User login
No link found between sleep position, pregnancy outcomes
such as stillbirth, small-for-gestational-age (SGA) newborns, and gestational hypertensive disorders. The finding, published in the October issue of Obstetrics & Gynecology, conflicts with previous retrospective case-control studies that suggested left-side sleeping may lead to reduced risk. The disagreement may be due to the prospective nature of the new study, which followed 8,706 women over 4 years.

Right-side or back sleeping had attracted suspicion because of its potential to compress uterine blood vessels and decrease uterine blood flow, and various public health campaigns urge pregnant women to sleep on their left side. Although case-control studies backed up those worries, those retrospective approaches can suffer from limitations including recall bias, in which grieving mothers overreport suspect sleeping behaviors, perhaps in search of an explanation for their loss.
Prospective analyses can counter some of those limitations. The researchers, led by Robert Silver, MD, of the University of Utah, Salt Lake City, conducted a secondary analysis of the nuMoM2b study, examining adverse pregnancy outcomes and risk factors. It was a multicenter observational cohort study of 8,706 nulliparous women with singleton gestations who completed two sleep questionnaires: one between 6 and 13 weeks of gestation, and one between 22 and 29 weeks.
Adverse outcomes occurred in 1,903 women, including 178 cases of both SGA and hypertensive disorders, 8 with SGA plus stillbirth, 3 with hypertensive disorders plus stillbirth, and 2 cases with all three complications.
The researchers found no association between any adverse outcomes and sleep position either at the first visit in early pregnancy (adjusted odds ratio, 1.00; 95% confidence interval, 0.89-1.14) or the third visit in midpregnancy (aOR, 0.99; 95% CI, 0.89-1.11). Propensity score matching to adjust non-left lateral positioning to the composite outcome also showed no association.
In midpregnancy, there was an association between non-left lateral sleeping and reduced risk of stillbirth (aOR, 0.27; 95% CI, 0.09-0.75). “This observation is likely spurious owing to small numbers of stillbirths, Dr. Silver and associates said.
A post hoc analysis indicated that the study was sufficiently powered to detect clinically meaningful risks; ORs of 1.2 for hypertensive disorders, 1.23 for SGA, 2.4 for stillbirth, and 1.2 for the composite outcome.
Let sleeping mothers lie
Pregnant women have enough on their minds. They shouldn’t have to worry about sleep position as well, according to Nathan Fox, MD, professor of obstetrics, gynecology, and reproductive science at Icahn School of Medicine at Mount Sinai, New York, and Emily Oster, PhD, economist at Brown University, Providence, R.I., who wrote an accompanying editorial.
It may seem harmless to direct women to sleep on their left side even if it has no benefit, but restricting a woman’s sleep options may leave her less well rested at a time when she is about to enter a period of sleep deprivation, as well as contribute to general discomfort, in their opinion.
Also, in the rare cases of a bad outcome, advice based on limited or poor quality evidence contributes to “devastating and unwarranted feelings of responsibility and guilt, and this harm to women already suffering from sadness and despair must not be minimized,” they said.
The study received funding from the Eunice Kennedy Shriver National Institute of Child Health and Human Development and grants from multiple universities. One author received research funding from Kyndermed through her institution. Another receives royalties from UpToDate.com. Dr. Fox and Dr. Oster have no relevant financial disclosures.
SOURCES: Silver RM et al. Obstet Gynecol. 2019. doi: 10.1097/AOG.0000000000003458; Fox N and Oster E. Obstet. Gynecol. 2019 Oct. doi: 10.1097/AOG.0000000000003466
such as stillbirth, small-for-gestational-age (SGA) newborns, and gestational hypertensive disorders. The finding, published in the October issue of Obstetrics & Gynecology, conflicts with previous retrospective case-control studies that suggested left-side sleeping may lead to reduced risk. The disagreement may be due to the prospective nature of the new study, which followed 8,706 women over 4 years.
Right-side or back sleeping had attracted suspicion because of its potential to compress uterine blood vessels and decrease uterine blood flow, and various public health campaigns urge pregnant women to sleep on their left side. Although case-control studies backed up those worries, those retrospective approaches can suffer from limitations including recall bias, in which grieving mothers overreport suspect sleeping behaviors, perhaps in search of an explanation for their loss.
Prospective analyses can counter some of those limitations. The researchers, led by Robert Silver, MD, of the University of Utah, Salt Lake City, conducted a secondary analysis of the nuMoM2b study, examining adverse pregnancy outcomes and risk factors. It was a multicenter observational cohort study of 8,706 nulliparous women with singleton gestations who completed two sleep questionnaires: one between 6 and 13 weeks of gestation, and one between 22 and 29 weeks.
Adverse outcomes occurred in 1,903 women, including 178 cases of both SGA and hypertensive disorders, 8 with SGA plus stillbirth, 3 with hypertensive disorders plus stillbirth, and 2 cases with all three complications.
The researchers found no association between any adverse outcomes and sleep position either at the first visit in early pregnancy (adjusted odds ratio, 1.00; 95% confidence interval, 0.89-1.14) or the third visit in midpregnancy (aOR, 0.99; 95% CI, 0.89-1.11). Propensity score matching to adjust non-left lateral positioning to the composite outcome also showed no association.
In midpregnancy, there was an association between non-left lateral sleeping and reduced risk of stillbirth (aOR, 0.27; 95% CI, 0.09-0.75). “This observation is likely spurious owing to small numbers of stillbirths, Dr. Silver and associates said.
A post hoc analysis indicated that the study was sufficiently powered to detect clinically meaningful risks; ORs of 1.2 for hypertensive disorders, 1.23 for SGA, 2.4 for stillbirth, and 1.2 for the composite outcome.
Let sleeping mothers lie
Pregnant women have enough on their minds. They shouldn’t have to worry about sleep position as well, according to Nathan Fox, MD, professor of obstetrics, gynecology, and reproductive science at Icahn School of Medicine at Mount Sinai, New York, and Emily Oster, PhD, economist at Brown University, Providence, R.I., who wrote an accompanying editorial.
It may seem harmless to direct women to sleep on their left side even if it has no benefit, but restricting a woman’s sleep options may leave her less well rested at a time when she is about to enter a period of sleep deprivation, as well as contribute to general discomfort, in their opinion.
Also, in the rare cases of a bad outcome, advice based on limited or poor quality evidence contributes to “devastating and unwarranted feelings of responsibility and guilt, and this harm to women already suffering from sadness and despair must not be minimized,” they said.
The study received funding from the Eunice Kennedy Shriver National Institute of Child Health and Human Development and grants from multiple universities. One author received research funding from Kyndermed through her institution. Another receives royalties from UpToDate.com. Dr. Fox and Dr. Oster have no relevant financial disclosures.
SOURCES: Silver RM et al. Obstet Gynecol. 2019. doi: 10.1097/AOG.0000000000003458; Fox N and Oster E. Obstet. Gynecol. 2019 Oct. doi: 10.1097/AOG.0000000000003466
such as stillbirth, small-for-gestational-age (SGA) newborns, and gestational hypertensive disorders. The finding, published in the October issue of Obstetrics & Gynecology, conflicts with previous retrospective case-control studies that suggested left-side sleeping may lead to reduced risk. The disagreement may be due to the prospective nature of the new study, which followed 8,706 women over 4 years.
Right-side or back sleeping had attracted suspicion because of its potential to compress uterine blood vessels and decrease uterine blood flow, and various public health campaigns urge pregnant women to sleep on their left side. Although case-control studies backed up those worries, those retrospective approaches can suffer from limitations including recall bias, in which grieving mothers overreport suspect sleeping behaviors, perhaps in search of an explanation for their loss.
Prospective analyses can counter some of those limitations. The researchers, led by Robert Silver, MD, of the University of Utah, Salt Lake City, conducted a secondary analysis of the nuMoM2b study, examining adverse pregnancy outcomes and risk factors. It was a multicenter observational cohort study of 8,706 nulliparous women with singleton gestations who completed two sleep questionnaires: one between 6 and 13 weeks of gestation, and one between 22 and 29 weeks.
Adverse outcomes occurred in 1,903 women, including 178 cases of both SGA and hypertensive disorders, 8 with SGA plus stillbirth, 3 with hypertensive disorders plus stillbirth, and 2 cases with all three complications.
The researchers found no association between any adverse outcomes and sleep position either at the first visit in early pregnancy (adjusted odds ratio, 1.00; 95% confidence interval, 0.89-1.14) or the third visit in midpregnancy (aOR, 0.99; 95% CI, 0.89-1.11). Propensity score matching to adjust non-left lateral positioning to the composite outcome also showed no association.
In midpregnancy, there was an association between non-left lateral sleeping and reduced risk of stillbirth (aOR, 0.27; 95% CI, 0.09-0.75). “This observation is likely spurious owing to small numbers of stillbirths, Dr. Silver and associates said.
A post hoc analysis indicated that the study was sufficiently powered to detect clinically meaningful risks; ORs of 1.2 for hypertensive disorders, 1.23 for SGA, 2.4 for stillbirth, and 1.2 for the composite outcome.
Let sleeping mothers lie
Pregnant women have enough on their minds. They shouldn’t have to worry about sleep position as well, according to Nathan Fox, MD, professor of obstetrics, gynecology, and reproductive science at Icahn School of Medicine at Mount Sinai, New York, and Emily Oster, PhD, economist at Brown University, Providence, R.I., who wrote an accompanying editorial.
It may seem harmless to direct women to sleep on their left side even if it has no benefit, but restricting a woman’s sleep options may leave her less well rested at a time when she is about to enter a period of sleep deprivation, as well as contribute to general discomfort, in their opinion.
Also, in the rare cases of a bad outcome, advice based on limited or poor quality evidence contributes to “devastating and unwarranted feelings of responsibility and guilt, and this harm to women already suffering from sadness and despair must not be minimized,” they said.
The study received funding from the Eunice Kennedy Shriver National Institute of Child Health and Human Development and grants from multiple universities. One author received research funding from Kyndermed through her institution. Another receives royalties from UpToDate.com. Dr. Fox and Dr. Oster have no relevant financial disclosures.
SOURCES: Silver RM et al. Obstet Gynecol. 2019. doi: 10.1097/AOG.0000000000003458; Fox N and Oster E. Obstet. Gynecol. 2019 Oct. doi: 10.1097/AOG.0000000000003466
FROM OBSTETRICS & GYNECOLOGY
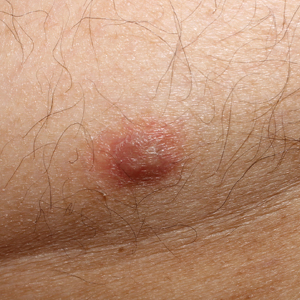
Staging PET/CT better defines extent of mantle cell lymphoma
Use of staging PET/CT better defines the extent of mantle cell lymphoma (MCL) in certain compartments, adding important information for treatment decisions, a cohort study suggests. However, patient selection and evaluation criteria are important for its utility.
“A correct and early identification of initial disease could be crucial because it could affect patient management and therapeutic choice,” Domenico Albano, MD, a nuclear medicine physician at the University of Brescia (Italy) and Spedali Civili Brescia, and colleagues wrote in Clinical Lymphoma, Myeloma, & Leukemia.
Using retrospective data from two centers and 122 patients with MCL, the investigators compared the utility of staging 18F-fluorodeoxyglucose (18F-FDG) PET/CT with that of other modalities used for detecting disease in specific compartments. They also assessed its impact on patient management.
The study results showed that, with the exception of a single patient having bone marrow involvement, all patients had positive PET/CT results, with detection of at least one hypermetabolic lesion.
For assessing nodal involvement, compared with CT alone, PET/CT detected additional lesions in 21% of patients. Splenic lesions were detected by PET/CT in 49% of patients and by CT alone in 47%.
For assessing bone marrow involvement, compared with bone marrow biopsy, PET/CT had a sensitivity of 52%, a specificity of 98%, a positive predictive value of 97%, a negative predictive value of 65%, and an overall accuracy of 74%.
For gastrointestinal involvement, compared with endoscopy, PET/CT had a sensitivity of 64% (78% after excluding diabetic patients taking metformin), a specificity of 91% (92%), a positive predictive value of 69% (72%), a negative predictive value of 90% (94%), and an overall accuracy of 85% (89%).
Ultimately, relative to CT alone, PET/CT altered stage and management in 19% of patients. Specifically, this imaging led to up-staging in 17% of patients, prompting clinicians to switch to more-aggressive chemotherapy, and down-staging in 2% of patients, prompting clinicians to skip unnecessary invasive therapies.
“We demonstrated that 18F-FDG pathologic uptake in MCL occurred in almost all patients, with excellent detection rate in nodal and splenic disease – indeed, better than CT,” the researchers wrote. “When we analyzed bone marrow and gastrointestinal involvement, the selection of patients excluding all conditions potentially affecting organ uptake and the use of specific criteria for the evaluation of these organs seemed to be crucial. Within these caveats, PET/CT showed good specificity for bone marrow and gastrointestinal evaluation.”
Study funding was not disclosed. Dr. Albano reported having no relevant conflicts of interest.
SOURCE: Albano D et al. Clin Lymphoma Myeloma Leuk. 2019 Aug;19(8):e457-64.
Use of staging PET/CT better defines the extent of mantle cell lymphoma (MCL) in certain compartments, adding important information for treatment decisions, a cohort study suggests. However, patient selection and evaluation criteria are important for its utility.
“A correct and early identification of initial disease could be crucial because it could affect patient management and therapeutic choice,” Domenico Albano, MD, a nuclear medicine physician at the University of Brescia (Italy) and Spedali Civili Brescia, and colleagues wrote in Clinical Lymphoma, Myeloma, & Leukemia.
Using retrospective data from two centers and 122 patients with MCL, the investigators compared the utility of staging 18F-fluorodeoxyglucose (18F-FDG) PET/CT with that of other modalities used for detecting disease in specific compartments. They also assessed its impact on patient management.
The study results showed that, with the exception of a single patient having bone marrow involvement, all patients had positive PET/CT results, with detection of at least one hypermetabolic lesion.
For assessing nodal involvement, compared with CT alone, PET/CT detected additional lesions in 21% of patients. Splenic lesions were detected by PET/CT in 49% of patients and by CT alone in 47%.
For assessing bone marrow involvement, compared with bone marrow biopsy, PET/CT had a sensitivity of 52%, a specificity of 98%, a positive predictive value of 97%, a negative predictive value of 65%, and an overall accuracy of 74%.
For gastrointestinal involvement, compared with endoscopy, PET/CT had a sensitivity of 64% (78% after excluding diabetic patients taking metformin), a specificity of 91% (92%), a positive predictive value of 69% (72%), a negative predictive value of 90% (94%), and an overall accuracy of 85% (89%).
Ultimately, relative to CT alone, PET/CT altered stage and management in 19% of patients. Specifically, this imaging led to up-staging in 17% of patients, prompting clinicians to switch to more-aggressive chemotherapy, and down-staging in 2% of patients, prompting clinicians to skip unnecessary invasive therapies.
“We demonstrated that 18F-FDG pathologic uptake in MCL occurred in almost all patients, with excellent detection rate in nodal and splenic disease – indeed, better than CT,” the researchers wrote. “When we analyzed bone marrow and gastrointestinal involvement, the selection of patients excluding all conditions potentially affecting organ uptake and the use of specific criteria for the evaluation of these organs seemed to be crucial. Within these caveats, PET/CT showed good specificity for bone marrow and gastrointestinal evaluation.”
Study funding was not disclosed. Dr. Albano reported having no relevant conflicts of interest.
SOURCE: Albano D et al. Clin Lymphoma Myeloma Leuk. 2019 Aug;19(8):e457-64.
Use of staging PET/CT better defines the extent of mantle cell lymphoma (MCL) in certain compartments, adding important information for treatment decisions, a cohort study suggests. However, patient selection and evaluation criteria are important for its utility.
“A correct and early identification of initial disease could be crucial because it could affect patient management and therapeutic choice,” Domenico Albano, MD, a nuclear medicine physician at the University of Brescia (Italy) and Spedali Civili Brescia, and colleagues wrote in Clinical Lymphoma, Myeloma, & Leukemia.
Using retrospective data from two centers and 122 patients with MCL, the investigators compared the utility of staging 18F-fluorodeoxyglucose (18F-FDG) PET/CT with that of other modalities used for detecting disease in specific compartments. They also assessed its impact on patient management.
The study results showed that, with the exception of a single patient having bone marrow involvement, all patients had positive PET/CT results, with detection of at least one hypermetabolic lesion.
For assessing nodal involvement, compared with CT alone, PET/CT detected additional lesions in 21% of patients. Splenic lesions were detected by PET/CT in 49% of patients and by CT alone in 47%.
For assessing bone marrow involvement, compared with bone marrow biopsy, PET/CT had a sensitivity of 52%, a specificity of 98%, a positive predictive value of 97%, a negative predictive value of 65%, and an overall accuracy of 74%.
For gastrointestinal involvement, compared with endoscopy, PET/CT had a sensitivity of 64% (78% after excluding diabetic patients taking metformin), a specificity of 91% (92%), a positive predictive value of 69% (72%), a negative predictive value of 90% (94%), and an overall accuracy of 85% (89%).
Ultimately, relative to CT alone, PET/CT altered stage and management in 19% of patients. Specifically, this imaging led to up-staging in 17% of patients, prompting clinicians to switch to more-aggressive chemotherapy, and down-staging in 2% of patients, prompting clinicians to skip unnecessary invasive therapies.
“We demonstrated that 18F-FDG pathologic uptake in MCL occurred in almost all patients, with excellent detection rate in nodal and splenic disease – indeed, better than CT,” the researchers wrote. “When we analyzed bone marrow and gastrointestinal involvement, the selection of patients excluding all conditions potentially affecting organ uptake and the use of specific criteria for the evaluation of these organs seemed to be crucial. Within these caveats, PET/CT showed good specificity for bone marrow and gastrointestinal evaluation.”
Study funding was not disclosed. Dr. Albano reported having no relevant conflicts of interest.
SOURCE: Albano D et al. Clin Lymphoma Myeloma Leuk. 2019 Aug;19(8):e457-64.
FROM CLINICAL LYMPHOMA, MYELOMA, & LEUKEMIA
Baby beverage 101: New recommendations on what young children should drink
entirely. A coalition of health and nutrition organizations offered all this and more in a new set of sometimes surprising recommendations about beverage consumption in children from birth to age 5 years.

The recommendations “are based on the best available evidence, combined with sound expert judgment, and provide consistent messages that can be used by a variety of stakeholders to improve the beverage intake patterns of young children,” the report authors wrote. “It is imperative to capitalize on early childhood as a critical window of opportunity during which dietary patterns are both impressionable and capable of setting the stage for lifelong eating behaviors.”
The Academy of Nutrition and Dietetics, the American Academy of Pediatric Dentistry, the American Academy of Pediatrics, and the American Heart Association released their recommendations titled “Healthy Beverage Consumption in Early Childhood” in a report published Sept. 18.
The organizations convened expert panels and analyzed research to develop consensus beverage recommendations. Here are some highlights from the report, the writing of which was led by Megan Lott, MPH, RDN, of Healthy Eating Research and Duke Global Health Institute at Duke University, Durham, N.C.
- Breast milk or formula. This is what babies should be drinking for the first year of life.
- Plain drinking water. None is needed in the first 6 months of life. Introduce 0.5-1.0 cups/day over the next 6 months in cups and during meals when solid food is introduced. Then 1-4 cups/day are recommended from age 1-3 years, then 1.5-5 cups by age 4-5 years.
