User login
One Heck of a Check-up
ANSWER
The correct interpretation includes sinus bradycardia with marked sinus arrhythmia and junctional escape beats, and an intraventricular conduction delay.
This type of ECG is not ideal for calculating the ventricular rate via the 30/150/100 method. An easier method is to multiply the number of QRS complexes in the rhythm strip by six (an ECG at standard paper speed takes 10 s; 6 × 10 s = 60 s). In the absence of a permanent pacemaker, variation of a few beats/min from the computer reading is acceptable. In this case, multiplying 9 x 6 yields a rate of 54 beats/min (close to the computer reading of 55) and reveals sinus bradycardia.
Looking at the lead I rhythm strip, notice that while the QRS complexes in the fourth and eighth beats look similar to the others, they are not preceded by P waves; the T waves of these two beats are also not similar to the others. These represent junctional escape beats, with a possible retrograde P wave in the T-wave complex. The long pauses and the absence of a P wave prior to the fourth and eighth beats make this a sinus arrhythmia.
The diagnosis of an intraventricular conduction delay can be made by the duration of the QRS complex (122 ms), which is above normal limits. This ECG does not meet the clear criteria for a right bundle branch block (QRS ≥ 120 ms, terminal broad S wave in lead I, RSR’ in V1) or left bundle branch block (QRS ≥ 120 ms, ST depressions and inverted T waves, particularly in I, aVL, V5, and V6).
ANSWER
The correct interpretation includes sinus bradycardia with marked sinus arrhythmia and junctional escape beats, and an intraventricular conduction delay.
This type of ECG is not ideal for calculating the ventricular rate via the 30/150/100 method. An easier method is to multiply the number of QRS complexes in the rhythm strip by six (an ECG at standard paper speed takes 10 s; 6 × 10 s = 60 s). In the absence of a permanent pacemaker, variation of a few beats/min from the computer reading is acceptable. In this case, multiplying 9 x 6 yields a rate of 54 beats/min (close to the computer reading of 55) and reveals sinus bradycardia.
Looking at the lead I rhythm strip, notice that while the QRS complexes in the fourth and eighth beats look similar to the others, they are not preceded by P waves; the T waves of these two beats are also not similar to the others. These represent junctional escape beats, with a possible retrograde P wave in the T-wave complex. The long pauses and the absence of a P wave prior to the fourth and eighth beats make this a sinus arrhythmia.
The diagnosis of an intraventricular conduction delay can be made by the duration of the QRS complex (122 ms), which is above normal limits. This ECG does not meet the clear criteria for a right bundle branch block (QRS ≥ 120 ms, terminal broad S wave in lead I, RSR’ in V1) or left bundle branch block (QRS ≥ 120 ms, ST depressions and inverted T waves, particularly in I, aVL, V5, and V6).
ANSWER
The correct interpretation includes sinus bradycardia with marked sinus arrhythmia and junctional escape beats, and an intraventricular conduction delay.
This type of ECG is not ideal for calculating the ventricular rate via the 30/150/100 method. An easier method is to multiply the number of QRS complexes in the rhythm strip by six (an ECG at standard paper speed takes 10 s; 6 × 10 s = 60 s). In the absence of a permanent pacemaker, variation of a few beats/min from the computer reading is acceptable. In this case, multiplying 9 x 6 yields a rate of 54 beats/min (close to the computer reading of 55) and reveals sinus bradycardia.
Looking at the lead I rhythm strip, notice that while the QRS complexes in the fourth and eighth beats look similar to the others, they are not preceded by P waves; the T waves of these two beats are also not similar to the others. These represent junctional escape beats, with a possible retrograde P wave in the T-wave complex. The long pauses and the absence of a P wave prior to the fourth and eighth beats make this a sinus arrhythmia.
The diagnosis of an intraventricular conduction delay can be made by the duration of the QRS complex (122 ms), which is above normal limits. This ECG does not meet the clear criteria for a right bundle branch block (QRS ≥ 120 ms, terminal broad S wave in lead I, RSR’ in V1) or left bundle branch block (QRS ≥ 120 ms, ST depressions and inverted T waves, particularly in I, aVL, V5, and V6).

He remained stable for six years, but then his exertional angina returned. Repeat catheterization showed progressive disease in the right coronary artery (RCA) with new disease in an obtuse marginal (OM) branch of the circumflex artery. His LVEF had also diminished to 38%. Stents were placed in the RCA and OM arteries, and his ß-blocker dose was increased.
Today, he reports that over the past six months, his heart rate has been slow and often skips beats. He stopped taking his ß-blocker, hoping it would help speed up his heart; it didn’t. He says he feels fine right now (although his heart continues to skip beats). He says he can climb a flight of stairs without difficulty and denies chest pain, dyspnea, dizziness, or syncope. At night, he sleeps on one or two pillows. He denies orthopnea, paroxysmal nocturnal dyspnea, or peripheral edema.
Medical history includes type 2 diabetes (controlled with diet and exercise), degenerative joint disease, and hyperlipidemia. Surgical history includes a tonsillectomy and trigger finger repair of the left third digit. His current medications include clopidogrel, losartan, pravastatin, spironolactone, and aspirin. He has no known drug allergies.
The patient used to be a heavy smoker (> 2 packs/d) but quit after his first stent was placed. He drinks one to two glasses of wine per week and denies recreational drug use.
Family history is positive for coronary artery disease and stroke. His father died of CAD at age 60, and his mother of heart failure at 58.
A review of systems is noncontributory. Vital signs include a blood pressure of 118/80 mm Hg; pulse, 53 beats/min; temperature, 37°C; respiratory rate, 14 breaths/min-1; and O2 saturation, 96% on room air. His weight is 214 lb and his height, 70 in.
Physical exam reveals an alert, well-kept male in no distress. Pertinent findings include a regularly irregular pulse and normal S1 and S2, with no murmurs, gallops, or rubs. The lungs are clear bilaterally. The abdomen is soft and benign with no organomegaly. Peripheral pulses are 2+ bilaterally, and there is no peripheral edema or calf tenderness. Neurologic exam shows a grossly intact sensory and motor system with no focal signs.
An ECG reveals a ventricular rate of 55 beats/min; PR interval, 146 ms; QRS duration, 122 ms; QT/QTc interval, 424/405 ms; P axis, 60°; R axis, 38°; and T axis, 29°. What is your interpretation?

A clinical pathway to standardize use of maintenance IV fluids
Clinical question
Can an evidence-based clinical pathway improve adherence to recent recommendations to use isotonic solutions for maintenance intravenous fluids in hospitalized children?
Background
The traditional teaching regarding composition of maintenance intravenous fluids (IVF) in children has been based on the Holliday-Segar method.1 Since its publication in Pediatrics in 1957, concerns have been raised regarding the risk of iatrogenic hyponatremia caused by giving hypotonic fluids determined by this method,2 especially in patients with an elevated risk of increased antidiuretic hormone (ADH) secretion.3 Multiple recent systematic reviews and meta-analyses have confirmed that isotonic IVF reduces the risk of hyponatremia in hospitalized children.4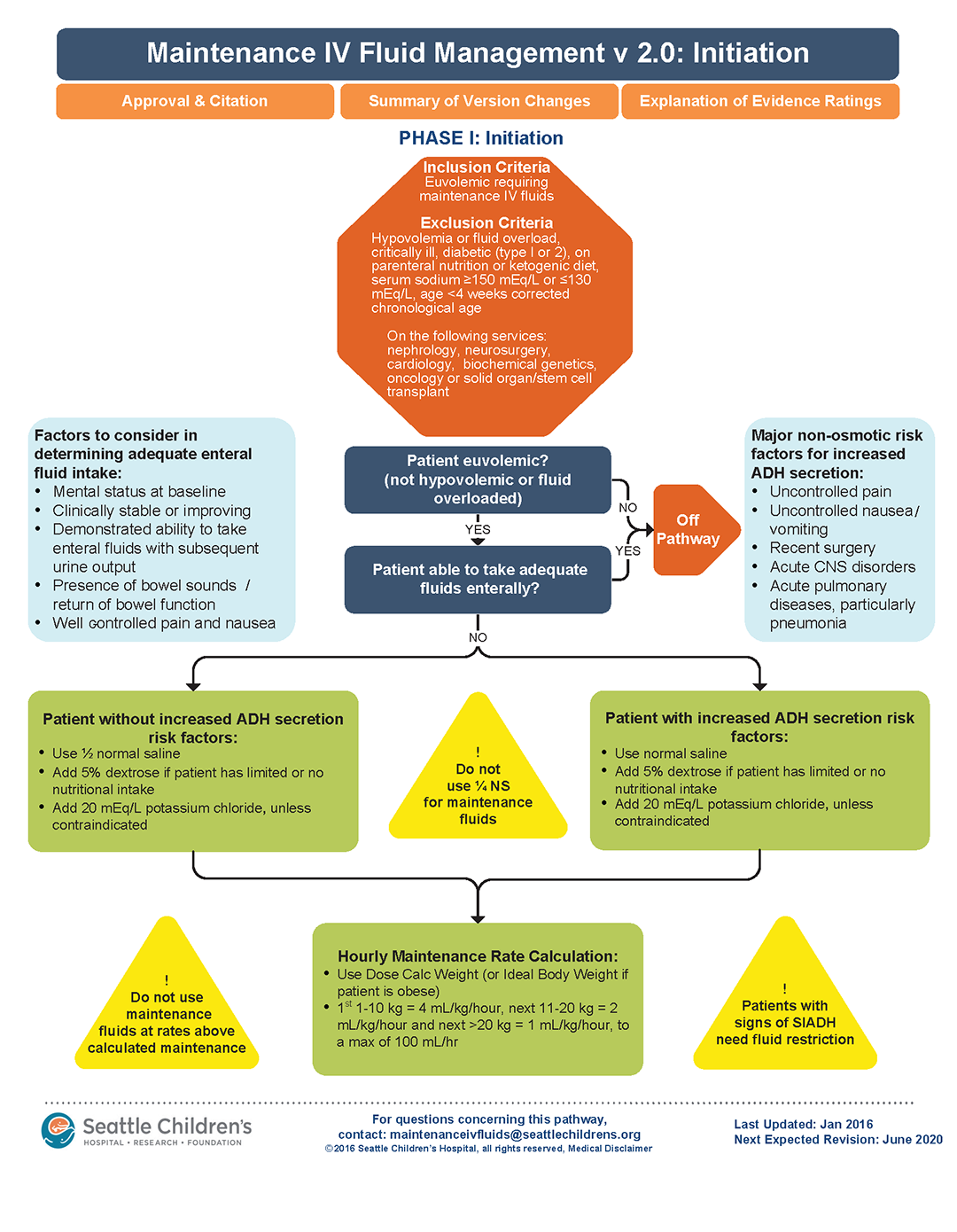
Study design
Interrupted time series analysis before and after pathway implementation.
Setting
370-bed tertiary care free-standing children’s hospital.
Synopsis
A multidisciplinary team was assembled, comprising physicians and nurses in hospital medicine, general pediatrics, emergency medicine, and nephrology. After a systematic review of the recent literature, a clinical algorithm and web-based training module were developed. Faculty in general pediatrics, hospital medicine, and emergency medicine were required to complete the module, while medical and surgical residents were encouraged but not required to complete the module. A maintenance IVF order set was created and embedded into all order sets previously containing IVF orders and was also available in stand-alone form.
Inclusion criteria (“pathway eligible”) included being euvolemic and requiring IVF. Exclusion criteria included fluid status derangements, critical illness, severe serum sodium abnormalities (serum sodium ≥150 mEq/L or ≤130 mEq/L) use of TPN or ketogenic diet. In the order set, IVF composition was determined based on risk factors for increased ADH secretion. Inclusion of potassium in IVF was also determined by the pathway.
Over the 1-year study period, 11,602 pathway-eligible encounters in 10,287 patients were reviewed. Use of isotonic maintenance IVF increased significantly from 9.3% to 50.6%, while use of hypotonic fluids decreased from 94.2% to 56.6%. Use of potassium-containing IVF increased from 52.9% to 75.3%. Dysnatremia continued to occur due to hypotonic IVF use.
Bottom line
A combined clinical pathway and training module to standardize the composition of IVF is feasible, and results in increased use of isotonic and potassium-containing fluids.
Citation
Rooholamini S, Clifton H, Haaland W, et al. Outcomes of a clinical pathway to standardize use of maintenance intravenous fluids. Hosp Pediatr. 2017 Dec;7(12):703-9.

Dr. Chang is a pediatric hospitalist at Baystate Children’s Hospital in Springfield, Mass., and is the pediatric editor of The Hospitalist.
References
1. Holliday MA et al. The maintenance need for water in parenteral fluid therapy. Pediatrics 1957;19:823-32.
2. Friedman JN et al. Comparison of isotonic and hypotonic intravenous maintenance fluids: a randomized clinical trial. JAMA Pediatr. 2015;169:445-51.
3. Fuchs J et al. Current Issues in Intravenous Fluid Use in Hospitalized Children. Rev Recent Clin Trials. 2017;12:284-9.
4. McNab S et al. Isotonic versus hypotonic solutions for maintenance intravenous fluid administration in children. Cochrane Database. Syst Rev 2014:CD009457.
Clinical question
Can an evidence-based clinical pathway improve adherence to recent recommendations to use isotonic solutions for maintenance intravenous fluids in hospitalized children?
Background
The traditional teaching regarding composition of maintenance intravenous fluids (IVF) in children has been based on the Holliday-Segar method.1 Since its publication in Pediatrics in 1957, concerns have been raised regarding the risk of iatrogenic hyponatremia caused by giving hypotonic fluids determined by this method,2 especially in patients with an elevated risk of increased antidiuretic hormone (ADH) secretion.3 Multiple recent systematic reviews and meta-analyses have confirmed that isotonic IVF reduces the risk of hyponatremia in hospitalized children.4
Study design
Interrupted time series analysis before and after pathway implementation.
Setting
370-bed tertiary care free-standing children’s hospital.
Synopsis
A multidisciplinary team was assembled, comprising physicians and nurses in hospital medicine, general pediatrics, emergency medicine, and nephrology. After a systematic review of the recent literature, a clinical algorithm and web-based training module were developed. Faculty in general pediatrics, hospital medicine, and emergency medicine were required to complete the module, while medical and surgical residents were encouraged but not required to complete the module. A maintenance IVF order set was created and embedded into all order sets previously containing IVF orders and was also available in stand-alone form.
Inclusion criteria (“pathway eligible”) included being euvolemic and requiring IVF. Exclusion criteria included fluid status derangements, critical illness, severe serum sodium abnormalities (serum sodium ≥150 mEq/L or ≤130 mEq/L) use of TPN or ketogenic diet. In the order set, IVF composition was determined based on risk factors for increased ADH secretion. Inclusion of potassium in IVF was also determined by the pathway.
Over the 1-year study period, 11,602 pathway-eligible encounters in 10,287 patients were reviewed. Use of isotonic maintenance IVF increased significantly from 9.3% to 50.6%, while use of hypotonic fluids decreased from 94.2% to 56.6%. Use of potassium-containing IVF increased from 52.9% to 75.3%. Dysnatremia continued to occur due to hypotonic IVF use.
Bottom line
A combined clinical pathway and training module to standardize the composition of IVF is feasible, and results in increased use of isotonic and potassium-containing fluids.
Citation
Rooholamini S, Clifton H, Haaland W, et al. Outcomes of a clinical pathway to standardize use of maintenance intravenous fluids. Hosp Pediatr. 2017 Dec;7(12):703-9.
Dr. Chang is a pediatric hospitalist at Baystate Children’s Hospital in Springfield, Mass., and is the pediatric editor of The Hospitalist.
References
1. Holliday MA et al. The maintenance need for water in parenteral fluid therapy. Pediatrics 1957;19:823-32.
2. Friedman JN et al. Comparison of isotonic and hypotonic intravenous maintenance fluids: a randomized clinical trial. JAMA Pediatr. 2015;169:445-51.
3. Fuchs J et al. Current Issues in Intravenous Fluid Use in Hospitalized Children. Rev Recent Clin Trials. 2017;12:284-9.
4. McNab S et al. Isotonic versus hypotonic solutions for maintenance intravenous fluid administration in children. Cochrane Database. Syst Rev 2014:CD009457.
Clinical question
Can an evidence-based clinical pathway improve adherence to recent recommendations to use isotonic solutions for maintenance intravenous fluids in hospitalized children?
Background
The traditional teaching regarding composition of maintenance intravenous fluids (IVF) in children has been based on the Holliday-Segar method.1 Since its publication in Pediatrics in 1957, concerns have been raised regarding the risk of iatrogenic hyponatremia caused by giving hypotonic fluids determined by this method,2 especially in patients with an elevated risk of increased antidiuretic hormone (ADH) secretion.3 Multiple recent systematic reviews and meta-analyses have confirmed that isotonic IVF reduces the risk of hyponatremia in hospitalized children.4
Study design
Interrupted time series analysis before and after pathway implementation.
Setting
370-bed tertiary care free-standing children’s hospital.
Synopsis
A multidisciplinary team was assembled, comprising physicians and nurses in hospital medicine, general pediatrics, emergency medicine, and nephrology. After a systematic review of the recent literature, a clinical algorithm and web-based training module were developed. Faculty in general pediatrics, hospital medicine, and emergency medicine were required to complete the module, while medical and surgical residents were encouraged but not required to complete the module. A maintenance IVF order set was created and embedded into all order sets previously containing IVF orders and was also available in stand-alone form.
Inclusion criteria (“pathway eligible”) included being euvolemic and requiring IVF. Exclusion criteria included fluid status derangements, critical illness, severe serum sodium abnormalities (serum sodium ≥150 mEq/L or ≤130 mEq/L) use of TPN or ketogenic diet. In the order set, IVF composition was determined based on risk factors for increased ADH secretion. Inclusion of potassium in IVF was also determined by the pathway.
Over the 1-year study period, 11,602 pathway-eligible encounters in 10,287 patients were reviewed. Use of isotonic maintenance IVF increased significantly from 9.3% to 50.6%, while use of hypotonic fluids decreased from 94.2% to 56.6%. Use of potassium-containing IVF increased from 52.9% to 75.3%. Dysnatremia continued to occur due to hypotonic IVF use.
Bottom line
A combined clinical pathway and training module to standardize the composition of IVF is feasible, and results in increased use of isotonic and potassium-containing fluids.
Citation
Rooholamini S, Clifton H, Haaland W, et al. Outcomes of a clinical pathway to standardize use of maintenance intravenous fluids. Hosp Pediatr. 2017 Dec;7(12):703-9.
Dr. Chang is a pediatric hospitalist at Baystate Children’s Hospital in Springfield, Mass., and is the pediatric editor of The Hospitalist.
References
1. Holliday MA et al. The maintenance need for water in parenteral fluid therapy. Pediatrics 1957;19:823-32.
2. Friedman JN et al. Comparison of isotonic and hypotonic intravenous maintenance fluids: a randomized clinical trial. JAMA Pediatr. 2015;169:445-51.
3. Fuchs J et al. Current Issues in Intravenous Fluid Use in Hospitalized Children. Rev Recent Clin Trials. 2017;12:284-9.
4. McNab S et al. Isotonic versus hypotonic solutions for maintenance intravenous fluid administration in children. Cochrane Database. Syst Rev 2014:CD009457.
Emicizumab gets priority review for hemophilia A without inhibitors
The Food and Drug Administration has granted priority review to Roche’s emicizumab-kxwh (Hemlibra) for the treatment of adults and children with hemophilia A without factor VIII inhibitors.
The agency is scheduled to make a decision on approval in October 2018.
Among patients aged 12 years and older without factor VIII inhibitors, emicizumab-kxwh prophylaxis every week reduced treated bleeds by 96% (P less than .0001) and treated bleeds were reduced by 97% (P less than .0001) in patients who were treated every 2 weeks, according to Roche. The drug-treated group was compared with patients who received no prophylaxis. Another arm of the study examined patients who had previously received factor VIII prophylaxis and then switched to emicizumab-kxwh prophylaxis. In an intrapatient comparison, emicizumab-kxwh showed a 68% reduction in treated bleeds, which was statistically significant and demonstrated superior efficacy to factor VIII prophylaxis.
Emicizumab-kxwh was approved by FDA in November 2017 for routine prophylaxis for adults and children with hemophilia A with factor VIII inhibitors. That approval was based on results from the HAVEN 1 and HAVEN 2 studies.
The Food and Drug Administration has granted priority review to Roche’s emicizumab-kxwh (Hemlibra) for the treatment of adults and children with hemophilia A without factor VIII inhibitors.
The agency is scheduled to make a decision on approval in October 2018.
Among patients aged 12 years and older without factor VIII inhibitors, emicizumab-kxwh prophylaxis every week reduced treated bleeds by 96% (P less than .0001) and treated bleeds were reduced by 97% (P less than .0001) in patients who were treated every 2 weeks, according to Roche. The drug-treated group was compared with patients who received no prophylaxis. Another arm of the study examined patients who had previously received factor VIII prophylaxis and then switched to emicizumab-kxwh prophylaxis. In an intrapatient comparison, emicizumab-kxwh showed a 68% reduction in treated bleeds, which was statistically significant and demonstrated superior efficacy to factor VIII prophylaxis.
Emicizumab-kxwh was approved by FDA in November 2017 for routine prophylaxis for adults and children with hemophilia A with factor VIII inhibitors. That approval was based on results from the HAVEN 1 and HAVEN 2 studies.
The Food and Drug Administration has granted priority review to Roche’s emicizumab-kxwh (Hemlibra) for the treatment of adults and children with hemophilia A without factor VIII inhibitors.
The agency is scheduled to make a decision on approval in October 2018.
Among patients aged 12 years and older without factor VIII inhibitors, emicizumab-kxwh prophylaxis every week reduced treated bleeds by 96% (P less than .0001) and treated bleeds were reduced by 97% (P less than .0001) in patients who were treated every 2 weeks, according to Roche. The drug-treated group was compared with patients who received no prophylaxis. Another arm of the study examined patients who had previously received factor VIII prophylaxis and then switched to emicizumab-kxwh prophylaxis. In an intrapatient comparison, emicizumab-kxwh showed a 68% reduction in treated bleeds, which was statistically significant and demonstrated superior efficacy to factor VIII prophylaxis.
Emicizumab-kxwh was approved by FDA in November 2017 for routine prophylaxis for adults and children with hemophilia A with factor VIII inhibitors. That approval was based on results from the HAVEN 1 and HAVEN 2 studies.
Medication management
This is the tenth in a series of articles from the National Center for Excellence in Primary Care Research (NCEPCR) in the Agency for Healthcare Research and Quality (AHRQ). This series introduces sets of tools and resources designed to help your practice.
An important part of self-management (last month’s article) is medication management, often augmented by the use of a portal and always cognizant of the importance of medication reconciliation and drug interactions. These latter issues can be addressed with health information technology (health IT), which will be discussed the next two columns. This month, we examine some of AHRQ’s other tools and resources to assist with medication management.
Patient understanding of the medications and medication schedule is important, and therefore health literacy key. The AHRQ Health Literacy Universal Precautions Toolkit – 2nd edition can help primary care practices reduce the complexity of health care, increase patient understanding of health information, and enhance support for patients of all health literacy levels. Also available are the companion guide, Implementing the AHRQ Health Literacy Universal Precautions Toolkit: Practical Ideas for Primary Care Practices, and a crosswalk showing how implementing health literacy tools can help meet standards for patient-centered medical home certification or recognition or meet Accreditation Canada standards.

Finally, How to Create a Pill Card helps users create an easy-to-use “pill card” for anyone who has a hard time keeping track of their medicines. Step-by-step instructions, sample clip art, and suggestions for design and use will help to customize a reminder card.
These and other tools can be found at the NCEPCR Web site: www.ahrq.gov/ncepcr.
Dr. Ganiats is director of the National Center for Excellence in Primary Care Research at AHRQ, Rockville, Md.
This is the tenth in a series of articles from the National Center for Excellence in Primary Care Research (NCEPCR) in the Agency for Healthcare Research and Quality (AHRQ). This series introduces sets of tools and resources designed to help your practice.
An important part of self-management (last month’s article) is medication management, often augmented by the use of a portal and always cognizant of the importance of medication reconciliation and drug interactions. These latter issues can be addressed with health information technology (health IT), which will be discussed the next two columns. This month, we examine some of AHRQ’s other tools and resources to assist with medication management.
Patient understanding of the medications and medication schedule is important, and therefore health literacy key. The AHRQ Health Literacy Universal Precautions Toolkit – 2nd edition can help primary care practices reduce the complexity of health care, increase patient understanding of health information, and enhance support for patients of all health literacy levels. Also available are the companion guide, Implementing the AHRQ Health Literacy Universal Precautions Toolkit: Practical Ideas for Primary Care Practices, and a crosswalk showing how implementing health literacy tools can help meet standards for patient-centered medical home certification or recognition or meet Accreditation Canada standards.
