User login
Rapid EGFR testing reduces time to therapy for patients with NSCLC
compared with current next-generation sequencing (NGS) work flows, according to a recent study.
Rapid testing maintained concordance with NGS while cutting median turnaround time in half. In addition, an ultrarapid, next-day work flow allowed several highly symptomatic patients with NSCLC to start therapy within 1 week of biopsy, reported Ibiayi Dagogo-Jack, MD, of the Massachusetts General Hospital Waltham Cancer Center and her coauthors.
Molecular testing for patients with NSCLC is necessary to select appropriate therapies; however, “genotyping by NGS requires complex bioinformatics that can create treatment delays,” the investigators wrote in JCO Precision Oncology. “Some patients with NSCLC present with symptomatic disease that requires initiation of treatment before molecular testing results are available. To our knowledge, the impact of molecular testing turnaround time on clinical decision making has not been formally assessed in NSCLC.”
For rapid testing, biopsies were collected from 243 hospitalized patients with newly diagnosed, metastatic NSCLC and were then fixed in formalin. An EGFR-specific polymerase chain reaction assay was then used to identify exon 19 deletions and L858R mutations. The ultrarapid testing phase of the study involved eight highly symptomatic patients with newly diagnosed NSCLC. Biopsy samples in this group were frozen instead of fixed. Both the rapid and ultrarapid testing results were compared with 121 historical biopsies that had been genotyped by NGS.
The rapid testing’s work flow reduced TTI from 37 days to 22 days (P = .01) while maintaining high concordance with NGS (sensitivity, 98%; specificity, 100%). Median turnaround time dropped in half from 14 days to 7 days with rapid testing (P less than .001); ultrarapid testing had a turnaround time of approximately 1 day. The median TTI for the ultrarapid group was 9 days, and several patients were able to begin therapy within 1 week.
The investigators concluded that rapid and ultrarapid testing are a cost-effective, reliable means for decreasing turnaround time and TTI for patients with newly diagnosed, metastatic NSCLC. However, concurrent NGS genotyping should still be performed because NGS results may influence future therapy sequencing. With the ultrarapid work flow, additional sampling is recommended to offset the risk of tissue failure. The investigators noted that “the growing number of actionable targets substantiates the need for diagnostic strategies that expedite molecular analysis.”
The study was funded in part by the National Institutes of Health. Authors reported compensation from Merck, Boehringer Ingelheim, Takeda, and others.
SOURCE: Dagogo-Jack et al. JCO Precis Oncol. 2018 Jul 24. doi: 10.1200/PO.17.00299.
compared with current next-generation sequencing (NGS) work flows, according to a recent study.
Rapid testing maintained concordance with NGS while cutting median turnaround time in half. In addition, an ultrarapid, next-day work flow allowed several highly symptomatic patients with NSCLC to start therapy within 1 week of biopsy, reported Ibiayi Dagogo-Jack, MD, of the Massachusetts General Hospital Waltham Cancer Center and her coauthors.
Molecular testing for patients with NSCLC is necessary to select appropriate therapies; however, “genotyping by NGS requires complex bioinformatics that can create treatment delays,” the investigators wrote in JCO Precision Oncology. “Some patients with NSCLC present with symptomatic disease that requires initiation of treatment before molecular testing results are available. To our knowledge, the impact of molecular testing turnaround time on clinical decision making has not been formally assessed in NSCLC.”
For rapid testing, biopsies were collected from 243 hospitalized patients with newly diagnosed, metastatic NSCLC and were then fixed in formalin. An EGFR-specific polymerase chain reaction assay was then used to identify exon 19 deletions and L858R mutations. The ultrarapid testing phase of the study involved eight highly symptomatic patients with newly diagnosed NSCLC. Biopsy samples in this group were frozen instead of fixed. Both the rapid and ultrarapid testing results were compared with 121 historical biopsies that had been genotyped by NGS.
The rapid testing’s work flow reduced TTI from 37 days to 22 days (P = .01) while maintaining high concordance with NGS (sensitivity, 98%; specificity, 100%). Median turnaround time dropped in half from 14 days to 7 days with rapid testing (P less than .001); ultrarapid testing had a turnaround time of approximately 1 day. The median TTI for the ultrarapid group was 9 days, and several patients were able to begin therapy within 1 week.
The investigators concluded that rapid and ultrarapid testing are a cost-effective, reliable means for decreasing turnaround time and TTI for patients with newly diagnosed, metastatic NSCLC. However, concurrent NGS genotyping should still be performed because NGS results may influence future therapy sequencing. With the ultrarapid work flow, additional sampling is recommended to offset the risk of tissue failure. The investigators noted that “the growing number of actionable targets substantiates the need for diagnostic strategies that expedite molecular analysis.”
The study was funded in part by the National Institutes of Health. Authors reported compensation from Merck, Boehringer Ingelheim, Takeda, and others.
SOURCE: Dagogo-Jack et al. JCO Precis Oncol. 2018 Jul 24. doi: 10.1200/PO.17.00299.
compared with current next-generation sequencing (NGS) work flows, according to a recent study.
Rapid testing maintained concordance with NGS while cutting median turnaround time in half. In addition, an ultrarapid, next-day work flow allowed several highly symptomatic patients with NSCLC to start therapy within 1 week of biopsy, reported Ibiayi Dagogo-Jack, MD, of the Massachusetts General Hospital Waltham Cancer Center and her coauthors.
Molecular testing for patients with NSCLC is necessary to select appropriate therapies; however, “genotyping by NGS requires complex bioinformatics that can create treatment delays,” the investigators wrote in JCO Precision Oncology. “Some patients with NSCLC present with symptomatic disease that requires initiation of treatment before molecular testing results are available. To our knowledge, the impact of molecular testing turnaround time on clinical decision making has not been formally assessed in NSCLC.”
For rapid testing, biopsies were collected from 243 hospitalized patients with newly diagnosed, metastatic NSCLC and were then fixed in formalin. An EGFR-specific polymerase chain reaction assay was then used to identify exon 19 deletions and L858R mutations. The ultrarapid testing phase of the study involved eight highly symptomatic patients with newly diagnosed NSCLC. Biopsy samples in this group were frozen instead of fixed. Both the rapid and ultrarapid testing results were compared with 121 historical biopsies that had been genotyped by NGS.
The rapid testing’s work flow reduced TTI from 37 days to 22 days (P = .01) while maintaining high concordance with NGS (sensitivity, 98%; specificity, 100%). Median turnaround time dropped in half from 14 days to 7 days with rapid testing (P less than .001); ultrarapid testing had a turnaround time of approximately 1 day. The median TTI for the ultrarapid group was 9 days, and several patients were able to begin therapy within 1 week.
The investigators concluded that rapid and ultrarapid testing are a cost-effective, reliable means for decreasing turnaround time and TTI for patients with newly diagnosed, metastatic NSCLC. However, concurrent NGS genotyping should still be performed because NGS results may influence future therapy sequencing. With the ultrarapid work flow, additional sampling is recommended to offset the risk of tissue failure. The investigators noted that “the growing number of actionable targets substantiates the need for diagnostic strategies that expedite molecular analysis.”
The study was funded in part by the National Institutes of Health. Authors reported compensation from Merck, Boehringer Ingelheim, Takeda, and others.
SOURCE: Dagogo-Jack et al. JCO Precis Oncol. 2018 Jul 24. doi: 10.1200/PO.17.00299.
FROM JCO PRECISION ONCOLOGY
Key clinical point: Rapid EGFR-specific genotyping for patients with NSCLC reduced time to initiation of targeted therapy while maintaining concordance with next-generation sequencing (NGS).
Major finding: Rapid EGFR-specific testing in patients with NSCLC reduced median time to initiation (TTI) of therapy from 37 days to 22 days (P =.01).
Study details: A multiphase, prospective and retrospective study comparing rapid EGFR-specific testing (n = 243), ultrarapid EGFR-specific testing (n = 8), and standard NGS (n = 121) for patients with NSCLC.
Disclosures: The study was funded in part by the National Institutes of Health. Authors reported compensation from Merck, Boehringer Ingelheim, Takeda, and others.
Source: Dagogo-Jack I et al. JCO Precis Oncol. 2018 Jul 24. doi: 10.1200/PO.17.00299.
Preventative Care in Orthopedics: Treating Injuries Before They Happen
By 2025, it is estimated that the annual cost of treating osteoporosis-related fractures in the United States will be 25 billion dollars, which is 10 billion dollars more than was spent in 2010.1 As healthcare costs in the United States continue to skyrocket, it is imperative that orthopedic surgeons take an active role in avoiding preventable injury and disease. For orthopedic surgeons, preventative medicine will include promoting bone health and educating patients on injury prevention. By incorporating these principles into residency and fellowship education, and by leveraging the electronic medical record to support preventive care through systematic reminders, orthopedic surgeons have a critical opportunity to take a leading role in promoting prevention to our patients.
In 2009, the American Orthopaedic Association (AOA) launched a “Own the Bone” campaign, a national quality improvement program designed to optimize the treatment of osteoporosis.2 This program came about following the Surgeon General’s call, in 2004, for orthopedic surgeons to take a more active role in treating osteoporosis. The program primarily aims to improve treatment of osteoporosis after a fragility fracture in an inpatient setting. Early results from a 2010 follow-up study showed that the new emphasis on prevention inspired by this program is effective. As compared with patients who had osteoporosis work-up and treatment initiated during their hospital admission, the group of patients who were referred for osteoporosis treatment after discharge were found to have a significantly lower rate of diagnosis and treatment.3 The loss of aftercare for patients who do not obtain immediate diagnosis and treatment for osteoporosis can and should be avoided. Many hospitals now have hip fracture services with multidisciplinary input. The successful outcomes of these programs include shorter times to the operating room, shorter hospital stays, decreased readmission, and decreased 30-day mortality.4-6 These services provide an excellent opportunity to ensure that each patient has initiated management of osteoporosis before discharge. Ideally, patients would be scheduled for bone mineral density testing prior to leaving the hospital, when applicable, and would begin calcium and vitamin D supplementation or bisphosphonate treatment in the hospital, when appropriate. As part of these hip fracture services, a goal of clearly initiating or managing treatment for osteoporosis should be routinely addressed.
While patients presenting with hip fractures are an easily identifiable high-risk population, other patients present in an outpatient setting following fragility fractures, such as distal radius or vertebral compression fractures. These patients should be considered for osteoporosis work-up and counseled accordingly. A recent study compared the efficacy of the orthopedic surgeon initiating bone mineral density testing after a distal radius fracture, compared with referring the patient back to their primary care physician for testing. The study found a significantly higher rate of patients going on to bone mineral density testing when the surgeon initiated this process.7 In the era of improved digital communication, the outpatient setting offers an opportunity for clinicians to communicate with patients’ primary care physicians and initiate a multidisciplinary approach to bone health and prevention. In the outpatient setting, the orthopedist can address nutritional issues and screening on a repeated basis. Studies have demonstrated that physician counseling can be very effective in changing behavior and helping patients to stop using tobacco.8 In this vein, efforts by the physician to encourage calcium and vitamin D intake and weight-bearing exercise have the potential to be very effective.
Programs such as “Own the Bone” are crucial to orthopedists’ treatment of osteoporosis, but prevention of bone disease and fragility fracture must extend even further. Individual practitioners must be cognizant that many patients may benefit from outpatient diagnosis of osteoporosis and initiation of appropriate treatment, before fragility fractures occur. Moreover, although patients at high risk include post-menopausal women, orthopedists need to be consistently aware of osteoporosis as a disease of both genders. An estimated 2.8 million men in the United States have osteoporosis.9 A 2012 study published out of Washington, DC found a significant disparity in the rate of osteoporosis screening between men and women. Among the elderly men and women in their patient population, 60% of women underwent screening compared with only 18.4% of men.10 This gender disparity potentially represents significant physician bias regarding the risk of osteoporosis and offers an important opportunity for orthopedic surgeons to improve preventative care for this population.
Preventative care in terms of advocating for bone health should not be limited to patients presenting with fragility fractures. Education regarding smoking cessation, resistance exercise, and calcium intake are relevant to many orthopedic patients. With the advent of the electronic medical record system, a simple intervention could easily ensure that patients report on their calcium intake. A trial published in 2006 found that a simple reminder from the electronic medical record improved osteoporosis management following a fragility fracture.11 This type of intervention could certainly be expanded to include counseling on calcium and vitamin D for any orthopedic patient.
Another area in which orthopedic surgeons have an opportunity to practice good preventative care is injury prevention. Several studies examining fall prevention among the elderly have shown that physical therapy or exercise may decrease the rate of falls.12 Promotion of activity and therapy among high-risk patients by orthopedic surgeons may help to reduce fracture incidence. Injury prevention is also relevant to young, healthy patients. It is well established that neuromuscular training helps to prevent anterior cruciate ligament injuries.13 Orthopedic surgeons have an opportunity during sports physicals or as team physicians to help promote injury prevention strategies. Discussion of training regimens may prevent overuse injuries among athletes. Moreover, faced with many patients who present with significant musculoskeletal trauma, orthopedic surgeons have the opportunity to offer education regarding motorcycle helmets, seatbelt use, and avoidance of drunk driving.
New orthopedic residency educational goals were recently published to include core competencies in resident education. Among these goals is to educate residents on care of a patient with hip fracture, including counseling and management of osteoporosis.14 These milestones could be expanded to include a thorough understanding of bone health. Residents should be able to make nutritional recommendations for any patient seen as an inpatient or outpatient, identify when a referral to an endocrinologist is needed, and educate patients regarding injury and fall prevention.
As healthcare expenditures rise, so does the impetus for physicians to work to improve efficiency in the healthcare system. Furthermore, the best possible care for our patients is to prevent injury and disability before it arises, rather than to depend on our ability to intervene after the fact. Residencies and training programs should work to incorporate preventative strategies into trainee education. Hospitals and outpatient settings should include a basic bone health questionnaire in the electronic medical record. The identification and management of risk factors for injury has the potential to help our patients and to help our healthcare system, but such intervention needs to start with the clinician.
- Burge R, Dawson-Hughes B, Solomon DH, Wong JB, King A, Tosteson A. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005-2025. J Bone Miner Res. 2007;22(3):465-475. doi:10.1359/jbmr.061113.
- Bunta AD. It is time for everyone to own the bone. Osteoporos Int. 2011;22 Suppl 3:477-482. doi:10.1007/s00198-011-1704-0.
- Edwards BJ, Koval K, Bunta AD, et al. Addressing secondary prevention of osteoporosis in fracture care: follow-up to “own the bone.” J Bone Joint Surg Am. 2011;93(15):e87. doi:10.2106/JBJS.I.00540.
- Sivakumar BS, McDermott LM, Bell JJ, Pulle CR, Jayamaha S, Ottley MC. Dedicated hip fracture service: implementing a novel model of care. ANZ J Surg. 2013;83(7-8):559-563. doi:10.1111/j.1445-2197.2012.06201.x.
- Khasraghi FA, Christmas C, Lee EJ, Mears SC, Wenz JF Sr. Effectiveness of a multidisciplinary team approach to hip fracture management. J Surg Orthop Adv. 2005;14(1):27-31.
- Vidan M, Serra JA, Moreno C, Riquelme G, Ortiz J. Efficacy of a comprehensive geriatric intervention in older patients hospitalized for hip fracture: a randomized, controlled trial. J Am Geriatr Soc. 2005;53(9):1476-1482. doi:10.1111/j.1532-5415.2005.53466.x.
- Rozental TD, Makhni EC, Day CS, Bouxsein ML. Improving evaluation and treatment for osteoporosis following distal radial fractures. A prospective randomized intervention. J Bone Joint Surg Am. 2008;90(5):953-961. doi:10.2106/JBJS.G.01121.
- Gorin SS, Heck JE. Meta-analysis of the efficacy of tobacco counseling by health care providers. Cancer Epidemiol Biomarkers Prev. 2004;13(12):2012-2022.
- Cawthon PM. Gender differences in osteoporosis and fractures. Clin Orthop Relat Res. 2011;469(7):1900-1905. doi:10.1007/s11999-011-1780-7.
- Alswat K, Adler SM. Gender differences in osteoporosis screening: retrospective analysis. Arch Osteoporos. 2012;7:311-313. doi:10.1007/s11657-012-0113-0.
- Feldstein A, Elmer PJ, Smith DH, et al. Electronic medical record reminder improves osteoporosis management after a fracture: a randomized, controlled trial. J Am Geriatr Soc. 2006;54(3):450-457. doi:10.1111/j.1532-5415.2005.00618.x.
- Suzuki T, Kim H, Yoshida H, Ishizaki T. Randomized controlled trial of exercise intervention for the prevention of falls in community-dwelling elderly Japanese women. J Bone Miner Metab. 2004;22(6):602-611. doi:10.1007/s00774-004-0530-2.
- Hewett TE, Ford KR, Myer GD. Anterior cruciate ligament injuries in female athletes: Part 2, a meta-analysis of neuromuscular interventions aimed at injury prevention. Am J Sports Med. 2006;34(3):490-498. doi:10.1177/0363546505282619.
