User login
CANVAS program analysis: Canagliflozin benefits patients with T2DM and poor kidney function
ORLANDO – according to an analysis of data from the CANVAS program.
The findings suggest that it may be time to reconsider limitations on usage of the sodium-glucose transporter 2 inhibitor in patients with poor kidney function, Dick de Zeeuw, MD, PhD, said at the annual scientific sessions of the American Diabetes Association.
Despite the smaller glycemic effects in patients with reduced estimated glomerular filtration rate (eGFR) seen in CANVAS (CANagliflozin cardioVascular Assessment Study), the relative effects on the primary – and most other – cardiovascular outcomes in CANVAS were similar in patients with low (less than 60 mL/min per 1.72 m2 [2,029 patients]) versus high (greater than 60 mL/min per 1.72 m2 [8,101 patients]) eGFR, and across four eGFR subgroups (less than 45 mL/min per 1.72 m2 [554 patients], 45-60 mL/min per 1.72 m2 [1,485 patients], 60-90 mL/min per1.72 m2 [5,625 patients], and 90 or greater mL/min per 1.72 m2 [2,684 patients]), reported Dr. de Zeeuw of the University Medical Center Groningen, the Netherlands.
An exception was a possible finding of heterogeneity of treatment effect for the exploratory outcome of stroke, he noted.
As expected, the effects of canagliflozin versus placebo on the surrogate marker of HbA1c was significantly smaller with lower renal function, with reductions of 0.35%, 0.45%, 0.57%, and 0.76% across the four eGFR subgroups, respectively.
“But most interestingly, the three other cardiovascular risk markers show no difference in the effect of canagliflozin when we compare them for the four different eGFR groups,” he said, explaining that reductions in systolic blood pressure, body weight, and albuminuria were “very consistent” across the groups.
For the CANVAS 3-point major cardiac adverse event primary endpoint, which included a composite of cardiovascular death, nonfatal MI, or nonfatal stroke (and which was reduced by 14% [hazard ratio, 0.86] with canagliflozin treatment in the primary CANVAS analysis), no significant heterogeneity for treatment effect was noted between the high versus low eGFR groups or the four eGFR groups (P = .08 and .33, respectively), Dr. de Zeeuw said.
“If anything, there was more effect in the low eGFR group than in the high eGFR group [HRs, 0.92 and 0.70, respectively; 0.65, 0.71, 0.95, and 0.84 for the four eGFR groups, respectively]”, he added, noting that the same was true for the cardiovascular death and MI components of the primary endpoint.
For stroke, however, possible heterogeneity by baseline kidney function was noted. In the primary CANVAS analysis, canagliflozin was associated with a nonsignificant 13% reduction in stroke risk (HR, 0.87), and the hazard ratios for low versus high eGFR in the current analysis were 0.50 and 1.01, respectively, and in the four eGFR groups they were 0.32, 0.56, 0.89, and 1.42 , respectively (P = .01 for both comparisons).
For the secondary endpoints of hospitalizations for heart failure and renal function (a composite of eGFR, end-stage kidney disease, or renal death), which both were important outcomes in the CANVAS program with each showing highly significant reductions with canagliflozin treatment versus placebo in the primary analysis (HRs, 0.67 and 0.60), no heterogeneity by eGFR was seen in the current analysis.
As for safety outcomes, which included serious adverse events, adverse events leading to treatment discontinuation, amputation, fractures, and renal safety (serious renal-related adverse events, serious acute kidney injury, and serious hyperkalemia), no heterogeneity was seen in the outcomes across eGFR groups, he said.
“The CANVAS program ... is a composite of two cardiovascular safety trials that have been conducted over the past [8+] years,” Dr. de Zeeuw said, noting that participants included patients with type 2 diabetes mellitus and cardiovascular disease or high cardiovascular risk who were treated with either 300 mg or 100 mg of canagliflozin or with placebo after a 2-week placebo run-in.
The outcomes favored canagliflozin across all endpoints, and particularly for the secondary endpoints of hospitalization for heart failure and the composite of renal outcomes.
“The interest [in terms of the current analysis] is that this drug supposedly works better in those that have a functioning kidney and a filtering kidney, where the tubule is actually the target of this drug. It was anticipated that there would be less effect of this drug on low renal functions,” he said, adding that although the effect on HbA1c is progressively attenuated with declining renal function, as expected, the effects on other cardiovascular risk markers were, surprisingly, very comparable across the eGFR subgroups.
“And with that, more importantly, the effect on the hard outcomes – like the primary outcomes, most of the CV outcomes [maybe except stroke], the renal and safety outcomes – are very consistent across the different kidney function levels,” he said, adding that these beneficial findings in patients with 30-45 eGFR who are at high cardiovascular and renal risk suggest that reconsideration of the current limitation on the use of canagliflozin inhibitors to patients with eGFR above 45 is warranted.
The CANVAS program was sponsored by Janssen Research & Development. Dr. de Zeeuw is a consultant and/or speaker for AbbVie, Astellas, Bayer, Boehringer Ingelheim, Fresenius, Janssen, and Mitsubishi Tanabe.
SOURCE: de Zeeuw D et al. ADA 2018, Abstract 258-OR.
ORLANDO – according to an analysis of data from the CANVAS program.
The findings suggest that it may be time to reconsider limitations on usage of the sodium-glucose transporter 2 inhibitor in patients with poor kidney function, Dick de Zeeuw, MD, PhD, said at the annual scientific sessions of the American Diabetes Association.
Despite the smaller glycemic effects in patients with reduced estimated glomerular filtration rate (eGFR) seen in CANVAS (CANagliflozin cardioVascular Assessment Study), the relative effects on the primary – and most other – cardiovascular outcomes in CANVAS were similar in patients with low (less than 60 mL/min per 1.72 m2 [2,029 patients]) versus high (greater than 60 mL/min per 1.72 m2 [8,101 patients]) eGFR, and across four eGFR subgroups (less than 45 mL/min per 1.72 m2 [554 patients], 45-60 mL/min per 1.72 m2 [1,485 patients], 60-90 mL/min per1.72 m2 [5,625 patients], and 90 or greater mL/min per 1.72 m2 [2,684 patients]), reported Dr. de Zeeuw of the University Medical Center Groningen, the Netherlands.
An exception was a possible finding of heterogeneity of treatment effect for the exploratory outcome of stroke, he noted.
As expected, the effects of canagliflozin versus placebo on the surrogate marker of HbA1c was significantly smaller with lower renal function, with reductions of 0.35%, 0.45%, 0.57%, and 0.76% across the four eGFR subgroups, respectively.
“But most interestingly, the three other cardiovascular risk markers show no difference in the effect of canagliflozin when we compare them for the four different eGFR groups,” he said, explaining that reductions in systolic blood pressure, body weight, and albuminuria were “very consistent” across the groups.
For the CANVAS 3-point major cardiac adverse event primary endpoint, which included a composite of cardiovascular death, nonfatal MI, or nonfatal stroke (and which was reduced by 14% [hazard ratio, 0.86] with canagliflozin treatment in the primary CANVAS analysis), no significant heterogeneity for treatment effect was noted between the high versus low eGFR groups or the four eGFR groups (P = .08 and .33, respectively), Dr. de Zeeuw said.
“If anything, there was more effect in the low eGFR group than in the high eGFR group [HRs, 0.92 and 0.70, respectively; 0.65, 0.71, 0.95, and 0.84 for the four eGFR groups, respectively]”, he added, noting that the same was true for the cardiovascular death and MI components of the primary endpoint.
For stroke, however, possible heterogeneity by baseline kidney function was noted. In the primary CANVAS analysis, canagliflozin was associated with a nonsignificant 13% reduction in stroke risk (HR, 0.87), and the hazard ratios for low versus high eGFR in the current analysis were 0.50 and 1.01, respectively, and in the four eGFR groups they were 0.32, 0.56, 0.89, and 1.42 , respectively (P = .01 for both comparisons).
For the secondary endpoints of hospitalizations for heart failure and renal function (a composite of eGFR, end-stage kidney disease, or renal death), which both were important outcomes in the CANVAS program with each showing highly significant reductions with canagliflozin treatment versus placebo in the primary analysis (HRs, 0.67 and 0.60), no heterogeneity by eGFR was seen in the current analysis.
As for safety outcomes, which included serious adverse events, adverse events leading to treatment discontinuation, amputation, fractures, and renal safety (serious renal-related adverse events, serious acute kidney injury, and serious hyperkalemia), no heterogeneity was seen in the outcomes across eGFR groups, he said.
“The CANVAS program ... is a composite of two cardiovascular safety trials that have been conducted over the past [8+] years,” Dr. de Zeeuw said, noting that participants included patients with type 2 diabetes mellitus and cardiovascular disease or high cardiovascular risk who were treated with either 300 mg or 100 mg of canagliflozin or with placebo after a 2-week placebo run-in.
The outcomes favored canagliflozin across all endpoints, and particularly for the secondary endpoints of hospitalization for heart failure and the composite of renal outcomes.
“The interest [in terms of the current analysis] is that this drug supposedly works better in those that have a functioning kidney and a filtering kidney, where the tubule is actually the target of this drug. It was anticipated that there would be less effect of this drug on low renal functions,” he said, adding that although the effect on HbA1c is progressively attenuated with declining renal function, as expected, the effects on other cardiovascular risk markers were, surprisingly, very comparable across the eGFR subgroups.
“And with that, more importantly, the effect on the hard outcomes – like the primary outcomes, most of the CV outcomes [maybe except stroke], the renal and safety outcomes – are very consistent across the different kidney function levels,” he said, adding that these beneficial findings in patients with 30-45 eGFR who are at high cardiovascular and renal risk suggest that reconsideration of the current limitation on the use of canagliflozin inhibitors to patients with eGFR above 45 is warranted.
The CANVAS program was sponsored by Janssen Research & Development. Dr. de Zeeuw is a consultant and/or speaker for AbbVie, Astellas, Bayer, Boehringer Ingelheim, Fresenius, Janssen, and Mitsubishi Tanabe.
SOURCE: de Zeeuw D et al. ADA 2018, Abstract 258-OR.
ORLANDO – according to an analysis of data from the CANVAS program.
The findings suggest that it may be time to reconsider limitations on usage of the sodium-glucose transporter 2 inhibitor in patients with poor kidney function, Dick de Zeeuw, MD, PhD, said at the annual scientific sessions of the American Diabetes Association.
Despite the smaller glycemic effects in patients with reduced estimated glomerular filtration rate (eGFR) seen in CANVAS (CANagliflozin cardioVascular Assessment Study), the relative effects on the primary – and most other – cardiovascular outcomes in CANVAS were similar in patients with low (less than 60 mL/min per 1.72 m2 [2,029 patients]) versus high (greater than 60 mL/min per 1.72 m2 [8,101 patients]) eGFR, and across four eGFR subgroups (less than 45 mL/min per 1.72 m2 [554 patients], 45-60 mL/min per 1.72 m2 [1,485 patients], 60-90 mL/min per1.72 m2 [5,625 patients], and 90 or greater mL/min per 1.72 m2 [2,684 patients]), reported Dr. de Zeeuw of the University Medical Center Groningen, the Netherlands.
An exception was a possible finding of heterogeneity of treatment effect for the exploratory outcome of stroke, he noted.
