User login
Verma unveils Medicaid scorecard but refuses to judge efforts
“This is about bringing a level of transparency and accountability to the Medicaid program that we have never had before,” said Seema Verma, administrator of the Centers for Medicare & Medicaid Services.
Yet in a meeting with reporters, Ms. Verma refused to discuss the findings in any detail or comment on any individual states that performed poorly or exceptionally.
“I will let you look at the data and make your own conclusions,” she told journalists a few minutes before the report was posted online.
When reporters pressed Ms. Verma to comment on the document, she refused to give an assessment of the Medicaid program, the federal-state health program for low-income residents. She has run Medicaid for the past 15 months.
“The idea here is to give you a sense of where states are on different areas,” she said. “The idea is to be used for best practices,” and it’s “an opportunity for us to identify” and have discussions with states that aren’t performing well.
Medicaid covers about 75 million people, about half of them children.
The report looked at how well states provide a wide variety of health services to children and adults. It also reviewed how quickly the federal government was approving state waiver requests to change their programs.
But not all states provided data for each service because sharing information was voluntary.
For example, half the states did not show how well they control Medicaid enrollees’ blood pressure.
The National Association of Medicaid Directors panned the scorecard. It acknowledged the need for a system to measure performance but said its members have concerns about its accuracy and usefulness.
“There are significant methodological issues with the underlying data, including completeness, timeliness, and quality,” the association said in a statement. It noted that most of the data come from 2015.
As expected, the data showed great variation in how states provide care, including immunizing teenagers and getting dental care to children. A big reason is that state Medicaid benefits and payments to doctors vary dramatically, the Medicaid directors said, so that “it will not be possible to make apples-to-apples comparisons between states.”
In her first public speech, Ms. Verma promised last November to release a Medicaid scorecard. She said states won’t immediately face any consequences for poor performance – but that could change.
“The data … begins to offer taxpayers insights into how their dollars are being spent and the impact those dollars have on health outcomes,” Ms. Verma said on June 4.
Sara Rosenbaum, a professor of health law and policy at George Washington University in Washington, who previously led a congressional advisory board on Medicaid, suggested that the information is still too incomplete to be of great value.
“It is amazing to me that in 2018 this is all we have when trying to understand how the nation’s largest insurer performs for its poorest and most vulnerable residents,” she said.
KHN’s coverage of children’s health care issues is supported in part by the Heising-Simons Foundation. Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente.
“This is about bringing a level of transparency and accountability to the Medicaid program that we have never had before,” said Seema Verma, administrator of the Centers for Medicare & Medicaid Services.
Yet in a meeting with reporters, Ms. Verma refused to discuss the findings in any detail or comment on any individual states that performed poorly or exceptionally.
“I will let you look at the data and make your own conclusions,” she told journalists a few minutes before the report was posted online.
When reporters pressed Ms. Verma to comment on the document, she refused to give an assessment of the Medicaid program, the federal-state health program for low-income residents. She has run Medicaid for the past 15 months.
“The idea here is to give you a sense of where states are on different areas,” she said. “The idea is to be used for best practices,” and it’s “an opportunity for us to identify” and have discussions with states that aren’t performing well.
Medicaid covers about 75 million people, about half of them children.
The report looked at how well states provide a wide variety of health services to children and adults. It also reviewed how quickly the federal government was approving state waiver requests to change their programs.
But not all states provided data for each service because sharing information was voluntary.
For example, half the states did not show how well they control Medicaid enrollees’ blood pressure.
The National Association of Medicaid Directors panned the scorecard. It acknowledged the need for a system to measure performance but said its members have concerns about its accuracy and usefulness.
“There are significant methodological issues with the underlying data, including completeness, timeliness, and quality,” the association said in a statement. It noted that most of the data come from 2015.
As expected, the data showed great variation in how states provide care, including immunizing teenagers and getting dental care to children. A big reason is that state Medicaid benefits and payments to doctors vary dramatically, the Medicaid directors said, so that “it will not be possible to make apples-to-apples comparisons between states.”
In her first public speech, Ms. Verma promised last November to release a Medicaid scorecard. She said states won’t immediately face any consequences for poor performance – but that could change.
“The data … begins to offer taxpayers insights into how their dollars are being spent and the impact those dollars have on health outcomes,” Ms. Verma said on June 4.
Sara Rosenbaum, a professor of health law and policy at George Washington University in Washington, who previously led a congressional advisory board on Medicaid, suggested that the information is still too incomplete to be of great value.
“It is amazing to me that in 2018 this is all we have when trying to understand how the nation’s largest insurer performs for its poorest and most vulnerable residents,” she said.
KHN’s coverage of children’s health care issues is supported in part by the Heising-Simons Foundation. Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente.
“This is about bringing a level of transparency and accountability to the Medicaid program that we have never had before,” said Seema Verma, administrator of the Centers for Medicare & Medicaid Services.
Yet in a meeting with reporters, Ms. Verma refused to discuss the findings in any detail or comment on any individual states that performed poorly or exceptionally.
“I will let you look at the data and make your own conclusions,” she told journalists a few minutes before the report was posted online.
When reporters pressed Ms. Verma to comment on the document, she refused to give an assessment of the Medicaid program, the federal-state health program for low-income residents. She has run Medicaid for the past 15 months.
“The idea here is to give you a sense of where states are on different areas,” she said. “The idea is to be used for best practices,” and it’s “an opportunity for us to identify” and have discussions with states that aren’t performing well.
Medicaid covers about 75 million people, about half of them children.
The report looked at how well states provide a wide variety of health services to children and adults. It also reviewed how quickly the federal government was approving state waiver requests to change their programs.
But not all states provided data for each service because sharing information was voluntary.
For example, half the states did not show how well they control Medicaid enrollees’ blood pressure.
The National Association of Medicaid Directors panned the scorecard. It acknowledged the need for a system to measure performance but said its members have concerns about its accuracy and usefulness.
“There are significant methodological issues with the underlying data, including completeness, timeliness, and quality,” the association said in a statement. It noted that most of the data come from 2015.
As expected, the data showed great variation in how states provide care, including immunizing teenagers and getting dental care to children. A big reason is that state Medicaid benefits and payments to doctors vary dramatically, the Medicaid directors said, so that “it will not be possible to make apples-to-apples comparisons between states.”
In her first public speech, Ms. Verma promised last November to release a Medicaid scorecard. She said states won’t immediately face any consequences for poor performance – but that could change.
“The data … begins to offer taxpayers insights into how their dollars are being spent and the impact those dollars have on health outcomes,” Ms. Verma said on June 4.
Sara Rosenbaum, a professor of health law and policy at George Washington University in Washington, who previously led a congressional advisory board on Medicaid, suggested that the information is still too incomplete to be of great value.
“It is amazing to me that in 2018 this is all we have when trying to understand how the nation’s largest insurer performs for its poorest and most vulnerable residents,” she said.
KHN’s coverage of children’s health care issues is supported in part by the Heising-Simons Foundation. Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente.
FDA approves first biosimilar to pegfilgrastim
to decrease the chance of infection in patients with nonmyeloid cancer who are receiving myelosuppressive chemotherapy and are at risk of febrile neutropenia.
The approval is based on structural and functional characterization, animal study data, human pharmacokinetic and pharmacodynamic data, clinical immunogenicity data, and other clinical safety and effectiveness data that demonstrates pegfilgrastim-jmdb is biosimilar to pegfilgrastim, the FDA said in a statement.
The FDA warns that “patients with a history of serious allergic reactions to human granulocyte colony–stimulating factors such as pegfilgrastim or filgrastim products should not take pegfilgrastim-jmdb.”
This approval is part of the FDA’s efforts to “help promote competition that can reduce drug costs and promote access,” FDA commissioner Scott Gottlieb, MD, said in the statement. “This summer, we’ll release a comprehensive new plan to advance new policy efforts that promote biosimilar product development. Biologics represent some of the most clinically important, but also costliest products that patients use to promote their health. We want to make sure that the pathway for developing biosimilar versions of approved biologics is efficient and effective, so that patients benefit from competition to existing biologics once lawful intellectual property has lapsed on these products.”
Pegfilgrastim-jmdb will be marketed as Fulphila by Mylan GmbH.
to decrease the chance of infection in patients with nonmyeloid cancer who are receiving myelosuppressive chemotherapy and are at risk of febrile neutropenia.
The approval is based on structural and functional characterization, animal study data, human pharmacokinetic and pharmacodynamic data, clinical immunogenicity data, and other clinical safety and effectiveness data that demonstrates pegfilgrastim-jmdb is biosimilar to pegfilgrastim, the FDA said in a statement.
The FDA warns that “patients with a history of serious allergic reactions to human granulocyte colony–stimulating factors such as pegfilgrastim or filgrastim products should not take pegfilgrastim-jmdb.”
This approval is part of the FDA’s efforts to “help promote competition that can reduce drug costs and promote access,” FDA commissioner Scott Gottlieb, MD, said in the statement. “This summer, we’ll release a comprehensive new plan to advance new policy efforts that promote biosimilar product development. Biologics represent some of the most clinically important, but also costliest products that patients use to promote their health. We want to make sure that the pathway for developing biosimilar versions of approved biologics is efficient and effective, so that patients benefit from competition to existing biologics once lawful intellectual property has lapsed on these products.”
Pegfilgrastim-jmdb will be marketed as Fulphila by Mylan GmbH.
to decrease the chance of infection in patients with nonmyeloid cancer who are receiving myelosuppressive chemotherapy and are at risk of febrile neutropenia.
The approval is based on structural and functional characterization, animal study data, human pharmacokinetic and pharmacodynamic data, clinical immunogenicity data, and other clinical safety and effectiveness data that demonstrates pegfilgrastim-jmdb is biosimilar to pegfilgrastim, the FDA said in a statement.
The FDA warns that “patients with a history of serious allergic reactions to human granulocyte colony–stimulating factors such as pegfilgrastim or filgrastim products should not take pegfilgrastim-jmdb.”
This approval is part of the FDA’s efforts to “help promote competition that can reduce drug costs and promote access,” FDA commissioner Scott Gottlieb, MD, said in the statement. “This summer, we’ll release a comprehensive new plan to advance new policy efforts that promote biosimilar product development. Biologics represent some of the most clinically important, but also costliest products that patients use to promote their health. We want to make sure that the pathway for developing biosimilar versions of approved biologics is efficient and effective, so that patients benefit from competition to existing biologics once lawful intellectual property has lapsed on these products.”
Pegfilgrastim-jmdb will be marketed as Fulphila by Mylan GmbH.
TAVR for low-risk patients shines at 6 years in NOTION
PARIS – Follow-up data from a randomized trial of transcatheter versus surgical aortic valve replacement in low-surgical-risk patients with symptomatic severe aortic stenosis showed sustained superior hemodynamic valve performance and less structural valve deterioration in the transcatheter group through 6 years, Lars Sondergaard, MD, reported at the annual meeting of the European Association of Percutaneous Cardiovascular Interventions.
