User login
Is Sodium Oxybate Effective in Children With Narcolepsy?
The drug appears to reduce cataplexy and excessive sleepiness without raising new safety concerns.
LOS ANGELES—Sodium oxybate reduces cataplexy and excessive sleepiness in children with narcolepsy type 1, according to a study described at the 70th Annual Meeting of the American Academy of Neurology. The treatment’s safety profile in this population is similar to that in adults.
Although symptoms of narcolepsy often begin during childhood or adolescence, few studies have evaluated treatments for narcolepsy in pediatric patients. Sodium oxybate is approved for the treatment of cataplexy and excessive daytime sleepiness in adults with narcolepsy, but it had not previously been studied in a large pediatric narcolepsy trial. Chad Ruoff, MD, Clinical Assistant Professor of Psychiatry and Behavioral Sciences at the Stanford Center for Sleep Sciences and Medicine in California, and colleagues conducted a double-blind, placebo-controlled, randomized-withdrawal study to evaluate the efficacy and safety of sodium oxybate in pediatric patients with narcolepsy type 1.

A Randomized-Withdrawal Study
Eligible participants were children and adolescents between ages 7 and 16 who had been diagnosed with narcolepsy type 1 and had cataplexy. Patients who were on stable doses of sodium oxybate and patients who were sodium-oxybate-naïve were included. Patients with evidence of sleep-disordered breathing were excluded.
Sodium-oxybate-naïve participants were titrated to a stable dose. After a stable-dose period, all participants began a two-week, double-blind, placebo-controlled withdrawal period. The investigators randomized participants in equal groups to continue sodium oxybate or to be switched to placebo. At the end of the double-blind period, all participants received open-label sodium oxybate treatment. Efficacy assessments compared measurements during or at the end of the double-blind period with those taken the last two weeks of the stable-dose period. The study’s primary end point was change in weekly number of cataplexy attacks.
Study Was Terminated Early
Dr. Ruoff and colleagues randomized 63 participants. Approximately 41% of the population was between ages 7 and 11, 44% was female, and 38% was receiving sodium oxybate at baseline. A preplanned interim analysis of 35 participants indicated that sodium oxybate was effective, based on the primary end point result. The double-blind, randomized-withdrawal period thus was terminated early.
For the total group of 63 randomized participants, weekly cataplexy attacks were significantly increased in the placebo group (median, 12.7/week), compared with the sodium-oxybate-treated group (median, 0.3/week). Cataplexy severity, assessed using the Clinical Global Impression of Change (CGI-C), was worse in the placebo group than in the sodium-oxybate group. For 65% of participants in the placebo group, cataplexy was rated “much worse” or “very much worse,” compared with 17% of the sodium-oxybate group. Excessive sleepiness, assessed using the Epworth Sleepiness Scale for Children and Adolescents, also was worse in the placebo group (median increase, 3.0 points) than in the sodium-oxybate group (no change). In addition, the CGI-C for narcolepsy overall was worse in the placebo group.
Treatment-emergent adverse events occurring in more than 10% of the overall sample were enuresis, nausea, vomiting, headache, and decreased weight. These adverse events had been reported in previous trials of sodium oxybate in adults with narcolepsy.
The study was sponsored by Jazz Pharmaceuticals.
The drug appears to reduce cataplexy and excessive sleepiness without raising new safety concerns.
The drug appears to reduce cataplexy and excessive sleepiness without raising new safety concerns.
LOS ANGELES—Sodium oxybate reduces cataplexy and excessive sleepiness in children with narcolepsy type 1, according to a study described at the 70th Annual Meeting of the American Academy of Neurology. The treatment’s safety profile in this population is similar to that in adults.
Although symptoms of narcolepsy often begin during childhood or adolescence, few studies have evaluated treatments for narcolepsy in pediatric patients. Sodium oxybate is approved for the treatment of cataplexy and excessive daytime sleepiness in adults with narcolepsy, but it had not previously been studied in a large pediatric narcolepsy trial. Chad Ruoff, MD, Clinical Assistant Professor of Psychiatry and Behavioral Sciences at the Stanford Center for Sleep Sciences and Medicine in California, and colleagues conducted a double-blind, placebo-controlled, randomized-withdrawal study to evaluate the efficacy and safety of sodium oxybate in pediatric patients with narcolepsy type 1.
A Randomized-Withdrawal Study
Eligible participants were children and adolescents between ages 7 and 16 who had been diagnosed with narcolepsy type 1 and had cataplexy. Patients who were on stable doses of sodium oxybate and patients who were sodium-oxybate-naïve were included. Patients with evidence of sleep-disordered breathing were excluded.
Sodium-oxybate-naïve participants were titrated to a stable dose. After a stable-dose period, all participants began a two-week, double-blind, placebo-controlled withdrawal period. The investigators randomized participants in equal groups to continue sodium oxybate or to be switched to placebo. At the end of the double-blind period, all participants received open-label sodium oxybate treatment. Efficacy assessments compared measurements during or at the end of the double-blind period with those taken the last two weeks of the stable-dose period. The study’s primary end point was change in weekly number of cataplexy attacks.
Study Was Terminated Early
Dr. Ruoff and colleagues randomized 63 participants. Approximately 41% of the population was between ages 7 and 11, 44% was female, and 38% was receiving sodium oxybate at baseline. A preplanned interim analysis of 35 participants indicated that sodium oxybate was effective, based on the primary end point result. The double-blind, randomized-withdrawal period thus was terminated early.
For the total group of 63 randomized participants, weekly cataplexy attacks were significantly increased in the placebo group (median, 12.7/week), compared with the sodium-oxybate-treated group (median, 0.3/week). Cataplexy severity, assessed using the Clinical Global Impression of Change (CGI-C), was worse in the placebo group than in the sodium-oxybate group. For 65% of participants in the placebo group, cataplexy was rated “much worse” or “very much worse,” compared with 17% of the sodium-oxybate group. Excessive sleepiness, assessed using the Epworth Sleepiness Scale for Children and Adolescents, also was worse in the placebo group (median increase, 3.0 points) than in the sodium-oxybate group (no change). In addition, the CGI-C for narcolepsy overall was worse in the placebo group.
Treatment-emergent adverse events occurring in more than 10% of the overall sample were enuresis, nausea, vomiting, headache, and decreased weight. These adverse events had been reported in previous trials of sodium oxybate in adults with narcolepsy.
The study was sponsored by Jazz Pharmaceuticals.
LOS ANGELES—Sodium oxybate reduces cataplexy and excessive sleepiness in children with narcolepsy type 1, according to a study described at the 70th Annual Meeting of the American Academy of Neurology. The treatment’s safety profile in this population is similar to that in adults.
Although symptoms of narcolepsy often begin during childhood or adolescence, few studies have evaluated treatments for narcolepsy in pediatric patients. Sodium oxybate is approved for the treatment of cataplexy and excessive daytime sleepiness in adults with narcolepsy, but it had not previously been studied in a large pediatric narcolepsy trial. Chad Ruoff, MD, Clinical Assistant Professor of Psychiatry and Behavioral Sciences at the Stanford Center for Sleep Sciences and Medicine in California, and colleagues conducted a double-blind, placebo-controlled, randomized-withdrawal study to evaluate the efficacy and safety of sodium oxybate in pediatric patients with narcolepsy type 1.
A Randomized-Withdrawal Study
Eligible participants were children and adolescents between ages 7 and 16 who had been diagnosed with narcolepsy type 1 and had cataplexy. Patients who were on stable doses of sodium oxybate and patients who were sodium-oxybate-naïve were included. Patients with evidence of sleep-disordered breathing were excluded.
Sodium-oxybate-naïve participants were titrated to a stable dose. After a stable-dose period, all participants began a two-week, double-blind, placebo-controlled withdrawal period. The investigators randomized participants in equal groups to continue sodium oxybate or to be switched to placebo. At the end of the double-blind period, all participants received open-label sodium oxybate treatment. Efficacy assessments compared measurements during or at the end of the double-blind period with those taken the last two weeks of the stable-dose period. The study’s primary end point was change in weekly number of cataplexy attacks.
Study Was Terminated Early
Dr. Ruoff and colleagues randomized 63 participants. Approximately 41% of the population was between ages 7 and 11, 44% was female, and 38% was receiving sodium oxybate at baseline. A preplanned interim analysis of 35 participants indicated that sodium oxybate was effective, based on the primary end point result. The double-blind, randomized-withdrawal period thus was terminated early.
For the total group of 63 randomized participants, weekly cataplexy attacks were significantly increased in the placebo group (median, 12.7/week), compared with the sodium-oxybate-treated group (median, 0.3/week). Cataplexy severity, assessed using the Clinical Global Impression of Change (CGI-C), was worse in the placebo group than in the sodium-oxybate group. For 65% of participants in the placebo group, cataplexy was rated “much worse” or “very much worse,” compared with 17% of the sodium-oxybate group. Excessive sleepiness, assessed using the Epworth Sleepiness Scale for Children and Adolescents, also was worse in the placebo group (median increase, 3.0 points) than in the sodium-oxybate group (no change). In addition, the CGI-C for narcolepsy overall was worse in the placebo group.
Treatment-emergent adverse events occurring in more than 10% of the overall sample were enuresis, nausea, vomiting, headache, and decreased weight. These adverse events had been reported in previous trials of sodium oxybate in adults with narcolepsy.
The study was sponsored by Jazz Pharmaceuticals.
Current and future applications of telemedicine to optimize the delivery of care in chronic liver disease
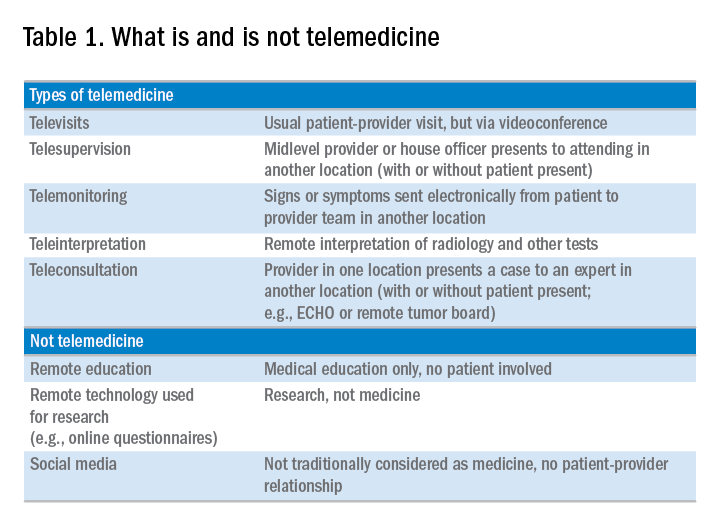
Telemedicine is defined broadly by the World Health Organization as the delivery of health care services at a distance using electronic means for “the diagnosis of treatment, and prevention of disease and injuries, research and evaluation, education of health care providers”1 to improve health. Although no single accepted definition exists, telehealth often is used as the umbrella term to encompass telemedicine (health care delivery) in addition to other activities such as education, research, health surveillance, and public health promotion.2 These various terms often are used interchangeably throughout the literature, leading to confusion.1,3 For the purpose of this review, we will use the term telemedicine to describe any care delivery model whereby patient care is provided at a distance using information technology such as cellphones, computers, or other electronic devices.
In the United States, the use of telemedicine is increasing. According to a 2017 survey of 184 health care executives conducted by the American Telemedicine Association, 88% believed that they would invest in telehealth in the near future, 98% believed that it offered a competitive advantage, with the caveat that 71% believed that lack of coverage and payments were barriers to implementation. Recent studies have shown that telehealth interventions are effective at improving clinical outcomes and decreasing inpatient utilization, with good patient satisfaction in the areas of mental health and chronic disease management. The Veterans Administration has emerged as an early telehealth adopter in chronic disease settings such as mental health, dermatology, hypertension, heart failure, and, as of 2016, has provided care to nearly 700,000 (12%) veterans since its inception.4-6 Despite the increased uptake, significant infrastructure and legal barriers to telemedicine remain and the literature regarding its utility in clinical practice continues to emerge.
Compared with other chronic diseases (e.g., heart failure, diabetes, mental illness) there is a dearth of literature on the use of telemedicine in liver disease. The first portion of this review synthesizes currently published literature of telemedicine/telehealth interventions to improve health care delivery and health outcomes in chronic liver disease including published peer-reviewed articles, abstracts, and ongoing clinical trials. The second portion discusses a framework for the future development of telemedicine and its integration into clinical practice by citing examples currently used throughout the country as well as ways to overcome implementation barriers.
