User login
Canagliflozin approved for cardiovascular event risk reduction
The Food and Drug Administration has approved canagliflozin (Invokana) as a way to reduce the risk of major adverse cardiovascular events in patients with type 2 diabetes and cardiovascular disease, according to Janssen Pharmaceuticals.

The sodium–glucose cotransporter 2 inhibitor was first approved in 2013 to improve glycemic control in adults with type 2 diabetes.
FDA approval was based on results from the CANVAS (Canagliflozin Cardiovascular Assessment Study) trial, which included more than 10,000 adults with type 2 diabetes who either had cardiovascular disease or were at risk for cardiovascular disease. Overall, patients who received canagliflozin had a 14% lower risk of experiencing a major cardiovascular event over the control group, and patients with established cardiovascular disease had an 18% lower risk.
The most common adverse events associated with canagliflozin include female genital mycotic infections, urinary tract infection, and increased urination. Notably, canagliflozin also increases the risk of lower-extremity amputation, especially in those with a history of amputation.
“Americans living with type 2 diabetes are two to three times more likely to die from heart disease than adults without diabetes. With this approval, Invokana now plays an even more important role in the overall treatment mix with its demonstrated ability to reduce the risk of potentially devastating cardiovascular events,” Ralph A. DeFronzo, MD, professor and division chief of medicine and diabetes at the University of Texas, San Antonio, said in the press release.
The new indication applies to all formulations of canagliflozin.
Find the full press release on the Janssen website.
The Food and Drug Administration has approved canagliflozin (Invokana) as a way to reduce the risk of major adverse cardiovascular events in patients with type 2 diabetes and cardiovascular disease, according to Janssen Pharmaceuticals.
The sodium–glucose cotransporter 2 inhibitor was first approved in 2013 to improve glycemic control in adults with type 2 diabetes.
FDA approval was based on results from the CANVAS (Canagliflozin Cardiovascular Assessment Study) trial, which included more than 10,000 adults with type 2 diabetes who either had cardiovascular disease or were at risk for cardiovascular disease. Overall, patients who received canagliflozin had a 14% lower risk of experiencing a major cardiovascular event over the control group, and patients with established cardiovascular disease had an 18% lower risk.
The most common adverse events associated with canagliflozin include female genital mycotic infections, urinary tract infection, and increased urination. Notably, canagliflozin also increases the risk of lower-extremity amputation, especially in those with a history of amputation.
“Americans living with type 2 diabetes are two to three times more likely to die from heart disease than adults without diabetes. With this approval, Invokana now plays an even more important role in the overall treatment mix with its demonstrated ability to reduce the risk of potentially devastating cardiovascular events,” Ralph A. DeFronzo, MD, professor and division chief of medicine and diabetes at the University of Texas, San Antonio, said in the press release.
The new indication applies to all formulations of canagliflozin.
Find the full press release on the Janssen website.
The Food and Drug Administration has approved canagliflozin (Invokana) as a way to reduce the risk of major adverse cardiovascular events in patients with type 2 diabetes and cardiovascular disease, according to Janssen Pharmaceuticals.
The sodium–glucose cotransporter 2 inhibitor was first approved in 2013 to improve glycemic control in adults with type 2 diabetes.
FDA approval was based on results from the CANVAS (Canagliflozin Cardiovascular Assessment Study) trial, which included more than 10,000 adults with type 2 diabetes who either had cardiovascular disease or were at risk for cardiovascular disease. Overall, patients who received canagliflozin had a 14% lower risk of experiencing a major cardiovascular event over the control group, and patients with established cardiovascular disease had an 18% lower risk.
The most common adverse events associated with canagliflozin include female genital mycotic infections, urinary tract infection, and increased urination. Notably, canagliflozin also increases the risk of lower-extremity amputation, especially in those with a history of amputation.
“Americans living with type 2 diabetes are two to three times more likely to die from heart disease than adults without diabetes. With this approval, Invokana now plays an even more important role in the overall treatment mix with its demonstrated ability to reduce the risk of potentially devastating cardiovascular events,” Ralph A. DeFronzo, MD, professor and division chief of medicine and diabetes at the University of Texas, San Antonio, said in the press release.
The new indication applies to all formulations of canagliflozin.
Find the full press release on the Janssen website.
Nivo + ipi shows durable activity against metastatic melanoma
MUNICH – Four years on, the combination of the immune checkpoint inhibitors nivolumab and ipilimumab as well as nivolumab alone continue to show benefit as first-line therapies for patients with advanced malignant melanoma, compared with ipilimumab monotherapy, reported investigators in the CheckMate O67 trial.

Among 945 patients with previously untreated and unresectable stage III or IV malignant melanoma, median overall survival at a minimum of 48 months follow-up had not been reached for patients assigned to the combination of nivolumab (Opdivo) and ipilimumab (Yervoy), compared with 36.9 months for patients assigned to nivolumab alone, and 19.9 months for patients assigned to ipilimumab alone, reported F. Stephen Hodi Jr., MD, of Dana-Farber Cancer Institute, Boston, and his colleagues.
“There’s a durable, sustained clinical benefit that can be achieved with first-line nivo plus ipi combination or nivo alone in patients with advanced melanoma,” he said at the European Society for Medical Oncology Congress. The study results were published online in The Lancet Oncology to coincide with the presentation.
The benefit of immunotherapy also was seen in patients whose tumors had BRAF mutations, and both the combination and nivolumab alone showed improved efficacy compared with ipilimumab alone regardless of tumor expression of the programmed death ligand-1 (PD-L1), the investigators reported.
As previously reported, investigators in CheckMate 067 randomly assigned 945 previously untreated patients with unresectable stage III or IV melanoma to nivolumab 3 mg/kg every 2 weeks or nivolumab 1 mg/kg plus ipilimumab 3 mg/kg every 3 weeks for four doses then nivolumab 3 mg/kg every 2 weeks, or ipilimumab 3 mg/kg every 3 weeks for four doses. Patients were stratified at baseline by PD-L1 expression, BRAF status, and American Joint Commission on Cancer M stage.
Earlier results from the trial, reported at the 2015 annual meeting of the American Society of Clinical Oncology, showed that after a minimum of 9 months follow-up, the risk of disease progression or death was reduced by 43% with nivolumab versus ipilimumab (hazard ratio, 0.57; P less than .001) and by 58% with nivolumab plus ipilimumab vs. ipilimumab (HR, 0.42; P less than .001).
At ESMO 2018, Dr. Hodi presented 4-year follow-up results from the trial, with the analysis conducted at a minimum of 4 years after randomization of the last patient to be enrolled.
Median follow-up was 46.9 months for the nivolumab-plus-ipilimumab arm, 36 months in the nivolumab arm, and 18.6 months in the ipilimumab arm.
Median overall survival in the intention-to-treat population, a coprimary endpoint with progression-free survival (PFS), was as noted before. The HR for death with the combination compared with ipilimumab was 0.54 (P less than .0001) and for nivolumab versus ipilimumab it was 0.65 (P less than .0001).
The 4-year OS rates were 53% in the combination arm, 46% in the nivolumab-alone arm, and 30% in the ipilimumab-alone arm.
Median PFS was 11.5 months with the checkpoint inhibitor combination, 6.9 months in the nivolumab-alone arm, and 2.9 months in the ipilimumab arm.
The HR for PFS with the combination compared with ipilimumab was 0.42 (P less than .0001), and for nivolumab versus ipilimumab it was 0.53 (P less than .0001).
The safety analysis, conducted in all patients who received at least one dose of study drugs, showed that 59% of patients treated with the nivolumab/ipilimumab combination had treatment-related grade 3 or 4 adverse events, compared with 22% for patients treated with nivolumab alone, and 28% of those who received ipilimumab alone.
The most common treatment-related grade 3 adverse events were diarrhea in the combination and nivolumab-alone arms, and colitis in the ipilimumab group. In all three study arms the most common grade 4 adverse event was increased lipase.
Over the 4 years of follow-up, four patients died from treatment-related causes: one patient from cardiomyopathy and one from liver necrosis in the combination group, one from neutropenia in the nivolumab group, and one from colon perforation in the ipilimumab group. All of the deaths occurred within the first 3 years of the follow-up.
The investigators did not report on serious adverse events in the current analysis.
Invited discussant Reinhard Dummer, MD, of University Hospital Zurich Skin Cancer Center in Switzerland, said that while the study shows improved response rates and duration of response and longer PFS and OS with the combination, it’s premature to state conclusively that the combination is superior, because the study was not powered to compare efficacy between the two nivolumab-containing arms.
“So unfortunately, we have results, but we are not really convinced that the combination is so much better,” he said.
He added that the 4-year overall survival results for each arm show a consistent difference in the curves between the nivolumab and ipilimumab-alone arms. He also pointed to encouraging data showing that among patients alive at 4 years, 71% in the combination group did not require subsequent therapy, compared with 50% in the nivolumab group, and 39% in the ipilimumab group.
Dr. Hodi has received grant/research support from, and is a nonpaid consultant to, Bristol-Myers Squibb, which supported Checkmate 067. Dr. Dummer reported advising/consulting roles with the company.
SOURCE: Hodi FS et al. Lancet Oncol. 2018 Oct 22. doi: 10.1016/S1470-2045(18)30700-9.
MUNICH – Four years on, the combination of the immune checkpoint inhibitors nivolumab and ipilimumab as well as nivolumab alone continue to show benefit as first-line therapies for patients with advanced malignant melanoma, compared with ipilimumab monotherapy, reported investigators in the CheckMate O67 trial.
Among 945 patients with previously untreated and unresectable stage III or IV malignant melanoma, median overall survival at a minimum of 48 months follow-up had not been reached for patients assigned to the combination of nivolumab (Opdivo) and ipilimumab (Yervoy), compared with 36.9 months for patients assigned to nivolumab alone, and 19.9 months for patients assigned to ipilimumab alone, reported F. Stephen Hodi Jr., MD, of Dana-Farber Cancer Institute, Boston, and his colleagues.
“There’s a durable, sustained clinical benefit that can be achieved with first-line nivo plus ipi combination or nivo alone in patients with advanced melanoma,” he said at the European Society for Medical Oncology Congress. The study results were published online in The Lancet Oncology to coincide with the presentation.
The benefit of immunotherapy also was seen in patients whose tumors had BRAF mutations, and both the combination and nivolumab alone showed improved efficacy compared with ipilimumab alone regardless of tumor expression of the programmed death ligand-1 (PD-L1), the investigators reported.
As previously reported, investigators in CheckMate 067 randomly assigned 945 previously untreated patients with unresectable stage III or IV melanoma to nivolumab 3 mg/kg every 2 weeks or nivolumab 1 mg/kg plus ipilimumab 3 mg/kg every 3 weeks for four doses then nivolumab 3 mg/kg every 2 weeks, or ipilimumab 3 mg/kg every 3 weeks for four doses. Patients were stratified at baseline by PD-L1 expression, BRAF status, and American Joint Commission on Cancer M stage.
Earlier results from the trial, reported at the 2015 annual meeting of the American Society of Clinical Oncology, showed that after a minimum of 9 months follow-up, the risk of disease progression or death was reduced by 43% with nivolumab versus ipilimumab (hazard ratio, 0.57; P less than .001) and by 58% with nivolumab plus ipilimumab vs. ipilimumab (HR, 0.42; P less than .001).
At ESMO 2018, Dr. Hodi presented 4-year follow-up results from the trial, with the analysis conducted at a minimum of 4 years after randomization of the last patient to be enrolled.
Median follow-up was 46.9 months for the nivolumab-plus-ipilimumab arm, 36 months in the nivolumab arm, and 18.6 months in the ipilimumab arm.
Median overall survival in the intention-to-treat population, a coprimary endpoint with progression-free survival (PFS), was as noted before. The HR for death with the combination compared with ipilimumab was 0.54 (P less than .0001) and for nivolumab versus ipilimumab it was 0.65 (P less than .0001).
The 4-year OS rates were 53% in the combination arm, 46% in the nivolumab-alone arm, and 30% in the ipilimumab-alone arm.
Median PFS was 11.5 months with the checkpoint inhibitor combination, 6.9 months in the nivolumab-alone arm, and 2.9 months in the ipilimumab arm.
The HR for PFS with the combination compared with ipilimumab was 0.42 (P less than .0001), and for nivolumab versus ipilimumab it was 0.53 (P less than .0001).
The safety analysis, conducted in all patients who received at least one dose of study drugs, showed that 59% of patients treated with the nivolumab/ipilimumab combination had treatment-related grade 3 or 4 adverse events, compared with 22% for patients treated with nivolumab alone, and 28% of those who received ipilimumab alone.
The most common treatment-related grade 3 adverse events were diarrhea in the combination and nivolumab-alone arms, and colitis in the ipilimumab group. In all three study arms the most common grade 4 adverse event was increased lipase.
Over the 4 years of follow-up, four patients died from treatment-related causes: one patient from cardiomyopathy and one from liver necrosis in the combination group, one from neutropenia in the nivolumab group, and one from colon perforation in the ipilimumab group. All of the deaths occurred within the first 3 years of the follow-up.
The investigators did not report on serious adverse events in the current analysis.
Invited discussant Reinhard Dummer, MD, of University Hospital Zurich Skin Cancer Center in Switzerland, said that while the study shows improved response rates and duration of response and longer PFS and OS with the combination, it’s premature to state conclusively that the combination is superior, because the study was not powered to compare efficacy between the two nivolumab-containing arms.
“So unfortunately, we have results, but we are not really convinced that the combination is so much better,” he said.
He added that the 4-year overall survival results for each arm show a consistent difference in the curves between the nivolumab and ipilimumab-alone arms. He also pointed to encouraging data showing that among patients alive at 4 years, 71% in the combination group did not require subsequent therapy, compared with 50% in the nivolumab group, and 39% in the ipilimumab group.
Dr. Hodi has received grant/research support from, and is a nonpaid consultant to, Bristol-Myers Squibb, which supported Checkmate 067. Dr. Dummer reported advising/consulting roles with the company.
SOURCE: Hodi FS et al. Lancet Oncol. 2018 Oct 22. doi: 10.1016/S1470-2045(18)30700-9.
MUNICH – Four years on, the combination of the immune checkpoint inhibitors nivolumab and ipilimumab as well as nivolumab alone continue to show benefit as first-line therapies for patients with advanced malignant melanoma, compared with ipilimumab monotherapy, reported investigators in the CheckMate O67 trial.
Among 945 patients with previously untreated and unresectable stage III or IV malignant melanoma, median overall survival at a minimum of 48 months follow-up had not been reached for patients assigned to the combination of nivolumab (Opdivo) and ipilimumab (Yervoy), compared with 36.9 months for patients assigned to nivolumab alone, and 19.9 months for patients assigned to ipilimumab alone, reported F. Stephen Hodi Jr., MD, of Dana-Farber Cancer Institute, Boston, and his colleagues.
“There’s a durable, sustained clinical benefit that can be achieved with first-line nivo plus ipi combination or nivo alone in patients with advanced melanoma,” he said at the European Society for Medical Oncology Congress. The study results were published online in The Lancet Oncology to coincide with the presentation.
The benefit of immunotherapy also was seen in patients whose tumors had BRAF mutations, and both the combination and nivolumab alone showed improved efficacy compared with ipilimumab alone regardless of tumor expression of the programmed death ligand-1 (PD-L1), the investigators reported.
As previously reported, investigators in CheckMate 067 randomly assigned 945 previously untreated patients with unresectable stage III or IV melanoma to nivolumab 3 mg/kg every 2 weeks or nivolumab 1 mg/kg plus ipilimumab 3 mg/kg every 3 weeks for four doses then nivolumab 3 mg/kg every 2 weeks, or ipilimumab 3 mg/kg every 3 weeks for four doses. Patients were stratified at baseline by PD-L1 expression, BRAF status, and American Joint Commission on Cancer M stage.
Earlier results from the trial, reported at the 2015 annual meeting of the American Society of Clinical Oncology, showed that after a minimum of 9 months follow-up, the risk of disease progression or death was reduced by 43% with nivolumab versus ipilimumab (hazard ratio, 0.57; P less than .001) and by 58% with nivolumab plus ipilimumab vs. ipilimumab (HR, 0.42; P less than .001).
At ESMO 2018, Dr. Hodi presented 4-year follow-up results from the trial, with the analysis conducted at a minimum of 4 years after randomization of the last patient to be enrolled.
Median follow-up was 46.9 months for the nivolumab-plus-ipilimumab arm, 36 months in the nivolumab arm, and 18.6 months in the ipilimumab arm.
Median overall survival in the intention-to-treat population, a coprimary endpoint with progression-free survival (PFS), was as noted before. The HR for death with the combination compared with ipilimumab was 0.54 (P less than .0001) and for nivolumab versus ipilimumab it was 0.65 (P less than .0001).
The 4-year OS rates were 53% in the combination arm, 46% in the nivolumab-alone arm, and 30% in the ipilimumab-alone arm.
Median PFS was 11.5 months with the checkpoint inhibitor combination, 6.9 months in the nivolumab-alone arm, and 2.9 months in the ipilimumab arm.
The HR for PFS with the combination compared with ipilimumab was 0.42 (P less than .0001), and for nivolumab versus ipilimumab it was 0.53 (P less than .0001).
The safety analysis, conducted in all patients who received at least one dose of study drugs, showed that 59% of patients treated with the nivolumab/ipilimumab combination had treatment-related grade 3 or 4 adverse events, compared with 22% for patients treated with nivolumab alone, and 28% of those who received ipilimumab alone.
The most common treatment-related grade 3 adverse events were diarrhea in the combination and nivolumab-alone arms, and colitis in the ipilimumab group. In all three study arms the most common grade 4 adverse event was increased lipase.
Over the 4 years of follow-up, four patients died from treatment-related causes: one patient from cardiomyopathy and one from liver necrosis in the combination group, one from neutropenia in the nivolumab group, and one from colon perforation in the ipilimumab group. All of the deaths occurred within the first 3 years of the follow-up.
The investigators did not report on serious adverse events in the current analysis.
