User login
Pembrolizumab extends survival of head and neck cancer
MUNICH – In patients with recurrent or metastatic head and neck squamous cell carcinoma expressing programmed death ligand-1 (PDL-1), the immune checkpoint inhibitor pembrolizumab alone or in combination with chemotherapy improved overall survival, compared with the EXTREME chemotherapy regimen, reported investigators in the Keynote 048 trial.

Overall survival (OS) among patients with a PD-L1 combined positive score (CPS) of 20 or greater treated with pembrolizumab (Keytruda) monotherapy was 14.9 months compared with 10.7 months for patients treated with the EXTREME regimen, a combination of cetuximab (Erbitux), carboplatin or cisplatin, and 5-fluorouracil.
A similar overall survival benefit was seen in patients with a CPS of 1 or greater, and in the total population of patients treated with pembrolizumab plus chemotherapy followed by pembrolizumab maintenance compared with EXTREME chemotherapy, Barbara Burtness, MD, of Yale Cancer Center, New Haven, Conn.
There were no differences in response rates between either pembrolizumab monotherapy or in combination compared with chemotherapy alone, but responses were more durable with the checkpoint inhibitor than with chemotherapy.
“Pembrolizumab alone and pembrolizumab given with platinum and 5-fluorouracil should represent new standards of care for the first-line treatment of metastatic head and neck carcinoma. Immune checkpoint monotherapy with pembrolizumab allows patients to live longer and has a better safety profile than the previous standard for those patients whose tumors express PD-L1,” she said at a briefing prior to her presentation of the data in a presidential symposium at the European Society for Medical Oncology Congress.
“This is the first time since 10 years that we show an improvement in survival for this group of patients,” said Jean-Pascal Machiels, MD, of University Clinic Saint-Luc, Brussels, the invited discussant for the briefing and the symposium.
The CPS is a ratio of PD-L1-positive tumor cells, lymphocytes, and macrophages to the total numbers of cells counted multiplied by 100. The investigators looked at progression-free survival (PFS) and OS in three cohorts of patients with squamous cell carcinomas of the oropharynx, oral cavity, hypopharynx, or larynx that were recurrent or metastatic and were incurable by local therapies. They compared pembrolizumab monotherapy with the EXTREME regimen, and pembrolizumab plus chemotherapy (as described in the following paragraph) with EXTREME.
A total of 882 patients were enrolled and stratified by PD-L1 expression (on 50% or greater of tumor cells, or less than 50%), p16 positive or negative status in the oropharynx, and Eastern Cooperative Oncology Group performance status of 0 or 1. The patients were then randomly assigned on a 1:1:1 basis to either pembrolizumab monotherapy at 200 mg every 3 weeks for up to 35 cycles, pembrolizumab plus a standard chemotherapy regimen (carboplatin to an area-under-the curve [AUC] of 5 or cisplatin 100 mg/m2 plus 5-FU 1000 mg/m2 per day for 4 days for six cycles, followed by pembrolizumab maintenance for up to 35 cycles or EXTREME (cetuximab at a loading dose of 400 mg/m2 followed by 250 mg/m2 once weekly plus the chemotherapy regimen described above, followed by maintenance cetuximab).
Pembrolizumab monotherapy vs. EXTREME
For the coprimary endpoint of OS in the CPS 20 or greater population, pembrolizumab was associated with significantly better survival than EXTREME at both the 12- and 24-month time points (56.9% vs. 44.9%, and 38.3% vs. 22,1%, respectively). After a minimum follow-up of 17 months, the median OS was 14.9 months with pembrolizumab, vs. 10.7 months for EXTREME. The hazard ratio (HR) for death with pembrolizumab was 0.61 (P = .0007).
The median OS in the CPS 1 or greater population was 12.3 months and 10.3 months, respectively (HR 0.78, P = .0086).
There were no differences between the arms in PFS, however, either in the CPS 20 or greater or CPS 1 or greater populations.
Although, as noted, response rates did not differ between the groups, the median duration of response was 20.9 months with pembrolizumab in both the CPS 20 and CPS 1 populations, compared with 4.2 and 4.5 months, respectively, for EXTREME.
Treatment-related adverse events of any grade occurred in 58% of patients in the monotherapy arm, vs. 96.9% in the EXTREME arm. Fatal adverse events occurred in 1% vs. 2.8%, and events leading to drug discontinuation occurred in 4.7% vs. 19.9%, respectively. There were more immune-mediated events in the pembrolizumab arm, including one death (from pneumonitis) vs. no deaths from immune-related causes in the EXTREME arm.
Pembrolizumab plus chemo vs. EXTREME
The combination of pembrolizumab was also superior to EXTREME in the total population, with 12- and 24-month OS rates of 53% vs. 43.9%, and 29% vs. 18.7%, respectively. The median OS was 13 months with the pembrolizumab/chemo combination, vs. 10.7 months for EXTREME, translating into an HR of 0.77 (P = .0034). In this analysis as well as in the pembrolizumab monotherapy combination, there was no difference in PFS or response rates, but responses in the pembrolizumab arm were more durable.
In this comparison, treatment-related adverse events were generally similar between the groups, although there were 10 treatment-related deaths with pembrolizumab, compared with eight in the EXTREME arm.
There was one immune-related death, from pneumonitis, in the pembrolizumab arm, vs. none in the EXTREME arm. Hypothyroidism, pneumonitis, hyperthyroidism and colitis were more frequent with pembrolizumab, whereas infusion reactions and severe skin reactions were more frequent with EXTREME.
“There are further analyses of biomarker and clinical predictors that will be forthcoming from this study, and these may eventually optimally guide the choice of whether to administer pembrolizumab alone or in the novel combination, Dr. Burtness said at the briefing.
“What’s extremely important also is that for a subgroup of patients with high expression of PD-L1, we can probably remove the cisplatin and have a good outcome with immunotherapy alone,” Dr. Machiels said.
He said that more work needs to be done to determine which patients are most likely to benefit from immunotherapy in the recurrent/metastatic setting, and “we have to see how we can now bring this active drug to the curative treatment of the patient, in combination with chemoradiation.”
The study was funded by Merck Sharp & Dohme. Dr. Burtness disclosed being and advisory board member and receiving travel expenses from MSD and others. Dr. Machiels disclosed speaker honoraria, travel expenses, and an uncompensated advisory role with MSD.
SOURCE: Burtness B et al. ESMO 2018. Abstract LBA8_PR.
MUNICH – In patients with recurrent or metastatic head and neck squamous cell carcinoma expressing programmed death ligand-1 (PDL-1), the immune checkpoint inhibitor pembrolizumab alone or in combination with chemotherapy improved overall survival, compared with the EXTREME chemotherapy regimen, reported investigators in the Keynote 048 trial.
Overall survival (OS) among patients with a PD-L1 combined positive score (CPS) of 20 or greater treated with pembrolizumab (Keytruda) monotherapy was 14.9 months compared with 10.7 months for patients treated with the EXTREME regimen, a combination of cetuximab (Erbitux), carboplatin or cisplatin, and 5-fluorouracil.
A similar overall survival benefit was seen in patients with a CPS of 1 or greater, and in the total population of patients treated with pembrolizumab plus chemotherapy followed by pembrolizumab maintenance compared with EXTREME chemotherapy, Barbara Burtness, MD, of Yale Cancer Center, New Haven, Conn.
There were no differences in response rates between either pembrolizumab monotherapy or in combination compared with chemotherapy alone, but responses were more durable with the checkpoint inhibitor than with chemotherapy.
“Pembrolizumab alone and pembrolizumab given with platinum and 5-fluorouracil should represent new standards of care for the first-line treatment of metastatic head and neck carcinoma. Immune checkpoint monotherapy with pembrolizumab allows patients to live longer and has a better safety profile than the previous standard for those patients whose tumors express PD-L1,” she said at a briefing prior to her presentation of the data in a presidential symposium at the European Society for Medical Oncology Congress.
“This is the first time since 10 years that we show an improvement in survival for this group of patients,” said Jean-Pascal Machiels, MD, of University Clinic Saint-Luc, Brussels, the invited discussant for the briefing and the symposium.
The CPS is a ratio of PD-L1-positive tumor cells, lymphocytes, and macrophages to the total numbers of cells counted multiplied by 100. The investigators looked at progression-free survival (PFS) and OS in three cohorts of patients with squamous cell carcinomas of the oropharynx, oral cavity, hypopharynx, or larynx that were recurrent or metastatic and were incurable by local therapies. They compared pembrolizumab monotherapy with the EXTREME regimen, and pembrolizumab plus chemotherapy (as described in the following paragraph) with EXTREME.
A total of 882 patients were enrolled and stratified by PD-L1 expression (on 50% or greater of tumor cells, or less than 50%), p16 positive or negative status in the oropharynx, and Eastern Cooperative Oncology Group performance status of 0 or 1. The patients were then randomly assigned on a 1:1:1 basis to either pembrolizumab monotherapy at 200 mg every 3 weeks for up to 35 cycles, pembrolizumab plus a standard chemotherapy regimen (carboplatin to an area-under-the curve [AUC] of 5 or cisplatin 100 mg/m2 plus 5-FU 1000 mg/m2 per day for 4 days for six cycles, followed by pembrolizumab maintenance for up to 35 cycles or EXTREME (cetuximab at a loading dose of 400 mg/m2 followed by 250 mg/m2 once weekly plus the chemotherapy regimen described above, followed by maintenance cetuximab).
Pembrolizumab monotherapy vs. EXTREME
For the coprimary endpoint of OS in the CPS 20 or greater population, pembrolizumab was associated with significantly better survival than EXTREME at both the 12- and 24-month time points (56.9% vs. 44.9%, and 38.3% vs. 22,1%, respectively). After a minimum follow-up of 17 months, the median OS was 14.9 months with pembrolizumab, vs. 10.7 months for EXTREME. The hazard ratio (HR) for death with pembrolizumab was 0.61 (P = .0007).
The median OS in the CPS 1 or greater population was 12.3 months and 10.3 months, respectively (HR 0.78, P = .0086).
There were no differences between the arms in PFS, however, either in the CPS 20 or greater or CPS 1 or greater populations.
Although, as noted, response rates did not differ between the groups, the median duration of response was 20.9 months with pembrolizumab in both the CPS 20 and CPS 1 populations, compared with 4.2 and 4.5 months, respectively, for EXTREME.
Treatment-related adverse events of any grade occurred in 58% of patients in the monotherapy arm, vs. 96.9% in the EXTREME arm. Fatal adverse events occurred in 1% vs. 2.8%, and events leading to drug discontinuation occurred in 4.7% vs. 19.9%, respectively. There were more immune-mediated events in the pembrolizumab arm, including one death (from pneumonitis) vs. no deaths from immune-related causes in the EXTREME arm.
Pembrolizumab plus chemo vs. EXTREME
The combination of pembrolizumab was also superior to EXTREME in the total population, with 12- and 24-month OS rates of 53% vs. 43.9%, and 29% vs. 18.7%, respectively. The median OS was 13 months with the pembrolizumab/chemo combination, vs. 10.7 months for EXTREME, translating into an HR of 0.77 (P = .0034). In this analysis as well as in the pembrolizumab monotherapy combination, there was no difference in PFS or response rates, but responses in the pembrolizumab arm were more durable.
In this comparison, treatment-related adverse events were generally similar between the groups, although there were 10 treatment-related deaths with pembrolizumab, compared with eight in the EXTREME arm.
There was one immune-related death, from pneumonitis, in the pembrolizumab arm, vs. none in the EXTREME arm. Hypothyroidism, pneumonitis, hyperthyroidism and colitis were more frequent with pembrolizumab, whereas infusion reactions and severe skin reactions were more frequent with EXTREME.
“There are further analyses of biomarker and clinical predictors that will be forthcoming from this study, and these may eventually optimally guide the choice of whether to administer pembrolizumab alone or in the novel combination, Dr. Burtness said at the briefing.
“What’s extremely important also is that for a subgroup of patients with high expression of PD-L1, we can probably remove the cisplatin and have a good outcome with immunotherapy alone,” Dr. Machiels said.
He said that more work needs to be done to determine which patients are most likely to benefit from immunotherapy in the recurrent/metastatic setting, and “we have to see how we can now bring this active drug to the curative treatment of the patient, in combination with chemoradiation.”
The study was funded by Merck Sharp & Dohme. Dr. Burtness disclosed being and advisory board member and receiving travel expenses from MSD and others. Dr. Machiels disclosed speaker honoraria, travel expenses, and an uncompensated advisory role with MSD.
SOURCE: Burtness B et al. ESMO 2018. Abstract LBA8_PR.
MUNICH – In patients with recurrent or metastatic head and neck squamous cell carcinoma expressing programmed death ligand-1 (PDL-1), the immune checkpoint inhibitor pembrolizumab alone or in combination with chemotherapy improved overall survival, compared with the EXTREME chemotherapy regimen, reported investigators in the Keynote 048 trial.
Overall survival (OS) among patients with a PD-L1 combined positive score (CPS) of 20 or greater treated with pembrolizumab (Keytruda) monotherapy was 14.9 months compared with 10.7 months for patients treated with the EXTREME regimen, a combination of cetuximab (Erbitux), carboplatin or cisplatin, and 5-fluorouracil.
A similar overall survival benefit was seen in patients with a CPS of 1 or greater, and in the total population of patients treated with pembrolizumab plus chemotherapy followed by pembrolizumab maintenance compared with EXTREME chemotherapy, Barbara Burtness, MD, of Yale Cancer Center, New Haven, Conn.
There were no differences in response rates between either pembrolizumab monotherapy or in combination compared with chemotherapy alone, but responses were more durable with the checkpoint inhibitor than with chemotherapy.
“Pembrolizumab alone and pembrolizumab given with platinum and 5-fluorouracil should represent new standards of care for the first-line treatment of metastatic head and neck carcinoma. Immune checkpoint monotherapy with pembrolizumab allows patients to live longer and has a better safety profile than the previous standard for those patients whose tumors express PD-L1,” she said at a briefing prior to her presentation of the data in a presidential symposium at the European Society for Medical Oncology Congress.
“This is the first time since 10 years that we show an improvement in survival for this group of patients,” said Jean-Pascal Machiels, MD, of University Clinic Saint-Luc, Brussels, the invited discussant for the briefing and the symposium.
The CPS is a ratio of PD-L1-positive tumor cells, lymphocytes, and macrophages to the total numbers of cells counted multiplied by 100. The investigators looked at progression-free survival (PFS) and OS in three cohorts of patients with squamous cell carcinomas of the oropharynx, oral cavity, hypopharynx, or larynx that were recurrent or metastatic and were incurable by local therapies. They compared pembrolizumab monotherapy with the EXTREME regimen, and pembrolizumab plus chemotherapy (as described in the following paragraph) with EXTREME.
A total of 882 patients were enrolled and stratified by PD-L1 expression (on 50% or greater of tumor cells, or less than 50%), p16 positive or negative status in the oropharynx, and Eastern Cooperative Oncology Group performance status of 0 or 1. The patients were then randomly assigned on a 1:1:1 basis to either pembrolizumab monotherapy at 200 mg every 3 weeks for up to 35 cycles, pembrolizumab plus a standard chemotherapy regimen (carboplatin to an area-under-the curve [AUC] of 5 or cisplatin 100 mg/m2 plus 5-FU 1000 mg/m2 per day for 4 days for six cycles, followed by pembrolizumab maintenance for up to 35 cycles or EXTREME (cetuximab at a loading dose of 400 mg/m2 followed by 250 mg/m2 once weekly plus the chemotherapy regimen described above, followed by maintenance cetuximab).
Pembrolizumab monotherapy vs. EXTREME
For the coprimary endpoint of OS in the CPS 20 or greater population, pembrolizumab was associated with significantly better survival than EXTREME at both the 12- and 24-month time points (56.9% vs. 44.9%, and 38.3% vs. 22,1%, respectively). After a minimum follow-up of 17 months, the median OS was 14.9 months with pembrolizumab, vs. 10.7 months for EXTREME. The hazard ratio (HR) for death with pembrolizumab was 0.61 (P = .0007).
The median OS in the CPS 1 or greater population was 12.3 months and 10.3 months, respectively (HR 0.78, P = .0086).
There were no differences between the arms in PFS, however, either in the CPS 20 or greater or CPS 1 or greater populations.
Although, as noted, response rates did not differ between the groups, the median duration of response was 20.9 months with pembrolizumab in both the CPS 20 and CPS 1 populations, compared with 4.2 and 4.5 months, respectively, for EXTREME.
Treatment-related adverse events of any grade occurred in 58% of patients in the monotherapy arm, vs. 96.9% in the EXTREME arm. Fatal adverse events occurred in 1% vs. 2.8%, and events leading to drug discontinuation occurred in 4.7% vs. 19.9%, respectively. There were more immune-mediated events in the pembrolizumab arm, including one death (from pneumonitis) vs. no deaths from immune-related causes in the EXTREME arm.
Pembrolizumab plus chemo vs. EXTREME
The combination of pembrolizumab was also superior to EXTREME in the total population, with 12- and 24-month OS rates of 53% vs. 43.9%, and 29% vs. 18.7%, respectively. The median OS was 13 months with the pembrolizumab/chemo combination, vs. 10.7 months for EXTREME, translating into an HR of 0.77 (P = .0034). In this analysis as well as in the pembrolizumab monotherapy combination, there was no difference in PFS or response rates, but responses in the pembrolizumab arm were more durable.
In this comparison, treatment-related adverse events were generally similar between the groups, although there were 10 treatment-related deaths with pembrolizumab, compared with eight in the EXTREME arm.
There was one immune-related death, from pneumonitis, in the pembrolizumab arm, vs. none in the EXTREME arm. Hypothyroidism, pneumonitis, hyperthyroidism and colitis were more frequent with pembrolizumab, whereas infusion reactions and severe skin reactions were more frequent with EXTREME.
“There are further analyses of biomarker and clinical predictors that will be forthcoming from this study, and these may eventually optimally guide the choice of whether to administer pembrolizumab alone or in the novel combination, Dr. Burtness said at the briefing.
“What’s extremely important also is that for a subgroup of patients with high expression of PD-L1, we can probably remove the cisplatin and have a good outcome with immunotherapy alone,” Dr. Machiels said.
He said that more work needs to be done to determine which patients are most likely to benefit from immunotherapy in the recurrent/metastatic setting, and “we have to see how we can now bring this active drug to the curative treatment of the patient, in combination with chemoradiation.”
The study was funded by Merck Sharp & Dohme. Dr. Burtness disclosed being and advisory board member and receiving travel expenses from MSD and others. Dr. Machiels disclosed speaker honoraria, travel expenses, and an uncompensated advisory role with MSD.
SOURCE: Burtness B et al. ESMO 2018. Abstract LBA8_PR.
REPORTING FROM ESMO 2018
Key clinical point: Pembrolizumab alone or in combination with chemotherapy was associated with better overall survival of squamous cell head and neck cancer, compared with the EXTREME chemotherapy regimen.
Major finding: Overall survival among patients with a PD-L1 combined positive score of 20 or greater treated with pembrolizumab (Keytruda) monotherapy was 14.9 compared with 10.7 months for patients treated with the EXTREME regimen.
Study details: Randomized phase 3 trial of 882 patients with recurrent or metastatic squamous cell carcinoma of the head and neck.
Disclosures: The study was funded by Merck Sharp & Dohme. Dr. Burtness disclosed being and advisory board member and receiving travel expenses from MSD and others. Dr. Machiels disclosed speaker honoraria, travel expenses, and an uncompensated advisory role with MSD.
Source: Burtness B et al. ESMO 2018. Abstract LBA8_PR.
Over one-third of psoriasis patients have PsA
Over one-third of psoriasis patients have PsA
About two-thirds of patients with psoriasis in a national registry also had psoriatic arthritis (PsA) and/or psoriasis in at least one challenging-to-treat (CTT) area, and one-quarter had both, according to Kristina Callis Duffin, MD, of the University of Utah, Salt Lake City, and her associates.
Their analysis included 2,042 psoriasis patients who were enrolled in the Corrona Psoriasis Registry between April 2015 and May 2018 and initiated biologic treatment during that time. The mean age was 49.6 years, 80% of the patients were white, and 51% were obese. Mean disease duration was 19.9 years and 89.2% of the patients had moderate to severe disease. CTT areas include the scalp, nails, and palmoplantar areas.
A total of 784 people in the cohort (38.4%) had PsA, 778 (38.1%) had scalp psoriasis, 326 (16.0%) had nail psoriasis, 223 (10.9%) had palmoplantar psoriasis, and 535 (26.2%) had both PsA and psoriasis in at least two CTT areas. The most common combinations were PsA plus scalp psoriasis and PsA plus nail and scalp psoriasis.
“These results indicate a need to further characterize patients with psoriasis who have PsA and CTT areas and evaluate the impact of these factors to better understand their treatment needs,” the investigators noted.
The Corrona registry has been supported by numerous pharmaceutical companies, and the study authors reported numerous financial relationships with industry; two authors are Novartis employees.
Secukinumab effective for slowing radiographic progression in active PsA
Treatment with secukinumab significantly reduced radiographic progression in patients with active PsA, according to Désirée van der Heijde, MD, PhD, professor of rheumatology at Leiden University Medical Center, and her associates.
The results come from an analysis of the FUTURE 5 trial, a study of 996 patients with active PsA despite previous NSAID treatment, disease-modifying antirheumatic drug treatment, or anti–tumor necrosis factor (TNF) therapy. Patients were randomized to receive 300 mg subcutaneous secukinumab with loading dose, 150 mg secukinumab with loading dose, 150 mg secukinumab without loading dose, or placebo, at baseline; weeks 1, 2, 3, and 4; then every 4 weeks.
After 24 weeks, the mean change in van der Heijde–modified Total Sharp Score for PsA was 0.08 for the 300-mg secukinumab group (P less than .01), 0.17 for the 150-mg secukinumab with loading dose group (P less than .05), a reduction of 0.09 for the 150-mg secukinumab without loading dose group (P less than .01), and 0.50 for the placebo group. Lower radiographic progression was seen regardless of prior anti-TNF or concomitant methotrexate treatment.
The study was funded by Novartis. The study authors reported financial disclosures with numerous companies; five authors are Novartis employees.
