User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
An Update on JAK Inhibitors in Skin Disease
Atopic dermatitis (AD) is a chronic inflammatory skin disorder affecting 7% of adults and 13% of children in the United States.1,2 Atopic dermatitis is characterized by pruritus, dry skin, and pain, all of which can negatively impact quality of life and put patients at higher risk for psychiatric comorbidities such as anxiety and depression.3 The pathogenesis of AD is multifactorial, involving genetics, epidermal barrier dysfunction, and immune dysregulation. Overactivation of helper T cell (TH2) pathway cytokines, including IL-4, IL-13, and IL-31, is thought to propagate both inflammation and pruritus, which are central to AD. The JAK-STAT signaling pathway plays a pivotal role in the immune system dysregulation and exaggeration of TH2 cell response, making JAK-STAT inhibitors (or JAK inhibitors) strong theoretical candidates for the treatment of AD.4 In humans, the Janus kinases are composed of 4 different members—JAK1, JAK2, JAK3, and tyrosine kinase 2—all of which can be targeted by JAK inhibitors.5
JAK inhibitors such as tofacitinib have already been approved by the US Food and Drug Administration (FDA) to treat various inflammatory conditions, including rheumatoid arthritis, ulcerative colitis, and psoriatic arthritis; other JAK inhibitors such as baricitinib are only approved for patients with rheumatoid arthritis.6,7 The success of these small molecule inhibitors in these immune-mediated conditions make them attractive candidates for the treatment of AD. Several JAK inhibitors are in phase 2 and phase 3 clinical trials as oral therapies (moderate to severe AD) or as topical treatments (mild to moderate AD). Currently, ruxolitinib (RUX) is the only topical JAK inhibitor that is FDA approved for the treatment of AD in the United States.8 In this editorial, we focus on recent trials of JAK inhibitors tested in patients with AD, including topical RUX, as well as oral abrocitinib, upadacitinib, and baricitinib.
Topical RUX in AD
Ruxolitinib is a topical JAK1/2 small molecule inhibitor approved by the FDA for the treatment of AD in 2021. In a randomized trial by Kim et al9 in 2020, all tested regimens of RUX demonstrated significant improvement in eczema area and severity index (EASI) scores vs vehicle; notably, RUX cream 1.5% applied twice daily achieved the greatest mean percentage change in baseline EASI score vs vehicle at 4 weeks (76.1% vs 15.5%; P<.0001). Ruxolitinib cream was well tolerated through week 8 of the trial, and all adverse events (AEs) were mild to moderate in severity and comparable to those in the vehicle group.9
Topical JAK inhibitors appear to be effective for mild to moderate AD and have had an acceptable safety profile in clinical trials thus far. Although topical corticosteroids and calcineurin inhibitors can have great clinical benefit in AD, they are recommended for short-term use given side effects such as thinning of the skin, burning, or telangiectasia formation.10,11 The hope is that topical JAK inhibitors may be an alternative to standard topical treatments for AD, as they can be used for longer periods due to a safer side-effect profile.
Oral JAK Inhibitors in AD
Several oral JAK inhibitors are undergoing investigation for the systemic treatment of moderate to severe AD. Abrocitinib is an oral JAK1 inhibitor that has demonstrated efficacy in several phase 3 trials in patients with moderate to severe AD. In a 2021 trial, patients were randomized in a 2:2:2:1 ratio to receive abrocitinib 200 mg daily, abrocitinib 100 mg daily, subcutaneous dupilumab 300 mg every other week, or placebo, respectively.12 Patients in both abrocitinib groups showed significant improvement in AD vs placebo, and EASI-75 response was achieved in 70.3%, 58.7%, 58.1%, and 27.1% of patients, respectively (P<.001 for both abrocitinib doses vs placebo). Adverse events occurred more frequently in the abrocitinib 200-mg group vs placebo. Nausea, acne, nasopharyngitis, and headache were the most frequently reported AEs with abrocitinib.12 Another phase 3 trial by Silverberg et al13 (N=391) had similar treatment results, with 38.1% of participants receiving abrocitinib 200 mg and 28.4% of participants receiving abrocitinib 100 mg achieving investigator global assessment scores of 0 (clear) or 1 (almost clear) vs 9.1% of participants receiving placebo (P<.001). Abrocitinib was well tolerated in this trial with few serious AEs (ie, herpangina [0.6%], pneumonia [0.6%]).13 In both trials, there were rare instances of laboratory values indicating thrombocytopenia with the 200-mg dose (0.9%12 and 3.2%13) without any clinical manifestations. Although a decrease in platelets was observed, no thrombocytopenia occurred in the abrocitinib 100-mg group in the latter trial.13
Baricitinib is another oral inhibitor of JAK1 and JAK2 with potential for the treatment of AD. One randomized trial (N=329) demonstrated its efficacy in combination with a topical corticosteroid (TCS). At 16 weeks, a higher number of participants treated with baricitinib and TCS achieved investigator global assessment scores of 0 (clear) or 1 (almost clear) compared to those who received placebo and TCS (31% with baricitinib 4 mg + TCS, 24% with baricitinib 2 mg + TCS, and 15% with placebo + TCS).14 Similarly, in BREEZE-AD5,another phase 3 trial (N=440), baricitinib monotherapy demonstrated a higher rate of treatment success vs placebo.15 Specifically, 13% of patients treated with baricitinib 1 mg and 30% of those treated with baricitinib 2 mg achieved 75% or greater reduction in EASI scores compared to 8% in the placebo group. The most common AEs associated with baricitinib were nasopharyngitis and headache. Adverse events occurred with similar frequency across both experimental and control groups.15 Reich et al14 demonstrated a higher overall rate of AEs—most commonly nasopharyngitis, upper respiratory tract infections, and folliculitis—in baricitinib-treated patients; however, serious AEs occurred with similar frequency across all groups, including the control group.
The selective JAK1 inhibitor upadacitinib also is undergoing testing in treating moderate to severe AD. In one trial, 167 patients were randomized to once daily oral upadacitinib 7.5 mg, 15 mg, or 30 mg or placebo.16 All doses of upadacitinib demonstrated considerably higher percentage improvements from baseline in EASI scores compared to placebo at 16 weeks with a clear dose-response relationship (39%, 62%, and 74% vs 23%, respectively). In this trial, there were no dose-limiting safety events. Serious AEs were infrequent, occurring in 4.8%, 2.4%, and 0% of upadacitinib groups vs 2.5% for placebo. The serious AEs observed with upadacitinib were 1 case of appendicitis, lower jaw pericoronitis in a patient with a history of repeated tooth infections, and an exacerbation of AD.16
Tofacitinib, another JAK inhibitor, has been shown to increase the risk for blood clots and death in a large trial in the treatment of rheumatoid arthritis. Following this study, the FDA is requiring black box warnings for tofacitinib and also for the 2 JAK inhibitors baricitinib and upadacitinib regarding the risks for heart-related events, cancer, blood clots, and death. Given that these medications share a similar mechanism of action to tofacitinib, they may have similar risks, though they have not yet been fully evaluated in large safety trials.17
With more recent investigation into novel therapeutics for AD, oral JAK inhibitors may play an important role in the future to treat patients with moderate to severe AD with inadequate response or contraindications to other systemic therapies. In trials thus far, oral JAK inhibitors have exhibited acceptable safety profiles and have demonstrated treatment success in AD. More randomized, controlled, phase 3 studies with larger patient populations are required to confirm their potential as effective treatments and elucidate their long-term safety.
Deucravacitinib in Psoriasis
Deucravacitinib is a first-in-class, oral, selective TYK2 inhibitor currently undergoing testing for the treatment of psoriasis. A randomized phase 2 trial (N=267) found that deucravacitinib was more effective than placebo in treating chronic plaque psoriasis at doses of 3 to 12 mg daily.18 The percentage of participants with a 75% or greater reduction from baseline in the psoriasis area and severity index score was 7% with placebo, 9% with deucravacitinib 3 mg every other day (P=.49 vs placebo), 39% with 3 mg once daily (P<.001 vs placebo), 69% with 3 mg twice daily (P<.001 vs placebo), 67% with 6 mg twice daily (P<.001 vs placebo), and 75% with 12 mg once daily (P<.001 vs placebo). The most commonly reported AEs were nasopharyngitis, headache, diarrhea, nausea, and upper respiratory tract infection. Adverse events occurred in 51% of participants in the control group and in 55% to 80% of those in the experimental groups. Additionally, there was 1 reported case of melanoma (stage 0) 96 days after the start of treatment in a patient in the 3-mg once-daily group. Serious AEs occurred in only 0% to 2% of participants who received deucravacitinib.18
Two phase 3 trials—POETYK PSO-1 and POETYK PSO-2 (N=1686)—found deucravacitinib to be notably more effective than both placebo and apremilast in treating psoriasis.19 Among participants receiving deucravacitinib 6 mg daily, 58.7% and 53.6% in the 2 respective trials achieved psoriasis area and severity index 75 response vs 12.7% and 9.4% receiving placebo and 35.1% and 40.2% receiving apremilast. Overall, the treatment was well tolerated, with a low rate of discontinuation of deucravacitinib due to AEs (2.4% of patients on deucravacitinib compared to 3.8% on placebo and 5.2% on apremilast). The most frequently observed AEs with deucravacitinib were nasopharyngitis and upper respiratory tract infection. The full results of these trials are expected to be published soon.19,20
Final Thoughts
Overall, JAK inhibitors are a novel class of therapeutics that may have further success in the treatment of other dermatologic conditions that negatively affect patients’ quality of life and productivity. We should look forward to additional successful trials with these promising medications.
- Chiesa Fuxench ZC, Block JK, Boguniewicz M, et al. Atopic dermatitis in America study: a cross-sectional study examining the prevalence and disease burden of atopic dermatitis in the US adult population. J Invest Dermatol. 2019;139:583-590.
- Silverberg JI , Simpson EL. Associations of childhood eczema severity: a US population-based study. Dermatitis. 2014;25:107-114.
- Schonmann Y, Mansfield KE, Hayes JF, et al. Atopic eczema in adulthood and risk of depression and anxiety: a population-based cohort study. J Allergy Clin Immunol Pract. 2020;8:248-257.e16.
- Bao L, Zhang H, Chan LS. The involvement of the JAK-STAT signaling pathway in chronic inflammatory skin disease atopic dermatitis. JAKSTAT. 2013;2:e24137.
- Villarino AV, Kanno Y, O’Shea JJ. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat Immunol. 2017;18:374-384.
- Xeljanz FDA approval history. Drugs.com website. Updated December 14, 2021. Accessed February 16, 2022. https://www.drugs.com/history/xeljanz.html
- Mullard A. FDA approves Eli Lilly’s baricitinib. Nat Rev Drug Discov. 2018;17:460.
- FDA approves Opzelura. Drugs.com website. Published September 2021. Accessed February 16, 2022. https://www.drugs.com/newdrugs/fda-approves-opzelura-ruxolitinib-cream-atopic-dermatitis-ad-5666.html
- Kim BS, Sun K, Papp K, et al. Effects of ruxolitinib cream on pruritus and quality of life in atopic dermatitis: results from a phase 2, randomized, dose-ranging, vehicle- and active-controlled study.J Am Acad Dermatol. 2020;82:1305-1313.
- Eichenfield LF, Tom WL, Berger TG, et al. Guidelines of care for the management of atopic dermatitis: section 2, management and treatment of atopic dermatitis with topical therapies. J Am Acad Dermatol. 2014;71:116-132.
- Wollenberg A, Barbarot S, Bieber T, et al. Consensus-based European guidelines for treatment of atopic eczema (atopic dermatitis) in adults and children: part I. J Eur Acad Dermatol Venereol. 2018;32:657-682.
- Bieber T, Simpson EL, Silverberg JI, et al. Abrocitinib versus placebo or dupilumab for atopic dermatitis. N Engl J Med. 2021;384:1101-1112.
- Silverberg JI, Simpson EL, Thyssen JP, et al. Efficacy and safety of abrocitinib in patients with moderate-to-severe atopic dermatitis: a randomized clinical trial. JAMA Dermatol. 2020;156:863-873.
- Reich K, Kabashima K, Peris K, et al. Efficacy and safety of baricitinib combined with topical corticosteroids for treatment of moderate to severe atopic dermatitis: a randomized clinical trial. JAMA Dermatol. 2020;156:1333-1343.
- Simpson EL, Forman S, Silverberg JI, et al. Baricitinib in patients with moderate-to-severe atopic dermatitis: results from a randomized monotherapy phase 3 trial in the United States and Canada (BREEZE-AD5). J Am Acad Dermatol. 2021;85:62-70.
- Guttman-Yassky E, Thaçi D, Pangan AL, et al. Upadacitinib in adults with moderate to severe atopic dermatitis: 16-week results from a randomized, placebo-controlled trial. J Allergy Clin Immunol. 2020;145:877-884.
- US Food and Drug Administration. FDA requires warnings about increased risk of serious heart-related events, cancer, blood clots, and death for JAK inhibitors that treat certain chronic inflammatory conditions. Published September 1, 2022. Accessed February 16, 2022. https://www.fda.gov/drugs/drug-safety-and-availability/fda-requires-warnings-about-increased-risk-serious-heart-related-events-cancer-blood-clots-and-death
- Papp K, Gordon K, Thaçi D, et al. Phase 2 trial of selective tyrosine kinase 2 inhibition in psoriasis. N Engl J Med. 2018;379:1313-1321.
- Bristol Myers Squibb presents positive data from two pivotal phase 3 psoriasis studies demonstrating superiority of deucravacitinib compared to placebo and Otezla® (apremilast). Press release. Bristol Meyers Squibb. April 23, 2021. Accessed February 16, 2022. https://news.bms.com/news/details/2021/Bristol-Myers-Squibb-Presents-Positive-Data-from-Two-Pivotal-Phase-3-Psoriasis-Studies-Demonstrating-Superiority-of-Deucravacitinib-Compared-to-Placebo-and-Otezla-apremilast/default.aspx
- Armstrong A, Gooderham M, Warren R, et al. Efficacy and safety of deucravacitinib, an oral, selective tyrosine kinase 2 (TYK2) inhibitor, compared with placebo and apremilast in moderate to severe plaque psoriasis: results from the POETYK PSO-1 study [abstract]. Abstract presented at: 2021 American Academy of Dermatology annual meeting; April 23-25, 2021; San Francisco, California.
Atopic dermatitis (AD) is a chronic inflammatory skin disorder affecting 7% of adults and 13% of children in the United States.1,2 Atopic dermatitis is characterized by pruritus, dry skin, and pain, all of which can negatively impact quality of life and put patients at higher risk for psychiatric comorbidities such as anxiety and depression.3 The pathogenesis of AD is multifactorial, involving genetics, epidermal barrier dysfunction, and immune dysregulation. Overactivation of helper T cell (TH2) pathway cytokines, including IL-4, IL-13, and IL-31, is thought to propagate both inflammation and pruritus, which are central to AD. The JAK-STAT signaling pathway plays a pivotal role in the immune system dysregulation and exaggeration of TH2 cell response, making JAK-STAT inhibitors (or JAK inhibitors) strong theoretical candidates for the treatment of AD.4 In humans, the Janus kinases are composed of 4 different members—JAK1, JAK2, JAK3, and tyrosine kinase 2—all of which can be targeted by JAK inhibitors.5
JAK inhibitors such as tofacitinib have already been approved by the US Food and Drug Administration (FDA) to treat various inflammatory conditions, including rheumatoid arthritis, ulcerative colitis, and psoriatic arthritis; other JAK inhibitors such as baricitinib are only approved for patients with rheumatoid arthritis.6,7 The success of these small molecule inhibitors in these immune-mediated conditions make them attractive candidates for the treatment of AD. Several JAK inhibitors are in phase 2 and phase 3 clinical trials as oral therapies (moderate to severe AD) or as topical treatments (mild to moderate AD). Currently, ruxolitinib (RUX) is the only topical JAK inhibitor that is FDA approved for the treatment of AD in the United States.8 In this editorial, we focus on recent trials of JAK inhibitors tested in patients with AD, including topical RUX, as well as oral abrocitinib, upadacitinib, and baricitinib.
Topical RUX in AD
Ruxolitinib is a topical JAK1/2 small molecule inhibitor approved by the FDA for the treatment of AD in 2021. In a randomized trial by Kim et al9 in 2020, all tested regimens of RUX demonstrated significant improvement in eczema area and severity index (EASI) scores vs vehicle; notably, RUX cream 1.5% applied twice daily achieved the greatest mean percentage change in baseline EASI score vs vehicle at 4 weeks (76.1% vs 15.5%; P<.0001). Ruxolitinib cream was well tolerated through week 8 of the trial, and all adverse events (AEs) were mild to moderate in severity and comparable to those in the vehicle group.9
Topical JAK inhibitors appear to be effective for mild to moderate AD and have had an acceptable safety profile in clinical trials thus far. Although topical corticosteroids and calcineurin inhibitors can have great clinical benefit in AD, they are recommended for short-term use given side effects such as thinning of the skin, burning, or telangiectasia formation.10,11 The hope is that topical JAK inhibitors may be an alternative to standard topical treatments for AD, as they can be used for longer periods due to a safer side-effect profile.
Oral JAK Inhibitors in AD
Several oral JAK inhibitors are undergoing investigation for the systemic treatment of moderate to severe AD. Abrocitinib is an oral JAK1 inhibitor that has demonstrated efficacy in several phase 3 trials in patients with moderate to severe AD. In a 2021 trial, patients were randomized in a 2:2:2:1 ratio to receive abrocitinib 200 mg daily, abrocitinib 100 mg daily, subcutaneous dupilumab 300 mg every other week, or placebo, respectively.12 Patients in both abrocitinib groups showed significant improvement in AD vs placebo, and EASI-75 response was achieved in 70.3%, 58.7%, 58.1%, and 27.1% of patients, respectively (P<.001 for both abrocitinib doses vs placebo). Adverse events occurred more frequently in the abrocitinib 200-mg group vs placebo. Nausea, acne, nasopharyngitis, and headache were the most frequently reported AEs with abrocitinib.12 Another phase 3 trial by Silverberg et al13 (N=391) had similar treatment results, with 38.1% of participants receiving abrocitinib 200 mg and 28.4% of participants receiving abrocitinib 100 mg achieving investigator global assessment scores of 0 (clear) or 1 (almost clear) vs 9.1% of participants receiving placebo (P<.001). Abrocitinib was well tolerated in this trial with few serious AEs (ie, herpangina [0.6%], pneumonia [0.6%]).13 In both trials, there were rare instances of laboratory values indicating thrombocytopenia with the 200-mg dose (0.9%12 and 3.2%13) without any clinical manifestations. Although a decrease in platelets was observed, no thrombocytopenia occurred in the abrocitinib 100-mg group in the latter trial.13
Baricitinib is another oral inhibitor of JAK1 and JAK2 with potential for the treatment of AD. One randomized trial (N=329) demonstrated its efficacy in combination with a topical corticosteroid (TCS). At 16 weeks, a higher number of participants treated with baricitinib and TCS achieved investigator global assessment scores of 0 (clear) or 1 (almost clear) compared to those who received placebo and TCS (31% with baricitinib 4 mg + TCS, 24% with baricitinib 2 mg + TCS, and 15% with placebo + TCS).14 Similarly, in BREEZE-AD5,another phase 3 trial (N=440), baricitinib monotherapy demonstrated a higher rate of treatment success vs placebo.15 Specifically, 13% of patients treated with baricitinib 1 mg and 30% of those treated with baricitinib 2 mg achieved 75% or greater reduction in EASI scores compared to 8% in the placebo group. The most common AEs associated with baricitinib were nasopharyngitis and headache. Adverse events occurred with similar frequency across both experimental and control groups.15 Reich et al14 demonstrated a higher overall rate of AEs—most commonly nasopharyngitis, upper respiratory tract infections, and folliculitis—in baricitinib-treated patients; however, serious AEs occurred with similar frequency across all groups, including the control group.
The selective JAK1 inhibitor upadacitinib also is undergoing testing in treating moderate to severe AD. In one trial, 167 patients were randomized to once daily oral upadacitinib 7.5 mg, 15 mg, or 30 mg or placebo.16 All doses of upadacitinib demonstrated considerably higher percentage improvements from baseline in EASI scores compared to placebo at 16 weeks with a clear dose-response relationship (39%, 62%, and 74% vs 23%, respectively). In this trial, there were no dose-limiting safety events. Serious AEs were infrequent, occurring in 4.8%, 2.4%, and 0% of upadacitinib groups vs 2.5% for placebo. The serious AEs observed with upadacitinib were 1 case of appendicitis, lower jaw pericoronitis in a patient with a history of repeated tooth infections, and an exacerbation of AD.16
Tofacitinib, another JAK inhibitor, has been shown to increase the risk for blood clots and death in a large trial in the treatment of rheumatoid arthritis. Following this study, the FDA is requiring black box warnings for tofacitinib and also for the 2 JAK inhibitors baricitinib and upadacitinib regarding the risks for heart-related events, cancer, blood clots, and death. Given that these medications share a similar mechanism of action to tofacitinib, they may have similar risks, though they have not yet been fully evaluated in large safety trials.17
With more recent investigation into novel therapeutics for AD, oral JAK inhibitors may play an important role in the future to treat patients with moderate to severe AD with inadequate response or contraindications to other systemic therapies. In trials thus far, oral JAK inhibitors have exhibited acceptable safety profiles and have demonstrated treatment success in AD. More randomized, controlled, phase 3 studies with larger patient populations are required to confirm their potential as effective treatments and elucidate their long-term safety.
Deucravacitinib in Psoriasis
Deucravacitinib is a first-in-class, oral, selective TYK2 inhibitor currently undergoing testing for the treatment of psoriasis. A randomized phase 2 trial (N=267) found that deucravacitinib was more effective than placebo in treating chronic plaque psoriasis at doses of 3 to 12 mg daily.18 The percentage of participants with a 75% or greater reduction from baseline in the psoriasis area and severity index score was 7% with placebo, 9% with deucravacitinib 3 mg every other day (P=.49 vs placebo), 39% with 3 mg once daily (P<.001 vs placebo), 69% with 3 mg twice daily (P<.001 vs placebo), 67% with 6 mg twice daily (P<.001 vs placebo), and 75% with 12 mg once daily (P<.001 vs placebo). The most commonly reported AEs were nasopharyngitis, headache, diarrhea, nausea, and upper respiratory tract infection. Adverse events occurred in 51% of participants in the control group and in 55% to 80% of those in the experimental groups. Additionally, there was 1 reported case of melanoma (stage 0) 96 days after the start of treatment in a patient in the 3-mg once-daily group. Serious AEs occurred in only 0% to 2% of participants who received deucravacitinib.18
Two phase 3 trials—POETYK PSO-1 and POETYK PSO-2 (N=1686)—found deucravacitinib to be notably more effective than both placebo and apremilast in treating psoriasis.19 Among participants receiving deucravacitinib 6 mg daily, 58.7% and 53.6% in the 2 respective trials achieved psoriasis area and severity index 75 response vs 12.7% and 9.4% receiving placebo and 35.1% and 40.2% receiving apremilast. Overall, the treatment was well tolerated, with a low rate of discontinuation of deucravacitinib due to AEs (2.4% of patients on deucravacitinib compared to 3.8% on placebo and 5.2% on apremilast). The most frequently observed AEs with deucravacitinib were nasopharyngitis and upper respiratory tract infection. The full results of these trials are expected to be published soon.19,20
Final Thoughts
Overall, JAK inhibitors are a novel class of therapeutics that may have further success in the treatment of other dermatologic conditions that negatively affect patients’ quality of life and productivity. We should look forward to additional successful trials with these promising medications.
Atopic dermatitis (AD) is a chronic inflammatory skin disorder affecting 7% of adults and 13% of children in the United States.1,2 Atopic dermatitis is characterized by pruritus, dry skin, and pain, all of which can negatively impact quality of life and put patients at higher risk for psychiatric comorbidities such as anxiety and depression.3 The pathogenesis of AD is multifactorial, involving genetics, epidermal barrier dysfunction, and immune dysregulation. Overactivation of helper T cell (TH2) pathway cytokines, including IL-4, IL-13, and IL-31, is thought to propagate both inflammation and pruritus, which are central to AD. The JAK-STAT signaling pathway plays a pivotal role in the immune system dysregulation and exaggeration of TH2 cell response, making JAK-STAT inhibitors (or JAK inhibitors) strong theoretical candidates for the treatment of AD.4 In humans, the Janus kinases are composed of 4 different members—JAK1, JAK2, JAK3, and tyrosine kinase 2—all of which can be targeted by JAK inhibitors.5
JAK inhibitors such as tofacitinib have already been approved by the US Food and Drug Administration (FDA) to treat various inflammatory conditions, including rheumatoid arthritis, ulcerative colitis, and psoriatic arthritis; other JAK inhibitors such as baricitinib are only approved for patients with rheumatoid arthritis.6,7 The success of these small molecule inhibitors in these immune-mediated conditions make them attractive candidates for the treatment of AD. Several JAK inhibitors are in phase 2 and phase 3 clinical trials as oral therapies (moderate to severe AD) or as topical treatments (mild to moderate AD). Currently, ruxolitinib (RUX) is the only topical JAK inhibitor that is FDA approved for the treatment of AD in the United States.8 In this editorial, we focus on recent trials of JAK inhibitors tested in patients with AD, including topical RUX, as well as oral abrocitinib, upadacitinib, and baricitinib.
Topical RUX in AD
Ruxolitinib is a topical JAK1/2 small molecule inhibitor approved by the FDA for the treatment of AD in 2021. In a randomized trial by Kim et al9 in 2020, all tested regimens of RUX demonstrated significant improvement in eczema area and severity index (EASI) scores vs vehicle; notably, RUX cream 1.5% applied twice daily achieved the greatest mean percentage change in baseline EASI score vs vehicle at 4 weeks (76.1% vs 15.5%; P<.0001). Ruxolitinib cream was well tolerated through week 8 of the trial, and all adverse events (AEs) were mild to moderate in severity and comparable to those in the vehicle group.9
Topical JAK inhibitors appear to be effective for mild to moderate AD and have had an acceptable safety profile in clinical trials thus far. Although topical corticosteroids and calcineurin inhibitors can have great clinical benefit in AD, they are recommended for short-term use given side effects such as thinning of the skin, burning, or telangiectasia formation.10,11 The hope is that topical JAK inhibitors may be an alternative to standard topical treatments for AD, as they can be used for longer periods due to a safer side-effect profile.
Oral JAK Inhibitors in AD
Several oral JAK inhibitors are undergoing investigation for the systemic treatment of moderate to severe AD. Abrocitinib is an oral JAK1 inhibitor that has demonstrated efficacy in several phase 3 trials in patients with moderate to severe AD. In a 2021 trial, patients were randomized in a 2:2:2:1 ratio to receive abrocitinib 200 mg daily, abrocitinib 100 mg daily, subcutaneous dupilumab 300 mg every other week, or placebo, respectively.12 Patients in both abrocitinib groups showed significant improvement in AD vs placebo, and EASI-75 response was achieved in 70.3%, 58.7%, 58.1%, and 27.1% of patients, respectively (P<.001 for both abrocitinib doses vs placebo). Adverse events occurred more frequently in the abrocitinib 200-mg group vs placebo. Nausea, acne, nasopharyngitis, and headache were the most frequently reported AEs with abrocitinib.12 Another phase 3 trial by Silverberg et al13 (N=391) had similar treatment results, with 38.1% of participants receiving abrocitinib 200 mg and 28.4% of participants receiving abrocitinib 100 mg achieving investigator global assessment scores of 0 (clear) or 1 (almost clear) vs 9.1% of participants receiving placebo (P<.001). Abrocitinib was well tolerated in this trial with few serious AEs (ie, herpangina [0.6%], pneumonia [0.6%]).13 In both trials, there were rare instances of laboratory values indicating thrombocytopenia with the 200-mg dose (0.9%12 and 3.2%13) without any clinical manifestations. Although a decrease in platelets was observed, no thrombocytopenia occurred in the abrocitinib 100-mg group in the latter trial.13
Baricitinib is another oral inhibitor of JAK1 and JAK2 with potential for the treatment of AD. One randomized trial (N=329) demonstrated its efficacy in combination with a topical corticosteroid (TCS). At 16 weeks, a higher number of participants treated with baricitinib and TCS achieved investigator global assessment scores of 0 (clear) or 1 (almost clear) compared to those who received placebo and TCS (31% with baricitinib 4 mg + TCS, 24% with baricitinib 2 mg + TCS, and 15% with placebo + TCS).14 Similarly, in BREEZE-AD5,another phase 3 trial (N=440), baricitinib monotherapy demonstrated a higher rate of treatment success vs placebo.15 Specifically, 13% of patients treated with baricitinib 1 mg and 30% of those treated with baricitinib 2 mg achieved 75% or greater reduction in EASI scores compared to 8% in the placebo group. The most common AEs associated with baricitinib were nasopharyngitis and headache. Adverse events occurred with similar frequency across both experimental and control groups.15 Reich et al14 demonstrated a higher overall rate of AEs—most commonly nasopharyngitis, upper respiratory tract infections, and folliculitis—in baricitinib-treated patients; however, serious AEs occurred with similar frequency across all groups, including the control group.
The selective JAK1 inhibitor upadacitinib also is undergoing testing in treating moderate to severe AD. In one trial, 167 patients were randomized to once daily oral upadacitinib 7.5 mg, 15 mg, or 30 mg or placebo.16 All doses of upadacitinib demonstrated considerably higher percentage improvements from baseline in EASI scores compared to placebo at 16 weeks with a clear dose-response relationship (39%, 62%, and 74% vs 23%, respectively). In this trial, there were no dose-limiting safety events. Serious AEs were infrequent, occurring in 4.8%, 2.4%, and 0% of upadacitinib groups vs 2.5% for placebo. The serious AEs observed with upadacitinib were 1 case of appendicitis, lower jaw pericoronitis in a patient with a history of repeated tooth infections, and an exacerbation of AD.16
Tofacitinib, another JAK inhibitor, has been shown to increase the risk for blood clots and death in a large trial in the treatment of rheumatoid arthritis. Following this study, the FDA is requiring black box warnings for tofacitinib and also for the 2 JAK inhibitors baricitinib and upadacitinib regarding the risks for heart-related events, cancer, blood clots, and death. Given that these medications share a similar mechanism of action to tofacitinib, they may have similar risks, though they have not yet been fully evaluated in large safety trials.17
With more recent investigation into novel therapeutics for AD, oral JAK inhibitors may play an important role in the future to treat patients with moderate to severe AD with inadequate response or contraindications to other systemic therapies. In trials thus far, oral JAK inhibitors have exhibited acceptable safety profiles and have demonstrated treatment success in AD. More randomized, controlled, phase 3 studies with larger patient populations are required to confirm their potential as effective treatments and elucidate their long-term safety.
Deucravacitinib in Psoriasis
Deucravacitinib is a first-in-class, oral, selective TYK2 inhibitor currently undergoing testing for the treatment of psoriasis. A randomized phase 2 trial (N=267) found that deucravacitinib was more effective than placebo in treating chronic plaque psoriasis at doses of 3 to 12 mg daily.18 The percentage of participants with a 75% or greater reduction from baseline in the psoriasis area and severity index score was 7% with placebo, 9% with deucravacitinib 3 mg every other day (P=.49 vs placebo), 39% with 3 mg once daily (P<.001 vs placebo), 69% with 3 mg twice daily (P<.001 vs placebo), 67% with 6 mg twice daily (P<.001 vs placebo), and 75% with 12 mg once daily (P<.001 vs placebo). The most commonly reported AEs were nasopharyngitis, headache, diarrhea, nausea, and upper respiratory tract infection. Adverse events occurred in 51% of participants in the control group and in 55% to 80% of those in the experimental groups. Additionally, there was 1 reported case of melanoma (stage 0) 96 days after the start of treatment in a patient in the 3-mg once-daily group. Serious AEs occurred in only 0% to 2% of participants who received deucravacitinib.18
Two phase 3 trials—POETYK PSO-1 and POETYK PSO-2 (N=1686)—found deucravacitinib to be notably more effective than both placebo and apremilast in treating psoriasis.19 Among participants receiving deucravacitinib 6 mg daily, 58.7% and 53.6% in the 2 respective trials achieved psoriasis area and severity index 75 response vs 12.7% and 9.4% receiving placebo and 35.1% and 40.2% receiving apremilast. Overall, the treatment was well tolerated, with a low rate of discontinuation of deucravacitinib due to AEs (2.4% of patients on deucravacitinib compared to 3.8% on placebo and 5.2% on apremilast). The most frequently observed AEs with deucravacitinib were nasopharyngitis and upper respiratory tract infection. The full results of these trials are expected to be published soon.19,20
Final Thoughts
Overall, JAK inhibitors are a novel class of therapeutics that may have further success in the treatment of other dermatologic conditions that negatively affect patients’ quality of life and productivity. We should look forward to additional successful trials with these promising medications.
- Chiesa Fuxench ZC, Block JK, Boguniewicz M, et al. Atopic dermatitis in America study: a cross-sectional study examining the prevalence and disease burden of atopic dermatitis in the US adult population. J Invest Dermatol. 2019;139:583-590.
- Silverberg JI , Simpson EL. Associations of childhood eczema severity: a US population-based study. Dermatitis. 2014;25:107-114.
- Schonmann Y, Mansfield KE, Hayes JF, et al. Atopic eczema in adulthood and risk of depression and anxiety: a population-based cohort study. J Allergy Clin Immunol Pract. 2020;8:248-257.e16.
- Bao L, Zhang H, Chan LS. The involvement of the JAK-STAT signaling pathway in chronic inflammatory skin disease atopic dermatitis. JAKSTAT. 2013;2:e24137.
- Villarino AV, Kanno Y, O’Shea JJ. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat Immunol. 2017;18:374-384.
- Xeljanz FDA approval history. Drugs.com website. Updated December 14, 2021. Accessed February 16, 2022. https://www.drugs.com/history/xeljanz.html
- Mullard A. FDA approves Eli Lilly’s baricitinib. Nat Rev Drug Discov. 2018;17:460.
- FDA approves Opzelura. Drugs.com website. Published September 2021. Accessed February 16, 2022. https://www.drugs.com/newdrugs/fda-approves-opzelura-ruxolitinib-cream-atopic-dermatitis-ad-5666.html
- Kim BS, Sun K, Papp K, et al. Effects of ruxolitinib cream on pruritus and quality of life in atopic dermatitis: results from a phase 2, randomized, dose-ranging, vehicle- and active-controlled study.J Am Acad Dermatol. 2020;82:1305-1313.
- Eichenfield LF, Tom WL, Berger TG, et al. Guidelines of care for the management of atopic dermatitis: section 2, management and treatment of atopic dermatitis with topical therapies. J Am Acad Dermatol. 2014;71:116-132.
- Wollenberg A, Barbarot S, Bieber T, et al. Consensus-based European guidelines for treatment of atopic eczema (atopic dermatitis) in adults and children: part I. J Eur Acad Dermatol Venereol. 2018;32:657-682.
- Bieber T, Simpson EL, Silverberg JI, et al. Abrocitinib versus placebo or dupilumab for atopic dermatitis. N Engl J Med. 2021;384:1101-1112.
- Silverberg JI, Simpson EL, Thyssen JP, et al. Efficacy and safety of abrocitinib in patients with moderate-to-severe atopic dermatitis: a randomized clinical trial. JAMA Dermatol. 2020;156:863-873.
- Reich K, Kabashima K, Peris K, et al. Efficacy and safety of baricitinib combined with topical corticosteroids for treatment of moderate to severe atopic dermatitis: a randomized clinical trial. JAMA Dermatol. 2020;156:1333-1343.
- Simpson EL, Forman S, Silverberg JI, et al. Baricitinib in patients with moderate-to-severe atopic dermatitis: results from a randomized monotherapy phase 3 trial in the United States and Canada (BREEZE-AD5). J Am Acad Dermatol. 2021;85:62-70.
- Guttman-Yassky E, Thaçi D, Pangan AL, et al. Upadacitinib in adults with moderate to severe atopic dermatitis: 16-week results from a randomized, placebo-controlled trial. J Allergy Clin Immunol. 2020;145:877-884.
- US Food and Drug Administration. FDA requires warnings about increased risk of serious heart-related events, cancer, blood clots, and death for JAK inhibitors that treat certain chronic inflammatory conditions. Published September 1, 2022. Accessed February 16, 2022. https://www.fda.gov/drugs/drug-safety-and-availability/fda-requires-warnings-about-increased-risk-serious-heart-related-events-cancer-blood-clots-and-death
- Papp K, Gordon K, Thaçi D, et al. Phase 2 trial of selective tyrosine kinase 2 inhibition in psoriasis. N Engl J Med. 2018;379:1313-1321.
- Bristol Myers Squibb presents positive data from two pivotal phase 3 psoriasis studies demonstrating superiority of deucravacitinib compared to placebo and Otezla® (apremilast). Press release. Bristol Meyers Squibb. April 23, 2021. Accessed February 16, 2022. https://news.bms.com/news/details/2021/Bristol-Myers-Squibb-Presents-Positive-Data-from-Two-Pivotal-Phase-3-Psoriasis-Studies-Demonstrating-Superiority-of-Deucravacitinib-Compared-to-Placebo-and-Otezla-apremilast/default.aspx
- Armstrong A, Gooderham M, Warren R, et al. Efficacy and safety of deucravacitinib, an oral, selective tyrosine kinase 2 (TYK2) inhibitor, compared with placebo and apremilast in moderate to severe plaque psoriasis: results from the POETYK PSO-1 study [abstract]. Abstract presented at: 2021 American Academy of Dermatology annual meeting; April 23-25, 2021; San Francisco, California.
- Chiesa Fuxench ZC, Block JK, Boguniewicz M, et al. Atopic dermatitis in America study: a cross-sectional study examining the prevalence and disease burden of atopic dermatitis in the US adult population. J Invest Dermatol. 2019;139:583-590.
- Silverberg JI , Simpson EL. Associations of childhood eczema severity: a US population-based study. Dermatitis. 2014;25:107-114.
- Schonmann Y, Mansfield KE, Hayes JF, et al. Atopic eczema in adulthood and risk of depression and anxiety: a population-based cohort study. J Allergy Clin Immunol Pract. 2020;8:248-257.e16.
- Bao L, Zhang H, Chan LS. The involvement of the JAK-STAT signaling pathway in chronic inflammatory skin disease atopic dermatitis. JAKSTAT. 2013;2:e24137.
- Villarino AV, Kanno Y, O’Shea JJ. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat Immunol. 2017;18:374-384.
- Xeljanz FDA approval history. Drugs.com website. Updated December 14, 2021. Accessed February 16, 2022. https://www.drugs.com/history/xeljanz.html
- Mullard A. FDA approves Eli Lilly’s baricitinib. Nat Rev Drug Discov. 2018;17:460.
- FDA approves Opzelura. Drugs.com website. Published September 2021. Accessed February 16, 2022. https://www.drugs.com/newdrugs/fda-approves-opzelura-ruxolitinib-cream-atopic-dermatitis-ad-5666.html
- Kim BS, Sun K, Papp K, et al. Effects of ruxolitinib cream on pruritus and quality of life in atopic dermatitis: results from a phase 2, randomized, dose-ranging, vehicle- and active-controlled study.J Am Acad Dermatol. 2020;82:1305-1313.
- Eichenfield LF, Tom WL, Berger TG, et al. Guidelines of care for the management of atopic dermatitis: section 2, management and treatment of atopic dermatitis with topical therapies. J Am Acad Dermatol. 2014;71:116-132.
- Wollenberg A, Barbarot S, Bieber T, et al. Consensus-based European guidelines for treatment of atopic eczema (atopic dermatitis) in adults and children: part I. J Eur Acad Dermatol Venereol. 2018;32:657-682.
- Bieber T, Simpson EL, Silverberg JI, et al. Abrocitinib versus placebo or dupilumab for atopic dermatitis. N Engl J Med. 2021;384:1101-1112.
- Silverberg JI, Simpson EL, Thyssen JP, et al. Efficacy and safety of abrocitinib in patients with moderate-to-severe atopic dermatitis: a randomized clinical trial. JAMA Dermatol. 2020;156:863-873.
- Reich K, Kabashima K, Peris K, et al. Efficacy and safety of baricitinib combined with topical corticosteroids for treatment of moderate to severe atopic dermatitis: a randomized clinical trial. JAMA Dermatol. 2020;156:1333-1343.
- Simpson EL, Forman S, Silverberg JI, et al. Baricitinib in patients with moderate-to-severe atopic dermatitis: results from a randomized monotherapy phase 3 trial in the United States and Canada (BREEZE-AD5). J Am Acad Dermatol. 2021;85:62-70.
- Guttman-Yassky E, Thaçi D, Pangan AL, et al. Upadacitinib in adults with moderate to severe atopic dermatitis: 16-week results from a randomized, placebo-controlled trial. J Allergy Clin Immunol. 2020;145:877-884.
- US Food and Drug Administration. FDA requires warnings about increased risk of serious heart-related events, cancer, blood clots, and death for JAK inhibitors that treat certain chronic inflammatory conditions. Published September 1, 2022. Accessed February 16, 2022. https://www.fda.gov/drugs/drug-safety-and-availability/fda-requires-warnings-about-increased-risk-serious-heart-related-events-cancer-blood-clots-and-death
- Papp K, Gordon K, Thaçi D, et al. Phase 2 trial of selective tyrosine kinase 2 inhibition in psoriasis. N Engl J Med. 2018;379:1313-1321.
- Bristol Myers Squibb presents positive data from two pivotal phase 3 psoriasis studies demonstrating superiority of deucravacitinib compared to placebo and Otezla® (apremilast). Press release. Bristol Meyers Squibb. April 23, 2021. Accessed February 16, 2022. https://news.bms.com/news/details/2021/Bristol-Myers-Squibb-Presents-Positive-Data-from-Two-Pivotal-Phase-3-Psoriasis-Studies-Demonstrating-Superiority-of-Deucravacitinib-Compared-to-Placebo-and-Otezla-apremilast/default.aspx
- Armstrong A, Gooderham M, Warren R, et al. Efficacy and safety of deucravacitinib, an oral, selective tyrosine kinase 2 (TYK2) inhibitor, compared with placebo and apremilast in moderate to severe plaque psoriasis: results from the POETYK PSO-1 study [abstract]. Abstract presented at: 2021 American Academy of Dermatology annual meeting; April 23-25, 2021; San Francisco, California.
Discoid Lupus