Invited discussant Reinhard Dummer, MD, of University Hospital Zurich Skin Cancer Center in Switzerland, said that while the study shows improved response rates and duration of response and longer PFS and OS with the combination, it’s premature to state conclusively that the combination is superior, because the study was not powered to compare efficacy between the two nivolumab-containing arms.
“So unfortunately, we have results, but we are not really convinced that the combination is so much better,” he said.
He added that the 4-year overall survival results for each arm show a consistent difference in the curves between the nivolumab and ipilimumab-alone arms. He also pointed to encouraging data showing that among patients alive at 4 years, 71% in the combination group did not require subsequent therapy, compared with 50% in the nivolumab group, and 39% in the ipilimumab group.
Dr. Hodi has received grant/research support from, and is a nonpaid consultant to, Bristol-Myers Squibb, which supported Checkmate 067. Dr. Dummer reported advising/consulting roles with the company.
SOURCE: Hodi FS et al. Lancet Oncol. 2018 Oct 22. doi: 10.1016/S1470-2045(18)30700-9.
REPORTING FROM ESMO 2018
Key clinical point: Nivolumab and ipilimumab combined provide superior progression-free and overall survival compared with nivolumab or ipilimumab alone.
Major finding: At 4-year minimum follow-up the median overall survival with the combination had not be reached, vs. 36.9 months for nivolumab and 19.9 months for ipilimumab.
Study details: Randomized phase 3 trial of 945 patients with previously untreated stage III or IV malignant melanoma.
Disclosures: Dr. Hodi has received grant/research support from, and is a nonpaid consultant to, Bristol-Myers Squibb, which supported Checkmate 067. Dr. Dummer reported advising/consulting roles with the company.
Source: Hodi FS et al. Lancet Oncol. 2018 Oct 22. doi: 10.1016/S1470-2045(18)30700-9.
Death of a sales pitch
The EHR and our troubled health care system, Part 1
In 2000, the Institute of Medicine published “To Err Is Human,” a landmark study that warned that as many as 98,000 people die annually as a result of medical errors. One conclusion of the report stated, “When patients see multiple providers in different settings, none of whom has access to complete information, it becomes easier for things to go wrong.” Government and public reaction to the study resulted in the rushed integration of electronic health records into the U.S. medical system. EHR vendors promised solutions that included a dramatic reduction of preventable errors, a simplified system of physician communication, and the consolidation of a patient’s salient medical information into a single, transferable file. Now, almost 20 years later, these promises remain mostly unfilled. How did we get here?

Systems of medical records have been in place since 1600 B.C. For thousands of years, they consisted mainly of the patient’s diagnosis and the physician’s treatment. In 1968, the New England Journal of Medicine published the special article “Medical Records That Guide and Teach” by Lawrence L. Weed, MD. In the report, Dr. Weed advocated for the organization of medical records by problems rather than by a single diagnosis. This was the birth of our modern system. Medical records would now include lists of symptoms, findings, and problems that would organize the physician’s planning and allow third parties to confirm the initial diagnosis. Nearly concurrent with this publication, the next major innovation was developing in a very unusual location.
In 1999, Fortune magazine labeled Jack Welch “Manager of the Century” for his innovative work as CEO of General Electric. His techniques involved cutting waste and streamlining his workforce. While these methods were somewhat controversial, GE’s market value increased dramatically under his watch. The publishers at Fortune became interested in finding similar innovators in other fields. In this pursuit, they sent journalist Philip Longman to find the “Jack Welch” of health care.
Mr. Longman had recently lost his wife to breast cancer and was becoming obsessed with medical errors and health care quality integration. He set out to discover the best health care system in the United States. After months of research, Mr. Longman reached a startling conclusion. By nearly every metric, the Veterans Affairs system produced the highest quality of care. The key factor in upholding that quality appeared to be the EHR system VistA (Veterans Information Systems and Technology Architecture).
The development of VistA was a grassroots effort begun in the 1970s. Using Tandy computers and Wang processors, the VA “hardhats” sought to develop an electronic system for medical records and communication. This effort was initially opposed and driven underground by the central bureaucracy. Laptops were confiscated, people were fired. Still, development continued, and in 1978, the Decentralized Hospital Computer Program was launched at 20 VA sites. The national rollout occurred in 1994 under the name VistA.
VistA was developed by doctors, for doctors, and routinely enjoys the highest satisfaction rates among all available EHRs. VistA also is an open source model; its code is readily available on the VA website. After seeing the evidence of VistA’s efficacy, Representative Pete Stark (D-CA) introduced HR 6898 on Sept. 15, 2008. The bill would establish a large federal open source health IT system that private hospitals could leverage. The bill also mandated that only open source solutions would receive federal funding. As opposed to proprietary systems, open source models allow for rapid innovation, easy personal configuration, and incorporation of open source apps from unlimited numbers of contributors.
HR 6898 never passed, despite initial bipartisan support. By relying on lobbyists, marketing, and money, the proprietary EHR vendors killed the Stark bill. After a 4-month scramble, the Health Information Technology for Economic and Clinical Health Act (HITECH) passed, with EHR vendor support. HITECH established a certification system for EHRs. While the Stark bill envisioned a single, open source network, there were soon hundreds of certified EHR systems in the United States.
Before the HITECH Act, many EHRs existed, but several barriers blocked full implementation. Early systems were essentially electronic filing cabinets. Their developers had not anticipated the lack of standardization among physicians and hospital systems. The need for custom EHR bases frustrated the vendors. The question of marketing was omnipresent. Who was the actual customer? An economic model developed in which clinicians would bear the time and even financial costs as the benefits would be passed on to insurers, hospitals, and, presumably, the patients.
EHRs needed to become practical, affordable, and interoperable, but who was demanding this? Where was the financial motivation? In the beginning, vendors of EHRs had to convince doctors, the public, and the government of their worth. Now, essentially mandated by the HITECH Act, they only had to sell themselves to hospital administrators, who often had a different motive. Profits.
Many of today’s EHRs are simply modified billing platforms, and doctors are paying the price. The Meaningful Use standards were meant to provide financial incentives for EHR adoption. Stage 2 required EHRs to be able to transport clinical information from one system to another. Looking at our actual practices can provide a master class in the gap between “be able to” and “actually doing.” Again, who does the EHR vendor see as the customer? Certainly not the physician. My patients can list every type of inferior vena cava filter (or at least those with pending legal action), but most of them have never heard of an EHR. Just like “service lines,” EHRs can make it very difficult for patients to seek care outside of their primary system. Who would see this barrier in communication as a perk and not a deficiency? Hospital administrators. The free transfer of medical records is bad for business. Therefore, hospitals don’t prioritize it in their EHRs. The EHR vendors also benefit since an easy transfer of records would simplify a hospital’s transition from one EHR to another. So, as with most deficiencies in the EHR, physicians are left to find ways around these problems. Sometimes, we need to go to comical lengths.
Two months ago, a patient pointed to a large machine behind our check-in desk. “What is that,” he asked incredulously; it was a fax machine. While my competence with this apparatus is marginal (my office staff has taken to yelling “doctor faxing!” to alert one another that I am about to inadvertently copy or scan my documents into oblivion), faxes remain a mainstay of medical care. Abandoned by modern business practices as a relic of the 1980s, why are we constantly faxing medical information? Because we are not the customer.

Disruption is now a favorable term in business. Doctors are busy people. BUSY people. Most of us walk a tightrope, a razor-thin timeline. Will we see the next patient in time, the next surgery? Will we get the medical records done today? Will we get the dictations done before being suspended? Will we make the committee meeting, the conference call, the next clinic across town? Will we have dinner with our spouse or see our kids today? Will we make it to the parent-teacher conference inexplicably scheduled for 10:45 a.m. on a Tuesday??!! When deciding between work commitments and family, we side with work overwhelmingly (and depressingly). Explaining this to a layperson is an impossible feat. I have stopped trying, stopped making excuses. Only we know how catastrophic “disruption” can be. Disruption in a 40-patient clinic. Disruption in the trauma bay. I have seen physicians reduced to tears by this disruption. Some activities need disruption. Typing with your back to the patient. Onerous documentation to facilitate billing. Faxing medical records. Will these be disrupted? Who is the customer?
In 1999, the Institute of Medicine started this process, telling us, “To err is human.” I now respond with another Alexander Pope quote, “The same ambition can destroy or save.” The money and influence of EHR vendors destroyed the chance to nationalize the most successful EHR our country has ever seen. What happens now? EHRs are incontrovertibly associated with burnout. Burnout is incontrovertibly associated with outcomes ranging from early retirement to suicide. EHRs cause physician harm. Major vendors can follow the Big Tobacco play book and deny the obvious, but the burden of proof is shifting to them. With their billions of dollars in profits, what have they done to study this problem? To help?
Who is their customer?
Dr. Sheahan is the Claude C. Craighead Jr. Professor and Chair, division of vascular and endovascular surgery, Louisiana State University Health Sciences Center, New Orleans.
References
Institute of Medicine (US) Committee on Quality of Health Care in America. 2000. To Err Is Human: Building a Safer Health System. Washington: The National Academies Press.
Weed LL. Medical records that guide and teach. N Engl J Med. 1968 Mar 14;278(11):593-600.
Longman P. “Best Care Anywhere: Why VA Health Care Is Better Than Yours.” (Oakland: Berrett-Koehler Publishers).
The EHR and our troubled health care system, Part 1
The EHR and our troubled health care system, Part 1
In 2000, the Institute of Medicine published “To Err Is Human,” a landmark study that warned that as many as 98,000 people die annually as a result of medical errors. One conclusion of the report stated, “When patients see multiple providers in different settings, none of whom has access to complete information, it becomes easier for things to go wrong.” Government and public reaction to the study resulted in the rushed integration of electronic health records into the U.S. medical system. EHR vendors promised solutions that included a dramatic reduction of preventable errors, a simplified system of physician communication, and the consolidation of a patient’s salient medical information into a single, transferable file. Now, almost 20 years later, these promises remain mostly unfilled. How did we get here?
Systems of medical records have been in place since 1600 B.C. For thousands of years, they consisted mainly of the patient’s diagnosis and the physician’s treatment. In 1968, the New England Journal of Medicine published the special article “Medical Records That Guide and Teach” by Lawrence L. Weed, MD. In the report, Dr. Weed advocated for the organization of medical records by problems rather than by a single diagnosis. This was the birth of our modern system. Medical records would now include lists of symptoms, findings, and problems that would organize the physician’s planning and allow third parties to confirm the initial diagnosis. Nearly concurrent with this publication, the next major innovation was developing in a very unusual location.
In 1999, Fortune magazine labeled Jack Welch “Manager of the Century” for his innovative work as CEO of General Electric. His techniques involved cutting waste and streamlining his workforce. While these methods were somewhat controversial, GE’s market value increased dramatically under his watch. The publishers at Fortune became interested in finding similar innovators in other fields. In this pursuit, they sent journalist Philip Longman to find the “Jack Welch” of health care.
Mr. Longman had recently lost his wife to breast cancer and was becoming obsessed with medical errors and health care quality integration. He set out to discover the best health care system in the United States. After months of research, Mr. Longman reached a startling conclusion. By nearly every metric, the Veterans Affairs system produced the highest quality of care. The key factor in upholding that quality appeared to be the EHR system VistA (Veterans Information Systems and Technology Architecture).
The development of VistA was a grassroots effort begun in the 1970s. Using Tandy computers and Wang processors, the VA “hardhats” sought to develop an electronic system for medical records and communication. This effort was initially opposed and driven underground by the central bureaucracy. Laptops were confiscated, people were fired. Still, development continued, and in 1978, the Decentralized Hospital Computer Program was launched at 20 VA sites. The national rollout occurred in 1994 under the name VistA.
VistA was developed by doctors, for doctors, and routinely enjoys the highest satisfaction rates among all available EHRs. VistA also is an open source model; its code is readily available on the VA website. After seeing the evidence of VistA’s efficacy, Representative Pete Stark (D-CA) introduced HR 6898 on Sept. 15, 2008. The bill would establish a large federal open source health IT system that private hospitals could leverage. The bill also mandated that only open source solutions would receive federal funding. As opposed to proprietary systems, open source models allow for rapid innovation, easy personal configuration, and incorporation of open source apps from unlimited numbers of contributors.
HR 6898 never passed, despite initial bipartisan support. By relying on lobbyists, marketing, and money, the proprietary EHR vendors killed the Stark bill. After a 4-month scramble, the Health Information Technology for Economic and Clinical Health Act (HITECH) passed, with EHR vendor support. HITECH established a certification system for EHRs. While the Stark bill envisioned a single, open source network, there were soon hundreds of certified EHR systems in the United States.
Before the HITECH Act, many EHRs existed, but several barriers blocked full implementation. Early systems were essentially electronic filing cabinets. Their developers had not anticipated the lack of standardization among physicians and hospital systems. The need for custom EHR bases frustrated the vendors. The question of marketing was omnipresent. Who was the actual customer? An economic model developed in which clinicians would bear the time and even financial costs as the benefits would be passed on to insurers, hospitals, and, presumably, the patients.
EHRs needed to become practical, affordable, and interoperable, but who was demanding this? Where was the financial motivation? In the beginning, vendors of EHRs had to convince doctors, the public, and the government of their worth. Now, essentially mandated by the HITECH Act, they only had to sell themselves to hospital administrators, who often had a different motive. Profits.
Many of today’s EHRs are simply modified billing platforms, and doctors are paying the price. The Meaningful Use standards were meant to provide financial incentives for EHR adoption. Stage 2 required EHRs to be able to transport clinical information from one system to another. Looking at our actual practices can provide a master class in the gap between “be able to” and “actually doing.” Again, who does the EHR vendor see as the customer? Certainly not the physician. My patients can list every type of inferior vena cava filter (or at least those with pending legal action), but most of them have never heard of an EHR. Just like “service lines,” EHRs can make it very difficult for patients to seek care outside of their primary system. Who would see this barrier in communication as a perk and not a deficiency? Hospital administrators. The free transfer of medical records is bad for business. Therefore, hospitals don’t prioritize it in their EHRs. The EHR vendors also benefit since an easy transfer of records would simplify a hospital’s transition from one EHR to another. So, as with most deficiencies in the EHR, physicians are left to find ways around these problems. Sometimes, we need to go to comical lengths.
Two months ago, a patient pointed to a large machine behind our check-in desk. “What is that,” he asked incredulously; it was a fax machine. While my competence with this apparatus is marginal (my office staff has taken to yelling “doctor faxing!” to alert one another that I am about to inadvertently copy or scan my documents into oblivion), faxes remain a mainstay of medical care. Abandoned by modern business practices as a relic of the 1980s, why are we constantly faxing medical information? Because we are not the customer.
Disruption is now a favorable term in business. Doctors are busy people. BUSY people. Most of us walk a tightrope, a razor-thin timeline. Will we see the next patient in time, the next surgery? Will we get the medical records done today? Will we get the dictations done before being suspended? Will we make the committee meeting, the conference call, the next clinic across town? Will we have dinner with our spouse or see our kids today? Will we make it to the parent-teacher conference inexplicably scheduled for 10:45 a.m. on a Tuesday??!! When deciding between work commitments and family, we side with work overwhelmingly (and depressingly). Explaining this to a layperson is an impossible feat. I have stopped trying, stopped making excuses. Only we know how catastrophic “disruption” can be. Disruption in a 40-patient clinic. Disruption in the trauma bay. I have seen physicians reduced to tears by this disruption. Some activities need disruption. Typing with your back to the patient. Onerous documentation to facilitate billing. Faxing medical records. Will these be disrupted? Who is the customer?
In 1999, the Institute of Medicine started this process, telling us, “To err is human.” I now respond with another Alexander Pope quote, “The same ambition can destroy or save.” The money and influence of EHR vendors destroyed the chance to nationalize the most successful EHR our country has ever seen. What happens now? EHRs are incontrovertibly associated with burnout. Burnout is incontrovertibly associated with outcomes ranging from early retirement to suicide. EHRs cause physician harm. Major vendors can follow the Big Tobacco play book and deny the obvious, but the burden of proof is shifting to them. With their billions of dollars in profits, what have they done to study this problem? To help?
Who is their customer?
Dr. Sheahan is the Claude C. Craighead Jr. Professor and Chair, division of vascular and endovascular surgery, Louisiana State University Health Sciences Center, New Orleans.
References
Institute of Medicine (US) Committee on Quality of Health Care in America. 2000. To Err Is Human: Building a Safer Health System. Washington: The National Academies Press.
Weed LL. Medical records that guide and teach. N Engl J Med. 1968 Mar 14;278(11):593-600.
Longman P. “Best Care Anywhere: Why VA Health Care Is Better Than Yours.” (Oakland: Berrett-Koehler Publishers).
In 2000, the Institute of Medicine published “To Err Is Human,” a landmark study that warned that as many as 98,000 people die annually as a result of medical errors. One conclusion of the report stated, “When patients see multiple providers in different settings, none of whom has access to complete information, it becomes easier for things to go wrong.” Government and public reaction to the study resulted in the rushed integration of electronic health records into the U.S. medical system. EHR vendors promised solutions that included a dramatic reduction of preventable errors, a simplified system of physician communication, and the consolidation of a patient’s salient medical information into a single, transferable file. Now, almost 20 years later, these promises remain mostly unfilled. How did we get here?
Systems of medical records have been in place since 1600 B.C. For thousands of years, they consisted mainly of the patient’s diagnosis and the physician’s treatment. In 1968, the New England Journal of Medicine published the special article “Medical Records That Guide and Teach” by Lawrence L. Weed, MD. In the report, Dr. Weed advocated for the organization of medical records by problems rather than by a single diagnosis. This was the birth of our modern system. Medical records would now include lists of symptoms, findings, and problems that would organize the physician’s planning and allow third parties to confirm the initial diagnosis. Nearly concurrent with this publication, the next major innovation was developing in a very unusual location.
In 1999, Fortune magazine labeled Jack Welch “Manager of the Century” for his innovative work as CEO of General Electric. His techniques involved cutting waste and streamlining his workforce. While these methods were somewhat controversial, GE’s market value increased dramatically under his watch. The publishers at Fortune became interested in finding similar innovators in other fields. In this pursuit, they sent journalist Philip Longman to find the “Jack Welch” of health care.