Tildrakizumab sustains efficacy in plaque psoriasis treatment after 1 year
Nearly all patients receiving the interleukin-23 inhibitor tildrakizumab for the treatment of moderate to severe plaque psoriasis maintained or improved their Psoriasis Area and Severity Index (PASI) response rate after 52 weeks of treatment, compared with their response after 28 weeks.
The analysis, conducted by Boni E. Elewski, MD, of the University of Alabama at Birmingham, and her associates, included 352 patients who received 100 mg tildrakizumab and 313 who received 200 mg tildrakizumab. Treatment was received at baseline, at 4 weeks, and then every 12 weeks afterward.
At week 28, the proportions of patients achieving PASI 100, PASI 90-99, PASI 75-89, and PASI 50-74 at week 28 were 25.9%, 38.4%, 25.3%, and 10.5%, respectively, among those treated with the 100-mg dose. The proportions were 24.6%, 24.3%, 19.5%, and 31.6%, respectively, among those treated with the 200-mg dose.
In patients who achieved at least PASI 90 on either dose at week 28, 88.9%-89.4% maintained that response at week 52. For patients with PASI 75-89, 39.3%-40.4% maintained that response and 33.7%-41.0% achieved a PASI 90 response. At week 52, in patients with PASI 50-74, 20.2%-29.7% achieved at least a PASI 90, 52.5%-64.9% achieved PASI 75, and only 2.6% of patients on either dose had fallen below PASI 50.
Four study authors reported being clinical investigators on studies sponsored by Merck and Sun Pharmaceuticals; five authors are employees of Sun Pharmaceuticals.
Halobetasol/tazarotene combination most effective for plaque psoriasis treatment
A fixed combination of halobetasol propionate 0.01% and tazarotene 0.045% lotion provided a synergistic effect over either component on its own for the treatment of plaque psoriasis, according to Leon H. Kircik, MD, of Indiana University, Indianapolis, and his associates.
The investigators performed a post hoc analysis of 212 patients with moderate to severe plaque psoriasis randomized to receive either the halobetasol/tazarotene combination, halobetasol only, tazarotene only, or vehicle only for 8 weeks, with follow-up at 12 weeks. Treatment success was based on the proportion of patients who achieved at least a 2-grade improvement in the Investigator Global Assessment (IGA) score, IGA scores of “clear” or “almost clear,” and percent change from baseline in IGA multiplied by Body Surface Area (BSA) composite score (IGAxBSA). “Synergy was calculated by summing up the contribution of the individual active ingredients (HP and TAZ) to overall efficacy and comparing to the efficacy achieved with HP/TAZ lotion relative to vehicle,” the authors explained.
Relative to vehicle, treatment success for halobetasol/tazarotene after 8 weeks was 42.8%, 23.6% for halobetasol alone, and 9.0% for tazarotene alone. After 12 weeks, the difference was 31.3%, 14.1%, and 5.9%, respectively. The percent change in IGAxBSA scores from baseline after 8 weeks, relative to vehicle, were 51.6%, 37.3%, and 3.3%, respectively. After 12 weeks, the change was 47.3%, 25.7%, and 8.6%, respectively.
After 8 weeks, the synergy ratio for treatment success and IGAxBSA scores for the halobetasol/tazarotene combination was 1.3. After 12 weeks, the synergy ratio for treatment success was 1.6 and the ratio for IGAxBSA scores was 1.4.
“By combining two agents into one once-daily formulation, this novel formulation reduces the number of product applications and may help patient adherence,” the study authors noted.
Dr. Kircik reported serving as a consultant and investigator for Valeant Pharmaceuticals. One study author is an employee of Bausch Health and Ortho Dermatologics, and another is an employee of Dow Pharmaceutical Sciences (a division of Valeant).
Brodalumab demonstrates low immunogenicity in moderate to severe psoriasis
The immunogenicity of brodalumab in patients with moderate to severe plaque psoriasis was low and did not compromise the efficacy or safety profile of the drug, according to Kristian Reich, MD, of Dermatologikum Berlin and SCIderm Research Institute in Hamburg, Germany, and his associates.
Data from a 12-week, phase 2 trial with a 352-week, open-label extension and three 52-week phase 3 trials were included in the analysis. Antidrug antibodies (ADAs) were tested, and positive samples were further analyzed for neutralizing ADAs by a cell-based assay.
Out of the 4,461 patients who received brodalumab, 122 (2.7%) were positive for ADAs after starting brodalumab. The incidence rate ranged from 1.9% to 3.4% between all dosing groups (140 mg, 210 mg, variable dosing, and 210 mg of brodalumab after ustekinumab). In 58 (1.4%) of patients, ADAs were transient. No patients had neutralizing ADAs, and no evidence of altered pharmacokinetics, loss of efficacy, or changes in the safety profile of brodalumab in subjects positive for ADAs was seen.
No significant difference was seen in the incidence rate of hypersensitivity or injection site reactions in brodalumab, compared with placebo or ustekinumab. The most common injection site reactions were injection site pain, erythema, and bruising.
The study was supported by Amgen. The study authors reported numerous disclosures. Two authors are employees of Leo Pharma, one author is a former employee of the company.
Secukinumab improves patient-reported outcomes in CTT psoriasis
Treatment with secukinumab significantly improved patient-reported outcomes such as fatigue, itch, pain, and quality of life measures in patients with CTT psoriasis after 6 months, according to Jerry Bagel, MD, of the Psoriasis Treatment Center of Central New Jersey, East Windsor, and his associates.
A total of 68 patients with psoriasis localized to at least one CTT area who were enrolled in the Corrona Psoriasis Registry from April 15, 2015, through May 10, 2018, and were receiving secukinumab for the entirety of the 6-month study period were included in the analysis. Patient-reported outcomes included in the analysis were fatigue, itch, pain, Dermatology Quality of Life Index (DLQI) score, and Work Productivity and Activity Impairment (WPAI) scale.
The mean age at enrollment was 51.2 years and almost 80% of patients were white. Mean psoriasis duration was 21.8 years and nearly half had PsA.
Visual analog scale scores improved over baseline for fatigue (mean, 23.2 vs. 33.2; P = .01), itch (20.9 vs. 49.6; P less than .0001), and pain (12.1 vs. 33.8; P less than .0001). DLQI scores also improved (2.9 vs. 8.1; P less than .0001), and the proportion of patients who reported that psoriasis had at least a moderate effect on their life was reduced after 6 months (22.1% vs. 59.7%; P less than .0001).
Based on WPAI results, patients experienced significant improvements in the percentage of daily activities impaired (mean, 9.5% vs 17.5%; P = .0075); of the 42 patients who were employed, both impairment percentage (3.7% vs. 11.2%; P = .0148) and percentage of work hours affected (4.9% vs. 11.9%; P = .0486) were reduced from baseline.
“These results are consistent with previous reports from secukinumab clinical trials; however, additional real-world studies are needed to evaluate the long-term effectiveness of secukinumab for improving [patient-reported outcomes] in patients with psoriasis in CTT areas,” the authors noted.
The Corrona registry has been supported by numerous pharmaceutical companies, and several study authors reported various disclosures with industry. Two authors are Novartis employees. The study was supported by Novartis; the company participated in the interpretation of data and review and approval of the abstract.
These posters were presented at Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar. SDEF and this news organization are owned by the same parent company.
Over one-third of psoriasis patients have PsA
About two-thirds of patients with psoriasis in a national registry also had psoriatic arthritis (PsA) and/or psoriasis in at least one challenging-to-treat (CTT) area, and one-quarter had both, according to Kristina Callis Duffin, MD, of the University of Utah, Salt Lake City, and her associates.
Their analysis included 2,042 psoriasis patients who were enrolled in the Corrona Psoriasis Registry between April 2015 and May 2018 and initiated biologic treatment during that time. The mean age was 49.6 years, 80% of the patients were white, and 51% were obese. Mean disease duration was 19.9 years and 89.2% of the patients had moderate to severe disease. CTT areas include the scalp, nails, and palmoplantar areas.
A total of 784 people in the cohort (38.4%) had PsA, 778 (38.1%) had scalp psoriasis, 326 (16.0%) had nail psoriasis, 223 (10.9%) had palmoplantar psoriasis, and 535 (26.2%) had both PsA and psoriasis in at least two CTT areas. The most common combinations were PsA plus scalp psoriasis and PsA plus nail and scalp psoriasis.
“These results indicate a need to further characterize patients with psoriasis who have PsA and CTT areas and evaluate the impact of these factors to better understand their treatment needs,” the investigators noted.
The Corrona registry has been supported by numerous pharmaceutical companies, and the study authors reported numerous financial relationships with industry; two authors are Novartis employees.
Secukinumab effective for slowing radiographic progression in active PsA
Treatment with secukinumab significantly reduced radiographic progression in patients with active PsA, according to Désirée van der Heijde, MD, PhD, professor of rheumatology at Leiden University Medical Center, and her associates.
The results come from an analysis of the FUTURE 5 trial, a study of 996 patients with active PsA despite previous NSAID treatment, disease-modifying antirheumatic drug treatment, or anti–tumor necrosis factor (TNF) therapy. Patients were randomized to receive 300 mg subcutaneous secukinumab with loading dose, 150 mg secukinumab with loading dose, 150 mg secukinumab without loading dose, or placebo, at baseline; weeks 1, 2, 3, and 4; then every 4 weeks.
After 24 weeks, the mean change in van der Heijde–modified Total Sharp Score for PsA was 0.08 for the 300-mg secukinumab group (P less than .01), 0.17 for the 150-mg secukinumab with loading dose group (P less than .05), a reduction of 0.09 for the 150-mg secukinumab without loading dose group (P less than .01), and 0.50 for the placebo group. Lower radiographic progression was seen regardless of prior anti-TNF or concomitant methotrexate treatment.
The study was funded by Novartis. The study authors reported financial disclosures with numerous companies; five authors are Novartis employees.
Tildrakizumab sustains efficacy in plaque psoriasis treatment after 1 year
Nearly all patients receiving the interleukin-23 inhibitor tildrakizumab for the treatment of moderate to severe plaque psoriasis maintained or improved their Psoriasis Area and Severity Index (PASI) response rate after 52 weeks of treatment, compared with their response after 28 weeks.
The analysis, conducted by Boni E. Elewski, MD, of the University of Alabama at Birmingham, and her associates, included 352 patients who received 100 mg tildrakizumab and 313 who received 200 mg tildrakizumab. Treatment was received at baseline, at 4 weeks, and then every 12 weeks afterward.
At week 28, the proportions of patients achieving PASI 100, PASI 90-99, PASI 75-89, and PASI 50-74 at week 28 were 25.9%, 38.4%, 25.3%, and 10.5%, respectively, among those treated with the 100-mg dose. The proportions were 24.6%, 24.3%, 19.5%, and 31.6%, respectively, among those treated with the 200-mg dose.
In patients who achieved at least PASI 90 on either dose at week 28, 88.9%-89.4% maintained that response at week 52. For patients with PASI 75-89, 39.3%-40.4% maintained that response and 33.7%-41.0% achieved a PASI 90 response. At week 52, in patients with PASI 50-74, 20.2%-29.7% achieved at least a PASI 90, 52.5%-64.9% achieved PASI 75, and only 2.6% of patients on either dose had fallen below PASI 50.
Four study authors reported being clinical investigators on studies sponsored by Merck and Sun Pharmaceuticals; five authors are employees of Sun Pharmaceuticals.
Halobetasol/tazarotene combination most effective for plaque psoriasis treatment
A fixed combination of halobetasol propionate 0.01% and tazarotene 0.045% lotion provided a synergistic effect over either component on its own for the treatment of plaque psoriasis, according to Leon H. Kircik, MD, of Indiana University, Indianapolis, and his associates.
The investigators performed a post hoc analysis of 212 patients with moderate to severe plaque psoriasis randomized to receive either the halobetasol/tazarotene combination, halobetasol only, tazarotene only, or vehicle only for 8 weeks, with follow-up at 12 weeks. Treatment success was based on the proportion of patients who achieved at least a 2-grade improvement in the Investigator Global Assessment (IGA) score, IGA scores of “clear” or “almost clear,” and percent change from baseline in IGA multiplied by Body Surface Area (BSA) composite score (IGAxBSA). “Synergy was calculated by summing up the contribution of the individual active ingredients (HP and TAZ) to overall efficacy and comparing to the efficacy achieved with HP/TAZ lotion relative to vehicle,” the authors explained.
Relative to vehicle, treatment success for halobetasol/tazarotene after 8 weeks was 42.8%, 23.6% for halobetasol alone, and 9.0% for tazarotene alone. After 12 weeks, the difference was 31.3%, 14.1%, and 5.9%, respectively. The percent change in IGAxBSA scores from baseline after 8 weeks, relative to vehicle, were 51.6%, 37.3%, and 3.3%, respectively. After 12 weeks, the change was 47.3%, 25.7%, and 8.6%, respectively.
After 8 weeks, the synergy ratio for treatment success and IGAxBSA scores for the halobetasol/tazarotene combination was 1.3. After 12 weeks, the synergy ratio for treatment success was 1.6 and the ratio for IGAxBSA scores was 1.4.
“By combining two agents into one once-daily formulation, this novel formulation reduces the number of product applications and may help patient adherence,” the study authors noted.
Dr. Kircik reported serving as a consultant and investigator for Valeant Pharmaceuticals. One study author is an employee of Bausch Health and Ortho Dermatologics, and another is an employee of Dow Pharmaceutical Sciences (a division of Valeant).
Brodalumab demonstrates low immunogenicity in moderate to severe psoriasis
The immunogenicity of brodalumab in patients with moderate to severe plaque psoriasis was low and did not compromise the efficacy or safety profile of the drug, according to Kristian Reich, MD, of Dermatologikum Berlin and SCIderm Research Institute in Hamburg, Germany, and his associates.
Data from a 12-week, phase 2 trial with a 352-week, open-label extension and three 52-week phase 3 trials were included in the analysis. Antidrug antibodies (ADAs) were tested, and positive samples were further analyzed for neutralizing ADAs by a cell-based assay.
Out of the 4,461 patients who received brodalumab, 122 (2.7%) were positive for ADAs after starting brodalumab. The incidence rate ranged from 1.9% to 3.4% between all dosing groups (140 mg, 210 mg, variable dosing, and 210 mg of brodalumab after ustekinumab). In 58 (1.4%) of patients, ADAs were transient. No patients had neutralizing ADAs, and no evidence of altered pharmacokinetics, loss of efficacy, or changes in the safety profile of brodalumab in subjects positive for ADAs was seen.
No significant difference was seen in the incidence rate of hypersensitivity or injection site reactions in brodalumab, compared with placebo or ustekinumab. The most common injection site reactions were injection site pain, erythema, and bruising.
The study was supported by Amgen. The study authors reported numerous disclosures. Two authors are employees of Leo Pharma, one author is a former employee of the company.
Secukinumab improves patient-reported outcomes in CTT psoriasis
Treatment with secukinumab significantly improved patient-reported outcomes such as fatigue, itch, pain, and quality of life measures in patients with CTT psoriasis after 6 months, according to Jerry Bagel, MD, of the Psoriasis Treatment Center of Central New Jersey, East Windsor, and his associates.
A total of 68 patients with psoriasis localized to at least one CTT area who were enrolled in the Corrona Psoriasis Registry from April 15, 2015, through May 10, 2018, and were receiving secukinumab for the entirety of the 6-month study period were included in the analysis. Patient-reported outcomes included in the analysis were fatigue, itch, pain, Dermatology Quality of Life Index (DLQI) score, and Work Productivity and Activity Impairment (WPAI) scale.
The mean age at enrollment was 51.2 years and almost 80% of patients were white. Mean psoriasis duration was 21.8 years and nearly half had PsA.
Visual analog scale scores improved over baseline for fatigue (mean, 23.2 vs. 33.2; P = .01), itch (20.9 vs. 49.6; P less than .0001), and pain (12.1 vs. 33.8; P less than .0001). DLQI scores also improved (2.9 vs. 8.1; P less than .0001), and the proportion of patients who reported that psoriasis had at least a moderate effect on their life was reduced after 6 months (22.1% vs. 59.7%; P less than .0001).
Based on WPAI results, patients experienced significant improvements in the percentage of daily activities impaired (mean, 9.5% vs 17.5%; P = .0075); of the 42 patients who were employed, both impairment percentage (3.7% vs. 11.2%; P = .0148) and percentage of work hours affected (4.9% vs. 11.9%; P = .0486) were reduced from baseline.
“These results are consistent with previous reports from secukinumab clinical trials; however, additional real-world studies are needed to evaluate the long-term effectiveness of secukinumab for improving [patient-reported outcomes] in patients with psoriasis in CTT areas,” the authors noted.
The Corrona registry has been supported by numerous pharmaceutical companies, and several study authors reported various disclosures with industry. Two authors are Novartis employees. The study was supported by Novartis; the company participated in the interpretation of data and review and approval of the abstract.
These posters were presented at Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar. SDEF and this news organization are owned by the same parent company.
Over one-third of psoriasis patients have PsA
About two-thirds of patients with psoriasis in a national registry also had psoriatic arthritis (PsA) and/or psoriasis in at least one challenging-to-treat (CTT) area, and one-quarter had both, according to Kristina Callis Duffin, MD, of the University of Utah, Salt Lake City, and her associates.
Their analysis included 2,042 psoriasis patients who were enrolled in the Corrona Psoriasis Registry between April 2015 and May 2018 and initiated biologic treatment during that time. The mean age was 49.6 years, 80% of the patients were white, and 51% were obese. Mean disease duration was 19.9 years and 89.2% of the patients had moderate to severe disease. CTT areas include the scalp, nails, and palmoplantar areas.
A total of 784 people in the cohort (38.4%) had PsA, 778 (38.1%) had scalp psoriasis, 326 (16.0%) had nail psoriasis, 223 (10.9%) had palmoplantar psoriasis, and 535 (26.2%) had both PsA and psoriasis in at least two CTT areas. The most common combinations were PsA plus scalp psoriasis and PsA plus nail and scalp psoriasis.
“These results indicate a need to further characterize patients with psoriasis who have PsA and CTT areas and evaluate the impact of these factors to better understand their treatment needs,” the investigators noted.
The Corrona registry has been supported by numerous pharmaceutical companies, and the study authors reported numerous financial relationships with industry; two authors are Novartis employees.
Secukinumab effective for slowing radiographic progression in active PsA
Treatment with secukinumab significantly reduced radiographic progression in patients with active PsA, according to Désirée van der Heijde, MD, PhD, professor of rheumatology at Leiden University Medical Center, and her associates.
The results come from an analysis of the FUTURE 5 trial, a study of 996 patients with active PsA despite previous NSAID treatment, disease-modifying antirheumatic drug treatment, or anti–tumor necrosis factor (TNF) therapy. Patients were randomized to receive 300 mg subcutaneous secukinumab with loading dose, 150 mg secukinumab with loading dose, 150 mg secukinumab without loading dose, or placebo, at baseline; weeks 1, 2, 3, and 4; then every 4 weeks.
After 24 weeks, the mean change in van der Heijde–modified Total Sharp Score for PsA was 0.08 for the 300-mg secukinumab group (P less than .01), 0.17 for the 150-mg secukinumab with loading dose group (P less than .05), a reduction of 0.09 for the 150-mg secukinumab without loading dose group (P less than .01), and 0.50 for the placebo group. Lower radiographic progression was seen regardless of prior anti-TNF or concomitant methotrexate treatment.
The study was funded by Novartis. The study authors reported financial disclosures with numerous companies; five authors are Novartis employees.
Tildrakizumab sustains efficacy in plaque psoriasis treatment after 1 year
Nearly all patients receiving the interleukin-23 inhibitor tildrakizumab for the treatment of moderate to severe plaque psoriasis maintained or improved their Psoriasis Area and Severity Index (PASI) response rate after 52 weeks of treatment, compared with their response after 28 weeks.
The analysis, conducted by Boni E. Elewski, MD, of the University of Alabama at Birmingham, and her associates, included 352 patients who received 100 mg tildrakizumab and 313 who received 200 mg tildrakizumab. Treatment was received at baseline, at 4 weeks, and then every 12 weeks afterward.
At week 28, the proportions of patients achieving PASI 100, PASI 90-99, PASI 75-89, and PASI 50-74 at week 28 were 25.9%, 38.4%, 25.3%, and 10.5%, respectively, among those treated with the 100-mg dose. The proportions were 24.6%, 24.3%, 19.5%, and 31.6%, respectively, among those treated with the 200-mg dose.
In patients who achieved at least PASI 90 on either dose at week 28, 88.9%-89.4% maintained that response at week 52. For patients with PASI 75-89, 39.3%-40.4% maintained that response and 33.7%-41.0% achieved a PASI 90 response. At week 52, in patients with PASI 50-74, 20.2%-29.7% achieved at least a PASI 90, 52.5%-64.9% achieved PASI 75, and only 2.6% of patients on either dose had fallen below PASI 50.
Four study authors reported being clinical investigators on studies sponsored by Merck and Sun Pharmaceuticals; five authors are employees of Sun Pharmaceuticals.
Halobetasol/tazarotene combination most effective for plaque psoriasis treatment
A fixed combination of halobetasol propionate 0.01% and tazarotene 0.045% lotion provided a synergistic effect over either component on its own for the treatment of plaque psoriasis, according to Leon H. Kircik, MD, of Indiana University, Indianapolis, and his associates.
The investigators performed a post hoc analysis of 212 patients with moderate to severe plaque psoriasis randomized to receive either the halobetasol/tazarotene combination, halobetasol only, tazarotene only, or vehicle only for 8 weeks, with follow-up at 12 weeks. Treatment success was based on the proportion of patients who achieved at least a 2-grade improvement in the Investigator Global Assessment (IGA) score, IGA scores of “clear” or “almost clear,” and percent change from baseline in IGA multiplied by Body Surface Area (BSA) composite score (IGAxBSA). “Synergy was calculated by summing up the contribution of the individual active ingredients (HP and TAZ) to overall efficacy and comparing to the efficacy achieved with HP/TAZ lotion relative to vehicle,” the authors explained.
Relative to vehicle, treatment success for halobetasol/tazarotene after 8 weeks was 42.8%, 23.6% for halobetasol alone, and 9.0% for tazarotene alone. After 12 weeks, the difference was 31.3%, 14.1%, and 5.9%, respectively. The percent change in IGAxBSA scores from baseline after 8 weeks, relative to vehicle, were 51.6%, 37.3%, and 3.3%, respectively. After 12 weeks, the change was 47.3%, 25.7%, and 8.6%, respectively.