Finally, How to Create a Pill Card helps users create an easy-to-use “pill card” for anyone who has a hard time keeping track of their medicines. Step-by-step instructions, sample clip art, and suggestions for design and use will help to customize a reminder card.
These and other tools can be found at the NCEPCR Web site: www.ahrq.gov/ncepcr.
Dr. Ganiats is director of the National Center for Excellence in Primary Care Research at AHRQ, Rockville, Md.
This is the tenth in a series of articles from the National Center for Excellence in Primary Care Research (NCEPCR) in the Agency for Healthcare Research and Quality (AHRQ). This series introduces sets of tools and resources designed to help your practice.
An important part of self-management (last month’s article) is medication management, often augmented by the use of a portal and always cognizant of the importance of medication reconciliation and drug interactions. These latter issues can be addressed with health information technology (health IT), which will be discussed the next two columns. This month, we examine some of AHRQ’s other tools and resources to assist with medication management.
Patient understanding of the medications and medication schedule is important, and therefore health literacy key. The AHRQ Health Literacy Universal Precautions Toolkit – 2nd edition can help primary care practices reduce the complexity of health care, increase patient understanding of health information, and enhance support for patients of all health literacy levels. Also available are the companion guide, Implementing the AHRQ Health Literacy Universal Precautions Toolkit: Practical Ideas for Primary Care Practices, and a crosswalk showing how implementing health literacy tools can help meet standards for patient-centered medical home certification or recognition or meet Accreditation Canada standards.
Finally, How to Create a Pill Card helps users create an easy-to-use “pill card” for anyone who has a hard time keeping track of their medicines. Step-by-step instructions, sample clip art, and suggestions for design and use will help to customize a reminder card.
These and other tools can be found at the NCEPCR Web site: www.ahrq.gov/ncepcr.
Dr. Ganiats is director of the National Center for Excellence in Primary Care Research at AHRQ, Rockville, Md.
Impact of Sagittal Rotation on Axial Glenoid Width Measurement in the Setting of Glenoid Bone Loss
ABSTRACT
Standard 2-dimensional (2-D) computed tomography (CT) scans of the shoulder are often aligned to the plane of the body as opposed to the plane of the scapula, which may challenge the ability to accurately measure glenoid width and glenoid bone loss (GBL). The purpose of this study is to determine the effect of sagittal rotation of the glenoid on axial anterior-posterior (AP) glenoid width measurements in the setting of anterior GBL.
Forty-three CT scans from consecutive patients with anterior GBL (minimum 10%) were reformatted utilizing open-source DICOM software (OsiriX MD). Patients were grouped according to extent of GBL: I, 10% to 14.9% (N = 12); II, 15% to 19.9% (N = 16); and III, >20% (N = 15). The uncorrected (UNCORR) and corrected (CORR) images were assessed in the axial plane at 5 standardized cuts and measured for AP glenoid width.
For groups I and III, UNCORR scans underestimated axial AP width (and thus overestimated anterior GBL) in cuts 1 and 2, while in cuts 3 to 5, the axial AP width was overestimated (GBL was underestimated). In Group II, axial AP width was underestimated (GBL was overestimated), while in cuts 2 to 5, the axial AP width was overestimated (GBL was underestimated). Overall, AP glenoid width was consistently underestimated in cut 1, the most caudal cut; while AP glenoid width was consistently overestimated in cuts 3 to 5, the more cephalad cuts.
UNCORR 2-D CT scans inaccurately estimated glenoid width and the degree of anterior GBL. This data suggests that corrected 2D CT scans or a 3-dimensional (3-D) reconstruction can help in accurately defining the anterior GBL in patients with shoulder instability.
The treatment of glenohumeral instability has substantially evolved over the past several decades. The understanding of glenoid bone loss (GBL), in particular, has advanced to such a level that we utilize the quantification of GBL for surgical decision-making. Unrecognized and/or untreated GBL is associated with recurrent instability, pain, and disability. Controversy exists, however, regarding the precise amount of anterior GBL that is significant enough to warrant surgical treatment. While historically, 25%1,2 of anterior GBL was thought to be the critical number required to warrant osseous augmentation, studies that are more recent have highlighted the need to perform osseous glenoid reconstruction with lesser degrees of GBL, particularly in the contact athlete.3-9 As small differences in the amount of GBL can change surgical decision-making from an all-soft tissue repair to an osseous reconstruction, it is paramount that we have accurate, valid, and reproducible methods for calculating GBL.
Continue to: Historically, plain radiographs...
Historically, plain radiographs have been the mainstay for evaluating the glenohumeral joint, including Grashey and axillary views, allowing clinicians to evaluate the congruency of the glenohumeral joint and to assess bone loss on both the glenoid and humeral head.1,10 While large, acute fractures of the glenoid are fairly evident on radiographs, including the Grashey view,11 shoulders with chronic and/or attritional anterior GBL are more difficult to evaluate, and often do not provide the information necessary to guide surgical decision-making.
Computed tomography (CT) of the shoulder has become the most commonly utilized imaging modality in the evaluation of patients with shoulder instability associated with GBL. Standard 2-dimensional (2-D) CT scans of the shoulder are often aligned to the plane of the body as opposed to the plane of the scapula/glenoid, as standard protocols often fail to account for the anterior sagittal rotation of the scapula/glenoid, similar to the disadvantage of standard radiographs. While 3-dimensional (3-D) CT reconstructions eliminate the effect of gantry angles, and thus allow for an en face view of the glenoid, 3-D reconstructions are not always available, and cannot always be measured.12-14 Thus, improved methodology for utilizing standard 2D scans is warranted, as the ability to correctly align the axial CT scan to the axis of the glenoid may allow for more accurate GBL measurements, which will ultimately impact surgical decision-making. Recently, Gross and colleagues15 reported the effect of sagittal rotation of the glenoid on axial measurements of anterior-posterior (AP) glenoid width and glenoid version in normal glenoids, without bone loss, and found that the mean angle of correction needed to align the sagittal plane was 20.1° ± 1.2° of rotation. To the authors’ knowledge, this same methodology has not been applied to patients with clinically meaningful anterior GBL. Given that the average glenoid width in human shoulders is 24.4 mm ± 2.9 mm,16 1 mm of glenoid bone loss (GBL) corresponds to approximately 4% of the glenoid width, and thus even subtle differences in the interpretation of GBL may have substantial clinical implications. Therefore, the purpose of this study is to determine the effect of sagittal rotation of the glenoid on axial AP glenoid width measurements in the setting of clinically significant anterior GBL.
METHODS
This study was approved by Massachusetts General Hospital Institutional Review Board. A retrospective review of consecutive patients with a diagnosis of anterior shoulder instability between 2009 and 2013 was conducted. Inclusion criteria comprised patients with a minimum of 10% anterior GBL, an available CT scan of the affected shoulder, and no history of prior ipsilateral surgeries. Exclusion criteria comprised evidence of degenerative changes to the glenoid and/or humeral head, as well as prior ipsilateral shoulder surgery. Sixty consecutive patients were originally identified as having anterior shoulder instability, and 17 were excluded based on the inclusion/exclusion criteria, leaving 43 patients (43 shoulders) available for inclusion. Shoulder CT scans from all 43 patients were reformatted utilizing open-source DICOM software (OsiriX MD, version 2.5.1 65-bit) multi-planar reconstruction (MPR).
CT PROTOCOL
All patients underwent a standard glenohumeral CT scan using a Siemens Sensation 64 Scanner (Siemens), a 64-detector scanner. Scans were acquired with 0.6 mm of collimation, 140 kV, and 300 mA-seconds. Slice thickness was set to 2 mm. All patient information was de-identified for analysis.
The uncorrected (UNCORR) scans were defined as the default orientation on the scanner. In the UNCORR scans, the axial, coronal, and sagittal views were oriented relative to the scanner gantry table, as opposed to the anatomy of the glenoid. The corrected (CORR) CT scans were aligned in all 3 planes relative to the glenoid face, and thus the cuts were perpendicular to the long axis of the glenoid.15 This resulted in sagittal cuts perpendicular to the 12-o’clock to 6-o’clock axis in the sagittal plane (Figure 1). 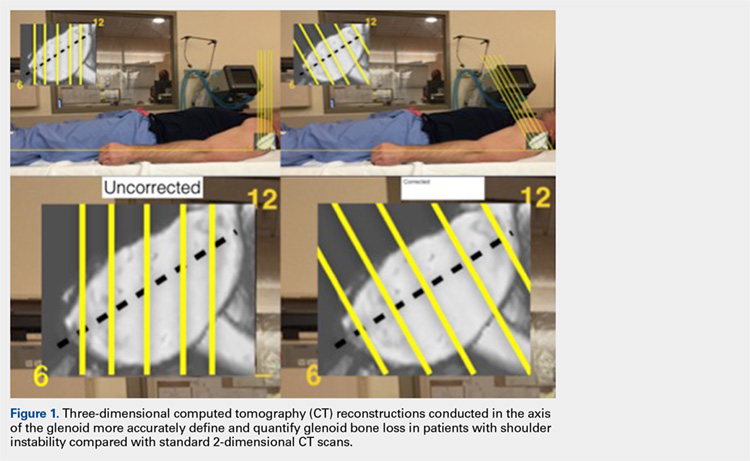
Continue to: In a de-identified fashion...
IMAGE ANALYSIS AND REFORMATTING
In a de-identified fashion, all CT scans were imported and analyzed using open-source Digital Imaging and Communications in Medicine (DICOM) software (OsiriX MD, version 2.5.1 64-bit). By following a previously developed method, CT scans were reformatted using OsiriX MPR. The OsiriX software has an MPR function that allows simultaneous manipulation of 2-D CT scans in 3 orthogonal planes: axial, sagittal, and coronal. In the MPR mode, the alternation of 1 plane directly affects the orientation of the remaining 2 planes. Thus, by using an MPR, one can analyze the impact that a default CT scan performed relative to the gantry of the table, UNCORR, has on the axial images.
First, the en face view was obtained via a 2-step process: alignment of the axial plane to account for the scapular angle, followed by alignment of the coronal plane to adjust for the glenoid inclination.15 These 2 adjustments provided a true en face sagittal glenoid view. The final adjustment step was a sagittal en face rotation of the glenoid such that the superior and inferior glenoid tubercles were placed on the 12-o’clock to 6-o’clock axis (CORR scan). Previous studies have identified a central longitudinal axis that was used in this method to align the supraglenoid tubercle with the 12-o’clock to 6-o’clock axis on the glenoid face.15,17,18 The standard error of mean was 1.21°. This new CORR view resulted in axial cuts through the glenoid that were oriented perpendicular to the 12-o’clock to 6-o’clock axis. The UNCORR and CORR images were assessed in the axial plane at 5 standardized cuts and measured for AP glenoid width by 2 independent observers in a blinded, randomized fashion. When the measured AP width of the UNCORR scan was less than that measured on the CORR scan, the AP width of the glenoid was considered underestimated, and the degree of GBL was considered overestimated (Figure 2). 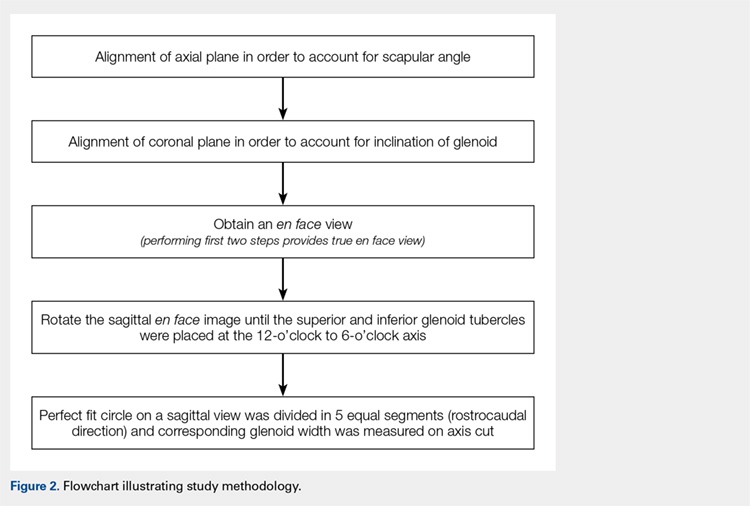
SCAPULAR ANGLE
Scapular angle measurements were performed on the axial view as the angle between a line through the long axis of the body of the scapula, and a line parallel to the CT gantry table.15,19 Subsequently, the axial plane was aligned to the glenoid surface.
CORONAL INCLINATION
Coronal inclination measurements were performed on the sagittal view as the angle between a line tangential to the face of the glenoid and a line perpendicular to the CT gantry table. Positive values represented superior inclination, while negative values represented inferior glenoid inclination.15
SAGITTAL ROTATION
Sagittal rotation measurements were performed using the built-in angle measurement tool in OsiriX in the sagittal plane since the degree of rotation required aligning the long axis of the glenoid to the 12-o’clock to 6-o’clock axis. The amount of rotation was defined as the rotation angle.15
Continue to: Similarly, as described by Gross...
GLENOID WIDTH
Similarly, as described by Gross and colleagues,15 the sagittal en face view was divided via 5 cuts, throughout a superimposed best-fit circle that closely represents the glenoid.9,15,20 For both the UNCORR and CORR, glenoid width (AP distance) was measured on the axial image at the widest point from AP cortex across the glenoid face.
PATIENT GROUPS
Utilizing the en face 3-D CT reconstruction view of the glenoid as the gold standard, patients were placed into 1 of 3 groups according to the degree of anterior GBL measured via the surface method.9,20 The groups were as follows:
I. 10% to 14.9% (N = 12)
II. 15% to 19.9% (N = 16)
III. >20% (N = 15)
STATISTICAL METHODS
Paired t-tests were used to compare all measurements between CORR and UNCORR scans for each of the 5 cuts. A P-value of .05 was used as the threshold for statistical significance in 2-tailed comparisons. Mean and standard errors are presented with standard deviations throughout the study. For interobserver reliability, the measurements between the observers, the intraclass correlation coefficient was calculated. All statistics were performed with SPSS (Version 22).
RESULTS
The study cohort was comprised of 19 left shoulders (44%) and 24 right shoulders (56%), including 36 male patients (84%) and 7 female patients (16%). The average age was 27.8 years (range, 21-40 years). The variability in measured difference, with respect to AP width, was 1.05 mm. The UNCORR CT scans required a mean correction for coronal inclination of 7.0° ± 5.8° (range, -8°-6°). The UNCORR CT scans required a mean correction for scapular angle of 30.2° ± 8.0° (range, 15°-49°). The mean angle of sagittal rotation required to align the glenoid face with the 12-o’clock to 6-o’clock axis was 24.2° ± 5.1 ° (range, 13°-30°). These results are summarized in Table 1.
Table 1. Mean Correction Values Required to Correct the Uncorrected Images to the Corrected Images | |||
Anatomic alignment | Mean (degrees) | Range (degrees) | SD (degrees) |
Scapular angle | 30.2 | 15-49 | 8.0 |
Coronal Inclination | 7.0 | -8-6 | 5.8 |
Sagittal rotation | 24.2 | 13-30 | 5.1 |
For all measurements, the intraclass correlation coefficient for independent observers for all cuts within the 3 groups was r >.900 in all cases.
On an optimized CT scan, over 5 standardized cuts across a best-fit circle of the inferior glenoid, there was a statistically significant absolute mean difference of 12.6% in axial AP glenoid width (2.86 mm ± 2.00 mm, P =.016) when compared with the UNCORR scan. This corresponds to a 3% to 21% error in measurement of the AP width of the glenoid.
Continue to: For the entire cohort...
For the entire cohort of 43 patients, the UNCORR scans underestimated the axial AP width (and thus overestimated GBL) in cut 1 (P =.003), and overestimated the axial AP width (and thus underestimated GBL) in cuts 3 to 5 (P < .001 for all) compared with that of the CORR scans. There was no significant difference between the UNCORR and CORR scans in cut 2 (P = .331).
For groups I (10%-14.9% GBL) and III (>20% GBL), the UNCORR scans underestimated the axial AP width (and thus overestimated anterior GBL) in cuts 1 and 2, while in cuts 3 to 5, the axial AP width was overestimated (GBL was underestimated) (Tables 2, 3). In Group II (15%-19.9% GBL), the axial AP width was underestimated (GBL was overestimated), while in cuts 2 to 5, the axial AP width was overestimated (GBL was underestimated). Overall, AP glenoid width was consistently underestimated in cut 1, the most caudal cut, while AP glenoid width was consistently overestimated in cuts 3 to 5, the more cephalad cuts.
Table 2. Absolute Mean Difference in Axial AP Width (mm) Between Corrected and Uncorrected Images (% difference) | |||||
Cut 1 (Caudal) | Cut 2 | Cut 3 (Center) | Cut 4 | Cut 5 (Cephalad) | |
Group I: 10%-14.9% GBL | 2.4 mm (15.3%) | 1.8 mm (9.0%) | 1.8 mm (7.7%) | 3.0 mm (11.7%) | 4.0 mm (16.8%) |
Group II: 15%-19.9% GBL | 1.8 mm (13.1%) | 1.7 mm (7.9%) | 2.8 mm (10.6%) | 4.1 mm (14.4%) | 4.8 mm (16.9%) |
Group III: >20% | 2.8 mm (16.1%) | 1.9 mm (8.0%) | 2.3 mm (10.3) | 4.4 mm (16.6%) | 5.2 mm (17.0%) |
Abbreviations: AP, anterior-posterior; GBL, glenoid bone loss.
Table 3. Mean AP Glenoid Width Based on CORR and UNCORR Images for the Entire Cohort of 43 Patients | |||||
Axial cut | Mean AP width (mm) | Mean AP width (mm) | Absolute mean AP width difference (mm) | Absolute mean AP width difference (%) | P value |
(Caudal) 1 | 16.6208 | 18.4958 | -1.875 | 14.7768 | .0029565 |
2 | 20.6558 | 21.3166 | -0.661 | 3.6137 | .3310965 |
3 | 24.2583 | 22.3125 | 1.946 | 7.8042 | <.0001 |
4 | 26.1291 | 21.8916 | 4.238 | 15.8449 | <.0001 |
(Rostral) 5 | 26.0875 | 20.4875 | 5.6 | 20.9717 | <.0001 |
Abbreviations: AP, anterior-posterior; CORR, corrected; UNCORR, uncorrected.
DISCUSSION
The principle findings of this study demonstrate that UNCORR conventional 2-D CT scans inaccurately estimate glenoid width as well as inaccurately quantify the degree of anterior GBL. Underestimations of GBL may lead to insufficient treatment of clinically meaningful GBL, thereby increasing the risk of instability recurrence; whereas overestimations of GBL may lead to unnecessary treatment, subjecting patients to increased surgical morbidity. Therefore, the authors recommend correcting the orientation of the scapula in cases wherein clinical decisions are entirely based on 2-D CT, or using alternative methods for quantifying GBL, specifically in the form of 3-D reconstructions.
The use of axial imaging, with CT scans and/or magnetic resonance imaging, is growing in popularity for evaluation of both glenoid anatomy and GBL. Nevertheless, despite our improved ability to critically evaluate the glenoid using these advanced imaging modalities, the images themselves require scrutiny by clinicians to determine if the images accurately depict the true anatomy of the glenoid. As demonstrated by Gross and colleagues,15 conventional 2D CT scan protocols are not optimized to the anatomy of the glenohumeral joint, even in patients without GBL. Due to the alignment of the image relative to the plane of the scapula as opposed to the plane of the glenoid, UNCORR scans result in significantly different measurements of glenoid version (2.0° ± 0.1°) and AP glenoid width (1.2 mm ± 0.42 mm) compared with corrected scans, requiring an average 20.1° ± 1.2° of correction to align the sagittal plane. In the present study involving the patients with GBL, we also found that conventional, UNCORR 2-D CT scan protocols inaccurately estimate glenoid width and the degree of anterior GBL. In particular, AP glenoid width was consistently underestimated in the more caudal cuts, while AP glenoid width was consistently overestimated in the more cephalad cuts. Thus, anterior GBL was overestimated (AP glenoid width was underestimated) in the more caudal cuts, whereas anterior GBL was underestimated in the more cranial cuts (AP glenoid width was overestimated). Given that approximately 1 mm of glenoid bone corresponds to approximately 4% of glenoid width,16 even subtle differences in the interpretation of GBL may lead to gross overestimation/underestimation of bone loss, with significant clinical implications.
In the anterior instability patient population, clinical decision-making is often based on the degree of GBL as determined by advanced imaging modalities. In addition to other patient-specific factors, including age, gender, activity level, type of sport, and number of prior dislocations and/or prior surgeries, the quantity of GBL will often determine which surgical procedure needs to be performed. Typically, patients with >20% to 25% anterior GBL are indicated for a glenoid reconstruction procedure, most commonly via the Latarjet procedure (coracoid transfer).21-27 The Latarjet procedure remains an excellent technique for appropriately indicated patients, with historically good clinical outcomes and low recurrence rates. Complications associated with the Latarjet procedure, however, are not uncommon, including devastating neuropraxia of the axillary and musculocutaneous nerves, and occasionally permanent neurologic deficits.28 Thus, it is critical to avoid overtreating patients with recurrent instability and GBL. As demonstrated by this study, depending on the cranial-to-caudal location on the glenoid, current 2-D CT techniques may underestimate AP glenoid width, resulting in an overestimation of GBL, potentially leading to the decision to proceed with glenoid bone reconstruction when such a procedure is not required. On the contrary, overestimation of AP glenoid width, which occurs in the more cephalad cuts of the glenoid, is perhaps more worrisome, as the resulting underestimation of GBL may lead to inadequate treatment of patients with recurrent instability. Certainly, one of the main risk factors for failed soft tissue shoulder stabilization is a failure to address GBL. If clinical decisions are made based on UNCORR 2-D CT scans, which are often inaccurate with respect to AP glenoid width by an average 2.86 mm ± 2.00 mm (equivalent to 12.6% ± 6.9% GBL) as determined in this study, patients who truly require osseous glenoid reconstructions may be indicated for only soft tissue stabilization, based on the underestimation of GBL.
Continue to: The current gold standard...
The current gold standard for GBL measurement is a perfect-fit circle performed on a 3-D CT scan.22 To that end, it would have been useful to measure the glenoids from this study on 3-D CT scans and compare the data with both UNCORR and CORR measurements. This would have provided a better understanding to what extent the CORR measurements on 2-D scans are relatable with the gold standard. As 3-D CT scans provide a better en face view of the glenoid, more accurate GBL measurements, and ease of 3-D manipulation, they have become more widely used across the country.29,30 Nevertheless, in situations where 3-D imaging is more challenging to obtain because of technology or cost limitations, having a strategy for ensuring proper orientation of 2-D scans would have a substantial impact on clinical decision-making. If such corrections are not made, the inaccuracy of current 2-D scanning protocols justifies the cost 3-D reconstruction protocols. The difference in GBL measurements are critical in cases of increasingly large degrees of GBL, as in these instances, the inferior glenoid becomes more of an inverted-pear shape as opposed to a perfect circle, and differences in CORR and UNCORR images are likely to be more profound.
LIMITATIONS
This study has limitations, such as the relatively small sample size and the selection bias by the reviewers with potential differences in interobserver reliability. Further, minor modifications during the reformatting process may be found with each attempt to manipulate the images and may result in minor, insignificant differences in AP width measurements. Performing 1 or more additional CT scans on the same cohort of patients would have been helpful; however, due to the increased risk of radiation exposure, this was not performed. Performing CT scans on cadaveric specimens with GBL and applying the study methodology would also have been helpful to provide independent verification of our clinical findings; however, specimens were not available for this study. Another limitation of this study is that we did not compare our findings with the findings of glenoid width, and bone loss, as determined using the circle method, which is commonly utilized when 3-D reconstructions are available. In this study, the purpose was to utilize only the 2-D reformatted images, with the assumption that 3-D reconstructions are not always available, and cannot always be measured. To minimize selection bias, the investigators measured the correction effects within groups of patients with similar degrees of GBL (10%-14.9%, 15%-19.9%, and >20%). In addition, not all the selected patients showed degenerative glenoid changes or irregular glenoid shape indicating previous bone augmentation.
CONCLUSIONS
UNCORR 2D CT scans inaccurately estimate glenoid width and the degree of anterior GBL. The clinical implications of these findings are profound and suggest corrected 2D CT scans or 3D reconstruction allow measurements to be taken in the axis of the glenoid to accurately define the anatomy and quantity of anterior GBL in patients with shoulder instability.