As expected, the effects of canagliflozin versus placebo on the surrogate marker of HbA1c was significantly smaller with lower renal function, with reductions of 0.35%, 0.45%, 0.57%, and 0.76% across the four eGFR subgroups, respectively.
“But most interestingly, the three other cardiovascular risk markers show no difference in the effect of canagliflozin when we compare them for the four different eGFR groups,” he said, explaining that reductions in systolic blood pressure, body weight, and albuminuria were “very consistent” across the groups.
For the CANVAS 3-point major cardiac adverse event primary endpoint, which included a composite of cardiovascular death, nonfatal MI, or nonfatal stroke (and which was reduced by 14% [hazard ratio, 0.86] with canagliflozin treatment in the primary CANVAS analysis), no significant heterogeneity for treatment effect was noted between the high versus low eGFR groups or the four eGFR groups (P = .08 and .33, respectively), Dr. de Zeeuw said.
“If anything, there was more effect in the low eGFR group than in the high eGFR group [HRs, 0.92 and 0.70, respectively; 0.65, 0.71, 0.95, and 0.84 for the four eGFR groups, respectively]”, he added, noting that the same was true for the cardiovascular death and MI components of the primary endpoint.
For stroke, however, possible heterogeneity by baseline kidney function was noted. In the primary CANVAS analysis, canagliflozin was associated with a nonsignificant 13% reduction in stroke risk (HR, 0.87), and the hazard ratios for low versus high eGFR in the current analysis were 0.50 and 1.01, respectively, and in the four eGFR groups they were 0.32, 0.56, 0.89, and 1.42 , respectively (P = .01 for both comparisons).
For the secondary endpoints of hospitalizations for heart failure and renal function (a composite of eGFR, end-stage kidney disease, or renal death), which both were important outcomes in the CANVAS program with each showing highly significant reductions with canagliflozin treatment versus placebo in the primary analysis (HRs, 0.67 and 0.60), no heterogeneity by eGFR was seen in the current analysis.
As for safety outcomes, which included serious adverse events, adverse events leading to treatment discontinuation, amputation, fractures, and renal safety (serious renal-related adverse events, serious acute kidney injury, and serious hyperkalemia), no heterogeneity was seen in the outcomes across eGFR groups, he said.
“The CANVAS program ... is a composite of two cardiovascular safety trials that have been conducted over the past [8+] years,” Dr. de Zeeuw said, noting that participants included patients with type 2 diabetes mellitus and cardiovascular disease or high cardiovascular risk who were treated with either 300 mg or 100 mg of canagliflozin or with placebo after a 2-week placebo run-in.
The outcomes favored canagliflozin across all endpoints, and particularly for the secondary endpoints of hospitalization for heart failure and the composite of renal outcomes.
“The interest [in terms of the current analysis] is that this drug supposedly works better in those that have a functioning kidney and a filtering kidney, where the tubule is actually the target of this drug. It was anticipated that there would be less effect of this drug on low renal functions,” he said, adding that although the effect on HbA1c is progressively attenuated with declining renal function, as expected, the effects on other cardiovascular risk markers were, surprisingly, very comparable across the eGFR subgroups.
“And with that, more importantly, the effect on the hard outcomes – like the primary outcomes, most of the CV outcomes [maybe except stroke], the renal and safety outcomes – are very consistent across the different kidney function levels,” he said, adding that these beneficial findings in patients with 30-45 eGFR who are at high cardiovascular and renal risk suggest that reconsideration of the current limitation on the use of canagliflozin inhibitors to patients with eGFR above 45 is warranted.
The CANVAS program was sponsored by Janssen Research & Development. Dr. de Zeeuw is a consultant and/or speaker for AbbVie, Astellas, Bayer, Boehringer Ingelheim, Fresenius, Janssen, and Mitsubishi Tanabe.
SOURCE: de Zeeuw D et al. ADA 2018, Abstract 258-OR.
REPORTING FROM ADA 2018
Key clinical point: Cardioprotective benefits of canagliflozin in patients with type 2 diabetes mellitus were apparent across varying levels of kidney function.
Major finding: No heterogeneity for treatment effect for the primary major cardiac adverse event endpoint was noted between the high versus low estimated glomerular filtration rate groups (P = .08).
Study details: An analysis of data from the CANVAS program studies involving 10,142 patients.
Disclosures: The CANVAS program was sponsored by Janssen Research & Development. Dr. de Zeeuw is a consultant and/or speaker for AbbVie, Astellas, Bayer, Boehringer Ingelheim, Fresenius, Janssen, and Mitsubishi Tanabe Pharma.
Source: de Zeeuw D et al. ADA 2018, Abstract 258-OR.
Light Exposure During Sleep May Increase Insulin Resistance
Healthy adults who slept with an overhead light on had significantly greater insulin resistance the next morning.
BALTIMORE—A single night of light exposure during sleep may acutely affect insulin resistance, according to research described at the 32nd Annual Meeting of the Associated Professional Sleep Societies. Studies have found that nighttime artificial light exposure affects circadian rhythms such as melatonin and sleep, and disturbances in these rhythms may affect cardiometabolic function.
To assess whether light exposure during sleep negatively affects cardiometabolic outcomes, possibly via disruptions to sleep architecture and melatonin profile, Ivy C. Mason, PhD, a research fellow at Harvard Medical School in Boston, and colleagues conducted a study in which they randomized 20 healthy adults ages 18 to 40 to different sleep conditions. The conditions were run in parallel for three days and two nights.
One group slept in the dark (ie, less than 3 lux) on the first night and in a lighted room (ie, with an overhead light of 100 lux) on the second night. The other group slept in the dark on both nights.
Participants had eight hours of sleep opportunity each night, starting at a habitual bedtime that was determined from one week of actigraphy with a sleep diary. On both nights, overnight polysomnography was performed and hourly blood samples were collected to measure melatonin.
Researchers performed oral glucose tolerance tests on both mornings and assessed changes from the first morning to the second morning. They compared the groups using repeated measures analysis of variance.
The dark–light group (n = 10; two males) had a mean age of 26.61 and a mean BMI of 23.25 kg/m2. The dark–dark group (n = 10; four males) had a mean age of 26.78 and a mean BMI of 24.25 kg/m2).
Homeostatic model assessments of insulin resistance values were significantly higher in the morning following sleep in the light in the dark–light group, compared with those values in the dark–dark group. This effect was primarily due to increased insulin levels in the group that slept in the light, said the researchers.
“Our preliminary findings show that a single night of light exposure during sleep acutely impacts measures of insulin resistance,” said Dr. Mason. “These results are important, given the increasingly widespread use of artificial light exposure, particularly at night.… More research is needed to determine if chronic overnight light exposure during sleep has long-term, cumulative effects on metabolic function.”
Healthy adults who slept with an overhead light on had significantly greater insulin resistance the next morning.
Healthy adults who slept with an overhead light on had significantly greater insulin resistance the next morning.
BALTIMORE—A single night of light exposure during sleep may acutely affect insulin resistance, according to research described at the 32nd Annual Meeting of the Associated Professional Sleep Societies. Studies have found that nighttime artificial light exposure affects circadian rhythms such as melatonin and sleep, and disturbances in these rhythms may affect cardiometabolic function.
To assess whether light exposure during sleep negatively affects cardiometabolic outcomes, possibly via disruptions to sleep architecture and melatonin profile, Ivy C. Mason, PhD, a research fellow at Harvard Medical School in Boston, and colleagues conducted a study in which they randomized 20 healthy adults ages 18 to 40 to different sleep conditions. The conditions were run in parallel for three days and two nights.
One group slept in the dark (ie, less than 3 lux) on the first night and in a lighted room (ie, with an overhead light of 100 lux) on the second night. The other group slept in the dark on both nights.
Participants had eight hours of sleep opportunity each night, starting at a habitual bedtime that was determined from one week of actigraphy with a sleep diary. On both nights, overnight polysomnography was performed and hourly blood samples were collected to measure melatonin.
Researchers performed oral glucose tolerance tests on both mornings and assessed changes from the first morning to the second morning. They compared the groups using repeated measures analysis of variance.
The dark–light group (n = 10; two males) had a mean age of 26.61 and a mean BMI of 23.25 kg/m2. The dark–dark group (n = 10; four males) had a mean age of 26.78 and a mean BMI of 24.25 kg/m2).
Homeostatic model assessments of insulin resistance values were significantly higher in the morning following sleep in the light in the dark–light group, compared with those values in the dark–dark group. This effect was primarily due to increased insulin levels in the group that slept in the light, said the researchers.
“Our preliminary findings show that a single night of light exposure during sleep acutely impacts measures of insulin resistance,” said Dr. Mason. “These results are important, given the increasingly widespread use of artificial light exposure, particularly at night.… More research is needed to determine if chronic overnight light exposure during sleep has long-term, cumulative effects on metabolic function.”
BALTIMORE—A single night of light exposure during sleep may acutely affect insulin resistance, according to research described at the 32nd Annual Meeting of the Associated Professional Sleep Societies. Studies have found that nighttime artificial light exposure affects circadian rhythms such as melatonin and sleep, and disturbances in these rhythms may affect cardiometabolic function.
To assess whether light exposure during sleep negatively affects cardiometabolic outcomes, possibly via disruptions to sleep architecture and melatonin profile, Ivy C. Mason, PhD, a research fellow at Harvard Medical School in Boston, and colleagues conducted a study in which they randomized 20 healthy adults ages 18 to 40 to different sleep conditions. The conditions were run in parallel for three days and two nights.
One group slept in the dark (ie, less than 3 lux) on the first night and in a lighted room (ie, with an overhead light of 100 lux) on the second night. The other group slept in the dark on both nights.
Participants had eight hours of sleep opportunity each night, starting at a habitual bedtime that was determined from one week of actigraphy with a sleep diary. On both nights, overnight polysomnography was performed and hourly blood samples were collected to measure melatonin.
Researchers performed oral glucose tolerance tests on both mornings and assessed changes from the first morning to the second morning. They compared the groups using repeated measures analysis of variance.
The dark–light group (n = 10; two males) had a mean age of 26.61 and a mean BMI of 23.25 kg/m2. The dark–dark group (n = 10; four males) had a mean age of 26.78 and a mean BMI of 24.25 kg/m2).
Homeostatic model assessments of insulin resistance values were significantly higher in the morning following sleep in the light in the dark–light group, compared with those values in the dark–dark group. This effect was primarily due to increased insulin levels in the group that slept in the light, said the researchers.
“Our preliminary findings show that a single night of light exposure during sleep acutely impacts measures of insulin resistance,” said Dr. Mason. “These results are important, given the increasingly widespread use of artificial light exposure, particularly at night.… More research is needed to determine if chronic overnight light exposure during sleep has long-term, cumulative effects on metabolic function.”
PSMA-targeting docetaxel nanoparticles active, safe for mCRPC
For men with chemotherapy-naive metastatic castration-resistant prostate cancer (mCRPC), a docetaxel-containing nanoparticle directed against prostate-specific membrane antigen (PSMA) was both active and well tolerated.
Among 42 men with mCRPC that had progressed after treatment with abiraterone acetate (Zytiga) and/or enzalutamide (Xtandi), the 6-month radiographic progression-free survival (PFS) rate, the primary endpoint, was 65%, and approximately one-third of evaluable patients had a prostate-specific antigen (PSA) response and measurable disease response, reported Karen A. Autio, MD, MSc, of Memorial Sloan Kettering Cancer Center, New York, and her colleagues.
