see tisagenlecleucel; Aug 30). The agency granted orphan drug designation and priority review to therapy for this indication.
- Abemaciclib (Verzenio, Eli Lilly; Sep 28) was approved in combination with fulvestrant for women with hormone receptor-positive, HER2-negative advanced or metastatic breast cancer with disease progression following endocrine therapy.
- Copanlisib (Aliqopa, Bayer; Sep 14) got accelerated approval for the treatment of adult patients with relapsed follicular lymphoma who have received at least two prior systemic therapies.
- Bevacizumab-awwb (Mvasi, Amgen; Sep 14) was approved as a biosimilar to bevacizumab (Avastin, Genentech) for treating multiple types of cancer. It was the first biosimilar approved in the US for the treatment of cancer.
- Gemtuzumab ozogamicin (Mylotarg, Pfizer; Sep 1) was approved for the treatment of newly diagnosed CD33-positive acute myeloid leukemia (AML) in adults and of relapsed/refractory CD33-positive AML in adults and pediatric patients aged 2 or older. It can be used in combination with daunorubicin and cytarabine for adults with newly diagnosed AML, or as a standalone treatment for certain adult and pediatric patients. The drug was originally approved in 2000 as a standalone treatment for CD33-positive AML in patients older than 60 years, but was withdrawn in 2010 because of safety concerns and postmarketing trials could not confirm benefit. The current approval is for a lower recommended dose and schedule.31
- Tisagenlecleucel (Kymriah, Novartis; Aug 30) was approved for the treatment of patients up to age 25 years with B-cell precursor acute lymphoblastic leukemia (ALL) that is refractory or in second or later relapse. It is the first CART immunotherapy approved by the agency.
- Inotuzumab ozogamicin (Besponsa, Wyeth; Aug 17) was approved for the treatment of adults with relapsed or refractory B-cell precursor ALL.
- A liposome-encapsulated combination of daunorubicin and cytarabine (Vyxeos, Jazz; Aug 3) was approved for the treatment of adults with newly diagnosed therapy-related AML (t-AML) or AML with myelodysplasia-related changes (AML-MRC), two types of AML having a poor prognosis.
- Enasidenib (Idhifa, Celgene; Aug 1) was approved for the treatment of adult patients with relapsed or refractory AML with an isocitrate dehydrogenase-2 mutation as detected by an FDA-approved test.
- Neratinib (Nerlynx, Puma; Jul 17) was approved as the first extended adjuvant therapy for adult patients with early stage HER2-overexpressed/amplified breast cancer, to follow adjuvant trastuzumab-based therapy.
- Blinatumomab (Blincyto, Amgen; Jul 11) was approved for the treatment of relapsed or refractory B-cell precursor acute lymphoblastic leukemia in adults and children.
- L-glutamine oral powder (Endari, Emmaus; Jul 7) was approved for oral administration to reduce the acute complications of sickle cell disease in adult and pediatric patients 5 years and older.
- Betrixaban (Bevyxxa, Portola; Jun 23) was approved for the prophylaxis of venous thromboembolism (VTE) in adult patients hospitalized for an acute medical illness who are at risk for thromboembolic complications because of moderate or severe restricted mobility and other risk factors for VTE.
- The combination of rituximab and hyaluronidase human (Rituxan Hycela, Genentech; Jun 22) was approved for adult patients with follicular lymphoma, DLBCL, and chronic lymphocytic leukemia. Hyaluronidase human is an enzyme that helps deliver the rituximab. This formulation allows subcutaneous administration of the combination, which will shorten patient visit times and potentially even allow at-home therapy delivery.
- Ceritinib (Zykadia, Novartis; May 26) was approved for patients with metastatic NSCLC whose tumors are anaplastic lymphoma kinase (ALK)-positive as detected by an FDA-approved test.
- Avelumab (Bavencio, EMD Serono; May 9) got accelerated approval for patients with locally advanced or metastatic urothelial carcinoma whose disease progressed during or following platinum-containing chemotherapy or within 12 months of neoadjuvant or adjuvant platinum-containing chemotherapy.
- Durvalumab (ImfinzI, AstraZeneca; May 1) got accelerated approval for the treatment of patients with locally advanced or metastatic urothelial carcinoma who have disease progression during or following platinum-containing chemotherapy or who have disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy.
- Brigatinib (Alunbrig tablets, Takeda through Ariad; Apr 28) got accelerated approval for the treatment of patients with metastatic anaplastic lymphoma kinase (ALK)-positive NSCLC who have progressed on or are intolerant to crizotinib.
- Midostaurin (Rydapt, Novartis; Apr 28) was approved for the treatment of adult patients with newly diagnosed AML who are FLT3 mutation-positive, as detected by an FDA-approved test, in combination with standard cytarabine and daunorubicin induction and cytarabine consolidation.
- Osimertinib (Tagrisso, AstraZeneca; Mar 30) got regular approval for the treatment of patients with metastatic epidermal growth factor receptor (EGFR) T790M mutation-positive NSCLC, as detected by an FDA-approved test, whose disease has progressed on or after EGFR tyrosine-kinase inhibitor therapy.
- Niraparib (Zejula, Tesaro; Mar 27), a poly ADP-ribose polymerase (PARP) inhibitor, was approved for the maintenance treatment of adult patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in complete or partial response to platinum-based chemotherapy.
- Avelumab (Mar 23), a PD-L1–blocking human IgG1 lambda monoclonal antibody, got accelerated approval for the treatment of patients 12 years and older with metastatic Merkel cell carcinoma. It is the first FDA-approved product to treat this type of cancer.
- Ribociclib (Kisqali, Novartis; Mar 13), a CDK4/6 inhibitor, was approved as a breakthrough therapy after priority review for use in combination with an aromatase inhibitor as initial endocrine-based therapy for the treatment of postmenopausal women with HR-positive, HER2-negative advanced or metastatic breast cancer.
Expanded/additional indications
- Nivolumab (Opdivo, Bristol-Myers Squibb; Sep 22) got accelerated expanded indication approval for treatment of hepatocellular carcinoma (HCC) in patients previously treated with sorafenib.
- Pembrolizumab (Keytruda, Merck; Sep 22) got accelerated expanded indication approval for recurrent locally advanced or metastatic gastric or gastroesophageal junction adenocarcinoma in patients whose tumors express PD-L1 as determined by an FDA-approved test.
- DigniCap Scalp Cooling System (Dignitana Inc; Jul 3) was cleared for expanded use for reducing hair loss during chemotherapy for all solid tumors. Marketing authorization for the cooling cap had been granted in 2015 for patients with breast cancer.
- Olaparib tablets (Lynparza, AstraZeneca; Aug 17) got approval for an expanded indication as maintenance treatment of adult patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer, who are in a complete or partial response to platinum-based chemotherapy.
- Ibrutinib (Imbruvica, Pharmacyclics; Aug 2) got expanded indication approval for the treatment of adult patients with chronic graft-versus-host disease (cGVHD) after failure of one or more lines of systemic therapy. It was the first FDA-approved therapy for the treatment of cGVHD. (Ibrutinib was previously approved for chronic lymphocytic leukemia/small lymphocytic lymphoma, chronic lymphocytic leukemia/small lymphocytic lymphoma with 17p deletion, Waldenström’s macroglobulinemia, marginal zone lymphoma, and mantle cell lymphoma).
- Nivolumab (Aug 2) got an accelerated expanded indication for the treatment of patients 12 years and older with mismatch repair deficient (dMMR) and microsatellite instability-high (MSI-H) metastatic colorectal cancer that has progressed after treatment with a fluoropyrimidine, oxaliplatin, and irinotecan.
- Dabrafenib and trametinib (Tafinlar and Mekinist, Novartis; Jun 22) were approved for the expanded indication in combination for patients with metastatic NSCLC with BRAF V600E mutation as detected by an FDA-approved test. The combination demonstrated superior efficacy compared with dabrafenib alone (overall response rate: 61% and 27%, respectively).32
- Pembrolizumab (May 23) got approved for expanded indication for adult and pediatric patients with unresectable or metastatic, MSI-H or dMMR solid tumors that have progressed after treatment and who have no satisfactory alternative treatment options or with MSI-H or dMMR colorectal cancer that has progressed after treatment with a fluoropyrimidine, oxaliplatin, and irinotecan.
- Pembrolizumab (May 18) got approval for expanded indication for patients with locally advanced or metastatic urothelial carcinoma who have disease progression during or following platinum-containing chemotherapy or within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy.
- Pembrolizumab (May 10) got accelerated expanded indication for use combination with pemetrexed and carboplatin for the treatment of patients with previously untreated metastatic NSCLC.
- Regorafenib (Stivarga, Bayer; Apr 27) got an additional indication for the treatment of patients with HCC who have been previously treated with sorafenib.
- Palbociclib (Ibrance, Pfizer; Mar 31) got an expanded indication that includes first-line therapy for the treatment of hormone receptor–positive, HER2-negative advanced or metastatic breast cancer in combination with an aromatase inhibitor as initial endocrine based therapy in postmenopausal women.
- Pembrolizumab (Mar 15) got an accelerated additional indication approval for treatment of adult and pediatric patients with refractory classical Hodgkin lymphoma, or those who have relapsed after three or more previous lines of therapy.
- Lenalidomide (Revlimid, Celgene; Feb 22) got an additional indication as maintenance therapy for patients with multiple myeloma following autologous stem cell transplant.
- Nivolumab (Feb 2) got an accelerated expanded indication for treatment of patients with locally advanced or metastatic urothelial carcinoma who have disease progression during or following platinum-containing chemotherapy or have disease progression within 12 months of neoadjuvant or adjuvant treatment with a platinum-containing chemotherapy.
Modified use
- Cabazitaxel (Jevtana, Sanofi-Aventis; Sep 14) in combination with prednisone was approved at a lower dose of 20 mg/m2 every 3 weeks for the treatment of patients with metastatic castration-resistant prostate cancer previously treated with a docetaxel-containing treatment regimen. It had been approved at 25 mg/m2 every 3 weeks for this indication in 2010.
Tests/diagnostics
- Marketing approval was given to the FoundationOne CDx (Foundation Medicine; Nov 30), an NGS-based in vitro diagnostic to detect genetic mutations in 324 genes and 2 genomic signatures in any solid tumor type.
- Marketing approval was given to the Praxis Extended RAS Panel (Illumina; Jun 29), a next generation sequencing test to detect certain genetic mutations in RAS genes in tumor samples of patients with metastatic colorectal cancer. The test is used to aid in the identification of patients who may be eligible for treatment with panitumumab (Vectibix, Amgen).
- Marketing was approved for ipsogen JAK2 RGQ PCR Kit (Qiagen ; Mar 27) to detect mutations affecting the Janus tyrosine kinase 2 gene. This is the first FDA-authorized test intended to help physicians in evaluating patients for suspected polycythemia vera.
Imaging and pathology aids
- Aminolevulinic acid hydrochloride, known as ALA HCl (Gleolan, NX; Jun 6) was approved as an optical imaging agent indicated in patients with gliomas (suspected World Health Organization grades III or IV on preoperative imaging) as an adjunct for the visualization of malignant tissue during surgery.
- Marketing was approved for the Philips IntelliSite Pathology Solution (PIPS, Philips Medical Systems Nederland; Apr 17), as an aid to the pathologist to review and interpret digital images of surgical pathology slides prepared from formalin-fixed paraffin embedded tissue.
Challenges and uncertainties
The current administration’s initiatives to reduce administrative burdens is underway with the Patients Over Paperwork initiative. Eliminating and streamlining regulations to increase efficiency and improve beneficiary experience could be helpful to both oncologists and patients. For now, the Medicare Access and CHIP Reauthorization Act (MACRA) program, allows you to “pick your pace” in the 2017 performance year and report on at least one measure to avoid a payment reduction penalty on your Medicare payments in 2019. In the final rule for 2018, the CMS finalized a proposal to apply the MIPS [Merit-based Incentive Payment System] adjustment to all Part B items and services, which will include Part B drugs. This would be unfair to oncologists who treat on the basis of evidence-based guidelines and pathways and have no control over the costs of the drugs they prescribe.
In addition, more requirements will be imposed in 2018 in a move toward full MACRA implementation. All four composite categories (Quality – 60% for 2017; Advancing Care Information (ACI, renamed from Meaningful Use) – 25% for 2017; Improvement Activities (IA) – 15% for 2017; and Cost – 0% for 2017, but weighted in the future) will be scored, including resource use (cost) at 10%. CMS will collect data to assess the total cost of care and the Medicare Spend per Beneficiary to assess use. Full program implementation, with cost being assessed at 30% of your score is expected in the 2019 performance year. ASCO’s clinical affairs and policy experts have studied the implications of Part B chemotherapy drugs being included in the cost component of the MIPS scoring and will continue advocating for policies that hold clinicians responsible only for the aspects of care they can control, such as providing high-quality care based on the patient’s disease, biomarkers, comorbidities, and preferences, and not the costs of the evidence-based therapies needed by patients.
Toward a better 2018 for ourselves and our patients
As an eternal optimist, I remain enthusiastic that despite the many challenges, we will find effective ways to bring standard as well as newer, cell-based and targeted therapies to our patients and cover the costs of highly effective therapies. I also remain hopeful that improving technological capabilities and payment reforms will be used by innovative clinical and administrative care teams to give clinicians more time to improve the care and health of patients while validating the methodologies so that real world data can help us further craft therapies to improve the health of each individual who needs our care. As we close this 15th year of our journal, we hope our presentations of practical science and implementation content has helped support your work while freeing some time for you to enjoy the journey. Our best wishes for a joyful holiday season celebrated with friends and family and the patients who entrust us to help them face and live beyond their cancer diagnoses.
1. US Food & Drug Administration. Biosimilar and interchangeable
products. https://www.fda.gov/Drugs/DevelopmentApprovalProcess/
HowDrugsareDevelopedandApproved/ApprovalApplications/
¬erapeuticBiologicApplications/Biosimilars/ucm580419.htm. Last
updated October 23, 2017. Accessed December 11, 2017.
2. ASCO CancerLinQ website. https://cancerlinq.org/. Publishing/
update information not available. Accessed November 3, 2017.
3. Bodenheimer T, Sinsky C. From triple to quadruple aim: care of the
patient requires care of the provider. https://www.ncbi.nlm.nih.gov/
pmc/articles/PMC4226781/. Published November 2014. Accessed
November 7.
4. Institute for Healthcare Improvement. http://www.ihi.org/Engage/
Initiatives/TripleAim/Pages/default.aspx. No update information
given. Accessed November 3, 2017.
5. Robinson JC. Value-based physician payment in oncology: public and
private insurer initiatives. Milbank Q. 2017;95(1);184-203.
6. Newcomer L. Oral communication: Washington State Medical
Oncology Society meeting, August 19, 2017.
7. Newcomer LN, Gould B, Page RD, Donelan SA, Perkins M.
Changing physician incentives for aordable, quality cancer
care: results of an episode payment model. J Oncol Pract.
2014;10(5):322-326.
8. US Department of Health and Human Services website. Hematology/
oncology (cancer) approvals & safety notications. https://www.fda.
gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm279174.htm.
Last updated December 1, 2017. Accessed December 3, 2017.
9. Hem-Onc Today website. CAR T-cell therapy approval huge
step for oncology, but only ‘beginning of story’. https://www.
healio.com/hematology-oncology/leukemia/news/print/hemonctoday/%
7B33119631-5996-45cf-9be6-8e36466ded9e%7D/car-tcell-
therapy-approval-huge-step-for-oncology-but-only-beginningof-
story. Published September 25, 2017. Accessed November 9, 2017.
10. Gauthier J, Yakoub-Agha I. Chimeric antigen-receptor T-cell therapy
for hematological malignancies and solid tumors: Clinical data
to date, current limitations and perspectives. Curr Res in Transl Med.
2017;65(3):93-102.
11. Martin M, Holmes FA, Ejlertsen B, et al. Neratinib after trastuzumab-
based adjuvant therapy in early-stage HER2+ breast cancer:
5-year analysis of the phase III ExteNET trial [ESMO oral presentation].
Ann Oncol. 2017;28(suppl 5):v43-v67.
12. Le Tourneau C, Delord JP, Goncalves A, et al. Molecularly targeted
therapy based on tumour molecular proling versus conventional
therapy for advanced cancer SHIVA): a multicentre, openlabel,
proof-of-concept, randomised, controlled phase 2 trial. Lancet
Oncol. 2015;16(13):1324-1334.
13. Forte V, Barrak DK, Elhodaky M, Tung L, Snow A, Lang JE. ¬e
potential for liquid biopsies in the precision medical treatment of
breast cancer. Cancer Biol Med. 2016;13(1):19-40.
14. Smerage JB, Barlow WE, Hortobagyi GN, et al. Circulating tumor
cells and response to chemotherapy in metastatic breast cancer:
SWOG S0500. J Clin Oncol. 2014;32(31):3483-3489.
15. US National Library of Medicine. TAPUR: testing the use of food
and drug administration (FDA) approved drugs that target a specific
abnormality in a tumor gene in people with advanced stage cancer
(TAPUR). https://clinicaltrials.gov/ct2/show/NCT02693535.
First posted February 26, 2016; last updated September 18, 2017.
Accessed November 10, 2017.
16. US National Library of Medicine. NCI-MATCH: Targeted therapy
directed by genetic testing in treating patients with advanced refractory
solid tumors, lymphomas, or multiple myeloma. https://clinicaltrials.
gov/ct2/show/NCT02465060. First posted June 8, 2015; last
updated November 9, 2017. Accessed November 10, 2017.
17. US National Library of Medicine. QUILT-3.039: NANT Pancreatic
cancer vaccine: combination immunotherapy in subjects with pancreatic
cancer who have progressed on or after standard-of-care therapy.
https://clinicaltrials.gov/ct2/show/NCT03136406. First posted May
2, 2017; last updated October 30, 2017. Accessed November 10,
2017.
18. Cunanan KM, Gonen M, Shen R, et al. Basket trials in oncology:
A trade-o between complexity and eciency. J Clin Oncol.
2017;35(3):271-273.
19. ASCO website. https://university.asco.org/motb. Last update
November 2017. Accessed November 10, 2017.
20. ASCO website. Molecular oncology tumor boards invite discussion
of growing eld in cancer care. http://www.ascopost.com/issues/
february-25-2015/molecular-oncology-tumor-boards-invite-discussion-
of-growing-eld-in-cancer-care/. Published February 25, 2017.
Accessed November 10, 2017.
21. de Lartigue J. Liquid gold: blood-based biopsies make headway.
JCSO 2017;15(1):49-54.
22. Cristofanilli M, Budd GT, Ellis MJ, et al. Circulating tumor cells,
disease progression, and survival in metastatic breast cancer. N Engl J
Med. 2004;351(8):781-791.
23. Lucci A, Hall CS, Lodhi AK, et al. Circulating tumour cells in
non-metastatic breast cancer: a prospective study. Lancet Oncol.
2012;13(7):688-695.
24. Dawson SJ, Tsui DW, Murtaza M, et al. Analysis of circulating
tumor DNA to monitor metastatic breast cancer. N Engl J Med.
2013;368(13):1199-1209.
25. Chandarlapaty S, Chen D, He W, et al. Prevalence of ESR1 mutations
in cell-free DNA and outcomes in metastatic breast cancer: a
secondary analysis of the BOLERO-2 clinical trial. JAMA Oncol.
2016;2(10):1310-1315.
26. Kim SB, Dent R, Wongchenko WJ, et al. Concordance between
plasma-based and tissue-based next-generation sequencing in
LOTUS [Correspondence]. http://www.thelancet.com/journals/
lanonc/article/PIIS1470-2045(17)30785-4/fulltext. Published
November 2017. Accessed December 3, 2017.
27. Adalsteinsson VA, Ha G, Freeman SS, et al. Scalable whole-exome
sequencing of cell-free DNA reveals high concordance with metastatic
tumors. https://www.nature.com/articles/s41467-017-00965-y.
Published online November 6, 2017. Accessed November 19, 2017.
28. Parsons DW, Roy A, Yang Y, et al. Clinical genomics for children
with solid tumors: current realities and future opportunities
[Abstract]. Clin Cancer Res. 2016;22(1 Suppl):abstract IA16.
29. ¬ompson JC, Yee SS, Troxel AB, et al. Detection of therapeutically
targetable driver and resistance mutations in lung cancer patients by
next-generation sequencing of cell-free circulating tumor DNA. Clin
Cancer Res. 2016;22(23):5772-5782.
30. Press release, FDA. FDA approves axicabtagene ciloleucel
for large B-cell lymphoma. https://www.fda.gov/Drugs/
InformationOnDrugs/ApprovedDrugs/ucm581296.htm. Last
updated October 25, 2017. Accessed November 6, 2017.
31. Press release, FDA. FDA Approves gemtuzumab ozogamicin
for CD33-positive AML. https://www.fda.gov/Drugs/
InformationOnDrugs/ApprovedDrugs/ucm574518.htm. Last
updated September 1, 2017. Accessed November 6, 2017.
32. Press release, FDA. FDA grants regular approval to dabrafenib and
trametinib combination for metastatic NSCLC with BRAF V600E
mutation. https://www.fda.gov/Drugs/InformationOnDrugs/
ApprovedDrugs/ucm564331.htm. Last updated June 22, 2017.
Accessed November 6, 2017.
1. US Food & Drug Administration. Biosimilar and interchangeable
products. https://www.fda.gov/Drugs/DevelopmentApprovalProcess/
HowDrugsareDevelopedandApproved/ApprovalApplications/
¬erapeuticBiologicApplications/Biosimilars/ucm580419.htm. Last
updated October 23, 2017. Accessed December 11, 2017.
2. ASCO CancerLinQ website. https://cancerlinq.org/. Publishing/
update information not available. Accessed November 3, 2017.
3. Bodenheimer T, Sinsky C. From triple to quadruple aim: care of the
patient requires care of the provider. https://www.ncbi.nlm.nih.gov/
pmc/articles/PMC4226781/. Published November 2014. Accessed
November 7.
4. Institute for Healthcare Improvement. http://www.ihi.org/Engage/
Initiatives/TripleAim/Pages/default.aspx. No update information
given. Accessed November 3, 2017.
5. Robinson JC. Value-based physician payment in oncology: public and
private insurer initiatives. Milbank Q. 2017;95(1);184-203.
6. Newcomer L. Oral communication: Washington State Medical
Oncology Society meeting, August 19, 2017.
7. Newcomer LN, Gould B, Page RD, Donelan SA, Perkins M.
Changing physician incentives for aordable, quality cancer
care: results of an episode payment model. J Oncol Pract.
2014;10(5):322-326.
8. US Department of Health and Human Services website. Hematology/
oncology (cancer) approvals & safety notications. https://www.fda.
gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm279174.htm.
Last updated December 1, 2017. Accessed December 3, 2017.
9. Hem-Onc Today website. CAR T-cell therapy approval huge
step for oncology, but only ‘beginning of story’. https://www.
healio.com/hematology-oncology/leukemia/news/print/hemonctoday/%
7B33119631-5996-45cf-9be6-8e36466ded9e%7D/car-tcell-
therapy-approval-huge-step-for-oncology-but-only-beginningof-
story. Published September 25, 2017. Accessed November 9, 2017.
10. Gauthier J, Yakoub-Agha I. Chimeric antigen-receptor T-cell therapy
for hematological malignancies and solid tumors: Clinical data
to date, current limitations and perspectives. Curr Res in Transl Med.
2017;65(3):93-102.
11. Martin M, Holmes FA, Ejlertsen B, et al. Neratinib after trastuzumab-
based adjuvant therapy in early-stage HER2+ breast cancer:
5-year analysis of the phase III ExteNET trial [ESMO oral presentation].
Ann Oncol. 2017;28(suppl 5):v43-v67.
12. Le Tourneau C, Delord JP, Goncalves A, et al. Molecularly targeted
therapy based on tumour molecular proling versus conventional
therapy for advanced cancer SHIVA): a multicentre, openlabel,
proof-of-concept, randomised, controlled phase 2 trial. Lancet
Oncol. 2015;16(13):1324-1334.
13. Forte V, Barrak DK, Elhodaky M, Tung L, Snow A, Lang JE. ¬e
potential for liquid biopsies in the precision medical treatment of
breast cancer. Cancer Biol Med. 2016;13(1):19-40.
14. Smerage JB, Barlow WE, Hortobagyi GN, et al. Circulating tumor
cells and response to chemotherapy in metastatic breast cancer:
SWOG S0500. J Clin Oncol. 2014;32(31):3483-3489.
15. US National Library of Medicine. TAPUR: testing the use of food
and drug administration (FDA) approved drugs that target a specific
abnormality in a tumor gene in people with advanced stage cancer
(TAPUR). https://clinicaltrials.gov/ct2/show/NCT02693535.
First posted February 26, 2016; last updated September 18, 2017.
Accessed November 10, 2017.
16. US National Library of Medicine. NCI-MATCH: Targeted therapy
directed by genetic testing in treating patients with advanced refractory
solid tumors, lymphomas, or multiple myeloma. https://clinicaltrials.
gov/ct2/show/NCT02465060. First posted June 8, 2015; last
updated November 9, 2017. Accessed November 10, 2017.
17. US National Library of Medicine. QUILT-3.039: NANT Pancreatic
cancer vaccine: combination immunotherapy in subjects with pancreatic
cancer who have progressed on or after standard-of-care therapy.
https://clinicaltrials.gov/ct2/show/NCT03136406. First posted May
2, 2017; last updated October 30, 2017. Accessed November 10,
2017.
