User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
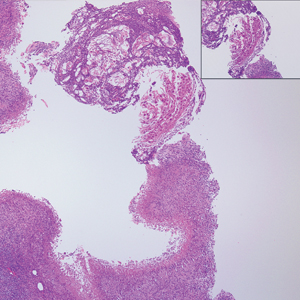
Solitary Nodule on the Thigh
The Diagnosis: Ruptured Molluscum
Molluscum contagiosum (MC) is caused by a DNA virus (MC virus) belonging to the poxvirus family. Molluscum contagiosum is common and predominantly seen in children and young adults. In sexually active adults, the lesions commonly occur in the genital region, abdomen, and inner thighs. In immunocompromised individuals, including those with AIDS, the lesions are more extensive and may cause disfigurement.1 Molluscum contagiosum involving epidermoid cysts has been reported.2
Histopathologically, MC can be classified as noninflammatory or inflammatory. In noninflamed lesions, multiple large, intracytoplasmic, eosinophilic inclusions (Henderson-Paterson bodies) appear within the lobulated endophytic and hyperplastic epidermis. Ultrastructurally, these bodies show membrane-bound collections of MC virus.1 Replicating Henderson-Paterson bodies can result in rupture and inflammation. This case demonstrates a palisading granuloma containing keratin with few Henderson-Paterson bodies (quiz image) due to prior rupture of a molluscum or molluscoid cyst.
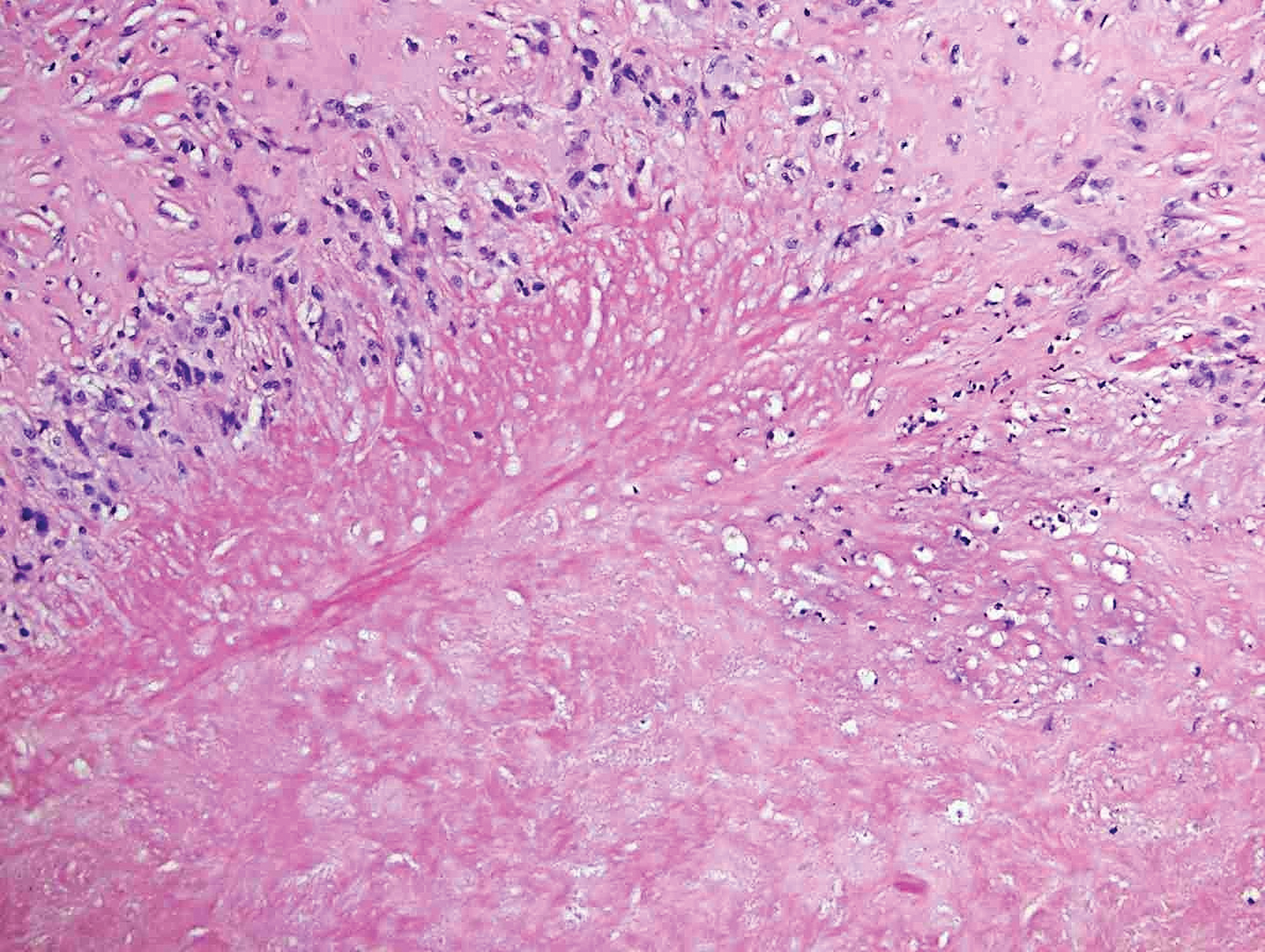
Rheumatoid nodules, the most characteristic histopathologic lesions of rheumatoid arthritis, are most commonly found in the subcutis at points of pressure and may occur in connective tissue of numerous organs. Rheumatoid nodules are firm, nontender, and mobile within the subcutaneous tissue but may be fixed to underlying structures including the periosteum, tendons, or bursae.3,4 Occasionally, superficial nodules may perforate the epidermis.5 The inner central necrobiotic zone appears as intensely eosinophilic, amorphous fibrin and other cellular debris. This central area is surrounded by histiocytes in a palisaded configuration (Figure 1). Multinucleated foreign body giant cells also may be present. Occasionally, mast cells, eosinophils, and neutrophils are present.6,7
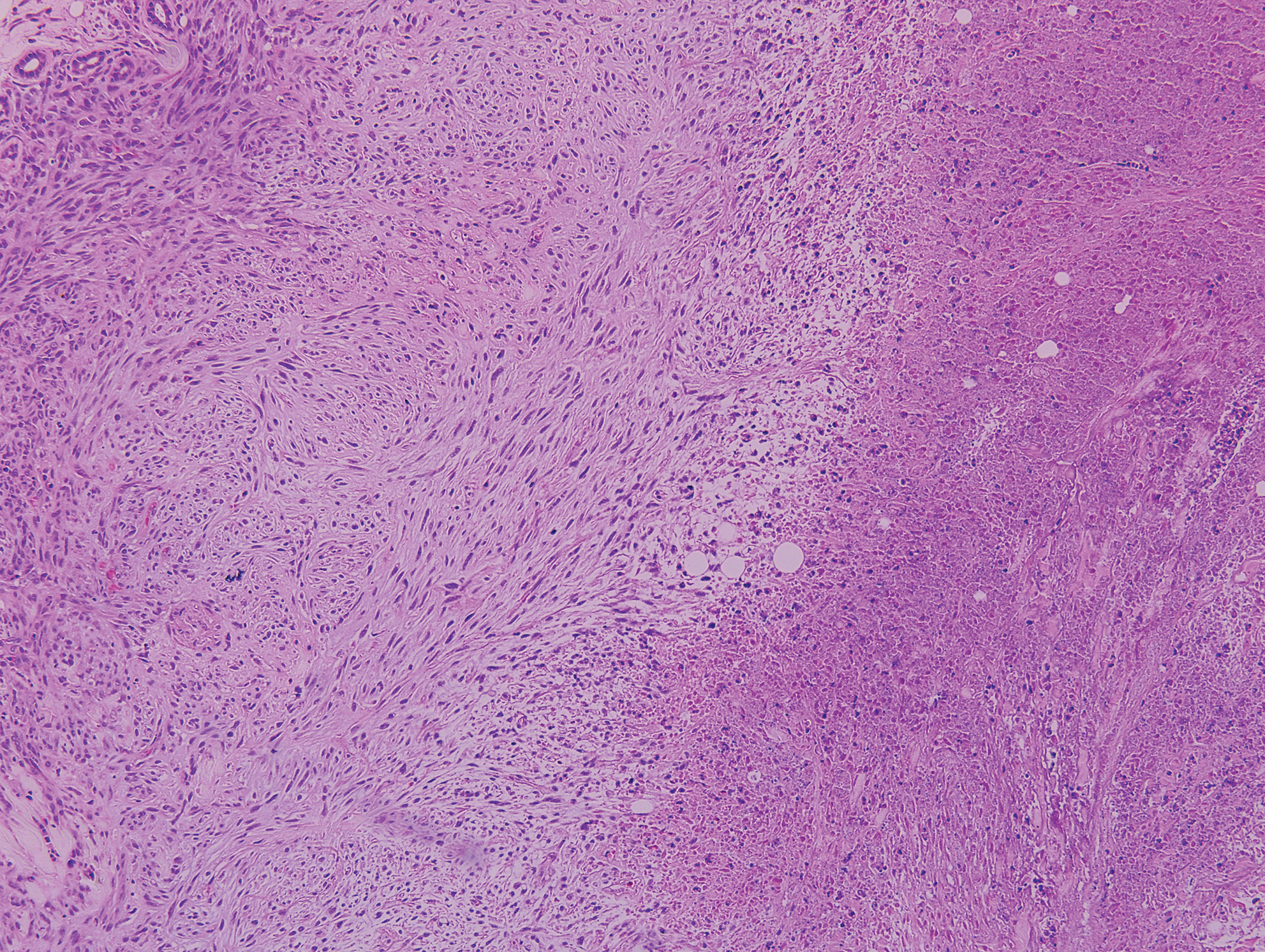
Lupus miliaris disseminatus faciei presents with multiple discrete, smooth, yellow-brown to red, dome-shaped papules. The lesions typically are located on the central and lateral sides of the face and infrequently involve the neck. Other sites including the axillae, arms, hands, legs, and groin occasionally can be involved. Diascopy may reveal an apple jelly color.8,9 The histopathologic hallmark of lupus miliaris disseminatus faciei is an epithelioid cell granuloma with central necrosis (Figure 2).

Epithelioid sarcoma (ES) is a soft tissue tumor with a known propensity for local recurrence, regional lymph node involvement, sporotrichoid spread, and distant metastases.10 The name was coined by Enzinger11 in 1970 during a review of 62 cases of a “peculiar form of sarcoma that has repeatedly been confused with a chronic inflammatory process, a necrotizing granuloma, and a squamous cell carcinoma.” Epithelioid sarcoma tends to grow slowly in a nodular or multinodular manner along fascial structures and tendons, often with central necrosis and ulceration of the overlying skin. Histopathologically, classic ES shows nodular masses of uniform plump epithelioid cells with abundant eosinophilic cytoplasm and prominent central necrosis. A biphasic pattern is typical with spindle cells merging with epithelioid cells. Cellular atypia is relatively mild and mitoses are rare (Figure 3). Recurrent or metastatic lesions can show a greater degree of pleomorphism.12 Given the low-grade atypia in early lesions, this sarcoma is easily misdiagnosed as granulomatous dermatitis. Immunohistochemically, the majority of ES cases are positive for cytokeratins and epithelial membrane antigen; SMARCB1/INI-1 expression is characteristically lost.13
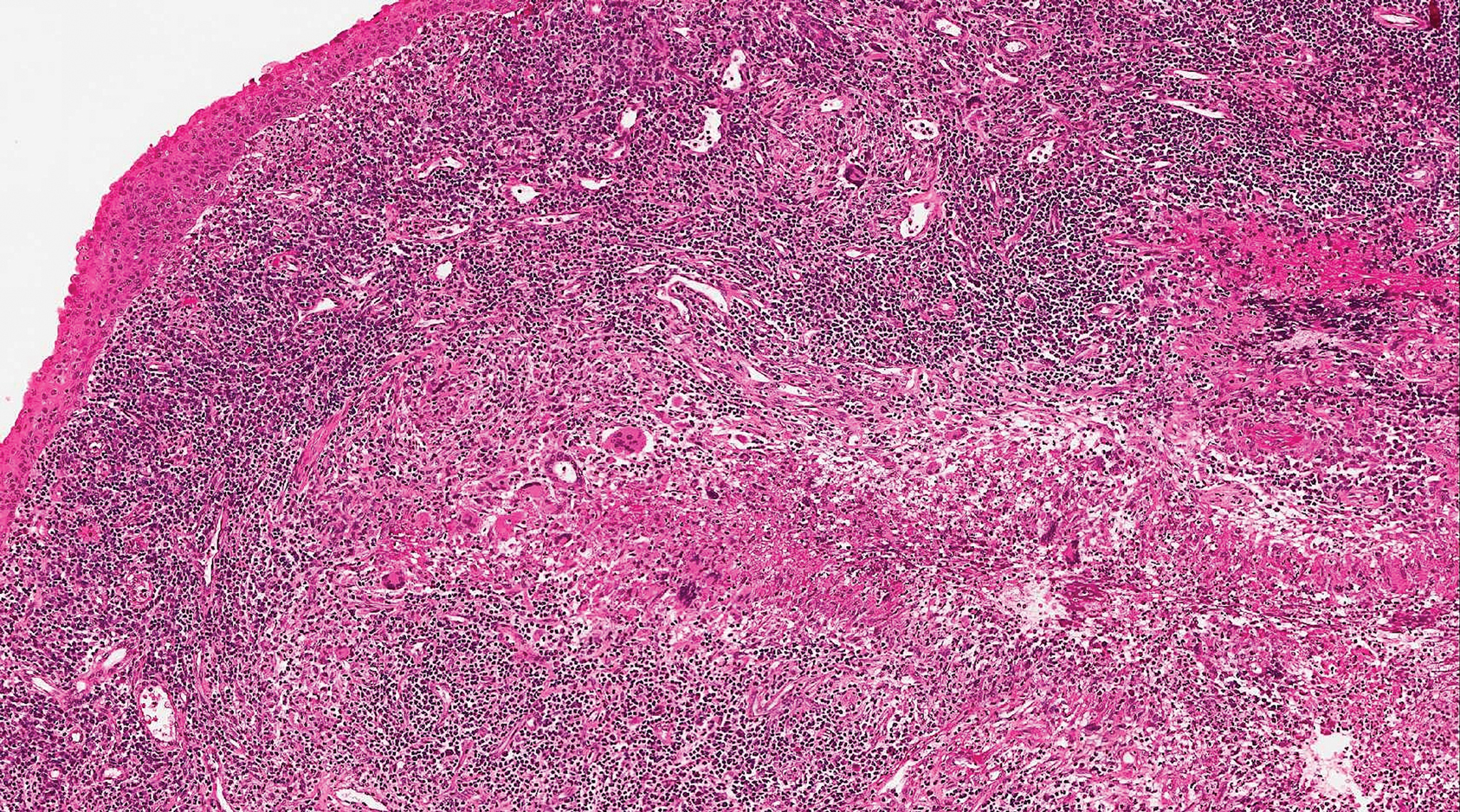
Granulomatosis with polyangiitis (formerly Wegener granulomatosis) is an autoimmune vasculitis highly associated with antineutrophil cytoplasmic antibodies. Clinical manifestations include systemic necrotizing vasculitis; necrotizing glomerulonephritis; and granulomatous inflammation, which predominantly involves the upper respiratory tract, skin, and mucosa.14,15 Skin involvement may be the initial manifestation of the disease and consists of palpable purpura, papules, ulcerations, vesicles, subcutaneous nodules, necrotizing ulcerations, papulonecrotic lesions, and petechiae. None of the findings are pathognomonic. The cutaneous histopathologic spectrum includes leukocytoclastic vasculitis, extravascular palisading granulomas, and granulomatous vasculitis.16 In the acute lesions of granulomatosis with polyangiitis, the predominant pattern of inflammation is not granulomatous but purulent with the appearance of an abscess. As it evolves, it develops a central zone of necrosis with extensive karyorrhectic debris and palisades of macrophages with scattered multinucleated giant cells (Figure 4).17
1. Nandhini G, Rajkumar K, Kanth KS, et al. Molluscum contagiosum in a 12-year-old child—report of a case and review of literature. J Int Oral Health. 2015;7:63-66.
2. Phelps A, Murphy M, Elaba Z, et al. Molluscum contagiosum virus infection in benign cutaneous epithelial cystic lesions-report of 2 cases with different pathogenesis? Am J Dermatopathol. 2010;32:740-742.
3. Sayah A, English JC 3rd. Rheumatoid arthritis: a review of the cutaneous manifestations. J Am Acad Dermatol. 2005;53:191-209; quiz 210-192.
4. Sibbitt WL Jr, Williams RC Jr. Cutaneous manifestations of rheumatoid arthritis. Int J Dermatol. 1982;21:563-572.
5. Barzilai A, Huszar M, Shpiro D, et al. Pseudorheumatoid nodules in adults: a juxta-articular form of nodular granuloma annulare. Am J Dermatopathol. 2005;27:1-5.
6. Garcia-Patos V. Rheumatoid nodule. Semin Cutan Med Surg. 2007;26:100-107.
7. Patterson JW. Rheumatoid nodule and subcutaneous granuloma annulare. a comparative histologic study. Am J Dermatopathol. 1988;10:1-8.
8. Sehgal VN, Srivastava G, Aggarwal AK, et al. Lupus miliaris disseminatus faciei part II: an overview. Skinmed. 2005;4:234-238.
9. Cymerman R, Rosenstein R, Shvartsbeyn M, et al. Lupus miliaris disseminatus faciei. Dermatol Online J. 2015;21. pii:13030/qt6b83q5gp.
10. Sobanko JF, Meijer L, Nigra TP. Epithelioid sarcoma: a review and update. J Clin Aesthet Dermatol. 2009;2:49-54.
11. Enzinger FM. Epitheloid sarcoma. a sarcoma simulating a granuloma or a carcinoma. Cancer. 1970;26:1029-1041.
12. Fisher C. Epithelioid sarcoma of Enzinger. Adv Anat Pathol. 2006;13:114-121.
13. Miettinen M, Fanburg-Smith JC, Virolainen M, et al. Epithelioid sarcoma: an immunohistochemical analysis of 112 classical and variant cases and a discussion of the differential diagnosis. Hum Pathol. 1999;30:934-942.
14. Lutalo PM, D’Cruz DP. Diagnosis and classification of granulomatosis with polyangiitis (aka Wegener’s granulomatosis)[published online January 29, 2014]. J Autoimmun. 2014;48-49:94-98.
15. Frances C, Du LT, Piette JC, et al. Wegener’s granulomatosis. dermatological manifestations in 75 cases with clinicopathologic correlation. Arch Dermatol. 1994;130:861-867.
16. Daoud MS, Gibson LE, DeRemee RA, et al. Cutaneous Wegener’s granulomatosis: clinical, histopathologic, and immunopathologic features of thirty patients. J Am Acad Dermatol. 1994;31:605-612.
17. Jennette JC. Nomenclature and classification of vasculitis: lessons learned from granulomatosis with polyangiitis (Wegener’s granulomatosis). Clin Exp Immunol. 2011;164 (suppl 1):7-10.
The Diagnosis: Ruptured Molluscum
Molluscum contagiosum (MC) is caused by a DNA virus (MC virus) belonging to the poxvirus family. Molluscum contagiosum is common and predominantly seen in children and young adults. In sexually active adults, the lesions commonly occur in the genital region, abdomen, and inner thighs. In immunocompromised individuals, including those with AIDS, the lesions are more extensive and may cause disfigurement.1 Molluscum contagiosum involving epidermoid cysts has been reported.2
Histopathologically, MC can be classified as noninflammatory or inflammatory. In noninflamed lesions, multiple large, intracytoplasmic, eosinophilic inclusions (Henderson-Paterson bodies) appear within the lobulated endophytic and hyperplastic epidermis. Ultrastructurally, these bodies show membrane-bound collections of MC virus.1 Replicating Henderson-Paterson bodies can result in rupture and inflammation. This case demonstrates a palisading granuloma containing keratin with few Henderson-Paterson bodies (quiz image) due to prior rupture of a molluscum or molluscoid cyst.
