User login
CMS seeks answers on prior authorization, other hassles to eliminate
Got an idea on how to reduce administrative burden to help reduce the cost of delivering health care? The Centers for Medicare & Medicaid Services wants to hear from you.
In a request for information published June 6, the agency seeks parties across the health care spectrum “to recommend further changes to rules, policies, and procedures that would shift more of clinicians’ time and our health care system’s resources from needless paperwork to high-quality care that improves patient health,” CMS officials said in a statement.
The request for information, part of the agency’s Patients Over Paperwork initiative, seeks suggestions on how to reduce hassles associated with reporting and documentation, coding, prior authorization, rural issues, dual eligible patients, enrollment/eligibility determination and the agency’s own process for issuing regulations and policies.
“Patients over Paperwork has made great inroads in clearing away needlessly complex, outdated, or duplicative requirements that drain clinicians’ time but contribute little to quality of care or patient health,” CMS Administrator Seema Verma said in a statement. “Our goal is to ensure that doctors are spending more time with their patients and less time in administrative tasks.”
The request for information is scheduled to published in the Federal Register on June 11. Comments are due to the agency on Aug. 12. Comments can be made at www.regulations.gov and should refer to file code CMS-6082-NC.
SOURCE: Federal Register, CMS-6082-NC, https://federalregister.gov/d/2019-12215.
Got an idea on how to reduce administrative burden to help reduce the cost of delivering health care? The Centers for Medicare & Medicaid Services wants to hear from you.
In a request for information published June 6, the agency seeks parties across the health care spectrum “to recommend further changes to rules, policies, and procedures that would shift more of clinicians’ time and our health care system’s resources from needless paperwork to high-quality care that improves patient health,” CMS officials said in a statement.
The request for information, part of the agency’s Patients Over Paperwork initiative, seeks suggestions on how to reduce hassles associated with reporting and documentation, coding, prior authorization, rural issues, dual eligible patients, enrollment/eligibility determination and the agency’s own process for issuing regulations and policies.
“Patients over Paperwork has made great inroads in clearing away needlessly complex, outdated, or duplicative requirements that drain clinicians’ time but contribute little to quality of care or patient health,” CMS Administrator Seema Verma said in a statement. “Our goal is to ensure that doctors are spending more time with their patients and less time in administrative tasks.”
The request for information is scheduled to published in the Federal Register on June 11. Comments are due to the agency on Aug. 12. Comments can be made at www.regulations.gov and should refer to file code CMS-6082-NC.
SOURCE: Federal Register, CMS-6082-NC, https://federalregister.gov/d/2019-12215.
Got an idea on how to reduce administrative burden to help reduce the cost of delivering health care? The Centers for Medicare & Medicaid Services wants to hear from you.
In a request for information published June 6, the agency seeks parties across the health care spectrum “to recommend further changes to rules, policies, and procedures that would shift more of clinicians’ time and our health care system’s resources from needless paperwork to high-quality care that improves patient health,” CMS officials said in a statement.
The request for information, part of the agency’s Patients Over Paperwork initiative, seeks suggestions on how to reduce hassles associated with reporting and documentation, coding, prior authorization, rural issues, dual eligible patients, enrollment/eligibility determination and the agency’s own process for issuing regulations and policies.
“Patients over Paperwork has made great inroads in clearing away needlessly complex, outdated, or duplicative requirements that drain clinicians’ time but contribute little to quality of care or patient health,” CMS Administrator Seema Verma said in a statement. “Our goal is to ensure that doctors are spending more time with their patients and less time in administrative tasks.”
The request for information is scheduled to published in the Federal Register on June 11. Comments are due to the agency on Aug. 12. Comments can be made at www.regulations.gov and should refer to file code CMS-6082-NC.
SOURCE: Federal Register, CMS-6082-NC, https://federalregister.gov/d/2019-12215.
Vitamin D did not reduce progression to type 2 diabetes in D2d trial
SAN FRANCISCO – Vitamin D supplementation did not significantly reduce the risk of progression from prediabetes to type 2 diabetes, according to the landmark D2d study.

A possible reason why the observed reduction was not statistically significant was that most participants already had acceptable levels of vitamin D. Still, the intervention “did not significantly reduce the risk [of diabetes],” Anastassios G. Pittas, MD, professor of medicine at Tufts Medical Center, Boston, said at the annual scientific sessions of the American Diabetes Association.
Vitamin D supplementation has been a hot topic on a range of medical fronts. As a 2016 report noted, “low vitamin D levels are also associated with hypertension, cancer, and cardiovascular disease. In addition, [diabetes] and chronic kidney disease (CKD) are also related to vitamin D levels. Vitamin D deficiency has been linked to onset and progression of [diabetes],” (World J Diabetes. 2016;7[5]:89-100).
However, as the report noted, “evidence regarding vitamin D levels and [diabetes] is contradictory, and well-controlled studies are needed.”
For the D2d study, which was coordinated out of the division of endocrinology at Tufts Medical Center, Dr. Pittas and associates recruited 2,423 patients who were considered to have prediabetes, with at least 2 of 3 ADA criteria: fasting plasma glucose level of 100-125 mg/dL; plasma glucose level 2 hours after a 75-g oral glucose load of 140-199 mg/dL; and hemoglobin A1c (HbA1c) level of 5.7%-6.4%.
All of the patients were at least 30 years old with the exception of American Indians, Alaska Natives, Native Hawaiians and other Pacific Islanders who were allowed to be aged 25-30 years. About 22% had low vitamin D levels.
The mean age of the patients was 60 years, mean body mass index was 32, 45% were women, and 33% were non-white. The trial was powered to show a reduction of 25% or more in diabetes risk with vitamin D.
The researchers randomly assigned 1,211 patients to take a once-daily capsule of vitamin D3 (cholecalciferol; 4,000 IU per day); 1,212 received a placebo.
Patients in the vitamin D group greatly boosted their mean serum 25-hydroxyvitamin D levels, from 27.7 ng/mL at baseline to 54.3 ng/mL at 24 months. In contrast, those in the placebo group saw little change, going from 28.2 ng/mL at baseline to 28.8 ng/mL at 24 months.
At a median follow-up of 2.5 years, with 99% of the study participants remaining in the trial, 616 patients developed diabetes (293 in the vitamin D group, 323 in the placebo group).
The risk was lower in the vitamin D group although the difference was not statistically significant. An analysis revealed no clear differences in any of the subgroups (race, age, body mass index, latitude-based geographic location, calcium supplement intake, and others).
However, a post hoc analysis of patients with vitamin D deficiency, which is defined by the National Academy of Medicine as having a 25-hydroxyvitamin D level of less than 12 ng/mL, showed that the vitamin D group had a 62% reduction in risk of diabetes, compared with placebo.
“Response to a nutritional intervention depends on nutritional status at baseline. Thus, if vitamin D has an effect on diabetes prevention, then people with lower levels of serum 25-hydroxyvitamin D would be expected to have a larger effect from supplementation than would those with higher baseline levels,” Dr. Pittas said.
He noted that two recent, similar trials (one in Norway and one in Japan) reported nearly identical, statistically significant risk reductions in the vitamin D group.
There was also some good news in the findings: Vitamin D supplementation “did not lead to significantly more kidney stones, high serum calcium, or low glomerular filtration rate,” Dr. Pittas said.
Although the study findings are disappointing, vitamin D supplementation is still crucial in patients who have low levels, Victor Lawrence Roberts, MD, an endocrinologist in private practice in Orlando, Fla., said in an interview.
“I diagnose at least three or four people a day with vitamin D deficiency,” Dr. Roberts said. “I’ve found if you replace vitamin D in diabetes – get it to 30 ng/ml or better – their diabetes may improve in some cases, although it may be that they’re paying more attention to their health.”
In the big picture, he said, “if people are vitamin D deficient, the vitamin should be replaced no matter what it does to their blood sugar.”
The study was published simultaneously in the New England Journal of Medicine (N Engl J Med. 2019 Jun 7. doi: 10.1056/NEJMoa1900906).
The study was funded by the National Institute of Diabetes and Digestive and Kidney Diseases, the NIH Office of Dietary Supplements, and American Diabetes Association. Dr. Pittas reports grants from the same institutions during the conduct of the study. Many coauthors disclosed relationships with multiple drug companies, but none relevant to the topic under study.
SOURCE: Pittas AG et al. ADA 2019
This article was updated on 6/18/2019.
SAN FRANCISCO – Vitamin D supplementation did not significantly reduce the risk of progression from prediabetes to type 2 diabetes, according to the landmark D2d study.
A possible reason why the observed reduction was not statistically significant was that most participants already had acceptable levels of vitamin D. Still, the intervention “did not significantly reduce the risk [of diabetes],” Anastassios G. Pittas, MD, professor of medicine at Tufts Medical Center, Boston, said at the annual scientific sessions of the American Diabetes Association.
Vitamin D supplementation has been a hot topic on a range of medical fronts. As a 2016 report noted, “low vitamin D levels are also associated with hypertension, cancer, and cardiovascular disease. In addition, [diabetes] and chronic kidney disease (CKD) are also related to vitamin D levels. Vitamin D deficiency has been linked to onset and progression of [diabetes],” (World J Diabetes. 2016;7[5]:89-100).
However, as the report noted, “evidence regarding vitamin D levels and [diabetes] is contradictory, and well-controlled studies are needed.”
For the D2d study, which was coordinated out of the division of endocrinology at Tufts Medical Center, Dr. Pittas and associates recruited 2,423 patients who were considered to have prediabetes, with at least 2 of 3 ADA criteria: fasting plasma glucose level of 100-125 mg/dL; plasma glucose level 2 hours after a 75-g oral glucose load of 140-199 mg/dL; and hemoglobin A1c (HbA1c) level of 5.7%-6.4%.
All of the patients were at least 30 years old with the exception of American Indians, Alaska Natives, Native Hawaiians and other Pacific Islanders who were allowed to be aged 25-30 years. About 22% had low vitamin D levels.
The mean age of the patients was 60 years, mean body mass index was 32, 45% were women, and 33% were non-white. The trial was powered to show a reduction of 25% or more in diabetes risk with vitamin D.
The researchers randomly assigned 1,211 patients to take a once-daily capsule of vitamin D3 (cholecalciferol; 4,000 IU per day); 1,212 received a placebo.
Patients in the vitamin D group greatly boosted their mean serum 25-hydroxyvitamin D levels, from 27.7 ng/mL at baseline to 54.3 ng/mL at 24 months. In contrast, those in the placebo group saw little change, going from 28.2 ng/mL at baseline to 28.8 ng/mL at 24 months.
At a median follow-up of 2.5 years, with 99% of the study participants remaining in the trial, 616 patients developed diabetes (293 in the vitamin D group, 323 in the placebo group).
The risk was lower in the vitamin D group although the difference was not statistically significant. An analysis revealed no clear differences in any of the subgroups (race, age, body mass index, latitude-based geographic location, calcium supplement intake, and others).
However, a post hoc analysis of patients with vitamin D deficiency, which is defined by the National Academy of Medicine as having a 25-hydroxyvitamin D level of less than 12 ng/mL, showed that the vitamin D group had a 62% reduction in risk of diabetes, compared with placebo.
“Response to a nutritional intervention depends on nutritional status at baseline. Thus, if vitamin D has an effect on diabetes prevention, then people with lower levels of serum 25-hydroxyvitamin D would be expected to have a larger effect from supplementation than would those with higher baseline levels,” Dr. Pittas said.
He noted that two recent, similar trials (one in Norway and one in Japan) reported nearly identical, statistically significant risk reductions in the vitamin D group.
There was also some good news in the findings: Vitamin D supplementation “did not lead to significantly more kidney stones, high serum calcium, or low glomerular filtration rate,” Dr. Pittas said.
