User login
Nonuremic Calciphylaxis Triggered by Rapid Weight Loss and Hypotension
Calciphylaxis, otherwise known as calcific uremic arteriolopathy, is characterized by calcification of the tunica media of the small- to medium-sized blood vessels of the dermis and subcutis, leading to ischemia and necrosis.1 It is a deadly disease with a 1-year mortality rate of more than 50%.2 End-stage renal disease (ESRD) is the most common risk factor for calciphylaxis, with a prevalence of 1% to 4% of hemodialysis patients with calciphylaxis in the United States.2-5 However, nonuremic calciphylaxis (NUC) has been increasingly reported in the literature and has risk factors other than ESRD, including but not limited to obesity, alcoholic liver disease, primary hyperparathyroidism, connective tissue disease, and underlying malignancy.3,6-9 Triggers for calciphylaxis in at-risk patients include use of corticosteroids or warfarin, iron or albumin infusions, and rapid weight loss.3,6,9-11 We report an unusual case of NUC that most likely was triggered by rapid weight loss and hypotension in a patient with multiple risk factors for calciphylaxis.
Case Report
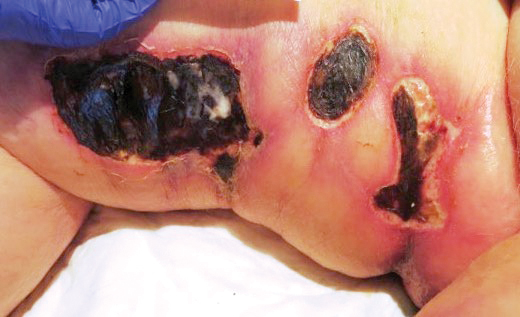
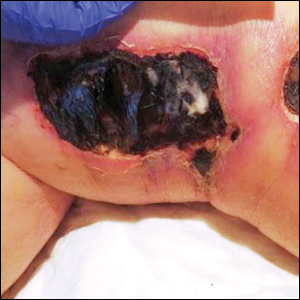
A 75-year-old white woman with history of morbid obesity (body mass index, 40 kg/m2), unexplained weight loss of 70 lb over the last year, and polymyalgia rheumatica requiring chronic prednisone therapy presented with painful lesions on the thighs, buttocks, and right shoulder of 4 months’ duration. She had multiple hospital admissions preceding the onset of lesions for severe infections resulting in sepsis with hypotension, including Enterococcus faecalis endocarditis, extended-spectrum beta-lactamase bacteremia, and Pseudomonas aeruginosa pneumonia. Physical examination revealed large well-demarcated ulcers and necrotic eschars with surrounding violaceous induration and stellate erythema on the anterior, medial, and posterior thighs and buttocks that were exquisitely tender (Figures 1 and 2).

Notable laboratory results included hypoalbuminemia (1.3 g/dL [reference range, 3.5–5.0 g/dL]) with normal renal function, a corrected calcium level of 9.7 mg/dL (reference range, 8.2–10.2 mg/dL), a serum phosphorus level of 3.5 mg/dL (reference range, 2.3–4.7 mg/dL), a calcium-phosphate product of 27.3 mg2/dL2 (reference range, <55 mg2/dL2), and a parathyroid hormone level of 49.3 pg/mL (reference range, 10–65 pg/mL). Antinuclear antibodies were negative. A hypercoagulability evaluation showed normal protein C and S levels, negative lupus anticoagulant, and negative anticardiolipin antibodies.
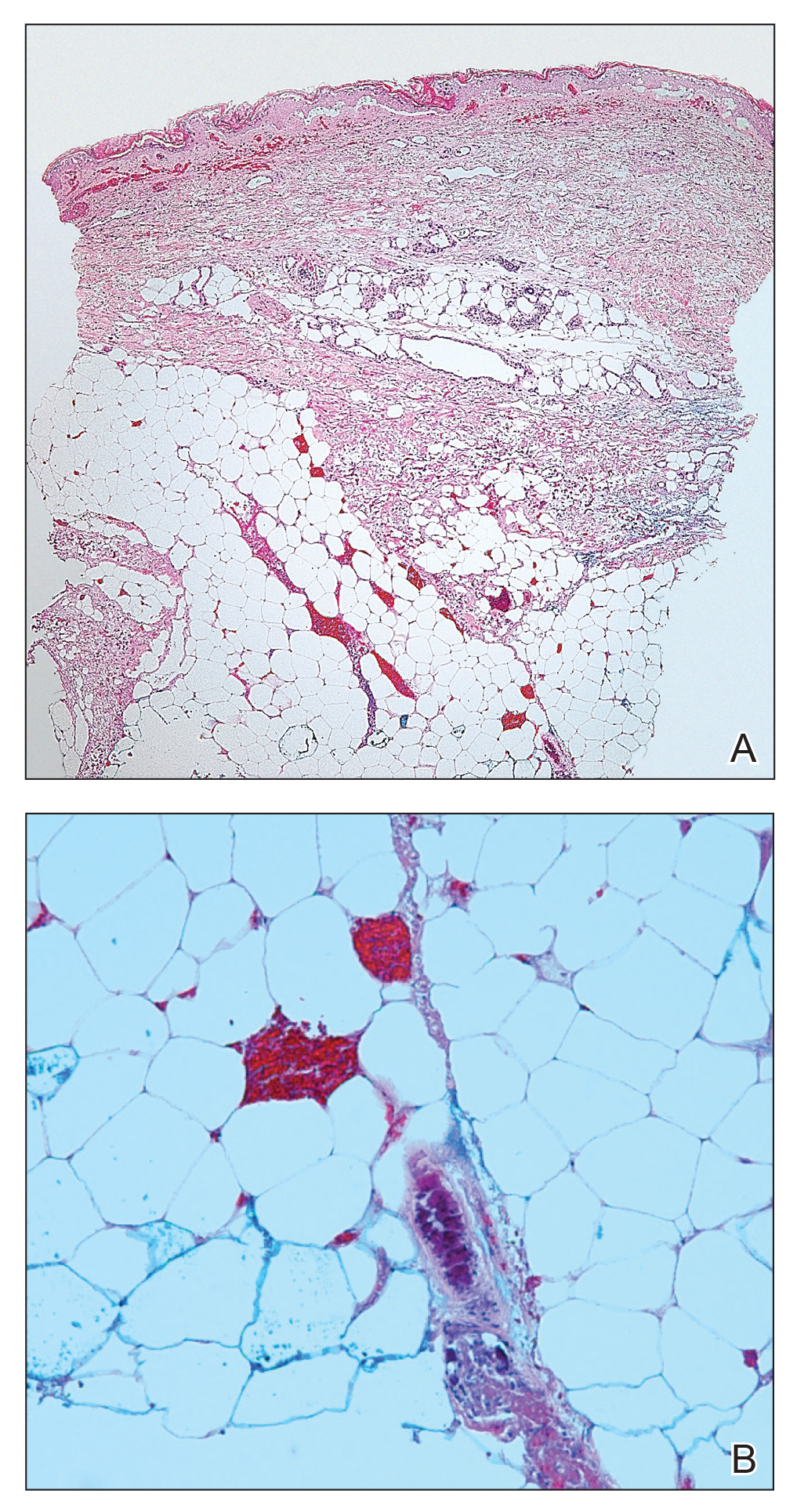
Telescoping punch biopsies of the indurated borders of the eschars showed prominent calcification of the small- and medium-sized vessels in the mid and deep dermis, intravascular thrombi, and necrosis of the epidermis and subcutaneous fat consistent with calciphylaxis (Figure 3).
After the diagnosis of calciphylaxis was made, the patient was treated with intravenous sodium thiosulfate 25 mg 3 times weekly and alendronate 70 mg weekly. Daily arterial blood gas studies did not detect metabolic acidosis during the patient’s sodium thiosulfate therapy. The wounds were debrided, and we attempted to slowly taper the patient off the oral prednisone. Unfortunately, her condition slowly deteriorated secondary to sepsis, resulting in septic shock. The patient died 3 weeks after the diagnosis of calciphylaxis was made. At the time of diagnosis, the patient had a poor prognosis and notable risk for sepsis due to the large eschars on the thighs and abdomen as well as her relative immunosuppression due to chronic prednisone use.
Comment
Background on Calciphylaxis
Calciphylaxis is a rare but deadly disease that affects both ESRD patients receiving dialysis and patients without ESRD who have known risk factors for calciphylaxis, including female gender, white race, obesity, alcoholic liver disease, primary hyperparathyroidism, connective tissue disease, underlying malignancy, protein C or S deficiency, corticosteroid use, warfarin use, diabetes, iron or albumin infusions, and rapid weight loss.3,6-9,11 Although the molecular pathogenesis of calciphylaxis is not completely understood, it is believed to be caused by local deposition of calcium in the tunica media of small- to medium-sized arterioles and venules in the skin.12 This deposition leads to intimal proliferation and progressive narrowing of the vessels with resultant thrombosis, ischemia, and necrosis. The cutaneous manifestations and histopathology of calciphylaxis classically follow its pathogenesis. Calciphylaxis typically presents with livedo reticularis as vessels narrow and then progresses to purpura, bullae, necrosis, and eschar formation with the onset of acute thrombosis and ischemia. Histopathology is characterized by small- and medium-sized vessel calcification and thrombus, dermal necrosis, and septal panniculitis, though the histology can be highly variable.12 Unfortunately, the already poor prognosis for calciphylaxis worsens when lesions become either ulcerative or present on the proximal extremities and trunk.4,13 Sepsis is the leading cause of death in calciphylaxis patients, affecting more than 50% of patients.2,3,14 The differential diagnoses for calciphylactic-appearing lesions include warfarin-induced skin necrosis, disseminated intravascular coagulation, pyoderma gangrenosum, cholesterol emboli, and various vasculitides and coagulopathies.
Risk Factors
Our case demonstrates the importance of risk factor minimization, trigger avoidance, and early intervention due to the high mortality rate of calciphylaxis. Selye et al15 coined the term calciphylaxis in 1961 based on experiments that induced calciphylaxis in rat models. Their research concluded that there were certain sensitizers (ie, risk factors) that predisposed patients to medial calcium deposition in blood vessels and other challengers (ie, triggers) that acted as inciting events to calcium deposition. Our patient presented with multiple known risk factors for calciphylaxis, including obesity (body mass index, 40 kg/m2), female gender, white race, hypoalbuminemia, and chronic corticosteroid use.16 In the presence of a milieu of risk factors, the patient’s rapid weight loss and episodes of hypotension likely were triggers for calciphylaxis.
Other case reports in the literature have suggested weight loss as a trigger for NUC. One morbidly obese patient with inactive rheumatoid arthritis had onset of calciphylaxis lesions after unintentional weight loss of approximately 50% body weight in 1 year17; however, the weight loss does not have to be drastic to trigger calciphylaxis. Another study of 16 patients with uremic calciphylaxis found that 7 of 16 (44%) patients lost 10 to 50 kg in the 6 months prior to calciphylaxis onset.14 One proposed mechanism by Munavalli et al10 is that elevated levels of matrix metalloproteinases during catabolic weight loss states enhance the deposition of calcium into elastic fibers of small vessels. The authors found elevated serum levels of matrix metalloproteinases in their patients with NUC induced by rapid weight loss.10
A meta-analysis by Nigwekar et al3 found a history of prior corticosteroid use in 61% (22/36) of NUC cases reviewed. However, it is unclear whether it is the use of corticosteroids or chronic inflammation that is implicated in NUC pathogenesis. Chronic inflammation causes downregulation of anticalcification signaling pathways.18-20 The role of 2 vascular calcification inhibitors has been evaluated in the pathogenesis of calciphylaxis: fetuin-A and matrix gla protein (MGP).21 The activity of these proteins is decreased not only in calciphylaxis but also in other inflammatory states and chronic renal failure.18-20 One study found lower fetuin-A levels in 312 hemodialysis patients compared to healthy controls and an association between low fetuin-A levels and increased C-reactive protein levels.22 Reduced fetuin-A and MGP levels may be the result of several calciphylaxis risk factors. Warfarin is believed to trigger calciphylaxis via inhibition of gamma-carboxylation of MGP, which is necessary for its anticalcification activity.23 Hypoalbuminemia and alcoholic liver disease also are risk factors that may be explained by the fact that fetuin-A is synthesized in the liver.24 Therefore, liver disease results in decreased production of fetuin-A that is permissive to vascular calcification in calciphylaxis patients.
There have been other reports of calciphylaxis patients who were originally hospitalized due to hypotension, which may serve as a trigger for calciphylaxis onset.25 Because calciphylaxis lesions are more likely to occur in the fatty areas of the abdomen and proximal thighs where blood flow is slower, hypotension likely accentuates the slowing of blood flow and subsequent blood vessel calcification. This theory is supported by studies showing that established calciphylactic lesions worsen more quickly in the presence of systemic hypotension.26 One patient with ESRD and calciphylaxis of the breasts had consistent systolic blood pressure readings in the high 60s to low 70s between dialysis sessions.27 Due to this association, we recommend that patients with calciphylaxis have close blood pressure monitoring to aid in preventing disease progression.28
Management
Calciphylaxis treatment has not yet been standardized, as it is an uncommon disease whose pathogenesis is not fully understood. Current management strategies aim to normalize metabolic abnormalities such as hypercalcemia if they are present and remove inciting agents such as warfarin and corticosteroids.29 Other medical treatments that have been successfully used include sodium thiosulfate, oral steroids, and adjunctive bisphosphonates.29-31 Sodium thiosulfate is known to cause metabolic acidosis by generating thiosulfuric acid in vivo in patients with or without renal disease; therefore, patients on sodium thiosulfate therapy should be monitored for development of metabolic acidosis and treated with oral sodium bicarbonate or dialysis as needed.30,32 Wound care also is an important element of calciphylaxis treatment; however, the debridement of wounds is controversial. Some argue that dry intact eschars serve to protect against sepsis, which is the leading cause of death in calciphylaxis.2,14,33 In contrast, a retrospective study of 63 calciphylaxis patients found a 1-year survival rate of 61.6% in 17 patients receiving wound debridement vs 27.4% in 46 patients who did not.2 The current consensus is that debridement should be considered on a case-by-case basis, factoring in the presence of wound infection, size of wounds, stability of eschars, and treatment goals of the patient.34 Future studies should be aimed at this issue, with special focus on how these factors and the decision to debride or not impact patient outcomes.
Conclusion
Calciphylaxis is a potentially fatal disease that impacts both patients with ESRD and those with nonuremic risk factors. The term calcific uremic arteriolopathy should be disregarded, as nonuremic causes are being reported with increased frequency in the literature. In such cases, patients often have multiple risk factors, including obesity, primary hyperparathyroidism, alcoholic liver disease, and underlying malignancy, among others. Certain triggers for onset of calciphylaxis should be avoided in at-risk patients, including the use of corticosteroids or warfarin; iron and albumin infusions; hypotension; and rapid weight loss. Our fatal case of NUC is a reminder to dermatologists treating at-risk patients to avoid these triggers and to keep calciphylaxis in the differential diagnosis when encountering early lesions such as livedo reticularis, as progression of these lesions has a 1-year mortality rate of more than 50% with the therapies being utilized at this time.
- Au S, Crawford RI. Three-dimensional analysis of a calciphylaxis plaque: clues to pathogenesis. J Am Acad Dermatol. 2007;47:53-57.
- Weenig RH, Sewell LD, Davis MD, et al. Calciphylaxis: natural history, risk factor analysis, and outcome. J Am Acad Dermatol. 2007;56:569-579.
- Nigwekar SU, Wolf M, Sterns RH, et al. Calciphylaxis from nonuremic causes: a systematic review. Clin J Am Soc Nephrol. 2008;3:1139-1143.
- Fine A, Zacharias J. Calciphylaxis is usually non-ulcerating: risk factors, outcome and therapy. Kidney Int. 2002;61:2210-2217.
- Angelis M, Wong LL, Myers SA, et al. Calciphylaxis in patients on hemodialysis: a prevalence study. Surgery. 1997;122:1083-1090.
- Chavel SM, Taraszka KS, Schaffer JV, et al. Calciphylaxis associated with acute, reversible renal failure in the setting of alcoholic cirrhosis. J Am Acad Dermatol. 2004;50:125-128.
- Bosler DS, Amin MB, Gulli F, et al. Unusual case of calciphylaxis associated with metastatic breast carcinoma. Am J Dermatopathol. 2007;29:400-403.
- Buxtorf K, Cerottini JP, Panizzon RG. Lower limb skin ulcerations, intravascular calcifications and sensorimotor polyneuropathy: calciphylaxis as part of a hyperparathyroidism? Dermatology. 1999;198:423-425.
- Brouns K, Verbeken E, Degreef H, et al. Fatal calciphylaxis in two patients with giant cell arteritis. Clin Rheumatol. 2007;26:836-840.
- Munavalli G, Reisenauer A, Moses M, et al. Weight loss-induced calciphylaxis: potential role of matrix metalloproteinases. J Dermatol. 2003;30:915-919.
- Bae GH, Nambudiri VE, Bach DQ, et al. Rapidly progressive nonuremic calciphylaxis in setting of warfarin. Am J Med. 2015;128:E19-E21.
- Essary LR, Wick MR. Cutaneous calciphylaxis. an underrecognized clinicopathologic entity. Am J Clin Pathol. 2000;113:280-287.
- Hafner J, Keusch G, Wahl C, et al. Uremic small-artery disease with medial calcification and intimal hyperplasia (so-called calciphylaxis): a complication of chronic renal failure and benefit from parathyroidectomy. J Am Acad Dermatol. 1995;33:954-962.
- Coates T, Kirkland GS, Dymock RB, et al. Cutaneous necrosis from calcific uremic arteriolopathy. Am J Kidney Dis. 1998;32:384-391.
- Selye H, Gentile G, Prioreschi P. Cutaneous molt induced by calciphylaxis in the rat. Science. 1961;134:1876-1877.
- Kalajian AH, Malhotra PS, Callen JP, et al. Calciphylaxis with normal renal and parathyroid function: not as rare as previously believed. Arch Dermatol. 2009;145:451-458.
- Malabu U, Roberts L, Sangla K. Calciphylaxis in a morbidly obese woman with rheumatoid arthritis presenting with severe weight loss and vitamin D deficiency. Endocr Pract. 2011;17:104-108.
- Schäfer C, Heiss A, Schwarz A, et al. The serum protein alpha 2–Heremans-Schmid glycoprotein/fetuin-A is a systemically acting inhibitor of ectopic calcification. J Clin Invest. 2003;112:357-366.
- Cozzolino M, Galassi A, Biondi ML, et al. Serum fetuin-A levels link inflammation and cardiovascular calcification in hemodialysis patients. Am J Nephrol. 2006;26:423-429.
- Luo G, Ducy P, McKee MD, et al. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997;386:78-81.
- Weenig RH. Pathogenesis of calciphylaxis: Hans Selye to nuclear factor kappa-B. J Am Acad Dermatol. 2008;58:458-471.
- Ketteler M, Bongartz P, Westenfeld R, et al. Association of low fetuin-A (AHSG) concentrations in serum with cardiovascular mortality in patients on dialysis: a cross-sectional study. Lancet. 2003;361:827-833.
- Wallin R, Cain D, Sane DC. Matrix Gla protein synthesis and gamma-carboxylation in the aortic vessel wall and proliferating vascular smooth muscle cells a cell system which resembles the system in bone cells. Thromb Haemost. 1999;82:1764-1767.
- Sowers KM, Hayden MR. Calcific uremic arteriolopathy: pathophysiology, reactive oxygen species and therapeutic approaches. Oxid Med Cell Longev. 2010;3:109-121.
- Allegretti AS, Nazarian RM, Goverman J, et al. Calciphylaxis: a rare but fatal delayed complication of Roux-en-Y gastric bypass surgery. Am J Kidney Dis. 2014;64:274-277.
- Wilmer WA, Magro CM. Calciphylaxis: emerging concepts in prevention, diagnosis, and treatment. Semin Dial. 2002;15:172-186.
- Gupta D, Tadros R, Mazumdar A, et al. Breast lesions with intractable pain in end-stage renal disease: calciphylaxis with chronic hypotensive dermatopathy related watershed breast lesions. J Palliat Med. 2013;16:551-554.
- Janigan DT, Hirsch DJ, Klassen GA, et al. Calcified subcutaneous arterioles with infarcts of the subcutis and skin (“calciphylaxis”) in chronic renal failure. Am J Kidney Dis. 2000;35:588-597.
- Jeong HS, Dominguez AR. Calciphylaxis: controversies in pathogenesis, diagnosis and treatment. Am J Med Sci. 2016;351:217-227.
- Bourgeois P, De Haes P. Sodium thiosulfate as a treatment for calciphylaxis: a case series. J Dermatolog Treat. 2016;27:520-524.
- Biswas A, Walsh NM, Tremaine R. A case of nonuremic calciphylaxis treated effectively with systemic corticosteroids. J Cutan Med Surg. 2016;20:275-278.
- Selk N, Rodby, RA. Unexpectedly severe metabolic acidosis associated with sodium thiosulfate therapy in a patient with calcific uremic arteriolopathy. Semin Dial. 2011;24:85-88.