18. Cunanan KM, Gonen M, Shen R, et al. Basket trials in oncology:
A trade-o between complexity and eciency. J Clin Oncol.
2017;35(3):271-273.
19. ASCO website. https://university.asco.org/motb. Last update
November 2017. Accessed November 10, 2017.
20. ASCO website. Molecular oncology tumor boards invite discussion
of growing eld in cancer care. http://www.ascopost.com/issues/
february-25-2015/molecular-oncology-tumor-boards-invite-discussion-
of-growing-eld-in-cancer-care/. Published February 25, 2017.
Accessed November 10, 2017.
21. de Lartigue J. Liquid gold: blood-based biopsies make headway.
JCSO 2017;15(1):49-54.
22. Cristofanilli M, Budd GT, Ellis MJ, et al. Circulating tumor cells,
disease progression, and survival in metastatic breast cancer. N Engl J
Med. 2004;351(8):781-791.
23. Lucci A, Hall CS, Lodhi AK, et al. Circulating tumour cells in
non-metastatic breast cancer: a prospective study. Lancet Oncol.
2012;13(7):688-695.
24. Dawson SJ, Tsui DW, Murtaza M, et al. Analysis of circulating
tumor DNA to monitor metastatic breast cancer. N Engl J Med.
2013;368(13):1199-1209.
25. Chandarlapaty S, Chen D, He W, et al. Prevalence of ESR1 mutations
in cell-free DNA and outcomes in metastatic breast cancer: a
secondary analysis of the BOLERO-2 clinical trial. JAMA Oncol.
2016;2(10):1310-1315.
26. Kim SB, Dent R, Wongchenko WJ, et al. Concordance between
plasma-based and tissue-based next-generation sequencing in
LOTUS [Correspondence]. http://www.thelancet.com/journals/
lanonc/article/PIIS1470-2045(17)30785-4/fulltext. Published
November 2017. Accessed December 3, 2017.
27. Adalsteinsson VA, Ha G, Freeman SS, et al. Scalable whole-exome
sequencing of cell-free DNA reveals high concordance with metastatic
tumors. https://www.nature.com/articles/s41467-017-00965-y.
Published online November 6, 2017. Accessed November 19, 2017.
28. Parsons DW, Roy A, Yang Y, et al. Clinical genomics for children
with solid tumors: current realities and future opportunities
[Abstract]. Clin Cancer Res. 2016;22(1 Suppl):abstract IA16.
29. ¬ompson JC, Yee SS, Troxel AB, et al. Detection of therapeutically
targetable driver and resistance mutations in lung cancer patients by
next-generation sequencing of cell-free circulating tumor DNA. Clin
Cancer Res. 2016;22(23):5772-5782.
30. Press release, FDA. FDA approves axicabtagene ciloleucel
for large B-cell lymphoma. https://www.fda.gov/Drugs/
InformationOnDrugs/ApprovedDrugs/ucm581296.htm. Last
updated October 25, 2017. Accessed November 6, 2017.
31. Press release, FDA. FDA Approves gemtuzumab ozogamicin
for CD33-positive AML. https://www.fda.gov/Drugs/
InformationOnDrugs/ApprovedDrugs/ucm574518.htm. Last
updated September 1, 2017. Accessed November 6, 2017.
32. Press release, FDA. FDA grants regular approval to dabrafenib and
trametinib combination for metastatic NSCLC with BRAF V600E
mutation. https://www.fda.gov/Drugs/InformationOnDrugs/
ApprovedDrugs/ucm564331.htm. Last updated June 22, 2017.
Accessed November 6, 2017.
Expanding treatment options for diverse neuroendocrine tumors
Neuroendocrine tumors (NETs) are an extremely diverse group of cancers that have steadily increased in incidence in recent years. They can prove challenging to treat but, as we discuss here, a steady evolution in our understanding of NETs has significantly expanded the scope of therapeutic options.
A unique tumor type
NETs arise from neuroendocrine cells – cells with features of both nerve and endocrine cells that have important physiological functions, including the production and release of hormones. These tumors were first recognized by a German pathologist in the mid-1800s and were initially referred to as carcinoids in reference to their carcinoma-like appearance but lack of other malignant features.1
Unlike other solid tumors, which are associated with a particular primary location, NETs can arise anywhere in the body where neuroendocrine cells are found. They are also unique in their ability to oversecrete bioactive substances that regulate bodily functions, which results in an associated clinical syndrome, known as carcinoid syndrome, in up to 35% of patients.2,3
Although they are considered to be a relatively rare type of tumor, the incidence of NETs has been increasing in recent years. According to data from the Surveillance, Epidemiology and End Results (SEER) program, the age-adjusted incidence of NETs increased more than two-and-a-half fold during 1973-2004 and the rise is predicted to continue at an accelerated rate.4
Historically, NETs have been thought of as relatively benign because of their slow-growing nature, but it is now widely appreciated that they often metastasize. Furthermore, many patients are not symptomatic at first, so around half of all cases are not diagnosed until they have reached this more aggressive stage.4
The challenge of NET diversity
The most common type of NETs are those that arise in the gastrointestinal tract (GI-NET), representing more than 65% of cases, and for which the “carcinoid” terminology often is still applied. GI-NETs most frequently arise in the small intestine (41.8%), rectum (27.4%), and stomach (8.7%).4,5
About a quarter of NETs originate in the bronchopulmonary system, including the lungs and the thymus. Thymic NETs are particularly aggressive and are associated with a poor prognosis. Pancreatic NETs (pNETs) make up the next largest group, although they represent less than 1% of total NETs. Compared with the most common type of pancreatic cancer, pancreatic ductal adenocarcinoma, they have a more favorable prognosis. pNETs are often grouped together with GI-NETs and referred to as gastroenteropancreatic NETs (GEP-NETs).3-5 Other rarer types of NET include Merkel cell carcinoma (a type of skin cancer) and medullary thyroid cancers.
The classification network
NETs are classified according to the anatomic site from which they originate, as well as their histology, grade, and stage. Another important consideration is their level of hormone secretion. “Functional” and “nonfunctional” NETs both produce hormones, but only the former cause related symptoms.3,4,6
Functionality plays a particularly important role in the subclassification of GEP-NETs. Functional pNETs, for instance, are further divided according to the clinical syndromes associated with the hormones they produce, as insulinomas, glucagonomas, gastrinomas, somatostatinomas, and VIPomas (producing vasoactive intestinal peptide).7,8
In 2010, the World Health Organization developed a classification system for GEP-NETs that categorized these tumors as well differentiated (grade 1 or 2, depending on their rate of proliferation) and poorly differentiated (grade 3).9 The WHO classification of bronchopulmonary NETs, published in 2015, is slightly different; broken down into 3 subgroups, typical carcinoid, atypical carcinoid (corresponding to grade 1 and 2 GEP-NETs), and large and small-cell NETs (equivalent to grade 3 GEP-NETs).10
Although NETs develop from the same cell type, they in fact comprise a spectrum of diseases that vary extensively in their underlying biology, histology, and clinical behavior. Both the diversity and unique nature of NETs have become increasingly evident in recent years with the application of next-generation sequencing technologies to this tumor type. In general, NETs seem to be more genetically stable than other tumor types from the same primary location, and have fewer somatic mutations. The classic tumor suppressors and oncogenes that drive other tumor types are not common in NETs.6,11
The diversity of NETs presents a diagnostic and therapeutic challenge and, until recently, there was a paucity of effective treatment options. In the past decade, an evolution in our understanding of the molecular mechanisms underlying these tumors has altered the treatment landscape for well-differentiated tumors as an expanding array of targeted therapies with proven efficacy have become available (Table 1).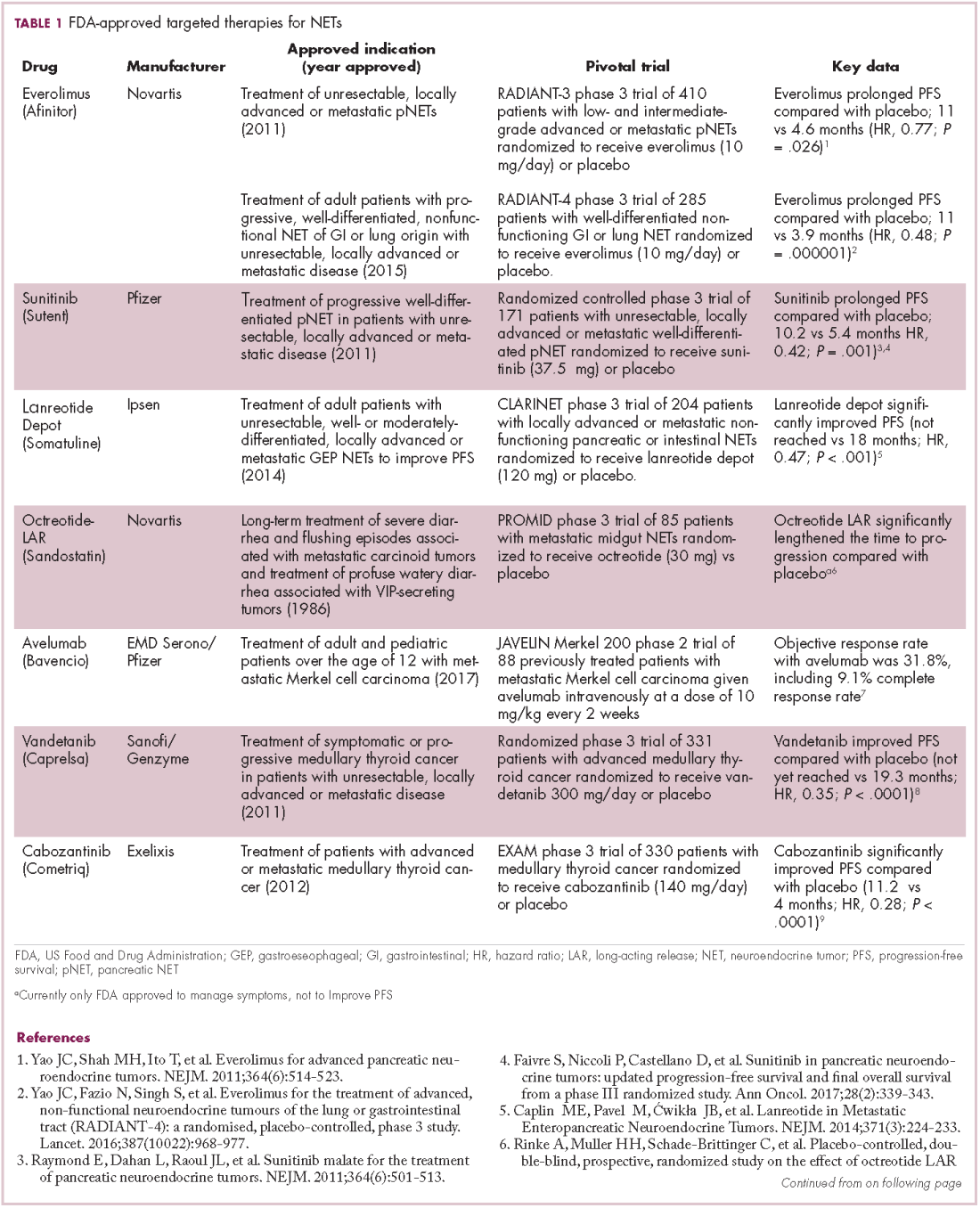

Their poorly differentiated counterparts, on the other hand, continue to present a significant unmet need.
Somatostatin analogs lead the charge
The fact that many NETs overexpress hormone receptors presents a significant therapeutic opportunity, and among the most successful targets to date are the somatostatin receptors (SSTRs). There are 5 main SSTRs that each bind to somatostatin with different effects on cell signaling and expression that varies according to the type of NET.
More stable synthetic analogs of the somatostatin hormone (somatostatin analogs [SSAs]), which has a very short half-life in the circulation, have been developed that act as SSTR agonists. Two long-acting SSAs, octreotide (Sandostatin LAR Depot) and lanreotide (Somatuline Depot), which bind SSTR2 and SSTR5, have been approved by the United States Food and Drug Administration (FDA), but were primarily used for the alleviation of the symptoms associated with NETs resulting from carcinoid syndrome.
In recent years, evidence has begun to emerge that SSAs also have an anti-tumor effect, which is thought to be both direct and indirect in nature. Direct effects result from the interaction between the SSA and SSTRs expressed on tumor cells, blocking the protumor cellular effects of SSTR signaling that are poorly understood but thought to involve the mitogen-activated protein kinase (MAPK) pathway. Indirect effects are fortuitous side effects mediated through off-target effects, such as the suppression of other cellular activities of SSTRs and the other growth factors that they bind to, which can impact processes such as angiogenesis and immune modulation.7,12
Several clinical trials have been designed to test the anti-tumor effects of NETs, including the PROMID trial of octreotide and the CLARINET trial of lanreotide, the latter leading to the 2014 approval of lanreotide for the improvement of progression-free survival (PFS) in patients with advanced GI- and pNETs.
The randomized phase 3 study compared lanreotide 120 mg with placebo in 204 patients with locally advanced or metastatic nonfunctioning pancreatic or intestinal NETs. Lanreotide treatment resulted in a significant improvement in PFS (Not yet reached vs 18 months for placebo; hazard ratio [HR], 0.47; P < .001).13
Meanwhile, the PROMID trial compared octreotide 30 mg with placebo in 85 patients with advanced midgut NETs and demonstrated that octreotide increased time to progression (TTP; 14.3 months vs 6 months for placebo; P = .000072) with no significant difference in side effects.14
Pasireotide is a second-generation SSA with improved binding affinity to SSTR1, 3, and 5. Despite its improved specificity, pasireotide has not proved more effective than other SSAs and its development for the treatment of NETs has been discontinued.
Coupling radioisotopes to SSAs provides another promising therapeutic option for NETs, known as peptide receptor radionuclide therapy, or PRRT, which uses SSAs to deliver therapeutic radiation directly to the tumor cells. Several variations have been studied with different radioactive isotopes, but most promising is lutetium-177 (177Lu). A 177Lu-labelled octreotide (177Lu-Dotatate) recently demonstrated significant efficacy in the phase 3 NETTER-1 clinical trial in patients with advanced stage NETs of the small bowel. The trial randomly assigned 229 patients who were progressing on an SSA to either 177Lu-Dotatate or high-dose octreotide LAR (long-acting release). There was a significant increase in PFS in the 177Lu-Dotatate arm (Not yet reached vs 8.4 months; P < .0001). There was also a trend toward improved overall survival (OS), and longer follow-up is eagerly anticipated for confirmation. 177Lu-Dotatate has been granted priority review by the FDA, and a decision on its approval is expected in the next few months.11,15-17
Molecularly and immune-targeted therapies continue to take aim
The mammalian target of rapamycin, or mTOR, is a serine/threonine kinase that sits at the confluence of a number of different upstream signaling pathways and mediates key cellular processes including cell proliferation and survival (Figure 1).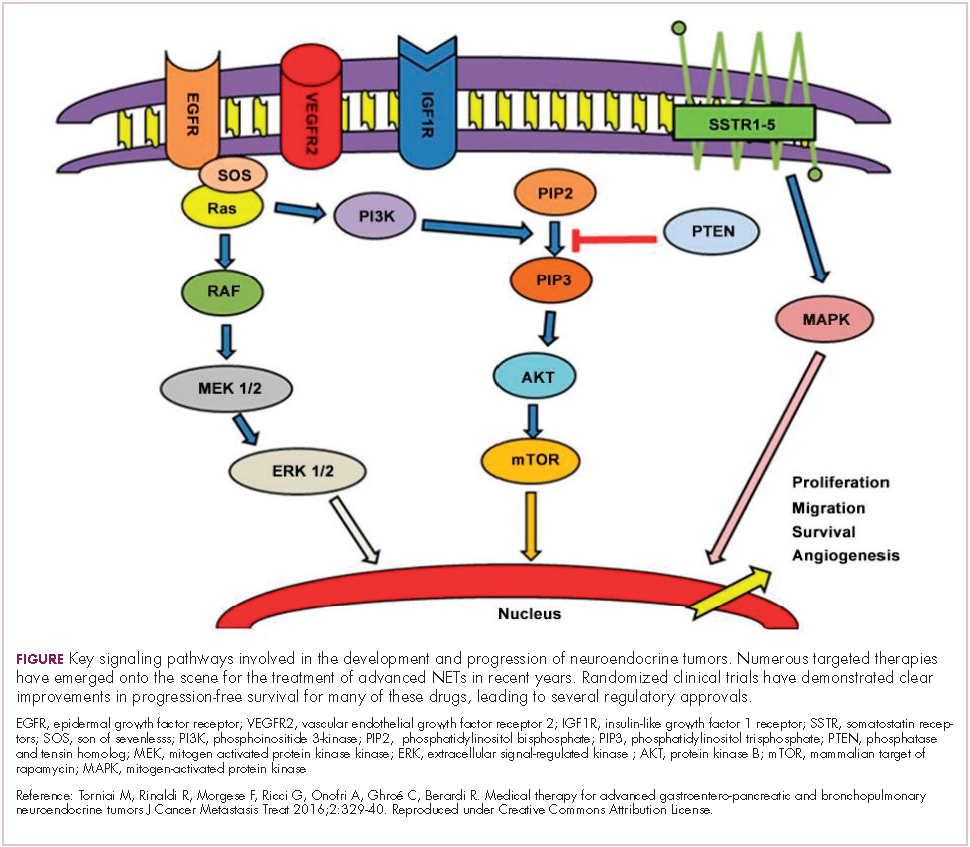
Alterations in nearly all members of the mTOR pathway, including upstream activators and downstream effectors, have been observed in NETs, in both sporadic disease and the genetic syndromes associated with the development of NETs.18
The involvement of the mTOR pathway in the pathogenesis of NETs first came into focus in pNETs and the mTOR inhibitor, everolimus (Afinitor) has been extensively studied in this indication, culminating in its regulatory approval in 2011. In the pivotal trial (RADIANT-3), everolimus monotherapy was compared with placebo in 410 patients with low- and intermediate-grade pNETs. There was a statistically significant improvement in PFS from 4.6 months to 11 months (HR, 0.77; P = .026).19 The final OS analysis for this trial also revealed a benefit of more than 6 months in the everolimus arm, although this was not statistically significant, which the study authors attribute to the high rate of crossover from the placebo arm after progression.20
More recently, the results of the RADIANT-4 trial, in which everolimus was compare with placebo in patients with advanced, well-differentiated, nonfunctioning NETs of the GI tract and lung, led to a new approved indication for the mTOR inhibitor and the first approved targeted therapy for advanced lung NETs. In the overall study population (n = 285), everolimus prolonged PFS by more than 7 months (11 months vs 3.9 months for placebo; HR, 0.48; P = .000001), corresponding to a 52% reduction in the risk of disease progression or death.21,22
Everolimus continues to be evaluated, with a particular focus on combination therapy to overcome the resistance that commonly occurs after treatment with molecularly targeted drugs (Table 2). For example, preclinical studies suggested that mTOR inhibitors and SSAs may have synergistic activity owing to combined inhibition of the mTOR and insulin-like growth factor pathways. In a phase 1 study, the combination of pasireotide and everolimus was found to be safe and to have preliminary anti-tumor activity. However, the subsequent phase 2 COOPERATE-2 study failed to show improved PFS.23,24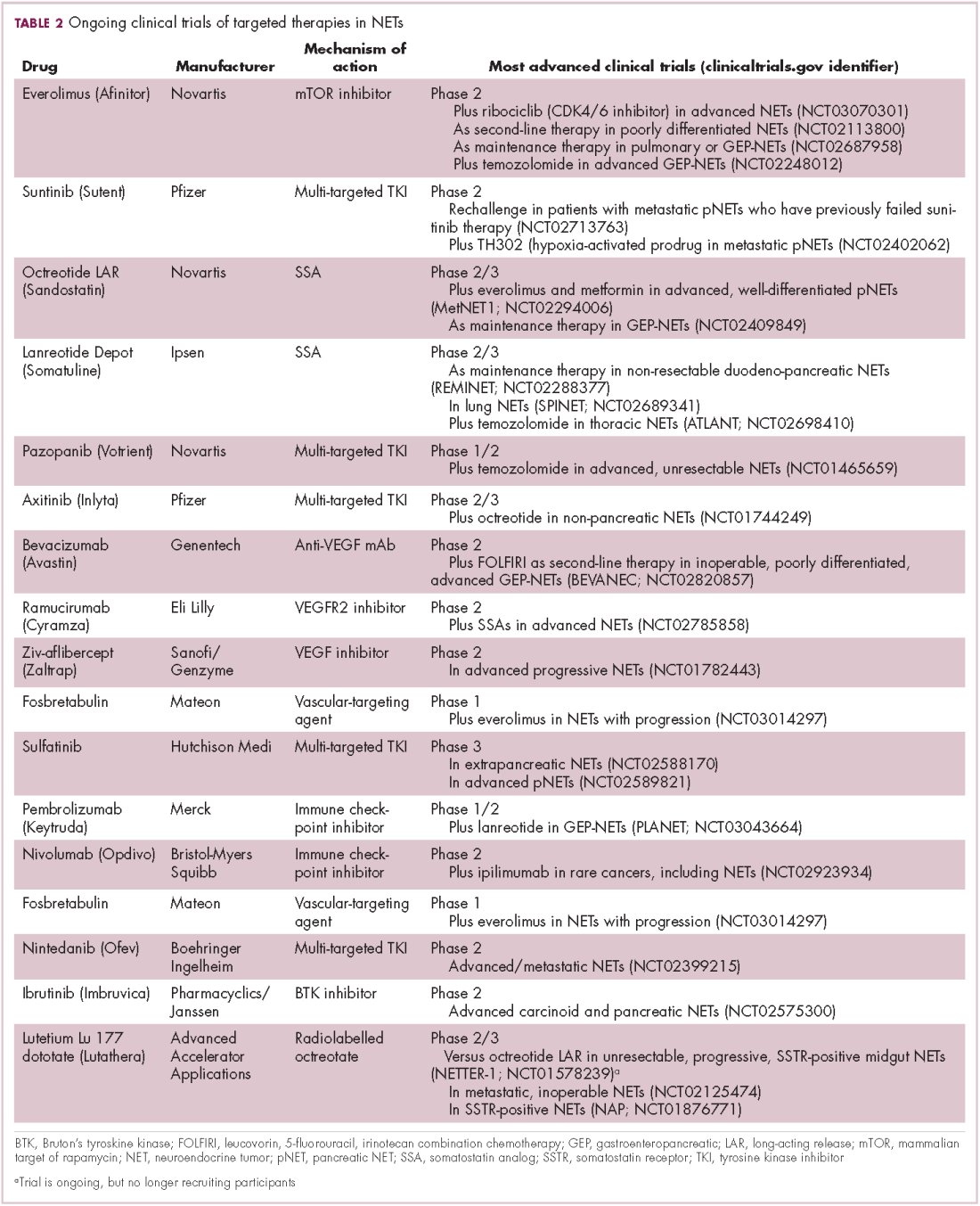
The observation that NETs are highly vascularized and frequently express vascular endothelial growth factor (VEGF) and its receptor (VEGFR), which play a key role in coordinating angiogenesis, led to the pursuit of anti-angiogenic therapies in NETs. Both the anti-VEGF monoclonal antibody bevacizumab and small molecule tyrosine kinase inhibitors that include among their targets VEGFRs and other receptors involved in angiogenesis, such as platelet-derived growth factor receptor, have been tested.
Sunitinib was approved for the treatment of pNETs in 2011, making it a banner year for this tumor type. Approval was granted on the basis of significantly improved PFS in the sunitinib arm of a phase 3 randomized trial, but long-term follow-up suggested that sunitinib also improved OS by 10 months. Like everolimus, the OS benefit was not statistically significant, and again this was thought to be the result of extensive crossover.
Two other multikinase inhibitors have received regulatory approval for a much rarer form of NET, medullary thyroid cancer. Vandetanib and cabozantinib were approved for this indication in 2011 and 2012, respectively. Early in 2017, the results of a single-arm phase 2 trial of cabozantinib suggested that this drug may also have significant activity in other types of NET. In patients with advanced carcinoid and pNETs who received cabozantinib at 60 mg/day orally, partial responses were observed in 15% of patients and the median PFS was 21.8 months in the pNET cohort and >30 months in the carcinoid tumor cohort.25 Confirmatory phase 3 trials are planned but not currently underway.
Sulfatinib is a novel kinase inhibitor that targets the VEGFRs and fibroblast growth factor receptor 1. It has recently shown significant promise in the treatment of patients with advanced NETs. According to data presented at this year’s annual conference of the European Neuroendocrine Tumor Society in Barcelona, sulfatinib demonstrated an overall response rate of 17.1% in pancreatic NETs and 15% in extra-pancreatic NETs, with an overall disease control rate of 91.4%, and was well tolerated.26 Based on these and other promising phase 1 and 2 data, 2 phase 3 trials are ongoing.