Although the study findings are disappointing, vitamin D supplementation is still crucial in patients who have low levels, Victor Lawrence Roberts, MD, an endocrinologist in private practice in Orlando, Fla., said in an interview.
“I diagnose at least three or four people a day with vitamin D deficiency,” Dr. Roberts said. “I’ve found if you replace vitamin D in diabetes – get it to 30 ng/ml or better – their diabetes may improve in some cases, although it may be that they’re paying more attention to their health.”
In the big picture, he said, “if people are vitamin D deficient, the vitamin should be replaced no matter what it does to their blood sugar.”
The study was published simultaneously in the New England Journal of Medicine (N Engl J Med. 2019 Jun 7. doi: 10.1056/NEJMoa1900906).
The study was funded by the National Institute of Diabetes and Digestive and Kidney Diseases, the NIH Office of Dietary Supplements, and American Diabetes Association. Dr. Pittas reports grants from the same institutions during the conduct of the study. Many coauthors disclosed relationships with multiple drug companies, but none relevant to the topic under study.
SOURCE: Pittas AG et al. ADA 2019
This article was updated on 6/18/2019.
SAN FRANCISCO – Vitamin D supplementation did not significantly reduce the risk of progression from prediabetes to type 2 diabetes, according to the landmark D2d study.
A possible reason why the observed reduction was not statistically significant was that most participants already had acceptable levels of vitamin D. Still, the intervention “did not significantly reduce the risk [of diabetes],” Anastassios G. Pittas, MD, professor of medicine at Tufts Medical Center, Boston, said at the annual scientific sessions of the American Diabetes Association.
Vitamin D supplementation has been a hot topic on a range of medical fronts. As a 2016 report noted, “low vitamin D levels are also associated with hypertension, cancer, and cardiovascular disease. In addition, [diabetes] and chronic kidney disease (CKD) are also related to vitamin D levels. Vitamin D deficiency has been linked to onset and progression of [diabetes],” (World J Diabetes. 2016;7[5]:89-100).
However, as the report noted, “evidence regarding vitamin D levels and [diabetes] is contradictory, and well-controlled studies are needed.”
For the D2d study, which was coordinated out of the division of endocrinology at Tufts Medical Center, Dr. Pittas and associates recruited 2,423 patients who were considered to have prediabetes, with at least 2 of 3 ADA criteria: fasting plasma glucose level of 100-125 mg/dL; plasma glucose level 2 hours after a 75-g oral glucose load of 140-199 mg/dL; and hemoglobin A1c (HbA1c) level of 5.7%-6.4%.
All of the patients were at least 30 years old with the exception of American Indians, Alaska Natives, Native Hawaiians and other Pacific Islanders who were allowed to be aged 25-30 years. About 22% had low vitamin D levels.
The mean age of the patients was 60 years, mean body mass index was 32, 45% were women, and 33% were non-white. The trial was powered to show a reduction of 25% or more in diabetes risk with vitamin D.
The researchers randomly assigned 1,211 patients to take a once-daily capsule of vitamin D3 (cholecalciferol; 4,000 IU per day); 1,212 received a placebo.
Patients in the vitamin D group greatly boosted their mean serum 25-hydroxyvitamin D levels, from 27.7 ng/mL at baseline to 54.3 ng/mL at 24 months. In contrast, those in the placebo group saw little change, going from 28.2 ng/mL at baseline to 28.8 ng/mL at 24 months.
At a median follow-up of 2.5 years, with 99% of the study participants remaining in the trial, 616 patients developed diabetes (293 in the vitamin D group, 323 in the placebo group).
The risk was lower in the vitamin D group although the difference was not statistically significant. An analysis revealed no clear differences in any of the subgroups (race, age, body mass index, latitude-based geographic location, calcium supplement intake, and others).
However, a post hoc analysis of patients with vitamin D deficiency, which is defined by the National Academy of Medicine as having a 25-hydroxyvitamin D level of less than 12 ng/mL, showed that the vitamin D group had a 62% reduction in risk of diabetes, compared with placebo.
“Response to a nutritional intervention depends on nutritional status at baseline. Thus, if vitamin D has an effect on diabetes prevention, then people with lower levels of serum 25-hydroxyvitamin D would be expected to have a larger effect from supplementation than would those with higher baseline levels,” Dr. Pittas said.
He noted that two recent, similar trials (one in Norway and one in Japan) reported nearly identical, statistically significant risk reductions in the vitamin D group.
There was also some good news in the findings: Vitamin D supplementation “did not lead to significantly more kidney stones, high serum calcium, or low glomerular filtration rate,” Dr. Pittas said.
Although the study findings are disappointing, vitamin D supplementation is still crucial in patients who have low levels, Victor Lawrence Roberts, MD, an endocrinologist in private practice in Orlando, Fla., said in an interview.
“I diagnose at least three or four people a day with vitamin D deficiency,” Dr. Roberts said. “I’ve found if you replace vitamin D in diabetes – get it to 30 ng/ml or better – their diabetes may improve in some cases, although it may be that they’re paying more attention to their health.”
In the big picture, he said, “if people are vitamin D deficient, the vitamin should be replaced no matter what it does to their blood sugar.”
The study was published simultaneously in the New England Journal of Medicine (N Engl J Med. 2019 Jun 7. doi: 10.1056/NEJMoa1900906).
The study was funded by the National Institute of Diabetes and Digestive and Kidney Diseases, the NIH Office of Dietary Supplements, and American Diabetes Association. Dr. Pittas reports grants from the same institutions during the conduct of the study. Many coauthors disclosed relationships with multiple drug companies, but none relevant to the topic under study.
SOURCE: Pittas AG et al. ADA 2019
This article was updated on 6/18/2019.
REPORTING FROM ADA 2019
Key clinical point: Vitamin D supplementation did significantly lower the risk of diabetes.
Major finding: Progression to diabetes occurred in 293 on vitamin D and 323 on placebo.
Study details: Randomized placebo controlled trial of 2,423 patients with prediabetes.
Disclosures: The study was funded by the NIDDK, NIH Office of Dietary Supplements, and American Diabetes Association. Dr. Pittas reports grants from the same institutions during the conduct of the study. Many coauthors disclosed relationships with multiple drug companies.
Source: Pittas AG et al. ADA 2019
National Survey Examines the Extent, Effects of Pain in Vascular Surgeons
Work-related pain and disability have been reported in the literature among various surgical specialties and can influence surgeon productivity and burnout. In Friday’s Scientific Session 4, Max Wohlauer, MD, of the University of Colorado, Denver, will report on a study that he and his colleagues performed to identify the prevalence and severity of pain symptoms in vascular surgeons.

Dr. Wohlauer will report on their survey, which was emailed to 2,910 members of the Society for Vascular Surgery. Pain was reported using the 0–10 Borg’s CR-10 scale, with 0 = no pain, 3 = moderate, 4 = somewhat strong, 5 = strong, and 10 = maximum pain.
Dr. Wohlauer and his colleagues received responses from 775 (26.6%) of the vascular surgeons; with retirees excluded from the study. Among those actively working, the mean age was 51.4 years, and the surgeons had a mean of 17.2 years in practice; 83.6% of the respondents were men.
According to the survey, after a full day of open surgery, the majority of vascular surgeons reported being in a somewhat strong amount of pain (mean score 4.4), while after a full day of endovascular procedures, most vascular surgeons reported being in a moderate amount of pain (mean score 3.9).
Pain following a day of open surgery was highest in the neck (45%) and lower back (39%);after endovascular procedures, respondents reported pain to be most severe in the lower back (44%) and neck (24%). Surgeons performing endovenous procedures report the lowest pain scores (mean 2.0).
In terms of treatment, 242 (36.9%) vascular surgeons reported having sought medical care for work-related issues, with 61 (8.3%) taking time away from the operating room. A total of 72 surgeons (10%) reported requiring surgery and other procedures (including traction), and 22 (3%) had been placed on short- or long-term disability.
In total, 193 (26.2%) of surgeons report pain severe enough that it interfered with sleep, with 9 (1.2%) leaving their career because of disability from work-related pain, and high work-related physical discomfort was significantly associated with Maslach Burnout Inventory single-item measure of burnout for open surgery, endovascular, and endovenous procedures, according to Dr. Wohlauer.
Altogether, 334 (50.6%) of the vascular surgeons surveyed reported that physical discomfort will affect the longevity of their career. This is borne out by the fact that, of the 39 respondents no longer practicing surgery, 26% (10) retired because of disability from work-related pain.
“Our study shows that the majority of practicing vascular surgeons are in pain after a day of operating. Work-related disability is significantly diminishing the workforce. Addressing work-related pain serves to improve the lives and careers of vascular surgeons, while enhancing surgical longevity would help address the current national workforce shortage,” Dr. Wohlauer concluded.
Friday
8-9:30 a.m.
Gaylor National, Potomac A/B
S4: Scientific Session 4: SS12
Work-related pain and disability have been reported in the literature among various surgical specialties and can influence surgeon productivity and burnout. In Friday’s Scientific Session 4, Max Wohlauer, MD, of the University of Colorado, Denver, will report on a study that he and his colleagues performed to identify the prevalence and severity of pain symptoms in vascular surgeons.
Dr. Wohlauer will report on their survey, which was emailed to 2,910 members of the Society for Vascular Surgery. Pain was reported using the 0–10 Borg’s CR-10 scale, with 0 = no pain, 3 = moderate, 4 = somewhat strong, 5 = strong, and 10 = maximum pain.
Dr. Wohlauer and his colleagues received responses from 775 (26.6%) of the vascular surgeons; with retirees excluded from the study. Among those actively working, the mean age was 51.4 years, and the surgeons had a mean of 17.2 years in practice; 83.6% of the respondents were men.
According to the survey, after a full day of open surgery, the majority of vascular surgeons reported being in a somewhat strong amount of pain (mean score 4.4), while after a full day of endovascular procedures, most vascular surgeons reported being in a moderate amount of pain (mean score 3.9).
Pain following a day of open surgery was highest in the neck (45%) and lower back (39%);after endovascular procedures, respondents reported pain to be most severe in the lower back (44%) and neck (24%). Surgeons performing endovenous procedures report the lowest pain scores (mean 2.0).
In terms of treatment, 242 (36.9%) vascular surgeons reported having sought medical care for work-related issues, with 61 (8.3%) taking time away from the operating room. A total of 72 surgeons (10%) reported requiring surgery and other procedures (including traction), and 22 (3%) had been placed on short- or long-term disability.
In total, 193 (26.2%) of surgeons report pain severe enough that it interfered with sleep, with 9 (1.2%) leaving their career because of disability from work-related pain, and high work-related physical discomfort was significantly associated with Maslach Burnout Inventory single-item measure of burnout for open surgery, endovascular, and endovenous procedures, according to Dr. Wohlauer.
Altogether, 334 (50.6%) of the vascular surgeons surveyed reported that physical discomfort will affect the longevity of their career. This is borne out by the fact that, of the 39 respondents no longer practicing surgery, 26% (10) retired because of disability from work-related pain.
“Our study shows that the majority of practicing vascular surgeons are in pain after a day of operating. Work-related disability is significantly diminishing the workforce. Addressing work-related pain serves to improve the lives and careers of vascular surgeons, while enhancing surgical longevity would help address the current national workforce shortage,” Dr. Wohlauer concluded.
Friday
8-9:30 a.m.
Gaylor National, Potomac A/B
S4: Scientific Session 4: SS12
Work-related pain and disability have been reported in the literature among various surgical specialties and can influence surgeon productivity and burnout. In Friday’s Scientific Session 4, Max Wohlauer, MD, of the University of Colorado, Denver, will report on a study that he and his colleagues performed to identify the prevalence and severity of pain symptoms in vascular surgeons.