- Martin R. Mysterious calciphylaxis: wounds with eschar—to debride or not to debride? Ostomy Wound Manage. 2004:50:64-66, 68-70.
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146.
Calciphylaxis, otherwise known as calcific uremic arteriolopathy, is characterized by calcification of the tunica media of the small- to medium-sized blood vessels of the dermis and subcutis, leading to ischemia and necrosis.1 It is a deadly disease with a 1-year mortality rate of more than 50%.2 End-stage renal disease (ESRD) is the most common risk factor for calciphylaxis, with a prevalence of 1% to 4% of hemodialysis patients with calciphylaxis in the United States.2-5 However, nonuremic calciphylaxis (NUC) has been increasingly reported in the literature and has risk factors other than ESRD, including but not limited to obesity, alcoholic liver disease, primary hyperparathyroidism, connective tissue disease, and underlying malignancy.3,6-9 Triggers for calciphylaxis in at-risk patients include use of corticosteroids or warfarin, iron or albumin infusions, and rapid weight loss.3,6,9-11 We report an unusual case of NUC that most likely was triggered by rapid weight loss and hypotension in a patient with multiple risk factors for calciphylaxis.
Case Report
A 75-year-old white woman with history of morbid obesity (body mass index, 40 kg/m2), unexplained weight loss of 70 lb over the last year, and polymyalgia rheumatica requiring chronic prednisone therapy presented with painful lesions on the thighs, buttocks, and right shoulder of 4 months’ duration. She had multiple hospital admissions preceding the onset of lesions for severe infections resulting in sepsis with hypotension, including Enterococcus faecalis endocarditis, extended-spectrum beta-lactamase bacteremia, and Pseudomonas aeruginosa pneumonia. Physical examination revealed large well-demarcated ulcers and necrotic eschars with surrounding violaceous induration and stellate erythema on the anterior, medial, and posterior thighs and buttocks that were exquisitely tender (Figures 1 and 2).
Notable laboratory results included hypoalbuminemia (1.3 g/dL [reference range, 3.5–5.0 g/dL]) with normal renal function, a corrected calcium level of 9.7 mg/dL (reference range, 8.2–10.2 mg/dL), a serum phosphorus level of 3.5 mg/dL (reference range, 2.3–4.7 mg/dL), a calcium-phosphate product of 27.3 mg2/dL2 (reference range, <55 mg2/dL2), and a parathyroid hormone level of 49.3 pg/mL (reference range, 10–65 pg/mL). Antinuclear antibodies were negative. A hypercoagulability evaluation showed normal protein C and S levels, negative lupus anticoagulant, and negative anticardiolipin antibodies.
Telescoping punch biopsies of the indurated borders of the eschars showed prominent calcification of the small- and medium-sized vessels in the mid and deep dermis, intravascular thrombi, and necrosis of the epidermis and subcutaneous fat consistent with calciphylaxis (Figure 3).
After the diagnosis of calciphylaxis was made, the patient was treated with intravenous sodium thiosulfate 25 mg 3 times weekly and alendronate 70 mg weekly. Daily arterial blood gas studies did not detect metabolic acidosis during the patient’s sodium thiosulfate therapy. The wounds were debrided, and we attempted to slowly taper the patient off the oral prednisone. Unfortunately, her condition slowly deteriorated secondary to sepsis, resulting in septic shock. The patient died 3 weeks after the diagnosis of calciphylaxis was made. At the time of diagnosis, the patient had a poor prognosis and notable risk for sepsis due to the large eschars on the thighs and abdomen as well as her relative immunosuppression due to chronic prednisone use.
Comment
Background on Calciphylaxis
Calciphylaxis is a rare but deadly disease that affects both ESRD patients receiving dialysis and patients without ESRD who have known risk factors for calciphylaxis, including female gender, white race, obesity, alcoholic liver disease, primary hyperparathyroidism, connective tissue disease, underlying malignancy, protein C or S deficiency, corticosteroid use, warfarin use, diabetes, iron or albumin infusions, and rapid weight loss.3,6-9,11 Although the molecular pathogenesis of calciphylaxis is not completely understood, it is believed to be caused by local deposition of calcium in the tunica media of small- to medium-sized arterioles and venules in the skin.12 This deposition leads to intimal proliferation and progressive narrowing of the vessels with resultant thrombosis, ischemia, and necrosis. The cutaneous manifestations and histopathology of calciphylaxis classically follow its pathogenesis. Calciphylaxis typically presents with livedo reticularis as vessels narrow and then progresses to purpura, bullae, necrosis, and eschar formation with the onset of acute thrombosis and ischemia. Histopathology is characterized by small- and medium-sized vessel calcification and thrombus, dermal necrosis, and septal panniculitis, though the histology can be highly variable.12 Unfortunately, the already poor prognosis for calciphylaxis worsens when lesions become either ulcerative or present on the proximal extremities and trunk.4,13 Sepsis is the leading cause of death in calciphylaxis patients, affecting more than 50% of patients.2,3,14 The differential diagnoses for calciphylactic-appearing lesions include warfarin-induced skin necrosis, disseminated intravascular coagulation, pyoderma gangrenosum, cholesterol emboli, and various vasculitides and coagulopathies.
Risk Factors
Our case demonstrates the importance of risk factor minimization, trigger avoidance, and early intervention due to the high mortality rate of calciphylaxis. Selye et al15 coined the term calciphylaxis in 1961 based on experiments that induced calciphylaxis in rat models. Their research concluded that there were certain sensitizers (ie, risk factors) that predisposed patients to medial calcium deposition in blood vessels and other challengers (ie, triggers) that acted as inciting events to calcium deposition. Our patient presented with multiple known risk factors for calciphylaxis, including obesity (body mass index, 40 kg/m2), female gender, white race, hypoalbuminemia, and chronic corticosteroid use.16 In the presence of a milieu of risk factors, the patient’s rapid weight loss and episodes of hypotension likely were triggers for calciphylaxis.
Other case reports in the literature have suggested weight loss as a trigger for NUC. One morbidly obese patient with inactive rheumatoid arthritis had onset of calciphylaxis lesions after unintentional weight loss of approximately 50% body weight in 1 year17; however, the weight loss does not have to be drastic to trigger calciphylaxis. Another study of 16 patients with uremic calciphylaxis found that 7 of 16 (44%) patients lost 10 to 50 kg in the 6 months prior to calciphylaxis onset.14 One proposed mechanism by Munavalli et al10 is that elevated levels of matrix metalloproteinases during catabolic weight loss states enhance the deposition of calcium into elastic fibers of small vessels. The authors found elevated serum levels of matrix metalloproteinases in their patients with NUC induced by rapid weight loss.10
A meta-analysis by Nigwekar et al3 found a history of prior corticosteroid use in 61% (22/36) of NUC cases reviewed. However, it is unclear whether it is the use of corticosteroids or chronic inflammation that is implicated in NUC pathogenesis. Chronic inflammation causes downregulation of anticalcification signaling pathways.18-20 The role of 2 vascular calcification inhibitors has been evaluated in the pathogenesis of calciphylaxis: fetuin-A and matrix gla protein (MGP).21 The activity of these proteins is decreased not only in calciphylaxis but also in other inflammatory states and chronic renal failure.18-20 One study found lower fetuin-A levels in 312 hemodialysis patients compared to healthy controls and an association between low fetuin-A levels and increased C-reactive protein levels.22 Reduced fetuin-A and MGP levels may be the result of several calciphylaxis risk factors. Warfarin is believed to trigger calciphylaxis via inhibition of gamma-carboxylation of MGP, which is necessary for its anticalcification activity.23 Hypoalbuminemia and alcoholic liver disease also are risk factors that may be explained by the fact that fetuin-A is synthesized in the liver.24 Therefore, liver disease results in decreased production of fetuin-A that is permissive to vascular calcification in calciphylaxis patients.
There have been other reports of calciphylaxis patients who were originally hospitalized due to hypotension, which may serve as a trigger for calciphylaxis onset.25 Because calciphylaxis lesions are more likely to occur in the fatty areas of the abdomen and proximal thighs where blood flow is slower, hypotension likely accentuates the slowing of blood flow and subsequent blood vessel calcification. This theory is supported by studies showing that established calciphylactic lesions worsen more quickly in the presence of systemic hypotension.26 One patient with ESRD and calciphylaxis of the breasts had consistent systolic blood pressure readings in the high 60s to low 70s between dialysis sessions.27 Due to this association, we recommend that patients with calciphylaxis have close blood pressure monitoring to aid in preventing disease progression.28
Management
Calciphylaxis treatment has not yet been standardized, as it is an uncommon disease whose pathogenesis is not fully understood. Current management strategies aim to normalize metabolic abnormalities such as hypercalcemia if they are present and remove inciting agents such as warfarin and corticosteroids.29 Other medical treatments that have been successfully used include sodium thiosulfate, oral steroids, and adjunctive bisphosphonates.29-31 Sodium thiosulfate is known to cause metabolic acidosis by generating thiosulfuric acid in vivo in patients with or without renal disease; therefore, patients on sodium thiosulfate therapy should be monitored for development of metabolic acidosis and treated with oral sodium bicarbonate or dialysis as needed.30,32 Wound care also is an important element of calciphylaxis treatment; however, the debridement of wounds is controversial. Some argue that dry intact eschars serve to protect against sepsis, which is the leading cause of death in calciphylaxis.2,14,33 In contrast, a retrospective study of 63 calciphylaxis patients found a 1-year survival rate of 61.6% in 17 patients receiving wound debridement vs 27.4% in 46 patients who did not.2 The current consensus is that debridement should be considered on a case-by-case basis, factoring in the presence of wound infection, size of wounds, stability of eschars, and treatment goals of the patient.34 Future studies should be aimed at this issue, with special focus on how these factors and the decision to debride or not impact patient outcomes.
Conclusion
Calciphylaxis is a potentially fatal disease that impacts both patients with ESRD and those with nonuremic risk factors. The term calcific uremic arteriolopathy should be disregarded, as nonuremic causes are being reported with increased frequency in the literature. In such cases, patients often have multiple risk factors, including obesity, primary hyperparathyroidism, alcoholic liver disease, and underlying malignancy, among others. Certain triggers for onset of calciphylaxis should be avoided in at-risk patients, including the use of corticosteroids or warfarin; iron and albumin infusions; hypotension; and rapid weight loss. Our fatal case of NUC is a reminder to dermatologists treating at-risk patients to avoid these triggers and to keep calciphylaxis in the differential diagnosis when encountering early lesions such as livedo reticularis, as progression of these lesions has a 1-year mortality rate of more than 50% with the therapies being utilized at this time.
Calciphylaxis, otherwise known as calcific uremic arteriolopathy, is characterized by calcification of the tunica media of the small- to medium-sized blood vessels of the dermis and subcutis, leading to ischemia and necrosis.1 It is a deadly disease with a 1-year mortality rate of more than 50%.2 End-stage renal disease (ESRD) is the most common risk factor for calciphylaxis, with a prevalence of 1% to 4% of hemodialysis patients with calciphylaxis in the United States.2-5 However, nonuremic calciphylaxis (NUC) has been increasingly reported in the literature and has risk factors other than ESRD, including but not limited to obesity, alcoholic liver disease, primary hyperparathyroidism, connective tissue disease, and underlying malignancy.3,6-9 Triggers for calciphylaxis in at-risk patients include use of corticosteroids or warfarin, iron or albumin infusions, and rapid weight loss.3,6,9-11 We report an unusual case of NUC that most likely was triggered by rapid weight loss and hypotension in a patient with multiple risk factors for calciphylaxis.
Case Report
A 75-year-old white woman with history of morbid obesity (body mass index, 40 kg/m2), unexplained weight loss of 70 lb over the last year, and polymyalgia rheumatica requiring chronic prednisone therapy presented with painful lesions on the thighs, buttocks, and right shoulder of 4 months’ duration. She had multiple hospital admissions preceding the onset of lesions for severe infections resulting in sepsis with hypotension, including Enterococcus faecalis endocarditis, extended-spectrum beta-lactamase bacteremia, and Pseudomonas aeruginosa pneumonia. Physical examination revealed large well-demarcated ulcers and necrotic eschars with surrounding violaceous induration and stellate erythema on the anterior, medial, and posterior thighs and buttocks that were exquisitely tender (Figures 1 and 2).
Notable laboratory results included hypoalbuminemia (1.3 g/dL [reference range, 3.5–5.0 g/dL]) with normal renal function, a corrected calcium level of 9.7 mg/dL (reference range, 8.2–10.2 mg/dL), a serum phosphorus level of 3.5 mg/dL (reference range, 2.3–4.7 mg/dL), a calcium-phosphate product of 27.3 mg2/dL2 (reference range, <55 mg2/dL2), and a parathyroid hormone level of 49.3 pg/mL (reference range, 10–65 pg/mL). Antinuclear antibodies were negative. A hypercoagulability evaluation showed normal protein C and S levels, negative lupus anticoagulant, and negative anticardiolipin antibodies.
Telescoping punch biopsies of the indurated borders of the eschars showed prominent calcification of the small- and medium-sized vessels in the mid and deep dermis, intravascular thrombi, and necrosis of the epidermis and subcutaneous fat consistent with calciphylaxis (Figure 3).
After the diagnosis of calciphylaxis was made, the patient was treated with intravenous sodium thiosulfate 25 mg 3 times weekly and alendronate 70 mg weekly. Daily arterial blood gas studies did not detect metabolic acidosis during the patient’s sodium thiosulfate therapy. The wounds were debrided, and we attempted to slowly taper the patient off the oral prednisone. Unfortunately, her condition slowly deteriorated secondary to sepsis, resulting in septic shock. The patient died 3 weeks after the diagnosis of calciphylaxis was made. At the time of diagnosis, the patient had a poor prognosis and notable risk for sepsis due to the large eschars on the thighs and abdomen as well as her relative immunosuppression due to chronic prednisone use.
Comment
Background on Calciphylaxis
Calciphylaxis is a rare but deadly disease that affects both ESRD patients receiving dialysis and patients without ESRD who have known risk factors for calciphylaxis, including female gender, white race, obesity, alcoholic liver disease, primary hyperparathyroidism, connective tissue disease, underlying malignancy, protein C or S deficiency, corticosteroid use, warfarin use, diabetes, iron or albumin infusions, and rapid weight loss.3,6-9,11 Although the molecular pathogenesis of calciphylaxis is not completely understood, it is believed to be caused by local deposition of calcium in the tunica media of small- to medium-sized arterioles and venules in the skin.12 This deposition leads to intimal proliferation and progressive narrowing of the vessels with resultant thrombosis, ischemia, and necrosis. The cutaneous manifestations and histopathology of calciphylaxis classically follow its pathogenesis. Calciphylaxis typically presents with livedo reticularis as vessels narrow and then progresses to purpura, bullae, necrosis, and eschar formation with the onset of acute thrombosis and ischemia. Histopathology is characterized by small- and medium-sized vessel calcification and thrombus, dermal necrosis, and septal panniculitis, though the histology can be highly variable.12 Unfortunately, the already poor prognosis for calciphylaxis worsens when lesions become either ulcerative or present on the proximal extremities and trunk.4,13 Sepsis is the leading cause of death in calciphylaxis patients, affecting more than 50% of patients.2,3,14 The differential diagnoses for calciphylactic-appearing lesions include warfarin-induced skin necrosis, disseminated intravascular coagulation, pyoderma gangrenosum, cholesterol emboli, and various vasculitides and coagulopathies.
Risk Factors
Our case demonstrates the importance of risk factor minimization, trigger avoidance, and early intervention due to the high mortality rate of calciphylaxis. Selye et al15 coined the term calciphylaxis in 1961 based on experiments that induced calciphylaxis in rat models. Their research concluded that there were certain sensitizers (ie, risk factors) that predisposed patients to medial calcium deposition in blood vessels and other challengers (ie, triggers) that acted as inciting events to calcium deposition. Our patient presented with multiple known risk factors for calciphylaxis, including obesity (body mass index, 40 kg/m2), female gender, white race, hypoalbuminemia, and chronic corticosteroid use.16 In the presence of a milieu of risk factors, the patient’s rapid weight loss and episodes of hypotension likely were triggers for calciphylaxis.
Other case reports in the literature have suggested weight loss as a trigger for NUC. One morbidly obese patient with inactive rheumatoid arthritis had onset of calciphylaxis lesions after unintentional weight loss of approximately 50% body weight in 1 year17; however, the weight loss does not have to be drastic to trigger calciphylaxis. Another study of 16 patients with uremic calciphylaxis found that 7 of 16 (44%) patients lost 10 to 50 kg in the 6 months prior to calciphylaxis onset.14 One proposed mechanism by Munavalli et al10 is that elevated levels of matrix metalloproteinases during catabolic weight loss states enhance the deposition of calcium into elastic fibers of small vessels. The authors found elevated serum levels of matrix metalloproteinases in their patients with NUC induced by rapid weight loss.10
A meta-analysis by Nigwekar et al3 found a history of prior corticosteroid use in 61% (22/36) of NUC cases reviewed. However, it is unclear whether it is the use of corticosteroids or chronic inflammation that is implicated in NUC pathogenesis. Chronic inflammation causes downregulation of anticalcification signaling pathways.18-20 The role of 2 vascular calcification inhibitors has been evaluated in the pathogenesis of calciphylaxis: fetuin-A and matrix gla protein (MGP).21 The activity of these proteins is decreased not only in calciphylaxis but also in other inflammatory states and chronic renal failure.18-20 One study found lower fetuin-A levels in 312 hemodialysis patients compared to healthy controls and an association between low fetuin-A levels and increased C-reactive protein levels.22 Reduced fetuin-A and MGP levels may be the result of several calciphylaxis risk factors. Warfarin is believed to trigger calciphylaxis via inhibition of gamma-carboxylation of MGP, which is necessary for its anticalcification activity.23 Hypoalbuminemia and alcoholic liver disease also are risk factors that may be explained by the fact that fetuin-A is synthesized in the liver.24 Therefore, liver disease results in decreased production of fetuin-A that is permissive to vascular calcification in calciphylaxis patients.
There have been other reports of calciphylaxis patients who were originally hospitalized due to hypotension, which may serve as a trigger for calciphylaxis onset.25 Because calciphylaxis lesions are more likely to occur in the fatty areas of the abdomen and proximal thighs where blood flow is slower, hypotension likely accentuates the slowing of blood flow and subsequent blood vessel calcification. This theory is supported by studies showing that established calciphylactic lesions worsen more quickly in the presence of systemic hypotension.26 One patient with ESRD and calciphylaxis of the breasts had consistent systolic blood pressure readings in the high 60s to low 70s between dialysis sessions.27 Due to this association, we recommend that patients with calciphylaxis have close blood pressure monitoring to aid in preventing disease progression.28
Management
Calciphylaxis treatment has not yet been standardized, as it is an uncommon disease whose pathogenesis is not fully understood. Current management strategies aim to normalize metabolic abnormalities such as hypercalcemia if they are present and remove inciting agents such as warfarin and corticosteroids.29 Other medical treatments that have been successfully used include sodium thiosulfate, oral steroids, and adjunctive bisphosphonates.29-31 Sodium thiosulfate is known to cause metabolic acidosis by generating thiosulfuric acid in vivo in patients with or without renal disease; therefore, patients on sodium thiosulfate therapy should be monitored for development of metabolic acidosis and treated with oral sodium bicarbonate or dialysis as needed.30,32 Wound care also is an important element of calciphylaxis treatment; however, the debridement of wounds is controversial. Some argue that dry intact eschars serve to protect against sepsis, which is the leading cause of death in calciphylaxis.2,14,33 In contrast, a retrospective study of 63 calciphylaxis patients found a 1-year survival rate of 61.6% in 17 patients receiving wound debridement vs 27.4% in 46 patients who did not.2 The current consensus is that debridement should be considered on a case-by-case basis, factoring in the presence of wound infection, size of wounds, stability of eschars, and treatment goals of the patient.34 Future studies should be aimed at this issue, with special focus on how these factors and the decision to debride or not impact patient outcomes.
Conclusion
Calciphylaxis is a potentially fatal disease that impacts both patients with ESRD and those with nonuremic risk factors. The term calcific uremic arteriolopathy should be disregarded, as nonuremic causes are being reported with increased frequency in the literature. In such cases, patients often have multiple risk factors, including obesity, primary hyperparathyroidism, alcoholic liver disease, and underlying malignancy, among others. Certain triggers for onset of calciphylaxis should be avoided in at-risk patients, including the use of corticosteroids or warfarin; iron and albumin infusions; hypotension; and rapid weight loss. Our fatal case of NUC is a reminder to dermatologists treating at-risk patients to avoid these triggers and to keep calciphylaxis in the differential diagnosis when encountering early lesions such as livedo reticularis, as progression of these lesions has a 1-year mortality rate of more than 50% with the therapies being utilized at this time.
- Au S, Crawford RI. Three-dimensional analysis of a calciphylaxis plaque: clues to pathogenesis. J Am Acad Dermatol. 2007;47:53-57.
- Weenig RH, Sewell LD, Davis MD, et al. Calciphylaxis: natural history, risk factor analysis, and outcome. J Am Acad Dermatol. 2007;56:569-579.