Rheumatoid nodules, the most characteristic histopathologic lesions of rheumatoid arthritis, are most commonly found in the subcutis at points of pressure and may occur in connective tissue of numerous organs. Rheumatoid nodules are firm, nontender, and mobile within the subcutaneous tissue but may be fixed to underlying structures including the periosteum, tendons, or bursae.3,4 Occasionally, superficial nodules may perforate the epidermis.5 The inner central necrobiotic zone appears as intensely eosinophilic, amorphous fibrin and other cellular debris. This central area is surrounded by histiocytes in a palisaded configuration (Figure 1). Multinucleated foreign body giant cells also may be present. Occasionally, mast cells, eosinophils, and neutrophils are present.6,7
Lupus miliaris disseminatus faciei presents with multiple discrete, smooth, yellow-brown to red, dome-shaped papules. The lesions typically are located on the central and lateral sides of the face and infrequently involve the neck. Other sites including the axillae, arms, hands, legs, and groin occasionally can be involved. Diascopy may reveal an apple jelly color.8,9 The histopathologic hallmark of lupus miliaris disseminatus faciei is an epithelioid cell granuloma with central necrosis (Figure 2).
Epithelioid sarcoma (ES) is a soft tissue tumor with a known propensity for local recurrence, regional lymph node involvement, sporotrichoid spread, and distant metastases.10 The name was coined by Enzinger11 in 1970 during a review of 62 cases of a “peculiar form of sarcoma that has repeatedly been confused with a chronic inflammatory process, a necrotizing granuloma, and a squamous cell carcinoma.” Epithelioid sarcoma tends to grow slowly in a nodular or multinodular manner along fascial structures and tendons, often with central necrosis and ulceration of the overlying skin. Histopathologically, classic ES shows nodular masses of uniform plump epithelioid cells with abundant eosinophilic cytoplasm and prominent central necrosis. A biphasic pattern is typical with spindle cells merging with epithelioid cells. Cellular atypia is relatively mild and mitoses are rare (Figure 3). Recurrent or metastatic lesions can show a greater degree of pleomorphism.12 Given the low-grade atypia in early lesions, this sarcoma is easily misdiagnosed as granulomatous dermatitis. Immunohistochemically, the majority of ES cases are positive for cytokeratins and epithelial membrane antigen; SMARCB1/INI-1 expression is characteristically lost.13
Granulomatosis with polyangiitis (formerly Wegener granulomatosis) is an autoimmune vasculitis highly associated with antineutrophil cytoplasmic antibodies. Clinical manifestations include systemic necrotizing vasculitis; necrotizing glomerulonephritis; and granulomatous inflammation, which predominantly involves the upper respiratory tract, skin, and mucosa.14,15 Skin involvement may be the initial manifestation of the disease and consists of palpable purpura, papules, ulcerations, vesicles, subcutaneous nodules, necrotizing ulcerations, papulonecrotic lesions, and petechiae. None of the findings are pathognomonic. The cutaneous histopathologic spectrum includes leukocytoclastic vasculitis, extravascular palisading granulomas, and granulomatous vasculitis.16 In the acute lesions of granulomatosis with polyangiitis, the predominant pattern of inflammation is not granulomatous but purulent with the appearance of an abscess. As it evolves, it develops a central zone of necrosis with extensive karyorrhectic debris and palisades of macrophages with scattered multinucleated giant cells (Figure 4).17
The Diagnosis: Ruptured Molluscum
Molluscum contagiosum (MC) is caused by a DNA virus (MC virus) belonging to the poxvirus family. Molluscum contagiosum is common and predominantly seen in children and young adults. In sexually active adults, the lesions commonly occur in the genital region, abdomen, and inner thighs. In immunocompromised individuals, including those with AIDS, the lesions are more extensive and may cause disfigurement.1 Molluscum contagiosum involving epidermoid cysts has been reported.2
Histopathologically, MC can be classified as noninflammatory or inflammatory. In noninflamed lesions, multiple large, intracytoplasmic, eosinophilic inclusions (Henderson-Paterson bodies) appear within the lobulated endophytic and hyperplastic epidermis. Ultrastructurally, these bodies show membrane-bound collections of MC virus.1 Replicating Henderson-Paterson bodies can result in rupture and inflammation. This case demonstrates a palisading granuloma containing keratin with few Henderson-Paterson bodies (quiz image) due to prior rupture of a molluscum or molluscoid cyst.
Rheumatoid nodules, the most characteristic histopathologic lesions of rheumatoid arthritis, are most commonly found in the subcutis at points of pressure and may occur in connective tissue of numerous organs. Rheumatoid nodules are firm, nontender, and mobile within the subcutaneous tissue but may be fixed to underlying structures including the periosteum, tendons, or bursae.3,4 Occasionally, superficial nodules may perforate the epidermis.5 The inner central necrobiotic zone appears as intensely eosinophilic, amorphous fibrin and other cellular debris. This central area is surrounded by histiocytes in a palisaded configuration (Figure 1). Multinucleated foreign body giant cells also may be present. Occasionally, mast cells, eosinophils, and neutrophils are present.6,7
Lupus miliaris disseminatus faciei presents with multiple discrete, smooth, yellow-brown to red, dome-shaped papules. The lesions typically are located on the central and lateral sides of the face and infrequently involve the neck. Other sites including the axillae, arms, hands, legs, and groin occasionally can be involved. Diascopy may reveal an apple jelly color.8,9 The histopathologic hallmark of lupus miliaris disseminatus faciei is an epithelioid cell granuloma with central necrosis (Figure 2).
Epithelioid sarcoma (ES) is a soft tissue tumor with a known propensity for local recurrence, regional lymph node involvement, sporotrichoid spread, and distant metastases.10 The name was coined by Enzinger11 in 1970 during a review of 62 cases of a “peculiar form of sarcoma that has repeatedly been confused with a chronic inflammatory process, a necrotizing granuloma, and a squamous cell carcinoma.” Epithelioid sarcoma tends to grow slowly in a nodular or multinodular manner along fascial structures and tendons, often with central necrosis and ulceration of the overlying skin. Histopathologically, classic ES shows nodular masses of uniform plump epithelioid cells with abundant eosinophilic cytoplasm and prominent central necrosis. A biphasic pattern is typical with spindle cells merging with epithelioid cells. Cellular atypia is relatively mild and mitoses are rare (Figure 3). Recurrent or metastatic lesions can show a greater degree of pleomorphism.12 Given the low-grade atypia in early lesions, this sarcoma is easily misdiagnosed as granulomatous dermatitis. Immunohistochemically, the majority of ES cases are positive for cytokeratins and epithelial membrane antigen; SMARCB1/INI-1 expression is characteristically lost.13
Granulomatosis with polyangiitis (formerly Wegener granulomatosis) is an autoimmune vasculitis highly associated with antineutrophil cytoplasmic antibodies. Clinical manifestations include systemic necrotizing vasculitis; necrotizing glomerulonephritis; and granulomatous inflammation, which predominantly involves the upper respiratory tract, skin, and mucosa.14,15 Skin involvement may be the initial manifestation of the disease and consists of palpable purpura, papules, ulcerations, vesicles, subcutaneous nodules, necrotizing ulcerations, papulonecrotic lesions, and petechiae. None of the findings are pathognomonic. The cutaneous histopathologic spectrum includes leukocytoclastic vasculitis, extravascular palisading granulomas, and granulomatous vasculitis.16 In the acute lesions of granulomatosis with polyangiitis, the predominant pattern of inflammation is not granulomatous but purulent with the appearance of an abscess. As it evolves, it develops a central zone of necrosis with extensive karyorrhectic debris and palisades of macrophages with scattered multinucleated giant cells (Figure 4).17
1. Nandhini G, Rajkumar K, Kanth KS, et al. Molluscum contagiosum in a 12-year-old child—report of a case and review of literature. J Int Oral Health. 2015;7:63-66.
2. Phelps A, Murphy M, Elaba Z, et al. Molluscum contagiosum virus infection in benign cutaneous epithelial cystic lesions-report of 2 cases with different pathogenesis? Am J Dermatopathol. 2010;32:740-742.
3. Sayah A, English JC 3rd. Rheumatoid arthritis: a review of the cutaneous manifestations. J Am Acad Dermatol. 2005;53:191-209; quiz 210-192.
4. Sibbitt WL Jr, Williams RC Jr. Cutaneous manifestations of rheumatoid arthritis. Int J Dermatol. 1982;21:563-572.
5. Barzilai A, Huszar M, Shpiro D, et al. Pseudorheumatoid nodules in adults: a juxta-articular form of nodular granuloma annulare. Am J Dermatopathol. 2005;27:1-5.
6. Garcia-Patos V. Rheumatoid nodule. Semin Cutan Med Surg. 2007;26:100-107.
7. Patterson JW. Rheumatoid nodule and subcutaneous granuloma annulare. a comparative histologic study. Am J Dermatopathol. 1988;10:1-8.
8. Sehgal VN, Srivastava G, Aggarwal AK, et al. Lupus miliaris disseminatus faciei part II: an overview. Skinmed. 2005;4:234-238.
9. Cymerman R, Rosenstein R, Shvartsbeyn M, et al. Lupus miliaris disseminatus faciei. Dermatol Online J. 2015;21. pii:13030/qt6b83q5gp.
10. Sobanko JF, Meijer L, Nigra TP. Epithelioid sarcoma: a review and update. J Clin Aesthet Dermatol. 2009;2:49-54.
11. Enzinger FM. Epitheloid sarcoma. a sarcoma simulating a granuloma or a carcinoma. Cancer. 1970;26:1029-1041.
12. Fisher C. Epithelioid sarcoma of Enzinger. Adv Anat Pathol. 2006;13:114-121.
13. Miettinen M, Fanburg-Smith JC, Virolainen M, et al. Epithelioid sarcoma: an immunohistochemical analysis of 112 classical and variant cases and a discussion of the differential diagnosis. Hum Pathol. 1999;30:934-942.
14. Lutalo PM, D’Cruz DP. Diagnosis and classification of granulomatosis with polyangiitis (aka Wegener’s granulomatosis)[published online January 29, 2014]. J Autoimmun. 2014;48-49:94-98.
15. Frances C, Du LT, Piette JC, et al. Wegener’s granulomatosis. dermatological manifestations in 75 cases with clinicopathologic correlation. Arch Dermatol. 1994;130:861-867.
16. Daoud MS, Gibson LE, DeRemee RA, et al. Cutaneous Wegener’s granulomatosis: clinical, histopathologic, and immunopathologic features of thirty patients. J Am Acad Dermatol. 1994;31:605-612.
17. Jennette JC. Nomenclature and classification of vasculitis: lessons learned from granulomatosis with polyangiitis (Wegener’s granulomatosis). Clin Exp Immunol. 2011;164 (suppl 1):7-10.
1. Nandhini G, Rajkumar K, Kanth KS, et al. Molluscum contagiosum in a 12-year-old child—report of a case and review of literature. J Int Oral Health. 2015;7:63-66.
2. Phelps A, Murphy M, Elaba Z, et al. Molluscum contagiosum virus infection in benign cutaneous epithelial cystic lesions-report of 2 cases with different pathogenesis? Am J Dermatopathol. 2010;32:740-742.
3. Sayah A, English JC 3rd. Rheumatoid arthritis: a review of the cutaneous manifestations. J Am Acad Dermatol. 2005;53:191-209; quiz 210-192.
4. Sibbitt WL Jr, Williams RC Jr. Cutaneous manifestations of rheumatoid arthritis. Int J Dermatol. 1982;21:563-572.
5. Barzilai A, Huszar M, Shpiro D, et al. Pseudorheumatoid nodules in adults: a juxta-articular form of nodular granuloma annulare. Am J Dermatopathol. 2005;27:1-5.
6. Garcia-Patos V. Rheumatoid nodule. Semin Cutan Med Surg. 2007;26:100-107.
7. Patterson JW. Rheumatoid nodule and subcutaneous granuloma annulare. a comparative histologic study. Am J Dermatopathol. 1988;10:1-8.
8. Sehgal VN, Srivastava G, Aggarwal AK, et al. Lupus miliaris disseminatus faciei part II: an overview. Skinmed. 2005;4:234-238.
9. Cymerman R, Rosenstein R, Shvartsbeyn M, et al. Lupus miliaris disseminatus faciei. Dermatol Online J. 2015;21. pii:13030/qt6b83q5gp.
10. Sobanko JF, Meijer L, Nigra TP. Epithelioid sarcoma: a review and update. J Clin Aesthet Dermatol. 2009;2:49-54.
11. Enzinger FM. Epitheloid sarcoma. a sarcoma simulating a granuloma or a carcinoma. Cancer. 1970;26:1029-1041.
12. Fisher C. Epithelioid sarcoma of Enzinger. Adv Anat Pathol. 2006;13:114-121.
13. Miettinen M, Fanburg-Smith JC, Virolainen M, et al. Epithelioid sarcoma: an immunohistochemical analysis of 112 classical and variant cases and a discussion of the differential diagnosis. Hum Pathol. 1999;30:934-942.
14. Lutalo PM, D’Cruz DP. Diagnosis and classification of granulomatosis with polyangiitis (aka Wegener’s granulomatosis)[published online January 29, 2014]. J Autoimmun. 2014;48-49:94-98.
15. Frances C, Du LT, Piette JC, et al. Wegener’s granulomatosis. dermatological manifestations in 75 cases with clinicopathologic correlation. Arch Dermatol. 1994;130:861-867.
16. Daoud MS, Gibson LE, DeRemee RA, et al. Cutaneous Wegener’s granulomatosis: clinical, histopathologic, and immunopathologic features of thirty patients. J Am Acad Dermatol. 1994;31:605-612.
17. Jennette JC. Nomenclature and classification of vasculitis: lessons learned from granulomatosis with polyangiitis (Wegener’s granulomatosis). Clin Exp Immunol. 2011;164 (suppl 1):7-10.
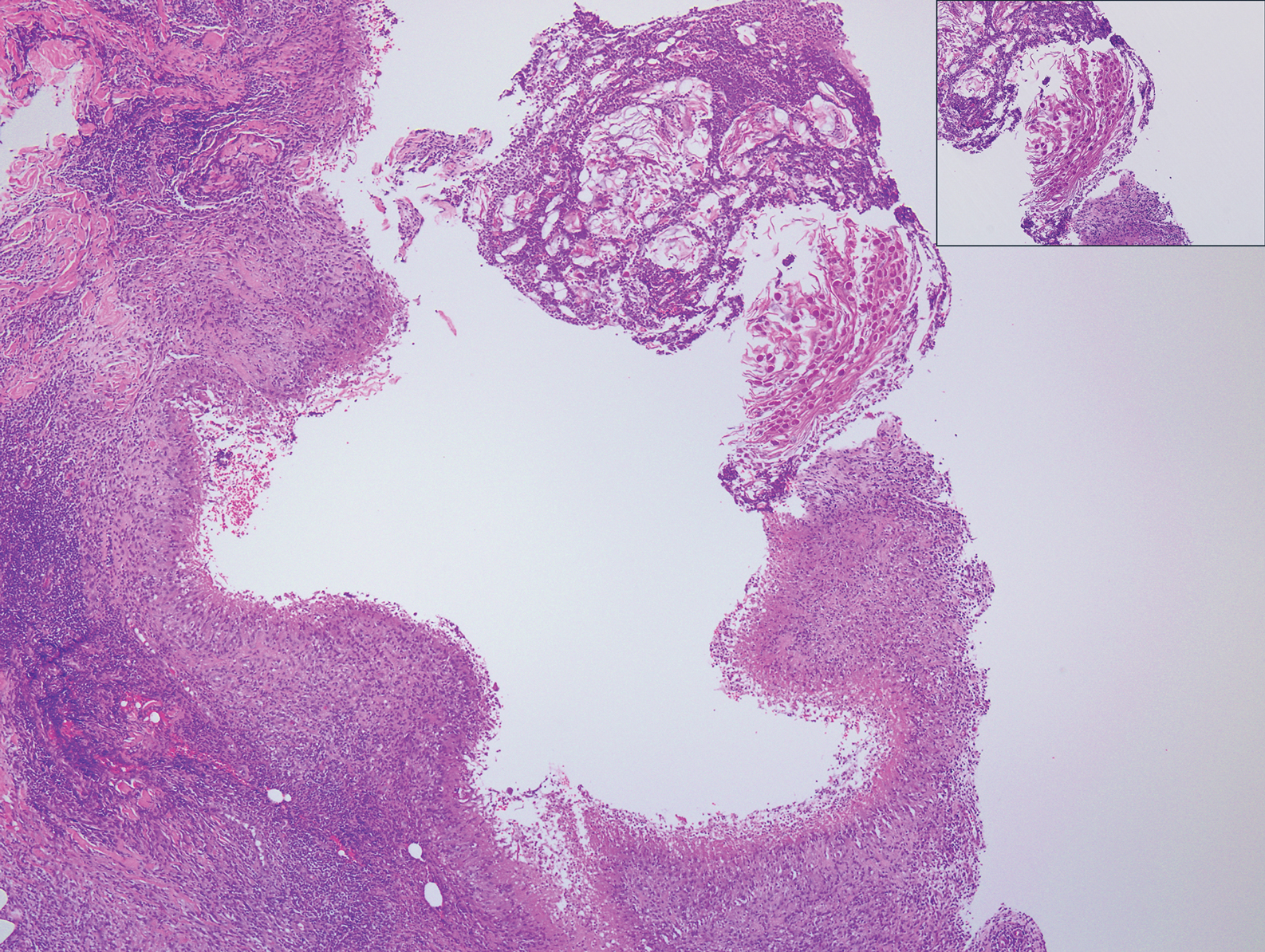
A 17-year-old adolescent girl presented with a discrete nodule on the thigh.
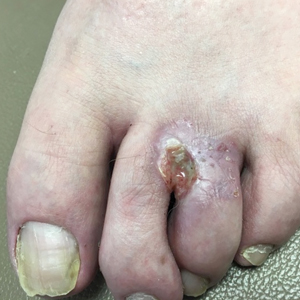
Nonhealing Eroded Plaque on an Interdigital Web Space of the Foot
The Diagnosis: Basal Cell Nevus Syndrome
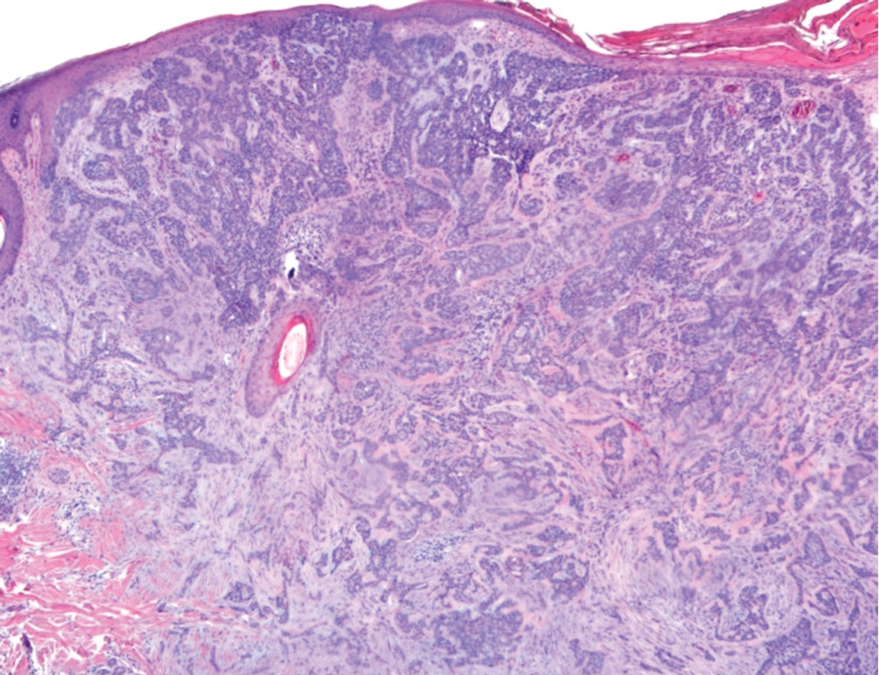
Given the patient’s history of numerous basal cell carcinomas (BCCs), odontogenic keratocysts, palmar pits, and a nonhealing ulcer, the clinical presentation was highly suggestive of interdigital BCC in the setting of basal cell nevus syndrome (BCNS). A shave biopsy was performed revealing islands of basaloid cells with peripheral palisading and a retraction artifact surrounded by fibromyxoid stroma, consistent with nodular and infiltrative BCC (Figure 1).