Mr. Longman had recently lost his wife to breast cancer and was becoming obsessed with medical errors and health care quality integration. He set out to discover the best health care system in the United States. After months of research, Mr. Longman reached a startling conclusion. By nearly every metric, the Veterans Affairs system produced the highest quality of care. The key factor in upholding that quality appeared to be the EHR system VistA (Veterans Information Systems and Technology Architecture).
The development of VistA was a grassroots effort begun in the 1970s. Using Tandy computers and Wang processors, the VA “hardhats” sought to develop an electronic system for medical records and communication. This effort was initially opposed and driven underground by the central bureaucracy. Laptops were confiscated, people were fired. Still, development continued, and in 1978, the Decentralized Hospital Computer Program was launched at 20 VA sites. The national rollout occurred in 1994 under the name VistA.
VistA was developed by doctors, for doctors, and routinely enjoys the highest satisfaction rates among all available EHRs. VistA also is an open source model; its code is readily available on the VA website. After seeing the evidence of VistA’s efficacy, Representative Pete Stark (D-CA) introduced HR 6898 on Sept. 15, 2008. The bill would establish a large federal open source health IT system that private hospitals could leverage. The bill also mandated that only open source solutions would receive federal funding. As opposed to proprietary systems, open source models allow for rapid innovation, easy personal configuration, and incorporation of open source apps from unlimited numbers of contributors.
HR 6898 never passed, despite initial bipartisan support. By relying on lobbyists, marketing, and money, the proprietary EHR vendors killed the Stark bill. After a 4-month scramble, the Health Information Technology for Economic and Clinical Health Act (HITECH) passed, with EHR vendor support. HITECH established a certification system for EHRs. While the Stark bill envisioned a single, open source network, there were soon hundreds of certified EHR systems in the United States.
Before the HITECH Act, many EHRs existed, but several barriers blocked full implementation. Early systems were essentially electronic filing cabinets. Their developers had not anticipated the lack of standardization among physicians and hospital systems. The need for custom EHR bases frustrated the vendors. The question of marketing was omnipresent. Who was the actual customer? An economic model developed in which clinicians would bear the time and even financial costs as the benefits would be passed on to insurers, hospitals, and, presumably, the patients.
EHRs needed to become practical, affordable, and interoperable, but who was demanding this? Where was the financial motivation? In the beginning, vendors of EHRs had to convince doctors, the public, and the government of their worth. Now, essentially mandated by the HITECH Act, they only had to sell themselves to hospital administrators, who often had a different motive. Profits.
Many of today’s EHRs are simply modified billing platforms, and doctors are paying the price. The Meaningful Use standards were meant to provide financial incentives for EHR adoption. Stage 2 required EHRs to be able to transport clinical information from one system to another. Looking at our actual practices can provide a master class in the gap between “be able to” and “actually doing.” Again, who does the EHR vendor see as the customer? Certainly not the physician. My patients can list every type of inferior vena cava filter (or at least those with pending legal action), but most of them have never heard of an EHR. Just like “service lines,” EHRs can make it very difficult for patients to seek care outside of their primary system. Who would see this barrier in communication as a perk and not a deficiency? Hospital administrators. The free transfer of medical records is bad for business. Therefore, hospitals don’t prioritize it in their EHRs. The EHR vendors also benefit since an easy transfer of records would simplify a hospital’s transition from one EHR to another. So, as with most deficiencies in the EHR, physicians are left to find ways around these problems. Sometimes, we need to go to comical lengths.
Two months ago, a patient pointed to a large machine behind our check-in desk. “What is that,” he asked incredulously; it was a fax machine. While my competence with this apparatus is marginal (my office staff has taken to yelling “doctor faxing!” to alert one another that I am about to inadvertently copy or scan my documents into oblivion), faxes remain a mainstay of medical care. Abandoned by modern business practices as a relic of the 1980s, why are we constantly faxing medical information? Because we are not the customer.
Disruption is now a favorable term in business. Doctors are busy people. BUSY people. Most of us walk a tightrope, a razor-thin timeline. Will we see the next patient in time, the next surgery? Will we get the medical records done today? Will we get the dictations done before being suspended? Will we make the committee meeting, the conference call, the next clinic across town? Will we have dinner with our spouse or see our kids today? Will we make it to the parent-teacher conference inexplicably scheduled for 10:45 a.m. on a Tuesday??!! When deciding between work commitments and family, we side with work overwhelmingly (and depressingly). Explaining this to a layperson is an impossible feat. I have stopped trying, stopped making excuses. Only we know how catastrophic “disruption” can be. Disruption in a 40-patient clinic. Disruption in the trauma bay. I have seen physicians reduced to tears by this disruption. Some activities need disruption. Typing with your back to the patient. Onerous documentation to facilitate billing. Faxing medical records. Will these be disrupted? Who is the customer?
In 1999, the Institute of Medicine started this process, telling us, “To err is human.” I now respond with another Alexander Pope quote, “The same ambition can destroy or save.” The money and influence of EHR vendors destroyed the chance to nationalize the most successful EHR our country has ever seen. What happens now? EHRs are incontrovertibly associated with burnout. Burnout is incontrovertibly associated with outcomes ranging from early retirement to suicide. EHRs cause physician harm. Major vendors can follow the Big Tobacco play book and deny the obvious, but the burden of proof is shifting to them. With their billions of dollars in profits, what have they done to study this problem? To help?
Who is their customer?
Dr. Sheahan is the Claude C. Craighead Jr. Professor and Chair, division of vascular and endovascular surgery, Louisiana State University Health Sciences Center, New Orleans.
References
Institute of Medicine (US) Committee on Quality of Health Care in America. 2000. To Err Is Human: Building a Safer Health System. Washington: The National Academies Press.
Weed LL. Medical records that guide and teach. N Engl J Med. 1968 Mar 14;278(11):593-600.
Longman P. “Best Care Anywhere: Why VA Health Care Is Better Than Yours.” (Oakland: Berrett-Koehler Publishers).
Constipation because of deportation-related trauma
I recently saw Anaeli (not her real name), an 8-year-old Mexican American girl, in clinic for worsening constipation. Her mother brought her in because of a year’s worth of increasingly irregular bowel movements. Looking through her chart, it was easy to find the starting point of Anaeli’s constipation – it aligned with her father’s deportation. U.S. Immigration and Customs Enforcement had arrested him while he was dropping Anaeli off at school.

Family separation at the border has reignited awareness of the effects of adverse childhood events. As a young pediatrician training in San Diego, I see both the impact of immigration policies on children and the resulting need for trauma-informed care. We need coordinated efforts in homes, schools, and hospitals to effectively treat affected kids.
For the past year, Anaeli’s caregivers have struggled to do so. She has been acting out, frequently crying and throwing fits about going to school. Anaeli has missed about 30 days of school because of behavioral issues.
What does 30 fewer days of first grade look like? Anaeli’s language skills are at a standstill. She cannot follow complex directions like her peers. Because of her academic shortcomings, Anaeli earned an individualized education plan and a teacher’s aide to help her focus. This aide has adopted a “tough love” attitude. Anaeli’s mom reports that she is often disciplined by long time-outs in the classroom bathroom and worries that this discipline is causing Anaeli to withhold stool to a point of loosing control and soiling herself. Since working with the aide, Anaeli has been having daily “accidents,” stooling in her pants, despite being toilet trained for years.
After the appointment, I called the school three times and was finally able to get in touch with Anaeli’s aide. She expressed frustration over Anaeli’s “lack of trying” and “meltdown” reaction to discipline. She said Anaeli’s mom was not enforcing limits at home. She told me she had successfully used time-outs in the bathroom with her own children. When I reviewed the impact of childhood trauma and more appropriate approaches to discipline, the aide grew defensive and challenged me by asking if I have kids of my own.
While I disagreed with the aide’s methods, I understood her frustration. Anaeli is not easy to help. But she is just one of a generation of children affected by the deportation of a family member. Like them, Anaeli’s health is deeply affected by stress in a way that she many not be able to verbalize.
Trauma-informed care should be an essential lens for caregivers of children who have been separated from their family. Resolving Anaeli’s constipation will require a concerted effort by her mom, health providers, teachers, and aides to encourage good behavior, use measured disciplinary tactics, and consume a high-fiber diet. In doing so, we can provide children like her with the appropriate environment to build resilience.
Dr. Parekh is a pediatrician in San Diego. Email her at [email protected].
I recently saw Anaeli (not her real name), an 8-year-old Mexican American girl, in clinic for worsening constipation. Her mother brought her in because of a year’s worth of increasingly irregular bowel movements. Looking through her chart, it was easy to find the starting point of Anaeli’s constipation – it aligned with her father’s deportation. U.S. Immigration and Customs Enforcement had arrested him while he was dropping Anaeli off at school.
Family separation at the border has reignited awareness of the effects of adverse childhood events. As a young pediatrician training in San Diego, I see both the impact of immigration policies on children and the resulting need for trauma-informed care. We need coordinated efforts in homes, schools, and hospitals to effectively treat affected kids.
For the past year, Anaeli’s caregivers have struggled to do so. She has been acting out, frequently crying and throwing fits about going to school. Anaeli has missed about 30 days of school because of behavioral issues.
What does 30 fewer days of first grade look like? Anaeli’s language skills are at a standstill. She cannot follow complex directions like her peers. Because of her academic shortcomings, Anaeli earned an individualized education plan and a teacher’s aide to help her focus. This aide has adopted a “tough love” attitude. Anaeli’s mom reports that she is often disciplined by long time-outs in the classroom bathroom and worries that this discipline is causing Anaeli to withhold stool to a point of loosing control and soiling herself. Since working with the aide, Anaeli has been having daily “accidents,” stooling in her pants, despite being toilet trained for years.
After the appointment, I called the school three times and was finally able to get in touch with Anaeli’s aide. She expressed frustration over Anaeli’s “lack of trying” and “meltdown” reaction to discipline. She said Anaeli’s mom was not enforcing limits at home. She told me she had successfully used time-outs in the bathroom with her own children. When I reviewed the impact of childhood trauma and more appropriate approaches to discipline, the aide grew defensive and challenged me by asking if I have kids of my own.
While I disagreed with the aide’s methods, I understood her frustration. Anaeli is not easy to help. But she is just one of a generation of children affected by the deportation of a family member. Like them, Anaeli’s health is deeply affected by stress in a way that she many not be able to verbalize.
Trauma-informed care should be an essential lens for caregivers of children who have been separated from their family. Resolving Anaeli’s constipation will require a concerted effort by her mom, health providers, teachers, and aides to encourage good behavior, use measured disciplinary tactics, and consume a high-fiber diet. In doing so, we can provide children like her with the appropriate environment to build resilience.
Dr. Parekh is a pediatrician in San Diego. Email her at [email protected].
I recently saw Anaeli (not her real name), an 8-year-old Mexican American girl, in clinic for worsening constipation. Her mother brought her in because of a year’s worth of increasingly irregular bowel movements. Looking through her chart, it was easy to find the starting point of Anaeli’s constipation – it aligned with her father’s deportation. U.S. Immigration and Customs Enforcement had arrested him while he was dropping Anaeli off at school.
Family separation at the border has reignited awareness of the effects of adverse childhood events. As a young pediatrician training in San Diego, I see both the impact of immigration policies on children and the resulting need for trauma-informed care. We need coordinated efforts in homes, schools, and hospitals to effectively treat affected kids.
For the past year, Anaeli’s caregivers have struggled to do so. She has been acting out, frequently crying and throwing fits about going to school. Anaeli has missed about 30 days of school because of behavioral issues.
What does 30 fewer days of first grade look like? Anaeli’s language skills are at a standstill. She cannot follow complex directions like her peers. Because of her academic shortcomings, Anaeli earned an individualized education plan and a teacher’s aide to help her focus. This aide has adopted a “tough love” attitude. Anaeli’s mom reports that she is often disciplined by long time-outs in the classroom bathroom and worries that this discipline is causing Anaeli to withhold stool to a point of loosing control and soiling herself. Since working with the aide, Anaeli has been having daily “accidents,” stooling in her pants, despite being toilet trained for years.
After the appointment, I called the school three times and was finally able to get in touch with Anaeli’s aide. She expressed frustration over Anaeli’s “lack of trying” and “meltdown” reaction to discipline. She said Anaeli’s mom was not enforcing limits at home. She told me she had successfully used time-outs in the bathroom with her own children. When I reviewed the impact of childhood trauma and more appropriate approaches to discipline, the aide grew defensive and challenged me by asking if I have kids of my own.
While I disagreed with the aide’s methods, I understood her frustration. Anaeli is not easy to help. But she is just one of a generation of children affected by the deportation of a family member. Like them, Anaeli’s health is deeply affected by stress in a way that she many not be able to verbalize.
Trauma-informed care should be an essential lens for caregivers of children who have been separated from their family. Resolving Anaeli’s constipation will require a concerted effort by her mom, health providers, teachers, and aides to encourage good behavior, use measured disciplinary tactics, and consume a high-fiber diet. In doing so, we can provide children like her with the appropriate environment to build resilience.
Dr. Parekh is a pediatrician in San Diego. Email her at [email protected].
Thiopurines linked to zoster in IBD patients
For patients with inflammatory bowel disease (IBD), thiopurine exposure was associated with a significantly increased risk of herpes zoster, compared with 5-aminosalicylic acid (5-ASA) monotherapy, according to the results of two large retrospective cohort studies.
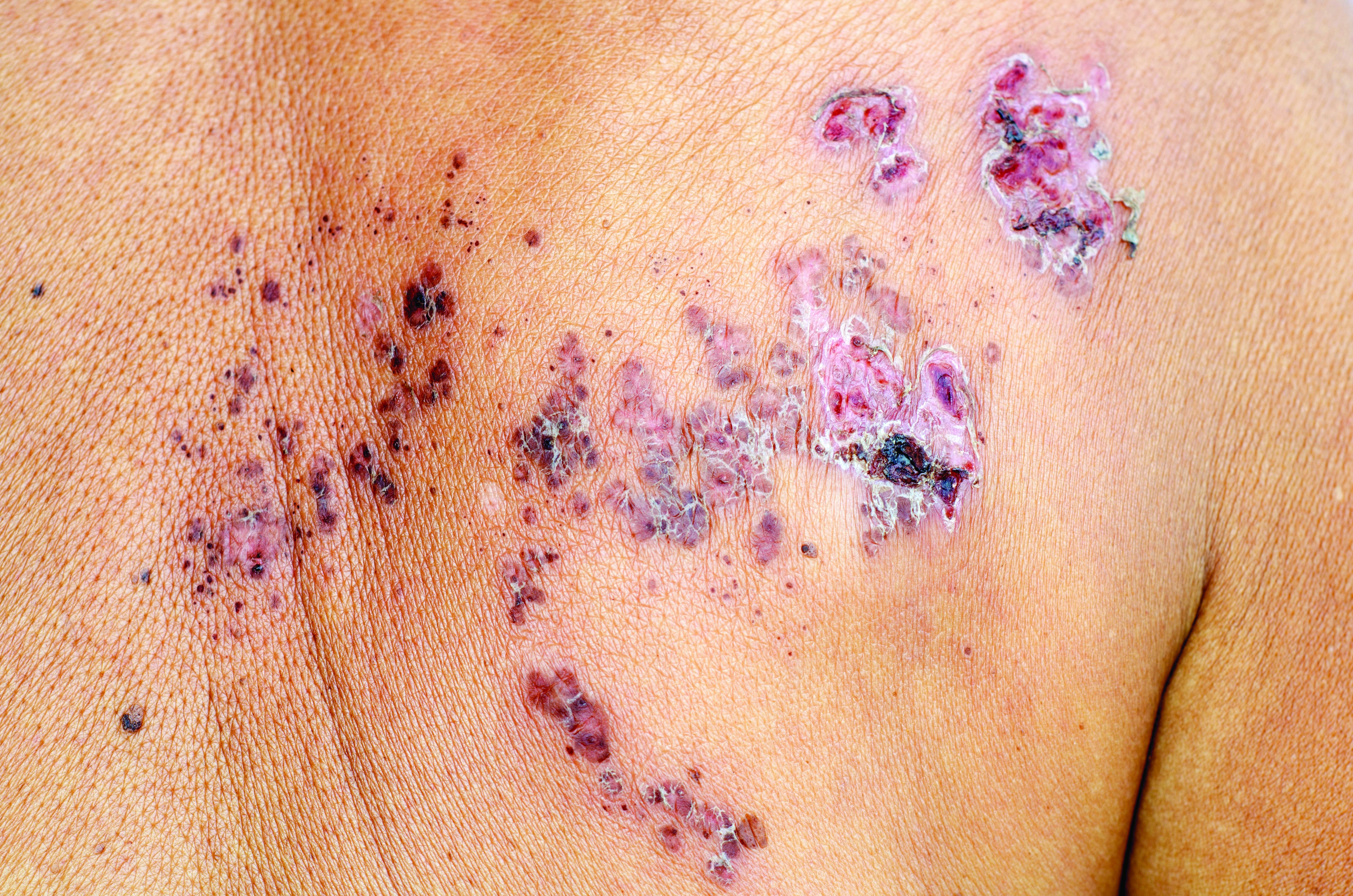
In the multivariable analysis, thiopurine monotherapy was linked to about a 47% increase in the risk of herpes zoster, compared with 5-ASA monotherapy (adjusted hazard ratio, 1.47; 95% confidence interval, 1.31-1.65; P less than .001). Combination therapy with thiopurines and tumor necrosis factor antagonists conferred about a 65% increase in zoster risk (aHR, 1.65; 95% CI, 1.22-2.23; P = .001). However, tumor necrosis factor–antagonist monotherapy did not appear to significantly increase the risk of zoster when compared with 5-ASA monotherapy, reported Nabeel Khan, MD, of the University of Pennsylvania in Philadelphia, and his associates.
