User login
No benefit of direct stenting in PCI
A patient-level analysis drawn from three randomized, controlled trials finds no evidence that direct stenting improved myocardial reperfusion or clinical outcomes in ST-segment elevation myocardial infarction (STEMI) patients undergoing percutaneous coronary intervention.
Patients who underwent thrombus aspiration were more likely to receive direct stenting than conventional stenting, but there was no interaction between thrombus aspiration and outcomes, Karim D. Mahmoud, MD, reported in the European Heart Journal.
Direct stenting – stent implantation without balloon predilatation – has been widely adopted in an effort to improve PCI outcomes in STEMI patients, though no formal guidelines call for it. Small trials have suggested a benefit, but no large, definitive trials have been conducted.
Three previous trials have looked at thrombus aspiration in STEMI patients: TAPAS (the Thrombus Aspiration During Percutaneous Coronary Intervention in Acute Myocardial Infarction Study) found that routine manual thrombus aspiration led to better myocardial reperfusion and lower 1-year cardiac mortality (N Engl J Med. 2008 Feb 7;358:557-67). Two larger trials – TASTE (Thrombus Aspiration in ST Elevation Myocardial Infarction in Scandinavia; N Engl J Med. 2013 Oct 24;369[17]:1587-97) and TOTAL (Trial of Routine Aspiration Thrombectomy with PCI vs. PCI Alone in Patients with STEMI; N Engl J Med. 2015 Apr 9;372[15]:1389-98) – both failed to show any benefit of routine thrombus aspiration. As a result, international guidelines recommended use of thrombus aspiration only in selected patients.
The TASTE and TOTAL trials suggested that thrombus aspiration may have led to higher rates of direct stenting, but those rates were lower in TAPAS. Some have suggested that a synergistic effect between thrombus aspiration and DS could explain the positive finding in TAPAS, compared with negative findings in TASTE and TOTAL, said Dr. Mahmoud of Erasmus Medical Center, Rotterdam, the Netherlands.
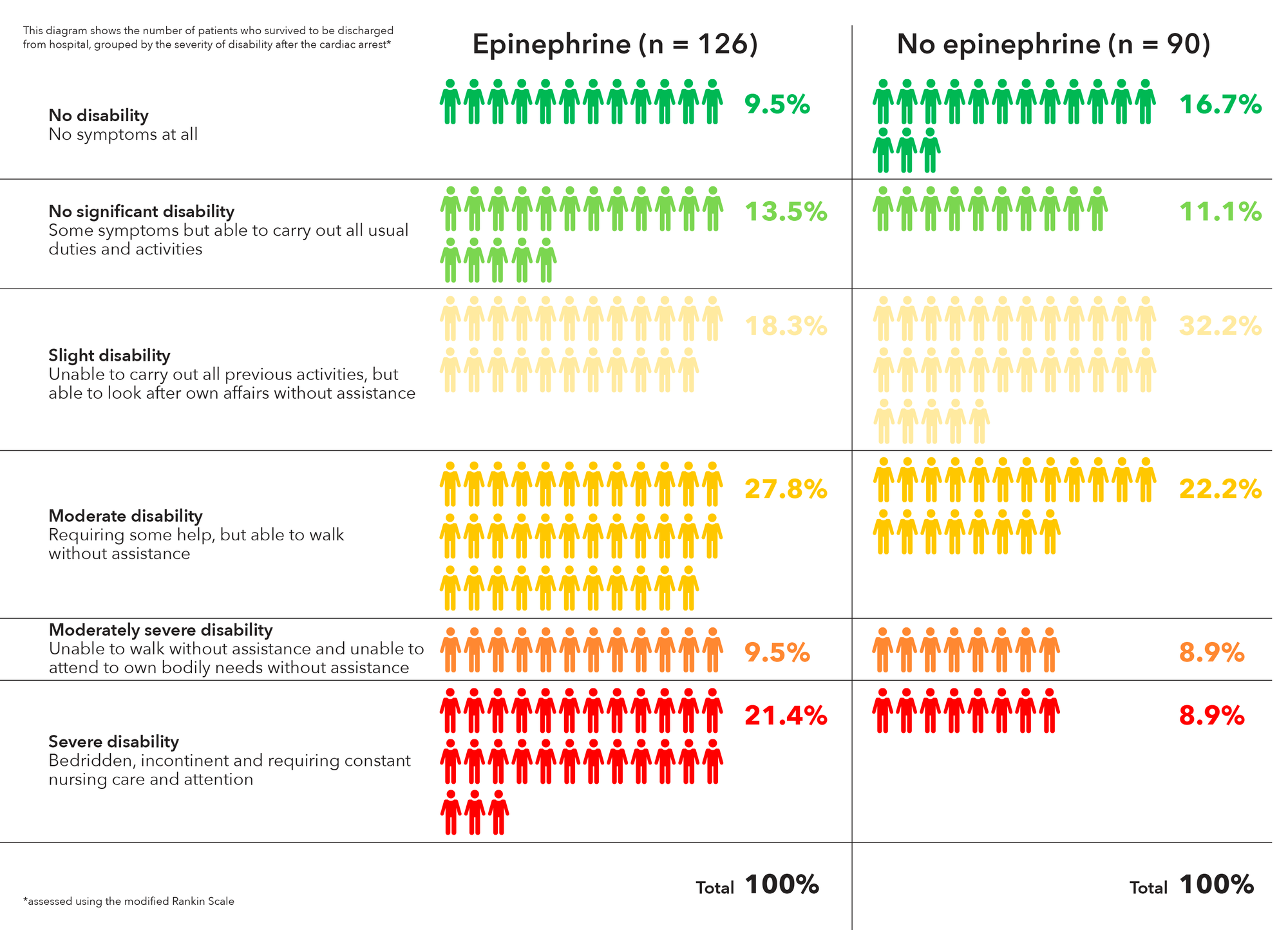
The researchers tested this idea by pooling patient-level data from the more than 17,000 participants in the three studies, 32% of whom underwent direct stenting. Patients who were randomized to undergo thrombus aspiration were nearly twice as likely to undergo direct stenting (41% vs. 22%, P less than .001). When the researchers 1:1 propensity matched direct stenting versus conventional stenting, 30-day cardiovascular death rates were similar between direct (1.7%) and conventional stenting (1.9%), and there was no interaction between direct stenting and thrombus aspiration. The latter result suggested that there is no synergistic effect. Similar results were found at 1-year follow-up, and with respect to 30-day stroke or transient ischemic attack.
One of the study authors received funding and or honoraria from Bayer, Medtronic, Vascular Solutions, Terumo, Boston Scientific, Abbott Vascular, AstraZeneca, and the Medicines Company.
SOURCE: Mahmoud KD et al. Eur Heart J. 2018;39:2472-9.
A patient-level analysis drawn from three randomized, controlled trials finds no evidence that direct stenting improved myocardial reperfusion or clinical outcomes in ST-segment elevation myocardial infarction (STEMI) patients undergoing percutaneous coronary intervention.
Patients who underwent thrombus aspiration were more likely to receive direct stenting than conventional stenting, but there was no interaction between thrombus aspiration and outcomes, Karim D. Mahmoud, MD, reported in the European Heart Journal.
Direct stenting – stent implantation without balloon predilatation – has been widely adopted in an effort to improve PCI outcomes in STEMI patients, though no formal guidelines call for it. Small trials have suggested a benefit, but no large, definitive trials have been conducted.
Three previous trials have looked at thrombus aspiration in STEMI patients: TAPAS (the Thrombus Aspiration During Percutaneous Coronary Intervention in Acute Myocardial Infarction Study) found that routine manual thrombus aspiration led to better myocardial reperfusion and lower 1-year cardiac mortality (N Engl J Med. 2008 Feb 7;358:557-67). Two larger trials – TASTE (Thrombus Aspiration in ST Elevation Myocardial Infarction in Scandinavia; N Engl J Med. 2013 Oct 24;369[17]:1587-97) and TOTAL (Trial of Routine Aspiration Thrombectomy with PCI vs. PCI Alone in Patients with STEMI; N Engl J Med. 2015 Apr 9;372[15]:1389-98) – both failed to show any benefit of routine thrombus aspiration. As a result, international guidelines recommended use of thrombus aspiration only in selected patients.
The TASTE and TOTAL trials suggested that thrombus aspiration may have led to higher rates of direct stenting, but those rates were lower in TAPAS. Some have suggested that a synergistic effect between thrombus aspiration and DS could explain the positive finding in TAPAS, compared with negative findings in TASTE and TOTAL, said Dr. Mahmoud of Erasmus Medical Center, Rotterdam, the Netherlands.
The researchers tested this idea by pooling patient-level data from the more than 17,000 participants in the three studies, 32% of whom underwent direct stenting. Patients who were randomized to undergo thrombus aspiration were nearly twice as likely to undergo direct stenting (41% vs. 22%, P less than .001). When the researchers 1:1 propensity matched direct stenting versus conventional stenting, 30-day cardiovascular death rates were similar between direct (1.7%) and conventional stenting (1.9%), and there was no interaction between direct stenting and thrombus aspiration. The latter result suggested that there is no synergistic effect. Similar results were found at 1-year follow-up, and with respect to 30-day stroke or transient ischemic attack.
One of the study authors received funding and or honoraria from Bayer, Medtronic, Vascular Solutions, Terumo, Boston Scientific, Abbott Vascular, AstraZeneca, and the Medicines Company.
SOURCE: Mahmoud KD et al. Eur Heart J. 2018;39:2472-9.
A patient-level analysis drawn from three randomized, controlled trials finds no evidence that direct stenting improved myocardial reperfusion or clinical outcomes in ST-segment elevation myocardial infarction (STEMI) patients undergoing percutaneous coronary intervention.
Patients who underwent thrombus aspiration were more likely to receive direct stenting than conventional stenting, but there was no interaction between thrombus aspiration and outcomes, Karim D. Mahmoud, MD, reported in the European Heart Journal.
Direct stenting – stent implantation without balloon predilatation – has been widely adopted in an effort to improve PCI outcomes in STEMI patients, though no formal guidelines call for it. Small trials have suggested a benefit, but no large, definitive trials have been conducted.
Three previous trials have looked at thrombus aspiration in STEMI patients: TAPAS (the Thrombus Aspiration During Percutaneous Coronary Intervention in Acute Myocardial Infarction Study) found that routine manual thrombus aspiration led to better myocardial reperfusion and lower 1-year cardiac mortality (N Engl J Med. 2008 Feb 7;358:557-67). Two larger trials – TASTE (Thrombus Aspiration in ST Elevation Myocardial Infarction in Scandinavia; N Engl J Med. 2013 Oct 24;369[17]:1587-97) and TOTAL (Trial of Routine Aspiration Thrombectomy with PCI vs. PCI Alone in Patients with STEMI; N Engl J Med. 2015 Apr 9;372[15]:1389-98) – both failed to show any benefit of routine thrombus aspiration. As a result, international guidelines recommended use of thrombus aspiration only in selected patients.
The TASTE and TOTAL trials suggested that thrombus aspiration may have led to higher rates of direct stenting, but those rates were lower in TAPAS. Some have suggested that a synergistic effect between thrombus aspiration and DS could explain the positive finding in TAPAS, compared with negative findings in TASTE and TOTAL, said Dr. Mahmoud of Erasmus Medical Center, Rotterdam, the Netherlands.
The researchers tested this idea by pooling patient-level data from the more than 17,000 participants in the three studies, 32% of whom underwent direct stenting. Patients who were randomized to undergo thrombus aspiration were nearly twice as likely to undergo direct stenting (41% vs. 22%, P less than .001). When the researchers 1:1 propensity matched direct stenting versus conventional stenting, 30-day cardiovascular death rates were similar between direct (1.7%) and conventional stenting (1.9%), and there was no interaction between direct stenting and thrombus aspiration. The latter result suggested that there is no synergistic effect. Similar results were found at 1-year follow-up, and with respect to 30-day stroke or transient ischemic attack.
One of the study authors received funding and or honoraria from Bayer, Medtronic, Vascular Solutions, Terumo, Boston Scientific, Abbott Vascular, AstraZeneca, and the Medicines Company.
SOURCE: Mahmoud KD et al. Eur Heart J. 2018;39:2472-9.
FROM EUROPEAN HEART JOURNAL
Key clinical point:
Major finding: Thirty-day cardiovascular death rates were similar between direct stenting (1.7%) and conventional stenting (1.9%).
Study details: Propensity-matched analysis of patient data from three previous trials (n = 17,329).
Disclosures: One of the study authors received funding and or honoraria from Bayer, Medtronic, Vascular Solutions, Terumo, Boston Scientific, Abbott Vascular, AstraZeneca, and the Medicines Company.
Source: Mahmoud KD et al. Eur Heart J. 2018;39:2472-9.
Neuroimaging may often be unneeded in ED seizure treatment
Neuroimaging may be appropriate only for specific types of patients with recurrent seizures who present at emergency departments because the scans are otherwise unlikely to prompt acute changes in treatment, a new multicenter study suggests. 
“Going forward, our results should help ED providers determine which patients are more likely to derive benefit from neuroimaging and which patients are not likely to benefit,” lead author Martin Salinsky, MD, of Oregon Health & Science University, Portland, said in an interview. “They can be more selective in ordering scans and reduce the total number obtained.”
As the study authors noted in their report, published July 18 in Epilepsia, “head CT is generally considered a benign procedure. However, overuse is problematic.”
The scans are costly and expose patients to radiation equivalent to 10 chest x-rays, the authors wrote. Scans can complicate care by clogging ED work flow and producing false positives, and patients often seize while undergoing scans, creating even more complications, they added.
“There is very little information in the medical literature that would help guide ED providers in their decision of whether to obtain neuroimaging on a patient who presents with a recurrent seizure,” Dr. Salinsky said. “Without this information, the tendency is to be cautious and obtain scans in more patients.”
For the study, the researchers tracked 822 consecutive ED visits for nonindex – recurrent – epileptic seizures at Oregon Health & Science University and VA Portland Health Care medical centers. (Nonindex seizures accounted for 78% of the total seizures that prompted ED care.)
The study subjects were adults treated for seizure as the main complaint. Patients who had a history of seizures but hadn’t had one for at least 5 years were excluded.
Of the total nonindex seizures, 46% of those resulted in neuroimaging.
“The overall yield of neuroimaging in this patient group was 2%-3%,” Dr. Salinsky said, referring to the percentage of patients whose scans resulted in an acute change in management.