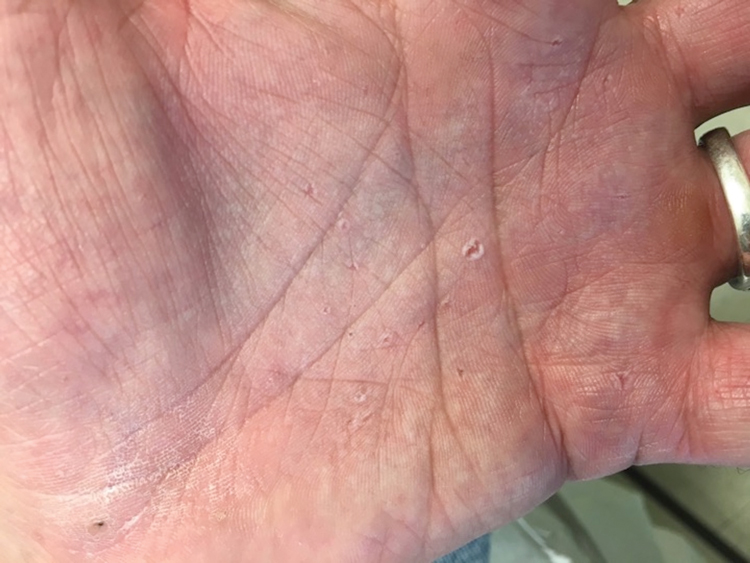
Basal cell nevus syndrome (also known as Gorlin syndrome) is a rare neurocutaneous syndrome that manifests with multiple BCCs; palmar and plantar pits (Figure 2); central nervous system tumors; and skeletal anomalies including jaw cysts, macrocephaly, frontal bossing, and bifid ribs.1 It is an autosomal-dominant condition caused by mutations in the PTCH1 gene, a tumor suppressor gene involved in the Hedgehog signaling pathway.2 Basal cell carcinoma is the most distinctive feature of BCNS, causing notable morbidity. Tumors typically present between puberty and 35 years of age, and patients can have anywhere from a few to thousands of tumors. They rarely become locally aggressive; however, with radiation therapy, proliferation and local invasion may occur within a few years. Therefore, radiotherapy should be avoided in these patients.1
Although the most common sites for BCCs in BCNS are the head, neck, and back, there is a higher rate of occurrence on sun-protected areas in BCNS compared to the general population.3 Our patient presented with interdigital BCC of the foot, which is an extremely rare occurrence. PubMed and Ovid searches using the terms basal cell carcinoma, BCC, foot, interdigital, and nonmelanoma skin cancer revealed only 3 cases of interdigital BCC of the foot. One case was associated with prior surgical trauma, the second presented as a junctional nevus, and the third did not appear to have any associated inciting factors.4-6 Dermatologists need to have a low threshold for biopsy for any unusual nonhealing lesions, especially in the setting of BCNS. Basal cell carcinomas in BCNS cannot be histologically differentiated from sporadic BCCs, and management largely depends on the size, location, recurrence, and number of lesions. Treatment methods range from topical agents to Mohs micrographic surgery.1
Nonhealing lesions of the foot may give an initial clinical impression of infection overlying peripheral vascular disease or diabetes mellitus with the possibility of associated osteomyelitis. Our patient had no clinical history to suggest peripheral vascular disease or diabetes mellitus, and he had palpable dorsalis pedis pulses as well as a normal neurologic examination. Clinicians also may consider fungal infection in the differential diagnosis. Erosio interdigitalis blastomycetica is a superficial yeast infection described as a well-defined, red, shiny plaque found in chronically wet areas, usually affecting the third or fourth interdigital spaces of the fingers.7 However, the lack of improvement with antibiotics and antifungals argued against bacterial or fungal infection in our patient. Although BCC also is a common feature of Bazex Dupré-Christol syndrome, it also is characterized by follicular atrophoderma, milia, hypohidrosis, and hypotrichosis,8 which were not evident in our patient. Pseudomonas hot foot syndrome is characterized by painful, plantar, erythematous nodules after exposure to ontaminated water that typically is self-limited but does respond to antibiotics for Pseudomonas.9
Our patient underwent Mohs micrographic surgery with a complex repair utilizing a full-thickness skin graft. There were no signs of recurrence at 3-month follow-up, and he was counseled on the importance of sun-protective behaviors along with regular dermatologic follow-up.
1. Gorlin RJ. Nevoid basal cell (Gorlin) syndrome. Genet Med. 2004; 6:530-539.
2. Bale A. The nevoid basal cell carcinoma syndrome: genetics and mechanism of carcinogenesis. Cancer Invest. 1997;15:180-186.
3. Goldstein AM, Bale SJ, Peck GL, et al. Sun exposure and basal cell carcinomas in nevoid basal cell carcinoma syndrome. J Am Acad Dermatol. 1993;29:34-41.
4. Silvers SH. Interdigital pedal basal cell carcinoma. Cutis. 1983;31:199-200.
5. Weitzner S. Basal cell carcinoma of toeweb presenting as a junctional nevus. Southwest Med. 1968;49:175.
6. Niwa A, Pimentel E. Basal cell carcinoma in unusual locations. An Bras Dermatol. 2006;81:281-284.
7. Mitchell JH. Erosio interdigitalis blastomycetica. Arch Derm Syphilol. 1922;6:675-679.
8. Kidd A, Carson L, Gregory DW, et al. A Scottish family with Bazex-Dupré-Christol syndrome: follicular atrophoderma, congenital hypotrichosis, and basal cell carcinoma. J Med Genet. 1996;33:493-497.
9. Yu Y, Cheng AS, Wang L, et al. Hot tub folliculitis or hot hand-foot syndrome caused by Pseudomonas aeruginosa. J Am Acad Dermatol. 2007;57:596-600.
The Diagnosis: Basal Cell Nevus Syndrome
Given the patient’s history of numerous basal cell carcinomas (BCCs), odontogenic keratocysts, palmar pits, and a nonhealing ulcer, the clinical presentation was highly suggestive of interdigital BCC in the setting of basal cell nevus syndrome (BCNS). A shave biopsy was performed revealing islands of basaloid cells with peripheral palisading and a retraction artifact surrounded by fibromyxoid stroma, consistent with nodular and infiltrative BCC (Figure 1).
Basal cell nevus syndrome (also known as Gorlin syndrome) is a rare neurocutaneous syndrome that manifests with multiple BCCs; palmar and plantar pits (Figure 2); central nervous system tumors; and skeletal anomalies including jaw cysts, macrocephaly, frontal bossing, and bifid ribs.1 It is an autosomal-dominant condition caused by mutations in the PTCH1 gene, a tumor suppressor gene involved in the Hedgehog signaling pathway.2 Basal cell carcinoma is the most distinctive feature of BCNS, causing notable morbidity. Tumors typically present between puberty and 35 years of age, and patients can have anywhere from a few to thousands of tumors. They rarely become locally aggressive; however, with radiation therapy, proliferation and local invasion may occur within a few years. Therefore, radiotherapy should be avoided in these patients.1
Although the most common sites for BCCs in BCNS are the head, neck, and back, there is a higher rate of occurrence on sun-protected areas in BCNS compared to the general population.3 Our patient presented with interdigital BCC of the foot, which is an extremely rare occurrence. PubMed and Ovid searches using the terms basal cell carcinoma, BCC, foot, interdigital, and nonmelanoma skin cancer revealed only 3 cases of interdigital BCC of the foot. One case was associated with prior surgical trauma, the second presented as a junctional nevus, and the third did not appear to have any associated inciting factors.4-6 Dermatologists need to have a low threshold for biopsy for any unusual nonhealing lesions, especially in the setting of BCNS. Basal cell carcinomas in BCNS cannot be histologically differentiated from sporadic BCCs, and management largely depends on the size, location, recurrence, and number of lesions. Treatment methods range from topical agents to Mohs micrographic surgery.1
Nonhealing lesions of the foot may give an initial clinical impression of infection overlying peripheral vascular disease or diabetes mellitus with the possibility of associated osteomyelitis. Our patient had no clinical history to suggest peripheral vascular disease or diabetes mellitus, and he had palpable dorsalis pedis pulses as well as a normal neurologic examination. Clinicians also may consider fungal infection in the differential diagnosis. Erosio interdigitalis blastomycetica is a superficial yeast infection described as a well-defined, red, shiny plaque found in chronically wet areas, usually affecting the third or fourth interdigital spaces of the fingers.7 However, the lack of improvement with antibiotics and antifungals argued against bacterial or fungal infection in our patient. Although BCC also is a common feature of Bazex Dupré-Christol syndrome, it also is characterized by follicular atrophoderma, milia, hypohidrosis, and hypotrichosis,8 which were not evident in our patient. Pseudomonas hot foot syndrome is characterized by painful, plantar, erythematous nodules after exposure to ontaminated water that typically is self-limited but does respond to antibiotics for Pseudomonas.9
Our patient underwent Mohs micrographic surgery with a complex repair utilizing a full-thickness skin graft. There were no signs of recurrence at 3-month follow-up, and he was counseled on the importance of sun-protective behaviors along with regular dermatologic follow-up.
The Diagnosis: Basal Cell Nevus Syndrome
Given the patient’s history of numerous basal cell carcinomas (BCCs), odontogenic keratocysts, palmar pits, and a nonhealing ulcer, the clinical presentation was highly suggestive of interdigital BCC in the setting of basal cell nevus syndrome (BCNS). A shave biopsy was performed revealing islands of basaloid cells with peripheral palisading and a retraction artifact surrounded by fibromyxoid stroma, consistent with nodular and infiltrative BCC (Figure 1).
Basal cell nevus syndrome (also known as Gorlin syndrome) is a rare neurocutaneous syndrome that manifests with multiple BCCs; palmar and plantar pits (Figure 2); central nervous system tumors; and skeletal anomalies including jaw cysts, macrocephaly, frontal bossing, and bifid ribs.1 It is an autosomal-dominant condition caused by mutations in the PTCH1 gene, a tumor suppressor gene involved in the Hedgehog signaling pathway.2 Basal cell carcinoma is the most distinctive feature of BCNS, causing notable morbidity. Tumors typically present between puberty and 35 years of age, and patients can have anywhere from a few to thousands of tumors. They rarely become locally aggressive; however, with radiation therapy, proliferation and local invasion may occur within a few years. Therefore, radiotherapy should be avoided in these patients.1
Although the most common sites for BCCs in BCNS are the head, neck, and back, there is a higher rate of occurrence on sun-protected areas in BCNS compared to the general population.3 Our patient presented with interdigital BCC of the foot, which is an extremely rare occurrence. PubMed and Ovid searches using the terms basal cell carcinoma, BCC, foot, interdigital, and nonmelanoma skin cancer revealed only 3 cases of interdigital BCC of the foot. One case was associated with prior surgical trauma, the second presented as a junctional nevus, and the third did not appear to have any associated inciting factors.4-6 Dermatologists need to have a low threshold for biopsy for any unusual nonhealing lesions, especially in the setting of BCNS. Basal cell carcinomas in BCNS cannot be histologically differentiated from sporadic BCCs, and management largely depends on the size, location, recurrence, and number of lesions. Treatment methods range from topical agents to Mohs micrographic surgery.1
Nonhealing lesions of the foot may give an initial clinical impression of infection overlying peripheral vascular disease or diabetes mellitus with the possibility of associated osteomyelitis. Our patient had no clinical history to suggest peripheral vascular disease or diabetes mellitus, and he had palpable dorsalis pedis pulses as well as a normal neurologic examination. Clinicians also may consider fungal infection in the differential diagnosis. Erosio interdigitalis blastomycetica is a superficial yeast infection described as a well-defined, red, shiny plaque found in chronically wet areas, usually affecting the third or fourth interdigital spaces of the fingers.7 However, the lack of improvement with antibiotics and antifungals argued against bacterial or fungal infection in our patient. Although BCC also is a common feature of Bazex Dupré-Christol syndrome, it also is characterized by follicular atrophoderma, milia, hypohidrosis, and hypotrichosis,8 which were not evident in our patient. Pseudomonas hot foot syndrome is characterized by painful, plantar, erythematous nodules after exposure to ontaminated water that typically is self-limited but does respond to antibiotics for Pseudomonas.9
Our patient underwent Mohs micrographic surgery with a complex repair utilizing a full-thickness skin graft. There were no signs of recurrence at 3-month follow-up, and he was counseled on the importance of sun-protective behaviors along with regular dermatologic follow-up.
1. Gorlin RJ. Nevoid basal cell (Gorlin) syndrome. Genet Med. 2004; 6:530-539.
2. Bale A. The nevoid basal cell carcinoma syndrome: genetics and mechanism of carcinogenesis. Cancer Invest. 1997;15:180-186.
3. Goldstein AM, Bale SJ, Peck GL, et al. Sun exposure and basal cell carcinomas in nevoid basal cell carcinoma syndrome. J Am Acad Dermatol. 1993;29:34-41.
4. Silvers SH. Interdigital pedal basal cell carcinoma. Cutis. 1983;31:199-200.
5. Weitzner S. Basal cell carcinoma of toeweb presenting as a junctional nevus. Southwest Med. 1968;49:175.
6. Niwa A, Pimentel E. Basal cell carcinoma in unusual locations. An Bras Dermatol. 2006;81:281-284.
7. Mitchell JH. Erosio interdigitalis blastomycetica. Arch Derm Syphilol. 1922;6:675-679.
8. Kidd A, Carson L, Gregory DW, et al. A Scottish family with Bazex-Dupré-Christol syndrome: follicular atrophoderma, congenital hypotrichosis, and basal cell carcinoma. J Med Genet. 1996;33:493-497.
9. Yu Y, Cheng AS, Wang L, et al. Hot tub folliculitis or hot hand-foot syndrome caused by Pseudomonas aeruginosa. J Am Acad Dermatol. 2007;57:596-600.
1. Gorlin RJ. Nevoid basal cell (Gorlin) syndrome. Genet Med. 2004; 6:530-539.
2. Bale A. The nevoid basal cell carcinoma syndrome: genetics and mechanism of carcinogenesis. Cancer Invest. 1997;15:180-186.
3. Goldstein AM, Bale SJ, Peck GL, et al. Sun exposure and basal cell carcinomas in nevoid basal cell carcinoma syndrome. J Am Acad Dermatol. 1993;29:34-41.
4. Silvers SH. Interdigital pedal basal cell carcinoma. Cutis. 1983;31:199-200.
5. Weitzner S. Basal cell carcinoma of toeweb presenting as a junctional nevus. Southwest Med. 1968;49:175.
6. Niwa A, Pimentel E. Basal cell carcinoma in unusual locations. An Bras Dermatol. 2006;81:281-284.
7. Mitchell JH. Erosio interdigitalis blastomycetica. Arch Derm Syphilol. 1922;6:675-679.
8. Kidd A, Carson L, Gregory DW, et al. A Scottish family with Bazex-Dupré-Christol syndrome: follicular atrophoderma, congenital hypotrichosis, and basal cell carcinoma. J Med Genet. 1996;33:493-497.
9. Yu Y, Cheng AS, Wang L, et al. Hot tub folliculitis or hot hand-foot syndrome caused by Pseudomonas aeruginosa. J Am Acad Dermatol. 2007;57:596-600.
A 53-year-old man with a history of numerous basal cell carcinomas and odontogenic keratocysts presented with a nonhealing erosion between the left second and third toes of several months’ duration. He was treated empirically with multiple courses of topical and systemic antibiotics as well as antifungals with minimal improvement. Physical examination revealed a 1.2×0.6-cm eroded plaque with rolled borders on the left second toe web; bilateral palmar pits; diffuse actinic damage; and several well-healed surgical scars on the head, neck, and back. Neurologic examination was normal, and dorsalis pedis pulses were equal and palpable bilaterally.
Ascending Erythematous Nodules on the Arm
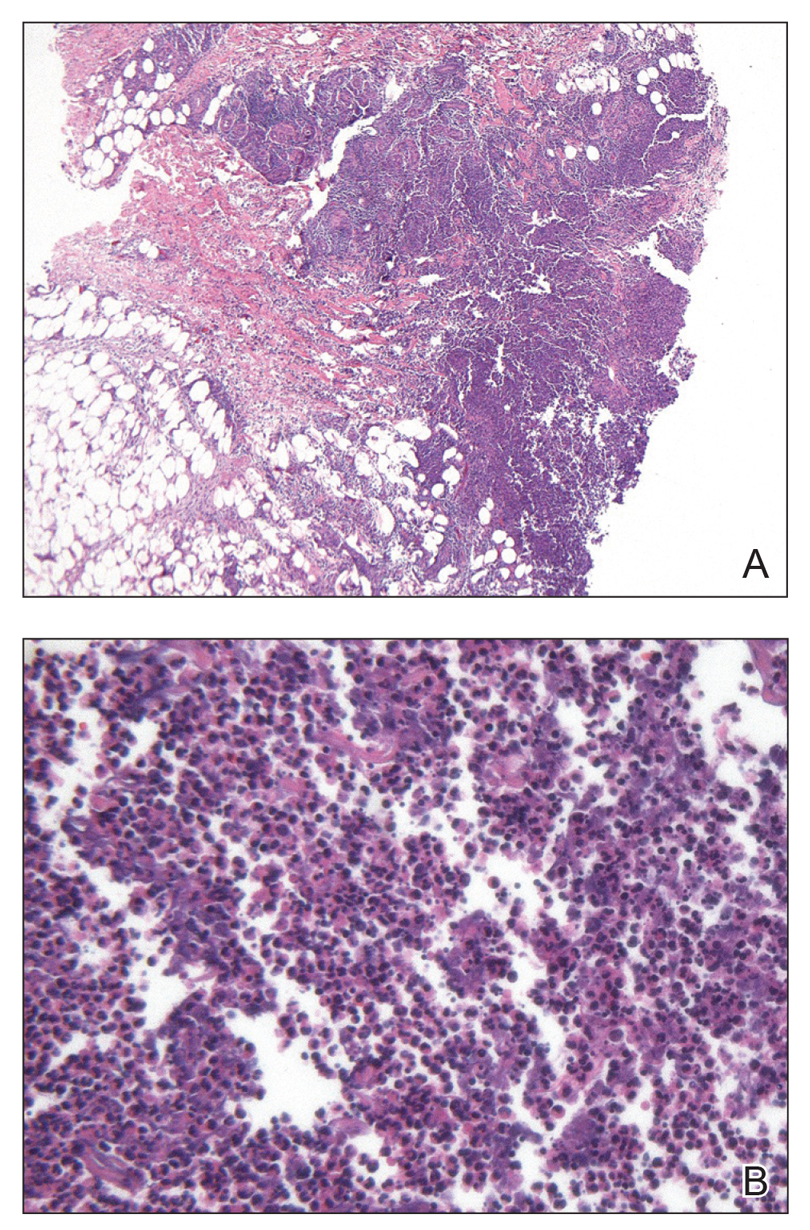
The Diagnosis: Primary Cutaneous Nocardiosis
Comprehensive metabolic panel and complete blood cell count were unremarkable; human immunodeficiency virus screening was nonreactive. Punch biopsies were obtained for histopathology, as well as bacterial, fungal, and mycobacterial cultures. Histopathologic examination of a 4-mm punch biopsy of the forearm nodule showed a dermal abscess with neutrophilic infiltration in the dermis (Figure 1). No organisms were seen on Gram, methenamine-silver, periodic acid–Schiff, or acid-fast bacteria stains. Given the clinical suspicion for lymphocutaneous sporotrichosis, the patient was started on itraconazole. She reported modest improvement but subsequently developed a morbilliform eruption necessitating medication discontinuation.
