User login
In the U.S., breastfeeding starts out strong, but drops off fast
More than half the infants born in the United States in 2015 (57.6%) were still breastfeeding at 6 months old – an improvement over 2014 survey results, and another step toward the 61% goal set forth in Healthy People 2020.

The CDC Breastfeeding Report Card for 2018 found that about 83% of infants born in 2015 started to breastfeed at birth. Further, five of eight Healthy People 2020 breastfeeding goals were met in 2015.
Yet, the numbers also paint a picture familiar to many clinicians and families: By 1 year, only 36% of infants were still breastfeeding, and just 25% of infants were exclusively breastfed through 6 months, as the American Academy of Pediatrics recommends. This puts the U.S. on the lower end of the global breastfeeding scale, with most countries reporting rates of 30% and higher, according to World Health Organization data. Globally, 41% of mothers breastfeed exclusively through 6 months, according to the latest UNICEF data.
The report concludes that U.S. mothers may not be getting the support they need from health care providers, family members, and employers to meet their breastfeeding goals.
“High breastfeeding initiation rates show that most mothers in the United States want to breastfeed and start out doing so,” the report states. “However, despite the recommendation to breastfeed exclusively for about the first 6 months, less than 50% of infants were exclusively breastfed through 3 months and about 25% were exclusively breastfed through 6 months.”
The CDC Breastfeeding Report Card provides national- and state-level data to help clinicians, child care providers, and families promote breastfeeding and support the women who choose it. In addition to providing an in-depth look at the numbers, the report compares current findings to the breastfeeding goals outlined in Healthy People 2020. This year, it looked at data on breastfeeding practices and support in all 50 states, the District of Columbia, Puerto Rico, the Virgin Islands, and – for the first time – Guam.
The report is published every 2 years, but the data comprising it are updated annually. The latest rates reflect breastfeeding practices among U.S. infants born in 2015 and are based on results of the 2016 and 2017 U.S. National Immunization Surveys.
Since the first report card in 2007, rates of exclusive breastfeeding at 3 and 6 months have increased. However, although rates for any breastfeeding at 6 and 12 months increased in 2015 from 2014 rates, there was no appreciable increase in exclusive breastfeeding at 3 and 6 months, the report noted. And about 17% of infants who were breastfed at birth still got some formula supplementation in the first 2 days of life.
Recognizing that breastfeeding support at the birth facility is a key driver of success, Healthy People 2020 tracks the proportion of live births in facilities that provided the recommended support. These “baby-friendly hospitals” receive a special designation from the WHO/UNICEF Baby-Friendly Hospital Initiative, and their numbers are increasing, the report noted. In 12 states, more than 40% of births occurred in such facilities, comprising more than 1 million infants in 2015 (26%), and far exceeding the HP2020 8% goal.
In a nation in which many new mothers must return to employment outside the home, breastfeeding support at work is crucial. Just 49% of employers provide breastfeeding facilities for these women, the report found. While this may seem less than optimal, it still exceeds the HP2020 goal of 38%.
“All sectors of society – family and friends, hospitals, health care offices/clinics, childcare facilities, community-based organizations, and workplaces – can play a role in improving the health of families by supporting breastfeeding,” the report said. “To reach their breastfeeding goals, mothers need continuity of care, which is achieved by consistent, collaborative, and high-quality breastfeeding services and support.”
SOURCE: CDC Breastfeeding Report Card
More than half the infants born in the United States in 2015 (57.6%) were still breastfeeding at 6 months old – an improvement over 2014 survey results, and another step toward the 61% goal set forth in Healthy People 2020.
The CDC Breastfeeding Report Card for 2018 found that about 83% of infants born in 2015 started to breastfeed at birth. Further, five of eight Healthy People 2020 breastfeeding goals were met in 2015.
Yet, the numbers also paint a picture familiar to many clinicians and families: By 1 year, only 36% of infants were still breastfeeding, and just 25% of infants were exclusively breastfed through 6 months, as the American Academy of Pediatrics recommends. This puts the U.S. on the lower end of the global breastfeeding scale, with most countries reporting rates of 30% and higher, according to World Health Organization data. Globally, 41% of mothers breastfeed exclusively through 6 months, according to the latest UNICEF data.
The report concludes that U.S. mothers may not be getting the support they need from health care providers, family members, and employers to meet their breastfeeding goals.
“High breastfeeding initiation rates show that most mothers in the United States want to breastfeed and start out doing so,” the report states. “However, despite the recommendation to breastfeed exclusively for about the first 6 months, less than 50% of infants were exclusively breastfed through 3 months and about 25% were exclusively breastfed through 6 months.”
The CDC Breastfeeding Report Card provides national- and state-level data to help clinicians, child care providers, and families promote breastfeeding and support the women who choose it. In addition to providing an in-depth look at the numbers, the report compares current findings to the breastfeeding goals outlined in Healthy People 2020. This year, it looked at data on breastfeeding practices and support in all 50 states, the District of Columbia, Puerto Rico, the Virgin Islands, and – for the first time – Guam.
The report is published every 2 years, but the data comprising it are updated annually. The latest rates reflect breastfeeding practices among U.S. infants born in 2015 and are based on results of the 2016 and 2017 U.S. National Immunization Surveys.
Since the first report card in 2007, rates of exclusive breastfeeding at 3 and 6 months have increased. However, although rates for any breastfeeding at 6 and 12 months increased in 2015 from 2014 rates, there was no appreciable increase in exclusive breastfeeding at 3 and 6 months, the report noted. And about 17% of infants who were breastfed at birth still got some formula supplementation in the first 2 days of life.
Recognizing that breastfeeding support at the birth facility is a key driver of success, Healthy People 2020 tracks the proportion of live births in facilities that provided the recommended support. These “baby-friendly hospitals” receive a special designation from the WHO/UNICEF Baby-Friendly Hospital Initiative, and their numbers are increasing, the report noted. In 12 states, more than 40% of births occurred in such facilities, comprising more than 1 million infants in 2015 (26%), and far exceeding the HP2020 8% goal.
In a nation in which many new mothers must return to employment outside the home, breastfeeding support at work is crucial. Just 49% of employers provide breastfeeding facilities for these women, the report found. While this may seem less than optimal, it still exceeds the HP2020 goal of 38%.
“All sectors of society – family and friends, hospitals, health care offices/clinics, childcare facilities, community-based organizations, and workplaces – can play a role in improving the health of families by supporting breastfeeding,” the report said. “To reach their breastfeeding goals, mothers need continuity of care, which is achieved by consistent, collaborative, and high-quality breastfeeding services and support.”
SOURCE: CDC Breastfeeding Report Card
More than half the infants born in the United States in 2015 (57.6%) were still breastfeeding at 6 months old – an improvement over 2014 survey results, and another step toward the 61% goal set forth in Healthy People 2020.
The CDC Breastfeeding Report Card for 2018 found that about 83% of infants born in 2015 started to breastfeed at birth. Further, five of eight Healthy People 2020 breastfeeding goals were met in 2015.
Yet, the numbers also paint a picture familiar to many clinicians and families: By 1 year, only 36% of infants were still breastfeeding, and just 25% of infants were exclusively breastfed through 6 months, as the American Academy of Pediatrics recommends. This puts the U.S. on the lower end of the global breastfeeding scale, with most countries reporting rates of 30% and higher, according to World Health Organization data. Globally, 41% of mothers breastfeed exclusively through 6 months, according to the latest UNICEF data.
The report concludes that U.S. mothers may not be getting the support they need from health care providers, family members, and employers to meet their breastfeeding goals.
“High breastfeeding initiation rates show that most mothers in the United States want to breastfeed and start out doing so,” the report states. “However, despite the recommendation to breastfeed exclusively for about the first 6 months, less than 50% of infants were exclusively breastfed through 3 months and about 25% were exclusively breastfed through 6 months.”
The CDC Breastfeeding Report Card provides national- and state-level data to help clinicians, child care providers, and families promote breastfeeding and support the women who choose it. In addition to providing an in-depth look at the numbers, the report compares current findings to the breastfeeding goals outlined in Healthy People 2020. This year, it looked at data on breastfeeding practices and support in all 50 states, the District of Columbia, Puerto Rico, the Virgin Islands, and – for the first time – Guam.
The report is published every 2 years, but the data comprising it are updated annually. The latest rates reflect breastfeeding practices among U.S. infants born in 2015 and are based on results of the 2016 and 2017 U.S. National Immunization Surveys.
Since the first report card in 2007, rates of exclusive breastfeeding at 3 and 6 months have increased. However, although rates for any breastfeeding at 6 and 12 months increased in 2015 from 2014 rates, there was no appreciable increase in exclusive breastfeeding at 3 and 6 months, the report noted. And about 17% of infants who were breastfed at birth still got some formula supplementation in the first 2 days of life.
Recognizing that breastfeeding support at the birth facility is a key driver of success, Healthy People 2020 tracks the proportion of live births in facilities that provided the recommended support. These “baby-friendly hospitals” receive a special designation from the WHO/UNICEF Baby-Friendly Hospital Initiative, and their numbers are increasing, the report noted. In 12 states, more than 40% of births occurred in such facilities, comprising more than 1 million infants in 2015 (26%), and far exceeding the HP2020 8% goal.
In a nation in which many new mothers must return to employment outside the home, breastfeeding support at work is crucial. Just 49% of employers provide breastfeeding facilities for these women, the report found. While this may seem less than optimal, it still exceeds the HP2020 goal of 38%.
“All sectors of society – family and friends, hospitals, health care offices/clinics, childcare facilities, community-based organizations, and workplaces – can play a role in improving the health of families by supporting breastfeeding,” the report said. “To reach their breastfeeding goals, mothers need continuity of care, which is achieved by consistent, collaborative, and high-quality breastfeeding services and support.”
SOURCE: CDC Breastfeeding Report Card
Better adherence, shorter course with rifampin for tuberculosis
Four months of rifampin is effective in prevention of active tuberculosis, with significantly higher adherence rates versus 9 months of isoniazid in adults and children, a pair of recent studies suggest.
In one randomized, open-label trial that included adults with latent Mycobacterium tuberculosis infection, the 4-month rifampin regimen was not inferior to the 9-month isoniazid regimen in preventing active tuberculosis, had better safety, and had a rate of treatment completion 15.1 percentage points higher than the comparator.
“This trial adds to the mounting evidence of benefits of rifamycin-containing regimens of 3 or 4 months’ duration,” investigators reported in the New England Journal of Medicine.
Similarly, in an open-label study in children with latent M. tuberculosis infection, the shorter rifampin regimen had comparable efficacy and safety, according to investigators, along with a rate of treatment completion 13.4 percentage points higher than the longer isoniazid regimen.
“Rifampin has the advantage of being a single-drug regimen with existing palatable formulations for children,” reported authors of this companion study, also published in the journal.
Treatment challenges
Treating latent tuberculosis infection is central to the World Health Organization End TB Strategy and other tuberculosis elimination plans. An estimated 1.7 billion individuals, or about one-quarter of the global population, harbor latent tuberculosis infection, according to one recent estimate.
The WHO recommends treatment of latent tuberculosis infection, as well as for children under 5 years of age who are household contacts of individuals with tuberculosis. The recommended treatment is 6 or 9 months of isoniazid, with the longer duration being associated with better efficacy, previous studies have shown.
However, isoniazid treatment has been associated with low rates of regimen completion because of the hepatotoxic effects, according to authors of the current studies comparing isoniazid to rifampin.
The 4-month daily rifampin regimen has been associated with superior treatment adherence rates and fewer hepatotoxic effects, compared with the 9-month isoniazid regimen in previous observational studies. Moreover, an earlier randomized trial including 679 men in Hong Kong demonstrated that 3 months of rifampin was superior to placebo and comparable to 6 months of isoniazid as tuberculosis prophylaxis.
Rifampin: Latest data
The adult trial just published in the New England Journal of Medicine demonstrates the efficacy and real-world effectiveness of the 4-month rifampin regimen versus the 9-month isoniazid regimen for prevention of active tuberculosis, according to lead author Dick Menzies, MD, of the Montreal Chest Institute at McGill University Health Centre.
The 4-month rifampin regimen is a “fundamental game-changer in TB prevention” based on its comparable efficacy is adults, along with improved safety and acceptability, Dr. Menzies said in a recent press release.
Dr. Menzies and his colleagues reported on 6,063 adults (aged 18 years or older) randomized to the 4-month rifampin or 9-month isoniazid regimen at trial sites in Australia, Benin, Brazil, Canada, Ghana, Guinea, Indonesia, Saudi Arabia, and South Korea.
Treatment was completed by 78.8% of individuals in the rifampin arm, compared with 63.2% of patients in the isoniazid arm, for a difference of 15.1 percentage points (95% confidence interval, 12.7-17.4; P less than .001), the researchers reported.
Rifampin was not inferior to isoniazid in preventing tuberculosis, according to the report. In the per-protocol analysis, there were a total of five confirmed or clinically diagnosed cases of active tuberculosis in each of the trial arms. All active cases were treated successfully, including two cases that had demonstrated drug resistance, investigators added.
