User login
Less is more: Nanotechnology enhances antifungal’s efficacy
The use of nanotechnology significantly reduced the amount of efinaconazole needed to effectively treat nail fungus in a study that pitted nitric oxide–releasing nanoparticles combined with the antifungal against reference strains of Trichophyton rubrum.
Efinaconazole has demonstrated effectiveness as a topical treatment for T. rubrum, but treatment can be expensive, with a single 4-mL bottle costing $691 at a major chain pharmacy, wrote Caroline B. Costa-Orlandi, PhD, of Universidade Estadual Paulista, Sao Paulo, Brazil, and her colleagues.
In a study published in the Journal of Drugs in Dermatology, an international research team evaluated topical efinaconazole and topical terbinafine, each combined with previously characterized, nitric oxide–releasing nanoparticles (NO-np) in a checkerboard design, to attack two reference strains of T. rubrum, ATCC MYA-4438 and ATCC 28189. NO-np was combined with 10% efinaconazole or with terbinafine.
The combination of NO-np and efinaconazole reduced the minimum inhibitory concentration (MIC) of efinaconazole by 16 times compared with treatment alone against ATCC MYA-4438; by 4 times when combined against ATCC 28189. With NO-np plus terbinafine, MICs against ATCC 28189 and ATCC MYA-4438 were reduced by four- and twofold, respectively, when compared with terbinafine alone. These data follow recently published findings in a study cited by the authors that demonstrated that NO-np is superior to topical terbinafine 1% cream in clearing infection in a mouse model of deep dermal dermatophytosis, suggesting that the combination may be even more effective (Nanomedicine. 2017 Oct;13[7]:2267-70).
“What we found was that we could impart the same antifungal activity at the highest concentrations tested of either alone by combining them at a fraction of these concentrations,” corresponding author Adam Friedman, MD, professor of dermatology, George Washington University, Washington, said in a press release issued by the university. The impact of this combination, “which we visualized using electron microscopy as compared to either product alone, highlighted their synergistic damaging effects at concentrations that would be completely safe to human cells,” he added.
Other benefits of NO-np include low cost, safety, ease of use, reduced likelihood for the development of antimicrobial resistance, and proven efficacy against other dermatophyte infections, the researchers noted.
The findings support the potential value of further research to evaluate nanoparticles combined with topical antifungals in a clinical setting, they said.
Dr. Costa-Orlandi had no financial conflicts to disclose. Authors Adam Friedman, MD, and Joel Friedman, MD, are coinventors of the nitric oxide–releasing nanoparticles used in the study. Dr. Adam Friedman is on the advisory board of Dermatology News.
SOURCE: Costa-Orlandi C et al. J Drugs Dermatol. 2018;17(7):717-20.
The use of nanotechnology significantly reduced the amount of efinaconazole needed to effectively treat nail fungus in a study that pitted nitric oxide–releasing nanoparticles combined with the antifungal against reference strains of Trichophyton rubrum.
Efinaconazole has demonstrated effectiveness as a topical treatment for T. rubrum, but treatment can be expensive, with a single 4-mL bottle costing $691 at a major chain pharmacy, wrote Caroline B. Costa-Orlandi, PhD, of Universidade Estadual Paulista, Sao Paulo, Brazil, and her colleagues.
In a study published in the Journal of Drugs in Dermatology, an international research team evaluated topical efinaconazole and topical terbinafine, each combined with previously characterized, nitric oxide–releasing nanoparticles (NO-np) in a checkerboard design, to attack two reference strains of T. rubrum, ATCC MYA-4438 and ATCC 28189. NO-np was combined with 10% efinaconazole or with terbinafine.
The combination of NO-np and efinaconazole reduced the minimum inhibitory concentration (MIC) of efinaconazole by 16 times compared with treatment alone against ATCC MYA-4438; by 4 times when combined against ATCC 28189. With NO-np plus terbinafine, MICs against ATCC 28189 and ATCC MYA-4438 were reduced by four- and twofold, respectively, when compared with terbinafine alone. These data follow recently published findings in a study cited by the authors that demonstrated that NO-np is superior to topical terbinafine 1% cream in clearing infection in a mouse model of deep dermal dermatophytosis, suggesting that the combination may be even more effective (Nanomedicine. 2017 Oct;13[7]:2267-70).
“What we found was that we could impart the same antifungal activity at the highest concentrations tested of either alone by combining them at a fraction of these concentrations,” corresponding author Adam Friedman, MD, professor of dermatology, George Washington University, Washington, said in a press release issued by the university. The impact of this combination, “which we visualized using electron microscopy as compared to either product alone, highlighted their synergistic damaging effects at concentrations that would be completely safe to human cells,” he added.
Other benefits of NO-np include low cost, safety, ease of use, reduced likelihood for the development of antimicrobial resistance, and proven efficacy against other dermatophyte infections, the researchers noted.
The findings support the potential value of further research to evaluate nanoparticles combined with topical antifungals in a clinical setting, they said.
Dr. Costa-Orlandi had no financial conflicts to disclose. Authors Adam Friedman, MD, and Joel Friedman, MD, are coinventors of the nitric oxide–releasing nanoparticles used in the study. Dr. Adam Friedman is on the advisory board of Dermatology News.
SOURCE: Costa-Orlandi C et al. J Drugs Dermatol. 2018;17(7):717-20.
The use of nanotechnology significantly reduced the amount of efinaconazole needed to effectively treat nail fungus in a study that pitted nitric oxide–releasing nanoparticles combined with the antifungal against reference strains of Trichophyton rubrum.
Efinaconazole has demonstrated effectiveness as a topical treatment for T. rubrum, but treatment can be expensive, with a single 4-mL bottle costing $691 at a major chain pharmacy, wrote Caroline B. Costa-Orlandi, PhD, of Universidade Estadual Paulista, Sao Paulo, Brazil, and her colleagues.
In a study published in the Journal of Drugs in Dermatology, an international research team evaluated topical efinaconazole and topical terbinafine, each combined with previously characterized, nitric oxide–releasing nanoparticles (NO-np) in a checkerboard design, to attack two reference strains of T. rubrum, ATCC MYA-4438 and ATCC 28189. NO-np was combined with 10% efinaconazole or with terbinafine.
The combination of NO-np and efinaconazole reduced the minimum inhibitory concentration (MIC) of efinaconazole by 16 times compared with treatment alone against ATCC MYA-4438; by 4 times when combined against ATCC 28189. With NO-np plus terbinafine, MICs against ATCC 28189 and ATCC MYA-4438 were reduced by four- and twofold, respectively, when compared with terbinafine alone. These data follow recently published findings in a study cited by the authors that demonstrated that NO-np is superior to topical terbinafine 1% cream in clearing infection in a mouse model of deep dermal dermatophytosis, suggesting that the combination may be even more effective (Nanomedicine. 2017 Oct;13[7]:2267-70).
“What we found was that we could impart the same antifungal activity at the highest concentrations tested of either alone by combining them at a fraction of these concentrations,” corresponding author Adam Friedman, MD, professor of dermatology, George Washington University, Washington, said in a press release issued by the university. The impact of this combination, “which we visualized using electron microscopy as compared to either product alone, highlighted their synergistic damaging effects at concentrations that would be completely safe to human cells,” he added.
Other benefits of NO-np include low cost, safety, ease of use, reduced likelihood for the development of antimicrobial resistance, and proven efficacy against other dermatophyte infections, the researchers noted.
The findings support the potential value of further research to evaluate nanoparticles combined with topical antifungals in a clinical setting, they said.
Dr. Costa-Orlandi had no financial conflicts to disclose. Authors Adam Friedman, MD, and Joel Friedman, MD, are coinventors of the nitric oxide–releasing nanoparticles used in the study. Dr. Adam Friedman is on the advisory board of Dermatology News.
SOURCE: Costa-Orlandi C et al. J Drugs Dermatol. 2018;17(7):717-20.
FROM JOURNAL OF DRUGS IN DERMATOLOGY
Key clinical point: Adding nanoparticles to antifungal medication improved the drug’s effectiveness and reduced the amount needed.
Major finding: Efinaconazole combined with nitric oxide–releasing nanoparticles reduced the antifungal’s minimum inhibitory concentration 16-fold, compared with the antifungal alone against T. rubrum reference strains.
Study details: The data come from an in vitro analysis of nanoparticle-enhanced efinaconazole or terbinafine against T. rubrum.
Disclosures: Dr. Costa-Orlandi had no financial conflicts to disclose. Coauthors Dr. Adam Friedman and Dr. Joel Friedman are coinventors of the nitric oxide–releasing nanoparticles used in the study.
Source: Costa-Orlandi C et al. J Drugs Dermatol. 2018;17(7):717-20.
Real-time microarrays can simultaneously detect HCV and HIV-1, -2 infections
The use of TaqMan Array Card (TAC) microarrays has been extended to permit simultaneous detection of HIV-1, HIV-2, and five hepatitis viruses from a small amount of extracted nucleic acid, according to a study by Timothy C. Granade, MD, and his colleagues at the Centers for Disease Control and Prevention, Atlanta.
This is particularly important for dealing with HIV-infected individuals, because HIV-1 and HIV-2 require different treatment interventions, and approximately one-third of HIV-infected patients have been found to be coinfected with hepatitis C or hepatitis B, according to the study report, published in the Journal of Virological Methods (J Virol Methods. 2018 Sep;259:60-5).
HIV-1-positive plasma samples from a variety of subtypes as well as whole blood specimens were confirmed for HIV-1-infection serologically or by nucleic amplification methods. HIV-2 whole blood and plasma specimens were also obtained.
TAC cards contained one positive control, one negative control, three HIV-1 replicates, and two HIV-2 replicates. In addition, the five common hepatitis viruses (A-E) were each replicated three times on each card. The cards were used to test the RNA isolates obtained from the various samples.
Ninety-five of the 104 known HIV-1-positive specimens were assayed positive using TAC; 23 of 26 HIV-2-seeded specimens were detectable using TAC and no cross-reactivity was seen between HIV-1-positive and HIV-2-positive specimens.
Eighteen of the HIV-1-positive specimens were also reactive in triplicate for HCV; three of the HIV-1-positive specimens were reactive to HBV and one specimen was reactive to HIV-1, HBV, and HCV.
“The TAC assay could be invaluable in large-scale screening environments or in surveying local outbreaks such as the recent HIV cluster found in Indiana. Many of these individuals were later determined to be infected with hepatitis C. The use of TAC could shorten the time to identifying and confirming such cases and permit the detection of multiple blood-borne infections in a single test. Application of TAC technology to general population surveillance could identify problem areas for both HIV prevention and intervention efforts in a variety of global environs,” the researchers concluded.
The authors were employed by the Centers for Disease Control and Prevention, Atlanta, which funded the study.
The use of TaqMan Array Card (TAC) microarrays has been extended to permit simultaneous detection of HIV-1, HIV-2, and five hepatitis viruses from a small amount of extracted nucleic acid, according to a study by Timothy C. Granade, MD, and his colleagues at the Centers for Disease Control and Prevention, Atlanta.
This is particularly important for dealing with HIV-infected individuals, because HIV-1 and HIV-2 require different treatment interventions, and approximately one-third of HIV-infected patients have been found to be coinfected with hepatitis C or hepatitis B, according to the study report, published in the Journal of Virological Methods (J Virol Methods. 2018 Sep;259:60-5).
HIV-1-positive plasma samples from a variety of subtypes as well as whole blood specimens were confirmed for HIV-1-infection serologically or by nucleic amplification methods. HIV-2 whole blood and plasma specimens were also obtained.
TAC cards contained one positive control, one negative control, three HIV-1 replicates, and two HIV-2 replicates. In addition, the five common hepatitis viruses (A-E) were each replicated three times on each card. The cards were used to test the RNA isolates obtained from the various samples.