- Plain, pasteurized milk. None during the first year, then 2-3 cups a day of whole milk at age 12-24 months; if weight gain is excessive or family history is positive for obesity, dyslipidemia, or other cardiovascular disease, the pediatrician may recommend skim or lowfat milk at 12-24 months. Then provide up to 2 cups (age 2-3 years) then 2.5 cups (age 4-5 years) per day of skim /fat-free milk or low fat/1% milk
- 100% juice. None until 12 months. Then it can be provided, although whole fruit is preferred. No more than 0.5 cup per day until age 4-5 years, when no more than 0.5-0.75 cup per day is recommended.
- Plant-based milks and non-dairy beverages. None from age 0-1 year. And at ages 1-5 years, they only should be consumed when medically indicated, such as when a child has an allergy or per a preferred diet such as a vegan one. The panel didn’t recommend them as full replacements for dairy milk from age 12-24 months, and it noted that “consumption of these beverages as a full replacement for dairy milk should be undertaken in consultation with a health care provider so that adequate intake of key nutrients commonly obtained from dairy milk can be considered in dietary planning.”
- Other beverages. Flavored milk (like chocolate milk), “toddler milk,” sugar-sweetened drinks (including soft drinks, flavored water, and various fruit drinks), low-calorie sweetened drinks, and caffeinated drinks are not recommended at any age from 0-5 years. Toddler milk products “offer no unique nutritional value beyond what a nutritionally adequate diet provides and may contribute added sugars to the diet and undermine sustained breastfeeding,” the report authors said.

Lillian M. Beard, MD, said in an interview, “Pediatricians and other child health providers have major roles of influence in parents’ choices of foods and beverages for their infants and young children.
“Following these first-ever consensus recommendations on healthy drinks will alter family grocery shopping patterns, and not only improve the overall health of children under age 5 years, but also may improve the health outcomes of everyone in the household.” Dr. Beard is a clinical professor of pediatrics at George Washington University, Washington. She was not one of the report authors and was asked to comment on the report.
The report was supported by the Healthy Eating Research, a national program of the Robert Wood Johnson Foundation.
SOURCE: Lott M et al. Healthy Beverage Consumption in Early Childhood: Recommendations from Key National Health and Nutrition Organizations. Technical Scientific Report. (Durham, NC: Healthy Eating Research, 2019).
entirely. A coalition of health and nutrition organizations offered all this and more in a new set of sometimes surprising recommendations about beverage consumption in children from birth to age 5 years.
The recommendations “are based on the best available evidence, combined with sound expert judgment, and provide consistent messages that can be used by a variety of stakeholders to improve the beverage intake patterns of young children,” the report authors wrote. “It is imperative to capitalize on early childhood as a critical window of opportunity during which dietary patterns are both impressionable and capable of setting the stage for lifelong eating behaviors.”
The Academy of Nutrition and Dietetics, the American Academy of Pediatric Dentistry, the American Academy of Pediatrics, and the American Heart Association released their recommendations titled “Healthy Beverage Consumption in Early Childhood” in a report published Sept. 18.
The organizations convened expert panels and analyzed research to develop consensus beverage recommendations. Here are some highlights from the report, the writing of which was led by Megan Lott, MPH, RDN, of Healthy Eating Research and Duke Global Health Institute at Duke University, Durham, N.C.
- Breast milk or formula. This is what babies should be drinking for the first year of life.
- Plain drinking water. None is needed in the first 6 months of life. Introduce 0.5-1.0 cups/day over the next 6 months in cups and during meals when solid food is introduced. Then 1-4 cups/day are recommended from age 1-3 years, then 1.5-5 cups by age 4-5 years.
- Plain, pasteurized milk. None during the first year, then 2-3 cups a day of whole milk at age 12-24 months; if weight gain is excessive or family history is positive for obesity, dyslipidemia, or other cardiovascular disease, the pediatrician may recommend skim or lowfat milk at 12-24 months. Then provide up to 2 cups (age 2-3 years) then 2.5 cups (age 4-5 years) per day of skim /fat-free milk or low fat/1% milk
- 100% juice. None until 12 months. Then it can be provided, although whole fruit is preferred. No more than 0.5 cup per day until age 4-5 years, when no more than 0.5-0.75 cup per day is recommended.
- Plant-based milks and non-dairy beverages. None from age 0-1 year. And at ages 1-5 years, they only should be consumed when medically indicated, such as when a child has an allergy or per a preferred diet such as a vegan one. The panel didn’t recommend them as full replacements for dairy milk from age 12-24 months, and it noted that “consumption of these beverages as a full replacement for dairy milk should be undertaken in consultation with a health care provider so that adequate intake of key nutrients commonly obtained from dairy milk can be considered in dietary planning.”
- Other beverages. Flavored milk (like chocolate milk), “toddler milk,” sugar-sweetened drinks (including soft drinks, flavored water, and various fruit drinks), low-calorie sweetened drinks, and caffeinated drinks are not recommended at any age from 0-5 years. Toddler milk products “offer no unique nutritional value beyond what a nutritionally adequate diet provides and may contribute added sugars to the diet and undermine sustained breastfeeding,” the report authors said.
Lillian M. Beard, MD, said in an interview, “Pediatricians and other child health providers have major roles of influence in parents’ choices of foods and beverages for their infants and young children.
“Following these first-ever consensus recommendations on healthy drinks will alter family grocery shopping patterns, and not only improve the overall health of children under age 5 years, but also may improve the health outcomes of everyone in the household.” Dr. Beard is a clinical professor of pediatrics at George Washington University, Washington. She was not one of the report authors and was asked to comment on the report.
The report was supported by the Healthy Eating Research, a national program of the Robert Wood Johnson Foundation.
SOURCE: Lott M et al. Healthy Beverage Consumption in Early Childhood: Recommendations from Key National Health and Nutrition Organizations. Technical Scientific Report. (Durham, NC: Healthy Eating Research, 2019).
entirely. A coalition of health and nutrition organizations offered all this and more in a new set of sometimes surprising recommendations about beverage consumption in children from birth to age 5 years.
The recommendations “are based on the best available evidence, combined with sound expert judgment, and provide consistent messages that can be used by a variety of stakeholders to improve the beverage intake patterns of young children,” the report authors wrote. “It is imperative to capitalize on early childhood as a critical window of opportunity during which dietary patterns are both impressionable and capable of setting the stage for lifelong eating behaviors.”
The Academy of Nutrition and Dietetics, the American Academy of Pediatric Dentistry, the American Academy of Pediatrics, and the American Heart Association released their recommendations titled “Healthy Beverage Consumption in Early Childhood” in a report published Sept. 18.
The organizations convened expert panels and analyzed research to develop consensus beverage recommendations. Here are some highlights from the report, the writing of which was led by Megan Lott, MPH, RDN, of Healthy Eating Research and Duke Global Health Institute at Duke University, Durham, N.C.
- Breast milk or formula. This is what babies should be drinking for the first year of life.
- Plain drinking water. None is needed in the first 6 months of life. Introduce 0.5-1.0 cups/day over the next 6 months in cups and during meals when solid food is introduced. Then 1-4 cups/day are recommended from age 1-3 years, then 1.5-5 cups by age 4-5 years.
- Plain, pasteurized milk. None during the first year, then 2-3 cups a day of whole milk at age 12-24 months; if weight gain is excessive or family history is positive for obesity, dyslipidemia, or other cardiovascular disease, the pediatrician may recommend skim or lowfat milk at 12-24 months. Then provide up to 2 cups (age 2-3 years) then 2.5 cups (age 4-5 years) per day of skim /fat-free milk or low fat/1% milk
- 100% juice. None until 12 months. Then it can be provided, although whole fruit is preferred. No more than 0.5 cup per day until age 4-5 years, when no more than 0.5-0.75 cup per day is recommended.
- Plant-based milks and non-dairy beverages. None from age 0-1 year. And at ages 1-5 years, they only should be consumed when medically indicated, such as when a child has an allergy or per a preferred diet such as a vegan one. The panel didn’t recommend them as full replacements for dairy milk from age 12-24 months, and it noted that “consumption of these beverages as a full replacement for dairy milk should be undertaken in consultation with a health care provider so that adequate intake of key nutrients commonly obtained from dairy milk can be considered in dietary planning.”
- Other beverages. Flavored milk (like chocolate milk), “toddler milk,” sugar-sweetened drinks (including soft drinks, flavored water, and various fruit drinks), low-calorie sweetened drinks, and caffeinated drinks are not recommended at any age from 0-5 years. Toddler milk products “offer no unique nutritional value beyond what a nutritionally adequate diet provides and may contribute added sugars to the diet and undermine sustained breastfeeding,” the report authors said.
Lillian M. Beard, MD, said in an interview, “Pediatricians and other child health providers have major roles of influence in parents’ choices of foods and beverages for their infants and young children.
“Following these first-ever consensus recommendations on healthy drinks will alter family grocery shopping patterns, and not only improve the overall health of children under age 5 years, but also may improve the health outcomes of everyone in the household.” Dr. Beard is a clinical professor of pediatrics at George Washington University, Washington. She was not one of the report authors and was asked to comment on the report.
The report was supported by the Healthy Eating Research, a national program of the Robert Wood Johnson Foundation.
SOURCE: Lott M et al. Healthy Beverage Consumption in Early Childhood: Recommendations from Key National Health and Nutrition Organizations. Technical Scientific Report. (Durham, NC: Healthy Eating Research, 2019).

Secondary prevention of osteoporotic fractures lacking
The report by independent actuarial firm Milliman examined the economic and clinical burden of new osteoporotic fractures in 2015 in the Medicare fee-for-service population, with data from a large medical claims database.
More than 10 million adults aged 50 years and older in the United States are thought to have osteoporosis, and 43.9% of adults are affected by low bone mass.
This report found that about 1.4 million Medicare fee-for-service beneficiaries experienced more than 1.6 million osteoporotic fractures in that year, which if extrapolated to include Medicare Advantage beneficiaries would increase to a total of 2.3 million fractures in 2 million individuals.
The most common types of fractures were of the spine (23%) and hip (17%), although the authors noted that the spinal fracture figure did not account for potential underdiagnosis of vertebral fractures.
Women had a 79% higher rate of osteoporotic fractures than that of men, and one-third of people who experienced at least one osteoporotic fracture were aged 74-85 years.
Dane Hansen and colleagues from Milliman from drew particular attention to the lack of secondary prevention in people who had experienced a first osteoporotic fracture. They estimated that 15% of those who had a new osteoporotic fracture experienced one or more subsequent fractures within 12 months, yet only 9% of women received a bone mineral density test within 6 months to evaluate them for osteoporosis.
Overall, 21% of individuals who had a new osteoporotic fracture underwent bone mineral density testing during the fracture episode.
The authors pointed out that their analysis wasn’t able to look at pharmaceutical treatment, and so did not present “a full picture of the overall rate of BMD [bone mineral density] testing and appropriate treatment after a fracture for surviving patients.”
Nearly one in five Medicare beneficiaries experienced at least one new pressure ulcer during the fracture episode, and beneficiaries with osteoporotic fracture were two times more likely than were other Medicare beneficiaries to experience pressure ulcers. “This is significant because research has found that pressure ulcers are clinically difficult and expensive to manage,” the authors wrote. They also saw that nearly 20% of Medicare beneficiaries who experienced an osteoporotic fracture died within 12 months, with the highest mortality (30%) seen in those with hip fracture.
Osteoporotic fractures presented a significant cost burden, with 45% of beneficiaries having at least one acute inpatient hospital stay within 30 days of having a new osteoporotic fracture. The hospitalization rate was as high as 92% for individuals with hip fracture, while 11% of those with wrist fractures were hospitalized within 7 days of the fracture.
The annual allowed medical costs in the 12 months after a new fracture were more than twice the costs of the 12-month period before the fracture in the same individual, and each new fracture was associated with an incremental annual medical cost greater than $21,800.
“An osteoporotic fracture is a sentinel event that should trigger appropriate clinical attention directed to reducing the risk of future subsequent fractures,” the authors said. “Therefore, the months following an osteoporotic fracture, in which the risk of a subsequent fracture is high, provide an important opportunity to identify and treat osteoporosis and to perform other interventions, such as patient education and care coordination, in order to reduce the individual’s risk of a subsequent fracture.”
The report estimated that preventing 5% of subsequent osteoporotic fractures could have saved the Medicare program $310 million just in the 2-3 years after a new fracture, while preventing 20% of subsequent fractures could have saved $1,230 million. These figures included the cost of the additional bone mineral density testing, but did not account for the increased costs of treatment or fracture prevention.
“In future analysis, it will be important to net the total cost of the intervention and additional pharmaceutical treatment for osteoporosis against Medicare savings from avoided subsequent fractures to comprehensively measure the savings from secondary fracture prevention initiatives.”
SOURCE: Milliman Research Report, “Medicare Report of Osteoporotic Fractures,” August 2019.
The report by independent actuarial firm Milliman examined the economic and clinical burden of new osteoporotic fractures in 2015 in the Medicare fee-for-service population, with data from a large medical claims database.
More than 10 million adults aged 50 years and older in the United States are thought to have osteoporosis, and 43.9% of adults are affected by low bone mass.
This report found that about 1.4 million Medicare fee-for-service beneficiaries experienced more than 1.6 million osteoporotic fractures in that year, which if extrapolated to include Medicare Advantage beneficiaries would increase to a total of 2.3 million fractures in 2 million individuals.
The most common types of fractures were of the spine (23%) and hip (17%), although the authors noted that the spinal fracture figure did not account for potential underdiagnosis of vertebral fractures.
Women had a 79% higher rate of osteoporotic fractures than that of men, and one-third of people who experienced at least one osteoporotic fracture were aged 74-85 years.
Dane Hansen and colleagues from Milliman from drew particular attention to the lack of secondary prevention in people who had experienced a first osteoporotic fracture. They estimated that 15% of those who had a new osteoporotic fracture experienced one or more subsequent fractures within 12 months, yet only 9% of women received a bone mineral density test within 6 months to evaluate them for osteoporosis.
Overall, 21% of individuals who had a new osteoporotic fracture underwent bone mineral density testing during the fracture episode.
The authors pointed out that their analysis wasn’t able to look at pharmaceutical treatment, and so did not present “a full picture of the overall rate of BMD [bone mineral density] testing and appropriate treatment after a fracture for surviving patients.”
Nearly one in five Medicare beneficiaries experienced at least one new pressure ulcer during the fracture episode, and beneficiaries with osteoporotic fracture were two times more likely than were other Medicare beneficiaries to experience pressure ulcers. “This is significant because research has found that pressure ulcers are clinically difficult and expensive to manage,” the authors wrote. They also saw that nearly 20% of Medicare beneficiaries who experienced an osteoporotic fracture died within 12 months, with the highest mortality (30%) seen in those with hip fracture.
Osteoporotic fractures presented a significant cost burden, with 45% of beneficiaries having at least one acute inpatient hospital stay within 30 days of having a new osteoporotic fracture. The hospitalization rate was as high as 92% for individuals with hip fracture, while 11% of those with wrist fractures were hospitalized within 7 days of the fracture.
The annual allowed medical costs in the 12 months after a new fracture were more than twice the costs of the 12-month period before the fracture in the same individual, and each new fracture was associated with an incremental annual medical cost greater than $21,800.
“An osteoporotic fracture is a sentinel event that should trigger appropriate clinical attention directed to reducing the risk of future subsequent fractures,” the authors said. “Therefore, the months following an osteoporotic fracture, in which the risk of a subsequent fracture is high, provide an important opportunity to identify and treat osteoporosis and to perform other interventions, such as patient education and care coordination, in order to reduce the individual’s risk of a subsequent fracture.”
The report estimated that preventing 5% of subsequent osteoporotic fractures could have saved the Medicare program $310 million just in the 2-3 years after a new fracture, while preventing 20% of subsequent fractures could have saved $1,230 million. These figures included the cost of the additional bone mineral density testing, but did not account for the increased costs of treatment or fracture prevention.
“In future analysis, it will be important to net the total cost of the intervention and additional pharmaceutical treatment for osteoporosis against Medicare savings from avoided subsequent fractures to comprehensively measure the savings from secondary fracture prevention initiatives.”
SOURCE: Milliman Research Report, “Medicare Report of Osteoporotic Fractures,” August 2019.
The report by independent actuarial firm Milliman examined the economic and clinical burden of new osteoporotic fractures in 2015 in the Medicare fee-for-service population, with data from a large medical claims database.
More than 10 million adults aged 50 years and older in the United States are thought to have osteoporosis, and 43.9% of adults are affected by low bone mass.
This report found that about 1.4 million Medicare fee-for-service beneficiaries experienced more than 1.6 million osteoporotic fractures in that year, which if extrapolated to include Medicare Advantage beneficiaries would increase to a total of 2.3 million fractures in 2 million individuals.
The most common types of fractures were of the spine (23%) and hip (17%), although the authors noted that the spinal fracture figure did not account for potential underdiagnosis of vertebral fractures.
Women had a 79% higher rate of osteoporotic fractures than that of men, and one-third of people who experienced at least one osteoporotic fracture were aged 74-85 years.
Dane Hansen and colleagues from Milliman from drew particular attention to the lack of secondary prevention in people who had experienced a first osteoporotic fracture. They estimated that 15% of those who had a new osteoporotic fracture experienced one or more subsequent fractures within 12 months, yet only 9% of women received a bone mineral density test within 6 months to evaluate them for osteoporosis.
Overall, 21% of individuals who had a new osteoporotic fracture underwent bone mineral density testing during the fracture episode.
The authors pointed out that their analysis wasn’t able to look at pharmaceutical treatment, and so did not present “a full picture of the overall rate of BMD [bone mineral density] testing and appropriate treatment after a fracture for surviving patients.”
Nearly one in five Medicare beneficiaries experienced at least one new pressure ulcer during the fracture episode, and beneficiaries with osteoporotic fracture were two times more likely than were other Medicare beneficiaries to experience pressure ulcers. “This is significant because research has found that pressure ulcers are clinically difficult and expensive to manage,” the authors wrote. They also saw that nearly 20% of Medicare beneficiaries who experienced an osteoporotic fracture died within 12 months, with the highest mortality (30%) seen in those with hip fracture.
Osteoporotic fractures presented a significant cost burden, with 45% of beneficiaries having at least one acute inpatient hospital stay within 30 days of having a new osteoporotic fracture. The hospitalization rate was as high as 92% for individuals with hip fracture, while 11% of those with wrist fractures were hospitalized within 7 days of the fracture.
The annual allowed medical costs in the 12 months after a new fracture were more than twice the costs of the 12-month period before the fracture in the same individual, and each new fracture was associated with an incremental annual medical cost greater than $21,800.
“An osteoporotic fracture is a sentinel event that should trigger appropriate clinical attention directed to reducing the risk of future subsequent fractures,” the authors said. “Therefore, the months following an osteoporotic fracture, in which the risk of a subsequent fracture is high, provide an important opportunity to identify and treat osteoporosis and to perform other interventions, such as patient education and care coordination, in order to reduce the individual’s risk of a subsequent fracture.”
The report estimated that preventing 5% of subsequent osteoporotic fractures could have saved the Medicare program $310 million just in the 2-3 years after a new fracture, while preventing 20% of subsequent fractures could have saved $1,230 million. These figures included the cost of the additional bone mineral density testing, but did not account for the increased costs of treatment or fracture prevention.