After 8 weeks, the synergy ratio for treatment success and IGAxBSA scores for the halobetasol/tazarotene combination was 1.3. After 12 weeks, the synergy ratio for treatment success was 1.6 and the ratio for IGAxBSA scores was 1.4.
“By combining two agents into one once-daily formulation, this novel formulation reduces the number of product applications and may help patient adherence,” the study authors noted.
Dr. Kircik reported serving as a consultant and investigator for Valeant Pharmaceuticals. One study author is an employee of Bausch Health and Ortho Dermatologics, and another is an employee of Dow Pharmaceutical Sciences (a division of Valeant).
Brodalumab demonstrates low immunogenicity in moderate to severe psoriasis
The immunogenicity of brodalumab in patients with moderate to severe plaque psoriasis was low and did not compromise the efficacy or safety profile of the drug, according to Kristian Reich, MD, of Dermatologikum Berlin and SCIderm Research Institute in Hamburg, Germany, and his associates.
Data from a 12-week, phase 2 trial with a 352-week, open-label extension and three 52-week phase 3 trials were included in the analysis. Antidrug antibodies (ADAs) were tested, and positive samples were further analyzed for neutralizing ADAs by a cell-based assay.
Out of the 4,461 patients who received brodalumab, 122 (2.7%) were positive for ADAs after starting brodalumab. The incidence rate ranged from 1.9% to 3.4% between all dosing groups (140 mg, 210 mg, variable dosing, and 210 mg of brodalumab after ustekinumab). In 58 (1.4%) of patients, ADAs were transient. No patients had neutralizing ADAs, and no evidence of altered pharmacokinetics, loss of efficacy, or changes in the safety profile of brodalumab in subjects positive for ADAs was seen.
No significant difference was seen in the incidence rate of hypersensitivity or injection site reactions in brodalumab, compared with placebo or ustekinumab. The most common injection site reactions were injection site pain, erythema, and bruising.
The study was supported by Amgen. The study authors reported numerous disclosures. Two authors are employees of Leo Pharma, one author is a former employee of the company.
Secukinumab improves patient-reported outcomes in CTT psoriasis
Treatment with secukinumab significantly improved patient-reported outcomes such as fatigue, itch, pain, and quality of life measures in patients with CTT psoriasis after 6 months, according to Jerry Bagel, MD, of the Psoriasis Treatment Center of Central New Jersey, East Windsor, and his associates.
A total of 68 patients with psoriasis localized to at least one CTT area who were enrolled in the Corrona Psoriasis Registry from April 15, 2015, through May 10, 2018, and were receiving secukinumab for the entirety of the 6-month study period were included in the analysis. Patient-reported outcomes included in the analysis were fatigue, itch, pain, Dermatology Quality of Life Index (DLQI) score, and Work Productivity and Activity Impairment (WPAI) scale.
The mean age at enrollment was 51.2 years and almost 80% of patients were white. Mean psoriasis duration was 21.8 years and nearly half had PsA.
Visual analog scale scores improved over baseline for fatigue (mean, 23.2 vs. 33.2; P = .01), itch (20.9 vs. 49.6; P less than .0001), and pain (12.1 vs. 33.8; P less than .0001). DLQI scores also improved (2.9 vs. 8.1; P less than .0001), and the proportion of patients who reported that psoriasis had at least a moderate effect on their life was reduced after 6 months (22.1% vs. 59.7%; P less than .0001).
Based on WPAI results, patients experienced significant improvements in the percentage of daily activities impaired (mean, 9.5% vs 17.5%; P = .0075); of the 42 patients who were employed, both impairment percentage (3.7% vs. 11.2%; P = .0148) and percentage of work hours affected (4.9% vs. 11.9%; P = .0486) were reduced from baseline.
“These results are consistent with previous reports from secukinumab clinical trials; however, additional real-world studies are needed to evaluate the long-term effectiveness of secukinumab for improving [patient-reported outcomes] in patients with psoriasis in CTT areas,” the authors noted.
The Corrona registry has been supported by numerous pharmaceutical companies, and several study authors reported various disclosures with industry. Two authors are Novartis employees. The study was supported by Novartis; the company participated in the interpretation of data and review and approval of the abstract.
These posters were presented at Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar. SDEF and this news organization are owned by the same parent company.
FROM SDEF LAS VEGAS DERMATOLOGY SEMINAR
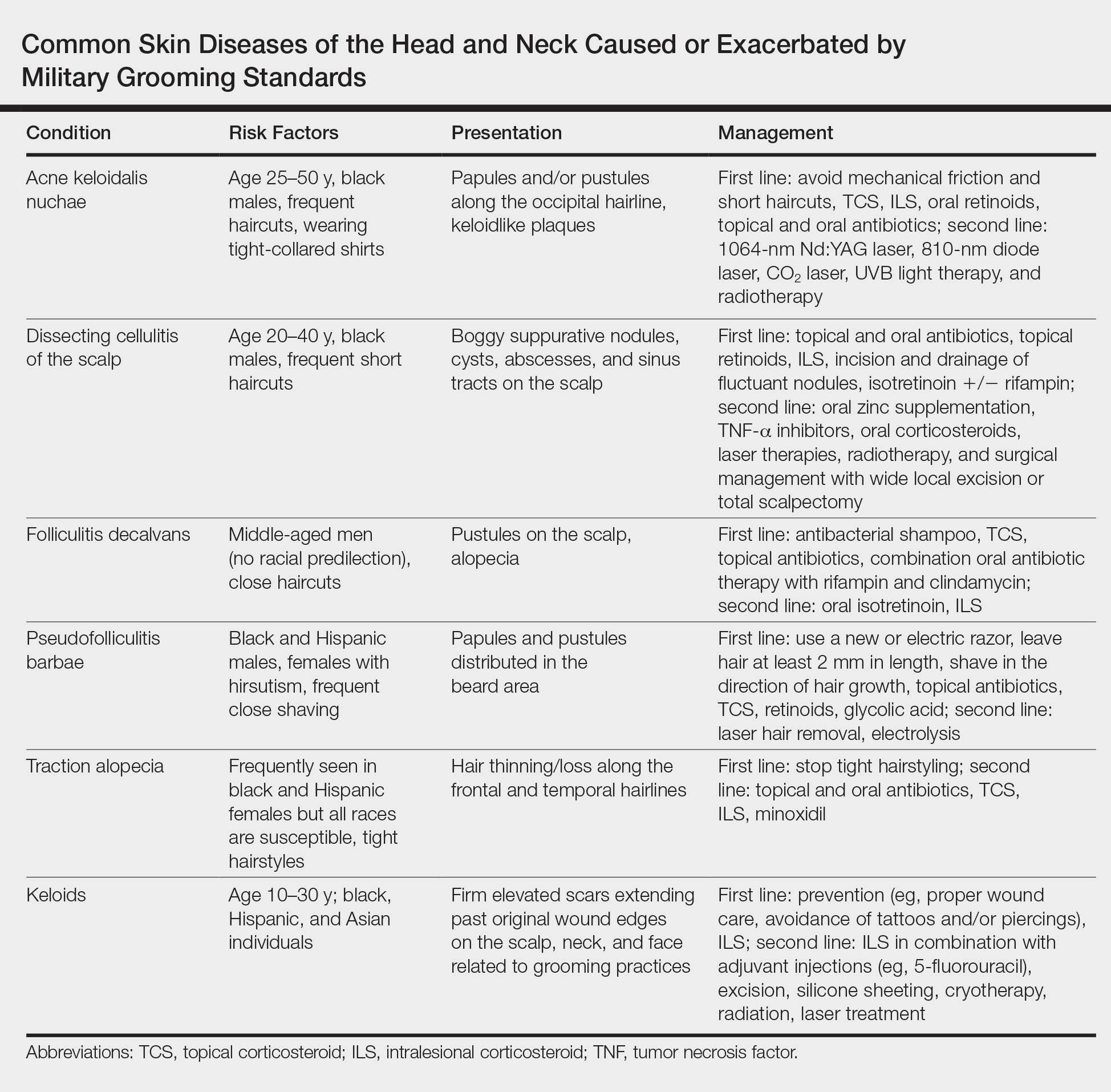
IV Dihydroergotamine Is Associated With Chest Pain in Pediatric Patients With Headache
Patients who continue DHE despite chest pain are more likely than patients who stop DHE to experience acute headache resolution.
CHICAGO—Among pediatric patients who receive IV dihydroergotamine (DHE) for headache, chest pain is a common side effect and reason for early cessation of DHE, according to a study presented at the 47th Annual Meeting of the Child Neurology Society. Chest pain may not represent a serious cardiovascular problem, and patients who continue DHE despite chest pain have better chances of acute headache resolution, compared with patients who stop DHE, said Sara Fridinger, MD, a fellow with the Division of Neurology at Children’s Hospital of Philadelphia.
IV DHE is an effective headache treatment for children, but it has many side effects, including chest pain. Chest pain in pediatric patients who receive IV DHE may result from esophageal spasms, but it raises concerns about myocardial ischemia because of the drug’s vasospastic qualities, the researchers said.
To determine the incidence and significance of chest pain among pediatric patients who received IV DHE for headache, Dr. Fridinger and Christina Szperka, MD, Director of the Pediatric Headache Program at Children’s Hospital of Philadelphia, conducted a retrospective chart review. They examined data from pediatric patients at their hospital who received IV DHE between January 2014 and July 2016. They excluded patients who received DHE for secondary headache. Data from 183 patients (median age, 15.7; 81% female) were included in their analysis, including reports of chest pain and other side effects, EKG data, and cardiac enzymes.
Chest pain occurred in 27% (n = 49) of patients who received DHE. Chest pain occurred after the first dose in 33% of patients and after the second dose in 61%. All patients received premedication before the dose that caused chest pain, and metoclopramide was used as premedication in 80% of cases. No patients with chest pain had elevated troponin. Of the 31% of patients with chest pain who had EKG abnormalities, the abnormalities were either unchanged from baseline or deemed not clinically significant. Of patients with chest pain, 39% stopped DHE due to chest pain, whereas 61% continued with the DHE protocol.
Thirty-seven percent of patients who stopped DHE due to chest pain and 50% of those who continued DHE despite chest pain achieved resolution of the acute headache.
“It is reassuring that no patients were found to have elevated cardiac enzymes and no patients had frankly abnormal EKGs,” said Drs. Fridinger and Szperka.
Patients who continue DHE despite chest pain are more likely than patients who stop DHE to experience acute headache resolution.
Patients who continue DHE despite chest pain are more likely than patients who stop DHE to experience acute headache resolution.
CHICAGO—Among pediatric patients who receive IV dihydroergotamine (DHE) for headache, chest pain is a common side effect and reason for early cessation of DHE, according to a study presented at the 47th Annual Meeting of the Child Neurology Society. Chest pain may not represent a serious cardiovascular problem, and patients who continue DHE despite chest pain have better chances of acute headache resolution, compared with patients who stop DHE, said Sara Fridinger, MD, a fellow with the Division of Neurology at Children’s Hospital of Philadelphia.
IV DHE is an effective headache treatment for children, but it has many side effects, including chest pain. Chest pain in pediatric patients who receive IV DHE may result from esophageal spasms, but it raises concerns about myocardial ischemia because of the drug’s vasospastic qualities, the researchers said.
To determine the incidence and significance of chest pain among pediatric patients who received IV DHE for headache, Dr. Fridinger and Christina Szperka, MD, Director of the Pediatric Headache Program at Children’s Hospital of Philadelphia, conducted a retrospective chart review. They examined data from pediatric patients at their hospital who received IV DHE between January 2014 and July 2016. They excluded patients who received DHE for secondary headache. Data from 183 patients (median age, 15.7; 81% female) were included in their analysis, including reports of chest pain and other side effects, EKG data, and cardiac enzymes.
Chest pain occurred in 27% (n = 49) of patients who received DHE. Chest pain occurred after the first dose in 33% of patients and after the second dose in 61%. All patients received premedication before the dose that caused chest pain, and metoclopramide was used as premedication in 80% of cases. No patients with chest pain had elevated troponin. Of the 31% of patients with chest pain who had EKG abnormalities, the abnormalities were either unchanged from baseline or deemed not clinically significant. Of patients with chest pain, 39% stopped DHE due to chest pain, whereas 61% continued with the DHE protocol.
Thirty-seven percent of patients who stopped DHE due to chest pain and 50% of those who continued DHE despite chest pain achieved resolution of the acute headache.
“It is reassuring that no patients were found to have elevated cardiac enzymes and no patients had frankly abnormal EKGs,” said Drs. Fridinger and Szperka.
CHICAGO—Among pediatric patients who receive IV dihydroergotamine (DHE) for headache, chest pain is a common side effect and reason for early cessation of DHE, according to a study presented at the 47th Annual Meeting of the Child Neurology Society. Chest pain may not represent a serious cardiovascular problem, and patients who continue DHE despite chest pain have better chances of acute headache resolution, compared with patients who stop DHE, said Sara Fridinger, MD, a fellow with the Division of Neurology at Children’s Hospital of Philadelphia.
IV DHE is an effective headache treatment for children, but it has many side effects, including chest pain. Chest pain in pediatric patients who receive IV DHE may result from esophageal spasms, but it raises concerns about myocardial ischemia because of the drug’s vasospastic qualities, the researchers said.
To determine the incidence and significance of chest pain among pediatric patients who received IV DHE for headache, Dr. Fridinger and Christina Szperka, MD, Director of the Pediatric Headache Program at Children’s Hospital of Philadelphia, conducted a retrospective chart review. They examined data from pediatric patients at their hospital who received IV DHE between January 2014 and July 2016. They excluded patients who received DHE for secondary headache. Data from 183 patients (median age, 15.7; 81% female) were included in their analysis, including reports of chest pain and other side effects, EKG data, and cardiac enzymes.
Chest pain occurred in 27% (n = 49) of patients who received DHE. Chest pain occurred after the first dose in 33% of patients and after the second dose in 61%. All patients received premedication before the dose that caused chest pain, and metoclopramide was used as premedication in 80% of cases. No patients with chest pain had elevated troponin. Of the 31% of patients with chest pain who had EKG abnormalities, the abnormalities were either unchanged from baseline or deemed not clinically significant. Of patients with chest pain, 39% stopped DHE due to chest pain, whereas 61% continued with the DHE protocol.
Thirty-seven percent of patients who stopped DHE due to chest pain and 50% of those who continued DHE despite chest pain achieved resolution of the acute headache.
“It is reassuring that no patients were found to have elevated cardiac enzymes and no patients had frankly abnormal EKGs,” said Drs. Fridinger and Szperka.
Military Grooming Standards and Their Impact on Skin Diseases of the Head and Neck
The US military enforces grooming standards to ensure the professional appearance and serviceability of soldiers in all operational settings. Although most individuals are able to uphold these regulations without incident, there is a growing cohort of servicemembers with skin diseases that were exacerbated or even initiated by haircuts, hairstyling, and shaving required to conform to these grooming standards. These skin diseases, which can affect both sexes and may not be appreciated until years into a soldier's service commitment, can have consequences related to individual morbidity and medical readiness for deployment, making it an important issue for medical practitioners to recognize and manage in servicemembers.
This review highlights several disorders of the pilosebaceous unit of the head and neck that can be caused or exacerbated by military grooming standards, including inflammatory hair disorders, traction alopecia, and pseudofolliculitis barbae. Discussion of each entity will include a review of susceptibility and causality as well as initial treatment options to consider (Table).
Inflammatory Hair Disorders
The proper appearance of servicemembers in uniform represents self-discipline and conformity to the high standards of the military. This transition occurs as a rite of passage for many new male recruits who receive shaved haircuts during their first days of basic training. Thereafter, male servicemembers are required to maintain a tapered appearance of the hair per military regulations.1 Clipping hair closely to the scalp or shaving the head entirely are authorized and often encouraged; therefore, high and tight haircuts and buzz cuts are popular among male soldiers due to the general ease of care and ability to maintain the haircut themselves. Conversely, these styles require servicemembers to get weekly or biweekly haircuts that in turn can lead to chronic trauma and irritation. In more susceptible populations, inflammatory hair disorders such as acne keloidalis nuchae (AKN), dissecting cellulitis of the scalp, and folliculitis decalvans may be incited.
Acne Keloidalis Nuchae
Acne keloidalis nuchae, also called folliculitis keloidalis, is a chronic scarring folliculitis presenting with papules and plaques on the occiput and nape of the neck that may merge to form hypertrophic scars or keloids. This disorder most commonly develops in young black men but also can be seen in black females and white patients of both sexes.2 Acne keloidalis nuchae shares many histologic features with central centrifugal cicatricial alopecia, which may suggest a similar pathogenesis. Apart from frequent haircuts, tight-collared shirts, such as those on military service uniforms, also have been associated with AKN. Because of these suspected etiologies, first-line treatment focuses on preventing further trauma by avoiding mechanical irritation and short haircuts, which may be difficult in the military setting. For earlier disease stages, topical and intralesional corticosteroids, oral retinoids, and topical and oral antibiotics are used for their anti-inflammatory properties.3 In refractory cases, surgical excision with healing by secondary intention may be attempted.4 Additional treatment options include the 1064-nm Nd:YAG and 810-nm diode lasers,3 UVB light therapy, CO2 laser, and radiotherapy.
Dissecting Cellulitis of the Scalp
Similar to AKN, dissecting cellulitis of the scalp is another inflammatory hair disorder that is worsened by frequent short haircuts.5 Dissecting cellulitis of the scalp is a primary cicatricial alopecia proposed to be secondary to follicular occlusion. It often is seen in black males aged 20 to 40 years and is characterized by boggy suppurative nodules and cysts with draining sinus tracts, abscesses, and resultant scarring alopecia. Dissecting cellulitis of the scalp is part of the follicular occlusion tetrad, which also includes hidradenitis suppurativa, acne conglobata, and pilonidal cysts. First-line therapies include topical and oral antibiotics, topical retinoids, intralesional corticosteroids, incision and drainage of fluctuant nodules, and oral isotretinoin with or without rifampin. Alternative treatments include oral zinc supplementation, oral corticosteroids, tumor necrosis factor α inhibitors, laser therapies, radiotherapy, and surgical management with wide local excision or total scalpectomy.6,7
Folliculitis Decalvans
Folliculitis decalvans is a primary cicatricial alopecia of the scalp that most commonly presents in middle-aged men without racial predilection.8 Folliculitis decalvans presents with multiple pustules, crusts, tufted hairs, and perifollicular hyperkeratosis, leading to scarring of the scalp, which often is most severe on the posterior vertex. Staphylococcus aureus is a presumed player in the pathogenesis of folliculitis decalvans with superantigens causing release of cytokines stimulating follicular destruction. Close haircuts in conformation with military grooming standards can contribute to this condition due to mechanical trauma and subsequent inflammation. It typically is diagnosed clinically, but if histologic confirmation is desired, a sample from the periphery of early lesions is preferred.9 Initial treatment consists of antibacterial shampoos, topical corticosteroids, topical antibiotics, and combination oral antibiotic therapy with rifampin and clindamycin. Studies using oral isotretinoin have shown variable results,10,11 and the most effective treatment of recalcitrant lesions appears to be intralesional corticosteroids.12
Follicular and Scarring Disorders
In addition to inflammatory hair disorders, military grooming standards have been linked to the pathogenesis of diseases such as pseudofolliculitis barbae, traction alopecia, and keloids, specifically through irritation of the face, neck, and scalp, as well as damage to the follicular unit.5 These conditions develop because grooming regulations necessitate certain hair practices such as close shaving of facial and neck hair and keeping long hair secured relatively tightly to the scalp.
Pseudofolliculitis Barbae
Males in the military are obligated to keep their faces clean-shaven.1 They may acquire a medical waiver for a specified beard length if deemed appropriate by the treating physician,1 which often leads to the need for continual waiver renewal and also may warrant possible negative perception from peers, subordinates, and leadership. One of the most prevalent conditions that is closely associated with shaving is pseudofolliculitis barbae. The combination of close shaving and tightly coiled hairs causes the hairs to grow toward and penetrate the skin, particularly on the neck.13 In some cases, the hairs never actually exit the skin and simply curl within the superficial epidermis. A foreign body reaction often arises, leading to inflamed follicular papules and pustules. Affected individuals may experience pain, pruritus, and secondary infections. Postinflammatory hyperpigmentation, hypertrophic scarring, and keloid formation are common sequelae in cases of untreated disease. Pseudofolliculitis barbae also is exacerbated by pulling the skin taut and shaving against the grain, making behavioral interventions a key component in management of this condition. Preliminary recommendations include using a new or electric razor, leaving hair at least 2 mm in length, and shaving in the direction of hair growth. Other treatment options with varying effectiveness include daily alternation of a mild topical corticosteroid and one of the following: a topical retinoid, topical antibiotics, or glycolic acid. The only treatments that approach definitive cure are laser hair removal and electrolysis for which patient skin type plays an important role in laser selection.5
Traction Alopecia
Similar to their male counterparts, female military members must also present a conservative professional appearance, including hair that is neatly groomed.1 If the length of the hair extends beyond the uniform collar, it must be inconspicuously fastened or pinned above the collar. As a result, loosely tied hair is unauthorized, and females with long hair must secure their hair tightly on a daily basis. Traction alopecia results from tight hairstyling over a prolonged period and commonly affects female soldiers. The etiology is presumed to be mechanical loosening of hair within the follicles, leading to inflammation. Although traditionally seen in black women along the frontal and temporal hairlines, traction alopecia has been identified in individuals of all races and can occur anywhere on the scalp.5 Perifollicular erythema may be the first sign, and papules and pustules may be visible. Although the hair loss in traction alopecia usually is reversible if the traction is ceased, end-stage disease may be permanent.6 Halting traction-inducing practices is paramount, and other treatment options that may slow progression include topical or oral antibiotics and topical or intralesional corticosteroids. Recovery of hair loss also may be aided by topical minoxidil.5
Keloids
Keloid formation is an important pathology to address, as it may result from several of the aforementioned conditions. Keloids are most commonly seen in black individuals but also can occur in Hispanic and Asian patients. The cause has not been fully elucidated but is thought to be a combination of dysfunctional fibroblasts with a genetic component based on racial predilection and twin concordance studies.5 The chest, shoulders, upper back, neck, and earlobes are particularly susceptible to keloid formation, which can appear from 1 to 24 years following dermal trauma.5 Unlike hypertrophic scars, keloids generally do not regress and frequently cause discomfort, pruritus, and emotional distress. They also can hinder wearing a military uniform. Sustained remission is problematic, making prevention a first-line approach, including proper care of wounds when they occur and avoiding elective procedures such as piercings and tattoos. Intralesional corticosteroids, adjuvant injections (eg, 5-fluorouracil), silicone sheeting, cryotherapy, radiation, laser therapy, and excision are some of the treatment options when keloids have formed.5
Final Comment
It is important to recognize military grooming standards as a cause or contributor to several diseases of the head and neck in military servicemembers. Specifically, frequent haircuts in male soldiers are associated with several inflammatory hair disorders, including AKN, dissecting cellulitis of the scalp, and folliculitis decalvans, while daily shaving predisposes individuals to pseudofolliculitis barbae with possible keloid formation. Females may develop traction alopecia from chronically tight, pulled back hairstyles. All of these conditions have health implications for the affected individuals and can compromise the military mission. Awareness, prevention, and recognition are key along with the knowledge base to provide anticipatory avoidance and initiate appropriate treatments, thereby mitigating these potential consequences.