Treatment with the PSMA-directed docetaxel-containing nanoparticle, called BIND-014, was also associated in some patients with a rapid and robust decline in PSMA-positive circulating tumor cells (CTCs), the investigators noted. The report was published in JAMA Oncology.
“Many aspects of our study – reductions in PSA, radiographically confirmed disease control in bone and visceral metastatic disease, favorable CTC conversions, and an acceptable adverse effect profile – were promising for BIND-014. However, standard therapy with docetaxel is widely used and effective in treating this disease, and as a natural comparator for a phase 3 randomized clinical trial with BIND-014, it sets a high bar for efficacy in an unselected population,” they wrote.
Their findings suggest that patients with PSMA-positive CTCs might be good candidates for treatment with BIND-014, which is associated with lower toxicities than standard docetaxel, the investigators said.
In the open-label phase 2 trial, 42 men (median age 66) with chemotherapy-naive mCRPC that had progressed on abiraterone and/or enzalutamide were treated with intravenous BIND-014 at dosages of 60 mg/m2 on the first day of each 21-day cycle, plus prednisone 5 mg twice daily, until disease progression, intolerable toxicity, or treatment discontinuation at the treating physician’s discretion.
The median number of doses delivered was six.
Of 40 evaluable patients, 12 (30%) had a PSA response, defined as a decrease of 50% or greater from baseline. Among 19 patients with disease measurable according to Response Evaluation Criteria in Solid Tumors, version 1.1, six (32%) had responses, including one complete response and five partial responses. Nine other patients had stable disease.
The median PFS was 9.9 months. As noted, the radiographic PFS rate at 6 months was 65% (26 of 40 patients).
CTC enumeration was performed on samples from 39 patients, of whom 67% had unfavorable counts at baseline, defined as 5 or more CTCs per 7.5 mL of blood.
After treatment, 13 of the patients had a conversion to a favorable count, including 6 whose CTCs became undetectable.
The most common treatment-related adverse events were fatigue, occurring in 69% of patients, nausea in 55%, diarrhea in 45%, and patient-reported neuropathy in 33%. Most toxicities were grade 1 or 2.
Grade 3 or 4 hematological toxicities included lymphopenia in five patients, anemia in three, neutropenia in one, leukopenia in one, and febrile neutropenia in one.
Nonhematological grade 3 or 4 events included fatigue and nausea in two patients each, and dyspnea and decreased appetite in one patient each.
“In this trial, two principal lessons are worth highlighting: the reduction in both total CTCs, which is a marker of clinical benefit, and specifically PSMA-positive CTCs, suggests that the detection of PSMA-positive CTCs before treatment can be used to identify patients who are most likely to benefit in addition to serving as a pharmacodynamic measure,” the investigators wrote.
“Second, there was marked intrapatient and interpatient heterogeneity of PSMA expression on CTCs. This underscores the complexity of targeting tumors with single cell-surface markers,” they wrote.
SOURCE: Autio KA et al. JAMA Oncol. 2018 July 5 doi: 10.1001/jamaoncol.2018.2168.
For men with chemotherapy-naive metastatic castration-resistant prostate cancer (mCRPC), a docetaxel-containing nanoparticle directed against prostate-specific membrane antigen (PSMA) was both active and well tolerated.
Among 42 men with mCRPC that had progressed after treatment with abiraterone acetate (Zytiga) and/or enzalutamide (Xtandi), the 6-month radiographic progression-free survival (PFS) rate, the primary endpoint, was 65%, and approximately one-third of evaluable patients had a prostate-specific antigen (PSA) response and measurable disease response, reported Karen A. Autio, MD, MSc, of Memorial Sloan Kettering Cancer Center, New York, and her colleagues.
Treatment with the PSMA-directed docetaxel-containing nanoparticle, called BIND-014, was also associated in some patients with a rapid and robust decline in PSMA-positive circulating tumor cells (CTCs), the investigators noted. The report was published in JAMA Oncology.
“Many aspects of our study – reductions in PSA, radiographically confirmed disease control in bone and visceral metastatic disease, favorable CTC conversions, and an acceptable adverse effect profile – were promising for BIND-014. However, standard therapy with docetaxel is widely used and effective in treating this disease, and as a natural comparator for a phase 3 randomized clinical trial with BIND-014, it sets a high bar for efficacy in an unselected population,” they wrote.
Their findings suggest that patients with PSMA-positive CTCs might be good candidates for treatment with BIND-014, which is associated with lower toxicities than standard docetaxel, the investigators said.
In the open-label phase 2 trial, 42 men (median age 66) with chemotherapy-naive mCRPC that had progressed on abiraterone and/or enzalutamide were treated with intravenous BIND-014 at dosages of 60 mg/m2 on the first day of each 21-day cycle, plus prednisone 5 mg twice daily, until disease progression, intolerable toxicity, or treatment discontinuation at the treating physician’s discretion.
The median number of doses delivered was six.
Of 40 evaluable patients, 12 (30%) had a PSA response, defined as a decrease of 50% or greater from baseline. Among 19 patients with disease measurable according to Response Evaluation Criteria in Solid Tumors, version 1.1, six (32%) had responses, including one complete response and five partial responses. Nine other patients had stable disease.
The median PFS was 9.9 months. As noted, the radiographic PFS rate at 6 months was 65% (26 of 40 patients).
CTC enumeration was performed on samples from 39 patients, of whom 67% had unfavorable counts at baseline, defined as 5 or more CTCs per 7.5 mL of blood.
After treatment, 13 of the patients had a conversion to a favorable count, including 6 whose CTCs became undetectable.
The most common treatment-related adverse events were fatigue, occurring in 69% of patients, nausea in 55%, diarrhea in 45%, and patient-reported neuropathy in 33%. Most toxicities were grade 1 or 2.
Grade 3 or 4 hematological toxicities included lymphopenia in five patients, anemia in three, neutropenia in one, leukopenia in one, and febrile neutropenia in one.
Nonhematological grade 3 or 4 events included fatigue and nausea in two patients each, and dyspnea and decreased appetite in one patient each.
“In this trial, two principal lessons are worth highlighting: the reduction in both total CTCs, which is a marker of clinical benefit, and specifically PSMA-positive CTCs, suggests that the detection of PSMA-positive CTCs before treatment can be used to identify patients who are most likely to benefit in addition to serving as a pharmacodynamic measure,” the investigators wrote.
“Second, there was marked intrapatient and interpatient heterogeneity of PSMA expression on CTCs. This underscores the complexity of targeting tumors with single cell-surface markers,” they wrote.
SOURCE: Autio KA et al. JAMA Oncol. 2018 July 5 doi: 10.1001/jamaoncol.2018.2168.
For men with chemotherapy-naive metastatic castration-resistant prostate cancer (mCRPC), a docetaxel-containing nanoparticle directed against prostate-specific membrane antigen (PSMA) was both active and well tolerated.
Among 42 men with mCRPC that had progressed after treatment with abiraterone acetate (Zytiga) and/or enzalutamide (Xtandi), the 6-month radiographic progression-free survival (PFS) rate, the primary endpoint, was 65%, and approximately one-third of evaluable patients had a prostate-specific antigen (PSA) response and measurable disease response, reported Karen A. Autio, MD, MSc, of Memorial Sloan Kettering Cancer Center, New York, and her colleagues.
Treatment with the PSMA-directed docetaxel-containing nanoparticle, called BIND-014, was also associated in some patients with a rapid and robust decline in PSMA-positive circulating tumor cells (CTCs), the investigators noted. The report was published in JAMA Oncology.
“Many aspects of our study – reductions in PSA, radiographically confirmed disease control in bone and visceral metastatic disease, favorable CTC conversions, and an acceptable adverse effect profile – were promising for BIND-014. However, standard therapy with docetaxel is widely used and effective in treating this disease, and as a natural comparator for a phase 3 randomized clinical trial with BIND-014, it sets a high bar for efficacy in an unselected population,” they wrote.
Their findings suggest that patients with PSMA-positive CTCs might be good candidates for treatment with BIND-014, which is associated with lower toxicities than standard docetaxel, the investigators said.
In the open-label phase 2 trial, 42 men (median age 66) with chemotherapy-naive mCRPC that had progressed on abiraterone and/or enzalutamide were treated with intravenous BIND-014 at dosages of 60 mg/m2 on the first day of each 21-day cycle, plus prednisone 5 mg twice daily, until disease progression, intolerable toxicity, or treatment discontinuation at the treating physician’s discretion.
The median number of doses delivered was six.
Of 40 evaluable patients, 12 (30%) had a PSA response, defined as a decrease of 50% or greater from baseline. Among 19 patients with disease measurable according to Response Evaluation Criteria in Solid Tumors, version 1.1, six (32%) had responses, including one complete response and five partial responses. Nine other patients had stable disease.
The median PFS was 9.9 months. As noted, the radiographic PFS rate at 6 months was 65% (26 of 40 patients).
CTC enumeration was performed on samples from 39 patients, of whom 67% had unfavorable counts at baseline, defined as 5 or more CTCs per 7.5 mL of blood.
After treatment, 13 of the patients had a conversion to a favorable count, including 6 whose CTCs became undetectable.
The most common treatment-related adverse events were fatigue, occurring in 69% of patients, nausea in 55%, diarrhea in 45%, and patient-reported neuropathy in 33%. Most toxicities were grade 1 or 2.
Grade 3 or 4 hematological toxicities included lymphopenia in five patients, anemia in three, neutropenia in one, leukopenia in one, and febrile neutropenia in one.
Nonhematological grade 3 or 4 events included fatigue and nausea in two patients each, and dyspnea and decreased appetite in one patient each.
“In this trial, two principal lessons are worth highlighting: the reduction in both total CTCs, which is a marker of clinical benefit, and specifically PSMA-positive CTCs, suggests that the detection of PSMA-positive CTCs before treatment can be used to identify patients who are most likely to benefit in addition to serving as a pharmacodynamic measure,” the investigators wrote.
“Second, there was marked intrapatient and interpatient heterogeneity of PSMA expression on CTCs. This underscores the complexity of targeting tumors with single cell-surface markers,” they wrote.
SOURCE: Autio KA et al. JAMA Oncol. 2018 July 5 doi: 10.1001/jamaoncol.2018.2168.
FROM JAMA ONCOLOGY
Key clinical point: A docetaxel-encapsulating nanoparticle targeted against PSMA was safe and showed good activity.
Major finding: The 6-month PFS rate, the primary endpoint, was 65%.
Study details: Open label phase 2 trial of 42 men with mCRPC that progressed on abiraterone acetate and/or enzalutamide.
Disclosures: The study was funded by BIND Therapeutics and by grants from the National Institutes of Health, Sidney Kimmel Center for Prostate and Urologic Cancers, and the David H. Koch Prostate Cancer Research Fund. Dr. Autio had no disclosures. Several coauthors are employees of BIND Therapeutics, and others are employed by Epic Sciences, maker of the CTC assay used in the study. Other coauthors reported advising/consulting, research support, and speakers bureau activities for multiple companies.
Source: Autio KA et al. JAMA Oncol. 2018 July 5. doi: 10.1001/jamaoncol.2018.2168.
Ankylosing spondylitis progression slowed when NSAIDs added to TNFi
AMSTERDAM – When combined with a tumor necrosis factor inhibitor, NSAIDs provide protection against long-term radiographic progression in patients with ankylosing spondylitis, according to an analysis of more than 500 patients presented at the European Congress of Rheumatology.