“Compared to [patients without] IBD, ulcerative colitis (UC) and Crohn’s disease (CD) each were associated with significantly increased risk of herpes zoster infection,” the researchers wrote online in Clinical Gastroenterology and Hepatology. “With the approval of a new and potentially safer vaccine for herpes zoster, the effects of immunization of patients with IBD should be investigated.”
Past studies have linked IBD with a 1.2- to 1.8-fold increase in the risk of zoster, but these studies date to the prebiologic era or excluded patients who were in their midsixties or older, the researchers wrote. “Additionally, these prior studies have not assessed the validity of the codes used to identify herpes zoster and also did not account for the impact of vaccination,” they added. “They also did not take into consideration the severity of the disease or degree of steroid exposure.”
Therefore, the researchers conducted two retrospective cohort studies of patients in the United States Department of Veterans Affairs between 2000 and 2016. The first cohort study compared the incidence of herpes zoster among patients with IBD who received 5-ASA alone with matched patients without IBD. The second cohort study measured the incidence of herpes zoster in patients with IBD who received various medications and combination regimen. “The VA has a predominantly older population, which makes it an ideal cohort to study herpes zoster incidence in a high-risk population,” the investigators noted. “Unlike insurance databases, the VA database can be validated internally and vaccination records are documented.”
After adjusting for age, race, sex, geographic region, disease flare, corticosteroid use, and baseline comorbidities, the estimated hazard of developing herpes zoster was 1.81 (95% confidence interval, 1.56-2.11) among patients with ulcerative colitis and 1.56 (95% CI, 1.28-1.91) among patients with Crohn’s disease, as compared with patients without IBD. Regardless of their age or the medications they were receiving, patients with IBD had a higher incidence of zoster than the oldest group of patients without IBD (older than 60 years), regardless of age or medication. “The highest risk of herpes zoster was observed in patients with IBD who were less than 60 years of age and on combination therapy,” the investigators wrote. “Patients with IBD younger than 50 years who were on combination therapy had higher risk of herpes zoster, compared with patients with IBD older than 60 years of age who were not on immunosuppressive therapy.” Based on the findings, they recommended studying the efficacy of widespread use of the new herpes zoster vaccine in patients with IBD.
Pfizer provided unrestricted research funding but was not otherwise involved in the study. One coinvestigator disclosed ties to Pfizer and several other pharmaceutical companies. The remaining investigators reported having no conflicts of interest.
SOURCE: Khan N et al. Clin Gastroenterol Hepatol. 2018 Jan 5. doi: 10.1016/j.cgh.2017.12.052.
Patients with inflammatory bowel disease are thought to have altered immune regulation, which may increase the risk of systemic complications including infections like herpes zoster. Many of the prior studies assessing the risk of herpes zoster in IBD patients were done before the advent of biologics and excluded older patients, thereby limiting their utility. This study by Khan et al. aimed to better estimate the incidence and risk factors for development of herpes zoster and to determine the effect of immunosuppressant use on this risk. In two large, retrospective cohort studies they found that, compared with patients without IBD, patients with IBD had a significantly increased risk of developing herpes zoster. Furthermore, this risk was higher in those with recent or cumulative steroid use and in those treated with thiopurines (as monotherapy or in combination with anti-TNF agents). Interestingly, exposure to TNF antagonists alone was not associated with an increased risk of herpes zoster infection.

Richa Shukla, MD, assistant professor, section of gastroenterology and hepatology, Baylor College of Medicine, Houston.
Patients with inflammatory bowel disease are thought to have altered immune regulation, which may increase the risk of systemic complications including infections like herpes zoster. Many of the prior studies assessing the risk of herpes zoster in IBD patients were done before the advent of biologics and excluded older patients, thereby limiting their utility. This study by Khan et al. aimed to better estimate the incidence and risk factors for development of herpes zoster and to determine the effect of immunosuppressant use on this risk. In two large, retrospective cohort studies they found that, compared with patients without IBD, patients with IBD had a significantly increased risk of developing herpes zoster. Furthermore, this risk was higher in those with recent or cumulative steroid use and in those treated with thiopurines (as monotherapy or in combination with anti-TNF agents). Interestingly, exposure to TNF antagonists alone was not associated with an increased risk of herpes zoster infection.
Richa Shukla, MD, assistant professor, section of gastroenterology and hepatology, Baylor College of Medicine, Houston.
Patients with inflammatory bowel disease are thought to have altered immune regulation, which may increase the risk of systemic complications including infections like herpes zoster. Many of the prior studies assessing the risk of herpes zoster in IBD patients were done before the advent of biologics and excluded older patients, thereby limiting their utility. This study by Khan et al. aimed to better estimate the incidence and risk factors for development of herpes zoster and to determine the effect of immunosuppressant use on this risk. In two large, retrospective cohort studies they found that, compared with patients without IBD, patients with IBD had a significantly increased risk of developing herpes zoster. Furthermore, this risk was higher in those with recent or cumulative steroid use and in those treated with thiopurines (as monotherapy or in combination with anti-TNF agents). Interestingly, exposure to TNF antagonists alone was not associated with an increased risk of herpes zoster infection.
Richa Shukla, MD, assistant professor, section of gastroenterology and hepatology, Baylor College of Medicine, Houston.
For patients with inflammatory bowel disease (IBD), thiopurine exposure was associated with a significantly increased risk of herpes zoster, compared with 5-aminosalicylic acid (5-ASA) monotherapy, according to the results of two large retrospective cohort studies.
In the multivariable analysis, thiopurine monotherapy was linked to about a 47% increase in the risk of herpes zoster, compared with 5-ASA monotherapy (adjusted hazard ratio, 1.47; 95% confidence interval, 1.31-1.65; P less than .001). Combination therapy with thiopurines and tumor necrosis factor antagonists conferred about a 65% increase in zoster risk (aHR, 1.65; 95% CI, 1.22-2.23; P = .001). However, tumor necrosis factor–antagonist monotherapy did not appear to significantly increase the risk of zoster when compared with 5-ASA monotherapy, reported Nabeel Khan, MD, of the University of Pennsylvania in Philadelphia, and his associates.
“Compared to [patients without] IBD, ulcerative colitis (UC) and Crohn’s disease (CD) each were associated with significantly increased risk of herpes zoster infection,” the researchers wrote online in Clinical Gastroenterology and Hepatology. “With the approval of a new and potentially safer vaccine for herpes zoster, the effects of immunization of patients with IBD should be investigated.”
Past studies have linked IBD with a 1.2- to 1.8-fold increase in the risk of zoster, but these studies date to the prebiologic era or excluded patients who were in their midsixties or older, the researchers wrote. “Additionally, these prior studies have not assessed the validity of the codes used to identify herpes zoster and also did not account for the impact of vaccination,” they added. “They also did not take into consideration the severity of the disease or degree of steroid exposure.”
Therefore, the researchers conducted two retrospective cohort studies of patients in the United States Department of Veterans Affairs between 2000 and 2016. The first cohort study compared the incidence of herpes zoster among patients with IBD who received 5-ASA alone with matched patients without IBD. The second cohort study measured the incidence of herpes zoster in patients with IBD who received various medications and combination regimen. “The VA has a predominantly older population, which makes it an ideal cohort to study herpes zoster incidence in a high-risk population,” the investigators noted. “Unlike insurance databases, the VA database can be validated internally and vaccination records are documented.”
After adjusting for age, race, sex, geographic region, disease flare, corticosteroid use, and baseline comorbidities, the estimated hazard of developing herpes zoster was 1.81 (95% confidence interval, 1.56-2.11) among patients with ulcerative colitis and 1.56 (95% CI, 1.28-1.91) among patients with Crohn’s disease, as compared with patients without IBD. Regardless of their age or the medications they were receiving, patients with IBD had a higher incidence of zoster than the oldest group of patients without IBD (older than 60 years), regardless of age or medication. “The highest risk of herpes zoster was observed in patients with IBD who were less than 60 years of age and on combination therapy,” the investigators wrote. “Patients with IBD younger than 50 years who were on combination therapy had higher risk of herpes zoster, compared with patients with IBD older than 60 years of age who were not on immunosuppressive therapy.” Based on the findings, they recommended studying the efficacy of widespread use of the new herpes zoster vaccine in patients with IBD.
Pfizer provided unrestricted research funding but was not otherwise involved in the study. One coinvestigator disclosed ties to Pfizer and several other pharmaceutical companies. The remaining investigators reported having no conflicts of interest.
SOURCE: Khan N et al. Clin Gastroenterol Hepatol. 2018 Jan 5. doi: 10.1016/j.cgh.2017.12.052.
For patients with inflammatory bowel disease (IBD), thiopurine exposure was associated with a significantly increased risk of herpes zoster, compared with 5-aminosalicylic acid (5-ASA) monotherapy, according to the results of two large retrospective cohort studies.
In the multivariable analysis, thiopurine monotherapy was linked to about a 47% increase in the risk of herpes zoster, compared with 5-ASA monotherapy (adjusted hazard ratio, 1.47; 95% confidence interval, 1.31-1.65; P less than .001). Combination therapy with thiopurines and tumor necrosis factor antagonists conferred about a 65% increase in zoster risk (aHR, 1.65; 95% CI, 1.22-2.23; P = .001). However, tumor necrosis factor–antagonist monotherapy did not appear to significantly increase the risk of zoster when compared with 5-ASA monotherapy, reported Nabeel Khan, MD, of the University of Pennsylvania in Philadelphia, and his associates.
“Compared to [patients without] IBD, ulcerative colitis (UC) and Crohn’s disease (CD) each were associated with significantly increased risk of herpes zoster infection,” the researchers wrote online in Clinical Gastroenterology and Hepatology. “With the approval of a new and potentially safer vaccine for herpes zoster, the effects of immunization of patients with IBD should be investigated.”
Past studies have linked IBD with a 1.2- to 1.8-fold increase in the risk of zoster, but these studies date to the prebiologic era or excluded patients who were in their midsixties or older, the researchers wrote. “Additionally, these prior studies have not assessed the validity of the codes used to identify herpes zoster and also did not account for the impact of vaccination,” they added. “They also did not take into consideration the severity of the disease or degree of steroid exposure.”
Therefore, the researchers conducted two retrospective cohort studies of patients in the United States Department of Veterans Affairs between 2000 and 2016. The first cohort study compared the incidence of herpes zoster among patients with IBD who received 5-ASA alone with matched patients without IBD. The second cohort study measured the incidence of herpes zoster in patients with IBD who received various medications and combination regimen. “The VA has a predominantly older population, which makes it an ideal cohort to study herpes zoster incidence in a high-risk population,” the investigators noted. “Unlike insurance databases, the VA database can be validated internally and vaccination records are documented.”
After adjusting for age, race, sex, geographic region, disease flare, corticosteroid use, and baseline comorbidities, the estimated hazard of developing herpes zoster was 1.81 (95% confidence interval, 1.56-2.11) among patients with ulcerative colitis and 1.56 (95% CI, 1.28-1.91) among patients with Crohn’s disease, as compared with patients without IBD. Regardless of their age or the medications they were receiving, patients with IBD had a higher incidence of zoster than the oldest group of patients without IBD (older than 60 years), regardless of age or medication. “The highest risk of herpes zoster was observed in patients with IBD who were less than 60 years of age and on combination therapy,” the investigators wrote. “Patients with IBD younger than 50 years who were on combination therapy had higher risk of herpes zoster, compared with patients with IBD older than 60 years of age who were not on immunosuppressive therapy.” Based on the findings, they recommended studying the efficacy of widespread use of the new herpes zoster vaccine in patients with IBD.
Pfizer provided unrestricted research funding but was not otherwise involved in the study. One coinvestigator disclosed ties to Pfizer and several other pharmaceutical companies. The remaining investigators reported having no conflicts of interest.
SOURCE: Khan N et al. Clin Gastroenterol Hepatol. 2018 Jan 5. doi: 10.1016/j.cgh.2017.12.052.
FROM CLINICAL GASTROENTEROLOGY AND HEPATOLOGY
Key clinical point: For patients with inflammatory bowel disease, thiopurine exposure was associated with a significantly increased risk of herpes zoster, compared with 5-aminosalicylic acid monotherapy.
Major finding: The adjusted hazard ratio was 1.47 (95% confidence interval, 1.31-1.65; P less than .001).
Study details: Two large retrospective cohort studies of veterans with and without inflammatory bowel disease.
Disclosures: Pfizer provided unrestricted research funding but was not otherwise involved in the study. One coinvestigator disclosed ties to Pfizer and several other pharmaceutical companies. The remaining investigators reported having no conflicts of interest.
Source: Khan N et al. Clin Gastroenterol Hepatol. 2018 Jan 5. doi: 10.1016/j.cgh.2017.12.052.
What Are the Clinical, Laboratory, and Electrodiagnostic Features of Zinc Deficiency-Induced Peripheral Neuropathy?
Reduced tendon reflexes and an abnormal Romberg test may be common in patients with this disorder.
WASHINGTON, DC—Patients with zinc deficiency-induced peripheral neuropathy may present with paresthesia, gait abnormalities, sensory deficits, reduced tendon reflexes, an abnormal Romberg test, and increased CSF protein, according to a study presented at the 2018 Annual Meeting of the American Association of Neuromuscular & Electrodiagnostic Medicine.
Recognition of the features of zinc deficiency-induced peripheral neuropathy may help neurologists diagnose the disorder and manage patients, researchers said.
“Zinc, an essential trace element, plays a critical role in maintaining normal structural and functional conditions in the body,” said lead author Favio C. Bumanlag, Chief Technologist in the Department of Neurology at the Lewis Katz School of Medicine at Temple University in Philadelphia. “Peripheral nerves are susceptible to damage when zinc deficiency occurs.... Recognition of [zinc deficiency-induced peripheral neuropathy] will help physicians and technologists effectively manage patients.”
To study the clinical and electrophysiologic features of zinc deficiency-induced peripheral neuropathy, Mr. Bumanlag and Jin Luo, MD, PhD, Professor of Neurology and Pharmacology at Temple University, retrospectively reviewed charts in their neuromuscular clinic and EMG laboratory database to identify patients with peripheral neuropathy and zinc deficiency. They included charts from between January 1, 2015, and December 31, 2017, in their review. They excluded patients with abnormal copper levels.
Mr. Bumanlag and Dr. Luo obtained information about patients’ clinical presentations, past medical histories, BMI, neurologic examinations, and laboratory results. They also examined patients’ needle electromyograms and nerve conduction studies.
In all, they identified 12 patients with peripheral neuropathy and zinc deficiency. Patients had a mean age of 55.1. Six were female. Patients’ mean zinc level was 52.5 μg/dL, with a range of 37 μg/dL to 58 μg/dL (reference, 56–134 μg/dL). Mean copper level was 107.6 μg/dL, with a range of 84 μg/dL to 173 μg/dL (reference, 72–166 μg/dL). Eleven of the 12 patients had received an electrophysiologic evaluation.
Notable findings in presentation included paresthesia in 75 and gait abnormalities in 42%. One patient was obese (8%), and three patients had diarrhea (25%). Neurologic examination showed sensory deficits in 83%, reduced tendon reflexes in 67%, and an abnormal Romberg test in 67%. Four of five patients had increased CSF protein. Electrophysiologic evaluations showed features of demyelinating peripheral neuropathy (28%) and distally active denervation in the lower extremities.
“Zinc participates in more than 200 enzymatic reactions,” said the researchers. “Unfortunately, zinc deficiency-induced peripheral neuropathy is often misdiagnosed or delayed in diagnosis. Literature on zinc deficiency-induced peripheral neuropathy is sparse.”
Reduced tendon reflexes and an abnormal Romberg test may be common in patients with this disorder.
Reduced tendon reflexes and an abnormal Romberg test may be common in patients with this disorder.
WASHINGTON, DC—Patients with zinc deficiency-induced peripheral neuropathy may present with paresthesia, gait abnormalities, sensory deficits, reduced tendon reflexes, an abnormal Romberg test, and increased CSF protein, according to a study presented at the 2018 Annual Meeting of the American Association of Neuromuscular & Electrodiagnostic Medicine.
Recognition of the features of zinc deficiency-induced peripheral neuropathy may help neurologists diagnose the disorder and manage patients, researchers said.
“Zinc, an essential trace element, plays a critical role in maintaining normal structural and functional conditions in the body,” said lead author Favio C. Bumanlag, Chief Technologist in the Department of Neurology at the Lewis Katz School of Medicine at Temple University in Philadelphia. “Peripheral nerves are susceptible to damage when zinc deficiency occurs.... Recognition of [zinc deficiency-induced peripheral neuropathy] will help physicians and technologists effectively manage patients.”
To study the clinical and electrophysiologic features of zinc deficiency-induced peripheral neuropathy, Mr. Bumanlag and Jin Luo, MD, PhD, Professor of Neurology and Pharmacology at Temple University, retrospectively reviewed charts in their neuromuscular clinic and EMG laboratory database to identify patients with peripheral neuropathy and zinc deficiency. They included charts from between January 1, 2015, and December 31, 2017, in their review. They excluded patients with abnormal copper levels.
Mr. Bumanlag and Dr. Luo obtained information about patients’ clinical presentations, past medical histories, BMI, neurologic examinations, and laboratory results. They also examined patients’ needle electromyograms and nerve conduction studies.
In all, they identified 12 patients with peripheral neuropathy and zinc deficiency. Patients had a mean age of 55.1. Six were female. Patients’ mean zinc level was 52.5 μg/dL, with a range of 37 μg/dL to 58 μg/dL (reference, 56–134 μg/dL). Mean copper level was 107.6 μg/dL, with a range of 84 μg/dL to 173 μg/dL (reference, 72–166 μg/dL). Eleven of the 12 patients had received an electrophysiologic evaluation.
Notable findings in presentation included paresthesia in 75 and gait abnormalities in 42%. One patient was obese (8%), and three patients had diarrhea (25%). Neurologic examination showed sensory deficits in 83%, reduced tendon reflexes in 67%, and an abnormal Romberg test in 67%. Four of five patients had increased CSF protein. Electrophysiologic evaluations showed features of demyelinating peripheral neuropathy (28%) and distally active denervation in the lower extremities.