THE COMPARISON
A Multicolored (pink, brown, and white) indurated plaques in a butterfly distribution on the face of a 30-year-old woman with a darker skin tone.
B Pink, elevated, indurated plaques with hypopigmentation in a butterfly distribution on the face of a 19-year-old woman with a lighter skin tone.
Cutaneous lupus erythematosus may occur with or without systemic lupus erythematosus. Discoid lupus erythematosus (DLE), a form of chronic cutaneous lupus, is most commonly found on the scalp, face, and ears.1
Epidemiology
Discoid lupus erythematosus is most common in adult women (age range, 20–40 years).2 It occurs more frequently in women of African descent.3,4
Key clinical features in people with darker skin tones:
Clinical features of DLE lesions include erythema, induration, follicular plugging, dyspigmentation, and scarring alopecia.1 In patients of African descent, lesions may be annular and hypopigmented to depigmented centrally with a border of hyperpigmentation. Active lesions may be painful and/or pruritic.2
Discoid lupus erythematosus lesions occur in photodistributed areas, although not exclusively. Photoprotective clothing and sunscreen are an important part of the treatment plan.1 Although sunscreen is recommended for patients with DLE, those with darker skin tones may find some sunscreens cosmetically unappealing due to a mismatch with their normal skin color.5 Tinted sunscreens may be beneficial additions.
Worth noting
Approximately 5% to 25% of patients with cutaneous lupus go on to develop systemic lupus erythematosus.6
Health disparity highlight
Discoid lesions may cause cutaneous scars that are quite disfiguring and may negatively impact quality of life. Some patients may have a few scattered lesions, whereas others have extensive disease covering most of the scalp. Discoid lupus erythematosus lesions of the scalp have classic clinical features including hair loss, erythema, hypopigmentation, and hyperpigmentation. The clinician’s comfort with performing a scalp examination with cultural humility is an important acquired skill and is especially important when the examination is performed on patients with more tightly coiled hair.7 For example, physicians may adopt the “compliment, discuss, and suggest” method when counseling patients.8
- Bolognia JL, Jorizzo JJ, Schaffer JV, et al. Dermatology. 3rd ed. Elsevier; 2012.
- Otberg N, Wu W-Y, McElwee KJ, et al. Diagnosis and management of primary cicatricial alopecia: part I. Skinmed. 2008;7:19-26. doi:10.1111/j.1540-9740.2007.07163.x
- Callen JP. Chronic cutaneous lupus erythematosus. clinical, laboratory, therapeutic, and prognostic examination of 62 patients. Arch Dermatol. 1982;118:412-416. doi:10.1001/archderm.118.6.412
- McCarty DJ, Manzi S, Medsger TA Jr, et al. Incidence of systemic lupus erythematosus. race and gender differences. Arthritis Rheum. 1995;38:1260-1270. doi:10.1002/art.1780380914
- Morquette AJ, Waples ER, Heath CR. The importance of cosmetically elegant sunscreen in skin of color populations. J Cosmet Dermatol. In press.
- Zhou W, Wu H, Zhao M, et al. New insights into the progression from cutaneous lupus to systemic lupus erythematosus. Expert Rev Clin Immunol. 2020;16:829-837. doi:10.1080/17446 66X.2020.1805316
- Grayson C, Heath C. An approach to examining tightly coiled hair among patients with hair loss in race-discordant patientphysician interactions. JAMA Dermatol. 2021;157:505-506. doi:10.1001/jamadermatol.2021.0338
- Grayson C, Heath CR. Counseling about traction alopecia: a “compliment, discuss, and suggest” method. Cutis. 2021;108:20-22.
THE COMPARISON
A Multicolored (pink, brown, and white) indurated plaques in a butterfly distribution on the face of a 30-year-old woman with a darker skin tone.
B Pink, elevated, indurated plaques with hypopigmentation in a butterfly distribution on the face of a 19-year-old woman with a lighter skin tone.
Cutaneous lupus erythematosus may occur with or without systemic lupus erythematosus. Discoid lupus erythematosus (DLE), a form of chronic cutaneous lupus, is most commonly found on the scalp, face, and ears.1
Epidemiology
Discoid lupus erythematosus is most common in adult women (age range, 20–40 years).2 It occurs more frequently in women of African descent.3,4
Key clinical features in people with darker skin tones:
Clinical features of DLE lesions include erythema, induration, follicular plugging, dyspigmentation, and scarring alopecia.1 In patients of African descent, lesions may be annular and hypopigmented to depigmented centrally with a border of hyperpigmentation. Active lesions may be painful and/or pruritic.2
Discoid lupus erythematosus lesions occur in photodistributed areas, although not exclusively. Photoprotective clothing and sunscreen are an important part of the treatment plan.1 Although sunscreen is recommended for patients with DLE, those with darker skin tones may find some sunscreens cosmetically unappealing due to a mismatch with their normal skin color.5 Tinted sunscreens may be beneficial additions.
Worth noting
Approximately 5% to 25% of patients with cutaneous lupus go on to develop systemic lupus erythematosus.6
Health disparity highlight
Discoid lesions may cause cutaneous scars that are quite disfiguring and may negatively impact quality of life. Some patients may have a few scattered lesions, whereas others have extensive disease covering most of the scalp. Discoid lupus erythematosus lesions of the scalp have classic clinical features including hair loss, erythema, hypopigmentation, and hyperpigmentation. The clinician’s comfort with performing a scalp examination with cultural humility is an important acquired skill and is especially important when the examination is performed on patients with more tightly coiled hair.7 For example, physicians may adopt the “compliment, discuss, and suggest” method when counseling patients.8
THE COMPARISON
A Multicolored (pink, brown, and white) indurated plaques in a butterfly distribution on the face of a 30-year-old woman with a darker skin tone.
B Pink, elevated, indurated plaques with hypopigmentation in a butterfly distribution on the face of a 19-year-old woman with a lighter skin tone.
Cutaneous lupus erythematosus may occur with or without systemic lupus erythematosus. Discoid lupus erythematosus (DLE), a form of chronic cutaneous lupus, is most commonly found on the scalp, face, and ears.1
Epidemiology
Discoid lupus erythematosus is most common in adult women (age range, 20–40 years).2 It occurs more frequently in women of African descent.3,4
Key clinical features in people with darker skin tones:
Clinical features of DLE lesions include erythema, induration, follicular plugging, dyspigmentation, and scarring alopecia.1 In patients of African descent, lesions may be annular and hypopigmented to depigmented centrally with a border of hyperpigmentation. Active lesions may be painful and/or pruritic.2
Discoid lupus erythematosus lesions occur in photodistributed areas, although not exclusively. Photoprotective clothing and sunscreen are an important part of the treatment plan.1 Although sunscreen is recommended for patients with DLE, those with darker skin tones may find some sunscreens cosmetically unappealing due to a mismatch with their normal skin color.5 Tinted sunscreens may be beneficial additions.
Worth noting
Approximately 5% to 25% of patients with cutaneous lupus go on to develop systemic lupus erythematosus.6
Health disparity highlight
Discoid lesions may cause cutaneous scars that are quite disfiguring and may negatively impact quality of life. Some patients may have a few scattered lesions, whereas others have extensive disease covering most of the scalp. Discoid lupus erythematosus lesions of the scalp have classic clinical features including hair loss, erythema, hypopigmentation, and hyperpigmentation. The clinician’s comfort with performing a scalp examination with cultural humility is an important acquired skill and is especially important when the examination is performed on patients with more tightly coiled hair.7 For example, physicians may adopt the “compliment, discuss, and suggest” method when counseling patients.8
- Bolognia JL, Jorizzo JJ, Schaffer JV, et al. Dermatology. 3rd ed. Elsevier; 2012.
- Otberg N, Wu W-Y, McElwee KJ, et al. Diagnosis and management of primary cicatricial alopecia: part I. Skinmed. 2008;7:19-26. doi:10.1111/j.1540-9740.2007.07163.x
- Callen JP. Chronic cutaneous lupus erythematosus. clinical, laboratory, therapeutic, and prognostic examination of 62 patients. Arch Dermatol. 1982;118:412-416. doi:10.1001/archderm.118.6.412
- McCarty DJ, Manzi S, Medsger TA Jr, et al. Incidence of systemic lupus erythematosus. race and gender differences. Arthritis Rheum. 1995;38:1260-1270. doi:10.1002/art.1780380914
- Morquette AJ, Waples ER, Heath CR. The importance of cosmetically elegant sunscreen in skin of color populations. J Cosmet Dermatol. In press.
- Zhou W, Wu H, Zhao M, et al. New insights into the progression from cutaneous lupus to systemic lupus erythematosus. Expert Rev Clin Immunol. 2020;16:829-837. doi:10.1080/17446 66X.2020.1805316
- Grayson C, Heath C. An approach to examining tightly coiled hair among patients with hair loss in race-discordant patientphysician interactions. JAMA Dermatol. 2021;157:505-506. doi:10.1001/jamadermatol.2021.0338
- Grayson C, Heath CR. Counseling about traction alopecia: a “compliment, discuss, and suggest” method. Cutis. 2021;108:20-22.
- Bolognia JL, Jorizzo JJ, Schaffer JV, et al. Dermatology. 3rd ed. Elsevier; 2012.
- Otberg N, Wu W-Y, McElwee KJ, et al. Diagnosis and management of primary cicatricial alopecia: part I. Skinmed. 2008;7:19-26. doi:10.1111/j.1540-9740.2007.07163.x
- Callen JP. Chronic cutaneous lupus erythematosus. clinical, laboratory, therapeutic, and prognostic examination of 62 patients. Arch Dermatol. 1982;118:412-416. doi:10.1001/archderm.118.6.412
- McCarty DJ, Manzi S, Medsger TA Jr, et al. Incidence of systemic lupus erythematosus. race and gender differences. Arthritis Rheum. 1995;38:1260-1270. doi:10.1002/art.1780380914
- Morquette AJ, Waples ER, Heath CR. The importance of cosmetically elegant sunscreen in skin of color populations. J Cosmet Dermatol. In press.
- Zhou W, Wu H, Zhao M, et al. New insights into the progression from cutaneous lupus to systemic lupus erythematosus. Expert Rev Clin Immunol. 2020;16:829-837. doi:10.1080/17446 66X.2020.1805316
- Grayson C, Heath C. An approach to examining tightly coiled hair among patients with hair loss in race-discordant patientphysician interactions. JAMA Dermatol. 2021;157:505-506. doi:10.1001/jamadermatol.2021.0338
- Grayson C, Heath CR. Counseling about traction alopecia: a “compliment, discuss, and suggest” method. Cutis. 2021;108:20-22.
 indurated plaques in a butterfly distribution on the face of a 30-year-old woman with a darker skin tone")
Leukemia Cutis Manifesting as Nonpalpable Purpura
To the Editor:
A 72-year-old man presented with symptomatic anemia and nonpalpable purpura of the legs, abdomen, and arms of 2 weeks’ duration (Figure 1). There were no associated perifollicular papules. Physical examination of the hair and gingiva were normal.