1. Cerciello S, Edwards TB, Walch G. Chronic anterior glenohumeral instability in soccer players: results for a series of 28 shoulders treated with the Latarjet procedure. J Orthop Traumatol. 2012;13(4):197-202. doi:10.1007/s10195-012-0201-3.
2. Itoi E, Lee SB, Berglund LJ, Berge LL, An KN. The effect of a glenoid defect on anteroinferior stability of the shoulder after Bankart repair: a cadaveric study. J Bone Joint Surg Am. 2000;82(1):35-46.
3. Bhatia S, Ghodadra NS, Romeo AA, et al. The importance of the recognition and treatment of glenoid bone loss in an athletic population. Sports Health. 2011;3(5):435-440. doi:10.1177/1941738111414126.
4. Lo IK, Parten PM, Burkhart SS. The inverted pear glenoid: an indicator of significant glenoid bone loss. Arthroscopy. 2004;20(2):169-174. doi:10.1016/j.arthro.2003.11.036.
5. Mologne TS, Provencher MT, Menzel KA, Vachon TA, Dewing CB. Arthroscopic stabilization in patients with an inverted pear glenoid: results in patients with bone loss of the anterior glenoid. Am J Sports Med. 2007;35(8):1276-1283. doi:10.1177/0363546507300262.
6. Piasecki DP, Verma NN, Romeo AA, Levine WN, Bach BR Jr, Provencher MT. Glenoid bone deficiency in recurrent anterior shoulder instability: diagnosis and management. J Am Acad Orthop Surg. 2009;17(8):482-493.
7. Provencher MT, Bhatia S, Ghodadra NS, et al. Recurrent shoulder instability: current concepts for evaluation and management of glenoid bone loss. J Bone Joint Surg Am. 2010;92(suppl 2):133-151. doi:10.2106/JBJS.J.00906.
8. Rowe CR, Zarins B, Ciullo JV. Recurrent anterior dislocation of the shoulder after surgical repair. Apparent causes of failure and treatment. J Bone Joint Surg Am. 1984;66(2):159-168.
9. Sugaya H, Moriishi J, Dohi M, Kon Y, Tsuchiya A. Glenoid rim morphology in recurrent anterior glenohumeral instability. J Bone Joint Surg Am. 2003;85-A(5):878-884.
10. Edwards TB, Boulahia A, Walch G. Radiographic analysis of bone defects in chronic anterior shoulder instability. Arthroscopy. 2003;19(7):732-739.
11. Jankauskas L, Rudiger HA, Pfirrmann CW, Jost B, Gerber C. Loss of the sclerotic line of the glenoid on anteroposterior radiographs of the shoulder: a diagnostic sign for an osseous defect of the anterior glenoid rim. J Shoulder Elbow Surg. 2010;19(1):151-156. doi:10.1016/j.jse.2009.04.013.
12. Altan E, Ozbaydar MU, Tonbul M, Yalcin L. Comparison of two different measurement methods to determine glenoid bone defects: area or width? J Shoulder Elbow Surg. 2014;23(8):1215-1222. doi:10.1016/j.jse.2013.11.029.
13. Bishop JY, Jones GL, Rerko MA, Donaldson C, Group MS. 3-D CT is the most reliable imaging modality when quantifying glenoid bone loss. Clin Orthop Relat Res. 2013;471(4):1251-1256. doi:10.1007/s11999-012-2607-x.
14. Chuang TY, Adams CR, Burkhart SS. Use of preoperative three-dimensional computed tomography to quantify glenoid bone loss in shoulder instability. Arthroscopy. 2008; 24(4):376-382. doi:10.1016/j.arthro.2007.10.008.
15. Gross DJ, Golijanin P, Dumont GD, et al. The effect of sagittal rotation of the glenoid on axial glenoid width and glenoid version in computed tomography scan imaging. J Shoulder Elbow Surg. 2016;25(1):61-68. doi:10.1016/j.jse.2015.06.017.
16. Lenart BA, Freedman R, Van Thiel GS, et al. Magnetic resonance imaging evaluation of normal glenoid length and width: an anatomic study. Arthroscopy. 2014;30(8):915-920. doi:10.1016/j.arthro.2014.03.006.
17. Bois AJ, Fening SD, Polster J, Jones MH, Miniaci A. Quantifying glenoid bone loss in anterior shoulder instability: reliability and accuracy of 2-dimensional and 3-dimensional computed tomography measurement techniques. Am J Sports Med. 2012;40(11):2569-2577. doi:10.1177/0363546512458247.
18. Griffith JF, Antonio GE, Tong CW, Ming CK. Anterior shoulder dislocation: quantification of glenoid bone loss with CT. AJR Am J Roentgenol. 2003;180(5):1423-1430. doi:10.2214/ajr.180.5.1801423.
19. Hoenecke HR Jr, Hermida JC, Flores-Hernandez C, D'Lima DD. Accuracy of CT-based measurements of glenoid version for total shoulder arthroplasty. J Shoulder Elbow Surg. 2010;19(2):166-171. doi:10.1016/j.jse.2009.08.009.
20. Huijsmans PE, de Witte PB, de Villiers RV, et al. Recurrent anterior shoulder instability: accuracy of estimations of glenoid bone loss with computed tomography is insufficient for therapeutic decision-making. Skeletal Radiol. 2011;40(10):1329-1334. doi:10.1007/s00256-011-1184-5.
21. Bhatia S, Frank RM, Ghodadra NS, et al. The outcomes and surgical techniques of the latarjet procedure. Arthroscopy. 2014;30(2):227-235. doi:10.1016/j.arthro.2013.10.013.
22. Cunningham G, Benchouk S, Kherad O, Ladermann A. Comparison of arthroscopic and open Latarjet with a learning curve analysis. Knee Surg Sports Traumatol Arthrosc. 2015;24(2):540-545. doi:10.1007/s00167-015-3910-3.
23. Fedorka CJ, Mulcahey MK. Recurrent anterior shoulder instability: a review of the Latarjet procedure and its postoperative rehabilitation. Phys Sportsmed. 2015;43(1):73-79. doi:10.1080/00913847.2015.1005543.
24. Flinkkila T, Sirniö K. Open Latarjet procedure for failed arthroscopic Bankart repair. Orthop Traumatol Surg Res. 2015;101(1):35-38. doi:10.1016/j.otsr.2014.11.005.
25. Hovelius L, Sandström B, Saebö M. One hundred eighteen Bristow-Latarjet repairs for recurrent anterior dislocation of the shoulder prospectively followed for fifteen years: study II-the evolution of dislocation arthropathy. J Shoulder Elbow Surg. 2006;15(3):279-289. doi:10.1016/j.jse.2005.09.014.
26. Hovelius L, Sandström B, Sundgren K, Saebö M. One hundred eighteen Bristow-Latarjet repairs for recurrent anterior dislocation of the shoulder prospectively followed for fifteen years: study I--clinical results. J Shoulder Elbow Surg. 2004;13(5):509-516. doi:10.1016/S1058274604000916.
27. Hovelius L, Vikerfors O, Olofsson A, Svensson O, Rahme H. Bristow-Latarjet and Bankart: a comparative study of shoulder stabilization in 185 shoulders during a seventeen-year follow-up. J Shoulder Elbow Surg. 2011;20(7):1095-1101. doi:10.1016/j.jse.2011.02.005.
28. Gupta A, Delaney R, Petkin K, Lafosse L. Complications of the Latarjet procedure. Curr Rev Musculoskelet Med. 2015;8(1):59-66. doi:10.1007/s12178-015-9258-y.
29. Kwon YW, Powell KA, Yum JK, Brems JJ, Iannotti JP. Use of three-dimensional computed tomography for the analysis of the glenoid anatomy. J Shoulder Elbow Surg. 2005;14(1):85-90. doi:10.1016/j.jse.2004.04.011.
30. Saito H, Itoi E, Sugaya H, Minagawa H, Yamamoto N, Tuoheti Y. Location of the glenoid defect in shoulders with recurrent anterior dislocation. Am J Sports Med. 2005;33(6):889-893. doi:10.1177/0363546504271521.
ABSTRACT
Standard 2-dimensional (2-D) computed tomography (CT) scans of the shoulder are often aligned to the plane of the body as opposed to the plane of the scapula, which may challenge the ability to accurately measure glenoid width and glenoid bone loss (GBL). The purpose of this study is to determine the effect of sagittal rotation of the glenoid on axial anterior-posterior (AP) glenoid width measurements in the setting of anterior GBL.
Forty-three CT scans from consecutive patients with anterior GBL (minimum 10%) were reformatted utilizing open-source DICOM software (OsiriX MD). Patients were grouped according to extent of GBL: I, 10% to 14.9% (N = 12); II, 15% to 19.9% (N = 16); and III, >20% (N = 15). The uncorrected (UNCORR) and corrected (CORR) images were assessed in the axial plane at 5 standardized cuts and measured for AP glenoid width.
For groups I and III, UNCORR scans underestimated axial AP width (and thus overestimated anterior GBL) in cuts 1 and 2, while in cuts 3 to 5, the axial AP width was overestimated (GBL was underestimated). In Group II, axial AP width was underestimated (GBL was overestimated), while in cuts 2 to 5, the axial AP width was overestimated (GBL was underestimated). Overall, AP glenoid width was consistently underestimated in cut 1, the most caudal cut; while AP glenoid width was consistently overestimated in cuts 3 to 5, the more cephalad cuts.
UNCORR 2-D CT scans inaccurately estimated glenoid width and the degree of anterior GBL. This data suggests that corrected 2D CT scans or a 3-dimensional (3-D) reconstruction can help in accurately defining the anterior GBL in patients with shoulder instability.
The treatment of glenohumeral instability has substantially evolved over the past several decades. The understanding of glenoid bone loss (GBL), in particular, has advanced to such a level that we utilize the quantification of GBL for surgical decision-making. Unrecognized and/or untreated GBL is associated with recurrent instability, pain, and disability. Controversy exists, however, regarding the precise amount of anterior GBL that is significant enough to warrant surgical treatment. While historically, 25%1,2 of anterior GBL was thought to be the critical number required to warrant osseous augmentation, studies that are more recent have highlighted the need to perform osseous glenoid reconstruction with lesser degrees of GBL, particularly in the contact athlete.3-9 As small differences in the amount of GBL can change surgical decision-making from an all-soft tissue repair to an osseous reconstruction, it is paramount that we have accurate, valid, and reproducible methods for calculating GBL.
Continue to: Historically, plain radiographs...
Historically, plain radiographs have been the mainstay for evaluating the glenohumeral joint, including Grashey and axillary views, allowing clinicians to evaluate the congruency of the glenohumeral joint and to assess bone loss on both the glenoid and humeral head.1,10 While large, acute fractures of the glenoid are fairly evident on radiographs, including the Grashey view,11 shoulders with chronic and/or attritional anterior GBL are more difficult to evaluate, and often do not provide the information necessary to guide surgical decision-making.
Computed tomography (CT) of the shoulder has become the most commonly utilized imaging modality in the evaluation of patients with shoulder instability associated with GBL. Standard 2-dimensional (2-D) CT scans of the shoulder are often aligned to the plane of the body as opposed to the plane of the scapula/glenoid, as standard protocols often fail to account for the anterior sagittal rotation of the scapula/glenoid, similar to the disadvantage of standard radiographs. While 3-dimensional (3-D) CT reconstructions eliminate the effect of gantry angles, and thus allow for an en face view of the glenoid, 3-D reconstructions are not always available, and cannot always be measured.12-14 Thus, improved methodology for utilizing standard 2D scans is warranted, as the ability to correctly align the axial CT scan to the axis of the glenoid may allow for more accurate GBL measurements, which will ultimately impact surgical decision-making. Recently, Gross and colleagues15 reported the effect of sagittal rotation of the glenoid on axial measurements of anterior-posterior (AP) glenoid width and glenoid version in normal glenoids, without bone loss, and found that the mean angle of correction needed to align the sagittal plane was 20.1° ± 1.2° of rotation. To the authors’ knowledge, this same methodology has not been applied to patients with clinically meaningful anterior GBL. Given that the average glenoid width in human shoulders is 24.4 mm ± 2.9 mm,16 1 mm of glenoid bone loss (GBL) corresponds to approximately 4% of the glenoid width, and thus even subtle differences in the interpretation of GBL may have substantial clinical implications. Therefore, the purpose of this study is to determine the effect of sagittal rotation of the glenoid on axial AP glenoid width measurements in the setting of clinically significant anterior GBL.
METHODS
This study was approved by Massachusetts General Hospital Institutional Review Board. A retrospective review of consecutive patients with a diagnosis of anterior shoulder instability between 2009 and 2013 was conducted. Inclusion criteria comprised patients with a minimum of 10% anterior GBL, an available CT scan of the affected shoulder, and no history of prior ipsilateral surgeries. Exclusion criteria comprised evidence of degenerative changes to the glenoid and/or humeral head, as well as prior ipsilateral shoulder surgery. Sixty consecutive patients were originally identified as having anterior shoulder instability, and 17 were excluded based on the inclusion/exclusion criteria, leaving 43 patients (43 shoulders) available for inclusion. Shoulder CT scans from all 43 patients were reformatted utilizing open-source DICOM software (OsiriX MD, version 2.5.1 65-bit) multi-planar reconstruction (MPR).
CT PROTOCOL
All patients underwent a standard glenohumeral CT scan using a Siemens Sensation 64 Scanner (Siemens), a 64-detector scanner. Scans were acquired with 0.6 mm of collimation, 140 kV, and 300 mA-seconds. Slice thickness was set to 2 mm. All patient information was de-identified for analysis.
The uncorrected (UNCORR) scans were defined as the default orientation on the scanner. In the UNCORR scans, the axial, coronal, and sagittal views were oriented relative to the scanner gantry table, as opposed to the anatomy of the glenoid. The corrected (CORR) CT scans were aligned in all 3 planes relative to the glenoid face, and thus the cuts were perpendicular to the long axis of the glenoid.15 This resulted in sagittal cuts perpendicular to the 12-o’clock to 6-o’clock axis in the sagittal plane (Figure 1).
Continue to: In a de-identified fashion...
IMAGE ANALYSIS AND REFORMATTING
In a de-identified fashion, all CT scans were imported and analyzed using open-source Digital Imaging and Communications in Medicine (DICOM) software (OsiriX MD, version 2.5.1 64-bit). By following a previously developed method, CT scans were reformatted using OsiriX MPR. The OsiriX software has an MPR function that allows simultaneous manipulation of 2-D CT scans in 3 orthogonal planes: axial, sagittal, and coronal. In the MPR mode, the alternation of 1 plane directly affects the orientation of the remaining 2 planes. Thus, by using an MPR, one can analyze the impact that a default CT scan performed relative to the gantry of the table, UNCORR, has on the axial images.
First, the en face view was obtained via a 2-step process: alignment of the axial plane to account for the scapular angle, followed by alignment of the coronal plane to adjust for the glenoid inclination.15 These 2 adjustments provided a true en face sagittal glenoid view. The final adjustment step was a sagittal en face rotation of the glenoid such that the superior and inferior glenoid tubercles were placed on the 12-o’clock to 6-o’clock axis (CORR scan). Previous studies have identified a central longitudinal axis that was used in this method to align the supraglenoid tubercle with the 12-o’clock to 6-o’clock axis on the glenoid face.15,17,18 The standard error of mean was 1.21°. This new CORR view resulted in axial cuts through the glenoid that were oriented perpendicular to the 12-o’clock to 6-o’clock axis. The UNCORR and CORR images were assessed in the axial plane at 5 standardized cuts and measured for AP glenoid width by 2 independent observers in a blinded, randomized fashion. When the measured AP width of the UNCORR scan was less than that measured on the CORR scan, the AP width of the glenoid was considered underestimated, and the degree of GBL was considered overestimated (Figure 2).
SCAPULAR ANGLE
Scapular angle measurements were performed on the axial view as the angle between a line through the long axis of the body of the scapula, and a line parallel to the CT gantry table.15,19 Subsequently, the axial plane was aligned to the glenoid surface.
CORONAL INCLINATION
Coronal inclination measurements were performed on the sagittal view as the angle between a line tangential to the face of the glenoid and a line perpendicular to the CT gantry table. Positive values represented superior inclination, while negative values represented inferior glenoid inclination.15
SAGITTAL ROTATION
Sagittal rotation measurements were performed using the built-in angle measurement tool in OsiriX in the sagittal plane since the degree of rotation required aligning the long axis of the glenoid to the 12-o’clock to 6-o’clock axis. The amount of rotation was defined as the rotation angle.15
Continue to: Similarly, as described by Gross...
GLENOID WIDTH
Similarly, as described by Gross and colleagues,15 the sagittal en face view was divided via 5 cuts, throughout a superimposed best-fit circle that closely represents the glenoid.9,15,20 For both the UNCORR and CORR, glenoid width (AP distance) was measured on the axial image at the widest point from AP cortex across the glenoid face.
PATIENT GROUPS
Utilizing the en face 3-D CT reconstruction view of the glenoid as the gold standard, patients were placed into 1 of 3 groups according to the degree of anterior GBL measured via the surface method.9,20 The groups were as follows:
I. 10% to 14.9% (N = 12)
II. 15% to 19.9% (N = 16)
III. >20% (N = 15)
STATISTICAL METHODS
Paired t-tests were used to compare all measurements between CORR and UNCORR scans for each of the 5 cuts. A P-value of .05 was used as the threshold for statistical significance in 2-tailed comparisons. Mean and standard errors are presented with standard deviations throughout the study. For interobserver reliability, the measurements between the observers, the intraclass correlation coefficient was calculated. All statistics were performed with SPSS (Version 22).
RESULTS
The study cohort was comprised of 19 left shoulders (44%) and 24 right shoulders (56%), including 36 male patients (84%) and 7 female patients (16%). The average age was 27.8 years (range, 21-40 years). The variability in measured difference, with respect to AP width, was 1.05 mm. The UNCORR CT scans required a mean correction for coronal inclination of 7.0° ± 5.8° (range, -8°-6°). The UNCORR CT scans required a mean correction for scapular angle of 30.2° ± 8.0° (range, 15°-49°). The mean angle of sagittal rotation required to align the glenoid face with the 12-o’clock to 6-o’clock axis was 24.2° ± 5.1 ° (range, 13°-30°). These results are summarized in Table 1.
Table 1. Mean Correction Values Required to Correct the Uncorrected Images to the Corrected Images | |||
Anatomic alignment | Mean (degrees) | Range (degrees) | SD (degrees) |
Scapular angle | 30.2 | 15-49 | 8.0 |
Coronal Inclination | 7.0 | -8-6 | 5.8 |
Sagittal rotation | 24.2 | 13-30 | 5.1 |
For all measurements, the intraclass correlation coefficient for independent observers for all cuts within the 3 groups was r >.900 in all cases.
On an optimized CT scan, over 5 standardized cuts across a best-fit circle of the inferior glenoid, there was a statistically significant absolute mean difference of 12.6% in axial AP glenoid width (2.86 mm ± 2.00 mm, P =.016) when compared with the UNCORR scan. This corresponds to a 3% to 21% error in measurement of the AP width of the glenoid.
Continue to: For the entire cohort...
For the entire cohort of 43 patients, the UNCORR scans underestimated the axial AP width (and thus overestimated GBL) in cut 1 (P =.003), and overestimated the axial AP width (and thus underestimated GBL) in cuts 3 to 5 (P < .001 for all) compared with that of the CORR scans. There was no significant difference between the UNCORR and CORR scans in cut 2 (P = .331).
For groups I (10%-14.9% GBL) and III (>20% GBL), the UNCORR scans underestimated the axial AP width (and thus overestimated anterior GBL) in cuts 1 and 2, while in cuts 3 to 5, the axial AP width was overestimated (GBL was underestimated) (Tables 2, 3). In Group II (15%-19.9% GBL), the axial AP width was underestimated (GBL was overestimated), while in cuts 2 to 5, the axial AP width was overestimated (GBL was underestimated). Overall, AP glenoid width was consistently underestimated in cut 1, the most caudal cut, while AP glenoid width was consistently overestimated in cuts 3 to 5, the more cephalad cuts.
Table 2. Absolute Mean Difference in Axial AP Width (mm) Between Corrected and Uncorrected Images (% difference) | |||||
Cut 1 (Caudal) | Cut 2 | Cut 3 (Center) | Cut 4 | Cut 5 (Cephalad) | |
Group I: 10%-14.9% GBL | 2.4 mm (15.3%) | 1.8 mm (9.0%) | 1.8 mm (7.7%) | 3.0 mm (11.7%) | 4.0 mm (16.8%) |
Group II: 15%-19.9% GBL | 1.8 mm (13.1%) | 1.7 mm (7.9%) | 2.8 mm (10.6%) | 4.1 mm (14.4%) | 4.8 mm (16.9%) |
Group III: >20% | 2.8 mm (16.1%) | 1.9 mm (8.0%) | 2.3 mm (10.3) | 4.4 mm (16.6%) | 5.2 mm (17.0%) |
Abbreviations: AP, anterior-posterior; GBL, glenoid bone loss.
Table 3. Mean AP Glenoid Width Based on CORR and UNCORR Images for the Entire Cohort of 43 Patients | |||||
Axial cut | Mean AP width (mm) | Mean AP width (mm) | Absolute mean AP width difference (mm) | Absolute mean AP width difference (%) | P value |
(Caudal) 1 | 16.6208 | 18.4958 | -1.875 | 14.7768 | .0029565 |
2 | 20.6558 | 21.3166 | -0.661 | 3.6137 | .3310965 |
3 | 24.2583 | 22.3125 | 1.946 | 7.8042 | <.0001 |
4 | 26.1291 | 21.8916 | 4.238 | 15.8449 | <.0001 |
(Rostral) 5 | 26.0875 | 20.4875 | 5.6 | 20.9717 | <.0001 |
Abbreviations: AP, anterior-posterior; CORR, corrected; UNCORR, uncorrected.
DISCUSSION
The principle findings of this study demonstrate that UNCORR conventional 2-D CT scans inaccurately estimate glenoid width as well as inaccurately quantify the degree of anterior GBL. Underestimations of GBL may lead to insufficient treatment of clinically meaningful GBL, thereby increasing the risk of instability recurrence; whereas overestimations of GBL may lead to unnecessary treatment, subjecting patients to increased surgical morbidity. Therefore, the authors recommend correcting the orientation of the scapula in cases wherein clinical decisions are entirely based on 2-D CT, or using alternative methods for quantifying GBL, specifically in the form of 3-D reconstructions.
The use of axial imaging, with CT scans and/or magnetic resonance imaging, is growing in popularity for evaluation of both glenoid anatomy and GBL. Nevertheless, despite our improved ability to critically evaluate the glenoid using these advanced imaging modalities, the images themselves require scrutiny by clinicians to determine if the images accurately depict the true anatomy of the glenoid. As demonstrated by Gross and colleagues,15 conventional 2D CT scan protocols are not optimized to the anatomy of the glenohumeral joint, even in patients without GBL. Due to the alignment of the image relative to the plane of the scapula as opposed to the plane of the glenoid, UNCORR scans result in significantly different measurements of glenoid version (2.0° ± 0.1°) and AP glenoid width (1.2 mm ± 0.42 mm) compared with corrected scans, requiring an average 20.1° ± 1.2° of correction to align the sagittal plane. In the present study involving the patients with GBL, we also found that conventional, UNCORR 2-D CT scan protocols inaccurately estimate glenoid width and the degree of anterior GBL. In particular, AP glenoid width was consistently underestimated in the more caudal cuts, while AP glenoid width was consistently overestimated in the more cephalad cuts. Thus, anterior GBL was overestimated (AP glenoid width was underestimated) in the more caudal cuts, whereas anterior GBL was underestimated in the more cranial cuts (AP glenoid width was overestimated). Given that approximately 1 mm of glenoid bone corresponds to approximately 4% of glenoid width,16 even subtle differences in the interpretation of GBL may lead to gross overestimation/underestimation of bone loss, with significant clinical implications.