Use of telemedicine in chronic liver disease: A literature review
We performed a systematic review of telemedicine in chronic liver disease. In consultation with a biomedical librarian, we searched for English-language articles for relevant studies with adult participants from July 1984 to May 2017 in PubMed, OVID Medline, American Association for the Study of Liver Disease, EMBASE, Web of Science, ClinicalTrials.gov, Elsevier/Science Direct, and the Cochrane Library (the search strategy is shown in the Supplementary Material at https://doi.org/10.1016/j.cgh.2017.10.004). The references of original publications and of review articles additionally were screened for potentially relevant studies. Abstracts that later resulted in no publications and studies in which telemedicine was used to deliver care, but was neither an exposure nor outcome, were excluded. Social media studies were not considered telemedicine if no patient care was involved. Studies of purely medical education interventions or those that evaluated the accuracy of technology to aid in diagnosis also were excluded.
Supplementary Table 1 (https://doi.org/10.1016/j.cgh.2017.10.004) shows the 20 published articles of telemedicine studies. Among these, there were 9 prospective trials, 3 retrospective studies, 2 case reports, and 6 small case series. One of the studies was randomized prospectively and 10 were uncontrolled.
Telemedicine in hepatitis C treatment

Telemedicine to aid in procedural/surgical management
A few reports have been published in the use of synchronous video and digital technology to aid in periprocedural management in liver disease. A case report highlighted a successful example of gastroenterologist-led teleproctoring using basic video technology to enable a surgeon to perform sclerotherapy for hemostasis in the setting of a variceal bleed.9 Another case report described the transmission of smart phone images from surgical trainees to an attending physician to make a real-time decision regarding a possibly questionable liver procurement, which took place 545 km away from the university hospital.10 A retrospective case series described the feasibility and successful use of high-resolution digital macroscopic photography and electronic transmission between liver transplant centers in the United Kingdom to increase the utilization of split liver transplantation, a setting in which detailed knowledge of vessel anatomy is needed for advanced surgical planning.11 Similarly, an uncontrolled case series from Greece reported on the feasibility and reliability of macroscopic image transmission to aid in the evaluation of liver grafts for transplantation.12
Telemedicine to support evaluation and management of hepatocellular carcinoma
One recent abstract reported on the use of asynchronous store-and-forward telemedicine for screening and management of hepatocellular carcinoma and evaluated process outcomes of specialty care access for newly diagnosed patients.13 A multifaceted approach included live video teleconferencing and centralized radiology review, which was conducted by a multidisciplinary tumor board at an expert hub site, which provided expert opinion and subsequent care (e.g., locoregional therapy, liver transplant evaluation) to spoke sites. As a result of the initiative, the time to specialty evaluation and receipt of hepatocellular carcinoma therapy decreased by 23 and 25 days, respectively.
Remote monitoring interventions

Proposed framework for advancing telemedicine in liver disease: The case for more research and policy changes
Telemedicine can serve two main goals in liver disease: improve access to specialty care, and improve care between visits. For the first goal, the technology is straightforward and limited research is required; the main barriers are regulatory and reimbursement. As an example, one of the authors (M.L.V.) uses telemedicine to perform liver transplant evaluations in Las Vegas, N.V., a state without a liver transplant program. Patients are seen initially by a nurse practitioner who resides in Las Vegas, and those patients needing transplant evaluation are scheduled for a video visit with the attending physician who is physically in California. This works well and patients love it; however, the business model is dependent on the downstream financial incentive of transplantation. In addition, various regulatory requirements must be satisfied such as monthly in-person visits. For the second goal, a number of exciting possibilities exist such as remote monitoring and patient disease management, but more research is needed.
Research
According to the Pew Research Center, 95% of American adults own a cellphone and 77% own a smartphone. These devices passively gather an extraordinary amount of data that could be harnessed to identify early warning signs of complications (remote monitoring). Another potentially fruitful area of research is patient disease management. This includes using technology (e.g., reminder texts) to effect behavior change such as with medication adherence, lifestyle modification, education, or peer mentoring. As an example, the coauthor (M.S.) is leading a study to promote physical activity among liver transplant recipients by using an online web portal developed by researchers at the University of Pennsylvania (Way to Health), which interfaces with patient cell phones and digital accelerometer devices. Participants receive daily feedback through text messages with their step counts, and small financial incentives are provided for adequate levels of physical activity. Technology also can facilitate the development of disease management platforms, which could improve both access and in-between visit monitoring, especially in remote areas. One of the authors (M.L.V.) currently is leading the development of a remote disease management program with funding from the American Association for the Study of Liver Diseases.
Despite the tremendous promise, traditional research methods in telemedicine may be challenging given the rapid and increasing uptake of health technology among patients and health systems. As such, the classic paradigm of randomized controlled trials to evaluate the success of an intervention or change in care delivery often is not feasible. We believe there is a need to recalibrate the definition of what constitutes a high-quality telemedicine study. For example, pragmatic trials and those designed within an implementation science framework that evaluate feasibility, scalability, and cost, in parallel with traditional clinical outcomes, may be better suited and should be accepted more widely.17
Policy
Even when the technology is available and research shows efficacy, the implementation of telemedicine in clinical practice faces regulatory and reimbursement barriers. The first regulatory question is whether a patient–provider relationship is being established (with the exception of limited provider–provider curbside consultation, the answer usually is yes). If so, the practice then is subject to all the usual regulatory concerns. The provider needs to be licensed at the site of origin (where the patient is located) and hold malpractice coverage for that location, and the video and medical record transmission should be compliant with the Health Insurance Portability and Accountability Act. The next challenge is reimbursement. Medicare only pays for video consultation if the patient lives in a designated rural Health Professional Shortage Area (www.cms.gov), and reimbursement by private payers varies. Even this is dependent on ever-changing state laws. Reimbursement for remote patient monitoring is even more limited (the National Telehealth Policy Research Center publishes a useful handbook: http://www.cchpca.org/sites/default/files/resources/50%20State%20FINAL%20April%202016.pdf). Absent a bipartisan Congressional effort to remedy this situation, the best hope for removing reimbursement barriers lies with payment reform. The Medicare Access and CHIP Reauthorization Act of 2015 mandates that the Centers for Medicare and Medicaid Services shift from fee-for-service to alternative payment models in the coming years. In these alternate payment models, providers are responsible for the overall quality and total cost of care for a population of patients. In this scenario, there may be a financial incentive for telemedicine, especially remote monitoring, to keep patients out of the hospital. Until then, under current payment models, reimbursement is limited and the barriers to widespread implementation are high.
Conclusions
Telemedicine has continued to increase in uptake and shows tremendous promise in expanding access to health care, promoting patient disease management, and facilitating in-between health care visit monitoring. Although the future is bright, more research is needed to determine optimal ways to integrate telemedicine — especially remote monitoring — into routine clinical care. We call on our specialty societies to send a clear political advocacy message that policy changes are needed to overcome regulatory and reimbursement challenges.
Acknowledgments
The authors would like to thank Lauren Jones and Mackenzie McDougal for their assistance with the literature review.
Supplementary materials and methods
The telemedicine interventions PubMed literature search strategy was as follows: ((“liver diseases”[MeSH Terms] OR (“liver”[All Fields] AND “diseases”[All Fields]) OR “liver diseases”[All Fields] OR (“liver”[All Fields] AND “disease”[All Fields]) OR “liver disease”[All Fields] OR liver dysfunction OR liver dysfunctions)) OR “liver transplantation”[MeSH Terms] OR “liver transplantation” [All Fields] AND (((“telemedicine”[MeSH Terms] OR “telemedicine”[All Fields] OR mobile health OR mhealth OR telehealth OR mhealth)) OR (videoconferencing OR videoconference)).
References
1. Kirsh S., Su G.L., Sales A., et al. Access to outpatient specialty care: solutions from an integrated health care system. Am J Med Qual. 2015;30:88-90.
2. Wilson L.S., Maeder A.J. recent directions in telemedicine: review of trends in research and practice. Healthc Inform Res. 2015;21:213-22.
3. Cross R.K., Kane, S. Integration of telemedicine into clinical gastroenterology and hepatology practice. Clin Gastroenterol Hepatol. 2017;15:175-81.
4. Darkins A., Ryan P., Kobb R., et al. Care coordination/home telehealth: the systematic implementation of health informatics, home telehealth, and disease management to support the care of veteran patients with chronic conditions. Telemed J E Health. 2008;14:1118-26.
5. Tuerk P.W., Fortney J., Bosworth H.B., et al. Toward the development of national telehealth services: the role of Veterans Health Administration and future directions for research. Telemed J E Health. 2010;16:115-7.
6. VA Press Release. Available: https://www.va.gov/opa/pressrel/includes/viewPDF.cfm?id=2789. Accessed: July 27, 2017.
7. Arora S., Kalishman S., Thornton K., et al. Expanding access to hepatitis C virus treatment-extension for Community Healthcare Outcomes (ECHO) project: disruptive innovation in specialty care. Hepatology. 2010;52:1124-33.
8. Mitruka K., Thornton K., Cusick S., et al. Expanding primary care capacity to treat hepatitis C virus infection through an evidence-based care model–Arizona and Utah, 2012-2014. MMWR Morb Mortal Wkly Rep. 2014;63:393-8.
9. Ahmed A., Slosberg E., Prasad P., et al. The successful use of telemedicine in acute variceal hemorrhage. J Clin Gastroenterol. 1999;29:212-3.
10. Croome K.P., Shum J., Al-Basheer M.A., et al. The benefit of smart phone usage in liver organ procurement. J Telemed Telecare. 2011;17:158-60.
11. Bhati C.S., Wigmore S.J., Reddy S., et al. Web-based image transmission: a novel approach to aid communication in split liver transplantation. Clin Transplant. 2010;24:98-103.
12. Mammas C.S., Geropoulos S., Saatsakis G., et al. Telepathology as a method to optimize quality in organ transplantation: a feasibility and reliability study of the virtual benching of liver graft. Stud Health Technol Inform. 2013;190:276-8.
13. Egert E.M., et al. A regional multidisciplinary liver tumor board improves access to hepatocellular carcinoma treatment for patients geographically distant from tertiary medical center. Hepatology. 2015;62:469A
14. Thomson M., Volk M., Kim H.M., et al. An automated telephone monitoring system to identify patients with cirrhosis at risk of re-hospitalization. Dig Dis Sci. 2015;60:3563-9.
15. Ertel A.E., Kaiser T.E., Abbott D.E., et al. Use of video-based education and tele-health home monitoring after liver transplantation: results of a novel pilot study. Surgery. 2016;160:869-76.
16. Thygesen G.B., Andersen H., Damsgaard B.S. et al. The effect of nurse performed telemedical video consultations for patients suffering from alcohol-related liver cirrhosis. J Hepatol. 2017;66:S349
17. Proctor E., Silmere H., Raghavan R. et al. Outcomes for implementation research: conceptual distinctions, measurement challenges, and research agenda. Adm Policy Ment Health. 2011;38:65-76.
Dr. Serper is in the division of gastroenterology, University of Pennsylvania Perelman School of Medicine, Philadelphia, and the department of medicine, Corporal Michael J. Crescenz VA Medical Center, Philadelphia; Dr. Volk is in the division of gastroenterology and Transplantation Institute, Loma Linda University, Loma Linda, Calif. The authors disclose no conflicts.
Telemedicine is defined broadly by the World Health Organization as the delivery of health care services at a distance using electronic means for “the diagnosis of treatment, and prevention of disease and injuries, research and evaluation, education of health care providers”1 to improve health. Although no single accepted definition exists, telehealth often is used as the umbrella term to encompass telemedicine (health care delivery) in addition to other activities such as education, research, health surveillance, and public health promotion.2 These various terms often are used interchangeably throughout the literature, leading to confusion.1,3 For the purpose of this review, we will use the term telemedicine to describe any care delivery model whereby patient care is provided at a distance using information technology such as cellphones, computers, or other electronic devices.
In the United States, the use of telemedicine is increasing. According to a 2017 survey of 184 health care executives conducted by the American Telemedicine Association, 88% believed that they would invest in telehealth in the near future, 98% believed that it offered a competitive advantage, with the caveat that 71% believed that lack of coverage and payments were barriers to implementation. Recent studies have shown that telehealth interventions are effective at improving clinical outcomes and decreasing inpatient utilization, with good patient satisfaction in the areas of mental health and chronic disease management. The Veterans Administration has emerged as an early telehealth adopter in chronic disease settings such as mental health, dermatology, hypertension, heart failure, and, as of 2016, has provided care to nearly 700,000 (12%) veterans since its inception.4-6 Despite the increased uptake, significant infrastructure and legal barriers to telemedicine remain and the literature regarding its utility in clinical practice continues to emerge.
Compared with other chronic diseases (e.g., heart failure, diabetes, mental illness) there is a dearth of literature on the use of telemedicine in liver disease. The first portion of this review synthesizes currently published literature of telemedicine/telehealth interventions to improve health care delivery and health outcomes in chronic liver disease including published peer-reviewed articles, abstracts, and ongoing clinical trials. The second portion discusses a framework for the future development of telemedicine and its integration into clinical practice by citing examples currently used throughout the country as well as ways to overcome implementation barriers.
