User login
Arthroscopically-Guided, Cannulated, Headless Compression Screw Fixation of the Symptomatic Os Acromiale
ABSTRACT
Os acromiale is a failure of fusion between 1 or more ossification centers of the scapula and the acromion process. Pain can be caused by motion and impingement of the unfused segment. Several methods for the management of os acromiale have been described. Internal fixation is the most common surgical technique, followed by excision and acromioplasty. We present a novel technique for treatment of symptomatic os acromiale using arthroscopically-guided headless compression screws. This is a viable technique in the management of symptomatic os acromiale due to preservation of the periosteal blood supply and less concern for symptomatic hardware.
Continue to: Os acromiale results from a failure of...
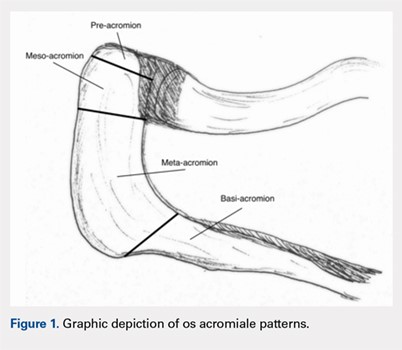
Os acromiale results from a failure of fusion between 1 or more ossification centers and the acromion process.1 The acromion consists of 4 different ossification centers, which appear by 14 years of age and fuse by age 25 years. The 4 ossification centers are the basi-acromion, meta-acromion, mesoacromion, and pre-acromion (Figure 1). Formation of an os acromiale occurs most often due to failure of fusion between the meta-acromion and mesoacromion. Os acromiale appears to occur in approximately 8% of the population, according to cadaveric studies.2 This anatomic variant occurs more commonly in African-Americans than Caucasians, and shows a preponderance for males over females.3
Plain radiographs are usually adequate for diagnosis. Axillary views are most sensitive for detection, which can be difficult to see on anteroposterior radiographs.4 In os acromiale, the unfused segment is connected to the acromioclavicular joint and the coracoid, which can lead to motion of the segment and impingement of the rotator cuff.2-4 Patients frequently experience localized tenderness and symptomatic pain with signs and symptoms of impingement. Rotator cuff tears may occur secondary to chronic impingement.5
Various forms of repair have been described. A recent meta-analysis showed that internal fixation (60%) was the most common surgical technique reported, followed by excision (27%) and acromioplasty (13%).6 Rotator cuff repair is a common concurrent surgical procedure.7-11 The available literature favors internal fixation through an open technique with or without bone grafting.5,7,8,12-15 Various forms of fixation have been presented in the literature, including Kirschner wire fixation, cannulated screw fixation alone, cannulated screw fixation with FiberWire Suture (Arthrex), and cannulated screw fixation with a stainless steel wire tension band technique. Based on the results of the meta-analysis, surgical fixation with cannulated screws has been shown to lead to a significantly greater rate of radiographic healing (23/24 patients) compared to Kirschner wire fixation (31/49 patients).6 Further, radiographic healing is significantly associated with improved clinical outcomes.12 Removal of symptomatic internal fixation hardware is significantly more common after Kirschner wire fixation cases (88%; 43/49) compared to cannulated screw fixation cases (38%; 9/24). However, hardware issues may also be encountered with screw fixation, with 1 case series reporting a 25% rate of hardware complication.16 The patient provided written informed consent for print and electronic publication of this case report.
CASE REPORT
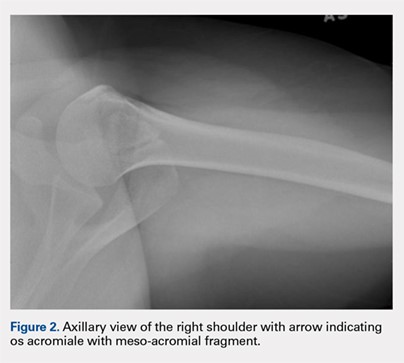
The patient is a 19-year-old right-hand-dominant woman who injured her right shoulder while diving into the bleachers during a volleyball game 4 years prior to presentation. She suffered a direct blow to her shoulder and immediately became symptomatic. She underwent a long period of nonoperative management, which included physical therapy, strengthening, nonsteroidal anti-inflammatory drug (NSAID) therapy, and narcotic pain medications. Her primary complaints upon presentation were pain with lifting, as well as mechanical symptoms. On examination, the patient had moderate tenderness directly over the acromion. She also had evidence of mild impingement symptoms. Plain radiographs revealed a mesoacromial-type os acromiale clearly seen on the axillary lateral film (Figure 2). She underwent magnetic resonance imaging, which suggested rotator cuff tendinosis and evidence of edema at the os acromiale site. She underwent a diagnostic injection directly into the site of maximal tenderness at the os, which provided complete transient relief of her pain. Despite the transient pain relief, the patient continued to be symptomatic after the local anesthetic effect wore off. Surgical options were then discussed with the patient.
Continue to: SURGICAL TECHNIQUE...
SURGICAL TECHNIQUE
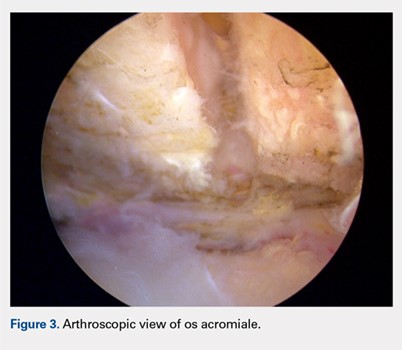
A standard diagnostic shoulder arthroscopy was performed using anterior, posterior and direct lateral portals. The rotator cuff was evaluated, and no evidence of a tear was found. The undersurface of the acromion was exposed, and the os acromiale was identified arthroscopically (Figure 3). This was found to be unstable under direct digital pressure.
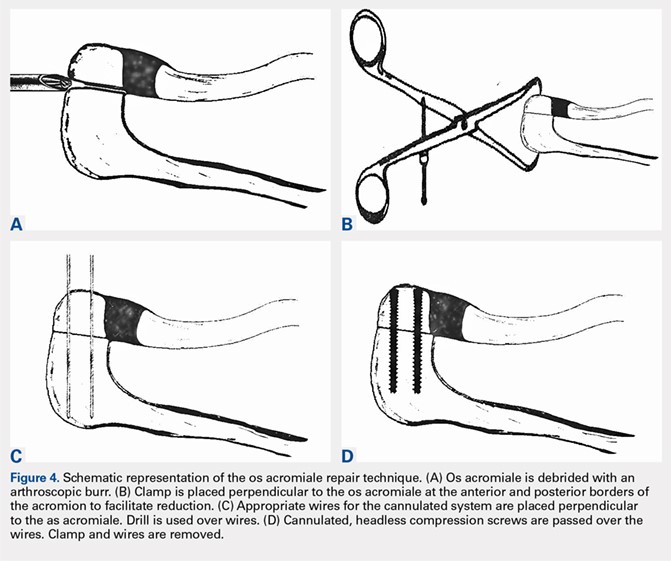
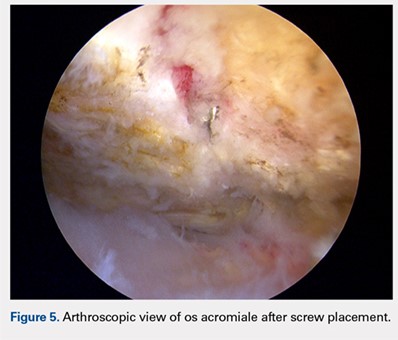
We then elected to repair the unstable fibrous os acromiale (Figures 4A-4D). The fibrous nonunion was first debrided to bleeding bone with a 4.0-mm round burr aligned with the os using the direct lateral portal (Smith & Nephew Endoscopy). Through the anterior portal, two AcutrakTM guide wires (Acumed) were placed under arthroscopic visualization from the anterior margin of the acromion, across the os site, and into the posterior acromion. A 1-cm counter incision was made at the level of the posterior acromion to allow confirmation of the guide wire position and to permit placement of a large, pointed reduction clamp, used to reduce the mesoacromial fragment to the stable portion of the acromion. The calibrated, cannulated drill bit was passed over each guide wire to a depth of 34 mm, according to standard technique, and viewed arthroscopically from the subacromial space. Two 34-mm AcutrakTM cannulated headless compression screws (Acumed) were then placed across the defect. Direct arthroscopic visualization confirmed reduction and complete intraosseous placement of the screws (Figure 5). Screw position was also assessed with image intensification. Fluoroscopic views showed the repair to be stable when the shoulder was taken through range of motion. The os site was never exposed directly through an incision. The surgery was performed on an outpatient basis.
POSTOPERATIVE COURSE
The patient was maintained in a sling and small abduction pillow (Ultrasling IIITM, DonJoy). She was kept non-weight-bearing but was permitted unrestricted motion through the elbow, wrist, and hand for the first 6 weeks. She was permitted supine passive external rotation of the shoulder to 30° and forward flexion to 45° for the first 2 weeks, and 90° through 6 weeks. At her initial postoperative visit 2 weeks later, she noted minimal pain in the shoulder, much improved from her preoperative pain. She was no longer taking any pain medicine, including NSAIDs. Radiographs showed no change in fixation.
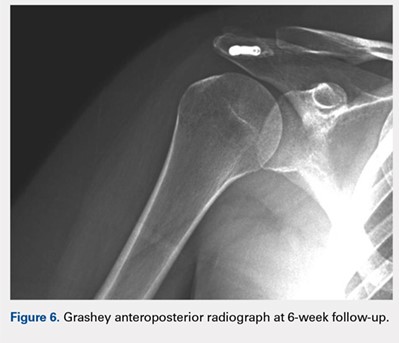
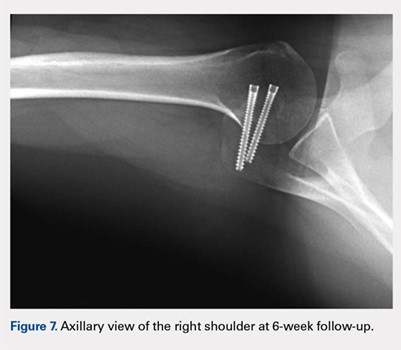
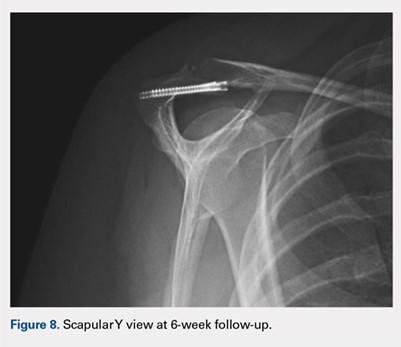
At her second visit (6 weeks), she was completely pain free. Clinical examination showed no tenderness at the acromion, healed incisions, and pain-free passive ROM. Radiographs demonstrated early evidence of consolidation and no sign of fixation failure (Figures 6-8). Her Single Assessment Numeric Evaluation (SANE) score was 85%, and her Simple Shoulder Test (SST) score was 3/12. She was permitted to discontinue the sling, to begin using the arm actively at the side, and progress with unloaded use above shoulder height over the next 6 weeks.
She was seen in follow-up at 4 months, where she was found to have no pain but had not yet returned to sports. At her 6-month follow-up, she showed continued improvement with no limitation of activity. At 1-year follow-up, her SANE score improved from 85% at 6 weeks postoperatively to 100%, and her SST improved from 3/12 at 6 weeks to 12/12. She demonstrated full function of her shoulder with no evidence of hardware loosening. At that time, her os acromiale had completely fused radiographically.
Continue to: DISCUSSION...
DISCUSSION
A variety of methods for the management of os acromiale have been described in the literature. Internal fixation is reported as the most common surgical technique, followed by excision and acromioplasty.6 Surgical fixation with cannulated screws is effective at achieving radiographic union.5,9,12,13,15
Excision is also an option in cases where there is a symptomatic pre-acromion with a relatively small fragment. In the case of a larger fragment, techniques that preserve the vascularity of the os acromiale appear more likely to be successful than excision.17 While excision can be performed arthroscopically to preserve the blood supply, a recent report showed that 35% of patients still had residual pain.18 Another study suggests that protecting the vascular supply with an arthroscopic technique would be a better option to promote healing to union.19
Given that removal of symptomatic internal fixation hardware is significantly more common after Kirschner wire fixation (88%; 43/49) than after cannulated screw fixation (38%; 9/24),6 and given that significant hardware complications can arise from screw tips,16 we chose headless, cannulated Acutrak compression screws for arthroscopic-assisted fixation. Performing the operation arthroscopically minimized soft-tissue violation, allowing us to directly visualize the reduction and also allowing confirmation that the screws were not at risk for impingement of the rotator cuff. The tapered nature of the Acutrak screws allowed for excellent compression at the reduction site without a prominent screw head.
CONCLUSION
Arthroscopic management of the symptomatic os acromiale has been documented in the literature. Cannulated screw fixation has shown to lead to a higher rate of radiographic union than Kirschner wire fixation. Arthroscopically guided placement of headless, cannulated compression screw fixation may be a viable repair alternative in the management of the symptomatic os acromiale with less concern for symptomatic hardware.6,20-27
1. Barbier O, Block D, Dezaly C, Sirveaux F, Mole D. Os acromiale, a cause of shoulder pain, not to be overlooked. Orthop Traumatol Surg Res. 2013;99(4):465-472. doi: 10.1016/j.otsr.2012.10.020.
2. Swain RA, Wilson FD, Harsha DM. The os acromiale: another cause of impingement. Med Sci Sports Exerc. 1996;28(12):1459-1462. doi:10.1097/00005768-199612000-00003.
3. Kurtz CA, Humble BJ, Rodosky MW, Sekiya JK. Symptomatic os acromiale. J Am Acad Orthop Surg. 2006;14(1):12-19. doi:10.5435/00124635-200601000-00004.
4. Buss DD, Freehill MQ, Marra G. Typical and atypical shoulder impingement syndrome: diagnosis, treatment, and pitfalls. Instr Course Lect. 2009;58:447-457.
5. Warner JJ, Beim GM, Higgins L. The treatment of symptomatic os acromiale. J Bone Joint Surg Am. 1998;80(9):1320-1326. doi:10.2106/00004623-199809000-00011.
6. Harris JD, Griesser MJ, Jones GL. Systematic review of the surgical treatment for symptomatic os acromiale. Int J Shoulder Surg. 2011;5(1):9-16. doi:10.4103/0973-6042.80461.
7. Abboud JA, Silverberg D, Pepe M, et al. Surgical treatment of os acromiale with and without associated rotator cuff tears. J Shoulder Elbow Surg. 2006;15(3):265-270. doi:10.1016/j.jse.2005.08.024.