False positives due to imaging artifacts were subsequently discovered in 3 of the 11 patients whose neuroimaging prompted an acute change in management. When the false positives were removed, the yield of acute management changes prompted by neuroimaging decreased to 2.1% overall.
“Three clinical factors – acute head trauma, prolonged alteration of consciousness, and focal neurological examination [at presentation] – were associated with an increased yield of imaging,” he said. “Absent all three factors, the yield in our patients was zero.”
At the two medical centers, the percentages of patients with acute head trauma were 10% and 15%. Prolonged alteration of consciousness occurred in 6% at both centers, and focal neurological examination at presentation was observed in 12% and 14%.
A fourth factor, presentation with status epilepticus/acute repetitive seizures, “bordered on statistical significance and might have reached significance in a larger series,” the authors wrote.
As they put it, “these results support a more conservative use of ED neuroimaging for nonindex seizures, based on clinical factors at the time of presentation. ... without specific indications, ED neuroimaging for nonindex seizures is unlikely to result in an acute change in care.”
The study authors estimated that hundreds of millions of dollars could be saved annually in the United States if neuroimaging in these ED patients could be cut in half.
No study funding was reported, and the authors reported no relevant disclosures.
SOURCE: Salinsky M et al. Epilepsia. 2018 July 18. doi: 10.1111/epi.14518
Neuroimaging may be appropriate only for specific types of patients with recurrent seizures who present at emergency departments because the scans are otherwise unlikely to prompt acute changes in treatment, a new multicenter study suggests.
“Going forward, our results should help ED providers determine which patients are more likely to derive benefit from neuroimaging and which patients are not likely to benefit,” lead author Martin Salinsky, MD, of Oregon Health & Science University, Portland, said in an interview. “They can be more selective in ordering scans and reduce the total number obtained.”
As the study authors noted in their report, published July 18 in Epilepsia, “head CT is generally considered a benign procedure. However, overuse is problematic.”
The scans are costly and expose patients to radiation equivalent to 10 chest x-rays, the authors wrote. Scans can complicate care by clogging ED work flow and producing false positives, and patients often seize while undergoing scans, creating even more complications, they added.
“There is very little information in the medical literature that would help guide ED providers in their decision of whether to obtain neuroimaging on a patient who presents with a recurrent seizure,” Dr. Salinsky said. “Without this information, the tendency is to be cautious and obtain scans in more patients.”
For the study, the researchers tracked 822 consecutive ED visits for nonindex – recurrent – epileptic seizures at Oregon Health & Science University and VA Portland Health Care medical centers. (Nonindex seizures accounted for 78% of the total seizures that prompted ED care.)
The study subjects were adults treated for seizure as the main complaint. Patients who had a history of seizures but hadn’t had one for at least 5 years were excluded.
Of the total nonindex seizures, 46% of those resulted in neuroimaging.
“The overall yield of neuroimaging in this patient group was 2%-3%,” Dr. Salinsky said, referring to the percentage of patients whose scans resulted in an acute change in management.
False positives due to imaging artifacts were subsequently discovered in 3 of the 11 patients whose neuroimaging prompted an acute change in management. When the false positives were removed, the yield of acute management changes prompted by neuroimaging decreased to 2.1% overall.
“Three clinical factors – acute head trauma, prolonged alteration of consciousness, and focal neurological examination [at presentation] – were associated with an increased yield of imaging,” he said. “Absent all three factors, the yield in our patients was zero.”
At the two medical centers, the percentages of patients with acute head trauma were 10% and 15%. Prolonged alteration of consciousness occurred in 6% at both centers, and focal neurological examination at presentation was observed in 12% and 14%.
A fourth factor, presentation with status epilepticus/acute repetitive seizures, “bordered on statistical significance and might have reached significance in a larger series,” the authors wrote.
As they put it, “these results support a more conservative use of ED neuroimaging for nonindex seizures, based on clinical factors at the time of presentation. ... without specific indications, ED neuroimaging for nonindex seizures is unlikely to result in an acute change in care.”
The study authors estimated that hundreds of millions of dollars could be saved annually in the United States if neuroimaging in these ED patients could be cut in half.
No study funding was reported, and the authors reported no relevant disclosures.
SOURCE: Salinsky M et al. Epilepsia. 2018 July 18. doi: 10.1111/epi.14518
Neuroimaging may be appropriate only for specific types of patients with recurrent seizures who present at emergency departments because the scans are otherwise unlikely to prompt acute changes in treatment, a new multicenter study suggests.
“Going forward, our results should help ED providers determine which patients are more likely to derive benefit from neuroimaging and which patients are not likely to benefit,” lead author Martin Salinsky, MD, of Oregon Health & Science University, Portland, said in an interview. “They can be more selective in ordering scans and reduce the total number obtained.”
As the study authors noted in their report, published July 18 in Epilepsia, “head CT is generally considered a benign procedure. However, overuse is problematic.”
The scans are costly and expose patients to radiation equivalent to 10 chest x-rays, the authors wrote. Scans can complicate care by clogging ED work flow and producing false positives, and patients often seize while undergoing scans, creating even more complications, they added.
“There is very little information in the medical literature that would help guide ED providers in their decision of whether to obtain neuroimaging on a patient who presents with a recurrent seizure,” Dr. Salinsky said. “Without this information, the tendency is to be cautious and obtain scans in more patients.”
For the study, the researchers tracked 822 consecutive ED visits for nonindex – recurrent – epileptic seizures at Oregon Health & Science University and VA Portland Health Care medical centers. (Nonindex seizures accounted for 78% of the total seizures that prompted ED care.)
The study subjects were adults treated for seizure as the main complaint. Patients who had a history of seizures but hadn’t had one for at least 5 years were excluded.
Of the total nonindex seizures, 46% of those resulted in neuroimaging.
“The overall yield of neuroimaging in this patient group was 2%-3%,” Dr. Salinsky said, referring to the percentage of patients whose scans resulted in an acute change in management.
False positives due to imaging artifacts were subsequently discovered in 3 of the 11 patients whose neuroimaging prompted an acute change in management. When the false positives were removed, the yield of acute management changes prompted by neuroimaging decreased to 2.1% overall.
“Three clinical factors – acute head trauma, prolonged alteration of consciousness, and focal neurological examination [at presentation] – were associated with an increased yield of imaging,” he said. “Absent all three factors, the yield in our patients was zero.”
At the two medical centers, the percentages of patients with acute head trauma were 10% and 15%. Prolonged alteration of consciousness occurred in 6% at both centers, and focal neurological examination at presentation was observed in 12% and 14%.
A fourth factor, presentation with status epilepticus/acute repetitive seizures, “bordered on statistical significance and might have reached significance in a larger series,” the authors wrote.
As they put it, “these results support a more conservative use of ED neuroimaging for nonindex seizures, based on clinical factors at the time of presentation. ... without specific indications, ED neuroimaging for nonindex seizures is unlikely to result in an acute change in care.”
The study authors estimated that hundreds of millions of dollars could be saved annually in the United States if neuroimaging in these ED patients could be cut in half.
No study funding was reported, and the authors reported no relevant disclosures.
SOURCE: Salinsky M et al. Epilepsia. 2018 July 18. doi: 10.1111/epi.14518
FROM EPILEPSIA
Key clinical point: In emergency departments, patients with seizure disorders and nonindex seizures may need neuroimaging only if they have acute head trauma, prolonged alteration of consciousness, or focal neurological examination at presentation.
Major finding: Absent the three factors above, neuroimaging did not prompt any acute changes in management.
Study details: Retrospective examination of 822 consecutive ED visits for nonindex seizures in patients with seizure disorders at two medical centers.
Disclosures: No study funding was reported, and the study authors reported no relevant disclosures.
Source: Salinsky M et al. Epilepsia. 2018 Jul 18. doi: 10.1111/epi.14518.
Lipid Metabolism May Be a Therapeutic Target in MS
NASHVILLE—Multiple sclerosis (MS) traditionally has been considered a chronic inflammatory autoimmune disease, but inflammation decreases as the disease progresses. Many other biologic processes are dysregulated in MS, such as myelin signal transport, mitochondrial function, and iron metabolism. Lipid metabolism affects all of these processes, including inflammation, and thus could be a valuable therapeutic target, according to research presented at the 2018 CMSC Annual Meeting.

“MS is not an inflammatory disease,” said John D. Nieland, PhD, Associate Professor of Health Science and Technology at Aalborg University in Denmark. “The inflammatory response is important, but it is not the only component. If you do not focus on the other components, you will never be able to treat the disease.”
The Role of Lipids in the CNS
Healthy brains have a high amount of glucose metabolism, but glucose metabolism is reduced in MS and other neurologic disorders such as Parkinson’s disease and Alzheimer’s disease. “If glucose metabolism is downregulated, something else has to be taking over,” said Dr. Nieland. He and his colleagues hypothesize that lipid metabolism replaces glucose metabolism in MS. They further hypothesize that MS fundamentally is a dysfunction of lipid metabolism.
Lipids have an essential role in the CNS. Proper signal transduction requires lipids to be bound to the myelin sheath. The proteins that compose myelin sheaths are highly immunogenic, and lipids shield them from exposure to the immune system. The half-life of lipids attached to the myelin sheath is three days, so these lipids must be replaced constantly. In addition, lipids are essential for the function of glutamate, cannabinoid, and insulin receptors.
An increase in lipid metabolism decreases glucose metabolism and induces the production of prostaglandin E2, which is a key molecule in the inflammatory response. In the early stages of MS, inflammation attacks the myelin sheath and other brain proteins. Increased lipid metabolism decreases lipid concentrations in the CNS, including around myelin. When lipids are removed from the myelin sheath, they expose the immunogenic proteins that compose it, thus provoking an immune response. Dysregulated lipid metabolism also contributes to oxidative stress, mitochondrial dysfunction, demyelination, and neuronal loss.
Chemical Inhibition of Lipid Metabolism
Dr. Nieland and colleagues hypothesized that blocking lipid metabolism would reverse the inflammatory response and other harmful processes that occur in MS. Previous research by Shriver and colleagues indicated that inhibition of carnitine palmitoyltransferase 1 (CPT1), a molecule essential to lipid metabolism, in encephalitogenic T cells increases apoptosis and reduces the production of inflammatory cytokines. Two of the molecule’s three isoforms, CPT1A and CPT1C, are upregulated in MS. Stress prompts an increase in CPT1 expression, which spurs a shift to lipid metabolism. “If you block CPT1, you jam lipid metabolism,” Dr. Nieland said. “There is no way around it.” Dr. Nieland’s group thus chose CPT1 as its target.
The investigators first conducted studies using etomoxir, which inhibits CPT1 and blocks long-chain fatty acids from entering the mitochondria for beta oxidation. Through these effects, etomoxir causes cells to shift to glucose metabolism.
The researchers immunized 42 mice with myelin oligodendrocyte glycoprotein (MOG35–55) to induce experimental autoimmune encephalopathy (EAE). When the animals first exhibited symptoms at Day 10, they were randomized to receive subcutaneous etomoxir or placebo daily. Disease score decreased significantly in the treated animals, compared with the control animals. On Day 24, more than 50% of the treated mice exhibited normal behavior, compared with approximately 20% of control mice.
In another study, the investigators immunized 47 rats with myelin basic protein to induce EAE. The animals began having symptoms at Day 7, and the investigators randomized them to daily treatment with subcutaneous etomoxir or placebo. At Day 11, disease score was significantly lower among treated animals, compared with control animals. Body weight was significantly higher among rats that received etomoxir, compared with controls, at that time point. Also, 25% of treated animals exhibited normal behavior, but no controls did.
In a third study, the investigators compared etomoxir, interferon beta, and placebo in a rat model of EAE. Each treatment group included 10 rats, and etomoxir had superior effects on disease score and body weight, compared with interferon beta and placebo. When the investigators examined the rats’ serum, they found that levels of antibodies against brain antigens common in EAE were lower in rats treated with etomoxir, compared with those treated with interferon beta or placebo.
Biologic Inhibition of Lipid Metabolism
In addition to pharmacologic treatment, genetic mutations affect CPT1 function. The Hutterites, an ethnoreligious group in Canada, have a mutation in CPT1A that almost completely blocks the molecule’s activity. Similarly, the Inuit have a mutation in CPT1A that reduces its activity to approximately 22%. The prevalence of MS is one in 1,100 among the Hutterites and one in 50,000 among the Inuit, compared with one in 350 in the Canadian population. These observations suggest that gene therapy could be another way to block CPT1.
Dr. Nieland’s group collaborated with the Netherlands Cancer Institute to develop mouse strains with two distinct mutations in CPT1A. The first mutation mimics that found among the Hutterites, and the other mimics that found among the Inuit. In a preliminary study, the investigators induced EAE in three wild-type mice and two mice with the CPT1A mutation similar to that of the Inuit. The mice were 10 weeks old at the time of immunization.
At 24 days, disease score was lower in the CPT1A mutant mice, compared with the wild-type mice. Furthermore, body weight was higher in the mutant mice, compared with the wild-type mice. The investigators also measured the mice’s grip strength at Day 2 and Day 24. Grip strength decreased in the wild-type mice but remained the same in the mutant mice. At Day 24, grip strength was significantly higher among mutant mice than among wild-type mice.
“These results indicate an interaction of the lipid metabolism in the brain and in the immune system, which supports our hypothesis regarding MS pathology,” said Anne Skøttrup Mørkholt, a doctoral student at Aalborg University, who collaborated with Dr. Nieland on these animal studies. “MS is not a disease of the immune system, but a systemic disease with dysregulation of multiple components.”

Lipid Metabolism in Other Neurologic Diseases
Data suggest that lipid metabolism may contribute to other neurologic diseases such as amyotrophic lateral sclerosis (ALS) as well. Huang et al found a correlation between serum triglyceride levels and the development of ALS. Dupuis et al observed upregulated lipid metabolism and downregulated glucose metabolism in SOD1 mouse models of the disease, as did subsequent researchers. In addition, a 2015 study by Palamiuc et al found that CPT1B was significantly increased in the muscle tissue of SOD1 mice.
To investigate whether suppressing lipid metabolism affected ALS, Dr. Nieland’s group examined a SOD1 mouse model of the disease. The mice developed symptoms at Day 70 and were randomized at Day 100 to etomoxir or placebo. Etomoxir was associated with less weight loss and better neurologic score, compared with placebo. Etomoxir also was associated with better performance on the wire hanging and rotarod tests, compared with placebo. “It seems like etomoxir was able to slow down the disease progression,” said Michael Sloth Trabjerg, MD, a doctoral student at Aalborg University.