“In future analysis, it will be important to net the total cost of the intervention and additional pharmaceutical treatment for osteoporosis against Medicare savings from avoided subsequent fractures to comprehensively measure the savings from secondary fracture prevention initiatives.”
SOURCE: Milliman Research Report, “Medicare Report of Osteoporotic Fractures,” August 2019.
FROM THE NATIONAL OSTEOPOROSIS FOUNDATION
Increased levels of a bacterial strain may cause nonalcoholic fatty liver disease
A new study involving both human patients and mice has confirmed a long-believed association between nonalcoholic fatty liver disease (NAFLD) and an alteration in the gut microbiome that produces high levels of alcohol.
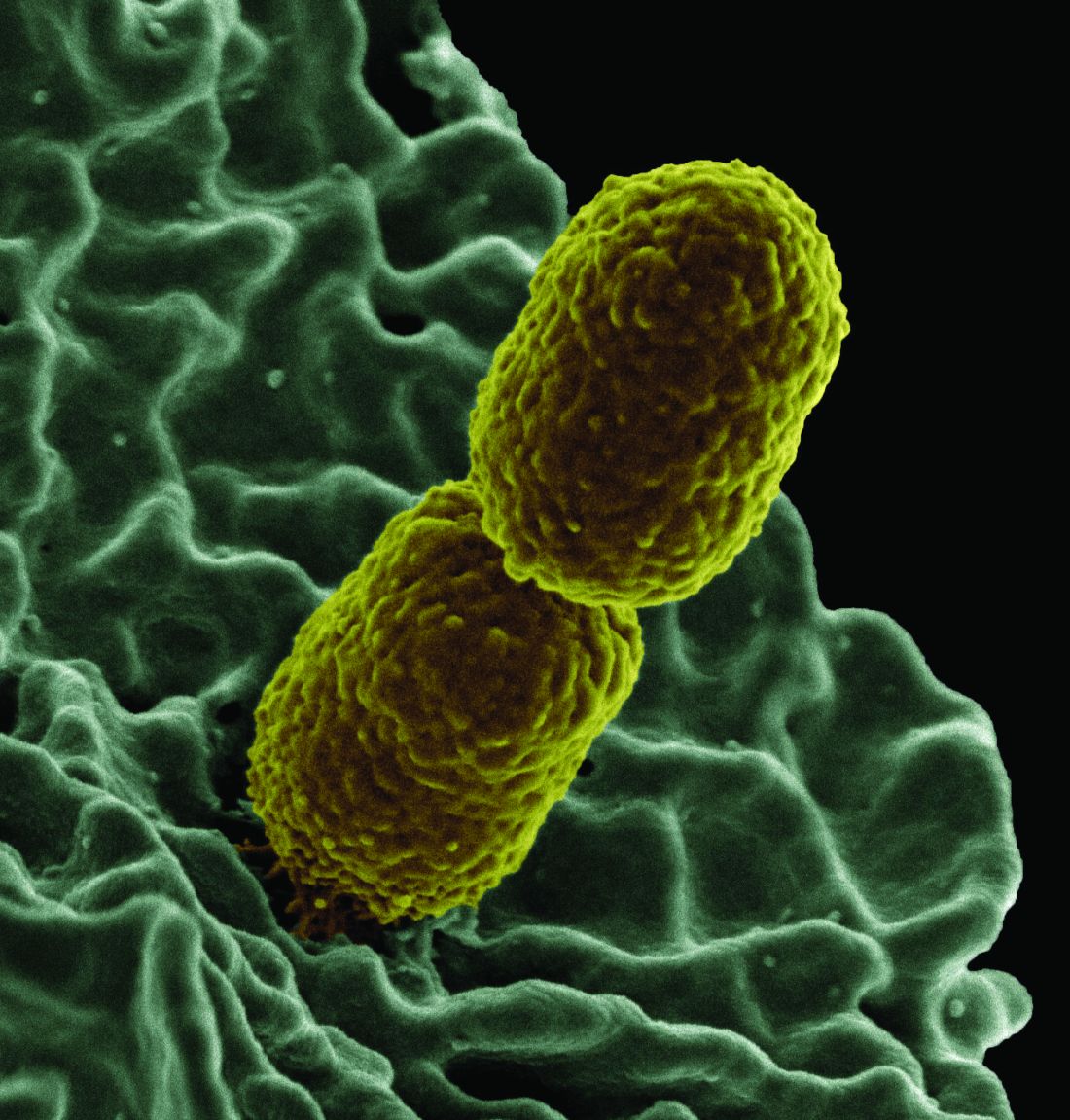
The study was initiated after the treatment of a rare case: a patient who presented with severe nonalcoholic steatohepatitis (NASH) plus auto-brewery syndrome. The patient had a very high blood alcohol concentration but an alcohol-free, high-carbohydrate diet. It was determined that strains of high alcohol-producing Klebsiella pneumoniae (HiAlc Kpn) rather than a fungal infection were the catalyst for the high blood alcohol level. As such, Jing Yuan of the Capital Institute of Pediatrics in Beijing and coauthors attempted to “connect these commensal HiAlc Kpn to the pathogenesis of hepatic damage” through this study, which was published in Cell Metabolism.
The researchers began by examining 43 patients with NAFLD and 48 healthy controls. Among the patients with NAFLD, 11 had nonalcoholic fatty liver and 32 had NASH. Specifically, they analyzed the presence and effects of HiAlc Kpn, determining that the abundance of Klebsiella pneumoniae was slightly higher in the feces of NAFLD patients, compared with healthy patients, but that their alcohol-producing ability in NAFLD patients was significantly stronger. Of the patients with NAFLD, 61% carried HiAlc and medium-alcohol-producing Kpn, compared with 6.25% of the controls.
Another phase of their study involved feeding specific pathogen-free mice either HiAlc Kpn, ethanol, or yeast extract peptone dextrose medium (pair-fed) for 4, 6, and 8 weeks. The mice that were fed HiAlc Kpn or ethanol showed clear microsteatosis and macrosteatosis in their livers at 4 and 8 weeks, compared with the pair-fed mice. In addition, the HiAlc-Kpn-fed and ethanol-fed mice had increased levels of aspartate transaminase and alanine transaminase in their serum and increased levels of triglycerides and thiobarbituric acid reactive substances in their liver. The results overall indicated that the HiAlc-Kpn-fed mice had developed hepatic steatosis.
An additional phase included the intestinal flora from a NASH patient with a specific Kpn strain being fed to germ-free mice. At the same time, two types of intestinal flora from mice with NAFLD were transplanted into healthy mice: one induced by two other specific Kpn strains and one in which those strains had been selectively eliminated. The results saw obvious steatosis in the mice who received the flora from either mice with NAFLD induced by Kpn or the NASH patient at 4 weeks and 8 weeks, respectively. The mice who received the flora win which Kpn had been eliminated saw no fat-related changes in the liver. “These results further suggest that HiAlc Kpn might be one of the major causes of NAFLD development,” the researchers wrote.
The authors acknowledged the study’s limitations, chiefly including the lack of a clinical cohort of individuals with auto brewery syndrome but without NAFLD that could be used as a control. However, they also noted a belief that “causality was shown by the transfer experiments of HiAlc Kpn” while adding that “the further analysis of the impact of ethanol in ABS [auto brewery syndrome] patients should be investigated.”
The study was funded by grants from the National Natural Science Foundation for Key Programs of China, the National Natural Science Foundation of China, Megaprojects of Science and Technology Research of China, and CAMS Innovation Fund for Medical Sciences. The authors reported no conflicts of interest.
SOURCE: Yuan J et al. Cell Metab. 2019 Sep 19. doi: 10.1016/j.cmet.2019.08.018.
A new study involving both human patients and mice has confirmed a long-believed association between nonalcoholic fatty liver disease (NAFLD) and an alteration in the gut microbiome that produces high levels of alcohol.
The study was initiated after the treatment of a rare case: a patient who presented with severe nonalcoholic steatohepatitis (NASH) plus auto-brewery syndrome. The patient had a very high blood alcohol concentration but an alcohol-free, high-carbohydrate diet. It was determined that strains of high alcohol-producing Klebsiella pneumoniae (HiAlc Kpn) rather than a fungal infection were the catalyst for the high blood alcohol level. As such, Jing Yuan of the Capital Institute of Pediatrics in Beijing and coauthors attempted to “connect these commensal HiAlc Kpn to the pathogenesis of hepatic damage” through this study, which was published in Cell Metabolism.
The researchers began by examining 43 patients with NAFLD and 48 healthy controls. Among the patients with NAFLD, 11 had nonalcoholic fatty liver and 32 had NASH. Specifically, they analyzed the presence and effects of HiAlc Kpn, determining that the abundance of Klebsiella pneumoniae was slightly higher in the feces of NAFLD patients, compared with healthy patients, but that their alcohol-producing ability in NAFLD patients was significantly stronger. Of the patients with NAFLD, 61% carried HiAlc and medium-alcohol-producing Kpn, compared with 6.25% of the controls.
Another phase of their study involved feeding specific pathogen-free mice either HiAlc Kpn, ethanol, or yeast extract peptone dextrose medium (pair-fed) for 4, 6, and 8 weeks. The mice that were fed HiAlc Kpn or ethanol showed clear microsteatosis and macrosteatosis in their livers at 4 and 8 weeks, compared with the pair-fed mice. In addition, the HiAlc-Kpn-fed and ethanol-fed mice had increased levels of aspartate transaminase and alanine transaminase in their serum and increased levels of triglycerides and thiobarbituric acid reactive substances in their liver. The results overall indicated that the HiAlc-Kpn-fed mice had developed hepatic steatosis.
An additional phase included the intestinal flora from a NASH patient with a specific Kpn strain being fed to germ-free mice. At the same time, two types of intestinal flora from mice with NAFLD were transplanted into healthy mice: one induced by two other specific Kpn strains and one in which those strains had been selectively eliminated. The results saw obvious steatosis in the mice who received the flora from either mice with NAFLD induced by Kpn or the NASH patient at 4 weeks and 8 weeks, respectively. The mice who received the flora win which Kpn had been eliminated saw no fat-related changes in the liver. “These results further suggest that HiAlc Kpn might be one of the major causes of NAFLD development,” the researchers wrote.
The authors acknowledged the study’s limitations, chiefly including the lack of a clinical cohort of individuals with auto brewery syndrome but without NAFLD that could be used as a control. However, they also noted a belief that “causality was shown by the transfer experiments of HiAlc Kpn” while adding that “the further analysis of the impact of ethanol in ABS [auto brewery syndrome] patients should be investigated.”
The study was funded by grants from the National Natural Science Foundation for Key Programs of China, the National Natural Science Foundation of China, Megaprojects of Science and Technology Research of China, and CAMS Innovation Fund for Medical Sciences. The authors reported no conflicts of interest.
SOURCE: Yuan J et al. Cell Metab. 2019 Sep 19. doi: 10.1016/j.cmet.2019.08.018.
A new study involving both human patients and mice has confirmed a long-believed association between nonalcoholic fatty liver disease (NAFLD) and an alteration in the gut microbiome that produces high levels of alcohol.
The study was initiated after the treatment of a rare case: a patient who presented with severe nonalcoholic steatohepatitis (NASH) plus auto-brewery syndrome. The patient had a very high blood alcohol concentration but an alcohol-free, high-carbohydrate diet. It was determined that strains of high alcohol-producing Klebsiella pneumoniae (HiAlc Kpn) rather than a fungal infection were the catalyst for the high blood alcohol level. As such, Jing Yuan of the Capital Institute of Pediatrics in Beijing and coauthors attempted to “connect these commensal HiAlc Kpn to the pathogenesis of hepatic damage” through this study, which was published in Cell Metabolism.
The researchers began by examining 43 patients with NAFLD and 48 healthy controls. Among the patients with NAFLD, 11 had nonalcoholic fatty liver and 32 had NASH. Specifically, they analyzed the presence and effects of HiAlc Kpn, determining that the abundance of Klebsiella pneumoniae was slightly higher in the feces of NAFLD patients, compared with healthy patients, but that their alcohol-producing ability in NAFLD patients was significantly stronger. Of the patients with NAFLD, 61% carried HiAlc and medium-alcohol-producing Kpn, compared with 6.25% of the controls.
Another phase of their study involved feeding specific pathogen-free mice either HiAlc Kpn, ethanol, or yeast extract peptone dextrose medium (pair-fed) for 4, 6, and 8 weeks. The mice that were fed HiAlc Kpn or ethanol showed clear microsteatosis and macrosteatosis in their livers at 4 and 8 weeks, compared with the pair-fed mice. In addition, the HiAlc-Kpn-fed and ethanol-fed mice had increased levels of aspartate transaminase and alanine transaminase in their serum and increased levels of triglycerides and thiobarbituric acid reactive substances in their liver. The results overall indicated that the HiAlc-Kpn-fed mice had developed hepatic steatosis.
An additional phase included the intestinal flora from a NASH patient with a specific Kpn strain being fed to germ-free mice. At the same time, two types of intestinal flora from mice with NAFLD were transplanted into healthy mice: one induced by two other specific Kpn strains and one in which those strains had been selectively eliminated. The results saw obvious steatosis in the mice who received the flora from either mice with NAFLD induced by Kpn or the NASH patient at 4 weeks and 8 weeks, respectively. The mice who received the flora win which Kpn had been eliminated saw no fat-related changes in the liver. “These results further suggest that HiAlc Kpn might be one of the major causes of NAFLD development,” the researchers wrote.
The authors acknowledged the study’s limitations, chiefly including the lack of a clinical cohort of individuals with auto brewery syndrome but without NAFLD that could be used as a control. However, they also noted a belief that “causality was shown by the transfer experiments of HiAlc Kpn” while adding that “the further analysis of the impact of ethanol in ABS [auto brewery syndrome] patients should be investigated.”
The study was funded by grants from the National Natural Science Foundation for Key Programs of China, the National Natural Science Foundation of China, Megaprojects of Science and Technology Research of China, and CAMS Innovation Fund for Medical Sciences. The authors reported no conflicts of interest.
SOURCE: Yuan J et al. Cell Metab. 2019 Sep 19. doi: 10.1016/j.cmet.2019.08.018.
FROM CELL METABOLISM
Key clinical point: Nonalcoholic fatty liver disease can be caused or exacerbated by excess levels of a high-alcohol-producing bacterial strain.
Major finding: 61% of patients with NAFLD carried high alcohol- and medium alcohol-producing Klebsiella pneumoniae (HiAlc Kpn), compared to 6.25% of healthy controls.
Study details: A multiphase study that included analysis of a 43-patient cohort with nonalcoholic fatty liver disease as well as experiments with mice and HiAlc Kpn.
Disclosures: The study was funded by grants from the National Natural Science Foundation for Key Programs of China, the National Natural Science Foundation of China, Megaprojects of Science and Technology Research of China, and CAMS Innovation Fund for Medical Sciences. The authors reported no conflicts of interest.
Source: Yuan J et al. Cell Metab. 2019 Sep 19. doi: 10.1016/j.cmet.2019.08.018.
Helping patients find balance between self and other
Cultural considerations require careful assessments on therapists’ part
This column is dedicated to the late Carl C. Bell, MD.

When we work with patients and their families from cultures that are not the culture in which we ourselves were raised, we think more deeply about this balance. In this column, I offer a simple but solid framework for this inquiry.
The first family therapist to crystallize the dialectic between the self and its relationship to others was Murray Bowen, MD. He believed that the differentiation of self from the family was the major task of human development. Dr. Bowen worked in a time when vilification of the “other” was common practice in individual psychotherapies and the goal of individual psychotherapy was the development of a healthy sense of self rather than repairing or developing relationships.
When faced with patients from cultures that are unfamiliar to us, we are less confident about how to assess the balance between self and other. In many cultures, marriages are based on social class and perceived social opportunities and are arranged by the respective families. If you come from a collectivist culture, where the focus is on the belief that the group is more important than the individual, the focus is more on self in relation to a group, belonging to a group, and participating in a group than self-striving. This is most evident in the role of women in many families (as well as in other organizations), in which women shoulder the responsibility for keeping families functional and together.
American culture is focused on serious self-striving. From kindergarten, children are expected to excel and to become the best self that they can be – regardless of the toll this takes on relationships. Self-expression and self-actualization frequently are considered the pinnacle of a life’s achievement. Relationships may take a backseat, often being transitory or utilitarian. This leads to switching relationships, peer groups, and friends – and a strong emphasis on cultivating work relationships.
Exploring Dr. Bowen’s theories
Dr. Bowen posited that the family relational pull affects individual development in a negative way. Despite this, his model is considered one of the most comprehensive explanations for the development of psychological problems from a systemic, relational, and multigenerational perspective.1 He identified the basic self (B-self), which strives for differentiation in contrast to the false/pseudo/relational self (R-self), which strives to meet group or family norms.
Dr. Bowen was the oldest of three and grew up in a small town in Tennessee. His father was the mayor of the town and owned several properties, including the funeral home. Following medical training, Dr. Bowen served during World War II. He accepted a fellowship in surgery at the Mayo Clinic in Rochester, Minn., but his wartime experiences resulted in a change of interest to psychiatry. Dr. Bowen trained at the Menninger Clinic in Topeka, Kan., and in 1954 became the first director of the family division at the National Institute of Mental Health. He and his colleagues studied the families of patients with schizophrenia. They described eight fundamental concepts that supported the important aspects of individual growth. When he moved to Georgetown University in Washington, he developed the Bowen Family Systems Theory.2
Dr. Bowen’s eight concepts
1. Nuclear Family Emotional Process
2. Differentiation of self
3. Triangles
4. Emotional cutoff
5. Family projection process
6. Multigenerational transmission process
7. Sibling position
8. Societal Emotional Process
According to Dr. Bowen, the B-self makes decisions on facts, principles, and intrinsic motivation and decides what they are willing to do/not willing to do based on their own internal ethics. On the other hand, the R-self goes along with everybody else, even when the person internally disagrees. He considered the R-self as wanting acceptance in relationship, possibly changing beliefs to find approval, and striving to be liked. Carmen Knudson-Martin, PhD,3 explored the relationship between the B-self and the R-self and suggested that they exist along two dimensions, both of which are important. My contention is that the R-self is undertheorized and deserves much more exploration.
Developmental psychologists and psychiatrists have focused on understanding the process of psychological maturation of the individual throughout life. However, there is little study of the development of a healthy relationship between self and other. We have, instead, gathered examples and descriptors of the pathological examples of the “other.” We can readily call out enmeshment, the manipulations of the borderline personality disordered, the cold withholding mother – to name the most vilified. What do we know about the healthy R-self?
Measuring the relational self
We have understood the R-self mostly through the study of pathological relationships. For example, pathological parenting has been shown to “result” in individual pathology and as a factor in the development of psychiatric illness. The measurement of the relationship between patient and family member/partner is aimed at elucidating pathology. The supreme example is emotional overinvolvement (EOI).