Eighteen days after obtaining the tissue culture, acid-fast organisms grew in culture. These organisms were subcultured on Middlebrook 7H11 agar (Sigma-Aldrich) with growth noted at 30°C and 37°C. Gram stain revealed filamentous gram-variable bacteria (Figure 2) that were identified as Nocardia brasiliensis by 16S ribosomal DNA analysis. Given the patient’s sulfonamide allergy, she started oral minocycline 100 mg twice daily. She responded to the therapy and subsequent testing confirmed susceptibility.
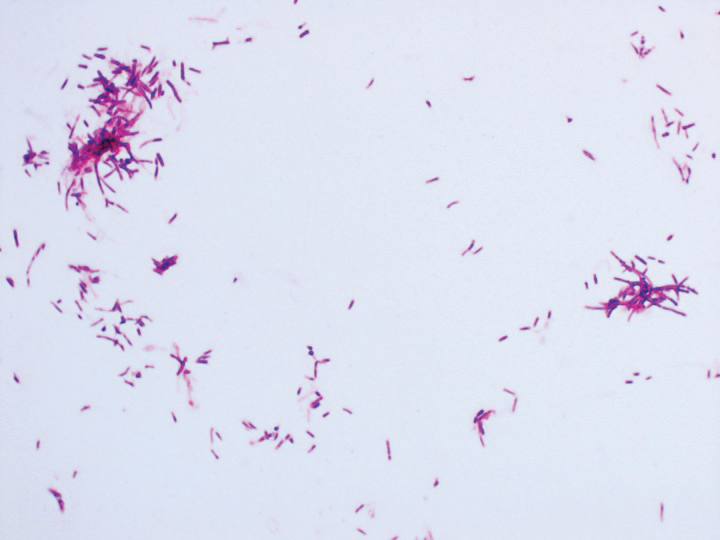
Nocardia brasiliensis, isolated from subculture on Gram stain (original
magnification ×1000).
The genus Nocardia consists of more than 50 species of gram-positive, weakly acid-fast, aerobic actinomycetes that can cause primary cutaneous infection via percutaneous inoculation. Nocardia brasiliensis is the leading cause (approximately 80% of cases) of primary cutaneous or subcutaneous nocardiosis and is found ubiquitously in soil and decaying vegetation.1 The clinical presentation varies, rendering definitive diagnosis a challenge without histopathologic and microbiologic testing.2 Patients presenting with nocardial cellulitis often are suspected to have Streptococcus pyogenes or Staphylococcus aureus infections. The differential diagnosis for patients presenting with nocardial nodular lymphangitis, also known as lymphocutaneous syndrome, includes atypical mycobacterial infections, leishmaniasis, and lymphocutaneous sporotrichosis.2
Histologic examination of nocardial nodules typically shows granulomatous or neutrophilic inflammation, and organisms may appear in small collections resembling sulfur granules.2 The organism itself is weakly positive on acid-fast stain, and useful stains include acid-fast bacteria, methenamine silver, and periodic acid–Schiff.2 Tissue culture often provides the definitive diagnosis, as the histology is nonspecific and organisms may not be visualized.
Oral trimethoprim-sulfamethoxazole 2.5 to 10 mg/kg and 12.5 to 50 mg/kg, respectively, twice daily is the treatment of choice for primary cutaneous nocardiosis. Minocycline 100 to 200 mg twice daily is an accepted alternative in case of sulfonamide allergy, as in our patient. Antibiotics should be tailored according to the susceptibility profile of the isolated organism.3
This case highlights the importance of forming a broad differential diagnosis for patients presenting with lymphocutaneous syndrome. The incidence and prevalence of N brasiliensis infection is difficult to determine due to its nonspecific clinical presentation and a lack of recent epidemiologic studies. Although primary cutaneous nocardiosis in the United States often is diagnosed in the South or Southwest, cases have been reported in other regions.4-6 Traumatic inoculation of contaminated soil, plants, and other organic matter, a well-known method of Sporothrix schenckii transmission, also is a method of N brasiliensis transmission. Because this organism may not be detected on histologic examination, empiric treatment should be considered if the diagnosis is suspected.
1. Brown-Eliot BA, Brown JM, Conville PS, et al. Clinical and laboratory features of the Nocardia spp. based on current molecular taxonomy. Clin Microbiol Rev. 2006;19:259-282.
2. Smego RA Jr, Castiglia M, Asperilla MO. Lymphocutaneous syndrome: a review of non-sporothrix causes. Medicine. 1999;78:38-63.
3. Lerner P. Nocardiosis. Clin Infect Dis. 1996;22:891-903.
4. Smego RA Jr, Gallis HA. The clinical spectrum of Nocardia brasiliensis infection in the United States. Rev Infect Dis. 1984;6:164-180.
5. Fukuda H, Saotome A, Usami N, et al. Lymphocutaneous type of nocardiosis caused by Nocardia brasiliensis: a case report and review of primary cutaneous nocardiosis caused by N. brasiliensis reported in Japan. J Dermatol. 2008;35:346-353.
6. Kil EH, Tsai CL, Kwark EH, et al. A case of nocardiosis with an uncharacteristically long incubation period. Cutis. 2005;76:33-36.
The Diagnosis: Primary Cutaneous Nocardiosis
Comprehensive metabolic panel and complete blood cell count were unremarkable; human immunodeficiency virus screening was nonreactive. Punch biopsies were obtained for histopathology, as well as bacterial, fungal, and mycobacterial cultures. Histopathologic examination of a 4-mm punch biopsy of the forearm nodule showed a dermal abscess with neutrophilic infiltration in the dermis (Figure 1). No organisms were seen on Gram, methenamine-silver, periodic acid–Schiff, or acid-fast bacteria stains. Given the clinical suspicion for lymphocutaneous sporotrichosis, the patient was started on itraconazole. She reported modest improvement but subsequently developed a morbilliform eruption necessitating medication discontinuation.
Eighteen days after obtaining the tissue culture, acid-fast organisms grew in culture. These organisms were subcultured on Middlebrook 7H11 agar (Sigma-Aldrich) with growth noted at 30°C and 37°C. Gram stain revealed filamentous gram-variable bacteria (Figure 2) that were identified as Nocardia brasiliensis by 16S ribosomal DNA analysis. Given the patient’s sulfonamide allergy, she started oral minocycline 100 mg twice daily. She responded to the therapy and subsequent testing confirmed susceptibility.
Nocardia brasiliensis, isolated from subculture on Gram stain (original
magnification ×1000).
The genus Nocardia consists of more than 50 species of gram-positive, weakly acid-fast, aerobic actinomycetes that can cause primary cutaneous infection via percutaneous inoculation. Nocardia brasiliensis is the leading cause (approximately 80% of cases) of primary cutaneous or subcutaneous nocardiosis and is found ubiquitously in soil and decaying vegetation.1 The clinical presentation varies, rendering definitive diagnosis a challenge without histopathologic and microbiologic testing.2 Patients presenting with nocardial cellulitis often are suspected to have Streptococcus pyogenes or Staphylococcus aureus infections. The differential diagnosis for patients presenting with nocardial nodular lymphangitis, also known as lymphocutaneous syndrome, includes atypical mycobacterial infections, leishmaniasis, and lymphocutaneous sporotrichosis.2
Histologic examination of nocardial nodules typically shows granulomatous or neutrophilic inflammation, and organisms may appear in small collections resembling sulfur granules.2 The organism itself is weakly positive on acid-fast stain, and useful stains include acid-fast bacteria, methenamine silver, and periodic acid–Schiff.2 Tissue culture often provides the definitive diagnosis, as the histology is nonspecific and organisms may not be visualized.
Oral trimethoprim-sulfamethoxazole 2.5 to 10 mg/kg and 12.5 to 50 mg/kg, respectively, twice daily is the treatment of choice for primary cutaneous nocardiosis. Minocycline 100 to 200 mg twice daily is an accepted alternative in case of sulfonamide allergy, as in our patient. Antibiotics should be tailored according to the susceptibility profile of the isolated organism.3
This case highlights the importance of forming a broad differential diagnosis for patients presenting with lymphocutaneous syndrome. The incidence and prevalence of N brasiliensis infection is difficult to determine due to its nonspecific clinical presentation and a lack of recent epidemiologic studies. Although primary cutaneous nocardiosis in the United States often is diagnosed in the South or Southwest, cases have been reported in other regions.4-6 Traumatic inoculation of contaminated soil, plants, and other organic matter, a well-known method of Sporothrix schenckii transmission, also is a method of N brasiliensis transmission. Because this organism may not be detected on histologic examination, empiric treatment should be considered if the diagnosis is suspected.
The Diagnosis: Primary Cutaneous Nocardiosis
Comprehensive metabolic panel and complete blood cell count were unremarkable; human immunodeficiency virus screening was nonreactive. Punch biopsies were obtained for histopathology, as well as bacterial, fungal, and mycobacterial cultures. Histopathologic examination of a 4-mm punch biopsy of the forearm nodule showed a dermal abscess with neutrophilic infiltration in the dermis (Figure 1). No organisms were seen on Gram, methenamine-silver, periodic acid–Schiff, or acid-fast bacteria stains. Given the clinical suspicion for lymphocutaneous sporotrichosis, the patient was started on itraconazole. She reported modest improvement but subsequently developed a morbilliform eruption necessitating medication discontinuation.
Eighteen days after obtaining the tissue culture, acid-fast organisms grew in culture. These organisms were subcultured on Middlebrook 7H11 agar (Sigma-Aldrich) with growth noted at 30°C and 37°C. Gram stain revealed filamentous gram-variable bacteria (Figure 2) that were identified as Nocardia brasiliensis by 16S ribosomal DNA analysis. Given the patient’s sulfonamide allergy, she started oral minocycline 100 mg twice daily. She responded to the therapy and subsequent testing confirmed susceptibility.
Nocardia brasiliensis, isolated from subculture on Gram stain (original
magnification ×1000).
The genus Nocardia consists of more than 50 species of gram-positive, weakly acid-fast, aerobic actinomycetes that can cause primary cutaneous infection via percutaneous inoculation. Nocardia brasiliensis is the leading cause (approximately 80% of cases) of primary cutaneous or subcutaneous nocardiosis and is found ubiquitously in soil and decaying vegetation.1 The clinical presentation varies, rendering definitive diagnosis a challenge without histopathologic and microbiologic testing.2 Patients presenting with nocardial cellulitis often are suspected to have Streptococcus pyogenes or Staphylococcus aureus infections. The differential diagnosis for patients presenting with nocardial nodular lymphangitis, also known as lymphocutaneous syndrome, includes atypical mycobacterial infections, leishmaniasis, and lymphocutaneous sporotrichosis.2
Histologic examination of nocardial nodules typically shows granulomatous or neutrophilic inflammation, and organisms may appear in small collections resembling sulfur granules.2 The organism itself is weakly positive on acid-fast stain, and useful stains include acid-fast bacteria, methenamine silver, and periodic acid–Schiff.2 Tissue culture often provides the definitive diagnosis, as the histology is nonspecific and organisms may not be visualized.
Oral trimethoprim-sulfamethoxazole 2.5 to 10 mg/kg and 12.5 to 50 mg/kg, respectively, twice daily is the treatment of choice for primary cutaneous nocardiosis. Minocycline 100 to 200 mg twice daily is an accepted alternative in case of sulfonamide allergy, as in our patient. Antibiotics should be tailored according to the susceptibility profile of the isolated organism.3
This case highlights the importance of forming a broad differential diagnosis for patients presenting with lymphocutaneous syndrome. The incidence and prevalence of N brasiliensis infection is difficult to determine due to its nonspecific clinical presentation and a lack of recent epidemiologic studies. Although primary cutaneous nocardiosis in the United States often is diagnosed in the South or Southwest, cases have been reported in other regions.4-6 Traumatic inoculation of contaminated soil, plants, and other organic matter, a well-known method of Sporothrix schenckii transmission, also is a method of N brasiliensis transmission. Because this organism may not be detected on histologic examination, empiric treatment should be considered if the diagnosis is suspected.
1. Brown-Eliot BA, Brown JM, Conville PS, et al. Clinical and laboratory features of the Nocardia spp. based on current molecular taxonomy. Clin Microbiol Rev. 2006;19:259-282.
2. Smego RA Jr, Castiglia M, Asperilla MO. Lymphocutaneous syndrome: a review of non-sporothrix causes. Medicine. 1999;78:38-63.
3. Lerner P. Nocardiosis. Clin Infect Dis. 1996;22:891-903.
4. Smego RA Jr, Gallis HA. The clinical spectrum of Nocardia brasiliensis infection in the United States. Rev Infect Dis. 1984;6:164-180.
5. Fukuda H, Saotome A, Usami N, et al. Lymphocutaneous type of nocardiosis caused by Nocardia brasiliensis: a case report and review of primary cutaneous nocardiosis caused by N. brasiliensis reported in Japan. J Dermatol. 2008;35:346-353.
6. Kil EH, Tsai CL, Kwark EH, et al. A case of nocardiosis with an uncharacteristically long incubation period. Cutis. 2005;76:33-36.
1. Brown-Eliot BA, Brown JM, Conville PS, et al. Clinical and laboratory features of the Nocardia spp. based on current molecular taxonomy. Clin Microbiol Rev. 2006;19:259-282.
2. Smego RA Jr, Castiglia M, Asperilla MO. Lymphocutaneous syndrome: a review of non-sporothrix causes. Medicine. 1999;78:38-63.
3. Lerner P. Nocardiosis. Clin Infect Dis. 1996;22:891-903.
4. Smego RA Jr, Gallis HA. The clinical spectrum of Nocardia brasiliensis infection in the United States. Rev Infect Dis. 1984;6:164-180.
5. Fukuda H, Saotome A, Usami N, et al. Lymphocutaneous type of nocardiosis caused by Nocardia brasiliensis: a case report and review of primary cutaneous nocardiosis caused by N. brasiliensis reported in Japan. J Dermatol. 2008;35:346-353.
6. Kil EH, Tsai CL, Kwark EH, et al. A case of nocardiosis with an uncharacteristically long incubation period. Cutis. 2005;76:33-36.
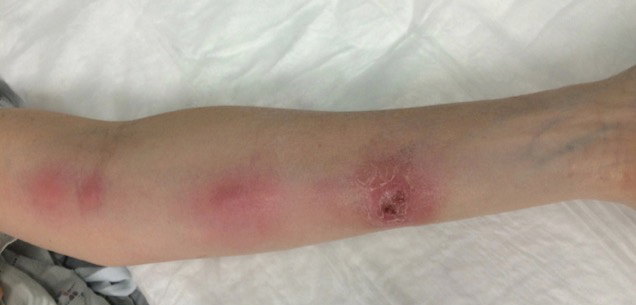
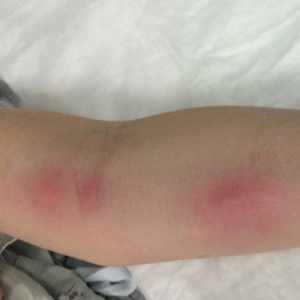
A 54-year-old woman called her primary care provider to report a painful pink nodule on the left wrist 1 week after sustaining thorn injuries while weeding in her garden. She started cephalexin and noted a pink streak with additional nodules extending up the arm over the next 2 days. She
was admitted to an outside hospital for incision and drainage of the wrist nodule and a 3-day course of intravenous vancomycin. Bacterial culture was negative, and she was discharged on oral clindamycin and doxycycline. Two days later, she presented to our emergency department with pain in the left axilla. Physical examination revealed 3 tender erythematous nodules in a linear distribution on the left arm with crusting at the incision and drainage site and painful left axillary lymphadenopathy. The patient was afebrile and otherwise asymptomatic.
Pityriasis Amiantacea Following Bone Marrow Transplant
Pityriasis amiantacea (PA) is characterized by adherence of hair shafts proximally.1 It has been associated with dermatologic conditions and rarely with medications. We describe a woman who developed PA following a bone marrow transplant with melphalan conditioning. We also review drug-induced PA and disorders that have been linked to this condition.
Case Report
A 67-year-old woman with a history of multiple myeloma was treated with 7 courses of chemotherapy (cyclophosphamide, bortezomib, prednisone). One month later, the patient underwent a bone marrow transplant with melphalan conditioning due to residual plasma cell myeloma. Following the transplant, she developed complete scalp alopecia. Prior to and following transplant, the patient’s hair care regimen included washing her hair and scalp every other day with over-the-counter “natural” shampoos. During drug-induced alopecia, the hair washing became less frequent.
The patient left the hospital 4 weeks posttransplant; her hair had started to regrow, but its appearance was altered. Posttransplant, the patient was maintained on bortezomib every other week and zoledronate once per month. She continued to develop multiple lesions in the scalp hairs during the following 4 months.
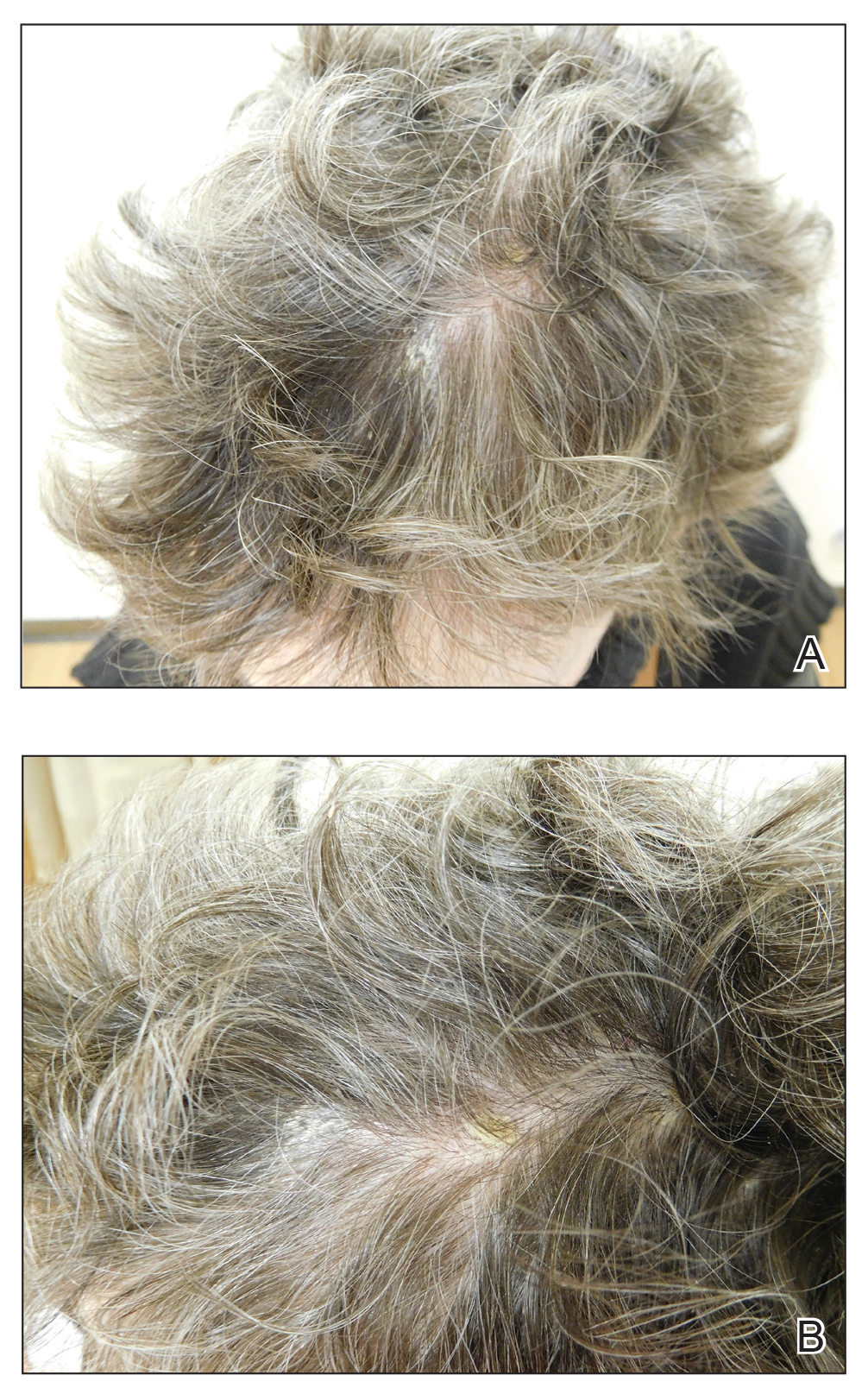
Eight months posttransplant she presented for evaluation of the scalp hair. Clinical examination showed hairs that were entwined together proximally, resulting in matting of the hair (Figure 1). A diagnosis of PA was established based on the clinical examination.