The rifampin group had consistently lower rates of grade 3-grade 5 adverse events, particularly hepatotoxic events, versus the isoniazid group, according to analyses outlined in the report.
“We believe this 4-month rifampin treatment should replace the 9 months on isoniazid for most people who need therapy for latent tuberculosis,” said Dr. Menzies, a respirologist with the Montreal Chest Institute and a professor of medicine, epidemiology and biostatistics at McGill University, also in Montreal.
Experience in children
In the related study, reported by lead author Thierno Diallo, MD, of Hôpital National Ignace Deen, in Conakry, Guinea, along with Dr. Menzies, and their coauthors, 829 children were randomized to 4 months of rifampin or 9 months of isoniazid.
The study population included 79 children under 2 years, the age group that has the highest risk of life-threatening TB, Dr. Diallo and his colleagues wrote in their report.
Treatment was completed in 86.5% of all children randomized to rifampin, compared with 77.1% in the isoniazid arm (difference of 13.6 percentage points; 95% confidence interval, 7.9-19.3; P less than .001), according to the investigators.
Two active tuberculosis cases were diagnosed in the isoniazid group over 542 person-years of follow-up, versus no cases in the rifampin group over a similar follow-up period.
“Although the only cases of active tuberculosis were diagnosed in the isoniazid group, we cannot conclude that 4 months of rifampin was either superior or noninferior to 9 months of isoniazid for the prevention of active tuberculosis,” the authors wrote.
“However, since there were no cases of active tuberculosis in the rifampin group in our trial or among 434 children who received 3 months of once-weekly isoniazid plus rifapentine in another trial, we suggest that these shorter rifamycin containing regimens are effective,” they added.
In contrast to the adult trial, safety profiles in this study were similar for rifampin and isoniazid, investigators said.
The lack of difference is side effects was possibly because of the differences in the pharmacokinetic activity of rifampin in younger patients, a topic that deserves further study, they concluded.
No potential conflicts of interest relevant to the studies were reported by Dr. Menzies, Dr. Diallo, or their coauthors.
Both studies were supported by grants from the Canadian Institutes of Health Research. The adult study was supported in part by a grant from the Australian National Health and Medical Research Council, while the companion study in children was supported in part by a grant from the Conselho Nacional de Pesquisa in Brazil.
SOURCES: Menzies D et al. N Engl J Med. 2018 Aug 2;379(5):440-53; Diallo T et al. N Engl J Med. 2018 Aug 2;379(5):454-63.
Four months of rifampin is effective in prevention of active tuberculosis, with significantly higher adherence rates versus 9 months of isoniazid in adults and children, a pair of recent studies suggest.
In one randomized, open-label trial that included adults with latent Mycobacterium tuberculosis infection, the 4-month rifampin regimen was not inferior to the 9-month isoniazid regimen in preventing active tuberculosis, had better safety, and had a rate of treatment completion 15.1 percentage points higher than the comparator.
“This trial adds to the mounting evidence of benefits of rifamycin-containing regimens of 3 or 4 months’ duration,” investigators reported in the New England Journal of Medicine.
Similarly, in an open-label study in children with latent M. tuberculosis infection, the shorter rifampin regimen had comparable efficacy and safety, according to investigators, along with a rate of treatment completion 13.4 percentage points higher than the longer isoniazid regimen.
“Rifampin has the advantage of being a single-drug regimen with existing palatable formulations for children,” reported authors of this companion study, also published in the journal.
Treatment challenges
Treating latent tuberculosis infection is central to the World Health Organization End TB Strategy and other tuberculosis elimination plans. An estimated 1.7 billion individuals, or about one-quarter of the global population, harbor latent tuberculosis infection, according to one recent estimate.
The WHO recommends treatment of latent tuberculosis infection, as well as for children under 5 years of age who are household contacts of individuals with tuberculosis. The recommended treatment is 6 or 9 months of isoniazid, with the longer duration being associated with better efficacy, previous studies have shown.
However, isoniazid treatment has been associated with low rates of regimen completion because of the hepatotoxic effects, according to authors of the current studies comparing isoniazid to rifampin.
The 4-month daily rifampin regimen has been associated with superior treatment adherence rates and fewer hepatotoxic effects, compared with the 9-month isoniazid regimen in previous observational studies. Moreover, an earlier randomized trial including 679 men in Hong Kong demonstrated that 3 months of rifampin was superior to placebo and comparable to 6 months of isoniazid as tuberculosis prophylaxis.
Rifampin: Latest data
The adult trial just published in the New England Journal of Medicine demonstrates the efficacy and real-world effectiveness of the 4-month rifampin regimen versus the 9-month isoniazid regimen for prevention of active tuberculosis, according to lead author Dick Menzies, MD, of the Montreal Chest Institute at McGill University Health Centre.
The 4-month rifampin regimen is a “fundamental game-changer in TB prevention” based on its comparable efficacy is adults, along with improved safety and acceptability, Dr. Menzies said in a recent press release.
Dr. Menzies and his colleagues reported on 6,063 adults (aged 18 years or older) randomized to the 4-month rifampin or 9-month isoniazid regimen at trial sites in Australia, Benin, Brazil, Canada, Ghana, Guinea, Indonesia, Saudi Arabia, and South Korea.
Treatment was completed by 78.8% of individuals in the rifampin arm, compared with 63.2% of patients in the isoniazid arm, for a difference of 15.1 percentage points (95% confidence interval, 12.7-17.4; P less than .001), the researchers reported.
Rifampin was not inferior to isoniazid in preventing tuberculosis, according to the report. In the per-protocol analysis, there were a total of five confirmed or clinically diagnosed cases of active tuberculosis in each of the trial arms. All active cases were treated successfully, including two cases that had demonstrated drug resistance, investigators added.
The rifampin group had consistently lower rates of grade 3-grade 5 adverse events, particularly hepatotoxic events, versus the isoniazid group, according to analyses outlined in the report.
“We believe this 4-month rifampin treatment should replace the 9 months on isoniazid for most people who need therapy for latent tuberculosis,” said Dr. Menzies, a respirologist with the Montreal Chest Institute and a professor of medicine, epidemiology and biostatistics at McGill University, also in Montreal.
Experience in children
In the related study, reported by lead author Thierno Diallo, MD, of Hôpital National Ignace Deen, in Conakry, Guinea, along with Dr. Menzies, and their coauthors, 829 children were randomized to 4 months of rifampin or 9 months of isoniazid.
The study population included 79 children under 2 years, the age group that has the highest risk of life-threatening TB, Dr. Diallo and his colleagues wrote in their report.
Treatment was completed in 86.5% of all children randomized to rifampin, compared with 77.1% in the isoniazid arm (difference of 13.6 percentage points; 95% confidence interval, 7.9-19.3; P less than .001), according to the investigators.
Two active tuberculosis cases were diagnosed in the isoniazid group over 542 person-years of follow-up, versus no cases in the rifampin group over a similar follow-up period.
“Although the only cases of active tuberculosis were diagnosed in the isoniazid group, we cannot conclude that 4 months of rifampin was either superior or noninferior to 9 months of isoniazid for the prevention of active tuberculosis,” the authors wrote.
“However, since there were no cases of active tuberculosis in the rifampin group in our trial or among 434 children who received 3 months of once-weekly isoniazid plus rifapentine in another trial, we suggest that these shorter rifamycin containing regimens are effective,” they added.
In contrast to the adult trial, safety profiles in this study were similar for rifampin and isoniazid, investigators said.
The lack of difference is side effects was possibly because of the differences in the pharmacokinetic activity of rifampin in younger patients, a topic that deserves further study, they concluded.
No potential conflicts of interest relevant to the studies were reported by Dr. Menzies, Dr. Diallo, or their coauthors.
Both studies were supported by grants from the Canadian Institutes of Health Research. The adult study was supported in part by a grant from the Australian National Health and Medical Research Council, while the companion study in children was supported in part by a grant from the Conselho Nacional de Pesquisa in Brazil.
SOURCES: Menzies D et al. N Engl J Med. 2018 Aug 2;379(5):440-53; Diallo T et al. N Engl J Med. 2018 Aug 2;379(5):454-63.
Four months of rifampin is effective in prevention of active tuberculosis, with significantly higher adherence rates versus 9 months of isoniazid in adults and children, a pair of recent studies suggest.
In one randomized, open-label trial that included adults with latent Mycobacterium tuberculosis infection, the 4-month rifampin regimen was not inferior to the 9-month isoniazid regimen in preventing active tuberculosis, had better safety, and had a rate of treatment completion 15.1 percentage points higher than the comparator.
“This trial adds to the mounting evidence of benefits of rifamycin-containing regimens of 3 or 4 months’ duration,” investigators reported in the New England Journal of Medicine.
Similarly, in an open-label study in children with latent M. tuberculosis infection, the shorter rifampin regimen had comparable efficacy and safety, according to investigators, along with a rate of treatment completion 13.4 percentage points higher than the longer isoniazid regimen.
“Rifampin has the advantage of being a single-drug regimen with existing palatable formulations for children,” reported authors of this companion study, also published in the journal.
Treatment challenges
Treating latent tuberculosis infection is central to the World Health Organization End TB Strategy and other tuberculosis elimination plans. An estimated 1.7 billion individuals, or about one-quarter of the global population, harbor latent tuberculosis infection, according to one recent estimate.
The WHO recommends treatment of latent tuberculosis infection, as well as for children under 5 years of age who are household contacts of individuals with tuberculosis. The recommended treatment is 6 or 9 months of isoniazid, with the longer duration being associated with better efficacy, previous studies have shown.
However, isoniazid treatment has been associated with low rates of regimen completion because of the hepatotoxic effects, according to authors of the current studies comparing isoniazid to rifampin.
The 4-month daily rifampin regimen has been associated with superior treatment adherence rates and fewer hepatotoxic effects, compared with the 9-month isoniazid regimen in previous observational studies. Moreover, an earlier randomized trial including 679 men in Hong Kong demonstrated that 3 months of rifampin was superior to placebo and comparable to 6 months of isoniazid as tuberculosis prophylaxis.
Rifampin: Latest data
The adult trial just published in the New England Journal of Medicine demonstrates the efficacy and real-world effectiveness of the 4-month rifampin regimen versus the 9-month isoniazid regimen for prevention of active tuberculosis, according to lead author Dick Menzies, MD, of the Montreal Chest Institute at McGill University Health Centre.
The 4-month rifampin regimen is a “fundamental game-changer in TB prevention” based on its comparable efficacy is adults, along with improved safety and acceptability, Dr. Menzies said in a recent press release.
Dr. Menzies and his colleagues reported on 6,063 adults (aged 18 years or older) randomized to the 4-month rifampin or 9-month isoniazid regimen at trial sites in Australia, Benin, Brazil, Canada, Ghana, Guinea, Indonesia, Saudi Arabia, and South Korea.
Treatment was completed by 78.8% of individuals in the rifampin arm, compared with 63.2% of patients in the isoniazid arm, for a difference of 15.1 percentage points (95% confidence interval, 12.7-17.4; P less than .001), the researchers reported.
Rifampin was not inferior to isoniazid in preventing tuberculosis, according to the report. In the per-protocol analysis, there were a total of five confirmed or clinically diagnosed cases of active tuberculosis in each of the trial arms. All active cases were treated successfully, including two cases that had demonstrated drug resistance, investigators added.
The rifampin group had consistently lower rates of grade 3-grade 5 adverse events, particularly hepatotoxic events, versus the isoniazid group, according to analyses outlined in the report.
“We believe this 4-month rifampin treatment should replace the 9 months on isoniazid for most people who need therapy for latent tuberculosis,” said Dr. Menzies, a respirologist with the Montreal Chest Institute and a professor of medicine, epidemiology and biostatistics at McGill University, also in Montreal.
Experience in children
In the related study, reported by lead author Thierno Diallo, MD, of Hôpital National Ignace Deen, in Conakry, Guinea, along with Dr. Menzies, and their coauthors, 829 children were randomized to 4 months of rifampin or 9 months of isoniazid.
The study population included 79 children under 2 years, the age group that has the highest risk of life-threatening TB, Dr. Diallo and his colleagues wrote in their report.
Treatment was completed in 86.5% of all children randomized to rifampin, compared with 77.1% in the isoniazid arm (difference of 13.6 percentage points; 95% confidence interval, 7.9-19.3; P less than .001), according to the investigators.
Two active tuberculosis cases were diagnosed in the isoniazid group over 542 person-years of follow-up, versus no cases in the rifampin group over a similar follow-up period.
“Although the only cases of active tuberculosis were diagnosed in the isoniazid group, we cannot conclude that 4 months of rifampin was either superior or noninferior to 9 months of isoniazid for the prevention of active tuberculosis,” the authors wrote.
“However, since there were no cases of active tuberculosis in the rifampin group in our trial or among 434 children who received 3 months of once-weekly isoniazid plus rifapentine in another trial, we suggest that these shorter rifamycin containing regimens are effective,” they added.
In contrast to the adult trial, safety profiles in this study were similar for rifampin and isoniazid, investigators said.
The lack of difference is side effects was possibly because of the differences in the pharmacokinetic activity of rifampin in younger patients, a topic that deserves further study, they concluded.