- Stern PJ, Albanese S, Bostrom M, et al. Orthopaedic surgery milestones. J Grad Med Educ. 2013;5(1 Suppl 1):36-58. doi:10.4300/JGME-05-01s1-05.
By 2025, it is estimated that the annual cost of treating osteoporosis-related fractures in the United States will be 25 billion dollars, which is 10 billion dollars more than was spent in 2010.1 As healthcare costs in the United States continue to skyrocket, it is imperative that orthopedic surgeons take an active role in avoiding preventable injury and disease. For orthopedic surgeons, preventative medicine will include promoting bone health and educating patients on injury prevention. By incorporating these principles into residency and fellowship education, and by leveraging the electronic medical record to support preventive care through systematic reminders, orthopedic surgeons have a critical opportunity to take a leading role in promoting prevention to our patients.
In 2009, the American Orthopaedic Association (AOA) launched a “Own the Bone” campaign, a national quality improvement program designed to optimize the treatment of osteoporosis.2 This program came about following the Surgeon General’s call, in 2004, for orthopedic surgeons to take a more active role in treating osteoporosis. The program primarily aims to improve treatment of osteoporosis after a fragility fracture in an inpatient setting. Early results from a 2010 follow-up study showed that the new emphasis on prevention inspired by this program is effective. As compared with patients who had osteoporosis work-up and treatment initiated during their hospital admission, the group of patients who were referred for osteoporosis treatment after discharge were found to have a significantly lower rate of diagnosis and treatment.3 The loss of aftercare for patients who do not obtain immediate diagnosis and treatment for osteoporosis can and should be avoided. Many hospitals now have hip fracture services with multidisciplinary input. The successful outcomes of these programs include shorter times to the operating room, shorter hospital stays, decreased readmission, and decreased 30-day mortality.4-6 These services provide an excellent opportunity to ensure that each patient has initiated management of osteoporosis before discharge. Ideally, patients would be scheduled for bone mineral density testing prior to leaving the hospital, when applicable, and would begin calcium and vitamin D supplementation or bisphosphonate treatment in the hospital, when appropriate. As part of these hip fracture services, a goal of clearly initiating or managing treatment for osteoporosis should be routinely addressed.
While patients presenting with hip fractures are an easily identifiable high-risk population, other patients present in an outpatient setting following fragility fractures, such as distal radius or vertebral compression fractures. These patients should be considered for osteoporosis work-up and counseled accordingly. A recent study compared the efficacy of the orthopedic surgeon initiating bone mineral density testing after a distal radius fracture, compared with referring the patient back to their primary care physician for testing. The study found a significantly higher rate of patients going on to bone mineral density testing when the surgeon initiated this process.7 In the era of improved digital communication, the outpatient setting offers an opportunity for clinicians to communicate with patients’ primary care physicians and initiate a multidisciplinary approach to bone health and prevention. In the outpatient setting, the orthopedist can address nutritional issues and screening on a repeated basis. Studies have demonstrated that physician counseling can be very effective in changing behavior and helping patients to stop using tobacco.8 In this vein, efforts by the physician to encourage calcium and vitamin D intake and weight-bearing exercise have the potential to be very effective.
Programs such as “Own the Bone” are crucial to orthopedists’ treatment of osteoporosis, but prevention of bone disease and fragility fracture must extend even further. Individual practitioners must be cognizant that many patients may benefit from outpatient diagnosis of osteoporosis and initiation of appropriate treatment, before fragility fractures occur. Moreover, although patients at high risk include post-menopausal women, orthopedists need to be consistently aware of osteoporosis as a disease of both genders. An estimated 2.8 million men in the United States have osteoporosis.9 A 2012 study published out of Washington, DC found a significant disparity in the rate of osteoporosis screening between men and women. Among the elderly men and women in their patient population, 60% of women underwent screening compared with only 18.4% of men.10 This gender disparity potentially represents significant physician bias regarding the risk of osteoporosis and offers an important opportunity for orthopedic surgeons to improve preventative care for this population.
Preventative care in terms of advocating for bone health should not be limited to patients presenting with fragility fractures. Education regarding smoking cessation, resistance exercise, and calcium intake are relevant to many orthopedic patients. With the advent of the electronic medical record system, a simple intervention could easily ensure that patients report on their calcium intake. A trial published in 2006 found that a simple reminder from the electronic medical record improved osteoporosis management following a fragility fracture.11 This type of intervention could certainly be expanded to include counseling on calcium and vitamin D for any orthopedic patient.
Another area in which orthopedic surgeons have an opportunity to practice good preventative care is injury prevention. Several studies examining fall prevention among the elderly have shown that physical therapy or exercise may decrease the rate of falls.12 Promotion of activity and therapy among high-risk patients by orthopedic surgeons may help to reduce fracture incidence. Injury prevention is also relevant to young, healthy patients. It is well established that neuromuscular training helps to prevent anterior cruciate ligament injuries.13 Orthopedic surgeons have an opportunity during sports physicals or as team physicians to help promote injury prevention strategies. Discussion of training regimens may prevent overuse injuries among athletes. Moreover, faced with many patients who present with significant musculoskeletal trauma, orthopedic surgeons have the opportunity to offer education regarding motorcycle helmets, seatbelt use, and avoidance of drunk driving.
New orthopedic residency educational goals were recently published to include core competencies in resident education. Among these goals is to educate residents on care of a patient with hip fracture, including counseling and management of osteoporosis.14 These milestones could be expanded to include a thorough understanding of bone health. Residents should be able to make nutritional recommendations for any patient seen as an inpatient or outpatient, identify when a referral to an endocrinologist is needed, and educate patients regarding injury and fall prevention.
As healthcare expenditures rise, so does the impetus for physicians to work to improve efficiency in the healthcare system. Furthermore, the best possible care for our patients is to prevent injury and disability before it arises, rather than to depend on our ability to intervene after the fact. Residencies and training programs should work to incorporate preventative strategies into trainee education. Hospitals and outpatient settings should include a basic bone health questionnaire in the electronic medical record. The identification and management of risk factors for injury has the potential to help our patients and to help our healthcare system, but such intervention needs to start with the clinician.
By 2025, it is estimated that the annual cost of treating osteoporosis-related fractures in the United States will be 25 billion dollars, which is 10 billion dollars more than was spent in 2010.1 As healthcare costs in the United States continue to skyrocket, it is imperative that orthopedic surgeons take an active role in avoiding preventable injury and disease. For orthopedic surgeons, preventative medicine will include promoting bone health and educating patients on injury prevention. By incorporating these principles into residency and fellowship education, and by leveraging the electronic medical record to support preventive care through systematic reminders, orthopedic surgeons have a critical opportunity to take a leading role in promoting prevention to our patients.
In 2009, the American Orthopaedic Association (AOA) launched a “Own the Bone” campaign, a national quality improvement program designed to optimize the treatment of osteoporosis.2 This program came about following the Surgeon General’s call, in 2004, for orthopedic surgeons to take a more active role in treating osteoporosis. The program primarily aims to improve treatment of osteoporosis after a fragility fracture in an inpatient setting. Early results from a 2010 follow-up study showed that the new emphasis on prevention inspired by this program is effective. As compared with patients who had osteoporosis work-up and treatment initiated during their hospital admission, the group of patients who were referred for osteoporosis treatment after discharge were found to have a significantly lower rate of diagnosis and treatment.3 The loss of aftercare for patients who do not obtain immediate diagnosis and treatment for osteoporosis can and should be avoided. Many hospitals now have hip fracture services with multidisciplinary input. The successful outcomes of these programs include shorter times to the operating room, shorter hospital stays, decreased readmission, and decreased 30-day mortality.4-6 These services provide an excellent opportunity to ensure that each patient has initiated management of osteoporosis before discharge. Ideally, patients would be scheduled for bone mineral density testing prior to leaving the hospital, when applicable, and would begin calcium and vitamin D supplementation or bisphosphonate treatment in the hospital, when appropriate. As part of these hip fracture services, a goal of clearly initiating or managing treatment for osteoporosis should be routinely addressed.
While patients presenting with hip fractures are an easily identifiable high-risk population, other patients present in an outpatient setting following fragility fractures, such as distal radius or vertebral compression fractures. These patients should be considered for osteoporosis work-up and counseled accordingly. A recent study compared the efficacy of the orthopedic surgeon initiating bone mineral density testing after a distal radius fracture, compared with referring the patient back to their primary care physician for testing. The study found a significantly higher rate of patients going on to bone mineral density testing when the surgeon initiated this process.7 In the era of improved digital communication, the outpatient setting offers an opportunity for clinicians to communicate with patients’ primary care physicians and initiate a multidisciplinary approach to bone health and prevention. In the outpatient setting, the orthopedist can address nutritional issues and screening on a repeated basis. Studies have demonstrated that physician counseling can be very effective in changing behavior and helping patients to stop using tobacco.8 In this vein, efforts by the physician to encourage calcium and vitamin D intake and weight-bearing exercise have the potential to be very effective.
Programs such as “Own the Bone” are crucial to orthopedists’ treatment of osteoporosis, but prevention of bone disease and fragility fracture must extend even further. Individual practitioners must be cognizant that many patients may benefit from outpatient diagnosis of osteoporosis and initiation of appropriate treatment, before fragility fractures occur. Moreover, although patients at high risk include post-menopausal women, orthopedists need to be consistently aware of osteoporosis as a disease of both genders. An estimated 2.8 million men in the United States have osteoporosis.9 A 2012 study published out of Washington, DC found a significant disparity in the rate of osteoporosis screening between men and women. Among the elderly men and women in their patient population, 60% of women underwent screening compared with only 18.4% of men.10 This gender disparity potentially represents significant physician bias regarding the risk of osteoporosis and offers an important opportunity for orthopedic surgeons to improve preventative care for this population.
Preventative care in terms of advocating for bone health should not be limited to patients presenting with fragility fractures. Education regarding smoking cessation, resistance exercise, and calcium intake are relevant to many orthopedic patients. With the advent of the electronic medical record system, a simple intervention could easily ensure that patients report on their calcium intake. A trial published in 2006 found that a simple reminder from the electronic medical record improved osteoporosis management following a fragility fracture.11 This type of intervention could certainly be expanded to include counseling on calcium and vitamin D for any orthopedic patient.
Another area in which orthopedic surgeons have an opportunity to practice good preventative care is injury prevention. Several studies examining fall prevention among the elderly have shown that physical therapy or exercise may decrease the rate of falls.12 Promotion of activity and therapy among high-risk patients by orthopedic surgeons may help to reduce fracture incidence. Injury prevention is also relevant to young, healthy patients. It is well established that neuromuscular training helps to prevent anterior cruciate ligament injuries.13 Orthopedic surgeons have an opportunity during sports physicals or as team physicians to help promote injury prevention strategies. Discussion of training regimens may prevent overuse injuries among athletes. Moreover, faced with many patients who present with significant musculoskeletal trauma, orthopedic surgeons have the opportunity to offer education regarding motorcycle helmets, seatbelt use, and avoidance of drunk driving.
New orthopedic residency educational goals were recently published to include core competencies in resident education. Among these goals is to educate residents on care of a patient with hip fracture, including counseling and management of osteoporosis.14 These milestones could be expanded to include a thorough understanding of bone health. Residents should be able to make nutritional recommendations for any patient seen as an inpatient or outpatient, identify when a referral to an endocrinologist is needed, and educate patients regarding injury and fall prevention.
As healthcare expenditures rise, so does the impetus for physicians to work to improve efficiency in the healthcare system. Furthermore, the best possible care for our patients is to prevent injury and disability before it arises, rather than to depend on our ability to intervene after the fact. Residencies and training programs should work to incorporate preventative strategies into trainee education. Hospitals and outpatient settings should include a basic bone health questionnaire in the electronic medical record. The identification and management of risk factors for injury has the potential to help our patients and to help our healthcare system, but such intervention needs to start with the clinician.
- Burge R, Dawson-Hughes B, Solomon DH, Wong JB, King A, Tosteson A. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005-2025. J Bone Miner Res. 2007;22(3):465-475. doi:10.1359/jbmr.061113.
- Bunta AD. It is time for everyone to own the bone. Osteoporos Int. 2011;22 Suppl 3:477-482. doi:10.1007/s00198-011-1704-0.
- Edwards BJ, Koval K, Bunta AD, et al. Addressing secondary prevention of osteoporosis in fracture care: follow-up to “own the bone.” J Bone Joint Surg Am. 2011;93(15):e87. doi:10.2106/JBJS.I.00540.
- Sivakumar BS, McDermott LM, Bell JJ, Pulle CR, Jayamaha S, Ottley MC. Dedicated hip fracture service: implementing a novel model of care. ANZ J Surg. 2013;83(7-8):559-563. doi:10.1111/j.1445-2197.2012.06201.x.
- Khasraghi FA, Christmas C, Lee EJ, Mears SC, Wenz JF Sr. Effectiveness of a multidisciplinary team approach to hip fracture management. J Surg Orthop Adv. 2005;14(1):27-31.
- Vidan M, Serra JA, Moreno C, Riquelme G, Ortiz J. Efficacy of a comprehensive geriatric intervention in older patients hospitalized for hip fracture: a randomized, controlled trial. J Am Geriatr Soc. 2005;53(9):1476-1482. doi:10.1111/j.1532-5415.2005.53466.x.
- Rozental TD, Makhni EC, Day CS, Bouxsein ML. Improving evaluation and treatment for osteoporosis following distal radial fractures. A prospective randomized intervention. J Bone Joint Surg Am. 2008;90(5):953-961. doi:10.2106/JBJS.G.01121.
- Gorin SS, Heck JE. Meta-analysis of the efficacy of tobacco counseling by health care providers. Cancer Epidemiol Biomarkers Prev. 2004;13(12):2012-2022.
- Cawthon PM. Gender differences in osteoporosis and fractures. Clin Orthop Relat Res. 2011;469(7):1900-1905. doi:10.1007/s11999-011-1780-7.
- Alswat K, Adler SM. Gender differences in osteoporosis screening: retrospective analysis. Arch Osteoporos. 2012;7:311-313. doi:10.1007/s11657-012-0113-0.
- Feldstein A, Elmer PJ, Smith DH, et al. Electronic medical record reminder improves osteoporosis management after a fracture: a randomized, controlled trial. J Am Geriatr Soc. 2006;54(3):450-457. doi:10.1111/j.1532-5415.2005.00618.x.
- Suzuki T, Kim H, Yoshida H, Ishizaki T. Randomized controlled trial of exercise intervention for the prevention of falls in community-dwelling elderly Japanese women. J Bone Miner Metab. 2004;22(6):602-611. doi:10.1007/s00774-004-0530-2.
- Hewett TE, Ford KR, Myer GD. Anterior cruciate ligament injuries in female athletes: Part 2, a meta-analysis of neuromuscular interventions aimed at injury prevention. Am J Sports Med. 2006;34(3):490-498. doi:10.1177/0363546505282619.
- Stern PJ, Albanese S, Bostrom M, et al. Orthopaedic surgery milestones. J Grad Med Educ. 2013;5(1 Suppl 1):36-58. doi:10.4300/JGME-05-01s1-05.
- Burge R, Dawson-Hughes B, Solomon DH, Wong JB, King A, Tosteson A. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005-2025. J Bone Miner Res. 2007;22(3):465-475. doi:10.1359/jbmr.061113.
- Bunta AD. It is time for everyone to own the bone. Osteoporos Int. 2011;22 Suppl 3:477-482. doi:10.1007/s00198-011-1704-0.
- Edwards BJ, Koval K, Bunta AD, et al. Addressing secondary prevention of osteoporosis in fracture care: follow-up to “own the bone.” J Bone Joint Surg Am. 2011;93(15):e87. doi:10.2106/JBJS.I.00540.
- Sivakumar BS, McDermott LM, Bell JJ, Pulle CR, Jayamaha S, Ottley MC. Dedicated hip fracture service: implementing a novel model of care. ANZ J Surg. 2013;83(7-8):559-563. doi:10.1111/j.1445-2197.2012.06201.x.
- Khasraghi FA, Christmas C, Lee EJ, Mears SC, Wenz JF Sr. Effectiveness of a multidisciplinary team approach to hip fracture management. J Surg Orthop Adv. 2005;14(1):27-31.
- Vidan M, Serra JA, Moreno C, Riquelme G, Ortiz J. Efficacy of a comprehensive geriatric intervention in older patients hospitalized for hip fracture: a randomized, controlled trial. J Am Geriatr Soc. 2005;53(9):1476-1482. doi:10.1111/j.1532-5415.2005.53466.x.
- Rozental TD, Makhni EC, Day CS, Bouxsein ML. Improving evaluation and treatment for osteoporosis following distal radial fractures. A prospective randomized intervention. J Bone Joint Surg Am. 2008;90(5):953-961. doi:10.2106/JBJS.G.01121.
- Gorin SS, Heck JE. Meta-analysis of the efficacy of tobacco counseling by health care providers. Cancer Epidemiol Biomarkers Prev. 2004;13(12):2012-2022.