Relative to TNFi alone, the addition of NSAIDs of any type provided protection at 4 years against radiographic progression as measured with the modified Stoke Ankylosing Spondylitis Spine Score (mSASSS). However, the protection associated with adding celecoxib was significant at 2 years and greater than that of adding nonselective NSAIDs at 4 years.
These data were drawn from 519 patients participating in the Prospective Study of Ankylosing Spondylitis study. All patients in this analysis were followed for at least 4 years. Radiographs were obtained every 6 months.
Although the study was a retrospective analysis of prospectively collected data, Dr. Gensler explained that control of variables such as disease and symptom duration with a technique called causal interference modeling “allows simulation of a randomized, controlled trial with observational data.”
Whether measured at 2 or 4 years, the reductions in mSASSS score for TNFi use versus no TNFi use were modest and did not reach statistical significance. However, exposure to NSAIDs plus TNFi did reach significance at 4 years, and the effect was dose dependent when patients taking a low-dose NSAID, defined as less than 50% of the index dose, were compared with those taking a higher dose. In this study, 70% were on chronic NSAID therapy, and these patients were divided relatively evenly between those on a low-dose or high-dose regimen.
At 2 years, relative radiographic protection for TNFi plus NSAIDs was not significantly greater than with TNFi alone, but at 4 years the median mSASSS score was 1.24 points lower (P less than .001) in those receiving low-dose NSAIDs, and 3.31 points lower (P less than .001) in those receiving high-dose NSAIDs.
In the subgroup of patients taking high-dose NSAIDs, the protection from progression was greatest among those receiving the selective COX2-inhibitor celecoxib. In these, the median 3.98 points lower mSASSS score (P less than .001) was already significant at 2 years. At 4 years, the median mSASSS score in those receiving TNFi plus celecoxib was 4.69 points lower (P less than .001).
Further evaluation suggested that the benefit from celecoxib plus TNFi was not just additive but synergistic, according to Dr. Gensler. She reported that neither TNFi nor celecoxib alone provided radiographic protection at 2 or 4 years.
Despite the modeling strategy employed to reduce the effect of bias, Dr. Gensler acknowledged that residual confounding is still possible. But she contended that “a large effect [from a such a variable] would be required to negate the findings.”
One of the messages from this study is that “not all NSAIDs are alike,” Dr. Gensler said. “Despite this, when I sit with a patient across from me, I will still treat the patient based on symptoms and disease activity first, though perhaps choose to be more NSAID selective if this is warranted and feasible.”
The next steps for research include a randomized, controlled trial combining TNFi and varying NSAIDs or different doses, Dr. Gensler said. In addition, “the development of newer imaging modalities will allow us to answer these questions in a more feasible time frame.”
The study was not industry funded. Dr. Gensler reported financial relationships with Amgen, AbbVie, Janssen, Eli Lilly, Novartis, and UCB.
SOURCE: Gensler L et al. Ann Rheum Dis. 2018;77(Suppl 2):148. Abstract OP0198.
AMSTERDAM – When combined with a tumor necrosis factor inhibitor, NSAIDs provide protection against long-term radiographic progression in patients with ankylosing spondylitis, according to an analysis of more than 500 patients presented at the European Congress of Rheumatology.
Relative to TNFi alone, the addition of NSAIDs of any type provided protection at 4 years against radiographic progression as measured with the modified Stoke Ankylosing Spondylitis Spine Score (mSASSS). However, the protection associated with adding celecoxib was significant at 2 years and greater than that of adding nonselective NSAIDs at 4 years.
These data were drawn from 519 patients participating in the Prospective Study of Ankylosing Spondylitis study. All patients in this analysis were followed for at least 4 years. Radiographs were obtained every 6 months.
Although the study was a retrospective analysis of prospectively collected data, Dr. Gensler explained that control of variables such as disease and symptom duration with a technique called causal interference modeling “allows simulation of a randomized, controlled trial with observational data.”
Whether measured at 2 or 4 years, the reductions in mSASSS score for TNFi use versus no TNFi use were modest and did not reach statistical significance. However, exposure to NSAIDs plus TNFi did reach significance at 4 years, and the effect was dose dependent when patients taking a low-dose NSAID, defined as less than 50% of the index dose, were compared with those taking a higher dose. In this study, 70% were on chronic NSAID therapy, and these patients were divided relatively evenly between those on a low-dose or high-dose regimen.
At 2 years, relative radiographic protection for TNFi plus NSAIDs was not significantly greater than with TNFi alone, but at 4 years the median mSASSS score was 1.24 points lower (P less than .001) in those receiving low-dose NSAIDs, and 3.31 points lower (P less than .001) in those receiving high-dose NSAIDs.
In the subgroup of patients taking high-dose NSAIDs, the protection from progression was greatest among those receiving the selective COX2-inhibitor celecoxib. In these, the median 3.98 points lower mSASSS score (P less than .001) was already significant at 2 years. At 4 years, the median mSASSS score in those receiving TNFi plus celecoxib was 4.69 points lower (P less than .001).
Further evaluation suggested that the benefit from celecoxib plus TNFi was not just additive but synergistic, according to Dr. Gensler. She reported that neither TNFi nor celecoxib alone provided radiographic protection at 2 or 4 years.
Despite the modeling strategy employed to reduce the effect of bias, Dr. Gensler acknowledged that residual confounding is still possible. But she contended that “a large effect [from a such a variable] would be required to negate the findings.”
One of the messages from this study is that “not all NSAIDs are alike,” Dr. Gensler said. “Despite this, when I sit with a patient across from me, I will still treat the patient based on symptoms and disease activity first, though perhaps choose to be more NSAID selective if this is warranted and feasible.”
The next steps for research include a randomized, controlled trial combining TNFi and varying NSAIDs or different doses, Dr. Gensler said. In addition, “the development of newer imaging modalities will allow us to answer these questions in a more feasible time frame.”
The study was not industry funded. Dr. Gensler reported financial relationships with Amgen, AbbVie, Janssen, Eli Lilly, Novartis, and UCB.
SOURCE: Gensler L et al. Ann Rheum Dis. 2018;77(Suppl 2):148. Abstract OP0198.
AMSTERDAM – When combined with a tumor necrosis factor inhibitor, NSAIDs provide protection against long-term radiographic progression in patients with ankylosing spondylitis, according to an analysis of more than 500 patients presented at the European Congress of Rheumatology.
Relative to TNFi alone, the addition of NSAIDs of any type provided protection at 4 years against radiographic progression as measured with the modified Stoke Ankylosing Spondylitis Spine Score (mSASSS). However, the protection associated with adding celecoxib was significant at 2 years and greater than that of adding nonselective NSAIDs at 4 years.
These data were drawn from 519 patients participating in the Prospective Study of Ankylosing Spondylitis study. All patients in this analysis were followed for at least 4 years. Radiographs were obtained every 6 months.
Although the study was a retrospective analysis of prospectively collected data, Dr. Gensler explained that control of variables such as disease and symptom duration with a technique called causal interference modeling “allows simulation of a randomized, controlled trial with observational data.”
Whether measured at 2 or 4 years, the reductions in mSASSS score for TNFi use versus no TNFi use were modest and did not reach statistical significance. However, exposure to NSAIDs plus TNFi did reach significance at 4 years, and the effect was dose dependent when patients taking a low-dose NSAID, defined as less than 50% of the index dose, were compared with those taking a higher dose. In this study, 70% were on chronic NSAID therapy, and these patients were divided relatively evenly between those on a low-dose or high-dose regimen.
At 2 years, relative radiographic protection for TNFi plus NSAIDs was not significantly greater than with TNFi alone, but at 4 years the median mSASSS score was 1.24 points lower (P less than .001) in those receiving low-dose NSAIDs, and 3.31 points lower (P less than .001) in those receiving high-dose NSAIDs.
In the subgroup of patients taking high-dose NSAIDs, the protection from progression was greatest among those receiving the selective COX2-inhibitor celecoxib. In these, the median 3.98 points lower mSASSS score (P less than .001) was already significant at 2 years. At 4 years, the median mSASSS score in those receiving TNFi plus celecoxib was 4.69 points lower (P less than .001).
Further evaluation suggested that the benefit from celecoxib plus TNFi was not just additive but synergistic, according to Dr. Gensler. She reported that neither TNFi nor celecoxib alone provided radiographic protection at 2 or 4 years.
Despite the modeling strategy employed to reduce the effect of bias, Dr. Gensler acknowledged that residual confounding is still possible. But she contended that “a large effect [from a such a variable] would be required to negate the findings.”
One of the messages from this study is that “not all NSAIDs are alike,” Dr. Gensler said. “Despite this, when I sit with a patient across from me, I will still treat the patient based on symptoms and disease activity first, though perhaps choose to be more NSAID selective if this is warranted and feasible.”
The next steps for research include a randomized, controlled trial combining TNFi and varying NSAIDs or different doses, Dr. Gensler said. In addition, “the development of newer imaging modalities will allow us to answer these questions in a more feasible time frame.”
The study was not industry funded. Dr. Gensler reported financial relationships with Amgen, AbbVie, Janssen, Eli Lilly, Novartis, and UCB.
SOURCE: Gensler L et al. Ann Rheum Dis. 2018;77(Suppl 2):148. Abstract OP0198.
REPORTING FROM THE EULAR 2018 CONGRESS
Key clinical point: Ankylosing spondylitis patients have less progression on a TNFi when they also receive an NSAID, especially celecoxib.
Major finding: At 4 years, the mSASSS score was 4.69 points (P less than .0001) lower on TNFi with celecoxib than on TNFi alone.
Study details: Retrospective study with causal interference modeling.
Disclosures: The study was not industry funded. Dr. Gensler reported financial relationships with Amgen, AbbVie, Janssen, Eli Lilly, Novartis, and UCB.
Source: Gensler L et al. Ann Rheum Dis. 2018;77(Suppl 2):148. Abstract OP0198.
Ultrasound aids treat-to-target approach for gout
AMSTERDAM – Results from the longitudinal NOR-GOUT study show that ultrasound can help visualize decreasing levels of uric acid deposits that occur during a treat-to-target approach with urate-lowering therapy.
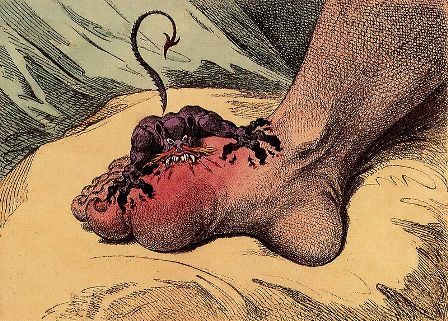
“Gout is a really painful disease when there are flares,” said Dr. Hammer, a senior consultant in the rheumatology department at Diakonhjemmet Hospital in Oslo. Ultrasound has been shown to be a sensitive method to detect uric monosodium urate (MSU) deposition and its use is included in the classification criteria for gout.
“MSU depositions are found in many different regions with some predilection sites,” Dr. Hammer noted. This led the OMERACT (Outcome Measures in Rheumatology) Ultrasound Group to develop three key definitions for MSU lesions: the “double contour sign” (DC), which occurs when urate crystals form on the surface of cartilage; tophus, which is where there is a larger, hypoechoic aggregation of crystals that are usually well delineated; and “aggregates,” which are small, hyperechoic deposits.