- Nigwekar SU, Wolf M, Sterns RH, et al. Calciphylaxis from nonuremic causes: a systematic review. Clin J Am Soc Nephrol. 2008;3:1139-1143.
- Fine A, Zacharias J. Calciphylaxis is usually non-ulcerating: risk factors, outcome and therapy. Kidney Int. 2002;61:2210-2217.
- Angelis M, Wong LL, Myers SA, et al. Calciphylaxis in patients on hemodialysis: a prevalence study. Surgery. 1997;122:1083-1090.
- Chavel SM, Taraszka KS, Schaffer JV, et al. Calciphylaxis associated with acute, reversible renal failure in the setting of alcoholic cirrhosis. J Am Acad Dermatol. 2004;50:125-128.
- Bosler DS, Amin MB, Gulli F, et al. Unusual case of calciphylaxis associated with metastatic breast carcinoma. Am J Dermatopathol. 2007;29:400-403.
- Buxtorf K, Cerottini JP, Panizzon RG. Lower limb skin ulcerations, intravascular calcifications and sensorimotor polyneuropathy: calciphylaxis as part of a hyperparathyroidism? Dermatology. 1999;198:423-425.
- Brouns K, Verbeken E, Degreef H, et al. Fatal calciphylaxis in two patients with giant cell arteritis. Clin Rheumatol. 2007;26:836-840.
- Munavalli G, Reisenauer A, Moses M, et al. Weight loss-induced calciphylaxis: potential role of matrix metalloproteinases. J Dermatol. 2003;30:915-919.
- Bae GH, Nambudiri VE, Bach DQ, et al. Rapidly progressive nonuremic calciphylaxis in setting of warfarin. Am J Med. 2015;128:E19-E21.
- Essary LR, Wick MR. Cutaneous calciphylaxis. an underrecognized clinicopathologic entity. Am J Clin Pathol. 2000;113:280-287.
- Hafner J, Keusch G, Wahl C, et al. Uremic small-artery disease with medial calcification and intimal hyperplasia (so-called calciphylaxis): a complication of chronic renal failure and benefit from parathyroidectomy. J Am Acad Dermatol. 1995;33:954-962.
- Coates T, Kirkland GS, Dymock RB, et al. Cutaneous necrosis from calcific uremic arteriolopathy. Am J Kidney Dis. 1998;32:384-391.
- Selye H, Gentile G, Prioreschi P. Cutaneous molt induced by calciphylaxis in the rat. Science. 1961;134:1876-1877.
- Kalajian AH, Malhotra PS, Callen JP, et al. Calciphylaxis with normal renal and parathyroid function: not as rare as previously believed. Arch Dermatol. 2009;145:451-458.
- Malabu U, Roberts L, Sangla K. Calciphylaxis in a morbidly obese woman with rheumatoid arthritis presenting with severe weight loss and vitamin D deficiency. Endocr Pract. 2011;17:104-108.
- Schäfer C, Heiss A, Schwarz A, et al. The serum protein alpha 2–Heremans-Schmid glycoprotein/fetuin-A is a systemically acting inhibitor of ectopic calcification. J Clin Invest. 2003;112:357-366.
- Cozzolino M, Galassi A, Biondi ML, et al. Serum fetuin-A levels link inflammation and cardiovascular calcification in hemodialysis patients. Am J Nephrol. 2006;26:423-429.
- Luo G, Ducy P, McKee MD, et al. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997;386:78-81.
- Weenig RH. Pathogenesis of calciphylaxis: Hans Selye to nuclear factor kappa-B. J Am Acad Dermatol. 2008;58:458-471.
- Ketteler M, Bongartz P, Westenfeld R, et al. Association of low fetuin-A (AHSG) concentrations in serum with cardiovascular mortality in patients on dialysis: a cross-sectional study. Lancet. 2003;361:827-833.
- Wallin R, Cain D, Sane DC. Matrix Gla protein synthesis and gamma-carboxylation in the aortic vessel wall and proliferating vascular smooth muscle cells a cell system which resembles the system in bone cells. Thromb Haemost. 1999;82:1764-1767.
- Sowers KM, Hayden MR. Calcific uremic arteriolopathy: pathophysiology, reactive oxygen species and therapeutic approaches. Oxid Med Cell Longev. 2010;3:109-121.
- Allegretti AS, Nazarian RM, Goverman J, et al. Calciphylaxis: a rare but fatal delayed complication of Roux-en-Y gastric bypass surgery. Am J Kidney Dis. 2014;64:274-277.
- Wilmer WA, Magro CM. Calciphylaxis: emerging concepts in prevention, diagnosis, and treatment. Semin Dial. 2002;15:172-186.
- Gupta D, Tadros R, Mazumdar A, et al. Breast lesions with intractable pain in end-stage renal disease: calciphylaxis with chronic hypotensive dermatopathy related watershed breast lesions. J Palliat Med. 2013;16:551-554.
- Janigan DT, Hirsch DJ, Klassen GA, et al. Calcified subcutaneous arterioles with infarcts of the subcutis and skin (“calciphylaxis”) in chronic renal failure. Am J Kidney Dis. 2000;35:588-597.
- Jeong HS, Dominguez AR. Calciphylaxis: controversies in pathogenesis, diagnosis and treatment. Am J Med Sci. 2016;351:217-227.
- Bourgeois P, De Haes P. Sodium thiosulfate as a treatment for calciphylaxis: a case series. J Dermatolog Treat. 2016;27:520-524.
- Biswas A, Walsh NM, Tremaine R. A case of nonuremic calciphylaxis treated effectively with systemic corticosteroids. J Cutan Med Surg. 2016;20:275-278.
- Selk N, Rodby, RA. Unexpectedly severe metabolic acidosis associated with sodium thiosulfate therapy in a patient with calcific uremic arteriolopathy. Semin Dial. 2011;24:85-88.
- Martin R. Mysterious calciphylaxis: wounds with eschar—to debride or not to debride? Ostomy Wound Manage. 2004:50:64-66, 68-70.
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146.
- Au S, Crawford RI. Three-dimensional analysis of a calciphylaxis plaque: clues to pathogenesis. J Am Acad Dermatol. 2007;47:53-57.
- Weenig RH, Sewell LD, Davis MD, et al. Calciphylaxis: natural history, risk factor analysis, and outcome. J Am Acad Dermatol. 2007;56:569-579.
- Nigwekar SU, Wolf M, Sterns RH, et al. Calciphylaxis from nonuremic causes: a systematic review. Clin J Am Soc Nephrol. 2008;3:1139-1143.
- Fine A, Zacharias J. Calciphylaxis is usually non-ulcerating: risk factors, outcome and therapy. Kidney Int. 2002;61:2210-2217.
- Angelis M, Wong LL, Myers SA, et al. Calciphylaxis in patients on hemodialysis: a prevalence study. Surgery. 1997;122:1083-1090.
- Chavel SM, Taraszka KS, Schaffer JV, et al. Calciphylaxis associated with acute, reversible renal failure in the setting of alcoholic cirrhosis. J Am Acad Dermatol. 2004;50:125-128.
- Bosler DS, Amin MB, Gulli F, et al. Unusual case of calciphylaxis associated with metastatic breast carcinoma. Am J Dermatopathol. 2007;29:400-403.
- Buxtorf K, Cerottini JP, Panizzon RG. Lower limb skin ulcerations, intravascular calcifications and sensorimotor polyneuropathy: calciphylaxis as part of a hyperparathyroidism? Dermatology. 1999;198:423-425.
- Brouns K, Verbeken E, Degreef H, et al. Fatal calciphylaxis in two patients with giant cell arteritis. Clin Rheumatol. 2007;26:836-840.
- Munavalli G, Reisenauer A, Moses M, et al. Weight loss-induced calciphylaxis: potential role of matrix metalloproteinases. J Dermatol. 2003;30:915-919.
- Bae GH, Nambudiri VE, Bach DQ, et al. Rapidly progressive nonuremic calciphylaxis in setting of warfarin. Am J Med. 2015;128:E19-E21.
- Essary LR, Wick MR. Cutaneous calciphylaxis. an underrecognized clinicopathologic entity. Am J Clin Pathol. 2000;113:280-287.
- Hafner J, Keusch G, Wahl C, et al. Uremic small-artery disease with medial calcification and intimal hyperplasia (so-called calciphylaxis): a complication of chronic renal failure and benefit from parathyroidectomy. J Am Acad Dermatol. 1995;33:954-962.
- Coates T, Kirkland GS, Dymock RB, et al. Cutaneous necrosis from calcific uremic arteriolopathy. Am J Kidney Dis. 1998;32:384-391.
- Selye H, Gentile G, Prioreschi P. Cutaneous molt induced by calciphylaxis in the rat. Science. 1961;134:1876-1877.
- Kalajian AH, Malhotra PS, Callen JP, et al. Calciphylaxis with normal renal and parathyroid function: not as rare as previously believed. Arch Dermatol. 2009;145:451-458.
- Malabu U, Roberts L, Sangla K. Calciphylaxis in a morbidly obese woman with rheumatoid arthritis presenting with severe weight loss and vitamin D deficiency. Endocr Pract. 2011;17:104-108.
- Schäfer C, Heiss A, Schwarz A, et al. The serum protein alpha 2–Heremans-Schmid glycoprotein/fetuin-A is a systemically acting inhibitor of ectopic calcification. J Clin Invest. 2003;112:357-366.
- Cozzolino M, Galassi A, Biondi ML, et al. Serum fetuin-A levels link inflammation and cardiovascular calcification in hemodialysis patients. Am J Nephrol. 2006;26:423-429.
- Luo G, Ducy P, McKee MD, et al. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997;386:78-81.
- Weenig RH. Pathogenesis of calciphylaxis: Hans Selye to nuclear factor kappa-B. J Am Acad Dermatol. 2008;58:458-471.
- Ketteler M, Bongartz P, Westenfeld R, et al. Association of low fetuin-A (AHSG) concentrations in serum with cardiovascular mortality in patients on dialysis: a cross-sectional study. Lancet. 2003;361:827-833.
- Wallin R, Cain D, Sane DC. Matrix Gla protein synthesis and gamma-carboxylation in the aortic vessel wall and proliferating vascular smooth muscle cells a cell system which resembles the system in bone cells. Thromb Haemost. 1999;82:1764-1767.
- Sowers KM, Hayden MR. Calcific uremic arteriolopathy: pathophysiology, reactive oxygen species and therapeutic approaches. Oxid Med Cell Longev. 2010;3:109-121.
- Allegretti AS, Nazarian RM, Goverman J, et al. Calciphylaxis: a rare but fatal delayed complication of Roux-en-Y gastric bypass surgery. Am J Kidney Dis. 2014;64:274-277.
- Wilmer WA, Magro CM. Calciphylaxis: emerging concepts in prevention, diagnosis, and treatment. Semin Dial. 2002;15:172-186.
- Gupta D, Tadros R, Mazumdar A, et al. Breast lesions with intractable pain in end-stage renal disease: calciphylaxis with chronic hypotensive dermatopathy related watershed breast lesions. J Palliat Med. 2013;16:551-554.
- Janigan DT, Hirsch DJ, Klassen GA, et al. Calcified subcutaneous arterioles with infarcts of the subcutis and skin (“calciphylaxis”) in chronic renal failure. Am J Kidney Dis. 2000;35:588-597.
- Jeong HS, Dominguez AR. Calciphylaxis: controversies in pathogenesis, diagnosis and treatment. Am J Med Sci. 2016;351:217-227.
- Bourgeois P, De Haes P. Sodium thiosulfate as a treatment for calciphylaxis: a case series. J Dermatolog Treat. 2016;27:520-524.
- Biswas A, Walsh NM, Tremaine R. A case of nonuremic calciphylaxis treated effectively with systemic corticosteroids. J Cutan Med Surg. 2016;20:275-278.
- Selk N, Rodby, RA. Unexpectedly severe metabolic acidosis associated with sodium thiosulfate therapy in a patient with calcific uremic arteriolopathy. Semin Dial. 2011;24:85-88.
- Martin R. Mysterious calciphylaxis: wounds with eschar—to debride or not to debride? Ostomy Wound Manage. 2004:50:64-66, 68-70.
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146.
Practice Points
- Calciphylaxis is a potentially fatal disease caused by metastatic calcification of cutaneous small- and medium-sized blood vessels leading to ischemia and necrosis.
- Calciphylaxis most commonly is seen in patients with renal disease requiring dialysis, but it also may be triggered by nonuremic causes in patients with known risk factors for calciphylaxis.
- Risk factors for calciphylaxis include female gender, white race, obesity, alcoholic liver disease, primary hyperparathyroidism, connective tissue disease, underlying malignancy, protein C or S deficiency, corticosteroid use, warfarin use, diabetes, iron or albumin infusions, and rapid weight loss.
- The term calcific uremic arteriolopathy should be disregarded, as nonuremic causes are being reported with increased frequency in the literature.
Antimalarial adherence is important for diabetes prevention in lupus
Adhering to antimalarial treatment offers some protection to patients with systemic lupus erythematosus (SLE) from developing type 2 diabetes mellitus (T2DM), according to new research.
Patients who took at least 90% of their prescribed antimalarial doses were 39% less likely to develop T2DM than patients who discontinued antimalarial therapy. Patients who took less than 90% of their prescribed doses but didn’t discontinue treatment were 22% less likely to develop T2DM.
“[O]ur study provides further support for the importance of adherence to antimalarials in SLE by demonstrating protective impacts on T2DM,” Shahrzad Salmasi, PhD, of the University of British Columbia, Vancouver, and colleagues wrote in Arthritis Care & Research.
Dr. Salmasi and colleagues conducted this retrospective study using administrative health data on patients in British Columbia. The researchers analyzed 1,498 patients with SLE. Their mean age was about 44 years, and 91% were women.
The researchers used data on prescription dates and days’ supply to establish antimalarial drug courses and gaps in treatment. A new treatment course occurred when a 90-day gap was exceeded between refills. The researchers calculated the proportion of days covered (PDC) – the total number of days with antimalarials divided by the length of the course – and separated patients into three categories:
- Adherent to treatment – PDC of 0.90 or greater
- Nonadherent – PDC greater than 0 but less than 0.90
- Discontinuer – PDC of 0
The patients had a mean of about 23 antimalarial prescriptions and a mean of about two courses. The mean course duration was 554 days.
At a median follow-up of 4.6 years, there were 140 incident cases of T2DM. The researchers calculated the risk of T2DM among adherent and nonadherent patients, comparing these groups with the discontinuers and adjusting for age, sex, comorbidities, and concomitant medications.
The adjusted hazard ratio for developing T2DM was 0.61 among adherent patients and 0.78 among nonadherent patients.
“This population-based study highlighted that taking less than 90% of the prescribed antimalarials compromises their effect in preventing T2DM in SLE patients,” Dr. Salmasi and colleagues wrote. “Our findings should be used to emphasize the importance of medication adherence in not only treating SLE but also preventing its complications.”
The researchers reported having no conflicts of interest.
SOURCE: Salmasi S et al. Arthritis Care Res. 2020 Jan 21. doi: 10.1002/acr.24147.
Adhering to antimalarial treatment offers some protection to patients with systemic lupus erythematosus (SLE) from developing type 2 diabetes mellitus (T2DM), according to new research.
Patients who took at least 90% of their prescribed antimalarial doses were 39% less likely to develop T2DM than patients who discontinued antimalarial therapy. Patients who took less than 90% of their prescribed doses but didn’t discontinue treatment were 22% less likely to develop T2DM.
“[O]ur study provides further support for the importance of adherence to antimalarials in SLE by demonstrating protective impacts on T2DM,” Shahrzad Salmasi, PhD, of the University of British Columbia, Vancouver, and colleagues wrote in Arthritis Care & Research.
Dr. Salmasi and colleagues conducted this retrospective study using administrative health data on patients in British Columbia. The researchers analyzed 1,498 patients with SLE. Their mean age was about 44 years, and 91% were women.
The researchers used data on prescription dates and days’ supply to establish antimalarial drug courses and gaps in treatment. A new treatment course occurred when a 90-day gap was exceeded between refills. The researchers calculated the proportion of days covered (PDC) – the total number of days with antimalarials divided by the length of the course – and separated patients into three categories:
- Adherent to treatment – PDC of 0.90 or greater
- Nonadherent – PDC greater than 0 but less than 0.90
- Discontinuer – PDC of 0
The patients had a mean of about 23 antimalarial prescriptions and a mean of about two courses. The mean course duration was 554 days.
At a median follow-up of 4.6 years, there were 140 incident cases of T2DM. The researchers calculated the risk of T2DM among adherent and nonadherent patients, comparing these groups with the discontinuers and adjusting for age, sex, comorbidities, and concomitant medications.
The adjusted hazard ratio for developing T2DM was 0.61 among adherent patients and 0.78 among nonadherent patients.
“This population-based study highlighted that taking less than 90% of the prescribed antimalarials compromises their effect in preventing T2DM in SLE patients,” Dr. Salmasi and colleagues wrote. “Our findings should be used to emphasize the importance of medication adherence in not only treating SLE but also preventing its complications.”
The researchers reported having no conflicts of interest.
SOURCE: Salmasi S et al. Arthritis Care Res. 2020 Jan 21. doi: 10.1002/acr.24147.
Adhering to antimalarial treatment offers some protection to patients with systemic lupus erythematosus (SLE) from developing type 2 diabetes mellitus (T2DM), according to new research.
Patients who took at least 90% of their prescribed antimalarial doses were 39% less likely to develop T2DM than patients who discontinued antimalarial therapy. Patients who took less than 90% of their prescribed doses but didn’t discontinue treatment were 22% less likely to develop T2DM.
“[O]ur study provides further support for the importance of adherence to antimalarials in SLE by demonstrating protective impacts on T2DM,” Shahrzad Salmasi, PhD, of the University of British Columbia, Vancouver, and colleagues wrote in Arthritis Care & Research.
Dr. Salmasi and colleagues conducted this retrospective study using administrative health data on patients in British Columbia. The researchers analyzed 1,498 patients with SLE. Their mean age was about 44 years, and 91% were women.
The researchers used data on prescription dates and days’ supply to establish antimalarial drug courses and gaps in treatment. A new treatment course occurred when a 90-day gap was exceeded between refills. The researchers calculated the proportion of days covered (PDC) – the total number of days with antimalarials divided by the length of the course – and separated patients into three categories:
- Adherent to treatment – PDC of 0.90 or greater
- Nonadherent – PDC greater than 0 but less than 0.90
- Discontinuer – PDC of 0
The patients had a mean of about 23 antimalarial prescriptions and a mean of about two courses. The mean course duration was 554 days.
At a median follow-up of 4.6 years, there were 140 incident cases of T2DM. The researchers calculated the risk of T2DM among adherent and nonadherent patients, comparing these groups with the discontinuers and adjusting for age, sex, comorbidities, and concomitant medications.
The adjusted hazard ratio for developing T2DM was 0.61 among adherent patients and 0.78 among nonadherent patients.
“This population-based study highlighted that taking less than 90% of the prescribed antimalarials compromises their effect in preventing T2DM in SLE patients,” Dr. Salmasi and colleagues wrote. “Our findings should be used to emphasize the importance of medication adherence in not only treating SLE but also preventing its complications.”
The researchers reported having no conflicts of interest.
SOURCE: Salmasi S et al. Arthritis Care Res. 2020 Jan 21. doi: 10.1002/acr.24147.
FROM ARTHRITIS CARE & RESEARCH
Core behaviors enhance communication about neonatal death
Clinicians can improve communications with parents during neonatal end-of-life situations by adopting key behaviors such as sitting down to talk to parents and using the infant’s name, according to data from a simulation study.
“Empirical evidence regarding communication with parents during and after a child’s critical instability or death is scarce,” wrote Marie-Hélène Lizotte, MD, of Centre Hospitalier Universitaire Sainte-Justine, Montréal, and colleagues. Noting that realistic simulation has been shown to help clinicians improve their communication skills, the investigators recruited clinicians to participate in a simulated unsuccessful neonatal resuscitation to identify behaviors associated with optimal parent communication.
Behaviors associated with high scores for clinicians deemed “good communicators” included introducing themselves to parents, using the infant’s name (if known), sitting down to speak to parents, not leaving the infant alone on the bed after death, and allowing time for silence, the researchers reported in Pediatrics.
The investigators presented the video simulations to evaluators, including clinicians and bereaved parents. In the simulation, a term infant was born after an emergency cesarean delivery for fetal distress and died after an unsuccessful attempt at resuscitation. A manikin infant was programmed to remain pulseless, and two actors played the roles of the parents in the video.
Evaluators scored the videos for overall performance and for communication with the parent actors during and after the resuscitation.
Overall, parent evaluators and parent actors agreed with clinicians on what actions exemplified optimal communication in about 81% of evaluations. Discrepancies were mainly related to the language participants used related to death, as some parent evaluators said they had trouble understanding certain sentences or found them insensitive, such as “her heart never came back” and “allowing natural death.”