Because research has found increased beta oxidation and decreased glucose metabolism in Parkinson’s disease, Dr. Nieland’s group studied the effect of CPT1 inhibition in a rotenone mouse model of the disease. They induced the disease in the mice for 32 days before randomizing them to placebo or etomoxir. For all mice, treatment alternated between rotenone on one day and placebo or etomoxir on the next day. The investigators observed significantly better sensorimotor performance among treated mice, compared with controls, on Day 47 and Day 56. At Day 60, mice who received etomoxir had significantly more muscle strength and longer latency to fall on the rotarod test, compared with mice that received placebo. These data are being prepared for publication.
The data suggest that dysregulated glucose metabolism and increased lipid metabolism play a role in ALS and Parkinson’s disease. “CPT1 seems to be a prominent target for moderating these diseases,” said Dr. Trabjerg.
—Erik Greb
Suggested Reading
Dodge JC, Treleaven CM, Fidler JA, et al. Metabolic signatures of amyotrophic lateral sclerosis reveal insights into disease pathogenesis. Proc Natl Acad Sci U S A. 2013;110(26):10812-10817.
Dupuis L, Oudart H, René F, et al. Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: benefit of a high-energy diet in a transgenic mouse model. Proc Natl Acad Sci U S A. 2004;101(30):11159-11164.
Huang R, Guo X, Chen X, et al. The serum lipid profiles of amyotrophic lateral sclerosis patients: A study from south-west China and a meta-analysis. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16(5-6):359-365.
Kim SM, Kim H, Kim JE, et al. Amyotrophic lateral sclerosis is associated with hypolipidemia at the presymptomatic stage in mice. PLoS One. 2011;6(3):e17985.
Lieury A, Chanal M, Androdias G, et al. Tissue remodeling in periplaque regions of multiple sclerosis spinal cord lesions. Glia. 2014;62(10):1645-1658.
Mørkholt AS, Kastaniegaard K, Trabjerg MS, et al. Identification of brain antigens recognized by autoantibodies in experimental autoimmune encephalomyelitis-induced animals treated with etomoxir or interferon-β. Sci Rep. 2018;8(1):7092.
Palamiuc L, Schlagowski A, Ngo ST, et al. A metabolic switch toward lipid use in glycolytic muscle is an early pathologic event in a mouse model of amyotrophic lateral sclerosis. EMBO Mol Med. 2015;7(5):526-546.
Shriver LP, Manchester M. Inhibition of fatty acid metabolism ameliorates disease activity in an animal model of multiple sclerosis. Sci Rep. 2011;1:79.
van der Windt GJ, Everts B, Chang CH, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. 2012;36(1):68-78.
NASHVILLE—Multiple sclerosis (MS) traditionally has been considered a chronic inflammatory autoimmune disease, but inflammation decreases as the disease progresses. Many other biologic processes are dysregulated in MS, such as myelin signal transport, mitochondrial function, and iron metabolism. Lipid metabolism affects all of these processes, including inflammation, and thus could be a valuable therapeutic target, according to research presented at the 2018 CMSC Annual Meeting.
“MS is not an inflammatory disease,” said John D. Nieland, PhD, Associate Professor of Health Science and Technology at Aalborg University in Denmark. “The inflammatory response is important, but it is not the only component. If you do not focus on the other components, you will never be able to treat the disease.”
The Role of Lipids in the CNS
Healthy brains have a high amount of glucose metabolism, but glucose metabolism is reduced in MS and other neurologic disorders such as Parkinson’s disease and Alzheimer’s disease. “If glucose metabolism is downregulated, something else has to be taking over,” said Dr. Nieland. He and his colleagues hypothesize that lipid metabolism replaces glucose metabolism in MS. They further hypothesize that MS fundamentally is a dysfunction of lipid metabolism.
Lipids have an essential role in the CNS. Proper signal transduction requires lipids to be bound to the myelin sheath. The proteins that compose myelin sheaths are highly immunogenic, and lipids shield them from exposure to the immune system. The half-life of lipids attached to the myelin sheath is three days, so these lipids must be replaced constantly. In addition, lipids are essential for the function of glutamate, cannabinoid, and insulin receptors.
An increase in lipid metabolism decreases glucose metabolism and induces the production of prostaglandin E2, which is a key molecule in the inflammatory response. In the early stages of MS, inflammation attacks the myelin sheath and other brain proteins. Increased lipid metabolism decreases lipid concentrations in the CNS, including around myelin. When lipids are removed from the myelin sheath, they expose the immunogenic proteins that compose it, thus provoking an immune response. Dysregulated lipid metabolism also contributes to oxidative stress, mitochondrial dysfunction, demyelination, and neuronal loss.
Chemical Inhibition of Lipid Metabolism
Dr. Nieland and colleagues hypothesized that blocking lipid metabolism would reverse the inflammatory response and other harmful processes that occur in MS. Previous research by Shriver and colleagues indicated that inhibition of carnitine palmitoyltransferase 1 (CPT1), a molecule essential to lipid metabolism, in encephalitogenic T cells increases apoptosis and reduces the production of inflammatory cytokines. Two of the molecule’s three isoforms, CPT1A and CPT1C, are upregulated in MS. Stress prompts an increase in CPT1 expression, which spurs a shift to lipid metabolism. “If you block CPT1, you jam lipid metabolism,” Dr. Nieland said. “There is no way around it.” Dr. Nieland’s group thus chose CPT1 as its target.
The investigators first conducted studies using etomoxir, which inhibits CPT1 and blocks long-chain fatty acids from entering the mitochondria for beta oxidation. Through these effects, etomoxir causes cells to shift to glucose metabolism.
The researchers immunized 42 mice with myelin oligodendrocyte glycoprotein (MOG35–55) to induce experimental autoimmune encephalopathy (EAE). When the animals first exhibited symptoms at Day 10, they were randomized to receive subcutaneous etomoxir or placebo daily. Disease score decreased significantly in the treated animals, compared with the control animals. On Day 24, more than 50% of the treated mice exhibited normal behavior, compared with approximately 20% of control mice.
In another study, the investigators immunized 47 rats with myelin basic protein to induce EAE. The animals began having symptoms at Day 7, and the investigators randomized them to daily treatment with subcutaneous etomoxir or placebo. At Day 11, disease score was significantly lower among treated animals, compared with control animals. Body weight was significantly higher among rats that received etomoxir, compared with controls, at that time point. Also, 25% of treated animals exhibited normal behavior, but no controls did.
In a third study, the investigators compared etomoxir, interferon beta, and placebo in a rat model of EAE. Each treatment group included 10 rats, and etomoxir had superior effects on disease score and body weight, compared with interferon beta and placebo. When the investigators examined the rats’ serum, they found that levels of antibodies against brain antigens common in EAE were lower in rats treated with etomoxir, compared with those treated with interferon beta or placebo.
Biologic Inhibition of Lipid Metabolism
In addition to pharmacologic treatment, genetic mutations affect CPT1 function. The Hutterites, an ethnoreligious group in Canada, have a mutation in CPT1A that almost completely blocks the molecule’s activity. Similarly, the Inuit have a mutation in CPT1A that reduces its activity to approximately 22%. The prevalence of MS is one in 1,100 among the Hutterites and one in 50,000 among the Inuit, compared with one in 350 in the Canadian population. These observations suggest that gene therapy could be another way to block CPT1.
Dr. Nieland’s group collaborated with the Netherlands Cancer Institute to develop mouse strains with two distinct mutations in CPT1A. The first mutation mimics that found among the Hutterites, and the other mimics that found among the Inuit. In a preliminary study, the investigators induced EAE in three wild-type mice and two mice with the CPT1A mutation similar to that of the Inuit. The mice were 10 weeks old at the time of immunization.
At 24 days, disease score was lower in the CPT1A mutant mice, compared with the wild-type mice. Furthermore, body weight was higher in the mutant mice, compared with the wild-type mice. The investigators also measured the mice’s grip strength at Day 2 and Day 24. Grip strength decreased in the wild-type mice but remained the same in the mutant mice. At Day 24, grip strength was significantly higher among mutant mice than among wild-type mice.
“These results indicate an interaction of the lipid metabolism in the brain and in the immune system, which supports our hypothesis regarding MS pathology,” said Anne Skøttrup Mørkholt, a doctoral student at Aalborg University, who collaborated with Dr. Nieland on these animal studies. “MS is not a disease of the immune system, but a systemic disease with dysregulation of multiple components.”
Lipid Metabolism in Other Neurologic Diseases
Data suggest that lipid metabolism may contribute to other neurologic diseases such as amyotrophic lateral sclerosis (ALS) as well. Huang et al found a correlation between serum triglyceride levels and the development of ALS. Dupuis et al observed upregulated lipid metabolism and downregulated glucose metabolism in SOD1 mouse models of the disease, as did subsequent researchers. In addition, a 2015 study by Palamiuc et al found that CPT1B was significantly increased in the muscle tissue of SOD1 mice.
To investigate whether suppressing lipid metabolism affected ALS, Dr. Nieland’s group examined a SOD1 mouse model of the disease. The mice developed symptoms at Day 70 and were randomized at Day 100 to etomoxir or placebo. Etomoxir was associated with less weight loss and better neurologic score, compared with placebo. Etomoxir also was associated with better performance on the wire hanging and rotarod tests, compared with placebo. “It seems like etomoxir was able to slow down the disease progression,” said Michael Sloth Trabjerg, MD, a doctoral student at Aalborg University.
Because research has found increased beta oxidation and decreased glucose metabolism in Parkinson’s disease, Dr. Nieland’s group studied the effect of CPT1 inhibition in a rotenone mouse model of the disease. They induced the disease in the mice for 32 days before randomizing them to placebo or etomoxir. For all mice, treatment alternated between rotenone on one day and placebo or etomoxir on the next day. The investigators observed significantly better sensorimotor performance among treated mice, compared with controls, on Day 47 and Day 56. At Day 60, mice who received etomoxir had significantly more muscle strength and longer latency to fall on the rotarod test, compared with mice that received placebo. These data are being prepared for publication.
The data suggest that dysregulated glucose metabolism and increased lipid metabolism play a role in ALS and Parkinson’s disease. “CPT1 seems to be a prominent target for moderating these diseases,” said Dr. Trabjerg.
—Erik Greb
Suggested Reading
Dodge JC, Treleaven CM, Fidler JA, et al. Metabolic signatures of amyotrophic lateral sclerosis reveal insights into disease pathogenesis. Proc Natl Acad Sci U S A. 2013;110(26):10812-10817.
Dupuis L, Oudart H, René F, et al. Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: benefit of a high-energy diet in a transgenic mouse model. Proc Natl Acad Sci U S A. 2004;101(30):11159-11164.
Huang R, Guo X, Chen X, et al. The serum lipid profiles of amyotrophic lateral sclerosis patients: A study from south-west China and a meta-analysis. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16(5-6):359-365.
Kim SM, Kim H, Kim JE, et al. Amyotrophic lateral sclerosis is associated with hypolipidemia at the presymptomatic stage in mice. PLoS One. 2011;6(3):e17985.
Lieury A, Chanal M, Androdias G, et al. Tissue remodeling in periplaque regions of multiple sclerosis spinal cord lesions. Glia. 2014;62(10):1645-1658.
Mørkholt AS, Kastaniegaard K, Trabjerg MS, et al. Identification of brain antigens recognized by autoantibodies in experimental autoimmune encephalomyelitis-induced animals treated with etomoxir or interferon-β. Sci Rep. 2018;8(1):7092.
Palamiuc L, Schlagowski A, Ngo ST, et al. A metabolic switch toward lipid use in glycolytic muscle is an early pathologic event in a mouse model of amyotrophic lateral sclerosis. EMBO Mol Med. 2015;7(5):526-546.
Shriver LP, Manchester M. Inhibition of fatty acid metabolism ameliorates disease activity in an animal model of multiple sclerosis. Sci Rep. 2011;1:79.
van der Windt GJ, Everts B, Chang CH, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. 2012;36(1):68-78.
NASHVILLE—Multiple sclerosis (MS) traditionally has been considered a chronic inflammatory autoimmune disease, but inflammation decreases as the disease progresses. Many other biologic processes are dysregulated in MS, such as myelin signal transport, mitochondrial function, and iron metabolism. Lipid metabolism affects all of these processes, including inflammation, and thus could be a valuable therapeutic target, according to research presented at the 2018 CMSC Annual Meeting.
“MS is not an inflammatory disease,” said John D. Nieland, PhD, Associate Professor of Health Science and Technology at Aalborg University in Denmark. “The inflammatory response is important, but it is not the only component. If you do not focus on the other components, you will never be able to treat the disease.”
The Role of Lipids in the CNS
Healthy brains have a high amount of glucose metabolism, but glucose metabolism is reduced in MS and other neurologic disorders such as Parkinson’s disease and Alzheimer’s disease. “If glucose metabolism is downregulated, something else has to be taking over,” said Dr. Nieland. He and his colleagues hypothesize that lipid metabolism replaces glucose metabolism in MS. They further hypothesize that MS fundamentally is a dysfunction of lipid metabolism.
Lipids have an essential role in the CNS. Proper signal transduction requires lipids to be bound to the myelin sheath. The proteins that compose myelin sheaths are highly immunogenic, and lipids shield them from exposure to the immune system. The half-life of lipids attached to the myelin sheath is three days, so these lipids must be replaced constantly. In addition, lipids are essential for the function of glutamate, cannabinoid, and insulin receptors.
An increase in lipid metabolism decreases glucose metabolism and induces the production of prostaglandin E2, which is a key molecule in the inflammatory response. In the early stages of MS, inflammation attacks the myelin sheath and other brain proteins. Increased lipid metabolism decreases lipid concentrations in the CNS, including around myelin. When lipids are removed from the myelin sheath, they expose the immunogenic proteins that compose it, thus provoking an immune response. Dysregulated lipid metabolism also contributes to oxidative stress, mitochondrial dysfunction, demyelination, and neuronal loss.
Chemical Inhibition of Lipid Metabolism
Dr. Nieland and colleagues hypothesized that blocking lipid metabolism would reverse the inflammatory response and other harmful processes that occur in MS. Previous research by Shriver and colleagues indicated that inhibition of carnitine palmitoyltransferase 1 (CPT1), a molecule essential to lipid metabolism, in encephalitogenic T cells increases apoptosis and reduces the production of inflammatory cytokines. Two of the molecule’s three isoforms, CPT1A and CPT1C, are upregulated in MS. Stress prompts an increase in CPT1 expression, which spurs a shift to lipid metabolism. “If you block CPT1, you jam lipid metabolism,” Dr. Nieland said. “There is no way around it.” Dr. Nieland’s group thus chose CPT1 as its target.