In the anterior instability patient population, clinical decision-making is often based on the degree of GBL as determined by advanced imaging modalities. In addition to other patient-specific factors, including age, gender, activity level, type of sport, and number of prior dislocations and/or prior surgeries, the quantity of GBL will often determine which surgical procedure needs to be performed. Typically, patients with >20% to 25% anterior GBL are indicated for a glenoid reconstruction procedure, most commonly via the Latarjet procedure (coracoid transfer).21-27 The Latarjet procedure remains an excellent technique for appropriately indicated patients, with historically good clinical outcomes and low recurrence rates. Complications associated with the Latarjet procedure, however, are not uncommon, including devastating neuropraxia of the axillary and musculocutaneous nerves, and occasionally permanent neurologic deficits.28 Thus, it is critical to avoid overtreating patients with recurrent instability and GBL. As demonstrated by this study, depending on the cranial-to-caudal location on the glenoid, current 2-D CT techniques may underestimate AP glenoid width, resulting in an overestimation of GBL, potentially leading to the decision to proceed with glenoid bone reconstruction when such a procedure is not required. On the contrary, overestimation of AP glenoid width, which occurs in the more cephalad cuts of the glenoid, is perhaps more worrisome, as the resulting underestimation of GBL may lead to inadequate treatment of patients with recurrent instability. Certainly, one of the main risk factors for failed soft tissue shoulder stabilization is a failure to address GBL. If clinical decisions are made based on UNCORR 2-D CT scans, which are often inaccurate with respect to AP glenoid width by an average 2.86 mm ± 2.00 mm (equivalent to 12.6% ± 6.9% GBL) as determined in this study, patients who truly require osseous glenoid reconstructions may be indicated for only soft tissue stabilization, based on the underestimation of GBL.
Continue to: The current gold standard...
The current gold standard for GBL measurement is a perfect-fit circle performed on a 3-D CT scan.22 To that end, it would have been useful to measure the glenoids from this study on 3-D CT scans and compare the data with both UNCORR and CORR measurements. This would have provided a better understanding to what extent the CORR measurements on 2-D scans are relatable with the gold standard. As 3-D CT scans provide a better en face view of the glenoid, more accurate GBL measurements, and ease of 3-D manipulation, they have become more widely used across the country.29,30 Nevertheless, in situations where 3-D imaging is more challenging to obtain because of technology or cost limitations, having a strategy for ensuring proper orientation of 2-D scans would have a substantial impact on clinical decision-making. If such corrections are not made, the inaccuracy of current 2-D scanning protocols justifies the cost 3-D reconstruction protocols. The difference in GBL measurements are critical in cases of increasingly large degrees of GBL, as in these instances, the inferior glenoid becomes more of an inverted-pear shape as opposed to a perfect circle, and differences in CORR and UNCORR images are likely to be more profound.
LIMITATIONS
This study has limitations, such as the relatively small sample size and the selection bias by the reviewers with potential differences in interobserver reliability. Further, minor modifications during the reformatting process may be found with each attempt to manipulate the images and may result in minor, insignificant differences in AP width measurements. Performing 1 or more additional CT scans on the same cohort of patients would have been helpful; however, due to the increased risk of radiation exposure, this was not performed. Performing CT scans on cadaveric specimens with GBL and applying the study methodology would also have been helpful to provide independent verification of our clinical findings; however, specimens were not available for this study. Another limitation of this study is that we did not compare our findings with the findings of glenoid width, and bone loss, as determined using the circle method, which is commonly utilized when 3-D reconstructions are available. In this study, the purpose was to utilize only the 2-D reformatted images, with the assumption that 3-D reconstructions are not always available, and cannot always be measured. To minimize selection bias, the investigators measured the correction effects within groups of patients with similar degrees of GBL (10%-14.9%, 15%-19.9%, and >20%). In addition, not all the selected patients showed degenerative glenoid changes or irregular glenoid shape indicating previous bone augmentation.
CONCLUSIONS
UNCORR 2D CT scans inaccurately estimate glenoid width and the degree of anterior GBL. The clinical implications of these findings are profound and suggest corrected 2D CT scans or 3D reconstruction allow measurements to be taken in the axis of the glenoid to accurately define the anatomy and quantity of anterior GBL in patients with shoulder instability.
ABSTRACT
Standard 2-dimensional (2-D) computed tomography (CT) scans of the shoulder are often aligned to the plane of the body as opposed to the plane of the scapula, which may challenge the ability to accurately measure glenoid width and glenoid bone loss (GBL). The purpose of this study is to determine the effect of sagittal rotation of the glenoid on axial anterior-posterior (AP) glenoid width measurements in the setting of anterior GBL.
Forty-three CT scans from consecutive patients with anterior GBL (minimum 10%) were reformatted utilizing open-source DICOM software (OsiriX MD). Patients were grouped according to extent of GBL: I, 10% to 14.9% (N = 12); II, 15% to 19.9% (N = 16); and III, >20% (N = 15). The uncorrected (UNCORR) and corrected (CORR) images were assessed in the axial plane at 5 standardized cuts and measured for AP glenoid width.
For groups I and III, UNCORR scans underestimated axial AP width (and thus overestimated anterior GBL) in cuts 1 and 2, while in cuts 3 to 5, the axial AP width was overestimated (GBL was underestimated). In Group II, axial AP width was underestimated (GBL was overestimated), while in cuts 2 to 5, the axial AP width was overestimated (GBL was underestimated). Overall, AP glenoid width was consistently underestimated in cut 1, the most caudal cut; while AP glenoid width was consistently overestimated in cuts 3 to 5, the more cephalad cuts.
UNCORR 2-D CT scans inaccurately estimated glenoid width and the degree of anterior GBL. This data suggests that corrected 2D CT scans or a 3-dimensional (3-D) reconstruction can help in accurately defining the anterior GBL in patients with shoulder instability.
The treatment of glenohumeral instability has substantially evolved over the past several decades. The understanding of glenoid bone loss (GBL), in particular, has advanced to such a level that we utilize the quantification of GBL for surgical decision-making. Unrecognized and/or untreated GBL is associated with recurrent instability, pain, and disability. Controversy exists, however, regarding the precise amount of anterior GBL that is significant enough to warrant surgical treatment. While historically, 25%1,2 of anterior GBL was thought to be the critical number required to warrant osseous augmentation, studies that are more recent have highlighted the need to perform osseous glenoid reconstruction with lesser degrees of GBL, particularly in the contact athlete.3-9 As small differences in the amount of GBL can change surgical decision-making from an all-soft tissue repair to an osseous reconstruction, it is paramount that we have accurate, valid, and reproducible methods for calculating GBL.
Continue to: Historically, plain radiographs...
Historically, plain radiographs have been the mainstay for evaluating the glenohumeral joint, including Grashey and axillary views, allowing clinicians to evaluate the congruency of the glenohumeral joint and to assess bone loss on both the glenoid and humeral head.1,10 While large, acute fractures of the glenoid are fairly evident on radiographs, including the Grashey view,11 shoulders with chronic and/or attritional anterior GBL are more difficult to evaluate, and often do not provide the information necessary to guide surgical decision-making.
Computed tomography (CT) of the shoulder has become the most commonly utilized imaging modality in the evaluation of patients with shoulder instability associated with GBL. Standard 2-dimensional (2-D) CT scans of the shoulder are often aligned to the plane of the body as opposed to the plane of the scapula/glenoid, as standard protocols often fail to account for the anterior sagittal rotation of the scapula/glenoid, similar to the disadvantage of standard radiographs. While 3-dimensional (3-D) CT reconstructions eliminate the effect of gantry angles, and thus allow for an en face view of the glenoid, 3-D reconstructions are not always available, and cannot always be measured.12-14 Thus, improved methodology for utilizing standard 2D scans is warranted, as the ability to correctly align the axial CT scan to the axis of the glenoid may allow for more accurate GBL measurements, which will ultimately impact surgical decision-making. Recently, Gross and colleagues15 reported the effect of sagittal rotation of the glenoid on axial measurements of anterior-posterior (AP) glenoid width and glenoid version in normal glenoids, without bone loss, and found that the mean angle of correction needed to align the sagittal plane was 20.1° ± 1.2° of rotation. To the authors’ knowledge, this same methodology has not been applied to patients with clinically meaningful anterior GBL. Given that the average glenoid width in human shoulders is 24.4 mm ± 2.9 mm,16 1 mm of glenoid bone loss (GBL) corresponds to approximately 4% of the glenoid width, and thus even subtle differences in the interpretation of GBL may have substantial clinical implications. Therefore, the purpose of this study is to determine the effect of sagittal rotation of the glenoid on axial AP glenoid width measurements in the setting of clinically significant anterior GBL.
METHODS
This study was approved by Massachusetts General Hospital Institutional Review Board. A retrospective review of consecutive patients with a diagnosis of anterior shoulder instability between 2009 and 2013 was conducted. Inclusion criteria comprised patients with a minimum of 10% anterior GBL, an available CT scan of the affected shoulder, and no history of prior ipsilateral surgeries. Exclusion criteria comprised evidence of degenerative changes to the glenoid and/or humeral head, as well as prior ipsilateral shoulder surgery. Sixty consecutive patients were originally identified as having anterior shoulder instability, and 17 were excluded based on the inclusion/exclusion criteria, leaving 43 patients (43 shoulders) available for inclusion. Shoulder CT scans from all 43 patients were reformatted utilizing open-source DICOM software (OsiriX MD, version 2.5.1 65-bit) multi-planar reconstruction (MPR).
CT PROTOCOL
All patients underwent a standard glenohumeral CT scan using a Siemens Sensation 64 Scanner (Siemens), a 64-detector scanner. Scans were acquired with 0.6 mm of collimation, 140 kV, and 300 mA-seconds. Slice thickness was set to 2 mm. All patient information was de-identified for analysis.
The uncorrected (UNCORR) scans were defined as the default orientation on the scanner. In the UNCORR scans, the axial, coronal, and sagittal views were oriented relative to the scanner gantry table, as opposed to the anatomy of the glenoid. The corrected (CORR) CT scans were aligned in all 3 planes relative to the glenoid face, and thus the cuts were perpendicular to the long axis of the glenoid.15 This resulted in sagittal cuts perpendicular to the 12-o’clock to 6-o’clock axis in the sagittal plane (Figure 1).
Continue to: In a de-identified fashion...
IMAGE ANALYSIS AND REFORMATTING
In a de-identified fashion, all CT scans were imported and analyzed using open-source Digital Imaging and Communications in Medicine (DICOM) software (OsiriX MD, version 2.5.1 64-bit). By following a previously developed method, CT scans were reformatted using OsiriX MPR. The OsiriX software has an MPR function that allows simultaneous manipulation of 2-D CT scans in 3 orthogonal planes: axial, sagittal, and coronal. In the MPR mode, the alternation of 1 plane directly affects the orientation of the remaining 2 planes. Thus, by using an MPR, one can analyze the impact that a default CT scan performed relative to the gantry of the table, UNCORR, has on the axial images.
First, the en face view was obtained via a 2-step process: alignment of the axial plane to account for the scapular angle, followed by alignment of the coronal plane to adjust for the glenoid inclination.15 These 2 adjustments provided a true en face sagittal glenoid view. The final adjustment step was a sagittal en face rotation of the glenoid such that the superior and inferior glenoid tubercles were placed on the 12-o’clock to 6-o’clock axis (CORR scan). Previous studies have identified a central longitudinal axis that was used in this method to align the supraglenoid tubercle with the 12-o’clock to 6-o’clock axis on the glenoid face.15,17,18 The standard error of mean was 1.21°. This new CORR view resulted in axial cuts through the glenoid that were oriented perpendicular to the 12-o’clock to 6-o’clock axis. The UNCORR and CORR images were assessed in the axial plane at 5 standardized cuts and measured for AP glenoid width by 2 independent observers in a blinded, randomized fashion. When the measured AP width of the UNCORR scan was less than that measured on the CORR scan, the AP width of the glenoid was considered underestimated, and the degree of GBL was considered overestimated (Figure 2).
SCAPULAR ANGLE
Scapular angle measurements were performed on the axial view as the angle between a line through the long axis of the body of the scapula, and a line parallel to the CT gantry table.15,19 Subsequently, the axial plane was aligned to the glenoid surface.
CORONAL INCLINATION
Coronal inclination measurements were performed on the sagittal view as the angle between a line tangential to the face of the glenoid and a line perpendicular to the CT gantry table. Positive values represented superior inclination, while negative values represented inferior glenoid inclination.15
SAGITTAL ROTATION
Sagittal rotation measurements were performed using the built-in angle measurement tool in OsiriX in the sagittal plane since the degree of rotation required aligning the long axis of the glenoid to the 12-o’clock to 6-o’clock axis. The amount of rotation was defined as the rotation angle.15
Continue to: Similarly, as described by Gross...
GLENOID WIDTH
Similarly, as described by Gross and colleagues,15 the sagittal en face view was divided via 5 cuts, throughout a superimposed best-fit circle that closely represents the glenoid.9,15,20 For both the UNCORR and CORR, glenoid width (AP distance) was measured on the axial image at the widest point from AP cortex across the glenoid face.
PATIENT GROUPS
Utilizing the en face 3-D CT reconstruction view of the glenoid as the gold standard, patients were placed into 1 of 3 groups according to the degree of anterior GBL measured via the surface method.9,20 The groups were as follows:
I. 10% to 14.9% (N = 12)
II. 15% to 19.9% (N = 16)
III. >20% (N = 15)
STATISTICAL METHODS
Paired t-tests were used to compare all measurements between CORR and UNCORR scans for each of the 5 cuts. A P-value of .05 was used as the threshold for statistical significance in 2-tailed comparisons. Mean and standard errors are presented with standard deviations throughout the study. For interobserver reliability, the measurements between the observers, the intraclass correlation coefficient was calculated. All statistics were performed with SPSS (Version 22).
RESULTS
The study cohort was comprised of 19 left shoulders (44%) and 24 right shoulders (56%), including 36 male patients (84%) and 7 female patients (16%). The average age was 27.8 years (range, 21-40 years). The variability in measured difference, with respect to AP width, was 1.05 mm. The UNCORR CT scans required a mean correction for coronal inclination of 7.0° ± 5.8° (range, -8°-6°). The UNCORR CT scans required a mean correction for scapular angle of 30.2° ± 8.0° (range, 15°-49°). The mean angle of sagittal rotation required to align the glenoid face with the 12-o’clock to 6-o’clock axis was 24.2° ± 5.1 ° (range, 13°-30°). These results are summarized in Table 1.
Table 1. Mean Correction Values Required to Correct the Uncorrected Images to the Corrected Images | |||
Anatomic alignment | Mean (degrees) | Range (degrees) | SD (degrees) |
Scapular angle | 30.2 | 15-49 | 8.0 |
Coronal Inclination | 7.0 | -8-6 | 5.8 |
Sagittal rotation | 24.2 | 13-30 | 5.1 |
For all measurements, the intraclass correlation coefficient for independent observers for all cuts within the 3 groups was r >.900 in all cases.
On an optimized CT scan, over 5 standardized cuts across a best-fit circle of the inferior glenoid, there was a statistically significant absolute mean difference of 12.6% in axial AP glenoid width (2.86 mm ± 2.00 mm, P =.016) when compared with the UNCORR scan. This corresponds to a 3% to 21% error in measurement of the AP width of the glenoid.
Continue to: For the entire cohort...
For the entire cohort of 43 patients, the UNCORR scans underestimated the axial AP width (and thus overestimated GBL) in cut 1 (P =.003), and overestimated the axial AP width (and thus underestimated GBL) in cuts 3 to 5 (P < .001 for all) compared with that of the CORR scans. There was no significant difference between the UNCORR and CORR scans in cut 2 (P = .331).
For groups I (10%-14.9% GBL) and III (>20% GBL), the UNCORR scans underestimated the axial AP width (and thus overestimated anterior GBL) in cuts 1 and 2, while in cuts 3 to 5, the axial AP width was overestimated (GBL was underestimated) (Tables 2, 3). In Group II (15%-19.9% GBL), the axial AP width was underestimated (GBL was overestimated), while in cuts 2 to 5, the axial AP width was overestimated (GBL was underestimated). Overall, AP glenoid width was consistently underestimated in cut 1, the most caudal cut, while AP glenoid width was consistently overestimated in cuts 3 to 5, the more cephalad cuts.
Table 2. Absolute Mean Difference in Axial AP Width (mm) Between Corrected and Uncorrected Images (% difference) | |||||
Cut 1 (Caudal) | Cut 2 | Cut 3 (Center) | Cut 4 | Cut 5 (Cephalad) | |
Group I: 10%-14.9% GBL | 2.4 mm (15.3%) | 1.8 mm (9.0%) | 1.8 mm (7.7%) | 3.0 mm (11.7%) | 4.0 mm (16.8%) |
Group II: 15%-19.9% GBL | 1.8 mm (13.1%) | 1.7 mm (7.9%) | 2.8 mm (10.6%) | 4.1 mm (14.4%) | 4.8 mm (16.9%) |
Group III: >20% | 2.8 mm (16.1%) | 1.9 mm (8.0%) | 2.3 mm (10.3) | 4.4 mm (16.6%) | 5.2 mm (17.0%) |
Abbreviations: AP, anterior-posterior; GBL, glenoid bone loss.
Table 3. Mean AP Glenoid Width Based on CORR and UNCORR Images for the Entire Cohort of 43 Patients | |||||
Axial cut | Mean AP width (mm) | Mean AP width (mm) | Absolute mean AP width difference (mm) | Absolute mean AP width difference (%) | P value |
(Caudal) 1 | 16.6208 | 18.4958 | -1.875 | 14.7768 | .0029565 |
2 | 20.6558 | 21.3166 | -0.661 | 3.6137 | .3310965 |
3 | 24.2583 | 22.3125 | 1.946 | 7.8042 | <.0001 |
4 | 26.1291 | 21.8916 | 4.238 | 15.8449 | <.0001 |
(Rostral) 5 | 26.0875 | 20.4875 | 5.6 | 20.9717 | <.0001 |
Abbreviations: AP, anterior-posterior; CORR, corrected; UNCORR, uncorrected.
DISCUSSION
The principle findings of this study demonstrate that UNCORR conventional 2-D CT scans inaccurately estimate glenoid width as well as inaccurately quantify the degree of anterior GBL. Underestimations of GBL may lead to insufficient treatment of clinically meaningful GBL, thereby increasing the risk of instability recurrence; whereas overestimations of GBL may lead to unnecessary treatment, subjecting patients to increased surgical morbidity. Therefore, the authors recommend correcting the orientation of the scapula in cases wherein clinical decisions are entirely based on 2-D CT, or using alternative methods for quantifying GBL, specifically in the form of 3-D reconstructions.
The use of axial imaging, with CT scans and/or magnetic resonance imaging, is growing in popularity for evaluation of both glenoid anatomy and GBL. Nevertheless, despite our improved ability to critically evaluate the glenoid using these advanced imaging modalities, the images themselves require scrutiny by clinicians to determine if the images accurately depict the true anatomy of the glenoid. As demonstrated by Gross and colleagues,15 conventional 2D CT scan protocols are not optimized to the anatomy of the glenohumeral joint, even in patients without GBL. Due to the alignment of the image relative to the plane of the scapula as opposed to the plane of the glenoid, UNCORR scans result in significantly different measurements of glenoid version (2.0° ± 0.1°) and AP glenoid width (1.2 mm ± 0.42 mm) compared with corrected scans, requiring an average 20.1° ± 1.2° of correction to align the sagittal plane. In the present study involving the patients with GBL, we also found that conventional, UNCORR 2-D CT scan protocols inaccurately estimate glenoid width and the degree of anterior GBL. In particular, AP glenoid width was consistently underestimated in the more caudal cuts, while AP glenoid width was consistently overestimated in the more cephalad cuts. Thus, anterior GBL was overestimated (AP glenoid width was underestimated) in the more caudal cuts, whereas anterior GBL was underestimated in the more cranial cuts (AP glenoid width was overestimated). Given that approximately 1 mm of glenoid bone corresponds to approximately 4% of glenoid width,16 even subtle differences in the interpretation of GBL may lead to gross overestimation/underestimation of bone loss, with significant clinical implications.
In the anterior instability patient population, clinical decision-making is often based on the degree of GBL as determined by advanced imaging modalities. In addition to other patient-specific factors, including age, gender, activity level, type of sport, and number of prior dislocations and/or prior surgeries, the quantity of GBL will often determine which surgical procedure needs to be performed. Typically, patients with >20% to 25% anterior GBL are indicated for a glenoid reconstruction procedure, most commonly via the Latarjet procedure (coracoid transfer).21-27 The Latarjet procedure remains an excellent technique for appropriately indicated patients, with historically good clinical outcomes and low recurrence rates. Complications associated with the Latarjet procedure, however, are not uncommon, including devastating neuropraxia of the axillary and musculocutaneous nerves, and occasionally permanent neurologic deficits.28 Thus, it is critical to avoid overtreating patients with recurrent instability and GBL. As demonstrated by this study, depending on the cranial-to-caudal location on the glenoid, current 2-D CT techniques may underestimate AP glenoid width, resulting in an overestimation of GBL, potentially leading to the decision to proceed with glenoid bone reconstruction when such a procedure is not required. On the contrary, overestimation of AP glenoid width, which occurs in the more cephalad cuts of the glenoid, is perhaps more worrisome, as the resulting underestimation of GBL may lead to inadequate treatment of patients with recurrent instability. Certainly, one of the main risk factors for failed soft tissue shoulder stabilization is a failure to address GBL. If clinical decisions are made based on UNCORR 2-D CT scans, which are often inaccurate with respect to AP glenoid width by an average 2.86 mm ± 2.00 mm (equivalent to 12.6% ± 6.9% GBL) as determined in this study, patients who truly require osseous glenoid reconstructions may be indicated for only soft tissue stabilization, based on the underestimation of GBL.
Continue to: The current gold standard...
The current gold standard for GBL measurement is a perfect-fit circle performed on a 3-D CT scan.22 To that end, it would have been useful to measure the glenoids from this study on 3-D CT scans and compare the data with both UNCORR and CORR measurements. This would have provided a better understanding to what extent the CORR measurements on 2-D scans are relatable with the gold standard. As 3-D CT scans provide a better en face view of the glenoid, more accurate GBL measurements, and ease of 3-D manipulation, they have become more widely used across the country.29,30 Nevertheless, in situations where 3-D imaging is more challenging to obtain because of technology or cost limitations, having a strategy for ensuring proper orientation of 2-D scans would have a substantial impact on clinical decision-making. If such corrections are not made, the inaccuracy of current 2-D scanning protocols justifies the cost 3-D reconstruction protocols. The difference in GBL measurements are critical in cases of increasingly large degrees of GBL, as in these instances, the inferior glenoid becomes more of an inverted-pear shape as opposed to a perfect circle, and differences in CORR and UNCORR images are likely to be more profound.
LIMITATIONS
This study has limitations, such as the relatively small sample size and the selection bias by the reviewers with potential differences in interobserver reliability. Further, minor modifications during the reformatting process may be found with each attempt to manipulate the images and may result in minor, insignificant differences in AP width measurements. Performing 1 or more additional CT scans on the same cohort of patients would have been helpful; however, due to the increased risk of radiation exposure, this was not performed. Performing CT scans on cadaveric specimens with GBL and applying the study methodology would also have been helpful to provide independent verification of our clinical findings; however, specimens were not available for this study. Another limitation of this study is that we did not compare our findings with the findings of glenoid width, and bone loss, as determined using the circle method, which is commonly utilized when 3-D reconstructions are available. In this study, the purpose was to utilize only the 2-D reformatted images, with the assumption that 3-D reconstructions are not always available, and cannot always be measured. To minimize selection bias, the investigators measured the correction effects within groups of patients with similar degrees of GBL (10%-14.9%, 15%-19.9%, and >20%). In addition, not all the selected patients showed degenerative glenoid changes or irregular glenoid shape indicating previous bone augmentation.
CONCLUSIONS
UNCORR 2D CT scans inaccurately estimate glenoid width and the degree of anterior GBL. The clinical implications of these findings are profound and suggest corrected 2D CT scans or 3D reconstruction allow measurements to be taken in the axis of the glenoid to accurately define the anatomy and quantity of anterior GBL in patients with shoulder instability.
1. Cerciello S, Edwards TB, Walch G. Chronic anterior glenohumeral instability in soccer players: results for a series of 28 shoulders treated with the Latarjet procedure. J Orthop Traumatol. 2012;13(4):197-202. doi:10.1007/s10195-012-0201-3.
2. Itoi E, Lee SB, Berglund LJ, Berge LL, An KN. The effect of a glenoid defect on anteroinferior stability of the shoulder after Bankart repair: a cadaveric study. J Bone Joint Surg Am. 2000;82(1):35-46.
3. Bhatia S, Ghodadra NS, Romeo AA, et al. The importance of the recognition and treatment of glenoid bone loss in an athletic population. Sports Health. 2011;3(5):435-440. doi:10.1177/1941738111414126.
4. Lo IK, Parten PM, Burkhart SS. The inverted pear glenoid: an indicator of significant glenoid bone loss. Arthroscopy. 2004;20(2):169-174. doi:10.1016/j.arthro.2003.11.036.