Dr. Wohlauer will report on their survey, which was emailed to 2,910 members of the Society for Vascular Surgery. Pain was reported using the 0–10 Borg’s CR-10 scale, with 0 = no pain, 3 = moderate, 4 = somewhat strong, 5 = strong, and 10 = maximum pain.
Dr. Wohlauer and his colleagues received responses from 775 (26.6%) of the vascular surgeons; with retirees excluded from the study. Among those actively working, the mean age was 51.4 years, and the surgeons had a mean of 17.2 years in practice; 83.6% of the respondents were men.
According to the survey, after a full day of open surgery, the majority of vascular surgeons reported being in a somewhat strong amount of pain (mean score 4.4), while after a full day of endovascular procedures, most vascular surgeons reported being in a moderate amount of pain (mean score 3.9).
Pain following a day of open surgery was highest in the neck (45%) and lower back (39%);after endovascular procedures, respondents reported pain to be most severe in the lower back (44%) and neck (24%). Surgeons performing endovenous procedures report the lowest pain scores (mean 2.0).
In terms of treatment, 242 (36.9%) vascular surgeons reported having sought medical care for work-related issues, with 61 (8.3%) taking time away from the operating room. A total of 72 surgeons (10%) reported requiring surgery and other procedures (including traction), and 22 (3%) had been placed on short- or long-term disability.
In total, 193 (26.2%) of surgeons report pain severe enough that it interfered with sleep, with 9 (1.2%) leaving their career because of disability from work-related pain, and high work-related physical discomfort was significantly associated with Maslach Burnout Inventory single-item measure of burnout for open surgery, endovascular, and endovenous procedures, according to Dr. Wohlauer.
Altogether, 334 (50.6%) of the vascular surgeons surveyed reported that physical discomfort will affect the longevity of their career. This is borne out by the fact that, of the 39 respondents no longer practicing surgery, 26% (10) retired because of disability from work-related pain.
“Our study shows that the majority of practicing vascular surgeons are in pain after a day of operating. Work-related disability is significantly diminishing the workforce. Addressing work-related pain serves to improve the lives and careers of vascular surgeons, while enhancing surgical longevity would help address the current national workforce shortage,” Dr. Wohlauer concluded.
Friday
8-9:30 a.m.
Gaylor National, Potomac A/B
S4: Scientific Session 4: SS12
From Our President
Society for Vascular Surgery President Michel S. Makaroun, MD, will reflect on his presidency during the 2019 Presidential Address, on Friday, June 14, from 11:15 a.m. to 12:15 p.m. President-Elect Kim Hodgson, MD, will introduce Dr. Makaroun, beginning at 11 a.m.

Dr. Makaroun is professor of surgery and clinical translational science at the University of Pittsburgh. He is the chair of vascular surgery and codirector of the University of Pittsburgh Medical Center Heart and Vascular Institute.
His address, “I am in Favor of Progress ... It is Change I Do Not Like,” will take place in Ballroom A/B.
Society for Vascular Surgery President Michel S. Makaroun, MD, will reflect on his presidency during the 2019 Presidential Address, on Friday, June 14, from 11:15 a.m. to 12:15 p.m. President-Elect Kim Hodgson, MD, will introduce Dr. Makaroun, beginning at 11 a.m.
Dr. Makaroun is professor of surgery and clinical translational science at the University of Pittsburgh. He is the chair of vascular surgery and codirector of the University of Pittsburgh Medical Center Heart and Vascular Institute.
His address, “I am in Favor of Progress ... It is Change I Do Not Like,” will take place in Ballroom A/B.
Society for Vascular Surgery President Michel S. Makaroun, MD, will reflect on his presidency during the 2019 Presidential Address, on Friday, June 14, from 11:15 a.m. to 12:15 p.m. President-Elect Kim Hodgson, MD, will introduce Dr. Makaroun, beginning at 11 a.m.
Dr. Makaroun is professor of surgery and clinical translational science at the University of Pittsburgh. He is the chair of vascular surgery and codirector of the University of Pittsburgh Medical Center Heart and Vascular Institute.
His address, “I am in Favor of Progress ... It is Change I Do Not Like,” will take place in Ballroom A/B.
Gun ownership practices linked to soldier suicide risk
U.S. soldiers who own firearms, store a loaded gun at home, or carry a gun publicly when not on duty are at significantly greater risk of suicide death, a case-control study of 135 U.S. Army soldiers who died by suicide shows.
“Our findings concurred with earlier studies by showing that factors beyond ownership of a firearm were associated with an increased risk of suicide,” wrote Catherine L. Dempsey, PhD, MPH, and her coauthors.
Since 2004, the rate of suicide deaths among Army soldiers has exceeded the rate of combat deaths each year, which prompted Dr. Dempsey, of the Center for the Study of Traumatic Stress at the Uniformed Services University of the Health Sciences, Bethesda, Md., and her coauthors to investigate whether increased access to firearms might be associated with an increased risk of suicide. The study was published by JAMA Network Open.
In the study, the researchers interviewed the next of kin or supervisors of 135 Army soldiers who had taken their own lives while on active duty between 2011 and 2013, 55% of whom used firearms to do so. They compared those findings with those 137 controls matched for suicide propensity based on sociodemographic and Army history risk factors, and 118 soldiers who had experienced suicidal ideation in the past year.
This analysis showed that soldiers who had stored a loaded gun with ammunition at home, or who had carried a personal gun in public had nearly fourfold higher odds of suicide (odds ratio, 3.9; P = .002), compared with propensity-matched controls.
Similarly, those who owned one or more handguns, stored a gun loaded with ammunition at home, and carried a personal gun in public had a greater than threefold odds of suicide.
, four times more likely to store that gun loaded with ammunition at home, and three times more likely to carry a gun in public, compared with controls.
There was the suggestion that the use of safety locks at home was protective, but this did not reach statistical significance.
However, the study did not find significant differences in firearm accessibility characteristics between the soldiers who died by suicide, and the controls with suicidal ideation.
“Some current theories of suicide (eg., the interpersonal theory of suicide) suggest that fatal suicidal behavior results require not only the presence of suicidal desire but also a developed capability or capacity for suicidal behavior,” the authors wrote. “According to the interpersonal theory of suicide, this capability for lethal self-injury is acquired through repeated exposure to painful and fear-inducing experiences, thus habituating an individual to the pain and fear required to enact a fatal suicide attempt.”
Dr. Dempsey and her coauthors argued that their study supported a continued focus on “means restriction” counseling; limiting or removing access to lethal methods for suicide; and “motivational interviewing.”
They cited the fairly small sample size and relatively small response rates to surveys as limitations. However, they wrote, the response rates “were high for multi-informant interviews conducted within a military population.”
The study was supported by the U.S. Department of the Army, U.S. Department of Defense, U.S. Department of Health & Human Services, National Institutes of Health, and National Institute of Mental Health. One author declared grants from the Military Suicide Research Consortium outside the submitted work, and one author declared support, consultancies, and advisory board positions with the pharmaceutical industry, and co-ownership of a mental health market research firm. No other conflicts of interest were declared.
SOURCE: Dempsey CL et al. JAMA Netw Open. 2019 Jun 7. doi: 10.1001/jamanetworkopen.2019.5383.
These findings add to the growing body of evidence that firearm-related behaviors, beyond just gun ownership, may influence suicide risk, and support the most evidence-based and conceptually sound recommendation for suicide prevention, which is to remove the firearm from the home.
There are some limitations to this study. For example, the validity of the “psychological autopsy” approach used in the study has not been determined, and there is also a question mark over the accuracy of family members’ reports about gun ownership and storage practices. Despite this, the study provides support for recommendations for a change in firearm behaviors to reduce the risk of suicide.
Joseph A. Simonetti, MD, MPH, is affiliated with the Rocky Mountain Mental Illness, Research, Education and Clinical Center for Suicide Prevention at the Rocky Mountain Regional VA Medical Center, Aurora, Colo. Ali Rowhani-Rahbar, MD, MPH, PhD, is affiliated with the Harborview Injury Prevention and Research Center at the University of Washington, Seattle. Those comments are adapted from an accompanying editorial (JAMA Netw Open. 2019. Jun 7. doi: 10.1001/jamanetworkopen.2019.5400). No conflicts of interest were declared.
These findings add to the growing body of evidence that firearm-related behaviors, beyond just gun ownership, may influence suicide risk, and support the most evidence-based and conceptually sound recommendation for suicide prevention, which is to remove the firearm from the home.
There are some limitations to this study. For example, the validity of the “psychological autopsy” approach used in the study has not been determined, and there is also a question mark over the accuracy of family members’ reports about gun ownership and storage practices. Despite this, the study provides support for recommendations for a change in firearm behaviors to reduce the risk of suicide.
Joseph A. Simonetti, MD, MPH, is affiliated with the Rocky Mountain Mental Illness, Research, Education and Clinical Center for Suicide Prevention at the Rocky Mountain Regional VA Medical Center, Aurora, Colo. Ali Rowhani-Rahbar, MD, MPH, PhD, is affiliated with the Harborview Injury Prevention and Research Center at the University of Washington, Seattle. Those comments are adapted from an accompanying editorial (JAMA Netw Open. 2019. Jun 7. doi: 10.1001/jamanetworkopen.2019.5400). No conflicts of interest were declared.
These findings add to the growing body of evidence that firearm-related behaviors, beyond just gun ownership, may influence suicide risk, and support the most evidence-based and conceptually sound recommendation for suicide prevention, which is to remove the firearm from the home.
There are some limitations to this study. For example, the validity of the “psychological autopsy” approach used in the study has not been determined, and there is also a question mark over the accuracy of family members’ reports about gun ownership and storage practices. Despite this, the study provides support for recommendations for a change in firearm behaviors to reduce the risk of suicide.
Joseph A. Simonetti, MD, MPH, is affiliated with the Rocky Mountain Mental Illness, Research, Education and Clinical Center for Suicide Prevention at the Rocky Mountain Regional VA Medical Center, Aurora, Colo. Ali Rowhani-Rahbar, MD, MPH, PhD, is affiliated with the Harborview Injury Prevention and Research Center at the University of Washington, Seattle. Those comments are adapted from an accompanying editorial (JAMA Netw Open. 2019. Jun 7. doi: 10.1001/jamanetworkopen.2019.5400). No conflicts of interest were declared.
U.S. soldiers who own firearms, store a loaded gun at home, or carry a gun publicly when not on duty are at significantly greater risk of suicide death, a case-control study of 135 U.S. Army soldiers who died by suicide shows.
“Our findings concurred with earlier studies by showing that factors beyond ownership of a firearm were associated with an increased risk of suicide,” wrote Catherine L. Dempsey, PhD, MPH, and her coauthors.
Since 2004, the rate of suicide deaths among Army soldiers has exceeded the rate of combat deaths each year, which prompted Dr. Dempsey, of the Center for the Study of Traumatic Stress at the Uniformed Services University of the Health Sciences, Bethesda, Md., and her coauthors to investigate whether increased access to firearms might be associated with an increased risk of suicide. The study was published by JAMA Network Open.
In the study, the researchers interviewed the next of kin or supervisors of 135 Army soldiers who had taken their own lives while on active duty between 2011 and 2013, 55% of whom used firearms to do so. They compared those findings with those 137 controls matched for suicide propensity based on sociodemographic and Army history risk factors, and 118 soldiers who had experienced suicidal ideation in the past year.
This analysis showed that soldiers who had stored a loaded gun with ammunition at home, or who had carried a personal gun in public had nearly fourfold higher odds of suicide (odds ratio, 3.9; P = .002), compared with propensity-matched controls.