“Zinc participates in more than 200 enzymatic reactions,” said the researchers. “Unfortunately, zinc deficiency-induced peripheral neuropathy is often misdiagnosed or delayed in diagnosis. Literature on zinc deficiency-induced peripheral neuropathy is sparse.”
WASHINGTON, DC—Patients with zinc deficiency-induced peripheral neuropathy may present with paresthesia, gait abnormalities, sensory deficits, reduced tendon reflexes, an abnormal Romberg test, and increased CSF protein, according to a study presented at the 2018 Annual Meeting of the American Association of Neuromuscular & Electrodiagnostic Medicine.
Recognition of the features of zinc deficiency-induced peripheral neuropathy may help neurologists diagnose the disorder and manage patients, researchers said.
“Zinc, an essential trace element, plays a critical role in maintaining normal structural and functional conditions in the body,” said lead author Favio C. Bumanlag, Chief Technologist in the Department of Neurology at the Lewis Katz School of Medicine at Temple University in Philadelphia. “Peripheral nerves are susceptible to damage when zinc deficiency occurs.... Recognition of [zinc deficiency-induced peripheral neuropathy] will help physicians and technologists effectively manage patients.”
To study the clinical and electrophysiologic features of zinc deficiency-induced peripheral neuropathy, Mr. Bumanlag and Jin Luo, MD, PhD, Professor of Neurology and Pharmacology at Temple University, retrospectively reviewed charts in their neuromuscular clinic and EMG laboratory database to identify patients with peripheral neuropathy and zinc deficiency. They included charts from between January 1, 2015, and December 31, 2017, in their review. They excluded patients with abnormal copper levels.
Mr. Bumanlag and Dr. Luo obtained information about patients’ clinical presentations, past medical histories, BMI, neurologic examinations, and laboratory results. They also examined patients’ needle electromyograms and nerve conduction studies.
In all, they identified 12 patients with peripheral neuropathy and zinc deficiency. Patients had a mean age of 55.1. Six were female. Patients’ mean zinc level was 52.5 μg/dL, with a range of 37 μg/dL to 58 μg/dL (reference, 56–134 μg/dL). Mean copper level was 107.6 μg/dL, with a range of 84 μg/dL to 173 μg/dL (reference, 72–166 μg/dL). Eleven of the 12 patients had received an electrophysiologic evaluation.
Notable findings in presentation included paresthesia in 75 and gait abnormalities in 42%. One patient was obese (8%), and three patients had diarrhea (25%). Neurologic examination showed sensory deficits in 83%, reduced tendon reflexes in 67%, and an abnormal Romberg test in 67%. Four of five patients had increased CSF protein. Electrophysiologic evaluations showed features of demyelinating peripheral neuropathy (28%) and distally active denervation in the lower extremities.
“Zinc participates in more than 200 enzymatic reactions,” said the researchers. “Unfortunately, zinc deficiency-induced peripheral neuropathy is often misdiagnosed or delayed in diagnosis. Literature on zinc deficiency-induced peripheral neuropathy is sparse.”
Does Thymectomy Benefit Patients With Anti-MuSK Myasthenia Gravis?
Favorable clinical outcomes are not more likely in patients with anti-MuSK myasthenia gravis who undergo thymectomy versus patients who do not.
WASHINGTON, DC—Among patients with anti-muscle-specific kinase (MuSK) myasthenia gravis, thymectomy is not associated with greater likelihood of clinical improvement, according to an analysis of data from a multicenter cohort study. The results were presented at the 2018 Annual Meeting of the American Association of Neuromuscular & Electrodiagnostic Medicine (AANEM).
Although a randomized trial has demonstrated benefit from thymectomy in nonthymomatous antiacetylcholine receptor (AChR) antibody positive generalized myasthenia gravis, observational studies suggest that thymectomy may not be efficacious in anti-MuSK myasthenia gravis. Histologic studies have found that patients with anti-MuSK myasthenia gravis have less hyperplastic thymic tissue, compared with patients with anti-AChR myasthenia gravis.
To evaluate the therapeutic impact of thymectomy in patients with anti-MuSK myasthenia gravis, Katherine Clifford, a medical student at the University of Vermont Larner College of Medicine in Burlington, and colleagues analyzed data from a multicenter, retrospective, blinded review of rituximab treatment in patients with anti-MuSK myasthenia gravis. The primary outcome was favorable outcome on the Myasthenia Gravis Foundation of America (MGFA) Post-Intervention Status (PIS). The researchers defined a favorable outcome as an MGFA PIS score of minimal manifestations or better.
Secondary outcomes included prednisone dose; use of other immunosuppressant medications, IV immunoglobulin (IVIG), or plasma exchange (PLEX) treatment; and Myasthenia Gravis Status and Treatment Intensity (MGSTI).
Baseline characteristics were similar between patients with anti-MuSK myasthenia gravis who underwent thymectomy (n = 26) and those who did not (n = 29), including treatment with rituximab (42% vs 45%). Median follow-up was more than three years.
At last visit, 35% (nine of 26) of patients who underwent thymectomy had a favorable outcome, compared with 55% (16 of 29) of patients who did not undergo thymectomy. In addition, 69% of patients who underwent thymectomy were taking prednisone, compared with 41% of patients who did not undergo thymectomy (median dose, 10 mg/day vs 0 mg/day).
“After controlling for rituximab, baseline prednisone, and final IVIG/PLEX treatment, thymectomy was not associated with greater likelihood of favorable clinical outcome, but broad confidence intervals cannot exclude therapeutic effect (odds ratio, 0.43),” the investigators reported.

“The recent MGTX trial clearly demonstrated the benefit of thymectomy for patients with AChR antibody positive myasthenia gravis,” said A. Gordon Smith, MD, Cochair of the AANEM Annual Meeting Program Committee. “Ms. Clifford and her colleagues now provide compelling data suggesting thymectomy may not be effective in MuSK-positive myasthenia gravis.”
The study’s follow-up is long enough for the findings to be clinically “relevant to all physicians treating myasthenia gravis,” said Robert W. Irwin, MD, Cochair of the AANEM Annual Meeting Program Committee.
Favorable clinical outcomes are not more likely in patients with anti-MuSK myasthenia gravis who undergo thymectomy versus patients who do not.
Favorable clinical outcomes are not more likely in patients with anti-MuSK myasthenia gravis who undergo thymectomy versus patients who do not.
WASHINGTON, DC—Among patients with anti-muscle-specific kinase (MuSK) myasthenia gravis, thymectomy is not associated with greater likelihood of clinical improvement, according to an analysis of data from a multicenter cohort study. The results were presented at the 2018 Annual Meeting of the American Association of Neuromuscular & Electrodiagnostic Medicine (AANEM).
Although a randomized trial has demonstrated benefit from thymectomy in nonthymomatous antiacetylcholine receptor (AChR) antibody positive generalized myasthenia gravis, observational studies suggest that thymectomy may not be efficacious in anti-MuSK myasthenia gravis. Histologic studies have found that patients with anti-MuSK myasthenia gravis have less hyperplastic thymic tissue, compared with patients with anti-AChR myasthenia gravis.
To evaluate the therapeutic impact of thymectomy in patients with anti-MuSK myasthenia gravis, Katherine Clifford, a medical student at the University of Vermont Larner College of Medicine in Burlington, and colleagues analyzed data from a multicenter, retrospective, blinded review of rituximab treatment in patients with anti-MuSK myasthenia gravis. The primary outcome was favorable outcome on the Myasthenia Gravis Foundation of America (MGFA) Post-Intervention Status (PIS). The researchers defined a favorable outcome as an MGFA PIS score of minimal manifestations or better.
Secondary outcomes included prednisone dose; use of other immunosuppressant medications, IV immunoglobulin (IVIG), or plasma exchange (PLEX) treatment; and Myasthenia Gravis Status and Treatment Intensity (MGSTI).
Baseline characteristics were similar between patients with anti-MuSK myasthenia gravis who underwent thymectomy (n = 26) and those who did not (n = 29), including treatment with rituximab (42% vs 45%). Median follow-up was more than three years.
At last visit, 35% (nine of 26) of patients who underwent thymectomy had a favorable outcome, compared with 55% (16 of 29) of patients who did not undergo thymectomy. In addition, 69% of patients who underwent thymectomy were taking prednisone, compared with 41% of patients who did not undergo thymectomy (median dose, 10 mg/day vs 0 mg/day).
“After controlling for rituximab, baseline prednisone, and final IVIG/PLEX treatment, thymectomy was not associated with greater likelihood of favorable clinical outcome, but broad confidence intervals cannot exclude therapeutic effect (odds ratio, 0.43),” the investigators reported.
“The recent MGTX trial clearly demonstrated the benefit of thymectomy for patients with AChR antibody positive myasthenia gravis,” said A. Gordon Smith, MD, Cochair of the AANEM Annual Meeting Program Committee. “Ms. Clifford and her colleagues now provide compelling data suggesting thymectomy may not be effective in MuSK-positive myasthenia gravis.”
The study’s follow-up is long enough for the findings to be clinically “relevant to all physicians treating myasthenia gravis,” said Robert W. Irwin, MD, Cochair of the AANEM Annual Meeting Program Committee.
WASHINGTON, DC—Among patients with anti-muscle-specific kinase (MuSK) myasthenia gravis, thymectomy is not associated with greater likelihood of clinical improvement, according to an analysis of data from a multicenter cohort study. The results were presented at the 2018 Annual Meeting of the American Association of Neuromuscular & Electrodiagnostic Medicine (AANEM).
Although a randomized trial has demonstrated benefit from thymectomy in nonthymomatous antiacetylcholine receptor (AChR) antibody positive generalized myasthenia gravis, observational studies suggest that thymectomy may not be efficacious in anti-MuSK myasthenia gravis. Histologic studies have found that patients with anti-MuSK myasthenia gravis have less hyperplastic thymic tissue, compared with patients with anti-AChR myasthenia gravis.
To evaluate the therapeutic impact of thymectomy in patients with anti-MuSK myasthenia gravis, Katherine Clifford, a medical student at the University of Vermont Larner College of Medicine in Burlington, and colleagues analyzed data from a multicenter, retrospective, blinded review of rituximab treatment in patients with anti-MuSK myasthenia gravis. The primary outcome was favorable outcome on the Myasthenia Gravis Foundation of America (MGFA) Post-Intervention Status (PIS). The researchers defined a favorable outcome as an MGFA PIS score of minimal manifestations or better.
Secondary outcomes included prednisone dose; use of other immunosuppressant medications, IV immunoglobulin (IVIG), or plasma exchange (PLEX) treatment; and Myasthenia Gravis Status and Treatment Intensity (MGSTI).
Baseline characteristics were similar between patients with anti-MuSK myasthenia gravis who underwent thymectomy (n = 26) and those who did not (n = 29), including treatment with rituximab (42% vs 45%). Median follow-up was more than three years.
At last visit, 35% (nine of 26) of patients who underwent thymectomy had a favorable outcome, compared with 55% (16 of 29) of patients who did not undergo thymectomy. In addition, 69% of patients who underwent thymectomy were taking prednisone, compared with 41% of patients who did not undergo thymectomy (median dose, 10 mg/day vs 0 mg/day).
“After controlling for rituximab, baseline prednisone, and final IVIG/PLEX treatment, thymectomy was not associated with greater likelihood of favorable clinical outcome, but broad confidence intervals cannot exclude therapeutic effect (odds ratio, 0.43),” the investigators reported.
“The recent MGTX trial clearly demonstrated the benefit of thymectomy for patients with AChR antibody positive myasthenia gravis,” said A. Gordon Smith, MD, Cochair of the AANEM Annual Meeting Program Committee. “Ms. Clifford and her colleagues now provide compelling data suggesting thymectomy may not be effective in MuSK-positive myasthenia gravis.”
The study’s follow-up is long enough for the findings to be clinically “relevant to all physicians treating myasthenia gravis,” said Robert W. Irwin, MD, Cochair of the AANEM Annual Meeting Program Committee.
Medical calculator apps allow point of care, rapid decision-making
The most useful applications (apps) for health care professionals and students? Medical calculator apps (along with drug reference and disease diagnosis apps), according to surveys of clinicians and students.1,2 The utility of calculator apps to these groups is not surprising; calculator apps fall in the category of clinical decision-making apps, which also includes decision support systems, clinical treatment guidelines, disease diagnosis aids, differential diagnosis aids, laboratory test ordering, laboratory test interpretation, and medical exams.3 Calculator apps obviously save time as most health care providers have not memorized the many medical formulas and do not have computational speed. I have previously discussed other, more ObGyn-specific calculators, such as due date calculators.4,5 In this App Review column, however, I would like to highlight 3 general calculator apps: Calculate by QxMD, CliniCalc Medical Calculator, and Medscape. Researchers found all 3 apps 100% accurate and contained the most functions desired by internists.6 The apps are available at no cost and include many unique calculators. My colleagues and I actually used Calculate by QxMD to verify calculations in a previous study.7
A clinical example for how to apply calculators in practice is as follows: A multiparous patient at term has undergone an unscheduled cesarean delivery for arrest of dilation and intra-amniotic infection. You need to decide if the patient requires anti‑coagulants for deep venous thrombosis (DVT) prophylaxis and her necessary daily dose for gentamicin for postpartum infection prophylaxis. You can use Medscape’s body mass index (BMI) calculator to find out that this patient’s BMI is 45 kg/m2 and that DVT prophylaxis is in fact indicated. You also can use QxMD’s ideal body weight calculator to get the patient’s weight and determine the appropriate daily dose for gentamicin.
The TABLE provides more information on the apps, with its inclusions based on a shortened version of the APPLICATIONS scoring system, APPLI (app comprehensiveness, price, platform, literature used, and important special features).7
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
1. Mosa AS, Yoo I, Sheets L. A systematic review of healthcare applications for smartphones. BMC Med Inform Decis Mak. 2012;12:67.
2. Payne KB, Wharrad H, Watts K. Smartphone and medical related App use among medical students and junior doctors in the United Kingdom (UK): a regional survey. BMC Med Inform Decis Mak. 2012;12:121.
3. Ventola CL. Mobile devices and apps for health care professionals: uses and benefits. P T. 2014;39:356-364.
4. Chen KT. Three good apps for calculating the date of delivery. OBG Manag. 2017;29:45-46.
5. Chen KT. ACOG app and applets: tools to augment your practice. OBG Manag. 2018;30:41-42.
6. Bierbrier R, Lo V, Wu RC. Evaluation of the accuracy of smartphone medical calculation apps. J Med Internet Res. 2014;16:e32.
7. Chyjek K, Farag S, Chen KT. Rating pregnancy wheel applications using the APPLICATIONS scoring system. Obstet Gynecol. 2015;125:1478-1483.

Dr. Chen is Professor of Obstetrics, Gynecology, and Reproductive Science and Medical Education, Vice-Chair of Ob-Gyn Education for the Mount Sinai Health System, Icahn School of Medicine, Mount Sinai, New York, New York. She is an OBG Management Contributing Editor.
The author reports being an advisory board member and receiving royalties from UpToDate, Inc.
Dr. Chen is Professor of Obstetrics, Gynecology, and Reproductive Science and Medical Education, Vice-Chair of Ob-Gyn Education for the Mount Sinai Health System, Icahn School of Medicine, Mount Sinai, New York, New York. She is an OBG Management Contributing Editor.
The author reports being an advisory board member and receiving royalties from UpToDate, Inc.
Dr. Chen is Professor of Obstetrics, Gynecology, and Reproductive Science and Medical Education, Vice-Chair of Ob-Gyn Education for the Mount Sinai Health System, Icahn School of Medicine, Mount Sinai, New York, New York. She is an OBG Management Contributing Editor.
The author reports being an advisory board member and receiving royalties from UpToDate, Inc.
The most useful applications (apps) for health care professionals and students? Medical calculator apps (along with drug reference and disease diagnosis apps), according to surveys of clinicians and students.1,2 The utility of calculator apps to these groups is not surprising; calculator apps fall in the category of clinical decision-making apps, which also includes decision support systems, clinical treatment guidelines, disease diagnosis aids, differential diagnosis aids, laboratory test ordering, laboratory test interpretation, and medical exams.3 Calculator apps obviously save time as most health care providers have not memorized the many medical formulas and do not have computational speed. I have previously discussed other, more ObGyn-specific calculators, such as due date calculators.4,5 In this App Review column, however, I would like to highlight 3 general calculator apps: Calculate by QxMD, CliniCalc Medical Calculator, and Medscape. Researchers found all 3 apps 100% accurate and contained the most functions desired by internists.6 The apps are available at no cost and include many unique calculators. My colleagues and I actually used Calculate by QxMD to verify calculations in a previous study.7
A clinical example for how to apply calculators in practice is as follows: A multiparous patient at term has undergone an unscheduled cesarean delivery for arrest of dilation and intra-amniotic infection. You need to decide if the patient requires anti‑coagulants for deep venous thrombosis (DVT) prophylaxis and her necessary daily dose for gentamicin for postpartum infection prophylaxis. You can use Medscape’s body mass index (BMI) calculator to find out that this patient’s BMI is 45 kg/m2 and that DVT prophylaxis is in fact indicated. You also can use QxMD’s ideal body weight calculator to get the patient’s weight and determine the appropriate daily dose for gentamicin.
The TABLE provides more information on the apps, with its inclusions based on a shortened version of the APPLICATIONS scoring system, APPLI (app comprehensiveness, price, platform, literature used, and important special features).7
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
The most useful applications (apps) for health care professionals and students? Medical calculator apps (along with drug reference and disease diagnosis apps), according to surveys of clinicians and students.1,2 The utility of calculator apps to these groups is not surprising; calculator apps fall in the category of clinical decision-making apps, which also includes decision support systems, clinical treatment guidelines, disease diagnosis aids, differential diagnosis aids, laboratory test ordering, laboratory test interpretation, and medical exams.3 Calculator apps obviously save time as most health care providers have not memorized the many medical formulas and do not have computational speed. I have previously discussed other, more ObGyn-specific calculators, such as due date calculators.4,5 In this App Review column, however, I would like to highlight 3 general calculator apps: Calculate by QxMD, CliniCalc Medical Calculator, and Medscape. Researchers found all 3 apps 100% accurate and contained the most functions desired by internists.6 The apps are available at no cost and include many unique calculators. My colleagues and I actually used Calculate by QxMD to verify calculations in a previous study.7
A clinical example for how to apply calculators in practice is as follows: A multiparous patient at term has undergone an unscheduled cesarean delivery for arrest of dilation and intra-amniotic infection. You need to decide if the patient requires anti‑coagulants for deep venous thrombosis (DVT) prophylaxis and her necessary daily dose for gentamicin for postpartum infection prophylaxis. You can use Medscape’s body mass index (BMI) calculator to find out that this patient’s BMI is 45 kg/m2 and that DVT prophylaxis is in fact indicated. You also can use QxMD’s ideal body weight calculator to get the patient’s weight and determine the appropriate daily dose for gentamicin.