Meanwhile, earlier this year, Mateon Therapeutics presented data from a phase 2 trial of a different kind of anti-angiogenic drug in patients with GI- or pNETs. Fosbretabulin is a vascular disrupting agent that targets the existing tumor vasculature rather than preventing the formation of new blood vessels. They do this via a number of different mechanisms, in the case of fosbretabulin it specifically targets endothelial cells and inhibits the assembly of microtubules and, hence, blocks mitosis. In 18 patients, fosbretabulin treatment resulted in 1 partial response and 7 patients who had stable disease; more than half of the patients reported improved quality of life.27 Fosbretabulin continues to be studied in NETs in combination with everolimus.
Finally, researchers are beginning to make a foray into the immunotherapy field that has revolutionized the treatment of many other tumor types. The immune checkpoint inhibitors nivolumab and pembrolizumab are being evaluated in ongoing phase 1 and 2 trials, while avelumab (Bavencio) was very recently approved by the FDA for the treatment of Merkel cell carcinoma.28,29
1. Pinchot SN, Holen K, Sippel RS, Chen H. Carcinoid tumors. Oncologist. 2008;13(12):1255-1269.
2. Rorstad O. Prognostic indicators for carcinoid neuroendocrine tumors of the gastrointestinal tract. J Surg Oncol. 2005;89(3):151-160.
3. The NET Alliance. Characterizing a challenging cancer. http://www.thenetalliance.com/hcp/facts-about-net/characterization/. Publishing date not provided. Accessed October 18, 2017.
4. Yao JC, Hassan M, Phan A, et al. One hundred years after ‘carcinoid’: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26(18):3063-3072.
5. Modlin IM, Lye KD, Kidd M. A 5-decade analysis of 13,715 carcinoid tumors. Cancer. 2003;97(4):934-959.
6. Spada F, Valente M. Review of recent advances in medical treatment for neuroendocrine neoplasms: somatostatin analogs and chemotherapy. J Cancer Metastasis Treat. 2016;2(8):313-320.
7. Kelgiorgi D, Dervenis C. Pancreatic neuroendocrine tumors: the basics, the gray zone, and the target. F1000Research. 2017;6:663.
8. Viudez A, De Jesus-Acosta A, Carvalho FL, Vera R, Martin-Algarra S, Ramirez N. Pancreatic neuroendocrine tumors: Challenges in an underestimated disease. Crit Rev Oncol Hematol. 2016;101:193-206.
9. World Health Organization, International Agency for Research on Cancer. Bosman FT, Carneiro F, Hruban RH, Theise ND (eds). WHO classification of tumours of the digestive system. 2010, 4th ed (vol 3).
10. Travis WD, Brambilla E, Nicholson AG, et al. The 2015 World Health Organization classification of lung tumors. J Thorac Oncol. 2015;10(9):1243-1260.
11. Lee A, Chan DL, Wong MH, et al. Systematic review of the role of targeted therapy in metastatic neuroendocrine tumors. Neuroendocrinology. 2017;104(3):209-222.
12. Theodoropoulou M, Stalla GK. Somatostatin receptors: from signaling to clinical practice. Front Neuroendocrinol. 2013;34(3):228-252.
13. Caplin ME, Pavel M, Cwikła JB, et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med. 2014;371(3):224-233.
14. Rinke A, Muller HH, Schade-Brittinger C, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol. 2009;27(28):4656-4663.
15. Strosberg J, El-Haddad G, Wolin E, et al. Phase 3 trial of 177Lu-Dotatate for midgut neuroendocrine tumors. N Engl J Med. 2017;376(2):125-135.
16. Falconi M, Partelli S. Neuroendocrine tumours in 2016: defining rules for increasingly personalized treatments. Nat Rev Clin Oncol. 2017;14(2):80-82.
17. Hutchinson L. Targeted therapies: widening the treatment NET. Nat Rev Clin Oncol. 2017;14(1):2-3.
18. Cingarlini S, Bonomi M, Corbo V, Scarpa A, Tortora G. Profiling mTOR pathway in neuroendocrine tumors. Target Oncol. 2012;7(3):183-188.
19. Yao JC, Shah MH, Ito T, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):514-523.
20. Yao JC, Pavel M, Lombard-Bohas C, et al. Everolimus for the treatment of advanced pancreatic neuroendocrine tumors: overall survival and circulating biomarkers from the randomized, phase III RADIANT-3 study. J Clin Oncol. http://ascopubs.org/ doi/abs/10.1200/JCO.2016.68.0702?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%3dpubmed. September 12, 2016. E-pub ahead of print.
21. Yao JC, Fazio N, Singh S, et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): a randomised, placebo-controlled, phase 3 study. Lancet. 2016;387(10022):968-977.
22. Gajate P, Martínez-Sáez O, Alonso-Gordoa T, Grande E. Emerging use of everolimus in the treatment of neuroendocrine tumors. Cancer Manage Res. 2017;9:215-224.
23. Chan JA, Ryan DP, Zhu AX, et al. Phase I study of pasireotide (SOM 230) and everolimus (RAD001) in advanced neuroendocrine tumors. Endocr Relat Cancer. 2012;19(5):615-623.
24. Kulke MH, Ruszniewski P, Van Cutsem E, et al. A randomized, open-label, phase 2 study of everolimus in combination with pasireotide LAR or everolimus alone in advanced, well-dierentiated, progressive pancreatic neuroendocrine tumors: COOPERATE-2 trial. Ann Oncol. 2017;28(6):1309-1315.
25. Chan JA, Faris JE, Murphy JE, et al. Phase II trial of cabozantinib in patients with carcinoid and pancreatic neuroendocrine tumors (pNET). J Clin Oncol. 2017;35(4 suppl):228-228.
26. Xu J, Li J, Bai CM, et al. An open-label phase Ib/II study of sulfatinib in patients with advanced neuroendocrine tumors (NCT02267967). Paper presented at the 14th Annual European Neuroendocrine Tumor Society Conference for the Diagnosis and Treatment of Neuroendocrine Tumor Disease; March 8-10, 2017, Barcelona, Spain.
27. Libutti SK, Anthony LB, Chaplin DJ, Sosa JA. A phase II study of combretastatin A4-phosphate (CA4P) in the treatment of well-differentiated, low- to intermediate-grade, unresectable, recurrent, or metastatic pancreatic, or GI neuroendocrine tumors/carcinoid (GI-NETs/PNETs) with elevated biomarkers. J Clin Oncol. 2017;35(4 suppl):432-432.
28. Cordes LM, Gulley JL. Avelumab for the treatment of metastatic Merkel cell carcinoma. Drugs Today (Barc). 2017;53(7):377-383.
29. Kaufman HL, Russell J, Hamid O, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016;17(10):1374-1385.
Neuroendocrine tumors (NETs) are an extremely diverse group of cancers that have steadily increased in incidence in recent years. They can prove challenging to treat but, as we discuss here, a steady evolution in our understanding of NETs has significantly expanded the scope of therapeutic options.
A unique tumor type
NETs arise from neuroendocrine cells – cells with features of both nerve and endocrine cells that have important physiological functions, including the production and release of hormones. These tumors were first recognized by a German pathologist in the mid-1800s and were initially referred to as carcinoids in reference to their carcinoma-like appearance but lack of other malignant features.1
Unlike other solid tumors, which are associated with a particular primary location, NETs can arise anywhere in the body where neuroendocrine cells are found. They are also unique in their ability to oversecrete bioactive substances that regulate bodily functions, which results in an associated clinical syndrome, known as carcinoid syndrome, in up to 35% of patients.2,3
Although they are considered to be a relatively rare type of tumor, the incidence of NETs has been increasing in recent years. According to data from the Surveillance, Epidemiology and End Results (SEER) program, the age-adjusted incidence of NETs increased more than two-and-a-half fold during 1973-2004 and the rise is predicted to continue at an accelerated rate.4
Historically, NETs have been thought of as relatively benign because of their slow-growing nature, but it is now widely appreciated that they often metastasize. Furthermore, many patients are not symptomatic at first, so around half of all cases are not diagnosed until they have reached this more aggressive stage.4
The challenge of NET diversity
The most common type of NETs are those that arise in the gastrointestinal tract (GI-NET), representing more than 65% of cases, and for which the “carcinoid” terminology often is still applied. GI-NETs most frequently arise in the small intestine (41.8%), rectum (27.4%), and stomach (8.7%).4,5
About a quarter of NETs originate in the bronchopulmonary system, including the lungs and the thymus. Thymic NETs are particularly aggressive and are associated with a poor prognosis. Pancreatic NETs (pNETs) make up the next largest group, although they represent less than 1% of total NETs. Compared with the most common type of pancreatic cancer, pancreatic ductal adenocarcinoma, they have a more favorable prognosis. pNETs are often grouped together with GI-NETs and referred to as gastroenteropancreatic NETs (GEP-NETs).3-5 Other rarer types of NET include Merkel cell carcinoma (a type of skin cancer) and medullary thyroid cancers.
The classification network
NETs are classified according to the anatomic site from which they originate, as well as their histology, grade, and stage. Another important consideration is their level of hormone secretion. “Functional” and “nonfunctional” NETs both produce hormones, but only the former cause related symptoms.3,4,6
Functionality plays a particularly important role in the subclassification of GEP-NETs. Functional pNETs, for instance, are further divided according to the clinical syndromes associated with the hormones they produce, as insulinomas, glucagonomas, gastrinomas, somatostatinomas, and VIPomas (producing vasoactive intestinal peptide).7,8
In 2010, the World Health Organization developed a classification system for GEP-NETs that categorized these tumors as well differentiated (grade 1 or 2, depending on their rate of proliferation) and poorly differentiated (grade 3).9 The WHO classification of bronchopulmonary NETs, published in 2015, is slightly different; broken down into 3 subgroups, typical carcinoid, atypical carcinoid (corresponding to grade 1 and 2 GEP-NETs), and large and small-cell NETs (equivalent to grade 3 GEP-NETs).10
Although NETs develop from the same cell type, they in fact comprise a spectrum of diseases that vary extensively in their underlying biology, histology, and clinical behavior. Both the diversity and unique nature of NETs have become increasingly evident in recent years with the application of next-generation sequencing technologies to this tumor type. In general, NETs seem to be more genetically stable than other tumor types from the same primary location, and have fewer somatic mutations. The classic tumor suppressors and oncogenes that drive other tumor types are not common in NETs.6,11
The diversity of NETs presents a diagnostic and therapeutic challenge and, until recently, there was a paucity of effective treatment options. In the past decade, an evolution in our understanding of the molecular mechanisms underlying these tumors has altered the treatment landscape for well-differentiated tumors as an expanding array of targeted therapies with proven efficacy have become available (Table 1).
Their poorly differentiated counterparts, on the other hand, continue to present a significant unmet need.
Somatostatin analogs lead the charge
The fact that many NETs overexpress hormone receptors presents a significant therapeutic opportunity, and among the most successful targets to date are the somatostatin receptors (SSTRs). There are 5 main SSTRs that each bind to somatostatin with different effects on cell signaling and expression that varies according to the type of NET.
More stable synthetic analogs of the somatostatin hormone (somatostatin analogs [SSAs]), which has a very short half-life in the circulation, have been developed that act as SSTR agonists. Two long-acting SSAs, octreotide (Sandostatin LAR Depot) and lanreotide (Somatuline Depot), which bind SSTR2 and SSTR5, have been approved by the United States Food and Drug Administration (FDA), but were primarily used for the alleviation of the symptoms associated with NETs resulting from carcinoid syndrome.
In recent years, evidence has begun to emerge that SSAs also have an anti-tumor effect, which is thought to be both direct and indirect in nature. Direct effects result from the interaction between the SSA and SSTRs expressed on tumor cells, blocking the protumor cellular effects of SSTR signaling that are poorly understood but thought to involve the mitogen-activated protein kinase (MAPK) pathway. Indirect effects are fortuitous side effects mediated through off-target effects, such as the suppression of other cellular activities of SSTRs and the other growth factors that they bind to, which can impact processes such as angiogenesis and immune modulation.7,12
Several clinical trials have been designed to test the anti-tumor effects of NETs, including the PROMID trial of octreotide and the CLARINET trial of lanreotide, the latter leading to the 2014 approval of lanreotide for the improvement of progression-free survival (PFS) in patients with advanced GI- and pNETs.
The randomized phase 3 study compared lanreotide 120 mg with placebo in 204 patients with locally advanced or metastatic nonfunctioning pancreatic or intestinal NETs. Lanreotide treatment resulted in a significant improvement in PFS (Not yet reached vs 18 months for placebo; hazard ratio [HR], 0.47; P < .001).13
Meanwhile, the PROMID trial compared octreotide 30 mg with placebo in 85 patients with advanced midgut NETs and demonstrated that octreotide increased time to progression (TTP; 14.3 months vs 6 months for placebo; P = .000072) with no significant difference in side effects.14
Pasireotide is a second-generation SSA with improved binding affinity to SSTR1, 3, and 5. Despite its improved specificity, pasireotide has not proved more effective than other SSAs and its development for the treatment of NETs has been discontinued.
Coupling radioisotopes to SSAs provides another promising therapeutic option for NETs, known as peptide receptor radionuclide therapy, or PRRT, which uses SSAs to deliver therapeutic radiation directly to the tumor cells. Several variations have been studied with different radioactive isotopes, but most promising is lutetium-177 (177Lu). A 177Lu-labelled octreotide (177Lu-Dotatate) recently demonstrated significant efficacy in the phase 3 NETTER-1 clinical trial in patients with advanced stage NETs of the small bowel. The trial randomly assigned 229 patients who were progressing on an SSA to either 177Lu-Dotatate or high-dose octreotide LAR (long-acting release). There was a significant increase in PFS in the 177Lu-Dotatate arm (Not yet reached vs 8.4 months; P < .0001). There was also a trend toward improved overall survival (OS), and longer follow-up is eagerly anticipated for confirmation. 177Lu-Dotatate has been granted priority review by the FDA, and a decision on its approval is expected in the next few months.11,15-17
Molecularly and immune-targeted therapies continue to take aim
The mammalian target of rapamycin, or mTOR, is a serine/threonine kinase that sits at the confluence of a number of different upstream signaling pathways and mediates key cellular processes including cell proliferation and survival (Figure 1).
Alterations in nearly all members of the mTOR pathway, including upstream activators and downstream effectors, have been observed in NETs, in both sporadic disease and the genetic syndromes associated with the development of NETs.18
The involvement of the mTOR pathway in the pathogenesis of NETs first came into focus in pNETs and the mTOR inhibitor, everolimus (Afinitor) has been extensively studied in this indication, culminating in its regulatory approval in 2011. In the pivotal trial (RADIANT-3), everolimus monotherapy was compared with placebo in 410 patients with low- and intermediate-grade pNETs. There was a statistically significant improvement in PFS from 4.6 months to 11 months (HR, 0.77; P = .026).19 The final OS analysis for this trial also revealed a benefit of more than 6 months in the everolimus arm, although this was not statistically significant, which the study authors attribute to the high rate of crossover from the placebo arm after progression.20
More recently, the results of the RADIANT-4 trial, in which everolimus was compare with placebo in patients with advanced, well-differentiated, nonfunctioning NETs of the GI tract and lung, led to a new approved indication for the mTOR inhibitor and the first approved targeted therapy for advanced lung NETs. In the overall study population (n = 285), everolimus prolonged PFS by more than 7 months (11 months vs 3.9 months for placebo; HR, 0.48; P = .000001), corresponding to a 52% reduction in the risk of disease progression or death.21,22
Everolimus continues to be evaluated, with a particular focus on combination therapy to overcome the resistance that commonly occurs after treatment with molecularly targeted drugs (Table 2). For example, preclinical studies suggested that mTOR inhibitors and SSAs may have synergistic activity owing to combined inhibition of the mTOR and insulin-like growth factor pathways. In a phase 1 study, the combination of pasireotide and everolimus was found to be safe and to have preliminary anti-tumor activity. However, the subsequent phase 2 COOPERATE-2 study failed to show improved PFS.23,24
The observation that NETs are highly vascularized and frequently express vascular endothelial growth factor (VEGF) and its receptor (VEGFR), which play a key role in coordinating angiogenesis, led to the pursuit of anti-angiogenic therapies in NETs. Both the anti-VEGF monoclonal antibody bevacizumab and small molecule tyrosine kinase inhibitors that include among their targets VEGFRs and other receptors involved in angiogenesis, such as platelet-derived growth factor receptor, have been tested.
Sunitinib was approved for the treatment of pNETs in 2011, making it a banner year for this tumor type. Approval was granted on the basis of significantly improved PFS in the sunitinib arm of a phase 3 randomized trial, but long-term follow-up suggested that sunitinib also improved OS by 10 months. Like everolimus, the OS benefit was not statistically significant, and again this was thought to be the result of extensive crossover.
Two other multikinase inhibitors have received regulatory approval for a much rarer form of NET, medullary thyroid cancer. Vandetanib and cabozantinib were approved for this indication in 2011 and 2012, respectively. Early in 2017, the results of a single-arm phase 2 trial of cabozantinib suggested that this drug may also have significant activity in other types of NET. In patients with advanced carcinoid and pNETs who received cabozantinib at 60 mg/day orally, partial responses were observed in 15% of patients and the median PFS was 21.8 months in the pNET cohort and >30 months in the carcinoid tumor cohort.25 Confirmatory phase 3 trials are planned but not currently underway.
Sulfatinib is a novel kinase inhibitor that targets the VEGFRs and fibroblast growth factor receptor 1. It has recently shown significant promise in the treatment of patients with advanced NETs. According to data presented at this year’s annual conference of the European Neuroendocrine Tumor Society in Barcelona, sulfatinib demonstrated an overall response rate of 17.1% in pancreatic NETs and 15% in extra-pancreatic NETs, with an overall disease control rate of 91.4%, and was well tolerated.26 Based on these and other promising phase 1 and 2 data, 2 phase 3 trials are ongoing.
Meanwhile, earlier this year, Mateon Therapeutics presented data from a phase 2 trial of a different kind of anti-angiogenic drug in patients with GI- or pNETs. Fosbretabulin is a vascular disrupting agent that targets the existing tumor vasculature rather than preventing the formation of new blood vessels. They do this via a number of different mechanisms, in the case of fosbretabulin it specifically targets endothelial cells and inhibits the assembly of microtubules and, hence, blocks mitosis. In 18 patients, fosbretabulin treatment resulted in 1 partial response and 7 patients who had stable disease; more than half of the patients reported improved quality of life.27 Fosbretabulin continues to be studied in NETs in combination with everolimus.
Finally, researchers are beginning to make a foray into the immunotherapy field that has revolutionized the treatment of many other tumor types. The immune checkpoint inhibitors nivolumab and pembrolizumab are being evaluated in ongoing phase 1 and 2 trials, while avelumab (Bavencio) was very recently approved by the FDA for the treatment of Merkel cell carcinoma.28,29
Neuroendocrine tumors (NETs) are an extremely diverse group of cancers that have steadily increased in incidence in recent years. They can prove challenging to treat but, as we discuss here, a steady evolution in our understanding of NETs has significantly expanded the scope of therapeutic options.
A unique tumor type
NETs arise from neuroendocrine cells – cells with features of both nerve and endocrine cells that have important physiological functions, including the production and release of hormones. These tumors were first recognized by a German pathologist in the mid-1800s and were initially referred to as carcinoids in reference to their carcinoma-like appearance but lack of other malignant features.1
Unlike other solid tumors, which are associated with a particular primary location, NETs can arise anywhere in the body where neuroendocrine cells are found. They are also unique in their ability to oversecrete bioactive substances that regulate bodily functions, which results in an associated clinical syndrome, known as carcinoid syndrome, in up to 35% of patients.2,3
Although they are considered to be a relatively rare type of tumor, the incidence of NETs has been increasing in recent years. According to data from the Surveillance, Epidemiology and End Results (SEER) program, the age-adjusted incidence of NETs increased more than two-and-a-half fold during 1973-2004 and the rise is predicted to continue at an accelerated rate.4
Historically, NETs have been thought of as relatively benign because of their slow-growing nature, but it is now widely appreciated that they often metastasize. Furthermore, many patients are not symptomatic at first, so around half of all cases are not diagnosed until they have reached this more aggressive stage.4
The challenge of NET diversity
The most common type of NETs are those that arise in the gastrointestinal tract (GI-NET), representing more than 65% of cases, and for which the “carcinoid” terminology often is still applied. GI-NETs most frequently arise in the small intestine (41.8%), rectum (27.4%), and stomach (8.7%).4,5
About a quarter of NETs originate in the bronchopulmonary system, including the lungs and the thymus. Thymic NETs are particularly aggressive and are associated with a poor prognosis. Pancreatic NETs (pNETs) make up the next largest group, although they represent less than 1% of total NETs. Compared with the most common type of pancreatic cancer, pancreatic ductal adenocarcinoma, they have a more favorable prognosis. pNETs are often grouped together with GI-NETs and referred to as gastroenteropancreatic NETs (GEP-NETs).3-5 Other rarer types of NET include Merkel cell carcinoma (a type of skin cancer) and medullary thyroid cancers.
The classification network
NETs are classified according to the anatomic site from which they originate, as well as their histology, grade, and stage. Another important consideration is their level of hormone secretion. “Functional” and “nonfunctional” NETs both produce hormones, but only the former cause related symptoms.3,4,6
Functionality plays a particularly important role in the subclassification of GEP-NETs. Functional pNETs, for instance, are further divided according to the clinical syndromes associated with the hormones they produce, as insulinomas, glucagonomas, gastrinomas, somatostatinomas, and VIPomas (producing vasoactive intestinal peptide).7,8
In 2010, the World Health Organization developed a classification system for GEP-NETs that categorized these tumors as well differentiated (grade 1 or 2, depending on their rate of proliferation) and poorly differentiated (grade 3).9 The WHO classification of bronchopulmonary NETs, published in 2015, is slightly different; broken down into 3 subgroups, typical carcinoid, atypical carcinoid (corresponding to grade 1 and 2 GEP-NETs), and large and small-cell NETs (equivalent to grade 3 GEP-NETs).10
Although NETs develop from the same cell type, they in fact comprise a spectrum of diseases that vary extensively in their underlying biology, histology, and clinical behavior. Both the diversity and unique nature of NETs have become increasingly evident in recent years with the application of next-generation sequencing technologies to this tumor type. In general, NETs seem to be more genetically stable than other tumor types from the same primary location, and have fewer somatic mutations. The classic tumor suppressors and oncogenes that drive other tumor types are not common in NETs.6,11
The diversity of NETs presents a diagnostic and therapeutic challenge and, until recently, there was a paucity of effective treatment options. In the past decade, an evolution in our understanding of the molecular mechanisms underlying these tumors has altered the treatment landscape for well-differentiated tumors as an expanding array of targeted therapies with proven efficacy have become available (Table 1).
Their poorly differentiated counterparts, on the other hand, continue to present a significant unmet need.
Somatostatin analogs lead the charge
The fact that many NETs overexpress hormone receptors presents a significant therapeutic opportunity, and among the most successful targets to date are the somatostatin receptors (SSTRs). There are 5 main SSTRs that each bind to somatostatin with different effects on cell signaling and expression that varies according to the type of NET.
More stable synthetic analogs of the somatostatin hormone (somatostatin analogs [SSAs]), which has a very short half-life in the circulation, have been developed that act as SSTR agonists. Two long-acting SSAs, octreotide (Sandostatin LAR Depot) and lanreotide (Somatuline Depot), which bind SSTR2 and SSTR5, have been approved by the United States Food and Drug Administration (FDA), but were primarily used for the alleviation of the symptoms associated with NETs resulting from carcinoid syndrome.
In recent years, evidence has begun to emerge that SSAs also have an anti-tumor effect, which is thought to be both direct and indirect in nature. Direct effects result from the interaction between the SSA and SSTRs expressed on tumor cells, blocking the protumor cellular effects of SSTR signaling that are poorly understood but thought to involve the mitogen-activated protein kinase (MAPK) pathway. Indirect effects are fortuitous side effects mediated through off-target effects, such as the suppression of other cellular activities of SSTRs and the other growth factors that they bind to, which can impact processes such as angiogenesis and immune modulation.7,12
Several clinical trials have been designed to test the anti-tumor effects of NETs, including the PROMID trial of octreotide and the CLARINET trial of lanreotide, the latter leading to the 2014 approval of lanreotide for the improvement of progression-free survival (PFS) in patients with advanced GI- and pNETs.
The randomized phase 3 study compared lanreotide 120 mg with placebo in 204 patients with locally advanced or metastatic nonfunctioning pancreatic or intestinal NETs. Lanreotide treatment resulted in a significant improvement in PFS (Not yet reached vs 18 months for placebo; hazard ratio [HR], 0.47; P < .001).13
Meanwhile, the PROMID trial compared octreotide 30 mg with placebo in 85 patients with advanced midgut NETs and demonstrated that octreotide increased time to progression (TTP; 14.3 months vs 6 months for placebo; P = .000072) with no significant difference in side effects.14
Pasireotide is a second-generation SSA with improved binding affinity to SSTR1, 3, and 5. Despite its improved specificity, pasireotide has not proved more effective than other SSAs and its development for the treatment of NETs has been discontinued.