Moreover, the rate of bioprosthetic valve failure as formally defined in a recent European consensus statement (Eur Heart J. 2017 Dec 1;38[45]:3382-90) was similarly low in the transcatheter aortic valve replacement (TAVR) and surgical aortic valve replacement (SAVR) arms at about 7%, in the randomized study known as NOTION (Nordic Aortic Valve Intervention), added Dr. Sondergaard, professor of cardiology at the University of Copenhagen, who was a coauthor of the consensus statement.

“As we look to expand TAVR to younger patients with longer life expectancy, durability, of course, becomes much more important,” the cardiologist observed.
NOTION was a pioneering prospective, multicenter, nonblinded, randomized trial of first-generation TAVR technology. The 280 participants, average age 79 years, were truly a low-surgical-risk population, with a mean Society of Thoracic Surgeons risk score of 3%.
NOTION is a small trial, but the results at 6 years of a planned 10-year follow-up augur well for TAVR success in the large, ongoing, definitive, randomized trials of TAVR versus SAVR in low-risk patients. That’s because NOTION participants were treated in 2009-2013, when the self-expanding TAVR CoreValve was implanted on the basis of aortic annulus measurements obtained via echocardiography, which is considerably less accurate than CT, the standard practice today. For this reason, it’s highly unlikely that the larger, ongoing trials, including PARTNER 3 and the Medtronic Evolut Transcatheter Aortic Valve Replacement in Low Risk Patients trial, will experience moderate paravalvular leak rates anything like the 20.9% rate seen in the TAVR group in NOTION, where the SAVR group’s rate was just 1.5%.
“I’m sure quite a few of the NOTION patients would have a larger TAVR valve prosthesis if they were treated today,” according to Dr. Sondergaard.
Valve function
The rate of moderate hemodynamic structural valve deterioration through 6 years of follow-up was 3.6% in the TAVR arm, compared with 23.7% in the SAVR group. The rate of nonstructural valve deterioration was 54.0% with TAVR and 57.8% with SAVR, and there were no cases of moderate or severe aortic regurgitation in either group.
The mean aortic valve gradient in the TAVR group went from 44.9 mm Hg at baseline to 12.2 mm Hg at 3 months and to 14.7 mm Hg at 6 years. In the SAVR group, the figures were 44.9 mm Hg, 8.3 mm Hg, and 9.9 mm Hg, respectively.
The effective orifice area in the TAVR group improved from 0.74 cm2 at baseline to 1.37 cm2 at 3 months and to 1.16 cm2 at 6 years. With SAVR, the effective orifice area was 0.74 cm2 at baseline, 1.66 cm2 at 3 months, and 1.53 cm2 at 6 years.
Clinical outcomes
Through 6 years of follow-up, there were no cases of thrombosis in either study arm, and the rate of endocarditis was just under 6% in each group through 6 years. All-cause mortality through 6 years was 42.5% in the TAVR group, not significantly different from the 37.7% rate in the SAVR arm.
The valve-related death rate was 5.0% in the TAVR arm and similar at 3.7% with SAVR. Reintervention occurred in 2.2% of TAVR patients and in 0.7% of SAVR patients. Severe hemodynamic structural valve deterioration was documented in 0.7% of TAVR patients and 3.0% of SAVR patients. The overall rate of bioprosthetic valve failure – a composite of these three endpoints – was 7.5% with TAVR and similar at 6.7% with SAVR.
Session cochair Alain G. Cribier, MD, a TAVR pioneer who is professor of medicine and director of cardiology at Charles Nicolle Hospital, University of Rouen (France), declared, “This is extremely encouraging.”
However, discussant Corrado Tamburino, MD, was more circumspect.
“If you look, there’s a trend for increased all-cause mortality in the TAVR versus SAVR group at 6 years. This could be related to the big difference in paravalvular leak. Do you think paravalvular leak could have a real impact on mortality?” asked Dr. Tamburino, professor of cardiology at the University of Catania, Italy.
Not in the NOTION study, where they looked for but didn’t find any such association, according to Dr. Sondergaard.
“If you look at the survival curves from the beginning out to 6 years, you’ll see that the lines cross each other several times, so this small difference right now could just be random. I don’t think we can say there’s a higher mortality rate with TAVR for the time being,” he added.
The NOTION trial was funded by the Danish Heart Foundation. Dr. Sondergaard reported receiving research grants from and/or serving as a consultant to Abbott, Boston Scientific, Edwards Lifesciences, Medtronic, and Symetis.
PARIS – Follow-up data from a randomized trial of transcatheter versus surgical aortic valve replacement in low-surgical-risk patients with symptomatic severe aortic stenosis showed sustained superior hemodynamic valve performance and less structural valve deterioration in the transcatheter group through 6 years, Lars Sondergaard, MD, reported at the annual meeting of the European Association of Percutaneous Cardiovascular Interventions.
Moreover, the rate of bioprosthetic valve failure as formally defined in a recent European consensus statement (Eur Heart J. 2017 Dec 1;38[45]:3382-90) was similarly low in the transcatheter aortic valve replacement (TAVR) and surgical aortic valve replacement (SAVR) arms at about 7%, in the randomized study known as NOTION (Nordic Aortic Valve Intervention), added Dr. Sondergaard, professor of cardiology at the University of Copenhagen, who was a coauthor of the consensus statement.
“As we look to expand TAVR to younger patients with longer life expectancy, durability, of course, becomes much more important,” the cardiologist observed.
NOTION was a pioneering prospective, multicenter, nonblinded, randomized trial of first-generation TAVR technology. The 280 participants, average age 79 years, were truly a low-surgical-risk population, with a mean Society of Thoracic Surgeons risk score of 3%.
NOTION is a small trial, but the results at 6 years of a planned 10-year follow-up augur well for TAVR success in the large, ongoing, definitive, randomized trials of TAVR versus SAVR in low-risk patients. That’s because NOTION participants were treated in 2009-2013, when the self-expanding TAVR CoreValve was implanted on the basis of aortic annulus measurements obtained via echocardiography, which is considerably less accurate than CT, the standard practice today. For this reason, it’s highly unlikely that the larger, ongoing trials, including PARTNER 3 and the Medtronic Evolut Transcatheter Aortic Valve Replacement in Low Risk Patients trial, will experience moderate paravalvular leak rates anything like the 20.9% rate seen in the TAVR group in NOTION, where the SAVR group’s rate was just 1.5%.
“I’m sure quite a few of the NOTION patients would have a larger TAVR valve prosthesis if they were treated today,” according to Dr. Sondergaard.
Valve function
The rate of moderate hemodynamic structural valve deterioration through 6 years of follow-up was 3.6% in the TAVR arm, compared with 23.7% in the SAVR group. The rate of nonstructural valve deterioration was 54.0% with TAVR and 57.8% with SAVR, and there were no cases of moderate or severe aortic regurgitation in either group.
The mean aortic valve gradient in the TAVR group went from 44.9 mm Hg at baseline to 12.2 mm Hg at 3 months and to 14.7 mm Hg at 6 years. In the SAVR group, the figures were 44.9 mm Hg, 8.3 mm Hg, and 9.9 mm Hg, respectively.
The effective orifice area in the TAVR group improved from 0.74 cm2 at baseline to 1.37 cm2 at 3 months and to 1.16 cm2 at 6 years. With SAVR, the effective orifice area was 0.74 cm2 at baseline, 1.66 cm2 at 3 months, and 1.53 cm2 at 6 years.
Clinical outcomes
Through 6 years of follow-up, there were no cases of thrombosis in either study arm, and the rate of endocarditis was just under 6% in each group through 6 years. All-cause mortality through 6 years was 42.5% in the TAVR group, not significantly different from the 37.7% rate in the SAVR arm.
The valve-related death rate was 5.0% in the TAVR arm and similar at 3.7% with SAVR. Reintervention occurred in 2.2% of TAVR patients and in 0.7% of SAVR patients. Severe hemodynamic structural valve deterioration was documented in 0.7% of TAVR patients and 3.0% of SAVR patients. The overall rate of bioprosthetic valve failure – a composite of these three endpoints – was 7.5% with TAVR and similar at 6.7% with SAVR.
Session cochair Alain G. Cribier, MD, a TAVR pioneer who is professor of medicine and director of cardiology at Charles Nicolle Hospital, University of Rouen (France), declared, “This is extremely encouraging.”
However, discussant Corrado Tamburino, MD, was more circumspect.
“If you look, there’s a trend for increased all-cause mortality in the TAVR versus SAVR group at 6 years. This could be related to the big difference in paravalvular leak. Do you think paravalvular leak could have a real impact on mortality?” asked Dr. Tamburino, professor of cardiology at the University of Catania, Italy.
Not in the NOTION study, where they looked for but didn’t find any such association, according to Dr. Sondergaard.
“If you look at the survival curves from the beginning out to 6 years, you’ll see that the lines cross each other several times, so this small difference right now could just be random. I don’t think we can say there’s a higher mortality rate with TAVR for the time being,” he added.
The NOTION trial was funded by the Danish Heart Foundation. Dr. Sondergaard reported receiving research grants from and/or serving as a consultant to Abbott, Boston Scientific, Edwards Lifesciences, Medtronic, and Symetis.
PARIS – Follow-up data from a randomized trial of transcatheter versus surgical aortic valve replacement in low-surgical-risk patients with symptomatic severe aortic stenosis showed sustained superior hemodynamic valve performance and less structural valve deterioration in the transcatheter group through 6 years, Lars Sondergaard, MD, reported at the annual meeting of the European Association of Percutaneous Cardiovascular Interventions.
Moreover, the rate of bioprosthetic valve failure as formally defined in a recent European consensus statement (Eur Heart J. 2017 Dec 1;38[45]:3382-90) was similarly low in the transcatheter aortic valve replacement (TAVR) and surgical aortic valve replacement (SAVR) arms at about 7%, in the randomized study known as NOTION (Nordic Aortic Valve Intervention), added Dr. Sondergaard, professor of cardiology at the University of Copenhagen, who was a coauthor of the consensus statement.
“As we look to expand TAVR to younger patients with longer life expectancy, durability, of course, becomes much more important,” the cardiologist observed.
NOTION was a pioneering prospective, multicenter, nonblinded, randomized trial of first-generation TAVR technology. The 280 participants, average age 79 years, were truly a low-surgical-risk population, with a mean Society of Thoracic Surgeons risk score of 3%.
NOTION is a small trial, but the results at 6 years of a planned 10-year follow-up augur well for TAVR success in the large, ongoing, definitive, randomized trials of TAVR versus SAVR in low-risk patients. That’s because NOTION participants were treated in 2009-2013, when the self-expanding TAVR CoreValve was implanted on the basis of aortic annulus measurements obtained via echocardiography, which is considerably less accurate than CT, the standard practice today. For this reason, it’s highly unlikely that the larger, ongoing trials, including PARTNER 3 and the Medtronic Evolut Transcatheter Aortic Valve Replacement in Low Risk Patients trial, will experience moderate paravalvular leak rates anything like the 20.9% rate seen in the TAVR group in NOTION, where the SAVR group’s rate was just 1.5%.
“I’m sure quite a few of the NOTION patients would have a larger TAVR valve prosthesis if they were treated today,” according to Dr. Sondergaard.