The investigators first conducted studies using etomoxir, which inhibits CPT1 and blocks long-chain fatty acids from entering the mitochondria for beta oxidation. Through these effects, etomoxir causes cells to shift to glucose metabolism.
The researchers immunized 42 mice with myelin oligodendrocyte glycoprotein (MOG35–55) to induce experimental autoimmune encephalopathy (EAE). When the animals first exhibited symptoms at Day 10, they were randomized to receive subcutaneous etomoxir or placebo daily. Disease score decreased significantly in the treated animals, compared with the control animals. On Day 24, more than 50% of the treated mice exhibited normal behavior, compared with approximately 20% of control mice.
In another study, the investigators immunized 47 rats with myelin basic protein to induce EAE. The animals began having symptoms at Day 7, and the investigators randomized them to daily treatment with subcutaneous etomoxir or placebo. At Day 11, disease score was significantly lower among treated animals, compared with control animals. Body weight was significantly higher among rats that received etomoxir, compared with controls, at that time point. Also, 25% of treated animals exhibited normal behavior, but no controls did.
In a third study, the investigators compared etomoxir, interferon beta, and placebo in a rat model of EAE. Each treatment group included 10 rats, and etomoxir had superior effects on disease score and body weight, compared with interferon beta and placebo. When the investigators examined the rats’ serum, they found that levels of antibodies against brain antigens common in EAE were lower in rats treated with etomoxir, compared with those treated with interferon beta or placebo.
Biologic Inhibition of Lipid Metabolism
In addition to pharmacologic treatment, genetic mutations affect CPT1 function. The Hutterites, an ethnoreligious group in Canada, have a mutation in CPT1A that almost completely blocks the molecule’s activity. Similarly, the Inuit have a mutation in CPT1A that reduces its activity to approximately 22%. The prevalence of MS is one in 1,100 among the Hutterites and one in 50,000 among the Inuit, compared with one in 350 in the Canadian population. These observations suggest that gene therapy could be another way to block CPT1.
Dr. Nieland’s group collaborated with the Netherlands Cancer Institute to develop mouse strains with two distinct mutations in CPT1A. The first mutation mimics that found among the Hutterites, and the other mimics that found among the Inuit. In a preliminary study, the investigators induced EAE in three wild-type mice and two mice with the CPT1A mutation similar to that of the Inuit. The mice were 10 weeks old at the time of immunization.
At 24 days, disease score was lower in the CPT1A mutant mice, compared with the wild-type mice. Furthermore, body weight was higher in the mutant mice, compared with the wild-type mice. The investigators also measured the mice’s grip strength at Day 2 and Day 24. Grip strength decreased in the wild-type mice but remained the same in the mutant mice. At Day 24, grip strength was significantly higher among mutant mice than among wild-type mice.
“These results indicate an interaction of the lipid metabolism in the brain and in the immune system, which supports our hypothesis regarding MS pathology,” said Anne Skøttrup Mørkholt, a doctoral student at Aalborg University, who collaborated with Dr. Nieland on these animal studies. “MS is not a disease of the immune system, but a systemic disease with dysregulation of multiple components.”
Lipid Metabolism in Other Neurologic Diseases
Data suggest that lipid metabolism may contribute to other neurologic diseases such as amyotrophic lateral sclerosis (ALS) as well. Huang et al found a correlation between serum triglyceride levels and the development of ALS. Dupuis et al observed upregulated lipid metabolism and downregulated glucose metabolism in SOD1 mouse models of the disease, as did subsequent researchers. In addition, a 2015 study by Palamiuc et al found that CPT1B was significantly increased in the muscle tissue of SOD1 mice.
To investigate whether suppressing lipid metabolism affected ALS, Dr. Nieland’s group examined a SOD1 mouse model of the disease. The mice developed symptoms at Day 70 and were randomized at Day 100 to etomoxir or placebo. Etomoxir was associated with less weight loss and better neurologic score, compared with placebo. Etomoxir also was associated with better performance on the wire hanging and rotarod tests, compared with placebo. “It seems like etomoxir was able to slow down the disease progression,” said Michael Sloth Trabjerg, MD, a doctoral student at Aalborg University.
Because research has found increased beta oxidation and decreased glucose metabolism in Parkinson’s disease, Dr. Nieland’s group studied the effect of CPT1 inhibition in a rotenone mouse model of the disease. They induced the disease in the mice for 32 days before randomizing them to placebo or etomoxir. For all mice, treatment alternated between rotenone on one day and placebo or etomoxir on the next day. The investigators observed significantly better sensorimotor performance among treated mice, compared with controls, on Day 47 and Day 56. At Day 60, mice who received etomoxir had significantly more muscle strength and longer latency to fall on the rotarod test, compared with mice that received placebo. These data are being prepared for publication.
The data suggest that dysregulated glucose metabolism and increased lipid metabolism play a role in ALS and Parkinson’s disease. “CPT1 seems to be a prominent target for moderating these diseases,” said Dr. Trabjerg.
—Erik Greb
Suggested Reading
Dodge JC, Treleaven CM, Fidler JA, et al. Metabolic signatures of amyotrophic lateral sclerosis reveal insights into disease pathogenesis. Proc Natl Acad Sci U S A. 2013;110(26):10812-10817.
Dupuis L, Oudart H, René F, et al. Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: benefit of a high-energy diet in a transgenic mouse model. Proc Natl Acad Sci U S A. 2004;101(30):11159-11164.
Huang R, Guo X, Chen X, et al. The serum lipid profiles of amyotrophic lateral sclerosis patients: A study from south-west China and a meta-analysis. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16(5-6):359-365.
Kim SM, Kim H, Kim JE, et al. Amyotrophic lateral sclerosis is associated with hypolipidemia at the presymptomatic stage in mice. PLoS One. 2011;6(3):e17985.
Lieury A, Chanal M, Androdias G, et al. Tissue remodeling in periplaque regions of multiple sclerosis spinal cord lesions. Glia. 2014;62(10):1645-1658.
Mørkholt AS, Kastaniegaard K, Trabjerg MS, et al. Identification of brain antigens recognized by autoantibodies in experimental autoimmune encephalomyelitis-induced animals treated with etomoxir or interferon-β. Sci Rep. 2018;8(1):7092.
Palamiuc L, Schlagowski A, Ngo ST, et al. A metabolic switch toward lipid use in glycolytic muscle is an early pathologic event in a mouse model of amyotrophic lateral sclerosis. EMBO Mol Med. 2015;7(5):526-546.
Shriver LP, Manchester M. Inhibition of fatty acid metabolism ameliorates disease activity in an animal model of multiple sclerosis. Sci Rep. 2011;1:79.
van der Windt GJ, Everts B, Chang CH, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. 2012;36(1):68-78.
Culotte stenting impresses in CELTIC Bifurcation Study
PARIS – Technical success rates were high and major adverse events impressively low with a two-stent culotte strategy using contemporary drug-eluting stents for coronary bifurcation lesions in the randomized CELTIC Bifurcation Study.

“We initiated this study because of a conviction that the story isn’t finished with bifurcation stenting. We’re very much under the impression that the accepted wisdom of a conservative approach is, we think, not correct, and the issue needs to be kept open,” said Dr. Foley, an interventional cardiologist at Beaumont Hospital in Dublin.
The widely accepted provisional single-stent strategy is based on early-days randomized trial evidence using first-generation drug-eluting stents and older techniques that are no longer relevant in contemporary practice. Moreover, this conservative single-stent approach doesn’t address the important issue of ischemia arising from large side branches, he asserted.
“I’ve always been fond of culotte stenting myself because I think it’s a very elegant, simple, repeatable strategy, and with modern stents it becomes easier for modest-volume operators to carry it out well. We’ve kept on trying to convert new colleagues and older colleagues who are set in their ways,” Dr. Foley said.
The CELTIC Bifurcation Study was an investigator-initiated trial in which 177 patients at nine centers in Ireland and the United Kingdom were randomized to culotte stenting using either two-connector, third-generation Synergy everolimus-eluting stents or the three-connector, second-generation Xience everolimus-eluting stents. All participants had Medina 1,1,1 coronary bifurcation lesions, which were left anterior descending/diagonal lesions in more than 80% of cases. A radial approach was used in more than 95% of the procedures. The indication for percutaneous coronary intervention was stable angina in more than 60% of cases. The rate of technical procedural success with final kissing balloon inflation exceeded 96%. The primary outcome – a MACCE (major adverse cardiovascular and cerebrovascular events) composite of death, MI, cerebrovascular accident, and target vessel revascularization over the course of 9 months – occurred in 5.9% of patients: 8.6% of the Synergy group and 3.7% with Xience stents, a nonsignificant difference. This MACCE rate was considerably lower than the 10% figure that the investigators had expected on the basis of published studies of PCI in these complex bifurcation lesions.
“The results were better than expected,” the cardiologist said. “We don’t get excited that easily, to be honest, but nonetheless we’re a little bit excited that the overall MACCE rate in this complex lesion presentation was 5.9%.”
Discussant Volker Schächinger, MD, director of cardiology at Fulda (Germany) Hospital, observed: “It’s always good to reassess what are believed to be answered questions when there are new devices available.” But why not compare culotte stenting to the provisional single-stent strategy? he asked.
“We think provisional versus culotte stenting has been thrashed to death already. And you’d need a bigger trial than we had funding for,” Dr. Foley replied.
“Many of us use the DK [double kissing] crush technique,” another panelist said. “It’s very popular. But if you look at bench testing, perhaps culotte is a better approach by many parameters. So I think it was important for you to highlight the value of culotte and how it can be done properly.”
Discussant James Nolan, MD, a cardiologist at the University Hospital of North Staffordshire (England), said, “The most critical thing with these bifurcation procedures is the operators and how they do it. So you have to do the culotte to the standard done in this trial. If you do a sloppy culotte, it’s not going to be great. It’s probably more important to deliver an excellently performed procedure, whatever it is. You’ll get a better result if you’re good at what you’re doing rather than selecting one procedure or another.”
Dr. Foley agreed, adding: “In some of the DK crush versus culotte randomized trials, I’m not convinced that culotte was done the way I would suggest it should be done.”
Operators in the CELTIC Bifurcation Study were asked to follow a standardized culotte procedure: predilate both limbs of the bifurcation, keep both wires in place, deploy the first stent in the side branch unless the main branch was awkwardly angulated, then cross by going from distally into the optimized first stent, and placing the second stent proximal to the first stent so that the two stents overlap in the proximal main vessel.
“We call that ‘nailing it down,’ ” he explained.
The procedure is completed by sequential high-pressure kissing balloon dilatation of both branches, with intravascular ultrasound or optical coherence tomography recommended but not required.
Simultaneously with this presentation, the study results were published online (EuroIntervention 2018 Jun 8;14[3]:e318-24).
The CELTIC Bifurcation Study was funded by an unrestricted grant from Boston Scientific. Dr. Foley reported having no financial conflicts of interest regarding the study.
PARIS – Technical success rates were high and major adverse events impressively low with a two-stent culotte strategy using contemporary drug-eluting stents for coronary bifurcation lesions in the randomized CELTIC Bifurcation Study.
“We initiated this study because of a conviction that the story isn’t finished with bifurcation stenting. We’re very much under the impression that the accepted wisdom of a conservative approach is, we think, not correct, and the issue needs to be kept open,” said Dr. Foley, an interventional cardiologist at Beaumont Hospital in Dublin.
The widely accepted provisional single-stent strategy is based on early-days randomized trial evidence using first-generation drug-eluting stents and older techniques that are no longer relevant in contemporary practice. Moreover, this conservative single-stent approach doesn’t address the important issue of ischemia arising from large side branches, he asserted.
“I’ve always been fond of culotte stenting myself because I think it’s a very elegant, simple, repeatable strategy, and with modern stents it becomes easier for modest-volume operators to carry it out well. We’ve kept on trying to convert new colleagues and older colleagues who are set in their ways,” Dr. Foley said.
The CELTIC Bifurcation Study was an investigator-initiated trial in which 177 patients at nine centers in Ireland and the United Kingdom were randomized to culotte stenting using either two-connector, third-generation Synergy everolimus-eluting stents or the three-connector, second-generation Xience everolimus-eluting stents. All participants had Medina 1,1,1 coronary bifurcation lesions, which were left anterior descending/diagonal lesions in more than 80% of cases. A radial approach was used in more than 95% of the procedures. The indication for percutaneous coronary intervention was stable angina in more than 60% of cases. The rate of technical procedural success with final kissing balloon inflation exceeded 96%. The primary outcome – a MACCE (major adverse cardiovascular and cerebrovascular events) composite of death, MI, cerebrovascular accident, and target vessel revascularization over the course of 9 months – occurred in 5.9% of patients: 8.6% of the Synergy group and 3.7% with Xience stents, a nonsignificant difference. This MACCE rate was considerably lower than the 10% figure that the investigators had expected on the basis of published studies of PCI in these complex bifurcation lesions.
“The results were better than expected,” the cardiologist said. “We don’t get excited that easily, to be honest, but nonetheless we’re a little bit excited that the overall MACCE rate in this complex lesion presentation was 5.9%.”
Discussant Volker Schächinger, MD, director of cardiology at Fulda (Germany) Hospital, observed: “It’s always good to reassess what are believed to be answered questions when there are new devices available.” But why not compare culotte stenting to the provisional single-stent strategy? he asked.
“We think provisional versus culotte stenting has been thrashed to death already. And you’d need a bigger trial than we had funding for,” Dr. Foley replied.
“Many of us use the DK [double kissing] crush technique,” another panelist said. “It’s very popular. But if you look at bench testing, perhaps culotte is a better approach by many parameters. So I think it was important for you to highlight the value of culotte and how it can be done properly.”
Discussant James Nolan, MD, a cardiologist at the University Hospital of North Staffordshire (England), said, “The most critical thing with these bifurcation procedures is the operators and how they do it. So you have to do the culotte to the standard done in this trial. If you do a sloppy culotte, it’s not going to be great. It’s probably more important to deliver an excellently performed procedure, whatever it is. You’ll get a better result if you’re good at what you’re doing rather than selecting one procedure or another.”
Dr. Foley agreed, adding: “In some of the DK crush versus culotte randomized trials, I’m not convinced that culotte was done the way I would suggest it should be done.”