5. Mologne TS, Provencher MT, Menzel KA, Vachon TA, Dewing CB. Arthroscopic stabilization in patients with an inverted pear glenoid: results in patients with bone loss of the anterior glenoid. Am J Sports Med. 2007;35(8):1276-1283. doi:10.1177/0363546507300262.
6. Piasecki DP, Verma NN, Romeo AA, Levine WN, Bach BR Jr, Provencher MT. Glenoid bone deficiency in recurrent anterior shoulder instability: diagnosis and management. J Am Acad Orthop Surg. 2009;17(8):482-493.
7. Provencher MT, Bhatia S, Ghodadra NS, et al. Recurrent shoulder instability: current concepts for evaluation and management of glenoid bone loss. J Bone Joint Surg Am. 2010;92(suppl 2):133-151. doi:10.2106/JBJS.J.00906.
8. Rowe CR, Zarins B, Ciullo JV. Recurrent anterior dislocation of the shoulder after surgical repair. Apparent causes of failure and treatment. J Bone Joint Surg Am. 1984;66(2):159-168.
9. Sugaya H, Moriishi J, Dohi M, Kon Y, Tsuchiya A. Glenoid rim morphology in recurrent anterior glenohumeral instability. J Bone Joint Surg Am. 2003;85-A(5):878-884.
10. Edwards TB, Boulahia A, Walch G. Radiographic analysis of bone defects in chronic anterior shoulder instability. Arthroscopy. 2003;19(7):732-739.
11. Jankauskas L, Rudiger HA, Pfirrmann CW, Jost B, Gerber C. Loss of the sclerotic line of the glenoid on anteroposterior radiographs of the shoulder: a diagnostic sign for an osseous defect of the anterior glenoid rim. J Shoulder Elbow Surg. 2010;19(1):151-156. doi:10.1016/j.jse.2009.04.013.
12. Altan E, Ozbaydar MU, Tonbul M, Yalcin L. Comparison of two different measurement methods to determine glenoid bone defects: area or width? J Shoulder Elbow Surg. 2014;23(8):1215-1222. doi:10.1016/j.jse.2013.11.029.
13. Bishop JY, Jones GL, Rerko MA, Donaldson C, Group MS. 3-D CT is the most reliable imaging modality when quantifying glenoid bone loss. Clin Orthop Relat Res. 2013;471(4):1251-1256. doi:10.1007/s11999-012-2607-x.
14. Chuang TY, Adams CR, Burkhart SS. Use of preoperative three-dimensional computed tomography to quantify glenoid bone loss in shoulder instability. Arthroscopy. 2008; 24(4):376-382. doi:10.1016/j.arthro.2007.10.008.
15. Gross DJ, Golijanin P, Dumont GD, et al. The effect of sagittal rotation of the glenoid on axial glenoid width and glenoid version in computed tomography scan imaging. J Shoulder Elbow Surg. 2016;25(1):61-68. doi:10.1016/j.jse.2015.06.017.
16. Lenart BA, Freedman R, Van Thiel GS, et al. Magnetic resonance imaging evaluation of normal glenoid length and width: an anatomic study. Arthroscopy. 2014;30(8):915-920. doi:10.1016/j.arthro.2014.03.006.
17. Bois AJ, Fening SD, Polster J, Jones MH, Miniaci A. Quantifying glenoid bone loss in anterior shoulder instability: reliability and accuracy of 2-dimensional and 3-dimensional computed tomography measurement techniques. Am J Sports Med. 2012;40(11):2569-2577. doi:10.1177/0363546512458247.
18. Griffith JF, Antonio GE, Tong CW, Ming CK. Anterior shoulder dislocation: quantification of glenoid bone loss with CT. AJR Am J Roentgenol. 2003;180(5):1423-1430. doi:10.2214/ajr.180.5.1801423.
19. Hoenecke HR Jr, Hermida JC, Flores-Hernandez C, D'Lima DD. Accuracy of CT-based measurements of glenoid version for total shoulder arthroplasty. J Shoulder Elbow Surg. 2010;19(2):166-171. doi:10.1016/j.jse.2009.08.009.
20. Huijsmans PE, de Witte PB, de Villiers RV, et al. Recurrent anterior shoulder instability: accuracy of estimations of glenoid bone loss with computed tomography is insufficient for therapeutic decision-making. Skeletal Radiol. 2011;40(10):1329-1334. doi:10.1007/s00256-011-1184-5.
21. Bhatia S, Frank RM, Ghodadra NS, et al. The outcomes and surgical techniques of the latarjet procedure. Arthroscopy. 2014;30(2):227-235. doi:10.1016/j.arthro.2013.10.013.
22. Cunningham G, Benchouk S, Kherad O, Ladermann A. Comparison of arthroscopic and open Latarjet with a learning curve analysis. Knee Surg Sports Traumatol Arthrosc. 2015;24(2):540-545. doi:10.1007/s00167-015-3910-3.
23. Fedorka CJ, Mulcahey MK. Recurrent anterior shoulder instability: a review of the Latarjet procedure and its postoperative rehabilitation. Phys Sportsmed. 2015;43(1):73-79. doi:10.1080/00913847.2015.1005543.
24. Flinkkila T, Sirniö K. Open Latarjet procedure for failed arthroscopic Bankart repair. Orthop Traumatol Surg Res. 2015;101(1):35-38. doi:10.1016/j.otsr.2014.11.005.
25. Hovelius L, Sandström B, Saebö M. One hundred eighteen Bristow-Latarjet repairs for recurrent anterior dislocation of the shoulder prospectively followed for fifteen years: study II-the evolution of dislocation arthropathy. J Shoulder Elbow Surg. 2006;15(3):279-289. doi:10.1016/j.jse.2005.09.014.
26. Hovelius L, Sandström B, Sundgren K, Saebö M. One hundred eighteen Bristow-Latarjet repairs for recurrent anterior dislocation of the shoulder prospectively followed for fifteen years: study I--clinical results. J Shoulder Elbow Surg. 2004;13(5):509-516. doi:10.1016/S1058274604000916.
27. Hovelius L, Vikerfors O, Olofsson A, Svensson O, Rahme H. Bristow-Latarjet and Bankart: a comparative study of shoulder stabilization in 185 shoulders during a seventeen-year follow-up. J Shoulder Elbow Surg. 2011;20(7):1095-1101. doi:10.1016/j.jse.2011.02.005.
28. Gupta A, Delaney R, Petkin K, Lafosse L. Complications of the Latarjet procedure. Curr Rev Musculoskelet Med. 2015;8(1):59-66. doi:10.1007/s12178-015-9258-y.
29. Kwon YW, Powell KA, Yum JK, Brems JJ, Iannotti JP. Use of three-dimensional computed tomography for the analysis of the glenoid anatomy. J Shoulder Elbow Surg. 2005;14(1):85-90. doi:10.1016/j.jse.2004.04.011.
30. Saito H, Itoi E, Sugaya H, Minagawa H, Yamamoto N, Tuoheti Y. Location of the glenoid defect in shoulders with recurrent anterior dislocation. Am J Sports Med. 2005;33(6):889-893. doi:10.1177/0363546504271521.
1. Cerciello S, Edwards TB, Walch G. Chronic anterior glenohumeral instability in soccer players: results for a series of 28 shoulders treated with the Latarjet procedure. J Orthop Traumatol. 2012;13(4):197-202. doi:10.1007/s10195-012-0201-3.
2. Itoi E, Lee SB, Berglund LJ, Berge LL, An KN. The effect of a glenoid defect on anteroinferior stability of the shoulder after Bankart repair: a cadaveric study. J Bone Joint Surg Am. 2000;82(1):35-46.
3. Bhatia S, Ghodadra NS, Romeo AA, et al. The importance of the recognition and treatment of glenoid bone loss in an athletic population. Sports Health. 2011;3(5):435-440. doi:10.1177/1941738111414126.
4. Lo IK, Parten PM, Burkhart SS. The inverted pear glenoid: an indicator of significant glenoid bone loss. Arthroscopy. 2004;20(2):169-174. doi:10.1016/j.arthro.2003.11.036.
5. Mologne TS, Provencher MT, Menzel KA, Vachon TA, Dewing CB. Arthroscopic stabilization in patients with an inverted pear glenoid: results in patients with bone loss of the anterior glenoid. Am J Sports Med. 2007;35(8):1276-1283. doi:10.1177/0363546507300262.
6. Piasecki DP, Verma NN, Romeo AA, Levine WN, Bach BR Jr, Provencher MT. Glenoid bone deficiency in recurrent anterior shoulder instability: diagnosis and management. J Am Acad Orthop Surg. 2009;17(8):482-493.
7. Provencher MT, Bhatia S, Ghodadra NS, et al. Recurrent shoulder instability: current concepts for evaluation and management of glenoid bone loss. J Bone Joint Surg Am. 2010;92(suppl 2):133-151. doi:10.2106/JBJS.J.00906.
8. Rowe CR, Zarins B, Ciullo JV. Recurrent anterior dislocation of the shoulder after surgical repair. Apparent causes of failure and treatment. J Bone Joint Surg Am. 1984;66(2):159-168.
9. Sugaya H, Moriishi J, Dohi M, Kon Y, Tsuchiya A. Glenoid rim morphology in recurrent anterior glenohumeral instability. J Bone Joint Surg Am. 2003;85-A(5):878-884.
10. Edwards TB, Boulahia A, Walch G. Radiographic analysis of bone defects in chronic anterior shoulder instability. Arthroscopy. 2003;19(7):732-739.
11. Jankauskas L, Rudiger HA, Pfirrmann CW, Jost B, Gerber C. Loss of the sclerotic line of the glenoid on anteroposterior radiographs of the shoulder: a diagnostic sign for an osseous defect of the anterior glenoid rim. J Shoulder Elbow Surg. 2010;19(1):151-156. doi:10.1016/j.jse.2009.04.013.
12. Altan E, Ozbaydar MU, Tonbul M, Yalcin L. Comparison of two different measurement methods to determine glenoid bone defects: area or width? J Shoulder Elbow Surg. 2014;23(8):1215-1222. doi:10.1016/j.jse.2013.11.029.
13. Bishop JY, Jones GL, Rerko MA, Donaldson C, Group MS. 3-D CT is the most reliable imaging modality when quantifying glenoid bone loss. Clin Orthop Relat Res. 2013;471(4):1251-1256. doi:10.1007/s11999-012-2607-x.
14. Chuang TY, Adams CR, Burkhart SS. Use of preoperative three-dimensional computed tomography to quantify glenoid bone loss in shoulder instability. Arthroscopy. 2008; 24(4):376-382. doi:10.1016/j.arthro.2007.10.008.
15. Gross DJ, Golijanin P, Dumont GD, et al. The effect of sagittal rotation of the glenoid on axial glenoid width and glenoid version in computed tomography scan imaging. J Shoulder Elbow Surg. 2016;25(1):61-68. doi:10.1016/j.jse.2015.06.017.
16. Lenart BA, Freedman R, Van Thiel GS, et al. Magnetic resonance imaging evaluation of normal glenoid length and width: an anatomic study. Arthroscopy. 2014;30(8):915-920. doi:10.1016/j.arthro.2014.03.006.
17. Bois AJ, Fening SD, Polster J, Jones MH, Miniaci A. Quantifying glenoid bone loss in anterior shoulder instability: reliability and accuracy of 2-dimensional and 3-dimensional computed tomography measurement techniques. Am J Sports Med. 2012;40(11):2569-2577. doi:10.1177/0363546512458247.
18. Griffith JF, Antonio GE, Tong CW, Ming CK. Anterior shoulder dislocation: quantification of glenoid bone loss with CT. AJR Am J Roentgenol. 2003;180(5):1423-1430. doi:10.2214/ajr.180.5.1801423.
19. Hoenecke HR Jr, Hermida JC, Flores-Hernandez C, D'Lima DD. Accuracy of CT-based measurements of glenoid version for total shoulder arthroplasty. J Shoulder Elbow Surg. 2010;19(2):166-171. doi:10.1016/j.jse.2009.08.009.
20. Huijsmans PE, de Witte PB, de Villiers RV, et al. Recurrent anterior shoulder instability: accuracy of estimations of glenoid bone loss with computed tomography is insufficient for therapeutic decision-making. Skeletal Radiol. 2011;40(10):1329-1334. doi:10.1007/s00256-011-1184-5.
21. Bhatia S, Frank RM, Ghodadra NS, et al. The outcomes and surgical techniques of the latarjet procedure. Arthroscopy. 2014;30(2):227-235. doi:10.1016/j.arthro.2013.10.013.
22. Cunningham G, Benchouk S, Kherad O, Ladermann A. Comparison of arthroscopic and open Latarjet with a learning curve analysis. Knee Surg Sports Traumatol Arthrosc. 2015;24(2):540-545. doi:10.1007/s00167-015-3910-3.
23. Fedorka CJ, Mulcahey MK. Recurrent anterior shoulder instability: a review of the Latarjet procedure and its postoperative rehabilitation. Phys Sportsmed. 2015;43(1):73-79. doi:10.1080/00913847.2015.1005543.
24. Flinkkila T, Sirniö K. Open Latarjet procedure for failed arthroscopic Bankart repair. Orthop Traumatol Surg Res. 2015;101(1):35-38. doi:10.1016/j.otsr.2014.11.005.
25. Hovelius L, Sandström B, Saebö M. One hundred eighteen Bristow-Latarjet repairs for recurrent anterior dislocation of the shoulder prospectively followed for fifteen years: study II-the evolution of dislocation arthropathy. J Shoulder Elbow Surg. 2006;15(3):279-289. doi:10.1016/j.jse.2005.09.014.
26. Hovelius L, Sandström B, Sundgren K, Saebö M. One hundred eighteen Bristow-Latarjet repairs for recurrent anterior dislocation of the shoulder prospectively followed for fifteen years: study I--clinical results. J Shoulder Elbow Surg. 2004;13(5):509-516. doi:10.1016/S1058274604000916.
27. Hovelius L, Vikerfors O, Olofsson A, Svensson O, Rahme H. Bristow-Latarjet and Bankart: a comparative study of shoulder stabilization in 185 shoulders during a seventeen-year follow-up. J Shoulder Elbow Surg. 2011;20(7):1095-1101. doi:10.1016/j.jse.2011.02.005.
28. Gupta A, Delaney R, Petkin K, Lafosse L. Complications of the Latarjet procedure. Curr Rev Musculoskelet Med. 2015;8(1):59-66. doi:10.1007/s12178-015-9258-y.
29. Kwon YW, Powell KA, Yum JK, Brems JJ, Iannotti JP. Use of three-dimensional computed tomography for the analysis of the glenoid anatomy. J Shoulder Elbow Surg. 2005;14(1):85-90. doi:10.1016/j.jse.2004.04.011.
30. Saito H, Itoi E, Sugaya H, Minagawa H, Yamamoto N, Tuoheti Y. Location of the glenoid defect in shoulders with recurrent anterior dislocation. Am J Sports Med. 2005;33(6):889-893. doi:10.1177/0363546504271521.
TAKE-HOME POINTS
- Standard 2-D CT scans of the shoulder are often aligned to the plane of the body as opposed to the plane of the scapula, which may challenge the ability to accurately measure glenoid width and GBL.
- Underestimations of GBL may lead to insufficient treatment of clinically meaningful GBL, thereby increasing the risk of instability recurrence; whereas overestimations of GBL may lead to unnecessary treatment, subjecting patients to increased surgical morbidity.
- AP glenoid width was consistently underestimated in uncorrected axial cut 1, the most caudal cut.
- AP glenoid width was consistently overestimated in uncorrected axial cuts 3 to 5, the more cephalad cuts.
- CORR 2-D CT scans or a 3-D reconstruction can help in accurately defining the anterior GBL in patients with shoulder instability.
New strategies eliminate cardiac device transvenous leads
reflecting a keen interest in getting leads out of the vascular space and potentially cutting risks for infections, pneumothorax, hematoma, and the possible need for future lead extraction.
Leads are “the Achilles heel” of pacemakers and defibrillators; “there are too many places where the wire can break,” John D. Day, MD, said in an interview during the annual scientific sessions of the Heart Rhythm Society. Eliminating transvenous leads, or any type of lead for that matter, “solves a lot of problems” that currently occur with implanted cardiac devices, said Dr. Day, a cardiac electrophysiologist with Intermountain Health in Murray, Utah.
The Acute Extravascular Defibrillation, Pacing, and Electrogram (ASD2) study tested a substernal lead in 79 patients enrolled at multiple centers in the United States and elsewhere. The lead’s design uses a minimally invasive subxiphoid approach with substernal lead advancement using a blunt tunneling rod, explained Lucas V.A. Boersma, MD, a professor of cardiology at the Academic Medical Center University in Amsterdam. Placed between the sternum and heart, the lead sits just beside the ventricles to provide both ventricular pacing and defibrillation, unlike existing subcutaneously placed leads that do not allow pacing and require high energy for defibrillation. It took a median of 12 minutes to place the lead, Dr. Boersma said.
Testing demonstrated successful ventricular pacing capture in 76 of 78 patients (97%) who underwent this testing, he reported. A single 30-joule shock delivered via the substernal lead resulted in successful defibrillation of induced fibrillation in 102 of 123 fibrillation events (83%). Six patients had seven adverse events that resolved without sequelae for all but two events. The two events with greater clinical impact included one patient with asystolic cardiac arrest 36 hours after lead placement who developed decompensated heart failure and required medical management, and one patient with pericardial effusion and tamponade who required supportive care.

“We are very used to working in the transvenous space” for lead placement, “and leaving that space is outside the comfort zone” for many clinicians, Dr. Boersma noted. “It will take time to adopt these new technologies, and obviously we need more proof” of efficacy and safety.
The second study examined a novel approach that modified the control algorithm of the Micra leadless single-chamber pacing device, approved for U.S. marketing in 2016, so that it used information collected by a built-in accelerometer to detect atrial contractions and produce atrial-ventricular (AV) synchrony in patients with AV block.
The MARVEL (Micra Atrial Tracking Using a Ventricular Accelerometer) study enrolled 70 patients at 12 centers worldwide and collected evaluable data from 64 patients. The average time spent in AV synchrony using this AV pacing was 87% in all patients, with an 80% average rate of AV synchrony in the 33 enrolled patients who had high-degree block, said Larry A. Chinitz, MD, professor of medicine and director of the Heart Rhythm Center at New York University Langone Health. Concurrently with his report at the meeting, the results also appeared in an article online (Heart Rhythm. 2018 May 11. doi: 10.1016/j.hrthm.2018.05.004).

The current leadless pacemaker is very limited as a single-chamber pacing device because it can only help patients who have permanent atrial fibrillation and a slow ventricular response and hence only need single-chamber pacing, about 14% of all patients who need cardiac pacing, said Dr. Chinitz. Using the accelerometer information appears to make the Micra device suitable for the much larger number of patients who have AV block. “You effectively have dual-chamber pacing but with a single pellet. This is a simple and attractive way to do it,” Dr. Day commented. “You get a two-for-one” that potentially could triple the number of patients who could benefit from the Micra leadless device, he estimated.
[email protected]
On Twitter @mitchelzoler
SOURCE: Boersma L et al. Heart Rhythm 2018, Abstract B-LBCT03-03; Chinitz L et al. Heart Rhythm 2018, Abstract B-LBCT03-04.
reflecting a keen interest in getting leads out of the vascular space and potentially cutting risks for infections, pneumothorax, hematoma, and the possible need for future lead extraction.
Leads are “the Achilles heel” of pacemakers and defibrillators; “there are too many places where the wire can break,” John D. Day, MD, said in an interview during the annual scientific sessions of the Heart Rhythm Society. Eliminating transvenous leads, or any type of lead for that matter, “solves a lot of problems” that currently occur with implanted cardiac devices, said Dr. Day, a cardiac electrophysiologist with Intermountain Health in Murray, Utah.
The Acute Extravascular Defibrillation, Pacing, and Electrogram (ASD2) study tested a substernal lead in 79 patients enrolled at multiple centers in the United States and elsewhere. The lead’s design uses a minimally invasive subxiphoid approach with substernal lead advancement using a blunt tunneling rod, explained Lucas V.A. Boersma, MD, a professor of cardiology at the Academic Medical Center University in Amsterdam. Placed between the sternum and heart, the lead sits just beside the ventricles to provide both ventricular pacing and defibrillation, unlike existing subcutaneously placed leads that do not allow pacing and require high energy for defibrillation. It took a median of 12 minutes to place the lead, Dr. Boersma said.
Testing demonstrated successful ventricular pacing capture in 76 of 78 patients (97%) who underwent this testing, he reported. A single 30-joule shock delivered via the substernal lead resulted in successful defibrillation of induced fibrillation in 102 of 123 fibrillation events (83%). Six patients had seven adverse events that resolved without sequelae for all but two events. The two events with greater clinical impact included one patient with asystolic cardiac arrest 36 hours after lead placement who developed decompensated heart failure and required medical management, and one patient with pericardial effusion and tamponade who required supportive care.
“We are very used to working in the transvenous space” for lead placement, “and leaving that space is outside the comfort zone” for many clinicians, Dr. Boersma noted. “It will take time to adopt these new technologies, and obviously we need more proof” of efficacy and safety.
The second study examined a novel approach that modified the control algorithm of the Micra leadless single-chamber pacing device, approved for U.S. marketing in 2016, so that it used information collected by a built-in accelerometer to detect atrial contractions and produce atrial-ventricular (AV) synchrony in patients with AV block.
The MARVEL (Micra Atrial Tracking Using a Ventricular Accelerometer) study enrolled 70 patients at 12 centers worldwide and collected evaluable data from 64 patients. The average time spent in AV synchrony using this AV pacing was 87% in all patients, with an 80% average rate of AV synchrony in the 33 enrolled patients who had high-degree block, said Larry A. Chinitz, MD, professor of medicine and director of the Heart Rhythm Center at New York University Langone Health. Concurrently with his report at the meeting, the results also appeared in an article online (Heart Rhythm. 2018 May 11. doi: 10.1016/j.hrthm.2018.05.004).
The current leadless pacemaker is very limited as a single-chamber pacing device because it can only help patients who have permanent atrial fibrillation and a slow ventricular response and hence only need single-chamber pacing, about 14% of all patients who need cardiac pacing, said Dr. Chinitz. Using the accelerometer information appears to make the Micra device suitable for the much larger number of patients who have AV block. “You effectively have dual-chamber pacing but with a single pellet. This is a simple and attractive way to do it,” Dr. Day commented. “You get a two-for-one” that potentially could triple the number of patients who could benefit from the Micra leadless device, he estimated.
[email protected]
On Twitter @mitchelzoler
SOURCE: Boersma L et al. Heart Rhythm 2018, Abstract B-LBCT03-03; Chinitz L et al. Heart Rhythm 2018, Abstract B-LBCT03-04.
reflecting a keen interest in getting leads out of the vascular space and potentially cutting risks for infections, pneumothorax, hematoma, and the possible need for future lead extraction.
Leads are “the Achilles heel” of pacemakers and defibrillators; “there are too many places where the wire can break,” John D. Day, MD, said in an interview during the annual scientific sessions of the Heart Rhythm Society. Eliminating transvenous leads, or any type of lead for that matter, “solves a lot of problems” that currently occur with implanted cardiac devices, said Dr. Day, a cardiac electrophysiologist with Intermountain Health in Murray, Utah.
The Acute Extravascular Defibrillation, Pacing, and Electrogram (ASD2) study tested a substernal lead in 79 patients enrolled at multiple centers in the United States and elsewhere. The lead’s design uses a minimally invasive subxiphoid approach with substernal lead advancement using a blunt tunneling rod, explained Lucas V.A. Boersma, MD, a professor of cardiology at the Academic Medical Center University in Amsterdam. Placed between the sternum and heart, the lead sits just beside the ventricles to provide both ventricular pacing and defibrillation, unlike existing subcutaneously placed leads that do not allow pacing and require high energy for defibrillation. It took a median of 12 minutes to place the lead, Dr. Boersma said.
Testing demonstrated successful ventricular pacing capture in 76 of 78 patients (97%) who underwent this testing, he reported. A single 30-joule shock delivered via the substernal lead resulted in successful defibrillation of induced fibrillation in 102 of 123 fibrillation events (83%). Six patients had seven adverse events that resolved without sequelae for all but two events. The two events with greater clinical impact included one patient with asystolic cardiac arrest 36 hours after lead placement who developed decompensated heart failure and required medical management, and one patient with pericardial effusion and tamponade who required supportive care.
“We are very used to working in the transvenous space” for lead placement, “and leaving that space is outside the comfort zone” for many clinicians, Dr. Boersma noted. “It will take time to adopt these new technologies, and obviously we need more proof” of efficacy and safety.
