User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Mohs Micrographic Surgery Overlying a Pacemaker
To the Editor:
Pacemakers and defibrillators are common in patients presenting for cutaneous surgery. The use and application of electrosurgery in this patient population has been reviewed extensively.1 The presence of a cardiac device immediately below a cutaneous surgical site presents as a potentially more complex surgical procedure. Damage to and/or manipulation of the cardiac device could activate the device and/or require subsequent repair of the unit. We present the case of a basal cell carcinoma (BCC) overlying a pacemaker along with a brief review of the literature.
An 89-year-old man presented to our Mohs surgical unit for treatment of a long-standing BCC on the left upper chest (Figure, A) via Mohs micrographic surgery (MMS), which was utilized due to the infiltrative nature of the tumor and its close proximity to the cardiac device. He had a history of heart disease including paroxysmal atrial fibrillation, first-degree atrioventricular block, and sick sinus syndrome, and a pacemaker had been placed 5 years prior. The tumor was located on the skin directly above the pacemaker. The pacemaker and associated lead wires were easily palpable to touch. Prior to the procedure, treatment options were discussed with the patient’s cardiologist. Due to the size of the tumor (21×22 mm) and more importantly its location directly above the pacemaker, the BCC was treated with a single stage of MMS (Figure, B). In an effort to minimize potential exposure of the pacemaker, the surgical site was infiltrated with additional local anesthesia, which created a temporary edematous thickening to provide an increased barrier between the surgical site and pacemaker. Hemostasis was achieved with thermocautery, and a fusiform repair was completed without consequence (Figure, C). There were no postoperative changes or concerns, and preoperative and postoperative electrocardiograms reviewed by the patient’s cardiologist revealed no change.

Treatment of cutaneous lesions near pacemakers or defibrillators requires caution, both in avoidance of the device itself as well as electrocautery interference.1-4 There are multiple treatment options available, including MMS, excision, curettage and desiccation, topical therapies, and radiation therapy. The benefits of MMS for cutaneous tumors overlying cardiac devices include decreased risk of damaging the underlying pacemaker by minimizing surgical depth of the defect, minimizing the risk of recurrence and hence any additional procedures, and minimizing the risk of surgical complications via a smaller surgical defect.4 Monopolar electrosurgery is associated with the risk of interfering with pacemaker function; however, the use of bipolar electrocoagulation has been shown to be safer.1,3,4 Additionally, thermocautery carries the least risk because it involves heat only.2,5
Awareness of the cardiac device location, communication with the patient’s cardiologist, use of local anesthesia infiltrates to maximize distance between the surgical site and cardiac device, and appropriate hemostasis methods offer the most effective and safest means for surgical removal of tumors overlying cardiac devices.
- El-Gamal HM, Dufresne RG, Saddler K. Electrosurgery, pacemakers and ICDs: a survey of precautions and complications experienced by cutaneous surgeons. Dermatol Surg. 2001;27:385-390.
- Chapas AM, Lee D, Rogers GS. Excision of malignant melanoma overlying a pacemaker. Dermatol Surg. 2005;31:112-114.
- Matzke TJ, Christenson LJ, Christenson SD, et al. Pacemakers and implantable cardiac defibrillators in dermatologic surgery. Dermatol Surg. 2006;32:1155-1162.
- Herrmann JL, Mishra V, Greenway HT. Basal cell carcinoma overlying a cardiac pacemaker successfully treated using Mohs micrographic surgery. 2014;4:474-477.
- Lane JE, O’Brien EM, Kent DE. Optimization of thermocautery in excisional dermatologic surgery. Dermatol Surg. 2006;32:669-675.
To the Editor:
Pacemakers and defibrillators are common in patients presenting for cutaneous surgery. The use and application of electrosurgery in this patient population has been reviewed extensively.1 The presence of a cardiac device immediately below a cutaneous surgical site presents as a potentially more complex surgical procedure. Damage to and/or manipulation of the cardiac device could activate the device and/or require subsequent repair of the unit. We present the case of a basal cell carcinoma (BCC) overlying a pacemaker along with a brief review of the literature.
An 89-year-old man presented to our Mohs surgical unit for treatment of a long-standing BCC on the left upper chest (Figure, A) via Mohs micrographic surgery (MMS), which was utilized due to the infiltrative nature of the tumor and its close proximity to the cardiac device. He had a history of heart disease including paroxysmal atrial fibrillation, first-degree atrioventricular block, and sick sinus syndrome, and a pacemaker had been placed 5 years prior. The tumor was located on the skin directly above the pacemaker. The pacemaker and associated lead wires were easily palpable to touch. Prior to the procedure, treatment options were discussed with the patient’s cardiologist. Due to the size of the tumor (21×22 mm) and more importantly its location directly above the pacemaker, the BCC was treated with a single stage of MMS (Figure, B). In an effort to minimize potential exposure of the pacemaker, the surgical site was infiltrated with additional local anesthesia, which created a temporary edematous thickening to provide an increased barrier between the surgical site and pacemaker. Hemostasis was achieved with thermocautery, and a fusiform repair was completed without consequence (Figure, C). There were no postoperative changes or concerns, and preoperative and postoperative electrocardiograms reviewed by the patient’s cardiologist revealed no change.
Treatment of cutaneous lesions near pacemakers or defibrillators requires caution, both in avoidance of the device itself as well as electrocautery interference.1-4 There are multiple treatment options available, including MMS, excision, curettage and desiccation, topical therapies, and radiation therapy. The benefits of MMS for cutaneous tumors overlying cardiac devices include decreased risk of damaging the underlying pacemaker by minimizing surgical depth of the defect, minimizing the risk of recurrence and hence any additional procedures, and minimizing the risk of surgical complications via a smaller surgical defect.4 Monopolar electrosurgery is associated with the risk of interfering with pacemaker function; however, the use of bipolar electrocoagulation has been shown to be safer.1,3,4 Additionally, thermocautery carries the least risk because it involves heat only.2,5
Awareness of the cardiac device location, communication with the patient’s cardiologist, use of local anesthesia infiltrates to maximize distance between the surgical site and cardiac device, and appropriate hemostasis methods offer the most effective and safest means for surgical removal of tumors overlying cardiac devices.
To the Editor:
Pacemakers and defibrillators are common in patients presenting for cutaneous surgery. The use and application of electrosurgery in this patient population has been reviewed extensively.1 The presence of a cardiac device immediately below a cutaneous surgical site presents as a potentially more complex surgical procedure. Damage to and/or manipulation of the cardiac device could activate the device and/or require subsequent repair of the unit. We present the case of a basal cell carcinoma (BCC) overlying a pacemaker along with a brief review of the literature.
An 89-year-old man presented to our Mohs surgical unit for treatment of a long-standing BCC on the left upper chest (Figure, A) via Mohs micrographic surgery (MMS), which was utilized due to the infiltrative nature of the tumor and its close proximity to the cardiac device. He had a history of heart disease including paroxysmal atrial fibrillation, first-degree atrioventricular block, and sick sinus syndrome, and a pacemaker had been placed 5 years prior. The tumor was located on the skin directly above the pacemaker. The pacemaker and associated lead wires were easily palpable to touch. Prior to the procedure, treatment options were discussed with the patient’s cardiologist. Due to the size of the tumor (21×22 mm) and more importantly its location directly above the pacemaker, the BCC was treated with a single stage of MMS (Figure, B). In an effort to minimize potential exposure of the pacemaker, the surgical site was infiltrated with additional local anesthesia, which created a temporary edematous thickening to provide an increased barrier between the surgical site and pacemaker. Hemostasis was achieved with thermocautery, and a fusiform repair was completed without consequence (Figure, C). There were no postoperative changes or concerns, and preoperative and postoperative electrocardiograms reviewed by the patient’s cardiologist revealed no change.
Treatment of cutaneous lesions near pacemakers or defibrillators requires caution, both in avoidance of the device itself as well as electrocautery interference.1-4 There are multiple treatment options available, including MMS, excision, curettage and desiccation, topical therapies, and radiation therapy. The benefits of MMS for cutaneous tumors overlying cardiac devices include decreased risk of damaging the underlying pacemaker by minimizing surgical depth of the defect, minimizing the risk of recurrence and hence any additional procedures, and minimizing the risk of surgical complications via a smaller surgical defect.4 Monopolar electrosurgery is associated with the risk of interfering with pacemaker function; however, the use of bipolar electrocoagulation has been shown to be safer.1,3,4 Additionally, thermocautery carries the least risk because it involves heat only.2,5
Awareness of the cardiac device location, communication with the patient’s cardiologist, use of local anesthesia infiltrates to maximize distance between the surgical site and cardiac device, and appropriate hemostasis methods offer the most effective and safest means for surgical removal of tumors overlying cardiac devices.
- El-Gamal HM, Dufresne RG, Saddler K. Electrosurgery, pacemakers and ICDs: a survey of precautions and complications experienced by cutaneous surgeons. Dermatol Surg. 2001;27:385-390.
- Chapas AM, Lee D, Rogers GS. Excision of malignant melanoma overlying a pacemaker. Dermatol Surg. 2005;31:112-114.
- Matzke TJ, Christenson LJ, Christenson SD, et al. Pacemakers and implantable cardiac defibrillators in dermatologic surgery. Dermatol Surg. 2006;32:1155-1162.
- Herrmann JL, Mishra V, Greenway HT. Basal cell carcinoma overlying a cardiac pacemaker successfully treated using Mohs micrographic surgery. 2014;4:474-477.
- Lane JE, O’Brien EM, Kent DE. Optimization of thermocautery in excisional dermatologic surgery. Dermatol Surg. 2006;32:669-675.
- El-Gamal HM, Dufresne RG, Saddler K. Electrosurgery, pacemakers and ICDs: a survey of precautions and complications experienced by cutaneous surgeons. Dermatol Surg. 2001;27:385-390.
- Chapas AM, Lee D, Rogers GS. Excision of malignant melanoma overlying a pacemaker. Dermatol Surg. 2005;31:112-114.
- Matzke TJ, Christenson LJ, Christenson SD, et al. Pacemakers and implantable cardiac defibrillators in dermatologic surgery. Dermatol Surg. 2006;32:1155-1162.
- Herrmann JL, Mishra V, Greenway HT. Basal cell carcinoma overlying a cardiac pacemaker successfully treated using Mohs micrographic surgery. 2014;4:474-477.
- Lane JE, O’Brien EM, Kent DE. Optimization of thermocautery in excisional dermatologic surgery. Dermatol Surg. 2006;32:669-675.
Practice Points
- Surgical treatment of a cutaneous lesion overlying a cardiac device requires caution, both in avoidance of the device itself as well as electrocautery interference.
- Local anesthesia infiltrates can be used to create a temporary edematous thickening to minimize potential exposure of the device during the procedure.
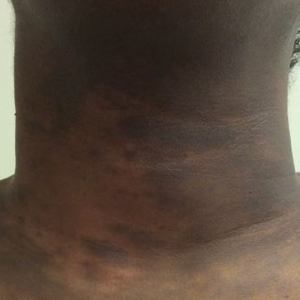
Military Grooming Standards and Their Impact on Skin Diseases of the Head and Neck
The US military enforces grooming standards to ensure the professional appearance and serviceability of soldiers in all operational settings. Although most individuals are able to uphold these regulations without incident, there is a growing cohort of servicemembers with skin diseases that were exacerbated or even initiated by haircuts, hairstyling, and shaving required to conform to these grooming standards. These skin diseases, which can affect both sexes and may not be appreciated until years into a soldier's service commitment, can have consequences related to individual morbidity and medical readiness for deployment, making it an important issue for medical practitioners to recognize and manage in servicemembers.
This review highlights several disorders of the pilosebaceous unit of the head and neck that can be caused or exacerbated by military grooming standards, including inflammatory hair disorders, traction alopecia, and pseudofolliculitis barbae. Discussion of each entity will include a review of susceptibility and causality as well as initial treatment options to consider (Table).
Inflammatory Hair Disorders
The proper appearance of servicemembers in uniform represents self-discipline and conformity to the high standards of the military. This transition occurs as a rite of passage for many new male recruits who receive shaved haircuts during their first days of basic training. Thereafter, male servicemembers are required to maintain a tapered appearance of the hair per military regulations.1 Clipping hair closely to the scalp or shaving the head entirely are authorized and often encouraged; therefore, high and tight haircuts and buzz cuts are popular among male soldiers due to the general ease of care and ability to maintain the haircut themselves. Conversely, these styles require servicemembers to get weekly or biweekly haircuts that in turn can lead to chronic trauma and irritation. In more susceptible populations, inflammatory hair disorders such as acne keloidalis nuchae (AKN), dissecting cellulitis of the scalp, and folliculitis decalvans may be incited.
Acne Keloidalis Nuchae
Acne keloidalis nuchae, also called folliculitis keloidalis, is a chronic scarring folliculitis presenting with papules and plaques on the occiput and nape of the neck that may merge to form hypertrophic scars or keloids. This disorder most commonly develops in young black men but also can be seen in black females and white patients of both sexes.2 Acne keloidalis nuchae shares many histologic features with central centrifugal cicatricial alopecia, which may suggest a similar pathogenesis. Apart from frequent haircuts, tight-collared shirts, such as those on military service uniforms, also have been associated with AKN. Because of these suspected etiologies, first-line treatment focuses on preventing further trauma by avoiding mechanical irritation and short haircuts, which may be difficult in the military setting. For earlier disease stages, topical and intralesional corticosteroids, oral retinoids, and topical and oral antibiotics are used for their anti-inflammatory properties.3 In refractory cases, surgical excision with healing by secondary intention may be attempted.4 Additional treatment options include the 1064-nm Nd:YAG and 810-nm diode lasers,3 UVB light therapy, CO2 laser, and radiotherapy.
Dissecting Cellulitis of the Scalp
Similar to AKN, dissecting cellulitis of the scalp is another inflammatory hair disorder that is worsened by frequent short haircuts.5 Dissecting cellulitis of the scalp is a primary cicatricial alopecia proposed to be secondary to follicular occlusion. It often is seen in black males aged 20 to 40 years and is characterized by boggy suppurative nodules and cysts with draining sinus tracts, abscesses, and resultant scarring alopecia. Dissecting cellulitis of the scalp is part of the follicular occlusion tetrad, which also includes hidradenitis suppurativa, acne conglobata, and pilonidal cysts. First-line therapies include topical and oral antibiotics, topical retinoids, intralesional corticosteroids, incision and drainage of fluctuant nodules, and oral isotretinoin with or without rifampin. Alternative treatments include oral zinc supplementation, oral corticosteroids, tumor necrosis factor α inhibitors, laser therapies, radiotherapy, and surgical management with wide local excision or total scalpectomy.6,7
Folliculitis Decalvans
Folliculitis decalvans is a primary cicatricial alopecia of the scalp that most commonly presents in middle-aged men without racial predilection.8 Folliculitis decalvans presents with multiple pustules, crusts, tufted hairs, and perifollicular hyperkeratosis, leading to scarring of the scalp, which often is most severe on the posterior vertex. Staphylococcus aureus is a presumed player in the pathogenesis of folliculitis decalvans with superantigens causing release of cytokines stimulating follicular destruction. Close haircuts in conformation with military grooming standards can contribute to this condition due to mechanical trauma and subsequent inflammation. It typically is diagnosed clinically, but if histologic confirmation is desired, a sample from the periphery of early lesions is preferred.9 Initial treatment consists of antibacterial shampoos, topical corticosteroids, topical antibiotics, and combination oral antibiotic therapy with rifampin and clindamycin. Studies using oral isotretinoin have shown variable results,10,11 and the most effective treatment of recalcitrant lesions appears to be intralesional corticosteroids.12
Follicular and Scarring Disorders
In addition to inflammatory hair disorders, military grooming standards have been linked to the pathogenesis of diseases such as pseudofolliculitis barbae, traction alopecia, and keloids, specifically through irritation of the face, neck, and scalp, as well as damage to the follicular unit.5 These conditions develop because grooming regulations necessitate certain hair practices such as close shaving of facial and neck hair and keeping long hair secured relatively tightly to the scalp.
Pseudofolliculitis Barbae
Males in the military are obligated to keep their faces clean-shaven.1 They may acquire a medical waiver for a specified beard length if deemed appropriate by the treating physician,1 which often leads to the need for continual waiver renewal and also may warrant possible negative perception from peers, subordinates, and leadership. One of the most prevalent conditions that is closely associated with shaving is pseudofolliculitis barbae. The combination of close shaving and tightly coiled hairs causes the hairs to grow toward and penetrate the skin, particularly on the neck.13 In some cases, the hairs never actually exit the skin and simply curl within the superficial epidermis. A foreign body reaction often arises, leading to inflamed follicular papules and pustules. Affected individuals may experience pain, pruritus, and secondary infections. Postinflammatory hyperpigmentation, hypertrophic scarring, and keloid formation are common sequelae in cases of untreated disease. Pseudofolliculitis barbae also is exacerbated by pulling the skin taut and shaving against the grain, making behavioral interventions a key component in management of this condition. Preliminary recommendations include using a new or electric razor, leaving hair at least 2 mm in length, and shaving in the direction of hair growth. Other treatment options with varying effectiveness include daily alternation of a mild topical corticosteroid and one of the following: a topical retinoid, topical antibiotics, or glycolic acid. The only treatments that approach definitive cure are laser hair removal and electrolysis for which patient skin type plays an important role in laser selection.5
Traction Alopecia
Similar to their male counterparts, female military members must also present a conservative professional appearance, including hair that is neatly groomed.1 If the length of the hair extends beyond the uniform collar, it must be inconspicuously fastened or pinned above the collar. As a result, loosely tied hair is unauthorized, and females with long hair must secure their hair tightly on a daily basis. Traction alopecia results from tight hairstyling over a prolonged period and commonly affects female soldiers. The etiology is presumed to be mechanical loosening of hair within the follicles, leading to inflammation. Although traditionally seen in black women along the frontal and temporal hairlines, traction alopecia has been identified in individuals of all races and can occur anywhere on the scalp.5 Perifollicular erythema may be the first sign, and papules and pustules may be visible. Although the hair loss in traction alopecia usually is reversible if the traction is ceased, end-stage disease may be permanent.6 Halting traction-inducing practices is paramount, and other treatment options that may slow progression include topical or oral antibiotics and topical or intralesional corticosteroids. Recovery of hair loss also may be aided by topical minoxidil.5
Keloids
Keloid formation is an important pathology to address, as it may result from several of the aforementioned conditions. Keloids are most commonly seen in black individuals but also can occur in Hispanic and Asian patients. The cause has not been fully elucidated but is thought to be a combination of dysfunctional fibroblasts with a genetic component based on racial predilection and twin concordance studies.5 The chest, shoulders, upper back, neck, and earlobes are particularly susceptible to keloid formation, which can appear from 1 to 24 years following dermal trauma.5 Unlike hypertrophic scars, keloids generally do not regress and frequently cause discomfort, pruritus, and emotional distress. They also can hinder wearing a military uniform. Sustained remission is problematic, making prevention a first-line approach, including proper care of wounds when they occur and avoiding elective procedures such as piercings and tattoos. Intralesional corticosteroids, adjuvant injections (eg, 5-fluorouracil), silicone sheeting, cryotherapy, radiation, laser therapy, and excision are some of the treatment options when keloids have formed.5
Final Comment
It is important to recognize military grooming standards as a cause or contributor to several diseases of the head and neck in military servicemembers. Specifically, frequent haircuts in male soldiers are associated with several inflammatory hair disorders, including AKN, dissecting cellulitis of the scalp, and folliculitis decalvans, while daily shaving predisposes individuals to pseudofolliculitis barbae with possible keloid formation. Females may develop traction alopecia from chronically tight, pulled back hairstyles. All of these conditions have health implications for the affected individuals and can compromise the military mission. Awareness, prevention, and recognition are key along with the knowledge base to provide anticipatory avoidance and initiate appropriate treatments, thereby mitigating these potential consequences.
- US Department of the Army. Wear and Appearance of Army Uniforms and Insignia: Army Regulation 670-1. Washington, DC: Department of the Army; 2017. https://history.army.mil/html/forcestruc/docs/AR670-1.pdf. Accessed October 11, 2018.
- East-Innis AD, Stylianou K, Paolino A, et al. Acne keloidalis nuchae: risk factors and associated disorders--a retrospective study. Int J Dermatol. 2017;56:828-832.
- Maranda EL, Simmons BJ, Nguyen AH, et al. Treatment of acne keloidalis nuchae: a systematic review of the literature. Dermatol Ther (Heidelb). 2016;6:363-378.
- Glenn MJ, Bennett RG, Kelly AP. Acne keloidalis nuchae: treatment with excision and second-intention healing. J Am Acad Dermatol. 1995;33:243-246.
- Madu P, Kundu RV. Follicular and scarring disorders in skin of color: presentation and management. Am J Clin Dermatol. 2014;15:307-321.
- Rodney IJ, Onwudiwe OC. Hair and scalp disorders in ethnic populations. J Drugs Dermatol. 2013;12:420-427.
- Lindsey SF, Tosti A. Ethnic hair disorders. Curr Probl Dermatol. 2015;47:139-148.
- Whiting DA. Cicatricial alopecia: clinico-pathological findings and treatment. Clin Dermatol. 2001;19:211-225.
- Sperling LC, Cowper SE, Knopp EA. An Atlas of Hair Pathology with Clinical Correlations. 2nd ed. Boca Raton, FL: CRC Press; 2012.
- Gemmeke A, Wollina U. Folliculitis decalvans of the scalp: response to triple therapy with isotretinoin, clindamycin, and prednisolone. Acta Dermatovenerol Alp Pannonica Adriat. 2006;15:184-186.
- Hallai N, Thompson I, Williams P, et al. Folliculitis spinulosa decalvans: failure to respond to oral isotretinoin. J Eur Acad Dermatol Venereol. 2006;20:223-224.
- Bolduc C, Sperling LC, Shapiro J. Primary cicatricial alopecia. J Am Acad Dermatol. 2016;75:101-117.
- Perry PK, Cook-Bolden FE, Rahman Z, et al. Defining pseudofolliculitis barbae in 2001: a review of the literature and current trends. J Am Acad Dermatol. 2002;46(2 suppl):S113-S119.
The US military enforces grooming standards to ensure the professional appearance and serviceability of soldiers in all operational settings. Although most individuals are able to uphold these regulations without incident, there is a growing cohort of servicemembers with skin diseases that were exacerbated or even initiated by haircuts, hairstyling, and shaving required to conform to these grooming standards. These skin diseases, which can affect both sexes and may not be appreciated until years into a soldier's service commitment, can have consequences related to individual morbidity and medical readiness for deployment, making it an important issue for medical practitioners to recognize and manage in servicemembers.
This review highlights several disorders of the pilosebaceous unit of the head and neck that can be caused or exacerbated by military grooming standards, including inflammatory hair disorders, traction alopecia, and pseudofolliculitis barbae. Discussion of each entity will include a review of susceptibility and causality as well as initial treatment options to consider (Table).
Inflammatory Hair Disorders
The proper appearance of servicemembers in uniform represents self-discipline and conformity to the high standards of the military. This transition occurs as a rite of passage for many new male recruits who receive shaved haircuts during their first days of basic training. Thereafter, male servicemembers are required to maintain a tapered appearance of the hair per military regulations.1 Clipping hair closely to the scalp or shaving the head entirely are authorized and often encouraged; therefore, high and tight haircuts and buzz cuts are popular among male soldiers due to the general ease of care and ability to maintain the haircut themselves. Conversely, these styles require servicemembers to get weekly or biweekly haircuts that in turn can lead to chronic trauma and irritation. In more susceptible populations, inflammatory hair disorders such as acne keloidalis nuchae (AKN), dissecting cellulitis of the scalp, and folliculitis decalvans may be incited.
Acne Keloidalis Nuchae
Acne keloidalis nuchae, also called folliculitis keloidalis, is a chronic scarring folliculitis presenting with papules and plaques on the occiput and nape of the neck that may merge to form hypertrophic scars or keloids. This disorder most commonly develops in young black men but also can be seen in black females and white patients of both sexes.2 Acne keloidalis nuchae shares many histologic features with central centrifugal cicatricial alopecia, which may suggest a similar pathogenesis. Apart from frequent haircuts, tight-collared shirts, such as those on military service uniforms, also have been associated with AKN. Because of these suspected etiologies, first-line treatment focuses on preventing further trauma by avoiding mechanical irritation and short haircuts, which may be difficult in the military setting. For earlier disease stages, topical and intralesional corticosteroids, oral retinoids, and topical and oral antibiotics are used for their anti-inflammatory properties.3 In refractory cases, surgical excision with healing by secondary intention may be attempted.4 Additional treatment options include the 1064-nm Nd:YAG and 810-nm diode lasers,3 UVB light therapy, CO2 laser, and radiotherapy.
Dissecting Cellulitis of the Scalp
Similar to AKN, dissecting cellulitis of the scalp is another inflammatory hair disorder that is worsened by frequent short haircuts.5 Dissecting cellulitis of the scalp is a primary cicatricial alopecia proposed to be secondary to follicular occlusion. It often is seen in black males aged 20 to 40 years and is characterized by boggy suppurative nodules and cysts with draining sinus tracts, abscesses, and resultant scarring alopecia. Dissecting cellulitis of the scalp is part of the follicular occlusion tetrad, which also includes hidradenitis suppurativa, acne conglobata, and pilonidal cysts. First-line therapies include topical and oral antibiotics, topical retinoids, intralesional corticosteroids, incision and drainage of fluctuant nodules, and oral isotretinoin with or without rifampin. Alternative treatments include oral zinc supplementation, oral corticosteroids, tumor necrosis factor α inhibitors, laser therapies, radiotherapy, and surgical management with wide local excision or total scalpectomy.6,7
Folliculitis Decalvans
Folliculitis decalvans is a primary cicatricial alopecia of the scalp that most commonly presents in middle-aged men without racial predilection.8 Folliculitis decalvans presents with multiple pustules, crusts, tufted hairs, and perifollicular hyperkeratosis, leading to scarring of the scalp, which often is most severe on the posterior vertex. Staphylococcus aureus is a presumed player in the pathogenesis of folliculitis decalvans with superantigens causing release of cytokines stimulating follicular destruction. Close haircuts in conformation with military grooming standards can contribute to this condition due to mechanical trauma and subsequent inflammation. It typically is diagnosed clinically, but if histologic confirmation is desired, a sample from the periphery of early lesions is preferred.9 Initial treatment consists of antibacterial shampoos, topical corticosteroids, topical antibiotics, and combination oral antibiotic therapy with rifampin and clindamycin. Studies using oral isotretinoin have shown variable results,10,11 and the most effective treatment of recalcitrant lesions appears to be intralesional corticosteroids.12
Follicular and Scarring Disorders
In addition to inflammatory hair disorders, military grooming standards have been linked to the pathogenesis of diseases such as pseudofolliculitis barbae, traction alopecia, and keloids, specifically through irritation of the face, neck, and scalp, as well as damage to the follicular unit.5 These conditions develop because grooming regulations necessitate certain hair practices such as close shaving of facial and neck hair and keeping long hair secured relatively tightly to the scalp.
Pseudofolliculitis Barbae
Males in the military are obligated to keep their faces clean-shaven.1 They may acquire a medical waiver for a specified beard length if deemed appropriate by the treating physician,1 which often leads to the need for continual waiver renewal and also may warrant possible negative perception from peers, subordinates, and leadership. One of the most prevalent conditions that is closely associated with shaving is pseudofolliculitis barbae. The combination of close shaving and tightly coiled hairs causes the hairs to grow toward and penetrate the skin, particularly on the neck.13 In some cases, the hairs never actually exit the skin and simply curl within the superficial epidermis. A foreign body reaction often arises, leading to inflamed follicular papules and pustules. Affected individuals may experience pain, pruritus, and secondary infections. Postinflammatory hyperpigmentation, hypertrophic scarring, and keloid formation are common sequelae in cases of untreated disease. Pseudofolliculitis barbae also is exacerbated by pulling the skin taut and shaving against the grain, making behavioral interventions a key component in management of this condition. Preliminary recommendations include using a new or electric razor, leaving hair at least 2 mm in length, and shaving in the direction of hair growth. Other treatment options with varying effectiveness include daily alternation of a mild topical corticosteroid and one of the following: a topical retinoid, topical antibiotics, or glycolic acid. The only treatments that approach definitive cure are laser hair removal and electrolysis for which patient skin type plays an important role in laser selection.5
Traction Alopecia
Similar to their male counterparts, female military members must also present a conservative professional appearance, including hair that is neatly groomed.1 If the length of the hair extends beyond the uniform collar, it must be inconspicuously fastened or pinned above the collar. As a result, loosely tied hair is unauthorized, and females with long hair must secure their hair tightly on a daily basis. Traction alopecia results from tight hairstyling over a prolonged period and commonly affects female soldiers. The etiology is presumed to be mechanical loosening of hair within the follicles, leading to inflammation. Although traditionally seen in black women along the frontal and temporal hairlines, traction alopecia has been identified in individuals of all races and can occur anywhere on the scalp.5 Perifollicular erythema may be the first sign, and papules and pustules may be visible. Although the hair loss in traction alopecia usually is reversible if the traction is ceased, end-stage disease may be permanent.6 Halting traction-inducing practices is paramount, and other treatment options that may slow progression include topical or oral antibiotics and topical or intralesional corticosteroids. Recovery of hair loss also may be aided by topical minoxidil.5
Keloids
Keloid formation is an important pathology to address, as it may result from several of the aforementioned conditions. Keloids are most commonly seen in black individuals but also can occur in Hispanic and Asian patients. The cause has not been fully elucidated but is thought to be a combination of dysfunctional fibroblasts with a genetic component based on racial predilection and twin concordance studies.5 The chest, shoulders, upper back, neck, and earlobes are particularly susceptible to keloid formation, which can appear from 1 to 24 years following dermal trauma.5 Unlike hypertrophic scars, keloids generally do not regress and frequently cause discomfort, pruritus, and emotional distress. They also can hinder wearing a military uniform. Sustained remission is problematic, making prevention a first-line approach, including proper care of wounds when they occur and avoiding elective procedures such as piercings and tattoos. Intralesional corticosteroids, adjuvant injections (eg, 5-fluorouracil), silicone sheeting, cryotherapy, radiation, laser therapy, and excision are some of the treatment options when keloids have formed.5
Final Comment
It is important to recognize military grooming standards as a cause or contributor to several diseases of the head and neck in military servicemembers. Specifically, frequent haircuts in male soldiers are associated with several inflammatory hair disorders, including AKN, dissecting cellulitis of the scalp, and folliculitis decalvans, while daily shaving predisposes individuals to pseudofolliculitis barbae with possible keloid formation. Females may develop traction alopecia from chronically tight, pulled back hairstyles. All of these conditions have health implications for the affected individuals and can compromise the military mission. Awareness, prevention, and recognition are key along with the knowledge base to provide anticipatory avoidance and initiate appropriate treatments, thereby mitigating these potential consequences.
The US military enforces grooming standards to ensure the professional appearance and serviceability of soldiers in all operational settings. Although most individuals are able to uphold these regulations without incident, there is a growing cohort of servicemembers with skin diseases that were exacerbated or even initiated by haircuts, hairstyling, and shaving required to conform to these grooming standards. These skin diseases, which can affect both sexes and may not be appreciated until years into a soldier's service commitment, can have consequences related to individual morbidity and medical readiness for deployment, making it an important issue for medical practitioners to recognize and manage in servicemembers.
This review highlights several disorders of the pilosebaceous unit of the head and neck that can be caused or exacerbated by military grooming standards, including inflammatory hair disorders, traction alopecia, and pseudofolliculitis barbae. Discussion of each entity will include a review of susceptibility and causality as well as initial treatment options to consider (Table).
Inflammatory Hair Disorders
The proper appearance of servicemembers in uniform represents self-discipline and conformity to the high standards of the military. This transition occurs as a rite of passage for many new male recruits who receive shaved haircuts during their first days of basic training. Thereafter, male servicemembers are required to maintain a tapered appearance of the hair per military regulations.1 Clipping hair closely to the scalp or shaving the head entirely are authorized and often encouraged; therefore, high and tight haircuts and buzz cuts are popular among male soldiers due to the general ease of care and ability to maintain the haircut themselves. Conversely, these styles require servicemembers to get weekly or biweekly haircuts that in turn can lead to chronic trauma and irritation. In more susceptible populations, inflammatory hair disorders such as acne keloidalis nuchae (AKN), dissecting cellulitis of the scalp, and folliculitis decalvans may be incited.
Acne Keloidalis Nuchae
Acne keloidalis nuchae, also called folliculitis keloidalis, is a chronic scarring folliculitis presenting with papules and plaques on the occiput and nape of the neck that may merge to form hypertrophic scars or keloids. This disorder most commonly develops in young black men but also can be seen in black females and white patients of both sexes.2 Acne keloidalis nuchae shares many histologic features with central centrifugal cicatricial alopecia, which may suggest a similar pathogenesis. Apart from frequent haircuts, tight-collared shirts, such as those on military service uniforms, also have been associated with AKN. Because of these suspected etiologies, first-line treatment focuses on preventing further trauma by avoiding mechanical irritation and short haircuts, which may be difficult in the military setting. For earlier disease stages, topical and intralesional corticosteroids, oral retinoids, and topical and oral antibiotics are used for their anti-inflammatory properties.3 In refractory cases, surgical excision with healing by secondary intention may be attempted.4 Additional treatment options include the 1064-nm Nd:YAG and 810-nm diode lasers,3 UVB light therapy, CO2 laser, and radiotherapy.
Dissecting Cellulitis of the Scalp
Similar to AKN, dissecting cellulitis of the scalp is another inflammatory hair disorder that is worsened by frequent short haircuts.5 Dissecting cellulitis of the scalp is a primary cicatricial alopecia proposed to be secondary to follicular occlusion. It often is seen in black males aged 20 to 40 years and is characterized by boggy suppurative nodules and cysts with draining sinus tracts, abscesses, and resultant scarring alopecia. Dissecting cellulitis of the scalp is part of the follicular occlusion tetrad, which also includes hidradenitis suppurativa, acne conglobata, and pilonidal cysts. First-line therapies include topical and oral antibiotics, topical retinoids, intralesional corticosteroids, incision and drainage of fluctuant nodules, and oral isotretinoin with or without rifampin. Alternative treatments include oral zinc supplementation, oral corticosteroids, tumor necrosis factor α inhibitors, laser therapies, radiotherapy, and surgical management with wide local excision or total scalpectomy.6,7
Folliculitis Decalvans
Folliculitis decalvans is a primary cicatricial alopecia of the scalp that most commonly presents in middle-aged men without racial predilection.8 Folliculitis decalvans presents with multiple pustules, crusts, tufted hairs, and perifollicular hyperkeratosis, leading to scarring of the scalp, which often is most severe on the posterior vertex. Staphylococcus aureus is a presumed player in the pathogenesis of folliculitis decalvans with superantigens causing release of cytokines stimulating follicular destruction. Close haircuts in conformation with military grooming standards can contribute to this condition due to mechanical trauma and subsequent inflammation. It typically is diagnosed clinically, but if histologic confirmation is desired, a sample from the periphery of early lesions is preferred.9 Initial treatment consists of antibacterial shampoos, topical corticosteroids, topical antibiotics, and combination oral antibiotic therapy with rifampin and clindamycin. Studies using oral isotretinoin have shown variable results,10,11 and the most effective treatment of recalcitrant lesions appears to be intralesional corticosteroids.12
Follicular and Scarring Disorders
In addition to inflammatory hair disorders, military grooming standards have been linked to the pathogenesis of diseases such as pseudofolliculitis barbae, traction alopecia, and keloids, specifically through irritation of the face, neck, and scalp, as well as damage to the follicular unit.5 These conditions develop because grooming regulations necessitate certain hair practices such as close shaving of facial and neck hair and keeping long hair secured relatively tightly to the scalp.