The patient’s medical history was notable for a poorly differentiated pancreatic adenocarcinoma (pT3N1M0) resected 7 months prior using a Whipple operation (pancreaticoduodenectomy). Adjuvant therapy consisted of 5 cycles of intravenous gemcitabine and paclitaxel. Treatment was discontinued 1 month prior due to progressive weight loss and the presence of new liver metastases on computed tomography. There was no recent history of corticosteroid, antiplatelet, or anticoagulant use. The patient had no known history of trauma at the affected sites.
The patient’s laboratory workup revealed the following results: hemoglobin, 5.5 g/dL (reference range, 13–18 g/dL); platelets, 128×109/L (reference range, 150–400×109/L); total white blood cell count (24.0×109/L [reference range, 4.0–11.0×109/L]), consisting of neutrophils (2.4×109/L [reference range, 2.0–7.5×109/L]), lymphocytes (3.1×109/L [reference range, 1.5–4.0×109/L]), and monocytes (18.5×109/L [reference range, 0.2–0.8×109/L]). Fibrinogen, activated partial thromboplastin time, and prothrombin time were within reference range. Results of a bone marrow biopsy showed 64% blasts. The lactate dehydrogenase level was 286 U/L (reference range, 135–220 U/L) and CA-19-9 antigen was 238 U/mL (reference range, 0–39 U/mL).

Results from a skin punch biopsy from the right leg showed a normal epidermis and papillary dermis. The reticular dermis was expanded by a diffuse cellular infiltrate with dermal edema and separation of collagen bundles (Figure 2), which consisted of small cells with irregular, cleaved, and notched nuclei, containing a variable amount of eosinophilic cytoplasm. Mitotic figures were present (Figure 3). There was no evidence of vasculitis, and Congo red stain for amyloid was negative. These atypical cells were positive for the leukocyte common antigen, favoring a hematopoietic infiltrate (Figure 4). Other positive markers included CD4 (associated with helper T cells, and mature and immature monocytes), CD68 (a monocyte/macrophage marker), and CD56 (associated with natural killer cells, myeloma, acute myeloid leukemia [AML], and neuroendocrine tumors). The cells were negative for CD3 (T-cell lineage–specific antigen), CD5 (marker of T cells and a subset of IgM-secreting B cells), CD34 (early hematopoietic marker), and CD20 (B-cell marker). Other negative myeloid markers included myeloperoxidase, CD117, and CD138. These findings suggested leukemic cell recruitment at the site of a reactive infiltrate. The patient completed 2 cycles of intravenous azacitidine with little response and subsequently opted for palliative measures.
 and numerous notched nuclei")
Nonpalpable purpura has a broad differential diagnosis including primary and secondary thrombocytopenia; coagulopathies, including vitamin K deficiency, specific clotting factor deficiencies, and amyloid-related purpura; genetic or acquired collagen disorders, including vitamin C deficiency; and eruptions induced by drugs and herbal remedies.

Leukemia cutis is a relatively rare cause of purpura and is defined as cutaneous infiltration by neoplastic leucocytes.1 It most commonly is associated with AML and complicates approximately 5% to 15%of all adult cases.2 Cutaneous involvement occurs predominantly in monocytic variants; acute myelomonocytic leukemia and acute monocytic leukemia may arise in up to 50% of these cases.3 The clinical presentation may vary from papules, nodules, and plaques to rarer manifestations including purpura. A leukemic infiltrate often is associated with sites of inflammation, such as infection or ulceration,4 though there was no reported history of any known triggering events in our patient. Lesions usually involve the legs, followed by the arms, back, chest, scalp, and face.4 One-third of cases coincide with systemic symptoms, and approximately 10% precede bone marrow or peripheral blood involvement, referred to as aleukemic leukemia. The remainder of cases arise following an established diagnosis of systemic leukemia.5 Leukemia cutis is considered a marker of poor prognosis in AML,4,6 requiring treatment for the underlying systemic disease. Our case also was complicated by a concurrent pancreatic malignancy and relatively advanced age, which limited the feasibility of further treatment.
- Strutton G. Cutaneous infiltrates: lymphomatous and leukemic. In: Weedon D, ed. Skin Pathology. 2nd ed. Churchill Livingstone; 2002:1118-1120.
- Cho-Vega JH, Medeiros LJ, Prieto VG, et al. Leukemia cutis. Am J Clin Pathol. 2008;129:130-142.
- Kaddu S, Zenahlik P, Beham-Schmid C, et al. Specific cutaneous infiltrates in patients with myelogenous leukemia: a clinicopathologic study of 26 patients with assessment of diagnostic criteria. J Am Acad Dermatol. 1999;40:966-978.
- Paydas S, Zorludemir S. Leukaemia cutis and leukaemic vasculitis. Br J Dermatol. 2000;143:773-779.
- Shaikh BS, Frantz E, Lookingbill DP. Histologically proven leukemia cutis carries a poor prognosis in acute nonlymphocytic leukemia. Cutis. 1987;39:57-60.
- Su WP. Clinical, histopathologic, and immunohistochemical correlations in leukemia cutis. Semin Dermatol. 1994;13:223-230.
To the Editor:
A 72-year-old man presented with symptomatic anemia and nonpalpable purpura of the legs, abdomen, and arms of 2 weeks’ duration (Figure 1). There were no associated perifollicular papules. Physical examination of the hair and gingiva were normal.
The patient’s medical history was notable for a poorly differentiated pancreatic adenocarcinoma (pT3N1M0) resected 7 months prior using a Whipple operation (pancreaticoduodenectomy). Adjuvant therapy consisted of 5 cycles of intravenous gemcitabine and paclitaxel. Treatment was discontinued 1 month prior due to progressive weight loss and the presence of new liver metastases on computed tomography. There was no recent history of corticosteroid, antiplatelet, or anticoagulant use. The patient had no known history of trauma at the affected sites.
The patient’s laboratory workup revealed the following results: hemoglobin, 5.5 g/dL (reference range, 13–18 g/dL); platelets, 128×109/L (reference range, 150–400×109/L); total white blood cell count (24.0×109/L [reference range, 4.0–11.0×109/L]), consisting of neutrophils (2.4×109/L [reference range, 2.0–7.5×109/L]), lymphocytes (3.1×109/L [reference range, 1.5–4.0×109/L]), and monocytes (18.5×109/L [reference range, 0.2–0.8×109/L]). Fibrinogen, activated partial thromboplastin time, and prothrombin time were within reference range. Results of a bone marrow biopsy showed 64% blasts. The lactate dehydrogenase level was 286 U/L (reference range, 135–220 U/L) and CA-19-9 antigen was 238 U/mL (reference range, 0–39 U/mL).
Results from a skin punch biopsy from the right leg showed a normal epidermis and papillary dermis. The reticular dermis was expanded by a diffuse cellular infiltrate with dermal edema and separation of collagen bundles (Figure 2), which consisted of small cells with irregular, cleaved, and notched nuclei, containing a variable amount of eosinophilic cytoplasm. Mitotic figures were present (Figure 3). There was no evidence of vasculitis, and Congo red stain for amyloid was negative. These atypical cells were positive for the leukocyte common antigen, favoring a hematopoietic infiltrate (Figure 4). Other positive markers included CD4 (associated with helper T cells, and mature and immature monocytes), CD68 (a monocyte/macrophage marker), and CD56 (associated with natural killer cells, myeloma, acute myeloid leukemia [AML], and neuroendocrine tumors). The cells were negative for CD3 (T-cell lineage–specific antigen), CD5 (marker of T cells and a subset of IgM-secreting B cells), CD34 (early hematopoietic marker), and CD20 (B-cell marker). Other negative myeloid markers included myeloperoxidase, CD117, and CD138. These findings suggested leukemic cell recruitment at the site of a reactive infiltrate. The patient completed 2 cycles of intravenous azacitidine with little response and subsequently opted for palliative measures.
Nonpalpable purpura has a broad differential diagnosis including primary and secondary thrombocytopenia; coagulopathies, including vitamin K deficiency, specific clotting factor deficiencies, and amyloid-related purpura; genetic or acquired collagen disorders, including vitamin C deficiency; and eruptions induced by drugs and herbal remedies.
Leukemia cutis is a relatively rare cause of purpura and is defined as cutaneous infiltration by neoplastic leucocytes.1 It most commonly is associated with AML and complicates approximately 5% to 15%of all adult cases.2 Cutaneous involvement occurs predominantly in monocytic variants; acute myelomonocytic leukemia and acute monocytic leukemia may arise in up to 50% of these cases.3 The clinical presentation may vary from papules, nodules, and plaques to rarer manifestations including purpura. A leukemic infiltrate often is associated with sites of inflammation, such as infection or ulceration,4 though there was no reported history of any known triggering events in our patient. Lesions usually involve the legs, followed by the arms, back, chest, scalp, and face.4 One-third of cases coincide with systemic symptoms, and approximately 10% precede bone marrow or peripheral blood involvement, referred to as aleukemic leukemia. The remainder of cases arise following an established diagnosis of systemic leukemia.5 Leukemia cutis is considered a marker of poor prognosis in AML,4,6 requiring treatment for the underlying systemic disease. Our case also was complicated by a concurrent pancreatic malignancy and relatively advanced age, which limited the feasibility of further treatment.
To the Editor:
A 72-year-old man presented with symptomatic anemia and nonpalpable purpura of the legs, abdomen, and arms of 2 weeks’ duration (Figure 1). There were no associated perifollicular papules. Physical examination of the hair and gingiva were normal.
The patient’s medical history was notable for a poorly differentiated pancreatic adenocarcinoma (pT3N1M0) resected 7 months prior using a Whipple operation (pancreaticoduodenectomy). Adjuvant therapy consisted of 5 cycles of intravenous gemcitabine and paclitaxel. Treatment was discontinued 1 month prior due to progressive weight loss and the presence of new liver metastases on computed tomography. There was no recent history of corticosteroid, antiplatelet, or anticoagulant use. The patient had no known history of trauma at the affected sites.
The patient’s laboratory workup revealed the following results: hemoglobin, 5.5 g/dL (reference range, 13–18 g/dL); platelets, 128×109/L (reference range, 150–400×109/L); total white blood cell count (24.0×109/L [reference range, 4.0–11.0×109/L]), consisting of neutrophils (2.4×109/L [reference range, 2.0–7.5×109/L]), lymphocytes (3.1×109/L [reference range, 1.5–4.0×109/L]), and monocytes (18.5×109/L [reference range, 0.2–0.8×109/L]). Fibrinogen, activated partial thromboplastin time, and prothrombin time were within reference range. Results of a bone marrow biopsy showed 64% blasts. The lactate dehydrogenase level was 286 U/L (reference range, 135–220 U/L) and CA-19-9 antigen was 238 U/mL (reference range, 0–39 U/mL).
Results from a skin punch biopsy from the right leg showed a normal epidermis and papillary dermis. The reticular dermis was expanded by a diffuse cellular infiltrate with dermal edema and separation of collagen bundles (Figure 2), which consisted of small cells with irregular, cleaved, and notched nuclei, containing a variable amount of eosinophilic cytoplasm. Mitotic figures were present (Figure 3). There was no evidence of vasculitis, and Congo red stain for amyloid was negative. These atypical cells were positive for the leukocyte common antigen, favoring a hematopoietic infiltrate (Figure 4). Other positive markers included CD4 (associated with helper T cells, and mature and immature monocytes), CD68 (a monocyte/macrophage marker), and CD56 (associated with natural killer cells, myeloma, acute myeloid leukemia [AML], and neuroendocrine tumors). The cells were negative for CD3 (T-cell lineage–specific antigen), CD5 (marker of T cells and a subset of IgM-secreting B cells), CD34 (early hematopoietic marker), and CD20 (B-cell marker). Other negative myeloid markers included myeloperoxidase, CD117, and CD138. These findings suggested leukemic cell recruitment at the site of a reactive infiltrate. The patient completed 2 cycles of intravenous azacitidine with little response and subsequently opted for palliative measures.
Nonpalpable purpura has a broad differential diagnosis including primary and secondary thrombocytopenia; coagulopathies, including vitamin K deficiency, specific clotting factor deficiencies, and amyloid-related purpura; genetic or acquired collagen disorders, including vitamin C deficiency; and eruptions induced by drugs and herbal remedies.
Leukemia cutis is a relatively rare cause of purpura and is defined as cutaneous infiltration by neoplastic leucocytes.1 It most commonly is associated with AML and complicates approximately 5% to 15%of all adult cases.2 Cutaneous involvement occurs predominantly in monocytic variants; acute myelomonocytic leukemia and acute monocytic leukemia may arise in up to 50% of these cases.3 The clinical presentation may vary from papules, nodules, and plaques to rarer manifestations including purpura. A leukemic infiltrate often is associated with sites of inflammation, such as infection or ulceration,4 though there was no reported history of any known triggering events in our patient. Lesions usually involve the legs, followed by the arms, back, chest, scalp, and face.4 One-third of cases coincide with systemic symptoms, and approximately 10% precede bone marrow or peripheral blood involvement, referred to as aleukemic leukemia. The remainder of cases arise following an established diagnosis of systemic leukemia.5 Leukemia cutis is considered a marker of poor prognosis in AML,4,6 requiring treatment for the underlying systemic disease. Our case also was complicated by a concurrent pancreatic malignancy and relatively advanced age, which limited the feasibility of further treatment.
- Strutton G. Cutaneous infiltrates: lymphomatous and leukemic. In: Weedon D, ed. Skin Pathology. 2nd ed. Churchill Livingstone; 2002:1118-1120.
- Cho-Vega JH, Medeiros LJ, Prieto VG, et al. Leukemia cutis. Am J Clin Pathol. 2008;129:130-142.
- Kaddu S, Zenahlik P, Beham-Schmid C, et al. Specific cutaneous infiltrates in patients with myelogenous leukemia: a clinicopathologic study of 26 patients with assessment of diagnostic criteria. J Am Acad Dermatol. 1999;40:966-978.
- Paydas S, Zorludemir S. Leukaemia cutis and leukaemic vasculitis. Br J Dermatol. 2000;143:773-779.
- Shaikh BS, Frantz E, Lookingbill DP. Histologically proven leukemia cutis carries a poor prognosis in acute nonlymphocytic leukemia. Cutis. 1987;39:57-60.
- Su WP. Clinical, histopathologic, and immunohistochemical correlations in leukemia cutis. Semin Dermatol. 1994;13:223-230.
- Strutton G. Cutaneous infiltrates: lymphomatous and leukemic. In: Weedon D, ed. Skin Pathology. 2nd ed. Churchill Livingstone; 2002:1118-1120.
- Cho-Vega JH, Medeiros LJ, Prieto VG, et al. Leukemia cutis. Am J Clin Pathol. 2008;129:130-142.
- Kaddu S, Zenahlik P, Beham-Schmid C, et al. Specific cutaneous infiltrates in patients with myelogenous leukemia: a clinicopathologic study of 26 patients with assessment of diagnostic criteria. J Am Acad Dermatol. 1999;40:966-978.
- Paydas S, Zorludemir S. Leukaemia cutis and leukaemic vasculitis. Br J Dermatol. 2000;143:773-779.
- Shaikh BS, Frantz E, Lookingbill DP. Histologically proven leukemia cutis carries a poor prognosis in acute nonlymphocytic leukemia. Cutis. 1987;39:57-60.
- Su WP. Clinical, histopathologic, and immunohistochemical correlations in leukemia cutis. Semin Dermatol. 1994;13:223-230.
Practice Points
- Leukemia cutis complicates 5% to 15% of all cases of acute myeloid leukemia (AML) in adults.
- The appearance of leukemia cutis may be highly variable. Therefore, it should be included in the differential diagnosis for any cutaneous presentation in patients with an existing diagnosis or high likelihood of AML.
- Leukemic infiltrates are associated with sites of inflammation.

Treatment of Elephantiasic Pretibial Myxedema With Rituximab Therapy
To the Editor:
Pretibial myxedema (PTM) is bilateral, nonpitting, scaly thickening and induration of the skin that most commonly occurs on the anterior aspects of the legs and feet. Pretibial myxedema occurs in approximately 0.5% to 4.3% of patients with hyperthyroidism.1 Thyroid dermopathy often is thought of as the classic nonpitting PTM with skin induration and color change. However, rarer forms of PTM, including plaque, nodular, and elephantiasic, also are important to note.2
Elephantiasic PTM is extremely rare, occurring in less than 1% of patients with PTM.2 Elephantiasic PTM is characterized by the persistent swelling of 1 or both legs; thickening of the skin overlying the dorsum of the feet, ankles, and toes; and verrucous irregular plaques that often are fleshy and flattened. The clinical differential diagnosis of elephantiasic PTM includes elephantiasis nostra verrucosa, a late-stage complication of chronic lymphedema that can be related to a variety of infectious or noninfectious obstructive processes. Few effective therapeutic modalities exist in the treatment of elephantiasic PTM. We present a case of elephantiasic PTM.
A 59-year-old man presented to dermatology with leonine facies with pronounced glabellar creases and indentations of the earlobes. He had diffuse woody induration, hyperpigmentation, and nonpitting edema of the lower extremities as well as several flesh-colored exophytic nodules scattered throughout the anterior shins and dorsal feet (Figure 1). On the left posterior calf, there was a large, 3-cm, exophytic, firm, flesh-colored nodule. Examination of the hands revealed mild hyperpigmentation of the distal digits, clubbing of the distal phalanges, and cheiroarthropathy.