Ninety-five of the 104 known HIV-1-positive specimens were assayed positive using TAC; 23 of 26 HIV-2-seeded specimens were detectable using TAC and no cross-reactivity was seen between HIV-1-positive and HIV-2-positive specimens.
Eighteen of the HIV-1-positive specimens were also reactive in triplicate for HCV; three of the HIV-1-positive specimens were reactive to HBV and one specimen was reactive to HIV-1, HBV, and HCV.
“The TAC assay could be invaluable in large-scale screening environments or in surveying local outbreaks such as the recent HIV cluster found in Indiana. Many of these individuals were later determined to be infected with hepatitis C. The use of TAC could shorten the time to identifying and confirming such cases and permit the detection of multiple blood-borne infections in a single test. Application of TAC technology to general population surveillance could identify problem areas for both HIV prevention and intervention efforts in a variety of global environs,” the researchers concluded.
The authors were employed by the Centers for Disease Control and Prevention, Atlanta, which funded the study.
The use of TaqMan Array Card (TAC) microarrays has been extended to permit simultaneous detection of HIV-1, HIV-2, and five hepatitis viruses from a small amount of extracted nucleic acid, according to a study by Timothy C. Granade, MD, and his colleagues at the Centers for Disease Control and Prevention, Atlanta.
This is particularly important for dealing with HIV-infected individuals, because HIV-1 and HIV-2 require different treatment interventions, and approximately one-third of HIV-infected patients have been found to be coinfected with hepatitis C or hepatitis B, according to the study report, published in the Journal of Virological Methods (J Virol Methods. 2018 Sep;259:60-5).
HIV-1-positive plasma samples from a variety of subtypes as well as whole blood specimens were confirmed for HIV-1-infection serologically or by nucleic amplification methods. HIV-2 whole blood and plasma specimens were also obtained.
TAC cards contained one positive control, one negative control, three HIV-1 replicates, and two HIV-2 replicates. In addition, the five common hepatitis viruses (A-E) were each replicated three times on each card. The cards were used to test the RNA isolates obtained from the various samples.
Ninety-five of the 104 known HIV-1-positive specimens were assayed positive using TAC; 23 of 26 HIV-2-seeded specimens were detectable using TAC and no cross-reactivity was seen between HIV-1-positive and HIV-2-positive specimens.
Eighteen of the HIV-1-positive specimens were also reactive in triplicate for HCV; three of the HIV-1-positive specimens were reactive to HBV and one specimen was reactive to HIV-1, HBV, and HCV.
“The TAC assay could be invaluable in large-scale screening environments or in surveying local outbreaks such as the recent HIV cluster found in Indiana. Many of these individuals were later determined to be infected with hepatitis C. The use of TAC could shorten the time to identifying and confirming such cases and permit the detection of multiple blood-borne infections in a single test. Application of TAC technology to general population surveillance could identify problem areas for both HIV prevention and intervention efforts in a variety of global environs,” the researchers concluded.
The authors were employed by the Centers for Disease Control and Prevention, Atlanta, which funded the study.
FROM THE JOURNAL OF VIROLOGICAL METHODS
Federal Health Care Data Trends: Respiratory Disorders
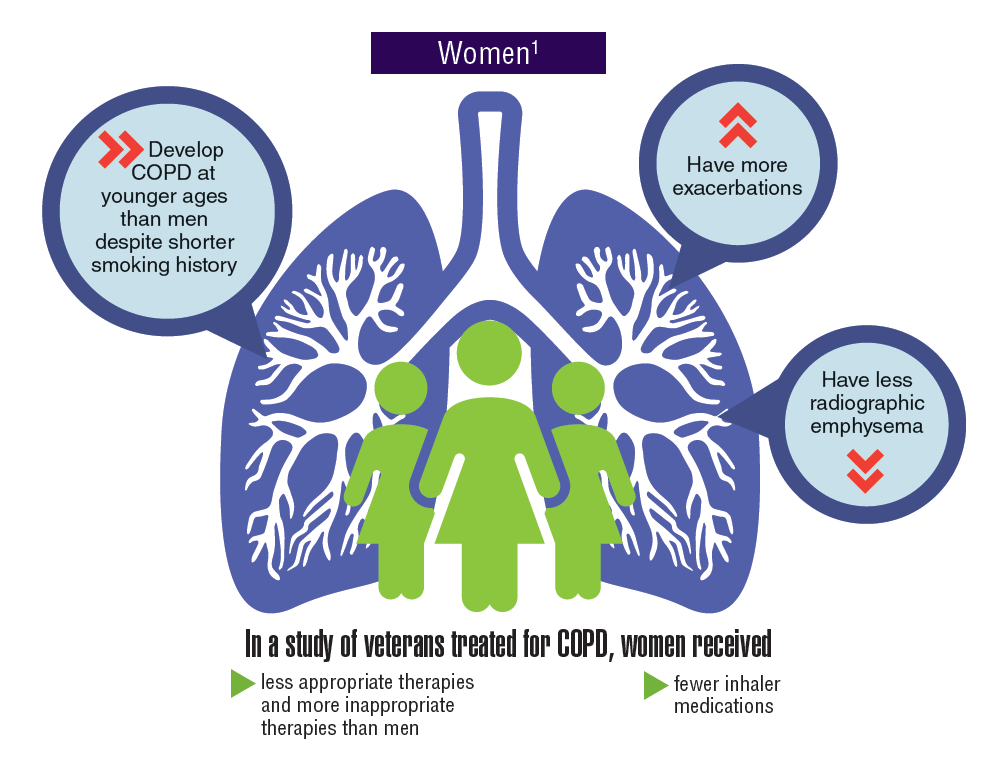
Asthma and chronic obstructive pulmonary disease (COPD), which comprises a combination of chronic and slowly progressive respiratory disorders, including emphysema and chronic bronchitis, are prevalent respiratory disorders in the active-duty and veteran populations. Although chronic and manageable, asthma, COPD, and other respiratory diseases represent a significant disease burden. Women tend to develop COPD at younger ages, have more exacerbations, and yet received fewer inhaler medications and less appropriate therapies. Not only do many respiratory diseases present a risk of mortality, but evidence suggests that there is increased risk of developing lung cancer.
Click here to continue reading.
Asthma and chronic obstructive pulmonary disease (COPD), which comprises a combination of chronic and slowly progressive respiratory disorders, including emphysema and chronic bronchitis, are prevalent respiratory disorders in the active-duty and veteran populations. Although chronic and manageable, asthma, COPD, and other respiratory diseases represent a significant disease burden. Women tend to develop COPD at younger ages, have more exacerbations, and yet received fewer inhaler medications and less appropriate therapies. Not only do many respiratory diseases present a risk of mortality, but evidence suggests that there is increased risk of developing lung cancer.
Click here to continue reading.
Asthma and chronic obstructive pulmonary disease (COPD), which comprises a combination of chronic and slowly progressive respiratory disorders, including emphysema and chronic bronchitis, are prevalent respiratory disorders in the active-duty and veteran populations. Although chronic and manageable, asthma, COPD, and other respiratory diseases represent a significant disease burden. Women tend to develop COPD at younger ages, have more exacerbations, and yet received fewer inhaler medications and less appropriate therapies. Not only do many respiratory diseases present a risk of mortality, but evidence suggests that there is increased risk of developing lung cancer.
Click here to continue reading.
Treatments, disease affect spermatogonia in boys

Alkylating agents, hydroxyurea (HU), and certain non-malignant diseases can significantly deplete spermatogonial cell counts in young boys, according to research published in Human Reproduction.
Boys who received alkylating agents to treat cancer had significantly lower spermatogonial cell counts than control subjects or boys with malignant/nonmalignant diseases treated with non-alkylating agents.
Five of 6 SCD patients treated with HU had a totally depleted spermatogonial pool, and the remaining patient had a low spermatogonial cell count.
Five boys with non-malignant diseases who were not exposed to chemotherapy had significantly lower spermatogonial cell counts than controls.
“Our findings of a dramatic decrease in germ cell numbers in boys treated with alkylating agents and in sickle cell disease patients treated with hydroxyurea suggest that storing frozen testicular tissue from these boys should be performed before these treatments are initiated,” said study author Cecilia Petersen, MD, PhD, of Karolinska Institutet and University Hospital in Stockholm, Sweden.
“This needs to be communicated to physicians as well as patients and their parents or carers. However, until sperm that are able to fertilize eggs are produced from stored testicular tissue, we cannot confirm that germ cell quantity might determine the success of transplantation of the tissue in adulthood. Further research on this is needed to establish a realistic fertility preservation technique.”
Dr Petersen and her colleagues also noted that preserving testicular tissue may not be a viable option for boys who have low spermatogonial cell counts prior to treatment.
Patients and controls
For this study, the researchers analyzed testicular tissue from 32 boys facing treatments that carried a high risk of infertility—testicular irradiation, chemotherapy, or radiotherapy in advance of stem cell transplant.
Twenty boys had the tissue taken after initial chemotherapy, and 12 had it taken before starting any treatment.1
Eight patients had received chemotherapy with non-alkylating agents, 6 (all with malignancies) had received alkylating agents, and 6 (all with SCD) had received HU.
Diseases included acute lymphoblastic leukemia (n=6), SCD (n=6), acute myeloid leukemia (n=3), thalassemia major (n=3), neuroblastoma (n=2), juvenile myelomonocytic leukemia (n=2), myelodysplastic syndromes (n=2), primary immunodeficiency (n=2), Wilms tumor (n=1), adrenoleukodystrophy (n=1), hepatoblastoma (n=1), primitive neuroectodermal tumor (n=1), severe aplastic anemia (n=1), and Fanconi anemia (n=1).
The researchers compared samples from these 32 patients to 14 healthy testicular tissue samples stored in the biobank at the Karolinska University Hospital.
For both sample types, the team counted the number of spermatogonial cells found in a cross-section of seminiferous tubules.
“We could compare the number of spermatogonia with those found in the healthy boys as a way to estimate the effect of medical treatment or the disease itself on the future fertility of a patient,” explained study author Jan-Bernd Stukenborg, PhD, of Karolinska Institutet and University Hospital.
Impact of treatment
There was no significant difference in the mean quantity of spermatogonia per transverse tubular cross-section (S/T) between patients exposed to non-alkylating agents (1.7 ± 1.0, n=8) and biobank controls (4.1 ± 4.6, n=14).
However, samples from patients who received alkylating agents had a significantly lower mean S/T value (0.2 ± 0.3, n=6) than samples from patients treated with non-alkylating agents (P=0.003) and biobank controls (P<0.001).
“We found that the numbers of germ cells present in the cross-sections of the seminiferous tubules were significantly depleted and close to 0 in patients treated with alkylating agents,” Dr Stukenborg said.
Samples from the SCD patients also had a significantly lower mean S/T value (0.3 ± 0.6, n=6) than biobank controls (P=0.003).
Dr Stukenborg noted that the germ cell pool was totally depleted in 5 of the boys with SCD, and the pool was “very low” in the sixth SCD patient.
“This was not seen in patients who had not started treatment or were treated with non-alkylating agents or in the biobank tissues,” Dr Stukenborg said.2
He and his colleagues noted that it is possible for germ cells to recover to normal levels after treatment that is highly toxic to the testes, but high doses of alkylating agents and radiotherapy to the testicles are strongly associated with permanent or long-term infertility.
“The first group of boys who received bone marrow transplants are now reaching their thirties,” said study author Kirsi Jahnukainen, MD, PhD, of Helsinki University Central Hospital in Finland.
“Recent data suggest they may have a high chance of their sperm production recovering, even if they received high-dose alkylating therapies, so long as they had no testicular irradiation.”
Impact of disease
The researchers also found evidence to suggest that, for some boys, their disease may have affected spermatogonial cell counts before any treatment began.
Five patients with non-malignant disease who had not been exposed to chemotherapy (3 with thalassemia major, 1 with Fanconi anemia, and 1 with primary immunodeficiency) had a significantly lower mean S/T value (0.4 ± 0.5) than controls (P=0.006).
