User login
Psoriasiform Drug Eruption Secondary to Sorafenib: Case Series and Review of the Literature
The expanded use of targeted anticancer agents such as sorafenib has revealed a growing spectrum of adverse cutaneous eruptions. We describe 3 patients with sorafenib-induced psoriasiform dermatitis and review the literature of only 10 other similar reported cases based on a search of PubMed, Web of Science, and American Society of Clinical Oncology abstracts using the terms psoriasis or psoriasiform dermatitis and sorafenib.1-10 We seek to increase awareness of this particular drug eruption in response to sorafenib and to describe potential effective treatment options, especially when sorafenib cannot be discontinued.
Case Reports
Patient 1
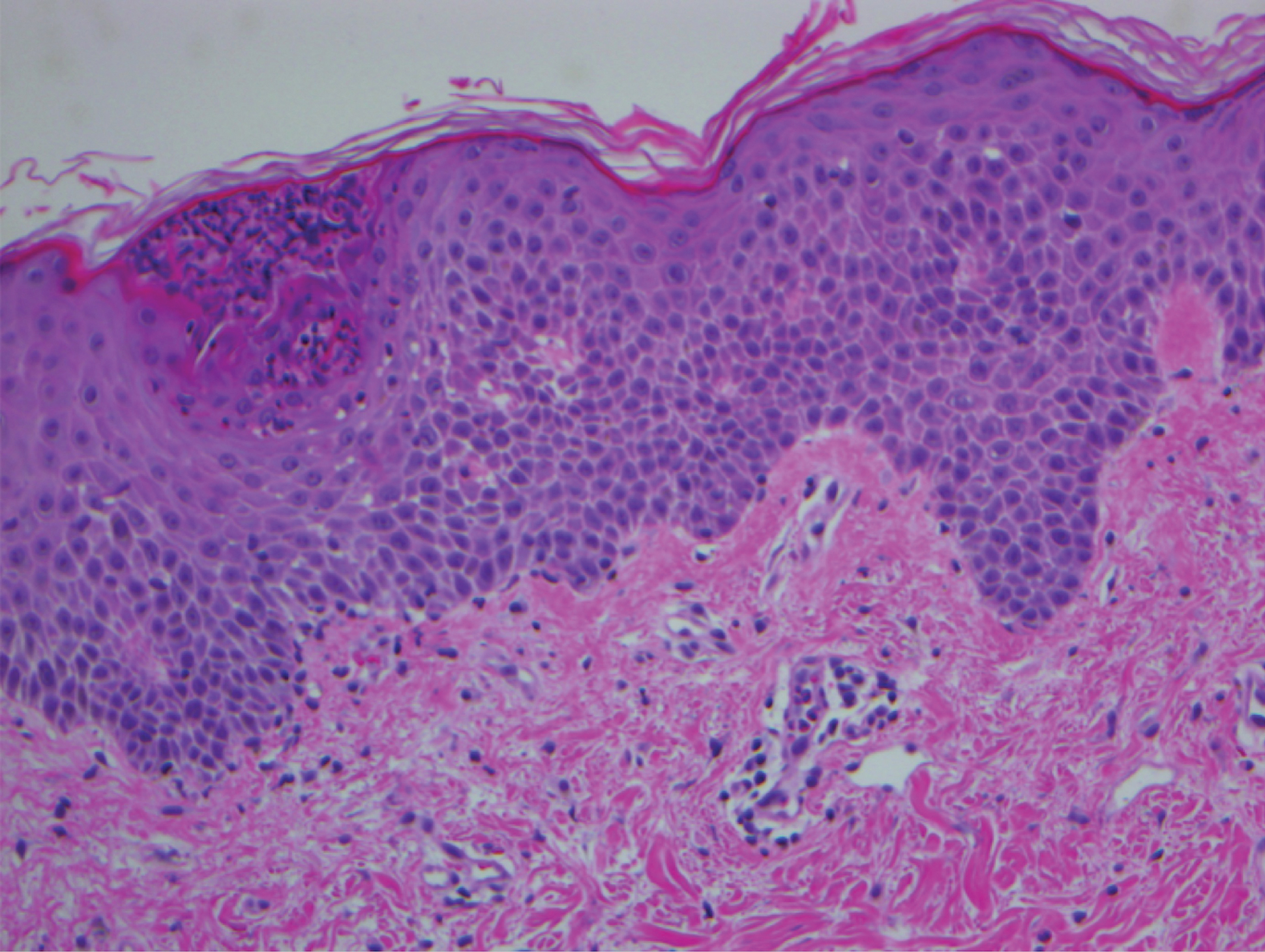
A 68-year-old man with chronic hepatitis B infection and hepatocellular carcinoma (HCC) was started on sorafenib 400 mg daily. After 2 months of treatment, he developed painful hyperkeratotic lesions on the bilateral palms and soles with formation of calluses and superficial blisters on an erythematous base that was consistent with hand-foot skin reaction (HFSR). He also had numerous erythematous thin papules and plaques with adherent white scale and yellow crust on the bilateral thighs, lower legs, forearms, dorsal hands, abdomen, back, and buttocks (Figure 1). He had no personal or family history of psoriasis, and blood tests were unremarkable. Histologic analysis of punch biopsies from the buttocks and right leg revealed focal parakeratosis with neutrophils and serous crust, acanthosis, mild spongiosis, and lymphocytes at the dermoepidermal junction and surrounding dermal vessels, consistent with psoriasiform dermatitis (Figure 2). Sorafenib was discontinued and the eruption began to resolve within a week. A lower dose of sorafenib (200 mg daily) was attempted and the psoriasiform eruption recurred.
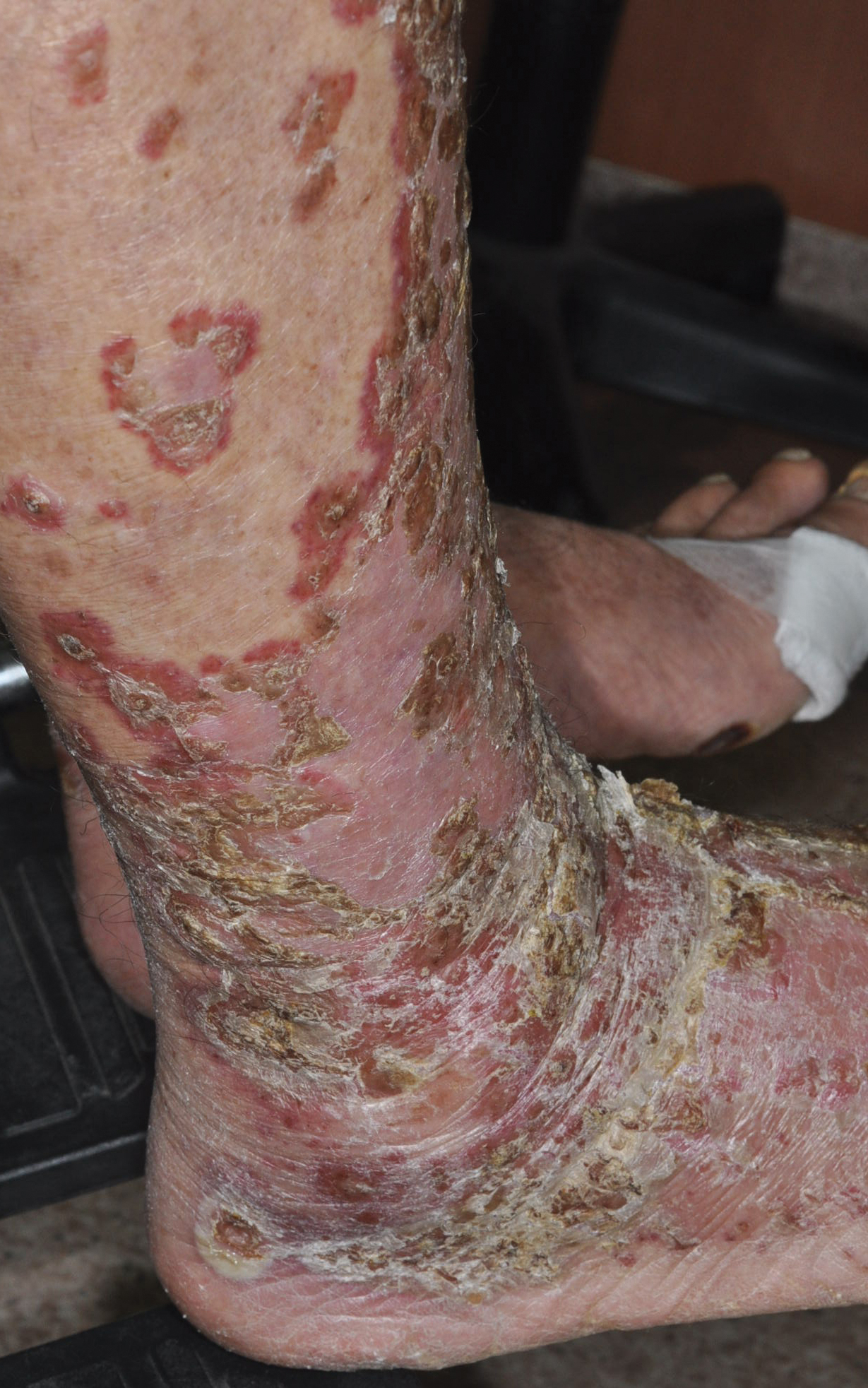
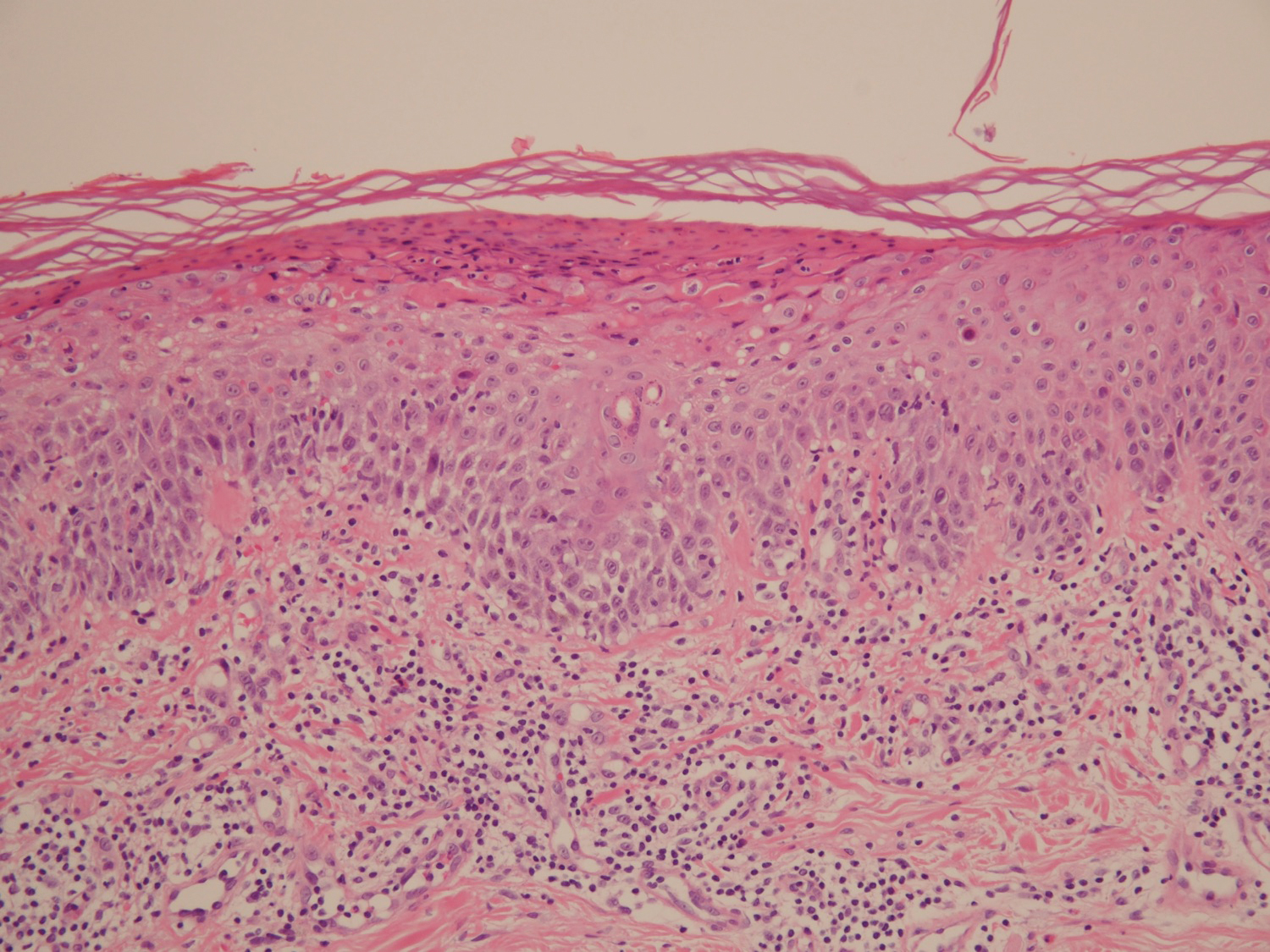
Patient 2
An 82-year-old man with chronic hepatitis B infection and HCC with lung metastasis was treated with sorafenib 400 mg daily. One week after treatment, he developed painful, thick, erythematous lesions on acral surfaces, consistent with HFSR. The sorafenib dose was decreased to 200 mg daily and HFSR resolved. Four months later, he developed well-demarcated, erythematous, scaly plaques with peripheral pustules on the right thigh (Figure 3) and right shin. He had no personal or family history of psoriasis, and blood tests were unremarkable. Samples from the pustules were taken for bacterial culture and fungal stain, but both were negative. Histologic analysis of a punch biopsy from the right thigh revealed necrotic parakeratosis, spongiform pustules, mild acanthosis, and a perivascular lymphocytic infiltrate with many neutrophils in the dermis. These findings suggested a diagnosis of pustular psoriasis, pustular drug eruption, or acute generalized exanthematous pustulosis. Treatment was initiated with mometasone cream. The patient subsequently developed hemoptysis and ascites from sorafenib. Sorafenib was discontinued and his skin eruption gradually resolved.
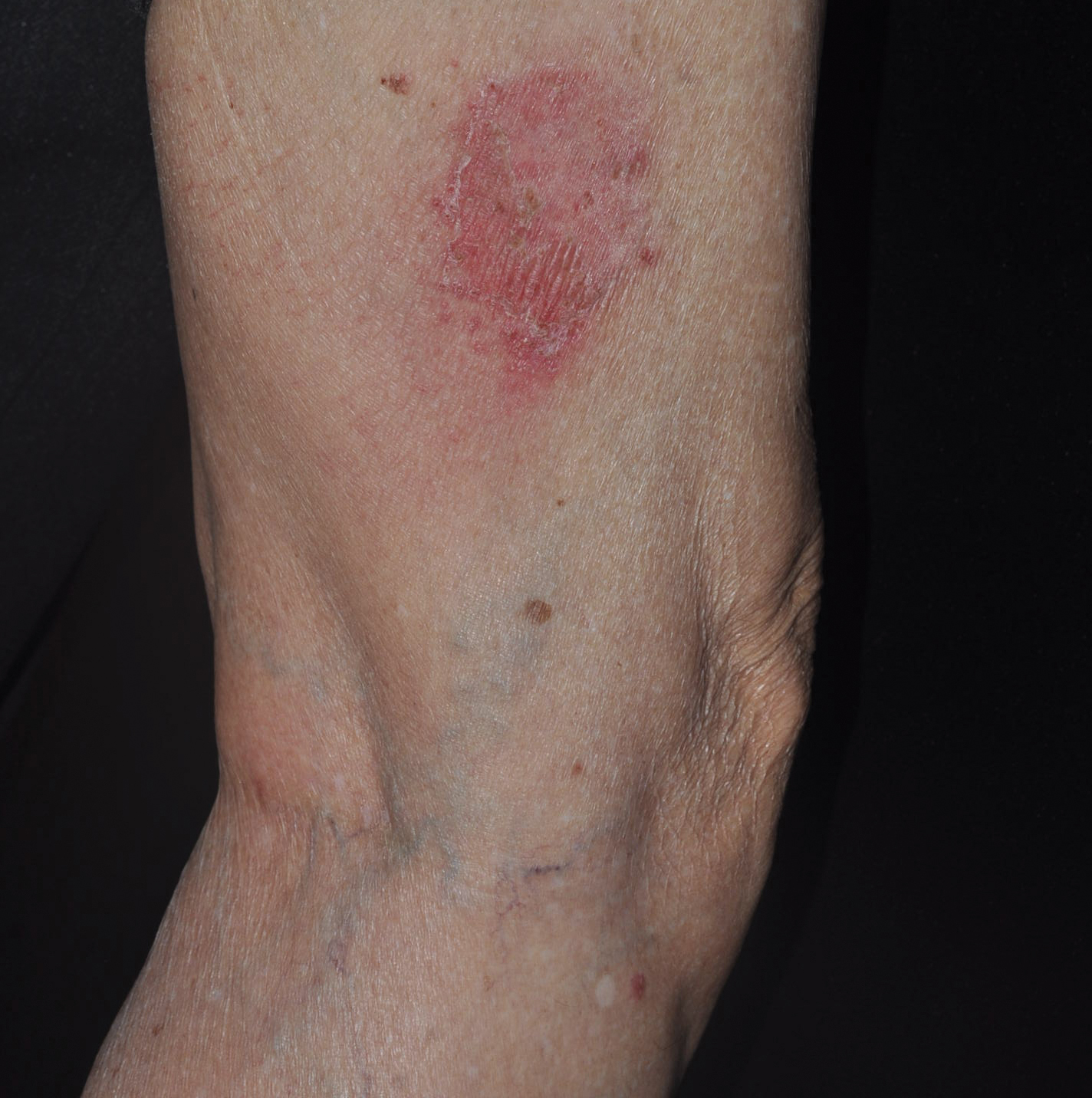
Patient 3
A 45-year-old woman with history of acute myeloid leukemia (AML) was started on sorafenib 200 mg twice daily as part of a clinical pilot study to maintain remission following an allogeneic bone marrow transplant. Four months after beginning sorafenib, the patient developed multiple well-defined, erythematous, thin papules and plaques with overlying flaky white scale on the bilateral upper extremities and trunk and scattered on the bilateral upper thighs (Figure 4) along with abdominal pain. Her other medical history, physical findings, and laboratory results were unremarkable, and there was no personal or family history of psoriasis. Her oncologist suspected that the eruption and symptoms were due to sorafenib and reduced the dose to 200 mg daily. Histologic analysis of a punch biopsy specimen revealed subcorneal neutrophilic collections with mild spongiosis and mild perivascular inflammatory infiltrate composed of lymphocytes and neutrophils (Figure 5). Direct immunofluorescence was negative for antibody or complement deposition. A bone marrow biopsy was negative for AML recurrence. The patient was continued on sorafenib to prevent AML recurrence, and she was started on triamcinolone cream 0.1% twice daily. Two weeks later, the eruption worsened and the patient was started on oral hydroxyzine for pruritus and narrowband UVB (NB-UVB) phototherapy 3 times a week. After 9 applications of NB-UVB phototherapy, there was complete resolution of the eruption.