Similarly, those who owned one or more handguns, stored a gun loaded with ammunition at home, and carried a personal gun in public had a greater than threefold odds of suicide.
, four times more likely to store that gun loaded with ammunition at home, and three times more likely to carry a gun in public, compared with controls.
There was the suggestion that the use of safety locks at home was protective, but this did not reach statistical significance.
However, the study did not find significant differences in firearm accessibility characteristics between the soldiers who died by suicide, and the controls with suicidal ideation.
“Some current theories of suicide (eg., the interpersonal theory of suicide) suggest that fatal suicidal behavior results require not only the presence of suicidal desire but also a developed capability or capacity for suicidal behavior,” the authors wrote. “According to the interpersonal theory of suicide, this capability for lethal self-injury is acquired through repeated exposure to painful and fear-inducing experiences, thus habituating an individual to the pain and fear required to enact a fatal suicide attempt.”
Dr. Dempsey and her coauthors argued that their study supported a continued focus on “means restriction” counseling; limiting or removing access to lethal methods for suicide; and “motivational interviewing.”
They cited the fairly small sample size and relatively small response rates to surveys as limitations. However, they wrote, the response rates “were high for multi-informant interviews conducted within a military population.”
The study was supported by the U.S. Department of the Army, U.S. Department of Defense, U.S. Department of Health & Human Services, National Institutes of Health, and National Institute of Mental Health. One author declared grants from the Military Suicide Research Consortium outside the submitted work, and one author declared support, consultancies, and advisory board positions with the pharmaceutical industry, and co-ownership of a mental health market research firm. No other conflicts of interest were declared.
SOURCE: Dempsey CL et al. JAMA Netw Open. 2019 Jun 7. doi: 10.1001/jamanetworkopen.2019.5383.
U.S. soldiers who own firearms, store a loaded gun at home, or carry a gun publicly when not on duty are at significantly greater risk of suicide death, a case-control study of 135 U.S. Army soldiers who died by suicide shows.
“Our findings concurred with earlier studies by showing that factors beyond ownership of a firearm were associated with an increased risk of suicide,” wrote Catherine L. Dempsey, PhD, MPH, and her coauthors.
Since 2004, the rate of suicide deaths among Army soldiers has exceeded the rate of combat deaths each year, which prompted Dr. Dempsey, of the Center for the Study of Traumatic Stress at the Uniformed Services University of the Health Sciences, Bethesda, Md., and her coauthors to investigate whether increased access to firearms might be associated with an increased risk of suicide. The study was published by JAMA Network Open.
In the study, the researchers interviewed the next of kin or supervisors of 135 Army soldiers who had taken their own lives while on active duty between 2011 and 2013, 55% of whom used firearms to do so. They compared those findings with those 137 controls matched for suicide propensity based on sociodemographic and Army history risk factors, and 118 soldiers who had experienced suicidal ideation in the past year.
This analysis showed that soldiers who had stored a loaded gun with ammunition at home, or who had carried a personal gun in public had nearly fourfold higher odds of suicide (odds ratio, 3.9; P = .002), compared with propensity-matched controls.
Similarly, those who owned one or more handguns, stored a gun loaded with ammunition at home, and carried a personal gun in public had a greater than threefold odds of suicide.
, four times more likely to store that gun loaded with ammunition at home, and three times more likely to carry a gun in public, compared with controls.
There was the suggestion that the use of safety locks at home was protective, but this did not reach statistical significance.
However, the study did not find significant differences in firearm accessibility characteristics between the soldiers who died by suicide, and the controls with suicidal ideation.
“Some current theories of suicide (eg., the interpersonal theory of suicide) suggest that fatal suicidal behavior results require not only the presence of suicidal desire but also a developed capability or capacity for suicidal behavior,” the authors wrote. “According to the interpersonal theory of suicide, this capability for lethal self-injury is acquired through repeated exposure to painful and fear-inducing experiences, thus habituating an individual to the pain and fear required to enact a fatal suicide attempt.”
Dr. Dempsey and her coauthors argued that their study supported a continued focus on “means restriction” counseling; limiting or removing access to lethal methods for suicide; and “motivational interviewing.”
They cited the fairly small sample size and relatively small response rates to surveys as limitations. However, they wrote, the response rates “were high for multi-informant interviews conducted within a military population.”
The study was supported by the U.S. Department of the Army, U.S. Department of Defense, U.S. Department of Health & Human Services, National Institutes of Health, and National Institute of Mental Health. One author declared grants from the Military Suicide Research Consortium outside the submitted work, and one author declared support, consultancies, and advisory board positions with the pharmaceutical industry, and co-ownership of a mental health market research firm. No other conflicts of interest were declared.
SOURCE: Dempsey CL et al. JAMA Netw Open. 2019 Jun 7. doi: 10.1001/jamanetworkopen.2019.5383.
FROM JAMA NETWORK OPEN
NSCLC: Local consolidative therapy in oligometastatic disease and immunotherapy in EGFR mutations
In this edition of “How I will treat my next patient,” I take a look at two recent trials in non–small cell lung cancer (NSCLC). One summarizes a late analysis of a previously published randomized trial in stage IV NSCLC with three or fewer sites of metastasis – oligometastatic disease. The other reviews deidentified patient data to discern whether immune-targeted treatment might be valuable in particular subsets of NSCLC patients with EGFR mutations.

Local consolidative therapy
Daniel R. Gomez, MD, and colleagues published an updated analysis of progression-free survival (PFS) and an initial analysis of overall survival (OS) data in a randomized phase 2 trial in oligometastatic NSCLC. As originally published, patients were randomized to local consolidative treatment (LCT) versus standard maintenance therapy or observation (MT/O). Patients were required to have responding or stable disease after first-line systemic therapy prior to randomization.
Among the 49 patients who received LCT, there was a clear benefit of LCT (PFS of 14.2 months vs. 4.4 months for MT/O; P = .022; and median OS 41.2 months vs. 17.0 months; P = .017). The OS benefit was seen despite allowing crossover to LCT for patients who demonstrated disease progression in the MT/O arm.
What this means in practice
These data are exciting and move clinical research forward – if not, at this time, clinical practice. They support the ongoing clinical trials in NSCLC (NRG LU002) and breast cancer (NRG BR002) investigating the role of LCT in the oligometastatic setting.
For patients who are not candidates for (or choose not to participate in) these important phase 2R/3 trials, I believe that LCT should be discussed with all of the caveats that the authors appropriately mention, from the small number of patients because of the premature closure of the trial, to heterogeneous systemic regimens, to the lack of clarity on whether newer systemic therapies are better.
Immune checkpoint blockade
Historically, EGFR-mutated NSCLCs have not derived comparable benefit to EGFR-wild type (WT) tumors from checkpoint inhibitors. For that reason, in EGFR-mutated tumors, guidelines from the National Comprehensive Cancer Network (NCCN) suggest immune-targeted treatment should be used only on clinical trials or after receipt of EGFR-targeted tyrosine kinase inhibitors and cytotoxic chemotherapy. Several recent studies (IMpower and ATLANTIC), however, have suggested that selected EGFR-mutated patients can benefit from immune-targeted treatment.
Katherine Hastings, PhD, of Yale University, New Haven, Conn., and associates found, in a multi-institution clinical-molecular data review, that among the 44 of 171 EGFR-mutated tumors with L858R mutations, benefit from checkpoint inhibitors was comparable to WT tumors with regard to overall response rate and OS, but not PFS. Additionally, tumors with the EGFR T790M mutation demonstrated similar benefit from checkpoint inhibitors as in WT tumors, L858R-mutated tumors (but not exon 19 deleted tumors) had high tumor mutation burden, and PD-L1 expression did not influence outcome from immunotherapy.
What this means in practice
I agree with the modesty of the authors’ conclusion that these findings should not change clinical practice but rather should encourage further research into which patients with EGFR-mutant disease might benefit from immune-targeted therapy. For now, outside of a clinical trial, in EGFR-mutated patients, I will follow NCCN guidelines, using immune-targeted therapy off-study only with attentiveness to the particular immunotherapy regimens that have shown promise in the literature – and later, not earlier.
Dr. Lyss has been a community-based medical oncologist and clinical researcher for more than 35 years, practicing in St. Louis. His clinical and research interests are in the prevention, diagnosis, and treatment of breast and lung cancers and in expanding access to clinical trials to medically underserved populations.
In this edition of “How I will treat my next patient,” I take a look at two recent trials in non–small cell lung cancer (NSCLC). One summarizes a late analysis of a previously published randomized trial in stage IV NSCLC with three or fewer sites of metastasis – oligometastatic disease. The other reviews deidentified patient data to discern whether immune-targeted treatment might be valuable in particular subsets of NSCLC patients with EGFR mutations.
Local consolidative therapy
Daniel R. Gomez, MD, and colleagues published an updated analysis of progression-free survival (PFS) and an initial analysis of overall survival (OS) data in a randomized phase 2 trial in oligometastatic NSCLC. As originally published, patients were randomized to local consolidative treatment (LCT) versus standard maintenance therapy or observation (MT/O). Patients were required to have responding or stable disease after first-line systemic therapy prior to randomization.
Among the 49 patients who received LCT, there was a clear benefit of LCT (PFS of 14.2 months vs. 4.4 months for MT/O; P = .022; and median OS 41.2 months vs. 17.0 months; P = .017). The OS benefit was seen despite allowing crossover to LCT for patients who demonstrated disease progression in the MT/O arm.
What this means in practice
These data are exciting and move clinical research forward – if not, at this time, clinical practice. They support the ongoing clinical trials in NSCLC (NRG LU002) and breast cancer (NRG BR002) investigating the role of LCT in the oligometastatic setting.
For patients who are not candidates for (or choose not to participate in) these important phase 2R/3 trials, I believe that LCT should be discussed with all of the caveats that the authors appropriately mention, from the small number of patients because of the premature closure of the trial, to heterogeneous systemic regimens, to the lack of clarity on whether newer systemic therapies are better.
Immune checkpoint blockade
Historically, EGFR-mutated NSCLCs have not derived comparable benefit to EGFR-wild type (WT) tumors from checkpoint inhibitors. For that reason, in EGFR-mutated tumors, guidelines from the National Comprehensive Cancer Network (NCCN) suggest immune-targeted treatment should be used only on clinical trials or after receipt of EGFR-targeted tyrosine kinase inhibitors and cytotoxic chemotherapy. Several recent studies (IMpower and ATLANTIC), however, have suggested that selected EGFR-mutated patients can benefit from immune-targeted treatment.
Katherine Hastings, PhD, of Yale University, New Haven, Conn., and associates found, in a multi-institution clinical-molecular data review, that among the 44 of 171 EGFR-mutated tumors with L858R mutations, benefit from checkpoint inhibitors was comparable to WT tumors with regard to overall response rate and OS, but not PFS. Additionally, tumors with the EGFR T790M mutation demonstrated similar benefit from checkpoint inhibitors as in WT tumors, L858R-mutated tumors (but not exon 19 deleted tumors) had high tumor mutation burden, and PD-L1 expression did not influence outcome from immunotherapy.
What this means in practice
I agree with the modesty of the authors’ conclusion that these findings should not change clinical practice but rather should encourage further research into which patients with EGFR-mutant disease might benefit from immune-targeted therapy. For now, outside of a clinical trial, in EGFR-mutated patients, I will follow NCCN guidelines, using immune-targeted therapy off-study only with attentiveness to the particular immunotherapy regimens that have shown promise in the literature – and later, not earlier.
Dr. Lyss has been a community-based medical oncologist and clinical researcher for more than 35 years, practicing in St. Louis. His clinical and research interests are in the prevention, diagnosis, and treatment of breast and lung cancers and in expanding access to clinical trials to medically underserved populations.
