User login
Digital CBT improves health, well-being, quality of life in randomized trial
Digitally delivered cognitive-behavioral therapy not only improved insomnia, but also provided “around-the-clock” health, psychological, and quality-of-life improvements, according to investigators.

Compared with control subjects, patients who used a cognitive-behavioral therapy (CBT) program and associated iPhone app had small improvements in functional health and psychological well-being and large improvements in sleep-related quality of life, the investigators reported.
Those changes were mediated by a large improvement in insomnia, according to Colin A. Espie, PhD, professor of sleep medicine at the University of Oxford (England), and his colleagues.
“These findings indicate that digital CBT improves both daytime and nighttime aspects of insomnia, lending further weight to the clinical guideline recommendation of CBT as the treatment of choice for insomnia,” Dr. Espie and his colleagues reported in JAMA Psychiatry.
Their study included 1,711 adults with symptoms of insomnia that were self-reported and a score of 16 or less on the Sleep Condition Indicator (SCI), which has a range of 0-32. A total of 853 were randomized to receive digital CBT, of whom 413 completed six scheduled 20-minute sessions; an additional 276 adults completed at least one session. The control arm included 858 individuals randomized to sleep hygiene education, of whom 759 went on to receive that intervention.
Small but significant improvements were seen in self-reported measures of functional health, Dr. Espie and his colleagues reported. The adjusted differences in mean scores on the Patient-Reported Outcomes Measurement Information System: Global Health Scale were 0.90 in a midtreatment evaluation at 4 weeks after baseline, 1.76 in a posttreatment evaluation at 8 weeks, and 1.76 at a 24-week follow-up assessment (P less than 0.001 for all comparisons).
Likewise, adjusted differences in the Warwick-Edinburgh Mental Wellbeing Scale were 1.04, 2.68, and 2.95 at 4, 8, and 24 weeks, respectively.
Adjusted differences in the Glasgow Sleep Impact Index, which measures sleep-related quality of life, were –8.76, –17.60, and –18.72 at 4, 8, and 24 weeks, respectively, indicating a benefit of the digital CBT intervention over the control intervention, the investigators said.
About 45%-84% of these effects were attributable to changes in insomnia symptoms, results of a mediation analysis showed.
Insomnia scores as measured by the eight-item SCI scale were 6.5 and 6.6 at baseline for the digital CBT and control groups, respectively. By week 4, SCI scores were 9.96 and 13.00 for the two groups (P less than 0.001), with significant differences also reported at the 8- and 24-week evaluations.
The investigators noted that their findings might not be generalizable because participants were not drawn from patient populations. In addition, although 58% of the participants completed 4 weeks or more of the digital CBT sessions, the dropout rate was “substantial.”
Nevertheless, they wrote, these findings, together with results of a recently reported parallel study looking at the effects of sleep on mental health, affirm CBT as the treatment of choice for insomnia. “The mediation analyses in these two studies, with a total of 5,466 participants, provide novel and convincing evidence that insomnia may be a legitimate and important target for mental health and well-being.”
Dr. Espie reported that he is cofounder and chief medical officer of Big Health. He is a shareholder and receives a salary from the company, which was involved in the design and conduct of the study, interpretation of data, and development of the manuscript appearing in JAMA Psychiatry. The study was funded by Big Health and other sources, including the National Institute for Health Research Oxford Biomedical Research Centre.
SOURCE: Espie CA et al. JAMA Psychiatry. 2018 Sep 25. doi: 10.1001/jamapsychiatry.2018.2745.
Digitally delivered cognitive-behavioral therapy not only improved insomnia, but also provided “around-the-clock” health, psychological, and quality-of-life improvements, according to investigators.
Compared with control subjects, patients who used a cognitive-behavioral therapy (CBT) program and associated iPhone app had small improvements in functional health and psychological well-being and large improvements in sleep-related quality of life, the investigators reported.
Those changes were mediated by a large improvement in insomnia, according to Colin A. Espie, PhD, professor of sleep medicine at the University of Oxford (England), and his colleagues.
“These findings indicate that digital CBT improves both daytime and nighttime aspects of insomnia, lending further weight to the clinical guideline recommendation of CBT as the treatment of choice for insomnia,” Dr. Espie and his colleagues reported in JAMA Psychiatry.
Their study included 1,711 adults with symptoms of insomnia that were self-reported and a score of 16 or less on the Sleep Condition Indicator (SCI), which has a range of 0-32. A total of 853 were randomized to receive digital CBT, of whom 413 completed six scheduled 20-minute sessions; an additional 276 adults completed at least one session. The control arm included 858 individuals randomized to sleep hygiene education, of whom 759 went on to receive that intervention.
Small but significant improvements were seen in self-reported measures of functional health, Dr. Espie and his colleagues reported. The adjusted differences in mean scores on the Patient-Reported Outcomes Measurement Information System: Global Health Scale were 0.90 in a midtreatment evaluation at 4 weeks after baseline, 1.76 in a posttreatment evaluation at 8 weeks, and 1.76 at a 24-week follow-up assessment (P less than 0.001 for all comparisons).
Likewise, adjusted differences in the Warwick-Edinburgh Mental Wellbeing Scale were 1.04, 2.68, and 2.95 at 4, 8, and 24 weeks, respectively.
Adjusted differences in the Glasgow Sleep Impact Index, which measures sleep-related quality of life, were –8.76, –17.60, and –18.72 at 4, 8, and 24 weeks, respectively, indicating a benefit of the digital CBT intervention over the control intervention, the investigators said.
About 45%-84% of these effects were attributable to changes in insomnia symptoms, results of a mediation analysis showed.
Insomnia scores as measured by the eight-item SCI scale were 6.5 and 6.6 at baseline for the digital CBT and control groups, respectively. By week 4, SCI scores were 9.96 and 13.00 for the two groups (P less than 0.001), with significant differences also reported at the 8- and 24-week evaluations.
The investigators noted that their findings might not be generalizable because participants were not drawn from patient populations. In addition, although 58% of the participants completed 4 weeks or more of the digital CBT sessions, the dropout rate was “substantial.”
Nevertheless, they wrote, these findings, together with results of a recently reported parallel study looking at the effects of sleep on mental health, affirm CBT as the treatment of choice for insomnia. “The mediation analyses in these two studies, with a total of 5,466 participants, provide novel and convincing evidence that insomnia may be a legitimate and important target for mental health and well-being.”
Dr. Espie reported that he is cofounder and chief medical officer of Big Health. He is a shareholder and receives a salary from the company, which was involved in the design and conduct of the study, interpretation of data, and development of the manuscript appearing in JAMA Psychiatry. The study was funded by Big Health and other sources, including the National Institute for Health Research Oxford Biomedical Research Centre.
SOURCE: Espie CA et al. JAMA Psychiatry. 2018 Sep 25. doi: 10.1001/jamapsychiatry.2018.2745.
Digitally delivered cognitive-behavioral therapy not only improved insomnia, but also provided “around-the-clock” health, psychological, and quality-of-life improvements, according to investigators.
Compared with control subjects, patients who used a cognitive-behavioral therapy (CBT) program and associated iPhone app had small improvements in functional health and psychological well-being and large improvements in sleep-related quality of life, the investigators reported.
Those changes were mediated by a large improvement in insomnia, according to Colin A. Espie, PhD, professor of sleep medicine at the University of Oxford (England), and his colleagues.
“These findings indicate that digital CBT improves both daytime and nighttime aspects of insomnia, lending further weight to the clinical guideline recommendation of CBT as the treatment of choice for insomnia,” Dr. Espie and his colleagues reported in JAMA Psychiatry.
Their study included 1,711 adults with symptoms of insomnia that were self-reported and a score of 16 or less on the Sleep Condition Indicator (SCI), which has a range of 0-32. A total of 853 were randomized to receive digital CBT, of whom 413 completed six scheduled 20-minute sessions; an additional 276 adults completed at least one session. The control arm included 858 individuals randomized to sleep hygiene education, of whom 759 went on to receive that intervention.
Small but significant improvements were seen in self-reported measures of functional health, Dr. Espie and his colleagues reported. The adjusted differences in mean scores on the Patient-Reported Outcomes Measurement Information System: Global Health Scale were 0.90 in a midtreatment evaluation at 4 weeks after baseline, 1.76 in a posttreatment evaluation at 8 weeks, and 1.76 at a 24-week follow-up assessment (P less than 0.001 for all comparisons).
Likewise, adjusted differences in the Warwick-Edinburgh Mental Wellbeing Scale were 1.04, 2.68, and 2.95 at 4, 8, and 24 weeks, respectively.
Adjusted differences in the Glasgow Sleep Impact Index, which measures sleep-related quality of life, were –8.76, –17.60, and –18.72 at 4, 8, and 24 weeks, respectively, indicating a benefit of the digital CBT intervention over the control intervention, the investigators said.
About 45%-84% of these effects were attributable to changes in insomnia symptoms, results of a mediation analysis showed.
Insomnia scores as measured by the eight-item SCI scale were 6.5 and 6.6 at baseline for the digital CBT and control groups, respectively. By week 4, SCI scores were 9.96 and 13.00 for the two groups (P less than 0.001), with significant differences also reported at the 8- and 24-week evaluations.
The investigators noted that their findings might not be generalizable because participants were not drawn from patient populations. In addition, although 58% of the participants completed 4 weeks or more of the digital CBT sessions, the dropout rate was “substantial.”
Nevertheless, they wrote, these findings, together with results of a recently reported parallel study looking at the effects of sleep on mental health, affirm CBT as the treatment of choice for insomnia. “The mediation analyses in these two studies, with a total of 5,466 participants, provide novel and convincing evidence that insomnia may be a legitimate and important target for mental health and well-being.”
Dr. Espie reported that he is cofounder and chief medical officer of Big Health. He is a shareholder and receives a salary from the company, which was involved in the design and conduct of the study, interpretation of data, and development of the manuscript appearing in JAMA Psychiatry. The study was funded by Big Health and other sources, including the National Institute for Health Research Oxford Biomedical Research Centre.
SOURCE: Espie CA et al. JAMA Psychiatry. 2018 Sep 25. doi: 10.1001/jamapsychiatry.2018.2745.
FROM JAMA PSYCHIATRY
Key clinical point: Individuals who used a cognitive-behavioral therapy program and app had significant health, psychological, and quality-of-life improvements, compared with controls.
Major finding: Significant improvements were seen in self-reported measures of functional health, psychological well-being, and sleep-related quality of life were reported. About 45%-84% of the effects were mediated by improvements in insomnia symptoms.
Study details: An online, randomized, parallel-group trial including 1,711 adults with self-reported insomnia symptoms who received digital cognitive-behavioral therapy or sleep hygiene education.
Disclosures: The senior study author is the cofounder and chief medical officer of Big Health, which was involved in the design and conduct of the study, interpretation of data, and development of the manuscript. The study was funded by Big Health and other sources, including the National Institute for Health Research Oxford Biomedical Research Centre.
Source: Espie CA et al. JAMA Psychiatry. 2018 Sep 25. doi: 10.1001/jamapsychiatry.2018.2745.
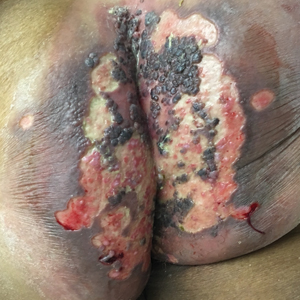
Perianal Ulceration and Verrucous Papules
The Diagnosis: Herpes Simplex Virus Infection
Viral culture of the ulcer was positive for herpes simplex virus type 2 (HHV-2). Bacterial culture grew enteric flora. The patient was started on intravenous acyclovir 5 mg/kg every 8 hours for 7 days and then transitioned to oral acyclovir for chronic suppressive therapy. One month later, there was near-complete reepithelialization with 2 remaining 1-cm shallow ulcers. The verrucous lesions had dried up and were flaking off (Figure). At 6-month follow-up, the ulcers and verrucous lesions had completely resolved.