The patient was diagnosed with Graves disease after experiencing the classic symptoms of hyperthyroidism, including heat intolerance, tremor, palpitations, and anxiety. He received thyroid ablation and subsequently was supplemented with levothyroxine 75 mg daily. Twelve years later, he was diagnosed with Graves ophthalmopathy with ocular proptosis requiring multiple courses of retro-orbital irradiation and surgical procedures for decompression. Approximately 1 year later, he noted increased swelling, firmness, and darkening of the pretibial surfaces. Initially, he was referred to vascular surgery and underwent bilateral saphenous vein ablation. He also was referred to a lymphedema specialist, and workup revealed an unremarkable lymphatic system. Minimal improvement was noted following the saphenous vein ablation, and he subsequently was referred to dermatology for further workup.
At the current presentation, laboratory analysis revealed a low thyrotropin level (0.03 mIU/L [reference range, 0.4–4.2 mIU/L]), and free thyroxine was within reference range. Radiography of the chest was unremarkable; however, radiography of the hand demonstrated arthrosis of the left fifth proximal interphalangeal joint. Nuclear medicine lymphoscintigraphy and lower extremity ultrasonography were unremarkable. Punch biopsies were performed of the left lateral leg and posterior calf. Hematoxylin and eosin staining demonstrated marked mucin deposition extending to the deep dermis along with deep fibroplasia and was read as consistent with PTM. Colloidal iron highlighted prominent mucin within the dermis (Figure 2).
. B, Colloidal iron staining highlighted the prominent mucin within the dermis")
The patient’s medical history, physical examination, laboratory analysis, imaging, and biopsies were considered, and a diagnosis of elephantiasic PTM was made. Minimal improvement was noted with initial therapeutic interventions including compression therapy and application of super high–potency topical corticosteroids. After further evaluation in our multidisciplinary rheumatology-dermatology clinic, the decision was made to initiate rituximab infusions.
Two months after 1 course of rituximab consisting of two 1000-mg infusions separated by 2 weeks, the patient showed substantial clinical improvement. There was striking improvement of the pretibial surfaces with resolution of the exophytic nodules and improvement of the induration (Figure 3). In addition, there was decreased induration of the glabella and earlobes and decreased fullness of the digital pulp on the hands. The patient also reported subjective improvements in mobility.
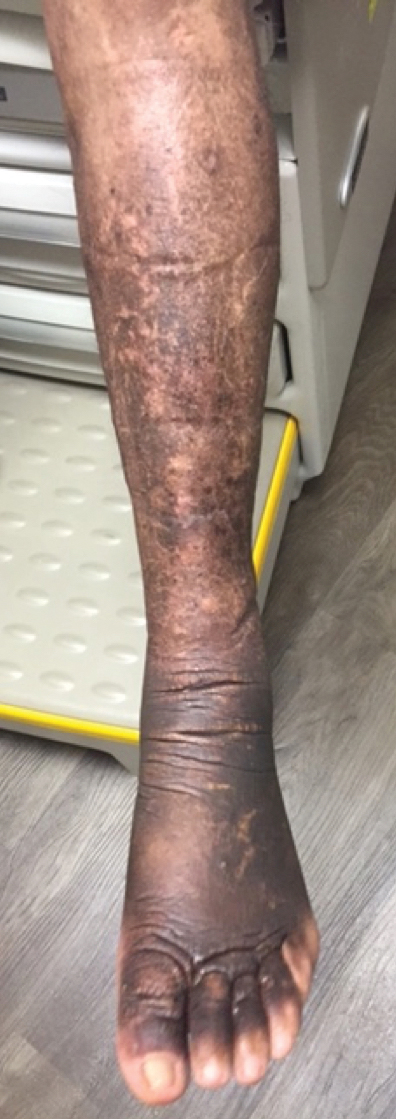
Our patient demonstrated all 3 aspects of the Diamond triad: PTM, exophthalmos, and acropachy. Patients present with all 3 features in less than 1% of reported cases of Graves disease.3 Although all 3 features are seen together infrequently, thyroid dermopathy and acropachy often are markers of severe Graves ophthalmopathy. In a study of 114 patients with Graves ophthalmopathy, patients who also had dermopathy and acropachy were more likely to have optic neuropathy or require orbital decompression.4
After overcoming the diagnostic dilemma that the elephantiasic presentation of PTM can present, therapeutic management remains a challenge. Heyes et al5 documented the successful treatment of highly recalcitrant elephantiasic PTM with rituximab and plasmapheresis therapy. In this case, a 44-year-old woman with an 11-year history of Graves disease and elephantiasic PTM received 29 rituximab infusions and 241 plasmapheresis treatments over the course of 3.5 years. Her elephantiasic PTM clinically resolved, and she was able to resume daily activities and wear normal shoes after being nonambulatory for years.5
Rituximab is a monoclonal antibody against CD20, a protein found primarily on the surface of B-cell lymphocytes. Although rituximab initially was approved by the US Food and Drug administration for the treatment of malignant lymphoma, it has had an increasing role in the treatment of autoimmune disorders such as rheumatoid arthritis. Rituximab is postulated to target B lymphocytes and halt their progression to plasma cells. By limiting the population of long-lasting, antibody-producing plasma cells and decreasing the autoantibodies that cause many of the symptoms in Graves disease, rituximab may be an effective therapy to consider in the treatment of elephantiasic PTM.6
Although the exact mechanism is poorly understood, PTM likely is a sequela of hyperthyroidism because of the expression of thyroid-stimulating hormone receptor proteins found on normal dermal fibroblasts. Thyroid-stimulating hormone receptor autoantibodies are thought to stimulate these fibroblasts to produce glycosaminoglycans. Histopathologically, accumulation of glycosaminoglycans deposited in the reticular dermis with high concentrations of hyaluronic acid is observed in PTM.7
Treatment of elephantiasic PTM remains a therapeutic challenge. Given the rarity of the disease process and limited information on effective therapeutic modalities, rituximab should be viewed as a viable treatment option in the management of recalcitrant elephantiasic PTM.
- Schwartz KM, Fatourechi V, Ahmed DDF, et al. Dermopathy of Graves’ disease (pretibial myxedema): long-term outcome. J Clin Endocrinol Metab. 2002;87:438-446.
- Kakati S, Doley B, Pal S, et al. Elephantiasis nostras verrucosa: a rare thyroid dermopathy in Graves’ disease. J Assoc Physicians India. 2005;53:571-572.
- Anderson CK, Miller OF 3rd. Triad of exophthalmos, pretibial myxedema, and acropachy in a patient with Graves’ disease. J Am Acad Dermatol. 2003;48:970-972.
- Fatourechi V, Bartley GB, Eghbali-Fatourechi GZ, et al. Graves’ dermopathy and acropachy are markers of severe Graves’ ophthalmopathy. Thyroid. 2003;13:1141-1144.
- Heyes C, Nolan R, Leahy M, et al. Treatment‐resistant elephantiasic thyroid dermopathy responding to rituximab and plasmapheresis. Australas J Dermatol. 2012;53:E1-E4.
- Salvi M, Vannucchi G, Campi I, et al. Treatment of Graves’ disease and associated ophthalmopathy with the anti-CD20 monoclonal antibody rituximab: an open study. Eur J Endocrinol. 2007;156:33-40.
- Heufelder AE, Dutton CM, Sarkar G, et al. Detection of TSH receptor RNA in cultured fibroblasts from patients with Graves’ ophthalmopathy and pretibial dermopathy. Thyroid. 1993;3:297-300.
To the Editor:
Pretibial myxedema (PTM) is bilateral, nonpitting, scaly thickening and induration of the skin that most commonly occurs on the anterior aspects of the legs and feet. Pretibial myxedema occurs in approximately 0.5% to 4.3% of patients with hyperthyroidism.1 Thyroid dermopathy often is thought of as the classic nonpitting PTM with skin induration and color change. However, rarer forms of PTM, including plaque, nodular, and elephantiasic, also are important to note.2
Elephantiasic PTM is extremely rare, occurring in less than 1% of patients with PTM.2 Elephantiasic PTM is characterized by the persistent swelling of 1 or both legs; thickening of the skin overlying the dorsum of the feet, ankles, and toes; and verrucous irregular plaques that often are fleshy and flattened. The clinical differential diagnosis of elephantiasic PTM includes elephantiasis nostra verrucosa, a late-stage complication of chronic lymphedema that can be related to a variety of infectious or noninfectious obstructive processes. Few effective therapeutic modalities exist in the treatment of elephantiasic PTM. We present a case of elephantiasic PTM.
A 59-year-old man presented to dermatology with leonine facies with pronounced glabellar creases and indentations of the earlobes. He had diffuse woody induration, hyperpigmentation, and nonpitting edema of the lower extremities as well as several flesh-colored exophytic nodules scattered throughout the anterior shins and dorsal feet (Figure 1). On the left posterior calf, there was a large, 3-cm, exophytic, firm, flesh-colored nodule. Examination of the hands revealed mild hyperpigmentation of the distal digits, clubbing of the distal phalanges, and cheiroarthropathy.
The patient was diagnosed with Graves disease after experiencing the classic symptoms of hyperthyroidism, including heat intolerance, tremor, palpitations, and anxiety. He received thyroid ablation and subsequently was supplemented with levothyroxine 75 mg daily. Twelve years later, he was diagnosed with Graves ophthalmopathy with ocular proptosis requiring multiple courses of retro-orbital irradiation and surgical procedures for decompression. Approximately 1 year later, he noted increased swelling, firmness, and darkening of the pretibial surfaces. Initially, he was referred to vascular surgery and underwent bilateral saphenous vein ablation. He also was referred to a lymphedema specialist, and workup revealed an unremarkable lymphatic system. Minimal improvement was noted following the saphenous vein ablation, and he subsequently was referred to dermatology for further workup.
At the current presentation, laboratory analysis revealed a low thyrotropin level (0.03 mIU/L [reference range, 0.4–4.2 mIU/L]), and free thyroxine was within reference range. Radiography of the chest was unremarkable; however, radiography of the hand demonstrated arthrosis of the left fifth proximal interphalangeal joint. Nuclear medicine lymphoscintigraphy and lower extremity ultrasonography were unremarkable. Punch biopsies were performed of the left lateral leg and posterior calf. Hematoxylin and eosin staining demonstrated marked mucin deposition extending to the deep dermis along with deep fibroplasia and was read as consistent with PTM. Colloidal iron highlighted prominent mucin within the dermis (Figure 2).
The patient’s medical history, physical examination, laboratory analysis, imaging, and biopsies were considered, and a diagnosis of elephantiasic PTM was made. Minimal improvement was noted with initial therapeutic interventions including compression therapy and application of super high–potency topical corticosteroids. After further evaluation in our multidisciplinary rheumatology-dermatology clinic, the decision was made to initiate rituximab infusions.
Two months after 1 course of rituximab consisting of two 1000-mg infusions separated by 2 weeks, the patient showed substantial clinical improvement. There was striking improvement of the pretibial surfaces with resolution of the exophytic nodules and improvement of the induration (Figure 3). In addition, there was decreased induration of the glabella and earlobes and decreased fullness of the digital pulp on the hands. The patient also reported subjective improvements in mobility.
Our patient demonstrated all 3 aspects of the Diamond triad: PTM, exophthalmos, and acropachy. Patients present with all 3 features in less than 1% of reported cases of Graves disease.3 Although all 3 features are seen together infrequently, thyroid dermopathy and acropachy often are markers of severe Graves ophthalmopathy. In a study of 114 patients with Graves ophthalmopathy, patients who also had dermopathy and acropachy were more likely to have optic neuropathy or require orbital decompression.4
After overcoming the diagnostic dilemma that the elephantiasic presentation of PTM can present, therapeutic management remains a challenge. Heyes et al5 documented the successful treatment of highly recalcitrant elephantiasic PTM with rituximab and plasmapheresis therapy. In this case, a 44-year-old woman with an 11-year history of Graves disease and elephantiasic PTM received 29 rituximab infusions and 241 plasmapheresis treatments over the course of 3.5 years. Her elephantiasic PTM clinically resolved, and she was able to resume daily activities and wear normal shoes after being nonambulatory for years.5
Rituximab is a monoclonal antibody against CD20, a protein found primarily on the surface of B-cell lymphocytes. Although rituximab initially was approved by the US Food and Drug administration for the treatment of malignant lymphoma, it has had an increasing role in the treatment of autoimmune disorders such as rheumatoid arthritis. Rituximab is postulated to target B lymphocytes and halt their progression to plasma cells. By limiting the population of long-lasting, antibody-producing plasma cells and decreasing the autoantibodies that cause many of the symptoms in Graves disease, rituximab may be an effective therapy to consider in the treatment of elephantiasic PTM.6
Although the exact mechanism is poorly understood, PTM likely is a sequela of hyperthyroidism because of the expression of thyroid-stimulating hormone receptor proteins found on normal dermal fibroblasts. Thyroid-stimulating hormone receptor autoantibodies are thought to stimulate these fibroblasts to produce glycosaminoglycans. Histopathologically, accumulation of glycosaminoglycans deposited in the reticular dermis with high concentrations of hyaluronic acid is observed in PTM.7
Treatment of elephantiasic PTM remains a therapeutic challenge. Given the rarity of the disease process and limited information on effective therapeutic modalities, rituximab should be viewed as a viable treatment option in the management of recalcitrant elephantiasic PTM.
To the Editor:
Pretibial myxedema (PTM) is bilateral, nonpitting, scaly thickening and induration of the skin that most commonly occurs on the anterior aspects of the legs and feet. Pretibial myxedema occurs in approximately 0.5% to 4.3% of patients with hyperthyroidism.1 Thyroid dermopathy often is thought of as the classic nonpitting PTM with skin induration and color change. However, rarer forms of PTM, including plaque, nodular, and elephantiasic, also are important to note.2
Elephantiasic PTM is extremely rare, occurring in less than 1% of patients with PTM.2 Elephantiasic PTM is characterized by the persistent swelling of 1 or both legs; thickening of the skin overlying the dorsum of the feet, ankles, and toes; and verrucous irregular plaques that often are fleshy and flattened. The clinical differential diagnosis of elephantiasic PTM includes elephantiasis nostra verrucosa, a late-stage complication of chronic lymphedema that can be related to a variety of infectious or noninfectious obstructive processes. Few effective therapeutic modalities exist in the treatment of elephantiasic PTM. We present a case of elephantiasic PTM.
A 59-year-old man presented to dermatology with leonine facies with pronounced glabellar creases and indentations of the earlobes. He had diffuse woody induration, hyperpigmentation, and nonpitting edema of the lower extremities as well as several flesh-colored exophytic nodules scattered throughout the anterior shins and dorsal feet (Figure 1). On the left posterior calf, there was a large, 3-cm, exophytic, firm, flesh-colored nodule. Examination of the hands revealed mild hyperpigmentation of the distal digits, clubbing of the distal phalanges, and cheiroarthropathy.
The patient was diagnosed with Graves disease after experiencing the classic symptoms of hyperthyroidism, including heat intolerance, tremor, palpitations, and anxiety. He received thyroid ablation and subsequently was supplemented with levothyroxine 75 mg daily. Twelve years later, he was diagnosed with Graves ophthalmopathy with ocular proptosis requiring multiple courses of retro-orbital irradiation and surgical procedures for decompression. Approximately 1 year later, he noted increased swelling, firmness, and darkening of the pretibial surfaces. Initially, he was referred to vascular surgery and underwent bilateral saphenous vein ablation. He also was referred to a lymphedema specialist, and workup revealed an unremarkable lymphatic system. Minimal improvement was noted following the saphenous vein ablation, and he subsequently was referred to dermatology for further workup.
At the current presentation, laboratory analysis revealed a low thyrotropin level (0.03 mIU/L [reference range, 0.4–4.2 mIU/L]), and free thyroxine was within reference range. Radiography of the chest was unremarkable; however, radiography of the hand demonstrated arthrosis of the left fifth proximal interphalangeal joint. Nuclear medicine lymphoscintigraphy and lower extremity ultrasonography were unremarkable. Punch biopsies were performed of the left lateral leg and posterior calf. Hematoxylin and eosin staining demonstrated marked mucin deposition extending to the deep dermis along with deep fibroplasia and was read as consistent with PTM. Colloidal iron highlighted prominent mucin within the dermis (Figure 2).
The patient’s medical history, physical examination, laboratory analysis, imaging, and biopsies were considered, and a diagnosis of elephantiasic PTM was made. Minimal improvement was noted with initial therapeutic interventions including compression therapy and application of super high–potency topical corticosteroids. After further evaluation in our multidisciplinary rheumatology-dermatology clinic, the decision was made to initiate rituximab infusions.
Two months after 1 course of rituximab consisting of two 1000-mg infusions separated by 2 weeks, the patient showed substantial clinical improvement. There was striking improvement of the pretibial surfaces with resolution of the exophytic nodules and improvement of the induration (Figure 3). In addition, there was decreased induration of the glabella and earlobes and decreased fullness of the digital pulp on the hands. The patient also reported subjective improvements in mobility.
Our patient demonstrated all 3 aspects of the Diamond triad: PTM, exophthalmos, and acropachy. Patients present with all 3 features in less than 1% of reported cases of Graves disease.3 Although all 3 features are seen together infrequently, thyroid dermopathy and acropachy often are markers of severe Graves ophthalmopathy. In a study of 114 patients with Graves ophthalmopathy, patients who also had dermopathy and acropachy were more likely to have optic neuropathy or require orbital decompression.4
After overcoming the diagnostic dilemma that the elephantiasic presentation of PTM can present, therapeutic management remains a challenge. Heyes et al5 documented the successful treatment of highly recalcitrant elephantiasic PTM with rituximab and plasmapheresis therapy. In this case, a 44-year-old woman with an 11-year history of Graves disease and elephantiasic PTM received 29 rituximab infusions and 241 plasmapheresis treatments over the course of 3.5 years. Her elephantiasic PTM clinically resolved, and she was able to resume daily activities and wear normal shoes after being nonambulatory for years.5
Rituximab is a monoclonal antibody against CD20, a protein found primarily on the surface of B-cell lymphocytes. Although rituximab initially was approved by the US Food and Drug administration for the treatment of malignant lymphoma, it has had an increasing role in the treatment of autoimmune disorders such as rheumatoid arthritis. Rituximab is postulated to target B lymphocytes and halt their progression to plasma cells. By limiting the population of long-lasting, antibody-producing plasma cells and decreasing the autoantibodies that cause many of the symptoms in Graves disease, rituximab may be an effective therapy to consider in the treatment of elephantiasic PTM.6
Although the exact mechanism is poorly understood, PTM likely is a sequela of hyperthyroidism because of the expression of thyroid-stimulating hormone receptor proteins found on normal dermal fibroblasts. Thyroid-stimulating hormone receptor autoantibodies are thought to stimulate these fibroblasts to produce glycosaminoglycans. Histopathologically, accumulation of glycosaminoglycans deposited in the reticular dermis with high concentrations of hyaluronic acid is observed in PTM.7
Treatment of elephantiasic PTM remains a therapeutic challenge. Given the rarity of the disease process and limited information on effective therapeutic modalities, rituximab should be viewed as a viable treatment option in the management of recalcitrant elephantiasic PTM.
- Schwartz KM, Fatourechi V, Ahmed DDF, et al. Dermopathy of Graves’ disease (pretibial myxedema): long-term outcome. J Clin Endocrinol Metab. 2002;87:438-446.
- Kakati S, Doley B, Pal S, et al. Elephantiasis nostras verrucosa: a rare thyroid dermopathy in Graves’ disease. J Assoc Physicians India. 2005;53:571-572.
- Anderson CK, Miller OF 3rd. Triad of exophthalmos, pretibial myxedema, and acropachy in a patient with Graves’ disease. J Am Acad Dermatol. 2003;48:970-972.
- Fatourechi V, Bartley GB, Eghbali-Fatourechi GZ, et al. Graves’ dermopathy and acropachy are markers of severe Graves’ ophthalmopathy. Thyroid. 2003;13:1141-1144.
- Heyes C, Nolan R, Leahy M, et al. Treatment‐resistant elephantiasic thyroid dermopathy responding to rituximab and plasmapheresis. Australas J Dermatol. 2012;53:E1-E4.
- Salvi M, Vannucchi G, Campi I, et al. Treatment of Graves’ disease and associated ophthalmopathy with the anti-CD20 monoclonal antibody rituximab: an open study. Eur J Endocrinol. 2007;156:33-40.
- Heufelder AE, Dutton CM, Sarkar G, et al. Detection of TSH receptor RNA in cultured fibroblasts from patients with Graves’ ophthalmopathy and pretibial dermopathy. Thyroid. 1993;3:297-300.
- Schwartz KM, Fatourechi V, Ahmed DDF, et al. Dermopathy of Graves’ disease (pretibial myxedema): long-term outcome. J Clin Endocrinol Metab. 2002;87:438-446.
- Kakati S, Doley B, Pal S, et al. Elephantiasis nostras verrucosa: a rare thyroid dermopathy in Graves’ disease. J Assoc Physicians India. 2005;53:571-572.
- Anderson CK, Miller OF 3rd. Triad of exophthalmos, pretibial myxedema, and acropachy in a patient with Graves’ disease. J Am Acad Dermatol. 2003;48:970-972.
- Fatourechi V, Bartley GB, Eghbali-Fatourechi GZ, et al. Graves’ dermopathy and acropachy are markers of severe Graves’ ophthalmopathy. Thyroid. 2003;13:1141-1144.
- Heyes C, Nolan R, Leahy M, et al. Treatment‐resistant elephantiasic thyroid dermopathy responding to rituximab and plasmapheresis. Australas J Dermatol. 2012;53:E1-E4.
- Salvi M, Vannucchi G, Campi I, et al. Treatment of Graves’ disease and associated ophthalmopathy with the anti-CD20 monoclonal antibody rituximab: an open study. Eur J Endocrinol. 2007;156:33-40.
- Heufelder AE, Dutton CM, Sarkar G, et al. Detection of TSH receptor RNA in cultured fibroblasts from patients with Graves’ ophthalmopathy and pretibial dermopathy. Thyroid. 1993;3:297-300.
Practice Points
- Pretibial myxedema (PTM) is bilateral, nonpitting, scaly thickening and induration of the skin that most commonly occurs on the anterior aspects of the legs and feet.
- Although many therapeutic modalities have been described for the management of the elephantiasis variant of PTM, few treatments have shown notable efficacy.
- Rituximab may be an effective therapy to consider in the treatment of elephantiasic PTM.

Slow-Growing Pink Nodule in an Active-Duty Service Member
The Diagnosis: Leishmaniasis
Hematoxylin and eosin staining of the tissue specimen revealed a dense histiocytic infiltrate with scattered lymphocytes and neutrophils. There were round to oval basophilic structures within the macrophages consistent with amastigotes. Giemsa staining was not necessary to visualize the organisms. The infiltrate abutted the overlying epidermis, which was acanthotic with pseudoepitheliomatous hyperplasia. There were collections of neutrophils, parakeratosis, and a serum crust overlying the epidermis (Figure). Clinical and histologic findings, as well as travel history, led to a diagnosis of cutaneous leishmaniasis (CL).