A total of 31 participants were recruited for the simulation, including 15 pediatric residents, 5 neonatal fellows, 3 neonatologists, 3 neonatal nurse practitioners, and 5 transport and resuscitation team providers. Videos of the simulations were examined by 21 evaluators, including bereaved parents, the parent actors, a neonatologist, a maternal-fetal medicine specialist, a psychologist, and a respiratory therapist. There were 651 evaluations.
The study findings were limited by several factors including the use of a single center, use of videos for evaluations, and the use of a single infant-resuscitation scenario, the researchers noted. The results were strengthened, however, by the large number of evaluations, and they support the core behaviors as “a skeleton on which to build additional skills with practice and training” with attention to cultural differences in their application, such as recognizing that infants are not named until after birth in some cultures, they said.
The existing literature on strategies for providing empathy and support to parents facing the death of a child is limited, but this simulation study provides a design model to help address this issue, Chris Feudtner, MD, of the Children’s Hospital of Philadelphia wrote in an accompanying editorial. “Overall, this study, in terms of design and methodologic rigor, is a great advance toward answering our key question: how to best support parents in such circumstances,” he said.
Dr. Feudtner said that he would divide the clinician behaviors into two groups. The first, “Calm kind politeness,” includes acknowledging the parents, introducing themselves, using the infant’s name, and remaining calm. The second set of behaviors, which he called “Skillful situational leadership,” includes preparing parents for the resuscitation activities and providing verbal milestones that prepared them for the fatal outcome.
“Picking up on a metaphor offered by the authors of the study, training and repetitive drills on these specific behaviors cannot be emphasized enough because they are not only the skeleton of excellent communication; they are likely also the muscles, the heart, and even the soul,” he concluded.
The study was supported by a grant from the Fonds de Recherche en Santé du Québec and the Medical Education Grant from Centre Hospitalier Universitaire Sainte-Justine. The researchers and Dr. Feudtner reported no financial conflicts.
SOURCE: Lizotte M-H et al. Pediatrics. 2020 Jan 27. doi: 10.1542/peds.2019-1925; Feudtner C. Pediatrics. 2020 Jan 27. doi: 10.1542/peds.2019-3116.
Clinicians can improve communications with parents during neonatal end-of-life situations by adopting key behaviors such as sitting down to talk to parents and using the infant’s name, according to data from a simulation study.
“Empirical evidence regarding communication with parents during and after a child’s critical instability or death is scarce,” wrote Marie-Hélène Lizotte, MD, of Centre Hospitalier Universitaire Sainte-Justine, Montréal, and colleagues. Noting that realistic simulation has been shown to help clinicians improve their communication skills, the investigators recruited clinicians to participate in a simulated unsuccessful neonatal resuscitation to identify behaviors associated with optimal parent communication.
Behaviors associated with high scores for clinicians deemed “good communicators” included introducing themselves to parents, using the infant’s name (if known), sitting down to speak to parents, not leaving the infant alone on the bed after death, and allowing time for silence, the researchers reported in Pediatrics.
The investigators presented the video simulations to evaluators, including clinicians and bereaved parents. In the simulation, a term infant was born after an emergency cesarean delivery for fetal distress and died after an unsuccessful attempt at resuscitation. A manikin infant was programmed to remain pulseless, and two actors played the roles of the parents in the video.
Evaluators scored the videos for overall performance and for communication with the parent actors during and after the resuscitation.
Overall, parent evaluators and parent actors agreed with clinicians on what actions exemplified optimal communication in about 81% of evaluations. Discrepancies were mainly related to the language participants used related to death, as some parent evaluators said they had trouble understanding certain sentences or found them insensitive, such as “her heart never came back” and “allowing natural death.”
A total of 31 participants were recruited for the simulation, including 15 pediatric residents, 5 neonatal fellows, 3 neonatologists, 3 neonatal nurse practitioners, and 5 transport and resuscitation team providers. Videos of the simulations were examined by 21 evaluators, including bereaved parents, the parent actors, a neonatologist, a maternal-fetal medicine specialist, a psychologist, and a respiratory therapist. There were 651 evaluations.
The study findings were limited by several factors including the use of a single center, use of videos for evaluations, and the use of a single infant-resuscitation scenario, the researchers noted. The results were strengthened, however, by the large number of evaluations, and they support the core behaviors as “a skeleton on which to build additional skills with practice and training” with attention to cultural differences in their application, such as recognizing that infants are not named until after birth in some cultures, they said.
The existing literature on strategies for providing empathy and support to parents facing the death of a child is limited, but this simulation study provides a design model to help address this issue, Chris Feudtner, MD, of the Children’s Hospital of Philadelphia wrote in an accompanying editorial. “Overall, this study, in terms of design and methodologic rigor, is a great advance toward answering our key question: how to best support parents in such circumstances,” he said.
Dr. Feudtner said that he would divide the clinician behaviors into two groups. The first, “Calm kind politeness,” includes acknowledging the parents, introducing themselves, using the infant’s name, and remaining calm. The second set of behaviors, which he called “Skillful situational leadership,” includes preparing parents for the resuscitation activities and providing verbal milestones that prepared them for the fatal outcome.
“Picking up on a metaphor offered by the authors of the study, training and repetitive drills on these specific behaviors cannot be emphasized enough because they are not only the skeleton of excellent communication; they are likely also the muscles, the heart, and even the soul,” he concluded.
The study was supported by a grant from the Fonds de Recherche en Santé du Québec and the Medical Education Grant from Centre Hospitalier Universitaire Sainte-Justine. The researchers and Dr. Feudtner reported no financial conflicts.
SOURCE: Lizotte M-H et al. Pediatrics. 2020 Jan 27. doi: 10.1542/peds.2019-1925; Feudtner C. Pediatrics. 2020 Jan 27. doi: 10.1542/peds.2019-3116.
Clinicians can improve communications with parents during neonatal end-of-life situations by adopting key behaviors such as sitting down to talk to parents and using the infant’s name, according to data from a simulation study.
“Empirical evidence regarding communication with parents during and after a child’s critical instability or death is scarce,” wrote Marie-Hélène Lizotte, MD, of Centre Hospitalier Universitaire Sainte-Justine, Montréal, and colleagues. Noting that realistic simulation has been shown to help clinicians improve their communication skills, the investigators recruited clinicians to participate in a simulated unsuccessful neonatal resuscitation to identify behaviors associated with optimal parent communication.
Behaviors associated with high scores for clinicians deemed “good communicators” included introducing themselves to parents, using the infant’s name (if known), sitting down to speak to parents, not leaving the infant alone on the bed after death, and allowing time for silence, the researchers reported in Pediatrics.
The investigators presented the video simulations to evaluators, including clinicians and bereaved parents. In the simulation, a term infant was born after an emergency cesarean delivery for fetal distress and died after an unsuccessful attempt at resuscitation. A manikin infant was programmed to remain pulseless, and two actors played the roles of the parents in the video.
Evaluators scored the videos for overall performance and for communication with the parent actors during and after the resuscitation.
Overall, parent evaluators and parent actors agreed with clinicians on what actions exemplified optimal communication in about 81% of evaluations. Discrepancies were mainly related to the language participants used related to death, as some parent evaluators said they had trouble understanding certain sentences or found them insensitive, such as “her heart never came back” and “allowing natural death.”
A total of 31 participants were recruited for the simulation, including 15 pediatric residents, 5 neonatal fellows, 3 neonatologists, 3 neonatal nurse practitioners, and 5 transport and resuscitation team providers. Videos of the simulations were examined by 21 evaluators, including bereaved parents, the parent actors, a neonatologist, a maternal-fetal medicine specialist, a psychologist, and a respiratory therapist. There were 651 evaluations.
The study findings were limited by several factors including the use of a single center, use of videos for evaluations, and the use of a single infant-resuscitation scenario, the researchers noted. The results were strengthened, however, by the large number of evaluations, and they support the core behaviors as “a skeleton on which to build additional skills with practice and training” with attention to cultural differences in their application, such as recognizing that infants are not named until after birth in some cultures, they said.
The existing literature on strategies for providing empathy and support to parents facing the death of a child is limited, but this simulation study provides a design model to help address this issue, Chris Feudtner, MD, of the Children’s Hospital of Philadelphia wrote in an accompanying editorial. “Overall, this study, in terms of design and methodologic rigor, is a great advance toward answering our key question: how to best support parents in such circumstances,” he said.
Dr. Feudtner said that he would divide the clinician behaviors into two groups. The first, “Calm kind politeness,” includes acknowledging the parents, introducing themselves, using the infant’s name, and remaining calm. The second set of behaviors, which he called “Skillful situational leadership,” includes preparing parents for the resuscitation activities and providing verbal milestones that prepared them for the fatal outcome.
“Picking up on a metaphor offered by the authors of the study, training and repetitive drills on these specific behaviors cannot be emphasized enough because they are not only the skeleton of excellent communication; they are likely also the muscles, the heart, and even the soul,” he concluded.
The study was supported by a grant from the Fonds de Recherche en Santé du Québec and the Medical Education Grant from Centre Hospitalier Universitaire Sainte-Justine. The researchers and Dr. Feudtner reported no financial conflicts.
SOURCE: Lizotte M-H et al. Pediatrics. 2020 Jan 27. doi: 10.1542/peds.2019-1925; Feudtner C. Pediatrics. 2020 Jan 27. doi: 10.1542/peds.2019-3116.
FROM PEDIATRICS
Key clinical point: Clinicians who took steps such as sitting down and using the infant’s name were seen by parents as good communicators.
Major finding: Evaluators of a simulation agreed 81% of the time on defining optimal communication.
Study details: The data come from a simulation study of 31 participants and 21 evaluators and a total of 651 evaluations.
Disclosures: The study was supported in part by the Fonds de Recherche en Santé du Québec and the Medical Education Grant from Centre Hospitalier Universitaire Sainte-Justine. The researchers and editorialist said they had no financial conflicts.
Source: Lizotte M-H et al. Pediatrics. 2020 Jan 27. doi: 10.1542/peds.2019-1925; Feudtner C. Pediatrics. 2020 Jan 27. doi: 10.1542/peds.2019-3116.
Cannabis for sleep: Short-term benefit, long-term disruption?
, new research shows.
Investigators found whole-plant medical cannabis use was associated with fewer problems with respect to waking up at night, but they also found that frequent medical cannabis use was associated with more problems initiating and maintaining sleep.

“Cannabis may improve overall sleep in the short term,” study investigator Sharon Sznitman, PhD, University of Haifa (Israel) Faculty of Social Welfare and Health Sciences, said in an interview. “But it’s also very interesting that when we looked at frequency of use in the group that used medical cannabis, individuals who had more frequent use also had poorer sleep in the long term.
“This suggests that while cannabis may improve overall sleep, it’s also possible that there is a tolerance that develops with either very frequent or long-term use,” she added.
The study was published online Jan. 20 in BMJ Supportive and Palliative Care.
A common problem
Estimates suggest chronic pain affects up to 37% of adults in the developed world. Individuals who suffer chronic pain often experience comorbid insomnia, which includes difficulty initiating sleep, sleep disruption, and early morning wakening.
For its part, medical cannabis to treat chronic pain symptoms and manage sleep problems has been widely reported as a prime motivation for medical cannabis use. Indeed, previous studies have concluded that the endocannabinoid system plays a role in sleep regulation, including sleep promotion and maintenance.
In recent years, investigators have reported the beneficial effects of medical cannabis for sleep. Nevertheless, some preclinical research has also concluded that chronic administration of tetrahydrocannabinol may result in tolerance to the sleep-enhancing effects of cannabis.
With that in mind, the researchers set out to examine the potential impact of whole-plant medicinal cannabis on sleep problems experienced by middle-aged patients suffering from chronic pain.
“People are self-reporting that they’re using cannabis for sleep and that it helps, but as we know, just because people are reporting that it works doesn’t mean that it will hold up in research,” Dr. Sznitman said.
The study included 128 individuals (mean age, 61±6 years; 51% females) with chronic neuropathic pain: 66 were medical cannabis users and 62 were not.
Three indicators of insomnia were measured using the 7-point Likert scale to assess issues with sleep initiation and maintenance.
In addition, investigators collected sociodemographic information, as well as data on daily consumption of tobacco, frequency of alcohol use, and pain severity. Finally, they collected patient data on the use of sleep-aid medications during the past month as well as tricyclic antidepressant use.
Frequent use, more sleep problems?
On average, medical cannabis users were 3 years younger than their nonusing counterparts (mean age, 60±6 vs. 63±6 years, respectively, P = .003) and more likely to be male (58% vs 40%, respectively, P = .038). Otherwise, the two groups were comparable.
Medical cannabis users reported taking the drug for an average of 4 years, at an average quantity of 31 g per month. The primary mode of administration was smoking (68.6%), followed by oil extracts (21.4%) and vaporization (20%).
Results showed that, of the total sample, 24.1% reported always waking up early and not falling back to sleep, 20.2% reported always having difficulty falling asleep, and 27.2% reported always waking up during the night.
After adjusting for patient age, sex, pain level, and use of sleep medications and antidepressants, medical cannabis use was associated with fewer problems with waking up at night, compared with nonmedical cannabis use. No differences were found between groups with respect to problems falling asleep or waking up early without being able to fall back to sleep, Dr. Sznitman and associates reported.
The final analysis of a subsample of patients that only included medical cannabis users showed frequency of medical cannabis use was associated with sleep problems, they said.
Specifically, more frequent cannabis use was associated with more problems related to waking up at night, as well as problems falling asleep.
Sleep problems associated with frequent medical cannabis use may signal the development of tolerance to the agent. However, frequent users of medical cannabis also maybsuffer pain or other comorbidities, which, in turn, may be linked to more sleep problems.
Either way, Dr. Sznitman said the study might open the door to another treatment option for patients suffering from chronic pain who struggle with sleep.
“If future research shows that the effect of medical cannabis on sleep is a consistent one, then we may be adding a new therapy for sleep problems, which are huge in society and especially in chronic pain patients,” she said.
Early days
Commenting on the findings in an interview, Ryan G. Vandrey, PhD, who was not involved in the study, said the findings are in line with previous research.
“I think the results make sense with respect to the data I’ve collected and from what I’ve seen,” said Dr. Vandrey, associate professor of psychiatry and behavioral sciences at Johns Hopkins Medicine in Baltimore.
“We typically only want to use sleep medications for short periods of time,” he continued. “When you think about recommended prescribing practices for any hypnotic medication, it’s usually short term, 2 weeks or less. Longer-term use often leads to tolerance, dependence, and withdrawal symptoms when the medication is stopped, which leads to an exacerbation of disordered sleep,” Dr. Vandrey said.
Nevertheless, he urged caution when interpreting the results.
“I think the study warrants caution about long-term daily use of cannabinoids with respect to sleep,” he said. “But we need more detailed evaluations, as the trial wasn’t testing a defined product, specific dose, or dose regimen.
“In addition, this was all done in the context of people with chronic pain and not treating disordered sleep or insomnia, but the study highlights the importance of recognizing that long-term chronic use of cannabis is not likely to fully resolve sleep problems.”
Dr. Sznitman agreed that the research is still in its very early stages.
“We’re still far from saying we have the evidence to support the use of medical cannabis for sleep,” she said. “For in the end it was just a cross-sectional, observational study, so we cannot say anything about cause and effect. But if these results pan out, they could be far-reaching and exciting.”
The study was funded by the University of Haifa and Rambam Hospital in Israel, and by the Evelyn Lipper Foundation. Dr. Sznitman and Dr. Vandrey have disclosed no relevant financial relationships.
This article first appeared on Medscape.com.
, new research shows.
Investigators found whole-plant medical cannabis use was associated with fewer problems with respect to waking up at night, but they also found that frequent medical cannabis use was associated with more problems initiating and maintaining sleep.
“Cannabis may improve overall sleep in the short term,” study investigator Sharon Sznitman, PhD, University of Haifa (Israel) Faculty of Social Welfare and Health Sciences, said in an interview. “But it’s also very interesting that when we looked at frequency of use in the group that used medical cannabis, individuals who had more frequent use also had poorer sleep in the long term.
“This suggests that while cannabis may improve overall sleep, it’s also possible that there is a tolerance that develops with either very frequent or long-term use,” she added.
The study was published online Jan. 20 in BMJ Supportive and Palliative Care.
A common problem
Estimates suggest chronic pain affects up to 37% of adults in the developed world. Individuals who suffer chronic pain often experience comorbid insomnia, which includes difficulty initiating sleep, sleep disruption, and early morning wakening.
For its part, medical cannabis to treat chronic pain symptoms and manage sleep problems has been widely reported as a prime motivation for medical cannabis use. Indeed, previous studies have concluded that the endocannabinoid system plays a role in sleep regulation, including sleep promotion and maintenance.
In recent years, investigators have reported the beneficial effects of medical cannabis for sleep. Nevertheless, some preclinical research has also concluded that chronic administration of tetrahydrocannabinol may result in tolerance to the sleep-enhancing effects of cannabis.
With that in mind, the researchers set out to examine the potential impact of whole-plant medicinal cannabis on sleep problems experienced by middle-aged patients suffering from chronic pain.
“People are self-reporting that they’re using cannabis for sleep and that it helps, but as we know, just because people are reporting that it works doesn’t mean that it will hold up in research,” Dr. Sznitman said.
The study included 128 individuals (mean age, 61±6 years; 51% females) with chronic neuropathic pain: 66 were medical cannabis users and 62 were not.
Three indicators of insomnia were measured using the 7-point Likert scale to assess issues with sleep initiation and maintenance.
In addition, investigators collected sociodemographic information, as well as data on daily consumption of tobacco, frequency of alcohol use, and pain severity. Finally, they collected patient data on the use of sleep-aid medications during the past month as well as tricyclic antidepressant use.
Frequent use, more sleep problems?
On average, medical cannabis users were 3 years younger than their nonusing counterparts (mean age, 60±6 vs. 63±6 years, respectively, P = .003) and more likely to be male (58% vs 40%, respectively, P = .038). Otherwise, the two groups were comparable.
Medical cannabis users reported taking the drug for an average of 4 years, at an average quantity of 31 g per month. The primary mode of administration was smoking (68.6%), followed by oil extracts (21.4%) and vaporization (20%).
Results showed that, of the total sample, 24.1% reported always waking up early and not falling back to sleep, 20.2% reported always having difficulty falling asleep, and 27.2% reported always waking up during the night.
After adjusting for patient age, sex, pain level, and use of sleep medications and antidepressants, medical cannabis use was associated with fewer problems with waking up at night, compared with nonmedical cannabis use. No differences were found between groups with respect to problems falling asleep or waking up early without being able to fall back to sleep, Dr. Sznitman and associates reported.
The final analysis of a subsample of patients that only included medical cannabis users showed frequency of medical cannabis use was associated with sleep problems, they said.
Specifically, more frequent cannabis use was associated with more problems related to waking up at night, as well as problems falling asleep.
Sleep problems associated with frequent medical cannabis use may signal the development of tolerance to the agent. However, frequent users of medical cannabis also maybsuffer pain or other comorbidities, which, in turn, may be linked to more sleep problems.
Either way, Dr. Sznitman said the study might open the door to another treatment option for patients suffering from chronic pain who struggle with sleep.
“If future research shows that the effect of medical cannabis on sleep is a consistent one, then we may be adding a new therapy for sleep problems, which are huge in society and especially in chronic pain patients,” she said.
Early days
Commenting on the findings in an interview, Ryan G. Vandrey, PhD, who was not involved in the study, said the findings are in line with previous research.
“I think the results make sense with respect to the data I’ve collected and from what I’ve seen,” said Dr. Vandrey, associate professor of psychiatry and behavioral sciences at Johns Hopkins Medicine in Baltimore.
“We typically only want to use sleep medications for short periods of time,” he continued. “When you think about recommended prescribing practices for any hypnotic medication, it’s usually short term, 2 weeks or less. Longer-term use often leads to tolerance, dependence, and withdrawal symptoms when the medication is stopped, which leads to an exacerbation of disordered sleep,” Dr. Vandrey said.
Nevertheless, he urged caution when interpreting the results.
“I think the study warrants caution about long-term daily use of cannabinoids with respect to sleep,” he said. “But we need more detailed evaluations, as the trial wasn’t testing a defined product, specific dose, or dose regimen.
“In addition, this was all done in the context of people with chronic pain and not treating disordered sleep or insomnia, but the study highlights the importance of recognizing that long-term chronic use of cannabis is not likely to fully resolve sleep problems.”
Dr. Sznitman agreed that the research is still in its very early stages.
“We’re still far from saying we have the evidence to support the use of medical cannabis for sleep,” she said. “For in the end it was just a cross-sectional, observational study, so we cannot say anything about cause and effect. But if these results pan out, they could be far-reaching and exciting.”
The study was funded by the University of Haifa and Rambam Hospital in Israel, and by the Evelyn Lipper Foundation. Dr. Sznitman and Dr. Vandrey have disclosed no relevant financial relationships.
This article first appeared on Medscape.com.
, new research shows.
Investigators found whole-plant medical cannabis use was associated with fewer problems with respect to waking up at night, but they also found that frequent medical cannabis use was associated with more problems initiating and maintaining sleep.