8. Boehm TD, Matzer M, Brazda D, Gohlke FE. Os acromiale associated with tear of the rotator cuff treated operatively Review of 33 patients. J Bone Joint Surg Br. 2003;85(4):545-549. doi:10.1302/0301-620X.85B4.13634.
9. Boehm TD, Rolf O, Martetschlaeger F, Kenn W, Gohlke F. Rotator cuff tears associated with os acromiale. Acta Orthop. 2005;76(2):241-244. doi:10.1080/00016470510030643.
10. Barbiera F, Bellissima G, Iovane A, De Maria M. OS acromiale producing rotator cuff impingement and rupture. A case report. Radiol Med. 2002;104(4):359-362.
11. Neer CS 2nd. Rotator cuff tears associated with os acromiale. J Bone Joint Surg Am. 1984;66(8):1320-1321.
12. Hertel R, Windisch W, Schuster A, Ballmer FT. Transacromial approach to obtain fusion of unstable os acromiale. J Shoulder Elbow Surg. 1998;7(6):606-609. doi:10.1016/S1058-2746(98)90008-8.
13. Ozbaydar MU, Keriş I, Altun M, Yalaman O. Results of the surgical treatment for symptomatic mesoacromion. Acta Orthop Traumatol Turc. 2006;40(2):123-129.
14. Satterlee CC. Successful osteosynthesis of an unstable mesoacromion in 6 shoulders: a new technique. J Shoulder Elbow Surg. 1999;8(2):125-129. doi:10.1016/S1058-2746(99)90004-6.
15. Ryu RK, Fan RS, Dunbar WHt. The treatment of symptomatic os acromiale. Orthopedics. 1999;22(3):325-328.
16. Atoun E, van Tongel A, Narvani A, Rath E, Sforza G, Levy O. Arthroscopically assisted internal fixation of the symptomatic unstable os acromiale with absorbable screws. J Shoulder Elbow Surg. 2012;21(12):1740-1745. doi:10.1016/j.jse.2011.12.011.
17. Johnston PS, Paxton ES, Gordon V, Kraeutler MJ, Abboud JA, Williams GR. Os acromiale: a review and an introduction of a new surgical technique for management. Orthop Clin North Am. 2013;44(4):635-644. doi:10.1016/j.ocl.2013.06.015.
18. Campbell PT, Nizlan NM, Skirving AP. Arthroscopic excision of os acromiale: effects on deltoid function and strength. Orthopedics. 2012;35(11):e1601-e1605. doi:10.3928/01477447-20121023-16.
19. Yepes H, Al-Hibshi A, Tang M, Morris SF, Stanish WD. Vascular anatomy of the subacromial space: a map of bleeding points for the arthroscopic surgeon. Arthroscopy. 2007;23(9):978-984. doi:10.1016/j.arthro.2007.03.093.
20. Kummer FJ, Van Gelderen J, Meislin RJ. Two-screw, arthroscopic fixation of os acromiale compared to a similar, open procedure incorporating a tension band: a laboratory study. Shoulder Elbow. 2011;3(2):85-87. doi:10.1111/j.1758-5740.2011.00115.x.
21. Wright RW, Heller MA, Quick DC, Buss DD. Arthroscopic decompression for impingement syndrome secondary to an unstable os acromiale. Arthroscopy. 2000;16(6):595-599. doi:10.1053/jars.2000.9239.
22. Edelson JG, Zuckerman J, Hershkovitz I. Os acromiale: anatomy and surgical implications. J Bone Joint Surg Br. 1993;75(4):551-555. doi:10.1302/0301-620X.75B4.8331108.
23. Fery A, Sommelet J. Os acromiale: significance--diagnosis--pathology Apropos of 28 cases including 2 with fracture separation. Rev Chir Orthop Reparatrice Appar Mot. 1988;74(2):160-172.
24. Lee DH. The double-density sign: a radiographic finding suggestive of an os acromiale. J Bone Joint Surg Am. 2004;86-A(12):2666-2670. doi:10.2106/00004623-200412000-00012.
25. Ortiguera CJ, Buss DD. Surgical management of the symptomatic os acromiale. J Shoulder Elbow Surg. 2002;11(5):521-528. doi:10.1067/mse.2002.122227.
26. Peckett WR, Gunther SB, Harper GD, Hughes JS, Sonnabend DH. Internal fixation of symptomatic os acromiale: a series of twenty-six cases. J Shoulder Elbow Surg. 2004;13(4):381-385. doi:10.1016/S1058274604000400.
27. Sahajpal D, Strauss EJ, Ishak C, Keyes JM, Joseph G, Jazrawi LM. Surgical management of os acromiale: a case report and review of the literature. Bull NYU Hosp Jt Dis. 2007;65(4):312-316.
ABSTRACT
Os acromiale is a failure of fusion between 1 or more ossification centers of the scapula and the acromion process. Pain can be caused by motion and impingement of the unfused segment. Several methods for the management of os acromiale have been described. Internal fixation is the most common surgical technique, followed by excision and acromioplasty. We present a novel technique for treatment of symptomatic os acromiale using arthroscopically-guided headless compression screws. This is a viable technique in the management of symptomatic os acromiale due to preservation of the periosteal blood supply and less concern for symptomatic hardware.
Continue to: Os acromiale results from a failure of...
Os acromiale results from a failure of fusion between 1 or more ossification centers and the acromion process.1 The acromion consists of 4 different ossification centers, which appear by 14 years of age and fuse by age 25 years. The 4 ossification centers are the basi-acromion, meta-acromion, mesoacromion, and pre-acromion (Figure 1). Formation of an os acromiale occurs most often due to failure of fusion between the meta-acromion and mesoacromion. Os acromiale appears to occur in approximately 8% of the population, according to cadaveric studies.2 This anatomic variant occurs more commonly in African-Americans than Caucasians, and shows a preponderance for males over females.3
Plain radiographs are usually adequate for diagnosis. Axillary views are most sensitive for detection, which can be difficult to see on anteroposterior radiographs.4 In os acromiale, the unfused segment is connected to the acromioclavicular joint and the coracoid, which can lead to motion of the segment and impingement of the rotator cuff.2-4 Patients frequently experience localized tenderness and symptomatic pain with signs and symptoms of impingement. Rotator cuff tears may occur secondary to chronic impingement.5
Various forms of repair have been described. A recent meta-analysis showed that internal fixation (60%) was the most common surgical technique reported, followed by excision (27%) and acromioplasty (13%).6 Rotator cuff repair is a common concurrent surgical procedure.7-11 The available literature favors internal fixation through an open technique with or without bone grafting.5,7,8,12-15 Various forms of fixation have been presented in the literature, including Kirschner wire fixation, cannulated screw fixation alone, cannulated screw fixation with FiberWire Suture (Arthrex), and cannulated screw fixation with a stainless steel wire tension band technique. Based on the results of the meta-analysis, surgical fixation with cannulated screws has been shown to lead to a significantly greater rate of radiographic healing (23/24 patients) compared to Kirschner wire fixation (31/49 patients).6 Further, radiographic healing is significantly associated with improved clinical outcomes.12 Removal of symptomatic internal fixation hardware is significantly more common after Kirschner wire fixation cases (88%; 43/49) compared to cannulated screw fixation cases (38%; 9/24). However, hardware issues may also be encountered with screw fixation, with 1 case series reporting a 25% rate of hardware complication.16 The patient provided written informed consent for print and electronic publication of this case report.
CASE REPORT
The patient is a 19-year-old right-hand-dominant woman who injured her right shoulder while diving into the bleachers during a volleyball game 4 years prior to presentation. She suffered a direct blow to her shoulder and immediately became symptomatic. She underwent a long period of nonoperative management, which included physical therapy, strengthening, nonsteroidal anti-inflammatory drug (NSAID) therapy, and narcotic pain medications. Her primary complaints upon presentation were pain with lifting, as well as mechanical symptoms. On examination, the patient had moderate tenderness directly over the acromion. She also had evidence of mild impingement symptoms. Plain radiographs revealed a mesoacromial-type os acromiale clearly seen on the axillary lateral film (Figure 2). She underwent magnetic resonance imaging, which suggested rotator cuff tendinosis and evidence of edema at the os acromiale site. She underwent a diagnostic injection directly into the site of maximal tenderness at the os, which provided complete transient relief of her pain. Despite the transient pain relief, the patient continued to be symptomatic after the local anesthetic effect wore off. Surgical options were then discussed with the patient.
Continue to: SURGICAL TECHNIQUE...
SURGICAL TECHNIQUE
A standard diagnostic shoulder arthroscopy was performed using anterior, posterior and direct lateral portals. The rotator cuff was evaluated, and no evidence of a tear was found. The undersurface of the acromion was exposed, and the os acromiale was identified arthroscopically (Figure 3). This was found to be unstable under direct digital pressure.
We then elected to repair the unstable fibrous os acromiale (Figures 4A-4D). The fibrous nonunion was first debrided to bleeding bone with a 4.0-mm round burr aligned with the os using the direct lateral portal (Smith & Nephew Endoscopy). Through the anterior portal, two AcutrakTM guide wires (Acumed) were placed under arthroscopic visualization from the anterior margin of the acromion, across the os site, and into the posterior acromion. A 1-cm counter incision was made at the level of the posterior acromion to allow confirmation of the guide wire position and to permit placement of a large, pointed reduction clamp, used to reduce the mesoacromial fragment to the stable portion of the acromion. The calibrated, cannulated drill bit was passed over each guide wire to a depth of 34 mm, according to standard technique, and viewed arthroscopically from the subacromial space. Two 34-mm AcutrakTM cannulated headless compression screws (Acumed) were then placed across the defect. Direct arthroscopic visualization confirmed reduction and complete intraosseous placement of the screws (Figure 5). Screw position was also assessed with image intensification. Fluoroscopic views showed the repair to be stable when the shoulder was taken through range of motion. The os site was never exposed directly through an incision. The surgery was performed on an outpatient basis.
POSTOPERATIVE COURSE
The patient was maintained in a sling and small abduction pillow (Ultrasling IIITM, DonJoy). She was kept non-weight-bearing but was permitted unrestricted motion through the elbow, wrist, and hand for the first 6 weeks. She was permitted supine passive external rotation of the shoulder to 30° and forward flexion to 45° for the first 2 weeks, and 90° through 6 weeks. At her initial postoperative visit 2 weeks later, she noted minimal pain in the shoulder, much improved from her preoperative pain. She was no longer taking any pain medicine, including NSAIDs. Radiographs showed no change in fixation.
At her second visit (6 weeks), she was completely pain free. Clinical examination showed no tenderness at the acromion, healed incisions, and pain-free passive ROM. Radiographs demonstrated early evidence of consolidation and no sign of fixation failure (Figures 6-8). Her Single Assessment Numeric Evaluation (SANE) score was 85%, and her Simple Shoulder Test (SST) score was 3/12. She was permitted to discontinue the sling, to begin using the arm actively at the side, and progress with unloaded use above shoulder height over the next 6 weeks.
She was seen in follow-up at 4 months, where she was found to have no pain but had not yet returned to sports. At her 6-month follow-up, she showed continued improvement with no limitation of activity. At 1-year follow-up, her SANE score improved from 85% at 6 weeks postoperatively to 100%, and her SST improved from 3/12 at 6 weeks to 12/12. She demonstrated full function of her shoulder with no evidence of hardware loosening. At that time, her os acromiale had completely fused radiographically.
Continue to: DISCUSSION...
DISCUSSION
A variety of methods for the management of os acromiale have been described in the literature. Internal fixation is reported as the most common surgical technique, followed by excision and acromioplasty.6 Surgical fixation with cannulated screws is effective at achieving radiographic union.5,9,12,13,15
Excision is also an option in cases where there is a symptomatic pre-acromion with a relatively small fragment. In the case of a larger fragment, techniques that preserve the vascularity of the os acromiale appear more likely to be successful than excision.17 While excision can be performed arthroscopically to preserve the blood supply, a recent report showed that 35% of patients still had residual pain.18 Another study suggests that protecting the vascular supply with an arthroscopic technique would be a better option to promote healing to union.19
Given that removal of symptomatic internal fixation hardware is significantly more common after Kirschner wire fixation (88%; 43/49) than after cannulated screw fixation (38%; 9/24),6 and given that significant hardware complications can arise from screw tips,16 we chose headless, cannulated Acutrak compression screws for arthroscopic-assisted fixation. Performing the operation arthroscopically minimized soft-tissue violation, allowing us to directly visualize the reduction and also allowing confirmation that the screws were not at risk for impingement of the rotator cuff. The tapered nature of the Acutrak screws allowed for excellent compression at the reduction site without a prominent screw head.
CONCLUSION
Arthroscopic management of the symptomatic os acromiale has been documented in the literature. Cannulated screw fixation has shown to lead to a higher rate of radiographic union than Kirschner wire fixation. Arthroscopically guided placement of headless, cannulated compression screw fixation may be a viable repair alternative in the management of the symptomatic os acromiale with less concern for symptomatic hardware.6,20-27
ABSTRACT
Os acromiale is a failure of fusion between 1 or more ossification centers of the scapula and the acromion process. Pain can be caused by motion and impingement of the unfused segment. Several methods for the management of os acromiale have been described. Internal fixation is the most common surgical technique, followed by excision and acromioplasty. We present a novel technique for treatment of symptomatic os acromiale using arthroscopically-guided headless compression screws. This is a viable technique in the management of symptomatic os acromiale due to preservation of the periosteal blood supply and less concern for symptomatic hardware.
Continue to: Os acromiale results from a failure of...