Use of telemedicine in chronic liver disease: A literature review
We performed a systematic review of telemedicine in chronic liver disease. In consultation with a biomedical librarian, we searched for English-language articles for relevant studies with adult participants from July 1984 to May 2017 in PubMed, OVID Medline, American Association for the Study of Liver Disease, EMBASE, Web of Science, ClinicalTrials.gov, Elsevier/Science Direct, and the Cochrane Library (the search strategy is shown in the Supplementary Material at https://doi.org/10.1016/j.cgh.2017.10.004). The references of original publications and of review articles additionally were screened for potentially relevant studies. Abstracts that later resulted in no publications and studies in which telemedicine was used to deliver care, but was neither an exposure nor outcome, were excluded. Social media studies were not considered telemedicine if no patient care was involved. Studies of purely medical education interventions or those that evaluated the accuracy of technology to aid in diagnosis also were excluded.
Supplementary Table 1 (https://doi.org/10.1016/j.cgh.2017.10.004) shows the 20 published articles of telemedicine studies. Among these, there were 9 prospective trials, 3 retrospective studies, 2 case reports, and 6 small case series. One of the studies was randomized prospectively and 10 were uncontrolled.
Telemedicine in hepatitis C treatment
Telemedicine to aid in procedural/surgical management
A few reports have been published in the use of synchronous video and digital technology to aid in periprocedural management in liver disease. A case report highlighted a successful example of gastroenterologist-led teleproctoring using basic video technology to enable a surgeon to perform sclerotherapy for hemostasis in the setting of a variceal bleed.9 Another case report described the transmission of smart phone images from surgical trainees to an attending physician to make a real-time decision regarding a possibly questionable liver procurement, which took place 545 km away from the university hospital.10 A retrospective case series described the feasibility and successful use of high-resolution digital macroscopic photography and electronic transmission between liver transplant centers in the United Kingdom to increase the utilization of split liver transplantation, a setting in which detailed knowledge of vessel anatomy is needed for advanced surgical planning.11 Similarly, an uncontrolled case series from Greece reported on the feasibility and reliability of macroscopic image transmission to aid in the evaluation of liver grafts for transplantation.12
Telemedicine to support evaluation and management of hepatocellular carcinoma
One recent abstract reported on the use of asynchronous store-and-forward telemedicine for screening and management of hepatocellular carcinoma and evaluated process outcomes of specialty care access for newly diagnosed patients.13 A multifaceted approach included live video teleconferencing and centralized radiology review, which was conducted by a multidisciplinary tumor board at an expert hub site, which provided expert opinion and subsequent care (e.g., locoregional therapy, liver transplant evaluation) to spoke sites. As a result of the initiative, the time to specialty evaluation and receipt of hepatocellular carcinoma therapy decreased by 23 and 25 days, respectively.
Remote monitoring interventions
Proposed framework for advancing telemedicine in liver disease: The case for more research and policy changes
Telemedicine can serve two main goals in liver disease: improve access to specialty care, and improve care between visits. For the first goal, the technology is straightforward and limited research is required; the main barriers are regulatory and reimbursement. As an example, one of the authors (M.L.V.) uses telemedicine to perform liver transplant evaluations in Las Vegas, N.V., a state without a liver transplant program. Patients are seen initially by a nurse practitioner who resides in Las Vegas, and those patients needing transplant evaluation are scheduled for a video visit with the attending physician who is physically in California. This works well and patients love it; however, the business model is dependent on the downstream financial incentive of transplantation. In addition, various regulatory requirements must be satisfied such as monthly in-person visits. For the second goal, a number of exciting possibilities exist such as remote monitoring and patient disease management, but more research is needed.
Research
According to the Pew Research Center, 95% of American adults own a cellphone and 77% own a smartphone. These devices passively gather an extraordinary amount of data that could be harnessed to identify early warning signs of complications (remote monitoring). Another potentially fruitful area of research is patient disease management. This includes using technology (e.g., reminder texts) to effect behavior change such as with medication adherence, lifestyle modification, education, or peer mentoring. As an example, the coauthor (M.S.) is leading a study to promote physical activity among liver transplant recipients by using an online web portal developed by researchers at the University of Pennsylvania (Way to Health), which interfaces with patient cell phones and digital accelerometer devices. Participants receive daily feedback through text messages with their step counts, and small financial incentives are provided for adequate levels of physical activity. Technology also can facilitate the development of disease management platforms, which could improve both access and in-between visit monitoring, especially in remote areas. One of the authors (M.L.V.) currently is leading the development of a remote disease management program with funding from the American Association for the Study of Liver Diseases.
Despite the tremendous promise, traditional research methods in telemedicine may be challenging given the rapid and increasing uptake of health technology among patients and health systems. As such, the classic paradigm of randomized controlled trials to evaluate the success of an intervention or change in care delivery often is not feasible. We believe there is a need to recalibrate the definition of what constitutes a high-quality telemedicine study. For example, pragmatic trials and those designed within an implementation science framework that evaluate feasibility, scalability, and cost, in parallel with traditional clinical outcomes, may be better suited and should be accepted more widely.17
Policy
Even when the technology is available and research shows efficacy, the implementation of telemedicine in clinical practice faces regulatory and reimbursement barriers. The first regulatory question is whether a patient–provider relationship is being established (with the exception of limited provider–provider curbside consultation, the answer usually is yes). If so, the practice then is subject to all the usual regulatory concerns. The provider needs to be licensed at the site of origin (where the patient is located) and hold malpractice coverage for that location, and the video and medical record transmission should be compliant with the Health Insurance Portability and Accountability Act. The next challenge is reimbursement. Medicare only pays for video consultation if the patient lives in a designated rural Health Professional Shortage Area (www.cms.gov), and reimbursement by private payers varies. Even this is dependent on ever-changing state laws. Reimbursement for remote patient monitoring is even more limited (the National Telehealth Policy Research Center publishes a useful handbook: http://www.cchpca.org/sites/default/files/resources/50%20State%20FINAL%20April%202016.pdf). Absent a bipartisan Congressional effort to remedy this situation, the best hope for removing reimbursement barriers lies with payment reform. The Medicare Access and CHIP Reauthorization Act of 2015 mandates that the Centers for Medicare and Medicaid Services shift from fee-for-service to alternative payment models in the coming years. In these alternate payment models, providers are responsible for the overall quality and total cost of care for a population of patients. In this scenario, there may be a financial incentive for telemedicine, especially remote monitoring, to keep patients out of the hospital. Until then, under current payment models, reimbursement is limited and the barriers to widespread implementation are high.
Conclusions
Telemedicine has continued to increase in uptake and shows tremendous promise in expanding access to health care, promoting patient disease management, and facilitating in-between health care visit monitoring. Although the future is bright, more research is needed to determine optimal ways to integrate telemedicine — especially remote monitoring — into routine clinical care. We call on our specialty societies to send a clear political advocacy message that policy changes are needed to overcome regulatory and reimbursement challenges.
Acknowledgments
The authors would like to thank Lauren Jones and Mackenzie McDougal for their assistance with the literature review.
Supplementary materials and methods
The telemedicine interventions PubMed literature search strategy was as follows: ((“liver diseases”[MeSH Terms] OR (“liver”[All Fields] AND “diseases”[All Fields]) OR “liver diseases”[All Fields] OR (“liver”[All Fields] AND “disease”[All Fields]) OR “liver disease”[All Fields] OR liver dysfunction OR liver dysfunctions)) OR “liver transplantation”[MeSH Terms] OR “liver transplantation” [All Fields] AND (((“telemedicine”[MeSH Terms] OR “telemedicine”[All Fields] OR mobile health OR mhealth OR telehealth OR mhealth)) OR (videoconferencing OR videoconference)).
References
1. Kirsh S., Su G.L., Sales A., et al. Access to outpatient specialty care: solutions from an integrated health care system. Am J Med Qual. 2015;30:88-90.
2. Wilson L.S., Maeder A.J. recent directions in telemedicine: review of trends in research and practice. Healthc Inform Res. 2015;21:213-22.
3. Cross R.K., Kane, S. Integration of telemedicine into clinical gastroenterology and hepatology practice. Clin Gastroenterol Hepatol. 2017;15:175-81.
4. Darkins A., Ryan P., Kobb R., et al. Care coordination/home telehealth: the systematic implementation of health informatics, home telehealth, and disease management to support the care of veteran patients with chronic conditions. Telemed J E Health. 2008;14:1118-26.
5. Tuerk P.W., Fortney J., Bosworth H.B., et al. Toward the development of national telehealth services: the role of Veterans Health Administration and future directions for research. Telemed J E Health. 2010;16:115-7.
6. VA Press Release. Available: https://www.va.gov/opa/pressrel/includes/viewPDF.cfm?id=2789. Accessed: July 27, 2017.
7. Arora S., Kalishman S., Thornton K., et al. Expanding access to hepatitis C virus treatment-extension for Community Healthcare Outcomes (ECHO) project: disruptive innovation in specialty care. Hepatology. 2010;52:1124-33.
8. Mitruka K., Thornton K., Cusick S., et al. Expanding primary care capacity to treat hepatitis C virus infection through an evidence-based care model–Arizona and Utah, 2012-2014. MMWR Morb Mortal Wkly Rep. 2014;63:393-8.
9. Ahmed A., Slosberg E., Prasad P., et al. The successful use of telemedicine in acute variceal hemorrhage. J Clin Gastroenterol. 1999;29:212-3.
10. Croome K.P., Shum J., Al-Basheer M.A., et al. The benefit of smart phone usage in liver organ procurement. J Telemed Telecare. 2011;17:158-60.
11. Bhati C.S., Wigmore S.J., Reddy S., et al. Web-based image transmission: a novel approach to aid communication in split liver transplantation. Clin Transplant. 2010;24:98-103.
12. Mammas C.S., Geropoulos S., Saatsakis G., et al. Telepathology as a method to optimize quality in organ transplantation: a feasibility and reliability study of the virtual benching of liver graft. Stud Health Technol Inform. 2013;190:276-8.
13. Egert E.M., et al. A regional multidisciplinary liver tumor board improves access to hepatocellular carcinoma treatment for patients geographically distant from tertiary medical center. Hepatology. 2015;62:469A
14. Thomson M., Volk M., Kim H.M., et al. An automated telephone monitoring system to identify patients with cirrhosis at risk of re-hospitalization. Dig Dis Sci. 2015;60:3563-9.
15. Ertel A.E., Kaiser T.E., Abbott D.E., et al. Use of video-based education and tele-health home monitoring after liver transplantation: results of a novel pilot study. Surgery. 2016;160:869-76.
16. Thygesen G.B., Andersen H., Damsgaard B.S. et al. The effect of nurse performed telemedical video consultations for patients suffering from alcohol-related liver cirrhosis. J Hepatol. 2017;66:S349
17. Proctor E., Silmere H., Raghavan R. et al. Outcomes for implementation research: conceptual distinctions, measurement challenges, and research agenda. Adm Policy Ment Health. 2011;38:65-76.
Dr. Serper is in the division of gastroenterology, University of Pennsylvania Perelman School of Medicine, Philadelphia, and the department of medicine, Corporal Michael J. Crescenz VA Medical Center, Philadelphia; Dr. Volk is in the division of gastroenterology and Transplantation Institute, Loma Linda University, Loma Linda, Calif. The authors disclose no conflicts.
Telemedicine is defined broadly by the World Health Organization as the delivery of health care services at a distance using electronic means for “the diagnosis of treatment, and prevention of disease and injuries, research and evaluation, education of health care providers”1 to improve health. Although no single accepted definition exists, telehealth often is used as the umbrella term to encompass telemedicine (health care delivery) in addition to other activities such as education, research, health surveillance, and public health promotion.2 These various terms often are used interchangeably throughout the literature, leading to confusion.1,3 For the purpose of this review, we will use the term telemedicine to describe any care delivery model whereby patient care is provided at a distance using information technology such as cellphones, computers, or other electronic devices.
In the United States, the use of telemedicine is increasing. According to a 2017 survey of 184 health care executives conducted by the American Telemedicine Association, 88% believed that they would invest in telehealth in the near future, 98% believed that it offered a competitive advantage, with the caveat that 71% believed that lack of coverage and payments were barriers to implementation. Recent studies have shown that telehealth interventions are effective at improving clinical outcomes and decreasing inpatient utilization, with good patient satisfaction in the areas of mental health and chronic disease management. The Veterans Administration has emerged as an early telehealth adopter in chronic disease settings such as mental health, dermatology, hypertension, heart failure, and, as of 2016, has provided care to nearly 700,000 (12%) veterans since its inception.4-6 Despite the increased uptake, significant infrastructure and legal barriers to telemedicine remain and the literature regarding its utility in clinical practice continues to emerge.
Compared with other chronic diseases (e.g., heart failure, diabetes, mental illness) there is a dearth of literature on the use of telemedicine in liver disease. The first portion of this review synthesizes currently published literature of telemedicine/telehealth interventions to improve health care delivery and health outcomes in chronic liver disease including published peer-reviewed articles, abstracts, and ongoing clinical trials. The second portion discusses a framework for the future development of telemedicine and its integration into clinical practice by citing examples currently used throughout the country as well as ways to overcome implementation barriers.