Pseudofolliculitis Barbae
Males in the military are obligated to keep their faces clean-shaven.1 They may acquire a medical waiver for a specified beard length if deemed appropriate by the treating physician,1 which often leads to the need for continual waiver renewal and also may warrant possible negative perception from peers, subordinates, and leadership. One of the most prevalent conditions that is closely associated with shaving is pseudofolliculitis barbae. The combination of close shaving and tightly coiled hairs causes the hairs to grow toward and penetrate the skin, particularly on the neck.13 In some cases, the hairs never actually exit the skin and simply curl within the superficial epidermis. A foreign body reaction often arises, leading to inflamed follicular papules and pustules. Affected individuals may experience pain, pruritus, and secondary infections. Postinflammatory hyperpigmentation, hypertrophic scarring, and keloid formation are common sequelae in cases of untreated disease. Pseudofolliculitis barbae also is exacerbated by pulling the skin taut and shaving against the grain, making behavioral interventions a key component in management of this condition. Preliminary recommendations include using a new or electric razor, leaving hair at least 2 mm in length, and shaving in the direction of hair growth. Other treatment options with varying effectiveness include daily alternation of a mild topical corticosteroid and one of the following: a topical retinoid, topical antibiotics, or glycolic acid. The only treatments that approach definitive cure are laser hair removal and electrolysis for which patient skin type plays an important role in laser selection.5
Traction Alopecia
Similar to their male counterparts, female military members must also present a conservative professional appearance, including hair that is neatly groomed.1 If the length of the hair extends beyond the uniform collar, it must be inconspicuously fastened or pinned above the collar. As a result, loosely tied hair is unauthorized, and females with long hair must secure their hair tightly on a daily basis. Traction alopecia results from tight hairstyling over a prolonged period and commonly affects female soldiers. The etiology is presumed to be mechanical loosening of hair within the follicles, leading to inflammation. Although traditionally seen in black women along the frontal and temporal hairlines, traction alopecia has been identified in individuals of all races and can occur anywhere on the scalp.5 Perifollicular erythema may be the first sign, and papules and pustules may be visible. Although the hair loss in traction alopecia usually is reversible if the traction is ceased, end-stage disease may be permanent.6 Halting traction-inducing practices is paramount, and other treatment options that may slow progression include topical or oral antibiotics and topical or intralesional corticosteroids. Recovery of hair loss also may be aided by topical minoxidil.5
Keloids
Keloid formation is an important pathology to address, as it may result from several of the aforementioned conditions. Keloids are most commonly seen in black individuals but also can occur in Hispanic and Asian patients. The cause has not been fully elucidated but is thought to be a combination of dysfunctional fibroblasts with a genetic component based on racial predilection and twin concordance studies.5 The chest, shoulders, upper back, neck, and earlobes are particularly susceptible to keloid formation, which can appear from 1 to 24 years following dermal trauma.5 Unlike hypertrophic scars, keloids generally do not regress and frequently cause discomfort, pruritus, and emotional distress. They also can hinder wearing a military uniform. Sustained remission is problematic, making prevention a first-line approach, including proper care of wounds when they occur and avoiding elective procedures such as piercings and tattoos. Intralesional corticosteroids, adjuvant injections (eg, 5-fluorouracil), silicone sheeting, cryotherapy, radiation, laser therapy, and excision are some of the treatment options when keloids have formed.5
Final Comment
It is important to recognize military grooming standards as a cause or contributor to several diseases of the head and neck in military servicemembers. Specifically, frequent haircuts in male soldiers are associated with several inflammatory hair disorders, including AKN, dissecting cellulitis of the scalp, and folliculitis decalvans, while daily shaving predisposes individuals to pseudofolliculitis barbae with possible keloid formation. Females may develop traction alopecia from chronically tight, pulled back hairstyles. All of these conditions have health implications for the affected individuals and can compromise the military mission. Awareness, prevention, and recognition are key along with the knowledge base to provide anticipatory avoidance and initiate appropriate treatments, thereby mitigating these potential consequences.
- US Department of the Army. Wear and Appearance of Army Uniforms and Insignia: Army Regulation 670-1. Washington, DC: Department of the Army; 2017. https://history.army.mil/html/forcestruc/docs/AR670-1.pdf. Accessed October 11, 2018.
- East-Innis AD, Stylianou K, Paolino A, et al. Acne keloidalis nuchae: risk factors and associated disorders--a retrospective study. Int J Dermatol. 2017;56:828-832.
- Maranda EL, Simmons BJ, Nguyen AH, et al. Treatment of acne keloidalis nuchae: a systematic review of the literature. Dermatol Ther (Heidelb). 2016;6:363-378.
- Glenn MJ, Bennett RG, Kelly AP. Acne keloidalis nuchae: treatment with excision and second-intention healing. J Am Acad Dermatol. 1995;33:243-246.
- Madu P, Kundu RV. Follicular and scarring disorders in skin of color: presentation and management. Am J Clin Dermatol. 2014;15:307-321.
- Rodney IJ, Onwudiwe OC. Hair and scalp disorders in ethnic populations. J Drugs Dermatol. 2013;12:420-427.
- Lindsey SF, Tosti A. Ethnic hair disorders. Curr Probl Dermatol. 2015;47:139-148.
- Whiting DA. Cicatricial alopecia: clinico-pathological findings and treatment. Clin Dermatol. 2001;19:211-225.
- Sperling LC, Cowper SE, Knopp EA. An Atlas of Hair Pathology with Clinical Correlations. 2nd ed. Boca Raton, FL: CRC Press; 2012.
- Gemmeke A, Wollina U. Folliculitis decalvans of the scalp: response to triple therapy with isotretinoin, clindamycin, and prednisolone. Acta Dermatovenerol Alp Pannonica Adriat. 2006;15:184-186.
- Hallai N, Thompson I, Williams P, et al. Folliculitis spinulosa decalvans: failure to respond to oral isotretinoin. J Eur Acad Dermatol Venereol. 2006;20:223-224.
- Bolduc C, Sperling LC, Shapiro J. Primary cicatricial alopecia. J Am Acad Dermatol. 2016;75:101-117.
- Perry PK, Cook-Bolden FE, Rahman Z, et al. Defining pseudofolliculitis barbae in 2001: a review of the literature and current trends. J Am Acad Dermatol. 2002;46(2 suppl):S113-S119.
- US Department of the Army. Wear and Appearance of Army Uniforms and Insignia: Army Regulation 670-1. Washington, DC: Department of the Army; 2017. https://history.army.mil/html/forcestruc/docs/AR670-1.pdf. Accessed October 11, 2018.
- East-Innis AD, Stylianou K, Paolino A, et al. Acne keloidalis nuchae: risk factors and associated disorders--a retrospective study. Int J Dermatol. 2017;56:828-832.
- Maranda EL, Simmons BJ, Nguyen AH, et al. Treatment of acne keloidalis nuchae: a systematic review of the literature. Dermatol Ther (Heidelb). 2016;6:363-378.
- Glenn MJ, Bennett RG, Kelly AP. Acne keloidalis nuchae: treatment with excision and second-intention healing. J Am Acad Dermatol. 1995;33:243-246.
- Madu P, Kundu RV. Follicular and scarring disorders in skin of color: presentation and management. Am J Clin Dermatol. 2014;15:307-321.
- Rodney IJ, Onwudiwe OC. Hair and scalp disorders in ethnic populations. J Drugs Dermatol. 2013;12:420-427.
- Lindsey SF, Tosti A. Ethnic hair disorders. Curr Probl Dermatol. 2015;47:139-148.
- Whiting DA. Cicatricial alopecia: clinico-pathological findings and treatment. Clin Dermatol. 2001;19:211-225.
- Sperling LC, Cowper SE, Knopp EA. An Atlas of Hair Pathology with Clinical Correlations. 2nd ed. Boca Raton, FL: CRC Press; 2012.
- Gemmeke A, Wollina U. Folliculitis decalvans of the scalp: response to triple therapy with isotretinoin, clindamycin, and prednisolone. Acta Dermatovenerol Alp Pannonica Adriat. 2006;15:184-186.
- Hallai N, Thompson I, Williams P, et al. Folliculitis spinulosa decalvans: failure to respond to oral isotretinoin. J Eur Acad Dermatol Venereol. 2006;20:223-224.
- Bolduc C, Sperling LC, Shapiro J. Primary cicatricial alopecia. J Am Acad Dermatol. 2016;75:101-117.
- Perry PK, Cook-Bolden FE, Rahman Z, et al. Defining pseudofolliculitis barbae in 2001: a review of the literature and current trends. J Am Acad Dermatol. 2002;46(2 suppl):S113-S119.
Practice Points
- The short frequent haircuts required to maintain a tapered appearance of the hair per US military regulations may lead to inflammatory hair disorders such as acne keloidalis nuchae, dissecting cellulitis of the scalp, and folliculitis decalvans.
- The mainstay of prevention for these conditions is avoidance of inciting factors such as short haircuts, tight-collared shirts, frequent shaving, or tight hairstyles.
- Early identification and treatment of inflammatory follicular and scarring disorders can prevent further scarring, pigmentation changes, and/or disfigurement.
Tinea Incognito in an Urban Pediatric Population
Tinea incognito (TI) describes a dermatophytosis with often atypical clinical features attributed to prior use of topical corticosteroids or other immunomodulating agents. Tinea incognito may lack the scale and elevated margin typical of cutaneous dermatophytoses and can be mistaken for other pediatric cutaneous diseases, particularly atopic dermatitis. 1 Given the prevalence of TI and its susceptibility to misdiagnosis, we conducted a retrospective medical record review of cases of pediatric dermatophytosis presenting from 2005 to 2016.
Methods
We reviewed medical records for patients younger than 18 years who had been seen at the Faculty Group Practice of the Ronald O. Perelman Department of Dermatology, New York University School of Medicine (New York, New York), between January 1, 2005, and October 21, 2016, using International Classification of Diseases, Ninth Revision (ICD-9) codes 110.0 (tinea capitis), 110.1 (onychomycosis/tinea unguium), 110.3 (tinea cruris), 110.4 (tinea pedis), 110.5 (tinea corporis), and 110.9 (tinea, unspecified site). Cases were included in this study if there was documentation of dermatophytosis previously treated with topical corticosteroids or calcineurin inhibitors as well as positive potassium hydroxide (KOH) preparation or fungal culture with dermatophyte growth obtained from lesions satisfying the first criterion. This study was approved by the New York University School of Medicine institutional review board (study no. S15-01388).
Statistical analyses were conducted in SPSS 19.0 for Windows. Categorical variables were assessed using the χ2 test for independence and the Fisher exact test.
Results
A total of 464 cases were reviewed. A positive KOH preparation or dermatophyte fungal culture was documented in 83 cases. Of them, 29 (34.9%) were treated with topical steroids and/or calcineurin inhibitors prior to presentation to dermatology (Table). The mean age at presentation was 8 years. Duration of symptoms prior to presentation was recorded for 23 of 29 patients (79.3%). Of them, 6 (26.1%) experienced symptoms for 1 month or less, 12 (52.2%) for 1 to 6 months, and 5 (21.7%) for 6 months to 1 year.

Physical examination findings (Figure) were documented in all 29 cases. Annular lesions were noted in 24 patients (82.8%). Pustules were present in 5 patients (17.2%) and papules in 11 patients (37.9%). Fourteen patients (48.3%) had involvement of the face, 14 (48.3%) of the body (ie, trunk, extremities, or groin), and 3 (10.3%) of the scalp. Six patients (20.7%) demonstrated findings at more than one body site.
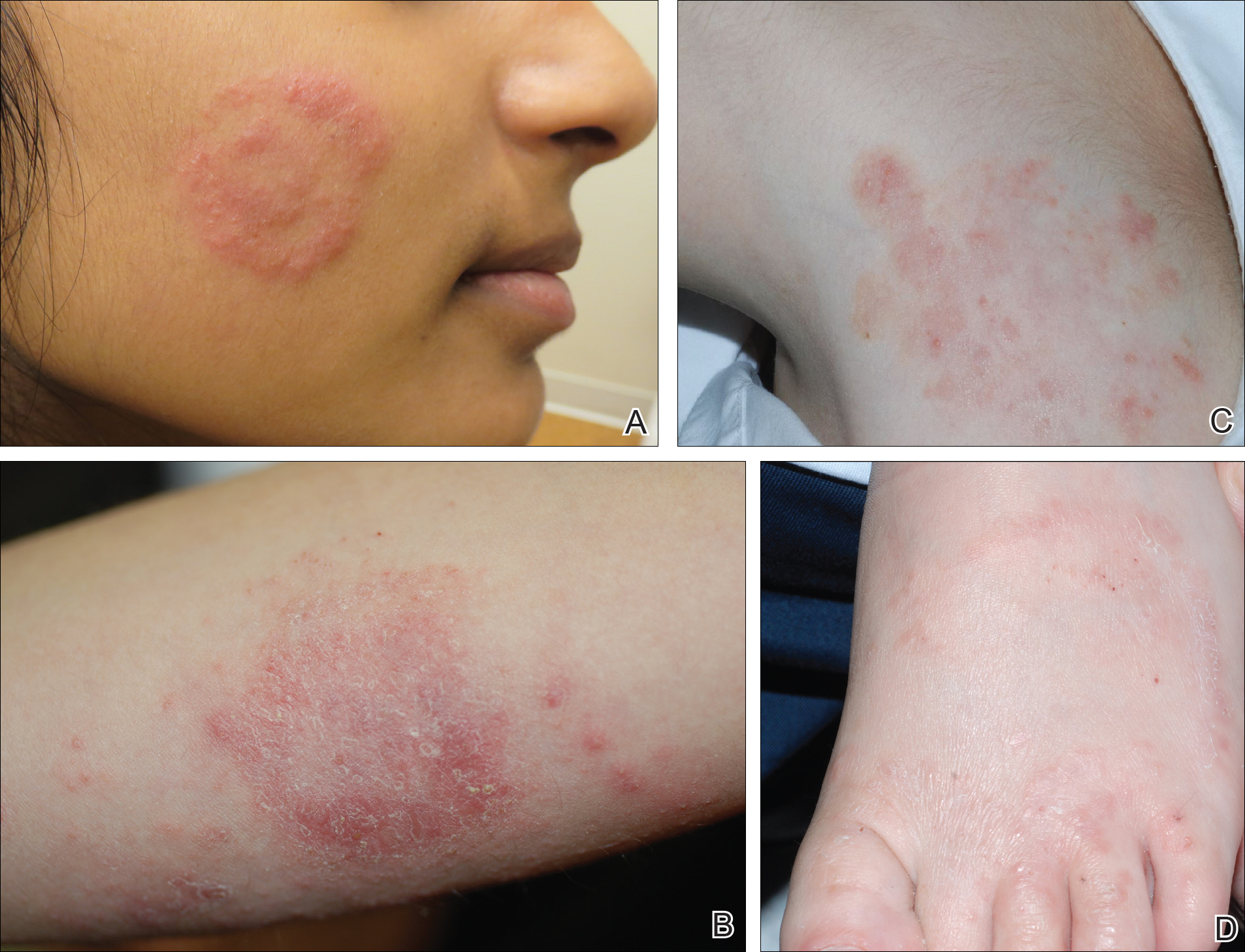
Females were more likely to demonstrate facial lesions (P=.02), while males were more likely to present with body lesions (P=.04). Of 26 patients diagnosed via fungal culture, 16 (55.2%) grew Trichophyton tonsurans, 4 (13.8%) grew Trichophyton rubrum, 3 (10.3%) grew Trichophyton mentagrophytes, 2 (6.9%) grew Microsporum canis, and 1 (3.4%) grew Microsporum gypseum. Treatment entailed oral medication in 18 cases (62.1%). Of them, 13 (72.2%) were treated with griseofulvin, 3 (16.7%) with fluconazole, and 2 (11.1%) with terbinafine. Topical antifungals were prescribed in the remaining 11 cases (37.9%); no further treatment was documented.
Comment
Since the initial description of TI, approximately 60 case reports and small series as well as several larger observational studies describing TI have been published. In our series of pediatric patients, 29 of 83 culture- or KOH-confirmed dermatophytosis cases (34.9%) were considered to be TI due to treatment with topical corticosteroids and/or calcineurin inhibitors prior to presentation. This high prevalence contrasts with the 5.6% prevalence reported in the only prior large case series examining TI in childhood.2 These authors further reported that in their pediatric population, TI was significantly (odds ratio, 8.7; 95% CI, 4.7-16.1) more likely to occur on the face relative to other dermatophytoses and significantly (odds ratio, 0.014; 95% CI, 0.002-0.099) less likely to occur on the scalp.2 We noted a significant association between female gender and facial symptoms as well as between male gender and truncal symptoms. Taken together, these findings suggest an increased likelihood of pediatric tinea faciei to be inappropriately treated, particularly in females.
Although TI treated with topical corticosteroids or calcineurin inhibitors can mimic other skin diseases, a majority of patients in our series demonstrated findings associated with classic tinea, such as annularity and scale. Further, we found that T tonsurans was the causative organism in most cases with T rubrum uncommonly seen, though it is the most prevalent dermatophyte observed worldwide and in 2 large TI case series.3,4 Regional variation in dermatophytes may account for these differences. In our study, griseofulvin was used most frequently in TI treatment, though a systematic review of oral antifungals in tinea capitis supported terbinafine’s greater efficacy in patients infected with T tonsurans.5
Conclusion
Our case series demonstrated a 35% prevalence of TI cases in a population of children with confirmed dermatophytosis presenting to dermatologists at an American academic medical center. We hope that noting the high prevalence and manifold presentations of this disease will aid practitioners in maintaining clinical suspicion for dermatophytosis and thereby facilitate appropriate identification and treatment of TI.
- Paloni G, Valerio E, Berti I, et al. Tinea incognito [published online September 28, 2015]. J Pediatr. 2015;167:1450-e2.
- del Boz J, Crespo V, Rivas‐Ruiz F, et al. Tinea incognito in children: 54 cases. Mycoses. 2011;54:254-258.
- Romano C, Maritati E, Gianni C. Tinea incognito in Italy: a 15-year survey. Mycoses. 2006;49:383-387.
- Kim WJ, Kim TW, Mun JH, et al. Tinea incognito in Korea and itsrisk factors: nine-year multicenter survey. J Korean Med Sci. 2013;28:145-151.
- Chen X, Jiang X, Yang M, et al. Systemic antifungal therapy for tinea capitis in children: an abridged Cochrane review. J Am Acad Dermatol. 2017;76:368-374.
Tinea incognito (TI) describes a dermatophytosis with often atypical clinical features attributed to prior use of topical corticosteroids or other immunomodulating agents. Tinea incognito may lack the scale and elevated margin typical of cutaneous dermatophytoses and can be mistaken for other pediatric cutaneous diseases, particularly atopic dermatitis. 1 Given the prevalence of TI and its susceptibility to misdiagnosis, we conducted a retrospective medical record review of cases of pediatric dermatophytosis presenting from 2005 to 2016.
Methods
We reviewed medical records for patients younger than 18 years who had been seen at the Faculty Group Practice of the Ronald O. Perelman Department of Dermatology, New York University School of Medicine (New York, New York), between January 1, 2005, and October 21, 2016, using International Classification of Diseases, Ninth Revision (ICD-9) codes 110.0 (tinea capitis), 110.1 (onychomycosis/tinea unguium), 110.3 (tinea cruris), 110.4 (tinea pedis), 110.5 (tinea corporis), and 110.9 (tinea, unspecified site). Cases were included in this study if there was documentation of dermatophytosis previously treated with topical corticosteroids or calcineurin inhibitors as well as positive potassium hydroxide (KOH) preparation or fungal culture with dermatophyte growth obtained from lesions satisfying the first criterion. This study was approved by the New York University School of Medicine institutional review board (study no. S15-01388).
Statistical analyses were conducted in SPSS 19.0 for Windows. Categorical variables were assessed using the χ2 test for independence and the Fisher exact test.
Results
A total of 464 cases were reviewed. A positive KOH preparation or dermatophyte fungal culture was documented in 83 cases. Of them, 29 (34.9%) were treated with topical steroids and/or calcineurin inhibitors prior to presentation to dermatology (Table). The mean age at presentation was 8 years. Duration of symptoms prior to presentation was recorded for 23 of 29 patients (79.3%). Of them, 6 (26.1%) experienced symptoms for 1 month or less, 12 (52.2%) for 1 to 6 months, and 5 (21.7%) for 6 months to 1 year.
Physical examination findings (Figure) were documented in all 29 cases. Annular lesions were noted in 24 patients (82.8%). Pustules were present in 5 patients (17.2%) and papules in 11 patients (37.9%). Fourteen patients (48.3%) had involvement of the face, 14 (48.3%) of the body (ie, trunk, extremities, or groin), and 3 (10.3%) of the scalp. Six patients (20.7%) demonstrated findings at more than one body site.
Females were more likely to demonstrate facial lesions (P=.02), while males were more likely to present with body lesions (P=.04). Of 26 patients diagnosed via fungal culture, 16 (55.2%) grew Trichophyton tonsurans, 4 (13.8%) grew Trichophyton rubrum, 3 (10.3%) grew Trichophyton mentagrophytes, 2 (6.9%) grew Microsporum canis, and 1 (3.4%) grew Microsporum gypseum. Treatment entailed oral medication in 18 cases (62.1%). Of them, 13 (72.2%) were treated with griseofulvin, 3 (16.7%) with fluconazole, and 2 (11.1%) with terbinafine. Topical antifungals were prescribed in the remaining 11 cases (37.9%); no further treatment was documented.
Comment
Since the initial description of TI, approximately 60 case reports and small series as well as several larger observational studies describing TI have been published. In our series of pediatric patients, 29 of 83 culture- or KOH-confirmed dermatophytosis cases (34.9%) were considered to be TI due to treatment with topical corticosteroids and/or calcineurin inhibitors prior to presentation. This high prevalence contrasts with the 5.6% prevalence reported in the only prior large case series examining TI in childhood.2 These authors further reported that in their pediatric population, TI was significantly (odds ratio, 8.7; 95% CI, 4.7-16.1) more likely to occur on the face relative to other dermatophytoses and significantly (odds ratio, 0.014; 95% CI, 0.002-0.099) less likely to occur on the scalp.2 We noted a significant association between female gender and facial symptoms as well as between male gender and truncal symptoms. Taken together, these findings suggest an increased likelihood of pediatric tinea faciei to be inappropriately treated, particularly in females.
Although TI treated with topical corticosteroids or calcineurin inhibitors can mimic other skin diseases, a majority of patients in our series demonstrated findings associated with classic tinea, such as annularity and scale. Further, we found that T tonsurans was the causative organism in most cases with T rubrum uncommonly seen, though it is the most prevalent dermatophyte observed worldwide and in 2 large TI case series.3,4 Regional variation in dermatophytes may account for these differences. In our study, griseofulvin was used most frequently in TI treatment, though a systematic review of oral antifungals in tinea capitis supported terbinafine’s greater efficacy in patients infected with T tonsurans.5
Conclusion
Our case series demonstrated a 35% prevalence of TI cases in a population of children with confirmed dermatophytosis presenting to dermatologists at an American academic medical center. We hope that noting the high prevalence and manifold presentations of this disease will aid practitioners in maintaining clinical suspicion for dermatophytosis and thereby facilitate appropriate identification and treatment of TI.
Tinea incognito (TI) describes a dermatophytosis with often atypical clinical features attributed to prior use of topical corticosteroids or other immunomodulating agents. Tinea incognito may lack the scale and elevated margin typical of cutaneous dermatophytoses and can be mistaken for other pediatric cutaneous diseases, particularly atopic dermatitis. 1 Given the prevalence of TI and its susceptibility to misdiagnosis, we conducted a retrospective medical record review of cases of pediatric dermatophytosis presenting from 2005 to 2016.
Methods
We reviewed medical records for patients younger than 18 years who had been seen at the Faculty Group Practice of the Ronald O. Perelman Department of Dermatology, New York University School of Medicine (New York, New York), between January 1, 2005, and October 21, 2016, using International Classification of Diseases, Ninth Revision (ICD-9) codes 110.0 (tinea capitis), 110.1 (onychomycosis/tinea unguium), 110.3 (tinea cruris), 110.4 (tinea pedis), 110.5 (tinea corporis), and 110.9 (tinea, unspecified site). Cases were included in this study if there was documentation of dermatophytosis previously treated with topical corticosteroids or calcineurin inhibitors as well as positive potassium hydroxide (KOH) preparation or fungal culture with dermatophyte growth obtained from lesions satisfying the first criterion. This study was approved by the New York University School of Medicine institutional review board (study no. S15-01388).
Statistical analyses were conducted in SPSS 19.0 for Windows. Categorical variables were assessed using the χ2 test for independence and the Fisher exact test.
Results
A total of 464 cases were reviewed. A positive KOH preparation or dermatophyte fungal culture was documented in 83 cases. Of them, 29 (34.9%) were treated with topical steroids and/or calcineurin inhibitors prior to presentation to dermatology (Table). The mean age at presentation was 8 years. Duration of symptoms prior to presentation was recorded for 23 of 29 patients (79.3%). Of them, 6 (26.1%) experienced symptoms for 1 month or less, 12 (52.2%) for 1 to 6 months, and 5 (21.7%) for 6 months to 1 year.
Physical examination findings (Figure) were documented in all 29 cases. Annular lesions were noted in 24 patients (82.8%). Pustules were present in 5 patients (17.2%) and papules in 11 patients (37.9%). Fourteen patients (48.3%) had involvement of the face, 14 (48.3%) of the body (ie, trunk, extremities, or groin), and 3 (10.3%) of the scalp. Six patients (20.7%) demonstrated findings at more than one body site.
Females were more likely to demonstrate facial lesions (P=.02), while males were more likely to present with body lesions (P=.04). Of 26 patients diagnosed via fungal culture, 16 (55.2%) grew Trichophyton tonsurans, 4 (13.8%) grew Trichophyton rubrum, 3 (10.3%) grew Trichophyton mentagrophytes, 2 (6.9%) grew Microsporum canis, and 1 (3.4%) grew Microsporum gypseum. Treatment entailed oral medication in 18 cases (62.1%). Of them, 13 (72.2%) were treated with griseofulvin, 3 (16.7%) with fluconazole, and 2 (11.1%) with terbinafine. Topical antifungals were prescribed in the remaining 11 cases (37.9%); no further treatment was documented.
Comment
Since the initial description of TI, approximately 60 case reports and small series as well as several larger observational studies describing TI have been published. In our series of pediatric patients, 29 of 83 culture- or KOH-confirmed dermatophytosis cases (34.9%) were considered to be TI due to treatment with topical corticosteroids and/or calcineurin inhibitors prior to presentation. This high prevalence contrasts with the 5.6% prevalence reported in the only prior large case series examining TI in childhood.2 These authors further reported that in their pediatric population, TI was significantly (odds ratio, 8.7; 95% CI, 4.7-16.1) more likely to occur on the face relative to other dermatophytoses and significantly (odds ratio, 0.014; 95% CI, 0.002-0.099) less likely to occur on the scalp.2 We noted a significant association between female gender and facial symptoms as well as between male gender and truncal symptoms. Taken together, these findings suggest an increased likelihood of pediatric tinea faciei to be inappropriately treated, particularly in females.
Although TI treated with topical corticosteroids or calcineurin inhibitors can mimic other skin diseases, a majority of patients in our series demonstrated findings associated with classic tinea, such as annularity and scale. Further, we found that T tonsurans was the causative organism in most cases with T rubrum uncommonly seen, though it is the most prevalent dermatophyte observed worldwide and in 2 large TI case series.3,4 Regional variation in dermatophytes may account for these differences. In our study, griseofulvin was used most frequently in TI treatment, though a systematic review of oral antifungals in tinea capitis supported terbinafine’s greater efficacy in patients infected with T tonsurans.5
Conclusion
Our case series demonstrated a 35% prevalence of TI cases in a population of children with confirmed dermatophytosis presenting to dermatologists at an American academic medical center. We hope that noting the high prevalence and manifold presentations of this disease will aid practitioners in maintaining clinical suspicion for dermatophytosis and thereby facilitate appropriate identification and treatment of TI.
- Paloni G, Valerio E, Berti I, et al. Tinea incognito [published online September 28, 2015]. J Pediatr. 2015;167:1450-e2.
- del Boz J, Crespo V, Rivas‐Ruiz F, et al. Tinea incognito in children: 54 cases. Mycoses. 2011;54:254-258.
- Romano C, Maritati E, Gianni C. Tinea incognito in Italy: a 15-year survey. Mycoses. 2006;49:383-387.
- Kim WJ, Kim TW, Mun JH, et al. Tinea incognito in Korea and itsrisk factors: nine-year multicenter survey. J Korean Med Sci. 2013;28:145-151.
- Chen X, Jiang X, Yang M, et al. Systemic antifungal therapy for tinea capitis in children: an abridged Cochrane review. J Am Acad Dermatol. 2017;76:368-374.
- Paloni G, Valerio E, Berti I, et al. Tinea incognito [published online September 28, 2015]. J Pediatr. 2015;167:1450-e2.
- del Boz J, Crespo V, Rivas‐Ruiz F, et al. Tinea incognito in children: 54 cases. Mycoses. 2011;54:254-258.
- Romano C, Maritati E, Gianni C. Tinea incognito in Italy: a 15-year survey. Mycoses. 2006;49:383-387.
- Kim WJ, Kim TW, Mun JH, et al. Tinea incognito in Korea and itsrisk factors: nine-year multicenter survey. J Korean Med Sci. 2013;28:145-151.
- Chen X, Jiang X, Yang M, et al. Systemic antifungal therapy for tinea capitis in children: an abridged Cochrane review. J Am Acad Dermatol. 2017;76:368-374.
Practice Points
- Within our pediatric study population of microbiologically confirmed tinea cases at an American academic center, we found a 35% prevalence of tinea incognito (TI).
- Unlike investigations of TI in other countries, Trichophyton tonsurans was found to be the most common causative dermatophyte.
- Our data suggest that facial tinea may be more likely to be improperly treated in females and likewise tinea of the trunk or extremities in males.
Eruptive Vellus Hair Cysts in Identical Triplets With Dermoscopic Findings
Case Report
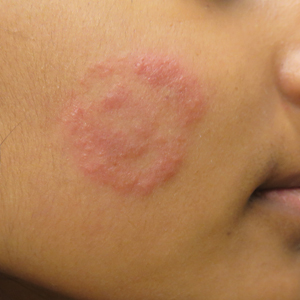
Four-year-old identical triplet girls with numerous asymptomatic scattered papules on the chest of 4 months’ duration were referred to a dermatologist by their pediatrician for molluscum contagiosum. The patients’ father reported that there was no history of trauma, irritation, or manipulation to the affected area. Their medical history was notable for prematurity at 32 weeks’ gestation and congenital dermal melanocytosis. Family history was notable for their father having acne and similar papules on the chest during adolescence that resolved with isotretinoin therapy.
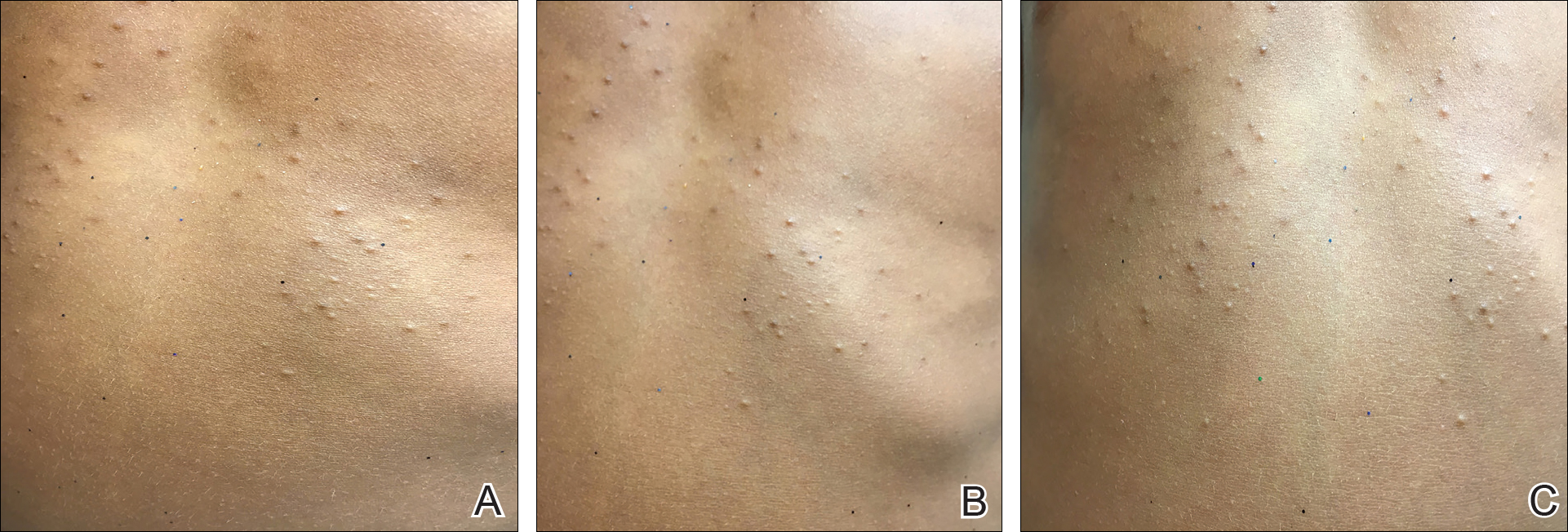
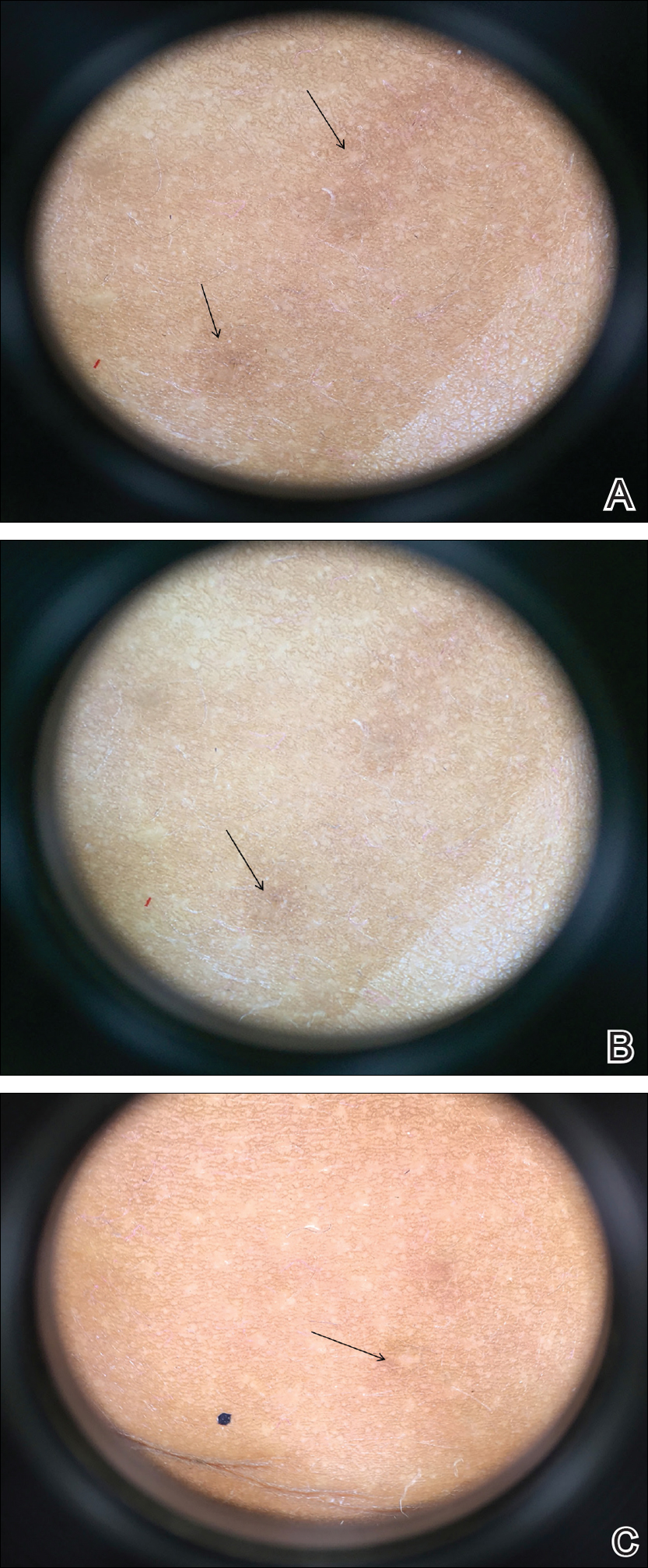
On physical examination there were multiple smooth, hyperpigmented to erythematous, comedonal, 1- to 2-mm papules dispersed on the anterior central chest of all 3 patients (Figure 1). Clinically, these lesions were fairly indistinguishable from other common dermatologic conditions such as acne or milia. Dermoscopic examination revealed homogenous yellow-white areas surrounded by light brown to erythematous halos (Figure 2). Histopathologic examination was not performed given the benign clinical diagnosis and avoidance of biopsy in pediatric populations. Based on dermoscopic features and history, a diagnosis of eruptive vellus hair cysts (EVHCs) in identical triplets was made.