In this edition of “How I will treat my next patient,” I take a look at two recent trials in non–small cell lung cancer (NSCLC). One summarizes a late analysis of a previously published randomized trial in stage IV NSCLC with three or fewer sites of metastasis – oligometastatic disease. The other reviews deidentified patient data to discern whether immune-targeted treatment might be valuable in particular subsets of NSCLC patients with EGFR mutations.
Local consolidative therapy
Daniel R. Gomez, MD, and colleagues published an updated analysis of progression-free survival (PFS) and an initial analysis of overall survival (OS) data in a randomized phase 2 trial in oligometastatic NSCLC. As originally published, patients were randomized to local consolidative treatment (LCT) versus standard maintenance therapy or observation (MT/O). Patients were required to have responding or stable disease after first-line systemic therapy prior to randomization.
Among the 49 patients who received LCT, there was a clear benefit of LCT (PFS of 14.2 months vs. 4.4 months for MT/O; P = .022; and median OS 41.2 months vs. 17.0 months; P = .017). The OS benefit was seen despite allowing crossover to LCT for patients who demonstrated disease progression in the MT/O arm.
What this means in practice
These data are exciting and move clinical research forward – if not, at this time, clinical practice. They support the ongoing clinical trials in NSCLC (NRG LU002) and breast cancer (NRG BR002) investigating the role of LCT in the oligometastatic setting.
For patients who are not candidates for (or choose not to participate in) these important phase 2R/3 trials, I believe that LCT should be discussed with all of the caveats that the authors appropriately mention, from the small number of patients because of the premature closure of the trial, to heterogeneous systemic regimens, to the lack of clarity on whether newer systemic therapies are better.
Immune checkpoint blockade
Historically, EGFR-mutated NSCLCs have not derived comparable benefit to EGFR-wild type (WT) tumors from checkpoint inhibitors. For that reason, in EGFR-mutated tumors, guidelines from the National Comprehensive Cancer Network (NCCN) suggest immune-targeted treatment should be used only on clinical trials or after receipt of EGFR-targeted tyrosine kinase inhibitors and cytotoxic chemotherapy. Several recent studies (IMpower and ATLANTIC), however, have suggested that selected EGFR-mutated patients can benefit from immune-targeted treatment.
Katherine Hastings, PhD, of Yale University, New Haven, Conn., and associates found, in a multi-institution clinical-molecular data review, that among the 44 of 171 EGFR-mutated tumors with L858R mutations, benefit from checkpoint inhibitors was comparable to WT tumors with regard to overall response rate and OS, but not PFS. Additionally, tumors with the EGFR T790M mutation demonstrated similar benefit from checkpoint inhibitors as in WT tumors, L858R-mutated tumors (but not exon 19 deleted tumors) had high tumor mutation burden, and PD-L1 expression did not influence outcome from immunotherapy.
What this means in practice
I agree with the modesty of the authors’ conclusion that these findings should not change clinical practice but rather should encourage further research into which patients with EGFR-mutant disease might benefit from immune-targeted therapy. For now, outside of a clinical trial, in EGFR-mutated patients, I will follow NCCN guidelines, using immune-targeted therapy off-study only with attentiveness to the particular immunotherapy regimens that have shown promise in the literature – and later, not earlier.
Dr. Lyss has been a community-based medical oncologist and clinical researcher for more than 35 years, practicing in St. Louis. His clinical and research interests are in the prevention, diagnosis, and treatment of breast and lung cancers and in expanding access to clinical trials to medically underserved populations.
FDA approves IB-Stim device for abdominal pain in adolescents with IBS
The IB-Stim device has been approved to aid in the reduction of functional abdominal pain in patients 11-18 years of age with irritable bowel syndrome (IBS), according to the U.S. Food and Drug Administration.

“This device offers a safe option for treatment of adolescents experiencing pain from IBS through the use of mild nerve stimulation,” Carlos Peña, PhD, director of the Office of Neurological and Physical Medicine Devices in the FDA’s Center for Devices and Radiological Health, said in a press release.
The prescription-only device has a single-use electrical nerve stimulator that is placed behind the patient’s ear. Stimulating nerve bundles in and around the ear is thought to provide pain relief. The battery-powered chip of the device emits low-frequency electrical pulses continuously for 5 days, at which time it is replaced. Patients can use the device for up to 3 consecutive weeks to reduce functional abdominal pain associated with IBS.
The FDA reviewed data from 50 patients, aged 11-18 years, with IBS; 27 patients were treated with the device and 23 patients received a placebo device. The study measured change from baseline to the end of the third week in worst abdominal pain, usual pain, and Pain Frequency Severity Duration (PFSD) scores. Patients were allowed to continue stable doses of medication to treat chronic abdominal pain.
IB-Stim treatment resulted in at least a 30% decrease in usual pain at the end of 3 weeks in 52% of treated patients, compared with 30% of patients who received the placebo, and at least a 30% decrease in worst pain in 59% of treated patients, compared with 26% of patients who received the placebo.
The treatment group also had greater changes in composite PFSD scores at the end of three weeks. During the study, six patients reported mild ear discomfort, and three patients reported adhesive allergy at the site of application, according to the press release.
The device is contraindicated for patients with hemophilia, patients with cardiac pacemakers, or those diagnosed with psoriasis vulgaris.
The FDA granted marketing authorization of the IB-Stim to Innovative Health Solutions.
The IB-Stim device has been approved to aid in the reduction of functional abdominal pain in patients 11-18 years of age with irritable bowel syndrome (IBS), according to the U.S. Food and Drug Administration.
“This device offers a safe option for treatment of adolescents experiencing pain from IBS through the use of mild nerve stimulation,” Carlos Peña, PhD, director of the Office of Neurological and Physical Medicine Devices in the FDA’s Center for Devices and Radiological Health, said in a press release.
The prescription-only device has a single-use electrical nerve stimulator that is placed behind the patient’s ear. Stimulating nerve bundles in and around the ear is thought to provide pain relief. The battery-powered chip of the device emits low-frequency electrical pulses continuously for 5 days, at which time it is replaced. Patients can use the device for up to 3 consecutive weeks to reduce functional abdominal pain associated with IBS.
The FDA reviewed data from 50 patients, aged 11-18 years, with IBS; 27 patients were treated with the device and 23 patients received a placebo device. The study measured change from baseline to the end of the third week in worst abdominal pain, usual pain, and Pain Frequency Severity Duration (PFSD) scores. Patients were allowed to continue stable doses of medication to treat chronic abdominal pain.
IB-Stim treatment resulted in at least a 30% decrease in usual pain at the end of 3 weeks in 52% of treated patients, compared with 30% of patients who received the placebo, and at least a 30% decrease in worst pain in 59% of treated patients, compared with 26% of patients who received the placebo.
The treatment group also had greater changes in composite PFSD scores at the end of three weeks. During the study, six patients reported mild ear discomfort, and three patients reported adhesive allergy at the site of application, according to the press release.
The device is contraindicated for patients with hemophilia, patients with cardiac pacemakers, or those diagnosed with psoriasis vulgaris.
The FDA granted marketing authorization of the IB-Stim to Innovative Health Solutions.
The IB-Stim device has been approved to aid in the reduction of functional abdominal pain in patients 11-18 years of age with irritable bowel syndrome (IBS), according to the U.S. Food and Drug Administration.
“This device offers a safe option for treatment of adolescents experiencing pain from IBS through the use of mild nerve stimulation,” Carlos Peña, PhD, director of the Office of Neurological and Physical Medicine Devices in the FDA’s Center for Devices and Radiological Health, said in a press release.
The prescription-only device has a single-use electrical nerve stimulator that is placed behind the patient’s ear. Stimulating nerve bundles in and around the ear is thought to provide pain relief. The battery-powered chip of the device emits low-frequency electrical pulses continuously for 5 days, at which time it is replaced. Patients can use the device for up to 3 consecutive weeks to reduce functional abdominal pain associated with IBS.
The FDA reviewed data from 50 patients, aged 11-18 years, with IBS; 27 patients were treated with the device and 23 patients received a placebo device. The study measured change from baseline to the end of the third week in worst abdominal pain, usual pain, and Pain Frequency Severity Duration (PFSD) scores. Patients were allowed to continue stable doses of medication to treat chronic abdominal pain.
IB-Stim treatment resulted in at least a 30% decrease in usual pain at the end of 3 weeks in 52% of treated patients, compared with 30% of patients who received the placebo, and at least a 30% decrease in worst pain in 59% of treated patients, compared with 26% of patients who received the placebo.
The treatment group also had greater changes in composite PFSD scores at the end of three weeks. During the study, six patients reported mild ear discomfort, and three patients reported adhesive allergy at the site of application, according to the press release.
The device is contraindicated for patients with hemophilia, patients with cardiac pacemakers, or those diagnosed with psoriasis vulgaris.
The FDA granted marketing authorization of the IB-Stim to Innovative Health Solutions.
Chemo-free Smart Start regimen looks promising in poor-prognosis DLBCL
CHICAGO – A chemotherapy-free regimen has produced promising early response and survival outcomes in patients with a particularly poor-prognosis subtype of diffuse large B-cell lymphoma, an investigator reported at the annual meeting of the American Society of Clinical Oncology.
The overall response rate was 86% after two cycles of combined rituximab, lenalidomide, and ibrutinib – or RLI – in patients with newly diagnosed diffuse large B-cell lymphoma (DLBCL) of the non–germinal center (non-GCB) subtype, said Jason Westin, MD, of the department of lymphoma/myeloma at the University of Texas MD Anderson Cancer Center in Houston.
The response rate increased to 96% after subsequent cycles of RLI plus standard chemotherapy, said Dr.Westin, who added that the rates of progression-free and overall survival at 1 year were also 96% in the investigator initiated, single-arm, open-label, phase 2 study, called Smart Start.
That looks quite favorable, compared with what’s been achieved in previous studies in this poor-prognosis group of patients, Dr. Westin said during a podium presentation of Smart Start data, though he cautioned against direct comparison to historical studies and added that further follow-up is needed.
“Our survival outcomes appear excellent with about a year’s worth of follow-up,” he said during his presentation. “I’d say the novel/novel combinations, with and without chemotherapy, are feasible for large cell, and next step studies are warranted.”
Jasmine M. Zain, MD, of City of Hope Comprehensive Cancer Center, said these results so far raise the possibility of an effective chemotherapy-free treatment regimen for aggressive lymphomas.
“This regimen, particularly for non-GCB subtypes, is extremely promising,” Dr. Zain said during a podium discussion of the study. “I think we were all oohing and aahing over the results, and it could possibly even be practice changing.”
Moving to a nonchemotherapy regimen could raise new questions for treatment of non-GCB and possibly also GCB subtypes of DLBCL, such as when the treatments could be stopped, or whether a maintenance approach would be useful, she added.
The Smart Start study enrolled a total of 60 patients with non-GCB DLBCL. The patients received RLI for two 21-day cycles, followed by another six cycles of RLI plus chemotherapy, which was either EPOCH or CHOP, at the investigators’ discretion.
“With a median follow-up of 362 days, we’ve had three progression events,” Dr. Westin said in his discussion of the preliminary efficacy results.
Adverse events were similar to what would be expected for standard chemotherapy, according to Dr. Westin, except for rash, which was seen mainly in cycles one and two.
There were two deaths on study protocol, including one fatal fungal infection that investigators attributed to high dose corticosteroids and RLI. There were no subsequent fungal infections after a protocol amendment prohibiting corticosteroids during the RLI-only cycles, according to the investigators’ report.