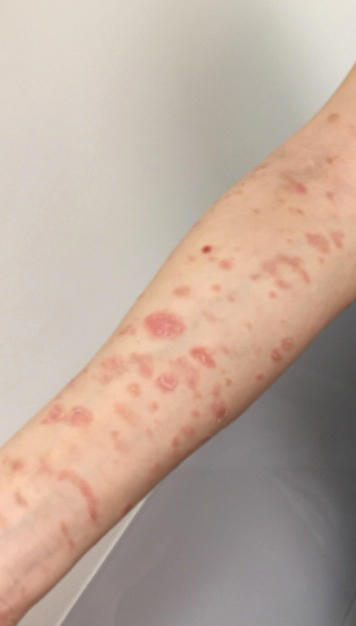
Comment
Sorafenib is an oral tyrosine kinase inhibitor that blocks tumor cell proliferation and angiogenesis due to its activity against vascular endothelial growth factor (VEGF) receptor, platelet-derived growth factor receptor, stem cell growth factor receptor, and rapidly accelerated fibrosarcoma kinases.11 It is primarily used for the treatment of solid tumors, such as advanced renal cell carcinoma, unresectable HCC, and thyroid carcinoma, and more recently has been expanded for treatment of AML due to potential inhibition of FMS-like tyrosine kinase 3 receptor. Although dermatologic toxicity is a common adverse event during treatment with sorafenib,11 reports of psoriasiform drug eruptions are rare.
Review of Cases
Based on our literature search, there are 10 previously reported cases of psoriasiform drug eruption secondary to sorafenib. Of the 13 total cases (including the 3 patients in this report), 7 patients had a history of psoriasis; most were middle-aged men; and the treatment with sorafenib was for solid tumors, primarily HCC with the exception of patient 3 from the current report who was treated for AML (Table). In all cases, the dose of sorafenib ranged from 200 to 800 mg daily. In 5 cases, HFSR preceded (as with patient 2 in the current report) or presented concurrently (as with patient 1 in the current report) with the onset of psoriasiform rash.1,3,5
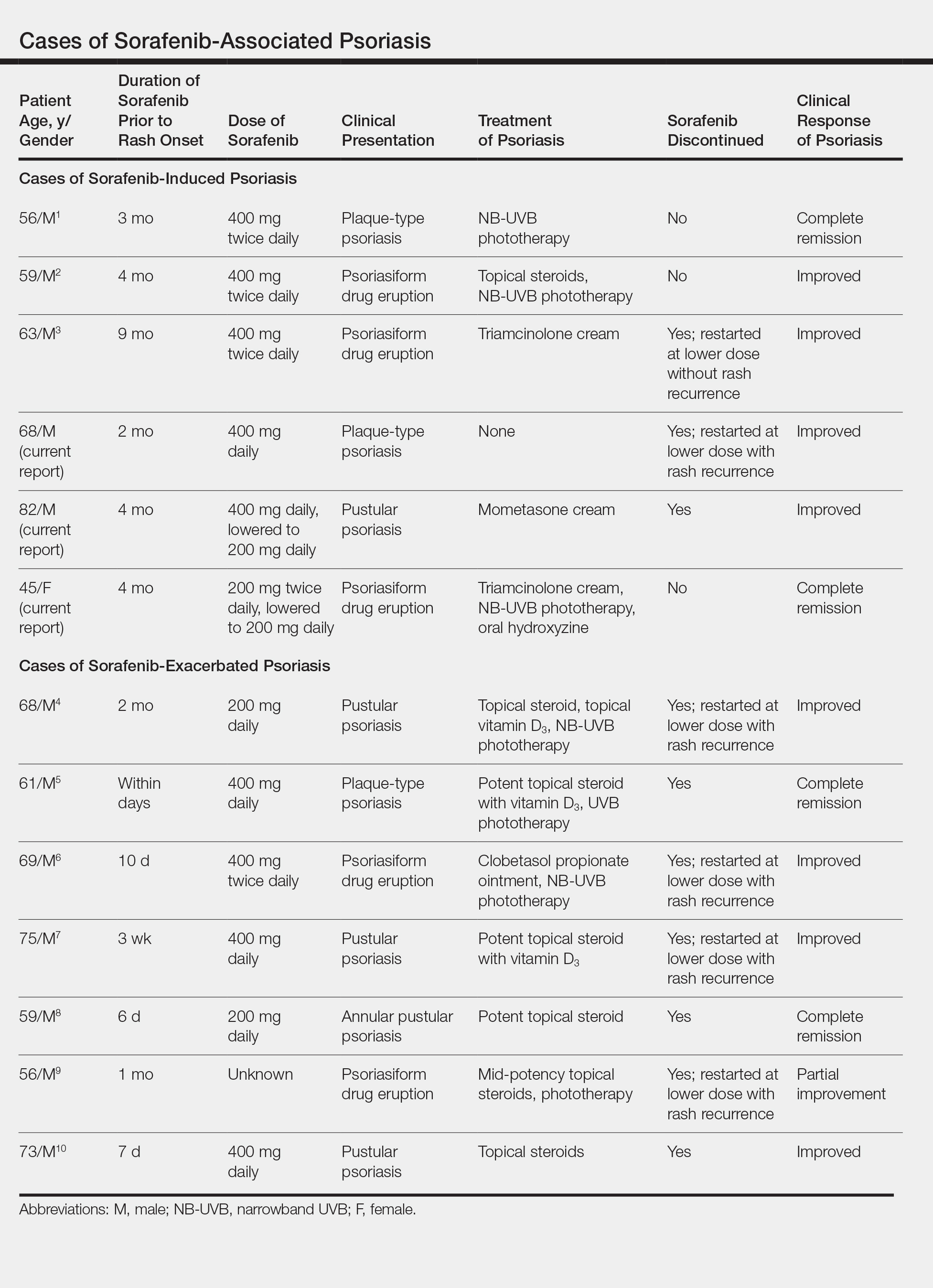
Of the 13 total cases, patients with a history of psoriasis generally developed the eruption in a shorter period of time after starting sorafenib (eg, days to 2 months) compared to those without a history of psoriasis (eg, 2 to 9 months)(Table), suggesting that patients with preexisting psoriasis more rapidly developed the drug eruption than patients without a history. In these patients with a history of psoriasis, all had long-standing mild to moderate stable plaque psoriasis, with the exception of 1 case in which the type of psoriasis was not described (Table).7 The presentation of the drug eruption following sorafenib varied from psoriasiform drug eruption (5 patients, including patient 3),2,3,6,9 pustular psoriasis (5 patients, including patient 2),4,7,8,10 and plaque psoriasis (3 patients, including patient 1).1,5 Interestingly, 5 of 6 patients with a history of plaque psoriasis presented with pustular psoriasis or psoriasiform drug eruption after treatment with sorafenib.4-6,8-10 These results suggest a causal relationship between sorafenib and exacerbation of preexisting psoriasis.
In the 13 total cases, treatments included mid- to high-potency topical steroids (10 cases), UVB or NB-UVB phototherapy (7 cases), and discontinuation of sorafenib (10 cases)(Table). All of these treatments led to improvement of the eruption with the exception of 1 case in which hand involvement was recalcitrant to therapy.9 Of the 10 cases in which sorafenib was discontinued, rechallenge at a lower dose was performed in 6 cases (including patient 1)3,4,6,7,9 with recurrence of psoriasiform rash seen in 5 cases (including patient 1)(Table).4,6,7,9 These data strongly implicate sorafenib as the direct cause of these psoriasiform eruptions. In the 3 cases in which sorafenib was not discontinued (including patient 3), there was notable improvement of the eruption with NB-UVB phototherapy.1,2
Vascular endothelial growth factor is overexpressed on psoriatic keratinocytes, contributes to epidermal hyperplasia, and induces angiogenesis in the dermis.12 The development of psoriasiform eruptions in patients treated with sorafenib seems paradoxical, as this drug has been considered as potential therapy for psoriasis due to its ability to block VEGF receptor signaling. Indeed, an improvement of psoriasis has been reported in 1 case of a patient treated with sorafenib13 and in multiple patients with psoriasis treated with other VEGF antagonists (eg, bevacizumab).14 The underlying mechanisms by which sorafenib induced or exacerbated psoriasis are not entirely clear. Palmoplantar hyperkeratosis, keratosis pilaris–like eruption, multiple cysts, eruptive keratoacanthomas, and squamous cell carcinoma have been described in patients treated with sorafenib, supporting the hypothesis that treatment with sorafenib alters keratinocyte proliferation and differentiation.15 In addition, B-Raf inhibitors such as imatinib are known to induce or exacerbate psoriasiform dermatitis.16 The activity of sorafenib resulting in psoriasis may be specific to RAF kinase inhibition, as there are no reports in the literature that describe psoriasiform dermatitis with agents that preferentially block other sorafenib targets such as VEGF receptor, stem cell growth factor receptor, or platelet-derived growth factor receptor. Future studies are needed to fully elucidate the underlying mechanisms by which sorafenib induces or exacerbates psoriasiform dermatitis and whether the severity of the drug eruption correlates with the antitumor efficacy of sorafenib.
Conclusion
Although psoriasiform drug eruptions secondary to sorafenib are not life-threatening, they impact quality of life with associated pain, pruritus, infection, and limitation of daily activities. Dose reduction or discontinuation of sorafenib resulted in resolution of the psoriasiform dermatitis; however, as demonstrated in 3 cases (including patient 3),1,2 psoriasiform dermatitis can be managed while maintaining the patient on sorafenib so that treatment of the malignancy is not compromised.
- Hung CT, Chiang CP, Wu BY. Sorafenib-induced psoriasis and hand-foot skin reaction responded dramatically to systemic narrowband ultraviolet B phototherapy. J Dermatol. 2012;39:1076-1077.
- González-López M, Yáñez S, Val-Bernal JF, et al. Psoriasiform skin eruption associated with sorafenib therapy. Indian J Dermatol Venereol Leprol. 2011;77:614-615.
- Diamantis ML, Chon SY. Sorafenib-induced psoriasiform eruption in a patient with metastatic thyroid carcinoma. J Drugs Dermatol. 2010;9:169-171.
- Hsu MC, Chen CC. Psoriasis flare-ups following sorafenib therapy: a rare case. Dermatologica Sin. 2016;34:148-150.
- Yiu ZZ, Ali FR, Griffiths CE. Paradoxical exacerbation of chronic plaque psoriasis by sorafenib. Clin Exp Dermatol. 2016;41:407-409.
- I˙lknur T, Akarsu S, Çarsanbali S, et al. Sorafenib-associated psoriasiform eruption in a patient with hepatocellular carcinoma. J Drugs Dermatol. 2014;13:899-900.
- Maki N, Komine M, Takatsuka Y, et al. Pustular eruption induced by sorafenib in a case of psoriasis vulgaris. J Dermatol. 2013;40:299-300.
- Du-Thanh A, Girard C, Pageaux GP, et al. Sorafenib-induced annular pustular psoriasis (Milian-Katchoura type). Eur J Dermatol. 2013;23:900-901.
- Laquer V, Saedi N, Dann F, et al. Sorafenib-associated psoriasiform skin changes. Cutis. 2010;85:301-302.
- Ohashi T, Yamamoto T. Exacerbation of psoriasis with pustulation by sorafenib in a patient with metastatic hepatocellular carcinoma. Indian J Dermatol. 2019;64:75-77.
- Chu D, Lacouture ME, Fillos T, et al. Risk of hand-foot skin reaction with sorafenib: a systematic review and meta-analysis. Acta Oncol (Madr). 2008;47:176-186.
- Canavese M, Altruda F, Ruzicka T, et al. Vascular endothelial growth factor (VEGF) in the pathogenesis of psoriasis--a possible target for novel therapies? J Dermatol Sci. 2010;58:171-176.
- Fournier C, Tisman G. Sorafenib-associated remission of psoriasis in hypernephroma: case report. Dermatol Online J. 2010;16:17.
- Akman A, Yilmaz E, Mutlu H, et al. Complete remission of psoriasis following bevacizumab therapy for colon cancer. Clin Exp Dermatol. 2009;34:E202-E204.
- Kong HH, Turner ML. Array of cutaneous adverse effects associated with sorafenib. J Am Acad Dermatol. 2009;61:360-361.
- Atalay F, Kızılkılıç E, Ada RS. Imatinib-induced psoriasis. Turk J Haematol. 2013;30:216-218.
The expanded use of targeted anticancer agents such as sorafenib has revealed a growing spectrum of adverse cutaneous eruptions. We describe 3 patients with sorafenib-induced psoriasiform dermatitis and review the literature of only 10 other similar reported cases based on a search of PubMed, Web of Science, and American Society of Clinical Oncology abstracts using the terms psoriasis or psoriasiform dermatitis and sorafenib.1-10 We seek to increase awareness of this particular drug eruption in response to sorafenib and to describe potential effective treatment options, especially when sorafenib cannot be discontinued.
Case Reports
Patient 1
A 68-year-old man with chronic hepatitis B infection and hepatocellular carcinoma (HCC) was started on sorafenib 400 mg daily. After 2 months of treatment, he developed painful hyperkeratotic lesions on the bilateral palms and soles with formation of calluses and superficial blisters on an erythematous base that was consistent with hand-foot skin reaction (HFSR). He also had numerous erythematous thin papules and plaques with adherent white scale and yellow crust on the bilateral thighs, lower legs, forearms, dorsal hands, abdomen, back, and buttocks (Figure 1). He had no personal or family history of psoriasis, and blood tests were unremarkable. Histologic analysis of punch biopsies from the buttocks and right leg revealed focal parakeratosis with neutrophils and serous crust, acanthosis, mild spongiosis, and lymphocytes at the dermoepidermal junction and surrounding dermal vessels, consistent with psoriasiform dermatitis (Figure 2). Sorafenib was discontinued and the eruption began to resolve within a week. A lower dose of sorafenib (200 mg daily) was attempted and the psoriasiform eruption recurred.
Patient 2
An 82-year-old man with chronic hepatitis B infection and HCC with lung metastasis was treated with sorafenib 400 mg daily. One week after treatment, he developed painful, thick, erythematous lesions on acral surfaces, consistent with HFSR. The sorafenib dose was decreased to 200 mg daily and HFSR resolved. Four months later, he developed well-demarcated, erythematous, scaly plaques with peripheral pustules on the right thigh (Figure 3) and right shin. He had no personal or family history of psoriasis, and blood tests were unremarkable. Samples from the pustules were taken for bacterial culture and fungal stain, but both were negative. Histologic analysis of a punch biopsy from the right thigh revealed necrotic parakeratosis, spongiform pustules, mild acanthosis, and a perivascular lymphocytic infiltrate with many neutrophils in the dermis. These findings suggested a diagnosis of pustular psoriasis, pustular drug eruption, or acute generalized exanthematous pustulosis. Treatment was initiated with mometasone cream. The patient subsequently developed hemoptysis and ascites from sorafenib. Sorafenib was discontinued and his skin eruption gradually resolved.
Patient 3
A 45-year-old woman with history of acute myeloid leukemia (AML) was started on sorafenib 200 mg twice daily as part of a clinical pilot study to maintain remission following an allogeneic bone marrow transplant. Four months after beginning sorafenib, the patient developed multiple well-defined, erythematous, thin papules and plaques with overlying flaky white scale on the bilateral upper extremities and trunk and scattered on the bilateral upper thighs (Figure 4) along with abdominal pain. Her other medical history, physical findings, and laboratory results were unremarkable, and there was no personal or family history of psoriasis. Her oncologist suspected that the eruption and symptoms were due to sorafenib and reduced the dose to 200 mg daily. Histologic analysis of a punch biopsy specimen revealed subcorneal neutrophilic collections with mild spongiosis and mild perivascular inflammatory infiltrate composed of lymphocytes and neutrophils (Figure 5). Direct immunofluorescence was negative for antibody or complement deposition. A bone marrow biopsy was negative for AML recurrence. The patient was continued on sorafenib to prevent AML recurrence, and she was started on triamcinolone cream 0.1% twice daily. Two weeks later, the eruption worsened and the patient was started on oral hydroxyzine for pruritus and narrowband UVB (NB-UVB) phototherapy 3 times a week. After 9 applications of NB-UVB phototherapy, there was complete resolution of the eruption.
Comment
Sorafenib is an oral tyrosine kinase inhibitor that blocks tumor cell proliferation and angiogenesis due to its activity against vascular endothelial growth factor (VEGF) receptor, platelet-derived growth factor receptor, stem cell growth factor receptor, and rapidly accelerated fibrosarcoma kinases.11 It is primarily used for the treatment of solid tumors, such as advanced renal cell carcinoma, unresectable HCC, and thyroid carcinoma, and more recently has been expanded for treatment of AML due to potential inhibition of FMS-like tyrosine kinase 3 receptor. Although dermatologic toxicity is a common adverse event during treatment with sorafenib,11 reports of psoriasiform drug eruptions are rare.