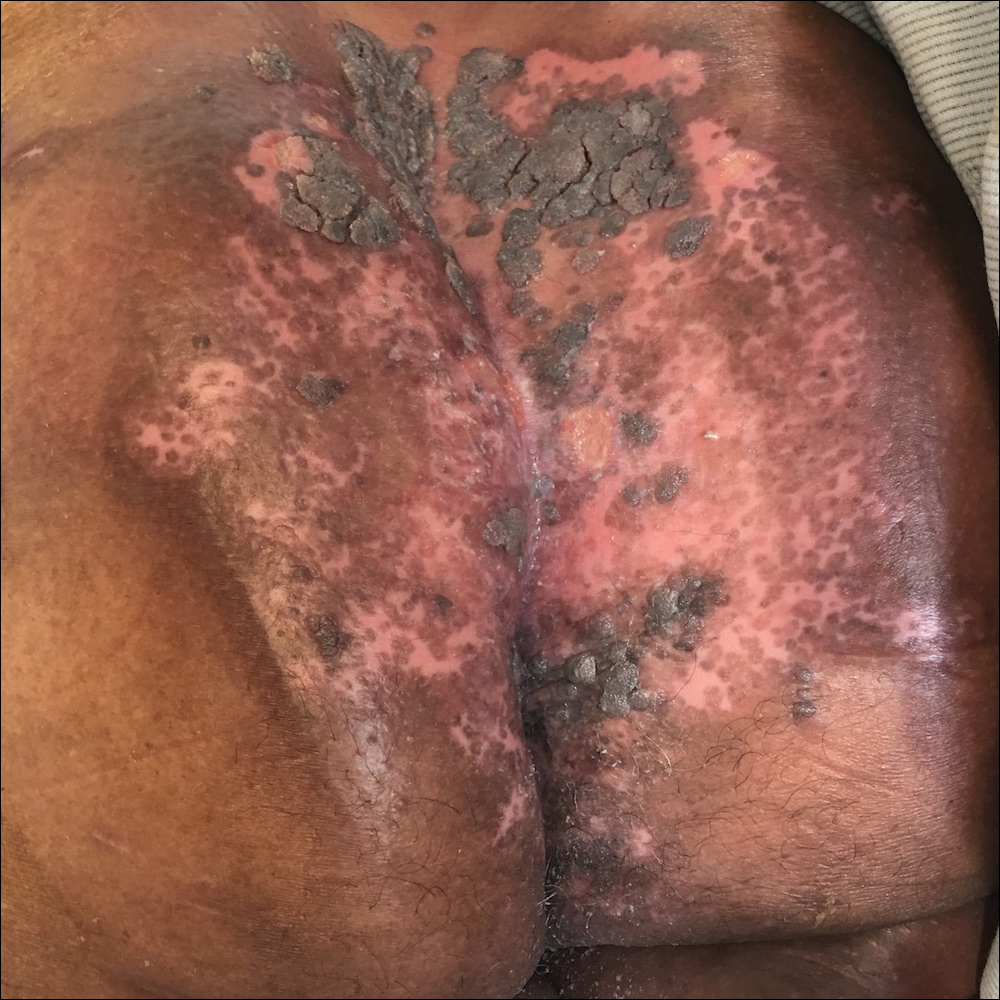
Herpes simplex virus type 2 is the most common cause of genital and perianal ulcers in immunocompromised individuals.1 Patients classically present with painful grouped vesicles followed by painful superficial ulcers that may rapidly progress to extensive confluent ulceration. A hypertrophic variant of genital herpes characterized by anogenital verrucous lesions, similar to condyloma acuminata, also can be seen in immunocompromised individuals.2 This form has almost exclusively been observed in patients with human immunodeficiency virus and may occur in isolation or together with the ulcerative form.1-5 A case of vegetative HHV infection of the genital area in a patient with common variable immunodeficiency has been reported.6 Verrucous lesions of the mouth secondary to HHV have been observed in Hodgkin lymphoma, acute myelogenous leukemia, and individuals on immunosuppressive medications.7-10
Perianal involvement of Crohn disease typically presents with fistulas, ulcers, abscesses, strictures, and skin tags in some cases. Invasive squamous cell carcinoma may arise within a chronic ulcer of the anogenital area or may itself manifest as an ulcer or anal fissure. Perianal ulcerative skin tuberculosis has been reported in the literature as a rare manifestation of extrapulmonary tuberculosis and should be considered in a patient with an appropriate clinical history. Pyoderma gangrenosum classically pre-sents as a large ulcer with irregular rolled borders, though a rare variant of vegetative pyoderma gangrenosum may manifest as a nodular or verrucous plaque.
Studies to diagnose HHV include viral cell culture, HHV polymerase chain reaction testing, HHV serology, and direct fluorescent antibody testing. Skin biopsy may be necessary to rule out underlying malignancy. Treatment of perianal HHV infection includes acyclovir, valacyclovir, or famciclovir.1,5,6 Hypertrophic lesions often are refractory to first-line antiviral therapy and may require surgical resection or treatment with alternative medications such as imiquimod, a topical immunomodulator.3,5,6,11
- Ranu H, Lee J, Chio M, et al. Tumour-like presentations of anogenital herpes simplex in HIV-positive patients. Int J STD AIDS. 2011;22:181-186.
- Tong P, Mutasim DF. Herpes simplex virus infection masquerading as condylomata accuminata in a patient with HIV disease. Br J Dermatol. 1996;134:797-800.
- Mosunjac M, Park J, Wang W, et al. Genital and perianal herpes simplex simulating neoplasia in patients with AIDS. AIDS Patient Care STDS. 2009;23:153-158.
- Gubinelli E, Cocuroccia B, Lazzarotto T, et al. Nodular perianal herpes simplex with prominent plasma cell infiltration. Sexually Transm Dis. 2003;30:157-159.
- Nadal SR, Calore EE, Manzione CR, et al. Hypertrophic herpes simplex simulating anal neoplasia in AIDS patients: report of five cases. Dis Colon Rectum. 2005;48:2289-2293.
- Beasley KL, Cooley GE, Kao GF, et al. Herpes simplex vegetans: atypical genital herpes infection in a patient with common variable immunodeficiency. J Am Acad Dermatol. 1997;37(5, pt 2):860-863.
- Burke EM, Karp DL, Wu TC, et al. Atypical oral presentation of herpes simplex virus infection in a patient after orthotopic liver transplantation. Eur Arch Otorhinolaryngol. 1994;251:301-303.
- Tabaee A, Saltman B, Shutter J, et al. Recurrent oral herpes simplex virus infection presenting as a tongue mass. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2004;97:376-380.
- Leming PD, Martin SE, Zwelling LA. Atypical herpes simplex (HSV) infection in a patient with Hodgkin's disease. Cancer. 1984;54:3043-3047.
- Burgoyne M, Burke W. Atypical herpes simplex infection in patients with acute myelogenous leukemia recovering from chemotherapy. J Am Acad Dermatol. 1989;20:1125-1126.
- Deza G, Martin-Ezquerra G, Curto-Barredo L, et al. Successful treatment of hypertrophic herpes simplex genitalis in HIV-infected patient with topical imiquimod. J Dermatol. 2015;42:1176-1178.
The Diagnosis: Herpes Simplex Virus Infection
Viral culture of the ulcer was positive for herpes simplex virus type 2 (HHV-2). Bacterial culture grew enteric flora. The patient was started on intravenous acyclovir 5 mg/kg every 8 hours for 7 days and then transitioned to oral acyclovir for chronic suppressive therapy. One month later, there was near-complete reepithelialization with 2 remaining 1-cm shallow ulcers. The verrucous lesions had dried up and were flaking off (Figure). At 6-month follow-up, the ulcers and verrucous lesions had completely resolved.
Herpes simplex virus type 2 is the most common cause of genital and perianal ulcers in immunocompromised individuals.1 Patients classically present with painful grouped vesicles followed by painful superficial ulcers that may rapidly progress to extensive confluent ulceration. A hypertrophic variant of genital herpes characterized by anogenital verrucous lesions, similar to condyloma acuminata, also can be seen in immunocompromised individuals.2 This form has almost exclusively been observed in patients with human immunodeficiency virus and may occur in isolation or together with the ulcerative form.1-5 A case of vegetative HHV infection of the genital area in a patient with common variable immunodeficiency has been reported.6 Verrucous lesions of the mouth secondary to HHV have been observed in Hodgkin lymphoma, acute myelogenous leukemia, and individuals on immunosuppressive medications.7-10
Perianal involvement of Crohn disease typically presents with fistulas, ulcers, abscesses, strictures, and skin tags in some cases. Invasive squamous cell carcinoma may arise within a chronic ulcer of the anogenital area or may itself manifest as an ulcer or anal fissure. Perianal ulcerative skin tuberculosis has been reported in the literature as a rare manifestation of extrapulmonary tuberculosis and should be considered in a patient with an appropriate clinical history. Pyoderma gangrenosum classically pre-sents as a large ulcer with irregular rolled borders, though a rare variant of vegetative pyoderma gangrenosum may manifest as a nodular or verrucous plaque.
Studies to diagnose HHV include viral cell culture, HHV polymerase chain reaction testing, HHV serology, and direct fluorescent antibody testing. Skin biopsy may be necessary to rule out underlying malignancy. Treatment of perianal HHV infection includes acyclovir, valacyclovir, or famciclovir.1,5,6 Hypertrophic lesions often are refractory to first-line antiviral therapy and may require surgical resection or treatment with alternative medications such as imiquimod, a topical immunomodulator.3,5,6,11
The Diagnosis: Herpes Simplex Virus Infection
Viral culture of the ulcer was positive for herpes simplex virus type 2 (HHV-2). Bacterial culture grew enteric flora. The patient was started on intravenous acyclovir 5 mg/kg every 8 hours for 7 days and then transitioned to oral acyclovir for chronic suppressive therapy. One month later, there was near-complete reepithelialization with 2 remaining 1-cm shallow ulcers. The verrucous lesions had dried up and were flaking off (Figure). At 6-month follow-up, the ulcers and verrucous lesions had completely resolved.
Herpes simplex virus type 2 is the most common cause of genital and perianal ulcers in immunocompromised individuals.1 Patients classically present with painful grouped vesicles followed by painful superficial ulcers that may rapidly progress to extensive confluent ulceration. A hypertrophic variant of genital herpes characterized by anogenital verrucous lesions, similar to condyloma acuminata, also can be seen in immunocompromised individuals.2 This form has almost exclusively been observed in patients with human immunodeficiency virus and may occur in isolation or together with the ulcerative form.1-5 A case of vegetative HHV infection of the genital area in a patient with common variable immunodeficiency has been reported.6 Verrucous lesions of the mouth secondary to HHV have been observed in Hodgkin lymphoma, acute myelogenous leukemia, and individuals on immunosuppressive medications.7-10
Perianal involvement of Crohn disease typically presents with fistulas, ulcers, abscesses, strictures, and skin tags in some cases. Invasive squamous cell carcinoma may arise within a chronic ulcer of the anogenital area or may itself manifest as an ulcer or anal fissure. Perianal ulcerative skin tuberculosis has been reported in the literature as a rare manifestation of extrapulmonary tuberculosis and should be considered in a patient with an appropriate clinical history. Pyoderma gangrenosum classically pre-sents as a large ulcer with irregular rolled borders, though a rare variant of vegetative pyoderma gangrenosum may manifest as a nodular or verrucous plaque.
Studies to diagnose HHV include viral cell culture, HHV polymerase chain reaction testing, HHV serology, and direct fluorescent antibody testing. Skin biopsy may be necessary to rule out underlying malignancy. Treatment of perianal HHV infection includes acyclovir, valacyclovir, or famciclovir.1,5,6 Hypertrophic lesions often are refractory to first-line antiviral therapy and may require surgical resection or treatment with alternative medications such as imiquimod, a topical immunomodulator.3,5,6,11
- Ranu H, Lee J, Chio M, et al. Tumour-like presentations of anogenital herpes simplex in HIV-positive patients. Int J STD AIDS. 2011;22:181-186.
- Tong P, Mutasim DF. Herpes simplex virus infection masquerading as condylomata accuminata in a patient with HIV disease. Br J Dermatol. 1996;134:797-800.
- Mosunjac M, Park J, Wang W, et al. Genital and perianal herpes simplex simulating neoplasia in patients with AIDS. AIDS Patient Care STDS. 2009;23:153-158.
- Gubinelli E, Cocuroccia B, Lazzarotto T, et al. Nodular perianal herpes simplex with prominent plasma cell infiltration. Sexually Transm Dis. 2003;30:157-159.
- Nadal SR, Calore EE, Manzione CR, et al. Hypertrophic herpes simplex simulating anal neoplasia in AIDS patients: report of five cases. Dis Colon Rectum. 2005;48:2289-2293.
- Beasley KL, Cooley GE, Kao GF, et al. Herpes simplex vegetans: atypical genital herpes infection in a patient with common variable immunodeficiency. J Am Acad Dermatol. 1997;37(5, pt 2):860-863.
- Burke EM, Karp DL, Wu TC, et al. Atypical oral presentation of herpes simplex virus infection in a patient after orthotopic liver transplantation. Eur Arch Otorhinolaryngol. 1994;251:301-303.
- Tabaee A, Saltman B, Shutter J, et al. Recurrent oral herpes simplex virus infection presenting as a tongue mass. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2004;97:376-380.
- Leming PD, Martin SE, Zwelling LA. Atypical herpes simplex (HSV) infection in a patient with Hodgkin's disease. Cancer. 1984;54:3043-3047.
- Burgoyne M, Burke W. Atypical herpes simplex infection in patients with acute myelogenous leukemia recovering from chemotherapy. J Am Acad Dermatol. 1989;20:1125-1126.
- Deza G, Martin-Ezquerra G, Curto-Barredo L, et al. Successful treatment of hypertrophic herpes simplex genitalis in HIV-infected patient with topical imiquimod. J Dermatol. 2015;42:1176-1178.
- Ranu H, Lee J, Chio M, et al. Tumour-like presentations of anogenital herpes simplex in HIV-positive patients. Int J STD AIDS. 2011;22:181-186.
- Tong P, Mutasim DF. Herpes simplex virus infection masquerading as condylomata accuminata in a patient with HIV disease. Br J Dermatol. 1996;134:797-800.
- Mosunjac M, Park J, Wang W, et al. Genital and perianal herpes simplex simulating neoplasia in patients with AIDS. AIDS Patient Care STDS. 2009;23:153-158.
- Gubinelli E, Cocuroccia B, Lazzarotto T, et al. Nodular perianal herpes simplex with prominent plasma cell infiltration. Sexually Transm Dis. 2003;30:157-159.
- Nadal SR, Calore EE, Manzione CR, et al. Hypertrophic herpes simplex simulating anal neoplasia in AIDS patients: report of five cases. Dis Colon Rectum. 2005;48:2289-2293.
- Beasley KL, Cooley GE, Kao GF, et al. Herpes simplex vegetans: atypical genital herpes infection in a patient with common variable immunodeficiency. J Am Acad Dermatol. 1997;37(5, pt 2):860-863.
- Burke EM, Karp DL, Wu TC, et al. Atypical oral presentation of herpes simplex virus infection in a patient after orthotopic liver transplantation. Eur Arch Otorhinolaryngol. 1994;251:301-303.
- Tabaee A, Saltman B, Shutter J, et al. Recurrent oral herpes simplex virus infection presenting as a tongue mass. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2004;97:376-380.
- Leming PD, Martin SE, Zwelling LA. Atypical herpes simplex (HSV) infection in a patient with Hodgkin's disease. Cancer. 1984;54:3043-3047.
- Burgoyne M, Burke W. Atypical herpes simplex infection in patients with acute myelogenous leukemia recovering from chemotherapy. J Am Acad Dermatol. 1989;20:1125-1126.
- Deza G, Martin-Ezquerra G, Curto-Barredo L, et al. Successful treatment of hypertrophic herpes simplex genitalis in HIV-infected patient with topical imiquimod. J Dermatol. 2015;42:1176-1178.