Use of telemedicine in chronic liver disease: A literature review
We performed a systematic review of telemedicine in chronic liver disease. In consultation with a biomedical librarian, we searched for English-language articles for relevant studies with adult participants from July 1984 to May 2017 in PubMed, OVID Medline, American Association for the Study of Liver Disease, EMBASE, Web of Science, ClinicalTrials.gov, Elsevier/Science Direct, and the Cochrane Library (the search strategy is shown in the Supplementary Material at https://doi.org/10.1016/j.cgh.2017.10.004). The references of original publications and of review articles additionally were screened for potentially relevant studies. Abstracts that later resulted in no publications and studies in which telemedicine was used to deliver care, but was neither an exposure nor outcome, were excluded. Social media studies were not considered telemedicine if no patient care was involved. Studies of purely medical education interventions or those that evaluated the accuracy of technology to aid in diagnosis also were excluded.
Supplementary Table 1 (https://doi.org/10.1016/j.cgh.2017.10.004) shows the 20 published articles of telemedicine studies. Among these, there were 9 prospective trials, 3 retrospective studies, 2 case reports, and 6 small case series. One of the studies was randomized prospectively and 10 were uncontrolled.
Telemedicine in hepatitis C treatment
Telemedicine to aid in procedural/surgical management
A few reports have been published in the use of synchronous video and digital technology to aid in periprocedural management in liver disease. A case report highlighted a successful example of gastroenterologist-led teleproctoring using basic video technology to enable a surgeon to perform sclerotherapy for hemostasis in the setting of a variceal bleed.9 Another case report described the transmission of smart phone images from surgical trainees to an attending physician to make a real-time decision regarding a possibly questionable liver procurement, which took place 545 km away from the university hospital.10 A retrospective case series described the feasibility and successful use of high-resolution digital macroscopic photography and electronic transmission between liver transplant centers in the United Kingdom to increase the utilization of split liver transplantation, a setting in which detailed knowledge of vessel anatomy is needed for advanced surgical planning.11 Similarly, an uncontrolled case series from Greece reported on the feasibility and reliability of macroscopic image transmission to aid in the evaluation of liver grafts for transplantation.12
Telemedicine to support evaluation and management of hepatocellular carcinoma
One recent abstract reported on the use of asynchronous store-and-forward telemedicine for screening and management of hepatocellular carcinoma and evaluated process outcomes of specialty care access for newly diagnosed patients.13 A multifaceted approach included live video teleconferencing and centralized radiology review, which was conducted by a multidisciplinary tumor board at an expert hub site, which provided expert opinion and subsequent care (e.g., locoregional therapy, liver transplant evaluation) to spoke sites. As a result of the initiative, the time to specialty evaluation and receipt of hepatocellular carcinoma therapy decreased by 23 and 25 days, respectively.
Remote monitoring interventions
Proposed framework for advancing telemedicine in liver disease: The case for more research and policy changes
Telemedicine can serve two main goals in liver disease: improve access to specialty care, and improve care between visits. For the first goal, the technology is straightforward and limited research is required; the main barriers are regulatory and reimbursement. As an example, one of the authors (M.L.V.) uses telemedicine to perform liver transplant evaluations in Las Vegas, N.V., a state without a liver transplant program. Patients are seen initially by a nurse practitioner who resides in Las Vegas, and those patients needing transplant evaluation are scheduled for a video visit with the attending physician who is physically in California. This works well and patients love it; however, the business model is dependent on the downstream financial incentive of transplantation. In addition, various regulatory requirements must be satisfied such as monthly in-person visits. For the second goal, a number of exciting possibilities exist such as remote monitoring and patient disease management, but more research is needed.
Research
According to the Pew Research Center, 95% of American adults own a cellphone and 77% own a smartphone. These devices passively gather an extraordinary amount of data that could be harnessed to identify early warning signs of complications (remote monitoring). Another potentially fruitful area of research is patient disease management. This includes using technology (e.g., reminder texts) to effect behavior change such as with medication adherence, lifestyle modification, education, or peer mentoring. As an example, the coauthor (M.S.) is leading a study to promote physical activity among liver transplant recipients by using an online web portal developed by researchers at the University of Pennsylvania (Way to Health), which interfaces with patient cell phones and digital accelerometer devices. Participants receive daily feedback through text messages with their step counts, and small financial incentives are provided for adequate levels of physical activity. Technology also can facilitate the development of disease management platforms, which could improve both access and in-between visit monitoring, especially in remote areas. One of the authors (M.L.V.) currently is leading the development of a remote disease management program with funding from the American Association for the Study of Liver Diseases.
Despite the tremendous promise, traditional research methods in telemedicine may be challenging given the rapid and increasing uptake of health technology among patients and health systems. As such, the classic paradigm of randomized controlled trials to evaluate the success of an intervention or change in care delivery often is not feasible. We believe there is a need to recalibrate the definition of what constitutes a high-quality telemedicine study. For example, pragmatic trials and those designed within an implementation science framework that evaluate feasibility, scalability, and cost, in parallel with traditional clinical outcomes, may be better suited and should be accepted more widely.17
Policy
Even when the technology is available and research shows efficacy, the implementation of telemedicine in clinical practice faces regulatory and reimbursement barriers. The first regulatory question is whether a patient–provider relationship is being established (with the exception of limited provider–provider curbside consultation, the answer usually is yes). If so, the practice then is subject to all the usual regulatory concerns. The provider needs to be licensed at the site of origin (where the patient is located) and hold malpractice coverage for that location, and the video and medical record transmission should be compliant with the Health Insurance Portability and Accountability Act. The next challenge is reimbursement. Medicare only pays for video consultation if the patient lives in a designated rural Health Professional Shortage Area (www.cms.gov), and reimbursement by private payers varies. Even this is dependent on ever-changing state laws. Reimbursement for remote patient monitoring is even more limited (the National Telehealth Policy Research Center publishes a useful handbook: http://www.cchpca.org/sites/default/files/resources/50%20State%20FINAL%20April%202016.pdf). Absent a bipartisan Congressional effort to remedy this situation, the best hope for removing reimbursement barriers lies with payment reform. The Medicare Access and CHIP Reauthorization Act of 2015 mandates that the Centers for Medicare and Medicaid Services shift from fee-for-service to alternative payment models in the coming years. In these alternate payment models, providers are responsible for the overall quality and total cost of care for a population of patients. In this scenario, there may be a financial incentive for telemedicine, especially remote monitoring, to keep patients out of the hospital. Until then, under current payment models, reimbursement is limited and the barriers to widespread implementation are high.
Conclusions
Telemedicine has continued to increase in uptake and shows tremendous promise in expanding access to health care, promoting patient disease management, and facilitating in-between health care visit monitoring. Although the future is bright, more research is needed to determine optimal ways to integrate telemedicine — especially remote monitoring — into routine clinical care. We call on our specialty societies to send a clear political advocacy message that policy changes are needed to overcome regulatory and reimbursement challenges.
Acknowledgments
The authors would like to thank Lauren Jones and Mackenzie McDougal for their assistance with the literature review.
Supplementary materials and methods
The telemedicine interventions PubMed literature search strategy was as follows: ((“liver diseases”[MeSH Terms] OR (“liver”[All Fields] AND “diseases”[All Fields]) OR “liver diseases”[All Fields] OR (“liver”[All Fields] AND “disease”[All Fields]) OR “liver disease”[All Fields] OR liver dysfunction OR liver dysfunctions)) OR “liver transplantation”[MeSH Terms] OR “liver transplantation” [All Fields] AND (((“telemedicine”[MeSH Terms] OR “telemedicine”[All Fields] OR mobile health OR mhealth OR telehealth OR mhealth)) OR (videoconferencing OR videoconference)).
References
1. Kirsh S., Su G.L., Sales A., et al. Access to outpatient specialty care: solutions from an integrated health care system. Am J Med Qual. 2015;30:88-90.
2. Wilson L.S., Maeder A.J. recent directions in telemedicine: review of trends in research and practice. Healthc Inform Res. 2015;21:213-22.
3. Cross R.K., Kane, S. Integration of telemedicine into clinical gastroenterology and hepatology practice. Clin Gastroenterol Hepatol. 2017;15:175-81.
4. Darkins A., Ryan P., Kobb R., et al. Care coordination/home telehealth: the systematic implementation of health informatics, home telehealth, and disease management to support the care of veteran patients with chronic conditions. Telemed J E Health. 2008;14:1118-26.
5. Tuerk P.W., Fortney J., Bosworth H.B., et al. Toward the development of national telehealth services: the role of Veterans Health Administration and future directions for research. Telemed J E Health. 2010;16:115-7.
6. VA Press Release. Available: https://www.va.gov/opa/pressrel/includes/viewPDF.cfm?id=2789. Accessed: July 27, 2017.
7. Arora S., Kalishman S., Thornton K., et al. Expanding access to hepatitis C virus treatment-extension for Community Healthcare Outcomes (ECHO) project: disruptive innovation in specialty care. Hepatology. 2010;52:1124-33.
8. Mitruka K., Thornton K., Cusick S., et al. Expanding primary care capacity to treat hepatitis C virus infection through an evidence-based care model–Arizona and Utah, 2012-2014. MMWR Morb Mortal Wkly Rep. 2014;63:393-8.
9. Ahmed A., Slosberg E., Prasad P., et al. The successful use of telemedicine in acute variceal hemorrhage. J Clin Gastroenterol. 1999;29:212-3.
10. Croome K.P., Shum J., Al-Basheer M.A., et al. The benefit of smart phone usage in liver organ procurement. J Telemed Telecare. 2011;17:158-60.
11. Bhati C.S., Wigmore S.J., Reddy S., et al. Web-based image transmission: a novel approach to aid communication in split liver transplantation. Clin Transplant. 2010;24:98-103.
12. Mammas C.S., Geropoulos S., Saatsakis G., et al. Telepathology as a method to optimize quality in organ transplantation: a feasibility and reliability study of the virtual benching of liver graft. Stud Health Technol Inform. 2013;190:276-8.
13. Egert E.M., et al. A regional multidisciplinary liver tumor board improves access to hepatocellular carcinoma treatment for patients geographically distant from tertiary medical center. Hepatology. 2015;62:469A
14. Thomson M., Volk M., Kim H.M., et al. An automated telephone monitoring system to identify patients with cirrhosis at risk of re-hospitalization. Dig Dis Sci. 2015;60:3563-9.
15. Ertel A.E., Kaiser T.E., Abbott D.E., et al. Use of video-based education and tele-health home monitoring after liver transplantation: results of a novel pilot study. Surgery. 2016;160:869-76.
16. Thygesen G.B., Andersen H., Damsgaard B.S. et al. The effect of nurse performed telemedical video consultations for patients suffering from alcohol-related liver cirrhosis. J Hepatol. 2017;66:S349
17. Proctor E., Silmere H., Raghavan R. et al. Outcomes for implementation research: conceptual distinctions, measurement challenges, and research agenda. Adm Policy Ment Health. 2011;38:65-76.
Dr. Serper is in the division of gastroenterology, University of Pennsylvania Perelman School of Medicine, Philadelphia, and the department of medicine, Corporal Michael J. Crescenz VA Medical Center, Philadelphia; Dr. Volk is in the division of gastroenterology and Transplantation Institute, Loma Linda University, Loma Linda, Calif. The authors disclose no conflicts.
Eating Fish May Be Associated With a Reduced Risk of MS
Omega-3 fatty acids, combined with a specific genetic profile, may modulate MS risk.
LOS ANGELES—Eating fish at least once per week or eating fish one to three times per month, in addition to taking daily fish oil supplements, may be associated with a reduced risk of multiple sclerosis (MS), according to a preliminary study presented at the American Academy of Neurology’s 70th Annual Meeting. These findings suggest that the omega-3 fatty acids found in fish may be associated with lowering the risk of developing MS.

“Consuming fish that contain omega-3 fatty acids has been shown to have a variety of health benefits, so we wanted to see if this simple lifestyle modification, regularly eating fish and taking fish oil supplements, could reduce the risk of MS,” said lead study author, Regional Lead for Clinical and Translational Neuroscience for the Southern California Permanente Medical Group in Pasadena, and Clinical Assistant Professor at the Keck School of Medicine of the University of Southern California in Los Angeles.
For this study, researchers examined the diets of 1,153 people (average, age 36) from the MS Sunshine Study, a multiethnic matched case–control study of incident MS or clinically isolated syndrome (CIS), recruited from Kaiser Permanente Southern California.
Researchers queried participants about how much fish they consumed regularly. Investigators also examined 13 single nucleotide polymorphisms (SNPs) in FADS1, FADS2, and ELOV2, which regulate fatty acid biosynthesis.