Valve function
The rate of moderate hemodynamic structural valve deterioration through 6 years of follow-up was 3.6% in the TAVR arm, compared with 23.7% in the SAVR group. The rate of nonstructural valve deterioration was 54.0% with TAVR and 57.8% with SAVR, and there were no cases of moderate or severe aortic regurgitation in either group.
The mean aortic valve gradient in the TAVR group went from 44.9 mm Hg at baseline to 12.2 mm Hg at 3 months and to 14.7 mm Hg at 6 years. In the SAVR group, the figures were 44.9 mm Hg, 8.3 mm Hg, and 9.9 mm Hg, respectively.
The effective orifice area in the TAVR group improved from 0.74 cm2 at baseline to 1.37 cm2 at 3 months and to 1.16 cm2 at 6 years. With SAVR, the effective orifice area was 0.74 cm2 at baseline, 1.66 cm2 at 3 months, and 1.53 cm2 at 6 years.
Clinical outcomes
Through 6 years of follow-up, there were no cases of thrombosis in either study arm, and the rate of endocarditis was just under 6% in each group through 6 years. All-cause mortality through 6 years was 42.5% in the TAVR group, not significantly different from the 37.7% rate in the SAVR arm.
The valve-related death rate was 5.0% in the TAVR arm and similar at 3.7% with SAVR. Reintervention occurred in 2.2% of TAVR patients and in 0.7% of SAVR patients. Severe hemodynamic structural valve deterioration was documented in 0.7% of TAVR patients and 3.0% of SAVR patients. The overall rate of bioprosthetic valve failure – a composite of these three endpoints – was 7.5% with TAVR and similar at 6.7% with SAVR.
Session cochair Alain G. Cribier, MD, a TAVR pioneer who is professor of medicine and director of cardiology at Charles Nicolle Hospital, University of Rouen (France), declared, “This is extremely encouraging.”
However, discussant Corrado Tamburino, MD, was more circumspect.
“If you look, there’s a trend for increased all-cause mortality in the TAVR versus SAVR group at 6 years. This could be related to the big difference in paravalvular leak. Do you think paravalvular leak could have a real impact on mortality?” asked Dr. Tamburino, professor of cardiology at the University of Catania, Italy.
Not in the NOTION study, where they looked for but didn’t find any such association, according to Dr. Sondergaard.
“If you look at the survival curves from the beginning out to 6 years, you’ll see that the lines cross each other several times, so this small difference right now could just be random. I don’t think we can say there’s a higher mortality rate with TAVR for the time being,” he added.
The NOTION trial was funded by the Danish Heart Foundation. Dr. Sondergaard reported receiving research grants from and/or serving as a consultant to Abbott, Boston Scientific, Edwards Lifesciences, Medtronic, and Symetis.
REPORTING FROM EUROPCR 2018
Key clinical point: TAVR provided superior hemodynamic valve performance and less structural valve deterioration than SAVR in low-surgical-risk patients through 6 years of follow-up.
Major finding: The rate of moderate hemodynamic structural valve deterioration through 6 years was 3.6% with TAVR and 23.7% with SAVR.
Study details: An analysis of 6-year follow-up data from NOTION, a prospective, multicenter, randomized trial in 280 low-surgical-risk patients.
Disclosures: NOTION was funded by the Danish Heart Foundation. The presenter reported receiving research grants from and/or serving as a consultant to several medical device companies.
AMA: Opioid prescriptions down since 2013
Opioid prescriptions dropped by 22% over the last 5 years, with decreases seen in all 50 states, according to a new report from the American Medical Association’s opioid task force.
“The largest decrease in opioid prescriptions in 25 years reflects the fact that physicians and other health care professionals are increasingly judicious when prescribing opioids,” Patrice A. Harris, MD, chair of the task force, said in the report.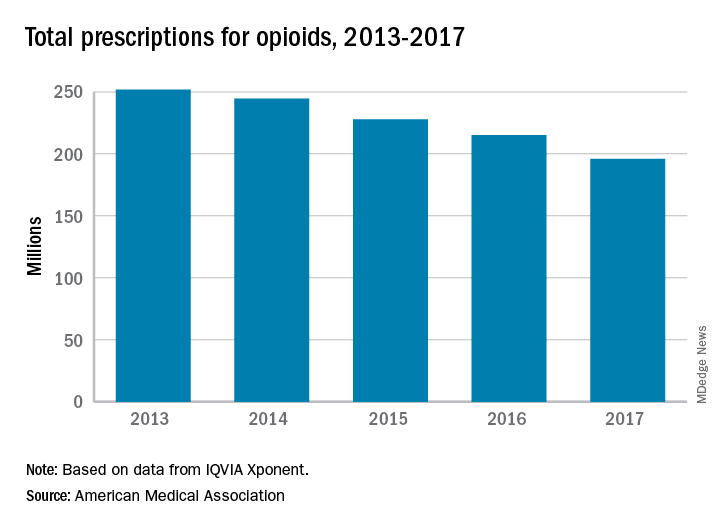
Other signs of progress against the opioid epidemic include the increasing number of health care professionals registered for prescription drug monitoring programs: It has more than tripled, from 472,000 in 2014 to 1.55 million in 2017. Furthermore, the number of naloxone prescriptions in 2017 had more than doubled, from about 3,500 per week to 8,000, the task force reported.
“Unfortunately, deaths related to heroin and illicit fentanyl, and to prescription opioids, continue to rise. We need well-designed initiatives that bring together public and private insurers, policymakers, public health infrastructure, and communities with the shared goal to improve access and coverage for comprehensive pain management and treatment for substance use disorders,” Dr. Harris said.
Opioid prescriptions dropped by 22% over the last 5 years, with decreases seen in all 50 states, according to a new report from the American Medical Association’s opioid task force.
“The largest decrease in opioid prescriptions in 25 years reflects the fact that physicians and other health care professionals are increasingly judicious when prescribing opioids,” Patrice A. Harris, MD, chair of the task force, said in the report.
Other signs of progress against the opioid epidemic include the increasing number of health care professionals registered for prescription drug monitoring programs: It has more than tripled, from 472,000 in 2014 to 1.55 million in 2017. Furthermore, the number of naloxone prescriptions in 2017 had more than doubled, from about 3,500 per week to 8,000, the task force reported.
“Unfortunately, deaths related to heroin and illicit fentanyl, and to prescription opioids, continue to rise. We need well-designed initiatives that bring together public and private insurers, policymakers, public health infrastructure, and communities with the shared goal to improve access and coverage for comprehensive pain management and treatment for substance use disorders,” Dr. Harris said.
Opioid prescriptions dropped by 22% over the last 5 years, with decreases seen in all 50 states, according to a new report from the American Medical Association’s opioid task force.
“The largest decrease in opioid prescriptions in 25 years reflects the fact that physicians and other health care professionals are increasingly judicious when prescribing opioids,” Patrice A. Harris, MD, chair of the task force, said in the report.
Other signs of progress against the opioid epidemic include the increasing number of health care professionals registered for prescription drug monitoring programs: It has more than tripled, from 472,000 in 2014 to 1.55 million in 2017. Furthermore, the number of naloxone prescriptions in 2017 had more than doubled, from about 3,500 per week to 8,000, the task force reported.
“Unfortunately, deaths related to heroin and illicit fentanyl, and to prescription opioids, continue to rise. We need well-designed initiatives that bring together public and private insurers, policymakers, public health infrastructure, and communities with the shared goal to improve access and coverage for comprehensive pain management and treatment for substance use disorders,” Dr. Harris said.
Checkpoint inhibitor shows promise in advanced squamous-cell carcinoma
An immune checkpoint inhibitor that targets the PD-1 receptor has shown “robust” efficacy among patients with advanced cutaneous squamous-cell carcinoma, according to researchers.
A combined phase 1/phase 2 study, published in the New England Journal of Medicine and presented simultaneously at the annual meeting of the American Society of Clinical Oncology, looked at the effect of monoclonal antibody cemiplimab in an expansion cohort of 26 patients with locally-advanced or metastatic cutaneous squamous-cell carcinoma who were not eligible for surgery. The phase 2 component involved 59 patients with metastatic disease.
Patients were treated with intravenous cemiplimab every 2 weeks for 48 weeks in the phase 1 study, and up to 96 weeks – or until unacceptable toxicity or disease progression – in the phase 2 study.
In the phase 1 study, researchers saw a response rate of 50% and a 65% rate of durable disease control, after a median follow-up of 11 months (1.1-17). The median time to response was 2.3 months, and more than half the patients (54%) who showed a response maintained that response past 6 months.
In the phase 2 study in patients with metastatic disease, 47% responded to the treatment – 24 patients showed a partial response and 4 showed a complete response. Of those who responded, 61% showed durable disease control after a median follow-up of 7.9 months.
The median time to response in this group of patients was 1.9 months, and 57% of those who did respond still showed a response at 6 months. However neither median progression-free survival nor median overall survival had been reached at the point of data cut-off.
The treatment showed similar effects in patients with regional and distant metastatic disease.
Advanced cutaneous squamous-cell carcinoma was thought to be an ideal target for immunotherapy because the high mutation burden in the tumor meant it would be sensitive to effector T cell attack, wrote Michael R. Migden, MD, of the University of Texas MD Anderson Cancer Center, Houston, and his coauthors.
“In addition, the dramatically increased risk of cutaneous squamous-cell carcinoma among people with immunosuppression pointed to an important role for immune surveillance with this cancer,” the authors wrote.
In the phase 2 study, 29% of patients experienced a serious adverse event – including two cases of pneumonitis – and three patients (5%) discontinued treatment. There were three deaths due to adverse events: One patient died from pneumonia complications, one died in his sleep, and one patient died following hypercalcemia and deep vein thrombosis.
Aside from these, most adverse events were grade 1 or 2. Around one-quarter of patients experienced diarrhea (27%) or fatigue (24%), while the other most common adverse events were nausea (17%), constipation (15%) and rash (15%). The authors noted that these adverse events were similar to those seen in other PD-1 inhibitors.
“Our results are consistent with an emerging theme regarding the high efficacy of immune checkpoint blockade for the treatment of hypermutated cancers, since the mutation burden of cutaneous squamous-cell carcinoma is similar to that reported for advanced solid tumors with microsatellite instability,” the authors wrote.
Cemiplimab is now being tested in a phase 2 trial in patients with advanced basal cell carcinoma.
The study was supported by Regeneron Pharmaceuticals and Sanofi. Eight authors declared funding from Regeneron to conduct the trial. Ten authors were employees of Regeneron. Fifteen authors also declared funding and payments from pharmaceutical companies outside the submitted work. Four had nothing to disclose.
SOURCE: Migden M et al. NEJM, 2018; June 4. doi: 10.1056/NEJMoa1805131.
An immune checkpoint inhibitor that targets the PD-1 receptor has shown “robust” efficacy among patients with advanced cutaneous squamous-cell carcinoma, according to researchers.
