User login
EULAR nears first recommendations for managing Sjögren’s syndrome
AMSTERDAM – , and they divide the treatment targets into sicca syndrome and systemic manifestations of the disease.
“In Sjögren’s, we always have two subtypes of patients: those who have sicca syndrome only, and those with sicca syndrome plus systemic disease,” explained Soledad Retamozo, MD, who presented the current version of the recommendations at the European Congress of Rheumatology. “We wanted to highlight that there are two types of patients,” said Dr. Retamozo, a rheumatologist at the University of Córdoba (Argentina). “It’s hard to treat patients with sicca syndrome plus fatigue and pain because there is no high-level evidence on how to do this; all we have is expert opinion,” Dr. Retamozo said in an interview.

In fact, roughly half of the recommendations have no supporting evidence base, as presented by Dr. Retamozo. That starts with all three general recommendations she presented:
• Patients with Sjögren’s should be managed at a center of expertise using a multidisciplinary approach, which she said should include ophthalmologists and dentists to help address the mouth and ocular manifestations of sicca syndrome.
• Patients with sicca syndrome should receive symptomatic relief with topical treatments.
• Systemic treatments – glucocorticoids, immunosuppressants, and biologicals – can be considered for patients with active systemic disease.
The statement’s specific recommendations start with managing oral dryness, an intervention that should begin by measuring salivary gland (SG) dysfunction. The document next recommends nonpharmacologic interventions for mild SG dysfunction, pharmacological stimulation for moderate SG dysfunction, and a saliva substitute for severe SG dysfunction. All three recommendations are evidence based, relying on results from either randomized trials or controlled studies.
The second target for topical treatments is ocular dryness, which starts with artificial tears, or ocular gels or ointments, recommendations based on randomized trials. Refractory or severe ocular dryness should receive eye drops that contain a nonsteroidal anti-inflammatory drug or a glucocorticoid, based on controlled study results, or autologous serum eye drops, a strategy tested in a randomized trial.
The recommendations then shift to dealing with systemic manifestations, starting with fatigue and pain, offering the expert recommendation to evaluate the contribution of comorbid diseases and assess their severity with tools such as the Eular Sjögren’s Syndrome Patient-Reported Index (ESSPRI) (Ann Rheum Dis. 2011 June;70[6]:968-72), the Profile of Fatigue, and the Brief Pain Inventory.
Using evidence from randomized trials, the recommendations tell clinicians to consider treatment with analgesics or pain-modifying agents for musculoskeletal pain by weighing the potential benefits and adverse effects from this treatment.
For other forms of systemic disease, the recommendations offer the expert opinion to tailor treatment to the organ-specific severity using the ESSPRI definitions. If using glucocorticoids to treat systemic disease, they should be given at the minimum effective dose and for the shortest period of time needed to control active systemic disease, a recommendation based on retrospective or descriptive studies. Expert opinion called for using immunosuppressive treatments as glucocorticoid-sparing options for systemic disease, and this recommendation added that no particular immunosuppressive agent stands out as best compared with all available agents. In more than 95% of reported cases of systemic disease treatment in Sjögren’s patients, clinicians used the immunosuppressive drugs in association with glucocorticoids, Dr. Retamozo noted.
Finally, for systemic disease the recommendations cited evidence from controlled studies that B-cell targeted therapies, such as rituximab (Rituxan) and belimumab (Benlysta), may be considered in patients with severe, refractory systemic disease. An additional expert opinion was that the systemic, organ-specific approach should sequence treatments by using glucocorticoids first, followed by immunosuppressants, and finally biological drugs.
The recommendations finish with an entry that treatment of B-cell lymphoma be individualized based on the specific histopathologic subtype involved and the level of disease extension, an approach based on results from retrospective or descriptive studies.
The recommendations must still undergo final EULAR review and endorsement, with publication on track to occur before the end of 2018, Dr. Retamozo said.
She had no disclosures.
SOURCE: Retamozo S al. Ann Rheum Dis. 2018;77(Suppl 2):42. Abstract SP0159.
AMSTERDAM – , and they divide the treatment targets into sicca syndrome and systemic manifestations of the disease.
“In Sjögren’s, we always have two subtypes of patients: those who have sicca syndrome only, and those with sicca syndrome plus systemic disease,” explained Soledad Retamozo, MD, who presented the current version of the recommendations at the European Congress of Rheumatology. “We wanted to highlight that there are two types of patients,” said Dr. Retamozo, a rheumatologist at the University of Córdoba (Argentina). “It’s hard to treat patients with sicca syndrome plus fatigue and pain because there is no high-level evidence on how to do this; all we have is expert opinion,” Dr. Retamozo said in an interview.
In fact, roughly half of the recommendations have no supporting evidence base, as presented by Dr. Retamozo. That starts with all three general recommendations she presented:
• Patients with Sjögren’s should be managed at a center of expertise using a multidisciplinary approach, which she said should include ophthalmologists and dentists to help address the mouth and ocular manifestations of sicca syndrome.
• Patients with sicca syndrome should receive symptomatic relief with topical treatments.
• Systemic treatments – glucocorticoids, immunosuppressants, and biologicals – can be considered for patients with active systemic disease.
The statement’s specific recommendations start with managing oral dryness, an intervention that should begin by measuring salivary gland (SG) dysfunction. The document next recommends nonpharmacologic interventions for mild SG dysfunction, pharmacological stimulation for moderate SG dysfunction, and a saliva substitute for severe SG dysfunction. All three recommendations are evidence based, relying on results from either randomized trials or controlled studies.
The second target for topical treatments is ocular dryness, which starts with artificial tears, or ocular gels or ointments, recommendations based on randomized trials. Refractory or severe ocular dryness should receive eye drops that contain a nonsteroidal anti-inflammatory drug or a glucocorticoid, based on controlled study results, or autologous serum eye drops, a strategy tested in a randomized trial.
The recommendations then shift to dealing with systemic manifestations, starting with fatigue and pain, offering the expert recommendation to evaluate the contribution of comorbid diseases and assess their severity with tools such as the Eular Sjögren’s Syndrome Patient-Reported Index (ESSPRI) (Ann Rheum Dis. 2011 June;70[6]:968-72), the Profile of Fatigue, and the Brief Pain Inventory.
Using evidence from randomized trials, the recommendations tell clinicians to consider treatment with analgesics or pain-modifying agents for musculoskeletal pain by weighing the potential benefits and adverse effects from this treatment.
For other forms of systemic disease, the recommendations offer the expert opinion to tailor treatment to the organ-specific severity using the ESSPRI definitions. If using glucocorticoids to treat systemic disease, they should be given at the minimum effective dose and for the shortest period of time needed to control active systemic disease, a recommendation based on retrospective or descriptive studies. Expert opinion called for using immunosuppressive treatments as glucocorticoid-sparing options for systemic disease, and this recommendation added that no particular immunosuppressive agent stands out as best compared with all available agents. In more than 95% of reported cases of systemic disease treatment in Sjögren’s patients, clinicians used the immunosuppressive drugs in association with glucocorticoids, Dr. Retamozo noted.
Finally, for systemic disease the recommendations cited evidence from controlled studies that B-cell targeted therapies, such as rituximab (Rituxan) and belimumab (Benlysta), may be considered in patients with severe, refractory systemic disease. An additional expert opinion was that the systemic, organ-specific approach should sequence treatments by using glucocorticoids first, followed by immunosuppressants, and finally biological drugs.
The recommendations finish with an entry that treatment of B-cell lymphoma be individualized based on the specific histopathologic subtype involved and the level of disease extension, an approach based on results from retrospective or descriptive studies.
The recommendations must still undergo final EULAR review and endorsement, with publication on track to occur before the end of 2018, Dr. Retamozo said.
She had no disclosures.
SOURCE: Retamozo S al. Ann Rheum Dis. 2018;77(Suppl 2):42. Abstract SP0159.
AMSTERDAM – , and they divide the treatment targets into sicca syndrome and systemic manifestations of the disease.
“In Sjögren’s, we always have two subtypes of patients: those who have sicca syndrome only, and those with sicca syndrome plus systemic disease,” explained Soledad Retamozo, MD, who presented the current version of the recommendations at the European Congress of Rheumatology. “We wanted to highlight that there are two types of patients,” said Dr. Retamozo, a rheumatologist at the University of Córdoba (Argentina). “It’s hard to treat patients with sicca syndrome plus fatigue and pain because there is no high-level evidence on how to do this; all we have is expert opinion,” Dr. Retamozo said in an interview.
In fact, roughly half of the recommendations have no supporting evidence base, as presented by Dr. Retamozo. That starts with all three general recommendations she presented:
• Patients with Sjögren’s should be managed at a center of expertise using a multidisciplinary approach, which she said should include ophthalmologists and dentists to help address the mouth and ocular manifestations of sicca syndrome.
• Patients with sicca syndrome should receive symptomatic relief with topical treatments.
• Systemic treatments – glucocorticoids, immunosuppressants, and biologicals – can be considered for patients with active systemic disease.
The statement’s specific recommendations start with managing oral dryness, an intervention that should begin by measuring salivary gland (SG) dysfunction. The document next recommends nonpharmacologic interventions for mild SG dysfunction, pharmacological stimulation for moderate SG dysfunction, and a saliva substitute for severe SG dysfunction. All three recommendations are evidence based, relying on results from either randomized trials or controlled studies.
The second target for topical treatments is ocular dryness, which starts with artificial tears, or ocular gels or ointments, recommendations based on randomized trials. Refractory or severe ocular dryness should receive eye drops that contain a nonsteroidal anti-inflammatory drug or a glucocorticoid, based on controlled study results, or autologous serum eye drops, a strategy tested in a randomized trial.
The recommendations then shift to dealing with systemic manifestations, starting with fatigue and pain, offering the expert recommendation to evaluate the contribution of comorbid diseases and assess their severity with tools such as the Eular Sjögren’s Syndrome Patient-Reported Index (ESSPRI) (Ann Rheum Dis. 2011 June;70[6]:968-72), the Profile of Fatigue, and the Brief Pain Inventory.
Using evidence from randomized trials, the recommendations tell clinicians to consider treatment with analgesics or pain-modifying agents for musculoskeletal pain by weighing the potential benefits and adverse effects from this treatment.
For other forms of systemic disease, the recommendations offer the expert opinion to tailor treatment to the organ-specific severity using the ESSPRI definitions. If using glucocorticoids to treat systemic disease, they should be given at the minimum effective dose and for the shortest period of time needed to control active systemic disease, a recommendation based on retrospective or descriptive studies. Expert opinion called for using immunosuppressive treatments as glucocorticoid-sparing options for systemic disease, and this recommendation added that no particular immunosuppressive agent stands out as best compared with all available agents. In more than 95% of reported cases of systemic disease treatment in Sjögren’s patients, clinicians used the immunosuppressive drugs in association with glucocorticoids, Dr. Retamozo noted.
Finally, for systemic disease the recommendations cited evidence from controlled studies that B-cell targeted therapies, such as rituximab (Rituxan) and belimumab (Benlysta), may be considered in patients with severe, refractory systemic disease. An additional expert opinion was that the systemic, organ-specific approach should sequence treatments by using glucocorticoids first, followed by immunosuppressants, and finally biological drugs.
The recommendations finish with an entry that treatment of B-cell lymphoma be individualized based on the specific histopathologic subtype involved and the level of disease extension, an approach based on results from retrospective or descriptive studies.
The recommendations must still undergo final EULAR review and endorsement, with publication on track to occur before the end of 2018, Dr. Retamozo said.
She had no disclosures.
SOURCE: Retamozo S al. Ann Rheum Dis. 2018;77(Suppl 2):42. Abstract SP0159.
REPORTING FROM THE EULAR 2018 CONGRESS
Trastuzumab biosimilar is equivalent on central review
CHICAGO – according to a new analysis of the phase 3 LILAC trial.
The 725 women in the multinational trial received run-in, anthracycline-based chemotherapy and were then evenly randomized to receive ABP 980 or trastuzumab, each with paclitaxel, followed by surgery.

The difference in pathologic complete response (pCR) rate assessed by local pathologists has been previously reported (Lancet Oncol. 2018 Jun 4. doi: 10.1016/S1470-2045(18)30241-9); those findings established non-inferiority of the biosimilar but left the matter of non-superiority inconclusive. However, in the new analysis, reported in a poster session at the ASCO Annual Meeting, the difference in pCR rate when instead assessed by a central pathologist fell within all bounds for equivalence.
“This is part of the totality of evidence in the course of approval of ABP 980,” lead author Hans-Christian Kolberg, MD, head of the department of gynecology and obstetrics of the Breast Cancer Center of the Gynecologic Cancer Center at Marien Hospital Bottrop (Germany), commented in an interview.
The new data prompted European regulators to authorize marketing of the biosimilar (branded as Kanjinti) for HER2+ early breast cancer and metastatic breast cancer, as well as HER2+ metastatic gastric cancer. (In the United States, the Food and Drug Administration recently rejected the application for ABP 980 market approval.)
“Breast cancer therapy is getting more and more expensive, and we somehow have to raise the money to pay for it. If we have a chance to make an antibody that is 20%-30% cheaper, which is what we hope it will be in Europe, we have that money for other things,” Dr. Kolberg said, reflecting on the bigger picture.
“I am also a visiting professor at a university in China, where patients who are HER2+ don’t get Herceptin because they can’t afford it. We always have to remember that in Europe and the U.S., we are kind of living on an island. If you look at Africa, Asia, and South America, making things affordable is important,” he added. “I hope and believe that this is just the beginning of the price fight. I hope that the biosimilar companies really will fight to see who will have the lowest price because that will be good for the patients. The lower the price, the better for the patients.”
Study details
Research leading up to the LILAC trial established that ABP 980 had analytic characteristics, nonclinical attributes, and pharmacokinetics similar to those of trastuzumab. The trial, conducted in 97 centers in 20 countries in western Europe, eastern Europe, and other world regions, assessed clinical similarity.
“I think central review was done in the study because we had so many centers all over the world that it was questionable as to how we could monitor the quality in dozens and dozens of pathology labs,” Dr. Kolberg explained. “So the idea was that we make it a little bit more difficult, a little bit more expensive, but more reliable if we use one pathologist.”
The central review was not without logistical issues, he acknowledged. In particular, it was challenging to ensure that all centers – including some doing so for the first time – followed a standardized procedure for sending tissue to the central lab.
The previously reported locally assessed pCR rates in breast tissue and axillary lymph nodes were 48.0% with ABP 980 and 40.5% with trastuzumab. The risk difference was 7.3% (90% confidence interval, 1.2%-13.4%) and the risk ratio was 1.188 (90% CI, 1.033-1.366), with the upper bounds of the confidence intervals exceeding the predefined equivalence margins of 13% and 1.318, respectively.
The centrally assessed pCR rates were 47.8% with ABP 980 and 41.8% with trastuzumab. The risk difference was 5.8% (90% CI, –0.5% to 12.0%), and the risk ratio was 1.14 (90% CI, 0.993 to 1.312), with the upper bounds of the confidence intervals now falling within the equivalence margins.
“This is the first study ever that used central pathology review for pCR in a neoadjuvant breast cancer study. We were really skeptical at the beginning as to whether that would work because we had a lot of centers all over the world, from Russia, Brazil, the U.S., Germany,” Dr. Kolberg commented.