- Cawthon PM. Gender differences in osteoporosis and fractures. Clin Orthop Relat Res. 2011;469(7):1900-1905. doi:10.1007/s11999-011-1780-7.
- Alswat K, Adler SM. Gender differences in osteoporosis screening: retrospective analysis. Arch Osteoporos. 2012;7:311-313. doi:10.1007/s11657-012-0113-0.
- Feldstein A, Elmer PJ, Smith DH, et al. Electronic medical record reminder improves osteoporosis management after a fracture: a randomized, controlled trial. J Am Geriatr Soc. 2006;54(3):450-457. doi:10.1111/j.1532-5415.2005.00618.x.
- Suzuki T, Kim H, Yoshida H, Ishizaki T. Randomized controlled trial of exercise intervention for the prevention of falls in community-dwelling elderly Japanese women. J Bone Miner Metab. 2004;22(6):602-611. doi:10.1007/s00774-004-0530-2.
- Hewett TE, Ford KR, Myer GD. Anterior cruciate ligament injuries in female athletes: Part 2, a meta-analysis of neuromuscular interventions aimed at injury prevention. Am J Sports Med. 2006;34(3):490-498. doi:10.1177/0363546505282619.
- Stern PJ, Albanese S, Bostrom M, et al. Orthopaedic surgery milestones. J Grad Med Educ. 2013;5(1 Suppl 1):36-58. doi:10.4300/JGME-05-01s1-05.
Novel HIV vaccine induces durable immune responses
AMSTERDAM – that had identified the most effective dosing strategy for the vaccine.
The 96-week follow-up data showed durable humoral and cellular immunity induction by the vaccine and its associated booster, a durable breadth of immune responses to the multiple HIV clades that the vaccine targets, and no serious or grade 3 or 4 adverse effects in the 393 people who participated in the early-phase study, Frank L. Tomaka, MD, said at the 22nd International AIDS Conference.
The 96-week results he reported came from follow-up of the 393 people who received one of several different dosing regimens for an engineered, mosaic HIV vaccine. The vaccine incorporates genes for three different HIV envelope antigens that contain components drawn from several different HIV clades (to induce more broadly protective immunity) into a serotype 26 adenovirus. The immunization regimen also includes treatment with HIV glycoprotein 140 as a booster agent. Dr. Tomaka and his associates reported the primary endpoints from this placebo-controlled study, APPROACH, measured just after the fourth and final immunizing regimen at 48 weeks after the first treatment in a recently published article (Lancet. 2018 Jul 21;392[10143]:232-43).
As part of the study, the investigators administered the vaccine and booster to rhesus monkeys and found that the regimens produced a pattern of immune responses in the monkeys similar to that seen in people. When the monkeys that received the regimen that performed best in people received six monthly challenges with a simian-human immunodeficiency virus that’s related to HIV, the researchers found a 67% efficacy for protection against infection. These “very encouraging” findings led the company developing the vaccine to launch in November 2017 a phase IIb trial, named Imbokodo, in five African countries, with a plan to enroll 2,600 people, Dr. Tomaka said.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
“What’s unique and very exciting” about the APPROACH findings are the nonhuman primate findings, and the fact that the vaccine was designed to provide protection against several different HIV clades, Dr. Tomaka said in a video interview. The 67% level of protection against viral challenge in the monkeys is at a level that would be clinically meaningful if replicated in people. A vaccine and booster regimen that provided something in the range of 50%-60% protection or better might be an attractive option in a region with relatively low resources, while in a more developed country a vaccine with a protective efficacy of 70% or better would also likely be seen as an attractive intervention, said Dr. Tomaka, clinical leader for HIV/STI vaccines for Janssen in Titusville, N.J.
SOURCE: Tomaka FL et al. AIDS 2018, Abstract TUAA0104.
The 1-year results for this new HIV vaccine and now the 1-year follow-up results appear very promising for its future prospects in wider clinical testing.
What’s especially interesting about the data reported for this vaccine so far is that the developers also tested the vaccine in rhesus monkeys and showed similar immunologic induction and a 67% efficacy for protecting against repeated challenges with a simian-human immunodeficiency virus. This level of protection against infection and the similar cellular and antibody responses to the vaccine and booster in the animal model and in people is encouraging. The vaccine protected monkeys. Now we need to find out whether it will protect humans.

R. Brad Jones, PhD , is an immunologist at Cornell University, New York. He had no disclosures. He made these comments during a talk at the conference.
The 1-year results for this new HIV vaccine and now the 1-year follow-up results appear very promising for its future prospects in wider clinical testing.
What’s especially interesting about the data reported for this vaccine so far is that the developers also tested the vaccine in rhesus monkeys and showed similar immunologic induction and a 67% efficacy for protecting against repeated challenges with a simian-human immunodeficiency virus. This level of protection against infection and the similar cellular and antibody responses to the vaccine and booster in the animal model and in people is encouraging. The vaccine protected monkeys. Now we need to find out whether it will protect humans.
R. Brad Jones, PhD , is an immunologist at Cornell University, New York. He had no disclosures. He made these comments during a talk at the conference.
The 1-year results for this new HIV vaccine and now the 1-year follow-up results appear very promising for its future prospects in wider clinical testing.
What’s especially interesting about the data reported for this vaccine so far is that the developers also tested the vaccine in rhesus monkeys and showed similar immunologic induction and a 67% efficacy for protecting against repeated challenges with a simian-human immunodeficiency virus. This level of protection against infection and the similar cellular and antibody responses to the vaccine and booster in the animal model and in people is encouraging. The vaccine protected monkeys. Now we need to find out whether it will protect humans.
R. Brad Jones, PhD , is an immunologist at Cornell University, New York. He had no disclosures. He made these comments during a talk at the conference.
AMSTERDAM – that had identified the most effective dosing strategy for the vaccine.
The 96-week follow-up data showed durable humoral and cellular immunity induction by the vaccine and its associated booster, a durable breadth of immune responses to the multiple HIV clades that the vaccine targets, and no serious or grade 3 or 4 adverse effects in the 393 people who participated in the early-phase study, Frank L. Tomaka, MD, said at the 22nd International AIDS Conference.
The 96-week results he reported came from follow-up of the 393 people who received one of several different dosing regimens for an engineered, mosaic HIV vaccine. The vaccine incorporates genes for three different HIV envelope antigens that contain components drawn from several different HIV clades (to induce more broadly protective immunity) into a serotype 26 adenovirus. The immunization regimen also includes treatment with HIV glycoprotein 140 as a booster agent. Dr. Tomaka and his associates reported the primary endpoints from this placebo-controlled study, APPROACH, measured just after the fourth and final immunizing regimen at 48 weeks after the first treatment in a recently published article (Lancet. 2018 Jul 21;392[10143]:232-43).
As part of the study, the investigators administered the vaccine and booster to rhesus monkeys and found that the regimens produced a pattern of immune responses in the monkeys similar to that seen in people. When the monkeys that received the regimen that performed best in people received six monthly challenges with a simian-human immunodeficiency virus that’s related to HIV, the researchers found a 67% efficacy for protection against infection. These “very encouraging” findings led the company developing the vaccine to launch in November 2017 a phase IIb trial, named Imbokodo, in five African countries, with a plan to enroll 2,600 people, Dr. Tomaka said.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
“What’s unique and very exciting” about the APPROACH findings are the nonhuman primate findings, and the fact that the vaccine was designed to provide protection against several different HIV clades, Dr. Tomaka said in a video interview. The 67% level of protection against viral challenge in the monkeys is at a level that would be clinically meaningful if replicated in people. A vaccine and booster regimen that provided something in the range of 50%-60% protection or better might be an attractive option in a region with relatively low resources, while in a more developed country a vaccine with a protective efficacy of 70% or better would also likely be seen as an attractive intervention, said Dr. Tomaka, clinical leader for HIV/STI vaccines for Janssen in Titusville, N.J.
SOURCE: Tomaka FL et al. AIDS 2018, Abstract TUAA0104.
AMSTERDAM – that had identified the most effective dosing strategy for the vaccine.
The 96-week follow-up data showed durable humoral and cellular immunity induction by the vaccine and its associated booster, a durable breadth of immune responses to the multiple HIV clades that the vaccine targets, and no serious or grade 3 or 4 adverse effects in the 393 people who participated in the early-phase study, Frank L. Tomaka, MD, said at the 22nd International AIDS Conference.
The 96-week results he reported came from follow-up of the 393 people who received one of several different dosing regimens for an engineered, mosaic HIV vaccine. The vaccine incorporates genes for three different HIV envelope antigens that contain components drawn from several different HIV clades (to induce more broadly protective immunity) into a serotype 26 adenovirus. The immunization regimen also includes treatment with HIV glycoprotein 140 as a booster agent. Dr. Tomaka and his associates reported the primary endpoints from this placebo-controlled study, APPROACH, measured just after the fourth and final immunizing regimen at 48 weeks after the first treatment in a recently published article (Lancet. 2018 Jul 21;392[10143]:232-43).
As part of the study, the investigators administered the vaccine and booster to rhesus monkeys and found that the regimens produced a pattern of immune responses in the monkeys similar to that seen in people. When the monkeys that received the regimen that performed best in people received six monthly challenges with a simian-human immunodeficiency virus that’s related to HIV, the researchers found a 67% efficacy for protection against infection. These “very encouraging” findings led the company developing the vaccine to launch in November 2017 a phase IIb trial, named Imbokodo, in five African countries, with a plan to enroll 2,600 people, Dr. Tomaka said.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
“What’s unique and very exciting” about the APPROACH findings are the nonhuman primate findings, and the fact that the vaccine was designed to provide protection against several different HIV clades, Dr. Tomaka said in a video interview. The 67% level of protection against viral challenge in the monkeys is at a level that would be clinically meaningful if replicated in people. A vaccine and booster regimen that provided something in the range of 50%-60% protection or better might be an attractive option in a region with relatively low resources, while in a more developed country a vaccine with a protective efficacy of 70% or better would also likely be seen as an attractive intervention, said Dr. Tomaka, clinical leader for HIV/STI vaccines for Janssen in Titusville, N.J.
SOURCE: Tomaka FL et al. AIDS 2018, Abstract TUAA0104.
REPORTING FROM AIDS 2018
Key clinical point: An investigational HIV vaccine and booster showed durable safety and anti-HIV immune effects.
Major finding: One year follow-up after the final dosage showed no serious or grade 3 or 4 adverse effects and durable immune responses.
Study details: The APPROACH study, a phase I/IIa study with 393 participants.
Disclosures: APPROACH was sponsored by Janssen, the company developing the vaccine. Dr. Tomaka is a Janssen employee.
Source: Tomaka FL et al. AIDS 2018, Abstract TUAA0104.
Total Joint Arthroplasty Quality Ratings: How Are They Similar and How Are They Different?
ABSTRACT
A patient’s perception of hospital or provider quality can have far-reaching effects, as it can impact reimbursement, patient selection of a surgeon, and healthcare competition. A variety of organizations offer quality designations for orthopedic surgery and its subspecialties. Our goal is to compare total joint arthroplasty (TJA) quality designation methodology across key quality rating organizations. One researcher conducted an initial Google search to determine organizations providing quality designations for hospitals and surgeons providing orthopedic procedures with a focus on TJA. Organizations that offer quality designation specific to TJA were determined. Organizations that provided general orthopedic surgery or only surgeon-specific quality designation were excluded from the analysis. The senior author confirmed the inclusion of the final organizations. Seven organizations fit our inclusion criteria. Only the private payers and The Joint Commission required hospital accreditation to meet quality designation criteria. Total arthroplasty volume was considered in 86% of the organizations’ methodologies, and 57% of organizations utilized process measurements such as antibiotic prophylaxis and care pathways. In addition, 57% of organizations included patient experience in their methodologies. Only 29% of organizations included a cost element in their methodology. All organizations utilized outcome data and publicly reported all hospitals receiving their quality designation. Hospital quality designation methodologies are inconsistent in the context of TJA. All stakeholders (ie, providers, payers, and patients) should be involved in deciding the definition of quality.
Continue to: Healthcare in the United States...
Healthcare in the United States has begun to move toward a system focused on value for patients, defined as health outcome per dollar expended.1 Indeed, an estimated 30% of Medicare payments are now made using the so-called alternative payment models (eg, bundled payments),2 and there is an expectation that consumerism in medicine will continue to expand.3 In addition, although there is a continuing debate regarding the benefits and pitfalls of hospital mergers, there is no question whether provider consolidation has increased dramatically in recent years.4 At the core of many of these changes is the push to improve healthcare quality and reduce costs.
Quality has the ability to affect payment, patient selection of providers, and hospital competition. Patients (ie, healthcare consumers) are increasingly using the Internet to find a variety of health information.5 Accessible provider quality information online would allow patients to make more informed decisions about where to seek care. In addition, the development of transparent quality ratings could assist payers in driving beneficiaries to higher quality and better value providers, which could mean more business for the highest quality physicians and better patient outcomes with fewer complications. Some payers such as the Centers for Medicare and Medicaid Services (CMS) have already started using quality measures as part of their reimbursement strategy.6 Because CMS is the largest payer in the United States, private insurers tend to follow their lead; thus, quality measurements will become even more common as a factor in reimbursement over the coming years.
To make quality ratings useful, “quality” must be clearly defined. Clarity around which factors are considered in a quality designation will create transparency for patients and allow providers to understand how their performance is being measured so that they focus on improving outcomes for their patients. Numerous organizations, including private payers, public payers, and both not-for-profit and for-profit entities, have created quality designation programs to rate providers. However, within orthopedics and several other medical specialties, there has been an ongoing debate about what measures best reflect quality.7 Although inconsistencies in quality ratings in arthroplasty care have been noted,8 it remains unknown how each quality designation program compares with the others in terms of the factors considered in deciding quality designations.
The purpose of this study is to evaluate publicly available information from key quality designation programs for total joint arthroplasty (TJA) providers to determine what factors are considered by each organization in awarding quality designations; what similarities and differences in quality designations exist across the different organizations; and how many of the organizations publish their quality designation methodologies and final rating results.
MATERIALS AND METHODS
A directed Google search was conducted to determine organizations (ie, payers, independent firms, and government entities) that rate hospitals and/or surgeons in orthopedic surgery. The identified organizations were then examined to determine whether they provided hospital ratings for total hip and/or knee arthroplasty. Entities were included if they provided quality designations for hospitals specifically addressing TJA. Organizations that provided only general hospital, other surgical procedures, orthopedic surgery, or orthopedic surgeon-specific quality designations were excluded. A list of all organizations determined to fit the inclusion criteria was then reviewed for completeness and approved by the senior author.
Continue to: One investigator reviewed the website of each organization...
One investigator reviewed the website of each organization fitting the inclusion criteria to determine the full rating methodology in 1 sitting on July 2, 2016. Detailed notes were taken on each program using publicly available information. For organizations that used proprietary criteria for quality designation (eg, The Joint Commission [TJC]), only publicly available information was used in the analysis. Therefore, the information reported is solely based on data available online to the public.
Detailed quality designation criteria were condensed into broader categories (accreditation, volume, structural, process, outcomes, patient experience, and cost/efficiency) to capture differences between each organization reviewed. In addition, we recorded whether each organization published a list of providers that received its quality designation.
RESULTS
A total of 7 organizations fit our inclusion criteria9-15 (Table). Of these 7 organizations, 3 were private payers (Aetna, UnitedHealth, and Blue Cross Blue Shield [BCBS]), 2 were nongovernmental not-for-profit organizations (TJC and Consumer Reports), and 2 were consumer-based and/or for-profit organizations (HealthGrades and US News & World Report [USNWR]). There were no government agencies that fit our inclusion criteria. BCBS had the following 2 separate quality designations: BCBS Blue Distinction and BCBS Blue Distinction+. The only difference between the 2 BCBS ratings is that BCBS Blue Distinction+ includes cost efficiency ratings, whereas BCBS Blue Distinction does not.
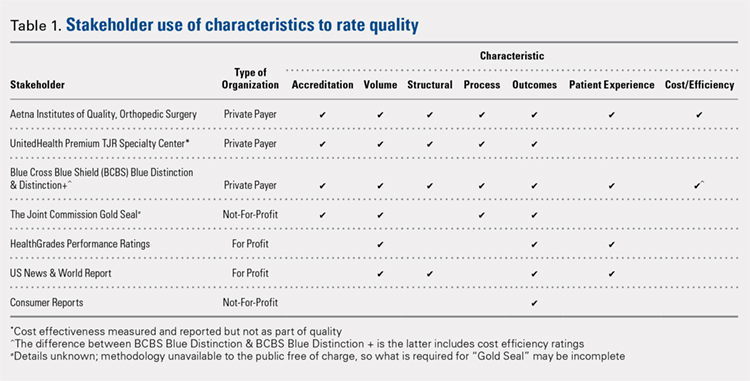
Only the 3 private payers and TJC, the primary hospital accreditation body in the United States, required accreditation as part of its quality designation criteria. TJC requires its own accreditation for quality designation consideration, whereas the 3 private payers allow accreditation from one of a variety of sources. Aetna Institutes of Quality for Orthopedic Surgery requires accreditation by TJC, Healthcare Facilities Accreditation Program, American Osteopathic Association, National Integrated Accreditation for Healthcare Organizations, or Det Norske Veritas Healthcare. UnitedHealth Premium Total Joint Replacement (TJR) Specialty Center requires accreditation by TJC and/or equivalent of TJC accreditation. However, TJC accreditation equivalents are not noted in the UnitedHealth handbook. BCBS Blue Distinction and Distinction+ require accreditation by TJC, Healthcare Facilities Accreditation Program, National Integrated Accreditation for Healthcare Organizations, or Center for Improvement in Healthcare Quality. In addition, BCBS is willing to consider alternative accreditations that are at least as stringent as the national alternatives noted. However, no detailed criteria that must be met to be equivalent to the national standards are noted in the relevant quality designation handbook.