Comment
Pathogenesis
Eruptive vellus hair cysts, first introduced by Esterly et al1 in 1977, are uncommon benign lesions presumed to be caused by an abnormal development of the infundibular portion of the hair follicle.2 They are usually 1- to 3-mm, reddish brown, monomorphous papules overlapping with pilosebaceous and apocrine units.3 Although the lesions typically are located on the chest and extremities, they may occur on the face, abdomen, axillae, buttocks, or genital area.1,3 The inheritance of EVHCs is unclear. The majority of reported cases are sporadic; however, the literature mentions 19 families affected by autosomal-dominant EVHCs based on phylogeny.3 In 2015, EVHCs were reported in identical twins, further supporting the case for a genetic mutation.4 We augment this autosomal-dominant inheritance pattern by presenting a case of identical triplets with EVHCs. The patients’ father reported similar lesions in childhood, further underscoring a genetic basis.
The pathogenesis of EVHC is uncertain, with 2 main theories. Some propose retention of vellus hair and keratin in a cavity formed by an abnormal vellus hair follicle causing infundibular occlusion. Others consider the growth of benign follicular hamartomas that differentiate to become vellus hairs.1
Clinical Presentation
The sporadic form of EVHCs is noted to be more common and clinically presents later, with an average age at onset of 16 years and an average age at diagnosis of 24 years.3 The sporadic form occurs without trauma or manipulation as a precursor. Less commonly, lesions present at birth or in early infancy and may show an autosomal-dominant inheritance pattern with a similar distribution across relatives.3
Other variants of EVHCs have been described. Late-onset EVHC usually occurs at 35 years or older (average age, 57 years), with a female to male predominance of 2.5 to 1.3 This late onset may be attributed to proliferation of ductal follicular keratinocytes or loss of perifollicular elastic fibers exacerbated by exogenous factors such as manipulation, UV rays, or trauma.5
For unilesional EVHC, the average age at diagnosis is 27 years.3 Some of these lesions may be pedunculated and greater than 8 mm. There is a female to male predominance of 2 to 1. Eruptive vellus hair cysts with steatocystoma multiplex can be seen with an average age at onset of 19 years and a female to male predominance of 0.2 to 1. There may be a family history of this subset, as reported in 3 patients with this pattern.3
Diagnosis
The recommended workup for EVHCs varies by patient and age. Eruptive vellus hair cysts present an opportunity to utilize noninvasive diagnostic procedures, especially for the pediatric population, to avoid scarring and pain from manipulation or biopsies. Although many practitioners may comfortably diagnose EVHCs clinically, 6 cases were misdiagnosed as steatocystoma multiplex, keratosis pilaris, or milia prior to histopathology revealing vellus hair cysts.6
Dermoscopy presents as a useful diagnostic aid. Eruptive vellus hair cysts exhibit light yellow homogenous circular structures with a maroon or erythematous halo.2,7 A central gray-blue color point may be seen due to melanin in the pigmented hair shaft.7 A dermoscopy review of EVHCs reported radiating capillaries.2 Occasionally, nonfollicular homogenous blue pigmentation may be seen due to a connection to atrophic hair follicles in the mid dermis and no normal hair follicle around the cysts.8 In comparison, dermoscopic characteristics of molluscum contagiosum demonstrated a polylobular, white-yellow, amorphous structure at the center with a hardened central umbilicated core and a crown of hairpin vessels at the periphery. Additionally, comedonal acne, commonly mistaken for EVHCs, reveals a brown-yellow hard central plug with sparse inflammation under dermoscopy.2 Thus, differentiation of these entities with dermoscopy should be highly prioritized to better aid in the diagnosis of pediatric dermatologic conditions using painless noninvasive techniques.
Treatment
The main indication for treatment of EVHCs is cosmetic concern. Twenty-five percent of EVHCs spontaneously resolve with transepidermal hair elimination or a granulomatous reaction.4,5 A case report of 4 siblings with congenital EVHCs also described a mother with similar lesions that resolved spontaneously in early adulthood,3 as our patients’ father also noted. Treatment modalities including topical keratolytic agents such as urea 10%, retinoic acid 0.05%, tazarotene cream 0.1%, and lactic acid 12%; incision and drainage; CO2 laser; or erbium-doped YAG laser ablation have been tried with minimal improvement.9 Of note, tazarotene cream 0.1% has demonstrated better results than both erbium-doped YAG laser and drainage and incision of EVHCs.4 Additionally, another report evidenced partial improvement with calcipotriene within 2 months with some lesions completely resolved and others flattened, which may be attributed to the antiproliferative and prodifferentiating effects on the ductal follicular keratinocytes by calcipotriene.5 Lastly, an additional study indicated that isotretinoin and vitamin A derivatives were ineffective for clearing EVHCs.10
Conclusion
We presented 3 identical triplets with the classic pediatric onset and dermoscopic findings of EVHCs on the trunk. Although the definitive diagnosis of EVHCs relies on histopathology, we argue that their unique dermoscopic findings combined with a thorough clinical examination is sufficient to recognize this benign condition and avoid painful procedures in the pediatric population.
- Esterly NB, Fretzin DF, Pinkus H. Eruptive vellus hair cysts. Arch Dermatol. 1977;113:500-503.
- Alfaro-Castellón P, Mejía-Rodríguez SA, Valencia-Herrera A, et al. Dermoscopy distinction of eruptive vellus hair cysts with molluscum contagiosum and acne lesions. Pediatr Dermatol. 2012;29:772-773.
- Torchia D, Vega J, Schachner LA. Eruptive vellus hair cysts: a systematic review. Am J Clin Dermatol. 2012;13:19-28.
- Pauline G, Alain H, Jean-Jaques R, et al. Eruptive vellus hair cysts: an original case occurring in twins [published online July 11, 2014]. Int J Dermatol. 2015;54:E209-E212.
- Erkek E, Kurtipek GS, Duman D, et al. Eruptive vellus hair cysts: report of a pediatric case with partial response to calcipotriene therapy. Cutis. 2009;84:295-298.
- Shi G, Zhou Y, Cai YX, et al. Clinicopathological features and expression of four keratins (K10, K14, K17 and K19) in six cases of eruptive vellus hair cysts. Clin Exp Dermatol. 2014;39:496-499.
- Panchaprateep R, Tanus A, Tosti A. Clinical, dermoscopic, and histopathologic features of body hair disorders. J Am Acad Dermatol. 2015;72:890-900.
- Takada S, Togawa Y, Wakabayashii S, et al. Dermoscopic findings in eruptive vellus hair cysts: a case report. Austin J Dermatol. 2014;1:1004.
- Khatu S, Vasani R, Amin S. Eruptive vellus hair cyst presenting as asymptomatic follicular papules on extremities. Indian Dermatol Online J. 2013;4:213-215.
- Urbina-Gonzalez F, Aguilar-Martinez A, Cristobal-Gil M, et al. The treatment of eruptive vellus hair cysts with isotretinoin. Br J Dermatol. 1987;116:465-466.
Case Report
Four-year-old identical triplet girls with numerous asymptomatic scattered papules on the chest of 4 months’ duration were referred to a dermatologist by their pediatrician for molluscum contagiosum. The patients’ father reported that there was no history of trauma, irritation, or manipulation to the affected area. Their medical history was notable for prematurity at 32 weeks’ gestation and congenital dermal melanocytosis. Family history was notable for their father having acne and similar papules on the chest during adolescence that resolved with isotretinoin therapy.
On physical examination there were multiple smooth, hyperpigmented to erythematous, comedonal, 1- to 2-mm papules dispersed on the anterior central chest of all 3 patients (Figure 1). Clinically, these lesions were fairly indistinguishable from other common dermatologic conditions such as acne or milia. Dermoscopic examination revealed homogenous yellow-white areas surrounded by light brown to erythematous halos (Figure 2). Histopathologic examination was not performed given the benign clinical diagnosis and avoidance of biopsy in pediatric populations. Based on dermoscopic features and history, a diagnosis of eruptive vellus hair cysts (EVHCs) in identical triplets was made.
Comment
Pathogenesis
Eruptive vellus hair cysts, first introduced by Esterly et al1 in 1977, are uncommon benign lesions presumed to be caused by an abnormal development of the infundibular portion of the hair follicle.2 They are usually 1- to 3-mm, reddish brown, monomorphous papules overlapping with pilosebaceous and apocrine units.3 Although the lesions typically are located on the chest and extremities, they may occur on the face, abdomen, axillae, buttocks, or genital area.1,3 The inheritance of EVHCs is unclear. The majority of reported cases are sporadic; however, the literature mentions 19 families affected by autosomal-dominant EVHCs based on phylogeny.3 In 2015, EVHCs were reported in identical twins, further supporting the case for a genetic mutation.4 We augment this autosomal-dominant inheritance pattern by presenting a case of identical triplets with EVHCs. The patients’ father reported similar lesions in childhood, further underscoring a genetic basis.
The pathogenesis of EVHC is uncertain, with 2 main theories. Some propose retention of vellus hair and keratin in a cavity formed by an abnormal vellus hair follicle causing infundibular occlusion. Others consider the growth of benign follicular hamartomas that differentiate to become vellus hairs.1
Clinical Presentation
The sporadic form of EVHCs is noted to be more common and clinically presents later, with an average age at onset of 16 years and an average age at diagnosis of 24 years.3 The sporadic form occurs without trauma or manipulation as a precursor. Less commonly, lesions present at birth or in early infancy and may show an autosomal-dominant inheritance pattern with a similar distribution across relatives.3
Other variants of EVHCs have been described. Late-onset EVHC usually occurs at 35 years or older (average age, 57 years), with a female to male predominance of 2.5 to 1.3 This late onset may be attributed to proliferation of ductal follicular keratinocytes or loss of perifollicular elastic fibers exacerbated by exogenous factors such as manipulation, UV rays, or trauma.5
For unilesional EVHC, the average age at diagnosis is 27 years.3 Some of these lesions may be pedunculated and greater than 8 mm. There is a female to male predominance of 2 to 1. Eruptive vellus hair cysts with steatocystoma multiplex can be seen with an average age at onset of 19 years and a female to male predominance of 0.2 to 1. There may be a family history of this subset, as reported in 3 patients with this pattern.3
Diagnosis
The recommended workup for EVHCs varies by patient and age. Eruptive vellus hair cysts present an opportunity to utilize noninvasive diagnostic procedures, especially for the pediatric population, to avoid scarring and pain from manipulation or biopsies. Although many practitioners may comfortably diagnose EVHCs clinically, 6 cases were misdiagnosed as steatocystoma multiplex, keratosis pilaris, or milia prior to histopathology revealing vellus hair cysts.6
Dermoscopy presents as a useful diagnostic aid. Eruptive vellus hair cysts exhibit light yellow homogenous circular structures with a maroon or erythematous halo.2,7 A central gray-blue color point may be seen due to melanin in the pigmented hair shaft.7 A dermoscopy review of EVHCs reported radiating capillaries.2 Occasionally, nonfollicular homogenous blue pigmentation may be seen due to a connection to atrophic hair follicles in the mid dermis and no normal hair follicle around the cysts.8 In comparison, dermoscopic characteristics of molluscum contagiosum demonstrated a polylobular, white-yellow, amorphous structure at the center with a hardened central umbilicated core and a crown of hairpin vessels at the periphery. Additionally, comedonal acne, commonly mistaken for EVHCs, reveals a brown-yellow hard central plug with sparse inflammation under dermoscopy.2 Thus, differentiation of these entities with dermoscopy should be highly prioritized to better aid in the diagnosis of pediatric dermatologic conditions using painless noninvasive techniques.
Treatment
The main indication for treatment of EVHCs is cosmetic concern. Twenty-five percent of EVHCs spontaneously resolve with transepidermal hair elimination or a granulomatous reaction.4,5 A case report of 4 siblings with congenital EVHCs also described a mother with similar lesions that resolved spontaneously in early adulthood,3 as our patients’ father also noted. Treatment modalities including topical keratolytic agents such as urea 10%, retinoic acid 0.05%, tazarotene cream 0.1%, and lactic acid 12%; incision and drainage; CO2 laser; or erbium-doped YAG laser ablation have been tried with minimal improvement.9 Of note, tazarotene cream 0.1% has demonstrated better results than both erbium-doped YAG laser and drainage and incision of EVHCs.4 Additionally, another report evidenced partial improvement with calcipotriene within 2 months with some lesions completely resolved and others flattened, which may be attributed to the antiproliferative and prodifferentiating effects on the ductal follicular keratinocytes by calcipotriene.5 Lastly, an additional study indicated that isotretinoin and vitamin A derivatives were ineffective for clearing EVHCs.10
Conclusion
We presented 3 identical triplets with the classic pediatric onset and dermoscopic findings of EVHCs on the trunk. Although the definitive diagnosis of EVHCs relies on histopathology, we argue that their unique dermoscopic findings combined with a thorough clinical examination is sufficient to recognize this benign condition and avoid painful procedures in the pediatric population.
Case Report
Four-year-old identical triplet girls with numerous asymptomatic scattered papules on the chest of 4 months’ duration were referred to a dermatologist by their pediatrician for molluscum contagiosum. The patients’ father reported that there was no history of trauma, irritation, or manipulation to the affected area. Their medical history was notable for prematurity at 32 weeks’ gestation and congenital dermal melanocytosis. Family history was notable for their father having acne and similar papules on the chest during adolescence that resolved with isotretinoin therapy.
On physical examination there were multiple smooth, hyperpigmented to erythematous, comedonal, 1- to 2-mm papules dispersed on the anterior central chest of all 3 patients (Figure 1). Clinically, these lesions were fairly indistinguishable from other common dermatologic conditions such as acne or milia. Dermoscopic examination revealed homogenous yellow-white areas surrounded by light brown to erythematous halos (Figure 2). Histopathologic examination was not performed given the benign clinical diagnosis and avoidance of biopsy in pediatric populations. Based on dermoscopic features and history, a diagnosis of eruptive vellus hair cysts (EVHCs) in identical triplets was made.
Comment
Pathogenesis
Eruptive vellus hair cysts, first introduced by Esterly et al1 in 1977, are uncommon benign lesions presumed to be caused by an abnormal development of the infundibular portion of the hair follicle.2 They are usually 1- to 3-mm, reddish brown, monomorphous papules overlapping with pilosebaceous and apocrine units.3 Although the lesions typically are located on the chest and extremities, they may occur on the face, abdomen, axillae, buttocks, or genital area.1,3 The inheritance of EVHCs is unclear. The majority of reported cases are sporadic; however, the literature mentions 19 families affected by autosomal-dominant EVHCs based on phylogeny.3 In 2015, EVHCs were reported in identical twins, further supporting the case for a genetic mutation.4 We augment this autosomal-dominant inheritance pattern by presenting a case of identical triplets with EVHCs. The patients’ father reported similar lesions in childhood, further underscoring a genetic basis.
The pathogenesis of EVHC is uncertain, with 2 main theories. Some propose retention of vellus hair and keratin in a cavity formed by an abnormal vellus hair follicle causing infundibular occlusion. Others consider the growth of benign follicular hamartomas that differentiate to become vellus hairs.1
Clinical Presentation
The sporadic form of EVHCs is noted to be more common and clinically presents later, with an average age at onset of 16 years and an average age at diagnosis of 24 years.3 The sporadic form occurs without trauma or manipulation as a precursor. Less commonly, lesions present at birth or in early infancy and may show an autosomal-dominant inheritance pattern with a similar distribution across relatives.3
Other variants of EVHCs have been described. Late-onset EVHC usually occurs at 35 years or older (average age, 57 years), with a female to male predominance of 2.5 to 1.3 This late onset may be attributed to proliferation of ductal follicular keratinocytes or loss of perifollicular elastic fibers exacerbated by exogenous factors such as manipulation, UV rays, or trauma.5
For unilesional EVHC, the average age at diagnosis is 27 years.3 Some of these lesions may be pedunculated and greater than 8 mm. There is a female to male predominance of 2 to 1. Eruptive vellus hair cysts with steatocystoma multiplex can be seen with an average age at onset of 19 years and a female to male predominance of 0.2 to 1. There may be a family history of this subset, as reported in 3 patients with this pattern.3
Diagnosis
The recommended workup for EVHCs varies by patient and age. Eruptive vellus hair cysts present an opportunity to utilize noninvasive diagnostic procedures, especially for the pediatric population, to avoid scarring and pain from manipulation or biopsies. Although many practitioners may comfortably diagnose EVHCs clinically, 6 cases were misdiagnosed as steatocystoma multiplex, keratosis pilaris, or milia prior to histopathology revealing vellus hair cysts.6
Dermoscopy presents as a useful diagnostic aid. Eruptive vellus hair cysts exhibit light yellow homogenous circular structures with a maroon or erythematous halo.2,7 A central gray-blue color point may be seen due to melanin in the pigmented hair shaft.7 A dermoscopy review of EVHCs reported radiating capillaries.2 Occasionally, nonfollicular homogenous blue pigmentation may be seen due to a connection to atrophic hair follicles in the mid dermis and no normal hair follicle around the cysts.8 In comparison, dermoscopic characteristics of molluscum contagiosum demonstrated a polylobular, white-yellow, amorphous structure at the center with a hardened central umbilicated core and a crown of hairpin vessels at the periphery. Additionally, comedonal acne, commonly mistaken for EVHCs, reveals a brown-yellow hard central plug with sparse inflammation under dermoscopy.2 Thus, differentiation of these entities with dermoscopy should be highly prioritized to better aid in the diagnosis of pediatric dermatologic conditions using painless noninvasive techniques.
Treatment
The main indication for treatment of EVHCs is cosmetic concern. Twenty-five percent of EVHCs spontaneously resolve with transepidermal hair elimination or a granulomatous reaction.4,5 A case report of 4 siblings with congenital EVHCs also described a mother with similar lesions that resolved spontaneously in early adulthood,3 as our patients’ father also noted. Treatment modalities including topical keratolytic agents such as urea 10%, retinoic acid 0.05%, tazarotene cream 0.1%, and lactic acid 12%; incision and drainage; CO2 laser; or erbium-doped YAG laser ablation have been tried with minimal improvement.9 Of note, tazarotene cream 0.1% has demonstrated better results than both erbium-doped YAG laser and drainage and incision of EVHCs.4 Additionally, another report evidenced partial improvement with calcipotriene within 2 months with some lesions completely resolved and others flattened, which may be attributed to the antiproliferative and prodifferentiating effects on the ductal follicular keratinocytes by calcipotriene.5 Lastly, an additional study indicated that isotretinoin and vitamin A derivatives were ineffective for clearing EVHCs.10
Conclusion
We presented 3 identical triplets with the classic pediatric onset and dermoscopic findings of EVHCs on the trunk. Although the definitive diagnosis of EVHCs relies on histopathology, we argue that their unique dermoscopic findings combined with a thorough clinical examination is sufficient to recognize this benign condition and avoid painful procedures in the pediatric population.
- Esterly NB, Fretzin DF, Pinkus H. Eruptive vellus hair cysts. Arch Dermatol. 1977;113:500-503.
- Alfaro-Castellón P, Mejía-Rodríguez SA, Valencia-Herrera A, et al. Dermoscopy distinction of eruptive vellus hair cysts with molluscum contagiosum and acne lesions. Pediatr Dermatol. 2012;29:772-773.
- Torchia D, Vega J, Schachner LA. Eruptive vellus hair cysts: a systematic review. Am J Clin Dermatol. 2012;13:19-28.
- Pauline G, Alain H, Jean-Jaques R, et al. Eruptive vellus hair cysts: an original case occurring in twins [published online July 11, 2014]. Int J Dermatol. 2015;54:E209-E212.
- Erkek E, Kurtipek GS, Duman D, et al. Eruptive vellus hair cysts: report of a pediatric case with partial response to calcipotriene therapy. Cutis. 2009;84:295-298.
- Shi G, Zhou Y, Cai YX, et al. Clinicopathological features and expression of four keratins (K10, K14, K17 and K19) in six cases of eruptive vellus hair cysts. Clin Exp Dermatol. 2014;39:496-499.
- Panchaprateep R, Tanus A, Tosti A. Clinical, dermoscopic, and histopathologic features of body hair disorders. J Am Acad Dermatol. 2015;72:890-900.
- Takada S, Togawa Y, Wakabayashii S, et al. Dermoscopic findings in eruptive vellus hair cysts: a case report. Austin J Dermatol. 2014;1:1004.
- Khatu S, Vasani R, Amin S. Eruptive vellus hair cyst presenting as asymptomatic follicular papules on extremities. Indian Dermatol Online J. 2013;4:213-215.
- Urbina-Gonzalez F, Aguilar-Martinez A, Cristobal-Gil M, et al. The treatment of eruptive vellus hair cysts with isotretinoin. Br J Dermatol. 1987;116:465-466.
- Esterly NB, Fretzin DF, Pinkus H. Eruptive vellus hair cysts. Arch Dermatol. 1977;113:500-503.
- Alfaro-Castellón P, Mejía-Rodríguez SA, Valencia-Herrera A, et al. Dermoscopy distinction of eruptive vellus hair cysts with molluscum contagiosum and acne lesions. Pediatr Dermatol. 2012;29:772-773.
- Torchia D, Vega J, Schachner LA. Eruptive vellus hair cysts: a systematic review. Am J Clin Dermatol. 2012;13:19-28.
- Pauline G, Alain H, Jean-Jaques R, et al. Eruptive vellus hair cysts: an original case occurring in twins [published online July 11, 2014]. Int J Dermatol. 2015;54:E209-E212.
- Erkek E, Kurtipek GS, Duman D, et al. Eruptive vellus hair cysts: report of a pediatric case with partial response to calcipotriene therapy. Cutis. 2009;84:295-298.
- Shi G, Zhou Y, Cai YX, et al. Clinicopathological features and expression of four keratins (K10, K14, K17 and K19) in six cases of eruptive vellus hair cysts. Clin Exp Dermatol. 2014;39:496-499.
- Panchaprateep R, Tanus A, Tosti A. Clinical, dermoscopic, and histopathologic features of body hair disorders. J Am Acad Dermatol. 2015;72:890-900.
- Takada S, Togawa Y, Wakabayashii S, et al. Dermoscopic findings in eruptive vellus hair cysts: a case report. Austin J Dermatol. 2014;1:1004.
- Khatu S, Vasani R, Amin S. Eruptive vellus hair cyst presenting as asymptomatic follicular papules on extremities. Indian Dermatol Online J. 2013;4:213-215.
- Urbina-Gonzalez F, Aguilar-Martinez A, Cristobal-Gil M, et al. The treatment of eruptive vellus hair cysts with isotretinoin. Br J Dermatol. 1987;116:465-466.
Practice Points
- Eruptive vellus hair cysts (EVHCs) are 1- to 3-mm round, dome-shaped, flesh-colored, asymptomatic, benign papules typically occurring on the chest and extremities.
- Pathogenesis and inheritance are unclear. Although the majority of EVHC cases are sporadic, the strong influence of genes is indicated by numerous reports of families in whom 2 or more members were affected.
- Dermoscopy is a noninvasive diagnostic procedure that should be utilized to diagnose EVHCs in the pediatric population; specifically, EVHCs exhibit light yellow, homogenous, circular structures with a maroon or erythematous halo.
- The main indication for treatment of EVHCs is cosmetic concern; however, one-quarter of cases may resolve spontaneously.
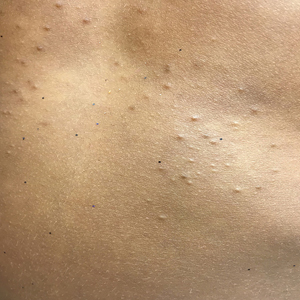
Pediatric Primary Cutaneous Blastomycosis Clinically Responsive to Itraconazole
Blastomycosis is a polymorphic disease caused by the thermally dimorphic fungus Blastomyces dermatitidis, which is naturally occurring worldwide but particularly prominent in the Great Lakes, Mississippi, and Ohio River areas of the United States. The disease was first described by Thomas Caspar Gilchrist in 1894 and historically has been referred to as Gilchrist disease, North American blastomycosis, or Chicago disease.1,2 Cutaneous blastomycosis can occur by dissemination of yeast to the skin from systemic and pulmonary disease or rarely via direct inoculation of the skin resulting in primary cutaneous disease. Clinically, the lesions are polymorphic and may appear as well-demarcated verrucous plaques containing foci of pustules or ulcerations. Lesions typically heal centrifugally with a cribriform scar.3
We describe an adolescent with a unique history of inoculation 2 weeks prior to the development of a biopsy-confirmed lesion of cutaneous blastomycosis on the left chest wall that clinically resolved following 6 months of itraconazole.
Case Report
A 16-year-old adolescent boy with a history of morbid obesity, asthma, and seasonal allergies presented for evaluation of a painful, slowly enlarging skin lesion on the left chest wall of 2 months’ duration. According to the patient, a “small pimple” appeared at the site of impact 2 weeks following a fall into a muddy flowerbed in Madison, Wisconsin. The patient recalled that although he had soiled his clothing, there was no identifiable puncture of the skin. Despite daily application of hydrogen peroxide and a 1-week course of trimethoprim-sulfamethoxazole, the lesion gradually enlarged. Complete review of systems as well as exposure and travel history were otherwise negative.
Physical examination revealed a 5.0×2.5-cm exophytic, firm, well-circumscribed plaque with a papillated crusted surface on the left side of the chest near the posterior axillary line (Figure 1). There was no palpable regional lymphadenopathy. Pulmonary examination was unremarkable. Diagnostic workup, including complete blood cell count with differential, hemoglobin A1c, human immunodeficiency virus antibody/antigen testing, interferon-gamma release assay, and chest radiograph were all within normal limits.
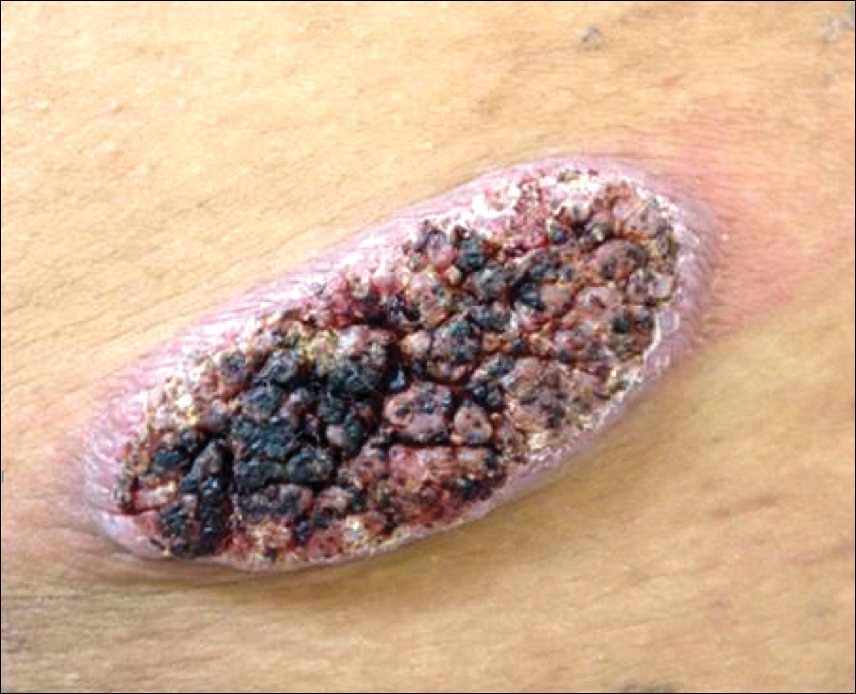
Histologic examination of a biopsy specimen showed pseudoepitheliomatous hyperplasia of the epidermis with a brisk mixed inflammatory infiltrate (Figure 2). Displayed in Figure 3 is the Grocott-Gomori methenamine-silver stain that highlighted the thick double-contoured wall-budding yeasts.
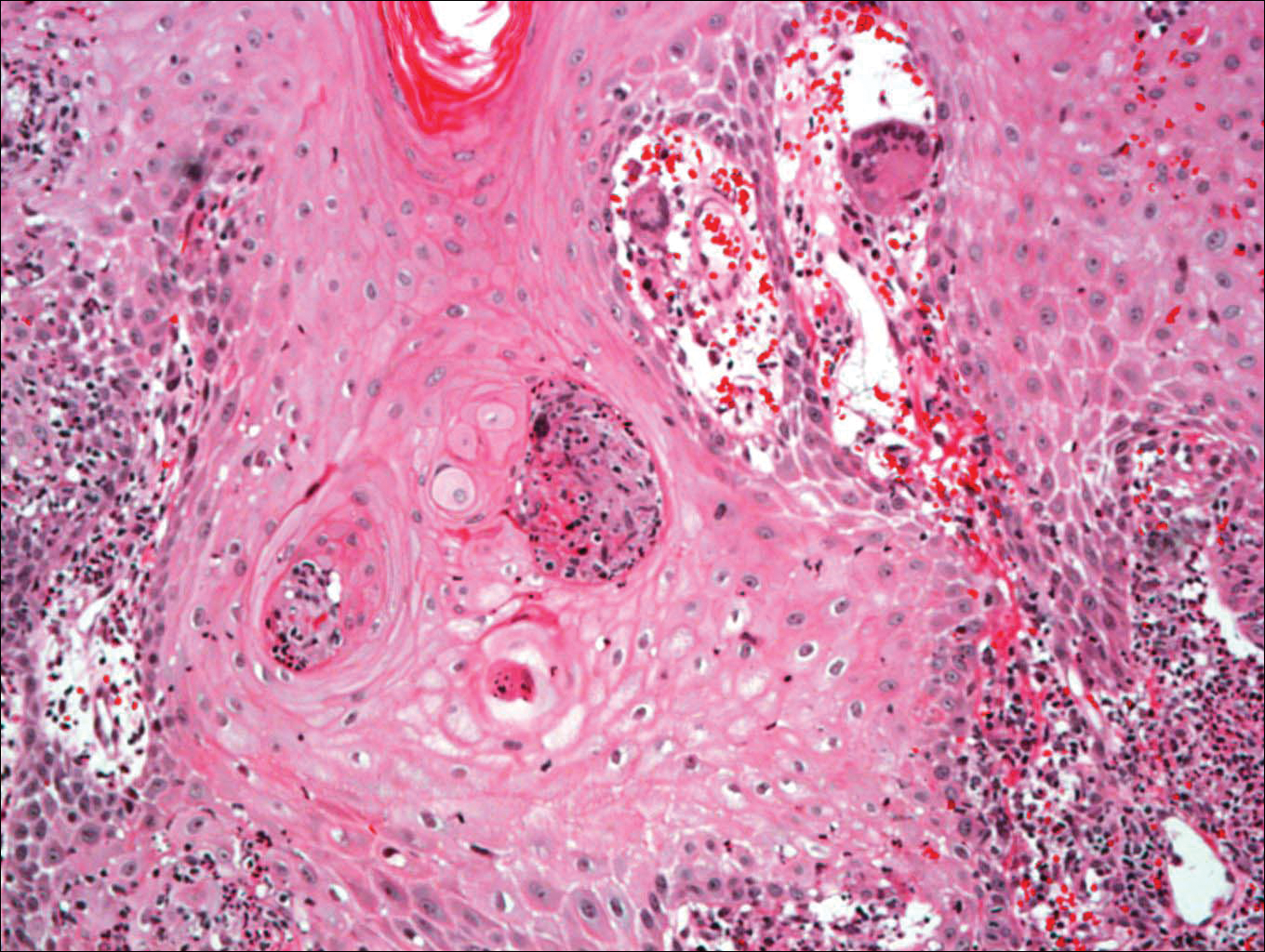
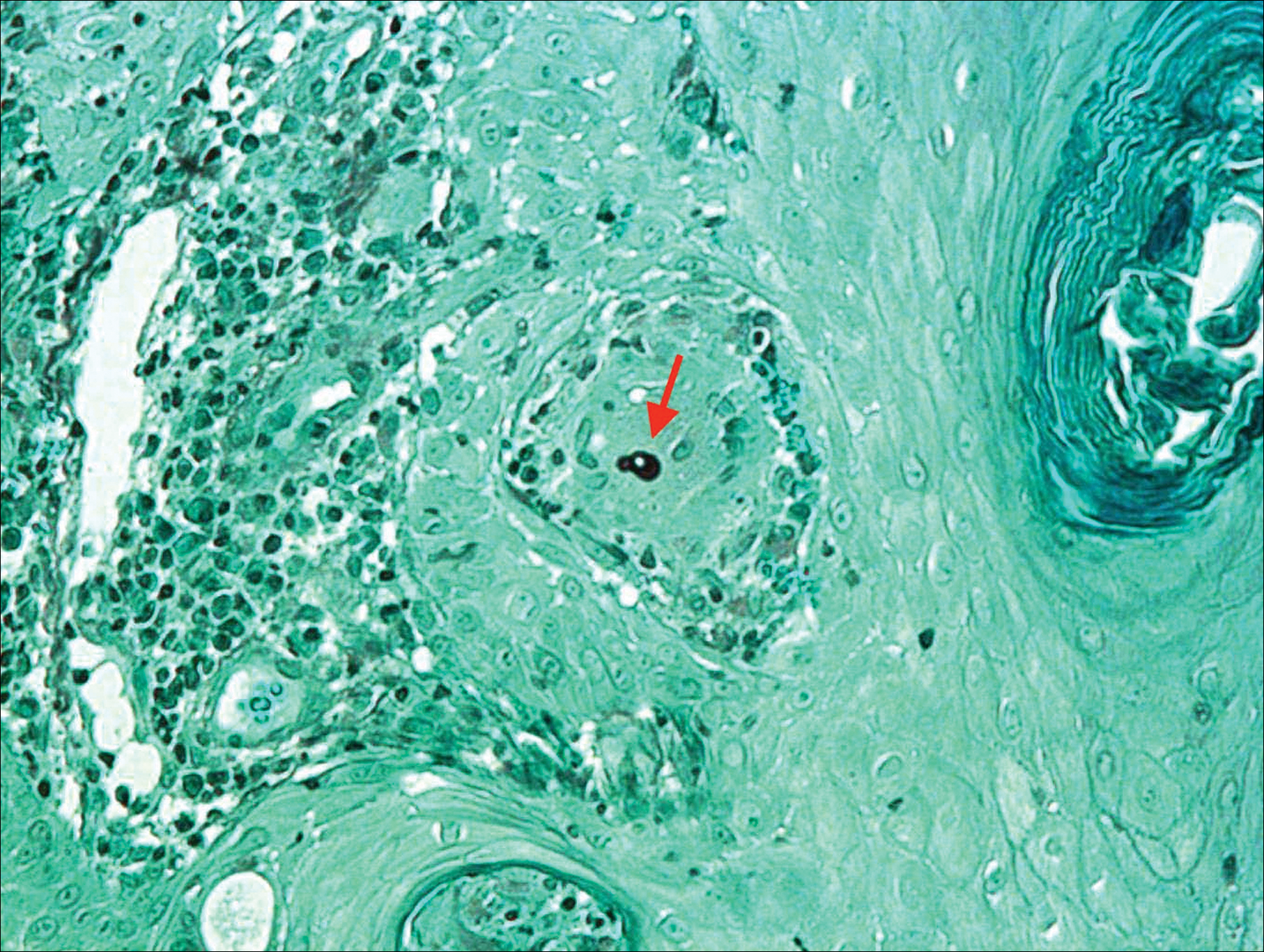
The patient was diagnosed with primary cutaneous blastomycosis. Treatment was initiated with itraconazole 200 mg 3 times daily for 3 days, followed by 200 mg 2 times daily for 6 months. Following 3 months of therapy, the lesion had markedly improved with violaceous dyschromia and no residual surface changes. After 5 months of itraconazole, the patient stopped taking the medication for 2 months due to pharmacy issues and then resumed. After 6 total months of therapy, the lesion healed with only residual dyschromia and itraconazole was discontinued.