EOI is an integral part of the construct called expressed emotion and is often measured using the Camberwell Family Interview.4 High EOI has been identified routinely as predictive of worsening of psychiatric illness.5 However, exceptions are found (when you look for them)! In African American families, for example, high EOI is predictive of better outcomes in patients with schizophrenia.6 Jill M. Hooley, DPhil, also has identified that patients with borderline personality disorders do better in families with high EOI.7
A shorter equivalent research tool is the 5-minute speech sample (FMSS). The FMSS analyses 5 minutes of the speech of a parent/family member who is asked to describe the identified patient. EOI is identified by expressions of excessive worry or concern, self-sacrifice, or exaggerated praise. In a study of 223 child-mother dyads, 56.5% of which were Hispanic, use of the FMSS found high EOI predicted externalizing behaviors.8
More recently, psychiatry has sought to identify and measure positive factors, such as family warmth. In Puerto Rican children, high parental warmth was found to be protective against psychiatric disorders.9 In a study of Burmese migrant families from 20 communities in Thailand (513 caregivers and 479 patients with schizophrenia, aged 7-15 years), families were randomized to a waitlist or a 12-week family intervention that promoted warmth.10 The family intervention resulted in increased parental warmth and affection and increased family well-being.
Applying the theories to practice
An adolescent, Jan, does not speak when her mother is in the room. Jan has a small B-self, and her mother has a large B-self. Not only does Jan have to develop a strong B, but she also has to change how she is in relation – she has to change her R-self. For Jan, individual therapy supports the development of a stronger B-self. Working with the patient and her mother, the balance between both B-selves and the joined R-self can be reworked. In essence, the therapist encourages Jan to speak and helps the mother keep her own counsel. This is a situation in which the individual and family intervention are best implemented by the same therapist.
Systemic family therapy, a specific type of family intervention, focuses on how all the R-selves in a family work together as a unit called the family, or F-self. The F-self also has its own family history, as relationship patterns are transmitted and played out through families and play out through subsequent generations. A new type of family therapy called family constellation therapy (FCT) focuses on the F-self as a collection of ancestral selves. This resonates strongly with families who have experienced significant trauma, such as war and Holocaust survivors. FCT is popular in collectivist cultures, where there is a strong belief in the power and influence of ancestors and where the self is understood as an “assemblage of ancestral relationships that often creates problems in the present day.”11 Dr. Bowen recognized this multigenerational pattern as one of his eight fundamental principles.
The patients whom we see often have failing or fractured relationships. They might be stuck in dysfunctional transactional patterns with intimate partners, or they might fail to find a suitable intimate partner. We recognize relational dysfunction such as “codependency,” “symbiosis,” and “enmeshment.” We recognize too much distance, identifying family cutoffs. We still have a long way to go before clinical practice incorporates the importance of assessment and development of healthy relationships in a deep way. A typical question heard across all clinics: Is your partner/family supportive? Not much else is asked in regard to relationships, unless the answer is no. We have yet to develop a good set of inquiring questions that focus on the assessment of healthy relationships.
What can the therapist do to help the patient manage this continual dialectic? The therapist can ask the questions: How important is your B-self versus your R-self? What is the balance between your B-self and your R-self? What do you know about your family or F-self? Is your F-self important to you?
References
1. Nichols MP and Davis S. Family Therapy: Concepts & Methods, 8th ed. (Boston: Allyn & Bacon, 2008).
2. The Bowen Center for the Study of the Family.
3. Knudson-Martin C. Fam J. 1996 Jul 1. doi: 1066480796043002.
4. Leff J and Vaughn C. Expressed Emotion in Families. (New York: The Guilford Press, 1985).
5. Breitborde NJK et al. J Nerv Ment Dis. 2013 Oct;201(10):833-40.
6. Gurak K and de Mamani AW. Fam Process. 2017;56(2):476-86.
7. Hooley JM et al. J Clin Psychiatry. 2010 Aug;71(8):1017-24.
8. Khafi TY et al. J Fam Psychol. 2015 Aug;29(4):585-94.
9. Santesteban-Echarr et al. J Psychiatr Res. 2017 Apr;87:30-6.
10. Puffer ES et al. PLoS One. 2017 Mar 28;12(3):e0172611.
11. Pritzker SE and WL Duncan. Cult Med Psychiatry. 2019 Sep;43(3):468-95.
Dr. Heru is professor of psychiatry at the University of Colorado at Denver, Aurora. She is editor of Working With Families in Family Settings: A Multidisciplinary Guide for Psychiatrists and Other Health Professionals (New York: Routledge, 2013). She has no conflicts of interest.
Cultural considerations require careful assessments on therapists’ part
Cultural considerations require careful assessments on therapists’ part
This column is dedicated to the late Carl C. Bell, MD.
When we work with patients and their families from cultures that are not the culture in which we ourselves were raised, we think more deeply about this balance. In this column, I offer a simple but solid framework for this inquiry.
The first family therapist to crystallize the dialectic between the self and its relationship to others was Murray Bowen, MD. He believed that the differentiation of self from the family was the major task of human development. Dr. Bowen worked in a time when vilification of the “other” was common practice in individual psychotherapies and the goal of individual psychotherapy was the development of a healthy sense of self rather than repairing or developing relationships.
When faced with patients from cultures that are unfamiliar to us, we are less confident about how to assess the balance between self and other. In many cultures, marriages are based on social class and perceived social opportunities and are arranged by the respective families. If you come from a collectivist culture, where the focus is on the belief that the group is more important than the individual, the focus is more on self in relation to a group, belonging to a group, and participating in a group than self-striving. This is most evident in the role of women in many families (as well as in other organizations), in which women shoulder the responsibility for keeping families functional and together.
American culture is focused on serious self-striving. From kindergarten, children are expected to excel and to become the best self that they can be – regardless of the toll this takes on relationships. Self-expression and self-actualization frequently are considered the pinnacle of a life’s achievement. Relationships may take a backseat, often being transitory or utilitarian. This leads to switching relationships, peer groups, and friends – and a strong emphasis on cultivating work relationships.
Exploring Dr. Bowen’s theories
Dr. Bowen posited that the family relational pull affects individual development in a negative way. Despite this, his model is considered one of the most comprehensive explanations for the development of psychological problems from a systemic, relational, and multigenerational perspective.1 He identified the basic self (B-self), which strives for differentiation in contrast to the false/pseudo/relational self (R-self), which strives to meet group or family norms.
Dr. Bowen was the oldest of three and grew up in a small town in Tennessee. His father was the mayor of the town and owned several properties, including the funeral home. Following medical training, Dr. Bowen served during World War II. He accepted a fellowship in surgery at the Mayo Clinic in Rochester, Minn., but his wartime experiences resulted in a change of interest to psychiatry. Dr. Bowen trained at the Menninger Clinic in Topeka, Kan., and in 1954 became the first director of the family division at the National Institute of Mental Health. He and his colleagues studied the families of patients with schizophrenia. They described eight fundamental concepts that supported the important aspects of individual growth. When he moved to Georgetown University in Washington, he developed the Bowen Family Systems Theory.2
Dr. Bowen’s eight concepts
1. Nuclear Family Emotional Process
2. Differentiation of self
3. Triangles
4. Emotional cutoff
5. Family projection process
6. Multigenerational transmission process
7. Sibling position
8. Societal Emotional Process
According to Dr. Bowen, the B-self makes decisions on facts, principles, and intrinsic motivation and decides what they are willing to do/not willing to do based on their own internal ethics. On the other hand, the R-self goes along with everybody else, even when the person internally disagrees. He considered the R-self as wanting acceptance in relationship, possibly changing beliefs to find approval, and striving to be liked. Carmen Knudson-Martin, PhD,3 explored the relationship between the B-self and the R-self and suggested that they exist along two dimensions, both of which are important. My contention is that the R-self is undertheorized and deserves much more exploration.
Developmental psychologists and psychiatrists have focused on understanding the process of psychological maturation of the individual throughout life. However, there is little study of the development of a healthy relationship between self and other. We have, instead, gathered examples and descriptors of the pathological examples of the “other.” We can readily call out enmeshment, the manipulations of the borderline personality disordered, the cold withholding mother – to name the most vilified. What do we know about the healthy R-self?
Measuring the relational self
We have understood the R-self mostly through the study of pathological relationships. For example, pathological parenting has been shown to “result” in individual pathology and as a factor in the development of psychiatric illness. The measurement of the relationship between patient and family member/partner is aimed at elucidating pathology. The supreme example is emotional overinvolvement (EOI).
EOI is an integral part of the construct called expressed emotion and is often measured using the Camberwell Family Interview.4 High EOI has been identified routinely as predictive of worsening of psychiatric illness.5 However, exceptions are found (when you look for them)! In African American families, for example, high EOI is predictive of better outcomes in patients with schizophrenia.6 Jill M. Hooley, DPhil, also has identified that patients with borderline personality disorders do better in families with high EOI.7
A shorter equivalent research tool is the 5-minute speech sample (FMSS). The FMSS analyses 5 minutes of the speech of a parent/family member who is asked to describe the identified patient. EOI is identified by expressions of excessive worry or concern, self-sacrifice, or exaggerated praise. In a study of 223 child-mother dyads, 56.5% of which were Hispanic, use of the FMSS found high EOI predicted externalizing behaviors.8
More recently, psychiatry has sought to identify and measure positive factors, such as family warmth. In Puerto Rican children, high parental warmth was found to be protective against psychiatric disorders.9 In a study of Burmese migrant families from 20 communities in Thailand (513 caregivers and 479 patients with schizophrenia, aged 7-15 years), families were randomized to a waitlist or a 12-week family intervention that promoted warmth.10 The family intervention resulted in increased parental warmth and affection and increased family well-being.
Applying the theories to practice
An adolescent, Jan, does not speak when her mother is in the room. Jan has a small B-self, and her mother has a large B-self. Not only does Jan have to develop a strong B, but she also has to change how she is in relation – she has to change her R-self. For Jan, individual therapy supports the development of a stronger B-self. Working with the patient and her mother, the balance between both B-selves and the joined R-self can be reworked. In essence, the therapist encourages Jan to speak and helps the mother keep her own counsel. This is a situation in which the individual and family intervention are best implemented by the same therapist.
Systemic family therapy, a specific type of family intervention, focuses on how all the R-selves in a family work together as a unit called the family, or F-self. The F-self also has its own family history, as relationship patterns are transmitted and played out through families and play out through subsequent generations. A new type of family therapy called family constellation therapy (FCT) focuses on the F-self as a collection of ancestral selves. This resonates strongly with families who have experienced significant trauma, such as war and Holocaust survivors. FCT is popular in collectivist cultures, where there is a strong belief in the power and influence of ancestors and where the self is understood as an “assemblage of ancestral relationships that often creates problems in the present day.”11 Dr. Bowen recognized this multigenerational pattern as one of his eight fundamental principles.
The patients whom we see often have failing or fractured relationships. They might be stuck in dysfunctional transactional patterns with intimate partners, or they might fail to find a suitable intimate partner. We recognize relational dysfunction such as “codependency,” “symbiosis,” and “enmeshment.” We recognize too much distance, identifying family cutoffs. We still have a long way to go before clinical practice incorporates the importance of assessment and development of healthy relationships in a deep way. A typical question heard across all clinics: Is your partner/family supportive? Not much else is asked in regard to relationships, unless the answer is no. We have yet to develop a good set of inquiring questions that focus on the assessment of healthy relationships.
What can the therapist do to help the patient manage this continual dialectic? The therapist can ask the questions: How important is your B-self versus your R-self? What is the balance between your B-self and your R-self? What do you know about your family or F-self? Is your F-self important to you?
References
1. Nichols MP and Davis S. Family Therapy: Concepts & Methods, 8th ed. (Boston: Allyn & Bacon, 2008).
2. The Bowen Center for the Study of the Family.
3. Knudson-Martin C. Fam J. 1996 Jul 1. doi: 1066480796043002.
4. Leff J and Vaughn C. Expressed Emotion in Families. (New York: The Guilford Press, 1985).
5. Breitborde NJK et al. J Nerv Ment Dis. 2013 Oct;201(10):833-40.
6. Gurak K and de Mamani AW. Fam Process. 2017;56(2):476-86.
7. Hooley JM et al. J Clin Psychiatry. 2010 Aug;71(8):1017-24.
8. Khafi TY et al. J Fam Psychol. 2015 Aug;29(4):585-94.
9. Santesteban-Echarr et al. J Psychiatr Res. 2017 Apr;87:30-6.
10. Puffer ES et al. PLoS One. 2017 Mar 28;12(3):e0172611.
11. Pritzker SE and WL Duncan. Cult Med Psychiatry. 2019 Sep;43(3):468-95.
Dr. Heru is professor of psychiatry at the University of Colorado at Denver, Aurora. She is editor of Working With Families in Family Settings: A Multidisciplinary Guide for Psychiatrists and Other Health Professionals (New York: Routledge, 2013). She has no conflicts of interest.
This column is dedicated to the late Carl C. Bell, MD.
When we work with patients and their families from cultures that are not the culture in which we ourselves were raised, we think more deeply about this balance. In this column, I offer a simple but solid framework for this inquiry.
The first family therapist to crystallize the dialectic between the self and its relationship to others was Murray Bowen, MD. He believed that the differentiation of self from the family was the major task of human development. Dr. Bowen worked in a time when vilification of the “other” was common practice in individual psychotherapies and the goal of individual psychotherapy was the development of a healthy sense of self rather than repairing or developing relationships.
When faced with patients from cultures that are unfamiliar to us, we are less confident about how to assess the balance between self and other. In many cultures, marriages are based on social class and perceived social opportunities and are arranged by the respective families. If you come from a collectivist culture, where the focus is on the belief that the group is more important than the individual, the focus is more on self in relation to a group, belonging to a group, and participating in a group than self-striving. This is most evident in the role of women in many families (as well as in other organizations), in which women shoulder the responsibility for keeping families functional and together.
American culture is focused on serious self-striving. From kindergarten, children are expected to excel and to become the best self that they can be – regardless of the toll this takes on relationships. Self-expression and self-actualization frequently are considered the pinnacle of a life’s achievement. Relationships may take a backseat, often being transitory or utilitarian. This leads to switching relationships, peer groups, and friends – and a strong emphasis on cultivating work relationships.
Exploring Dr. Bowen’s theories
Dr. Bowen posited that the family relational pull affects individual development in a negative way. Despite this, his model is considered one of the most comprehensive explanations for the development of psychological problems from a systemic, relational, and multigenerational perspective.1 He identified the basic self (B-self), which strives for differentiation in contrast to the false/pseudo/relational self (R-self), which strives to meet group or family norms.
Dr. Bowen was the oldest of three and grew up in a small town in Tennessee. His father was the mayor of the town and owned several properties, including the funeral home. Following medical training, Dr. Bowen served during World War II. He accepted a fellowship in surgery at the Mayo Clinic in Rochester, Minn., but his wartime experiences resulted in a change of interest to psychiatry. Dr. Bowen trained at the Menninger Clinic in Topeka, Kan., and in 1954 became the first director of the family division at the National Institute of Mental Health. He and his colleagues studied the families of patients with schizophrenia. They described eight fundamental concepts that supported the important aspects of individual growth. When he moved to Georgetown University in Washington, he developed the Bowen Family Systems Theory.2
Dr. Bowen’s eight concepts
1. Nuclear Family Emotional Process
2. Differentiation of self
3. Triangles
4. Emotional cutoff
5. Family projection process
6. Multigenerational transmission process
7. Sibling position
8. Societal Emotional Process
According to Dr. Bowen, the B-self makes decisions on facts, principles, and intrinsic motivation and decides what they are willing to do/not willing to do based on their own internal ethics. On the other hand, the R-self goes along with everybody else, even when the person internally disagrees. He considered the R-self as wanting acceptance in relationship, possibly changing beliefs to find approval, and striving to be liked. Carmen Knudson-Martin, PhD,3 explored the relationship between the B-self and the R-self and suggested that they exist along two dimensions, both of which are important. My contention is that the R-self is undertheorized and deserves much more exploration.
Developmental psychologists and psychiatrists have focused on understanding the process of psychological maturation of the individual throughout life. However, there is little study of the development of a healthy relationship between self and other. We have, instead, gathered examples and descriptors of the pathological examples of the “other.” We can readily call out enmeshment, the manipulations of the borderline personality disordered, the cold withholding mother – to name the most vilified. What do we know about the healthy R-self?
Measuring the relational self
We have understood the R-self mostly through the study of pathological relationships. For example, pathological parenting has been shown to “result” in individual pathology and as a factor in the development of psychiatric illness. The measurement of the relationship between patient and family member/partner is aimed at elucidating pathology. The supreme example is emotional overinvolvement (EOI).
EOI is an integral part of the construct called expressed emotion and is often measured using the Camberwell Family Interview.4 High EOI has been identified routinely as predictive of worsening of psychiatric illness.5 However, exceptions are found (when you look for them)! In African American families, for example, high EOI is predictive of better outcomes in patients with schizophrenia.6 Jill M. Hooley, DPhil, also has identified that patients with borderline personality disorders do better in families with high EOI.7
A shorter equivalent research tool is the 5-minute speech sample (FMSS). The FMSS analyses 5 minutes of the speech of a parent/family member who is asked to describe the identified patient. EOI is identified by expressions of excessive worry or concern, self-sacrifice, or exaggerated praise. In a study of 223 child-mother dyads, 56.5% of which were Hispanic, use of the FMSS found high EOI predicted externalizing behaviors.8
More recently, psychiatry has sought to identify and measure positive factors, such as family warmth. In Puerto Rican children, high parental warmth was found to be protective against psychiatric disorders.9 In a study of Burmese migrant families from 20 communities in Thailand (513 caregivers and 479 patients with schizophrenia, aged 7-15 years), families were randomized to a waitlist or a 12-week family intervention that promoted warmth.10 The family intervention resulted in increased parental warmth and affection and increased family well-being.
Applying the theories to practice
An adolescent, Jan, does not speak when her mother is in the room. Jan has a small B-self, and her mother has a large B-self. Not only does Jan have to develop a strong B, but she also has to change how she is in relation – she has to change her R-self. For Jan, individual therapy supports the development of a stronger B-self. Working with the patient and her mother, the balance between both B-selves and the joined R-self can be reworked. In essence, the therapist encourages Jan to speak and helps the mother keep her own counsel. This is a situation in which the individual and family intervention are best implemented by the same therapist.
Systemic family therapy, a specific type of family intervention, focuses on how all the R-selves in a family work together as a unit called the family, or F-self. The F-self also has its own family history, as relationship patterns are transmitted and played out through families and play out through subsequent generations. A new type of family therapy called family constellation therapy (FCT) focuses on the F-self as a collection of ancestral selves. This resonates strongly with families who have experienced significant trauma, such as war and Holocaust survivors. FCT is popular in collectivist cultures, where there is a strong belief in the power and influence of ancestors and where the self is understood as an “assemblage of ancestral relationships that often creates problems in the present day.”11 Dr. Bowen recognized this multigenerational pattern as one of his eight fundamental principles.
The patients whom we see often have failing or fractured relationships. They might be stuck in dysfunctional transactional patterns with intimate partners, or they might fail to find a suitable intimate partner. We recognize relational dysfunction such as “codependency,” “symbiosis,” and “enmeshment.” We recognize too much distance, identifying family cutoffs. We still have a long way to go before clinical practice incorporates the importance of assessment and development of healthy relationships in a deep way. A typical question heard across all clinics: Is your partner/family supportive? Not much else is asked in regard to relationships, unless the answer is no. We have yet to develop a good set of inquiring questions that focus on the assessment of healthy relationships.
What can the therapist do to help the patient manage this continual dialectic? The therapist can ask the questions: How important is your B-self versus your R-self? What is the balance between your B-self and your R-self? What do you know about your family or F-self? Is your F-self important to you?