- US Department of the Army. Wear and Appearance of Army Uniforms and Insignia: Army Regulation 670-1. Washington, DC: Department of the Army; 2017. https://history.army.mil/html/forcestruc/docs/AR670-1.pdf. Accessed October 11, 2018.
- East-Innis AD, Stylianou K, Paolino A, et al. Acne keloidalis nuchae: risk factors and associated disorders--a retrospective study. Int J Dermatol. 2017;56:828-832.
- Maranda EL, Simmons BJ, Nguyen AH, et al. Treatment of acne keloidalis nuchae: a systematic review of the literature. Dermatol Ther (Heidelb). 2016;6:363-378.
- Glenn MJ, Bennett RG, Kelly AP. Acne keloidalis nuchae: treatment with excision and second-intention healing. J Am Acad Dermatol. 1995;33:243-246.
- Madu P, Kundu RV. Follicular and scarring disorders in skin of color: presentation and management. Am J Clin Dermatol. 2014;15:307-321.
- Rodney IJ, Onwudiwe OC. Hair and scalp disorders in ethnic populations. J Drugs Dermatol. 2013;12:420-427.
- Lindsey SF, Tosti A. Ethnic hair disorders. Curr Probl Dermatol. 2015;47:139-148.
- Whiting DA. Cicatricial alopecia: clinico-pathological findings and treatment. Clin Dermatol. 2001;19:211-225.
- Sperling LC, Cowper SE, Knopp EA. An Atlas of Hair Pathology with Clinical Correlations. 2nd ed. Boca Raton, FL: CRC Press; 2012.
- Gemmeke A, Wollina U. Folliculitis decalvans of the scalp: response to triple therapy with isotretinoin, clindamycin, and prednisolone. Acta Dermatovenerol Alp Pannonica Adriat. 2006;15:184-186.
- Hallai N, Thompson I, Williams P, et al. Folliculitis spinulosa decalvans: failure to respond to oral isotretinoin. J Eur Acad Dermatol Venereol. 2006;20:223-224.
- Bolduc C, Sperling LC, Shapiro J. Primary cicatricial alopecia. J Am Acad Dermatol. 2016;75:101-117.
- Perry PK, Cook-Bolden FE, Rahman Z, et al. Defining pseudofolliculitis barbae in 2001: a review of the literature and current trends. J Am Acad Dermatol. 2002;46(2 suppl):S113-S119.
The US military enforces grooming standards to ensure the professional appearance and serviceability of soldiers in all operational settings. Although most individuals are able to uphold these regulations without incident, there is a growing cohort of servicemembers with skin diseases that were exacerbated or even initiated by haircuts, hairstyling, and shaving required to conform to these grooming standards. These skin diseases, which can affect both sexes and may not be appreciated until years into a soldier's service commitment, can have consequences related to individual morbidity and medical readiness for deployment, making it an important issue for medical practitioners to recognize and manage in servicemembers.
This review highlights several disorders of the pilosebaceous unit of the head and neck that can be caused or exacerbated by military grooming standards, including inflammatory hair disorders, traction alopecia, and pseudofolliculitis barbae. Discussion of each entity will include a review of susceptibility and causality as well as initial treatment options to consider (Table).
Inflammatory Hair Disorders
The proper appearance of servicemembers in uniform represents self-discipline and conformity to the high standards of the military. This transition occurs as a rite of passage for many new male recruits who receive shaved haircuts during their first days of basic training. Thereafter, male servicemembers are required to maintain a tapered appearance of the hair per military regulations.1 Clipping hair closely to the scalp or shaving the head entirely are authorized and often encouraged; therefore, high and tight haircuts and buzz cuts are popular among male soldiers due to the general ease of care and ability to maintain the haircut themselves. Conversely, these styles require servicemembers to get weekly or biweekly haircuts that in turn can lead to chronic trauma and irritation. In more susceptible populations, inflammatory hair disorders such as acne keloidalis nuchae (AKN), dissecting cellulitis of the scalp, and folliculitis decalvans may be incited.
Acne Keloidalis Nuchae
Acne keloidalis nuchae, also called folliculitis keloidalis, is a chronic scarring folliculitis presenting with papules and plaques on the occiput and nape of the neck that may merge to form hypertrophic scars or keloids. This disorder most commonly develops in young black men but also can be seen in black females and white patients of both sexes.2 Acne keloidalis nuchae shares many histologic features with central centrifugal cicatricial alopecia, which may suggest a similar pathogenesis. Apart from frequent haircuts, tight-collared shirts, such as those on military service uniforms, also have been associated with AKN. Because of these suspected etiologies, first-line treatment focuses on preventing further trauma by avoiding mechanical irritation and short haircuts, which may be difficult in the military setting. For earlier disease stages, topical and intralesional corticosteroids, oral retinoids, and topical and oral antibiotics are used for their anti-inflammatory properties.3 In refractory cases, surgical excision with healing by secondary intention may be attempted.4 Additional treatment options include the 1064-nm Nd:YAG and 810-nm diode lasers,3 UVB light therapy, CO2 laser, and radiotherapy.
Dissecting Cellulitis of the Scalp
Similar to AKN, dissecting cellulitis of the scalp is another inflammatory hair disorder that is worsened by frequent short haircuts.5 Dissecting cellulitis of the scalp is a primary cicatricial alopecia proposed to be secondary to follicular occlusion. It often is seen in black males aged 20 to 40 years and is characterized by boggy suppurative nodules and cysts with draining sinus tracts, abscesses, and resultant scarring alopecia. Dissecting cellulitis of the scalp is part of the follicular occlusion tetrad, which also includes hidradenitis suppurativa, acne conglobata, and pilonidal cysts. First-line therapies include topical and oral antibiotics, topical retinoids, intralesional corticosteroids, incision and drainage of fluctuant nodules, and oral isotretinoin with or without rifampin. Alternative treatments include oral zinc supplementation, oral corticosteroids, tumor necrosis factor α inhibitors, laser therapies, radiotherapy, and surgical management with wide local excision or total scalpectomy.6,7
Folliculitis Decalvans
Folliculitis decalvans is a primary cicatricial alopecia of the scalp that most commonly presents in middle-aged men without racial predilection.8 Folliculitis decalvans presents with multiple pustules, crusts, tufted hairs, and perifollicular hyperkeratosis, leading to scarring of the scalp, which often is most severe on the posterior vertex. Staphylococcus aureus is a presumed player in the pathogenesis of folliculitis decalvans with superantigens causing release of cytokines stimulating follicular destruction. Close haircuts in conformation with military grooming standards can contribute to this condition due to mechanical trauma and subsequent inflammation. It typically is diagnosed clinically, but if histologic confirmation is desired, a sample from the periphery of early lesions is preferred.9 Initial treatment consists of antibacterial shampoos, topical corticosteroids, topical antibiotics, and combination oral antibiotic therapy with rifampin and clindamycin. Studies using oral isotretinoin have shown variable results,10,11 and the most effective treatment of recalcitrant lesions appears to be intralesional corticosteroids.12
Follicular and Scarring Disorders
In addition to inflammatory hair disorders, military grooming standards have been linked to the pathogenesis of diseases such as pseudofolliculitis barbae, traction alopecia, and keloids, specifically through irritation of the face, neck, and scalp, as well as damage to the follicular unit.5 These conditions develop because grooming regulations necessitate certain hair practices such as close shaving of facial and neck hair and keeping long hair secured relatively tightly to the scalp.
Pseudofolliculitis Barbae
Males in the military are obligated to keep their faces clean-shaven.1 They may acquire a medical waiver for a specified beard length if deemed appropriate by the treating physician,1 which often leads to the need for continual waiver renewal and also may warrant possible negative perception from peers, subordinates, and leadership. One of the most prevalent conditions that is closely associated with shaving is pseudofolliculitis barbae. The combination of close shaving and tightly coiled hairs causes the hairs to grow toward and penetrate the skin, particularly on the neck.13 In some cases, the hairs never actually exit the skin and simply curl within the superficial epidermis. A foreign body reaction often arises, leading to inflamed follicular papules and pustules. Affected individuals may experience pain, pruritus, and secondary infections. Postinflammatory hyperpigmentation, hypertrophic scarring, and keloid formation are common sequelae in cases of untreated disease. Pseudofolliculitis barbae also is exacerbated by pulling the skin taut and shaving against the grain, making behavioral interventions a key component in management of this condition. Preliminary recommendations include using a new or electric razor, leaving hair at least 2 mm in length, and shaving in the direction of hair growth. Other treatment options with varying effectiveness include daily alternation of a mild topical corticosteroid and one of the following: a topical retinoid, topical antibiotics, or glycolic acid. The only treatments that approach definitive cure are laser hair removal and electrolysis for which patient skin type plays an important role in laser selection.5
Traction Alopecia
Similar to their male counterparts, female military members must also present a conservative professional appearance, including hair that is neatly groomed.1 If the length of the hair extends beyond the uniform collar, it must be inconspicuously fastened or pinned above the collar. As a result, loosely tied hair is unauthorized, and females with long hair must secure their hair tightly on a daily basis. Traction alopecia results from tight hairstyling over a prolonged period and commonly affects female soldiers. The etiology is presumed to be mechanical loosening of hair within the follicles, leading to inflammation. Although traditionally seen in black women along the frontal and temporal hairlines, traction alopecia has been identified in individuals of all races and can occur anywhere on the scalp.5 Perifollicular erythema may be the first sign, and papules and pustules may be visible. Although the hair loss in traction alopecia usually is reversible if the traction is ceased, end-stage disease may be permanent.6 Halting traction-inducing practices is paramount, and other treatment options that may slow progression include topical or oral antibiotics and topical or intralesional corticosteroids. Recovery of hair loss also may be aided by topical minoxidil.5
Keloids
Keloid formation is an important pathology to address, as it may result from several of the aforementioned conditions. Keloids are most commonly seen in black individuals but also can occur in Hispanic and Asian patients. The cause has not been fully elucidated but is thought to be a combination of dysfunctional fibroblasts with a genetic component based on racial predilection and twin concordance studies.5 The chest, shoulders, upper back, neck, and earlobes are particularly susceptible to keloid formation, which can appear from 1 to 24 years following dermal trauma.5 Unlike hypertrophic scars, keloids generally do not regress and frequently cause discomfort, pruritus, and emotional distress. They also can hinder wearing a military uniform. Sustained remission is problematic, making prevention a first-line approach, including proper care of wounds when they occur and avoiding elective procedures such as piercings and tattoos. Intralesional corticosteroids, adjuvant injections (eg, 5-fluorouracil), silicone sheeting, cryotherapy, radiation, laser therapy, and excision are some of the treatment options when keloids have formed.5
Final Comment
It is important to recognize military grooming standards as a cause or contributor to several diseases of the head and neck in military servicemembers. Specifically, frequent haircuts in male soldiers are associated with several inflammatory hair disorders, including AKN, dissecting cellulitis of the scalp, and folliculitis decalvans, while daily shaving predisposes individuals to pseudofolliculitis barbae with possible keloid formation. Females may develop traction alopecia from chronically tight, pulled back hairstyles. All of these conditions have health implications for the affected individuals and can compromise the military mission. Awareness, prevention, and recognition are key along with the knowledge base to provide anticipatory avoidance and initiate appropriate treatments, thereby mitigating these potential consequences.
The US military enforces grooming standards to ensure the professional appearance and serviceability of soldiers in all operational settings. Although most individuals are able to uphold these regulations without incident, there is a growing cohort of servicemembers with skin diseases that were exacerbated or even initiated by haircuts, hairstyling, and shaving required to conform to these grooming standards. These skin diseases, which can affect both sexes and may not be appreciated until years into a soldier's service commitment, can have consequences related to individual morbidity and medical readiness for deployment, making it an important issue for medical practitioners to recognize and manage in servicemembers.
This review highlights several disorders of the pilosebaceous unit of the head and neck that can be caused or exacerbated by military grooming standards, including inflammatory hair disorders, traction alopecia, and pseudofolliculitis barbae. Discussion of each entity will include a review of susceptibility and causality as well as initial treatment options to consider (Table).
Inflammatory Hair Disorders
The proper appearance of servicemembers in uniform represents self-discipline and conformity to the high standards of the military. This transition occurs as a rite of passage for many new male recruits who receive shaved haircuts during their first days of basic training. Thereafter, male servicemembers are required to maintain a tapered appearance of the hair per military regulations.1 Clipping hair closely to the scalp or shaving the head entirely are authorized and often encouraged; therefore, high and tight haircuts and buzz cuts are popular among male soldiers due to the general ease of care and ability to maintain the haircut themselves. Conversely, these styles require servicemembers to get weekly or biweekly haircuts that in turn can lead to chronic trauma and irritation. In more susceptible populations, inflammatory hair disorders such as acne keloidalis nuchae (AKN), dissecting cellulitis of the scalp, and folliculitis decalvans may be incited.
Acne Keloidalis Nuchae
Acne keloidalis nuchae, also called folliculitis keloidalis, is a chronic scarring folliculitis presenting with papules and plaques on the occiput and nape of the neck that may merge to form hypertrophic scars or keloids. This disorder most commonly develops in young black men but also can be seen in black females and white patients of both sexes.2 Acne keloidalis nuchae shares many histologic features with central centrifugal cicatricial alopecia, which may suggest a similar pathogenesis. Apart from frequent haircuts, tight-collared shirts, such as those on military service uniforms, also have been associated with AKN. Because of these suspected etiologies, first-line treatment focuses on preventing further trauma by avoiding mechanical irritation and short haircuts, which may be difficult in the military setting. For earlier disease stages, topical and intralesional corticosteroids, oral retinoids, and topical and oral antibiotics are used for their anti-inflammatory properties.3 In refractory cases, surgical excision with healing by secondary intention may be attempted.4 Additional treatment options include the 1064-nm Nd:YAG and 810-nm diode lasers,3 UVB light therapy, CO2 laser, and radiotherapy.
Dissecting Cellulitis of the Scalp
Similar to AKN, dissecting cellulitis of the scalp is another inflammatory hair disorder that is worsened by frequent short haircuts.5 Dissecting cellulitis of the scalp is a primary cicatricial alopecia proposed to be secondary to follicular occlusion. It often is seen in black males aged 20 to 40 years and is characterized by boggy suppurative nodules and cysts with draining sinus tracts, abscesses, and resultant scarring alopecia. Dissecting cellulitis of the scalp is part of the follicular occlusion tetrad, which also includes hidradenitis suppurativa, acne conglobata, and pilonidal cysts. First-line therapies include topical and oral antibiotics, topical retinoids, intralesional corticosteroids, incision and drainage of fluctuant nodules, and oral isotretinoin with or without rifampin. Alternative treatments include oral zinc supplementation, oral corticosteroids, tumor necrosis factor α inhibitors, laser therapies, radiotherapy, and surgical management with wide local excision or total scalpectomy.6,7
Folliculitis Decalvans
Folliculitis decalvans is a primary cicatricial alopecia of the scalp that most commonly presents in middle-aged men without racial predilection.8 Folliculitis decalvans presents with multiple pustules, crusts, tufted hairs, and perifollicular hyperkeratosis, leading to scarring of the scalp, which often is most severe on the posterior vertex. Staphylococcus aureus is a presumed player in the pathogenesis of folliculitis decalvans with superantigens causing release of cytokines stimulating follicular destruction. Close haircuts in conformation with military grooming standards can contribute to this condition due to mechanical trauma and subsequent inflammation. It typically is diagnosed clinically, but if histologic confirmation is desired, a sample from the periphery of early lesions is preferred.9 Initial treatment consists of antibacterial shampoos, topical corticosteroids, topical antibiotics, and combination oral antibiotic therapy with rifampin and clindamycin. Studies using oral isotretinoin have shown variable results,10,11 and the most effective treatment of recalcitrant lesions appears to be intralesional corticosteroids.12
Follicular and Scarring Disorders
In addition to inflammatory hair disorders, military grooming standards have been linked to the pathogenesis of diseases such as pseudofolliculitis barbae, traction alopecia, and keloids, specifically through irritation of the face, neck, and scalp, as well as damage to the follicular unit.5 These conditions develop because grooming regulations necessitate certain hair practices such as close shaving of facial and neck hair and keeping long hair secured relatively tightly to the scalp.
Pseudofolliculitis Barbae
Males in the military are obligated to keep their faces clean-shaven.1 They may acquire a medical waiver for a specified beard length if deemed appropriate by the treating physician,1 which often leads to the need for continual waiver renewal and also may warrant possible negative perception from peers, subordinates, and leadership. One of the most prevalent conditions that is closely associated with shaving is pseudofolliculitis barbae. The combination of close shaving and tightly coiled hairs causes the hairs to grow toward and penetrate the skin, particularly on the neck.13 In some cases, the hairs never actually exit the skin and simply curl within the superficial epidermis. A foreign body reaction often arises, leading to inflamed follicular papules and pustules. Affected individuals may experience pain, pruritus, and secondary infections. Postinflammatory hyperpigmentation, hypertrophic scarring, and keloid formation are common sequelae in cases of untreated disease. Pseudofolliculitis barbae also is exacerbated by pulling the skin taut and shaving against the grain, making behavioral interventions a key component in management of this condition. Preliminary recommendations include using a new or electric razor, leaving hair at least 2 mm in length, and shaving in the direction of hair growth. Other treatment options with varying effectiveness include daily alternation of a mild topical corticosteroid and one of the following: a topical retinoid, topical antibiotics, or glycolic acid. The only treatments that approach definitive cure are laser hair removal and electrolysis for which patient skin type plays an important role in laser selection.5
Traction Alopecia
Similar to their male counterparts, female military members must also present a conservative professional appearance, including hair that is neatly groomed.1 If the length of the hair extends beyond the uniform collar, it must be inconspicuously fastened or pinned above the collar. As a result, loosely tied hair is unauthorized, and females with long hair must secure their hair tightly on a daily basis. Traction alopecia results from tight hairstyling over a prolonged period and commonly affects female soldiers. The etiology is presumed to be mechanical loosening of hair within the follicles, leading to inflammation. Although traditionally seen in black women along the frontal and temporal hairlines, traction alopecia has been identified in individuals of all races and can occur anywhere on the scalp.5 Perifollicular erythema may be the first sign, and papules and pustules may be visible. Although the hair loss in traction alopecia usually is reversible if the traction is ceased, end-stage disease may be permanent.6 Halting traction-inducing practices is paramount, and other treatment options that may slow progression include topical or oral antibiotics and topical or intralesional corticosteroids. Recovery of hair loss also may be aided by topical minoxidil.5
Keloids
Keloid formation is an important pathology to address, as it may result from several of the aforementioned conditions. Keloids are most commonly seen in black individuals but also can occur in Hispanic and Asian patients. The cause has not been fully elucidated but is thought to be a combination of dysfunctional fibroblasts with a genetic component based on racial predilection and twin concordance studies.5 The chest, shoulders, upper back, neck, and earlobes are particularly susceptible to keloid formation, which can appear from 1 to 24 years following dermal trauma.5 Unlike hypertrophic scars, keloids generally do not regress and frequently cause discomfort, pruritus, and emotional distress. They also can hinder wearing a military uniform. Sustained remission is problematic, making prevention a first-line approach, including proper care of wounds when they occur and avoiding elective procedures such as piercings and tattoos. Intralesional corticosteroids, adjuvant injections (eg, 5-fluorouracil), silicone sheeting, cryotherapy, radiation, laser therapy, and excision are some of the treatment options when keloids have formed.5
Final Comment
It is important to recognize military grooming standards as a cause or contributor to several diseases of the head and neck in military servicemembers. Specifically, frequent haircuts in male soldiers are associated with several inflammatory hair disorders, including AKN, dissecting cellulitis of the scalp, and folliculitis decalvans, while daily shaving predisposes individuals to pseudofolliculitis barbae with possible keloid formation. Females may develop traction alopecia from chronically tight, pulled back hairstyles. All of these conditions have health implications for the affected individuals and can compromise the military mission. Awareness, prevention, and recognition are key along with the knowledge base to provide anticipatory avoidance and initiate appropriate treatments, thereby mitigating these potential consequences.
- US Department of the Army. Wear and Appearance of Army Uniforms and Insignia: Army Regulation 670-1. Washington, DC: Department of the Army; 2017. https://history.army.mil/html/forcestruc/docs/AR670-1.pdf. Accessed October 11, 2018.
- East-Innis AD, Stylianou K, Paolino A, et al. Acne keloidalis nuchae: risk factors and associated disorders--a retrospective study. Int J Dermatol. 2017;56:828-832.
- Maranda EL, Simmons BJ, Nguyen AH, et al. Treatment of acne keloidalis nuchae: a systematic review of the literature. Dermatol Ther (Heidelb). 2016;6:363-378.
- Glenn MJ, Bennett RG, Kelly AP. Acne keloidalis nuchae: treatment with excision and second-intention healing. J Am Acad Dermatol. 1995;33:243-246.