Os acromiale results from a failure of fusion between 1 or more ossification centers and the acromion process.1 The acromion consists of 4 different ossification centers, which appear by 14 years of age and fuse by age 25 years. The 4 ossification centers are the basi-acromion, meta-acromion, mesoacromion, and pre-acromion (Figure 1). Formation of an os acromiale occurs most often due to failure of fusion between the meta-acromion and mesoacromion. Os acromiale appears to occur in approximately 8% of the population, according to cadaveric studies.2 This anatomic variant occurs more commonly in African-Americans than Caucasians, and shows a preponderance for males over females.3
Plain radiographs are usually adequate for diagnosis. Axillary views are most sensitive for detection, which can be difficult to see on anteroposterior radiographs.4 In os acromiale, the unfused segment is connected to the acromioclavicular joint and the coracoid, which can lead to motion of the segment and impingement of the rotator cuff.2-4 Patients frequently experience localized tenderness and symptomatic pain with signs and symptoms of impingement. Rotator cuff tears may occur secondary to chronic impingement.5
Various forms of repair have been described. A recent meta-analysis showed that internal fixation (60%) was the most common surgical technique reported, followed by excision (27%) and acromioplasty (13%).6 Rotator cuff repair is a common concurrent surgical procedure.7-11 The available literature favors internal fixation through an open technique with or without bone grafting.5,7,8,12-15 Various forms of fixation have been presented in the literature, including Kirschner wire fixation, cannulated screw fixation alone, cannulated screw fixation with FiberWire Suture (Arthrex), and cannulated screw fixation with a stainless steel wire tension band technique. Based on the results of the meta-analysis, surgical fixation with cannulated screws has been shown to lead to a significantly greater rate of radiographic healing (23/24 patients) compared to Kirschner wire fixation (31/49 patients).6 Further, radiographic healing is significantly associated with improved clinical outcomes.12 Removal of symptomatic internal fixation hardware is significantly more common after Kirschner wire fixation cases (88%; 43/49) compared to cannulated screw fixation cases (38%; 9/24). However, hardware issues may also be encountered with screw fixation, with 1 case series reporting a 25% rate of hardware complication.16 The patient provided written informed consent for print and electronic publication of this case report.
CASE REPORT
The patient is a 19-year-old right-hand-dominant woman who injured her right shoulder while diving into the bleachers during a volleyball game 4 years prior to presentation. She suffered a direct blow to her shoulder and immediately became symptomatic. She underwent a long period of nonoperative management, which included physical therapy, strengthening, nonsteroidal anti-inflammatory drug (NSAID) therapy, and narcotic pain medications. Her primary complaints upon presentation were pain with lifting, as well as mechanical symptoms. On examination, the patient had moderate tenderness directly over the acromion. She also had evidence of mild impingement symptoms. Plain radiographs revealed a mesoacromial-type os acromiale clearly seen on the axillary lateral film (Figure 2). She underwent magnetic resonance imaging, which suggested rotator cuff tendinosis and evidence of edema at the os acromiale site. She underwent a diagnostic injection directly into the site of maximal tenderness at the os, which provided complete transient relief of her pain. Despite the transient pain relief, the patient continued to be symptomatic after the local anesthetic effect wore off. Surgical options were then discussed with the patient.
Continue to: SURGICAL TECHNIQUE...
SURGICAL TECHNIQUE
A standard diagnostic shoulder arthroscopy was performed using anterior, posterior and direct lateral portals. The rotator cuff was evaluated, and no evidence of a tear was found. The undersurface of the acromion was exposed, and the os acromiale was identified arthroscopically (Figure 3). This was found to be unstable under direct digital pressure.
We then elected to repair the unstable fibrous os acromiale (Figures 4A-4D). The fibrous nonunion was first debrided to bleeding bone with a 4.0-mm round burr aligned with the os using the direct lateral portal (Smith & Nephew Endoscopy). Through the anterior portal, two AcutrakTM guide wires (Acumed) were placed under arthroscopic visualization from the anterior margin of the acromion, across the os site, and into the posterior acromion. A 1-cm counter incision was made at the level of the posterior acromion to allow confirmation of the guide wire position and to permit placement of a large, pointed reduction clamp, used to reduce the mesoacromial fragment to the stable portion of the acromion. The calibrated, cannulated drill bit was passed over each guide wire to a depth of 34 mm, according to standard technique, and viewed arthroscopically from the subacromial space. Two 34-mm AcutrakTM cannulated headless compression screws (Acumed) were then placed across the defect. Direct arthroscopic visualization confirmed reduction and complete intraosseous placement of the screws (Figure 5). Screw position was also assessed with image intensification. Fluoroscopic views showed the repair to be stable when the shoulder was taken through range of motion. The os site was never exposed directly through an incision. The surgery was performed on an outpatient basis.
POSTOPERATIVE COURSE
The patient was maintained in a sling and small abduction pillow (Ultrasling IIITM, DonJoy). She was kept non-weight-bearing but was permitted unrestricted motion through the elbow, wrist, and hand for the first 6 weeks. She was permitted supine passive external rotation of the shoulder to 30° and forward flexion to 45° for the first 2 weeks, and 90° through 6 weeks. At her initial postoperative visit 2 weeks later, she noted minimal pain in the shoulder, much improved from her preoperative pain. She was no longer taking any pain medicine, including NSAIDs. Radiographs showed no change in fixation.
At her second visit (6 weeks), she was completely pain free. Clinical examination showed no tenderness at the acromion, healed incisions, and pain-free passive ROM. Radiographs demonstrated early evidence of consolidation and no sign of fixation failure (Figures 6-8). Her Single Assessment Numeric Evaluation (SANE) score was 85%, and her Simple Shoulder Test (SST) score was 3/12. She was permitted to discontinue the sling, to begin using the arm actively at the side, and progress with unloaded use above shoulder height over the next 6 weeks.
She was seen in follow-up at 4 months, where she was found to have no pain but had not yet returned to sports. At her 6-month follow-up, she showed continued improvement with no limitation of activity. At 1-year follow-up, her SANE score improved from 85% at 6 weeks postoperatively to 100%, and her SST improved from 3/12 at 6 weeks to 12/12. She demonstrated full function of her shoulder with no evidence of hardware loosening. At that time, her os acromiale had completely fused radiographically.
Continue to: DISCUSSION...
DISCUSSION
A variety of methods for the management of os acromiale have been described in the literature. Internal fixation is reported as the most common surgical technique, followed by excision and acromioplasty.6 Surgical fixation with cannulated screws is effective at achieving radiographic union.5,9,12,13,15
Excision is also an option in cases where there is a symptomatic pre-acromion with a relatively small fragment. In the case of a larger fragment, techniques that preserve the vascularity of the os acromiale appear more likely to be successful than excision.17 While excision can be performed arthroscopically to preserve the blood supply, a recent report showed that 35% of patients still had residual pain.18 Another study suggests that protecting the vascular supply with an arthroscopic technique would be a better option to promote healing to union.19
Given that removal of symptomatic internal fixation hardware is significantly more common after Kirschner wire fixation (88%; 43/49) than after cannulated screw fixation (38%; 9/24),6 and given that significant hardware complications can arise from screw tips,16 we chose headless, cannulated Acutrak compression screws for arthroscopic-assisted fixation. Performing the operation arthroscopically minimized soft-tissue violation, allowing us to directly visualize the reduction and also allowing confirmation that the screws were not at risk for impingement of the rotator cuff. The tapered nature of the Acutrak screws allowed for excellent compression at the reduction site without a prominent screw head.
CONCLUSION
Arthroscopic management of the symptomatic os acromiale has been documented in the literature. Cannulated screw fixation has shown to lead to a higher rate of radiographic union than Kirschner wire fixation. Arthroscopically guided placement of headless, cannulated compression screw fixation may be a viable repair alternative in the management of the symptomatic os acromiale with less concern for symptomatic hardware.6,20-27
1. Barbier O, Block D, Dezaly C, Sirveaux F, Mole D. Os acromiale, a cause of shoulder pain, not to be overlooked. Orthop Traumatol Surg Res. 2013;99(4):465-472. doi: 10.1016/j.otsr.2012.10.020.
2. Swain RA, Wilson FD, Harsha DM. The os acromiale: another cause of impingement. Med Sci Sports Exerc. 1996;28(12):1459-1462. doi:10.1097/00005768-199612000-00003.
3. Kurtz CA, Humble BJ, Rodosky MW, Sekiya JK. Symptomatic os acromiale. J Am Acad Orthop Surg. 2006;14(1):12-19. doi:10.5435/00124635-200601000-00004.
4. Buss DD, Freehill MQ, Marra G. Typical and atypical shoulder impingement syndrome: diagnosis, treatment, and pitfalls. Instr Course Lect. 2009;58:447-457.
5. Warner JJ, Beim GM, Higgins L. The treatment of symptomatic os acromiale. J Bone Joint Surg Am. 1998;80(9):1320-1326. doi:10.2106/00004623-199809000-00011.
6. Harris JD, Griesser MJ, Jones GL. Systematic review of the surgical treatment for symptomatic os acromiale. Int J Shoulder Surg. 2011;5(1):9-16. doi:10.4103/0973-6042.80461.
7. Abboud JA, Silverberg D, Pepe M, et al. Surgical treatment of os acromiale with and without associated rotator cuff tears. J Shoulder Elbow Surg. 2006;15(3):265-270. doi:10.1016/j.jse.2005.08.024.
8. Boehm TD, Matzer M, Brazda D, Gohlke FE. Os acromiale associated with tear of the rotator cuff treated operatively Review of 33 patients. J Bone Joint Surg Br. 2003;85(4):545-549. doi:10.1302/0301-620X.85B4.13634.
9. Boehm TD, Rolf O, Martetschlaeger F, Kenn W, Gohlke F. Rotator cuff tears associated with os acromiale. Acta Orthop. 2005;76(2):241-244. doi:10.1080/00016470510030643.
10. Barbiera F, Bellissima G, Iovane A, De Maria M. OS acromiale producing rotator cuff impingement and rupture. A case report. Radiol Med. 2002;104(4):359-362.
11. Neer CS 2nd. Rotator cuff tears associated with os acromiale. J Bone Joint Surg Am. 1984;66(8):1320-1321.
12. Hertel R, Windisch W, Schuster A, Ballmer FT. Transacromial approach to obtain fusion of unstable os acromiale. J Shoulder Elbow Surg. 1998;7(6):606-609. doi:10.1016/S1058-2746(98)90008-8.
13. Ozbaydar MU, Keriş I, Altun M, Yalaman O. Results of the surgical treatment for symptomatic mesoacromion. Acta Orthop Traumatol Turc. 2006;40(2):123-129.
14. Satterlee CC. Successful osteosynthesis of an unstable mesoacromion in 6 shoulders: a new technique. J Shoulder Elbow Surg. 1999;8(2):125-129. doi:10.1016/S1058-2746(99)90004-6.
15. Ryu RK, Fan RS, Dunbar WHt. The treatment of symptomatic os acromiale. Orthopedics. 1999;22(3):325-328.
16. Atoun E, van Tongel A, Narvani A, Rath E, Sforza G, Levy O. Arthroscopically assisted internal fixation of the symptomatic unstable os acromiale with absorbable screws. J Shoulder Elbow Surg. 2012;21(12):1740-1745. doi:10.1016/j.jse.2011.12.011.
17. Johnston PS, Paxton ES, Gordon V, Kraeutler MJ, Abboud JA, Williams GR. Os acromiale: a review and an introduction of a new surgical technique for management. Orthop Clin North Am. 2013;44(4):635-644. doi:10.1016/j.ocl.2013.06.015.
18. Campbell PT, Nizlan NM, Skirving AP. Arthroscopic excision of os acromiale: effects on deltoid function and strength. Orthopedics. 2012;35(11):e1601-e1605. doi:10.3928/01477447-20121023-16.
19. Yepes H, Al-Hibshi A, Tang M, Morris SF, Stanish WD. Vascular anatomy of the subacromial space: a map of bleeding points for the arthroscopic surgeon. Arthroscopy. 2007;23(9):978-984. doi:10.1016/j.arthro.2007.03.093.
20. Kummer FJ, Van Gelderen J, Meislin RJ. Two-screw, arthroscopic fixation of os acromiale compared to a similar, open procedure incorporating a tension band: a laboratory study. Shoulder Elbow. 2011;3(2):85-87. doi:10.1111/j.1758-5740.2011.00115.x.
21. Wright RW, Heller MA, Quick DC, Buss DD. Arthroscopic decompression for impingement syndrome secondary to an unstable os acromiale. Arthroscopy. 2000;16(6):595-599. doi:10.1053/jars.2000.9239.
22. Edelson JG, Zuckerman J, Hershkovitz I. Os acromiale: anatomy and surgical implications. J Bone Joint Surg Br. 1993;75(4):551-555. doi:10.1302/0301-620X.75B4.8331108.
23. Fery A, Sommelet J. Os acromiale: significance--diagnosis--pathology Apropos of 28 cases including 2 with fracture separation. Rev Chir Orthop Reparatrice Appar Mot. 1988;74(2):160-172.
24. Lee DH. The double-density sign: a radiographic finding suggestive of an os acromiale. J Bone Joint Surg Am. 2004;86-A(12):2666-2670. doi:10.2106/00004623-200412000-00012.
25. Ortiguera CJ, Buss DD. Surgical management of the symptomatic os acromiale. J Shoulder Elbow Surg. 2002;11(5):521-528. doi:10.1067/mse.2002.122227.
26. Peckett WR, Gunther SB, Harper GD, Hughes JS, Sonnabend DH. Internal fixation of symptomatic os acromiale: a series of twenty-six cases. J Shoulder Elbow Surg. 2004;13(4):381-385. doi:10.1016/S1058274604000400.
27. Sahajpal D, Strauss EJ, Ishak C, Keyes JM, Joseph G, Jazrawi LM. Surgical management of os acromiale: a case report and review of the literature. Bull NYU Hosp Jt Dis. 2007;65(4):312-316.
1. Barbier O, Block D, Dezaly C, Sirveaux F, Mole D. Os acromiale, a cause of shoulder pain, not to be overlooked. Orthop Traumatol Surg Res. 2013;99(4):465-472. doi: 10.1016/j.otsr.2012.10.020.
2. Swain RA, Wilson FD, Harsha DM. The os acromiale: another cause of impingement. Med Sci Sports Exerc. 1996;28(12):1459-1462. doi:10.1097/00005768-199612000-00003.
3. Kurtz CA, Humble BJ, Rodosky MW, Sekiya JK. Symptomatic os acromiale. J Am Acad Orthop Surg. 2006;14(1):12-19. doi:10.5435/00124635-200601000-00004.
4. Buss DD, Freehill MQ, Marra G. Typical and atypical shoulder impingement syndrome: diagnosis, treatment, and pitfalls. Instr Course Lect. 2009;58:447-457.
5. Warner JJ, Beim GM, Higgins L. The treatment of symptomatic os acromiale. J Bone Joint Surg Am. 1998;80(9):1320-1326. doi:10.2106/00004623-199809000-00011.
6. Harris JD, Griesser MJ, Jones GL. Systematic review of the surgical treatment for symptomatic os acromiale. Int J Shoulder Surg. 2011;5(1):9-16. doi:10.4103/0973-6042.80461.
7. Abboud JA, Silverberg D, Pepe M, et al. Surgical treatment of os acromiale with and without associated rotator cuff tears. J Shoulder Elbow Surg. 2006;15(3):265-270. doi:10.1016/j.jse.2005.08.024.