High fish intake was defined as either eating one serving of fish per week or eating one to three servings per month in addition to taking daily fish oil supplements. Low intake was defined as less than one serving of fish per month and no fish oil supplements.
High fish intake was associated with a 45% reduced risk of MS or CIS, when compared with those who ate fish less than once a month and did not take fish oil supplements. A total of 180 of participants with MS had high fish intake, compared with 251 of the healthy controls.
In addition, two SNPs, rs174611 and rs174618, in FADS2 were independently associated with a lower risk of MS, even after accounting for high fish intake. This result suggests that some people may have a genetic advantage when it comes to regulating fatty acid levels, the researchers noted.
The study suggests that omega-3 fatty acids and how they are processed by the body may play an important role in reducing MS risk. Dr. Langer-Gould and colleagues emphasized that their findings show an association, and not cause and effect. More research is needed to confirm the findings and to examine how omega-3 fatty acids may affect inflammation, metabolism, and nerve function.
Omega-3 fatty acids, combined with a specific genetic profile, may modulate MS risk.
Omega-3 fatty acids, combined with a specific genetic profile, may modulate MS risk.
LOS ANGELES—Eating fish at least once per week or eating fish one to three times per month, in addition to taking daily fish oil supplements, may be associated with a reduced risk of multiple sclerosis (MS), according to a preliminary study presented at the American Academy of Neurology’s 70th Annual Meeting. These findings suggest that the omega-3 fatty acids found in fish may be associated with lowering the risk of developing MS.
“Consuming fish that contain omega-3 fatty acids has been shown to have a variety of health benefits, so we wanted to see if this simple lifestyle modification, regularly eating fish and taking fish oil supplements, could reduce the risk of MS,” said lead study author, Regional Lead for Clinical and Translational Neuroscience for the Southern California Permanente Medical Group in Pasadena, and Clinical Assistant Professor at the Keck School of Medicine of the University of Southern California in Los Angeles.
For this study, researchers examined the diets of 1,153 people (average, age 36) from the MS Sunshine Study, a multiethnic matched case–control study of incident MS or clinically isolated syndrome (CIS), recruited from Kaiser Permanente Southern California.
Researchers queried participants about how much fish they consumed regularly. Investigators also examined 13 single nucleotide polymorphisms (SNPs) in FADS1, FADS2, and ELOV2, which regulate fatty acid biosynthesis.
High fish intake was defined as either eating one serving of fish per week or eating one to three servings per month in addition to taking daily fish oil supplements. Low intake was defined as less than one serving of fish per month and no fish oil supplements.
High fish intake was associated with a 45% reduced risk of MS or CIS, when compared with those who ate fish less than once a month and did not take fish oil supplements. A total of 180 of participants with MS had high fish intake, compared with 251 of the healthy controls.
In addition, two SNPs, rs174611 and rs174618, in FADS2 were independently associated with a lower risk of MS, even after accounting for high fish intake. This result suggests that some people may have a genetic advantage when it comes to regulating fatty acid levels, the researchers noted.
The study suggests that omega-3 fatty acids and how they are processed by the body may play an important role in reducing MS risk. Dr. Langer-Gould and colleagues emphasized that their findings show an association, and not cause and effect. More research is needed to confirm the findings and to examine how omega-3 fatty acids may affect inflammation, metabolism, and nerve function.
LOS ANGELES—Eating fish at least once per week or eating fish one to three times per month, in addition to taking daily fish oil supplements, may be associated with a reduced risk of multiple sclerosis (MS), according to a preliminary study presented at the American Academy of Neurology’s 70th Annual Meeting. These findings suggest that the omega-3 fatty acids found in fish may be associated with lowering the risk of developing MS.
“Consuming fish that contain omega-3 fatty acids has been shown to have a variety of health benefits, so we wanted to see if this simple lifestyle modification, regularly eating fish and taking fish oil supplements, could reduce the risk of MS,” said lead study author, Regional Lead for Clinical and Translational Neuroscience for the Southern California Permanente Medical Group in Pasadena, and Clinical Assistant Professor at the Keck School of Medicine of the University of Southern California in Los Angeles.
For this study, researchers examined the diets of 1,153 people (average, age 36) from the MS Sunshine Study, a multiethnic matched case–control study of incident MS or clinically isolated syndrome (CIS), recruited from Kaiser Permanente Southern California.
Researchers queried participants about how much fish they consumed regularly. Investigators also examined 13 single nucleotide polymorphisms (SNPs) in FADS1, FADS2, and ELOV2, which regulate fatty acid biosynthesis.
High fish intake was defined as either eating one serving of fish per week or eating one to three servings per month in addition to taking daily fish oil supplements. Low intake was defined as less than one serving of fish per month and no fish oil supplements.
High fish intake was associated with a 45% reduced risk of MS or CIS, when compared with those who ate fish less than once a month and did not take fish oil supplements. A total of 180 of participants with MS had high fish intake, compared with 251 of the healthy controls.
In addition, two SNPs, rs174611 and rs174618, in FADS2 were independently associated with a lower risk of MS, even after accounting for high fish intake. This result suggests that some people may have a genetic advantage when it comes to regulating fatty acid levels, the researchers noted.
The study suggests that omega-3 fatty acids and how they are processed by the body may play an important role in reducing MS risk. Dr. Langer-Gould and colleagues emphasized that their findings show an association, and not cause and effect. More research is needed to confirm the findings and to examine how omega-3 fatty acids may affect inflammation, metabolism, and nerve function.
Concomitant drugs may explain PEG-ASP liver toxicities in ALL
STOCKHOLM – Liver toxicities in adults with acute lymphoblastic leukemia (ALL) treated with a pediatric-type regimen containing pegylated asparaginase (PEG-ASP) may be related to concomitant use of other hepatotoxic drugs, investigators cautioned.

A retrospective review of records on 26 adult ALL patients treated with PEG-ASP since 2013 showed that concomitant use of vincristine, idarubicin, and vancomycin was associated with an increased risk for grade 3 or 4 hepatotoxicity, reported Fabio Guolo, MD, from the University of Genoa (Italy) and his colleagues.
In contrast, patients who received other chemotherapy drugs or antimicrobial agents did not have significant liver toxicities, Dr. Guolo said in an interview at the annual congress of the European Hematology Association.
“Increased toxicity from therapy prevents delivery of the most active therapy, and asparaginase is one of the keys to the success of pediatric trials in ALL, so we have tried to push the dose of asparaginase as high as we could in adult patients,” he said.
“We asked why some patients will develop toxicity while receiving a relatively low dose of asparaginase, whereas other patients who received higher doses did not,” Dr. Guolo added.
In recent years, investigators have found that adults with ALL tend to have better outcomes when they were treated with standard pediatric ALL regimens, which includes high-dose PEG-ASP.
To identify factors related to potential PEG-ASP toxicity in adults with ALL, the investigators combed through records of 26 adults patients, 19 of whom had received PEG-ASP in the frontline setting, and 7 of whom received it during treatment of relapsed or refractory disease.
The investigators looked at each course of PEG-ASP as an independent event (51 total episodes), paying special attention to concomitant chemotherapy and the use of both antimycotic and antibiotic agents.
Five of the patients had grade 3 hepatotoxicity, and three had grade 4 hepatotoxicity. The patients with grade 4 events had unexplained severe weight gain and painful hepatomegaly. Ultrasonography in these patients revealed acute steatosis similar to that seen with sinusoidal occlusive disease. All three patients had received concomitant idarubicin, vincristine, and vancomycin.
In univariate analysis, neither being older than 45 years, administration of PEG-ASP during an active leukemia phase, nor having a body mass index greater than 25 kg/m2 were significantly associated with increased incidence of grade 3 or 4 hepatotoxicity.
When the investigators looked at concomitant chemotherapy drugs, however, they found that liver toxicity was significantly higher with idarubicin cumulative doses of 20 mg/m2 or greater (hazard ratio, 1.49; P = .047) and that vincristine doses of 2 mg/m2 or greater were associated with a borderline increase in risk (HR, 4.75; P = .055).
There was no increased risk for liver toxicities with either steroids, daunorubicin, cyclophosphamide, cytarabine, methotrexate, or 6-mercaptopurine.
Additionally, concomitant vancomycin was also linked to increased hepatotoxicity (HR, 1.86; P =.009). In contrast, neither carbapenem-class anti-infectives nor azole were significantly associated with liver toxicities.
“Notably, none of the patients undergoing full pediatric induction, which contains higher cumulative doses of PEG-ASP, experienced grade 4 hepatotoxicity regardless of age,” Dr. Guolo and his colleagues wrote in their poster presentation.
In multivariate analysis controlling for age, BMI, drug regimen, and concomitant therapies, idarubicin remained a significant risk factor for severe hepatotoxicity (P = .004), and vancomycin remained as a borderline risk (P = .054).
Dr. Guolo acknowledged that the investigators could not account for the potential contribution of over-the-counter medications with known risk for hepatotoxicity, such as acetaminophen.
He noted that in his group’s experience, the toxicity profile of PEG-ASP in adults, including high-dose regimens, was manageable without excess toxicities as long as clinicians paid close attention to the use of concomitant agents.
The study was internally funded. The authors reported having no relevant conflicts of interest.
SOURCE: Minetto P et al. EHA Congress, Abstract PS934.
STOCKHOLM – Liver toxicities in adults with acute lymphoblastic leukemia (ALL) treated with a pediatric-type regimen containing pegylated asparaginase (PEG-ASP) may be related to concomitant use of other hepatotoxic drugs, investigators cautioned.
A retrospective review of records on 26 adult ALL patients treated with PEG-ASP since 2013 showed that concomitant use of vincristine, idarubicin, and vancomycin was associated with an increased risk for grade 3 or 4 hepatotoxicity, reported Fabio Guolo, MD, from the University of Genoa (Italy) and his colleagues.
In contrast, patients who received other chemotherapy drugs or antimicrobial agents did not have significant liver toxicities, Dr. Guolo said in an interview at the annual congress of the European Hematology Association.
“Increased toxicity from therapy prevents delivery of the most active therapy, and asparaginase is one of the keys to the success of pediatric trials in ALL, so we have tried to push the dose of asparaginase as high as we could in adult patients,” he said.
“We asked why some patients will develop toxicity while receiving a relatively low dose of asparaginase, whereas other patients who received higher doses did not,” Dr. Guolo added.
In recent years, investigators have found that adults with ALL tend to have better outcomes when they were treated with standard pediatric ALL regimens, which includes high-dose PEG-ASP.
To identify factors related to potential PEG-ASP toxicity in adults with ALL, the investigators combed through records of 26 adults patients, 19 of whom had received PEG-ASP in the frontline setting, and 7 of whom received it during treatment of relapsed or refractory disease.
The investigators looked at each course of PEG-ASP as an independent event (51 total episodes), paying special attention to concomitant chemotherapy and the use of both antimycotic and antibiotic agents.
Five of the patients had grade 3 hepatotoxicity, and three had grade 4 hepatotoxicity. The patients with grade 4 events had unexplained severe weight gain and painful hepatomegaly. Ultrasonography in these patients revealed acute steatosis similar to that seen with sinusoidal occlusive disease. All three patients had received concomitant idarubicin, vincristine, and vancomycin.
In univariate analysis, neither being older than 45 years, administration of PEG-ASP during an active leukemia phase, nor having a body mass index greater than 25 kg/m2 were significantly associated with increased incidence of grade 3 or 4 hepatotoxicity.
When the investigators looked at concomitant chemotherapy drugs, however, they found that liver toxicity was significantly higher with idarubicin cumulative doses of 20 mg/m2 or greater (hazard ratio, 1.49; P = .047) and that vincristine doses of 2 mg/m2 or greater were associated with a borderline increase in risk (HR, 4.75; P = .055).
There was no increased risk for liver toxicities with either steroids, daunorubicin, cyclophosphamide, cytarabine, methotrexate, or 6-mercaptopurine.
Additionally, concomitant vancomycin was also linked to increased hepatotoxicity (HR, 1.86; P =.009). In contrast, neither carbapenem-class anti-infectives nor azole were significantly associated with liver toxicities.
“Notably, none of the patients undergoing full pediatric induction, which contains higher cumulative doses of PEG-ASP, experienced grade 4 hepatotoxicity regardless of age,” Dr. Guolo and his colleagues wrote in their poster presentation.
In multivariate analysis controlling for age, BMI, drug regimen, and concomitant therapies, idarubicin remained a significant risk factor for severe hepatotoxicity (P = .004), and vancomycin remained as a borderline risk (P = .054).
Dr. Guolo acknowledged that the investigators could not account for the potential contribution of over-the-counter medications with known risk for hepatotoxicity, such as acetaminophen.
He noted that in his group’s experience, the toxicity profile of PEG-ASP in adults, including high-dose regimens, was manageable without excess toxicities as long as clinicians paid close attention to the use of concomitant agents.
The study was internally funded. The authors reported having no relevant conflicts of interest.