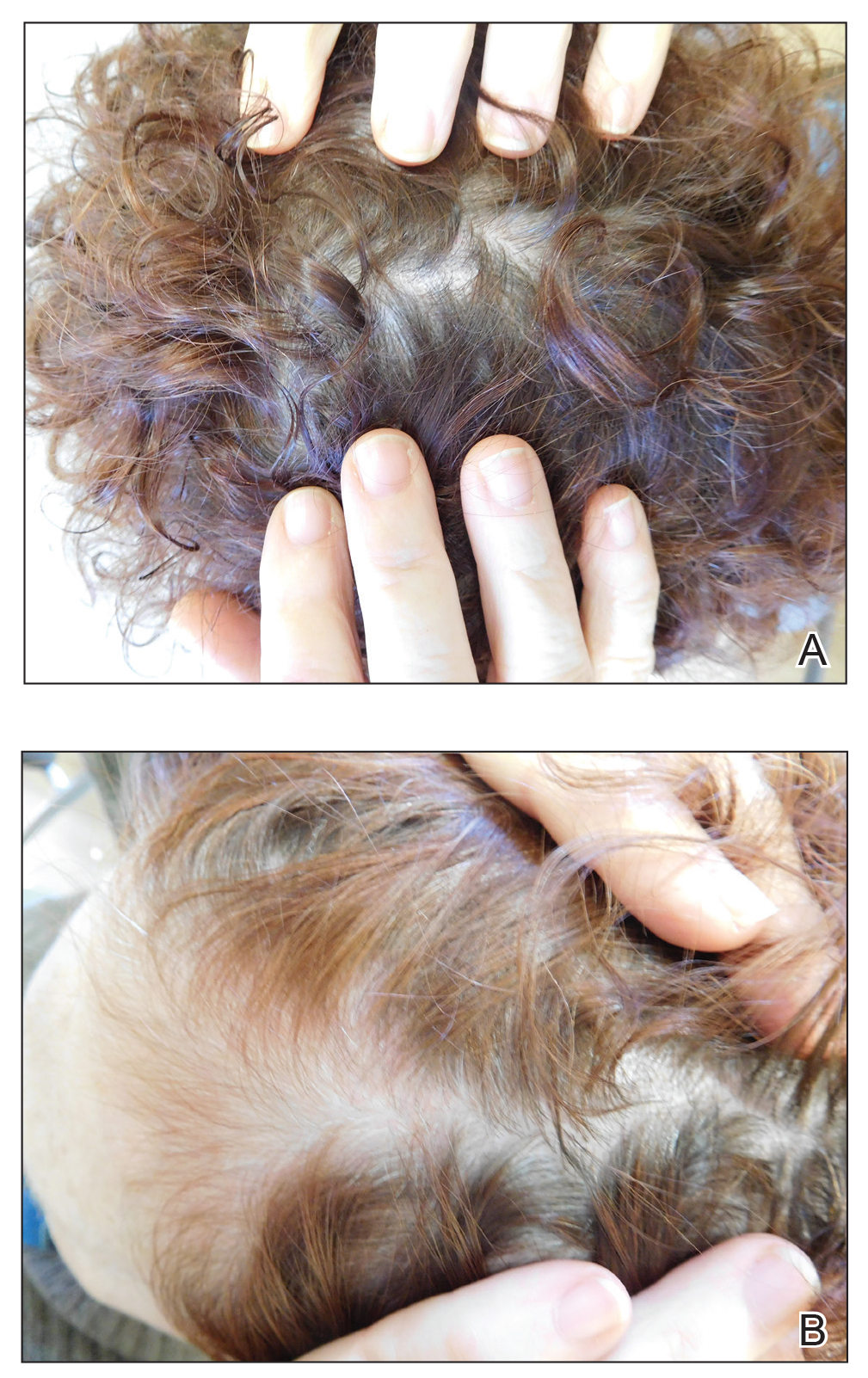
Treatment included mineral oil application to the scalp under occlusion each evening, followed by morning washing with coal tar 0.5%, salicylic acid 6%, or ketoconazole 2% shampoo in a repeating sequential manner. Within 1 month there was complete resolution of the scalp condition (Figure 2).
Comment
Clinical Presentation
Pityriasis amiantacea is characterized by thick excessive scale of the scalp1; it was initially described by Alibert2 in 1832. He described the gross appearance of the scales as resembling the feathers of young birds, which naturalists dub “amiante” or asbestoslike.1,2 In 1917, Gougerot3 explored infectious etiologies of this condition by describing cases of impetigo that transitioned into PA.1 Later, in 1929, Photinos4 described fungal origins of PA, giving credence to “tinea amiantacea.”1 However, more recent analyses failed to isolate fungus.5-7 As such, pityriasis (scaling) amiantacea is the more appropriate term, as emphasized by Brown8 in 1948. The cause of PA remains unclear; it is hypothesized that the condition is a reaction to underlying inflammatory dermatoses, though concurrent bacterial or fungal infection may be present.5,9
Prevalence
Pityriasis amiantacea is considered to be most prevalent in pediatric patients and young adults; it is more common in females.1,9,10 In a review of 85 PA patients, more than 80% were women (n=69), and the mean age at presentation was 23.8 years. Approximately half of these patients had widespread scalp lesions (n=42); however, focal localized lesions were common.9 No hereditary patterns have been described, though 3 pairs of the 10 patients with PA in Ring and Kaplan’s7 review were siblings.
Clinical Findings
Clinically, lesions of PA present as matted hairs.1 Thick scales encompass multiple hair shafts, binding down tufts of hair.1,6,11 Patients are asymptomatic, though the lesions may be accompanied by pruritus. The hairs enclosed by the scales in some cases may be easily pulled out.6 Notably, alopecia often accompanies PA; it often is reversible, but in some cases, it is permanent and can lead to scarring.9,12
Histopathology
Submission of hair specimens to histopathology usually is not performed since the diagnosis often is established based on the clinical presentation.5 However, submitted specimens have demonstrated spongiosis and parakeratosis along with reduction in the size of the sebaceous glands.1,9 Additionally, follicular keratosis that surrounds the hair shafts with a sheath of horn is present.9 Acanthosis and migration of lymphocytes into the epidermis also have been found.1 Often, Staphylococcus aureus isolates are detected.9,13
Differential Diagnosis
The clinical differential diagnosis of PA includes hair casts,11 pediculosis,14 and tinea capitis.12 In PA, thick scales surround hair shafts and thus bind down tufts of hair.9 In patients with pediculosis, nits are attached to the hair shaft at an angle and do not entirely envelop the hair shaft.14 In addition, PA may be complicated by impetiginization; bacteria often are found in the keratin surrounding the hair shaft and represent either normal flora or secondary infection.1,15 It has been speculated that microbial biofilms from S aureus and Staphylococcus epidermidis promote agglomeration of hair shafts and adherent scale.16 Bona fide dermatophyte infection of the scalp also may be concurrently present.12
Treatment
Our treatment included occlusion with mineral oil to loosen the scales from the scalp in tandem with shampoos traditionally used in patients with seborrheic dermatitis or psoriasis. Timely treatment is important to prevent scarring alopecia.13,17 Pityriasis amiantacea may be treatment resistant, and there are no specific therapeutic guidelines; rather, therapy should be targeted at the suspected underlying condition.17 Treatment generally includes keratolytic agents, such as salicylic acid.18 These agents allow enhanced penetration of other topical agents.19 Topical antifungal shampoos such as ketoconazole and ciclopirox are recommended,18 though other topical agents, such as coal tar and zinc pyrithione, also may benefit patients.13 Topical corticosteroids may be used if the condition is linked with psoriasis.13 Systemic antibiotics are added if S aureus superinfection is suspected.9
A single report described successful management of a patient with severe refractory PA who was treated with the tumor necrosis factor (TNF) α inhibitor infliximab.13 A 47-year-old woman presented with thick adherent scale on the scalp. She was treated with coal tar for 18 months but showed no improvement; the patient was subsequently prescribed salicylic acid 10%, clobetasol solution, and coal tar shampoo. After 3 months, when no improvement was observed, the patient was offered infliximab but declined. For 6 years the patient was treated with salicylic acid 20%, clobetasol (foam, lotion, shampoo, and solution), and coal tar shampoo without improvement. She then consented to infliximab therapy; after 3 infusions at weeks 0, 2, and 6, she demonstrated notable improvement. The patient was maintained on infliximab every 8 weeks.13
Pathogenesis
The pathogenesis of PA has yet to be definitively established, and the condition is usually idiopathic. In addition to bacterial or fungal etiologies,3,4 PA has been linked to medications (Table 1)16,20,21 and systemic conditions (Table 2).1,3,5,7-10,12,22-25
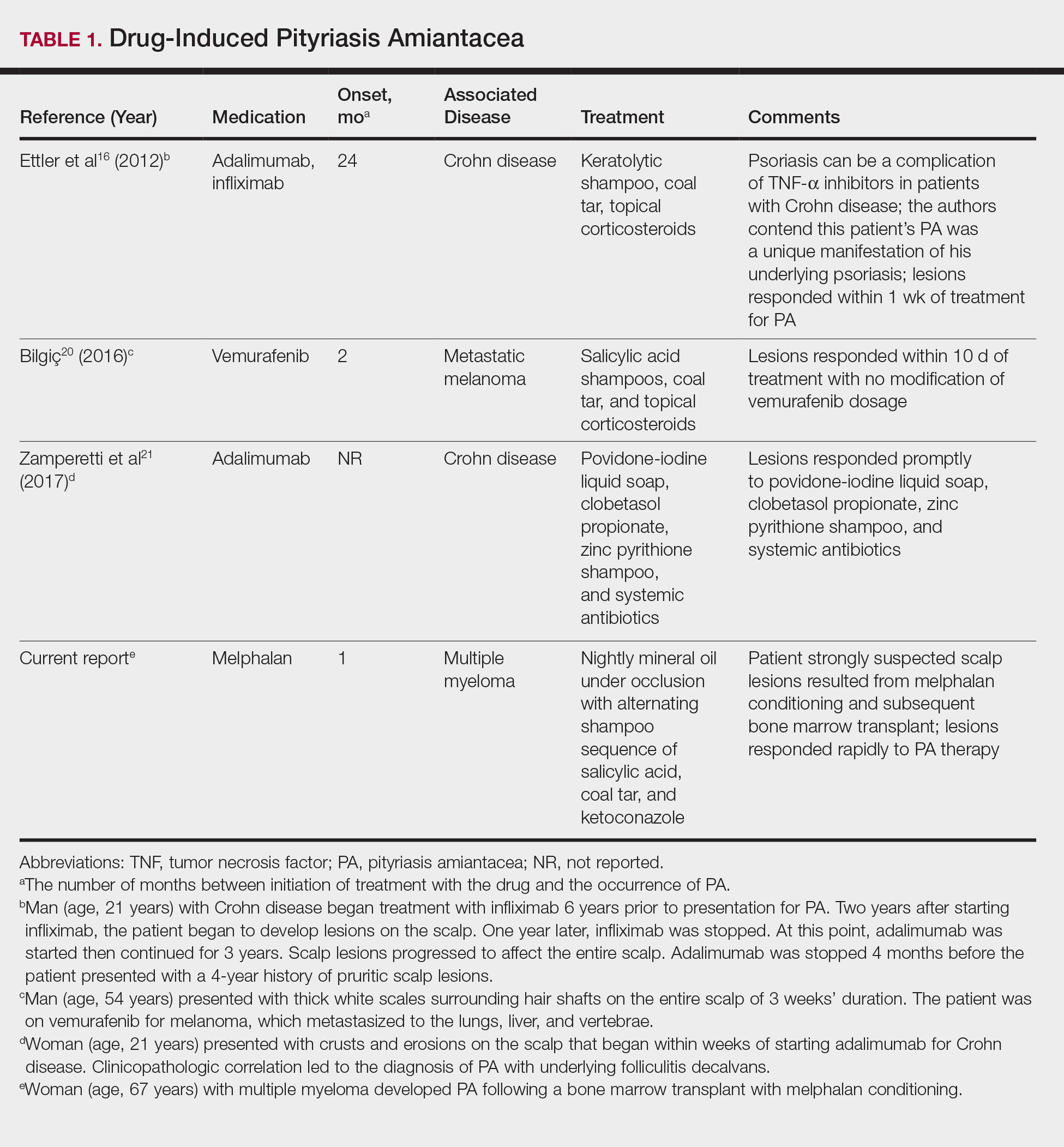
A PubMed search of articles indexed for MEDLINE using the search terms amiantacea, bone, drug, hair marrow, malignancy, melphalan, pityriasis, tinea, and transplant yielded 4 patients—2 men and 2 women (including our patient)—with possible drug-induced PA (Table 1)16,20,21; however, the onset after 2 years of medication (TNF-α inhibitors) or resolution while still receiving the agent (vemurafenib) makes the drug-induced linkage weak. The patients ranged in age from 21 to 67 years, with the median age being 37.5 years. Medications included melphalan, TNF-α inhibitors (adalimumab, infliximab),16,21 and vemurafenib20; it is interesting that infliximab was the medication associated with eliciting PA in 1 patient yet was an effective therapy in another patient with treatment-resistant PA. The onset of PA occurred between 1 month (melphalan) and 24 months (TNF-α inhibitors) after drug initiation. The patients’ associated diseases included Crohn disease,16,21 metastatic melanoma,20 and multiple myeloma.
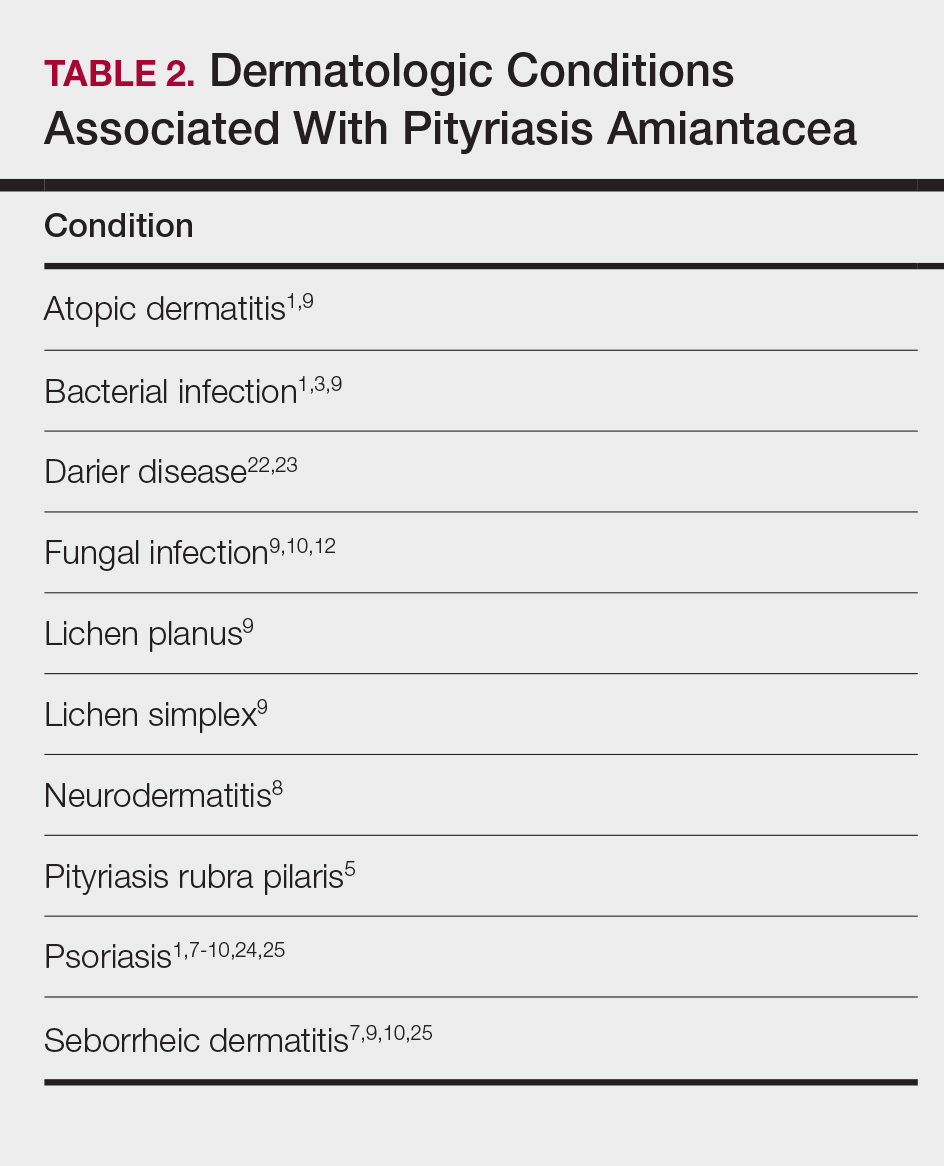
Other conditions have been described in patients with PA (Table 2). Indeed, PA may be a manifestation of an underlying inflammatory skin disease.9 In addition to dermatologic conditions, procedures or malignancy may be associated with the disease, as demonstrated in our patient. Most commonly, PA is seen in association with psoriasis and seborrheic dermatitis; atopic dermatitis, bacterial infection, fungal infection, lichen planus, and neurodermatitis also have been associated with PA.1,3,5,7-10,12,18,22-25
Conclusion
Pityriasis amiantacea is a benign condition affecting the scalp hair. Albeit uncommon, it may appear in patients treated with medications such as melphalan, TNF-α inhibitors, and vemurafenib. In addition, it has been described in individuals with dermatologic conditions, systemic procedures, or underlying malignancy. Our patient developed PA following a bone marrow transplant after receiving conditioning with melphalan.
- Knight AG. Pityriasis amiantacea: a clinical and histopathological investigation. Clin Exp Dermatol. 1977;2:137-143.
- Alibert JL. De la porrigine amiantacée. In: Monographie des Dermatoses. Paris, France: Baillère; 1832:293-295.
- Gougerot H. La teigne amiantacee D’Alibert. Progres Medical. 1917;13:101-104.
- Photinos P. Recherches sur la fausse teigne amiantacée. Ann Dermatol Syphiligr. 1929;10:743-758.
- Verardino GC, Azulay-Abulafia L, Macedo PM, et al. Pityriasis amiantacea: clinical-dermatoscopic features and microscopy of hair tufts. An Bras Dermatol. 2012;87:142-145.
- Keipert JA. Greasy scaling pityriasis amiantacea and alopecia: a syndrome in search of a cause. Australas J Dermatol. 1985;26:41-44.
- Ring DS, Kaplan DL. Pityriasis amiantacea: a report of 10 cases. Arch Dermatol. 1993;129:913-914.
- Brown WH. Some observations on neurodermatitis of the scalp, with particular reference to tinea amiantacea. Br J Dermatol Syph. 1948;60:81-90.