“Among patients who had not been treated previously with chemotherapy, there were several boys with a low number of germ cells for their age,” Dr Jahnukainen said.
“This suggests that some non-malignant diseases that require bone marrow transplants may affect the fertility of young boys even before exposure to therapy that is toxic for the testes.”
The researchers noted that a limitation of this study was that biobank samples had no detailed information regarding previous medical treatments and testicular volumes.
1. Testicular tissue is taken from patients under general anesthesia. The surgeon removes approximately 20% of the tissue from the testicular capsule in one of the testicles. For this study, a third of the tissue was taken to the Karolinska Institutet for analysis.
2. A recent meta-analysis showed that normal testicular tissue samples of newborns contain approximately 2.5 germ cells per tubular cross-section. This number decreases to approximately 1.2 within the first 3 years of age, followed by an increase up to 2.6 germ cells per tubular cross-section at 6 to 7 years, reaching a plateau until the age of 11. At the onset of puberty, an increase of up to 7 spermatogonia per tubular cross-section could be observed.
Alkylating agents, hydroxyurea (HU), and certain non-malignant diseases can significantly deplete spermatogonial cell counts in young boys, according to research published in Human Reproduction.
Boys who received alkylating agents to treat cancer had significantly lower spermatogonial cell counts than control subjects or boys with malignant/nonmalignant diseases treated with non-alkylating agents.
Five of 6 SCD patients treated with HU had a totally depleted spermatogonial pool, and the remaining patient had a low spermatogonial cell count.
Five boys with non-malignant diseases who were not exposed to chemotherapy had significantly lower spermatogonial cell counts than controls.
“Our findings of a dramatic decrease in germ cell numbers in boys treated with alkylating agents and in sickle cell disease patients treated with hydroxyurea suggest that storing frozen testicular tissue from these boys should be performed before these treatments are initiated,” said study author Cecilia Petersen, MD, PhD, of Karolinska Institutet and University Hospital in Stockholm, Sweden.
“This needs to be communicated to physicians as well as patients and their parents or carers. However, until sperm that are able to fertilize eggs are produced from stored testicular tissue, we cannot confirm that germ cell quantity might determine the success of transplantation of the tissue in adulthood. Further research on this is needed to establish a realistic fertility preservation technique.”
Dr Petersen and her colleagues also noted that preserving testicular tissue may not be a viable option for boys who have low spermatogonial cell counts prior to treatment.
Patients and controls
For this study, the researchers analyzed testicular tissue from 32 boys facing treatments that carried a high risk of infertility—testicular irradiation, chemotherapy, or radiotherapy in advance of stem cell transplant.
Twenty boys had the tissue taken after initial chemotherapy, and 12 had it taken before starting any treatment.1
Eight patients had received chemotherapy with non-alkylating agents, 6 (all with malignancies) had received alkylating agents, and 6 (all with SCD) had received HU.
Diseases included acute lymphoblastic leukemia (n=6), SCD (n=6), acute myeloid leukemia (n=3), thalassemia major (n=3), neuroblastoma (n=2), juvenile myelomonocytic leukemia (n=2), myelodysplastic syndromes (n=2), primary immunodeficiency (n=2), Wilms tumor (n=1), adrenoleukodystrophy (n=1), hepatoblastoma (n=1), primitive neuroectodermal tumor (n=1), severe aplastic anemia (n=1), and Fanconi anemia (n=1).
The researchers compared samples from these 32 patients to 14 healthy testicular tissue samples stored in the biobank at the Karolinska University Hospital.
For both sample types, the team counted the number of spermatogonial cells found in a cross-section of seminiferous tubules.
“We could compare the number of spermatogonia with those found in the healthy boys as a way to estimate the effect of medical treatment or the disease itself on the future fertility of a patient,” explained study author Jan-Bernd Stukenborg, PhD, of Karolinska Institutet and University Hospital.
Impact of treatment
There was no significant difference in the mean quantity of spermatogonia per transverse tubular cross-section (S/T) between patients exposed to non-alkylating agents (1.7 ± 1.0, n=8) and biobank controls (4.1 ± 4.6, n=14).
However, samples from patients who received alkylating agents had a significantly lower mean S/T value (0.2 ± 0.3, n=6) than samples from patients treated with non-alkylating agents (P=0.003) and biobank controls (P<0.001).
“We found that the numbers of germ cells present in the cross-sections of the seminiferous tubules were significantly depleted and close to 0 in patients treated with alkylating agents,” Dr Stukenborg said.
Samples from the SCD patients also had a significantly lower mean S/T value (0.3 ± 0.6, n=6) than biobank controls (P=0.003).
Dr Stukenborg noted that the germ cell pool was totally depleted in 5 of the boys with SCD, and the pool was “very low” in the sixth SCD patient.
“This was not seen in patients who had not started treatment or were treated with non-alkylating agents or in the biobank tissues,” Dr Stukenborg said.2
He and his colleagues noted that it is possible for germ cells to recover to normal levels after treatment that is highly toxic to the testes, but high doses of alkylating agents and radiotherapy to the testicles are strongly associated with permanent or long-term infertility.
“The first group of boys who received bone marrow transplants are now reaching their thirties,” said study author Kirsi Jahnukainen, MD, PhD, of Helsinki University Central Hospital in Finland.
“Recent data suggest they may have a high chance of their sperm production recovering, even if they received high-dose alkylating therapies, so long as they had no testicular irradiation.”
Impact of disease
The researchers also found evidence to suggest that, for some boys, their disease may have affected spermatogonial cell counts before any treatment began.
Five patients with non-malignant disease who had not been exposed to chemotherapy (3 with thalassemia major, 1 with Fanconi anemia, and 1 with primary immunodeficiency) had a significantly lower mean S/T value (0.4 ± 0.5) than controls (P=0.006).
“Among patients who had not been treated previously with chemotherapy, there were several boys with a low number of germ cells for their age,” Dr Jahnukainen said.
“This suggests that some non-malignant diseases that require bone marrow transplants may affect the fertility of young boys even before exposure to therapy that is toxic for the testes.”
The researchers noted that a limitation of this study was that biobank samples had no detailed information regarding previous medical treatments and testicular volumes.
1. Testicular tissue is taken from patients under general anesthesia. The surgeon removes approximately 20% of the tissue from the testicular capsule in one of the testicles. For this study, a third of the tissue was taken to the Karolinska Institutet for analysis.
2. A recent meta-analysis showed that normal testicular tissue samples of newborns contain approximately 2.5 germ cells per tubular cross-section. This number decreases to approximately 1.2 within the first 3 years of age, followed by an increase up to 2.6 germ cells per tubular cross-section at 6 to 7 years, reaching a plateau until the age of 11. At the onset of puberty, an increase of up to 7 spermatogonia per tubular cross-section could be observed.
Alkylating agents, hydroxyurea (HU), and certain non-malignant diseases can significantly deplete spermatogonial cell counts in young boys, according to research published in Human Reproduction.
Boys who received alkylating agents to treat cancer had significantly lower spermatogonial cell counts than control subjects or boys with malignant/nonmalignant diseases treated with non-alkylating agents.
Five of 6 SCD patients treated with HU had a totally depleted spermatogonial pool, and the remaining patient had a low spermatogonial cell count.
Five boys with non-malignant diseases who were not exposed to chemotherapy had significantly lower spermatogonial cell counts than controls.
“Our findings of a dramatic decrease in germ cell numbers in boys treated with alkylating agents and in sickle cell disease patients treated with hydroxyurea suggest that storing frozen testicular tissue from these boys should be performed before these treatments are initiated,” said study author Cecilia Petersen, MD, PhD, of Karolinska Institutet and University Hospital in Stockholm, Sweden.
“This needs to be communicated to physicians as well as patients and their parents or carers. However, until sperm that are able to fertilize eggs are produced from stored testicular tissue, we cannot confirm that germ cell quantity might determine the success of transplantation of the tissue in adulthood. Further research on this is needed to establish a realistic fertility preservation technique.”
Dr Petersen and her colleagues also noted that preserving testicular tissue may not be a viable option for boys who have low spermatogonial cell counts prior to treatment.
Patients and controls
For this study, the researchers analyzed testicular tissue from 32 boys facing treatments that carried a high risk of infertility—testicular irradiation, chemotherapy, or radiotherapy in advance of stem cell transplant.
Twenty boys had the tissue taken after initial chemotherapy, and 12 had it taken before starting any treatment.1
Eight patients had received chemotherapy with non-alkylating agents, 6 (all with malignancies) had received alkylating agents, and 6 (all with SCD) had received HU.
Diseases included acute lymphoblastic leukemia (n=6), SCD (n=6), acute myeloid leukemia (n=3), thalassemia major (n=3), neuroblastoma (n=2), juvenile myelomonocytic leukemia (n=2), myelodysplastic syndromes (n=2), primary immunodeficiency (n=2), Wilms tumor (n=1), adrenoleukodystrophy (n=1), hepatoblastoma (n=1), primitive neuroectodermal tumor (n=1), severe aplastic anemia (n=1), and Fanconi anemia (n=1).
The researchers compared samples from these 32 patients to 14 healthy testicular tissue samples stored in the biobank at the Karolinska University Hospital.
For both sample types, the team counted the number of spermatogonial cells found in a cross-section of seminiferous tubules.
“We could compare the number of spermatogonia with those found in the healthy boys as a way to estimate the effect of medical treatment or the disease itself on the future fertility of a patient,” explained study author Jan-Bernd Stukenborg, PhD, of Karolinska Institutet and University Hospital.
Impact of treatment
There was no significant difference in the mean quantity of spermatogonia per transverse tubular cross-section (S/T) between patients exposed to non-alkylating agents (1.7 ± 1.0, n=8) and biobank controls (4.1 ± 4.6, n=14).
However, samples from patients who received alkylating agents had a significantly lower mean S/T value (0.2 ± 0.3, n=6) than samples from patients treated with non-alkylating agents (P=0.003) and biobank controls (P<0.001).
“We found that the numbers of germ cells present in the cross-sections of the seminiferous tubules were significantly depleted and close to 0 in patients treated with alkylating agents,” Dr Stukenborg said.
Samples from the SCD patients also had a significantly lower mean S/T value (0.3 ± 0.6, n=6) than biobank controls (P=0.003).
Dr Stukenborg noted that the germ cell pool was totally depleted in 5 of the boys with SCD, and the pool was “very low” in the sixth SCD patient.
“This was not seen in patients who had not started treatment or were treated with non-alkylating agents or in the biobank tissues,” Dr Stukenborg said.2
He and his colleagues noted that it is possible for germ cells to recover to normal levels after treatment that is highly toxic to the testes, but high doses of alkylating agents and radiotherapy to the testicles are strongly associated with permanent or long-term infertility.
“The first group of boys who received bone marrow transplants are now reaching their thirties,” said study author Kirsi Jahnukainen, MD, PhD, of Helsinki University Central Hospital in Finland.
“Recent data suggest they may have a high chance of their sperm production recovering, even if they received high-dose alkylating therapies, so long as they had no testicular irradiation.”
Impact of disease
The researchers also found evidence to suggest that, for some boys, their disease may have affected spermatogonial cell counts before any treatment began.
Five patients with non-malignant disease who had not been exposed to chemotherapy (3 with thalassemia major, 1 with Fanconi anemia, and 1 with primary immunodeficiency) had a significantly lower mean S/T value (0.4 ± 0.5) than controls (P=0.006).
“Among patients who had not been treated previously with chemotherapy, there were several boys with a low number of germ cells for their age,” Dr Jahnukainen said.
“This suggests that some non-malignant diseases that require bone marrow transplants may affect the fertility of young boys even before exposure to therapy that is toxic for the testes.”