A combined phase 1/phase 2 study, published in the New England Journal of Medicine and presented simultaneously at the annual meeting of the American Society of Clinical Oncology, looked at the effect of monoclonal antibody cemiplimab in an expansion cohort of 26 patients with locally-advanced or metastatic cutaneous squamous-cell carcinoma who were not eligible for surgery. The phase 2 component involved 59 patients with metastatic disease.
Patients were treated with intravenous cemiplimab every 2 weeks for 48 weeks in the phase 1 study, and up to 96 weeks – or until unacceptable toxicity or disease progression – in the phase 2 study.
In the phase 1 study, researchers saw a response rate of 50% and a 65% rate of durable disease control, after a median follow-up of 11 months (1.1-17). The median time to response was 2.3 months, and more than half the patients (54%) who showed a response maintained that response past 6 months.
In the phase 2 study in patients with metastatic disease, 47% responded to the treatment – 24 patients showed a partial response and 4 showed a complete response. Of those who responded, 61% showed durable disease control after a median follow-up of 7.9 months.
The median time to response in this group of patients was 1.9 months, and 57% of those who did respond still showed a response at 6 months. However neither median progression-free survival nor median overall survival had been reached at the point of data cut-off.
The treatment showed similar effects in patients with regional and distant metastatic disease.
Advanced cutaneous squamous-cell carcinoma was thought to be an ideal target for immunotherapy because the high mutation burden in the tumor meant it would be sensitive to effector T cell attack, wrote Michael R. Migden, MD, of the University of Texas MD Anderson Cancer Center, Houston, and his coauthors.
“In addition, the dramatically increased risk of cutaneous squamous-cell carcinoma among people with immunosuppression pointed to an important role for immune surveillance with this cancer,” the authors wrote.
In the phase 2 study, 29% of patients experienced a serious adverse event – including two cases of pneumonitis – and three patients (5%) discontinued treatment. There were three deaths due to adverse events: One patient died from pneumonia complications, one died in his sleep, and one patient died following hypercalcemia and deep vein thrombosis.
Aside from these, most adverse events were grade 1 or 2. Around one-quarter of patients experienced diarrhea (27%) or fatigue (24%), while the other most common adverse events were nausea (17%), constipation (15%) and rash (15%). The authors noted that these adverse events were similar to those seen in other PD-1 inhibitors.
“Our results are consistent with an emerging theme regarding the high efficacy of immune checkpoint blockade for the treatment of hypermutated cancers, since the mutation burden of cutaneous squamous-cell carcinoma is similar to that reported for advanced solid tumors with microsatellite instability,” the authors wrote.
Cemiplimab is now being tested in a phase 2 trial in patients with advanced basal cell carcinoma.
The study was supported by Regeneron Pharmaceuticals and Sanofi. Eight authors declared funding from Regeneron to conduct the trial. Ten authors were employees of Regeneron. Fifteen authors also declared funding and payments from pharmaceutical companies outside the submitted work. Four had nothing to disclose.
SOURCE: Migden M et al. NEJM, 2018; June 4. doi: 10.1056/NEJMoa1805131.
An immune checkpoint inhibitor that targets the PD-1 receptor has shown “robust” efficacy among patients with advanced cutaneous squamous-cell carcinoma, according to researchers.
A combined phase 1/phase 2 study, published in the New England Journal of Medicine and presented simultaneously at the annual meeting of the American Society of Clinical Oncology, looked at the effect of monoclonal antibody cemiplimab in an expansion cohort of 26 patients with locally-advanced or metastatic cutaneous squamous-cell carcinoma who were not eligible for surgery. The phase 2 component involved 59 patients with metastatic disease.
Patients were treated with intravenous cemiplimab every 2 weeks for 48 weeks in the phase 1 study, and up to 96 weeks – or until unacceptable toxicity or disease progression – in the phase 2 study.
In the phase 1 study, researchers saw a response rate of 50% and a 65% rate of durable disease control, after a median follow-up of 11 months (1.1-17). The median time to response was 2.3 months, and more than half the patients (54%) who showed a response maintained that response past 6 months.
In the phase 2 study in patients with metastatic disease, 47% responded to the treatment – 24 patients showed a partial response and 4 showed a complete response. Of those who responded, 61% showed durable disease control after a median follow-up of 7.9 months.
The median time to response in this group of patients was 1.9 months, and 57% of those who did respond still showed a response at 6 months. However neither median progression-free survival nor median overall survival had been reached at the point of data cut-off.
The treatment showed similar effects in patients with regional and distant metastatic disease.
Advanced cutaneous squamous-cell carcinoma was thought to be an ideal target for immunotherapy because the high mutation burden in the tumor meant it would be sensitive to effector T cell attack, wrote Michael R. Migden, MD, of the University of Texas MD Anderson Cancer Center, Houston, and his coauthors.
“In addition, the dramatically increased risk of cutaneous squamous-cell carcinoma among people with immunosuppression pointed to an important role for immune surveillance with this cancer,” the authors wrote.
In the phase 2 study, 29% of patients experienced a serious adverse event – including two cases of pneumonitis – and three patients (5%) discontinued treatment. There were three deaths due to adverse events: One patient died from pneumonia complications, one died in his sleep, and one patient died following hypercalcemia and deep vein thrombosis.
Aside from these, most adverse events were grade 1 or 2. Around one-quarter of patients experienced diarrhea (27%) or fatigue (24%), while the other most common adverse events were nausea (17%), constipation (15%) and rash (15%). The authors noted that these adverse events were similar to those seen in other PD-1 inhibitors.
“Our results are consistent with an emerging theme regarding the high efficacy of immune checkpoint blockade for the treatment of hypermutated cancers, since the mutation burden of cutaneous squamous-cell carcinoma is similar to that reported for advanced solid tumors with microsatellite instability,” the authors wrote.
Cemiplimab is now being tested in a phase 2 trial in patients with advanced basal cell carcinoma.
The study was supported by Regeneron Pharmaceuticals and Sanofi. Eight authors declared funding from Regeneron to conduct the trial. Ten authors were employees of Regeneron. Fifteen authors also declared funding and payments from pharmaceutical companies outside the submitted work. Four had nothing to disclose.
SOURCE: Migden M et al. NEJM, 2018; June 4. doi: 10.1056/NEJMoa1805131.
FROM NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: PD-1 inhibitor cemiplimab shows significant response in advanced squamous-cell carcinoma.
Major finding: Around half of patients with advanced squamous-cell carcinoma responded to checkpoint inhibitor cemiplimab.
Study details: Phase 1 expanded cohort study of 26 patients with advanced cutaneous squamous-cell carcinoma and phase 2 study of 59 patients with metastatic squamous-cell carcinoma.
Disclosures: The study was supported by Regeneron Pharmaceuticals and Sanofi. Eight authors declared funding from Regeneron to conduct the trial. Ten authors were employees of Regeneron. Fifteen authors declared funding and payments from pharmaceutical companies outside the submitted work. Four had nothing to disclose.
Source: Migden M et al. N Engl J Med. 2018 June 4. doi: 10.1056/NEJMoa1805131.
FDA alerts clinicians to gastric balloon deaths
according to an alert from the Food and Drug Administration issued on June 4.
Seven of these deaths occurred in patients in the United States; four involved the ORBERA Intragastric Balloon System, and three involved the ReShape Integrated Dual Balloon System.
The FDA has approved updated labeling for the ORBERA and ReShape balloon systems in the United States. The labels contain more information about possible death associated with the use of these devices in the United States. The manufacturers’ sites, Apollo Endosurgery and ReShape Lifesciences, provide more details about the new labeling.
In a letter to health care providers, the FDA advised clinicians to educate bariatric surgery patients about the symptoms of complications from balloon procedures, including not only gastric perforation but also esophageal perforation, balloon deflation, gastrointestinal obstruction, and ulceration. In addition, the FDA reminded clinicians to monitor patients during the entire course of treatment for additional complications, including acute pancreatitis and spontaneous hyperinflation.
Any adverse events involving intragastric balloon systems should be reported to the FDA through MedWatch, the FDA Safety Information and Adverse Event Reporting program.
according to an alert from the Food and Drug Administration issued on June 4.
Seven of these deaths occurred in patients in the United States; four involved the ORBERA Intragastric Balloon System, and three involved the ReShape Integrated Dual Balloon System.
The FDA has approved updated labeling for the ORBERA and ReShape balloon systems in the United States. The labels contain more information about possible death associated with the use of these devices in the United States. The manufacturers’ sites, Apollo Endosurgery and ReShape Lifesciences, provide more details about the new labeling.
In a letter to health care providers, the FDA advised clinicians to educate bariatric surgery patients about the symptoms of complications from balloon procedures, including not only gastric perforation but also esophageal perforation, balloon deflation, gastrointestinal obstruction, and ulceration. In addition, the FDA reminded clinicians to monitor patients during the entire course of treatment for additional complications, including acute pancreatitis and spontaneous hyperinflation.
Any adverse events involving intragastric balloon systems should be reported to the FDA through MedWatch, the FDA Safety Information and Adverse Event Reporting program.
according to an alert from the Food and Drug Administration issued on June 4.
Seven of these deaths occurred in patients in the United States; four involved the ORBERA Intragastric Balloon System, and three involved the ReShape Integrated Dual Balloon System.
The FDA has approved updated labeling for the ORBERA and ReShape balloon systems in the United States. The labels contain more information about possible death associated with the use of these devices in the United States. The manufacturers’ sites, Apollo Endosurgery and ReShape Lifesciences, provide more details about the new labeling.
In a letter to health care providers, the FDA advised clinicians to educate bariatric surgery patients about the symptoms of complications from balloon procedures, including not only gastric perforation but also esophageal perforation, balloon deflation, gastrointestinal obstruction, and ulceration. In addition, the FDA reminded clinicians to monitor patients during the entire course of treatment for additional complications, including acute pancreatitis and spontaneous hyperinflation.
Any adverse events involving intragastric balloon systems should be reported to the FDA through MedWatch, the FDA Safety Information and Adverse Event Reporting program.
MRD-negative status signals better outcomes in CAR T–treated ALL
CHICAGO – Minimal residual disease (MRD)–negative complete remission was strongly associated with improved survival outcomes in patients with B-cell acute lymphocytic leukemia (ALL) who received CD19 chimeric antigen receptor (CAR) T cells, results of a retrospective study showed.
Allogeneic hematopoietic stem cell transplant (HSCT) appeared to improve both disease-free and overall survival in those patients who had achieved MRD-negative complete remission, according to results of the study, which were presented at the annual meeting of the American Society of Clinical Oncology.
“Based upon our interaction testing, the potential benefit [of transplant] appears to exist in both good-risk and bad-risk patients as identified through multivariate modeling,” said study investigator Kevin Anthony Hay, MD, of Fred Hutchinson Cancer Research Center, Seattle.
In a comment on the results, Sarah Cooley, MD, noted that the benefits of allogeneic transplant were apparent regardless of whether the patients met criteria for the good-risk subgroup, which was defined by levels of lactate dehydrogenase (LDH) and platelets along with exposure to fludarabine as part of the conditioning regimen.