The high response rates following the initial lead-in phase made investigators wonder what would happen without subsequent chemotherapy, Dr. Westin told attendees during his oral presentation. In one case, a 74-year-old man did complete the two lead-in cycles of RLI and declined further therapy.
“He’s now nearly 2 years out, without any additional therapy, and has not relapsed to date,” Dr. Westin said. “This is, again, with 6 weeks worth of RLI therapy.”
Final results and minimal residual disease data from the Smart Start study will be presented at a conference later in 2019, Dr. Westin said.
The study received research support and funding from the ASCO Conquer Cancer Foundation. The trial drug and support were provided by Celgene and Janssen. Dr. Westin reported disclosures related to Celgene, Genentech/Abbvie, Kite Pharma, Kite/Gilead, Novartis, ProNAi, Spectrum Pharmaceuticals, Bristol-Myers Squibb, Janssen, and Karyopharm Therapeutics.
SOURCE: Westin J et al. ASCO 2019, Abstract 7508.
CHICAGO – A chemotherapy-free regimen has produced promising early response and survival outcomes in patients with a particularly poor-prognosis subtype of diffuse large B-cell lymphoma, an investigator reported at the annual meeting of the American Society of Clinical Oncology.
The overall response rate was 86% after two cycles of combined rituximab, lenalidomide, and ibrutinib – or RLI – in patients with newly diagnosed diffuse large B-cell lymphoma (DLBCL) of the non–germinal center (non-GCB) subtype, said Jason Westin, MD, of the department of lymphoma/myeloma at the University of Texas MD Anderson Cancer Center in Houston.
The response rate increased to 96% after subsequent cycles of RLI plus standard chemotherapy, said Dr.Westin, who added that the rates of progression-free and overall survival at 1 year were also 96% in the investigator initiated, single-arm, open-label, phase 2 study, called Smart Start.
That looks quite favorable, compared with what’s been achieved in previous studies in this poor-prognosis group of patients, Dr. Westin said during a podium presentation of Smart Start data, though he cautioned against direct comparison to historical studies and added that further follow-up is needed.
“Our survival outcomes appear excellent with about a year’s worth of follow-up,” he said during his presentation. “I’d say the novel/novel combinations, with and without chemotherapy, are feasible for large cell, and next step studies are warranted.”
Jasmine M. Zain, MD, of City of Hope Comprehensive Cancer Center, said these results so far raise the possibility of an effective chemotherapy-free treatment regimen for aggressive lymphomas.
“This regimen, particularly for non-GCB subtypes, is extremely promising,” Dr. Zain said during a podium discussion of the study. “I think we were all oohing and aahing over the results, and it could possibly even be practice changing.”
Moving to a nonchemotherapy regimen could raise new questions for treatment of non-GCB and possibly also GCB subtypes of DLBCL, such as when the treatments could be stopped, or whether a maintenance approach would be useful, she added.
The Smart Start study enrolled a total of 60 patients with non-GCB DLBCL. The patients received RLI for two 21-day cycles, followed by another six cycles of RLI plus chemotherapy, which was either EPOCH or CHOP, at the investigators’ discretion.
“With a median follow-up of 362 days, we’ve had three progression events,” Dr. Westin said in his discussion of the preliminary efficacy results.
Adverse events were similar to what would be expected for standard chemotherapy, according to Dr. Westin, except for rash, which was seen mainly in cycles one and two.
There were two deaths on study protocol, including one fatal fungal infection that investigators attributed to high dose corticosteroids and RLI. There were no subsequent fungal infections after a protocol amendment prohibiting corticosteroids during the RLI-only cycles, according to the investigators’ report.
The high response rates following the initial lead-in phase made investigators wonder what would happen without subsequent chemotherapy, Dr. Westin told attendees during his oral presentation. In one case, a 74-year-old man did complete the two lead-in cycles of RLI and declined further therapy.
“He’s now nearly 2 years out, without any additional therapy, and has not relapsed to date,” Dr. Westin said. “This is, again, with 6 weeks worth of RLI therapy.”
Final results and minimal residual disease data from the Smart Start study will be presented at a conference later in 2019, Dr. Westin said.
The study received research support and funding from the ASCO Conquer Cancer Foundation. The trial drug and support were provided by Celgene and Janssen. Dr. Westin reported disclosures related to Celgene, Genentech/Abbvie, Kite Pharma, Kite/Gilead, Novartis, ProNAi, Spectrum Pharmaceuticals, Bristol-Myers Squibb, Janssen, and Karyopharm Therapeutics.
SOURCE: Westin J et al. ASCO 2019, Abstract 7508.
CHICAGO – A chemotherapy-free regimen has produced promising early response and survival outcomes in patients with a particularly poor-prognosis subtype of diffuse large B-cell lymphoma, an investigator reported at the annual meeting of the American Society of Clinical Oncology.
The overall response rate was 86% after two cycles of combined rituximab, lenalidomide, and ibrutinib – or RLI – in patients with newly diagnosed diffuse large B-cell lymphoma (DLBCL) of the non–germinal center (non-GCB) subtype, said Jason Westin, MD, of the department of lymphoma/myeloma at the University of Texas MD Anderson Cancer Center in Houston.
The response rate increased to 96% after subsequent cycles of RLI plus standard chemotherapy, said Dr.Westin, who added that the rates of progression-free and overall survival at 1 year were also 96% in the investigator initiated, single-arm, open-label, phase 2 study, called Smart Start.
That looks quite favorable, compared with what’s been achieved in previous studies in this poor-prognosis group of patients, Dr. Westin said during a podium presentation of Smart Start data, though he cautioned against direct comparison to historical studies and added that further follow-up is needed.
“Our survival outcomes appear excellent with about a year’s worth of follow-up,” he said during his presentation. “I’d say the novel/novel combinations, with and without chemotherapy, are feasible for large cell, and next step studies are warranted.”
Jasmine M. Zain, MD, of City of Hope Comprehensive Cancer Center, said these results so far raise the possibility of an effective chemotherapy-free treatment regimen for aggressive lymphomas.
“This regimen, particularly for non-GCB subtypes, is extremely promising,” Dr. Zain said during a podium discussion of the study. “I think we were all oohing and aahing over the results, and it could possibly even be practice changing.”
Moving to a nonchemotherapy regimen could raise new questions for treatment of non-GCB and possibly also GCB subtypes of DLBCL, such as when the treatments could be stopped, or whether a maintenance approach would be useful, she added.
The Smart Start study enrolled a total of 60 patients with non-GCB DLBCL. The patients received RLI for two 21-day cycles, followed by another six cycles of RLI plus chemotherapy, which was either EPOCH or CHOP, at the investigators’ discretion.
“With a median follow-up of 362 days, we’ve had three progression events,” Dr. Westin said in his discussion of the preliminary efficacy results.
Adverse events were similar to what would be expected for standard chemotherapy, according to Dr. Westin, except for rash, which was seen mainly in cycles one and two.
There were two deaths on study protocol, including one fatal fungal infection that investigators attributed to high dose corticosteroids and RLI. There were no subsequent fungal infections after a protocol amendment prohibiting corticosteroids during the RLI-only cycles, according to the investigators’ report.
The high response rates following the initial lead-in phase made investigators wonder what would happen without subsequent chemotherapy, Dr. Westin told attendees during his oral presentation. In one case, a 74-year-old man did complete the two lead-in cycles of RLI and declined further therapy.
“He’s now nearly 2 years out, without any additional therapy, and has not relapsed to date,” Dr. Westin said. “This is, again, with 6 weeks worth of RLI therapy.”
Final results and minimal residual disease data from the Smart Start study will be presented at a conference later in 2019, Dr. Westin said.
The study received research support and funding from the ASCO Conquer Cancer Foundation. The trial drug and support were provided by Celgene and Janssen. Dr. Westin reported disclosures related to Celgene, Genentech/Abbvie, Kite Pharma, Kite/Gilead, Novartis, ProNAi, Spectrum Pharmaceuticals, Bristol-Myers Squibb, Janssen, and Karyopharm Therapeutics.
SOURCE: Westin J et al. ASCO 2019, Abstract 7508.
REPORTING FROM ASCO 2019
The costs of surviving cancer
Cancer survivors have significantly higher out-of-pocket medical costs than those with no history of cancer, and a quarter of those survivors have some type of material hardship related to their diagnosis, according to the Centers for Disease Control and Prevention.
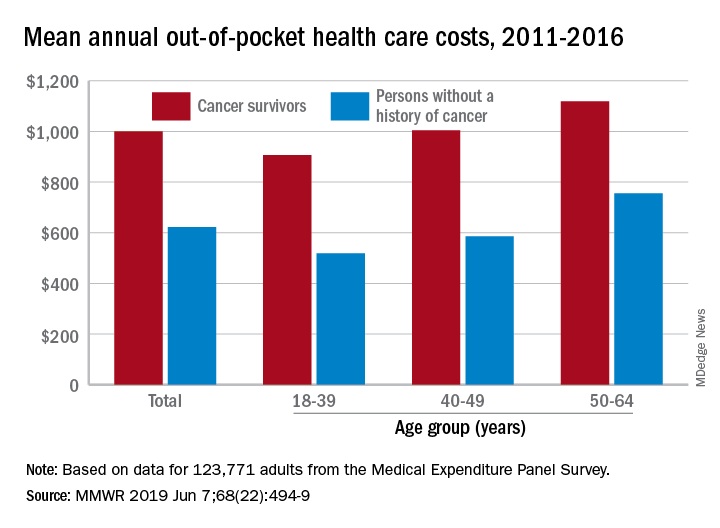
Along with those material financial hardships – the need to borrow money, go into debt, or declare bankruptcy – more than 34% of cancer survivors aged 18-64 years experienced psychological financial hardship, defined as worry about large medical bills, in 2011 and 2016, Donatus U. Ekwueme, PhD, and his associates reported in the Morbidity and Mortality Weekly Report.
Cancer survivors spend 60% more out of pocket than those with no cancer history: $1,000 a year from 2011 to 2016, compared with $622 for adults without a history of cancer. Spending was lowest among younger people (18-39 years) and increased with age, but the prevalence of both material and psychological hardships was highest in the middle age group (40-49 years) and lowest in the oldest group (50-64 years), they said.
Women had higher out-of-pocket costs than men, although the difference was smaller for those with cancer ($1,023 vs. $976) than for those without ($721 vs. $519). Material and psychological hardships were both more common among women, said Dr. Ekwueme of the CDC’s National Center for Chronic Disease Prevention and Health Promotion, Atlanta, and his associates.
Mean out-of-pocket spending was much higher for cancer survivors with private health insurance ($1,114) than for survivors with public insurance ($471), but material hardship was much more prevalent among those with public insurance (33.1% vs. 21.9%). Rates of psychological hardship, however, were much closer: 35.9% for those with public insurance and 32.5% for those with private insurance, the investigators said.
“The number of Americans with a history of cancer is projected to increase in the next decade, and the economic burden associated with living with a cancer diagnosis will likely increase as well,” they wrote, and interventions such as “systematic screening for financial hardship at cancer diagnosis and throughout the cancer care trajectory [are needed] to minimize financial hardship for cancer survivors.”
The analysis was based on data for 123,771 adults aged 18-64 years from the Medical Expenditure Panel Survey. Out-of-pocket costs were calculated using data from 2011 to 2016, with all costs adjusted to 2016 dollars, but the hardship calculations involved data from only 2011 and 2016.
SOURCE: Ekwueme DU et al. MMWR 2019 Jun 7;68(22):494-9.
Cancer survivors have significantly higher out-of-pocket medical costs than those with no history of cancer, and a quarter of those survivors have some type of material hardship related to their diagnosis, according to the Centers for Disease Control and Prevention.