- Abdel-Hamid IA, Agha SA, Moustafa YM, et al. Pityriasis amiantacea: a clinical and etiopathologic study of 85 patients. Int J Dermatol. 2003;42:260-264.
- Becker SW, Muir KB. Tinea amiantacea. Arch Dermatol Syphil. 1929;20:45-53.
- Dawber RP. Hair casts. Br J Dermatol. 1979;100:417-421.
- Ginarte M, Pereiro M, Fernández-Redondo V, et al. Case reports. pityriasis amiantacea as manifestation of tinea capitis due to Microsporum canis. Mycoses. 2000;43:93-96.
- Pham RK, Chan CS, Hsu S. Treatment of pityriasis amiantacea with infliximab. Dermatol Online J. 2009;15:13.
- Roberts RJ. Clinical practice. Head lice. N Engl J Med. 2002;346:1645-1650.
- Mcginley KJ, Leyden JJ, Marples RR, et al. Quantitative microbiology of the scalp in non-dandruff, dandruff, and seborrheic dermatitis. J Invest Dermatol. 1975;64:401-405.
- Ettler J, Wetter DA, Pittelkow MR. Pityriasis amiantacea: a distinctive presentation of psoriasis associated with tumour necrosis factor-α inhibitor therapy. Clin Exp Dermatol. 2012;37:639-641.
- Mannino G, McCaughey C, Vanness E. A case of pityriasis amiantacea with rapid response to treatment. WMJ. 2014;113:119-120.
- Jamil A, Muthupalaniappen L. Scales on the scalp. Malays Fam Physician. 2013;8:48-49.
- Gupta LK, Khare AK, Masatkar V, et al. Pityriasis amiantacea. Indian Dermatol Online J. 2014;5(suppl 1):S63-S64.
- Bilgiç Ö. Vemurafenib-induced pityriasis amiantacea: a case report. Cutan Ocul Toxicol. 2016;35:329-331.
- Zamperetti M, Zelger B, Höpfl R. Pityriasis amiantacea and folliculitis decalvans: an unusual manifestation associated with antitumor necrosis factor-α therapy. Hautarzt. 2017;68:1007-1010.
- Udayashankar C, Nath AK, Anuradha P. Extensive Darier’s disease with pityriasis amiantacea, alopecia and congenital facial nerve palsy. Dermatol Online J. 2013;19:18574.
- Hussain W, Coulson IH, Salman WD. Pityriasis amiantacea as the sole manifestation of Darier’s disease. Clin Exp Dermatol. 2009;34:554-556.
- Hansted B, Lindskov R. Pityriasis amiantacea and psoriasis. a follow-up study. Dermatologica. 1983;166:314-315.
- Hersle K, Lindholm A, Mobacken H, et al. Relationship of pityriasis amiantacea to psoriasis. a follow-up study. Dermatologica. 1979;159:245-250.
Pityriasis amiantacea (PA) is characterized by adherence of hair shafts proximally.1 It has been associated with dermatologic conditions and rarely with medications. We describe a woman who developed PA following a bone marrow transplant with melphalan conditioning. We also review drug-induced PA and disorders that have been linked to this condition.
Case Report
A 67-year-old woman with a history of multiple myeloma was treated with 7 courses of chemotherapy (cyclophosphamide, bortezomib, prednisone). One month later, the patient underwent a bone marrow transplant with melphalan conditioning due to residual plasma cell myeloma. Following the transplant, she developed complete scalp alopecia. Prior to and following transplant, the patient’s hair care regimen included washing her hair and scalp every other day with over-the-counter “natural” shampoos. During drug-induced alopecia, the hair washing became less frequent.
The patient left the hospital 4 weeks posttransplant; her hair had started to regrow, but its appearance was altered. Posttransplant, the patient was maintained on bortezomib every other week and zoledronate once per month. She continued to develop multiple lesions in the scalp hairs during the following 4 months.
Eight months posttransplant she presented for evaluation of the scalp hair. Clinical examination showed hairs that were entwined together proximally, resulting in matting of the hair (Figure 1). A diagnosis of PA was established based on the clinical examination.
Treatment included mineral oil application to the scalp under occlusion each evening, followed by morning washing with coal tar 0.5%, salicylic acid 6%, or ketoconazole 2% shampoo in a repeating sequential manner. Within 1 month there was complete resolution of the scalp condition (Figure 2).
Comment
Clinical Presentation
Pityriasis amiantacea is characterized by thick excessive scale of the scalp1; it was initially described by Alibert2 in 1832. He described the gross appearance of the scales as resembling the feathers of young birds, which naturalists dub “amiante” or asbestoslike.1,2 In 1917, Gougerot3 explored infectious etiologies of this condition by describing cases of impetigo that transitioned into PA.1 Later, in 1929, Photinos4 described fungal origins of PA, giving credence to “tinea amiantacea.”1 However, more recent analyses failed to isolate fungus.5-7 As such, pityriasis (scaling) amiantacea is the more appropriate term, as emphasized by Brown8 in 1948. The cause of PA remains unclear; it is hypothesized that the condition is a reaction to underlying inflammatory dermatoses, though concurrent bacterial or fungal infection may be present.5,9
Prevalence
Pityriasis amiantacea is considered to be most prevalent in pediatric patients and young adults; it is more common in females.1,9,10 In a review of 85 PA patients, more than 80% were women (n=69), and the mean age at presentation was 23.8 years. Approximately half of these patients had widespread scalp lesions (n=42); however, focal localized lesions were common.9 No hereditary patterns have been described, though 3 pairs of the 10 patients with PA in Ring and Kaplan’s7 review were siblings.
Clinical Findings
Clinically, lesions of PA present as matted hairs.1 Thick scales encompass multiple hair shafts, binding down tufts of hair.1,6,11 Patients are asymptomatic, though the lesions may be accompanied by pruritus. The hairs enclosed by the scales in some cases may be easily pulled out.6 Notably, alopecia often accompanies PA; it often is reversible, but in some cases, it is permanent and can lead to scarring.9,12
Histopathology
Submission of hair specimens to histopathology usually is not performed since the diagnosis often is established based on the clinical presentation.5 However, submitted specimens have demonstrated spongiosis and parakeratosis along with reduction in the size of the sebaceous glands.1,9 Additionally, follicular keratosis that surrounds the hair shafts with a sheath of horn is present.9 Acanthosis and migration of lymphocytes into the epidermis also have been found.1 Often, Staphylococcus aureus isolates are detected.9,13
Differential Diagnosis
The clinical differential diagnosis of PA includes hair casts,11 pediculosis,14 and tinea capitis.12 In PA, thick scales surround hair shafts and thus bind down tufts of hair.9 In patients with pediculosis, nits are attached to the hair shaft at an angle and do not entirely envelop the hair shaft.14 In addition, PA may be complicated by impetiginization; bacteria often are found in the keratin surrounding the hair shaft and represent either normal flora or secondary infection.1,15 It has been speculated that microbial biofilms from S aureus and Staphylococcus epidermidis promote agglomeration of hair shafts and adherent scale.16 Bona fide dermatophyte infection of the scalp also may be concurrently present.12
Treatment
Our treatment included occlusion with mineral oil to loosen the scales from the scalp in tandem with shampoos traditionally used in patients with seborrheic dermatitis or psoriasis. Timely treatment is important to prevent scarring alopecia.13,17 Pityriasis amiantacea may be treatment resistant, and there are no specific therapeutic guidelines; rather, therapy should be targeted at the suspected underlying condition.17 Treatment generally includes keratolytic agents, such as salicylic acid.18 These agents allow enhanced penetration of other topical agents.19 Topical antifungal shampoos such as ketoconazole and ciclopirox are recommended,18 though other topical agents, such as coal tar and zinc pyrithione, also may benefit patients.13 Topical corticosteroids may be used if the condition is linked with psoriasis.13 Systemic antibiotics are added if S aureus superinfection is suspected.9
A single report described successful management of a patient with severe refractory PA who was treated with the tumor necrosis factor (TNF) α inhibitor infliximab.13 A 47-year-old woman presented with thick adherent scale on the scalp. She was treated with coal tar for 18 months but showed no improvement; the patient was subsequently prescribed salicylic acid 10%, clobetasol solution, and coal tar shampoo. After 3 months, when no improvement was observed, the patient was offered infliximab but declined. For 6 years the patient was treated with salicylic acid 20%, clobetasol (foam, lotion, shampoo, and solution), and coal tar shampoo without improvement. She then consented to infliximab therapy; after 3 infusions at weeks 0, 2, and 6, she demonstrated notable improvement. The patient was maintained on infliximab every 8 weeks.13
Pathogenesis
The pathogenesis of PA has yet to be definitively established, and the condition is usually idiopathic. In addition to bacterial or fungal etiologies,3,4 PA has been linked to medications (Table 1)16,20,21 and systemic conditions (Table 2).1,3,5,7-10,12,22-25
A PubMed search of articles indexed for MEDLINE using the search terms amiantacea, bone, drug, hair marrow, malignancy, melphalan, pityriasis, tinea, and transplant yielded 4 patients—2 men and 2 women (including our patient)—with possible drug-induced PA (Table 1)16,20,21; however, the onset after 2 years of medication (TNF-α inhibitors) or resolution while still receiving the agent (vemurafenib) makes the drug-induced linkage weak. The patients ranged in age from 21 to 67 years, with the median age being 37.5 years. Medications included melphalan, TNF-α inhibitors (adalimumab, infliximab),16,21 and vemurafenib20; it is interesting that infliximab was the medication associated with eliciting PA in 1 patient yet was an effective therapy in another patient with treatment-resistant PA. The onset of PA occurred between 1 month (melphalan) and 24 months (TNF-α inhibitors) after drug initiation. The patients’ associated diseases included Crohn disease,16,21 metastatic melanoma,20 and multiple myeloma.
Other conditions have been described in patients with PA (Table 2). Indeed, PA may be a manifestation of an underlying inflammatory skin disease.9 In addition to dermatologic conditions, procedures or malignancy may be associated with the disease, as demonstrated in our patient. Most commonly, PA is seen in association with psoriasis and seborrheic dermatitis; atopic dermatitis, bacterial infection, fungal infection, lichen planus, and neurodermatitis also have been associated with PA.1,3,5,7-10,12,18,22-25
Conclusion
Pityriasis amiantacea is a benign condition affecting the scalp hair. Albeit uncommon, it may appear in patients treated with medications such as melphalan, TNF-α inhibitors, and vemurafenib. In addition, it has been described in individuals with dermatologic conditions, systemic procedures, or underlying malignancy. Our patient developed PA following a bone marrow transplant after receiving conditioning with melphalan.
Pityriasis amiantacea (PA) is characterized by adherence of hair shafts proximally.1 It has been associated with dermatologic conditions and rarely with medications. We describe a woman who developed PA following a bone marrow transplant with melphalan conditioning. We also review drug-induced PA and disorders that have been linked to this condition.
Case Report
A 67-year-old woman with a history of multiple myeloma was treated with 7 courses of chemotherapy (cyclophosphamide, bortezomib, prednisone). One month later, the patient underwent a bone marrow transplant with melphalan conditioning due to residual plasma cell myeloma. Following the transplant, she developed complete scalp alopecia. Prior to and following transplant, the patient’s hair care regimen included washing her hair and scalp every other day with over-the-counter “natural” shampoos. During drug-induced alopecia, the hair washing became less frequent.
The patient left the hospital 4 weeks posttransplant; her hair had started to regrow, but its appearance was altered. Posttransplant, the patient was maintained on bortezomib every other week and zoledronate once per month. She continued to develop multiple lesions in the scalp hairs during the following 4 months.
Eight months posttransplant she presented for evaluation of the scalp hair. Clinical examination showed hairs that were entwined together proximally, resulting in matting of the hair (Figure 1). A diagnosis of PA was established based on the clinical examination.
Treatment included mineral oil application to the scalp under occlusion each evening, followed by morning washing with coal tar 0.5%, salicylic acid 6%, or ketoconazole 2% shampoo in a repeating sequential manner. Within 1 month there was complete resolution of the scalp condition (Figure 2).
Comment
Clinical Presentation
Pityriasis amiantacea is characterized by thick excessive scale of the scalp1; it was initially described by Alibert2 in 1832. He described the gross appearance of the scales as resembling the feathers of young birds, which naturalists dub “amiante” or asbestoslike.1,2 In 1917, Gougerot3 explored infectious etiologies of this condition by describing cases of impetigo that transitioned into PA.1 Later, in 1929, Photinos4 described fungal origins of PA, giving credence to “tinea amiantacea.”1 However, more recent analyses failed to isolate fungus.5-7 As such, pityriasis (scaling) amiantacea is the more appropriate term, as emphasized by Brown8 in 1948. The cause of PA remains unclear; it is hypothesized that the condition is a reaction to underlying inflammatory dermatoses, though concurrent bacterial or fungal infection may be present.5,9
Prevalence
Pityriasis amiantacea is considered to be most prevalent in pediatric patients and young adults; it is more common in females.1,9,10 In a review of 85 PA patients, more than 80% were women (n=69), and the mean age at presentation was 23.8 years. Approximately half of these patients had widespread scalp lesions (n=42); however, focal localized lesions were common.9 No hereditary patterns have been described, though 3 pairs of the 10 patients with PA in Ring and Kaplan’s7 review were siblings.
Clinical Findings
Clinically, lesions of PA present as matted hairs.1 Thick scales encompass multiple hair shafts, binding down tufts of hair.1,6,11 Patients are asymptomatic, though the lesions may be accompanied by pruritus. The hairs enclosed by the scales in some cases may be easily pulled out.6 Notably, alopecia often accompanies PA; it often is reversible, but in some cases, it is permanent and can lead to scarring.9,12
Histopathology
Submission of hair specimens to histopathology usually is not performed since the diagnosis often is established based on the clinical presentation.5 However, submitted specimens have demonstrated spongiosis and parakeratosis along with reduction in the size of the sebaceous glands.1,9 Additionally, follicular keratosis that surrounds the hair shafts with a sheath of horn is present.9 Acanthosis and migration of lymphocytes into the epidermis also have been found.1 Often, Staphylococcus aureus isolates are detected.9,13
Differential Diagnosis
The clinical differential diagnosis of PA includes hair casts,11 pediculosis,14 and tinea capitis.12 In PA, thick scales surround hair shafts and thus bind down tufts of hair.9 In patients with pediculosis, nits are attached to the hair shaft at an angle and do not entirely envelop the hair shaft.14 In addition, PA may be complicated by impetiginization; bacteria often are found in the keratin surrounding the hair shaft and represent either normal flora or secondary infection.1,15 It has been speculated that microbial biofilms from S aureus and Staphylococcus epidermidis promote agglomeration of hair shafts and adherent scale.16 Bona fide dermatophyte infection of the scalp also may be concurrently present.12
Treatment
Our treatment included occlusion with mineral oil to loosen the scales from the scalp in tandem with shampoos traditionally used in patients with seborrheic dermatitis or psoriasis. Timely treatment is important to prevent scarring alopecia.13,17 Pityriasis amiantacea may be treatment resistant, and there are no specific therapeutic guidelines; rather, therapy should be targeted at the suspected underlying condition.17 Treatment generally includes keratolytic agents, such as salicylic acid.18 These agents allow enhanced penetration of other topical agents.19 Topical antifungal shampoos such as ketoconazole and ciclopirox are recommended,18 though other topical agents, such as coal tar and zinc pyrithione, also may benefit patients.13 Topical corticosteroids may be used if the condition is linked with psoriasis.13 Systemic antibiotics are added if S aureus superinfection is suspected.9
A single report described successful management of a patient with severe refractory PA who was treated with the tumor necrosis factor (TNF) α inhibitor infliximab.13 A 47-year-old woman presented with thick adherent scale on the scalp. She was treated with coal tar for 18 months but showed no improvement; the patient was subsequently prescribed salicylic acid 10%, clobetasol solution, and coal tar shampoo. After 3 months, when no improvement was observed, the patient was offered infliximab but declined. For 6 years the patient was treated with salicylic acid 20%, clobetasol (foam, lotion, shampoo, and solution), and coal tar shampoo without improvement. She then consented to infliximab therapy; after 3 infusions at weeks 0, 2, and 6, she demonstrated notable improvement. The patient was maintained on infliximab every 8 weeks.13
Pathogenesis
The pathogenesis of PA has yet to be definitively established, and the condition is usually idiopathic. In addition to bacterial or fungal etiologies,3,4 PA has been linked to medications (Table 1)16,20,21 and systemic conditions (Table 2).1,3,5,7-10,12,22-25
A PubMed search of articles indexed for MEDLINE using the search terms amiantacea, bone, drug, hair marrow, malignancy, melphalan, pityriasis, tinea, and transplant yielded 4 patients—2 men and 2 women (including our patient)—with possible drug-induced PA (Table 1)16,20,21; however, the onset after 2 years of medication (TNF-α inhibitors) or resolution while still receiving the agent (vemurafenib) makes the drug-induced linkage weak. The patients ranged in age from 21 to 67 years, with the median age being 37.5 years. Medications included melphalan, TNF-α inhibitors (adalimumab, infliximab),16,21 and vemurafenib20; it is interesting that infliximab was the medication associated with eliciting PA in 1 patient yet was an effective therapy in another patient with treatment-resistant PA. The onset of PA occurred between 1 month (melphalan) and 24 months (TNF-α inhibitors) after drug initiation. The patients’ associated diseases included Crohn disease,16,21 metastatic melanoma,20 and multiple myeloma.
Other conditions have been described in patients with PA (Table 2). Indeed, PA may be a manifestation of an underlying inflammatory skin disease.9 In addition to dermatologic conditions, procedures or malignancy may be associated with the disease, as demonstrated in our patient. Most commonly, PA is seen in association with psoriasis and seborrheic dermatitis; atopic dermatitis, bacterial infection, fungal infection, lichen planus, and neurodermatitis also have been associated with PA.1,3,5,7-10,12,18,22-25
Conclusion
Pityriasis amiantacea is a benign condition affecting the scalp hair. Albeit uncommon, it may appear in patients treated with medications such as melphalan, TNF-α inhibitors, and vemurafenib. In addition, it has been described in individuals with dermatologic conditions, systemic procedures, or underlying malignancy. Our patient developed PA following a bone marrow transplant after receiving conditioning with melphalan.
- Knight AG. Pityriasis amiantacea: a clinical and histopathological investigation. Clin Exp Dermatol. 1977;2:137-143.
- Alibert JL. De la porrigine amiantacée. In: Monographie des Dermatoses. Paris, France: Baillère; 1832:293-295.
- Gougerot H. La teigne amiantacee D’Alibert. Progres Medical. 1917;13:101-104.
- Photinos P. Recherches sur la fausse teigne amiantacée. Ann Dermatol Syphiligr. 1929;10:743-758.
- Verardino GC, Azulay-Abulafia L, Macedo PM, et al. Pityriasis amiantacea: clinical-dermatoscopic features and microscopy of hair tufts. An Bras Dermatol. 2012;87:142-145.
- Keipert JA. Greasy scaling pityriasis amiantacea and alopecia: a syndrome in search of a cause. Australas J Dermatol. 1985;26:41-44.
- Ring DS, Kaplan DL. Pityriasis amiantacea: a report of 10 cases. Arch Dermatol. 1993;129:913-914.
- Brown WH. Some observations on neurodermatitis of the scalp, with particular reference to tinea amiantacea. Br J Dermatol Syph. 1948;60:81-90.
- Abdel-Hamid IA, Agha SA, Moustafa YM, et al. Pityriasis amiantacea: a clinical and etiopathologic study of 85 patients. Int J Dermatol. 2003;42:260-264.
- Becker SW, Muir KB. Tinea amiantacea. Arch Dermatol Syphil. 1929;20:45-53.
- Dawber RP. Hair casts. Br J Dermatol. 1979;100:417-421.