The second study examined a novel approach that modified the control algorithm of the Micra leadless single-chamber pacing device, approved for U.S. marketing in 2016, so that it used information collected by a built-in accelerometer to detect atrial contractions and produce atrial-ventricular (AV) synchrony in patients with AV block.
The MARVEL (Micra Atrial Tracking Using a Ventricular Accelerometer) study enrolled 70 patients at 12 centers worldwide and collected evaluable data from 64 patients. The average time spent in AV synchrony using this AV pacing was 87% in all patients, with an 80% average rate of AV synchrony in the 33 enrolled patients who had high-degree block, said Larry A. Chinitz, MD, professor of medicine and director of the Heart Rhythm Center at New York University Langone Health. Concurrently with his report at the meeting, the results also appeared in an article online (Heart Rhythm. 2018 May 11. doi: 10.1016/j.hrthm.2018.05.004).
The current leadless pacemaker is very limited as a single-chamber pacing device because it can only help patients who have permanent atrial fibrillation and a slow ventricular response and hence only need single-chamber pacing, about 14% of all patients who need cardiac pacing, said Dr. Chinitz. Using the accelerometer information appears to make the Micra device suitable for the much larger number of patients who have AV block. “You effectively have dual-chamber pacing but with a single pellet. This is a simple and attractive way to do it,” Dr. Day commented. “You get a two-for-one” that potentially could triple the number of patients who could benefit from the Micra leadless device, he estimated.
[email protected]
On Twitter @mitchelzoler
SOURCE: Boersma L et al. Heart Rhythm 2018, Abstract B-LBCT03-03; Chinitz L et al. Heart Rhythm 2018, Abstract B-LBCT03-04.
REPORTING FROM HEART RHYTHM 2018
Key clinical point: Results from pilot studies showed promise for two different approaches to eliminating transvenous leads from cardiac devices.
Major finding: A substernal lead produced ventricular capture pacing in 97% of patients. Atrial syncing with an accelerometer produced AV synchrony in 87% of patients.
Study details: The ASD2 multicenter study enrolled 79 patients. The MARVEL multicenter study enrolled 64 evaluable patients.
Disclosures: The ASD2 and MARVEL studies were both funded by Medtronic, the company developing the tested devices. Dr. Boersma has been a consultant to Medtronic and Boston Scientific. Dr. Chinitz has been a consultant to Medtronic and several other device companies. Dr. Day has been a consultant to Boston Scientific, Biotronik, and St. Jude.
Source: Boersma L et al. Heart Rhythm 2018, Abstract B-LBCT03-03; Chinitz L et al. Heart Rhythm 2018, Abstract B-LBCT03-04.
What’s Eating You? Clothes Moths (Tineola Species)
Clothes moths are common pests found inside buildings such as homes, stores, and museums. The most common species of importance include the webbing clothes moth (Tineola bisselliella)(Figure) and the casemaking clothes moth (Tineola pellionella). Both species target textiles such as wool, rugs, feathers, felts, hair, furs, and even grains.1,2 They avoid synthetics and plant materials such as cottons.1
 larva.")
Characteristics
Adult clothes moths extend 7 to 8 mm and are a golden (T bisselliella) to brown (T pellionella) color with fringed wings and a tuft on their heads.1,3 Adults do not eat; females die within a few days of laying eggs, while males live approximately 1 month. Once laid, eggs hatch within 4 to 10 days.1,3 The larvae (caterpillars) incur damage to clothes and other household goods. Fully mature larvae are 12- to 13-mm long, and the Tineola species have white- to cream-colored bodies with brown heads. The webbing clothes moth larva lacks ocelli (eyes), while the casemaking moth larva has a singular ocellus.1
Transmission
An infestation is evidenced by woolen items that have furrows or holes in them. Pheromone traps also can expose an active infestation.3 The webbing moth larvae can be found beneath a self-spun silken mat on the food source that offers the insect protection and camouflage while it eats; the mat collects frass (feces) and clothes particles.1,3,4 The casemaking moth larvae drag around a portable silken bag that takes on the color of the fabric being eaten and serves as a refuge when disturbed.1,3,4 Both adult and larval stages prefer low light conditions. The total time of development from caterpillar to adult varies depending on the temperature and humidity of the environment, but most clothes moths complete their life cycle within 1 to 3 months.1
Management of an Infestation
Multiple infestation treatment options exist should a patient present with a clothes moth infestation. Infested clothing articles or small blankets and rugs can be dry-cleaned or laundered. Any items not in use should be laundered before being sealed in airtight storage containers. Mothball vapor at appropriate concentrations is lethal to the moths, and when possible, clothing should be stored with mothballs or flakes at the concentration recommended by the manufacturer.4 Individuals should avoid application of household insecticides to clothing or bedding, which may be poisonous to people.1,4 Freezing, heating, and dry ice fumigation techniques also can be used to treat infested products.3 Cedarwood usually is insufficient to deter an infestation, as the oil vapor rarely reaches an effective concentration to repel or harm the insects.3,4 Strict housekeeping with attention to vacuuming carpets, baseboards, closets, and laundering all linens and furniture covers can further reduce an infestation.4 Clothes items can be set in the sunlight and brushed to help loosen the pests, as they dislike direct light and may fall from the garments.3 Dust insecticides also can be used per the manufacturer label to treat crevices and baseboards in an active area of infestation that may otherwise be difficult to clean.3 If an extensive infestation exists or larger items are infested, then a professional pest control agency should be employed for proper eradication.
Conclusion
Understanding the life cycle and basic biology of clothes moths and other common household pests will help the clinician identify an infestation and counsel patients if an insect is a true ectoparasite. Clothes moth larvae are not parasites but can be found on clothing and can be confused with myiasis or true parasites.
- Jacobs S. Clothes moth. Penn State Extension website. http://ento.psu.edu/extension/factsheets/clothes-moth. Updated January 2013. Accessed May 14, 2018.
- Querner P. Insect pests and integrated pest management in museums, libraries and historic buildings. Insects. 2015;6:595-607.
- Choe D-H. Pest notes: clothes moths (publication 7435). University of California Agriculture & Natural Resources website. http://ipm.ucanr.edu/PMG/PESTNOTES/pn7435.html. Updated March 2013. Accessed May 14, 2018.
- Potter M. Entfact-609: clothes moths. Entomology at the University of Kentucky website. https://entomology.ca.uky.edu/ef609. Updated October 2001. Accessed May 14, 2018.
Clothes moths are common pests found inside buildings such as homes, stores, and museums. The most common species of importance include the webbing clothes moth (Tineola bisselliella)(Figure) and the casemaking clothes moth (Tineola pellionella). Both species target textiles such as wool, rugs, feathers, felts, hair, furs, and even grains.1,2 They avoid synthetics and plant materials such as cottons.1
Characteristics
Adult clothes moths extend 7 to 8 mm and are a golden (T bisselliella) to brown (T pellionella) color with fringed wings and a tuft on their heads.1,3 Adults do not eat; females die within a few days of laying eggs, while males live approximately 1 month. Once laid, eggs hatch within 4 to 10 days.1,3 The larvae (caterpillars) incur damage to clothes and other household goods. Fully mature larvae are 12- to 13-mm long, and the Tineola species have white- to cream-colored bodies with brown heads. The webbing clothes moth larva lacks ocelli (eyes), while the casemaking moth larva has a singular ocellus.1
Transmission
An infestation is evidenced by woolen items that have furrows or holes in them. Pheromone traps also can expose an active infestation.3 The webbing moth larvae can be found beneath a self-spun silken mat on the food source that offers the insect protection and camouflage while it eats; the mat collects frass (feces) and clothes particles.1,3,4 The casemaking moth larvae drag around a portable silken bag that takes on the color of the fabric being eaten and serves as a refuge when disturbed.1,3,4 Both adult and larval stages prefer low light conditions. The total time of development from caterpillar to adult varies depending on the temperature and humidity of the environment, but most clothes moths complete their life cycle within 1 to 3 months.1
Management of an Infestation
Multiple infestation treatment options exist should a patient present with a clothes moth infestation. Infested clothing articles or small blankets and rugs can be dry-cleaned or laundered. Any items not in use should be laundered before being sealed in airtight storage containers. Mothball vapor at appropriate concentrations is lethal to the moths, and when possible, clothing should be stored with mothballs or flakes at the concentration recommended by the manufacturer.4 Individuals should avoid application of household insecticides to clothing or bedding, which may be poisonous to people.1,4 Freezing, heating, and dry ice fumigation techniques also can be used to treat infested products.3 Cedarwood usually is insufficient to deter an infestation, as the oil vapor rarely reaches an effective concentration to repel or harm the insects.3,4 Strict housekeeping with attention to vacuuming carpets, baseboards, closets, and laundering all linens and furniture covers can further reduce an infestation.4 Clothes items can be set in the sunlight and brushed to help loosen the pests, as they dislike direct light and may fall from the garments.3 Dust insecticides also can be used per the manufacturer label to treat crevices and baseboards in an active area of infestation that may otherwise be difficult to clean.3 If an extensive infestation exists or larger items are infested, then a professional pest control agency should be employed for proper eradication.
Conclusion
Understanding the life cycle and basic biology of clothes moths and other common household pests will help the clinician identify an infestation and counsel patients if an insect is a true ectoparasite. Clothes moth larvae are not parasites but can be found on clothing and can be confused with myiasis or true parasites.
Clothes moths are common pests found inside buildings such as homes, stores, and museums. The most common species of importance include the webbing clothes moth (Tineola bisselliella)(Figure) and the casemaking clothes moth (Tineola pellionella). Both species target textiles such as wool, rugs, feathers, felts, hair, furs, and even grains.1,2 They avoid synthetics and plant materials such as cottons.1
Characteristics
Adult clothes moths extend 7 to 8 mm and are a golden (T bisselliella) to brown (T pellionella) color with fringed wings and a tuft on their heads.1,3 Adults do not eat; females die within a few days of laying eggs, while males live approximately 1 month. Once laid, eggs hatch within 4 to 10 days.1,3 The larvae (caterpillars) incur damage to clothes and other household goods. Fully mature larvae are 12- to 13-mm long, and the Tineola species have white- to cream-colored bodies with brown heads. The webbing clothes moth larva lacks ocelli (eyes), while the casemaking moth larva has a singular ocellus.1
Transmission
An infestation is evidenced by woolen items that have furrows or holes in them. Pheromone traps also can expose an active infestation.3 The webbing moth larvae can be found beneath a self-spun silken mat on the food source that offers the insect protection and camouflage while it eats; the mat collects frass (feces) and clothes particles.1,3,4 The casemaking moth larvae drag around a portable silken bag that takes on the color of the fabric being eaten and serves as a refuge when disturbed.1,3,4 Both adult and larval stages prefer low light conditions. The total time of development from caterpillar to adult varies depending on the temperature and humidity of the environment, but most clothes moths complete their life cycle within 1 to 3 months.1
Management of an Infestation
Multiple infestation treatment options exist should a patient present with a clothes moth infestation. Infested clothing articles or small blankets and rugs can be dry-cleaned or laundered. Any items not in use should be laundered before being sealed in airtight storage containers. Mothball vapor at appropriate concentrations is lethal to the moths, and when possible, clothing should be stored with mothballs or flakes at the concentration recommended by the manufacturer.4 Individuals should avoid application of household insecticides to clothing or bedding, which may be poisonous to people.1,4 Freezing, heating, and dry ice fumigation techniques also can be used to treat infested products.3 Cedarwood usually is insufficient to deter an infestation, as the oil vapor rarely reaches an effective concentration to repel or harm the insects.3,4 Strict housekeeping with attention to vacuuming carpets, baseboards, closets, and laundering all linens and furniture covers can further reduce an infestation.4 Clothes items can be set in the sunlight and brushed to help loosen the pests, as they dislike direct light and may fall from the garments.3 Dust insecticides also can be used per the manufacturer label to treat crevices and baseboards in an active area of infestation that may otherwise be difficult to clean.3 If an extensive infestation exists or larger items are infested, then a professional pest control agency should be employed for proper eradication.
Conclusion
Understanding the life cycle and basic biology of clothes moths and other common household pests will help the clinician identify an infestation and counsel patients if an insect is a true ectoparasite. Clothes moth larvae are not parasites but can be found on clothing and can be confused with myiasis or true parasites.
- Jacobs S. Clothes moth. Penn State Extension website. http://ento.psu.edu/extension/factsheets/clothes-moth. Updated January 2013. Accessed May 14, 2018.
- Querner P. Insect pests and integrated pest management in museums, libraries and historic buildings. Insects. 2015;6:595-607.
- Choe D-H. Pest notes: clothes moths (publication 7435). University of California Agriculture & Natural Resources website. http://ipm.ucanr.edu/PMG/PESTNOTES/pn7435.html. Updated March 2013. Accessed May 14, 2018.
- Potter M. Entfact-609: clothes moths. Entomology at the University of Kentucky website. https://entomology.ca.uky.edu/ef609. Updated October 2001. Accessed May 14, 2018.
- Jacobs S. Clothes moth. Penn State Extension website. http://ento.psu.edu/extension/factsheets/clothes-moth. Updated January 2013. Accessed May 14, 2018.
- Querner P. Insect pests and integrated pest management in museums, libraries and historic buildings. Insects. 2015;6:595-607.
- Choe D-H. Pest notes: clothes moths (publication 7435). University of California Agriculture & Natural Resources website. http://ipm.ucanr.edu/PMG/PESTNOTES/pn7435.html. Updated March 2013. Accessed May 14, 2018.
- Potter M. Entfact-609: clothes moths. Entomology at the University of Kentucky website. https://entomology.ca.uky.edu/ef609. Updated October 2001. Accessed May 14, 2018.
Practice Points
- Clothes moth larvae are common household pests that may be misidentified as a parasitic infection such as myiasis when found on a person.
- Understanding the basic biology of clothes moths will help the clinician identify an infestation and appropriately counsel patients that clothes moths do not pose a considerable health risk.

Modifier -25 Victory, But the Battle Is Not Over
On February 23, 2018, Anthem Insurance Companies, Inc, announced the reversal of its proposed policy to reduce reimbursement for evaluation and management (E/M) services billed using modifier -25.1 This win for physicians was the result of a broad-based, multipronged advocacy campaign, and the American Academy of Dermatology Association (AADA) was critical to this victory.
Dermatology Took the Lead in Opposing Policy That Would Reduce Reimbursement
Dermatology was the first to trumpet the dangers of modifier -25 reduction policies and explain why other specialty societies should care. The AADA took the lead in assembling a coalition of physician groups to oppose the proposed policy by sharing language for opposition letters and meeting talking points with many societies outside of dermatology as well as producing the first draft for the American Medical Association (AMA) House of Delegates’ resolution that spurred action against Anthem’s proposed policy.2 Members of the AADA attended numerous conference calls and in-person meetings with health insurance officials to urge them to reverse the policy and helped coordinate opposition from state and national specialty societies. The influence and advocacy of the AMA was critical in reversing the proposed policy, but dermatology started the opposition and organized the players to bring the fight against Anthem.
AADA Continues to Fight
Despite the victory against Anthem, challenges to fair reimbursement of modifier -25 claims are ongoing. Two insurers recently announced implementation of new modifier -25 reduction policies.3,4 Moreover, 4 other insurers have existing modifier -25 reduction policies in place.5-8 The AADA has engaged, and will continue engaging, each of these insurers with the message that the ability to perform procedures and distinct E/M services on the same day is integral to efficient, patient-centered care of dermatologic diseases. The AADA argues that insurers’ rationale (overlapping indirect practice expenses) for payment reduction is improper and that appropriately documented modifier -25 claims should be reimbursed at 100% of allowable charges.
In addition to the existing insurer policies affecting modifier -25, Anthem has announced that it plans to conduct audits of modifier -25 claims with recoupments of inappropriate charges.1 Some Medicare Administrative Contractors also have cited modifier -25 as an area of concern and issued guidelines for reporting modifier -25, which frequently precede audits.9 The AADA is concerned that if Anthem follows through with its aggressive audits to recoup money, which can result in substantial take-backs, and finds a high error rate in modifier -25 claims, it also may consider revisiting its reduction policy. Moreover, it is likely that other insurers will use failed modifier -25 audits as an excuse to continue, expand, or adopt modifier -25 reduction policies. It is clear that blanket reduction in payment is much easier and less expensive for insurers than auditing medical records and not paying those who abuse modifier -25. As long as insurers are under financial pressure, modifier -25 will be a tempting target for reducing health care costs.
How to Use Modifier -25 Correctly
In addition to opposing inappropriate reimbursement of modifier -25, the AADA is committed to educating its members and insurers on the correct use and documentation of modifier -25. In December 2017, Mollie A. MacCormack, MD (Nashua, New Hampshire), and I led an educational webinar on modifier -25 for more than 1100 attendees.10 We discussed the performance standards and documentation requirements of modifier -25. It was clear that dermatologists were interested not only in the correct coding of modifier -25 claims but also avoiding the consequences of audits.
My peers frequently ask, “What can we [dermatologists] do to prepare for potential modifier -25 audits?” My advice is always, “Physician audit thyself.” I recommend self-auditing to make sure you and your practice are in compliance with modifier -25 documentation requirements. In a March 2017 Cutis column on modifier -25, I discussed what constitutes separate and distinct E/M services and what is included in a procedure’s global surgical package.11 A self-audit is as simple as pulling 10 to 20 medical records in which a same-day procedure and E/M service were billed. Cross out any information in the note included in the procedure’s global surgical package including history associated with establishing the diagnosis, physical examination of the procedure area(s), and discussion of treatment options. If complete documentation for an E/M is still present after removing the procedure and associated evaluation, you have passed the self-audit. If not, consider changing your coding to better reflect the documentation in your records. In my experience, insurance auditors are not physicians and often are not even medical professionals. Clear documentation and clear existence of a separate, distinct, and medically necessary E/M service is needed to succeed in a modifier -25 audit.
Final Thoughts
Dermatologists should rejoice that Anthem decided to rescind its modifier -25 policy. If this policy had gone into effect, the modifier -25 reduction would likely have spread to most other insurers as industry standard. This victory certainly shows what can be accomplished when organized medicine works together. Your state dermatology and medical societies, national societies, and the AMA collaborated on a critical existential threat to cost-effective and efficient patient care and won. These organizations deserve your membership and support as well as your thanks; however, our celebration must be short-lived, as there still are other insurers with modifier -25 reductions in place, and audits have been promised. We must continue to focus on proper use and documentation of modifier -25. This effort will not only help dermatologists decrease the risk of large recoupments from audits but also help the AADA continue to oppose inappropriate payment policies.
- Anthem Insurance Companies, Inc. Network Update. April 2018. https://www11.anthem.com/provider/ct/f5/s1/t0/pw_g334020.pdf?refer=ahpprovider&state=ct. Accessed May 25, 2018.
- American Academy of Dermatology, American Society for Dermatologic Surgery Association, American College of Mohs Surgery, American Society of Dermatopathology, Society for Investigative Dermatology. Opposition to reduced payment for the 25 modifier. https://www.ama-assn.org/sites/default/files/media-browser/public/hod/i17-808.pdf. Accessed May 14, 2018.
- We’re changing the payment policy when evaluation and management services are billed with surgery. Blue Cross Blue Shield of Michigan website. https://www.bcbsm.com/newsletter/therecord/2018/record_0418/Record_0418u.shtml. Published April 2018. Accessed May 14, 2018.
- Centene Corporation. Payment policy: problem oriented visits billed with surgical procedures. https://www.superiorhealthplan.com/content/dam/centene/Superior/Provider/PDFs/Problem%20Oriented%20Visits%20Billed
%20with%20Surgical%20Procedures%20(Ambetter%20MMP
%20and%20Medicare%20Advantage%20Only).pdf. Accessed May 14, 2018. - Blue Cross Blue Shield of Rhode Island. Payment Policy: Coding and Payment Guidelines. August 16, 2016. https://www.bcbsri.com/sites/default/files/polices/Coding-and-Payment-Guidelines-Oct2016.pdf. Accessed May 9, 2018.
- Modifier payment policy. Tufts Health Plan website. https://tuftshealthplan.com/documents/providers/payment-policies/modifier-payment-policy. Updated November 2017. Accessed May 14, 2018.
- Harvard Pilgrim Health Care. Payment Policies: Evaluation and Management. February 2018. https://www.harvardpilgrim.org/pls/portal/docs/PAGE/PROVIDERS/MANUALS/PAYMENT%20POLICIES/H-2%20EVALUATION-MGT_02
0118.PDF. Accessed May 25, 2018. - Updates to the policies on modifier 25 reporting and reimbursement. Independence Blue Cross website. http://provcomm.ibx.com/ProvComm/ProvComm.nsf/4bcc623b93e226638525792c00575962/bbe9e72728cc01e285258167005c629d!OpenDocument. Published July 24, 2017. Accessed May 14, 2018.
- Centers for Medicare & Medicaid Services. Payment for evaluation and management services provided during global period of surgery. MLN Matters. May 19, 2006. https://www.cms.gov/Outreach-and-Education/Medicare-Learning-Network-MLN/MLNMattersArticles/downloads/MM5025.pdf. Accessed May 14, 2018.
- Audits on modifier 25 are coming—complimentary live webinar. American Academy of Dermatology website. https://store.aad.org/products/11928. Accessed May 9, 2018.
- Rogers HW. One diagnosis and modifier -25: appropriate or audit target? Cutis. 2017;99:165-166.
On February 23, 2018, Anthem Insurance Companies, Inc, announced the reversal of its proposed policy to reduce reimbursement for evaluation and management (E/M) services billed using modifier -25.1 This win for physicians was the result of a broad-based, multipronged advocacy campaign, and the American Academy of Dermatology Association (AADA) was critical to this victory.
Dermatology Took the Lead in Opposing Policy That Would Reduce Reimbursement
Dermatology was the first to trumpet the dangers of modifier -25 reduction policies and explain why other specialty societies should care. The AADA took the lead in assembling a coalition of physician groups to oppose the proposed policy by sharing language for opposition letters and meeting talking points with many societies outside of dermatology as well as producing the first draft for the American Medical Association (AMA) House of Delegates’ resolution that spurred action against Anthem’s proposed policy.2 Members of the AADA attended numerous conference calls and in-person meetings with health insurance officials to urge them to reverse the policy and helped coordinate opposition from state and national specialty societies. The influence and advocacy of the AMA was critical in reversing the proposed policy, but dermatology started the opposition and organized the players to bring the fight against Anthem.
AADA Continues to Fight
Despite the victory against Anthem, challenges to fair reimbursement of modifier -25 claims are ongoing. Two insurers recently announced implementation of new modifier -25 reduction policies.3,4 Moreover, 4 other insurers have existing modifier -25 reduction policies in place.5-8 The AADA has engaged, and will continue engaging, each of these insurers with the message that the ability to perform procedures and distinct E/M services on the same day is integral to efficient, patient-centered care of dermatologic diseases. The AADA argues that insurers’ rationale (overlapping indirect practice expenses) for payment reduction is improper and that appropriately documented modifier -25 claims should be reimbursed at 100% of allowable charges.
In addition to the existing insurer policies affecting modifier -25, Anthem has announced that it plans to conduct audits of modifier -25 claims with recoupments of inappropriate charges.1 Some Medicare Administrative Contractors also have cited modifier -25 as an area of concern and issued guidelines for reporting modifier -25, which frequently precede audits.9 The AADA is concerned that if Anthem follows through with its aggressive audits to recoup money, which can result in substantial take-backs, and finds a high error rate in modifier -25 claims, it also may consider revisiting its reduction policy. Moreover, it is likely that other insurers will use failed modifier -25 audits as an excuse to continue, expand, or adopt modifier -25 reduction policies. It is clear that blanket reduction in payment is much easier and less expensive for insurers than auditing medical records and not paying those who abuse modifier -25. As long as insurers are under financial pressure, modifier -25 will be a tempting target for reducing health care costs.
How to Use Modifier -25 Correctly
In addition to opposing inappropriate reimbursement of modifier -25, the AADA is committed to educating its members and insurers on the correct use and documentation of modifier -25. In December 2017, Mollie A. MacCormack, MD (Nashua, New Hampshire), and I led an educational webinar on modifier -25 for more than 1100 attendees.10 We discussed the performance standards and documentation requirements of modifier -25. It was clear that dermatologists were interested not only in the correct coding of modifier -25 claims but also avoiding the consequences of audits.