The volume of completed total hip and knee arthroplasty procedures was considered in 6 of the organizations’ quality ratings methodologies. Of those 6, all private payers, TJC (not-for-profit), and 2 for-profit rating agencies were included. Surgeon specialization in TJA was only explicitly noted as a factor considered in UnitedHealth Premium TJR Specialty Center criteria; however, the requirements for surgeon specialization were not clearly defined. In addition, the presence of a multidisciplinary clinical pathway was only explicitly considered for Aetna Institutes of Quality for Orthopedic Surgery.
Structural requirements (eg, use of electronic health records [EHR], staffing levels, etc.) were taken into account in private payer and USNWR quality methodologies. Process measures (eg, antibiotic prophylaxis and other care pathways) were considered for the private payers and TJC but not for USNWR quality designation. Cost and/or efficiency measures were factors in the quality formula for Aetna Institutes of Quality for Orthopedic Surgery and BCBS Distinction+. Aetna utilizes its own cost data and risk-adjusts using a product known as Symmetry Episode Risk Groups to determine cost-effectiveness, while BCBS uses its own Composite Facility Cost Index. Patient experience (eg, Hospital Consumer Assessment of Healthcare Providers and Systems [HCAHPS]) was incorporated into the quality formulas for 4 of the 7 quality designation programs examined.
Continue to: All of the 7 quality designation programs included...
All of the 7 quality designation programs included outcomes (ie, readmission rates and/or mortality rates) and publicly reported the hospitals receiving their quality designation. In contrast, only Aetna explicitly included the presence of multidisciplinary clinical care pathways as part of their quality designation criteria. In addition, only UnitedHealth included surgeon specialization in joint arthroplasty as a factor for quality consideration for its quality designation program. BCBS Distinction+ and Aetna Institutes of Quality for Orthopedic Surgery were the only 2 quality designations that included at least 1 variable that fit into each of the 7 characteristics considered (accreditation, volume, structural, process, outcomes, patient experience, and cost/efficiency).
DISCUSSION
As healthcare continues to shift toward value-based delivery and payment models, quality becomes a critical factor in reimbursement and provider rankings. However, quality is a vague term. Several providers probably do not know what is required to be designated as high quality by a particular rating agency. Moreover, there are multiple quality designation programs, all using distinct criteria to determine “quality,” which further complicates the matter. Our objective was to determine the key stakeholders that provide quality designations in TJA and what criteria each organization uses in assessing quality.
Our idea of comprehensive quality is based on Avedis Donabedian’s enduring framework for healthcare quality focused on structure, process, and outcome.16 We expanded on these 3 areas and analyzed quality designations based on variables fitting into the following categories: accreditation, volume, structural, process, outcomes, patient experience, and cost/efficiency. We believe that these categories encompass a comprehensive rating system that addresses key elements of patient care. However, our results suggest that only 2 major quality designations (BCBS Distinction+ and Aetna Institutes of Quality for Orthopedic Surgery) take all such variables into account.
All quality designation programs that we analyzed required outcome data (ie, readmission and/or mortality rates within 30 days); however, only 2 programs utilized cost in their quality designation criteria (BCBS Distinction+ and Aetna Institutes of Quality for Orthopedic Surgery). Aetna Institutes of Quality for Orthopedic Surgery risk-adjusted for its cost-effectiveness calculations based on age, sex, and other unspecified conditions using a product known as Symmetry Episode Risk Groups. However, the organization also noted that although it did risk-adjust for inpatient mortality, it did not do so for pulmonary embolism or deep vein thrombosis. BCBS Distinction+ also utilized risk adjustment for its cost efficiency measure, and its step-by-step methodology is available online. Further, Consumer Reports does risk-adjust using logistic regression models in their quality analysis, but the description provided is minimal; it is noted that such risk adjustments are already completed by CMS prior to Consumer Reports acquiring the data. The CMS Compare model information is available on the CMS website. The data utilized by several organizations and presented on CMS Compare are already risk-adjusted using CMS’ approach. In contrast, UnitedHealth Premium TJR Specialty Center gathers its own data from providers and does not describe a risk adjustment methodology. Risk adjustment is important because the lack of risk adjustment may lead to physicians “cherry-picking” easy cases to boost positive outcomes, leading to increased financial benefits and higher quality ratings. Having a consistent risk adjustment formula will ensure accurate comparisons across outcomes and cost-effectiveness measures used by quality designation programs.
Factors considered for quality designation varied greatly from one organization to the other. The range of categories of factors considered varied from 1 (Consumer Reports only considered outcome data) to all 7 categories (BCBS Distinction+ and Aetna Institutes of Quality for Orthopedic Surgery). Our findings are consistent with the work by Keswani and colleagues,8 which showed that there is likely variation in factors considered when rating hospital quality more broadly. Our work suggests that quality designation formulas do not appear to get more consistent when focused on TJA.
We found that all organizations in our analysis published the providers earning their quality designation. However, TJC does not provide publicly a detailed methodology on how to qualify for its quality designation. The price to purchase the necessary manual for this information is $146.00 for accredited organizations and $186.00 for all others.17 For large healthcare providers, this is not a large sum of money. Nonetheless, this provides an additional hurdle for stakeholders to gain a full understanding of the requirements to receive a TJC Gold Seal for Orthopedics.
Previous work has evaluated the consistency of and the variety of means of gauging healthcare quality. Previous work by Rothberg and colleagues18 comparing hospital rankings across 5 common consumer-oriented websites found disagreement on hospital rankings within any diagnosis and even among metrics such as mortality. Another study by Halasyamani and Davis19 found that CMS Compare and USNWR rankings were dissimilar and the authors attributed the discrepancy to different methodologies. In addition, a study by Krumholz and colleagues20 focused on Internet report cards, which measured the appropriate use of select medications and mortality rates for acute myocardial infarction as the quality metrics. The authors found that, in aggregate, there was a clear difference in quality of care and outcomes but that comparisons between 2 hospitals provided poor discrimination.20 Other work has analyzed the increasing trend of online ratings of orthopedic surgeons by patients.21 However, there remains no agreed-upon definition of quality. Thus, the use of the term “quality” in several studies may be misleading.
Our results must be interpreted keeping the limitations of our work in mind. First, we used expert knowledge and a public search engine to develop our list of organizations that provide TJA quality designations. However, there is a possibility that we did not include all relevant organizations. Second, although all authors reviewed the final data, it is possible that there was human error in the analysis of each organization’s quality designation criteria.
CONCLUSION
As healthcare progresses further toward a system that rewards providers for delivering value to patients, accurately defining and measuring quality becomes critical because it can be suggestive of value to patients, payers, and providers. Furthermore, it gives providers a goal to focus on as they strive to improve the value of care they deliver to patients. Measuring healthcare quality is currently a novel, imperfect science,22 and there continues to be a debate about what factors should be included in a quality designation formula. Nonetheless, more and more quality designations and performance measurements are being created for orthopedic care, including total hip and total knee arthroplasty. In fact, in 2016, The Leapfrog Group added readmission for patients undergoing TJA to its survey.23 Consensus on a quality definition may facilitate the movement toward a value-based healthcare system. Future research should evaluate strategies for gaining consensus among stakeholders for a universal quality metric in TJA. Surgeons, hospitals, payers, and most importantly patients should play critical roles in defining quality.
- Porter ME. A strategy for health care reform--toward a value-based system. N Engl J Med. 2009;361(2):109-112. doi:10.1056/NEJMp0904131.
- Obama B. United States health care reform: progress to date and next steps. JAMA. 2016;316(5):525-532. doi:10.1001/jama.2016.9797.
- Mulvany C. The march to consumerism the evolution from patient to active shopper continues. Healthc Financ Manage. 2014;68(2):36-38.
- Tsai TC, Jha AK. Hospital consolidation, competition, and quality: is bigger necessarily better? JAMA. 2014;312(1):29-30. doi:10.1001/jama.2014.4692.
- Cline RJ, Haynes KM. Consumer health information seeking on the Internet: the state of the art. Health Educ Res. 2001;16(6):671-692. doi:10.1093/her/16.6.671.
- Werner RM, Kolstad JT, Stuart EA, Polsky D. The effect of pay-for-performance in hospitals: lessons for quality improvement. Health Aff (Millwood). 2011;30(4):690-698. doi:10.1377/hlthaff.2010.1277.
- Birkmeyer JD, Dimick JB, Birkmeyer NJ. Measuring the quality of surgical care: structure, process, or outcomes? J Am Coll Surg. 2004;198(4):626-632. doi:10.1016/j.jamcollsurg.2003.11.017.
- Keswani A, Uhler LM, Bozic KJ. What quality metrics is my hospital being evaluated on and what are the consequences? J Arthroplast. 2016;31(6):1139-1143. doi:10.1016/j.arth.2016.01.075.
- Aetna Inc. Aetna Institutes of Quality® facilities fact book. A comprehensive reference guide for Aetna members, doctors and health care professionals. http://www.aetna.com/individuals-families-health-insurance/document-libr.... Accessed July 2, 2016.
- United HealthCare. UnitedHealth Premium® Program. https://www.uhcprovider.com/en/reports-quality-programs/premium-designation.html. Accessed July 2, 2016.
- 11. Blue Cross Blue Shield. Association. Blue Distinction Specialty Care. Selection criteria and program documentation: knee and hip replacement and spine surgery. https://www.bcbs.com/sites/default/files/fileattachments/page/KneeHip.SelectionCriteria_0.pdf. Published October 2015. Accessed July 2, 2016.
- The Joint Commission. Advanced certification for total hip and total knee replacement eligibility. https://www.jointcommission.org/advanced_certification_for_total_hip_and.... Published December 10, 2015. Accessed July 2, 2016.
- Healthgrades Operating Company. Healthgrades methodology: anatomy of a rating. https://www.healthgrades.com/quality/ratings-awards/methodology. Accessed July 2, 2016.
- Comarow A, Harder B; Dr. Foster Project Team. Methodology: U.S. News & World Report best hospitals for common care. U.S. News & World Report Web site. http://www.usnews.com/pubfiles/BHCC_MethReport_2015.pdf. Published May 20, 2015. Accessed July 2, 2016.
- Consumer Reports. How we rate hospitals. http://static3.consumerreportscdn.org/content/dam/cro/news_articles/heal.... Accessed July 2, 2016.
- Ayanian JZ, Markel H. Donabedian’s lasting framework for health care quality. N Engl J Med. 2016;375(3):205-207. doi:10.1056/NEJMp1605101.
- The Joint Commission. 2016 Certification Manuals. 2016; http://www.jcrinc.com/2016-certification-manuals/. Accessed July 2, 2016.
- Rothberg MB, Morsi E, Benjamin EM, Pekow PS, Lindenauer PK. Choosing the best hospital: the limitations of public quality reporting. Health Aff (Millwood). 2008;27(6):1680-1687. doi:10.1377/hlthaff.27.6.1680.
- Halasyamani LK, Davis MM. Conflicting measures of hospital quality: ratings from "Hospital Compare" versus "Best Hospitals". J Hosp Med. 2007;2(3):128-134. doi:10.1002/jhm.176.
- Krumholz HM, Rathore SS, Chen J, Wang Y, Radford MJ. Evaluation of a consumer-oriented internet health care report card: the risk of quality ratings based on mortality data. JAMA. 2002;287(10):1277-1287.
- Frost C, Mesfin A. Online reviews of orthopedic surgeons: an emerging trend. Orthopedics. 2015;38(4):e257-e262. doi:10.3928/01477447-20150402-52.
- Harder B, Comarow A. Hospital Quality reporting by US News & World Report: why, how, and what's ahead. JAMA. 2015;313(19):1903-1904. doi:10.1001/jama.2015.4566.
- The Leapfrog Group. New in 2016. http://www.leapfroggroup.org/ratings-reports/new-2016. Accessed July 2, 2016.
ABSTRACT
A patient’s perception of hospital or provider quality can have far-reaching effects, as it can impact reimbursement, patient selection of a surgeon, and healthcare competition. A variety of organizations offer quality designations for orthopedic surgery and its subspecialties. Our goal is to compare total joint arthroplasty (TJA) quality designation methodology across key quality rating organizations. One researcher conducted an initial Google search to determine organizations providing quality designations for hospitals and surgeons providing orthopedic procedures with a focus on TJA. Organizations that offer quality designation specific to TJA were determined. Organizations that provided general orthopedic surgery or only surgeon-specific quality designation were excluded from the analysis. The senior author confirmed the inclusion of the final organizations. Seven organizations fit our inclusion criteria. Only the private payers and The Joint Commission required hospital accreditation to meet quality designation criteria. Total arthroplasty volume was considered in 86% of the organizations’ methodologies, and 57% of organizations utilized process measurements such as antibiotic prophylaxis and care pathways. In addition, 57% of organizations included patient experience in their methodologies. Only 29% of organizations included a cost element in their methodology. All organizations utilized outcome data and publicly reported all hospitals receiving their quality designation. Hospital quality designation methodologies are inconsistent in the context of TJA. All stakeholders (ie, providers, payers, and patients) should be involved in deciding the definition of quality.
Continue to: Healthcare in the United States...
Healthcare in the United States has begun to move toward a system focused on value for patients, defined as health outcome per dollar expended.1 Indeed, an estimated 30% of Medicare payments are now made using the so-called alternative payment models (eg, bundled payments),2 and there is an expectation that consumerism in medicine will continue to expand.3 In addition, although there is a continuing debate regarding the benefits and pitfalls of hospital mergers, there is no question whether provider consolidation has increased dramatically in recent years.4 At the core of many of these changes is the push to improve healthcare quality and reduce costs.
Quality has the ability to affect payment, patient selection of providers, and hospital competition. Patients (ie, healthcare consumers) are increasingly using the Internet to find a variety of health information.5 Accessible provider quality information online would allow patients to make more informed decisions about where to seek care. In addition, the development of transparent quality ratings could assist payers in driving beneficiaries to higher quality and better value providers, which could mean more business for the highest quality physicians and better patient outcomes with fewer complications. Some payers such as the Centers for Medicare and Medicaid Services (CMS) have already started using quality measures as part of their reimbursement strategy.6 Because CMS is the largest payer in the United States, private insurers tend to follow their lead; thus, quality measurements will become even more common as a factor in reimbursement over the coming years.
To make quality ratings useful, “quality” must be clearly defined. Clarity around which factors are considered in a quality designation will create transparency for patients and allow providers to understand how their performance is being measured so that they focus on improving outcomes for their patients. Numerous organizations, including private payers, public payers, and both not-for-profit and for-profit entities, have created quality designation programs to rate providers. However, within orthopedics and several other medical specialties, there has been an ongoing debate about what measures best reflect quality.7 Although inconsistencies in quality ratings in arthroplasty care have been noted,8 it remains unknown how each quality designation program compares with the others in terms of the factors considered in deciding quality designations.
The purpose of this study is to evaluate publicly available information from key quality designation programs for total joint arthroplasty (TJA) providers to determine what factors are considered by each organization in awarding quality designations; what similarities and differences in quality designations exist across the different organizations; and how many of the organizations publish their quality designation methodologies and final rating results.
MATERIALS AND METHODS
A directed Google search was conducted to determine organizations (ie, payers, independent firms, and government entities) that rate hospitals and/or surgeons in orthopedic surgery. The identified organizations were then examined to determine whether they provided hospital ratings for total hip and/or knee arthroplasty. Entities were included if they provided quality designations for hospitals specifically addressing TJA. Organizations that provided only general hospital, other surgical procedures, orthopedic surgery, or orthopedic surgeon-specific quality designations were excluded. A list of all organizations determined to fit the inclusion criteria was then reviewed for completeness and approved by the senior author.
Continue to: One investigator reviewed the website of each organization...
One investigator reviewed the website of each organization fitting the inclusion criteria to determine the full rating methodology in 1 sitting on July 2, 2016. Detailed notes were taken on each program using publicly available information. For organizations that used proprietary criteria for quality designation (eg, The Joint Commission [TJC]), only publicly available information was used in the analysis. Therefore, the information reported is solely based on data available online to the public.
Detailed quality designation criteria were condensed into broader categories (accreditation, volume, structural, process, outcomes, patient experience, and cost/efficiency) to capture differences between each organization reviewed. In addition, we recorded whether each organization published a list of providers that received its quality designation.