Review of Cases
Based on our literature search, there are 10 previously reported cases of psoriasiform drug eruption secondary to sorafenib. Of the 13 total cases (including the 3 patients in this report), 7 patients had a history of psoriasis; most were middle-aged men; and the treatment with sorafenib was for solid tumors, primarily HCC with the exception of patient 3 from the current report who was treated for AML (Table). In all cases, the dose of sorafenib ranged from 200 to 800 mg daily. In 5 cases, HFSR preceded (as with patient 2 in the current report) or presented concurrently (as with patient 1 in the current report) with the onset of psoriasiform rash.1,3,5
Of the 13 total cases, patients with a history of psoriasis generally developed the eruption in a shorter period of time after starting sorafenib (eg, days to 2 months) compared to those without a history of psoriasis (eg, 2 to 9 months)(Table), suggesting that patients with preexisting psoriasis more rapidly developed the drug eruption than patients without a history. In these patients with a history of psoriasis, all had long-standing mild to moderate stable plaque psoriasis, with the exception of 1 case in which the type of psoriasis was not described (Table).7 The presentation of the drug eruption following sorafenib varied from psoriasiform drug eruption (5 patients, including patient 3),2,3,6,9 pustular psoriasis (5 patients, including patient 2),4,7,8,10 and plaque psoriasis (3 patients, including patient 1).1,5 Interestingly, 5 of 6 patients with a history of plaque psoriasis presented with pustular psoriasis or psoriasiform drug eruption after treatment with sorafenib.4-6,8-10 These results suggest a causal relationship between sorafenib and exacerbation of preexisting psoriasis.
In the 13 total cases, treatments included mid- to high-potency topical steroids (10 cases), UVB or NB-UVB phototherapy (7 cases), and discontinuation of sorafenib (10 cases)(Table). All of these treatments led to improvement of the eruption with the exception of 1 case in which hand involvement was recalcitrant to therapy.9 Of the 10 cases in which sorafenib was discontinued, rechallenge at a lower dose was performed in 6 cases (including patient 1)3,4,6,7,9 with recurrence of psoriasiform rash seen in 5 cases (including patient 1)(Table).4,6,7,9 These data strongly implicate sorafenib as the direct cause of these psoriasiform eruptions. In the 3 cases in which sorafenib was not discontinued (including patient 3), there was notable improvement of the eruption with NB-UVB phototherapy.1,2
Vascular endothelial growth factor is overexpressed on psoriatic keratinocytes, contributes to epidermal hyperplasia, and induces angiogenesis in the dermis.12 The development of psoriasiform eruptions in patients treated with sorafenib seems paradoxical, as this drug has been considered as potential therapy for psoriasis due to its ability to block VEGF receptor signaling. Indeed, an improvement of psoriasis has been reported in 1 case of a patient treated with sorafenib13 and in multiple patients with psoriasis treated with other VEGF antagonists (eg, bevacizumab).14 The underlying mechanisms by which sorafenib induced or exacerbated psoriasis are not entirely clear. Palmoplantar hyperkeratosis, keratosis pilaris–like eruption, multiple cysts, eruptive keratoacanthomas, and squamous cell carcinoma have been described in patients treated with sorafenib, supporting the hypothesis that treatment with sorafenib alters keratinocyte proliferation and differentiation.15 In addition, B-Raf inhibitors such as imatinib are known to induce or exacerbate psoriasiform dermatitis.16 The activity of sorafenib resulting in psoriasis may be specific to RAF kinase inhibition, as there are no reports in the literature that describe psoriasiform dermatitis with agents that preferentially block other sorafenib targets such as VEGF receptor, stem cell growth factor receptor, or platelet-derived growth factor receptor. Future studies are needed to fully elucidate the underlying mechanisms by which sorafenib induces or exacerbates psoriasiform dermatitis and whether the severity of the drug eruption correlates with the antitumor efficacy of sorafenib.
Conclusion
Although psoriasiform drug eruptions secondary to sorafenib are not life-threatening, they impact quality of life with associated pain, pruritus, infection, and limitation of daily activities. Dose reduction or discontinuation of sorafenib resulted in resolution of the psoriasiform dermatitis; however, as demonstrated in 3 cases (including patient 3),1,2 psoriasiform dermatitis can be managed while maintaining the patient on sorafenib so that treatment of the malignancy is not compromised.
The expanded use of targeted anticancer agents such as sorafenib has revealed a growing spectrum of adverse cutaneous eruptions. We describe 3 patients with sorafenib-induced psoriasiform dermatitis and review the literature of only 10 other similar reported cases based on a search of PubMed, Web of Science, and American Society of Clinical Oncology abstracts using the terms psoriasis or psoriasiform dermatitis and sorafenib.1-10 We seek to increase awareness of this particular drug eruption in response to sorafenib and to describe potential effective treatment options, especially when sorafenib cannot be discontinued.
Case Reports
Patient 1
A 68-year-old man with chronic hepatitis B infection and hepatocellular carcinoma (HCC) was started on sorafenib 400 mg daily. After 2 months of treatment, he developed painful hyperkeratotic lesions on the bilateral palms and soles with formation of calluses and superficial blisters on an erythematous base that was consistent with hand-foot skin reaction (HFSR). He also had numerous erythematous thin papules and plaques with adherent white scale and yellow crust on the bilateral thighs, lower legs, forearms, dorsal hands, abdomen, back, and buttocks (Figure 1). He had no personal or family history of psoriasis, and blood tests were unremarkable. Histologic analysis of punch biopsies from the buttocks and right leg revealed focal parakeratosis with neutrophils and serous crust, acanthosis, mild spongiosis, and lymphocytes at the dermoepidermal junction and surrounding dermal vessels, consistent with psoriasiform dermatitis (Figure 2). Sorafenib was discontinued and the eruption began to resolve within a week. A lower dose of sorafenib (200 mg daily) was attempted and the psoriasiform eruption recurred.
Patient 2
An 82-year-old man with chronic hepatitis B infection and HCC with lung metastasis was treated with sorafenib 400 mg daily. One week after treatment, he developed painful, thick, erythematous lesions on acral surfaces, consistent with HFSR. The sorafenib dose was decreased to 200 mg daily and HFSR resolved. Four months later, he developed well-demarcated, erythematous, scaly plaques with peripheral pustules on the right thigh (Figure 3) and right shin. He had no personal or family history of psoriasis, and blood tests were unremarkable. Samples from the pustules were taken for bacterial culture and fungal stain, but both were negative. Histologic analysis of a punch biopsy from the right thigh revealed necrotic parakeratosis, spongiform pustules, mild acanthosis, and a perivascular lymphocytic infiltrate with many neutrophils in the dermis. These findings suggested a diagnosis of pustular psoriasis, pustular drug eruption, or acute generalized exanthematous pustulosis. Treatment was initiated with mometasone cream. The patient subsequently developed hemoptysis and ascites from sorafenib. Sorafenib was discontinued and his skin eruption gradually resolved.
Patient 3
A 45-year-old woman with history of acute myeloid leukemia (AML) was started on sorafenib 200 mg twice daily as part of a clinical pilot study to maintain remission following an allogeneic bone marrow transplant. Four months after beginning sorafenib, the patient developed multiple well-defined, erythematous, thin papules and plaques with overlying flaky white scale on the bilateral upper extremities and trunk and scattered on the bilateral upper thighs (Figure 4) along with abdominal pain. Her other medical history, physical findings, and laboratory results were unremarkable, and there was no personal or family history of psoriasis. Her oncologist suspected that the eruption and symptoms were due to sorafenib and reduced the dose to 200 mg daily. Histologic analysis of a punch biopsy specimen revealed subcorneal neutrophilic collections with mild spongiosis and mild perivascular inflammatory infiltrate composed of lymphocytes and neutrophils (Figure 5). Direct immunofluorescence was negative for antibody or complement deposition. A bone marrow biopsy was negative for AML recurrence. The patient was continued on sorafenib to prevent AML recurrence, and she was started on triamcinolone cream 0.1% twice daily. Two weeks later, the eruption worsened and the patient was started on oral hydroxyzine for pruritus and narrowband UVB (NB-UVB) phototherapy 3 times a week. After 9 applications of NB-UVB phototherapy, there was complete resolution of the eruption.
Comment
Sorafenib is an oral tyrosine kinase inhibitor that blocks tumor cell proliferation and angiogenesis due to its activity against vascular endothelial growth factor (VEGF) receptor, platelet-derived growth factor receptor, stem cell growth factor receptor, and rapidly accelerated fibrosarcoma kinases.11 It is primarily used for the treatment of solid tumors, such as advanced renal cell carcinoma, unresectable HCC, and thyroid carcinoma, and more recently has been expanded for treatment of AML due to potential inhibition of FMS-like tyrosine kinase 3 receptor. Although dermatologic toxicity is a common adverse event during treatment with sorafenib,11 reports of psoriasiform drug eruptions are rare.
Review of Cases
Based on our literature search, there are 10 previously reported cases of psoriasiform drug eruption secondary to sorafenib. Of the 13 total cases (including the 3 patients in this report), 7 patients had a history of psoriasis; most were middle-aged men; and the treatment with sorafenib was for solid tumors, primarily HCC with the exception of patient 3 from the current report who was treated for AML (Table). In all cases, the dose of sorafenib ranged from 200 to 800 mg daily. In 5 cases, HFSR preceded (as with patient 2 in the current report) or presented concurrently (as with patient 1 in the current report) with the onset of psoriasiform rash.1,3,5
Of the 13 total cases, patients with a history of psoriasis generally developed the eruption in a shorter period of time after starting sorafenib (eg, days to 2 months) compared to those without a history of psoriasis (eg, 2 to 9 months)(Table), suggesting that patients with preexisting psoriasis more rapidly developed the drug eruption than patients without a history. In these patients with a history of psoriasis, all had long-standing mild to moderate stable plaque psoriasis, with the exception of 1 case in which the type of psoriasis was not described (Table).7 The presentation of the drug eruption following sorafenib varied from psoriasiform drug eruption (5 patients, including patient 3),2,3,6,9 pustular psoriasis (5 patients, including patient 2),4,7,8,10 and plaque psoriasis (3 patients, including patient 1).1,5 Interestingly, 5 of 6 patients with a history of plaque psoriasis presented with pustular psoriasis or psoriasiform drug eruption after treatment with sorafenib.4-6,8-10 These results suggest a causal relationship between sorafenib and exacerbation of preexisting psoriasis.
In the 13 total cases, treatments included mid- to high-potency topical steroids (10 cases), UVB or NB-UVB phototherapy (7 cases), and discontinuation of sorafenib (10 cases)(Table). All of these treatments led to improvement of the eruption with the exception of 1 case in which hand involvement was recalcitrant to therapy.9 Of the 10 cases in which sorafenib was discontinued, rechallenge at a lower dose was performed in 6 cases (including patient 1)3,4,6,7,9 with recurrence of psoriasiform rash seen in 5 cases (including patient 1)(Table).4,6,7,9 These data strongly implicate sorafenib as the direct cause of these psoriasiform eruptions. In the 3 cases in which sorafenib was not discontinued (including patient 3), there was notable improvement of the eruption with NB-UVB phototherapy.1,2
Vascular endothelial growth factor is overexpressed on psoriatic keratinocytes, contributes to epidermal hyperplasia, and induces angiogenesis in the dermis.12 The development of psoriasiform eruptions in patients treated with sorafenib seems paradoxical, as this drug has been considered as potential therapy for psoriasis due to its ability to block VEGF receptor signaling. Indeed, an improvement of psoriasis has been reported in 1 case of a patient treated with sorafenib13 and in multiple patients with psoriasis treated with other VEGF antagonists (eg, bevacizumab).14 The underlying mechanisms by which sorafenib induced or exacerbated psoriasis are not entirely clear. Palmoplantar hyperkeratosis, keratosis pilaris–like eruption, multiple cysts, eruptive keratoacanthomas, and squamous cell carcinoma have been described in patients treated with sorafenib, supporting the hypothesis that treatment with sorafenib alters keratinocyte proliferation and differentiation.15 In addition, B-Raf inhibitors such as imatinib are known to induce or exacerbate psoriasiform dermatitis.16 The activity of sorafenib resulting in psoriasis may be specific to RAF kinase inhibition, as there are no reports in the literature that describe psoriasiform dermatitis with agents that preferentially block other sorafenib targets such as VEGF receptor, stem cell growth factor receptor, or platelet-derived growth factor receptor. Future studies are needed to fully elucidate the underlying mechanisms by which sorafenib induces or exacerbates psoriasiform dermatitis and whether the severity of the drug eruption correlates with the antitumor efficacy of sorafenib.
Conclusion
Although psoriasiform drug eruptions secondary to sorafenib are not life-threatening, they impact quality of life with associated pain, pruritus, infection, and limitation of daily activities. Dose reduction or discontinuation of sorafenib resulted in resolution of the psoriasiform dermatitis; however, as demonstrated in 3 cases (including patient 3),1,2 psoriasiform dermatitis can be managed while maintaining the patient on sorafenib so that treatment of the malignancy is not compromised.
- Hung CT, Chiang CP, Wu BY. Sorafenib-induced psoriasis and hand-foot skin reaction responded dramatically to systemic narrowband ultraviolet B phototherapy. J Dermatol. 2012;39:1076-1077.
- González-López M, Yáñez S, Val-Bernal JF, et al. Psoriasiform skin eruption associated with sorafenib therapy. Indian J Dermatol Venereol Leprol. 2011;77:614-615.
- Diamantis ML, Chon SY. Sorafenib-induced psoriasiform eruption in a patient with metastatic thyroid carcinoma. J Drugs Dermatol. 2010;9:169-171.
- Hsu MC, Chen CC. Psoriasis flare-ups following sorafenib therapy: a rare case. Dermatologica Sin. 2016;34:148-150.
- Yiu ZZ, Ali FR, Griffiths CE. Paradoxical exacerbation of chronic plaque psoriasis by sorafenib. Clin Exp Dermatol. 2016;41:407-409.
- I˙lknur T, Akarsu S, Çarsanbali S, et al. Sorafenib-associated psoriasiform eruption in a patient with hepatocellular carcinoma. J Drugs Dermatol. 2014;13:899-900.
- Maki N, Komine M, Takatsuka Y, et al. Pustular eruption induced by sorafenib in a case of psoriasis vulgaris. J Dermatol. 2013;40:299-300.
- Du-Thanh A, Girard C, Pageaux GP, et al. Sorafenib-induced annular pustular psoriasis (Milian-Katchoura type). Eur J Dermatol. 2013;23:900-901.
- Laquer V, Saedi N, Dann F, et al. Sorafenib-associated psoriasiform skin changes. Cutis. 2010;85:301-302.
- Ohashi T, Yamamoto T. Exacerbation of psoriasis with pustulation by sorafenib in a patient with metastatic hepatocellular carcinoma. Indian J Dermatol. 2019;64:75-77.
- Chu D, Lacouture ME, Fillos T, et al. Risk of hand-foot skin reaction with sorafenib: a systematic review and meta-analysis. Acta Oncol (Madr). 2008;47:176-186.
- Canavese M, Altruda F, Ruzicka T, et al. Vascular endothelial growth factor (VEGF) in the pathogenesis of psoriasis--a possible target for novel therapies? J Dermatol Sci. 2010;58:171-176.
- Fournier C, Tisman G. Sorafenib-associated remission of psoriasis in hypernephroma: case report. Dermatol Online J. 2010;16:17.
- Akman A, Yilmaz E, Mutlu H, et al. Complete remission of psoriasis following bevacizumab therapy for colon cancer. Clin Exp Dermatol. 2009;34:E202-E204.
- Kong HH, Turner ML. Array of cutaneous adverse effects associated with sorafenib. J Am Acad Dermatol. 2009;61:360-361.
- Atalay F, Kızılkılıç E, Ada RS. Imatinib-induced psoriasis. Turk J Haematol. 2013;30:216-218.
- Hung CT, Chiang CP, Wu BY. Sorafenib-induced psoriasis and hand-foot skin reaction responded dramatically to systemic narrowband ultraviolet B phototherapy. J Dermatol. 2012;39:1076-1077.
- González-López M, Yáñez S, Val-Bernal JF, et al. Psoriasiform skin eruption associated with sorafenib therapy. Indian J Dermatol Venereol Leprol. 2011;77:614-615.
- Diamantis ML, Chon SY. Sorafenib-induced psoriasiform eruption in a patient with metastatic thyroid carcinoma. J Drugs Dermatol. 2010;9:169-171.
- Hsu MC, Chen CC. Psoriasis flare-ups following sorafenib therapy: a rare case. Dermatologica Sin. 2016;34:148-150.
- Yiu ZZ, Ali FR, Griffiths CE. Paradoxical exacerbation of chronic plaque psoriasis by sorafenib. Clin Exp Dermatol. 2016;41:407-409.
- I˙lknur T, Akarsu S, Çarsanbali S, et al. Sorafenib-associated psoriasiform eruption in a patient with hepatocellular carcinoma. J Drugs Dermatol. 2014;13:899-900.
- Maki N, Komine M, Takatsuka Y, et al. Pustular eruption induced by sorafenib in a case of psoriasis vulgaris. J Dermatol. 2013;40:299-300.
- Du-Thanh A, Girard C, Pageaux GP, et al. Sorafenib-induced annular pustular psoriasis (Milian-Katchoura type). Eur J Dermatol. 2013;23:900-901.
- Laquer V, Saedi N, Dann F, et al. Sorafenib-associated psoriasiform skin changes. Cutis. 2010;85:301-302.
- Ohashi T, Yamamoto T. Exacerbation of psoriasis with pustulation by sorafenib in a patient with metastatic hepatocellular carcinoma. Indian J Dermatol. 2019;64:75-77.
- Chu D, Lacouture ME, Fillos T, et al. Risk of hand-foot skin reaction with sorafenib: a systematic review and meta-analysis. Acta Oncol (Madr). 2008;47:176-186.
- Canavese M, Altruda F, Ruzicka T, et al. Vascular endothelial growth factor (VEGF) in the pathogenesis of psoriasis--a possible target for novel therapies? J Dermatol Sci. 2010;58:171-176.