No potential conflicts of interest relevant to the studies were reported by Dr. Menzies, Dr. Diallo, or their coauthors.
Both studies were supported by grants from the Canadian Institutes of Health Research. The adult study was supported in part by a grant from the Australian National Health and Medical Research Council, while the companion study in children was supported in part by a grant from the Conselho Nacional de Pesquisa in Brazil.
SOURCES: Menzies D et al. N Engl J Med. 2018 Aug 2;379(5):440-53; Diallo T et al. N Engl J Med. 2018 Aug 2;379(5):454-63.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: Four months of rifampin is effective in prevention of active tuberculosis, with significantly higher adherence rates versus 9 months of isoniazid in adults and children.
Major finding: Treatment was completed by 78.8% and 63.2% of adults randomized to rifampin or isoniazid, respectively.
Study details: A randomized, open-label trial including 6,063 randomized adults, and a companion study including 829 children, with latent Mycobacterium tuberculosis infection.
Disclosures: Authors reported no potential conflicts of interest relevant to the research. Support for the research came from the Canadian Institutes of Health Research, the Australian National Health and Medical Research Council, and the Conselho Nacional de Pesquisa in Brazil.
Sources: Menzies D et al. N Engl J Med. 2018 Aug 2;379(5):440-53; Diallo T, et al. N Engl J Med. 2018 Aug 2;379(5):454-63.
Longitudinal melanonychia: the good, the bad, and the confusing
CHICAGO – A discolored nail can give even seasoned dermatologists pause: Is the cause exogenous? Fungal or bacterial, perhaps? Could it be a subungual melanoma? Should it be followed, clipped, or biopsied? of the American Academy of Dermatology summer meeting.

The session came after a recent nationwide survey performed by Dr. Lipner and her collaborators who asked dermatologists at different practice stages how confident they were in the diagnosis and management of melanonychia. “On the whole, they were not very confident at all,” said Dr. Lipner, director of the nail division at Cornell University, New York.
Of 142 dermatology residents, as well as 58 junior and 199 senior attending dermatologists, just 18.2% performed nail exams at each visit, and most (58%) only looked at nails during the total body skin exam. Over half (62%) of resident physicians reported feeling not confident about melanonychia diagnosis and management, while that figure dropped to 8.6% for senior attending physicians. Still, most senior physicians (64.3%) were just “fairly confident” in their melanonychia skills (J Am Acad Dermatol. 2017 May;76[5]:994-6).
Tools of the trade
Dermoscopy can be an invaluable tool for determining the cause of longitudinal melanonychia (LM). “Contact dermoscopy is helpful, so I always have ultrasound gel available,” Dr. Lipner said. “The gel makes the nail more of a flat surface,” which makes accurate viewing easier. Other useful tools include a double-action nail clipper, which, she said, is a worthwhile investment.
Because patients who are concerned about one of their nails will often come to their appointment with nail polish still on the other nails, Dr. Lipner always has polish remover pads available in the office. It’s important to be able to see all nails, she said, but she and her collaborators, including first author Pierre Halteh, MD, who was then a medical student at Cornell, discovered from their survey that “few physicians (32/402; 8%) asked their patients to remove nail polish at every visit.”
Nonmelanocytic causes of LM
Longitudinal melanonychias can have a nonmelanocytic etiology, which can range from subungual hematomas to pseudomonas and fungal infections to exogenous pigment.
Overall, subungual hematomas are the most common cause of melanonychia, although longitudinal hematomas are not commonly seen. The more remote the causative trauma, the darker the subungual discoloration, Dr. Lipner said. “Dermoscopy is very helpful” for subungual hematomas, which will usually show a homogeneous pattern, although “you can also see peripheral fadings, streaks, and periungual hemorrhages,” she added.
It is important to monitor these patients “because melanomas can bleed,” she said. In-office photography, or even pictures taken by patients, can be used to track the hematoma to resolution.
When thinking about exogenous sources of pigment, in addition to clues from the history, a tip-off can be that the proximal nail fold is also discolored, Dr. Lipner pointed out. A wide variety of common and less-common culprits may crop up, including from tar, tobacco, henna and other hair dyes, potassium permanganate, and even newspaper print, she said. With an exogenous source, careful clinical and dermoscopic examination may show that the pigment does not extend all the way proximally to the lunula, although it may follow the outline of the proximal nail fold.
When fungus is the cause of LM, the band is often wider proximally and tapers distally, Dr. Lipner noted. While Trichophyton rubrum var. nigricans is a known culprit, nondermatophytes, such as Neoscytalidium dimidiatum, can also cause an LM that often runs along the proximal and lateral nail folds. “To make the diagnosis, sending a clipping to the dermatopathologist is helpful,” she said. Hyphae can often be seen on staining and culture, she said. Polymerase chain reaction “is also possible and very helpful for these nondermatophytes.”
Bacterial colonization of the nail bed can be a cause of LM. Pathogens can include Pseudomonas aeruginosa, which will often show the characteristic greenish tint. Klebsiella and Proteus species may result in more of a grayish-black discoloration. A history of wet work, such as farming and other agricultural and dairy occupations, as well as housekeeping work, increases the risk for bacterial colonization.
Commonly, a bacterial etiology will result in discoloration beginning at the lateral nail fold or at the juncture of the proximal and lateral nail folds. Dermoscopy will show irregular fading of the discoloration toward the medial aspect of the nail, and gram staining of affected clippings will show gram-negative rods.
Melanocytic causes of longitudinal melanonychia
The melanotic macule, sometimes called melanocytic activation, is the most common subtype of melanin-derived LM in adults, Dr. Lipner said. This benign condition results from increased melanin synthesis without an increase in the number of melanocytes, which will be evident on histopathologic examination of the nail bed. Any of a variety of triggers can provoke the increased pigment, which can range from endocrine disruptions to inflammatory conditions, such as psoriasis, to trauma (including nail biting or habit tics).
Pregnancy, normal ethnic variation, and chemotherapy administration are all also associated with melanotic macules. In any case, dermoscopy will show an LM characterized by a grayish background that contains darker grayish lines.
Melanocyte hyperplasia can also cause melanonychia, in which case the trick is sorting out which cases are benign and which are malignant, Dr. Lipner noted. And getting the diagnosis right in a timely fashion matters: “Ideally, we want to catch these melanomas in in situ stages where we can preserve the digit,” she said. “It’s been shown that there is no survival benefit for amputation versus en bloc excision for nail melanomas in situ.”
Nail matrix nevi are the most common cause of LM in children, Dr. Lipner said. Here, dermoscopy shows a brown background with brown lines, with regular color, thickness, and spacing.
On examination of a nail with a melanoma, “typically, we see features suggestive of melanoma but really no pathognomonic features,” she commented. Some signs that should prompt concern and a more thorough investigation, she said, include a dark brown or black band of LM; lack of homogeneity, such as the presence of lines of different colors; blurring of the borders of the pigmentation; and a triangular or wavering outline. Changes in the nail, such as fissuring or splitting, also are worrying, as is any associated discoloration of the periungual skin.
Dermoscopy may confirm the irregularity of the pigmentation pattern and show irregularly colored and spaced lines of varying thicknesses within the pigmented band. An LM caused by melanoma may also be marked by loss of parallelism within the pigmented band.
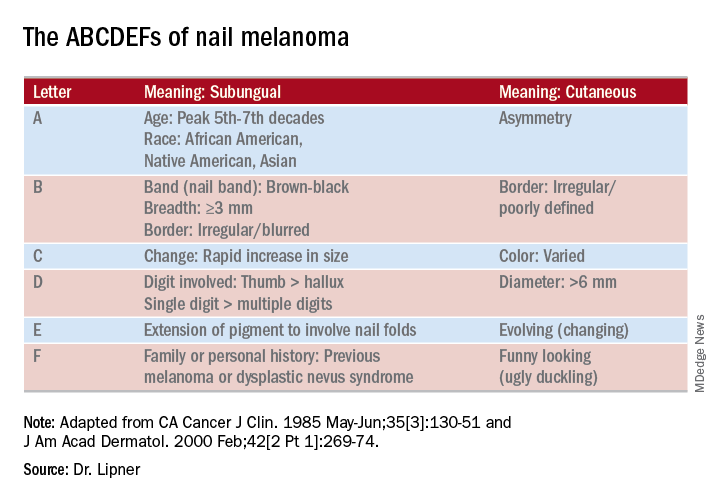
She pointed out that these concerning characteristics have been encapsulated in a mnemonic, first created in 2000, that’s meant to mirror the ABCDs of nonnail melanoma detection (J Am Acad Dermatol. Feb 2000;42[2 Pt 1]:269-74). Her survey found that overall, just one in four (24.8%) of respondents knew of the mnemonic for subungual melanomas.
Dr. Lipner reported that she has received research support from MOE Medical Devices and has served as a consultant to BAKO Therapeutics.
SOURCE: Lipner S. Summer AAD 2018, Presentation F004.
CHICAGO – A discolored nail can give even seasoned dermatologists pause: Is the cause exogenous? Fungal or bacterial, perhaps? Could it be a subungual melanoma? Should it be followed, clipped, or biopsied? of the American Academy of Dermatology summer meeting.
The session came after a recent nationwide survey performed by Dr. Lipner and her collaborators who asked dermatologists at different practice stages how confident they were in the diagnosis and management of melanonychia. “On the whole, they were not very confident at all,” said Dr. Lipner, director of the nail division at Cornell University, New York.
Of 142 dermatology residents, as well as 58 junior and 199 senior attending dermatologists, just 18.2% performed nail exams at each visit, and most (58%) only looked at nails during the total body skin exam. Over half (62%) of resident physicians reported feeling not confident about melanonychia diagnosis and management, while that figure dropped to 8.6% for senior attending physicians. Still, most senior physicians (64.3%) were just “fairly confident” in their melanonychia skills (J Am Acad Dermatol. 2017 May;76[5]:994-6).
Tools of the trade
Dermoscopy can be an invaluable tool for determining the cause of longitudinal melanonychia (LM). “Contact dermoscopy is helpful, so I always have ultrasound gel available,” Dr. Lipner said. “The gel makes the nail more of a flat surface,” which makes accurate viewing easier. Other useful tools include a double-action nail clipper, which, she said, is a worthwhile investment.
Because patients who are concerned about one of their nails will often come to their appointment with nail polish still on the other nails, Dr. Lipner always has polish remover pads available in the office. It’s important to be able to see all nails, she said, but she and her collaborators, including first author Pierre Halteh, MD, who was then a medical student at Cornell, discovered from their survey that “few physicians (32/402; 8%) asked their patients to remove nail polish at every visit.”
Nonmelanocytic causes of LM
Longitudinal melanonychias can have a nonmelanocytic etiology, which can range from subungual hematomas to pseudomonas and fungal infections to exogenous pigment.
Overall, subungual hematomas are the most common cause of melanonychia, although longitudinal hematomas are not commonly seen. The more remote the causative trauma, the darker the subungual discoloration, Dr. Lipner said. “Dermoscopy is very helpful” for subungual hematomas, which will usually show a homogeneous pattern, although “you can also see peripheral fadings, streaks, and periungual hemorrhages,” she added.
It is important to monitor these patients “because melanomas can bleed,” she said. In-office photography, or even pictures taken by patients, can be used to track the hematoma to resolution.
When thinking about exogenous sources of pigment, in addition to clues from the history, a tip-off can be that the proximal nail fold is also discolored, Dr. Lipner pointed out. A wide variety of common and less-common culprits may crop up, including from tar, tobacco, henna and other hair dyes, potassium permanganate, and even newspaper print, she said. With an exogenous source, careful clinical and dermoscopic examination may show that the pigment does not extend all the way proximally to the lunula, although it may follow the outline of the proximal nail fold.
When fungus is the cause of LM, the band is often wider proximally and tapers distally, Dr. Lipner noted. While Trichophyton rubrum var. nigricans is a known culprit, nondermatophytes, such as Neoscytalidium dimidiatum, can also cause an LM that often runs along the proximal and lateral nail folds. “To make the diagnosis, sending a clipping to the dermatopathologist is helpful,” she said. Hyphae can often be seen on staining and culture, she said. Polymerase chain reaction “is also possible and very helpful for these nondermatophytes.”
Bacterial colonization of the nail bed can be a cause of LM. Pathogens can include Pseudomonas aeruginosa, which will often show the characteristic greenish tint. Klebsiella and Proteus species may result in more of a grayish-black discoloration. A history of wet work, such as farming and other agricultural and dairy occupations, as well as housekeeping work, increases the risk for bacterial colonization.
Commonly, a bacterial etiology will result in discoloration beginning at the lateral nail fold or at the juncture of the proximal and lateral nail folds. Dermoscopy will show irregular fading of the discoloration toward the medial aspect of the nail, and gram staining of affected clippings will show gram-negative rods.
Melanocytic causes of longitudinal melanonychia
The melanotic macule, sometimes called melanocytic activation, is the most common subtype of melanin-derived LM in adults, Dr. Lipner said. This benign condition results from increased melanin synthesis without an increase in the number of melanocytes, which will be evident on histopathologic examination of the nail bed. Any of a variety of triggers can provoke the increased pigment, which can range from endocrine disruptions to inflammatory conditions, such as psoriasis, to trauma (including nail biting or habit tics).