- Madu P, Kundu RV. Follicular and scarring disorders in skin of color: presentation and management. Am J Clin Dermatol. 2014;15:307-321.
- Rodney IJ, Onwudiwe OC. Hair and scalp disorders in ethnic populations. J Drugs Dermatol. 2013;12:420-427.
- Lindsey SF, Tosti A. Ethnic hair disorders. Curr Probl Dermatol. 2015;47:139-148.
- Whiting DA. Cicatricial alopecia: clinico-pathological findings and treatment. Clin Dermatol. 2001;19:211-225.
- Sperling LC, Cowper SE, Knopp EA. An Atlas of Hair Pathology with Clinical Correlations. 2nd ed. Boca Raton, FL: CRC Press; 2012.
- Gemmeke A, Wollina U. Folliculitis decalvans of the scalp: response to triple therapy with isotretinoin, clindamycin, and prednisolone. Acta Dermatovenerol Alp Pannonica Adriat. 2006;15:184-186.
- Hallai N, Thompson I, Williams P, et al. Folliculitis spinulosa decalvans: failure to respond to oral isotretinoin. J Eur Acad Dermatol Venereol. 2006;20:223-224.
- Bolduc C, Sperling LC, Shapiro J. Primary cicatricial alopecia. J Am Acad Dermatol. 2016;75:101-117.
- Perry PK, Cook-Bolden FE, Rahman Z, et al. Defining pseudofolliculitis barbae in 2001: a review of the literature and current trends. J Am Acad Dermatol. 2002;46(2 suppl):S113-S119.
- US Department of the Army. Wear and Appearance of Army Uniforms and Insignia: Army Regulation 670-1. Washington, DC: Department of the Army; 2017. https://history.army.mil/html/forcestruc/docs/AR670-1.pdf. Accessed October 11, 2018.
- East-Innis AD, Stylianou K, Paolino A, et al. Acne keloidalis nuchae: risk factors and associated disorders--a retrospective study. Int J Dermatol. 2017;56:828-832.
- Maranda EL, Simmons BJ, Nguyen AH, et al. Treatment of acne keloidalis nuchae: a systematic review of the literature. Dermatol Ther (Heidelb). 2016;6:363-378.
- Glenn MJ, Bennett RG, Kelly AP. Acne keloidalis nuchae: treatment with excision and second-intention healing. J Am Acad Dermatol. 1995;33:243-246.
- Madu P, Kundu RV. Follicular and scarring disorders in skin of color: presentation and management. Am J Clin Dermatol. 2014;15:307-321.
- Rodney IJ, Onwudiwe OC. Hair and scalp disorders in ethnic populations. J Drugs Dermatol. 2013;12:420-427.
- Lindsey SF, Tosti A. Ethnic hair disorders. Curr Probl Dermatol. 2015;47:139-148.
- Whiting DA. Cicatricial alopecia: clinico-pathological findings and treatment. Clin Dermatol. 2001;19:211-225.
- Sperling LC, Cowper SE, Knopp EA. An Atlas of Hair Pathology with Clinical Correlations. 2nd ed. Boca Raton, FL: CRC Press; 2012.
- Gemmeke A, Wollina U. Folliculitis decalvans of the scalp: response to triple therapy with isotretinoin, clindamycin, and prednisolone. Acta Dermatovenerol Alp Pannonica Adriat. 2006;15:184-186.
- Hallai N, Thompson I, Williams P, et al. Folliculitis spinulosa decalvans: failure to respond to oral isotretinoin. J Eur Acad Dermatol Venereol. 2006;20:223-224.
- Bolduc C, Sperling LC, Shapiro J. Primary cicatricial alopecia. J Am Acad Dermatol. 2016;75:101-117.
- Perry PK, Cook-Bolden FE, Rahman Z, et al. Defining pseudofolliculitis barbae in 2001: a review of the literature and current trends. J Am Acad Dermatol. 2002;46(2 suppl):S113-S119.
Practice Points
- The short frequent haircuts required to maintain a tapered appearance of the hair per US military regulations may lead to inflammatory hair disorders such as acne keloidalis nuchae, dissecting cellulitis of the scalp, and folliculitis decalvans.
- The mainstay of prevention for these conditions is avoidance of inciting factors such as short haircuts, tight-collared shirts, frequent shaving, or tight hairstyles.
- Early identification and treatment of inflammatory follicular and scarring disorders can prevent further scarring, pigmentation changes, and/or disfigurement.
Smartphone Versus Holter Monitoring for Poststroke Atrial Fibrillation Detection
Although guidelines recommend Holter monitoring for all patients with ischemic stroke or TIA, comparatively few receive it.
MONTREAL—In patients hospitalized for a recent acute ischemic stroke or transient ischemic attack (TIA), a smartphone-based method identified three times more patients with atrial fibrillation than did 24-hour Holter monitoring after discharge, according to a study presented at the 11th World Stroke Congress.
This high level of atrial fibrillation detection suggests that this relatively cheap and noninvasive device is a good complement to conventional monitoring by a 24-hour Holter recording or an implanted loop recorder in patients with recent stroke. The device may thus satisfy the requirements of current guidelines from the world’s cardiology societies.
In-Hospital and Postdischarge Monitoring
In the study, 294 of 1,079 patients with acute ischemic stroke or TIA underwent serial, 30-second monitoring with the AliveCor device while hospitalized. The device was designed for smartphone-enabled ECG measurement. After discharge, the same patients underwent Holter monitoring. The latter technique identified eight patients (3%) with atrial fibrillation, compared with 25 patients (9%) who were identified using the AliveCor device, said Bernard Yan, MD, a consultant neurologist and endovascular neurointerventionist in the Comprehensive Stroke Center of the Royal Melbourne Hospital. Seven of the eight patients identified with atrial fibrillation by Holter monitoring were also found to have atrial fibrillation by the AliveCor device.

Dr. Yan attributed the higher in-hospital detection rate for atrial fibrillation to the timing of screening, which occurred within days of the stroke or TIA, rather than after the patient had left the hospital. “The difference may be because we monitored patients [with the AliveCor device] much earlier, during their hot period right after their stroke.”
Practice Does Not Match Recommendations
The trial, which was called SPOT-AF, was conducted at several centers in Australia, China, and Hong Kong. All patients underwent AliveCor monitoring during their stay in the hospital, which lasted for a median of four days. Patients performed a 30-second heart rhythm check every time a nurse saw them for a routine vital-sign examination, which usually was three or four times per day. The current analysis focused on the 294 patients (27% of the 1,079 patients) who also underwent 24-hour Holter monitoring following hospital discharge when ordered by their personal physician.
This 27% rate of postdischarge Holter monitoring was consistent with that of a 2016 review of more than 17,000 patients with stroke or TIA in Canada. That study found that 31% of participants underwent 24-hour Holter monitoring for atrial fibrillation during the 30 days following their index event. Guidelines, however, call for atrial fibrillation screening in all patients with recent ischemic stroke and TIA.
Although screening for atrial fibrillation with a smartphone-based device is inexpensive and easy, Dr. Yan did not suggest that this approach could replace a Holter monitor or an implanted loop recorder, which is what current guidelines recommend. “To change the guidelines, we need a different study that compares these approaches head to head.”
SPOT-AF received partial funding from Boehringer Ingelheim. Dr. Yan has spoken on behalf of Bayer, Boehringer Ingelheim, Pfizer, and Stryker.
—Mitchel L. Zoler
Suggested Reading
Edwards JD, Kapral MK, Fang J, et al. Underutilization of ambulatory ECG monitoring after stroke and transient ischemic attack: missed opportunities for atrial fibrillation detection. Stroke. 2016;47(8):1982-1989.
Tu HT, Chen Z, Swift C, et al. Smartphone electrographic monitoring for atrial fibrillation in acute ischemic stroke and transient ischemic attack. Int J Stroke. 2017;12(7):786-789.
Although guidelines recommend Holter monitoring for all patients with ischemic stroke or TIA, comparatively few receive it.
Although guidelines recommend Holter monitoring for all patients with ischemic stroke or TIA, comparatively few receive it.
MONTREAL—In patients hospitalized for a recent acute ischemic stroke or transient ischemic attack (TIA), a smartphone-based method identified three times more patients with atrial fibrillation than did 24-hour Holter monitoring after discharge, according to a study presented at the 11th World Stroke Congress.
This high level of atrial fibrillation detection suggests that this relatively cheap and noninvasive device is a good complement to conventional monitoring by a 24-hour Holter recording or an implanted loop recorder in patients with recent stroke. The device may thus satisfy the requirements of current guidelines from the world’s cardiology societies.
In-Hospital and Postdischarge Monitoring
In the study, 294 of 1,079 patients with acute ischemic stroke or TIA underwent serial, 30-second monitoring with the AliveCor device while hospitalized. The device was designed for smartphone-enabled ECG measurement. After discharge, the same patients underwent Holter monitoring. The latter technique identified eight patients (3%) with atrial fibrillation, compared with 25 patients (9%) who were identified using the AliveCor device, said Bernard Yan, MD, a consultant neurologist and endovascular neurointerventionist in the Comprehensive Stroke Center of the Royal Melbourne Hospital. Seven of the eight patients identified with atrial fibrillation by Holter monitoring were also found to have atrial fibrillation by the AliveCor device.
Dr. Yan attributed the higher in-hospital detection rate for atrial fibrillation to the timing of screening, which occurred within days of the stroke or TIA, rather than after the patient had left the hospital. “The difference may be because we monitored patients [with the AliveCor device] much earlier, during their hot period right after their stroke.”
Practice Does Not Match Recommendations
The trial, which was called SPOT-AF, was conducted at several centers in Australia, China, and Hong Kong. All patients underwent AliveCor monitoring during their stay in the hospital, which lasted for a median of four days. Patients performed a 30-second heart rhythm check every time a nurse saw them for a routine vital-sign examination, which usually was three or four times per day. The current analysis focused on the 294 patients (27% of the 1,079 patients) who also underwent 24-hour Holter monitoring following hospital discharge when ordered by their personal physician.
This 27% rate of postdischarge Holter monitoring was consistent with that of a 2016 review of more than 17,000 patients with stroke or TIA in Canada. That study found that 31% of participants underwent 24-hour Holter monitoring for atrial fibrillation during the 30 days following their index event. Guidelines, however, call for atrial fibrillation screening in all patients with recent ischemic stroke and TIA.
Although screening for atrial fibrillation with a smartphone-based device is inexpensive and easy, Dr. Yan did not suggest that this approach could replace a Holter monitor or an implanted loop recorder, which is what current guidelines recommend. “To change the guidelines, we need a different study that compares these approaches head to head.”
SPOT-AF received partial funding from Boehringer Ingelheim. Dr. Yan has spoken on behalf of Bayer, Boehringer Ingelheim, Pfizer, and Stryker.
—Mitchel L. Zoler
Suggested Reading
Edwards JD, Kapral MK, Fang J, et al. Underutilization of ambulatory ECG monitoring after stroke and transient ischemic attack: missed opportunities for atrial fibrillation detection. Stroke. 2016;47(8):1982-1989.
Tu HT, Chen Z, Swift C, et al. Smartphone electrographic monitoring for atrial fibrillation in acute ischemic stroke and transient ischemic attack. Int J Stroke. 2017;12(7):786-789.
MONTREAL—In patients hospitalized for a recent acute ischemic stroke or transient ischemic attack (TIA), a smartphone-based method identified three times more patients with atrial fibrillation than did 24-hour Holter monitoring after discharge, according to a study presented at the 11th World Stroke Congress.
This high level of atrial fibrillation detection suggests that this relatively cheap and noninvasive device is a good complement to conventional monitoring by a 24-hour Holter recording or an implanted loop recorder in patients with recent stroke. The device may thus satisfy the requirements of current guidelines from the world’s cardiology societies.
In-Hospital and Postdischarge Monitoring
In the study, 294 of 1,079 patients with acute ischemic stroke or TIA underwent serial, 30-second monitoring with the AliveCor device while hospitalized. The device was designed for smartphone-enabled ECG measurement. After discharge, the same patients underwent Holter monitoring. The latter technique identified eight patients (3%) with atrial fibrillation, compared with 25 patients (9%) who were identified using the AliveCor device, said Bernard Yan, MD, a consultant neurologist and endovascular neurointerventionist in the Comprehensive Stroke Center of the Royal Melbourne Hospital. Seven of the eight patients identified with atrial fibrillation by Holter monitoring were also found to have atrial fibrillation by the AliveCor device.
Dr. Yan attributed the higher in-hospital detection rate for atrial fibrillation to the timing of screening, which occurred within days of the stroke or TIA, rather than after the patient had left the hospital. “The difference may be because we monitored patients [with the AliveCor device] much earlier, during their hot period right after their stroke.”
Practice Does Not Match Recommendations
The trial, which was called SPOT-AF, was conducted at several centers in Australia, China, and Hong Kong. All patients underwent AliveCor monitoring during their stay in the hospital, which lasted for a median of four days. Patients performed a 30-second heart rhythm check every time a nurse saw them for a routine vital-sign examination, which usually was three or four times per day. The current analysis focused on the 294 patients (27% of the 1,079 patients) who also underwent 24-hour Holter monitoring following hospital discharge when ordered by their personal physician.
This 27% rate of postdischarge Holter monitoring was consistent with that of a 2016 review of more than 17,000 patients with stroke or TIA in Canada. That study found that 31% of participants underwent 24-hour Holter monitoring for atrial fibrillation during the 30 days following their index event. Guidelines, however, call for atrial fibrillation screening in all patients with recent ischemic stroke and TIA.
Although screening for atrial fibrillation with a smartphone-based device is inexpensive and easy, Dr. Yan did not suggest that this approach could replace a Holter monitor or an implanted loop recorder, which is what current guidelines recommend. “To change the guidelines, we need a different study that compares these approaches head to head.”
SPOT-AF received partial funding from Boehringer Ingelheim. Dr. Yan has spoken on behalf of Bayer, Boehringer Ingelheim, Pfizer, and Stryker.
—Mitchel L. Zoler
Suggested Reading
Edwards JD, Kapral MK, Fang J, et al. Underutilization of ambulatory ECG monitoring after stroke and transient ischemic attack: missed opportunities for atrial fibrillation detection. Stroke. 2016;47(8):1982-1989.
Tu HT, Chen Z, Swift C, et al. Smartphone electrographic monitoring for atrial fibrillation in acute ischemic stroke and transient ischemic attack. Int J Stroke. 2017;12(7):786-789.
'Liver first' for select stage IV colon cancer gaining traction
BOSTON –
It’s an alternative to usual care, meaning simultaneous bowel and liver resection or bowel resection with liver surgery later on.
Systemic chemotherapy comes first, followed by liver resection. If margins are microscopically negative, the patient gets another round of chemotherapy. If no additional lesions emerge, the primary tumor is taken out. The entire process can take up to a year.
The approach was developed in the Netherlands for rectal cancer with advanced liver metastases. The idea was to get the liver lesions out before they became unresectable, then remove the primary tumor later on. It’s gaining traction now for colon cancer, and beginning to trickle into the United States at a few academic medical centers.
It comes down to what’s more dangerous, the metastases or the primary tumor? Tumor science hasn’t answered that question yet. There’s general agreement that metastases are what kill people with cancer, but it’s not known if they come mostly from previous metastases or from the primary tumor. The liver-first approach assumes the former.

Liver-first is “extremely controversial. For older surgeons who are not in tertiary care centers, liver-first doesn’t make sense, and it doesn’t seem to make sense to patients. They wonder why you would go after the liver when they were diagnosed with a colon tumor,” said Janice Rafferty, MD, FACS, professor of surgery at the University of Cincinnati, at the annual clinical congress of the American College of Surgeons.
“Well, it’s because the primary tumor doesn’t limit your life,” she continued. “The life-limiting disease is in the liver, not the colon. If you explain it to them that way, it makes sense. If we cannot get an R0 resection on the liver, it doesn’t make sense to go after the primary, unless it’s symptomatic with obstruction, bleeding, or fistula.”
There have been about 10 attempts at a randomized trial of this approach versus usual care, but they were not successful because of the difficulty of recruiting patients. Patients – and no doubt, some surgeons – may have some resistance to the logic of going after metastases first.
Dr. Rafferty moderated a review of research from Yale University, New Haven, Conn., that attempted to plug the evidence gap. The Yale investigators “presented really interesting data that shows that liver-first has improved survival,” she said.
The Yale team used the National Cancer Database to compare 2010-2015 outcomes from liver-first patients with patients who had simultaneous or bowel-first resections, followed by later liver resections. The database didn’t allow them to tease out simultaneous from bowel-first cases, so they lumped them together as usual care. To avoid confounding, rectal carcinomas and metastases to the lung, brain, and other organs were excluded.
Median survival was 34 months among 358 liver-first patients versus 24 months among 18,042 usual care patients in an intention-to-treat analysis. Among patients who completed their resections, median survival was 57 months among 140 liver-first patients versus 36 months with usual care in 3,988.
The benefit held after adjustment for patient and tumor characteristics (hazard ratio for death 0.77 in favor of liver first). When further adjusted for chemotherapy timing, there was a strong trend for liver-first but it was not statistically significant, suggesting that up-front chemotherapy contributed to the results (HR, 0.88; 95% confidence interval, 0.75-1.01; P = .09).
There were many caveats. The liver-first patients were younger, with over half under the age of 60 years versus just over 40% in usual care. They were also healthier based on Charlson comorbidity scores and more likely to have upfront chemotherapy and be treated at an academic center.

So, what should surgeons make of these findings? Lead investigator Vadim Kurbatov, MD, a Yale surgery resident, argued that, at the very least, they suggest that liver-first is a viable option for stage IV colon cancer with isolated liver metastases. Going further, they suggest that liver first may be the right way to go for younger, healthier patients at academic centers.
For sicker stage IV patients, however, the role of liver-first is unclear. “We really do need a randomized trial,” he said.
Dr. Kurbatov and Dr. Rafferty had no relevant disclosures to report. The work was funded in part by the National Institutes of Health.
BOSTON –
It’s an alternative to usual care, meaning simultaneous bowel and liver resection or bowel resection with liver surgery later on.
Systemic chemotherapy comes first, followed by liver resection. If margins are microscopically negative, the patient gets another round of chemotherapy. If no additional lesions emerge, the primary tumor is taken out. The entire process can take up to a year.
The approach was developed in the Netherlands for rectal cancer with advanced liver metastases. The idea was to get the liver lesions out before they became unresectable, then remove the primary tumor later on. It’s gaining traction now for colon cancer, and beginning to trickle into the United States at a few academic medical centers.
It comes down to what’s more dangerous, the metastases or the primary tumor? Tumor science hasn’t answered that question yet. There’s general agreement that metastases are what kill people with cancer, but it’s not known if they come mostly from previous metastases or from the primary tumor. The liver-first approach assumes the former.
Liver-first is “extremely controversial. For older surgeons who are not in tertiary care centers, liver-first doesn’t make sense, and it doesn’t seem to make sense to patients. They wonder why you would go after the liver when they were diagnosed with a colon tumor,” said Janice Rafferty, MD, FACS, professor of surgery at the University of Cincinnati, at the annual clinical congress of the American College of Surgeons.
“Well, it’s because the primary tumor doesn’t limit your life,” she continued. “The life-limiting disease is in the liver, not the colon. If you explain it to them that way, it makes sense. If we cannot get an R0 resection on the liver, it doesn’t make sense to go after the primary, unless it’s symptomatic with obstruction, bleeding, or fistula.”
There have been about 10 attempts at a randomized trial of this approach versus usual care, but they were not successful because of the difficulty of recruiting patients. Patients – and no doubt, some surgeons – may have some resistance to the logic of going after metastases first.
Dr. Rafferty moderated a review of research from Yale University, New Haven, Conn., that attempted to plug the evidence gap. The Yale investigators “presented really interesting data that shows that liver-first has improved survival,” she said.
The Yale team used the National Cancer Database to compare 2010-2015 outcomes from liver-first patients with patients who had simultaneous or bowel-first resections, followed by later liver resections. The database didn’t allow them to tease out simultaneous from bowel-first cases, so they lumped them together as usual care. To avoid confounding, rectal carcinomas and metastases to the lung, brain, and other organs were excluded.
Median survival was 34 months among 358 liver-first patients versus 24 months among 18,042 usual care patients in an intention-to-treat analysis. Among patients who completed their resections, median survival was 57 months among 140 liver-first patients versus 36 months with usual care in 3,988.
The benefit held after adjustment for patient and tumor characteristics (hazard ratio for death 0.77 in favor of liver first). When further adjusted for chemotherapy timing, there was a strong trend for liver-first but it was not statistically significant, suggesting that up-front chemotherapy contributed to the results (HR, 0.88; 95% confidence interval, 0.75-1.01; P = .09).
There were many caveats. The liver-first patients were younger, with over half under the age of 60 years versus just over 40% in usual care. They were also healthier based on Charlson comorbidity scores and more likely to have upfront chemotherapy and be treated at an academic center.
So, what should surgeons make of these findings? Lead investigator Vadim Kurbatov, MD, a Yale surgery resident, argued that, at the very least, they suggest that liver-first is a viable option for stage IV colon cancer with isolated liver metastases. Going further, they suggest that liver first may be the right way to go for younger, healthier patients at academic centers.
For sicker stage IV patients, however, the role of liver-first is unclear. “We really do need a randomized trial,” he said.
Dr. Kurbatov and Dr. Rafferty had no relevant disclosures to report. The work was funded in part by the National Institutes of Health.
BOSTON –
It’s an alternative to usual care, meaning simultaneous bowel and liver resection or bowel resection with liver surgery later on.
Systemic chemotherapy comes first, followed by liver resection. If margins are microscopically negative, the patient gets another round of chemotherapy. If no additional lesions emerge, the primary tumor is taken out. The entire process can take up to a year.
The approach was developed in the Netherlands for rectal cancer with advanced liver metastases. The idea was to get the liver lesions out before they became unresectable, then remove the primary tumor later on. It’s gaining traction now for colon cancer, and beginning to trickle into the United States at a few academic medical centers.
It comes down to what’s more dangerous, the metastases or the primary tumor? Tumor science hasn’t answered that question yet. There’s general agreement that metastases are what kill people with cancer, but it’s not known if they come mostly from previous metastases or from the primary tumor. The liver-first approach assumes the former.