My peers frequently ask, “What can we [dermatologists] do to prepare for potential modifier -25 audits?” My advice is always, “Physician audit thyself.” I recommend self-auditing to make sure you and your practice are in compliance with modifier -25 documentation requirements. In a March 2017 Cutis column on modifier -25, I discussed what constitutes separate and distinct E/M services and what is included in a procedure’s global surgical package.11 A self-audit is as simple as pulling 10 to 20 medical records in which a same-day procedure and E/M service were billed. Cross out any information in the note included in the procedure’s global surgical package including history associated with establishing the diagnosis, physical examination of the procedure area(s), and discussion of treatment options. If complete documentation for an E/M is still present after removing the procedure and associated evaluation, you have passed the self-audit. If not, consider changing your coding to better reflect the documentation in your records. In my experience, insurance auditors are not physicians and often are not even medical professionals. Clear documentation and clear existence of a separate, distinct, and medically necessary E/M service is needed to succeed in a modifier -25 audit.
Final Thoughts
Dermatologists should rejoice that Anthem decided to rescind its modifier -25 policy. If this policy had gone into effect, the modifier -25 reduction would likely have spread to most other insurers as industry standard. This victory certainly shows what can be accomplished when organized medicine works together. Your state dermatology and medical societies, national societies, and the AMA collaborated on a critical existential threat to cost-effective and efficient patient care and won. These organizations deserve your membership and support as well as your thanks; however, our celebration must be short-lived, as there still are other insurers with modifier -25 reductions in place, and audits have been promised. We must continue to focus on proper use and documentation of modifier -25. This effort will not only help dermatologists decrease the risk of large recoupments from audits but also help the AADA continue to oppose inappropriate payment policies.
On February 23, 2018, Anthem Insurance Companies, Inc, announced the reversal of its proposed policy to reduce reimbursement for evaluation and management (E/M) services billed using modifier -25.1 This win for physicians was the result of a broad-based, multipronged advocacy campaign, and the American Academy of Dermatology Association (AADA) was critical to this victory.
Dermatology Took the Lead in Opposing Policy That Would Reduce Reimbursement
Dermatology was the first to trumpet the dangers of modifier -25 reduction policies and explain why other specialty societies should care. The AADA took the lead in assembling a coalition of physician groups to oppose the proposed policy by sharing language for opposition letters and meeting talking points with many societies outside of dermatology as well as producing the first draft for the American Medical Association (AMA) House of Delegates’ resolution that spurred action against Anthem’s proposed policy.2 Members of the AADA attended numerous conference calls and in-person meetings with health insurance officials to urge them to reverse the policy and helped coordinate opposition from state and national specialty societies. The influence and advocacy of the AMA was critical in reversing the proposed policy, but dermatology started the opposition and organized the players to bring the fight against Anthem.
AADA Continues to Fight
Despite the victory against Anthem, challenges to fair reimbursement of modifier -25 claims are ongoing. Two insurers recently announced implementation of new modifier -25 reduction policies.3,4 Moreover, 4 other insurers have existing modifier -25 reduction policies in place.5-8 The AADA has engaged, and will continue engaging, each of these insurers with the message that the ability to perform procedures and distinct E/M services on the same day is integral to efficient, patient-centered care of dermatologic diseases. The AADA argues that insurers’ rationale (overlapping indirect practice expenses) for payment reduction is improper and that appropriately documented modifier -25 claims should be reimbursed at 100% of allowable charges.
In addition to the existing insurer policies affecting modifier -25, Anthem has announced that it plans to conduct audits of modifier -25 claims with recoupments of inappropriate charges.1 Some Medicare Administrative Contractors also have cited modifier -25 as an area of concern and issued guidelines for reporting modifier -25, which frequently precede audits.9 The AADA is concerned that if Anthem follows through with its aggressive audits to recoup money, which can result in substantial take-backs, and finds a high error rate in modifier -25 claims, it also may consider revisiting its reduction policy. Moreover, it is likely that other insurers will use failed modifier -25 audits as an excuse to continue, expand, or adopt modifier -25 reduction policies. It is clear that blanket reduction in payment is much easier and less expensive for insurers than auditing medical records and not paying those who abuse modifier -25. As long as insurers are under financial pressure, modifier -25 will be a tempting target for reducing health care costs.
How to Use Modifier -25 Correctly
In addition to opposing inappropriate reimbursement of modifier -25, the AADA is committed to educating its members and insurers on the correct use and documentation of modifier -25. In December 2017, Mollie A. MacCormack, MD (Nashua, New Hampshire), and I led an educational webinar on modifier -25 for more than 1100 attendees.10 We discussed the performance standards and documentation requirements of modifier -25. It was clear that dermatologists were interested not only in the correct coding of modifier -25 claims but also avoiding the consequences of audits.
My peers frequently ask, “What can we [dermatologists] do to prepare for potential modifier -25 audits?” My advice is always, “Physician audit thyself.” I recommend self-auditing to make sure you and your practice are in compliance with modifier -25 documentation requirements. In a March 2017 Cutis column on modifier -25, I discussed what constitutes separate and distinct E/M services and what is included in a procedure’s global surgical package.11 A self-audit is as simple as pulling 10 to 20 medical records in which a same-day procedure and E/M service were billed. Cross out any information in the note included in the procedure’s global surgical package including history associated with establishing the diagnosis, physical examination of the procedure area(s), and discussion of treatment options. If complete documentation for an E/M is still present after removing the procedure and associated evaluation, you have passed the self-audit. If not, consider changing your coding to better reflect the documentation in your records. In my experience, insurance auditors are not physicians and often are not even medical professionals. Clear documentation and clear existence of a separate, distinct, and medically necessary E/M service is needed to succeed in a modifier -25 audit.
Final Thoughts
Dermatologists should rejoice that Anthem decided to rescind its modifier -25 policy. If this policy had gone into effect, the modifier -25 reduction would likely have spread to most other insurers as industry standard. This victory certainly shows what can be accomplished when organized medicine works together. Your state dermatology and medical societies, national societies, and the AMA collaborated on a critical existential threat to cost-effective and efficient patient care and won. These organizations deserve your membership and support as well as your thanks; however, our celebration must be short-lived, as there still are other insurers with modifier -25 reductions in place, and audits have been promised. We must continue to focus on proper use and documentation of modifier -25. This effort will not only help dermatologists decrease the risk of large recoupments from audits but also help the AADA continue to oppose inappropriate payment policies.
- Anthem Insurance Companies, Inc. Network Update. April 2018. https://www11.anthem.com/provider/ct/f5/s1/t0/pw_g334020.pdf?refer=ahpprovider&state=ct. Accessed May 25, 2018.
- American Academy of Dermatology, American Society for Dermatologic Surgery Association, American College of Mohs Surgery, American Society of Dermatopathology, Society for Investigative Dermatology. Opposition to reduced payment for the 25 modifier. https://www.ama-assn.org/sites/default/files/media-browser/public/hod/i17-808.pdf. Accessed May 14, 2018.
- We’re changing the payment policy when evaluation and management services are billed with surgery. Blue Cross Blue Shield of Michigan website. https://www.bcbsm.com/newsletter/therecord/2018/record_0418/Record_0418u.shtml. Published April 2018. Accessed May 14, 2018.
- Centene Corporation. Payment policy: problem oriented visits billed with surgical procedures. https://www.superiorhealthplan.com/content/dam/centene/Superior/Provider/PDFs/Problem%20Oriented%20Visits%20Billed
%20with%20Surgical%20Procedures%20(Ambetter%20MMP
%20and%20Medicare%20Advantage%20Only).pdf. Accessed May 14, 2018. - Blue Cross Blue Shield of Rhode Island. Payment Policy: Coding and Payment Guidelines. August 16, 2016. https://www.bcbsri.com/sites/default/files/polices/Coding-and-Payment-Guidelines-Oct2016.pdf. Accessed May 9, 2018.
- Modifier payment policy. Tufts Health Plan website. https://tuftshealthplan.com/documents/providers/payment-policies/modifier-payment-policy. Updated November 2017. Accessed May 14, 2018.
- Harvard Pilgrim Health Care. Payment Policies: Evaluation and Management. February 2018. https://www.harvardpilgrim.org/pls/portal/docs/PAGE/PROVIDERS/MANUALS/PAYMENT%20POLICIES/H-2%20EVALUATION-MGT_02
0118.PDF. Accessed May 25, 2018. - Updates to the policies on modifier 25 reporting and reimbursement. Independence Blue Cross website. http://provcomm.ibx.com/ProvComm/ProvComm.nsf/4bcc623b93e226638525792c00575962/bbe9e72728cc01e285258167005c629d!OpenDocument. Published July 24, 2017. Accessed May 14, 2018.
- Centers for Medicare & Medicaid Services. Payment for evaluation and management services provided during global period of surgery. MLN Matters. May 19, 2006. https://www.cms.gov/Outreach-and-Education/Medicare-Learning-Network-MLN/MLNMattersArticles/downloads/MM5025.pdf. Accessed May 14, 2018.
- Audits on modifier 25 are coming—complimentary live webinar. American Academy of Dermatology website. https://store.aad.org/products/11928. Accessed May 9, 2018.
- Rogers HW. One diagnosis and modifier -25: appropriate or audit target? Cutis. 2017;99:165-166.
- Anthem Insurance Companies, Inc. Network Update. April 2018. https://www11.anthem.com/provider/ct/f5/s1/t0/pw_g334020.pdf?refer=ahpprovider&state=ct. Accessed May 25, 2018.
- American Academy of Dermatology, American Society for Dermatologic Surgery Association, American College of Mohs Surgery, American Society of Dermatopathology, Society for Investigative Dermatology. Opposition to reduced payment for the 25 modifier. https://www.ama-assn.org/sites/default/files/media-browser/public/hod/i17-808.pdf. Accessed May 14, 2018.
- We’re changing the payment policy when evaluation and management services are billed with surgery. Blue Cross Blue Shield of Michigan website. https://www.bcbsm.com/newsletter/therecord/2018/record_0418/Record_0418u.shtml. Published April 2018. Accessed May 14, 2018.
- Centene Corporation. Payment policy: problem oriented visits billed with surgical procedures. https://www.superiorhealthplan.com/content/dam/centene/Superior/Provider/PDFs/Problem%20Oriented%20Visits%20Billed
%20with%20Surgical%20Procedures%20(Ambetter%20MMP
%20and%20Medicare%20Advantage%20Only).pdf. Accessed May 14, 2018. - Blue Cross Blue Shield of Rhode Island. Payment Policy: Coding and Payment Guidelines. August 16, 2016. https://www.bcbsri.com/sites/default/files/polices/Coding-and-Payment-Guidelines-Oct2016.pdf. Accessed May 9, 2018.
- Modifier payment policy. Tufts Health Plan website. https://tuftshealthplan.com/documents/providers/payment-policies/modifier-payment-policy. Updated November 2017. Accessed May 14, 2018.
- Harvard Pilgrim Health Care. Payment Policies: Evaluation and Management. February 2018. https://www.harvardpilgrim.org/pls/portal/docs/PAGE/PROVIDERS/MANUALS/PAYMENT%20POLICIES/H-2%20EVALUATION-MGT_02
0118.PDF. Accessed May 25, 2018. - Updates to the policies on modifier 25 reporting and reimbursement. Independence Blue Cross website. http://provcomm.ibx.com/ProvComm/ProvComm.nsf/4bcc623b93e226638525792c00575962/bbe9e72728cc01e285258167005c629d!OpenDocument. Published July 24, 2017. Accessed May 14, 2018.
- Centers for Medicare & Medicaid Services. Payment for evaluation and management services provided during global period of surgery. MLN Matters. May 19, 2006. https://www.cms.gov/Outreach-and-Education/Medicare-Learning-Network-MLN/MLNMattersArticles/downloads/MM5025.pdf. Accessed May 14, 2018.
- Audits on modifier 25 are coming—complimentary live webinar. American Academy of Dermatology website. https://store.aad.org/products/11928. Accessed May 9, 2018.
- Rogers HW. One diagnosis and modifier -25: appropriate or audit target? Cutis. 2017;99:165-166.
Practice Points
- Insurers are increasingly targeting modifier -25 for audits and payment reductions.
- Physicians should understand modifier -25 documentation requirements and self-audit to ensure compliance.

Updated Guidelines on Peanut Allergy Prevention in Infants With Atopic Dermatitis
It has been said that “extraordinary claims require extraordinary evidence.”1 In the pursuit of evidence-based medicine, we are encouraged to follow a similar standard, with an emphasis on waiting for multiple studies with good-quality data and high levels of agreement before changing any aspect of our clinical practice. The ostensible purpose is that studies can be flawed, conclusions can be incorrect, or biases can be overlooked. In such cases, acting on questionable results could imperil patients. It is for this reason that so many review articles sometimes frustratingly seem to conclude that further evidence is needed.2
Based on this standard, recently published addendum guidelines from the National Institute of Allergy and Infectious Diseases for prevention of peanut allergy in the United States3 are somewhat striking in that they make fairly bold recommendations based on results from the 2015 Learning Early about Peanut Allergy (LEAP) study,4 a randomized trial evaluating early peanut introduction as a preventive strategy for peanut allergy. Of note, this study was not placebo controlled, was conducted at only 1 site in the United Kingdom, and only included 640 children, though the number of participants was admittedly large for this type of study.4 Arguably, the LEAP study alone does not provide enough evidence upon which to base what essentially amounts to an about-face in the official recommendations for prevention of peanut and other food allergies, which emphasized delayed introduction of high-risk foods, especially in high-risk individuals.5-7 To better understand this shift, we need to briefly explore the context of the addendum guidelines.
As many as one-third of pediatric patients with atopic dermatitis (AD) have food allergies, thus diet often is invoked by patients and providers alike as an underlying cause of the disease.8 Many patients in my practice are so focused on potential food allergies that actual treatment of the affected skin is marginalized and often dismissed as a stopgap that does not address the root of the problem. A 2004 study of 100 children with AD found that diet was manipulated by the parents in 75% of patients in an attempt to manage the disease.9
Patients are not the only ones who consider food allergies to be a driving force in AD. The medical literature indicates that this theory has existed for centuries; for instance, with regard to the relationship between diet and AD, the author of an article from 1830 quipped, “There is probably no subject in which more deeply rooted convictions have been held . . . than the connection between diet and disease, both as regards the causation and treatment of the latter . . .”10 More apropos perhaps is a statement from the 2010 National Institute of Allergy and Infectious Diseases guidelines on food allergy management, which noted that while the expert group “does not mean to imply that AD results from [food allergies], the role of [food allergies] in the pathogenesis and severity of this condition remains controversial.”11
Prior to the LEAP study, food allergy recommendations for clinical practice in the United Kingdom in 199812 and the United States in 200013 recommended excluding allergenic foods (eg, peanuts, tree nuts, soy, milk, eggs) from the diet in infants with a family history of atopy until 3 years of age. However, those recommendations did not seem to be working, when in fact just the opposite was happening. From 1997 until the LEAP study was conducted in 2015, the prevalence of peanut allergy more than quadrupled and became the leading cause of anaphylaxis and death related to food allergy.14 Additionally, study after study concluded that elimination diets did not seem to help most patients with AD.15 As is required in good scientific thinking, when a hypothesis is proven false, other approaches must be considered.
The idea arose that perhaps delaying introduction of allergenic foods was the opposite of the answer.4 The LEAP study tested the notion that peanut allergies are rare in countries where peanuts are introduced early and if telling families to delay introduction of peanuts in infants might actually be causing development of a peanut allergy, and the tests bore fruit. It was found that giving infants peanut-containing foods resulted in a more than 80% reduction in peanut allergy at 5 years of age (P<.001).4 What was perhaps even more interesting was the connection between AD and peanut allergy. An important idea articulated in the LEAP study is in some ways revolutionary: Rather than foods causing AD, it could be that “early environmental exposure (through the skin) to peanut may account for early sensitization, whereas early oral exposure may lead to immune tolerance.”4 This concept—that impaired eczematous skin may actually lead to the development of food allergies—turns the whole thing upside down.
What do these updated guidelines actually suggest? The first guideline focuses on infants with severe AD, egg allergy, or both, who therefore are thought to be at the highest risk for developing peanut allergy.3 Because of the higher baseline risk in this subgroup, measurement of the peanut-specific IgE (peanut sIgE) level, skin prick testing (SPT), or both is strongly recommended before introducing peanut protein into the diet. This testing can be performed by qualified providers as a screening measure, but if positive (≥0.35 kUA/L for peanut sIgE or >2 mm on the peanut SPT), referral to an allergy specialist is warranted. If these studies are negative, it is thought the likelihood of peanut allergy is low, and it is recommended that caregivers introduce age-appropriate peanut-containing foods (eg, peanut butter snack puffs, diluted peanut butter) as early as 4 to 6 months of age. The second guideline recommends that peanut-containing foods should be introduced into the diets of infants with mild or moderate AD at approximately 6 months of age without the need for prior screening via peanut sIgE or SPT. Lastly, the third guideline recommends that caregivers freely introduce peanut-containing foods together with other solid foods in infants without AD or food allergies in accordance with family preference.3
The results of the LEAP study are certainly exciting, and although the theoretical basis makes good scientific sense and the updated guidelines truly address an important and growing problem, there are several issues with this update that are worth considering. Given the constraints of the LEAP study, it certainly seems possible that the results will not be applicable to all populations or foods. More research is needed to ensure that this robust finding applies to other children and to explore the introduction of other allergenic foods, which the LEAP study investigators also emphasized.4
In fairness, the updated guidelines clearly state the quality of evidence of their recommendations and make it clear that expert opinion is playing a large role.3 For the first guideline regarding recommendations for those with severe AD and/or egg allergy, the quality of evidence is deemed moderate, while the contribution of expert opinion is listed as significant. For the second and third guidelines regarding recommendations for mild to moderate AD and those without AD, respectively, the quality of evidence is low and expert opinion is again listed as significant.3
Importantly, delineating severe AD from moderate disease—which is necessary because only severe AD warrants evaluation with peanut sIgE and/or SPT—can be difficult, as the distinction relies on a degree of subjectivity that may vary between specialists. Indeed, 2 publications suggest extending the definition of severe AD to include infants with early-onset AD (<3 months of age) and those with moderate AD not responding to treatment.16,17
Despite these reservations, the updated guidelines represent a breakthrough in understanding in an area truly in need of advancement. Although the evidence may not be exactly extraordinary, the context for these developments and our deeper understanding suggest that we do indeed live in extraordinary times.
- Encyclopaedia Galactica [television transcript]. Cosmos: A Personal Voyage. Public Broadcasting Service. December 14, 1980.
- Ezzo J, Bausell B, Moerman DE, et al. Reviewing the reviews: how strong is the evidence? how clear are the conclusions? Int J Technol Assess Health Care. 2001;17:457-466.
- Togias A, Cooper SF, Acebal ML, et al. Addendum guidelines for the prevention of peanut allergy in the United States: report of the National Institute of Allergy and Infectious Diseases–sponsored expert panel.J Allergy Clin Immunol. 2017;139:29-44.
- Du Toit G, Roberts G, Sayre PH, et al. Randomized trial of peanut consumption in infants at risk for peanut allergy. N Engl J Med. 2015;372:803-813.
- Høst A, Koletzko B, Dreborg S, et al. Dietary products used in infants for treatment and prevention of food allergy. joint statement of the European Society for Paediatric Allergology and Clinical Immunology (ESPACI) Committee on Hypoallergenic Formulas and the European Society for Paediatric Gastroenterology, Hepatology and Nutrition (ESPGHAN) Committee on Nutrition. Arch Dis Child. 1999;81:80-84.
- American Academy of Pediatrics. Committee on Nutrition. hypoallergenic infant formulas. Pediatrics. 2000;106(2, pt 1):346-349.
- Fiocchi A, Assa’ad A, Bahna S; Adverse Reactions to Foods Committee; American College of Allergy, Asthma and Immunology. Food allergy and the introduction of solid foods to infants: a consensus document. Ann Allergy Asthma Immunol. 2006;97:10-20.
- Thompson MM, Hanifin JM. Effective therapy of childhood atopic dermatitis allays food allergy concerns. J Am Acad Dermatol. 2005;53(2 suppl 2):S214-S219.
- Johnston GA, Bilbao RM, Graham-Brown RA. The use of dietary manipulation by parents of children with atopic dermatitis. Br J Dermatol. 2004;150:1186-1189.
- Mackenzie S. The inaugural address on the advantages to be derived from the study of dermatology. BMJ. 1830;1:193-197.
- Boyce JA, Assa’ad A, Burks AW, et al; NIAID-Sponsored Expert Panel. Guidelines for the diagnosis and management of food allergy in the United States: report of the NIAID-sponsored expert panel. J Allergy Clin Immunol. 2010;126(6 suppl):S1-S58.
- Committee on Toxicity of Chemicals in Food, Consumer Products and the Environment. Peanut Allergy. London, England: Department of Health; 1998.
- American Academy of Pediatrics Committee on Nutrition. Hypoallergenic infant formulas. Pediatrics. 2000;106(2, pt 1):346-349.
- Gruchalla RS, Sampson HA. Preventing peanut allergy through early consumption—ready for prime time? N Engl J Med. 2015;372:875-877.
- Lim NR, Lohman ME, Lio PA. The role of elimination diets in atopic dermatitis: a comprehensive review. Pediatr Dermatol. 2017;34:516-527.
- Wong CC, Allen KJ, Orchard D. Changes to infant feeding guidelines: relevance to dermatologists. Australas J Dermatol. 2017;58:e171-e175.
- Martin PE, Eckert JK, Koplin JJ, et al. Which infants with eczema are at risk of food allergy? results from a population-based cohort. Clin Exp Allergy. 2015;45:255-264.
It has been said that “extraordinary claims require extraordinary evidence.”1 In the pursuit of evidence-based medicine, we are encouraged to follow a similar standard, with an emphasis on waiting for multiple studies with good-quality data and high levels of agreement before changing any aspect of our clinical practice. The ostensible purpose is that studies can be flawed, conclusions can be incorrect, or biases can be overlooked. In such cases, acting on questionable results could imperil patients. It is for this reason that so many review articles sometimes frustratingly seem to conclude that further evidence is needed.2
Based on this standard, recently published addendum guidelines from the National Institute of Allergy and Infectious Diseases for prevention of peanut allergy in the United States3 are somewhat striking in that they make fairly bold recommendations based on results from the 2015 Learning Early about Peanut Allergy (LEAP) study,4 a randomized trial evaluating early peanut introduction as a preventive strategy for peanut allergy. Of note, this study was not placebo controlled, was conducted at only 1 site in the United Kingdom, and only included 640 children, though the number of participants was admittedly large for this type of study.4 Arguably, the LEAP study alone does not provide enough evidence upon which to base what essentially amounts to an about-face in the official recommendations for prevention of peanut and other food allergies, which emphasized delayed introduction of high-risk foods, especially in high-risk individuals.5-7 To better understand this shift, we need to briefly explore the context of the addendum guidelines.
As many as one-third of pediatric patients with atopic dermatitis (AD) have food allergies, thus diet often is invoked by patients and providers alike as an underlying cause of the disease.8 Many patients in my practice are so focused on potential food allergies that actual treatment of the affected skin is marginalized and often dismissed as a stopgap that does not address the root of the problem. A 2004 study of 100 children with AD found that diet was manipulated by the parents in 75% of patients in an attempt to manage the disease.9
Patients are not the only ones who consider food allergies to be a driving force in AD. The medical literature indicates that this theory has existed for centuries; for instance, with regard to the relationship between diet and AD, the author of an article from 1830 quipped, “There is probably no subject in which more deeply rooted convictions have been held . . . than the connection between diet and disease, both as regards the causation and treatment of the latter . . .”10 More apropos perhaps is a statement from the 2010 National Institute of Allergy and Infectious Diseases guidelines on food allergy management, which noted that while the expert group “does not mean to imply that AD results from [food allergies], the role of [food allergies] in the pathogenesis and severity of this condition remains controversial.”11
Prior to the LEAP study, food allergy recommendations for clinical practice in the United Kingdom in 199812 and the United States in 200013 recommended excluding allergenic foods (eg, peanuts, tree nuts, soy, milk, eggs) from the diet in infants with a family history of atopy until 3 years of age. However, those recommendations did not seem to be working, when in fact just the opposite was happening. From 1997 until the LEAP study was conducted in 2015, the prevalence of peanut allergy more than quadrupled and became the leading cause of anaphylaxis and death related to food allergy.14 Additionally, study after study concluded that elimination diets did not seem to help most patients with AD.15 As is required in good scientific thinking, when a hypothesis is proven false, other approaches must be considered.