Operators in the CELTIC Bifurcation Study were asked to follow a standardized culotte procedure: predilate both limbs of the bifurcation, keep both wires in place, deploy the first stent in the side branch unless the main branch was awkwardly angulated, then cross by going from distally into the optimized first stent, and placing the second stent proximal to the first stent so that the two stents overlap in the proximal main vessel.
“We call that ‘nailing it down,’ ” he explained.
The procedure is completed by sequential high-pressure kissing balloon dilatation of both branches, with intravascular ultrasound or optical coherence tomography recommended but not required.
Simultaneously with this presentation, the study results were published online (EuroIntervention 2018 Jun 8;14[3]:e318-24).
The CELTIC Bifurcation Study was funded by an unrestricted grant from Boston Scientific. Dr. Foley reported having no financial conflicts of interest regarding the study.
PARIS – Technical success rates were high and major adverse events impressively low with a two-stent culotte strategy using contemporary drug-eluting stents for coronary bifurcation lesions in the randomized CELTIC Bifurcation Study.
“We initiated this study because of a conviction that the story isn’t finished with bifurcation stenting. We’re very much under the impression that the accepted wisdom of a conservative approach is, we think, not correct, and the issue needs to be kept open,” said Dr. Foley, an interventional cardiologist at Beaumont Hospital in Dublin.
The widely accepted provisional single-stent strategy is based on early-days randomized trial evidence using first-generation drug-eluting stents and older techniques that are no longer relevant in contemporary practice. Moreover, this conservative single-stent approach doesn’t address the important issue of ischemia arising from large side branches, he asserted.
“I’ve always been fond of culotte stenting myself because I think it’s a very elegant, simple, repeatable strategy, and with modern stents it becomes easier for modest-volume operators to carry it out well. We’ve kept on trying to convert new colleagues and older colleagues who are set in their ways,” Dr. Foley said.
The CELTIC Bifurcation Study was an investigator-initiated trial in which 177 patients at nine centers in Ireland and the United Kingdom were randomized to culotte stenting using either two-connector, third-generation Synergy everolimus-eluting stents or the three-connector, second-generation Xience everolimus-eluting stents. All participants had Medina 1,1,1 coronary bifurcation lesions, which were left anterior descending/diagonal lesions in more than 80% of cases. A radial approach was used in more than 95% of the procedures. The indication for percutaneous coronary intervention was stable angina in more than 60% of cases. The rate of technical procedural success with final kissing balloon inflation exceeded 96%. The primary outcome – a MACCE (major adverse cardiovascular and cerebrovascular events) composite of death, MI, cerebrovascular accident, and target vessel revascularization over the course of 9 months – occurred in 5.9% of patients: 8.6% of the Synergy group and 3.7% with Xience stents, a nonsignificant difference. This MACCE rate was considerably lower than the 10% figure that the investigators had expected on the basis of published studies of PCI in these complex bifurcation lesions.
“The results were better than expected,” the cardiologist said. “We don’t get excited that easily, to be honest, but nonetheless we’re a little bit excited that the overall MACCE rate in this complex lesion presentation was 5.9%.”
Discussant Volker Schächinger, MD, director of cardiology at Fulda (Germany) Hospital, observed: “It’s always good to reassess what are believed to be answered questions when there are new devices available.” But why not compare culotte stenting to the provisional single-stent strategy? he asked.
“We think provisional versus culotte stenting has been thrashed to death already. And you’d need a bigger trial than we had funding for,” Dr. Foley replied.
“Many of us use the DK [double kissing] crush technique,” another panelist said. “It’s very popular. But if you look at bench testing, perhaps culotte is a better approach by many parameters. So I think it was important for you to highlight the value of culotte and how it can be done properly.”
Discussant James Nolan, MD, a cardiologist at the University Hospital of North Staffordshire (England), said, “The most critical thing with these bifurcation procedures is the operators and how they do it. So you have to do the culotte to the standard done in this trial. If you do a sloppy culotte, it’s not going to be great. It’s probably more important to deliver an excellently performed procedure, whatever it is. You’ll get a better result if you’re good at what you’re doing rather than selecting one procedure or another.”
Dr. Foley agreed, adding: “In some of the DK crush versus culotte randomized trials, I’m not convinced that culotte was done the way I would suggest it should be done.”
Operators in the CELTIC Bifurcation Study were asked to follow a standardized culotte procedure: predilate both limbs of the bifurcation, keep both wires in place, deploy the first stent in the side branch unless the main branch was awkwardly angulated, then cross by going from distally into the optimized first stent, and placing the second stent proximal to the first stent so that the two stents overlap in the proximal main vessel.
“We call that ‘nailing it down,’ ” he explained.
The procedure is completed by sequential high-pressure kissing balloon dilatation of both branches, with intravascular ultrasound or optical coherence tomography recommended but not required.
Simultaneously with this presentation, the study results were published online (EuroIntervention 2018 Jun 8;14[3]:e318-24).
The CELTIC Bifurcation Study was funded by an unrestricted grant from Boston Scientific. Dr. Foley reported having no financial conflicts of interest regarding the study.
REPORTING FROM EUROPCR 2018
Key clinical point: .
Major finding: The 9-month MACCE rate following culotte stenting for bifurcation lesions was 5.9%, with no significant difference between patients randomized to the Xience or Synergy stents.
Study details: This multicenter randomized trial comprised 177 patients with coronary bifurcation lesions who underwent culotte stenting with either Xience or Synergy everolimus-eluting stents.
Disclosures: The presenter reported having no financial conflicts regarding the study, funded by an unrestricted grant from Boston Scientific.
For men with SCD and priapism, hypoxia may prompt RBC adhesion
WASHINGTON – For male patients with sickle cell disease, priapism can be more than just painful and embarrassing. The prolonged erections prompted by vasoocclusive events in the penis may lead to irreversible impotence, but little is known about risk factors for priapism, which remains a difficult-to-treat complication of the disease.
In males with HbSS sickle cell disease (SCD) and priapism, RBC adhesion is increased in hypoxic conditions, according to preliminary findings from work using a newly developed biochip that mimics microvascular conditions in SCD. This significant level of adhesion prompted by hypoxia was not seen in men who did not have priapism, according to study coauthor Erina Quinn, a research assistant in hematology and oncology at Case Western Reserve University, Cleveland, who presented the results at the annual meeting of the Foundation for Sickle Cell Disease Research.
When hemoglobin desaturation occurs, polymerization can be increased, leading to increased end-organ damage, Ms. Quinn said. The biochip is “an effort to measure cellular adhesion in a clinically meaningful way.” The tool can detect hemoglobin phenotype, differentiating among HbSS, HbSbeta+, and HbSC. It can also measure the degree of hemolysis and RBC deformability.
The biochip “mimics postcapillary flow conditions in microchannels,” Ms. Quinn said. The device forces blood samples through microchannels that are at the diameter of smaller venules, approximately 50 mcm, and at a physiological flow rate ranging from 1-13 mm/sec. The microfluidic channels are coated with laminin, a subendothelial matrix protein implicated in RBC adhesion. A second microfluidic biochip mimics hypoxic conditions.
The study enrolled 26 men with the HbSS genotype, 14 of whom reported priapism, and assessed RBC adhesion in blood samples run though both the SCD-modeled biochip and the hypoxia biochip. Investigators also assessed contemporaneous in vivo hemoglobin desaturation, and looked for associations with the in vitro biochip findings.
Of the 26 participants, 16 also had either nocturnal or exertional hemoglobin desaturation. In addition, 10 participants had both priapism and desaturations. These data were collected by retrospective chart review and patient survey.
Patients with priapism were a mean age of 34 years, compared with a mean age of 29 years for the other participants, a nonsignificant difference. There were no significant differences in mean hemoglobin or bilirubin levels, or in reticulocyte counts, between the two groups.
However, white blood count, absolute neutrophil count, and lactate dehydrogenase levels were significantly higher for men with priapism (P = .022, .037, and .008, respectively). Ferritin levels were higher as well, at a mean 2,433 (plus or minus 2,234) mcg/L for those with priapism, compared with a mean 269 (plus or minus 3,015) mcg/L for those without priapism (P = .031).
When absolute reticulocyte count was mapped against lactate dehydrogenase levels to create a measure of degree of hemolysis, “individuals with priapism had a more hemolytic lab profile,” said Ms. Quinn (P = .0186).
Though 10 of 14 men with priapism had hemoglobin desaturation, compared with 5 of 12 who did not have priapism, the difference was not statistically significant.
When the researchers compared microchip analysis of RBC adhesion, though, they found marked differences in RBC adhesion in hypoxic versus nonhypoxic conditions. Significantly more RBCs were adherent under hypoxic conditions – in the hypoxic biochip – for the patients with priapism than for patients without priapism (mean, 529 vs. 3,268 adherent cells; P = .016).
Though numbers were small, RBCs from patients with reported priapism and hemoglobin desaturation in vivo showed increased hypoxia enhanced adhesion in vitro (P = .013), Ms. Quinn said. These was no significant difference between adhesion in normoxic and hypoxic conditions for the patients without priapism.
Future directions of work with the biochip include prospective identification of desaturation events and better characterization of nocturnal symptoms, Ms. Quinn said. The investigators also plan to see whether treatment with supplemental oxygen affects RBC adhesion.
The research was supported by the National Institutes of Health, the Doris Duke Charitable Foundation, and the National Science Foundation. Two coauthors have filed an international patent for the biochip technology.
WASHINGTON – For male patients with sickle cell disease, priapism can be more than just painful and embarrassing. The prolonged erections prompted by vasoocclusive events in the penis may lead to irreversible impotence, but little is known about risk factors for priapism, which remains a difficult-to-treat complication of the disease.
In males with HbSS sickle cell disease (SCD) and priapism, RBC adhesion is increased in hypoxic conditions, according to preliminary findings from work using a newly developed biochip that mimics microvascular conditions in SCD. This significant level of adhesion prompted by hypoxia was not seen in men who did not have priapism, according to study coauthor Erina Quinn, a research assistant in hematology and oncology at Case Western Reserve University, Cleveland, who presented the results at the annual meeting of the Foundation for Sickle Cell Disease Research.
When hemoglobin desaturation occurs, polymerization can be increased, leading to increased end-organ damage, Ms. Quinn said. The biochip is “an effort to measure cellular adhesion in a clinically meaningful way.” The tool can detect hemoglobin phenotype, differentiating among HbSS, HbSbeta+, and HbSC. It can also measure the degree of hemolysis and RBC deformability.
The biochip “mimics postcapillary flow conditions in microchannels,” Ms. Quinn said. The device forces blood samples through microchannels that are at the diameter of smaller venules, approximately 50 mcm, and at a physiological flow rate ranging from 1-13 mm/sec. The microfluidic channels are coated with laminin, a subendothelial matrix protein implicated in RBC adhesion. A second microfluidic biochip mimics hypoxic conditions.
The study enrolled 26 men with the HbSS genotype, 14 of whom reported priapism, and assessed RBC adhesion in blood samples run though both the SCD-modeled biochip and the hypoxia biochip. Investigators also assessed contemporaneous in vivo hemoglobin desaturation, and looked for associations with the in vitro biochip findings.
Of the 26 participants, 16 also had either nocturnal or exertional hemoglobin desaturation. In addition, 10 participants had both priapism and desaturations. These data were collected by retrospective chart review and patient survey.
Patients with priapism were a mean age of 34 years, compared with a mean age of 29 years for the other participants, a nonsignificant difference. There were no significant differences in mean hemoglobin or bilirubin levels, or in reticulocyte counts, between the two groups.
However, white blood count, absolute neutrophil count, and lactate dehydrogenase levels were significantly higher for men with priapism (P = .022, .037, and .008, respectively). Ferritin levels were higher as well, at a mean 2,433 (plus or minus 2,234) mcg/L for those with priapism, compared with a mean 269 (plus or minus 3,015) mcg/L for those without priapism (P = .031).
When absolute reticulocyte count was mapped against lactate dehydrogenase levels to create a measure of degree of hemolysis, “individuals with priapism had a more hemolytic lab profile,” said Ms. Quinn (P = .0186).
Though 10 of 14 men with priapism had hemoglobin desaturation, compared with 5 of 12 who did not have priapism, the difference was not statistically significant.
When the researchers compared microchip analysis of RBC adhesion, though, they found marked differences in RBC adhesion in hypoxic versus nonhypoxic conditions. Significantly more RBCs were adherent under hypoxic conditions – in the hypoxic biochip – for the patients with priapism than for patients without priapism (mean, 529 vs. 3,268 adherent cells; P = .016).
Though numbers were small, RBCs from patients with reported priapism and hemoglobin desaturation in vivo showed increased hypoxia enhanced adhesion in vitro (P = .013), Ms. Quinn said. These was no significant difference between adhesion in normoxic and hypoxic conditions for the patients without priapism.
Future directions of work with the biochip include prospective identification of desaturation events and better characterization of nocturnal symptoms, Ms. Quinn said. The investigators also plan to see whether treatment with supplemental oxygen affects RBC adhesion.
The research was supported by the National Institutes of Health, the Doris Duke Charitable Foundation, and the National Science Foundation. Two coauthors have filed an international patent for the biochip technology.
WASHINGTON – For male patients with sickle cell disease, priapism can be more than just painful and embarrassing. The prolonged erections prompted by vasoocclusive events in the penis may lead to irreversible impotence, but little is known about risk factors for priapism, which remains a difficult-to-treat complication of the disease.
In males with HbSS sickle cell disease (SCD) and priapism, RBC adhesion is increased in hypoxic conditions, according to preliminary findings from work using a newly developed biochip that mimics microvascular conditions in SCD. This significant level of adhesion prompted by hypoxia was not seen in men who did not have priapism, according to study coauthor Erina Quinn, a research assistant in hematology and oncology at Case Western Reserve University, Cleveland, who presented the results at the annual meeting of the Foundation for Sickle Cell Disease Research.
When hemoglobin desaturation occurs, polymerization can be increased, leading to increased end-organ damage, Ms. Quinn said. The biochip is “an effort to measure cellular adhesion in a clinically meaningful way.” The tool can detect hemoglobin phenotype, differentiating among HbSS, HbSbeta+, and HbSC. It can also measure the degree of hemolysis and RBC deformability.
The biochip “mimics postcapillary flow conditions in microchannels,” Ms. Quinn said. The device forces blood samples through microchannels that are at the diameter of smaller venules, approximately 50 mcm, and at a physiological flow rate ranging from 1-13 mm/sec. The microfluidic channels are coated with laminin, a subendothelial matrix protein implicated in RBC adhesion. A second microfluidic biochip mimics hypoxic conditions.