Liver-first is “extremely controversial. For older surgeons who are not in tertiary care centers, liver-first doesn’t make sense, and it doesn’t seem to make sense to patients. They wonder why you would go after the liver when they were diagnosed with a colon tumor,” said Janice Rafferty, MD, FACS, professor of surgery at the University of Cincinnati, at the annual clinical congress of the American College of Surgeons.
“Well, it’s because the primary tumor doesn’t limit your life,” she continued. “The life-limiting disease is in the liver, not the colon. If you explain it to them that way, it makes sense. If we cannot get an R0 resection on the liver, it doesn’t make sense to go after the primary, unless it’s symptomatic with obstruction, bleeding, or fistula.”
There have been about 10 attempts at a randomized trial of this approach versus usual care, but they were not successful because of the difficulty of recruiting patients. Patients – and no doubt, some surgeons – may have some resistance to the logic of going after metastases first.
Dr. Rafferty moderated a review of research from Yale University, New Haven, Conn., that attempted to plug the evidence gap. The Yale investigators “presented really interesting data that shows that liver-first has improved survival,” she said.
The Yale team used the National Cancer Database to compare 2010-2015 outcomes from liver-first patients with patients who had simultaneous or bowel-first resections, followed by later liver resections. The database didn’t allow them to tease out simultaneous from bowel-first cases, so they lumped them together as usual care. To avoid confounding, rectal carcinomas and metastases to the lung, brain, and other organs were excluded.
Median survival was 34 months among 358 liver-first patients versus 24 months among 18,042 usual care patients in an intention-to-treat analysis. Among patients who completed their resections, median survival was 57 months among 140 liver-first patients versus 36 months with usual care in 3,988.
The benefit held after adjustment for patient and tumor characteristics (hazard ratio for death 0.77 in favor of liver first). When further adjusted for chemotherapy timing, there was a strong trend for liver-first but it was not statistically significant, suggesting that up-front chemotherapy contributed to the results (HR, 0.88; 95% confidence interval, 0.75-1.01; P = .09).
There were many caveats. The liver-first patients were younger, with over half under the age of 60 years versus just over 40% in usual care. They were also healthier based on Charlson comorbidity scores and more likely to have upfront chemotherapy and be treated at an academic center.
So, what should surgeons make of these findings? Lead investigator Vadim Kurbatov, MD, a Yale surgery resident, argued that, at the very least, they suggest that liver-first is a viable option for stage IV colon cancer with isolated liver metastases. Going further, they suggest that liver first may be the right way to go for younger, healthier patients at academic centers.
For sicker stage IV patients, however, the role of liver-first is unclear. “We really do need a randomized trial,” he said.
Dr. Kurbatov and Dr. Rafferty had no relevant disclosures to report. The work was funded in part by the National Institutes of Health.
REPORTING FROM THE ACS CLINICAL CONGRESS
Key clinical point: The liver-first approach may be appropriate for younger, healthier patients at academic centers.
Major finding: Median survival was 34 months among 358 liver-first patients versus 24 months among 18,042 usual care patients in an intention-to-treat analysis.
Study details: A review of over 18,000 patients in the National Cancer Database
Disclosures: The lead investigator had no disclosures to report. The work was funded in part by the National Institutes of Health.
Tinea Incognito in an Urban Pediatric Population
Tinea incognito (TI) describes a dermatophytosis with often atypical clinical features attributed to prior use of topical corticosteroids or other immunomodulating agents. Tinea incognito may lack the scale and elevated margin typical of cutaneous dermatophytoses and can be mistaken for other pediatric cutaneous diseases, particularly atopic dermatitis. 1 Given the prevalence of TI and its susceptibility to misdiagnosis, we conducted a retrospective medical record review of cases of pediatric dermatophytosis presenting from 2005 to 2016.
Methods
We reviewed medical records for patients younger than 18 years who had been seen at the Faculty Group Practice of the Ronald O. Perelman Department of Dermatology, New York University School of Medicine (New York, New York), between January 1, 2005, and October 21, 2016, using International Classification of Diseases, Ninth Revision (ICD-9) codes 110.0 (tinea capitis), 110.1 (onychomycosis/tinea unguium), 110.3 (tinea cruris), 110.4 (tinea pedis), 110.5 (tinea corporis), and 110.9 (tinea, unspecified site). Cases were included in this study if there was documentation of dermatophytosis previously treated with topical corticosteroids or calcineurin inhibitors as well as positive potassium hydroxide (KOH) preparation or fungal culture with dermatophyte growth obtained from lesions satisfying the first criterion. This study was approved by the New York University School of Medicine institutional review board (study no. S15-01388).
Statistical analyses were conducted in SPSS 19.0 for Windows. Categorical variables were assessed using the χ2 test for independence and the Fisher exact test.
Results
A total of 464 cases were reviewed. A positive KOH preparation or dermatophyte fungal culture was documented in 83 cases. Of them, 29 (34.9%) were treated with topical steroids and/or calcineurin inhibitors prior to presentation to dermatology (Table). The mean age at presentation was 8 years. Duration of symptoms prior to presentation was recorded for 23 of 29 patients (79.3%). Of them, 6 (26.1%) experienced symptoms for 1 month or less, 12 (52.2%) for 1 to 6 months, and 5 (21.7%) for 6 months to 1 year.

Physical examination findings (Figure) were documented in all 29 cases. Annular lesions were noted in 24 patients (82.8%). Pustules were present in 5 patients (17.2%) and papules in 11 patients (37.9%). Fourteen patients (48.3%) had involvement of the face, 14 (48.3%) of the body (ie, trunk, extremities, or groin), and 3 (10.3%) of the scalp. Six patients (20.7%) demonstrated findings at more than one body site.
Females were more likely to demonstrate facial lesions (P=.02), while males were more likely to present with body lesions (P=.04). Of 26 patients diagnosed via fungal culture, 16 (55.2%) grew Trichophyton tonsurans, 4 (13.8%) grew Trichophyton rubrum, 3 (10.3%) grew Trichophyton mentagrophytes, 2 (6.9%) grew Microsporum canis, and 1 (3.4%) grew Microsporum gypseum. Treatment entailed oral medication in 18 cases (62.1%). Of them, 13 (72.2%) were treated with griseofulvin, 3 (16.7%) with fluconazole, and 2 (11.1%) with terbinafine. Topical antifungals were prescribed in the remaining 11 cases (37.9%); no further treatment was documented.
Comment
Since the initial description of TI, approximately 60 case reports and small series as well as several larger observational studies describing TI have been published. In our series of pediatric patients, 29 of 83 culture- or KOH-confirmed dermatophytosis cases (34.9%) were considered to be TI due to treatment with topical corticosteroids and/or calcineurin inhibitors prior to presentation. This high prevalence contrasts with the 5.6% prevalence reported in the only prior large case series examining TI in childhood.2 These authors further reported that in their pediatric population, TI was significantly (odds ratio, 8.7; 95% CI, 4.7-16.1) more likely to occur on the face relative to other dermatophytoses and significantly (odds ratio, 0.014; 95% CI, 0.002-0.099) less likely to occur on the scalp.2 We noted a significant association between female gender and facial symptoms as well as between male gender and truncal symptoms. Taken together, these findings suggest an increased likelihood of pediatric tinea faciei to be inappropriately treated, particularly in females.
Although TI treated with topical corticosteroids or calcineurin inhibitors can mimic other skin diseases, a majority of patients in our series demonstrated findings associated with classic tinea, such as annularity and scale. Further, we found that T tonsurans was the causative organism in most cases with T rubrum uncommonly seen, though it is the most prevalent dermatophyte observed worldwide and in 2 large TI case series.3,4 Regional variation in dermatophytes may account for these differences. In our study, griseofulvin was used most frequently in TI treatment, though a systematic review of oral antifungals in tinea capitis supported terbinafine’s greater efficacy in patients infected with T tonsurans.5
Conclusion
Our case series demonstrated a 35% prevalence of TI cases in a population of children with confirmed dermatophytosis presenting to dermatologists at an American academic medical center. We hope that noting the high prevalence and manifold presentations of this disease will aid practitioners in maintaining clinical suspicion for dermatophytosis and thereby facilitate appropriate identification and treatment of TI.
- Paloni G, Valerio E, Berti I, et al. Tinea incognito [published online September 28, 2015]. J Pediatr. 2015;167:1450-e2.
- del Boz J, Crespo V, Rivas‐Ruiz F, et al. Tinea incognito in children: 54 cases. Mycoses. 2011;54:254-258.
- Romano C, Maritati E, Gianni C. Tinea incognito in Italy: a 15-year survey. Mycoses. 2006;49:383-387.
- Kim WJ, Kim TW, Mun JH, et al. Tinea incognito in Korea and itsrisk factors: nine-year multicenter survey. J Korean Med Sci. 2013;28:145-151.
- Chen X, Jiang X, Yang M, et al. Systemic antifungal therapy for tinea capitis in children: an abridged Cochrane review. J Am Acad Dermatol. 2017;76:368-374.
Tinea incognito (TI) describes a dermatophytosis with often atypical clinical features attributed to prior use of topical corticosteroids or other immunomodulating agents. Tinea incognito may lack the scale and elevated margin typical of cutaneous dermatophytoses and can be mistaken for other pediatric cutaneous diseases, particularly atopic dermatitis. 1 Given the prevalence of TI and its susceptibility to misdiagnosis, we conducted a retrospective medical record review of cases of pediatric dermatophytosis presenting from 2005 to 2016.
Methods
We reviewed medical records for patients younger than 18 years who had been seen at the Faculty Group Practice of the Ronald O. Perelman Department of Dermatology, New York University School of Medicine (New York, New York), between January 1, 2005, and October 21, 2016, using International Classification of Diseases, Ninth Revision (ICD-9) codes 110.0 (tinea capitis), 110.1 (onychomycosis/tinea unguium), 110.3 (tinea cruris), 110.4 (tinea pedis), 110.5 (tinea corporis), and 110.9 (tinea, unspecified site). Cases were included in this study if there was documentation of dermatophytosis previously treated with topical corticosteroids or calcineurin inhibitors as well as positive potassium hydroxide (KOH) preparation or fungal culture with dermatophyte growth obtained from lesions satisfying the first criterion. This study was approved by the New York University School of Medicine institutional review board (study no. S15-01388).
Statistical analyses were conducted in SPSS 19.0 for Windows. Categorical variables were assessed using the χ2 test for independence and the Fisher exact test.
Results
A total of 464 cases were reviewed. A positive KOH preparation or dermatophyte fungal culture was documented in 83 cases. Of them, 29 (34.9%) were treated with topical steroids and/or calcineurin inhibitors prior to presentation to dermatology (Table). The mean age at presentation was 8 years. Duration of symptoms prior to presentation was recorded for 23 of 29 patients (79.3%). Of them, 6 (26.1%) experienced symptoms for 1 month or less, 12 (52.2%) for 1 to 6 months, and 5 (21.7%) for 6 months to 1 year.
Physical examination findings (Figure) were documented in all 29 cases. Annular lesions were noted in 24 patients (82.8%). Pustules were present in 5 patients (17.2%) and papules in 11 patients (37.9%). Fourteen patients (48.3%) had involvement of the face, 14 (48.3%) of the body (ie, trunk, extremities, or groin), and 3 (10.3%) of the scalp. Six patients (20.7%) demonstrated findings at more than one body site.
Females were more likely to demonstrate facial lesions (P=.02), while males were more likely to present with body lesions (P=.04). Of 26 patients diagnosed via fungal culture, 16 (55.2%) grew Trichophyton tonsurans, 4 (13.8%) grew Trichophyton rubrum, 3 (10.3%) grew Trichophyton mentagrophytes, 2 (6.9%) grew Microsporum canis, and 1 (3.4%) grew Microsporum gypseum. Treatment entailed oral medication in 18 cases (62.1%). Of them, 13 (72.2%) were treated with griseofulvin, 3 (16.7%) with fluconazole, and 2 (11.1%) with terbinafine. Topical antifungals were prescribed in the remaining 11 cases (37.9%); no further treatment was documented.
Comment
Since the initial description of TI, approximately 60 case reports and small series as well as several larger observational studies describing TI have been published. In our series of pediatric patients, 29 of 83 culture- or KOH-confirmed dermatophytosis cases (34.9%) were considered to be TI due to treatment with topical corticosteroids and/or calcineurin inhibitors prior to presentation. This high prevalence contrasts with the 5.6% prevalence reported in the only prior large case series examining TI in childhood.2 These authors further reported that in their pediatric population, TI was significantly (odds ratio, 8.7; 95% CI, 4.7-16.1) more likely to occur on the face relative to other dermatophytoses and significantly (odds ratio, 0.014; 95% CI, 0.002-0.099) less likely to occur on the scalp.2 We noted a significant association between female gender and facial symptoms as well as between male gender and truncal symptoms. Taken together, these findings suggest an increased likelihood of pediatric tinea faciei to be inappropriately treated, particularly in females.
Although TI treated with topical corticosteroids or calcineurin inhibitors can mimic other skin diseases, a majority of patients in our series demonstrated findings associated with classic tinea, such as annularity and scale. Further, we found that T tonsurans was the causative organism in most cases with T rubrum uncommonly seen, though it is the most prevalent dermatophyte observed worldwide and in 2 large TI case series.3,4 Regional variation in dermatophytes may account for these differences. In our study, griseofulvin was used most frequently in TI treatment, though a systematic review of oral antifungals in tinea capitis supported terbinafine’s greater efficacy in patients infected with T tonsurans.5
Conclusion
Our case series demonstrated a 35% prevalence of TI cases in a population of children with confirmed dermatophytosis presenting to dermatologists at an American academic medical center. We hope that noting the high prevalence and manifold presentations of this disease will aid practitioners in maintaining clinical suspicion for dermatophytosis and thereby facilitate appropriate identification and treatment of TI.
Tinea incognito (TI) describes a dermatophytosis with often atypical clinical features attributed to prior use of topical corticosteroids or other immunomodulating agents. Tinea incognito may lack the scale and elevated margin typical of cutaneous dermatophytoses and can be mistaken for other pediatric cutaneous diseases, particularly atopic dermatitis. 1 Given the prevalence of TI and its susceptibility to misdiagnosis, we conducted a retrospective medical record review of cases of pediatric dermatophytosis presenting from 2005 to 2016.
Methods
We reviewed medical records for patients younger than 18 years who had been seen at the Faculty Group Practice of the Ronald O. Perelman Department of Dermatology, New York University School of Medicine (New York, New York), between January 1, 2005, and October 21, 2016, using International Classification of Diseases, Ninth Revision (ICD-9) codes 110.0 (tinea capitis), 110.1 (onychomycosis/tinea unguium), 110.3 (tinea cruris), 110.4 (tinea pedis), 110.5 (tinea corporis), and 110.9 (tinea, unspecified site). Cases were included in this study if there was documentation of dermatophytosis previously treated with topical corticosteroids or calcineurin inhibitors as well as positive potassium hydroxide (KOH) preparation or fungal culture with dermatophyte growth obtained from lesions satisfying the first criterion. This study was approved by the New York University School of Medicine institutional review board (study no. S15-01388).
Statistical analyses were conducted in SPSS 19.0 for Windows. Categorical variables were assessed using the χ2 test for independence and the Fisher exact test.
Results
A total of 464 cases were reviewed. A positive KOH preparation or dermatophyte fungal culture was documented in 83 cases. Of them, 29 (34.9%) were treated with topical steroids and/or calcineurin inhibitors prior to presentation to dermatology (Table). The mean age at presentation was 8 years. Duration of symptoms prior to presentation was recorded for 23 of 29 patients (79.3%). Of them, 6 (26.1%) experienced symptoms for 1 month or less, 12 (52.2%) for 1 to 6 months, and 5 (21.7%) for 6 months to 1 year.
Physical examination findings (Figure) were documented in all 29 cases. Annular lesions were noted in 24 patients (82.8%). Pustules were present in 5 patients (17.2%) and papules in 11 patients (37.9%). Fourteen patients (48.3%) had involvement of the face, 14 (48.3%) of the body (ie, trunk, extremities, or groin), and 3 (10.3%) of the scalp. Six patients (20.7%) demonstrated findings at more than one body site.
Females were more likely to demonstrate facial lesions (P=.02), while males were more likely to present with body lesions (P=.04). Of 26 patients diagnosed via fungal culture, 16 (55.2%) grew Trichophyton tonsurans, 4 (13.8%) grew Trichophyton rubrum, 3 (10.3%) grew Trichophyton mentagrophytes, 2 (6.9%) grew Microsporum canis, and 1 (3.4%) grew Microsporum gypseum. Treatment entailed oral medication in 18 cases (62.1%). Of them, 13 (72.2%) were treated with griseofulvin, 3 (16.7%) with fluconazole, and 2 (11.1%) with terbinafine. Topical antifungals were prescribed in the remaining 11 cases (37.9%); no further treatment was documented.
Comment
Since the initial description of TI, approximately 60 case reports and small series as well as several larger observational studies describing TI have been published. In our series of pediatric patients, 29 of 83 culture- or KOH-confirmed dermatophytosis cases (34.9%) were considered to be TI due to treatment with topical corticosteroids and/or calcineurin inhibitors prior to presentation. This high prevalence contrasts with the 5.6% prevalence reported in the only prior large case series examining TI in childhood.2 These authors further reported that in their pediatric population, TI was significantly (odds ratio, 8.7; 95% CI, 4.7-16.1) more likely to occur on the face relative to other dermatophytoses and significantly (odds ratio, 0.014; 95% CI, 0.002-0.099) less likely to occur on the scalp.2 We noted a significant association between female gender and facial symptoms as well as between male gender and truncal symptoms. Taken together, these findings suggest an increased likelihood of pediatric tinea faciei to be inappropriately treated, particularly in females.
Although TI treated with topical corticosteroids or calcineurin inhibitors can mimic other skin diseases, a majority of patients in our series demonstrated findings associated with classic tinea, such as annularity and scale. Further, we found that T tonsurans was the causative organism in most cases with T rubrum uncommonly seen, though it is the most prevalent dermatophyte observed worldwide and in 2 large TI case series.3,4 Regional variation in dermatophytes may account for these differences. In our study, griseofulvin was used most frequently in TI treatment, though a systematic review of oral antifungals in tinea capitis supported terbinafine’s greater efficacy in patients infected with T tonsurans.5
Conclusion
Our case series demonstrated a 35% prevalence of TI cases in a population of children with confirmed dermatophytosis presenting to dermatologists at an American academic medical center. We hope that noting the high prevalence and manifold presentations of this disease will aid practitioners in maintaining clinical suspicion for dermatophytosis and thereby facilitate appropriate identification and treatment of TI.
- Paloni G, Valerio E, Berti I, et al. Tinea incognito [published online September 28, 2015]. J Pediatr. 2015;167:1450-e2.
- del Boz J, Crespo V, Rivas‐Ruiz F, et al. Tinea incognito in children: 54 cases. Mycoses. 2011;54:254-258.
- Romano C, Maritati E, Gianni C. Tinea incognito in Italy: a 15-year survey. Mycoses. 2006;49:383-387.
- Kim WJ, Kim TW, Mun JH, et al. Tinea incognito in Korea and itsrisk factors: nine-year multicenter survey. J Korean Med Sci. 2013;28:145-151.
- Chen X, Jiang X, Yang M, et al. Systemic antifungal therapy for tinea capitis in children: an abridged Cochrane review. J Am Acad Dermatol. 2017;76:368-374.
- Paloni G, Valerio E, Berti I, et al. Tinea incognito [published online September 28, 2015]. J Pediatr. 2015;167:1450-e2.
- del Boz J, Crespo V, Rivas‐Ruiz F, et al. Tinea incognito in children: 54 cases. Mycoses. 2011;54:254-258.
- Romano C, Maritati E, Gianni C. Tinea incognito in Italy: a 15-year survey. Mycoses. 2006;49:383-387.
- Kim WJ, Kim TW, Mun JH, et al. Tinea incognito in Korea and itsrisk factors: nine-year multicenter survey. J Korean Med Sci. 2013;28:145-151.
- Chen X, Jiang X, Yang M, et al. Systemic antifungal therapy for tinea capitis in children: an abridged Cochrane review. J Am Acad Dermatol. 2017;76:368-374.
Practice Points
- Within our pediatric study population of microbiologically confirmed tinea cases at an American academic center, we found a 35% prevalence of tinea incognito (TI).
- Unlike investigations of TI in other countries, Trichophyton tonsurans was found to be the most common causative dermatophyte.
- Our data suggest that facial tinea may be more likely to be improperly treated in females and likewise tinea of the trunk or extremities in males.
Researchers Develop Guidelines for Evaluating Cognitive and Behavioral Syndromes in Adults
A care partner almost always should be involved in the evaluation, the guidelines advise.
CHICAGO—An Alzheimer’s Association workgroup has developed 20 recommendations for the clinical evaluation of patients with cognitive or behavioral complaints. All middle-aged or older individuals who report or whose care partner or clinician reports cognitive, behavioral, or functional changes should undergo a timely evaluation, the guidelines advise. A care partner almost always should be involved the evaluation, according to the guidelines.
The recommendations cover the recognition and evaluation of symptoms, selection of brain imaging and other tests, and communication with and support of affected individuals and their caregivers.

Alireza Atri, MD, PhD, cochair of the workgroup, presented the recommendations at AAIC 2018. The authors plan to finalize and publish the guidelines in 2018.
“Until now, we have not had highly specific and multispecialty US national guidelines that can inform the diagnostic process across all care settings and that provide standards meant to improve patient autonomy, care, and outcomes,” said Dr. Atri, Director of the Banner Sun Health Research Institute in Sun City, Arizona, and Lecturer in Neurology at the Center for Brain/Mind Medicine at Brigham and Women’s Hospital and Harvard Medical School in Boston.
Cognitive Behavioral Syndromes
The clinical practice guidelines recognize a broad category of cognitive behavioral syndromes marked by memory and thinking symptoms as well as changes in sleep, anxiety, personality, and relationships.
The Alzheimer’s Association in 2017 convened a Diagnostic Evaluation Clinical Practice Guideline workgroup to develop evidence-based guidelines. The group includes experts in medical, neuropsychologic, and nursing specialties. The members conducted a systematic review of the literature and made recommendations using a modified Delphi consensus process. They graded the recommendations as “A” (must be done; will improve outcomes in almost all cases), “B” (should be done), and “C” (may be done).