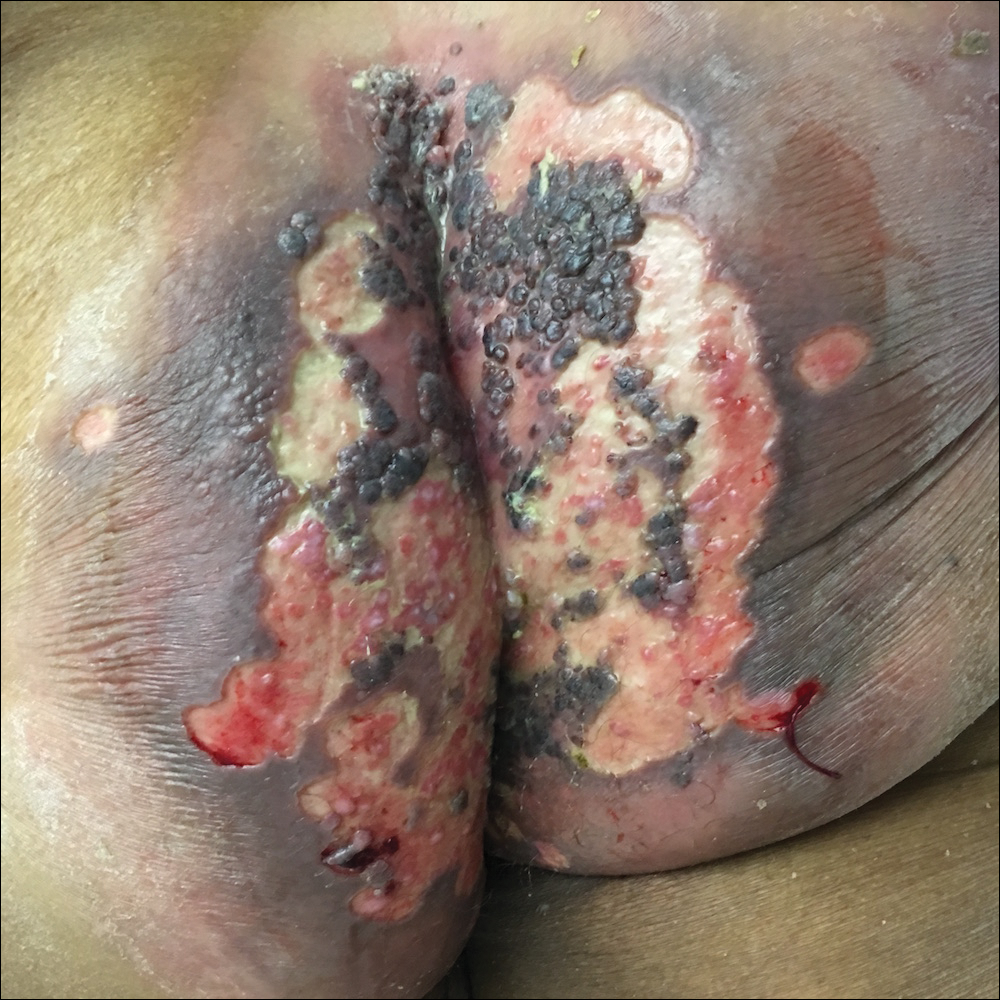
A 75-year-old woman with chronic lymphocytic leukemia undergoing ibrutinib targeted therapy presented to the emergency department with fever and perianal pain of 4 months' duration. The patient denied history of genital or perianal ulcers, warts, masses, bedsores, prolonged immobilization, anal surgeries, or recent travel. She had not been previously treated for the perianal pain. On physical examination there was an 18×15-cm shallow ulceration with rolled borders involving the intergluteal cleft and perianal area. There were numerous hyperpigmented verrucous papules clustered in the center of the ulceration. No vesicles or bullae were present. Laboratory results were pertinent for a white blood cell count of 3600/µL (reference range, 4500-11,000/µL) and absolute neutrophil count of 1300/µL (reference range, 1900-8000/µL). Human immunodeficiency virus testing was negative.
Signs point to growing abuse of gabapentinoids in the U.S.
LAS VEGAS – Up to 22% of opioid abusers also abuse gabapentin, taking high doses of the antiepileptic for its psychoactive effects, or to potentiate the effect of opioids.
The drug – the 10th most commonly prescribed in the United States – is increasingly implicated in overdose deaths. In response, several states have recently reclassified the antiepileptic as a Schedule V controlled substance. Other states have declined to go that far, but have added gabapentin (Neurontin and others) to the drugs that must be reported to state prescription monitoring programs.
“Gabapentin and pregabalin [Lyrica] are versatile and important drugs that are widely prescribed and used off label for a number of conditions from seizure disorder to fibromyalgia,” said Joseph Pergolizzi Jr., MD, who discussed the issue during the annual PAINWeek. “Abuse patterns differ somewhat from abuse patterns with prescription opioids. People who misuse gabapentinoids tend to already use other drugs inappropriately. It’s rare to find a person who only takes them recreationally, but it is increasingly common to find polydrug abusers who take gabapentinoids.”
Gabapentin abuse appears to be more common in Europe than in the United States, where it’s just beginning to emerge, Dr. Pergolizzi said. And the picture is more nuanced than it might first appear: Many of those who are misusing the drug are using it as a “do-it-yourself” opioid withdrawal aid, he said in an interview.
At the meeting, he presented a literature review comprising 46 papers on pregabalin abuse and 263 on gabapentin abuse. Several important themes arose from these papers, said Dr. Pergolizzi, cofounder and chief operating officer of NEMA Research Inc., Naples, Fla.:
- Gabapentin and pregabalin are being prescribed off label for numerous conditions, including bipolar disorder, neuropathic pain, diabetic neuropathy, complex regional pain syndrome, attention deficit disorder, restless leg syndrome, trigeminal neuralgia, periodic limb movement disorder of sleep, migraine, drug and alcohol withdrawal seizures, chronic low back pain, and even menopausal symptoms.
- About a one-third of users experience withdrawal symptoms with sudden discontinuation. Symptoms include disorientation, anxiety, insomnia, heart palpitations, diaphoresis, and abdominal cramps.
- Risk factors for abuse are emerging. These include opioid use disorders, mental illnesses, and a history of taking supratherapeutic doses of the drugs. Age and sex don’t seem to be a consistent risk factor for abuse.
- Abusers can obtain nonprescription gabapentinoids more easily each year, including street sales and online orders, Dr. Pergolizzi said. “A Google search conducted in July for ‘buy gabapentin without a prescription’ yielded 4.48 million results and ‘buy pregabalin without a prescription,’ 622,000. A similar search conducted in September 2017 yielded 1.19 million and 352,000 results, respectively.”
- Few urine drug assays screen for gabapentinoids, which makes them easy to conceal in random drug testing.
Reports of gabapentin-involved drug overdoses and even deaths have recently emerged in the United States, particularly in the opioid abuse-plagued Appalachian states. An article in May in Drug and Alcohol Dependence examined the prevalence of gabapentin in postmortem toxicology in drug overdose deaths in four Appalachian states in 2015 (Drug Alcohol Depend. 2018;186:80-5). Rates were 4% in eastern Tennessee, 15% in West Virginia, 20% in North Carolina, and 41% in Kentucky.
Three states have now added gabapentin to their list of Schedule V controlled substances: Kentucky in 2017, West Virginia this May, and Tennessee in July.
Ohio, Minnesota, Virginia, and Massachusetts have taken a different tack to controlling dispensing. In those states, all pharmacies, prescribers, and wholesalers must report all dispensing and sales of gabapentin to their prescription monitoring databases.
Dr. Pergolizzi disclosed financial relationships with numerous pharmaceutical companies.
LAS VEGAS – Up to 22% of opioid abusers also abuse gabapentin, taking high doses of the antiepileptic for its psychoactive effects, or to potentiate the effect of opioids.
The drug – the 10th most commonly prescribed in the United States – is increasingly implicated in overdose deaths. In response, several states have recently reclassified the antiepileptic as a Schedule V controlled substance. Other states have declined to go that far, but have added gabapentin (Neurontin and others) to the drugs that must be reported to state prescription monitoring programs.
“Gabapentin and pregabalin [Lyrica] are versatile and important drugs that are widely prescribed and used off label for a number of conditions from seizure disorder to fibromyalgia,” said Joseph Pergolizzi Jr., MD, who discussed the issue during the annual PAINWeek. “Abuse patterns differ somewhat from abuse patterns with prescription opioids. People who misuse gabapentinoids tend to already use other drugs inappropriately. It’s rare to find a person who only takes them recreationally, but it is increasingly common to find polydrug abusers who take gabapentinoids.”
Gabapentin abuse appears to be more common in Europe than in the United States, where it’s just beginning to emerge, Dr. Pergolizzi said. And the picture is more nuanced than it might first appear: Many of those who are misusing the drug are using it as a “do-it-yourself” opioid withdrawal aid, he said in an interview.
At the meeting, he presented a literature review comprising 46 papers on pregabalin abuse and 263 on gabapentin abuse. Several important themes arose from these papers, said Dr. Pergolizzi, cofounder and chief operating officer of NEMA Research Inc., Naples, Fla.:
- Gabapentin and pregabalin are being prescribed off label for numerous conditions, including bipolar disorder, neuropathic pain, diabetic neuropathy, complex regional pain syndrome, attention deficit disorder, restless leg syndrome, trigeminal neuralgia, periodic limb movement disorder of sleep, migraine, drug and alcohol withdrawal seizures, chronic low back pain, and even menopausal symptoms.
- About a one-third of users experience withdrawal symptoms with sudden discontinuation. Symptoms include disorientation, anxiety, insomnia, heart palpitations, diaphoresis, and abdominal cramps.
- Risk factors for abuse are emerging. These include opioid use disorders, mental illnesses, and a history of taking supratherapeutic doses of the drugs. Age and sex don’t seem to be a consistent risk factor for abuse.
- Abusers can obtain nonprescription gabapentinoids more easily each year, including street sales and online orders, Dr. Pergolizzi said. “A Google search conducted in July for ‘buy gabapentin without a prescription’ yielded 4.48 million results and ‘buy pregabalin without a prescription,’ 622,000. A similar search conducted in September 2017 yielded 1.19 million and 352,000 results, respectively.”
- Few urine drug assays screen for gabapentinoids, which makes them easy to conceal in random drug testing.
Reports of gabapentin-involved drug overdoses and even deaths have recently emerged in the United States, particularly in the opioid abuse-plagued Appalachian states. An article in May in Drug and Alcohol Dependence examined the prevalence of gabapentin in postmortem toxicology in drug overdose deaths in four Appalachian states in 2015 (Drug Alcohol Depend. 2018;186:80-5). Rates were 4% in eastern Tennessee, 15% in West Virginia, 20% in North Carolina, and 41% in Kentucky.
Three states have now added gabapentin to their list of Schedule V controlled substances: Kentucky in 2017, West Virginia this May, and Tennessee in July.
Ohio, Minnesota, Virginia, and Massachusetts have taken a different tack to controlling dispensing. In those states, all pharmacies, prescribers, and wholesalers must report all dispensing and sales of gabapentin to their prescription monitoring databases.
Dr. Pergolizzi disclosed financial relationships with numerous pharmaceutical companies.
LAS VEGAS – Up to 22% of opioid abusers also abuse gabapentin, taking high doses of the antiepileptic for its psychoactive effects, or to potentiate the effect of opioids.
The drug – the 10th most commonly prescribed in the United States – is increasingly implicated in overdose deaths. In response, several states have recently reclassified the antiepileptic as a Schedule V controlled substance. Other states have declined to go that far, but have added gabapentin (Neurontin and others) to the drugs that must be reported to state prescription monitoring programs.
“Gabapentin and pregabalin [Lyrica] are versatile and important drugs that are widely prescribed and used off label for a number of conditions from seizure disorder to fibromyalgia,” said Joseph Pergolizzi Jr., MD, who discussed the issue during the annual PAINWeek. “Abuse patterns differ somewhat from abuse patterns with prescription opioids. People who misuse gabapentinoids tend to already use other drugs inappropriately. It’s rare to find a person who only takes them recreationally, but it is increasingly common to find polydrug abusers who take gabapentinoids.”
Gabapentin abuse appears to be more common in Europe than in the United States, where it’s just beginning to emerge, Dr. Pergolizzi said. And the picture is more nuanced than it might first appear: Many of those who are misusing the drug are using it as a “do-it-yourself” opioid withdrawal aid, he said in an interview.
At the meeting, he presented a literature review comprising 46 papers on pregabalin abuse and 263 on gabapentin abuse. Several important themes arose from these papers, said Dr. Pergolizzi, cofounder and chief operating officer of NEMA Research Inc., Naples, Fla.:
- Gabapentin and pregabalin are being prescribed off label for numerous conditions, including bipolar disorder, neuropathic pain, diabetic neuropathy, complex regional pain syndrome, attention deficit disorder, restless leg syndrome, trigeminal neuralgia, periodic limb movement disorder of sleep, migraine, drug and alcohol withdrawal seizures, chronic low back pain, and even menopausal symptoms.
- About a one-third of users experience withdrawal symptoms with sudden discontinuation. Symptoms include disorientation, anxiety, insomnia, heart palpitations, diaphoresis, and abdominal cramps.
- Risk factors for abuse are emerging. These include opioid use disorders, mental illnesses, and a history of taking supratherapeutic doses of the drugs. Age and sex don’t seem to be a consistent risk factor for abuse.
- Abusers can obtain nonprescription gabapentinoids more easily each year, including street sales and online orders, Dr. Pergolizzi said. “A Google search conducted in July for ‘buy gabapentin without a prescription’ yielded 4.48 million results and ‘buy pregabalin without a prescription,’ 622,000. A similar search conducted in September 2017 yielded 1.19 million and 352,000 results, respectively.”
- Few urine drug assays screen for gabapentinoids, which makes them easy to conceal in random drug testing.
Reports of gabapentin-involved drug overdoses and even deaths have recently emerged in the United States, particularly in the opioid abuse-plagued Appalachian states. An article in May in Drug and Alcohol Dependence examined the prevalence of gabapentin in postmortem toxicology in drug overdose deaths in four Appalachian states in 2015 (Drug Alcohol Depend. 2018;186:80-5). Rates were 4% in eastern Tennessee, 15% in West Virginia, 20% in North Carolina, and 41% in Kentucky.