8. Boehm TD, Matzer M, Brazda D, Gohlke FE. Os acromiale associated with tear of the rotator cuff treated operatively Review of 33 patients. J Bone Joint Surg Br. 2003;85(4):545-549. doi:10.1302/0301-620X.85B4.13634.
9. Boehm TD, Rolf O, Martetschlaeger F, Kenn W, Gohlke F. Rotator cuff tears associated with os acromiale. Acta Orthop. 2005;76(2):241-244. doi:10.1080/00016470510030643.
10. Barbiera F, Bellissima G, Iovane A, De Maria M. OS acromiale producing rotator cuff impingement and rupture. A case report. Radiol Med. 2002;104(4):359-362.
11. Neer CS 2nd. Rotator cuff tears associated with os acromiale. J Bone Joint Surg Am. 1984;66(8):1320-1321.
12. Hertel R, Windisch W, Schuster A, Ballmer FT. Transacromial approach to obtain fusion of unstable os acromiale. J Shoulder Elbow Surg. 1998;7(6):606-609. doi:10.1016/S1058-2746(98)90008-8.
13. Ozbaydar MU, Keriş I, Altun M, Yalaman O. Results of the surgical treatment for symptomatic mesoacromion. Acta Orthop Traumatol Turc. 2006;40(2):123-129.
14. Satterlee CC. Successful osteosynthesis of an unstable mesoacromion in 6 shoulders: a new technique. J Shoulder Elbow Surg. 1999;8(2):125-129. doi:10.1016/S1058-2746(99)90004-6.
15. Ryu RK, Fan RS, Dunbar WHt. The treatment of symptomatic os acromiale. Orthopedics. 1999;22(3):325-328.
16. Atoun E, van Tongel A, Narvani A, Rath E, Sforza G, Levy O. Arthroscopically assisted internal fixation of the symptomatic unstable os acromiale with absorbable screws. J Shoulder Elbow Surg. 2012;21(12):1740-1745. doi:10.1016/j.jse.2011.12.011.
17. Johnston PS, Paxton ES, Gordon V, Kraeutler MJ, Abboud JA, Williams GR. Os acromiale: a review and an introduction of a new surgical technique for management. Orthop Clin North Am. 2013;44(4):635-644. doi:10.1016/j.ocl.2013.06.015.
18. Campbell PT, Nizlan NM, Skirving AP. Arthroscopic excision of os acromiale: effects on deltoid function and strength. Orthopedics. 2012;35(11):e1601-e1605. doi:10.3928/01477447-20121023-16.
19. Yepes H, Al-Hibshi A, Tang M, Morris SF, Stanish WD. Vascular anatomy of the subacromial space: a map of bleeding points for the arthroscopic surgeon. Arthroscopy. 2007;23(9):978-984. doi:10.1016/j.arthro.2007.03.093.
20. Kummer FJ, Van Gelderen J, Meislin RJ. Two-screw, arthroscopic fixation of os acromiale compared to a similar, open procedure incorporating a tension band: a laboratory study. Shoulder Elbow. 2011;3(2):85-87. doi:10.1111/j.1758-5740.2011.00115.x.
21. Wright RW, Heller MA, Quick DC, Buss DD. Arthroscopic decompression for impingement syndrome secondary to an unstable os acromiale. Arthroscopy. 2000;16(6):595-599. doi:10.1053/jars.2000.9239.
22. Edelson JG, Zuckerman J, Hershkovitz I. Os acromiale: anatomy and surgical implications. J Bone Joint Surg Br. 1993;75(4):551-555. doi:10.1302/0301-620X.75B4.8331108.
23. Fery A, Sommelet J. Os acromiale: significance--diagnosis--pathology Apropos of 28 cases including 2 with fracture separation. Rev Chir Orthop Reparatrice Appar Mot. 1988;74(2):160-172.
24. Lee DH. The double-density sign: a radiographic finding suggestive of an os acromiale. J Bone Joint Surg Am. 2004;86-A(12):2666-2670. doi:10.2106/00004623-200412000-00012.
25. Ortiguera CJ, Buss DD. Surgical management of the symptomatic os acromiale. J Shoulder Elbow Surg. 2002;11(5):521-528. doi:10.1067/mse.2002.122227.
26. Peckett WR, Gunther SB, Harper GD, Hughes JS, Sonnabend DH. Internal fixation of symptomatic os acromiale: a series of twenty-six cases. J Shoulder Elbow Surg. 2004;13(4):381-385. doi:10.1016/S1058274604000400.
27. Sahajpal D, Strauss EJ, Ishak C, Keyes JM, Joseph G, Jazrawi LM. Surgical management of os acromiale: a case report and review of the literature. Bull NYU Hosp Jt Dis. 2007;65(4):312-316.
TAKE-HOME POINTS
- Os acromiale is a failure of acromial ossification centers to fuse, and occurs in 8% of the population.
- Symptomatic os acromiale can be treated with repair, or sometimes excision or acromioplasty.
- Repair preserves the anterior deltoid origin and can result in less pain than excision of the fragment.
- Repair of larger fragments can be completed with cannulated screws to reliably achieve union.
- The arthroscope-assisted repair technique described in this article preserves vascularity and can reduce the risk of hardware-related complaints.
CAR T-cell studies dominate ongoing cellular therapy trials
NEW YORK – The cell therapy landscape increasingly involves strategies beyond chimeric antigen receptor (CAR) T-cell therapy, but those studies still predominate among investigational trials, according to Frederick L. Locke, MD, of Moffitt Cancer Center in Tampa.
Researchers are looking at CAR T-cell therapy for earlier lines of treatment, especially in patients with aggressive lymphomas, Dr. Locke said at the annual congress on Hematologic Malignancies held by the National Comprehensive Cancer Network.
Of 753 trials examining cell therapies and listed at ClinicalTrials.gov as of March 30, 2018, about half (404) were CAR T-cell therapies. The others included T-cell receptor therapies, tumor infiltrating lymphocyte therapies, dendritic cell vaccines, and natural killer cell–based therapies, according to an article in Nature Reviews.
“The development isn’t just here in the United States,” Dr. Locke said. “It’s really global. We see a lot of activity in Europe, but also in China. We’re seeing medical advances across the world through molecular biology and gene engineering of T cells and other immune cells which can be adoptively transferred into patients.”
That activity includes studies seeking to move CAR T-cell therapy earlier in the treatment paradigm for some diseases, he added. “CAR T-cell therapy in non-Hodgkin lymphoma is really beginning a paradigm shift, at least in my mind.”
Several large, randomized trials that are now comparing CD19 CAR T-cell therapy with second-line standard-of-care therapies for patients with aggressive B-cell lymphomas. Among those trials is ZUMA-7, a phase 3, randomized trial comparing axicabtagene ciloleucel with standard-of-care treatment in patients with relapsed or refractory diffuse large B-cell lymphoma.
While prognosis remains poor for relapsed or progressing aggressive B-cell lymphomas treated with chemotherapy, data to date suggest CAR T-cell therapy produces durable, long-term remissions in about 40% of patients at “a year out and counting,” Dr. Locke said.
He presented a proposed treatment algorithm that included R-CHOP chemotherapy up front and CAR T-cell therapy in later lines of treatment, an approach that Dr. Locke speculated could result in a cure rate of perhaps 80% in large-cell lymphomas.
Encouraging longer-term data is emerging, with some patients with aggressive T-cell lymphomas now without recurrence for 5 years or more following a single infusion of CAR T-cell therapy, he said.
Dr. Locke reported a financial disclosure related to Cellular Biomedicine Group.
NEW YORK – The cell therapy landscape increasingly involves strategies beyond chimeric antigen receptor (CAR) T-cell therapy, but those studies still predominate among investigational trials, according to Frederick L. Locke, MD, of Moffitt Cancer Center in Tampa.
Researchers are looking at CAR T-cell therapy for earlier lines of treatment, especially in patients with aggressive lymphomas, Dr. Locke said at the annual congress on Hematologic Malignancies held by the National Comprehensive Cancer Network.
Of 753 trials examining cell therapies and listed at ClinicalTrials.gov as of March 30, 2018, about half (404) were CAR T-cell therapies. The others included T-cell receptor therapies, tumor infiltrating lymphocyte therapies, dendritic cell vaccines, and natural killer cell–based therapies, according to an article in Nature Reviews.
“The development isn’t just here in the United States,” Dr. Locke said. “It’s really global. We see a lot of activity in Europe, but also in China. We’re seeing medical advances across the world through molecular biology and gene engineering of T cells and other immune cells which can be adoptively transferred into patients.”
That activity includes studies seeking to move CAR T-cell therapy earlier in the treatment paradigm for some diseases, he added. “CAR T-cell therapy in non-Hodgkin lymphoma is really beginning a paradigm shift, at least in my mind.”
Several large, randomized trials that are now comparing CD19 CAR T-cell therapy with second-line standard-of-care therapies for patients with aggressive B-cell lymphomas. Among those trials is ZUMA-7, a phase 3, randomized trial comparing axicabtagene ciloleucel with standard-of-care treatment in patients with relapsed or refractory diffuse large B-cell lymphoma.
While prognosis remains poor for relapsed or progressing aggressive B-cell lymphomas treated with chemotherapy, data to date suggest CAR T-cell therapy produces durable, long-term remissions in about 40% of patients at “a year out and counting,” Dr. Locke said.
He presented a proposed treatment algorithm that included R-CHOP chemotherapy up front and CAR T-cell therapy in later lines of treatment, an approach that Dr. Locke speculated could result in a cure rate of perhaps 80% in large-cell lymphomas.
Encouraging longer-term data is emerging, with some patients with aggressive T-cell lymphomas now without recurrence for 5 years or more following a single infusion of CAR T-cell therapy, he said.
Dr. Locke reported a financial disclosure related to Cellular Biomedicine Group.
NEW YORK – The cell therapy landscape increasingly involves strategies beyond chimeric antigen receptor (CAR) T-cell therapy, but those studies still predominate among investigational trials, according to Frederick L. Locke, MD, of Moffitt Cancer Center in Tampa.
Researchers are looking at CAR T-cell therapy for earlier lines of treatment, especially in patients with aggressive lymphomas, Dr. Locke said at the annual congress on Hematologic Malignancies held by the National Comprehensive Cancer Network.
Of 753 trials examining cell therapies and listed at ClinicalTrials.gov as of March 30, 2018, about half (404) were CAR T-cell therapies. The others included T-cell receptor therapies, tumor infiltrating lymphocyte therapies, dendritic cell vaccines, and natural killer cell–based therapies, according to an article in Nature Reviews.
“The development isn’t just here in the United States,” Dr. Locke said. “It’s really global. We see a lot of activity in Europe, but also in China. We’re seeing medical advances across the world through molecular biology and gene engineering of T cells and other immune cells which can be adoptively transferred into patients.”
That activity includes studies seeking to move CAR T-cell therapy earlier in the treatment paradigm for some diseases, he added. “CAR T-cell therapy in non-Hodgkin lymphoma is really beginning a paradigm shift, at least in my mind.”
Several large, randomized trials that are now comparing CD19 CAR T-cell therapy with second-line standard-of-care therapies for patients with aggressive B-cell lymphomas. Among those trials is ZUMA-7, a phase 3, randomized trial comparing axicabtagene ciloleucel with standard-of-care treatment in patients with relapsed or refractory diffuse large B-cell lymphoma.
While prognosis remains poor for relapsed or progressing aggressive B-cell lymphomas treated with chemotherapy, data to date suggest CAR T-cell therapy produces durable, long-term remissions in about 40% of patients at “a year out and counting,” Dr. Locke said.
He presented a proposed treatment algorithm that included R-CHOP chemotherapy up front and CAR T-cell therapy in later lines of treatment, an approach that Dr. Locke speculated could result in a cure rate of perhaps 80% in large-cell lymphomas.
Encouraging longer-term data is emerging, with some patients with aggressive T-cell lymphomas now without recurrence for 5 years or more following a single infusion of CAR T-cell therapy, he said.
Dr. Locke reported a financial disclosure related to Cellular Biomedicine Group.
EXPERT ANALYSIS FROM THE NCCN HEMATOLOGIC MALIGNANCIES CONGRESS
ADHD: The big picture
Also today, prosthesis-patient mismatch post TAVR ups the risk of death by 19%, residents help to curb the overuse of IV antibiotics in children, and both age and other risk factors ought to guide chlamydia and gonorrhea screening for women infected with HIV.
Amazon Alexa
Apple Podcasts
Spotify
Also today, prosthesis-patient mismatch post TAVR ups the risk of death by 19%, residents help to curb the overuse of IV antibiotics in children, and both age and other risk factors ought to guide chlamydia and gonorrhea screening for women infected with HIV.
Amazon Alexa
Apple Podcasts
Spotify
Also today, prosthesis-patient mismatch post TAVR ups the risk of death by 19%, residents help to curb the overuse of IV antibiotics in children, and both age and other risk factors ought to guide chlamydia and gonorrhea screening for women infected with HIV.
Amazon Alexa
Apple Podcasts
Spotify

Educate your adolescent patients about herpes
We are all familiar with the line, “Herpes lasts forever.” There is no cure for infection with a herpes virus, whether it is herpes simplex 1 (HSV-1) or herpes simplex 2 (HSV-2).