Pregnancy, normal ethnic variation, and chemotherapy administration are all also associated with melanotic macules. In any case, dermoscopy will show an LM characterized by a grayish background that contains darker grayish lines.
Melanocyte hyperplasia can also cause melanonychia, in which case the trick is sorting out which cases are benign and which are malignant, Dr. Lipner noted. And getting the diagnosis right in a timely fashion matters: “Ideally, we want to catch these melanomas in in situ stages where we can preserve the digit,” she said. “It’s been shown that there is no survival benefit for amputation versus en bloc excision for nail melanomas in situ.”
Nail matrix nevi are the most common cause of LM in children, Dr. Lipner said. Here, dermoscopy shows a brown background with brown lines, with regular color, thickness, and spacing.
On examination of a nail with a melanoma, “typically, we see features suggestive of melanoma but really no pathognomonic features,” she commented. Some signs that should prompt concern and a more thorough investigation, she said, include a dark brown or black band of LM; lack of homogeneity, such as the presence of lines of different colors; blurring of the borders of the pigmentation; and a triangular or wavering outline. Changes in the nail, such as fissuring or splitting, also are worrying, as is any associated discoloration of the periungual skin.
Dermoscopy may confirm the irregularity of the pigmentation pattern and show irregularly colored and spaced lines of varying thicknesses within the pigmented band. An LM caused by melanoma may also be marked by loss of parallelism within the pigmented band.
She pointed out that these concerning characteristics have been encapsulated in a mnemonic, first created in 2000, that’s meant to mirror the ABCDs of nonnail melanoma detection (J Am Acad Dermatol. Feb 2000;42[2 Pt 1]:269-74). Her survey found that overall, just one in four (24.8%) of respondents knew of the mnemonic for subungual melanomas.
Dr. Lipner reported that she has received research support from MOE Medical Devices and has served as a consultant to BAKO Therapeutics.
SOURCE: Lipner S. Summer AAD 2018, Presentation F004.
CHICAGO – A discolored nail can give even seasoned dermatologists pause: Is the cause exogenous? Fungal or bacterial, perhaps? Could it be a subungual melanoma? Should it be followed, clipped, or biopsied? of the American Academy of Dermatology summer meeting.
The session came after a recent nationwide survey performed by Dr. Lipner and her collaborators who asked dermatologists at different practice stages how confident they were in the diagnosis and management of melanonychia. “On the whole, they were not very confident at all,” said Dr. Lipner, director of the nail division at Cornell University, New York.
Of 142 dermatology residents, as well as 58 junior and 199 senior attending dermatologists, just 18.2% performed nail exams at each visit, and most (58%) only looked at nails during the total body skin exam. Over half (62%) of resident physicians reported feeling not confident about melanonychia diagnosis and management, while that figure dropped to 8.6% for senior attending physicians. Still, most senior physicians (64.3%) were just “fairly confident” in their melanonychia skills (J Am Acad Dermatol. 2017 May;76[5]:994-6).
Tools of the trade
Dermoscopy can be an invaluable tool for determining the cause of longitudinal melanonychia (LM). “Contact dermoscopy is helpful, so I always have ultrasound gel available,” Dr. Lipner said. “The gel makes the nail more of a flat surface,” which makes accurate viewing easier. Other useful tools include a double-action nail clipper, which, she said, is a worthwhile investment.
Because patients who are concerned about one of their nails will often come to their appointment with nail polish still on the other nails, Dr. Lipner always has polish remover pads available in the office. It’s important to be able to see all nails, she said, but she and her collaborators, including first author Pierre Halteh, MD, who was then a medical student at Cornell, discovered from their survey that “few physicians (32/402; 8%) asked their patients to remove nail polish at every visit.”
Nonmelanocytic causes of LM
Longitudinal melanonychias can have a nonmelanocytic etiology, which can range from subungual hematomas to pseudomonas and fungal infections to exogenous pigment.
Overall, subungual hematomas are the most common cause of melanonychia, although longitudinal hematomas are not commonly seen. The more remote the causative trauma, the darker the subungual discoloration, Dr. Lipner said. “Dermoscopy is very helpful” for subungual hematomas, which will usually show a homogeneous pattern, although “you can also see peripheral fadings, streaks, and periungual hemorrhages,” she added.
It is important to monitor these patients “because melanomas can bleed,” she said. In-office photography, or even pictures taken by patients, can be used to track the hematoma to resolution.
When thinking about exogenous sources of pigment, in addition to clues from the history, a tip-off can be that the proximal nail fold is also discolored, Dr. Lipner pointed out. A wide variety of common and less-common culprits may crop up, including from tar, tobacco, henna and other hair dyes, potassium permanganate, and even newspaper print, she said. With an exogenous source, careful clinical and dermoscopic examination may show that the pigment does not extend all the way proximally to the lunula, although it may follow the outline of the proximal nail fold.
When fungus is the cause of LM, the band is often wider proximally and tapers distally, Dr. Lipner noted. While Trichophyton rubrum var. nigricans is a known culprit, nondermatophytes, such as Neoscytalidium dimidiatum, can also cause an LM that often runs along the proximal and lateral nail folds. “To make the diagnosis, sending a clipping to the dermatopathologist is helpful,” she said. Hyphae can often be seen on staining and culture, she said. Polymerase chain reaction “is also possible and very helpful for these nondermatophytes.”
Bacterial colonization of the nail bed can be a cause of LM. Pathogens can include Pseudomonas aeruginosa, which will often show the characteristic greenish tint. Klebsiella and Proteus species may result in more of a grayish-black discoloration. A history of wet work, such as farming and other agricultural and dairy occupations, as well as housekeeping work, increases the risk for bacterial colonization.
Commonly, a bacterial etiology will result in discoloration beginning at the lateral nail fold or at the juncture of the proximal and lateral nail folds. Dermoscopy will show irregular fading of the discoloration toward the medial aspect of the nail, and gram staining of affected clippings will show gram-negative rods.
Melanocytic causes of longitudinal melanonychia
The melanotic macule, sometimes called melanocytic activation, is the most common subtype of melanin-derived LM in adults, Dr. Lipner said. This benign condition results from increased melanin synthesis without an increase in the number of melanocytes, which will be evident on histopathologic examination of the nail bed. Any of a variety of triggers can provoke the increased pigment, which can range from endocrine disruptions to inflammatory conditions, such as psoriasis, to trauma (including nail biting or habit tics).
Pregnancy, normal ethnic variation, and chemotherapy administration are all also associated with melanotic macules. In any case, dermoscopy will show an LM characterized by a grayish background that contains darker grayish lines.
Melanocyte hyperplasia can also cause melanonychia, in which case the trick is sorting out which cases are benign and which are malignant, Dr. Lipner noted. And getting the diagnosis right in a timely fashion matters: “Ideally, we want to catch these melanomas in in situ stages where we can preserve the digit,” she said. “It’s been shown that there is no survival benefit for amputation versus en bloc excision for nail melanomas in situ.”
Nail matrix nevi are the most common cause of LM in children, Dr. Lipner said. Here, dermoscopy shows a brown background with brown lines, with regular color, thickness, and spacing.
On examination of a nail with a melanoma, “typically, we see features suggestive of melanoma but really no pathognomonic features,” she commented. Some signs that should prompt concern and a more thorough investigation, she said, include a dark brown or black band of LM; lack of homogeneity, such as the presence of lines of different colors; blurring of the borders of the pigmentation; and a triangular or wavering outline. Changes in the nail, such as fissuring or splitting, also are worrying, as is any associated discoloration of the periungual skin.
Dermoscopy may confirm the irregularity of the pigmentation pattern and show irregularly colored and spaced lines of varying thicknesses within the pigmented band. An LM caused by melanoma may also be marked by loss of parallelism within the pigmented band.
She pointed out that these concerning characteristics have been encapsulated in a mnemonic, first created in 2000, that’s meant to mirror the ABCDs of nonnail melanoma detection (J Am Acad Dermatol. Feb 2000;42[2 Pt 1]:269-74). Her survey found that overall, just one in four (24.8%) of respondents knew of the mnemonic for subungual melanomas.
Dr. Lipner reported that she has received research support from MOE Medical Devices and has served as a consultant to BAKO Therapeutics.
SOURCE: Lipner S. Summer AAD 2018, Presentation F004.
EXPERT ANALYSIS FROM SUMMER AAD 2018
Treating the effects of bruxism with botulinum toxin
Bruxism is grinding and clenching of the teeth with unconscious contractions of the temporal and masseter muscles while awake or during sleep. Bruxism occurs in 8%-16% of the population and is often an underdiagnosed condition that not only leads to dental problems but also to pain in the teeth, jaw, temporomandibular joint, and neck; headaches; and potentially, to tooth loss.

Although the pathogenesis of bruxism remains unclear, multiple factors, such as physical or psychological stress, malocclusion, sleep disorders and medication side effects, can cause bruxism. Treatment can be difficult given psychogenic and neurogenic components, and bruxism can be resistant to medical and behavioral therapy. There are various treatment options for bruxism, including oral splints; medications, such as muscle relaxants; antidepressants; and botulinum toxin. Multiple studies have shown that botulinum toxin injections into the masseter and temporalis muscles result in relaxation of the muscles and improvement of bruxism and the pain associated with chronic clenching and grinding.
In a recent study by Al-Wayli, 50 subjects who reported nocturnal bruxism were randomized to receive botulinum toxin versus conventional treatment (pharmacotherapy or oral splints). After 3 weeks, 2 months, 6 months, and 1 year, patients who received botulinum toxin had significantly less pain after only one treatment than did the traditional treatment group. Similarly, in a study by Lee et al., subjects randomized to receive botulinum toxin versus a placebo saline injections showed not only decreased pain but also decreased bruxism seen with nocturnal electromyography.
In our clinic, Botulinum toxin when injected into the temporalis and masseter muscles also helps with tension headaches and migraines related to clenching of the jaw. Albeit effective, the dose of botulinum toxin used in the aforementioned studies ranged between 25 U and 40 U of botulinum toxin and were lower than what we have found to be effective. Our patients receive 50 U botulinum toxin in each masseter muscle (100 U total). In a small minority of our patients, the temporalis muscle also needed 15-20 U per side as well. Clinical improvement starts within 3-5 days, and patients can expect to have relaxation of the muscle and decreased pain for 6 months. Side effects include mild swelling and bruising. Rarely, if the injection is not performed properly, the risorius muscle may be paralyzed, leading to an asymmetric smile. In addition, if the botulinum toxin is underdosed, the pain may not completely subside and the patient may report some symptoms returning within a couple of weeks of the initial treatment. Most patients also report thinning of the face and jaw, which is a much anticipated and appreciated result. Masseter hypertrophy with and without bruxism is treated similarly with botulinum toxin to sculpt the lower face.

Bruxism is a growing problem leading to facial pain, headaches, migraines, and significant dental pathology. Traditional treatments have been ineffective at treating the pain and masseter hypertrophy associated with chronic grinding and clenching. Botulinum toxin is a safe, effective, treatment with little downtime or side effects for treating both the neurogenic and muscular components of bruxism.
Dr. Lily Talakoub and Dr. Naissan Wesley and are co-contributors to this column. Dr. Talakoub is in private practice in McLean, Va. This month’s column is by Dr. Talakoub. Dr. Wesley practices dermatology in Beverly Hills, Calif. Write to them at [email protected]. They had no relevant disclosures.
References
Al-Wayli H. J Clin Exp Dent. 2017 Jan 1;9(1):e112-e117.
Asutay F et al. Pain Res Manag. 2017;2017:6264146. doi: 10.1155/2017/6264146.
Lee SJ et al. Am J Phys Med Rehabil. 2010 Jan;89(1):16-23.
Santamato A et al. J Chiropr Med. 2010 Sep;9(3):132-7.
Shetty S et al. J Indian Prosthodont Soc. 2010 Sep;10(3):141-8.
Tan EK et al. J Am Dent Assoc. 2000 Feb;131(2):211-6.
Bruxism is grinding and clenching of the teeth with unconscious contractions of the temporal and masseter muscles while awake or during sleep. Bruxism occurs in 8%-16% of the population and is often an underdiagnosed condition that not only leads to dental problems but also to pain in the teeth, jaw, temporomandibular joint, and neck; headaches; and potentially, to tooth loss.
Although the pathogenesis of bruxism remains unclear, multiple factors, such as physical or psychological stress, malocclusion, sleep disorders and medication side effects, can cause bruxism. Treatment can be difficult given psychogenic and neurogenic components, and bruxism can be resistant to medical and behavioral therapy. There are various treatment options for bruxism, including oral splints; medications, such as muscle relaxants; antidepressants; and botulinum toxin. Multiple studies have shown that botulinum toxin injections into the masseter and temporalis muscles result in relaxation of the muscles and improvement of bruxism and the pain associated with chronic clenching and grinding.