Three states have now added gabapentin to their list of Schedule V controlled substances: Kentucky in 2017, West Virginia this May, and Tennessee in July.
Ohio, Minnesota, Virginia, and Massachusetts have taken a different tack to controlling dispensing. In those states, all pharmacies, prescribers, and wholesalers must report all dispensing and sales of gabapentin to their prescription monitoring databases.
Dr. Pergolizzi disclosed financial relationships with numerous pharmaceutical companies.
REPORTING FROM PAINWEEK 2018
Key clinical point:
Major finding: Up to 22% of opioid users are also using gabapentin.
Study details: A literature review of 46 papers on pregabalin abuse and 263 on gabapentin abuse.
Disclosures: Dr. Pergolizzi disclosed financial relationships with numerous pharmaceutical companies.
Source: Pergolizzi J et al. PAINWeek 2018, Poster 55.
Decision aid aims to make lupus nephritis treatment decisions more understandable
A computer-based decision aid aims to facilitate the complex discussions and decisions that face patients with lupus nephritis.
Under development at the University of Alabama at Birmingham, the Shared Decision-Making in Lupus Electronic tool (SMILE) describes in easy-to-understand modules the pathology of the disease, and what can happen if it progresses untreated. The tool also identifies treatment options and discusses the potential benefits of each one, as well as the risks. It can be folded into office visits, accessed during waiting times, or viewed at the patient’s leisure. Its creators hope that patients will experience more fruitful discussions with their physicians by learning more about these complicated concepts as they co-navigate the difficult decisions both must make.

“Our intent was to figure out how best to offer the education about lupus and its treatments that women need when coming into a busy clinic,” said Jasvinder Singh, MD, who is leading the developmental team. “Everyone’s time is limited, and patients want information delivered quickly, but in a way they can understand.”
SMILE is the first tool of its kind for patients with lupus nephritis, said Alexa Meara, MD, one of the rheumatologists investigating it.
“There is just nothing out there like this for either lupus, or lupus nephritis,” said Dr. Meara, a rheumatologist at Ohio State University, Columbus. “Lupus nephritis is a heterogeneous relapsing-remitting disease with complex medical regimens. It can have cognitive impacts and affect fertility, but these are things that may happen over decades. Patients may not be ready to talk about them early on, but they must understand them. We would like to enable the conversation between physician and patient, so they’re on the same page with everyone’s values and concerns addressed on both sides.”
But getting everyone on the same page at the same time is challenging, said Dr. Singh, a rheumatologist and professor of medicine and epidemiology at the University of Alabama at Birmingham. “This is a bit of a dilemma and a challenge. What’s great about the lupus decision aid is that it is culturally and linguistically appropriate for patients of all backgrounds, races, ages, literacy levels, and socioeconomic statuses. This levels the playing field for patients who are trying to understand their diagnosis and options to the best of their abilities.”

Three years in the making, SMILE comprises several modules addressing different facets of lupus nephritis. In simple words and concepts, the tool informs patients about what lupus is and what it can do to the kidneys, the information gleaned from blood and urine tests and what it means, and the potential consequences of not treating lupus kidney disease. The heart of the program is its breakdown of treatment options.
“It helps patients understand why they may be getting these cancer medications, and what their side effects are,” Dr. Singh said. “The tool also walks them through a comparison of the medications, with one slide on each medication and its risks and benefits. We really focus on that – about 60%-70% of the tool is comparing the drugs.”
The decision aid also contains optional modules that patients can bypass or explore. “There are separate sections on the risks and benefits of corticosteroids, on pregnancy and lactation, and on preserving fertility.”
A complete click-through of the basic information takes about 20 minutes, Dr. Singh said. An ideal time for a patient to do so would be before the initial visit to discuss treatment options, which can be an overwhelming experience. The tool could be presented in several ways. In the office, preloaded iPads could be one vehicle. These could be standalone, or linked to the patient’s electronic health record, so that any entered information could be directly transferred. At home, patients could navigate to a web link for an online version.
The first visit is a crucial moment, he said, where patients can evolve into management partners, or leave even more confused than when they arrived.
“We aim to motivate and provide tools to the patient, so that the conversation isn’t just, ‘You have the disease and we can put you on this or that drug,’ ” Dr. Singh said. “You can lose the patient right away in this scenario, while you’re talking about so many things the patient isn’t even hearing you, because she’s thinking about other issues that are important in her life, completely overwhelmed with the shock of the diagnosis and with fear.”
The Patient-Centered Outcomes Research Institute (PCORI) has funded much of the work getting SMILE up and running, including a soon-to-be-published trial that favorably compared it against the educational standard: a paper pamphlet on lupus nephritis prepared by the American College of Rheumatology.
In this study, 301 high‐risk women with lupus kidney disease, including racial/ethnic minorities with low socioeconomic status, either received the pamphlet or the SMILE tool. The group was demographically diverse: 47% black, 26% Hispanic, 15% white, and 7% Asian. Other groups made up the remainder.
More patients rated the information in the decision aid to be excellent for understanding the impact of lupus (49% vs. 33%), risk factors (43% vs. 27%), medication options (50% vs. 33%), and evidence about medications (47% vs. 24%). About half of the tool users rated the ease of use of materials as excellent (51% vs. 38%). Women who used the tool also reported much less decisional conflict for immunosuppressive drugs and were much more likely to choose the treatment option most consistent with their values, having viewed information that mattered the most for the treatment decision.
PCORI continues to support the project, too. Dr. Singh just received a $2.2 million grant to investigate the tool in 16 clinics. The 2-year observational study has three aims:
• Identify physician attitudes and patient barriers toward the tool, to guide decisions about how to best implement it in its final form.
• Track changes in subjective and objective measures of implementation effectiveness.
• Identify opportunities for disseminating the tool and develop a step‐by‐step implementation guide for incorporating the decision aid tool into regular lupus clinic visits and care pathways.
The study will also look at some patient‐centered outcomes, including decision conflict, emergency department and inpatient visits, patient‐physician communication, and implementation success. Some practice-level outcomes will be assessed, including potential barriers to implementation. “The team will focus on context and strategy as facilitators and barriers to effective implementation of the lupus decision aid in busy, clinical practices of various types – for example, private versus academic, urban versus suburban, large versus small group practice,” according to the PCORI study description.
“This study can’t solve the entire problem about patients choosing treatment or no treatment, but it can provide a start to the conversation,” Dr. Singh said. “And my hope is that once it’s available, people will modify it to fit their needs, make adaptations and modifications to keep it relevant and valuable.”
A computer-based decision aid aims to facilitate the complex discussions and decisions that face patients with lupus nephritis.
Under development at the University of Alabama at Birmingham, the Shared Decision-Making in Lupus Electronic tool (SMILE) describes in easy-to-understand modules the pathology of the disease, and what can happen if it progresses untreated. The tool also identifies treatment options and discusses the potential benefits of each one, as well as the risks. It can be folded into office visits, accessed during waiting times, or viewed at the patient’s leisure. Its creators hope that patients will experience more fruitful discussions with their physicians by learning more about these complicated concepts as they co-navigate the difficult decisions both must make.
“Our intent was to figure out how best to offer the education about lupus and its treatments that women need when coming into a busy clinic,” said Jasvinder Singh, MD, who is leading the developmental team. “Everyone’s time is limited, and patients want information delivered quickly, but in a way they can understand.”
SMILE is the first tool of its kind for patients with lupus nephritis, said Alexa Meara, MD, one of the rheumatologists investigating it.
“There is just nothing out there like this for either lupus, or lupus nephritis,” said Dr. Meara, a rheumatologist at Ohio State University, Columbus. “Lupus nephritis is a heterogeneous relapsing-remitting disease with complex medical regimens. It can have cognitive impacts and affect fertility, but these are things that may happen over decades. Patients may not be ready to talk about them early on, but they must understand them. We would like to enable the conversation between physician and patient, so they’re on the same page with everyone’s values and concerns addressed on both sides.”
But getting everyone on the same page at the same time is challenging, said Dr. Singh, a rheumatologist and professor of medicine and epidemiology at the University of Alabama at Birmingham. “This is a bit of a dilemma and a challenge. What’s great about the lupus decision aid is that it is culturally and linguistically appropriate for patients of all backgrounds, races, ages, literacy levels, and socioeconomic statuses. This levels the playing field for patients who are trying to understand their diagnosis and options to the best of their abilities.”
Three years in the making, SMILE comprises several modules addressing different facets of lupus nephritis. In simple words and concepts, the tool informs patients about what lupus is and what it can do to the kidneys, the information gleaned from blood and urine tests and what it means, and the potential consequences of not treating lupus kidney disease. The heart of the program is its breakdown of treatment options.
“It helps patients understand why they may be getting these cancer medications, and what their side effects are,” Dr. Singh said. “The tool also walks them through a comparison of the medications, with one slide on each medication and its risks and benefits. We really focus on that – about 60%-70% of the tool is comparing the drugs.”
The decision aid also contains optional modules that patients can bypass or explore. “There are separate sections on the risks and benefits of corticosteroids, on pregnancy and lactation, and on preserving fertility.”
A complete click-through of the basic information takes about 20 minutes, Dr. Singh said. An ideal time for a patient to do so would be before the initial visit to discuss treatment options, which can be an overwhelming experience. The tool could be presented in several ways. In the office, preloaded iPads could be one vehicle. These could be standalone, or linked to the patient’s electronic health record, so that any entered information could be directly transferred. At home, patients could navigate to a web link for an online version.
The first visit is a crucial moment, he said, where patients can evolve into management partners, or leave even more confused than when they arrived.
“We aim to motivate and provide tools to the patient, so that the conversation isn’t just, ‘You have the disease and we can put you on this or that drug,’ ” Dr. Singh said. “You can lose the patient right away in this scenario, while you’re talking about so many things the patient isn’t even hearing you, because she’s thinking about other issues that are important in her life, completely overwhelmed with the shock of the diagnosis and with fear.”
The Patient-Centered Outcomes Research Institute (PCORI) has funded much of the work getting SMILE up and running, including a soon-to-be-published trial that favorably compared it against the educational standard: a paper pamphlet on lupus nephritis prepared by the American College of Rheumatology.
In this study, 301 high‐risk women with lupus kidney disease, including racial/ethnic minorities with low socioeconomic status, either received the pamphlet or the SMILE tool. The group was demographically diverse: 47% black, 26% Hispanic, 15% white, and 7% Asian. Other groups made up the remainder.
More patients rated the information in the decision aid to be excellent for understanding the impact of lupus (49% vs. 33%), risk factors (43% vs. 27%), medication options (50% vs. 33%), and evidence about medications (47% vs. 24%). About half of the tool users rated the ease of use of materials as excellent (51% vs. 38%). Women who used the tool also reported much less decisional conflict for immunosuppressive drugs and were much more likely to choose the treatment option most consistent with their values, having viewed information that mattered the most for the treatment decision.
PCORI continues to support the project, too. Dr. Singh just received a $2.2 million grant to investigate the tool in 16 clinics. The 2-year observational study has three aims:
• Identify physician attitudes and patient barriers toward the tool, to guide decisions about how to best implement it in its final form.
• Track changes in subjective and objective measures of implementation effectiveness.
• Identify opportunities for disseminating the tool and develop a step‐by‐step implementation guide for incorporating the decision aid tool into regular lupus clinic visits and care pathways.
The study will also look at some patient‐centered outcomes, including decision conflict, emergency department and inpatient visits, patient‐physician communication, and implementation success. Some practice-level outcomes will be assessed, including potential barriers to implementation. “The team will focus on context and strategy as facilitators and barriers to effective implementation of the lupus decision aid in busy, clinical practices of various types – for example, private versus academic, urban versus suburban, large versus small group practice,” according to the PCORI study description.
“This study can’t solve the entire problem about patients choosing treatment or no treatment, but it can provide a start to the conversation,” Dr. Singh said. “And my hope is that once it’s available, people will modify it to fit their needs, make adaptations and modifications to keep it relevant and valuable.”
A computer-based decision aid aims to facilitate the complex discussions and decisions that face patients with lupus nephritis.
Under development at the University of Alabama at Birmingham, the Shared Decision-Making in Lupus Electronic tool (SMILE) describes in easy-to-understand modules the pathology of the disease, and what can happen if it progresses untreated. The tool also identifies treatment options and discusses the potential benefits of each one, as well as the risks. It can be folded into office visits, accessed during waiting times, or viewed at the patient’s leisure. Its creators hope that patients will experience more fruitful discussions with their physicians by learning more about these complicated concepts as they co-navigate the difficult decisions both must make.
“Our intent was to figure out how best to offer the education about lupus and its treatments that women need when coming into a busy clinic,” said Jasvinder Singh, MD, who is leading the developmental team. “Everyone’s time is limited, and patients want information delivered quickly, but in a way they can understand.”
SMILE is the first tool of its kind for patients with lupus nephritis, said Alexa Meara, MD, one of the rheumatologists investigating it.
“There is just nothing out there like this for either lupus, or lupus nephritis,” said Dr. Meara, a rheumatologist at Ohio State University, Columbus. “Lupus nephritis is a heterogeneous relapsing-remitting disease with complex medical regimens. It can have cognitive impacts and affect fertility, but these are things that may happen over decades. Patients may not be ready to talk about them early on, but they must understand them. We would like to enable the conversation between physician and patient, so they’re on the same page with everyone’s values and concerns addressed on both sides.”
But getting everyone on the same page at the same time is challenging, said Dr. Singh, a rheumatologist and professor of medicine and epidemiology at the University of Alabama at Birmingham. “This is a bit of a dilemma and a challenge. What’s great about the lupus decision aid is that it is culturally and linguistically appropriate for patients of all backgrounds, races, ages, literacy levels, and socioeconomic statuses. This levels the playing field for patients who are trying to understand their diagnosis and options to the best of their abilities.”
Three years in the making, SMILE comprises several modules addressing different facets of lupus nephritis. In simple words and concepts, the tool informs patients about what lupus is and what it can do to the kidneys, the information gleaned from blood and urine tests and what it means, and the potential consequences of not treating lupus kidney disease. The heart of the program is its breakdown of treatment options.