“I think this suggests that the goal at this point is to get patients to an MRD-negative state and to potentially curative transplant,” said Dr. Cooley, director of investigator-initiated research at Masonic Medical Center at the University of Minnesota, Minneapolis.
The retrospective analysis by Dr. Hay and his colleagues included 53 adults with relapsed or refractory ALL who had bone marrow or extramedullary disease at baseline and had received CD19 CAR T cells at or under the maximum tolerated dose at least 1 year prior to this analysis. Of that group, 45 (85%) achieved MRD-negative complete remission.
Those patients who did achieve MRD-negative complete remission had an improved median disease-free survival at 7.6 months versus 0.8 months (P less than .0001) and improved overall survival at 20.0 months versus 5.0 months (P = 0.014).
Most of the MRD-negative patients who relapsed did so within the first 6 months, an observation that led investigators to consider whether factors exist that could predict better outcomes.
In a multivariate analysis, they found three variables associated with disease free survival: higher LDH prior to lymphodepletion (hazard ratio, 1.39), along with higher platelet count prior to lymphodepletion and incorporation of fludarabine into the regimen, with hazard ratios of 0.65 and 0.34, respectively.
Using those three characteristics, investigators grouped patients as “good risk” if they had normal LDH, platelet count at or above 100 prior to lymphodepletion that included fludarabine. The 24-month disease-free survival for good-risk patients was 78%, and overall survival was 86%.
The role of allogeneic HSCT after ALL patients achieved MRD-negative complete remission with CAR T-cell therapy was one of the “major questions in the field,” Dr. Hay said.
In this analysis, Dr. Hay and colleagues found that patients who underwent transplant in MRD-negative complete remission had a 24-month disease free survival and overall survival of 61% and 72%, respectively, both of which were significantly higher than in patients with MRD-negative complete remission who had no transplant.
The disease-free survival benefit was not specific to the good-risk group, according to Dr. Hay, who said an interaction test demonstrated no significant interaction between risk group and allogeneic HSCT after CAR T-cell infusion (P = 0.53).
“This is a very important finding that should be further [studied] in an appropriately designed clinical trial,” Dr. Hay said during an oral presentation of the study results.
Dr. Hay and several coauthors reported financial disclosures related to Juno Therapeutics. Other disclosures reported by study coauthors included Cell Medica, Celgene, Eureka Therapeutics, Genentech/Roche, Gilead Sciences, Kite Pharma, Novartis, and others.
SOURCE: Hay KA. ASCO 2018, Abstract 7005.
CHICAGO – Minimal residual disease (MRD)–negative complete remission was strongly associated with improved survival outcomes in patients with B-cell acute lymphocytic leukemia (ALL) who received CD19 chimeric antigen receptor (CAR) T cells, results of a retrospective study showed.
Allogeneic hematopoietic stem cell transplant (HSCT) appeared to improve both disease-free and overall survival in those patients who had achieved MRD-negative complete remission, according to results of the study, which were presented at the annual meeting of the American Society of Clinical Oncology.
“Based upon our interaction testing, the potential benefit [of transplant] appears to exist in both good-risk and bad-risk patients as identified through multivariate modeling,” said study investigator Kevin Anthony Hay, MD, of Fred Hutchinson Cancer Research Center, Seattle.
In a comment on the results, Sarah Cooley, MD, noted that the benefits of allogeneic transplant were apparent regardless of whether the patients met criteria for the good-risk subgroup, which was defined by levels of lactate dehydrogenase (LDH) and platelets along with exposure to fludarabine as part of the conditioning regimen.
“I think this suggests that the goal at this point is to get patients to an MRD-negative state and to potentially curative transplant,” said Dr. Cooley, director of investigator-initiated research at Masonic Medical Center at the University of Minnesota, Minneapolis.
The retrospective analysis by Dr. Hay and his colleagues included 53 adults with relapsed or refractory ALL who had bone marrow or extramedullary disease at baseline and had received CD19 CAR T cells at or under the maximum tolerated dose at least 1 year prior to this analysis. Of that group, 45 (85%) achieved MRD-negative complete remission.
Those patients who did achieve MRD-negative complete remission had an improved median disease-free survival at 7.6 months versus 0.8 months (P less than .0001) and improved overall survival at 20.0 months versus 5.0 months (P = 0.014).
Most of the MRD-negative patients who relapsed did so within the first 6 months, an observation that led investigators to consider whether factors exist that could predict better outcomes.
In a multivariate analysis, they found three variables associated with disease free survival: higher LDH prior to lymphodepletion (hazard ratio, 1.39), along with higher platelet count prior to lymphodepletion and incorporation of fludarabine into the regimen, with hazard ratios of 0.65 and 0.34, respectively.
Using those three characteristics, investigators grouped patients as “good risk” if they had normal LDH, platelet count at or above 100 prior to lymphodepletion that included fludarabine. The 24-month disease-free survival for good-risk patients was 78%, and overall survival was 86%.
The role of allogeneic HSCT after ALL patients achieved MRD-negative complete remission with CAR T-cell therapy was one of the “major questions in the field,” Dr. Hay said.
In this analysis, Dr. Hay and colleagues found that patients who underwent transplant in MRD-negative complete remission had a 24-month disease free survival and overall survival of 61% and 72%, respectively, both of which were significantly higher than in patients with MRD-negative complete remission who had no transplant.
The disease-free survival benefit was not specific to the good-risk group, according to Dr. Hay, who said an interaction test demonstrated no significant interaction between risk group and allogeneic HSCT after CAR T-cell infusion (P = 0.53).
“This is a very important finding that should be further [studied] in an appropriately designed clinical trial,” Dr. Hay said during an oral presentation of the study results.
Dr. Hay and several coauthors reported financial disclosures related to Juno Therapeutics. Other disclosures reported by study coauthors included Cell Medica, Celgene, Eureka Therapeutics, Genentech/Roche, Gilead Sciences, Kite Pharma, Novartis, and others.
SOURCE: Hay KA. ASCO 2018, Abstract 7005.
CHICAGO – Minimal residual disease (MRD)–negative complete remission was strongly associated with improved survival outcomes in patients with B-cell acute lymphocytic leukemia (ALL) who received CD19 chimeric antigen receptor (CAR) T cells, results of a retrospective study showed.
Allogeneic hematopoietic stem cell transplant (HSCT) appeared to improve both disease-free and overall survival in those patients who had achieved MRD-negative complete remission, according to results of the study, which were presented at the annual meeting of the American Society of Clinical Oncology.
“Based upon our interaction testing, the potential benefit [of transplant] appears to exist in both good-risk and bad-risk patients as identified through multivariate modeling,” said study investigator Kevin Anthony Hay, MD, of Fred Hutchinson Cancer Research Center, Seattle.
In a comment on the results, Sarah Cooley, MD, noted that the benefits of allogeneic transplant were apparent regardless of whether the patients met criteria for the good-risk subgroup, which was defined by levels of lactate dehydrogenase (LDH) and platelets along with exposure to fludarabine as part of the conditioning regimen.
“I think this suggests that the goal at this point is to get patients to an MRD-negative state and to potentially curative transplant,” said Dr. Cooley, director of investigator-initiated research at Masonic Medical Center at the University of Minnesota, Minneapolis.
The retrospective analysis by Dr. Hay and his colleagues included 53 adults with relapsed or refractory ALL who had bone marrow or extramedullary disease at baseline and had received CD19 CAR T cells at or under the maximum tolerated dose at least 1 year prior to this analysis. Of that group, 45 (85%) achieved MRD-negative complete remission.
Those patients who did achieve MRD-negative complete remission had an improved median disease-free survival at 7.6 months versus 0.8 months (P less than .0001) and improved overall survival at 20.0 months versus 5.0 months (P = 0.014).
Most of the MRD-negative patients who relapsed did so within the first 6 months, an observation that led investigators to consider whether factors exist that could predict better outcomes.
In a multivariate analysis, they found three variables associated with disease free survival: higher LDH prior to lymphodepletion (hazard ratio, 1.39), along with higher platelet count prior to lymphodepletion and incorporation of fludarabine into the regimen, with hazard ratios of 0.65 and 0.34, respectively.
Using those three characteristics, investigators grouped patients as “good risk” if they had normal LDH, platelet count at or above 100 prior to lymphodepletion that included fludarabine. The 24-month disease-free survival for good-risk patients was 78%, and overall survival was 86%.
The role of allogeneic HSCT after ALL patients achieved MRD-negative complete remission with CAR T-cell therapy was one of the “major questions in the field,” Dr. Hay said.
In this analysis, Dr. Hay and colleagues found that patients who underwent transplant in MRD-negative complete remission had a 24-month disease free survival and overall survival of 61% and 72%, respectively, both of which were significantly higher than in patients with MRD-negative complete remission who had no transplant.
The disease-free survival benefit was not specific to the good-risk group, according to Dr. Hay, who said an interaction test demonstrated no significant interaction between risk group and allogeneic HSCT after CAR T-cell infusion (P = 0.53).
“This is a very important finding that should be further [studied] in an appropriately designed clinical trial,” Dr. Hay said during an oral presentation of the study results.
Dr. Hay and several coauthors reported financial disclosures related to Juno Therapeutics. Other disclosures reported by study coauthors included Cell Medica, Celgene, Eureka Therapeutics, Genentech/Roche, Gilead Sciences, Kite Pharma, Novartis, and others.
SOURCE: Hay KA. ASCO 2018, Abstract 7005.
REPORTING FROM ASCO 2018
Key clinical point:
Major finding: Patients who achieved MRD-negative complete remission had an improved median disease-free survival at 7.6 months versus 0.8 months (P less than .0001)
Study details: A retrospective analysis including 53 patients with ALL who had bone marrow or extramedullary disease at baseline and had received CD19 CAR T cells at or under the maximum tolerated dose at least 1 year prior to this analysis.
Disclosures: Researchers reported financial ties to Juno Therapeutics, Cell Medica, Celgene, Eureka Therapeutics, Genentech/Roche, Gilead Sciences, Kite Pharma, Novartis, and others.
Source: Hay KA. ASCO 2018, Abstract 7005.
Dr. William J. Gradishar shares breast cancer take-aways from ASCO 2018
CHICAGO – William J. Gradishar, MD, discussed the clinical impact of breast cancer research presented at the annual meeting of the American Society of Clinical Oncology.
In a video interview, Dr. Gradishar, the Betsy Bramsen Professor of Breast Oncology at Northwestern University, Chicago, said TAILORx was a “big win” in that it has no doubt diminished the number of women with early-stage breast cancer who will require chemotherapy. However, although the trial has provided some clarity, it also has left some questions open, particularly for patients under 50 years of age, he said.
Dr. Gradishar also discussed the results of combination trials of targeted therapy with either endocrine therapy or chemotherapy. In discussing SANDPIPER, which evaluated whether a phosphoinositide 3-kinase inhibitor could enhance the effect of anti-hormonal therapy, he said that although it was a positive trial, “from a clinician’s standpoint, it’s probably not sufficient in my mind to get really excited about.”
CHICAGO – William J. Gradishar, MD, discussed the clinical impact of breast cancer research presented at the annual meeting of the American Society of Clinical Oncology.