“There are, up until now, only a few smaller studies that have explored the decrease of depositions during uric acid–lowering treatment,” Dr. Hammer observed.
NOR-GOUT was a prospective, observational study of 161 consecutively-recruited patients with urate crystal–proven gout who needed treatment with urate-lowering therapy. Patients were included if they had a recent gout flare and had serum urate levels of more than 360 micromol/L (6.0 mg/dL) and had no contraindication to urate-lowering therapy.
“We used a treat-to-target approach with the medication,” Dr. Hammer explained. The aim was to get uric acid levels to 360 micromol/L or lower, or to less than 300 micromol/L (5.0 mg/dL) if clinical tophi were present. “The medication was optimized by monthly follow-up by a study nurse until the treatment target was met,” she added.
Patients underwent an extensive ultrasound assessment at study entry and again after 3, 6, and 12 months of urate-lowering therapy. This included bilateral assessment of all relevant joints and the presence of crystals semiquantitatively scored from 0 to 3, the latter signifying many deposits. The sum of scores for the three key OMERACT definitions were calculated each time the patients were assessed, with a total score for all three also calculated.
Mean serum urate levels dropped from a baseline of 487 to 312 micromol/L (from 8.2 to 5.3 mg/dL) at 12 months (P less than .001), Dr. Hammer reported. The percentage of patients achieving a urate target of less than 360 micromol/L increased from 71% at 3 months to 81% at 6 months and to 84% at 12 months, she said.
Ultrasound scores decreased with decreasing urate levels at 3, 6, and 12 months, with the highest numeric difference from baseline seen at 12 months for DC (3.1, 2.3, and 1.2; all P less than .001 vs. baseline of 4.2). The respective values for tophi were 6.3, 5.4, and 4.2 versus a baseline of 6.5; for aggregates, the values were 8.8, 7.9, and 6.7 versus a baseline of 9.1.
Standardized Response Mean values from baseline to 3, 6, and 12 months showed that DC was the most sensitive for change, with a respective 0.73, 1.02, and 1.26 in ultrasound scores. Values for tophi were 0.06, 0.57, and 0.91, and 0.20, 0.51 and 0.66 for aggregates.
“Not all patients had reached 12 months of follow-up when we made these calculations,” Dr. Hammer said, noting the limitations of the study. Nevertheless, these interim findings suggest that ultrasound is a valuable tool that can help see how patients fare on a treat-to-target approach, she concluded.
Dr. Hammer had no conflicts of interest to disclose.
SOURCE: Hammer HB et al. Ann Rheum Dis. 2018;77(Suppl 2):154-5. Abstract OP0211.
AMSTERDAM – Results from the longitudinal NOR-GOUT study show that ultrasound can help visualize decreasing levels of uric acid deposits that occur during a treat-to-target approach with urate-lowering therapy.
“Gout is a really painful disease when there are flares,” said Dr. Hammer, a senior consultant in the rheumatology department at Diakonhjemmet Hospital in Oslo. Ultrasound has been shown to be a sensitive method to detect uric monosodium urate (MSU) deposition and its use is included in the classification criteria for gout.
“MSU depositions are found in many different regions with some predilection sites,” Dr. Hammer noted. This led the OMERACT (Outcome Measures in Rheumatology) Ultrasound Group to develop three key definitions for MSU lesions: the “double contour sign” (DC), which occurs when urate crystals form on the surface of cartilage; tophus, which is where there is a larger, hypoechoic aggregation of crystals that are usually well delineated; and “aggregates,” which are small, hyperechoic deposits.
“There are, up until now, only a few smaller studies that have explored the decrease of depositions during uric acid–lowering treatment,” Dr. Hammer observed.
NOR-GOUT was a prospective, observational study of 161 consecutively-recruited patients with urate crystal–proven gout who needed treatment with urate-lowering therapy. Patients were included if they had a recent gout flare and had serum urate levels of more than 360 micromol/L (6.0 mg/dL) and had no contraindication to urate-lowering therapy.
“We used a treat-to-target approach with the medication,” Dr. Hammer explained. The aim was to get uric acid levels to 360 micromol/L or lower, or to less than 300 micromol/L (5.0 mg/dL) if clinical tophi were present. “The medication was optimized by monthly follow-up by a study nurse until the treatment target was met,” she added.
Patients underwent an extensive ultrasound assessment at study entry and again after 3, 6, and 12 months of urate-lowering therapy. This included bilateral assessment of all relevant joints and the presence of crystals semiquantitatively scored from 0 to 3, the latter signifying many deposits. The sum of scores for the three key OMERACT definitions were calculated each time the patients were assessed, with a total score for all three also calculated.
Mean serum urate levels dropped from a baseline of 487 to 312 micromol/L (from 8.2 to 5.3 mg/dL) at 12 months (P less than .001), Dr. Hammer reported. The percentage of patients achieving a urate target of less than 360 micromol/L increased from 71% at 3 months to 81% at 6 months and to 84% at 12 months, she said.
Ultrasound scores decreased with decreasing urate levels at 3, 6, and 12 months, with the highest numeric difference from baseline seen at 12 months for DC (3.1, 2.3, and 1.2; all P less than .001 vs. baseline of 4.2). The respective values for tophi were 6.3, 5.4, and 4.2 versus a baseline of 6.5; for aggregates, the values were 8.8, 7.9, and 6.7 versus a baseline of 9.1.
Standardized Response Mean values from baseline to 3, 6, and 12 months showed that DC was the most sensitive for change, with a respective 0.73, 1.02, and 1.26 in ultrasound scores. Values for tophi were 0.06, 0.57, and 0.91, and 0.20, 0.51 and 0.66 for aggregates.
“Not all patients had reached 12 months of follow-up when we made these calculations,” Dr. Hammer said, noting the limitations of the study. Nevertheless, these interim findings suggest that ultrasound is a valuable tool that can help see how patients fare on a treat-to-target approach, she concluded.
Dr. Hammer had no conflicts of interest to disclose.
SOURCE: Hammer HB et al. Ann Rheum Dis. 2018;77(Suppl 2):154-5. Abstract OP0211.
AMSTERDAM – Results from the longitudinal NOR-GOUT study show that ultrasound can help visualize decreasing levels of uric acid deposits that occur during a treat-to-target approach with urate-lowering therapy.
“Gout is a really painful disease when there are flares,” said Dr. Hammer, a senior consultant in the rheumatology department at Diakonhjemmet Hospital in Oslo. Ultrasound has been shown to be a sensitive method to detect uric monosodium urate (MSU) deposition and its use is included in the classification criteria for gout.
“MSU depositions are found in many different regions with some predilection sites,” Dr. Hammer noted. This led the OMERACT (Outcome Measures in Rheumatology) Ultrasound Group to develop three key definitions for MSU lesions: the “double contour sign” (DC), which occurs when urate crystals form on the surface of cartilage; tophus, which is where there is a larger, hypoechoic aggregation of crystals that are usually well delineated; and “aggregates,” which are small, hyperechoic deposits.
“There are, up until now, only a few smaller studies that have explored the decrease of depositions during uric acid–lowering treatment,” Dr. Hammer observed.
NOR-GOUT was a prospective, observational study of 161 consecutively-recruited patients with urate crystal–proven gout who needed treatment with urate-lowering therapy. Patients were included if they had a recent gout flare and had serum urate levels of more than 360 micromol/L (6.0 mg/dL) and had no contraindication to urate-lowering therapy.
“We used a treat-to-target approach with the medication,” Dr. Hammer explained. The aim was to get uric acid levels to 360 micromol/L or lower, or to less than 300 micromol/L (5.0 mg/dL) if clinical tophi were present. “The medication was optimized by monthly follow-up by a study nurse until the treatment target was met,” she added.
Patients underwent an extensive ultrasound assessment at study entry and again after 3, 6, and 12 months of urate-lowering therapy. This included bilateral assessment of all relevant joints and the presence of crystals semiquantitatively scored from 0 to 3, the latter signifying many deposits. The sum of scores for the three key OMERACT definitions were calculated each time the patients were assessed, with a total score for all three also calculated.
Mean serum urate levels dropped from a baseline of 487 to 312 micromol/L (from 8.2 to 5.3 mg/dL) at 12 months (P less than .001), Dr. Hammer reported. The percentage of patients achieving a urate target of less than 360 micromol/L increased from 71% at 3 months to 81% at 6 months and to 84% at 12 months, she said.
Ultrasound scores decreased with decreasing urate levels at 3, 6, and 12 months, with the highest numeric difference from baseline seen at 12 months for DC (3.1, 2.3, and 1.2; all P less than .001 vs. baseline of 4.2). The respective values for tophi were 6.3, 5.4, and 4.2 versus a baseline of 6.5; for aggregates, the values were 8.8, 7.9, and 6.7 versus a baseline of 9.1.
Standardized Response Mean values from baseline to 3, 6, and 12 months showed that DC was the most sensitive for change, with a respective 0.73, 1.02, and 1.26 in ultrasound scores. Values for tophi were 0.06, 0.57, and 0.91, and 0.20, 0.51 and 0.66 for aggregates.
“Not all patients had reached 12 months of follow-up when we made these calculations,” Dr. Hammer said, noting the limitations of the study. Nevertheless, these interim findings suggest that ultrasound is a valuable tool that can help see how patients fare on a treat-to-target approach, she concluded.
Dr. Hammer had no conflicts of interest to disclose.
SOURCE: Hammer HB et al. Ann Rheum Dis. 2018;77(Suppl 2):154-5. Abstract OP0211.
REPORTING FROM THE EULAR 2018 CONGRESS
Key clinical point: Deceasing uric acid load during a treat-to-target approach can be seen on ultrasound.
Major finding: Ultrasound scores decreased with decreasing urate levels at 3, 6 and 12 month, with the highest numeric difference from baseline seen at 12 months for double contour sign (3.1, 2.3, and 1.2; all P less than .001 vs. baseline of 4.2).
Study details: A prospective, observational study of 161 patients with urate crystal–proven gout treated with urate-lowering therapy.
Disclosures: Dr. Hammer had no conflicts of interest to disclose.
Sources: Hammer HB et al. Ann Rheum Dis. 2018;77(Suppl 2):154-5. Abstract OP0211.
Hydroxyurea well tolerated in longitudinal European study
WASHINGTON – European patients with sickle cell disease (SCD) are tolerating hydroxyurea treatment well, 9 years into a 10-year prospective cohort study.

ESCORT-HU (European Sickle Cell Disease Cohort – Hydroxyurea) is slated to enroll 2,000 children and adults over the study period to assess the long-term safety and efficacy of hydroxyurea (HU) as a treatment for SCD.
“Hydroxyurea is still underused in sickle cell disease” both in Europe and the United States, said Bernard Dauvergne, PharmD, executive director of Addmedica, which markets hydroxyurea as Siklos. In part, fears about toxicity and side effects contribute to this underuse, Dr. Dauvergne said at a top abstracts session of the annual meeting of the Foundation for Sickle Cell Disease Research.
ESCORT-HU was designed as a purely observational, noninterventional study of patients treated with HU. It was initiated as part of the European Medicines Agency risk management plan, conceived at the time of the drug’s approval for SCD, explained Dr. Dauvergne.
“This prospective cohort study could enable the identification of delayed rare adverse events in real life, occurring in between 1 in 10,000 and 1 in 1,000 patients,” Dr. Dauvergne said. However, “no new safety information has been detected ... and no secondary cancer has been reported.”