“It helps patients understand why they may be getting these cancer medications, and what their side effects are,” Dr. Singh said. “The tool also walks them through a comparison of the medications, with one slide on each medication and its risks and benefits. We really focus on that – about 60%-70% of the tool is comparing the drugs.”
The decision aid also contains optional modules that patients can bypass or explore. “There are separate sections on the risks and benefits of corticosteroids, on pregnancy and lactation, and on preserving fertility.”
A complete click-through of the basic information takes about 20 minutes, Dr. Singh said. An ideal time for a patient to do so would be before the initial visit to discuss treatment options, which can be an overwhelming experience. The tool could be presented in several ways. In the office, preloaded iPads could be one vehicle. These could be standalone, or linked to the patient’s electronic health record, so that any entered information could be directly transferred. At home, patients could navigate to a web link for an online version.
The first visit is a crucial moment, he said, where patients can evolve into management partners, or leave even more confused than when they arrived.
“We aim to motivate and provide tools to the patient, so that the conversation isn’t just, ‘You have the disease and we can put you on this or that drug,’ ” Dr. Singh said. “You can lose the patient right away in this scenario, while you’re talking about so many things the patient isn’t even hearing you, because she’s thinking about other issues that are important in her life, completely overwhelmed with the shock of the diagnosis and with fear.”
The Patient-Centered Outcomes Research Institute (PCORI) has funded much of the work getting SMILE up and running, including a soon-to-be-published trial that favorably compared it against the educational standard: a paper pamphlet on lupus nephritis prepared by the American College of Rheumatology.
In this study, 301 high‐risk women with lupus kidney disease, including racial/ethnic minorities with low socioeconomic status, either received the pamphlet or the SMILE tool. The group was demographically diverse: 47% black, 26% Hispanic, 15% white, and 7% Asian. Other groups made up the remainder.
More patients rated the information in the decision aid to be excellent for understanding the impact of lupus (49% vs. 33%), risk factors (43% vs. 27%), medication options (50% vs. 33%), and evidence about medications (47% vs. 24%). About half of the tool users rated the ease of use of materials as excellent (51% vs. 38%). Women who used the tool also reported much less decisional conflict for immunosuppressive drugs and were much more likely to choose the treatment option most consistent with their values, having viewed information that mattered the most for the treatment decision.
PCORI continues to support the project, too. Dr. Singh just received a $2.2 million grant to investigate the tool in 16 clinics. The 2-year observational study has three aims:
• Identify physician attitudes and patient barriers toward the tool, to guide decisions about how to best implement it in its final form.
• Track changes in subjective and objective measures of implementation effectiveness.
• Identify opportunities for disseminating the tool and develop a step‐by‐step implementation guide for incorporating the decision aid tool into regular lupus clinic visits and care pathways.
The study will also look at some patient‐centered outcomes, including decision conflict, emergency department and inpatient visits, patient‐physician communication, and implementation success. Some practice-level outcomes will be assessed, including potential barriers to implementation. “The team will focus on context and strategy as facilitators and barriers to effective implementation of the lupus decision aid in busy, clinical practices of various types – for example, private versus academic, urban versus suburban, large versus small group practice,” according to the PCORI study description.
“This study can’t solve the entire problem about patients choosing treatment or no treatment, but it can provide a start to the conversation,” Dr. Singh said. “And my hope is that once it’s available, people will modify it to fit their needs, make adaptations and modifications to keep it relevant and valuable.”
Proposal to change Part B drug WAC-based reimbursement draws criticism
Some physician groups are pushing back on a proposal to lower the add-on percentage for reimbursement of new Part B drugs paid using the wholesale acquisition cost (WAC) when an average sales price (ASP) has not yet been established.

Under current regulation, physicians are reimbursed at WAC plus 6% for newly approved drugs. The Centers for Medicare & Medicaid Services is looking to reduce the add-on to 3% in the proposed update to the physician fee schedule for 2019.
In Sept. 6 comments to the agency, the American College of Rheumatology took on a neutral stance to the proposal as a whole, stating only that it appreciates “that the proposed rule does clarify that the change in reimbursement would not apply to new biosimilars, whose reimbursement would remain at the drug’s WAC plus 6% of the reference drug’s ASP.”
The American Society of Clinical Oncology took a more hard-line stance.
“CMS should not finalize the proposed reduction in the add-on rate for Part B drugs subject to payment through the wholesale acquisition cost methodology and should instead focus on pursuing comprehensive solutions that drive value-based cancer care,” ASCO said in Sept. 10 comments to the agency. It acknowledged CMS’ pursuit to lower drug spending, but suggested this will not have any meaningful impact “since most drugs are paid through a WAC-based methodology on a temporary basis only.”
The American Medical Association argued in Sept. 10 comments to the agency that, when accounting for the budget sequester that is in effect, physicians would only be getting reimbursed with a 1.4% add-on and that enactment of this proposal “would trigger reimbursement cuts for new drugs that will preclude their use in most physician offices and hinder Medicare patients’ access to new and innovated therapies that are more effective and/or less debilitating than existing drugs. AMA strongly believes that this proposal should not be finalized.”
Some physician groups are pushing back on a proposal to lower the add-on percentage for reimbursement of new Part B drugs paid using the wholesale acquisition cost (WAC) when an average sales price (ASP) has not yet been established.
Under current regulation, physicians are reimbursed at WAC plus 6% for newly approved drugs. The Centers for Medicare & Medicaid Services is looking to reduce the add-on to 3% in the proposed update to the physician fee schedule for 2019.
In Sept. 6 comments to the agency, the American College of Rheumatology took on a neutral stance to the proposal as a whole, stating only that it appreciates “that the proposed rule does clarify that the change in reimbursement would not apply to new biosimilars, whose reimbursement would remain at the drug’s WAC plus 6% of the reference drug’s ASP.”
The American Society of Clinical Oncology took a more hard-line stance.
“CMS should not finalize the proposed reduction in the add-on rate for Part B drugs subject to payment through the wholesale acquisition cost methodology and should instead focus on pursuing comprehensive solutions that drive value-based cancer care,” ASCO said in Sept. 10 comments to the agency. It acknowledged CMS’ pursuit to lower drug spending, but suggested this will not have any meaningful impact “since most drugs are paid through a WAC-based methodology on a temporary basis only.”
The American Medical Association argued in Sept. 10 comments to the agency that, when accounting for the budget sequester that is in effect, physicians would only be getting reimbursed with a 1.4% add-on and that enactment of this proposal “would trigger reimbursement cuts for new drugs that will preclude their use in most physician offices and hinder Medicare patients’ access to new and innovated therapies that are more effective and/or less debilitating than existing drugs. AMA strongly believes that this proposal should not be finalized.”
Some physician groups are pushing back on a proposal to lower the add-on percentage for reimbursement of new Part B drugs paid using the wholesale acquisition cost (WAC) when an average sales price (ASP) has not yet been established.
Under current regulation, physicians are reimbursed at WAC plus 6% for newly approved drugs. The Centers for Medicare & Medicaid Services is looking to reduce the add-on to 3% in the proposed update to the physician fee schedule for 2019.
In Sept. 6 comments to the agency, the American College of Rheumatology took on a neutral stance to the proposal as a whole, stating only that it appreciates “that the proposed rule does clarify that the change in reimbursement would not apply to new biosimilars, whose reimbursement would remain at the drug’s WAC plus 6% of the reference drug’s ASP.”
The American Society of Clinical Oncology took a more hard-line stance.
“CMS should not finalize the proposed reduction in the add-on rate for Part B drugs subject to payment through the wholesale acquisition cost methodology and should instead focus on pursuing comprehensive solutions that drive value-based cancer care,” ASCO said in Sept. 10 comments to the agency. It acknowledged CMS’ pursuit to lower drug spending, but suggested this will not have any meaningful impact “since most drugs are paid through a WAC-based methodology on a temporary basis only.”
The American Medical Association argued in Sept. 10 comments to the agency that, when accounting for the budget sequester that is in effect, physicians would only be getting reimbursed with a 1.4% add-on and that enactment of this proposal “would trigger reimbursement cuts for new drugs that will preclude their use in most physician offices and hinder Medicare patients’ access to new and innovated therapies that are more effective and/or less debilitating than existing drugs. AMA strongly believes that this proposal should not be finalized.”
Peripheral opioid blocker eases opioid-induced constipation without inducing withdrawal
LAS VEGAS – .
In three placebo-controlled randomized studies, the drug increased the number of spontaneous bowel movements and improved stool consistency without inducing opioid withdrawal symptoms.
Naldemedine (Symproic) received FDA approval last year based on COMPOSE 1 and COMPOSE 2, which were published in Lancet Gastroenterology and Hepatology last year (2017 Aug 2[8]:555-64). James E. Wild, MD, of Upstate Clinical Research Associates, in Williamsville, N.Y., reported these in a poster presented at the annual PAINWeek.
The year-long COMPOSE 3 trial was not presented at the meeting, but appeared in the journal Pain (2018 May;5:987-94).
Opioids not only affect the central nervous system but also bind to mu-opioid receptors in the gut, decreasing intestinal motility. Naldemedine blocks these receptors from opioid binding but cannot cross the blood-brain barrier. Its peripheral action blocks gut opioid binding, side-stepping the motility problem without inducing any opioid withdrawal symptoms.
COMPOSE 1 (C1) and COMPOSE 2 (C2) comprised a total of 1,095 subjects with chronic, noncancer pain. They were randomized to either naldemedine 0.2 mg daily or placebo for 12 weeks.
Patients were a median of 53 years with a mean opioid use of 61 months. About 60% had a mean daily morphine equivalent of 30-100 mg, and 40% a mean dose of more than 100 mg. At baseline, they had a mean of one spontaneous bowel movement (BM) per week, with a mean of 0.4 deemed “complete.” All the BMs were accompanied by straining.
Response was defined as a patient who had at least three spontaneous BMs per week and an increase of at least one for that week, for at least 9 of the 12 treatment weeks and at least 3 of the last 4 weeks of the trial.
In both studies, the responder rate was significantly higher in the naldemedine group than in the placebo group (C1 48% vs. 34%; C2 53% vs. 33%). Those taking naldemedine had a mean of two more spontaneous BMs per week than did those taking placebo, and significantly more of those were accomplished without straining.
The most common treatment-emergent adverse effects were diarrhea (about 8% vs. 3%) and abdominal pain (about 6% vs. 1%).
Three patients in C1 experienced at least one event of opioid withdrawal (two taking the study drug and one taking placebo). There were no confirmed withdrawal events in C2, but seven patients (five taking the study drug and two taking placebo) experienced possible gastrointestinal withdrawal symptoms.
COMPOSE 3 demonstrated naldemedine’s lasting benefit in this population. It randomized 1,246 patients to 52 weeks of placebo or the 0.2 mg/day dose. Patient demographics were similar to the earlier COMPOSE studies.
The primary endpoint was treatment-emergent adverse events. Additional endpoints included opioid withdrawal, pain intensity, frequency of bowel movements, and constipation-related symptoms.
There was a significant and sustained increase from baseline in the frequency of bowel movements with naldemedine, increasing from about two to four each week. Constipation symptoms and quality of life scores both improved significantly, relative to placebo.
Again, the most common adverse event was diarrhea (11% naldemedine vs. 5% placebo). The drug was not associated with any opioid withdrawal symptoms, nor did it interfere with a patient’s pain control.
LAS VEGAS – .
In three placebo-controlled randomized studies, the drug increased the number of spontaneous bowel movements and improved stool consistency without inducing opioid withdrawal symptoms.
Naldemedine (Symproic) received FDA approval last year based on COMPOSE 1 and COMPOSE 2, which were published in Lancet Gastroenterology and Hepatology last year (2017 Aug 2[8]:555-64). James E. Wild, MD, of Upstate Clinical Research Associates, in Williamsville, N.Y., reported these in a poster presented at the annual PAINWeek.
The year-long COMPOSE 3 trial was not presented at the meeting, but appeared in the journal Pain (2018 May;5:987-94).
Opioids not only affect the central nervous system but also bind to mu-opioid receptors in the gut, decreasing intestinal motility. Naldemedine blocks these receptors from opioid binding but cannot cross the blood-brain barrier. Its peripheral action blocks gut opioid binding, side-stepping the motility problem without inducing any opioid withdrawal symptoms.
COMPOSE 1 (C1) and COMPOSE 2 (C2) comprised a total of 1,095 subjects with chronic, noncancer pain. They were randomized to either naldemedine 0.2 mg daily or placebo for 12 weeks.
Patients were a median of 53 years with a mean opioid use of 61 months. About 60% had a mean daily morphine equivalent of 30-100 mg, and 40% a mean dose of more than 100 mg. At baseline, they had a mean of one spontaneous bowel movement (BM) per week, with a mean of 0.4 deemed “complete.” All the BMs were accompanied by straining.
Response was defined as a patient who had at least three spontaneous BMs per week and an increase of at least one for that week, for at least 9 of the 12 treatment weeks and at least 3 of the last 4 weeks of the trial.
In both studies, the responder rate was significantly higher in the naldemedine group than in the placebo group (C1 48% vs. 34%; C2 53% vs. 33%). Those taking naldemedine had a mean of two more spontaneous BMs per week than did those taking placebo, and significantly more of those were accomplished without straining.
The most common treatment-emergent adverse effects were diarrhea (about 8% vs. 3%) and abdominal pain (about 6% vs. 1%).
Three patients in C1 experienced at least one event of opioid withdrawal (two taking the study drug and one taking placebo). There were no confirmed withdrawal events in C2, but seven patients (five taking the study drug and two taking placebo) experienced possible gastrointestinal withdrawal symptoms.
COMPOSE 3 demonstrated naldemedine’s lasting benefit in this population. It randomized 1,246 patients to 52 weeks of placebo or the 0.2 mg/day dose. Patient demographics were similar to the earlier COMPOSE studies.
The primary endpoint was treatment-emergent adverse events. Additional endpoints included opioid withdrawal, pain intensity, frequency of bowel movements, and constipation-related symptoms.