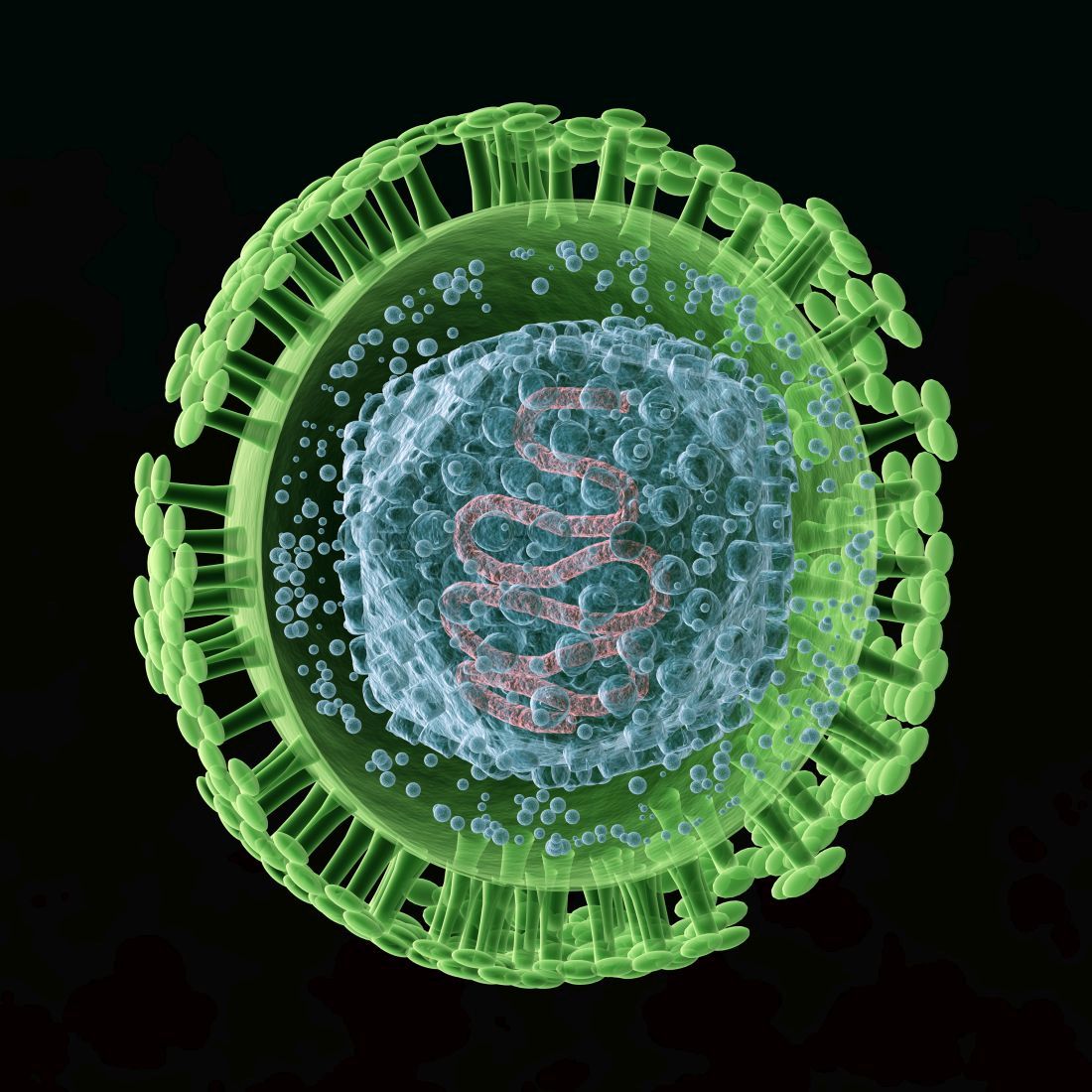
There are antivirals to reduce the length and severity of flare-ups, and continued therapy can suppress the virus, which reduces shedding. Both HSV-1 and HSV-2 can cause genital herpes and oral herpes, i.e. cold sores. HSV-1 has a milder initial episode and fewer flareups, whereas HSV-2 can have a more severe initial episode and frequent flareups.1
According to data from the National Health and Nutrition Examination Survey (NHANES) for 2015-2016, HSV-1 prevalence was 48% among 14- to 19-year-olds and HSV-2 prevalence was 12% in the same age group. Overall, age-adjusted HSV-1 prevalence was higher in females (51%) than in males (45%) in persons aged 14-49 years.2
The reality is that most people with HSV-1 or HSV-2 don’t even know they have it, as both tend to be asymptomatic. Therefore, all reported statistics are grossly underrepresenting the prevalence of the disease.
HSV is a common disease. Regardless of symptoms, shedding occurs. Although condoms reduce the risk of spread, using one doesn’t eliminate it because of the possibility of contact beyond the area covered by the condom and the ability of HSV to be passed through oral sex. The only true prevention is abstinence.
Herpes simplex virus is a sexually transmitted infection that is lifelong. Its presence can increase the risk of contracting HIV. If it is contracted in the third trimester of pregnancy or if a breakout occurs during the third trimester, risk of transmitting to the infant can occur, with devastating neurological impact. Despite the seriousness and longevity of the virus, the vast majority of people with the virus have it unknowingly, and live normal healthy lives.
It is just as important that we educate them that, if they contract herpes, it is not end of their ability to have intimate relationships. Debunking the myth that HSV-2 is a worse disease to have than HSV-1 can significantly reduce the psychological burden caused by this disease, and encourage patients to be more honest about their diagnosis. This not only will assist people in seeking medical advice if they have concerns, but it will encourage conversations about HSV, which hopefully will reduce spread of the virus.

Dr. Pearce is a pediatrician in Frankfort, Ill. She said she had no relevant financial disclosures. Email her at [email protected].
References
1. J Infect Dis. 2014 Feb. doi: 10.1093/infdis/jit458.
2. NCHS Data Brief, no 304. 2018 Feb.
We are all familiar with the line, “Herpes lasts forever.” There is no cure for infection with a herpes virus, whether it is herpes simplex 1 (HSV-1) or herpes simplex 2 (HSV-2).
There are antivirals to reduce the length and severity of flare-ups, and continued therapy can suppress the virus, which reduces shedding. Both HSV-1 and HSV-2 can cause genital herpes and oral herpes, i.e. cold sores. HSV-1 has a milder initial episode and fewer flareups, whereas HSV-2 can have a more severe initial episode and frequent flareups.1
According to data from the National Health and Nutrition Examination Survey (NHANES) for 2015-2016, HSV-1 prevalence was 48% among 14- to 19-year-olds and HSV-2 prevalence was 12% in the same age group. Overall, age-adjusted HSV-1 prevalence was higher in females (51%) than in males (45%) in persons aged 14-49 years.2
The reality is that most people with HSV-1 or HSV-2 don’t even know they have it, as both tend to be asymptomatic. Therefore, all reported statistics are grossly underrepresenting the prevalence of the disease.
HSV is a common disease. Regardless of symptoms, shedding occurs. Although condoms reduce the risk of spread, using one doesn’t eliminate it because of the possibility of contact beyond the area covered by the condom and the ability of HSV to be passed through oral sex. The only true prevention is abstinence.
Herpes simplex virus is a sexually transmitted infection that is lifelong. Its presence can increase the risk of contracting HIV. If it is contracted in the third trimester of pregnancy or if a breakout occurs during the third trimester, risk of transmitting to the infant can occur, with devastating neurological impact. Despite the seriousness and longevity of the virus, the vast majority of people with the virus have it unknowingly, and live normal healthy lives.
It is just as important that we educate them that, if they contract herpes, it is not end of their ability to have intimate relationships. Debunking the myth that HSV-2 is a worse disease to have than HSV-1 can significantly reduce the psychological burden caused by this disease, and encourage patients to be more honest about their diagnosis. This not only will assist people in seeking medical advice if they have concerns, but it will encourage conversations about HSV, which hopefully will reduce spread of the virus.
Dr. Pearce is a pediatrician in Frankfort, Ill. She said she had no relevant financial disclosures. Email her at [email protected].
References
1. J Infect Dis. 2014 Feb. doi: 10.1093/infdis/jit458.
2. NCHS Data Brief, no 304. 2018 Feb.
We are all familiar with the line, “Herpes lasts forever.” There is no cure for infection with a herpes virus, whether it is herpes simplex 1 (HSV-1) or herpes simplex 2 (HSV-2).
There are antivirals to reduce the length and severity of flare-ups, and continued therapy can suppress the virus, which reduces shedding. Both HSV-1 and HSV-2 can cause genital herpes and oral herpes, i.e. cold sores. HSV-1 has a milder initial episode and fewer flareups, whereas HSV-2 can have a more severe initial episode and frequent flareups.1
According to data from the National Health and Nutrition Examination Survey (NHANES) for 2015-2016, HSV-1 prevalence was 48% among 14- to 19-year-olds and HSV-2 prevalence was 12% in the same age group. Overall, age-adjusted HSV-1 prevalence was higher in females (51%) than in males (45%) in persons aged 14-49 years.2
The reality is that most people with HSV-1 or HSV-2 don’t even know they have it, as both tend to be asymptomatic. Therefore, all reported statistics are grossly underrepresenting the prevalence of the disease.
HSV is a common disease. Regardless of symptoms, shedding occurs. Although condoms reduce the risk of spread, using one doesn’t eliminate it because of the possibility of contact beyond the area covered by the condom and the ability of HSV to be passed through oral sex. The only true prevention is abstinence.
Herpes simplex virus is a sexually transmitted infection that is lifelong. Its presence can increase the risk of contracting HIV. If it is contracted in the third trimester of pregnancy or if a breakout occurs during the third trimester, risk of transmitting to the infant can occur, with devastating neurological impact. Despite the seriousness and longevity of the virus, the vast majority of people with the virus have it unknowingly, and live normal healthy lives.
It is just as important that we educate them that, if they contract herpes, it is not end of their ability to have intimate relationships. Debunking the myth that HSV-2 is a worse disease to have than HSV-1 can significantly reduce the psychological burden caused by this disease, and encourage patients to be more honest about their diagnosis. This not only will assist people in seeking medical advice if they have concerns, but it will encourage conversations about HSV, which hopefully will reduce spread of the virus.
Dr. Pearce is a pediatrician in Frankfort, Ill. She said she had no relevant financial disclosures. Email her at [email protected].
References
1. J Infect Dis. 2014 Feb. doi: 10.1093/infdis/jit458.
2. NCHS Data Brief, no 304. 2018 Feb.
Lorenzo Norris: Solo

Topical cyclosporine safely tamed atopic dermatitis in 4-week study
PARIS – A first-of-its-kind Ana M. Giménez-Arnau, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.

The 5% cyclosporine topical spray, known as Cyclatop, showed significantly better results across the board than its vehicle, even during the first week of treatment in the 4-week, multicenter, Spanish, double-blind, randomized trial, which included 44 patients with mild or moderate atopic dermatitis (AD), according to Dr. Giménez-Arnau, a dermatologist at Hospital del Mar and the Autonomous University of Barcelona.
“Besides the clinical efficacy, the study also demonstrated that, when cyclosporine was detectable in the blood, the highest blood level was at least 200-fold less than after systemic administration of cyclosporine at therapeutic doses,” she noted.
The motivation to develop a topical formulation of cyclosporine stemmed from the need to find substitutes for topical corticosteroids, especially in the pediatric population, where steroid phobia is rampant among parents. And while systemic cyclosporine is approved by European regulators for treatment of difficult cases of AD and is widely utilized off label for this purpose in the United States, the fact is that it is an immunosuppressant that paints with a broad brush and is best utilized for a matter of weeks as induction therapy.
But developing a topical formulation of cyclosporine suitable for long-term use posed many challenges. Lack of stability in cream and ointment formulations was a recurring issue. “Cyclosporine is a very big molecule, which is not easy to work with topically,” she explained. “The challenge was to find a stable formulation with good skin penetration, but without systemic absorption.”
Indeed, researchers at Barcelona-based Spherium Biomed evaluated more than 100 prototype compounds in animal models before settling on a proprietary oil emulsion formulation of 5% cyclosporine delivered via a spray without propellant gas.
Key study findings
The 44 study participants had a mean baseline of 8.3% body surface area involvement. As a condition of participation, they needed to have similar lesional areas bilaterally. They treated involved areas on one side of the body twice daily with Cyclatop, while they sprayed those on the opposite side with its vehicle.
From a mean baseline Eczema Area and Severity Index (EASI) score of 5.5, EASI scores improved by an average of 3.2 points after 28 days of cyclosporine spray, compared with 1.7 points with vehicle. Atopic Dermatitis Area and Severity Index (ADSI) scores improved from a mean baseline of 6.5 by 3.6 points with topical cyclosporine versus 2.4 points with vehicle.
At week 3, an EASI 75 response – that is, at least a 75% reduction from baseline EASI scores – was achieved at 44.4% of actively treated sites, compared with 25.9% of control sites. ADSI 75 rates at 3 weeks were 33.3% and 11.1%, respectively. An Investigator’s Global Assessment of clear or almost clear was reached at week 4 at 61.5% of active treatment sites, compared with 42.3% of vehicle-treated sites.
Itching responded dramatically to topical cyclosporine. From a mean baseline score of 3.3 on a standard 10-point pruritus visual analog scale, cyclosporine spray–treated areas showed a mean 1.2-point decrease at week 4, compared with a 0.4-point reduction at vehicle-treated areas. About 50% of the reduction in pruritus scores was achieved within the first week of active treatment. Moreover, among patients with moderate as opposed to mild itching scores at baseline, who had a mean pruritus score of 5.6, topical cyclosporine spray resulted in a mean 3.3-point reduction at week 4, Dr. Giménez-Arnau continued.
No safety signals emerged in this initial study. Side effects associated with the cyclosporine spray were the same as with its vehicle, and in exit interviews, more than 85% of patients indicated they were satisfied with the comfort and practicality of topical cyclosporine.
Session chair DeDee Murrell, MD, professor of dermatology at the University of New South Wales, Sydney, noted that the study was restricted to patients with less than 10% body surface area of involvement.
“Are you concerned that if you use this product over widespread areas, as is quite common in eczema, that you might get positive blood levels?” she asked.
“We don’t know. We should check that,” Dr. Giménez-Arnau replied. She added that more studies need to be done before cyclosporine spray is ready for the market. These studies will need to address the optimal dosing schedule and duration, the spectrum of disease severity where the topical spray works best, and other key issues.
Cyclatop is being developed by Spherium Biomed, which sponsored the study. Dr. Giménez-Arnau reported receiving research grants from and/or serving as a consultant to roughly half a dozen pharmaceutical companies.
PARIS – A first-of-its-kind Ana M. Giménez-Arnau, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
The 5% cyclosporine topical spray, known as Cyclatop, showed significantly better results across the board than its vehicle, even during the first week of treatment in the 4-week, multicenter, Spanish, double-blind, randomized trial, which included 44 patients with mild or moderate atopic dermatitis (AD), according to Dr. Giménez-Arnau, a dermatologist at Hospital del Mar and the Autonomous University of Barcelona.
“Besides the clinical efficacy, the study also demonstrated that, when cyclosporine was detectable in the blood, the highest blood level was at least 200-fold less than after systemic administration of cyclosporine at therapeutic doses,” she noted.
The motivation to develop a topical formulation of cyclosporine stemmed from the need to find substitutes for topical corticosteroids, especially in the pediatric population, where steroid phobia is rampant among parents. And while systemic cyclosporine is approved by European regulators for treatment of difficult cases of AD and is widely utilized off label for this purpose in the United States, the fact is that it is an immunosuppressant that paints with a broad brush and is best utilized for a matter of weeks as induction therapy.