The TABLE provides more information on the apps, with its inclusions based on a shortened version of the APPLICATIONS scoring system, APPLI (app comprehensiveness, price, platform, literature used, and important special features).7
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
1. Mosa AS, Yoo I, Sheets L. A systematic review of healthcare applications for smartphones. BMC Med Inform Decis Mak. 2012;12:67.
2. Payne KB, Wharrad H, Watts K. Smartphone and medical related App use among medical students and junior doctors in the United Kingdom (UK): a regional survey. BMC Med Inform Decis Mak. 2012;12:121.
3. Ventola CL. Mobile devices and apps for health care professionals: uses and benefits. P T. 2014;39:356-364.
4. Chen KT. Three good apps for calculating the date of delivery. OBG Manag. 2017;29:45-46.
5. Chen KT. ACOG app and applets: tools to augment your practice. OBG Manag. 2018;30:41-42.
6. Bierbrier R, Lo V, Wu RC. Evaluation of the accuracy of smartphone medical calculation apps. J Med Internet Res. 2014;16:e32.
7. Chyjek K, Farag S, Chen KT. Rating pregnancy wheel applications using the APPLICATIONS scoring system. Obstet Gynecol. 2015;125:1478-1483.
1. Mosa AS, Yoo I, Sheets L. A systematic review of healthcare applications for smartphones. BMC Med Inform Decis Mak. 2012;12:67.
2. Payne KB, Wharrad H, Watts K. Smartphone and medical related App use among medical students and junior doctors in the United Kingdom (UK): a regional survey. BMC Med Inform Decis Mak. 2012;12:121.
3. Ventola CL. Mobile devices and apps for health care professionals: uses and benefits. P T. 2014;39:356-364.
4. Chen KT. Three good apps for calculating the date of delivery. OBG Manag. 2017;29:45-46.
5. Chen KT. ACOG app and applets: tools to augment your practice. OBG Manag. 2018;30:41-42.
6. Bierbrier R, Lo V, Wu RC. Evaluation of the accuracy of smartphone medical calculation apps. J Med Internet Res. 2014;16:e32.
7. Chyjek K, Farag S, Chen KT. Rating pregnancy wheel applications using the APPLICATIONS scoring system. Obstet Gynecol. 2015;125:1478-1483.

Collaboration is key to bridging the AYA cancer care divide
Survival gains among adolescents and young adults (AYAs) with cancer continue to lag behind outcomes for children and older adult patients. It’s a trend that spans decades, but clinicians and researchers are finally getting serious about trying to understand the underlying causes and are re-examining prevailing practices in an effort to address the discrepancies.
“This is a very heterogeneous group of disorders,” Rabi Hanna, MD, a pediatric hematologist and oncologist at Cleveland Clinic Children’s Hospital, Ohio, said in an interview. He’s specifically referring to the cancers that affect AYAs, who are broadly defined as patients aged 15 through 39 years. “A few cancers, such as [acute lymphoblastic leukemia], are more common in children, and others, such as breast cancer, are more common in adults. The biology may be different in the adolescent and young adult patients, which may lead to different outcomes.”
In addition, the psychosocial needs in this age group differ vastly from those in other groups. “Many of these patients are in college or have just started their families, so we have to pay more attention to [issues related to] financial toxicity and fertility, for example,” said Dr Hanna, who is the director of pediatric bone marrow transplantation at the clinic. (The term “financial toxicity” describes the cumulative negative impact of the high cost of care, lost work time, and delays in reaching educational and career goals on patients with cancer and their families.)
Another factor that likely contributes to the outcome disparities between AYAs and other populations with cancer is the relative lack of clinical trial involvement among AYAs.
A recent series of articles published in the journal Blood addressed these and other issues, among them, whether AYAs with acute lymphoblastic leukemia (ALL)1 or aggressive B-cell non-Hodgkin lymphomas (NHLs) 2 should be treated as children or adults; treatment strategies for those with acute myeloid leukemias (AMLs); 3 management of Hodgkin lymphoma;4 and psychosocial challenges and health-related quality of life (QoL) in AYAs with hematologic malignancies.5
In the introduction to the series, Jorge Cortes, MD, an assistant editor on the journal, wrote that hematologic malignancies in AYAs “represent a unique challenge because of their special biological features and distinctive therapeutic requirements, as well as the unique medical, social, and psychological characteristics of this patient population.”6
He noted, however, that “not much has been done to explore unique molecular and biological features of AYA hematologic malignancies. The discussion on the management of AYAs often centers on whether these patients should be treated in a pediatric setting or an adult setting, or with regimens designed for children or for adults,” noted Dr Cortes, professor and chair of the chronic myeloid leukemia section in the department of leukemia at the University of Texas MD Anderson Cancer Center, Houston.
Therapeutic options: pediatric or adult protocols?
In their article on ALL in AYAs, Nicolas Boissel, MD, and André Baruchel, MD, note that the use of “fully pediatric protocols” in patients aged 15 through 20 years is supported by findings from numerous studies. In young adults, evidence increasingly supports “pediatric-inspired or even fully pediatric approaches” because they have been shown to significantly improve outcomes, with long-term survival rates nearing 70%.1 Patients in these age groups require specific programs that factor in access to care and to trials, an increased risk of acute toxicities, and treatment adherence, which can be particularly problematic in AYAs, they concluded.
However, Kristen O’Dwyer, MD, and colleagues, argue in an article on AML treatment in AYAs that neither the pediatric nor adult approaches are ideally suited for AYAs because of the “distinguishing characteristics of AYAs with AML.” Rather, they conclude that AYA-specific approaches merit consideration.3
Similarly, Kieron Dunleavy, MD, and Thomas G Gross, MD, note in an article on managing aggressive B-cell NHLs in AYAs that there is a “remarkable divide” in the treatment of patients younger than 18 years with lymphoma compared with their young adult counterparts, and that it underscores the need for collaboration in developing consensus regarding treatment of AYAs.2
Clinical setting: pediatric or adult?
Consideration is also being given to the clinical setting in which AYA patients receive their treatment. Lori Muffly, MD, MS, and colleagues have reported that survival was superior for AYA patients with ALL who were treated in pediatric cancer settings,7 and other researchers have reported similar findings.
However, those improved outcomes in the pediatric setting might be offset by a higher use of resources and therefore higher costs, based on recent findings in a Canadian study by Paul C Nathan, MD, and colleagues.8 Among 1,356 patients aged 15-17 years who were diagnosed with cancer between 1996 and 2010, the authors found that the cost of care was higher when treatment took place in a pediatric setting compared with in an adult institution, and that it was driven in part by higher hospitalization rates and longer hospital stays. These findings were true across different diagnoses, including leukemias, lymphomas, sarcomas, and germ cell tumors, but only during the initial treatment phase.
In an accompanying editorial, Helen M Parsons, PhD, and her co-authors wrote that adolescents who receive treatment in the pediatric setting “tended to seek more [emergency department (ED)] care immediately before diagnosis and during the initial treatment phase; these adolescents also used more home care services during initial treatment and survivorship.9 They pointed out that the findings of higher inpatient days in the pediatric setting was not surprising given that induction therapies for pediatric ALL tend to be more complex and intensive than therapies commonly used in adults with ALL, and that pediatric cancer hospitals tend to have a wider array of services, including psychosocial and family support services.
“What is less clear is why individuals seen in pediatric settings have higher rates of ED care directly before diagnosis and during the initial treatment phase,” they wrote, adding that further investigation was needed on this topic to better understand those trends. “The finding that adolescents treated in pediatric institutions had higher resource use across diagnostic groups demonstrates that resource utilization may be driven just as much by care setting as diagnosis.” 9
The authors of the editorial emphasized that because of the differences in health care delivery and payment structures between the United States and Canada, where the Nathan study was done, it was important that similar studies are done in the United States to confirm these findings.
Disease and developmental biology
As Dr Hanna noted, biological differences and changes over time suggest that different age groups need varying approaches to treatment and that they may have different outcomes with the same treatments.
For example, the biology of AML is known to change with age, Dr O'Dwyer and her colleagues noted,3 citing a recent European study of 5,564 patients with de novo AML that showed that the frequency of favorable cytogenetics was low in infants (13.7%), increased in children (25%) and young adults (44%), and decreased again in middle age and older patients.10
“Most unfavorable cytogenetic abnormalities are rare across all age groups, though complex cytogenetics are relatively more frequent in infants, decrease in frequency in AYAs, and then increase in frequency beyond AYA,” Dr O'Dwyer and her colleagues wrote.3 It was also becoming more apparent that age influences the presence of AML-related molecular abnormalities, and recognition of age-related differences in disease biology “will provide the best opportunity to improve the clinical outcomes that have been static for decades.”
Dr Boissel and Dr Baruchel also noted in their report that light was finally being shed on the “black hole” of understanding ALL biology in AYAs, and research has shown that there is a continuum between childhood and adult ALL.1 They concluded that “risk stratification based on recent biology findings and sequential [minimum residual disease] evaluations should now be implemented, as well as new therapeutic options including immunotherapy and targeted therapies, at best within the setting of integrated pediatric and AYA protocols.”
Psychosocial factors
“Cancer is a non-normative event for AYAs. It is extremely disruptive to them physically, psychologically, and vocationally ... and this poses significant challenges,” John Salsman, PhD, director of clinical research in AYA oncology at Wake Forest University, Winston-Salem, NC, said in an interview.
These patients have 5-year survival rates that haven’t improved in tandem with those in pediatric and adult populations over the last 3 decades, and in addition to the financial toxicity and strain, they also have higher rates of depression and anxiety, including fear of recurrence, he added. “Quality of life is incredibly important, and these things need to be addressed because of the developmental changes AYAs are navigating; there are issues of positive body image, family and career decisions ... these are challenging for anyone, and when you throw a cancer diagnosis into the mix they become disproportionately so.”
In a 2014 study, Dr Salsman and his colleagues found that AYAs with cancer had poorer physical and emotional quality of life when compared with matched controls, but better social quality of life.11 The latter finding was surprising and highlights the importance of the social dimension in the lives of AYAs. “Patient after patient will say ‘I found out who my real friends are,’ ” he said. “There’s this refinement and deepening of the social network among some posttreatment survivors.”
Dr Salsman and his colleagues are using those findings to develop interventions that can maximize self-care in posttreatment survivorship – a time when AYAs may feel they have a new lease on life and may be more motivated to adhere to recommendations and take care of themselves. For example, a randomized controlled pilot study that incorporates social media apps and other technologies to build on the positive social components of their lives in promoting physical activity interventions is underway.
Another intervention targets emotional well-being through the use of web-based tools to increase positive affect. A proof-of-concept study showed that the approach was feasible and well received, and a larger-scale randomized controlled trial is being planned, he said.
Dr Salsman also praised the PRISM (Promoting Resilience in Stress Management) tool developed by researchers at Seattle Children’s Hospital. It was created to help AYAs with cancer and other illnesses learn coping skills to manage stress after their diagnosis and to boost quality of life beyond treatment. A digital app has also been developed to be used in conjunction with the program.
Trial enrollment
In his editorial introducing the Blood series on AYAs and cancer, Dr Cortes noted a paucity of clinical trials specifically designed for this population. “At the time of this writing, I could identify four therapeutic trials registered at www.clinicaltrials.gov that appeared to be somewhat specifically designed for AYAs (some included children also),” he wrote, describing AYA enrollment in clinical trials in cancer as “suboptimal at best.”6
Dr Salsman said these dismal enrolment numbers could in part be related to treatment setting. Data suggest that most AYAs with cancer are treated in community-based practices rather than comprehensive cancer centers where the bulk of research is being done, he explained.
Dr Hanna agreed that more research involving AYAs was needed as is a better understanding of why enrollment is so much lower in this population. He pointed out that in 2017 the American Society of Clinical Oncology and Friends of Cancer Research released a statement recommending that pediatric patients be considered for enrollment in later-phase trials for cancer types that span both adults and children.12 The organizations said that individuals aged 12 years and older should routinely be included in such trials because their drug metabolism is similar to adults, and inclusion of younger patients may also be appropriate if they are part of the population affected by the disease, depending on specific disease biology, action of the drug, and available safety information.
Officials at the Food and Drug Administration are considering that possibility, Dr Hanna said.
Dr Salsman added there has been an increase in recent years in the attention paid to disparities in survival improvements and trial involvement among AYAs with cancer, compared with other age groups. For example, about 5 years ago, the National Clinical Trials Network formed a working group that developed a number of specific objectives for incorporating more AYAs into cancer trials and finding better ways to study this population;13 the Institute of Medicine held a forum on the care of AYAs with cancer;14 and the National Cancer Institute held a state-of-the-science meeting that focused on identifying strategic priorities for AYA oncology,15 he noted.
Dr Hanna added that “scientific groups such as Southwest Oncology Group (SWOG) and Children’s Oncology Group (COG) also have AYA committees now. One of the success stories of working together between SWOG and COG was the intergroup study C10403 for patients with ALL. And now there are efforts for an intergroup AYA-AML task force to include representatives from each of the cooperative groups that historically co-ordinated myeloid disease clinical trials – COG, SWOG, Alliance, and ECOG-ACRIN,” he said.
In fact, all of the National Clinical Trials Network groups have some initiative in place to address AYA concerns, said Dr Salsman, who chairs the ECOG-ACRIN AYA oncology subcommittee.
Despite these efforts, and many others, long-term survival improvements among AYAs with cancer still fall short, compared with those of other age groups.16
Next steps
Among the recommendations from authors in the AYA series in Blood is a call for assessing AYA-specific therapy in future clinical trials, as well as improved collaboration between adult and pediatric teams and the involvement of multidisciplinary teams in care for this population.
Many centers are already working on models for collaborative care, Dr Salsman said, citing the Fort Worth AYA Oncology Coalition led by medical director Karen Albritton, MD, as an example of a program that has been successful in helping clinical and supportive caregivers and their AYA patients “have a shared vision” as they work to maximize improvements in outcomes.
Patients are also taking the lead in demanding better care and attention to their psychosocial needs, Dr Hanna said. In the case of the community-powered advocacy organization Critical Mass, members have succeeded in getting lawmakers to introduce a bill in the US House of Representatives that would allow college students to defer loan payments while undergoing cancer treatment.
1. Boissel N, Baruchel A. Acute lymphoblastic leukemia in adolescent and young adults: treat as adults or as children? Blood. 2018;132:351-361.
2. Dunleavy K, Gross TG. Management of aggressive B-cell NHLs in the AYA population: an adult vs pediatric perspective. Blood. 2018;132:369-375.
3. O’Dwyer K, Freyer DR, Horan JT. Treatment strategies for adolescent and young adult patients with acute myeloid leukemia. Blood. 2018;132:362-368.
4. Flerlage JE, Metzger ML, Bhakta N. The management of Hodgkin lymphoma in adolescents and young adults: burden of disease or burden of choice? Blood. 2018;132:376-384.
5. Husson O, Huijgens PC, van der Graaf WTA. Psychosocial challenges and health-related quality of life of adolescents and young adults with hematologic malignancies. Blood. 2018;132:385-392.
6. Cortes J. Introduction to a review series on adolescent and young adult malignant hematology. Blood. 2018;132:345-346.
7. Muffly L, Alvarez E, Lichtensztajn D, Abrahão R, Gomez SL, Keegan T. Patterns of care and outcomes in adolescent and young adult acute lymphoblastic leukemia: a population-based study. Blood Adv. 2018;2(8):895-903.
8. Nathan PC, Bremner KE, Liu N, et al. Resource utilization and costs in adolescents treated for cancer in pediatric vs adult institutions. J Natl Cancer Inst. July 19, 2018. [Epub ahead of print.]
9. Parsons HM, Muffly L, Alvarez EM, Keegan THM. Does treatment setting matter? Evaluating resource utilization for adolescents treated in pediatric vs adult cancer institutions. https://academic.oup.com/jnci/advance-article/doi/10.1093/jnci/djy123/5056313?searchresult=1. Published July 19, 2018. Last accessed October 12, 2018.
10. Creutzig U, Zimmermann M, Reinhardt D, et al. Changes in cytogenetics and molecular genetics in acute myeloid leukemia from childhood to adult age groups. Cancer. 2016;122(24):3821-3830.
11. Salsman JM, Garcia SF, Yanez B, et al. Physical, emotional, and social health differences between posttreatment young adults with cancer and matched healthy controls. Cancer. 2014;120(15):2247-2254.
12. Kim ES, Bruinooge SS, Roberts S, et al. Broadening eligibility criteria to make clinical trials more representative: American Society of Clinical Oncology and Friends of Cancer Research joint research statement. J Clin Oncol. 2017;35(33):3737-3744.
13. Freyer DR, Seibel NL. The clinical trials gap for adolescents and young adults with cancer: recent progress and conceptual framework for continued research. Curr Pediatr Rep. Published online February 18, 2015. DOI 10.1007/s40124-015-0075-y.
14. Nass SJ, Beaupin LK, Demark-Wahnefried W, et al. Identifying and addressing the needs of adolescents and young adults with cancer: summary of an Institute of Medicine workshop. Oncologist. 2015;20(2):186-195.
15. Wilder Smith A, Seibel NL, Lewis DR, et al. Next steps for adolescent and young adult oncology workshop: An update on progress and recommendations for the future. Cancer. 2016;122(7):988-999.