RESULTS
A total of 7 organizations fit our inclusion criteria9-15 (Table). Of these 7 organizations, 3 were private payers (Aetna, UnitedHealth, and Blue Cross Blue Shield [BCBS]), 2 were nongovernmental not-for-profit organizations (TJC and Consumer Reports), and 2 were consumer-based and/or for-profit organizations (HealthGrades and US News & World Report [USNWR]). There were no government agencies that fit our inclusion criteria. BCBS had the following 2 separate quality designations: BCBS Blue Distinction and BCBS Blue Distinction+. The only difference between the 2 BCBS ratings is that BCBS Blue Distinction+ includes cost efficiency ratings, whereas BCBS Blue Distinction does not.
Only the 3 private payers and TJC, the primary hospital accreditation body in the United States, required accreditation as part of its quality designation criteria. TJC requires its own accreditation for quality designation consideration, whereas the 3 private payers allow accreditation from one of a variety of sources. Aetna Institutes of Quality for Orthopedic Surgery requires accreditation by TJC, Healthcare Facilities Accreditation Program, American Osteopathic Association, National Integrated Accreditation for Healthcare Organizations, or Det Norske Veritas Healthcare. UnitedHealth Premium Total Joint Replacement (TJR) Specialty Center requires accreditation by TJC and/or equivalent of TJC accreditation. However, TJC accreditation equivalents are not noted in the UnitedHealth handbook. BCBS Blue Distinction and Distinction+ require accreditation by TJC, Healthcare Facilities Accreditation Program, National Integrated Accreditation for Healthcare Organizations, or Center for Improvement in Healthcare Quality. In addition, BCBS is willing to consider alternative accreditations that are at least as stringent as the national alternatives noted. However, no detailed criteria that must be met to be equivalent to the national standards are noted in the relevant quality designation handbook.
The volume of completed total hip and knee arthroplasty procedures was considered in 6 of the organizations’ quality ratings methodologies. Of those 6, all private payers, TJC (not-for-profit), and 2 for-profit rating agencies were included. Surgeon specialization in TJA was only explicitly noted as a factor considered in UnitedHealth Premium TJR Specialty Center criteria; however, the requirements for surgeon specialization were not clearly defined. In addition, the presence of a multidisciplinary clinical pathway was only explicitly considered for Aetna Institutes of Quality for Orthopedic Surgery.
Structural requirements (eg, use of electronic health records [EHR], staffing levels, etc.) were taken into account in private payer and USNWR quality methodologies. Process measures (eg, antibiotic prophylaxis and other care pathways) were considered for the private payers and TJC but not for USNWR quality designation. Cost and/or efficiency measures were factors in the quality formula for Aetna Institutes of Quality for Orthopedic Surgery and BCBS Distinction+. Aetna utilizes its own cost data and risk-adjusts using a product known as Symmetry Episode Risk Groups to determine cost-effectiveness, while BCBS uses its own Composite Facility Cost Index. Patient experience (eg, Hospital Consumer Assessment of Healthcare Providers and Systems [HCAHPS]) was incorporated into the quality formulas for 4 of the 7 quality designation programs examined.
Continue to: All of the 7 quality designation programs included...
All of the 7 quality designation programs included outcomes (ie, readmission rates and/or mortality rates) and publicly reported the hospitals receiving their quality designation. In contrast, only Aetna explicitly included the presence of multidisciplinary clinical care pathways as part of their quality designation criteria. In addition, only UnitedHealth included surgeon specialization in joint arthroplasty as a factor for quality consideration for its quality designation program. BCBS Distinction+ and Aetna Institutes of Quality for Orthopedic Surgery were the only 2 quality designations that included at least 1 variable that fit into each of the 7 characteristics considered (accreditation, volume, structural, process, outcomes, patient experience, and cost/efficiency).
DISCUSSION
As healthcare continues to shift toward value-based delivery and payment models, quality becomes a critical factor in reimbursement and provider rankings. However, quality is a vague term. Several providers probably do not know what is required to be designated as high quality by a particular rating agency. Moreover, there are multiple quality designation programs, all using distinct criteria to determine “quality,” which further complicates the matter. Our objective was to determine the key stakeholders that provide quality designations in TJA and what criteria each organization uses in assessing quality.
Our idea of comprehensive quality is based on Avedis Donabedian’s enduring framework for healthcare quality focused on structure, process, and outcome.16 We expanded on these 3 areas and analyzed quality designations based on variables fitting into the following categories: accreditation, volume, structural, process, outcomes, patient experience, and cost/efficiency. We believe that these categories encompass a comprehensive rating system that addresses key elements of patient care. However, our results suggest that only 2 major quality designations (BCBS Distinction+ and Aetna Institutes of Quality for Orthopedic Surgery) take all such variables into account.
All quality designation programs that we analyzed required outcome data (ie, readmission and/or mortality rates within 30 days); however, only 2 programs utilized cost in their quality designation criteria (BCBS Distinction+ and Aetna Institutes of Quality for Orthopedic Surgery). Aetna Institutes of Quality for Orthopedic Surgery risk-adjusted for its cost-effectiveness calculations based on age, sex, and other unspecified conditions using a product known as Symmetry Episode Risk Groups. However, the organization also noted that although it did risk-adjust for inpatient mortality, it did not do so for pulmonary embolism or deep vein thrombosis. BCBS Distinction+ also utilized risk adjustment for its cost efficiency measure, and its step-by-step methodology is available online. Further, Consumer Reports does risk-adjust using logistic regression models in their quality analysis, but the description provided is minimal; it is noted that such risk adjustments are already completed by CMS prior to Consumer Reports acquiring the data. The CMS Compare model information is available on the CMS website. The data utilized by several organizations and presented on CMS Compare are already risk-adjusted using CMS’ approach. In contrast, UnitedHealth Premium TJR Specialty Center gathers its own data from providers and does not describe a risk adjustment methodology. Risk adjustment is important because the lack of risk adjustment may lead to physicians “cherry-picking” easy cases to boost positive outcomes, leading to increased financial benefits and higher quality ratings. Having a consistent risk adjustment formula will ensure accurate comparisons across outcomes and cost-effectiveness measures used by quality designation programs.
Factors considered for quality designation varied greatly from one organization to the other. The range of categories of factors considered varied from 1 (Consumer Reports only considered outcome data) to all 7 categories (BCBS Distinction+ and Aetna Institutes of Quality for Orthopedic Surgery). Our findings are consistent with the work by Keswani and colleagues,8 which showed that there is likely variation in factors considered when rating hospital quality more broadly. Our work suggests that quality designation formulas do not appear to get more consistent when focused on TJA.
We found that all organizations in our analysis published the providers earning their quality designation. However, TJC does not provide publicly a detailed methodology on how to qualify for its quality designation. The price to purchase the necessary manual for this information is $146.00 for accredited organizations and $186.00 for all others.17 For large healthcare providers, this is not a large sum of money. Nonetheless, this provides an additional hurdle for stakeholders to gain a full understanding of the requirements to receive a TJC Gold Seal for Orthopedics.
Previous work has evaluated the consistency of and the variety of means of gauging healthcare quality. Previous work by Rothberg and colleagues18 comparing hospital rankings across 5 common consumer-oriented websites found disagreement on hospital rankings within any diagnosis and even among metrics such as mortality. Another study by Halasyamani and Davis19 found that CMS Compare and USNWR rankings were dissimilar and the authors attributed the discrepancy to different methodologies. In addition, a study by Krumholz and colleagues20 focused on Internet report cards, which measured the appropriate use of select medications and mortality rates for acute myocardial infarction as the quality metrics. The authors found that, in aggregate, there was a clear difference in quality of care and outcomes but that comparisons between 2 hospitals provided poor discrimination.20 Other work has analyzed the increasing trend of online ratings of orthopedic surgeons by patients.21 However, there remains no agreed-upon definition of quality. Thus, the use of the term “quality” in several studies may be misleading.
Our results must be interpreted keeping the limitations of our work in mind. First, we used expert knowledge and a public search engine to develop our list of organizations that provide TJA quality designations. However, there is a possibility that we did not include all relevant organizations. Second, although all authors reviewed the final data, it is possible that there was human error in the analysis of each organization’s quality designation criteria.
CONCLUSION
As healthcare progresses further toward a system that rewards providers for delivering value to patients, accurately defining and measuring quality becomes critical because it can be suggestive of value to patients, payers, and providers. Furthermore, it gives providers a goal to focus on as they strive to improve the value of care they deliver to patients. Measuring healthcare quality is currently a novel, imperfect science,22 and there continues to be a debate about what factors should be included in a quality designation formula. Nonetheless, more and more quality designations and performance measurements are being created for orthopedic care, including total hip and total knee arthroplasty. In fact, in 2016, The Leapfrog Group added readmission for patients undergoing TJA to its survey.23 Consensus on a quality definition may facilitate the movement toward a value-based healthcare system. Future research should evaluate strategies for gaining consensus among stakeholders for a universal quality metric in TJA. Surgeons, hospitals, payers, and most importantly patients should play critical roles in defining quality.
ABSTRACT
A patient’s perception of hospital or provider quality can have far-reaching effects, as it can impact reimbursement, patient selection of a surgeon, and healthcare competition. A variety of organizations offer quality designations for orthopedic surgery and its subspecialties. Our goal is to compare total joint arthroplasty (TJA) quality designation methodology across key quality rating organizations. One researcher conducted an initial Google search to determine organizations providing quality designations for hospitals and surgeons providing orthopedic procedures with a focus on TJA. Organizations that offer quality designation specific to TJA were determined. Organizations that provided general orthopedic surgery or only surgeon-specific quality designation were excluded from the analysis. The senior author confirmed the inclusion of the final organizations. Seven organizations fit our inclusion criteria. Only the private payers and The Joint Commission required hospital accreditation to meet quality designation criteria. Total arthroplasty volume was considered in 86% of the organizations’ methodologies, and 57% of organizations utilized process measurements such as antibiotic prophylaxis and care pathways. In addition, 57% of organizations included patient experience in their methodologies. Only 29% of organizations included a cost element in their methodology. All organizations utilized outcome data and publicly reported all hospitals receiving their quality designation. Hospital quality designation methodologies are inconsistent in the context of TJA. All stakeholders (ie, providers, payers, and patients) should be involved in deciding the definition of quality.
Continue to: Healthcare in the United States...
Healthcare in the United States has begun to move toward a system focused on value for patients, defined as health outcome per dollar expended.1 Indeed, an estimated 30% of Medicare payments are now made using the so-called alternative payment models (eg, bundled payments),2 and there is an expectation that consumerism in medicine will continue to expand.3 In addition, although there is a continuing debate regarding the benefits and pitfalls of hospital mergers, there is no question whether provider consolidation has increased dramatically in recent years.4 At the core of many of these changes is the push to improve healthcare quality and reduce costs.
Quality has the ability to affect payment, patient selection of providers, and hospital competition. Patients (ie, healthcare consumers) are increasingly using the Internet to find a variety of health information.5 Accessible provider quality information online would allow patients to make more informed decisions about where to seek care. In addition, the development of transparent quality ratings could assist payers in driving beneficiaries to higher quality and better value providers, which could mean more business for the highest quality physicians and better patient outcomes with fewer complications. Some payers such as the Centers for Medicare and Medicaid Services (CMS) have already started using quality measures as part of their reimbursement strategy.6 Because CMS is the largest payer in the United States, private insurers tend to follow their lead; thus, quality measurements will become even more common as a factor in reimbursement over the coming years.
To make quality ratings useful, “quality” must be clearly defined. Clarity around which factors are considered in a quality designation will create transparency for patients and allow providers to understand how their performance is being measured so that they focus on improving outcomes for their patients. Numerous organizations, including private payers, public payers, and both not-for-profit and for-profit entities, have created quality designation programs to rate providers. However, within orthopedics and several other medical specialties, there has been an ongoing debate about what measures best reflect quality.7 Although inconsistencies in quality ratings in arthroplasty care have been noted,8 it remains unknown how each quality designation program compares with the others in terms of the factors considered in deciding quality designations.
The purpose of this study is to evaluate publicly available information from key quality designation programs for total joint arthroplasty (TJA) providers to determine what factors are considered by each organization in awarding quality designations; what similarities and differences in quality designations exist across the different organizations; and how many of the organizations publish their quality designation methodologies and final rating results.
MATERIALS AND METHODS
A directed Google search was conducted to determine organizations (ie, payers, independent firms, and government entities) that rate hospitals and/or surgeons in orthopedic surgery. The identified organizations were then examined to determine whether they provided hospital ratings for total hip and/or knee arthroplasty. Entities were included if they provided quality designations for hospitals specifically addressing TJA. Organizations that provided only general hospital, other surgical procedures, orthopedic surgery, or orthopedic surgeon-specific quality designations were excluded. A list of all organizations determined to fit the inclusion criteria was then reviewed for completeness and approved by the senior author.
Continue to: One investigator reviewed the website of each organization...
One investigator reviewed the website of each organization fitting the inclusion criteria to determine the full rating methodology in 1 sitting on July 2, 2016. Detailed notes were taken on each program using publicly available information. For organizations that used proprietary criteria for quality designation (eg, The Joint Commission [TJC]), only publicly available information was used in the analysis. Therefore, the information reported is solely based on data available online to the public.
Detailed quality designation criteria were condensed into broader categories (accreditation, volume, structural, process, outcomes, patient experience, and cost/efficiency) to capture differences between each organization reviewed. In addition, we recorded whether each organization published a list of providers that received its quality designation.
RESULTS
A total of 7 organizations fit our inclusion criteria9-15 (Table). Of these 7 organizations, 3 were private payers (Aetna, UnitedHealth, and Blue Cross Blue Shield [BCBS]), 2 were nongovernmental not-for-profit organizations (TJC and Consumer Reports), and 2 were consumer-based and/or for-profit organizations (HealthGrades and US News & World Report [USNWR]). There were no government agencies that fit our inclusion criteria. BCBS had the following 2 separate quality designations: BCBS Blue Distinction and BCBS Blue Distinction+. The only difference between the 2 BCBS ratings is that BCBS Blue Distinction+ includes cost efficiency ratings, whereas BCBS Blue Distinction does not.
Only the 3 private payers and TJC, the primary hospital accreditation body in the United States, required accreditation as part of its quality designation criteria. TJC requires its own accreditation for quality designation consideration, whereas the 3 private payers allow accreditation from one of a variety of sources. Aetna Institutes of Quality for Orthopedic Surgery requires accreditation by TJC, Healthcare Facilities Accreditation Program, American Osteopathic Association, National Integrated Accreditation for Healthcare Organizations, or Det Norske Veritas Healthcare. UnitedHealth Premium Total Joint Replacement (TJR) Specialty Center requires accreditation by TJC and/or equivalent of TJC accreditation. However, TJC accreditation equivalents are not noted in the UnitedHealth handbook. BCBS Blue Distinction and Distinction+ require accreditation by TJC, Healthcare Facilities Accreditation Program, National Integrated Accreditation for Healthcare Organizations, or Center for Improvement in Healthcare Quality. In addition, BCBS is willing to consider alternative accreditations that are at least as stringent as the national alternatives noted. However, no detailed criteria that must be met to be equivalent to the national standards are noted in the relevant quality designation handbook.
The volume of completed total hip and knee arthroplasty procedures was considered in 6 of the organizations’ quality ratings methodologies. Of those 6, all private payers, TJC (not-for-profit), and 2 for-profit rating agencies were included. Surgeon specialization in TJA was only explicitly noted as a factor considered in UnitedHealth Premium TJR Specialty Center criteria; however, the requirements for surgeon specialization were not clearly defined. In addition, the presence of a multidisciplinary clinical pathway was only explicitly considered for Aetna Institutes of Quality for Orthopedic Surgery.
Structural requirements (eg, use of electronic health records [EHR], staffing levels, etc.) were taken into account in private payer and USNWR quality methodologies. Process measures (eg, antibiotic prophylaxis and other care pathways) were considered for the private payers and TJC but not for USNWR quality designation. Cost and/or efficiency measures were factors in the quality formula for Aetna Institutes of Quality for Orthopedic Surgery and BCBS Distinction+. Aetna utilizes its own cost data and risk-adjusts using a product known as Symmetry Episode Risk Groups to determine cost-effectiveness, while BCBS uses its own Composite Facility Cost Index. Patient experience (eg, Hospital Consumer Assessment of Healthcare Providers and Systems [HCAHPS]) was incorporated into the quality formulas for 4 of the 7 quality designation programs examined.
Continue to: All of the 7 quality designation programs included...
All of the 7 quality designation programs included outcomes (ie, readmission rates and/or mortality rates) and publicly reported the hospitals receiving their quality designation. In contrast, only Aetna explicitly included the presence of multidisciplinary clinical care pathways as part of their quality designation criteria. In addition, only UnitedHealth included surgeon specialization in joint arthroplasty as a factor for quality consideration for its quality designation program. BCBS Distinction+ and Aetna Institutes of Quality for Orthopedic Surgery were the only 2 quality designations that included at least 1 variable that fit into each of the 7 characteristics considered (accreditation, volume, structural, process, outcomes, patient experience, and cost/efficiency).