“It worked, and we were very lucky that it worked because in the local review, we did not reach our biosimilar margins, our equivalence margins. In the central review, we were well within the margins,” he said. “So if we had not in the beginning planned a coprimary endpoint with local and central pathology review, the medication would never have been approved.”
Dr. Kolberg disclosed that he is a consultant for Amgen, Carl Zeiss Meditec, Genomic Health, GlaxoSmithKline, Janssen, LIV Pharma, Novartis, Pfizer, Roche, SurgVision, Teva Pharmaceutical Industries, and Theraclion. The trial was sponsored by Amgen.
SOURCE: Kolberg HC et al. ASCO Annual Meeting, Abstract 583.
CHICAGO – according to a new analysis of the phase 3 LILAC trial.
The 725 women in the multinational trial received run-in, anthracycline-based chemotherapy and were then evenly randomized to receive ABP 980 or trastuzumab, each with paclitaxel, followed by surgery.
The difference in pathologic complete response (pCR) rate assessed by local pathologists has been previously reported (Lancet Oncol. 2018 Jun 4. doi: 10.1016/S1470-2045(18)30241-9); those findings established non-inferiority of the biosimilar but left the matter of non-superiority inconclusive. However, in the new analysis, reported in a poster session at the ASCO Annual Meeting, the difference in pCR rate when instead assessed by a central pathologist fell within all bounds for equivalence.
“This is part of the totality of evidence in the course of approval of ABP 980,” lead author Hans-Christian Kolberg, MD, head of the department of gynecology and obstetrics of the Breast Cancer Center of the Gynecologic Cancer Center at Marien Hospital Bottrop (Germany), commented in an interview.
The new data prompted European regulators to authorize marketing of the biosimilar (branded as Kanjinti) for HER2+ early breast cancer and metastatic breast cancer, as well as HER2+ metastatic gastric cancer. (In the United States, the Food and Drug Administration recently rejected the application for ABP 980 market approval.)
“Breast cancer therapy is getting more and more expensive, and we somehow have to raise the money to pay for it. If we have a chance to make an antibody that is 20%-30% cheaper, which is what we hope it will be in Europe, we have that money for other things,” Dr. Kolberg said, reflecting on the bigger picture.
“I am also a visiting professor at a university in China, where patients who are HER2+ don’t get Herceptin because they can’t afford it. We always have to remember that in Europe and the U.S., we are kind of living on an island. If you look at Africa, Asia, and South America, making things affordable is important,” he added. “I hope and believe that this is just the beginning of the price fight. I hope that the biosimilar companies really will fight to see who will have the lowest price because that will be good for the patients. The lower the price, the better for the patients.”
Study details
Research leading up to the LILAC trial established that ABP 980 had analytic characteristics, nonclinical attributes, and pharmacokinetics similar to those of trastuzumab. The trial, conducted in 97 centers in 20 countries in western Europe, eastern Europe, and other world regions, assessed clinical similarity.
“I think central review was done in the study because we had so many centers all over the world that it was questionable as to how we could monitor the quality in dozens and dozens of pathology labs,” Dr. Kolberg explained. “So the idea was that we make it a little bit more difficult, a little bit more expensive, but more reliable if we use one pathologist.”
The central review was not without logistical issues, he acknowledged. In particular, it was challenging to ensure that all centers – including some doing so for the first time – followed a standardized procedure for sending tissue to the central lab.
The previously reported locally assessed pCR rates in breast tissue and axillary lymph nodes were 48.0% with ABP 980 and 40.5% with trastuzumab. The risk difference was 7.3% (90% confidence interval, 1.2%-13.4%) and the risk ratio was 1.188 (90% CI, 1.033-1.366), with the upper bounds of the confidence intervals exceeding the predefined equivalence margins of 13% and 1.318, respectively.
The centrally assessed pCR rates were 47.8% with ABP 980 and 41.8% with trastuzumab. The risk difference was 5.8% (90% CI, –0.5% to 12.0%), and the risk ratio was 1.14 (90% CI, 0.993 to 1.312), with the upper bounds of the confidence intervals now falling within the equivalence margins.
“This is the first study ever that used central pathology review for pCR in a neoadjuvant breast cancer study. We were really skeptical at the beginning as to whether that would work because we had a lot of centers all over the world, from Russia, Brazil, the U.S., Germany,” Dr. Kolberg commented.
“It worked, and we were very lucky that it worked because in the local review, we did not reach our biosimilar margins, our equivalence margins. In the central review, we were well within the margins,” he said. “So if we had not in the beginning planned a coprimary endpoint with local and central pathology review, the medication would never have been approved.”
Dr. Kolberg disclosed that he is a consultant for Amgen, Carl Zeiss Meditec, Genomic Health, GlaxoSmithKline, Janssen, LIV Pharma, Novartis, Pfizer, Roche, SurgVision, Teva Pharmaceutical Industries, and Theraclion. The trial was sponsored by Amgen.
SOURCE: Kolberg HC et al. ASCO Annual Meeting, Abstract 583.
CHICAGO – according to a new analysis of the phase 3 LILAC trial.
The 725 women in the multinational trial received run-in, anthracycline-based chemotherapy and were then evenly randomized to receive ABP 980 or trastuzumab, each with paclitaxel, followed by surgery.
The difference in pathologic complete response (pCR) rate assessed by local pathologists has been previously reported (Lancet Oncol. 2018 Jun 4. doi: 10.1016/S1470-2045(18)30241-9); those findings established non-inferiority of the biosimilar but left the matter of non-superiority inconclusive. However, in the new analysis, reported in a poster session at the ASCO Annual Meeting, the difference in pCR rate when instead assessed by a central pathologist fell within all bounds for equivalence.
“This is part of the totality of evidence in the course of approval of ABP 980,” lead author Hans-Christian Kolberg, MD, head of the department of gynecology and obstetrics of the Breast Cancer Center of the Gynecologic Cancer Center at Marien Hospital Bottrop (Germany), commented in an interview.
The new data prompted European regulators to authorize marketing of the biosimilar (branded as Kanjinti) for HER2+ early breast cancer and metastatic breast cancer, as well as HER2+ metastatic gastric cancer. (In the United States, the Food and Drug Administration recently rejected the application for ABP 980 market approval.)
“Breast cancer therapy is getting more and more expensive, and we somehow have to raise the money to pay for it. If we have a chance to make an antibody that is 20%-30% cheaper, which is what we hope it will be in Europe, we have that money for other things,” Dr. Kolberg said, reflecting on the bigger picture.
“I am also a visiting professor at a university in China, where patients who are HER2+ don’t get Herceptin because they can’t afford it. We always have to remember that in Europe and the U.S., we are kind of living on an island. If you look at Africa, Asia, and South America, making things affordable is important,” he added. “I hope and believe that this is just the beginning of the price fight. I hope that the biosimilar companies really will fight to see who will have the lowest price because that will be good for the patients. The lower the price, the better for the patients.”
Study details
Research leading up to the LILAC trial established that ABP 980 had analytic characteristics, nonclinical attributes, and pharmacokinetics similar to those of trastuzumab. The trial, conducted in 97 centers in 20 countries in western Europe, eastern Europe, and other world regions, assessed clinical similarity.
“I think central review was done in the study because we had so many centers all over the world that it was questionable as to how we could monitor the quality in dozens and dozens of pathology labs,” Dr. Kolberg explained. “So the idea was that we make it a little bit more difficult, a little bit more expensive, but more reliable if we use one pathologist.”
The central review was not without logistical issues, he acknowledged. In particular, it was challenging to ensure that all centers – including some doing so for the first time – followed a standardized procedure for sending tissue to the central lab.
The previously reported locally assessed pCR rates in breast tissue and axillary lymph nodes were 48.0% with ABP 980 and 40.5% with trastuzumab. The risk difference was 7.3% (90% confidence interval, 1.2%-13.4%) and the risk ratio was 1.188 (90% CI, 1.033-1.366), with the upper bounds of the confidence intervals exceeding the predefined equivalence margins of 13% and 1.318, respectively.
The centrally assessed pCR rates were 47.8% with ABP 980 and 41.8% with trastuzumab. The risk difference was 5.8% (90% CI, –0.5% to 12.0%), and the risk ratio was 1.14 (90% CI, 0.993 to 1.312), with the upper bounds of the confidence intervals now falling within the equivalence margins.
“This is the first study ever that used central pathology review for pCR in a neoadjuvant breast cancer study. We were really skeptical at the beginning as to whether that would work because we had a lot of centers all over the world, from Russia, Brazil, the U.S., Germany,” Dr. Kolberg commented.
“It worked, and we were very lucky that it worked because in the local review, we did not reach our biosimilar margins, our equivalence margins. In the central review, we were well within the margins,” he said. “So if we had not in the beginning planned a coprimary endpoint with local and central pathology review, the medication would never have been approved.”
Dr. Kolberg disclosed that he is a consultant for Amgen, Carl Zeiss Meditec, Genomic Health, GlaxoSmithKline, Janssen, LIV Pharma, Novartis, Pfizer, Roche, SurgVision, Teva Pharmaceutical Industries, and Theraclion. The trial was sponsored by Amgen.
SOURCE: Kolberg HC et al. ASCO Annual Meeting, Abstract 583.
REPORTING FROM THE ASCO ANNUAL MEETING
Key clinical point: Central review determined that ABP 980 was neither inferior nor superior to trastuzumab in breast cancer patients.
Major finding: The centrally determined pCR rates were 47.8% with ABP 980 and 41.8% with trastuzumab, with bounds of the confidence intervals for risk difference and for risk ratio falling within the predefined equivalence margins.
Study details: An analysis of a phase 3 randomized controlled trial of neoadjuvant (and adjuvant) therapy among 725 patients with HER2+ early breast cancer (LILAC trial).
Disclosures: Dr. Kolberg disclosed that he is a consultant for Amgen, Carl Zeiss Meditec, Genomic Health, GlaxoSmithKline, Janssen, LIV Pharma, Novartis, Pfizer, Roche, SurgVision, Teva Pharmaceutical Industries, and Theraclion. The trial was sponsored by Amgen.
Source: Kolberg HC et al. ASCO Annual Meeting, Abstract 583.
My experience with the 2017 Gastroenterology Editorial Fellowship
When I entered the Gastroenterology Editorial Fellowship last year, many of my cofellows asked, “What exactly is an editorial fellowship?” After completing the program, I can now reflect on what was truly a fantastic year-long experience that complemented my final year of fellowship training.
Manuscripts certainly don’t review and accept themselves into journals, and fellowship training usually gives little insight into how manuscripts move through the submission, peer review, and production processes. What happens while authors wait for an editorial decision?

Since its first publication in 1943, Gastroenterology has importantly affected clinical care and the direction of research in our field. The quality of the American Gastroenterological Association’s flagship journal is derived from the sweat and muscle put in daily by gastroenterology-oriented and hepatology-oriented professionals who strive to transform the steady stream of cutting-edge manuscript submissions into an influential monthly publication read by a broad audience of clinicians, trainees, academic researchers, and policy makers. Without a doubt, this fellowship provided me with a sincere appreciation for the dedication that the board of editors puts into the peer review process and into maintaining the quality of monthly publications.
Near the beginning of my editorial fellowship, I spent a week at Vanderbilt University with the on-site editors. This was an irreplaceable opportunity for a trainee like myself to meet with both clinical- and research-oriented academic gastroenterologists who integrate demanding editorial roles into busy and fulfilling professional careers. Throughout my week there, I met with editors and staff who held various roles within the journal. Overall, this experience taught me about what metrics the journal uses to ensure quality, how manuscripts move from submission to publication, and how the direction and content of the journal is directed toward both AGA members and a broader readership.
At its core, the fellowship was focused on teaching the fundamental process of peer review. High-quality reviews for Gastroenterology provide consultative content and methodological expertise to editors who can then provide direction and make editorial recommendations to the authors. During my fellowship, I learned how to write a structured and nuanced review on the basis of novelty, clinical relevance and effects, and methodological rigor. I was paired with one of the associate editors on the basis of my primary content area of interest and regularly provided reviews for original article submissions. As the year progressed, I become more comfortable with reviewing beyond my immediate knowledge base. I also became more adept at providing detailed comments that would be insightful and accessible to both authors and editors.
Each week, I participated in a phone call with the board of editors, which was composed of thought leaders with content expertise in both gastroenterology and hepatology. During the call, we would thoughtfully critique some of the most cutting-edge research in our field; each manuscript often represented the culmination of years of meticulous work by research groups and multinational collaborations. From a fellow’s perspective, these calls gave me access to what may be the most insightful discussions taking place in our field, discussions which could have potential implications on future disease management principles and clinical practice guidelines. Through our meetings, it became apparent how much work goes into finding quality reviewers and how much time goes into assimilating the resulting recommendations into a cohesive discussion. This was an opportunity to learn how associate editors walk the entire board through a manuscript: from a basis of current knowledge and practice, through the conduct and findings of a particular study, and ultimately, to how study findings might affect the field.
What I came away with the most from the Gastroenterology Editorial Fellowship was an appreciation for the importance of the editorial and peer review process in maintaining the integrity and detail needed in high-quality research. Ultimately, this fellowship gave me a meaningful and immediate way to give back to the field that I can continue over the course of my professional career. I am certain that this unique program will continue to give future editorial fellows the skills and motivation they need to become actively involved in the editorial and peer review processes when they are beginning their independent careers.
Dr. Shah, MD, MBA, is an assistant professor; he is also the director of the Center for Gastrointestinal Motility in the division of gastroenterology in the department of internal medicine at Dartmouth-Hitchcock Medical Center, Lebanon, N.H.
When I entered the Gastroenterology Editorial Fellowship last year, many of my cofellows asked, “What exactly is an editorial fellowship?” After completing the program, I can now reflect on what was truly a fantastic year-long experience that complemented my final year of fellowship training.
Manuscripts certainly don’t review and accept themselves into journals, and fellowship training usually gives little insight into how manuscripts move through the submission, peer review, and production processes. What happens while authors wait for an editorial decision?
Since its first publication in 1943, Gastroenterology has importantly affected clinical care and the direction of research in our field. The quality of the American Gastroenterological Association’s flagship journal is derived from the sweat and muscle put in daily by gastroenterology-oriented and hepatology-oriented professionals who strive to transform the steady stream of cutting-edge manuscript submissions into an influential monthly publication read by a broad audience of clinicians, trainees, academic researchers, and policy makers. Without a doubt, this fellowship provided me with a sincere appreciation for the dedication that the board of editors puts into the peer review process and into maintaining the quality of monthly publications.
Near the beginning of my editorial fellowship, I spent a week at Vanderbilt University with the on-site editors. This was an irreplaceable opportunity for a trainee like myself to meet with both clinical- and research-oriented academic gastroenterologists who integrate demanding editorial roles into busy and fulfilling professional careers. Throughout my week there, I met with editors and staff who held various roles within the journal. Overall, this experience taught me about what metrics the journal uses to ensure quality, how manuscripts move from submission to publication, and how the direction and content of the journal is directed toward both AGA members and a broader readership.