- Fournier C, Tisman G. Sorafenib-associated remission of psoriasis in hypernephroma: case report. Dermatol Online J. 2010;16:17.
- Akman A, Yilmaz E, Mutlu H, et al. Complete remission of psoriasis following bevacizumab therapy for colon cancer. Clin Exp Dermatol. 2009;34:E202-E204.
- Kong HH, Turner ML. Array of cutaneous adverse effects associated with sorafenib. J Am Acad Dermatol. 2009;61:360-361.
- Atalay F, Kızılkılıç E, Ada RS. Imatinib-induced psoriasis. Turk J Haematol. 2013;30:216-218.
Practice Points
- The use of targeted anticancer agents continues to expand. With this expansion, the number and type of cutaneous adverse events continues to increase.
- Although sorafenib is known to cause various dermatologic side effects, there are few reports of psoriasiform dermatitis.
- Increased awareness of sorafenib-induced psoriasiform dermatitis and its management is vital to prevent discontinuation of potentially life-saving anticancer therapy.
Gastrostomy tube placement associated with higher pneumonia recurrence in children with neurologic impairment
according to findings published in Pediatrics.
Five of the remaining seven strategies – gastrostomy tube placement, chest physiotherapy, outpatient antibiotics before hospitalization, and clinic visit before and after index hospitalization – were associated with increased recurrence, Jody L. Lin, MD, of the department of pediatrics at Stanford (Calif.) University, and colleagues reported. Oral secretion management and gastric acid suppression were associated with increased risk, but to a lesser extent.
The researchers examined the outcomes of the prevention strategies because, although children with neurologic impairment are more susceptible to community-acquired pneumonia, current guidelines are based mostly on expert opinion. The study included 3,632 children aged 21 years or younger with neurologic impairment and at least one hospitalization for pneumonia, who were enrolled in the California Children’s Services program between July 1, 2009, and June 30, 2014.
Propensity-score matching based on factors such as age, sex, household income, as well as characteristics of index hospitalization, showed decreased odds of recurrence only with receipt of dental care (adjusted odds ratio, 0.64; 95% confidence interval, 0.49-0.85), whereas increased odds were seen with other recommended prevention strategies, such as chest physiotherapy (aOR, 2.03; 95% CI, 1.29-3.20), receipt of antibiotics before hospitalization (aOR, 1.42; 95% CI, 1.06-1.92), and clinic visit before (aOR, 1.30; 95% CI, 1.11-1.52) and after index hospitalization (aOR, 1.72; 95% CI, 1.35-2.20).
The greatest increased odds, however, were seen with new gastrostomy tube placement (aOR, 2.15; 95% CI, 1.63-2.85).
The investigators noted that the biggest limitation of this study was the potential for residual confounding by indication even after adjustment, whereby certain interventions were provided to patients deemed more clinically severe to begin with. A strength of the study is its longitudinal nature.
“Our results suggest that more attention should be paid to dental health for children with [neurologic impairment],” the researchers wrote, although they noted that dental care “remains the most common unmet health care need” for children with special health care needs.
The findings also “support a clinical trial of dental care for prevention of severe pneumonia in children with [neurologic impairment] and do not support the widespread use of gastrostomy tubes for that purpose,” they added.
The study was funded by the National Institutes of Health. Dr. Lin received support from the NIH and the Clinical Excellence Research Center. The authors reported that they had no conflicts of interest.
[email protected]
SOURCE: Lin JL et al. Pediatrics. 2019 Sep 19. doi: 10.1542/peds.2019-0543.
according to findings published in Pediatrics.
Five of the remaining seven strategies – gastrostomy tube placement, chest physiotherapy, outpatient antibiotics before hospitalization, and clinic visit before and after index hospitalization – were associated with increased recurrence, Jody L. Lin, MD, of the department of pediatrics at Stanford (Calif.) University, and colleagues reported. Oral secretion management and gastric acid suppression were associated with increased risk, but to a lesser extent.
The researchers examined the outcomes of the prevention strategies because, although children with neurologic impairment are more susceptible to community-acquired pneumonia, current guidelines are based mostly on expert opinion. The study included 3,632 children aged 21 years or younger with neurologic impairment and at least one hospitalization for pneumonia, who were enrolled in the California Children’s Services program between July 1, 2009, and June 30, 2014.
Propensity-score matching based on factors such as age, sex, household income, as well as characteristics of index hospitalization, showed decreased odds of recurrence only with receipt of dental care (adjusted odds ratio, 0.64; 95% confidence interval, 0.49-0.85), whereas increased odds were seen with other recommended prevention strategies, such as chest physiotherapy (aOR, 2.03; 95% CI, 1.29-3.20), receipt of antibiotics before hospitalization (aOR, 1.42; 95% CI, 1.06-1.92), and clinic visit before (aOR, 1.30; 95% CI, 1.11-1.52) and after index hospitalization (aOR, 1.72; 95% CI, 1.35-2.20).
The greatest increased odds, however, were seen with new gastrostomy tube placement (aOR, 2.15; 95% CI, 1.63-2.85).
The investigators noted that the biggest limitation of this study was the potential for residual confounding by indication even after adjustment, whereby certain interventions were provided to patients deemed more clinically severe to begin with. A strength of the study is its longitudinal nature.
“Our results suggest that more attention should be paid to dental health for children with [neurologic impairment],” the researchers wrote, although they noted that dental care “remains the most common unmet health care need” for children with special health care needs.
The findings also “support a clinical trial of dental care for prevention of severe pneumonia in children with [neurologic impairment] and do not support the widespread use of gastrostomy tubes for that purpose,” they added.
The study was funded by the National Institutes of Health. Dr. Lin received support from the NIH and the Clinical Excellence Research Center. The authors reported that they had no conflicts of interest.
[email protected]
SOURCE: Lin JL et al. Pediatrics. 2019 Sep 19. doi: 10.1542/peds.2019-0543.
according to findings published in Pediatrics.
Five of the remaining seven strategies – gastrostomy tube placement, chest physiotherapy, outpatient antibiotics before hospitalization, and clinic visit before and after index hospitalization – were associated with increased recurrence, Jody L. Lin, MD, of the department of pediatrics at Stanford (Calif.) University, and colleagues reported. Oral secretion management and gastric acid suppression were associated with increased risk, but to a lesser extent.
The researchers examined the outcomes of the prevention strategies because, although children with neurologic impairment are more susceptible to community-acquired pneumonia, current guidelines are based mostly on expert opinion. The study included 3,632 children aged 21 years or younger with neurologic impairment and at least one hospitalization for pneumonia, who were enrolled in the California Children’s Services program between July 1, 2009, and June 30, 2014.
Propensity-score matching based on factors such as age, sex, household income, as well as characteristics of index hospitalization, showed decreased odds of recurrence only with receipt of dental care (adjusted odds ratio, 0.64; 95% confidence interval, 0.49-0.85), whereas increased odds were seen with other recommended prevention strategies, such as chest physiotherapy (aOR, 2.03; 95% CI, 1.29-3.20), receipt of antibiotics before hospitalization (aOR, 1.42; 95% CI, 1.06-1.92), and clinic visit before (aOR, 1.30; 95% CI, 1.11-1.52) and after index hospitalization (aOR, 1.72; 95% CI, 1.35-2.20).
The greatest increased odds, however, were seen with new gastrostomy tube placement (aOR, 2.15; 95% CI, 1.63-2.85).
The investigators noted that the biggest limitation of this study was the potential for residual confounding by indication even after adjustment, whereby certain interventions were provided to patients deemed more clinically severe to begin with. A strength of the study is its longitudinal nature.
“Our results suggest that more attention should be paid to dental health for children with [neurologic impairment],” the researchers wrote, although they noted that dental care “remains the most common unmet health care need” for children with special health care needs.
The findings also “support a clinical trial of dental care for prevention of severe pneumonia in children with [neurologic impairment] and do not support the widespread use of gastrostomy tubes for that purpose,” they added.
The study was funded by the National Institutes of Health. Dr. Lin received support from the NIH and the Clinical Excellence Research Center. The authors reported that they had no conflicts of interest.
[email protected]
SOURCE: Lin JL et al. Pediatrics. 2019 Sep 19. doi: 10.1542/peds.2019-0543.
FROM PEDIATRICS
Key clinical point: Gastrostomy tube placement is associated with higher pneumonia recurrence in children with neurologic impairment, and dental care is linked to decreased recurrence.
Major finding: There was an increased odds of pneumonia recurrence with new gastrostomy tube placement (adjusted odds ratio, 2.15; 95% confidence interval, 1.63-2.85) and decreased odds with dental care (aOR, 0.64; 95% CI, 0.49-0.85).
Study details: A comparative effectiveness study of a retrospective cohort of 3,632 children with neurologic impairment and at least one hospitalization for pneumonia, enrolled in California Children’s Services from July 1, 2009, to June 30, 2014.
Disclosures: The study was funded by the National Institutes of Health. Dr. Lin received support from the NIH and the Clinical Excellence Research Center. The authors reported that they had no conflicts of interest.
Source: Lin JL et al. Pediatrics. 2019 Sep 19. doi: 10.1542/peds.2019-0543.
Rash on lower legs and abdomen

The FP suspected leukocytoclastic vasculitis (LCV) and, with the patient’s consent, performed a 4-mm punch biopsy on a well-developed lesion on the abdomen. Biopsies on the abdomen heal faster than the legs and may provide a better specimen to the pathologist. (See the Watch & Learn video on “Punch biopsy.”)
The biopsy confirmed the diagnosis of LCV. This is the most commonly seen form of small vessel vasculitis. LCV causes acute inflammation and necrosis of venules in the dermis. The term leukocytoclastic vasculitis describes the histologic pattern produced when leukocytes break apart into fragments. The purpura begins as asymptomatic localized areas of cutaneous hemorrhage that become palpable.
Discrete lesions are most commonly seen on the lower extremities, but they may occur on any dependent area. Small lesions may itch and be painful, but nodules, ulcers, and bullae may be more painful. Lesions appear in crops, last for 1 to 4 weeks, and may heal with residual scarring and hyperpigmentation. Patients may experience a single episode caused by a drug reaction or viral infection or have multiple episodes associated with rheumatologic diseases. LCV usually is self-limited and confined to the skin.
To make the diagnosis, look for the presence of 3 or more of the following:
- age > 16 years;
- use of a possible offending drug in temporal relation to the symptoms;
- palpable purpura;
- maculopapular rash; and
- neutrophils around an arteriole or venule in a biopsy of a skin lesion.
In this case, the use of ibuprofen was the most likely precipitating event. Blood and urine tests did not show any renal or other organ system involvement. The patient was warned to not use ibuprofen in the future and that acetaminophen is a safer option for him. He was given topical triamcinolone cream 0.1% to apply twice daily for symptomatic relief. In this case, oral prednisone was not prescribed because the numerous potential adverse effects of prednisone outweighed the benefits. The vasculitis resolved in 4 weeks without any sequelae.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Mayeaux EJ, Usatine R, Martin N, et al. Vasculitis. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas and Synopsis of Family Medicine. 3rd ed. New York, NY: McGraw-Hill; 2019:1169-1173.
To learn more about the newest 3rd edition of the Color Atlas and Synopsis of Family Medicine, see: https://www.amazon.com/Color-Atlas-Synopsis-Family-Medicine/dp/1259862046/
You can get the 3rd edition of the Color Atlas and Synopsis of Family Medicine as an app by clicking on this link: https://usatinemedia.com/app/color-atlas-of-family-medicine/
The FP suspected leukocytoclastic vasculitis (LCV) and, with the patient’s consent, performed a 4-mm punch biopsy on a well-developed lesion on the abdomen. Biopsies on the abdomen heal faster than the legs and may provide a better specimen to the pathologist. (See the Watch & Learn video on “Punch biopsy.”)
The biopsy confirmed the diagnosis of LCV. This is the most commonly seen form of small vessel vasculitis. LCV causes acute inflammation and necrosis of venules in the dermis. The term leukocytoclastic vasculitis describes the histologic pattern produced when leukocytes break apart into fragments. The purpura begins as asymptomatic localized areas of cutaneous hemorrhage that become palpable.
Discrete lesions are most commonly seen on the lower extremities, but they may occur on any dependent area. Small lesions may itch and be painful, but nodules, ulcers, and bullae may be more painful. Lesions appear in crops, last for 1 to 4 weeks, and may heal with residual scarring and hyperpigmentation. Patients may experience a single episode caused by a drug reaction or viral infection or have multiple episodes associated with rheumatologic diseases. LCV usually is self-limited and confined to the skin.
To make the diagnosis, look for the presence of 3 or more of the following:
- age > 16 years;
- use of a possible offending drug in temporal relation to the symptoms;
- palpable purpura;
- maculopapular rash; and
- neutrophils around an arteriole or venule in a biopsy of a skin lesion.
In this case, the use of ibuprofen was the most likely precipitating event. Blood and urine tests did not show any renal or other organ system involvement. The patient was warned to not use ibuprofen in the future and that acetaminophen is a safer option for him. He was given topical triamcinolone cream 0.1% to apply twice daily for symptomatic relief. In this case, oral prednisone was not prescribed because the numerous potential adverse effects of prednisone outweighed the benefits. The vasculitis resolved in 4 weeks without any sequelae.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Mayeaux EJ, Usatine R, Martin N, et al. Vasculitis. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas and Synopsis of Family Medicine. 3rd ed. New York, NY: McGraw-Hill; 2019:1169-1173.
To learn more about the newest 3rd edition of the Color Atlas and Synopsis of Family Medicine, see: https://www.amazon.com/Color-Atlas-Synopsis-Family-Medicine/dp/1259862046/
You can get the 3rd edition of the Color Atlas and Synopsis of Family Medicine as an app by clicking on this link: https://usatinemedia.com/app/color-atlas-of-family-medicine/
The FP suspected leukocytoclastic vasculitis (LCV) and, with the patient’s consent, performed a 4-mm punch biopsy on a well-developed lesion on the abdomen. Biopsies on the abdomen heal faster than the legs and may provide a better specimen to the pathologist. (See the Watch & Learn video on “Punch biopsy.”)
The biopsy confirmed the diagnosis of LCV. This is the most commonly seen form of small vessel vasculitis. LCV causes acute inflammation and necrosis of venules in the dermis. The term leukocytoclastic vasculitis describes the histologic pattern produced when leukocytes break apart into fragments. The purpura begins as asymptomatic localized areas of cutaneous hemorrhage that become palpable.
Discrete lesions are most commonly seen on the lower extremities, but they may occur on any dependent area. Small lesions may itch and be painful, but nodules, ulcers, and bullae may be more painful. Lesions appear in crops, last for 1 to 4 weeks, and may heal with residual scarring and hyperpigmentation. Patients may experience a single episode caused by a drug reaction or viral infection or have multiple episodes associated with rheumatologic diseases. LCV usually is self-limited and confined to the skin.
To make the diagnosis, look for the presence of 3 or more of the following:
- age > 16 years;
- use of a possible offending drug in temporal relation to the symptoms;
- palpable purpura;
- maculopapular rash; and
- neutrophils around an arteriole or venule in a biopsy of a skin lesion.
In this case, the use of ibuprofen was the most likely precipitating event. Blood and urine tests did not show any renal or other organ system involvement. The patient was warned to not use ibuprofen in the future and that acetaminophen is a safer option for him. He was given topical triamcinolone cream 0.1% to apply twice daily for symptomatic relief. In this case, oral prednisone was not prescribed because the numerous potential adverse effects of prednisone outweighed the benefits. The vasculitis resolved in 4 weeks without any sequelae.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Mayeaux EJ, Usatine R, Martin N, et al. Vasculitis. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas and Synopsis of Family Medicine. 3rd ed. New York, NY: McGraw-Hill; 2019:1169-1173.
To learn more about the newest 3rd edition of the Color Atlas and Synopsis of Family Medicine, see: https://www.amazon.com/Color-Atlas-Synopsis-Family-Medicine/dp/1259862046/
You can get the 3rd edition of the Color Atlas and Synopsis of Family Medicine as an app by clicking on this link: https://usatinemedia.com/app/color-atlas-of-family-medicine/

Growing vaping habit may lead to nicotine addiction in adolescents
and in 2019 almost 12% of high school seniors reported that they were vaping every day, according to data from the Monitoring the Future surveys.
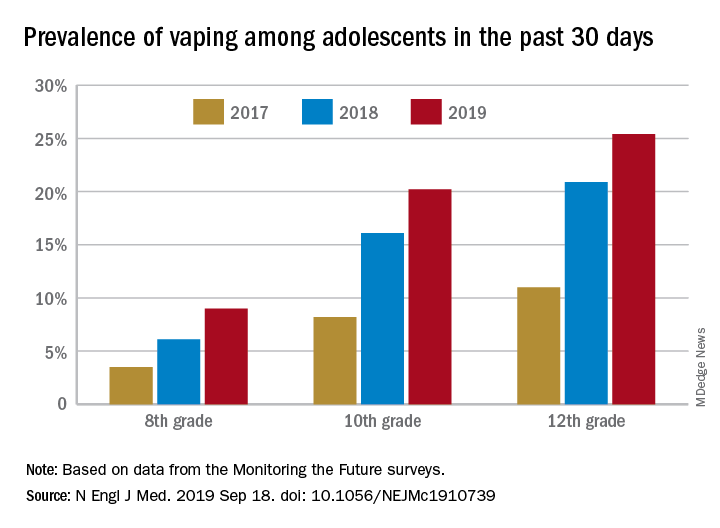
Daily use – defined as vaping on 20 or more of the previous 30 days – was reported by 6.9% of 10th-grade and 1.9% of 8th-grade respondents in the 2019 survey, which was the first time use in these age groups was assessed. “The substantial levels of daily vaping suggest the development of nicotine addiction,” Richard Miech, PhD, and associates said Sept. 18 in the New England Journal of Medicine.
From 2017 to 2019, e-cigarette use over the previous 30 days increased from 11.0% to 25.4% among 12th graders, from 8.2% to 20.2% in 10th graders, and from 3.5% to 9.0% of 8th graders, suggesting that “current efforts by the vaping industry, government agencies, and schools have thus far proved insufficient to stop the rapid spread of nicotine vaping among adolescents,” the investigators wrote.