The researchers noted that a limitation of this study was that biobank samples had no detailed information regarding previous medical treatments and testicular volumes.
1. Testicular tissue is taken from patients under general anesthesia. The surgeon removes approximately 20% of the tissue from the testicular capsule in one of the testicles. For this study, a third of the tissue was taken to the Karolinska Institutet for analysis.
2. A recent meta-analysis showed that normal testicular tissue samples of newborns contain approximately 2.5 germ cells per tubular cross-section. This number decreases to approximately 1.2 within the first 3 years of age, followed by an increase up to 2.6 germ cells per tubular cross-section at 6 to 7 years, reaching a plateau until the age of 11. At the onset of puberty, an increase of up to 7 spermatogonia per tubular cross-section could be observed.
Fitness trackers help monitor cancer patients
A small study suggests fitness trackers can be used to assess the quality of life and daily functioning of cancer patients during treatment.
Results indicated that objective data collected from these wearable activity monitors can supplement current assessments of health status and physical function.
This is important because current assessments are limited by their subjectivity and potential for bias, according to Gillian Gresham, PhD, of Cedars-Sinai Medical Center in Los Angeles, California.
Dr Gresham and her colleagues conducted this study and reported the results in npj Digital Medicine.
“One of the challenges in treating patients with advanced cancer is obtaining ongoing, timely, objective data about their physical status during therapy,” said study author Andrew Hendifar, MD, of Cedars-Sinai.
“After all, patients typically spend most of their time at home or work, not in a clinic, and their health statuses change from day to day.”
With this in mind, the researchers studied 37 patients undergoing treatment for advanced cancer at Cedars-Sinai.
The patients wore wrist-mounted fitness trackers throughout the study except when showering or swimming. These devices log the wearer’s step counts, stairs climbed, calories, heart rate, and sleep.
Sets of activity data were collected for 3 consecutive visits during treatment. After the final clinical visit, patients were followed for 6 months to gather additional clinical and survival outcomes.
The researchers compared data from the trackers with patients’ assessments of their own symptoms, including pain, fatigue, and sleep quality, as collected from a National Institutes of Health questionnaire.
These data sets were also compared with Eastern Cooperative Oncology Group Performance Status (ECOG-PS) and Karnofsky Performance Status (KPS) scores.
Results
Patients had a median age of 62 (range, 34-81), about 54% were male, and most (73%) had pancreatic cancer. On average, the patients walked 3700 steps (1.7 miles) per day, climbed 3 flights of stairs per day, and slept 8 hours per night.
The researchers found that activity metrics correlated with ECOG-PS and KPS scores. As scores increased, daily steps and flights of stairs decreased.
The team said the largest correlation coefficients (r) were observed between average steps and increasing ECOG-PS (r=0.63, P<0.01) and KPS (r=0.69, P<0.01) scores.
Patient-reported outcomes also correlated with activity metrics. Average steps were significantly (P<0.05 for all) associated with physical functioning (r=0.57), pain (r=—0.46), and fatigue (r=—0.53). There were significant associations for distance walked and stairs climbed as well.
Finally, the researchers observed an association between activity and grade 3/4 adverse events, hospitalizations, and survival.
An increase of 1000 steps per day, on average, was associated with significantly lower odds of hospitalization (odds ratio: 0.21, 95% CI 0.56, 0.79) and grade 3/4 adverse events (odds ratio: 0.34, 95% CI 0.13, 0.94) as well as increased survival (hazard ratio: 0.48, 95% CI 0.28, 0.83).
“Data gathered through advancements in technology has the potential to help physicians measure the impact of a particular treatment on a patient’s daily functioning,” Dr Gresham said. “Furthermore, continuous activity monitoring may help predict and monitor treatment complications and allow for more timely and appropriate interventions.”
As a next step, the researchers plan to study long-term use of activity monitors in a larger, more diverse group of advanced cancer patients and correlate that data with clinical and self-reported outcomes.
“Our hope is that findings from future studies with wearable activity monitors could lead to development of individualized treatment and exercise plans that may result in increased treatment tolerability and improved survival outcomes for patients,” Dr Hendifar said.
A small study suggests fitness trackers can be used to assess the quality of life and daily functioning of cancer patients during treatment.
Results indicated that objective data collected from these wearable activity monitors can supplement current assessments of health status and physical function.
This is important because current assessments are limited by their subjectivity and potential for bias, according to Gillian Gresham, PhD, of Cedars-Sinai Medical Center in Los Angeles, California.
Dr Gresham and her colleagues conducted this study and reported the results in npj Digital Medicine.
“One of the challenges in treating patients with advanced cancer is obtaining ongoing, timely, objective data about their physical status during therapy,” said study author Andrew Hendifar, MD, of Cedars-Sinai.
“After all, patients typically spend most of their time at home or work, not in a clinic, and their health statuses change from day to day.”
With this in mind, the researchers studied 37 patients undergoing treatment for advanced cancer at Cedars-Sinai.
The patients wore wrist-mounted fitness trackers throughout the study except when showering or swimming. These devices log the wearer’s step counts, stairs climbed, calories, heart rate, and sleep.
Sets of activity data were collected for 3 consecutive visits during treatment. After the final clinical visit, patients were followed for 6 months to gather additional clinical and survival outcomes.
The researchers compared data from the trackers with patients’ assessments of their own symptoms, including pain, fatigue, and sleep quality, as collected from a National Institutes of Health questionnaire.
These data sets were also compared with Eastern Cooperative Oncology Group Performance Status (ECOG-PS) and Karnofsky Performance Status (KPS) scores.
Results
Patients had a median age of 62 (range, 34-81), about 54% were male, and most (73%) had pancreatic cancer. On average, the patients walked 3700 steps (1.7 miles) per day, climbed 3 flights of stairs per day, and slept 8 hours per night.
The researchers found that activity metrics correlated with ECOG-PS and KPS scores. As scores increased, daily steps and flights of stairs decreased.
The team said the largest correlation coefficients (r) were observed between average steps and increasing ECOG-PS (r=0.63, P<0.01) and KPS (r=0.69, P<0.01) scores.
Patient-reported outcomes also correlated with activity metrics. Average steps were significantly (P<0.05 for all) associated with physical functioning (r=0.57), pain (r=—0.46), and fatigue (r=—0.53). There were significant associations for distance walked and stairs climbed as well.
Finally, the researchers observed an association between activity and grade 3/4 adverse events, hospitalizations, and survival.
An increase of 1000 steps per day, on average, was associated with significantly lower odds of hospitalization (odds ratio: 0.21, 95% CI 0.56, 0.79) and grade 3/4 adverse events (odds ratio: 0.34, 95% CI 0.13, 0.94) as well as increased survival (hazard ratio: 0.48, 95% CI 0.28, 0.83).
“Data gathered through advancements in technology has the potential to help physicians measure the impact of a particular treatment on a patient’s daily functioning,” Dr Gresham said. “Furthermore, continuous activity monitoring may help predict and monitor treatment complications and allow for more timely and appropriate interventions.”
As a next step, the researchers plan to study long-term use of activity monitors in a larger, more diverse group of advanced cancer patients and correlate that data with clinical and self-reported outcomes.
“Our hope is that findings from future studies with wearable activity monitors could lead to development of individualized treatment and exercise plans that may result in increased treatment tolerability and improved survival outcomes for patients,” Dr Hendifar said.
A small study suggests fitness trackers can be used to assess the quality of life and daily functioning of cancer patients during treatment.
Results indicated that objective data collected from these wearable activity monitors can supplement current assessments of health status and physical function.
This is important because current assessments are limited by their subjectivity and potential for bias, according to Gillian Gresham, PhD, of Cedars-Sinai Medical Center in Los Angeles, California.
Dr Gresham and her colleagues conducted this study and reported the results in npj Digital Medicine.
“One of the challenges in treating patients with advanced cancer is obtaining ongoing, timely, objective data about their physical status during therapy,” said study author Andrew Hendifar, MD, of Cedars-Sinai.
“After all, patients typically spend most of their time at home or work, not in a clinic, and their health statuses change from day to day.”
With this in mind, the researchers studied 37 patients undergoing treatment for advanced cancer at Cedars-Sinai.
The patients wore wrist-mounted fitness trackers throughout the study except when showering or swimming. These devices log the wearer’s step counts, stairs climbed, calories, heart rate, and sleep.
Sets of activity data were collected for 3 consecutive visits during treatment. After the final clinical visit, patients were followed for 6 months to gather additional clinical and survival outcomes.
The researchers compared data from the trackers with patients’ assessments of their own symptoms, including pain, fatigue, and sleep quality, as collected from a National Institutes of Health questionnaire.
These data sets were also compared with Eastern Cooperative Oncology Group Performance Status (ECOG-PS) and Karnofsky Performance Status (KPS) scores.
Results
Patients had a median age of 62 (range, 34-81), about 54% were male, and most (73%) had pancreatic cancer. On average, the patients walked 3700 steps (1.7 miles) per day, climbed 3 flights of stairs per day, and slept 8 hours per night.
The researchers found that activity metrics correlated with ECOG-PS and KPS scores. As scores increased, daily steps and flights of stairs decreased.
The team said the largest correlation coefficients (r) were observed between average steps and increasing ECOG-PS (r=0.63, P<0.01) and KPS (r=0.69, P<0.01) scores.
Patient-reported outcomes also correlated with activity metrics. Average steps were significantly (P<0.05 for all) associated with physical functioning (r=0.57), pain (r=—0.46), and fatigue (r=—0.53). There were significant associations for distance walked and stairs climbed as well.
Finally, the researchers observed an association between activity and grade 3/4 adverse events, hospitalizations, and survival.
An increase of 1000 steps per day, on average, was associated with significantly lower odds of hospitalization (odds ratio: 0.21, 95% CI 0.56, 0.79) and grade 3/4 adverse events (odds ratio: 0.34, 95% CI 0.13, 0.94) as well as increased survival (hazard ratio: 0.48, 95% CI 0.28, 0.83).
“Data gathered through advancements in technology has the potential to help physicians measure the impact of a particular treatment on a patient’s daily functioning,” Dr Gresham said. “Furthermore, continuous activity monitoring may help predict and monitor treatment complications and allow for more timely and appropriate interventions.”
As a next step, the researchers plan to study long-term use of activity monitors in a larger, more diverse group of advanced cancer patients and correlate that data with clinical and self-reported outcomes.
“Our hope is that findings from future studies with wearable activity monitors could lead to development of individualized treatment and exercise plans that may result in increased treatment tolerability and improved survival outcomes for patients,” Dr Hendifar said.
NIH aims to improve access to cloud computing

The National Institutes of Health (NIH) is attempting to improve biomedical researchers’ access to cloud computing.
With its new STRIDES* initiative, the NIH intends to establish partnerships with commercial cloud service providers (CSPs) to reduce economic and technological barriers to accessing and computing on large biomedical data sets.
The CSPs will work with the NIH and its funded researchers to develop and test new ways to make large data sets and associated computational tools available to wider audiences.
The NIH’s initial efforts with the STRIDES initiative will focus on making NIH high-value data sets more accessible through the cloud, leveraging partnerships with CSPs to take advantage of data-related innovations such as machine learning and artificial intelligence, and experimenting with new ways to optimize technology-intensive research.
The goals of the STRIDES initiative are to:
- Support researchers’ transition to conducting biomedical research using commercial cloud technologies through cost-effective storage and computing arrangements with CSPs
- Provide NIH researchers access to and training on new and emerging cloud-based tools and services
- Facilitate researchers’ access to and use of high-value NIH research data that are currently stored on, or will be moved into, cloud environments
- Enable the formation of an interconnected ecosystem that breaks down silos related to generating, analyzing, and sharing research data.
The NIH has already partnered with Google Cloud for the STRIDES initiative, but the agency hopes to create partnerships with other CSPs as well.