At its core, the fellowship was focused on teaching the fundamental process of peer review. High-quality reviews for Gastroenterology provide consultative content and methodological expertise to editors who can then provide direction and make editorial recommendations to the authors. During my fellowship, I learned how to write a structured and nuanced review on the basis of novelty, clinical relevance and effects, and methodological rigor. I was paired with one of the associate editors on the basis of my primary content area of interest and regularly provided reviews for original article submissions. As the year progressed, I become more comfortable with reviewing beyond my immediate knowledge base. I also became more adept at providing detailed comments that would be insightful and accessible to both authors and editors.
Each week, I participated in a phone call with the board of editors, which was composed of thought leaders with content expertise in both gastroenterology and hepatology. During the call, we would thoughtfully critique some of the most cutting-edge research in our field; each manuscript often represented the culmination of years of meticulous work by research groups and multinational collaborations. From a fellow’s perspective, these calls gave me access to what may be the most insightful discussions taking place in our field, discussions which could have potential implications on future disease management principles and clinical practice guidelines. Through our meetings, it became apparent how much work goes into finding quality reviewers and how much time goes into assimilating the resulting recommendations into a cohesive discussion. This was an opportunity to learn how associate editors walk the entire board through a manuscript: from a basis of current knowledge and practice, through the conduct and findings of a particular study, and ultimately, to how study findings might affect the field.
What I came away with the most from the Gastroenterology Editorial Fellowship was an appreciation for the importance of the editorial and peer review process in maintaining the integrity and detail needed in high-quality research. Ultimately, this fellowship gave me a meaningful and immediate way to give back to the field that I can continue over the course of my professional career. I am certain that this unique program will continue to give future editorial fellows the skills and motivation they need to become actively involved in the editorial and peer review processes when they are beginning their independent careers.
Dr. Shah, MD, MBA, is an assistant professor; he is also the director of the Center for Gastrointestinal Motility in the division of gastroenterology in the department of internal medicine at Dartmouth-Hitchcock Medical Center, Lebanon, N.H.
When I entered the Gastroenterology Editorial Fellowship last year, many of my cofellows asked, “What exactly is an editorial fellowship?” After completing the program, I can now reflect on what was truly a fantastic year-long experience that complemented my final year of fellowship training.
Manuscripts certainly don’t review and accept themselves into journals, and fellowship training usually gives little insight into how manuscripts move through the submission, peer review, and production processes. What happens while authors wait for an editorial decision?
Since its first publication in 1943, Gastroenterology has importantly affected clinical care and the direction of research in our field. The quality of the American Gastroenterological Association’s flagship journal is derived from the sweat and muscle put in daily by gastroenterology-oriented and hepatology-oriented professionals who strive to transform the steady stream of cutting-edge manuscript submissions into an influential monthly publication read by a broad audience of clinicians, trainees, academic researchers, and policy makers. Without a doubt, this fellowship provided me with a sincere appreciation for the dedication that the board of editors puts into the peer review process and into maintaining the quality of monthly publications.
Near the beginning of my editorial fellowship, I spent a week at Vanderbilt University with the on-site editors. This was an irreplaceable opportunity for a trainee like myself to meet with both clinical- and research-oriented academic gastroenterologists who integrate demanding editorial roles into busy and fulfilling professional careers. Throughout my week there, I met with editors and staff who held various roles within the journal. Overall, this experience taught me about what metrics the journal uses to ensure quality, how manuscripts move from submission to publication, and how the direction and content of the journal is directed toward both AGA members and a broader readership.
At its core, the fellowship was focused on teaching the fundamental process of peer review. High-quality reviews for Gastroenterology provide consultative content and methodological expertise to editors who can then provide direction and make editorial recommendations to the authors. During my fellowship, I learned how to write a structured and nuanced review on the basis of novelty, clinical relevance and effects, and methodological rigor. I was paired with one of the associate editors on the basis of my primary content area of interest and regularly provided reviews for original article submissions. As the year progressed, I become more comfortable with reviewing beyond my immediate knowledge base. I also became more adept at providing detailed comments that would be insightful and accessible to both authors and editors.
Each week, I participated in a phone call with the board of editors, which was composed of thought leaders with content expertise in both gastroenterology and hepatology. During the call, we would thoughtfully critique some of the most cutting-edge research in our field; each manuscript often represented the culmination of years of meticulous work by research groups and multinational collaborations. From a fellow’s perspective, these calls gave me access to what may be the most insightful discussions taking place in our field, discussions which could have potential implications on future disease management principles and clinical practice guidelines. Through our meetings, it became apparent how much work goes into finding quality reviewers and how much time goes into assimilating the resulting recommendations into a cohesive discussion. This was an opportunity to learn how associate editors walk the entire board through a manuscript: from a basis of current knowledge and practice, through the conduct and findings of a particular study, and ultimately, to how study findings might affect the field.
What I came away with the most from the Gastroenterology Editorial Fellowship was an appreciation for the importance of the editorial and peer review process in maintaining the integrity and detail needed in high-quality research. Ultimately, this fellowship gave me a meaningful and immediate way to give back to the field that I can continue over the course of my professional career. I am certain that this unique program will continue to give future editorial fellows the skills and motivation they need to become actively involved in the editorial and peer review processes when they are beginning their independent careers.
Dr. Shah, MD, MBA, is an assistant professor; he is also the director of the Center for Gastrointestinal Motility in the division of gastroenterology in the department of internal medicine at Dartmouth-Hitchcock Medical Center, Lebanon, N.H.
Female authorship trends in academic gastroenterology over 4 decades
WASHINGTON – Gastroenterology is still a majority male specialty, although women are entering the field at higher and higher rates. Female first authorship tripled from 1995 to 2010 (from 11% to 32%) and female senior authorship tripled from 2000 to 2010 (from 7% to 24%), but gains have not been equal in all areas and have not continued in all areas.
Eileen J. Benz, MD, of Beaumont Hospital, Dublin, described in a video interview at the annual Digestive Disease Week® a study she and her colleagues conducted to analyze published research in the journal Gastroenterology for the changing prevalence of female authorship over 4 decades.
The researchers reviewed all research published in the January and July issues of Gastroenterology during 1971-2010 (865 abstracts); animal trials were excluded. The sex of the first author and the last author (considered the senior author) of each paper was recorded, as was the type of study (basic science, clinical trials, or epidemiologic research). The increase in female senior authorship lagged behind the increase in first authorship, which likely reflects the promotion of female gastroenterologists over time into senior academic positions.
Also noted was that basic science and epidemiology research have the highest number of female authors overall, and these areas seem to continue to add female authors, whereas the number of female authors in clinical trials research seems to have stagnated since 1996. Dr. Benz hypothesizes that both bench science and epidemiology have research time built in, but that for a physician who may have other demands on her time, clinical trials research is an add-on for which there may not be protected time.
WASHINGTON – Gastroenterology is still a majority male specialty, although women are entering the field at higher and higher rates. Female first authorship tripled from 1995 to 2010 (from 11% to 32%) and female senior authorship tripled from 2000 to 2010 (from 7% to 24%), but gains have not been equal in all areas and have not continued in all areas.
Eileen J. Benz, MD, of Beaumont Hospital, Dublin, described in a video interview at the annual Digestive Disease Week® a study she and her colleagues conducted to analyze published research in the journal Gastroenterology for the changing prevalence of female authorship over 4 decades.
The researchers reviewed all research published in the January and July issues of Gastroenterology during 1971-2010 (865 abstracts); animal trials were excluded. The sex of the first author and the last author (considered the senior author) of each paper was recorded, as was the type of study (basic science, clinical trials, or epidemiologic research). The increase in female senior authorship lagged behind the increase in first authorship, which likely reflects the promotion of female gastroenterologists over time into senior academic positions.
Also noted was that basic science and epidemiology research have the highest number of female authors overall, and these areas seem to continue to add female authors, whereas the number of female authors in clinical trials research seems to have stagnated since 1996. Dr. Benz hypothesizes that both bench science and epidemiology have research time built in, but that for a physician who may have other demands on her time, clinical trials research is an add-on for which there may not be protected time.
WASHINGTON – Gastroenterology is still a majority male specialty, although women are entering the field at higher and higher rates. Female first authorship tripled from 1995 to 2010 (from 11% to 32%) and female senior authorship tripled from 2000 to 2010 (from 7% to 24%), but gains have not been equal in all areas and have not continued in all areas.
Eileen J. Benz, MD, of Beaumont Hospital, Dublin, described in a video interview at the annual Digestive Disease Week® a study she and her colleagues conducted to analyze published research in the journal Gastroenterology for the changing prevalence of female authorship over 4 decades.
The researchers reviewed all research published in the January and July issues of Gastroenterology during 1971-2010 (865 abstracts); animal trials were excluded. The sex of the first author and the last author (considered the senior author) of each paper was recorded, as was the type of study (basic science, clinical trials, or epidemiologic research). The increase in female senior authorship lagged behind the increase in first authorship, which likely reflects the promotion of female gastroenterologists over time into senior academic positions.
Also noted was that basic science and epidemiology research have the highest number of female authors overall, and these areas seem to continue to add female authors, whereas the number of female authors in clinical trials research seems to have stagnated since 1996. Dr. Benz hypothesizes that both bench science and epidemiology have research time built in, but that for a physician who may have other demands on her time, clinical trials research is an add-on for which there may not be protected time.
Reporting from DDW 2018

Clinical Pearl: Mohs Cantaloupe Analogy for the Dermatology Resident
Practice Gap
Mohs micrographic surgery (MMS) is a highly curative tissue-sparing skin cancer treatment1 and is a required component of dermatology residency training. According to the Accreditation Council for Graduate Medical Education, residents must have exposure “either through direct observation or as an assistant in Mohs micrographic surgery, and reconstruction of these defects, to include flaps and grafts.”2 The MMS technique allows for complete circumferential peripheral and deep margin assessment of excised specimens; however, the conformation of a 3-dimensional gross tissue specimen into a 2-dimensional specimen as represented on a microscope slide is challenging to conceptualize.
Behavioral science research has shown that analogies and metaphors help integrate topics into a memorable format and produce deeper comprehension.3 As such, analogies can aid in the visualization of these complex spatial concepts. The MMS tissue-processing technique has been compared to flattening a pie pan.4 More recently, a peanut butter cup analogy was described as a visualization tool for explaining the various steps of MMS to patients.5 Although these analogies may help elucidate certain aspects of the MMS technique, none adequately account for the multilayered anatomy of the skin.
The Technique
To address this need, we developed the cantaloupe analogy, which provides visual representation of the 3 basic skin layers: (1) the rind represents the epidermis; (2) the flesh represents the dermis, and (3) the seed cavity represents the subcutaneous layer (Figures 1 and 2).
, as well as the epidermal, dermal, and subcutaneous layers.")
, flesh (dermis), and seed cavity (subcutaneous layer)...")
In MMS tissue processing, the peripheral margin of the ovoid excised skin specimen is pressed down into the same plane as the deepest layer through a process called relaxation.4 The cantaloupe represents the dome shape of the relaxed tissue, which is then serially sectioned in horizontal layers from deep to superficial (Figure 2). The first slice represents the deepest subcutaneous layer and most peripheral dermal and epidermal layers of the specimen (Figure 3). Using the cantaloupe analogy, subsequent stages (if warranted) would be guided by the location of the residual skin cancer. If the skin cancer is in the epidermis (rind) or dermis (flesh), then a skin specimen from the perimeter of the defect would be indicated. Residual skin cancer extending into the subcutaneous layer (seed cavity) would require a deeper resection.

Practice Implications
The cantaloupe provides a simple analogy to conceptualize the transition from the multilayered 3-dimensional skin tissue specimen to the 2-dimensional histologic slide specimen. Use of this cantaloupe analogy will aid dermatology residents and others interested in gaining a clearer understanding of MMS.
- Semkova K, Mallipeddi R, Robson A, et al. Mohs micrographic surgery concordance between Mohs surgeons and dermatopathologists. Dermatol Surg. 2013;39:1648-1652.
- ACGME program requirements for graduate medical education in dermatology. Accreditation Council for Graduate Medical Education website. https://www.acgme.org/Portals/0/PFAssets/ProgramRequirements/080_dermatology_2017-07-01.pdf. Updated July 1, 2017. Accessed June 6, 2018.
- Wolfe CR. Plant a tree in cyberspace: metaphor and analogy as design elements in Web-based learning environments. CyberPsychol Behav. 2001;4:67-76.
- Beck B, Peters SR. Frozen section techniques used in Mohs micrographic surgery. In: Peters SR, ed. A Practical Guide to Frozen Section Technique. New York, NY: Springer; 2010:151-170.
- Lee E, Wolverton JE, Somani AK. A simple, effective analogy to elucidate the Mohs micrographic surgery procedure—the peanut butter cup. JAMA Dermatol. 2017;153:743-744.
Practice Gap
Mohs micrographic surgery (MMS) is a highly curative tissue-sparing skin cancer treatment1 and is a required component of dermatology residency training. According to the Accreditation Council for Graduate Medical Education, residents must have exposure “either through direct observation or as an assistant in Mohs micrographic surgery, and reconstruction of these defects, to include flaps and grafts.”2 The MMS technique allows for complete circumferential peripheral and deep margin assessment of excised specimens; however, the conformation of a 3-dimensional gross tissue specimen into a 2-dimensional specimen as represented on a microscope slide is challenging to conceptualize.
Behavioral science research has shown that analogies and metaphors help integrate topics into a memorable format and produce deeper comprehension.3 As such, analogies can aid in the visualization of these complex spatial concepts. The MMS tissue-processing technique has been compared to flattening a pie pan.4 More recently, a peanut butter cup analogy was described as a visualization tool for explaining the various steps of MMS to patients.5 Although these analogies may help elucidate certain aspects of the MMS technique, none adequately account for the multilayered anatomy of the skin.
The Technique
To address this need, we developed the cantaloupe analogy, which provides visual representation of the 3 basic skin layers: (1) the rind represents the epidermis; (2) the flesh represents the dermis, and (3) the seed cavity represents the subcutaneous layer (Figures 1 and 2).
In MMS tissue processing, the peripheral margin of the ovoid excised skin specimen is pressed down into the same plane as the deepest layer through a process called relaxation.4 The cantaloupe represents the dome shape of the relaxed tissue, which is then serially sectioned in horizontal layers from deep to superficial (Figure 2). The first slice represents the deepest subcutaneous layer and most peripheral dermal and epidermal layers of the specimen (Figure 3). Using the cantaloupe analogy, subsequent stages (if warranted) would be guided by the location of the residual skin cancer. If the skin cancer is in the epidermis (rind) or dermis (flesh), then a skin specimen from the perimeter of the defect would be indicated. Residual skin cancer extending into the subcutaneous layer (seed cavity) would require a deeper resection.
Practice Implications
The cantaloupe provides a simple analogy to conceptualize the transition from the multilayered 3-dimensional skin tissue specimen to the 2-dimensional histologic slide specimen. Use of this cantaloupe analogy will aid dermatology residents and others interested in gaining a clearer understanding of MMS.
Practice Gap
Mohs micrographic surgery (MMS) is a highly curative tissue-sparing skin cancer treatment1 and is a required component of dermatology residency training. According to the Accreditation Council for Graduate Medical Education, residents must have exposure “either through direct observation or as an assistant in Mohs micrographic surgery, and reconstruction of these defects, to include flaps and grafts.”2 The MMS technique allows for complete circumferential peripheral and deep margin assessment of excised specimens; however, the conformation of a 3-dimensional gross tissue specimen into a 2-dimensional specimen as represented on a microscope slide is challenging to conceptualize.