References
1. Nichols MP and Davis S. Family Therapy: Concepts & Methods, 8th ed. (Boston: Allyn & Bacon, 2008).
2. The Bowen Center for the Study of the Family.
3. Knudson-Martin C. Fam J. 1996 Jul 1. doi: 1066480796043002.
4. Leff J and Vaughn C. Expressed Emotion in Families. (New York: The Guilford Press, 1985).
5. Breitborde NJK et al. J Nerv Ment Dis. 2013 Oct;201(10):833-40.
6. Gurak K and de Mamani AW. Fam Process. 2017;56(2):476-86.
7. Hooley JM et al. J Clin Psychiatry. 2010 Aug;71(8):1017-24.
8. Khafi TY et al. J Fam Psychol. 2015 Aug;29(4):585-94.
9. Santesteban-Echarr et al. J Psychiatr Res. 2017 Apr;87:30-6.
10. Puffer ES et al. PLoS One. 2017 Mar 28;12(3):e0172611.
11. Pritzker SE and WL Duncan. Cult Med Psychiatry. 2019 Sep;43(3):468-95.
Dr. Heru is professor of psychiatry at the University of Colorado at Denver, Aurora. She is editor of Working With Families in Family Settings: A Multidisciplinary Guide for Psychiatrists and Other Health Professionals (New York: Routledge, 2013). She has no conflicts of interest.
Melflufen-dexamethasone active in patients with relapsed/refractory myeloma and EMD
BOSTON – Melflufen plus dexamethasone is active in patients with relapsed/refractory multiple myeloma, whether or not they have extramedullary disease (EMD), a phase 2 trial suggests.

In the HORIZON trial, melflufen-dexamethasone produced an overall response rate of 23% in patients with EMD and 27% in those without EMD.
Paul G. Richardson, MD, of Harvard Medical School and the Dana-Farber Cancer Institute, both in Boston, presented these results as a late-breaking abstract at the International Myeloma Workshop, held by the International Myeloma Society.
As of July 30, 2019, 136 patients had been treated on the HORIZON trial. The trial is enrolling patients with relapsed/refractory multiple myeloma refractory to pomalidomide, an anti-CD38 monoclonal antibody (mAb), or both. The patients must have received at least two prior lines of therapy, including a proteasome inhibitor (PI) and an immunomodulatory agent (IMiD).
Dr. Richardson presented results for 130 patients, 44 with EMD and 86 without it. The median age at baseline was 64 years in the EMD and non-EMD groups (overall range, 35-86 years). More than half of patients had high-risk cytogenetics (52% in the EMD group and 57% in the non-EMD group).
The median number of prior therapies was five in both the EMD and non-EMD groups. Most patients had received at least one prior transplant (73% in the EMD group and 69% in the non-EMD group). Most patients in both groups were refractory to an anti-CD38 mAb (93% EMD and 72% non-EMD); an IMiD and a PI (93% EMD and 90% non-EMD); an IMiD, a PI, and an anti-CD38 mAb (91% EMD and 63% non-EMD); and their last therapy (100% EMD and 95% non-EMD).
Dr. Richardson pointed out that the incidence of EMD in this trial is higher than has been reported previously. Of the 44 EMD patients, 26 had soft-tissue EMD, and 18 had bone-related EMD. Five patients had CNS involvement.
Another key finding, according to Dr. Richardson, was that EMD appeared to be associated with prior anti-CD38 therapy. Specifically, 40% of patients exposed to an anti-CD38 mAb had EMD, compared with 11% of patients who had not received an anti-CD38 mAb (P = .01).
“I don’t for a minute want to say that CD38-targeted therapy engenders extramedullary disease,” Dr. Richardson said. “I think what we can say, though, is that, once CD38 treatment fails a patient, extramedullary disease … is a very real challenge. Therefore, we need rationally targeted approaches, ideally in combination, to meet that challenge.”
The overall response rate was similar for EMD and non-EMD patients – 23% and 27%, respectively. The median duration of response was 3.4 months in the EMD patients and 4.4 months in the non-EMD group. The clinical benefit rate was 30% and 45%, respectively.
In the EMD group, the overall response rate was 19% in patients with soft-tissue EMD and 28% in bone-related EMD. None of the patients with CNS disease responded.
The median progression-free survival was 2.9 months for patients with EMD and 4.6 months for those without EMD. The median overall survival was 5.8 month and 11.6 months, respectively.
The median overall survival was 18.5 months in EMD responders and 17.2 months in non-EMD responders. The median overall survival was 5.1 months in EMD nonresponders and 8.5 months in non-EMD nonresponders.
In all, 54% of patients received subsequent therapy. There were no significant differences in outcomes between EMD and non-EMD patients.
The safety profiles were similar for EMD and non-EMD patients, Dr. Richardson said. Melflufen-dexamethasone was considered well tolerated overall, and there were no treatment-related deaths.
The most common treatment-emergent adverse events (grade 3 and 4, respectively) were thrombocytopenia (22% and 46%), neutropenia (32% and 35%), anemia (35% and 1%), white blood cell count decrease (10% and 7%), and pneumonia (7% and 1%).
This trial is sponsored by Oncopeptides. Dr. Richardson reported an advisory role and research funding from Oncopeptides.
SOURCE: Richardson PG et al. IMW 2019, Abstract OAB-086.
BOSTON – Melflufen plus dexamethasone is active in patients with relapsed/refractory multiple myeloma, whether or not they have extramedullary disease (EMD), a phase 2 trial suggests.
In the HORIZON trial, melflufen-dexamethasone produced an overall response rate of 23% in patients with EMD and 27% in those without EMD.
Paul G. Richardson, MD, of Harvard Medical School and the Dana-Farber Cancer Institute, both in Boston, presented these results as a late-breaking abstract at the International Myeloma Workshop, held by the International Myeloma Society.
As of July 30, 2019, 136 patients had been treated on the HORIZON trial. The trial is enrolling patients with relapsed/refractory multiple myeloma refractory to pomalidomide, an anti-CD38 monoclonal antibody (mAb), or both. The patients must have received at least two prior lines of therapy, including a proteasome inhibitor (PI) and an immunomodulatory agent (IMiD).
Dr. Richardson presented results for 130 patients, 44 with EMD and 86 without it. The median age at baseline was 64 years in the EMD and non-EMD groups (overall range, 35-86 years). More than half of patients had high-risk cytogenetics (52% in the EMD group and 57% in the non-EMD group).
The median number of prior therapies was five in both the EMD and non-EMD groups. Most patients had received at least one prior transplant (73% in the EMD group and 69% in the non-EMD group). Most patients in both groups were refractory to an anti-CD38 mAb (93% EMD and 72% non-EMD); an IMiD and a PI (93% EMD and 90% non-EMD); an IMiD, a PI, and an anti-CD38 mAb (91% EMD and 63% non-EMD); and their last therapy (100% EMD and 95% non-EMD).
Dr. Richardson pointed out that the incidence of EMD in this trial is higher than has been reported previously. Of the 44 EMD patients, 26 had soft-tissue EMD, and 18 had bone-related EMD. Five patients had CNS involvement.
Another key finding, according to Dr. Richardson, was that EMD appeared to be associated with prior anti-CD38 therapy. Specifically, 40% of patients exposed to an anti-CD38 mAb had EMD, compared with 11% of patients who had not received an anti-CD38 mAb (P = .01).
“I don’t for a minute want to say that CD38-targeted therapy engenders extramedullary disease,” Dr. Richardson said. “I think what we can say, though, is that, once CD38 treatment fails a patient, extramedullary disease … is a very real challenge. Therefore, we need rationally targeted approaches, ideally in combination, to meet that challenge.”
The overall response rate was similar for EMD and non-EMD patients – 23% and 27%, respectively. The median duration of response was 3.4 months in the EMD patients and 4.4 months in the non-EMD group. The clinical benefit rate was 30% and 45%, respectively.
In the EMD group, the overall response rate was 19% in patients with soft-tissue EMD and 28% in bone-related EMD. None of the patients with CNS disease responded.
The median progression-free survival was 2.9 months for patients with EMD and 4.6 months for those without EMD. The median overall survival was 5.8 month and 11.6 months, respectively.
The median overall survival was 18.5 months in EMD responders and 17.2 months in non-EMD responders. The median overall survival was 5.1 months in EMD nonresponders and 8.5 months in non-EMD nonresponders.
In all, 54% of patients received subsequent therapy. There were no significant differences in outcomes between EMD and non-EMD patients.
The safety profiles were similar for EMD and non-EMD patients, Dr. Richardson said. Melflufen-dexamethasone was considered well tolerated overall, and there were no treatment-related deaths.
The most common treatment-emergent adverse events (grade 3 and 4, respectively) were thrombocytopenia (22% and 46%), neutropenia (32% and 35%), anemia (35% and 1%), white blood cell count decrease (10% and 7%), and pneumonia (7% and 1%).
This trial is sponsored by Oncopeptides. Dr. Richardson reported an advisory role and research funding from Oncopeptides.
SOURCE: Richardson PG et al. IMW 2019, Abstract OAB-086.
BOSTON – Melflufen plus dexamethasone is active in patients with relapsed/refractory multiple myeloma, whether or not they have extramedullary disease (EMD), a phase 2 trial suggests.
In the HORIZON trial, melflufen-dexamethasone produced an overall response rate of 23% in patients with EMD and 27% in those without EMD.
Paul G. Richardson, MD, of Harvard Medical School and the Dana-Farber Cancer Institute, both in Boston, presented these results as a late-breaking abstract at the International Myeloma Workshop, held by the International Myeloma Society.
As of July 30, 2019, 136 patients had been treated on the HORIZON trial. The trial is enrolling patients with relapsed/refractory multiple myeloma refractory to pomalidomide, an anti-CD38 monoclonal antibody (mAb), or both. The patients must have received at least two prior lines of therapy, including a proteasome inhibitor (PI) and an immunomodulatory agent (IMiD).
Dr. Richardson presented results for 130 patients, 44 with EMD and 86 without it. The median age at baseline was 64 years in the EMD and non-EMD groups (overall range, 35-86 years). More than half of patients had high-risk cytogenetics (52% in the EMD group and 57% in the non-EMD group).
The median number of prior therapies was five in both the EMD and non-EMD groups. Most patients had received at least one prior transplant (73% in the EMD group and 69% in the non-EMD group). Most patients in both groups were refractory to an anti-CD38 mAb (93% EMD and 72% non-EMD); an IMiD and a PI (93% EMD and 90% non-EMD); an IMiD, a PI, and an anti-CD38 mAb (91% EMD and 63% non-EMD); and their last therapy (100% EMD and 95% non-EMD).
Dr. Richardson pointed out that the incidence of EMD in this trial is higher than has been reported previously. Of the 44 EMD patients, 26 had soft-tissue EMD, and 18 had bone-related EMD. Five patients had CNS involvement.
Another key finding, according to Dr. Richardson, was that EMD appeared to be associated with prior anti-CD38 therapy. Specifically, 40% of patients exposed to an anti-CD38 mAb had EMD, compared with 11% of patients who had not received an anti-CD38 mAb (P = .01).
“I don’t for a minute want to say that CD38-targeted therapy engenders extramedullary disease,” Dr. Richardson said. “I think what we can say, though, is that, once CD38 treatment fails a patient, extramedullary disease … is a very real challenge. Therefore, we need rationally targeted approaches, ideally in combination, to meet that challenge.”
The overall response rate was similar for EMD and non-EMD patients – 23% and 27%, respectively. The median duration of response was 3.4 months in the EMD patients and 4.4 months in the non-EMD group. The clinical benefit rate was 30% and 45%, respectively.
In the EMD group, the overall response rate was 19% in patients with soft-tissue EMD and 28% in bone-related EMD. None of the patients with CNS disease responded.
The median progression-free survival was 2.9 months for patients with EMD and 4.6 months for those without EMD. The median overall survival was 5.8 month and 11.6 months, respectively.
The median overall survival was 18.5 months in EMD responders and 17.2 months in non-EMD responders. The median overall survival was 5.1 months in EMD nonresponders and 8.5 months in non-EMD nonresponders.
In all, 54% of patients received subsequent therapy. There were no significant differences in outcomes between EMD and non-EMD patients.
The safety profiles were similar for EMD and non-EMD patients, Dr. Richardson said. Melflufen-dexamethasone was considered well tolerated overall, and there were no treatment-related deaths.
The most common treatment-emergent adverse events (grade 3 and 4, respectively) were thrombocytopenia (22% and 46%), neutropenia (32% and 35%), anemia (35% and 1%), white blood cell count decrease (10% and 7%), and pneumonia (7% and 1%).
This trial is sponsored by Oncopeptides. Dr. Richardson reported an advisory role and research funding from Oncopeptides.
SOURCE: Richardson PG et al. IMW 2019, Abstract OAB-086.
REPORTING FROM IMW 2019
Subcutaneous vedolizumab effective for maintenance in subset of UC patients
, results from a phase 3, double-blind trial demonstrated.

“The route of drug administration can be an important determinant of a patient’s treatment experience, particularly for chronic diseases such as UC [ulcerative colitis],” investigators led by William J. Sandborn, MD, of the division of gastroenterology and hepatology at the University of California, San Diego, wrote in a study published online in Gastroenterology (doi: 10/1053/j.gastro.2019.08.027). “Intravenous administration of a biologic treatment requires the patient to set time aside and travel to a treatment center for an infusion. In addition, the greater use of a health care facility increases the direct costs of care. Some studies show that even with the option of self-injection some patients may still prefer an IV route of administration for the reassurance provided by the opportunity for interacting with a health care professional or because they are averse to self-injection. The availability of both an SC and IV injection of vedolizumab will enable patients to choose the route of administration for maintenance treatment.”
Between Dec. 18, 2015, and Aug. 21, 2018, Dr. Sandborn and colleagues at 141 sites in 29 countries enrolled 353 patients with moderate to severely active UC to receive IV vedolizumab 300 mg at weeks 0 and 2. At week 6, 216 patients who demonstrated clinical response were randomly assigned to maintenance treatment: 106 to SC vedolizumab 108 mg every 2 weeks, 54 to IV vedolizumab 300 mg every 8 weeks, and 56 to placebo. The study’s primary endpoint was clinical remission at week 52, which was defined as a total Mayo score of 2 or lower and no subscore greater than 1.
The mean age of patients was 40 years and 60% were male, and they had UC for a mean of 8 years. At week 52, the researchers found that clinical remission was achieved by 46.2% of patients in the SC vedolizumab group, compared with 42.6% of patients in the IV vedolizumab group and 14.3% of patients in the placebo group. In addition, patients in the SC vedolizumab group experienced significantly greater rates of endoscopic improvement and durable clinical response compared with those in the placebo group (P less than .001).
In terms of safety, injection-site reactions were noted by 10.4% of patients in the SC vedolizumab group (mostly rash, swelling, erythema, and pruritus), compared with 1.9% of patients in the IV vedolizumab group and in no patients in the placebo group. “No serious cases were reported for the AEs of special interest: hypersensitivity (including injection site reactions or infusion-related AEs), malignancies, and liver injury,” the researchers wrote. “There were no cases of PML [progressive multifocal leukoencephalopathy] and no deaths.” They acknowledged that the study’s sample size was smaller than the previous GEMINI pivotal trial for vedolizumab IV in ulcerative colitis (N Engl J Med 2013;369:699-710). “This limitation may have contributed to the findings of numerically greater but not statistically significant differences between treatment arms for some secondary endpoints such as durable clinical remission and corticosteroid-free clinical remission,” they wrote.
Takeda sponsored the study. Dr. Sandborn and coauthors reported having numerous financial ties to industry.
SOURCE: Sandborn WJ et al. Gastroenterol 2019 Aug. 27. doi: 10/1053/j.gastro.2019.08.027.
, results from a phase 3, double-blind trial demonstrated.
“The route of drug administration can be an important determinant of a patient’s treatment experience, particularly for chronic diseases such as UC [ulcerative colitis],” investigators led by William J. Sandborn, MD, of the division of gastroenterology and hepatology at the University of California, San Diego, wrote in a study published online in Gastroenterology (doi: 10/1053/j.gastro.2019.08.027). “Intravenous administration of a biologic treatment requires the patient to set time aside and travel to a treatment center for an infusion. In addition, the greater use of a health care facility increases the direct costs of care. Some studies show that even with the option of self-injection some patients may still prefer an IV route of administration for the reassurance provided by the opportunity for interacting with a health care professional or because they are averse to self-injection. The availability of both an SC and IV injection of vedolizumab will enable patients to choose the route of administration for maintenance treatment.”
Between Dec. 18, 2015, and Aug. 21, 2018, Dr. Sandborn and colleagues at 141 sites in 29 countries enrolled 353 patients with moderate to severely active UC to receive IV vedolizumab 300 mg at weeks 0 and 2. At week 6, 216 patients who demonstrated clinical response were randomly assigned to maintenance treatment: 106 to SC vedolizumab 108 mg every 2 weeks, 54 to IV vedolizumab 300 mg every 8 weeks, and 56 to placebo. The study’s primary endpoint was clinical remission at week 52, which was defined as a total Mayo score of 2 or lower and no subscore greater than 1.
The mean age of patients was 40 years and 60% were male, and they had UC for a mean of 8 years. At week 52, the researchers found that clinical remission was achieved by 46.2% of patients in the SC vedolizumab group, compared with 42.6% of patients in the IV vedolizumab group and 14.3% of patients in the placebo group. In addition, patients in the SC vedolizumab group experienced significantly greater rates of endoscopic improvement and durable clinical response compared with those in the placebo group (P less than .001).
In terms of safety, injection-site reactions were noted by 10.4% of patients in the SC vedolizumab group (mostly rash, swelling, erythema, and pruritus), compared with 1.9% of patients in the IV vedolizumab group and in no patients in the placebo group. “No serious cases were reported for the AEs of special interest: hypersensitivity (including injection site reactions or infusion-related AEs), malignancies, and liver injury,” the researchers wrote. “There were no cases of PML [progressive multifocal leukoencephalopathy] and no deaths.” They acknowledged that the study’s sample size was smaller than the previous GEMINI pivotal trial for vedolizumab IV in ulcerative colitis (N Engl J Med 2013;369:699-710). “This limitation may have contributed to the findings of numerically greater but not statistically significant differences between treatment arms for some secondary endpoints such as durable clinical remission and corticosteroid-free clinical remission,” they wrote.
Takeda sponsored the study. Dr. Sandborn and coauthors reported having numerous financial ties to industry.
SOURCE: Sandborn WJ et al. Gastroenterol 2019 Aug. 27. doi: 10/1053/j.gastro.2019.08.027.
, results from a phase 3, double-blind trial demonstrated.
“The route of drug administration can be an important determinant of a patient’s treatment experience, particularly for chronic diseases such as UC [ulcerative colitis],” investigators led by William J. Sandborn, MD, of the division of gastroenterology and hepatology at the University of California, San Diego, wrote in a study published online in Gastroenterology (doi: 10/1053/j.gastro.2019.08.027). “Intravenous administration of a biologic treatment requires the patient to set time aside and travel to a treatment center for an infusion. In addition, the greater use of a health care facility increases the direct costs of care. Some studies show that even with the option of self-injection some patients may still prefer an IV route of administration for the reassurance provided by the opportunity for interacting with a health care professional or because they are averse to self-injection. The availability of both an SC and IV injection of vedolizumab will enable patients to choose the route of administration for maintenance treatment.”