16. Keegan THM, Ries LAG, Barr RD, et al. Comparison of cancer survival trends in the United States of adolescents and young adults with those in children and older adults. Cancer. 2016;122(7):1009-1016.
Survival gains among adolescents and young adults (AYAs) with cancer continue to lag behind outcomes for children and older adult patients. It’s a trend that spans decades, but clinicians and researchers are finally getting serious about trying to understand the underlying causes and are re-examining prevailing practices in an effort to address the discrepancies.
“This is a very heterogeneous group of disorders,” Rabi Hanna, MD, a pediatric hematologist and oncologist at Cleveland Clinic Children’s Hospital, Ohio, said in an interview. He’s specifically referring to the cancers that affect AYAs, who are broadly defined as patients aged 15 through 39 years. “A few cancers, such as [acute lymphoblastic leukemia], are more common in children, and others, such as breast cancer, are more common in adults. The biology may be different in the adolescent and young adult patients, which may lead to different outcomes.”
In addition, the psychosocial needs in this age group differ vastly from those in other groups. “Many of these patients are in college or have just started their families, so we have to pay more attention to [issues related to] financial toxicity and fertility, for example,” said Dr Hanna, who is the director of pediatric bone marrow transplantation at the clinic. (The term “financial toxicity” describes the cumulative negative impact of the high cost of care, lost work time, and delays in reaching educational and career goals on patients with cancer and their families.)
Another factor that likely contributes to the outcome disparities between AYAs and other populations with cancer is the relative lack of clinical trial involvement among AYAs.
A recent series of articles published in the journal Blood addressed these and other issues, among them, whether AYAs with acute lymphoblastic leukemia (ALL)1 or aggressive B-cell non-Hodgkin lymphomas (NHLs) 2 should be treated as children or adults; treatment strategies for those with acute myeloid leukemias (AMLs); 3 management of Hodgkin lymphoma;4 and psychosocial challenges and health-related quality of life (QoL) in AYAs with hematologic malignancies.5
In the introduction to the series, Jorge Cortes, MD, an assistant editor on the journal, wrote that hematologic malignancies in AYAs “represent a unique challenge because of their special biological features and distinctive therapeutic requirements, as well as the unique medical, social, and psychological characteristics of this patient population.”6
He noted, however, that “not much has been done to explore unique molecular and biological features of AYA hematologic malignancies. The discussion on the management of AYAs often centers on whether these patients should be treated in a pediatric setting or an adult setting, or with regimens designed for children or for adults,” noted Dr Cortes, professor and chair of the chronic myeloid leukemia section in the department of leukemia at the University of Texas MD Anderson Cancer Center, Houston.
Therapeutic options: pediatric or adult protocols?
In their article on ALL in AYAs, Nicolas Boissel, MD, and André Baruchel, MD, note that the use of “fully pediatric protocols” in patients aged 15 through 20 years is supported by findings from numerous studies. In young adults, evidence increasingly supports “pediatric-inspired or even fully pediatric approaches” because they have been shown to significantly improve outcomes, with long-term survival rates nearing 70%.1 Patients in these age groups require specific programs that factor in access to care and to trials, an increased risk of acute toxicities, and treatment adherence, which can be particularly problematic in AYAs, they concluded.
However, Kristen O’Dwyer, MD, and colleagues, argue in an article on AML treatment in AYAs that neither the pediatric nor adult approaches are ideally suited for AYAs because of the “distinguishing characteristics of AYAs with AML.” Rather, they conclude that AYA-specific approaches merit consideration.3
Similarly, Kieron Dunleavy, MD, and Thomas G Gross, MD, note in an article on managing aggressive B-cell NHLs in AYAs that there is a “remarkable divide” in the treatment of patients younger than 18 years with lymphoma compared with their young adult counterparts, and that it underscores the need for collaboration in developing consensus regarding treatment of AYAs.2
Clinical setting: pediatric or adult?
Consideration is also being given to the clinical setting in which AYA patients receive their treatment. Lori Muffly, MD, MS, and colleagues have reported that survival was superior for AYA patients with ALL who were treated in pediatric cancer settings,7 and other researchers have reported similar findings.
However, those improved outcomes in the pediatric setting might be offset by a higher use of resources and therefore higher costs, based on recent findings in a Canadian study by Paul C Nathan, MD, and colleagues.8 Among 1,356 patients aged 15-17 years who were diagnosed with cancer between 1996 and 2010, the authors found that the cost of care was higher when treatment took place in a pediatric setting compared with in an adult institution, and that it was driven in part by higher hospitalization rates and longer hospital stays. These findings were true across different diagnoses, including leukemias, lymphomas, sarcomas, and germ cell tumors, but only during the initial treatment phase.
In an accompanying editorial, Helen M Parsons, PhD, and her co-authors wrote that adolescents who receive treatment in the pediatric setting “tended to seek more [emergency department (ED)] care immediately before diagnosis and during the initial treatment phase; these adolescents also used more home care services during initial treatment and survivorship.9 They pointed out that the findings of higher inpatient days in the pediatric setting was not surprising given that induction therapies for pediatric ALL tend to be more complex and intensive than therapies commonly used in adults with ALL, and that pediatric cancer hospitals tend to have a wider array of services, including psychosocial and family support services.
“What is less clear is why individuals seen in pediatric settings have higher rates of ED care directly before diagnosis and during the initial treatment phase,” they wrote, adding that further investigation was needed on this topic to better understand those trends. “The finding that adolescents treated in pediatric institutions had higher resource use across diagnostic groups demonstrates that resource utilization may be driven just as much by care setting as diagnosis.” 9
The authors of the editorial emphasized that because of the differences in health care delivery and payment structures between the United States and Canada, where the Nathan study was done, it was important that similar studies are done in the United States to confirm these findings.
Disease and developmental biology
As Dr Hanna noted, biological differences and changes over time suggest that different age groups need varying approaches to treatment and that they may have different outcomes with the same treatments.
For example, the biology of AML is known to change with age, Dr O'Dwyer and her colleagues noted,3 citing a recent European study of 5,564 patients with de novo AML that showed that the frequency of favorable cytogenetics was low in infants (13.7%), increased in children (25%) and young adults (44%), and decreased again in middle age and older patients.10
“Most unfavorable cytogenetic abnormalities are rare across all age groups, though complex cytogenetics are relatively more frequent in infants, decrease in frequency in AYAs, and then increase in frequency beyond AYA,” Dr O'Dwyer and her colleagues wrote.3 It was also becoming more apparent that age influences the presence of AML-related molecular abnormalities, and recognition of age-related differences in disease biology “will provide the best opportunity to improve the clinical outcomes that have been static for decades.”
Dr Boissel and Dr Baruchel also noted in their report that light was finally being shed on the “black hole” of understanding ALL biology in AYAs, and research has shown that there is a continuum between childhood and adult ALL.1 They concluded that “risk stratification based on recent biology findings and sequential [minimum residual disease] evaluations should now be implemented, as well as new therapeutic options including immunotherapy and targeted therapies, at best within the setting of integrated pediatric and AYA protocols.”
Psychosocial factors
“Cancer is a non-normative event for AYAs. It is extremely disruptive to them physically, psychologically, and vocationally ... and this poses significant challenges,” John Salsman, PhD, director of clinical research in AYA oncology at Wake Forest University, Winston-Salem, NC, said in an interview.
These patients have 5-year survival rates that haven’t improved in tandem with those in pediatric and adult populations over the last 3 decades, and in addition to the financial toxicity and strain, they also have higher rates of depression and anxiety, including fear of recurrence, he added. “Quality of life is incredibly important, and these things need to be addressed because of the developmental changes AYAs are navigating; there are issues of positive body image, family and career decisions ... these are challenging for anyone, and when you throw a cancer diagnosis into the mix they become disproportionately so.”
In a 2014 study, Dr Salsman and his colleagues found that AYAs with cancer had poorer physical and emotional quality of life when compared with matched controls, but better social quality of life.11 The latter finding was surprising and highlights the importance of the social dimension in the lives of AYAs. “Patient after patient will say ‘I found out who my real friends are,’ ” he said. “There’s this refinement and deepening of the social network among some posttreatment survivors.”
Dr Salsman and his colleagues are using those findings to develop interventions that can maximize self-care in posttreatment survivorship – a time when AYAs may feel they have a new lease on life and may be more motivated to adhere to recommendations and take care of themselves. For example, a randomized controlled pilot study that incorporates social media apps and other technologies to build on the positive social components of their lives in promoting physical activity interventions is underway.
Another intervention targets emotional well-being through the use of web-based tools to increase positive affect. A proof-of-concept study showed that the approach was feasible and well received, and a larger-scale randomized controlled trial is being planned, he said.
Dr Salsman also praised the PRISM (Promoting Resilience in Stress Management) tool developed by researchers at Seattle Children’s Hospital. It was created to help AYAs with cancer and other illnesses learn coping skills to manage stress after their diagnosis and to boost quality of life beyond treatment. A digital app has also been developed to be used in conjunction with the program.
Trial enrollment
In his editorial introducing the Blood series on AYAs and cancer, Dr Cortes noted a paucity of clinical trials specifically designed for this population. “At the time of this writing, I could identify four therapeutic trials registered at www.clinicaltrials.gov that appeared to be somewhat specifically designed for AYAs (some included children also),” he wrote, describing AYA enrollment in clinical trials in cancer as “suboptimal at best.”6
Dr Salsman said these dismal enrolment numbers could in part be related to treatment setting. Data suggest that most AYAs with cancer are treated in community-based practices rather than comprehensive cancer centers where the bulk of research is being done, he explained.
Dr Hanna agreed that more research involving AYAs was needed as is a better understanding of why enrollment is so much lower in this population. He pointed out that in 2017 the American Society of Clinical Oncology and Friends of Cancer Research released a statement recommending that pediatric patients be considered for enrollment in later-phase trials for cancer types that span both adults and children.12 The organizations said that individuals aged 12 years and older should routinely be included in such trials because their drug metabolism is similar to adults, and inclusion of younger patients may also be appropriate if they are part of the population affected by the disease, depending on specific disease biology, action of the drug, and available safety information.
Officials at the Food and Drug Administration are considering that possibility, Dr Hanna said.
Dr Salsman added there has been an increase in recent years in the attention paid to disparities in survival improvements and trial involvement among AYAs with cancer, compared with other age groups. For example, about 5 years ago, the National Clinical Trials Network formed a working group that developed a number of specific objectives for incorporating more AYAs into cancer trials and finding better ways to study this population;13 the Institute of Medicine held a forum on the care of AYAs with cancer;14 and the National Cancer Institute held a state-of-the-science meeting that focused on identifying strategic priorities for AYA oncology,15 he noted.
Dr Hanna added that “scientific groups such as Southwest Oncology Group (SWOG) and Children’s Oncology Group (COG) also have AYA committees now. One of the success stories of working together between SWOG and COG was the intergroup study C10403 for patients with ALL. And now there are efforts for an intergroup AYA-AML task force to include representatives from each of the cooperative groups that historically co-ordinated myeloid disease clinical trials – COG, SWOG, Alliance, and ECOG-ACRIN,” he said.
In fact, all of the National Clinical Trials Network groups have some initiative in place to address AYA concerns, said Dr Salsman, who chairs the ECOG-ACRIN AYA oncology subcommittee.
Despite these efforts, and many others, long-term survival improvements among AYAs with cancer still fall short, compared with those of other age groups.16
Next steps
Among the recommendations from authors in the AYA series in Blood is a call for assessing AYA-specific therapy in future clinical trials, as well as improved collaboration between adult and pediatric teams and the involvement of multidisciplinary teams in care for this population.
Many centers are already working on models for collaborative care, Dr Salsman said, citing the Fort Worth AYA Oncology Coalition led by medical director Karen Albritton, MD, as an example of a program that has been successful in helping clinical and supportive caregivers and their AYA patients “have a shared vision” as they work to maximize improvements in outcomes.
Patients are also taking the lead in demanding better care and attention to their psychosocial needs, Dr Hanna said. In the case of the community-powered advocacy organization Critical Mass, members have succeeded in getting lawmakers to introduce a bill in the US House of Representatives that would allow college students to defer loan payments while undergoing cancer treatment.
Survival gains among adolescents and young adults (AYAs) with cancer continue to lag behind outcomes for children and older adult patients. It’s a trend that spans decades, but clinicians and researchers are finally getting serious about trying to understand the underlying causes and are re-examining prevailing practices in an effort to address the discrepancies.
“This is a very heterogeneous group of disorders,” Rabi Hanna, MD, a pediatric hematologist and oncologist at Cleveland Clinic Children’s Hospital, Ohio, said in an interview. He’s specifically referring to the cancers that affect AYAs, who are broadly defined as patients aged 15 through 39 years. “A few cancers, such as [acute lymphoblastic leukemia], are more common in children, and others, such as breast cancer, are more common in adults. The biology may be different in the adolescent and young adult patients, which may lead to different outcomes.”
In addition, the psychosocial needs in this age group differ vastly from those in other groups. “Many of these patients are in college or have just started their families, so we have to pay more attention to [issues related to] financial toxicity and fertility, for example,” said Dr Hanna, who is the director of pediatric bone marrow transplantation at the clinic. (The term “financial toxicity” describes the cumulative negative impact of the high cost of care, lost work time, and delays in reaching educational and career goals on patients with cancer and their families.)
Another factor that likely contributes to the outcome disparities between AYAs and other populations with cancer is the relative lack of clinical trial involvement among AYAs.
A recent series of articles published in the journal Blood addressed these and other issues, among them, whether AYAs with acute lymphoblastic leukemia (ALL)1 or aggressive B-cell non-Hodgkin lymphomas (NHLs) 2 should be treated as children or adults; treatment strategies for those with acute myeloid leukemias (AMLs); 3 management of Hodgkin lymphoma;4 and psychosocial challenges and health-related quality of life (QoL) in AYAs with hematologic malignancies.5
In the introduction to the series, Jorge Cortes, MD, an assistant editor on the journal, wrote that hematologic malignancies in AYAs “represent a unique challenge because of their special biological features and distinctive therapeutic requirements, as well as the unique medical, social, and psychological characteristics of this patient population.”6
He noted, however, that “not much has been done to explore unique molecular and biological features of AYA hematologic malignancies. The discussion on the management of AYAs often centers on whether these patients should be treated in a pediatric setting or an adult setting, or with regimens designed for children or for adults,” noted Dr Cortes, professor and chair of the chronic myeloid leukemia section in the department of leukemia at the University of Texas MD Anderson Cancer Center, Houston.
Therapeutic options: pediatric or adult protocols?
In their article on ALL in AYAs, Nicolas Boissel, MD, and André Baruchel, MD, note that the use of “fully pediatric protocols” in patients aged 15 through 20 years is supported by findings from numerous studies. In young adults, evidence increasingly supports “pediatric-inspired or even fully pediatric approaches” because they have been shown to significantly improve outcomes, with long-term survival rates nearing 70%.1 Patients in these age groups require specific programs that factor in access to care and to trials, an increased risk of acute toxicities, and treatment adherence, which can be particularly problematic in AYAs, they concluded.
However, Kristen O’Dwyer, MD, and colleagues, argue in an article on AML treatment in AYAs that neither the pediatric nor adult approaches are ideally suited for AYAs because of the “distinguishing characteristics of AYAs with AML.” Rather, they conclude that AYA-specific approaches merit consideration.3
Similarly, Kieron Dunleavy, MD, and Thomas G Gross, MD, note in an article on managing aggressive B-cell NHLs in AYAs that there is a “remarkable divide” in the treatment of patients younger than 18 years with lymphoma compared with their young adult counterparts, and that it underscores the need for collaboration in developing consensus regarding treatment of AYAs.2
Clinical setting: pediatric or adult?
Consideration is also being given to the clinical setting in which AYA patients receive their treatment. Lori Muffly, MD, MS, and colleagues have reported that survival was superior for AYA patients with ALL who were treated in pediatric cancer settings,7 and other researchers have reported similar findings.
However, those improved outcomes in the pediatric setting might be offset by a higher use of resources and therefore higher costs, based on recent findings in a Canadian study by Paul C Nathan, MD, and colleagues.8 Among 1,356 patients aged 15-17 years who were diagnosed with cancer between 1996 and 2010, the authors found that the cost of care was higher when treatment took place in a pediatric setting compared with in an adult institution, and that it was driven in part by higher hospitalization rates and longer hospital stays. These findings were true across different diagnoses, including leukemias, lymphomas, sarcomas, and germ cell tumors, but only during the initial treatment phase.
In an accompanying editorial, Helen M Parsons, PhD, and her co-authors wrote that adolescents who receive treatment in the pediatric setting “tended to seek more [emergency department (ED)] care immediately before diagnosis and during the initial treatment phase; these adolescents also used more home care services during initial treatment and survivorship.9 They pointed out that the findings of higher inpatient days in the pediatric setting was not surprising given that induction therapies for pediatric ALL tend to be more complex and intensive than therapies commonly used in adults with ALL, and that pediatric cancer hospitals tend to have a wider array of services, including psychosocial and family support services.
“What is less clear is why individuals seen in pediatric settings have higher rates of ED care directly before diagnosis and during the initial treatment phase,” they wrote, adding that further investigation was needed on this topic to better understand those trends. “The finding that adolescents treated in pediatric institutions had higher resource use across diagnostic groups demonstrates that resource utilization may be driven just as much by care setting as diagnosis.” 9
The authors of the editorial emphasized that because of the differences in health care delivery and payment structures between the United States and Canada, where the Nathan study was done, it was important that similar studies are done in the United States to confirm these findings.
Disease and developmental biology
As Dr Hanna noted, biological differences and changes over time suggest that different age groups need varying approaches to treatment and that they may have different outcomes with the same treatments.