There was a significant and sustained increase from baseline in the frequency of bowel movements with naldemedine, increasing from about two to four each week. Constipation symptoms and quality of life scores both improved significantly, relative to placebo.
Again, the most common adverse event was diarrhea (11% naldemedine vs. 5% placebo). The drug was not associated with any opioid withdrawal symptoms, nor did it interfere with a patient’s pain control.
LAS VEGAS – .
In three placebo-controlled randomized studies, the drug increased the number of spontaneous bowel movements and improved stool consistency without inducing opioid withdrawal symptoms.
Naldemedine (Symproic) received FDA approval last year based on COMPOSE 1 and COMPOSE 2, which were published in Lancet Gastroenterology and Hepatology last year (2017 Aug 2[8]:555-64). James E. Wild, MD, of Upstate Clinical Research Associates, in Williamsville, N.Y., reported these in a poster presented at the annual PAINWeek.
The year-long COMPOSE 3 trial was not presented at the meeting, but appeared in the journal Pain (2018 May;5:987-94).
Opioids not only affect the central nervous system but also bind to mu-opioid receptors in the gut, decreasing intestinal motility. Naldemedine blocks these receptors from opioid binding but cannot cross the blood-brain barrier. Its peripheral action blocks gut opioid binding, side-stepping the motility problem without inducing any opioid withdrawal symptoms.
COMPOSE 1 (C1) and COMPOSE 2 (C2) comprised a total of 1,095 subjects with chronic, noncancer pain. They were randomized to either naldemedine 0.2 mg daily or placebo for 12 weeks.
Patients were a median of 53 years with a mean opioid use of 61 months. About 60% had a mean daily morphine equivalent of 30-100 mg, and 40% a mean dose of more than 100 mg. At baseline, they had a mean of one spontaneous bowel movement (BM) per week, with a mean of 0.4 deemed “complete.” All the BMs were accompanied by straining.
Response was defined as a patient who had at least three spontaneous BMs per week and an increase of at least one for that week, for at least 9 of the 12 treatment weeks and at least 3 of the last 4 weeks of the trial.
In both studies, the responder rate was significantly higher in the naldemedine group than in the placebo group (C1 48% vs. 34%; C2 53% vs. 33%). Those taking naldemedine had a mean of two more spontaneous BMs per week than did those taking placebo, and significantly more of those were accomplished without straining.
The most common treatment-emergent adverse effects were diarrhea (about 8% vs. 3%) and abdominal pain (about 6% vs. 1%).
Three patients in C1 experienced at least one event of opioid withdrawal (two taking the study drug and one taking placebo). There were no confirmed withdrawal events in C2, but seven patients (five taking the study drug and two taking placebo) experienced possible gastrointestinal withdrawal symptoms.
COMPOSE 3 demonstrated naldemedine’s lasting benefit in this population. It randomized 1,246 patients to 52 weeks of placebo or the 0.2 mg/day dose. Patient demographics were similar to the earlier COMPOSE studies.
The primary endpoint was treatment-emergent adverse events. Additional endpoints included opioid withdrawal, pain intensity, frequency of bowel movements, and constipation-related symptoms.
There was a significant and sustained increase from baseline in the frequency of bowel movements with naldemedine, increasing from about two to four each week. Constipation symptoms and quality of life scores both improved significantly, relative to placebo.
Again, the most common adverse event was diarrhea (11% naldemedine vs. 5% placebo). The drug was not associated with any opioid withdrawal symptoms, nor did it interfere with a patient’s pain control.
AT PAINWEEK 2018
Key clinical point: Naldemedine improves symptoms of opioid-induced constipation over 12 and 52 weeks.
Major finding: The drug doubled the frequency of bowel movements in both 12- and 52-week studies.
Study details: Altogether, COMPOSE 1, 2, and 3 randomized 2,300 patients to placebo or 0.2 mg/day naldemedine.
Disclosures: The studies were sponsored by Shionogi. Dr. Wild is a clinical trialist with Upstate Clinical Research Associates, Williamsville, N.Y.
Source: Wild JE et al. PAINWeek 2018, Abstract 34
New Edition of the ‘Go-To’ Book on Diabetes Available
“Diabetes in America was written to serve as the go-to book for anything you ever wanted to know about diabetes,” says Catherine Cowie, PhD, editor and senior advisor for the National Institute of Diabetes and Digestive and Kidney Diseases’ Diabetes Epidemiology Program. “It’s a resource for everyone, because diabetes affects just about everyone.”
Written by recognized experts who “represent every facet of diabetes,” the book covers relevant research, data and trends, complications and related conditions, and prevention and medical care. It is “rich in data,” says Dr. Cowie, and includes cross-sectional national data, as well as smaller geographic community and longitudinal studies. This edition includes both published and unpublished data that were specifically analyzed for the book.
Clinical trial data are summarized to show the strongest evidence available for the effectiveness of interventions, but the book also emphasizes “points of hope” found through research: For example, people at high risk can prevent or delay type 2 diabetes by losing a modest amount of weight, and rates of some complications, such as lower extremity amputations, are on the decline.
Cowie says Diabetes in America is designed to be useful to a variety of readers. Patients can use it to better understand their condition or risk factors; practitioners can use it to assess patients’ risk of diabetes and associated complications; health policy makers who need “sound quantitative knowledge” can use it to guide decision making; scientists can use it to help identify areas of needed research.
To download, visit: https://www.niddk.nih.gov/about-niddk/strategic-plans-reports/diabetes-in-america-3rd-edition.
“Diabetes in America was written to serve as the go-to book for anything you ever wanted to know about diabetes,” says Catherine Cowie, PhD, editor and senior advisor for the National Institute of Diabetes and Digestive and Kidney Diseases’ Diabetes Epidemiology Program. “It’s a resource for everyone, because diabetes affects just about everyone.”
Written by recognized experts who “represent every facet of diabetes,” the book covers relevant research, data and trends, complications and related conditions, and prevention and medical care. It is “rich in data,” says Dr. Cowie, and includes cross-sectional national data, as well as smaller geographic community and longitudinal studies. This edition includes both published and unpublished data that were specifically analyzed for the book.
Clinical trial data are summarized to show the strongest evidence available for the effectiveness of interventions, but the book also emphasizes “points of hope” found through research: For example, people at high risk can prevent or delay type 2 diabetes by losing a modest amount of weight, and rates of some complications, such as lower extremity amputations, are on the decline.
Cowie says Diabetes in America is designed to be useful to a variety of readers. Patients can use it to better understand their condition or risk factors; practitioners can use it to assess patients’ risk of diabetes and associated complications; health policy makers who need “sound quantitative knowledge” can use it to guide decision making; scientists can use it to help identify areas of needed research.
To download, visit: https://www.niddk.nih.gov/about-niddk/strategic-plans-reports/diabetes-in-america-3rd-edition.
“Diabetes in America was written to serve as the go-to book for anything you ever wanted to know about diabetes,” says Catherine Cowie, PhD, editor and senior advisor for the National Institute of Diabetes and Digestive and Kidney Diseases’ Diabetes Epidemiology Program. “It’s a resource for everyone, because diabetes affects just about everyone.”
Written by recognized experts who “represent every facet of diabetes,” the book covers relevant research, data and trends, complications and related conditions, and prevention and medical care. It is “rich in data,” says Dr. Cowie, and includes cross-sectional national data, as well as smaller geographic community and longitudinal studies. This edition includes both published and unpublished data that were specifically analyzed for the book.
Clinical trial data are summarized to show the strongest evidence available for the effectiveness of interventions, but the book also emphasizes “points of hope” found through research: For example, people at high risk can prevent or delay type 2 diabetes by losing a modest amount of weight, and rates of some complications, such as lower extremity amputations, are on the decline.
Cowie says Diabetes in America is designed to be useful to a variety of readers. Patients can use it to better understand their condition or risk factors; practitioners can use it to assess patients’ risk of diabetes and associated complications; health policy makers who need “sound quantitative knowledge” can use it to guide decision making; scientists can use it to help identify areas of needed research.
To download, visit: https://www.niddk.nih.gov/about-niddk/strategic-plans-reports/diabetes-in-america-3rd-edition.
Novel agents changing treatment algorithm in AML

NEW YORK—Recent drug approvals for acute myeloid leukemia (AML) have greatly expanded options for treating patients, according to a presentation at the NCCN 13th Annual Congress: Hematologic Malignancies.
Richard M. Stone, MD, of the Dana-Farber Cancer Institute in Boston, M.A., gave this presentation, providing some guidance for how to incorporate newly approved AML drugs into practice.
Dr. Stone also discussed therapies and treatment strategies that are under investigation.
Midostaurin
Midostaurin was approved by the U.S. Food and Drug Administration (FDA) in 2017. It is approved for use in combination with standard cytarabine and daunorubicin induction, followed by cytarabine consolidation, in adults with newly diagnosed AML who are FLT3-mutation-positive, as detected by an FDA-approved test.
Even though FLT3-ITD mutation confers a bad prognosis in AML, midostaurin capitalizes on the activated enzyme by inhibiting it.
The RATIFY trial (CALGB 10603) enrolled only FLT3-mutated AML patients younger than 60 and randomized them to induction and consolidation with or without midostaurin.
“Bottom line, being on midostaurin was a good thing,” Dr. Stone said.
Midostaurin led to a 23% reduction in the risk of death, and the 4-year survival rate was improved by about 7%.
“What is meaningful, if you happen to get a transplant in CR1 [first complete remission], you may be going to transplant with lower disease burden,” Dr. Stone explained. “Adding midostaurin is probably a good thing for mutated FLT3.”
The addition of midostaurin to induction may also be appropriate for fit older adults with FLT3 mutations, Dr. Stone said.
Gemtuzumab ozogamicin
The antibody-drug conjugate gemtuzumab ozogamicin has been around for almost 20 years and has an “interesting” history, according to Dr. Stone.
Gemtuzumab ozogamicin was first approved by the FDA in 2000, based on a 30% remission rate in relapsed AML.
However, the agent was voluntarily withdrawn from the market in 2010 because it was used in the upfront setting with chemotherapy and didn’t show a benefit.
Recent studies of the agent suggested use of the agent should be revisited but with lower doses.
Data from the ALFA 0701 study was key for the FDA’s re-approval of gemtuzumab in 2017.
According to Dr. Stone, the most important finding of this trial was the major event-free survival benefit for those on gemtuzumab, which was 41% at 2 years, compared to 17% for the control group.
Overall survival, however, was not significantly superior with gemtuzumab.
A meta-analysis of 5 trials showed that adding gemtuzumab to treatment of patients with favorable and intermediate risk conferred a survival advantage, but this was not the case in patients with adverse-risk cytogenetics.
Fit older adults with CBF mutation may benefit from the addition of gemtuzumab, Dr. Stone said.
He also pointed out that gemtuzumab has a big “booby prize,” which is veno-occlusive disease, shown to be a problem with high doses of gemtuzumab and transplant.
CPX-351
CPX-351 is a liposomal co-formulation of cytarabine and daunorubicin, the two drugs delivered separately in the standard induction chemotherapy referred to as 7+3.
Last year, the FDA approved CPX-351 to treat adults who have AML with myelodysplasia-related changes or newly diagnosed, therapy-related AML.
In a phase 3 trial, CPX-351 conferred a survival advantage over standard 7+3. There was a 31% reduction in the risk of death with CPX-351.
Patients who went on to transplant after CPX-351 did much better than patients who received 7+3 and transplant, with a hazard ratio of 0.51.
Dr. Stone noted that minimal residual disease was not measured, “but it’s quite possible that patients who had CPX went into transplant with a lower level of disease burden.”
Dr. Stone also said CPX-351 may be added to induction for fit older patients with secondary AML.
Enasidenib and ivosidenib
In relapsed AML, the treatment approach is to induce a second complete remission and then proceed to allogeneic stem cell transplant.
Traditionally, FLAG-IDA (fludarabine, cytarabine, idarubicin, and G-CSF) and MEC (mitoxantrone, etoposide, and cytarabine) have been used as salvage, and another course of 7+3 is an option if the patient has been disease-free for over a year.
Now, however, enasidenib and ivosidenib may be an option for IDH2- and IDH1-mutated patients, respectively.
The FDA approved enasidenib in 2017 to treat adults with relapsed or refractory AML and an IDH2 mutation, as detected by an FDA-approved test.
Ivosidenib was approved by the FDA this year to treat adults with relapsed or refractory AML who have an IDH1 mutation, as detected by an FDA-approved test.
New approaches on the horizon
Dr. Stone noted that gilteritinib and quizartinib are currently in development for FLT3-mutated patients, and he anticipates these therapies will be approved by the FDA in 2019.
Dr. Stone also touched on other new approaches under investigation, such as hedgehog pathway inhibition, dysregulation of the spliceosome complex, inhibiting MDM2, and strengthening the immune system.
However, he believes the most important is BCL-2 inhibition with venetoclax.
Venetoclax combined with a hypomethylator (azacitidine or decitabine) produced a response rate of 75% with azacitidine, double what one would expect with azacitidine or decitabine alone, Dr. Stone noted.
And venetoclax with low-dose cytarabine may enable elderly good-risk patients to avoid 7+3.
Dr. Stone’s presentation also covered genes commonly mutated in AML, the increasing scrutiny of complete remission, and minimal residual disease assessment. An account of this part of the presentation can be found here: “Current management of AML patients.”
NEW YORK—Recent drug approvals for acute myeloid leukemia (AML) have greatly expanded options for treating patients, according to a presentation at the NCCN 13th Annual Congress: Hematologic Malignancies.
Richard M. Stone, MD, of the Dana-Farber Cancer Institute in Boston, M.A., gave this presentation, providing some guidance for how to incorporate newly approved AML drugs into practice.
Dr. Stone also discussed therapies and treatment strategies that are under investigation.
Midostaurin
Midostaurin was approved by the U.S. Food and Drug Administration (FDA) in 2017. It is approved for use in combination with standard cytarabine and daunorubicin induction, followed by cytarabine consolidation, in adults with newly diagnosed AML who are FLT3-mutation-positive, as detected by an FDA-approved test.
Even though FLT3-ITD mutation confers a bad prognosis in AML, midostaurin capitalizes on the activated enzyme by inhibiting it.