By 2019, over 40% of 12th-grade students reported ever using e-cigarettes, along with more than 36% of 10th graders and almost 21% of 8th graders. Corresponding figures for past 12-month use were 35.1%, 31.1%, and 16.1%, they reported.
“New efforts are needed to protect youth from using nicotine during adolescence, when the developing brain is particularly susceptible to permanent changes from nicotine use and when almost all nicotine addiction is established,” the investigators wrote.
The analysis was funded by a grant from the National Institute on Drug Abuse to Dr. Miech.
SOURCE: Miech R et al. N Engl J Med. 2019 Sep 18. doi: 10.1056/NEJMc1910739.
and in 2019 almost 12% of high school seniors reported that they were vaping every day, according to data from the Monitoring the Future surveys.
Daily use – defined as vaping on 20 or more of the previous 30 days – was reported by 6.9% of 10th-grade and 1.9% of 8th-grade respondents in the 2019 survey, which was the first time use in these age groups was assessed. “The substantial levels of daily vaping suggest the development of nicotine addiction,” Richard Miech, PhD, and associates said Sept. 18 in the New England Journal of Medicine.
From 2017 to 2019, e-cigarette use over the previous 30 days increased from 11.0% to 25.4% among 12th graders, from 8.2% to 20.2% in 10th graders, and from 3.5% to 9.0% of 8th graders, suggesting that “current efforts by the vaping industry, government agencies, and schools have thus far proved insufficient to stop the rapid spread of nicotine vaping among adolescents,” the investigators wrote.
By 2019, over 40% of 12th-grade students reported ever using e-cigarettes, along with more than 36% of 10th graders and almost 21% of 8th graders. Corresponding figures for past 12-month use were 35.1%, 31.1%, and 16.1%, they reported.
“New efforts are needed to protect youth from using nicotine during adolescence, when the developing brain is particularly susceptible to permanent changes from nicotine use and when almost all nicotine addiction is established,” the investigators wrote.
The analysis was funded by a grant from the National Institute on Drug Abuse to Dr. Miech.
SOURCE: Miech R et al. N Engl J Med. 2019 Sep 18. doi: 10.1056/NEJMc1910739.
and in 2019 almost 12% of high school seniors reported that they were vaping every day, according to data from the Monitoring the Future surveys.
Daily use – defined as vaping on 20 or more of the previous 30 days – was reported by 6.9% of 10th-grade and 1.9% of 8th-grade respondents in the 2019 survey, which was the first time use in these age groups was assessed. “The substantial levels of daily vaping suggest the development of nicotine addiction,” Richard Miech, PhD, and associates said Sept. 18 in the New England Journal of Medicine.
From 2017 to 2019, e-cigarette use over the previous 30 days increased from 11.0% to 25.4% among 12th graders, from 8.2% to 20.2% in 10th graders, and from 3.5% to 9.0% of 8th graders, suggesting that “current efforts by the vaping industry, government agencies, and schools have thus far proved insufficient to stop the rapid spread of nicotine vaping among adolescents,” the investigators wrote.
By 2019, over 40% of 12th-grade students reported ever using e-cigarettes, along with more than 36% of 10th graders and almost 21% of 8th graders. Corresponding figures for past 12-month use were 35.1%, 31.1%, and 16.1%, they reported.
“New efforts are needed to protect youth from using nicotine during adolescence, when the developing brain is particularly susceptible to permanent changes from nicotine use and when almost all nicotine addiction is established,” the investigators wrote.
The analysis was funded by a grant from the National Institute on Drug Abuse to Dr. Miech.
SOURCE: Miech R et al. N Engl J Med. 2019 Sep 18. doi: 10.1056/NEJMc1910739.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: Adolescents who use e-cigarettes every day may be developing nicotine addiction.
Major finding: In 2019, almost 12% of high school seniors were vaping every day.
Study details: Monitoring the Future surveys nationally representative samples of 8th-, 10th-, and 12th-grade students each year.
Disclosures: The analysis was funded by a grant from the National Institute on Drug Abuse to Dr. Miech.
Source: Miech R et al. N Engl J Med. 2019 Sep 18. doi: 10.1056/NEJMc1910739.
Daily polypill lowers BP, cholesterol in underserved population
A daily polypill regimen improved cardiovascular risk factors in a socioeconomically vulnerable minority population, in a randomized controlled trial.
Patients at a federally qualified community health center in Alabama who received treatment with a combination pill for 1 year had greater reductions in systolic blood pressure and LDL cholesterol than did patients who received usual care, according to results published online on Sept. 19 in the New England Journal of Medicine.
“The simplicity and low cost of the polypill regimen make this approach attractive” when barriers such as lack of income, underinsurance, and difficulty attending clinic visits are common, said first author Daniel Muñoz, MD, of Vanderbilt University in Nashville, and coinvestigators. The investigators obtained the pills at a cost of $26 per month per participant.
People with low socioeconomic status and those who are nonwhite have high cardiovascular mortality, and the southeastern United States and rural areas have disproportionately high levels of cardiovascular disease burden, according to the investigators. The rates at which people with low socioeconomic status receive treatment for hypertension and hypercholesterolemia – leading cardiovascular disease risk factors – “are strikingly low,” Dr. Muñoz and colleagues said.
To assess the effectiveness of a polypill-based strategy in an underserved population with low socioeconomic status, the researchers conducted the randomized trial.
They enrolled 303 adults without cardiovascular disease, and 148 of the patients were randomized to receive the polypill, which contained generic versions of atorvastatin (10 mg), amlodipine (2.5 mg), losartan (25 mg), and hydrochlorothiazide (12.5 mg). The remaining 155 patients received usual care. All participants scheduled 2-month and 12-month follow-up visits.
The participants had an average age of 56 years, 60% were women, and more than 95% were black. More than 70% had an annual household income of less than $15,000. Baseline characteristics of the treatment groups did not significantly differ.
At baseline, the average BP was 140/83 mm Hg, and the average LDL cholesterol level was 113 mg/dL.
In all, 91% of the participants completed the 12-month trial visit. Average systolic BP decreased by 9 mm Hg in the group that received the polypill, compared with 2 mm Hg in the group that received usual care. Average LDL cholesterol level decreased by 15 mg/dL in the polypill group, versus 4 mg/dL in the usual-care group.
Changes in other medications
Clinicians discontinued or reduced doses of other antihypertensive or lipid-lowering medications in 44% of the patients in the polypill group and none in the usual-care group. Clinicians escalated therapy in 2% of the participants in the polypill group and in 10% of the usual-care group.
Side effects in participants who received the polypill included a 1% incidence of myalgias and a 1% incidence of hypotension or light-headedness. Liver function test results were normal.
Five serious adverse events that occurred during the trial – two in the polypill group and three in the usual-care group – were judged to be unrelated to the trial by a data and safety monitoring board.
The authors noted that limitations of the trial include its open-label design and that it was conducted at a single center.
“It is important to emphasize that use of the polypill does not preclude individualized, add-on therapies for residual elevations in blood-pressure or cholesterol levels, as judged by a patient’s physician,” said Dr. Muñoz and colleagues. “We recognize that a ‘one size fits all’ approach to cardiovascular disease prevention runs counter to current trends in precision medicine, in which clinical, genomic, and lifestyle factors are used for the development of individualized treatment strategies. Although the precision approach has clear virtues, a broader approach may benefit patients who face barriers to accessing the full advantages of precision medicine.”
The study was supported by grants from the American Heart Association Strategically Focused Prevention Research Network and the National Institutes of Health. One author disclosed personal fees from Novartis outside the study.
SOURCE: Muñoz D et al. N Engl J Med. 2019 Sep 18;381(12):1114-23. doi: 10.1056/NEJMoa1815359.
A daily polypill regimen improved cardiovascular risk factors in a socioeconomically vulnerable minority population, in a randomized controlled trial.
Patients at a federally qualified community health center in Alabama who received treatment with a combination pill for 1 year had greater reductions in systolic blood pressure and LDL cholesterol than did patients who received usual care, according to results published online on Sept. 19 in the New England Journal of Medicine.
“The simplicity and low cost of the polypill regimen make this approach attractive” when barriers such as lack of income, underinsurance, and difficulty attending clinic visits are common, said first author Daniel Muñoz, MD, of Vanderbilt University in Nashville, and coinvestigators. The investigators obtained the pills at a cost of $26 per month per participant.
People with low socioeconomic status and those who are nonwhite have high cardiovascular mortality, and the southeastern United States and rural areas have disproportionately high levels of cardiovascular disease burden, according to the investigators. The rates at which people with low socioeconomic status receive treatment for hypertension and hypercholesterolemia – leading cardiovascular disease risk factors – “are strikingly low,” Dr. Muñoz and colleagues said.
To assess the effectiveness of a polypill-based strategy in an underserved population with low socioeconomic status, the researchers conducted the randomized trial.
They enrolled 303 adults without cardiovascular disease, and 148 of the patients were randomized to receive the polypill, which contained generic versions of atorvastatin (10 mg), amlodipine (2.5 mg), losartan (25 mg), and hydrochlorothiazide (12.5 mg). The remaining 155 patients received usual care. All participants scheduled 2-month and 12-month follow-up visits.
The participants had an average age of 56 years, 60% were women, and more than 95% were black. More than 70% had an annual household income of less than $15,000. Baseline characteristics of the treatment groups did not significantly differ.
At baseline, the average BP was 140/83 mm Hg, and the average LDL cholesterol level was 113 mg/dL.
In all, 91% of the participants completed the 12-month trial visit. Average systolic BP decreased by 9 mm Hg in the group that received the polypill, compared with 2 mm Hg in the group that received usual care. Average LDL cholesterol level decreased by 15 mg/dL in the polypill group, versus 4 mg/dL in the usual-care group.
Changes in other medications
Clinicians discontinued or reduced doses of other antihypertensive or lipid-lowering medications in 44% of the patients in the polypill group and none in the usual-care group. Clinicians escalated therapy in 2% of the participants in the polypill group and in 10% of the usual-care group.
Side effects in participants who received the polypill included a 1% incidence of myalgias and a 1% incidence of hypotension or light-headedness. Liver function test results were normal.
Five serious adverse events that occurred during the trial – two in the polypill group and three in the usual-care group – were judged to be unrelated to the trial by a data and safety monitoring board.
The authors noted that limitations of the trial include its open-label design and that it was conducted at a single center.
“It is important to emphasize that use of the polypill does not preclude individualized, add-on therapies for residual elevations in blood-pressure or cholesterol levels, as judged by a patient’s physician,” said Dr. Muñoz and colleagues. “We recognize that a ‘one size fits all’ approach to cardiovascular disease prevention runs counter to current trends in precision medicine, in which clinical, genomic, and lifestyle factors are used for the development of individualized treatment strategies. Although the precision approach has clear virtues, a broader approach may benefit patients who face barriers to accessing the full advantages of precision medicine.”
The study was supported by grants from the American Heart Association Strategically Focused Prevention Research Network and the National Institutes of Health. One author disclosed personal fees from Novartis outside the study.
SOURCE: Muñoz D et al. N Engl J Med. 2019 Sep 18;381(12):1114-23. doi: 10.1056/NEJMoa1815359.
A daily polypill regimen improved cardiovascular risk factors in a socioeconomically vulnerable minority population, in a randomized controlled trial.
Patients at a federally qualified community health center in Alabama who received treatment with a combination pill for 1 year had greater reductions in systolic blood pressure and LDL cholesterol than did patients who received usual care, according to results published online on Sept. 19 in the New England Journal of Medicine.
“The simplicity and low cost of the polypill regimen make this approach attractive” when barriers such as lack of income, underinsurance, and difficulty attending clinic visits are common, said first author Daniel Muñoz, MD, of Vanderbilt University in Nashville, and coinvestigators. The investigators obtained the pills at a cost of $26 per month per participant.
People with low socioeconomic status and those who are nonwhite have high cardiovascular mortality, and the southeastern United States and rural areas have disproportionately high levels of cardiovascular disease burden, according to the investigators. The rates at which people with low socioeconomic status receive treatment for hypertension and hypercholesterolemia – leading cardiovascular disease risk factors – “are strikingly low,” Dr. Muñoz and colleagues said.
To assess the effectiveness of a polypill-based strategy in an underserved population with low socioeconomic status, the researchers conducted the randomized trial.
They enrolled 303 adults without cardiovascular disease, and 148 of the patients were randomized to receive the polypill, which contained generic versions of atorvastatin (10 mg), amlodipine (2.5 mg), losartan (25 mg), and hydrochlorothiazide (12.5 mg). The remaining 155 patients received usual care. All participants scheduled 2-month and 12-month follow-up visits.
The participants had an average age of 56 years, 60% were women, and more than 95% were black. More than 70% had an annual household income of less than $15,000. Baseline characteristics of the treatment groups did not significantly differ.
At baseline, the average BP was 140/83 mm Hg, and the average LDL cholesterol level was 113 mg/dL.
In all, 91% of the participants completed the 12-month trial visit. Average systolic BP decreased by 9 mm Hg in the group that received the polypill, compared with 2 mm Hg in the group that received usual care. Average LDL cholesterol level decreased by 15 mg/dL in the polypill group, versus 4 mg/dL in the usual-care group.
Changes in other medications
Clinicians discontinued or reduced doses of other antihypertensive or lipid-lowering medications in 44% of the patients in the polypill group and none in the usual-care group. Clinicians escalated therapy in 2% of the participants in the polypill group and in 10% of the usual-care group.
Side effects in participants who received the polypill included a 1% incidence of myalgias and a 1% incidence of hypotension or light-headedness. Liver function test results were normal.
Five serious adverse events that occurred during the trial – two in the polypill group and three in the usual-care group – were judged to be unrelated to the trial by a data and safety monitoring board.
The authors noted that limitations of the trial include its open-label design and that it was conducted at a single center.
“It is important to emphasize that use of the polypill does not preclude individualized, add-on therapies for residual elevations in blood-pressure or cholesterol levels, as judged by a patient’s physician,” said Dr. Muñoz and colleagues. “We recognize that a ‘one size fits all’ approach to cardiovascular disease prevention runs counter to current trends in precision medicine, in which clinical, genomic, and lifestyle factors are used for the development of individualized treatment strategies. Although the precision approach has clear virtues, a broader approach may benefit patients who face barriers to accessing the full advantages of precision medicine.”
The study was supported by grants from the American Heart Association Strategically Focused Prevention Research Network and the National Institutes of Health. One author disclosed personal fees from Novartis outside the study.
SOURCE: Muñoz D et al. N Engl J Med. 2019 Sep 18;381(12):1114-23. doi: 10.1056/NEJMoa1815359.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: A daily polypill regimen may improve cardiovascular disease prevention in underserved populations.
Major finding: Mean LDL cholesterol levels decreased by 15 mg/dL in the polypill group, vs. 4 mg/dL in the usual-care group.
Study details: An open-label, randomized trial that enrolled 303 adults without cardiovascular disease at a federally qualified community health center in Alabama.
Disclosures: The study was supported by grants from the American Heart Association Strategically Focused Prevention Research Network and the National Institutes of Health. One author disclosed personal fees from Novartis outside the study.
Source: Muñoz D et al. N Engl J Med. 2019;381(12):1114-23. doi: 10.1056/NEJMoa1815359.
Obesity, moderate drinking linked to psoriatic arthritis
a study has found.
Around one in five people with psoriasis will develop psoriatic arthritis (PsA), wrote Amelia Green of the University of Bath (England) and coauthors in the British Journal of Dermatology.
Previous studies have explored possible links between obesity, alcohol consumption, or smoking, and an increased risk of developing psoriatic arthritis. However, some of these studies found conflicting results or had limitations such as measuring only a single exposure.
In a cohort study, the Ms. Green and her colleagues examined data from the U.K. Clinical Practice Research Datalink for 90,189 individuals with psoriasis, 1,409 of whom were subsequently also diagnosed with psoriatic arthritis.
The analysis showed a significant association between increasing body mass index (BMI) and increasing odds of developing psoriatic arthritis. Compared with individuals with a BMI below 25 kg/m2, those with a BMI of 25.0-29.9 had a 79% greater odds of psoriatic arthritis, those with a BMI of 30.0-34.9 had a 2.10-fold greater odds, and those with a BMI at or above 35 had a 2.68-fold greater odds of developing psoriatic arthritis (P for trend less than .001). Adjustment for potential confounders such as sex, age, duration and severity of psoriasis, diabetes, smoking, and alcohol use slightly attenuated the association, but it remained statistically significant.
Researchers also examined the cumulative effect of lower BMIs over time, and found that over a 10-year period, reductions in BMI were associated with reductions in the risk of developing PsA, compared with remaining at the same BMI over that time.
“Here we have shown for the first time that losing weight over time could reduce the risk of developing PsA in a population with documented psoriasis,” the authors wrote. “As the effect of obesity on the risk of developing PsA may in fact occur with some delay and change over time, our analysis took into account both updated BMI measurements over time and the possible nonlinear and cumulative effects of BMI, which have not previously been investigated.”
Commenting on the mechanisms underlying the association between obesity and the development of PsA, the authors noted that adipose tissue is a source of inflammatory mediators such as adipokines and proinflammatory cytokines, which could lead to the development of PsA. Increasing body weight also could cause microtraumas of the connective tissue between tendon and bone, which may act as an initiating pathogenic event for PsA.
Moderate drinkers – defined as 0.1–3.0 drinks per day – had 57% higher odds of developing PsA when compared with nondrinkers, but former drinkers or heavy drinkers did not have an increased risk.
The study also didn’t see any effect of either past or current smoking on the risk of PsA, although there was a nonsignificant interaction with obesity that hinted at increased odds.
“While we found no association between smoking status and the development of PsA in people with psoriasis, further analysis revealed that the effect of smoking on the risk of PsA was possibly mediated through the effect of BMI on PsA; in other words, the protective effect of smoking may be associated with lower BMI among smokers,” the authors wrote.
Patients who developed PsA were also more likely to be younger (mean age of 44.7 years vs. 48.5 years), have severe psoriasis, and have had the disease for a shorter duration.