DISCUSSION
As healthcare continues to shift toward value-based delivery and payment models, quality becomes a critical factor in reimbursement and provider rankings. However, quality is a vague term. Several providers probably do not know what is required to be designated as high quality by a particular rating agency. Moreover, there are multiple quality designation programs, all using distinct criteria to determine “quality,” which further complicates the matter. Our objective was to determine the key stakeholders that provide quality designations in TJA and what criteria each organization uses in assessing quality.
Our idea of comprehensive quality is based on Avedis Donabedian’s enduring framework for healthcare quality focused on structure, process, and outcome.16 We expanded on these 3 areas and analyzed quality designations based on variables fitting into the following categories: accreditation, volume, structural, process, outcomes, patient experience, and cost/efficiency. We believe that these categories encompass a comprehensive rating system that addresses key elements of patient care. However, our results suggest that only 2 major quality designations (BCBS Distinction+ and Aetna Institutes of Quality for Orthopedic Surgery) take all such variables into account.
All quality designation programs that we analyzed required outcome data (ie, readmission and/or mortality rates within 30 days); however, only 2 programs utilized cost in their quality designation criteria (BCBS Distinction+ and Aetna Institutes of Quality for Orthopedic Surgery). Aetna Institutes of Quality for Orthopedic Surgery risk-adjusted for its cost-effectiveness calculations based on age, sex, and other unspecified conditions using a product known as Symmetry Episode Risk Groups. However, the organization also noted that although it did risk-adjust for inpatient mortality, it did not do so for pulmonary embolism or deep vein thrombosis. BCBS Distinction+ also utilized risk adjustment for its cost efficiency measure, and its step-by-step methodology is available online. Further, Consumer Reports does risk-adjust using logistic regression models in their quality analysis, but the description provided is minimal; it is noted that such risk adjustments are already completed by CMS prior to Consumer Reports acquiring the data. The CMS Compare model information is available on the CMS website. The data utilized by several organizations and presented on CMS Compare are already risk-adjusted using CMS’ approach. In contrast, UnitedHealth Premium TJR Specialty Center gathers its own data from providers and does not describe a risk adjustment methodology. Risk adjustment is important because the lack of risk adjustment may lead to physicians “cherry-picking” easy cases to boost positive outcomes, leading to increased financial benefits and higher quality ratings. Having a consistent risk adjustment formula will ensure accurate comparisons across outcomes and cost-effectiveness measures used by quality designation programs.
Factors considered for quality designation varied greatly from one organization to the other. The range of categories of factors considered varied from 1 (Consumer Reports only considered outcome data) to all 7 categories (BCBS Distinction+ and Aetna Institutes of Quality for Orthopedic Surgery). Our findings are consistent with the work by Keswani and colleagues,8 which showed that there is likely variation in factors considered when rating hospital quality more broadly. Our work suggests that quality designation formulas do not appear to get more consistent when focused on TJA.
We found that all organizations in our analysis published the providers earning their quality designation. However, TJC does not provide publicly a detailed methodology on how to qualify for its quality designation. The price to purchase the necessary manual for this information is $146.00 for accredited organizations and $186.00 for all others.17 For large healthcare providers, this is not a large sum of money. Nonetheless, this provides an additional hurdle for stakeholders to gain a full understanding of the requirements to receive a TJC Gold Seal for Orthopedics.
Previous work has evaluated the consistency of and the variety of means of gauging healthcare quality. Previous work by Rothberg and colleagues18 comparing hospital rankings across 5 common consumer-oriented websites found disagreement on hospital rankings within any diagnosis and even among metrics such as mortality. Another study by Halasyamani and Davis19 found that CMS Compare and USNWR rankings were dissimilar and the authors attributed the discrepancy to different methodologies. In addition, a study by Krumholz and colleagues20 focused on Internet report cards, which measured the appropriate use of select medications and mortality rates for acute myocardial infarction as the quality metrics. The authors found that, in aggregate, there was a clear difference in quality of care and outcomes but that comparisons between 2 hospitals provided poor discrimination.20 Other work has analyzed the increasing trend of online ratings of orthopedic surgeons by patients.21 However, there remains no agreed-upon definition of quality. Thus, the use of the term “quality” in several studies may be misleading.
Our results must be interpreted keeping the limitations of our work in mind. First, we used expert knowledge and a public search engine to develop our list of organizations that provide TJA quality designations. However, there is a possibility that we did not include all relevant organizations. Second, although all authors reviewed the final data, it is possible that there was human error in the analysis of each organization’s quality designation criteria.
CONCLUSION
As healthcare progresses further toward a system that rewards providers for delivering value to patients, accurately defining and measuring quality becomes critical because it can be suggestive of value to patients, payers, and providers. Furthermore, it gives providers a goal to focus on as they strive to improve the value of care they deliver to patients. Measuring healthcare quality is currently a novel, imperfect science,22 and there continues to be a debate about what factors should be included in a quality designation formula. Nonetheless, more and more quality designations and performance measurements are being created for orthopedic care, including total hip and total knee arthroplasty. In fact, in 2016, The Leapfrog Group added readmission for patients undergoing TJA to its survey.23 Consensus on a quality definition may facilitate the movement toward a value-based healthcare system. Future research should evaluate strategies for gaining consensus among stakeholders for a universal quality metric in TJA. Surgeons, hospitals, payers, and most importantly patients should play critical roles in defining quality.
- Porter ME. A strategy for health care reform--toward a value-based system. N Engl J Med. 2009;361(2):109-112. doi:10.1056/NEJMp0904131.
- Obama B. United States health care reform: progress to date and next steps. JAMA. 2016;316(5):525-532. doi:10.1001/jama.2016.9797.
- Mulvany C. The march to consumerism the evolution from patient to active shopper continues. Healthc Financ Manage. 2014;68(2):36-38.
- Tsai TC, Jha AK. Hospital consolidation, competition, and quality: is bigger necessarily better? JAMA. 2014;312(1):29-30. doi:10.1001/jama.2014.4692.
- Cline RJ, Haynes KM. Consumer health information seeking on the Internet: the state of the art. Health Educ Res. 2001;16(6):671-692. doi:10.1093/her/16.6.671.
- Werner RM, Kolstad JT, Stuart EA, Polsky D. The effect of pay-for-performance in hospitals: lessons for quality improvement. Health Aff (Millwood). 2011;30(4):690-698. doi:10.1377/hlthaff.2010.1277.
- Birkmeyer JD, Dimick JB, Birkmeyer NJ. Measuring the quality of surgical care: structure, process, or outcomes? J Am Coll Surg. 2004;198(4):626-632. doi:10.1016/j.jamcollsurg.2003.11.017.
- Keswani A, Uhler LM, Bozic KJ. What quality metrics is my hospital being evaluated on and what are the consequences? J Arthroplast. 2016;31(6):1139-1143. doi:10.1016/j.arth.2016.01.075.
- Aetna Inc. Aetna Institutes of Quality® facilities fact book. A comprehensive reference guide for Aetna members, doctors and health care professionals. http://www.aetna.com/individuals-families-health-insurance/document-libr.... Accessed July 2, 2016.
- United HealthCare. UnitedHealth Premium® Program. https://www.uhcprovider.com/en/reports-quality-programs/premium-designation.html. Accessed July 2, 2016.
- 11. Blue Cross Blue Shield. Association. Blue Distinction Specialty Care. Selection criteria and program documentation: knee and hip replacement and spine surgery. https://www.bcbs.com/sites/default/files/fileattachments/page/KneeHip.SelectionCriteria_0.pdf. Published October 2015. Accessed July 2, 2016.
- The Joint Commission. Advanced certification for total hip and total knee replacement eligibility. https://www.jointcommission.org/advanced_certification_for_total_hip_and.... Published December 10, 2015. Accessed July 2, 2016.
- Healthgrades Operating Company. Healthgrades methodology: anatomy of a rating. https://www.healthgrades.com/quality/ratings-awards/methodology. Accessed July 2, 2016.
- Comarow A, Harder B; Dr. Foster Project Team. Methodology: U.S. News & World Report best hospitals for common care. U.S. News & World Report Web site. http://www.usnews.com/pubfiles/BHCC_MethReport_2015.pdf. Published May 20, 2015. Accessed July 2, 2016.
- Consumer Reports. How we rate hospitals. http://static3.consumerreportscdn.org/content/dam/cro/news_articles/heal.... Accessed July 2, 2016.
- Ayanian JZ, Markel H. Donabedian’s lasting framework for health care quality. N Engl J Med. 2016;375(3):205-207. doi:10.1056/NEJMp1605101.
- The Joint Commission. 2016 Certification Manuals. 2016; http://www.jcrinc.com/2016-certification-manuals/. Accessed July 2, 2016.
- Rothberg MB, Morsi E, Benjamin EM, Pekow PS, Lindenauer PK. Choosing the best hospital: the limitations of public quality reporting. Health Aff (Millwood). 2008;27(6):1680-1687. doi:10.1377/hlthaff.27.6.1680.
- Halasyamani LK, Davis MM. Conflicting measures of hospital quality: ratings from "Hospital Compare" versus "Best Hospitals". J Hosp Med. 2007;2(3):128-134. doi:10.1002/jhm.176.
- Krumholz HM, Rathore SS, Chen J, Wang Y, Radford MJ. Evaluation of a consumer-oriented internet health care report card: the risk of quality ratings based on mortality data. JAMA. 2002;287(10):1277-1287.
- Frost C, Mesfin A. Online reviews of orthopedic surgeons: an emerging trend. Orthopedics. 2015;38(4):e257-e262. doi:10.3928/01477447-20150402-52.
- Harder B, Comarow A. Hospital Quality reporting by US News & World Report: why, how, and what's ahead. JAMA. 2015;313(19):1903-1904. doi:10.1001/jama.2015.4566.
- The Leapfrog Group. New in 2016. http://www.leapfroggroup.org/ratings-reports/new-2016. Accessed July 2, 2016.
- Porter ME. A strategy for health care reform--toward a value-based system. N Engl J Med. 2009;361(2):109-112. doi:10.1056/NEJMp0904131.
- Obama B. United States health care reform: progress to date and next steps. JAMA. 2016;316(5):525-532. doi:10.1001/jama.2016.9797.
- Mulvany C. The march to consumerism the evolution from patient to active shopper continues. Healthc Financ Manage. 2014;68(2):36-38.
- Tsai TC, Jha AK. Hospital consolidation, competition, and quality: is bigger necessarily better? JAMA. 2014;312(1):29-30. doi:10.1001/jama.2014.4692.
- Cline RJ, Haynes KM. Consumer health information seeking on the Internet: the state of the art. Health Educ Res. 2001;16(6):671-692. doi:10.1093/her/16.6.671.
- Werner RM, Kolstad JT, Stuart EA, Polsky D. The effect of pay-for-performance in hospitals: lessons for quality improvement. Health Aff (Millwood). 2011;30(4):690-698. doi:10.1377/hlthaff.2010.1277.
- Birkmeyer JD, Dimick JB, Birkmeyer NJ. Measuring the quality of surgical care: structure, process, or outcomes? J Am Coll Surg. 2004;198(4):626-632. doi:10.1016/j.jamcollsurg.2003.11.017.
- Keswani A, Uhler LM, Bozic KJ. What quality metrics is my hospital being evaluated on and what are the consequences? J Arthroplast. 2016;31(6):1139-1143. doi:10.1016/j.arth.2016.01.075.
- Aetna Inc. Aetna Institutes of Quality® facilities fact book. A comprehensive reference guide for Aetna members, doctors and health care professionals. http://www.aetna.com/individuals-families-health-insurance/document-libr.... Accessed July 2, 2016.
- United HealthCare. UnitedHealth Premium® Program. https://www.uhcprovider.com/en/reports-quality-programs/premium-designation.html. Accessed July 2, 2016.
- 11. Blue Cross Blue Shield. Association. Blue Distinction Specialty Care. Selection criteria and program documentation: knee and hip replacement and spine surgery. https://www.bcbs.com/sites/default/files/fileattachments/page/KneeHip.SelectionCriteria_0.pdf. Published October 2015. Accessed July 2, 2016.
- The Joint Commission. Advanced certification for total hip and total knee replacement eligibility. https://www.jointcommission.org/advanced_certification_for_total_hip_and.... Published December 10, 2015. Accessed July 2, 2016.
- Healthgrades Operating Company. Healthgrades methodology: anatomy of a rating. https://www.healthgrades.com/quality/ratings-awards/methodology. Accessed July 2, 2016.
- Comarow A, Harder B; Dr. Foster Project Team. Methodology: U.S. News & World Report best hospitals for common care. U.S. News & World Report Web site. http://www.usnews.com/pubfiles/BHCC_MethReport_2015.pdf. Published May 20, 2015. Accessed July 2, 2016.
- Consumer Reports. How we rate hospitals. http://static3.consumerreportscdn.org/content/dam/cro/news_articles/heal.... Accessed July 2, 2016.
- Ayanian JZ, Markel H. Donabedian’s lasting framework for health care quality. N Engl J Med. 2016;375(3):205-207. doi:10.1056/NEJMp1605101.
- The Joint Commission. 2016 Certification Manuals. 2016; http://www.jcrinc.com/2016-certification-manuals/. Accessed July 2, 2016.
- Rothberg MB, Morsi E, Benjamin EM, Pekow PS, Lindenauer PK. Choosing the best hospital: the limitations of public quality reporting. Health Aff (Millwood). 2008;27(6):1680-1687. doi:10.1377/hlthaff.27.6.1680.
- Halasyamani LK, Davis MM. Conflicting measures of hospital quality: ratings from "Hospital Compare" versus "Best Hospitals". J Hosp Med. 2007;2(3):128-134. doi:10.1002/jhm.176.
- Krumholz HM, Rathore SS, Chen J, Wang Y, Radford MJ. Evaluation of a consumer-oriented internet health care report card: the risk of quality ratings based on mortality data. JAMA. 2002;287(10):1277-1287.
- Frost C, Mesfin A. Online reviews of orthopedic surgeons: an emerging trend. Orthopedics. 2015;38(4):e257-e262. doi:10.3928/01477447-20150402-52.
- Harder B, Comarow A. Hospital Quality reporting by US News & World Report: why, how, and what's ahead. JAMA. 2015;313(19):1903-1904. doi:10.1001/jama.2015.4566.
- The Leapfrog Group. New in 2016. http://www.leapfroggroup.org/ratings-reports/new-2016. Accessed July 2, 2016.
TAKE-HOME POINTS
- TJA quality designation methodologies differ substantially across rating organizations.
- Only 29% of TJA quality rating methodologies evaluated include a cost element.
- Only 57% of TJA quality rating methodologies evaluated include patient experience.
- Only 57% of TJA quality rating methodologies evaluated include process measurements, including antibiotic prophylaxis and standardized care pathways.
- There is a need for consistent definitions of quality as healthcare stakeholders continue to shift focus from volume to value.
Heather Snyder, PhD

World off track for meeting 2020 HIV prevention, mortality goals
Globally, new HIV infections have declined by just 18% in the past 7 years, but the decline is not quick enough to reach the United Nations’ target goal of reduced deaths and infections by 2020, according to a warning issued by UNAIDS in its July 2018 AIDS update report, Miles to Go: Closing gaps, breaking barriers, righting injustices.
“We are sounding the alarm. Entire regions are falling behind, the huge gains we made for children are not being sustained, women are still most affected, resources are still not matching political commitments, and key populations continue to be ignored,” Michel Sidibé, executive director of UNAIDS, said in a press release.
Even though almost 60% of the 36.9 million people living with HIV were on treatment in 2017, to reach the 30 million target an annual increase of 2.8 million people is required, and indications are that the rate of scale-up is slowing down, according to the report. 
Not meeting the 2020 United Nations self-imposed fast-track deadline for reducing HIV infections and AIDS-related deaths by 5 years (moving it to 2025), will mean an additional 2.1 million infections and 1.0 million more AIDS-related deaths.
Part of the problem is localization of new therapies for prevention. For example, two-thirds of the estimated 350,000 people globally who have ever started pre-exposure prophylaxis (PrEP) are in the United States.
In Africa, little progress in preventing new infections has been achieved outside of the sub-Saharan region, and in eastern Europe and central Asia, the annual number of new HIV infections approximately doubled since 2000, according to the report.
Other portions of the report deal with the impact of law and politics, including the stigmatization and criminalization around the world of men who have sex with men, and the role of poverty and inequality as it affects the scope of the HIV goals, especially the problem of food insecurity, and how women and children are put at special risk by socioeconomic factors.
Globally, new HIV infections have declined by just 18% in the past 7 years, but the decline is not quick enough to reach the United Nations’ target goal of reduced deaths and infections by 2020, according to a warning issued by UNAIDS in its July 2018 AIDS update report, Miles to Go: Closing gaps, breaking barriers, righting injustices.
“We are sounding the alarm. Entire regions are falling behind, the huge gains we made for children are not being sustained, women are still most affected, resources are still not matching political commitments, and key populations continue to be ignored,” Michel Sidibé, executive director of UNAIDS, said in a press release.