“Cannabis may improve overall sleep in the short term,” study investigator Sharon Sznitman, PhD, University of Haifa (Israel) Faculty of Social Welfare and Health Sciences, said in an interview. “But it’s also very interesting that when we looked at frequency of use in the group that used medical cannabis, individuals who had more frequent use also had poorer sleep in the long term.
“This suggests that while cannabis may improve overall sleep, it’s also possible that there is a tolerance that develops with either very frequent or long-term use,” she added.
The study was published online Jan. 20 in BMJ Supportive and Palliative Care.
A common problem
Estimates suggest chronic pain affects up to 37% of adults in the developed world. Individuals who suffer chronic pain often experience comorbid insomnia, which includes difficulty initiating sleep, sleep disruption, and early morning wakening.
For its part, medical cannabis to treat chronic pain symptoms and manage sleep problems has been widely reported as a prime motivation for medical cannabis use. Indeed, previous studies have concluded that the endocannabinoid system plays a role in sleep regulation, including sleep promotion and maintenance.
In recent years, investigators have reported the beneficial effects of medical cannabis for sleep. Nevertheless, some preclinical research has also concluded that chronic administration of tetrahydrocannabinol may result in tolerance to the sleep-enhancing effects of cannabis.
With that in mind, the researchers set out to examine the potential impact of whole-plant medicinal cannabis on sleep problems experienced by middle-aged patients suffering from chronic pain.
“People are self-reporting that they’re using cannabis for sleep and that it helps, but as we know, just because people are reporting that it works doesn’t mean that it will hold up in research,” Dr. Sznitman said.
The study included 128 individuals (mean age, 61±6 years; 51% females) with chronic neuropathic pain: 66 were medical cannabis users and 62 were not.
Three indicators of insomnia were measured using the 7-point Likert scale to assess issues with sleep initiation and maintenance.
In addition, investigators collected sociodemographic information, as well as data on daily consumption of tobacco, frequency of alcohol use, and pain severity. Finally, they collected patient data on the use of sleep-aid medications during the past month as well as tricyclic antidepressant use.
Frequent use, more sleep problems?
On average, medical cannabis users were 3 years younger than their nonusing counterparts (mean age, 60±6 vs. 63±6 years, respectively, P = .003) and more likely to be male (58% vs 40%, respectively, P = .038). Otherwise, the two groups were comparable.
Medical cannabis users reported taking the drug for an average of 4 years, at an average quantity of 31 g per month. The primary mode of administration was smoking (68.6%), followed by oil extracts (21.4%) and vaporization (20%).
Results showed that, of the total sample, 24.1% reported always waking up early and not falling back to sleep, 20.2% reported always having difficulty falling asleep, and 27.2% reported always waking up during the night.
After adjusting for patient age, sex, pain level, and use of sleep medications and antidepressants, medical cannabis use was associated with fewer problems with waking up at night, compared with nonmedical cannabis use. No differences were found between groups with respect to problems falling asleep or waking up early without being able to fall back to sleep, Dr. Sznitman and associates reported.
The final analysis of a subsample of patients that only included medical cannabis users showed frequency of medical cannabis use was associated with sleep problems, they said.
Specifically, more frequent cannabis use was associated with more problems related to waking up at night, as well as problems falling asleep.
Sleep problems associated with frequent medical cannabis use may signal the development of tolerance to the agent. However, frequent users of medical cannabis also maybsuffer pain or other comorbidities, which, in turn, may be linked to more sleep problems.
Either way, Dr. Sznitman said the study might open the door to another treatment option for patients suffering from chronic pain who struggle with sleep.
“If future research shows that the effect of medical cannabis on sleep is a consistent one, then we may be adding a new therapy for sleep problems, which are huge in society and especially in chronic pain patients,” she said.
Early days
Commenting on the findings in an interview, Ryan G. Vandrey, PhD, who was not involved in the study, said the findings are in line with previous research.
“I think the results make sense with respect to the data I’ve collected and from what I’ve seen,” said Dr. Vandrey, associate professor of psychiatry and behavioral sciences at Johns Hopkins Medicine in Baltimore.
“We typically only want to use sleep medications for short periods of time,” he continued. “When you think about recommended prescribing practices for any hypnotic medication, it’s usually short term, 2 weeks or less. Longer-term use often leads to tolerance, dependence, and withdrawal symptoms when the medication is stopped, which leads to an exacerbation of disordered sleep,” Dr. Vandrey said.
Nevertheless, he urged caution when interpreting the results.
“I think the study warrants caution about long-term daily use of cannabinoids with respect to sleep,” he said. “But we need more detailed evaluations, as the trial wasn’t testing a defined product, specific dose, or dose regimen.
“In addition, this was all done in the context of people with chronic pain and not treating disordered sleep or insomnia, but the study highlights the importance of recognizing that long-term chronic use of cannabis is not likely to fully resolve sleep problems.”
Dr. Sznitman agreed that the research is still in its very early stages.
“We’re still far from saying we have the evidence to support the use of medical cannabis for sleep,” she said. “For in the end it was just a cross-sectional, observational study, so we cannot say anything about cause and effect. But if these results pan out, they could be far-reaching and exciting.”
The study was funded by the University of Haifa and Rambam Hospital in Israel, and by the Evelyn Lipper Foundation. Dr. Sznitman and Dr. Vandrey have disclosed no relevant financial relationships.
This article first appeared on Medscape.com.
FROM BMJ SUPPORTIVE AND PALLIATIVE CARE
IBD: Inpatient opioids linked with outpatient use
, based on a retrospective analysis of more than 800 patients.
Awareness of this dose-dependent relationship and IBD-related risks of opioid use should encourage physicians to consider alternative analgesics, according to lead author Rahul S. Dalal, MD, of Brigham and Women’s Hospital, Boston, and colleagues.
“Recent evidence has demonstrated that opioid use is associated with severe infections and increased mortality among IBD patients,” the investigators wrote in Clinical Gastroenterology and Hepatology. “Despite these concerns, opioids are commonly prescribed to IBD patients in the outpatient setting and to as many as 70% of IBD patients who are hospitalized.”
To look for a possible relationship between inpatient and outpatient opioid use, the investigators reviewed electronic medical records of 862 IBD patients who were treated at three urban hospitals in the University of Pennsylvania Health System. The primary outcome was opioid prescription within 12 months of discharge, including prescriptions at time of hospital dismissal.
During hospitalization, about two-thirds (67.6%) of patients received intravenous opioids. Of the total population, slightly more than half (54.6%) received intravenous hydromorphone and about one-quarter (25.9%) received intravenous morphine. Following discharge, almost half of the population (44.7%) was prescribed opioids, and about 3 out of 4 patients (77.9%) received an additional opioid prescription within the same year.
After accounting for confounders such as IBD severity, preadmission opioid use, pain scores, and psychiatric conditions, data analysis showed that inpatients who received intravenous opioids had a threefold (odds ratio [OR], 3.3) increased likelihood of receiving postdischarge opioid prescription, compared with patients who received no opioids while hospitalized. This association was stronger among those who had IBD flares (OR, 5.4). Furthermore, intravenous dose was positively correlated with postdischarge opioid prescription.
Avoiding intravenous opioids had no impact on the relationship between inpatient and outpatient opioid use. Among inpatients who received only oral or transdermal opioids, a similarly increased likelihood of postdischarge opioid prescription was observed (OR, 4.2), although this was a small cohort (n = 67).
Compared with other physicians, gastroenterologists were the least likely to prescribe opioids. Considering that gastroenterologists were also most likely aware of IBD-related risks of opioid use, the investigators concluded that more interdisciplinary communication and education are needed.
“Alternative analgesics such as acetaminophen, dicyclomine, hyoscyamine, and celecoxib could be advised, as many of these therapies have been deemed relatively safe and effective in this population,” they wrote.The investigators disclosed relationships with Abbott, Gilead, Romark, and others.
SOURCE: Dalal RS et al. Clin Gastro Hepatol. 2019 Dec 27. doi: 10.1016/j.cgh.2019.12.024.
, based on a retrospective analysis of more than 800 patients.
Awareness of this dose-dependent relationship and IBD-related risks of opioid use should encourage physicians to consider alternative analgesics, according to lead author Rahul S. Dalal, MD, of Brigham and Women’s Hospital, Boston, and colleagues.
“Recent evidence has demonstrated that opioid use is associated with severe infections and increased mortality among IBD patients,” the investigators wrote in Clinical Gastroenterology and Hepatology. “Despite these concerns, opioids are commonly prescribed to IBD patients in the outpatient setting and to as many as 70% of IBD patients who are hospitalized.”
To look for a possible relationship between inpatient and outpatient opioid use, the investigators reviewed electronic medical records of 862 IBD patients who were treated at three urban hospitals in the University of Pennsylvania Health System. The primary outcome was opioid prescription within 12 months of discharge, including prescriptions at time of hospital dismissal.
During hospitalization, about two-thirds (67.6%) of patients received intravenous opioids. Of the total population, slightly more than half (54.6%) received intravenous hydromorphone and about one-quarter (25.9%) received intravenous morphine. Following discharge, almost half of the population (44.7%) was prescribed opioids, and about 3 out of 4 patients (77.9%) received an additional opioid prescription within the same year.
After accounting for confounders such as IBD severity, preadmission opioid use, pain scores, and psychiatric conditions, data analysis showed that inpatients who received intravenous opioids had a threefold (odds ratio [OR], 3.3) increased likelihood of receiving postdischarge opioid prescription, compared with patients who received no opioids while hospitalized. This association was stronger among those who had IBD flares (OR, 5.4). Furthermore, intravenous dose was positively correlated with postdischarge opioid prescription.
Avoiding intravenous opioids had no impact on the relationship between inpatient and outpatient opioid use. Among inpatients who received only oral or transdermal opioids, a similarly increased likelihood of postdischarge opioid prescription was observed (OR, 4.2), although this was a small cohort (n = 67).
Compared with other physicians, gastroenterologists were the least likely to prescribe opioids. Considering that gastroenterologists were also most likely aware of IBD-related risks of opioid use, the investigators concluded that more interdisciplinary communication and education are needed.
“Alternative analgesics such as acetaminophen, dicyclomine, hyoscyamine, and celecoxib could be advised, as many of these therapies have been deemed relatively safe and effective in this population,” they wrote.The investigators disclosed relationships with Abbott, Gilead, Romark, and others.
SOURCE: Dalal RS et al. Clin Gastro Hepatol. 2019 Dec 27. doi: 10.1016/j.cgh.2019.12.024.
, based on a retrospective analysis of more than 800 patients.
Awareness of this dose-dependent relationship and IBD-related risks of opioid use should encourage physicians to consider alternative analgesics, according to lead author Rahul S. Dalal, MD, of Brigham and Women’s Hospital, Boston, and colleagues.
“Recent evidence has demonstrated that opioid use is associated with severe infections and increased mortality among IBD patients,” the investigators wrote in Clinical Gastroenterology and Hepatology. “Despite these concerns, opioids are commonly prescribed to IBD patients in the outpatient setting and to as many as 70% of IBD patients who are hospitalized.”
To look for a possible relationship between inpatient and outpatient opioid use, the investigators reviewed electronic medical records of 862 IBD patients who were treated at three urban hospitals in the University of Pennsylvania Health System. The primary outcome was opioid prescription within 12 months of discharge, including prescriptions at time of hospital dismissal.
During hospitalization, about two-thirds (67.6%) of patients received intravenous opioids. Of the total population, slightly more than half (54.6%) received intravenous hydromorphone and about one-quarter (25.9%) received intravenous morphine. Following discharge, almost half of the population (44.7%) was prescribed opioids, and about 3 out of 4 patients (77.9%) received an additional opioid prescription within the same year.
After accounting for confounders such as IBD severity, preadmission opioid use, pain scores, and psychiatric conditions, data analysis showed that inpatients who received intravenous opioids had a threefold (odds ratio [OR], 3.3) increased likelihood of receiving postdischarge opioid prescription, compared with patients who received no opioids while hospitalized. This association was stronger among those who had IBD flares (OR, 5.4). Furthermore, intravenous dose was positively correlated with postdischarge opioid prescription.
Avoiding intravenous opioids had no impact on the relationship between inpatient and outpatient opioid use. Among inpatients who received only oral or transdermal opioids, a similarly increased likelihood of postdischarge opioid prescription was observed (OR, 4.2), although this was a small cohort (n = 67).
Compared with other physicians, gastroenterologists were the least likely to prescribe opioids. Considering that gastroenterologists were also most likely aware of IBD-related risks of opioid use, the investigators concluded that more interdisciplinary communication and education are needed.
“Alternative analgesics such as acetaminophen, dicyclomine, hyoscyamine, and celecoxib could be advised, as many of these therapies have been deemed relatively safe and effective in this population,” they wrote.The investigators disclosed relationships with Abbott, Gilead, Romark, and others.
SOURCE: Dalal RS et al. Clin Gastro Hepatol. 2019 Dec 27. doi: 10.1016/j.cgh.2019.12.024.
FROM CLINICAL GASTROENTEROLOGY AND HEPATOLOGY
Key clinical point: Patients with inflammatory bowel disease (IBD) who receive opioids while hospitalized are three times as likely to be prescribed opioids after discharge.
Major finding: Patients who were given intravenous opioids while hospitalized were three times as likely to receive a postdischarge opioid prescription, compared with patients who did not receive inpatient intravenous opioids (odds ratio, 3.3).
Study details: A retrospective cohort study involving 862 patients with inflammatory bowel disease.
Disclosures: The investigators disclosed relationships Abbott, Gilead, Romark, and others.
Source: Dalal RS et al. Clin Gastro Hepatol. 2019 Dec 27. doi: 10.1016/j.cgh.2019.12.024.
Score predicts surgery’s benefits for obesity, diabetes
LAS VEGAS –

The Individualized Diabetes Complications risk score “can provide personalized, evidence-based risk information for patients with type 2 diabetes and obesity about their future cardiovascular disease outcomes and mortality with and without metabolic surgery,” Ali Aminian, MD, said at a meeting presented by the Obesity Society and the American Society for Metabolic and Bariatric Surgery.
Although the calculator needs validation in a prospective, randomized study to document its impact on practice, it is now available on two separate websites and as a downloadable app, said Dr. Aminian, a surgeon at the Cleveland Clinic.
The calculator inputs data for 26 distinct, “readily available” demographic and clinical entries, and based on that, estimates the patient’s 10-year risk for all-cause death, diabetic kidney disease, cerebrovascular disease, heart failure, and coronary artery disease if no surgery occurs or after some type of metabolic or bariatric surgery. The calculator does not currently have the ability to individualize predicted risks based on the specific type of metabolic surgery performed, but that is planned as a future refinement of the score.
“We validated the model in the nonsurgical patients, which showed it was very accurate. The next step is to run a randomized trial to see how useful the calculator is” for assisting in patients’ decision making, Dr. Aminian said.
The data for deriving the risk calculator, and for a preliminary validation of it, came from 13,722 patients with obesity (body mass index, 30 kg/m2 or greater) and type 2 diabetes, who were managed at the Cleveland Clinic during 1998-2017, drawn from more than 287,000 such patients in the clinic’s database. The study focused on 2,287 patients who underwent metabolic (bariatric) surgery and 11,435 patients from the same database who did not have surgery and matched by propensity scoring on a 5:1 basis with those who had surgery. The two cohorts this created matched well for age (about 54 years), sex (about two-thirds women), BMI (about 44 kg/m2), and the prevalence of various comorbidities at baseline.
Dr. Aminian and associates then analyzed the incidence of all-cause mortality and various cardiovascular disease endpoints, as well as nephropathy during follow-up, through December 2018. Patients who had undergone metabolic surgery showed statistically significant reductions in the incidence of each of those events, compared with patients who did not have surgery (JAMA. 2019;322[13]:1271-82).The investigators used these findings to create their model for calculating a patient’s risk score. For example, to calculate an estimate for the 10-year risk from all-cause mortality, the results showed that the most powerful risk factors were age, baseline body mass index, heart failure, need for insulin, and smoking status. For the endpoint of nephropathy, the most important factors were estimated glomerular filtration rate at baseline and age. Identified risk factors could account for about 80% of the 10-year risk for all-cause death and for about 75% of the risk for developing nephropathy during 10 years, based on the area-under-the-curve values the model produced.
The calculator is available at a website maintained by the Cleveland Clinic, at a site of the American Society for Metabolic and Bariatric Surgery, and in app stores, he said.
The work was partially funded by Medtronic. Dr. Aminian has received grants from Medtronic.
The AGA Practice guide on Obesity and Weight management, Education and Resources (POWER) white paper provides physicians with a comprehensive, multidisciplinary process to guide and personalize innovative obesity care for safe and effective weight management. Learn more at www.gastro.org/obesity.
SOURCE: Aminian A et al. Obesity Week 2019, Abstract A101.
LAS VEGAS –
The Individualized Diabetes Complications risk score “can provide personalized, evidence-based risk information for patients with type 2 diabetes and obesity about their future cardiovascular disease outcomes and mortality with and without metabolic surgery,” Ali Aminian, MD, said at a meeting presented by the Obesity Society and the American Society for Metabolic and Bariatric Surgery.
Although the calculator needs validation in a prospective, randomized study to document its impact on practice, it is now available on two separate websites and as a downloadable app, said Dr. Aminian, a surgeon at the Cleveland Clinic.
The calculator inputs data for 26 distinct, “readily available” demographic and clinical entries, and based on that, estimates the patient’s 10-year risk for all-cause death, diabetic kidney disease, cerebrovascular disease, heart failure, and coronary artery disease if no surgery occurs or after some type of metabolic or bariatric surgery. The calculator does not currently have the ability to individualize predicted risks based on the specific type of metabolic surgery performed, but that is planned as a future refinement of the score.
“We validated the model in the nonsurgical patients, which showed it was very accurate. The next step is to run a randomized trial to see how useful the calculator is” for assisting in patients’ decision making, Dr. Aminian said.
The data for deriving the risk calculator, and for a preliminary validation of it, came from 13,722 patients with obesity (body mass index, 30 kg/m2 or greater) and type 2 diabetes, who were managed at the Cleveland Clinic during 1998-2017, drawn from more than 287,000 such patients in the clinic’s database. The study focused on 2,287 patients who underwent metabolic (bariatric) surgery and 11,435 patients from the same database who did not have surgery and matched by propensity scoring on a 5:1 basis with those who had surgery. The two cohorts this created matched well for age (about 54 years), sex (about two-thirds women), BMI (about 44 kg/m2), and the prevalence of various comorbidities at baseline.
Dr. Aminian and associates then analyzed the incidence of all-cause mortality and various cardiovascular disease endpoints, as well as nephropathy during follow-up, through December 2018. Patients who had undergone metabolic surgery showed statistically significant reductions in the incidence of each of those events, compared with patients who did not have surgery (JAMA. 2019;322[13]:1271-82).The investigators used these findings to create their model for calculating a patient’s risk score. For example, to calculate an estimate for the 10-year risk from all-cause mortality, the results showed that the most powerful risk factors were age, baseline body mass index, heart failure, need for insulin, and smoking status. For the endpoint of nephropathy, the most important factors were estimated glomerular filtration rate at baseline and age. Identified risk factors could account for about 80% of the 10-year risk for all-cause death and for about 75% of the risk for developing nephropathy during 10 years, based on the area-under-the-curve values the model produced.
The calculator is available at a website maintained by the Cleveland Clinic, at a site of the American Society for Metabolic and Bariatric Surgery, and in app stores, he said.
The work was partially funded by Medtronic. Dr. Aminian has received grants from Medtronic.
The AGA Practice guide on Obesity and Weight management, Education and Resources (POWER) white paper provides physicians with a comprehensive, multidisciplinary process to guide and personalize innovative obesity care for safe and effective weight management. Learn more at www.gastro.org/obesity.
SOURCE: Aminian A et al. Obesity Week 2019, Abstract A101.
LAS VEGAS –
The Individualized Diabetes Complications risk score “can provide personalized, evidence-based risk information for patients with type 2 diabetes and obesity about their future cardiovascular disease outcomes and mortality with and without metabolic surgery,” Ali Aminian, MD, said at a meeting presented by the Obesity Society and the American Society for Metabolic and Bariatric Surgery.
Although the calculator needs validation in a prospective, randomized study to document its impact on practice, it is now available on two separate websites and as a downloadable app, said Dr. Aminian, a surgeon at the Cleveland Clinic.
The calculator inputs data for 26 distinct, “readily available” demographic and clinical entries, and based on that, estimates the patient’s 10-year risk for all-cause death, diabetic kidney disease, cerebrovascular disease, heart failure, and coronary artery disease if no surgery occurs or after some type of metabolic or bariatric surgery. The calculator does not currently have the ability to individualize predicted risks based on the specific type of metabolic surgery performed, but that is planned as a future refinement of the score.
“We validated the model in the nonsurgical patients, which showed it was very accurate. The next step is to run a randomized trial to see how useful the calculator is” for assisting in patients’ decision making, Dr. Aminian said.