The idea arose that perhaps delaying introduction of allergenic foods was the opposite of the answer.4 The LEAP study tested the notion that peanut allergies are rare in countries where peanuts are introduced early and if telling families to delay introduction of peanuts in infants might actually be causing development of a peanut allergy, and the tests bore fruit. It was found that giving infants peanut-containing foods resulted in a more than 80% reduction in peanut allergy at 5 years of age (P<.001).4 What was perhaps even more interesting was the connection between AD and peanut allergy. An important idea articulated in the LEAP study is in some ways revolutionary: Rather than foods causing AD, it could be that “early environmental exposure (through the skin) to peanut may account for early sensitization, whereas early oral exposure may lead to immune tolerance.”4 This concept—that impaired eczematous skin may actually lead to the development of food allergies—turns the whole thing upside down.
What do these updated guidelines actually suggest? The first guideline focuses on infants with severe AD, egg allergy, or both, who therefore are thought to be at the highest risk for developing peanut allergy.3 Because of the higher baseline risk in this subgroup, measurement of the peanut-specific IgE (peanut sIgE) level, skin prick testing (SPT), or both is strongly recommended before introducing peanut protein into the diet. This testing can be performed by qualified providers as a screening measure, but if positive (≥0.35 kUA/L for peanut sIgE or >2 mm on the peanut SPT), referral to an allergy specialist is warranted. If these studies are negative, it is thought the likelihood of peanut allergy is low, and it is recommended that caregivers introduce age-appropriate peanut-containing foods (eg, peanut butter snack puffs, diluted peanut butter) as early as 4 to 6 months of age. The second guideline recommends that peanut-containing foods should be introduced into the diets of infants with mild or moderate AD at approximately 6 months of age without the need for prior screening via peanut sIgE or SPT. Lastly, the third guideline recommends that caregivers freely introduce peanut-containing foods together with other solid foods in infants without AD or food allergies in accordance with family preference.3
The results of the LEAP study are certainly exciting, and although the theoretical basis makes good scientific sense and the updated guidelines truly address an important and growing problem, there are several issues with this update that are worth considering. Given the constraints of the LEAP study, it certainly seems possible that the results will not be applicable to all populations or foods. More research is needed to ensure that this robust finding applies to other children and to explore the introduction of other allergenic foods, which the LEAP study investigators also emphasized.4
In fairness, the updated guidelines clearly state the quality of evidence of their recommendations and make it clear that expert opinion is playing a large role.3 For the first guideline regarding recommendations for those with severe AD and/or egg allergy, the quality of evidence is deemed moderate, while the contribution of expert opinion is listed as significant. For the second and third guidelines regarding recommendations for mild to moderate AD and those without AD, respectively, the quality of evidence is low and expert opinion is again listed as significant.3
Importantly, delineating severe AD from moderate disease—which is necessary because only severe AD warrants evaluation with peanut sIgE and/or SPT—can be difficult, as the distinction relies on a degree of subjectivity that may vary between specialists. Indeed, 2 publications suggest extending the definition of severe AD to include infants with early-onset AD (<3 months of age) and those with moderate AD not responding to treatment.16,17
Despite these reservations, the updated guidelines represent a breakthrough in understanding in an area truly in need of advancement. Although the evidence may not be exactly extraordinary, the context for these developments and our deeper understanding suggest that we do indeed live in extraordinary times.
It has been said that “extraordinary claims require extraordinary evidence.”1 In the pursuit of evidence-based medicine, we are encouraged to follow a similar standard, with an emphasis on waiting for multiple studies with good-quality data and high levels of agreement before changing any aspect of our clinical practice. The ostensible purpose is that studies can be flawed, conclusions can be incorrect, or biases can be overlooked. In such cases, acting on questionable results could imperil patients. It is for this reason that so many review articles sometimes frustratingly seem to conclude that further evidence is needed.2
Based on this standard, recently published addendum guidelines from the National Institute of Allergy and Infectious Diseases for prevention of peanut allergy in the United States3 are somewhat striking in that they make fairly bold recommendations based on results from the 2015 Learning Early about Peanut Allergy (LEAP) study,4 a randomized trial evaluating early peanut introduction as a preventive strategy for peanut allergy. Of note, this study was not placebo controlled, was conducted at only 1 site in the United Kingdom, and only included 640 children, though the number of participants was admittedly large for this type of study.4 Arguably, the LEAP study alone does not provide enough evidence upon which to base what essentially amounts to an about-face in the official recommendations for prevention of peanut and other food allergies, which emphasized delayed introduction of high-risk foods, especially in high-risk individuals.5-7 To better understand this shift, we need to briefly explore the context of the addendum guidelines.
As many as one-third of pediatric patients with atopic dermatitis (AD) have food allergies, thus diet often is invoked by patients and providers alike as an underlying cause of the disease.8 Many patients in my practice are so focused on potential food allergies that actual treatment of the affected skin is marginalized and often dismissed as a stopgap that does not address the root of the problem. A 2004 study of 100 children with AD found that diet was manipulated by the parents in 75% of patients in an attempt to manage the disease.9
Patients are not the only ones who consider food allergies to be a driving force in AD. The medical literature indicates that this theory has existed for centuries; for instance, with regard to the relationship between diet and AD, the author of an article from 1830 quipped, “There is probably no subject in which more deeply rooted convictions have been held . . . than the connection between diet and disease, both as regards the causation and treatment of the latter . . .”10 More apropos perhaps is a statement from the 2010 National Institute of Allergy and Infectious Diseases guidelines on food allergy management, which noted that while the expert group “does not mean to imply that AD results from [food allergies], the role of [food allergies] in the pathogenesis and severity of this condition remains controversial.”11
Prior to the LEAP study, food allergy recommendations for clinical practice in the United Kingdom in 199812 and the United States in 200013 recommended excluding allergenic foods (eg, peanuts, tree nuts, soy, milk, eggs) from the diet in infants with a family history of atopy until 3 years of age. However, those recommendations did not seem to be working, when in fact just the opposite was happening. From 1997 until the LEAP study was conducted in 2015, the prevalence of peanut allergy more than quadrupled and became the leading cause of anaphylaxis and death related to food allergy.14 Additionally, study after study concluded that elimination diets did not seem to help most patients with AD.15 As is required in good scientific thinking, when a hypothesis is proven false, other approaches must be considered.
The idea arose that perhaps delaying introduction of allergenic foods was the opposite of the answer.4 The LEAP study tested the notion that peanut allergies are rare in countries where peanuts are introduced early and if telling families to delay introduction of peanuts in infants might actually be causing development of a peanut allergy, and the tests bore fruit. It was found that giving infants peanut-containing foods resulted in a more than 80% reduction in peanut allergy at 5 years of age (P<.001).4 What was perhaps even more interesting was the connection between AD and peanut allergy. An important idea articulated in the LEAP study is in some ways revolutionary: Rather than foods causing AD, it could be that “early environmental exposure (through the skin) to peanut may account for early sensitization, whereas early oral exposure may lead to immune tolerance.”4 This concept—that impaired eczematous skin may actually lead to the development of food allergies—turns the whole thing upside down.
What do these updated guidelines actually suggest? The first guideline focuses on infants with severe AD, egg allergy, or both, who therefore are thought to be at the highest risk for developing peanut allergy.3 Because of the higher baseline risk in this subgroup, measurement of the peanut-specific IgE (peanut sIgE) level, skin prick testing (SPT), or both is strongly recommended before introducing peanut protein into the diet. This testing can be performed by qualified providers as a screening measure, but if positive (≥0.35 kUA/L for peanut sIgE or >2 mm on the peanut SPT), referral to an allergy specialist is warranted. If these studies are negative, it is thought the likelihood of peanut allergy is low, and it is recommended that caregivers introduce age-appropriate peanut-containing foods (eg, peanut butter snack puffs, diluted peanut butter) as early as 4 to 6 months of age. The second guideline recommends that peanut-containing foods should be introduced into the diets of infants with mild or moderate AD at approximately 6 months of age without the need for prior screening via peanut sIgE or SPT. Lastly, the third guideline recommends that caregivers freely introduce peanut-containing foods together with other solid foods in infants without AD or food allergies in accordance with family preference.3
The results of the LEAP study are certainly exciting, and although the theoretical basis makes good scientific sense and the updated guidelines truly address an important and growing problem, there are several issues with this update that are worth considering. Given the constraints of the LEAP study, it certainly seems possible that the results will not be applicable to all populations or foods. More research is needed to ensure that this robust finding applies to other children and to explore the introduction of other allergenic foods, which the LEAP study investigators also emphasized.4
In fairness, the updated guidelines clearly state the quality of evidence of their recommendations and make it clear that expert opinion is playing a large role.3 For the first guideline regarding recommendations for those with severe AD and/or egg allergy, the quality of evidence is deemed moderate, while the contribution of expert opinion is listed as significant. For the second and third guidelines regarding recommendations for mild to moderate AD and those without AD, respectively, the quality of evidence is low and expert opinion is again listed as significant.3
Importantly, delineating severe AD from moderate disease—which is necessary because only severe AD warrants evaluation with peanut sIgE and/or SPT—can be difficult, as the distinction relies on a degree of subjectivity that may vary between specialists. Indeed, 2 publications suggest extending the definition of severe AD to include infants with early-onset AD (<3 months of age) and those with moderate AD not responding to treatment.16,17
Despite these reservations, the updated guidelines represent a breakthrough in understanding in an area truly in need of advancement. Although the evidence may not be exactly extraordinary, the context for these developments and our deeper understanding suggest that we do indeed live in extraordinary times.
- Encyclopaedia Galactica [television transcript]. Cosmos: A Personal Voyage. Public Broadcasting Service. December 14, 1980.
- Ezzo J, Bausell B, Moerman DE, et al. Reviewing the reviews: how strong is the evidence? how clear are the conclusions? Int J Technol Assess Health Care. 2001;17:457-466.
- Togias A, Cooper SF, Acebal ML, et al. Addendum guidelines for the prevention of peanut allergy in the United States: report of the National Institute of Allergy and Infectious Diseases–sponsored expert panel.J Allergy Clin Immunol. 2017;139:29-44.
- Du Toit G, Roberts G, Sayre PH, et al. Randomized trial of peanut consumption in infants at risk for peanut allergy. N Engl J Med. 2015;372:803-813.
- Høst A, Koletzko B, Dreborg S, et al. Dietary products used in infants for treatment and prevention of food allergy. joint statement of the European Society for Paediatric Allergology and Clinical Immunology (ESPACI) Committee on Hypoallergenic Formulas and the European Society for Paediatric Gastroenterology, Hepatology and Nutrition (ESPGHAN) Committee on Nutrition. Arch Dis Child. 1999;81:80-84.
- American Academy of Pediatrics. Committee on Nutrition. hypoallergenic infant formulas. Pediatrics. 2000;106(2, pt 1):346-349.
- Fiocchi A, Assa’ad A, Bahna S; Adverse Reactions to Foods Committee; American College of Allergy, Asthma and Immunology. Food allergy and the introduction of solid foods to infants: a consensus document. Ann Allergy Asthma Immunol. 2006;97:10-20.
- Thompson MM, Hanifin JM. Effective therapy of childhood atopic dermatitis allays food allergy concerns. J Am Acad Dermatol. 2005;53(2 suppl 2):S214-S219.
- Johnston GA, Bilbao RM, Graham-Brown RA. The use of dietary manipulation by parents of children with atopic dermatitis. Br J Dermatol. 2004;150:1186-1189.
- Mackenzie S. The inaugural address on the advantages to be derived from the study of dermatology. BMJ. 1830;1:193-197.
- Boyce JA, Assa’ad A, Burks AW, et al; NIAID-Sponsored Expert Panel. Guidelines for the diagnosis and management of food allergy in the United States: report of the NIAID-sponsored expert panel. J Allergy Clin Immunol. 2010;126(6 suppl):S1-S58.
- Committee on Toxicity of Chemicals in Food, Consumer Products and the Environment. Peanut Allergy. London, England: Department of Health; 1998.
- American Academy of Pediatrics Committee on Nutrition. Hypoallergenic infant formulas. Pediatrics. 2000;106(2, pt 1):346-349.
- Gruchalla RS, Sampson HA. Preventing peanut allergy through early consumption—ready for prime time? N Engl J Med. 2015;372:875-877.
- Lim NR, Lohman ME, Lio PA. The role of elimination diets in atopic dermatitis: a comprehensive review. Pediatr Dermatol. 2017;34:516-527.
- Wong CC, Allen KJ, Orchard D. Changes to infant feeding guidelines: relevance to dermatologists. Australas J Dermatol. 2017;58:e171-e175.
- Martin PE, Eckert JK, Koplin JJ, et al. Which infants with eczema are at risk of food allergy? results from a population-based cohort. Clin Exp Allergy. 2015;45:255-264.
- Encyclopaedia Galactica [television transcript]. Cosmos: A Personal Voyage. Public Broadcasting Service. December 14, 1980.
- Ezzo J, Bausell B, Moerman DE, et al. Reviewing the reviews: how strong is the evidence? how clear are the conclusions? Int J Technol Assess Health Care. 2001;17:457-466.
- Togias A, Cooper SF, Acebal ML, et al. Addendum guidelines for the prevention of peanut allergy in the United States: report of the National Institute of Allergy and Infectious Diseases–sponsored expert panel.J Allergy Clin Immunol. 2017;139:29-44.
- Du Toit G, Roberts G, Sayre PH, et al. Randomized trial of peanut consumption in infants at risk for peanut allergy. N Engl J Med. 2015;372:803-813.
- Høst A, Koletzko B, Dreborg S, et al. Dietary products used in infants for treatment and prevention of food allergy. joint statement of the European Society for Paediatric Allergology and Clinical Immunology (ESPACI) Committee on Hypoallergenic Formulas and the European Society for Paediatric Gastroenterology, Hepatology and Nutrition (ESPGHAN) Committee on Nutrition. Arch Dis Child. 1999;81:80-84.
- American Academy of Pediatrics. Committee on Nutrition. hypoallergenic infant formulas. Pediatrics. 2000;106(2, pt 1):346-349.
- Fiocchi A, Assa’ad A, Bahna S; Adverse Reactions to Foods Committee; American College of Allergy, Asthma and Immunology. Food allergy and the introduction of solid foods to infants: a consensus document. Ann Allergy Asthma Immunol. 2006;97:10-20.
- Thompson MM, Hanifin JM. Effective therapy of childhood atopic dermatitis allays food allergy concerns. J Am Acad Dermatol. 2005;53(2 suppl 2):S214-S219.
- Johnston GA, Bilbao RM, Graham-Brown RA. The use of dietary manipulation by parents of children with atopic dermatitis. Br J Dermatol. 2004;150:1186-1189.
- Mackenzie S. The inaugural address on the advantages to be derived from the study of dermatology. BMJ. 1830;1:193-197.
- Boyce JA, Assa’ad A, Burks AW, et al; NIAID-Sponsored Expert Panel. Guidelines for the diagnosis and management of food allergy in the United States: report of the NIAID-sponsored expert panel. J Allergy Clin Immunol. 2010;126(6 suppl):S1-S58.
- Committee on Toxicity of Chemicals in Food, Consumer Products and the Environment. Peanut Allergy. London, England: Department of Health; 1998.
- American Academy of Pediatrics Committee on Nutrition. Hypoallergenic infant formulas. Pediatrics. 2000;106(2, pt 1):346-349.
- Gruchalla RS, Sampson HA. Preventing peanut allergy through early consumption—ready for prime time? N Engl J Med. 2015;372:875-877.
- Lim NR, Lohman ME, Lio PA. The role of elimination diets in atopic dermatitis: a comprehensive review. Pediatr Dermatol. 2017;34:516-527.
- Wong CC, Allen KJ, Orchard D. Changes to infant feeding guidelines: relevance to dermatologists. Australas J Dermatol. 2017;58:e171-e175.
- Martin PE, Eckert JK, Koplin JJ, et al. Which infants with eczema are at risk of food allergy? results from a population-based cohort. Clin Exp Allergy. 2015;45:255-264.

Statin effect in prostate cancer may be caused by reduced inflammation
SAN FRANCISCO – according to the results of a new genetic analysis of prostate cancers in men in the Health Professionals Follow Up Study.
A previous analysis from the Health Professionals Follow Up Study published in 2015 showed no difference in the risk of lethal prostate cancer for those who began using statins after diagnosis of their tumors.

A genetic analysis of these lethal cases revealed that patients taking long-term statins had a lower incidence of phosphatase and tensin homolog (PTEN)–null cancers, which are associated with worse outcomes. Integrating molecular and epidemiologic data, PTEN and PI3K (phosphatidylinositol) signaling and inflammation and immune activation appear to be two potential mechanisms contributing to this association.
Genetic analysis of normal prostate tissue also showed unique traits among statin users. “We found that the top ten pathways that came up were almost all involved in inflammation or immune activation, and we found those differences only in tumor and adjacent normal prostate tissue. We didn’t see any pathways that were differentially expressed by statin use within the tumor tissue itself,” said Emma Allott, PhD, who presented the research at a poster session at the annual meeting of the American Urological Association.
An association between statin use and improved survival was first described in a 2006 study based on results from the Health Professionals Follow Up Study. Since then, “we’ve been generating molecular data on cancers that developed in statin users and nonusers,” said Dr. Allott of the University of North Carolina, Chapel Hill.
Of the 5,792 prostate cancer diagnoses, 17% were advanced cases, defined as stage T3b or more, having spread to lymph nodes or metastasized, or lethal; 13% were lethal, 46% were positive for the ERG oncogene, and 14% were PTEN-null. “Statin use was associated with a lower risk of PTEN-null prostate cancer, so that seems to drive some of the reduced association with lethal disease,” said Dr. Allott.
There was no association between lethality and ERG-positive or ERG-negative status. Those who used statins for more than 5 years were less likely to have a PTEN-null tumor (hazard ratio, 0.42; 95% confidence interval, 0.20-0.90) but not more likely to have a PTEN-positive tumor (HR, 1.18; 95% CI, 0.95-1.46).
Compared with never users, long-term statin users also were less likely to have advanced prostate cancer (multivariate analysis, HR, 0.62; 95% CI, 0.45-0.85) as well as lethal prostate cancer (HR, 0.52; 95% CI, 0.35-0.78).
The researchers conducted a gene set enrichment analysis in statin users and found an enrichment of T-cell, B-cell, and PI3K signaling in tumor-adjacent normal prostate tissue, as well as other changes. “We think maybe there’s a microenvironment inflammation component to the mechanism through which statins are associated with lower risk of lethal prostate cancer,” said Dr. Allott.
The molecular data could identify patient subgroups that could benefit from statins. Dr. Allott said that is the goal, but it will take time. “That’s more obviously translatable to the clinic, but we don’t yet have enough data in this cohort to look at that.”
SOURCE: Allott E et al. AUA 2018, Abstract MP21-01.
SAN FRANCISCO – according to the results of a new genetic analysis of prostate cancers in men in the Health Professionals Follow Up Study.
A previous analysis from the Health Professionals Follow Up Study published in 2015 showed no difference in the risk of lethal prostate cancer for those who began using statins after diagnosis of their tumors.
A genetic analysis of these lethal cases revealed that patients taking long-term statins had a lower incidence of phosphatase and tensin homolog (PTEN)–null cancers, which are associated with worse outcomes. Integrating molecular and epidemiologic data, PTEN and PI3K (phosphatidylinositol) signaling and inflammation and immune activation appear to be two potential mechanisms contributing to this association.
Genetic analysis of normal prostate tissue also showed unique traits among statin users. “We found that the top ten pathways that came up were almost all involved in inflammation or immune activation, and we found those differences only in tumor and adjacent normal prostate tissue. We didn’t see any pathways that were differentially expressed by statin use within the tumor tissue itself,” said Emma Allott, PhD, who presented the research at a poster session at the annual meeting of the American Urological Association.
An association between statin use and improved survival was first described in a 2006 study based on results from the Health Professionals Follow Up Study. Since then, “we’ve been generating molecular data on cancers that developed in statin users and nonusers,” said Dr. Allott of the University of North Carolina, Chapel Hill.
Of the 5,792 prostate cancer diagnoses, 17% were advanced cases, defined as stage T3b or more, having spread to lymph nodes or metastasized, or lethal; 13% were lethal, 46% were positive for the ERG oncogene, and 14% were PTEN-null. “Statin use was associated with a lower risk of PTEN-null prostate cancer, so that seems to drive some of the reduced association with lethal disease,” said Dr. Allott.
There was no association between lethality and ERG-positive or ERG-negative status. Those who used statins for more than 5 years were less likely to have a PTEN-null tumor (hazard ratio, 0.42; 95% confidence interval, 0.20-0.90) but not more likely to have a PTEN-positive tumor (HR, 1.18; 95% CI, 0.95-1.46).
Compared with never users, long-term statin users also were less likely to have advanced prostate cancer (multivariate analysis, HR, 0.62; 95% CI, 0.45-0.85) as well as lethal prostate cancer (HR, 0.52; 95% CI, 0.35-0.78).
The researchers conducted a gene set enrichment analysis in statin users and found an enrichment of T-cell, B-cell, and PI3K signaling in tumor-adjacent normal prostate tissue, as well as other changes. “We think maybe there’s a microenvironment inflammation component to the mechanism through which statins are associated with lower risk of lethal prostate cancer,” said Dr. Allott.
The molecular data could identify patient subgroups that could benefit from statins. Dr. Allott said that is the goal, but it will take time. “That’s more obviously translatable to the clinic, but we don’t yet have enough data in this cohort to look at that.”
SOURCE: Allott E et al. AUA 2018, Abstract MP21-01.
SAN FRANCISCO – according to the results of a new genetic analysis of prostate cancers in men in the Health Professionals Follow Up Study.
A previous analysis from the Health Professionals Follow Up Study published in 2015 showed no difference in the risk of lethal prostate cancer for those who began using statins after diagnosis of their tumors.
A genetic analysis of these lethal cases revealed that patients taking long-term statins had a lower incidence of phosphatase and tensin homolog (PTEN)–null cancers, which are associated with worse outcomes. Integrating molecular and epidemiologic data, PTEN and PI3K (phosphatidylinositol) signaling and inflammation and immune activation appear to be two potential mechanisms contributing to this association.
Genetic analysis of normal prostate tissue also showed unique traits among statin users. “We found that the top ten pathways that came up were almost all involved in inflammation or immune activation, and we found those differences only in tumor and adjacent normal prostate tissue. We didn’t see any pathways that were differentially expressed by statin use within the tumor tissue itself,” said Emma Allott, PhD, who presented the research at a poster session at the annual meeting of the American Urological Association.
An association between statin use and improved survival was first described in a 2006 study based on results from the Health Professionals Follow Up Study. Since then, “we’ve been generating molecular data on cancers that developed in statin users and nonusers,” said Dr. Allott of the University of North Carolina, Chapel Hill.
Of the 5,792 prostate cancer diagnoses, 17% were advanced cases, defined as stage T3b or more, having spread to lymph nodes or metastasized, or lethal; 13% were lethal, 46% were positive for the ERG oncogene, and 14% were PTEN-null. “Statin use was associated with a lower risk of PTEN-null prostate cancer, so that seems to drive some of the reduced association with lethal disease,” said Dr. Allott.
There was no association between lethality and ERG-positive or ERG-negative status. Those who used statins for more than 5 years were less likely to have a PTEN-null tumor (hazard ratio, 0.42; 95% confidence interval, 0.20-0.90) but not more likely to have a PTEN-positive tumor (HR, 1.18; 95% CI, 0.95-1.46).
Compared with never users, long-term statin users also were less likely to have advanced prostate cancer (multivariate analysis, HR, 0.62; 95% CI, 0.45-0.85) as well as lethal prostate cancer (HR, 0.52; 95% CI, 0.35-0.78).
The researchers conducted a gene set enrichment analysis in statin users and found an enrichment of T-cell, B-cell, and PI3K signaling in tumor-adjacent normal prostate tissue, as well as other changes. “We think maybe there’s a microenvironment inflammation component to the mechanism through which statins are associated with lower risk of lethal prostate cancer,” said Dr. Allott.
The molecular data could identify patient subgroups that could benefit from statins. Dr. Allott said that is the goal, but it will take time. “That’s more obviously translatable to the clinic, but we don’t yet have enough data in this cohort to look at that.”
SOURCE: Allott E et al. AUA 2018, Abstract MP21-01.
REPORTING FROM THE AUA ANNUAL MEETING
Key clinical point: Researchers hope that genetic analyses could eventually point to prostate cancer patients who might benefit from statin use.
Major finding: Long-term statin users had lower odds of having a pTEN-null tumor (hazard ratio, 0.42), which is associated with worse outcomes.
Study details: Retrospective analysis of 5,792 diagnoses of prostate cancer among 44,076 men.
Disclosures: The study was funded by the Irish Cancer Society, the John Fitzpatrick Fellowship, and the National Cancer Institute. Dr. Allott reported no relevant financial relationships.
Source: Allott E et al. AUA 2018, Abstract MP21-01.