Even though almost 60% of the 36.9 million people living with HIV were on treatment in 2017, to reach the 30 million target an annual increase of 2.8 million people is required, and indications are that the rate of scale-up is slowing down, according to the report.
Not meeting the 2020 United Nations self-imposed fast-track deadline for reducing HIV infections and AIDS-related deaths by 5 years (moving it to 2025), will mean an additional 2.1 million infections and 1.0 million more AIDS-related deaths.
Part of the problem is localization of new therapies for prevention. For example, two-thirds of the estimated 350,000 people globally who have ever started pre-exposure prophylaxis (PrEP) are in the United States.
In Africa, little progress in preventing new infections has been achieved outside of the sub-Saharan region, and in eastern Europe and central Asia, the annual number of new HIV infections approximately doubled since 2000, according to the report.
Other portions of the report deal with the impact of law and politics, including the stigmatization and criminalization around the world of men who have sex with men, and the role of poverty and inequality as it affects the scope of the HIV goals, especially the problem of food insecurity, and how women and children are put at special risk by socioeconomic factors.
Globally, new HIV infections have declined by just 18% in the past 7 years, but the decline is not quick enough to reach the United Nations’ target goal of reduced deaths and infections by 2020, according to a warning issued by UNAIDS in its July 2018 AIDS update report, Miles to Go: Closing gaps, breaking barriers, righting injustices.
“We are sounding the alarm. Entire regions are falling behind, the huge gains we made for children are not being sustained, women are still most affected, resources are still not matching political commitments, and key populations continue to be ignored,” Michel Sidibé, executive director of UNAIDS, said in a press release.
Even though almost 60% of the 36.9 million people living with HIV were on treatment in 2017, to reach the 30 million target an annual increase of 2.8 million people is required, and indications are that the rate of scale-up is slowing down, according to the report.
Not meeting the 2020 United Nations self-imposed fast-track deadline for reducing HIV infections and AIDS-related deaths by 5 years (moving it to 2025), will mean an additional 2.1 million infections and 1.0 million more AIDS-related deaths.
Part of the problem is localization of new therapies for prevention. For example, two-thirds of the estimated 350,000 people globally who have ever started pre-exposure prophylaxis (PrEP) are in the United States.
In Africa, little progress in preventing new infections has been achieved outside of the sub-Saharan region, and in eastern Europe and central Asia, the annual number of new HIV infections approximately doubled since 2000, according to the report.
Other portions of the report deal with the impact of law and politics, including the stigmatization and criminalization around the world of men who have sex with men, and the role of poverty and inequality as it affects the scope of the HIV goals, especially the problem of food insecurity, and how women and children are put at special risk by socioeconomic factors.
Psoriasis, Etanercept, and Myelodysplasia: Looking for Connections
Physicians from Menoufia University and Cairo University in Egypt, and Al Hada Armed Forces Hospital in Saudi Arabia report on a patient who developed myelodysplasia with excess blasts 1 year after he started on the tumor necrosis factor-alpha blocker etanercept for psoriasis. The patient, a 76-year-old man, arrived at the emergency department (ED) with ecchymosis and recurrent epistaxis. He had a critically low platelet count, anemia, and normal leukocyte count. The reticulocyte index, serum ferritin, and folate levels indicated ineffective erythropoiesis. Bone marrow aspirate and biopsy confirmed a diagnosis of myelodysplastic syndrome.
The physicians stopped the etanercept and administered 2 cycles of azacitidine and folic acid supplementation, but the response was minima,l and the patient platelet count worsened. While waiting for the third cycle, the patient was readmitted to the ED with lower gastrointestinal bleeding, epistaxis, and shock. He died of cardiopulmonary arrest.
The physicians note that immune dysregulation and altered T-cell hemostasis are essential to the development of myelodysplastic syndrome. They also note that nonspecific activation and proliferation of T lymphocytes has been documented as promoting epidermal growth in genetically susceptible psoriasis patients.
Myelodysplastic syndrome has been associated with psoriasis in about 7% of cases, and researchers have found a higher incidence of leukemia and laryngeal cancer in families of psoriasis patients. There also have been reports of leukemia in psoriasis patients on systemic immunosuppressives. Etanercept has various hematologic adverse effects, including pancytopenia and aplastic anemia.
However, only 4 cases (including this one) have been reported of myelodysplastic syndrome in psoriasis patients. Taken together, the cases add to the growing evidence that suggests a link between myelodysplastic syndrome and etanercept treatment for psoriasis. Those patients, the physicians caution, should be considered at dual risk from treatment and disease. The physicians also recommend regular routine blood counts and discontinuing etanercept at onset of any cytopenias.
Source:
Dawoud NM, Ayoub OH, Essa ES, Dawoud DM. Indian J Dermatol Venereol Leprol. 2018;84(4):463-465.
doi: 10.4103/ijdvl.IJDVL_463_17
Physicians from Menoufia University and Cairo University in Egypt, and Al Hada Armed Forces Hospital in Saudi Arabia report on a patient who developed myelodysplasia with excess blasts 1 year after he started on the tumor necrosis factor-alpha blocker etanercept for psoriasis. The patient, a 76-year-old man, arrived at the emergency department (ED) with ecchymosis and recurrent epistaxis. He had a critically low platelet count, anemia, and normal leukocyte count. The reticulocyte index, serum ferritin, and folate levels indicated ineffective erythropoiesis. Bone marrow aspirate and biopsy confirmed a diagnosis of myelodysplastic syndrome.
The physicians stopped the etanercept and administered 2 cycles of azacitidine and folic acid supplementation, but the response was minima,l and the patient platelet count worsened. While waiting for the third cycle, the patient was readmitted to the ED with lower gastrointestinal bleeding, epistaxis, and shock. He died of cardiopulmonary arrest.
The physicians note that immune dysregulation and altered T-cell hemostasis are essential to the development of myelodysplastic syndrome. They also note that nonspecific activation and proliferation of T lymphocytes has been documented as promoting epidermal growth in genetically susceptible psoriasis patients.
Myelodysplastic syndrome has been associated with psoriasis in about 7% of cases, and researchers have found a higher incidence of leukemia and laryngeal cancer in families of psoriasis patients. There also have been reports of leukemia in psoriasis patients on systemic immunosuppressives. Etanercept has various hematologic adverse effects, including pancytopenia and aplastic anemia.
However, only 4 cases (including this one) have been reported of myelodysplastic syndrome in psoriasis patients. Taken together, the cases add to the growing evidence that suggests a link between myelodysplastic syndrome and etanercept treatment for psoriasis. Those patients, the physicians caution, should be considered at dual risk from treatment and disease. The physicians also recommend regular routine blood counts and discontinuing etanercept at onset of any cytopenias.
Source:
Dawoud NM, Ayoub OH, Essa ES, Dawoud DM. Indian J Dermatol Venereol Leprol. 2018;84(4):463-465.
doi: 10.4103/ijdvl.IJDVL_463_17
Physicians from Menoufia University and Cairo University in Egypt, and Al Hada Armed Forces Hospital in Saudi Arabia report on a patient who developed myelodysplasia with excess blasts 1 year after he started on the tumor necrosis factor-alpha blocker etanercept for psoriasis. The patient, a 76-year-old man, arrived at the emergency department (ED) with ecchymosis and recurrent epistaxis. He had a critically low platelet count, anemia, and normal leukocyte count. The reticulocyte index, serum ferritin, and folate levels indicated ineffective erythropoiesis. Bone marrow aspirate and biopsy confirmed a diagnosis of myelodysplastic syndrome.
The physicians stopped the etanercept and administered 2 cycles of azacitidine and folic acid supplementation, but the response was minima,l and the patient platelet count worsened. While waiting for the third cycle, the patient was readmitted to the ED with lower gastrointestinal bleeding, epistaxis, and shock. He died of cardiopulmonary arrest.
The physicians note that immune dysregulation and altered T-cell hemostasis are essential to the development of myelodysplastic syndrome. They also note that nonspecific activation and proliferation of T lymphocytes has been documented as promoting epidermal growth in genetically susceptible psoriasis patients.
Myelodysplastic syndrome has been associated with psoriasis in about 7% of cases, and researchers have found a higher incidence of leukemia and laryngeal cancer in families of psoriasis patients. There also have been reports of leukemia in psoriasis patients on systemic immunosuppressives. Etanercept has various hematologic adverse effects, including pancytopenia and aplastic anemia.
However, only 4 cases (including this one) have been reported of myelodysplastic syndrome in psoriasis patients. Taken together, the cases add to the growing evidence that suggests a link between myelodysplastic syndrome and etanercept treatment for psoriasis. Those patients, the physicians caution, should be considered at dual risk from treatment and disease. The physicians also recommend regular routine blood counts and discontinuing etanercept at onset of any cytopenias.
Source:
Dawoud NM, Ayoub OH, Essa ES, Dawoud DM. Indian J Dermatol Venereol Leprol. 2018;84(4):463-465.
doi: 10.4103/ijdvl.IJDVL_463_17
VA Funds Intimate Partner Violence Programs
In the US, 36% of women and 29% of men have experienced rape, physical violence, or stalking by an intimate partner. Research suggests that veterans may be at greater risk for intimate partner violence than civilian counterparts, given the unique stressors posed by military life, such as military deployments that result in family separation, reintegration issues, and combat-related health issues, including PTSD and TBI. According to the VA’s Domestic Violence Task Force, the overall 12-month prevalence of inmate partner violence (IPV) perpetration among active duty service members was 22%, and victimization was 30%.
To help address this problem, the VA launched the IPV Assistance Program in 2014 and has since established coordinators at more than 115 facilities. The program coordinators use resources from mental health, primary care, women’s health, veterans’ justice outreach, and employee occupational health and assistance programs. The program also offers intervention through VA and community partnerships that address housing, education, and employment needs.
The program takes a holistic approach, focusing on developing a culture of safety, the VA says, with the goal of understanding, recognizing and responding to the effects of all types of trauma, including physical, sexual, and psychological. “We are giving careful attention to this program,” says Acting VA Secretary Peter O’Rourke, “ensuring it is integrated into clinical care and workplace safety.”
In the US, 36% of women and 29% of men have experienced rape, physical violence, or stalking by an intimate partner. Research suggests that veterans may be at greater risk for intimate partner violence than civilian counterparts, given the unique stressors posed by military life, such as military deployments that result in family separation, reintegration issues, and combat-related health issues, including PTSD and TBI. According to the VA’s Domestic Violence Task Force, the overall 12-month prevalence of inmate partner violence (IPV) perpetration among active duty service members was 22%, and victimization was 30%.
To help address this problem, the VA launched the IPV Assistance Program in 2014 and has since established coordinators at more than 115 facilities. The program coordinators use resources from mental health, primary care, women’s health, veterans’ justice outreach, and employee occupational health and assistance programs. The program also offers intervention through VA and community partnerships that address housing, education, and employment needs.
The program takes a holistic approach, focusing on developing a culture of safety, the VA says, with the goal of understanding, recognizing and responding to the effects of all types of trauma, including physical, sexual, and psychological. “We are giving careful attention to this program,” says Acting VA Secretary Peter O’Rourke, “ensuring it is integrated into clinical care and workplace safety.”
In the US, 36% of women and 29% of men have experienced rape, physical violence, or stalking by an intimate partner. Research suggests that veterans may be at greater risk for intimate partner violence than civilian counterparts, given the unique stressors posed by military life, such as military deployments that result in family separation, reintegration issues, and combat-related health issues, including PTSD and TBI. According to the VA’s Domestic Violence Task Force, the overall 12-month prevalence of inmate partner violence (IPV) perpetration among active duty service members was 22%, and victimization was 30%.
To help address this problem, the VA launched the IPV Assistance Program in 2014 and has since established coordinators at more than 115 facilities. The program coordinators use resources from mental health, primary care, women’s health, veterans’ justice outreach, and employee occupational health and assistance programs. The program also offers intervention through VA and community partnerships that address housing, education, and employment needs.
The program takes a holistic approach, focusing on developing a culture of safety, the VA says, with the goal of understanding, recognizing and responding to the effects of all types of trauma, including physical, sexual, and psychological. “We are giving careful attention to this program,” says Acting VA Secretary Peter O’Rourke, “ensuring it is integrated into clinical care and workplace safety.”
Team creates tool to assess frailty in MM

Researchers say they have developed a frailty index that can predict overall survival (OS) in patients newly diagnosed with multiple myeloma (MM).
An increasing frailty index score was significantly associated with an increased risk of death in these patients, and frailty retained a significant association with OS even after the researchers controlled for patients’ chronological age.
“Our goal was to create a tool that could be widely applied using data sources at hand and that helps doctors provide better informed treatment recommendations for their patients,” said Tanya S. Wildes, MD, of the Washington University School of Medicine in St Louis, Missouri.
“Our results demonstrate that, for patients with multiple myeloma, chronological age alone is not a good measure for assessing overall health.”
Dr Wildes and her colleagues reported these results in JCO Clinical Cancer Informatics.
Creating the index
The researchers began this study with data from 2,692,361 patients without cancer who were older than 66 years of age. The data were collected from the Medicare Health Outcomes Survey (MHOS), which is used to annually collect self-reported symptoms, functional status, and health-related quality of life data from Medicare beneficiaries enrolled in Medicare Advantage plans.
The researchers used the MHOS data to create a deficit accumulation frailty index made up of a 25-item scale and scoring system. The index includes criteria in 5 categories for scoring frailty:
- Activities of daily living (eg, difficulty dressing or eating)
- Chronic health conditions
- Functioning (eg, difficulty walking or climbing several sets of stairs)
- General health
- Mental health.
Patients whose scores exceed a certain threshold on the scale are classified as frail.
Applying the index
The researchers applied their frailty index to 305 patients with newly diagnosed MM. Data from these patients were obtained from the Surveillance, Epidemiology, and End Results (SEER)-MHOS linked database. In this dataset, data from MHOS are linked to demographics, tumor characteristics, and survival for individuals with a cancer diagnosis who reside in the coverage area of the 14 registries participating in the SEER-MHOS linkage.
The researchers compared findings in the MM patients to findings in the patients without cancer.
In the non-cancer patients, the median age was 74, and the mean frailty score was 0.23. In the MM patients, the median age was 76, and the mean frailty score was 0.28.
Chronological age was weakly correlated with a higher frailty score in MM patients. However, for non-cancer patients, an increase in chronological age was strongly correlated with a higher frailty score.
Among non-cancer patients, each 10% increase in frailty score was associated with a 40% increased risk for death (adjusted hazard ratio, 1.397; P<0.001).
Among MM patients, each 10% increase in frailty score was associated with a 16% increased risk of death (adjusted hazard ratio, 1.159; P<0.001).
The median OS was 33 months for the entire MM cohort, 26.8 months for frail MM patients, and 43.7 months for non-frail MM patients (P=0.015 for the frail to non-frail comparison).
“These findings underscore the need to place more consideration on biological age versus chronological age in multiple myeloma, recognizing that frailty is dynamic and encompasses many factors beyond the disease itself,” Dr Wildes said.
“Ultimately, the hope is that this tool will help us to better personalize care based on a fuller picture of our patients’ health so that we are not under-treating an older adult who can tolerate a more intense therapy or over-treating one who’s going to be vulnerable to the toxicities of therapy.”
Limitations and next steps
The researchers believe there are several options for optimizing the data in the frailty index, including turning it into a computerized program and examining patients who are not newly diagnosed and have subsequent relapses, disease burden, and treatment toxicities.
This study is limited in the fact that researchers only assessed OS and not progression-free survival, chemotherapy toxicity, or hospitalization rates.
Additionally, the MM data was derived from patients enrolled in the Medicare Advantage program, which may have contributed to selecting participants who are, overall, lower-risk due to the way the program incentivizes lower-cost enrollees.
Researchers say they have developed a frailty index that can predict overall survival (OS) in patients newly diagnosed with multiple myeloma (MM).
An increasing frailty index score was significantly associated with an increased risk of death in these patients, and frailty retained a significant association with OS even after the researchers controlled for patients’ chronological age.
“Our goal was to create a tool that could be widely applied using data sources at hand and that helps doctors provide better informed treatment recommendations for their patients,” said Tanya S. Wildes, MD, of the Washington University School of Medicine in St Louis, Missouri.
“Our results demonstrate that, for patients with multiple myeloma, chronological age alone is not a good measure for assessing overall health.”
Dr Wildes and her colleagues reported these results in JCO Clinical Cancer Informatics.
Creating the index
The researchers began this study with data from 2,692,361 patients without cancer who were older than 66 years of age. The data were collected from the Medicare Health Outcomes Survey (MHOS), which is used to annually collect self-reported symptoms, functional status, and health-related quality of life data from Medicare beneficiaries enrolled in Medicare Advantage plans.
The researchers used the MHOS data to create a deficit accumulation frailty index made up of a 25-item scale and scoring system. The index includes criteria in 5 categories for scoring frailty:
- Activities of daily living (eg, difficulty dressing or eating)
- Chronic health conditions
- Functioning (eg, difficulty walking or climbing several sets of stairs)
- General health
- Mental health.
Patients whose scores exceed a certain threshold on the scale are classified as frail.