Between Dec. 18, 2015, and Aug. 21, 2018, Dr. Sandborn and colleagues at 141 sites in 29 countries enrolled 353 patients with moderate to severely active UC to receive IV vedolizumab 300 mg at weeks 0 and 2. At week 6, 216 patients who demonstrated clinical response were randomly assigned to maintenance treatment: 106 to SC vedolizumab 108 mg every 2 weeks, 54 to IV vedolizumab 300 mg every 8 weeks, and 56 to placebo. The study’s primary endpoint was clinical remission at week 52, which was defined as a total Mayo score of 2 or lower and no subscore greater than 1.
The mean age of patients was 40 years and 60% were male, and they had UC for a mean of 8 years. At week 52, the researchers found that clinical remission was achieved by 46.2% of patients in the SC vedolizumab group, compared with 42.6% of patients in the IV vedolizumab group and 14.3% of patients in the placebo group. In addition, patients in the SC vedolizumab group experienced significantly greater rates of endoscopic improvement and durable clinical response compared with those in the placebo group (P less than .001).
In terms of safety, injection-site reactions were noted by 10.4% of patients in the SC vedolizumab group (mostly rash, swelling, erythema, and pruritus), compared with 1.9% of patients in the IV vedolizumab group and in no patients in the placebo group. “No serious cases were reported for the AEs of special interest: hypersensitivity (including injection site reactions or infusion-related AEs), malignancies, and liver injury,” the researchers wrote. “There were no cases of PML [progressive multifocal leukoencephalopathy] and no deaths.” They acknowledged that the study’s sample size was smaller than the previous GEMINI pivotal trial for vedolizumab IV in ulcerative colitis (N Engl J Med 2013;369:699-710). “This limitation may have contributed to the findings of numerically greater but not statistically significant differences between treatment arms for some secondary endpoints such as durable clinical remission and corticosteroid-free clinical remission,” they wrote.
Takeda sponsored the study. Dr. Sandborn and coauthors reported having numerous financial ties to industry.
SOURCE: Sandborn WJ et al. Gastroenterol 2019 Aug. 27. doi: 10/1053/j.gastro.2019.08.027.
FROM GASTROENTEROLOGY

First data VERIFY value of early combination therapy in type 2 diabetes
BARCELONA – Upfront use of a dual combination of vildagliptin (Galvus) and metformin was associated with better and more durable glycemic control than metformin alone in patients with newly diagnosed type 2 diabetes, according to findings reported at the annual meeting of the European Association for the Study of Diabetes.

Fewer patients treated with the combination than with metformin monotherapy experienced “treatment failure” (43.6% vs. 62.1%, respectively) during the initial study period. The time-to-treatment failure, which was defined as an hemoglobin A1c of at least 7% (53 mmol/L) or higher on two occasions 3 months apart, was estimated to be beyond the study’s duration, at 61·9 months, for the combination and a median of 36.1 months in the monotherapy group.
Moreover, there was a significant (P less than .0001) 49% reduction in the relative risk for the time-to-initial-treatment failure in the early combination treatment group, compared with the monotherapy group, during the 5-year study period. The time-to-second-treatment failure was also longer in patients who received initial combination therapy (hazard ratio, 0.74; P less than .0001).
These results of the VERIFY (Vildagliptin Efficacy in Combination With Metformin for Early Treatment of Type 2 Diabetes) study, which were published simultaneously in the Lancet, provide the first real evidence to support the use of combination therapy rather than the current standard of metformin alone in the initial treatment of type 2 diabetes.
VERIFY was a phase 4, randomized, parallel-group study designed to compare the durability of glycemic control achieved with a combination of vildagliptin plus metformin or metformin alone in treatment-naive patients with type 2 diabetes.
At a press briefing, three members of the VERITY steering committee explained the rationale, design, results, and implications of the study.
EASD president David R. Matthews, DPhil, FRCP, who is emeritus professor at the Oxford Centre for Diabetes, Endocrinology and Metabolism at the University of Oxford (England), observed that the study aimed to answer three important questions: Do patients with type 2 diabetes benefit from having combination treatment from the start of their pharmacologic management, and if so, is this more beneficial than a step-up approach, and ultimately, “does it really matter?”

A typical cohort of patients was included, said Michael Stumvoll, MD, of the University Hospital Leipzig (Germany). Patients had to be aged between 18 and 70 years, have a body mass index of 22-40 kg/m2, and an hemoglobin A1c level of 6.5%-7.5%. This “rather narrow range” was decided “on purpose to really fulfill the idea of having newly diagnosed [type 2 diabetes]”, Dr. Stumvoll noted. In addition, patients had to have adequate renal function, have been diagnosed with type 2 diabetes in the past 2 years, and be drug naive or have received no more than 4 weeks of metformin.
In all, 2,001 patients from 254 centers in 34 countries were included, with 998 randomized to initial treatment with vildagliptin and metformin and 1,003 to receive metformin alone after an initial run-in phase during which the dose of metformin was up-titrated from 500 to 1,500 mg/day. The study ran for 5 years, with treatment intensified if there was a loss of glycemic control at the discretion of the study investigators – first vildagliptin was added to patients taking metformin monotherapy, then insulin, if needed.
There were no safety concerns: A similar percentage of patients in the early combination and initial monotherapy arms experienced an adverse event (83.5% vs. 83.2%, respectively), a serious adverse event (16.6% vs. 18.3%), a drug-related adverse event (15.9% vs. 14.3%), a severe adverse event (10.5% vs. 10.6%), and adverse events leading to discontinuation of treatment (4.1% vs. 5.3%) or death (13 vs. 9 patients). There was no difference in the change in body weight, and rates of hypoglycemia were 1.3% and 0.9%, respectively.
Adjudication and an independent data-monitoring committee were set up after cardiovascular events occurred in a few patients, although this was not a cardiovascular outcomes trials, Dr. Matthews stressed. There were fewer absolute cumulative adjudicated events in the early combination arm, compared with the initial monotherapy arm (30 vs. 44, respectively), and the time to the first adjudicated macrovascular event favored early combination over initial monotherapy (2.4% vs. 3.3%; HR, 0.71).
“There is a big caveat here,” said Dr. Matthews, “these are very small numbers and wide confidence intervals and the P value is .194.” Although “it is not a significant finding, and it was never intended to be a significant finding,” it gives “an indication that we absolutely should be looking at this.”

Stefano Del Prato, MD, of the University of Pisa (Italy), noted that “there has been a lot of discussion around initial combination therapy for type 2 diabetes,” and although there was a realization that multiple treatment might be necessary, there was no evidence for that. The results of the VERIFY trial, however, now provide some of the proof that this approach may be of benefit. Patients “benefit twice as much” with the combination therapy as they do with the monotherapy, Dr. Del Prato said. “There are twice as many patients retained under control with an early combination, compared with the monotherapy.” That means no longer “running after the patient losing control” he said, but “being proactive” and with a very low risk of hypoglycemia. The clinical implication is that there is now evidence for combination therapy as an initial approach for managing type 2 diabetes.
Novartis funded the study. Dr. Matthews has served on advisory boards or as a consultant for, and has given lectures for, Novartis and numerous other companies not related to the study. He is currently the president of the European Association for the Study of Diabetes. Dr. Stumvoll has received speaker's honoraria and consulting fees from Novartis and other companies. Dr. Del Prato serves or has served on advisory boards and speakers bureaus for, and received research support from, Novartis and numerous other companies.
SOURCE: Matthews DR et al. Lancet. 2019 Sept 18. doi: 10.1016/ S0140-6736(19)32131-2.
This article was updated on 9/19/2019.
BARCELONA – Upfront use of a dual combination of vildagliptin (Galvus) and metformin was associated with better and more durable glycemic control than metformin alone in patients with newly diagnosed type 2 diabetes, according to findings reported at the annual meeting of the European Association for the Study of Diabetes.
Fewer patients treated with the combination than with metformin monotherapy experienced “treatment failure” (43.6% vs. 62.1%, respectively) during the initial study period. The time-to-treatment failure, which was defined as an hemoglobin A1c of at least 7% (53 mmol/L) or higher on two occasions 3 months apart, was estimated to be beyond the study’s duration, at 61·9 months, for the combination and a median of 36.1 months in the monotherapy group.
Moreover, there was a significant (P less than .0001) 49% reduction in the relative risk for the time-to-initial-treatment failure in the early combination treatment group, compared with the monotherapy group, during the 5-year study period. The time-to-second-treatment failure was also longer in patients who received initial combination therapy (hazard ratio, 0.74; P less than .0001).
These results of the VERIFY (Vildagliptin Efficacy in Combination With Metformin for Early Treatment of Type 2 Diabetes) study, which were published simultaneously in the Lancet, provide the first real evidence to support the use of combination therapy rather than the current standard of metformin alone in the initial treatment of type 2 diabetes.
VERIFY was a phase 4, randomized, parallel-group study designed to compare the durability of glycemic control achieved with a combination of vildagliptin plus metformin or metformin alone in treatment-naive patients with type 2 diabetes.
At a press briefing, three members of the VERITY steering committee explained the rationale, design, results, and implications of the study.
EASD president David R. Matthews, DPhil, FRCP, who is emeritus professor at the Oxford Centre for Diabetes, Endocrinology and Metabolism at the University of Oxford (England), observed that the study aimed to answer three important questions: Do patients with type 2 diabetes benefit from having combination treatment from the start of their pharmacologic management, and if so, is this more beneficial than a step-up approach, and ultimately, “does it really matter?”
A typical cohort of patients was included, said Michael Stumvoll, MD, of the University Hospital Leipzig (Germany). Patients had to be aged between 18 and 70 years, have a body mass index of 22-40 kg/m2, and an hemoglobin A1c level of 6.5%-7.5%. This “rather narrow range” was decided “on purpose to really fulfill the idea of having newly diagnosed [type 2 diabetes]”, Dr. Stumvoll noted. In addition, patients had to have adequate renal function, have been diagnosed with type 2 diabetes in the past 2 years, and be drug naive or have received no more than 4 weeks of metformin.
In all, 2,001 patients from 254 centers in 34 countries were included, with 998 randomized to initial treatment with vildagliptin and metformin and 1,003 to receive metformin alone after an initial run-in phase during which the dose of metformin was up-titrated from 500 to 1,500 mg/day. The study ran for 5 years, with treatment intensified if there was a loss of glycemic control at the discretion of the study investigators – first vildagliptin was added to patients taking metformin monotherapy, then insulin, if needed.
There were no safety concerns: A similar percentage of patients in the early combination and initial monotherapy arms experienced an adverse event (83.5% vs. 83.2%, respectively), a serious adverse event (16.6% vs. 18.3%), a drug-related adverse event (15.9% vs. 14.3%), a severe adverse event (10.5% vs. 10.6%), and adverse events leading to discontinuation of treatment (4.1% vs. 5.3%) or death (13 vs. 9 patients). There was no difference in the change in body weight, and rates of hypoglycemia were 1.3% and 0.9%, respectively.
Adjudication and an independent data-monitoring committee were set up after cardiovascular events occurred in a few patients, although this was not a cardiovascular outcomes trials, Dr. Matthews stressed. There were fewer absolute cumulative adjudicated events in the early combination arm, compared with the initial monotherapy arm (30 vs. 44, respectively), and the time to the first adjudicated macrovascular event favored early combination over initial monotherapy (2.4% vs. 3.3%; HR, 0.71).
“There is a big caveat here,” said Dr. Matthews, “these are very small numbers and wide confidence intervals and the P value is .194.” Although “it is not a significant finding, and it was never intended to be a significant finding,” it gives “an indication that we absolutely should be looking at this.”
Stefano Del Prato, MD, of the University of Pisa (Italy), noted that “there has been a lot of discussion around initial combination therapy for type 2 diabetes,” and although there was a realization that multiple treatment might be necessary, there was no evidence for that. The results of the VERIFY trial, however, now provide some of the proof that this approach may be of benefit. Patients “benefit twice as much” with the combination therapy as they do with the monotherapy, Dr. Del Prato said. “There are twice as many patients retained under control with an early combination, compared with the monotherapy.” That means no longer “running after the patient losing control” he said, but “being proactive” and with a very low risk of hypoglycemia. The clinical implication is that there is now evidence for combination therapy as an initial approach for managing type 2 diabetes.
Novartis funded the study. Dr. Matthews has served on advisory boards or as a consultant for, and has given lectures for, Novartis and numerous other companies not related to the study. He is currently the president of the European Association for the Study of Diabetes. Dr. Stumvoll has received speaker's honoraria and consulting fees from Novartis and other companies. Dr. Del Prato serves or has served on advisory boards and speakers bureaus for, and received research support from, Novartis and numerous other companies.
SOURCE: Matthews DR et al. Lancet. 2019 Sept 18. doi: 10.1016/ S0140-6736(19)32131-2.
This article was updated on 9/19/2019.
BARCELONA – Upfront use of a dual combination of vildagliptin (Galvus) and metformin was associated with better and more durable glycemic control than metformin alone in patients with newly diagnosed type 2 diabetes, according to findings reported at the annual meeting of the European Association for the Study of Diabetes.
Fewer patients treated with the combination than with metformin monotherapy experienced “treatment failure” (43.6% vs. 62.1%, respectively) during the initial study period. The time-to-treatment failure, which was defined as an hemoglobin A1c of at least 7% (53 mmol/L) or higher on two occasions 3 months apart, was estimated to be beyond the study’s duration, at 61·9 months, for the combination and a median of 36.1 months in the monotherapy group.
Moreover, there was a significant (P less than .0001) 49% reduction in the relative risk for the time-to-initial-treatment failure in the early combination treatment group, compared with the monotherapy group, during the 5-year study period. The time-to-second-treatment failure was also longer in patients who received initial combination therapy (hazard ratio, 0.74; P less than .0001).
These results of the VERIFY (Vildagliptin Efficacy in Combination With Metformin for Early Treatment of Type 2 Diabetes) study, which were published simultaneously in the Lancet, provide the first real evidence to support the use of combination therapy rather than the current standard of metformin alone in the initial treatment of type 2 diabetes.
VERIFY was a phase 4, randomized, parallel-group study designed to compare the durability of glycemic control achieved with a combination of vildagliptin plus metformin or metformin alone in treatment-naive patients with type 2 diabetes.
At a press briefing, three members of the VERITY steering committee explained the rationale, design, results, and implications of the study.
EASD president David R. Matthews, DPhil, FRCP, who is emeritus professor at the Oxford Centre for Diabetes, Endocrinology and Metabolism at the University of Oxford (England), observed that the study aimed to answer three important questions: Do patients with type 2 diabetes benefit from having combination treatment from the start of their pharmacologic management, and if so, is this more beneficial than a step-up approach, and ultimately, “does it really matter?”
A typical cohort of patients was included, said Michael Stumvoll, MD, of the University Hospital Leipzig (Germany). Patients had to be aged between 18 and 70 years, have a body mass index of 22-40 kg/m2, and an hemoglobin A1c level of 6.5%-7.5%. This “rather narrow range” was decided “on purpose to really fulfill the idea of having newly diagnosed [type 2 diabetes]”, Dr. Stumvoll noted. In addition, patients had to have adequate renal function, have been diagnosed with type 2 diabetes in the past 2 years, and be drug naive or have received no more than 4 weeks of metformin.
In all, 2,001 patients from 254 centers in 34 countries were included, with 998 randomized to initial treatment with vildagliptin and metformin and 1,003 to receive metformin alone after an initial run-in phase during which the dose of metformin was up-titrated from 500 to 1,500 mg/day. The study ran for 5 years, with treatment intensified if there was a loss of glycemic control at the discretion of the study investigators – first vildagliptin was added to patients taking metformin monotherapy, then insulin, if needed.
There were no safety concerns: A similar percentage of patients in the early combination and initial monotherapy arms experienced an adverse event (83.5% vs. 83.2%, respectively), a serious adverse event (16.6% vs. 18.3%), a drug-related adverse event (15.9% vs. 14.3%), a severe adverse event (10.5% vs. 10.6%), and adverse events leading to discontinuation of treatment (4.1% vs. 5.3%) or death (13 vs. 9 patients). There was no difference in the change in body weight, and rates of hypoglycemia were 1.3% and 0.9%, respectively.
Adjudication and an independent data-monitoring committee were set up after cardiovascular events occurred in a few patients, although this was not a cardiovascular outcomes trials, Dr. Matthews stressed. There were fewer absolute cumulative adjudicated events in the early combination arm, compared with the initial monotherapy arm (30 vs. 44, respectively), and the time to the first adjudicated macrovascular event favored early combination over initial monotherapy (2.4% vs. 3.3%; HR, 0.71).
“There is a big caveat here,” said Dr. Matthews, “these are very small numbers and wide confidence intervals and the P value is .194.” Although “it is not a significant finding, and it was never intended to be a significant finding,” it gives “an indication that we absolutely should be looking at this.”
Stefano Del Prato, MD, of the University of Pisa (Italy), noted that “there has been a lot of discussion around initial combination therapy for type 2 diabetes,” and although there was a realization that multiple treatment might be necessary, there was no evidence for that. The results of the VERIFY trial, however, now provide some of the proof that this approach may be of benefit. Patients “benefit twice as much” with the combination therapy as they do with the monotherapy, Dr. Del Prato said. “There are twice as many patients retained under control with an early combination, compared with the monotherapy.” That means no longer “running after the patient losing control” he said, but “being proactive” and with a very low risk of hypoglycemia. The clinical implication is that there is now evidence for combination therapy as an initial approach for managing type 2 diabetes.
Novartis funded the study. Dr. Matthews has served on advisory boards or as a consultant for, and has given lectures for, Novartis and numerous other companies not related to the study. He is currently the president of the European Association for the Study of Diabetes. Dr. Stumvoll has received speaker's honoraria and consulting fees from Novartis and other companies. Dr. Del Prato serves or has served on advisory boards and speakers bureaus for, and received research support from, Novartis and numerous other companies.
SOURCE: Matthews DR et al. Lancet. 2019 Sept 18. doi: 10.1016/ S0140-6736(19)32131-2.
This article was updated on 9/19/2019.
REPORTING FROM EASD 2019
Mycobacterium haemophilum: A Challenging Treatment Dilemma in an Immunocompromised Patient
To the Editor:
The increase in nontuberculous mycobacteria (NTM) infections over the last 3 decades likely is multifaceted, including increased clinical awareness, improved laboratory diagnostics, growing numbers of immunocompromised patients, and an aging population.1,2 Historically, the majority of mycobacteria-related diseases are due to Mycobacterium tuberculosis, Mycobacterium bovis, and Mycobacterium leprae.3
Mycobacterium haemophilum is a slow-growing acid-fast bacillus (AFB) that differs from other Mycobacterium species in that it requires iron-supplemented media and incubation temperatures of 30°C to 32°C for culture. As these requirements for growth are not standard for AFB cultures, M haemophilum infection may be underrecognized and underreported.3Mycobacterium haemophilum infections largely are cutaneous and generally are seen in AIDS patients and bone marrow transplant recipients who are iatrogenically immunosuppressed.4,5 No species-specific treatment guidelines exist2; however, triple-drug therapy combining a macrolide, rifamycin, and a quinolone for a minimum of 12 months often is recommended.
A 64-year-old man with a history of coronary artery disease, hypertension, hyperlipidemia, and acute myelogenous leukemia (AML) underwent allogenic stem cell transplantation. His posttransplant course was complicated by multiple deep vein thromboses, hypogammaglobulinemia, and graft-vs-host disease (GVHD) of the skin and gastrointestinal tract that manifested as chronic diarrhea, which was managed with chronic prednisone. Thirteen months after the transplant, the patient presented to his outpatient oncologist (M.K.) for evaluation of painless, nonpruritic, erythematous papules and nodules that had emerged on the right side of the chest, right arm, and left leg of approximately 2 weeks’ duration.