In a recent study by Al-Wayli, 50 subjects who reported nocturnal bruxism were randomized to receive botulinum toxin versus conventional treatment (pharmacotherapy or oral splints). After 3 weeks, 2 months, 6 months, and 1 year, patients who received botulinum toxin had significantly less pain after only one treatment than did the traditional treatment group. Similarly, in a study by Lee et al., subjects randomized to receive botulinum toxin versus a placebo saline injections showed not only decreased pain but also decreased bruxism seen with nocturnal electromyography.
In our clinic, Botulinum toxin when injected into the temporalis and masseter muscles also helps with tension headaches and migraines related to clenching of the jaw. Albeit effective, the dose of botulinum toxin used in the aforementioned studies ranged between 25 U and 40 U of botulinum toxin and were lower than what we have found to be effective. Our patients receive 50 U botulinum toxin in each masseter muscle (100 U total). In a small minority of our patients, the temporalis muscle also needed 15-20 U per side as well. Clinical improvement starts within 3-5 days, and patients can expect to have relaxation of the muscle and decreased pain for 6 months. Side effects include mild swelling and bruising. Rarely, if the injection is not performed properly, the risorius muscle may be paralyzed, leading to an asymmetric smile. In addition, if the botulinum toxin is underdosed, the pain may not completely subside and the patient may report some symptoms returning within a couple of weeks of the initial treatment. Most patients also report thinning of the face and jaw, which is a much anticipated and appreciated result. Masseter hypertrophy with and without bruxism is treated similarly with botulinum toxin to sculpt the lower face.
Bruxism is a growing problem leading to facial pain, headaches, migraines, and significant dental pathology. Traditional treatments have been ineffective at treating the pain and masseter hypertrophy associated with chronic grinding and clenching. Botulinum toxin is a safe, effective, treatment with little downtime or side effects for treating both the neurogenic and muscular components of bruxism.
Dr. Lily Talakoub and Dr. Naissan Wesley and are co-contributors to this column. Dr. Talakoub is in private practice in McLean, Va. This month’s column is by Dr. Talakoub. Dr. Wesley practices dermatology in Beverly Hills, Calif. Write to them at [email protected]. They had no relevant disclosures.
References
Al-Wayli H. J Clin Exp Dent. 2017 Jan 1;9(1):e112-e117.
Asutay F et al. Pain Res Manag. 2017;2017:6264146. doi: 10.1155/2017/6264146.
Lee SJ et al. Am J Phys Med Rehabil. 2010 Jan;89(1):16-23.
Santamato A et al. J Chiropr Med. 2010 Sep;9(3):132-7.
Shetty S et al. J Indian Prosthodont Soc. 2010 Sep;10(3):141-8.
Tan EK et al. J Am Dent Assoc. 2000 Feb;131(2):211-6.
Bruxism is grinding and clenching of the teeth with unconscious contractions of the temporal and masseter muscles while awake or during sleep. Bruxism occurs in 8%-16% of the population and is often an underdiagnosed condition that not only leads to dental problems but also to pain in the teeth, jaw, temporomandibular joint, and neck; headaches; and potentially, to tooth loss.
Although the pathogenesis of bruxism remains unclear, multiple factors, such as physical or psychological stress, malocclusion, sleep disorders and medication side effects, can cause bruxism. Treatment can be difficult given psychogenic and neurogenic components, and bruxism can be resistant to medical and behavioral therapy. There are various treatment options for bruxism, including oral splints; medications, such as muscle relaxants; antidepressants; and botulinum toxin. Multiple studies have shown that botulinum toxin injections into the masseter and temporalis muscles result in relaxation of the muscles and improvement of bruxism and the pain associated with chronic clenching and grinding.
In a recent study by Al-Wayli, 50 subjects who reported nocturnal bruxism were randomized to receive botulinum toxin versus conventional treatment (pharmacotherapy or oral splints). After 3 weeks, 2 months, 6 months, and 1 year, patients who received botulinum toxin had significantly less pain after only one treatment than did the traditional treatment group. Similarly, in a study by Lee et al., subjects randomized to receive botulinum toxin versus a placebo saline injections showed not only decreased pain but also decreased bruxism seen with nocturnal electromyography.
In our clinic, Botulinum toxin when injected into the temporalis and masseter muscles also helps with tension headaches and migraines related to clenching of the jaw. Albeit effective, the dose of botulinum toxin used in the aforementioned studies ranged between 25 U and 40 U of botulinum toxin and were lower than what we have found to be effective. Our patients receive 50 U botulinum toxin in each masseter muscle (100 U total). In a small minority of our patients, the temporalis muscle also needed 15-20 U per side as well. Clinical improvement starts within 3-5 days, and patients can expect to have relaxation of the muscle and decreased pain for 6 months. Side effects include mild swelling and bruising. Rarely, if the injection is not performed properly, the risorius muscle may be paralyzed, leading to an asymmetric smile. In addition, if the botulinum toxin is underdosed, the pain may not completely subside and the patient may report some symptoms returning within a couple of weeks of the initial treatment. Most patients also report thinning of the face and jaw, which is a much anticipated and appreciated result. Masseter hypertrophy with and without bruxism is treated similarly with botulinum toxin to sculpt the lower face.
Bruxism is a growing problem leading to facial pain, headaches, migraines, and significant dental pathology. Traditional treatments have been ineffective at treating the pain and masseter hypertrophy associated with chronic grinding and clenching. Botulinum toxin is a safe, effective, treatment with little downtime or side effects for treating both the neurogenic and muscular components of bruxism.
Dr. Lily Talakoub and Dr. Naissan Wesley and are co-contributors to this column. Dr. Talakoub is in private practice in McLean, Va. This month’s column is by Dr. Talakoub. Dr. Wesley practices dermatology in Beverly Hills, Calif. Write to them at [email protected]. They had no relevant disclosures.
References
Al-Wayli H. J Clin Exp Dent. 2017 Jan 1;9(1):e112-e117.
Asutay F et al. Pain Res Manag. 2017;2017:6264146. doi: 10.1155/2017/6264146.
Lee SJ et al. Am J Phys Med Rehabil. 2010 Jan;89(1):16-23.
Santamato A et al. J Chiropr Med. 2010 Sep;9(3):132-7.
Shetty S et al. J Indian Prosthodont Soc. 2010 Sep;10(3):141-8.
Tan EK et al. J Am Dent Assoc. 2000 Feb;131(2):211-6.
FDA grants accelerated approval for Opdivo in metastatic SCLC
The Food and Drug Administration has granted accelerated approval to nivolumab (Opdivo) for treating patients with metastatic small-cell lung cancer (SCLC) whose cancer has progressed after platinum-based chemotherapy and at least one other line of therapy.
Approval was based on an overall response rate and duration of response in the monotherapy arm of the multicohort phase 1/2 CheckMate 032 trial. Of 109 patients with SCLC and progression after at least one previous platinum-containing regimen, 12% responded to monotherapy treatment with nivolumab regardless of PD-L1 status, 12 patients had a partial response, and one patient had a complete response.
Among responders, the median duration of response was 17.9 months. Results of the trial were presented at the ASCO annual meeting and published online in The Lancet Oncology in 2016.
Serious adverse reactions occurred in 45% of patients. The most frequent serious adverse reactions were pneumonia, dyspnea, pneumonitis, pleural effusion, and dehydration.
The approved dose is 240 milligrams administered every 2 weeks by intravenous infusion until disease progression or unacceptable toxicity, the company said in a press statement announcing the approval.
The checkpoint inhibitor was approved for treating patients with metastatic non–small-cell lung cancer with progression on or after platinum-based chemotherapy in 2015.
The Food and Drug Administration has granted accelerated approval to nivolumab (Opdivo) for treating patients with metastatic small-cell lung cancer (SCLC) whose cancer has progressed after platinum-based chemotherapy and at least one other line of therapy.
Approval was based on an overall response rate and duration of response in the monotherapy arm of the multicohort phase 1/2 CheckMate 032 trial. Of 109 patients with SCLC and progression after at least one previous platinum-containing regimen, 12% responded to monotherapy treatment with nivolumab regardless of PD-L1 status, 12 patients had a partial response, and one patient had a complete response.
Among responders, the median duration of response was 17.9 months. Results of the trial were presented at the ASCO annual meeting and published online in The Lancet Oncology in 2016.
Serious adverse reactions occurred in 45% of patients. The most frequent serious adverse reactions were pneumonia, dyspnea, pneumonitis, pleural effusion, and dehydration.
The approved dose is 240 milligrams administered every 2 weeks by intravenous infusion until disease progression or unacceptable toxicity, the company said in a press statement announcing the approval.
The checkpoint inhibitor was approved for treating patients with metastatic non–small-cell lung cancer with progression on or after platinum-based chemotherapy in 2015.
The Food and Drug Administration has granted accelerated approval to nivolumab (Opdivo) for treating patients with metastatic small-cell lung cancer (SCLC) whose cancer has progressed after platinum-based chemotherapy and at least one other line of therapy.
Approval was based on an overall response rate and duration of response in the monotherapy arm of the multicohort phase 1/2 CheckMate 032 trial. Of 109 patients with SCLC and progression after at least one previous platinum-containing regimen, 12% responded to monotherapy treatment with nivolumab regardless of PD-L1 status, 12 patients had a partial response, and one patient had a complete response.
Among responders, the median duration of response was 17.9 months. Results of the trial were presented at the ASCO annual meeting and published online in The Lancet Oncology in 2016.
Serious adverse reactions occurred in 45% of patients. The most frequent serious adverse reactions were pneumonia, dyspnea, pneumonitis, pleural effusion, and dehydration.
The approved dose is 240 milligrams administered every 2 weeks by intravenous infusion until disease progression or unacceptable toxicity, the company said in a press statement announcing the approval.
The checkpoint inhibitor was approved for treating patients with metastatic non–small-cell lung cancer with progression on or after platinum-based chemotherapy in 2015.
Nighttime media use threatens teen sleep
Nighttime media use was associated with less sleep, as well as self-reported anxiety and depression, in teens with attention-deficit/hyperactivity disorder, based on data from 81 adolescents.
“This is the first study to document an association between nighttime media use and more sleep problems and internalizing symptoms in adolescents diagnosed with ADHD,” wrote Stephen P. Becker, PhD, of the University of Cincinnati and colleagues.
Although previous research has addressed the impact of screen time on sleep, anxiety, and depression in children and teens, the impact on adolescents with conditions such as ADHD has not been well studied, the researchers noted.
In a study published in Sleep Medicine, the researchers conducted a study of 81 adolescents aged 13-17 years who met diagnostic criteria for ADHD. The School Sleep Habits Survey (SSHS) was used to measure sleep patterns based on self-reports, and parents reported on teens’ sleep using the Sleep Disturbance Scale for Children. In addition, several other tools that measured ADHD symptoms, daytime sleepiness, anxiety, and depression were administered to both the teens and their parents.
The researchers assessed the number of technologies in each participant’s bedroom and the total hours of electronic media use at night, defined as after 9:00 p.m.
Overall, approximately 60% of the teens in the sample reported more than 4 hours of nighttime media use; 63% reported less than 8 hours of sleep on school nights, but this figure reached 76% when parent reports of teens’ sleep was used. When the teens’ self-reports were used, media use was not significantly different between those who had less than 8 hours of sleep vs. those who had 8 hours or more (5.85 vs. 4.39 hours of nighttime media use). But their parents’ reports told another story. In the parent reports, media use was significantly higher in the short sleepers vs. long sleepers (6.12 vs. 2.65 hours of nighttime media use).
After controlling for factors including age, sex, pubertal stage, use of stimulant medication, and severity of ADHD symptoms, nighttime media use was significantly associated with shorter sleep duration, the researchers said.
Nighttime media use also was significantly associated with greater adolescent-reported depressive symptoms, total anxiety symptoms overall, and panic symptoms in particular, as well as with parent-reported generalized anxiety symptoms.
The study findings were limited by several factors including the cross-sectional design, the lack of an objective sleep measure, and the lack of non-ADHD controls, the researchers noted. Also, the researchers were unable to measure parental control over teen media use or to examine different types of media use, including media multitasking (such as texting while video gaming).
However, the findings “suggest that it is important for clinicians to consider nighttime media use when assessing and treating adolescent ADHD, specifically regarding sleep issues and co-occurring depression and anxiety,” they said.
The researchers had no financial conflicts to disclose. The study was funded by grants from the National Institutes of Mental Health.
SOURCE: Becker S et al. Sleep Med. 2018. doi: 10.1016/ j.sleep.2018.06.021.
Nighttime media use was associated with less sleep, as well as self-reported anxiety and depression, in teens with attention-deficit/hyperactivity disorder, based on data from 81 adolescents.
“This is the first study to document an association between nighttime media use and more sleep problems and internalizing symptoms in adolescents diagnosed with ADHD,” wrote Stephen P. Becker, PhD, of the University of Cincinnati and colleagues.
Although previous research has addressed the impact of screen time on sleep, anxiety, and depression in children and teens, the impact on adolescents with conditions such as ADHD has not been well studied, the researchers noted.