Leishmaniases is a group of diseases caused by a parasitic infection with flagellated protozoa of the genus Leishmania. There are more than 20 different Leishmania species that are pathogenic to humans, all presenting with cutaneous findings. The presentation depends on the inoculating species and the host cellular immune response and includes cutaneous, mucosal, and visceral involvement. The disease is transmitted via the bite of an infected bloodsucking female sand fly.1 There are approximately 30 different species of sand flies that are proven to be vectors of the disease, with up to 40 more suspected of involvement in transmission, predominantly from the genera Phlebotomus (Old World) and Lutzomyia (New World).1,2 There are an estimated 1 to 2 million new cases of cutaneous leishmaniasis diagnosed annually in 70 endemic countries of the tropics, subtropics, and southern Europe.1,3,4
The differential diagnosis included cutaneous tuberculosis, which can have a similar progression and clinical appearance. Cutaneous tuberculosis starts as firm, reddish-brown, painless papules that slowly enlarge and ulcerate.5 It may be further differentiated on histopathology by the presence of tuberculoid granulomas, caseating necrosis, and acid-fast bacilli, which are easily detected in early lesions but are less prevalent after the granuloma develops.6 Sporotrichosis presents as a nodule, which may or may not ulcerate, on the extremities. However, the classic morphology is a sporotrichoid pattern, which describes the initial lesion plus subcutaneous nodular spread along the lymphatics.7 On histology, sporotrichosis has a characteristic “sporotrichoid asteroid” comprised of the yeast form surrounded by eosinophilic hyaline material in raylike processes that are found in the center of suppurative granulomas or foci.8
Atypical mycobacteria, principally Mycobacterium marinum (swimming pool granuloma) and Mycobacterium ulcerans (Buruli ulcer), are capable of causing cutaneous infections. They may be differentiated histologically by a neutrophilic infiltrate of poorly formed granulomas without caseation and extensive coagulative necrosis with little cellular infiltrate, respectively.6 Histoplasma capsulatum also infects histiocytes and may appear similar in size and shape; however, histoplasmosis is surrounded by a pseudocapsule and evenly spaced.8
Conversely, the histology of leishmaniasis lacks a pseudocapsule. The amastigotes may form the classic marquee sign by lining the periphery of the macrophage or they can be randomly spaced. Classically, the epidermis shows hyperkeratosis and acanthosis. Sometimes atrophy, ulceration, or intraepidermal abscesses also can be observed. Pseudoepitheliomatous hyperplasia can be seen in some long-standing lesions.1,4 Many of these findings were observed on hematoxylin and eosin staining from a punch biopsy obtained from the center of the lesion in our patient. For further delineation, a speciation kit was obtained from Walter Reed National Military Medical Center (Bethesda, Maryland). A second punch biopsy was obtained from the lesion edge, sectioned into 4 individual pieces, and placed in Schneider tissue culture medium. It was sent for tissue culture, polymerase chain reaction, and histology. Polymerase chain reaction analysis was positive for Leishmania, which was further identified as Leishmania tropica by tissue culture.
Leishmania tropica (Old World CL) commonly causes CL and is endemic to Central Asia, the Middle East, parts of North Africa, and Southeast Asia. Old and New World CL start as a small erythematous papule after a bite from an infected female sand fly. The papule develops into a nodule over weeks to months. The lesion may ulcerate and typically heals leaving an atrophic scar in months to years.1 Speciation of CL is important to guide therapy.
Leishmania mexicana, a New World species that commonly causes CL, classically is found in Central and South America, but there also have been documented cases in Texas. A 2008 case series identified 9 cases in northern Texas in residents without a travel history to endemic locations.9 Similarly, a cross-sectional study identified 41 locally endemic cases of CL over a 10-year period (2007-2017) in Texas; 22 of these cases had speciation by polymerase chain reaction, and all cases were attributed to L mexicana.10
In the United States, CL classically has been associated with travelers and military personnel returning from the Middle East; however, a growing body of literature suggests that it may be endemic to Texas, where it is now a reportable disease. Physicians should have an increased awareness of this entity and a high index of suspicion when treating patients with nonhealing cutaneous lesions.
- Bravo F. Protozoa and worms. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. WB Saunders Co; 2018:1470-1502.
- Killick-Kendrick R. The biology and control of phlebotomine sandflies. Clin Dermatol. 1999;17:279-289.
- Reithinger R, Dujardin JC, Louzir H, et al. Cutaneous leishmaniasis. Lancet Infect Dis. 2007;7:581-596.
- Patterson J. Protozoal infections. Weedon’s Skin Pathology. 5th ed. Elsevier; 2021:787-795.
- Ramos-e-Silva M, Ribeiro de Castro MC. Mycobacterial infections. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. WB Saunders Co; 2018:1296-1318.
- Patterson J. Bacterial and rickettsial infections. Weedon’s Skin Pathology. 5th ed. Elsevier; 2021:673-709.
- Elewski B, Hughey L, Hunt K, et al. Fungal diseases. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. WB Saunders Co; 2018:1329-1363.
- Patterson J. Mycoses and algal infections. Weedon’s Skin Pathology. 5th ed. Elsevier; 2021:721-755.
- Wright NA, Davis LE, Aftergut KS, et al. Cutaneous leishmaniasis in Texas: a northern spread of endemic areas [published online February 4, 2008]. J Am Acad Dermatol. 2008;58:650-652. doi:10.1016/j .jaad.2007.11.008.
- McIlwee BE, Weis SE, Hosler GA. Incidence of endemic human cutaneous leishmaniasis in the United States. JAMA Dermatol. 2018;154:1032-1039. doi:10.1001/jamadermatol.2018.2133
The Diagnosis: Leishmaniasis
Hematoxylin and eosin staining of the tissue specimen revealed a dense histiocytic infiltrate with scattered lymphocytes and neutrophils. There were round to oval basophilic structures within the macrophages consistent with amastigotes. Giemsa staining was not necessary to visualize the organisms. The infiltrate abutted the overlying epidermis, which was acanthotic with pseudoepitheliomatous hyperplasia. There were collections of neutrophils, parakeratosis, and a serum crust overlying the epidermis (Figure). Clinical and histologic findings, as well as travel history, led to a diagnosis of cutaneous leishmaniasis (CL).
Leishmaniases is a group of diseases caused by a parasitic infection with flagellated protozoa of the genus Leishmania. There are more than 20 different Leishmania species that are pathogenic to humans, all presenting with cutaneous findings. The presentation depends on the inoculating species and the host cellular immune response and includes cutaneous, mucosal, and visceral involvement. The disease is transmitted via the bite of an infected bloodsucking female sand fly.1 There are approximately 30 different species of sand flies that are proven to be vectors of the disease, with up to 40 more suspected of involvement in transmission, predominantly from the genera Phlebotomus (Old World) and Lutzomyia (New World).1,2 There are an estimated 1 to 2 million new cases of cutaneous leishmaniasis diagnosed annually in 70 endemic countries of the tropics, subtropics, and southern Europe.1,3,4
The differential diagnosis included cutaneous tuberculosis, which can have a similar progression and clinical appearance. Cutaneous tuberculosis starts as firm, reddish-brown, painless papules that slowly enlarge and ulcerate.5 It may be further differentiated on histopathology by the presence of tuberculoid granulomas, caseating necrosis, and acid-fast bacilli, which are easily detected in early lesions but are less prevalent after the granuloma develops.6 Sporotrichosis presents as a nodule, which may or may not ulcerate, on the extremities. However, the classic morphology is a sporotrichoid pattern, which describes the initial lesion plus subcutaneous nodular spread along the lymphatics.7 On histology, sporotrichosis has a characteristic “sporotrichoid asteroid” comprised of the yeast form surrounded by eosinophilic hyaline material in raylike processes that are found in the center of suppurative granulomas or foci.8
Atypical mycobacteria, principally Mycobacterium marinum (swimming pool granuloma) and Mycobacterium ulcerans (Buruli ulcer), are capable of causing cutaneous infections. They may be differentiated histologically by a neutrophilic infiltrate of poorly formed granulomas without caseation and extensive coagulative necrosis with little cellular infiltrate, respectively.6 Histoplasma capsulatum also infects histiocytes and may appear similar in size and shape; however, histoplasmosis is surrounded by a pseudocapsule and evenly spaced.8
Conversely, the histology of leishmaniasis lacks a pseudocapsule. The amastigotes may form the classic marquee sign by lining the periphery of the macrophage or they can be randomly spaced. Classically, the epidermis shows hyperkeratosis and acanthosis. Sometimes atrophy, ulceration, or intraepidermal abscesses also can be observed. Pseudoepitheliomatous hyperplasia can be seen in some long-standing lesions.1,4 Many of these findings were observed on hematoxylin and eosin staining from a punch biopsy obtained from the center of the lesion in our patient. For further delineation, a speciation kit was obtained from Walter Reed National Military Medical Center (Bethesda, Maryland). A second punch biopsy was obtained from the lesion edge, sectioned into 4 individual pieces, and placed in Schneider tissue culture medium. It was sent for tissue culture, polymerase chain reaction, and histology. Polymerase chain reaction analysis was positive for Leishmania, which was further identified as Leishmania tropica by tissue culture.
Leishmania tropica (Old World CL) commonly causes CL and is endemic to Central Asia, the Middle East, parts of North Africa, and Southeast Asia. Old and New World CL start as a small erythematous papule after a bite from an infected female sand fly. The papule develops into a nodule over weeks to months. The lesion may ulcerate and typically heals leaving an atrophic scar in months to years.1 Speciation of CL is important to guide therapy.
Leishmania mexicana, a New World species that commonly causes CL, classically is found in Central and South America, but there also have been documented cases in Texas. A 2008 case series identified 9 cases in northern Texas in residents without a travel history to endemic locations.9 Similarly, a cross-sectional study identified 41 locally endemic cases of CL over a 10-year period (2007-2017) in Texas; 22 of these cases had speciation by polymerase chain reaction, and all cases were attributed to L mexicana.10
In the United States, CL classically has been associated with travelers and military personnel returning from the Middle East; however, a growing body of literature suggests that it may be endemic to Texas, where it is now a reportable disease. Physicians should have an increased awareness of this entity and a high index of suspicion when treating patients with nonhealing cutaneous lesions.
The Diagnosis: Leishmaniasis
Hematoxylin and eosin staining of the tissue specimen revealed a dense histiocytic infiltrate with scattered lymphocytes and neutrophils. There were round to oval basophilic structures within the macrophages consistent with amastigotes. Giemsa staining was not necessary to visualize the organisms. The infiltrate abutted the overlying epidermis, which was acanthotic with pseudoepitheliomatous hyperplasia. There were collections of neutrophils, parakeratosis, and a serum crust overlying the epidermis (Figure). Clinical and histologic findings, as well as travel history, led to a diagnosis of cutaneous leishmaniasis (CL).
Leishmaniases is a group of diseases caused by a parasitic infection with flagellated protozoa of the genus Leishmania. There are more than 20 different Leishmania species that are pathogenic to humans, all presenting with cutaneous findings. The presentation depends on the inoculating species and the host cellular immune response and includes cutaneous, mucosal, and visceral involvement. The disease is transmitted via the bite of an infected bloodsucking female sand fly.1 There are approximately 30 different species of sand flies that are proven to be vectors of the disease, with up to 40 more suspected of involvement in transmission, predominantly from the genera Phlebotomus (Old World) and Lutzomyia (New World).1,2 There are an estimated 1 to 2 million new cases of cutaneous leishmaniasis diagnosed annually in 70 endemic countries of the tropics, subtropics, and southern Europe.1,3,4
The differential diagnosis included cutaneous tuberculosis, which can have a similar progression and clinical appearance. Cutaneous tuberculosis starts as firm, reddish-brown, painless papules that slowly enlarge and ulcerate.5 It may be further differentiated on histopathology by the presence of tuberculoid granulomas, caseating necrosis, and acid-fast bacilli, which are easily detected in early lesions but are less prevalent after the granuloma develops.6 Sporotrichosis presents as a nodule, which may or may not ulcerate, on the extremities. However, the classic morphology is a sporotrichoid pattern, which describes the initial lesion plus subcutaneous nodular spread along the lymphatics.7 On histology, sporotrichosis has a characteristic “sporotrichoid asteroid” comprised of the yeast form surrounded by eosinophilic hyaline material in raylike processes that are found in the center of suppurative granulomas or foci.8
Atypical mycobacteria, principally Mycobacterium marinum (swimming pool granuloma) and Mycobacterium ulcerans (Buruli ulcer), are capable of causing cutaneous infections. They may be differentiated histologically by a neutrophilic infiltrate of poorly formed granulomas without caseation and extensive coagulative necrosis with little cellular infiltrate, respectively.6 Histoplasma capsulatum also infects histiocytes and may appear similar in size and shape; however, histoplasmosis is surrounded by a pseudocapsule and evenly spaced.8
Conversely, the histology of leishmaniasis lacks a pseudocapsule. The amastigotes may form the classic marquee sign by lining the periphery of the macrophage or they can be randomly spaced. Classically, the epidermis shows hyperkeratosis and acanthosis. Sometimes atrophy, ulceration, or intraepidermal abscesses also can be observed. Pseudoepitheliomatous hyperplasia can be seen in some long-standing lesions.1,4 Many of these findings were observed on hematoxylin and eosin staining from a punch biopsy obtained from the center of the lesion in our patient. For further delineation, a speciation kit was obtained from Walter Reed National Military Medical Center (Bethesda, Maryland). A second punch biopsy was obtained from the lesion edge, sectioned into 4 individual pieces, and placed in Schneider tissue culture medium. It was sent for tissue culture, polymerase chain reaction, and histology. Polymerase chain reaction analysis was positive for Leishmania, which was further identified as Leishmania tropica by tissue culture.
Leishmania tropica (Old World CL) commonly causes CL and is endemic to Central Asia, the Middle East, parts of North Africa, and Southeast Asia. Old and New World CL start as a small erythematous papule after a bite from an infected female sand fly. The papule develops into a nodule over weeks to months. The lesion may ulcerate and typically heals leaving an atrophic scar in months to years.1 Speciation of CL is important to guide therapy.
Leishmania mexicana, a New World species that commonly causes CL, classically is found in Central and South America, but there also have been documented cases in Texas. A 2008 case series identified 9 cases in northern Texas in residents without a travel history to endemic locations.9 Similarly, a cross-sectional study identified 41 locally endemic cases of CL over a 10-year period (2007-2017) in Texas; 22 of these cases had speciation by polymerase chain reaction, and all cases were attributed to L mexicana.10
In the United States, CL classically has been associated with travelers and military personnel returning from the Middle East; however, a growing body of literature suggests that it may be endemic to Texas, where it is now a reportable disease. Physicians should have an increased awareness of this entity and a high index of suspicion when treating patients with nonhealing cutaneous lesions.
- Bravo F. Protozoa and worms. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. WB Saunders Co; 2018:1470-1502.
- Killick-Kendrick R. The biology and control of phlebotomine sandflies. Clin Dermatol. 1999;17:279-289.
- Reithinger R, Dujardin JC, Louzir H, et al. Cutaneous leishmaniasis. Lancet Infect Dis. 2007;7:581-596.
- Patterson J. Protozoal infections. Weedon’s Skin Pathology. 5th ed. Elsevier; 2021:787-795.
- Ramos-e-Silva M, Ribeiro de Castro MC. Mycobacterial infections. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. WB Saunders Co; 2018:1296-1318.
- Patterson J. Bacterial and rickettsial infections. Weedon’s Skin Pathology. 5th ed. Elsevier; 2021:673-709.
- Elewski B, Hughey L, Hunt K, et al. Fungal diseases. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. WB Saunders Co; 2018:1329-1363.
- Patterson J. Mycoses and algal infections. Weedon’s Skin Pathology. 5th ed. Elsevier; 2021:721-755.
- Wright NA, Davis LE, Aftergut KS, et al. Cutaneous leishmaniasis in Texas: a northern spread of endemic areas [published online February 4, 2008]. J Am Acad Dermatol. 2008;58:650-652. doi:10.1016/j .jaad.2007.11.008.
- McIlwee BE, Weis SE, Hosler GA. Incidence of endemic human cutaneous leishmaniasis in the United States. JAMA Dermatol. 2018;154:1032-1039. doi:10.1001/jamadermatol.2018.2133
- Bravo F. Protozoa and worms. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. WB Saunders Co; 2018:1470-1502.
- Killick-Kendrick R. The biology and control of phlebotomine sandflies. Clin Dermatol. 1999;17:279-289.
- Reithinger R, Dujardin JC, Louzir H, et al. Cutaneous leishmaniasis. Lancet Infect Dis. 2007;7:581-596.
- Patterson J. Protozoal infections. Weedon’s Skin Pathology. 5th ed. Elsevier; 2021:787-795.
- Ramos-e-Silva M, Ribeiro de Castro MC. Mycobacterial infections. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. WB Saunders Co; 2018:1296-1318.
- Patterson J. Bacterial and rickettsial infections. Weedon’s Skin Pathology. 5th ed. Elsevier; 2021:673-709.
- Elewski B, Hughey L, Hunt K, et al. Fungal diseases. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. WB Saunders Co; 2018:1329-1363.
- Patterson J. Mycoses and algal infections. Weedon’s Skin Pathology. 5th ed. Elsevier; 2021:721-755.
- Wright NA, Davis LE, Aftergut KS, et al. Cutaneous leishmaniasis in Texas: a northern spread of endemic areas [published online February 4, 2008]. J Am Acad Dermatol. 2008;58:650-652. doi:10.1016/j .jaad.2007.11.008.
- McIlwee BE, Weis SE, Hosler GA. Incidence of endemic human cutaneous leishmaniasis in the United States. JAMA Dermatol. 2018;154:1032-1039. doi:10.1001/jamadermatol.2018.2133
A 36-year-old active-duty male service member with no notable medical history presented to the dermatology clinic with an asymptomatic nodule on the right forearm that he initially noticed approximately 1 year prior while deployed in Syria and thought that it was a mosquito bite; it continued to enlarge slowly since that time. He attempted self-extraction but was only able to express a small amount of clear fluid. No other therapies had been used. He denied any other symptoms on a review of systems and was not taking any medications. Physical examination revealed a 1.5-cm, erythematous, nonulcerated, pink nodule on the right distal volar forearm without other cutaneous findings. A 4-mm punch biopsy was performed.


Fungating Mass on the Abdominal Wall
The Diagnosis: Basal Cell Carcinoma
Histopathology was consistent with fungating basal cell carcinoma (BCC). The nodules were comprised of syncytial basaloid cells with high nuclear to cytoplasmic ratios, numerous mitotic figures, fibromyxoid stroma, and peripheral nuclear palisading (Figure). Fortunately, no perineural or lymphovascular invasion was identified, and the margins of the specimen were negative. Despite the high-risk nature of giant BCC, the mass was solitary without notable local invasion, leaving it amendable to surgery. On follow-up, the patient has remained recurrence free, and her hemoglobin level has since stabilized.