The data for deriving the risk calculator, and for a preliminary validation of it, came from 13,722 patients with obesity (body mass index, 30 kg/m2 or greater) and type 2 diabetes, who were managed at the Cleveland Clinic during 1998-2017, drawn from more than 287,000 such patients in the clinic’s database. The study focused on 2,287 patients who underwent metabolic (bariatric) surgery and 11,435 patients from the same database who did not have surgery and matched by propensity scoring on a 5:1 basis with those who had surgery. The two cohorts this created matched well for age (about 54 years), sex (about two-thirds women), BMI (about 44 kg/m2), and the prevalence of various comorbidities at baseline.
Dr. Aminian and associates then analyzed the incidence of all-cause mortality and various cardiovascular disease endpoints, as well as nephropathy during follow-up, through December 2018. Patients who had undergone metabolic surgery showed statistically significant reductions in the incidence of each of those events, compared with patients who did not have surgery (JAMA. 2019;322[13]:1271-82).The investigators used these findings to create their model for calculating a patient’s risk score. For example, to calculate an estimate for the 10-year risk from all-cause mortality, the results showed that the most powerful risk factors were age, baseline body mass index, heart failure, need for insulin, and smoking status. For the endpoint of nephropathy, the most important factors were estimated glomerular filtration rate at baseline and age. Identified risk factors could account for about 80% of the 10-year risk for all-cause death and for about 75% of the risk for developing nephropathy during 10 years, based on the area-under-the-curve values the model produced.
The calculator is available at a website maintained by the Cleveland Clinic, at a site of the American Society for Metabolic and Bariatric Surgery, and in app stores, he said.
The work was partially funded by Medtronic. Dr. Aminian has received grants from Medtronic.
The AGA Practice guide on Obesity and Weight management, Education and Resources (POWER) white paper provides physicians with a comprehensive, multidisciplinary process to guide and personalize innovative obesity care for safe and effective weight management. Learn more at www.gastro.org/obesity.
SOURCE: Aminian A et al. Obesity Week 2019, Abstract A101.
Bariatric surgery lacks impact on teens’ long-term mental health
Young people treated with bariatric surgery for severe obesity did not experience better mental health in the 5 years following their procedures, Swedish researchers said, and indeed fared worse than their nontreated peers on certain measures.
The results of this study do not necessarily argue “that metabolic and bariatric surgery during adolescence causes mental health problems,” the investigators wrote in the Lancet Child and Adolescent Health, but “it is reasonable to conclude that metabolic and bariatric surgery does not result in a substantial alleviation of mental health problems in adolescents with severe obesity,” and that “long-term mental health support should be required in programs providing adolescent metabolic and bariatric surgery.”
Kajsa Järvholm, PhD, of Skåne University Hospital, in Malmö, Sweden, and colleagues reported results from a prospective nonrandomized study that recruited 81 adolescents in Sweden aged 13-18 years (mean age, 16.5) who had a body mass index of 40 or higher, or BMI of 35 with obesity-related comorbidities and who underwent Roux-en-Y gastric bypass for weight loss. Subjects were matched by age, sex, and BMI to 80 controls (mean age, 15.8 years) who were assigned to conventional nonsurgical treatment. All patients were assessed at 1, 2, and 5 years.
Although mental health treatment, including use of psychiatric drugs, did not differ between the groups at baseline, during the follow-up period the subjects who underwent surgery saw 15% more impatient and outpatient mental health treatment, compared with controls, a significant difference. About a quarter of patients in the surgically treated group required specialized mental health treatment for the first time after their surgeries.
Though the surgical group lost much more weight – mean BMI was 32.3 at 5 years, compared with 41.7 for controls – none of the mental health changes from baseline were significantly associated with percentage change of BMI at 5 years.
The findings from the study are consistent with results from studies in adults in which bariatric surgery improves many health outcomes but does not alter the need for mental health treatment. Although 5 years is a longer follow-up than in previous studies in young patients – a key strength of the study – Dr. Järvholm and colleagues acknowledged some weaknesses, including a nonrandomized design, lack of a comparison group of nonobese youths for mental health measures, a small sample size, and a surgical procedure that is now out of favor in adolescents.
The study was funded by Swedish government and health foundations. Dr. Järvholm disclosed pharmaceutical industry funding not related to the study, and three coauthors also disclosed industry relationships.
SOURCE: Järvholm K et al. Lancet Child Adolesc Health. 2020. doi: 10.1016/s2352-4642(20)30024-9.
Young people treated with bariatric surgery for severe obesity did not experience better mental health in the 5 years following their procedures, Swedish researchers said, and indeed fared worse than their nontreated peers on certain measures.
The results of this study do not necessarily argue “that metabolic and bariatric surgery during adolescence causes mental health problems,” the investigators wrote in the Lancet Child and Adolescent Health, but “it is reasonable to conclude that metabolic and bariatric surgery does not result in a substantial alleviation of mental health problems in adolescents with severe obesity,” and that “long-term mental health support should be required in programs providing adolescent metabolic and bariatric surgery.”
Kajsa Järvholm, PhD, of Skåne University Hospital, in Malmö, Sweden, and colleagues reported results from a prospective nonrandomized study that recruited 81 adolescents in Sweden aged 13-18 years (mean age, 16.5) who had a body mass index of 40 or higher, or BMI of 35 with obesity-related comorbidities and who underwent Roux-en-Y gastric bypass for weight loss. Subjects were matched by age, sex, and BMI to 80 controls (mean age, 15.8 years) who were assigned to conventional nonsurgical treatment. All patients were assessed at 1, 2, and 5 years.
Although mental health treatment, including use of psychiatric drugs, did not differ between the groups at baseline, during the follow-up period the subjects who underwent surgery saw 15% more impatient and outpatient mental health treatment, compared with controls, a significant difference. About a quarter of patients in the surgically treated group required specialized mental health treatment for the first time after their surgeries.
Though the surgical group lost much more weight – mean BMI was 32.3 at 5 years, compared with 41.7 for controls – none of the mental health changes from baseline were significantly associated with percentage change of BMI at 5 years.
The findings from the study are consistent with results from studies in adults in which bariatric surgery improves many health outcomes but does not alter the need for mental health treatment. Although 5 years is a longer follow-up than in previous studies in young patients – a key strength of the study – Dr. Järvholm and colleagues acknowledged some weaknesses, including a nonrandomized design, lack of a comparison group of nonobese youths for mental health measures, a small sample size, and a surgical procedure that is now out of favor in adolescents.
The study was funded by Swedish government and health foundations. Dr. Järvholm disclosed pharmaceutical industry funding not related to the study, and three coauthors also disclosed industry relationships.
SOURCE: Järvholm K et al. Lancet Child Adolesc Health. 2020. doi: 10.1016/s2352-4642(20)30024-9.
Young people treated with bariatric surgery for severe obesity did not experience better mental health in the 5 years following their procedures, Swedish researchers said, and indeed fared worse than their nontreated peers on certain measures.
The results of this study do not necessarily argue “that metabolic and bariatric surgery during adolescence causes mental health problems,” the investigators wrote in the Lancet Child and Adolescent Health, but “it is reasonable to conclude that metabolic and bariatric surgery does not result in a substantial alleviation of mental health problems in adolescents with severe obesity,” and that “long-term mental health support should be required in programs providing adolescent metabolic and bariatric surgery.”
Kajsa Järvholm, PhD, of Skåne University Hospital, in Malmö, Sweden, and colleagues reported results from a prospective nonrandomized study that recruited 81 adolescents in Sweden aged 13-18 years (mean age, 16.5) who had a body mass index of 40 or higher, or BMI of 35 with obesity-related comorbidities and who underwent Roux-en-Y gastric bypass for weight loss. Subjects were matched by age, sex, and BMI to 80 controls (mean age, 15.8 years) who were assigned to conventional nonsurgical treatment. All patients were assessed at 1, 2, and 5 years.
Although mental health treatment, including use of psychiatric drugs, did not differ between the groups at baseline, during the follow-up period the subjects who underwent surgery saw 15% more impatient and outpatient mental health treatment, compared with controls, a significant difference. About a quarter of patients in the surgically treated group required specialized mental health treatment for the first time after their surgeries.
Though the surgical group lost much more weight – mean BMI was 32.3 at 5 years, compared with 41.7 for controls – none of the mental health changes from baseline were significantly associated with percentage change of BMI at 5 years.
The findings from the study are consistent with results from studies in adults in which bariatric surgery improves many health outcomes but does not alter the need for mental health treatment. Although 5 years is a longer follow-up than in previous studies in young patients – a key strength of the study – Dr. Järvholm and colleagues acknowledged some weaknesses, including a nonrandomized design, lack of a comparison group of nonobese youths for mental health measures, a small sample size, and a surgical procedure that is now out of favor in adolescents.
The study was funded by Swedish government and health foundations. Dr. Järvholm disclosed pharmaceutical industry funding not related to the study, and three coauthors also disclosed industry relationships.
SOURCE: Järvholm K et al. Lancet Child Adolesc Health. 2020. doi: 10.1016/s2352-4642(20)30024-9.
FROM THE LANCET CHILD & ADOLESCENT HEALTH
Key clinical point: Bariatric surgery was not associated with improvement in obese adolescents’ long-term mental health, despite significant weight loss.
Major finding: During 5 years of follow up, surgically treated patients experienced 15% more mental health care usage than controls.
Study details: A prospective, nonrandomized study involving 161 adolescents with a BMI of 40 or greater (or 35 with comorbidities).
Disclosures: The Swedish government and Swedish health research foundations sponsored the study.
Source: Järvholm K et al. Lancet Child Adolesc Health. 2020. doi: 10.1016/s2352-4642(20)30024-9.
Quality reporting of improvement activities in 2020
2020 has begun and therefore so has a new year of quality reporting requirements. Quality reporting under the Centers for Medicare and Medicaid Services (CMS) Merit-based Incentive Payment System (MIPS) may seem like a burden, but it doesn’t need to be. You can likely get credit for the things you are already doing in your practice with little to no augmentation needed.

First, there are a few pieces of information to keep in mind when tracking your data and preparing your staff for their 2020 strategy.
1. Group Participation – For 2020 there is an increase in the MIPS participation threshold for those participating as part of a group. At least 50% of the MIPS eligible clinicians in the reporting group must participate in the same continuous 90-day period to receive credit for a quality improvement activity. That’s a significant increase from 2019 when only one (1) MIPS eligible clinician in a group was required to participate. Connect with your staff now to make sure your group meets the new 50% participation requirement.
2. Improvement Activities for Group Participation – Improvement Activities that are approved for credit by CMS are given a weight based on their requirements. Approved activities are weighted as either medium or high, and this impacts how many activities a practitioner must report on. In 2020, CMS increased the participation threshold for group reporting from a single clinician to 50% of the clinicians in the practice for the Improvement Activities category along with other changes such as modifying the definition of rural area to mean a ZIP code designated as rural by the Federal Office of Rural Health Policy using the most recent file available, updating the improvement activities and removing some criteria for Patient-Centered Medical Home designation. Work with your staff now to make sure at least 50% of the MIPS-eligible clinicians in your group are participating in the same Improvement Activities.
3. Quantity of Improvement Activities Required – CMS requires most individuals or groups report on any of the following options during any continuous 90-day period (or as specified in the activity description) in the same performance year, provided that all participating clinicians are reporting on the same activities:
a. 2 high-weighted activities, or
b. 1 high-weighted and 2 medium-weighted activities, or
c. 4 medium-weighted activities
Be sure to pay attention to the weight of the activity you (if you’re reporting as an individual) or your group is reporting so you don’t have any surprises at the end of the reporting period.
There are a variety of options for activities you can report on and some may be a lower lift than you expect.
Does your practice treat Medicaid patients? If so, do you know their average wait time for an initial visit? If that number is 10 days or less, you can report on this activity. If you aren’t quite hitting this benchmark, then consider implementing a scheduling protocol for this population of your patients in the new year.
Engagement of new Medicaid patients and follow-up
Seeing new and follow-up Medicaid patients in a timely manner, including individuals dually eligible for Medicaid and Medicare. A timely manner is defined as within 10 business days for this activity.
Subcategory name: Achieving Health Equity
Activity weighting: High
Are you responsible for onboarding and training new clinicians to your rural practice? If so, you could report on the next activity. Eligible clinicians would be responsible for training of new clinicians including physicians, advanced practice providers and clinical nursing specialists. These clinicians must practice in small, underserved, or rural areas. What is considered a small, rural, or underserved practice for the purpose of MIPS?
Small practice
- Defined as a practice with 15 or fewer eligible clinicians-based billing under the same TIN
Rural/underserved practice
- Defined as a practice in a zip code included in the most recent set of Health Professional Shortage Areas (HPSAs), as determined by the Health Resources and Services Administration (HRSA).
- HPSAs are designations that indicate health care provider shortages in primary care, dental health or mental health can be geographic population based or facility based.
Provide education opportunities for new clinicians
MIPS-eligible clinicians acting as a preceptor for clinicians-in-training (such as medical residents/fellows, medical students, physician assistants, nurse practitioners, or clinical nurse specialists) and accepting such clinicians for clinical rotations in community practices in small, underserved, or rural areas.
Subcategory name: Achieving Health Equity
Activity weighting: High
There are also activities you can report on under the beneficiary engagement category that you may already be doing in your practice. First, the collection of patient experience and satisfaction data and the development of an improvement plan as necessary counts as one activity. Second, the engagement of the patient’s support team in the development of a plan of care, which needs to include goals and be documented in the electronic health record.
Collection and follow-up on patient experience and satisfaction data on beneficiary engagement
Collection and follow-up on patient experience and satisfaction data on beneficiary engagement, including development of improvement plan.
Subcategory name: Beneficiary Engagement
Activity weighting: High
Engagement of Patients, Family, and Caregivers in Developing a Plan of Care
Engage patients, family, and caregivers in developing a plan of care and prioritizing their goals for action, documented in the electronic health record (EHR) technology.
Subcategory name: Beneficiary Engagement
Activity weighting: Medium
Another data collection category is patient access to care. If you collect and use patient data on their satisfaction and experience related to access to care and commit to developing an improvement plan as necessary, you can receive credit for this reporting category.
Collection and use of patient experience and satisfaction data on access
Collection of patient experience and satisfaction data on access to care and development of an improvement plan, such as outlining steps for improving communications with patients to help understanding of urgent access needs.
Subcategory name: Expanded Practice Access
Activity weighting: Medium
One of the hallmarks of a medical practice in any specialty is improvement. We are always striving to improve something, whether it be the patient experience, patient outcomes, the bottom line, or the education of clinical staff. You can leverage the practice improvement plans you have put into place for credit.
Leadership engagement in regular guidance and demonstrated commitment for implementing practice improvement changes
Ensure full engagement of clinical and administrative leadership in practice improvement that could include one or more of the following: Make responsibility for guidance of practice change a component of clinical and administrative leadership roles; Allocate time for clinical and administrative leadership for practice improvement efforts, including participation in regular team meetings; and/or Incorporate population health, quality and patient experience metrics in regular reviews of practice performance.
Subcategory name: Patient Safety and Practice Assessment
Activity weighting: Medium
Prescription drug use is a topic on every providers’ radar right now. Proper prescribing and monitoring of patients are crucial to their safety and quality of care. In the field of gastroenterology, step-therapy adds a new level of complication to the use of prescription drugs. Ensuring the proper medication protocols allows you to provide appropriate and timely treatment for your patients.
Annual registration in the Prescription Drug Monitoring Program
Annual registration by eligible clinician or group in the prescription drug–monitoring program of the state where they practice. Activities that simply involve registration are not sufficient. MIPS-eligible clinicians and groups must participate for a minimum of 6 months.
Subcategory name: Patient Safety and Practice Assessment
Activity weighting: Medium
As you can see, there are a variety of improvement activities that you can report on for 2020. This article has outlined several of them that you may already be doing in your practice, but many more can be found by visiting https://qpp.cms.gov/mips/improvement-activities?py=2020 along with information on how to report and the necessary forms for submission.
Dr. Shah is associate professor, Mount Sinai Medical Center, New York, member of the AGA Quality Leadership Council. He has no disclosures.
2020 has begun and therefore so has a new year of quality reporting requirements. Quality reporting under the Centers for Medicare and Medicaid Services (CMS) Merit-based Incentive Payment System (MIPS) may seem like a burden, but it doesn’t need to be. You can likely get credit for the things you are already doing in your practice with little to no augmentation needed.
First, there are a few pieces of information to keep in mind when tracking your data and preparing your staff for their 2020 strategy.
1. Group Participation – For 2020 there is an increase in the MIPS participation threshold for those participating as part of a group. At least 50% of the MIPS eligible clinicians in the reporting group must participate in the same continuous 90-day period to receive credit for a quality improvement activity. That’s a significant increase from 2019 when only one (1) MIPS eligible clinician in a group was required to participate. Connect with your staff now to make sure your group meets the new 50% participation requirement.
2. Improvement Activities for Group Participation – Improvement Activities that are approved for credit by CMS are given a weight based on their requirements. Approved activities are weighted as either medium or high, and this impacts how many activities a practitioner must report on. In 2020, CMS increased the participation threshold for group reporting from a single clinician to 50% of the clinicians in the practice for the Improvement Activities category along with other changes such as modifying the definition of rural area to mean a ZIP code designated as rural by the Federal Office of Rural Health Policy using the most recent file available, updating the improvement activities and removing some criteria for Patient-Centered Medical Home designation. Work with your staff now to make sure at least 50% of the MIPS-eligible clinicians in your group are participating in the same Improvement Activities.
3. Quantity of Improvement Activities Required – CMS requires most individuals or groups report on any of the following options during any continuous 90-day period (or as specified in the activity description) in the same performance year, provided that all participating clinicians are reporting on the same activities:
a. 2 high-weighted activities, or
b. 1 high-weighted and 2 medium-weighted activities, or
c. 4 medium-weighted activities
Be sure to pay attention to the weight of the activity you (if you’re reporting as an individual) or your group is reporting so you don’t have any surprises at the end of the reporting period.
There are a variety of options for activities you can report on and some may be a lower lift than you expect.
Does your practice treat Medicaid patients? If so, do you know their average wait time for an initial visit? If that number is 10 days or less, you can report on this activity. If you aren’t quite hitting this benchmark, then consider implementing a scheduling protocol for this population of your patients in the new year.
Engagement of new Medicaid patients and follow-up
Seeing new and follow-up Medicaid patients in a timely manner, including individuals dually eligible for Medicaid and Medicare. A timely manner is defined as within 10 business days for this activity.
Subcategory name: Achieving Health Equity
Activity weighting: High
Are you responsible for onboarding and training new clinicians to your rural practice? If so, you could report on the next activity. Eligible clinicians would be responsible for training of new clinicians including physicians, advanced practice providers and clinical nursing specialists. These clinicians must practice in small, underserved, or rural areas. What is considered a small, rural, or underserved practice for the purpose of MIPS?
Small practice
- Defined as a practice with 15 or fewer eligible clinicians-based billing under the same TIN
Rural/underserved practice
- Defined as a practice in a zip code included in the most recent set of Health Professional Shortage Areas (HPSAs), as determined by the Health Resources and Services Administration (HRSA).
- HPSAs are designations that indicate health care provider shortages in primary care, dental health or mental health can be geographic population based or facility based.
Provide education opportunities for new clinicians
MIPS-eligible clinicians acting as a preceptor for clinicians-in-training (such as medical residents/fellows, medical students, physician assistants, nurse practitioners, or clinical nurse specialists) and accepting such clinicians for clinical rotations in community practices in small, underserved, or rural areas.
Subcategory name: Achieving Health Equity
Activity weighting: High
There are also activities you can report on under the beneficiary engagement category that you may already be doing in your practice. First, the collection of patient experience and satisfaction data and the development of an improvement plan as necessary counts as one activity. Second, the engagement of the patient’s support team in the development of a plan of care, which needs to include goals and be documented in the electronic health record.
Collection and follow-up on patient experience and satisfaction data on beneficiary engagement
Collection and follow-up on patient experience and satisfaction data on beneficiary engagement, including development of improvement plan.
Subcategory name: Beneficiary Engagement
Activity weighting: High
Engagement of Patients, Family, and Caregivers in Developing a Plan of Care
Engage patients, family, and caregivers in developing a plan of care and prioritizing their goals for action, documented in the electronic health record (EHR) technology.
Subcategory name: Beneficiary Engagement
Activity weighting: Medium
Another data collection category is patient access to care. If you collect and use patient data on their satisfaction and experience related to access to care and commit to developing an improvement plan as necessary, you can receive credit for this reporting category.
Collection and use of patient experience and satisfaction data on access
Collection of patient experience and satisfaction data on access to care and development of an improvement plan, such as outlining steps for improving communications with patients to help understanding of urgent access needs.
Subcategory name: Expanded Practice Access
Activity weighting: Medium
One of the hallmarks of a medical practice in any specialty is improvement. We are always striving to improve something, whether it be the patient experience, patient outcomes, the bottom line, or the education of clinical staff. You can leverage the practice improvement plans you have put into place for credit.
Leadership engagement in regular guidance and demonstrated commitment for implementing practice improvement changes
Ensure full engagement of clinical and administrative leadership in practice improvement that could include one or more of the following: Make responsibility for guidance of practice change a component of clinical and administrative leadership roles; Allocate time for clinical and administrative leadership for practice improvement efforts, including participation in regular team meetings; and/or Incorporate population health, quality and patient experience metrics in regular reviews of practice performance.