For example, the biology of AML is known to change with age, Dr O'Dwyer and her colleagues noted,3 citing a recent European study of 5,564 patients with de novo AML that showed that the frequency of favorable cytogenetics was low in infants (13.7%), increased in children (25%) and young adults (44%), and decreased again in middle age and older patients.10
“Most unfavorable cytogenetic abnormalities are rare across all age groups, though complex cytogenetics are relatively more frequent in infants, decrease in frequency in AYAs, and then increase in frequency beyond AYA,” Dr O'Dwyer and her colleagues wrote.3 It was also becoming more apparent that age influences the presence of AML-related molecular abnormalities, and recognition of age-related differences in disease biology “will provide the best opportunity to improve the clinical outcomes that have been static for decades.”
Dr Boissel and Dr Baruchel also noted in their report that light was finally being shed on the “black hole” of understanding ALL biology in AYAs, and research has shown that there is a continuum between childhood and adult ALL.1 They concluded that “risk stratification based on recent biology findings and sequential [minimum residual disease] evaluations should now be implemented, as well as new therapeutic options including immunotherapy and targeted therapies, at best within the setting of integrated pediatric and AYA protocols.”
Psychosocial factors
“Cancer is a non-normative event for AYAs. It is extremely disruptive to them physically, psychologically, and vocationally ... and this poses significant challenges,” John Salsman, PhD, director of clinical research in AYA oncology at Wake Forest University, Winston-Salem, NC, said in an interview.
These patients have 5-year survival rates that haven’t improved in tandem with those in pediatric and adult populations over the last 3 decades, and in addition to the financial toxicity and strain, they also have higher rates of depression and anxiety, including fear of recurrence, he added. “Quality of life is incredibly important, and these things need to be addressed because of the developmental changes AYAs are navigating; there are issues of positive body image, family and career decisions ... these are challenging for anyone, and when you throw a cancer diagnosis into the mix they become disproportionately so.”
In a 2014 study, Dr Salsman and his colleagues found that AYAs with cancer had poorer physical and emotional quality of life when compared with matched controls, but better social quality of life.11 The latter finding was surprising and highlights the importance of the social dimension in the lives of AYAs. “Patient after patient will say ‘I found out who my real friends are,’ ” he said. “There’s this refinement and deepening of the social network among some posttreatment survivors.”
Dr Salsman and his colleagues are using those findings to develop interventions that can maximize self-care in posttreatment survivorship – a time when AYAs may feel they have a new lease on life and may be more motivated to adhere to recommendations and take care of themselves. For example, a randomized controlled pilot study that incorporates social media apps and other technologies to build on the positive social components of their lives in promoting physical activity interventions is underway.
Another intervention targets emotional well-being through the use of web-based tools to increase positive affect. A proof-of-concept study showed that the approach was feasible and well received, and a larger-scale randomized controlled trial is being planned, he said.
Dr Salsman also praised the PRISM (Promoting Resilience in Stress Management) tool developed by researchers at Seattle Children’s Hospital. It was created to help AYAs with cancer and other illnesses learn coping skills to manage stress after their diagnosis and to boost quality of life beyond treatment. A digital app has also been developed to be used in conjunction with the program.
Trial enrollment
In his editorial introducing the Blood series on AYAs and cancer, Dr Cortes noted a paucity of clinical trials specifically designed for this population. “At the time of this writing, I could identify four therapeutic trials registered at www.clinicaltrials.gov that appeared to be somewhat specifically designed for AYAs (some included children also),” he wrote, describing AYA enrollment in clinical trials in cancer as “suboptimal at best.”6
Dr Salsman said these dismal enrolment numbers could in part be related to treatment setting. Data suggest that most AYAs with cancer are treated in community-based practices rather than comprehensive cancer centers where the bulk of research is being done, he explained.
Dr Hanna agreed that more research involving AYAs was needed as is a better understanding of why enrollment is so much lower in this population. He pointed out that in 2017 the American Society of Clinical Oncology and Friends of Cancer Research released a statement recommending that pediatric patients be considered for enrollment in later-phase trials for cancer types that span both adults and children.12 The organizations said that individuals aged 12 years and older should routinely be included in such trials because their drug metabolism is similar to adults, and inclusion of younger patients may also be appropriate if they are part of the population affected by the disease, depending on specific disease biology, action of the drug, and available safety information.
Officials at the Food and Drug Administration are considering that possibility, Dr Hanna said.
Dr Salsman added there has been an increase in recent years in the attention paid to disparities in survival improvements and trial involvement among AYAs with cancer, compared with other age groups. For example, about 5 years ago, the National Clinical Trials Network formed a working group that developed a number of specific objectives for incorporating more AYAs into cancer trials and finding better ways to study this population;13 the Institute of Medicine held a forum on the care of AYAs with cancer;14 and the National Cancer Institute held a state-of-the-science meeting that focused on identifying strategic priorities for AYA oncology,15 he noted.
Dr Hanna added that “scientific groups such as Southwest Oncology Group (SWOG) and Children’s Oncology Group (COG) also have AYA committees now. One of the success stories of working together between SWOG and COG was the intergroup study C10403 for patients with ALL. And now there are efforts for an intergroup AYA-AML task force to include representatives from each of the cooperative groups that historically co-ordinated myeloid disease clinical trials – COG, SWOG, Alliance, and ECOG-ACRIN,” he said.
In fact, all of the National Clinical Trials Network groups have some initiative in place to address AYA concerns, said Dr Salsman, who chairs the ECOG-ACRIN AYA oncology subcommittee.
Despite these efforts, and many others, long-term survival improvements among AYAs with cancer still fall short, compared with those of other age groups.16
Next steps
Among the recommendations from authors in the AYA series in Blood is a call for assessing AYA-specific therapy in future clinical trials, as well as improved collaboration between adult and pediatric teams and the involvement of multidisciplinary teams in care for this population.
Many centers are already working on models for collaborative care, Dr Salsman said, citing the Fort Worth AYA Oncology Coalition led by medical director Karen Albritton, MD, as an example of a program that has been successful in helping clinical and supportive caregivers and their AYA patients “have a shared vision” as they work to maximize improvements in outcomes.
Patients are also taking the lead in demanding better care and attention to their psychosocial needs, Dr Hanna said. In the case of the community-powered advocacy organization Critical Mass, members have succeeded in getting lawmakers to introduce a bill in the US House of Representatives that would allow college students to defer loan payments while undergoing cancer treatment.
1. Boissel N, Baruchel A. Acute lymphoblastic leukemia in adolescent and young adults: treat as adults or as children? Blood. 2018;132:351-361.
2. Dunleavy K, Gross TG. Management of aggressive B-cell NHLs in the AYA population: an adult vs pediatric perspective. Blood. 2018;132:369-375.
3. O’Dwyer K, Freyer DR, Horan JT. Treatment strategies for adolescent and young adult patients with acute myeloid leukemia. Blood. 2018;132:362-368.
4. Flerlage JE, Metzger ML, Bhakta N. The management of Hodgkin lymphoma in adolescents and young adults: burden of disease or burden of choice? Blood. 2018;132:376-384.
5. Husson O, Huijgens PC, van der Graaf WTA. Psychosocial challenges and health-related quality of life of adolescents and young adults with hematologic malignancies. Blood. 2018;132:385-392.
6. Cortes J. Introduction to a review series on adolescent and young adult malignant hematology. Blood. 2018;132:345-346.
7. Muffly L, Alvarez E, Lichtensztajn D, Abrahão R, Gomez SL, Keegan T. Patterns of care and outcomes in adolescent and young adult acute lymphoblastic leukemia: a population-based study. Blood Adv. 2018;2(8):895-903.
8. Nathan PC, Bremner KE, Liu N, et al. Resource utilization and costs in adolescents treated for cancer in pediatric vs adult institutions. J Natl Cancer Inst. July 19, 2018. [Epub ahead of print.]
9. Parsons HM, Muffly L, Alvarez EM, Keegan THM. Does treatment setting matter? Evaluating resource utilization for adolescents treated in pediatric vs adult cancer institutions. https://academic.oup.com/jnci/advance-article/doi/10.1093/jnci/djy123/5056313?searchresult=1. Published July 19, 2018. Last accessed October 12, 2018.
10. Creutzig U, Zimmermann M, Reinhardt D, et al. Changes in cytogenetics and molecular genetics in acute myeloid leukemia from childhood to adult age groups. Cancer. 2016;122(24):3821-3830.
11. Salsman JM, Garcia SF, Yanez B, et al. Physical, emotional, and social health differences between posttreatment young adults with cancer and matched healthy controls. Cancer. 2014;120(15):2247-2254.
12. Kim ES, Bruinooge SS, Roberts S, et al. Broadening eligibility criteria to make clinical trials more representative: American Society of Clinical Oncology and Friends of Cancer Research joint research statement. J Clin Oncol. 2017;35(33):3737-3744.
13. Freyer DR, Seibel NL. The clinical trials gap for adolescents and young adults with cancer: recent progress and conceptual framework for continued research. Curr Pediatr Rep. Published online February 18, 2015. DOI 10.1007/s40124-015-0075-y.
14. Nass SJ, Beaupin LK, Demark-Wahnefried W, et al. Identifying and addressing the needs of adolescents and young adults with cancer: summary of an Institute of Medicine workshop. Oncologist. 2015;20(2):186-195.
15. Wilder Smith A, Seibel NL, Lewis DR, et al. Next steps for adolescent and young adult oncology workshop: An update on progress and recommendations for the future. Cancer. 2016;122(7):988-999.
16. Keegan THM, Ries LAG, Barr RD, et al. Comparison of cancer survival trends in the United States of adolescents and young adults with those in children and older adults. Cancer. 2016;122(7):1009-1016.
1. Boissel N, Baruchel A. Acute lymphoblastic leukemia in adolescent and young adults: treat as adults or as children? Blood. 2018;132:351-361.
2. Dunleavy K, Gross TG. Management of aggressive B-cell NHLs in the AYA population: an adult vs pediatric perspective. Blood. 2018;132:369-375.
3. O’Dwyer K, Freyer DR, Horan JT. Treatment strategies for adolescent and young adult patients with acute myeloid leukemia. Blood. 2018;132:362-368.
4. Flerlage JE, Metzger ML, Bhakta N. The management of Hodgkin lymphoma in adolescents and young adults: burden of disease or burden of choice? Blood. 2018;132:376-384.
5. Husson O, Huijgens PC, van der Graaf WTA. Psychosocial challenges and health-related quality of life of adolescents and young adults with hematologic malignancies. Blood. 2018;132:385-392.
6. Cortes J. Introduction to a review series on adolescent and young adult malignant hematology. Blood. 2018;132:345-346.
7. Muffly L, Alvarez E, Lichtensztajn D, Abrahão R, Gomez SL, Keegan T. Patterns of care and outcomes in adolescent and young adult acute lymphoblastic leukemia: a population-based study. Blood Adv. 2018;2(8):895-903.
8. Nathan PC, Bremner KE, Liu N, et al. Resource utilization and costs in adolescents treated for cancer in pediatric vs adult institutions. J Natl Cancer Inst. July 19, 2018. [Epub ahead of print.]
9. Parsons HM, Muffly L, Alvarez EM, Keegan THM. Does treatment setting matter? Evaluating resource utilization for adolescents treated in pediatric vs adult cancer institutions. https://academic.oup.com/jnci/advance-article/doi/10.1093/jnci/djy123/5056313?searchresult=1. Published July 19, 2018. Last accessed October 12, 2018.
10. Creutzig U, Zimmermann M, Reinhardt D, et al. Changes in cytogenetics and molecular genetics in acute myeloid leukemia from childhood to adult age groups. Cancer. 2016;122(24):3821-3830.
11. Salsman JM, Garcia SF, Yanez B, et al. Physical, emotional, and social health differences between posttreatment young adults with cancer and matched healthy controls. Cancer. 2014;120(15):2247-2254.
12. Kim ES, Bruinooge SS, Roberts S, et al. Broadening eligibility criteria to make clinical trials more representative: American Society of Clinical Oncology and Friends of Cancer Research joint research statement. J Clin Oncol. 2017;35(33):3737-3744.
13. Freyer DR, Seibel NL. The clinical trials gap for adolescents and young adults with cancer: recent progress and conceptual framework for continued research. Curr Pediatr Rep. Published online February 18, 2015. DOI 10.1007/s40124-015-0075-y.
14. Nass SJ, Beaupin LK, Demark-Wahnefried W, et al. Identifying and addressing the needs of adolescents and young adults with cancer: summary of an Institute of Medicine workshop. Oncologist. 2015;20(2):186-195.
15. Wilder Smith A, Seibel NL, Lewis DR, et al. Next steps for adolescent and young adult oncology workshop: An update on progress and recommendations for the future. Cancer. 2016;122(7):988-999.
16. Keegan THM, Ries LAG, Barr RD, et al. Comparison of cancer survival trends in the United States of adolescents and young adults with those in children and older adults. Cancer. 2016;122(7):1009-1016.
NIH Program Enhances Diversity Among Researchers
The NIH has chosen 13 researchers for the inaugural class of the Distinguished Scholars Program (DSP), launched earlier this year to build diversity within the NIH Intramural Research Program. “Nurturing diversity in the NIH Intramural Research Program is paramount to upholding our mission,” said NIH Director Francis Collins, MD, PhD. Research has shown that a “diversity of perspectives” is vital to the improved quality and number of discoveries, he adds.
The DSP aims to facilitate hiring and career progression of tenure-track investigators who have demonstrated commitment to promoting diversity and inclusion in the biomedical research workforce, according to the NIH. The DSP is unique in its focus on early-stage investigators, says Hannah A. Valantine, MD, NIH Chief Officer for Scientific Workforce Diversity. She says that is the “major point where we lose underrepresented groups from scientific careers.”
Dr. Collins says the DSP can serve as a model for universities to prevent the attrition of underrepresented groups, including women, blacks, Hispanics or Latinos, American Indians and Alaska Natives, Native Hawaiians and other Pacific Islanders, individuals with disabilities, and individuals from disadvantaged backgrounds.
The pilot program will fund 3 cohorts of up to 15 scholars each. Nominees are chosen for their scientific excellence and commitment to diversity and inclusion, shown through participation in activities, such as mentoring programs.
Scholars will receive 4 years of research support of up to $2.35 million from the DSP; their nominating institute or center will continue to fund their research throughout their tenure track. Each scholar also will be mentored by a highly experienced NIH senior investigator and receive professional leadership training, workshops on management skills, and networking opportunities with NIH leadership.
Source:
NIH selects first scholars in pioneering program to enhance diversity within inhouse research program [news release]. Bethesda, MD: National Institutes of Health; October 23, 2018. https://www.nih.gov/news-events/news-releases/nih-selects-first-scholars-pioneering-program-enhance-diversity-within-house-research-program . Accessed October 31, 2018.
The NIH has chosen 13 researchers for the inaugural class of the Distinguished Scholars Program (DSP), launched earlier this year to build diversity within the NIH Intramural Research Program. “Nurturing diversity in the NIH Intramural Research Program is paramount to upholding our mission,” said NIH Director Francis Collins, MD, PhD. Research has shown that a “diversity of perspectives” is vital to the improved quality and number of discoveries, he adds.
The DSP aims to facilitate hiring and career progression of tenure-track investigators who have demonstrated commitment to promoting diversity and inclusion in the biomedical research workforce, according to the NIH. The DSP is unique in its focus on early-stage investigators, says Hannah A. Valantine, MD, NIH Chief Officer for Scientific Workforce Diversity. She says that is the “major point where we lose underrepresented groups from scientific careers.”
Dr. Collins says the DSP can serve as a model for universities to prevent the attrition of underrepresented groups, including women, blacks, Hispanics or Latinos, American Indians and Alaska Natives, Native Hawaiians and other Pacific Islanders, individuals with disabilities, and individuals from disadvantaged backgrounds.
The pilot program will fund 3 cohorts of up to 15 scholars each. Nominees are chosen for their scientific excellence and commitment to diversity and inclusion, shown through participation in activities, such as mentoring programs.
Scholars will receive 4 years of research support of up to $2.35 million from the DSP; their nominating institute or center will continue to fund their research throughout their tenure track. Each scholar also will be mentored by a highly experienced NIH senior investigator and receive professional leadership training, workshops on management skills, and networking opportunities with NIH leadership.
Source:
NIH selects first scholars in pioneering program to enhance diversity within inhouse research program [news release]. Bethesda, MD: National Institutes of Health; October 23, 2018. https://www.nih.gov/news-events/news-releases/nih-selects-first-scholars-pioneering-program-enhance-diversity-within-house-research-program . Accessed October 31, 2018.
The NIH has chosen 13 researchers for the inaugural class of the Distinguished Scholars Program (DSP), launched earlier this year to build diversity within the NIH Intramural Research Program. “Nurturing diversity in the NIH Intramural Research Program is paramount to upholding our mission,” said NIH Director Francis Collins, MD, PhD. Research has shown that a “diversity of perspectives” is vital to the improved quality and number of discoveries, he adds.
The DSP aims to facilitate hiring and career progression of tenure-track investigators who have demonstrated commitment to promoting diversity and inclusion in the biomedical research workforce, according to the NIH. The DSP is unique in its focus on early-stage investigators, says Hannah A. Valantine, MD, NIH Chief Officer for Scientific Workforce Diversity. She says that is the “major point where we lose underrepresented groups from scientific careers.”
Dr. Collins says the DSP can serve as a model for universities to prevent the attrition of underrepresented groups, including women, blacks, Hispanics or Latinos, American Indians and Alaska Natives, Native Hawaiians and other Pacific Islanders, individuals with disabilities, and individuals from disadvantaged backgrounds.
The pilot program will fund 3 cohorts of up to 15 scholars each. Nominees are chosen for their scientific excellence and commitment to diversity and inclusion, shown through participation in activities, such as mentoring programs.
Scholars will receive 4 years of research support of up to $2.35 million from the DSP; their nominating institute or center will continue to fund their research throughout their tenure track. Each scholar also will be mentored by a highly experienced NIH senior investigator and receive professional leadership training, workshops on management skills, and networking opportunities with NIH leadership.
Source:
NIH selects first scholars in pioneering program to enhance diversity within inhouse research program [news release]. Bethesda, MD: National Institutes of Health; October 23, 2018. https://www.nih.gov/news-events/news-releases/nih-selects-first-scholars-pioneering-program-enhance-diversity-within-house-research-program . Accessed October 31, 2018.