Along with those material financial hardships – the need to borrow money, go into debt, or declare bankruptcy – more than 34% of cancer survivors aged 18-64 years experienced psychological financial hardship, defined as worry about large medical bills, in 2011 and 2016, Donatus U. Ekwueme, PhD, and his associates reported in the Morbidity and Mortality Weekly Report.
Cancer survivors spend 60% more out of pocket than those with no cancer history: $1,000 a year from 2011 to 2016, compared with $622 for adults without a history of cancer. Spending was lowest among younger people (18-39 years) and increased with age, but the prevalence of both material and psychological hardships was highest in the middle age group (40-49 years) and lowest in the oldest group (50-64 years), they said.
Women had higher out-of-pocket costs than men, although the difference was smaller for those with cancer ($1,023 vs. $976) than for those without ($721 vs. $519). Material and psychological hardships were both more common among women, said Dr. Ekwueme of the CDC’s National Center for Chronic Disease Prevention and Health Promotion, Atlanta, and his associates.
Mean out-of-pocket spending was much higher for cancer survivors with private health insurance ($1,114) than for survivors with public insurance ($471), but material hardship was much more prevalent among those with public insurance (33.1% vs. 21.9%). Rates of psychological hardship, however, were much closer: 35.9% for those with public insurance and 32.5% for those with private insurance, the investigators said.
“The number of Americans with a history of cancer is projected to increase in the next decade, and the economic burden associated with living with a cancer diagnosis will likely increase as well,” they wrote, and interventions such as “systematic screening for financial hardship at cancer diagnosis and throughout the cancer care trajectory [are needed] to minimize financial hardship for cancer survivors.”
The analysis was based on data for 123,771 adults aged 18-64 years from the Medical Expenditure Panel Survey. Out-of-pocket costs were calculated using data from 2011 to 2016, with all costs adjusted to 2016 dollars, but the hardship calculations involved data from only 2011 and 2016.
SOURCE: Ekwueme DU et al. MMWR 2019 Jun 7;68(22):494-9.
Cancer survivors have significantly higher out-of-pocket medical costs than those with no history of cancer, and a quarter of those survivors have some type of material hardship related to their diagnosis, according to the Centers for Disease Control and Prevention.
Along with those material financial hardships – the need to borrow money, go into debt, or declare bankruptcy – more than 34% of cancer survivors aged 18-64 years experienced psychological financial hardship, defined as worry about large medical bills, in 2011 and 2016, Donatus U. Ekwueme, PhD, and his associates reported in the Morbidity and Mortality Weekly Report.
Cancer survivors spend 60% more out of pocket than those with no cancer history: $1,000 a year from 2011 to 2016, compared with $622 for adults without a history of cancer. Spending was lowest among younger people (18-39 years) and increased with age, but the prevalence of both material and psychological hardships was highest in the middle age group (40-49 years) and lowest in the oldest group (50-64 years), they said.
Women had higher out-of-pocket costs than men, although the difference was smaller for those with cancer ($1,023 vs. $976) than for those without ($721 vs. $519). Material and psychological hardships were both more common among women, said Dr. Ekwueme of the CDC’s National Center for Chronic Disease Prevention and Health Promotion, Atlanta, and his associates.
Mean out-of-pocket spending was much higher for cancer survivors with private health insurance ($1,114) than for survivors with public insurance ($471), but material hardship was much more prevalent among those with public insurance (33.1% vs. 21.9%). Rates of psychological hardship, however, were much closer: 35.9% for those with public insurance and 32.5% for those with private insurance, the investigators said.
“The number of Americans with a history of cancer is projected to increase in the next decade, and the economic burden associated with living with a cancer diagnosis will likely increase as well,” they wrote, and interventions such as “systematic screening for financial hardship at cancer diagnosis and throughout the cancer care trajectory [are needed] to minimize financial hardship for cancer survivors.”
The analysis was based on data for 123,771 adults aged 18-64 years from the Medical Expenditure Panel Survey. Out-of-pocket costs were calculated using data from 2011 to 2016, with all costs adjusted to 2016 dollars, but the hardship calculations involved data from only 2011 and 2016.
SOURCE: Ekwueme DU et al. MMWR 2019 Jun 7;68(22):494-9.
FROM MMWR
A large employer ‘frames’ the Medicare-for-all debate
EASTON, PA. — Walk into a big-box retailer such as Walmart or Michaels and you’re likely to see MCS Industries’ picture frames, decorative mirrors, or kitschy wall décor.
Adjacent to a dairy farm a few miles west of downtown Easton, MCS is the nation’s largest maker of such household products. But MCS doesn’t actually make anything here anymore. It has moved its manufacturing operations to Mexico and China, with the last manufacturing jobs departing this city along the Delaware River in 2005. MCS now has about 175 U.S. employees and 600 people overseas.
“We were going to lose the business because we were no longer competitive,” CEO Richard Master explained. And one of the biggest impediments to keeping labor costs in line, he said, has been the increasing expense of health coverage in the United States.
Today, he’s at the vanguard of a small but growing group of business executives who are lining up to support a Medicare-for-all national health program.
In February, Master stood with Rep. Pramila Jayapal (D-Wash.) outside the Capitol after she introduced her Medicare-for-all bill. “This bill removes an albatross from the neck of American business, puts more money in consumer products and will boost our economy,” he said.
As health costs continue to grow, straining employer budgets and slowing wage growth, others in the business community are beginning to take the option more seriously.
While the influential U.S. Chamber of Commerce and other large business lobbying groups strongly oppose increased government involvement in health care, the resolve of many in the business community – especially among smaller firms – may be shifting.
“There is growing momentum among employers supporting single-payer,” said Dan Geiger, codirector of the Business Alliance for a Healthy California, which has sought to generate business support for a universal health care program in California. About 300 mostly small employers have signed on.
“Businesses are really angry about the system, and there is a lot of frustration with its rising costs and dysfunction,” he said.
Mr. Geiger acknowledged the effort still lacks support from any Fortune 500 company CEOs. He said large businesses are hesitant to get involved in this political debate and many don’t want to lose the ability to attract workers with generous health benefits. “There is also a lingering distrust of the government, and they think they can offer coverage better than the government,” he said.
In addition, some in the business community are hesitant to sign on to Medicare-for-all with many details missing, such as how much it would increase taxes, said Ellen Kelsay, chief strategy officer for the National Business Group on Health, a leading business group focused on health benefits.
Democrats propel the debate
For decades, a government-run health plan was considered too radical an idea for serious consideration. But Medicare-for-all has been garnering more political support in recent months, especially after a progressive wave helped Democrats take control of the House this year. Several 2020 Democratic presidential candidates, including Sens. Bernie Sanders and Elizabeth Warren, strongly back it.
The labor unions and consumer groups that have long endorsed a single-payer health system hope that the embrace of it by employers such as Mr. Master marks another turning point for the movement.
Supporters of the concept say the health system overall would see savings from a coordinated effort to bring down prices and the elimination of many administrative costs or insurance company profits.
“It’s critical for our success to engage employers, particularly because our current system is hurting employers almost as much as it is patients,” said Melinda St. Louis, campaign director of Medicare-for-all at Public Citizen, a consumer-rights group based in Washington.
Mr. Master, a former Washington lawyer, worked on Democratic Sen. George McGovern’s presidential campaign before returning to Pennsylvania in 1973 to take over his father’s company, which made rigid paper boxes. In 1980, he founded MCS, which pioneered the popular front-loading picture frame and steamless fog-free mirrors for bathrooms. The company has grown into a $250 million corporation.
Mr. Master frequently travels to Washington and around the country to talk with business leaders as he seeks to build political support for a single-payer health system.
In the past 4 years, he has produced several documentary videos on the topic. In 2018, he formed the Business Initiative for Health Policy, a nonprofit group of business leaders, economists, and health policy experts trying to explain the financial benefits of a single-payer system.
Dan Wolf, CEO of Cape Air, a Hyannis, Mass.–based regional airline that employs 800 people calls himself “a free-market guy.” But he also supports Medicare-for-all. He said Mr. Master helps turn the political argument over single-payer into a practical one.
“It’s about good business sense and about caring for his employees and their well-being,” he said, adding that employers should no longer be straddled with the cost and complexity of health care.
“It makes no more sense for an airline to understand health policy for the bulk of its workers than for a health facility to have to supply all the air transportation for its employees,” he said.
Employers also are an important voice in the debate because 156 million Americans get employer-paid health care, making it by far the single-largest form of coverage.
Mr. Master said his company has tried various methods to control costs with little success, including high deductibles, narrow networks of providers and wellness plans that emphasize preventive medicine.
Insurers who are supposed to negotiate lower rates from hospitals and doctors have failed, he added, and too many premium dollars go to covering administrative costs. Only by having the federal government set rates can the United States control costs of drugs, hospitals, and other health services, he said.
“Insurance companies are not watching the store and don’t have incentives to hold down costs in the current system,” he said.
Glad the boss is trying to make a difference
What’s left of MCS in Pennsylvania is a spacious corporate office building housing administrative staff, designers, and a giant distribution center piled high with carton boxes from floor to ceiling.
MCS pays an average of $1,260 per month for each employee’s health care, up from $716 in 2009, the company said. In recent years, the company has reduced out-of-pocket costs for employees by covering most of their deductibles.
Medicare-for-all would require several new taxes to raise money, but Mr. Master said such a plan would mean savings for his company and employees.
MCS employees largely support Mr. Master’s attempt to fix the health system even if they are not all on board with a Medicare-for-all approach, according to interviews with several workers in Easton.
“I think it’s a good idea,” said Faith Wildrick, a shipper at MCS who has worked for the company 26 years. “If the other countries are doing it and it is working for them, why can’t it work for us?”
Ms. Wildrick said that even with insurance her family struggles with health costs as her husband, Bill, a former MCS employee, deals with liver disease and needs many diagnostic tests and prescription medications. Their annual deductible has swung from $4,000 several years ago to $500 this year as the company has worked to lower employees’ out-of-pocket costs.
“I’m really glad someone is fighting for this and trying to make a difference,” said Ms. Wildrick.
Jessica Ehrhardt, the human resources manager at MCS, said the effort to reduce employees’ out-of-pocket health costs means the company must pay higher health costs. That results in less money for salary increases and other benefits, she added.
Asked about Medicare-for-all, Ms. Ehrhardt said, “It’s a drastic solution, but something needs to happen.”
For too long, Mr. Master said, the push for a single-payer health system has been about ideology.
“The movement has been about making health care a human right and that we have a right to universal health care,” he said. “What I am saying is this is prudent for our economy and am trying to make the business and economic case.”
Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente.
EASTON, PA. — Walk into a big-box retailer such as Walmart or Michaels and you’re likely to see MCS Industries’ picture frames, decorative mirrors, or kitschy wall décor.
Adjacent to a dairy farm a few miles west of downtown Easton, MCS is the nation’s largest maker of such household products. But MCS doesn’t actually make anything here anymore. It has moved its manufacturing operations to Mexico and China, with the last manufacturing jobs departing this city along the Delaware River in 2005. MCS now has about 175 U.S. employees and 600 people overseas.
“We were going to lose the business because we were no longer competitive,” CEO Richard Master explained. And one of the biggest impediments to keeping labor costs in line, he said, has been the increasing expense of health coverage in the United States.
Today, he’s at the vanguard of a small but growing group of business executives who are lining up to support a Medicare-for-all national health program.
In February, Master stood with Rep. Pramila Jayapal (D-Wash.) outside the Capitol after she introduced her Medicare-for-all bill. “This bill removes an albatross from the neck of American business, puts more money in consumer products and will boost our economy,” he said.
As health costs continue to grow, straining employer budgets and slowing wage growth, others in the business community are beginning to take the option more seriously.