SOURCE: Minetto P et al. EHA Congress, Abstract PS934.
STOCKHOLM – Liver toxicities in adults with acute lymphoblastic leukemia (ALL) treated with a pediatric-type regimen containing pegylated asparaginase (PEG-ASP) may be related to concomitant use of other hepatotoxic drugs, investigators cautioned.
A retrospective review of records on 26 adult ALL patients treated with PEG-ASP since 2013 showed that concomitant use of vincristine, idarubicin, and vancomycin was associated with an increased risk for grade 3 or 4 hepatotoxicity, reported Fabio Guolo, MD, from the University of Genoa (Italy) and his colleagues.
In contrast, patients who received other chemotherapy drugs or antimicrobial agents did not have significant liver toxicities, Dr. Guolo said in an interview at the annual congress of the European Hematology Association.
“Increased toxicity from therapy prevents delivery of the most active therapy, and asparaginase is one of the keys to the success of pediatric trials in ALL, so we have tried to push the dose of asparaginase as high as we could in adult patients,” he said.
“We asked why some patients will develop toxicity while receiving a relatively low dose of asparaginase, whereas other patients who received higher doses did not,” Dr. Guolo added.
In recent years, investigators have found that adults with ALL tend to have better outcomes when they were treated with standard pediatric ALL regimens, which includes high-dose PEG-ASP.
To identify factors related to potential PEG-ASP toxicity in adults with ALL, the investigators combed through records of 26 adults patients, 19 of whom had received PEG-ASP in the frontline setting, and 7 of whom received it during treatment of relapsed or refractory disease.
The investigators looked at each course of PEG-ASP as an independent event (51 total episodes), paying special attention to concomitant chemotherapy and the use of both antimycotic and antibiotic agents.
Five of the patients had grade 3 hepatotoxicity, and three had grade 4 hepatotoxicity. The patients with grade 4 events had unexplained severe weight gain and painful hepatomegaly. Ultrasonography in these patients revealed acute steatosis similar to that seen with sinusoidal occlusive disease. All three patients had received concomitant idarubicin, vincristine, and vancomycin.
In univariate analysis, neither being older than 45 years, administration of PEG-ASP during an active leukemia phase, nor having a body mass index greater than 25 kg/m2 were significantly associated with increased incidence of grade 3 or 4 hepatotoxicity.
When the investigators looked at concomitant chemotherapy drugs, however, they found that liver toxicity was significantly higher with idarubicin cumulative doses of 20 mg/m2 or greater (hazard ratio, 1.49; P = .047) and that vincristine doses of 2 mg/m2 or greater were associated with a borderline increase in risk (HR, 4.75; P = .055).
There was no increased risk for liver toxicities with either steroids, daunorubicin, cyclophosphamide, cytarabine, methotrexate, or 6-mercaptopurine.
Additionally, concomitant vancomycin was also linked to increased hepatotoxicity (HR, 1.86; P =.009). In contrast, neither carbapenem-class anti-infectives nor azole were significantly associated with liver toxicities.
“Notably, none of the patients undergoing full pediatric induction, which contains higher cumulative doses of PEG-ASP, experienced grade 4 hepatotoxicity regardless of age,” Dr. Guolo and his colleagues wrote in their poster presentation.
In multivariate analysis controlling for age, BMI, drug regimen, and concomitant therapies, idarubicin remained a significant risk factor for severe hepatotoxicity (P = .004), and vancomycin remained as a borderline risk (P = .054).
Dr. Guolo acknowledged that the investigators could not account for the potential contribution of over-the-counter medications with known risk for hepatotoxicity, such as acetaminophen.
He noted that in his group’s experience, the toxicity profile of PEG-ASP in adults, including high-dose regimens, was manageable without excess toxicities as long as clinicians paid close attention to the use of concomitant agents.
The study was internally funded. The authors reported having no relevant conflicts of interest.
SOURCE: Minetto P et al. EHA Congress, Abstract PS934.
REPORTING FROM EHA CONGRESS
Key clinical point:
Major finding: Idarubicin was associated with a higher risk of grade 3 or 4 hepatotoxicity, and vincristine was associated with a borderline risk.
Study details: Retrospective review of 51 PEG-ASP dosing events in 26 adult patients with ALL.
Disclosures: The study was internally funded. The authors reported having no relevant conflicts of interest.
Source: Minetto P et al. EHA Congress, Abstract PS934.
What Is the Prevalence of Sleep Disorders in Neurologic Populations?
A retrospective study finds that insomnia may be associated with worse neurologic status in patients with movement disorders and patients with epilepsy.
LOS ANGELES—About one-third of neurologic patients has a high risk of obstructive sleep apnea (OSA), and approximately one-quarter has significant symptoms of insomnia, according to data presented at the 70th Annual Meeting of the American Academy of Neurology. The presence of insomnia symptoms is associated with worse neurologic status in movement disorders and epilepsy populations, researchers said.
“Given the high prevalence of sleep disorder symptoms, further investigation into the role of sleep therapies on disease-specific outcomes in neurologic populations is warranted,” said Thapanee Somboon, MD, a neurologist at Prasat Neurological Institute in Bangkok, Thailand, and research fellow at the Cleveland Clinic Sleep Disorders Center, and colleagues.

Analyzing STOP and Insomnia Severity Index Scores
OSA and insomnia are highly prevalent in the general population and may be more common in patients with neurologic conditions. To examine the association between sleep instrument scores and disease-specific outcomes in neurologic populations, Dr. Somboon and colleagues conducted a retrospective analysis of data from 19,052 adult initial visits to the psychiatry, brain tumor, movement disorders, cerebrovascular, and epilepsy centers at the Cleveland Clinic between March 2015 and October 2016.
In all, 7,762 patients had completed the snoring, tiredness, observed apnea, and high blood pressure (STOP) questionnaire, and 8,530 patients had completed the Insomnia Severity Index. A STOP score of 2 or greater predicted a high risk of OSA, and an Insomnia Severity Index score of 15 or greater indicated significant insomnia symptoms.
The crude prevalence of high-risk OSA was 47.9% in the cerebrovascular center, 44.1% in the movement disorders center, 34% in the brain tumor center, 33% in the epilepsy center, 29.8% in the psychiatry center, and 36.7% overall.
The crude prevalence of significant insomnia symptoms was 33.6% in the psychiatry center, 26.1% in the epilepsy center, 20.7% in the brain tumor center, 20% in the movement disorders center, 19.5% in the cerebrovascular center, and 25.5% overall.
Disease-Specific Outcomes
The researchers used regression models to adjust for patients’ age, sex, race, marital status, self-reported sleep duration, income, tobacco use, and comorbid conditions. Multivariate models evaluated the associations between abnormal sleep scores and scores on the Patient Health Questionnaire-9 (PHQ-9; from all centers), Karnofsky Performance Status (from the brain tumor center), Unified Parkinson’s Disease Rating Scale (UPDRS II; from the movement disorders center), modified Rankin Scale (from the cerebrovascular center), and Liverpool Seizure Severity Scale (from the epilepsy center).
Patients with a STOP score of 2 or greater were older, more likely to be male, more likely to be a current or former smoker, had greater PHQ-9 scores, and had more comorbidities.
Patients with Insomnia Severity Index scores of 15 or greater were younger, more likely to be female, more likely to be a current or former smoker, and had a higher prevalence of depression.
OSA and insomnia were significantly associated with PHQ-9 scores. In addition, insomnia symptoms were significantly associated with Liverpool Seizure Severity Scale and UPDRS II scores.
—Jake Remaly
A retrospective study finds that insomnia may be associated with worse neurologic status in patients with movement disorders and patients with epilepsy.
A retrospective study finds that insomnia may be associated with worse neurologic status in patients with movement disorders and patients with epilepsy.
LOS ANGELES—About one-third of neurologic patients has a high risk of obstructive sleep apnea (OSA), and approximately one-quarter has significant symptoms of insomnia, according to data presented at the 70th Annual Meeting of the American Academy of Neurology. The presence of insomnia symptoms is associated with worse neurologic status in movement disorders and epilepsy populations, researchers said.
“Given the high prevalence of sleep disorder symptoms, further investigation into the role of sleep therapies on disease-specific outcomes in neurologic populations is warranted,” said Thapanee Somboon, MD, a neurologist at Prasat Neurological Institute in Bangkok, Thailand, and research fellow at the Cleveland Clinic Sleep Disorders Center, and colleagues.
Analyzing STOP and Insomnia Severity Index Scores
OSA and insomnia are highly prevalent in the general population and may be more common in patients with neurologic conditions. To examine the association between sleep instrument scores and disease-specific outcomes in neurologic populations, Dr. Somboon and colleagues conducted a retrospective analysis of data from 19,052 adult initial visits to the psychiatry, brain tumor, movement disorders, cerebrovascular, and epilepsy centers at the Cleveland Clinic between March 2015 and October 2016.
In all, 7,762 patients had completed the snoring, tiredness, observed apnea, and high blood pressure (STOP) questionnaire, and 8,530 patients had completed the Insomnia Severity Index. A STOP score of 2 or greater predicted a high risk of OSA, and an Insomnia Severity Index score of 15 or greater indicated significant insomnia symptoms.
The crude prevalence of high-risk OSA was 47.9% in the cerebrovascular center, 44.1% in the movement disorders center, 34% in the brain tumor center, 33% in the epilepsy center, 29.8% in the psychiatry center, and 36.7% overall.
The crude prevalence of significant insomnia symptoms was 33.6% in the psychiatry center, 26.1% in the epilepsy center, 20.7% in the brain tumor center, 20% in the movement disorders center, 19.5% in the cerebrovascular center, and 25.5% overall.
Disease-Specific Outcomes
The researchers used regression models to adjust for patients’ age, sex, race, marital status, self-reported sleep duration, income, tobacco use, and comorbid conditions. Multivariate models evaluated the associations between abnormal sleep scores and scores on the Patient Health Questionnaire-9 (PHQ-9; from all centers), Karnofsky Performance Status (from the brain tumor center), Unified Parkinson’s Disease Rating Scale (UPDRS II; from the movement disorders center), modified Rankin Scale (from the cerebrovascular center), and Liverpool Seizure Severity Scale (from the epilepsy center).
Patients with a STOP score of 2 or greater were older, more likely to be male, more likely to be a current or former smoker, had greater PHQ-9 scores, and had more comorbidities.
Patients with Insomnia Severity Index scores of 15 or greater were younger, more likely to be female, more likely to be a current or former smoker, and had a higher prevalence of depression.
OSA and insomnia were significantly associated with PHQ-9 scores. In addition, insomnia symptoms were significantly associated with Liverpool Seizure Severity Scale and UPDRS II scores.
—Jake Remaly
LOS ANGELES—About one-third of neurologic patients has a high risk of obstructive sleep apnea (OSA), and approximately one-quarter has significant symptoms of insomnia, according to data presented at the 70th Annual Meeting of the American Academy of Neurology. The presence of insomnia symptoms is associated with worse neurologic status in movement disorders and epilepsy populations, researchers said.
“Given the high prevalence of sleep disorder symptoms, further investigation into the role of sleep therapies on disease-specific outcomes in neurologic populations is warranted,” said Thapanee Somboon, MD, a neurologist at Prasat Neurological Institute in Bangkok, Thailand, and research fellow at the Cleveland Clinic Sleep Disorders Center, and colleagues.
Analyzing STOP and Insomnia Severity Index Scores
OSA and insomnia are highly prevalent in the general population and may be more common in patients with neurologic conditions. To examine the association between sleep instrument scores and disease-specific outcomes in neurologic populations, Dr. Somboon and colleagues conducted a retrospective analysis of data from 19,052 adult initial visits to the psychiatry, brain tumor, movement disorders, cerebrovascular, and epilepsy centers at the Cleveland Clinic between March 2015 and October 2016.
In all, 7,762 patients had completed the snoring, tiredness, observed apnea, and high blood pressure (STOP) questionnaire, and 8,530 patients had completed the Insomnia Severity Index. A STOP score of 2 or greater predicted a high risk of OSA, and an Insomnia Severity Index score of 15 or greater indicated significant insomnia symptoms.
The crude prevalence of high-risk OSA was 47.9% in the cerebrovascular center, 44.1% in the movement disorders center, 34% in the brain tumor center, 33% in the epilepsy center, 29.8% in the psychiatry center, and 36.7% overall.
The crude prevalence of significant insomnia symptoms was 33.6% in the psychiatry center, 26.1% in the epilepsy center, 20.7% in the brain tumor center, 20% in the movement disorders center, 19.5% in the cerebrovascular center, and 25.5% overall.
Disease-Specific Outcomes
The researchers used regression models to adjust for patients’ age, sex, race, marital status, self-reported sleep duration, income, tobacco use, and comorbid conditions. Multivariate models evaluated the associations between abnormal sleep scores and scores on the Patient Health Questionnaire-9 (PHQ-9; from all centers), Karnofsky Performance Status (from the brain tumor center), Unified Parkinson’s Disease Rating Scale (UPDRS II; from the movement disorders center), modified Rankin Scale (from the cerebrovascular center), and Liverpool Seizure Severity Scale (from the epilepsy center).