At the last data analysis cutpoint of June 2017, 1,921 patients had been enrolled. Patients have come from 63 centers in France, Germany, Greece, and Italy, Dr. Dauvergne reported.
Of the 1,829 patients with data available for analysis, 804 were under the age of 18. The pediatric patients were a mean 9 years of age, and the adult patients were a mean 33 years old at enrollment. Most enrollees (84%) have the HbSS genotype.
At baseline, the pediatric patients were dosed at a mean 17.2 mg/kg per day of HU; for adult patients, the figure was 15.54 mg/kg per day. More than one-third (35.4%) of pediatric patients had been on HU at the time of study enrollment, for a mean of 4.5 years. In contrast, more than half (60.4%) of adults had been taking hydroxyurea for a mean of 6.25 years at enrollment.
The 1,002 adult patients with recently reported safety data were on a mean HU dose of 20.2 mg/kg per day. About 40% of adults reported at least one adverse event (AE), and a total of 334 AEs in 16.3% of adult patients were judged to be related to HU.
Skin and subcutaneous tissue reactions occurred in about 1 in 10 patients, with 153 reported incidences of skin dryness, skin reactions, alopecia, or changes in skin pigmentation. The next most common adverse events were blood and lymphatic system disorders, mostly neutropenia or thrombocytopenia, occurring in 9.6% of patients.
Discontinuation rates have been low, with an overall dropout rate of 3.8%.
The ESCORT-HU data “confirm that HU is an efficient and well-tolerated drug in both children and adults,” Dr. Dauvergne said. Over the short term, adverse events are related to the expected dose dependent myelosuppression that’s key to the drug’s mechanism of action. This makes sense given the narrow therapeutic index, with a tight range between an efficacious dose and a cytotoxic one, he said. “And the lower the [patient] weight is, the more sensitive the dose variations become,” Dr. Dauvergne said.
Being flexible about fine-tuning doses is key to finding the balance between tolerability and efficacy, “and consequently, for compliance,” he said.
Data collection for the study will end in 2018, with safety and efficacy data expected in 2019.
WASHINGTON – European patients with sickle cell disease (SCD) are tolerating hydroxyurea treatment well, 9 years into a 10-year prospective cohort study.
ESCORT-HU (European Sickle Cell Disease Cohort – Hydroxyurea) is slated to enroll 2,000 children and adults over the study period to assess the long-term safety and efficacy of hydroxyurea (HU) as a treatment for SCD.
“Hydroxyurea is still underused in sickle cell disease” both in Europe and the United States, said Bernard Dauvergne, PharmD, executive director of Addmedica, which markets hydroxyurea as Siklos. In part, fears about toxicity and side effects contribute to this underuse, Dr. Dauvergne said at a top abstracts session of the annual meeting of the Foundation for Sickle Cell Disease Research.
ESCORT-HU was designed as a purely observational, noninterventional study of patients treated with HU. It was initiated as part of the European Medicines Agency risk management plan, conceived at the time of the drug’s approval for SCD, explained Dr. Dauvergne.
“This prospective cohort study could enable the identification of delayed rare adverse events in real life, occurring in between 1 in 10,000 and 1 in 1,000 patients,” Dr. Dauvergne said. However, “no new safety information has been detected ... and no secondary cancer has been reported.”
At the last data analysis cutpoint of June 2017, 1,921 patients had been enrolled. Patients have come from 63 centers in France, Germany, Greece, and Italy, Dr. Dauvergne reported.
Of the 1,829 patients with data available for analysis, 804 were under the age of 18. The pediatric patients were a mean 9 years of age, and the adult patients were a mean 33 years old at enrollment. Most enrollees (84%) have the HbSS genotype.
At baseline, the pediatric patients were dosed at a mean 17.2 mg/kg per day of HU; for adult patients, the figure was 15.54 mg/kg per day. More than one-third (35.4%) of pediatric patients had been on HU at the time of study enrollment, for a mean of 4.5 years. In contrast, more than half (60.4%) of adults had been taking hydroxyurea for a mean of 6.25 years at enrollment.
The 1,002 adult patients with recently reported safety data were on a mean HU dose of 20.2 mg/kg per day. About 40% of adults reported at least one adverse event (AE), and a total of 334 AEs in 16.3% of adult patients were judged to be related to HU.
Skin and subcutaneous tissue reactions occurred in about 1 in 10 patients, with 153 reported incidences of skin dryness, skin reactions, alopecia, or changes in skin pigmentation. The next most common adverse events were blood and lymphatic system disorders, mostly neutropenia or thrombocytopenia, occurring in 9.6% of patients.
Discontinuation rates have been low, with an overall dropout rate of 3.8%.
The ESCORT-HU data “confirm that HU is an efficient and well-tolerated drug in both children and adults,” Dr. Dauvergne said. Over the short term, adverse events are related to the expected dose dependent myelosuppression that’s key to the drug’s mechanism of action. This makes sense given the narrow therapeutic index, with a tight range between an efficacious dose and a cytotoxic one, he said. “And the lower the [patient] weight is, the more sensitive the dose variations become,” Dr. Dauvergne said.
Being flexible about fine-tuning doses is key to finding the balance between tolerability and efficacy, “and consequently, for compliance,” he said.
Data collection for the study will end in 2018, with safety and efficacy data expected in 2019.
WASHINGTON – European patients with sickle cell disease (SCD) are tolerating hydroxyurea treatment well, 9 years into a 10-year prospective cohort study.
ESCORT-HU (European Sickle Cell Disease Cohort – Hydroxyurea) is slated to enroll 2,000 children and adults over the study period to assess the long-term safety and efficacy of hydroxyurea (HU) as a treatment for SCD.
“Hydroxyurea is still underused in sickle cell disease” both in Europe and the United States, said Bernard Dauvergne, PharmD, executive director of Addmedica, which markets hydroxyurea as Siklos. In part, fears about toxicity and side effects contribute to this underuse, Dr. Dauvergne said at a top abstracts session of the annual meeting of the Foundation for Sickle Cell Disease Research.
ESCORT-HU was designed as a purely observational, noninterventional study of patients treated with HU. It was initiated as part of the European Medicines Agency risk management plan, conceived at the time of the drug’s approval for SCD, explained Dr. Dauvergne.
“This prospective cohort study could enable the identification of delayed rare adverse events in real life, occurring in between 1 in 10,000 and 1 in 1,000 patients,” Dr. Dauvergne said. However, “no new safety information has been detected ... and no secondary cancer has been reported.”
At the last data analysis cutpoint of June 2017, 1,921 patients had been enrolled. Patients have come from 63 centers in France, Germany, Greece, and Italy, Dr. Dauvergne reported.
Of the 1,829 patients with data available for analysis, 804 were under the age of 18. The pediatric patients were a mean 9 years of age, and the adult patients were a mean 33 years old at enrollment. Most enrollees (84%) have the HbSS genotype.
At baseline, the pediatric patients were dosed at a mean 17.2 mg/kg per day of HU; for adult patients, the figure was 15.54 mg/kg per day. More than one-third (35.4%) of pediatric patients had been on HU at the time of study enrollment, for a mean of 4.5 years. In contrast, more than half (60.4%) of adults had been taking hydroxyurea for a mean of 6.25 years at enrollment.
The 1,002 adult patients with recently reported safety data were on a mean HU dose of 20.2 mg/kg per day. About 40% of adults reported at least one adverse event (AE), and a total of 334 AEs in 16.3% of adult patients were judged to be related to HU.
Skin and subcutaneous tissue reactions occurred in about 1 in 10 patients, with 153 reported incidences of skin dryness, skin reactions, alopecia, or changes in skin pigmentation. The next most common adverse events were blood and lymphatic system disorders, mostly neutropenia or thrombocytopenia, occurring in 9.6% of patients.
Discontinuation rates have been low, with an overall dropout rate of 3.8%.
The ESCORT-HU data “confirm that HU is an efficient and well-tolerated drug in both children and adults,” Dr. Dauvergne said. Over the short term, adverse events are related to the expected dose dependent myelosuppression that’s key to the drug’s mechanism of action. This makes sense given the narrow therapeutic index, with a tight range between an efficacious dose and a cytotoxic one, he said. “And the lower the [patient] weight is, the more sensitive the dose variations become,” Dr. Dauvergne said.
Being flexible about fine-tuning doses is key to finding the balance between tolerability and efficacy, “and consequently, for compliance,” he said.
Data collection for the study will end in 2018, with safety and efficacy data expected in 2019.
REPORTING FROM FSCDR 2018
Key clinical point:
Major finding: Of adult participants, 16.9% have experienced hydroxyurea-related adverse events.
Study details: Observational 10-year study of 2,000 adults and children with SCD who were taking hydroxyurea.
Disclosures: The study was sponsored by Addmedica, which markets hydroxyurea as Siklos. Dr. Dauvergne is employed by Addmedica.
Source: Dauvergne B et al. FSCDR 2018, presentation JSCDH-D-18-00052.
PET-driven chemo strategy helps reduce toxicity in Hodgkin lymphoma
CHICAGO – Positron emission tomography (PET) performed after two cycles of BEACOPP could help identify a subset of advanced-stage Hodgkin lymphoma patients who can receive de-escalated treatment without compromising disease control, results of a phase 3 randomized trial show.
Five-year progression-free survival (PFS) exceeded 85% not only for patients receiving six cycles of escalated BEACOPP (bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, and prednisone) but also for patients who were de-escalated to ABVD (doxorubicin, bleomycin, vinblastine, dacarbazine) chemotherapy based on negative PET results, according to the final analysis of the AHL2011 LYSA study, presented at the annual meeting of the American Society of Clinical Oncology.
“This approach allows us to significantly reduce the treatment-related toxicity in most patients, and provides similar patient outcomes compared to standard BEACOPP escalated treatment,” said Olivier Casasnovas, MD, of the University Hospital Le Bocage and Inserm, Dijon, France.
Previous studies have shown that BEACOPP may improve PFS, compared with ABVD, but is more toxic and associated with a higher risk of infertility and myelodysplasia or acute leukemia. Dr. Casasnovas and his colleagues sought to evaluate whether upfront BEACOPP followed by de-escalation to ABVD, when warranted by negative PET results, would improve outcomes without compromising efficacy.
The AHL2011 LYSA study included 823 patients (median age, 30 years; 63% male) with previously untreated advanced classical Hodgkin lymphoma. All patients received PET at baseline, after cycle two of chemotherapy, and again after cycle four.
Patients were randomized either to six cycles of escalated BEACOPP, or to an experimental arm in which patients started with BEACOPP but were de-escalated to ABVD if PET results were negative after two or four cycles of treatment.
PET results after cycle two were negative for 87% of patients in the experimental arm, so on an intent-to-treat basis, 84% of them received two cycles of BEACOPP and four cycles of ABVD, Dr. Casasnovas reported.
PFS, with a median follow-up of 50.4 months, was not significantly different for the standard versus the experimental arm (hazard ratio, 1.084; 95% confidence interval, 0.73-1.59; P = .68). Five-year PFS was 85.7% in the experimental treatment de-escalation arm, compared with 86.2% in the standard arm.
Overall survival was likewise similar between arms, with 5-year overall survival exceeding 95% in both groups, Dr. Casasnovas said.