Coupling radioisotopes to SSAs provides another promising therapeutic option for NETs, known as peptide receptor radionuclide therapy, or PRRT, which uses SSAs to deliver therapeutic radiation directly to the tumor cells. Several variations have been studied with different radioactive isotopes, but most promising is lutetium-177 (177Lu). A 177Lu-labelled octreotide (177Lu-Dotatate) recently demonstrated significant efficacy in the phase 3 NETTER-1 clinical trial in patients with advanced stage NETs of the small bowel. The trial randomly assigned 229 patients who were progressing on an SSA to either 177Lu-Dotatate or high-dose octreotide LAR (long-acting release). There was a significant increase in PFS in the 177Lu-Dotatate arm (Not yet reached vs 8.4 months; P < .0001). There was also a trend toward improved overall survival (OS), and longer follow-up is eagerly anticipated for confirmation. 177Lu-Dotatate has been granted priority review by the FDA, and a decision on its approval is expected in the next few months.11,15-17
Molecularly and immune-targeted therapies continue to take aim
The mammalian target of rapamycin, or mTOR, is a serine/threonine kinase that sits at the confluence of a number of different upstream signaling pathways and mediates key cellular processes including cell proliferation and survival (Figure 1).
Alterations in nearly all members of the mTOR pathway, including upstream activators and downstream effectors, have been observed in NETs, in both sporadic disease and the genetic syndromes associated with the development of NETs.18
The involvement of the mTOR pathway in the pathogenesis of NETs first came into focus in pNETs and the mTOR inhibitor, everolimus (Afinitor) has been extensively studied in this indication, culminating in its regulatory approval in 2011. In the pivotal trial (RADIANT-3), everolimus monotherapy was compared with placebo in 410 patients with low- and intermediate-grade pNETs. There was a statistically significant improvement in PFS from 4.6 months to 11 months (HR, 0.77; P = .026).19 The final OS analysis for this trial also revealed a benefit of more than 6 months in the everolimus arm, although this was not statistically significant, which the study authors attribute to the high rate of crossover from the placebo arm after progression.20
More recently, the results of the RADIANT-4 trial, in which everolimus was compare with placebo in patients with advanced, well-differentiated, nonfunctioning NETs of the GI tract and lung, led to a new approved indication for the mTOR inhibitor and the first approved targeted therapy for advanced lung NETs. In the overall study population (n = 285), everolimus prolonged PFS by more than 7 months (11 months vs 3.9 months for placebo; HR, 0.48; P = .000001), corresponding to a 52% reduction in the risk of disease progression or death.21,22
Everolimus continues to be evaluated, with a particular focus on combination therapy to overcome the resistance that commonly occurs after treatment with molecularly targeted drugs (Table 2). For example, preclinical studies suggested that mTOR inhibitors and SSAs may have synergistic activity owing to combined inhibition of the mTOR and insulin-like growth factor pathways. In a phase 1 study, the combination of pasireotide and everolimus was found to be safe and to have preliminary anti-tumor activity. However, the subsequent phase 2 COOPERATE-2 study failed to show improved PFS.23,24
The observation that NETs are highly vascularized and frequently express vascular endothelial growth factor (VEGF) and its receptor (VEGFR), which play a key role in coordinating angiogenesis, led to the pursuit of anti-angiogenic therapies in NETs. Both the anti-VEGF monoclonal antibody bevacizumab and small molecule tyrosine kinase inhibitors that include among their targets VEGFRs and other receptors involved in angiogenesis, such as platelet-derived growth factor receptor, have been tested.
Sunitinib was approved for the treatment of pNETs in 2011, making it a banner year for this tumor type. Approval was granted on the basis of significantly improved PFS in the sunitinib arm of a phase 3 randomized trial, but long-term follow-up suggested that sunitinib also improved OS by 10 months. Like everolimus, the OS benefit was not statistically significant, and again this was thought to be the result of extensive crossover.
Two other multikinase inhibitors have received regulatory approval for a much rarer form of NET, medullary thyroid cancer. Vandetanib and cabozantinib were approved for this indication in 2011 and 2012, respectively. Early in 2017, the results of a single-arm phase 2 trial of cabozantinib suggested that this drug may also have significant activity in other types of NET. In patients with advanced carcinoid and pNETs who received cabozantinib at 60 mg/day orally, partial responses were observed in 15% of patients and the median PFS was 21.8 months in the pNET cohort and >30 months in the carcinoid tumor cohort.25 Confirmatory phase 3 trials are planned but not currently underway.
Sulfatinib is a novel kinase inhibitor that targets the VEGFRs and fibroblast growth factor receptor 1. It has recently shown significant promise in the treatment of patients with advanced NETs. According to data presented at this year’s annual conference of the European Neuroendocrine Tumor Society in Barcelona, sulfatinib demonstrated an overall response rate of 17.1% in pancreatic NETs and 15% in extra-pancreatic NETs, with an overall disease control rate of 91.4%, and was well tolerated.26 Based on these and other promising phase 1 and 2 data, 2 phase 3 trials are ongoing.
Meanwhile, earlier this year, Mateon Therapeutics presented data from a phase 2 trial of a different kind of anti-angiogenic drug in patients with GI- or pNETs. Fosbretabulin is a vascular disrupting agent that targets the existing tumor vasculature rather than preventing the formation of new blood vessels. They do this via a number of different mechanisms, in the case of fosbretabulin it specifically targets endothelial cells and inhibits the assembly of microtubules and, hence, blocks mitosis. In 18 patients, fosbretabulin treatment resulted in 1 partial response and 7 patients who had stable disease; more than half of the patients reported improved quality of life.27 Fosbretabulin continues to be studied in NETs in combination with everolimus.
Finally, researchers are beginning to make a foray into the immunotherapy field that has revolutionized the treatment of many other tumor types. The immune checkpoint inhibitors nivolumab and pembrolizumab are being evaluated in ongoing phase 1 and 2 trials, while avelumab (Bavencio) was very recently approved by the FDA for the treatment of Merkel cell carcinoma.28,29
1. Pinchot SN, Holen K, Sippel RS, Chen H. Carcinoid tumors. Oncologist. 2008;13(12):1255-1269.
2. Rorstad O. Prognostic indicators for carcinoid neuroendocrine tumors of the gastrointestinal tract. J Surg Oncol. 2005;89(3):151-160.
3. The NET Alliance. Characterizing a challenging cancer. http://www.thenetalliance.com/hcp/facts-about-net/characterization/. Publishing date not provided. Accessed October 18, 2017.
4. Yao JC, Hassan M, Phan A, et al. One hundred years after ‘carcinoid’: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26(18):3063-3072.
5. Modlin IM, Lye KD, Kidd M. A 5-decade analysis of 13,715 carcinoid tumors. Cancer. 2003;97(4):934-959.
6. Spada F, Valente M. Review of recent advances in medical treatment for neuroendocrine neoplasms: somatostatin analogs and chemotherapy. J Cancer Metastasis Treat. 2016;2(8):313-320.
7. Kelgiorgi D, Dervenis C. Pancreatic neuroendocrine tumors: the basics, the gray zone, and the target. F1000Research. 2017;6:663.
8. Viudez A, De Jesus-Acosta A, Carvalho FL, Vera R, Martin-Algarra S, Ramirez N. Pancreatic neuroendocrine tumors: Challenges in an underestimated disease. Crit Rev Oncol Hematol. 2016;101:193-206.
9. World Health Organization, International Agency for Research on Cancer. Bosman FT, Carneiro F, Hruban RH, Theise ND (eds). WHO classification of tumours of the digestive system. 2010, 4th ed (vol 3).
10. Travis WD, Brambilla E, Nicholson AG, et al. The 2015 World Health Organization classification of lung tumors. J Thorac Oncol. 2015;10(9):1243-1260.
11. Lee A, Chan DL, Wong MH, et al. Systematic review of the role of targeted therapy in metastatic neuroendocrine tumors. Neuroendocrinology. 2017;104(3):209-222.
12. Theodoropoulou M, Stalla GK. Somatostatin receptors: from signaling to clinical practice. Front Neuroendocrinol. 2013;34(3):228-252.
13. Caplin ME, Pavel M, Cwikła JB, et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med. 2014;371(3):224-233.
14. Rinke A, Muller HH, Schade-Brittinger C, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol. 2009;27(28):4656-4663.
15. Strosberg J, El-Haddad G, Wolin E, et al. Phase 3 trial of 177Lu-Dotatate for midgut neuroendocrine tumors. N Engl J Med. 2017;376(2):125-135.
16. Falconi M, Partelli S. Neuroendocrine tumours in 2016: defining rules for increasingly personalized treatments. Nat Rev Clin Oncol. 2017;14(2):80-82.
17. Hutchinson L. Targeted therapies: widening the treatment NET. Nat Rev Clin Oncol. 2017;14(1):2-3.
18. Cingarlini S, Bonomi M, Corbo V, Scarpa A, Tortora G. Profiling mTOR pathway in neuroendocrine tumors. Target Oncol. 2012;7(3):183-188.
19. Yao JC, Shah MH, Ito T, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):514-523.
20. Yao JC, Pavel M, Lombard-Bohas C, et al. Everolimus for the treatment of advanced pancreatic neuroendocrine tumors: overall survival and circulating biomarkers from the randomized, phase III RADIANT-3 study. J Clin Oncol. http://ascopubs.org/ doi/abs/10.1200/JCO.2016.68.0702?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%3dpubmed. September 12, 2016. E-pub ahead of print.
21. Yao JC, Fazio N, Singh S, et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): a randomised, placebo-controlled, phase 3 study. Lancet. 2016;387(10022):968-977.
22. Gajate P, Martínez-Sáez O, Alonso-Gordoa T, Grande E. Emerging use of everolimus in the treatment of neuroendocrine tumors. Cancer Manage Res. 2017;9:215-224.
23. Chan JA, Ryan DP, Zhu AX, et al. Phase I study of pasireotide (SOM 230) and everolimus (RAD001) in advanced neuroendocrine tumors. Endocr Relat Cancer. 2012;19(5):615-623.
24. Kulke MH, Ruszniewski P, Van Cutsem E, et al. A randomized, open-label, phase 2 study of everolimus in combination with pasireotide LAR or everolimus alone in advanced, well-dierentiated, progressive pancreatic neuroendocrine tumors: COOPERATE-2 trial. Ann Oncol. 2017;28(6):1309-1315.
25. Chan JA, Faris JE, Murphy JE, et al. Phase II trial of cabozantinib in patients with carcinoid and pancreatic neuroendocrine tumors (pNET). J Clin Oncol. 2017;35(4 suppl):228-228.
26. Xu J, Li J, Bai CM, et al. An open-label phase Ib/II study of sulfatinib in patients with advanced neuroendocrine tumors (NCT02267967). Paper presented at the 14th Annual European Neuroendocrine Tumor Society Conference for the Diagnosis and Treatment of Neuroendocrine Tumor Disease; March 8-10, 2017, Barcelona, Spain.
27. Libutti SK, Anthony LB, Chaplin DJ, Sosa JA. A phase II study of combretastatin A4-phosphate (CA4P) in the treatment of well-differentiated, low- to intermediate-grade, unresectable, recurrent, or metastatic pancreatic, or GI neuroendocrine tumors/carcinoid (GI-NETs/PNETs) with elevated biomarkers. J Clin Oncol. 2017;35(4 suppl):432-432.
28. Cordes LM, Gulley JL. Avelumab for the treatment of metastatic Merkel cell carcinoma. Drugs Today (Barc). 2017;53(7):377-383.
29. Kaufman HL, Russell J, Hamid O, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016;17(10):1374-1385.
1. Pinchot SN, Holen K, Sippel RS, Chen H. Carcinoid tumors. Oncologist. 2008;13(12):1255-1269.
2. Rorstad O. Prognostic indicators for carcinoid neuroendocrine tumors of the gastrointestinal tract. J Surg Oncol. 2005;89(3):151-160.
3. The NET Alliance. Characterizing a challenging cancer. http://www.thenetalliance.com/hcp/facts-about-net/characterization/. Publishing date not provided. Accessed October 18, 2017.
4. Yao JC, Hassan M, Phan A, et al. One hundred years after ‘carcinoid’: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26(18):3063-3072.
5. Modlin IM, Lye KD, Kidd M. A 5-decade analysis of 13,715 carcinoid tumors. Cancer. 2003;97(4):934-959.
6. Spada F, Valente M. Review of recent advances in medical treatment for neuroendocrine neoplasms: somatostatin analogs and chemotherapy. J Cancer Metastasis Treat. 2016;2(8):313-320.
7. Kelgiorgi D, Dervenis C. Pancreatic neuroendocrine tumors: the basics, the gray zone, and the target. F1000Research. 2017;6:663.
8. Viudez A, De Jesus-Acosta A, Carvalho FL, Vera R, Martin-Algarra S, Ramirez N. Pancreatic neuroendocrine tumors: Challenges in an underestimated disease. Crit Rev Oncol Hematol. 2016;101:193-206.
9. World Health Organization, International Agency for Research on Cancer. Bosman FT, Carneiro F, Hruban RH, Theise ND (eds). WHO classification of tumours of the digestive system. 2010, 4th ed (vol 3).
10. Travis WD, Brambilla E, Nicholson AG, et al. The 2015 World Health Organization classification of lung tumors. J Thorac Oncol. 2015;10(9):1243-1260.
11. Lee A, Chan DL, Wong MH, et al. Systematic review of the role of targeted therapy in metastatic neuroendocrine tumors. Neuroendocrinology. 2017;104(3):209-222.
12. Theodoropoulou M, Stalla GK. Somatostatin receptors: from signaling to clinical practice. Front Neuroendocrinol. 2013;34(3):228-252.
13. Caplin ME, Pavel M, Cwikła JB, et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med. 2014;371(3):224-233.
14. Rinke A, Muller HH, Schade-Brittinger C, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol. 2009;27(28):4656-4663.
15. Strosberg J, El-Haddad G, Wolin E, et al. Phase 3 trial of 177Lu-Dotatate for midgut neuroendocrine tumors. N Engl J Med. 2017;376(2):125-135.
16. Falconi M, Partelli S. Neuroendocrine tumours in 2016: defining rules for increasingly personalized treatments. Nat Rev Clin Oncol. 2017;14(2):80-82.
17. Hutchinson L. Targeted therapies: widening the treatment NET. Nat Rev Clin Oncol. 2017;14(1):2-3.
18. Cingarlini S, Bonomi M, Corbo V, Scarpa A, Tortora G. Profiling mTOR pathway in neuroendocrine tumors. Target Oncol. 2012;7(3):183-188.
19. Yao JC, Shah MH, Ito T, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):514-523.
20. Yao JC, Pavel M, Lombard-Bohas C, et al. Everolimus for the treatment of advanced pancreatic neuroendocrine tumors: overall survival and circulating biomarkers from the randomized, phase III RADIANT-3 study. J Clin Oncol. http://ascopubs.org/ doi/abs/10.1200/JCO.2016.68.0702?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%3dpubmed. September 12, 2016. E-pub ahead of print.
21. Yao JC, Fazio N, Singh S, et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): a randomised, placebo-controlled, phase 3 study. Lancet. 2016;387(10022):968-977.
22. Gajate P, Martínez-Sáez O, Alonso-Gordoa T, Grande E. Emerging use of everolimus in the treatment of neuroendocrine tumors. Cancer Manage Res. 2017;9:215-224.
23. Chan JA, Ryan DP, Zhu AX, et al. Phase I study of pasireotide (SOM 230) and everolimus (RAD001) in advanced neuroendocrine tumors. Endocr Relat Cancer. 2012;19(5):615-623.
24. Kulke MH, Ruszniewski P, Van Cutsem E, et al. A randomized, open-label, phase 2 study of everolimus in combination with pasireotide LAR or everolimus alone in advanced, well-dierentiated, progressive pancreatic neuroendocrine tumors: COOPERATE-2 trial. Ann Oncol. 2017;28(6):1309-1315.
25. Chan JA, Faris JE, Murphy JE, et al. Phase II trial of cabozantinib in patients with carcinoid and pancreatic neuroendocrine tumors (pNET). J Clin Oncol. 2017;35(4 suppl):228-228.
26. Xu J, Li J, Bai CM, et al. An open-label phase Ib/II study of sulfatinib in patients with advanced neuroendocrine tumors (NCT02267967). Paper presented at the 14th Annual European Neuroendocrine Tumor Society Conference for the Diagnosis and Treatment of Neuroendocrine Tumor Disease; March 8-10, 2017, Barcelona, Spain.
27. Libutti SK, Anthony LB, Chaplin DJ, Sosa JA. A phase II study of combretastatin A4-phosphate (CA4P) in the treatment of well-differentiated, low- to intermediate-grade, unresectable, recurrent, or metastatic pancreatic, or GI neuroendocrine tumors/carcinoid (GI-NETs/PNETs) with elevated biomarkers. J Clin Oncol. 2017;35(4 suppl):432-432.
28. Cordes LM, Gulley JL. Avelumab for the treatment of metastatic Merkel cell carcinoma. Drugs Today (Barc). 2017;53(7):377-383.
29. Kaufman HL, Russell J, Hamid O, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016;17(10):1374-1385.
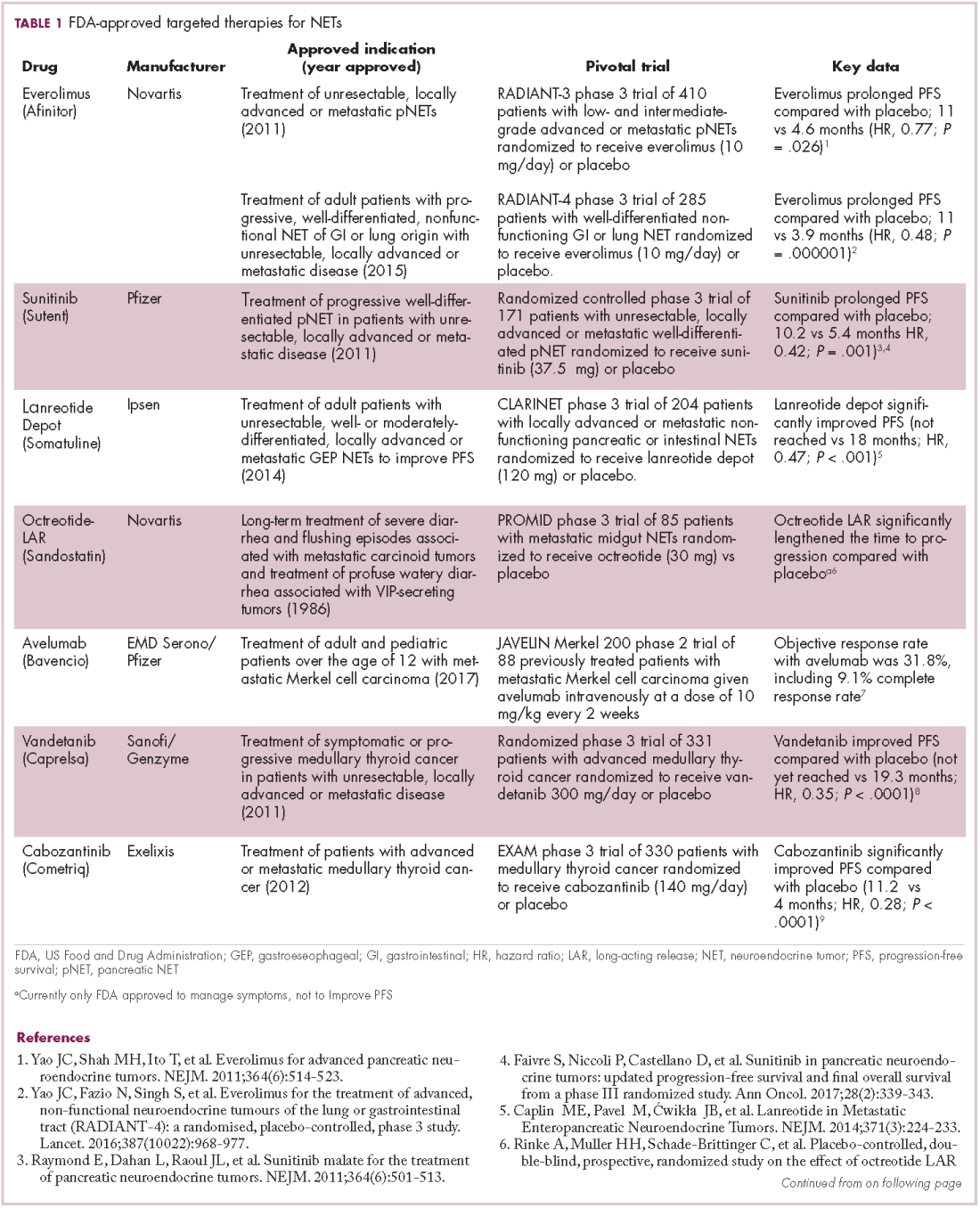
Who’s Smoking What?
About 1 in 5 U.S. adults used tobacco in 2015 every day or on some days, according to the CDC and the FDA. The most common tobacco product was cigarettes (15%). About 9.5 million adults used ≥ 2 tobacco products.
This is the first time the CDC and FDA have used the National Health Interview Survey to assess the range of tobacco products used. Survey questions asked about current cigarette smoking since 1965, but only recently began to track other tobacco products.
About 42 million adults, or > 87% of the nearly 49 million tobacco product users in the U.S., reported using cigarettes, cigars, or pipes; the remaining tobacco users reported using e-cigarettes or smokeless tobacco products (eg, chewing tobacco and snuff).
The survey also found that men were more likely than women to use tobacco products (25% vs 15%). Adults aged 25 to 44 years were more likely than those aged ≥ 65 years to use tobacco products (23% vs 11%). By race and ethnicity, tobacco product use ranged from 9% among Asians to 27% among American Indians/Alaska Natives.
Using tobacco products was more common among adults in the Midwest; people with annual incomes less than $35,000; the uninsured or Medicaid insured; those with a disability; and those who are lesbian, gay, or bisexual. Adults with serious psychological distress were more than 2 times as likely to use tobacco as those who reported no serious psychological distress (47% vs 19%).
Cigarette smoking remains the leading preventable cause of death and disease in the U.S. The CDC urges full implementation of comprehensive state tobacco control programs along with FDA regulation of tobacco products and targeted interventions to reach subpopulations with the greatest burden of use.
About 1 in 5 U.S. adults used tobacco in 2015 every day or on some days, according to the CDC and the FDA. The most common tobacco product was cigarettes (15%). About 9.5 million adults used ≥ 2 tobacco products.
This is the first time the CDC and FDA have used the National Health Interview Survey to assess the range of tobacco products used. Survey questions asked about current cigarette smoking since 1965, but only recently began to track other tobacco products.
About 42 million adults, or > 87% of the nearly 49 million tobacco product users in the U.S., reported using cigarettes, cigars, or pipes; the remaining tobacco users reported using e-cigarettes or smokeless tobacco products (eg, chewing tobacco and snuff).
The survey also found that men were more likely than women to use tobacco products (25% vs 15%). Adults aged 25 to 44 years were more likely than those aged ≥ 65 years to use tobacco products (23% vs 11%). By race and ethnicity, tobacco product use ranged from 9% among Asians to 27% among American Indians/Alaska Natives.
Using tobacco products was more common among adults in the Midwest; people with annual incomes less than $35,000; the uninsured or Medicaid insured; those with a disability; and those who are lesbian, gay, or bisexual. Adults with serious psychological distress were more than 2 times as likely to use tobacco as those who reported no serious psychological distress (47% vs 19%).
Cigarette smoking remains the leading preventable cause of death and disease in the U.S. The CDC urges full implementation of comprehensive state tobacco control programs along with FDA regulation of tobacco products and targeted interventions to reach subpopulations with the greatest burden of use.
About 1 in 5 U.S. adults used tobacco in 2015 every day or on some days, according to the CDC and the FDA. The most common tobacco product was cigarettes (15%). About 9.5 million adults used ≥ 2 tobacco products.
This is the first time the CDC and FDA have used the National Health Interview Survey to assess the range of tobacco products used. Survey questions asked about current cigarette smoking since 1965, but only recently began to track other tobacco products.
About 42 million adults, or > 87% of the nearly 49 million tobacco product users in the U.S., reported using cigarettes, cigars, or pipes; the remaining tobacco users reported using e-cigarettes or smokeless tobacco products (eg, chewing tobacco and snuff).
The survey also found that men were more likely than women to use tobacco products (25% vs 15%). Adults aged 25 to 44 years were more likely than those aged ≥ 65 years to use tobacco products (23% vs 11%). By race and ethnicity, tobacco product use ranged from 9% among Asians to 27% among American Indians/Alaska Natives.
Using tobacco products was more common among adults in the Midwest; people with annual incomes less than $35,000; the uninsured or Medicaid insured; those with a disability; and those who are lesbian, gay, or bisexual. Adults with serious psychological distress were more than 2 times as likely to use tobacco as those who reported no serious psychological distress (47% vs 19%).
Cigarette smoking remains the leading preventable cause of death and disease in the U.S. The CDC urges full implementation of comprehensive state tobacco control programs along with FDA regulation of tobacco products and targeted interventions to reach subpopulations with the greatest burden of use.
Supportive medications and interventions received by prostate cancer survivors: results from the PiCTure study
Prostate cancer treatments are associated with various physical after-effects, including urinary, sexual, and bowel symptoms.1 These after-effects can have an impact on survivors’ health-related quality of life (HRQoL).2 Pharmaceutical and surgical interventions are available to manage or ameliorate many of these after-effects (eg, sildenafil citrate taken during and after radiotherapy improves sexual function),3 and their receipt has a positive impact on HRQoL.4
However, studies of clinicians suggest that such interventions may not be used widely.5,6 Patient-reported data on this topic is lacking. Therefore, we investigated the use of supportive medications and interventions in this population-based study of prostate cancer survivors.