“NIH is in a unique position to bring together academic and innovation industry partners to create a biomedical data ecosystem that maximizes the use of NIH-supported biomedical research data for the greatest benefit to human health,” said NIH Principal Deputy Director Lawrence A. Tabak, DDS, PhD.
The NIH says its agreement with Google Cloud creates a cost-efficient framework for NIH researchers, as well as researchers receiving NIH support, to make use of Google Cloud’s storage, computing, and machine learning technologies.
The partnership will also enable the creation of training programs for researchers at NIH-funded institutions on how to use the Google Cloud platform. And the partnership will involve collaboration with NIH’s Data Commons Pilot—a group of projects testing new tools and methods for working with and sharing data in the cloud.
“Through our partnership with NIH, we are bringing the power of data and the cloud to the biomedical research community globally,” said Gregory Moore, MD, PhD, vice-president of healthcare at Google Cloud.
“Together, we are making it easier for scientists and physicians to access and garner insights from NIH-funded data sets with appropriate privacy protections, which will ultimately accelerate biomedical research progress toward finding treatments and cures for the most devastating diseases of our time.”
A central tenet of STRIDES is that data made available through these partnerships will incorporate standards endorsed by the biomedical research community to make data findable, accessible, interoperable, and reusable.
*Science and Technology Research Infrastructure for Discovery, Experimentation, and Sustainability
The National Institutes of Health (NIH) is attempting to improve biomedical researchers’ access to cloud computing.
With its new STRIDES* initiative, the NIH intends to establish partnerships with commercial cloud service providers (CSPs) to reduce economic and technological barriers to accessing and computing on large biomedical data sets.
The CSPs will work with the NIH and its funded researchers to develop and test new ways to make large data sets and associated computational tools available to wider audiences.
The NIH’s initial efforts with the STRIDES initiative will focus on making NIH high-value data sets more accessible through the cloud, leveraging partnerships with CSPs to take advantage of data-related innovations such as machine learning and artificial intelligence, and experimenting with new ways to optimize technology-intensive research.
The goals of the STRIDES initiative are to:
- Support researchers’ transition to conducting biomedical research using commercial cloud technologies through cost-effective storage and computing arrangements with CSPs
- Provide NIH researchers access to and training on new and emerging cloud-based tools and services
- Facilitate researchers’ access to and use of high-value NIH research data that are currently stored on, or will be moved into, cloud environments
- Enable the formation of an interconnected ecosystem that breaks down silos related to generating, analyzing, and sharing research data.
The NIH has already partnered with Google Cloud for the STRIDES initiative, but the agency hopes to create partnerships with other CSPs as well.
“NIH is in a unique position to bring together academic and innovation industry partners to create a biomedical data ecosystem that maximizes the use of NIH-supported biomedical research data for the greatest benefit to human health,” said NIH Principal Deputy Director Lawrence A. Tabak, DDS, PhD.
The NIH says its agreement with Google Cloud creates a cost-efficient framework for NIH researchers, as well as researchers receiving NIH support, to make use of Google Cloud’s storage, computing, and machine learning technologies.
The partnership will also enable the creation of training programs for researchers at NIH-funded institutions on how to use the Google Cloud platform. And the partnership will involve collaboration with NIH’s Data Commons Pilot—a group of projects testing new tools and methods for working with and sharing data in the cloud.
“Through our partnership with NIH, we are bringing the power of data and the cloud to the biomedical research community globally,” said Gregory Moore, MD, PhD, vice-president of healthcare at Google Cloud.
“Together, we are making it easier for scientists and physicians to access and garner insights from NIH-funded data sets with appropriate privacy protections, which will ultimately accelerate biomedical research progress toward finding treatments and cures for the most devastating diseases of our time.”
A central tenet of STRIDES is that data made available through these partnerships will incorporate standards endorsed by the biomedical research community to make data findable, accessible, interoperable, and reusable.
*Science and Technology Research Infrastructure for Discovery, Experimentation, and Sustainability
The National Institutes of Health (NIH) is attempting to improve biomedical researchers’ access to cloud computing.
With its new STRIDES* initiative, the NIH intends to establish partnerships with commercial cloud service providers (CSPs) to reduce economic and technological barriers to accessing and computing on large biomedical data sets.
The CSPs will work with the NIH and its funded researchers to develop and test new ways to make large data sets and associated computational tools available to wider audiences.
The NIH’s initial efforts with the STRIDES initiative will focus on making NIH high-value data sets more accessible through the cloud, leveraging partnerships with CSPs to take advantage of data-related innovations such as machine learning and artificial intelligence, and experimenting with new ways to optimize technology-intensive research.
The goals of the STRIDES initiative are to:
- Support researchers’ transition to conducting biomedical research using commercial cloud technologies through cost-effective storage and computing arrangements with CSPs
- Provide NIH researchers access to and training on new and emerging cloud-based tools and services
- Facilitate researchers’ access to and use of high-value NIH research data that are currently stored on, or will be moved into, cloud environments
- Enable the formation of an interconnected ecosystem that breaks down silos related to generating, analyzing, and sharing research data.
The NIH has already partnered with Google Cloud for the STRIDES initiative, but the agency hopes to create partnerships with other CSPs as well.
“NIH is in a unique position to bring together academic and innovation industry partners to create a biomedical data ecosystem that maximizes the use of NIH-supported biomedical research data for the greatest benefit to human health,” said NIH Principal Deputy Director Lawrence A. Tabak, DDS, PhD.
The NIH says its agreement with Google Cloud creates a cost-efficient framework for NIH researchers, as well as researchers receiving NIH support, to make use of Google Cloud’s storage, computing, and machine learning technologies.
The partnership will also enable the creation of training programs for researchers at NIH-funded institutions on how to use the Google Cloud platform. And the partnership will involve collaboration with NIH’s Data Commons Pilot—a group of projects testing new tools and methods for working with and sharing data in the cloud.
“Through our partnership with NIH, we are bringing the power of data and the cloud to the biomedical research community globally,” said Gregory Moore, MD, PhD, vice-president of healthcare at Google Cloud.
“Together, we are making it easier for scientists and physicians to access and garner insights from NIH-funded data sets with appropriate privacy protections, which will ultimately accelerate biomedical research progress toward finding treatments and cures for the most devastating diseases of our time.”
A central tenet of STRIDES is that data made available through these partnerships will incorporate standards endorsed by the biomedical research community to make data findable, accessible, interoperable, and reusable.
*Science and Technology Research Infrastructure for Discovery, Experimentation, and Sustainability
Psychological screening integration improves quality of care for children with abdominal pain
according to results from a recent report published in Pediatrics.
Natoshia R. Cunningham, PhD, from the divisions of behavioral medicine and clinical psychology and the department of pediatrics at the University of Cincinnati and her colleagues tested their screening process with a pilot study in a single clinic between August 2015 and October 2016. Researchers assessed patients with FAPD using the Screen for Child Anxiety Related Disorders (SCARED)–Child Report and Functional Disability Inventory (FDI)–Child Version. Clinically significant anxiety was defined as a score of at least 25 (range, 0-82) for the SCARED; clinical cutoffs for minimal, moderate, and severe disability in the child version of the FDI were defined as scores of 0-12, 13-29, and greater than 30, respectively. After fine-tuning the screening process in the pilot, the researchers scaled the effort to six different clinics within a large gastroenterology division at a Midwest urban medical center.
“Children with FAPD who are at the greatest risk for persistent functional disability (i.e., those with clinical elevations in all three risk areas) are now being immediately identified and managed as part of routine care,” Dr. Cunningham and colleagues wrote in their study.
Of 6,744 eligible children (mean age, 13.34 years; 58% female; 87.6% non-Hispanic white), 5,221 children completed the screening, with 1,291 of 1,369 children completing both the screening process and reporting abdominal pain as a presenting complaint. Researchers found 43.1% of children showed clinically significant anxiety under SCARED, with a mean SCARED score of 24.3. Children had a mean FDI score of 13.7, and nearly half of the children had functional disability that was moderate (34.2%) or severe (10.8%), with 61.5% overall reporting a pain level of least 4 out of 10 during the week.
There were 21.1% of children with “clinical elevations” in pain, disability, and anxiety, with researchers noting that, compared with patients without clinical anxiety, those with anxiety had significantly higher FDI scores (mean, 16.29 vs. 11.54) and higher pain levels (mean, 4.63 vs. 3.72).
Among those referred to psychological services, the number of referrals after implementing psychological screening nearly doubled to 15.2 patients per 1,000 per month between March 2017 and September 2017, compared with baseline referrals the previous year (8.3 per 1,000, March 2016 to September 2016).
The researchers suggested future work in psychological screening for children with FAPD should consider shortening screening time by using different outcome measures to lessen the burden on clinical staff; they noted it should also consider applying psychological screening in a telehealth, primary care, or school setting to increase access to care.
“Even in patients with all three risk factors, the decision to refer to psychological providers remains an individualized process driven by clinical judgement,” Dr. Cunningham and her colleagues wrote. “Many factors, including provider practice patterns and other considerations (e.g., patient and family interest, provider availability, distance to care, and insurance coverage) guide these decisions.”
The authors report no relevant financial disclosures.
SOURCE: Cunningham NR et al. Pediatrics. 2018 Jul 25. doi: 10.1542/peds.2017-2876.
according to results from a recent report published in Pediatrics.
Natoshia R. Cunningham, PhD, from the divisions of behavioral medicine and clinical psychology and the department of pediatrics at the University of Cincinnati and her colleagues tested their screening process with a pilot study in a single clinic between August 2015 and October 2016. Researchers assessed patients with FAPD using the Screen for Child Anxiety Related Disorders (SCARED)–Child Report and Functional Disability Inventory (FDI)–Child Version. Clinically significant anxiety was defined as a score of at least 25 (range, 0-82) for the SCARED; clinical cutoffs for minimal, moderate, and severe disability in the child version of the FDI were defined as scores of 0-12, 13-29, and greater than 30, respectively. After fine-tuning the screening process in the pilot, the researchers scaled the effort to six different clinics within a large gastroenterology division at a Midwest urban medical center.
“Children with FAPD who are at the greatest risk for persistent functional disability (i.e., those with clinical elevations in all three risk areas) are now being immediately identified and managed as part of routine care,” Dr. Cunningham and colleagues wrote in their study.
Of 6,744 eligible children (mean age, 13.34 years; 58% female; 87.6% non-Hispanic white), 5,221 children completed the screening, with 1,291 of 1,369 children completing both the screening process and reporting abdominal pain as a presenting complaint. Researchers found 43.1% of children showed clinically significant anxiety under SCARED, with a mean SCARED score of 24.3. Children had a mean FDI score of 13.7, and nearly half of the children had functional disability that was moderate (34.2%) or severe (10.8%), with 61.5% overall reporting a pain level of least 4 out of 10 during the week.
There were 21.1% of children with “clinical elevations” in pain, disability, and anxiety, with researchers noting that, compared with patients without clinical anxiety, those with anxiety had significantly higher FDI scores (mean, 16.29 vs. 11.54) and higher pain levels (mean, 4.63 vs. 3.72).
Among those referred to psychological services, the number of referrals after implementing psychological screening nearly doubled to 15.2 patients per 1,000 per month between March 2017 and September 2017, compared with baseline referrals the previous year (8.3 per 1,000, March 2016 to September 2016).
The researchers suggested future work in psychological screening for children with FAPD should consider shortening screening time by using different outcome measures to lessen the burden on clinical staff; they noted it should also consider applying psychological screening in a telehealth, primary care, or school setting to increase access to care.
“Even in patients with all three risk factors, the decision to refer to psychological providers remains an individualized process driven by clinical judgement,” Dr. Cunningham and her colleagues wrote. “Many factors, including provider practice patterns and other considerations (e.g., patient and family interest, provider availability, distance to care, and insurance coverage) guide these decisions.”
The authors report no relevant financial disclosures.