- Ginarte M, Pereiro M, Fernández-Redondo V, et al. Case reports. pityriasis amiantacea as manifestation of tinea capitis due to Microsporum canis. Mycoses. 2000;43:93-96.
- Pham RK, Chan CS, Hsu S. Treatment of pityriasis amiantacea with infliximab. Dermatol Online J. 2009;15:13.
- Roberts RJ. Clinical practice. Head lice. N Engl J Med. 2002;346:1645-1650.
- Mcginley KJ, Leyden JJ, Marples RR, et al. Quantitative microbiology of the scalp in non-dandruff, dandruff, and seborrheic dermatitis. J Invest Dermatol. 1975;64:401-405.
- Ettler J, Wetter DA, Pittelkow MR. Pityriasis amiantacea: a distinctive presentation of psoriasis associated with tumour necrosis factor-α inhibitor therapy. Clin Exp Dermatol. 2012;37:639-641.
- Mannino G, McCaughey C, Vanness E. A case of pityriasis amiantacea with rapid response to treatment. WMJ. 2014;113:119-120.
- Jamil A, Muthupalaniappen L. Scales on the scalp. Malays Fam Physician. 2013;8:48-49.
- Gupta LK, Khare AK, Masatkar V, et al. Pityriasis amiantacea. Indian Dermatol Online J. 2014;5(suppl 1):S63-S64.
- Bilgiç Ö. Vemurafenib-induced pityriasis amiantacea: a case report. Cutan Ocul Toxicol. 2016;35:329-331.
- Zamperetti M, Zelger B, Höpfl R. Pityriasis amiantacea and folliculitis decalvans: an unusual manifestation associated with antitumor necrosis factor-α therapy. Hautarzt. 2017;68:1007-1010.
- Udayashankar C, Nath AK, Anuradha P. Extensive Darier’s disease with pityriasis amiantacea, alopecia and congenital facial nerve palsy. Dermatol Online J. 2013;19:18574.
- Hussain W, Coulson IH, Salman WD. Pityriasis amiantacea as the sole manifestation of Darier’s disease. Clin Exp Dermatol. 2009;34:554-556.
- Hansted B, Lindskov R. Pityriasis amiantacea and psoriasis. a follow-up study. Dermatologica. 1983;166:314-315.
- Hersle K, Lindholm A, Mobacken H, et al. Relationship of pityriasis amiantacea to psoriasis. a follow-up study. Dermatologica. 1979;159:245-250.
- Knight AG. Pityriasis amiantacea: a clinical and histopathological investigation. Clin Exp Dermatol. 1977;2:137-143.
- Alibert JL. De la porrigine amiantacée. In: Monographie des Dermatoses. Paris, France: Baillère; 1832:293-295.
- Gougerot H. La teigne amiantacee D’Alibert. Progres Medical. 1917;13:101-104.
- Photinos P. Recherches sur la fausse teigne amiantacée. Ann Dermatol Syphiligr. 1929;10:743-758.
- Verardino GC, Azulay-Abulafia L, Macedo PM, et al. Pityriasis amiantacea: clinical-dermatoscopic features and microscopy of hair tufts. An Bras Dermatol. 2012;87:142-145.
- Keipert JA. Greasy scaling pityriasis amiantacea and alopecia: a syndrome in search of a cause. Australas J Dermatol. 1985;26:41-44.
- Ring DS, Kaplan DL. Pityriasis amiantacea: a report of 10 cases. Arch Dermatol. 1993;129:913-914.
- Brown WH. Some observations on neurodermatitis of the scalp, with particular reference to tinea amiantacea. Br J Dermatol Syph. 1948;60:81-90.
- Abdel-Hamid IA, Agha SA, Moustafa YM, et al. Pityriasis amiantacea: a clinical and etiopathologic study of 85 patients. Int J Dermatol. 2003;42:260-264.
- Becker SW, Muir KB. Tinea amiantacea. Arch Dermatol Syphil. 1929;20:45-53.
- Dawber RP. Hair casts. Br J Dermatol. 1979;100:417-421.
- Ginarte M, Pereiro M, Fernández-Redondo V, et al. Case reports. pityriasis amiantacea as manifestation of tinea capitis due to Microsporum canis. Mycoses. 2000;43:93-96.
- Pham RK, Chan CS, Hsu S. Treatment of pityriasis amiantacea with infliximab. Dermatol Online J. 2009;15:13.
- Roberts RJ. Clinical practice. Head lice. N Engl J Med. 2002;346:1645-1650.
- Mcginley KJ, Leyden JJ, Marples RR, et al. Quantitative microbiology of the scalp in non-dandruff, dandruff, and seborrheic dermatitis. J Invest Dermatol. 1975;64:401-405.
- Ettler J, Wetter DA, Pittelkow MR. Pityriasis amiantacea: a distinctive presentation of psoriasis associated with tumour necrosis factor-α inhibitor therapy. Clin Exp Dermatol. 2012;37:639-641.
- Mannino G, McCaughey C, Vanness E. A case of pityriasis amiantacea with rapid response to treatment. WMJ. 2014;113:119-120.
- Jamil A, Muthupalaniappen L. Scales on the scalp. Malays Fam Physician. 2013;8:48-49.
- Gupta LK, Khare AK, Masatkar V, et al. Pityriasis amiantacea. Indian Dermatol Online J. 2014;5(suppl 1):S63-S64.
- Bilgiç Ö. Vemurafenib-induced pityriasis amiantacea: a case report. Cutan Ocul Toxicol. 2016;35:329-331.
- Zamperetti M, Zelger B, Höpfl R. Pityriasis amiantacea and folliculitis decalvans: an unusual manifestation associated with antitumor necrosis factor-α therapy. Hautarzt. 2017;68:1007-1010.
- Udayashankar C, Nath AK, Anuradha P. Extensive Darier’s disease with pityriasis amiantacea, alopecia and congenital facial nerve palsy. Dermatol Online J. 2013;19:18574.
- Hussain W, Coulson IH, Salman WD. Pityriasis amiantacea as the sole manifestation of Darier’s disease. Clin Exp Dermatol. 2009;34:554-556.
- Hansted B, Lindskov R. Pityriasis amiantacea and psoriasis. a follow-up study. Dermatologica. 1983;166:314-315.
- Hersle K, Lindholm A, Mobacken H, et al. Relationship of pityriasis amiantacea to psoriasis. a follow-up study. Dermatologica. 1979;159:245-250.
Practice Points
- Pityriasis amiantacea (PA) is associated with several dermatologic conditions, including atopic dermatitis, bacterial and fungal infections, psoriasis, and seborrheic dermatitis.
- Drug-induced PA is rare, but the condition has been reported in the context of treatment with tumor necrosis factor Symbol Stdα inhibitors and vemurafenib.
- Our report suggests that PA may be associated with either melphalan conditioning, bone marrow transplant, or both.
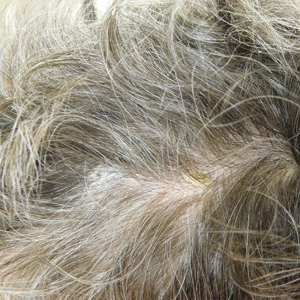
Digital Revolution: Dermatology Is on the Edge
The Digital Revolution has invaded the House of Medicine, which is not really news since the invasion has been long-standing, but it seems to be generating more interest and concern in recent months. The American Medical Association recently created the Digital Health Implementation Playbook to lend guidance in developing technologies that are fundamentally altering the manner in which patients interact with health care providers.1 The playbook lays out specific steps for developing and implementing digital health technologies such as remote patient monitoring devices. The goal of the playbook is to make certain that such devices are accurate, reliable, and validated as valuable additions to high-quality patient care.1
In the February 2018 issue of Cutis, Masud et al2 evaluated 44 patient-directed mobile applications (apps) for dermatologic conditions and developed a schematic for evaluating their value in providing valid usable information for patients. They found that most of the apps failed to live up to their purported usefulness in patient education.2 I am certain we have all seen numerous examples on the Internet, many times brought to us by patients, of fallacious and inaccurate information about the diagnosis and treatment of dermatologic conditions that are actually harmful to the care of our patients.
A more upsetting trend in recent years is the proliferation of open-access journals. Although such digital journals can result in more rapid dissemination of valid scientific information, many of them do not follow a true peer-review process. So-called predatory journals from for-profit unethical publishers are increasing at an alarming rate.3
Furthermore, there is a need to present data more accurately and in formats that provide more meaningful interpretation, according to a recent Letter From the Editor in the Journal of the American Academy of Dermatology.4 Elston4 wrote: “Be honest about your data and the limitations of the study design.” He cautioned further about the proper use of statistical analysis.
As dermatologists, how do we make certain that the Digital Revolution results in better care of our patients? The answer, of course, is in education of the practitioner and our young colleagues in training. Although most Cutis readers still access the print version of our journal, more and more readers are accessing our digital format. Online we are able to offer readers the peer-reviewed content you have known to trust and rely on to improve your care of patients as well as other educational tools. Furthermore, we can provide readers access to additional charts and tables pertaining to research published in the print journal.
In January 2019 the Cutis website will merge with Dermatology News, our sister news publication, to become MDedge Dermatology (www.mdedge.com/dermatology). This site will be your new one-stop destination for timely news and clinical content you can trust from both publications. This interactive site is designed to help clinicians quickly find the information they need to improve the treatment and care of patients with conditions affecting the hair, skin, and nails. You will have free access to digital resources such as procedural videos, podcasts, image quizzes, board review, and resident resources, as well as an archive of Cutis content dating back to 2000.
As we at Cutis broaden our digital footprint, we look forward to providing our readers with a larger volume of clinically relevant content in more easily accessed formats while maintaining our commitment to valid trustworthy information. In the coming months we look forward to joining with you in this new digital endeavor, and as always, we appreciate the input of our readers during this process.
1. AMA announces playbook to successfully adopt digital health [press release]. Boston, MA: American Medical Association; October 16, 2018. https://www.ama-assn.org/press-center/press-releases/ama-announces-playbook-successfully-adopt-digital-health. Accessed December 14, 2018.
2. Masud A, Shafi S, Rao BK. Mobile medical apps for patient education: a graded review of available dermatology apps. Cutis. 2018;101:141-144.
3. Shahrivar N, Grant-Kels JM, Payette MJ. Predatory journals: how to recognize and avoid the threat of involvement with these unethical “publishers.” J Am Acad Dermatol. 2016;75:658-659.
4. Elston DM. Presentation of data. J Am Acad Dermatol. 2019;80:55.
The Digital Revolution has invaded the House of Medicine, which is not really news since the invasion has been long-standing, but it seems to be generating more interest and concern in recent months. The American Medical Association recently created the Digital Health Implementation Playbook to lend guidance in developing technologies that are fundamentally altering the manner in which patients interact with health care providers.1 The playbook lays out specific steps for developing and implementing digital health technologies such as remote patient monitoring devices. The goal of the playbook is to make certain that such devices are accurate, reliable, and validated as valuable additions to high-quality patient care.1
In the February 2018 issue of Cutis, Masud et al2 evaluated 44 patient-directed mobile applications (apps) for dermatologic conditions and developed a schematic for evaluating their value in providing valid usable information for patients. They found that most of the apps failed to live up to their purported usefulness in patient education.2 I am certain we have all seen numerous examples on the Internet, many times brought to us by patients, of fallacious and inaccurate information about the diagnosis and treatment of dermatologic conditions that are actually harmful to the care of our patients.
A more upsetting trend in recent years is the proliferation of open-access journals. Although such digital journals can result in more rapid dissemination of valid scientific information, many of them do not follow a true peer-review process. So-called predatory journals from for-profit unethical publishers are increasing at an alarming rate.3
Furthermore, there is a need to present data more accurately and in formats that provide more meaningful interpretation, according to a recent Letter From the Editor in the Journal of the American Academy of Dermatology.4 Elston4 wrote: “Be honest about your data and the limitations of the study design.” He cautioned further about the proper use of statistical analysis.
As dermatologists, how do we make certain that the Digital Revolution results in better care of our patients? The answer, of course, is in education of the practitioner and our young colleagues in training. Although most Cutis readers still access the print version of our journal, more and more readers are accessing our digital format. Online we are able to offer readers the peer-reviewed content you have known to trust and rely on to improve your care of patients as well as other educational tools. Furthermore, we can provide readers access to additional charts and tables pertaining to research published in the print journal.
In January 2019 the Cutis website will merge with Dermatology News, our sister news publication, to become MDedge Dermatology (www.mdedge.com/dermatology). This site will be your new one-stop destination for timely news and clinical content you can trust from both publications. This interactive site is designed to help clinicians quickly find the information they need to improve the treatment and care of patients with conditions affecting the hair, skin, and nails. You will have free access to digital resources such as procedural videos, podcasts, image quizzes, board review, and resident resources, as well as an archive of Cutis content dating back to 2000.
As we at Cutis broaden our digital footprint, we look forward to providing our readers with a larger volume of clinically relevant content in more easily accessed formats while maintaining our commitment to valid trustworthy information. In the coming months we look forward to joining with you in this new digital endeavor, and as always, we appreciate the input of our readers during this process.
The Digital Revolution has invaded the House of Medicine, which is not really news since the invasion has been long-standing, but it seems to be generating more interest and concern in recent months. The American Medical Association recently created the Digital Health Implementation Playbook to lend guidance in developing technologies that are fundamentally altering the manner in which patients interact with health care providers.1 The playbook lays out specific steps for developing and implementing digital health technologies such as remote patient monitoring devices. The goal of the playbook is to make certain that such devices are accurate, reliable, and validated as valuable additions to high-quality patient care.1
In the February 2018 issue of Cutis, Masud et al2 evaluated 44 patient-directed mobile applications (apps) for dermatologic conditions and developed a schematic for evaluating their value in providing valid usable information for patients. They found that most of the apps failed to live up to their purported usefulness in patient education.2 I am certain we have all seen numerous examples on the Internet, many times brought to us by patients, of fallacious and inaccurate information about the diagnosis and treatment of dermatologic conditions that are actually harmful to the care of our patients.
A more upsetting trend in recent years is the proliferation of open-access journals. Although such digital journals can result in more rapid dissemination of valid scientific information, many of them do not follow a true peer-review process. So-called predatory journals from for-profit unethical publishers are increasing at an alarming rate.3
Furthermore, there is a need to present data more accurately and in formats that provide more meaningful interpretation, according to a recent Letter From the Editor in the Journal of the American Academy of Dermatology.4 Elston4 wrote: “Be honest about your data and the limitations of the study design.” He cautioned further about the proper use of statistical analysis.
As dermatologists, how do we make certain that the Digital Revolution results in better care of our patients? The answer, of course, is in education of the practitioner and our young colleagues in training. Although most Cutis readers still access the print version of our journal, more and more readers are accessing our digital format. Online we are able to offer readers the peer-reviewed content you have known to trust and rely on to improve your care of patients as well as other educational tools. Furthermore, we can provide readers access to additional charts and tables pertaining to research published in the print journal.
In January 2019 the Cutis website will merge with Dermatology News, our sister news publication, to become MDedge Dermatology (www.mdedge.com/dermatology). This site will be your new one-stop destination for timely news and clinical content you can trust from both publications. This interactive site is designed to help clinicians quickly find the information they need to improve the treatment and care of patients with conditions affecting the hair, skin, and nails. You will have free access to digital resources such as procedural videos, podcasts, image quizzes, board review, and resident resources, as well as an archive of Cutis content dating back to 2000.
As we at Cutis broaden our digital footprint, we look forward to providing our readers with a larger volume of clinically relevant content in more easily accessed formats while maintaining our commitment to valid trustworthy information. In the coming months we look forward to joining with you in this new digital endeavor, and as always, we appreciate the input of our readers during this process.
1. AMA announces playbook to successfully adopt digital health [press release]. Boston, MA: American Medical Association; October 16, 2018. https://www.ama-assn.org/press-center/press-releases/ama-announces-playbook-successfully-adopt-digital-health. Accessed December 14, 2018.
2. Masud A, Shafi S, Rao BK. Mobile medical apps for patient education: a graded review of available dermatology apps. Cutis. 2018;101:141-144.
3. Shahrivar N, Grant-Kels JM, Payette MJ. Predatory journals: how to recognize and avoid the threat of involvement with these unethical “publishers.” J Am Acad Dermatol. 2016;75:658-659.
4. Elston DM. Presentation of data. J Am Acad Dermatol. 2019;80:55.
1. AMA announces playbook to successfully adopt digital health [press release]. Boston, MA: American Medical Association; October 16, 2018. https://www.ama-assn.org/press-center/press-releases/ama-announces-playbook-successfully-adopt-digital-health. Accessed December 14, 2018.
2. Masud A, Shafi S, Rao BK. Mobile medical apps for patient education: a graded review of available dermatology apps. Cutis. 2018;101:141-144.
3. Shahrivar N, Grant-Kels JM, Payette MJ. Predatory journals: how to recognize and avoid the threat of involvement with these unethical “publishers.” J Am Acad Dermatol. 2016;75:658-659.
4. Elston DM. Presentation of data. J Am Acad Dermatol. 2019;80:55.

Blanchable Erythematous Patches on the Fingers
The Diagnosis: Irritant Contact Dermatitis
The diagnosis of irritant contact dermatitis secondary to skateboarding is similar to pool palms, a benign, self-limiting irritant contact dermatitis.1 We propose that contact with concrete surfaces during skateboarding can lead to a presentation similar to pool palms. In our case, it was likely that the finger pulpitis noted in the physical examination was due to daily skateboarding rather than once-weekly swimming. Furthermore, the fingertip contact with concrete in pool palms is similar to the rough surface exposure on the skateboard.
Pool palms is more commonly reported in children due to their participation in sports and other activities with recent exposure to rough surfaces, most commonly the floor of swimming pools.2 The condition resolves after eliminating exposures.3 The frequency and duration of exposure to rough surfaces in swimming pools leading to development of this condition is unknown.
There have been mixed reports on the pathogenesis of pool palms. Some literature supports the idea that it is a wet dermatitis, a combination of prolonged water contact, friction, chemicals, and microbes leading to a chronic dermatitis. This theory states that the primary factor influencing the development of erythematous patches on the fingers, palms, and soles is the hyperhydration of the corneal layer at these sites.4 A different theory attributes pool palms to a mechanical origin, such as repeated microtrauma from contact with the rough concrete surfaces of swimming pools.5 This theory further states that the chemicals in pool water, such as chlorine and sodium hypochlorite, rarely produce irritant, allergic, or urticarial reactions.3
Based on these theories, we hypothesized that fingertip pulpitis can result from activities other than swimming (eg, skateboarding). Our case supports the latter theory on fingertip pulpitis in pool palms being a result of frictional dermatitis rather than wet dermatitis because we attributed our patient’s findings to contact with rough surfaces during skateboarding. Although the patient did swim, he only did so once weekly in the summer months, and the lesions had been persistent for 2 years consistently. His skateboarding hobby was more frequent, and he endorsed contact of the pads of the bilateral second to fifth fingers to the rough surfaces of the road and skateboard. The patient did not have lesions on the toes, further supporting the hypothesis that skateboarding led to the current presentation.
In children, hand-foot-and-mouth disease classically presents with oval-shaped, erythematous vesicles on the palmar surfaces of the hands and feet and generally is accompanied by fever and sore throat.6 Furthermore, unlike in our case, the viral exanthem usually would be present for up to 3 weeks and would not persist for more than 2 years. Erythema multiforme has an erythematous color and can present on the palms; however, the lesions have a classic targetoid appearance. It would be unique for erythema multiforme to present only on the fingertips rather than more diffusely on the palms or in other areas such as the face.7 Limited cutaneous sclerosis (scleroderma) initially can present with edematous pitted scars on the digital tips; however, with time the fingers will have a taut, white, shiny appearance that can develop into contractures and debilitating ulcerations.8 In our patient, the plaques did not advance to any further disease. Lastly, in contrast to our patient, punctate palmoplantar keratoderma presents as hyperkeratotic, firm, translucent, or opaque papules on the palms and soles. Over time, the papules can appear verrucous or callouslike.9 In our case, the plaques on the fingertips were erythematous rather than translucent or opaque papules.