In a study published in Sleep Medicine, the researchers conducted a study of 81 adolescents aged 13-17 years who met diagnostic criteria for ADHD. The School Sleep Habits Survey (SSHS) was used to measure sleep patterns based on self-reports, and parents reported on teens’ sleep using the Sleep Disturbance Scale for Children. In addition, several other tools that measured ADHD symptoms, daytime sleepiness, anxiety, and depression were administered to both the teens and their parents.
The researchers assessed the number of technologies in each participant’s bedroom and the total hours of electronic media use at night, defined as after 9:00 p.m.
Overall, approximately 60% of the teens in the sample reported more than 4 hours of nighttime media use; 63% reported less than 8 hours of sleep on school nights, but this figure reached 76% when parent reports of teens’ sleep was used. When the teens’ self-reports were used, media use was not significantly different between those who had less than 8 hours of sleep vs. those who had 8 hours or more (5.85 vs. 4.39 hours of nighttime media use). But their parents’ reports told another story. In the parent reports, media use was significantly higher in the short sleepers vs. long sleepers (6.12 vs. 2.65 hours of nighttime media use).
After controlling for factors including age, sex, pubertal stage, use of stimulant medication, and severity of ADHD symptoms, nighttime media use was significantly associated with shorter sleep duration, the researchers said.
Nighttime media use also was significantly associated with greater adolescent-reported depressive symptoms, total anxiety symptoms overall, and panic symptoms in particular, as well as with parent-reported generalized anxiety symptoms.
The study findings were limited by several factors including the cross-sectional design, the lack of an objective sleep measure, and the lack of non-ADHD controls, the researchers noted. Also, the researchers were unable to measure parental control over teen media use or to examine different types of media use, including media multitasking (such as texting while video gaming).
However, the findings “suggest that it is important for clinicians to consider nighttime media use when assessing and treating adolescent ADHD, specifically regarding sleep issues and co-occurring depression and anxiety,” they said.
The researchers had no financial conflicts to disclose. The study was funded by grants from the National Institutes of Mental Health.
SOURCE: Becker S et al. Sleep Med. 2018. doi: 10.1016/ j.sleep.2018.06.021.
Nighttime media use was associated with less sleep, as well as self-reported anxiety and depression, in teens with attention-deficit/hyperactivity disorder, based on data from 81 adolescents.
“This is the first study to document an association between nighttime media use and more sleep problems and internalizing symptoms in adolescents diagnosed with ADHD,” wrote Stephen P. Becker, PhD, of the University of Cincinnati and colleagues.
Although previous research has addressed the impact of screen time on sleep, anxiety, and depression in children and teens, the impact on adolescents with conditions such as ADHD has not been well studied, the researchers noted.
In a study published in Sleep Medicine, the researchers conducted a study of 81 adolescents aged 13-17 years who met diagnostic criteria for ADHD. The School Sleep Habits Survey (SSHS) was used to measure sleep patterns based on self-reports, and parents reported on teens’ sleep using the Sleep Disturbance Scale for Children. In addition, several other tools that measured ADHD symptoms, daytime sleepiness, anxiety, and depression were administered to both the teens and their parents.
The researchers assessed the number of technologies in each participant’s bedroom and the total hours of electronic media use at night, defined as after 9:00 p.m.
Overall, approximately 60% of the teens in the sample reported more than 4 hours of nighttime media use; 63% reported less than 8 hours of sleep on school nights, but this figure reached 76% when parent reports of teens’ sleep was used. When the teens’ self-reports were used, media use was not significantly different between those who had less than 8 hours of sleep vs. those who had 8 hours or more (5.85 vs. 4.39 hours of nighttime media use). But their parents’ reports told another story. In the parent reports, media use was significantly higher in the short sleepers vs. long sleepers (6.12 vs. 2.65 hours of nighttime media use).
After controlling for factors including age, sex, pubertal stage, use of stimulant medication, and severity of ADHD symptoms, nighttime media use was significantly associated with shorter sleep duration, the researchers said.
Nighttime media use also was significantly associated with greater adolescent-reported depressive symptoms, total anxiety symptoms overall, and panic symptoms in particular, as well as with parent-reported generalized anxiety symptoms.
The study findings were limited by several factors including the cross-sectional design, the lack of an objective sleep measure, and the lack of non-ADHD controls, the researchers noted. Also, the researchers were unable to measure parental control over teen media use or to examine different types of media use, including media multitasking (such as texting while video gaming).
However, the findings “suggest that it is important for clinicians to consider nighttime media use when assessing and treating adolescent ADHD, specifically regarding sleep issues and co-occurring depression and anxiety,” they said.
The researchers had no financial conflicts to disclose. The study was funded by grants from the National Institutes of Mental Health.
SOURCE: Becker S et al. Sleep Med. 2018. doi: 10.1016/ j.sleep.2018.06.021.
FROM SLEEP MEDICINE
Key clinical point: Among teens with ADHD, those who had significantly more nighttime media use tended to get less than 8 hours of sleep at night.
Major finding: About 60% of the teens reported more than 4 hours of nighttime media use; 63% reported less than 8 hours of sleep on school nights.
Study details: The data come from a range of research tools used to gather sleep and media use data on 81 adolescents with ADHD, aged 13-17 years.
Disclosures: The researchers had no financial conflicts to disclose. The study was funded by grants from the National Institutes of Mental Health.
Source: Becker S et al. Sleep Med. 2018. doi: 10.1016/ j.sleep.2018.06.021.
Cosmetic surgery patients want beauty ... and more
but motives involving mental and social well-being and physical health are common as well, according to a prospective national study involving more than 500 patients.
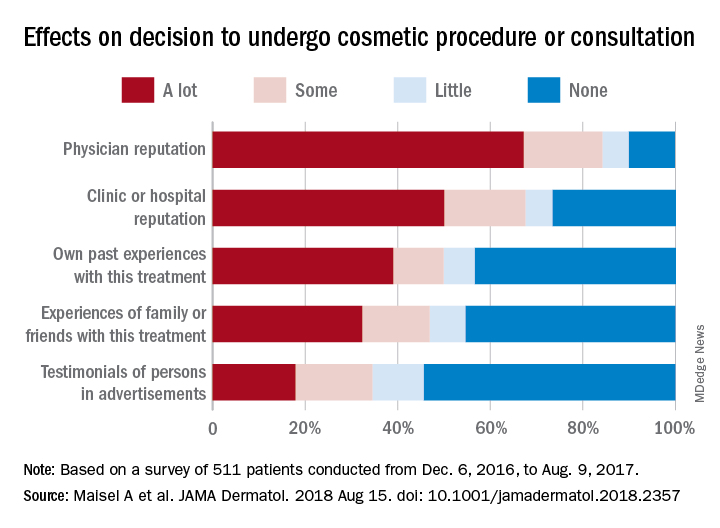
“Cosmetic procedures may also be necessary to correct significant physical disfigurement interfering with work or daily living. Most patients were concerned with how they looked at work and in protecting their physical health, and for some, this motive was the most important. Together, these data add to the growing body of evidence that treatments aimed at improving physical appearance can treat significant physical and psychological illness,” Amanda L. Maisel, of Northwestern University, Chicago, and her associates wrote in JAMA Dermatology.
Among the motives related to appearance, 88.5% of patients said that they “wanted to look better, prettier, or more attractive for themselves,” compared with 64.4% who wanted to look good for others. Patients also were interested in “looking younger or fresher” (83.4%) and having “clear-looking or beautiful skin” (81.4%), the investigators said.
The most common mental or emotional motive was increased self-confidence (69.5%), followed by the desire to “feel happier or better overall or improve quality of life” at 67.2% and to “treat oneself, feel rewarded, or celebrate” at 61.3%. As for social well-being, 56.6% of patients “reported wanting to look good when running into people they knew” and 50.3% reported that they wanted “to feel less self-conscious around others.”
The leading motive involving physical health was “preventing worsening of their condition/symptoms,” which was reported by 53.3% of patients, the investigators reported.
They also examined how reputation and experience influenced patients’ decision to have cosmetic surgery. When asked to what degree a physician’s reputation affected their decision, 67.2% of respondents said a lot, 17% said that it had some effect, 5.7% said it had little effect, and 10% said none. Half of the patients surveyed said the clinic or hospital’s reputation had a lot of influence on their decision, compared with 39% for their own past experiences, 32.2% for experiences of family or friends, and 17.9% for testimonials of persons in advertisements, the researchers said.
The survey was conducted from Dec. 4, 2016, to Aug. 9, 2017, at 2 academic and 11 private dermatology practices. A total of 511 patients were involved, although not all individuals answered every question, so sample sizes varied. The study was supported by a research grant from the American Society for Dermatologic Surgery. The senior investigator reported consulting for Pulse Biosciences that was unrelated to this research and being principal investigator for studies funded in part by Regeneron.
SOURCE: Maisel A et al. JAMA Dermatol. 2018 Aug 15. doi: 10.1001/jamadermatol.2018.2357.
but motives involving mental and social well-being and physical health are common as well, according to a prospective national study involving more than 500 patients.
“Cosmetic procedures may also be necessary to correct significant physical disfigurement interfering with work or daily living. Most patients were concerned with how they looked at work and in protecting their physical health, and for some, this motive was the most important. Together, these data add to the growing body of evidence that treatments aimed at improving physical appearance can treat significant physical and psychological illness,” Amanda L. Maisel, of Northwestern University, Chicago, and her associates wrote in JAMA Dermatology.
Among the motives related to appearance, 88.5% of patients said that they “wanted to look better, prettier, or more attractive for themselves,” compared with 64.4% who wanted to look good for others. Patients also were interested in “looking younger or fresher” (83.4%) and having “clear-looking or beautiful skin” (81.4%), the investigators said.
The most common mental or emotional motive was increased self-confidence (69.5%), followed by the desire to “feel happier or better overall or improve quality of life” at 67.2% and to “treat oneself, feel rewarded, or celebrate” at 61.3%. As for social well-being, 56.6% of patients “reported wanting to look good when running into people they knew” and 50.3% reported that they wanted “to feel less self-conscious around others.”
The leading motive involving physical health was “preventing worsening of their condition/symptoms,” which was reported by 53.3% of patients, the investigators reported.
They also examined how reputation and experience influenced patients’ decision to have cosmetic surgery. When asked to what degree a physician’s reputation affected their decision, 67.2% of respondents said a lot, 17% said that it had some effect, 5.7% said it had little effect, and 10% said none. Half of the patients surveyed said the clinic or hospital’s reputation had a lot of influence on their decision, compared with 39% for their own past experiences, 32.2% for experiences of family or friends, and 17.9% for testimonials of persons in advertisements, the researchers said.
The survey was conducted from Dec. 4, 2016, to Aug. 9, 2017, at 2 academic and 11 private dermatology practices. A total of 511 patients were involved, although not all individuals answered every question, so sample sizes varied. The study was supported by a research grant from the American Society for Dermatologic Surgery. The senior investigator reported consulting for Pulse Biosciences that was unrelated to this research and being principal investigator for studies funded in part by Regeneron.
SOURCE: Maisel A et al. JAMA Dermatol. 2018 Aug 15. doi: 10.1001/jamadermatol.2018.2357.
but motives involving mental and social well-being and physical health are common as well, according to a prospective national study involving more than 500 patients.
“Cosmetic procedures may also be necessary to correct significant physical disfigurement interfering with work or daily living. Most patients were concerned with how they looked at work and in protecting their physical health, and for some, this motive was the most important. Together, these data add to the growing body of evidence that treatments aimed at improving physical appearance can treat significant physical and psychological illness,” Amanda L. Maisel, of Northwestern University, Chicago, and her associates wrote in JAMA Dermatology.
Among the motives related to appearance, 88.5% of patients said that they “wanted to look better, prettier, or more attractive for themselves,” compared with 64.4% who wanted to look good for others. Patients also were interested in “looking younger or fresher” (83.4%) and having “clear-looking or beautiful skin” (81.4%), the investigators said.
The most common mental or emotional motive was increased self-confidence (69.5%), followed by the desire to “feel happier or better overall or improve quality of life” at 67.2% and to “treat oneself, feel rewarded, or celebrate” at 61.3%. As for social well-being, 56.6% of patients “reported wanting to look good when running into people they knew” and 50.3% reported that they wanted “to feel less self-conscious around others.”
The leading motive involving physical health was “preventing worsening of their condition/symptoms,” which was reported by 53.3% of patients, the investigators reported.
They also examined how reputation and experience influenced patients’ decision to have cosmetic surgery. When asked to what degree a physician’s reputation affected their decision, 67.2% of respondents said a lot, 17% said that it had some effect, 5.7% said it had little effect, and 10% said none. Half of the patients surveyed said the clinic or hospital’s reputation had a lot of influence on their decision, compared with 39% for their own past experiences, 32.2% for experiences of family or friends, and 17.9% for testimonials of persons in advertisements, the researchers said.
The survey was conducted from Dec. 4, 2016, to Aug. 9, 2017, at 2 academic and 11 private dermatology practices. A total of 511 patients were involved, although not all individuals answered every question, so sample sizes varied. The study was supported by a research grant from the American Society for Dermatologic Surgery. The senior investigator reported consulting for Pulse Biosciences that was unrelated to this research and being principal investigator for studies funded in part by Regeneron.