But developing a topical formulation of cyclosporine suitable for long-term use posed many challenges. Lack of stability in cream and ointment formulations was a recurring issue. “Cyclosporine is a very big molecule, which is not easy to work with topically,” she explained. “The challenge was to find a stable formulation with good skin penetration, but without systemic absorption.”
Indeed, researchers at Barcelona-based Spherium Biomed evaluated more than 100 prototype compounds in animal models before settling on a proprietary oil emulsion formulation of 5% cyclosporine delivered via a spray without propellant gas.
Key study findings
The 44 study participants had a mean baseline of 8.3% body surface area involvement. As a condition of participation, they needed to have similar lesional areas bilaterally. They treated involved areas on one side of the body twice daily with Cyclatop, while they sprayed those on the opposite side with its vehicle.
From a mean baseline Eczema Area and Severity Index (EASI) score of 5.5, EASI scores improved by an average of 3.2 points after 28 days of cyclosporine spray, compared with 1.7 points with vehicle. Atopic Dermatitis Area and Severity Index (ADSI) scores improved from a mean baseline of 6.5 by 3.6 points with topical cyclosporine versus 2.4 points with vehicle.
At week 3, an EASI 75 response – that is, at least a 75% reduction from baseline EASI scores – was achieved at 44.4% of actively treated sites, compared with 25.9% of control sites. ADSI 75 rates at 3 weeks were 33.3% and 11.1%, respectively. An Investigator’s Global Assessment of clear or almost clear was reached at week 4 at 61.5% of active treatment sites, compared with 42.3% of vehicle-treated sites.
Itching responded dramatically to topical cyclosporine. From a mean baseline score of 3.3 on a standard 10-point pruritus visual analog scale, cyclosporine spray–treated areas showed a mean 1.2-point decrease at week 4, compared with a 0.4-point reduction at vehicle-treated areas. About 50% of the reduction in pruritus scores was achieved within the first week of active treatment. Moreover, among patients with moderate as opposed to mild itching scores at baseline, who had a mean pruritus score of 5.6, topical cyclosporine spray resulted in a mean 3.3-point reduction at week 4, Dr. Giménez-Arnau continued.
No safety signals emerged in this initial study. Side effects associated with the cyclosporine spray were the same as with its vehicle, and in exit interviews, more than 85% of patients indicated they were satisfied with the comfort and practicality of topical cyclosporine.
Session chair DeDee Murrell, MD, professor of dermatology at the University of New South Wales, Sydney, noted that the study was restricted to patients with less than 10% body surface area of involvement.
“Are you concerned that if you use this product over widespread areas, as is quite common in eczema, that you might get positive blood levels?” she asked.
“We don’t know. We should check that,” Dr. Giménez-Arnau replied. She added that more studies need to be done before cyclosporine spray is ready for the market. These studies will need to address the optimal dosing schedule and duration, the spectrum of disease severity where the topical spray works best, and other key issues.
Cyclatop is being developed by Spherium Biomed, which sponsored the study. Dr. Giménez-Arnau reported receiving research grants from and/or serving as a consultant to roughly half a dozen pharmaceutical companies.
PARIS – A first-of-its-kind Ana M. Giménez-Arnau, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
The 5% cyclosporine topical spray, known as Cyclatop, showed significantly better results across the board than its vehicle, even during the first week of treatment in the 4-week, multicenter, Spanish, double-blind, randomized trial, which included 44 patients with mild or moderate atopic dermatitis (AD), according to Dr. Giménez-Arnau, a dermatologist at Hospital del Mar and the Autonomous University of Barcelona.
“Besides the clinical efficacy, the study also demonstrated that, when cyclosporine was detectable in the blood, the highest blood level was at least 200-fold less than after systemic administration of cyclosporine at therapeutic doses,” she noted.
The motivation to develop a topical formulation of cyclosporine stemmed from the need to find substitutes for topical corticosteroids, especially in the pediatric population, where steroid phobia is rampant among parents. And while systemic cyclosporine is approved by European regulators for treatment of difficult cases of AD and is widely utilized off label for this purpose in the United States, the fact is that it is an immunosuppressant that paints with a broad brush and is best utilized for a matter of weeks as induction therapy.
But developing a topical formulation of cyclosporine suitable for long-term use posed many challenges. Lack of stability in cream and ointment formulations was a recurring issue. “Cyclosporine is a very big molecule, which is not easy to work with topically,” she explained. “The challenge was to find a stable formulation with good skin penetration, but without systemic absorption.”
Indeed, researchers at Barcelona-based Spherium Biomed evaluated more than 100 prototype compounds in animal models before settling on a proprietary oil emulsion formulation of 5% cyclosporine delivered via a spray without propellant gas.
Key study findings
The 44 study participants had a mean baseline of 8.3% body surface area involvement. As a condition of participation, they needed to have similar lesional areas bilaterally. They treated involved areas on one side of the body twice daily with Cyclatop, while they sprayed those on the opposite side with its vehicle.
From a mean baseline Eczema Area and Severity Index (EASI) score of 5.5, EASI scores improved by an average of 3.2 points after 28 days of cyclosporine spray, compared with 1.7 points with vehicle. Atopic Dermatitis Area and Severity Index (ADSI) scores improved from a mean baseline of 6.5 by 3.6 points with topical cyclosporine versus 2.4 points with vehicle.
At week 3, an EASI 75 response – that is, at least a 75% reduction from baseline EASI scores – was achieved at 44.4% of actively treated sites, compared with 25.9% of control sites. ADSI 75 rates at 3 weeks were 33.3% and 11.1%, respectively. An Investigator’s Global Assessment of clear or almost clear was reached at week 4 at 61.5% of active treatment sites, compared with 42.3% of vehicle-treated sites.
Itching responded dramatically to topical cyclosporine. From a mean baseline score of 3.3 on a standard 10-point pruritus visual analog scale, cyclosporine spray–treated areas showed a mean 1.2-point decrease at week 4, compared with a 0.4-point reduction at vehicle-treated areas. About 50% of the reduction in pruritus scores was achieved within the first week of active treatment. Moreover, among patients with moderate as opposed to mild itching scores at baseline, who had a mean pruritus score of 5.6, topical cyclosporine spray resulted in a mean 3.3-point reduction at week 4, Dr. Giménez-Arnau continued.
No safety signals emerged in this initial study. Side effects associated with the cyclosporine spray were the same as with its vehicle, and in exit interviews, more than 85% of patients indicated they were satisfied with the comfort and practicality of topical cyclosporine.
Session chair DeDee Murrell, MD, professor of dermatology at the University of New South Wales, Sydney, noted that the study was restricted to patients with less than 10% body surface area of involvement.
“Are you concerned that if you use this product over widespread areas, as is quite common in eczema, that you might get positive blood levels?” she asked.
“We don’t know. We should check that,” Dr. Giménez-Arnau replied. She added that more studies need to be done before cyclosporine spray is ready for the market. These studies will need to address the optimal dosing schedule and duration, the spectrum of disease severity where the topical spray works best, and other key issues.
Cyclatop is being developed by Spherium Biomed, which sponsored the study. Dr. Giménez-Arnau reported receiving research grants from and/or serving as a consultant to roughly half a dozen pharmaceutical companies.
REPORTING FROM THE EADV CONGRESS
Key clinical point: Cyclosporine 5% topical spray shows promise for atopic dermatitis.
Major finding: About 62% of patients with mild to moderate atopic dermatitis were clear or almost clear after 4 weeks of twice-daily cyclosporine 5% topical spray.
Study details: This prospective, multicenter, double-blind, vehicle-controlled study included 44 children and adults with mild or moderate atopic dermatitis.
Disclosures: The study was sponsored by Spherium Biomed. The presenter reported receiving research grants from and/or serving as a consultant to that and roughly half a dozen other pharmaceutical companies.
NHLBI commits to a sickle cell cure

“We have new exigency and intensity of effort to enable curative strategies for sickle cell disease to move forward,” said W. Keith Hoots, MD, the director of the division of blood diseases at NHLBI.
The key word in the cure effort is partnership – whether it’s among federal agencies, with public and private organizations, or with patients and families.
“Developmental strategies are built on partnerships to enhance care and accelerate cure for sickle cell disease in the U.S. and worldwide,” Dr. Hoots said at the 12th annual symposium of the Foundation for Sickle Cell Disease Research in Washington.
The reach also extends internationally. Supporting research in sub-Saharan Africa has promised to accelerate the clinical trial process by bringing advanced research capabilities to a region with a very high per capita rate of SCD. While in the United States, infrastructure is being built for a future research network, with the goal of developing a secure database of shared elements that harmonize and unite existing data.
Future cohort studies, enhanced newborn screening, and higher uptake of hydroxyurea will all be supported as part of this effort, Dr. Hoots said.
In the United States, patients can participate in a meaningful way as citizen-scientists, as new technology makes it possible to crowdsource high-quality data collection securely.
And including both community organizations and primary care providers in the “circle of partners” means not only that advances are brought out to patients expeditiously but also that the voices of patients and families have a clear channel back to researchers and policy makers through formal patient engagement and lay participation at all levels, Dr. Hoots said.
“The number of presently interested partners may surprise you,” Dr. Hoots said.
This multifaceted approach allows for “multiple shots on goal, with the acceptance that there could potentially be some failures,” Dr. Hoots said. Keeping all players better connected, though, should allow efforts to be redirected when needed, with a particular focus on accelerating work toward genetic therapies for SCD.
Perhaps the flagship effort is the Cure Sickle Cell Disease Initiative, a new partnership focused on accelerating cure-focused SCD research by filling in gaps left in the network of other funding strategies.
NHLBI named Edward J. Benz Jr., MD, the president and CEO emeritus of Boston’s Dana-Farber Cancer Institute, as the executive director and the Emmes Corporation, a contract research organization with expertise in clinical trials, as the coordinating center.
Traveling the last mile
New strategies also need to focus on how to boost uptake of such currently available best practices in SCD treatment as hydroxyurea use. To that end, Dr. Hoots said, NHLBI is drawing on implementation science, a discipline that, in a medical setting, can help solve such “last-mile” problems as bringing best practices in SCD treatment to patients.
In clinical practice, this might look like solving transportation issues for family members so that appointments aren’t missed and hydroxyurea prescriptions are filled. For researchers, implementation science can help with thorny details of participant recruitment and retention.
Established in 2016, the Sickle Cell Disease Implementation Consortium comprises nine U.S. research centers and NHLBI, which are each seeking to recruit at least 300 participants with SCD, aged 15-45 years, to study effective identification of barriers to care, and the best means to overcome them.
However, Dr. Hoots said, NHLBI also will continue funding SCD research through the traditional investigator-initiated application process, in conjunction with “a suite of specialized programs that can support translational and clinical research in SCD.”
Some of the features rolling out within the Cure SCD Initiative are included in direct response to stakeholder feedback about pressing needs and top priorities. For example, an economic case needed to be made in order for insurance companies, public and private alike, to reimburse for genetic SCD treatments. This requires an understanding of the lifetime cost burden of SCD, as well as determining what the long-term follow-up of costs of gene therapy will be.
Patients, family members and those providing primary care for SCD patients all agreed that clinical trials should have endpoints that reflect meaningful outcomes for patients and should be designed with the input of both patients and providers.
When queried, sickle cell disease researchers expressed a need to identify common data elements in SCD research, and wished for a secure yet accessible national data warehouse for data from gene and cell therapy trials.
At present, there are three clinical trials of curative stem cell approaches for SCD registered with the Blood and Marrow Transplant Clinical Trials Network and several more early phase clinical trials underway, Dr. Hoots said. A primary focus is the use of autologous cells for genomic editing, gene therapy, and erythroid-specific vectors.
Genetic research
As an example of the new collaboration, research centers and biotechnology companies sent their cell and genetic therapy experts to an NIH-sponsored gathering in March 2017. By pooling expertise in this way, the group was able to “identify some unprecedented opportunities, as well as some necessary barriers to overcome,” he said. These players continue to collaborate in the ongoing clinical trials of novel – and potentially curative – SCD therapies.
The TOPMed (Trans-Omics for Precision Medicine) program is a key mechanism to support SCD-related genetic research. For example, Dr. Hoots said, TOPMed is being used in support of whole-genome sequencing in a longitudinal cohort of patients with SCD who receive transfusion care at four large centers in Brazil.
These renewed efforts, set against the backdrop of paradigm-shifting genetic therapies, represent new promise for a generation of individuals with SCD, Dr. Hoots said. “It takes all of us to address the SCD challenge.”
ASH initiatives
NHLBI isn’t alone in making SCD a priority. The American Society of Hematology also is putting a spotlight on the condition.
The ASH multifaceted sickle cell disease (SCD) initiative addresses the disease burden both within the United States and globally, said LaTasha Lee, PhD, senior manager of sickle cell disease policy and programs for ASH.
Speaking at the 12th annual symposium of the Foundation for Sickle Cell Disease Research, Dr. Lee said that four prongs make up the initiative: disease research, attention to global issues, a renewed focus on access to care in the United States, and work to develop ASH’s new SCD guidelines.
New guidelines on the management of acute and chronic complications of SCD are in the works, with an anticipated 2019 date for publication of five separate guidelines. Topics covered in the guidelines will include pain, cerebrovascular disease, cardiopulmonary and kidney disease, transfusion support, and stem cell transplantation.
“We have new exigency and intensity of effort to enable curative strategies for sickle cell disease to move forward,” said W. Keith Hoots, MD, the director of the division of blood diseases at NHLBI.
The key word in the cure effort is partnership – whether it’s among federal agencies, with public and private organizations, or with patients and families.
“Developmental strategies are built on partnerships to enhance care and accelerate cure for sickle cell disease in the U.S. and worldwide,” Dr. Hoots said at the 12th annual symposium of the Foundation for Sickle Cell Disease Research in Washington.
The reach also extends internationally. Supporting research in sub-Saharan Africa has promised to accelerate the clinical trial process by bringing advanced research capabilities to a region with a very high per capita rate of SCD. While in the United States, infrastructure is being built for a future research network, with the goal of developing a secure database of shared elements that harmonize and unite existing data.
Future cohort studies, enhanced newborn screening, and higher uptake of hydroxyurea will all be supported as part of this effort, Dr. Hoots said.
In the United States, patients can participate in a meaningful way as citizen-scientists, as new technology makes it possible to crowdsource high-quality data collection securely.
And including both community organizations and primary care providers in the “circle of partners” means not only that advances are brought out to patients expeditiously but also that the voices of patients and families have a clear channel back to researchers and policy makers through formal patient engagement and lay participation at all levels, Dr. Hoots said.