Comment
Epidemiology
Blastomycosis is a polymorphic pyogranulomatous disease caused by the dimorphic fungus B dermatitidis, naturally occurring in the soil with a worldwide distribution.4 Individuals affected by the disease often reside in locations where the fungus is endemic, specifically in areas that border the Mississippi and Ohio rivers, the Great Lakes, and Canadian provinces near the Saint Lawrence Seaway. More recently there has been an increased incidence of blastomycosis, with the highest proportion found in Wisconsin and Michigan.1,2 Exposures often are associated with recreational and occupational activities near streams or rivers where there may be decaying vegetation.1 Despite the ubiquitous presence of B dermatitidis in regions where the species is endemic, it is likely that many individuals who are exposed to the organism do not develop infection.
Pathogenesis
The exact pathogenesis for the development of disease in a particular individual remains unclear. Immunosuppression is not a prerequisite for susceptibility, as evidenced by a review of 123 cases of blastomycosis in which a preceding immunodepressive disorder was present in only 25% of patients. The same study found that it was almost equally common as diabetes mellitus and present in 22% of patients.5 The organism is considered a true pathogen given its ability to affect healthy individuals and the presence of a newly identified novel 120-kD glycoprotein antigen (WI-1) on the cell wall that may confer virulence via extracellular matrix and macrophage binding. Intact cell-mediated immunity that prevents the conversion of conidia (the infectious agent) to yeast (the form that exists at body temperature) plays a key role in conferring natural resistance.6,7
Cutaneous infection may occur by either dissemination of yeast to the skin from systemic disease or less commonly via direct inoculation of the skin, resulting in primary cutaneous disease. With respect to systemic disease, infection occurs through inhalation of conidia from moist soil containing organic debris, with an incubation period of 4 to 6 weeks. In the lungs, in a process largely dependent on host cell-mediated immunity, the mold quickly converts to yeast and may then either multiply or be phagocytized.2,6,7 Transmission does not occur from person to person.7 Asymptomatic infection may occur in at least 50% of patients, often leading to a delay in diagnosis. Symptomatic pulmonary disease may range from mild flulike symptoms to overt pneumonia, clinically indistinguishable from community-acquired bacterial pneumonia, tuberculosis, other fungal infections, and cancer. Of patients with primary pulmonary disease, 25% to 80% have been reported to develop secondary organ involvement via lymphohematogenous spread most commonly to the skin, followed respectively by the skeletal, genitourinary, and central nervous systems. Currently, there are 54 documented cases of secondary disseminated cutaneous blastomycosis in children reported in the literature.3,8-14
Presentation
Primary cutaneous disease resulting from direct cutaneous inoculation is rare, especially among children.14 Of 28 cases of isolated cutaneous blastomycosis reported in the literature, 12 (42%) were pediatric.3,8-21 Inoculation blastomycosis typically presents as a papule that expands to a well-demarcated verrucous plaque, often up to several centimeters in diameter, and is located on the skin at the site of contact. The lesion may exhibit a myriad of features ranging from pustules or nodules to focal ulcerations, either present centrally or within raised borders that ultimately may communicate via sinus tracking.7 Lesions that are purely pustular in morphology also have been reported. Healing typically begins centrally and expands centrifugally, often with cribriform scarring.2,4,22 Histologic features of primary and secondary blastomycosis include pseudoepitheliomatous hyperplasia, intraepidermal microabscesses, and dermal suppurative granulomatous inflammation.4 Classically, broad-based budding yeast are identified with a doubly refractile cell wall that is best visualized on periodic acid–Schiff staining.2
Diagnosis
In approximately 50% of patients with cutaneous blastomycosis resulting from secondary spread, there may be an absence of clinically active pulmonary disease, posing a diagnostic dilemma when differentiating from primary cutaneous disease.1,2,4 Furthermore, the skin findings exhibited in primary and secondary cutaneous blastomycosis cannot be distinguished by clinical inspection.19 To fulfill the criteria for diagnosis of primary cutaneous blastomycosis, there must be an identifiable source of infection from the environment, a lesion at the site of contact, a proven absence of systemic infection, and visualization and/or isolation of fungus from the lesion.4,12 The incubation period of lesions is shorter in primary cutaneous disease (2 weeks) and may aid in its differentiation from secondary disease, which typically is longer with lesions presenting 4 to 6 weeks following initial exposure.4
Treatment
Under the current 2015 guidelines from the American Academy of Pediatrics Committee on Infectious Diseases, 6 to 12 months of itraconazole is the treatment recommendation for mild to moderate pulmonary systemic disease without central nervous system involvement.7 Central nervous system disease and moderate to severe pulmonary and systemic disease are treated with intravenous amphotericin B followed by 12 months of oral itraconazole.1,7 Primary cutaneous disease, unlike secondary disease, may self-resolve; however, primary cutaneous disease usually is treated with 6 months of itraconazole, though successful therapy with surgical excision, radiation therapy, and incision and drainage have been reported.19
Unlike secondary cutaneous blastomycosis, primary inoculation disease may be self-limited; however, as treatment with antifungal therapy has become the standard of care, the disease’s propensity to self-resolve has not been well studied.4 Oral itraconazole for 6 to 12 months is the treatment of choice for mild to moderate cutaneous disease.1,22 Effective treatment duration may be difficult to definitively assess because of the self-limited nature of the disease. Our patient showed marked improvement after 3 months and resolution of the skin lesion following 6 months of itraconazole therapy. Our findings support the previously documented observation that systemic therapy might potentially be needed only for the time required to eliminate the clinical evidence of cutaneous disease.19 Our patient received the full 6 months of treatment according to current guidelines. Among a review of 22 cases of primary inoculation blastomycosis, the 5 patients who were treated with an azole agent alone showed disease clearance with an average treatment course of 3.2 months, ranging from 1 to 6 months.19 Further studies that assess the time to clearance with antifungal therapy and subsequent recurrence rates may be warranted.
Conclusion
Pediatric primary cutaneous blastomycosis is a rare cutaneous disease. Identifying sources of probable inoculation from the environment for this patient was unique in that the patient fell into a muddy puddle within a flowerbed. Given the patient’s atopic history, a predominance of humoral over cell-mediated immunity may have placed him at risk. He responded well to 6 months of oral itraconazole and there was no ulceration or scar formation. An increased awareness of this infection, particularly in geographic areas where its reported incidence is on the rise, could be helpful in reducing delays in diagnosis and treatment.
Acknowledgments
We thank Wenhua Liu, MD (Libertyville, Illinois), for reviewing the pathology and Pravin Muniyappa, MD (Chicago, Illinois), for referring the case.
- Chapman SW, Dismukes WE, Proia LA, et al. Clinical practice guidelines for the management of blastomycosis: 2008 update by the Infectious Diseases Society of America. Clin Infect Dis. 2008;46:1801-1812.
- Smith JA, Riddell Jt, Kauffman CA. Cutaneous manifestations of endemic mycoses. Curr Infect Dis Rep. 2013;15:440-449.
- Fisher KR, Baselski V, Beard G, et al. Pustular blastomycosis. J Am Acad Dermatol. 2009;6:355-358.
- Mason AR, Cortes GY, Cook J, et al. Cutaneous blastomycosis: a diagnostic challenge. Int J Dermatol. 2008;47:824-830.
- Lemos LB, Baliga M, Guo M. Blastomycosis: the great pretender can also be an opportunist. initial clinical diagnosis and underlying diseases in 123 patients. Ann Diagn Pathol. 2002;6:194-203.
- Bradsher RW, Chapman SW, Pappas PG. Blastomycosis. Infect Dis Clin North Am. 2003;17:21-40, vii.
- Blastomycosis. In: Kimberlin DW, ed. Red Book: 2015 Report of the Committee on Infectious Diseases. 30th ed. Elk Grove Village, IL: American Academy of Pediatrics; 2015:263-264.
- Brick KE, Drolet BA, Lyon VB, et al. Cutaneous and disseminated blastomycosis: a pediatric case series. Pediatr Dermatol. 2013;30:23-28.
- Fanella S, Skinner S, Trepman E, et al. Blastomycosis in children and adolescents: a 30-year experience from Manitoba. Med Mycol. 2011;49:627-632.
- Frost HM, Anderson J, Ivacic L, et al. Blastomycosis in children: an analysis of clinical, epidemiologic, and genetic features. J Pediatr Infect Dis Soc. 2017;6:49-56.
- Shukla S, Singh S, Jain M, et al. Paediatric cutaneous blastomycosis: a rare case diagnosed on FNAC. Diagn Cytopathol. 2009;37:119-121.
- Smith RJ, Boos MD, Burnham JM, et al. Atypical cutaneous blastomycosis in a child with juvenile idiopathic arthritis on infliximab. Pediatrics. 2015;136:E1386-E1389.
- Wilson JW, Cawley EP, Weidman FD, et al. Primary cutaneous North American blastomycosis. AMA Arch Derm. 1955;71:39-45.
- Zampogna JC, Hoy MJ, Ramos-Caro FA. Primary cutaneous north american blastomycosis in an immunosuppressed child. Pediatr Dermatol. 2003;20:128-130.
- Balasaraswathy P, Theerthanath. Cutaneous blastomycosis presenting as non-healing ulcer and responding to oral ketoconazole. Dermatol Online J. 2003;9:19.
- Bonifaz A, Morales D, Morales N, et al. Cutaneous blastomycosis. an imported case with good response to itraconazole. Rev Iberoam Micol. 2016;33:51-54.
- Clinton TS, Timko AL. Cutaneous blastomycosis without evidence of pulmonary involvement. Mil Med. 2003;168:651-653.
- Dhamija A, D’Souza P, Salgia P, et al. Blastomycosis presenting as solitary nodule: a rare presentation. Indian J Dermatol. 2012;57:133-135.
- Gray NA, Baddour LM. Cutaneous inoculation blastomycosis. Clin Infect Dis. 2002;34:E44-E49.
- Motswaledi HM, Monyemangene FM, Maloba BR, et al. Blastomycosis: a case report and review of the literature. Int J Dermatol. 2012;51:1090-1093.
- Rodríguez-Mena A, Mayorga J, Solís-Ledesma G, et al. Blastomycosis: report of an imported case in Mexico, with only cutaneous lesions [in Spanish]. Rev Iberoam Micol. 2010;27:210-212.
- Saccente M, Woods GL. Clinical and laboratory update on blastomycosis. Clin Microbiol Rev. 2010;23:367-381.
Blastomycosis is a polymorphic disease caused by the thermally dimorphic fungus Blastomyces dermatitidis, which is naturally occurring worldwide but particularly prominent in the Great Lakes, Mississippi, and Ohio River areas of the United States. The disease was first described by Thomas Caspar Gilchrist in 1894 and historically has been referred to as Gilchrist disease, North American blastomycosis, or Chicago disease.1,2 Cutaneous blastomycosis can occur by dissemination of yeast to the skin from systemic and pulmonary disease or rarely via direct inoculation of the skin resulting in primary cutaneous disease. Clinically, the lesions are polymorphic and may appear as well-demarcated verrucous plaques containing foci of pustules or ulcerations. Lesions typically heal centrifugally with a cribriform scar.3
We describe an adolescent with a unique history of inoculation 2 weeks prior to the development of a biopsy-confirmed lesion of cutaneous blastomycosis on the left chest wall that clinically resolved following 6 months of itraconazole.
Case Report
A 16-year-old adolescent boy with a history of morbid obesity, asthma, and seasonal allergies presented for evaluation of a painful, slowly enlarging skin lesion on the left chest wall of 2 months’ duration. According to the patient, a “small pimple” appeared at the site of impact 2 weeks following a fall into a muddy flowerbed in Madison, Wisconsin. The patient recalled that although he had soiled his clothing, there was no identifiable puncture of the skin. Despite daily application of hydrogen peroxide and a 1-week course of trimethoprim-sulfamethoxazole, the lesion gradually enlarged. Complete review of systems as well as exposure and travel history were otherwise negative.
Physical examination revealed a 5.0×2.5-cm exophytic, firm, well-circumscribed plaque with a papillated crusted surface on the left side of the chest near the posterior axillary line (Figure 1). There was no palpable regional lymphadenopathy. Pulmonary examination was unremarkable. Diagnostic workup, including complete blood cell count with differential, hemoglobin A1c, human immunodeficiency virus antibody/antigen testing, interferon-gamma release assay, and chest radiograph were all within normal limits.
Histologic examination of a biopsy specimen showed pseudoepitheliomatous hyperplasia of the epidermis with a brisk mixed inflammatory infiltrate (Figure 2). Displayed in Figure 3 is the Grocott-Gomori methenamine-silver stain that highlighted the thick double-contoured wall-budding yeasts.
The patient was diagnosed with primary cutaneous blastomycosis. Treatment was initiated with itraconazole 200 mg 3 times daily for 3 days, followed by 200 mg 2 times daily for 6 months. Following 3 months of therapy, the lesion had markedly improved with violaceous dyschromia and no residual surface changes. After 5 months of itraconazole, the patient stopped taking the medication for 2 months due to pharmacy issues and then resumed. After 6 total months of therapy, the lesion healed with only residual dyschromia and itraconazole was discontinued.
Comment
Epidemiology
Blastomycosis is a polymorphic pyogranulomatous disease caused by the dimorphic fungus B dermatitidis, naturally occurring in the soil with a worldwide distribution.4 Individuals affected by the disease often reside in locations where the fungus is endemic, specifically in areas that border the Mississippi and Ohio rivers, the Great Lakes, and Canadian provinces near the Saint Lawrence Seaway. More recently there has been an increased incidence of blastomycosis, with the highest proportion found in Wisconsin and Michigan.1,2 Exposures often are associated with recreational and occupational activities near streams or rivers where there may be decaying vegetation.1 Despite the ubiquitous presence of B dermatitidis in regions where the species is endemic, it is likely that many individuals who are exposed to the organism do not develop infection.
Pathogenesis
The exact pathogenesis for the development of disease in a particular individual remains unclear. Immunosuppression is not a prerequisite for susceptibility, as evidenced by a review of 123 cases of blastomycosis in which a preceding immunodepressive disorder was present in only 25% of patients. The same study found that it was almost equally common as diabetes mellitus and present in 22% of patients.5 The organism is considered a true pathogen given its ability to affect healthy individuals and the presence of a newly identified novel 120-kD glycoprotein antigen (WI-1) on the cell wall that may confer virulence via extracellular matrix and macrophage binding. Intact cell-mediated immunity that prevents the conversion of conidia (the infectious agent) to yeast (the form that exists at body temperature) plays a key role in conferring natural resistance.6,7
Cutaneous infection may occur by either dissemination of yeast to the skin from systemic disease or less commonly via direct inoculation of the skin, resulting in primary cutaneous disease. With respect to systemic disease, infection occurs through inhalation of conidia from moist soil containing organic debris, with an incubation period of 4 to 6 weeks. In the lungs, in a process largely dependent on host cell-mediated immunity, the mold quickly converts to yeast and may then either multiply or be phagocytized.2,6,7 Transmission does not occur from person to person.7 Asymptomatic infection may occur in at least 50% of patients, often leading to a delay in diagnosis. Symptomatic pulmonary disease may range from mild flulike symptoms to overt pneumonia, clinically indistinguishable from community-acquired bacterial pneumonia, tuberculosis, other fungal infections, and cancer. Of patients with primary pulmonary disease, 25% to 80% have been reported to develop secondary organ involvement via lymphohematogenous spread most commonly to the skin, followed respectively by the skeletal, genitourinary, and central nervous systems. Currently, there are 54 documented cases of secondary disseminated cutaneous blastomycosis in children reported in the literature.3,8-14
Presentation
Primary cutaneous disease resulting from direct cutaneous inoculation is rare, especially among children.14 Of 28 cases of isolated cutaneous blastomycosis reported in the literature, 12 (42%) were pediatric.3,8-21 Inoculation blastomycosis typically presents as a papule that expands to a well-demarcated verrucous plaque, often up to several centimeters in diameter, and is located on the skin at the site of contact. The lesion may exhibit a myriad of features ranging from pustules or nodules to focal ulcerations, either present centrally or within raised borders that ultimately may communicate via sinus tracking.7 Lesions that are purely pustular in morphology also have been reported. Healing typically begins centrally and expands centrifugally, often with cribriform scarring.2,4,22 Histologic features of primary and secondary blastomycosis include pseudoepitheliomatous hyperplasia, intraepidermal microabscesses, and dermal suppurative granulomatous inflammation.4 Classically, broad-based budding yeast are identified with a doubly refractile cell wall that is best visualized on periodic acid–Schiff staining.2
Diagnosis
In approximately 50% of patients with cutaneous blastomycosis resulting from secondary spread, there may be an absence of clinically active pulmonary disease, posing a diagnostic dilemma when differentiating from primary cutaneous disease.1,2,4 Furthermore, the skin findings exhibited in primary and secondary cutaneous blastomycosis cannot be distinguished by clinical inspection.19 To fulfill the criteria for diagnosis of primary cutaneous blastomycosis, there must be an identifiable source of infection from the environment, a lesion at the site of contact, a proven absence of systemic infection, and visualization and/or isolation of fungus from the lesion.4,12 The incubation period of lesions is shorter in primary cutaneous disease (2 weeks) and may aid in its differentiation from secondary disease, which typically is longer with lesions presenting 4 to 6 weeks following initial exposure.4
Treatment
Under the current 2015 guidelines from the American Academy of Pediatrics Committee on Infectious Diseases, 6 to 12 months of itraconazole is the treatment recommendation for mild to moderate pulmonary systemic disease without central nervous system involvement.7 Central nervous system disease and moderate to severe pulmonary and systemic disease are treated with intravenous amphotericin B followed by 12 months of oral itraconazole.1,7 Primary cutaneous disease, unlike secondary disease, may self-resolve; however, primary cutaneous disease usually is treated with 6 months of itraconazole, though successful therapy with surgical excision, radiation therapy, and incision and drainage have been reported.19
Unlike secondary cutaneous blastomycosis, primary inoculation disease may be self-limited; however, as treatment with antifungal therapy has become the standard of care, the disease’s propensity to self-resolve has not been well studied.4 Oral itraconazole for 6 to 12 months is the treatment of choice for mild to moderate cutaneous disease.1,22 Effective treatment duration may be difficult to definitively assess because of the self-limited nature of the disease. Our patient showed marked improvement after 3 months and resolution of the skin lesion following 6 months of itraconazole therapy. Our findings support the previously documented observation that systemic therapy might potentially be needed only for the time required to eliminate the clinical evidence of cutaneous disease.19 Our patient received the full 6 months of treatment according to current guidelines. Among a review of 22 cases of primary inoculation blastomycosis, the 5 patients who were treated with an azole agent alone showed disease clearance with an average treatment course of 3.2 months, ranging from 1 to 6 months.19 Further studies that assess the time to clearance with antifungal therapy and subsequent recurrence rates may be warranted.
Conclusion
Pediatric primary cutaneous blastomycosis is a rare cutaneous disease. Identifying sources of probable inoculation from the environment for this patient was unique in that the patient fell into a muddy puddle within a flowerbed. Given the patient’s atopic history, a predominance of humoral over cell-mediated immunity may have placed him at risk. He responded well to 6 months of oral itraconazole and there was no ulceration or scar formation. An increased awareness of this infection, particularly in geographic areas where its reported incidence is on the rise, could be helpful in reducing delays in diagnosis and treatment.
Acknowledgments
We thank Wenhua Liu, MD (Libertyville, Illinois), for reviewing the pathology and Pravin Muniyappa, MD (Chicago, Illinois), for referring the case.
Blastomycosis is a polymorphic disease caused by the thermally dimorphic fungus Blastomyces dermatitidis, which is naturally occurring worldwide but particularly prominent in the Great Lakes, Mississippi, and Ohio River areas of the United States. The disease was first described by Thomas Caspar Gilchrist in 1894 and historically has been referred to as Gilchrist disease, North American blastomycosis, or Chicago disease.1,2 Cutaneous blastomycosis can occur by dissemination of yeast to the skin from systemic and pulmonary disease or rarely via direct inoculation of the skin resulting in primary cutaneous disease. Clinically, the lesions are polymorphic and may appear as well-demarcated verrucous plaques containing foci of pustules or ulcerations. Lesions typically heal centrifugally with a cribriform scar.3
We describe an adolescent with a unique history of inoculation 2 weeks prior to the development of a biopsy-confirmed lesion of cutaneous blastomycosis on the left chest wall that clinically resolved following 6 months of itraconazole.
Case Report
A 16-year-old adolescent boy with a history of morbid obesity, asthma, and seasonal allergies presented for evaluation of a painful, slowly enlarging skin lesion on the left chest wall of 2 months’ duration. According to the patient, a “small pimple” appeared at the site of impact 2 weeks following a fall into a muddy flowerbed in Madison, Wisconsin. The patient recalled that although he had soiled his clothing, there was no identifiable puncture of the skin. Despite daily application of hydrogen peroxide and a 1-week course of trimethoprim-sulfamethoxazole, the lesion gradually enlarged. Complete review of systems as well as exposure and travel history were otherwise negative.
Physical examination revealed a 5.0×2.5-cm exophytic, firm, well-circumscribed plaque with a papillated crusted surface on the left side of the chest near the posterior axillary line (Figure 1). There was no palpable regional lymphadenopathy. Pulmonary examination was unremarkable. Diagnostic workup, including complete blood cell count with differential, hemoglobin A1c, human immunodeficiency virus antibody/antigen testing, interferon-gamma release assay, and chest radiograph were all within normal limits.
Histologic examination of a biopsy specimen showed pseudoepitheliomatous hyperplasia of the epidermis with a brisk mixed inflammatory infiltrate (Figure 2). Displayed in Figure 3 is the Grocott-Gomori methenamine-silver stain that highlighted the thick double-contoured wall-budding yeasts.
The patient was diagnosed with primary cutaneous blastomycosis. Treatment was initiated with itraconazole 200 mg 3 times daily for 3 days, followed by 200 mg 2 times daily for 6 months. Following 3 months of therapy, the lesion had markedly improved with violaceous dyschromia and no residual surface changes. After 5 months of itraconazole, the patient stopped taking the medication for 2 months due to pharmacy issues and then resumed. After 6 total months of therapy, the lesion healed with only residual dyschromia and itraconazole was discontinued.
Comment
Epidemiology
Blastomycosis is a polymorphic pyogranulomatous disease caused by the dimorphic fungus B dermatitidis, naturally occurring in the soil with a worldwide distribution.4 Individuals affected by the disease often reside in locations where the fungus is endemic, specifically in areas that border the Mississippi and Ohio rivers, the Great Lakes, and Canadian provinces near the Saint Lawrence Seaway. More recently there has been an increased incidence of blastomycosis, with the highest proportion found in Wisconsin and Michigan.1,2 Exposures often are associated with recreational and occupational activities near streams or rivers where there may be decaying vegetation.1 Despite the ubiquitous presence of B dermatitidis in regions where the species is endemic, it is likely that many individuals who are exposed to the organism do not develop infection.
Pathogenesis
The exact pathogenesis for the development of disease in a particular individual remains unclear. Immunosuppression is not a prerequisite for susceptibility, as evidenced by a review of 123 cases of blastomycosis in which a preceding immunodepressive disorder was present in only 25% of patients. The same study found that it was almost equally common as diabetes mellitus and present in 22% of patients.5 The organism is considered a true pathogen given its ability to affect healthy individuals and the presence of a newly identified novel 120-kD glycoprotein antigen (WI-1) on the cell wall that may confer virulence via extracellular matrix and macrophage binding. Intact cell-mediated immunity that prevents the conversion of conidia (the infectious agent) to yeast (the form that exists at body temperature) plays a key role in conferring natural resistance.6,7
Cutaneous infection may occur by either dissemination of yeast to the skin from systemic disease or less commonly via direct inoculation of the skin, resulting in primary cutaneous disease. With respect to systemic disease, infection occurs through inhalation of conidia from moist soil containing organic debris, with an incubation period of 4 to 6 weeks. In the lungs, in a process largely dependent on host cell-mediated immunity, the mold quickly converts to yeast and may then either multiply or be phagocytized.2,6,7 Transmission does not occur from person to person.7 Asymptomatic infection may occur in at least 50% of patients, often leading to a delay in diagnosis. Symptomatic pulmonary disease may range from mild flulike symptoms to overt pneumonia, clinically indistinguishable from community-acquired bacterial pneumonia, tuberculosis, other fungal infections, and cancer. Of patients with primary pulmonary disease, 25% to 80% have been reported to develop secondary organ involvement via lymphohematogenous spread most commonly to the skin, followed respectively by the skeletal, genitourinary, and central nervous systems. Currently, there are 54 documented cases of secondary disseminated cutaneous blastomycosis in children reported in the literature.3,8-14
Presentation
Primary cutaneous disease resulting from direct cutaneous inoculation is rare, especially among children.14 Of 28 cases of isolated cutaneous blastomycosis reported in the literature, 12 (42%) were pediatric.3,8-21 Inoculation blastomycosis typically presents as a papule that expands to a well-demarcated verrucous plaque, often up to several centimeters in diameter, and is located on the skin at the site of contact. The lesion may exhibit a myriad of features ranging from pustules or nodules to focal ulcerations, either present centrally or within raised borders that ultimately may communicate via sinus tracking.7 Lesions that are purely pustular in morphology also have been reported. Healing typically begins centrally and expands centrifugally, often with cribriform scarring.2,4,22 Histologic features of primary and secondary blastomycosis include pseudoepitheliomatous hyperplasia, intraepidermal microabscesses, and dermal suppurative granulomatous inflammation.4 Classically, broad-based budding yeast are identified with a doubly refractile cell wall that is best visualized on periodic acid–Schiff staining.2
Diagnosis
In approximately 50% of patients with cutaneous blastomycosis resulting from secondary spread, there may be an absence of clinically active pulmonary disease, posing a diagnostic dilemma when differentiating from primary cutaneous disease.1,2,4 Furthermore, the skin findings exhibited in primary and secondary cutaneous blastomycosis cannot be distinguished by clinical inspection.19 To fulfill the criteria for diagnosis of primary cutaneous blastomycosis, there must be an identifiable source of infection from the environment, a lesion at the site of contact, a proven absence of systemic infection, and visualization and/or isolation of fungus from the lesion.4,12 The incubation period of lesions is shorter in primary cutaneous disease (2 weeks) and may aid in its differentiation from secondary disease, which typically is longer with lesions presenting 4 to 6 weeks following initial exposure.4
Treatment
Under the current 2015 guidelines from the American Academy of Pediatrics Committee on Infectious Diseases, 6 to 12 months of itraconazole is the treatment recommendation for mild to moderate pulmonary systemic disease without central nervous system involvement.7 Central nervous system disease and moderate to severe pulmonary and systemic disease are treated with intravenous amphotericin B followed by 12 months of oral itraconazole.1,7 Primary cutaneous disease, unlike secondary disease, may self-resolve; however, primary cutaneous disease usually is treated with 6 months of itraconazole, though successful therapy with surgical excision, radiation therapy, and incision and drainage have been reported.19
Unlike secondary cutaneous blastomycosis, primary inoculation disease may be self-limited; however, as treatment with antifungal therapy has become the standard of care, the disease’s propensity to self-resolve has not been well studied.4 Oral itraconazole for 6 to 12 months is the treatment of choice for mild to moderate cutaneous disease.1,22 Effective treatment duration may be difficult to definitively assess because of the self-limited nature of the disease. Our patient showed marked improvement after 3 months and resolution of the skin lesion following 6 months of itraconazole therapy. Our findings support the previously documented observation that systemic therapy might potentially be needed only for the time required to eliminate the clinical evidence of cutaneous disease.19 Our patient received the full 6 months of treatment according to current guidelines. Among a review of 22 cases of primary inoculation blastomycosis, the 5 patients who were treated with an azole agent alone showed disease clearance with an average treatment course of 3.2 months, ranging from 1 to 6 months.19 Further studies that assess the time to clearance with antifungal therapy and subsequent recurrence rates may be warranted.
Conclusion
Pediatric primary cutaneous blastomycosis is a rare cutaneous disease. Identifying sources of probable inoculation from the environment for this patient was unique in that the patient fell into a muddy puddle within a flowerbed. Given the patient’s atopic history, a predominance of humoral over cell-mediated immunity may have placed him at risk. He responded well to 6 months of oral itraconazole and there was no ulceration or scar formation. An increased awareness of this infection, particularly in geographic areas where its reported incidence is on the rise, could be helpful in reducing delays in diagnosis and treatment.
Acknowledgments
We thank Wenhua Liu, MD (Libertyville, Illinois), for reviewing the pathology and Pravin Muniyappa, MD (Chicago, Illinois), for referring the case.
- Chapman SW, Dismukes WE, Proia LA, et al. Clinical practice guidelines for the management of blastomycosis: 2008 update by the Infectious Diseases Society of America. Clin Infect Dis. 2008;46:1801-1812.
- Smith JA, Riddell Jt, Kauffman CA. Cutaneous manifestations of endemic mycoses. Curr Infect Dis Rep. 2013;15:440-449.
- Fisher KR, Baselski V, Beard G, et al. Pustular blastomycosis. J Am Acad Dermatol. 2009;6:355-358.
- Mason AR, Cortes GY, Cook J, et al. Cutaneous blastomycosis: a diagnostic challenge. Int J Dermatol. 2008;47:824-830.
- Lemos LB, Baliga M, Guo M. Blastomycosis: the great pretender can also be an opportunist. initial clinical diagnosis and underlying diseases in 123 patients. Ann Diagn Pathol. 2002;6:194-203.
- Bradsher RW, Chapman SW, Pappas PG. Blastomycosis. Infect Dis Clin North Am. 2003;17:21-40, vii.
- Blastomycosis. In: Kimberlin DW, ed. Red Book: 2015 Report of the Committee on Infectious Diseases. 30th ed. Elk Grove Village, IL: American Academy of Pediatrics; 2015:263-264.
- Brick KE, Drolet BA, Lyon VB, et al. Cutaneous and disseminated blastomycosis: a pediatric case series. Pediatr Dermatol. 2013;30:23-28.
- Fanella S, Skinner S, Trepman E, et al. Blastomycosis in children and adolescents: a 30-year experience from Manitoba. Med Mycol. 2011;49:627-632.
- Frost HM, Anderson J, Ivacic L, et al. Blastomycosis in children: an analysis of clinical, epidemiologic, and genetic features. J Pediatr Infect Dis Soc. 2017;6:49-56.
- Shukla S, Singh S, Jain M, et al. Paediatric cutaneous blastomycosis: a rare case diagnosed on FNAC. Diagn Cytopathol. 2009;37:119-121.
- Smith RJ, Boos MD, Burnham JM, et al. Atypical cutaneous blastomycosis in a child with juvenile idiopathic arthritis on infliximab. Pediatrics. 2015;136:E1386-E1389.
- Wilson JW, Cawley EP, Weidman FD, et al. Primary cutaneous North American blastomycosis. AMA Arch Derm. 1955;71:39-45.
- Zampogna JC, Hoy MJ, Ramos-Caro FA. Primary cutaneous north american blastomycosis in an immunosuppressed child. Pediatr Dermatol. 2003;20:128-130.
- Balasaraswathy P, Theerthanath. Cutaneous blastomycosis presenting as non-healing ulcer and responding to oral ketoconazole. Dermatol Online J. 2003;9:19.
- Bonifaz A, Morales D, Morales N, et al. Cutaneous blastomycosis. an imported case with good response to itraconazole. Rev Iberoam Micol. 2016;33:51-54.
- Clinton TS, Timko AL. Cutaneous blastomycosis without evidence of pulmonary involvement. Mil Med. 2003;168:651-653.
- Dhamija A, D’Souza P, Salgia P, et al. Blastomycosis presenting as solitary nodule: a rare presentation. Indian J Dermatol. 2012;57:133-135.
- Gray NA, Baddour LM. Cutaneous inoculation blastomycosis. Clin Infect Dis. 2002;34:E44-E49.
- Motswaledi HM, Monyemangene FM, Maloba BR, et al. Blastomycosis: a case report and review of the literature. Int J Dermatol. 2012;51:1090-1093.
- Rodríguez-Mena A, Mayorga J, Solís-Ledesma G, et al. Blastomycosis: report of an imported case in Mexico, with only cutaneous lesions [in Spanish]. Rev Iberoam Micol. 2010;27:210-212.
- Saccente M, Woods GL. Clinical and laboratory update on blastomycosis. Clin Microbiol Rev. 2010;23:367-381.
- Chapman SW, Dismukes WE, Proia LA, et al. Clinical practice guidelines for the management of blastomycosis: 2008 update by the Infectious Diseases Society of America. Clin Infect Dis. 2008;46:1801-1812.
- Smith JA, Riddell Jt, Kauffman CA. Cutaneous manifestations of endemic mycoses. Curr Infect Dis Rep. 2013;15:440-449.
- Fisher KR, Baselski V, Beard G, et al. Pustular blastomycosis. J Am Acad Dermatol. 2009;6:355-358.
- Mason AR, Cortes GY, Cook J, et al. Cutaneous blastomycosis: a diagnostic challenge. Int J Dermatol. 2008;47:824-830.
- Lemos LB, Baliga M, Guo M. Blastomycosis: the great pretender can also be an opportunist. initial clinical diagnosis and underlying diseases in 123 patients. Ann Diagn Pathol. 2002;6:194-203.
- Bradsher RW, Chapman SW, Pappas PG. Blastomycosis. Infect Dis Clin North Am. 2003;17:21-40, vii.
- Blastomycosis. In: Kimberlin DW, ed. Red Book: 2015 Report of the Committee on Infectious Diseases. 30th ed. Elk Grove Village, IL: American Academy of Pediatrics; 2015:263-264.
- Brick KE, Drolet BA, Lyon VB, et al. Cutaneous and disseminated blastomycosis: a pediatric case series. Pediatr Dermatol. 2013;30:23-28.
- Fanella S, Skinner S, Trepman E, et al. Blastomycosis in children and adolescents: a 30-year experience from Manitoba. Med Mycol. 2011;49:627-632.
- Frost HM, Anderson J, Ivacic L, et al. Blastomycosis in children: an analysis of clinical, epidemiologic, and genetic features. J Pediatr Infect Dis Soc. 2017;6:49-56.
- Shukla S, Singh S, Jain M, et al. Paediatric cutaneous blastomycosis: a rare case diagnosed on FNAC. Diagn Cytopathol. 2009;37:119-121.
- Smith RJ, Boos MD, Burnham JM, et al. Atypical cutaneous blastomycosis in a child with juvenile idiopathic arthritis on infliximab. Pediatrics. 2015;136:E1386-E1389.
- Wilson JW, Cawley EP, Weidman FD, et al. Primary cutaneous North American blastomycosis. AMA Arch Derm. 1955;71:39-45.
- Zampogna JC, Hoy MJ, Ramos-Caro FA. Primary cutaneous north american blastomycosis in an immunosuppressed child. Pediatr Dermatol. 2003;20:128-130.
- Balasaraswathy P, Theerthanath. Cutaneous blastomycosis presenting as non-healing ulcer and responding to oral ketoconazole. Dermatol Online J. 2003;9:19.
- Bonifaz A, Morales D, Morales N, et al. Cutaneous blastomycosis. an imported case with good response to itraconazole. Rev Iberoam Micol. 2016;33:51-54.
- Clinton TS, Timko AL. Cutaneous blastomycosis without evidence of pulmonary involvement. Mil Med. 2003;168:651-653.
- Dhamija A, D’Souza P, Salgia P, et al. Blastomycosis presenting as solitary nodule: a rare presentation. Indian J Dermatol. 2012;57:133-135.
- Gray NA, Baddour LM. Cutaneous inoculation blastomycosis. Clin Infect Dis. 2002;34:E44-E49.
- Motswaledi HM, Monyemangene FM, Maloba BR, et al. Blastomycosis: a case report and review of the literature. Int J Dermatol. 2012;51:1090-1093.
- Rodríguez-Mena A, Mayorga J, Solís-Ledesma G, et al. Blastomycosis: report of an imported case in Mexico, with only cutaneous lesions [in Spanish]. Rev Iberoam Micol. 2010;27:210-212.
- Saccente M, Woods GL. Clinical and laboratory update on blastomycosis. Clin Microbiol Rev. 2010;23:367-381.
Practice Points
- Cutaneous blastomycosis can occur by dissemination of yeast to the skin from systemic and pulmonary disease or rarely via direct inoculation of the skin, resulting in primary cutaneous disease.
- Exposures often are associated with recreational and occupational activities near streams or rivers where there may be decaying vegetation.