Behavioral science research has shown that analogies and metaphors help integrate topics into a memorable format and produce deeper comprehension.3 As such, analogies can aid in the visualization of these complex spatial concepts. The MMS tissue-processing technique has been compared to flattening a pie pan.4 More recently, a peanut butter cup analogy was described as a visualization tool for explaining the various steps of MMS to patients.5 Although these analogies may help elucidate certain aspects of the MMS technique, none adequately account for the multilayered anatomy of the skin.
The Technique
To address this need, we developed the cantaloupe analogy, which provides visual representation of the 3 basic skin layers: (1) the rind represents the epidermis; (2) the flesh represents the dermis, and (3) the seed cavity represents the subcutaneous layer (Figures 1 and 2).
In MMS tissue processing, the peripheral margin of the ovoid excised skin specimen is pressed down into the same plane as the deepest layer through a process called relaxation.4 The cantaloupe represents the dome shape of the relaxed tissue, which is then serially sectioned in horizontal layers from deep to superficial (Figure 2). The first slice represents the deepest subcutaneous layer and most peripheral dermal and epidermal layers of the specimen (Figure 3). Using the cantaloupe analogy, subsequent stages (if warranted) would be guided by the location of the residual skin cancer. If the skin cancer is in the epidermis (rind) or dermis (flesh), then a skin specimen from the perimeter of the defect would be indicated. Residual skin cancer extending into the subcutaneous layer (seed cavity) would require a deeper resection.
Practice Implications
The cantaloupe provides a simple analogy to conceptualize the transition from the multilayered 3-dimensional skin tissue specimen to the 2-dimensional histologic slide specimen. Use of this cantaloupe analogy will aid dermatology residents and others interested in gaining a clearer understanding of MMS.
- Semkova K, Mallipeddi R, Robson A, et al. Mohs micrographic surgery concordance between Mohs surgeons and dermatopathologists. Dermatol Surg. 2013;39:1648-1652.
- ACGME program requirements for graduate medical education in dermatology. Accreditation Council for Graduate Medical Education website. https://www.acgme.org/Portals/0/PFAssets/ProgramRequirements/080_dermatology_2017-07-01.pdf. Updated July 1, 2017. Accessed June 6, 2018.
- Wolfe CR. Plant a tree in cyberspace: metaphor and analogy as design elements in Web-based learning environments. CyberPsychol Behav. 2001;4:67-76.
- Beck B, Peters SR. Frozen section techniques used in Mohs micrographic surgery. In: Peters SR, ed. A Practical Guide to Frozen Section Technique. New York, NY: Springer; 2010:151-170.
- Lee E, Wolverton JE, Somani AK. A simple, effective analogy to elucidate the Mohs micrographic surgery procedure—the peanut butter cup. JAMA Dermatol. 2017;153:743-744.
- Semkova K, Mallipeddi R, Robson A, et al. Mohs micrographic surgery concordance between Mohs surgeons and dermatopathologists. Dermatol Surg. 2013;39:1648-1652.
- ACGME program requirements for graduate medical education in dermatology. Accreditation Council for Graduate Medical Education website. https://www.acgme.org/Portals/0/PFAssets/ProgramRequirements/080_dermatology_2017-07-01.pdf. Updated July 1, 2017. Accessed June 6, 2018.
- Wolfe CR. Plant a tree in cyberspace: metaphor and analogy as design elements in Web-based learning environments. CyberPsychol Behav. 2001;4:67-76.
- Beck B, Peters SR. Frozen section techniques used in Mohs micrographic surgery. In: Peters SR, ed. A Practical Guide to Frozen Section Technique. New York, NY: Springer; 2010:151-170.
- Lee E, Wolverton JE, Somani AK. A simple, effective analogy to elucidate the Mohs micrographic surgery procedure—the peanut butter cup. JAMA Dermatol. 2017;153:743-744.

Research supports cannabis in MS, but legal, clinical pictures are murky
The medical marijuana landscape is changing so fast that Colorado Neurological Institute neurologist Allen C. Bowling, MD, PhD, already needs to update a presentation he gave about cannabis in multiple sclerosis in late May.

Since then, both those facts became history over a span of 2 days.
First, on June 25, the FDA announced its approval of Epidiolex (cannabidiol) for the treatment of seizures in two rare forms of epilepsy, Lennox-Gastaut syndrome and Dravet syndrome. It’s the first time the FDA has approved a drug with a purified ingredient – cannabidiol, a nonpsychoactive substance – that’s derived from marijuana.
Then, on June 26, voters in Oklahoma approved a ballot measure that allows the possession of marijuana for medical use; users must register with the state. Thirty states and the District of Columbia have made medical marijuana legal, according to the procon.org website, although the two newest ones (Oklahoma and West Virginia) are still developing procedures.
The laws vary widely. Some states don’t allow patients to smoke medical marijuana, and some don’t allow visitors to use out-of-state registry ID cards. And certain states limit the use of medical marijuana to specific conditions. Medical marijuana use by patients with MS is specifically allowed in many states, including Alaska, Arizona, Florida, Minnesota, and several others.
There’s another complexity: According to procon.org, 17 states have laws about the use of cannabidiol. In Georgia, for instance, the use of some cannabis oil is allowed for the treatment of MS and other conditions.
In the wake of the FDA ruling, Dr. Bowling spoke in an interview about cannabis, MS, and the questions that neurologists should be asking themselves.
Q: What are studies telling us about cannabis and MS?
A: There are lots of clinical studies – 19 randomized controlled trials. A consistent finding is that there’s benefit in terms of pain and people’s subjective sense of spasticity (Neurology. 2014 Apr 29;82(17):1556-63).
Q: During your CMSC presentation, you talked about how “fidelity” has been a problem in cannabis research. Could you elaborate on what you mean?
A: The products used in these studies are generally standardized, research-grade products that you can’t buy in any U.S. dispensary.
Cannabis is complex and contains more than 100 different potentially pharmacologically active molecules. You can’t conclude that if you see a product in clinical trials, you’ll then be able to walk into a dispensary for recreational or medical cannabis and get a product that produces the same effect.
Q: What have you seen in your own patient population in terms of cannabis use?
A: I find what’s been found with the studies: It helps with pain and people’s sense of muscle stiffness.
It’s especially helpful in people with pain and spasticity that breaks through in the late afternoon or at night when they’re trying to go to sleep. Just a little bit of cannabis can get them through those difficult times and improve their quality of life.
Q: What choices do patients make regarding whether to get high from the cannabis they use?
A: Some have absolutely zero interest in getting high, and they try to avoid the THC-containing products. Other like getting high in addition to getting help with pain and spasticity.
Q: Who should not use medical marijuana in the MS community?
A: Patients who don’t have symptoms that could respond.
I’m also very concerned about patients who are 25 years and younger because of the effects that cannabis can have on brain development out to age 25 and the higher risk of addiction in people who are younger.
Q: What do you think the future will hold on the cannabis front?
A: Now that it’s less of a taboo topic, there’s an ever-growing number of trials each year, including very high-quality studies.
Pharmaceutically produced, cannabis-based medicines will be a growing area. Epidiolex is a perfect example of that.
It’s important for physicians to know that the way cannabis-based medicine is produced by a pharmaceutical company is different in so many levels than the cannabis in states with recreational and medical marijuana.
Q: What are some ways that the pharmaceutical products are different?
A: The rigor of the production process, the standardization, the purity, the correct labeling and expiration dates. Plus, the lack of the use of pesticides and other contaminants. And they’re distributed by pharmacists.
Q: What should neurologists be thinking if they’re considering whether to recommend cannabis to their patients?
A: This is a very complex topic, and it’s not something that most of us have training in. You can’t sit down for 1 or 2 hours, get up to speed, and have your own well-informed opinion on it. You really need to put more time and effort.
Q: What are some issues that neurologists should consider?
A: You really need to find out what your state is doing about it and see how you feel about that.
How is your state administering medical and/or recreational marijuana? The administration of these programs is extremely different from state to state. Do these details satisfy you, and are you content having your patients interface with these programs?
Dr. Bowling reports no relevant disclosures.
The medical marijuana landscape is changing so fast that Colorado Neurological Institute neurologist Allen C. Bowling, MD, PhD, already needs to update a presentation he gave about cannabis in multiple sclerosis in late May.
Since then, both those facts became history over a span of 2 days.
First, on June 25, the FDA announced its approval of Epidiolex (cannabidiol) for the treatment of seizures in two rare forms of epilepsy, Lennox-Gastaut syndrome and Dravet syndrome. It’s the first time the FDA has approved a drug with a purified ingredient – cannabidiol, a nonpsychoactive substance – that’s derived from marijuana.
Then, on June 26, voters in Oklahoma approved a ballot measure that allows the possession of marijuana for medical use; users must register with the state. Thirty states and the District of Columbia have made medical marijuana legal, according to the procon.org website, although the two newest ones (Oklahoma and West Virginia) are still developing procedures.
The laws vary widely. Some states don’t allow patients to smoke medical marijuana, and some don’t allow visitors to use out-of-state registry ID cards. And certain states limit the use of medical marijuana to specific conditions. Medical marijuana use by patients with MS is specifically allowed in many states, including Alaska, Arizona, Florida, Minnesota, and several others.
There’s another complexity: According to procon.org, 17 states have laws about the use of cannabidiol. In Georgia, for instance, the use of some cannabis oil is allowed for the treatment of MS and other conditions.
In the wake of the FDA ruling, Dr. Bowling spoke in an interview about cannabis, MS, and the questions that neurologists should be asking themselves.
Q: What are studies telling us about cannabis and MS?
A: There are lots of clinical studies – 19 randomized controlled trials. A consistent finding is that there’s benefit in terms of pain and people’s subjective sense of spasticity (Neurology. 2014 Apr 29;82(17):1556-63).
Q: During your CMSC presentation, you talked about how “fidelity” has been a problem in cannabis research. Could you elaborate on what you mean?
A: The products used in these studies are generally standardized, research-grade products that you can’t buy in any U.S. dispensary.
Cannabis is complex and contains more than 100 different potentially pharmacologically active molecules. You can’t conclude that if you see a product in clinical trials, you’ll then be able to walk into a dispensary for recreational or medical cannabis and get a product that produces the same effect.
Q: What have you seen in your own patient population in terms of cannabis use?
A: I find what’s been found with the studies: It helps with pain and people’s sense of muscle stiffness.
It’s especially helpful in people with pain and spasticity that breaks through in the late afternoon or at night when they’re trying to go to sleep. Just a little bit of cannabis can get them through those difficult times and improve their quality of life.
Q: What choices do patients make regarding whether to get high from the cannabis they use?
A: Some have absolutely zero interest in getting high, and they try to avoid the THC-containing products. Other like getting high in addition to getting help with pain and spasticity.
Q: Who should not use medical marijuana in the MS community?
A: Patients who don’t have symptoms that could respond.
I’m also very concerned about patients who are 25 years and younger because of the effects that cannabis can have on brain development out to age 25 and the higher risk of addiction in people who are younger.
Q: What do you think the future will hold on the cannabis front?
A: Now that it’s less of a taboo topic, there’s an ever-growing number of trials each year, including very high-quality studies.
Pharmaceutically produced, cannabis-based medicines will be a growing area. Epidiolex is a perfect example of that.
It’s important for physicians to know that the way cannabis-based medicine is produced by a pharmaceutical company is different in so many levels than the cannabis in states with recreational and medical marijuana.
Q: What are some ways that the pharmaceutical products are different?
A: The rigor of the production process, the standardization, the purity, the correct labeling and expiration dates. Plus, the lack of the use of pesticides and other contaminants. And they’re distributed by pharmacists.
Q: What should neurologists be thinking if they’re considering whether to recommend cannabis to their patients?
A: This is a very complex topic, and it’s not something that most of us have training in. You can’t sit down for 1 or 2 hours, get up to speed, and have your own well-informed opinion on it. You really need to put more time and effort.
Q: What are some issues that neurologists should consider?
A: You really need to find out what your state is doing about it and see how you feel about that.
How is your state administering medical and/or recreational marijuana? The administration of these programs is extremely different from state to state. Do these details satisfy you, and are you content having your patients interface with these programs?
Dr. Bowling reports no relevant disclosures.
The medical marijuana landscape is changing so fast that Colorado Neurological Institute neurologist Allen C. Bowling, MD, PhD, already needs to update a presentation he gave about cannabis in multiple sclerosis in late May.
Since then, both those facts became history over a span of 2 days.
First, on June 25, the FDA announced its approval of Epidiolex (cannabidiol) for the treatment of seizures in two rare forms of epilepsy, Lennox-Gastaut syndrome and Dravet syndrome. It’s the first time the FDA has approved a drug with a purified ingredient – cannabidiol, a nonpsychoactive substance – that’s derived from marijuana.
Then, on June 26, voters in Oklahoma approved a ballot measure that allows the possession of marijuana for medical use; users must register with the state. Thirty states and the District of Columbia have made medical marijuana legal, according to the procon.org website, although the two newest ones (Oklahoma and West Virginia) are still developing procedures.
The laws vary widely. Some states don’t allow patients to smoke medical marijuana, and some don’t allow visitors to use out-of-state registry ID cards. And certain states limit the use of medical marijuana to specific conditions. Medical marijuana use by patients with MS is specifically allowed in many states, including Alaska, Arizona, Florida, Minnesota, and several others.
There’s another complexity: According to procon.org, 17 states have laws about the use of cannabidiol. In Georgia, for instance, the use of some cannabis oil is allowed for the treatment of MS and other conditions.
In the wake of the FDA ruling, Dr. Bowling spoke in an interview about cannabis, MS, and the questions that neurologists should be asking themselves.
Q: What are studies telling us about cannabis and MS?
A: There are lots of clinical studies – 19 randomized controlled trials. A consistent finding is that there’s benefit in terms of pain and people’s subjective sense of spasticity (Neurology. 2014 Apr 29;82(17):1556-63).
Q: During your CMSC presentation, you talked about how “fidelity” has been a problem in cannabis research. Could you elaborate on what you mean?
A: The products used in these studies are generally standardized, research-grade products that you can’t buy in any U.S. dispensary.
Cannabis is complex and contains more than 100 different potentially pharmacologically active molecules. You can’t conclude that if you see a product in clinical trials, you’ll then be able to walk into a dispensary for recreational or medical cannabis and get a product that produces the same effect.
Q: What have you seen in your own patient population in terms of cannabis use?
A: I find what’s been found with the studies: It helps with pain and people’s sense of muscle stiffness.
It’s especially helpful in people with pain and spasticity that breaks through in the late afternoon or at night when they’re trying to go to sleep. Just a little bit of cannabis can get them through those difficult times and improve their quality of life.
Q: What choices do patients make regarding whether to get high from the cannabis they use?
A: Some have absolutely zero interest in getting high, and they try to avoid the THC-containing products. Other like getting high in addition to getting help with pain and spasticity.
Q: Who should not use medical marijuana in the MS community?
A: Patients who don’t have symptoms that could respond.
I’m also very concerned about patients who are 25 years and younger because of the effects that cannabis can have on brain development out to age 25 and the higher risk of addiction in people who are younger.
Q: What do you think the future will hold on the cannabis front?