SOURCE: Cunningham NR et al. Pediatrics. 2018 Jul 25. doi: 10.1542/peds.2017-2876.
according to results from a recent report published in Pediatrics.
Natoshia R. Cunningham, PhD, from the divisions of behavioral medicine and clinical psychology and the department of pediatrics at the University of Cincinnati and her colleagues tested their screening process with a pilot study in a single clinic between August 2015 and October 2016. Researchers assessed patients with FAPD using the Screen for Child Anxiety Related Disorders (SCARED)–Child Report and Functional Disability Inventory (FDI)–Child Version. Clinically significant anxiety was defined as a score of at least 25 (range, 0-82) for the SCARED; clinical cutoffs for minimal, moderate, and severe disability in the child version of the FDI were defined as scores of 0-12, 13-29, and greater than 30, respectively. After fine-tuning the screening process in the pilot, the researchers scaled the effort to six different clinics within a large gastroenterology division at a Midwest urban medical center.
“Children with FAPD who are at the greatest risk for persistent functional disability (i.e., those with clinical elevations in all three risk areas) are now being immediately identified and managed as part of routine care,” Dr. Cunningham and colleagues wrote in their study.
Of 6,744 eligible children (mean age, 13.34 years; 58% female; 87.6% non-Hispanic white), 5,221 children completed the screening, with 1,291 of 1,369 children completing both the screening process and reporting abdominal pain as a presenting complaint. Researchers found 43.1% of children showed clinically significant anxiety under SCARED, with a mean SCARED score of 24.3. Children had a mean FDI score of 13.7, and nearly half of the children had functional disability that was moderate (34.2%) or severe (10.8%), with 61.5% overall reporting a pain level of least 4 out of 10 during the week.
There were 21.1% of children with “clinical elevations” in pain, disability, and anxiety, with researchers noting that, compared with patients without clinical anxiety, those with anxiety had significantly higher FDI scores (mean, 16.29 vs. 11.54) and higher pain levels (mean, 4.63 vs. 3.72).
Among those referred to psychological services, the number of referrals after implementing psychological screening nearly doubled to 15.2 patients per 1,000 per month between March 2017 and September 2017, compared with baseline referrals the previous year (8.3 per 1,000, March 2016 to September 2016).
The researchers suggested future work in psychological screening for children with FAPD should consider shortening screening time by using different outcome measures to lessen the burden on clinical staff; they noted it should also consider applying psychological screening in a telehealth, primary care, or school setting to increase access to care.
“Even in patients with all three risk factors, the decision to refer to psychological providers remains an individualized process driven by clinical judgement,” Dr. Cunningham and her colleagues wrote. “Many factors, including provider practice patterns and other considerations (e.g., patient and family interest, provider availability, distance to care, and insurance coverage) guide these decisions.”
The authors report no relevant financial disclosures.
SOURCE: Cunningham NR et al. Pediatrics. 2018 Jul 25. doi: 10.1542/peds.2017-2876.
FROM PEDIATRICS
Key clinical point: Implementing a screening system in clinical centers for pediatric patients with abdominal pain led to identification of anxiety and disability symptoms, additional tailored care, and psychological referrals for some patients with clinical anxiety.
Major finding: Of those who completed screening, 43.1% of children met criteria for clinically significant anxiety, 34.2% reported moderate disability, 10.8% reported severe functional disability, and 61.5% reported a pain level of at least 4 out of 10.
Study details: A report of 1,291 pediatric patients with abdominal pain screened at six clinics within a large gastroenterology division at a Midwest urban medical center.
Disclosures: The authors reported no relevant financial disclosures.
Source: Cunningham NR et al. Pediatrics. 2018 Jul 25. doi: 10.1542/peds.2017-2876.
Acute care prescriptions can be cut to minimize opioid exposure
ORLANDO – By cutting the number of pills prescribed after a surgical procedure, exposure to opioids can be minimized in a largely opioid-naive patient population at risk of new, persistent use, according to Michael J. Englesbe, MD, FACS, professor of surgery at the University of Michigan, Ann Arbor, who is leading a Michigan initiative to tailor acute care prescribing.

About 90% of surgically patients are opioid-naive, and of those, studies suggest about 6% may become new, persistent opioid users, according to Dr. Englesbe, codirector of the Michigan Opioid Prescribing and Engagement Network (Michigan-OPEN), a state-wide effort to transform acute pain prescribing across all surgical specialties.
“This is a very vulnerable population where their operation can lead to life-changing events way beyond their surgical outcomes,” Dr. Englesbe said in a presentation at the American College of Surgeons Quality and Safety Conference.
“We have to really worry about them,” he added. “It’s hard to identify who they are, and I think minimizing exposure to opioids is the best we have at this point.”
By following evidence-based prescribing guidelines after laparoscopic cholecystectomy, Dr. Englesbe and his colleagues were able to reduce prescription size by 63% with no increase in refills and no change in pain score, according to a research letter recently published in JAMA Surgery.
After adopting the guidelines, median postoperative opioid use dropped from 30 mg to 20 mg (P = .04), they reported.
Laparoscopic cholecystectomy patients could be prescribed as few as 10 5-mg tablets of oxycodone, according to recommendations developed by Michigan-OPEN that are published on opioidprescribing.info. Dr. Englesbe called the website figures “precise prescribing recommendations” that are still relatively generous, meeting or exceeding self-reported use for 75% of patients.
“I think this is an important template for change,” he said. “We’ve found the surgeons in the state very receptive, but more importantly, we’ve been able to partner with other really important stakeholders.” For example, one insurer in the state now aligns some hospital incentive reimbursement based on some of these prescribing methods, he added.
Dr. Englesbe reported no commercial disclosures related to his presentation.
ORLANDO – By cutting the number of pills prescribed after a surgical procedure, exposure to opioids can be minimized in a largely opioid-naive patient population at risk of new, persistent use, according to Michael J. Englesbe, MD, FACS, professor of surgery at the University of Michigan, Ann Arbor, who is leading a Michigan initiative to tailor acute care prescribing.
About 90% of surgically patients are opioid-naive, and of those, studies suggest about 6% may become new, persistent opioid users, according to Dr. Englesbe, codirector of the Michigan Opioid Prescribing and Engagement Network (Michigan-OPEN), a state-wide effort to transform acute pain prescribing across all surgical specialties.
“This is a very vulnerable population where their operation can lead to life-changing events way beyond their surgical outcomes,” Dr. Englesbe said in a presentation at the American College of Surgeons Quality and Safety Conference.
“We have to really worry about them,” he added. “It’s hard to identify who they are, and I think minimizing exposure to opioids is the best we have at this point.”
By following evidence-based prescribing guidelines after laparoscopic cholecystectomy, Dr. Englesbe and his colleagues were able to reduce prescription size by 63% with no increase in refills and no change in pain score, according to a research letter recently published in JAMA Surgery.
After adopting the guidelines, median postoperative opioid use dropped from 30 mg to 20 mg (P = .04), they reported.
Laparoscopic cholecystectomy patients could be prescribed as few as 10 5-mg tablets of oxycodone, according to recommendations developed by Michigan-OPEN that are published on opioidprescribing.info. Dr. Englesbe called the website figures “precise prescribing recommendations” that are still relatively generous, meeting or exceeding self-reported use for 75% of patients.
“I think this is an important template for change,” he said. “We’ve found the surgeons in the state very receptive, but more importantly, we’ve been able to partner with other really important stakeholders.” For example, one insurer in the state now aligns some hospital incentive reimbursement based on some of these prescribing methods, he added.
Dr. Englesbe reported no commercial disclosures related to his presentation.
ORLANDO – By cutting the number of pills prescribed after a surgical procedure, exposure to opioids can be minimized in a largely opioid-naive patient population at risk of new, persistent use, according to Michael J. Englesbe, MD, FACS, professor of surgery at the University of Michigan, Ann Arbor, who is leading a Michigan initiative to tailor acute care prescribing.
About 90% of surgically patients are opioid-naive, and of those, studies suggest about 6% may become new, persistent opioid users, according to Dr. Englesbe, codirector of the Michigan Opioid Prescribing and Engagement Network (Michigan-OPEN), a state-wide effort to transform acute pain prescribing across all surgical specialties.
“This is a very vulnerable population where their operation can lead to life-changing events way beyond their surgical outcomes,” Dr. Englesbe said in a presentation at the American College of Surgeons Quality and Safety Conference.
“We have to really worry about them,” he added. “It’s hard to identify who they are, and I think minimizing exposure to opioids is the best we have at this point.”
By following evidence-based prescribing guidelines after laparoscopic cholecystectomy, Dr. Englesbe and his colleagues were able to reduce prescription size by 63% with no increase in refills and no change in pain score, according to a research letter recently published in JAMA Surgery.
After adopting the guidelines, median postoperative opioid use dropped from 30 mg to 20 mg (P = .04), they reported.
Laparoscopic cholecystectomy patients could be prescribed as few as 10 5-mg tablets of oxycodone, according to recommendations developed by Michigan-OPEN that are published on opioidprescribing.info. Dr. Englesbe called the website figures “precise prescribing recommendations” that are still relatively generous, meeting or exceeding self-reported use for 75% of patients.
“I think this is an important template for change,” he said. “We’ve found the surgeons in the state very receptive, but more importantly, we’ve been able to partner with other really important stakeholders.” For example, one insurer in the state now aligns some hospital incentive reimbursement based on some of these prescribing methods, he added.
Dr. Englesbe reported no commercial disclosures related to his presentation.
EXPERT ANALYSIS FROM ACSQSC 2018
Hospitals gear up for new diagnosis: human trafficking
The woman arrived at the emergency department at Huntington Hospital on New York’s Long Island after she was hit by her boyfriend during an argument. Her situation raised concerns among the medical staff, which had recently been trained to be on the lookout for signs of sex trafficking.

An undocumented immigrant from El Salvador, she worked at a local “cantina” frequented by immigrants. Her job was to get patrons drinks and to dance with them, but many workers in those jobs are expected to offer sex, too. Her boyfriend didn’t want her to work there and that led to the fight, one doctor recalled.
As part of the intake process, the emergency staff asked the 36-year-old woman a series of questions about whether she’d ever had sex for money or whether she had to give someone else part of what she earned, among other things. The screening questions were part of a new program at Northwell Health, a 23-hospital system in the New York metro area that includes Huntington Hospital, to train staff and provide them with tools to identify and support victims of human trafficking.
There are no hard figures for how many people are involved in human trafficking, the term used when individuals are forced to work or have sex for someone else’s commercial benefit. Polaris, a Washington-based nonprofit that advocates for these people and runs help lines for them, said calls and texts to its national hotlines have steadily ticked up in recent years, increasing the number of cases 13% between 2016 and 2017, to 8,759.
But health care providers frequently fail to recognize these patients’ situation. According to a 2014 survey of about 100 survivors of sex trafficking, 88 percent said that while they were being trafficked they had contact with a health care provider, typically someone in an emergency department.
“When trafficking victims come through the health care system but we don’t identify them, it’s a big missed opportunity,” said Santhosh K. Paulus, MD, a family physician who is the site director of the Huntington Hospital’s family medicine residency program and who started the program at Northwell.
Northwell is one of a growing number of hospitals and health care systems that are putting such programs in place. They want to alert staff to be on the lookout for trafficking, much as they watch for signs of child abuse, domestic violence, and elder abuse.
Since last spring, nearly 300 staff members at Huntington Hospital and a family clinic have received training in how to spot trafficking victims and how to help them.
Training is given not only to doctors and nurses but also to registration and reception staff, social workers and security guards. Restore NYC, an organization that assists people caught up in sex trafficking, provided the initial training to key staff, and a hospital task force trains the others. During the next few years, similar efforts will be rolled out at all of Northwell’s 23 hospitals, Paulus said.
Identifying victims of trafficking is not unlike identifying victims of other forms of violence, said Wendy L. Macias-Konstantopoulos, MD, director of the human trafficking initiative at Massachusetts General Hospital in Boston.