Patients with a STOP score of 2 or greater were older, more likely to be male, more likely to be a current or former smoker, had greater PHQ-9 scores, and had more comorbidities.
Patients with Insomnia Severity Index scores of 15 or greater were younger, more likely to be female, more likely to be a current or former smoker, and had a higher prevalence of depression.
OSA and insomnia were significantly associated with PHQ-9 scores. In addition, insomnia symptoms were significantly associated with Liverpool Seizure Severity Scale and UPDRS II scores.
—Jake Remaly
Sleep Paralysis and Hallucinations Are Prevalent in Student Athletes
These symptoms may predict depression severity, independent of short sleep and insomnia.
BALTIMORE—Many student athletes have sleep paralysis and hypnagogic or hypnopompic hallucinations, according to research presented at the 32nd Annual Meeting of the Associated Professional Sleep Societies. In addition, these sleep symptoms are independently associated with symptoms of depression.
“These sleep symptoms are usually harmless on their own, but they can be a sign of more serious sleep problems,” said Serena Liu, a student research assistant in the Sleep and Health Research Program at the University of Arizona College of Medicine in Tucson. “The fact that they are so common among student athletes suggests that this is a group with some significant sleep problems that should be evaluated and dealt with.”
A Study of NCAA Athletes
Student athletes often have difficulty finding time to rest because of their busy schedules. Shorter sleep duration and poor sleep quality contribute to disordered sleep in many student athletes, and data indicate a high prevalence of common sleep symptoms in this group. Investigators have not studied the prevalence of less common symptoms (eg, sleep paralysis and hypnagogic and hypnopompic hallucinations, which are more prevalent in younger adults) in student athletes, however. Nor have they examined the potential role of these symptoms in mental health, independent of insufficient sleep duration or insomnia.
Ms. Liu and colleagues collected data from 189 student athletes in the National Collegiate Athletic Association’s Division I. The researchers asked the athletes how often they had sleep paralysis and hypnagogic or hypnopompic hallucinations. Responses were “never,” “rarely (once per month or less),” or “often (once or more per week).” Participants also gave information about sleep duration and underwent evaluation with the Insomnia Severity Index and the Centers for Epidemiological Studies Depression Scale. Ms. Liu and colleagues performed regression analyses to examine depression score as outcome and sleep symptoms as predictor in a model adjusted for age and sex and a model adjusted for age, sex, insomnia severity, and sleep duration.
Sleep Symptoms May Indicate Other Problems
About 18% of the sample reported having sleep paralysis rarely, and 7% reported having it often. In addition, 24% of the sample reported having hypnagogic or hypnopompic hallucinations rarely, and 11% reported having them often. In models adjusted for age and sex, reporting sleep paralysis rarely and often were associated with higher depression score, compared with reporting them never. Similarly, reporting hypnagogic and hypnopompic hallucinations rarely and often were associated with higher depression score. In models adjusted for insomnia and sleep duration, these relationships were attenuated, but remained significant.
These symptoms may predict depression severity, independent of short sleep and insomnia.
These symptoms may predict depression severity, independent of short sleep and insomnia.
BALTIMORE—Many student athletes have sleep paralysis and hypnagogic or hypnopompic hallucinations, according to research presented at the 32nd Annual Meeting of the Associated Professional Sleep Societies. In addition, these sleep symptoms are independently associated with symptoms of depression.
“These sleep symptoms are usually harmless on their own, but they can be a sign of more serious sleep problems,” said Serena Liu, a student research assistant in the Sleep and Health Research Program at the University of Arizona College of Medicine in Tucson. “The fact that they are so common among student athletes suggests that this is a group with some significant sleep problems that should be evaluated and dealt with.”
A Study of NCAA Athletes
Student athletes often have difficulty finding time to rest because of their busy schedules. Shorter sleep duration and poor sleep quality contribute to disordered sleep in many student athletes, and data indicate a high prevalence of common sleep symptoms in this group. Investigators have not studied the prevalence of less common symptoms (eg, sleep paralysis and hypnagogic and hypnopompic hallucinations, which are more prevalent in younger adults) in student athletes, however. Nor have they examined the potential role of these symptoms in mental health, independent of insufficient sleep duration or insomnia.
Ms. Liu and colleagues collected data from 189 student athletes in the National Collegiate Athletic Association’s Division I. The researchers asked the athletes how often they had sleep paralysis and hypnagogic or hypnopompic hallucinations. Responses were “never,” “rarely (once per month or less),” or “often (once or more per week).” Participants also gave information about sleep duration and underwent evaluation with the Insomnia Severity Index and the Centers for Epidemiological Studies Depression Scale. Ms. Liu and colleagues performed regression analyses to examine depression score as outcome and sleep symptoms as predictor in a model adjusted for age and sex and a model adjusted for age, sex, insomnia severity, and sleep duration.
Sleep Symptoms May Indicate Other Problems
About 18% of the sample reported having sleep paralysis rarely, and 7% reported having it often. In addition, 24% of the sample reported having hypnagogic or hypnopompic hallucinations rarely, and 11% reported having them often. In models adjusted for age and sex, reporting sleep paralysis rarely and often were associated with higher depression score, compared with reporting them never. Similarly, reporting hypnagogic and hypnopompic hallucinations rarely and often were associated with higher depression score. In models adjusted for insomnia and sleep duration, these relationships were attenuated, but remained significant.
BALTIMORE—Many student athletes have sleep paralysis and hypnagogic or hypnopompic hallucinations, according to research presented at the 32nd Annual Meeting of the Associated Professional Sleep Societies. In addition, these sleep symptoms are independently associated with symptoms of depression.
“These sleep symptoms are usually harmless on their own, but they can be a sign of more serious sleep problems,” said Serena Liu, a student research assistant in the Sleep and Health Research Program at the University of Arizona College of Medicine in Tucson. “The fact that they are so common among student athletes suggests that this is a group with some significant sleep problems that should be evaluated and dealt with.”
A Study of NCAA Athletes
Student athletes often have difficulty finding time to rest because of their busy schedules. Shorter sleep duration and poor sleep quality contribute to disordered sleep in many student athletes, and data indicate a high prevalence of common sleep symptoms in this group. Investigators have not studied the prevalence of less common symptoms (eg, sleep paralysis and hypnagogic and hypnopompic hallucinations, which are more prevalent in younger adults) in student athletes, however. Nor have they examined the potential role of these symptoms in mental health, independent of insufficient sleep duration or insomnia.
Ms. Liu and colleagues collected data from 189 student athletes in the National Collegiate Athletic Association’s Division I. The researchers asked the athletes how often they had sleep paralysis and hypnagogic or hypnopompic hallucinations. Responses were “never,” “rarely (once per month or less),” or “often (once or more per week).” Participants also gave information about sleep duration and underwent evaluation with the Insomnia Severity Index and the Centers for Epidemiological Studies Depression Scale. Ms. Liu and colleagues performed regression analyses to examine depression score as outcome and sleep symptoms as predictor in a model adjusted for age and sex and a model adjusted for age, sex, insomnia severity, and sleep duration.
Sleep Symptoms May Indicate Other Problems
About 18% of the sample reported having sleep paralysis rarely, and 7% reported having it often. In addition, 24% of the sample reported having hypnagogic or hypnopompic hallucinations rarely, and 11% reported having them often. In models adjusted for age and sex, reporting sleep paralysis rarely and often were associated with higher depression score, compared with reporting them never. Similarly, reporting hypnagogic and hypnopompic hallucinations rarely and often were associated with higher depression score. In models adjusted for insomnia and sleep duration, these relationships were attenuated, but remained significant.
High-Intensity Interval Exercise May Be Appropriate for Patients With MS
The activity is as enjoyable as continuous exercise for this population and could improve physiologic conditioning.
NASHVILLE—For patients with multiple sclerosis (MS) and walking impairments, high-intensity interval exercise is as enjoyable as traditional continuous exercise and does not induce sustained, harmful effects on mood, according to research presented at the 2018 CMSC Annual Meeting. This finding could influence evidence-based prescriptions for exercise that are appropriate for improving physiologic conditioning, according to the researchers.
MS often results in walking dysfunction and physiologic deconditioning. High-intensity interval exercise has induced significant improvements in physiologic conditioning in healthy and clinical populations, including patients with MS. Before integrating high-intensity interval exercise into an exercise prescription, and to increase the likelihood that participants will engage in and adhere to this exercise regimen, investigators thought it important to ascertain whether people with MS and mobility disability enjoy the high-intensity interval exercise and experience mood benefits.
Elizabeth Hubbard, PhD, Assistant Professor of Kinesiology at Berry College in Mount Berry, Georgia, conducted a study to examine the effects of single sessions of high-intensity interval exercise and continuous exercise on mood and enjoyment outcomes in persons with MS and mobility disability. In their study, 20 participants (median Expanded Disability Status Scale score, 5.8) underwent counterbalanced presentations of high-intensity interval exercise and continuous exercise bouts using a recumbent stepper. The high-intensity interval exercise bout was 20 minutes long and included 10 cycles of one-minute intervals at a wattage associated with 90% VO2 peak, followed by one-minute recovery intervals at 15 W. The continuous exercise bout lasted for 20 minutes at a wattage associated with 50% to 60% VO2 peak. The researchers administered the Profile of Mood States (POMS) survey before, immediately after, and 30 minutes after exercise. The Physical Activity Enjoyment Scale (PACES) questionnaire was administered immediately after exercise.

Dr. Hubbard and colleagues observed no significant condition-by-time interactions or main effects of condition on any POMS subscales. The data indicated large, statistically significant main effects of time on total mood disturbance, fatigue, and vigor. Specifically, mood worsened immediately after exercise, but rebounded after the 30-minute recovery period. The researchers saw no significant difference in scores for the PACES questionnaire between high-intensity interval exercise (mean, 95.5) and continuous exercise (mean, 97.7) conditions.
One weakness of the study was that the population was relatively active, although with a low level of fitness. In addition, the researchers did not control for age, sex, or disability status.
The high-intensity interval exercise and continuous exercise were feasible, safe, and enjoyable in people with MS who have mobility disability. “High-intensity interval exercise … might be particularly beneficial for improving fitness and functional capacity in persons with MS who have mobility disability and experience subsequent physiologic deconditioning,” said Dr. Hubbard.
The activity is as enjoyable as continuous exercise for this population and could improve physiologic conditioning.
The activity is as enjoyable as continuous exercise for this population and could improve physiologic conditioning.
NASHVILLE—For patients with multiple sclerosis (MS) and walking impairments, high-intensity interval exercise is as enjoyable as traditional continuous exercise and does not induce sustained, harmful effects on mood, according to research presented at the 2018 CMSC Annual Meeting. This finding could influence evidence-based prescriptions for exercise that are appropriate for improving physiologic conditioning, according to the researchers.
MS often results in walking dysfunction and physiologic deconditioning. High-intensity interval exercise has induced significant improvements in physiologic conditioning in healthy and clinical populations, including patients with MS. Before integrating high-intensity interval exercise into an exercise prescription, and to increase the likelihood that participants will engage in and adhere to this exercise regimen, investigators thought it important to ascertain whether people with MS and mobility disability enjoy the high-intensity interval exercise and experience mood benefits.
Elizabeth Hubbard, PhD, Assistant Professor of Kinesiology at Berry College in Mount Berry, Georgia, conducted a study to examine the effects of single sessions of high-intensity interval exercise and continuous exercise on mood and enjoyment outcomes in persons with MS and mobility disability. In their study, 20 participants (median Expanded Disability Status Scale score, 5.8) underwent counterbalanced presentations of high-intensity interval exercise and continuous exercise bouts using a recumbent stepper. The high-intensity interval exercise bout was 20 minutes long and included 10 cycles of one-minute intervals at a wattage associated with 90% VO2 peak, followed by one-minute recovery intervals at 15 W. The continuous exercise bout lasted for 20 minutes at a wattage associated with 50% to 60% VO2 peak. The researchers administered the Profile of Mood States (POMS) survey before, immediately after, and 30 minutes after exercise. The Physical Activity Enjoyment Scale (PACES) questionnaire was administered immediately after exercise.
Dr. Hubbard and colleagues observed no significant condition-by-time interactions or main effects of condition on any POMS subscales. The data indicated large, statistically significant main effects of time on total mood disturbance, fatigue, and vigor. Specifically, mood worsened immediately after exercise, but rebounded after the 30-minute recovery period. The researchers saw no significant difference in scores for the PACES questionnaire between high-intensity interval exercise (mean, 95.5) and continuous exercise (mean, 97.7) conditions.
One weakness of the study was that the population was relatively active, although with a low level of fitness. In addition, the researchers did not control for age, sex, or disability status.