The study enrolled 26 men with the HbSS genotype, 14 of whom reported priapism, and assessed RBC adhesion in blood samples run though both the SCD-modeled biochip and the hypoxia biochip. Investigators also assessed contemporaneous in vivo hemoglobin desaturation, and looked for associations with the in vitro biochip findings.
Of the 26 participants, 16 also had either nocturnal or exertional hemoglobin desaturation. In addition, 10 participants had both priapism and desaturations. These data were collected by retrospective chart review and patient survey.
Patients with priapism were a mean age of 34 years, compared with a mean age of 29 years for the other participants, a nonsignificant difference. There were no significant differences in mean hemoglobin or bilirubin levels, or in reticulocyte counts, between the two groups.
However, white blood count, absolute neutrophil count, and lactate dehydrogenase levels were significantly higher for men with priapism (P = .022, .037, and .008, respectively). Ferritin levels were higher as well, at a mean 2,433 (plus or minus 2,234) mcg/L for those with priapism, compared with a mean 269 (plus or minus 3,015) mcg/L for those without priapism (P = .031).
When absolute reticulocyte count was mapped against lactate dehydrogenase levels to create a measure of degree of hemolysis, “individuals with priapism had a more hemolytic lab profile,” said Ms. Quinn (P = .0186).
Though 10 of 14 men with priapism had hemoglobin desaturation, compared with 5 of 12 who did not have priapism, the difference was not statistically significant.
When the researchers compared microchip analysis of RBC adhesion, though, they found marked differences in RBC adhesion in hypoxic versus nonhypoxic conditions. Significantly more RBCs were adherent under hypoxic conditions – in the hypoxic biochip – for the patients with priapism than for patients without priapism (mean, 529 vs. 3,268 adherent cells; P = .016).
Though numbers were small, RBCs from patients with reported priapism and hemoglobin desaturation in vivo showed increased hypoxia enhanced adhesion in vitro (P = .013), Ms. Quinn said. These was no significant difference between adhesion in normoxic and hypoxic conditions for the patients without priapism.
Future directions of work with the biochip include prospective identification of desaturation events and better characterization of nocturnal symptoms, Ms. Quinn said. The investigators also plan to see whether treatment with supplemental oxygen affects RBC adhesion.
The research was supported by the National Institutes of Health, the Doris Duke Charitable Foundation, and the National Science Foundation. Two coauthors have filed an international patent for the biochip technology.
REPORTING FROM FSCDR 2018
Key clinical point: RBC adhesion was increased, but only in hypoxia, for men with sickle cell disease and priapism.
Major finding: Men who had desaturations and priapism had significantly higher RBC adhesion than those without priapism (P = .013).
Study details: An in vitro and in vivo study of 26 men with HbSS sickle cell disease, with and without priapism.
Disclosures: The study was funded by the National Institutes of Health, the Doris Duke Charitable Foundation, and the National Science Foundation. Two coauthors have filed an international patent for the biochip technology.
Nerve growth factor inhibitor shows phase 3 efficacy in osteoarthritis
Two subcutaneous dosages of the nerve growth factor–inhibitor tanezumab showed significant benefits in patients with osteoarthritic joint pain in a multicenter, randomized, phase 3 trial of 698 patients run primarily at U.S. centers.
The 16-week responses to two subcutaneous injections with tanezumab spaced 8 weeks apart showed statistically significant improvements in pain, physical function, and patient global self assessment, compared with placebo, the primary endpoints for the study, Pfizer and Lilly jointly reported. The two companies together are developing tanezumab for an indication for osteoarthritic pain, as well as for chronic lower back pain and pain from cancer metastases.
The company announcement said that patients showed good tolerance to the tanezumab treatments, with no new safety signals and no osteonecrosis seen. About 1% of patients on tanezumab stopped treatment because of an adverse effect, and less than 1.5% of patients on the drug had progressive osteoarthritis during treatment, compared with no patients in the placebo group.
The study enrolled patients at any one of 98 centers in the United States, Puerto Rico, or Canada with confirmed moderate or severe osteoarthritis of the knee or hip that either produced pain refractory to conventional pain medications or involved patients unable to take these medications. The researchers randomized patients to receive two 2.5-mg doses of tanezumab, a 2.5-mg dose followed 8 weeks later by a 5-mg dose, or two placebo doses. The primary outcomes were changes from baseline when measured 16 weeks after the start of treatment in the Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) pain subscale, the WOMAC physical function subscale, and patient’s global assessment of osteoarthritis. Both of the tested tanezumab regimens produced statistically significant improvements in each of the three measures, compared with the placebo control patients, the companies reported.
Tanezumab is a humanized monoclonal antibody that binds to and inhibits nerve growth factor. This inhibition may prevent pain signals from reaching the spinal cord and brain, according to the companies’ report. In June 2017, the two companies announced that development of tanezumab had received “Fast Track” designation from the Food and Drug Administration for the indications of treating chronic osteoarthritic pain and chronic lower back pain.
Two subcutaneous dosages of the nerve growth factor–inhibitor tanezumab showed significant benefits in patients with osteoarthritic joint pain in a multicenter, randomized, phase 3 trial of 698 patients run primarily at U.S. centers.
The 16-week responses to two subcutaneous injections with tanezumab spaced 8 weeks apart showed statistically significant improvements in pain, physical function, and patient global self assessment, compared with placebo, the primary endpoints for the study, Pfizer and Lilly jointly reported. The two companies together are developing tanezumab for an indication for osteoarthritic pain, as well as for chronic lower back pain and pain from cancer metastases.
The company announcement said that patients showed good tolerance to the tanezumab treatments, with no new safety signals and no osteonecrosis seen. About 1% of patients on tanezumab stopped treatment because of an adverse effect, and less than 1.5% of patients on the drug had progressive osteoarthritis during treatment, compared with no patients in the placebo group.
The study enrolled patients at any one of 98 centers in the United States, Puerto Rico, or Canada with confirmed moderate or severe osteoarthritis of the knee or hip that either produced pain refractory to conventional pain medications or involved patients unable to take these medications. The researchers randomized patients to receive two 2.5-mg doses of tanezumab, a 2.5-mg dose followed 8 weeks later by a 5-mg dose, or two placebo doses. The primary outcomes were changes from baseline when measured 16 weeks after the start of treatment in the Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) pain subscale, the WOMAC physical function subscale, and patient’s global assessment of osteoarthritis. Both of the tested tanezumab regimens produced statistically significant improvements in each of the three measures, compared with the placebo control patients, the companies reported.
Tanezumab is a humanized monoclonal antibody that binds to and inhibits nerve growth factor. This inhibition may prevent pain signals from reaching the spinal cord and brain, according to the companies’ report. In June 2017, the two companies announced that development of tanezumab had received “Fast Track” designation from the Food and Drug Administration for the indications of treating chronic osteoarthritic pain and chronic lower back pain.
Two subcutaneous dosages of the nerve growth factor–inhibitor tanezumab showed significant benefits in patients with osteoarthritic joint pain in a multicenter, randomized, phase 3 trial of 698 patients run primarily at U.S. centers.
The 16-week responses to two subcutaneous injections with tanezumab spaced 8 weeks apart showed statistically significant improvements in pain, physical function, and patient global self assessment, compared with placebo, the primary endpoints for the study, Pfizer and Lilly jointly reported. The two companies together are developing tanezumab for an indication for osteoarthritic pain, as well as for chronic lower back pain and pain from cancer metastases.
The company announcement said that patients showed good tolerance to the tanezumab treatments, with no new safety signals and no osteonecrosis seen. About 1% of patients on tanezumab stopped treatment because of an adverse effect, and less than 1.5% of patients on the drug had progressive osteoarthritis during treatment, compared with no patients in the placebo group.
The study enrolled patients at any one of 98 centers in the United States, Puerto Rico, or Canada with confirmed moderate or severe osteoarthritis of the knee or hip that either produced pain refractory to conventional pain medications or involved patients unable to take these medications. The researchers randomized patients to receive two 2.5-mg doses of tanezumab, a 2.5-mg dose followed 8 weeks later by a 5-mg dose, or two placebo doses. The primary outcomes were changes from baseline when measured 16 weeks after the start of treatment in the Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) pain subscale, the WOMAC physical function subscale, and patient’s global assessment of osteoarthritis. Both of the tested tanezumab regimens produced statistically significant improvements in each of the three measures, compared with the placebo control patients, the companies reported.
Tanezumab is a humanized monoclonal antibody that binds to and inhibits nerve growth factor. This inhibition may prevent pain signals from reaching the spinal cord and brain, according to the companies’ report. In June 2017, the two companies announced that development of tanezumab had received “Fast Track” designation from the Food and Drug Administration for the indications of treating chronic osteoarthritic pain and chronic lower back pain.
CARRA continues to lead the North American pediatric rheumatology research community
This year’s annual meeting of the Childhood Arthritis & Rheumatology Research Alliance (CARRA) saw a growing number of participants and new developments in collaborative research projects.

CARRA continues to work in unison with the Arthritis Foundation, which has supported an intramural grant program, a pilot program to support registry sites, and a project to develop a clinically useful electronic dashboard to allow patients and clinicians to coproduce care and make treatment decisions. CARRA and the Arthritis Foundation support an externally-led Food and Drug Administration initiative called the Patient-Focused Drug Development program to bring patient voices into the arena of drug development. The Patient-Centered Outcomes Research Institute (PCORI) is also funding development of a patient-centered learning health system within the Patients, Advocates and Rheumatology Teams Network for Research and Service (PARTNERS).

The meeting featured premeetings for patients and parents, fellows, the CARRA registry, and a research basics course. During the meeting, 81 abstracts based on work supported by CARRA were presented and were published in Pediatric Rheumatology (Pediatr Rheumatol. 2018;16[Suppl 1]:42. doi: 10.1186/s12969-018-0252-y). There were 57 workgroup/committee meetings with parents and patients, providing input on the research agenda. This included workgroups addressing juvenile dermatomyositis, JIA, pain, systemic lupus erythematosus, scleroderma, vasculitis, and autoinflammatory and rare diseases. There were also meetings for small centers, translational research, transition to adult care, and early investigators.
The next meeting will be held in Louisville, Ky., April 10-14, 2019.
Dr. Klein-Gitelman is professor of pediatrics at Northwestern University, Chicago, and is a pediatric rheumatologist at the Ann & Robert H. Lurie Children’s Hospital of Chicago. Dr. Kimura is past president of CARRA and is chief of pediatric rheumatology at the Joseph M. Sanzari Children’s Hospital at Hackensack (N.J.) University Medical Center.
This year’s annual meeting of the Childhood Arthritis & Rheumatology Research Alliance (CARRA) saw a growing number of participants and new developments in collaborative research projects.
CARRA continues to work in unison with the Arthritis Foundation, which has supported an intramural grant program, a pilot program to support registry sites, and a project to develop a clinically useful electronic dashboard to allow patients and clinicians to coproduce care and make treatment decisions. CARRA and the Arthritis Foundation support an externally-led Food and Drug Administration initiative called the Patient-Focused Drug Development program to bring patient voices into the arena of drug development. The Patient-Centered Outcomes Research Institute (PCORI) is also funding development of a patient-centered learning health system within the Patients, Advocates and Rheumatology Teams Network for Research and Service (PARTNERS).
The meeting featured premeetings for patients and parents, fellows, the CARRA registry, and a research basics course. During the meeting, 81 abstracts based on work supported by CARRA were presented and were published in Pediatric Rheumatology (Pediatr Rheumatol. 2018;16[Suppl 1]:42. doi: 10.1186/s12969-018-0252-y). There were 57 workgroup/committee meetings with parents and patients, providing input on the research agenda. This included workgroups addressing juvenile dermatomyositis, JIA, pain, systemic lupus erythematosus, scleroderma, vasculitis, and autoinflammatory and rare diseases. There were also meetings for small centers, translational research, transition to adult care, and early investigators.
The next meeting will be held in Louisville, Ky., April 10-14, 2019.
Dr. Klein-Gitelman is professor of pediatrics at Northwestern University, Chicago, and is a pediatric rheumatologist at the Ann & Robert H. Lurie Children’s Hospital of Chicago. Dr. Kimura is past president of CARRA and is chief of pediatric rheumatology at the Joseph M. Sanzari Children’s Hospital at Hackensack (N.J.) University Medical Center.
This year’s annual meeting of the Childhood Arthritis & Rheumatology Research Alliance (CARRA) saw a growing number of participants and new developments in collaborative research projects.
CARRA continues to work in unison with the Arthritis Foundation, which has supported an intramural grant program, a pilot program to support registry sites, and a project to develop a clinically useful electronic dashboard to allow patients and clinicians to coproduce care and make treatment decisions. CARRA and the Arthritis Foundation support an externally-led Food and Drug Administration initiative called the Patient-Focused Drug Development program to bring patient voices into the arena of drug development. The Patient-Centered Outcomes Research Institute (PCORI) is also funding development of a patient-centered learning health system within the Patients, Advocates and Rheumatology Teams Network for Research and Service (PARTNERS).
The meeting featured premeetings for patients and parents, fellows, the CARRA registry, and a research basics course. During the meeting, 81 abstracts based on work supported by CARRA were presented and were published in Pediatric Rheumatology (Pediatr Rheumatol. 2018;16[Suppl 1]:42. doi: 10.1186/s12969-018-0252-y). There were 57 workgroup/committee meetings with parents and patients, providing input on the research agenda. This included workgroups addressing juvenile dermatomyositis, JIA, pain, systemic lupus erythematosus, scleroderma, vasculitis, and autoinflammatory and rare diseases. There were also meetings for small centers, translational research, transition to adult care, and early investigators.
The next meeting will be held in Louisville, Ky., April 10-14, 2019.
Dr. Klein-Gitelman is professor of pediatrics at Northwestern University, Chicago, and is a pediatric rheumatologist at the Ann & Robert H. Lurie Children’s Hospital of Chicago. Dr. Kimura is past president of CARRA and is chief of pediatric rheumatology at the Joseph M. Sanzari Children’s Hospital at Hackensack (N.J.) University Medical Center.

FDA announces plan for biosimilar innovation and competition
Some of the actions include tools to enhance public information about the FDA’s evaluation of biosimilars, including more information about approved biological products in the Purple Book; exploring the potential for entering into new data sharing agreements with foreign regulators to facilitate the increased use of non–U.S.-licensed comparator products in certain studies to support a biosimilar application; releasing a series of videos that explain key concepts about biosimilar and interchangeable products; and requesting information from the public on additional policy steps the FDA should consider for enhancing the biosimilar program.