Methods
The PiCTure (Prostate Cancer Treatment, Your Experience) study methods have been described elsewhere.7 Briefly, 6,559 prostate cancer survivors 2-15 years after diagnosis (diagnosed during January 1, 1995-March 31, 2010, and alive in November 2011), identified from population-based cancer registries in the Republic of Ireland and Northern Ireland, were invited to complete a postal survey. Information was sought on after-effects (incontinence, impotence, gynaecomastia, hot flashes/sweats, bowel problems, depression) that had been experienced at any time after treatment. For each after-effect, men were asked if they had received any medication or interventions to alleviate symptoms, and, if so, what they had received; examples of common interventions were provided. Men were also asked if they had been told they may become infertile and, if so, whether they had preserved their sperm. The Decisional Regret Scale8 was used to measure survivors’ regret over their entire treatment experience. This 5-item scale, rated on a 5-point Likert scale from 1 (strongly agree) to 5 (strongly disagree) was summed and standardized to a value of 0-100, with higher scores reflecting higher levels of decisional regret. 8 This scale has good psychometric properties8 and strong reliability in our sample (Cronbach’s alpha = 0.85). Responders were categorized as having any regret (score ≥1) or no regret (score = 0).
The number of men who reported receiving an intervention was expressed as a percentage of survey responders and of men who reported ever having the relevant after-effect. Chi-square tests were used to investigate variations in receipt by: age at diagnosis (≤59, 60-69, ≥70 years); time since diagnosis (≤5, 5-10, >10 years); jurisdiction (Republic of Ireland, or Northern Ireland); and primary treatment(s) received (radical prostatectomy [RP], external beam radiotherapy [EBRT] with androgen deprivation therapy [ADT], EBRT without ADT, brachytherapy, ADT [without other therapies], and active surveillance/watchful waiting). Among survivors who ever experienced an after-effect, chi-square tests were used to investigate whether the percentage who reported decisional regret differed depending on whether or not they received the relevant supportive intervention.
Ethics approval was from the Irish College of General Practitioners (Republic of Ireland) and the Office for Research Ethics Committee Northern Ireland.
Results
In all, 3,348 survivors participated in the survey (adjusted response rate, 54%). Compared with nonresponders, responders were more often from the Republic of Ireland (P = .007), <70 years at diagnosis (P < .001), 5-10 years post diagnosis (P < .001), with low or medium Gleason grade (Gleason scores of ≤6 [good prognosis] and 7, respectively; P < .001), and clinical stage II-IV (P < .001; Table 1).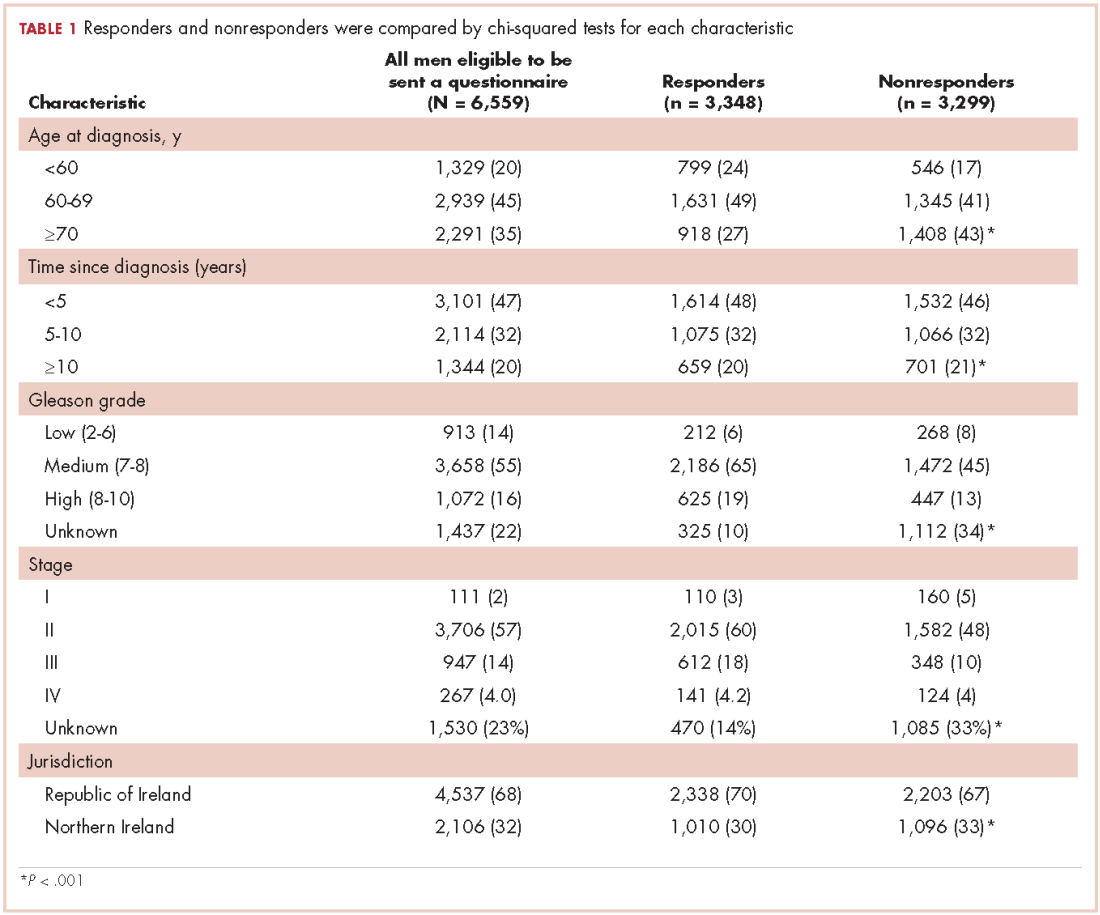
Impotence (70%) was the most commonly reported after-effect, followed by hot flashes/sweats (40%), incontinence (37%), bowel problems (23%), gynaecomastia (19%), and depression (18%; Table 2).
Of responders, 2% received an artificial sphincter, representing 6% of men who ever experienced incontinence post diagnosis (Table 2). This percentage was significantly higher in participants diagnosed longer ago, from the Republic of Ireland, and who received RP (Table 3).
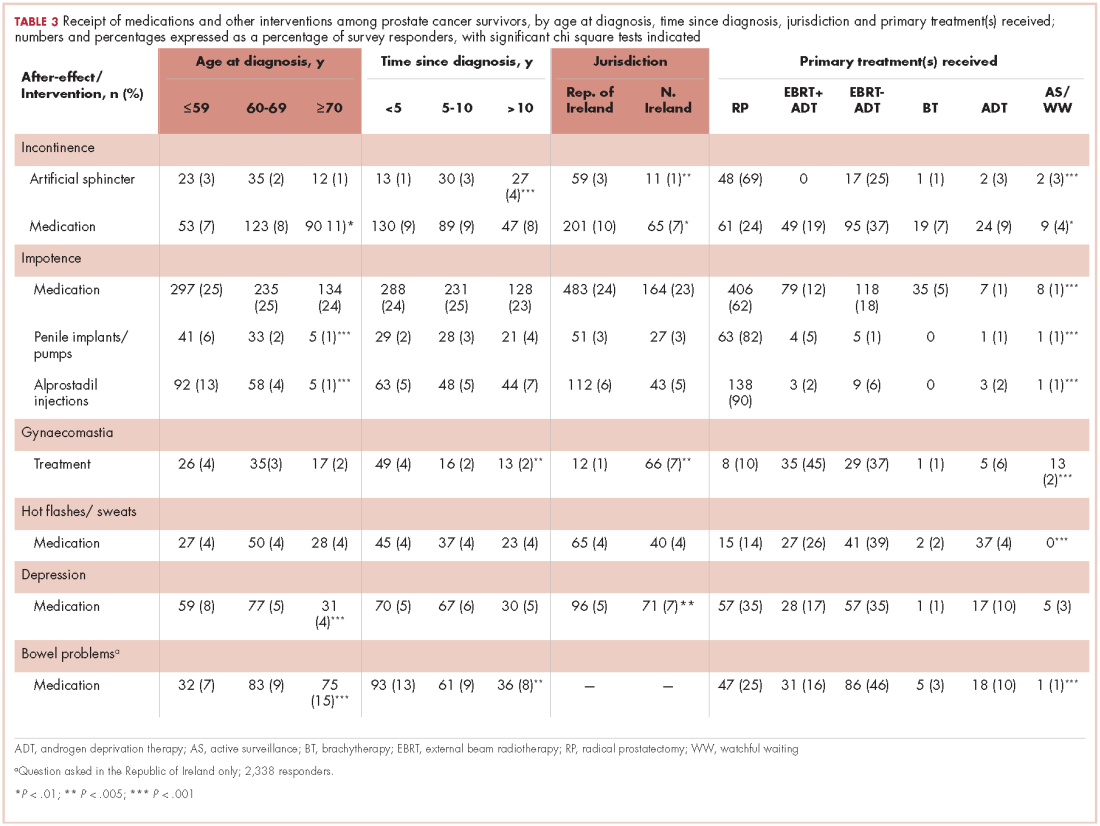
Incontinence medication was received by 8% of participants (21% of those who experienced incontinence). Use varied significantly by age, jurisdiction, and treatment. For impotence, medications were more commonly used (20% of participants; 28% with impotence) than were injections (5% and 7%, respectively) or penile implants/pumps (2% and 3%, respectively). Use of all 3 types of intervention was highest in men who had RP; injections and implants/pumps were significantly more common among younger men. Of those experiencing gynaecomastia, 13% received interventions; receipt was highest in men who had EBRT with ADT, were <5 years post diagnosis and from Northern Ireland. For hot flashes/sweats, 3% of participants (8% who experienced symptoms) received mediations; this was higher in men who had EBRT. Of those who reported depression, 28% received medication; receipt was highest in younger men and in Northern Ireland. Medication for bowel problems was used by 35% of men who experienced these; use was highest in older men, those diagnosed more recently, and those who had EBRT. Sixty percent of men reported having been told they would become infertile; 11 (0.3% of participants) preserved their sperm, 7 from the Republic of Ireland and 4 from Northern Ireland.
A total of 35.6% of survivors reported any decisional regret. Among survivors who ever had an after-effect, a higher percentage of those who used a supportive intervention reported decisional regret compared with those who did not; this was only statistically significant for those using medication or alprostadil injections for impotence (Table 2).
Discussion
This study documents, for the first time, population-based data on patient-reported use of supportive medications and interventions to alleviate adverse effects of prostate cancer and its treatment. Among survivors who experienced after-effects, use was highest for bowel problems, impotence, and depression, but even for those, only 28%-35% of men took medication. Although it is possible that some survivors declined medications or other interventions, these low levels of use strongly suggest that not all survivors who might benefit from supports receive them.
There was little evidence that utilisation was higher in survivors diagnosed more recently. This suggests that, although the number of prostate cancer survivors has grown, and there is greater focus on survivorship issues in clinical practice, this has not translated into more men receiving support to manage after-effects. Care is needed to ensure that the newer models of post-cancer follow-up being considered or adopted in many settings,9 do not exacerbate this issue.
As expected, patterns of utilisation varied by treatment(s) received. Higher use of surgical and pharmaceutical interventions to alleviate incontinence among survivors in the Republic of Ireland than in Northern Ireland is likely owing to the higher rate of radical prostatectomy in the Republic of Ireland, whereas greater use of treatments for gynaecomastia in Northern Ireland reflects higher use of hormone therapy there.10 Other variations in intervention use were more surprising. Younger men were significantly more likely to report using supportive interventions for depression and impotence, the latter finding being consistent with findings in a Swedish population-based study.11 Older men were significantly more likely to report interventions for incontinence and bowel problems. Although those trends could be explained by differences in treatment receipt by age, it is possible that men of different ages may be more likely to seek, or be offered, help for certain types of after-effects. With the exception of interventions for bowel problems, a higher percentage of men who received intervention(s) for an after-effect reported decisional regret. There are a number of possible explanations: these men may have experienced more severe after-effects, which required interventions; they may have been less satisfied with their posttreatment function and/or more proactive about recovering or treating their after-effects. This requires further investigation.
This is a large, international, population-based study, the first such study to describe patient-reported use of supportive care following a range of prostate cancer treatments. Although this study is novel, there are a number of limitations. It is a cross-sectional, descriptive study. We did not ask survivors whether the supportive interventions received matched their needs and wants, and whether they were satisfied with the supportive care received. Furthermore, although the response rate is comparable with other similar studies,12,13 it is possible that the supportive care of nonresponders was different to that of responders.
Our study included men from 2 jurisdictions with separate health care systems, suggesting that low use of supportive interventions may be common across systems. There is a need for further research into patient and health care system factors associated with the receipt of supportive interventions and how satisfied men are with these, in this and other health care settings. Presently, it is clear that more needs to be done in the clinical setting to support prostate cancer survivors manage treatment after-effects; this in turn could improve survivors’ HRQoL.
1. Drummond FJ, Kinnear H, O’Leary E, Donnelly, Gavin A, Sharp L. Long-term health-related quality of life of prostate cancer survivors varies by primary treatment. Results from the PiCTure (Prostate Cancer Treatment, your experience) study. J Cancer Surviv. 2015;9(2):361-72.
2. Smith DP, King MT, Egger S, et al. Quality of life three years after diagnosis of localised prostate cancer: population based cohort study. BMJ 2009; 339:b4817.
3. Zelefsky MJ, Shasha D, Branco RD, et al. Prophylactic sildenafil citrate improves select aspects of sexual function in men treated with radiotherapy for prostate cancer. J Urol. 2014;192(3):868-874.
4. Haab F, Trockman BA, Zimmern PE, Leach GE. Quality of life and continence assessment of the artificial urinary sphincter in men with minimum 3.5 years of follow-up. J Urol. 1997;158(2):435-439.
5. Tanvetyanon T. Physician practices of bone density testing and drug prescribing to prevent or treat osteoporosis during androgen deprivation therapy. Cancer. 2005;103(2):237-241.
6. Alibhai SM, Rahman S, Warde PR, Jewett MA, Jaffer T, Cheung AM. Prevention and management of osteoporosis in men receiving androgen deprivation therapy: a survey of urologists and radiation oncologists. Urology. 2006;68(1):126-131,
7. Drummond FJ, Kinnear H, Donnelly C, et al. Establishing a population-based patient reported outcomes study (PROMs) using national cancer registries across two jurisdictions: Prostate Cancer Treatment, your experience (PiCTure) Study. BMJ Open 2015;5:e006851.
8. Brehaut JC, O’Connor AM, Wood TJ, et al. Validation of a decision regret scale. Med Decis Making. 2003;23(4):281-92.
9. Howell D, Hack TF, Oliver et al. Models of care for post-treatment follow-up of adult cancer survivors: a systematic review and quality appraisal of the evidence. J Cancer Surviv. 2012;6(4):359-371.
10. Donnelly DW, Gavin AT, Comber H. Cancer in Ireland 1994-2004. A comprehensive report. Northern Ireland Cancer Registry/National Cancer Registry, Ireland, 2009.
11. Plym A, Folkvaljon Y, Garmo H, et al. Drug prescription for erectile dysfunction before and after diagnosis of localized prostate cancer. J Sex Med. 2014;11(8):2100-2108.
12. Hervouet S, Savard J, Simard S, et al. Psychological functioning associated with prostate cancer: cross-sectional comparison of patients treated with radiotherapy, brachytherapy, or surgery. J Pain Symptom Manage. 2005;30(5):474-484.
13. Glaser AW, Fraser LK, Corner J, et al. Patient-reported outcomes of cancer survivors in England 1-5 years after diagnosis: a cross-sectional survey. BMJ Open. 2013;3(4). pii: e002317.
Prostate cancer treatments are associated with various physical after-effects, including urinary, sexual, and bowel symptoms.1 These after-effects can have an impact on survivors’ health-related quality of life (HRQoL).2 Pharmaceutical and surgical interventions are available to manage or ameliorate many of these after-effects (eg, sildenafil citrate taken during and after radiotherapy improves sexual function),3 and their receipt has a positive impact on HRQoL.4
However, studies of clinicians suggest that such interventions may not be used widely.5,6 Patient-reported data on this topic is lacking. Therefore, we investigated the use of supportive medications and interventions in this population-based study of prostate cancer survivors.
Methods
The PiCTure (Prostate Cancer Treatment, Your Experience) study methods have been described elsewhere.7 Briefly, 6,559 prostate cancer survivors 2-15 years after diagnosis (diagnosed during January 1, 1995-March 31, 2010, and alive in November 2011), identified from population-based cancer registries in the Republic of Ireland and Northern Ireland, were invited to complete a postal survey. Information was sought on after-effects (incontinence, impotence, gynaecomastia, hot flashes/sweats, bowel problems, depression) that had been experienced at any time after treatment. For each after-effect, men were asked if they had received any medication or interventions to alleviate symptoms, and, if so, what they had received; examples of common interventions were provided. Men were also asked if they had been told they may become infertile and, if so, whether they had preserved their sperm. The Decisional Regret Scale8 was used to measure survivors’ regret over their entire treatment experience. This 5-item scale, rated on a 5-point Likert scale from 1 (strongly agree) to 5 (strongly disagree) was summed and standardized to a value of 0-100, with higher scores reflecting higher levels of decisional regret. 8 This scale has good psychometric properties8 and strong reliability in our sample (Cronbach’s alpha = 0.85). Responders were categorized as having any regret (score ≥1) or no regret (score = 0).
The number of men who reported receiving an intervention was expressed as a percentage of survey responders and of men who reported ever having the relevant after-effect. Chi-square tests were used to investigate variations in receipt by: age at diagnosis (≤59, 60-69, ≥70 years); time since diagnosis (≤5, 5-10, >10 years); jurisdiction (Republic of Ireland, or Northern Ireland); and primary treatment(s) received (radical prostatectomy [RP], external beam radiotherapy [EBRT] with androgen deprivation therapy [ADT], EBRT without ADT, brachytherapy, ADT [without other therapies], and active surveillance/watchful waiting). Among survivors who ever experienced an after-effect, chi-square tests were used to investigate whether the percentage who reported decisional regret differed depending on whether or not they received the relevant supportive intervention.
Ethics approval was from the Irish College of General Practitioners (Republic of Ireland) and the Office for Research Ethics Committee Northern Ireland.
Results
In all, 3,348 survivors participated in the survey (adjusted response rate, 54%). Compared with nonresponders, responders were more often from the Republic of Ireland (P = .007), <70 years at diagnosis (P < .001), 5-10 years post diagnosis (P < .001), with low or medium Gleason grade (Gleason scores of ≤6 [good prognosis] and 7, respectively; P < .001), and clinical stage II-IV (P < .001; Table 1).
Impotence (70%) was the most commonly reported after-effect, followed by hot flashes/sweats (40%), incontinence (37%), bowel problems (23%), gynaecomastia (19%), and depression (18%; Table 2).
Of responders, 2% received an artificial sphincter, representing 6% of men who ever experienced incontinence post diagnosis (Table 2). This percentage was significantly higher in participants diagnosed longer ago, from the Republic of Ireland, and who received RP (Table 3).
Incontinence medication was received by 8% of participants (21% of those who experienced incontinence). Use varied significantly by age, jurisdiction, and treatment. For impotence, medications were more commonly used (20% of participants; 28% with impotence) than were injections (5% and 7%, respectively) or penile implants/pumps (2% and 3%, respectively). Use of all 3 types of intervention was highest in men who had RP; injections and implants/pumps were significantly more common among younger men. Of those experiencing gynaecomastia, 13% received interventions; receipt was highest in men who had EBRT with ADT, were <5 years post diagnosis and from Northern Ireland. For hot flashes/sweats, 3% of participants (8% who experienced symptoms) received mediations; this was higher in men who had EBRT. Of those who reported depression, 28% received medication; receipt was highest in younger men and in Northern Ireland. Medication for bowel problems was used by 35% of men who experienced these; use was highest in older men, those diagnosed more recently, and those who had EBRT. Sixty percent of men reported having been told they would become infertile; 11 (0.3% of participants) preserved their sperm, 7 from the Republic of Ireland and 4 from Northern Ireland.
A total of 35.6% of survivors reported any decisional regret. Among survivors who ever had an after-effect, a higher percentage of those who used a supportive intervention reported decisional regret compared with those who did not; this was only statistically significant for those using medication or alprostadil injections for impotence (Table 2).
Discussion
This study documents, for the first time, population-based data on patient-reported use of supportive medications and interventions to alleviate adverse effects of prostate cancer and its treatment. Among survivors who experienced after-effects, use was highest for bowel problems, impotence, and depression, but even for those, only 28%-35% of men took medication. Although it is possible that some survivors declined medications or other interventions, these low levels of use strongly suggest that not all survivors who might benefit from supports receive them.
There was little evidence that utilisation was higher in survivors diagnosed more recently. This suggests that, although the number of prostate cancer survivors has grown, and there is greater focus on survivorship issues in clinical practice, this has not translated into more men receiving support to manage after-effects. Care is needed to ensure that the newer models of post-cancer follow-up being considered or adopted in many settings,9 do not exacerbate this issue.
As expected, patterns of utilisation varied by treatment(s) received. Higher use of surgical and pharmaceutical interventions to alleviate incontinence among survivors in the Republic of Ireland than in Northern Ireland is likely owing to the higher rate of radical prostatectomy in the Republic of Ireland, whereas greater use of treatments for gynaecomastia in Northern Ireland reflects higher use of hormone therapy there.10 Other variations in intervention use were more surprising. Younger men were significantly more likely to report using supportive interventions for depression and impotence, the latter finding being consistent with findings in a Swedish population-based study.11 Older men were significantly more likely to report interventions for incontinence and bowel problems. Although those trends could be explained by differences in treatment receipt by age, it is possible that men of different ages may be more likely to seek, or be offered, help for certain types of after-effects. With the exception of interventions for bowel problems, a higher percentage of men who received intervention(s) for an after-effect reported decisional regret. There are a number of possible explanations: these men may have experienced more severe after-effects, which required interventions; they may have been less satisfied with their posttreatment function and/or more proactive about recovering or treating their after-effects. This requires further investigation.
This is a large, international, population-based study, the first such study to describe patient-reported use of supportive care following a range of prostate cancer treatments. Although this study is novel, there are a number of limitations. It is a cross-sectional, descriptive study. We did not ask survivors whether the supportive interventions received matched their needs and wants, and whether they were satisfied with the supportive care received. Furthermore, although the response rate is comparable with other similar studies,12,13 it is possible that the supportive care of nonresponders was different to that of responders.
Our study included men from 2 jurisdictions with separate health care systems, suggesting that low use of supportive interventions may be common across systems. There is a need for further research into patient and health care system factors associated with the receipt of supportive interventions and how satisfied men are with these, in this and other health care settings. Presently, it is clear that more needs to be done in the clinical setting to support prostate cancer survivors manage treatment after-effects; this in turn could improve survivors’ HRQoL.
Prostate cancer treatments are associated with various physical after-effects, including urinary, sexual, and bowel symptoms.1 These after-effects can have an impact on survivors’ health-related quality of life (HRQoL).2 Pharmaceutical and surgical interventions are available to manage or ameliorate many of these after-effects (eg, sildenafil citrate taken during and after radiotherapy improves sexual function),3 and their receipt has a positive impact on HRQoL.4
However, studies of clinicians suggest that such interventions may not be used widely.5,6 Patient-reported data on this topic is lacking. Therefore, we investigated the use of supportive medications and interventions in this population-based study of prostate cancer survivors.
Methods
The PiCTure (Prostate Cancer Treatment, Your Experience) study methods have been described elsewhere.7 Briefly, 6,559 prostate cancer survivors 2-15 years after diagnosis (diagnosed during January 1, 1995-March 31, 2010, and alive in November 2011), identified from population-based cancer registries in the Republic of Ireland and Northern Ireland, were invited to complete a postal survey. Information was sought on after-effects (incontinence, impotence, gynaecomastia, hot flashes/sweats, bowel problems, depression) that had been experienced at any time after treatment. For each after-effect, men were asked if they had received any medication or interventions to alleviate symptoms, and, if so, what they had received; examples of common interventions were provided. Men were also asked if they had been told they may become infertile and, if so, whether they had preserved their sperm. The Decisional Regret Scale8 was used to measure survivors’ regret over their entire treatment experience. This 5-item scale, rated on a 5-point Likert scale from 1 (strongly agree) to 5 (strongly disagree) was summed and standardized to a value of 0-100, with higher scores reflecting higher levels of decisional regret. 8 This scale has good psychometric properties8 and strong reliability in our sample (Cronbach’s alpha = 0.85). Responders were categorized as having any regret (score ≥1) or no regret (score = 0).
The number of men who reported receiving an intervention was expressed as a percentage of survey responders and of men who reported ever having the relevant after-effect. Chi-square tests were used to investigate variations in receipt by: age at diagnosis (≤59, 60-69, ≥70 years); time since diagnosis (≤5, 5-10, >10 years); jurisdiction (Republic of Ireland, or Northern Ireland); and primary treatment(s) received (radical prostatectomy [RP], external beam radiotherapy [EBRT] with androgen deprivation therapy [ADT], EBRT without ADT, brachytherapy, ADT [without other therapies], and active surveillance/watchful waiting). Among survivors who ever experienced an after-effect, chi-square tests were used to investigate whether the percentage who reported decisional regret differed depending on whether or not they received the relevant supportive intervention.