Skin cancer is the most common malignancy worldwide, and BCC accounts for more than 80% of nonmelanoma skin cancers in the United States. The incidence is on the rise due to the aging population and increasing cumulative skin exposure.1 Risk factors include both individual physical characteristics and environmental exposures. Individuals with lighter skin tones, red and blonde hair, and blue and green eyes are at an increased risk.2 UV radiation exposure is the most important cause of BCC.3 Chronic immunosuppression and exposure to arsenic, ionizing radiation, and psoralen plus UVA radiation also have been linked to the development of BCC.4-6 Basal cell carcinomas most commonly arise on sun-exposed areas such as the face, though more than 10% of cases appear on the trunk.7 Lesions characteristically remain localized, and growth rate is variable; however, when left untreated, BCCs have the potential to become locally destructive and difficult to treat.
Advanced BCCs are tumors that penetrate deeply into the skin. They often are not amenable to traditional therapy and/ or metastasize. Those that grow to a diameter greater than 5 cm, as in our patient, are known as giant BCCs. Only 0.5% to 1% of BCCs are giant BCCs8 ; they typically are more aggressive in nature with higher rates of local recurrence and metastasis. Individuals who develop giant BCCs either have had a delay in access to medical care or a history of BCC that was inadequately managed.9,10 During the COVID-19 pandemic, patient access to health care was substantially impacted during lockdowns. As in our patient, skin neoplasms and other medical conditions may present in later stages due to medical neglect.11,12 Metastasis is rare, even in advanced BCCs. A review of the literature from 1984 estimated that the incidence of metastasis of BCCs is 1 in 1000 to 35,000. Metastasis portends a poor prognosis with a median overall survival of 8 to 14 months.13 An updated review in 2013 found similar outcomes.14
The choice of management for BCCs depends on the risk for recurrence as well as individual patient factors. Characteristics such as tumor size, location, histology, whether it is a primary or recurrent lesion, and the presence of chronic skin disease determine the recurrence rate.15 The management of advanced BCCs often requires a multidisciplinary approach, as these neoplasms may not be amenable to local therapy without causing substantial morbidity. Mohs micrographic surgery is the treatment of choice for BCCs at high risk for recurrence.16 Standard surgical excision with postoperative margin assessment is acceptable when Mohs micrographic surgery is not available.17 Radiation therapy is an alternative for patients who are not candidates for surgery.18
Recently, improved understanding of the molecular pathogenesis of BCCs has led to the development of novel systemic therapies. The Hedgehog signaling pathway has been found to play a critical role in the development of most BCCs.19 Vismodegib and sonidegib are small-molecule inhibitors of the Hedgehog signaling pathway approved for the treatment of locally advanced and metastatic BCCs that are not amenable to surgery or radiation. Approximately 50% of advanced BCCs respond to these therapies; however, long-term treatment may be limited by intolerable side effects and the development of resistance.20 Basal cell carcinomas that spread to lymph nodes or distant sites are treated with traditional systemic therapy. Historically, conventional cytotoxic chemotherapies, such as platinum-containing regimens, were employed with limited benefit and notable morbidity.21
The differential diagnosis for our patient included several other cutaneous neoplasms. Squamous cell carcinoma is the second most common type of skin cancer. Similar to BCC, it can reach a substantial size if left untreated. Risk factors include chronic inflammation, exposure to radiation or chemical carcinogens, burns, human papillomavirus, and other chronic infections. Giant squamous cell carcinomas have high malignant potential and require imaging to assess the extent of invasion and for metastasis. Surgery typically is necessary for both staging and treatment. Adjuvant therapy also may be necessary.22,23
Internal malignant neoplasms rarely present as cutaneous metastases. Breast cancer, melanoma, and cancers of the upper respiratory tract most frequently metastasize to the skin. Although colorectal cancer (CRC) rarely metastasizes to the skin, it is an important cause of cutaneous metastasis due to its high incidence in the general population. When it does spread to the skin, CRC preferentially affects the abdominal wall. Lesions typically resemble the primary tumor but may appear anaplastic. The occurrence of cutaneous metastasis suggests latestage disease and carries a poor prognosis.24
Merkel cell carcinoma and melanoma are aggressive skin cancers with high mortality rates. The former is rarer but more lethal. Merkel cell carcinomas typically occur in elderly white men on sun-exposed areas of the skin. Tumors present as asymptomatic, rapidly expanding, blue-red, firm nodules. Immunosuppression and UV light exposure are notable risk factors.25 Of the 4 major subtypes of cutaneous melanoma, superficial spreading is the most common, followed by nodular, lentigo maligna, and acral lentiginous.26 Superficial spreading melanoma characteristically presents as an expanding asymmetric macule or thin plaque with irregular borders and variation in size and color (black, brown, or red). Nodular melanoma usually presents as symmetric in shape and color (amelanotic, black, or brown). Early recognition by both the patient and clinician is essential in preventing tumor growth and progression.27
Our patient’s presentation was highly concerning for cutaneous metastasis given her history of CRC. Furthermore, the finding of severe anemia was atypical for skin cancer and more characteristic of the prior malignancy. Imaging revealed a locally confined mass with no evidence of extension, lymph node involvement, or additional lesions. The diagnosis was clinched with histopathologic examination.
- Rogers HW, Weinstock MA, Harris AR, et al. Incidence estimate of nonmelanoma skin cancer in the United States, 2006. Arch Dermatol. 2010;146:283-287.
- Lear JT, Tan BB, Smith AG, et al. Risk factors for basal cell carcinoma in the UK: case-control study in 806 patients. J R Soc Med. 1997; 90:371-374.
- Gallagher RP, Hill GB, Bajdik CD, et al. Sunlight exposure, pigmentary factors, and risk of nonmelanocytic skin cancer: I. basal cell carcinoma. Arch Dermatol. 1995;131:157-163.
- Guo HR, Yu HS, Hu H, et al. Arsenic in drinking water and skin cancers: cell-type specificity (Taiwan, ROC). Cancer Causes Control. 2001;12:909-916.
- Lichter MD, Karagas MR, Mott LA, et al; The New Hampshire Skin Cancer Study Group. Therapeutic ionizing radiation and the incidence of basal cell carcinoma and squamous cell carcinoma. Arch Dermatol. 2000;136:1007-1011.
- Nijsten TEC, Stern RS. The increased risk of skin cancer is persistent after discontinuation of psoralen plus ultraviolet A: a cohort study. J Invest Dermatol. 2003;121:252-258.
- Scrivener Y, Grosshans E, Cribier B. Variations of basal cell carcinomas according to gender, age, location and histopathological subtype. Br J Dermatol. 2002;147:41-47.
- Gualdi G, Monari P, Calzavara‐Pinton P, et al. When basal cell carcinomas became giant: an Italian multicenter study. Int J Dermatol. 2020;59:377-382.
- Randle HW, Roenigk RK, Brodland DG. Giant basal cell carcinoma (T3). who is at risk? Cancer. 1993;72:1624-1630.
- Archontaki M, Stavrianos SD, Korkolis DP, et al. Giant basal cell carcinoma: clinicopathological analysis of 51 cases and review of the literature. Anticancer Res. 2009;29:2655-2663.
- Shifat Ahmed SAK, Ajisola M, Azeem K, et al. Impact of the societal response to COVID-19 on access to healthcare for non-COVID-19 health issues in slum communities of Bangladesh, Kenya, Nigeria and Pakistan: results of pre-COVID and COVID-19 lockdown ssstakeholder engagements. BMJ Glob Health. 2020;5:E003042.
- Gomolin T, Cline A, Handler MZ. The danger of neglecting melanoma during the COVID-19 pandemic. J Dermatolog Treat. 2020;31:444-445.
- von Domarus H, Stevens PJ. Metastatic basal cell carcinoma. report of five cases and review of 170 cases in the literature. J Am Acad Dermatol. 1984;10:1043-1060.
- Wysong A, Aasi SZ, Tang JY. Update on metastatic basal cell carcinoma: a summary of published cases from 1981 through 2011. JAMA Dermatol. 2013;149:615-616.
- Bøgelund FS, Philipsen PA, Gniadecki R. Factors affecting the recurrence rate of basal cell carcinoma. Acta Derm Venereol. 2007;87:330-334.
- Mosterd K, Krekels GAM, Nieman FH, et al. Surgical excision versus Mohs’ micrographic surgery for primary and recurrent basal-cell carcinoma of the face: a prospective randomised controlled trial with 5-years’ follow-up. Lancet Oncol. 2008;9:1149-1156.
- Wetzig T, Woitek M, Eichhorn K, et al. Surgical excision of basal cell carcinoma with complete margin control: outcome at 5-year follow-up. Dermatology. 2010;220:363-369.
- Silverman MK, Kopf AW, Gladstein AH, et al. Recurrence rates of treated basal cell carcinomas. part 4: X-ray therapy. J Dermatol Surg Oncol. 1992;18:549-554.
- Tanese K, Emoto K, Kubota N, et al. Immunohistochemical visualization of the signature of activated Hedgehog signaling pathway in cutaneous epithelial tumors. J Dermatol. 2018;45:1181-1186.
- Basset-Séguin N, Hauschild A, Kunstfeld R, et al. Vismodegib in patients with advanced basal cell carcinoma: primary analysis of STEVIE, an international, open-label trial. Eur J Cancer. 2017;86:334-348.
- Carneiro BA, Watkin WG, Mehta UK, et al. Metastatic basal cell carcinoma: complete response to chemotherapy and associated pure red cell aplasia. Cancer Invest. 2006;24:396-400.
- Misiakos EP, Damaskou V, Koumarianou A, et al. A giant squamous cell carcinoma of the skin of the thoracic wall: a case report and review of the literature. J Med Case Rep. 2017;11:136.
- Wollina U, Bayyoud Y, Krönert C, et al. Giant epithelial malignancies (basal cell carcinoma, squamous cell carcinoma): a series of 20 tumors from a single center. J Cutan Aesthet Surg. 2012;5:12-19.
- Bittencourt MJS, Imbiriba AA, Oliveira OA, et al. Cutaneous metastasis of colorectal cancer. An Bras Dermatol. 2018;93:884-886.
- Heath M, Jaimes N, Lemos B, et al. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the AEIOU features. J Am Acad Dermatol. 2008;58:375-381.
- Buettner PG, Leiter U, Eigentler TK, et al. Development of prognostic factors and survival in cutaneous melanoma over 25 years: an analysis of the Central Malignant Melanoma Registry of the German Dermatological Society. Cancer. 2005;103:616-624.
- Klebanov N, Gunasekera N, Lin WM, et al. The clinical spectrum of cutaneous melanoma morphology. J Am Acad Dermatol. 2019; 80:178-188.e3.
The Diagnosis: Basal Cell Carcinoma
Histopathology was consistent with fungating basal cell carcinoma (BCC). The nodules were comprised of syncytial basaloid cells with high nuclear to cytoplasmic ratios, numerous mitotic figures, fibromyxoid stroma, and peripheral nuclear palisading (Figure). Fortunately, no perineural or lymphovascular invasion was identified, and the margins of the specimen were negative. Despite the high-risk nature of giant BCC, the mass was solitary without notable local invasion, leaving it amendable to surgery. On follow-up, the patient has remained recurrence free, and her hemoglobin level has since stabilized.
Skin cancer is the most common malignancy worldwide, and BCC accounts for more than 80% of nonmelanoma skin cancers in the United States. The incidence is on the rise due to the aging population and increasing cumulative skin exposure.1 Risk factors include both individual physical characteristics and environmental exposures. Individuals with lighter skin tones, red and blonde hair, and blue and green eyes are at an increased risk.2 UV radiation exposure is the most important cause of BCC.3 Chronic immunosuppression and exposure to arsenic, ionizing radiation, and psoralen plus UVA radiation also have been linked to the development of BCC.4-6 Basal cell carcinomas most commonly arise on sun-exposed areas such as the face, though more than 10% of cases appear on the trunk.7 Lesions characteristically remain localized, and growth rate is variable; however, when left untreated, BCCs have the potential to become locally destructive and difficult to treat.
Advanced BCCs are tumors that penetrate deeply into the skin. They often are not amenable to traditional therapy and/ or metastasize. Those that grow to a diameter greater than 5 cm, as in our patient, are known as giant BCCs. Only 0.5% to 1% of BCCs are giant BCCs8 ; they typically are more aggressive in nature with higher rates of local recurrence and metastasis. Individuals who develop giant BCCs either have had a delay in access to medical care or a history of BCC that was inadequately managed.9,10 During the COVID-19 pandemic, patient access to health care was substantially impacted during lockdowns. As in our patient, skin neoplasms and other medical conditions may present in later stages due to medical neglect.11,12 Metastasis is rare, even in advanced BCCs. A review of the literature from 1984 estimated that the incidence of metastasis of BCCs is 1 in 1000 to 35,000. Metastasis portends a poor prognosis with a median overall survival of 8 to 14 months.13 An updated review in 2013 found similar outcomes.14
The choice of management for BCCs depends on the risk for recurrence as well as individual patient factors. Characteristics such as tumor size, location, histology, whether it is a primary or recurrent lesion, and the presence of chronic skin disease determine the recurrence rate.15 The management of advanced BCCs often requires a multidisciplinary approach, as these neoplasms may not be amenable to local therapy without causing substantial morbidity. Mohs micrographic surgery is the treatment of choice for BCCs at high risk for recurrence.16 Standard surgical excision with postoperative margin assessment is acceptable when Mohs micrographic surgery is not available.17 Radiation therapy is an alternative for patients who are not candidates for surgery.18
Recently, improved understanding of the molecular pathogenesis of BCCs has led to the development of novel systemic therapies. The Hedgehog signaling pathway has been found to play a critical role in the development of most BCCs.19 Vismodegib and sonidegib are small-molecule inhibitors of the Hedgehog signaling pathway approved for the treatment of locally advanced and metastatic BCCs that are not amenable to surgery or radiation. Approximately 50% of advanced BCCs respond to these therapies; however, long-term treatment may be limited by intolerable side effects and the development of resistance.20 Basal cell carcinomas that spread to lymph nodes or distant sites are treated with traditional systemic therapy. Historically, conventional cytotoxic chemotherapies, such as platinum-containing regimens, were employed with limited benefit and notable morbidity.21
The differential diagnosis for our patient included several other cutaneous neoplasms. Squamous cell carcinoma is the second most common type of skin cancer. Similar to BCC, it can reach a substantial size if left untreated. Risk factors include chronic inflammation, exposure to radiation or chemical carcinogens, burns, human papillomavirus, and other chronic infections. Giant squamous cell carcinomas have high malignant potential and require imaging to assess the extent of invasion and for metastasis. Surgery typically is necessary for both staging and treatment. Adjuvant therapy also may be necessary.22,23
Internal malignant neoplasms rarely present as cutaneous metastases. Breast cancer, melanoma, and cancers of the upper respiratory tract most frequently metastasize to the skin. Although colorectal cancer (CRC) rarely metastasizes to the skin, it is an important cause of cutaneous metastasis due to its high incidence in the general population. When it does spread to the skin, CRC preferentially affects the abdominal wall. Lesions typically resemble the primary tumor but may appear anaplastic. The occurrence of cutaneous metastasis suggests latestage disease and carries a poor prognosis.24
Merkel cell carcinoma and melanoma are aggressive skin cancers with high mortality rates. The former is rarer but more lethal. Merkel cell carcinomas typically occur in elderly white men on sun-exposed areas of the skin. Tumors present as asymptomatic, rapidly expanding, blue-red, firm nodules. Immunosuppression and UV light exposure are notable risk factors.25 Of the 4 major subtypes of cutaneous melanoma, superficial spreading is the most common, followed by nodular, lentigo maligna, and acral lentiginous.26 Superficial spreading melanoma characteristically presents as an expanding asymmetric macule or thin plaque with irregular borders and variation in size and color (black, brown, or red). Nodular melanoma usually presents as symmetric in shape and color (amelanotic, black, or brown). Early recognition by both the patient and clinician is essential in preventing tumor growth and progression.27
Our patient’s presentation was highly concerning for cutaneous metastasis given her history of CRC. Furthermore, the finding of severe anemia was atypical for skin cancer and more characteristic of the prior malignancy. Imaging revealed a locally confined mass with no evidence of extension, lymph node involvement, or additional lesions. The diagnosis was clinched with histopathologic examination.
The Diagnosis: Basal Cell Carcinoma
Histopathology was consistent with fungating basal cell carcinoma (BCC). The nodules were comprised of syncytial basaloid cells with high nuclear to cytoplasmic ratios, numerous mitotic figures, fibromyxoid stroma, and peripheral nuclear palisading (Figure). Fortunately, no perineural or lymphovascular invasion was identified, and the margins of the specimen were negative. Despite the high-risk nature of giant BCC, the mass was solitary without notable local invasion, leaving it amendable to surgery. On follow-up, the patient has remained recurrence free, and her hemoglobin level has since stabilized.
Skin cancer is the most common malignancy worldwide, and BCC accounts for more than 80% of nonmelanoma skin cancers in the United States. The incidence is on the rise due to the aging population and increasing cumulative skin exposure.1 Risk factors include both individual physical characteristics and environmental exposures. Individuals with lighter skin tones, red and blonde hair, and blue and green eyes are at an increased risk.2 UV radiation exposure is the most important cause of BCC.3 Chronic immunosuppression and exposure to arsenic, ionizing radiation, and psoralen plus UVA radiation also have been linked to the development of BCC.4-6 Basal cell carcinomas most commonly arise on sun-exposed areas such as the face, though more than 10% of cases appear on the trunk.7 Lesions characteristically remain localized, and growth rate is variable; however, when left untreated, BCCs have the potential to become locally destructive and difficult to treat.
Advanced BCCs are tumors that penetrate deeply into the skin. They often are not amenable to traditional therapy and/ or metastasize. Those that grow to a diameter greater than 5 cm, as in our patient, are known as giant BCCs. Only 0.5% to 1% of BCCs are giant BCCs8 ; they typically are more aggressive in nature with higher rates of local recurrence and metastasis. Individuals who develop giant BCCs either have had a delay in access to medical care or a history of BCC that was inadequately managed.9,10 During the COVID-19 pandemic, patient access to health care was substantially impacted during lockdowns. As in our patient, skin neoplasms and other medical conditions may present in later stages due to medical neglect.11,12 Metastasis is rare, even in advanced BCCs. A review of the literature from 1984 estimated that the incidence of metastasis of BCCs is 1 in 1000 to 35,000. Metastasis portends a poor prognosis with a median overall survival of 8 to 14 months.13 An updated review in 2013 found similar outcomes.14
The choice of management for BCCs depends on the risk for recurrence as well as individual patient factors. Characteristics such as tumor size, location, histology, whether it is a primary or recurrent lesion, and the presence of chronic skin disease determine the recurrence rate.15 The management of advanced BCCs often requires a multidisciplinary approach, as these neoplasms may not be amenable to local therapy without causing substantial morbidity. Mohs micrographic surgery is the treatment of choice for BCCs at high risk for recurrence.16 Standard surgical excision with postoperative margin assessment is acceptable when Mohs micrographic surgery is not available.17 Radiation therapy is an alternative for patients who are not candidates for surgery.18
Recently, improved understanding of the molecular pathogenesis of BCCs has led to the development of novel systemic therapies. The Hedgehog signaling pathway has been found to play a critical role in the development of most BCCs.19 Vismodegib and sonidegib are small-molecule inhibitors of the Hedgehog signaling pathway approved for the treatment of locally advanced and metastatic BCCs that are not amenable to surgery or radiation. Approximately 50% of advanced BCCs respond to these therapies; however, long-term treatment may be limited by intolerable side effects and the development of resistance.20 Basal cell carcinomas that spread to lymph nodes or distant sites are treated with traditional systemic therapy. Historically, conventional cytotoxic chemotherapies, such as platinum-containing regimens, were employed with limited benefit and notable morbidity.21
The differential diagnosis for our patient included several other cutaneous neoplasms. Squamous cell carcinoma is the second most common type of skin cancer. Similar to BCC, it can reach a substantial size if left untreated. Risk factors include chronic inflammation, exposure to radiation or chemical carcinogens, burns, human papillomavirus, and other chronic infections. Giant squamous cell carcinomas have high malignant potential and require imaging to assess the extent of invasion and for metastasis. Surgery typically is necessary for both staging and treatment. Adjuvant therapy also may be necessary.22,23
Internal malignant neoplasms rarely present as cutaneous metastases. Breast cancer, melanoma, and cancers of the upper respiratory tract most frequently metastasize to the skin. Although colorectal cancer (CRC) rarely metastasizes to the skin, it is an important cause of cutaneous metastasis due to its high incidence in the general population. When it does spread to the skin, CRC preferentially affects the abdominal wall. Lesions typically resemble the primary tumor but may appear anaplastic. The occurrence of cutaneous metastasis suggests latestage disease and carries a poor prognosis.24
Merkel cell carcinoma and melanoma are aggressive skin cancers with high mortality rates. The former is rarer but more lethal. Merkel cell carcinomas typically occur in elderly white men on sun-exposed areas of the skin. Tumors present as asymptomatic, rapidly expanding, blue-red, firm nodules. Immunosuppression and UV light exposure are notable risk factors.25 Of the 4 major subtypes of cutaneous melanoma, superficial spreading is the most common, followed by nodular, lentigo maligna, and acral lentiginous.26 Superficial spreading melanoma characteristically presents as an expanding asymmetric macule or thin plaque with irregular borders and variation in size and color (black, brown, or red). Nodular melanoma usually presents as symmetric in shape and color (amelanotic, black, or brown). Early recognition by both the patient and clinician is essential in preventing tumor growth and progression.27
Our patient’s presentation was highly concerning for cutaneous metastasis given her history of CRC. Furthermore, the finding of severe anemia was atypical for skin cancer and more characteristic of the prior malignancy. Imaging revealed a locally confined mass with no evidence of extension, lymph node involvement, or additional lesions. The diagnosis was clinched with histopathologic examination.
- Rogers HW, Weinstock MA, Harris AR, et al. Incidence estimate of nonmelanoma skin cancer in the United States, 2006. Arch Dermatol. 2010;146:283-287.
- Lear JT, Tan BB, Smith AG, et al. Risk factors for basal cell carcinoma in the UK: case-control study in 806 patients. J R Soc Med. 1997; 90:371-374.
- Gallagher RP, Hill GB, Bajdik CD, et al. Sunlight exposure, pigmentary factors, and risk of nonmelanocytic skin cancer: I. basal cell carcinoma. Arch Dermatol. 1995;131:157-163.
- Guo HR, Yu HS, Hu H, et al. Arsenic in drinking water and skin cancers: cell-type specificity (Taiwan, ROC). Cancer Causes Control. 2001;12:909-916.
- Lichter MD, Karagas MR, Mott LA, et al; The New Hampshire Skin Cancer Study Group. Therapeutic ionizing radiation and the incidence of basal cell carcinoma and squamous cell carcinoma. Arch Dermatol. 2000;136:1007-1011.
- Nijsten TEC, Stern RS. The increased risk of skin cancer is persistent after discontinuation of psoralen plus ultraviolet A: a cohort study. J Invest Dermatol. 2003;121:252-258.
- Scrivener Y, Grosshans E, Cribier B. Variations of basal cell carcinomas according to gender, age, location and histopathological subtype. Br J Dermatol. 2002;147:41-47.
- Gualdi G, Monari P, Calzavara‐Pinton P, et al. When basal cell carcinomas became giant: an Italian multicenter study. Int J Dermatol. 2020;59:377-382.
- Randle HW, Roenigk RK, Brodland DG. Giant basal cell carcinoma (T3). who is at risk? Cancer. 1993;72:1624-1630.
- Archontaki M, Stavrianos SD, Korkolis DP, et al. Giant basal cell carcinoma: clinicopathological analysis of 51 cases and review of the literature. Anticancer Res. 2009;29:2655-2663.
- Shifat Ahmed SAK, Ajisola M, Azeem K, et al. Impact of the societal response to COVID-19 on access to healthcare for non-COVID-19 health issues in slum communities of Bangladesh, Kenya, Nigeria and Pakistan: results of pre-COVID and COVID-19 lockdown ssstakeholder engagements. BMJ Glob Health. 2020;5:E003042.
- Gomolin T, Cline A, Handler MZ. The danger of neglecting melanoma during the COVID-19 pandemic. J Dermatolog Treat. 2020;31:444-445.
- von Domarus H, Stevens PJ. Metastatic basal cell carcinoma. report of five cases and review of 170 cases in the literature. J Am Acad Dermatol. 1984;10:1043-1060.
- Wysong A, Aasi SZ, Tang JY. Update on metastatic basal cell carcinoma: a summary of published cases from 1981 through 2011. JAMA Dermatol. 2013;149:615-616.
- Bøgelund FS, Philipsen PA, Gniadecki R. Factors affecting the recurrence rate of basal cell carcinoma. Acta Derm Venereol. 2007;87:330-334.
- Mosterd K, Krekels GAM, Nieman FH, et al. Surgical excision versus Mohs’ micrographic surgery for primary and recurrent basal-cell carcinoma of the face: a prospective randomised controlled trial with 5-years’ follow-up. Lancet Oncol. 2008;9:1149-1156.
- Wetzig T, Woitek M, Eichhorn K, et al. Surgical excision of basal cell carcinoma with complete margin control: outcome at 5-year follow-up. Dermatology. 2010;220:363-369.
- Silverman MK, Kopf AW, Gladstein AH, et al. Recurrence rates of treated basal cell carcinomas. part 4: X-ray therapy. J Dermatol Surg Oncol. 1992;18:549-554.
- Tanese K, Emoto K, Kubota N, et al. Immunohistochemical visualization of the signature of activated Hedgehog signaling pathway in cutaneous epithelial tumors. J Dermatol. 2018;45:1181-1186.
- Basset-Séguin N, Hauschild A, Kunstfeld R, et al. Vismodegib in patients with advanced basal cell carcinoma: primary analysis of STEVIE, an international, open-label trial. Eur J Cancer. 2017;86:334-348.
- Carneiro BA, Watkin WG, Mehta UK, et al. Metastatic basal cell carcinoma: complete response to chemotherapy and associated pure red cell aplasia. Cancer Invest. 2006;24:396-400.
- Misiakos EP, Damaskou V, Koumarianou A, et al. A giant squamous cell carcinoma of the skin of the thoracic wall: a case report and review of the literature. J Med Case Rep. 2017;11:136.
- Wollina U, Bayyoud Y, Krönert C, et al. Giant epithelial malignancies (basal cell carcinoma, squamous cell carcinoma): a series of 20 tumors from a single center. J Cutan Aesthet Surg. 2012;5:12-19.
- Bittencourt MJS, Imbiriba AA, Oliveira OA, et al. Cutaneous metastasis of colorectal cancer. An Bras Dermatol. 2018;93:884-886.
- Heath M, Jaimes N, Lemos B, et al. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the AEIOU features. J Am Acad Dermatol. 2008;58:375-381.
- Buettner PG, Leiter U, Eigentler TK, et al. Development of prognostic factors and survival in cutaneous melanoma over 25 years: an analysis of the Central Malignant Melanoma Registry of the German Dermatological Society. Cancer. 2005;103:616-624.
- Klebanov N, Gunasekera N, Lin WM, et al. The clinical spectrum of cutaneous melanoma morphology. J Am Acad Dermatol. 2019; 80:178-188.e3.
- Rogers HW, Weinstock MA, Harris AR, et al. Incidence estimate of nonmelanoma skin cancer in the United States, 2006. Arch Dermatol. 2010;146:283-287.
- Lear JT, Tan BB, Smith AG, et al. Risk factors for basal cell carcinoma in the UK: case-control study in 806 patients. J R Soc Med. 1997; 90:371-374.
- Gallagher RP, Hill GB, Bajdik CD, et al. Sunlight exposure, pigmentary factors, and risk of nonmelanocytic skin cancer: I. basal cell carcinoma. Arch Dermatol. 1995;131:157-163.
- Guo HR, Yu HS, Hu H, et al. Arsenic in drinking water and skin cancers: cell-type specificity (Taiwan, ROC). Cancer Causes Control. 2001;12:909-916.
- Lichter MD, Karagas MR, Mott LA, et al; The New Hampshire Skin Cancer Study Group. Therapeutic ionizing radiation and the incidence of basal cell carcinoma and squamous cell carcinoma. Arch Dermatol. 2000;136:1007-1011.
- Nijsten TEC, Stern RS. The increased risk of skin cancer is persistent after discontinuation of psoralen plus ultraviolet A: a cohort study. J Invest Dermatol. 2003;121:252-258.
- Scrivener Y, Grosshans E, Cribier B. Variations of basal cell carcinomas according to gender, age, location and histopathological subtype. Br J Dermatol. 2002;147:41-47.
- Gualdi G, Monari P, Calzavara‐Pinton P, et al. When basal cell carcinomas became giant: an Italian multicenter study. Int J Dermatol. 2020;59:377-382.
- Randle HW, Roenigk RK, Brodland DG. Giant basal cell carcinoma (T3). who is at risk? Cancer. 1993;72:1624-1630.
- Archontaki M, Stavrianos SD, Korkolis DP, et al. Giant basal cell carcinoma: clinicopathological analysis of 51 cases and review of the literature. Anticancer Res. 2009;29:2655-2663.
- Shifat Ahmed SAK, Ajisola M, Azeem K, et al. Impact of the societal response to COVID-19 on access to healthcare for non-COVID-19 health issues in slum communities of Bangladesh, Kenya, Nigeria and Pakistan: results of pre-COVID and COVID-19 lockdown ssstakeholder engagements. BMJ Glob Health. 2020;5:E003042.
- Gomolin T, Cline A, Handler MZ. The danger of neglecting melanoma during the COVID-19 pandemic. J Dermatolog Treat. 2020;31:444-445.
- von Domarus H, Stevens PJ. Metastatic basal cell carcinoma. report of five cases and review of 170 cases in the literature. J Am Acad Dermatol. 1984;10:1043-1060.
- Wysong A, Aasi SZ, Tang JY. Update on metastatic basal cell carcinoma: a summary of published cases from 1981 through 2011. JAMA Dermatol. 2013;149:615-616.
- Bøgelund FS, Philipsen PA, Gniadecki R. Factors affecting the recurrence rate of basal cell carcinoma. Acta Derm Venereol. 2007;87:330-334.
- Mosterd K, Krekels GAM, Nieman FH, et al. Surgical excision versus Mohs’ micrographic surgery for primary and recurrent basal-cell carcinoma of the face: a prospective randomised controlled trial with 5-years’ follow-up. Lancet Oncol. 2008;9:1149-1156.
- Wetzig T, Woitek M, Eichhorn K, et al. Surgical excision of basal cell carcinoma with complete margin control: outcome at 5-year follow-up. Dermatology. 2010;220:363-369.
- Silverman MK, Kopf AW, Gladstein AH, et al. Recurrence rates of treated basal cell carcinomas. part 4: X-ray therapy. J Dermatol Surg Oncol. 1992;18:549-554.
- Tanese K, Emoto K, Kubota N, et al. Immunohistochemical visualization of the signature of activated Hedgehog signaling pathway in cutaneous epithelial tumors. J Dermatol. 2018;45:1181-1186.
- Basset-Séguin N, Hauschild A, Kunstfeld R, et al. Vismodegib in patients with advanced basal cell carcinoma: primary analysis of STEVIE, an international, open-label trial. Eur J Cancer. 2017;86:334-348.
- Carneiro BA, Watkin WG, Mehta UK, et al. Metastatic basal cell carcinoma: complete response to chemotherapy and associated pure red cell aplasia. Cancer Invest. 2006;24:396-400.
- Misiakos EP, Damaskou V, Koumarianou A, et al. A giant squamous cell carcinoma of the skin of the thoracic wall: a case report and review of the literature. J Med Case Rep. 2017;11:136.
- Wollina U, Bayyoud Y, Krönert C, et al. Giant epithelial malignancies (basal cell carcinoma, squamous cell carcinoma): a series of 20 tumors from a single center. J Cutan Aesthet Surg. 2012;5:12-19.
- Bittencourt MJS, Imbiriba AA, Oliveira OA, et al. Cutaneous metastasis of colorectal cancer. An Bras Dermatol. 2018;93:884-886.
- Heath M, Jaimes N, Lemos B, et al. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the AEIOU features. J Am Acad Dermatol. 2008;58:375-381.
- Buettner PG, Leiter U, Eigentler TK, et al. Development of prognostic factors and survival in cutaneous melanoma over 25 years: an analysis of the Central Malignant Melanoma Registry of the German Dermatological Society. Cancer. 2005;103:616-624.
- Klebanov N, Gunasekera N, Lin WM, et al. The clinical spectrum of cutaneous melanoma morphology. J Am Acad Dermatol. 2019; 80:178-188.e3.
A 77-year-old woman was admitted to the hospital with anemia (hemoglobin, 5.2 g/dL [reference range, 12.0–15.5 g/dL]) and a rapidly growing abdominal wall mass. She had a history of stage IIA colon cancer (T3N0M0) that was treated 5 years prior with a partial colon resection and adjuvant chemotherapy. She initially noticed a red scaly lesion developing around a scar from a prior surgery that had been stable for years. Over the last 2 months, the lesion rapidly expanded and would intermittently bleed. Physical examination revealed a 13×10×4.5-cm, pink-red, nodular, firm mass over the patient’s right upper quadrant. Computed tomography revealed a mass limited to the skin and superficial tissue. General surgery was consulted for excision of the mass.


Sarcoidosis Presenting as Telangiectatic Macules
To the Editor:
Sarcoidosis is a multisystem, noncaseating, granulomatous disorder thought to occur from a combination of immunologic, genetic, and environmental factors.1 Often referred to as the “great imitator,” the cutaneous manifestations of sarcoidosis encompass many morphologies, including papules, plaques, nodules, and scars.1 We report an unusual case of sarcoidosis presenting as telangiectatic macules on the lower extremities.
A woman in her early 30s presented with a burning, pruritic, erythematous, telangiectatic eruption on the lower extremities with concurrent ankle swelling of 4 weeks’ duration. The patient denied any fevers, chills, recent infections, or new medications. Evaluation by her primary care physician during the time of the eruption included unremarkable antinuclear antibodies, thyroid stimulating hormone level, complete blood cell count, comprehensive metabolic panel, urinalysis, chest radiography, and lower-extremity Doppler ultrasonography.
Physical examination at the current presentation revealed numerous scattered, faint, erythematous, blanchable macules on the lower extremities along with mild pitting edema (Figure 1). The patient’s current medications included cetirizine, which she had been taking for years, as well as an intrauterine device. A punch biopsy from the right lower leg revealed small, well-demarcated sarcoidal granulomatous inflammation surrounding vascular structures and skin appendages (Figure 2). No foreign bodies were observed with polarized light microscopy. Microscopic findings suggestive of an infection, including caseation necrosis and suppurative inflammation, also were absent. Angiotensin-converting enzyme levels were normal. Myeloperoxidase and proteinase 3 IgG antibody levels were evaluated due to potential vascular involvement but were negative. An infectious cause of the sarcoidal granulomas was unlikely given histopathologic findings and negative tuberculosis skin testing, which the patient underwent annually for her job, so a tissue culture was not performed. The patient was prescribed triamcinolone acetonide cream 0.1% for the itching and burning at the initial visit and was continued on this treatment after the diagnosis of sarcoidosis was made. At 2-month follow-up, the patient’s eruption had nearly resolved with topical therapy.