The recommendations emphasize obtaining a history from not only the patient, but also from someone who knows the patient well to establish the presence and characteristics of any substantial changes and to categorize the cognitive behavioral syndrome.
Other recommendations for evaluating patients with cognitive behavioral syndromes include the following:
- For patients with atypical or rapidly progressive cognitive behavioral symptoms, the clinician should expedite an evaluation and strongly consider referral to a specialist. (Level A)
- The evaluation process should use tiers of assessments and tests based on a patient’s presentation, risk factors, and profile. (Level A)
- The clinician should involve an informant to obtain reliable information about changes in cognition, activities of daily living, mood and other neuropsychiatric symptoms, and sensory and motor function. Use of structured instruments for assessing these domains is helpful. (Level A)
- Clinicians should use validated tools to assess cognition. (Level A)
- When office-based cognitive assessment is not sufficiently informative (eg, when interpretation of results is uncertain due to a complex clinical profile or confounding demographic characteristics), neuropsychologic evaluation is recommended. (Level A)
- The clinician should obtain MRI as a first-tier approach to aid in establishing etiology. If MRI is not available or is contraindicated, CT should be obtained. (Level B)
- If etiology remains uncertain after interpretation of structural imaging, a dementia specialist can obtain molecular imaging with FDG-PET to improve diagnostic accuracy. (Level B)
- In cases with continued diagnostic uncertainty, a dementia specialist can obtain CSF according to appropriate use criteria for analysis of aβ42 amyloid and tau/p-tau profiles to evaluate for Alzheimer’s disease pathology. (Level C)
- If diagnostic uncertainty remains after obtaining structural imaging and FDG-PET, and CSF aβ and tau/p-tau profiles are unavailable or uninterpretable, the dementia specialist can obtain an amyloid PET scan according to the appropriate use criteria. (Level C)
- In a patient with an established cognitive behavioral syndrome and a likely autosomal dominant family history, the dementia specialist should consider whether genetic testing is warranted. A genetic counselor should be involved throughout the process. (Level A)
A Tool for Medical Professionals
According to the workgroup, a timely and accurate diagnosis of Alzheimer’s disease and related dementias increases patient autonomy when he or she is most able to participate in goals of treatment and life and care decisions. It also allows for early intervention to maximize support opportunities and treatment outcomes.
“These new guidelines will provide an important new tool for medical professionals to more accurately diagnose Alzheimer’s [disease] and other dementias. As a result, people will get the right care and appropriate treatments; families will get the right support and be able to plan for the future,” said James Hendrix, PhD, Alzheimer’s Association Director of Global Science Initiatives and a member of the workgroup. “Too often, cognitive and behavioral symptoms due to Alzheimer’s disease and other dementias are unrecognized or attributed to something else.”
“The guidelines can empower patients, families, and clinicians to expect that symptoms will be evaluated in a patient-centered, structured, and collaborative manner,” Dr. Atri said. “In addition, they help to ensure that, regardless of the specific diagnosis, the results are communicated in a timely and compassionate way to help patients and families live the best lives possible.”
A care partner almost always should be involved in the evaluation, the guidelines advise.
A care partner almost always should be involved in the evaluation, the guidelines advise.
CHICAGO—An Alzheimer’s Association workgroup has developed 20 recommendations for the clinical evaluation of patients with cognitive or behavioral complaints. All middle-aged or older individuals who report or whose care partner or clinician reports cognitive, behavioral, or functional changes should undergo a timely evaluation, the guidelines advise. A care partner almost always should be involved the evaluation, according to the guidelines.
The recommendations cover the recognition and evaluation of symptoms, selection of brain imaging and other tests, and communication with and support of affected individuals and their caregivers.
Alireza Atri, MD, PhD, cochair of the workgroup, presented the recommendations at AAIC 2018. The authors plan to finalize and publish the guidelines in 2018.
“Until now, we have not had highly specific and multispecialty US national guidelines that can inform the diagnostic process across all care settings and that provide standards meant to improve patient autonomy, care, and outcomes,” said Dr. Atri, Director of the Banner Sun Health Research Institute in Sun City, Arizona, and Lecturer in Neurology at the Center for Brain/Mind Medicine at Brigham and Women’s Hospital and Harvard Medical School in Boston.
Cognitive Behavioral Syndromes
The clinical practice guidelines recognize a broad category of cognitive behavioral syndromes marked by memory and thinking symptoms as well as changes in sleep, anxiety, personality, and relationships.
The Alzheimer’s Association in 2017 convened a Diagnostic Evaluation Clinical Practice Guideline workgroup to develop evidence-based guidelines. The group includes experts in medical, neuropsychologic, and nursing specialties. The members conducted a systematic review of the literature and made recommendations using a modified Delphi consensus process. They graded the recommendations as “A” (must be done; will improve outcomes in almost all cases), “B” (should be done), and “C” (may be done).
The recommendations emphasize obtaining a history from not only the patient, but also from someone who knows the patient well to establish the presence and characteristics of any substantial changes and to categorize the cognitive behavioral syndrome.
Other recommendations for evaluating patients with cognitive behavioral syndromes include the following:
- For patients with atypical or rapidly progressive cognitive behavioral symptoms, the clinician should expedite an evaluation and strongly consider referral to a specialist. (Level A)
- The evaluation process should use tiers of assessments and tests based on a patient’s presentation, risk factors, and profile. (Level A)
- The clinician should involve an informant to obtain reliable information about changes in cognition, activities of daily living, mood and other neuropsychiatric symptoms, and sensory and motor function. Use of structured instruments for assessing these domains is helpful. (Level A)
- Clinicians should use validated tools to assess cognition. (Level A)
- When office-based cognitive assessment is not sufficiently informative (eg, when interpretation of results is uncertain due to a complex clinical profile or confounding demographic characteristics), neuropsychologic evaluation is recommended. (Level A)
- The clinician should obtain MRI as a first-tier approach to aid in establishing etiology. If MRI is not available or is contraindicated, CT should be obtained. (Level B)
- If etiology remains uncertain after interpretation of structural imaging, a dementia specialist can obtain molecular imaging with FDG-PET to improve diagnostic accuracy. (Level B)
- In cases with continued diagnostic uncertainty, a dementia specialist can obtain CSF according to appropriate use criteria for analysis of aβ42 amyloid and tau/p-tau profiles to evaluate for Alzheimer’s disease pathology. (Level C)
- If diagnostic uncertainty remains after obtaining structural imaging and FDG-PET, and CSF aβ and tau/p-tau profiles are unavailable or uninterpretable, the dementia specialist can obtain an amyloid PET scan according to the appropriate use criteria. (Level C)
- In a patient with an established cognitive behavioral syndrome and a likely autosomal dominant family history, the dementia specialist should consider whether genetic testing is warranted. A genetic counselor should be involved throughout the process. (Level A)
A Tool for Medical Professionals
According to the workgroup, a timely and accurate diagnosis of Alzheimer’s disease and related dementias increases patient autonomy when he or she is most able to participate in goals of treatment and life and care decisions. It also allows for early intervention to maximize support opportunities and treatment outcomes.
“These new guidelines will provide an important new tool for medical professionals to more accurately diagnose Alzheimer’s [disease] and other dementias. As a result, people will get the right care and appropriate treatments; families will get the right support and be able to plan for the future,” said James Hendrix, PhD, Alzheimer’s Association Director of Global Science Initiatives and a member of the workgroup. “Too often, cognitive and behavioral symptoms due to Alzheimer’s disease and other dementias are unrecognized or attributed to something else.”
“The guidelines can empower patients, families, and clinicians to expect that symptoms will be evaluated in a patient-centered, structured, and collaborative manner,” Dr. Atri said. “In addition, they help to ensure that, regardless of the specific diagnosis, the results are communicated in a timely and compassionate way to help patients and families live the best lives possible.”
CHICAGO—An Alzheimer’s Association workgroup has developed 20 recommendations for the clinical evaluation of patients with cognitive or behavioral complaints. All middle-aged or older individuals who report or whose care partner or clinician reports cognitive, behavioral, or functional changes should undergo a timely evaluation, the guidelines advise. A care partner almost always should be involved the evaluation, according to the guidelines.
The recommendations cover the recognition and evaluation of symptoms, selection of brain imaging and other tests, and communication with and support of affected individuals and their caregivers.
Alireza Atri, MD, PhD, cochair of the workgroup, presented the recommendations at AAIC 2018. The authors plan to finalize and publish the guidelines in 2018.
“Until now, we have not had highly specific and multispecialty US national guidelines that can inform the diagnostic process across all care settings and that provide standards meant to improve patient autonomy, care, and outcomes,” said Dr. Atri, Director of the Banner Sun Health Research Institute in Sun City, Arizona, and Lecturer in Neurology at the Center for Brain/Mind Medicine at Brigham and Women’s Hospital and Harvard Medical School in Boston.
Cognitive Behavioral Syndromes
The clinical practice guidelines recognize a broad category of cognitive behavioral syndromes marked by memory and thinking symptoms as well as changes in sleep, anxiety, personality, and relationships.
The Alzheimer’s Association in 2017 convened a Diagnostic Evaluation Clinical Practice Guideline workgroup to develop evidence-based guidelines. The group includes experts in medical, neuropsychologic, and nursing specialties. The members conducted a systematic review of the literature and made recommendations using a modified Delphi consensus process. They graded the recommendations as “A” (must be done; will improve outcomes in almost all cases), “B” (should be done), and “C” (may be done).
The recommendations emphasize obtaining a history from not only the patient, but also from someone who knows the patient well to establish the presence and characteristics of any substantial changes and to categorize the cognitive behavioral syndrome.
Other recommendations for evaluating patients with cognitive behavioral syndromes include the following:
- For patients with atypical or rapidly progressive cognitive behavioral symptoms, the clinician should expedite an evaluation and strongly consider referral to a specialist. (Level A)
- The evaluation process should use tiers of assessments and tests based on a patient’s presentation, risk factors, and profile. (Level A)
- The clinician should involve an informant to obtain reliable information about changes in cognition, activities of daily living, mood and other neuropsychiatric symptoms, and sensory and motor function. Use of structured instruments for assessing these domains is helpful. (Level A)
- Clinicians should use validated tools to assess cognition. (Level A)
- When office-based cognitive assessment is not sufficiently informative (eg, when interpretation of results is uncertain due to a complex clinical profile or confounding demographic characteristics), neuropsychologic evaluation is recommended. (Level A)
- The clinician should obtain MRI as a first-tier approach to aid in establishing etiology. If MRI is not available or is contraindicated, CT should be obtained. (Level B)
- If etiology remains uncertain after interpretation of structural imaging, a dementia specialist can obtain molecular imaging with FDG-PET to improve diagnostic accuracy. (Level B)
- In cases with continued diagnostic uncertainty, a dementia specialist can obtain CSF according to appropriate use criteria for analysis of aβ42 amyloid and tau/p-tau profiles to evaluate for Alzheimer’s disease pathology. (Level C)
- If diagnostic uncertainty remains after obtaining structural imaging and FDG-PET, and CSF aβ and tau/p-tau profiles are unavailable or uninterpretable, the dementia specialist can obtain an amyloid PET scan according to the appropriate use criteria. (Level C)
- In a patient with an established cognitive behavioral syndrome and a likely autosomal dominant family history, the dementia specialist should consider whether genetic testing is warranted. A genetic counselor should be involved throughout the process. (Level A)
A Tool for Medical Professionals
According to the workgroup, a timely and accurate diagnosis of Alzheimer’s disease and related dementias increases patient autonomy when he or she is most able to participate in goals of treatment and life and care decisions. It also allows for early intervention to maximize support opportunities and treatment outcomes.
“These new guidelines will provide an important new tool for medical professionals to more accurately diagnose Alzheimer’s [disease] and other dementias. As a result, people will get the right care and appropriate treatments; families will get the right support and be able to plan for the future,” said James Hendrix, PhD, Alzheimer’s Association Director of Global Science Initiatives and a member of the workgroup. “Too often, cognitive and behavioral symptoms due to Alzheimer’s disease and other dementias are unrecognized or attributed to something else.”
“The guidelines can empower patients, families, and clinicians to expect that symptoms will be evaluated in a patient-centered, structured, and collaborative manner,” Dr. Atri said. “In addition, they help to ensure that, regardless of the specific diagnosis, the results are communicated in a timely and compassionate way to help patients and families live the best lives possible.”
Eruptive Vellus Hair Cysts in Identical Triplets With Dermoscopic Findings
Case Report
Four-year-old identical triplet girls with numerous asymptomatic scattered papules on the chest of 4 months’ duration were referred to a dermatologist by their pediatrician for molluscum contagiosum. The patients’ father reported that there was no history of trauma, irritation, or manipulation to the affected area. Their medical history was notable for prematurity at 32 weeks’ gestation and congenital dermal melanocytosis. Family history was notable for their father having acne and similar papules on the chest during adolescence that resolved with isotretinoin therapy.
On physical examination there were multiple smooth, hyperpigmented to erythematous, comedonal, 1- to 2-mm papules dispersed on the anterior central chest of all 3 patients (Figure 1). Clinically, these lesions were fairly indistinguishable from other common dermatologic conditions such as acne or milia. Dermoscopic examination revealed homogenous yellow-white areas surrounded by light brown to erythematous halos (Figure 2). Histopathologic examination was not performed given the benign clinical diagnosis and avoidance of biopsy in pediatric populations. Based on dermoscopic features and history, a diagnosis of eruptive vellus hair cysts (EVHCs) in identical triplets was made.
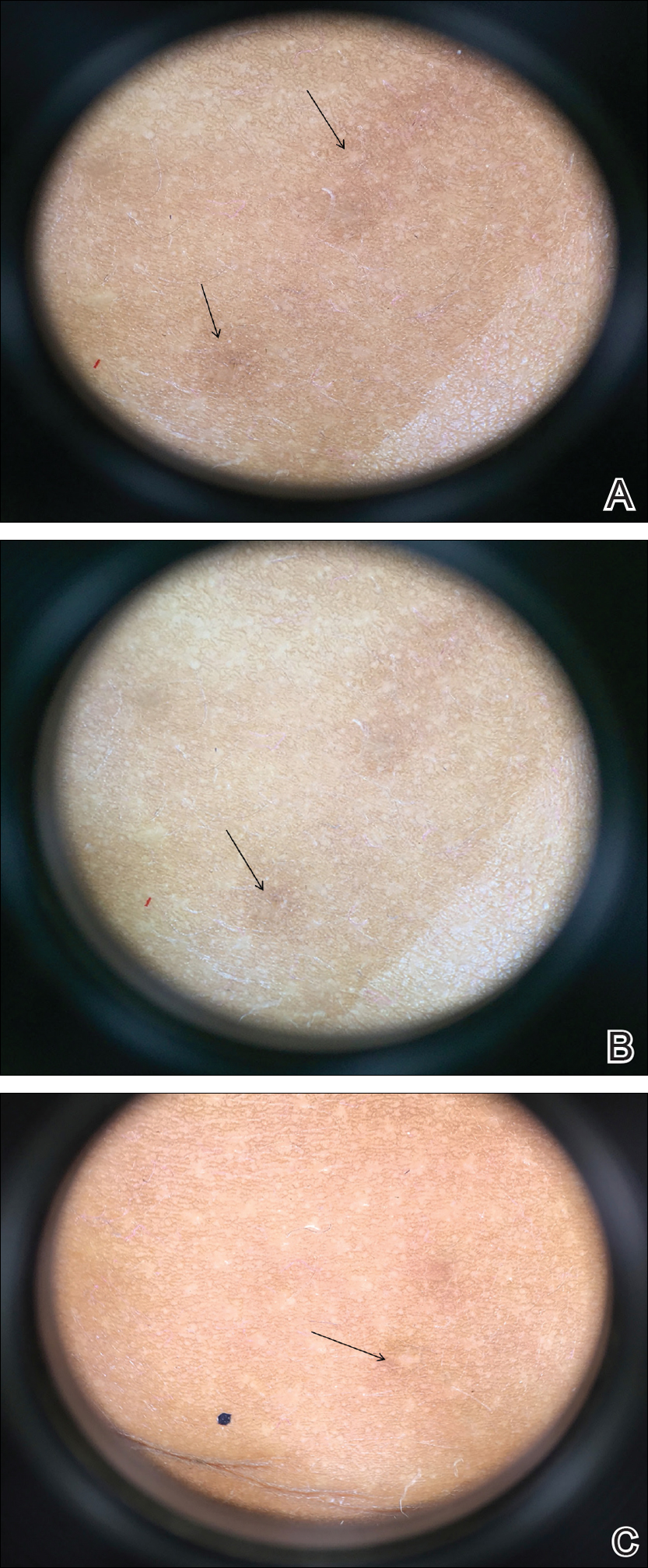
Comment
Pathogenesis
Eruptive vellus hair cysts, first introduced by Esterly et al1 in 1977, are uncommon benign lesions presumed to be caused by an abnormal development of the infundibular portion of the hair follicle.2 They are usually 1- to 3-mm, reddish brown, monomorphous papules overlapping with pilosebaceous and apocrine units.3 Although the lesions typically are located on the chest and extremities, they may occur on the face, abdomen, axillae, buttocks, or genital area.1,3 The inheritance of EVHCs is unclear. The majority of reported cases are sporadic; however, the literature mentions 19 families affected by autosomal-dominant EVHCs based on phylogeny.3 In 2015, EVHCs were reported in identical twins, further supporting the case for a genetic mutation.4 We augment this autosomal-dominant inheritance pattern by presenting a case of identical triplets with EVHCs. The patients’ father reported similar lesions in childhood, further underscoring a genetic basis.
The pathogenesis of EVHC is uncertain, with 2 main theories. Some propose retention of vellus hair and keratin in a cavity formed by an abnormal vellus hair follicle causing infundibular occlusion. Others consider the growth of benign follicular hamartomas that differentiate to become vellus hairs.1
Clinical Presentation
The sporadic form of EVHCs is noted to be more common and clinically presents later, with an average age at onset of 16 years and an average age at diagnosis of 24 years.3 The sporadic form occurs without trauma or manipulation as a precursor. Less commonly, lesions present at birth or in early infancy and may show an autosomal-dominant inheritance pattern with a similar distribution across relatives.3
Other variants of EVHCs have been described. Late-onset EVHC usually occurs at 35 years or older (average age, 57 years), with a female to male predominance of 2.5 to 1.3 This late onset may be attributed to proliferation of ductal follicular keratinocytes or loss of perifollicular elastic fibers exacerbated by exogenous factors such as manipulation, UV rays, or trauma.5
For unilesional EVHC, the average age at diagnosis is 27 years.3 Some of these lesions may be pedunculated and greater than 8 mm. There is a female to male predominance of 2 to 1. Eruptive vellus hair cysts with steatocystoma multiplex can be seen with an average age at onset of 19 years and a female to male predominance of 0.2 to 1. There may be a family history of this subset, as reported in 3 patients with this pattern.3
Diagnosis
The recommended workup for EVHCs varies by patient and age. Eruptive vellus hair cysts present an opportunity to utilize noninvasive diagnostic procedures, especially for the pediatric population, to avoid scarring and pain from manipulation or biopsies. Although many practitioners may comfortably diagnose EVHCs clinically, 6 cases were misdiagnosed as steatocystoma multiplex, keratosis pilaris, or milia prior to histopathology revealing vellus hair cysts.6
Dermoscopy presents as a useful diagnostic aid. Eruptive vellus hair cysts exhibit light yellow homogenous circular structures with a maroon or erythematous halo.2,7 A central gray-blue color point may be seen due to melanin in the pigmented hair shaft.7 A dermoscopy review of EVHCs reported radiating capillaries.2 Occasionally, nonfollicular homogenous blue pigmentation may be seen due to a connection to atrophic hair follicles in the mid dermis and no normal hair follicle around the cysts.8 In comparison, dermoscopic characteristics of molluscum contagiosum demonstrated a polylobular, white-yellow, amorphous structure at the center with a hardened central umbilicated core and a crown of hairpin vessels at the periphery. Additionally, comedonal acne, commonly mistaken for EVHCs, reveals a brown-yellow hard central plug with sparse inflammation under dermoscopy.2 Thus, differentiation of these entities with dermoscopy should be highly prioritized to better aid in the diagnosis of pediatric dermatologic conditions using painless noninvasive techniques.
Treatment
The main indication for treatment of EVHCs is cosmetic concern. Twenty-five percent of EVHCs spontaneously resolve with transepidermal hair elimination or a granulomatous reaction.4,5 A case report of 4 siblings with congenital EVHCs also described a mother with similar lesions that resolved spontaneously in early adulthood,3 as our patients’ father also noted. Treatment modalities including topical keratolytic agents such as urea 10%, retinoic acid 0.05%, tazarotene cream 0.1%, and lactic acid 12%; incision and drainage; CO2 laser; or erbium-doped YAG laser ablation have been tried with minimal improvement.9 Of note, tazarotene cream 0.1% has demonstrated better results than both erbium-doped YAG laser and drainage and incision of EVHCs.4 Additionally, another report evidenced partial improvement with calcipotriene within 2 months with some lesions completely resolved and others flattened, which may be attributed to the antiproliferative and prodifferentiating effects on the ductal follicular keratinocytes by calcipotriene.5 Lastly, an additional study indicated that isotretinoin and vitamin A derivatives were ineffective for clearing EVHCs.10
Conclusion
We presented 3 identical triplets with the classic pediatric onset and dermoscopic findings of EVHCs on the trunk. Although the definitive diagnosis of EVHCs relies on histopathology, we argue that their unique dermoscopic findings combined with a thorough clinical examination is sufficient to recognize this benign condition and avoid painful procedures in the pediatric population.
- Esterly NB, Fretzin DF, Pinkus H. Eruptive vellus hair cysts. Arch Dermatol. 1977;113:500-503.
- Alfaro-Castellón P, Mejía-Rodríguez SA, Valencia-Herrera A, et al. Dermoscopy distinction of eruptive vellus hair cysts with molluscum contagiosum and acne lesions. Pediatr Dermatol. 2012;29:772-773.
- Torchia D, Vega J, Schachner LA. Eruptive vellus hair cysts: a systematic review. Am J Clin Dermatol. 2012;13:19-28.