- Oral itraconazole for 6 to 12 months is the treatment of choice for mild to moderate cutaneous disease.
- Increased awareness of this rare infection, particularly in geographic areas where its reported incidence is on the rise, could be helpful in reducing delays in diagnosis and treatment.
Acute Hemorrhagic Edema of Infancy: Guide to Prevent Misdiagnosis
Acute hemorrhagic edema of infancy (AHEI) is an uncommon leukocytoclastic vasculitis affecting children aged 6 to 24 months; Henoch-Schönlein purpura (HSP) is the most common misdiagnosis. The 2 entities should be differentiated, as HSP may have renal and gastrointestinal (GI) comorbidities that need serial follow-up, whereas AHEI follows a benign course without systemic sequelae. Patient history and physical examination are the most important factors in differentiating the 2 diseases; histopathologic and direct immunofluorescence (DIF) analyses may lend further diagnostic confidence.
We report the case of a 10-month-old previously healthy boy who presented with acute rash, edema, and low-grade fever in the setting of recent diarrhea. We differentiate between AHEI and HSP to help prevent misdiagnosis by health care providers.
Case Report
A 10-month-old previously healthy boy presented to the emergency department (ED) for evaluation of a rash and swelling of 4 days’ duration. He had nonbloody diarrhea 1 week prior; soon after, he developed bilateral lower leg edema and rash. On evaluation in a different ED, he had a low-grade fever (rectal temperature, 38.0°C) but normal blood work, including complete blood cell count, basic metabolic panel, and coagulation studies. The patient was discharged to outpatient follow-up with his pediatrician who reported normal urinalysis.
Due to progression of the rash, the patient presented to our ED 3 days after his initial ED assessment. Dermatology was consulted. At the time of presentation, he was afebrile but with GI upset and fussiness. His parents denied additional symptoms or blood in urine or stool. Physical examination revealed a nontoxic-appearing infant with scattered palpable, annular, purpuric papules coalescing into plaques on both legs and feet (Figure 1), with sparse petechiae noted on the lower abdomen. The cheeks had scattered purpuric papules and plaques bilaterally, a few with a small central crust (Figure 2), and the right superior helix had a faint purpuric macule. The hands had a few pink edematous coalescing papules.
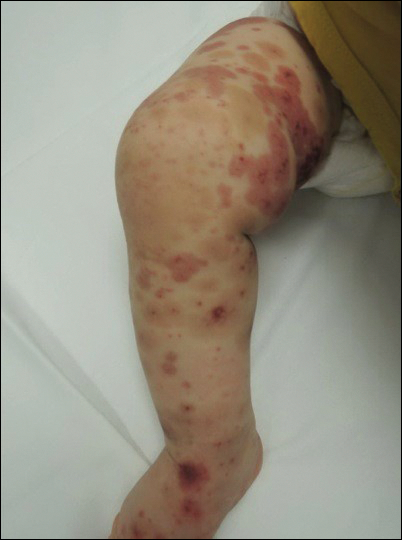
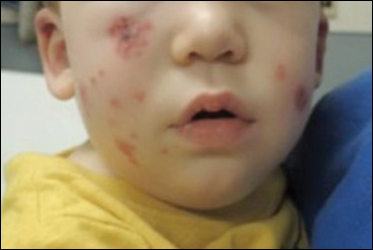
Histopathologic analyses with hematoxylin and eosin staining (Figure 3) and DIF (Figure 4) were performed from within a representative purpuric plaque on the right hip. Direct immunofluorescence was performed to evaluate for an IgA vasculitis versus an alternative type of vasculitis. The hematoxylin and eosin–stained specimen demonstrated a dermal perivascular infiltrate involving superficial and deep vessels with neutrophils, karyorrhexis, and erythrocyte extravasation. The endothelium was intact, with a mild suggestion of fibrinoid change of the blood vessel walls. Direct immunofluorescence revealed granular deposition of IgA, C3, and fibrinogen in multiple dermal blood vessels. Combined, the specimens were interpreted as evolving IgA-associated leukocytoclastic vasculitis.
The case was reviewed with our 2 department pediatric dermatologists; a diagnosis of AHEI was made based on the clinical and supportive histopathological presentations. The patient’s parents chose active treatment with a 2-week taper of oral prednisone because of the patient’s discomfort with edema. No GI or adverse renal sequelae, including findings on urinalysis, were reported at 1-month hospital follow-up with dermatology and pediatrics.
Comment
Incidence and Clinical Characteristics
Acute hemorrhagic edema of infancy is an uncommon leukocytoclastic vasculitis first described in the United States by Snow1 in 1913. Other names for the disorder include acute hemorrhagic edema of young children, cockade purpura and edema, Finkelstein disease, and Seidlmayer disease.2 Boys are affected more often than girls, with most children presenting at 6 to 24 months of age. Most affected children experience a prodrome of simple respiratory tract illness (most common), diarrhea (as in our case), or urinary tract infection.2 The exact pathophysiology behind AHEI is unknown, but it is thought to be an immune complex–mediated disease evidenced by the fact that infection, use of medication, or immunization precedes most cases.3,4
Diagnosis
Acute hemorrhagic edema of infancy is diagnosed clinically, with or without the support of skin biopsy. It should be differentiated from HSP because of renal and GI sequelae that HSP portends compared to the benign course of AHEI.2 Notably, some health care providers consider AHEI a benign variant of HSP.2,3
Characteristically, AHEI patients are nontoxic-appearing infants with a low-grade fever who develop relatively large (1–5 cm) targetoid purpuric lesions and indurated nonpitting edema of the extremities.2,5 Purpura in AHEI frequently occurs on the face, ears, and upper and lower extremities, whereas purpura in HSP most commonly presents on the buttocks and extensor legs with sparing of the face. Henoch-Schönlein purpura most often affects children aged 3 to 6 years compared to AHEI’s younger demographic (age <2 years).4,5 Clinically, HSP presents with palpable purpura and 1 or more of the following features: diffuse abdominal pain, arthritis/arthralgia, renal involvement, and skin or renal biopsy showing predominant IgA deposition.2,6
Both AHEI and HSP show leukocytoclastic vasculitis on histopathology.2-4,6,7 Positive perivascular IgA staining on DIF is strongly associated with HSP, but nearly one-quarter of AHEI cases also show this deposition pattern2,4,7; therefore, DIF alone cannot exclude a diagnosis of AHEI.
Differential Diagnosis
Alternative diagnoses to consider with AHEI include drug-induced vasculitis, erythema multiforme, HSP, Kawasaki disease, meningococcemia, nonaccidental skin bruising, Rocky Mountain spotted fever, septic vasculitis, and urticarial vasculitis (Table).2-4,6-8

Treatment
Acute hemorrhagic edema of infancy is self-limited, with only rare reports of extracutaneous involvement. Supportive treatment is indicated because spontaneous recovery without sequelae is expected within 21 days.2,3,6 If edema is symptomatic, as was the case with our patient, corticosteroids may shorten the disease course.3
Conclusion
Our case highlights the need to combine clinical history, physical examination, and histopathologic analysis to differentiate between AHEI and HSP, which is important for 2 reasons: (1) it helps with the decision to undertake active or observational treatment, and (2) it helps the clinician counsel the patient and guardians regarding potential associated renal and GI risks.
- Snow IM. Purpura, urticaria and angioneurotic edema of the hands and feet in a nursing baby. JAMA. 1913;61:18-19.
- Fiore E, Rizzi M, Ragazzi M, et al. Acute hemorrhagic edema of young children (cockade purpura and edema): a case series and systematic review. J Am Acad Dermatol. 2008;59:684-695.
- Freitas P, Bygum A. Visual impairment caused by periorbital edema in an infant with acute hemorrhagic edema of infancy. Pediatr Dermatol. 2013;30:e132-e135.
- Legrain V, Lejean S, Taïeb A, et al. Infantile acute hemorrhagic edema of the skin: study of ten cases. J Am Acad Dermatol. 1991;24:17-22.
- Breda L, Franchini S, Marzetti V, et al. Escherichia coli urinary infection as a cause of acute hemorrhagic edema in infancy. Pediatr Dermatol. 2015;32:e309-e311.
- Ozen S, Ruperto N, Dillon MJ, et al. EULAR/PReS endorsed consensus criteria for the classification of childhood vasculitides. Ann Rheum Dis. 2006;65:936-941.
- Saraclar Y, Tinaztepe K, Adalioğlu G, et al. Acute hemorrhagic edema of infancy (AHEI)—a variant of Henoch-Schönlein purpura or a distinct clinical entity? J Allergy Clin Immunol. 1990;86:473-483.
- Shinkai K, Fox L. Cutaneous vasculitis. In: Bolognia J, Jorizzo J, Schaffer J, eds. Dermatology. 3rd ed. China: Elsevier Limited; 2012:385-410.
Acute hemorrhagic edema of infancy (AHEI) is an uncommon leukocytoclastic vasculitis affecting children aged 6 to 24 months; Henoch-Schönlein purpura (HSP) is the most common misdiagnosis. The 2 entities should be differentiated, as HSP may have renal and gastrointestinal (GI) comorbidities that need serial follow-up, whereas AHEI follows a benign course without systemic sequelae. Patient history and physical examination are the most important factors in differentiating the 2 diseases; histopathologic and direct immunofluorescence (DIF) analyses may lend further diagnostic confidence.
We report the case of a 10-month-old previously healthy boy who presented with acute rash, edema, and low-grade fever in the setting of recent diarrhea. We differentiate between AHEI and HSP to help prevent misdiagnosis by health care providers.
Case Report
A 10-month-old previously healthy boy presented to the emergency department (ED) for evaluation of a rash and swelling of 4 days’ duration. He had nonbloody diarrhea 1 week prior; soon after, he developed bilateral lower leg edema and rash. On evaluation in a different ED, he had a low-grade fever (rectal temperature, 38.0°C) but normal blood work, including complete blood cell count, basic metabolic panel, and coagulation studies. The patient was discharged to outpatient follow-up with his pediatrician who reported normal urinalysis.
Due to progression of the rash, the patient presented to our ED 3 days after his initial ED assessment. Dermatology was consulted. At the time of presentation, he was afebrile but with GI upset and fussiness. His parents denied additional symptoms or blood in urine or stool. Physical examination revealed a nontoxic-appearing infant with scattered palpable, annular, purpuric papules coalescing into plaques on both legs and feet (Figure 1), with sparse petechiae noted on the lower abdomen. The cheeks had scattered purpuric papules and plaques bilaterally, a few with a small central crust (Figure 2), and the right superior helix had a faint purpuric macule. The hands had a few pink edematous coalescing papules.
Histopathologic analyses with hematoxylin and eosin staining (Figure 3) and DIF (Figure 4) were performed from within a representative purpuric plaque on the right hip. Direct immunofluorescence was performed to evaluate for an IgA vasculitis versus an alternative type of vasculitis. The hematoxylin and eosin–stained specimen demonstrated a dermal perivascular infiltrate involving superficial and deep vessels with neutrophils, karyorrhexis, and erythrocyte extravasation. The endothelium was intact, with a mild suggestion of fibrinoid change of the blood vessel walls. Direct immunofluorescence revealed granular deposition of IgA, C3, and fibrinogen in multiple dermal blood vessels. Combined, the specimens were interpreted as evolving IgA-associated leukocytoclastic vasculitis.
The case was reviewed with our 2 department pediatric dermatologists; a diagnosis of AHEI was made based on the clinical and supportive histopathological presentations. The patient’s parents chose active treatment with a 2-week taper of oral prednisone because of the patient’s discomfort with edema. No GI or adverse renal sequelae, including findings on urinalysis, were reported at 1-month hospital follow-up with dermatology and pediatrics.
Comment
Incidence and Clinical Characteristics
Acute hemorrhagic edema of infancy is an uncommon leukocytoclastic vasculitis first described in the United States by Snow1 in 1913. Other names for the disorder include acute hemorrhagic edema of young children, cockade purpura and edema, Finkelstein disease, and Seidlmayer disease.2 Boys are affected more often than girls, with most children presenting at 6 to 24 months of age. Most affected children experience a prodrome of simple respiratory tract illness (most common), diarrhea (as in our case), or urinary tract infection.2 The exact pathophysiology behind AHEI is unknown, but it is thought to be an immune complex–mediated disease evidenced by the fact that infection, use of medication, or immunization precedes most cases.3,4
Diagnosis
Acute hemorrhagic edema of infancy is diagnosed clinically, with or without the support of skin biopsy. It should be differentiated from HSP because of renal and GI sequelae that HSP portends compared to the benign course of AHEI.2 Notably, some health care providers consider AHEI a benign variant of HSP.2,3
Characteristically, AHEI patients are nontoxic-appearing infants with a low-grade fever who develop relatively large (1–5 cm) targetoid purpuric lesions and indurated nonpitting edema of the extremities.2,5 Purpura in AHEI frequently occurs on the face, ears, and upper and lower extremities, whereas purpura in HSP most commonly presents on the buttocks and extensor legs with sparing of the face. Henoch-Schönlein purpura most often affects children aged 3 to 6 years compared to AHEI’s younger demographic (age <2 years).4,5 Clinically, HSP presents with palpable purpura and 1 or more of the following features: diffuse abdominal pain, arthritis/arthralgia, renal involvement, and skin or renal biopsy showing predominant IgA deposition.2,6
Both AHEI and HSP show leukocytoclastic vasculitis on histopathology.2-4,6,7 Positive perivascular IgA staining on DIF is strongly associated with HSP, but nearly one-quarter of AHEI cases also show this deposition pattern2,4,7; therefore, DIF alone cannot exclude a diagnosis of AHEI.
Differential Diagnosis
Alternative diagnoses to consider with AHEI include drug-induced vasculitis, erythema multiforme, HSP, Kawasaki disease, meningococcemia, nonaccidental skin bruising, Rocky Mountain spotted fever, septic vasculitis, and urticarial vasculitis (Table).2-4,6-8
Treatment
Acute hemorrhagic edema of infancy is self-limited, with only rare reports of extracutaneous involvement. Supportive treatment is indicated because spontaneous recovery without sequelae is expected within 21 days.2,3,6 If edema is symptomatic, as was the case with our patient, corticosteroids may shorten the disease course.3
Conclusion
Our case highlights the need to combine clinical history, physical examination, and histopathologic analysis to differentiate between AHEI and HSP, which is important for 2 reasons: (1) it helps with the decision to undertake active or observational treatment, and (2) it helps the clinician counsel the patient and guardians regarding potential associated renal and GI risks.
Acute hemorrhagic edema of infancy (AHEI) is an uncommon leukocytoclastic vasculitis affecting children aged 6 to 24 months; Henoch-Schönlein purpura (HSP) is the most common misdiagnosis. The 2 entities should be differentiated, as HSP may have renal and gastrointestinal (GI) comorbidities that need serial follow-up, whereas AHEI follows a benign course without systemic sequelae. Patient history and physical examination are the most important factors in differentiating the 2 diseases; histopathologic and direct immunofluorescence (DIF) analyses may lend further diagnostic confidence.
We report the case of a 10-month-old previously healthy boy who presented with acute rash, edema, and low-grade fever in the setting of recent diarrhea. We differentiate between AHEI and HSP to help prevent misdiagnosis by health care providers.
Case Report
A 10-month-old previously healthy boy presented to the emergency department (ED) for evaluation of a rash and swelling of 4 days’ duration. He had nonbloody diarrhea 1 week prior; soon after, he developed bilateral lower leg edema and rash. On evaluation in a different ED, he had a low-grade fever (rectal temperature, 38.0°C) but normal blood work, including complete blood cell count, basic metabolic panel, and coagulation studies. The patient was discharged to outpatient follow-up with his pediatrician who reported normal urinalysis.
Due to progression of the rash, the patient presented to our ED 3 days after his initial ED assessment. Dermatology was consulted. At the time of presentation, he was afebrile but with GI upset and fussiness. His parents denied additional symptoms or blood in urine or stool. Physical examination revealed a nontoxic-appearing infant with scattered palpable, annular, purpuric papules coalescing into plaques on both legs and feet (Figure 1), with sparse petechiae noted on the lower abdomen. The cheeks had scattered purpuric papules and plaques bilaterally, a few with a small central crust (Figure 2), and the right superior helix had a faint purpuric macule. The hands had a few pink edematous coalescing papules.
Histopathologic analyses with hematoxylin and eosin staining (Figure 3) and DIF (Figure 4) were performed from within a representative purpuric plaque on the right hip. Direct immunofluorescence was performed to evaluate for an IgA vasculitis versus an alternative type of vasculitis. The hematoxylin and eosin–stained specimen demonstrated a dermal perivascular infiltrate involving superficial and deep vessels with neutrophils, karyorrhexis, and erythrocyte extravasation. The endothelium was intact, with a mild suggestion of fibrinoid change of the blood vessel walls. Direct immunofluorescence revealed granular deposition of IgA, C3, and fibrinogen in multiple dermal blood vessels. Combined, the specimens were interpreted as evolving IgA-associated leukocytoclastic vasculitis.
The case was reviewed with our 2 department pediatric dermatologists; a diagnosis of AHEI was made based on the clinical and supportive histopathological presentations. The patient’s parents chose active treatment with a 2-week taper of oral prednisone because of the patient’s discomfort with edema. No GI or adverse renal sequelae, including findings on urinalysis, were reported at 1-month hospital follow-up with dermatology and pediatrics.
Comment
Incidence and Clinical Characteristics
Acute hemorrhagic edema of infancy is an uncommon leukocytoclastic vasculitis first described in the United States by Snow1 in 1913. Other names for the disorder include acute hemorrhagic edema of young children, cockade purpura and edema, Finkelstein disease, and Seidlmayer disease.2 Boys are affected more often than girls, with most children presenting at 6 to 24 months of age. Most affected children experience a prodrome of simple respiratory tract illness (most common), diarrhea (as in our case), or urinary tract infection.2 The exact pathophysiology behind AHEI is unknown, but it is thought to be an immune complex–mediated disease evidenced by the fact that infection, use of medication, or immunization precedes most cases.3,4
Diagnosis
Acute hemorrhagic edema of infancy is diagnosed clinically, with or without the support of skin biopsy. It should be differentiated from HSP because of renal and GI sequelae that HSP portends compared to the benign course of AHEI.2 Notably, some health care providers consider AHEI a benign variant of HSP.2,3
Characteristically, AHEI patients are nontoxic-appearing infants with a low-grade fever who develop relatively large (1–5 cm) targetoid purpuric lesions and indurated nonpitting edema of the extremities.2,5 Purpura in AHEI frequently occurs on the face, ears, and upper and lower extremities, whereas purpura in HSP most commonly presents on the buttocks and extensor legs with sparing of the face. Henoch-Schönlein purpura most often affects children aged 3 to 6 years compared to AHEI’s younger demographic (age <2 years).4,5 Clinically, HSP presents with palpable purpura and 1 or more of the following features: diffuse abdominal pain, arthritis/arthralgia, renal involvement, and skin or renal biopsy showing predominant IgA deposition.2,6
Both AHEI and HSP show leukocytoclastic vasculitis on histopathology.2-4,6,7 Positive perivascular IgA staining on DIF is strongly associated with HSP, but nearly one-quarter of AHEI cases also show this deposition pattern2,4,7; therefore, DIF alone cannot exclude a diagnosis of AHEI.
Differential Diagnosis
Alternative diagnoses to consider with AHEI include drug-induced vasculitis, erythema multiforme, HSP, Kawasaki disease, meningococcemia, nonaccidental skin bruising, Rocky Mountain spotted fever, septic vasculitis, and urticarial vasculitis (Table).2-4,6-8
Treatment
Acute hemorrhagic edema of infancy is self-limited, with only rare reports of extracutaneous involvement. Supportive treatment is indicated because spontaneous recovery without sequelae is expected within 21 days.2,3,6 If edema is symptomatic, as was the case with our patient, corticosteroids may shorten the disease course.3
Conclusion
Our case highlights the need to combine clinical history, physical examination, and histopathologic analysis to differentiate between AHEI and HSP, which is important for 2 reasons: (1) it helps with the decision to undertake active or observational treatment, and (2) it helps the clinician counsel the patient and guardians regarding potential associated renal and GI risks.
- Snow IM. Purpura, urticaria and angioneurotic edema of the hands and feet in a nursing baby. JAMA. 1913;61:18-19.
- Fiore E, Rizzi M, Ragazzi M, et al. Acute hemorrhagic edema of young children (cockade purpura and edema): a case series and systematic review. J Am Acad Dermatol. 2008;59:684-695.
- Freitas P, Bygum A. Visual impairment caused by periorbital edema in an infant with acute hemorrhagic edema of infancy. Pediatr Dermatol. 2013;30:e132-e135.
- Legrain V, Lejean S, Taïeb A, et al. Infantile acute hemorrhagic edema of the skin: study of ten cases. J Am Acad Dermatol. 1991;24:17-22.
- Breda L, Franchini S, Marzetti V, et al. Escherichia coli urinary infection as a cause of acute hemorrhagic edema in infancy. Pediatr Dermatol. 2015;32:e309-e311.
- Ozen S, Ruperto N, Dillon MJ, et al. EULAR/PReS endorsed consensus criteria for the classification of childhood vasculitides. Ann Rheum Dis. 2006;65:936-941.
- Saraclar Y, Tinaztepe K, Adalioğlu G, et al. Acute hemorrhagic edema of infancy (AHEI)—a variant of Henoch-Schönlein purpura or a distinct clinical entity? J Allergy Clin Immunol. 1990;86:473-483.
- Shinkai K, Fox L. Cutaneous vasculitis. In: Bolognia J, Jorizzo J, Schaffer J, eds. Dermatology. 3rd ed. China: Elsevier Limited; 2012:385-410.
- Snow IM. Purpura, urticaria and angioneurotic edema of the hands and feet in a nursing baby. JAMA. 1913;61:18-19.
- Fiore E, Rizzi M, Ragazzi M, et al. Acute hemorrhagic edema of young children (cockade purpura and edema): a case series and systematic review. J Am Acad Dermatol. 2008;59:684-695.
- Freitas P, Bygum A. Visual impairment caused by periorbital edema in an infant with acute hemorrhagic edema of infancy. Pediatr Dermatol. 2013;30:e132-e135.
- Legrain V, Lejean S, Taïeb A, et al. Infantile acute hemorrhagic edema of the skin: study of ten cases. J Am Acad Dermatol. 1991;24:17-22.
- Breda L, Franchini S, Marzetti V, et al. Escherichia coli urinary infection as a cause of acute hemorrhagic edema in infancy. Pediatr Dermatol. 2015;32:e309-e311.
- Ozen S, Ruperto N, Dillon MJ, et al. EULAR/PReS endorsed consensus criteria for the classification of childhood vasculitides. Ann Rheum Dis. 2006;65:936-941.
- Saraclar Y, Tinaztepe K, Adalioğlu G, et al. Acute hemorrhagic edema of infancy (AHEI)—a variant of Henoch-Schönlein purpura or a distinct clinical entity? J Allergy Clin Immunol. 1990;86:473-483.
- Shinkai K, Fox L. Cutaneous vasculitis. In: Bolognia J, Jorizzo J, Schaffer J, eds. Dermatology. 3rd ed. China: Elsevier Limited; 2012:385-410.
Practice Points
- Acute hemorrhagic edema of infancy (AHEI) is an uncommon benign leukocytoclastic vasculitis of unknown precise pathophysiology that is thought be immune complex mediated.
- Clinical history, physical examination, and histopathologic analysis combine to allow the important differentiation between AHEI and Henoch-Schönlein purpura (HSP).
- Differentiation between AHEI and HSP determines treatment decisions and indicates the need for counseling on potential associated renal and gastrointestinal risks of HSP.
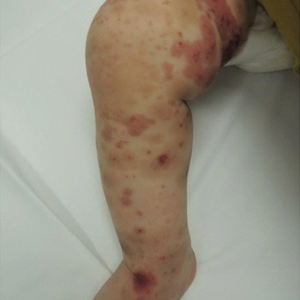
Hand-foot-and-mouth Disease Caused by Coxsackievirus A6 on the Rise
Hand-foot-and-mouth disease (HFMD) is a viral illness caused by several enteroviruses, most commonly coxsackievirus A16 (CVA16) and enterovirus 71 (EV71). The disease is generally seen in children younger than 5 years, characterized by lesions of the oral mucosa, palms, and soles, usually lasting 7 to 10 days. Other coxsackie type A viruses, including CVA6, CVA9, and CVA10, also are associated with HFMD.1-5 Although CVA16 has traditionally been the primary strain causing HFMD, CVA6 has become a major cause of HFMD outbreaks in the United States and worldwide in recent years.6-12 Interestingly, CVA6 also has been found to be associated with adult HFMD, which has increased in incidence. The CVA6 strain was first identified in association with the disease during HFMD outbreaks in Finland and Singapore in 2008,13,14 with similar strains detected in subsequent outbreaks in Taiwan, Japan, Spain, France, China, India, and the United States.12,15-25 Most cases took place in warmer months, with one winter outbreak in Massachusetts in 2012.24
Herein, we review the incidence of CVA6, as well as its atypical presentation, diagnosis, and treatment to aid dermatologists. Given the increasing incidence of HFMD caused by CVA6 and its often atypical presentation, it is important for dermatologists to be aware of this increasingly notable disease state and its viral cause.
Incidence of CVA6
Coxsackievirus A6 has been identified as the cause of many reported outbreaks of HFMD since it was first identified in 2008, and it is known to cause both pediatric and adult outbreaks.7-12 It may even be surpassing other strains in frequency in certain areas. In Tianjin, China, for example, EV71 and CVA16 were the most common serotypes causing HFMD from 2008 to 2012; however, in 2013, CVA6 was the most prevalent strain.26
The incidence of CVA6 also has been increasing in other areas.28
In 2015, an outbreak of HFMD took place at Lackland Air Force Base in Texas during a basic military training. Eight cases were confirmed and 45 cases were suspected. The rate of infection was 0.4% (50/12,270) among trainees and 0.3% (2/602) among instructors.7 Eight of 12 nasopharyngeal swabs tested positive for EV by way of local real-time reverse transcription–polymerase chain reaction (RT-PCR). Four nasopharyngeal swabs were sent to the CDC for evaluation and all were positive for CVA6.7
Presentation
Because the prevalence of CVA6 has increased, it is important to be able to identify the presentation of HFMD caused by this strain. Coxsackievirus A6 has been found to affect a broader demographic and cause more severe cases of HFMD with its unique constellation of findings compared to other known strains. Patients present with flulike symptoms; higher fever than present in typical HFMD; and a longer duration of disease, typically lasting 2 weeks. Patients also may present with more severe skin disease compared to classic HFMD, not only including vesicles but also large bullae, erosions, and ulcers on the dorsal and plantar feet (Figure 1).
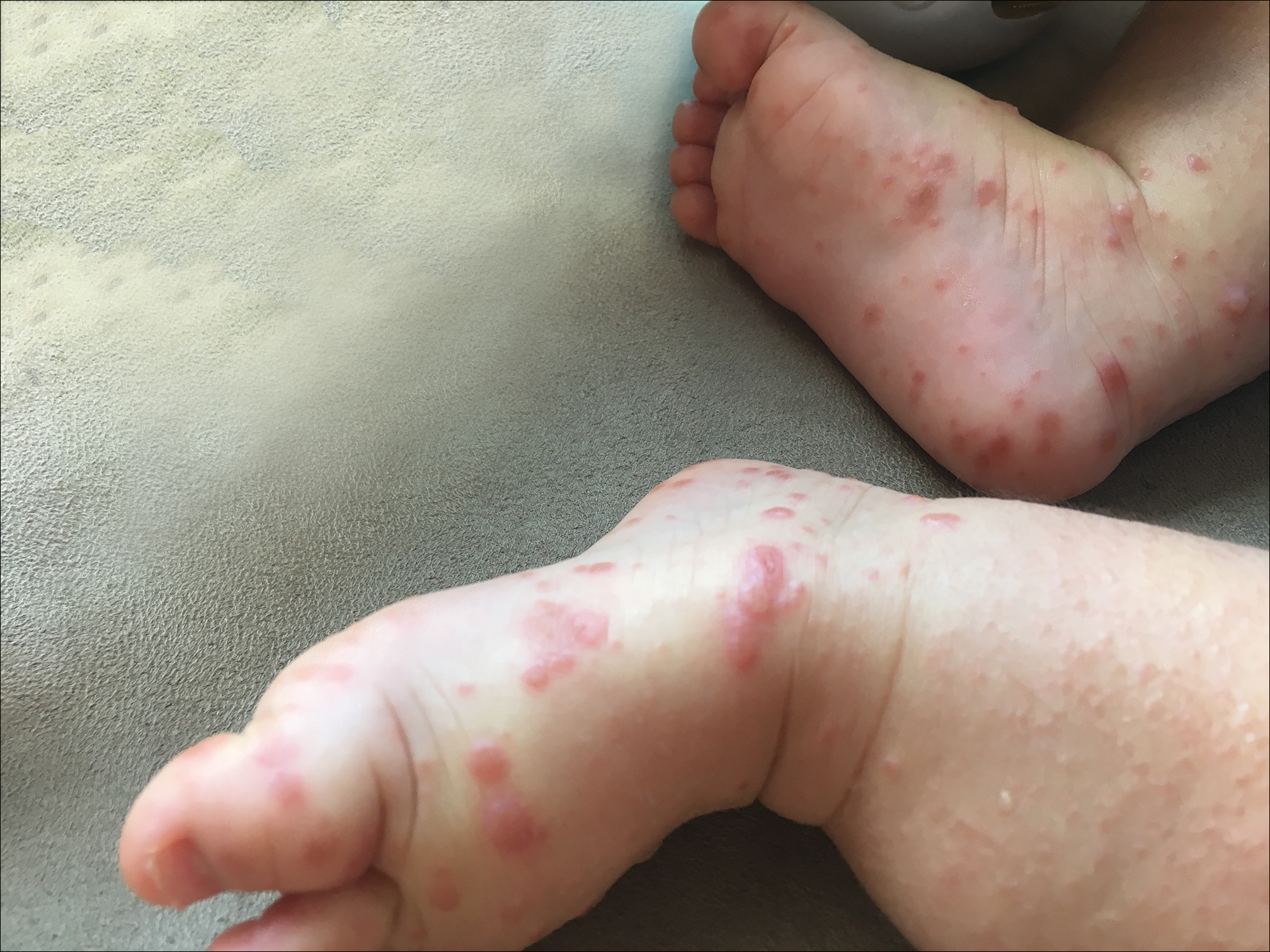
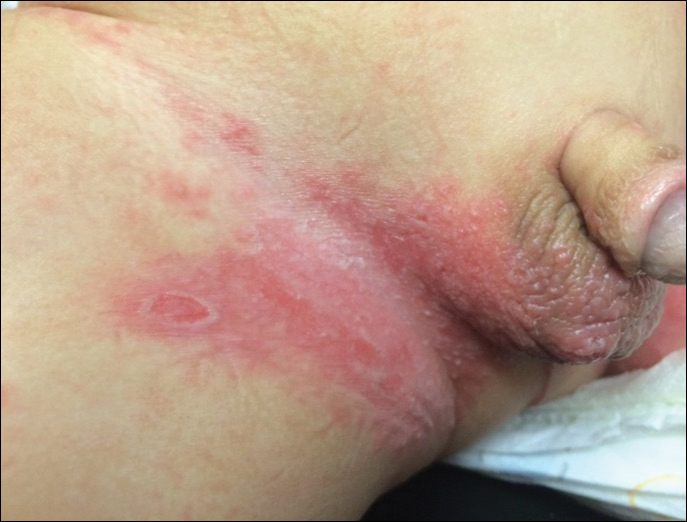
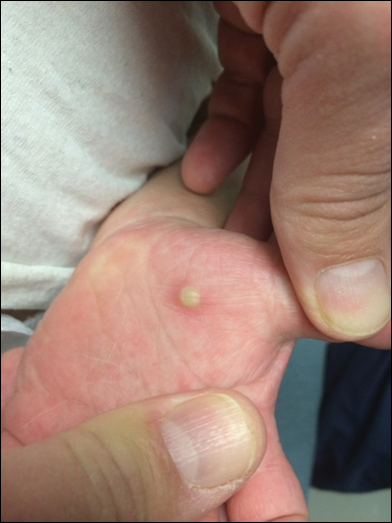
In patients with atopic dermatitis, CVA6 also shows a predilection to appear in areas of skin disease, such as the flexural regions of the arms and legs, and is referred to as eczema coxsackium.24,38,39 It can mimic eczema herpeticum or varicella superinfection, which are important considerations to include in the differential diagnosis. Additionally, CVA6-induced lesions often show up in previously irritated or traumatized areas such as sunburns, fungal infections, and diaper dermatitis in children. Lesions have been described to sometimes mimic Gianotti-Crosti syndrome, with involvement of the extensor surfaces, buttocks, and cheeks, and sparing of the trunk.24
Clinical Diagnosis
Because HFMD is uncommon and atypical in adults, skin biopsies may be used in the initial workup and evaluation of patients. It is important to understand the histologic features associated with HFMD, including spongiosis with exocytosis of neutrophils as well as keratinocyte necrosis and pallor with associated shadow cells.6 In one series, the most extensively involved areas of keratinocyte necrosis were the stratum granulosum and upper half of the stratum spinosum.40 In the dermis, vascular involvement may be present on a spectrum with the extravasation of red blood cells and leukocytoclasis or true leukocytoclastic vasculitis.6,40 Vesicular lesions show severe dermal edema and inflammatory infiltrate.6,41 CD3+ and CD8+ lymphocytes predominate. Cytotoxic T lymphocytes are present and express granzyme B and granulysin, both important mediators of apoptosis in virally infected keratinocytes.6
Adult HFMD primarily is a clinical diagnosis, and histopathologic analysis can be a useful tool in certain cases. Coxsackievirus A6 does not grow well on culture and is not detected by standard serologic testing laboratories, necessitating the use of quantitative RT-PCR analysis.41,42 In one study, culture was able to detect only 14% to 16% of samples that tested positive by quantitative RT-PCR.43 This form of PCR identifies viral subtype through amplification of enterovirus viral protein 1 capsid gene sequence.24 Unfortunately, this testing often is not offered in most readily available laboratories and often necessitates being sent out to more well-equipped laboratories.2,24
Treatment
Hand-foot-and-mouth disease is a self-limited illness and requires only supportive care with a focus on hydration and pain management. Lesions heal without scarring but may leave notable postinflammatory pigment alteration that may last months to years, depending on extent of disease and skin type. Secondarily infected individuals should be treated with appropriate antibiotics or antivirals depending on the infectious agent. Hand hygiene is of great importance, and hospitalized patients should be put on strict contact precautions. It also is important to isolate patients from vulnerable individuals, especially pregnant women, as coxsackievirus has been linked to intrauterine infections and loss of pregnancy.24
Genetic Analysis
Genetic studies of the virus have suggested that nonstructural genes may be playing an interesting role in clinical phenotypes and outcomes of CVA6 infection.44 These genetic studies also are being implemented into the understanding of the virus’ evolution as well as the construction of vaccinations.27,44
Conclusion
With the increasing prevalence of CVA6-associated HFMD, it is important to understand the clinical presentation and histologic findings associated with this atypical presentation of the disease as well as the changing epidemiology of the viral strains causing HFMD.
- Galen WK. Cutaneous manifestations of enterovirus infections. In: Tyring SK, ed. Mucocutaneous Manifestations of Viral Diseases. New York, NY: Marcel Dekker; 2002:455-467.
- Ramirez-Fort M, Downing C, Doan H, et al. Coxsackievirus A6 associated hand, foot and mouth disease in adults: clinical presentation and review of the literature. J Clin Virol. 2014;60:381-386.
- Khetsuriani N, Lamonte-Fowlkes A, Oberst S, et al. Enterovirus surveillance—United States, 1970-2005. MMWR Surveill Summ. 2006;55:1-20.
- Yang F, Zhang T, Hu Y, et al. Survey of enterovirus infections from hand, foot and mouth disease outbreak in China, 2009. Virol J. 2011;8:508.
- Ho M, Chen ER, Hsu KH, et al. An epidemic of enterovirus 71 infection in Taiwan. Taiwan Enterovirus Epidemic Working Group. N Engl J Med. 1999;341:929-935.
- Second J, Velter C, Calès S, et al. Clinicopathologic analysis of atypical hand, foot, and mouth disease in adult patients. J Am Acad Dermatol. 2016;76:722-729.