One of the big red flags is when people delay coming in for medical care, such as waiting weeks to come in to get an injured ankle or sexually transmitted infection checked out, Dr. Macias-Konstantopoulos said. Or it may be a pattern of injuries that don’t make sense. Sometimes people are reluctant to explain their injury, or they come in with someone who seems overbearing.
“Having a high index of suspicion is the first step,” she said. “If we’re not asking about it, we’re just not going to see it.”
Starting in October, health care providers can also use new diagnosis codes in their records that differentiate trafficking from other types of abuse. This will help track the number of victims and provide appropriate treatment.
Asking may not be enough, however. Depending on what’s going on in their lives, these patients may not be willing or ready to acknowledge that they need help, said Holly Gibbs, human trafficking response program director for Dignity Health, a health care system with nearly 40 hospitals in California, Nevada and Arizona.
Ms. Gibbs knows the issue well. She was forced briefly into prostitution in Atlantic City, N.J., after meeting a man at a shopping mall as a 14-year-old and running away with him. The man persuaded Ms. Gibbs to go with him with promises of a new, glamorous life as a musician or model. At the time, Ms. Gibbs said, she thought that what happened to her was her own fault, a result of choices she made. No health care or law enforcement professional connected her to social services that could have helped her understand otherwise. She was reunited with her family by law enforcement personnel, who arrested the man. He was later convicted.
Dignity Health has implemented a human trafficking response program in the emergency departments and labor and delivery areas of each of its hospitals. Now it’s rolling out the program at clinics and physicians’ offices as well.
A key priority is to help clinicians know how to talk to patients about any violence they may be facing and to connect the patients with outside sources of help.
But in the end, if these patients don’t want assistance, “you respect their wishes,” Ms. Gibbs said. “They may not be ready to accept help now, but you may plant seeds so they’ll be able to accept it later on.”
Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente.
The woman arrived at the emergency department at Huntington Hospital on New York’s Long Island after she was hit by her boyfriend during an argument. Her situation raised concerns among the medical staff, which had recently been trained to be on the lookout for signs of sex trafficking.
An undocumented immigrant from El Salvador, she worked at a local “cantina” frequented by immigrants. Her job was to get patrons drinks and to dance with them, but many workers in those jobs are expected to offer sex, too. Her boyfriend didn’t want her to work there and that led to the fight, one doctor recalled.
As part of the intake process, the emergency staff asked the 36-year-old woman a series of questions about whether she’d ever had sex for money or whether she had to give someone else part of what she earned, among other things. The screening questions were part of a new program at Northwell Health, a 23-hospital system in the New York metro area that includes Huntington Hospital, to train staff and provide them with tools to identify and support victims of human trafficking.
There are no hard figures for how many people are involved in human trafficking, the term used when individuals are forced to work or have sex for someone else’s commercial benefit. Polaris, a Washington-based nonprofit that advocates for these people and runs help lines for them, said calls and texts to its national hotlines have steadily ticked up in recent years, increasing the number of cases 13% between 2016 and 2017, to 8,759.
But health care providers frequently fail to recognize these patients’ situation. According to a 2014 survey of about 100 survivors of sex trafficking, 88 percent said that while they were being trafficked they had contact with a health care provider, typically someone in an emergency department.
“When trafficking victims come through the health care system but we don’t identify them, it’s a big missed opportunity,” said Santhosh K. Paulus, MD, a family physician who is the site director of the Huntington Hospital’s family medicine residency program and who started the program at Northwell.
Northwell is one of a growing number of hospitals and health care systems that are putting such programs in place. They want to alert staff to be on the lookout for trafficking, much as they watch for signs of child abuse, domestic violence, and elder abuse.
Since last spring, nearly 300 staff members at Huntington Hospital and a family clinic have received training in how to spot trafficking victims and how to help them.
Training is given not only to doctors and nurses but also to registration and reception staff, social workers and security guards. Restore NYC, an organization that assists people caught up in sex trafficking, provided the initial training to key staff, and a hospital task force trains the others. During the next few years, similar efforts will be rolled out at all of Northwell’s 23 hospitals, Paulus said.
Identifying victims of trafficking is not unlike identifying victims of other forms of violence, said Wendy L. Macias-Konstantopoulos, MD, director of the human trafficking initiative at Massachusetts General Hospital in Boston.
One of the big red flags is when people delay coming in for medical care, such as waiting weeks to come in to get an injured ankle or sexually transmitted infection checked out, Dr. Macias-Konstantopoulos said. Or it may be a pattern of injuries that don’t make sense. Sometimes people are reluctant to explain their injury, or they come in with someone who seems overbearing.
“Having a high index of suspicion is the first step,” she said. “If we’re not asking about it, we’re just not going to see it.”
Starting in October, health care providers can also use new diagnosis codes in their records that differentiate trafficking from other types of abuse. This will help track the number of victims and provide appropriate treatment.
Asking may not be enough, however. Depending on what’s going on in their lives, these patients may not be willing or ready to acknowledge that they need help, said Holly Gibbs, human trafficking response program director for Dignity Health, a health care system with nearly 40 hospitals in California, Nevada and Arizona.
Ms. Gibbs knows the issue well. She was forced briefly into prostitution in Atlantic City, N.J., after meeting a man at a shopping mall as a 14-year-old and running away with him. The man persuaded Ms. Gibbs to go with him with promises of a new, glamorous life as a musician or model. At the time, Ms. Gibbs said, she thought that what happened to her was her own fault, a result of choices she made. No health care or law enforcement professional connected her to social services that could have helped her understand otherwise. She was reunited with her family by law enforcement personnel, who arrested the man. He was later convicted.
Dignity Health has implemented a human trafficking response program in the emergency departments and labor and delivery areas of each of its hospitals. Now it’s rolling out the program at clinics and physicians’ offices as well.
A key priority is to help clinicians know how to talk to patients about any violence they may be facing and to connect the patients with outside sources of help.
But in the end, if these patients don’t want assistance, “you respect their wishes,” Ms. Gibbs said. “They may not be ready to accept help now, but you may plant seeds so they’ll be able to accept it later on.”
Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente.
The woman arrived at the emergency department at Huntington Hospital on New York’s Long Island after she was hit by her boyfriend during an argument. Her situation raised concerns among the medical staff, which had recently been trained to be on the lookout for signs of sex trafficking.
An undocumented immigrant from El Salvador, she worked at a local “cantina” frequented by immigrants. Her job was to get patrons drinks and to dance with them, but many workers in those jobs are expected to offer sex, too. Her boyfriend didn’t want her to work there and that led to the fight, one doctor recalled.
As part of the intake process, the emergency staff asked the 36-year-old woman a series of questions about whether she’d ever had sex for money or whether she had to give someone else part of what she earned, among other things. The screening questions were part of a new program at Northwell Health, a 23-hospital system in the New York metro area that includes Huntington Hospital, to train staff and provide them with tools to identify and support victims of human trafficking.
There are no hard figures for how many people are involved in human trafficking, the term used when individuals are forced to work or have sex for someone else’s commercial benefit. Polaris, a Washington-based nonprofit that advocates for these people and runs help lines for them, said calls and texts to its national hotlines have steadily ticked up in recent years, increasing the number of cases 13% between 2016 and 2017, to 8,759.
But health care providers frequently fail to recognize these patients’ situation. According to a 2014 survey of about 100 survivors of sex trafficking, 88 percent said that while they were being trafficked they had contact with a health care provider, typically someone in an emergency department.
“When trafficking victims come through the health care system but we don’t identify them, it’s a big missed opportunity,” said Santhosh K. Paulus, MD, a family physician who is the site director of the Huntington Hospital’s family medicine residency program and who started the program at Northwell.
Northwell is one of a growing number of hospitals and health care systems that are putting such programs in place. They want to alert staff to be on the lookout for trafficking, much as they watch for signs of child abuse, domestic violence, and elder abuse.
Since last spring, nearly 300 staff members at Huntington Hospital and a family clinic have received training in how to spot trafficking victims and how to help them.
Training is given not only to doctors and nurses but also to registration and reception staff, social workers and security guards. Restore NYC, an organization that assists people caught up in sex trafficking, provided the initial training to key staff, and a hospital task force trains the others. During the next few years, similar efforts will be rolled out at all of Northwell’s 23 hospitals, Paulus said.
Identifying victims of trafficking is not unlike identifying victims of other forms of violence, said Wendy L. Macias-Konstantopoulos, MD, director of the human trafficking initiative at Massachusetts General Hospital in Boston.
One of the big red flags is when people delay coming in for medical care, such as waiting weeks to come in to get an injured ankle or sexually transmitted infection checked out, Dr. Macias-Konstantopoulos said. Or it may be a pattern of injuries that don’t make sense. Sometimes people are reluctant to explain their injury, or they come in with someone who seems overbearing.
“Having a high index of suspicion is the first step,” she said. “If we’re not asking about it, we’re just not going to see it.”
Starting in October, health care providers can also use new diagnosis codes in their records that differentiate trafficking from other types of abuse. This will help track the number of victims and provide appropriate treatment.
Asking may not be enough, however. Depending on what’s going on in their lives, these patients may not be willing or ready to acknowledge that they need help, said Holly Gibbs, human trafficking response program director for Dignity Health, a health care system with nearly 40 hospitals in California, Nevada and Arizona.
Ms. Gibbs knows the issue well. She was forced briefly into prostitution in Atlantic City, N.J., after meeting a man at a shopping mall as a 14-year-old and running away with him. The man persuaded Ms. Gibbs to go with him with promises of a new, glamorous life as a musician or model. At the time, Ms. Gibbs said, she thought that what happened to her was her own fault, a result of choices she made. No health care or law enforcement professional connected her to social services that could have helped her understand otherwise. She was reunited with her family by law enforcement personnel, who arrested the man. He was later convicted.
Dignity Health has implemented a human trafficking response program in the emergency departments and labor and delivery areas of each of its hospitals. Now it’s rolling out the program at clinics and physicians’ offices as well.
A key priority is to help clinicians know how to talk to patients about any violence they may be facing and to connect the patients with outside sources of help.
But in the end, if these patients don’t want assistance, “you respect their wishes,” Ms. Gibbs said. “They may not be ready to accept help now, but you may plant seeds so they’ll be able to accept it later on.”
Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente.
Questions abound about availability, legality of new cannabis-derived epilepsy drug
The first-ever federal approval of a marijuana-derived drug, Epidiolex (cannabidiol), comes with a cloud of complications.

Epidiolex is an oral solution of purified cannabidiol (CBD), a component of the marijuana plant that does not make people high.
According to the results of a phase 3 trial in patients with Lennox-Gastaut syndrome published earlier this year, median monthly drop seizures fell by 43.9% in a Epidiolex group compared with 21.8% in a placebo group (Lancet. 2018;391[10125]:1085-96). A 2017 study found that patients with Dravet syndrome who took the drug had fewer median convulsive seizures per month, dropping from 12.4 to 5.9, while there was barely a difference for the placebo group (N Engl J Med. 2017;376:2011-20). In both trials, patients received Epidiolex as an add-on treatment.
“There definitely are side effects such as sleepiness, abnormal liver function values, and other things that people will have to watch out for, such as interactions with other drugs,” Jacqueline A. French, MD, professor of neurology at New York University and chief scientific officer of the Epilepsy Foundation, said in an interview. “But it will definitely be a great benefit to some and a benefit to many.”
It’s not clear how the drug works, she said.
Greenwich Biosciences, the U.S. subsidiary of British drug maker GW Pharmaceuticals, plans to market Epidiolex this fall, spokesman Steve Schultz said in an interview. First, however, the U.S. Drug Enforcement Agency must reschedule the drug so it doesn’t fall under federal antimarijuana laws. It has 90 days after the FDA approval to do so, and its rescheduling is expected.