Subcategory name: Patient Safety and Practice Assessment
Activity weighting: Medium
Prescription drug use is a topic on every providers’ radar right now. Proper prescribing and monitoring of patients are crucial to their safety and quality of care. In the field of gastroenterology, step-therapy adds a new level of complication to the use of prescription drugs. Ensuring the proper medication protocols allows you to provide appropriate and timely treatment for your patients.
Annual registration in the Prescription Drug Monitoring Program
Annual registration by eligible clinician or group in the prescription drug–monitoring program of the state where they practice. Activities that simply involve registration are not sufficient. MIPS-eligible clinicians and groups must participate for a minimum of 6 months.
Subcategory name: Patient Safety and Practice Assessment
Activity weighting: Medium
As you can see, there are a variety of improvement activities that you can report on for 2020. This article has outlined several of them that you may already be doing in your practice, but many more can be found by visiting https://qpp.cms.gov/mips/improvement-activities?py=2020 along with information on how to report and the necessary forms for submission.
Dr. Shah is associate professor, Mount Sinai Medical Center, New York, member of the AGA Quality Leadership Council. He has no disclosures.
2020 has begun and therefore so has a new year of quality reporting requirements. Quality reporting under the Centers for Medicare and Medicaid Services (CMS) Merit-based Incentive Payment System (MIPS) may seem like a burden, but it doesn’t need to be. You can likely get credit for the things you are already doing in your practice with little to no augmentation needed.
First, there are a few pieces of information to keep in mind when tracking your data and preparing your staff for their 2020 strategy.
1. Group Participation – For 2020 there is an increase in the MIPS participation threshold for those participating as part of a group. At least 50% of the MIPS eligible clinicians in the reporting group must participate in the same continuous 90-day period to receive credit for a quality improvement activity. That’s a significant increase from 2019 when only one (1) MIPS eligible clinician in a group was required to participate. Connect with your staff now to make sure your group meets the new 50% participation requirement.
2. Improvement Activities for Group Participation – Improvement Activities that are approved for credit by CMS are given a weight based on their requirements. Approved activities are weighted as either medium or high, and this impacts how many activities a practitioner must report on. In 2020, CMS increased the participation threshold for group reporting from a single clinician to 50% of the clinicians in the practice for the Improvement Activities category along with other changes such as modifying the definition of rural area to mean a ZIP code designated as rural by the Federal Office of Rural Health Policy using the most recent file available, updating the improvement activities and removing some criteria for Patient-Centered Medical Home designation. Work with your staff now to make sure at least 50% of the MIPS-eligible clinicians in your group are participating in the same Improvement Activities.
3. Quantity of Improvement Activities Required – CMS requires most individuals or groups report on any of the following options during any continuous 90-day period (or as specified in the activity description) in the same performance year, provided that all participating clinicians are reporting on the same activities:
a. 2 high-weighted activities, or
b. 1 high-weighted and 2 medium-weighted activities, or
c. 4 medium-weighted activities
Be sure to pay attention to the weight of the activity you (if you’re reporting as an individual) or your group is reporting so you don’t have any surprises at the end of the reporting period.
There are a variety of options for activities you can report on and some may be a lower lift than you expect.
Does your practice treat Medicaid patients? If so, do you know their average wait time for an initial visit? If that number is 10 days or less, you can report on this activity. If you aren’t quite hitting this benchmark, then consider implementing a scheduling protocol for this population of your patients in the new year.
Engagement of new Medicaid patients and follow-up
Seeing new and follow-up Medicaid patients in a timely manner, including individuals dually eligible for Medicaid and Medicare. A timely manner is defined as within 10 business days for this activity.
Subcategory name: Achieving Health Equity
Activity weighting: High
Are you responsible for onboarding and training new clinicians to your rural practice? If so, you could report on the next activity. Eligible clinicians would be responsible for training of new clinicians including physicians, advanced practice providers and clinical nursing specialists. These clinicians must practice in small, underserved, or rural areas. What is considered a small, rural, or underserved practice for the purpose of MIPS?
Small practice
- Defined as a practice with 15 or fewer eligible clinicians-based billing under the same TIN
Rural/underserved practice
- Defined as a practice in a zip code included in the most recent set of Health Professional Shortage Areas (HPSAs), as determined by the Health Resources and Services Administration (HRSA).
- HPSAs are designations that indicate health care provider shortages in primary care, dental health or mental health can be geographic population based or facility based.
Provide education opportunities for new clinicians
MIPS-eligible clinicians acting as a preceptor for clinicians-in-training (such as medical residents/fellows, medical students, physician assistants, nurse practitioners, or clinical nurse specialists) and accepting such clinicians for clinical rotations in community practices in small, underserved, or rural areas.
Subcategory name: Achieving Health Equity
Activity weighting: High
There are also activities you can report on under the beneficiary engagement category that you may already be doing in your practice. First, the collection of patient experience and satisfaction data and the development of an improvement plan as necessary counts as one activity. Second, the engagement of the patient’s support team in the development of a plan of care, which needs to include goals and be documented in the electronic health record.
Collection and follow-up on patient experience and satisfaction data on beneficiary engagement
Collection and follow-up on patient experience and satisfaction data on beneficiary engagement, including development of improvement plan.
Subcategory name: Beneficiary Engagement
Activity weighting: High
Engagement of Patients, Family, and Caregivers in Developing a Plan of Care
Engage patients, family, and caregivers in developing a plan of care and prioritizing their goals for action, documented in the electronic health record (EHR) technology.
Subcategory name: Beneficiary Engagement
Activity weighting: Medium
Another data collection category is patient access to care. If you collect and use patient data on their satisfaction and experience related to access to care and commit to developing an improvement plan as necessary, you can receive credit for this reporting category.
Collection and use of patient experience and satisfaction data on access
Collection of patient experience and satisfaction data on access to care and development of an improvement plan, such as outlining steps for improving communications with patients to help understanding of urgent access needs.
Subcategory name: Expanded Practice Access
Activity weighting: Medium
One of the hallmarks of a medical practice in any specialty is improvement. We are always striving to improve something, whether it be the patient experience, patient outcomes, the bottom line, or the education of clinical staff. You can leverage the practice improvement plans you have put into place for credit.
Leadership engagement in regular guidance and demonstrated commitment for implementing practice improvement changes
Ensure full engagement of clinical and administrative leadership in practice improvement that could include one or more of the following: Make responsibility for guidance of practice change a component of clinical and administrative leadership roles; Allocate time for clinical and administrative leadership for practice improvement efforts, including participation in regular team meetings; and/or Incorporate population health, quality and patient experience metrics in regular reviews of practice performance.
Subcategory name: Patient Safety and Practice Assessment
Activity weighting: Medium
Prescription drug use is a topic on every providers’ radar right now. Proper prescribing and monitoring of patients are crucial to their safety and quality of care. In the field of gastroenterology, step-therapy adds a new level of complication to the use of prescription drugs. Ensuring the proper medication protocols allows you to provide appropriate and timely treatment for your patients.
Annual registration in the Prescription Drug Monitoring Program
Annual registration by eligible clinician or group in the prescription drug–monitoring program of the state where they practice. Activities that simply involve registration are not sufficient. MIPS-eligible clinicians and groups must participate for a minimum of 6 months.
Subcategory name: Patient Safety and Practice Assessment
Activity weighting: Medium
As you can see, there are a variety of improvement activities that you can report on for 2020. This article has outlined several of them that you may already be doing in your practice, but many more can be found by visiting https://qpp.cms.gov/mips/improvement-activities?py=2020 along with information on how to report and the necessary forms for submission.
Dr. Shah is associate professor, Mount Sinai Medical Center, New York, member of the AGA Quality Leadership Council. He has no disclosures.
Wuhan virus: What clinicians need to know
As the Wuhan coronavirus story unfolds, , according to infectious disease experts.

“We are asking that of everyone with fever and respiratory symptoms who comes to our clinics, hospital, or emergency room. It’s a powerful screening tool,” said William Schaffner, MD, professor of preventive medicine and infectious diseases at Vanderbilt University Medical Center, Nashville, Tenn.
In addition to fever, common signs of infection include cough, shortness of breath, and breathing difficulties. Some patients have had diarrhea, vomiting, and other gastrointestinal symptoms. In more severe cases, infection can cause pneumonia, severe acute respiratory syndrome, kidney failure, and death. The incubation period appears to be up to 2 weeks, according to the World Health Organization (WHO).
If patients exhibit symptoms and either they or a close contact has returned from China recently, take standard airborne precautions and send specimens – a serum sample, oral and nasal pharyngeal swabs, and lower respiratory tract specimens if available – to the local health department, which will forward them to the Centers for Disease Control and Prevention (CDC) for testing. Turnaround time is 24-48 hours.

The 2019 Novel Coronavirus (2019-nCoV), identified as the cause of an outbreak of respiratory illness first detected in December in association with a live animal market in Wuhan, China, has been implicated in almost 2,000 cases and 56 deaths in that country. Cases have been reported in 13 countries besides China. Five cases of 2019-nCoV infection have been confirmed in the United States, all in people recently returned from Wuhan. As the virus spreads in China, however, it’s almost certain more cases will show up in the United States. Travel history is key, Dr. Schaffner and others said.
Plan and rehearse
The first step to prepare is to use the CDC’s Interim Guidance for Healthcare Professionals to make a written plan specific to your practice to respond to a potential case. The plan must include notifying the local health department, the CDC liaison for testing, and tracking down patient contacts.
“It’s not good enough to just download CDC’s guidance; use it to make your own local plan and know what to do 24/7,” said Daniel Lucey, MD, an infectious disease expert at Georgetown University Medical Center, Washington, D.C.
“Know who is on call at the health department on weekends and nights,” he said. Know where the patient is going to be isolated; figure out what to do if there’s more than one, and tests come back positive. Have masks on hand, and rehearse the response. “Make a coronavirus team, and absolutely have the nurses involved,” as well as other providers who may come into contact with a case, he added.

“You want to be able to do as well as your counterparts in Washington state and Chicago,” where the first two U.S. cases emerged. “They were prepared. They knew what to do,” Dr. Lucey said.
Those first two U.S. patients – a man in Everett, Wash., and a Chicago woman – developed symptoms after returning from Wuhan, a city of 11 million just over 400 miles inland from the port city of Shanghai. On Jan. 26 three more cases were confirmed by the CDC, two in California and one in Arizona, and each had recently traveled to Wuhan. All five patients remain hospitalized, and there’s no evidence they spread the infection further. There is also no evidence of human-to-human transmission of other cases exported from China to any other countries, according to the WHO.
WHO declined to declare a global health emergency – a Public Health Emergency of International Concern, in its parlance – on Jan. 23. The step would have triggered travel and trade restrictions in member states, including the United States. For now, at least, the group said it wasn’t warranted at this point.
Fatality rates
The focus right now is China. The outbreak has spread beyond Wuhan to other parts of the country, and there’s evidence of fourth-generation spread.

Transportation into and out of Wuhan and other cities has been curtailed, Lunar New Year festivals have been canceled, and the Shanghai Disneyland has been closed, among other measures taken by Chinese officials.
The government could be taking drastic measures in part to prevent the public criticism it took in the early 2000’s for the delayed response and lack of transparency during the global outbreak of another wildlife market coronavirus epidemic, severe acute respiratory syndrome (SARS). In a press conference Jan. 22, WHO officials commended the government’s containment efforts but did not say they recommended them.
According to WHO, serious cases in China have mostly been in people over 40 years old with significant comorbidities and have skewed towards men. Spread seems to be limited to family members, health care providers, and other close contacts, probably by respiratory droplets. If that pattern holds, WHO officials said, the outbreak is containable.
The fatality rate appears to be around 3%, a good deal lower than the 10% reported for SARS and much lower than the nearly 40% reported for Middle East respiratory syndrome (MERS), another recent coronavirus mutation from the animal trade.
The Wuhan virus fatality rate might drop as milder cases are detected and added to the denominator. “It definitely appears to be less severe than SARS and MERS,” said Amesh Adalja, MD, an infectious disease physician in Pittsburgh and emerging infectious disease researcher at Johns Hopkins University, Baltimore.
SARS: Lessons learned
In general, the world is much better equipped for coronavirus outbreaks than when SARS, in particular, emerged in 2003.

WHO officials in their press conference lauded China for it openness with the current outbreak, and for isolating and sequencing the virus immediately, which gave the world a diagnostic test in the first days of the outbreak, something that wasn’t available for SARS. China and other countries also are cooperating and working closely to contain the Wuhan virus.
“What we know today might change tomorrow, so we have to keep tuned in to new information, but we learned a lot from SARS,” Dr. Shaffner said. Overall, it’s likely “the impact on the United States of this new coronavirus is going to be trivial,” he predicted.
Dr. Lucey, however, recalled that the SARS outbreak in Toronto in 2003 started with one missed case. A woman returned asymptomatic from Hong Kong and spread the infection to her family members before she died. Her cause of death wasn’t immediately recognized, nor was the reason her family members were sick, since they hadn’t been to Hong Kong recently.
The infection ultimately spread to more than 200 people, about half of them health care workers. A few people died.
If a virus is sufficiently contagious, “it just takes one. You don’t want to be the one who misses that first patient,” Dr. Lucey said.
Currently, there are no antivirals or vaccines for coronaviruses; researchers are working on both, but for now, care is supportive.
This article was updated with new case numbers on 1/26/20.
As the Wuhan coronavirus story unfolds, , according to infectious disease experts.
“We are asking that of everyone with fever and respiratory symptoms who comes to our clinics, hospital, or emergency room. It’s a powerful screening tool,” said William Schaffner, MD, professor of preventive medicine and infectious diseases at Vanderbilt University Medical Center, Nashville, Tenn.
In addition to fever, common signs of infection include cough, shortness of breath, and breathing difficulties. Some patients have had diarrhea, vomiting, and other gastrointestinal symptoms. In more severe cases, infection can cause pneumonia, severe acute respiratory syndrome, kidney failure, and death. The incubation period appears to be up to 2 weeks, according to the World Health Organization (WHO).
If patients exhibit symptoms and either they or a close contact has returned from China recently, take standard airborne precautions and send specimens – a serum sample, oral and nasal pharyngeal swabs, and lower respiratory tract specimens if available – to the local health department, which will forward them to the Centers for Disease Control and Prevention (CDC) for testing. Turnaround time is 24-48 hours.
The 2019 Novel Coronavirus (2019-nCoV), identified as the cause of an outbreak of respiratory illness first detected in December in association with a live animal market in Wuhan, China, has been implicated in almost 2,000 cases and 56 deaths in that country. Cases have been reported in 13 countries besides China. Five cases of 2019-nCoV infection have been confirmed in the United States, all in people recently returned from Wuhan. As the virus spreads in China, however, it’s almost certain more cases will show up in the United States. Travel history is key, Dr. Schaffner and others said.
Plan and rehearse
The first step to prepare is to use the CDC’s Interim Guidance for Healthcare Professionals to make a written plan specific to your practice to respond to a potential case. The plan must include notifying the local health department, the CDC liaison for testing, and tracking down patient contacts.
“It’s not good enough to just download CDC’s guidance; use it to make your own local plan and know what to do 24/7,” said Daniel Lucey, MD, an infectious disease expert at Georgetown University Medical Center, Washington, D.C.
“Know who is on call at the health department on weekends and nights,” he said. Know where the patient is going to be isolated; figure out what to do if there’s more than one, and tests come back positive. Have masks on hand, and rehearse the response. “Make a coronavirus team, and absolutely have the nurses involved,” as well as other providers who may come into contact with a case, he added.
“You want to be able to do as well as your counterparts in Washington state and Chicago,” where the first two U.S. cases emerged. “They were prepared. They knew what to do,” Dr. Lucey said.
Those first two U.S. patients – a man in Everett, Wash., and a Chicago woman – developed symptoms after returning from Wuhan, a city of 11 million just over 400 miles inland from the port city of Shanghai. On Jan. 26 three more cases were confirmed by the CDC, two in California and one in Arizona, and each had recently traveled to Wuhan. All five patients remain hospitalized, and there’s no evidence they spread the infection further. There is also no evidence of human-to-human transmission of other cases exported from China to any other countries, according to the WHO.
WHO declined to declare a global health emergency – a Public Health Emergency of International Concern, in its parlance – on Jan. 23. The step would have triggered travel and trade restrictions in member states, including the United States. For now, at least, the group said it wasn’t warranted at this point.
Fatality rates
The focus right now is China. The outbreak has spread beyond Wuhan to other parts of the country, and there’s evidence of fourth-generation spread.
Transportation into and out of Wuhan and other cities has been curtailed, Lunar New Year festivals have been canceled, and the Shanghai Disneyland has been closed, among other measures taken by Chinese officials.
The government could be taking drastic measures in part to prevent the public criticism it took in the early 2000’s for the delayed response and lack of transparency during the global outbreak of another wildlife market coronavirus epidemic, severe acute respiratory syndrome (SARS). In a press conference Jan. 22, WHO officials commended the government’s containment efforts but did not say they recommended them.
According to WHO, serious cases in China have mostly been in people over 40 years old with significant comorbidities and have skewed towards men. Spread seems to be limited to family members, health care providers, and other close contacts, probably by respiratory droplets. If that pattern holds, WHO officials said, the outbreak is containable.
The fatality rate appears to be around 3%, a good deal lower than the 10% reported for SARS and much lower than the nearly 40% reported for Middle East respiratory syndrome (MERS), another recent coronavirus mutation from the animal trade.
The Wuhan virus fatality rate might drop as milder cases are detected and added to the denominator. “It definitely appears to be less severe than SARS and MERS,” said Amesh Adalja, MD, an infectious disease physician in Pittsburgh and emerging infectious disease researcher at Johns Hopkins University, Baltimore.
SARS: Lessons learned
In general, the world is much better equipped for coronavirus outbreaks than when SARS, in particular, emerged in 2003.
WHO officials in their press conference lauded China for it openness with the current outbreak, and for isolating and sequencing the virus immediately, which gave the world a diagnostic test in the first days of the outbreak, something that wasn’t available for SARS. China and other countries also are cooperating and working closely to contain the Wuhan virus.
“What we know today might change tomorrow, so we have to keep tuned in to new information, but we learned a lot from SARS,” Dr. Shaffner said. Overall, it’s likely “the impact on the United States of this new coronavirus is going to be trivial,” he predicted.
Dr. Lucey, however, recalled that the SARS outbreak in Toronto in 2003 started with one missed case. A woman returned asymptomatic from Hong Kong and spread the infection to her family members before she died. Her cause of death wasn’t immediately recognized, nor was the reason her family members were sick, since they hadn’t been to Hong Kong recently.
The infection ultimately spread to more than 200 people, about half of them health care workers. A few people died.
If a virus is sufficiently contagious, “it just takes one. You don’t want to be the one who misses that first patient,” Dr. Lucey said.
Currently, there are no antivirals or vaccines for coronaviruses; researchers are working on both, but for now, care is supportive.
This article was updated with new case numbers on 1/26/20.
As the Wuhan coronavirus story unfolds, , according to infectious disease experts.
“We are asking that of everyone with fever and respiratory symptoms who comes to our clinics, hospital, or emergency room. It’s a powerful screening tool,” said William Schaffner, MD, professor of preventive medicine and infectious diseases at Vanderbilt University Medical Center, Nashville, Tenn.
In addition to fever, common signs of infection include cough, shortness of breath, and breathing difficulties. Some patients have had diarrhea, vomiting, and other gastrointestinal symptoms. In more severe cases, infection can cause pneumonia, severe acute respiratory syndrome, kidney failure, and death. The incubation period appears to be up to 2 weeks, according to the World Health Organization (WHO).
If patients exhibit symptoms and either they or a close contact has returned from China recently, take standard airborne precautions and send specimens – a serum sample, oral and nasal pharyngeal swabs, and lower respiratory tract specimens if available – to the local health department, which will forward them to the Centers for Disease Control and Prevention (CDC) for testing. Turnaround time is 24-48 hours.
The 2019 Novel Coronavirus (2019-nCoV), identified as the cause of an outbreak of respiratory illness first detected in December in association with a live animal market in Wuhan, China, has been implicated in almost 2,000 cases and 56 deaths in that country. Cases have been reported in 13 countries besides China. Five cases of 2019-nCoV infection have been confirmed in the United States, all in people recently returned from Wuhan. As the virus spreads in China, however, it’s almost certain more cases will show up in the United States. Travel history is key, Dr. Schaffner and others said.
Plan and rehearse
The first step to prepare is to use the CDC’s Interim Guidance for Healthcare Professionals to make a written plan specific to your practice to respond to a potential case. The plan must include notifying the local health department, the CDC liaison for testing, and tracking down patient contacts.
“It’s not good enough to just download CDC’s guidance; use it to make your own local plan and know what to do 24/7,” said Daniel Lucey, MD, an infectious disease expert at Georgetown University Medical Center, Washington, D.C.
“Know who is on call at the health department on weekends and nights,” he said. Know where the patient is going to be isolated; figure out what to do if there’s more than one, and tests come back positive. Have masks on hand, and rehearse the response. “Make a coronavirus team, and absolutely have the nurses involved,” as well as other providers who may come into contact with a case, he added.