- Banta J, Lenz B, Pawlak M, et al. Notes from the field: outbreak of hand, foot, and mouth disease caused by coxsackievirus A6 among basic military trainees—Texas, 2015. MMWR Morb Mortal Wkly Rep. 2016;65.26:678-680.
- Bian L, Wang Y, Yao X, et al. Coxsackievirus A6: a new emerging pathogen causing hand, foot and mouth disease outbreaks worldwide. Expert Rev Anti Infect Ther. 2015;13:1061-1071.
- Buttery VW, Kenyon C, Grunewald S, et al. Notes from the field: atypical presentations of hand, foot, and mouth disease caused by coxsackievirus A6—Minnesota, 2014. MMWR Morb Mortal Wkly Rep. 2015;64:805.
- Puenpa J, Chieochansin T, Linsuwanon P, et al. Hand, foot, and mouth disease caused by coxsackievirus A6, Thailand, 2012. Emerg Infect Dis. 2013;19:641-643.
- Flett K, Youngster I, Huang J, et al. Hand, foot, and mouth disease caused by coxsackievirus A6. Emerg Infect Dis. 2012;18:1702-1704.
- Centers for Disease Control and Prevention. Notes from the field: severe hand, foot, and mouth disease associated with coxsackievirus A6—Alabama, Connecticut, California, and Nevada, November 2011-February 2012. MMWR Morb Mortal Wkly Rep. 2012;61:213-214.
- Blomqvist S, Klemola P, Kaijalainen S, et al. Co-circulation of coxsackieviruses A6 and A10 in hand, foot and mouth disease outbreak in Finland. J Clin Virol. 2010;48:49-54.
- Osterback R, Vuorinen T, Linna M, et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerg Infect Dis. 2009;15:1485-1488.
- Zeng H, Lu J, Zheng H, et al. The epidemiological study of coxsackievirus A6 revealing hand, foot and mouth disease epidemic patterns in Guandong, China. Sci Rep. 2015;5:10550.
- Mirand A, Henquell C, Archimbaud C, et al. Outbreak of hand, foot and mouth disease/herpangina associated with coxsackievirus A6 andA10 infections in 2010, France: a large citywide, prospective observational study. Clin Microbiol Infect. 2012;18:E110-E118.
- Wei SH, Huang YP, Liu MC, et al. An outbreak of coxsackievirus A6 hand, foot, and mouth disease associated with onychomadesis in Taiwan, 2010. BMC Infect Dis. 2011;11:346.
- Fujimoto T, Iizuka S, Enomoto M, et al. Hand, foot, and mouth disease caused by coxsackievirus A6, Japan, 2011. Emerg Infect Dis. 2012;18:337-339.
- Bracho MA, Gonzalez-Candelas F, Valero A, et al. Enterovirus co-infections and onychomadesis after hand, foot, and mouth disease, Spain, 2008. Emerg Infect Dis. 2011;17:2223-2231.
- Gopalkrishna V, Patil PR, Patil GP, et al. Circulation of multiple enterovirus serotypes causing hand, foot and mouth disease in India. J Med Microbiol. 2012;61:420-425.
- Lo SH, Huang YC, Huang CG, et al. Clinical and epidemiologic features of coxsackievirus A6 infection in children in northern Taiwan between 2004 and 2009. J Microbiol Immunol Infect. 2011;44:252-257.
- Lu QB, Zhang XA, Wo Y, et al. Circulation of coxsackievirus A10 and A6 in hand-foot-mouth disease in China, 2009-2011. PLoS One. 2012;7:E52073.
- Wu Y, Yeo A, Phoon MC, et al. The largest outbreak of hand; foot and mouth disease in Singapore in 2008: the role of enterovirus 71 and coxsackievirus A strains. Int J Infect Dis. 2010;14:E1076-E1081.
- Ventarola D, Bordone L, Silverberg N. Update on hand-foot-and-mouth disease. Clin Dermatol. 2015;33:340-346.
- Li Y, Chang Z, Wu P, et al. Emerging enteroviruses causing hand, foot and mouth disease, China. 2010-2016. Emerg Infect Dis. 2018;24:1902-1906.
- Tan X, Li L, Zhang B, et al. Molecular epidemiology of coxsackievirus A6 associated with outbreaks of hand, foot, and mouth disease in Tianjin, China, in 2013. Arch Virol. 2015;160:1097-1104.
- Li Y, Bao H, Zhang X, et al. Epidemiological and genetic analysis concerning the non-enterovirus 71 and non-coxsackievirus A16 causative agents related to hand, foot and mouth disease in Anyang City, Henan Province, China, from 2011 to 2015. J Med Virol. 2017;89:1749-1758.
- Guan H, Wang J, Wang C, et al. Etiology of multiple non-EV71 and non-CVA16 enteroviruses associated with hand, foot, and mouth disease in Jinan, China, 2009-2013. PLoS One. 2015;10:E0142733.
- Cabrerizo M, Tarrago´ D, Muñoz-Almagro C, et al. Mollecular epidemiology of enterovirus 71, coxsackievirus A16 and A6 associated with hand, foot and mouth disease in Spain. Clin Microbiol Infect. 2014;20:O150-O156.
- Lønnberg A, Elberling J, Fischer T, et al. Two cases of hand, foot, and mouth disease involving the scalp. Acta Derm Venereol. 2013;93:467-468.
- Lott JP, Liu K, Landry ML, et al. Atypical hand-foot-and-mouth disease associated with coxsackievirus A6 infection. J Am Acad Dermatol. 2013;69:736-741.
- Kaminska K, Martinetti G, Lucchini R, et al. Coxsackievirus A6 and hand, foot and mouth disease: three case reports of familial child-to-immunocompetent adult transmission and a literature review. Case Rep Dermatol. 2013;5:203-209.
- Shin JU, Oh SH, Lee JH. A case of hand-foot-mouth disease in an immunocompetent adult. Ann Dermatol. 2010;22:216-218.
- Osterback R, Vuorinen T, Linna M, et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerg Infect Dis. 2009;15:1485-1488.
- Feder HM, Bennett N, Modlin JF. Atypical hand, foot, and mouth disease: a vesiculobullous eruption caused by coxsackie virus A6. Lancet Infect Dis. 2014;14:83-86.
- Wei SH, Huang YP, Liu MC, et al. An outbreak of coxsackievirus A6 hand, foot, and mouth disease associated with onychomadesis in Taiwan, 2010. BMC Infect Dis. 2011;11:346.
- Kim M, Kim B, Byun S, et al. Beau’s lines and onychomadesis after hand-foot-mouth disease. Clin Pediatr Dermatol. 2015;1:1.
- Mathes EF, Oza V, Frieden IJ, et al. “Eczema coxsackium” and unusual cutaneous findings in an enterovirus outbreak. Pediatrics. 2013;132:E149-E157.
- Lynch M, Sears A, Cookson H, et al. Disseminated coxsackievirus A6 affecting children with atopic dermatitis. Clin Exp Dermatol. 2015;40:525-528.
- Laga A, Shroba S, Hanna J. Atypical hand, foot and mouth disease in adults associated with coxsackievirus A6: a clinicopathologic study. J Cutan Pathol. 2016;43:940-945.
- Schmidt NJ, Ho HH, Lennette EH. Propagation and isolation of group A coxsackieviruses in RD cells. J Clin Microbiol. 1975;2:183-185.
- Oberste MS, Penaranda S, Rogers SL, et al. Comparative evaluation of Taqman real-time PCR and semi-nested VP1 PCR for detection of enteroviruses in clinical specimens. J Clin Virol. 2010;49:73-74.
- Lee MK, Chan PK, Ho II, et al. Enterovirus infection among patients admitted to hospital in Hong Kong in 2010: epidemiology, clinical characteristics, and importance of molecular diagnosis. J Med Virol. 2013;85:1811-1817.
- Yee PTI, Laa Poh C. Impact of genetic changes, pathogenicity and antigenicity on enterovirus A71 vaccine development. Virology. 2017;506:121-129.
Hand-foot-and-mouth disease (HFMD) is a viral illness caused by several enteroviruses, most commonly coxsackievirus A16 (CVA16) and enterovirus 71 (EV71). The disease is generally seen in children younger than 5 years, characterized by lesions of the oral mucosa, palms, and soles, usually lasting 7 to 10 days. Other coxsackie type A viruses, including CVA6, CVA9, and CVA10, also are associated with HFMD.1-5 Although CVA16 has traditionally been the primary strain causing HFMD, CVA6 has become a major cause of HFMD outbreaks in the United States and worldwide in recent years.6-12 Interestingly, CVA6 also has been found to be associated with adult HFMD, which has increased in incidence. The CVA6 strain was first identified in association with the disease during HFMD outbreaks in Finland and Singapore in 2008,13,14 with similar strains detected in subsequent outbreaks in Taiwan, Japan, Spain, France, China, India, and the United States.12,15-25 Most cases took place in warmer months, with one winter outbreak in Massachusetts in 2012.24
Herein, we review the incidence of CVA6, as well as its atypical presentation, diagnosis, and treatment to aid dermatologists. Given the increasing incidence of HFMD caused by CVA6 and its often atypical presentation, it is important for dermatologists to be aware of this increasingly notable disease state and its viral cause.
Incidence of CVA6
Coxsackievirus A6 has been identified as the cause of many reported outbreaks of HFMD since it was first identified in 2008, and it is known to cause both pediatric and adult outbreaks.7-12 It may even be surpassing other strains in frequency in certain areas. In Tianjin, China, for example, EV71 and CVA16 were the most common serotypes causing HFMD from 2008 to 2012; however, in 2013, CVA6 was the most prevalent strain.26
The incidence of CVA6 also has been increasing in other areas.28
In 2015, an outbreak of HFMD took place at Lackland Air Force Base in Texas during a basic military training. Eight cases were confirmed and 45 cases were suspected. The rate of infection was 0.4% (50/12,270) among trainees and 0.3% (2/602) among instructors.7 Eight of 12 nasopharyngeal swabs tested positive for EV by way of local real-time reverse transcription–polymerase chain reaction (RT-PCR). Four nasopharyngeal swabs were sent to the CDC for evaluation and all were positive for CVA6.7
Presentation
Because the prevalence of CVA6 has increased, it is important to be able to identify the presentation of HFMD caused by this strain. Coxsackievirus A6 has been found to affect a broader demographic and cause more severe cases of HFMD with its unique constellation of findings compared to other known strains. Patients present with flulike symptoms; higher fever than present in typical HFMD; and a longer duration of disease, typically lasting 2 weeks. Patients also may present with more severe skin disease compared to classic HFMD, not only including vesicles but also large bullae, erosions, and ulcers on the dorsal and plantar feet (Figure 1).
In patients with atopic dermatitis, CVA6 also shows a predilection to appear in areas of skin disease, such as the flexural regions of the arms and legs, and is referred to as eczema coxsackium.24,38,39 It can mimic eczema herpeticum or varicella superinfection, which are important considerations to include in the differential diagnosis. Additionally, CVA6-induced lesions often show up in previously irritated or traumatized areas such as sunburns, fungal infections, and diaper dermatitis in children. Lesions have been described to sometimes mimic Gianotti-Crosti syndrome, with involvement of the extensor surfaces, buttocks, and cheeks, and sparing of the trunk.24
Clinical Diagnosis
Because HFMD is uncommon and atypical in adults, skin biopsies may be used in the initial workup and evaluation of patients. It is important to understand the histologic features associated with HFMD, including spongiosis with exocytosis of neutrophils as well as keratinocyte necrosis and pallor with associated shadow cells.6 In one series, the most extensively involved areas of keratinocyte necrosis were the stratum granulosum and upper half of the stratum spinosum.40 In the dermis, vascular involvement may be present on a spectrum with the extravasation of red blood cells and leukocytoclasis or true leukocytoclastic vasculitis.6,40 Vesicular lesions show severe dermal edema and inflammatory infiltrate.6,41 CD3+ and CD8+ lymphocytes predominate. Cytotoxic T lymphocytes are present and express granzyme B and granulysin, both important mediators of apoptosis in virally infected keratinocytes.6
Adult HFMD primarily is a clinical diagnosis, and histopathologic analysis can be a useful tool in certain cases. Coxsackievirus A6 does not grow well on culture and is not detected by standard serologic testing laboratories, necessitating the use of quantitative RT-PCR analysis.41,42 In one study, culture was able to detect only 14% to 16% of samples that tested positive by quantitative RT-PCR.43 This form of PCR identifies viral subtype through amplification of enterovirus viral protein 1 capsid gene sequence.24 Unfortunately, this testing often is not offered in most readily available laboratories and often necessitates being sent out to more well-equipped laboratories.2,24
Treatment
Hand-foot-and-mouth disease is a self-limited illness and requires only supportive care with a focus on hydration and pain management. Lesions heal without scarring but may leave notable postinflammatory pigment alteration that may last months to years, depending on extent of disease and skin type. Secondarily infected individuals should be treated with appropriate antibiotics or antivirals depending on the infectious agent. Hand hygiene is of great importance, and hospitalized patients should be put on strict contact precautions. It also is important to isolate patients from vulnerable individuals, especially pregnant women, as coxsackievirus has been linked to intrauterine infections and loss of pregnancy.24
Genetic Analysis
Genetic studies of the virus have suggested that nonstructural genes may be playing an interesting role in clinical phenotypes and outcomes of CVA6 infection.44 These genetic studies also are being implemented into the understanding of the virus’ evolution as well as the construction of vaccinations.27,44
Conclusion
With the increasing prevalence of CVA6-associated HFMD, it is important to understand the clinical presentation and histologic findings associated with this atypical presentation of the disease as well as the changing epidemiology of the viral strains causing HFMD.
Hand-foot-and-mouth disease (HFMD) is a viral illness caused by several enteroviruses, most commonly coxsackievirus A16 (CVA16) and enterovirus 71 (EV71). The disease is generally seen in children younger than 5 years, characterized by lesions of the oral mucosa, palms, and soles, usually lasting 7 to 10 days. Other coxsackie type A viruses, including CVA6, CVA9, and CVA10, also are associated with HFMD.1-5 Although CVA16 has traditionally been the primary strain causing HFMD, CVA6 has become a major cause of HFMD outbreaks in the United States and worldwide in recent years.6-12 Interestingly, CVA6 also has been found to be associated with adult HFMD, which has increased in incidence. The CVA6 strain was first identified in association with the disease during HFMD outbreaks in Finland and Singapore in 2008,13,14 with similar strains detected in subsequent outbreaks in Taiwan, Japan, Spain, France, China, India, and the United States.12,15-25 Most cases took place in warmer months, with one winter outbreak in Massachusetts in 2012.24
Herein, we review the incidence of CVA6, as well as its atypical presentation, diagnosis, and treatment to aid dermatologists. Given the increasing incidence of HFMD caused by CVA6 and its often atypical presentation, it is important for dermatologists to be aware of this increasingly notable disease state and its viral cause.
Incidence of CVA6
Coxsackievirus A6 has been identified as the cause of many reported outbreaks of HFMD since it was first identified in 2008, and it is known to cause both pediatric and adult outbreaks.7-12 It may even be surpassing other strains in frequency in certain areas. In Tianjin, China, for example, EV71 and CVA16 were the most common serotypes causing HFMD from 2008 to 2012; however, in 2013, CVA6 was the most prevalent strain.26
The incidence of CVA6 also has been increasing in other areas.28
In 2015, an outbreak of HFMD took place at Lackland Air Force Base in Texas during a basic military training. Eight cases were confirmed and 45 cases were suspected. The rate of infection was 0.4% (50/12,270) among trainees and 0.3% (2/602) among instructors.7 Eight of 12 nasopharyngeal swabs tested positive for EV by way of local real-time reverse transcription–polymerase chain reaction (RT-PCR). Four nasopharyngeal swabs were sent to the CDC for evaluation and all were positive for CVA6.7
Presentation
Because the prevalence of CVA6 has increased, it is important to be able to identify the presentation of HFMD caused by this strain. Coxsackievirus A6 has been found to affect a broader demographic and cause more severe cases of HFMD with its unique constellation of findings compared to other known strains. Patients present with flulike symptoms; higher fever than present in typical HFMD; and a longer duration of disease, typically lasting 2 weeks. Patients also may present with more severe skin disease compared to classic HFMD, not only including vesicles but also large bullae, erosions, and ulcers on the dorsal and plantar feet (Figure 1).
In patients with atopic dermatitis, CVA6 also shows a predilection to appear in areas of skin disease, such as the flexural regions of the arms and legs, and is referred to as eczema coxsackium.24,38,39 It can mimic eczema herpeticum or varicella superinfection, which are important considerations to include in the differential diagnosis. Additionally, CVA6-induced lesions often show up in previously irritated or traumatized areas such as sunburns, fungal infections, and diaper dermatitis in children. Lesions have been described to sometimes mimic Gianotti-Crosti syndrome, with involvement of the extensor surfaces, buttocks, and cheeks, and sparing of the trunk.24
Clinical Diagnosis
Because HFMD is uncommon and atypical in adults, skin biopsies may be used in the initial workup and evaluation of patients. It is important to understand the histologic features associated with HFMD, including spongiosis with exocytosis of neutrophils as well as keratinocyte necrosis and pallor with associated shadow cells.6 In one series, the most extensively involved areas of keratinocyte necrosis were the stratum granulosum and upper half of the stratum spinosum.40 In the dermis, vascular involvement may be present on a spectrum with the extravasation of red blood cells and leukocytoclasis or true leukocytoclastic vasculitis.6,40 Vesicular lesions show severe dermal edema and inflammatory infiltrate.6,41 CD3+ and CD8+ lymphocytes predominate. Cytotoxic T lymphocytes are present and express granzyme B and granulysin, both important mediators of apoptosis in virally infected keratinocytes.6
Adult HFMD primarily is a clinical diagnosis, and histopathologic analysis can be a useful tool in certain cases. Coxsackievirus A6 does not grow well on culture and is not detected by standard serologic testing laboratories, necessitating the use of quantitative RT-PCR analysis.41,42 In one study, culture was able to detect only 14% to 16% of samples that tested positive by quantitative RT-PCR.43 This form of PCR identifies viral subtype through amplification of enterovirus viral protein 1 capsid gene sequence.24 Unfortunately, this testing often is not offered in most readily available laboratories and often necessitates being sent out to more well-equipped laboratories.2,24
Treatment
Hand-foot-and-mouth disease is a self-limited illness and requires only supportive care with a focus on hydration and pain management. Lesions heal without scarring but may leave notable postinflammatory pigment alteration that may last months to years, depending on extent of disease and skin type. Secondarily infected individuals should be treated with appropriate antibiotics or antivirals depending on the infectious agent. Hand hygiene is of great importance, and hospitalized patients should be put on strict contact precautions. It also is important to isolate patients from vulnerable individuals, especially pregnant women, as coxsackievirus has been linked to intrauterine infections and loss of pregnancy.24
Genetic Analysis
Genetic studies of the virus have suggested that nonstructural genes may be playing an interesting role in clinical phenotypes and outcomes of CVA6 infection.44 These genetic studies also are being implemented into the understanding of the virus’ evolution as well as the construction of vaccinations.27,44
Conclusion
With the increasing prevalence of CVA6-associated HFMD, it is important to understand the clinical presentation and histologic findings associated with this atypical presentation of the disease as well as the changing epidemiology of the viral strains causing HFMD.
- Galen WK. Cutaneous manifestations of enterovirus infections. In: Tyring SK, ed. Mucocutaneous Manifestations of Viral Diseases. New York, NY: Marcel Dekker; 2002:455-467.
- Ramirez-Fort M, Downing C, Doan H, et al. Coxsackievirus A6 associated hand, foot and mouth disease in adults: clinical presentation and review of the literature. J Clin Virol. 2014;60:381-386.
- Khetsuriani N, Lamonte-Fowlkes A, Oberst S, et al. Enterovirus surveillance—United States, 1970-2005. MMWR Surveill Summ. 2006;55:1-20.
- Yang F, Zhang T, Hu Y, et al. Survey of enterovirus infections from hand, foot and mouth disease outbreak in China, 2009. Virol J. 2011;8:508.
- Ho M, Chen ER, Hsu KH, et al. An epidemic of enterovirus 71 infection in Taiwan. Taiwan Enterovirus Epidemic Working Group. N Engl J Med. 1999;341:929-935.
- Second J, Velter C, Calès S, et al. Clinicopathologic analysis of atypical hand, foot, and mouth disease in adult patients. J Am Acad Dermatol. 2016;76:722-729.
- Banta J, Lenz B, Pawlak M, et al. Notes from the field: outbreak of hand, foot, and mouth disease caused by coxsackievirus A6 among basic military trainees—Texas, 2015. MMWR Morb Mortal Wkly Rep. 2016;65.26:678-680.
- Bian L, Wang Y, Yao X, et al. Coxsackievirus A6: a new emerging pathogen causing hand, foot and mouth disease outbreaks worldwide. Expert Rev Anti Infect Ther. 2015;13:1061-1071.
- Buttery VW, Kenyon C, Grunewald S, et al. Notes from the field: atypical presentations of hand, foot, and mouth disease caused by coxsackievirus A6—Minnesota, 2014. MMWR Morb Mortal Wkly Rep. 2015;64:805.
- Puenpa J, Chieochansin T, Linsuwanon P, et al. Hand, foot, and mouth disease caused by coxsackievirus A6, Thailand, 2012. Emerg Infect Dis. 2013;19:641-643.
- Flett K, Youngster I, Huang J, et al. Hand, foot, and mouth disease caused by coxsackievirus A6. Emerg Infect Dis. 2012;18:1702-1704.
- Centers for Disease Control and Prevention. Notes from the field: severe hand, foot, and mouth disease associated with coxsackievirus A6—Alabama, Connecticut, California, and Nevada, November 2011-February 2012. MMWR Morb Mortal Wkly Rep. 2012;61:213-214.
- Blomqvist S, Klemola P, Kaijalainen S, et al. Co-circulation of coxsackieviruses A6 and A10 in hand, foot and mouth disease outbreak in Finland. J Clin Virol. 2010;48:49-54.
- Osterback R, Vuorinen T, Linna M, et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerg Infect Dis. 2009;15:1485-1488.
- Zeng H, Lu J, Zheng H, et al. The epidemiological study of coxsackievirus A6 revealing hand, foot and mouth disease epidemic patterns in Guandong, China. Sci Rep. 2015;5:10550.
- Mirand A, Henquell C, Archimbaud C, et al. Outbreak of hand, foot and mouth disease/herpangina associated with coxsackievirus A6 andA10 infections in 2010, France: a large citywide, prospective observational study. Clin Microbiol Infect. 2012;18:E110-E118.
- Wei SH, Huang YP, Liu MC, et al. An outbreak of coxsackievirus A6 hand, foot, and mouth disease associated with onychomadesis in Taiwan, 2010. BMC Infect Dis. 2011;11:346.
- Fujimoto T, Iizuka S, Enomoto M, et al. Hand, foot, and mouth disease caused by coxsackievirus A6, Japan, 2011. Emerg Infect Dis. 2012;18:337-339.
- Bracho MA, Gonzalez-Candelas F, Valero A, et al. Enterovirus co-infections and onychomadesis after hand, foot, and mouth disease, Spain, 2008. Emerg Infect Dis. 2011;17:2223-2231.
- Gopalkrishna V, Patil PR, Patil GP, et al. Circulation of multiple enterovirus serotypes causing hand, foot and mouth disease in India. J Med Microbiol. 2012;61:420-425.
- Lo SH, Huang YC, Huang CG, et al. Clinical and epidemiologic features of coxsackievirus A6 infection in children in northern Taiwan between 2004 and 2009. J Microbiol Immunol Infect. 2011;44:252-257.
- Lu QB, Zhang XA, Wo Y, et al. Circulation of coxsackievirus A10 and A6 in hand-foot-mouth disease in China, 2009-2011. PLoS One. 2012;7:E52073.
- Wu Y, Yeo A, Phoon MC, et al. The largest outbreak of hand; foot and mouth disease in Singapore in 2008: the role of enterovirus 71 and coxsackievirus A strains. Int J Infect Dis. 2010;14:E1076-E1081.
- Ventarola D, Bordone L, Silverberg N. Update on hand-foot-and-mouth disease. Clin Dermatol. 2015;33:340-346.
- Li Y, Chang Z, Wu P, et al. Emerging enteroviruses causing hand, foot and mouth disease, China. 2010-2016. Emerg Infect Dis. 2018;24:1902-1906.
- Tan X, Li L, Zhang B, et al. Molecular epidemiology of coxsackievirus A6 associated with outbreaks of hand, foot, and mouth disease in Tianjin, China, in 2013. Arch Virol. 2015;160:1097-1104.
- Li Y, Bao H, Zhang X, et al. Epidemiological and genetic analysis concerning the non-enterovirus 71 and non-coxsackievirus A16 causative agents related to hand, foot and mouth disease in Anyang City, Henan Province, China, from 2011 to 2015. J Med Virol. 2017;89:1749-1758.
- Guan H, Wang J, Wang C, et al. Etiology of multiple non-EV71 and non-CVA16 enteroviruses associated with hand, foot, and mouth disease in Jinan, China, 2009-2013. PLoS One. 2015;10:E0142733.
- Cabrerizo M, Tarrago´ D, Muñoz-Almagro C, et al. Mollecular epidemiology of enterovirus 71, coxsackievirus A16 and A6 associated with hand, foot and mouth disease in Spain. Clin Microbiol Infect. 2014;20:O150-O156.
- Lønnberg A, Elberling J, Fischer T, et al. Two cases of hand, foot, and mouth disease involving the scalp. Acta Derm Venereol. 2013;93:467-468.
- Lott JP, Liu K, Landry ML, et al. Atypical hand-foot-and-mouth disease associated with coxsackievirus A6 infection. J Am Acad Dermatol. 2013;69:736-741.
- Kaminska K, Martinetti G, Lucchini R, et al. Coxsackievirus A6 and hand, foot and mouth disease: three case reports of familial child-to-immunocompetent adult transmission and a literature review. Case Rep Dermatol. 2013;5:203-209.
- Shin JU, Oh SH, Lee JH. A case of hand-foot-mouth disease in an immunocompetent adult. Ann Dermatol. 2010;22:216-218.
- Osterback R, Vuorinen T, Linna M, et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerg Infect Dis. 2009;15:1485-1488.
- Feder HM, Bennett N, Modlin JF. Atypical hand, foot, and mouth disease: a vesiculobullous eruption caused by coxsackie virus A6. Lancet Infect Dis. 2014;14:83-86.
- Wei SH, Huang YP, Liu MC, et al. An outbreak of coxsackievirus A6 hand, foot, and mouth disease associated with onychomadesis in Taiwan, 2010. BMC Infect Dis. 2011;11:346.
- Kim M, Kim B, Byun S, et al. Beau’s lines and onychomadesis after hand-foot-mouth disease. Clin Pediatr Dermatol. 2015;1:1.
- Mathes EF, Oza V, Frieden IJ, et al. “Eczema coxsackium” and unusual cutaneous findings in an enterovirus outbreak. Pediatrics. 2013;132:E149-E157.
- Lynch M, Sears A, Cookson H, et al. Disseminated coxsackievirus A6 affecting children with atopic dermatitis. Clin Exp Dermatol. 2015;40:525-528.
- Laga A, Shroba S, Hanna J. Atypical hand, foot and mouth disease in adults associated with coxsackievirus A6: a clinicopathologic study. J Cutan Pathol. 2016;43:940-945.
- Schmidt NJ, Ho HH, Lennette EH. Propagation and isolation of group A coxsackieviruses in RD cells. J Clin Microbiol. 1975;2:183-185.
- Oberste MS, Penaranda S, Rogers SL, et al. Comparative evaluation of Taqman real-time PCR and semi-nested VP1 PCR for detection of enteroviruses in clinical specimens. J Clin Virol. 2010;49:73-74.
- Lee MK, Chan PK, Ho II, et al. Enterovirus infection among patients admitted to hospital in Hong Kong in 2010: epidemiology, clinical characteristics, and importance of molecular diagnosis. J Med Virol. 2013;85:1811-1817.
- Yee PTI, Laa Poh C. Impact of genetic changes, pathogenicity and antigenicity on enterovirus A71 vaccine development. Virology. 2017;506:121-129.
- Galen WK. Cutaneous manifestations of enterovirus infections. In: Tyring SK, ed. Mucocutaneous Manifestations of Viral Diseases. New York, NY: Marcel Dekker; 2002:455-467.
- Ramirez-Fort M, Downing C, Doan H, et al. Coxsackievirus A6 associated hand, foot and mouth disease in adults: clinical presentation and review of the literature. J Clin Virol. 2014;60:381-386.
- Khetsuriani N, Lamonte-Fowlkes A, Oberst S, et al. Enterovirus surveillance—United States, 1970-2005. MMWR Surveill Summ. 2006;55:1-20.
- Yang F, Zhang T, Hu Y, et al. Survey of enterovirus infections from hand, foot and mouth disease outbreak in China, 2009. Virol J. 2011;8:508.
- Ho M, Chen ER, Hsu KH, et al. An epidemic of enterovirus 71 infection in Taiwan. Taiwan Enterovirus Epidemic Working Group. N Engl J Med. 1999;341:929-935.
- Second J, Velter C, Calès S, et al. Clinicopathologic analysis of atypical hand, foot, and mouth disease in adult patients. J Am Acad Dermatol. 2016;76:722-729.
- Banta J, Lenz B, Pawlak M, et al. Notes from the field: outbreak of hand, foot, and mouth disease caused by coxsackievirus A6 among basic military trainees—Texas, 2015. MMWR Morb Mortal Wkly Rep. 2016;65.26:678-680.
- Bian L, Wang Y, Yao X, et al. Coxsackievirus A6: a new emerging pathogen causing hand, foot and mouth disease outbreaks worldwide. Expert Rev Anti Infect Ther. 2015;13:1061-1071.
- Buttery VW, Kenyon C, Grunewald S, et al. Notes from the field: atypical presentations of hand, foot, and mouth disease caused by coxsackievirus A6—Minnesota, 2014. MMWR Morb Mortal Wkly Rep. 2015;64:805.
- Puenpa J, Chieochansin T, Linsuwanon P, et al. Hand, foot, and mouth disease caused by coxsackievirus A6, Thailand, 2012. Emerg Infect Dis. 2013;19:641-643.
- Flett K, Youngster I, Huang J, et al. Hand, foot, and mouth disease caused by coxsackievirus A6. Emerg Infect Dis. 2012;18:1702-1704.
- Centers for Disease Control and Prevention. Notes from the field: severe hand, foot, and mouth disease associated with coxsackievirus A6—Alabama, Connecticut, California, and Nevada, November 2011-February 2012. MMWR Morb Mortal Wkly Rep. 2012;61:213-214.
- Blomqvist S, Klemola P, Kaijalainen S, et al. Co-circulation of coxsackieviruses A6 and A10 in hand, foot and mouth disease outbreak in Finland. J Clin Virol. 2010;48:49-54.
- Osterback R, Vuorinen T, Linna M, et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerg Infect Dis. 2009;15:1485-1488.
- Zeng H, Lu J, Zheng H, et al. The epidemiological study of coxsackievirus A6 revealing hand, foot and mouth disease epidemic patterns in Guandong, China. Sci Rep. 2015;5:10550.
- Mirand A, Henquell C, Archimbaud C, et al. Outbreak of hand, foot and mouth disease/herpangina associated with coxsackievirus A6 andA10 infections in 2010, France: a large citywide, prospective observational study. Clin Microbiol Infect. 2012;18:E110-E118.
- Wei SH, Huang YP, Liu MC, et al. An outbreak of coxsackievirus A6 hand, foot, and mouth disease associated with onychomadesis in Taiwan, 2010. BMC Infect Dis. 2011;11:346.
- Fujimoto T, Iizuka S, Enomoto M, et al. Hand, foot, and mouth disease caused by coxsackievirus A6, Japan, 2011. Emerg Infect Dis. 2012;18:337-339.
- Bracho MA, Gonzalez-Candelas F, Valero A, et al. Enterovirus co-infections and onychomadesis after hand, foot, and mouth disease, Spain, 2008. Emerg Infect Dis. 2011;17:2223-2231.
- Gopalkrishna V, Patil PR, Patil GP, et al. Circulation of multiple enterovirus serotypes causing hand, foot and mouth disease in India. J Med Microbiol. 2012;61:420-425.
- Lo SH, Huang YC, Huang CG, et al. Clinical and epidemiologic features of coxsackievirus A6 infection in children in northern Taiwan between 2004 and 2009. J Microbiol Immunol Infect. 2011;44:252-257.
- Lu QB, Zhang XA, Wo Y, et al. Circulation of coxsackievirus A10 and A6 in hand-foot-mouth disease in China, 2009-2011. PLoS One. 2012;7:E52073.
- Wu Y, Yeo A, Phoon MC, et al. The largest outbreak of hand; foot and mouth disease in Singapore in 2008: the role of enterovirus 71 and coxsackievirus A strains. Int J Infect Dis. 2010;14:E1076-E1081.
- Ventarola D, Bordone L, Silverberg N. Update on hand-foot-and-mouth disease. Clin Dermatol. 2015;33:340-346.
- Li Y, Chang Z, Wu P, et al. Emerging enteroviruses causing hand, foot and mouth disease, China. 2010-2016. Emerg Infect Dis. 2018;24:1902-1906.
- Tan X, Li L, Zhang B, et al. Molecular epidemiology of coxsackievirus A6 associated with outbreaks of hand, foot, and mouth disease in Tianjin, China, in 2013. Arch Virol. 2015;160:1097-1104.
- Li Y, Bao H, Zhang X, et al. Epidemiological and genetic analysis concerning the non-enterovirus 71 and non-coxsackievirus A16 causative agents related to hand, foot and mouth disease in Anyang City, Henan Province, China, from 2011 to 2015. J Med Virol. 2017;89:1749-1758.
- Guan H, Wang J, Wang C, et al. Etiology of multiple non-EV71 and non-CVA16 enteroviruses associated with hand, foot, and mouth disease in Jinan, China, 2009-2013. PLoS One. 2015;10:E0142733.
- Cabrerizo M, Tarrago´ D, Muñoz-Almagro C, et al. Mollecular epidemiology of enterovirus 71, coxsackievirus A16 and A6 associated with hand, foot and mouth disease in Spain. Clin Microbiol Infect. 2014;20:O150-O156.
- Lønnberg A, Elberling J, Fischer T, et al. Two cases of hand, foot, and mouth disease involving the scalp. Acta Derm Venereol. 2013;93:467-468.
- Lott JP, Liu K, Landry ML, et al. Atypical hand-foot-and-mouth disease associated with coxsackievirus A6 infection. J Am Acad Dermatol. 2013;69:736-741.
- Kaminska K, Martinetti G, Lucchini R, et al. Coxsackievirus A6 and hand, foot and mouth disease: three case reports of familial child-to-immunocompetent adult transmission and a literature review. Case Rep Dermatol. 2013;5:203-209.
- Shin JU, Oh SH, Lee JH. A case of hand-foot-mouth disease in an immunocompetent adult. Ann Dermatol. 2010;22:216-218.
- Osterback R, Vuorinen T, Linna M, et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerg Infect Dis. 2009;15:1485-1488.
- Feder HM, Bennett N, Modlin JF. Atypical hand, foot, and mouth disease: a vesiculobullous eruption caused by coxsackie virus A6. Lancet Infect Dis. 2014;14:83-86.
- Wei SH, Huang YP, Liu MC, et al. An outbreak of coxsackievirus A6 hand, foot, and mouth disease associated with onychomadesis in Taiwan, 2010. BMC Infect Dis. 2011;11:346.
- Kim M, Kim B, Byun S, et al. Beau’s lines and onychomadesis after hand-foot-mouth disease. Clin Pediatr Dermatol. 2015;1:1.
- Mathes EF, Oza V, Frieden IJ, et al. “Eczema coxsackium” and unusual cutaneous findings in an enterovirus outbreak. Pediatrics. 2013;132:E149-E157.
- Lynch M, Sears A, Cookson H, et al. Disseminated coxsackievirus A6 affecting children with atopic dermatitis. Clin Exp Dermatol. 2015;40:525-528.
- Laga A, Shroba S, Hanna J. Atypical hand, foot and mouth disease in adults associated with coxsackievirus A6: a clinicopathologic study. J Cutan Pathol. 2016;43:940-945.