“The number of presently interested partners may surprise you,” Dr. Hoots said.
This multifaceted approach allows for “multiple shots on goal, with the acceptance that there could potentially be some failures,” Dr. Hoots said. Keeping all players better connected, though, should allow efforts to be redirected when needed, with a particular focus on accelerating work toward genetic therapies for SCD.
Perhaps the flagship effort is the Cure Sickle Cell Disease Initiative, a new partnership focused on accelerating cure-focused SCD research by filling in gaps left in the network of other funding strategies.
NHLBI named Edward J. Benz Jr., MD, the president and CEO emeritus of Boston’s Dana-Farber Cancer Institute, as the executive director and the Emmes Corporation, a contract research organization with expertise in clinical trials, as the coordinating center.
Traveling the last mile
New strategies also need to focus on how to boost uptake of such currently available best practices in SCD treatment as hydroxyurea use. To that end, Dr. Hoots said, NHLBI is drawing on implementation science, a discipline that, in a medical setting, can help solve such “last-mile” problems as bringing best practices in SCD treatment to patients.
In clinical practice, this might look like solving transportation issues for family members so that appointments aren’t missed and hydroxyurea prescriptions are filled. For researchers, implementation science can help with thorny details of participant recruitment and retention.
Established in 2016, the Sickle Cell Disease Implementation Consortium comprises nine U.S. research centers and NHLBI, which are each seeking to recruit at least 300 participants with SCD, aged 15-45 years, to study effective identification of barriers to care, and the best means to overcome them.
However, Dr. Hoots said, NHLBI also will continue funding SCD research through the traditional investigator-initiated application process, in conjunction with “a suite of specialized programs that can support translational and clinical research in SCD.”
Some of the features rolling out within the Cure SCD Initiative are included in direct response to stakeholder feedback about pressing needs and top priorities. For example, an economic case needed to be made in order for insurance companies, public and private alike, to reimburse for genetic SCD treatments. This requires an understanding of the lifetime cost burden of SCD, as well as determining what the long-term follow-up of costs of gene therapy will be.
Patients, family members and those providing primary care for SCD patients all agreed that clinical trials should have endpoints that reflect meaningful outcomes for patients and should be designed with the input of both patients and providers.
When queried, sickle cell disease researchers expressed a need to identify common data elements in SCD research, and wished for a secure yet accessible national data warehouse for data from gene and cell therapy trials.
At present, there are three clinical trials of curative stem cell approaches for SCD registered with the Blood and Marrow Transplant Clinical Trials Network and several more early phase clinical trials underway, Dr. Hoots said. A primary focus is the use of autologous cells for genomic editing, gene therapy, and erythroid-specific vectors.
Genetic research
As an example of the new collaboration, research centers and biotechnology companies sent their cell and genetic therapy experts to an NIH-sponsored gathering in March 2017. By pooling expertise in this way, the group was able to “identify some unprecedented opportunities, as well as some necessary barriers to overcome,” he said. These players continue to collaborate in the ongoing clinical trials of novel – and potentially curative – SCD therapies.
The TOPMed (Trans-Omics for Precision Medicine) program is a key mechanism to support SCD-related genetic research. For example, Dr. Hoots said, TOPMed is being used in support of whole-genome sequencing in a longitudinal cohort of patients with SCD who receive transfusion care at four large centers in Brazil.
These renewed efforts, set against the backdrop of paradigm-shifting genetic therapies, represent new promise for a generation of individuals with SCD, Dr. Hoots said. “It takes all of us to address the SCD challenge.”
ASH initiatives
NHLBI isn’t alone in making SCD a priority. The American Society of Hematology also is putting a spotlight on the condition.
The ASH multifaceted sickle cell disease (SCD) initiative addresses the disease burden both within the United States and globally, said LaTasha Lee, PhD, senior manager of sickle cell disease policy and programs for ASH.
Speaking at the 12th annual symposium of the Foundation for Sickle Cell Disease Research, Dr. Lee said that four prongs make up the initiative: disease research, attention to global issues, a renewed focus on access to care in the United States, and work to develop ASH’s new SCD guidelines.
New guidelines on the management of acute and chronic complications of SCD are in the works, with an anticipated 2019 date for publication of five separate guidelines. Topics covered in the guidelines will include pain, cerebrovascular disease, cardiopulmonary and kidney disease, transfusion support, and stem cell transplantation.
“We have new exigency and intensity of effort to enable curative strategies for sickle cell disease to move forward,” said W. Keith Hoots, MD, the director of the division of blood diseases at NHLBI.
The key word in the cure effort is partnership – whether it’s among federal agencies, with public and private organizations, or with patients and families.
“Developmental strategies are built on partnerships to enhance care and accelerate cure for sickle cell disease in the U.S. and worldwide,” Dr. Hoots said at the 12th annual symposium of the Foundation for Sickle Cell Disease Research in Washington.
The reach also extends internationally. Supporting research in sub-Saharan Africa has promised to accelerate the clinical trial process by bringing advanced research capabilities to a region with a very high per capita rate of SCD. While in the United States, infrastructure is being built for a future research network, with the goal of developing a secure database of shared elements that harmonize and unite existing data.
Future cohort studies, enhanced newborn screening, and higher uptake of hydroxyurea will all be supported as part of this effort, Dr. Hoots said.
In the United States, patients can participate in a meaningful way as citizen-scientists, as new technology makes it possible to crowdsource high-quality data collection securely.
And including both community organizations and primary care providers in the “circle of partners” means not only that advances are brought out to patients expeditiously but also that the voices of patients and families have a clear channel back to researchers and policy makers through formal patient engagement and lay participation at all levels, Dr. Hoots said.
“The number of presently interested partners may surprise you,” Dr. Hoots said.
This multifaceted approach allows for “multiple shots on goal, with the acceptance that there could potentially be some failures,” Dr. Hoots said. Keeping all players better connected, though, should allow efforts to be redirected when needed, with a particular focus on accelerating work toward genetic therapies for SCD.
Perhaps the flagship effort is the Cure Sickle Cell Disease Initiative, a new partnership focused on accelerating cure-focused SCD research by filling in gaps left in the network of other funding strategies.
NHLBI named Edward J. Benz Jr., MD, the president and CEO emeritus of Boston’s Dana-Farber Cancer Institute, as the executive director and the Emmes Corporation, a contract research organization with expertise in clinical trials, as the coordinating center.
Traveling the last mile
New strategies also need to focus on how to boost uptake of such currently available best practices in SCD treatment as hydroxyurea use. To that end, Dr. Hoots said, NHLBI is drawing on implementation science, a discipline that, in a medical setting, can help solve such “last-mile” problems as bringing best practices in SCD treatment to patients.
In clinical practice, this might look like solving transportation issues for family members so that appointments aren’t missed and hydroxyurea prescriptions are filled. For researchers, implementation science can help with thorny details of participant recruitment and retention.
Established in 2016, the Sickle Cell Disease Implementation Consortium comprises nine U.S. research centers and NHLBI, which are each seeking to recruit at least 300 participants with SCD, aged 15-45 years, to study effective identification of barriers to care, and the best means to overcome them.
However, Dr. Hoots said, NHLBI also will continue funding SCD research through the traditional investigator-initiated application process, in conjunction with “a suite of specialized programs that can support translational and clinical research in SCD.”
Some of the features rolling out within the Cure SCD Initiative are included in direct response to stakeholder feedback about pressing needs and top priorities. For example, an economic case needed to be made in order for insurance companies, public and private alike, to reimburse for genetic SCD treatments. This requires an understanding of the lifetime cost burden of SCD, as well as determining what the long-term follow-up of costs of gene therapy will be.
Patients, family members and those providing primary care for SCD patients all agreed that clinical trials should have endpoints that reflect meaningful outcomes for patients and should be designed with the input of both patients and providers.
When queried, sickle cell disease researchers expressed a need to identify common data elements in SCD research, and wished for a secure yet accessible national data warehouse for data from gene and cell therapy trials.
At present, there are three clinical trials of curative stem cell approaches for SCD registered with the Blood and Marrow Transplant Clinical Trials Network and several more early phase clinical trials underway, Dr. Hoots said. A primary focus is the use of autologous cells for genomic editing, gene therapy, and erythroid-specific vectors.
Genetic research
As an example of the new collaboration, research centers and biotechnology companies sent their cell and genetic therapy experts to an NIH-sponsored gathering in March 2017. By pooling expertise in this way, the group was able to “identify some unprecedented opportunities, as well as some necessary barriers to overcome,” he said. These players continue to collaborate in the ongoing clinical trials of novel – and potentially curative – SCD therapies.
The TOPMed (Trans-Omics for Precision Medicine) program is a key mechanism to support SCD-related genetic research. For example, Dr. Hoots said, TOPMed is being used in support of whole-genome sequencing in a longitudinal cohort of patients with SCD who receive transfusion care at four large centers in Brazil.
These renewed efforts, set against the backdrop of paradigm-shifting genetic therapies, represent new promise for a generation of individuals with SCD, Dr. Hoots said. “It takes all of us to address the SCD challenge.”
ASH initiatives
NHLBI isn’t alone in making SCD a priority. The American Society of Hematology also is putting a spotlight on the condition.
The ASH multifaceted sickle cell disease (SCD) initiative addresses the disease burden both within the United States and globally, said LaTasha Lee, PhD, senior manager of sickle cell disease policy and programs for ASH.
Speaking at the 12th annual symposium of the Foundation for Sickle Cell Disease Research, Dr. Lee said that four prongs make up the initiative: disease research, attention to global issues, a renewed focus on access to care in the United States, and work to develop ASH’s new SCD guidelines.
New guidelines on the management of acute and chronic complications of SCD are in the works, with an anticipated 2019 date for publication of five separate guidelines. Topics covered in the guidelines will include pain, cerebrovascular disease, cardiopulmonary and kidney disease, transfusion support, and stem cell transplantation.
Taking a global perspective on infectious disease
The Infectious Diseases Society of America, which has its annual meeting as part of IDWeek, has a particular emphasis on global health. In fact, “IDSA prioritizes promoting sufficient U.S. investment in global infectious diseases responses and research, and in using the voices of physician scientists to inform multilateral and national global health policies. IDSA is a trusted leader in shaping and advancing international and national policies and investments in global HIV, TB, health security, and antimicrobial resistance,” according to the global health policy page of the organization’s website.
So it is no surprise that the global face of infectious disease is a prominent focus of IDWeek 2018 as exemplified by the Global ID track.
Although many of the overall sessions have a global ID component, with research from other countries prominent in IDWeek 2018 presentations across all topics, there are particular sessions that focus on more prominent global issues and diseases. For example, tuberculosis and HIV in a global context are strongly represented in many sessions.
But of unique interest also are emerging diseases and those that are, for now, primarily geographically localized. For example, the Global Health and Travel Medicine session on Thursday features presentations on a wide range of such diseases, including babseiosis, brucellosis, Burkholderia pseudomallei, Chagas disease, and chikungunya virus.
Similarly, the session titled Adventures with Globally Acquired Infections on Friday covers diseases that are comparatively rare in the United States, but endemic in many areas of the world, including Leishmania major infection, Lassa fever, and malaria.
Friday’s late-breaking oral abstracts feature Emerging Infections, including details on recent outbreaks of Andes virus, Enterovirus A71, and Shigella that have spread into the United States.
And uniquely, Global ID and Transplant ID merge in a special session titled When Transplant Tours the World featuring disease risk in transplantation from a global perspective, including the growing issue of “transplant tourism.”
The Infectious Diseases Society of America, which has its annual meeting as part of IDWeek, has a particular emphasis on global health. In fact, “IDSA prioritizes promoting sufficient U.S. investment in global infectious diseases responses and research, and in using the voices of physician scientists to inform multilateral and national global health policies. IDSA is a trusted leader in shaping and advancing international and national policies and investments in global HIV, TB, health security, and antimicrobial resistance,” according to the global health policy page of the organization’s website.
So it is no surprise that the global face of infectious disease is a prominent focus of IDWeek 2018 as exemplified by the Global ID track.
Although many of the overall sessions have a global ID component, with research from other countries prominent in IDWeek 2018 presentations across all topics, there are particular sessions that focus on more prominent global issues and diseases. For example, tuberculosis and HIV in a global context are strongly represented in many sessions.
But of unique interest also are emerging diseases and those that are, for now, primarily geographically localized. For example, the Global Health and Travel Medicine session on Thursday features presentations on a wide range of such diseases, including babseiosis, brucellosis, Burkholderia pseudomallei, Chagas disease, and chikungunya virus.
Similarly, the session titled Adventures with Globally Acquired Infections on Friday covers diseases that are comparatively rare in the United States, but endemic in many areas of the world, including Leishmania major infection, Lassa fever, and malaria.
Friday’s late-breaking oral abstracts feature Emerging Infections, including details on recent outbreaks of Andes virus, Enterovirus A71, and Shigella that have spread into the United States.
And uniquely, Global ID and Transplant ID merge in a special session titled When Transplant Tours the World featuring disease risk in transplantation from a global perspective, including the growing issue of “transplant tourism.”
The Infectious Diseases Society of America, which has its annual meeting as part of IDWeek, has a particular emphasis on global health. In fact, “IDSA prioritizes promoting sufficient U.S. investment in global infectious diseases responses and research, and in using the voices of physician scientists to inform multilateral and national global health policies. IDSA is a trusted leader in shaping and advancing international and national policies and investments in global HIV, TB, health security, and antimicrobial resistance,” according to the global health policy page of the organization’s website.
So it is no surprise that the global face of infectious disease is a prominent focus of IDWeek 2018 as exemplified by the Global ID track.
Although many of the overall sessions have a global ID component, with research from other countries prominent in IDWeek 2018 presentations across all topics, there are particular sessions that focus on more prominent global issues and diseases. For example, tuberculosis and HIV in a global context are strongly represented in many sessions.
But of unique interest also are emerging diseases and those that are, for now, primarily geographically localized. For example, the Global Health and Travel Medicine session on Thursday features presentations on a wide range of such diseases, including babseiosis, brucellosis, Burkholderia pseudomallei, Chagas disease, and chikungunya virus.
Similarly, the session titled Adventures with Globally Acquired Infections on Friday covers diseases that are comparatively rare in the United States, but endemic in many areas of the world, including Leishmania major infection, Lassa fever, and malaria.
Friday’s late-breaking oral abstracts feature Emerging Infections, including details on recent outbreaks of Andes virus, Enterovirus A71, and Shigella that have spread into the United States.