Cutaneous manifestation occurs in 20% to 35% of sarcoidosis cases and may develop in the presence or absence of systemic disease. Approximately 60% of individuals with cutaneous sarcoidosis are found to have systemic involvement; therefore, careful monitoring and diagnostic workup are important in the management of these patients.2 While most cases of cutaneous sarcoidosis are papular, it is important for clinicians to maintain a level of suspicion for sarcoidosis in any uncertain dermatologic presentation.1,2 Evidence of telangiectasias has been shown in rarer forms of sarcoidosis (eg, angiolupoid), but the lesions usually are confined to the face, ears, or neck.3 Granulomatous vasculitis has been reported in a small number of individuals with ulcerative sarcoidosis.4 In our case, no ulcerations were present, possibly indicating an early lesion or an entirely novel process. Lastly, although reticular dermal granulomas are found in drug-induced interstitial granulomatous dermatitis, these lesions often are dispersed interstitially amongst collagen bundles and are associated with necrobiosis of collagen and eosinophilic/neutrophilic infiltrates.5 The lack of these characteristic pathologic findings in our patient along with no known reported cases of cetirizine-induced granulomatous dermatitis led us to rule out reticular dermal granulomas as a diagnosis. We present our case as a reminder of the diversity of cutaneous sarcoidosis manifestations and the importance of early diagnosis of these lesions.
.")
- Haimovic A, Sanchez M, Judson MA, et al. Sarcoidosis: a comprehensive review and update for the dermatologist: part I. cutaneous disease. J Am Acad Dermatol. 2012;66:699.E1-E18.
- Yanardag H, Tetikkurt C, Bilir M, et al. Diagnosis of cutaneous sarcoidosis; clinical and the prognostic significance of skin lesions. Multidiscip Respir Med. 2013;8:26.
- Arias-Santiago S, Fernández-Pugnaire MA, Aneiros- Fernández J, et al. Recurrent telangiectasias on the cheek: angiolupoid sarcoidosis. Am J Med. 2010;123:E7-E8.
- Wei C-H, Huang Y-H, Shih Y-C, et al. Sarcoidosis with cutaneous granulomatous vasculitis. Australas J Dermatol. 2010;51:198-201.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
To the Editor:
Sarcoidosis is a multisystem, noncaseating, granulomatous disorder thought to occur from a combination of immunologic, genetic, and environmental factors.1 Often referred to as the “great imitator,” the cutaneous manifestations of sarcoidosis encompass many morphologies, including papules, plaques, nodules, and scars.1 We report an unusual case of sarcoidosis presenting as telangiectatic macules on the lower extremities.
A woman in her early 30s presented with a burning, pruritic, erythematous, telangiectatic eruption on the lower extremities with concurrent ankle swelling of 4 weeks’ duration. The patient denied any fevers, chills, recent infections, or new medications. Evaluation by her primary care physician during the time of the eruption included unremarkable antinuclear antibodies, thyroid stimulating hormone level, complete blood cell count, comprehensive metabolic panel, urinalysis, chest radiography, and lower-extremity Doppler ultrasonography.
Physical examination at the current presentation revealed numerous scattered, faint, erythematous, blanchable macules on the lower extremities along with mild pitting edema (Figure 1). The patient’s current medications included cetirizine, which she had been taking for years, as well as an intrauterine device. A punch biopsy from the right lower leg revealed small, well-demarcated sarcoidal granulomatous inflammation surrounding vascular structures and skin appendages (Figure 2). No foreign bodies were observed with polarized light microscopy. Microscopic findings suggestive of an infection, including caseation necrosis and suppurative inflammation, also were absent. Angiotensin-converting enzyme levels were normal. Myeloperoxidase and proteinase 3 IgG antibody levels were evaluated due to potential vascular involvement but were negative. An infectious cause of the sarcoidal granulomas was unlikely given histopathologic findings and negative tuberculosis skin testing, which the patient underwent annually for her job, so a tissue culture was not performed. The patient was prescribed triamcinolone acetonide cream 0.1% for the itching and burning at the initial visit and was continued on this treatment after the diagnosis of sarcoidosis was made. At 2-month follow-up, the patient’s eruption had nearly resolved with topical therapy.
Cutaneous manifestation occurs in 20% to 35% of sarcoidosis cases and may develop in the presence or absence of systemic disease. Approximately 60% of individuals with cutaneous sarcoidosis are found to have systemic involvement; therefore, careful monitoring and diagnostic workup are important in the management of these patients.2 While most cases of cutaneous sarcoidosis are papular, it is important for clinicians to maintain a level of suspicion for sarcoidosis in any uncertain dermatologic presentation.1,2 Evidence of telangiectasias has been shown in rarer forms of sarcoidosis (eg, angiolupoid), but the lesions usually are confined to the face, ears, or neck.3 Granulomatous vasculitis has been reported in a small number of individuals with ulcerative sarcoidosis.4 In our case, no ulcerations were present, possibly indicating an early lesion or an entirely novel process. Lastly, although reticular dermal granulomas are found in drug-induced interstitial granulomatous dermatitis, these lesions often are dispersed interstitially amongst collagen bundles and are associated with necrobiosis of collagen and eosinophilic/neutrophilic infiltrates.5 The lack of these characteristic pathologic findings in our patient along with no known reported cases of cetirizine-induced granulomatous dermatitis led us to rule out reticular dermal granulomas as a diagnosis. We present our case as a reminder of the diversity of cutaneous sarcoidosis manifestations and the importance of early diagnosis of these lesions.
To the Editor:
Sarcoidosis is a multisystem, noncaseating, granulomatous disorder thought to occur from a combination of immunologic, genetic, and environmental factors.1 Often referred to as the “great imitator,” the cutaneous manifestations of sarcoidosis encompass many morphologies, including papules, plaques, nodules, and scars.1 We report an unusual case of sarcoidosis presenting as telangiectatic macules on the lower extremities.
A woman in her early 30s presented with a burning, pruritic, erythematous, telangiectatic eruption on the lower extremities with concurrent ankle swelling of 4 weeks’ duration. The patient denied any fevers, chills, recent infections, or new medications. Evaluation by her primary care physician during the time of the eruption included unremarkable antinuclear antibodies, thyroid stimulating hormone level, complete blood cell count, comprehensive metabolic panel, urinalysis, chest radiography, and lower-extremity Doppler ultrasonography.
Physical examination at the current presentation revealed numerous scattered, faint, erythematous, blanchable macules on the lower extremities along with mild pitting edema (Figure 1). The patient’s current medications included cetirizine, which she had been taking for years, as well as an intrauterine device. A punch biopsy from the right lower leg revealed small, well-demarcated sarcoidal granulomatous inflammation surrounding vascular structures and skin appendages (Figure 2). No foreign bodies were observed with polarized light microscopy. Microscopic findings suggestive of an infection, including caseation necrosis and suppurative inflammation, also were absent. Angiotensin-converting enzyme levels were normal. Myeloperoxidase and proteinase 3 IgG antibody levels were evaluated due to potential vascular involvement but were negative. An infectious cause of the sarcoidal granulomas was unlikely given histopathologic findings and negative tuberculosis skin testing, which the patient underwent annually for her job, so a tissue culture was not performed. The patient was prescribed triamcinolone acetonide cream 0.1% for the itching and burning at the initial visit and was continued on this treatment after the diagnosis of sarcoidosis was made. At 2-month follow-up, the patient’s eruption had nearly resolved with topical therapy.
Cutaneous manifestation occurs in 20% to 35% of sarcoidosis cases and may develop in the presence or absence of systemic disease. Approximately 60% of individuals with cutaneous sarcoidosis are found to have systemic involvement; therefore, careful monitoring and diagnostic workup are important in the management of these patients.2 While most cases of cutaneous sarcoidosis are papular, it is important for clinicians to maintain a level of suspicion for sarcoidosis in any uncertain dermatologic presentation.1,2 Evidence of telangiectasias has been shown in rarer forms of sarcoidosis (eg, angiolupoid), but the lesions usually are confined to the face, ears, or neck.3 Granulomatous vasculitis has been reported in a small number of individuals with ulcerative sarcoidosis.4 In our case, no ulcerations were present, possibly indicating an early lesion or an entirely novel process. Lastly, although reticular dermal granulomas are found in drug-induced interstitial granulomatous dermatitis, these lesions often are dispersed interstitially amongst collagen bundles and are associated with necrobiosis of collagen and eosinophilic/neutrophilic infiltrates.5 The lack of these characteristic pathologic findings in our patient along with no known reported cases of cetirizine-induced granulomatous dermatitis led us to rule out reticular dermal granulomas as a diagnosis. We present our case as a reminder of the diversity of cutaneous sarcoidosis manifestations and the importance of early diagnosis of these lesions.
- Haimovic A, Sanchez M, Judson MA, et al. Sarcoidosis: a comprehensive review and update for the dermatologist: part I. cutaneous disease. J Am Acad Dermatol. 2012;66:699.E1-E18.
- Yanardag H, Tetikkurt C, Bilir M, et al. Diagnosis of cutaneous sarcoidosis; clinical and the prognostic significance of skin lesions. Multidiscip Respir Med. 2013;8:26.
- Arias-Santiago S, Fernández-Pugnaire MA, Aneiros- Fernández J, et al. Recurrent telangiectasias on the cheek: angiolupoid sarcoidosis. Am J Med. 2010;123:E7-E8.
- Wei C-H, Huang Y-H, Shih Y-C, et al. Sarcoidosis with cutaneous granulomatous vasculitis. Australas J Dermatol. 2010;51:198-201.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Haimovic A, Sanchez M, Judson MA, et al. Sarcoidosis: a comprehensive review and update for the dermatologist: part I. cutaneous disease. J Am Acad Dermatol. 2012;66:699.E1-E18.
- Yanardag H, Tetikkurt C, Bilir M, et al. Diagnosis of cutaneous sarcoidosis; clinical and the prognostic significance of skin lesions. Multidiscip Respir Med. 2013;8:26.
- Arias-Santiago S, Fernández-Pugnaire MA, Aneiros- Fernández J, et al. Recurrent telangiectasias on the cheek: angiolupoid sarcoidosis. Am J Med. 2010;123:E7-E8.
- Wei C-H, Huang Y-H, Shih Y-C, et al. Sarcoidosis with cutaneous granulomatous vasculitis. Australas J Dermatol. 2010;51:198-201.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
Practice Points
- Cutaneous manifestations of sarcoidosis can encompass numerous morphologies. A high degree of suspicion should be maintained for any uncertain dermatologic presentation.
- Although papular eruptions are the most common cutaneous findings in sarcoidosis, this case report illustrates a less common vascular-appearing presentation.
- A systemic workup is indicated in any presentation of sarcoidosis.

Graft-vs-host Disease and Toxic Epidermal Necrolysis Following Hematopoietic Stem Cell Transplantation
To the Editor:
Acute graft-vs-host disease (GVHD) remains a limitation to hematopoietic stem cell transplantation (HSCT) in 20% to 50% of patients after transplant. Furthermore, failed treatment with corticosteroids is frequent and portends a poor prognosis.1 Toxic epidermal necrolysis (TEN) is an epidermolytic skin disorder thought to represent an adverse drug reaction, though its pathogenesis remains unclear. Severe forms of acute GVHD can mimic TEN clinically and histologically. Both can present with widespread cutaneous and mucosal bullae, erosions, and desquamation. Toxic epidermal necrolysis in the context of allogeneic hematopoietic stem cell transplantation is extremely rare, with almost 100% mortality in adult patients. Features that favor acute GVHD over TEN include diarrhea, elevation in bilirubin level, and chimerism.2 However, these features might be absent, posing a therapeutic dilemma, as current treatment preferences for each of these entities differ.
Growing evidence supports the use of anti–tumor necrosis factor (TNF) α drugs for the treatment of TEN. Success has been reported with both anti–TNF-α monoclonal antibodies as well as the soluble fusion protein etanercept.3,4 The use of TNF-α inhibitors in acute GVHD remains anecdotal.

A 58-year-old man (patient 1) with a history of acute myelogenous leukemia presented with a pruritic morbilliform eruption 28 days after HSCT. There was no desquamation or mucosal involvement and the biopsy obtained was histologically suggestive of grade 2 acute GVHD. His immunosuppressive regimen included sirolimus and cyclophosphamide. He was receiving trimethoprim-sulfamethoxazole (TMP-SMX), voriconazole, and acyclovir for infectious prophylaxis. At the time of presentation, he was treated with high-dose systemic steroids (prednisone 2 mg/kg/d) for acute GVHD with partial improvement. Upon tapering of the steroids 3 weeks after initiating TMP-SMX and 1 week after initiating voriconazole, he developed painful desquamation and erosions involving 95% of the body surface area (BSA), necessitating admission to the local burn unit (Figure 1). Biopsies demonstrated full-thickness epidermal necrosis with subepidermal blistering and interface dermatitis (Figure 2). No gastrointestinal tract involvement of acute GVHD was noted. The patient was a 100% donor chimera, supporting the diagnosis of acute GVHD; however, the patient and donor carried the HLA-C*06:02 allele, which previously has been described in association with TMP-SMX–related Stevens-Johnson syndrome/TEN.5 In addition, causality assessment using the algorithm of drug causality for epidermal necrolysis indicated TMP-SMX as a probable cause and voriconazole as a possible cause. The diagnosis of TEN with a SCORe of Toxic Epidermal Necrosis (SCORTEN) of 4 in the setting of acute GVHD was favored, though grade 4 acute GVHD could not be excluded. Trimethoprim-sulfamethoxazole was discontinued, and voriconazole was changed to posaconazole. He received supportive care along with 1 dose of 25-mg subcutaneous etanercept and 3 days of intravenous immunoglobulin (IVIG). Skin re-epithelialization was complete by 3 weeks. At 4 weeks, the patient developed a new asymptomatic erythematous eruption. Biopsies demonstrated changes of acute and chronic GVHD (Figure 3) that resolved with up-titration of sirolimus. The patient remained hospitalized for 96 days and continued to follow up with his transplant team as well as ophthalmology and dermatology. He died 2 years after HSCT.

A 67-year-old woman (patient 2) with high-grade myelodysplastic syndrome presented with an erythematous morbilliform eruption on the torso on day 20 after a matched unrelated HSCT that histologically was consistent with grade 2 GVHD (Figure 4). She had been receiving sirolimus and tacrolimus for GVHD prophylaxis. Infectious prophylaxis included acyclovir, pentamidine, micafungin, and TMP-SMX. Despite high-dose systemic steroids, the rash progressed and ultimately involved 80% BSA. A positive Nikolsky sign was noted involving 21% BSA (Figure 5), in addition to oral and genital mucosal ulcers. She denied nausea, vomiting, fever, or diarrhea. Chimerism studies were negative. Trimethoprim-sulfamethoxazole was discontinued, and she was transferred to a burn unit. Biopsies showed full-thickness epidermal necrosis. A diagnosis of TEN with a SCORTEN of 4 in the setting of acute GVHD was favored; grade 4 acute GVHD could not be excluded. Steroids were discontinued. Because laboratory studies indicated IgA deficiency, IVIG was not considered as a systemic option for therapy. The patient received 1 dose of infliximab (5 mg/kg). Cyclophosphamide 1600 mg weekly was added for GVHD therapy. The wounds progressively healed, and 2 weeks into her admission she was noted to have only 3% BSA with denuded skin. The patient was transferred to the cancer treatment center for further management of the malignancy. Unfortunately, after 2 months she died due to ischemic colitis that was confirmed on autopsy.

Graft-vs-host disease and TEN are rare, life-threatening complications seen in patients with allogeneic HSCT.2 Graft-vs-host disease and TEN share clinicopathologic characteristics and effector immune mechanisms, largely the substantial role of T-cell activation and tissue destruction, which occur through mediators such as TNF-α.6-8

Given the sparse lymphocytic infiltrate, keratinocyte death in TEN is thought to result from soluble molecules, including TNF-α and TNF-related apoptosis-inducing ligand.9 Tumor necrosis factor α has been identified in blister fluid, biopsy specimens, and serum of patients with TEN. Tumor necrosis factor α increases the expression of keratinocyte-inducible nitric oxide synthase, which upregulates keratinocyte Fas ligand expression and subsequent Fas- and caspase-8–mediated keratinocyte cell death.10