- Pauline G, Alain H, Jean-Jaques R, et al. Eruptive vellus hair cysts: an original case occurring in twins [published online July 11, 2014]. Int J Dermatol. 2015;54:E209-E212.
- Erkek E, Kurtipek GS, Duman D, et al. Eruptive vellus hair cysts: report of a pediatric case with partial response to calcipotriene therapy. Cutis. 2009;84:295-298.
- Shi G, Zhou Y, Cai YX, et al. Clinicopathological features and expression of four keratins (K10, K14, K17 and K19) in six cases of eruptive vellus hair cysts. Clin Exp Dermatol. 2014;39:496-499.
- Panchaprateep R, Tanus A, Tosti A. Clinical, dermoscopic, and histopathologic features of body hair disorders. J Am Acad Dermatol. 2015;72:890-900.
- Takada S, Togawa Y, Wakabayashii S, et al. Dermoscopic findings in eruptive vellus hair cysts: a case report. Austin J Dermatol. 2014;1:1004.
- Khatu S, Vasani R, Amin S. Eruptive vellus hair cyst presenting as asymptomatic follicular papules on extremities. Indian Dermatol Online J. 2013;4:213-215.
- Urbina-Gonzalez F, Aguilar-Martinez A, Cristobal-Gil M, et al. The treatment of eruptive vellus hair cysts with isotretinoin. Br J Dermatol. 1987;116:465-466.
Case Report
Four-year-old identical triplet girls with numerous asymptomatic scattered papules on the chest of 4 months’ duration were referred to a dermatologist by their pediatrician for molluscum contagiosum. The patients’ father reported that there was no history of trauma, irritation, or manipulation to the affected area. Their medical history was notable for prematurity at 32 weeks’ gestation and congenital dermal melanocytosis. Family history was notable for their father having acne and similar papules on the chest during adolescence that resolved with isotretinoin therapy.
On physical examination there were multiple smooth, hyperpigmented to erythematous, comedonal, 1- to 2-mm papules dispersed on the anterior central chest of all 3 patients (Figure 1). Clinically, these lesions were fairly indistinguishable from other common dermatologic conditions such as acne or milia. Dermoscopic examination revealed homogenous yellow-white areas surrounded by light brown to erythematous halos (Figure 2). Histopathologic examination was not performed given the benign clinical diagnosis and avoidance of biopsy in pediatric populations. Based on dermoscopic features and history, a diagnosis of eruptive vellus hair cysts (EVHCs) in identical triplets was made.
Comment
Pathogenesis
Eruptive vellus hair cysts, first introduced by Esterly et al1 in 1977, are uncommon benign lesions presumed to be caused by an abnormal development of the infundibular portion of the hair follicle.2 They are usually 1- to 3-mm, reddish brown, monomorphous papules overlapping with pilosebaceous and apocrine units.3 Although the lesions typically are located on the chest and extremities, they may occur on the face, abdomen, axillae, buttocks, or genital area.1,3 The inheritance of EVHCs is unclear. The majority of reported cases are sporadic; however, the literature mentions 19 families affected by autosomal-dominant EVHCs based on phylogeny.3 In 2015, EVHCs were reported in identical twins, further supporting the case for a genetic mutation.4 We augment this autosomal-dominant inheritance pattern by presenting a case of identical triplets with EVHCs. The patients’ father reported similar lesions in childhood, further underscoring a genetic basis.
The pathogenesis of EVHC is uncertain, with 2 main theories. Some propose retention of vellus hair and keratin in a cavity formed by an abnormal vellus hair follicle causing infundibular occlusion. Others consider the growth of benign follicular hamartomas that differentiate to become vellus hairs.1
Clinical Presentation
The sporadic form of EVHCs is noted to be more common and clinically presents later, with an average age at onset of 16 years and an average age at diagnosis of 24 years.3 The sporadic form occurs without trauma or manipulation as a precursor. Less commonly, lesions present at birth or in early infancy and may show an autosomal-dominant inheritance pattern with a similar distribution across relatives.3
Other variants of EVHCs have been described. Late-onset EVHC usually occurs at 35 years or older (average age, 57 years), with a female to male predominance of 2.5 to 1.3 This late onset may be attributed to proliferation of ductal follicular keratinocytes or loss of perifollicular elastic fibers exacerbated by exogenous factors such as manipulation, UV rays, or trauma.5
For unilesional EVHC, the average age at diagnosis is 27 years.3 Some of these lesions may be pedunculated and greater than 8 mm. There is a female to male predominance of 2 to 1. Eruptive vellus hair cysts with steatocystoma multiplex can be seen with an average age at onset of 19 years and a female to male predominance of 0.2 to 1. There may be a family history of this subset, as reported in 3 patients with this pattern.3
Diagnosis
The recommended workup for EVHCs varies by patient and age. Eruptive vellus hair cysts present an opportunity to utilize noninvasive diagnostic procedures, especially for the pediatric population, to avoid scarring and pain from manipulation or biopsies. Although many practitioners may comfortably diagnose EVHCs clinically, 6 cases were misdiagnosed as steatocystoma multiplex, keratosis pilaris, or milia prior to histopathology revealing vellus hair cysts.6
Dermoscopy presents as a useful diagnostic aid. Eruptive vellus hair cysts exhibit light yellow homogenous circular structures with a maroon or erythematous halo.2,7 A central gray-blue color point may be seen due to melanin in the pigmented hair shaft.7 A dermoscopy review of EVHCs reported radiating capillaries.2 Occasionally, nonfollicular homogenous blue pigmentation may be seen due to a connection to atrophic hair follicles in the mid dermis and no normal hair follicle around the cysts.8 In comparison, dermoscopic characteristics of molluscum contagiosum demonstrated a polylobular, white-yellow, amorphous structure at the center with a hardened central umbilicated core and a crown of hairpin vessels at the periphery. Additionally, comedonal acne, commonly mistaken for EVHCs, reveals a brown-yellow hard central plug with sparse inflammation under dermoscopy.2 Thus, differentiation of these entities with dermoscopy should be highly prioritized to better aid in the diagnosis of pediatric dermatologic conditions using painless noninvasive techniques.
Treatment
The main indication for treatment of EVHCs is cosmetic concern. Twenty-five percent of EVHCs spontaneously resolve with transepidermal hair elimination or a granulomatous reaction.4,5 A case report of 4 siblings with congenital EVHCs also described a mother with similar lesions that resolved spontaneously in early adulthood,3 as our patients’ father also noted. Treatment modalities including topical keratolytic agents such as urea 10%, retinoic acid 0.05%, tazarotene cream 0.1%, and lactic acid 12%; incision and drainage; CO2 laser; or erbium-doped YAG laser ablation have been tried with minimal improvement.9 Of note, tazarotene cream 0.1% has demonstrated better results than both erbium-doped YAG laser and drainage and incision of EVHCs.4 Additionally, another report evidenced partial improvement with calcipotriene within 2 months with some lesions completely resolved and others flattened, which may be attributed to the antiproliferative and prodifferentiating effects on the ductal follicular keratinocytes by calcipotriene.5 Lastly, an additional study indicated that isotretinoin and vitamin A derivatives were ineffective for clearing EVHCs.10
Conclusion
We presented 3 identical triplets with the classic pediatric onset and dermoscopic findings of EVHCs on the trunk. Although the definitive diagnosis of EVHCs relies on histopathology, we argue that their unique dermoscopic findings combined with a thorough clinical examination is sufficient to recognize this benign condition and avoid painful procedures in the pediatric population.
Case Report
Four-year-old identical triplet girls with numerous asymptomatic scattered papules on the chest of 4 months’ duration were referred to a dermatologist by their pediatrician for molluscum contagiosum. The patients’ father reported that there was no history of trauma, irritation, or manipulation to the affected area. Their medical history was notable for prematurity at 32 weeks’ gestation and congenital dermal melanocytosis. Family history was notable for their father having acne and similar papules on the chest during adolescence that resolved with isotretinoin therapy.
On physical examination there were multiple smooth, hyperpigmented to erythematous, comedonal, 1- to 2-mm papules dispersed on the anterior central chest of all 3 patients (Figure 1). Clinically, these lesions were fairly indistinguishable from other common dermatologic conditions such as acne or milia. Dermoscopic examination revealed homogenous yellow-white areas surrounded by light brown to erythematous halos (Figure 2). Histopathologic examination was not performed given the benign clinical diagnosis and avoidance of biopsy in pediatric populations. Based on dermoscopic features and history, a diagnosis of eruptive vellus hair cysts (EVHCs) in identical triplets was made.
Comment
Pathogenesis
Eruptive vellus hair cysts, first introduced by Esterly et al1 in 1977, are uncommon benign lesions presumed to be caused by an abnormal development of the infundibular portion of the hair follicle.2 They are usually 1- to 3-mm, reddish brown, monomorphous papules overlapping with pilosebaceous and apocrine units.3 Although the lesions typically are located on the chest and extremities, they may occur on the face, abdomen, axillae, buttocks, or genital area.1,3 The inheritance of EVHCs is unclear. The majority of reported cases are sporadic; however, the literature mentions 19 families affected by autosomal-dominant EVHCs based on phylogeny.3 In 2015, EVHCs were reported in identical twins, further supporting the case for a genetic mutation.4 We augment this autosomal-dominant inheritance pattern by presenting a case of identical triplets with EVHCs. The patients’ father reported similar lesions in childhood, further underscoring a genetic basis.
The pathogenesis of EVHC is uncertain, with 2 main theories. Some propose retention of vellus hair and keratin in a cavity formed by an abnormal vellus hair follicle causing infundibular occlusion. Others consider the growth of benign follicular hamartomas that differentiate to become vellus hairs.1
Clinical Presentation
The sporadic form of EVHCs is noted to be more common and clinically presents later, with an average age at onset of 16 years and an average age at diagnosis of 24 years.3 The sporadic form occurs without trauma or manipulation as a precursor. Less commonly, lesions present at birth or in early infancy and may show an autosomal-dominant inheritance pattern with a similar distribution across relatives.3
Other variants of EVHCs have been described. Late-onset EVHC usually occurs at 35 years or older (average age, 57 years), with a female to male predominance of 2.5 to 1.3 This late onset may be attributed to proliferation of ductal follicular keratinocytes or loss of perifollicular elastic fibers exacerbated by exogenous factors such as manipulation, UV rays, or trauma.5
For unilesional EVHC, the average age at diagnosis is 27 years.3 Some of these lesions may be pedunculated and greater than 8 mm. There is a female to male predominance of 2 to 1. Eruptive vellus hair cysts with steatocystoma multiplex can be seen with an average age at onset of 19 years and a female to male predominance of 0.2 to 1. There may be a family history of this subset, as reported in 3 patients with this pattern.3
Diagnosis
The recommended workup for EVHCs varies by patient and age. Eruptive vellus hair cysts present an opportunity to utilize noninvasive diagnostic procedures, especially for the pediatric population, to avoid scarring and pain from manipulation or biopsies. Although many practitioners may comfortably diagnose EVHCs clinically, 6 cases were misdiagnosed as steatocystoma multiplex, keratosis pilaris, or milia prior to histopathology revealing vellus hair cysts.6
Dermoscopy presents as a useful diagnostic aid. Eruptive vellus hair cysts exhibit light yellow homogenous circular structures with a maroon or erythematous halo.2,7 A central gray-blue color point may be seen due to melanin in the pigmented hair shaft.7 A dermoscopy review of EVHCs reported radiating capillaries.2 Occasionally, nonfollicular homogenous blue pigmentation may be seen due to a connection to atrophic hair follicles in the mid dermis and no normal hair follicle around the cysts.8 In comparison, dermoscopic characteristics of molluscum contagiosum demonstrated a polylobular, white-yellow, amorphous structure at the center with a hardened central umbilicated core and a crown of hairpin vessels at the periphery. Additionally, comedonal acne, commonly mistaken for EVHCs, reveals a brown-yellow hard central plug with sparse inflammation under dermoscopy.2 Thus, differentiation of these entities with dermoscopy should be highly prioritized to better aid in the diagnosis of pediatric dermatologic conditions using painless noninvasive techniques.
Treatment
The main indication for treatment of EVHCs is cosmetic concern. Twenty-five percent of EVHCs spontaneously resolve with transepidermal hair elimination or a granulomatous reaction.4,5 A case report of 4 siblings with congenital EVHCs also described a mother with similar lesions that resolved spontaneously in early adulthood,3 as our patients’ father also noted. Treatment modalities including topical keratolytic agents such as urea 10%, retinoic acid 0.05%, tazarotene cream 0.1%, and lactic acid 12%; incision and drainage; CO2 laser; or erbium-doped YAG laser ablation have been tried with minimal improvement.9 Of note, tazarotene cream 0.1% has demonstrated better results than both erbium-doped YAG laser and drainage and incision of EVHCs.4 Additionally, another report evidenced partial improvement with calcipotriene within 2 months with some lesions completely resolved and others flattened, which may be attributed to the antiproliferative and prodifferentiating effects on the ductal follicular keratinocytes by calcipotriene.5 Lastly, an additional study indicated that isotretinoin and vitamin A derivatives were ineffective for clearing EVHCs.10
Conclusion
We presented 3 identical triplets with the classic pediatric onset and dermoscopic findings of EVHCs on the trunk. Although the definitive diagnosis of EVHCs relies on histopathology, we argue that their unique dermoscopic findings combined with a thorough clinical examination is sufficient to recognize this benign condition and avoid painful procedures in the pediatric population.
- Esterly NB, Fretzin DF, Pinkus H. Eruptive vellus hair cysts. Arch Dermatol. 1977;113:500-503.
- Alfaro-Castellón P, Mejía-Rodríguez SA, Valencia-Herrera A, et al. Dermoscopy distinction of eruptive vellus hair cysts with molluscum contagiosum and acne lesions. Pediatr Dermatol. 2012;29:772-773.
- Torchia D, Vega J, Schachner LA. Eruptive vellus hair cysts: a systematic review. Am J Clin Dermatol. 2012;13:19-28.
- Pauline G, Alain H, Jean-Jaques R, et al. Eruptive vellus hair cysts: an original case occurring in twins [published online July 11, 2014]. Int J Dermatol. 2015;54:E209-E212.
- Erkek E, Kurtipek GS, Duman D, et al. Eruptive vellus hair cysts: report of a pediatric case with partial response to calcipotriene therapy. Cutis. 2009;84:295-298.
- Shi G, Zhou Y, Cai YX, et al. Clinicopathological features and expression of four keratins (K10, K14, K17 and K19) in six cases of eruptive vellus hair cysts. Clin Exp Dermatol. 2014;39:496-499.
- Panchaprateep R, Tanus A, Tosti A. Clinical, dermoscopic, and histopathologic features of body hair disorders. J Am Acad Dermatol. 2015;72:890-900.
- Takada S, Togawa Y, Wakabayashii S, et al. Dermoscopic findings in eruptive vellus hair cysts: a case report. Austin J Dermatol. 2014;1:1004.
- Khatu S, Vasani R, Amin S. Eruptive vellus hair cyst presenting as asymptomatic follicular papules on extremities. Indian Dermatol Online J. 2013;4:213-215.
- Urbina-Gonzalez F, Aguilar-Martinez A, Cristobal-Gil M, et al. The treatment of eruptive vellus hair cysts with isotretinoin. Br J Dermatol. 1987;116:465-466.
- Esterly NB, Fretzin DF, Pinkus H. Eruptive vellus hair cysts. Arch Dermatol. 1977;113:500-503.
- Alfaro-Castellón P, Mejía-Rodríguez SA, Valencia-Herrera A, et al. Dermoscopy distinction of eruptive vellus hair cysts with molluscum contagiosum and acne lesions. Pediatr Dermatol. 2012;29:772-773.
- Torchia D, Vega J, Schachner LA. Eruptive vellus hair cysts: a systematic review. Am J Clin Dermatol. 2012;13:19-28.
- Pauline G, Alain H, Jean-Jaques R, et al. Eruptive vellus hair cysts: an original case occurring in twins [published online July 11, 2014]. Int J Dermatol. 2015;54:E209-E212.
- Erkek E, Kurtipek GS, Duman D, et al. Eruptive vellus hair cysts: report of a pediatric case with partial response to calcipotriene therapy. Cutis. 2009;84:295-298.
- Shi G, Zhou Y, Cai YX, et al. Clinicopathological features and expression of four keratins (K10, K14, K17 and K19) in six cases of eruptive vellus hair cysts. Clin Exp Dermatol. 2014;39:496-499.
- Panchaprateep R, Tanus A, Tosti A. Clinical, dermoscopic, and histopathologic features of body hair disorders. J Am Acad Dermatol. 2015;72:890-900.
- Takada S, Togawa Y, Wakabayashii S, et al. Dermoscopic findings in eruptive vellus hair cysts: a case report. Austin J Dermatol. 2014;1:1004.
- Khatu S, Vasani R, Amin S. Eruptive vellus hair cyst presenting as asymptomatic follicular papules on extremities. Indian Dermatol Online J. 2013;4:213-215.
- Urbina-Gonzalez F, Aguilar-Martinez A, Cristobal-Gil M, et al. The treatment of eruptive vellus hair cysts with isotretinoin. Br J Dermatol. 1987;116:465-466.
Practice Points
- Eruptive vellus hair cysts (EVHCs) are 1- to 3-mm round, dome-shaped, flesh-colored, asymptomatic, benign papules typically occurring on the chest and extremities.
- Pathogenesis and inheritance are unclear. Although the majority of EVHC cases are sporadic, the strong influence of genes is indicated by numerous reports of families in whom 2 or more members were affected.
- Dermoscopy is a noninvasive diagnostic procedure that should be utilized to diagnose EVHCs in the pediatric population; specifically, EVHCs exhibit light yellow, homogenous, circular structures with a maroon or erythematous halo.
- The main indication for treatment of EVHCs is cosmetic concern; however, one-quarter of cases may resolve spontaneously.
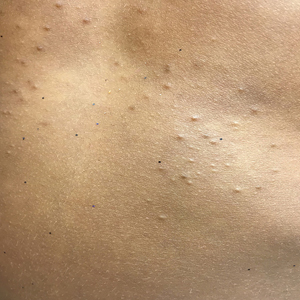
Growth on right cheek
Figure 1

The FP suspected that this was a nodular basal cell carcinoma (BCC) with pigmentation. The physical exam was suspicious because of the pearly appearance, superficial ulcerations, and presence of telangiectasias with a loss of the normal pore pattern. Dermoscopy gave further evidence for a nodular BCC by revealing arborizing “tree-like” telangiectasias, ulcerations, shiny white areas, and gray-blue globules. Skin cancers often produce their own vascular supply and also ulcerate. The shiny white areas (which are the result of collagen deposition and occur in many skin cancers) are best seen with polarized dermoscopy.
The FP recommended a shave biopsy and performed one immediately after obtaining patient consent. (See the Watch & Learn video on “Shave biopsy.”) Knowing that the BCC would be vascular, the FP injected 1% lidocaine with epinephrine and waited 15 minutes for the epinephrine to work.
After seeing another patient, he performed the shave biopsy with a Dermablade, and used a cotton-tipped applicator to vigorously apply the aluminum chloride to the site. He used a twisting motion and pressure to stop most of the bleeding and then used his electrosurgical instrument—with a sharp tipped electrode—to stop recalcitrant bleeders.
The patient was given a diagnosis of BCC on the follow-up visit. The FP referred the patient for Mohs surgery because of the large size and location of the tumor.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Karnes J, Usatine R. Basal cell carcinoma. In: Usatine R, Smith M, Mayeaux EJ, et al. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:989-998.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/.
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com.
Figure 1
The FP suspected that this was a nodular basal cell carcinoma (BCC) with pigmentation. The physical exam was suspicious because of the pearly appearance, superficial ulcerations, and presence of telangiectasias with a loss of the normal pore pattern. Dermoscopy gave further evidence for a nodular BCC by revealing arborizing “tree-like” telangiectasias, ulcerations, shiny white areas, and gray-blue globules. Skin cancers often produce their own vascular supply and also ulcerate. The shiny white areas (which are the result of collagen deposition and occur in many skin cancers) are best seen with polarized dermoscopy.
The FP recommended a shave biopsy and performed one immediately after obtaining patient consent. (See the Watch & Learn video on “Shave biopsy.”) Knowing that the BCC would be vascular, the FP injected 1% lidocaine with epinephrine and waited 15 minutes for the epinephrine to work.
After seeing another patient, he performed the shave biopsy with a Dermablade, and used a cotton-tipped applicator to vigorously apply the aluminum chloride to the site. He used a twisting motion and pressure to stop most of the bleeding and then used his electrosurgical instrument—with a sharp tipped electrode—to stop recalcitrant bleeders.
The patient was given a diagnosis of BCC on the follow-up visit. The FP referred the patient for Mohs surgery because of the large size and location of the tumor.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Karnes J, Usatine R. Basal cell carcinoma. In: Usatine R, Smith M, Mayeaux EJ, et al. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:989-998.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/.
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com.
Figure 1
The FP suspected that this was a nodular basal cell carcinoma (BCC) with pigmentation. The physical exam was suspicious because of the pearly appearance, superficial ulcerations, and presence of telangiectasias with a loss of the normal pore pattern. Dermoscopy gave further evidence for a nodular BCC by revealing arborizing “tree-like” telangiectasias, ulcerations, shiny white areas, and gray-blue globules. Skin cancers often produce their own vascular supply and also ulcerate. The shiny white areas (which are the result of collagen deposition and occur in many skin cancers) are best seen with polarized dermoscopy.
The FP recommended a shave biopsy and performed one immediately after obtaining patient consent. (See the Watch & Learn video on “Shave biopsy.”) Knowing that the BCC would be vascular, the FP injected 1% lidocaine with epinephrine and waited 15 minutes for the epinephrine to work.
After seeing another patient, he performed the shave biopsy with a Dermablade, and used a cotton-tipped applicator to vigorously apply the aluminum chloride to the site. He used a twisting motion and pressure to stop most of the bleeding and then used his electrosurgical instrument—with a sharp tipped electrode—to stop recalcitrant bleeders.
The patient was given a diagnosis of BCC on the follow-up visit. The FP referred the patient for Mohs surgery because of the large size and location of the tumor.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Karnes J, Usatine R. Basal cell carcinoma. In: Usatine R, Smith M, Mayeaux EJ, et al. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:989-998.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/.
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com.