On review of systems by oncology, the patient denied any fevers, chills, or night sweats but noted chronic loose nonbloody stools without abdominal pain, likely related to the GVHD. The patient’s medications included prednisone 20 mg once daily, fluconazole, amitriptyline, atovaquone, budesonide, dabigatran, metoprolol, pantoprazole, rosuvastatin, senna glycoside, spironolactone, tramadol, and valacyclovir.
Physical examination revealed multiple singular erythematous nodules on the right side of the chest (Figure 1A), right arm (Figure 1B), and left leg. There was no regional lymphadenopathy. The patient was afebrile and hemodynamically stable. A biopsy of the arm performed to rule out leukemia cutis revealed a granulomatous dermatitis with numerous AFB (Figures 2A and 2B), which were confirmed on Ziehl-Neelsen staining (Figures 2C and 2D). The presence of AFB raised concern for a disseminated mycobacterial infection. The patient was admitted to our institution approximately 1 week after the outpatient biopsy was performed. He was evaluated by infectious diseases (B.H.) and was recommended for repeat biopsy with AFB culture and for initiation of intravenous antibiotics.

The patient was evaluated by the dermatology consultation service on hospital day 1. At the time of consultation, the lesions were still painless but had enlarged. Two new satellite lesions were noted on his other extremities. Due to the widespread distribution of the lesions, there was concern for disseminated disease. The relatively rapid onset of new lesions increased concern for infection with rapid-growing mycobacteria, including Mycobacterium abscessus, Mycobacterium fortuitum, and Mycobacterium chelonae. A detailed history revealed that the patient’s wife had a fish tank, which supported the inclusion of Mycobacterium marinum in the differential; however, further questioning revealed that the patient never came in contact with the aquarium water. The initial outpatient biopsy had not been sent for culture. Following inpatient biopsy, the patient was initiated on empiric antimycobacterials, including imipenem, amikacin, clarithromycin, and levofloxacin. Computed tomography of the head was negative for cerebral involvement.
Acid-fast bacilli blood cultures were drawn per the recommendation from infectious diseases in an attempt to confirm disseminated disease; however, blood cultures remained negative. Tissue biopsy from the right arm was sent for AFB staining and culture. Many AFB were identified on microscopy, and growth was observed in the mycobacterial growth indicator tube after 6 days of incubation. The DNA probe was negative for M tuberculosis complex or Mycobacterium avium complex.
The patient was discharged on hospital day 6 on empiric therapy for rapid-growing mycobacteria while cultures were pending. The empiric regimen included intravenous imipenem 1 g every 6 hours, intravenous amikacin 1 g once daily, clarithromycin 500 mg every 12 hours, and levofloxacin 750 mg once daily. All solid media cultures were negative at the time of discharge.
The biopsy specimen proved difficult to culture on solid media using traditional methods. Three weeks after the inpatient biopsy, the microbiology laboratory reported that growth was observed on solid media that was incubated at 30°C and supplemented with iron. These findings were not characteristic of a rapidly growing mycobacteria (eg, M fortuitum, M chelonae, M abscessus) or M marinum but raised concern for infectionwith M haemophilum. Antimycobacterial treatment was adjusted to amikacin, clarithromycin, levofloxacin, and rifabutin.
Six weeks after the inpatient skin biopsy, final speciation confirmed infection with M haemophilum. The isolate proved susceptible to amikacin (minimal inhibitory concentration [MIC], 16), clarithromycin (MIC, 0.12), linezolid (MIC, <1), moxifloxacin (MIC, 0.5), rifabutin (MIC, <0.25), and trimethoprim-sulfamethoxazole (MIC, 0.5/9.5). The isolate was resistant to ciprofloxacin (MIC, 4), ethambutol (MIC, >16), and rifampin (MIC, 2). Based on these findings, an infectious disease specialist modified the treatment regimen to azithromycin 600 mg once daily, moxifloxacin 400 mg once daily, and rifabutin 300 mg once daily. Azithromycin was substituted for clarithromycin in an attempt to minimize the gastrointestinal side effects of the antibiotics. The infectious disease specialist was concerned that the clarithromycin could exacerbate the patient’s chronic GVHD-associated diarrhea, which posed a challenge to the oncologist, who was attempting to manage the patient’s GVHD and minimize the use of additional prednisone. At the time of this change, the patient was doing well clinically and denied any active skin lesions.
Four months later, he developed new left-sided neck swelling. Computed tomography revealed nonspecific enhancement involving the skin and superficial subcutaneous tissues in the left anterior neck. He was referred to otolaryngology given concern for recurrent infection vs leukemia cutis. He underwent excisional biopsy. Pathology was negative for malignancy but demonstrated subcutaneous necrotizing granulomatous inflammation with a positive AFB stain. Tissue AFB cultures revealed moderate AFB on direct stain, but there was no AFB growth at 12 weeks. Clarithromycin was restarted in place of azithromycin to increase the potency of the antimycobacterial regimen. Cultures from this neck biopsy were negative after 12 weeks of incubation.
In addition to this change in antibiotic coverage, the patient’s medical oncologist tapered the patient’s immunosuppression considerably. The patient subsequently completed 12 months of therapy with clarithromycin, moxifloxacin, and rifabutin starting from the time of the neck biopsy. He remained free of recurrence of mycobacterial infection for nearly 2 years until he died from an unrelated illness.
Nontuberculous mycobacteria are an ubiquitous environmental group.2 Sources include soil and natural water (M avium), fish tanks and swimming pools (M marinum), and tap water and occasionally domestic animals (Mycobacterium kansasii). Additionally, rapidly growing NTM such as M abscessus, M chelonae, and M fortuitum have been isolated from soil and natural water supplies.3
Mycobacterium haemophilum is a fastidious organism with a predilection for skin of the chest and extremities. Iatrogenically or inherently immunocompromised patients are most commonly affected6-11; however, there also have been reports in healthy patients.12,13 Infections typically present as painless erythematous papules or nodules that eventually suppurate, ulcerate, and become painful. Presentations involving Fitz-Hugh–Curtis syndrome,13 new B-cell lymphoma,10 and lymphadenitis12 also have been described. Beyond cutaneous involvement, M haemophilum has been cultured from bone, the synovium, the lungs, and the central nervous system.4,9 The majority of morbidities occur in patients with lung involvement.4 Therefore, even patients presenting with isolated cutaneous disease require close follow-up.
Mycobacterium haemophilum is a slowly proliferating organism that is unable to grow in standard egg-potato (Lowenstein-Jensen) medium or agar base (Middlebrook 7H10 or 7H11 agar) without iron supplementation (ferric ammonium citrate, hemin, or hemoglobin). It also requires temperatures of 30°C to 32°C for growth. Its iron requisite is unique, but species such as M marinum and Mycobacterium ulcerans also share reduced temperature requirements. Without a high index of suspicion, growth often is absent because standard Mycobacterium culture techniques will not foster organism growth. Our case demonstrated that special culture instructions must be relayed to the laboratory, even in the face of positive AFB smears. Failure to request hemin and modified incubation temperatures may have contributed to the negative AFB blood culture in our patient.
Due to the relatively rare incidence of M haemophilum infection, there are no known randomized controlled trials guiding antibiotic regimens. Infectious disease specialists often treat empirically with triple-drug therapy derived from locally reported species susceptibilities. The largest case series to date did not identify resistance to amikacin, ciprofloxacin, or clarithromycin.4 Our case identified a novel finding of ciprofloxacin and rifampin resistance, which may highlight the emergence of a newly resistant strain of M haemophilum. Of note, one case of rifampin resistance has been reported, but the culture was drawn from a postmortem specimen in the setting of previously rifampin-sensitive isolates.4 Empiric therapies should be guided by hospital susceptibility reports and expert consultation.
Coinfection with 2 or more NTM—including M tuberculosis, M leprae, and M fortuitum—has been reported.8,14 Temporally distinct coinfections with M leprae and M haemophilum also have been described.15 Thus, practitioners should have a low threshold for repeat cultures in the context of new cutaneous nodules or granulomas, not only to detect concomitant infections but also to identify resistance patterns that might explain recurrent or recalcitrant disease. Immune reconstitution inflammatory syndrome also must be considered with new or worsening lesions, especially in the first months of therapy, as this is a common occurrence when immunosuppressive regimens are tapered to help manage infections.
In conclusion, M haemophilum is an underrecognized infection that presents as cutaneous nodules or lymphadenitis in immunocompromised or healthy individuals. Diagnosis requires a high index of suspicion because its unique growth requirements necessitate special laboratory techniques. Our case represents a classic presentation of this NTM infection in a patient with AML following allogenic stem cell transplantation. Repeat cultures, workup of potentially disseminated infections, and close follow-up are requisite to minimizing morbidity and mortality. A multidisciplinary approach involving infectious disease, medical oncology, radiology, and dermatology best manages this type of infection.
- Sheu LC, Tran TM, Jarlsberg LG, et al. Non-tuberculous mycobacterial infections at San Francisco General Hospital. Clin Respir J. 2015;9:436-442.
- Knoll BM. Update on nontuberculous mycobacterial infections in solid organ and hematopoietic stem cell transplant recipients. Curr Infect Dis Rep. 2014;16:421.
- Diagnosis and treatment of disease caused by nontuberculous mycobacteria. this official statement of the American Thoracic Society was approved by the Board of Directors, March 1997. Medical Section of the American Lung Association. Am J Respir Crit Care Med. 1997;156(2 pt 2):S1-S25.
- Shah MK, Sebti A, Kiehn TE, et al. Mycobacterium haemophilum in immunocompromised patients. Clin Infect Dis. 2001;33:330-337.
- Griffiths DE, Aksamit T, Brown-Elliott BA. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416.
- Copeland NK, Arora NS, Ferguson TM. Mycobacterium haemophilum masquerading as leprosy in a renal transplant patient [published online November 28, 2013]. Case Rep Dermatol Med. 2013;2013:793127.
- Aslam A, Green RL, Motta L, et al. Cutaneous Mycobacterium haemophilum infection in a patient receiving infliximab for psoriasis. Br J Dermatol. 2013;168:446-447.
- Agrawal S, Sharma A. Dual mycobacterial infection in the setting of leflunomide treatment for rheumatoid arthritis. Ann Rheum Dis. 2007;66:277.
- Buppajarntham A, Apisarnthanarak A, Rutjanawech S, et al. Central nervous system infection due to Mycobacterium haemophilum in a patient with acquired immunodeficiency syndrome. Int J STD AIDS. 2015;26:288-290.
- Doherty T, Lynn M, Cavazza A, et al. Mycobacterium haemophilum as the initial presentation of a B-cell lymphoma in a liver transplant patient [published online January 12, 2014]. Case Rep Rheumatol. 2014;2014:742978.
- Ducharlet K, Murphy C, Tan SJ, et al. Recurrent Mycobacterium haemophilum in a renal transplant recipient. Nephrology (Carlton). 2014;(19 suppl 1):14-17.
- Dawson DJ, Blacklock ZM, Kane DW. Mycobacterium haemophilum causing lymphadenitis in an otherwise healthy child. Med J Aust. 1981;2:289-290.
- Jang HY, Burbelo PD, Chae YS, et al. Nontuberculous mycobacterial infection in a clinical presentation of Fitz-Hugh-Curtis syndrome: a case report with multigene diagnostic approach. BMC Womens Health. 2014;14:95.
- Scollard DM, Stryjewska BM, Prestigiacomo JF, et al. Hansen’s disease (leprosy) complicated by secondary mycobacterial infection. J Am Acad Dermatol. 2011;64:593-596.
- SoRelle JA, Beal SG, Scollard DM, et al. Mycobacterium leprae and Mycobacterium haemophilum co-infection in an iatrogenically immunosuppressed patient. Diagn Microbiol Infect Dis. 2014;78:494-496.
To the Editor:
The increase in nontuberculous mycobacteria (NTM) infections over the last 3 decades likely is multifaceted, including increased clinical awareness, improved laboratory diagnostics, growing numbers of immunocompromised patients, and an aging population.1,2 Historically, the majority of mycobacteria-related diseases are due to Mycobacterium tuberculosis, Mycobacterium bovis, and Mycobacterium leprae.3
Mycobacterium haemophilum is a slow-growing acid-fast bacillus (AFB) that differs from other Mycobacterium species in that it requires iron-supplemented media and incubation temperatures of 30°C to 32°C for culture. As these requirements for growth are not standard for AFB cultures, M haemophilum infection may be underrecognized and underreported.3Mycobacterium haemophilum infections largely are cutaneous and generally are seen in AIDS patients and bone marrow transplant recipients who are iatrogenically immunosuppressed.4,5 No species-specific treatment guidelines exist2; however, triple-drug therapy combining a macrolide, rifamycin, and a quinolone for a minimum of 12 months often is recommended.
A 64-year-old man with a history of coronary artery disease, hypertension, hyperlipidemia, and acute myelogenous leukemia (AML) underwent allogenic stem cell transplantation. His posttransplant course was complicated by multiple deep vein thromboses, hypogammaglobulinemia, and graft-vs-host disease (GVHD) of the skin and gastrointestinal tract that manifested as chronic diarrhea, which was managed with chronic prednisone. Thirteen months after the transplant, the patient presented to his outpatient oncologist (M.K.) for evaluation of painless, nonpruritic, erythematous papules and nodules that had emerged on the right side of the chest, right arm, and left leg of approximately 2 weeks’ duration.
On review of systems by oncology, the patient denied any fevers, chills, or night sweats but noted chronic loose nonbloody stools without abdominal pain, likely related to the GVHD. The patient’s medications included prednisone 20 mg once daily, fluconazole, amitriptyline, atovaquone, budesonide, dabigatran, metoprolol, pantoprazole, rosuvastatin, senna glycoside, spironolactone, tramadol, and valacyclovir.
Physical examination revealed multiple singular erythematous nodules on the right side of the chest (Figure 1A), right arm (Figure 1B), and left leg. There was no regional lymphadenopathy. The patient was afebrile and hemodynamically stable. A biopsy of the arm performed to rule out leukemia cutis revealed a granulomatous dermatitis with numerous AFB (Figures 2A and 2B), which were confirmed on Ziehl-Neelsen staining (Figures 2C and 2D). The presence of AFB raised concern for a disseminated mycobacterial infection. The patient was admitted to our institution approximately 1 week after the outpatient biopsy was performed. He was evaluated by infectious diseases (B.H.) and was recommended for repeat biopsy with AFB culture and for initiation of intravenous antibiotics.
The patient was evaluated by the dermatology consultation service on hospital day 1. At the time of consultation, the lesions were still painless but had enlarged. Two new satellite lesions were noted on his other extremities. Due to the widespread distribution of the lesions, there was concern for disseminated disease. The relatively rapid onset of new lesions increased concern for infection with rapid-growing mycobacteria, including Mycobacterium abscessus, Mycobacterium fortuitum, and Mycobacterium chelonae. A detailed history revealed that the patient’s wife had a fish tank, which supported the inclusion of Mycobacterium marinum in the differential; however, further questioning revealed that the patient never came in contact with the aquarium water. The initial outpatient biopsy had not been sent for culture. Following inpatient biopsy, the patient was initiated on empiric antimycobacterials, including imipenem, amikacin, clarithromycin, and levofloxacin. Computed tomography of the head was negative for cerebral involvement.
Acid-fast bacilli blood cultures were drawn per the recommendation from infectious diseases in an attempt to confirm disseminated disease; however, blood cultures remained negative. Tissue biopsy from the right arm was sent for AFB staining and culture. Many AFB were identified on microscopy, and growth was observed in the mycobacterial growth indicator tube after 6 days of incubation. The DNA probe was negative for M tuberculosis complex or Mycobacterium avium complex.
The patient was discharged on hospital day 6 on empiric therapy for rapid-growing mycobacteria while cultures were pending. The empiric regimen included intravenous imipenem 1 g every 6 hours, intravenous amikacin 1 g once daily, clarithromycin 500 mg every 12 hours, and levofloxacin 750 mg once daily. All solid media cultures were negative at the time of discharge.
The biopsy specimen proved difficult to culture on solid media using traditional methods. Three weeks after the inpatient biopsy, the microbiology laboratory reported that growth was observed on solid media that was incubated at 30°C and supplemented with iron. These findings were not characteristic of a rapidly growing mycobacteria (eg, M fortuitum, M chelonae, M abscessus) or M marinum but raised concern for infectionwith M haemophilum. Antimycobacterial treatment was adjusted to amikacin, clarithromycin, levofloxacin, and rifabutin.
Six weeks after the inpatient skin biopsy, final speciation confirmed infection with M haemophilum. The isolate proved susceptible to amikacin (minimal inhibitory concentration [MIC], 16), clarithromycin (MIC, 0.12), linezolid (MIC, <1), moxifloxacin (MIC, 0.5), rifabutin (MIC, <0.25), and trimethoprim-sulfamethoxazole (MIC, 0.5/9.5). The isolate was resistant to ciprofloxacin (MIC, 4), ethambutol (MIC, >16), and rifampin (MIC, 2). Based on these findings, an infectious disease specialist modified the treatment regimen to azithromycin 600 mg once daily, moxifloxacin 400 mg once daily, and rifabutin 300 mg once daily. Azithromycin was substituted for clarithromycin in an attempt to minimize the gastrointestinal side effects of the antibiotics. The infectious disease specialist was concerned that the clarithromycin could exacerbate the patient’s chronic GVHD-associated diarrhea, which posed a challenge to the oncologist, who was attempting to manage the patient’s GVHD and minimize the use of additional prednisone. At the time of this change, the patient was doing well clinically and denied any active skin lesions.
Four months later, he developed new left-sided neck swelling. Computed tomography revealed nonspecific enhancement involving the skin and superficial subcutaneous tissues in the left anterior neck. He was referred to otolaryngology given concern for recurrent infection vs leukemia cutis. He underwent excisional biopsy. Pathology was negative for malignancy but demonstrated subcutaneous necrotizing granulomatous inflammation with a positive AFB stain. Tissue AFB cultures revealed moderate AFB on direct stain, but there was no AFB growth at 12 weeks. Clarithromycin was restarted in place of azithromycin to increase the potency of the antimycobacterial regimen. Cultures from this neck biopsy were negative after 12 weeks of incubation.
In addition to this change in antibiotic coverage, the patient’s medical oncologist tapered the patient’s immunosuppression considerably. The patient subsequently completed 12 months of therapy with clarithromycin, moxifloxacin, and rifabutin starting from the time of the neck biopsy. He remained free of recurrence of mycobacterial infection for nearly 2 years until he died from an unrelated illness.
Nontuberculous mycobacteria are an ubiquitous environmental group.2 Sources include soil and natural water (M avium), fish tanks and swimming pools (M marinum), and tap water and occasionally domestic animals (Mycobacterium kansasii). Additionally, rapidly growing NTM such as M abscessus, M chelonae, and M fortuitum have been isolated from soil and natural water supplies.3
Mycobacterium haemophilum is a fastidious organism with a predilection for skin of the chest and extremities. Iatrogenically or inherently immunocompromised patients are most commonly affected6-11; however, there also have been reports in healthy patients.12,13 Infections typically present as painless erythematous papules or nodules that eventually suppurate, ulcerate, and become painful. Presentations involving Fitz-Hugh–Curtis syndrome,13 new B-cell lymphoma,10 and lymphadenitis12 also have been described. Beyond cutaneous involvement, M haemophilum has been cultured from bone, the synovium, the lungs, and the central nervous system.4,9 The majority of morbidities occur in patients with lung involvement.4 Therefore, even patients presenting with isolated cutaneous disease require close follow-up.