Acute GVHD results from donor lymphocyte activation after infusion into damaged recipient tissues that previously have been radiated or chemoablated. Mismatches in histocompatibility complexes between donor cells and recipient tissue antigens serve as the initial trigger for immune activation. Activation of antigen-presenting cells followed by activation, proliferation, differentiation, and migration of donor T cells ultimately results in destruction of the target tissue.11 Immune mediators, such as TNF-α and lymphotoxin α (another member of the TNF superfamily), play a nonredundant role in the pathogenesis of GVHD.12
Current treatment strategies for severe acute GVHD and TEN differ. In North America, high-dose IVIG frequently is used as first-line systemic therapy, while high-dose systemic corticosteroids rarely are used.13 Studies have demonstrated successful use of anti–TNF-α drugs for the treatment of TEN.3,4 Moreover, etanercept has shown to effectively inhibit lymphotoxin α.14 Similarly, TNF inhibition in the management of steroid-refractory acute GVHD has been successful.1 These studies coupled with the underlying immune mechanisms that both diseases share encouraged initiating a trial of anti–TNF-α therapy in our patients.
Patient 1 merits further discussion because he was both a 100% donor chimera as well as a carrier of an human leukocyte antigen susceptibility candidate allele to TMP-SMX. Historical features of his presentation are consistent with either steroid-refractory GVHD or TEN superimposed on acute GVHD. His initial presentation of the more typical macular exanthem of cutaneous acute GVHD was both biopsy proven and supported by clinical improvement with steroid therapy, which was later followed by a robust blistering mucocutaneous presentation approximately 3 weeks after the administration of TMP-SMX and 1 week after initiating voriconazole that improved with IVIG and etanercept.
It is difficult to determine if TEN represents a continuum or result of the underlying drivers of acute GVHD vs a drug reaction. Although there is insufficient evidence to establish a clear-cut diagnosis of TEN, these cases illustrate the need for better diagnostic techniques to allow differentiation between TEN and grade 4 acute GVHD, and in the context of uncertainty, TNF-α inhibition poses a viable therapeutic strategy for these 2 often lethal conditions. Our cases do unequivocally indicate the benefit of this therapeutic modality, add to the current body of literature supporting the use of TNF-α inhibitors in patients such as ours without an official TEN diagnosis, and may guide future investigative efforts.
- Couriel DR, Saliba R, de Lima M, et al. A phase III study of infliximab and corticosteroids for the initial treatment of acute graft-versus-host disease. Biol Blood Marrow Transplant. 2009;15:1555-1562.
- Jeanmonod P, Hubbuch M, Grünhage F, et al. Graft-versus-host disease or toxic epidermal necrolysis: diagnostic dilemma after liver transplantation. Transpl Infect Dis. 2012;14:422-426.
- Paradisi A, Abeni D, Bergamo F, et al. Etanercept therapy for toxic epidermal necrolysis. J Am Acad Dermatol. 2014;71:278-283.
- Scott-Lang V, Tidman M, McKay D. Toxic epidermal necrolysis in a child successfully treated with infliximab. Pediatr Dermatol. 2014;31:532-534.
- Kingpin T, Mahasirimongkol S, Konyoung P, et al. Candidate HLA genes for prediction of co-trimoxazole-induced severe cutaneous reactions. Pharmacogenet Genomics. 2015;25:402-411.
- Correia O, Delgado L, Barbosa IL, et al. Increased interleukin 10, tumor necrosis factor alpha, and interleukin 6 levels in blister fluid of toxic epidermal necrolysis. J Am Acad Dermatol. 2002;47:58-62.
- French LE, Tschopp J. Fas-mediated cell death in toxic epidermal necrolysis and graft-versus-host disease: potential for therapeutic inhibition. Schweiz Med Wochenschr. 2000;130:1656-1661.
- Downey A, Jackson C, Harun N, et al. Toxic epidermal necrolysis: review of pathogenesis and management. J Am Acad Dermatol. 2012;66:995-1003.
- de Araujo E, Dessirier V, Laprée G, et al. Death ligand TRAIL, secreted by CD1a+ and CD14+ cells in blister fluids, is involved in killing keratinocytes in toxic epidermal necrolysis. Exp Dermatol. 2011;20:107-112.
- Viard-Leveugle I, Gaide O, Jankovic D, et al. TNF-α and IFN-γ are potential inducers of Fas-mediated keratinocyte apoptosis through activation of inducible nitric oxide synthase in toxic epidermal necrolysis. J Invest Dermatol. 2013;133:489-498.
- Choi SW, Levine JE, Ferrara JL. Pathogenesis and management of graft-versus-host disease. Immunol Allergy Clin North Am. 2010;30:75-101.
- Markey KA, Burman AC, Banovic T, et al. Soluble lymphotoxin is an important effector molecule in GVHD and GVL. Blood. 2010;115:122-132.
- Dodiuk-Gad RP, Olteanu C, Jeschke MG, et al. Treatment of toxic epidermal necrolysis in North America. J Am Acad Dermatol. 2015;73:876-877.
- Tracey D, Klareskog L, Sasso EH, et al. Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther. 2008;117:244-279.
To the Editor:
Acute graft-vs-host disease (GVHD) remains a limitation to hematopoietic stem cell transplantation (HSCT) in 20% to 50% of patients after transplant. Furthermore, failed treatment with corticosteroids is frequent and portends a poor prognosis.1 Toxic epidermal necrolysis (TEN) is an epidermolytic skin disorder thought to represent an adverse drug reaction, though its pathogenesis remains unclear. Severe forms of acute GVHD can mimic TEN clinically and histologically. Both can present with widespread cutaneous and mucosal bullae, erosions, and desquamation. Toxic epidermal necrolysis in the context of allogeneic hematopoietic stem cell transplantation is extremely rare, with almost 100% mortality in adult patients. Features that favor acute GVHD over TEN include diarrhea, elevation in bilirubin level, and chimerism.2 However, these features might be absent, posing a therapeutic dilemma, as current treatment preferences for each of these entities differ.
Growing evidence supports the use of anti–tumor necrosis factor (TNF) α drugs for the treatment of TEN. Success has been reported with both anti–TNF-α monoclonal antibodies as well as the soluble fusion protein etanercept.3,4 The use of TNF-α inhibitors in acute GVHD remains anecdotal.
A 58-year-old man (patient 1) with a history of acute myelogenous leukemia presented with a pruritic morbilliform eruption 28 days after HSCT. There was no desquamation or mucosal involvement and the biopsy obtained was histologically suggestive of grade 2 acute GVHD. His immunosuppressive regimen included sirolimus and cyclophosphamide. He was receiving trimethoprim-sulfamethoxazole (TMP-SMX), voriconazole, and acyclovir for infectious prophylaxis. At the time of presentation, he was treated with high-dose systemic steroids (prednisone 2 mg/kg/d) for acute GVHD with partial improvement. Upon tapering of the steroids 3 weeks after initiating TMP-SMX and 1 week after initiating voriconazole, he developed painful desquamation and erosions involving 95% of the body surface area (BSA), necessitating admission to the local burn unit (Figure 1). Biopsies demonstrated full-thickness epidermal necrosis with subepidermal blistering and interface dermatitis (Figure 2). No gastrointestinal tract involvement of acute GVHD was noted. The patient was a 100% donor chimera, supporting the diagnosis of acute GVHD; however, the patient and donor carried the HLA-C*06:02 allele, which previously has been described in association with TMP-SMX–related Stevens-Johnson syndrome/TEN.5 In addition, causality assessment using the algorithm of drug causality for epidermal necrolysis indicated TMP-SMX as a probable cause and voriconazole as a possible cause. The diagnosis of TEN with a SCORe of Toxic Epidermal Necrosis (SCORTEN) of 4 in the setting of acute GVHD was favored, though grade 4 acute GVHD could not be excluded. Trimethoprim-sulfamethoxazole was discontinued, and voriconazole was changed to posaconazole. He received supportive care along with 1 dose of 25-mg subcutaneous etanercept and 3 days of intravenous immunoglobulin (IVIG). Skin re-epithelialization was complete by 3 weeks. At 4 weeks, the patient developed a new asymptomatic erythematous eruption. Biopsies demonstrated changes of acute and chronic GVHD (Figure 3) that resolved with up-titration of sirolimus. The patient remained hospitalized for 96 days and continued to follow up with his transplant team as well as ophthalmology and dermatology. He died 2 years after HSCT.
A 67-year-old woman (patient 2) with high-grade myelodysplastic syndrome presented with an erythematous morbilliform eruption on the torso on day 20 after a matched unrelated HSCT that histologically was consistent with grade 2 GVHD (Figure 4). She had been receiving sirolimus and tacrolimus for GVHD prophylaxis. Infectious prophylaxis included acyclovir, pentamidine, micafungin, and TMP-SMX. Despite high-dose systemic steroids, the rash progressed and ultimately involved 80% BSA. A positive Nikolsky sign was noted involving 21% BSA (Figure 5), in addition to oral and genital mucosal ulcers. She denied nausea, vomiting, fever, or diarrhea. Chimerism studies were negative. Trimethoprim-sulfamethoxazole was discontinued, and she was transferred to a burn unit. Biopsies showed full-thickness epidermal necrosis. A diagnosis of TEN with a SCORTEN of 4 in the setting of acute GVHD was favored; grade 4 acute GVHD could not be excluded. Steroids were discontinued. Because laboratory studies indicated IgA deficiency, IVIG was not considered as a systemic option for therapy. The patient received 1 dose of infliximab (5 mg/kg). Cyclophosphamide 1600 mg weekly was added for GVHD therapy. The wounds progressively healed, and 2 weeks into her admission she was noted to have only 3% BSA with denuded skin. The patient was transferred to the cancer treatment center for further management of the malignancy. Unfortunately, after 2 months she died due to ischemic colitis that was confirmed on autopsy.
Graft-vs-host disease and TEN are rare, life-threatening complications seen in patients with allogeneic HSCT.2 Graft-vs-host disease and TEN share clinicopathologic characteristics and effector immune mechanisms, largely the substantial role of T-cell activation and tissue destruction, which occur through mediators such as TNF-α.6-8
Given the sparse lymphocytic infiltrate, keratinocyte death in TEN is thought to result from soluble molecules, including TNF-α and TNF-related apoptosis-inducing ligand.9 Tumor necrosis factor α has been identified in blister fluid, biopsy specimens, and serum of patients with TEN. Tumor necrosis factor α increases the expression of keratinocyte-inducible nitric oxide synthase, which upregulates keratinocyte Fas ligand expression and subsequent Fas- and caspase-8–mediated keratinocyte cell death.10
Acute GVHD results from donor lymphocyte activation after infusion into damaged recipient tissues that previously have been radiated or chemoablated. Mismatches in histocompatibility complexes between donor cells and recipient tissue antigens serve as the initial trigger for immune activation. Activation of antigen-presenting cells followed by activation, proliferation, differentiation, and migration of donor T cells ultimately results in destruction of the target tissue.11 Immune mediators, such as TNF-α and lymphotoxin α (another member of the TNF superfamily), play a nonredundant role in the pathogenesis of GVHD.12
Current treatment strategies for severe acute GVHD and TEN differ. In North America, high-dose IVIG frequently is used as first-line systemic therapy, while high-dose systemic corticosteroids rarely are used.13 Studies have demonstrated successful use of anti–TNF-α drugs for the treatment of TEN.3,4 Moreover, etanercept has shown to effectively inhibit lymphotoxin α.14 Similarly, TNF inhibition in the management of steroid-refractory acute GVHD has been successful.1 These studies coupled with the underlying immune mechanisms that both diseases share encouraged initiating a trial of anti–TNF-α therapy in our patients.
Patient 1 merits further discussion because he was both a 100% donor chimera as well as a carrier of an human leukocyte antigen susceptibility candidate allele to TMP-SMX. Historical features of his presentation are consistent with either steroid-refractory GVHD or TEN superimposed on acute GVHD. His initial presentation of the more typical macular exanthem of cutaneous acute GVHD was both biopsy proven and supported by clinical improvement with steroid therapy, which was later followed by a robust blistering mucocutaneous presentation approximately 3 weeks after the administration of TMP-SMX and 1 week after initiating voriconazole that improved with IVIG and etanercept.
It is difficult to determine if TEN represents a continuum or result of the underlying drivers of acute GVHD vs a drug reaction. Although there is insufficient evidence to establish a clear-cut diagnosis of TEN, these cases illustrate the need for better diagnostic techniques to allow differentiation between TEN and grade 4 acute GVHD, and in the context of uncertainty, TNF-α inhibition poses a viable therapeutic strategy for these 2 often lethal conditions. Our cases do unequivocally indicate the benefit of this therapeutic modality, add to the current body of literature supporting the use of TNF-α inhibitors in patients such as ours without an official TEN diagnosis, and may guide future investigative efforts.
To the Editor:
Acute graft-vs-host disease (GVHD) remains a limitation to hematopoietic stem cell transplantation (HSCT) in 20% to 50% of patients after transplant. Furthermore, failed treatment with corticosteroids is frequent and portends a poor prognosis.1 Toxic epidermal necrolysis (TEN) is an epidermolytic skin disorder thought to represent an adverse drug reaction, though its pathogenesis remains unclear. Severe forms of acute GVHD can mimic TEN clinically and histologically. Both can present with widespread cutaneous and mucosal bullae, erosions, and desquamation. Toxic epidermal necrolysis in the context of allogeneic hematopoietic stem cell transplantation is extremely rare, with almost 100% mortality in adult patients. Features that favor acute GVHD over TEN include diarrhea, elevation in bilirubin level, and chimerism.2 However, these features might be absent, posing a therapeutic dilemma, as current treatment preferences for each of these entities differ.
Growing evidence supports the use of anti–tumor necrosis factor (TNF) α drugs for the treatment of TEN. Success has been reported with both anti–TNF-α monoclonal antibodies as well as the soluble fusion protein etanercept.3,4 The use of TNF-α inhibitors in acute GVHD remains anecdotal.
A 58-year-old man (patient 1) with a history of acute myelogenous leukemia presented with a pruritic morbilliform eruption 28 days after HSCT. There was no desquamation or mucosal involvement and the biopsy obtained was histologically suggestive of grade 2 acute GVHD. His immunosuppressive regimen included sirolimus and cyclophosphamide. He was receiving trimethoprim-sulfamethoxazole (TMP-SMX), voriconazole, and acyclovir for infectious prophylaxis. At the time of presentation, he was treated with high-dose systemic steroids (prednisone 2 mg/kg/d) for acute GVHD with partial improvement. Upon tapering of the steroids 3 weeks after initiating TMP-SMX and 1 week after initiating voriconazole, he developed painful desquamation and erosions involving 95% of the body surface area (BSA), necessitating admission to the local burn unit (Figure 1). Biopsies demonstrated full-thickness epidermal necrosis with subepidermal blistering and interface dermatitis (Figure 2). No gastrointestinal tract involvement of acute GVHD was noted. The patient was a 100% donor chimera, supporting the diagnosis of acute GVHD; however, the patient and donor carried the HLA-C*06:02 allele, which previously has been described in association with TMP-SMX–related Stevens-Johnson syndrome/TEN.5 In addition, causality assessment using the algorithm of drug causality for epidermal necrolysis indicated TMP-SMX as a probable cause and voriconazole as a possible cause. The diagnosis of TEN with a SCORe of Toxic Epidermal Necrosis (SCORTEN) of 4 in the setting of acute GVHD was favored, though grade 4 acute GVHD could not be excluded. Trimethoprim-sulfamethoxazole was discontinued, and voriconazole was changed to posaconazole. He received supportive care along with 1 dose of 25-mg subcutaneous etanercept and 3 days of intravenous immunoglobulin (IVIG). Skin re-epithelialization was complete by 3 weeks. At 4 weeks, the patient developed a new asymptomatic erythematous eruption. Biopsies demonstrated changes of acute and chronic GVHD (Figure 3) that resolved with up-titration of sirolimus. The patient remained hospitalized for 96 days and continued to follow up with his transplant team as well as ophthalmology and dermatology. He died 2 years after HSCT.
A 67-year-old woman (patient 2) with high-grade myelodysplastic syndrome presented with an erythematous morbilliform eruption on the torso on day 20 after a matched unrelated HSCT that histologically was consistent with grade 2 GVHD (Figure 4). She had been receiving sirolimus and tacrolimus for GVHD prophylaxis. Infectious prophylaxis included acyclovir, pentamidine, micafungin, and TMP-SMX. Despite high-dose systemic steroids, the rash progressed and ultimately involved 80% BSA. A positive Nikolsky sign was noted involving 21% BSA (Figure 5), in addition to oral and genital mucosal ulcers. She denied nausea, vomiting, fever, or diarrhea. Chimerism studies were negative. Trimethoprim-sulfamethoxazole was discontinued, and she was transferred to a burn unit. Biopsies showed full-thickness epidermal necrosis. A diagnosis of TEN with a SCORTEN of 4 in the setting of acute GVHD was favored; grade 4 acute GVHD could not be excluded. Steroids were discontinued. Because laboratory studies indicated IgA deficiency, IVIG was not considered as a systemic option for therapy. The patient received 1 dose of infliximab (5 mg/kg). Cyclophosphamide 1600 mg weekly was added for GVHD therapy. The wounds progressively healed, and 2 weeks into her admission she was noted to have only 3% BSA with denuded skin. The patient was transferred to the cancer treatment center for further management of the malignancy. Unfortunately, after 2 months she died due to ischemic colitis that was confirmed on autopsy.
Graft-vs-host disease and TEN are rare, life-threatening complications seen in patients with allogeneic HSCT.2 Graft-vs-host disease and TEN share clinicopathologic characteristics and effector immune mechanisms, largely the substantial role of T-cell activation and tissue destruction, which occur through mediators such as TNF-α.6-8
Given the sparse lymphocytic infiltrate, keratinocyte death in TEN is thought to result from soluble molecules, including TNF-α and TNF-related apoptosis-inducing ligand.9 Tumor necrosis factor α has been identified in blister fluid, biopsy specimens, and serum of patients with TEN. Tumor necrosis factor α increases the expression of keratinocyte-inducible nitric oxide synthase, which upregulates keratinocyte Fas ligand expression and subsequent Fas- and caspase-8–mediated keratinocyte cell death.10
Acute GVHD results from donor lymphocyte activation after infusion into damaged recipient tissues that previously have been radiated or chemoablated. Mismatches in histocompatibility complexes between donor cells and recipient tissue antigens serve as the initial trigger for immune activation. Activation of antigen-presenting cells followed by activation, proliferation, differentiation, and migration of donor T cells ultimately results in destruction of the target tissue.11 Immune mediators, such as TNF-α and lymphotoxin α (another member of the TNF superfamily), play a nonredundant role in the pathogenesis of GVHD.12
Current treatment strategies for severe acute GVHD and TEN differ. In North America, high-dose IVIG frequently is used as first-line systemic therapy, while high-dose systemic corticosteroids rarely are used.13 Studies have demonstrated successful use of anti–TNF-α drugs for the treatment of TEN.3,4 Moreover, etanercept has shown to effectively inhibit lymphotoxin α.14 Similarly, TNF inhibition in the management of steroid-refractory acute GVHD has been successful.1 These studies coupled with the underlying immune mechanisms that both diseases share encouraged initiating a trial of anti–TNF-α therapy in our patients.
Patient 1 merits further discussion because he was both a 100% donor chimera as well as a carrier of an human leukocyte antigen susceptibility candidate allele to TMP-SMX. Historical features of his presentation are consistent with either steroid-refractory GVHD or TEN superimposed on acute GVHD. His initial presentation of the more typical macular exanthem of cutaneous acute GVHD was both biopsy proven and supported by clinical improvement with steroid therapy, which was later followed by a robust blistering mucocutaneous presentation approximately 3 weeks after the administration of TMP-SMX and 1 week after initiating voriconazole that improved with IVIG and etanercept.
It is difficult to determine if TEN represents a continuum or result of the underlying drivers of acute GVHD vs a drug reaction. Although there is insufficient evidence to establish a clear-cut diagnosis of TEN, these cases illustrate the need for better diagnostic techniques to allow differentiation between TEN and grade 4 acute GVHD, and in the context of uncertainty, TNF-α inhibition poses a viable therapeutic strategy for these 2 often lethal conditions. Our cases do unequivocally indicate the benefit of this therapeutic modality, add to the current body of literature supporting the use of TNF-α inhibitors in patients such as ours without an official TEN diagnosis, and may guide future investigative efforts.
- Couriel DR, Saliba R, de Lima M, et al. A phase III study of infliximab and corticosteroids for the initial treatment of acute graft-versus-host disease. Biol Blood Marrow Transplant. 2009;15:1555-1562.
- Jeanmonod P, Hubbuch M, Grünhage F, et al. Graft-versus-host disease or toxic epidermal necrolysis: diagnostic dilemma after liver transplantation. Transpl Infect Dis. 2012;14:422-426.
- Paradisi A, Abeni D, Bergamo F, et al. Etanercept therapy for toxic epidermal necrolysis. J Am Acad Dermatol. 2014;71:278-283.
- Scott-Lang V, Tidman M, McKay D. Toxic epidermal necrolysis in a child successfully treated with infliximab. Pediatr Dermatol. 2014;31:532-534.
- Kingpin T, Mahasirimongkol S, Konyoung P, et al. Candidate HLA genes for prediction of co-trimoxazole-induced severe cutaneous reactions. Pharmacogenet Genomics. 2015;25:402-411.
- Correia O, Delgado L, Barbosa IL, et al. Increased interleukin 10, tumor necrosis factor alpha, and interleukin 6 levels in blister fluid of toxic epidermal necrolysis. J Am Acad Dermatol. 2002;47:58-62.
- French LE, Tschopp J. Fas-mediated cell death in toxic epidermal necrolysis and graft-versus-host disease: potential for therapeutic inhibition. Schweiz Med Wochenschr. 2000;130:1656-1661.
- Downey A, Jackson C, Harun N, et al. Toxic epidermal necrolysis: review of pathogenesis and management. J Am Acad Dermatol. 2012;66:995-1003.
- de Araujo E, Dessirier V, Laprée G, et al. Death ligand TRAIL, secreted by CD1a+ and CD14+ cells in blister fluids, is involved in killing keratinocytes in toxic epidermal necrolysis. Exp Dermatol. 2011;20:107-112.
- Viard-Leveugle I, Gaide O, Jankovic D, et al. TNF-α and IFN-γ are potential inducers of Fas-mediated keratinocyte apoptosis through activation of inducible nitric oxide synthase in toxic epidermal necrolysis. J Invest Dermatol. 2013;133:489-498.
- Choi SW, Levine JE, Ferrara JL. Pathogenesis and management of graft-versus-host disease. Immunol Allergy Clin North Am. 2010;30:75-101.
- Markey KA, Burman AC, Banovic T, et al. Soluble lymphotoxin is an important effector molecule in GVHD and GVL. Blood. 2010;115:122-132.
- Dodiuk-Gad RP, Olteanu C, Jeschke MG, et al. Treatment of toxic epidermal necrolysis in North America. J Am Acad Dermatol. 2015;73:876-877.
- Tracey D, Klareskog L, Sasso EH, et al. Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther. 2008;117:244-279.
- Couriel DR, Saliba R, de Lima M, et al. A phase III study of infliximab and corticosteroids for the initial treatment of acute graft-versus-host disease. Biol Blood Marrow Transplant. 2009;15:1555-1562.
- Jeanmonod P, Hubbuch M, Grünhage F, et al. Graft-versus-host disease or toxic epidermal necrolysis: diagnostic dilemma after liver transplantation. Transpl Infect Dis. 2012;14:422-426.
- Paradisi A, Abeni D, Bergamo F, et al. Etanercept therapy for toxic epidermal necrolysis. J Am Acad Dermatol. 2014;71:278-283.
- Scott-Lang V, Tidman M, McKay D. Toxic epidermal necrolysis in a child successfully treated with infliximab. Pediatr Dermatol. 2014;31:532-534.
- Kingpin T, Mahasirimongkol S, Konyoung P, et al. Candidate HLA genes for prediction of co-trimoxazole-induced severe cutaneous reactions. Pharmacogenet Genomics. 2015;25:402-411.
- Correia O, Delgado L, Barbosa IL, et al. Increased interleukin 10, tumor necrosis factor alpha, and interleukin 6 levels in blister fluid of toxic epidermal necrolysis. J Am Acad Dermatol. 2002;47:58-62.
- French LE, Tschopp J. Fas-mediated cell death in toxic epidermal necrolysis and graft-versus-host disease: potential for therapeutic inhibition. Schweiz Med Wochenschr. 2000;130:1656-1661.
- Downey A, Jackson C, Harun N, et al. Toxic epidermal necrolysis: review of pathogenesis and management. J Am Acad Dermatol. 2012;66:995-1003.
- de Araujo E, Dessirier V, Laprée G, et al. Death ligand TRAIL, secreted by CD1a+ and CD14+ cells in blister fluids, is involved in killing keratinocytes in toxic epidermal necrolysis. Exp Dermatol. 2011;20:107-112.
- Viard-Leveugle I, Gaide O, Jankovic D, et al. TNF-α and IFN-γ are potential inducers of Fas-mediated keratinocyte apoptosis through activation of inducible nitric oxide synthase in toxic epidermal necrolysis. J Invest Dermatol. 2013;133:489-498.
- Choi SW, Levine JE, Ferrara JL. Pathogenesis and management of graft-versus-host disease. Immunol Allergy Clin North Am. 2010;30:75-101.
- Markey KA, Burman AC, Banovic T, et al. Soluble lymphotoxin is an important effector molecule in GVHD and GVL. Blood. 2010;115:122-132.
- Dodiuk-Gad RP, Olteanu C, Jeschke MG, et al. Treatment of toxic epidermal necrolysis in North America. J Am Acad Dermatol. 2015;73:876-877.
- Tracey D, Klareskog L, Sasso EH, et al. Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther. 2008;117:244-279.
Practice Points
- Graft-vs-host disease (GVHD) and toxic epidermal necrolysis (TEN) are rare life-threatening complications seen in patients with allogeneic hematopoietic stem cell transplantation.
- Although mild acute GVHD easily is distinguished from TEN, severe acute GVHD and TEN share overlapping features and present a diagnostic challenge.
- Therapeutic decisions and associated outcomes hinge on accurate diagnosis, as high-dose systemic corticosteroids have been associated with higher mortality rates in TEN.

Phototoxic Contact Dermatitis From Over-the-counter 8-Methoxypsoralen
To the Editor:
A 71-year-old Hispanic man with a history of vitiligo presented with an acute-onset blistering rash on the face, arms, and hands. Physical examination demonstrated photodistributed erythematous plaques with overlying vesicles and erosions with hemorrhagic crust on the face, neck, dorsal aspects of the hands, and wrists (Figure). Further history revealed that the patient applied a new cream that was recommended to treat vitiligo the night before the rash onset; he obtained the cream from a Central American market without a prescription. He had gone running in the park without any form of sun protection and then developed the rash within several hours. He denied taking any other medications or supplements. The involvement of sun-protected areas (ie, upper eyelids, nasolabial folds, submental area) was explained when the patient further elaborated that he had performed supine exercises during his outdoor recreation. He brought his new cream into the clinic, which was found to contain prescription-strength methoxsalen (8-methoxypsoralen), confirming the diagnosis of acute phototoxic contact dermatitis. The acute reaction had subsided, and the patient already had discontinued the causative agent. He was counseled on further avoidance of the cream and sun-protective measures.

The photosensitizing properties of certain compounds have been harnessed for therapeutic purposes. For example, psoralen plus UVA therapy has been used for psoriasis and vitiligo and photodynamic therapy for actinic keratoses and superficial nonmelanoma skin cancers.1 However, these agents can induce severe phototoxicity if UV light exposure is not carefully monitored, as seen in our patient. This case is a classic example of phototoxic contact dermatitis and highlights the importance of obtaining a detailed patient history to allow for proper diagnosis and identification of the causative agent. Importantly, because prescription-strength topical medications are readily available over-the-counter, particularly in stores specializing in international goods, patients should be questioned about the use of all topical and systemic medications, both prescription and nonprescription.2
- Richard EG. The science and (lost) art of psoralen plus UVA phototherapy. Dermatol Clin. 2020;38:11-23. doi:10.1016/j.det.2019.08.002
- Kimyon RS, Schlarbaum JP, Liou YL, et al. Prescription-strengthtopical corticosteroids available over the counter: cross-sectional study of 80 stores in 13 United States cities. J Am Acad Dermatol. 2020;82:524-525. doi:10.1016/j.jaad.2019.10.035
To the Editor:
A 71-year-old Hispanic man with a history of vitiligo presented with an acute-onset blistering rash on the face, arms, and hands. Physical examination demonstrated photodistributed erythematous plaques with overlying vesicles and erosions with hemorrhagic crust on the face, neck, dorsal aspects of the hands, and wrists (Figure). Further history revealed that the patient applied a new cream that was recommended to treat vitiligo the night before the rash onset; he obtained the cream from a Central American market without a prescription. He had gone running in the park without any form of sun protection and then developed the rash within several hours. He denied taking any other medications or supplements. The involvement of sun-protected areas (ie, upper eyelids, nasolabial folds, submental area) was explained when the patient further elaborated that he had performed supine exercises during his outdoor recreation. He brought his new cream into the clinic, which was found to contain prescription-strength methoxsalen (8-methoxypsoralen), confirming the diagnosis of acute phototoxic contact dermatitis. The acute reaction had subsided, and the patient already had discontinued the causative agent. He was counseled on further avoidance of the cream and sun-protective measures.
The photosensitizing properties of certain compounds have been harnessed for therapeutic purposes. For example, psoralen plus UVA therapy has been used for psoriasis and vitiligo and photodynamic therapy for actinic keratoses and superficial nonmelanoma skin cancers.1 However, these agents can induce severe phototoxicity if UV light exposure is not carefully monitored, as seen in our patient. This case is a classic example of phototoxic contact dermatitis and highlights the importance of obtaining a detailed patient history to allow for proper diagnosis and identification of the causative agent. Importantly, because prescription-strength topical medications are readily available over-the-counter, particularly in stores specializing in international goods, patients should be questioned about the use of all topical and systemic medications, both prescription and nonprescription.2
To the Editor:
A 71-year-old Hispanic man with a history of vitiligo presented with an acute-onset blistering rash on the face, arms, and hands. Physical examination demonstrated photodistributed erythematous plaques with overlying vesicles and erosions with hemorrhagic crust on the face, neck, dorsal aspects of the hands, and wrists (Figure). Further history revealed that the patient applied a new cream that was recommended to treat vitiligo the night before the rash onset; he obtained the cream from a Central American market without a prescription. He had gone running in the park without any form of sun protection and then developed the rash within several hours. He denied taking any other medications or supplements. The involvement of sun-protected areas (ie, upper eyelids, nasolabial folds, submental area) was explained when the patient further elaborated that he had performed supine exercises during his outdoor recreation. He brought his new cream into the clinic, which was found to contain prescription-strength methoxsalen (8-methoxypsoralen), confirming the diagnosis of acute phototoxic contact dermatitis. The acute reaction had subsided, and the patient already had discontinued the causative agent. He was counseled on further avoidance of the cream and sun-protective measures.
The photosensitizing properties of certain compounds have been harnessed for therapeutic purposes. For example, psoralen plus UVA therapy has been used for psoriasis and vitiligo and photodynamic therapy for actinic keratoses and superficial nonmelanoma skin cancers.1 However, these agents can induce severe phototoxicity if UV light exposure is not carefully monitored, as seen in our patient. This case is a classic example of phototoxic contact dermatitis and highlights the importance of obtaining a detailed patient history to allow for proper diagnosis and identification of the causative agent. Importantly, because prescription-strength topical medications are readily available over-the-counter, particularly in stores specializing in international goods, patients should be questioned about the use of all topical and systemic medications, both prescription and nonprescription.2
- Richard EG. The science and (lost) art of psoralen plus UVA phototherapy. Dermatol Clin. 2020;38:11-23. doi:10.1016/j.det.2019.08.002
- Kimyon RS, Schlarbaum JP, Liou YL, et al. Prescription-strengthtopical corticosteroids available over the counter: cross-sectional study of 80 stores in 13 United States cities. J Am Acad Dermatol. 2020;82:524-525. doi:10.1016/j.jaad.2019.10.035
- Richard EG. The science and (lost) art of psoralen plus UVA phototherapy. Dermatol Clin. 2020;38:11-23. doi:10.1016/j.det.2019.08.002
- Kimyon RS, Schlarbaum JP, Liou YL, et al. Prescription-strengthtopical corticosteroids available over the counter: cross-sectional study of 80 stores in 13 United States cities. J Am Acad Dermatol. 2020;82:524-525. doi:10.1016/j.jaad.2019.10.035
Practice Points
- Phototoxic contact dermatitis is an irritant reaction resembling an exaggerated sunburn that occurs with the use of a photosensitizing agent and UV light exposure.
- A range of topical and systemic medications, plants, and natural products can elicit phototoxic reactions.
- With the wide availability of prescription-strength over-the-counter medications, a detailed history often is necessary to identify the causative agents of phototoxic contact dermatitis and ensure future avoidance.