In a video interview, Dr. Gradishar, the Betsy Bramsen Professor of Breast Oncology at Northwestern University, Chicago, said TAILORx was a “big win” in that it has no doubt diminished the number of women with early-stage breast cancer who will require chemotherapy. However, although the trial has provided some clarity, it also has left some questions open, particularly for patients under 50 years of age, he said.
Dr. Gradishar also discussed the results of combination trials of targeted therapy with either endocrine therapy or chemotherapy. In discussing SANDPIPER, which evaluated whether a phosphoinositide 3-kinase inhibitor could enhance the effect of anti-hormonal therapy, he said that although it was a positive trial, “from a clinician’s standpoint, it’s probably not sufficient in my mind to get really excited about.”
CHICAGO – William J. Gradishar, MD, discussed the clinical impact of breast cancer research presented at the annual meeting of the American Society of Clinical Oncology.
In a video interview, Dr. Gradishar, the Betsy Bramsen Professor of Breast Oncology at Northwestern University, Chicago, said TAILORx was a “big win” in that it has no doubt diminished the number of women with early-stage breast cancer who will require chemotherapy. However, although the trial has provided some clarity, it also has left some questions open, particularly for patients under 50 years of age, he said.
Dr. Gradishar also discussed the results of combination trials of targeted therapy with either endocrine therapy or chemotherapy. In discussing SANDPIPER, which evaluated whether a phosphoinositide 3-kinase inhibitor could enhance the effect of anti-hormonal therapy, he said that although it was a positive trial, “from a clinician’s standpoint, it’s probably not sufficient in my mind to get really excited about.”
REPORTING FROM ASCO 2018

Beltlike Lichen Planus Pigmentosus Complicated With Focal Amyloidosis
To the Editor:
A 68-year-old man presented with slightly itchy macules on the waist and abdomen of approximately 2 years’ duration. He reported that the initial lesions were dark red and subsequently coalesced to form a beltlike pigmentation on the abdomen. He denied any prior treatment, and the lesions did not spontaneously resolve. The patient was taking escitalopram oxalate, telmisartan, and aspirin for depression and cardiovascular disease that was diagnosed 3 years prior. He reported no exposure to UV radiation or a heat source. He denied use of any cosmetics on the body as well as a family history of similar symptoms.
Physical examination showed reticulate brown-purple macules with slight scale on the surface that had become confluent, forming a beltlike pigmentation on the waist and abdomen (Figure 1). Wickham striae were not seen. The oral mucosa and nails were not affected. Microscopic examination for fungal infections was negative.

Systematic physical and laboratory examinations revealed no abnormalities. A skin biopsy from a macule on the abdomen showed hyperkeratosis, thinned out stratum spinosum with flattening of rete ridges, hypergranulosis with vacuolar alteration of the basal cell layer, and bandlike infiltration of lymphocytes and melanophages with incontinence of pigment (Figure 2). Focalized purplish homogeneous deposits were observed in the upper dermis (Figure 3), of which positive crystal violet staining indicated amyloidosis (Figure 4). Congo red stain revealed amyloid deposition (Figure 5). Thus, the diagnosis of lichen planus pigmentosus (LPP) complicated with focal amyloidosis was made. The patient was treated with topical corticosteroids and tretinoin, and no notable therapeutic effects were observed at 3-month follow-up.

.")
.")
.")
Lichen planus pigmentosus, a variant of lichen planus, is a condition of unknown etiology exhibiting dark brown macules and/or papules and a long clinical course. The face, neck, trunk, arms, and legs are the most common areas of presentation, whereas involvement of the scalp, nails, or oral mucosa is relatively rare.
The first clinicohistopathological study with a large sample size was documented by Bhutani et al1 in 1974 who termed the currently recognized entity lichen planus pigmentosus. Lichen planus pigmentosus is a frequently encountered hyperpigmentation disorder in Indians, whereas sporadic cases also are reported in other regions and ethnicities.2 In cases of LPP, the pigmentation is symmetrical, and its pattern most often is diffuse, then reticular, blotchy, and perifollicular.3 Two unique patterns of LPP have been documented, including linear/blaschkoid LPP and zosteriform LPP.4,5 Our patient showed a unique beltlike distribution pattern.
The pathogenesis of LPP still is unclear, and several inciting factors such as mustard oil, gold therapy,6 and hepatitis C virus infection have been cited.7 Mancuso and Berdondini8 reported a case of LPP flaring immediately after relapse of nephrotic syndrome. It also has been considered as a paraneoplastic phenomen.9 No exact cause was found in our patient after a series of relative examinations.
The histopathologic changes associated with LPP consist of atrophic epidermis; bandlike lymphocytic infiltrate with vacuolar degeneration of the basal layer in the epidermis; and prominent melanin incontinence in the upper dermis, which can be diverse depending on different sites of skin biopsy and the phase of LPP. Histopathologic findings in our patient were consistent with LPP. The differential diagnosis for the reticulate pattern of pigmentation seen in our patient included confluent and reticulated papillomatosis and poikilodermalike cutaneous amyloidosis, both easily excluded with histopathologic confirmation.
Local amyloidosis also was confirmed by crystal violet staining in our case and its etiology was uncertain. Generalized and local amyloidosis has been reported in association with lichen planus. The diagnosis of lichen planus was followed by the diagnosis of amyloidosis, and the typical skin lesions of these 2 conditions were able to be differentiated in these reported cases.10,11 However, beltlike pigmentation was the only manifestation for our patient and we could not separate the 2 conditions with the naked eye.
Chronic irritation to the skin resulting in excessive production of degenerate keratins and their subsequent conversion into amyloid deposits has been proposed to be an etiologic factor of amyloidosis.11 Because of the distribution pattern in our case, we believe focal amyloidosis could be attributed to chronic friction and scratching.
- Bhutani LK, Bedi TR, Pandhi RK, et al. Lichen planus pigmentosus. Dermatologica. 1974;149:43-50.
- Kanwar AJ, Kaur S. Lichen planus pigmentosus. J Am Acad Dermatol. 1989;21(4, pt 1):815.
- Kanwar AJ, Dogra S, Handa S, et al. A study of 124 Indian patients with lichen planus pigmentosus. Clin Exp Dermatol. 2003;28:481-485.
- Akarsu S, Ilknur T, Özer E, et al. Lichen planus pigmentosus distributed along the lines of Blaschko. Int J Dermatol. 2013;52:253-254.
- Cho S, Whang KK. Lichen planus pigmentosus presenting in zosteriform pattern. J Dermatol. 1997;24:193-197.
- Ingber A, Weissmann-Katzenelson V, David M, et al. Lichen planus and lichen planus pigmentosus following gold therapy—case reports and review of the literature [in German]. Z Hautkr. 1986;61:315-319.
- Al-Mutairi N, El-Khalawany M. Clinicopathological characteristics of lichen planus pigmentosus and its response to tacrolimus ointment: an open label, non-randomized, prospective study. J Eur Acad Dermatol Venereol. 2010;24:535-540.
- Mancuso G, Berdondini RM. Coexistence of lichen planus pigmentosus and minimal change nephrotic syndrome. Eur J Dermatol. 2009;19:389-390.
- Sassolas B, Zagnoli A, Leroy JP, et al. Lichen planus pigmentosus associated with acrokeratosis of Bazex. Clin Exp Dermatol. 1994;19:70-73.
- Maeda H, Ohta S, Saito Y, et al. Epidermal origin of the amyloid in localized cutaneous amyloidosis. Br J Dermatol. 1982;106:345-351.
- Hongcharu W, Baldassano M, Gonzalez E. Generalized lichen amyloidosis associated with chronic lichen planus. J Am Acad Dermatol. 2000;43:346-348.
To the Editor:
A 68-year-old man presented with slightly itchy macules on the waist and abdomen of approximately 2 years’ duration. He reported that the initial lesions were dark red and subsequently coalesced to form a beltlike pigmentation on the abdomen. He denied any prior treatment, and the lesions did not spontaneously resolve. The patient was taking escitalopram oxalate, telmisartan, and aspirin for depression and cardiovascular disease that was diagnosed 3 years prior. He reported no exposure to UV radiation or a heat source. He denied use of any cosmetics on the body as well as a family history of similar symptoms.
Physical examination showed reticulate brown-purple macules with slight scale on the surface that had become confluent, forming a beltlike pigmentation on the waist and abdomen (Figure 1). Wickham striae were not seen. The oral mucosa and nails were not affected. Microscopic examination for fungal infections was negative.
Systematic physical and laboratory examinations revealed no abnormalities. A skin biopsy from a macule on the abdomen showed hyperkeratosis, thinned out stratum spinosum with flattening of rete ridges, hypergranulosis with vacuolar alteration of the basal cell layer, and bandlike infiltration of lymphocytes and melanophages with incontinence of pigment (Figure 2). Focalized purplish homogeneous deposits were observed in the upper dermis (Figure 3), of which positive crystal violet staining indicated amyloidosis (Figure 4). Congo red stain revealed amyloid deposition (Figure 5). Thus, the diagnosis of lichen planus pigmentosus (LPP) complicated with focal amyloidosis was made. The patient was treated with topical corticosteroids and tretinoin, and no notable therapeutic effects were observed at 3-month follow-up.
Lichen planus pigmentosus, a variant of lichen planus, is a condition of unknown etiology exhibiting dark brown macules and/or papules and a long clinical course. The face, neck, trunk, arms, and legs are the most common areas of presentation, whereas involvement of the scalp, nails, or oral mucosa is relatively rare.
The first clinicohistopathological study with a large sample size was documented by Bhutani et al1 in 1974 who termed the currently recognized entity lichen planus pigmentosus. Lichen planus pigmentosus is a frequently encountered hyperpigmentation disorder in Indians, whereas sporadic cases also are reported in other regions and ethnicities.2 In cases of LPP, the pigmentation is symmetrical, and its pattern most often is diffuse, then reticular, blotchy, and perifollicular.3 Two unique patterns of LPP have been documented, including linear/blaschkoid LPP and zosteriform LPP.4,5 Our patient showed a unique beltlike distribution pattern.
The pathogenesis of LPP still is unclear, and several inciting factors such as mustard oil, gold therapy,6 and hepatitis C virus infection have been cited.7 Mancuso and Berdondini8 reported a case of LPP flaring immediately after relapse of nephrotic syndrome. It also has been considered as a paraneoplastic phenomen.9 No exact cause was found in our patient after a series of relative examinations.
The histopathologic changes associated with LPP consist of atrophic epidermis; bandlike lymphocytic infiltrate with vacuolar degeneration of the basal layer in the epidermis; and prominent melanin incontinence in the upper dermis, which can be diverse depending on different sites of skin biopsy and the phase of LPP. Histopathologic findings in our patient were consistent with LPP. The differential diagnosis for the reticulate pattern of pigmentation seen in our patient included confluent and reticulated papillomatosis and poikilodermalike cutaneous amyloidosis, both easily excluded with histopathologic confirmation.