SOURCE: Maisel A et al. JAMA Dermatol. 2018 Aug 15. doi: 10.1001/jamadermatol.2018.2357.
FROM JAMA DERMATOLOGY
What is your treatment plan?
The treatment choice is oral terbinafine for tinea capitis with kerion, a scalp dermatophyte infection with a concurrent inflammatory process. Tinea capitis is a very common infection with the peak occurrence at age 3-7 years. Tinea capitis is caused by a variety of dermatophyte species, most commonly by Trichophyton tonsurans or Microsporum canis. T. tonsurans is an endothrix infection, invading the hair shaft and superficial hair while M. canis is an ectothrix infection. T. tonsurans has a person to person transmission; in contrast, M. canis is a zoonotic infection most commonly acquired from infected pets.1 The epidemiology of tinea capitis is affected by multiple factors, including immigration patterns. For example, in Montreal, a study showed a sixfold increase of African dermatophyte species, M. audouinii and T. soudanense.2 Similarly, global variation has increased prevalence of T. violaceum, more commonly found in Europe and Africa, which has been seen in immigrant populations in the United States.1 Kerion is thought to be a hypersensitivity reaction to dermatophytes. Often, misdiagnosis of kerion can result in unnecessary antibiotic prescription and delays in initiation of antifungal therapy.3,4
Tinea capitis can present with focal, “patchy,” well-demarcated hair loss and overlying scale, broken-off hairs at the scalp, and often with pustules. It may be associated with occipital or posterior cervical lymphadenopathy. which presents as a painful boggy scalp mass with or without purulent drainage. Kerions can have associated fever. It is important to differentiate tinea capitis with kerion from processes requiring a different treatment course.3,5 Bacterial folliculitis may have erythema and pustules but only rarely causes hair loss. Similarly, alopecia areata can present with focal alopecia but lacks the scalp inflammation and pustules. Scalp psoriasis is quite scaly, but is more thickened, and usually does not have pustules, nor would purulent drainage be evident. There is a broader differential for other inflammatory focal alopecias including discoid lupus, lichen planopilaris, and dissecting cellulitis of the scalp, which can have similar pus-filled lumps, and cause permanent hair loss, but may be differentiated by associated conditions such as acne conglobata, hidradenitis suppurativa, and pilonidal sinus.

While some clinicians advocate clinical diagnosis of tinea capitis, we advocate confirmation of infection by fungal culture, which can identify the causative organism and influence therapy selection.1 The presence of a fungal infection can be confirmed on potassium hydroxide wet mount prep if hyphae and small spores are seen.6,7 Wood lamp examination will fluoresce if there are ectothrix species, however a negative fluorescence does not differentiate between an endothrix species or lack of infection.
It is important that tinea capitis with kerion is treated with systemic antifungal treatments to allow resolution of the infection, recovery of hair growth, and to prevent or minimize scarring. Systemic antifungal options include terbinafine, griseofulvin, and azoles. Terbinafine is becoming the treatment of choice given shorter duration of treatment with similar efficacy.1T. tonsurans also is thought to respond better to terbinafine than to griseofulvin.8 Griseofulvin is the historical treatment of choice because of its history of clinical safety and no need for laboratory testing. It is important to note that higher doses of griseofulvin (20-25 mg/kg) are recommended, as older lower dose regimens have high rates of failure. Griseofulvin may be more effective for treatment of Microsporum spp than is terbinafine.8 Fluconazole is the only oral antifungal agent that is approved for patients younger than 2 years of age, however it has lower cure rates, compared with terbinafine and griseofulvin.1 Tinea capitis with kerion does not generally require antibiotics unless there is superimposed bacterial infection. Kerion also do not require incision or drainage, which may increase scarring and complicate the clinical course.
Management of tinea capitis should include evaluation of any other household members for coinfection. Depending on the dermatophyte involved there can be risk of person-to-person transmission.5 Families should be educated about fomite transmission via shared combs or hats. Additionally, if a zoophilic dermatophyte is suspected, pets also should be appropriately examined and treated. Topical antifungals are insufficient to eradicate tinea capitis but can be used as adjunctive therapy. Kerion can be treated with oral prednisone in addition to oral antifungals if the lesions are very painful, however there is limited data on this treatment option.9

Ms. Kaushik is a research fellow and Dr. Eichenfield is professor of dermatology and pediatrics in the division of pediatric and adolescent dermatology at Rady Children’s Hospital and the University of California, both in San Diego. They have no conflicts of interest or relevant financial disclosures.
References
1. “Red Book: Report of the Committee on Infectious Diseases,” 31st Edition, (Elk Grove Village, Ill.: American Academy of Pediatrics, 2018, p. 1264).
2. Pediatr Dermatol. 2018 May;35(3):323-8
3. Arch Dis Child. 2016 May;101(5):503.
4. IDCases. 2018 Jun 28;14:e00418.
5. Int J Dermatol. 2018 Jan;57(1):3-9.
6. Pediatr Rev. 2007 May;28(5):164-74.
7. Clin Cosmet Investig Dermatol. 2010;3:89-98.
8. Cochrane Database Syst Rev. 2016 May 12;(5):CD004685.
9. Med Mycol. 1999 Apr;37(2):97-9.
The treatment choice is oral terbinafine for tinea capitis with kerion, a scalp dermatophyte infection with a concurrent inflammatory process. Tinea capitis is a very common infection with the peak occurrence at age 3-7 years. Tinea capitis is caused by a variety of dermatophyte species, most commonly by Trichophyton tonsurans or Microsporum canis. T. tonsurans is an endothrix infection, invading the hair shaft and superficial hair while M. canis is an ectothrix infection. T. tonsurans has a person to person transmission; in contrast, M. canis is a zoonotic infection most commonly acquired from infected pets.1 The epidemiology of tinea capitis is affected by multiple factors, including immigration patterns. For example, in Montreal, a study showed a sixfold increase of African dermatophyte species, M. audouinii and T. soudanense.2 Similarly, global variation has increased prevalence of T. violaceum, more commonly found in Europe and Africa, which has been seen in immigrant populations in the United States.1 Kerion is thought to be a hypersensitivity reaction to dermatophytes. Often, misdiagnosis of kerion can result in unnecessary antibiotic prescription and delays in initiation of antifungal therapy.3,4
Tinea capitis can present with focal, “patchy,” well-demarcated hair loss and overlying scale, broken-off hairs at the scalp, and often with pustules. It may be associated with occipital or posterior cervical lymphadenopathy. which presents as a painful boggy scalp mass with or without purulent drainage. Kerions can have associated fever. It is important to differentiate tinea capitis with kerion from processes requiring a different treatment course.3,5 Bacterial folliculitis may have erythema and pustules but only rarely causes hair loss. Similarly, alopecia areata can present with focal alopecia but lacks the scalp inflammation and pustules. Scalp psoriasis is quite scaly, but is more thickened, and usually does not have pustules, nor would purulent drainage be evident. There is a broader differential for other inflammatory focal alopecias including discoid lupus, lichen planopilaris, and dissecting cellulitis of the scalp, which can have similar pus-filled lumps, and cause permanent hair loss, but may be differentiated by associated conditions such as acne conglobata, hidradenitis suppurativa, and pilonidal sinus.
While some clinicians advocate clinical diagnosis of tinea capitis, we advocate confirmation of infection by fungal culture, which can identify the causative organism and influence therapy selection.1 The presence of a fungal infection can be confirmed on potassium hydroxide wet mount prep if hyphae and small spores are seen.6,7 Wood lamp examination will fluoresce if there are ectothrix species, however a negative fluorescence does not differentiate between an endothrix species or lack of infection.
It is important that tinea capitis with kerion is treated with systemic antifungal treatments to allow resolution of the infection, recovery of hair growth, and to prevent or minimize scarring. Systemic antifungal options include terbinafine, griseofulvin, and azoles. Terbinafine is becoming the treatment of choice given shorter duration of treatment with similar efficacy.1T. tonsurans also is thought to respond better to terbinafine than to griseofulvin.8 Griseofulvin is the historical treatment of choice because of its history of clinical safety and no need for laboratory testing. It is important to note that higher doses of griseofulvin (20-25 mg/kg) are recommended, as older lower dose regimens have high rates of failure. Griseofulvin may be more effective for treatment of Microsporum spp than is terbinafine.8 Fluconazole is the only oral antifungal agent that is approved for patients younger than 2 years of age, however it has lower cure rates, compared with terbinafine and griseofulvin.1 Tinea capitis with kerion does not generally require antibiotics unless there is superimposed bacterial infection. Kerion also do not require incision or drainage, which may increase scarring and complicate the clinical course.
Management of tinea capitis should include evaluation of any other household members for coinfection. Depending on the dermatophyte involved there can be risk of person-to-person transmission.5 Families should be educated about fomite transmission via shared combs or hats. Additionally, if a zoophilic dermatophyte is suspected, pets also should be appropriately examined and treated. Topical antifungals are insufficient to eradicate tinea capitis but can be used as adjunctive therapy. Kerion can be treated with oral prednisone in addition to oral antifungals if the lesions are very painful, however there is limited data on this treatment option.9
Ms. Kaushik is a research fellow and Dr. Eichenfield is professor of dermatology and pediatrics in the division of pediatric and adolescent dermatology at Rady Children’s Hospital and the University of California, both in San Diego. They have no conflicts of interest or relevant financial disclosures.
References
1. “Red Book: Report of the Committee on Infectious Diseases,” 31st Edition, (Elk Grove Village, Ill.: American Academy of Pediatrics, 2018, p. 1264).
2. Pediatr Dermatol. 2018 May;35(3):323-8
3. Arch Dis Child. 2016 May;101(5):503.
4. IDCases. 2018 Jun 28;14:e00418.
5. Int J Dermatol. 2018 Jan;57(1):3-9.
6. Pediatr Rev. 2007 May;28(5):164-74.
7. Clin Cosmet Investig Dermatol. 2010;3:89-98.
8. Cochrane Database Syst Rev. 2016 May 12;(5):CD004685.
9. Med Mycol. 1999 Apr;37(2):97-9.
The treatment choice is oral terbinafine for tinea capitis with kerion, a scalp dermatophyte infection with a concurrent inflammatory process. Tinea capitis is a very common infection with the peak occurrence at age 3-7 years. Tinea capitis is caused by a variety of dermatophyte species, most commonly by Trichophyton tonsurans or Microsporum canis. T. tonsurans is an endothrix infection, invading the hair shaft and superficial hair while M. canis is an ectothrix infection. T. tonsurans has a person to person transmission; in contrast, M. canis is a zoonotic infection most commonly acquired from infected pets.1 The epidemiology of tinea capitis is affected by multiple factors, including immigration patterns. For example, in Montreal, a study showed a sixfold increase of African dermatophyte species, M. audouinii and T. soudanense.2 Similarly, global variation has increased prevalence of T. violaceum, more commonly found in Europe and Africa, which has been seen in immigrant populations in the United States.1 Kerion is thought to be a hypersensitivity reaction to dermatophytes. Often, misdiagnosis of kerion can result in unnecessary antibiotic prescription and delays in initiation of antifungal therapy.3,4
Tinea capitis can present with focal, “patchy,” well-demarcated hair loss and overlying scale, broken-off hairs at the scalp, and often with pustules. It may be associated with occipital or posterior cervical lymphadenopathy. which presents as a painful boggy scalp mass with or without purulent drainage. Kerions can have associated fever. It is important to differentiate tinea capitis with kerion from processes requiring a different treatment course.3,5 Bacterial folliculitis may have erythema and pustules but only rarely causes hair loss. Similarly, alopecia areata can present with focal alopecia but lacks the scalp inflammation and pustules. Scalp psoriasis is quite scaly, but is more thickened, and usually does not have pustules, nor would purulent drainage be evident. There is a broader differential for other inflammatory focal alopecias including discoid lupus, lichen planopilaris, and dissecting cellulitis of the scalp, which can have similar pus-filled lumps, and cause permanent hair loss, but may be differentiated by associated conditions such as acne conglobata, hidradenitis suppurativa, and pilonidal sinus.
While some clinicians advocate clinical diagnosis of tinea capitis, we advocate confirmation of infection by fungal culture, which can identify the causative organism and influence therapy selection.1 The presence of a fungal infection can be confirmed on potassium hydroxide wet mount prep if hyphae and small spores are seen.6,7 Wood lamp examination will fluoresce if there are ectothrix species, however a negative fluorescence does not differentiate between an endothrix species or lack of infection.
It is important that tinea capitis with kerion is treated with systemic antifungal treatments to allow resolution of the infection, recovery of hair growth, and to prevent or minimize scarring. Systemic antifungal options include terbinafine, griseofulvin, and azoles. Terbinafine is becoming the treatment of choice given shorter duration of treatment with similar efficacy.1T. tonsurans also is thought to respond better to terbinafine than to griseofulvin.8 Griseofulvin is the historical treatment of choice because of its history of clinical safety and no need for laboratory testing. It is important to note that higher doses of griseofulvin (20-25 mg/kg) are recommended, as older lower dose regimens have high rates of failure. Griseofulvin may be more effective for treatment of Microsporum spp than is terbinafine.8 Fluconazole is the only oral antifungal agent that is approved for patients younger than 2 years of age, however it has lower cure rates, compared with terbinafine and griseofulvin.1 Tinea capitis with kerion does not generally require antibiotics unless there is superimposed bacterial infection. Kerion also do not require incision or drainage, which may increase scarring and complicate the clinical course.