A: Now that it’s less of a taboo topic, there’s an ever-growing number of trials each year, including very high-quality studies.
Pharmaceutically produced, cannabis-based medicines will be a growing area. Epidiolex is a perfect example of that.
It’s important for physicians to know that the way cannabis-based medicine is produced by a pharmaceutical company is different in so many levels than the cannabis in states with recreational and medical marijuana.
Q: What are some ways that the pharmaceutical products are different?
A: The rigor of the production process, the standardization, the purity, the correct labeling and expiration dates. Plus, the lack of the use of pesticides and other contaminants. And they’re distributed by pharmacists.
Q: What should neurologists be thinking if they’re considering whether to recommend cannabis to their patients?
A: This is a very complex topic, and it’s not something that most of us have training in. You can’t sit down for 1 or 2 hours, get up to speed, and have your own well-informed opinion on it. You really need to put more time and effort.
Q: What are some issues that neurologists should consider?
A: You really need to find out what your state is doing about it and see how you feel about that.
How is your state administering medical and/or recreational marijuana? The administration of these programs is extremely different from state to state. Do these details satisfy you, and are you content having your patients interface with these programs?
Dr. Bowling reports no relevant disclosures.
Painless Ulcer on the Areola
The Diagnosis: Primary Syphilitic Chancre of the Nipple
Because laboratory investigation was negative, a primary syphilitic chancre was suspected based on clinical findings, which was confirmed by a positive rapid plasma reagin with a titer of 1:32 and a positive Treponema pallidum particle agglutination assay. Results were negative for human immunodeficiency virus. On further inquiry, the patient acknowledged that the right areola had been traumatized during sexual activity with his regular male partner 1 month prior. In the last year he reported having had 5 different male partners. He was treated with a single dose of 2.4 million IU of intramuscular benzathine penicillin. Screening for other sexually transmitted infections revealed concomitant gonococcal infection of the pharynx and chlamydia proctitis, both of which were subsequently treated. On follow-up 2 weeks after presentation the ulcer had resolved, and he currently is undergoing serial rapid plasma reagin titer monitoring.
Primary syphilitic chancres can occur at any mucocutaneous site of inoculation, most frequently on the genitalia.1 Classically, after an incubation period of 9 to 90 days, a painless indurated ulcer forms2 and heals spontaneously after 3 to 6 weeks if left untreated.3 Chancres at extragenital sites are uncommon, occurring in approximately 2% of patients with primary syphilis.1 Of them, common sites include the lips and mouth (40%-70%),4 with areolar involvement rarely being reported. A PubMed search of articles indexed for MEDLINE using the terms nipple and chancre revealed 9 case reports in the English-language literature, with the first 2 cases being reported by Lee et al5 in 2006. The characteristics of these cases and our patient are summarized in the Table.5-12
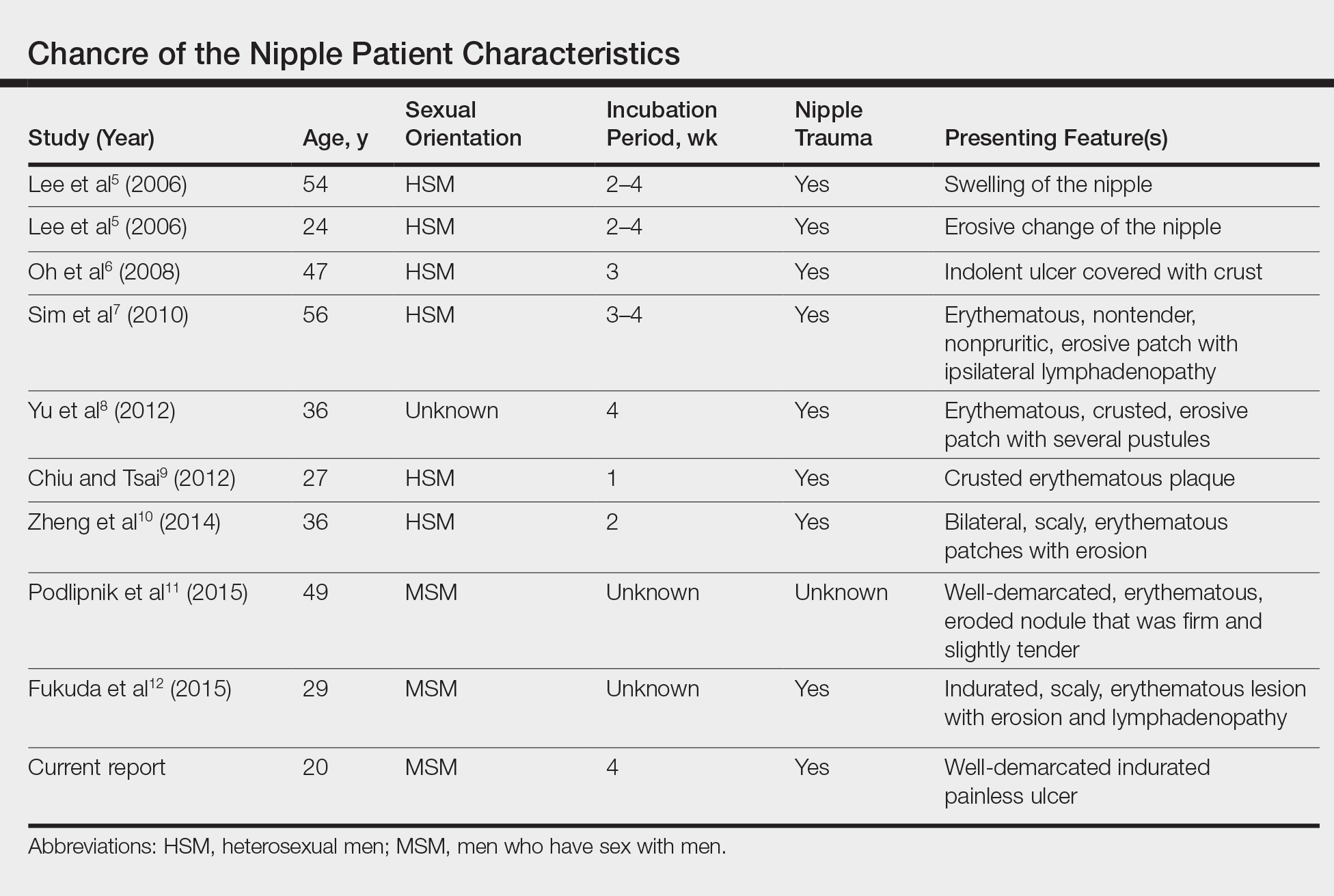
Oral contact or traumatization of the nipple by the patient's sexual partner was reported in all but one of these cases5-10,12; trauma was unknown in one case.11 Our patient reported a similar history of trauma to the nipple. It is known that transmission of syphilis can take place via kissing or oral contact, and it has been asserted that oral syphilitic lesions are highly infectious.13 Syphilis also can be transmitted by an already infected sexual partner sustaining minor trauma at the oral mucosa, allowing Treponema pallidum from the bloodstream to be inoculated onto the nipple. Another explanation for transmission could be the Koebner phenomenon, whereby trauma at the nipple of an already infected patient could lead to the formation of a chancre.6,8
The differential diagnosis includes erosive adenomatosis of the nipple, nipple eczema, Paget disease of the breast, and ulcerated basal cell carcinoma. Erosive adenomatosis of the nipple is a benign tumor of unilateral involvement that presents as an asymptomatic eroded/ulcerated papule. Clinically, it is similar to Paget disease of the breast. Eczema of the nipple usually is associated with pruritus and epidermal changes such as scaling.7,8 Paget disease of the breast arises from the extension of breast ductal carcinoma in situ onto the skin overlying the nipple. It can present as a unilateral nipple plaque with ulceration and bloody discharge. The diagnoses of erosive adenomatosis and Paget disease are confirmed with histologic examination. Basal cell carcinoma is the most common nonmelanoma skin cancer and can present as an ulcerated plaque, often with rolled borders, pearly edges, and overlying telangiectasia. It is known to be locally invasive. A punch biopsy and histopathologic examination would confirm the diagnosis of basal cell carcinoma.14
Extragenital chancres, especially those occurring at unusual sites, are uncommon. Therefore, a high index of suspicion is required to diagnose and initiate appropriate treatment for these patients.
- Mindel A, Tovey SJ, Timmins DJ, et al. Primary and secondary syphilis, 20 years' experience. 2. clinical features. Genitourin Med. 1989;65:1-3.
- Goh B. Syphilis in adults. Sex Transm Infect. 2005;81:448-452.
- Katz KA. Syphilis. In: Goldsmith LA, Katz SI, Gilchrest BA, eds. Fitzpatrick's Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill Medical; 2012:2471-2492.
- Singh AE, Romanowski B. Syphilis: review with emphasis on clinical, epidemiologic, and some biologic features. Clin Microbiol Rev. 1999;12:187-209.
- Lee JY, Lin MH, Jung YC. Extragenital syphilitic chancre manifesting as a solitary nodule of the nipple. J Eur Acad Dermatol Venereol. 2006;20:886-887.
- Oh Y, Ahn S, Hong SP, et al. A case of extragenital chancre on a nipple from a human bite during sexual intercourse. Int J Dermatol. 2008;47:978-980.
- Sim JH, Lee MG, In SI, et al. Erythematous erosive patch on the left nipple--quiz case. diagnosis: extragenital syphilitic chancres. Arch Dermatol. 2010;146:81-86.
- Yu M, Lee HR, Han TY, et al. A solitary erosive patch on the left nipple. extragenital syphilitic chancres. Int J Dermatol. 2012;51:27-28.
- Chiu HY, Tsai TF. A crusted plaque on the right nipple. JAMA. 2012;308:403-404.
- Zheng S, Liu J, Xu XG, et al. Primary syphilis presenting as bilateral nipple-areola eczematoid lesions. Acta Derm Venereol. 2014;94:617-618.
- Podlipnik S, Giavedoni P, Alsina M, et al. An erythematous nodule on the nipple: an unusual presentation of primary syphilis. J Cutan Pathol. 2015;42:239-243.
- Fukuda H, Takahashi M, Kato K, et al. Multiple primary syphilis on the lip, nipple-areola and penis: an immunohistochemical examination of Treponema pallidum localization using an anti-T. pallidum antibody. J Dermatol. 2015;42:515-517.
- Yu X, Zheng H. Syphilitic chancre of the lips transmitted by kissing. Medicine (Baltimore). 2016;95:E3303.
- Carucci JA, Leffell DJ, Pettersen JS. Basal cell carcinoma. In: Goldsmith LA, Katz SI, Gilchrest BA, eds. Fitzpatrick's Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill Medical; 2012:1294-1303.
The Diagnosis: Primary Syphilitic Chancre of the Nipple
Because laboratory investigation was negative, a primary syphilitic chancre was suspected based on clinical findings, which was confirmed by a positive rapid plasma reagin with a titer of 1:32 and a positive Treponema pallidum particle agglutination assay. Results were negative for human immunodeficiency virus. On further inquiry, the patient acknowledged that the right areola had been traumatized during sexual activity with his regular male partner 1 month prior. In the last year he reported having had 5 different male partners. He was treated with a single dose of 2.4 million IU of intramuscular benzathine penicillin. Screening for other sexually transmitted infections revealed concomitant gonococcal infection of the pharynx and chlamydia proctitis, both of which were subsequently treated. On follow-up 2 weeks after presentation the ulcer had resolved, and he currently is undergoing serial rapid plasma reagin titer monitoring.
Primary syphilitic chancres can occur at any mucocutaneous site of inoculation, most frequently on the genitalia.1 Classically, after an incubation period of 9 to 90 days, a painless indurated ulcer forms2 and heals spontaneously after 3 to 6 weeks if left untreated.3 Chancres at extragenital sites are uncommon, occurring in approximately 2% of patients with primary syphilis.1 Of them, common sites include the lips and mouth (40%-70%),4 with areolar involvement rarely being reported. A PubMed search of articles indexed for MEDLINE using the terms nipple and chancre revealed 9 case reports in the English-language literature, with the first 2 cases being reported by Lee et al5 in 2006. The characteristics of these cases and our patient are summarized in the Table.5-12
Oral contact or traumatization of the nipple by the patient's sexual partner was reported in all but one of these cases5-10,12; trauma was unknown in one case.11 Our patient reported a similar history of trauma to the nipple. It is known that transmission of syphilis can take place via kissing or oral contact, and it has been asserted that oral syphilitic lesions are highly infectious.13 Syphilis also can be transmitted by an already infected sexual partner sustaining minor trauma at the oral mucosa, allowing Treponema pallidum from the bloodstream to be inoculated onto the nipple. Another explanation for transmission could be the Koebner phenomenon, whereby trauma at the nipple of an already infected patient could lead to the formation of a chancre.6,8
The differential diagnosis includes erosive adenomatosis of the nipple, nipple eczema, Paget disease of the breast, and ulcerated basal cell carcinoma. Erosive adenomatosis of the nipple is a benign tumor of unilateral involvement that presents as an asymptomatic eroded/ulcerated papule. Clinically, it is similar to Paget disease of the breast. Eczema of the nipple usually is associated with pruritus and epidermal changes such as scaling.7,8 Paget disease of the breast arises from the extension of breast ductal carcinoma in situ onto the skin overlying the nipple. It can present as a unilateral nipple plaque with ulceration and bloody discharge. The diagnoses of erosive adenomatosis and Paget disease are confirmed with histologic examination. Basal cell carcinoma is the most common nonmelanoma skin cancer and can present as an ulcerated plaque, often with rolled borders, pearly edges, and overlying telangiectasia. It is known to be locally invasive. A punch biopsy and histopathologic examination would confirm the diagnosis of basal cell carcinoma.14
Extragenital chancres, especially those occurring at unusual sites, are uncommon. Therefore, a high index of suspicion is required to diagnose and initiate appropriate treatment for these patients.
The Diagnosis: Primary Syphilitic Chancre of the Nipple
Because laboratory investigation was negative, a primary syphilitic chancre was suspected based on clinical findings, which was confirmed by a positive rapid plasma reagin with a titer of 1:32 and a positive Treponema pallidum particle agglutination assay. Results were negative for human immunodeficiency virus. On further inquiry, the patient acknowledged that the right areola had been traumatized during sexual activity with his regular male partner 1 month prior. In the last year he reported having had 5 different male partners. He was treated with a single dose of 2.4 million IU of intramuscular benzathine penicillin. Screening for other sexually transmitted infections revealed concomitant gonococcal infection of the pharynx and chlamydia proctitis, both of which were subsequently treated. On follow-up 2 weeks after presentation the ulcer had resolved, and he currently is undergoing serial rapid plasma reagin titer monitoring.