Local amyloidosis also was confirmed by crystal violet staining in our case and its etiology was uncertain. Generalized and local amyloidosis has been reported in association with lichen planus. The diagnosis of lichen planus was followed by the diagnosis of amyloidosis, and the typical skin lesions of these 2 conditions were able to be differentiated in these reported cases.10,11 However, beltlike pigmentation was the only manifestation for our patient and we could not separate the 2 conditions with the naked eye.
Chronic irritation to the skin resulting in excessive production of degenerate keratins and their subsequent conversion into amyloid deposits has been proposed to be an etiologic factor of amyloidosis.11 Because of the distribution pattern in our case, we believe focal amyloidosis could be attributed to chronic friction and scratching.
To the Editor:
A 68-year-old man presented with slightly itchy macules on the waist and abdomen of approximately 2 years’ duration. He reported that the initial lesions were dark red and subsequently coalesced to form a beltlike pigmentation on the abdomen. He denied any prior treatment, and the lesions did not spontaneously resolve. The patient was taking escitalopram oxalate, telmisartan, and aspirin for depression and cardiovascular disease that was diagnosed 3 years prior. He reported no exposure to UV radiation or a heat source. He denied use of any cosmetics on the body as well as a family history of similar symptoms.
Physical examination showed reticulate brown-purple macules with slight scale on the surface that had become confluent, forming a beltlike pigmentation on the waist and abdomen (Figure 1). Wickham striae were not seen. The oral mucosa and nails were not affected. Microscopic examination for fungal infections was negative.
Systematic physical and laboratory examinations revealed no abnormalities. A skin biopsy from a macule on the abdomen showed hyperkeratosis, thinned out stratum spinosum with flattening of rete ridges, hypergranulosis with vacuolar alteration of the basal cell layer, and bandlike infiltration of lymphocytes and melanophages with incontinence of pigment (Figure 2). Focalized purplish homogeneous deposits were observed in the upper dermis (Figure 3), of which positive crystal violet staining indicated amyloidosis (Figure 4). Congo red stain revealed amyloid deposition (Figure 5). Thus, the diagnosis of lichen planus pigmentosus (LPP) complicated with focal amyloidosis was made. The patient was treated with topical corticosteroids and tretinoin, and no notable therapeutic effects were observed at 3-month follow-up.
Lichen planus pigmentosus, a variant of lichen planus, is a condition of unknown etiology exhibiting dark brown macules and/or papules and a long clinical course. The face, neck, trunk, arms, and legs are the most common areas of presentation, whereas involvement of the scalp, nails, or oral mucosa is relatively rare.
The first clinicohistopathological study with a large sample size was documented by Bhutani et al1 in 1974 who termed the currently recognized entity lichen planus pigmentosus. Lichen planus pigmentosus is a frequently encountered hyperpigmentation disorder in Indians, whereas sporadic cases also are reported in other regions and ethnicities.2 In cases of LPP, the pigmentation is symmetrical, and its pattern most often is diffuse, then reticular, blotchy, and perifollicular.3 Two unique patterns of LPP have been documented, including linear/blaschkoid LPP and zosteriform LPP.4,5 Our patient showed a unique beltlike distribution pattern.
The pathogenesis of LPP still is unclear, and several inciting factors such as mustard oil, gold therapy,6 and hepatitis C virus infection have been cited.7 Mancuso and Berdondini8 reported a case of LPP flaring immediately after relapse of nephrotic syndrome. It also has been considered as a paraneoplastic phenomen.9 No exact cause was found in our patient after a series of relative examinations.
The histopathologic changes associated with LPP consist of atrophic epidermis; bandlike lymphocytic infiltrate with vacuolar degeneration of the basal layer in the epidermis; and prominent melanin incontinence in the upper dermis, which can be diverse depending on different sites of skin biopsy and the phase of LPP. Histopathologic findings in our patient were consistent with LPP. The differential diagnosis for the reticulate pattern of pigmentation seen in our patient included confluent and reticulated papillomatosis and poikilodermalike cutaneous amyloidosis, both easily excluded with histopathologic confirmation.
Local amyloidosis also was confirmed by crystal violet staining in our case and its etiology was uncertain. Generalized and local amyloidosis has been reported in association with lichen planus. The diagnosis of lichen planus was followed by the diagnosis of amyloidosis, and the typical skin lesions of these 2 conditions were able to be differentiated in these reported cases.10,11 However, beltlike pigmentation was the only manifestation for our patient and we could not separate the 2 conditions with the naked eye.
Chronic irritation to the skin resulting in excessive production of degenerate keratins and their subsequent conversion into amyloid deposits has been proposed to be an etiologic factor of amyloidosis.11 Because of the distribution pattern in our case, we believe focal amyloidosis could be attributed to chronic friction and scratching.
- Bhutani LK, Bedi TR, Pandhi RK, et al. Lichen planus pigmentosus. Dermatologica. 1974;149:43-50.
- Kanwar AJ, Kaur S. Lichen planus pigmentosus. J Am Acad Dermatol. 1989;21(4, pt 1):815.
- Kanwar AJ, Dogra S, Handa S, et al. A study of 124 Indian patients with lichen planus pigmentosus. Clin Exp Dermatol. 2003;28:481-485.
- Akarsu S, Ilknur T, Özer E, et al. Lichen planus pigmentosus distributed along the lines of Blaschko. Int J Dermatol. 2013;52:253-254.
- Cho S, Whang KK. Lichen planus pigmentosus presenting in zosteriform pattern. J Dermatol. 1997;24:193-197.
- Ingber A, Weissmann-Katzenelson V, David M, et al. Lichen planus and lichen planus pigmentosus following gold therapy—case reports and review of the literature [in German]. Z Hautkr. 1986;61:315-319.
- Al-Mutairi N, El-Khalawany M. Clinicopathological characteristics of lichen planus pigmentosus and its response to tacrolimus ointment: an open label, non-randomized, prospective study. J Eur Acad Dermatol Venereol. 2010;24:535-540.
- Mancuso G, Berdondini RM. Coexistence of lichen planus pigmentosus and minimal change nephrotic syndrome. Eur J Dermatol. 2009;19:389-390.
- Sassolas B, Zagnoli A, Leroy JP, et al. Lichen planus pigmentosus associated with acrokeratosis of Bazex. Clin Exp Dermatol. 1994;19:70-73.
- Maeda H, Ohta S, Saito Y, et al. Epidermal origin of the amyloid in localized cutaneous amyloidosis. Br J Dermatol. 1982;106:345-351.
- Hongcharu W, Baldassano M, Gonzalez E. Generalized lichen amyloidosis associated with chronic lichen planus. J Am Acad Dermatol. 2000;43:346-348.
- Bhutani LK, Bedi TR, Pandhi RK, et al. Lichen planus pigmentosus. Dermatologica. 1974;149:43-50.
- Kanwar AJ, Kaur S. Lichen planus pigmentosus. J Am Acad Dermatol. 1989;21(4, pt 1):815.
- Kanwar AJ, Dogra S, Handa S, et al. A study of 124 Indian patients with lichen planus pigmentosus. Clin Exp Dermatol. 2003;28:481-485.
- Akarsu S, Ilknur T, Özer E, et al. Lichen planus pigmentosus distributed along the lines of Blaschko. Int J Dermatol. 2013;52:253-254.
- Cho S, Whang KK. Lichen planus pigmentosus presenting in zosteriform pattern. J Dermatol. 1997;24:193-197.
- Ingber A, Weissmann-Katzenelson V, David M, et al. Lichen planus and lichen planus pigmentosus following gold therapy—case reports and review of the literature [in German]. Z Hautkr. 1986;61:315-319.
- Al-Mutairi N, El-Khalawany M. Clinicopathological characteristics of lichen planus pigmentosus and its response to tacrolimus ointment: an open label, non-randomized, prospective study. J Eur Acad Dermatol Venereol. 2010;24:535-540.
- Mancuso G, Berdondini RM. Coexistence of lichen planus pigmentosus and minimal change nephrotic syndrome. Eur J Dermatol. 2009;19:389-390.
- Sassolas B, Zagnoli A, Leroy JP, et al. Lichen planus pigmentosus associated with acrokeratosis of Bazex. Clin Exp Dermatol. 1994;19:70-73.
- Maeda H, Ohta S, Saito Y, et al. Epidermal origin of the amyloid in localized cutaneous amyloidosis. Br J Dermatol. 1982;106:345-351.
- Hongcharu W, Baldassano M, Gonzalez E. Generalized lichen amyloidosis associated with chronic lichen planus. J Am Acad Dermatol. 2000;43:346-348.
Practice Points
- Lichen planus pigmentosus can present in a unique beltlike distribution pattern.
- Focal amyloidosis due to chronic friction and scratching cannot be excluded from the differential diagnosis.
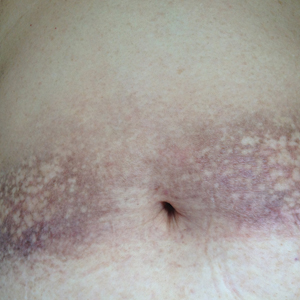
Metastatic lung cancer: Pembrolizumab plus chemo prolongs survival
, according to results from the phase 3 KEYNOTE-189 trial.
The researchers found that while patients in all subgroups benefited from the addition of pembrolizumab to chemotherapy, patients with a tumor proportion score of at least 50% benefited the most.
In 2017, accelerated approval was granted for pembrolizumab in combination with carboplatin and pemetrexed as first-line therapy for patients with NSCLC lacking EGFR and ALK mutations. This approval was based on response rates and progression-free survival from the KEYNOTE-024 phase 2 trial.
KEYNOTE-189 solidifies these findings, according to investigators. “Together with the results from KEYNOTE-024, the data from KEYNOTE-189 suggest that introducing immunotherapy as a first-line therapy may have a favorable long-term effect on outcomes,” wrote lead author Leena Gandhi, MD, director of the Perlmutter Cancer Center at New York University, and her colleagues. The report was published in the New England Journal of Medicine.
The double-blind, phase 3 trial assessed 616 treatment-naive patients with metastatic nonsquamous NSCLC without ALK or EGFR mutations. Patients received a platinum-based drug and pemetrexed plus placebo or 200 mg of pembrolizumab every 3 weeks for four cycles. After this, patients received placebo or pembrolizumab with pemetrexed maintenance therapy for up to 35 cycles. Primary endpoints were overall and progression-free survival, determined by a blinded radiologist.
At 12 months, the estimated overall survival was 69.2% for patients treated with pembrolizumab plus chemotherapy, compared with 49.4% for patients treated with chemotherapy alone. For patients treated with the pembrolizumab combination, median progression-free survival was 8.8 months, compared with 4.9 months for those treated with just chemotherapy. Median follow-up time was 10.5 months.
“The survival benefit associated with the pembrolizumab combination was observed in all subgroups of PD-L1 tumor proportion scores,” the authors wrote, “including patients with a score of less than 1%, a population for which single-agent PD-1 and PD-L1 inhibition have a small chance of benefit.” As with previous studies, the subgroup of patients with a tumor proportion score greater than or equal to 50% received the greatest benefit when pembrolizumab was added.