- Schmidt NJ, Ho HH, Lennette EH. Propagation and isolation of group A coxsackieviruses in RD cells. J Clin Microbiol. 1975;2:183-185.
- Oberste MS, Penaranda S, Rogers SL, et al. Comparative evaluation of Taqman real-time PCR and semi-nested VP1 PCR for detection of enteroviruses in clinical specimens. J Clin Virol. 2010;49:73-74.
- Lee MK, Chan PK, Ho II, et al. Enterovirus infection among patients admitted to hospital in Hong Kong in 2010: epidemiology, clinical characteristics, and importance of molecular diagnosis. J Med Virol. 2013;85:1811-1817.
- Yee PTI, Laa Poh C. Impact of genetic changes, pathogenicity and antigenicity on enterovirus A71 vaccine development. Virology. 2017;506:121-129.
Practice Points
- Coxsackievirus A6 is an increasingly more common cause of hand-foot-and-mouth disease (HFMD), often with atypical presentation, more severe disease, and association with HFMD in adults.
- Coxsackievirus A6 has become a major cause of HFMD outbreak in the United States and worldwide.
Practice Expense–Only Codes: No Physician Work, No Sweat
I have written previously about Current Procedural Terminology (CPT) procedure codes submitted on the same date of service as evaluation and management (E/M) services in the context of modifier -25.1 Billing same-day procedures and E/M services is under close scrutiny by insurers, and accurate and complete documentation is a must.2 An understanding of what aspects of evaluation are included in the global surgical package is critical in deciding whether a separate and distinct same-day evaluation was performed. In general, the decision to perform a procedure is included in the payment for the procedure itself, as is the examination of the body site in question, diagnosis of the medical condition, discussion of treatment options, and postoperative services related to the procedure. This is true for CPT codes that contain physician work, which constitute the majority of CPT codes reported by dermatologists.3
However, there is one set of codes where these principles do not apply: the practice expense (PE)–only codes, or no physician work codes. These codes are defined by CPT and the Relative Value Scale Update Committee (RUC) of the American Medical Association as containing no physician work. Their valuations are based only on staff/nursing time and the other aspects of direct and indirect practice costs included in providing the service, such as gauze, sutures, equipment, office rent, and utilities.4 Examples of PE-only codes include the nonphysician-performed photodynamic therapy code 96567; phototherapy codes 96900, 96910, and 96912; and patch testing and photopatch testing codes 95044, 95052, and 95056.
For PE-only codes, only the provision of the service by staff is included in the code reimbursement; there is no physician time or work built into these codes. Thus, neither the initial evaluation of the patient by the physician, the decision to perform the procedure, nor the evaluation of therapy effectiveness or side effects or interpretation of the results is included. Understanding that there is no physician involvement in PE-only codes is critical in deciding whether an E/M service should be billed on the same day as a PE-only code. To that end, although a physician does not actually have to personally evaluate the patient on the day of service to bill PE-only codes, the Centers for Medicare & Medicaid Services has indicated that a physician or qualified medical provider must be on premises.5 Billing for PE-only services when no provider is present will be interpreted as a false claim or fraudulent billing practice.
Because PE-only codes do not include physician work, an E/M service will be billed in addition to the treatment almost any time a same-day physician evaluation is performed. For example, if a patient presents with a changing mole that is evaluated on the same date of service as phototherapy for the treatment of psoriasis, that service is clearly reportable with an E/M code because the mole check is separate and distinct from the phototherapy treatment. A more common scenario is for the physician to see a patient with a rash consistent with an allergic contact dermatitis and the decision to perform same-day patch testing is made. In this circumstance, the E/M service is still reportable because the evaluation of the rash and the decision to perform patch testing are not included in this PE-only code.
Phototherapy typically is provided as a prolonged course of multiple treatments, and reporting of same-day E/M services during the course of therapy is common. Phototherapy must be monitored by the physician for clinical effectiveness, dose changes, and side effects, as well as to determine whether to continue therapy. A standard operating procedure should be created to document that the physician typically evaluates the patient’s progress at set intervals or as dictated by patient or staff concerns. Reporting an E/M service with every phototherapy session is not considered medically necessary. Moreover, a nurse evaluation of the patient prior to each phototherapy treatment, including questions on disease severity, how the patient did with the last treatment, and whether medications have changed, is included in the payment for the phototherapy codes. Only formal and medically necessary physician E/M services should be billed, not drive-by visits in which the physician pops in just to see how the patient is doing.
Final Thoughts
Practice expense–only codes include no payment for physician time or work but require the presence of a qualified health care provider on premises to bill. Medically necessary physician evaluations on the same day as PE-only services will typically result in both an E/M service and the procedure being reported. Understanding performance and documentation requirements of PE-only codes is critical for proper reimbursement for a dermatology practice.
- Rogers H. One diagnosis and modifier -25: appropriate or audit target? Cutis. 2017;99:165-166.
- Rogers H. Modifier -25 victory, but the battle is not over. Cutis. 2018;101:409-410.
- American Academy of Dermatology. Medicare update. Derm Coding Consult. March 2001;5:5-7. https://www.aad.org/File%20Library/Global%20navigation/Member%20tools%20and%20benefits/Publications/Derm%20Coding%20Consult%20archives/2001-spring.pdf.
- Current Procedural Terminology 2018, Professional Edition. Chicago, IL: American Medical Association; 2018.
- Determining who has the authority to bill. The Dermatologist. September 4, 2018. https://www.the-dermatologist.com/article/3006. Accessed October 25, 2018.
I have written previously about Current Procedural Terminology (CPT) procedure codes submitted on the same date of service as evaluation and management (E/M) services in the context of modifier -25.1 Billing same-day procedures and E/M services is under close scrutiny by insurers, and accurate and complete documentation is a must.2 An understanding of what aspects of evaluation are included in the global surgical package is critical in deciding whether a separate and distinct same-day evaluation was performed. In general, the decision to perform a procedure is included in the payment for the procedure itself, as is the examination of the body site in question, diagnosis of the medical condition, discussion of treatment options, and postoperative services related to the procedure. This is true for CPT codes that contain physician work, which constitute the majority of CPT codes reported by dermatologists.3
However, there is one set of codes where these principles do not apply: the practice expense (PE)–only codes, or no physician work codes. These codes are defined by CPT and the Relative Value Scale Update Committee (RUC) of the American Medical Association as containing no physician work. Their valuations are based only on staff/nursing time and the other aspects of direct and indirect practice costs included in providing the service, such as gauze, sutures, equipment, office rent, and utilities.4 Examples of PE-only codes include the nonphysician-performed photodynamic therapy code 96567; phototherapy codes 96900, 96910, and 96912; and patch testing and photopatch testing codes 95044, 95052, and 95056.
For PE-only codes, only the provision of the service by staff is included in the code reimbursement; there is no physician time or work built into these codes. Thus, neither the initial evaluation of the patient by the physician, the decision to perform the procedure, nor the evaluation of therapy effectiveness or side effects or interpretation of the results is included. Understanding that there is no physician involvement in PE-only codes is critical in deciding whether an E/M service should be billed on the same day as a PE-only code. To that end, although a physician does not actually have to personally evaluate the patient on the day of service to bill PE-only codes, the Centers for Medicare & Medicaid Services has indicated that a physician or qualified medical provider must be on premises.5 Billing for PE-only services when no provider is present will be interpreted as a false claim or fraudulent billing practice.
Because PE-only codes do not include physician work, an E/M service will be billed in addition to the treatment almost any time a same-day physician evaluation is performed. For example, if a patient presents with a changing mole that is evaluated on the same date of service as phototherapy for the treatment of psoriasis, that service is clearly reportable with an E/M code because the mole check is separate and distinct from the phototherapy treatment. A more common scenario is for the physician to see a patient with a rash consistent with an allergic contact dermatitis and the decision to perform same-day patch testing is made. In this circumstance, the E/M service is still reportable because the evaluation of the rash and the decision to perform patch testing are not included in this PE-only code.
Phototherapy typically is provided as a prolonged course of multiple treatments, and reporting of same-day E/M services during the course of therapy is common. Phototherapy must be monitored by the physician for clinical effectiveness, dose changes, and side effects, as well as to determine whether to continue therapy. A standard operating procedure should be created to document that the physician typically evaluates the patient’s progress at set intervals or as dictated by patient or staff concerns. Reporting an E/M service with every phototherapy session is not considered medically necessary. Moreover, a nurse evaluation of the patient prior to each phototherapy treatment, including questions on disease severity, how the patient did with the last treatment, and whether medications have changed, is included in the payment for the phototherapy codes. Only formal and medically necessary physician E/M services should be billed, not drive-by visits in which the physician pops in just to see how the patient is doing.
Final Thoughts
Practice expense–only codes include no payment for physician time or work but require the presence of a qualified health care provider on premises to bill. Medically necessary physician evaluations on the same day as PE-only services will typically result in both an E/M service and the procedure being reported. Understanding performance and documentation requirements of PE-only codes is critical for proper reimbursement for a dermatology practice.
I have written previously about Current Procedural Terminology (CPT) procedure codes submitted on the same date of service as evaluation and management (E/M) services in the context of modifier -25.1 Billing same-day procedures and E/M services is under close scrutiny by insurers, and accurate and complete documentation is a must.2 An understanding of what aspects of evaluation are included in the global surgical package is critical in deciding whether a separate and distinct same-day evaluation was performed. In general, the decision to perform a procedure is included in the payment for the procedure itself, as is the examination of the body site in question, diagnosis of the medical condition, discussion of treatment options, and postoperative services related to the procedure. This is true for CPT codes that contain physician work, which constitute the majority of CPT codes reported by dermatologists.3
However, there is one set of codes where these principles do not apply: the practice expense (PE)–only codes, or no physician work codes. These codes are defined by CPT and the Relative Value Scale Update Committee (RUC) of the American Medical Association as containing no physician work. Their valuations are based only on staff/nursing time and the other aspects of direct and indirect practice costs included in providing the service, such as gauze, sutures, equipment, office rent, and utilities.4 Examples of PE-only codes include the nonphysician-performed photodynamic therapy code 96567; phototherapy codes 96900, 96910, and 96912; and patch testing and photopatch testing codes 95044, 95052, and 95056.
For PE-only codes, only the provision of the service by staff is included in the code reimbursement; there is no physician time or work built into these codes. Thus, neither the initial evaluation of the patient by the physician, the decision to perform the procedure, nor the evaluation of therapy effectiveness or side effects or interpretation of the results is included. Understanding that there is no physician involvement in PE-only codes is critical in deciding whether an E/M service should be billed on the same day as a PE-only code. To that end, although a physician does not actually have to personally evaluate the patient on the day of service to bill PE-only codes, the Centers for Medicare & Medicaid Services has indicated that a physician or qualified medical provider must be on premises.5 Billing for PE-only services when no provider is present will be interpreted as a false claim or fraudulent billing practice.
Because PE-only codes do not include physician work, an E/M service will be billed in addition to the treatment almost any time a same-day physician evaluation is performed. For example, if a patient presents with a changing mole that is evaluated on the same date of service as phototherapy for the treatment of psoriasis, that service is clearly reportable with an E/M code because the mole check is separate and distinct from the phototherapy treatment. A more common scenario is for the physician to see a patient with a rash consistent with an allergic contact dermatitis and the decision to perform same-day patch testing is made. In this circumstance, the E/M service is still reportable because the evaluation of the rash and the decision to perform patch testing are not included in this PE-only code.
Phototherapy typically is provided as a prolonged course of multiple treatments, and reporting of same-day E/M services during the course of therapy is common. Phototherapy must be monitored by the physician for clinical effectiveness, dose changes, and side effects, as well as to determine whether to continue therapy. A standard operating procedure should be created to document that the physician typically evaluates the patient’s progress at set intervals or as dictated by patient or staff concerns. Reporting an E/M service with every phototherapy session is not considered medically necessary. Moreover, a nurse evaluation of the patient prior to each phototherapy treatment, including questions on disease severity, how the patient did with the last treatment, and whether medications have changed, is included in the payment for the phototherapy codes. Only formal and medically necessary physician E/M services should be billed, not drive-by visits in which the physician pops in just to see how the patient is doing.
Final Thoughts
Practice expense–only codes include no payment for physician time or work but require the presence of a qualified health care provider on premises to bill. Medically necessary physician evaluations on the same day as PE-only services will typically result in both an E/M service and the procedure being reported. Understanding performance and documentation requirements of PE-only codes is critical for proper reimbursement for a dermatology practice.
- Rogers H. One diagnosis and modifier -25: appropriate or audit target? Cutis. 2017;99:165-166.
- Rogers H. Modifier -25 victory, but the battle is not over. Cutis. 2018;101:409-410.
- American Academy of Dermatology. Medicare update. Derm Coding Consult. March 2001;5:5-7. https://www.aad.org/File%20Library/Global%20navigation/Member%20tools%20and%20benefits/Publications/Derm%20Coding%20Consult%20archives/2001-spring.pdf.
- Current Procedural Terminology 2018, Professional Edition. Chicago, IL: American Medical Association; 2018.
- Determining who has the authority to bill. The Dermatologist. September 4, 2018. https://www.the-dermatologist.com/article/3006. Accessed October 25, 2018.
- Rogers H. One diagnosis and modifier -25: appropriate or audit target? Cutis. 2017;99:165-166.
- Rogers H. Modifier -25 victory, but the battle is not over. Cutis. 2018;101:409-410.
- American Academy of Dermatology. Medicare update. Derm Coding Consult. March 2001;5:5-7. https://www.aad.org/File%20Library/Global%20navigation/Member%20tools%20and%20benefits/Publications/Derm%20Coding%20Consult%20archives/2001-spring.pdf.
- Current Procedural Terminology 2018, Professional Edition. Chicago, IL: American Medical Association; 2018.
- Determining who has the authority to bill. The Dermatologist. September 4, 2018. https://www.the-dermatologist.com/article/3006. Accessed October 25, 2018.
Practice Points
- Billing same-day procedures and evaluation and management services is under close scrutiny by insurers, and accurate and complete documentation is a must.
- For practice expense–only codes, only the provision of the service by staff is included in the code reimbursement; there is no physician time or work built into these codes.
- Practice expense–only codes require the presence of a qualified health care provider on premises to bill.
Acrokeratoelastoidosis and Knuckle Pads Coexisting in a Child
Case Report
An 11-year-old boy presented with atraumatic thickening of the skin on the bilateral distal and proximal interphalangeal joints of 1 year’s duration. The patient also noted small bumps of unknown duration across the bilateral palms and soles with prominence on the lateral aspects. The patient previously used over-the-counter topical wart removal treatment and topical salicylic acid with minimal improvement. The patient reported no pertinent medical or surgical history, although there was a family history of Alport syndrome, predominantly in male relatives. The patient’s father and paternal grandfather were noted to have similar lesions on the palms.
On physical examination, multiple pink to flesh-colored hyperkeratotic plaques were noted over the proximal and distal interphalangeal joints of the bilateral hands (Figure 1A). Upon close inspection, there were small flesh-colored and slightly translucent papules in a linear distribution on the palmar surfaces of the hands (Figure 2A) with predominance on the thenar and hypothenar eminences. The flexural creases of the bilateral wrists also revealed linear flesh-colored papules. The same small flesh-colored and translucent papules also were noted on the plantar surfaces of the bilateral feet (Figure 2B).
A biopsy was obtained from one of the small translucent papules on the left palm. Hematoxylin and eosin–stained sections revealed elevated compact orthokeratosis with an underlying central epidermal dell (Figure 3). A diagnosis of marginal papular keratoderma was made and further elastin staining was completed. Elastin stains showed marked thinning of the elastin fibers throughout the reticular dermis. Many elastin fibers in the reticular dermis demonstrated a fine arborizing pattern that normally is only evident in the papillary dermis (Figure 4). Acrokeratoelastoidosis (AKE) was diagnosed histopathologically, and knuckle pads were diagnosed clinically.
Because the patient was asymptomatic, he did not want treatment of AKE. He had marked improvement of the knuckle pads after 1 month with daily application of urea cream 10% (Figure 1B), and intermittent use was required for maintenance.
Comment
Etiology
Acrokeratoelastoidosis was first described in 1953 and is considered a type of palmoplantar marginal papular keratoderma.1 There is overlap within the marginal papular keratodermas that makes precise diagnosis difficult within this group. The marginal papular keratodermas on the palms and soles are a group of disorders that include AKE, focal acral hyperkeratosis (FAH), mosaic acral keratosis, degenerative collagenous plaques on the hands, and digital papular calcific elastosis. These diseases are similar in clinical and histopathological features; some argue these diseases are the same entity.2
Acrokeratoelastoidosis has been hypothesized to originate from altered elastic fiber synthesis from fibroblasts.3 Because AKE is rare, most cases of common knuckle pads do not coexist with AKE; therefore, it is unknown if the underlying etiology remains the same for both entities. Unlike AKE, knuckle pads are often associated with Dupuytren contractures, repetitive trauma, or friction to the area.1,2
Presentation
Acrokeratoelastoidosis is a rare disease with onset in childhood or young adulthood. Childhood cases are inherited in an autosomal-dominant fashion.1 Adulthood onset suggests a sporadic form of inheritance. Acrokeratoelastoidosis has no gender or racial predilection.4 It presents over the thenar and hypothenar eminences, as well as the lateral digits, calcaneal tendon, and dorsal digits.1 Most often, AKE occurs symmetrically along the border separating the ventral and dorsal aspects on the palms and soles. These lesions present as small, firm, translucent papules that align linearly on the ventral-dorsal palmoplantar junction in a pattern resembling paving stones.1 Coalescence of papules into plaques has been reported. Extension of lesions to the dorsal and palmar surfaces can occur. Small circumscribed callosities may develop over the metacarpophalangeal and interphalangeal joints resembling knuckle pads.2
Histopathology
Histopathologically, AKE is distinguished by elastorrhexis—thinning, fragmenting, and rarefaction of elastin fibers—in the epidermis and reticular dermis layers.3 Acrokeratoelastoidosis also presents with orthokeratosis overlying a cuplike epithelial depression and possible epithelial acanthosis.2,5 Many cases exhibit hypergranulosis at the base of the epidermal dell. Dense basophilic granules may be seen in the peripheral cytoplasm of fibroblast cells coming from the hypothesized defect in elastin secretion.1,3,4
Differential Diagnosis
The main differential diagnosis of AKE is FAH. Clinically and histopathologically they appear identical; both diseases have cuplike epidermal depressions with overlying orthohyperkeratosis and prominent hypergranulosis.5 The elastin stains, Verhoeff-van Gieson or acid orcein stain, are imperative for distinguishing these two diseases. Although AKE demonstrates elastorrhexis and reduced elastic fibers, FAH reveals no alteration of elastic fibers. It has been suggested that FAH is a clinical variant of AKE and should be titled AKE without elastorrhexis.1
Treatment
Acrokeratoelastoidosis is asymptomatic except for mild palmoplantar hyperhidrosis and typically does not require treatment4; however, the condition can be of cosmetic concern for patients. Lesions can be treated topically with keratolytics such as tretinoin and salicylic acid. A wide variety of systemic treatments including methotrexate, prednisolone, dapsone, and acitretin have been reported with variable clinical response.2-4 Copresenting knuckle pads can be treated with urea cream, salicylic acid cream, or intralesional corticosteroids.1
- Erkek E, Koçak M, Bozdog˘an O, et al. Focal acral hyperkeratosis: a rare cutaneous disorder within the spectrum are Costa acrokeratoelastoidosis. Pediatr Dermatol. 2004;21:128-130.
- Abulafia J, Vignale R. Degenerative collagenous plaques of the hands and acrokeratoelastoidosis: pathogenesis and relationship with knuckle pads. Int J Dermatol. 2000;39:424-432.
- Nelson-Adesokan P, Mallory SB, Leonardi CL, et al. Acrokeratoelastoidosis of Costa. Int J Dermatol. 1995;34:431-433.
- Shbaklo Z, Jamaleddine NF, Kibbi AG, et al. Acrokeratoelastoidosis. Int J Dermatol. 1990;29:333-336.
- Ming M. Papules overlying finger joints—diagnosis. Arch Dermatol. 2006;142:235-240.
Case Report
An 11-year-old boy presented with atraumatic thickening of the skin on the bilateral distal and proximal interphalangeal joints of 1 year’s duration. The patient also noted small bumps of unknown duration across the bilateral palms and soles with prominence on the lateral aspects. The patient previously used over-the-counter topical wart removal treatment and topical salicylic acid with minimal improvement. The patient reported no pertinent medical or surgical history, although there was a family history of Alport syndrome, predominantly in male relatives. The patient’s father and paternal grandfather were noted to have similar lesions on the palms.
On physical examination, multiple pink to flesh-colored hyperkeratotic plaques were noted over the proximal and distal interphalangeal joints of the bilateral hands (Figure 1A). Upon close inspection, there were small flesh-colored and slightly translucent papules in a linear distribution on the palmar surfaces of the hands (Figure 2A) with predominance on the thenar and hypothenar eminences. The flexural creases of the bilateral wrists also revealed linear flesh-colored papules. The same small flesh-colored and translucent papules also were noted on the plantar surfaces of the bilateral feet (Figure 2B).
A biopsy was obtained from one of the small translucent papules on the left palm. Hematoxylin and eosin–stained sections revealed elevated compact orthokeratosis with an underlying central epidermal dell (Figure 3). A diagnosis of marginal papular keratoderma was made and further elastin staining was completed. Elastin stains showed marked thinning of the elastin fibers throughout the reticular dermis. Many elastin fibers in the reticular dermis demonstrated a fine arborizing pattern that normally is only evident in the papillary dermis (Figure 4). Acrokeratoelastoidosis (AKE) was diagnosed histopathologically, and knuckle pads were diagnosed clinically.
Because the patient was asymptomatic, he did not want treatment of AKE. He had marked improvement of the knuckle pads after 1 month with daily application of urea cream 10% (Figure 1B), and intermittent use was required for maintenance.
Comment
Etiology
Acrokeratoelastoidosis was first described in 1953 and is considered a type of palmoplantar marginal papular keratoderma.1 There is overlap within the marginal papular keratodermas that makes precise diagnosis difficult within this group. The marginal papular keratodermas on the palms and soles are a group of disorders that include AKE, focal acral hyperkeratosis (FAH), mosaic acral keratosis, degenerative collagenous plaques on the hands, and digital papular calcific elastosis. These diseases are similar in clinical and histopathological features; some argue these diseases are the same entity.2
Acrokeratoelastoidosis has been hypothesized to originate from altered elastic fiber synthesis from fibroblasts.3 Because AKE is rare, most cases of common knuckle pads do not coexist with AKE; therefore, it is unknown if the underlying etiology remains the same for both entities. Unlike AKE, knuckle pads are often associated with Dupuytren contractures, repetitive trauma, or friction to the area.1,2
Presentation
Acrokeratoelastoidosis is a rare disease with onset in childhood or young adulthood. Childhood cases are inherited in an autosomal-dominant fashion.1 Adulthood onset suggests a sporadic form of inheritance. Acrokeratoelastoidosis has no gender or racial predilection.4 It presents over the thenar and hypothenar eminences, as well as the lateral digits, calcaneal tendon, and dorsal digits.1 Most often, AKE occurs symmetrically along the border separating the ventral and dorsal aspects on the palms and soles. These lesions present as small, firm, translucent papules that align linearly on the ventral-dorsal palmoplantar junction in a pattern resembling paving stones.1 Coalescence of papules into plaques has been reported. Extension of lesions to the dorsal and palmar surfaces can occur. Small circumscribed callosities may develop over the metacarpophalangeal and interphalangeal joints resembling knuckle pads.2
Histopathology
Histopathologically, AKE is distinguished by elastorrhexis—thinning, fragmenting, and rarefaction of elastin fibers—in the epidermis and reticular dermis layers.3 Acrokeratoelastoidosis also presents with orthokeratosis overlying a cuplike epithelial depression and possible epithelial acanthosis.2,5 Many cases exhibit hypergranulosis at the base of the epidermal dell. Dense basophilic granules may be seen in the peripheral cytoplasm of fibroblast cells coming from the hypothesized defect in elastin secretion.1,3,4
Differential Diagnosis
The main differential diagnosis of AKE is FAH. Clinically and histopathologically they appear identical; both diseases have cuplike epidermal depressions with overlying orthohyperkeratosis and prominent hypergranulosis.5 The elastin stains, Verhoeff-van Gieson or acid orcein stain, are imperative for distinguishing these two diseases. Although AKE demonstrates elastorrhexis and reduced elastic fibers, FAH reveals no alteration of elastic fibers. It has been suggested that FAH is a clinical variant of AKE and should be titled AKE without elastorrhexis.1
Treatment
Acrokeratoelastoidosis is asymptomatic except for mild palmoplantar hyperhidrosis and typically does not require treatment4; however, the condition can be of cosmetic concern for patients. Lesions can be treated topically with keratolytics such as tretinoin and salicylic acid. A wide variety of systemic treatments including methotrexate, prednisolone, dapsone, and acitretin have been reported with variable clinical response.2-4 Copresenting knuckle pads can be treated with urea cream, salicylic acid cream, or intralesional corticosteroids.1
Case Report
An 11-year-old boy presented with atraumatic thickening of the skin on the bilateral distal and proximal interphalangeal joints of 1 year’s duration. The patient also noted small bumps of unknown duration across the bilateral palms and soles with prominence on the lateral aspects. The patient previously used over-the-counter topical wart removal treatment and topical salicylic acid with minimal improvement. The patient reported no pertinent medical or surgical history, although there was a family history of Alport syndrome, predominantly in male relatives. The patient’s father and paternal grandfather were noted to have similar lesions on the palms.
On physical examination, multiple pink to flesh-colored hyperkeratotic plaques were noted over the proximal and distal interphalangeal joints of the bilateral hands (Figure 1A). Upon close inspection, there were small flesh-colored and slightly translucent papules in a linear distribution on the palmar surfaces of the hands (Figure 2A) with predominance on the thenar and hypothenar eminences. The flexural creases of the bilateral wrists also revealed linear flesh-colored papules. The same small flesh-colored and translucent papules also were noted on the plantar surfaces of the bilateral feet (Figure 2B).
A biopsy was obtained from one of the small translucent papules on the left palm. Hematoxylin and eosin–stained sections revealed elevated compact orthokeratosis with an underlying central epidermal dell (Figure 3). A diagnosis of marginal papular keratoderma was made and further elastin staining was completed. Elastin stains showed marked thinning of the elastin fibers throughout the reticular dermis. Many elastin fibers in the reticular dermis demonstrated a fine arborizing pattern that normally is only evident in the papillary dermis (Figure 4). Acrokeratoelastoidosis (AKE) was diagnosed histopathologically, and knuckle pads were diagnosed clinically.
Because the patient was asymptomatic, he did not want treatment of AKE. He had marked improvement of the knuckle pads after 1 month with daily application of urea cream 10% (Figure 1B), and intermittent use was required for maintenance.
Comment
Etiology
Acrokeratoelastoidosis was first described in 1953 and is considered a type of palmoplantar marginal papular keratoderma.1 There is overlap within the marginal papular keratodermas that makes precise diagnosis difficult within this group. The marginal papular keratodermas on the palms and soles are a group of disorders that include AKE, focal acral hyperkeratosis (FAH), mosaic acral keratosis, degenerative collagenous plaques on the hands, and digital papular calcific elastosis. These diseases are similar in clinical and histopathological features; some argue these diseases are the same entity.2
Acrokeratoelastoidosis has been hypothesized to originate from altered elastic fiber synthesis from fibroblasts.3 Because AKE is rare, most cases of common knuckle pads do not coexist with AKE; therefore, it is unknown if the underlying etiology remains the same for both entities. Unlike AKE, knuckle pads are often associated with Dupuytren contractures, repetitive trauma, or friction to the area.1,2
Presentation
Acrokeratoelastoidosis is a rare disease with onset in childhood or young adulthood. Childhood cases are inherited in an autosomal-dominant fashion.1 Adulthood onset suggests a sporadic form of inheritance. Acrokeratoelastoidosis has no gender or racial predilection.4 It presents over the thenar and hypothenar eminences, as well as the lateral digits, calcaneal tendon, and dorsal digits.1 Most often, AKE occurs symmetrically along the border separating the ventral and dorsal aspects on the palms and soles. These lesions present as small, firm, translucent papules that align linearly on the ventral-dorsal palmoplantar junction in a pattern resembling paving stones.1 Coalescence of papules into plaques has been reported. Extension of lesions to the dorsal and palmar surfaces can occur. Small circumscribed callosities may develop over the metacarpophalangeal and interphalangeal joints resembling knuckle pads.2
Histopathology
Histopathologically, AKE is distinguished by elastorrhexis—thinning, fragmenting, and rarefaction of elastin fibers—in the epidermis and reticular dermis layers.3 Acrokeratoelastoidosis also presents with orthokeratosis overlying a cuplike epithelial depression and possible epithelial acanthosis.2,5 Many cases exhibit hypergranulosis at the base of the epidermal dell. Dense basophilic granules may be seen in the peripheral cytoplasm of fibroblast cells coming from the hypothesized defect in elastin secretion.1,3,4
Differential Diagnosis
The main differential diagnosis of AKE is FAH. Clinically and histopathologically they appear identical; both diseases have cuplike epidermal depressions with overlying orthohyperkeratosis and prominent hypergranulosis.5 The elastin stains, Verhoeff-van Gieson or acid orcein stain, are imperative for distinguishing these two diseases. Although AKE demonstrates elastorrhexis and reduced elastic fibers, FAH reveals no alteration of elastic fibers. It has been suggested that FAH is a clinical variant of AKE and should be titled AKE without elastorrhexis.1
Treatment
Acrokeratoelastoidosis is asymptomatic except for mild palmoplantar hyperhidrosis and typically does not require treatment4; however, the condition can be of cosmetic concern for patients. Lesions can be treated topically with keratolytics such as tretinoin and salicylic acid. A wide variety of systemic treatments including methotrexate, prednisolone, dapsone, and acitretin have been reported with variable clinical response.2-4 Copresenting knuckle pads can be treated with urea cream, salicylic acid cream, or intralesional corticosteroids.1
- Erkek E, Koçak M, Bozdog˘an O, et al. Focal acral hyperkeratosis: a rare cutaneous disorder within the spectrum are Costa acrokeratoelastoidosis. Pediatr Dermatol. 2004;21:128-130.
- Abulafia J, Vignale R. Degenerative collagenous plaques of the hands and acrokeratoelastoidosis: pathogenesis and relationship with knuckle pads. Int J Dermatol. 2000;39:424-432.
- Nelson-Adesokan P, Mallory SB, Leonardi CL, et al. Acrokeratoelastoidosis of Costa. Int J Dermatol. 1995;34:431-433.
- Shbaklo Z, Jamaleddine NF, Kibbi AG, et al. Acrokeratoelastoidosis. Int J Dermatol. 1990;29:333-336.
- Ming M. Papules overlying finger joints—diagnosis. Arch Dermatol. 2006;142:235-240.
- Erkek E, Koçak M, Bozdog˘an O, et al. Focal acral hyperkeratosis: a rare cutaneous disorder within the spectrum are Costa acrokeratoelastoidosis. Pediatr Dermatol. 2004;21:128-130.
- Abulafia J, Vignale R. Degenerative collagenous plaques of the hands and acrokeratoelastoidosis: pathogenesis and relationship with knuckle pads. Int J Dermatol. 2000;39:424-432.
- Nelson-Adesokan P, Mallory SB, Leonardi CL, et al. Acrokeratoelastoidosis of Costa. Int J Dermatol. 1995;34:431-433.
- Shbaklo Z, Jamaleddine NF, Kibbi AG, et al. Acrokeratoelastoidosis. Int J Dermatol. 1990;29:333-336.
- Ming M. Papules overlying finger joints—diagnosis. Arch Dermatol. 2006;142:235-240.
Practice Points
- Acrokeratoelastoidosis presents as small, firm, translucent, linear papules on the ventral-dorsal palmoplantar junction.
- Acrokeratoelastoidosis does not require treatment but can be treated topically with keratolytics such as tretinoin and salicylic acid.
- Knuckle pads may respond to urea cream, salicylic acid cream, or intralesional corticosteroids.
Frontal Fibrosing Alopecia: Cutaneous Associations in Women With Skin of Color
Frontal fibrosing alopecia (FFA) has been reported in association with lichen planus pigmentosus (LPP) and facial papules.1-3 Lichen planus pigmentosus is a variant of lichen planus that causes hyperpigmentation of the face, neck, and/or intertriginous areas that may be useful as a clinical indicator in the development of FFA.1 Facial papules in association with FFA are secondary to fibrosed vellus hairs.2,3 Currently, reports of concomitant FFA, LPP, and facial papules in women with skin of color are limited in the literature. This case series includes 5 women of color (Hispanic and black) who presented to our clinic with FFA and various cutaneous associations. A review of the current literature on cutaneous associations of FFA also is provided.
Case Reports
Patient 1
A 50-year-old Hispanic woman who was previously presumed to have melasma by an outside physician presented with pruritus of the scalp and eyebrows of 1 month’s duration. Physical examination revealed decreased frontal scalp hair density with perifollicular erythema and scale with thinning of the lateral eyebrows. Hyperpigmented coalesced macules (Figure 1A) and erythematous perifollicular papules were noted along the temples and on the perioral skin. Depressed forehead and temporal veins also were noted (Figure 1B). A biopsy of the scalp demonstrated perifollicular and perivascular lymphocytic inflammation and fibrosed hair follicles, and a biopsy of the perioral skin demonstrated perivascular lymphocytic inflammation with melanophages in the papillary dermis. A diagnosis of FFA with LPP was established with these biopsies.
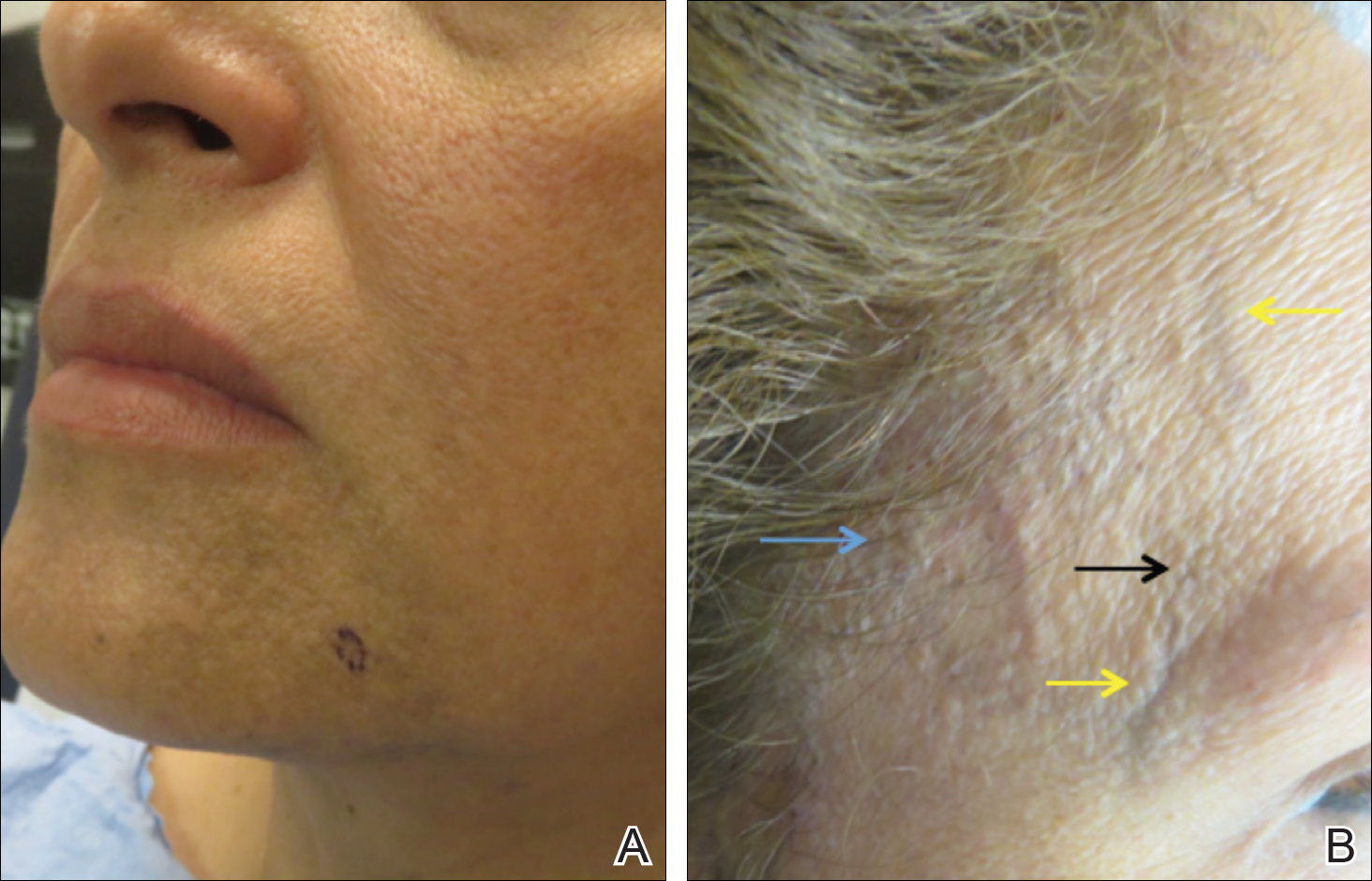
Patient 2
A 61-year-old black woman presented with asymptomatic hair loss along the frontal hairline for an unknown duration. On physical examination the frontal scalp and lateral eyebrows demonstrated decreased hair density with loss of follicular ostia. Fine, flesh-colored, monomorphic papules were scattered along the forehead and temples, and ill-defined brown pigmentation was present along the forehead, temples, and cheeks. Biopsy of the frontal scalp demonstrated patchy lichenoid inflammation with decreased number of follicles with replacement by follicular scars, confirming the diagnosis of FFA.