Although there was no significant difference overall in the incidence of adverse events, there was significantly less anemia, febrile neutropenia, thrombocytopenia, and sepsis in the PET-driven de-escalation arm. Overall, 27% of patients in the standard chemotherapy arm experienced at least one serious adverse event, compared with 17% in the PET-driven arm (P less than .002).
The incidence of second primary malignancies was numerically lower in the experimental arm (1.2% vs. 2.4%), though that finding did not reach statistical significance.
On multivariate analysis, interim PET positivity after cycle two was associated with increased risk of disease progression for patients in this study (HR, 3.316). Risk of progression was even higher for patients with PET positivity after four cycles (HR, 12.968), identifying a subset of patients with “particularly poor outcome” who could benefit from development of new treatment options, Dr. Casasnovas said.
Dr. Casasnovas reported financial ties to AbbVie, Bristol-Myers Squibb, Celgene, Gilead Sciences, Janssen, Merck, Roche/Genentech, Sanofi, and Takeda.
SOURCE: Casasnovas O et al. ASCO 2018, Abstract 7503.
CHICAGO – Positron emission tomography (PET) performed after two cycles of BEACOPP could help identify a subset of advanced-stage Hodgkin lymphoma patients who can receive de-escalated treatment without compromising disease control, results of a phase 3 randomized trial show.
Five-year progression-free survival (PFS) exceeded 85% not only for patients receiving six cycles of escalated BEACOPP (bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, and prednisone) but also for patients who were de-escalated to ABVD (doxorubicin, bleomycin, vinblastine, dacarbazine) chemotherapy based on negative PET results, according to the final analysis of the AHL2011 LYSA study, presented at the annual meeting of the American Society of Clinical Oncology.
“This approach allows us to significantly reduce the treatment-related toxicity in most patients, and provides similar patient outcomes compared to standard BEACOPP escalated treatment,” said Olivier Casasnovas, MD, of the University Hospital Le Bocage and Inserm, Dijon, France.
Previous studies have shown that BEACOPP may improve PFS, compared with ABVD, but is more toxic and associated with a higher risk of infertility and myelodysplasia or acute leukemia. Dr. Casasnovas and his colleagues sought to evaluate whether upfront BEACOPP followed by de-escalation to ABVD, when warranted by negative PET results, would improve outcomes without compromising efficacy.
The AHL2011 LYSA study included 823 patients (median age, 30 years; 63% male) with previously untreated advanced classical Hodgkin lymphoma. All patients received PET at baseline, after cycle two of chemotherapy, and again after cycle four.
Patients were randomized either to six cycles of escalated BEACOPP, or to an experimental arm in which patients started with BEACOPP but were de-escalated to ABVD if PET results were negative after two or four cycles of treatment.
PET results after cycle two were negative for 87% of patients in the experimental arm, so on an intent-to-treat basis, 84% of them received two cycles of BEACOPP and four cycles of ABVD, Dr. Casasnovas reported.
PFS, with a median follow-up of 50.4 months, was not significantly different for the standard versus the experimental arm (hazard ratio, 1.084; 95% confidence interval, 0.73-1.59; P = .68). Five-year PFS was 85.7% in the experimental treatment de-escalation arm, compared with 86.2% in the standard arm.
Overall survival was likewise similar between arms, with 5-year overall survival exceeding 95% in both groups, Dr. Casasnovas said.
Although there was no significant difference overall in the incidence of adverse events, there was significantly less anemia, febrile neutropenia, thrombocytopenia, and sepsis in the PET-driven de-escalation arm. Overall, 27% of patients in the standard chemotherapy arm experienced at least one serious adverse event, compared with 17% in the PET-driven arm (P less than .002).
The incidence of second primary malignancies was numerically lower in the experimental arm (1.2% vs. 2.4%), though that finding did not reach statistical significance.
On multivariate analysis, interim PET positivity after cycle two was associated with increased risk of disease progression for patients in this study (HR, 3.316). Risk of progression was even higher for patients with PET positivity after four cycles (HR, 12.968), identifying a subset of patients with “particularly poor outcome” who could benefit from development of new treatment options, Dr. Casasnovas said.
Dr. Casasnovas reported financial ties to AbbVie, Bristol-Myers Squibb, Celgene, Gilead Sciences, Janssen, Merck, Roche/Genentech, Sanofi, and Takeda.
SOURCE: Casasnovas O et al. ASCO 2018, Abstract 7503.
CHICAGO – Positron emission tomography (PET) performed after two cycles of BEACOPP could help identify a subset of advanced-stage Hodgkin lymphoma patients who can receive de-escalated treatment without compromising disease control, results of a phase 3 randomized trial show.
Five-year progression-free survival (PFS) exceeded 85% not only for patients receiving six cycles of escalated BEACOPP (bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, and prednisone) but also for patients who were de-escalated to ABVD (doxorubicin, bleomycin, vinblastine, dacarbazine) chemotherapy based on negative PET results, according to the final analysis of the AHL2011 LYSA study, presented at the annual meeting of the American Society of Clinical Oncology.
“This approach allows us to significantly reduce the treatment-related toxicity in most patients, and provides similar patient outcomes compared to standard BEACOPP escalated treatment,” said Olivier Casasnovas, MD, of the University Hospital Le Bocage and Inserm, Dijon, France.
Previous studies have shown that BEACOPP may improve PFS, compared with ABVD, but is more toxic and associated with a higher risk of infertility and myelodysplasia or acute leukemia. Dr. Casasnovas and his colleagues sought to evaluate whether upfront BEACOPP followed by de-escalation to ABVD, when warranted by negative PET results, would improve outcomes without compromising efficacy.
The AHL2011 LYSA study included 823 patients (median age, 30 years; 63% male) with previously untreated advanced classical Hodgkin lymphoma. All patients received PET at baseline, after cycle two of chemotherapy, and again after cycle four.
Patients were randomized either to six cycles of escalated BEACOPP, or to an experimental arm in which patients started with BEACOPP but were de-escalated to ABVD if PET results were negative after two or four cycles of treatment.
PET results after cycle two were negative for 87% of patients in the experimental arm, so on an intent-to-treat basis, 84% of them received two cycles of BEACOPP and four cycles of ABVD, Dr. Casasnovas reported.
PFS, with a median follow-up of 50.4 months, was not significantly different for the standard versus the experimental arm (hazard ratio, 1.084; 95% confidence interval, 0.73-1.59; P = .68). Five-year PFS was 85.7% in the experimental treatment de-escalation arm, compared with 86.2% in the standard arm.
Overall survival was likewise similar between arms, with 5-year overall survival exceeding 95% in both groups, Dr. Casasnovas said.
Although there was no significant difference overall in the incidence of adverse events, there was significantly less anemia, febrile neutropenia, thrombocytopenia, and sepsis in the PET-driven de-escalation arm. Overall, 27% of patients in the standard chemotherapy arm experienced at least one serious adverse event, compared with 17% in the PET-driven arm (P less than .002).
The incidence of second primary malignancies was numerically lower in the experimental arm (1.2% vs. 2.4%), though that finding did not reach statistical significance.
On multivariate analysis, interim PET positivity after cycle two was associated with increased risk of disease progression for patients in this study (HR, 3.316). Risk of progression was even higher for patients with PET positivity after four cycles (HR, 12.968), identifying a subset of patients with “particularly poor outcome” who could benefit from development of new treatment options, Dr. Casasnovas said.
Dr. Casasnovas reported financial ties to AbbVie, Bristol-Myers Squibb, Celgene, Gilead Sciences, Janssen, Merck, Roche/Genentech, Sanofi, and Takeda.
SOURCE: Casasnovas O et al. ASCO 2018, Abstract 7503.
REPORTING FROM ASCO 2018
Key clinical point:
Major finding: With a median follow-up of 50.4 months, PFS was not significantly different for escalated BEACOPP vs. the experimental arm (HR, 1.084; 95% CI, 0.73-1.59; P = .68).
Study details: Final analysis of AHL2011 LYSA, a randomized phase 3 study including 823 patients with advanced-stage Hodgkin lymphoma.
Disclosures: Dr. Casasnovas reported financial ties to AbbVie, Bristol-Myers Squibb, Celgene, Gilead Sciences, Janssen, Merck, Roche/Genentech, Sanofi, and Takeda.
Source: Casasnovas O et al. ASCO 2018, Abstract 7503.
EUS and MRCP as complementary studies in the etiologic diagnosis of idiopathic acute pancreatitis
Background: Approximately 10%-30% of patients with acute pancreatitis do not have an established etiology after routine investigation with imaging. These patients are classified as IAP. Less invasive tests, such as EUS, MRCP, and secretin stimulation MRCP (S-MRCP) have been used to further explore IAP, but their comparison in the etiologic diagnosis of idiopathic acute pancreatitis is lacking.
Study design: Meta-analysis involving 34 studies that investigated the etiology of IAP with MRCP and/or S-MRCP and/or EUS.
Setting: Brazil, Canada, China, France, Hong Kong, India, Italy, Korea, Spain, the United Kingdom, and the United States.
Synopsis: When EUS was compared with MRCP, the diagnostic yield of EUS (153/239 patients; 64%) was higher than that of MRCP (82/238 patients; 34%; P less than .01). Specifically, EUS seemed to have a significant benefit in detecting biliary disease, compared with MRCP. In the subgroup analysis, the diagnostic yield of EUS was higher than that of MRCP for detecting parenchymal changes suggestive of chronic pancreatitis (10% vs. 1%). S-MRCP was superior to EUS and MRCP (12% vs. 2% vs. 2%, respectively) in diagnosing pancreatic divisum, a congenital anomaly that is prevalent in 5%-14% of the population and an etiology of IAP.
A limitation in this meta-analysis was that EUS was used in seven times as many studies as was MRCP, which may have influenced the overall results.
Bottom line: Less invasive modalities, such as EUS and S-MRCP, used together could improve the diagnostic yield in evaluating the etiology of AIP.
Citation: Wang J et al. Comparison of EUS with MRCP in idiopathic acute pancreatitis: A systemic review and meta-analysis. Gastrointest Endosc. 2017 Dec 7. doi: 10.1016/j.gie.2017.11.028.

Dr. Farkhondehpour is a hospitalist at UC San Diego Health and an assistant clinical professor at the University of California, San Diego.
Background: Approximately 10%-30% of patients with acute pancreatitis do not have an established etiology after routine investigation with imaging. These patients are classified as IAP. Less invasive tests, such as EUS, MRCP, and secretin stimulation MRCP (S-MRCP) have been used to further explore IAP, but their comparison in the etiologic diagnosis of idiopathic acute pancreatitis is lacking.
Study design: Meta-analysis involving 34 studies that investigated the etiology of IAP with MRCP and/or S-MRCP and/or EUS.
Setting: Brazil, Canada, China, France, Hong Kong, India, Italy, Korea, Spain, the United Kingdom, and the United States.
Synopsis: When EUS was compared with MRCP, the diagnostic yield of EUS (153/239 patients; 64%) was higher than that of MRCP (82/238 patients; 34%; P less than .01). Specifically, EUS seemed to have a significant benefit in detecting biliary disease, compared with MRCP. In the subgroup analysis, the diagnostic yield of EUS was higher than that of MRCP for detecting parenchymal changes suggestive of chronic pancreatitis (10% vs. 1%). S-MRCP was superior to EUS and MRCP (12% vs. 2% vs. 2%, respectively) in diagnosing pancreatic divisum, a congenital anomaly that is prevalent in 5%-14% of the population and an etiology of IAP.