The FDA’s Biosimilar Action Plan is available here.
Some of the actions include tools to enhance public information about the FDA’s evaluation of biosimilars, including more information about approved biological products in the Purple Book; exploring the potential for entering into new data sharing agreements with foreign regulators to facilitate the increased use of non–U.S.-licensed comparator products in certain studies to support a biosimilar application; releasing a series of videos that explain key concepts about biosimilar and interchangeable products; and requesting information from the public on additional policy steps the FDA should consider for enhancing the biosimilar program.
The FDA’s Biosimilar Action Plan is available here.
Some of the actions include tools to enhance public information about the FDA’s evaluation of biosimilars, including more information about approved biological products in the Purple Book; exploring the potential for entering into new data sharing agreements with foreign regulators to facilitate the increased use of non–U.S.-licensed comparator products in certain studies to support a biosimilar application; releasing a series of videos that explain key concepts about biosimilar and interchangeable products; and requesting information from the public on additional policy steps the FDA should consider for enhancing the biosimilar program.
The FDA’s Biosimilar Action Plan is available here.
Deaths from liver disease surged in U.S. since 1999
Cirrhosis mortality showed a sharp rise beginning in 2009, with a 3.6% annual increase driven entirely by a surge in alcoholic cirrhosis among young people aged 25-34 years, Elliot B. Tapper, MD, and Neehar D. Parikh, MD, reported in the BMJ. The uptick in hepatocellular carcinoma, however, was gradual and consistent, with a 2% annual increase felt mostly in older people, wrote Dr. Tapper and Dr. Parikh, both at the University of Michigan, Ann Arbor.
“The increasing mortality due to cirrhosis and hepatocellular carcinoma speaks to the expanding socioeconomic impact of liver disease,” the colleagues wrote. “Adverse trends in liver-related mortality are particularly unfortunate given that in most cases the liver disease is preventable. Understanding the factors associated with mortality due to these conditions will inform how best to allocate resources.”
The study extracted its data from the Vital Statistic Cooperative and the Centers for Disease Control and Prevention. The investigators not only examined raw mortality numbers, but analyzed them for demographic and geographic trends, in analyses that controlled for age.
Cirrhosis
During the study period, 460,760 patients died from cirrhosis (20,661 in 1999 and 34,174 in 2016, an increase of 65.4%).
Men were twice as likely to die from cirrhosis. Young people aged 25-34 years had the highest rate of increase (3.7% over the entire period and 10.5% from 2009 to 2016). This was directly driven by parallel increases in both alcohol use disorder and alcohol-related liver diseases, which increased by about 16% and 10%, respectively, in this group.
Native Americans had the highest mortality rate (25.8 per 100,000) followed by whites (12.7 per 100,000). “Notably, by 2016, cirrhosis accounted for 6.3% [up from 4.3% in 2009] and 7% [up from 5.8% in 2009] of deaths for Native Americans aged 25-34 and 35 or more, respectively,” and 2.3% of all deaths among adults aged 25-34 years, the authors wrote.
The increases were largely felt in the southern and western states (about 13 per 100,000 in each region). The greatest increases occurred in Kentucky (6.8%), New Mexico (6%), Arkansas (5.7%), Indiana (5%), and Alabama (5%). There was a statistically significant 1.2% decrease in deaths from cirrhosis in Maryland.
Hepatocellular carcinoma
Hepatocellular carcinoma accounted for 136,442 deaths during the study period (5,112 in 1999 and 11,073 in 2016 – an increase of 116.6%). This represented an average annual increase of 2%.
Men were four times more likely to die from hepatocellular carcinoma. The increase manifested mostly in older people, decreasing in those younger than 55 years. Mortality was highest among Asians and Pacific Islanders (6 per 100,000), followed by blacks (4.94 per 100,000).
The increases were largely felt in western states, with an overall increase of 4.2 per 100,000.
“Many of the same states with worsening cirrhosis-related mortality also experienced worsening mortality from hepatocellular carcinoma, including Oregon and Iowa,” the authors wrote. But mortality from the disease also increased significantly in Arizona (5.1%), Kansas (4.3%), Kentucky (4%), and Washington (3.9%).
“Potential explanations supported by these data include increasing early detection of hepatocellular carcinoma, application of curative or locoregional therapies, and, because hepatitis B is the principal cause of hepatocellular carcinoma worldwide and among Asian Americans, effectiveness of vaccination programs and the efficacy of antiviral therapy for hepatitis B in preventing the development of hepatocellular carcinoma.”
However, they noted, “it is unclear how these trends are, or will be, affected by direct-acting antivirals for hepatitis C virus ... eradication of hepatitis C virus will prevent the development of cirrhosis and its complications, potentially changing these trends in the next 5-10 years. However, therapy for hepatitis C viral infection cannot modify the statistically significant trends observed related to alcohol or the expected increase in the burden of nonalcoholic fatty liver disease.”
Neither author had any financial disclosure relevant to the work.
SOURCE: Tapper EB et al. BMJ 2018;362:k2817.
Cirrhosis mortality showed a sharp rise beginning in 2009, with a 3.6% annual increase driven entirely by a surge in alcoholic cirrhosis among young people aged 25-34 years, Elliot B. Tapper, MD, and Neehar D. Parikh, MD, reported in the BMJ. The uptick in hepatocellular carcinoma, however, was gradual and consistent, with a 2% annual increase felt mostly in older people, wrote Dr. Tapper and Dr. Parikh, both at the University of Michigan, Ann Arbor.
“The increasing mortality due to cirrhosis and hepatocellular carcinoma speaks to the expanding socioeconomic impact of liver disease,” the colleagues wrote. “Adverse trends in liver-related mortality are particularly unfortunate given that in most cases the liver disease is preventable. Understanding the factors associated with mortality due to these conditions will inform how best to allocate resources.”
The study extracted its data from the Vital Statistic Cooperative and the Centers for Disease Control and Prevention. The investigators not only examined raw mortality numbers, but analyzed them for demographic and geographic trends, in analyses that controlled for age.
Cirrhosis
During the study period, 460,760 patients died from cirrhosis (20,661 in 1999 and 34,174 in 2016, an increase of 65.4%).
Men were twice as likely to die from cirrhosis. Young people aged 25-34 years had the highest rate of increase (3.7% over the entire period and 10.5% from 2009 to 2016). This was directly driven by parallel increases in both alcohol use disorder and alcohol-related liver diseases, which increased by about 16% and 10%, respectively, in this group.
Native Americans had the highest mortality rate (25.8 per 100,000) followed by whites (12.7 per 100,000). “Notably, by 2016, cirrhosis accounted for 6.3% [up from 4.3% in 2009] and 7% [up from 5.8% in 2009] of deaths for Native Americans aged 25-34 and 35 or more, respectively,” and 2.3% of all deaths among adults aged 25-34 years, the authors wrote.
The increases were largely felt in the southern and western states (about 13 per 100,000 in each region). The greatest increases occurred in Kentucky (6.8%), New Mexico (6%), Arkansas (5.7%), Indiana (5%), and Alabama (5%). There was a statistically significant 1.2% decrease in deaths from cirrhosis in Maryland.
Hepatocellular carcinoma
Hepatocellular carcinoma accounted for 136,442 deaths during the study period (5,112 in 1999 and 11,073 in 2016 – an increase of 116.6%). This represented an average annual increase of 2%.
Men were four times more likely to die from hepatocellular carcinoma. The increase manifested mostly in older people, decreasing in those younger than 55 years. Mortality was highest among Asians and Pacific Islanders (6 per 100,000), followed by blacks (4.94 per 100,000).
The increases were largely felt in western states, with an overall increase of 4.2 per 100,000.
“Many of the same states with worsening cirrhosis-related mortality also experienced worsening mortality from hepatocellular carcinoma, including Oregon and Iowa,” the authors wrote. But mortality from the disease also increased significantly in Arizona (5.1%), Kansas (4.3%), Kentucky (4%), and Washington (3.9%).
“Potential explanations supported by these data include increasing early detection of hepatocellular carcinoma, application of curative or locoregional therapies, and, because hepatitis B is the principal cause of hepatocellular carcinoma worldwide and among Asian Americans, effectiveness of vaccination programs and the efficacy of antiviral therapy for hepatitis B in preventing the development of hepatocellular carcinoma.”
However, they noted, “it is unclear how these trends are, or will be, affected by direct-acting antivirals for hepatitis C virus ... eradication of hepatitis C virus will prevent the development of cirrhosis and its complications, potentially changing these trends in the next 5-10 years. However, therapy for hepatitis C viral infection cannot modify the statistically significant trends observed related to alcohol or the expected increase in the burden of nonalcoholic fatty liver disease.”
Neither author had any financial disclosure relevant to the work.
SOURCE: Tapper EB et al. BMJ 2018;362:k2817.
Cirrhosis mortality showed a sharp rise beginning in 2009, with a 3.6% annual increase driven entirely by a surge in alcoholic cirrhosis among young people aged 25-34 years, Elliot B. Tapper, MD, and Neehar D. Parikh, MD, reported in the BMJ. The uptick in hepatocellular carcinoma, however, was gradual and consistent, with a 2% annual increase felt mostly in older people, wrote Dr. Tapper and Dr. Parikh, both at the University of Michigan, Ann Arbor.
“The increasing mortality due to cirrhosis and hepatocellular carcinoma speaks to the expanding socioeconomic impact of liver disease,” the colleagues wrote. “Adverse trends in liver-related mortality are particularly unfortunate given that in most cases the liver disease is preventable. Understanding the factors associated with mortality due to these conditions will inform how best to allocate resources.”
The study extracted its data from the Vital Statistic Cooperative and the Centers for Disease Control and Prevention. The investigators not only examined raw mortality numbers, but analyzed them for demographic and geographic trends, in analyses that controlled for age.
Cirrhosis
During the study period, 460,760 patients died from cirrhosis (20,661 in 1999 and 34,174 in 2016, an increase of 65.4%).
Men were twice as likely to die from cirrhosis. Young people aged 25-34 years had the highest rate of increase (3.7% over the entire period and 10.5% from 2009 to 2016). This was directly driven by parallel increases in both alcohol use disorder and alcohol-related liver diseases, which increased by about 16% and 10%, respectively, in this group.
Native Americans had the highest mortality rate (25.8 per 100,000) followed by whites (12.7 per 100,000). “Notably, by 2016, cirrhosis accounted for 6.3% [up from 4.3% in 2009] and 7% [up from 5.8% in 2009] of deaths for Native Americans aged 25-34 and 35 or more, respectively,” and 2.3% of all deaths among adults aged 25-34 years, the authors wrote.
The increases were largely felt in the southern and western states (about 13 per 100,000 in each region). The greatest increases occurred in Kentucky (6.8%), New Mexico (6%), Arkansas (5.7%), Indiana (5%), and Alabama (5%). There was a statistically significant 1.2% decrease in deaths from cirrhosis in Maryland.
Hepatocellular carcinoma
Hepatocellular carcinoma accounted for 136,442 deaths during the study period (5,112 in 1999 and 11,073 in 2016 – an increase of 116.6%). This represented an average annual increase of 2%.
Men were four times more likely to die from hepatocellular carcinoma. The increase manifested mostly in older people, decreasing in those younger than 55 years. Mortality was highest among Asians and Pacific Islanders (6 per 100,000), followed by blacks (4.94 per 100,000).
The increases were largely felt in western states, with an overall increase of 4.2 per 100,000.
“Many of the same states with worsening cirrhosis-related mortality also experienced worsening mortality from hepatocellular carcinoma, including Oregon and Iowa,” the authors wrote. But mortality from the disease also increased significantly in Arizona (5.1%), Kansas (4.3%), Kentucky (4%), and Washington (3.9%).
“Potential explanations supported by these data include increasing early detection of hepatocellular carcinoma, application of curative or locoregional therapies, and, because hepatitis B is the principal cause of hepatocellular carcinoma worldwide and among Asian Americans, effectiveness of vaccination programs and the efficacy of antiviral therapy for hepatitis B in preventing the development of hepatocellular carcinoma.”
However, they noted, “it is unclear how these trends are, or will be, affected by direct-acting antivirals for hepatitis C virus ... eradication of hepatitis C virus will prevent the development of cirrhosis and its complications, potentially changing these trends in the next 5-10 years. However, therapy for hepatitis C viral infection cannot modify the statistically significant trends observed related to alcohol or the expected increase in the burden of nonalcoholic fatty liver disease.”
Neither author had any financial disclosure relevant to the work.
SOURCE: Tapper EB et al. BMJ 2018;362:k2817.
FROM BMJ
Key clinical point: Deaths from cirrhosis and hepatocellular carcinoma have surged in the United States since 1999.
Major finding: Liver cirrhosis mortality increased by 65% and hepatocellular carcinoma mortality by about 117%.
Study details: The study extracted data from the National Vital Statistics database and the CDC.
Disclosures: Neither author had relevant financial disclosures.
Source: Tapper EB et al. BMJ 2018;362:k2817.
Epinephrine for cardiac arrest: Better survival, more brain damage
Using epinephrine for cardiac arrest improves 30-day survival by less than 1%, and nearly doubles the risk of severe brain damage among survivors, according to PARAMEDIC2, a randomized, double-blind trial in more than 8,000 patients in Great Britain.