Management of tinea capitis should include evaluation of any other household members for coinfection. Depending on the dermatophyte involved there can be risk of person-to-person transmission.5 Families should be educated about fomite transmission via shared combs or hats. Additionally, if a zoophilic dermatophyte is suspected, pets also should be appropriately examined and treated. Topical antifungals are insufficient to eradicate tinea capitis but can be used as adjunctive therapy. Kerion can be treated with oral prednisone in addition to oral antifungals if the lesions are very painful, however there is limited data on this treatment option.9
Ms. Kaushik is a research fellow and Dr. Eichenfield is professor of dermatology and pediatrics in the division of pediatric and adolescent dermatology at Rady Children’s Hospital and the University of California, both in San Diego. They have no conflicts of interest or relevant financial disclosures.
References
1. “Red Book: Report of the Committee on Infectious Diseases,” 31st Edition, (Elk Grove Village, Ill.: American Academy of Pediatrics, 2018, p. 1264).
2. Pediatr Dermatol. 2018 May;35(3):323-8
3. Arch Dis Child. 2016 May;101(5):503.
4. IDCases. 2018 Jun 28;14:e00418.
5. Int J Dermatol. 2018 Jan;57(1):3-9.
6. Pediatr Rev. 2007 May;28(5):164-74.
7. Clin Cosmet Investig Dermatol. 2010;3:89-98.
8. Cochrane Database Syst Rev. 2016 May 12;(5):CD004685.
9. Med Mycol. 1999 Apr;37(2):97-9.
A 10-year-old otherwise-healthy male presents for a progressing lesion on his scalp. One month prior to coming in, he developed some peeling and itch followed by loss of hair. This had worsened, becoming a painful and boggy mass on the back of his head with focal alopecia. He went to the local ED, where he had plain films of his skull, which were normal and was prescribed cephalexin. He has not shown any improvement after starting the antibiotics. He has had no fevers in this time, but the pain persists.
On physical exam, he is noted to have a hairless patch on a boggy left occipital mass, which is tender to palpation. There is a small amount of overlying honey-colored crusting. He has associated posterior occipital nontender lymphadenopathy.
The patient's older sister has a small area of scalp hair loss.
SMART Self-Management Program Enhances Epilepsy Care
A self-management program called SMART can help patients with epilepsy reduce the risk of negative health events according to researchers at Case Western Reserve University School of Medicine.
- A 6-month randomized controlled trial of the community-based program included 60 adult patients and was compared to 60 control patients on a waitlist.
- The experiment monitored a variety of events, including seizures, accidents, attempts at self-harm, emergency department visits, and hospitalizations.
- The average patient in this trial was about 41 years old, about 70% were African American, 74% were unemployed.
- Patients who were randomized to the SMART program had fewer negative health events by 6 months compared to controls.
- SMART was also linked to improved scores on the Patient Health Questionnaire (P=.002), the 10 item Quality of Life in Epilepsy Inventory, and the Short Form Health Survey.
Sajatovic M, Colon-Zimmermann K, Kahriman M, et al. A 6-month prospective randomized controlled trial of remotely delivered group format epilepsy self-management versus waitlist control for high-risk people with epilepsy. [Published online ahead of print August 10, 2018]. Epilepsia. https://doi.org/10.1111/epi.14527.
A self-management program called SMART can help patients with epilepsy reduce the risk of negative health events according to researchers at Case Western Reserve University School of Medicine.
- A 6-month randomized controlled trial of the community-based program included 60 adult patients and was compared to 60 control patients on a waitlist.
- The experiment monitored a variety of events, including seizures, accidents, attempts at self-harm, emergency department visits, and hospitalizations.
- The average patient in this trial was about 41 years old, about 70% were African American, 74% were unemployed.
- Patients who were randomized to the SMART program had fewer negative health events by 6 months compared to controls.
- SMART was also linked to improved scores on the Patient Health Questionnaire (P=.002), the 10 item Quality of Life in Epilepsy Inventory, and the Short Form Health Survey.
Sajatovic M, Colon-Zimmermann K, Kahriman M, et al. A 6-month prospective randomized controlled trial of remotely delivered group format epilepsy self-management versus waitlist control for high-risk people with epilepsy. [Published online ahead of print August 10, 2018]. Epilepsia. https://doi.org/10.1111/epi.14527.
A self-management program called SMART can help patients with epilepsy reduce the risk of negative health events according to researchers at Case Western Reserve University School of Medicine.
- A 6-month randomized controlled trial of the community-based program included 60 adult patients and was compared to 60 control patients on a waitlist.
- The experiment monitored a variety of events, including seizures, accidents, attempts at self-harm, emergency department visits, and hospitalizations.
- The average patient in this trial was about 41 years old, about 70% were African American, 74% were unemployed.
- Patients who were randomized to the SMART program had fewer negative health events by 6 months compared to controls.
- SMART was also linked to improved scores on the Patient Health Questionnaire (P=.002), the 10 item Quality of Life in Epilepsy Inventory, and the Short Form Health Survey.
Sajatovic M, Colon-Zimmermann K, Kahriman M, et al. A 6-month prospective randomized controlled trial of remotely delivered group format epilepsy self-management versus waitlist control for high-risk people with epilepsy. [Published online ahead of print August 10, 2018]. Epilepsia. https://doi.org/10.1111/epi.14527.
FDA puts partial hold on trial of vascular agent for AML, MDS
The Food and Drug Administration has placed a partial clinical hold on a phase 1b/2 study of OXi4503, a vascular disrupting agent.

In this trial (NCT02576301), researchers are evaluating OXi4503 alone and in combination with cytarabine in patients with relapsed/refractory acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS).
The partial clinical hold applies to the 12.2 mg/m2 dose of OXi4503. The FDA is allowing the continued treatment and enrollment of patients using a dose of 9.76 mg/m2. Additional data on patients receiving OXi4503 at 9.76 mg/m2 must be evaluated before dosing at 12.2 mg/m2 can be resumed.
The partial clinical hold is a result of two potential dose-limiting toxicities (DLTs) observed at the 12.2-mg/m2 dose level: hypotension, which occurred shortly after initial treatment with OXi4503, and acute hypoxic respiratory failure, which occurred approximately 2 weeks after receiving OXi4503 and cytarabine.
Both events were deemed “possibly related” to OXi4503, and both patients recovered following treatment.
“Although it is disappointing that we are not currently continuing with the higher dose of OXi4503, we look forward to gathering more safety and efficacy data at the previous dose level, where we observed 2 complete remissions in the 4 patients that we treated,” William D. Schwieterman, MD, chief executive officer of Mateon Therapeutics Inc., the company developing OXi4503, said in a statement.
In preclinical research, OXi4503 demonstrated activity against AML, both when given alone and in combination with bevacizumab. These results were published in Blood in 2010.
In a phase 1 trial (NCT01085656), researchers evaluated OXi4503 in patients with relapsed or refractory AML or MDS. OXi4503 demonstrated preliminary evidence of disease response in heavily pretreated, refractory AML and advanced MDS.
The maximum tolerated dose of OXi4503 was not identified, but adverse events attributable to the drug included hypertension, bone pain, fever, anemia, thrombocytopenia, and coagulopathies.
Results from this study were presented at the 2013 annual meeting of the American Society of Hematology.
In 2015, Mateon Therapeutics initiated the phase 1b/2 study of OXi4503 (NCT02576301) that is now on partial clinical hold.
The phase 1 portion of this study was designed to assess the safety, pharmacokinetics, pharmacodynamics, and preliminary efficacy of single-agent OXi4503 in patients with relapsed/refractory AML and MDS. It is also aimed at determining the safety, pharmacokinetics, and pharmacodynamics of OXi4503 plus intermediate-dose cytarabine.
The goal of the phase 2 portion is to assess the preliminary efficacy of OXi4503 and cytarabine in patients with AML and MDS.
The Food and Drug Administration has placed a partial clinical hold on a phase 1b/2 study of OXi4503, a vascular disrupting agent.
In this trial (NCT02576301), researchers are evaluating OXi4503 alone and in combination with cytarabine in patients with relapsed/refractory acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS).
The partial clinical hold applies to the 12.2 mg/m2 dose of OXi4503. The FDA is allowing the continued treatment and enrollment of patients using a dose of 9.76 mg/m2. Additional data on patients receiving OXi4503 at 9.76 mg/m2 must be evaluated before dosing at 12.2 mg/m2 can be resumed.
The partial clinical hold is a result of two potential dose-limiting toxicities (DLTs) observed at the 12.2-mg/m2 dose level: hypotension, which occurred shortly after initial treatment with OXi4503, and acute hypoxic respiratory failure, which occurred approximately 2 weeks after receiving OXi4503 and cytarabine.
Both events were deemed “possibly related” to OXi4503, and both patients recovered following treatment.
“Although it is disappointing that we are not currently continuing with the higher dose of OXi4503, we look forward to gathering more safety and efficacy data at the previous dose level, where we observed 2 complete remissions in the 4 patients that we treated,” William D. Schwieterman, MD, chief executive officer of Mateon Therapeutics Inc., the company developing OXi4503, said in a statement.
In preclinical research, OXi4503 demonstrated activity against AML, both when given alone and in combination with bevacizumab. These results were published in Blood in 2010.
In a phase 1 trial (NCT01085656), researchers evaluated OXi4503 in patients with relapsed or refractory AML or MDS. OXi4503 demonstrated preliminary evidence of disease response in heavily pretreated, refractory AML and advanced MDS.
The maximum tolerated dose of OXi4503 was not identified, but adverse events attributable to the drug included hypertension, bone pain, fever, anemia, thrombocytopenia, and coagulopathies.
Results from this study were presented at the 2013 annual meeting of the American Society of Hematology.
In 2015, Mateon Therapeutics initiated the phase 1b/2 study of OXi4503 (NCT02576301) that is now on partial clinical hold.
The phase 1 portion of this study was designed to assess the safety, pharmacokinetics, pharmacodynamics, and preliminary efficacy of single-agent OXi4503 in patients with relapsed/refractory AML and MDS. It is also aimed at determining the safety, pharmacokinetics, and pharmacodynamics of OXi4503 plus intermediate-dose cytarabine.
The goal of the phase 2 portion is to assess the preliminary efficacy of OXi4503 and cytarabine in patients with AML and MDS.
The Food and Drug Administration has placed a partial clinical hold on a phase 1b/2 study of OXi4503, a vascular disrupting agent.
In this trial (NCT02576301), researchers are evaluating OXi4503 alone and in combination with cytarabine in patients with relapsed/refractory acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS).
The partial clinical hold applies to the 12.2 mg/m2 dose of OXi4503. The FDA is allowing the continued treatment and enrollment of patients using a dose of 9.76 mg/m2. Additional data on patients receiving OXi4503 at 9.76 mg/m2 must be evaluated before dosing at 12.2 mg/m2 can be resumed.
The partial clinical hold is a result of two potential dose-limiting toxicities (DLTs) observed at the 12.2-mg/m2 dose level: hypotension, which occurred shortly after initial treatment with OXi4503, and acute hypoxic respiratory failure, which occurred approximately 2 weeks after receiving OXi4503 and cytarabine.
Both events were deemed “possibly related” to OXi4503, and both patients recovered following treatment.
“Although it is disappointing that we are not currently continuing with the higher dose of OXi4503, we look forward to gathering more safety and efficacy data at the previous dose level, where we observed 2 complete remissions in the 4 patients that we treated,” William D. Schwieterman, MD, chief executive officer of Mateon Therapeutics Inc., the company developing OXi4503, said in a statement.
In preclinical research, OXi4503 demonstrated activity against AML, both when given alone and in combination with bevacizumab. These results were published in Blood in 2010.
In a phase 1 trial (NCT01085656), researchers evaluated OXi4503 in patients with relapsed or refractory AML or MDS. OXi4503 demonstrated preliminary evidence of disease response in heavily pretreated, refractory AML and advanced MDS.
The maximum tolerated dose of OXi4503 was not identified, but adverse events attributable to the drug included hypertension, bone pain, fever, anemia, thrombocytopenia, and coagulopathies.
Results from this study were presented at the 2013 annual meeting of the American Society of Hematology.
In 2015, Mateon Therapeutics initiated the phase 1b/2 study of OXi4503 (NCT02576301) that is now on partial clinical hold.
The phase 1 portion of this study was designed to assess the safety, pharmacokinetics, pharmacodynamics, and preliminary efficacy of single-agent OXi4503 in patients with relapsed/refractory AML and MDS. It is also aimed at determining the safety, pharmacokinetics, and pharmacodynamics of OXi4503 plus intermediate-dose cytarabine.
The goal of the phase 2 portion is to assess the preliminary efficacy of OXi4503 and cytarabine in patients with AML and MDS.