Ethics approval was from the Irish College of General Practitioners (Republic of Ireland) and the Office for Research Ethics Committee Northern Ireland.
Results
In all, 3,348 survivors participated in the survey (adjusted response rate, 54%). Compared with nonresponders, responders were more often from the Republic of Ireland (P = .007), <70 years at diagnosis (P < .001), 5-10 years post diagnosis (P < .001), with low or medium Gleason grade (Gleason scores of ≤6 [good prognosis] and 7, respectively; P < .001), and clinical stage II-IV (P < .001; Table 1).
Impotence (70%) was the most commonly reported after-effect, followed by hot flashes/sweats (40%), incontinence (37%), bowel problems (23%), gynaecomastia (19%), and depression (18%; Table 2).
Of responders, 2% received an artificial sphincter, representing 6% of men who ever experienced incontinence post diagnosis (Table 2). This percentage was significantly higher in participants diagnosed longer ago, from the Republic of Ireland, and who received RP (Table 3).
Incontinence medication was received by 8% of participants (21% of those who experienced incontinence). Use varied significantly by age, jurisdiction, and treatment. For impotence, medications were more commonly used (20% of participants; 28% with impotence) than were injections (5% and 7%, respectively) or penile implants/pumps (2% and 3%, respectively). Use of all 3 types of intervention was highest in men who had RP; injections and implants/pumps were significantly more common among younger men. Of those experiencing gynaecomastia, 13% received interventions; receipt was highest in men who had EBRT with ADT, were <5 years post diagnosis and from Northern Ireland. For hot flashes/sweats, 3% of participants (8% who experienced symptoms) received mediations; this was higher in men who had EBRT. Of those who reported depression, 28% received medication; receipt was highest in younger men and in Northern Ireland. Medication for bowel problems was used by 35% of men who experienced these; use was highest in older men, those diagnosed more recently, and those who had EBRT. Sixty percent of men reported having been told they would become infertile; 11 (0.3% of participants) preserved their sperm, 7 from the Republic of Ireland and 4 from Northern Ireland.
A total of 35.6% of survivors reported any decisional regret. Among survivors who ever had an after-effect, a higher percentage of those who used a supportive intervention reported decisional regret compared with those who did not; this was only statistically significant for those using medication or alprostadil injections for impotence (Table 2).
Discussion
This study documents, for the first time, population-based data on patient-reported use of supportive medications and interventions to alleviate adverse effects of prostate cancer and its treatment. Among survivors who experienced after-effects, use was highest for bowel problems, impotence, and depression, but even for those, only 28%-35% of men took medication. Although it is possible that some survivors declined medications or other interventions, these low levels of use strongly suggest that not all survivors who might benefit from supports receive them.
There was little evidence that utilisation was higher in survivors diagnosed more recently. This suggests that, although the number of prostate cancer survivors has grown, and there is greater focus on survivorship issues in clinical practice, this has not translated into more men receiving support to manage after-effects. Care is needed to ensure that the newer models of post-cancer follow-up being considered or adopted in many settings,9 do not exacerbate this issue.
As expected, patterns of utilisation varied by treatment(s) received. Higher use of surgical and pharmaceutical interventions to alleviate incontinence among survivors in the Republic of Ireland than in Northern Ireland is likely owing to the higher rate of radical prostatectomy in the Republic of Ireland, whereas greater use of treatments for gynaecomastia in Northern Ireland reflects higher use of hormone therapy there.10 Other variations in intervention use were more surprising. Younger men were significantly more likely to report using supportive interventions for depression and impotence, the latter finding being consistent with findings in a Swedish population-based study.11 Older men were significantly more likely to report interventions for incontinence and bowel problems. Although those trends could be explained by differences in treatment receipt by age, it is possible that men of different ages may be more likely to seek, or be offered, help for certain types of after-effects. With the exception of interventions for bowel problems, a higher percentage of men who received intervention(s) for an after-effect reported decisional regret. There are a number of possible explanations: these men may have experienced more severe after-effects, which required interventions; they may have been less satisfied with their posttreatment function and/or more proactive about recovering or treating their after-effects. This requires further investigation.
This is a large, international, population-based study, the first such study to describe patient-reported use of supportive care following a range of prostate cancer treatments. Although this study is novel, there are a number of limitations. It is a cross-sectional, descriptive study. We did not ask survivors whether the supportive interventions received matched their needs and wants, and whether they were satisfied with the supportive care received. Furthermore, although the response rate is comparable with other similar studies,12,13 it is possible that the supportive care of nonresponders was different to that of responders.
Our study included men from 2 jurisdictions with separate health care systems, suggesting that low use of supportive interventions may be common across systems. There is a need for further research into patient and health care system factors associated with the receipt of supportive interventions and how satisfied men are with these, in this and other health care settings. Presently, it is clear that more needs to be done in the clinical setting to support prostate cancer survivors manage treatment after-effects; this in turn could improve survivors’ HRQoL.
1. Drummond FJ, Kinnear H, O’Leary E, Donnelly, Gavin A, Sharp L. Long-term health-related quality of life of prostate cancer survivors varies by primary treatment. Results from the PiCTure (Prostate Cancer Treatment, your experience) study. J Cancer Surviv. 2015;9(2):361-72.
2. Smith DP, King MT, Egger S, et al. Quality of life three years after diagnosis of localised prostate cancer: population based cohort study. BMJ 2009; 339:b4817.
3. Zelefsky MJ, Shasha D, Branco RD, et al. Prophylactic sildenafil citrate improves select aspects of sexual function in men treated with radiotherapy for prostate cancer. J Urol. 2014;192(3):868-874.
4. Haab F, Trockman BA, Zimmern PE, Leach GE. Quality of life and continence assessment of the artificial urinary sphincter in men with minimum 3.5 years of follow-up. J Urol. 1997;158(2):435-439.
5. Tanvetyanon T. Physician practices of bone density testing and drug prescribing to prevent or treat osteoporosis during androgen deprivation therapy. Cancer. 2005;103(2):237-241.
6. Alibhai SM, Rahman S, Warde PR, Jewett MA, Jaffer T, Cheung AM. Prevention and management of osteoporosis in men receiving androgen deprivation therapy: a survey of urologists and radiation oncologists. Urology. 2006;68(1):126-131,
7. Drummond FJ, Kinnear H, Donnelly C, et al. Establishing a population-based patient reported outcomes study (PROMs) using national cancer registries across two jurisdictions: Prostate Cancer Treatment, your experience (PiCTure) Study. BMJ Open 2015;5:e006851.
8. Brehaut JC, O’Connor AM, Wood TJ, et al. Validation of a decision regret scale. Med Decis Making. 2003;23(4):281-92.
9. Howell D, Hack TF, Oliver et al. Models of care for post-treatment follow-up of adult cancer survivors: a systematic review and quality appraisal of the evidence. J Cancer Surviv. 2012;6(4):359-371.
10. Donnelly DW, Gavin AT, Comber H. Cancer in Ireland 1994-2004. A comprehensive report. Northern Ireland Cancer Registry/National Cancer Registry, Ireland, 2009.
11. Plym A, Folkvaljon Y, Garmo H, et al. Drug prescription for erectile dysfunction before and after diagnosis of localized prostate cancer. J Sex Med. 2014;11(8):2100-2108.
12. Hervouet S, Savard J, Simard S, et al. Psychological functioning associated with prostate cancer: cross-sectional comparison of patients treated with radiotherapy, brachytherapy, or surgery. J Pain Symptom Manage. 2005;30(5):474-484.
13. Glaser AW, Fraser LK, Corner J, et al. Patient-reported outcomes of cancer survivors in England 1-5 years after diagnosis: a cross-sectional survey. BMJ Open. 2013;3(4). pii: e002317.
1. Drummond FJ, Kinnear H, O’Leary E, Donnelly, Gavin A, Sharp L. Long-term health-related quality of life of prostate cancer survivors varies by primary treatment. Results from the PiCTure (Prostate Cancer Treatment, your experience) study. J Cancer Surviv. 2015;9(2):361-72.
2. Smith DP, King MT, Egger S, et al. Quality of life three years after diagnosis of localised prostate cancer: population based cohort study. BMJ 2009; 339:b4817.
3. Zelefsky MJ, Shasha D, Branco RD, et al. Prophylactic sildenafil citrate improves select aspects of sexual function in men treated with radiotherapy for prostate cancer. J Urol. 2014;192(3):868-874.
4. Haab F, Trockman BA, Zimmern PE, Leach GE. Quality of life and continence assessment of the artificial urinary sphincter in men with minimum 3.5 years of follow-up. J Urol. 1997;158(2):435-439.
5. Tanvetyanon T. Physician practices of bone density testing and drug prescribing to prevent or treat osteoporosis during androgen deprivation therapy. Cancer. 2005;103(2):237-241.
6. Alibhai SM, Rahman S, Warde PR, Jewett MA, Jaffer T, Cheung AM. Prevention and management of osteoporosis in men receiving androgen deprivation therapy: a survey of urologists and radiation oncologists. Urology. 2006;68(1):126-131,
7. Drummond FJ, Kinnear H, Donnelly C, et al. Establishing a population-based patient reported outcomes study (PROMs) using national cancer registries across two jurisdictions: Prostate Cancer Treatment, your experience (PiCTure) Study. BMJ Open 2015;5:e006851.
8. Brehaut JC, O’Connor AM, Wood TJ, et al. Validation of a decision regret scale. Med Decis Making. 2003;23(4):281-92.
9. Howell D, Hack TF, Oliver et al. Models of care for post-treatment follow-up of adult cancer survivors: a systematic review and quality appraisal of the evidence. J Cancer Surviv. 2012;6(4):359-371.
10. Donnelly DW, Gavin AT, Comber H. Cancer in Ireland 1994-2004. A comprehensive report. Northern Ireland Cancer Registry/National Cancer Registry, Ireland, 2009.
11. Plym A, Folkvaljon Y, Garmo H, et al. Drug prescription for erectile dysfunction before and after diagnosis of localized prostate cancer. J Sex Med. 2014;11(8):2100-2108.
12. Hervouet S, Savard J, Simard S, et al. Psychological functioning associated with prostate cancer: cross-sectional comparison of patients treated with radiotherapy, brachytherapy, or surgery. J Pain Symptom Manage. 2005;30(5):474-484.
13. Glaser AW, Fraser LK, Corner J, et al. Patient-reported outcomes of cancer survivors in England 1-5 years after diagnosis: a cross-sectional survey. BMJ Open. 2013;3(4). pii: e002317.
Differences in psychosocial stressors between black and white cancer patients
For patients with cancer, acknowledgment of mental and emotional distress is critically important when developing and implementing a treatment plan. The psychosocial distress associated with cancer diagnosis and treatment can have an impact on a patient’s quality of life, influence a patient’s ability to adhere to treatment regimens, and increase cost of care.1-4 Rates of depression have been reported to range from 8%-36%, with a 29% risk of anxiety in cancer patients.5, 6 Emotional distress is linked to increased hopelessness about their cancer diagnosis, increased issues with chronic pain, and negative treatment outcomes.7 Timely screening of psychosocial distress at the first clinical visit enables providers to make appropriate referrals to resources early in their course of treatment; however, referrals to psychosocial interventions remain infrequent nationwide in the United States.8
There is some evidence of a differential impact of cancer on mental health diagnoses between racial/ethnic groups; however, results are not entirely consistent across studies. Using the Kessler Pyschological Distress Scale (K6) score, Alcala and colleagues found that cancer was more detrimental to mental health for black patients than for non-Hispanic white patients.9 Black breast cancer survivors have also been shown to be more likely to stop working during the early phases of their treatment, indicating that they and their physicians need to take steps to minimize long-term employment consequences.10 However, in a study of women with breast cancer, black women reported fewer depressive symptoms than did non-Hispanic whites.11
The American College of Surgeons’ Commission on Cancer (ACS CoC) developed a set of Continuum of Care standards in 2012, including the implementation of psychosocial distress screening for patients with cancer. Since 2015, all accredited cancer programs are now required to evaluate these patients for signs of distress during at least 1 pivotal physician visit.12 The National Comprehensive Cancer Network (NCCN) Clinical Practice Guidelines in Oncology has developed a tool that provides a mechanism for meeting the requirements of the ACS CoC accreditation requirements. The NCCN defines distress in cancer as “a multifactorial unpleasant emotional experience of a psychological (cognitive, behavioral, emotional), social, and/or spiritual nature that may interfere with the ability to cope effectively with cancer, its physical symptoms and its treatment.”13 The recommendation of the NCCN is to provide a brief screening for psychosocial distress to identify individuals in need of additional support and to provide referrals for patients at high risk of psychosocial distress. The NCCN Distress Thermometer screening tool has been widely accepted as an effective method of identifying and characterizing distress. The NCCN tool provides a visual analogue scale for patients to rate their current distress on a scale of 1-10, as well as a problem checklist. The problem checklist includes 22 stressors addressing the practical, spiritual/religious, emotional, and physical concerns of patients. Although the NCCN tool is used widely, differences in distress scores between black and white cancer patients have not been previously described. The purpose of the study was to compare the global distress screening scores of black and white patients at an academic comprehensive cancer center in the Midwest. A second objective was to examine the distribution of individual stressors between black and white women.
Methods
Study sample
The study included all cancer patients from a cancer center in the Midwest who completed the NCCN distress thermometer during January 1, 2015-February 19, 2016. The patient population for this cancer center was primarily non-Hispanic white and non-Hispanic black, therefore, only patients identifying as non-Hispanic white and non-Hispanic black are included in this analysis. As part of routine clinical care, patients are asked to complete the NCCN distress thermometer at their first visit to the center. All patients in this analytic sample were newly diagnosed patients. Some patients also completed the NCCN screening tool at additional appointments; therefore, for patients with more than 1 completed tool, only the first distress screening was used in this analysis. Overall scores and individual stressor scores were entered into the electronic medical record by clinic staff at the time the patients were roomed for their visit. Patient demographics were collected through a reporting mechanism within the electronic medical record that allows for monitoring of the psychosocial screening process.
Variables
Race was assessed through self-report and classified as non-Hispanic white and non-Hispanic black. There were not enough patients of any other racial/ethnic group to be included in this analysis. Age was categorized as 18-40 years, 41-60 years, 61-84 years, and 85 years and older. Cancer type was grouped as follows: head and neck cancer, gastrointestinal cancer (esophagus, stomach, small intestine, colon, rectum, anus), hepatobiliary (liver, gallbladder, pancreas), sarcoma (bone and soft tissue), melanoma, nonmelanoma skin cancer, breast cancer, genitourinary (prostate, kidney, bladder), hematologic, and brain.
Two primary outcomes were assessed: overall distress, and each individual problem indicator. Overall distress was assessed using the thermometer visual analog rating (the thermometer rating of the NCCN screening tool) where possible values range from 0 (no distress) to 10 (extreme distress). The overall distress score was categorized into low distress (<4) and high distress (≥4) for analysis. The response options for individual stressors on the problem list are Yes or No for each of 17 discrete stressors: child care, housing, insurance/financial, transportation, work/school, treatment decisions, dealing with children, dealing with partner, ability to have children, family health issues, depression, fears, nervousness, sadness, worry, loss of interest, and spiritual/religious concerns. Physical complaints were not assessed in this study. Comparisons were made between white and black patients on overall distress score as well as for each individual psychosocial stressor.
Data analysis
Descriptive statistics (counts and proportions or means and standard deviations) were calculated stratified by race. Categorical variables were compared by race using chi-square or Fisher exact test. Logistic regression was used to predict high distress by race adjusting for sex, age, and cancer type. All analyses were conducted using SAS 9.4 (Cary, NJ).
This study was reviewed and approved by the Saint Louis University Institutional Review Board (protocol number 26269).
Results
A total of 933 patients with cancer completed the NCCN distress screening tool. Of that total, 45 patients did not complete the overall distress score thermometer, but did complete the checklist of individual stressors. Those 45 patients were excluded from the logistic regression analysis for overall distress score, but included on comparisons of individual stressors. The distribution of overall distress scores by race can be seen in the Figure.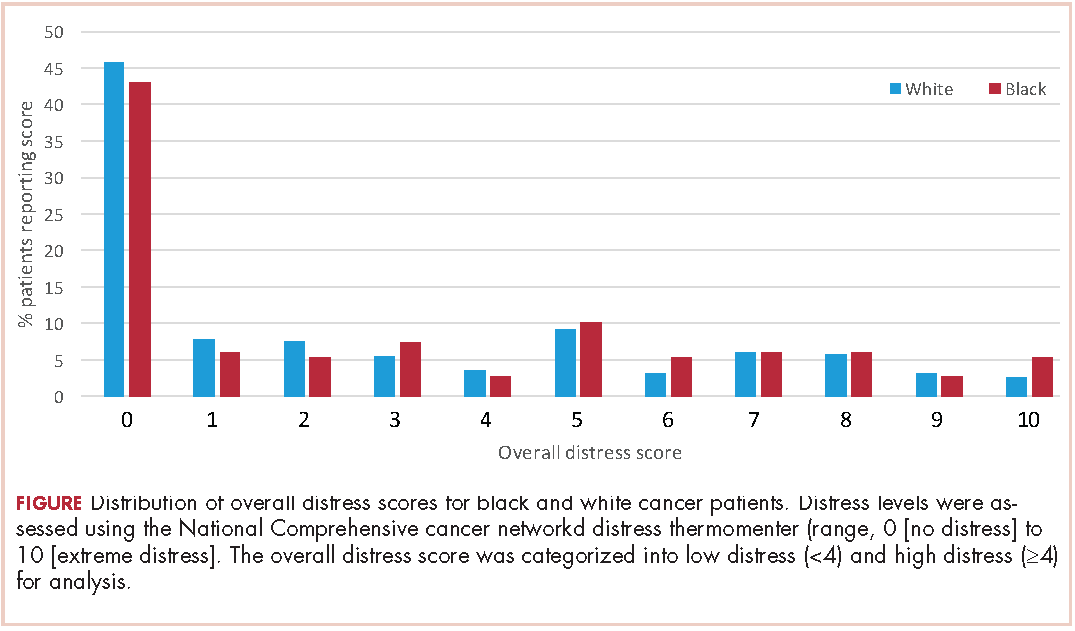
Briefly, the full sample was 16.9% black and 38.8% female. In all, 32.6% of the sample indicated high distress on the distress thermometer at their first visit. Demographics for the participants stratified by race are reported in Table 1 (see PDF).There was no statistically significant difference in the gender or age distribution between black and white patients. Cancer distribution did vary by race. Black patients were proportionally more represented in gastrointestinal cancers, hepatobiliary cancers, sarcomas, breast cancer, and genitourinary cancers. White patients were proportionally more represented in melanoma, nonmelanoma skin cancers, and hematologic cancers.
Table 2 presents bivariate comparisons on overall distress and individual stressors between black and white patients. There was no difference in the high distress between black and white patients in bivariate analysis (31.9% and 36.1%, respectively, P = .30). However, there were differences in the individual stressors identified for each racial group (Table 2). White patients, compared with black patients, more frequently identified treatment decisions (17.6% vs 10.1%, P = .02) and nervousness (26.8% vs 18.4%, P = .02) as sources of distress. Black patients, compared with white patients, more frequently identified housing (5.1% vs 1.7%, P = .009), the ability to have children (2.5% vs 0.4%, P =.02), and loss of interest (15.2% vs 8.9%, P = .02) as sources of distress. Distress scores did not differ between black and white patients for child care, insurance or financial issues, transportation, work or school, dealing with children, dealing with partners, family health issues, depression, fears, sadness, worry, or spiritual or religious concerns.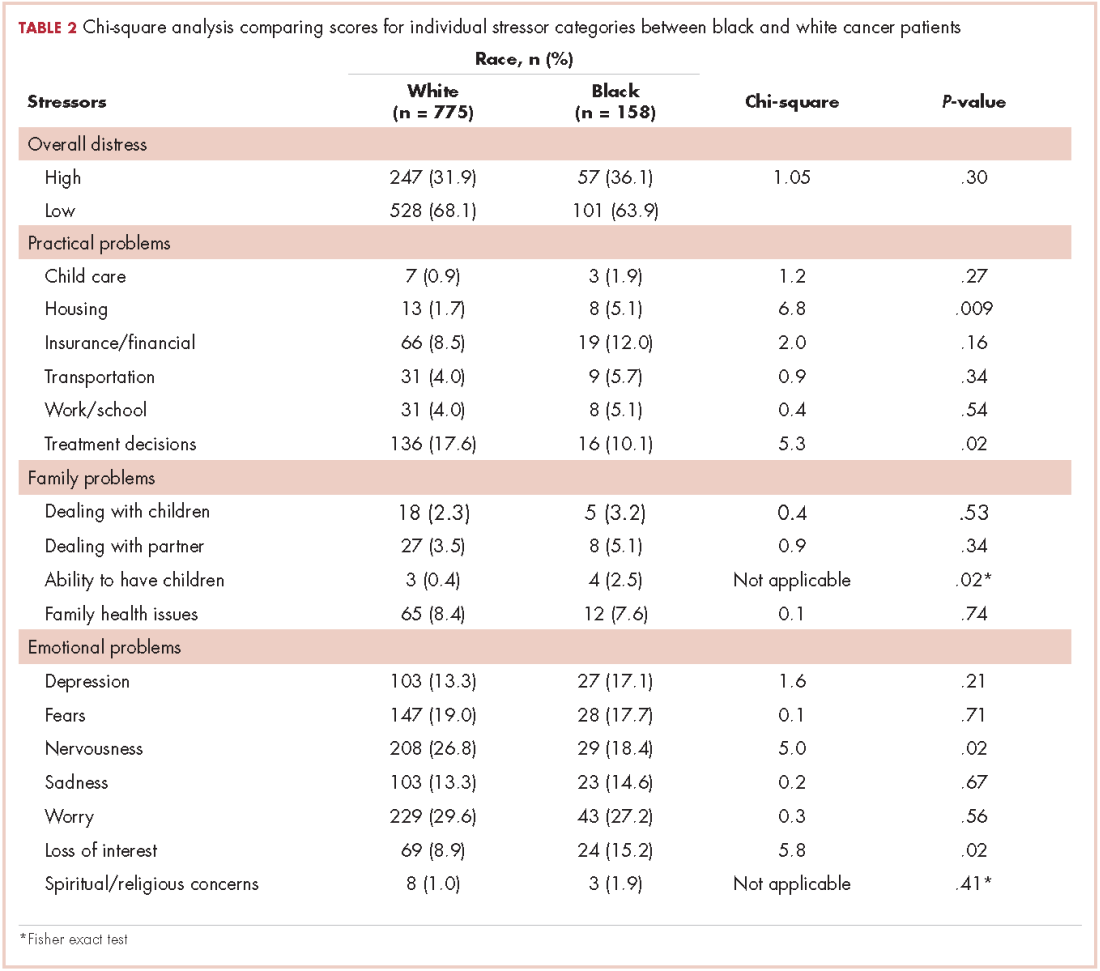
Table 3 presents the results from the logistic analysis predicting high distress. In adjusted analysis, black race did not predict high distress (OR, 0.94; 95% confidence interval [CI], 0.62-1.44). High distress was associated with sex, age, and some cancer categories. Women had 77% higher odds of high distress compared with men (OR, 1.77; 95% CI, 1.25-2.51).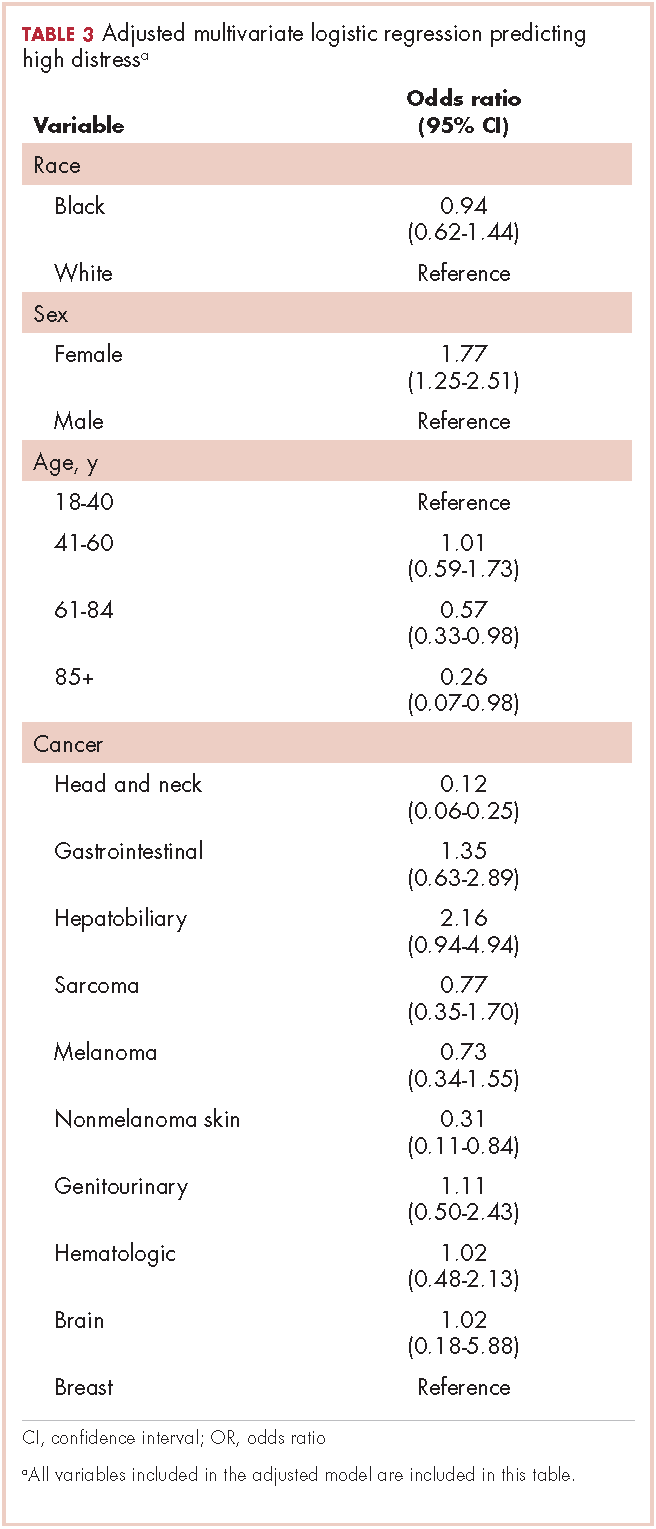
Compared with patients aged 18-44 years, patients aged 61-84 had 43% lower odds of high distress (OR, 0.57; 95% CI, 0.33-0.98), and patients aged 85 and older had 74% lower odds of high distress (OR, 0.26; 95% CI, 0.07-0.98). There was no statistically significant difference between patients aged 18-40 and those aged 41-60 for high distress (OR, 1.01; 95% CI, 0.59-1.73).