The RATIFY trial (CALGB 10603) enrolled only FLT3-mutated AML patients younger than 60 and randomized them to induction and consolidation with or without midostaurin.
“Bottom line, being on midostaurin was a good thing,” Dr. Stone said.
Midostaurin led to a 23% reduction in the risk of death, and the 4-year survival rate was improved by about 7%.
“What is meaningful, if you happen to get a transplant in CR1 [first complete remission], you may be going to transplant with lower disease burden,” Dr. Stone explained. “Adding midostaurin is probably a good thing for mutated FLT3.”
The addition of midostaurin to induction may also be appropriate for fit older adults with FLT3 mutations, Dr. Stone said.
Gemtuzumab ozogamicin
The antibody-drug conjugate gemtuzumab ozogamicin has been around for almost 20 years and has an “interesting” history, according to Dr. Stone.
Gemtuzumab ozogamicin was first approved by the FDA in 2000, based on a 30% remission rate in relapsed AML.
However, the agent was voluntarily withdrawn from the market in 2010 because it was used in the upfront setting with chemotherapy and didn’t show a benefit.
Recent studies of the agent suggested use of the agent should be revisited but with lower doses.
Data from the ALFA 0701 study was key for the FDA’s re-approval of gemtuzumab in 2017.
According to Dr. Stone, the most important finding of this trial was the major event-free survival benefit for those on gemtuzumab, which was 41% at 2 years, compared to 17% for the control group.
Overall survival, however, was not significantly superior with gemtuzumab.
A meta-analysis of 5 trials showed that adding gemtuzumab to treatment of patients with favorable and intermediate risk conferred a survival advantage, but this was not the case in patients with adverse-risk cytogenetics.
Fit older adults with CBF mutation may benefit from the addition of gemtuzumab, Dr. Stone said.
He also pointed out that gemtuzumab has a big “booby prize,” which is veno-occlusive disease, shown to be a problem with high doses of gemtuzumab and transplant.
CPX-351
CPX-351 is a liposomal co-formulation of cytarabine and daunorubicin, the two drugs delivered separately in the standard induction chemotherapy referred to as 7+3.
Last year, the FDA approved CPX-351 to treat adults who have AML with myelodysplasia-related changes or newly diagnosed, therapy-related AML.
In a phase 3 trial, CPX-351 conferred a survival advantage over standard 7+3. There was a 31% reduction in the risk of death with CPX-351.
Patients who went on to transplant after CPX-351 did much better than patients who received 7+3 and transplant, with a hazard ratio of 0.51.
Dr. Stone noted that minimal residual disease was not measured, “but it’s quite possible that patients who had CPX went into transplant with a lower level of disease burden.”
Dr. Stone also said CPX-351 may be added to induction for fit older patients with secondary AML.
Enasidenib and ivosidenib
In relapsed AML, the treatment approach is to induce a second complete remission and then proceed to allogeneic stem cell transplant.
Traditionally, FLAG-IDA (fludarabine, cytarabine, idarubicin, and G-CSF) and MEC (mitoxantrone, etoposide, and cytarabine) have been used as salvage, and another course of 7+3 is an option if the patient has been disease-free for over a year.
Now, however, enasidenib and ivosidenib may be an option for IDH2- and IDH1-mutated patients, respectively.
The FDA approved enasidenib in 2017 to treat adults with relapsed or refractory AML and an IDH2 mutation, as detected by an FDA-approved test.
Ivosidenib was approved by the FDA this year to treat adults with relapsed or refractory AML who have an IDH1 mutation, as detected by an FDA-approved test.
New approaches on the horizon
Dr. Stone noted that gilteritinib and quizartinib are currently in development for FLT3-mutated patients, and he anticipates these therapies will be approved by the FDA in 2019.
Dr. Stone also touched on other new approaches under investigation, such as hedgehog pathway inhibition, dysregulation of the spliceosome complex, inhibiting MDM2, and strengthening the immune system.
However, he believes the most important is BCL-2 inhibition with venetoclax.
Venetoclax combined with a hypomethylator (azacitidine or decitabine) produced a response rate of 75% with azacitidine, double what one would expect with azacitidine or decitabine alone, Dr. Stone noted.
And venetoclax with low-dose cytarabine may enable elderly good-risk patients to avoid 7+3.
Dr. Stone’s presentation also covered genes commonly mutated in AML, the increasing scrutiny of complete remission, and minimal residual disease assessment. An account of this part of the presentation can be found here: “Current management of AML patients.”
NEW YORK—Recent drug approvals for acute myeloid leukemia (AML) have greatly expanded options for treating patients, according to a presentation at the NCCN 13th Annual Congress: Hematologic Malignancies.
Richard M. Stone, MD, of the Dana-Farber Cancer Institute in Boston, M.A., gave this presentation, providing some guidance for how to incorporate newly approved AML drugs into practice.
Dr. Stone also discussed therapies and treatment strategies that are under investigation.
Midostaurin
Midostaurin was approved by the U.S. Food and Drug Administration (FDA) in 2017. It is approved for use in combination with standard cytarabine and daunorubicin induction, followed by cytarabine consolidation, in adults with newly diagnosed AML who are FLT3-mutation-positive, as detected by an FDA-approved test.
Even though FLT3-ITD mutation confers a bad prognosis in AML, midostaurin capitalizes on the activated enzyme by inhibiting it.
The RATIFY trial (CALGB 10603) enrolled only FLT3-mutated AML patients younger than 60 and randomized them to induction and consolidation with or without midostaurin.
“Bottom line, being on midostaurin was a good thing,” Dr. Stone said.
Midostaurin led to a 23% reduction in the risk of death, and the 4-year survival rate was improved by about 7%.
“What is meaningful, if you happen to get a transplant in CR1 [first complete remission], you may be going to transplant with lower disease burden,” Dr. Stone explained. “Adding midostaurin is probably a good thing for mutated FLT3.”
The addition of midostaurin to induction may also be appropriate for fit older adults with FLT3 mutations, Dr. Stone said.
Gemtuzumab ozogamicin
The antibody-drug conjugate gemtuzumab ozogamicin has been around for almost 20 years and has an “interesting” history, according to Dr. Stone.
Gemtuzumab ozogamicin was first approved by the FDA in 2000, based on a 30% remission rate in relapsed AML.
However, the agent was voluntarily withdrawn from the market in 2010 because it was used in the upfront setting with chemotherapy and didn’t show a benefit.
Recent studies of the agent suggested use of the agent should be revisited but with lower doses.
Data from the ALFA 0701 study was key for the FDA’s re-approval of gemtuzumab in 2017.
According to Dr. Stone, the most important finding of this trial was the major event-free survival benefit for those on gemtuzumab, which was 41% at 2 years, compared to 17% for the control group.
Overall survival, however, was not significantly superior with gemtuzumab.
A meta-analysis of 5 trials showed that adding gemtuzumab to treatment of patients with favorable and intermediate risk conferred a survival advantage, but this was not the case in patients with adverse-risk cytogenetics.
Fit older adults with CBF mutation may benefit from the addition of gemtuzumab, Dr. Stone said.
He also pointed out that gemtuzumab has a big “booby prize,” which is veno-occlusive disease, shown to be a problem with high doses of gemtuzumab and transplant.
CPX-351
CPX-351 is a liposomal co-formulation of cytarabine and daunorubicin, the two drugs delivered separately in the standard induction chemotherapy referred to as 7+3.
Last year, the FDA approved CPX-351 to treat adults who have AML with myelodysplasia-related changes or newly diagnosed, therapy-related AML.
In a phase 3 trial, CPX-351 conferred a survival advantage over standard 7+3. There was a 31% reduction in the risk of death with CPX-351.
Patients who went on to transplant after CPX-351 did much better than patients who received 7+3 and transplant, with a hazard ratio of 0.51.
Dr. Stone noted that minimal residual disease was not measured, “but it’s quite possible that patients who had CPX went into transplant with a lower level of disease burden.”
Dr. Stone also said CPX-351 may be added to induction for fit older patients with secondary AML.
Enasidenib and ivosidenib
In relapsed AML, the treatment approach is to induce a second complete remission and then proceed to allogeneic stem cell transplant.
Traditionally, FLAG-IDA (fludarabine, cytarabine, idarubicin, and G-CSF) and MEC (mitoxantrone, etoposide, and cytarabine) have been used as salvage, and another course of 7+3 is an option if the patient has been disease-free for over a year.
Now, however, enasidenib and ivosidenib may be an option for IDH2- and IDH1-mutated patients, respectively.
The FDA approved enasidenib in 2017 to treat adults with relapsed or refractory AML and an IDH2 mutation, as detected by an FDA-approved test.
Ivosidenib was approved by the FDA this year to treat adults with relapsed or refractory AML who have an IDH1 mutation, as detected by an FDA-approved test.
New approaches on the horizon
Dr. Stone noted that gilteritinib and quizartinib are currently in development for FLT3-mutated patients, and he anticipates these therapies will be approved by the FDA in 2019.
Dr. Stone also touched on other new approaches under investigation, such as hedgehog pathway inhibition, dysregulation of the spliceosome complex, inhibiting MDM2, and strengthening the immune system.
However, he believes the most important is BCL-2 inhibition with venetoclax.
Venetoclax combined with a hypomethylator (azacitidine or decitabine) produced a response rate of 75% with azacitidine, double what one would expect with azacitidine or decitabine alone, Dr. Stone noted.
And venetoclax with low-dose cytarabine may enable elderly good-risk patients to avoid 7+3.
Dr. Stone’s presentation also covered genes commonly mutated in AML, the increasing scrutiny of complete remission, and minimal residual disease assessment. An account of this part of the presentation can be found here: “Current management of AML patients.”
Current management of AML patients

NEW YORK—A presentation at the NCCN 13th Annual Congress: Hematologic Malignancies outlined current practices for managing patients with acute myeloid leukemia (AML).
Richard M. Stone, MD, of the Dana-Farber Cancer Institute in Boston, M.A., noted that management of AML now includes increased screening of commonly mutated genes, greater scrutiny of complete remission (CR), and a focus on minimal residual disease (MRD).
Genetics and cytogenetics
Genome sequencing projects in AML have revealed about 30 genes commonly mutated in AML patients, which can be further divided into 9 subcategories, Dr. Stone said.
He noted that the list of genes to screen for is getting longer every year, from FLT3-ITD, NPM1, and CEBPA last year, to RUNX1, TP53, ASXL1, KIT, and CBF mutations this year.
“Just looking at genetics, you can tell if you have CEBPA mutation, you are most likely going to be free of AML, and if you have TP53, you are in big trouble,” Dr. Stone said.
He added that the mutation status of IDH1/2, DNMT3A, and TET2 will be important in the future.
“So, basically, I like to have an NGS [next-generation sequencing] panel on all of our patients with AML today,” Dr. Stone said. “It’s probably going to be cheaper than trying to do the shotgun approach we were recently used to.”
Achieving CR
Dr. Stone noted that the first goal of AML treatment is to reduce the gross leukemia to undetectable levels with induction therapy and to achieve a CR.
“But there are real good remissions and not so good remissions,” he said. “But once we get into remission, that’s just the beginning of the curative approach.”
The second goal is to reduce patients’ leukemic cells still present at CR to a level low enough to achieve prolonged disease-free survival.
“CR is coming under more scrutiny,” according to Dr. Stone, because some patients with CR have low blasts, but the blood counts aren’t returning to normal.
“[T]here’s CRc, CRi, CRp, you name it, all the little subscripts, which mean the remission really isn’t as strong as it should be,” Dr. Stone said.
“But if you do have a complete remission with low blast counts in the marrow and blood and normal blood counts, you might have an MRD-negative remission, which is the goal.”
Quantitation techniques and MRD
The European LeukemiaNet recommends all AML patients have an MRD assessment at the time of remission by either multiparameter flow cytometry (MFC) or molecular means.
The most sensitive techniques to determine disease burden are MFC and polymerase chain reaction (PCR)-based methods, both of which are sensitive enough to find one leukemic cell in 10,000 cells (10-4).
NGS techniques are becoming more sensitive, Dr. Stone added, but probably not down to the level of 10-4.
In a study by Ivey et al, MRD levels based on PCR of NPM1-mutated patients after two rounds of chemotherapy could independently predict clinical outcome. Overall survival was 73% for MRD-negative patients and 24% for MRD-positive patients.
MFC and NGS data have been shown in a study by Jongen-Lavrencic et al to be mutually helpful. If a patient is positive by neither method, the relapse rate is low. If the patient is positive by both, the relapse rate is high. And if the patient is positive by one and negative by the other, the relapse rate is intermediate.
“So what do we do with this?” Dr. Stone asked. “The problem is, I don’t know what to do with that data except get worried.”
The dilemma is that if patients are MRD-positive in remission, they are probably not going to respond well to a transplant. And if they are MRD-negative, they don’t need a transplant.
A retrospective study by Milano et al indicated that MRD-positive patients seem to fare better than expected with a double umbilical cord blood transplant. However, Dr. Stone noted that this finding has not been confirmed prospectively.
The most exciting aspect of MRD in AML, Dr. Stone said, is it may allow us to get to new drugs quicker by revealing whether a drug is actually lowering MRD burden.
Dr. Stone also discussed newly approved drugs for AML and new treatment approaches under investigation. Details on that portion of his presentation are available here: “Novel agents changing treatment algorithm in AML.”
NEW YORK—A presentation at the NCCN 13th Annual Congress: Hematologic Malignancies outlined current practices for managing patients with acute myeloid leukemia (AML).
Richard M. Stone, MD, of the Dana-Farber Cancer Institute in Boston, M.A., noted that management of AML now includes increased screening of commonly mutated genes, greater scrutiny of complete remission (CR), and a focus on minimal residual disease (MRD).
Genetics and cytogenetics
Genome sequencing projects in AML have revealed about 30 genes commonly mutated in AML patients, which can be further divided into 9 subcategories, Dr. Stone said.
He noted that the list of genes to screen for is getting longer every year, from FLT3-ITD, NPM1, and CEBPA last year, to RUNX1, TP53, ASXL1, KIT, and CBF mutations this year.