The study was funded by the National Institute for Health Research, and the authors declared grants from the funder during the conduct of the study. No other conflicts of interest were declared.
SOURCE: Green A et al. Br J Dermatol. 2019 Jun 18. doi: 10.1111/bjd.18227
a study has found.
Around one in five people with psoriasis will develop psoriatic arthritis (PsA), wrote Amelia Green of the University of Bath (England) and coauthors in the British Journal of Dermatology.
Previous studies have explored possible links between obesity, alcohol consumption, or smoking, and an increased risk of developing psoriatic arthritis. However, some of these studies found conflicting results or had limitations such as measuring only a single exposure.
In a cohort study, the Ms. Green and her colleagues examined data from the U.K. Clinical Practice Research Datalink for 90,189 individuals with psoriasis, 1,409 of whom were subsequently also diagnosed with psoriatic arthritis.
The analysis showed a significant association between increasing body mass index (BMI) and increasing odds of developing psoriatic arthritis. Compared with individuals with a BMI below 25 kg/m2, those with a BMI of 25.0-29.9 had a 79% greater odds of psoriatic arthritis, those with a BMI of 30.0-34.9 had a 2.10-fold greater odds, and those with a BMI at or above 35 had a 2.68-fold greater odds of developing psoriatic arthritis (P for trend less than .001). Adjustment for potential confounders such as sex, age, duration and severity of psoriasis, diabetes, smoking, and alcohol use slightly attenuated the association, but it remained statistically significant.
Researchers also examined the cumulative effect of lower BMIs over time, and found that over a 10-year period, reductions in BMI were associated with reductions in the risk of developing PsA, compared with remaining at the same BMI over that time.
“Here we have shown for the first time that losing weight over time could reduce the risk of developing PsA in a population with documented psoriasis,” the authors wrote. “As the effect of obesity on the risk of developing PsA may in fact occur with some delay and change over time, our analysis took into account both updated BMI measurements over time and the possible nonlinear and cumulative effects of BMI, which have not previously been investigated.”
Commenting on the mechanisms underlying the association between obesity and the development of PsA, the authors noted that adipose tissue is a source of inflammatory mediators such as adipokines and proinflammatory cytokines, which could lead to the development of PsA. Increasing body weight also could cause microtraumas of the connective tissue between tendon and bone, which may act as an initiating pathogenic event for PsA.
Moderate drinkers – defined as 0.1–3.0 drinks per day – had 57% higher odds of developing PsA when compared with nondrinkers, but former drinkers or heavy drinkers did not have an increased risk.
The study also didn’t see any effect of either past or current smoking on the risk of PsA, although there was a nonsignificant interaction with obesity that hinted at increased odds.
“While we found no association between smoking status and the development of PsA in people with psoriasis, further analysis revealed that the effect of smoking on the risk of PsA was possibly mediated through the effect of BMI on PsA; in other words, the protective effect of smoking may be associated with lower BMI among smokers,” the authors wrote.
Patients who developed PsA were also more likely to be younger (mean age of 44.7 years vs. 48.5 years), have severe psoriasis, and have had the disease for a shorter duration.
The study was funded by the National Institute for Health Research, and the authors declared grants from the funder during the conduct of the study. No other conflicts of interest were declared.
SOURCE: Green A et al. Br J Dermatol. 2019 Jun 18. doi: 10.1111/bjd.18227
a study has found.
Around one in five people with psoriasis will develop psoriatic arthritis (PsA), wrote Amelia Green of the University of Bath (England) and coauthors in the British Journal of Dermatology.
Previous studies have explored possible links between obesity, alcohol consumption, or smoking, and an increased risk of developing psoriatic arthritis. However, some of these studies found conflicting results or had limitations such as measuring only a single exposure.
In a cohort study, the Ms. Green and her colleagues examined data from the U.K. Clinical Practice Research Datalink for 90,189 individuals with psoriasis, 1,409 of whom were subsequently also diagnosed with psoriatic arthritis.
The analysis showed a significant association between increasing body mass index (BMI) and increasing odds of developing psoriatic arthritis. Compared with individuals with a BMI below 25 kg/m2, those with a BMI of 25.0-29.9 had a 79% greater odds of psoriatic arthritis, those with a BMI of 30.0-34.9 had a 2.10-fold greater odds, and those with a BMI at or above 35 had a 2.68-fold greater odds of developing psoriatic arthritis (P for trend less than .001). Adjustment for potential confounders such as sex, age, duration and severity of psoriasis, diabetes, smoking, and alcohol use slightly attenuated the association, but it remained statistically significant.
Researchers also examined the cumulative effect of lower BMIs over time, and found that over a 10-year period, reductions in BMI were associated with reductions in the risk of developing PsA, compared with remaining at the same BMI over that time.
“Here we have shown for the first time that losing weight over time could reduce the risk of developing PsA in a population with documented psoriasis,” the authors wrote. “As the effect of obesity on the risk of developing PsA may in fact occur with some delay and change over time, our analysis took into account both updated BMI measurements over time and the possible nonlinear and cumulative effects of BMI, which have not previously been investigated.”
Commenting on the mechanisms underlying the association between obesity and the development of PsA, the authors noted that adipose tissue is a source of inflammatory mediators such as adipokines and proinflammatory cytokines, which could lead to the development of PsA. Increasing body weight also could cause microtraumas of the connective tissue between tendon and bone, which may act as an initiating pathogenic event for PsA.
Moderate drinkers – defined as 0.1–3.0 drinks per day – had 57% higher odds of developing PsA when compared with nondrinkers, but former drinkers or heavy drinkers did not have an increased risk.
The study also didn’t see any effect of either past or current smoking on the risk of PsA, although there was a nonsignificant interaction with obesity that hinted at increased odds.
“While we found no association between smoking status and the development of PsA in people with psoriasis, further analysis revealed that the effect of smoking on the risk of PsA was possibly mediated through the effect of BMI on PsA; in other words, the protective effect of smoking may be associated with lower BMI among smokers,” the authors wrote.
Patients who developed PsA were also more likely to be younger (mean age of 44.7 years vs. 48.5 years), have severe psoriasis, and have had the disease for a shorter duration.
The study was funded by the National Institute for Health Research, and the authors declared grants from the funder during the conduct of the study. No other conflicts of interest were declared.
SOURCE: Green A et al. Br J Dermatol. 2019 Jun 18. doi: 10.1111/bjd.18227
FROM THE BRITISH JOURNAL OF DERMATOLOGY
Cancer with meatballs and the unkindest frozen cut
Two great tastes that cause cancer together
Spaghetti and meatballs. They go together like chocolate and peanut butter. It almost feels wrong to eat one without the other; but if you’re worried about cancer, you may have to go meatless.

The latest blow to an enjoyable meal comes courtesy of a study published in Molecular Nutrition & Food Research, which tested how lycopene – a carotenoid found in tomatoes that has notable anticancer properties – is absorbed by the body when in the presence of iron, which meat contains plenty of. When the study subjects drank a tomato-based shake infused with iron, lycopene was far less present in the blood and digestive system than in subjects who drank an iron-free tomato shake.
The study authors claim that either the iron is oxidizing with the lycopene or that the iron turns the mix of tomato and fat into something like separated salad dressing, preventing everything from mixing together when it enters the body.
Tastes like chicken
It’s an enduring oncologic mystery: How can some cancer cells endure what should be a lethal therapeutic beating, only to bounce off the canvas after an eight-count to deliver a devastating relapse-counterpunch of their own?
A new study offers an unsavory answer: cannibalism.
Turns out that dining on one’s weaker cancer cell neighbors during a chemotherapy barrage provides just enough energy to rope-a-dope and stage a late-round comeback.
Breast cancer cells with wild-type TP53 genes are particularly prone to revival after taking a beating at the hands of doxorubicin or other chemotherapy drugs. Like many of their cancerous compatriots, they retreat to a corner of the therapy ring during chemo and go gloves up in a state of senescence.
But researchers at Tulane University noticed that, in the midst of that pharmaceutical pummeling, those senescent wild-type TP53 cells start doing something that their other senescent, cancerous neighbors don’t: They engulf other cancer cells. Why? Seems those breast cancer cells with the wild-type TP53 gloves are equipped with gene expression programs similar to macrophages.
What’s more, the cannibals’ appetite for fellow cells appears to confer a survival advantage when the chemo rounds end.
We at the Bureau of LOTME will resist the impulse to ring out this item with a tasteless Donner Party punchline. Instead, we’ll indulge our high-brow inner child by retooling an elementary school comedy classic.
Why don’t cancer-cell cannibals eat cancer-cell comedians? They taste funny.
Poop, what is it good for?
One thing you can cross off the list: Cutting meat.

That might seem pretty obvious, but there’s actually a bit of history here. In a book published in 1998, anthropologist Wade Davis shared an account of an elderly Inuit man trapped alone in a storm. He had no tools and no food, so he made a knife out of his own frozen stool and used it to kill and butcher a dog.
That story, which has since become something of an urban legend, directly inspired the career of another anthropologist, Metin Eren, PhD, of Kent State University in Ohio. As director of the school’s laboratory of experimental archaeology, Dr. Eren decided that the time had come to prove or disprove the poop-knife hypothesis.
First, he and his team had to make such a knife. To produce the needed raw materials, Dr. Eren went on an 8-day “Arctic diet” that included lots of beef, turkey, and salmon, with some applesauce and butternut squash risotto thrown in, while a colleague stuck to a more Western diet. Their samples were then frozen to –58° F and sharpened with metal files.
“I was surprised at how hard human feces could get when frozen,” Dr. Eren told Live Science. “I started to think, ‘Oh my gosh, this might actually work!’ ”
The team’s attempts to cut refrigerated pig hide, however, were not successful. “Like a crayon, it just left brown streaks on the meat – no slices at all,” he said.
Today’s lesson? Don’t meat your heroes or their poop knives; they’re sure to disappoint you.
Two great tastes that cause cancer together
Spaghetti and meatballs. They go together like chocolate and peanut butter. It almost feels wrong to eat one without the other; but if you’re worried about cancer, you may have to go meatless.
The latest blow to an enjoyable meal comes courtesy of a study published in Molecular Nutrition & Food Research, which tested how lycopene – a carotenoid found in tomatoes that has notable anticancer properties – is absorbed by the body when in the presence of iron, which meat contains plenty of. When the study subjects drank a tomato-based shake infused with iron, lycopene was far less present in the blood and digestive system than in subjects who drank an iron-free tomato shake.
The study authors claim that either the iron is oxidizing with the lycopene or that the iron turns the mix of tomato and fat into something like separated salad dressing, preventing everything from mixing together when it enters the body.
Tastes like chicken
It’s an enduring oncologic mystery: How can some cancer cells endure what should be a lethal therapeutic beating, only to bounce off the canvas after an eight-count to deliver a devastating relapse-counterpunch of their own?
A new study offers an unsavory answer: cannibalism.
Turns out that dining on one’s weaker cancer cell neighbors during a chemotherapy barrage provides just enough energy to rope-a-dope and stage a late-round comeback.
Breast cancer cells with wild-type TP53 genes are particularly prone to revival after taking a beating at the hands of doxorubicin or other chemotherapy drugs. Like many of their cancerous compatriots, they retreat to a corner of the therapy ring during chemo and go gloves up in a state of senescence.
But researchers at Tulane University noticed that, in the midst of that pharmaceutical pummeling, those senescent wild-type TP53 cells start doing something that their other senescent, cancerous neighbors don’t: They engulf other cancer cells. Why? Seems those breast cancer cells with the wild-type TP53 gloves are equipped with gene expression programs similar to macrophages.
What’s more, the cannibals’ appetite for fellow cells appears to confer a survival advantage when the chemo rounds end.
We at the Bureau of LOTME will resist the impulse to ring out this item with a tasteless Donner Party punchline. Instead, we’ll indulge our high-brow inner child by retooling an elementary school comedy classic.
Why don’t cancer-cell cannibals eat cancer-cell comedians? They taste funny.
Poop, what is it good for?
One thing you can cross off the list: Cutting meat.
That might seem pretty obvious, but there’s actually a bit of history here. In a book published in 1998, anthropologist Wade Davis shared an account of an elderly Inuit man trapped alone in a storm. He had no tools and no food, so he made a knife out of his own frozen stool and used it to kill and butcher a dog.
That story, which has since become something of an urban legend, directly inspired the career of another anthropologist, Metin Eren, PhD, of Kent State University in Ohio. As director of the school’s laboratory of experimental archaeology, Dr. Eren decided that the time had come to prove or disprove the poop-knife hypothesis.
First, he and his team had to make such a knife. To produce the needed raw materials, Dr. Eren went on an 8-day “Arctic diet” that included lots of beef, turkey, and salmon, with some applesauce and butternut squash risotto thrown in, while a colleague stuck to a more Western diet. Their samples were then frozen to –58° F and sharpened with metal files.
“I was surprised at how hard human feces could get when frozen,” Dr. Eren told Live Science. “I started to think, ‘Oh my gosh, this might actually work!’ ”
The team’s attempts to cut refrigerated pig hide, however, were not successful. “Like a crayon, it just left brown streaks on the meat – no slices at all,” he said.
Today’s lesson? Don’t meat your heroes or their poop knives; they’re sure to disappoint you.
Two great tastes that cause cancer together
Spaghetti and meatballs. They go together like chocolate and peanut butter. It almost feels wrong to eat one without the other; but if you’re worried about cancer, you may have to go meatless.
The latest blow to an enjoyable meal comes courtesy of a study published in Molecular Nutrition & Food Research, which tested how lycopene – a carotenoid found in tomatoes that has notable anticancer properties – is absorbed by the body when in the presence of iron, which meat contains plenty of. When the study subjects drank a tomato-based shake infused with iron, lycopene was far less present in the blood and digestive system than in subjects who drank an iron-free tomato shake.
The study authors claim that either the iron is oxidizing with the lycopene or that the iron turns the mix of tomato and fat into something like separated salad dressing, preventing everything from mixing together when it enters the body.
Tastes like chicken
It’s an enduring oncologic mystery: How can some cancer cells endure what should be a lethal therapeutic beating, only to bounce off the canvas after an eight-count to deliver a devastating relapse-counterpunch of their own?
A new study offers an unsavory answer: cannibalism.
Turns out that dining on one’s weaker cancer cell neighbors during a chemotherapy barrage provides just enough energy to rope-a-dope and stage a late-round comeback.
Breast cancer cells with wild-type TP53 genes are particularly prone to revival after taking a beating at the hands of doxorubicin or other chemotherapy drugs. Like many of their cancerous compatriots, they retreat to a corner of the therapy ring during chemo and go gloves up in a state of senescence.
But researchers at Tulane University noticed that, in the midst of that pharmaceutical pummeling, those senescent wild-type TP53 cells start doing something that their other senescent, cancerous neighbors don’t: They engulf other cancer cells. Why? Seems those breast cancer cells with the wild-type TP53 gloves are equipped with gene expression programs similar to macrophages.
What’s more, the cannibals’ appetite for fellow cells appears to confer a survival advantage when the chemo rounds end.
We at the Bureau of LOTME will resist the impulse to ring out this item with a tasteless Donner Party punchline. Instead, we’ll indulge our high-brow inner child by retooling an elementary school comedy classic.
Why don’t cancer-cell cannibals eat cancer-cell comedians? They taste funny.
Poop, what is it good for?
One thing you can cross off the list: Cutting meat.
That might seem pretty obvious, but there’s actually a bit of history here. In a book published in 1998, anthropologist Wade Davis shared an account of an elderly Inuit man trapped alone in a storm. He had no tools and no food, so he made a knife out of his own frozen stool and used it to kill and butcher a dog.
That story, which has since become something of an urban legend, directly inspired the career of another anthropologist, Metin Eren, PhD, of Kent State University in Ohio. As director of the school’s laboratory of experimental archaeology, Dr. Eren decided that the time had come to prove or disprove the poop-knife hypothesis.
First, he and his team had to make such a knife. To produce the needed raw materials, Dr. Eren went on an 8-day “Arctic diet” that included lots of beef, turkey, and salmon, with some applesauce and butternut squash risotto thrown in, while a colleague stuck to a more Western diet. Their samples were then frozen to –58° F and sharpened with metal files.
“I was surprised at how hard human feces could get when frozen,” Dr. Eren told Live Science. “I started to think, ‘Oh my gosh, this might actually work!’ ”
The team’s attempts to cut refrigerated pig hide, however, were not successful. “Like a crayon, it just left brown streaks on the meat – no slices at all,” he said.
Today’s lesson? Don’t meat your heroes or their poop knives; they’re sure to disappoint you.
Esketamine nasal spray may get expanded indication
COPENHAGEN – Esketamine nasal spray achieved rapid reduction of major depressive disorder symptoms in patients at imminent risk for suicide in a pair of pivotal phase 3 clinical trials known as ASPIRE-1 and ASPIRE-2, Carla M. Canuso, MD, reported at the annual congress of the European College of Neuropsychopharmacology.

These were groundbreaking studies, which addressed a major unmet need familiar to every mental health professional, given that more than 800,000 suicides per year occur worldwide. Standard antidepressants are of only limited value during the period of acute suicidal crisis because they take 4-6 weeks to work. Moreover, this population of seriously depressed and suicidal patients has been understudied.
“Patients with active suicidal ideation and intent are routinely excluded from antidepressant trials,” observed Dr. Canuso, a psychiatrist who serves as senior director of neuroscience clinical development at Janssen Research and Development in Titusville, N.J.
It’s very important for the field to know that we can actually study these patients safely and effectively in clinical trials,” she said.
ASPIRE-1 and -2 were identically designed, randomized, double-blind, placebo-controlled, multinational studies conducted in patients with moderate to severe major depressive disorder as evidenced by a baseline Montgomery-Åsberg Depression Rating Scale (MADRS) total score of about 40, along with moderate to extreme active suicidal ideation and intent as assessed using the Clinical Global Impression-Severity of Suicidality-Revised (CGI-SS-R).
“These were all patients in psychiatric crisis seeking clinical care,” according to Dr. Canuso.