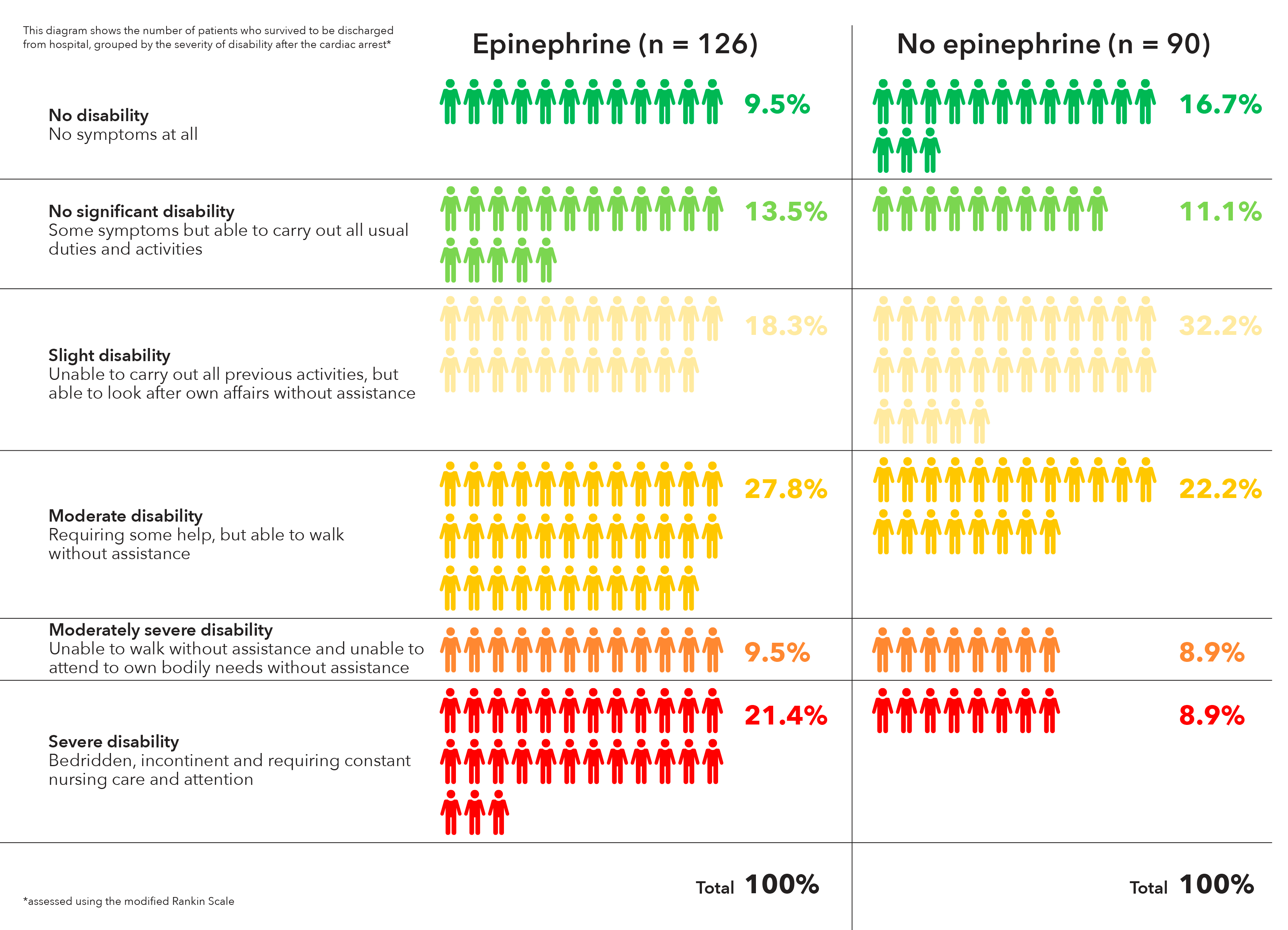
It’s clear what patients want. “Our own work with patients and the public before starting the trial identified survival without brain damage [as] more important to patients than survival alone. The findings of this trial will require careful consideration by the wider community and those responsible for clinical practice guidelines for cardiac arrest,” lead investigator Gavin D. Perkins, MD, professor of critical care medicine at the University of Warwick, Coventry, England, and lead author of the study published in the New England Journal of Medicine, wrote in a statement.
In PARAMEDIC2, after initial attempts with CPR and defibrillation failed, 4,012 patients were given epinephrine 1 mg by intravenous or intraosseous infusion every 3-5 minutes for a maximum of 10 doses, and 3,995 were given a saline placebo in the same fashion. The median time from emergency call to ambulance arrival was just over 6 minutes in both groups, with a further 14 minutes until drug administration.
The heart restarted in a higher proportion of epinephrine patients (36.3% vs. 11.7%), and 3.2% of epinephrine patients were alive at 30 days, versus 2.4% in the placebo arm, a 39% increase.
However, that slight benefit came at a significant cost. Of the 126 epinephrine patients who survived to hospital discharge, 39 (31%) had severe brain damage, compared with 16 (17.8%) among the 90 placebo survivors. Severe brain damage meant inability to walk and tend to bodily functions, or a persistent vegetative state (modified Rankin scale grade 4 or 5).
The trial addresses a long-standing question in resuscitation medicine, the role of epinephrine in cardiac arrest. It’s a devil’s bargain: Epinephrine increases blood flow to the heart, so helps with resuscitation, but it also reduces blood flow in the brain’s microvasculature, increasing the risk of brain damage.
“The benefit of epinephrine on survival demonstrated in this trial should be considered in comparison with other treatments in the chain of survival.” Early cardiac arrest recognition saves 1 in every 11 patients, bystander CPR saves 1 in every 15, and early defibrillation saves 1 in 5, the investigators noted.
The trial did not collect data on prearrest neurologic status, but the number of subjects with impaired function was probably very small and balanced between the groups, according to the report.
On average, patients were aged just under 70 years, 65% were men, and bystander CPR was performed in about 60% in both groups. They were enrolled by five ambulance services in England and Wales. Informed consent was obtained, when possible, after resuscitation.
The trial was funded by the U.K. National Institute for Health Research. The researchers had no relevant disclosures to report.
SOURCE: Perkins GD et al. N Engl J Med. 2018 Jul 18. doi:10.1056/NEJMoa1806842.
Epinephrine has been used in resuscitation efforts since the 1960s, yet no reliable evidence on the practice has been collected. Now, PARAMEDIC2 provides the most rigorous data on patient-centered outcomes with respect to epinephrine to date.
Epinephrine increased 30-day survival in patients with nonshockable rhythms by more than 100%, but the benefit was less clear in those with shockable rhythms. Shockable rhythms are more likely to occur in patients with cardiac or cardiovascular causes of arrest, which epinephrine may exacerbate. The results underscore the principle that drug administration should not compete with or delay defibrillation, and that epinephrine may have different effects in patients with different ECG rhythms.
The PARAMEDIC2 results leave us with several questions: Could other, additional treatments after a return of spontaneous circulation improve functional recovery, should drug use differ on the basis of cardiac rhythm, and would lower doses of epinephrine be superior to higher doses among patients with out-of-hospital cardiac arrest?
Clifton W. Callaway, MD, PhD, of the University of Pittsburgh, and Michael W. Donnino, MD, of Beth Israel Deaconess Medical Center, Boston, made these comments in an accompanying editorial (N Engl J Med. 2018 Jul 18. doi: 10.1056/NEJMe1808255). They had no relevant disclosures.
Epinephrine has been used in resuscitation efforts since the 1960s, yet no reliable evidence on the practice has been collected. Now, PARAMEDIC2 provides the most rigorous data on patient-centered outcomes with respect to epinephrine to date.
Epinephrine increased 30-day survival in patients with nonshockable rhythms by more than 100%, but the benefit was less clear in those with shockable rhythms. Shockable rhythms are more likely to occur in patients with cardiac or cardiovascular causes of arrest, which epinephrine may exacerbate. The results underscore the principle that drug administration should not compete with or delay defibrillation, and that epinephrine may have different effects in patients with different ECG rhythms.
The PARAMEDIC2 results leave us with several questions: Could other, additional treatments after a return of spontaneous circulation improve functional recovery, should drug use differ on the basis of cardiac rhythm, and would lower doses of epinephrine be superior to higher doses among patients with out-of-hospital cardiac arrest?
Clifton W. Callaway, MD, PhD, of the University of Pittsburgh, and Michael W. Donnino, MD, of Beth Israel Deaconess Medical Center, Boston, made these comments in an accompanying editorial (N Engl J Med. 2018 Jul 18. doi: 10.1056/NEJMe1808255). They had no relevant disclosures.
Epinephrine has been used in resuscitation efforts since the 1960s, yet no reliable evidence on the practice has been collected. Now, PARAMEDIC2 provides the most rigorous data on patient-centered outcomes with respect to epinephrine to date.
Epinephrine increased 30-day survival in patients with nonshockable rhythms by more than 100%, but the benefit was less clear in those with shockable rhythms. Shockable rhythms are more likely to occur in patients with cardiac or cardiovascular causes of arrest, which epinephrine may exacerbate. The results underscore the principle that drug administration should not compete with or delay defibrillation, and that epinephrine may have different effects in patients with different ECG rhythms.
The PARAMEDIC2 results leave us with several questions: Could other, additional treatments after a return of spontaneous circulation improve functional recovery, should drug use differ on the basis of cardiac rhythm, and would lower doses of epinephrine be superior to higher doses among patients with out-of-hospital cardiac arrest?
Clifton W. Callaway, MD, PhD, of the University of Pittsburgh, and Michael W. Donnino, MD, of Beth Israel Deaconess Medical Center, Boston, made these comments in an accompanying editorial (N Engl J Med. 2018 Jul 18. doi: 10.1056/NEJMe1808255). They had no relevant disclosures.
Using epinephrine for cardiac arrest improves 30-day survival by less than 1%, and nearly doubles the risk of severe brain damage among survivors, according to PARAMEDIC2, a randomized, double-blind trial in more than 8,000 patients in Great Britain.
It’s clear what patients want. “Our own work with patients and the public before starting the trial identified survival without brain damage [as] more important to patients than survival alone. The findings of this trial will require careful consideration by the wider community and those responsible for clinical practice guidelines for cardiac arrest,” lead investigator Gavin D. Perkins, MD, professor of critical care medicine at the University of Warwick, Coventry, England, and lead author of the study published in the New England Journal of Medicine, wrote in a statement.
In PARAMEDIC2, after initial attempts with CPR and defibrillation failed, 4,012 patients were given epinephrine 1 mg by intravenous or intraosseous infusion every 3-5 minutes for a maximum of 10 doses, and 3,995 were given a saline placebo in the same fashion. The median time from emergency call to ambulance arrival was just over 6 minutes in both groups, with a further 14 minutes until drug administration.
The heart restarted in a higher proportion of epinephrine patients (36.3% vs. 11.7%), and 3.2% of epinephrine patients were alive at 30 days, versus 2.4% in the placebo arm, a 39% increase.
However, that slight benefit came at a significant cost. Of the 126 epinephrine patients who survived to hospital discharge, 39 (31%) had severe brain damage, compared with 16 (17.8%) among the 90 placebo survivors. Severe brain damage meant inability to walk and tend to bodily functions, or a persistent vegetative state (modified Rankin scale grade 4 or 5).
The trial addresses a long-standing question in resuscitation medicine, the role of epinephrine in cardiac arrest. It’s a devil’s bargain: Epinephrine increases blood flow to the heart, so helps with resuscitation, but it also reduces blood flow in the brain’s microvasculature, increasing the risk of brain damage.
“The benefit of epinephrine on survival demonstrated in this trial should be considered in comparison with other treatments in the chain of survival.” Early cardiac arrest recognition saves 1 in every 11 patients, bystander CPR saves 1 in every 15, and early defibrillation saves 1 in 5, the investigators noted.
The trial did not collect data on prearrest neurologic status, but the number of subjects with impaired function was probably very small and balanced between the groups, according to the report.
On average, patients were aged just under 70 years, 65% were men, and bystander CPR was performed in about 60% in both groups. They were enrolled by five ambulance services in England and Wales. Informed consent was obtained, when possible, after resuscitation.
The trial was funded by the U.K. National Institute for Health Research. The researchers had no relevant disclosures to report.
SOURCE: Perkins GD et al. N Engl J Med. 2018 Jul 18. doi:10.1056/NEJMoa1806842.
Using epinephrine for cardiac arrest improves 30-day survival by less than 1%, and nearly doubles the risk of severe brain damage among survivors, according to PARAMEDIC2, a randomized, double-blind trial in more than 8,000 patients in Great Britain.
It’s clear what patients want. “Our own work with patients and the public before starting the trial identified survival without brain damage [as] more important to patients than survival alone. The findings of this trial will require careful consideration by the wider community and those responsible for clinical practice guidelines for cardiac arrest,” lead investigator Gavin D. Perkins, MD, professor of critical care medicine at the University of Warwick, Coventry, England, and lead author of the study published in the New England Journal of Medicine, wrote in a statement.
In PARAMEDIC2, after initial attempts with CPR and defibrillation failed, 4,012 patients were given epinephrine 1 mg by intravenous or intraosseous infusion every 3-5 minutes for a maximum of 10 doses, and 3,995 were given a saline placebo in the same fashion. The median time from emergency call to ambulance arrival was just over 6 minutes in both groups, with a further 14 minutes until drug administration.
The heart restarted in a higher proportion of epinephrine patients (36.3% vs. 11.7%), and 3.2% of epinephrine patients were alive at 30 days, versus 2.4% in the placebo arm, a 39% increase.
However, that slight benefit came at a significant cost. Of the 126 epinephrine patients who survived to hospital discharge, 39 (31%) had severe brain damage, compared with 16 (17.8%) among the 90 placebo survivors. Severe brain damage meant inability to walk and tend to bodily functions, or a persistent vegetative state (modified Rankin scale grade 4 or 5).
The trial addresses a long-standing question in resuscitation medicine, the role of epinephrine in cardiac arrest. It’s a devil’s bargain: Epinephrine increases blood flow to the heart, so helps with resuscitation, but it also reduces blood flow in the brain’s microvasculature, increasing the risk of brain damage.
“The benefit of epinephrine on survival demonstrated in this trial should be considered in comparison with other treatments in the chain of survival.” Early cardiac arrest recognition saves 1 in every 11 patients, bystander CPR saves 1 in every 15, and early defibrillation saves 1 in 5, the investigators noted.
The trial did not collect data on prearrest neurologic status, but the number of subjects with impaired function was probably very small and balanced between the groups, according to the report.
On average, patients were aged just under 70 years, 65% were men, and bystander CPR was performed in about 60% in both groups. They were enrolled by five ambulance services in England and Wales. Informed consent was obtained, when possible, after resuscitation.
The trial was funded by the U.K. National Institute for Health Research. The researchers had no relevant disclosures to report.
SOURCE: Perkins GD et al. N Engl J Med. 2018 Jul 18. doi:10.1056/NEJMoa1806842.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point:
Major finding: Of the 128 epinephrine patients who survived to hospital discharge, 39 (30.1%) had severe brain damage, compared with 16 (18.7%) among the 91 placebo survivors.
Study details: A randomized, double-blind trial of over 8,000 U.K. patients experiencing an out-of-hospital cardiac arrest.
Disclosures: The trial was funded by the U.K. National Institute for Health Research. The researchers had no relevant disclosures to report.
Source: Perkins GD et al. N Engl J Med. 2018 Jul 18. doi:10.1056/NEJMoa1806842.