And uniquely, Global ID and Transplant ID merge in a special session titled When Transplant Tours the World featuring disease risk in transplantation from a global perspective, including the growing issue of “transplant tourism.”
Getting the most out of IDWeek 2018
IDWeek, whose theme is Advancing Science, Improving Care, is the combined annual meeting of the Infectious Diseases Society of America (IDSA), the HIV Medicine Association (HIVMA), the Society for Healthcare Epidemiology of America (SHEA), and the Pediatric Infectious Diseases Society (PIDS). As always,

The organizers have provided an interactive planning tool for attendees to create their own schedule. Features of the personal planner include:
Browse/Search: Gives several options for searching or browsing the program, including searching to quickly find a specific author and/or topic of interest.
My Schedule: Displays the sessions or abstracts/posters that the user has chosen to attend concisely.
My Bookmarks: Allows the user to create a hyperlinked list of bookmarks for future reference, including sessions not attended.
Anyone who’s interested can see the entire program online here and can browse by date and by the program tracks that comprise the following: Adult ID, Epidemiology & Infection Control, Global ID, HIV-STD-TB, Investigative ID, Pediatric ID, Trainee, and Transplant ID.
According to the organizers, “with so many common issues and challenges cutting across our four disciplines, IDWeek provides an opportunity to learn from each other’s knowledge, experience and expertise, for the improvement of patient care and public health.”
IDWeek 2018 is being held at the Moscone Center in downtown San Francisco, with the official program beginning on Wed., Oct. 3, at 1:30 p.m., and ending on Sun., Oct. 7, at 10:45 a.m.
Valuable premeeting workshops will be offered on Tuesday, including sessions on antimicrobial stewardship and research training program and grant-writing strategies, and on Wednesday morning, when there will be workshops, including those on TB, hepatitis C, and how to use the Grading of Recommendation Assessment, Development and Evaluation framework to develop guidelines.
And while you are in San Francisco for the meeting, be sure to check out the various sights and entertainment opportunities available there. Many websites highlight the fun and interesting things to do while in the city and can help you plan your stay, including the comprehensive SanFranciscoTravel page.
IDWeek, whose theme is Advancing Science, Improving Care, is the combined annual meeting of the Infectious Diseases Society of America (IDSA), the HIV Medicine Association (HIVMA), the Society for Healthcare Epidemiology of America (SHEA), and the Pediatric Infectious Diseases Society (PIDS). As always,
The organizers have provided an interactive planning tool for attendees to create their own schedule. Features of the personal planner include:
Browse/Search: Gives several options for searching or browsing the program, including searching to quickly find a specific author and/or topic of interest.
My Schedule: Displays the sessions or abstracts/posters that the user has chosen to attend concisely.
My Bookmarks: Allows the user to create a hyperlinked list of bookmarks for future reference, including sessions not attended.
Anyone who’s interested can see the entire program online here and can browse by date and by the program tracks that comprise the following: Adult ID, Epidemiology & Infection Control, Global ID, HIV-STD-TB, Investigative ID, Pediatric ID, Trainee, and Transplant ID.
According to the organizers, “with so many common issues and challenges cutting across our four disciplines, IDWeek provides an opportunity to learn from each other’s knowledge, experience and expertise, for the improvement of patient care and public health.”
IDWeek 2018 is being held at the Moscone Center in downtown San Francisco, with the official program beginning on Wed., Oct. 3, at 1:30 p.m., and ending on Sun., Oct. 7, at 10:45 a.m.
Valuable premeeting workshops will be offered on Tuesday, including sessions on antimicrobial stewardship and research training program and grant-writing strategies, and on Wednesday morning, when there will be workshops, including those on TB, hepatitis C, and how to use the Grading of Recommendation Assessment, Development and Evaluation framework to develop guidelines.
And while you are in San Francisco for the meeting, be sure to check out the various sights and entertainment opportunities available there. Many websites highlight the fun and interesting things to do while in the city and can help you plan your stay, including the comprehensive SanFranciscoTravel page.
IDWeek, whose theme is Advancing Science, Improving Care, is the combined annual meeting of the Infectious Diseases Society of America (IDSA), the HIV Medicine Association (HIVMA), the Society for Healthcare Epidemiology of America (SHEA), and the Pediatric Infectious Diseases Society (PIDS). As always,
The organizers have provided an interactive planning tool for attendees to create their own schedule. Features of the personal planner include:
Browse/Search: Gives several options for searching or browsing the program, including searching to quickly find a specific author and/or topic of interest.
My Schedule: Displays the sessions or abstracts/posters that the user has chosen to attend concisely.
My Bookmarks: Allows the user to create a hyperlinked list of bookmarks for future reference, including sessions not attended.
Anyone who’s interested can see the entire program online here and can browse by date and by the program tracks that comprise the following: Adult ID, Epidemiology & Infection Control, Global ID, HIV-STD-TB, Investigative ID, Pediatric ID, Trainee, and Transplant ID.
According to the organizers, “with so many common issues and challenges cutting across our four disciplines, IDWeek provides an opportunity to learn from each other’s knowledge, experience and expertise, for the improvement of patient care and public health.”
IDWeek 2018 is being held at the Moscone Center in downtown San Francisco, with the official program beginning on Wed., Oct. 3, at 1:30 p.m., and ending on Sun., Oct. 7, at 10:45 a.m.
Valuable premeeting workshops will be offered on Tuesday, including sessions on antimicrobial stewardship and research training program and grant-writing strategies, and on Wednesday morning, when there will be workshops, including those on TB, hepatitis C, and how to use the Grading of Recommendation Assessment, Development and Evaluation framework to develop guidelines.
And while you are in San Francisco for the meeting, be sure to check out the various sights and entertainment opportunities available there. Many websites highlight the fun and interesting things to do while in the city and can help you plan your stay, including the comprehensive SanFranciscoTravel page.
FDA lifts partial hold on tazemetostat trials

The U.S. Food and Drug Administration (FDA) has lifted the partial clinical hold on trials of tazemetostat, an EZH2 inhibitor being developed to treat solid tumors and lymphomas.
The FDA had placed the hold in April, and this halted U.S.-based enrollment of new patients in tazemetostat clinical trials.
Now, Epizyme, Inc., the company developing tazemetostat, is in the process of reopening enrollment in all company-sponsored trials in the U.S.
The FDA had placed the partial hold on tazemetostat trials after an adverse event was observed in a pediatric patient on a phase 1 study.
The patient, who had advanced poorly differentiated chordoma, developed secondary T-cell lymphoblastic lymphoma (T-LBL) while taking tazemetostat. The patient has since discontinued tazemetostat and responded to treatment for T-LBL.
“This remains the only case of T-LBL we’ve seen in more than 750 patients treated with tazemetostat,” said Robert Bazemore, president and chief executive officer of Epizyme.
Due to this adverse event and the partial clinical hold, Epizyme began to assess the risk of T-LBL and other secondary malignancies potentially associated with tazemetostat.
The company also assessed the overall risks and benefits of tazemetostat treatment, conducting a review of the published literature and an examination of efficacy and safety data across all of its tazemetostat trials.
Epizyme concluded that the benefits of tazemetostat outweigh the risks, and the risk of T-LBL appears confined to pediatric patients who received higher doses of the drug. The phase 1 pediatric study in which the patient developed T-LBL included higher doses of tazemetostat than those used in the phase 2 adult studies.
Epizyme convened a panel of external scientific and medical experts who reviewed and validated the company’s findings.
“The team at Epizyme has worked diligently in collaboration with external experts and FDA over the past several months, culminating in decisions . . . to lift the partial clinical hold and allow re-opening of enrollment in our clinical trials,” Bazemore said.
He noted that the company is not making any substantial changes to trial designs or the patient populations involved in tazemetostat trials.
However, Epizyme is modifying dosing in the pediatric studies, improving patient monitoring, and making changes to exclusion criteria to reduce the potential risk of T-LBL and other secondary malignancies.
Bazemore said the lifting of the partial clinical hold allows Epizyme to turn its full attention to key priorities, including plans to submit a new drug application for tazemetostat in epithelioid sarcoma.
The company also plans to begin preparing for a potential new drug application for tazemetostat in follicular lymphoma.
Tazemetostat is currently under investigation as monotherapy in phase 2 trials of follicular lymphoma and solid tumor malignancies. The drug is also being studied as part of combination therapy for non-small cell lung cancer and diffuse large B-cell lymphoma (DLBCL).
In August, Epizyme announced its decision to stop developing tazemetostat for use as monotherapy or in combination with prednisolone for patients with DLBCL. However, tazemetostat is still under investigation as a potential treatment for DLBCL as part of other combination regimens.
Now that Epizyme has resolved the U.S. hold on tazemetostat trials, the company is working to resolve partial clinical holds placed in France and Germany to resume trial enrollment in those countries.
The U.S. Food and Drug Administration (FDA) has lifted the partial clinical hold on trials of tazemetostat, an EZH2 inhibitor being developed to treat solid tumors and lymphomas.
The FDA had placed the hold in April, and this halted U.S.-based enrollment of new patients in tazemetostat clinical trials.
Now, Epizyme, Inc., the company developing tazemetostat, is in the process of reopening enrollment in all company-sponsored trials in the U.S.
The FDA had placed the partial hold on tazemetostat trials after an adverse event was observed in a pediatric patient on a phase 1 study.
The patient, who had advanced poorly differentiated chordoma, developed secondary T-cell lymphoblastic lymphoma (T-LBL) while taking tazemetostat. The patient has since discontinued tazemetostat and responded to treatment for T-LBL.
“This remains the only case of T-LBL we’ve seen in more than 750 patients treated with tazemetostat,” said Robert Bazemore, president and chief executive officer of Epizyme.
Due to this adverse event and the partial clinical hold, Epizyme began to assess the risk of T-LBL and other secondary malignancies potentially associated with tazemetostat.
The company also assessed the overall risks and benefits of tazemetostat treatment, conducting a review of the published literature and an examination of efficacy and safety data across all of its tazemetostat trials.
Epizyme concluded that the benefits of tazemetostat outweigh the risks, and the risk of T-LBL appears confined to pediatric patients who received higher doses of the drug. The phase 1 pediatric study in which the patient developed T-LBL included higher doses of tazemetostat than those used in the phase 2 adult studies.
Epizyme convened a panel of external scientific and medical experts who reviewed and validated the company’s findings.
“The team at Epizyme has worked diligently in collaboration with external experts and FDA over the past several months, culminating in decisions . . . to lift the partial clinical hold and allow re-opening of enrollment in our clinical trials,” Bazemore said.
He noted that the company is not making any substantial changes to trial designs or the patient populations involved in tazemetostat trials.
However, Epizyme is modifying dosing in the pediatric studies, improving patient monitoring, and making changes to exclusion criteria to reduce the potential risk of T-LBL and other secondary malignancies.
Bazemore said the lifting of the partial clinical hold allows Epizyme to turn its full attention to key priorities, including plans to submit a new drug application for tazemetostat in epithelioid sarcoma.
The company also plans to begin preparing for a potential new drug application for tazemetostat in follicular lymphoma.
Tazemetostat is currently under investigation as monotherapy in phase 2 trials of follicular lymphoma and solid tumor malignancies. The drug is also being studied as part of combination therapy for non-small cell lung cancer and diffuse large B-cell lymphoma (DLBCL).
In August, Epizyme announced its decision to stop developing tazemetostat for use as monotherapy or in combination with prednisolone for patients with DLBCL. However, tazemetostat is still under investigation as a potential treatment for DLBCL as part of other combination regimens.
Now that Epizyme has resolved the U.S. hold on tazemetostat trials, the company is working to resolve partial clinical holds placed in France and Germany to resume trial enrollment in those countries.
The U.S. Food and Drug Administration (FDA) has lifted the partial clinical hold on trials of tazemetostat, an EZH2 inhibitor being developed to treat solid tumors and lymphomas.
The FDA had placed the hold in April, and this halted U.S.-based enrollment of new patients in tazemetostat clinical trials.
Now, Epizyme, Inc., the company developing tazemetostat, is in the process of reopening enrollment in all company-sponsored trials in the U.S.
The FDA had placed the partial hold on tazemetostat trials after an adverse event was observed in a pediatric patient on a phase 1 study.
The patient, who had advanced poorly differentiated chordoma, developed secondary T-cell lymphoblastic lymphoma (T-LBL) while taking tazemetostat. The patient has since discontinued tazemetostat and responded to treatment for T-LBL.
“This remains the only case of T-LBL we’ve seen in more than 750 patients treated with tazemetostat,” said Robert Bazemore, president and chief executive officer of Epizyme.
Due to this adverse event and the partial clinical hold, Epizyme began to assess the risk of T-LBL and other secondary malignancies potentially associated with tazemetostat.
The company also assessed the overall risks and benefits of tazemetostat treatment, conducting a review of the published literature and an examination of efficacy and safety data across all of its tazemetostat trials.
Epizyme concluded that the benefits of tazemetostat outweigh the risks, and the risk of T-LBL appears confined to pediatric patients who received higher doses of the drug. The phase 1 pediatric study in which the patient developed T-LBL included higher doses of tazemetostat than those used in the phase 2 adult studies.
Epizyme convened a panel of external scientific and medical experts who reviewed and validated the company’s findings.
“The team at Epizyme has worked diligently in collaboration with external experts and FDA over the past several months, culminating in decisions . . . to lift the partial clinical hold and allow re-opening of enrollment in our clinical trials,” Bazemore said.
He noted that the company is not making any substantial changes to trial designs or the patient populations involved in tazemetostat trials.
However, Epizyme is modifying dosing in the pediatric studies, improving patient monitoring, and making changes to exclusion criteria to reduce the potential risk of T-LBL and other secondary malignancies.
Bazemore said the lifting of the partial clinical hold allows Epizyme to turn its full attention to key priorities, including plans to submit a new drug application for tazemetostat in epithelioid sarcoma.
The company also plans to begin preparing for a potential new drug application for tazemetostat in follicular lymphoma.
Tazemetostat is currently under investigation as monotherapy in phase 2 trials of follicular lymphoma and solid tumor malignancies. The drug is also being studied as part of combination therapy for non-small cell lung cancer and diffuse large B-cell lymphoma (DLBCL).
In August, Epizyme announced its decision to stop developing tazemetostat for use as monotherapy or in combination with prednisolone for patients with DLBCL. However, tazemetostat is still under investigation as a potential treatment for DLBCL as part of other combination regimens.
Now that Epizyme has resolved the U.S. hold on tazemetostat trials, the company is working to resolve partial clinical holds placed in France and Germany to resume trial enrollment in those countries.