Then the focus will turn to the states, which have a crazy quilt of laws about marijuana and its medical use. “We’ve worked with the state legislators to enact laws or to modify laws to allow for our medicine to be made available,” Mr. Schultz said. “There may be one or two that we’re still working on.”
At issue: States and the District of Columbia have a wide variety of laws that appear to affect the sale of Epidiolex.
According to the website procon.org, 30 states and the District of Columbia allow medical marijuana, although the laws vary regarding what is allowed and do not address cannabis-derived pharmaceuticals.
Another 17 states have laws that address CBD specifically, often with language that allows its use to treat epilepsy in all or specific cases, according to procon.org.

In Oklahoma, an anticannabis law excludes “from the definition of marijuana ‘any federal Food and Drug Administration–approved cannabidiol drug or substance.’ ”
North Carolina attorney Rod Kight, who represents businesses in the cannabis industry, predicts that “once the federal government [via the DEA] changes the law, the states will fall in line. The only thing that would prevent them would be a lack of understanding of what’s going on.”
Meanwhile, many sellers of CBD-based products continue to promote their use as treatments for epilepsy and many other conditions.
Legal exceptions made by states for FDA-approved CBD-based drugs could affect access to unapproved CBD-based products. It’s also possible that Epidiolex could hurt sellers of cannabis products in other ways. “They’ll now have to compete with a drug made to a specific strength and purity, and that’s probably going to have some impact on their business,” William J. McNichol, adjunct professor at Rutgers Law School, Camden, N.J., said in an interview. “If you have a choice, are you going to choose aspirin I made myself in my artisan aspirin factory that the FDA never saw? Or aspirin made by Bayer?”

What’s next? For one, the FDA’s approval of Epidiolex seems likely to open the door for more cannabis-based medications. “The FDA is quite open to evaluating cannabinoid-based medicines as long as they go through their process,” said Mr. Schultz, the Greenwich Biosciences spokesman.
As for the legal front, Mr. Kight said potential changes in federal law could expand the legality of cannabis-based products by allowing them when they’re derived from “industrial hemp.” This could mean that patients with epilepsy will have more legal ways to buy CBD products.
Regardless of the legal situation, Mr. Kight said, “a physician can explain to patients across the board about how CBD might benefit them. The physician can say there are extract products that are out there and available over the counter.”
As for the maker of Epidiolex, GW Pharmaceuticals is recruiting for a phase 3 trial of a cannabinoid treatment for tuberous sclerosis complex, Mr. Schultz said. The results are due in the first half of 2019.
The company is also conducting cannabinoid research in the areas of autism and Rett syndrome, Mr. Schultz said, adding: “We also have done work in schizophrenia and in oncology and glioblastoma.”
Mr. Kight disclosed that he represents companies in the cannabis industry. Dr. French is president of the Epilepsy Study Consortium and disclosed working with multiple drug makers in the epilepsy field, including GW Pharmaceuticals. Dr. French receives fixed compensation by the consortium and is not paid by any pharmaceutical company. Mr. McNichol reported no relevant disclosures.
The first-ever federal approval of a marijuana-derived drug, Epidiolex (cannabidiol), comes with a cloud of complications.
Epidiolex is an oral solution of purified cannabidiol (CBD), a component of the marijuana plant that does not make people high.
According to the results of a phase 3 trial in patients with Lennox-Gastaut syndrome published earlier this year, median monthly drop seizures fell by 43.9% in a Epidiolex group compared with 21.8% in a placebo group (Lancet. 2018;391[10125]:1085-96). A 2017 study found that patients with Dravet syndrome who took the drug had fewer median convulsive seizures per month, dropping from 12.4 to 5.9, while there was barely a difference for the placebo group (N Engl J Med. 2017;376:2011-20). In both trials, patients received Epidiolex as an add-on treatment.
“There definitely are side effects such as sleepiness, abnormal liver function values, and other things that people will have to watch out for, such as interactions with other drugs,” Jacqueline A. French, MD, professor of neurology at New York University and chief scientific officer of the Epilepsy Foundation, said in an interview. “But it will definitely be a great benefit to some and a benefit to many.”
It’s not clear how the drug works, she said.
Greenwich Biosciences, the U.S. subsidiary of British drug maker GW Pharmaceuticals, plans to market Epidiolex this fall, spokesman Steve Schultz said in an interview. First, however, the U.S. Drug Enforcement Agency must reschedule the drug so it doesn’t fall under federal antimarijuana laws. It has 90 days after the FDA approval to do so, and its rescheduling is expected.
Then the focus will turn to the states, which have a crazy quilt of laws about marijuana and its medical use. “We’ve worked with the state legislators to enact laws or to modify laws to allow for our medicine to be made available,” Mr. Schultz said. “There may be one or two that we’re still working on.”
At issue: States and the District of Columbia have a wide variety of laws that appear to affect the sale of Epidiolex.
According to the website procon.org, 30 states and the District of Columbia allow medical marijuana, although the laws vary regarding what is allowed and do not address cannabis-derived pharmaceuticals.
Another 17 states have laws that address CBD specifically, often with language that allows its use to treat epilepsy in all or specific cases, according to procon.org.
In Oklahoma, an anticannabis law excludes “from the definition of marijuana ‘any federal Food and Drug Administration–approved cannabidiol drug or substance.’ ”
North Carolina attorney Rod Kight, who represents businesses in the cannabis industry, predicts that “once the federal government [via the DEA] changes the law, the states will fall in line. The only thing that would prevent them would be a lack of understanding of what’s going on.”
Meanwhile, many sellers of CBD-based products continue to promote their use as treatments for epilepsy and many other conditions.
Legal exceptions made by states for FDA-approved CBD-based drugs could affect access to unapproved CBD-based products. It’s also possible that Epidiolex could hurt sellers of cannabis products in other ways. “They’ll now have to compete with a drug made to a specific strength and purity, and that’s probably going to have some impact on their business,” William J. McNichol, adjunct professor at Rutgers Law School, Camden, N.J., said in an interview. “If you have a choice, are you going to choose aspirin I made myself in my artisan aspirin factory that the FDA never saw? Or aspirin made by Bayer?”
What’s next? For one, the FDA’s approval of Epidiolex seems likely to open the door for more cannabis-based medications. “The FDA is quite open to evaluating cannabinoid-based medicines as long as they go through their process,” said Mr. Schultz, the Greenwich Biosciences spokesman.
As for the legal front, Mr. Kight said potential changes in federal law could expand the legality of cannabis-based products by allowing them when they’re derived from “industrial hemp.” This could mean that patients with epilepsy will have more legal ways to buy CBD products.
Regardless of the legal situation, Mr. Kight said, “a physician can explain to patients across the board about how CBD might benefit them. The physician can say there are extract products that are out there and available over the counter.”
As for the maker of Epidiolex, GW Pharmaceuticals is recruiting for a phase 3 trial of a cannabinoid treatment for tuberous sclerosis complex, Mr. Schultz said. The results are due in the first half of 2019.
The company is also conducting cannabinoid research in the areas of autism and Rett syndrome, Mr. Schultz said, adding: “We also have done work in schizophrenia and in oncology and glioblastoma.”
Mr. Kight disclosed that he represents companies in the cannabis industry. Dr. French is president of the Epilepsy Study Consortium and disclosed working with multiple drug makers in the epilepsy field, including GW Pharmaceuticals. Dr. French receives fixed compensation by the consortium and is not paid by any pharmaceutical company. Mr. McNichol reported no relevant disclosures.
The first-ever federal approval of a marijuana-derived drug, Epidiolex (cannabidiol), comes with a cloud of complications.
Epidiolex is an oral solution of purified cannabidiol (CBD), a component of the marijuana plant that does not make people high.
According to the results of a phase 3 trial in patients with Lennox-Gastaut syndrome published earlier this year, median monthly drop seizures fell by 43.9% in a Epidiolex group compared with 21.8% in a placebo group (Lancet. 2018;391[10125]:1085-96). A 2017 study found that patients with Dravet syndrome who took the drug had fewer median convulsive seizures per month, dropping from 12.4 to 5.9, while there was barely a difference for the placebo group (N Engl J Med. 2017;376:2011-20). In both trials, patients received Epidiolex as an add-on treatment.
“There definitely are side effects such as sleepiness, abnormal liver function values, and other things that people will have to watch out for, such as interactions with other drugs,” Jacqueline A. French, MD, professor of neurology at New York University and chief scientific officer of the Epilepsy Foundation, said in an interview. “But it will definitely be a great benefit to some and a benefit to many.”
It’s not clear how the drug works, she said.
Greenwich Biosciences, the U.S. subsidiary of British drug maker GW Pharmaceuticals, plans to market Epidiolex this fall, spokesman Steve Schultz said in an interview. First, however, the U.S. Drug Enforcement Agency must reschedule the drug so it doesn’t fall under federal antimarijuana laws. It has 90 days after the FDA approval to do so, and its rescheduling is expected.
Then the focus will turn to the states, which have a crazy quilt of laws about marijuana and its medical use. “We’ve worked with the state legislators to enact laws or to modify laws to allow for our medicine to be made available,” Mr. Schultz said. “There may be one or two that we’re still working on.”
At issue: States and the District of Columbia have a wide variety of laws that appear to affect the sale of Epidiolex.
According to the website procon.org, 30 states and the District of Columbia allow medical marijuana, although the laws vary regarding what is allowed and do not address cannabis-derived pharmaceuticals.
Another 17 states have laws that address CBD specifically, often with language that allows its use to treat epilepsy in all or specific cases, according to procon.org.
In Oklahoma, an anticannabis law excludes “from the definition of marijuana ‘any federal Food and Drug Administration–approved cannabidiol drug or substance.’ ”
North Carolina attorney Rod Kight, who represents businesses in the cannabis industry, predicts that “once the federal government [via the DEA] changes the law, the states will fall in line. The only thing that would prevent them would be a lack of understanding of what’s going on.”
Meanwhile, many sellers of CBD-based products continue to promote their use as treatments for epilepsy and many other conditions.
Legal exceptions made by states for FDA-approved CBD-based drugs could affect access to unapproved CBD-based products. It’s also possible that Epidiolex could hurt sellers of cannabis products in other ways. “They’ll now have to compete with a drug made to a specific strength and purity, and that’s probably going to have some impact on their business,” William J. McNichol, adjunct professor at Rutgers Law School, Camden, N.J., said in an interview. “If you have a choice, are you going to choose aspirin I made myself in my artisan aspirin factory that the FDA never saw? Or aspirin made by Bayer?”
What’s next? For one, the FDA’s approval of Epidiolex seems likely to open the door for more cannabis-based medications. “The FDA is quite open to evaluating cannabinoid-based medicines as long as they go through their process,” said Mr. Schultz, the Greenwich Biosciences spokesman.
As for the legal front, Mr. Kight said potential changes in federal law could expand the legality of cannabis-based products by allowing them when they’re derived from “industrial hemp.” This could mean that patients with epilepsy will have more legal ways to buy CBD products.
Regardless of the legal situation, Mr. Kight said, “a physician can explain to patients across the board about how CBD might benefit them. The physician can say there are extract products that are out there and available over the counter.”
As for the maker of Epidiolex, GW Pharmaceuticals is recruiting for a phase 3 trial of a cannabinoid treatment for tuberous sclerosis complex, Mr. Schultz said. The results are due in the first half of 2019.
The company is also conducting cannabinoid research in the areas of autism and Rett syndrome, Mr. Schultz said, adding: “We also have done work in schizophrenia and in oncology and glioblastoma.”
Mr. Kight disclosed that he represents companies in the cannabis industry. Dr. French is president of the Epilepsy Study Consortium and disclosed working with multiple drug makers in the epilepsy field, including GW Pharmaceuticals. Dr. French receives fixed compensation by the consortium and is not paid by any pharmaceutical company. Mr. McNichol reported no relevant disclosures.