“You want to be able to do as well as your counterparts in Washington state and Chicago,” where the first two U.S. cases emerged. “They were prepared. They knew what to do,” Dr. Lucey said.
Those first two U.S. patients – a man in Everett, Wash., and a Chicago woman – developed symptoms after returning from Wuhan, a city of 11 million just over 400 miles inland from the port city of Shanghai. On Jan. 26 three more cases were confirmed by the CDC, two in California and one in Arizona, and each had recently traveled to Wuhan. All five patients remain hospitalized, and there’s no evidence they spread the infection further. There is also no evidence of human-to-human transmission of other cases exported from China to any other countries, according to the WHO.
WHO declined to declare a global health emergency – a Public Health Emergency of International Concern, in its parlance – on Jan. 23. The step would have triggered travel and trade restrictions in member states, including the United States. For now, at least, the group said it wasn’t warranted at this point.
Fatality rates
The focus right now is China. The outbreak has spread beyond Wuhan to other parts of the country, and there’s evidence of fourth-generation spread.
Transportation into and out of Wuhan and other cities has been curtailed, Lunar New Year festivals have been canceled, and the Shanghai Disneyland has been closed, among other measures taken by Chinese officials.
The government could be taking drastic measures in part to prevent the public criticism it took in the early 2000’s for the delayed response and lack of transparency during the global outbreak of another wildlife market coronavirus epidemic, severe acute respiratory syndrome (SARS). In a press conference Jan. 22, WHO officials commended the government’s containment efforts but did not say they recommended them.
According to WHO, serious cases in China have mostly been in people over 40 years old with significant comorbidities and have skewed towards men. Spread seems to be limited to family members, health care providers, and other close contacts, probably by respiratory droplets. If that pattern holds, WHO officials said, the outbreak is containable.
The fatality rate appears to be around 3%, a good deal lower than the 10% reported for SARS and much lower than the nearly 40% reported for Middle East respiratory syndrome (MERS), another recent coronavirus mutation from the animal trade.
The Wuhan virus fatality rate might drop as milder cases are detected and added to the denominator. “It definitely appears to be less severe than SARS and MERS,” said Amesh Adalja, MD, an infectious disease physician in Pittsburgh and emerging infectious disease researcher at Johns Hopkins University, Baltimore.
SARS: Lessons learned
In general, the world is much better equipped for coronavirus outbreaks than when SARS, in particular, emerged in 2003.
WHO officials in their press conference lauded China for it openness with the current outbreak, and for isolating and sequencing the virus immediately, which gave the world a diagnostic test in the first days of the outbreak, something that wasn’t available for SARS. China and other countries also are cooperating and working closely to contain the Wuhan virus.
“What we know today might change tomorrow, so we have to keep tuned in to new information, but we learned a lot from SARS,” Dr. Shaffner said. Overall, it’s likely “the impact on the United States of this new coronavirus is going to be trivial,” he predicted.
Dr. Lucey, however, recalled that the SARS outbreak in Toronto in 2003 started with one missed case. A woman returned asymptomatic from Hong Kong and spread the infection to her family members before she died. Her cause of death wasn’t immediately recognized, nor was the reason her family members were sick, since they hadn’t been to Hong Kong recently.
The infection ultimately spread to more than 200 people, about half of them health care workers. A few people died.
If a virus is sufficiently contagious, “it just takes one. You don’t want to be the one who misses that first patient,” Dr. Lucey said.
Currently, there are no antivirals or vaccines for coronaviruses; researchers are working on both, but for now, care is supportive.
This article was updated with new case numbers on 1/26/20.
Catheter cryoablation effective for persistent AFib in pivotal trial
NATIONAL HARBOR, MD. – , setting the stage for the device to become the first to receive U.S. labeling for catheter ablation in this atrial fibrillation population.
The Arctic Front Advance cryoballoon, used on 165 patients with persistent atrial fibrillation (AFib) enrolled in the trial, produced a 55% rate of treatment success, including freedom from recurrent AFib during 12 months of follow-up, and produced one prespecified serious adverse event in the primary safety endpoint.

Both results easily surpassed the prespecified performance goals set by negotiation with the FDA, Hugh Calkins, MD, said at the annual International AF Symposium. The trial design included no control group and instead assessed safety and efficacy against prespecified standards set by the regulatory agency.
The cryoballoon “showed excellent performance. I don’t see how this could possibly be turned down by the FDA,” said Dr. Calkins, professor of medicine and director of the cardiac arrhythmia service at Johns Hopkins Medicine in Baltimore.
Cardiac electrophysiologists have for years routinely performed catheter ablation procedures on patients with persistent AFib even though the devices, based on ablation by radiofrequency or by chilling, have been labeled for use only in treating patients with paroxysmal AFib. Although this off-label use has not resulted in any problems with health insurance coverage, Dr. Calkins said, it has kept manufacturers from marketing their ablation devices for use in persistent AFib patients.
If the reported data result in labeling for the tested cryoballoon for persistant AFib patients, “it will have a big impact,” he predicted. “People have used cryoballoons for ablating persistent AFib for years, but this would put more fuel in the fire, both the [very positive] safety and efficacy data, and getting an FDA label, which is worth a lot,” he said in an interview.
But Dr. Calkins stopped short of anticipating that the results would convince operators experienced and focused on performing radiofrequency ablation to switch to cryo devices for treating persistent AFib patients. “People are pretty stuck in their ways,” he noted, and reports are expected soon from pivotal trials that are now testing various radiofrequency devices, as well as other types of cryo devices, in persistent AFib patients, so the range of device options labeled for this population may soon grow even more.
The STOP Persistent AF trial ran at 25 sites in the United States, Canada, and Japan during March 2017-August 2019, and included 165 adults with symptomatic, persistent AFib who had not responded to at least one antiarrhythmic drug and had a history of AFib episodes lasting at least 7 days but with no episodes persisting for 6 months or longer. The study excluded patients with prior ablation or left atrial surgery, a recent cerebrovascular event, substantially reduced left ventricular function, or substantial left atrial enlargement.
The enrolled patients were an average 65 years of age and 70% were men. Patients had been diagnosed with paroxysmal AFib an average of 5 years before study entry and with persistent AFib a little over 6 months before entry. The most recent AFib episode of enrolled patients averaged about 60 days, on average they had been unsuccessfully treated with just over one antiarrhythmic drug, and on average they had previously undergone about two cardioversions, after which their arrhythmia recurred.
The primary efficacy endpoint – a 55% rate of acute procedural success plus freedom from AFib recurrence during the 9 months following a 90-day blanking period immediately after ablation plus no added or increased antiarrhythmic drugs – significantly exceeded the prespecified performance goal of a 40% rate, Dr. Calkins reported. The study used the standard measure of recurrence as any 30-second or longer AFib episode detected during a weekly ECG telemonitoring session or during 48-hour ambulatory ECG monitoring at 6- and 12-month follow-ups or during in-office 12-lead ECG assessment at 3-, 6-, and 12-month follow-up. Twelve-month follow-up occurred for 145 of the enrolled patients.
The only prespecified primary safety event was one episode of cardiac perforation, which occurred during a repeat procedure. This rate of one safety event among 165 patients (0.6%) fell well within the prespecified safety performance goal of no more than 13%. In addition to this perforation, five additional serious adverse events (3%) occurred that were attributable to the cryoballoon treatment, including two cases of vascular pseudoaneurysm, one puncture-site hematoma, one case of pericarditis, and one episode of atrial tachycardia. Four additional serious adverse events occurred that were attributable to the ablation procedure (one acute cardiac failure, one postprocedure ileus, one respiratory failure, and one urinary tract infection), for an overall serious event rate of 5%.
During follow-up, 13% of patients had a repeat ablation procedure, following an initial ablation limited to pulmonary-vein isolation (PVI). The overall rate of 1-year efficacy, including the relatively low rate of need for redo ablation, “are an impressive endorsement of a PVI-only strategy” for initial ablation, Dr. Calkins said. “I’m a strong believer in PVI only for the first ablation for both paroxysmal and persistent AFib.”
He also noted that the 30-second threshold for scoring recurrent arrhythmia episodes following the 90-day blanking period after ablation was a very conservative measure of treatment failure, but it continues to define recurrence in this and many other current AFib ablation studies because it is the historical criteria for measuring ablation success or failure. It is especially important to maintain this criteria in a study that relied on prespecified performance criteria rather than a control arm for judging efficacy, Dr. Calkins said.
The study also included three quality-of-life measures. Patient scores on the Atrial Fibrillation Effect on Quality-of-Life (AFEQT) questionnaire rose by an average of nearly 26 points from baseline to 12 months, a statistically significant and clinically meaningful increase. Scores on both the physical and mental domains of the Short Form-12 (SF-12) improved from baseline by an average of about five points on each subscale, also statistically significant and clinically meaningful changes. Patients also reported statistically significant and in some cases substantial reductions in the prevalence rates of each of five different AFib symptoms: dizziness, dyspnea, fatigue, palpitations, and rapid heartbeat.
The study was funded by Medtronic, the company that markets the tested cryoballoon (Arctic Front Advance). Dr. Calkins has been a consultant to and has received honoraria from Medtronic, as well as from Abbott, AtriCure, Boehringer Ingelheim, Boston Scientific, and Johnson & Johnson.
NATIONAL HARBOR, MD. – , setting the stage for the device to become the first to receive U.S. labeling for catheter ablation in this atrial fibrillation population.
The Arctic Front Advance cryoballoon, used on 165 patients with persistent atrial fibrillation (AFib) enrolled in the trial, produced a 55% rate of treatment success, including freedom from recurrent AFib during 12 months of follow-up, and produced one prespecified serious adverse event in the primary safety endpoint.
Both results easily surpassed the prespecified performance goals set by negotiation with the FDA, Hugh Calkins, MD, said at the annual International AF Symposium. The trial design included no control group and instead assessed safety and efficacy against prespecified standards set by the regulatory agency.
The cryoballoon “showed excellent performance. I don’t see how this could possibly be turned down by the FDA,” said Dr. Calkins, professor of medicine and director of the cardiac arrhythmia service at Johns Hopkins Medicine in Baltimore.
Cardiac electrophysiologists have for years routinely performed catheter ablation procedures on patients with persistent AFib even though the devices, based on ablation by radiofrequency or by chilling, have been labeled for use only in treating patients with paroxysmal AFib. Although this off-label use has not resulted in any problems with health insurance coverage, Dr. Calkins said, it has kept manufacturers from marketing their ablation devices for use in persistent AFib patients.
If the reported data result in labeling for the tested cryoballoon for persistant AFib patients, “it will have a big impact,” he predicted. “People have used cryoballoons for ablating persistent AFib for years, but this would put more fuel in the fire, both the [very positive] safety and efficacy data, and getting an FDA label, which is worth a lot,” he said in an interview.
But Dr. Calkins stopped short of anticipating that the results would convince operators experienced and focused on performing radiofrequency ablation to switch to cryo devices for treating persistent AFib patients. “People are pretty stuck in their ways,” he noted, and reports are expected soon from pivotal trials that are now testing various radiofrequency devices, as well as other types of cryo devices, in persistent AFib patients, so the range of device options labeled for this population may soon grow even more.
The STOP Persistent AF trial ran at 25 sites in the United States, Canada, and Japan during March 2017-August 2019, and included 165 adults with symptomatic, persistent AFib who had not responded to at least one antiarrhythmic drug and had a history of AFib episodes lasting at least 7 days but with no episodes persisting for 6 months or longer. The study excluded patients with prior ablation or left atrial surgery, a recent cerebrovascular event, substantially reduced left ventricular function, or substantial left atrial enlargement.
The enrolled patients were an average 65 years of age and 70% were men. Patients had been diagnosed with paroxysmal AFib an average of 5 years before study entry and with persistent AFib a little over 6 months before entry. The most recent AFib episode of enrolled patients averaged about 60 days, on average they had been unsuccessfully treated with just over one antiarrhythmic drug, and on average they had previously undergone about two cardioversions, after which their arrhythmia recurred.
The primary efficacy endpoint – a 55% rate of acute procedural success plus freedom from AFib recurrence during the 9 months following a 90-day blanking period immediately after ablation plus no added or increased antiarrhythmic drugs – significantly exceeded the prespecified performance goal of a 40% rate, Dr. Calkins reported. The study used the standard measure of recurrence as any 30-second or longer AFib episode detected during a weekly ECG telemonitoring session or during 48-hour ambulatory ECG monitoring at 6- and 12-month follow-ups or during in-office 12-lead ECG assessment at 3-, 6-, and 12-month follow-up. Twelve-month follow-up occurred for 145 of the enrolled patients.
The only prespecified primary safety event was one episode of cardiac perforation, which occurred during a repeat procedure. This rate of one safety event among 165 patients (0.6%) fell well within the prespecified safety performance goal of no more than 13%. In addition to this perforation, five additional serious adverse events (3%) occurred that were attributable to the cryoballoon treatment, including two cases of vascular pseudoaneurysm, one puncture-site hematoma, one case of pericarditis, and one episode of atrial tachycardia. Four additional serious adverse events occurred that were attributable to the ablation procedure (one acute cardiac failure, one postprocedure ileus, one respiratory failure, and one urinary tract infection), for an overall serious event rate of 5%.
During follow-up, 13% of patients had a repeat ablation procedure, following an initial ablation limited to pulmonary-vein isolation (PVI). The overall rate of 1-year efficacy, including the relatively low rate of need for redo ablation, “are an impressive endorsement of a PVI-only strategy” for initial ablation, Dr. Calkins said. “I’m a strong believer in PVI only for the first ablation for both paroxysmal and persistent AFib.”
He also noted that the 30-second threshold for scoring recurrent arrhythmia episodes following the 90-day blanking period after ablation was a very conservative measure of treatment failure, but it continues to define recurrence in this and many other current AFib ablation studies because it is the historical criteria for measuring ablation success or failure. It is especially important to maintain this criteria in a study that relied on prespecified performance criteria rather than a control arm for judging efficacy, Dr. Calkins said.
The study also included three quality-of-life measures. Patient scores on the Atrial Fibrillation Effect on Quality-of-Life (AFEQT) questionnaire rose by an average of nearly 26 points from baseline to 12 months, a statistically significant and clinically meaningful increase. Scores on both the physical and mental domains of the Short Form-12 (SF-12) improved from baseline by an average of about five points on each subscale, also statistically significant and clinically meaningful changes. Patients also reported statistically significant and in some cases substantial reductions in the prevalence rates of each of five different AFib symptoms: dizziness, dyspnea, fatigue, palpitations, and rapid heartbeat.
The study was funded by Medtronic, the company that markets the tested cryoballoon (Arctic Front Advance). Dr. Calkins has been a consultant to and has received honoraria from Medtronic, as well as from Abbott, AtriCure, Boehringer Ingelheim, Boston Scientific, and Johnson & Johnson.
NATIONAL HARBOR, MD. – , setting the stage for the device to become the first to receive U.S. labeling for catheter ablation in this atrial fibrillation population.
The Arctic Front Advance cryoballoon, used on 165 patients with persistent atrial fibrillation (AFib) enrolled in the trial, produced a 55% rate of treatment success, including freedom from recurrent AFib during 12 months of follow-up, and produced one prespecified serious adverse event in the primary safety endpoint.
Both results easily surpassed the prespecified performance goals set by negotiation with the FDA, Hugh Calkins, MD, said at the annual International AF Symposium. The trial design included no control group and instead assessed safety and efficacy against prespecified standards set by the regulatory agency.
The cryoballoon “showed excellent performance. I don’t see how this could possibly be turned down by the FDA,” said Dr. Calkins, professor of medicine and director of the cardiac arrhythmia service at Johns Hopkins Medicine in Baltimore.
Cardiac electrophysiologists have for years routinely performed catheter ablation procedures on patients with persistent AFib even though the devices, based on ablation by radiofrequency or by chilling, have been labeled for use only in treating patients with paroxysmal AFib. Although this off-label use has not resulted in any problems with health insurance coverage, Dr. Calkins said, it has kept manufacturers from marketing their ablation devices for use in persistent AFib patients.
If the reported data result in labeling for the tested cryoballoon for persistant AFib patients, “it will have a big impact,” he predicted. “People have used cryoballoons for ablating persistent AFib for years, but this would put more fuel in the fire, both the [very positive] safety and efficacy data, and getting an FDA label, which is worth a lot,” he said in an interview.
But Dr. Calkins stopped short of anticipating that the results would convince operators experienced and focused on performing radiofrequency ablation to switch to cryo devices for treating persistent AFib patients. “People are pretty stuck in their ways,” he noted, and reports are expected soon from pivotal trials that are now testing various radiofrequency devices, as well as other types of cryo devices, in persistent AFib patients, so the range of device options labeled for this population may soon grow even more.
The STOP Persistent AF trial ran at 25 sites in the United States, Canada, and Japan during March 2017-August 2019, and included 165 adults with symptomatic, persistent AFib who had not responded to at least one antiarrhythmic drug and had a history of AFib episodes lasting at least 7 days but with no episodes persisting for 6 months or longer. The study excluded patients with prior ablation or left atrial surgery, a recent cerebrovascular event, substantially reduced left ventricular function, or substantial left atrial enlargement.
The enrolled patients were an average 65 years of age and 70% were men. Patients had been diagnosed with paroxysmal AFib an average of 5 years before study entry and with persistent AFib a little over 6 months before entry. The most recent AFib episode of enrolled patients averaged about 60 days, on average they had been unsuccessfully treated with just over one antiarrhythmic drug, and on average they had previously undergone about two cardioversions, after which their arrhythmia recurred.
The primary efficacy endpoint – a 55% rate of acute procedural success plus freedom from AFib recurrence during the 9 months following a 90-day blanking period immediately after ablation plus no added or increased antiarrhythmic drugs – significantly exceeded the prespecified performance goal of a 40% rate, Dr. Calkins reported. The study used the standard measure of recurrence as any 30-second or longer AFib episode detected during a weekly ECG telemonitoring session or during 48-hour ambulatory ECG monitoring at 6- and 12-month follow-ups or during in-office 12-lead ECG assessment at 3-, 6-, and 12-month follow-up. Twelve-month follow-up occurred for 145 of the enrolled patients.
The only prespecified primary safety event was one episode of cardiac perforation, which occurred during a repeat procedure. This rate of one safety event among 165 patients (0.6%) fell well within the prespecified safety performance goal of no more than 13%. In addition to this perforation, five additional serious adverse events (3%) occurred that were attributable to the cryoballoon treatment, including two cases of vascular pseudoaneurysm, one puncture-site hematoma, one case of pericarditis, and one episode of atrial tachycardia. Four additional serious adverse events occurred that were attributable to the ablation procedure (one acute cardiac failure, one postprocedure ileus, one respiratory failure, and one urinary tract infection), for an overall serious event rate of 5%.
During follow-up, 13% of patients had a repeat ablation procedure, following an initial ablation limited to pulmonary-vein isolation (PVI). The overall rate of 1-year efficacy, including the relatively low rate of need for redo ablation, “are an impressive endorsement of a PVI-only strategy” for initial ablation, Dr. Calkins said. “I’m a strong believer in PVI only for the first ablation for both paroxysmal and persistent AFib.”
He also noted that the 30-second threshold for scoring recurrent arrhythmia episodes following the 90-day blanking period after ablation was a very conservative measure of treatment failure, but it continues to define recurrence in this and many other current AFib ablation studies because it is the historical criteria for measuring ablation success or failure. It is especially important to maintain this criteria in a study that relied on prespecified performance criteria rather than a control arm for judging efficacy, Dr. Calkins said.
The study also included three quality-of-life measures. Patient scores on the Atrial Fibrillation Effect on Quality-of-Life (AFEQT) questionnaire rose by an average of nearly 26 points from baseline to 12 months, a statistically significant and clinically meaningful increase. Scores on both the physical and mental domains of the Short Form-12 (SF-12) improved from baseline by an average of about five points on each subscale, also statistically significant and clinically meaningful changes. Patients also reported statistically significant and in some cases substantial reductions in the prevalence rates of each of five different AFib symptoms: dizziness, dyspnea, fatigue, palpitations, and rapid heartbeat.
The study was funded by Medtronic, the company that markets the tested cryoballoon (Arctic Front Advance). Dr. Calkins has been a consultant to and has received honoraria from Medtronic, as well as from Abbott, AtriCure, Boehringer Ingelheim, Boston Scientific, and Johnson & Johnson.
REPORTING FROM THE AF SYMPOSIUM 2020
Key clinical point: Catheter ablation using a cryoballoon was safe and effective for patients with persistent atrial fibrillation in a pivotal trial.
Major finding: The rate of freedom from treatment failure after 12 months was 55%, significantly exceeding the prespecified performance goal of 40%.
Study details: A multicenter, international study with 165 enrolled patients.
Disclosures: The study was funded by Medtronic, the company that markets the tested cryoballoon (Arctic Front Advance). Dr. Calkins has been a consultant to and has received honoraria from Medtronic, as well as from Abbott, AtriCure, Boehringer Ingelheim, Boston Scientific, and Johnson & Johnson.