All 456 participants in the two phase 3 studies underwent an initial 5-14 days of psychiatric hospitalization, during which they began treatment with esketamine nasal spray at 84 mg twice weekly or placebo coupled with comprehensive standard of care, which included a newly initiated and/or optimized oral antidepressant regimen.
The primary endpoint in the two clinical trials was the change in MADRS total score 24 hours after the first dose of study medication. The esketamine-treated patients demonstrated a mean reduction of 16.4 and 15.7 points, respectively, in the two trials, which was 3.82 points greater than in the pooled placebo group. This represents a clinically meaningful and statistically significant between-group difference.
The treatment effect size was even larger in some of the prespecified patient subgroups. Dr. Canuso drew attention to two key groups: In the roughly 60% of ASPIRE participants with a prior suicide attempt, esketamine resulted in a mean 4.81-point greater reduction in MADRS total score than placebo, and in the nearly 30% of participants with a suicide attempt during the past month, the difference was 5.22 points.
A word on the study design: Patients received intranasal esketamine at 84 mg per dose or placebo in double-blind fashion twice weekly for 4 weeks, thereby giving time for their oral antidepressant therapy to kick in, and were then followed on the conventional therapy out to 90 days.
A between-group difference in MADRS total score in favor of the esketamine group was evident as early as 4 hours after the first dose and continued through day 25, the end of the double-blind treatment period, at which point 54% and 47% of the esketamine-plus-conventional-antidepressant groups in the two trials had achieved remission as defined by a MADRS score of 12 or less, as had about one-third of the control group.
The prespecified key secondary efficacy endpoint in ASPIRE-1 and -2 was change in the CGI-SS-R 24 hours after the first dose. Both the esketamine and placebo-treated patients experienced significant improvement in this domain, with a disappointing absence of between-group statistical significance.
“We think that this may be due to the effect of acute hospitalization in diffusing the suicidal crisis,” Dr. Canuso said.
She noted, however, that other suicidality indices did show significant improvement in the esketamine-treated group during assessments at 4 hours, 24 hours, and 25 days after the first dose. For example, the double-blind esketamine-treated patients were 2.62-fold more likely than controls to show a significant improvement in MADRS Suicidal Thoughts at 4 hours after dose number one, and 6.15 times more likely to do so 4 hours after their day-25 dose. The CGI structured physician assessments of suicide risk and frequency of suicidal thinking, as well as patient-reported frequency of suicidal thinking, showed consistent favorable numeric trends for improvement with esketamine, with odds ratios of 1.46-2.11 from 4 hours through 25 days, although those results generally failed to achieve statistical significance.
In terms of safety, the rate of serious adverse events was just under 12% in both the esketamine and placebo arms. As in earlier studies, the most common adverse events associated with the novel antidepressant were dizziness, dissociation, nausea, and sleepiness, all several-fold more frequent than with placebo.
Esketamine is the S-enantiomer of racemic ketamine. It’s a noncompetitive N-methyl-D-aspartate receptor antagonist.
Janssen, which already markets intranasal esketamine as Spravato in the United States for treatment-resistant depression, plans to file for an expanded indication on the basis of the ASPIRE-1 and -2 results. The Food and Drug Administration already has granted Breakthrough Therapy designation for research on esketamine for reduction of major depression symptoms in patients with active suicidal ideation.
The ASPIRE studies were funded by Janssen.
COPENHAGEN – Esketamine nasal spray achieved rapid reduction of major depressive disorder symptoms in patients at imminent risk for suicide in a pair of pivotal phase 3 clinical trials known as ASPIRE-1 and ASPIRE-2, Carla M. Canuso, MD, reported at the annual congress of the European College of Neuropsychopharmacology.
These were groundbreaking studies, which addressed a major unmet need familiar to every mental health professional, given that more than 800,000 suicides per year occur worldwide. Standard antidepressants are of only limited value during the period of acute suicidal crisis because they take 4-6 weeks to work. Moreover, this population of seriously depressed and suicidal patients has been understudied.
“Patients with active suicidal ideation and intent are routinely excluded from antidepressant trials,” observed Dr. Canuso, a psychiatrist who serves as senior director of neuroscience clinical development at Janssen Research and Development in Titusville, N.J.
It’s very important for the field to know that we can actually study these patients safely and effectively in clinical trials,” she said.
ASPIRE-1 and -2 were identically designed, randomized, double-blind, placebo-controlled, multinational studies conducted in patients with moderate to severe major depressive disorder as evidenced by a baseline Montgomery-Åsberg Depression Rating Scale (MADRS) total score of about 40, along with moderate to extreme active suicidal ideation and intent as assessed using the Clinical Global Impression-Severity of Suicidality-Revised (CGI-SS-R).
“These were all patients in psychiatric crisis seeking clinical care,” according to Dr. Canuso.
All 456 participants in the two phase 3 studies underwent an initial 5-14 days of psychiatric hospitalization, during which they began treatment with esketamine nasal spray at 84 mg twice weekly or placebo coupled with comprehensive standard of care, which included a newly initiated and/or optimized oral antidepressant regimen.
The primary endpoint in the two clinical trials was the change in MADRS total score 24 hours after the first dose of study medication. The esketamine-treated patients demonstrated a mean reduction of 16.4 and 15.7 points, respectively, in the two trials, which was 3.82 points greater than in the pooled placebo group. This represents a clinically meaningful and statistically significant between-group difference.
The treatment effect size was even larger in some of the prespecified patient subgroups. Dr. Canuso drew attention to two key groups: In the roughly 60% of ASPIRE participants with a prior suicide attempt, esketamine resulted in a mean 4.81-point greater reduction in MADRS total score than placebo, and in the nearly 30% of participants with a suicide attempt during the past month, the difference was 5.22 points.
A word on the study design: Patients received intranasal esketamine at 84 mg per dose or placebo in double-blind fashion twice weekly for 4 weeks, thereby giving time for their oral antidepressant therapy to kick in, and were then followed on the conventional therapy out to 90 days.
A between-group difference in MADRS total score in favor of the esketamine group was evident as early as 4 hours after the first dose and continued through day 25, the end of the double-blind treatment period, at which point 54% and 47% of the esketamine-plus-conventional-antidepressant groups in the two trials had achieved remission as defined by a MADRS score of 12 or less, as had about one-third of the control group.
The prespecified key secondary efficacy endpoint in ASPIRE-1 and -2 was change in the CGI-SS-R 24 hours after the first dose. Both the esketamine and placebo-treated patients experienced significant improvement in this domain, with a disappointing absence of between-group statistical significance.
“We think that this may be due to the effect of acute hospitalization in diffusing the suicidal crisis,” Dr. Canuso said.
She noted, however, that other suicidality indices did show significant improvement in the esketamine-treated group during assessments at 4 hours, 24 hours, and 25 days after the first dose. For example, the double-blind esketamine-treated patients were 2.62-fold more likely than controls to show a significant improvement in MADRS Suicidal Thoughts at 4 hours after dose number one, and 6.15 times more likely to do so 4 hours after their day-25 dose. The CGI structured physician assessments of suicide risk and frequency of suicidal thinking, as well as patient-reported frequency of suicidal thinking, showed consistent favorable numeric trends for improvement with esketamine, with odds ratios of 1.46-2.11 from 4 hours through 25 days, although those results generally failed to achieve statistical significance.
In terms of safety, the rate of serious adverse events was just under 12% in both the esketamine and placebo arms. As in earlier studies, the most common adverse events associated with the novel antidepressant were dizziness, dissociation, nausea, and sleepiness, all several-fold more frequent than with placebo.
Esketamine is the S-enantiomer of racemic ketamine. It’s a noncompetitive N-methyl-D-aspartate receptor antagonist.
Janssen, which already markets intranasal esketamine as Spravato in the United States for treatment-resistant depression, plans to file for an expanded indication on the basis of the ASPIRE-1 and -2 results. The Food and Drug Administration already has granted Breakthrough Therapy designation for research on esketamine for reduction of major depression symptoms in patients with active suicidal ideation.
The ASPIRE studies were funded by Janssen.
COPENHAGEN – Esketamine nasal spray achieved rapid reduction of major depressive disorder symptoms in patients at imminent risk for suicide in a pair of pivotal phase 3 clinical trials known as ASPIRE-1 and ASPIRE-2, Carla M. Canuso, MD, reported at the annual congress of the European College of Neuropsychopharmacology.
These were groundbreaking studies, which addressed a major unmet need familiar to every mental health professional, given that more than 800,000 suicides per year occur worldwide. Standard antidepressants are of only limited value during the period of acute suicidal crisis because they take 4-6 weeks to work. Moreover, this population of seriously depressed and suicidal patients has been understudied.
“Patients with active suicidal ideation and intent are routinely excluded from antidepressant trials,” observed Dr. Canuso, a psychiatrist who serves as senior director of neuroscience clinical development at Janssen Research and Development in Titusville, N.J.
It’s very important for the field to know that we can actually study these patients safely and effectively in clinical trials,” she said.
ASPIRE-1 and -2 were identically designed, randomized, double-blind, placebo-controlled, multinational studies conducted in patients with moderate to severe major depressive disorder as evidenced by a baseline Montgomery-Åsberg Depression Rating Scale (MADRS) total score of about 40, along with moderate to extreme active suicidal ideation and intent as assessed using the Clinical Global Impression-Severity of Suicidality-Revised (CGI-SS-R).
“These were all patients in psychiatric crisis seeking clinical care,” according to Dr. Canuso.
All 456 participants in the two phase 3 studies underwent an initial 5-14 days of psychiatric hospitalization, during which they began treatment with esketamine nasal spray at 84 mg twice weekly or placebo coupled with comprehensive standard of care, which included a newly initiated and/or optimized oral antidepressant regimen.
The primary endpoint in the two clinical trials was the change in MADRS total score 24 hours after the first dose of study medication. The esketamine-treated patients demonstrated a mean reduction of 16.4 and 15.7 points, respectively, in the two trials, which was 3.82 points greater than in the pooled placebo group. This represents a clinically meaningful and statistically significant between-group difference.
The treatment effect size was even larger in some of the prespecified patient subgroups. Dr. Canuso drew attention to two key groups: In the roughly 60% of ASPIRE participants with a prior suicide attempt, esketamine resulted in a mean 4.81-point greater reduction in MADRS total score than placebo, and in the nearly 30% of participants with a suicide attempt during the past month, the difference was 5.22 points.
A word on the study design: Patients received intranasal esketamine at 84 mg per dose or placebo in double-blind fashion twice weekly for 4 weeks, thereby giving time for their oral antidepressant therapy to kick in, and were then followed on the conventional therapy out to 90 days.
A between-group difference in MADRS total score in favor of the esketamine group was evident as early as 4 hours after the first dose and continued through day 25, the end of the double-blind treatment period, at which point 54% and 47% of the esketamine-plus-conventional-antidepressant groups in the two trials had achieved remission as defined by a MADRS score of 12 or less, as had about one-third of the control group.
The prespecified key secondary efficacy endpoint in ASPIRE-1 and -2 was change in the CGI-SS-R 24 hours after the first dose. Both the esketamine and placebo-treated patients experienced significant improvement in this domain, with a disappointing absence of between-group statistical significance.
“We think that this may be due to the effect of acute hospitalization in diffusing the suicidal crisis,” Dr. Canuso said.
She noted, however, that other suicidality indices did show significant improvement in the esketamine-treated group during assessments at 4 hours, 24 hours, and 25 days after the first dose. For example, the double-blind esketamine-treated patients were 2.62-fold more likely than controls to show a significant improvement in MADRS Suicidal Thoughts at 4 hours after dose number one, and 6.15 times more likely to do so 4 hours after their day-25 dose. The CGI structured physician assessments of suicide risk and frequency of suicidal thinking, as well as patient-reported frequency of suicidal thinking, showed consistent favorable numeric trends for improvement with esketamine, with odds ratios of 1.46-2.11 from 4 hours through 25 days, although those results generally failed to achieve statistical significance.
In terms of safety, the rate of serious adverse events was just under 12% in both the esketamine and placebo arms. As in earlier studies, the most common adverse events associated with the novel antidepressant were dizziness, dissociation, nausea, and sleepiness, all several-fold more frequent than with placebo.
Esketamine is the S-enantiomer of racemic ketamine. It’s a noncompetitive N-methyl-D-aspartate receptor antagonist.
Janssen, which already markets intranasal esketamine as Spravato in the United States for treatment-resistant depression, plans to file for an expanded indication on the basis of the ASPIRE-1 and -2 results. The Food and Drug Administration already has granted Breakthrough Therapy designation for research on esketamine for reduction of major depression symptoms in patients with active suicidal ideation.
The ASPIRE studies were funded by Janssen.
REPORTING FROM ECNP 2019
Most practices not screening for five social needs
that are associated with health outcomes, a study found.

Lead author Taressa K. Fraze, PhD, of the Dartmouth Institute for Health Policy and Clinical Practice in Lebanon, N.H., and colleagues conducted a cross-sectional survey analysis of responses by physician practices and hospitals that participated in the 2017-2018 National Survey of Healthcare Organizations and Systems. The investigators evaluated how many practices and hospitals reported screening of patients for five social needs: food insecurity, housing instability, utility needs, transportation needs, and experience with interpersonal violence. The final analysis included 2,190 physician practices and 739 hospitals.
Of physician practices, 56% reported screening for interpersonal violence, 35% screened for transportation needs, 30% for food insecurity, 28% for housing instability, and 23% for utility needs, according to the study published in JAMA Network Open.
Among hospitals, 75% reported screening for interpersonal violence, 74% for transportation needs, 60% for housing instability, 40% for food insecurity, and 36% for utility needs. Only 16% of physician practices and 24% of hospitals screened for all five social needs, the study found, while 33% of physician practices and 8% of hospitals reported screening for no social needs. The majority of the overall screening activity was driven by interpersonal violence screenings.
Physician practices that served more disadvantaged patients, including federally qualified health centers and those with more Medicaid revenue were more likely to screen for all five social needs. Practices in Medicaid accountable care organization contracts and those in Medicaid expansion states also had higher screening rates. Regionally, practices in the West had the highest screening rates, while practices in the Midwest had the lowest rates.
Among hospitals, the investigators found few significant screening differences based on hospital characteristics. Ownership, critical access status, delivery reform participation, rural status, region, and Medicaid expansion had no significant effects on screening rates, although academic medical centers were more likely to screen patients for all needs compared with nonacademic medical centers.
The study authors wrote that doctors and hospitals may need more resources and additional processes to screen for and/or to address the social needs of patients. They noted that practices and hospitals that did not screen for social needs were more likely to report a lack of financial resources, time, and incentives as major barriers.
To implement better screening protocols and address patients’ needs, the investigators wrote that doctors and hospitals will need financial support. For example, the Centers for Medicare & Medicaid Services should consider expanding care management billing to include managing care for patients who are both at risk or have clinically complex conditions in addition to social needs.
Dr. Fraze and three coauthors reported receiving grants from the Agency for Healthcare Research and Quality during the conduct of the study. Dr. Fraze also reported receiving grants from the Robert Wood Johnson Foundation during the conduct of the study and receiving grants as an investigator from the 6 Foundation Collaborative, Commonwealth Fund, and Centers for Disease Control and Prevention. One coauthor reported receiving grants from the National Institute on Aging/National Institutes of Health during the conduct of the study.
SOURCE: Fraze TK et al. JAMA Netw Open. 2019 Sep 18. doi: 10.1001/jamanetworkopen.2019.11514.
While momentum for social risk screening is growing nationally, the recent study by Fraze et al. illustrates that screening across multiple domains is not yet common in clinical settings, wrote Rachel Gold, PhD, of Kaiser Permanente Center for Health Research Northwest in Portland, Ore.
In an editorial accompanying the study, Dr. Gold and coauthor Laura Gottlieb, MD, an associate professor of family and community medicine at the University of California, San Francisco, wrote that a critical finding of the study is that reimbursement is associated with uptake of social risk screening (JAMA Network Open. 2019 Sep 18. doi: 10.1001/jamanetworkopen.2019.11513). Specifically, the analysis found that screening for social risks is more common in care settings that receive some form of payment to support such efforts, directly or indirectly.
“This finding aligns with other research showing that altering incentive structures may enhance the adoption of social risk screening in health care settings,” Dr. Gold and Dr. Gottlieb wrote. “But these findings are just a beginning. Disseminating and sustaining social risk screening will require a deep understanding of how best to structure financial and other incentives to optimally support social risk screening; high-quality research is needed to help design reimbursement models that reliably influence adoption.”
Further research is needed not only to explain challenges to the implementation of social risk screening, but also to reveal the best evidence-based methods for overcoming them, the authors wrote. Such methods will likely require a range of support strategies targeted to the needs of various health care settings.
“Documenting social risk data in health care settings requires identifying ways to implement such screening effectively and sustainably,” Dr. Gold and Dr. Gottlieb wrote. “These findings underscore how much we still have to learn about the types of support needed to implement and sustain these practices.”
Dr. Gold reported receiving grants from the National Institutes of Health during the conduct of the study. Dr. Gottlieb reported receiving grants from the Robert Wood Johnson Foundation, the Commonwealth Fund, Kaiser Permanente, Episcopal Health Foundation, the Agency for Healthcare Research and Quality, St. David’s Foundation, the Pritzker Family Fund, and the Harvard Research Network on Toxic Stress outside the submitted work.
While momentum for social risk screening is growing nationally, the recent study by Fraze et al. illustrates that screening across multiple domains is not yet common in clinical settings, wrote Rachel Gold, PhD, of Kaiser Permanente Center for Health Research Northwest in Portland, Ore.
In an editorial accompanying the study, Dr. Gold and coauthor Laura Gottlieb, MD, an associate professor of family and community medicine at the University of California, San Francisco, wrote that a critical finding of the study is that reimbursement is associated with uptake of social risk screening (JAMA Network Open. 2019 Sep 18. doi: 10.1001/jamanetworkopen.2019.11513). Specifically, the analysis found that screening for social risks is more common in care settings that receive some form of payment to support such efforts, directly or indirectly.