“Just looking at genetics, you can tell if you have CEBPA mutation, you are most likely going to be free of AML, and if you have TP53, you are in big trouble,” Dr. Stone said.
He added that the mutation status of IDH1/2, DNMT3A, and TET2 will be important in the future.
“So, basically, I like to have an NGS [next-generation sequencing] panel on all of our patients with AML today,” Dr. Stone said. “It’s probably going to be cheaper than trying to do the shotgun approach we were recently used to.”
Achieving CR
Dr. Stone noted that the first goal of AML treatment is to reduce the gross leukemia to undetectable levels with induction therapy and to achieve a CR.
“But there are real good remissions and not so good remissions,” he said. “But once we get into remission, that’s just the beginning of the curative approach.”
The second goal is to reduce patients’ leukemic cells still present at CR to a level low enough to achieve prolonged disease-free survival.
“CR is coming under more scrutiny,” according to Dr. Stone, because some patients with CR have low blasts, but the blood counts aren’t returning to normal.
“[T]here’s CRc, CRi, CRp, you name it, all the little subscripts, which mean the remission really isn’t as strong as it should be,” Dr. Stone said.
“But if you do have a complete remission with low blast counts in the marrow and blood and normal blood counts, you might have an MRD-negative remission, which is the goal.”
Quantitation techniques and MRD
The European LeukemiaNet recommends all AML patients have an MRD assessment at the time of remission by either multiparameter flow cytometry (MFC) or molecular means.
The most sensitive techniques to determine disease burden are MFC and polymerase chain reaction (PCR)-based methods, both of which are sensitive enough to find one leukemic cell in 10,000 cells (10-4).
NGS techniques are becoming more sensitive, Dr. Stone added, but probably not down to the level of 10-4.
In a study by Ivey et al, MRD levels based on PCR of NPM1-mutated patients after two rounds of chemotherapy could independently predict clinical outcome. Overall survival was 73% for MRD-negative patients and 24% for MRD-positive patients.
MFC and NGS data have been shown in a study by Jongen-Lavrencic et al to be mutually helpful. If a patient is positive by neither method, the relapse rate is low. If the patient is positive by both, the relapse rate is high. And if the patient is positive by one and negative by the other, the relapse rate is intermediate.
“So what do we do with this?” Dr. Stone asked. “The problem is, I don’t know what to do with that data except get worried.”
The dilemma is that if patients are MRD-positive in remission, they are probably not going to respond well to a transplant. And if they are MRD-negative, they don’t need a transplant.
A retrospective study by Milano et al indicated that MRD-positive patients seem to fare better than expected with a double umbilical cord blood transplant. However, Dr. Stone noted that this finding has not been confirmed prospectively.
The most exciting aspect of MRD in AML, Dr. Stone said, is it may allow us to get to new drugs quicker by revealing whether a drug is actually lowering MRD burden.
Dr. Stone also discussed newly approved drugs for AML and new treatment approaches under investigation. Details on that portion of his presentation are available here: “Novel agents changing treatment algorithm in AML.”
NEW YORK—A presentation at the NCCN 13th Annual Congress: Hematologic Malignancies outlined current practices for managing patients with acute myeloid leukemia (AML).
Richard M. Stone, MD, of the Dana-Farber Cancer Institute in Boston, M.A., noted that management of AML now includes increased screening of commonly mutated genes, greater scrutiny of complete remission (CR), and a focus on minimal residual disease (MRD).
Genetics and cytogenetics
Genome sequencing projects in AML have revealed about 30 genes commonly mutated in AML patients, which can be further divided into 9 subcategories, Dr. Stone said.
He noted that the list of genes to screen for is getting longer every year, from FLT3-ITD, NPM1, and CEBPA last year, to RUNX1, TP53, ASXL1, KIT, and CBF mutations this year.
“Just looking at genetics, you can tell if you have CEBPA mutation, you are most likely going to be free of AML, and if you have TP53, you are in big trouble,” Dr. Stone said.
He added that the mutation status of IDH1/2, DNMT3A, and TET2 will be important in the future.
“So, basically, I like to have an NGS [next-generation sequencing] panel on all of our patients with AML today,” Dr. Stone said. “It’s probably going to be cheaper than trying to do the shotgun approach we were recently used to.”
Achieving CR
Dr. Stone noted that the first goal of AML treatment is to reduce the gross leukemia to undetectable levels with induction therapy and to achieve a CR.
“But there are real good remissions and not so good remissions,” he said. “But once we get into remission, that’s just the beginning of the curative approach.”
The second goal is to reduce patients’ leukemic cells still present at CR to a level low enough to achieve prolonged disease-free survival.
“CR is coming under more scrutiny,” according to Dr. Stone, because some patients with CR have low blasts, but the blood counts aren’t returning to normal.
“[T]here’s CRc, CRi, CRp, you name it, all the little subscripts, which mean the remission really isn’t as strong as it should be,” Dr. Stone said.
“But if you do have a complete remission with low blast counts in the marrow and blood and normal blood counts, you might have an MRD-negative remission, which is the goal.”
Quantitation techniques and MRD
The European LeukemiaNet recommends all AML patients have an MRD assessment at the time of remission by either multiparameter flow cytometry (MFC) or molecular means.
The most sensitive techniques to determine disease burden are MFC and polymerase chain reaction (PCR)-based methods, both of which are sensitive enough to find one leukemic cell in 10,000 cells (10-4).
NGS techniques are becoming more sensitive, Dr. Stone added, but probably not down to the level of 10-4.
In a study by Ivey et al, MRD levels based on PCR of NPM1-mutated patients after two rounds of chemotherapy could independently predict clinical outcome. Overall survival was 73% for MRD-negative patients and 24% for MRD-positive patients.
MFC and NGS data have been shown in a study by Jongen-Lavrencic et al to be mutually helpful. If a patient is positive by neither method, the relapse rate is low. If the patient is positive by both, the relapse rate is high. And if the patient is positive by one and negative by the other, the relapse rate is intermediate.
“So what do we do with this?” Dr. Stone asked. “The problem is, I don’t know what to do with that data except get worried.”
The dilemma is that if patients are MRD-positive in remission, they are probably not going to respond well to a transplant. And if they are MRD-negative, they don’t need a transplant.
A retrospective study by Milano et al indicated that MRD-positive patients seem to fare better than expected with a double umbilical cord blood transplant. However, Dr. Stone noted that this finding has not been confirmed prospectively.
The most exciting aspect of MRD in AML, Dr. Stone said, is it may allow us to get to new drugs quicker by revealing whether a drug is actually lowering MRD burden.
Dr. Stone also discussed newly approved drugs for AML and new treatment approaches under investigation. Details on that portion of his presentation are available here: “Novel agents changing treatment algorithm in AML.”
FLT3 inhibitor approved for rel/ref AML in Japan

The Japanese Ministry of Health, Labour and Welfare (MHLW) has approved the FLT3 inhibitor gilteritinib (Xospata®) to treat patients with FLT3-positive relapsed or refractory acute myeloid leukemia (AML).
Gilteritinib is available in 40 mg tablets. The usual recommended starting dose of gilteritinib for an adult is 120 mg once daily.
The dosage may be adjusted depending on the patient’s condition. However, the daily maximum dose should be 200 mg.
Gilteritinib has demonstrated inhibitory activity against FLT3 internal tandem duplication and FLT tyrosine kinase domain mutation. These two mutations are present in approximately one-third of AML patients.
The MHLW approval of gilteritinib is based on interim results from the phase 3 ADMIRAL study (NCT02421939).
ADIMRAL is designed to compare gilteritinib to salvage chemotherapy in adults who have AML with FLT3 mutations and have relapsed after or are refractory to frontline therapy.
Patients are randomized in a 2:1 ratio to receive gilteritinib (120 mg) or salvage chemotherapy, which may consist of low-dose cytarabine, azacitidine, MEC (mitoxantrone, etoposide, and cytarabine), or FLAG-IDA (fludarabine, cytarabine, and granulocyte colony-stimulating factor [G-CSF] with idarubicin).
Results from this trial have not yet been presented or published. However, a description of the trial was presented at the 2018 ASCO Annual Meeting (abstract TPS7075).
Results from a phase 1/2 study of gilteritinib in AML were published in The Lancet Oncology in 2017.
Orphan and SAKIGAKE designations
The MHLW previously granted SAKIGAKE designation and orphan drug designation to gilteritinib.
To receive orphan designation, a product (drug or medical device) must be intended for use in less than 50,000 patients in Japan. Furthermore, orphan products must be indicated for the treatment of serious diseases for which there are high medical needs.
Companies granted orphan designation can receive preferential tax treatment as well as subsidies through the National Institute of Biomedical Innovation (NIBIO) to reduce the financial burden of product development.
Companies with orphan designation can also receive guidance and consultation from the MHLW, the Pharmaceuticals and Medical Devices Agency (PMDA), and NIBIO on research and development activities.
Orphan designation also allows for priority review and an extension of the re-examination period—up to 10 years for drugs and up to 7 years for medical devices.
SAKIGAKE designation can shorten the review period for a product via prioritized consultation, substantial pre-application consultation, and prioritized review.
SAKIGAKE designation also helps promote development with the review partner system (to be conducted by the PMDA) and “substantial” post-marketing safety measures.
The Japanese Ministry of Health, Labour and Welfare (MHLW) has approved the FLT3 inhibitor gilteritinib (Xospata®) to treat patients with FLT3-positive relapsed or refractory acute myeloid leukemia (AML).
Gilteritinib is available in 40 mg tablets. The usual recommended starting dose of gilteritinib for an adult is 120 mg once daily.
The dosage may be adjusted depending on the patient’s condition. However, the daily maximum dose should be 200 mg.
Gilteritinib has demonstrated inhibitory activity against FLT3 internal tandem duplication and FLT tyrosine kinase domain mutation. These two mutations are present in approximately one-third of AML patients.
The MHLW approval of gilteritinib is based on interim results from the phase 3 ADMIRAL study (NCT02421939).
ADIMRAL is designed to compare gilteritinib to salvage chemotherapy in adults who have AML with FLT3 mutations and have relapsed after or are refractory to frontline therapy.
Patients are randomized in a 2:1 ratio to receive gilteritinib (120 mg) or salvage chemotherapy, which may consist of low-dose cytarabine, azacitidine, MEC (mitoxantrone, etoposide, and cytarabine), or FLAG-IDA (fludarabine, cytarabine, and granulocyte colony-stimulating factor [G-CSF] with idarubicin).
Results from this trial have not yet been presented or published. However, a description of the trial was presented at the 2018 ASCO Annual Meeting (abstract TPS7075).
Results from a phase 1/2 study of gilteritinib in AML were published in The Lancet Oncology in 2017.
Orphan and SAKIGAKE designations
The MHLW previously granted SAKIGAKE designation and orphan drug designation to gilteritinib.
To receive orphan designation, a product (drug or medical device) must be intended for use in less than 50,000 patients in Japan. Furthermore, orphan products must be indicated for the treatment of serious diseases for which there are high medical needs.
Companies granted orphan designation can receive preferential tax treatment as well as subsidies through the National Institute of Biomedical Innovation (NIBIO) to reduce the financial burden of product development.
Companies with orphan designation can also receive guidance and consultation from the MHLW, the Pharmaceuticals and Medical Devices Agency (PMDA), and NIBIO on research and development activities.
Orphan designation also allows for priority review and an extension of the re-examination period—up to 10 years for drugs and up to 7 years for medical devices.
SAKIGAKE designation can shorten the review period for a product via prioritized consultation, substantial pre-application consultation, and prioritized review.
SAKIGAKE designation also helps promote development with the review partner system (to be conducted by the PMDA) and “substantial” post-marketing safety measures.
The Japanese Ministry of Health, Labour and Welfare (MHLW) has approved the FLT3 inhibitor gilteritinib (Xospata®) to treat patients with FLT3-positive relapsed or refractory acute myeloid leukemia (AML).
Gilteritinib is available in 40 mg tablets. The usual recommended starting dose of gilteritinib for an adult is 120 mg once daily.
The dosage may be adjusted depending on the patient’s condition. However, the daily maximum dose should be 200 mg.
Gilteritinib has demonstrated inhibitory activity against FLT3 internal tandem duplication and FLT tyrosine kinase domain mutation. These two mutations are present in approximately one-third of AML patients.
The MHLW approval of gilteritinib is based on interim results from the phase 3 ADMIRAL study (NCT02421939).
ADIMRAL is designed to compare gilteritinib to salvage chemotherapy in adults who have AML with FLT3 mutations and have relapsed after or are refractory to frontline therapy.
Patients are randomized in a 2:1 ratio to receive gilteritinib (120 mg) or salvage chemotherapy, which may consist of low-dose cytarabine, azacitidine, MEC (mitoxantrone, etoposide, and cytarabine), or FLAG-IDA (fludarabine, cytarabine, and granulocyte colony-stimulating factor [G-CSF] with idarubicin).
Results from this trial have not yet been presented or published. However, a description of the trial was presented at the 2018 ASCO Annual Meeting (abstract TPS7075).
Results from a phase 1/2 study of gilteritinib in AML were published in The Lancet Oncology in 2017.
Orphan and SAKIGAKE designations
The MHLW previously granted SAKIGAKE designation and orphan drug designation to gilteritinib.
To receive orphan designation, a product (drug or medical device) must be intended for use in less than 50,000 patients in Japan. Furthermore, orphan products must be indicated for the treatment of serious diseases for which there are high medical needs.
Companies granted orphan designation can receive preferential tax treatment as well as subsidies through the National Institute of Biomedical Innovation (NIBIO) to reduce the financial burden of product development.
Companies with orphan designation can also receive guidance and consultation from the MHLW, the Pharmaceuticals and Medical Devices Agency (PMDA), and NIBIO on research and development activities.
Orphan designation also allows for priority review and an extension of the re-examination period—up to 10 years for drugs and up to 7 years for medical devices.
SAKIGAKE designation can shorten the review period for a product via prioritized consultation, substantial pre-application consultation, and prioritized review.
SAKIGAKE designation also helps promote development with the review partner system (to be conducted by the PMDA) and “substantial” post-marketing safety measures.