Discussion
Management of patients with cancer continues to evolve. Although a tremendous amount of importance is still placed on the pathophysiology of cancer and its prescribed treatments, more emphasis is being assigned to the physical and psychosocial effects of cancer on these patients. In 2008, the Institute of Medicine published a report that examined the psychosocial health of patients with cancer.14 The report recommended that all cancer care should ensure the provision of appropriate psychosocial health services by facilitating effective communication between patients and care providers, identifying each patient’s psychosocial health needs, coordinating referrals for psychosocial services and monitoring efficacy of psychosocial interventions. The inclusion of psychosocial distress screening in all cancer programs accredited by the ACS CoC helped to prioritize the identification and treatment of psychosocial issues for all cancer patients.
The present study is the first of its kind to compare the individual stressors identified through psychosocial distress screening between black and white cancer patients. In our sample, 304 of 933 patients (32%) reported high distress, with a total score of ≥4. Previous research on overall distress difference across race/ethnicity is mixed. VanHoose and colleagues found no difference in overall distress between racial groups,15 Alcala and colleagues found higher overall distress in black patients with cancer compared with white patients with cancer,9 and Culver and colleagues found black women with breast cancer had lower overall distress compared with white women.11 We found no difference in the presence of high distress between black and white patients at our cancer center in either crude or adjusted analysis. Differences in overall distress across studies may be owing to the timing of screening. Given that overall distress may vary across time16,17 and there is no current information on whether temporal variations in distress differ by race, it is possible that the time of distress assessment may influence demonstrated differences between racial groups. For example, if different stressors affect black and white women differentially, and those stressors are associated with different points across the cancer continuum, then we might see that the magnitude of racial differences in overall stress are time dependent. Alcala and colleagues examined any cancer diagnosis across the lifespan, whereas Culver and colleagues examined multiple time points across treatment for a small group of breast cancer patients. Badr and colleagues, in a sample of head and neck cancer patients, found that distress increased across the course of treatment;18 however they did not examine variations in type of stressors related to overall distress, nor did they examine racial differences in distress. Differences in results may also be the result of differences in measurement of distress. Culver and colleagues did not examine distress using the NCCN distress thermometer, rather psychological distress was measured by a scale rating a series of “mood-descriptive adjectives” (p. 497).11 Alcala used the K-6 as a measure of psychological distress;9 therefore, demonstrated differences in overall distress between white and black women may vary across studies because of differences in measurement of the underlying distress variable. The lack of racial differences in overall distress in our study is consistent with the findings of VanHoose and colleagues,15 who also examined distress near the start of treatment and also used the NCCN distress thermometer as the measure of psychosocial distress.
We did find differences in the individual stressors between racial groups, indicating that the source of distress does vary between black and white cancer patients. Black patients more frequently reported distress secondary to housing, loss of interest and their ability to have children than did white patients. By comparison, white patients more frequently reported distress secondary to nervousness and treatment decisions than black patients. Identified differences in individual stressors may be attributable to sociocultural differences or differences in external support. It is also possible that black patients are more likely to willingly report distress related to nonpsychological factors, whereas white patients are more willing to report factors, such as nervousness, that are related to psychological disorders. Although it has been suggested that black cancer patients have more concerns about finances and work than do white cancer patients,19 we did not identify a statistically significant difference in child care, insurance or financial issues, transportation, work, or school between these 2 cohorts. This may be because the psychosocial distress screening score included in this study was performed at the time of initial diagnosis, and not further into their prescribed treatment at which point the financial worries may be more realized. Psychosocial screening scores obtained at subsequent visits were not included in the analysis because they are not routinely collected as part of clinical care in the center where this study took place. Furthermore, it is impossible to identify where a specific patient is in their treatment regimen based on their demographic data or subsequent distress scores in our data extraction tool. Further investigation into the sources of distress at different time points along the continuum of care may shed more light on this topic.
Limitations
There are several limitations to this study. First, the method of data extraction from an electronic medical record report limited the capacity to explore possible differences between the patients in our sample, such as insurance status, level of education, available social support, current employment status, stage of disease, overall prognosis and prescribed treatment regimen.
Second, there were likely patients who either did not complete a psychosocial distress screening tool or whose data were not entered into the electronic medical record for inclusion in the analysis. The present study period took place during the implementation of the NCCN tool at the center. Although the policy was to screen all new patients as part of routine care; not all patients seen at the center received the NCCN screening tool at their first visit. Owing to the mechanisms for data entry and abstraction, only information from the patients who had a completed form was able to be accessed for this study, thus a statistical comparison between those who did and did not receive the NCCN tool cannot be made. During the timeframe for this study, the head and neck, breast, genitourinary, and hematologic services completed proportionally more NCCN screening of new patients than other services in the center. This is reflected in the distributional breakdown of cancer in the overall sample of this study. It is possible that the results are more representative of differences between black and white cancer patients in the services that were more likely to properly implement NCCN screening.
Third, our patient population was derived from an urban, academic medical center and the results may not be generalizable to other patient populations.
Fourth, the NCCN distress thermometer is a single-item rating of overall global distress that is not intended to be a diagnostic indicator of psychological comorbidity and, therefore, does not distinguish between common psychological diagnoses such as depression or anxiety. However, the usefulness of the tool is to provide an impetus for referral to services that may then encompass the evaluation and diagnosis of particular psychological conditions. Further, the distress thermometer tool is designed to identify stress relating to the social aspects of cancer diagnosis and treatment and is not limited to psychological distress alone.
Strengths
Despite the limitations, there are also significant strengths to this study. The NCCN tool is a widely accepted measure for the assessment of psychosocial distress in patients with cancer. The measure is a common and routine clinical instrument,20 and has also been used widely in research.18,21-24 Given the urban, academic environment of our clinical practice, our population is more racially diverse than other settings, allowing for initial examination of disparities between white and black cancer patients.
Clinical implications
Understanding differences in common psychosocial stressor between black and white cancer patients may allow for clinicians to strategically look for different types of stressors in order to facilitate faster referrals to appropriate services. It has been established in the literature that distress is correlated to cancer-related outcomes and distress screening is now considered standard of care when treating cancer patients. Identifying differences in psychosocial stressors among black and white cancer patients is paramount to ensuring that the appropriate resources are available to assist them through their cancer journey. The differences in type of stressor, may indicate fundamental differences in the way patients perceive their disease or the social and cultural implication of a cancer diagnosis. In this study, white patients were more likely to find distress in the psychological realm (nervousness, decision-making), whereas black patients were more likely to be distressed about social issues (housing, ability to have children, and loss of interest). The referral needs of patients may be quite different, even with similar levels of overall distress. More research is necessary to further characterize sources of distress for cancer patients, how this distress impacts a patient’s physical and emotional well-being and how health care providers can better identify these issues and make the necessary referrals to support the whole patient.
1. Holland JC, Reznik I. Pathways for psychosocial care of cancer survivors. Cancer. 2005;104(11 Suppl):2624-2637.
2. Strasser F, Sweeney C, Willey J, Benisch-Tolley S, Palmer L, Bruera E. Impact of a half-day multidisciplinary symptom control and palliative care outpatient clinic in a comprehensive cancer center on recommendations, symptom intensity, and patient satisfaction: a retrospective descriptive study. J Pain Symptom Manage. 2004;27(6):481-491.
3. Carlson LE, Bultz BD. Efficacy and medical cost offset of psychosocial interventions in cancer care: making the case for economic analyses. Psychooncology. 2004;13(12):837-849.
4. Holland J, Bultz BD. The NCCN Guideline for distress management: a case for making distress the sixth vital sign. J Natl Compr Canc Netw. 2007;5(1):3-7.
5. Krebber A, Buffart L, Kleijn G, et al. Prevalence of depression in cancer patients: a meta-analysis of diagnostic interviews and self-report instruments. Psychooncology. 2014;23(2):121-130.
6. Sharp L, Carsin AE , Timmons A. Associations between cancer-related financial stress and strain and psychological well-being among individuals living with cancer. Psychooncology. 2013;22(4):745-755.
7. Bruce J, Thornton AJ, Powell R, et al. Psychological, surgical, and sociodemographic predictors of pain outcomes after breast cancer surgery: a population-based cohort study. Pain. 2014;155(2):232-243.
8. Holland JC. Preliminary guidelines for the treatment of distress. Oncology. 1997;11(11A):109-114.
9. Alcala HE. Differential mental health impact of cancer across racial/ethnic groups: findings from a population-based study in California. BMC Public Health. 2014;14:930.
10. Bradley CJ, Wilk A. Racial differences in quality of life and employment outcomes in insured women with breast cancer. J Cancer Surviv. 2014;8(1):49-59.
11. Culver JL, Arena PL, Antoni MH, Carver CS. Coping and distress among women under treatment for early stage breast cancer: comparing African Americans, Hispanics and non-Hispanic whites. Psychooncology. 2002;11(6):495-504.
12. American College of Surgeons Commission on Cancer. ACSCC website. Cancer program standards: ensuring patient-centered care. 2016 edition. https://www.facs.org/quality-programs/cancer/coc/standards. Posted 2016. Accessed August 30, 2017.
13. National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology: Distress Management. National Comprehensive Cancer Network, 2014.https://www.nccn.org/store/login/login.aspx?ReturnURL=https://www.nccn.org/professionals/physician_gls/pdf/distress.pdf Accessed August 30, 2017.
14. Institute of Medicine. Cancer care for the whole patient: meeting psychosocial health needs. Washington, DC: The National Academies Press; 2008. https://doi.org/10.17226/11993. Accessed August 30, 2017.
15. VanHoose L, Black LL, Doty K, et al. An analysis of the distress thermometer problem list and distress in patients with cancer. Support Care Cancer. 2015;23(5):1225-1232.
16. Gessler S, Low J, Daniells E, et al. Screening for distress in cancer patients: is the distress thermometer a valid measure in the UK and does it measure change over time? A prospective validation study. Psychooncology. 2008;17(6):538-547.
17. Enns A, Waller A, Groff SL, Bultz BD, Fung T, Carlson LE. Risk factors for continuous distress over a 12-month period in newly diagnosed cancer outpatients. J Psychosoc Oncol. 2013;31(5):489-506.
18. Badr H, Gupta V, Sikora A, Posner M. Psychological distress in patients and caregivers over the course of radiotherapy for head and neck cancer. Oral Oncol. 2014;50(10):1005-1011.
19. Wang X, Cosby LG, Harris MG, Liu T. Major concerns and needs of breast cancer patients. Cancer Nurs. 1999;22(2):157-163.
20. Dabrowski M, Boucher K, Ward JH, et al. Clinical experience with the NCCN distress thermometer in breast cancer patients. J Natl Compr Canc Netw. 2007;5(1):104-11.
21. Buchmann L, Conlee J, Hunt J, Agarwal J, White S. Psychosocial distress in prevalent in head and neck cancer patients. Laryngoscope. 2013;123(6):1424-1429.
22. Agarwal J, Powers K, Pappas L, et al. Correlates of elevated distress thermometer scores in breast cancer patients. Support Care Cancer. 2013;21(8):2125-2136.
23. Johnson R, Gold MA, Wythe KF. Distress in women with gynecologic cancer. Psychooncology. 2010;19(6):665-668.
24. Kendall J, Glaze K, Oakland S, Hansen J, Parry C. What do 1281 distress screeners tell us about cancer patients in a community cancer center? Psychooncology. 2011;20(6):594-600.
For patients with cancer, acknowledgment of mental and emotional distress is critically important when developing and implementing a treatment plan. The psychosocial distress associated with cancer diagnosis and treatment can have an impact on a patient’s quality of life, influence a patient’s ability to adhere to treatment regimens, and increase cost of care.1-4 Rates of depression have been reported to range from 8%-36%, with a 29% risk of anxiety in cancer patients.5, 6 Emotional distress is linked to increased hopelessness about their cancer diagnosis, increased issues with chronic pain, and negative treatment outcomes.7 Timely screening of psychosocial distress at the first clinical visit enables providers to make appropriate referrals to resources early in their course of treatment; however, referrals to psychosocial interventions remain infrequent nationwide in the United States.8
There is some evidence of a differential impact of cancer on mental health diagnoses between racial/ethnic groups; however, results are not entirely consistent across studies. Using the Kessler Pyschological Distress Scale (K6) score, Alcala and colleagues found that cancer was more detrimental to mental health for black patients than for non-Hispanic white patients.9 Black breast cancer survivors have also been shown to be more likely to stop working during the early phases of their treatment, indicating that they and their physicians need to take steps to minimize long-term employment consequences.10 However, in a study of women with breast cancer, black women reported fewer depressive symptoms than did non-Hispanic whites.11
The American College of Surgeons’ Commission on Cancer (ACS CoC) developed a set of Continuum of Care standards in 2012, including the implementation of psychosocial distress screening for patients with cancer. Since 2015, all accredited cancer programs are now required to evaluate these patients for signs of distress during at least 1 pivotal physician visit.12 The National Comprehensive Cancer Network (NCCN) Clinical Practice Guidelines in Oncology has developed a tool that provides a mechanism for meeting the requirements of the ACS CoC accreditation requirements. The NCCN defines distress in cancer as “a multifactorial unpleasant emotional experience of a psychological (cognitive, behavioral, emotional), social, and/or spiritual nature that may interfere with the ability to cope effectively with cancer, its physical symptoms and its treatment.”13 The recommendation of the NCCN is to provide a brief screening for psychosocial distress to identify individuals in need of additional support and to provide referrals for patients at high risk of psychosocial distress. The NCCN Distress Thermometer screening tool has been widely accepted as an effective method of identifying and characterizing distress. The NCCN tool provides a visual analogue scale for patients to rate their current distress on a scale of 1-10, as well as a problem checklist. The problem checklist includes 22 stressors addressing the practical, spiritual/religious, emotional, and physical concerns of patients. Although the NCCN tool is used widely, differences in distress scores between black and white cancer patients have not been previously described. The purpose of the study was to compare the global distress screening scores of black and white patients at an academic comprehensive cancer center in the Midwest. A second objective was to examine the distribution of individual stressors between black and white women.
Methods
Study sample
The study included all cancer patients from a cancer center in the Midwest who completed the NCCN distress thermometer during January 1, 2015-February 19, 2016. The patient population for this cancer center was primarily non-Hispanic white and non-Hispanic black, therefore, only patients identifying as non-Hispanic white and non-Hispanic black are included in this analysis. As part of routine clinical care, patients are asked to complete the NCCN distress thermometer at their first visit to the center. All patients in this analytic sample were newly diagnosed patients. Some patients also completed the NCCN screening tool at additional appointments; therefore, for patients with more than 1 completed tool, only the first distress screening was used in this analysis. Overall scores and individual stressor scores were entered into the electronic medical record by clinic staff at the time the patients were roomed for their visit. Patient demographics were collected through a reporting mechanism within the electronic medical record that allows for monitoring of the psychosocial screening process.
Variables
Race was assessed through self-report and classified as non-Hispanic white and non-Hispanic black. There were not enough patients of any other racial/ethnic group to be included in this analysis. Age was categorized as 18-40 years, 41-60 years, 61-84 years, and 85 years and older. Cancer type was grouped as follows: head and neck cancer, gastrointestinal cancer (esophagus, stomach, small intestine, colon, rectum, anus), hepatobiliary (liver, gallbladder, pancreas), sarcoma (bone and soft tissue), melanoma, nonmelanoma skin cancer, breast cancer, genitourinary (prostate, kidney, bladder), hematologic, and brain.
Two primary outcomes were assessed: overall distress, and each individual problem indicator. Overall distress was assessed using the thermometer visual analog rating (the thermometer rating of the NCCN screening tool) where possible values range from 0 (no distress) to 10 (extreme distress). The overall distress score was categorized into low distress (<4) and high distress (≥4) for analysis. The response options for individual stressors on the problem list are Yes or No for each of 17 discrete stressors: child care, housing, insurance/financial, transportation, work/school, treatment decisions, dealing with children, dealing with partner, ability to have children, family health issues, depression, fears, nervousness, sadness, worry, loss of interest, and spiritual/religious concerns. Physical complaints were not assessed in this study. Comparisons were made between white and black patients on overall distress score as well as for each individual psychosocial stressor.
Data analysis
Descriptive statistics (counts and proportions or means and standard deviations) were calculated stratified by race. Categorical variables were compared by race using chi-square or Fisher exact test. Logistic regression was used to predict high distress by race adjusting for sex, age, and cancer type. All analyses were conducted using SAS 9.4 (Cary, NJ).
This study was reviewed and approved by the Saint Louis University Institutional Review Board (protocol number 26269).
Results
A total of 933 patients with cancer completed the NCCN distress screening tool. Of that total, 45 patients did not complete the overall distress score thermometer, but did complete the checklist of individual stressors. Those 45 patients were excluded from the logistic regression analysis for overall distress score, but included on comparisons of individual stressors. The distribution of overall distress scores by race can be seen in the Figure.
Briefly, the full sample was 16.9% black and 38.8% female. In all, 32.6% of the sample indicated high distress on the distress thermometer at their first visit. Demographics for the participants stratified by race are reported in Table 1 (see PDF).There was no statistically significant difference in the gender or age distribution between black and white patients. Cancer distribution did vary by race. Black patients were proportionally more represented in gastrointestinal cancers, hepatobiliary cancers, sarcomas, breast cancer, and genitourinary cancers. White patients were proportionally more represented in melanoma, nonmelanoma skin cancers, and hematologic cancers.
Table 2 presents bivariate comparisons on overall distress and individual stressors between black and white patients. There was no difference in the high distress between black and white patients in bivariate analysis (31.9% and 36.1%, respectively, P = .30). However, there were differences in the individual stressors identified for each racial group (Table 2). White patients, compared with black patients, more frequently identified treatment decisions (17.6% vs 10.1%, P = .02) and nervousness (26.8% vs 18.4%, P = .02) as sources of distress. Black patients, compared with white patients, more frequently identified housing (5.1% vs 1.7%, P = .009), the ability to have children (2.5% vs 0.4%, P =.02), and loss of interest (15.2% vs 8.9%, P = .02) as sources of distress. Distress scores did not differ between black and white patients for child care, insurance or financial issues, transportation, work or school, dealing with children, dealing with partners, family health issues, depression, fears, sadness, worry, or spiritual or religious concerns.
Table 3 presents the results from the logistic analysis predicting high distress. In adjusted analysis, black race did not predict high distress (OR, 0.94; 95% confidence interval [CI], 0.62-1.44). High distress was associated with sex, age, and some cancer categories. Women had 77% higher odds of high distress compared with men (OR, 1.77; 95% CI, 1.25-2.51).
Compared with patients aged 18-44 years, patients aged 61-84 had 43% lower odds of high distress (OR, 0.57; 95% CI, 0.33-0.98), and patients aged 85 and older had 74% lower odds of high distress (OR, 0.26; 95% CI, 0.07-0.98). There was no statistically significant difference between patients aged 18-40 and those aged 41-60 for high distress (OR, 1.01; 95% CI, 0.59-1.73).
Discussion
Management of patients with cancer continues to evolve. Although a tremendous amount of importance is still placed on the pathophysiology of cancer and its prescribed treatments, more emphasis is being assigned to the physical and psychosocial effects of cancer on these patients. In 2008, the Institute of Medicine published a report that examined the psychosocial health of patients with cancer.14 The report recommended that all cancer care should ensure the provision of appropriate psychosocial health services by facilitating effective communication between patients and care providers, identifying each patient’s psychosocial health needs, coordinating referrals for psychosocial services and monitoring efficacy of psychosocial interventions. The inclusion of psychosocial distress screening in all cancer programs accredited by the ACS CoC helped to prioritize the identification and treatment of psychosocial issues for all cancer patients.
The present study is the first of its kind to compare the individual stressors identified through psychosocial distress screening between black and white cancer patients. In our sample, 304 of 933 patients (32%) reported high distress, with a total score of ≥4. Previous research on overall distress difference across race/ethnicity is mixed. VanHoose and colleagues found no difference in overall distress between racial groups,15 Alcala and colleagues found higher overall distress in black patients with cancer compared with white patients with cancer,9 and Culver and colleagues found black women with breast cancer had lower overall distress compared with white women.11 We found no difference in the presence of high distress between black and white patients at our cancer center in either crude or adjusted analysis. Differences in overall distress across studies may be owing to the timing of screening. Given that overall distress may vary across time16,17 and there is no current information on whether temporal variations in distress differ by race, it is possible that the time of distress assessment may influence demonstrated differences between racial groups. For example, if different stressors affect black and white women differentially, and those stressors are associated with different points across the cancer continuum, then we might see that the magnitude of racial differences in overall stress are time dependent. Alcala and colleagues examined any cancer diagnosis across the lifespan, whereas Culver and colleagues examined multiple time points across treatment for a small group of breast cancer patients. Badr and colleagues, in a sample of head and neck cancer patients, found that distress increased across the course of treatment;18 however they did not examine variations in type of stressors related to overall distress, nor did they examine racial differences in distress. Differences in results may also be the result of differences in measurement of distress. Culver and colleagues did not examine distress using the NCCN distress thermometer, rather psychological distress was measured by a scale rating a series of “mood-descriptive adjectives” (p. 497).11 Alcala used the K-6 as a measure of psychological distress;9 therefore, demonstrated differences in overall distress between white and black women may vary across studies because of differences in measurement of the underlying distress variable. The lack of racial differences in overall distress in our study is consistent with the findings of VanHoose and colleagues,15 who also examined distress near the start of treatment and also used the NCCN distress thermometer as the measure of psychosocial distress.
We did find differences in the individual stressors between racial groups, indicating that the source of distress does vary between black and white cancer patients. Black patients more frequently reported distress secondary to housing, loss of interest and their ability to have children than did white patients. By comparison, white patients more frequently reported distress secondary to nervousness and treatment decisions than black patients. Identified differences in individual stressors may be attributable to sociocultural differences or differences in external support. It is also possible that black patients are more likely to willingly report distress related to nonpsychological factors, whereas white patients are more willing to report factors, such as nervousness, that are related to psychological disorders. Although it has been suggested that black cancer patients have more concerns about finances and work than do white cancer patients,19 we did not identify a statistically significant difference in child care, insurance or financial issues, transportation, work, or school between these 2 cohorts. This may be because the psychosocial distress screening score included in this study was performed at the time of initial diagnosis, and not further into their prescribed treatment at which point the financial worries may be more realized. Psychosocial screening scores obtained at subsequent visits were not included in the analysis because they are not routinely collected as part of clinical care in the center where this study took place. Furthermore, it is impossible to identify where a specific patient is in their treatment regimen based on their demographic data or subsequent distress scores in our data extraction tool. Further investigation into the sources of distress at different time points along the continuum of care may shed more light on this topic.
Limitations
There are several limitations to this study. First, the method of data extraction from an electronic medical record report limited the capacity to explore possible differences between the patients in our sample, such as insurance status, level of education, available social support, current employment status, stage of disease, overall prognosis and prescribed treatment regimen.
Second, there were likely patients who either did not complete a psychosocial distress screening tool or whose data were not entered into the electronic medical record for inclusion in the analysis. The present study period took place during the implementation of the NCCN tool at the center. Although the policy was to screen all new patients as part of routine care; not all patients seen at the center received the NCCN screening tool at their first visit. Owing to the mechanisms for data entry and abstraction, only information from the patients who had a completed form was able to be accessed for this study, thus a statistical comparison between those who did and did not receive the NCCN tool cannot be made. During the timeframe for this study, the head and neck, breast, genitourinary, and hematologic services completed proportionally more NCCN screening of new patients than other services in the center. This is reflected in the distributional breakdown of cancer in the overall sample of this study. It is possible that the results are more representative of differences between black and white cancer patients in the services that were more likely to properly implement NCCN screening.
Third, our patient population was derived from an urban, academic medical center and the results may not be generalizable to other patient populations.