Applying the index
The researchers applied their frailty index to 305 patients with newly diagnosed MM. Data from these patients were obtained from the Surveillance, Epidemiology, and End Results (SEER)-MHOS linked database. In this dataset, data from MHOS are linked to demographics, tumor characteristics, and survival for individuals with a cancer diagnosis who reside in the coverage area of the 14 registries participating in the SEER-MHOS linkage.
The researchers compared findings in the MM patients to findings in the patients without cancer.
In the non-cancer patients, the median age was 74, and the mean frailty score was 0.23. In the MM patients, the median age was 76, and the mean frailty score was 0.28.
Chronological age was weakly correlated with a higher frailty score in MM patients. However, for non-cancer patients, an increase in chronological age was strongly correlated with a higher frailty score.
Among non-cancer patients, each 10% increase in frailty score was associated with a 40% increased risk for death (adjusted hazard ratio, 1.397; P<0.001).
Among MM patients, each 10% increase in frailty score was associated with a 16% increased risk of death (adjusted hazard ratio, 1.159; P<0.001).
The median OS was 33 months for the entire MM cohort, 26.8 months for frail MM patients, and 43.7 months for non-frail MM patients (P=0.015 for the frail to non-frail comparison).
“These findings underscore the need to place more consideration on biological age versus chronological age in multiple myeloma, recognizing that frailty is dynamic and encompasses many factors beyond the disease itself,” Dr Wildes said.
“Ultimately, the hope is that this tool will help us to better personalize care based on a fuller picture of our patients’ health so that we are not under-treating an older adult who can tolerate a more intense therapy or over-treating one who’s going to be vulnerable to the toxicities of therapy.”
Limitations and next steps
The researchers believe there are several options for optimizing the data in the frailty index, including turning it into a computerized program and examining patients who are not newly diagnosed and have subsequent relapses, disease burden, and treatment toxicities.
This study is limited in the fact that researchers only assessed OS and not progression-free survival, chemotherapy toxicity, or hospitalization rates.
Additionally, the MM data was derived from patients enrolled in the Medicare Advantage program, which may have contributed to selecting participants who are, overall, lower-risk due to the way the program incentivizes lower-cost enrollees.
Researchers say they have developed a frailty index that can predict overall survival (OS) in patients newly diagnosed with multiple myeloma (MM).
An increasing frailty index score was significantly associated with an increased risk of death in these patients, and frailty retained a significant association with OS even after the researchers controlled for patients’ chronological age.
“Our goal was to create a tool that could be widely applied using data sources at hand and that helps doctors provide better informed treatment recommendations for their patients,” said Tanya S. Wildes, MD, of the Washington University School of Medicine in St Louis, Missouri.
“Our results demonstrate that, for patients with multiple myeloma, chronological age alone is not a good measure for assessing overall health.”
Dr Wildes and her colleagues reported these results in JCO Clinical Cancer Informatics.
Creating the index
The researchers began this study with data from 2,692,361 patients without cancer who were older than 66 years of age. The data were collected from the Medicare Health Outcomes Survey (MHOS), which is used to annually collect self-reported symptoms, functional status, and health-related quality of life data from Medicare beneficiaries enrolled in Medicare Advantage plans.
The researchers used the MHOS data to create a deficit accumulation frailty index made up of a 25-item scale and scoring system. The index includes criteria in 5 categories for scoring frailty:
- Activities of daily living (eg, difficulty dressing or eating)
- Chronic health conditions
- Functioning (eg, difficulty walking or climbing several sets of stairs)
- General health
- Mental health.
Patients whose scores exceed a certain threshold on the scale are classified as frail.
Applying the index
The researchers applied their frailty index to 305 patients with newly diagnosed MM. Data from these patients were obtained from the Surveillance, Epidemiology, and End Results (SEER)-MHOS linked database. In this dataset, data from MHOS are linked to demographics, tumor characteristics, and survival for individuals with a cancer diagnosis who reside in the coverage area of the 14 registries participating in the SEER-MHOS linkage.
The researchers compared findings in the MM patients to findings in the patients without cancer.
In the non-cancer patients, the median age was 74, and the mean frailty score was 0.23. In the MM patients, the median age was 76, and the mean frailty score was 0.28.
Chronological age was weakly correlated with a higher frailty score in MM patients. However, for non-cancer patients, an increase in chronological age was strongly correlated with a higher frailty score.
Among non-cancer patients, each 10% increase in frailty score was associated with a 40% increased risk for death (adjusted hazard ratio, 1.397; P<0.001).
Among MM patients, each 10% increase in frailty score was associated with a 16% increased risk of death (adjusted hazard ratio, 1.159; P<0.001).
The median OS was 33 months for the entire MM cohort, 26.8 months for frail MM patients, and 43.7 months for non-frail MM patients (P=0.015 for the frail to non-frail comparison).
“These findings underscore the need to place more consideration on biological age versus chronological age in multiple myeloma, recognizing that frailty is dynamic and encompasses many factors beyond the disease itself,” Dr Wildes said.
“Ultimately, the hope is that this tool will help us to better personalize care based on a fuller picture of our patients’ health so that we are not under-treating an older adult who can tolerate a more intense therapy or over-treating one who’s going to be vulnerable to the toxicities of therapy.”
Limitations and next steps
The researchers believe there are several options for optimizing the data in the frailty index, including turning it into a computerized program and examining patients who are not newly diagnosed and have subsequent relapses, disease burden, and treatment toxicities.
This study is limited in the fact that researchers only assessed OS and not progression-free survival, chemotherapy toxicity, or hospitalization rates.
Additionally, the MM data was derived from patients enrolled in the Medicare Advantage program, which may have contributed to selecting participants who are, overall, lower-risk due to the way the program incentivizes lower-cost enrollees.
Plasma transfusion during air transport can reduce mortality

Receiving a plasma transfusion during emergency air transport can improve survival for trauma patients, according to a study published in NEJM.
Trauma patients with severe bleeding had a significant decrease in 30-day mortality when they received a plasma transfusion while being airlifted to a hospital.
Transfusion-related reactions and allergic reactions were more common among plasma recipients than patients who only received standard care.
However, this difference was not significant, and most of these reactions were considered minor.
“These results have the power to significantly alter trauma resuscitation, and their importance to the trauma community cannot be overstated,” said study author Jason Sperry, MD, of the University of Pittsburgh School of Medicine in Pennsylvania.
“This is the first trial in a quarter century to have the potential to alter prehospital care so considerably.”
Patients and intervention
This trial, known as PAMPer (Prehospital Air Medical Plasma), was a phase 3, randomized study enrolling 501 trauma patients at risk of hemorrhagic shock.
Most patients were male (72.7%), and most had suffered blunt trauma (82.4%). About half of patients (51.1%) had prehospital intubation, and more than a third (34.7%) received a prehospital transfusion of red blood cells.
Air medical bases participating in this study were randomized to administer plasma or standard care to eligible patients for 1-month intervals. When the air transport teams were in their plasma interval, they’d begin administering 2 units of thawed plasma to a patient as soon as trial eligibility was confirmed.
If the 2 units were completed during the flight, the team would revert to standard care. If the transfusions weren’t completed, the plasma would continue to be administered when the patient arrived at the trauma center.
The teams administered the assigned treatment 99% of the time (496/501).
In the plasma group, there were 205 patients (89.1%) who received 2 units of plasma, 21 (9.1%) who received 1 unit, and 4 patients (1.7%) who did not receive plasma due to logistical challenges.
In 84.4% of the patients, the plasma infusion was completed during air transport. The remaining patients completed their plasma transfusions at the trauma center.
There was 1 patient (0.4%) in the standard-care group who received plasma before transport began.
Primary outcome
The study’s primary outcome was 30-day mortality. Ninety-six percent of patients (n=481) had data for this outcome—220 patients in the plasma group and 261 in the standard-care group.
Thirty-day mortality was significantly lower in the plasma group than the standard-care group—23.2% and 33.0%, respectively (P=0.03).
In an adjusted analysis, the administration of prehospital plasma was associated with a 39% lower risk for 30-day mortality than standard care (adjusted odds ratio, 0.61; P=0.02).
Secondary outcomes
Initially, there were significant differences between the plasma (n=230) and standard-care groups (n=271) when it came to:
- Mortality at 24 hours—13.9% and 22.1%, respectively (P=0.02)
- In-hospital mortality—22.2% and 32.5%, respectively (P=0.01)
- Median volume of blood components transfused in the first 24 hours—3 and 4 units, respectively (P=0.02)
- Median volume of red cells transfused in the first 24 hours—3 and 4 units, respectively (P=0.03).
- Median prothrombin-time ratio at first blood sampling—1.2 and 1.3, respectively (P<0.001).
When the researchers adjusted P values for multiple comparisons, the between-group difference in prothrombin-time ratio remained significant (P<0.001).
However, the differences in 24-hour mortality (P=0.55), in-hospital mortality (P=0.33), blood components transfused (P=0.41), and red cells transfused (P=0.69) did not retain significance.
Likewise, there were no significant between-group differences (in adjusted or unadjusted analyses) when it came to multi-organ failure, acute lung injury/acute respiratory distress syndrome, nosocomial infections, or allergic/transfusion-related reactions.
There were 10 adverse events (AEs) considered related to the trial regimen. In the standard-care group, the 4 AEs were sepsis (a serious AE), adult respiratory distress syndrome (a serious AE), fever, and pain.
In the plasma group, the 6 AEs were 2 allergic reactions, 1 case of anaphylaxis, 1 case of hypotension, 1 case of urticaria, and 1 transfusion-related reaction (a serious AE).
Receiving a plasma transfusion during emergency air transport can improve survival for trauma patients, according to a study published in NEJM.
Trauma patients with severe bleeding had a significant decrease in 30-day mortality when they received a plasma transfusion while being airlifted to a hospital.
Transfusion-related reactions and allergic reactions were more common among plasma recipients than patients who only received standard care.
However, this difference was not significant, and most of these reactions were considered minor.
“These results have the power to significantly alter trauma resuscitation, and their importance to the trauma community cannot be overstated,” said study author Jason Sperry, MD, of the University of Pittsburgh School of Medicine in Pennsylvania.
“This is the first trial in a quarter century to have the potential to alter prehospital care so considerably.”
Patients and intervention
This trial, known as PAMPer (Prehospital Air Medical Plasma), was a phase 3, randomized study enrolling 501 trauma patients at risk of hemorrhagic shock.
Most patients were male (72.7%), and most had suffered blunt trauma (82.4%). About half of patients (51.1%) had prehospital intubation, and more than a third (34.7%) received a prehospital transfusion of red blood cells.
Air medical bases participating in this study were randomized to administer plasma or standard care to eligible patients for 1-month intervals. When the air transport teams were in their plasma interval, they’d begin administering 2 units of thawed plasma to a patient as soon as trial eligibility was confirmed.
If the 2 units were completed during the flight, the team would revert to standard care. If the transfusions weren’t completed, the plasma would continue to be administered when the patient arrived at the trauma center.
The teams administered the assigned treatment 99% of the time (496/501).
In the plasma group, there were 205 patients (89.1%) who received 2 units of plasma, 21 (9.1%) who received 1 unit, and 4 patients (1.7%) who did not receive plasma due to logistical challenges.
In 84.4% of the patients, the plasma infusion was completed during air transport. The remaining patients completed their plasma transfusions at the trauma center.
There was 1 patient (0.4%) in the standard-care group who received plasma before transport began.
Primary outcome
The study’s primary outcome was 30-day mortality. Ninety-six percent of patients (n=481) had data for this outcome—220 patients in the plasma group and 261 in the standard-care group.
Thirty-day mortality was significantly lower in the plasma group than the standard-care group—23.2% and 33.0%, respectively (P=0.03).
In an adjusted analysis, the administration of prehospital plasma was associated with a 39% lower risk for 30-day mortality than standard care (adjusted odds ratio, 0.61; P=0.02).
Secondary outcomes
Initially, there were significant differences between the plasma (n=230) and standard-care groups (n=271) when it came to:
- Mortality at 24 hours—13.9% and 22.1%, respectively (P=0.02)
- In-hospital mortality—22.2% and 32.5%, respectively (P=0.01)
- Median volume of blood components transfused in the first 24 hours—3 and 4 units, respectively (P=0.02)
- Median volume of red cells transfused in the first 24 hours—3 and 4 units, respectively (P=0.03).
- Median prothrombin-time ratio at first blood sampling—1.2 and 1.3, respectively (P<0.001).
When the researchers adjusted P values for multiple comparisons, the between-group difference in prothrombin-time ratio remained significant (P<0.001).
However, the differences in 24-hour mortality (P=0.55), in-hospital mortality (P=0.33), blood components transfused (P=0.41), and red cells transfused (P=0.69) did not retain significance.
Likewise, there were no significant between-group differences (in adjusted or unadjusted analyses) when it came to multi-organ failure, acute lung injury/acute respiratory distress syndrome, nosocomial infections, or allergic/transfusion-related reactions.
There were 10 adverse events (AEs) considered related to the trial regimen. In the standard-care group, the 4 AEs were sepsis (a serious AE), adult respiratory distress syndrome (a serious AE), fever, and pain.
In the plasma group, the 6 AEs were 2 allergic reactions, 1 case of anaphylaxis, 1 case of hypotension, 1 case of urticaria, and 1 transfusion-related reaction (a serious AE).
Receiving a plasma transfusion during emergency air transport can improve survival for trauma patients, according to a study published in NEJM.
Trauma patients with severe bleeding had a significant decrease in 30-day mortality when they received a plasma transfusion while being airlifted to a hospital.
Transfusion-related reactions and allergic reactions were more common among plasma recipients than patients who only received standard care.
However, this difference was not significant, and most of these reactions were considered minor.
“These results have the power to significantly alter trauma resuscitation, and their importance to the trauma community cannot be overstated,” said study author Jason Sperry, MD, of the University of Pittsburgh School of Medicine in Pennsylvania.
“This is the first trial in a quarter century to have the potential to alter prehospital care so considerably.”
Patients and intervention
This trial, known as PAMPer (Prehospital Air Medical Plasma), was a phase 3, randomized study enrolling 501 trauma patients at risk of hemorrhagic shock.
Most patients were male (72.7%), and most had suffered blunt trauma (82.4%). About half of patients (51.1%) had prehospital intubation, and more than a third (34.7%) received a prehospital transfusion of red blood cells.
Air medical bases participating in this study were randomized to administer plasma or standard care to eligible patients for 1-month intervals. When the air transport teams were in their plasma interval, they’d begin administering 2 units of thawed plasma to a patient as soon as trial eligibility was confirmed.
If the 2 units were completed during the flight, the team would revert to standard care. If the transfusions weren’t completed, the plasma would continue to be administered when the patient arrived at the trauma center.
The teams administered the assigned treatment 99% of the time (496/501).
In the plasma group, there were 205 patients (89.1%) who received 2 units of plasma, 21 (9.1%) who received 1 unit, and 4 patients (1.7%) who did not receive plasma due to logistical challenges.
In 84.4% of the patients, the plasma infusion was completed during air transport. The remaining patients completed their plasma transfusions at the trauma center.
There was 1 patient (0.4%) in the standard-care group who received plasma before transport began.
Primary outcome
The study’s primary outcome was 30-day mortality. Ninety-six percent of patients (n=481) had data for this outcome—220 patients in the plasma group and 261 in the standard-care group.
Thirty-day mortality was significantly lower in the plasma group than the standard-care group—23.2% and 33.0%, respectively (P=0.03).
In an adjusted analysis, the administration of prehospital plasma was associated with a 39% lower risk for 30-day mortality than standard care (adjusted odds ratio, 0.61; P=0.02).
Secondary outcomes
Initially, there were significant differences between the plasma (n=230) and standard-care groups (n=271) when it came to:
- Mortality at 24 hours—13.9% and 22.1%, respectively (P=0.02)
- In-hospital mortality—22.2% and 32.5%, respectively (P=0.01)
- Median volume of blood components transfused in the first 24 hours—3 and 4 units, respectively (P=0.02)
- Median volume of red cells transfused in the first 24 hours—3 and 4 units, respectively (P=0.03).
- Median prothrombin-time ratio at first blood sampling—1.2 and 1.3, respectively (P<0.001).
When the researchers adjusted P values for multiple comparisons, the between-group difference in prothrombin-time ratio remained significant (P<0.001).
However, the differences in 24-hour mortality (P=0.55), in-hospital mortality (P=0.33), blood components transfused (P=0.41), and red cells transfused (P=0.69) did not retain significance.
Likewise, there were no significant between-group differences (in adjusted or unadjusted analyses) when it came to multi-organ failure, acute lung injury/acute respiratory distress syndrome, nosocomial infections, or allergic/transfusion-related reactions.
There were 10 adverse events (AEs) considered related to the trial regimen. In the standard-care group, the 4 AEs were sepsis (a serious AE), adult respiratory distress syndrome (a serious AE), fever, and pain.
In the plasma group, the 6 AEs were 2 allergic reactions, 1 case of anaphylaxis, 1 case of hypotension, 1 case of urticaria, and 1 transfusion-related reaction (a serious AE).