Mycobacterium haemophilum is a slowly proliferating organism that is unable to grow in standard egg-potato (Lowenstein-Jensen) medium or agar base (Middlebrook 7H10 or 7H11 agar) without iron supplementation (ferric ammonium citrate, hemin, or hemoglobin). It also requires temperatures of 30°C to 32°C for growth. Its iron requisite is unique, but species such as M marinum and Mycobacterium ulcerans also share reduced temperature requirements. Without a high index of suspicion, growth often is absent because standard Mycobacterium culture techniques will not foster organism growth. Our case demonstrated that special culture instructions must be relayed to the laboratory, even in the face of positive AFB smears. Failure to request hemin and modified incubation temperatures may have contributed to the negative AFB blood culture in our patient.
Due to the relatively rare incidence of M haemophilum infection, there are no known randomized controlled trials guiding antibiotic regimens. Infectious disease specialists often treat empirically with triple-drug therapy derived from locally reported species susceptibilities. The largest case series to date did not identify resistance to amikacin, ciprofloxacin, or clarithromycin.4 Our case identified a novel finding of ciprofloxacin and rifampin resistance, which may highlight the emergence of a newly resistant strain of M haemophilum. Of note, one case of rifampin resistance has been reported, but the culture was drawn from a postmortem specimen in the setting of previously rifampin-sensitive isolates.4 Empiric therapies should be guided by hospital susceptibility reports and expert consultation.
Coinfection with 2 or more NTM—including M tuberculosis, M leprae, and M fortuitum—has been reported.8,14 Temporally distinct coinfections with M leprae and M haemophilum also have been described.15 Thus, practitioners should have a low threshold for repeat cultures in the context of new cutaneous nodules or granulomas, not only to detect concomitant infections but also to identify resistance patterns that might explain recurrent or recalcitrant disease. Immune reconstitution inflammatory syndrome also must be considered with new or worsening lesions, especially in the first months of therapy, as this is a common occurrence when immunosuppressive regimens are tapered to help manage infections.
In conclusion, M haemophilum is an underrecognized infection that presents as cutaneous nodules or lymphadenitis in immunocompromised or healthy individuals. Diagnosis requires a high index of suspicion because its unique growth requirements necessitate special laboratory techniques. Our case represents a classic presentation of this NTM infection in a patient with AML following allogenic stem cell transplantation. Repeat cultures, workup of potentially disseminated infections, and close follow-up are requisite to minimizing morbidity and mortality. A multidisciplinary approach involving infectious disease, medical oncology, radiology, and dermatology best manages this type of infection.
To the Editor:
The increase in nontuberculous mycobacteria (NTM) infections over the last 3 decades likely is multifaceted, including increased clinical awareness, improved laboratory diagnostics, growing numbers of immunocompromised patients, and an aging population.1,2 Historically, the majority of mycobacteria-related diseases are due to Mycobacterium tuberculosis, Mycobacterium bovis, and Mycobacterium leprae.3
Mycobacterium haemophilum is a slow-growing acid-fast bacillus (AFB) that differs from other Mycobacterium species in that it requires iron-supplemented media and incubation temperatures of 30°C to 32°C for culture. As these requirements for growth are not standard for AFB cultures, M haemophilum infection may be underrecognized and underreported.3Mycobacterium haemophilum infections largely are cutaneous and generally are seen in AIDS patients and bone marrow transplant recipients who are iatrogenically immunosuppressed.4,5 No species-specific treatment guidelines exist2; however, triple-drug therapy combining a macrolide, rifamycin, and a quinolone for a minimum of 12 months often is recommended.
A 64-year-old man with a history of coronary artery disease, hypertension, hyperlipidemia, and acute myelogenous leukemia (AML) underwent allogenic stem cell transplantation. His posttransplant course was complicated by multiple deep vein thromboses, hypogammaglobulinemia, and graft-vs-host disease (GVHD) of the skin and gastrointestinal tract that manifested as chronic diarrhea, which was managed with chronic prednisone. Thirteen months after the transplant, the patient presented to his outpatient oncologist (M.K.) for evaluation of painless, nonpruritic, erythematous papules and nodules that had emerged on the right side of the chest, right arm, and left leg of approximately 2 weeks’ duration.
On review of systems by oncology, the patient denied any fevers, chills, or night sweats but noted chronic loose nonbloody stools without abdominal pain, likely related to the GVHD. The patient’s medications included prednisone 20 mg once daily, fluconazole, amitriptyline, atovaquone, budesonide, dabigatran, metoprolol, pantoprazole, rosuvastatin, senna glycoside, spironolactone, tramadol, and valacyclovir.
Physical examination revealed multiple singular erythematous nodules on the right side of the chest (Figure 1A), right arm (Figure 1B), and left leg. There was no regional lymphadenopathy. The patient was afebrile and hemodynamically stable. A biopsy of the arm performed to rule out leukemia cutis revealed a granulomatous dermatitis with numerous AFB (Figures 2A and 2B), which were confirmed on Ziehl-Neelsen staining (Figures 2C and 2D). The presence of AFB raised concern for a disseminated mycobacterial infection. The patient was admitted to our institution approximately 1 week after the outpatient biopsy was performed. He was evaluated by infectious diseases (B.H.) and was recommended for repeat biopsy with AFB culture and for initiation of intravenous antibiotics.
The patient was evaluated by the dermatology consultation service on hospital day 1. At the time of consultation, the lesions were still painless but had enlarged. Two new satellite lesions were noted on his other extremities. Due to the widespread distribution of the lesions, there was concern for disseminated disease. The relatively rapid onset of new lesions increased concern for infection with rapid-growing mycobacteria, including Mycobacterium abscessus, Mycobacterium fortuitum, and Mycobacterium chelonae. A detailed history revealed that the patient’s wife had a fish tank, which supported the inclusion of Mycobacterium marinum in the differential; however, further questioning revealed that the patient never came in contact with the aquarium water. The initial outpatient biopsy had not been sent for culture. Following inpatient biopsy, the patient was initiated on empiric antimycobacterials, including imipenem, amikacin, clarithromycin, and levofloxacin. Computed tomography of the head was negative for cerebral involvement.
Acid-fast bacilli blood cultures were drawn per the recommendation from infectious diseases in an attempt to confirm disseminated disease; however, blood cultures remained negative. Tissue biopsy from the right arm was sent for AFB staining and culture. Many AFB were identified on microscopy, and growth was observed in the mycobacterial growth indicator tube after 6 days of incubation. The DNA probe was negative for M tuberculosis complex or Mycobacterium avium complex.
The patient was discharged on hospital day 6 on empiric therapy for rapid-growing mycobacteria while cultures were pending. The empiric regimen included intravenous imipenem 1 g every 6 hours, intravenous amikacin 1 g once daily, clarithromycin 500 mg every 12 hours, and levofloxacin 750 mg once daily. All solid media cultures were negative at the time of discharge.
The biopsy specimen proved difficult to culture on solid media using traditional methods. Three weeks after the inpatient biopsy, the microbiology laboratory reported that growth was observed on solid media that was incubated at 30°C and supplemented with iron. These findings were not characteristic of a rapidly growing mycobacteria (eg, M fortuitum, M chelonae, M abscessus) or M marinum but raised concern for infectionwith M haemophilum. Antimycobacterial treatment was adjusted to amikacin, clarithromycin, levofloxacin, and rifabutin.
Six weeks after the inpatient skin biopsy, final speciation confirmed infection with M haemophilum. The isolate proved susceptible to amikacin (minimal inhibitory concentration [MIC], 16), clarithromycin (MIC, 0.12), linezolid (MIC, <1), moxifloxacin (MIC, 0.5), rifabutin (MIC, <0.25), and trimethoprim-sulfamethoxazole (MIC, 0.5/9.5). The isolate was resistant to ciprofloxacin (MIC, 4), ethambutol (MIC, >16), and rifampin (MIC, 2). Based on these findings, an infectious disease specialist modified the treatment regimen to azithromycin 600 mg once daily, moxifloxacin 400 mg once daily, and rifabutin 300 mg once daily. Azithromycin was substituted for clarithromycin in an attempt to minimize the gastrointestinal side effects of the antibiotics. The infectious disease specialist was concerned that the clarithromycin could exacerbate the patient’s chronic GVHD-associated diarrhea, which posed a challenge to the oncologist, who was attempting to manage the patient’s GVHD and minimize the use of additional prednisone. At the time of this change, the patient was doing well clinically and denied any active skin lesions.
Four months later, he developed new left-sided neck swelling. Computed tomography revealed nonspecific enhancement involving the skin and superficial subcutaneous tissues in the left anterior neck. He was referred to otolaryngology given concern for recurrent infection vs leukemia cutis. He underwent excisional biopsy. Pathology was negative for malignancy but demonstrated subcutaneous necrotizing granulomatous inflammation with a positive AFB stain. Tissue AFB cultures revealed moderate AFB on direct stain, but there was no AFB growth at 12 weeks. Clarithromycin was restarted in place of azithromycin to increase the potency of the antimycobacterial regimen. Cultures from this neck biopsy were negative after 12 weeks of incubation.
In addition to this change in antibiotic coverage, the patient’s medical oncologist tapered the patient’s immunosuppression considerably. The patient subsequently completed 12 months of therapy with clarithromycin, moxifloxacin, and rifabutin starting from the time of the neck biopsy. He remained free of recurrence of mycobacterial infection for nearly 2 years until he died from an unrelated illness.
Nontuberculous mycobacteria are an ubiquitous environmental group.2 Sources include soil and natural water (M avium), fish tanks and swimming pools (M marinum), and tap water and occasionally domestic animals (Mycobacterium kansasii). Additionally, rapidly growing NTM such as M abscessus, M chelonae, and M fortuitum have been isolated from soil and natural water supplies.3
Mycobacterium haemophilum is a fastidious organism with a predilection for skin of the chest and extremities. Iatrogenically or inherently immunocompromised patients are most commonly affected6-11; however, there also have been reports in healthy patients.12,13 Infections typically present as painless erythematous papules or nodules that eventually suppurate, ulcerate, and become painful. Presentations involving Fitz-Hugh–Curtis syndrome,13 new B-cell lymphoma,10 and lymphadenitis12 also have been described. Beyond cutaneous involvement, M haemophilum has been cultured from bone, the synovium, the lungs, and the central nervous system.4,9 The majority of morbidities occur in patients with lung involvement.4 Therefore, even patients presenting with isolated cutaneous disease require close follow-up.
Mycobacterium haemophilum is a slowly proliferating organism that is unable to grow in standard egg-potato (Lowenstein-Jensen) medium or agar base (Middlebrook 7H10 or 7H11 agar) without iron supplementation (ferric ammonium citrate, hemin, or hemoglobin). It also requires temperatures of 30°C to 32°C for growth. Its iron requisite is unique, but species such as M marinum and Mycobacterium ulcerans also share reduced temperature requirements. Without a high index of suspicion, growth often is absent because standard Mycobacterium culture techniques will not foster organism growth. Our case demonstrated that special culture instructions must be relayed to the laboratory, even in the face of positive AFB smears. Failure to request hemin and modified incubation temperatures may have contributed to the negative AFB blood culture in our patient.
Due to the relatively rare incidence of M haemophilum infection, there are no known randomized controlled trials guiding antibiotic regimens. Infectious disease specialists often treat empirically with triple-drug therapy derived from locally reported species susceptibilities. The largest case series to date did not identify resistance to amikacin, ciprofloxacin, or clarithromycin.4 Our case identified a novel finding of ciprofloxacin and rifampin resistance, which may highlight the emergence of a newly resistant strain of M haemophilum. Of note, one case of rifampin resistance has been reported, but the culture was drawn from a postmortem specimen in the setting of previously rifampin-sensitive isolates.4 Empiric therapies should be guided by hospital susceptibility reports and expert consultation.
Coinfection with 2 or more NTM—including M tuberculosis, M leprae, and M fortuitum—has been reported.8,14 Temporally distinct coinfections with M leprae and M haemophilum also have been described.15 Thus, practitioners should have a low threshold for repeat cultures in the context of new cutaneous nodules or granulomas, not only to detect concomitant infections but also to identify resistance patterns that might explain recurrent or recalcitrant disease. Immune reconstitution inflammatory syndrome also must be considered with new or worsening lesions, especially in the first months of therapy, as this is a common occurrence when immunosuppressive regimens are tapered to help manage infections.
In conclusion, M haemophilum is an underrecognized infection that presents as cutaneous nodules or lymphadenitis in immunocompromised or healthy individuals. Diagnosis requires a high index of suspicion because its unique growth requirements necessitate special laboratory techniques. Our case represents a classic presentation of this NTM infection in a patient with AML following allogenic stem cell transplantation. Repeat cultures, workup of potentially disseminated infections, and close follow-up are requisite to minimizing morbidity and mortality. A multidisciplinary approach involving infectious disease, medical oncology, radiology, and dermatology best manages this type of infection.
- Sheu LC, Tran TM, Jarlsberg LG, et al. Non-tuberculous mycobacterial infections at San Francisco General Hospital. Clin Respir J. 2015;9:436-442.
- Knoll BM. Update on nontuberculous mycobacterial infections in solid organ and hematopoietic stem cell transplant recipients. Curr Infect Dis Rep. 2014;16:421.
- Diagnosis and treatment of disease caused by nontuberculous mycobacteria. this official statement of the American Thoracic Society was approved by the Board of Directors, March 1997. Medical Section of the American Lung Association. Am J Respir Crit Care Med. 1997;156(2 pt 2):S1-S25.
- Shah MK, Sebti A, Kiehn TE, et al. Mycobacterium haemophilum in immunocompromised patients. Clin Infect Dis. 2001;33:330-337.
- Griffiths DE, Aksamit T, Brown-Elliott BA. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416.
- Copeland NK, Arora NS, Ferguson TM. Mycobacterium haemophilum masquerading as leprosy in a renal transplant patient [published online November 28, 2013]. Case Rep Dermatol Med. 2013;2013:793127.
- Aslam A, Green RL, Motta L, et al. Cutaneous Mycobacterium haemophilum infection in a patient receiving infliximab for psoriasis. Br J Dermatol. 2013;168:446-447.
- Agrawal S, Sharma A. Dual mycobacterial infection in the setting of leflunomide treatment for rheumatoid arthritis. Ann Rheum Dis. 2007;66:277.
- Buppajarntham A, Apisarnthanarak A, Rutjanawech S, et al. Central nervous system infection due to Mycobacterium haemophilum in a patient with acquired immunodeficiency syndrome. Int J STD AIDS. 2015;26:288-290.
- Doherty T, Lynn M, Cavazza A, et al. Mycobacterium haemophilum as the initial presentation of a B-cell lymphoma in a liver transplant patient [published online January 12, 2014]. Case Rep Rheumatol. 2014;2014:742978.
- Ducharlet K, Murphy C, Tan SJ, et al. Recurrent Mycobacterium haemophilum in a renal transplant recipient. Nephrology (Carlton). 2014;(19 suppl 1):14-17.
- Dawson DJ, Blacklock ZM, Kane DW. Mycobacterium haemophilum causing lymphadenitis in an otherwise healthy child. Med J Aust. 1981;2:289-290.
- Jang HY, Burbelo PD, Chae YS, et al. Nontuberculous mycobacterial infection in a clinical presentation of Fitz-Hugh-Curtis syndrome: a case report with multigene diagnostic approach. BMC Womens Health. 2014;14:95.
- Scollard DM, Stryjewska BM, Prestigiacomo JF, et al. Hansen’s disease (leprosy) complicated by secondary mycobacterial infection. J Am Acad Dermatol. 2011;64:593-596.
- SoRelle JA, Beal SG, Scollard DM, et al. Mycobacterium leprae and Mycobacterium haemophilum co-infection in an iatrogenically immunosuppressed patient. Diagn Microbiol Infect Dis. 2014;78:494-496.
- Sheu LC, Tran TM, Jarlsberg LG, et al. Non-tuberculous mycobacterial infections at San Francisco General Hospital. Clin Respir J. 2015;9:436-442.
- Knoll BM. Update on nontuberculous mycobacterial infections in solid organ and hematopoietic stem cell transplant recipients. Curr Infect Dis Rep. 2014;16:421.
- Diagnosis and treatment of disease caused by nontuberculous mycobacteria. this official statement of the American Thoracic Society was approved by the Board of Directors, March 1997. Medical Section of the American Lung Association. Am J Respir Crit Care Med. 1997;156(2 pt 2):S1-S25.
- Shah MK, Sebti A, Kiehn TE, et al. Mycobacterium haemophilum in immunocompromised patients. Clin Infect Dis. 2001;33:330-337.
- Griffiths DE, Aksamit T, Brown-Elliott BA. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416.
- Copeland NK, Arora NS, Ferguson TM. Mycobacterium haemophilum masquerading as leprosy in a renal transplant patient [published online November 28, 2013]. Case Rep Dermatol Med. 2013;2013:793127.
- Aslam A, Green RL, Motta L, et al. Cutaneous Mycobacterium haemophilum infection in a patient receiving infliximab for psoriasis. Br J Dermatol. 2013;168:446-447.
- Agrawal S, Sharma A. Dual mycobacterial infection in the setting of leflunomide treatment for rheumatoid arthritis. Ann Rheum Dis. 2007;66:277.
- Buppajarntham A, Apisarnthanarak A, Rutjanawech S, et al. Central nervous system infection due to Mycobacterium haemophilum in a patient with acquired immunodeficiency syndrome. Int J STD AIDS. 2015;26:288-290.
- Doherty T, Lynn M, Cavazza A, et al. Mycobacterium haemophilum as the initial presentation of a B-cell lymphoma in a liver transplant patient [published online January 12, 2014]. Case Rep Rheumatol. 2014;2014:742978.
- Ducharlet K, Murphy C, Tan SJ, et al. Recurrent Mycobacterium haemophilum in a renal transplant recipient. Nephrology (Carlton). 2014;(19 suppl 1):14-17.
- Dawson DJ, Blacklock ZM, Kane DW. Mycobacterium haemophilum causing lymphadenitis in an otherwise healthy child. Med J Aust. 1981;2:289-290.
- Jang HY, Burbelo PD, Chae YS, et al. Nontuberculous mycobacterial infection in a clinical presentation of Fitz-Hugh-Curtis syndrome: a case report with multigene diagnostic approach. BMC Womens Health. 2014;14:95.
- Scollard DM, Stryjewska BM, Prestigiacomo JF, et al. Hansen’s disease (leprosy) complicated by secondary mycobacterial infection. J Am Acad Dermatol. 2011;64:593-596.
- SoRelle JA, Beal SG, Scollard DM, et al. Mycobacterium leprae and Mycobacterium haemophilum co-infection in an iatrogenically immunosuppressed patient. Diagn Microbiol Infect Dis. 2014;78:494-496.
Practice Points
- Mycobacterium haemophilum is a slow-growing acid-fast bacillus that requires iron-supplemented media and incubation temperatures of 30°C to 32°C for culture. Because these requirements for growth are not standard for acid-fast bacteria cultures, M haemophilum infection may be underrecognized and underreported.
- There are no species-specific treatment guidelines, but extended course of treatment with multiple active antibacterials typically is recommended.