Primary syphilitic chancres can occur at any mucocutaneous site of inoculation, most frequently on the genitalia.1 Classically, after an incubation period of 9 to 90 days, a painless indurated ulcer forms2 and heals spontaneously after 3 to 6 weeks if left untreated.3 Chancres at extragenital sites are uncommon, occurring in approximately 2% of patients with primary syphilis.1 Of them, common sites include the lips and mouth (40%-70%),4 with areolar involvement rarely being reported. A PubMed search of articles indexed for MEDLINE using the terms nipple and chancre revealed 9 case reports in the English-language literature, with the first 2 cases being reported by Lee et al5 in 2006. The characteristics of these cases and our patient are summarized in the Table.5-12
Oral contact or traumatization of the nipple by the patient's sexual partner was reported in all but one of these cases5-10,12; trauma was unknown in one case.11 Our patient reported a similar history of trauma to the nipple. It is known that transmission of syphilis can take place via kissing or oral contact, and it has been asserted that oral syphilitic lesions are highly infectious.13 Syphilis also can be transmitted by an already infected sexual partner sustaining minor trauma at the oral mucosa, allowing Treponema pallidum from the bloodstream to be inoculated onto the nipple. Another explanation for transmission could be the Koebner phenomenon, whereby trauma at the nipple of an already infected patient could lead to the formation of a chancre.6,8
The differential diagnosis includes erosive adenomatosis of the nipple, nipple eczema, Paget disease of the breast, and ulcerated basal cell carcinoma. Erosive adenomatosis of the nipple is a benign tumor of unilateral involvement that presents as an asymptomatic eroded/ulcerated papule. Clinically, it is similar to Paget disease of the breast. Eczema of the nipple usually is associated with pruritus and epidermal changes such as scaling.7,8 Paget disease of the breast arises from the extension of breast ductal carcinoma in situ onto the skin overlying the nipple. It can present as a unilateral nipple plaque with ulceration and bloody discharge. The diagnoses of erosive adenomatosis and Paget disease are confirmed with histologic examination. Basal cell carcinoma is the most common nonmelanoma skin cancer and can present as an ulcerated plaque, often with rolled borders, pearly edges, and overlying telangiectasia. It is known to be locally invasive. A punch biopsy and histopathologic examination would confirm the diagnosis of basal cell carcinoma.14
Extragenital chancres, especially those occurring at unusual sites, are uncommon. Therefore, a high index of suspicion is required to diagnose and initiate appropriate treatment for these patients.
- Mindel A, Tovey SJ, Timmins DJ, et al. Primary and secondary syphilis, 20 years' experience. 2. clinical features. Genitourin Med. 1989;65:1-3.
- Goh B. Syphilis in adults. Sex Transm Infect. 2005;81:448-452.
- Katz KA. Syphilis. In: Goldsmith LA, Katz SI, Gilchrest BA, eds. Fitzpatrick's Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill Medical; 2012:2471-2492.
- Singh AE, Romanowski B. Syphilis: review with emphasis on clinical, epidemiologic, and some biologic features. Clin Microbiol Rev. 1999;12:187-209.
- Lee JY, Lin MH, Jung YC. Extragenital syphilitic chancre manifesting as a solitary nodule of the nipple. J Eur Acad Dermatol Venereol. 2006;20:886-887.
- Oh Y, Ahn S, Hong SP, et al. A case of extragenital chancre on a nipple from a human bite during sexual intercourse. Int J Dermatol. 2008;47:978-980.
- Sim JH, Lee MG, In SI, et al. Erythematous erosive patch on the left nipple--quiz case. diagnosis: extragenital syphilitic chancres. Arch Dermatol. 2010;146:81-86.
- Yu M, Lee HR, Han TY, et al. A solitary erosive patch on the left nipple. extragenital syphilitic chancres. Int J Dermatol. 2012;51:27-28.
- Chiu HY, Tsai TF. A crusted plaque on the right nipple. JAMA. 2012;308:403-404.
- Zheng S, Liu J, Xu XG, et al. Primary syphilis presenting as bilateral nipple-areola eczematoid lesions. Acta Derm Venereol. 2014;94:617-618.
- Podlipnik S, Giavedoni P, Alsina M, et al. An erythematous nodule on the nipple: an unusual presentation of primary syphilis. J Cutan Pathol. 2015;42:239-243.
- Fukuda H, Takahashi M, Kato K, et al. Multiple primary syphilis on the lip, nipple-areola and penis: an immunohistochemical examination of Treponema pallidum localization using an anti-T. pallidum antibody. J Dermatol. 2015;42:515-517.
- Yu X, Zheng H. Syphilitic chancre of the lips transmitted by kissing. Medicine (Baltimore). 2016;95:E3303.
- Carucci JA, Leffell DJ, Pettersen JS. Basal cell carcinoma. In: Goldsmith LA, Katz SI, Gilchrest BA, eds. Fitzpatrick's Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill Medical; 2012:1294-1303.
- Mindel A, Tovey SJ, Timmins DJ, et al. Primary and secondary syphilis, 20 years' experience. 2. clinical features. Genitourin Med. 1989;65:1-3.
- Goh B. Syphilis in adults. Sex Transm Infect. 2005;81:448-452.
- Katz KA. Syphilis. In: Goldsmith LA, Katz SI, Gilchrest BA, eds. Fitzpatrick's Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill Medical; 2012:2471-2492.
- Singh AE, Romanowski B. Syphilis: review with emphasis on clinical, epidemiologic, and some biologic features. Clin Microbiol Rev. 1999;12:187-209.
- Lee JY, Lin MH, Jung YC. Extragenital syphilitic chancre manifesting as a solitary nodule of the nipple. J Eur Acad Dermatol Venereol. 2006;20:886-887.
- Oh Y, Ahn S, Hong SP, et al. A case of extragenital chancre on a nipple from a human bite during sexual intercourse. Int J Dermatol. 2008;47:978-980.
- Sim JH, Lee MG, In SI, et al. Erythematous erosive patch on the left nipple--quiz case. diagnosis: extragenital syphilitic chancres. Arch Dermatol. 2010;146:81-86.
- Yu M, Lee HR, Han TY, et al. A solitary erosive patch on the left nipple. extragenital syphilitic chancres. Int J Dermatol. 2012;51:27-28.
- Chiu HY, Tsai TF. A crusted plaque on the right nipple. JAMA. 2012;308:403-404.
- Zheng S, Liu J, Xu XG, et al. Primary syphilis presenting as bilateral nipple-areola eczematoid lesions. Acta Derm Venereol. 2014;94:617-618.
- Podlipnik S, Giavedoni P, Alsina M, et al. An erythematous nodule on the nipple: an unusual presentation of primary syphilis. J Cutan Pathol. 2015;42:239-243.
- Fukuda H, Takahashi M, Kato K, et al. Multiple primary syphilis on the lip, nipple-areola and penis: an immunohistochemical examination of Treponema pallidum localization using an anti-T. pallidum antibody. J Dermatol. 2015;42:515-517.
- Yu X, Zheng H. Syphilitic chancre of the lips transmitted by kissing. Medicine (Baltimore). 2016;95:E3303.
- Carucci JA, Leffell DJ, Pettersen JS. Basal cell carcinoma. In: Goldsmith LA, Katz SI, Gilchrest BA, eds. Fitzpatrick's Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill Medical; 2012:1294-1303.
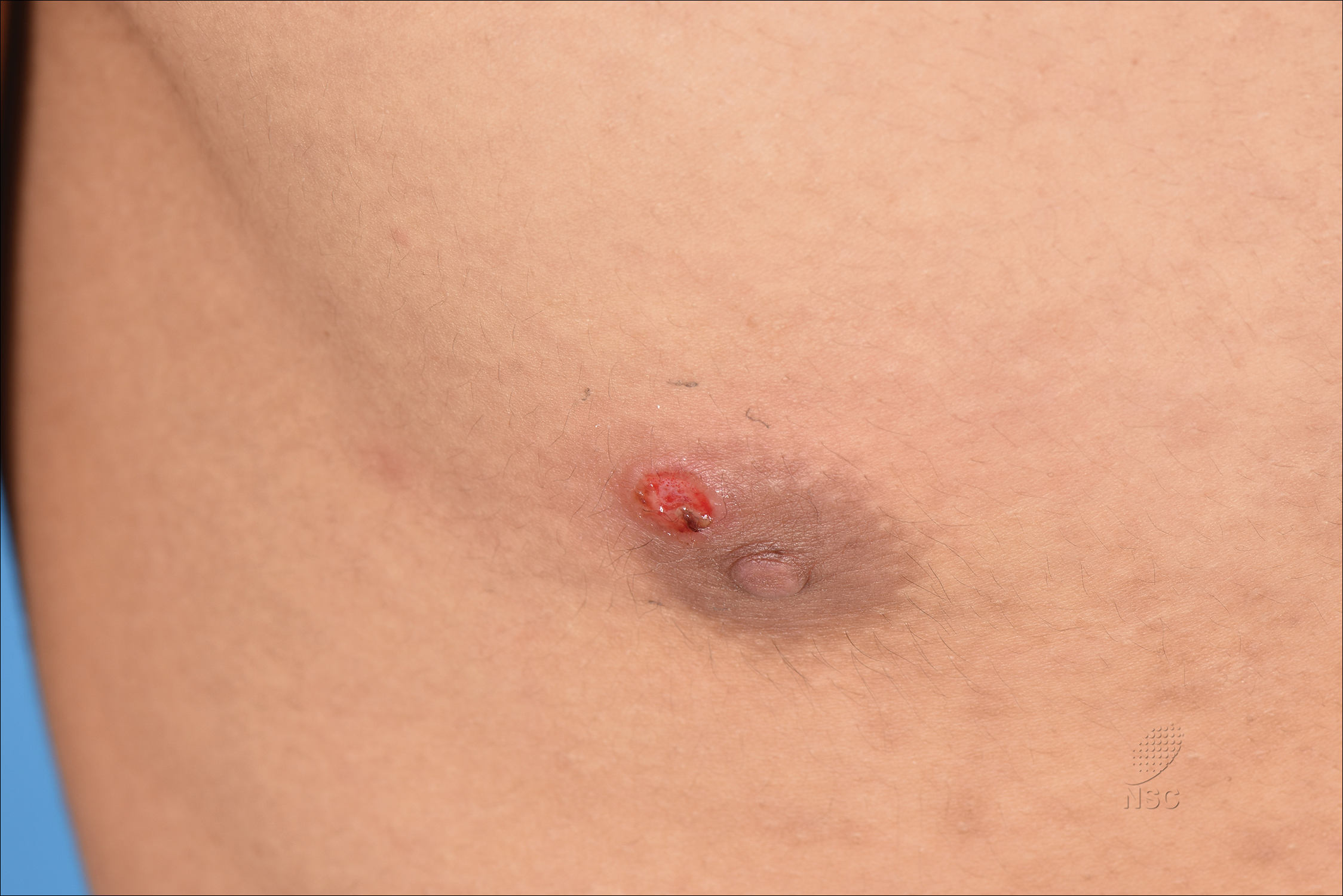
A previously healthy 20-year-old Chinese man presented to our dermatology outpatient clinic with a solitary painless ulcer on the right areola of 1 week's duration. Examination showed a small, slightly indurated ulcer with well-defined borders. No lesions were noted elsewhere. Swabs for pyogenic culture and herpes simplex virus polymerase chain reaction tests were sent, and he was treated empirically with oral cephalexin and tetracycline ointment 3%. At 1-week follow-up the ulcer had dried up and begun to heal, and results from the laboratory investigations were negative.
New insights into sleep, pregnancy weight gain
BALTIMORE – Pregnant women who are overweight and obese are like the general population in that the less they sleep, the more weight they gain, particularly in the first half of pregnancy. However, unlike in the larger adult population, prolonged daily total eating time was not associated with gestational weight gain in these women, particularly early in pregnancy, according to findings from a small study presented at the Associated Professional Sleep Societies annual meeting.
Those findings point to a need to further study the , said Rachel P. Kolko, PhD, a postdoctoral scholar at the University of Pittsburgh, Western Psychiatric Institute and Clinic.
“The association with total sleep time was found to be significant, such that if you had less sleep, you had higher amounts of weight gain; we did not find a significant relation with our eating window variable,” Dr. Kolko said.
She reported on research involving 62 pregnant women, 53% of whom were overweight with a body mass index of 25-29.9 kg/m2 and 47% of whom were obese with BMI greater than 30. Forty-seven percent of the study population was nonwhite.
The research grew out of a need to identify potentially modifiable factors to curtail excessive gestational weight gain during pregnancy, she said. The study hypotheses were that both shorter total sleep time and longer total eating time would lead to higher gestational weight gain, but the study confirmed only the former as a contributing factor.

The women in the study were at 12-20 weeks of pregnancy. Gestational weight gain was calculated as the difference between self-reported prepregnancy weight and current weight. Total sleep time was based on the Pittsburgh Sleep Quality Index, and total eating time was calculated as the time difference between the day’s first meal or snack of more than 50 calories and the last, as self-reported.
Average total sleep time was 7.8 hours, with total eating time spanning 10.8 hours. On average, study participants gained 9.7 pounds through the first half of pregnancy, Dr. Kolko said. She noted that the Institute of Medicine, now known as the National Academy of Medicine, recommends that women who are overweight women gain 15-25 pounds during pregnancy and women who are obese gain 11-20 pounds (JAMA. 2017;317:2207-25). “Already about 20% of our sample has gained that amount of weight within the first half of pregnancy,” she said.
“Total sleep time was related to a higher early gestational weight in women with overweight and obesity, and it’s possible that addressing this may affect and hopefully improve women’s weight gain during early pregnancy to fit within those guidelines,” she said.
Future research should look at the entire gestational period – possibly targeting sleep patterns during pregnancy – and should expand to include women who are not overweight or obese, Dr. Kolko noted.
Dr. Kolko reported having no financial relationships to disclose.
SOURCE: Kolko RP, et al., SLEEP 2018, Abstract 0692.
BALTIMORE – Pregnant women who are overweight and obese are like the general population in that the less they sleep, the more weight they gain, particularly in the first half of pregnancy. However, unlike in the larger adult population, prolonged daily total eating time was not associated with gestational weight gain in these women, particularly early in pregnancy, according to findings from a small study presented at the Associated Professional Sleep Societies annual meeting.
Those findings point to a need to further study the , said Rachel P. Kolko, PhD, a postdoctoral scholar at the University of Pittsburgh, Western Psychiatric Institute and Clinic.
“The association with total sleep time was found to be significant, such that if you had less sleep, you had higher amounts of weight gain; we did not find a significant relation with our eating window variable,” Dr. Kolko said.
She reported on research involving 62 pregnant women, 53% of whom were overweight with a body mass index of 25-29.9 kg/m2 and 47% of whom were obese with BMI greater than 30. Forty-seven percent of the study population was nonwhite.
The research grew out of a need to identify potentially modifiable factors to curtail excessive gestational weight gain during pregnancy, she said. The study hypotheses were that both shorter total sleep time and longer total eating time would lead to higher gestational weight gain, but the study confirmed only the former as a contributing factor.
The women in the study were at 12-20 weeks of pregnancy. Gestational weight gain was calculated as the difference between self-reported prepregnancy weight and current weight. Total sleep time was based on the Pittsburgh Sleep Quality Index, and total eating time was calculated as the time difference between the day’s first meal or snack of more than 50 calories and the last, as self-reported.
Average total sleep time was 7.8 hours, with total eating time spanning 10.8 hours. On average, study participants gained 9.7 pounds through the first half of pregnancy, Dr. Kolko said. She noted that the Institute of Medicine, now known as the National Academy of Medicine, recommends that women who are overweight women gain 15-25 pounds during pregnancy and women who are obese gain 11-20 pounds (JAMA. 2017;317:2207-25). “Already about 20% of our sample has gained that amount of weight within the first half of pregnancy,” she said.
“Total sleep time was related to a higher early gestational weight in women with overweight and obesity, and it’s possible that addressing this may affect and hopefully improve women’s weight gain during early pregnancy to fit within those guidelines,” she said.
Future research should look at the entire gestational period – possibly targeting sleep patterns during pregnancy – and should expand to include women who are not overweight or obese, Dr. Kolko noted.
Dr. Kolko reported having no financial relationships to disclose.
SOURCE: Kolko RP, et al., SLEEP 2018, Abstract 0692.
BALTIMORE – Pregnant women who are overweight and obese are like the general population in that the less they sleep, the more weight they gain, particularly in the first half of pregnancy. However, unlike in the larger adult population, prolonged daily total eating time was not associated with gestational weight gain in these women, particularly early in pregnancy, according to findings from a small study presented at the Associated Professional Sleep Societies annual meeting.
Those findings point to a need to further study the , said Rachel P. Kolko, PhD, a postdoctoral scholar at the University of Pittsburgh, Western Psychiatric Institute and Clinic.
“The association with total sleep time was found to be significant, such that if you had less sleep, you had higher amounts of weight gain; we did not find a significant relation with our eating window variable,” Dr. Kolko said.
She reported on research involving 62 pregnant women, 53% of whom were overweight with a body mass index of 25-29.9 kg/m2 and 47% of whom were obese with BMI greater than 30. Forty-seven percent of the study population was nonwhite.
The research grew out of a need to identify potentially modifiable factors to curtail excessive gestational weight gain during pregnancy, she said. The study hypotheses were that both shorter total sleep time and longer total eating time would lead to higher gestational weight gain, but the study confirmed only the former as a contributing factor.
The women in the study were at 12-20 weeks of pregnancy. Gestational weight gain was calculated as the difference between self-reported prepregnancy weight and current weight. Total sleep time was based on the Pittsburgh Sleep Quality Index, and total eating time was calculated as the time difference between the day’s first meal or snack of more than 50 calories and the last, as self-reported.
Average total sleep time was 7.8 hours, with total eating time spanning 10.8 hours. On average, study participants gained 9.7 pounds through the first half of pregnancy, Dr. Kolko said. She noted that the Institute of Medicine, now known as the National Academy of Medicine, recommends that women who are overweight women gain 15-25 pounds during pregnancy and women who are obese gain 11-20 pounds (JAMA. 2017;317:2207-25). “Already about 20% of our sample has gained that amount of weight within the first half of pregnancy,” she said.
“Total sleep time was related to a higher early gestational weight in women with overweight and obesity, and it’s possible that addressing this may affect and hopefully improve women’s weight gain during early pregnancy to fit within those guidelines,” she said.
Future research should look at the entire gestational period – possibly targeting sleep patterns during pregnancy – and should expand to include women who are not overweight or obese, Dr. Kolko noted.
Dr. Kolko reported having no financial relationships to disclose.
SOURCE: Kolko RP, et al., SLEEP 2018, Abstract 0692.
REPORTING FROM SLEEP 2018
Key clinical point: In overweight/obese women, shorter sleep times are linked to early gestational weight gain.
Major finding: Overweight/obese women slept 30% less and had higher gestational weight gain in early pregnancy.
Study details: Study of 62 women between 12 and 20 weeks’ gestation with prepregnancy BMI greater than 25 kg/m2.
Disclosures: Dr. Kolko reported having no financial relationships.
Source: Kolko RP et al. SLEEP 2018, Abstract 0692.
Pediatric dermatology admissions: Crunching the numbers
according to data from the Agency for Healthcare Research and Quality.
There were 74,229 such admissions in the United States that year – all others totaled 1.77 million – and the children at the highest risk for dermatology hospitalization were those living in communities with the lowest household incomes, the uninsured and those on Medicaid, and those living in the South, Justin D. Arnold of George Washington University, Washington, and his associates said in Pediatric Dermatology.
“Individuals from communities of low socioeconomic status may be more likely to be hospitalized because of gaps in insurance coverage, difficulty with transportation, or inconsistent access to preventative medical care, which for skin disease, would include access to an outpatient pediatric dermatologist,” they wrote.
All those admissions for skin diseases cost the health care system $379.8 million in 2012, or 1.9% of the $20.3 billion spent on all pediatric hospitalizations, excluding those related to pregnancy or childbirth. The mean cost of a skin disease admission was $5,211 for a child aged less than 18 years, compared with $11,409 for nondermatology admissions, according to data from the 2012 Kids’ Inpatient Database, which includes records of pediatric discharges from 44 states.
Cutaneous lymphoma was the most expensive skin disease per admission at a mean cost of $58,294, with congenital skin abnormalities second at $24,186, and ulcers third at $17,064. Bacterial skin infections and infestations were only the 19th most expensive admission at $4,135, but it was by far the most common (59,115 admissions) and the most expensive overall, with a total cost of $240 million. The second most common condition was viral diseases with 3,812 admissions and the next most expensive total was $33.5 million for connective tissue disorders, Mr. Arnold and his associates said.
Multivariate models that adjusted for such factors as age, sex, and race revealed that “the risk of hospitalization for skin disease increased as the median income of one’s zip code declined,” the investigators noted. The adjusted odds ratio for hospitalization in the lowest-income quartile (less than $39,000) was 1.22, compared with the highest-income quartile.
Insurance status also affected hospitalization, putting children from families with no insurance (aOR, 1.35) and those on Medicaid (aOR, 1.17) at a disadvantage, compared with those who had private insurance. “Policy makers should consider increasing Medicaid reimbursement rates to outpatient dermatologists, which might encourage more clinicians to accept this form of insurance and thereby expand access to preventative skin care,” they said.
Regional differences also were observed, which put children from the southern states at the highest risk (aOR, 1.32), compared with those in the West, which could be related to access issues. “In 2016, 4 of the 10 communities in the United States with the lowest density of dermatologists were in the South, suggesting that the high rate of hospitalizations there may also be partially attributed to lack of access to dermatologists,” Mr. Arnold and his associates wrote.
The investigators did not report funding or disclose conflicts of interest.
SOURCE: Arnold JD et al. Pediatr Dermatol. 2018 Jul 1. doi: 10.1111/pde.13549.
according to data from the Agency for Healthcare Research and Quality.
There were 74,229 such admissions in the United States that year – all others totaled 1.77 million – and the children at the highest risk for dermatology hospitalization were those living in communities with the lowest household incomes, the uninsured and those on Medicaid, and those living in the South, Justin D. Arnold of George Washington University, Washington, and his associates said in Pediatric Dermatology.
“Individuals from communities of low socioeconomic status may be more likely to be hospitalized because of gaps in insurance coverage, difficulty with transportation, or inconsistent access to preventative medical care, which for skin disease, would include access to an outpatient pediatric dermatologist,” they wrote.
All those admissions for skin diseases cost the health care system $379.8 million in 2012, or 1.9% of the $20.3 billion spent on all pediatric hospitalizations, excluding those related to pregnancy or childbirth. The mean cost of a skin disease admission was $5,211 for a child aged less than 18 years, compared with $11,409 for nondermatology admissions, according to data from the 2012 Kids’ Inpatient Database, which includes records of pediatric discharges from 44 states.
Cutaneous lymphoma was the most expensive skin disease per admission at a mean cost of $58,294, with congenital skin abnormalities second at $24,186, and ulcers third at $17,064. Bacterial skin infections and infestations were only the 19th most expensive admission at $4,135, but it was by far the most common (59,115 admissions) and the most expensive overall, with a total cost of $240 million. The second most common condition was viral diseases with 3,812 admissions and the next most expensive total was $33.5 million for connective tissue disorders, Mr. Arnold and his associates said.
Multivariate models that adjusted for such factors as age, sex, and race revealed that “the risk of hospitalization for skin disease increased as the median income of one’s zip code declined,” the investigators noted. The adjusted odds ratio for hospitalization in the lowest-income quartile (less than $39,000) was 1.22, compared with the highest-income quartile.
Insurance status also affected hospitalization, putting children from families with no insurance (aOR, 1.35) and those on Medicaid (aOR, 1.17) at a disadvantage, compared with those who had private insurance. “Policy makers should consider increasing Medicaid reimbursement rates to outpatient dermatologists, which might encourage more clinicians to accept this form of insurance and thereby expand access to preventative skin care,” they said.
Regional differences also were observed, which put children from the southern states at the highest risk (aOR, 1.32), compared with those in the West, which could be related to access issues. “In 2016, 4 of the 10 communities in the United States with the lowest density of dermatologists were in the South, suggesting that the high rate of hospitalizations there may also be partially attributed to lack of access to dermatologists,” Mr. Arnold and his associates wrote.
The investigators did not report funding or disclose conflicts of interest.
SOURCE: Arnold JD et al. Pediatr Dermatol. 2018 Jul 1. doi: 10.1111/pde.13549.
according to data from the Agency for Healthcare Research and Quality.
There were 74,229 such admissions in the United States that year – all others totaled 1.77 million – and the children at the highest risk for dermatology hospitalization were those living in communities with the lowest household incomes, the uninsured and those on Medicaid, and those living in the South, Justin D. Arnold of George Washington University, Washington, and his associates said in Pediatric Dermatology.
“Individuals from communities of low socioeconomic status may be more likely to be hospitalized because of gaps in insurance coverage, difficulty with transportation, or inconsistent access to preventative medical care, which for skin disease, would include access to an outpatient pediatric dermatologist,” they wrote.
All those admissions for skin diseases cost the health care system $379.8 million in 2012, or 1.9% of the $20.3 billion spent on all pediatric hospitalizations, excluding those related to pregnancy or childbirth. The mean cost of a skin disease admission was $5,211 for a child aged less than 18 years, compared with $11,409 for nondermatology admissions, according to data from the 2012 Kids’ Inpatient Database, which includes records of pediatric discharges from 44 states.
Cutaneous lymphoma was the most expensive skin disease per admission at a mean cost of $58,294, with congenital skin abnormalities second at $24,186, and ulcers third at $17,064. Bacterial skin infections and infestations were only the 19th most expensive admission at $4,135, but it was by far the most common (59,115 admissions) and the most expensive overall, with a total cost of $240 million. The second most common condition was viral diseases with 3,812 admissions and the next most expensive total was $33.5 million for connective tissue disorders, Mr. Arnold and his associates said.
Multivariate models that adjusted for such factors as age, sex, and race revealed that “the risk of hospitalization for skin disease increased as the median income of one’s zip code declined,” the investigators noted. The adjusted odds ratio for hospitalization in the lowest-income quartile (less than $39,000) was 1.22, compared with the highest-income quartile.
Insurance status also affected hospitalization, putting children from families with no insurance (aOR, 1.35) and those on Medicaid (aOR, 1.17) at a disadvantage, compared with those who had private insurance. “Policy makers should consider increasing Medicaid reimbursement rates to outpatient dermatologists, which might encourage more clinicians to accept this form of insurance and thereby expand access to preventative skin care,” they said.
Regional differences also were observed, which put children from the southern states at the highest risk (aOR, 1.32), compared with those in the West, which could be related to access issues. “In 2016, 4 of the 10 communities in the United States with the lowest density of dermatologists were in the South, suggesting that the high rate of hospitalizations there may also be partially attributed to lack of access to dermatologists,” Mr. Arnold and his associates wrote.
The investigators did not report funding or disclose conflicts of interest.
SOURCE: Arnold JD et al. Pediatr Dermatol. 2018 Jul 1. doi: 10.1111/pde.13549.
FROM PEDIATRIC DERMATOLOGY
Key clinical point: Children at the highest risk for dermatology hospitalization lived in communities with the lowest household incomes, were uninsured or on Medicaid, or lived in the South.
Major finding: Admissions for skin diseases cost $379.8 million in 2012, or 1.9% of all spending on pediatric hospitalizations.
Study details: A statistical analysis of the 2012 Kids’ Inpatient Database.
Disclosures: The investigators did not report funding or disclose conflicts of interest.
Source: Arnold JD et al. Pediatr Dermatol. 2018 Jul 1. doi: 10.1111/pde.13549.
Family separations and the intergenerational transmission of trauma
Editor’s Note: Alison M. Heru, MD, the Families in Psychiatry columnist, invited Dr. Reinstein to address this topic.
Growing up, I was always intrigued by the strong emotions that even the mildest separation evoked within me. As a psychiatrist, I now believe that these emotions were related to my family’s difficulty with separation, a concept likely transmitted from my grandmother’s sudden separation as a child.
The political circumstances of the “Kindertransport” and the recent family separation at the southern U.S. border differ, but the Kindertransport is a model for studying the effects of forced parent-child separation and its intergenerational transmission. As a rescue operation that took place immediately before World War II, the Kindertransport was the emigration of approximately 10,000 German Jewish children from Germany and Nazi-occupied countries to England. Even though they were not literally forced, the parents involved were compelled to separate from their children to give their children a chance to survive.

In a recent letter1 published in The New York Times, Eva Yachnes, herself a Kindertransport participant, reflected on the current situation at the southern U.S. border. She alluded to the lifelong effects of her own separation at the age of 6 when emigrating from Vienna to Germany. Personally, as a granddaughter of a Kindertransport participant, I am particularly concerned about the intergenerational transmission of the trauma of family separation.
Since early May 2018, more than 2,000 children have been forcibly separated from their parents after illegally crossing the border. As part of a “zero tolerance policy,” the separation was characterized by the Trump administration as a deterrent to illegal border crossings. Although many Americans are horrified by the news reports about family separation, psychiatrists in particular have expressed concern about this trauma. The American Psychiatric Association issued a statement2 warning that “any forced separation is highly stressful for children and can cause lifelong trauma.” Several weeks later, the APA joined several other mental health organizations in a letter3 calling for the immediate end of enforcement of those policies. In that letter, the organizations said forced separations can cause “an increased risk of ... mental illnesses such as depression, anxiety, and posttraumatic stress disorder.”
What must psychiatrists understand about the impact of childhood trauma? How can psychiatrists approach treatment of children separated from their parents? Are there ways to minimize the risk for intergenerational impact of this trauma?
What do we know?
Childhood trauma is influenced by multiple factors and can be expressed in several ways. According to research,4 the age of the child at the time of traumatic event, the frequency of traumatic experiences, and the degree to which the child’s caretakers were involved in the trauma are factors that influence the extent of psychological damage. This research also suggests that childhood trauma is associated with emotional dysregulation, aggression against self and others, difficulties in attention and dissociation, medical problems, and difficulty with navigating adult interpersonal relationships.
When viewed through the lens of attachment theory, the forced separation of a child from its caretakers is a potent form of childhood trauma. Joanna E. Chambers, MD, summarizes and explains John Bowlby’s attachment theory as a neurobiological system originating from an infant’s connection to the primary caretaker. This connection becomes a lifelong model for all subsequent relationships.
Any traumatic disruptions in the development of this system puts the child at risk of developing “insecure attachment.” This insecure attachment can lead to lifelong emotional problems for the child, affecting the quality of subsequent marital relationships, relationships to children, and the development of personality disorders.
In addition, it correlates to the development of psychiatric illnesses, specifically depression and anxiety. There is also a plausible biological basis for attachment theory. Both oxytocin, a hormone released in human bonding, and social interaction itself have been shown to decrease cortisol levels. Elevated cortisol has been found to negatively affect infant brain development, Dr. Chambers argued.5
Given those significant effects of childhood trauma, it is understandable that there exists a concept of “intergenerational transmission of trauma.” Originating from studies of Holocaust survivors and their descendants, researchers Amy Lehrner, PhD, and Rachel Yehuda, PhD, conceptualize intergenerational transmission of trauma as the intergenerational impact of prenatal PTSD.6 This impact is expressed as a predisposition in the offspring of Holocaust survivors to developing PTSD, difficulties in individuation and separation, higher rates of mood and anxiety disorders, and higher rates of physical health issues.
Although they are complex and clearly multidetermined, Dr. Yehuda and Dr. Lehrner also summarize plausible biological theories for the intergenerational transmission of trauma. Epigenetic differences in the hypothalamic-pituitary-adrenal axis, circadian rhythm, urinary and plasma cortisol levels, glucocorticoid sensitivity, and regulation of the glucocorticoid receptor gene all have been found in Holocaust offspring with parental PTSD, in contrast to offspring without parental PTSD.6
What can we as psychiatrists do?
We are uniquely equipped to take several concrete steps to help mitigate the effects of these traumatic events. Among them, we can:
- Provide opportunities for the child and the family to process their experience. This can be profoundly healing and can help minimize the devastating psychological effects of this separation.
- Become acquainted with the concept of intergenerational transmission of resilience.
- Work with trauma survivors to develop their own personal narratives and cultural rituals surrounding the trauma.6
- Encourage second- and third-generation descendants to engage in artistic expression of the trauma, visit places of importance to their parents, and engage in social and political activism. These are all expressions of resilience in the offspring of trauma victims.6
In summary, recent U.S. political events have caused thousands of children to be forcibly separated from their parents. Those separations are traumatic and can have lifelong psychological implications for the children and their offspring. It is important to provide quality mental health treatment to these children with a specific focus on treating PTSD and processing the traumatic experience. Psychological treatment can help mitigate the effects of the traumatic separation and create a sense of resiliency.
References
1. New York Times. June 20, 2018. “My separation trauma.”
2. American Psychiatric Association statement, May 30, 2018.
3. Letter to the departments of Justice, Health & Human Services, and Homeland Security, June 20, 2018.
4. Child Adolesc Psychiatric Clin N Am. 2003;(12.2):293-318.
5. Psychodynamic Psychiatry. 2017 Dec;45(4):542-63.
6. Psychological Trauma: Theory, Research, Practice, and Policy. 2018 Jan;10(1):22-9.
Dr. Reinstein is a psychiatry attending at Zucker Hillside Hospital of the Northwell Health System, Glen Oaks, New York. Her clinical interests include reproductive psychiatry and family therapy, with a specific focus on maternal mental health. Dr. Reinstein completed her adult psychiatry residency training at Montefiore Hospital/Albert Einstein College of Medicine after graduating from the Albert Einstein College of Medicine and Yeshiva University with a B.A. in biology. She is one of the recipients of the 4th Annual Resident Recognition Award for Excellence in Family Oriented Care.
Editor’s Note: Alison M. Heru, MD, the Families in Psychiatry columnist, invited Dr. Reinstein to address this topic.
Growing up, I was always intrigued by the strong emotions that even the mildest separation evoked within me. As a psychiatrist, I now believe that these emotions were related to my family’s difficulty with separation, a concept likely transmitted from my grandmother’s sudden separation as a child.
The political circumstances of the “Kindertransport” and the recent family separation at the southern U.S. border differ, but the Kindertransport is a model for studying the effects of forced parent-child separation and its intergenerational transmission. As a rescue operation that took place immediately before World War II, the Kindertransport was the emigration of approximately 10,000 German Jewish children from Germany and Nazi-occupied countries to England. Even though they were not literally forced, the parents involved were compelled to separate from their children to give their children a chance to survive.
In a recent letter1 published in The New York Times, Eva Yachnes, herself a Kindertransport participant, reflected on the current situation at the southern U.S. border. She alluded to the lifelong effects of her own separation at the age of 6 when emigrating from Vienna to Germany. Personally, as a granddaughter of a Kindertransport participant, I am particularly concerned about the intergenerational transmission of the trauma of family separation.
Since early May 2018, more than 2,000 children have been forcibly separated from their parents after illegally crossing the border. As part of a “zero tolerance policy,” the separation was characterized by the Trump administration as a deterrent to illegal border crossings. Although many Americans are horrified by the news reports about family separation, psychiatrists in particular have expressed concern about this trauma. The American Psychiatric Association issued a statement2 warning that “any forced separation is highly stressful for children and can cause lifelong trauma.” Several weeks later, the APA joined several other mental health organizations in a letter3 calling for the immediate end of enforcement of those policies. In that letter, the organizations said forced separations can cause “an increased risk of ... mental illnesses such as depression, anxiety, and posttraumatic stress disorder.”
What must psychiatrists understand about the impact of childhood trauma? How can psychiatrists approach treatment of children separated from their parents? Are there ways to minimize the risk for intergenerational impact of this trauma?
What do we know?
Childhood trauma is influenced by multiple factors and can be expressed in several ways. According to research,4 the age of the child at the time of traumatic event, the frequency of traumatic experiences, and the degree to which the child’s caretakers were involved in the trauma are factors that influence the extent of psychological damage. This research also suggests that childhood trauma is associated with emotional dysregulation, aggression against self and others, difficulties in attention and dissociation, medical problems, and difficulty with navigating adult interpersonal relationships.
When viewed through the lens of attachment theory, the forced separation of a child from its caretakers is a potent form of childhood trauma. Joanna E. Chambers, MD, summarizes and explains John Bowlby’s attachment theory as a neurobiological system originating from an infant’s connection to the primary caretaker. This connection becomes a lifelong model for all subsequent relationships.
Any traumatic disruptions in the development of this system puts the child at risk of developing “insecure attachment.” This insecure attachment can lead to lifelong emotional problems for the child, affecting the quality of subsequent marital relationships, relationships to children, and the development of personality disorders.
In addition, it correlates to the development of psychiatric illnesses, specifically depression and anxiety. There is also a plausible biological basis for attachment theory. Both oxytocin, a hormone released in human bonding, and social interaction itself have been shown to decrease cortisol levels. Elevated cortisol has been found to negatively affect infant brain development, Dr. Chambers argued.5
Given those significant effects of childhood trauma, it is understandable that there exists a concept of “intergenerational transmission of trauma.” Originating from studies of Holocaust survivors and their descendants, researchers Amy Lehrner, PhD, and Rachel Yehuda, PhD, conceptualize intergenerational transmission of trauma as the intergenerational impact of prenatal PTSD.6 This impact is expressed as a predisposition in the offspring of Holocaust survivors to developing PTSD, difficulties in individuation and separation, higher rates of mood and anxiety disorders, and higher rates of physical health issues.
Although they are complex and clearly multidetermined, Dr. Yehuda and Dr. Lehrner also summarize plausible biological theories for the intergenerational transmission of trauma. Epigenetic differences in the hypothalamic-pituitary-adrenal axis, circadian rhythm, urinary and plasma cortisol levels, glucocorticoid sensitivity, and regulation of the glucocorticoid receptor gene all have been found in Holocaust offspring with parental PTSD, in contrast to offspring without parental PTSD.6
What can we as psychiatrists do?
We are uniquely equipped to take several concrete steps to help mitigate the effects of these traumatic events. Among them, we can:
- Provide opportunities for the child and the family to process their experience. This can be profoundly healing and can help minimize the devastating psychological effects of this separation.
- Become acquainted with the concept of intergenerational transmission of resilience.
- Work with trauma survivors to develop their own personal narratives and cultural rituals surrounding the trauma.6
- Encourage second- and third-generation descendants to engage in artistic expression of the trauma, visit places of importance to their parents, and engage in social and political activism. These are all expressions of resilience in the offspring of trauma victims.6
In summary, recent U.S. political events have caused thousands of children to be forcibly separated from their parents. Those separations are traumatic and can have lifelong psychological implications for the children and their offspring. It is important to provide quality mental health treatment to these children with a specific focus on treating PTSD and processing the traumatic experience. Psychological treatment can help mitigate the effects of the traumatic separation and create a sense of resiliency.
References
1. New York Times. June 20, 2018. “My separation trauma.”
2. American Psychiatric Association statement, May 30, 2018.
3. Letter to the departments of Justice, Health & Human Services, and Homeland Security, June 20, 2018.
4. Child Adolesc Psychiatric Clin N Am. 2003;(12.2):293-318.
5. Psychodynamic Psychiatry. 2017 Dec;45(4):542-63.
6. Psychological Trauma: Theory, Research, Practice, and Policy. 2018 Jan;10(1):22-9.
Dr. Reinstein is a psychiatry attending at Zucker Hillside Hospital of the Northwell Health System, Glen Oaks, New York. Her clinical interests include reproductive psychiatry and family therapy, with a specific focus on maternal mental health. Dr. Reinstein completed her adult psychiatry residency training at Montefiore Hospital/Albert Einstein College of Medicine after graduating from the Albert Einstein College of Medicine and Yeshiva University with a B.A. in biology. She is one of the recipients of the 4th Annual Resident Recognition Award for Excellence in Family Oriented Care.
Editor’s Note: Alison M. Heru, MD, the Families in Psychiatry columnist, invited Dr. Reinstein to address this topic.
Growing up, I was always intrigued by the strong emotions that even the mildest separation evoked within me. As a psychiatrist, I now believe that these emotions were related to my family’s difficulty with separation, a concept likely transmitted from my grandmother’s sudden separation as a child.
The political circumstances of the “Kindertransport” and the recent family separation at the southern U.S. border differ, but the Kindertransport is a model for studying the effects of forced parent-child separation and its intergenerational transmission. As a rescue operation that took place immediately before World War II, the Kindertransport was the emigration of approximately 10,000 German Jewish children from Germany and Nazi-occupied countries to England. Even though they were not literally forced, the parents involved were compelled to separate from their children to give their children a chance to survive.
In a recent letter1 published in The New York Times, Eva Yachnes, herself a Kindertransport participant, reflected on the current situation at the southern U.S. border. She alluded to the lifelong effects of her own separation at the age of 6 when emigrating from Vienna to Germany. Personally, as a granddaughter of a Kindertransport participant, I am particularly concerned about the intergenerational transmission of the trauma of family separation.
Since early May 2018, more than 2,000 children have been forcibly separated from their parents after illegally crossing the border. As part of a “zero tolerance policy,” the separation was characterized by the Trump administration as a deterrent to illegal border crossings. Although many Americans are horrified by the news reports about family separation, psychiatrists in particular have expressed concern about this trauma. The American Psychiatric Association issued a statement2 warning that “any forced separation is highly stressful for children and can cause lifelong trauma.” Several weeks later, the APA joined several other mental health organizations in a letter3 calling for the immediate end of enforcement of those policies. In that letter, the organizations said forced separations can cause “an increased risk of ... mental illnesses such as depression, anxiety, and posttraumatic stress disorder.”
What must psychiatrists understand about the impact of childhood trauma? How can psychiatrists approach treatment of children separated from their parents? Are there ways to minimize the risk for intergenerational impact of this trauma?
What do we know?
Childhood trauma is influenced by multiple factors and can be expressed in several ways. According to research,4 the age of the child at the time of traumatic event, the frequency of traumatic experiences, and the degree to which the child’s caretakers were involved in the trauma are factors that influence the extent of psychological damage. This research also suggests that childhood trauma is associated with emotional dysregulation, aggression against self and others, difficulties in attention and dissociation, medical problems, and difficulty with navigating adult interpersonal relationships.
When viewed through the lens of attachment theory, the forced separation of a child from its caretakers is a potent form of childhood trauma. Joanna E. Chambers, MD, summarizes and explains John Bowlby’s attachment theory as a neurobiological system originating from an infant’s connection to the primary caretaker. This connection becomes a lifelong model for all subsequent relationships.
Any traumatic disruptions in the development of this system puts the child at risk of developing “insecure attachment.” This insecure attachment can lead to lifelong emotional problems for the child, affecting the quality of subsequent marital relationships, relationships to children, and the development of personality disorders.
In addition, it correlates to the development of psychiatric illnesses, specifically depression and anxiety. There is also a plausible biological basis for attachment theory. Both oxytocin, a hormone released in human bonding, and social interaction itself have been shown to decrease cortisol levels. Elevated cortisol has been found to negatively affect infant brain development, Dr. Chambers argued.5
Given those significant effects of childhood trauma, it is understandable that there exists a concept of “intergenerational transmission of trauma.” Originating from studies of Holocaust survivors and their descendants, researchers Amy Lehrner, PhD, and Rachel Yehuda, PhD, conceptualize intergenerational transmission of trauma as the intergenerational impact of prenatal PTSD.6 This impact is expressed as a predisposition in the offspring of Holocaust survivors to developing PTSD, difficulties in individuation and separation, higher rates of mood and anxiety disorders, and higher rates of physical health issues.
Although they are complex and clearly multidetermined, Dr. Yehuda and Dr. Lehrner also summarize plausible biological theories for the intergenerational transmission of trauma. Epigenetic differences in the hypothalamic-pituitary-adrenal axis, circadian rhythm, urinary and plasma cortisol levels, glucocorticoid sensitivity, and regulation of the glucocorticoid receptor gene all have been found in Holocaust offspring with parental PTSD, in contrast to offspring without parental PTSD.6
What can we as psychiatrists do?
We are uniquely equipped to take several concrete steps to help mitigate the effects of these traumatic events. Among them, we can:
- Provide opportunities for the child and the family to process their experience. This can be profoundly healing and can help minimize the devastating psychological effects of this separation.
- Become acquainted with the concept of intergenerational transmission of resilience.
- Work with trauma survivors to develop their own personal narratives and cultural rituals surrounding the trauma.6
- Encourage second- and third-generation descendants to engage in artistic expression of the trauma, visit places of importance to their parents, and engage in social and political activism. These are all expressions of resilience in the offspring of trauma victims.6
In summary, recent U.S. political events have caused thousands of children to be forcibly separated from their parents. Those separations are traumatic and can have lifelong psychological implications for the children and their offspring. It is important to provide quality mental health treatment to these children with a specific focus on treating PTSD and processing the traumatic experience. Psychological treatment can help mitigate the effects of the traumatic separation and create a sense of resiliency.
References
1. New York Times. June 20, 2018. “My separation trauma.”
2. American Psychiatric Association statement, May 30, 2018.
3. Letter to the departments of Justice, Health & Human Services, and Homeland Security, June 20, 2018.
4. Child Adolesc Psychiatric Clin N Am. 2003;(12.2):293-318.
5. Psychodynamic Psychiatry. 2017 Dec;45(4):542-63.
6. Psychological Trauma: Theory, Research, Practice, and Policy. 2018 Jan;10(1):22-9.
Dr. Reinstein is a psychiatry attending at Zucker Hillside Hospital of the Northwell Health System, Glen Oaks, New York. Her clinical interests include reproductive psychiatry and family therapy, with a specific focus on maternal mental health. Dr. Reinstein completed her adult psychiatry residency training at Montefiore Hospital/Albert Einstein College of Medicine after graduating from the Albert Einstein College of Medicine and Yeshiva University with a B.A. in biology. She is one of the recipients of the 4th Annual Resident Recognition Award for Excellence in Family Oriented Care.