Fourth, the NCCN distress thermometer is a single-item rating of overall global distress that is not intended to be a diagnostic indicator of psychological comorbidity and, therefore, does not distinguish between common psychological diagnoses such as depression or anxiety. However, the usefulness of the tool is to provide an impetus for referral to services that may then encompass the evaluation and diagnosis of particular psychological conditions. Further, the distress thermometer tool is designed to identify stress relating to the social aspects of cancer diagnosis and treatment and is not limited to psychological distress alone.
Strengths
Despite the limitations, there are also significant strengths to this study. The NCCN tool is a widely accepted measure for the assessment of psychosocial distress in patients with cancer. The measure is a common and routine clinical instrument,20 and has also been used widely in research.18,21-24 Given the urban, academic environment of our clinical practice, our population is more racially diverse than other settings, allowing for initial examination of disparities between white and black cancer patients.
Clinical implications
Understanding differences in common psychosocial stressor between black and white cancer patients may allow for clinicians to strategically look for different types of stressors in order to facilitate faster referrals to appropriate services. It has been established in the literature that distress is correlated to cancer-related outcomes and distress screening is now considered standard of care when treating cancer patients. Identifying differences in psychosocial stressors among black and white cancer patients is paramount to ensuring that the appropriate resources are available to assist them through their cancer journey. The differences in type of stressor, may indicate fundamental differences in the way patients perceive their disease or the social and cultural implication of a cancer diagnosis. In this study, white patients were more likely to find distress in the psychological realm (nervousness, decision-making), whereas black patients were more likely to be distressed about social issues (housing, ability to have children, and loss of interest). The referral needs of patients may be quite different, even with similar levels of overall distress. More research is necessary to further characterize sources of distress for cancer patients, how this distress impacts a patient’s physical and emotional well-being and how health care providers can better identify these issues and make the necessary referrals to support the whole patient.
For patients with cancer, acknowledgment of mental and emotional distress is critically important when developing and implementing a treatment plan. The psychosocial distress associated with cancer diagnosis and treatment can have an impact on a patient’s quality of life, influence a patient’s ability to adhere to treatment regimens, and increase cost of care.1-4 Rates of depression have been reported to range from 8%-36%, with a 29% risk of anxiety in cancer patients.5, 6 Emotional distress is linked to increased hopelessness about their cancer diagnosis, increased issues with chronic pain, and negative treatment outcomes.7 Timely screening of psychosocial distress at the first clinical visit enables providers to make appropriate referrals to resources early in their course of treatment; however, referrals to psychosocial interventions remain infrequent nationwide in the United States.8
There is some evidence of a differential impact of cancer on mental health diagnoses between racial/ethnic groups; however, results are not entirely consistent across studies. Using the Kessler Pyschological Distress Scale (K6) score, Alcala and colleagues found that cancer was more detrimental to mental health for black patients than for non-Hispanic white patients.9 Black breast cancer survivors have also been shown to be more likely to stop working during the early phases of their treatment, indicating that they and their physicians need to take steps to minimize long-term employment consequences.10 However, in a study of women with breast cancer, black women reported fewer depressive symptoms than did non-Hispanic whites.11
The American College of Surgeons’ Commission on Cancer (ACS CoC) developed a set of Continuum of Care standards in 2012, including the implementation of psychosocial distress screening for patients with cancer. Since 2015, all accredited cancer programs are now required to evaluate these patients for signs of distress during at least 1 pivotal physician visit.12 The National Comprehensive Cancer Network (NCCN) Clinical Practice Guidelines in Oncology has developed a tool that provides a mechanism for meeting the requirements of the ACS CoC accreditation requirements. The NCCN defines distress in cancer as “a multifactorial unpleasant emotional experience of a psychological (cognitive, behavioral, emotional), social, and/or spiritual nature that may interfere with the ability to cope effectively with cancer, its physical symptoms and its treatment.”13 The recommendation of the NCCN is to provide a brief screening for psychosocial distress to identify individuals in need of additional support and to provide referrals for patients at high risk of psychosocial distress. The NCCN Distress Thermometer screening tool has been widely accepted as an effective method of identifying and characterizing distress. The NCCN tool provides a visual analogue scale for patients to rate their current distress on a scale of 1-10, as well as a problem checklist. The problem checklist includes 22 stressors addressing the practical, spiritual/religious, emotional, and physical concerns of patients. Although the NCCN tool is used widely, differences in distress scores between black and white cancer patients have not been previously described. The purpose of the study was to compare the global distress screening scores of black and white patients at an academic comprehensive cancer center in the Midwest. A second objective was to examine the distribution of individual stressors between black and white women.
Methods
Study sample
The study included all cancer patients from a cancer center in the Midwest who completed the NCCN distress thermometer during January 1, 2015-February 19, 2016. The patient population for this cancer center was primarily non-Hispanic white and non-Hispanic black, therefore, only patients identifying as non-Hispanic white and non-Hispanic black are included in this analysis. As part of routine clinical care, patients are asked to complete the NCCN distress thermometer at their first visit to the center. All patients in this analytic sample were newly diagnosed patients. Some patients also completed the NCCN screening tool at additional appointments; therefore, for patients with more than 1 completed tool, only the first distress screening was used in this analysis. Overall scores and individual stressor scores were entered into the electronic medical record by clinic staff at the time the patients were roomed for their visit. Patient demographics were collected through a reporting mechanism within the electronic medical record that allows for monitoring of the psychosocial screening process.
Variables
Race was assessed through self-report and classified as non-Hispanic white and non-Hispanic black. There were not enough patients of any other racial/ethnic group to be included in this analysis. Age was categorized as 18-40 years, 41-60 years, 61-84 years, and 85 years and older. Cancer type was grouped as follows: head and neck cancer, gastrointestinal cancer (esophagus, stomach, small intestine, colon, rectum, anus), hepatobiliary (liver, gallbladder, pancreas), sarcoma (bone and soft tissue), melanoma, nonmelanoma skin cancer, breast cancer, genitourinary (prostate, kidney, bladder), hematologic, and brain.
Two primary outcomes were assessed: overall distress, and each individual problem indicator. Overall distress was assessed using the thermometer visual analog rating (the thermometer rating of the NCCN screening tool) where possible values range from 0 (no distress) to 10 (extreme distress). The overall distress score was categorized into low distress (<4) and high distress (≥4) for analysis. The response options for individual stressors on the problem list are Yes or No for each of 17 discrete stressors: child care, housing, insurance/financial, transportation, work/school, treatment decisions, dealing with children, dealing with partner, ability to have children, family health issues, depression, fears, nervousness, sadness, worry, loss of interest, and spiritual/religious concerns. Physical complaints were not assessed in this study. Comparisons were made between white and black patients on overall distress score as well as for each individual psychosocial stressor.
Data analysis
Descriptive statistics (counts and proportions or means and standard deviations) were calculated stratified by race. Categorical variables were compared by race using chi-square or Fisher exact test. Logistic regression was used to predict high distress by race adjusting for sex, age, and cancer type. All analyses were conducted using SAS 9.4 (Cary, NJ).
This study was reviewed and approved by the Saint Louis University Institutional Review Board (protocol number 26269).
Results
A total of 933 patients with cancer completed the NCCN distress screening tool. Of that total, 45 patients did not complete the overall distress score thermometer, but did complete the checklist of individual stressors. Those 45 patients were excluded from the logistic regression analysis for overall distress score, but included on comparisons of individual stressors. The distribution of overall distress scores by race can be seen in the Figure.
Briefly, the full sample was 16.9% black and 38.8% female. In all, 32.6% of the sample indicated high distress on the distress thermometer at their first visit. Demographics for the participants stratified by race are reported in Table 1 (see PDF).There was no statistically significant difference in the gender or age distribution between black and white patients. Cancer distribution did vary by race. Black patients were proportionally more represented in gastrointestinal cancers, hepatobiliary cancers, sarcomas, breast cancer, and genitourinary cancers. White patients were proportionally more represented in melanoma, nonmelanoma skin cancers, and hematologic cancers.
Table 2 presents bivariate comparisons on overall distress and individual stressors between black and white patients. There was no difference in the high distress between black and white patients in bivariate analysis (31.9% and 36.1%, respectively, P = .30). However, there were differences in the individual stressors identified for each racial group (Table 2). White patients, compared with black patients, more frequently identified treatment decisions (17.6% vs 10.1%, P = .02) and nervousness (26.8% vs 18.4%, P = .02) as sources of distress. Black patients, compared with white patients, more frequently identified housing (5.1% vs 1.7%, P = .009), the ability to have children (2.5% vs 0.4%, P =.02), and loss of interest (15.2% vs 8.9%, P = .02) as sources of distress. Distress scores did not differ between black and white patients for child care, insurance or financial issues, transportation, work or school, dealing with children, dealing with partners, family health issues, depression, fears, sadness, worry, or spiritual or religious concerns.
Table 3 presents the results from the logistic analysis predicting high distress. In adjusted analysis, black race did not predict high distress (OR, 0.94; 95% confidence interval [CI], 0.62-1.44). High distress was associated with sex, age, and some cancer categories. Women had 77% higher odds of high distress compared with men (OR, 1.77; 95% CI, 1.25-2.51).
Compared with patients aged 18-44 years, patients aged 61-84 had 43% lower odds of high distress (OR, 0.57; 95% CI, 0.33-0.98), and patients aged 85 and older had 74% lower odds of high distress (OR, 0.26; 95% CI, 0.07-0.98). There was no statistically significant difference between patients aged 18-40 and those aged 41-60 for high distress (OR, 1.01; 95% CI, 0.59-1.73).
Discussion
Management of patients with cancer continues to evolve. Although a tremendous amount of importance is still placed on the pathophysiology of cancer and its prescribed treatments, more emphasis is being assigned to the physical and psychosocial effects of cancer on these patients. In 2008, the Institute of Medicine published a report that examined the psychosocial health of patients with cancer.14 The report recommended that all cancer care should ensure the provision of appropriate psychosocial health services by facilitating effective communication between patients and care providers, identifying each patient’s psychosocial health needs, coordinating referrals for psychosocial services and monitoring efficacy of psychosocial interventions. The inclusion of psychosocial distress screening in all cancer programs accredited by the ACS CoC helped to prioritize the identification and treatment of psychosocial issues for all cancer patients.
The present study is the first of its kind to compare the individual stressors identified through psychosocial distress screening between black and white cancer patients. In our sample, 304 of 933 patients (32%) reported high distress, with a total score of ≥4. Previous research on overall distress difference across race/ethnicity is mixed. VanHoose and colleagues found no difference in overall distress between racial groups,15 Alcala and colleagues found higher overall distress in black patients with cancer compared with white patients with cancer,9 and Culver and colleagues found black women with breast cancer had lower overall distress compared with white women.11 We found no difference in the presence of high distress between black and white patients at our cancer center in either crude or adjusted analysis. Differences in overall distress across studies may be owing to the timing of screening. Given that overall distress may vary across time16,17 and there is no current information on whether temporal variations in distress differ by race, it is possible that the time of distress assessment may influence demonstrated differences between racial groups. For example, if different stressors affect black and white women differentially, and those stressors are associated with different points across the cancer continuum, then we might see that the magnitude of racial differences in overall stress are time dependent. Alcala and colleagues examined any cancer diagnosis across the lifespan, whereas Culver and colleagues examined multiple time points across treatment for a small group of breast cancer patients. Badr and colleagues, in a sample of head and neck cancer patients, found that distress increased across the course of treatment;18 however they did not examine variations in type of stressors related to overall distress, nor did they examine racial differences in distress. Differences in results may also be the result of differences in measurement of distress. Culver and colleagues did not examine distress using the NCCN distress thermometer, rather psychological distress was measured by a scale rating a series of “mood-descriptive adjectives” (p. 497).11 Alcala used the K-6 as a measure of psychological distress;9 therefore, demonstrated differences in overall distress between white and black women may vary across studies because of differences in measurement of the underlying distress variable. The lack of racial differences in overall distress in our study is consistent with the findings of VanHoose and colleagues,15 who also examined distress near the start of treatment and also used the NCCN distress thermometer as the measure of psychosocial distress.
We did find differences in the individual stressors between racial groups, indicating that the source of distress does vary between black and white cancer patients. Black patients more frequently reported distress secondary to housing, loss of interest and their ability to have children than did white patients. By comparison, white patients more frequently reported distress secondary to nervousness and treatment decisions than black patients. Identified differences in individual stressors may be attributable to sociocultural differences or differences in external support. It is also possible that black patients are more likely to willingly report distress related to nonpsychological factors, whereas white patients are more willing to report factors, such as nervousness, that are related to psychological disorders. Although it has been suggested that black cancer patients have more concerns about finances and work than do white cancer patients,19 we did not identify a statistically significant difference in child care, insurance or financial issues, transportation, work, or school between these 2 cohorts. This may be because the psychosocial distress screening score included in this study was performed at the time of initial diagnosis, and not further into their prescribed treatment at which point the financial worries may be more realized. Psychosocial screening scores obtained at subsequent visits were not included in the analysis because they are not routinely collected as part of clinical care in the center where this study took place. Furthermore, it is impossible to identify where a specific patient is in their treatment regimen based on their demographic data or subsequent distress scores in our data extraction tool. Further investigation into the sources of distress at different time points along the continuum of care may shed more light on this topic.
Limitations
There are several limitations to this study. First, the method of data extraction from an electronic medical record report limited the capacity to explore possible differences between the patients in our sample, such as insurance status, level of education, available social support, current employment status, stage of disease, overall prognosis and prescribed treatment regimen.
Second, there were likely patients who either did not complete a psychosocial distress screening tool or whose data were not entered into the electronic medical record for inclusion in the analysis. The present study period took place during the implementation of the NCCN tool at the center. Although the policy was to screen all new patients as part of routine care; not all patients seen at the center received the NCCN screening tool at their first visit. Owing to the mechanisms for data entry and abstraction, only information from the patients who had a completed form was able to be accessed for this study, thus a statistical comparison between those who did and did not receive the NCCN tool cannot be made. During the timeframe for this study, the head and neck, breast, genitourinary, and hematologic services completed proportionally more NCCN screening of new patients than other services in the center. This is reflected in the distributional breakdown of cancer in the overall sample of this study. It is possible that the results are more representative of differences between black and white cancer patients in the services that were more likely to properly implement NCCN screening.
Third, our patient population was derived from an urban, academic medical center and the results may not be generalizable to other patient populations.
Fourth, the NCCN distress thermometer is a single-item rating of overall global distress that is not intended to be a diagnostic indicator of psychological comorbidity and, therefore, does not distinguish between common psychological diagnoses such as depression or anxiety. However, the usefulness of the tool is to provide an impetus for referral to services that may then encompass the evaluation and diagnosis of particular psychological conditions. Further, the distress thermometer tool is designed to identify stress relating to the social aspects of cancer diagnosis and treatment and is not limited to psychological distress alone.
Strengths
Despite the limitations, there are also significant strengths to this study. The NCCN tool is a widely accepted measure for the assessment of psychosocial distress in patients with cancer. The measure is a common and routine clinical instrument,20 and has also been used widely in research.18,21-24 Given the urban, academic environment of our clinical practice, our population is more racially diverse than other settings, allowing for initial examination of disparities between white and black cancer patients.
Clinical implications
Understanding differences in common psychosocial stressor between black and white cancer patients may allow for clinicians to strategically look for different types of stressors in order to facilitate faster referrals to appropriate services. It has been established in the literature that distress is correlated to cancer-related outcomes and distress screening is now considered standard of care when treating cancer patients. Identifying differences in psychosocial stressors among black and white cancer patients is paramount to ensuring that the appropriate resources are available to assist them through their cancer journey. The differences in type of stressor, may indicate fundamental differences in the way patients perceive their disease or the social and cultural implication of a cancer diagnosis. In this study, white patients were more likely to find distress in the psychological realm (nervousness, decision-making), whereas black patients were more likely to be distressed about social issues (housing, ability to have children, and loss of interest). The referral needs of patients may be quite different, even with similar levels of overall distress. More research is necessary to further characterize sources of distress for cancer patients, how this distress impacts a patient’s physical and emotional well-being and how health care providers can better identify these issues and make the necessary referrals to support the whole patient.
1. Holland JC, Reznik I. Pathways for psychosocial care of cancer survivors. Cancer. 2005;104(11 Suppl):2624-2637.
2. Strasser F, Sweeney C, Willey J, Benisch-Tolley S, Palmer L, Bruera E. Impact of a half-day multidisciplinary symptom control and palliative care outpatient clinic in a comprehensive cancer center on recommendations, symptom intensity, and patient satisfaction: a retrospective descriptive study. J Pain Symptom Manage. 2004;27(6):481-491.
3. Carlson LE, Bultz BD. Efficacy and medical cost offset of psychosocial interventions in cancer care: making the case for economic analyses. Psychooncology. 2004;13(12):837-849.
4. Holland J, Bultz BD. The NCCN Guideline for distress management: a case for making distress the sixth vital sign. J Natl Compr Canc Netw. 2007;5(1):3-7.
5. Krebber A, Buffart L, Kleijn G, et al. Prevalence of depression in cancer patients: a meta-analysis of diagnostic interviews and self-report instruments. Psychooncology. 2014;23(2):121-130.
6. Sharp L, Carsin AE , Timmons A. Associations between cancer-related financial stress and strain and psychological well-being among individuals living with cancer. Psychooncology. 2013;22(4):745-755.
7. Bruce J, Thornton AJ, Powell R, et al. Psychological, surgical, and sociodemographic predictors of pain outcomes after breast cancer surgery: a population-based cohort study. Pain. 2014;155(2):232-243.
8. Holland JC. Preliminary guidelines for the treatment of distress. Oncology. 1997;11(11A):109-114.
9. Alcala HE. Differential mental health impact of cancer across racial/ethnic groups: findings from a population-based study in California. BMC Public Health. 2014;14:930.
10. Bradley CJ, Wilk A. Racial differences in quality of life and employment outcomes in insured women with breast cancer. J Cancer Surviv. 2014;8(1):49-59.
11. Culver JL, Arena PL, Antoni MH, Carver CS. Coping and distress among women under treatment for early stage breast cancer: comparing African Americans, Hispanics and non-Hispanic whites. Psychooncology. 2002;11(6):495-504.
12. American College of Surgeons Commission on Cancer. ACSCC website. Cancer program standards: ensuring patient-centered care. 2016 edition. https://www.facs.org/quality-programs/cancer/coc/standards. Posted 2016. Accessed August 30, 2017.
13. National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology: Distress Management. National Comprehensive Cancer Network, 2014.https://www.nccn.org/store/login/login.aspx?ReturnURL=https://www.nccn.org/professionals/physician_gls/pdf/distress.pdf Accessed August 30, 2017.
14. Institute of Medicine. Cancer care for the whole patient: meeting psychosocial health needs. Washington, DC: The National Academies Press; 2008. https://doi.org/10.17226/11993. Accessed August 30, 2017.
15. VanHoose L, Black LL, Doty K, et al. An analysis of the distress thermometer problem list and distress in patients with cancer. Support Care Cancer. 2015;23(5):1225-1232.
16. Gessler S, Low J, Daniells E, et al. Screening for distress in cancer patients: is the distress thermometer a valid measure in the UK and does it measure change over time? A prospective validation study. Psychooncology. 2008;17(6):538-547.
17. Enns A, Waller A, Groff SL, Bultz BD, Fung T, Carlson LE. Risk factors for continuous distress over a 12-month period in newly diagnosed cancer outpatients. J Psychosoc Oncol. 2013;31(5):489-506.
18. Badr H, Gupta V, Sikora A, Posner M. Psychological distress in patients and caregivers over the course of radiotherapy for head and neck cancer. Oral Oncol. 2014;50(10):1005-1011.
19. Wang X, Cosby LG, Harris MG, Liu T. Major concerns and needs of breast cancer patients. Cancer Nurs. 1999;22(2):157-163.
20. Dabrowski M, Boucher K, Ward JH, et al. Clinical experience with the NCCN distress thermometer in breast cancer patients. J Natl Compr Canc Netw. 2007;5(1):104-11.
21. Buchmann L, Conlee J, Hunt J, Agarwal J, White S. Psychosocial distress in prevalent in head and neck cancer patients. Laryngoscope. 2013;123(6):1424-1429.
22. Agarwal J, Powers K, Pappas L, et al. Correlates of elevated distress thermometer scores in breast cancer patients. Support Care Cancer. 2013;21(8):2125-2136.
23. Johnson R, Gold MA, Wythe KF. Distress in women with gynecologic cancer. Psychooncology. 2010;19(6):665-668.
24. Kendall J, Glaze K, Oakland S, Hansen J, Parry C. What do 1281 distress screeners tell us about cancer patients in a community cancer center? Psychooncology. 2011;20(6):594-600.
1. Holland JC, Reznik I. Pathways for psychosocial care of cancer survivors. Cancer. 2005;104(11 Suppl):2624-2637.
2. Strasser F, Sweeney C, Willey J, Benisch-Tolley S, Palmer L, Bruera E. Impact of a half-day multidisciplinary symptom control and palliative care outpatient clinic in a comprehensive cancer center on recommendations, symptom intensity, and patient satisfaction: a retrospective descriptive study. J Pain Symptom Manage. 2004;27(6):481-491.
3. Carlson LE, Bultz BD. Efficacy and medical cost offset of psychosocial interventions in cancer care: making the case for economic analyses. Psychooncology. 2004;13(12):837-849.
4. Holland J, Bultz BD. The NCCN Guideline for distress management: a case for making distress the sixth vital sign. J Natl Compr Canc Netw. 2007;5(1):3-7.
5. Krebber A, Buffart L, Kleijn G, et al. Prevalence of depression in cancer patients: a meta-analysis of diagnostic interviews and self-report instruments. Psychooncology. 2014;23(2):121-130.
6. Sharp L, Carsin AE , Timmons A. Associations between cancer-related financial stress and strain and psychological well-being among individuals living with cancer. Psychooncology. 2013;22(4):745-755.
7. Bruce J, Thornton AJ, Powell R, et al. Psychological, surgical, and sociodemographic predictors of pain outcomes after breast cancer surgery: a population-based cohort study. Pain. 2014;155(2):232-243.
8. Holland JC. Preliminary guidelines for the treatment of distress. Oncology. 1997;11(11A):109-114.
9. Alcala HE. Differential mental health impact of cancer across racial/ethnic groups: findings from a population-based study in California. BMC Public Health. 2014;14:930.
10. Bradley CJ, Wilk A. Racial differences in quality of life and employment outcomes in insured women with breast cancer. J Cancer Surviv. 2014;8(1):49-59.
11. Culver JL, Arena PL, Antoni MH, Carver CS. Coping and distress among women under treatment for early stage breast cancer: comparing African Americans, Hispanics and non-Hispanic whites. Psychooncology. 2002;11(6):495-504.
12. American College of Surgeons Commission on Cancer. ACSCC website. Cancer program standards: ensuring patient-centered care. 2016 edition. https://www.facs.org/quality-programs/cancer/coc/standards. Posted 2016. Accessed August 30, 2017.
13. National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology: Distress Management. National Comprehensive Cancer Network, 2014.https://www.nccn.org/store/login/login.aspx?ReturnURL=https://www.nccn.org/professionals/physician_gls/pdf/distress.pdf Accessed August 30, 2017.
14. Institute of Medicine. Cancer care for the whole patient: meeting psychosocial health needs. Washington, DC: The National Academies Press; 2008. https://doi.org/10.17226/11993. Accessed August 30, 2017.
15. VanHoose L, Black LL, Doty K, et al. An analysis of the distress thermometer problem list and distress in patients with cancer. Support Care Cancer. 2015;23(5):1225-1232.
16. Gessler S, Low J, Daniells E, et al. Screening for distress in cancer patients: is the distress thermometer a valid measure in the UK and does it measure change over time? A prospective validation study. Psychooncology. 2008;17(6):538-547.
17. Enns A, Waller A, Groff SL, Bultz BD, Fung T, Carlson LE. Risk factors for continuous distress over a 12-month period in newly diagnosed cancer outpatients. J Psychosoc Oncol. 2013;31(5):489-506.
18. Badr H, Gupta V, Sikora A, Posner M. Psychological distress in patients and caregivers over the course of radiotherapy for head and neck cancer. Oral Oncol. 2014;50(10):1005-1011.
19. Wang X, Cosby LG, Harris MG, Liu T. Major concerns and needs of breast cancer patients. Cancer Nurs. 1999;22(2):157-163.
20. Dabrowski M, Boucher K, Ward JH, et al. Clinical experience with the NCCN distress thermometer in breast cancer patients. J Natl Compr Canc Netw. 2007;5(1):104-11.
21. Buchmann L, Conlee J, Hunt J, Agarwal J, White S. Psychosocial distress in prevalent in head and neck cancer patients. Laryngoscope. 2013;123(6):1424-1429.
22. Agarwal J, Powers K, Pappas L, et al. Correlates of elevated distress thermometer scores in breast cancer patients. Support Care Cancer. 2013;21(8):2125-2136.
23. Johnson R, Gold MA, Wythe KF. Distress in women with gynecologic cancer. Psychooncology. 2010;19(6):665-668.
24. Kendall J, Glaze K, Oakland S, Hansen J, Parry C. What do 1281 distress screeners tell us about cancer patients in a community cancer center? Psychooncology. 2011;20(6):594-600.