Patient 3
A 47-year-old Hispanic woman presented with hair loss of the frontal scalp and bilateral eyebrows with associated burning of 2 years’ duration. Physical examination demonstrated recession of the frontotemporal hairline with scattered lone hairs and thinning of the eyebrows. Innumerable flesh-colored papules were present on the forehead and temples (Figure 2A). Glabellar and eyebrow erythema was noted (Figure 2B). Biopsy of the frontal scalp demonstrated decreased terminal anagen hair follicles with perifollicular lymphoid infiltrate and fibrosis, consistent with a diagnosis of FFA. The patient was started on oral hydroxychloroquine 400 mg once daily, and 3 months later hyperpigmentation of the forehead and perioral skin was noted. The patient reported that she had facial hyperpigmentation prior to starting hydroxychloroquine and declined a biopsy.
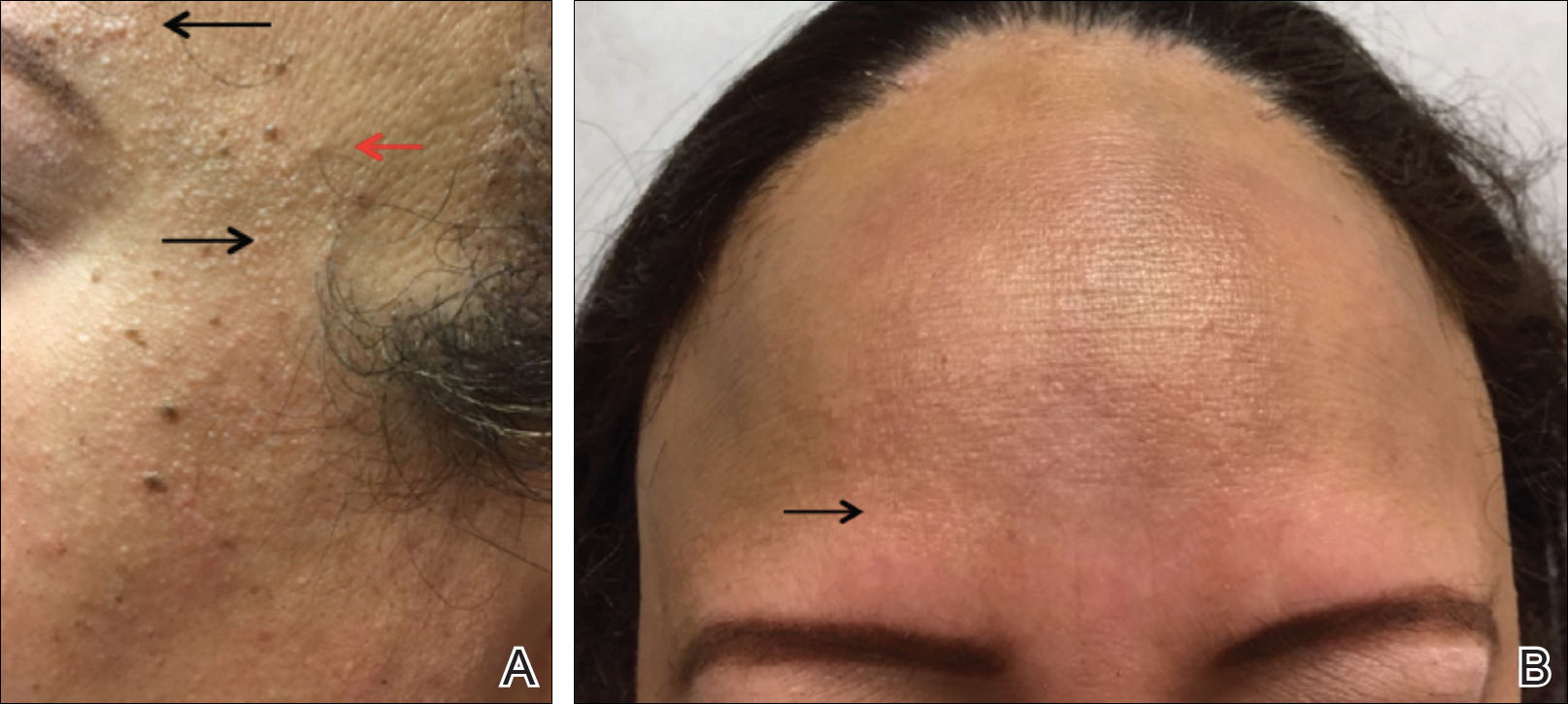
Patient 4
A 40-year-old black woman presented with brown pruritic macles of the face, neck, arms, and forearms of 4 years’ duration. She also reported hair loss on the frontal and occipital scalp, eyebrows, and arms. On physical examination, ill-defined brown macules and patches were noted on the neck (Figure 3), face, arms, and forearms. Decreased hair density was noted on the frontal and occipital scalp with follicular dropout and perifollicular hyperpigmentation. Biopsy of the scalp demonstrated perivascular lymphocytic inflammation with sparse anagen follicles and fibrous tracts, and biopsy of the neck revealed superficial perivascular inflammation with numerous melanophages in the upper dermis; these histopathologic findings were consistent with FFA and LPP, respectively.
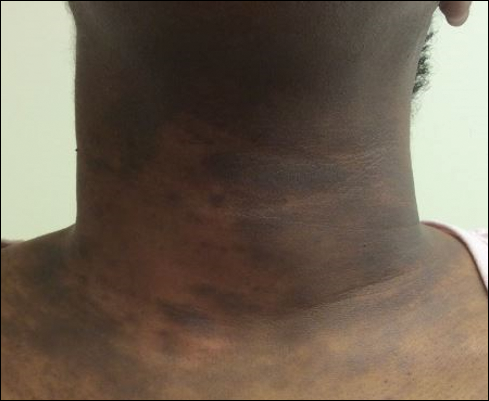
Patient 5
A 46-year-old black woman with history of hair loss presented with hyperpigmentation of the face and neck of 2 years’ duration. On physical examination decreased hair density of the frontal and vertex scalp and lateral eyebrows was noted. Flesh-colored papules were noted on the forehead and cheeks, and confluent dark brown patches were present on the temples and neck. Three punch biopsies were performed. Biopsy of the scalp revealed lymphocytic inflammation with surrounding fibroplasia with overlapping features of FFA and central centrifugal cicatricial alopecia (Figure 4). Biopsy of the neck revealed vacuolar interface dermatitis. Additionally, biopsy of a facial papule revealed lichenoid inflammation involving a vellus hair follicle. Clinical and histopathological correlation confirmed the diagnosis of FFA with LPP and facial papules.
Comment
Current understanding of FFA as a progressive, lymphocytic, scarring alopecia has expanded in recent years. Clinical observation suggests that the incidence of FFA is increasing4; however more epidemiologic data are needed. Frontal fibrosing alopecia presents clinically with symmetrical frontotemporal hair loss with lone hairs. Trichoscopy reveals perifollicular hyperkeratosis, perifollicular erythema, and follicular plugging in 72%, 66%, and 44% of cases, respectively.5 In one study (N=242), patients were classified into 3 clinical patterns of FFA: pattern I (linear) showed bandlike loss of frontal hair with normal density directly behind the hairline; pattern II (diffuse) showed loss of density behind the frontal hairline; and pattern III (double line) showed a pseudo–“fringe sign” appearance. The majority of patients were classified as either pattern I or II, with pattern II predicting a poorer prognosis.6
rontal fibrosing alopecia is increasingly recognized in men, with prevalence as high as 5%.1 Facial hair involvement, particularly of the upper lip and sideburns, is an important consideration in men.7 Most studies suggest that 80% to 90% of affected women are postmenopausal,8 though a case series presented by Dlova1 identified 27% of affected women as postmenopausal. The coexistence of premature menopause and hysterectomy in FFA patients suggests a hormonal contribution, but this association is still poorly understood.8 Epidemiologic data on ethnicity in FFA are sparse but suggest that white individuals are more likely to be affected. Frontal fibrosing alopecia also may be misdiagnosed as traction alopecia in Hispanic and black patients.8
It is prudent for physicians to assess for and recognize clinical clues to severe forms of FFA. A 2014 multicenter review of 355 patients identified 3 clinical entities that predicted more severe forms of FFA: eyelash loss (madarosis), loss of body hair, and facial papules.8 Madarosis occurs due to perifollicular inflammation and fibrosis of eyelash hair follicles. Similarly, perifollicular inflammation of body hair was present in 24% of patients (N=86), most commonly of the axillary and pubic hair. Facial papules form due to facial vellus hair inflammation and fibrosis and were identified in 14% of patients (N=49).8 These clinical findings may allow providers to predict more extensive clinical involvement of FFA.
Frontal fibrosing alopecia and LPP occur concomitantly in up 54% of patients, more commonly in darker-skinned patients.1,9,10 Lichen planus pigmentosus frequently occurs on the face and neck, most commonly in a diffuse pattern, though reticulated and macular patterns also have been identified.11 In some patients, LPP precedes the development of FFA and may be useful as a herald sign1; therefore, it is important for dermatologists to evaluate for signs of FFA when evaluating those with LPP. Thorough evaluation in patients with skin of color also is important because FFA may be misdiagnosed as traction alopecia.
Additional cutaneous associations of FFA include eyebrow loss, glabellar red dots, and prominent frontal veins. Eyebrow loss occurs secondary to fibrosis of eyebrow hair follicles and has been found in 40% to 80% of patients with FFA; it is thought to be associated with milder forms of FFA.8 Glabellar red dots correlate with histopathologic lymphocytic inflammation of vellus hair follicles.12 Additionally, frontal vein prominence has been described in FFA and is thought to be secondary to atrophy in this scarring process, perhaps worsened by local steroid treatments.13 Mucocutaneous lichen planus, rosacea, thyroid disease, vitiligo, and other autoimmune disorders also have been reported in patients with FFA.14
Conclusion
Concomitant FFA, LPP, and facial papules have been rarely reported and exemplify the spectrum of cutaneous associations with FFA, particularly in individuals with skin of color. Clinical variants and associations of FFA are broad, including predictors of poorer prognosis such as eyelash loss and vellus hair involvement seen as facial papules. Lichen planus pigmentosus is well described in association with FFA and may serve as a herald sign that frontal hair loss should not be mistaken for traction alopecia in early stages. Eyebrow loss is thought to represent milder disease. It is important for dermatologists to be aware of these findings to understand the breadth of this disease and for appropriate evaluation and management of patients with FFA.
- Dlova NC. Frontal fibrosing alopecia and lichen planus pigmentosus: is there a link? Br J Dermatol. 2013;168:439-432.
- Donati A, Molina L, Doche I, et al. Facial papules in frontal fibrosing alopecia: evidence of vellus follicle involvement. Arch Dermatol. 2011;147:1424-1427.
- Tan KT, Messenger AG. Frontal fibrosing alopecia: clinical presentations and prognosis. Br J Dermatol. 2009;160:75-79.
- Rudnicka L, Rakowska A. The increasing incidence of frontal fibrosing alopecia. in search of triggering factors. J Eur Acad Dermatol Venereol. 2017;31:1579-1580.
- Toledo-Pastrana T, Hernández MJ, Camacho Martínez FM. Perifollicular erythema as a trichoscopy sign of progression in frontal fibrosing alopecia. Int J Trichology. 2013;5:151-153.
- Moreno-Arrones OM, Saceda-Corralo D, Fonda-Pascual P, et al. Frontal fibrosing alopecia: clinical and prognostic classification. J Eur Acad Dermatol Venereol. 2017;31:1739-1745.
- Tolkachjov SN, Chaudhry HM, Camilleri MJ, et al. Frontal fibrosing alopecia among men: a clinicopathologic study of 7 cases. J Am Acad Dermatol. 2017;77:683-690.e2.
- Vañó-Galván S, Molina-Ruiz AM, Serrano-Falcón C, et al. Frontal fibrosing alopecia: a multicenter review of 355 patients. J Am Acad Dermatol. 2014;70:670-678.
- Berliner JG, McCalmont TH, Price VH, et al. Frontal fibrosing alopecia and lichen planus pigmentosus. J Am Acad Dermatol. 2014;71:E26-E27.
- Rao R, Sarda A, Khanna R, et al. Coexistence of frontal fibrosing alopecia with lichen planus pigmentosus. Int J Dermatol. 2014;53:622-624.
- Pirmez R, Duque-Estrada B, Donati A, et al. Clinical and dermoscopic features of lichen planus pigmentosus in 37 patients with frontal fibrosing alopecia. Br J Dermatol. 2016;175:1387-1390.
- Pirmez R, Donati A, Valente NS, et al. Glabellar red dots in frontal fibrosing alopecia: a further clinical sign of vellus follicle involvement. Br J Dermatol. 2014;170:745-746.
- Vañó-Galván S, Rodrigues-Barata AR, Urech M, et al. Depression of the frontal veins: a new clinical sign of frontal fibrosing alopecia. J Am Acad Dermatol. 2015;72:1087-1088.
- Pindado-Ortega C, Saceda-Corralo D, Buendía-Castaño D, et al. Frontal fibrosing alopecia and cutaneous comorbidities: a potential relationship with rosacea. J Am Acad Dermatol. 2018;78:596-597.e1.
Frontal fibrosing alopecia (FFA) has been reported in association with lichen planus pigmentosus (LPP) and facial papules.1-3 Lichen planus pigmentosus is a variant of lichen planus that causes hyperpigmentation of the face, neck, and/or intertriginous areas that may be useful as a clinical indicator in the development of FFA.1 Facial papules in association with FFA are secondary to fibrosed vellus hairs.2,3 Currently, reports of concomitant FFA, LPP, and facial papules in women with skin of color are limited in the literature. This case series includes 5 women of color (Hispanic and black) who presented to our clinic with FFA and various cutaneous associations. A review of the current literature on cutaneous associations of FFA also is provided.
Case Reports
Patient 1
A 50-year-old Hispanic woman who was previously presumed to have melasma by an outside physician presented with pruritus of the scalp and eyebrows of 1 month’s duration. Physical examination revealed decreased frontal scalp hair density with perifollicular erythema and scale with thinning of the lateral eyebrows. Hyperpigmented coalesced macules (Figure 1A) and erythematous perifollicular papules were noted along the temples and on the perioral skin. Depressed forehead and temporal veins also were noted (Figure 1B). A biopsy of the scalp demonstrated perifollicular and perivascular lymphocytic inflammation and fibrosed hair follicles, and a biopsy of the perioral skin demonstrated perivascular lymphocytic inflammation with melanophages in the papillary dermis. A diagnosis of FFA with LPP was established with these biopsies.
Patient 2
A 61-year-old black woman presented with asymptomatic hair loss along the frontal hairline for an unknown duration. On physical examination the frontal scalp and lateral eyebrows demonstrated decreased hair density with loss of follicular ostia. Fine, flesh-colored, monomorphic papules were scattered along the forehead and temples, and ill-defined brown pigmentation was present along the forehead, temples, and cheeks. Biopsy of the frontal scalp demonstrated patchy lichenoid inflammation with decreased number of follicles with replacement by follicular scars, confirming the diagnosis of FFA.
Patient 3
A 47-year-old Hispanic woman presented with hair loss of the frontal scalp and bilateral eyebrows with associated burning of 2 years’ duration. Physical examination demonstrated recession of the frontotemporal hairline with scattered lone hairs and thinning of the eyebrows. Innumerable flesh-colored papules were present on the forehead and temples (Figure 2A). Glabellar and eyebrow erythema was noted (Figure 2B). Biopsy of the frontal scalp demonstrated decreased terminal anagen hair follicles with perifollicular lymphoid infiltrate and fibrosis, consistent with a diagnosis of FFA. The patient was started on oral hydroxychloroquine 400 mg once daily, and 3 months later hyperpigmentation of the forehead and perioral skin was noted. The patient reported that she had facial hyperpigmentation prior to starting hydroxychloroquine and declined a biopsy.
Patient 4
A 40-year-old black woman presented with brown pruritic macles of the face, neck, arms, and forearms of 4 years’ duration. She also reported hair loss on the frontal and occipital scalp, eyebrows, and arms. On physical examination, ill-defined brown macules and patches were noted on the neck (Figure 3), face, arms, and forearms. Decreased hair density was noted on the frontal and occipital scalp with follicular dropout and perifollicular hyperpigmentation. Biopsy of the scalp demonstrated perivascular lymphocytic inflammation with sparse anagen follicles and fibrous tracts, and biopsy of the neck revealed superficial perivascular inflammation with numerous melanophages in the upper dermis; these histopathologic findings were consistent with FFA and LPP, respectively.
Patient 5
A 46-year-old black woman with history of hair loss presented with hyperpigmentation of the face and neck of 2 years’ duration. On physical examination decreased hair density of the frontal and vertex scalp and lateral eyebrows was noted. Flesh-colored papules were noted on the forehead and cheeks, and confluent dark brown patches were present on the temples and neck. Three punch biopsies were performed. Biopsy of the scalp revealed lymphocytic inflammation with surrounding fibroplasia with overlapping features of FFA and central centrifugal cicatricial alopecia (Figure 4). Biopsy of the neck revealed vacuolar interface dermatitis. Additionally, biopsy of a facial papule revealed lichenoid inflammation involving a vellus hair follicle. Clinical and histopathological correlation confirmed the diagnosis of FFA with LPP and facial papules.
Comment
Current understanding of FFA as a progressive, lymphocytic, scarring alopecia has expanded in recent years. Clinical observation suggests that the incidence of FFA is increasing4; however more epidemiologic data are needed. Frontal fibrosing alopecia presents clinically with symmetrical frontotemporal hair loss with lone hairs. Trichoscopy reveals perifollicular hyperkeratosis, perifollicular erythema, and follicular plugging in 72%, 66%, and 44% of cases, respectively.5 In one study (N=242), patients were classified into 3 clinical patterns of FFA: pattern I (linear) showed bandlike loss of frontal hair with normal density directly behind the hairline; pattern II (diffuse) showed loss of density behind the frontal hairline; and pattern III (double line) showed a pseudo–“fringe sign” appearance. The majority of patients were classified as either pattern I or II, with pattern II predicting a poorer prognosis.6
rontal fibrosing alopecia is increasingly recognized in men, with prevalence as high as 5%.1 Facial hair involvement, particularly of the upper lip and sideburns, is an important consideration in men.7 Most studies suggest that 80% to 90% of affected women are postmenopausal,8 though a case series presented by Dlova1 identified 27% of affected women as postmenopausal. The coexistence of premature menopause and hysterectomy in FFA patients suggests a hormonal contribution, but this association is still poorly understood.8 Epidemiologic data on ethnicity in FFA are sparse but suggest that white individuals are more likely to be affected. Frontal fibrosing alopecia also may be misdiagnosed as traction alopecia in Hispanic and black patients.8
It is prudent for physicians to assess for and recognize clinical clues to severe forms of FFA. A 2014 multicenter review of 355 patients identified 3 clinical entities that predicted more severe forms of FFA: eyelash loss (madarosis), loss of body hair, and facial papules.8 Madarosis occurs due to perifollicular inflammation and fibrosis of eyelash hair follicles. Similarly, perifollicular inflammation of body hair was present in 24% of patients (N=86), most commonly of the axillary and pubic hair. Facial papules form due to facial vellus hair inflammation and fibrosis and were identified in 14% of patients (N=49).8 These clinical findings may allow providers to predict more extensive clinical involvement of FFA.
Frontal fibrosing alopecia and LPP occur concomitantly in up 54% of patients, more commonly in darker-skinned patients.1,9,10 Lichen planus pigmentosus frequently occurs on the face and neck, most commonly in a diffuse pattern, though reticulated and macular patterns also have been identified.11 In some patients, LPP precedes the development of FFA and may be useful as a herald sign1; therefore, it is important for dermatologists to evaluate for signs of FFA when evaluating those with LPP. Thorough evaluation in patients with skin of color also is important because FFA may be misdiagnosed as traction alopecia.
Additional cutaneous associations of FFA include eyebrow loss, glabellar red dots, and prominent frontal veins. Eyebrow loss occurs secondary to fibrosis of eyebrow hair follicles and has been found in 40% to 80% of patients with FFA; it is thought to be associated with milder forms of FFA.8 Glabellar red dots correlate with histopathologic lymphocytic inflammation of vellus hair follicles.12 Additionally, frontal vein prominence has been described in FFA and is thought to be secondary to atrophy in this scarring process, perhaps worsened by local steroid treatments.13 Mucocutaneous lichen planus, rosacea, thyroid disease, vitiligo, and other autoimmune disorders also have been reported in patients with FFA.14
Conclusion
Concomitant FFA, LPP, and facial papules have been rarely reported and exemplify the spectrum of cutaneous associations with FFA, particularly in individuals with skin of color. Clinical variants and associations of FFA are broad, including predictors of poorer prognosis such as eyelash loss and vellus hair involvement seen as facial papules. Lichen planus pigmentosus is well described in association with FFA and may serve as a herald sign that frontal hair loss should not be mistaken for traction alopecia in early stages. Eyebrow loss is thought to represent milder disease. It is important for dermatologists to be aware of these findings to understand the breadth of this disease and for appropriate evaluation and management of patients with FFA.
Frontal fibrosing alopecia (FFA) has been reported in association with lichen planus pigmentosus (LPP) and facial papules.1-3 Lichen planus pigmentosus is a variant of lichen planus that causes hyperpigmentation of the face, neck, and/or intertriginous areas that may be useful as a clinical indicator in the development of FFA.1 Facial papules in association with FFA are secondary to fibrosed vellus hairs.2,3 Currently, reports of concomitant FFA, LPP, and facial papules in women with skin of color are limited in the literature. This case series includes 5 women of color (Hispanic and black) who presented to our clinic with FFA and various cutaneous associations. A review of the current literature on cutaneous associations of FFA also is provided.
Case Reports
Patient 1
A 50-year-old Hispanic woman who was previously presumed to have melasma by an outside physician presented with pruritus of the scalp and eyebrows of 1 month’s duration. Physical examination revealed decreased frontal scalp hair density with perifollicular erythema and scale with thinning of the lateral eyebrows. Hyperpigmented coalesced macules (Figure 1A) and erythematous perifollicular papules were noted along the temples and on the perioral skin. Depressed forehead and temporal veins also were noted (Figure 1B). A biopsy of the scalp demonstrated perifollicular and perivascular lymphocytic inflammation and fibrosed hair follicles, and a biopsy of the perioral skin demonstrated perivascular lymphocytic inflammation with melanophages in the papillary dermis. A diagnosis of FFA with LPP was established with these biopsies.
Patient 2
A 61-year-old black woman presented with asymptomatic hair loss along the frontal hairline for an unknown duration. On physical examination the frontal scalp and lateral eyebrows demonstrated decreased hair density with loss of follicular ostia. Fine, flesh-colored, monomorphic papules were scattered along the forehead and temples, and ill-defined brown pigmentation was present along the forehead, temples, and cheeks. Biopsy of the frontal scalp demonstrated patchy lichenoid inflammation with decreased number of follicles with replacement by follicular scars, confirming the diagnosis of FFA.
Patient 3
A 47-year-old Hispanic woman presented with hair loss of the frontal scalp and bilateral eyebrows with associated burning of 2 years’ duration. Physical examination demonstrated recession of the frontotemporal hairline with scattered lone hairs and thinning of the eyebrows. Innumerable flesh-colored papules were present on the forehead and temples (Figure 2A). Glabellar and eyebrow erythema was noted (Figure 2B). Biopsy of the frontal scalp demonstrated decreased terminal anagen hair follicles with perifollicular lymphoid infiltrate and fibrosis, consistent with a diagnosis of FFA. The patient was started on oral hydroxychloroquine 400 mg once daily, and 3 months later hyperpigmentation of the forehead and perioral skin was noted. The patient reported that she had facial hyperpigmentation prior to starting hydroxychloroquine and declined a biopsy.
Patient 4
A 40-year-old black woman presented with brown pruritic macles of the face, neck, arms, and forearms of 4 years’ duration. She also reported hair loss on the frontal and occipital scalp, eyebrows, and arms. On physical examination, ill-defined brown macules and patches were noted on the neck (Figure 3), face, arms, and forearms. Decreased hair density was noted on the frontal and occipital scalp with follicular dropout and perifollicular hyperpigmentation. Biopsy of the scalp demonstrated perivascular lymphocytic inflammation with sparse anagen follicles and fibrous tracts, and biopsy of the neck revealed superficial perivascular inflammation with numerous melanophages in the upper dermis; these histopathologic findings were consistent with FFA and LPP, respectively.
Patient 5
A 46-year-old black woman with history of hair loss presented with hyperpigmentation of the face and neck of 2 years’ duration. On physical examination decreased hair density of the frontal and vertex scalp and lateral eyebrows was noted. Flesh-colored papules were noted on the forehead and cheeks, and confluent dark brown patches were present on the temples and neck. Three punch biopsies were performed. Biopsy of the scalp revealed lymphocytic inflammation with surrounding fibroplasia with overlapping features of FFA and central centrifugal cicatricial alopecia (Figure 4). Biopsy of the neck revealed vacuolar interface dermatitis. Additionally, biopsy of a facial papule revealed lichenoid inflammation involving a vellus hair follicle. Clinical and histopathological correlation confirmed the diagnosis of FFA with LPP and facial papules.
Comment
Current understanding of FFA as a progressive, lymphocytic, scarring alopecia has expanded in recent years. Clinical observation suggests that the incidence of FFA is increasing4; however more epidemiologic data are needed. Frontal fibrosing alopecia presents clinically with symmetrical frontotemporal hair loss with lone hairs. Trichoscopy reveals perifollicular hyperkeratosis, perifollicular erythema, and follicular plugging in 72%, 66%, and 44% of cases, respectively.5 In one study (N=242), patients were classified into 3 clinical patterns of FFA: pattern I (linear) showed bandlike loss of frontal hair with normal density directly behind the hairline; pattern II (diffuse) showed loss of density behind the frontal hairline; and pattern III (double line) showed a pseudo–“fringe sign” appearance. The majority of patients were classified as either pattern I or II, with pattern II predicting a poorer prognosis.6
rontal fibrosing alopecia is increasingly recognized in men, with prevalence as high as 5%.1 Facial hair involvement, particularly of the upper lip and sideburns, is an important consideration in men.7 Most studies suggest that 80% to 90% of affected women are postmenopausal,8 though a case series presented by Dlova1 identified 27% of affected women as postmenopausal. The coexistence of premature menopause and hysterectomy in FFA patients suggests a hormonal contribution, but this association is still poorly understood.8 Epidemiologic data on ethnicity in FFA are sparse but suggest that white individuals are more likely to be affected. Frontal fibrosing alopecia also may be misdiagnosed as traction alopecia in Hispanic and black patients.8
It is prudent for physicians to assess for and recognize clinical clues to severe forms of FFA. A 2014 multicenter review of 355 patients identified 3 clinical entities that predicted more severe forms of FFA: eyelash loss (madarosis), loss of body hair, and facial papules.8 Madarosis occurs due to perifollicular inflammation and fibrosis of eyelash hair follicles. Similarly, perifollicular inflammation of body hair was present in 24% of patients (N=86), most commonly of the axillary and pubic hair. Facial papules form due to facial vellus hair inflammation and fibrosis and were identified in 14% of patients (N=49).8 These clinical findings may allow providers to predict more extensive clinical involvement of FFA.
Frontal fibrosing alopecia and LPP occur concomitantly in up 54% of patients, more commonly in darker-skinned patients.1,9,10 Lichen planus pigmentosus frequently occurs on the face and neck, most commonly in a diffuse pattern, though reticulated and macular patterns also have been identified.11 In some patients, LPP precedes the development of FFA and may be useful as a herald sign1; therefore, it is important for dermatologists to evaluate for signs of FFA when evaluating those with LPP. Thorough evaluation in patients with skin of color also is important because FFA may be misdiagnosed as traction alopecia.
Additional cutaneous associations of FFA include eyebrow loss, glabellar red dots, and prominent frontal veins. Eyebrow loss occurs secondary to fibrosis of eyebrow hair follicles and has been found in 40% to 80% of patients with FFA; it is thought to be associated with milder forms of FFA.8 Glabellar red dots correlate with histopathologic lymphocytic inflammation of vellus hair follicles.12 Additionally, frontal vein prominence has been described in FFA and is thought to be secondary to atrophy in this scarring process, perhaps worsened by local steroid treatments.13 Mucocutaneous lichen planus, rosacea, thyroid disease, vitiligo, and other autoimmune disorders also have been reported in patients with FFA.14
Conclusion
Concomitant FFA, LPP, and facial papules have been rarely reported and exemplify the spectrum of cutaneous associations with FFA, particularly in individuals with skin of color. Clinical variants and associations of FFA are broad, including predictors of poorer prognosis such as eyelash loss and vellus hair involvement seen as facial papules. Lichen planus pigmentosus is well described in association with FFA and may serve as a herald sign that frontal hair loss should not be mistaken for traction alopecia in early stages. Eyebrow loss is thought to represent milder disease. It is important for dermatologists to be aware of these findings to understand the breadth of this disease and for appropriate evaluation and management of patients with FFA.
- Dlova NC. Frontal fibrosing alopecia and lichen planus pigmentosus: is there a link? Br J Dermatol. 2013;168:439-432.
- Donati A, Molina L, Doche I, et al. Facial papules in frontal fibrosing alopecia: evidence of vellus follicle involvement. Arch Dermatol. 2011;147:1424-1427.
- Tan KT, Messenger AG. Frontal fibrosing alopecia: clinical presentations and prognosis. Br J Dermatol. 2009;160:75-79.
- Rudnicka L, Rakowska A. The increasing incidence of frontal fibrosing alopecia. in search of triggering factors. J Eur Acad Dermatol Venereol. 2017;31:1579-1580.
- Toledo-Pastrana T, Hernández MJ, Camacho Martínez FM. Perifollicular erythema as a trichoscopy sign of progression in frontal fibrosing alopecia. Int J Trichology. 2013;5:151-153.
- Moreno-Arrones OM, Saceda-Corralo D, Fonda-Pascual P, et al. Frontal fibrosing alopecia: clinical and prognostic classification. J Eur Acad Dermatol Venereol. 2017;31:1739-1745.
- Tolkachjov SN, Chaudhry HM, Camilleri MJ, et al. Frontal fibrosing alopecia among men: a clinicopathologic study of 7 cases. J Am Acad Dermatol. 2017;77:683-690.e2.
- Vañó-Galván S, Molina-Ruiz AM, Serrano-Falcón C, et al. Frontal fibrosing alopecia: a multicenter review of 355 patients. J Am Acad Dermatol. 2014;70:670-678.
- Berliner JG, McCalmont TH, Price VH, et al. Frontal fibrosing alopecia and lichen planus pigmentosus. J Am Acad Dermatol. 2014;71:E26-E27.
- Rao R, Sarda A, Khanna R, et al. Coexistence of frontal fibrosing alopecia with lichen planus pigmentosus. Int J Dermatol. 2014;53:622-624.
- Pirmez R, Duque-Estrada B, Donati A, et al. Clinical and dermoscopic features of lichen planus pigmentosus in 37 patients with frontal fibrosing alopecia. Br J Dermatol. 2016;175:1387-1390.
- Pirmez R, Donati A, Valente NS, et al. Glabellar red dots in frontal fibrosing alopecia: a further clinical sign of vellus follicle involvement. Br J Dermatol. 2014;170:745-746.
- Vañó-Galván S, Rodrigues-Barata AR, Urech M, et al. Depression of the frontal veins: a new clinical sign of frontal fibrosing alopecia. J Am Acad Dermatol. 2015;72:1087-1088.
- Pindado-Ortega C, Saceda-Corralo D, Buendía-Castaño D, et al. Frontal fibrosing alopecia and cutaneous comorbidities: a potential relationship with rosacea. J Am Acad Dermatol. 2018;78:596-597.e1.
- Dlova NC. Frontal fibrosing alopecia and lichen planus pigmentosus: is there a link? Br J Dermatol. 2013;168:439-432.
- Donati A, Molina L, Doche I, et al. Facial papules in frontal fibrosing alopecia: evidence of vellus follicle involvement. Arch Dermatol. 2011;147:1424-1427.
- Tan KT, Messenger AG. Frontal fibrosing alopecia: clinical presentations and prognosis. Br J Dermatol. 2009;160:75-79.
- Rudnicka L, Rakowska A. The increasing incidence of frontal fibrosing alopecia. in search of triggering factors. J Eur Acad Dermatol Venereol. 2017;31:1579-1580.
- Toledo-Pastrana T, Hernández MJ, Camacho Martínez FM. Perifollicular erythema as a trichoscopy sign of progression in frontal fibrosing alopecia. Int J Trichology. 2013;5:151-153.
- Moreno-Arrones OM, Saceda-Corralo D, Fonda-Pascual P, et al. Frontal fibrosing alopecia: clinical and prognostic classification. J Eur Acad Dermatol Venereol. 2017;31:1739-1745.
- Tolkachjov SN, Chaudhry HM, Camilleri MJ, et al. Frontal fibrosing alopecia among men: a clinicopathologic study of 7 cases. J Am Acad Dermatol. 2017;77:683-690.e2.
- Vañó-Galván S, Molina-Ruiz AM, Serrano-Falcón C, et al. Frontal fibrosing alopecia: a multicenter review of 355 patients. J Am Acad Dermatol. 2014;70:670-678.
- Berliner JG, McCalmont TH, Price VH, et al. Frontal fibrosing alopecia and lichen planus pigmentosus. J Am Acad Dermatol. 2014;71:E26-E27.
- Rao R, Sarda A, Khanna R, et al. Coexistence of frontal fibrosing alopecia with lichen planus pigmentosus. Int J Dermatol. 2014;53:622-624.
- Pirmez R, Duque-Estrada B, Donati A, et al. Clinical and dermoscopic features of lichen planus pigmentosus in 37 patients with frontal fibrosing alopecia. Br J Dermatol. 2016;175:1387-1390.
- Pirmez R, Donati A, Valente NS, et al. Glabellar red dots in frontal fibrosing alopecia: a further clinical sign of vellus follicle involvement. Br J Dermatol. 2014;170:745-746.
- Vañó-Galván S, Rodrigues-Barata AR, Urech M, et al. Depression of the frontal veins: a new clinical sign of frontal fibrosing alopecia. J Am Acad Dermatol. 2015;72:1087-1088.
- Pindado-Ortega C, Saceda-Corralo D, Buendía-Castaño D, et al. Frontal fibrosing alopecia and cutaneous comorbidities: a potential relationship with rosacea. J Am Acad Dermatol. 2018;78:596-597.e1.
Practice Points
- Frontal fibrosing alopecia (FFA) is associated with lichen planus pigmentosus, especially in patients with skin of color.
- Patients with FFA should be evaluated for additional cutaneous features including facial papules, glabellar red dots, and depressed frontal veins.