The high-intensity interval exercise and continuous exercise were feasible, safe, and enjoyable in people with MS who have mobility disability. “High-intensity interval exercise … might be particularly beneficial for improving fitness and functional capacity in persons with MS who have mobility disability and experience subsequent physiologic deconditioning,” said Dr. Hubbard.
NASHVILLE—For patients with multiple sclerosis (MS) and walking impairments, high-intensity interval exercise is as enjoyable as traditional continuous exercise and does not induce sustained, harmful effects on mood, according to research presented at the 2018 CMSC Annual Meeting. This finding could influence evidence-based prescriptions for exercise that are appropriate for improving physiologic conditioning, according to the researchers.
MS often results in walking dysfunction and physiologic deconditioning. High-intensity interval exercise has induced significant improvements in physiologic conditioning in healthy and clinical populations, including patients with MS. Before integrating high-intensity interval exercise into an exercise prescription, and to increase the likelihood that participants will engage in and adhere to this exercise regimen, investigators thought it important to ascertain whether people with MS and mobility disability enjoy the high-intensity interval exercise and experience mood benefits.
Elizabeth Hubbard, PhD, Assistant Professor of Kinesiology at Berry College in Mount Berry, Georgia, conducted a study to examine the effects of single sessions of high-intensity interval exercise and continuous exercise on mood and enjoyment outcomes in persons with MS and mobility disability. In their study, 20 participants (median Expanded Disability Status Scale score, 5.8) underwent counterbalanced presentations of high-intensity interval exercise and continuous exercise bouts using a recumbent stepper. The high-intensity interval exercise bout was 20 minutes long and included 10 cycles of one-minute intervals at a wattage associated with 90% VO2 peak, followed by one-minute recovery intervals at 15 W. The continuous exercise bout lasted for 20 minutes at a wattage associated with 50% to 60% VO2 peak. The researchers administered the Profile of Mood States (POMS) survey before, immediately after, and 30 minutes after exercise. The Physical Activity Enjoyment Scale (PACES) questionnaire was administered immediately after exercise.
Dr. Hubbard and colleagues observed no significant condition-by-time interactions or main effects of condition on any POMS subscales. The data indicated large, statistically significant main effects of time on total mood disturbance, fatigue, and vigor. Specifically, mood worsened immediately after exercise, but rebounded after the 30-minute recovery period. The researchers saw no significant difference in scores for the PACES questionnaire between high-intensity interval exercise (mean, 95.5) and continuous exercise (mean, 97.7) conditions.
One weakness of the study was that the population was relatively active, although with a low level of fitness. In addition, the researchers did not control for age, sex, or disability status.
The high-intensity interval exercise and continuous exercise were feasible, safe, and enjoyable in people with MS who have mobility disability. “High-intensity interval exercise … might be particularly beneficial for improving fitness and functional capacity in persons with MS who have mobility disability and experience subsequent physiologic deconditioning,” said Dr. Hubbard.
Introducing the 2018 class of AGA Research Foundation awardees
The American Gastroenterological Association (AGA) and the AGA Research Foundation are pleased to award 41 investigators with more than $2 million in research funding in the 2018 award year.
“We were impressed by the quality of applications received in 2018,” said Robert S. Sandler, MD, MPH, AGAF, chair, AGA Research Foundation. “The AGA Research Foundation is excited to add 41 investigators into the AGA Research Foundation awards family and we look forward to seeing the results of their research. Based on the proposals, we are confident that the newest class of awardees will continue to push gastroenterology and hepatology research forward and contribute to the next big discoveries in our field.”
The AGA Research Foundation Awards Program recruits, retains, and supports the most promising investigators in gastroenterology and hepatology. With AGA Research Foundation funding, recipients have protected time to continue their fundamental research into causes and treatments for digestive disorders. AGA grants have launched the careers of investigators doing important work that translates to new patient care tools for clinicians and better outcomes for patients. To view the list of recipients go to https://www.gastro.org/press-release/introducing-the-2018-class-of-aga-research-foundation-awardees.
The awards program is made possible thanks to generous donors and funders contributing to the AGA Research Foundation. Learn more about the AGA Research Foundation at www.gastro.org/foundation.
The American Gastroenterological Association (AGA) and the AGA Research Foundation are pleased to award 41 investigators with more than $2 million in research funding in the 2018 award year.
“We were impressed by the quality of applications received in 2018,” said Robert S. Sandler, MD, MPH, AGAF, chair, AGA Research Foundation. “The AGA Research Foundation is excited to add 41 investigators into the AGA Research Foundation awards family and we look forward to seeing the results of their research. Based on the proposals, we are confident that the newest class of awardees will continue to push gastroenterology and hepatology research forward and contribute to the next big discoveries in our field.”
The AGA Research Foundation Awards Program recruits, retains, and supports the most promising investigators in gastroenterology and hepatology. With AGA Research Foundation funding, recipients have protected time to continue their fundamental research into causes and treatments for digestive disorders. AGA grants have launched the careers of investigators doing important work that translates to new patient care tools for clinicians and better outcomes for patients. To view the list of recipients go to https://www.gastro.org/press-release/introducing-the-2018-class-of-aga-research-foundation-awardees.
The awards program is made possible thanks to generous donors and funders contributing to the AGA Research Foundation. Learn more about the AGA Research Foundation at www.gastro.org/foundation.
The American Gastroenterological Association (AGA) and the AGA Research Foundation are pleased to award 41 investigators with more than $2 million in research funding in the 2018 award year.
“We were impressed by the quality of applications received in 2018,” said Robert S. Sandler, MD, MPH, AGAF, chair, AGA Research Foundation. “The AGA Research Foundation is excited to add 41 investigators into the AGA Research Foundation awards family and we look forward to seeing the results of their research. Based on the proposals, we are confident that the newest class of awardees will continue to push gastroenterology and hepatology research forward and contribute to the next big discoveries in our field.”
The AGA Research Foundation Awards Program recruits, retains, and supports the most promising investigators in gastroenterology and hepatology. With AGA Research Foundation funding, recipients have protected time to continue their fundamental research into causes and treatments for digestive disorders. AGA grants have launched the careers of investigators doing important work that translates to new patient care tools for clinicians and better outcomes for patients. To view the list of recipients go to https://www.gastro.org/press-release/introducing-the-2018-class-of-aga-research-foundation-awardees.
The awards program is made possible thanks to generous donors and funders contributing to the AGA Research Foundation. Learn more about the AGA Research Foundation at www.gastro.org/foundation.
AGA announces its newest class of Fellows
AGA Fellowship status is an honor awarded to members who demonstrate a personal commitment to the field of gastroenterology, as well as professional achievement in clinical private or academic practice and in basic or clinical research.
The most recent inductees into the AGA Fellows Program were recognized at Digestive Disease Week® (DDW) 2018 and received a digital ribbon in their AGA Community profile. The 2018 class of AGA Fellows includes 112 members, who added the designation “AGAF” in their professional activities.
Join the AGA Fellowship Recognition Panel in congratulating these distinguished members and view the 2018 class of AGA Fellows in the AGA Community forum, community.gastro.org
Learn more about joining this international community of excellence. Applications for the 2019 cohort are now being accepted. Those in clinical private or academic practice and in basic or clinical research who meet the AGAF criteria are invited to apply. Applications are due Aug. 27, 2018. Learn more at gastro.org/fellowship.
AGA Fellowship status is an honor awarded to members who demonstrate a personal commitment to the field of gastroenterology, as well as professional achievement in clinical private or academic practice and in basic or clinical research.
The most recent inductees into the AGA Fellows Program were recognized at Digestive Disease Week® (DDW) 2018 and received a digital ribbon in their AGA Community profile. The 2018 class of AGA Fellows includes 112 members, who added the designation “AGAF” in their professional activities.
Join the AGA Fellowship Recognition Panel in congratulating these distinguished members and view the 2018 class of AGA Fellows in the AGA Community forum, community.gastro.org
Learn more about joining this international community of excellence. Applications for the 2019 cohort are now being accepted. Those in clinical private or academic practice and in basic or clinical research who meet the AGAF criteria are invited to apply. Applications are due Aug. 27, 2018. Learn more at gastro.org/fellowship.
AGA Fellowship status is an honor awarded to members who demonstrate a personal commitment to the field of gastroenterology, as well as professional achievement in clinical private or academic practice and in basic or clinical research.
The most recent inductees into the AGA Fellows Program were recognized at Digestive Disease Week® (DDW) 2018 and received a digital ribbon in their AGA Community profile. The 2018 class of AGA Fellows includes 112 members, who added the designation “AGAF” in their professional activities.
Join the AGA Fellowship Recognition Panel in congratulating these distinguished members and view the 2018 class of AGA Fellows in the AGA Community forum, community.gastro.org
Learn more about joining this international community of excellence. Applications for the 2019 cohort are now being accepted. Those in clinical private or academic practice and in basic or clinical research who meet the AGAF criteria are invited to apply. Applications are due Aug. 27, 2018. Learn more at gastro.org/fellowship.
How does the Quality Payment Program affect you?
AGA asks Congress and CMS to continue to implement the Quality Payment Program (QPP) in a way that maximizes flexibility and success for you and your Medicare patients.
Most gastroenterologists participate in the Merit-Based Incentive Payment System (MIPS), which means how the QPP is implemented impacts the entire GI profession. The QPP replaced the sustainable growth rate (SGR) formula in 2015 when the Medicare Access and CHIP Reauthorization Act (MACRA) was signed into law. The QPP is comprised of two tracks: MIPS and Advanced Alternative Payment Models (Advanced APMs).
CMS has designated 2017 and 2018 as transition years to allow providers to learn about the QPP and to gradually increase their preparedness for MIPS.
Congress also recently acted to provide CMS additional flexibility with respect to QPP and MIPS implementation, including:
• Excluding Medicare Part B drug costs from MIPS payment adjustments.
• Eliminating improvement scoring for the cost performance category for the second through fifth years of MIPS.
• Allowing CMS to weight the cost performance category at less than 30 percent, but not less than 10 percent for the second through fifth years of MIPS.
• Allowing CMS flexibility in setting the performance threshold for MIPS in years two through five to ensure a gradual and incremental transition to the performance threshold set at the mean or median for the sixth year.
QPP implementation is a top priority for AGA to ensure that the value of specialty care is recognized. Learn more on our website www.gastro.org/QPP.
AGA asks Congress and CMS to continue to implement the Quality Payment Program (QPP) in a way that maximizes flexibility and success for you and your Medicare patients.
Most gastroenterologists participate in the Merit-Based Incentive Payment System (MIPS), which means how the QPP is implemented impacts the entire GI profession. The QPP replaced the sustainable growth rate (SGR) formula in 2015 when the Medicare Access and CHIP Reauthorization Act (MACRA) was signed into law. The QPP is comprised of two tracks: MIPS and Advanced Alternative Payment Models (Advanced APMs).
CMS has designated 2017 and 2018 as transition years to allow providers to learn about the QPP and to gradually increase their preparedness for MIPS.
Congress also recently acted to provide CMS additional flexibility with respect to QPP and MIPS implementation, including:
• Excluding Medicare Part B drug costs from MIPS payment adjustments.
• Eliminating improvement scoring for the cost performance category for the second through fifth years of MIPS.
• Allowing CMS to weight the cost performance category at less than 30 percent, but not less than 10 percent for the second through fifth years of MIPS.
• Allowing CMS flexibility in setting the performance threshold for MIPS in years two through five to ensure a gradual and incremental transition to the performance threshold set at the mean or median for the sixth year.
QPP implementation is a top priority for AGA to ensure that the value of specialty care is recognized. Learn more on our website www.gastro.org/QPP.
AGA asks Congress and CMS to continue to implement the Quality Payment Program (QPP) in a way that maximizes flexibility and success for you and your Medicare patients.
Most gastroenterologists participate in the Merit-Based Incentive Payment System (MIPS), which means how the QPP is implemented impacts the entire GI profession. The QPP replaced the sustainable growth rate (SGR) formula in 2015 when the Medicare Access and CHIP Reauthorization Act (MACRA) was signed into law. The QPP is comprised of two tracks: MIPS and Advanced Alternative Payment Models (Advanced APMs).
CMS has designated 2017 and 2018 as transition years to allow providers to learn about the QPP and to gradually increase their preparedness for MIPS.
Congress also recently acted to provide CMS additional flexibility with respect to QPP and MIPS implementation, including:
• Excluding Medicare Part B drug costs from MIPS payment adjustments.
• Eliminating improvement scoring for the cost performance category for the second through fifth years of MIPS.
• Allowing CMS to weight the cost performance category at less than 30 percent, but not less than 10 percent for the second through fifth years of MIPS.
• Allowing CMS flexibility in setting the performance threshold for MIPS in years two through five to ensure a gradual and incremental transition to the performance threshold set at the mean or median for the sixth year.
QPP implementation is a top priority for AGA to ensure that the value of specialty care is recognized. Learn more on our website www.gastro.org/QPP.