While the influential U.S. Chamber of Commerce and other large business lobbying groups strongly oppose increased government involvement in health care, the resolve of many in the business community – especially among smaller firms – may be shifting.
“There is growing momentum among employers supporting single-payer,” said Dan Geiger, codirector of the Business Alliance for a Healthy California, which has sought to generate business support for a universal health care program in California. About 300 mostly small employers have signed on.
“Businesses are really angry about the system, and there is a lot of frustration with its rising costs and dysfunction,” he said.
Mr. Geiger acknowledged the effort still lacks support from any Fortune 500 company CEOs. He said large businesses are hesitant to get involved in this political debate and many don’t want to lose the ability to attract workers with generous health benefits. “There is also a lingering distrust of the government, and they think they can offer coverage better than the government,” he said.
In addition, some in the business community are hesitant to sign on to Medicare-for-all with many details missing, such as how much it would increase taxes, said Ellen Kelsay, chief strategy officer for the National Business Group on Health, a leading business group focused on health benefits.
Democrats propel the debate
For decades, a government-run health plan was considered too radical an idea for serious consideration. But Medicare-for-all has been garnering more political support in recent months, especially after a progressive wave helped Democrats take control of the House this year. Several 2020 Democratic presidential candidates, including Sens. Bernie Sanders and Elizabeth Warren, strongly back it.
The labor unions and consumer groups that have long endorsed a single-payer health system hope that the embrace of it by employers such as Mr. Master marks another turning point for the movement.
Supporters of the concept say the health system overall would see savings from a coordinated effort to bring down prices and the elimination of many administrative costs or insurance company profits.
“It’s critical for our success to engage employers, particularly because our current system is hurting employers almost as much as it is patients,” said Melinda St. Louis, campaign director of Medicare-for-all at Public Citizen, a consumer-rights group based in Washington.
Mr. Master, a former Washington lawyer, worked on Democratic Sen. George McGovern’s presidential campaign before returning to Pennsylvania in 1973 to take over his father’s company, which made rigid paper boxes. In 1980, he founded MCS, which pioneered the popular front-loading picture frame and steamless fog-free mirrors for bathrooms. The company has grown into a $250 million corporation.
Mr. Master frequently travels to Washington and around the country to talk with business leaders as he seeks to build political support for a single-payer health system.
In the past 4 years, he has produced several documentary videos on the topic. In 2018, he formed the Business Initiative for Health Policy, a nonprofit group of business leaders, economists, and health policy experts trying to explain the financial benefits of a single-payer system.
Dan Wolf, CEO of Cape Air, a Hyannis, Mass.–based regional airline that employs 800 people calls himself “a free-market guy.” But he also supports Medicare-for-all. He said Mr. Master helps turn the political argument over single-payer into a practical one.
“It’s about good business sense and about caring for his employees and their well-being,” he said, adding that employers should no longer be straddled with the cost and complexity of health care.
“It makes no more sense for an airline to understand health policy for the bulk of its workers than for a health facility to have to supply all the air transportation for its employees,” he said.
Employers also are an important voice in the debate because 156 million Americans get employer-paid health care, making it by far the single-largest form of coverage.
Mr. Master said his company has tried various methods to control costs with little success, including high deductibles, narrow networks of providers and wellness plans that emphasize preventive medicine.
Insurers who are supposed to negotiate lower rates from hospitals and doctors have failed, he added, and too many premium dollars go to covering administrative costs. Only by having the federal government set rates can the United States control costs of drugs, hospitals, and other health services, he said.
“Insurance companies are not watching the store and don’t have incentives to hold down costs in the current system,” he said.
Glad the boss is trying to make a difference
What’s left of MCS in Pennsylvania is a spacious corporate office building housing administrative staff, designers, and a giant distribution center piled high with carton boxes from floor to ceiling.
MCS pays an average of $1,260 per month for each employee’s health care, up from $716 in 2009, the company said. In recent years, the company has reduced out-of-pocket costs for employees by covering most of their deductibles.
Medicare-for-all would require several new taxes to raise money, but Mr. Master said such a plan would mean savings for his company and employees.
MCS employees largely support Mr. Master’s attempt to fix the health system even if they are not all on board with a Medicare-for-all approach, according to interviews with several workers in Easton.
“I think it’s a good idea,” said Faith Wildrick, a shipper at MCS who has worked for the company 26 years. “If the other countries are doing it and it is working for them, why can’t it work for us?”
Ms. Wildrick said that even with insurance her family struggles with health costs as her husband, Bill, a former MCS employee, deals with liver disease and needs many diagnostic tests and prescription medications. Their annual deductible has swung from $4,000 several years ago to $500 this year as the company has worked to lower employees’ out-of-pocket costs.
“I’m really glad someone is fighting for this and trying to make a difference,” said Ms. Wildrick.
Jessica Ehrhardt, the human resources manager at MCS, said the effort to reduce employees’ out-of-pocket health costs means the company must pay higher health costs. That results in less money for salary increases and other benefits, she added.
Asked about Medicare-for-all, Ms. Ehrhardt said, “It’s a drastic solution, but something needs to happen.”
For too long, Mr. Master said, the push for a single-payer health system has been about ideology.
“The movement has been about making health care a human right and that we have a right to universal health care,” he said. “What I am saying is this is prudent for our economy and am trying to make the business and economic case.”
Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente.
EASTON, PA. — Walk into a big-box retailer such as Walmart or Michaels and you’re likely to see MCS Industries’ picture frames, decorative mirrors, or kitschy wall décor.
Adjacent to a dairy farm a few miles west of downtown Easton, MCS is the nation’s largest maker of such household products. But MCS doesn’t actually make anything here anymore. It has moved its manufacturing operations to Mexico and China, with the last manufacturing jobs departing this city along the Delaware River in 2005. MCS now has about 175 U.S. employees and 600 people overseas.
“We were going to lose the business because we were no longer competitive,” CEO Richard Master explained. And one of the biggest impediments to keeping labor costs in line, he said, has been the increasing expense of health coverage in the United States.
Today, he’s at the vanguard of a small but growing group of business executives who are lining up to support a Medicare-for-all national health program.
In February, Master stood with Rep. Pramila Jayapal (D-Wash.) outside the Capitol after she introduced her Medicare-for-all bill. “This bill removes an albatross from the neck of American business, puts more money in consumer products and will boost our economy,” he said.
As health costs continue to grow, straining employer budgets and slowing wage growth, others in the business community are beginning to take the option more seriously.
While the influential U.S. Chamber of Commerce and other large business lobbying groups strongly oppose increased government involvement in health care, the resolve of many in the business community – especially among smaller firms – may be shifting.
“There is growing momentum among employers supporting single-payer,” said Dan Geiger, codirector of the Business Alliance for a Healthy California, which has sought to generate business support for a universal health care program in California. About 300 mostly small employers have signed on.
“Businesses are really angry about the system, and there is a lot of frustration with its rising costs and dysfunction,” he said.
Mr. Geiger acknowledged the effort still lacks support from any Fortune 500 company CEOs. He said large businesses are hesitant to get involved in this political debate and many don’t want to lose the ability to attract workers with generous health benefits. “There is also a lingering distrust of the government, and they think they can offer coverage better than the government,” he said.
In addition, some in the business community are hesitant to sign on to Medicare-for-all with many details missing, such as how much it would increase taxes, said Ellen Kelsay, chief strategy officer for the National Business Group on Health, a leading business group focused on health benefits.
Democrats propel the debate
For decades, a government-run health plan was considered too radical an idea for serious consideration. But Medicare-for-all has been garnering more political support in recent months, especially after a progressive wave helped Democrats take control of the House this year. Several 2020 Democratic presidential candidates, including Sens. Bernie Sanders and Elizabeth Warren, strongly back it.
The labor unions and consumer groups that have long endorsed a single-payer health system hope that the embrace of it by employers such as Mr. Master marks another turning point for the movement.
Supporters of the concept say the health system overall would see savings from a coordinated effort to bring down prices and the elimination of many administrative costs or insurance company profits.
“It’s critical for our success to engage employers, particularly because our current system is hurting employers almost as much as it is patients,” said Melinda St. Louis, campaign director of Medicare-for-all at Public Citizen, a consumer-rights group based in Washington.
Mr. Master, a former Washington lawyer, worked on Democratic Sen. George McGovern’s presidential campaign before returning to Pennsylvania in 1973 to take over his father’s company, which made rigid paper boxes. In 1980, he founded MCS, which pioneered the popular front-loading picture frame and steamless fog-free mirrors for bathrooms. The company has grown into a $250 million corporation.
Mr. Master frequently travels to Washington and around the country to talk with business leaders as he seeks to build political support for a single-payer health system.
In the past 4 years, he has produced several documentary videos on the topic. In 2018, he formed the Business Initiative for Health Policy, a nonprofit group of business leaders, economists, and health policy experts trying to explain the financial benefits of a single-payer system.
Dan Wolf, CEO of Cape Air, a Hyannis, Mass.–based regional airline that employs 800 people calls himself “a free-market guy.” But he also supports Medicare-for-all. He said Mr. Master helps turn the political argument over single-payer into a practical one.
“It’s about good business sense and about caring for his employees and their well-being,” he said, adding that employers should no longer be straddled with the cost and complexity of health care.
“It makes no more sense for an airline to understand health policy for the bulk of its workers than for a health facility to have to supply all the air transportation for its employees,” he said.
Employers also are an important voice in the debate because 156 million Americans get employer-paid health care, making it by far the single-largest form of coverage.
Mr. Master said his company has tried various methods to control costs with little success, including high deductibles, narrow networks of providers and wellness plans that emphasize preventive medicine.
Insurers who are supposed to negotiate lower rates from hospitals and doctors have failed, he added, and too many premium dollars go to covering administrative costs. Only by having the federal government set rates can the United States control costs of drugs, hospitals, and other health services, he said.
“Insurance companies are not watching the store and don’t have incentives to hold down costs in the current system,” he said.
Glad the boss is trying to make a difference
What’s left of MCS in Pennsylvania is a spacious corporate office building housing administrative staff, designers, and a giant distribution center piled high with carton boxes from floor to ceiling.
MCS pays an average of $1,260 per month for each employee’s health care, up from $716 in 2009, the company said. In recent years, the company has reduced out-of-pocket costs for employees by covering most of their deductibles.
Medicare-for-all would require several new taxes to raise money, but Mr. Master said such a plan would mean savings for his company and employees.
MCS employees largely support Mr. Master’s attempt to fix the health system even if they are not all on board with a Medicare-for-all approach, according to interviews with several workers in Easton.
“I think it’s a good idea,” said Faith Wildrick, a shipper at MCS who has worked for the company 26 years. “If the other countries are doing it and it is working for them, why can’t it work for us?”
Ms. Wildrick said that even with insurance her family struggles with health costs as her husband, Bill, a former MCS employee, deals with liver disease and needs many diagnostic tests and prescription medications. Their annual deductible has swung from $4,000 several years ago to $500 this year as the company has worked to lower employees’ out-of-pocket costs.
“I’m really glad someone is fighting for this and trying to make a difference,” said Ms. Wildrick.
Jessica Ehrhardt, the human resources manager at MCS, said the effort to reduce employees’ out-of-pocket health costs means the company must pay higher health costs. That results in less money for salary increases and other benefits, she added.
Asked about Medicare-for-all, Ms. Ehrhardt said, “It’s a drastic solution, but something needs to happen.”
For too long, Mr. Master said, the push for a single-payer health system has been about ideology.
“The movement has been about making health care a human right and that we have a right to universal health care,” he said. “What I am saying is this is prudent for our economy and am trying to make the business and economic case.”
Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente.