User login
Greening intervention tied to self-reported improved mental health
Living close to an urban greening program improved mental health in low-income city dwellers, according to results from a randomized trial, in which the greening intervention consisted of removing trash from vacant lots, grading the land, planting new grass and trees, installing wooden fences, and performing maintenance.

Options for depression treatment are limited, and this has led researchers to consider environmental factors that might contribute to the condition. Vacant or dilapidated neighborhood spaces are a known factor for depression.
The researchers performed a citywide cluster randomized trial in Philadelphia, which included 541 vacant lots in 110 clusters. A total of 37 clusters were assigned to the greening intervention, 36 to the trash cleanup intervention, and 37 clusters were left alone. The researchers interviewed local residents near each cluster, using the short-form Kessler-6 Psychological Distress Scale to assess mental health outcomes, wrote Dr. South, of the University of Pennsylvania, Philadelphia.
The researchers interviewed 442 participants (mean age 44.6 years, 59.7% female) in the preintervention period, and 342 (77.4%) during the postintervention period. The researchers conducted a neighborhood poverty subset analysis that included 139 participants.
Intention-to-treat analyses showed that, compared with no intervention, those in the greening intervention experienced a significant decrease in reporting feeling depressed (–41.5%; 95% confidence interval, –63.6 to –5.9; P = .03) and feeling worthless (–50.9%; 95% CI, –74.7 to –4.7; P = .04). The investigators also found a trend toward a reduction in participants who self-reported being in poor mental health (–62.8%; 95% CI, –86.5 to –27.5; P = .051).
When the analysis was restricted to neighborhoods below the poverty line, the greening intervention led to a significant decrease in participants feeling depressed (–68.7%; 95% CI, –86.5 to –27.5; P =.007), but no statistically significant difference was found in participants with self-reported poor mental health.
The trash cleanup intervention had no significant associations with changes in mental health outcomes, compared with no intervention.
Dr. South reported having no disclosures. The study was funded by the National Institutes of Health, and the Centers for Disease Control and Prevention.
SOURCE: South EC et al. JAMA Network Open. 2018 Jul 20. doi: 10.1001/jamanetworkopen.2018.0298.
Living close to an urban greening program improved mental health in low-income city dwellers, according to results from a randomized trial, in which the greening intervention consisted of removing trash from vacant lots, grading the land, planting new grass and trees, installing wooden fences, and performing maintenance.
Options for depression treatment are limited, and this has led researchers to consider environmental factors that might contribute to the condition. Vacant or dilapidated neighborhood spaces are a known factor for depression.
The researchers performed a citywide cluster randomized trial in Philadelphia, which included 541 vacant lots in 110 clusters. A total of 37 clusters were assigned to the greening intervention, 36 to the trash cleanup intervention, and 37 clusters were left alone. The researchers interviewed local residents near each cluster, using the short-form Kessler-6 Psychological Distress Scale to assess mental health outcomes, wrote Dr. South, of the University of Pennsylvania, Philadelphia.
The researchers interviewed 442 participants (mean age 44.6 years, 59.7% female) in the preintervention period, and 342 (77.4%) during the postintervention period. The researchers conducted a neighborhood poverty subset analysis that included 139 participants.
Intention-to-treat analyses showed that, compared with no intervention, those in the greening intervention experienced a significant decrease in reporting feeling depressed (–41.5%; 95% confidence interval, –63.6 to –5.9; P = .03) and feeling worthless (–50.9%; 95% CI, –74.7 to –4.7; P = .04). The investigators also found a trend toward a reduction in participants who self-reported being in poor mental health (–62.8%; 95% CI, –86.5 to –27.5; P = .051).
When the analysis was restricted to neighborhoods below the poverty line, the greening intervention led to a significant decrease in participants feeling depressed (–68.7%; 95% CI, –86.5 to –27.5; P =.007), but no statistically significant difference was found in participants with self-reported poor mental health.
The trash cleanup intervention had no significant associations with changes in mental health outcomes, compared with no intervention.
Dr. South reported having no disclosures. The study was funded by the National Institutes of Health, and the Centers for Disease Control and Prevention.
SOURCE: South EC et al. JAMA Network Open. 2018 Jul 20. doi: 10.1001/jamanetworkopen.2018.0298.
Living close to an urban greening program improved mental health in low-income city dwellers, according to results from a randomized trial, in which the greening intervention consisted of removing trash from vacant lots, grading the land, planting new grass and trees, installing wooden fences, and performing maintenance.
Options for depression treatment are limited, and this has led researchers to consider environmental factors that might contribute to the condition. Vacant or dilapidated neighborhood spaces are a known factor for depression.
The researchers performed a citywide cluster randomized trial in Philadelphia, which included 541 vacant lots in 110 clusters. A total of 37 clusters were assigned to the greening intervention, 36 to the trash cleanup intervention, and 37 clusters were left alone. The researchers interviewed local residents near each cluster, using the short-form Kessler-6 Psychological Distress Scale to assess mental health outcomes, wrote Dr. South, of the University of Pennsylvania, Philadelphia.
The researchers interviewed 442 participants (mean age 44.6 years, 59.7% female) in the preintervention period, and 342 (77.4%) during the postintervention period. The researchers conducted a neighborhood poverty subset analysis that included 139 participants.
Intention-to-treat analyses showed that, compared with no intervention, those in the greening intervention experienced a significant decrease in reporting feeling depressed (–41.5%; 95% confidence interval, –63.6 to –5.9; P = .03) and feeling worthless (–50.9%; 95% CI, –74.7 to –4.7; P = .04). The investigators also found a trend toward a reduction in participants who self-reported being in poor mental health (–62.8%; 95% CI, –86.5 to –27.5; P = .051).
When the analysis was restricted to neighborhoods below the poverty line, the greening intervention led to a significant decrease in participants feeling depressed (–68.7%; 95% CI, –86.5 to –27.5; P =.007), but no statistically significant difference was found in participants with self-reported poor mental health.
The trash cleanup intervention had no significant associations with changes in mental health outcomes, compared with no intervention.
Dr. South reported having no disclosures. The study was funded by the National Institutes of Health, and the Centers for Disease Control and Prevention.
SOURCE: South EC et al. JAMA Network Open. 2018 Jul 20. doi: 10.1001/jamanetworkopen.2018.0298.
FROM JAMA NETWORK OPEN
Key clinical point: Treating blighted physical environments in addition to traditional patient treatments may help lift self-reported depression.
Major finding: Low-income residents reported a 68% reduction in feeling depressed.
Study details: Randomized, controlled trial of 442 individuals living near vacant lots.
Disclosures: Dr. South reported having no disclosures. The study was funded by the National Institutes of Health, and the Centers for Disease Control and Prevention.
Source: South EC et al. JAMA Network Open. 2018 Jul 20. doi: 10.1001/jamanetworkopen.2018.0298.
FDA approves Nivestym, second biosimilar to Neupogen
Nivestym (filgrastim-aafi), a biosimilar to Neupogen (filgrastim) was approved July 20 by the Food and Drug Administration, according to a statement provided by the agency. Nivestym is the second biosimilar to Neupogen to be approved in the United States.

- Patients with cancer receiving myelosuppressive chemotherapy.
- Patients with acute myeloid leukemia receiving induction or consolidation chemotherapy.
- Patients with cancer undergoing bone marrow transplantation.
- Patients undergoing autologous peripheral blood progenitor cell collection and therapy.
- Patients with severe chronic neutropenia.
According to a press release from Pfizer, the manufacturer of the biosimilar, Nivestym is expected to be available in the United States at a significant discount to the current wholesale acquisition cost of Neupogen, which is not inclusive of discounts to payers, providers, distributors, and other purchasing organizations.
The FDA statement notes that a biosimilar is approved based on a showing that it is highly similar to an already approved biologic product, known as a reference product. The biosimilar also must be shown to have no clinically meaningful differences in terms of safety and effectiveness from the reference product. Only minor differences in clinically inactive components are allowable in biosimilar products.
Prescribing information is available here.
Nivestym (filgrastim-aafi), a biosimilar to Neupogen (filgrastim) was approved July 20 by the Food and Drug Administration, according to a statement provided by the agency. Nivestym is the second biosimilar to Neupogen to be approved in the United States.
- Patients with cancer receiving myelosuppressive chemotherapy.
- Patients with acute myeloid leukemia receiving induction or consolidation chemotherapy.
- Patients with cancer undergoing bone marrow transplantation.
- Patients undergoing autologous peripheral blood progenitor cell collection and therapy.
- Patients with severe chronic neutropenia.
According to a press release from Pfizer, the manufacturer of the biosimilar, Nivestym is expected to be available in the United States at a significant discount to the current wholesale acquisition cost of Neupogen, which is not inclusive of discounts to payers, providers, distributors, and other purchasing organizations.
The FDA statement notes that a biosimilar is approved based on a showing that it is highly similar to an already approved biologic product, known as a reference product. The biosimilar also must be shown to have no clinically meaningful differences in terms of safety and effectiveness from the reference product. Only minor differences in clinically inactive components are allowable in biosimilar products.
Prescribing information is available here.
Nivestym (filgrastim-aafi), a biosimilar to Neupogen (filgrastim) was approved July 20 by the Food and Drug Administration, according to a statement provided by the agency. Nivestym is the second biosimilar to Neupogen to be approved in the United States.
- Patients with cancer receiving myelosuppressive chemotherapy.
- Patients with acute myeloid leukemia receiving induction or consolidation chemotherapy.
- Patients with cancer undergoing bone marrow transplantation.
- Patients undergoing autologous peripheral blood progenitor cell collection and therapy.
- Patients with severe chronic neutropenia.
According to a press release from Pfizer, the manufacturer of the biosimilar, Nivestym is expected to be available in the United States at a significant discount to the current wholesale acquisition cost of Neupogen, which is not inclusive of discounts to payers, providers, distributors, and other purchasing organizations.
The FDA statement notes that a biosimilar is approved based on a showing that it is highly similar to an already approved biologic product, known as a reference product. The biosimilar also must be shown to have no clinically meaningful differences in terms of safety and effectiveness from the reference product. Only minor differences in clinically inactive components are allowable in biosimilar products.
Prescribing information is available here.
Researchers identify potential sickle cell disease target
Researchers have identified a repressor of fetal hemoglobin (HbF), which they assert could offer a therapeutic target for patients with sickle cell disease (SCD) and some forms of beta thalassemia, according to a new paper published in Science.
Strategies to boost HbF have so far been limited to gene therapy and hydroxyurea, which has been shown to have limited efficacy.
The finding was made using a tailored CRISPR screen of adult human erythroid cells; the CRISPR screen targeted protein kinases, which are controllable by small molecules, which makes them more feasible for treatment therapeutically, researchers wrote.
“Using an improved CRISPR-Cas9 domain-focused screening approach, we identified the erythroid-specific kinase HRI [heme-regulated inhibitor] as a potentially druggable target that is involved in HbF silencing,” wrote Gerd A. Blobel, MD, PhD, of Children’s Hospital of Philadelphia and his colleagues.
HRI is an erythroid-specific kinase that controls protein translation. Once identified, depleting HRI resulted in an increase of HbF production and reduced sickling in cultured erythroid cells. Researchers said that diminished expression of the transcription factor BCL11A, an HbF repressor, accounted for the effects of HRI depletion.
The results with erythroid cell cultures suggest that HRI loss is well tolerated, but the mechanism of inducing fetal hemoglobin is still not fully understood, the researchers noted. Also, while HRI may be targetable, the extent of the benefits of this targeting are not yet known.
“It remains to be seen whether HRI inhibition in SCD patients would elevate HbF levels sufficiently to improve outcomes,” the researchers wrote. “HRI inhibition elevated HbF levels to a point at which it reduced cell sickling in culture, suggesting that pharmacologic HRI inhibitors may provide clinical benefit in SCD patients. Moreover, in light of our results, combining HRI inhibition with an additional pharmacologic HbF inducer may improve the therapeutic index. "Funding was provided by NIH training grants and Cold Spring Harbor Laboratory. Three of the authors reported that they are inventors on a patent submitted by the Children’s Hospital of Philadelphia that covers the therapeutic targeting of HRI for hemoglobinopathies.
SOURCE: Grevet J et al. Science. 2018 Jul 20. doi: 10.1126/science.aao0932.
Researchers have identified a repressor of fetal hemoglobin (HbF), which they assert could offer a therapeutic target for patients with sickle cell disease (SCD) and some forms of beta thalassemia, according to a new paper published in Science.
Strategies to boost HbF have so far been limited to gene therapy and hydroxyurea, which has been shown to have limited efficacy.
The finding was made using a tailored CRISPR screen of adult human erythroid cells; the CRISPR screen targeted protein kinases, which are controllable by small molecules, which makes them more feasible for treatment therapeutically, researchers wrote.
“Using an improved CRISPR-Cas9 domain-focused screening approach, we identified the erythroid-specific kinase HRI [heme-regulated inhibitor] as a potentially druggable target that is involved in HbF silencing,” wrote Gerd A. Blobel, MD, PhD, of Children’s Hospital of Philadelphia and his colleagues.
HRI is an erythroid-specific kinase that controls protein translation. Once identified, depleting HRI resulted in an increase of HbF production and reduced sickling in cultured erythroid cells. Researchers said that diminished expression of the transcription factor BCL11A, an HbF repressor, accounted for the effects of HRI depletion.
The results with erythroid cell cultures suggest that HRI loss is well tolerated, but the mechanism of inducing fetal hemoglobin is still not fully understood, the researchers noted. Also, while HRI may be targetable, the extent of the benefits of this targeting are not yet known.
“It remains to be seen whether HRI inhibition in SCD patients would elevate HbF levels sufficiently to improve outcomes,” the researchers wrote. “HRI inhibition elevated HbF levels to a point at which it reduced cell sickling in culture, suggesting that pharmacologic HRI inhibitors may provide clinical benefit in SCD patients. Moreover, in light of our results, combining HRI inhibition with an additional pharmacologic HbF inducer may improve the therapeutic index. "Funding was provided by NIH training grants and Cold Spring Harbor Laboratory. Three of the authors reported that they are inventors on a patent submitted by the Children’s Hospital of Philadelphia that covers the therapeutic targeting of HRI for hemoglobinopathies.
SOURCE: Grevet J et al. Science. 2018 Jul 20. doi: 10.1126/science.aao0932.
Researchers have identified a repressor of fetal hemoglobin (HbF), which they assert could offer a therapeutic target for patients with sickle cell disease (SCD) and some forms of beta thalassemia, according to a new paper published in Science.
Strategies to boost HbF have so far been limited to gene therapy and hydroxyurea, which has been shown to have limited efficacy.
The finding was made using a tailored CRISPR screen of adult human erythroid cells; the CRISPR screen targeted protein kinases, which are controllable by small molecules, which makes them more feasible for treatment therapeutically, researchers wrote.
“Using an improved CRISPR-Cas9 domain-focused screening approach, we identified the erythroid-specific kinase HRI [heme-regulated inhibitor] as a potentially druggable target that is involved in HbF silencing,” wrote Gerd A. Blobel, MD, PhD, of Children’s Hospital of Philadelphia and his colleagues.
HRI is an erythroid-specific kinase that controls protein translation. Once identified, depleting HRI resulted in an increase of HbF production and reduced sickling in cultured erythroid cells. Researchers said that diminished expression of the transcription factor BCL11A, an HbF repressor, accounted for the effects of HRI depletion.
The results with erythroid cell cultures suggest that HRI loss is well tolerated, but the mechanism of inducing fetal hemoglobin is still not fully understood, the researchers noted. Also, while HRI may be targetable, the extent of the benefits of this targeting are not yet known.
“It remains to be seen whether HRI inhibition in SCD patients would elevate HbF levels sufficiently to improve outcomes,” the researchers wrote. “HRI inhibition elevated HbF levels to a point at which it reduced cell sickling in culture, suggesting that pharmacologic HRI inhibitors may provide clinical benefit in SCD patients. Moreover, in light of our results, combining HRI inhibition with an additional pharmacologic HbF inducer may improve the therapeutic index. "Funding was provided by NIH training grants and Cold Spring Harbor Laboratory. Three of the authors reported that they are inventors on a patent submitted by the Children’s Hospital of Philadelphia that covers the therapeutic targeting of HRI for hemoglobinopathies.
SOURCE: Grevet J et al. Science. 2018 Jul 20. doi: 10.1126/science.aao0932.
REPORTING FROM SCIENCE
CDC reports Salmonella outbreak
A total of 90 people in 26 states have been infected with multidrug-resistant Salmonella in an outbreak linked to raw turkey products, according to the Centers for Disease Control and Prevention.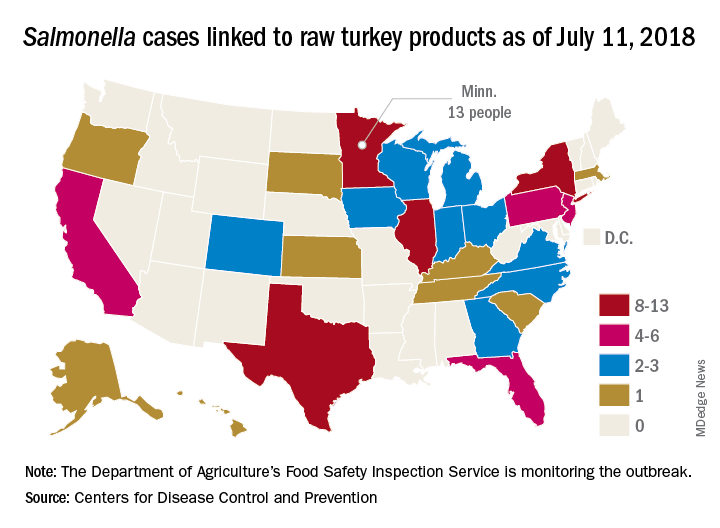
As of July 11, 2018, 40 of the 78 people with available information who were infected with the outbreak strain of Salmonella Reading have been hospitalized, but no deaths have been reported. Of the 61 ill people who have been interviewed, most have reported preparing or eating turkey products from a number of sources, although two lived in households where raw turkey was given to pets: No common supplier has been identified, the CDC reported in an investigation notice posted July 19.
The first illness in this outbreak started on Nov. 20, 2017, and the most recent one started on June 29, 2018. The U.S. Department of Agriculture’s Food Safety Inspection Service is monitoring the outbreak, and public health and regulatory agency efforts are being coordinated by the CDC through its PulseNet national subtyping network. DNA fingerprinting “performed on Salmonella from ill people in this outbreak showed that they are closely related genetically. This means that the ill people are more likely to share a common source of infection,” the CDC said.
Consumers should handle raw turkey carefully and cook it thoroughly to prevent Salmonella, the CDC advised. Raw food of any type should not be given to pets. At this time, the CDC said that it is “not advising that consumers avoid eating properly cooked turkey products, or that retailers stop selling raw turkey products.”
A total of 90 people in 26 states have been infected with multidrug-resistant Salmonella in an outbreak linked to raw turkey products, according to the Centers for Disease Control and Prevention.
As of July 11, 2018, 40 of the 78 people with available information who were infected with the outbreak strain of Salmonella Reading have been hospitalized, but no deaths have been reported. Of the 61 ill people who have been interviewed, most have reported preparing or eating turkey products from a number of sources, although two lived in households where raw turkey was given to pets: No common supplier has been identified, the CDC reported in an investigation notice posted July 19.
The first illness in this outbreak started on Nov. 20, 2017, and the most recent one started on June 29, 2018. The U.S. Department of Agriculture’s Food Safety Inspection Service is monitoring the outbreak, and public health and regulatory agency efforts are being coordinated by the CDC through its PulseNet national subtyping network. DNA fingerprinting “performed on Salmonella from ill people in this outbreak showed that they are closely related genetically. This means that the ill people are more likely to share a common source of infection,” the CDC said.
Consumers should handle raw turkey carefully and cook it thoroughly to prevent Salmonella, the CDC advised. Raw food of any type should not be given to pets. At this time, the CDC said that it is “not advising that consumers avoid eating properly cooked turkey products, or that retailers stop selling raw turkey products.”
A total of 90 people in 26 states have been infected with multidrug-resistant Salmonella in an outbreak linked to raw turkey products, according to the Centers for Disease Control and Prevention.
As of July 11, 2018, 40 of the 78 people with available information who were infected with the outbreak strain of Salmonella Reading have been hospitalized, but no deaths have been reported. Of the 61 ill people who have been interviewed, most have reported preparing or eating turkey products from a number of sources, although two lived in households where raw turkey was given to pets: No common supplier has been identified, the CDC reported in an investigation notice posted July 19.
The first illness in this outbreak started on Nov. 20, 2017, and the most recent one started on June 29, 2018. The U.S. Department of Agriculture’s Food Safety Inspection Service is monitoring the outbreak, and public health and regulatory agency efforts are being coordinated by the CDC through its PulseNet national subtyping network. DNA fingerprinting “performed on Salmonella from ill people in this outbreak showed that they are closely related genetically. This means that the ill people are more likely to share a common source of infection,” the CDC said.
Consumers should handle raw turkey carefully and cook it thoroughly to prevent Salmonella, the CDC advised. Raw food of any type should not be given to pets. At this time, the CDC said that it is “not advising that consumers avoid eating properly cooked turkey products, or that retailers stop selling raw turkey products.”
Meet our 2018 AGA Research Scholar Award Recipients
In 2018, the AGA Research Foundation was proud to provide more than $2 million in research funding to 41 investigators.
AGA’s flagship award, the AGA Research Scholar Award, was given to five exceptional early-career investigators who represent the future of GI research. In addition, one researcher was awarded the AGA-Takeda Pharmaceuticals Research Scholar Award in Inflammatory Bowel Disease. Read about the 2018 awardees’ research projects below.
Sarah Andres, PhD
University of Pennsylvania, Philadelphia
Project title: The mRNA-binding protein IMP1 regulates intestinal epithelial exosome biology during homeostasis and metastasis

Dr. Andres will use this award to delve more deeply into understanding how RNA-binding proteins regulate exosomes within the intestinal and colonic epithelium and how this plays a part in health and disease. RNA-binding proteins provide an exquisite layer of biological regulation to gene expression and downstream cellular processes, which is only beginning to be appreciated. Dr. Andres’ long-term hope is that her work will improve the diagnosis, treatment and ultimately survival of patients with colon cancer.
Swathi Eluri, MD, MSCR
University North Carolina at Chapel Hill
Project title: Improving Barrett’s esophagus screening practices in primary care

Dr. Eluri’s AGA-funded project will gather data to develop and test a multilevel screening intervention for Barrett’s esophagus to be implemented in primary care. The ultimate goal of her work is to improve esophageal adenocarcinoma detection. Given our highly effective endoscopic therapies for early neoplasia in Barrett’s esophagus, early detection has the potential to yield substantial benefits for patients.
Jill Hoffman, PhD
University of California, Los Angeles
AGA-Takeda Pharmaceuticals Research Scholar Award in Inflammatory Bowel Disease
Project title: Characterization of CRHR2-mediated enteric glial cell function during colitis

Dr. Hoffman will use her AGA-Takeda funding to define a role for corticotropin-releasing hormone (CRH) signaling in enteric glial cell function and determine CRHR2-dependent crosstalk between enteric glial cells and the intestinal epithelium during inflammation. Through research aimed at understanding the basic mechanisms of cell-to-cell signaling during intestinal inflammation, Dr. Hoffman hopes to determine how to harness these pathways to limit inflammation and promote repair in patients with IBD.
Elizabeth Jensen, MPH, PhD
Wake Forest University, Winston-Salem, N.C.
Project title: Early-life factors, gene-environment interaction and eosinophilic esophagitis (EoE)

With this funding, Dr. Jensen will conduct the largest study to date on early-life factors and EoE, using data that have been collected prospectively through population-based registries in Denmark. Ultimately, Dr. Jensen hopes her research will lead to advancements in our understanding of etiologic factors for development of immune-mediated GI diseases, such as EoE, and will lead to the identification of modifiable factors for disease prevention.
Sumera Rizvi, MD
Mayo Clinic, Rochester, Minn.
Project title: Necrosis enhances tumor immunogenicity and augments cholangiocarcinoma tumor suppression in combination with PD-L1 blockade

Dr. Rizvi’s research is focused on elucidating immunogenic cell death mechanisms and exploring novel, immune-mediated therapeutic approaches in cholangiocarcinoma. This work has the potential to open novel therapeutic avenues for treatment of cholangiocarcinoma, which will ultimately improve the outcomes of patients with this devastating malignancy.
Niels Vande Casteele, PhD
University of California, San Diego
Project title: Identifying optimal thresholds & personalized dosing regimens of infliximab to maximize endoscopic remission rates in patients with ulcerative colitis

Dr. Vande Casteele’s research project is all about determining the right drug for the right patient at the right time using the right dose. By studying optimal thresholds and personalized dosing regimens of infliximab, Dr. Vande Casteele will build the basis for exposure-based dosing regimens that can be applied to other anti-TNF antibodies and antibodies with other targets used in the treatment of patients with IBD, as well as other chronic inflammatory diseases and/or oncology. Dr. Vande Casteele’s goal is for his work to have a direct impact on patients by allowing us to achieve better treatment outcomes with minimal side effects.
View the 2019 AGA research funding opportunities. Please review the deadlines as application deadlines have shifted. Research Scholar Award applications open Sept. 7, 2018.
In 2018, the AGA Research Foundation was proud to provide more than $2 million in research funding to 41 investigators.
AGA’s flagship award, the AGA Research Scholar Award, was given to five exceptional early-career investigators who represent the future of GI research. In addition, one researcher was awarded the AGA-Takeda Pharmaceuticals Research Scholar Award in Inflammatory Bowel Disease. Read about the 2018 awardees’ research projects below.
Sarah Andres, PhD
University of Pennsylvania, Philadelphia
Project title: The mRNA-binding protein IMP1 regulates intestinal epithelial exosome biology during homeostasis and metastasis
Dr. Andres will use this award to delve more deeply into understanding how RNA-binding proteins regulate exosomes within the intestinal and colonic epithelium and how this plays a part in health and disease. RNA-binding proteins provide an exquisite layer of biological regulation to gene expression and downstream cellular processes, which is only beginning to be appreciated. Dr. Andres’ long-term hope is that her work will improve the diagnosis, treatment and ultimately survival of patients with colon cancer.
Swathi Eluri, MD, MSCR
University North Carolina at Chapel Hill
Project title: Improving Barrett’s esophagus screening practices in primary care
Dr. Eluri’s AGA-funded project will gather data to develop and test a multilevel screening intervention for Barrett’s esophagus to be implemented in primary care. The ultimate goal of her work is to improve esophageal adenocarcinoma detection. Given our highly effective endoscopic therapies for early neoplasia in Barrett’s esophagus, early detection has the potential to yield substantial benefits for patients.
Jill Hoffman, PhD
University of California, Los Angeles
AGA-Takeda Pharmaceuticals Research Scholar Award in Inflammatory Bowel Disease
Project title: Characterization of CRHR2-mediated enteric glial cell function during colitis
Dr. Hoffman will use her AGA-Takeda funding to define a role for corticotropin-releasing hormone (CRH) signaling in enteric glial cell function and determine CRHR2-dependent crosstalk between enteric glial cells and the intestinal epithelium during inflammation. Through research aimed at understanding the basic mechanisms of cell-to-cell signaling during intestinal inflammation, Dr. Hoffman hopes to determine how to harness these pathways to limit inflammation and promote repair in patients with IBD.
Elizabeth Jensen, MPH, PhD
Wake Forest University, Winston-Salem, N.C.
Project title: Early-life factors, gene-environment interaction and eosinophilic esophagitis (EoE)
With this funding, Dr. Jensen will conduct the largest study to date on early-life factors and EoE, using data that have been collected prospectively through population-based registries in Denmark. Ultimately, Dr. Jensen hopes her research will lead to advancements in our understanding of etiologic factors for development of immune-mediated GI diseases, such as EoE, and will lead to the identification of modifiable factors for disease prevention.
Sumera Rizvi, MD
Mayo Clinic, Rochester, Minn.
Project title: Necrosis enhances tumor immunogenicity and augments cholangiocarcinoma tumor suppression in combination with PD-L1 blockade
Dr. Rizvi’s research is focused on elucidating immunogenic cell death mechanisms and exploring novel, immune-mediated therapeutic approaches in cholangiocarcinoma. This work has the potential to open novel therapeutic avenues for treatment of cholangiocarcinoma, which will ultimately improve the outcomes of patients with this devastating malignancy.
Niels Vande Casteele, PhD
University of California, San Diego
Project title: Identifying optimal thresholds & personalized dosing regimens of infliximab to maximize endoscopic remission rates in patients with ulcerative colitis
Dr. Vande Casteele’s research project is all about determining the right drug for the right patient at the right time using the right dose. By studying optimal thresholds and personalized dosing regimens of infliximab, Dr. Vande Casteele will build the basis for exposure-based dosing regimens that can be applied to other anti-TNF antibodies and antibodies with other targets used in the treatment of patients with IBD, as well as other chronic inflammatory diseases and/or oncology. Dr. Vande Casteele’s goal is for his work to have a direct impact on patients by allowing us to achieve better treatment outcomes with minimal side effects.
View the 2019 AGA research funding opportunities. Please review the deadlines as application deadlines have shifted. Research Scholar Award applications open Sept. 7, 2018.
In 2018, the AGA Research Foundation was proud to provide more than $2 million in research funding to 41 investigators.
AGA’s flagship award, the AGA Research Scholar Award, was given to five exceptional early-career investigators who represent the future of GI research. In addition, one researcher was awarded the AGA-Takeda Pharmaceuticals Research Scholar Award in Inflammatory Bowel Disease. Read about the 2018 awardees’ research projects below.
Sarah Andres, PhD
University of Pennsylvania, Philadelphia
Project title: The mRNA-binding protein IMP1 regulates intestinal epithelial exosome biology during homeostasis and metastasis
Dr. Andres will use this award to delve more deeply into understanding how RNA-binding proteins regulate exosomes within the intestinal and colonic epithelium and how this plays a part in health and disease. RNA-binding proteins provide an exquisite layer of biological regulation to gene expression and downstream cellular processes, which is only beginning to be appreciated. Dr. Andres’ long-term hope is that her work will improve the diagnosis, treatment and ultimately survival of patients with colon cancer.
Swathi Eluri, MD, MSCR
University North Carolina at Chapel Hill
Project title: Improving Barrett’s esophagus screening practices in primary care
Dr. Eluri’s AGA-funded project will gather data to develop and test a multilevel screening intervention for Barrett’s esophagus to be implemented in primary care. The ultimate goal of her work is to improve esophageal adenocarcinoma detection. Given our highly effective endoscopic therapies for early neoplasia in Barrett’s esophagus, early detection has the potential to yield substantial benefits for patients.
Jill Hoffman, PhD
University of California, Los Angeles
AGA-Takeda Pharmaceuticals Research Scholar Award in Inflammatory Bowel Disease
Project title: Characterization of CRHR2-mediated enteric glial cell function during colitis
Dr. Hoffman will use her AGA-Takeda funding to define a role for corticotropin-releasing hormone (CRH) signaling in enteric glial cell function and determine CRHR2-dependent crosstalk between enteric glial cells and the intestinal epithelium during inflammation. Through research aimed at understanding the basic mechanisms of cell-to-cell signaling during intestinal inflammation, Dr. Hoffman hopes to determine how to harness these pathways to limit inflammation and promote repair in patients with IBD.
Elizabeth Jensen, MPH, PhD
Wake Forest University, Winston-Salem, N.C.
Project title: Early-life factors, gene-environment interaction and eosinophilic esophagitis (EoE)
With this funding, Dr. Jensen will conduct the largest study to date on early-life factors and EoE, using data that have been collected prospectively through population-based registries in Denmark. Ultimately, Dr. Jensen hopes her research will lead to advancements in our understanding of etiologic factors for development of immune-mediated GI diseases, such as EoE, and will lead to the identification of modifiable factors for disease prevention.
Sumera Rizvi, MD
Mayo Clinic, Rochester, Minn.
Project title: Necrosis enhances tumor immunogenicity and augments cholangiocarcinoma tumor suppression in combination with PD-L1 blockade
Dr. Rizvi’s research is focused on elucidating immunogenic cell death mechanisms and exploring novel, immune-mediated therapeutic approaches in cholangiocarcinoma. This work has the potential to open novel therapeutic avenues for treatment of cholangiocarcinoma, which will ultimately improve the outcomes of patients with this devastating malignancy.
Niels Vande Casteele, PhD
University of California, San Diego
Project title: Identifying optimal thresholds & personalized dosing regimens of infliximab to maximize endoscopic remission rates in patients with ulcerative colitis
Dr. Vande Casteele’s research project is all about determining the right drug for the right patient at the right time using the right dose. By studying optimal thresholds and personalized dosing regimens of infliximab, Dr. Vande Casteele will build the basis for exposure-based dosing regimens that can be applied to other anti-TNF antibodies and antibodies with other targets used in the treatment of patients with IBD, as well as other chronic inflammatory diseases and/or oncology. Dr. Vande Casteele’s goal is for his work to have a direct impact on patients by allowing us to achieve better treatment outcomes with minimal side effects.
View the 2019 AGA research funding opportunities. Please review the deadlines as application deadlines have shifted. Research Scholar Award applications open Sept. 7, 2018.
Opioid crisis brings bonanza for headache research
SAN FRANCISCO – Congressional funding of the National Institutes of Health’s HEAL Initiative represents an unprecedented golden opportunity for research funding on headache and other pain conditions, according to Michael L. Oshinsky, PhD, a neurobiologist who serves as program director for pain and migraine at the National Institute of Neurological Disorders and Stroke (NINDS).

“This is a once-in-a-lifetime opportunity for physician researchers and basic science researchers to identify projects and request funding from NIH. This is an amount of money that doesn’t come along very often. It’s really a once in a lifetime situation, and I implore you to take advantage of this tremendous opportunity,” he urged attendees at the annual meeting of the American Headache Society.
Congress gave the NIH $500 million for the HEAL (Helping to End Addiction Long-term) Initiative to be spent over the course of fiscal years 2018 and 2019. The money is to be split equally on the initiative’s two components: opioid use disorder and pain management.
“We’re going to spend $250 million on pain, be sure of it,” Dr. Oshinsky promised.
Pain management issues cut across all 27 NIH institutes and centers. However, the biggest chunk of the money goes to NINDS, which leads the NIH Pain Consortium, charged with enhancing pain research and promoting collaboration across NIH.
Within NINDS, funding for migraine research has been formally designated as an HPP, or high program priority, according to Dr. Oshinsky.
“Headache research gets funded. It really does. I want to dispel the myth that there isn’t an interest in headache research or migraine research at NIH. Currently, at NINDS, we have more than 60 funded projects that specifically address scientific questions about migraine and headache. That’s more than any other government in the world,” he said.
And it’s not all basic science research related to headache that NINDS is interested in fostering, either. The institute is eager to fund large clinical trials, pediatric headache research, studies of device therapies, and a wealth of other projects.
Among the HEAL initiatives that have already been approved is a project aimed at identifying and validating new pain targets. Another is focused upon finding risk factors with clinically meaningful predictive value for transition from acute to chronic pain, be they biomarkers, imaging findings, or genetic characteristics. Yet another program is an NIH-industry collaboration that aims to identify new treatments for pain that are nonaddictive, nonrewarding, nonsedating, and safe across age groups; candidate biologics, small molecules, devices, and natural products will be screened in vitro and in animal models, with the successful ones moving on to clinical trials.
Funding opportunity announcements are imminent for creation of a planned NIH clinical trials network for pain research, which will conduct phase 2 trials in specialized pain centers. The focus will be on well-characterized pain conditions with high unmet medical need, including headache as well as orofacial, pelvic, gastrointestinal, and cancer pain.
Dr. Oshinsky encouraged academicians to have their fellows apply for the NIH early career development awards known as K awards.
“They are much easier to get than R01 awards and they provide a tremendous opportunity for jump-starting the career of new scientists in the headache field. Have them reach out to me. I can help them through the process, help them develop their project, and get them funded with a K award. And the transition from a K award to an R01 is a much smoother transition,” he said.
Research funding proposals should use the NINDS Common Data Elements for headache, recently updated by more than 40 experts in headache medicine and research.
Dr. Oshinsky said most of the new pain management programs will be funded in fiscal year 2019. These novel programs have to be approved by oversight committees, and that takes time. The reason that the bulk of HEAL Initiative funding in fiscal year 2018 is going for opioid use disorder is that much of that money is being spent on expansion of existing programs, which can quickly be ramped up without the same degree of oversight required for new programs.
SAN FRANCISCO – Congressional funding of the National Institutes of Health’s HEAL Initiative represents an unprecedented golden opportunity for research funding on headache and other pain conditions, according to Michael L. Oshinsky, PhD, a neurobiologist who serves as program director for pain and migraine at the National Institute of Neurological Disorders and Stroke (NINDS).
“This is a once-in-a-lifetime opportunity for physician researchers and basic science researchers to identify projects and request funding from NIH. This is an amount of money that doesn’t come along very often. It’s really a once in a lifetime situation, and I implore you to take advantage of this tremendous opportunity,” he urged attendees at the annual meeting of the American Headache Society.
Congress gave the NIH $500 million for the HEAL (Helping to End Addiction Long-term) Initiative to be spent over the course of fiscal years 2018 and 2019. The money is to be split equally on the initiative’s two components: opioid use disorder and pain management.
“We’re going to spend $250 million on pain, be sure of it,” Dr. Oshinsky promised.
Pain management issues cut across all 27 NIH institutes and centers. However, the biggest chunk of the money goes to NINDS, which leads the NIH Pain Consortium, charged with enhancing pain research and promoting collaboration across NIH.
Within NINDS, funding for migraine research has been formally designated as an HPP, or high program priority, according to Dr. Oshinsky.
“Headache research gets funded. It really does. I want to dispel the myth that there isn’t an interest in headache research or migraine research at NIH. Currently, at NINDS, we have more than 60 funded projects that specifically address scientific questions about migraine and headache. That’s more than any other government in the world,” he said.
And it’s not all basic science research related to headache that NINDS is interested in fostering, either. The institute is eager to fund large clinical trials, pediatric headache research, studies of device therapies, and a wealth of other projects.
Among the HEAL initiatives that have already been approved is a project aimed at identifying and validating new pain targets. Another is focused upon finding risk factors with clinically meaningful predictive value for transition from acute to chronic pain, be they biomarkers, imaging findings, or genetic characteristics. Yet another program is an NIH-industry collaboration that aims to identify new treatments for pain that are nonaddictive, nonrewarding, nonsedating, and safe across age groups; candidate biologics, small molecules, devices, and natural products will be screened in vitro and in animal models, with the successful ones moving on to clinical trials.
Funding opportunity announcements are imminent for creation of a planned NIH clinical trials network for pain research, which will conduct phase 2 trials in specialized pain centers. The focus will be on well-characterized pain conditions with high unmet medical need, including headache as well as orofacial, pelvic, gastrointestinal, and cancer pain.
Dr. Oshinsky encouraged academicians to have their fellows apply for the NIH early career development awards known as K awards.
“They are much easier to get than R01 awards and they provide a tremendous opportunity for jump-starting the career of new scientists in the headache field. Have them reach out to me. I can help them through the process, help them develop their project, and get them funded with a K award. And the transition from a K award to an R01 is a much smoother transition,” he said.
Research funding proposals should use the NINDS Common Data Elements for headache, recently updated by more than 40 experts in headache medicine and research.
Dr. Oshinsky said most of the new pain management programs will be funded in fiscal year 2019. These novel programs have to be approved by oversight committees, and that takes time. The reason that the bulk of HEAL Initiative funding in fiscal year 2018 is going for opioid use disorder is that much of that money is being spent on expansion of existing programs, which can quickly be ramped up without the same degree of oversight required for new programs.
SAN FRANCISCO – Congressional funding of the National Institutes of Health’s HEAL Initiative represents an unprecedented golden opportunity for research funding on headache and other pain conditions, according to Michael L. Oshinsky, PhD, a neurobiologist who serves as program director for pain and migraine at the National Institute of Neurological Disorders and Stroke (NINDS).
“This is a once-in-a-lifetime opportunity for physician researchers and basic science researchers to identify projects and request funding from NIH. This is an amount of money that doesn’t come along very often. It’s really a once in a lifetime situation, and I implore you to take advantage of this tremendous opportunity,” he urged attendees at the annual meeting of the American Headache Society.
Congress gave the NIH $500 million for the HEAL (Helping to End Addiction Long-term) Initiative to be spent over the course of fiscal years 2018 and 2019. The money is to be split equally on the initiative’s two components: opioid use disorder and pain management.
“We’re going to spend $250 million on pain, be sure of it,” Dr. Oshinsky promised.
Pain management issues cut across all 27 NIH institutes and centers. However, the biggest chunk of the money goes to NINDS, which leads the NIH Pain Consortium, charged with enhancing pain research and promoting collaboration across NIH.
Within NINDS, funding for migraine research has been formally designated as an HPP, or high program priority, according to Dr. Oshinsky.
“Headache research gets funded. It really does. I want to dispel the myth that there isn’t an interest in headache research or migraine research at NIH. Currently, at NINDS, we have more than 60 funded projects that specifically address scientific questions about migraine and headache. That’s more than any other government in the world,” he said.
And it’s not all basic science research related to headache that NINDS is interested in fostering, either. The institute is eager to fund large clinical trials, pediatric headache research, studies of device therapies, and a wealth of other projects.
Among the HEAL initiatives that have already been approved is a project aimed at identifying and validating new pain targets. Another is focused upon finding risk factors with clinically meaningful predictive value for transition from acute to chronic pain, be they biomarkers, imaging findings, or genetic characteristics. Yet another program is an NIH-industry collaboration that aims to identify new treatments for pain that are nonaddictive, nonrewarding, nonsedating, and safe across age groups; candidate biologics, small molecules, devices, and natural products will be screened in vitro and in animal models, with the successful ones moving on to clinical trials.
Funding opportunity announcements are imminent for creation of a planned NIH clinical trials network for pain research, which will conduct phase 2 trials in specialized pain centers. The focus will be on well-characterized pain conditions with high unmet medical need, including headache as well as orofacial, pelvic, gastrointestinal, and cancer pain.
Dr. Oshinsky encouraged academicians to have their fellows apply for the NIH early career development awards known as K awards.
“They are much easier to get than R01 awards and they provide a tremendous opportunity for jump-starting the career of new scientists in the headache field. Have them reach out to me. I can help them through the process, help them develop their project, and get them funded with a K award. And the transition from a K award to an R01 is a much smoother transition,” he said.
Research funding proposals should use the NINDS Common Data Elements for headache, recently updated by more than 40 experts in headache medicine and research.
Dr. Oshinsky said most of the new pain management programs will be funded in fiscal year 2019. These novel programs have to be approved by oversight committees, and that takes time. The reason that the bulk of HEAL Initiative funding in fiscal year 2018 is going for opioid use disorder is that much of that money is being spent on expansion of existing programs, which can quickly be ramped up without the same degree of oversight required for new programs.
REPORTING FROM THE AHS ANNUAL MEETING
Don’t lose your access to essential resources
If you are a current AGA trainee, medical resident, or student member, please renew your membership today to ensure the continuation of your career-enhancing benefits for the upcoming membership year. Prepare for your next chapter with the latest news and breakthroughs in the field, as well as access to educational programs, events and much more.
While renewing, please update your member profile at My AGA for news and resources tailored to your professional interests. The deadline to renew is Aug. 31, 2018.
If you have any questions, please contact AGA Member Relations at [email protected] or 301-941-2651.
If you are a current AGA trainee, medical resident, or student member, please renew your membership today to ensure the continuation of your career-enhancing benefits for the upcoming membership year. Prepare for your next chapter with the latest news and breakthroughs in the field, as well as access to educational programs, events and much more.
While renewing, please update your member profile at My AGA for news and resources tailored to your professional interests. The deadline to renew is Aug. 31, 2018.
If you have any questions, please contact AGA Member Relations at [email protected] or 301-941-2651.
If you are a current AGA trainee, medical resident, or student member, please renew your membership today to ensure the continuation of your career-enhancing benefits for the upcoming membership year. Prepare for your next chapter with the latest news and breakthroughs in the field, as well as access to educational programs, events and much more.
While renewing, please update your member profile at My AGA for news and resources tailored to your professional interests. The deadline to renew is Aug. 31, 2018.
If you have any questions, please contact AGA Member Relations at [email protected] or 301-941-2651.
FDA approves IDH1 inhibitor for relapsed/refractory AML
The Food and Drug Administration has approved ivosidenib (Tibsovo) as the first treatment of adult patients with relapsed/refractory acute myeloid leukemia (AML) and an isocitrate dehydrogenase-1 (IDH1) mutation.
More specifically, the oral treatment has been approved for patients whose mutations have been identified by the Abbott RealTime IDH1 assay, a companion diagnostic test.
The approval was based on results from a phase 1, open-label, single-arm, multicenter, dose-escalation and expansion trial of adult patients in this AML population. The primary end point was combined complete remission and complete remission with partial hematologic improvement; this combined rate was 32.8%, and the median duration of this remission was 8.2 months.
The most serious adverse events included differentiation syndrome, QTc prolongation, and Guillain-Barré syndrome. Other adverse reactions included fatigue, leukocytosis, arthralgia, diarrhea, dyspnea, edema, and constipation.
Ivosidenib is marketed as Tibsovo by Agios Pharmaceuticals. The RealTime IDH1 Assay is marketed by Abbott Laboratories.
The Food and Drug Administration has approved ivosidenib (Tibsovo) as the first treatment of adult patients with relapsed/refractory acute myeloid leukemia (AML) and an isocitrate dehydrogenase-1 (IDH1) mutation.
More specifically, the oral treatment has been approved for patients whose mutations have been identified by the Abbott RealTime IDH1 assay, a companion diagnostic test.
The approval was based on results from a phase 1, open-label, single-arm, multicenter, dose-escalation and expansion trial of adult patients in this AML population. The primary end point was combined complete remission and complete remission with partial hematologic improvement; this combined rate was 32.8%, and the median duration of this remission was 8.2 months.
The most serious adverse events included differentiation syndrome, QTc prolongation, and Guillain-Barré syndrome. Other adverse reactions included fatigue, leukocytosis, arthralgia, diarrhea, dyspnea, edema, and constipation.
Ivosidenib is marketed as Tibsovo by Agios Pharmaceuticals. The RealTime IDH1 Assay is marketed by Abbott Laboratories.
The Food and Drug Administration has approved ivosidenib (Tibsovo) as the first treatment of adult patients with relapsed/refractory acute myeloid leukemia (AML) and an isocitrate dehydrogenase-1 (IDH1) mutation.
More specifically, the oral treatment has been approved for patients whose mutations have been identified by the Abbott RealTime IDH1 assay, a companion diagnostic test.
The approval was based on results from a phase 1, open-label, single-arm, multicenter, dose-escalation and expansion trial of adult patients in this AML population. The primary end point was combined complete remission and complete remission with partial hematologic improvement; this combined rate was 32.8%, and the median duration of this remission was 8.2 months.
The most serious adverse events included differentiation syndrome, QTc prolongation, and Guillain-Barré syndrome. Other adverse reactions included fatigue, leukocytosis, arthralgia, diarrhea, dyspnea, edema, and constipation.
Ivosidenib is marketed as Tibsovo by Agios Pharmaceuticals. The RealTime IDH1 Assay is marketed by Abbott Laboratories.
A successful career starts with taking charge
Successful careers don’t just happen. They are made by individuals who take charge and build their own success. The alternative is burnout.
“Part of burnout is feeling overburdened, overworked, and out of control,” said Barbara Jung, MD, AGAF, professor and chief of gastroenterology and hepatology at the University of Illinois at Chicago, during Strategies for a Successful Career: Wellness, Empowerment, Leadership and Resilience at Digestive Disease Week® (DDW) 2018. “If somebody is staying until 9 or 10 o’clock [at night] to finish notes, I have a discussion with them. It is good to be done at 5 p.m. and go home. It is all about setting your own priorities and not letting the job take over your life.”
Associations have a role to play, too. The ASGE Technology Committee reported in 2010 that up to 89% of GIs suffer musculoskeletal injuries from manipulating scopes. Colonoscopist’s thumb (left thumb tendonitis) and metacarpophalangeal joint strain were the most common injuries.
“Risk factors are part of our work,” said Mehnaz Shafi, MD, AGAF, professor of gastroenterology, hepatology and nutrition at the University of Texas MD Anderson Cancer Center, Houston. “Pinching, pushing, pulling, and awkward positions are part of what we do. This is an injury with consequences.”
Dr. Shafi chairs the AGA Task Force on Ergonomics. The group has recommended changes to endoscopic work stations that minimize injury. The most important changes include mounting monitors on flexible stands to accommodate GIs of all heights, adding straps to the control head to allow the fingers to relax, providing ergonomic training to all GIs and using patient beds that can be raised and lowered to accommodate both tall and short GIs.
“Shaping your career is one of the key principles in preventing burnout,” said Arthur DeCross, MD, AGAF, professor of medicine, gastroenterology and hepatology at the University of Rochester Medical Center, N.Y. “We know that more than half of gastroenterologists self-identify as being burned out. And one of the major contributors to burnout is lack of control over your work environment, your career, your colleagues. Taking control of your career can make a difference.”
Taking control can be particularly important for women. An AGA burnout survey in 2015 found that 51% of male GIs reported burnout versus 62% of female GIs.
One reason is women’s tendency to negotiate poorly on their own behalf, said Marie-Pier Tétreault, PhD, assistant professor of gastroenterology and hepatology at Northwestern University Feinberg School of Medicine, Chicago.
“It’s a matter of attitude,” she explained. “Men tend to believe they can and should make life happen. Women tend to believe that what you see is what you get. Even when women do negotiate, they tend to ask for 15%-30% less than their male colleagues. If you don’t ask, you won’t get.”
Simply taking the lead in negotiations can improve the outcome, she continued. Network with colleagues and mentors to find the appropriate ranges for salaries, benefits, and perks such as parking, spousal job opportunities, facilities and space, teaching expectations, administrative support, and more.
Successful careers don’t just happen. They are made by individuals who take charge and build their own success. The alternative is burnout.
“Part of burnout is feeling overburdened, overworked, and out of control,” said Barbara Jung, MD, AGAF, professor and chief of gastroenterology and hepatology at the University of Illinois at Chicago, during Strategies for a Successful Career: Wellness, Empowerment, Leadership and Resilience at Digestive Disease Week® (DDW) 2018. “If somebody is staying until 9 or 10 o’clock [at night] to finish notes, I have a discussion with them. It is good to be done at 5 p.m. and go home. It is all about setting your own priorities and not letting the job take over your life.”
Associations have a role to play, too. The ASGE Technology Committee reported in 2010 that up to 89% of GIs suffer musculoskeletal injuries from manipulating scopes. Colonoscopist’s thumb (left thumb tendonitis) and metacarpophalangeal joint strain were the most common injuries.
“Risk factors are part of our work,” said Mehnaz Shafi, MD, AGAF, professor of gastroenterology, hepatology and nutrition at the University of Texas MD Anderson Cancer Center, Houston. “Pinching, pushing, pulling, and awkward positions are part of what we do. This is an injury with consequences.”
Dr. Shafi chairs the AGA Task Force on Ergonomics. The group has recommended changes to endoscopic work stations that minimize injury. The most important changes include mounting monitors on flexible stands to accommodate GIs of all heights, adding straps to the control head to allow the fingers to relax, providing ergonomic training to all GIs and using patient beds that can be raised and lowered to accommodate both tall and short GIs.
“Shaping your career is one of the key principles in preventing burnout,” said Arthur DeCross, MD, AGAF, professor of medicine, gastroenterology and hepatology at the University of Rochester Medical Center, N.Y. “We know that more than half of gastroenterologists self-identify as being burned out. And one of the major contributors to burnout is lack of control over your work environment, your career, your colleagues. Taking control of your career can make a difference.”
Taking control can be particularly important for women. An AGA burnout survey in 2015 found that 51% of male GIs reported burnout versus 62% of female GIs.
One reason is women’s tendency to negotiate poorly on their own behalf, said Marie-Pier Tétreault, PhD, assistant professor of gastroenterology and hepatology at Northwestern University Feinberg School of Medicine, Chicago.
“It’s a matter of attitude,” she explained. “Men tend to believe they can and should make life happen. Women tend to believe that what you see is what you get. Even when women do negotiate, they tend to ask for 15%-30% less than their male colleagues. If you don’t ask, you won’t get.”
Simply taking the lead in negotiations can improve the outcome, she continued. Network with colleagues and mentors to find the appropriate ranges for salaries, benefits, and perks such as parking, spousal job opportunities, facilities and space, teaching expectations, administrative support, and more.
Successful careers don’t just happen. They are made by individuals who take charge and build their own success. The alternative is burnout.
“Part of burnout is feeling overburdened, overworked, and out of control,” said Barbara Jung, MD, AGAF, professor and chief of gastroenterology and hepatology at the University of Illinois at Chicago, during Strategies for a Successful Career: Wellness, Empowerment, Leadership and Resilience at Digestive Disease Week® (DDW) 2018. “If somebody is staying until 9 or 10 o’clock [at night] to finish notes, I have a discussion with them. It is good to be done at 5 p.m. and go home. It is all about setting your own priorities and not letting the job take over your life.”
Associations have a role to play, too. The ASGE Technology Committee reported in 2010 that up to 89% of GIs suffer musculoskeletal injuries from manipulating scopes. Colonoscopist’s thumb (left thumb tendonitis) and metacarpophalangeal joint strain were the most common injuries.
“Risk factors are part of our work,” said Mehnaz Shafi, MD, AGAF, professor of gastroenterology, hepatology and nutrition at the University of Texas MD Anderson Cancer Center, Houston. “Pinching, pushing, pulling, and awkward positions are part of what we do. This is an injury with consequences.”
Dr. Shafi chairs the AGA Task Force on Ergonomics. The group has recommended changes to endoscopic work stations that minimize injury. The most important changes include mounting monitors on flexible stands to accommodate GIs of all heights, adding straps to the control head to allow the fingers to relax, providing ergonomic training to all GIs and using patient beds that can be raised and lowered to accommodate both tall and short GIs.
“Shaping your career is one of the key principles in preventing burnout,” said Arthur DeCross, MD, AGAF, professor of medicine, gastroenterology and hepatology at the University of Rochester Medical Center, N.Y. “We know that more than half of gastroenterologists self-identify as being burned out. And one of the major contributors to burnout is lack of control over your work environment, your career, your colleagues. Taking control of your career can make a difference.”
Taking control can be particularly important for women. An AGA burnout survey in 2015 found that 51% of male GIs reported burnout versus 62% of female GIs.
One reason is women’s tendency to negotiate poorly on their own behalf, said Marie-Pier Tétreault, PhD, assistant professor of gastroenterology and hepatology at Northwestern University Feinberg School of Medicine, Chicago.
“It’s a matter of attitude,” she explained. “Men tend to believe they can and should make life happen. Women tend to believe that what you see is what you get. Even when women do negotiate, they tend to ask for 15%-30% less than their male colleagues. If you don’t ask, you won’t get.”
Simply taking the lead in negotiations can improve the outcome, she continued. Network with colleagues and mentors to find the appropriate ranges for salaries, benefits, and perks such as parking, spousal job opportunities, facilities and space, teaching expectations, administrative support, and more.
Essure sales to halt in U.S. by end of 2018
The Essure permanent birth control device will no longer be sold or distributed after Dec. 31, 2018, in the United States.
Bayer, the manufacturer of Essure, notified the Food and Drug Administration of its decision to halt U.S. sales of the device, Commissioner Scott Gottlieb, MD, announced July 20 in a press release. Dr. Gottlieb added that the agency would continue its commitment to postmarketing review of Essure. “We expect Bayer to meet its postmarket obligations concerning this device.”
Since its approval, Essure is estimated to have been used by more than 750,000 patients worldwide, the FDA release stated. The device has been associated with serious risks, including persistent pain, perforation of the uterus and fallopian tubes, and migration of the coils into the pelvis or abdomen.
“In April, when the FDA became aware that many patients were not being adequately counseled, we required a restriction, which limits the sale and distribution of the device to only health care providers and facilities that provide information to patients about the risks and benefits of this device and gives patients the opportunity to sign an acknowledgment that they fully understood these potential risks before having the device implanted. Since the FDA ordered Bayer to conduct the postmarket study and then to add a boxed warning and a Patient Decision Checklist to the labeling, there has been an approximate 70% decline in sales of Essure in the United States,” Dr. Gottlieb said in the FDA press release. The company stated its decision to halt sales and distribution of the device was because of commercial reasons.
“Numerous adverse events ... were reported to the FDA, including a significant collection of recent reports that have mentioned issues involving surgery to remove the device. We’re continuing our evaluation of these reports to better understand reasons for the device removal. The agency is committed to continuing to provide updates on our evaluation of this data as the information is collected and we develop new findings about the device.”
In September 2015, the FDA convened an expert panel to examine and follow up on complaints from Essure users that included abdominal pain, abnormal uterine bleeding, and device migration. In February 2016, the FDA ordered Bayer to conduct a postmarket (522) study to better evaluate the safety profile of the device when used in the real world. The following October, the agency issued the final guidance, “Labeling for Permanent Hysteroscopically Placed Tubal Implants Intended for Sterilization.” Soon thereafter, the FDA approved updated labeling for Essure that added a boxed warning and a Patient Decision Checklist.
In March 2018, the FDA reported a rise in new medical device reports submitted to the agency’s public database in 2017, with more than 90% of the reports involving potential device removal. The April restriction of sales and distribution was in response to concerns that not every patient was receiving adequate risk information.
“I want to stress that, even when Essure is no longer sold, the FDA will remain vigilant in protecting patients who’ve already had this device implanted. We’ll continue to monitor adverse events reported to our database, as well as other data sources. And we’ll communicate publicly on any new findings or concerns. The restriction on sale and distribution will remain in place. Regarding the postmarket 522 study, Bayer will continue to enroll new participants. Each study participant will be followed for a total of 3 years, and the company will continue to submit reports to the FDA on the study’s progress and results. Since Bayer will not be able to meet its expected enrollment numbers for this study that relied on enrolling patients who were newly implanted with Essure, we’ll be working with the company to best determine how to move forward to answer the critical questions we posed concerning certain patient complications that may be experienced by patients who have Essure,” Dr. Gottlieb stated.
He added that women who are using Essure successfully to prevent pregnancy should continue to do so, as “device removal has its own risks.”
The Essure permanent birth control device will no longer be sold or distributed after Dec. 31, 2018, in the United States.
Bayer, the manufacturer of Essure, notified the Food and Drug Administration of its decision to halt U.S. sales of the device, Commissioner Scott Gottlieb, MD, announced July 20 in a press release. Dr. Gottlieb added that the agency would continue its commitment to postmarketing review of Essure. “We expect Bayer to meet its postmarket obligations concerning this device.”
Since its approval, Essure is estimated to have been used by more than 750,000 patients worldwide, the FDA release stated. The device has been associated with serious risks, including persistent pain, perforation of the uterus and fallopian tubes, and migration of the coils into the pelvis or abdomen.
“In April, when the FDA became aware that many patients were not being adequately counseled, we required a restriction, which limits the sale and distribution of the device to only health care providers and facilities that provide information to patients about the risks and benefits of this device and gives patients the opportunity to sign an acknowledgment that they fully understood these potential risks before having the device implanted. Since the FDA ordered Bayer to conduct the postmarket study and then to add a boxed warning and a Patient Decision Checklist to the labeling, there has been an approximate 70% decline in sales of Essure in the United States,” Dr. Gottlieb said in the FDA press release. The company stated its decision to halt sales and distribution of the device was because of commercial reasons.
“Numerous adverse events ... were reported to the FDA, including a significant collection of recent reports that have mentioned issues involving surgery to remove the device. We’re continuing our evaluation of these reports to better understand reasons for the device removal. The agency is committed to continuing to provide updates on our evaluation of this data as the information is collected and we develop new findings about the device.”
In September 2015, the FDA convened an expert panel to examine and follow up on complaints from Essure users that included abdominal pain, abnormal uterine bleeding, and device migration. In February 2016, the FDA ordered Bayer to conduct a postmarket (522) study to better evaluate the safety profile of the device when used in the real world. The following October, the agency issued the final guidance, “Labeling for Permanent Hysteroscopically Placed Tubal Implants Intended for Sterilization.” Soon thereafter, the FDA approved updated labeling for Essure that added a boxed warning and a Patient Decision Checklist.
In March 2018, the FDA reported a rise in new medical device reports submitted to the agency’s public database in 2017, with more than 90% of the reports involving potential device removal. The April restriction of sales and distribution was in response to concerns that not every patient was receiving adequate risk information.
“I want to stress that, even when Essure is no longer sold, the FDA will remain vigilant in protecting patients who’ve already had this device implanted. We’ll continue to monitor adverse events reported to our database, as well as other data sources. And we’ll communicate publicly on any new findings or concerns. The restriction on sale and distribution will remain in place. Regarding the postmarket 522 study, Bayer will continue to enroll new participants. Each study participant will be followed for a total of 3 years, and the company will continue to submit reports to the FDA on the study’s progress and results. Since Bayer will not be able to meet its expected enrollment numbers for this study that relied on enrolling patients who were newly implanted with Essure, we’ll be working with the company to best determine how to move forward to answer the critical questions we posed concerning certain patient complications that may be experienced by patients who have Essure,” Dr. Gottlieb stated.
He added that women who are using Essure successfully to prevent pregnancy should continue to do so, as “device removal has its own risks.”
The Essure permanent birth control device will no longer be sold or distributed after Dec. 31, 2018, in the United States.
Bayer, the manufacturer of Essure, notified the Food and Drug Administration of its decision to halt U.S. sales of the device, Commissioner Scott Gottlieb, MD, announced July 20 in a press release. Dr. Gottlieb added that the agency would continue its commitment to postmarketing review of Essure. “We expect Bayer to meet its postmarket obligations concerning this device.”
Since its approval, Essure is estimated to have been used by more than 750,000 patients worldwide, the FDA release stated. The device has been associated with serious risks, including persistent pain, perforation of the uterus and fallopian tubes, and migration of the coils into the pelvis or abdomen.
“In April, when the FDA became aware that many patients were not being adequately counseled, we required a restriction, which limits the sale and distribution of the device to only health care providers and facilities that provide information to patients about the risks and benefits of this device and gives patients the opportunity to sign an acknowledgment that they fully understood these potential risks before having the device implanted. Since the FDA ordered Bayer to conduct the postmarket study and then to add a boxed warning and a Patient Decision Checklist to the labeling, there has been an approximate 70% decline in sales of Essure in the United States,” Dr. Gottlieb said in the FDA press release. The company stated its decision to halt sales and distribution of the device was because of commercial reasons.
“Numerous adverse events ... were reported to the FDA, including a significant collection of recent reports that have mentioned issues involving surgery to remove the device. We’re continuing our evaluation of these reports to better understand reasons for the device removal. The agency is committed to continuing to provide updates on our evaluation of this data as the information is collected and we develop new findings about the device.”
In September 2015, the FDA convened an expert panel to examine and follow up on complaints from Essure users that included abdominal pain, abnormal uterine bleeding, and device migration. In February 2016, the FDA ordered Bayer to conduct a postmarket (522) study to better evaluate the safety profile of the device when used in the real world. The following October, the agency issued the final guidance, “Labeling for Permanent Hysteroscopically Placed Tubal Implants Intended for Sterilization.” Soon thereafter, the FDA approved updated labeling for Essure that added a boxed warning and a Patient Decision Checklist.
In March 2018, the FDA reported a rise in new medical device reports submitted to the agency’s public database in 2017, with more than 90% of the reports involving potential device removal. The April restriction of sales and distribution was in response to concerns that not every patient was receiving adequate risk information.
“I want to stress that, even when Essure is no longer sold, the FDA will remain vigilant in protecting patients who’ve already had this device implanted. We’ll continue to monitor adverse events reported to our database, as well as other data sources. And we’ll communicate publicly on any new findings or concerns. The restriction on sale and distribution will remain in place. Regarding the postmarket 522 study, Bayer will continue to enroll new participants. Each study participant will be followed for a total of 3 years, and the company will continue to submit reports to the FDA on the study’s progress and results. Since Bayer will not be able to meet its expected enrollment numbers for this study that relied on enrolling patients who were newly implanted with Essure, we’ll be working with the company to best determine how to move forward to answer the critical questions we posed concerning certain patient complications that may be experienced by patients who have Essure,” Dr. Gottlieb stated.
He added that women who are using Essure successfully to prevent pregnancy should continue to do so, as “device removal has its own risks.”