“This finding aligns with other research showing that altering incentive structures may enhance the adoption of social risk screening in health care settings,” Dr. Gold and Dr. Gottlieb wrote. “But these findings are just a beginning. Disseminating and sustaining social risk screening will require a deep understanding of how best to structure financial and other incentives to optimally support social risk screening; high-quality research is needed to help design reimbursement models that reliably influence adoption.”
Further research is needed not only to explain challenges to the implementation of social risk screening, but also to reveal the best evidence-based methods for overcoming them, the authors wrote. Such methods will likely require a range of support strategies targeted to the needs of various health care settings.
“Documenting social risk data in health care settings requires identifying ways to implement such screening effectively and sustainably,” Dr. Gold and Dr. Gottlieb wrote. “These findings underscore how much we still have to learn about the types of support needed to implement and sustain these practices.”
Dr. Gold reported receiving grants from the National Institutes of Health during the conduct of the study. Dr. Gottlieb reported receiving grants from the Robert Wood Johnson Foundation, the Commonwealth Fund, Kaiser Permanente, Episcopal Health Foundation, the Agency for Healthcare Research and Quality, St. David’s Foundation, the Pritzker Family Fund, and the Harvard Research Network on Toxic Stress outside the submitted work.
While momentum for social risk screening is growing nationally, the recent study by Fraze et al. illustrates that screening across multiple domains is not yet common in clinical settings, wrote Rachel Gold, PhD, of Kaiser Permanente Center for Health Research Northwest in Portland, Ore.
In an editorial accompanying the study, Dr. Gold and coauthor Laura Gottlieb, MD, an associate professor of family and community medicine at the University of California, San Francisco, wrote that a critical finding of the study is that reimbursement is associated with uptake of social risk screening (JAMA Network Open. 2019 Sep 18. doi: 10.1001/jamanetworkopen.2019.11513). Specifically, the analysis found that screening for social risks is more common in care settings that receive some form of payment to support such efforts, directly or indirectly.
“This finding aligns with other research showing that altering incentive structures may enhance the adoption of social risk screening in health care settings,” Dr. Gold and Dr. Gottlieb wrote. “But these findings are just a beginning. Disseminating and sustaining social risk screening will require a deep understanding of how best to structure financial and other incentives to optimally support social risk screening; high-quality research is needed to help design reimbursement models that reliably influence adoption.”
Further research is needed not only to explain challenges to the implementation of social risk screening, but also to reveal the best evidence-based methods for overcoming them, the authors wrote. Such methods will likely require a range of support strategies targeted to the needs of various health care settings.
“Documenting social risk data in health care settings requires identifying ways to implement such screening effectively and sustainably,” Dr. Gold and Dr. Gottlieb wrote. “These findings underscore how much we still have to learn about the types of support needed to implement and sustain these practices.”
Dr. Gold reported receiving grants from the National Institutes of Health during the conduct of the study. Dr. Gottlieb reported receiving grants from the Robert Wood Johnson Foundation, the Commonwealth Fund, Kaiser Permanente, Episcopal Health Foundation, the Agency for Healthcare Research and Quality, St. David’s Foundation, the Pritzker Family Fund, and the Harvard Research Network on Toxic Stress outside the submitted work.
that are associated with health outcomes, a study found.
Lead author Taressa K. Fraze, PhD, of the Dartmouth Institute for Health Policy and Clinical Practice in Lebanon, N.H., and colleagues conducted a cross-sectional survey analysis of responses by physician practices and hospitals that participated in the 2017-2018 National Survey of Healthcare Organizations and Systems. The investigators evaluated how many practices and hospitals reported screening of patients for five social needs: food insecurity, housing instability, utility needs, transportation needs, and experience with interpersonal violence. The final analysis included 2,190 physician practices and 739 hospitals.
Of physician practices, 56% reported screening for interpersonal violence, 35% screened for transportation needs, 30% for food insecurity, 28% for housing instability, and 23% for utility needs, according to the study published in JAMA Network Open.
Among hospitals, 75% reported screening for interpersonal violence, 74% for transportation needs, 60% for housing instability, 40% for food insecurity, and 36% for utility needs. Only 16% of physician practices and 24% of hospitals screened for all five social needs, the study found, while 33% of physician practices and 8% of hospitals reported screening for no social needs. The majority of the overall screening activity was driven by interpersonal violence screenings.
Physician practices that served more disadvantaged patients, including federally qualified health centers and those with more Medicaid revenue were more likely to screen for all five social needs. Practices in Medicaid accountable care organization contracts and those in Medicaid expansion states also had higher screening rates. Regionally, practices in the West had the highest screening rates, while practices in the Midwest had the lowest rates.
Among hospitals, the investigators found few significant screening differences based on hospital characteristics. Ownership, critical access status, delivery reform participation, rural status, region, and Medicaid expansion had no significant effects on screening rates, although academic medical centers were more likely to screen patients for all needs compared with nonacademic medical centers.
The study authors wrote that doctors and hospitals may need more resources and additional processes to screen for and/or to address the social needs of patients. They noted that practices and hospitals that did not screen for social needs were more likely to report a lack of financial resources, time, and incentives as major barriers.
To implement better screening protocols and address patients’ needs, the investigators wrote that doctors and hospitals will need financial support. For example, the Centers for Medicare & Medicaid Services should consider expanding care management billing to include managing care for patients who are both at risk or have clinically complex conditions in addition to social needs.
Dr. Fraze and three coauthors reported receiving grants from the Agency for Healthcare Research and Quality during the conduct of the study. Dr. Fraze also reported receiving grants from the Robert Wood Johnson Foundation during the conduct of the study and receiving grants as an investigator from the 6 Foundation Collaborative, Commonwealth Fund, and Centers for Disease Control and Prevention. One coauthor reported receiving grants from the National Institute on Aging/National Institutes of Health during the conduct of the study.
SOURCE: Fraze TK et al. JAMA Netw Open. 2019 Sep 18. doi: 10.1001/jamanetworkopen.2019.11514.
that are associated with health outcomes, a study found.
Lead author Taressa K. Fraze, PhD, of the Dartmouth Institute for Health Policy and Clinical Practice in Lebanon, N.H., and colleagues conducted a cross-sectional survey analysis of responses by physician practices and hospitals that participated in the 2017-2018 National Survey of Healthcare Organizations and Systems. The investigators evaluated how many practices and hospitals reported screening of patients for five social needs: food insecurity, housing instability, utility needs, transportation needs, and experience with interpersonal violence. The final analysis included 2,190 physician practices and 739 hospitals.
Of physician practices, 56% reported screening for interpersonal violence, 35% screened for transportation needs, 30% for food insecurity, 28% for housing instability, and 23% for utility needs, according to the study published in JAMA Network Open.
Among hospitals, 75% reported screening for interpersonal violence, 74% for transportation needs, 60% for housing instability, 40% for food insecurity, and 36% for utility needs. Only 16% of physician practices and 24% of hospitals screened for all five social needs, the study found, while 33% of physician practices and 8% of hospitals reported screening for no social needs. The majority of the overall screening activity was driven by interpersonal violence screenings.
Physician practices that served more disadvantaged patients, including federally qualified health centers and those with more Medicaid revenue were more likely to screen for all five social needs. Practices in Medicaid accountable care organization contracts and those in Medicaid expansion states also had higher screening rates. Regionally, practices in the West had the highest screening rates, while practices in the Midwest had the lowest rates.
Among hospitals, the investigators found few significant screening differences based on hospital characteristics. Ownership, critical access status, delivery reform participation, rural status, region, and Medicaid expansion had no significant effects on screening rates, although academic medical centers were more likely to screen patients for all needs compared with nonacademic medical centers.
The study authors wrote that doctors and hospitals may need more resources and additional processes to screen for and/or to address the social needs of patients. They noted that practices and hospitals that did not screen for social needs were more likely to report a lack of financial resources, time, and incentives as major barriers.
To implement better screening protocols and address patients’ needs, the investigators wrote that doctors and hospitals will need financial support. For example, the Centers for Medicare & Medicaid Services should consider expanding care management billing to include managing care for patients who are both at risk or have clinically complex conditions in addition to social needs.
Dr. Fraze and three coauthors reported receiving grants from the Agency for Healthcare Research and Quality during the conduct of the study. Dr. Fraze also reported receiving grants from the Robert Wood Johnson Foundation during the conduct of the study and receiving grants as an investigator from the 6 Foundation Collaborative, Commonwealth Fund, and Centers for Disease Control and Prevention. One coauthor reported receiving grants from the National Institute on Aging/National Institutes of Health during the conduct of the study.
SOURCE: Fraze TK et al. JAMA Netw Open. 2019 Sep 18. doi: 10.1001/jamanetworkopen.2019.11514.
FROM JAMA NETWORK OPEN
New engineered HIV-1 vaccine candidate shows improved immunogenicity in early trial
ALVAC-HIV vaccine showed immunogenicity across several HIV clades in an early trial involving 100 healthy patients at low risk of HIV infection, according to a study by Glenda E. Gray, MBBCH, FCPaed, of the University of the Witwatersrand, Johannesburg, South Africa, and colleagues that was published online in the Sep. 18 issue of Science Translational Medicine.

ALVAC-HIV (vCP1521) is a live attenuated recombinant canarypox-derived virus that expresses gene products from the HIV-1 gp120 (92TH023/clade E), Gag (clade B), and Pro (clade B) that is cultured in chicken embryo fibroblast cells.
Four injections of ALVAC-HIV were given at months 0, 1, 3, and 6. At months 3 and 6, two booster injections were given of AIDSVAX/BE, a bivalent HIV glycoprotein 120 (gp120) that was previously studied in the RV144 trial. The HVTN 097 trial examined primary immunogenicity endpoints including the frequency and magnitude of IgG and IgG3 antibody binding, measured in serum specimens obtained at baseline, at a peak time point (2 weeks after second ALVAC/AIDSVAX vaccination), a durability time point (6 months after second ALVAC/AIDSVAX vaccination), and the response rates and magnitudes of CD4+ and CD8+ T-cell responses at the baseline, peak, and durability time points. One hundred healthy adults at low risk for HIV infection were randomized in 3:1:1 ratio to group T1 (HIV vaccines, tetanus vaccine, and hepatitis B vaccine), group T2 (HIV vaccine only), and the placebo group T3 (tetanus vaccine and hepatitis B vaccine). There were no meaningful differences in HIV immune responses between the HIV vaccine recipients with or without the tetanus and hepatitis B vaccines, so the researchers pooled the data from groups T1 and T2 in their analysis.
At the peak immunogenicity time point, the vaccine schedule predominantly induced CD4+ T cells directed to HIV-1 Env; this was measured by expression of interleukin-2 and/or interferon-gamma. The Env-specific CD4+ T-cell response rate was significantly higher in HVTN 097 vaccine recipients than it was in those in the RV144 trial (51.9% vs. 36.4%; P = .043). The HVTN 097 trial also showed significantly higher response rates for CD40L(59.3% for HVTN 097 vs. 33.7% for RV144; P less than .001) and for interferon-gamma (42.6% in HVTN 097 vs. 19.5% in RV144; P = .001).
However, durability at 6 months after the second vaccine injection remained an issue, with the frequency of circulating Env-specific CD4+ T-cell responses among vaccine recipients declining significantly; the response rate dropped from 70.8% to 36.1%.
“These data may indicate that cross-clade immune responses, especially to non-neutralizing epitopes correlated with decreased HIV-1 risk, can be achieved for a globally effective vaccine by using unique HIV Env strains,” Dr. Gray and associates concluded.
The authors declared that they had no competing interests.
SOURCE: Gray GE et al. Sci. Transl. Med. 2019 Sep 18. doi: 10.1126/scitranslmed.aax1880..
ALVAC-HIV vaccine showed immunogenicity across several HIV clades in an early trial involving 100 healthy patients at low risk of HIV infection, according to a study by Glenda E. Gray, MBBCH, FCPaed, of the University of the Witwatersrand, Johannesburg, South Africa, and colleagues that was published online in the Sep. 18 issue of Science Translational Medicine.
ALVAC-HIV (vCP1521) is a live attenuated recombinant canarypox-derived virus that expresses gene products from the HIV-1 gp120 (92TH023/clade E), Gag (clade B), and Pro (clade B) that is cultured in chicken embryo fibroblast cells.
Four injections of ALVAC-HIV were given at months 0, 1, 3, and 6. At months 3 and 6, two booster injections were given of AIDSVAX/BE, a bivalent HIV glycoprotein 120 (gp120) that was previously studied in the RV144 trial. The HVTN 097 trial examined primary immunogenicity endpoints including the frequency and magnitude of IgG and IgG3 antibody binding, measured in serum specimens obtained at baseline, at a peak time point (2 weeks after second ALVAC/AIDSVAX vaccination), a durability time point (6 months after second ALVAC/AIDSVAX vaccination), and the response rates and magnitudes of CD4+ and CD8+ T-cell responses at the baseline, peak, and durability time points. One hundred healthy adults at low risk for HIV infection were randomized in 3:1:1 ratio to group T1 (HIV vaccines, tetanus vaccine, and hepatitis B vaccine), group T2 (HIV vaccine only), and the placebo group T3 (tetanus vaccine and hepatitis B vaccine). There were no meaningful differences in HIV immune responses between the HIV vaccine recipients with or without the tetanus and hepatitis B vaccines, so the researchers pooled the data from groups T1 and T2 in their analysis.
At the peak immunogenicity time point, the vaccine schedule predominantly induced CD4+ T cells directed to HIV-1 Env; this was measured by expression of interleukin-2 and/or interferon-gamma. The Env-specific CD4+ T-cell response rate was significantly higher in HVTN 097 vaccine recipients than it was in those in the RV144 trial (51.9% vs. 36.4%; P = .043). The HVTN 097 trial also showed significantly higher response rates for CD40L(59.3% for HVTN 097 vs. 33.7% for RV144; P less than .001) and for interferon-gamma (42.6% in HVTN 097 vs. 19.5% in RV144; P = .001).
However, durability at 6 months after the second vaccine injection remained an issue, with the frequency of circulating Env-specific CD4+ T-cell responses among vaccine recipients declining significantly; the response rate dropped from 70.8% to 36.1%.
“These data may indicate that cross-clade immune responses, especially to non-neutralizing epitopes correlated with decreased HIV-1 risk, can be achieved for a globally effective vaccine by using unique HIV Env strains,” Dr. Gray and associates concluded.
The authors declared that they had no competing interests.
SOURCE: Gray GE et al. Sci. Transl. Med. 2019 Sep 18. doi: 10.1126/scitranslmed.aax1880..
ALVAC-HIV vaccine showed immunogenicity across several HIV clades in an early trial involving 100 healthy patients at low risk of HIV infection, according to a study by Glenda E. Gray, MBBCH, FCPaed, of the University of the Witwatersrand, Johannesburg, South Africa, and colleagues that was published online in the Sep. 18 issue of Science Translational Medicine.
ALVAC-HIV (vCP1521) is a live attenuated recombinant canarypox-derived virus that expresses gene products from the HIV-1 gp120 (92TH023/clade E), Gag (clade B), and Pro (clade B) that is cultured in chicken embryo fibroblast cells.
Four injections of ALVAC-HIV were given at months 0, 1, 3, and 6. At months 3 and 6, two booster injections were given of AIDSVAX/BE, a bivalent HIV glycoprotein 120 (gp120) that was previously studied in the RV144 trial. The HVTN 097 trial examined primary immunogenicity endpoints including the frequency and magnitude of IgG and IgG3 antibody binding, measured in serum specimens obtained at baseline, at a peak time point (2 weeks after second ALVAC/AIDSVAX vaccination), a durability time point (6 months after second ALVAC/AIDSVAX vaccination), and the response rates and magnitudes of CD4+ and CD8+ T-cell responses at the baseline, peak, and durability time points. One hundred healthy adults at low risk for HIV infection were randomized in 3:1:1 ratio to group T1 (HIV vaccines, tetanus vaccine, and hepatitis B vaccine), group T2 (HIV vaccine only), and the placebo group T3 (tetanus vaccine and hepatitis B vaccine). There were no meaningful differences in HIV immune responses between the HIV vaccine recipients with or without the tetanus and hepatitis B vaccines, so the researchers pooled the data from groups T1 and T2 in their analysis.
At the peak immunogenicity time point, the vaccine schedule predominantly induced CD4+ T cells directed to HIV-1 Env; this was measured by expression of interleukin-2 and/or interferon-gamma. The Env-specific CD4+ T-cell response rate was significantly higher in HVTN 097 vaccine recipients than it was in those in the RV144 trial (51.9% vs. 36.4%; P = .043). The HVTN 097 trial also showed significantly higher response rates for CD40L(59.3% for HVTN 097 vs. 33.7% for RV144; P less than .001) and for interferon-gamma (42.6% in HVTN 097 vs. 19.5% in RV144; P = .001).
However, durability at 6 months after the second vaccine injection remained an issue, with the frequency of circulating Env-specific CD4+ T-cell responses among vaccine recipients declining significantly; the response rate dropped from 70.8% to 36.1%.
“These data may indicate that cross-clade immune responses, especially to non-neutralizing epitopes correlated with decreased HIV-1 risk, can be achieved for a globally effective vaccine by using unique HIV Env strains,” Dr. Gray and associates concluded.
The authors declared that they had no competing interests.
SOURCE: Gray GE et al. Sci. Transl. Med. 2019 Sep 18. doi: 10.1126/scitranslmed.aax1880..
FROM SCIENCE TRANSLATIONAL MEDICINE
Key clinical point: “These data may indicate that cross-clade immune responses ... can be achieved for a globally effective vaccine by using unique HIV Env strains.”
Major finding: At the peak immunogenicity time point, the vaccine schedule predominantly induced CD4+ T cells directed to HIV-1 Env .
Study details: A phase 1b randomized, double-blind, placebo-controlled trial to assess the safety and immunogenicity of the ALVAC-HIV vaccine in 100 healthy patients at low risk of HIV infection.
Disclosures: The study was supported by the National Institute of Allergy and Infectious Diseases and other global health agencies. The authors declared that they had no competing interests.
Source: Gray GE et al. Sci Transl Med. 2019 Sep 18. doi: 10.1126/scitranslmed.aax1880.