User login
What’s New in Diabetes Management: Psychosocial Care
The wide array of comorbidities and treatment variables can make diabetes a difficult disease to manage—and to live with. Providers must be equipped to address the complexities and complications that affect patients with diabetes. In December 2016, the American Diabetes Association published a position statement recognizing the psychosocial factors (environmental, social, behavioral, and emotional) that affect medical outcomes and psychological well-being in persons with diabetes. These include self-management, diabetes distress, psychological comorbidities, and life-course considerations.1
SELF-MANAGEMENT
A patient’s perception of his or her ability to self-manage diabetes is an important psychosocial factor in treatment and management outcomes. Training patients with diabetes in self-care skills and the use of technologies—at the time of diagnosis, annually, and/or when complications or transitions in care occur—can empower patients to assume an active role in their daily management. These interventions can be tailored to address specific, individualized problems that contribute to suboptimal glycemic outcomes, such as issues in numeracy or coping, food insecurity, or lack of support. Employing a nonjudgmental approach that normalizes periodic lapses in self-management may help encourage patients and minimize their resistance to self-management.
DIABETES DISTRESS
The frustration, worry, anger, guilt, and burnout imposed by diabetes and its management (via glucose monitoring, medication dosing, and insulin titration) is known as diabetes distress. With a reported prevalence of 18% to 45%, this disease burden is quite common.2 Because high levels of diabetes distress are associated with low self-efficacy, poor glycemic outcomes, and suboptimal exercise/dietary habits, referral for counseling should be considered if a patient expresses feelings of distress.
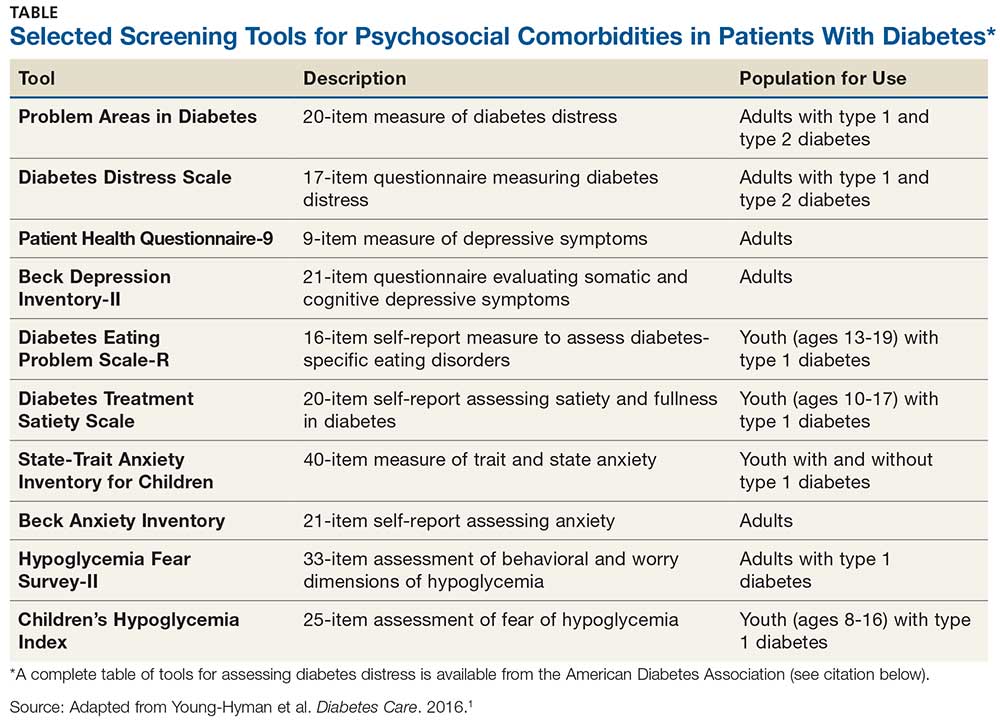
Use of validated screening tools, such as Problem Areas in Diabetes (PAID)3,4 or the Diabetes Distress Scale (DDS)5, can aid in routine monitoring for diabetes distress. (See the Table for more information.) If distress is identified in specific self-care areas, further patient education on self-management is appropriate.
PSYCHOLOGICAL COMORBIDITIES
Depression, anxiety, disordered eating, and serious mental illness (eg, schizophrenia) are known psychological comorbidities of diabetes. Screening for symptoms using patient-appropriate, standardized/validated tools should occur at initial visit, at periodic intervals, and when there is a change in disease, treatment, or life circumstance.
Depression
Patients with diabetes should be screened for depression when medical status worsens or when complications occur; it is recommended to include caregivers and family members in this assessment. Patients who screen positive for depression should be referred to mental health providers who have experience with cognitive behavioral therapy, interpersonal therapy, or other evidence-based treatment approaches, and who can provide collaborative care alongside the diabetes treatment team. Once diagnosed with depression, patients should be screened annually.
Anxiety
Expression of fear, dread, or irrational thoughts, avoidant and/or repetitious behaviors, and social withdrawal are signs of anxiety that should prompt screening. Consider screening for anxiety in patients who express worry about diabetes complications, insulin injections or infusion, taking medications, and/or hypoglycemia that interferes with self-management behaviors.
Continue to: Patients with hypoglycemia unawareness...
Patients with hypoglycemia unawareness, which can co-occur with fear of hypoglycemia, can increase self-monitoring of glucose with a glucometer or continuous glucose monitor. Blood Glucose Awareness Training (or other similar evidence-based intervention) can be used to help reestablish awareness of hypoglycemia and reduce fear of hypoglycemia.6-8 Providers can deliver hypoglycemia awareness education in the clinic.
Disordered Eating
When hyperglycemia and weight loss are unexplained by self-reported medication dosing, diet, and exercise, consider screening for disordered or disrupted eating (see Table for screening tools). In addition, reviewing the medical regimen is recommended to identify potential treatment-related effects on hunger/caloric intake.
Cognitive Impairment
Since research has shown significantly increased rates of diabetes among persons with serious mental illness (eg, schizophrenia), annual screening for prediabetes and diabetes is recommended for those taking atypical antipsychotic medications. Furthermore, some of the effects of serious mental illness—such as disordered thinking and impaired judgment—make it difficult for a patient to engage in risk-reducing behaviors or (if diagnosed) to manage diabetes. Therefore, monitoring of diabetes self-care activities should be incorporated into treatment goals for persons with these comorbid conditions.
Continue to: CONCLUSION
CONCLUSION
As providers, we should be familiar with the evidence-based, validated tools available to identify the psychosocial comorbidities of diabetes. Screening and assessing patients for psychosocial/behavioral challenges should be performed at an initial visit, at periodic intervals, and whenever there is a change in disease, treatment, or life circumstances.
Health care alliances with behavioral/mental health providers who are knowledgeable about diabetes treatment and the psychosocial aspects of diabetes are key. Patient-centered care is essential to promote optimal medical outcomes and psychological well-being. As members of the health care team, we must be respectful and responsive to patient preferences, needs, and values; clinical decisions should be guided by patient values. If A1C is not at goal despite maximized medication therapy and lifestyle modification, consider identifying and addressing any psychosocial factors that may be involved.
1. Young-Hyman D, de Groot M, Hill-Briggs F, et al. Psychosocial care for people with diabetes: a position statement of the American Diabetes Association. Diabetes Care. 2016;39(12):2126-2140.
2. Aikens JE. Prospective associations between emotional distress and poor outcomes in type 2 diabetes. Diabetes Care. 2012;35(12):2472-2478.
3. Polonsky WH, Anderson BJ, Lohrer PA, et al. Assessment of diabetes-related distress. Diabetes Care. 1995;18(6):754-760.
4. Welch G, Weinger K, Anderson B, Polonsky WH. Responsiveness of the Problem Areas in Diabetes (PAID) questionnaire. Diabet Med. 2003;20(1):69-72.
5. Polonsky WH, Fisher L, Earles J, et al. Assessing psychosocial stress in diabetes: development of the Diabetes Distress Scale. Diabetes Care. 2005;28(3):626-631.
6. Cox DJ, Gonder-Frederick L, Polonsky W, et al. Blood Glucose Awareness Training (BGAT-2). Diabetes Care. 2001;24(4):637-642.
7. Fisher L, Hessler DM, Polonsky WH, Mullan J. When is diabetes distress clinically meaningful? Establishing cut points for the Diabetes Distress Scale. Diabetes Care. 2012;35(2):259-264.
8. Gonder-Frederick LA, Schmidt KM, Vajda KA, et al. Psychometric properties of the Hypoglycemia Fear Survey-II for adults with type 1 diabetes. Diabetes Care. 2011;34(4):801-806.
Clinician Reviews in partnership with
Amy Butts, Billy Collins, and Joy Dugan are members of the American Diabetes Association's Primary Care Advisory Group. Amy Butts is in private practice in Weirton, West Virginia. Billy Collins is a Health Services Officer for the Indian Health Service on the Alabama-Coushatta Tribe of Texas reservation in Polk County. Joy Dugan is an Adjunct Assistant Professor in the Joint Master of Physician Assistant and Master of Public Health Program at Touro University California in Vallejo.
Amy Butts serves on the speakers' bureau for Janssen Pharmaceuticals. Billy Collins and Joy Dugan have no potential conflicts of interest to report.
Clinician Reviews in partnership with
Amy Butts, Billy Collins, and Joy Dugan are members of the American Diabetes Association's Primary Care Advisory Group. Amy Butts is in private practice in Weirton, West Virginia. Billy Collins is a Health Services Officer for the Indian Health Service on the Alabama-Coushatta Tribe of Texas reservation in Polk County. Joy Dugan is an Adjunct Assistant Professor in the Joint Master of Physician Assistant and Master of Public Health Program at Touro University California in Vallejo.
Amy Butts serves on the speakers' bureau for Janssen Pharmaceuticals. Billy Collins and Joy Dugan have no potential conflicts of interest to report.
Clinician Reviews in partnership with
Amy Butts, Billy Collins, and Joy Dugan are members of the American Diabetes Association's Primary Care Advisory Group. Amy Butts is in private practice in Weirton, West Virginia. Billy Collins is a Health Services Officer for the Indian Health Service on the Alabama-Coushatta Tribe of Texas reservation in Polk County. Joy Dugan is an Adjunct Assistant Professor in the Joint Master of Physician Assistant and Master of Public Health Program at Touro University California in Vallejo.
Amy Butts serves on the speakers' bureau for Janssen Pharmaceuticals. Billy Collins and Joy Dugan have no potential conflicts of interest to report.
The wide array of comorbidities and treatment variables can make diabetes a difficult disease to manage—and to live with. Providers must be equipped to address the complexities and complications that affect patients with diabetes. In December 2016, the American Diabetes Association published a position statement recognizing the psychosocial factors (environmental, social, behavioral, and emotional) that affect medical outcomes and psychological well-being in persons with diabetes. These include self-management, diabetes distress, psychological comorbidities, and life-course considerations.1
SELF-MANAGEMENT
A patient’s perception of his or her ability to self-manage diabetes is an important psychosocial factor in treatment and management outcomes. Training patients with diabetes in self-care skills and the use of technologies—at the time of diagnosis, annually, and/or when complications or transitions in care occur—can empower patients to assume an active role in their daily management. These interventions can be tailored to address specific, individualized problems that contribute to suboptimal glycemic outcomes, such as issues in numeracy or coping, food insecurity, or lack of support. Employing a nonjudgmental approach that normalizes periodic lapses in self-management may help encourage patients and minimize their resistance to self-management.
DIABETES DISTRESS
The frustration, worry, anger, guilt, and burnout imposed by diabetes and its management (via glucose monitoring, medication dosing, and insulin titration) is known as diabetes distress. With a reported prevalence of 18% to 45%, this disease burden is quite common.2 Because high levels of diabetes distress are associated with low self-efficacy, poor glycemic outcomes, and suboptimal exercise/dietary habits, referral for counseling should be considered if a patient expresses feelings of distress.
Use of validated screening tools, such as Problem Areas in Diabetes (PAID)3,4 or the Diabetes Distress Scale (DDS)5, can aid in routine monitoring for diabetes distress. (See the Table for more information.) If distress is identified in specific self-care areas, further patient education on self-management is appropriate.
PSYCHOLOGICAL COMORBIDITIES
Depression, anxiety, disordered eating, and serious mental illness (eg, schizophrenia) are known psychological comorbidities of diabetes. Screening for symptoms using patient-appropriate, standardized/validated tools should occur at initial visit, at periodic intervals, and when there is a change in disease, treatment, or life circumstance.
Depression
Patients with diabetes should be screened for depression when medical status worsens or when complications occur; it is recommended to include caregivers and family members in this assessment. Patients who screen positive for depression should be referred to mental health providers who have experience with cognitive behavioral therapy, interpersonal therapy, or other evidence-based treatment approaches, and who can provide collaborative care alongside the diabetes treatment team. Once diagnosed with depression, patients should be screened annually.
Anxiety
Expression of fear, dread, or irrational thoughts, avoidant and/or repetitious behaviors, and social withdrawal are signs of anxiety that should prompt screening. Consider screening for anxiety in patients who express worry about diabetes complications, insulin injections or infusion, taking medications, and/or hypoglycemia that interferes with self-management behaviors.
Continue to: Patients with hypoglycemia unawareness...
Patients with hypoglycemia unawareness, which can co-occur with fear of hypoglycemia, can increase self-monitoring of glucose with a glucometer or continuous glucose monitor. Blood Glucose Awareness Training (or other similar evidence-based intervention) can be used to help reestablish awareness of hypoglycemia and reduce fear of hypoglycemia.6-8 Providers can deliver hypoglycemia awareness education in the clinic.
Disordered Eating
When hyperglycemia and weight loss are unexplained by self-reported medication dosing, diet, and exercise, consider screening for disordered or disrupted eating (see Table for screening tools). In addition, reviewing the medical regimen is recommended to identify potential treatment-related effects on hunger/caloric intake.
Cognitive Impairment
Since research has shown significantly increased rates of diabetes among persons with serious mental illness (eg, schizophrenia), annual screening for prediabetes and diabetes is recommended for those taking atypical antipsychotic medications. Furthermore, some of the effects of serious mental illness—such as disordered thinking and impaired judgment—make it difficult for a patient to engage in risk-reducing behaviors or (if diagnosed) to manage diabetes. Therefore, monitoring of diabetes self-care activities should be incorporated into treatment goals for persons with these comorbid conditions.
Continue to: CONCLUSION
CONCLUSION
As providers, we should be familiar with the evidence-based, validated tools available to identify the psychosocial comorbidities of diabetes. Screening and assessing patients for psychosocial/behavioral challenges should be performed at an initial visit, at periodic intervals, and whenever there is a change in disease, treatment, or life circumstances.
Health care alliances with behavioral/mental health providers who are knowledgeable about diabetes treatment and the psychosocial aspects of diabetes are key. Patient-centered care is essential to promote optimal medical outcomes and psychological well-being. As members of the health care team, we must be respectful and responsive to patient preferences, needs, and values; clinical decisions should be guided by patient values. If A1C is not at goal despite maximized medication therapy and lifestyle modification, consider identifying and addressing any psychosocial factors that may be involved.
The wide array of comorbidities and treatment variables can make diabetes a difficult disease to manage—and to live with. Providers must be equipped to address the complexities and complications that affect patients with diabetes. In December 2016, the American Diabetes Association published a position statement recognizing the psychosocial factors (environmental, social, behavioral, and emotional) that affect medical outcomes and psychological well-being in persons with diabetes. These include self-management, diabetes distress, psychological comorbidities, and life-course considerations.1
SELF-MANAGEMENT
A patient’s perception of his or her ability to self-manage diabetes is an important psychosocial factor in treatment and management outcomes. Training patients with diabetes in self-care skills and the use of technologies—at the time of diagnosis, annually, and/or when complications or transitions in care occur—can empower patients to assume an active role in their daily management. These interventions can be tailored to address specific, individualized problems that contribute to suboptimal glycemic outcomes, such as issues in numeracy or coping, food insecurity, or lack of support. Employing a nonjudgmental approach that normalizes periodic lapses in self-management may help encourage patients and minimize their resistance to self-management.
DIABETES DISTRESS
The frustration, worry, anger, guilt, and burnout imposed by diabetes and its management (via glucose monitoring, medication dosing, and insulin titration) is known as diabetes distress. With a reported prevalence of 18% to 45%, this disease burden is quite common.2 Because high levels of diabetes distress are associated with low self-efficacy, poor glycemic outcomes, and suboptimal exercise/dietary habits, referral for counseling should be considered if a patient expresses feelings of distress.
Use of validated screening tools, such as Problem Areas in Diabetes (PAID)3,4 or the Diabetes Distress Scale (DDS)5, can aid in routine monitoring for diabetes distress. (See the Table for more information.) If distress is identified in specific self-care areas, further patient education on self-management is appropriate.
PSYCHOLOGICAL COMORBIDITIES
Depression, anxiety, disordered eating, and serious mental illness (eg, schizophrenia) are known psychological comorbidities of diabetes. Screening for symptoms using patient-appropriate, standardized/validated tools should occur at initial visit, at periodic intervals, and when there is a change in disease, treatment, or life circumstance.
Depression
Patients with diabetes should be screened for depression when medical status worsens or when complications occur; it is recommended to include caregivers and family members in this assessment. Patients who screen positive for depression should be referred to mental health providers who have experience with cognitive behavioral therapy, interpersonal therapy, or other evidence-based treatment approaches, and who can provide collaborative care alongside the diabetes treatment team. Once diagnosed with depression, patients should be screened annually.
Anxiety
Expression of fear, dread, or irrational thoughts, avoidant and/or repetitious behaviors, and social withdrawal are signs of anxiety that should prompt screening. Consider screening for anxiety in patients who express worry about diabetes complications, insulin injections or infusion, taking medications, and/or hypoglycemia that interferes with self-management behaviors.
Continue to: Patients with hypoglycemia unawareness...
Patients with hypoglycemia unawareness, which can co-occur with fear of hypoglycemia, can increase self-monitoring of glucose with a glucometer or continuous glucose monitor. Blood Glucose Awareness Training (or other similar evidence-based intervention) can be used to help reestablish awareness of hypoglycemia and reduce fear of hypoglycemia.6-8 Providers can deliver hypoglycemia awareness education in the clinic.
Disordered Eating
When hyperglycemia and weight loss are unexplained by self-reported medication dosing, diet, and exercise, consider screening for disordered or disrupted eating (see Table for screening tools). In addition, reviewing the medical regimen is recommended to identify potential treatment-related effects on hunger/caloric intake.
Cognitive Impairment
Since research has shown significantly increased rates of diabetes among persons with serious mental illness (eg, schizophrenia), annual screening for prediabetes and diabetes is recommended for those taking atypical antipsychotic medications. Furthermore, some of the effects of serious mental illness—such as disordered thinking and impaired judgment—make it difficult for a patient to engage in risk-reducing behaviors or (if diagnosed) to manage diabetes. Therefore, monitoring of diabetes self-care activities should be incorporated into treatment goals for persons with these comorbid conditions.
Continue to: CONCLUSION
CONCLUSION
As providers, we should be familiar with the evidence-based, validated tools available to identify the psychosocial comorbidities of diabetes. Screening and assessing patients for psychosocial/behavioral challenges should be performed at an initial visit, at periodic intervals, and whenever there is a change in disease, treatment, or life circumstances.
Health care alliances with behavioral/mental health providers who are knowledgeable about diabetes treatment and the psychosocial aspects of diabetes are key. Patient-centered care is essential to promote optimal medical outcomes and psychological well-being. As members of the health care team, we must be respectful and responsive to patient preferences, needs, and values; clinical decisions should be guided by patient values. If A1C is not at goal despite maximized medication therapy and lifestyle modification, consider identifying and addressing any psychosocial factors that may be involved.
1. Young-Hyman D, de Groot M, Hill-Briggs F, et al. Psychosocial care for people with diabetes: a position statement of the American Diabetes Association. Diabetes Care. 2016;39(12):2126-2140.
2. Aikens JE. Prospective associations between emotional distress and poor outcomes in type 2 diabetes. Diabetes Care. 2012;35(12):2472-2478.
3. Polonsky WH, Anderson BJ, Lohrer PA, et al. Assessment of diabetes-related distress. Diabetes Care. 1995;18(6):754-760.
4. Welch G, Weinger K, Anderson B, Polonsky WH. Responsiveness of the Problem Areas in Diabetes (PAID) questionnaire. Diabet Med. 2003;20(1):69-72.
5. Polonsky WH, Fisher L, Earles J, et al. Assessing psychosocial stress in diabetes: development of the Diabetes Distress Scale. Diabetes Care. 2005;28(3):626-631.
6. Cox DJ, Gonder-Frederick L, Polonsky W, et al. Blood Glucose Awareness Training (BGAT-2). Diabetes Care. 2001;24(4):637-642.
7. Fisher L, Hessler DM, Polonsky WH, Mullan J. When is diabetes distress clinically meaningful? Establishing cut points for the Diabetes Distress Scale. Diabetes Care. 2012;35(2):259-264.
8. Gonder-Frederick LA, Schmidt KM, Vajda KA, et al. Psychometric properties of the Hypoglycemia Fear Survey-II for adults with type 1 diabetes. Diabetes Care. 2011;34(4):801-806.
1. Young-Hyman D, de Groot M, Hill-Briggs F, et al. Psychosocial care for people with diabetes: a position statement of the American Diabetes Association. Diabetes Care. 2016;39(12):2126-2140.
2. Aikens JE. Prospective associations between emotional distress and poor outcomes in type 2 diabetes. Diabetes Care. 2012;35(12):2472-2478.
3. Polonsky WH, Anderson BJ, Lohrer PA, et al. Assessment of diabetes-related distress. Diabetes Care. 1995;18(6):754-760.
4. Welch G, Weinger K, Anderson B, Polonsky WH. Responsiveness of the Problem Areas in Diabetes (PAID) questionnaire. Diabet Med. 2003;20(1):69-72.
5. Polonsky WH, Fisher L, Earles J, et al. Assessing psychosocial stress in diabetes: development of the Diabetes Distress Scale. Diabetes Care. 2005;28(3):626-631.
6. Cox DJ, Gonder-Frederick L, Polonsky W, et al. Blood Glucose Awareness Training (BGAT-2). Diabetes Care. 2001;24(4):637-642.
7. Fisher L, Hessler DM, Polonsky WH, Mullan J. When is diabetes distress clinically meaningful? Establishing cut points for the Diabetes Distress Scale. Diabetes Care. 2012;35(2):259-264.
8. Gonder-Frederick LA, Schmidt KM, Vajda KA, et al. Psychometric properties of the Hypoglycemia Fear Survey-II for adults with type 1 diabetes. Diabetes Care. 2011;34(4):801-806.
Where the latest HCV drug combos fit in
MAUI, HAWAII – The addition of the two latest treatment regimens to receive approval for hepatitis C essentially closes the circle on treatment of this disease, Steven L. Flamm, MD, declared at the Gastroenterology Updates, IBD, Liver Disease meeting.
“We now have good options available for all the hepatitis C scenarios you will ever see in your practice,” said Dr. Flamm, professor of medicine and chief of the hepatology program at Northwestern University, Chicago.

“We’re already seeing a decline in the number of patients who are listed for liver transplantation with hepatitis C as the indication with UNOS [the United Organ Sharing database],” the gastroenterologist noted, citing a study presented at the 2017 annual meeting of the American Association for the Study of Liver Disease that showed that the proportion of patients who join the transplant wait-list with hepatitis C as their qualifying diagnosis has fallen by 35% since approval of the direct-acting antiviral (DAA) regimens in late 2013.
What’s special about the two newest DAA treatment regimens – sofosbuvir/velpatasvir/voxilaprevir (Vosevi) and glecaprevir/pibrentasvir (Mavyret) – is that they are pangenotypic, they are effective in prior treatment failures, they don’t need to be accompanied by ribavirin, and there is no need for baseline pretreatment resistance-associated substitution testing, Dr. Flamm noted.
“So if you have a patient sitting in front of you with any genotype of hepatitis C infection who has failed on NS5a-inhibitor therapy, you can tell them in general their chance of getting an SVR [sustained viral response] with sofosbuvir/velpatasvir/voxilaprevir is about 97%. And you can give it without worrying about what resistances they might have to begin with,” he said.
His copanelist Norah Terrault, MD, agreed that these two regimens are important additions.
“Glecaprevir/pibrentasvir is the first pangenic 8-week regimen for noncirrhotics. This is a major advance. And now having sofosbuvir/velpatasvir/voxilaprevir for treatment-experienced patients, that’s another strong advance,” commented Dr. Terrault, professor of medicine and director of the Viral Hepatitis Center at the University of California, San Francisco.
Dr. Flamm said the biggest remaining challenge in the treatment of hepatitis C is to gain improved access to therapy.
“The public-aid patients make up 30%-35% of patients with hepatitis C in my part of the country, and they still can’t get therapy unless they have cirrhosis. We can’t even treat people who have stage 2 fibrosis if they’re public-aid patients in Illinois. So we can’t achieve the goal of eliminating hepatitis C,” Dr. Flamm said.
He reported having no financial conflicts regarding his presentation.
MAUI, HAWAII – The addition of the two latest treatment regimens to receive approval for hepatitis C essentially closes the circle on treatment of this disease, Steven L. Flamm, MD, declared at the Gastroenterology Updates, IBD, Liver Disease meeting.
“We now have good options available for all the hepatitis C scenarios you will ever see in your practice,” said Dr. Flamm, professor of medicine and chief of the hepatology program at Northwestern University, Chicago.
“We’re already seeing a decline in the number of patients who are listed for liver transplantation with hepatitis C as the indication with UNOS [the United Organ Sharing database],” the gastroenterologist noted, citing a study presented at the 2017 annual meeting of the American Association for the Study of Liver Disease that showed that the proportion of patients who join the transplant wait-list with hepatitis C as their qualifying diagnosis has fallen by 35% since approval of the direct-acting antiviral (DAA) regimens in late 2013.
What’s special about the two newest DAA treatment regimens – sofosbuvir/velpatasvir/voxilaprevir (Vosevi) and glecaprevir/pibrentasvir (Mavyret) – is that they are pangenotypic, they are effective in prior treatment failures, they don’t need to be accompanied by ribavirin, and there is no need for baseline pretreatment resistance-associated substitution testing, Dr. Flamm noted.
“So if you have a patient sitting in front of you with any genotype of hepatitis C infection who has failed on NS5a-inhibitor therapy, you can tell them in general their chance of getting an SVR [sustained viral response] with sofosbuvir/velpatasvir/voxilaprevir is about 97%. And you can give it without worrying about what resistances they might have to begin with,” he said.
His copanelist Norah Terrault, MD, agreed that these two regimens are important additions.
“Glecaprevir/pibrentasvir is the first pangenic 8-week regimen for noncirrhotics. This is a major advance. And now having sofosbuvir/velpatasvir/voxilaprevir for treatment-experienced patients, that’s another strong advance,” commented Dr. Terrault, professor of medicine and director of the Viral Hepatitis Center at the University of California, San Francisco.
Dr. Flamm said the biggest remaining challenge in the treatment of hepatitis C is to gain improved access to therapy.
“The public-aid patients make up 30%-35% of patients with hepatitis C in my part of the country, and they still can’t get therapy unless they have cirrhosis. We can’t even treat people who have stage 2 fibrosis if they’re public-aid patients in Illinois. So we can’t achieve the goal of eliminating hepatitis C,” Dr. Flamm said.
He reported having no financial conflicts regarding his presentation.
MAUI, HAWAII – The addition of the two latest treatment regimens to receive approval for hepatitis C essentially closes the circle on treatment of this disease, Steven L. Flamm, MD, declared at the Gastroenterology Updates, IBD, Liver Disease meeting.
“We now have good options available for all the hepatitis C scenarios you will ever see in your practice,” said Dr. Flamm, professor of medicine and chief of the hepatology program at Northwestern University, Chicago.
“We’re already seeing a decline in the number of patients who are listed for liver transplantation with hepatitis C as the indication with UNOS [the United Organ Sharing database],” the gastroenterologist noted, citing a study presented at the 2017 annual meeting of the American Association for the Study of Liver Disease that showed that the proportion of patients who join the transplant wait-list with hepatitis C as their qualifying diagnosis has fallen by 35% since approval of the direct-acting antiviral (DAA) regimens in late 2013.
What’s special about the two newest DAA treatment regimens – sofosbuvir/velpatasvir/voxilaprevir (Vosevi) and glecaprevir/pibrentasvir (Mavyret) – is that they are pangenotypic, they are effective in prior treatment failures, they don’t need to be accompanied by ribavirin, and there is no need for baseline pretreatment resistance-associated substitution testing, Dr. Flamm noted.
“So if you have a patient sitting in front of you with any genotype of hepatitis C infection who has failed on NS5a-inhibitor therapy, you can tell them in general their chance of getting an SVR [sustained viral response] with sofosbuvir/velpatasvir/voxilaprevir is about 97%. And you can give it without worrying about what resistances they might have to begin with,” he said.
His copanelist Norah Terrault, MD, agreed that these two regimens are important additions.
“Glecaprevir/pibrentasvir is the first pangenic 8-week regimen for noncirrhotics. This is a major advance. And now having sofosbuvir/velpatasvir/voxilaprevir for treatment-experienced patients, that’s another strong advance,” commented Dr. Terrault, professor of medicine and director of the Viral Hepatitis Center at the University of California, San Francisco.
Dr. Flamm said the biggest remaining challenge in the treatment of hepatitis C is to gain improved access to therapy.
“The public-aid patients make up 30%-35% of patients with hepatitis C in my part of the country, and they still can’t get therapy unless they have cirrhosis. We can’t even treat people who have stage 2 fibrosis if they’re public-aid patients in Illinois. So we can’t achieve the goal of eliminating hepatitis C,” Dr. Flamm said.
He reported having no financial conflicts regarding his presentation.
Don’t shorten therapy for older, sicker cellulitis patients
MADRID – An attempt to balance effective treatment with good antibiotic stewardship fell short when patients with cellulitis who got 6 days of flucloxacillin relapsed significantly sooner and more frequently than did those who received the standard 12 days of treatment.
While cellulitis cure rates at 14 and 28 days were similar between the two groups, 90-day relapse rates were significantly higher for those who took the 6-day course (23.5% vs. 6%), Duncan R. Cranendonk, MD, said at the European Congress of Clinical Microbiology and Infectious Diseases annual conference. The cohort demographics perhaps played into this finding: Most of the group was elderly, hospitalized, and had comorbid conditions.

“However, this is the population clinicians are most likely to see,” said Dr Cranendonk of the University of Amsterdam. “It appears that therapy cannot be safely shortened in this population.”
In light of recent antibiotic trials showing that shorter courses can be as effective as prolonged treatment, Dr. Cranendonk and his colleagues conducted the DANCE (Duration of Antibiotic Therapy for Cellulitis) trial. The study investigated the efficacy of an abbreviated course of intravenous flucloxacillin among 248 patients with cellulitis admitted to 11 Dutch hospitals. At treatment day 6, those who had clinically improved after their initial treatment were randomized to 6 additional days of IV flucloxacillin or to placebo. The primary outcome was cure by day 14 without relapse by day 28.
A 2004 study successfully paved the way for DANCE, Dr. Cranendonk noted. That trial examined 5 versus 10 days of levofloxacin 500 mg for uncomplicated cellulitis in 87 patients. The outcome was positive: There was no significant difference in clinical outcome between the two arms, with a 98% cure rate in both groups.
However, Dr. Cranendonk noted, there were some important differences between the patients in that study and the DANCE cohort. They were, on the whole, younger and generally in better overall health. Also, only 15% of those patients were hospitalized for their infections, while all of the DANCE subjects were treated in the hospital.
Patients enrolled in DANCE were a mean of 62 years old, with a median 28 kg/m2 body mass index. About 40% had experienced cellulitis before, and 25% had diabetes. Most infections were on the leg (84%) and involved the lower leg or the lower leg and the foot. Fever was present in half of the group, lymphadenopathy in a third, and leukocytosis in 70%.
Upon enrollment, all 248 patients received 6 days of 1,000 mg/day IV flucloxacillin, with the option of a step-down to oral treatment (500 mg four times per day) at the treating physician’s discretion. At day 6, patients who were clinically improved (afebrile, no need to an antibiotic switch, no growth in blood culture, and improved symptoms of pain, ulceration, discharge, and fluctuance) were randomized to either another 6 days of flucloxacillin or placebo.
The primary endpoint was cure by day 14, with no relapse and no need for new antibiotics by day 28. The secondary endpoint was relapse by 90 days after initial cure.
After initial treatment, 151 patients entered the randomization phase. At 28 days, relapse-free cure rates were nearly identical: 49% of the 12-day group and 50% of the 6-day group. However, by 90 days, a significant difference became apparent: Patients who had received the 6-day course of flucloxacillin were significantly more likely to have experienced a relapse of cellulitis in the same region (23.5% vs. 6% in the 12-day group). A Kaplan-Meier analysis showed that these patients began to relapse as early as 35 days after the end of therapy. Most relapses occurred during days 60-90. The few relapses in the 12-day group occurred toward the end of the follow-up period, from day 75 onward.
Dr. Cranendonk said the investigation shows that older, less-healthy cellulitis patients can probably benefit from the longer course of antibiotics. “Short-term outcomes aren’t everything,” he noted.
He had no financial disclosures.
A video interview of Dr. Cranendock by ECCMID 2018 is available.
SOURCE: Cranendonk et al. ECCMID 2018, Abstract O1122
MADRID – An attempt to balance effective treatment with good antibiotic stewardship fell short when patients with cellulitis who got 6 days of flucloxacillin relapsed significantly sooner and more frequently than did those who received the standard 12 days of treatment.
While cellulitis cure rates at 14 and 28 days were similar between the two groups, 90-day relapse rates were significantly higher for those who took the 6-day course (23.5% vs. 6%), Duncan R. Cranendonk, MD, said at the European Congress of Clinical Microbiology and Infectious Diseases annual conference. The cohort demographics perhaps played into this finding: Most of the group was elderly, hospitalized, and had comorbid conditions.
“However, this is the population clinicians are most likely to see,” said Dr Cranendonk of the University of Amsterdam. “It appears that therapy cannot be safely shortened in this population.”
In light of recent antibiotic trials showing that shorter courses can be as effective as prolonged treatment, Dr. Cranendonk and his colleagues conducted the DANCE (Duration of Antibiotic Therapy for Cellulitis) trial. The study investigated the efficacy of an abbreviated course of intravenous flucloxacillin among 248 patients with cellulitis admitted to 11 Dutch hospitals. At treatment day 6, those who had clinically improved after their initial treatment were randomized to 6 additional days of IV flucloxacillin or to placebo. The primary outcome was cure by day 14 without relapse by day 28.
A 2004 study successfully paved the way for DANCE, Dr. Cranendonk noted. That trial examined 5 versus 10 days of levofloxacin 500 mg for uncomplicated cellulitis in 87 patients. The outcome was positive: There was no significant difference in clinical outcome between the two arms, with a 98% cure rate in both groups.
However, Dr. Cranendonk noted, there were some important differences between the patients in that study and the DANCE cohort. They were, on the whole, younger and generally in better overall health. Also, only 15% of those patients were hospitalized for their infections, while all of the DANCE subjects were treated in the hospital.
Patients enrolled in DANCE were a mean of 62 years old, with a median 28 kg/m2 body mass index. About 40% had experienced cellulitis before, and 25% had diabetes. Most infections were on the leg (84%) and involved the lower leg or the lower leg and the foot. Fever was present in half of the group, lymphadenopathy in a third, and leukocytosis in 70%.
Upon enrollment, all 248 patients received 6 days of 1,000 mg/day IV flucloxacillin, with the option of a step-down to oral treatment (500 mg four times per day) at the treating physician’s discretion. At day 6, patients who were clinically improved (afebrile, no need to an antibiotic switch, no growth in blood culture, and improved symptoms of pain, ulceration, discharge, and fluctuance) were randomized to either another 6 days of flucloxacillin or placebo.
The primary endpoint was cure by day 14, with no relapse and no need for new antibiotics by day 28. The secondary endpoint was relapse by 90 days after initial cure.
After initial treatment, 151 patients entered the randomization phase. At 28 days, relapse-free cure rates were nearly identical: 49% of the 12-day group and 50% of the 6-day group. However, by 90 days, a significant difference became apparent: Patients who had received the 6-day course of flucloxacillin were significantly more likely to have experienced a relapse of cellulitis in the same region (23.5% vs. 6% in the 12-day group). A Kaplan-Meier analysis showed that these patients began to relapse as early as 35 days after the end of therapy. Most relapses occurred during days 60-90. The few relapses in the 12-day group occurred toward the end of the follow-up period, from day 75 onward.
Dr. Cranendonk said the investigation shows that older, less-healthy cellulitis patients can probably benefit from the longer course of antibiotics. “Short-term outcomes aren’t everything,” he noted.
He had no financial disclosures.
A video interview of Dr. Cranendock by ECCMID 2018 is available.
SOURCE: Cranendonk et al. ECCMID 2018, Abstract O1122
MADRID – An attempt to balance effective treatment with good antibiotic stewardship fell short when patients with cellulitis who got 6 days of flucloxacillin relapsed significantly sooner and more frequently than did those who received the standard 12 days of treatment.
While cellulitis cure rates at 14 and 28 days were similar between the two groups, 90-day relapse rates were significantly higher for those who took the 6-day course (23.5% vs. 6%), Duncan R. Cranendonk, MD, said at the European Congress of Clinical Microbiology and Infectious Diseases annual conference. The cohort demographics perhaps played into this finding: Most of the group was elderly, hospitalized, and had comorbid conditions.
“However, this is the population clinicians are most likely to see,” said Dr Cranendonk of the University of Amsterdam. “It appears that therapy cannot be safely shortened in this population.”
In light of recent antibiotic trials showing that shorter courses can be as effective as prolonged treatment, Dr. Cranendonk and his colleagues conducted the DANCE (Duration of Antibiotic Therapy for Cellulitis) trial. The study investigated the efficacy of an abbreviated course of intravenous flucloxacillin among 248 patients with cellulitis admitted to 11 Dutch hospitals. At treatment day 6, those who had clinically improved after their initial treatment were randomized to 6 additional days of IV flucloxacillin or to placebo. The primary outcome was cure by day 14 without relapse by day 28.
A 2004 study successfully paved the way for DANCE, Dr. Cranendonk noted. That trial examined 5 versus 10 days of levofloxacin 500 mg for uncomplicated cellulitis in 87 patients. The outcome was positive: There was no significant difference in clinical outcome between the two arms, with a 98% cure rate in both groups.
However, Dr. Cranendonk noted, there were some important differences between the patients in that study and the DANCE cohort. They were, on the whole, younger and generally in better overall health. Also, only 15% of those patients were hospitalized for their infections, while all of the DANCE subjects were treated in the hospital.
Patients enrolled in DANCE were a mean of 62 years old, with a median 28 kg/m2 body mass index. About 40% had experienced cellulitis before, and 25% had diabetes. Most infections were on the leg (84%) and involved the lower leg or the lower leg and the foot. Fever was present in half of the group, lymphadenopathy in a third, and leukocytosis in 70%.
Upon enrollment, all 248 patients received 6 days of 1,000 mg/day IV flucloxacillin, with the option of a step-down to oral treatment (500 mg four times per day) at the treating physician’s discretion. At day 6, patients who were clinically improved (afebrile, no need to an antibiotic switch, no growth in blood culture, and improved symptoms of pain, ulceration, discharge, and fluctuance) were randomized to either another 6 days of flucloxacillin or placebo.
The primary endpoint was cure by day 14, with no relapse and no need for new antibiotics by day 28. The secondary endpoint was relapse by 90 days after initial cure.
After initial treatment, 151 patients entered the randomization phase. At 28 days, relapse-free cure rates were nearly identical: 49% of the 12-day group and 50% of the 6-day group. However, by 90 days, a significant difference became apparent: Patients who had received the 6-day course of flucloxacillin were significantly more likely to have experienced a relapse of cellulitis in the same region (23.5% vs. 6% in the 12-day group). A Kaplan-Meier analysis showed that these patients began to relapse as early as 35 days after the end of therapy. Most relapses occurred during days 60-90. The few relapses in the 12-day group occurred toward the end of the follow-up period, from day 75 onward.
Dr. Cranendonk said the investigation shows that older, less-healthy cellulitis patients can probably benefit from the longer course of antibiotics. “Short-term outcomes aren’t everything,” he noted.
He had no financial disclosures.
A video interview of Dr. Cranendock by ECCMID 2018 is available.
SOURCE: Cranendonk et al. ECCMID 2018, Abstract O1122
REPORTING FROM ECCMID 2018
Key clinical point: Elderly patients with cellulitis and comorbid conditions probably need a full 12-day course of treatment.
Major finding: Three-month relapse rates were significantly higher in those who received 6 days of flucloxacillin than they were among those who received 12 days (23.5% vs. 6%).
Study details: Patients who improved on 6 days of treatment were randomized to either placebo or another 6 days of therapy.
Disclosures: Dr. Cranendonk had no financial disclosures.
Source: Cranendonk DR et al. ECCMID 2018, Abstract O1122
Inadequate antibiotic use persists in gonorrhea
Inappropriate treatment of gonorrhea persists despite growing antibiotic resistance, investigators reported in the Morbidity and Mortality Weekly Report.
In 2016, about 19% of gonorrhea cases diagnosed in the United States were not treated according to recommendations from the Centers for Disease Control and Prevention, wrote Emily J. Weston, MPH, and her associates. They recommended additional training and education on the need for providers to follow treatment recommendations.
![]()
To assess adherence to this recommendation, Ms. Weston and her associates analyzed data for 3,213 gonorrhea cases with complete treatment data reported in the United States in 2016. The cases spanned seven CDC sentinel surveillance jurisdictions, including California (excluding San Francisco), Florida, Massachusetts, Minnesota, Philadelphia (Pa.), Baltimore (Md.), and Multnomah County (Oregon).
In all, 18.7% of patients did not receive CDC-recommended treatment, the investigators reported. Inappropriate treatment most often consisted of ceftriaxone (250 mg) alone (5.9%), ceftriaxone with doxycycline (4.4%), azithromycin only (3.1%), ceftriaxone with azithromycin of other or unknown dosages (2.1%), or doxycycline only (1.2%).
Rates of appropriate treatment varied from 79% to 83% among individual jurisdictions and were unrelated to patient race, ethnicity, sex, or age, the researchers found. Men who had sex with men were more likely to receive recommended treatment (85%) than were heterosexual men and women (79%). Patients also were more likely to receive appropriate treatment at family planning, reproductive health, or sexually transmitted disease clinics than in other health care settings.
The results highlight the need for state and local departments to identify and educate providers who are inadequately treating gonorrhea, the researchers concluded. “State and local health departments should continue to work with providers and patients to assure timely detection and treatment of gonorrhea according to current CDC treatment recommendations.”
The study received no external funding. The investigators reported having no conflicts of interest.
SOURCE: Weston EJ et al. MMWR. 2018 Apr 27. doi: 10.15585/mmwr.mm6716a4
Inappropriate treatment of gonorrhea persists despite growing antibiotic resistance, investigators reported in the Morbidity and Mortality Weekly Report.
In 2016, about 19% of gonorrhea cases diagnosed in the United States were not treated according to recommendations from the Centers for Disease Control and Prevention, wrote Emily J. Weston, MPH, and her associates. They recommended additional training and education on the need for providers to follow treatment recommendations.
![]()
To assess adherence to this recommendation, Ms. Weston and her associates analyzed data for 3,213 gonorrhea cases with complete treatment data reported in the United States in 2016. The cases spanned seven CDC sentinel surveillance jurisdictions, including California (excluding San Francisco), Florida, Massachusetts, Minnesota, Philadelphia (Pa.), Baltimore (Md.), and Multnomah County (Oregon).
In all, 18.7% of patients did not receive CDC-recommended treatment, the investigators reported. Inappropriate treatment most often consisted of ceftriaxone (250 mg) alone (5.9%), ceftriaxone with doxycycline (4.4%), azithromycin only (3.1%), ceftriaxone with azithromycin of other or unknown dosages (2.1%), or doxycycline only (1.2%).
Rates of appropriate treatment varied from 79% to 83% among individual jurisdictions and were unrelated to patient race, ethnicity, sex, or age, the researchers found. Men who had sex with men were more likely to receive recommended treatment (85%) than were heterosexual men and women (79%). Patients also were more likely to receive appropriate treatment at family planning, reproductive health, or sexually transmitted disease clinics than in other health care settings.
The results highlight the need for state and local departments to identify and educate providers who are inadequately treating gonorrhea, the researchers concluded. “State and local health departments should continue to work with providers and patients to assure timely detection and treatment of gonorrhea according to current CDC treatment recommendations.”
The study received no external funding. The investigators reported having no conflicts of interest.
SOURCE: Weston EJ et al. MMWR. 2018 Apr 27. doi: 10.15585/mmwr.mm6716a4
Inappropriate treatment of gonorrhea persists despite growing antibiotic resistance, investigators reported in the Morbidity and Mortality Weekly Report.
In 2016, about 19% of gonorrhea cases diagnosed in the United States were not treated according to recommendations from the Centers for Disease Control and Prevention, wrote Emily J. Weston, MPH, and her associates. They recommended additional training and education on the need for providers to follow treatment recommendations.
![]()
To assess adherence to this recommendation, Ms. Weston and her associates analyzed data for 3,213 gonorrhea cases with complete treatment data reported in the United States in 2016. The cases spanned seven CDC sentinel surveillance jurisdictions, including California (excluding San Francisco), Florida, Massachusetts, Minnesota, Philadelphia (Pa.), Baltimore (Md.), and Multnomah County (Oregon).
In all, 18.7% of patients did not receive CDC-recommended treatment, the investigators reported. Inappropriate treatment most often consisted of ceftriaxone (250 mg) alone (5.9%), ceftriaxone with doxycycline (4.4%), azithromycin only (3.1%), ceftriaxone with azithromycin of other or unknown dosages (2.1%), or doxycycline only (1.2%).
Rates of appropriate treatment varied from 79% to 83% among individual jurisdictions and were unrelated to patient race, ethnicity, sex, or age, the researchers found. Men who had sex with men were more likely to receive recommended treatment (85%) than were heterosexual men and women (79%). Patients also were more likely to receive appropriate treatment at family planning, reproductive health, or sexually transmitted disease clinics than in other health care settings.
The results highlight the need for state and local departments to identify and educate providers who are inadequately treating gonorrhea, the researchers concluded. “State and local health departments should continue to work with providers and patients to assure timely detection and treatment of gonorrhea according to current CDC treatment recommendations.”
The study received no external funding. The investigators reported having no conflicts of interest.
SOURCE: Weston EJ et al. MMWR. 2018 Apr 27. doi: 10.15585/mmwr.mm6716a4
Key clinical point: Inadequate treatment of gonorrhea persists despite its growing antibiotic resistance.
Major finding: In all, 18.7% patients did not receive CDC-recommended treatment.
Study details: Analyses of 3,213 cases from seven U.S. jurisdictions.
Disclosures: The investigators reported no external funding sources. They reported having no conflicts of interest.
Source: Weston EJ et al. MMWR. 2018 Apr 27. doi: 10.15585/mmwr.mm6716a4
Endovascular interventions associated with large benefits in peripheral artery disease
WASHINGTON – An all-comer observational study associated endovascular treatment of lower limb peripheral artery disease (PAD) with low event rates and substantial improvements in quality of life at 18 months, even in Rutherford stage 6 patients.
Although the proportion of patients with Rutherford stage 6 PAD was relatively small, the study results showed that peripheral vascular intervention “can be successful in this patient population as evidenced by a high freedom from major amputation,” reported William Gray, MD, system chief of the division of cardiovascular disease at the Lankenau Heart Group, Wynnewood, Pa., at CRT 2018, sponsored by the Cardiovascular Research Institute at Washington Hospital Center.

“Many of the patients in this trial, particularly the Rutherford 6 patients, would never be included in the pivotal trial for endovascular devices,” Dr. Gray said. He called this “a unique study” in that it had almost no exclusions.
The study enrolled 1,204 patients with peripheral artery disease at 51 participating sites. After 18 months, follow-up data were available on 793 patients. These were divided by Rutherford classifications into three groups: 374 patients in the combined Rutherford 2 and 3 classifications (R2/3); 371 in the combined Rutherford 4 and 5 classifications (R4/5); and 48 in the Rutherford 6 classification (R6). Patients treated with any Food and Drug Administration–approved technology for treatment of claudication and critical limb ischemia for PAD were eligible.
The endpoints considered at 18 months included procedural and lesion success, major adverse events, and quality of life. Four core laboratories were responsible for an independent analysis of outcomes. A follow-up of 5 years is planned and will include an economic analysis.
The procedural success rates for were 84.4% for the R2/3 group, 76.9% for the R4/5 group, and 70.2% for the R6 group. Almost all of those in the R2/3 and R4/5 groups were discharged immediately after treatment. In the R6 group, approximately 25% of patients were held for complications or additional care.
At 18 months of follow-up, freedom from major adverse events, defined as death, major amputation, or a target vessel revascularization, was achieved by 76.9% of those in the R2/3 group, 68.2% of those in the R4/5 group, and 52.8% of those in the R6 group. The analysis also looked at specific events: The rates for freedom from amputation were 99.3%, 95.3%, and 81.7% in the R2/3, R4/5, and R6 groups, respectively; the freedom from death was 93.9%, 85.5%, and 76.2%; and the freedom from target vessel revascularization was 77.5%, 70.6%, ad 65.7%.
Those in R2/3 maintained the improvement in Rutherford classification observed at 30 days for the subsequent 12 months. Those in R2/3 and R4/5 showed continued improvement in Rutherford classification. For example, R4 represented approximately 50% of the patients in R3/4 classification at baseline but less than 20% of this group at 18 months.
The change in Rutherford classification was reflected in quality of life (QOL) analyses. As far as total QOL scores, the R6 group, which had lower scores at baseline, was no longer significantly different at 18 months from the R4/5 group. On the pain subdomain QOL score, which was incrementally worse at baseline for increased PAD severity, there were no differences at 18 months after improvements in all groups.
Overall, the LIBERTY 360 study “supports aggressive management” with endovascular procedures in symptomatic patients with PAD. This is important because PAD often is inadequately treated or left untreated, according to Dr. Gray. He cited data suggesting that up to 50% of patients who undergo amputation because of lower limb claudication never even undergo a vascular evaluation.
Although there was no control group to evaluate outcomes in patients not treated or treated with another intervention, such as surgery, Dr. Gray suggested that there are encouraging results in a study that was conducted to enroll patients “with as many confounders as possible.”
Dr. Gray reported financial relationships with Abbott Vascular, Cordis, Medtronic, WL Gore, and a number of other device manufacturers.
WASHINGTON – An all-comer observational study associated endovascular treatment of lower limb peripheral artery disease (PAD) with low event rates and substantial improvements in quality of life at 18 months, even in Rutherford stage 6 patients.
Although the proportion of patients with Rutherford stage 6 PAD was relatively small, the study results showed that peripheral vascular intervention “can be successful in this patient population as evidenced by a high freedom from major amputation,” reported William Gray, MD, system chief of the division of cardiovascular disease at the Lankenau Heart Group, Wynnewood, Pa., at CRT 2018, sponsored by the Cardiovascular Research Institute at Washington Hospital Center.
“Many of the patients in this trial, particularly the Rutherford 6 patients, would never be included in the pivotal trial for endovascular devices,” Dr. Gray said. He called this “a unique study” in that it had almost no exclusions.
The study enrolled 1,204 patients with peripheral artery disease at 51 participating sites. After 18 months, follow-up data were available on 793 patients. These were divided by Rutherford classifications into three groups: 374 patients in the combined Rutherford 2 and 3 classifications (R2/3); 371 in the combined Rutherford 4 and 5 classifications (R4/5); and 48 in the Rutherford 6 classification (R6). Patients treated with any Food and Drug Administration–approved technology for treatment of claudication and critical limb ischemia for PAD were eligible.
The endpoints considered at 18 months included procedural and lesion success, major adverse events, and quality of life. Four core laboratories were responsible for an independent analysis of outcomes. A follow-up of 5 years is planned and will include an economic analysis.
The procedural success rates for were 84.4% for the R2/3 group, 76.9% for the R4/5 group, and 70.2% for the R6 group. Almost all of those in the R2/3 and R4/5 groups were discharged immediately after treatment. In the R6 group, approximately 25% of patients were held for complications or additional care.
At 18 months of follow-up, freedom from major adverse events, defined as death, major amputation, or a target vessel revascularization, was achieved by 76.9% of those in the R2/3 group, 68.2% of those in the R4/5 group, and 52.8% of those in the R6 group. The analysis also looked at specific events: The rates for freedom from amputation were 99.3%, 95.3%, and 81.7% in the R2/3, R4/5, and R6 groups, respectively; the freedom from death was 93.9%, 85.5%, and 76.2%; and the freedom from target vessel revascularization was 77.5%, 70.6%, ad 65.7%.
Those in R2/3 maintained the improvement in Rutherford classification observed at 30 days for the subsequent 12 months. Those in R2/3 and R4/5 showed continued improvement in Rutherford classification. For example, R4 represented approximately 50% of the patients in R3/4 classification at baseline but less than 20% of this group at 18 months.
The change in Rutherford classification was reflected in quality of life (QOL) analyses. As far as total QOL scores, the R6 group, which had lower scores at baseline, was no longer significantly different at 18 months from the R4/5 group. On the pain subdomain QOL score, which was incrementally worse at baseline for increased PAD severity, there were no differences at 18 months after improvements in all groups.
Overall, the LIBERTY 360 study “supports aggressive management” with endovascular procedures in symptomatic patients with PAD. This is important because PAD often is inadequately treated or left untreated, according to Dr. Gray. He cited data suggesting that up to 50% of patients who undergo amputation because of lower limb claudication never even undergo a vascular evaluation.
Although there was no control group to evaluate outcomes in patients not treated or treated with another intervention, such as surgery, Dr. Gray suggested that there are encouraging results in a study that was conducted to enroll patients “with as many confounders as possible.”
Dr. Gray reported financial relationships with Abbott Vascular, Cordis, Medtronic, WL Gore, and a number of other device manufacturers.
WASHINGTON – An all-comer observational study associated endovascular treatment of lower limb peripheral artery disease (PAD) with low event rates and substantial improvements in quality of life at 18 months, even in Rutherford stage 6 patients.
Although the proportion of patients with Rutherford stage 6 PAD was relatively small, the study results showed that peripheral vascular intervention “can be successful in this patient population as evidenced by a high freedom from major amputation,” reported William Gray, MD, system chief of the division of cardiovascular disease at the Lankenau Heart Group, Wynnewood, Pa., at CRT 2018, sponsored by the Cardiovascular Research Institute at Washington Hospital Center.
“Many of the patients in this trial, particularly the Rutherford 6 patients, would never be included in the pivotal trial for endovascular devices,” Dr. Gray said. He called this “a unique study” in that it had almost no exclusions.
The study enrolled 1,204 patients with peripheral artery disease at 51 participating sites. After 18 months, follow-up data were available on 793 patients. These were divided by Rutherford classifications into three groups: 374 patients in the combined Rutherford 2 and 3 classifications (R2/3); 371 in the combined Rutherford 4 and 5 classifications (R4/5); and 48 in the Rutherford 6 classification (R6). Patients treated with any Food and Drug Administration–approved technology for treatment of claudication and critical limb ischemia for PAD were eligible.
The endpoints considered at 18 months included procedural and lesion success, major adverse events, and quality of life. Four core laboratories were responsible for an independent analysis of outcomes. A follow-up of 5 years is planned and will include an economic analysis.
The procedural success rates for were 84.4% for the R2/3 group, 76.9% for the R4/5 group, and 70.2% for the R6 group. Almost all of those in the R2/3 and R4/5 groups were discharged immediately after treatment. In the R6 group, approximately 25% of patients were held for complications or additional care.
At 18 months of follow-up, freedom from major adverse events, defined as death, major amputation, or a target vessel revascularization, was achieved by 76.9% of those in the R2/3 group, 68.2% of those in the R4/5 group, and 52.8% of those in the R6 group. The analysis also looked at specific events: The rates for freedom from amputation were 99.3%, 95.3%, and 81.7% in the R2/3, R4/5, and R6 groups, respectively; the freedom from death was 93.9%, 85.5%, and 76.2%; and the freedom from target vessel revascularization was 77.5%, 70.6%, ad 65.7%.
Those in R2/3 maintained the improvement in Rutherford classification observed at 30 days for the subsequent 12 months. Those in R2/3 and R4/5 showed continued improvement in Rutherford classification. For example, R4 represented approximately 50% of the patients in R3/4 classification at baseline but less than 20% of this group at 18 months.
The change in Rutherford classification was reflected in quality of life (QOL) analyses. As far as total QOL scores, the R6 group, which had lower scores at baseline, was no longer significantly different at 18 months from the R4/5 group. On the pain subdomain QOL score, which was incrementally worse at baseline for increased PAD severity, there were no differences at 18 months after improvements in all groups.
Overall, the LIBERTY 360 study “supports aggressive management” with endovascular procedures in symptomatic patients with PAD. This is important because PAD often is inadequately treated or left untreated, according to Dr. Gray. He cited data suggesting that up to 50% of patients who undergo amputation because of lower limb claudication never even undergo a vascular evaluation.
Although there was no control group to evaluate outcomes in patients not treated or treated with another intervention, such as surgery, Dr. Gray suggested that there are encouraging results in a study that was conducted to enroll patients “with as many confounders as possible.”
Dr. Gray reported financial relationships with Abbott Vascular, Cordis, Medtronic, WL Gore, and a number of other device manufacturers.
Key clinical point: An observational study supports endovascular strategies even in the most advanced cases of peripheral artery disease (PAD).
Major finding: In Rutherford stage 6 patients, low amputation rates and large improvements in quality of life were observed at 18 months of follow-up.
Data source: Prospective, observational multicenter study.
Disclosures: Dr. Gray reports financial relationships with Abbott Vascular, BioCardia, Boston Scientific, Cardiovascular Systems, Coherex Medical, Contego Cook, Cordis, Medtronic, Shockwave Medical, Silk Road Medical, and WL Gore.
Safety and support experiences of transgender and gender nonconforming youth explored
TORONTO – Youth with nonbinary identities – those that are beyond or outside of the categories of male/man and female/woman – feel significantly safer and more supported at school, compared with their transgender and gender nonconforming peers with binary identities, results from a survey of more than 300 youth showed.
“There has been little research specifically about the experiences of transgender and gender nonconforming youth that have nonbinary identities,” lead study author Brittany Allen, MD, said in an interview in advance of the Pediatric Academic Societies meeting.

Dr. Allen, a pediatrician at the University of Wisconsin–Madison, and her associates conducted an online survey of 311 transgender, nonbinary, and gender nonconforming youth in the state who ranged aged 12-22 years. Study participants were asked about their school safety and support experiences, and the researchers used Wilcoxon rank-sum tests to compare Likert scale responses among youth who reported nonbinary identities with those who reported binary identities. On the 1-5 scale, 1 meant “strongly agree” while 5 meant “strongly disagree.”
Dr. Allen, who is also comedical director of the Pediatric and Adolescent Transgender Health Clinic at American Family Children’s Hospital, Madison, reported that 311 young people completed more than 70% of the survey. Of those, 287 identified as having either binary (164; 57%) or nonbinary (123; 43%) gender identities. That percentage of those reporting nonbinary identities “is striking,” she said, and is “a much higher percentage than seen in adult studies of transgender and gender nonconforming people.”
Compared with respondents with binary identities, those with nonbinary identities were more often Caucasian/White (81% vs. 65% for those with binary identities; P = .003) and less likely to qualify for free lunch (28% vs. 55%; P = .001). Both binary and nonbinary groups reported similar school attendance and belonging. However, compared with the binary group, the nonbinary group reported significantly higher ratings of school safety (Likert score of 2.62 vs. 2.96, respectively; P = .0078) and peer support (Likert score of 2.54 vs. 2.87; P = .0139) and also were more likely to report being able to access adult support at school if needed (Likert score of 2.31 vs. 2.66; P = .0085).
“The primary message is that many transgender or gender nonconforming youth have identities outside of a gender binary and that transgender and gender nonconforming youth with different gender identities may have different strengths and challenges in different settings,” Dr. Allen said.
“Our work shows that nonbinary youth have relative safety and support at school compared to their transgender and gender nonconforming peers with binary identities – though it’s important to note that transgender and gender nonconforming youth overall are still at high risk of school harassment and violence. Interventions to promote school safety for youth of all gender identities should consider that transgender and gender nonconforming youth with different gender identities have different risks related to school safety and support.”
She acknowledged certain limitations of the study, including the fact that the researchers used a convenience sample to recruit participants, “which means that we may not have reached transgender and gender nonconforming youth that were less connected to support services or transgender and gender nonconforming peers,” Dr. Allen said. “This study also specifically assessed transgender and gender nonconforming youth; we did not have a comparison group of cisgender participants for comparison due to our study design.”
The study was funded by the National Institutes of Health, the Baldwin Wisconsin Ideas Endowment, the University of Wisconsin Advancing Health Equity and Diversity initiative, and the Wisconsin Partnership Program. Dr. Allen reported having no financial disclosures.
TORONTO – Youth with nonbinary identities – those that are beyond or outside of the categories of male/man and female/woman – feel significantly safer and more supported at school, compared with their transgender and gender nonconforming peers with binary identities, results from a survey of more than 300 youth showed.
“There has been little research specifically about the experiences of transgender and gender nonconforming youth that have nonbinary identities,” lead study author Brittany Allen, MD, said in an interview in advance of the Pediatric Academic Societies meeting.
Dr. Allen, a pediatrician at the University of Wisconsin–Madison, and her associates conducted an online survey of 311 transgender, nonbinary, and gender nonconforming youth in the state who ranged aged 12-22 years. Study participants were asked about their school safety and support experiences, and the researchers used Wilcoxon rank-sum tests to compare Likert scale responses among youth who reported nonbinary identities with those who reported binary identities. On the 1-5 scale, 1 meant “strongly agree” while 5 meant “strongly disagree.”
Dr. Allen, who is also comedical director of the Pediatric and Adolescent Transgender Health Clinic at American Family Children’s Hospital, Madison, reported that 311 young people completed more than 70% of the survey. Of those, 287 identified as having either binary (164; 57%) or nonbinary (123; 43%) gender identities. That percentage of those reporting nonbinary identities “is striking,” she said, and is “a much higher percentage than seen in adult studies of transgender and gender nonconforming people.”
Compared with respondents with binary identities, those with nonbinary identities were more often Caucasian/White (81% vs. 65% for those with binary identities; P = .003) and less likely to qualify for free lunch (28% vs. 55%; P = .001). Both binary and nonbinary groups reported similar school attendance and belonging. However, compared with the binary group, the nonbinary group reported significantly higher ratings of school safety (Likert score of 2.62 vs. 2.96, respectively; P = .0078) and peer support (Likert score of 2.54 vs. 2.87; P = .0139) and also were more likely to report being able to access adult support at school if needed (Likert score of 2.31 vs. 2.66; P = .0085).
“The primary message is that many transgender or gender nonconforming youth have identities outside of a gender binary and that transgender and gender nonconforming youth with different gender identities may have different strengths and challenges in different settings,” Dr. Allen said.
“Our work shows that nonbinary youth have relative safety and support at school compared to their transgender and gender nonconforming peers with binary identities – though it’s important to note that transgender and gender nonconforming youth overall are still at high risk of school harassment and violence. Interventions to promote school safety for youth of all gender identities should consider that transgender and gender nonconforming youth with different gender identities have different risks related to school safety and support.”
She acknowledged certain limitations of the study, including the fact that the researchers used a convenience sample to recruit participants, “which means that we may not have reached transgender and gender nonconforming youth that were less connected to support services or transgender and gender nonconforming peers,” Dr. Allen said. “This study also specifically assessed transgender and gender nonconforming youth; we did not have a comparison group of cisgender participants for comparison due to our study design.”
The study was funded by the National Institutes of Health, the Baldwin Wisconsin Ideas Endowment, the University of Wisconsin Advancing Health Equity and Diversity initiative, and the Wisconsin Partnership Program. Dr. Allen reported having no financial disclosures.
TORONTO – Youth with nonbinary identities – those that are beyond or outside of the categories of male/man and female/woman – feel significantly safer and more supported at school, compared with their transgender and gender nonconforming peers with binary identities, results from a survey of more than 300 youth showed.
“There has been little research specifically about the experiences of transgender and gender nonconforming youth that have nonbinary identities,” lead study author Brittany Allen, MD, said in an interview in advance of the Pediatric Academic Societies meeting.
Dr. Allen, a pediatrician at the University of Wisconsin–Madison, and her associates conducted an online survey of 311 transgender, nonbinary, and gender nonconforming youth in the state who ranged aged 12-22 years. Study participants were asked about their school safety and support experiences, and the researchers used Wilcoxon rank-sum tests to compare Likert scale responses among youth who reported nonbinary identities with those who reported binary identities. On the 1-5 scale, 1 meant “strongly agree” while 5 meant “strongly disagree.”
Dr. Allen, who is also comedical director of the Pediatric and Adolescent Transgender Health Clinic at American Family Children’s Hospital, Madison, reported that 311 young people completed more than 70% of the survey. Of those, 287 identified as having either binary (164; 57%) or nonbinary (123; 43%) gender identities. That percentage of those reporting nonbinary identities “is striking,” she said, and is “a much higher percentage than seen in adult studies of transgender and gender nonconforming people.”
Compared with respondents with binary identities, those with nonbinary identities were more often Caucasian/White (81% vs. 65% for those with binary identities; P = .003) and less likely to qualify for free lunch (28% vs. 55%; P = .001). Both binary and nonbinary groups reported similar school attendance and belonging. However, compared with the binary group, the nonbinary group reported significantly higher ratings of school safety (Likert score of 2.62 vs. 2.96, respectively; P = .0078) and peer support (Likert score of 2.54 vs. 2.87; P = .0139) and also were more likely to report being able to access adult support at school if needed (Likert score of 2.31 vs. 2.66; P = .0085).
“The primary message is that many transgender or gender nonconforming youth have identities outside of a gender binary and that transgender and gender nonconforming youth with different gender identities may have different strengths and challenges in different settings,” Dr. Allen said.
“Our work shows that nonbinary youth have relative safety and support at school compared to their transgender and gender nonconforming peers with binary identities – though it’s important to note that transgender and gender nonconforming youth overall are still at high risk of school harassment and violence. Interventions to promote school safety for youth of all gender identities should consider that transgender and gender nonconforming youth with different gender identities have different risks related to school safety and support.”
She acknowledged certain limitations of the study, including the fact that the researchers used a convenience sample to recruit participants, “which means that we may not have reached transgender and gender nonconforming youth that were less connected to support services or transgender and gender nonconforming peers,” Dr. Allen said. “This study also specifically assessed transgender and gender nonconforming youth; we did not have a comparison group of cisgender participants for comparison due to our study design.”
The study was funded by the National Institutes of Health, the Baldwin Wisconsin Ideas Endowment, the University of Wisconsin Advancing Health Equity and Diversity initiative, and the Wisconsin Partnership Program. Dr. Allen reported having no financial disclosures.
Key clinical point: Nonbinary youth felt significantly safer and more supported at school, compared with transgender peers with binary identities.
Major finding: Compared with the binary group, the nonbinary group reported significantly higher ratings of school safety (Likert score of 2.62 vs. 2.96, respectively) and peer support (Likert score of 2.54 vs. 2.87).
Study details: An online survey of 311 transgender, nonbinary, and gender nonconforming youth in Wisconsin who aged 12-22 years.
Disclosures: The study was funded by the National Institutes of Health, the Baldwin Wisconsin Ideas Endowment, the University of Wisconsin Advancing Health Equity and Diversity initiative, and the Wisconsin Partnership Program. Dr. Allen reported having no financial disclosures.
Relapse rate drives stem cell transplant failure in pediatric ALL patients
Relapse was the main impediment to successful hematopoietic stem cell transplant (HSCT) in high-risk pediatric acute lymphocytic leukemia (ALL), according to one of the largest single-center experiences reported to date.
The effects of relapse were especially evident for patients with haploidentical donors; these patients had a 3-year cumulative relapse incidence of 47% and event-free survival rate of 35%, both significantly higher than what was seen in other transplant recipients treated at the same center.
The findings, recently published in the journal Biology of Blood and Marrow Transplantation, suggest a substantial unmet need in the treatment of high-risk patients in first remission.
“Newer methods to improve graft-versus-leukemia effect are being tested and will need to be incorporated into the management of high-risk patients,” Asaf D. Yanir, MD, and coauthors at Baylor College of Medicine in Houston said in the report.
Dr. Yanir and colleagues reported recent outcomes for 124 patients who had undergone HSCT for ALL at their center during 2008-2016. That group included 20 haploidentical transplant recipients, 48 patients with matched sibling donors, and 56 with unrelated matched donors.
The 3-year cumulative incidence of relapse was 47% for haploidentical recipients, compared with 20% for matched sibling donors recipients and 24% for unrelated matched donors recipients (P = .02), according to their findings.
The main cause of HSCT failure was relapse, occurring in 47% of haploidentical transplant recipients, compared with 20% for those with matched sibling donors and 24% for those with unrelated matched donors (P = .02).
Those findings are in line with other studies showing inferior outcomes following haploidentical donor HSCT. However, in contrast to those studies, Dr. Yanir and colleagues did find a rate of treatment-related mortality comparable with other transplant approaches. The 3-year incidence of treatment-related mortality was 15% for the haploidentical group and, similarly, 17% in the matched sibling donor group and 16% in the unrelated matched donor group.
That lower rate of treatment-related mortality in the haploidentical group may be caused by improvements in procedures and supportive care. “Unfortunately, the benefits gained by reducing treatment-related mortality were offset by the high rate of relapse, which remains the main obstacle to successful haploidentical donor HSCT,” Dr. Yanir and coauthors wrote in their report.
New strategies are being studied to retain the graft-versus-leukemia effect or enhance it in patients who’ve undergone haploidentical HSCT, such as selectively depleting alloreactive T cells while sparing other immune effectors, investigators wrote.
“Given evolving practices, it is important to continually evaluate the projected event-free survival for pediatric ALL following HSCT, based on the donor type used,” they wrote.
Dr. Yanir and coauthors had no financial disclosures or conflicts of interest to report.
SOURCE: Yanir AD et al. Biol Blood Marrow Transplant. 2018 Mar 14. doi: 10.1016/j.bbmt.2018.03.001.
Relapse was the main impediment to successful hematopoietic stem cell transplant (HSCT) in high-risk pediatric acute lymphocytic leukemia (ALL), according to one of the largest single-center experiences reported to date.
The effects of relapse were especially evident for patients with haploidentical donors; these patients had a 3-year cumulative relapse incidence of 47% and event-free survival rate of 35%, both significantly higher than what was seen in other transplant recipients treated at the same center.
The findings, recently published in the journal Biology of Blood and Marrow Transplantation, suggest a substantial unmet need in the treatment of high-risk patients in first remission.
“Newer methods to improve graft-versus-leukemia effect are being tested and will need to be incorporated into the management of high-risk patients,” Asaf D. Yanir, MD, and coauthors at Baylor College of Medicine in Houston said in the report.
Dr. Yanir and colleagues reported recent outcomes for 124 patients who had undergone HSCT for ALL at their center during 2008-2016. That group included 20 haploidentical transplant recipients, 48 patients with matched sibling donors, and 56 with unrelated matched donors.
The 3-year cumulative incidence of relapse was 47% for haploidentical recipients, compared with 20% for matched sibling donors recipients and 24% for unrelated matched donors recipients (P = .02), according to their findings.
The main cause of HSCT failure was relapse, occurring in 47% of haploidentical transplant recipients, compared with 20% for those with matched sibling donors and 24% for those with unrelated matched donors (P = .02).
Those findings are in line with other studies showing inferior outcomes following haploidentical donor HSCT. However, in contrast to those studies, Dr. Yanir and colleagues did find a rate of treatment-related mortality comparable with other transplant approaches. The 3-year incidence of treatment-related mortality was 15% for the haploidentical group and, similarly, 17% in the matched sibling donor group and 16% in the unrelated matched donor group.
That lower rate of treatment-related mortality in the haploidentical group may be caused by improvements in procedures and supportive care. “Unfortunately, the benefits gained by reducing treatment-related mortality were offset by the high rate of relapse, which remains the main obstacle to successful haploidentical donor HSCT,” Dr. Yanir and coauthors wrote in their report.
New strategies are being studied to retain the graft-versus-leukemia effect or enhance it in patients who’ve undergone haploidentical HSCT, such as selectively depleting alloreactive T cells while sparing other immune effectors, investigators wrote.
“Given evolving practices, it is important to continually evaluate the projected event-free survival for pediatric ALL following HSCT, based on the donor type used,” they wrote.
Dr. Yanir and coauthors had no financial disclosures or conflicts of interest to report.
SOURCE: Yanir AD et al. Biol Blood Marrow Transplant. 2018 Mar 14. doi: 10.1016/j.bbmt.2018.03.001.
Relapse was the main impediment to successful hematopoietic stem cell transplant (HSCT) in high-risk pediatric acute lymphocytic leukemia (ALL), according to one of the largest single-center experiences reported to date.
The effects of relapse were especially evident for patients with haploidentical donors; these patients had a 3-year cumulative relapse incidence of 47% and event-free survival rate of 35%, both significantly higher than what was seen in other transplant recipients treated at the same center.
The findings, recently published in the journal Biology of Blood and Marrow Transplantation, suggest a substantial unmet need in the treatment of high-risk patients in first remission.
“Newer methods to improve graft-versus-leukemia effect are being tested and will need to be incorporated into the management of high-risk patients,” Asaf D. Yanir, MD, and coauthors at Baylor College of Medicine in Houston said in the report.
Dr. Yanir and colleagues reported recent outcomes for 124 patients who had undergone HSCT for ALL at their center during 2008-2016. That group included 20 haploidentical transplant recipients, 48 patients with matched sibling donors, and 56 with unrelated matched donors.
The 3-year cumulative incidence of relapse was 47% for haploidentical recipients, compared with 20% for matched sibling donors recipients and 24% for unrelated matched donors recipients (P = .02), according to their findings.
The main cause of HSCT failure was relapse, occurring in 47% of haploidentical transplant recipients, compared with 20% for those with matched sibling donors and 24% for those with unrelated matched donors (P = .02).
Those findings are in line with other studies showing inferior outcomes following haploidentical donor HSCT. However, in contrast to those studies, Dr. Yanir and colleagues did find a rate of treatment-related mortality comparable with other transplant approaches. The 3-year incidence of treatment-related mortality was 15% for the haploidentical group and, similarly, 17% in the matched sibling donor group and 16% in the unrelated matched donor group.
That lower rate of treatment-related mortality in the haploidentical group may be caused by improvements in procedures and supportive care. “Unfortunately, the benefits gained by reducing treatment-related mortality were offset by the high rate of relapse, which remains the main obstacle to successful haploidentical donor HSCT,” Dr. Yanir and coauthors wrote in their report.
New strategies are being studied to retain the graft-versus-leukemia effect or enhance it in patients who’ve undergone haploidentical HSCT, such as selectively depleting alloreactive T cells while sparing other immune effectors, investigators wrote.
“Given evolving practices, it is important to continually evaluate the projected event-free survival for pediatric ALL following HSCT, based on the donor type used,” they wrote.
Dr. Yanir and coauthors had no financial disclosures or conflicts of interest to report.
SOURCE: Yanir AD et al. Biol Blood Marrow Transplant. 2018 Mar 14. doi: 10.1016/j.bbmt.2018.03.001.
FROM BIOLOGY OF BLOOD AND MARROW TRANSPLANTATION
Key clinical point: In pediatric acute lymphocytic leukemia (ALL) patients, relapse is the main barrier to successful hematopoietic stem cell transplant (HSCT), especially for those who receive haploidentical donor grafts.
Major finding: The main cause of HSCT failure was relapse, occurring in 47% of haploidentical transplant recipients, compared with 20% for those with matched sibling donors and 24% for those with unrelated matched donors (P = .02).
Study details: A retrospective analysis of 124 transplants performed at a single center during 2008-2016.
Disclosures: Authors had no financial disclosures or conflicts of interest to report.
Source: Yanir AD et al. Biol Blood Marrow Transplant. doi: 10.1016/j.bbmt.2018.03.001.
Meta-analyses clarify roles for gabapentin, naltrexone, and psychotherapy in alcohol use disorder
NEW YORK – A meta-analysis of 10 studies provides at least preliminary support for the use of gabapentin for the treatment of alcohol cravings and withdrawal.
In this video interview, Ali Mahmood Khan, MD, of Kings County Hospital, New York, discusses the findings – presented in a poster at the annual meeting of the American Psychiatric Association – which show that patients treated with gabapentin have significantly reduced alcohol craving and withdrawal. While the findings require further study, he said.
Gapapentin also can be used in combination with naltrexone, which was shown in a separate poster presented by Dr. Khan and his colleagues to be useful for treating alcohol use disorders – but mainly through reducing consumption rather than cravings.
In that meta-analysis of 30 studies, no significant added benefit was seen when psychotherapy was combined with naltrexone, he said, noting, however, that additional study is warranted given that various psychotherapies were used across the studies.
Dr. Khan reported having no disclosures.
NEW YORK – A meta-analysis of 10 studies provides at least preliminary support for the use of gabapentin for the treatment of alcohol cravings and withdrawal.
In this video interview, Ali Mahmood Khan, MD, of Kings County Hospital, New York, discusses the findings – presented in a poster at the annual meeting of the American Psychiatric Association – which show that patients treated with gabapentin have significantly reduced alcohol craving and withdrawal. While the findings require further study, he said.
Gapapentin also can be used in combination with naltrexone, which was shown in a separate poster presented by Dr. Khan and his colleagues to be useful for treating alcohol use disorders – but mainly through reducing consumption rather than cravings.
In that meta-analysis of 30 studies, no significant added benefit was seen when psychotherapy was combined with naltrexone, he said, noting, however, that additional study is warranted given that various psychotherapies were used across the studies.
Dr. Khan reported having no disclosures.
NEW YORK – A meta-analysis of 10 studies provides at least preliminary support for the use of gabapentin for the treatment of alcohol cravings and withdrawal.
In this video interview, Ali Mahmood Khan, MD, of Kings County Hospital, New York, discusses the findings – presented in a poster at the annual meeting of the American Psychiatric Association – which show that patients treated with gabapentin have significantly reduced alcohol craving and withdrawal. While the findings require further study, he said.
Gapapentin also can be used in combination with naltrexone, which was shown in a separate poster presented by Dr. Khan and his colleagues to be useful for treating alcohol use disorders – but mainly through reducing consumption rather than cravings.
In that meta-analysis of 30 studies, no significant added benefit was seen when psychotherapy was combined with naltrexone, he said, noting, however, that additional study is warranted given that various psychotherapies were used across the studies.
Dr. Khan reported having no disclosures.
REPORTING FROM APA
Class III obesity increases risk of acute on chronic liver failure in cirrhotic patients
Class III obesity was significantly, independently associated with acute on chronic liver failure (ACLF) in patients with decompensated cirrhosis, and patients with both class III obesity and acute on chronic liver failure also had a significant risk of renal failure, according to a recent retrospective analysis of two databases publised in the Journal of Hepatology.
Vinay Sundaram, MD, from Cedars-Sinai Medical Center in Los Angeles, and his colleagues evaluated 387,884 patients who were in the United Network for Organ Sharing (UNOS) during 2005-2016; were class I or II obese (body mass index 30-39 kg/m2), class III obese (BMI greater than or equal to 40), or not obese (BMI less than 30); and were on a wait list for liver transplantation.
They used the definition of ACLF outlined in the CANONIC (Consortium Acute on Chronic Liver Failure in Cirrhosis) study, which defined it as having “a single hepatic decompensation, such as ascites, hepatic encephalopathy, variceal bleed, or bacterial infection, and one of the following organ failures: single renal failure, single nonrenal organ failure with renal dysfunction or hepatic encephalopathy, or two nonrenal organ failures,” and confirmed the results in the Nationwide Inpatient Sample (NIS) databases by using diagnostic coding algorithms to identify factors such as hepatic decompensation, obesity, and ACLF in that study population.
Dr. Sundarem and his colleagues identified 116,704 patients (30.1%) with acute on chronic liver failure in both the UNOS and NIS databases. At the time of liver transplantation, there was a significant association between ACLF and class I and class II obesity (hazard ratio, 1.12; 95% confidence interval, 1.05-1.19; P less than .001) and class III obesity (HR, 1.24; 95% CI, 1.09-1.41; P less than .001). Other predictors of ACLF in this population were increased age (HR, 1.01 per year; 95% CI, 1.00-1.01; P = .037), hepatitis C virus (HR, 1.25; 95% CI, 1.16-1.35; P less than .001) and hepatitis C combined with alcoholic liver disease (HR, 1.18; 95% CI, 1.06-1.30; P = .002). Regarding organ failure, “renal insufficiency was similar among the three groups,” with increasing obesity class associated with a greater prevalence of renal failure.
“Given the heightened risk of renal failure among obese patients with cirrhosis, we suggest particularly careful management of this fragile population regarding diuretic usage, avoidance of nephrotoxic agents, and administration of an adequate albumin challenge in the setting of acute kidney injury,” the researchers wrote.
The researchers encouraged “an even greater emphasis on weight reduction” for class III obese patients. They noted the association between class III obesity and ACLF is likely caused by an “obesity-related chronic inflammatory state” and said future prospective studies should seek to describe the inflammatory pathways for each condition to predict risk of ACLF in these patients.
The authors reported having no financial disclosures.
SOURCE: Sundarem V et al. J Hepatol. 2018 April 27. doi: 10.1016/j.jhep.2018.04.016.
Class III obesity was significantly, independently associated with acute on chronic liver failure (ACLF) in patients with decompensated cirrhosis, and patients with both class III obesity and acute on chronic liver failure also had a significant risk of renal failure, according to a recent retrospective analysis of two databases publised in the Journal of Hepatology.
Vinay Sundaram, MD, from Cedars-Sinai Medical Center in Los Angeles, and his colleagues evaluated 387,884 patients who were in the United Network for Organ Sharing (UNOS) during 2005-2016; were class I or II obese (body mass index 30-39 kg/m2), class III obese (BMI greater than or equal to 40), or not obese (BMI less than 30); and were on a wait list for liver transplantation.
They used the definition of ACLF outlined in the CANONIC (Consortium Acute on Chronic Liver Failure in Cirrhosis) study, which defined it as having “a single hepatic decompensation, such as ascites, hepatic encephalopathy, variceal bleed, or bacterial infection, and one of the following organ failures: single renal failure, single nonrenal organ failure with renal dysfunction or hepatic encephalopathy, or two nonrenal organ failures,” and confirmed the results in the Nationwide Inpatient Sample (NIS) databases by using diagnostic coding algorithms to identify factors such as hepatic decompensation, obesity, and ACLF in that study population.
Dr. Sundarem and his colleagues identified 116,704 patients (30.1%) with acute on chronic liver failure in both the UNOS and NIS databases. At the time of liver transplantation, there was a significant association between ACLF and class I and class II obesity (hazard ratio, 1.12; 95% confidence interval, 1.05-1.19; P less than .001) and class III obesity (HR, 1.24; 95% CI, 1.09-1.41; P less than .001). Other predictors of ACLF in this population were increased age (HR, 1.01 per year; 95% CI, 1.00-1.01; P = .037), hepatitis C virus (HR, 1.25; 95% CI, 1.16-1.35; P less than .001) and hepatitis C combined with alcoholic liver disease (HR, 1.18; 95% CI, 1.06-1.30; P = .002). Regarding organ failure, “renal insufficiency was similar among the three groups,” with increasing obesity class associated with a greater prevalence of renal failure.
“Given the heightened risk of renal failure among obese patients with cirrhosis, we suggest particularly careful management of this fragile population regarding diuretic usage, avoidance of nephrotoxic agents, and administration of an adequate albumin challenge in the setting of acute kidney injury,” the researchers wrote.
The researchers encouraged “an even greater emphasis on weight reduction” for class III obese patients. They noted the association between class III obesity and ACLF is likely caused by an “obesity-related chronic inflammatory state” and said future prospective studies should seek to describe the inflammatory pathways for each condition to predict risk of ACLF in these patients.
The authors reported having no financial disclosures.
SOURCE: Sundarem V et al. J Hepatol. 2018 April 27. doi: 10.1016/j.jhep.2018.04.016.
Class III obesity was significantly, independently associated with acute on chronic liver failure (ACLF) in patients with decompensated cirrhosis, and patients with both class III obesity and acute on chronic liver failure also had a significant risk of renal failure, according to a recent retrospective analysis of two databases publised in the Journal of Hepatology.
Vinay Sundaram, MD, from Cedars-Sinai Medical Center in Los Angeles, and his colleagues evaluated 387,884 patients who were in the United Network for Organ Sharing (UNOS) during 2005-2016; were class I or II obese (body mass index 30-39 kg/m2), class III obese (BMI greater than or equal to 40), or not obese (BMI less than 30); and were on a wait list for liver transplantation.
They used the definition of ACLF outlined in the CANONIC (Consortium Acute on Chronic Liver Failure in Cirrhosis) study, which defined it as having “a single hepatic decompensation, such as ascites, hepatic encephalopathy, variceal bleed, or bacterial infection, and one of the following organ failures: single renal failure, single nonrenal organ failure with renal dysfunction or hepatic encephalopathy, or two nonrenal organ failures,” and confirmed the results in the Nationwide Inpatient Sample (NIS) databases by using diagnostic coding algorithms to identify factors such as hepatic decompensation, obesity, and ACLF in that study population.
Dr. Sundarem and his colleagues identified 116,704 patients (30.1%) with acute on chronic liver failure in both the UNOS and NIS databases. At the time of liver transplantation, there was a significant association between ACLF and class I and class II obesity (hazard ratio, 1.12; 95% confidence interval, 1.05-1.19; P less than .001) and class III obesity (HR, 1.24; 95% CI, 1.09-1.41; P less than .001). Other predictors of ACLF in this population were increased age (HR, 1.01 per year; 95% CI, 1.00-1.01; P = .037), hepatitis C virus (HR, 1.25; 95% CI, 1.16-1.35; P less than .001) and hepatitis C combined with alcoholic liver disease (HR, 1.18; 95% CI, 1.06-1.30; P = .002). Regarding organ failure, “renal insufficiency was similar among the three groups,” with increasing obesity class associated with a greater prevalence of renal failure.
“Given the heightened risk of renal failure among obese patients with cirrhosis, we suggest particularly careful management of this fragile population regarding diuretic usage, avoidance of nephrotoxic agents, and administration of an adequate albumin challenge in the setting of acute kidney injury,” the researchers wrote.
The researchers encouraged “an even greater emphasis on weight reduction” for class III obese patients. They noted the association between class III obesity and ACLF is likely caused by an “obesity-related chronic inflammatory state” and said future prospective studies should seek to describe the inflammatory pathways for each condition to predict risk of ACLF in these patients.
The authors reported having no financial disclosures.
SOURCE: Sundarem V et al. J Hepatol. 2018 April 27. doi: 10.1016/j.jhep.2018.04.016.
FROM THE JOURNAL OF HEPATOLOGY
Key clinical point: Patients with a BMI greater than or equal to 40 kg/m2 with decompensated cirrhosis are at greater risk of developing acute on chronic liver failure.
Major finding: Class III obesity carried a hazard ratio of 1.24 in the UNOS database and an odds ratio of 1.30 in the NIS database at the time of liver transplantation.
Data source: A retrospective cohort database study of 116,704 patients with acute on chronic liver failure listed during 2005-2016.
Disclosures: The authors reported having no financial disclosures.
Source: Sundaram V et al. J Hepatol. 2018 Apr 27. doi: 10.1016/j.jhep.2018.04.016.
Very few infants born to HCV-infected mothers receive testing
Despite the increasing prevalence of hepatitis C virus (HCV) infection in pregnant women, infants exposed to the disease are screened at a very low rate, Catherine A. Chappell, MD, and her associates wrote in Pediatrics.
During 2006-2014, 87,924 women gave birth at the Magee-Womens Hospital at the University of Pittsburgh Medical Center, of whom 1,043 had HCV. Over this time, the HCV prevalence rate increased 60%, from 1,026 cases per 100,000 women to 1,637 cases per 100,000 women. Women with HCV were more likely to be white, have Medicaid, have opiate use disorder, have other substance use disorders, and be under the age of 30 years.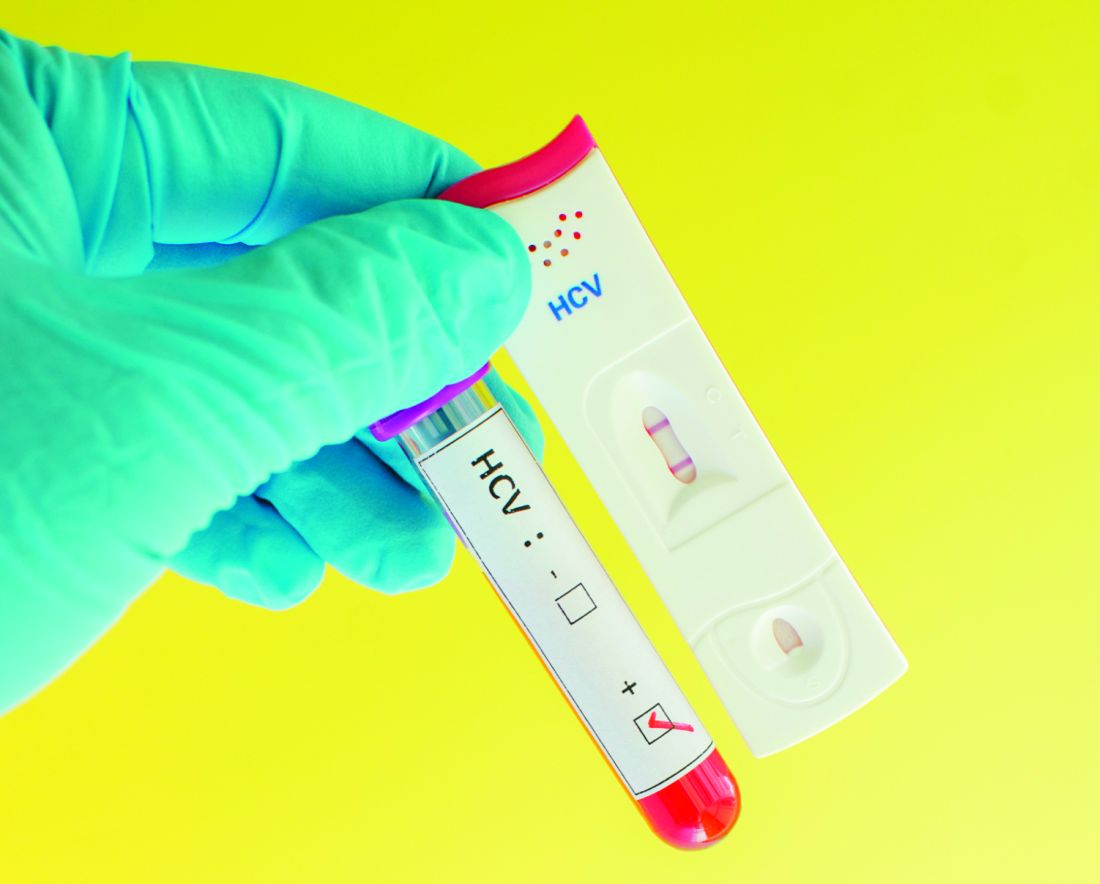
Infants born to HCV-infected women are significantly more likely to be preterm and of low birth weight.
An additional 32 infants who did not receive well child care did receive HCV testing. A total of nine infants, seven in the well child group and two in the non-well child group, tested positive for HCV.
“Of the infants tested with conclusive results, the HCV transmission rate was 8.4%, with 7.2% having chronic HCV infection,” which is in line with previous reports, according to the researchers.
“Because of the poor rates of pediatric HCV screening described, future researchers should focus on interventions to increase screening in infants who are at risk for perinatal HCV acquisition by including technology to improve the transfer of maternal HCV status to the pediatric record and increase pediatric provider awareness regarding HCV screening guidelines,” the investigators concluded.
SOURCE: Chappell CA et al. Pediatrics. 2018 May 2. doi: 10.1542/peds.2017-3273.
Despite the increasing prevalence of hepatitis C virus (HCV) infection in pregnant women, infants exposed to the disease are screened at a very low rate, Catherine A. Chappell, MD, and her associates wrote in Pediatrics.
During 2006-2014, 87,924 women gave birth at the Magee-Womens Hospital at the University of Pittsburgh Medical Center, of whom 1,043 had HCV. Over this time, the HCV prevalence rate increased 60%, from 1,026 cases per 100,000 women to 1,637 cases per 100,000 women. Women with HCV were more likely to be white, have Medicaid, have opiate use disorder, have other substance use disorders, and be under the age of 30 years.
Infants born to HCV-infected women are significantly more likely to be preterm and of low birth weight.
An additional 32 infants who did not receive well child care did receive HCV testing. A total of nine infants, seven in the well child group and two in the non-well child group, tested positive for HCV.
“Of the infants tested with conclusive results, the HCV transmission rate was 8.4%, with 7.2% having chronic HCV infection,” which is in line with previous reports, according to the researchers.
“Because of the poor rates of pediatric HCV screening described, future researchers should focus on interventions to increase screening in infants who are at risk for perinatal HCV acquisition by including technology to improve the transfer of maternal HCV status to the pediatric record and increase pediatric provider awareness regarding HCV screening guidelines,” the investigators concluded.
SOURCE: Chappell CA et al. Pediatrics. 2018 May 2. doi: 10.1542/peds.2017-3273.
Despite the increasing prevalence of hepatitis C virus (HCV) infection in pregnant women, infants exposed to the disease are screened at a very low rate, Catherine A. Chappell, MD, and her associates wrote in Pediatrics.
During 2006-2014, 87,924 women gave birth at the Magee-Womens Hospital at the University of Pittsburgh Medical Center, of whom 1,043 had HCV. Over this time, the HCV prevalence rate increased 60%, from 1,026 cases per 100,000 women to 1,637 cases per 100,000 women. Women with HCV were more likely to be white, have Medicaid, have opiate use disorder, have other substance use disorders, and be under the age of 30 years.
Infants born to HCV-infected women are significantly more likely to be preterm and of low birth weight.
An additional 32 infants who did not receive well child care did receive HCV testing. A total of nine infants, seven in the well child group and two in the non-well child group, tested positive for HCV.
“Of the infants tested with conclusive results, the HCV transmission rate was 8.4%, with 7.2% having chronic HCV infection,” which is in line with previous reports, according to the researchers.
“Because of the poor rates of pediatric HCV screening described, future researchers should focus on interventions to increase screening in infants who are at risk for perinatal HCV acquisition by including technology to improve the transfer of maternal HCV status to the pediatric record and increase pediatric provider awareness regarding HCV screening guidelines,” the investigators concluded.
SOURCE: Chappell CA et al. Pediatrics. 2018 May 2. doi: 10.1542/peds.2017-3273.
FROM PEDIATRICS