A limitation in this meta-analysis was that EUS was used in seven times as many studies as was MRCP, which may have influenced the overall results.
Bottom line: Less invasive modalities, such as EUS and S-MRCP, used together could improve the diagnostic yield in evaluating the etiology of AIP.
Citation: Wang J et al. Comparison of EUS with MRCP in idiopathic acute pancreatitis: A systemic review and meta-analysis. Gastrointest Endosc. 2017 Dec 7. doi: 10.1016/j.gie.2017.11.028.
Dr. Farkhondehpour is a hospitalist at UC San Diego Health and an assistant clinical professor at the University of California, San Diego.
Background: Approximately 10%-30% of patients with acute pancreatitis do not have an established etiology after routine investigation with imaging. These patients are classified as IAP. Less invasive tests, such as EUS, MRCP, and secretin stimulation MRCP (S-MRCP) have been used to further explore IAP, but their comparison in the etiologic diagnosis of idiopathic acute pancreatitis is lacking.
Study design: Meta-analysis involving 34 studies that investigated the etiology of IAP with MRCP and/or S-MRCP and/or EUS.
Setting: Brazil, Canada, China, France, Hong Kong, India, Italy, Korea, Spain, the United Kingdom, and the United States.
Synopsis: When EUS was compared with MRCP, the diagnostic yield of EUS (153/239 patients; 64%) was higher than that of MRCP (82/238 patients; 34%; P less than .01). Specifically, EUS seemed to have a significant benefit in detecting biliary disease, compared with MRCP. In the subgroup analysis, the diagnostic yield of EUS was higher than that of MRCP for detecting parenchymal changes suggestive of chronic pancreatitis (10% vs. 1%). S-MRCP was superior to EUS and MRCP (12% vs. 2% vs. 2%, respectively) in diagnosing pancreatic divisum, a congenital anomaly that is prevalent in 5%-14% of the population and an etiology of IAP.
A limitation in this meta-analysis was that EUS was used in seven times as many studies as was MRCP, which may have influenced the overall results.
Bottom line: Less invasive modalities, such as EUS and S-MRCP, used together could improve the diagnostic yield in evaluating the etiology of AIP.
Citation: Wang J et al. Comparison of EUS with MRCP in idiopathic acute pancreatitis: A systemic review and meta-analysis. Gastrointest Endosc. 2017 Dec 7. doi: 10.1016/j.gie.2017.11.028.
Dr. Farkhondehpour is a hospitalist at UC San Diego Health and an assistant clinical professor at the University of California, San Diego.
Patients With Concussions Aren’t Getting Enough Follow-Up
Many patients with traumatic brain injury (TBI) may not be receiving follow-up care, according to findings from Transforming Research and Clinical Knowledge in Traumatic Brain Injury (TRACK-TBI), a long-term NIH-funded study.
Of 831 patients who completed questionnaires 2 weeks and 3 months after sustaining TBI, 44% reported seeing a health care provider within 3 months. Of those, 15% visited a clinic that specialized in head injury. Approximately half saw a general practitioner; close to a third reported seeing ≥ 1 type of doctor.
Among the 279 patients with ≥ 3 symptoms of moderate to severe postconcussion, 41% had not had a follow-up visit at 3 months. Moreover, half of the patients were discharged without TBI educational materials.
Rates and components of follow-up care varied widely from institution to institution even among patients with the same initial degree of injury.
Many patients with traumatic brain injury (TBI) may not be receiving follow-up care, according to findings from Transforming Research and Clinical Knowledge in Traumatic Brain Injury (TRACK-TBI), a long-term NIH-funded study.
Of 831 patients who completed questionnaires 2 weeks and 3 months after sustaining TBI, 44% reported seeing a health care provider within 3 months. Of those, 15% visited a clinic that specialized in head injury. Approximately half saw a general practitioner; close to a third reported seeing ≥ 1 type of doctor.
Among the 279 patients with ≥ 3 symptoms of moderate to severe postconcussion, 41% had not had a follow-up visit at 3 months. Moreover, half of the patients were discharged without TBI educational materials.
Rates and components of follow-up care varied widely from institution to institution even among patients with the same initial degree of injury.
Many patients with traumatic brain injury (TBI) may not be receiving follow-up care, according to findings from Transforming Research and Clinical Knowledge in Traumatic Brain Injury (TRACK-TBI), a long-term NIH-funded study.
Of 831 patients who completed questionnaires 2 weeks and 3 months after sustaining TBI, 44% reported seeing a health care provider within 3 months. Of those, 15% visited a clinic that specialized in head injury. Approximately half saw a general practitioner; close to a third reported seeing ≥ 1 type of doctor.
Among the 279 patients with ≥ 3 symptoms of moderate to severe postconcussion, 41% had not had a follow-up visit at 3 months. Moreover, half of the patients were discharged without TBI educational materials.
Rates and components of follow-up care varied widely from institution to institution even among patients with the same initial degree of injury.
Talk Therapy—Without Words
Many forms of therapy rely on the patient being able to verbally communicate thoughts and feelings. Arts-based therapies, however, can help people explore sensitive and controversial topics that might be hard to talk about.
Body mapping, which has been in use for more than 30 years, is an interesting, revealing, and productive way of using art to help patients “talk” about their thoughts and feelings. Several studies have used body maps as therapy for patients with HIV/AIDS, but researchers from University of New South Wales, Sydney, theorized that it could be particularly helpful for patients with cognitive disability who have complex support needs as well as those who are socially marginalized.
In body-mapping sessions, participants trace outlines of their bodies and then “populate” the outlines with drawings, magazine photos, symbols, words, and other visual representations of the experience they are investigating. It is a form of storytelling that allows the participant to engage physically, visually, verbally, and relationally (through dialogue and interaction with the researcher).
The researchers used body mapping in 2 studies, first with 29 adults with cognitive disability and complex support needs, such as mental illness and sensory impairment, to explore experiences of support planning. In the second study, one of the researchers used body mapping with 13 teens and young adults with complex support needs (eg, drug and alcohol misuse) to explore support they received during a life transition.
The body-mapping technique, the researchers say, shifts the power balance between researcher and participant, because the patient is in control of the images used and where they are placed on the map. Patients could decide not only how they spoke about the topic, but also which topic they spoke about. The researchers say, “we were often taken to surprising places” that might not have come up in an interview, as when participants used images to reveal aspects of cultural heritage and sexual orientation that had not come up in conversation. For example, a transsexual woman in the second study was uncomfortable with the process until she covered her incorrectly gendered body with another piece of paper on which she could redraw her body as she wished.
Body mapping does not suit everyone, the researchers acknowledge. Participants need to be able to engage in a level of abstraction and reflection about personal experiences. It is important to have other methods available for those who do not want to take part. The researchers say one way they protected patients was by recruiting through service providers so support could be “embedded in existing relationships.” The potential vulnerabilities of the patients mean researchers need to be flexible, they add, and allow the method to evolve, much like the patients’ personal stories.
Source:
Dew A, Smith L, Collings S, Savage ID. FQS. 2018;19(2).
doi: http://dx.doi.org/10.17169/fqs-19.2.2929.
Many forms of therapy rely on the patient being able to verbally communicate thoughts and feelings. Arts-based therapies, however, can help people explore sensitive and controversial topics that might be hard to talk about.
Body mapping, which has been in use for more than 30 years, is an interesting, revealing, and productive way of using art to help patients “talk” about their thoughts and feelings. Several studies have used body maps as therapy for patients with HIV/AIDS, but researchers from University of New South Wales, Sydney, theorized that it could be particularly helpful for patients with cognitive disability who have complex support needs as well as those who are socially marginalized.
In body-mapping sessions, participants trace outlines of their bodies and then “populate” the outlines with drawings, magazine photos, symbols, words, and other visual representations of the experience they are investigating. It is a form of storytelling that allows the participant to engage physically, visually, verbally, and relationally (through dialogue and interaction with the researcher).
The researchers used body mapping in 2 studies, first with 29 adults with cognitive disability and complex support needs, such as mental illness and sensory impairment, to explore experiences of support planning. In the second study, one of the researchers used body mapping with 13 teens and young adults with complex support needs (eg, drug and alcohol misuse) to explore support they received during a life transition.
The body-mapping technique, the researchers say, shifts the power balance between researcher and participant, because the patient is in control of the images used and where they are placed on the map. Patients could decide not only how they spoke about the topic, but also which topic they spoke about. The researchers say, “we were often taken to surprising places” that might not have come up in an interview, as when participants used images to reveal aspects of cultural heritage and sexual orientation that had not come up in conversation. For example, a transsexual woman in the second study was uncomfortable with the process until she covered her incorrectly gendered body with another piece of paper on which she could redraw her body as she wished.
Body mapping does not suit everyone, the researchers acknowledge. Participants need to be able to engage in a level of abstraction and reflection about personal experiences. It is important to have other methods available for those who do not want to take part. The researchers say one way they protected patients was by recruiting through service providers so support could be “embedded in existing relationships.” The potential vulnerabilities of the patients mean researchers need to be flexible, they add, and allow the method to evolve, much like the patients’ personal stories.
Source:
Dew A, Smith L, Collings S, Savage ID. FQS. 2018;19(2).
doi: http://dx.doi.org/10.17169/fqs-19.2.2929.
Many forms of therapy rely on the patient being able to verbally communicate thoughts and feelings. Arts-based therapies, however, can help people explore sensitive and controversial topics that might be hard to talk about.
Body mapping, which has been in use for more than 30 years, is an interesting, revealing, and productive way of using art to help patients “talk” about their thoughts and feelings. Several studies have used body maps as therapy for patients with HIV/AIDS, but researchers from University of New South Wales, Sydney, theorized that it could be particularly helpful for patients with cognitive disability who have complex support needs as well as those who are socially marginalized.
In body-mapping sessions, participants trace outlines of their bodies and then “populate” the outlines with drawings, magazine photos, symbols, words, and other visual representations of the experience they are investigating. It is a form of storytelling that allows the participant to engage physically, visually, verbally, and relationally (through dialogue and interaction with the researcher).
The researchers used body mapping in 2 studies, first with 29 adults with cognitive disability and complex support needs, such as mental illness and sensory impairment, to explore experiences of support planning. In the second study, one of the researchers used body mapping with 13 teens and young adults with complex support needs (eg, drug and alcohol misuse) to explore support they received during a life transition.
The body-mapping technique, the researchers say, shifts the power balance between researcher and participant, because the patient is in control of the images used and where they are placed on the map. Patients could decide not only how they spoke about the topic, but also which topic they spoke about. The researchers say, “we were often taken to surprising places” that might not have come up in an interview, as when participants used images to reveal aspects of cultural heritage and sexual orientation that had not come up in conversation. For example, a transsexual woman in the second study was uncomfortable with the process until she covered her incorrectly gendered body with another piece of paper on which she could redraw her body as she wished.
Body mapping does not suit everyone, the researchers acknowledge. Participants need to be able to engage in a level of abstraction and reflection about personal experiences. It is important to have other methods available for those who do not want to take part. The researchers say one way they protected patients was by recruiting through service providers so support could be “embedded in existing relationships.” The potential vulnerabilities of the patients mean researchers need to be flexible, they add, and allow the method to evolve, much like the patients’ personal stories.
Source:
Dew A, Smith L, Collings S, Savage ID. FQS. 2018;19(2).
doi: http://dx.doi.org/10.17169/fqs-19.2.2929.