The authors noted that further research is needed to determine whether patients with high PD-L1 expression (TMS ≥ 50%) would benefit more from pembrolizumab combination therapy compared with pembrolizumab monotherapy, which is currently indicated.
SOURCE: Gandhi et al. N Engl J Med. 2018 May 31. doi: 10.1056/NEJMoa1801005.
The recent phase 3 KEYNOTE-189 trial by Gandhi and her colleagues solidifies a shift in first-line standard therapy for some lung cancer patients, according to Joan H. Schiller, MD.
“This trial illustrates that PD-1 pathway inhibitors can be successfully combined with chemotherapy,” Dr. Schiller wrote in an editorial in the New England Journal of Medicine.
In the study, patients treated with carboplatin-pemetrexed-pembrolizumab therapy had longer overall and progression-free survival compared with patients treated with just chemotherapy.
“The magnitude of benefit is impressive,” Dr. Schiller wrote, “with a hazard ratio for death of 0.49 and 12-month overall survival of 69.2% in the pembrolizumab-combination group.”
However, “many unanswered questions remain,” Dr. Schiller said, particularly concerning the utility of the commonly used PD-L1 tumor proportion score, which “appears to be problematic, with some randomized trials showing benefit only with a score of 50% or more and others showing benefit at other cutoff points, including less than 1%.”
In the current study, all subgroups benefited from the addition of pembrolizumab, including patients with a score of less than 1%.
Despite the need for more research, Dr. Schiller said she believes that existing findings have revealed unprecedented benefits. “Is carboplatin-pemetrexed-pembrolizumab now the first-line standard therapy for patients with nonsquamous NSCLC lacking targetable mutations? In my opinion, the answer is yes.”
Dr. Schiller is with the Inova Schar Cancer Institute. These comments are adapted from an editorial (N Eng J Med. 2018 May 31. doi: 10.1056/NEJMe1804364 ). The author reported financial support from Merck, Lily, AstraZeneca, Genentech/Roche, OncoGenex, Halozyme, Synta, Vertex, Bristol-Myers Squibb, Clovis, Xcovery, AbbVie, Astex, Janssen, and Free to Breathe.
The recent phase 3 KEYNOTE-189 trial by Gandhi and her colleagues solidifies a shift in first-line standard therapy for some lung cancer patients, according to Joan H. Schiller, MD.
“This trial illustrates that PD-1 pathway inhibitors can be successfully combined with chemotherapy,” Dr. Schiller wrote in an editorial in the New England Journal of Medicine.
In the study, patients treated with carboplatin-pemetrexed-pembrolizumab therapy had longer overall and progression-free survival compared with patients treated with just chemotherapy.
“The magnitude of benefit is impressive,” Dr. Schiller wrote, “with a hazard ratio for death of 0.49 and 12-month overall survival of 69.2% in the pembrolizumab-combination group.”
However, “many unanswered questions remain,” Dr. Schiller said, particularly concerning the utility of the commonly used PD-L1 tumor proportion score, which “appears to be problematic, with some randomized trials showing benefit only with a score of 50% or more and others showing benefit at other cutoff points, including less than 1%.”
In the current study, all subgroups benefited from the addition of pembrolizumab, including patients with a score of less than 1%.
Despite the need for more research, Dr. Schiller said she believes that existing findings have revealed unprecedented benefits. “Is carboplatin-pemetrexed-pembrolizumab now the first-line standard therapy for patients with nonsquamous NSCLC lacking targetable mutations? In my opinion, the answer is yes.”
Dr. Schiller is with the Inova Schar Cancer Institute. These comments are adapted from an editorial (N Eng J Med. 2018 May 31. doi: 10.1056/NEJMe1804364 ). The author reported financial support from Merck, Lily, AstraZeneca, Genentech/Roche, OncoGenex, Halozyme, Synta, Vertex, Bristol-Myers Squibb, Clovis, Xcovery, AbbVie, Astex, Janssen, and Free to Breathe.
The recent phase 3 KEYNOTE-189 trial by Gandhi and her colleagues solidifies a shift in first-line standard therapy for some lung cancer patients, according to Joan H. Schiller, MD.
“This trial illustrates that PD-1 pathway inhibitors can be successfully combined with chemotherapy,” Dr. Schiller wrote in an editorial in the New England Journal of Medicine.
In the study, patients treated with carboplatin-pemetrexed-pembrolizumab therapy had longer overall and progression-free survival compared with patients treated with just chemotherapy.
“The magnitude of benefit is impressive,” Dr. Schiller wrote, “with a hazard ratio for death of 0.49 and 12-month overall survival of 69.2% in the pembrolizumab-combination group.”
However, “many unanswered questions remain,” Dr. Schiller said, particularly concerning the utility of the commonly used PD-L1 tumor proportion score, which “appears to be problematic, with some randomized trials showing benefit only with a score of 50% or more and others showing benefit at other cutoff points, including less than 1%.”
In the current study, all subgroups benefited from the addition of pembrolizumab, including patients with a score of less than 1%.
Despite the need for more research, Dr. Schiller said she believes that existing findings have revealed unprecedented benefits. “Is carboplatin-pemetrexed-pembrolizumab now the first-line standard therapy for patients with nonsquamous NSCLC lacking targetable mutations? In my opinion, the answer is yes.”
Dr. Schiller is with the Inova Schar Cancer Institute. These comments are adapted from an editorial (N Eng J Med. 2018 May 31. doi: 10.1056/NEJMe1804364 ). The author reported financial support from Merck, Lily, AstraZeneca, Genentech/Roche, OncoGenex, Halozyme, Synta, Vertex, Bristol-Myers Squibb, Clovis, Xcovery, AbbVie, Astex, Janssen, and Free to Breathe.
, according to results from the phase 3 KEYNOTE-189 trial.
The researchers found that while patients in all subgroups benefited from the addition of pembrolizumab to chemotherapy, patients with a tumor proportion score of at least 50% benefited the most.
In 2017, accelerated approval was granted for pembrolizumab in combination with carboplatin and pemetrexed as first-line therapy for patients with NSCLC lacking EGFR and ALK mutations. This approval was based on response rates and progression-free survival from the KEYNOTE-024 phase 2 trial.
KEYNOTE-189 solidifies these findings, according to investigators. “Together with the results from KEYNOTE-024, the data from KEYNOTE-189 suggest that introducing immunotherapy as a first-line therapy may have a favorable long-term effect on outcomes,” wrote lead author Leena Gandhi, MD, director of the Perlmutter Cancer Center at New York University, and her colleagues. The report was published in the New England Journal of Medicine.
The double-blind, phase 3 trial assessed 616 treatment-naive patients with metastatic nonsquamous NSCLC without ALK or EGFR mutations. Patients received a platinum-based drug and pemetrexed plus placebo or 200 mg of pembrolizumab every 3 weeks for four cycles. After this, patients received placebo or pembrolizumab with pemetrexed maintenance therapy for up to 35 cycles. Primary endpoints were overall and progression-free survival, determined by a blinded radiologist.
At 12 months, the estimated overall survival was 69.2% for patients treated with pembrolizumab plus chemotherapy, compared with 49.4% for patients treated with chemotherapy alone. For patients treated with the pembrolizumab combination, median progression-free survival was 8.8 months, compared with 4.9 months for those treated with just chemotherapy. Median follow-up time was 10.5 months.
“The survival benefit associated with the pembrolizumab combination was observed in all subgroups of PD-L1 tumor proportion scores,” the authors wrote, “including patients with a score of less than 1%, a population for which single-agent PD-1 and PD-L1 inhibition have a small chance of benefit.” As with previous studies, the subgroup of patients with a tumor proportion score greater than or equal to 50% received the greatest benefit when pembrolizumab was added.
The authors noted that further research is needed to determine whether patients with high PD-L1 expression (TMS ≥ 50%) would benefit more from pembrolizumab combination therapy compared with pembrolizumab monotherapy, which is currently indicated.
SOURCE: Gandhi et al. N Engl J Med. 2018 May 31. doi: 10.1056/NEJMoa1801005.
, according to results from the phase 3 KEYNOTE-189 trial.
The researchers found that while patients in all subgroups benefited from the addition of pembrolizumab to chemotherapy, patients with a tumor proportion score of at least 50% benefited the most.
In 2017, accelerated approval was granted for pembrolizumab in combination with carboplatin and pemetrexed as first-line therapy for patients with NSCLC lacking EGFR and ALK mutations. This approval was based on response rates and progression-free survival from the KEYNOTE-024 phase 2 trial.
KEYNOTE-189 solidifies these findings, according to investigators. “Together with the results from KEYNOTE-024, the data from KEYNOTE-189 suggest that introducing immunotherapy as a first-line therapy may have a favorable long-term effect on outcomes,” wrote lead author Leena Gandhi, MD, director of the Perlmutter Cancer Center at New York University, and her colleagues. The report was published in the New England Journal of Medicine.
The double-blind, phase 3 trial assessed 616 treatment-naive patients with metastatic nonsquamous NSCLC without ALK or EGFR mutations. Patients received a platinum-based drug and pemetrexed plus placebo or 200 mg of pembrolizumab every 3 weeks for four cycles. After this, patients received placebo or pembrolizumab with pemetrexed maintenance therapy for up to 35 cycles. Primary endpoints were overall and progression-free survival, determined by a blinded radiologist.
At 12 months, the estimated overall survival was 69.2% for patients treated with pembrolizumab plus chemotherapy, compared with 49.4% for patients treated with chemotherapy alone. For patients treated with the pembrolizumab combination, median progression-free survival was 8.8 months, compared with 4.9 months for those treated with just chemotherapy. Median follow-up time was 10.5 months.
“The survival benefit associated with the pembrolizumab combination was observed in all subgroups of PD-L1 tumor proportion scores,” the authors wrote, “including patients with a score of less than 1%, a population for which single-agent PD-1 and PD-L1 inhibition have a small chance of benefit.” As with previous studies, the subgroup of patients with a tumor proportion score greater than or equal to 50% received the greatest benefit when pembrolizumab was added.
The authors noted that further research is needed to determine whether patients with high PD-L1 expression (TMS ≥ 50%) would benefit more from pembrolizumab combination therapy compared with pembrolizumab monotherapy, which is currently indicated.
SOURCE: Gandhi et al. N Engl J Med. 2018 May 31. doi: 10.1056/NEJMoa1801005.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: In patients with advanced NSCLC without targetable mutations, the addition of pembrolizumab to standard combination chemotherapy improves overall survival and progression-free survival.
Major finding: The estimated rate of overall survival at 12 months in patients treated with pembrolizumab-combination therapy was 69.2% (95% CI, 64.1-73.8) compared with 49.4% (95% CI, 42.1-56.2) in patients treated with placebo-combination therapy.
Study details: A double-blind, phase 3 trial of 616 patients with metastatic NSCLC lacking ALK or EGFR mutations (KEYNOTE-189).
Disclosures: Merck sponsored the study. Researchers reported financial support from Genentech/Roche, Pfizer, Ignyta, AbbVie, Eli Lilly, and other companies.
Source: Gandhi et al. N Engl J Med. 2018 May 31. doi: 10.1056/NEJMoa1801005.