Our case raises questions on whether prior reports of pool palms can be attributed to other activities involving contact with rough surfaces. More research is needed on the frequency and duration of rough surface exposure resulting in fingertip pulpitis.
- Lopez-Neyra A, Vano-Galvan S, Alvarez-Twose I, et al. Pool palms [in Spanish]. Dermatol Online J. 2009;15:17.
- Wong LC, Rogers M. Pool palms. Pediatr Dermatol. 2007;24:95.
- Mandojana RM. Pool palms. J Am Acad Dermatol. 1993;28(2 pt 1):280-281.
- Novoa A, Klear S. Pool palms [published online September 30, 2015]. Arch Dis Child. 2016;101:41.
- Martín JM, Martín JM, Ricart JM. Erythematous-violaceous lesions on the palms [in Spanish]. Actas Dermosifiliogr. 2009;100:507-508.
- Marcini AJ, Shani-Adir A. Other viral diseases. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Ltd; 2012:1345-1366.
- French LE, Prins C. Erythema multiforme, Stevens-Johnson syndrome, and toxic epidermal necrosis. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Ltd; 2012:319-334.
- Connoly MK. Systemic sclerosis (scleroderma) and related disorders. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Ltd; 2012:643-646.
- Krol AL, Siegel D. Keratodermas. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Ltd; 2012:871-886.
The Diagnosis: Irritant Contact Dermatitis
The diagnosis of irritant contact dermatitis secondary to skateboarding is similar to pool palms, a benign, self-limiting irritant contact dermatitis.1 We propose that contact with concrete surfaces during skateboarding can lead to a presentation similar to pool palms. In our case, it was likely that the finger pulpitis noted in the physical examination was due to daily skateboarding rather than once-weekly swimming. Furthermore, the fingertip contact with concrete in pool palms is similar to the rough surface exposure on the skateboard.
Pool palms is more commonly reported in children due to their participation in sports and other activities with recent exposure to rough surfaces, most commonly the floor of swimming pools.2 The condition resolves after eliminating exposures.3 The frequency and duration of exposure to rough surfaces in swimming pools leading to development of this condition is unknown.
There have been mixed reports on the pathogenesis of pool palms. Some literature supports the idea that it is a wet dermatitis, a combination of prolonged water contact, friction, chemicals, and microbes leading to a chronic dermatitis. This theory states that the primary factor influencing the development of erythematous patches on the fingers, palms, and soles is the hyperhydration of the corneal layer at these sites.4 A different theory attributes pool palms to a mechanical origin, such as repeated microtrauma from contact with the rough concrete surfaces of swimming pools.5 This theory further states that the chemicals in pool water, such as chlorine and sodium hypochlorite, rarely produce irritant, allergic, or urticarial reactions.3
Based on these theories, we hypothesized that fingertip pulpitis can result from activities other than swimming (eg, skateboarding). Our case supports the latter theory on fingertip pulpitis in pool palms being a result of frictional dermatitis rather than wet dermatitis because we attributed our patient’s findings to contact with rough surfaces during skateboarding. Although the patient did swim, he only did so once weekly in the summer months, and the lesions had been persistent for 2 years consistently. His skateboarding hobby was more frequent, and he endorsed contact of the pads of the bilateral second to fifth fingers to the rough surfaces of the road and skateboard. The patient did not have lesions on the toes, further supporting the hypothesis that skateboarding led to the current presentation.
In children, hand-foot-and-mouth disease classically presents with oval-shaped, erythematous vesicles on the palmar surfaces of the hands and feet and generally is accompanied by fever and sore throat.6 Furthermore, unlike in our case, the viral exanthem usually would be present for up to 3 weeks and would not persist for more than 2 years. Erythema multiforme has an erythematous color and can present on the palms; however, the lesions have a classic targetoid appearance. It would be unique for erythema multiforme to present only on the fingertips rather than more diffusely on the palms or in other areas such as the face.7 Limited cutaneous sclerosis (scleroderma) initially can present with edematous pitted scars on the digital tips; however, with time the fingers will have a taut, white, shiny appearance that can develop into contractures and debilitating ulcerations.8 In our patient, the plaques did not advance to any further disease. Lastly, in contrast to our patient, punctate palmoplantar keratoderma presents as hyperkeratotic, firm, translucent, or opaque papules on the palms and soles. Over time, the papules can appear verrucous or callouslike.9 In our case, the plaques on the fingertips were erythematous rather than translucent or opaque papules.
Our case raises questions on whether prior reports of pool palms can be attributed to other activities involving contact with rough surfaces. More research is needed on the frequency and duration of rough surface exposure resulting in fingertip pulpitis.
The Diagnosis: Irritant Contact Dermatitis
The diagnosis of irritant contact dermatitis secondary to skateboarding is similar to pool palms, a benign, self-limiting irritant contact dermatitis.1 We propose that contact with concrete surfaces during skateboarding can lead to a presentation similar to pool palms. In our case, it was likely that the finger pulpitis noted in the physical examination was due to daily skateboarding rather than once-weekly swimming. Furthermore, the fingertip contact with concrete in pool palms is similar to the rough surface exposure on the skateboard.
Pool palms is more commonly reported in children due to their participation in sports and other activities with recent exposure to rough surfaces, most commonly the floor of swimming pools.2 The condition resolves after eliminating exposures.3 The frequency and duration of exposure to rough surfaces in swimming pools leading to development of this condition is unknown.
There have been mixed reports on the pathogenesis of pool palms. Some literature supports the idea that it is a wet dermatitis, a combination of prolonged water contact, friction, chemicals, and microbes leading to a chronic dermatitis. This theory states that the primary factor influencing the development of erythematous patches on the fingers, palms, and soles is the hyperhydration of the corneal layer at these sites.4 A different theory attributes pool palms to a mechanical origin, such as repeated microtrauma from contact with the rough concrete surfaces of swimming pools.5 This theory further states that the chemicals in pool water, such as chlorine and sodium hypochlorite, rarely produce irritant, allergic, or urticarial reactions.3
Based on these theories, we hypothesized that fingertip pulpitis can result from activities other than swimming (eg, skateboarding). Our case supports the latter theory on fingertip pulpitis in pool palms being a result of frictional dermatitis rather than wet dermatitis because we attributed our patient’s findings to contact with rough surfaces during skateboarding. Although the patient did swim, he only did so once weekly in the summer months, and the lesions had been persistent for 2 years consistently. His skateboarding hobby was more frequent, and he endorsed contact of the pads of the bilateral second to fifth fingers to the rough surfaces of the road and skateboard. The patient did not have lesions on the toes, further supporting the hypothesis that skateboarding led to the current presentation.
In children, hand-foot-and-mouth disease classically presents with oval-shaped, erythematous vesicles on the palmar surfaces of the hands and feet and generally is accompanied by fever and sore throat.6 Furthermore, unlike in our case, the viral exanthem usually would be present for up to 3 weeks and would not persist for more than 2 years. Erythema multiforme has an erythematous color and can present on the palms; however, the lesions have a classic targetoid appearance. It would be unique for erythema multiforme to present only on the fingertips rather than more diffusely on the palms or in other areas such as the face.7 Limited cutaneous sclerosis (scleroderma) initially can present with edematous pitted scars on the digital tips; however, with time the fingers will have a taut, white, shiny appearance that can develop into contractures and debilitating ulcerations.8 In our patient, the plaques did not advance to any further disease. Lastly, in contrast to our patient, punctate palmoplantar keratoderma presents as hyperkeratotic, firm, translucent, or opaque papules on the palms and soles. Over time, the papules can appear verrucous or callouslike.9 In our case, the plaques on the fingertips were erythematous rather than translucent or opaque papules.
Our case raises questions on whether prior reports of pool palms can be attributed to other activities involving contact with rough surfaces. More research is needed on the frequency and duration of rough surface exposure resulting in fingertip pulpitis.
- Lopez-Neyra A, Vano-Galvan S, Alvarez-Twose I, et al. Pool palms [in Spanish]. Dermatol Online J. 2009;15:17.
- Wong LC, Rogers M. Pool palms. Pediatr Dermatol. 2007;24:95.
- Mandojana RM. Pool palms. J Am Acad Dermatol. 1993;28(2 pt 1):280-281.
- Novoa A, Klear S. Pool palms [published online September 30, 2015]. Arch Dis Child. 2016;101:41.
- Martín JM, Martín JM, Ricart JM. Erythematous-violaceous lesions on the palms [in Spanish]. Actas Dermosifiliogr. 2009;100:507-508.
- Marcini AJ, Shani-Adir A. Other viral diseases. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Ltd; 2012:1345-1366.
- French LE, Prins C. Erythema multiforme, Stevens-Johnson syndrome, and toxic epidermal necrosis. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Ltd; 2012:319-334.
- Connoly MK. Systemic sclerosis (scleroderma) and related disorders. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Ltd; 2012:643-646.
- Krol AL, Siegel D. Keratodermas. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Ltd; 2012:871-886.
- Lopez-Neyra A, Vano-Galvan S, Alvarez-Twose I, et al. Pool palms [in Spanish]. Dermatol Online J. 2009;15:17.
- Wong LC, Rogers M. Pool palms. Pediatr Dermatol. 2007;24:95.
- Mandojana RM. Pool palms. J Am Acad Dermatol. 1993;28(2 pt 1):280-281.
- Novoa A, Klear S. Pool palms [published online September 30, 2015]. Arch Dis Child. 2016;101:41.
- Martín JM, Martín JM, Ricart JM. Erythematous-violaceous lesions on the palms [in Spanish]. Actas Dermosifiliogr. 2009;100:507-508.
- Marcini AJ, Shani-Adir A. Other viral diseases. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Ltd; 2012:1345-1366.
- French LE, Prins C. Erythema multiforme, Stevens-Johnson syndrome, and toxic epidermal necrosis. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Ltd; 2012:319-334.
- Connoly MK. Systemic sclerosis (scleroderma) and related disorders. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Ltd; 2012:643-646.
- Krol AL, Siegel D. Keratodermas. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Ltd; 2012:871-886.

A 12-year-old boy presented with well-defined, blanchable, erythematous patches on the distal bilateral palmar aspects of the second to fifth fingers of 2 years’ duration. The patient stated that he skateboarded daily throughout the year and swam once weekly in the summer months. Furthermore, the patient cited frequent contact with the rough undersurface of the skateboard and concrete road surfaces while skateboarding. He stated that the lesions were always present and worsened in the summer months. The lesions had an occasional burning sensation when they were more prominently erythematous, and the patient denied any pattern of exacerbation, numbness, bleeding, or itching. There was no notable family history or evidence of systemic disease.

Lipoblastoma of the Scalp in a Child
To the Editor:
A 2-year-old boy was referred to our pediatric dermatology clinic by his pediatrician for an enlarging mass on the mid frontal scalp. The lesion had been present since birth and slowly enlarged. His parents thought the lesion was mostly asymptomatic; however, if it was irritated, the child would cry. He was otherwise healthy and had no history of skin conditions. There was no family history of skin conditions, birthmarks, or vascular malformations. On physical examination, we observed an isolated, approximately 3-cm, well-circumscribed, mobile, flesh-colored and violaceous nodule on the mid frontal scalp (Figure 1). At that time our differential diagnosis included a complex hemangioma or other vascular proliferation, nevus lipomatosis, or even a soft-tissue malignancy such as a sarcoma. Prior to biopsy, we ordered magnetic resonance imaging (MRI) to evaluate for intracranial extension of the lesion. The MRI revealed a 3.2-cm frontal midline scalp mass with complex imaging characteristics, and the radiologist gave a differential diagnosis of hemangioma, teratoma, or less likely liposarcoma (Figure 2). Fortunately, there was no evidence of central nervous system or intracranial invasion. We then proceeded with excisional biopsy, which grossly revealed a nodular, well-circumscribed, yellow mass (Figure 3). The wound was closed with primary closure. Histologically, there was a lobulated tumor with thin, well-vascularized connective tissue septa within a myxoid stroma (Figure 4A). The tumor was composed of lipocytes in varying stages of maturity without obvious nuclear atypia (Figures 4B and 4C), leading to a diagnosis of lipoblastoma.

Lipoblastoma (also known as an embryonic lipoma) is a rare variant of lipoma. It is a benign neoplasm of immature white fat cells primarily seen in children younger than 3 years. It is reportedly twice as common in boys versus girls. Lipoblastomas present as enlarging, soft, mobile, painless nodules, usually 3 to 5 cm in diameter. The extremities are the favored location, but they also have been described on the head, neck, and trunk. Additionally, mediastinal and retroperitoneal lipoblastomas have been documented.1 The tumors may be symptomatic, particularly when involving the neck or mediastinum. In rare instances, they may present with respiratory distress.2 Multiple cases of head and neck lipoblastomas have been published in the English-language literature.3 Growing evidence supports that a chromosomal breakpoint abnormality at 8q11-q13 may be implicated in the pathogenesis.4

mass without intracranial extension.
Most lipoblastomas are well circumscribed, encapsulated, and limited to the subcutis. However, lipoblastomatosis is the diffuse counterpart to lipoblastoma, affecting deeper soft tissue and often infiltrating adjacent skeletal muscle.1

Diagnosis is made by histologic evaluation. Lipoblastoma appears as immature fat cells in varying stages of maturity with septa separating them into lobules. There should not be nuclear atypia.5 The histologic differential diagnosis includes other adipose tumors, most chiefly myxoid liposarcoma. These tumors have a lobular pattern without prominent septae and contain nuclear atypia with atypical mitotic figures. Myxoid liposarcoma has an infiltrating pattern similar to lipoblastomatosis and has a metastatic rate up to 60%.6 Imaging studies such as MRI are helpful in diagnosis, particularly in head and neck or visceral cases.3 The treatment of choice of lipoblastoma is wide excision. With complete removal, tumors rarely recur. Recurrences are more common in lipoblastomatosis or with incompletely excised primary lesions.3 A 14% to 24% recurrence rate has been recognized. Cytogenic analysis of lipomatous tumors has begun to reveal translocations in chromosome 8q11-13 region breakpoint abnormalities and translocations, specifically involving the pleomorphic adenoma gene 1, PLAG1, as the oncogenic target in lipoblastoma.6 Identification of these molecular mutations may provide aid in differentiating histologically similar-appearing tumors in the future.
 (H&E, original magnification ×2). Higher-power view showed mature fat cells intermingled with spindle-shaped mesenchymal cells and lipobl")
This case illustrates a rare benign childhood tumor that can be difficult to diagnose prior to histologic examination. Our patient did not fit the typical description of a lipoblastoma, as his tumor was axially located as opposed to the more common peripheral presentation. We aim to raise awareness of this diagnosis as more cases are being recognized.
- Kaddu S, Kohler S. Muscle, adipose, and cartilage neoplasms. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. St. Louis, MO: Mosby; 2003:1988-1989.
- Benato C, Falezza G, Lonardoni A, et al. Acute respiratory distress caused by a giant mediastinal lipoblastoma in a 16-month-old boy. Ann Thorac Surg. 2011;92:119-120.
- Pham NS, Poirier B, Fuller SC, et al. Pediatric lipoblastoma in the head and neck: a systematic review of 48 reported cases. Int J Pediatr Otorhinolaryngol. 2010;74:723-728.
- Chen Z, Coffin CM, Scott S, et al. Evidence by spectral karyotyping that 8q11.2 is nonrandomly involved in lipoblastoma. J Mol Diagn. 2000;2:73-77.
- Weedon, D. Weedon’s Skin Pathology. 3rd ed. China: Churchill Livingstone; 2010.
- Hicks J, Dilley A, Patal D. Lipoblastoma and lipoblastomatosis in infancy and childhood: histopathologic, ultrastructural, and cytogenetic features. Ultrastruct Pathol. 2001;25:321-333.
To the Editor:
A 2-year-old boy was referred to our pediatric dermatology clinic by his pediatrician for an enlarging mass on the mid frontal scalp. The lesion had been present since birth and slowly enlarged. His parents thought the lesion was mostly asymptomatic; however, if it was irritated, the child would cry. He was otherwise healthy and had no history of skin conditions. There was no family history of skin conditions, birthmarks, or vascular malformations. On physical examination, we observed an isolated, approximately 3-cm, well-circumscribed, mobile, flesh-colored and violaceous nodule on the mid frontal scalp (Figure 1). At that time our differential diagnosis included a complex hemangioma or other vascular proliferation, nevus lipomatosis, or even a soft-tissue malignancy such as a sarcoma. Prior to biopsy, we ordered magnetic resonance imaging (MRI) to evaluate for intracranial extension of the lesion. The MRI revealed a 3.2-cm frontal midline scalp mass with complex imaging characteristics, and the radiologist gave a differential diagnosis of hemangioma, teratoma, or less likely liposarcoma (Figure 2). Fortunately, there was no evidence of central nervous system or intracranial invasion. We then proceeded with excisional biopsy, which grossly revealed a nodular, well-circumscribed, yellow mass (Figure 3). The wound was closed with primary closure. Histologically, there was a lobulated tumor with thin, well-vascularized connective tissue septa within a myxoid stroma (Figure 4A). The tumor was composed of lipocytes in varying stages of maturity without obvious nuclear atypia (Figures 4B and 4C), leading to a diagnosis of lipoblastoma.
Lipoblastoma (also known as an embryonic lipoma) is a rare variant of lipoma. It is a benign neoplasm of immature white fat cells primarily seen in children younger than 3 years. It is reportedly twice as common in boys versus girls. Lipoblastomas present as enlarging, soft, mobile, painless nodules, usually 3 to 5 cm in diameter. The extremities are the favored location, but they also have been described on the head, neck, and trunk. Additionally, mediastinal and retroperitoneal lipoblastomas have been documented.1 The tumors may be symptomatic, particularly when involving the neck or mediastinum. In rare instances, they may present with respiratory distress.2 Multiple cases of head and neck lipoblastomas have been published in the English-language literature.3 Growing evidence supports that a chromosomal breakpoint abnormality at 8q11-q13 may be implicated in the pathogenesis.4
mass without intracranial extension.
Most lipoblastomas are well circumscribed, encapsulated, and limited to the subcutis. However, lipoblastomatosis is the diffuse counterpart to lipoblastoma, affecting deeper soft tissue and often infiltrating adjacent skeletal muscle.1
Diagnosis is made by histologic evaluation. Lipoblastoma appears as immature fat cells in varying stages of maturity with septa separating them into lobules. There should not be nuclear atypia.5 The histologic differential diagnosis includes other adipose tumors, most chiefly myxoid liposarcoma. These tumors have a lobular pattern without prominent septae and contain nuclear atypia with atypical mitotic figures. Myxoid liposarcoma has an infiltrating pattern similar to lipoblastomatosis and has a metastatic rate up to 60%.6 Imaging studies such as MRI are helpful in diagnosis, particularly in head and neck or visceral cases.3 The treatment of choice of lipoblastoma is wide excision. With complete removal, tumors rarely recur. Recurrences are more common in lipoblastomatosis or with incompletely excised primary lesions.3 A 14% to 24% recurrence rate has been recognized. Cytogenic analysis of lipomatous tumors has begun to reveal translocations in chromosome 8q11-13 region breakpoint abnormalities and translocations, specifically involving the pleomorphic adenoma gene 1, PLAG1, as the oncogenic target in lipoblastoma.6 Identification of these molecular mutations may provide aid in differentiating histologically similar-appearing tumors in the future.
This case illustrates a rare benign childhood tumor that can be difficult to diagnose prior to histologic examination. Our patient did not fit the typical description of a lipoblastoma, as his tumor was axially located as opposed to the more common peripheral presentation. We aim to raise awareness of this diagnosis as more cases are being recognized.
To the Editor:
A 2-year-old boy was referred to our pediatric dermatology clinic by his pediatrician for an enlarging mass on the mid frontal scalp. The lesion had been present since birth and slowly enlarged. His parents thought the lesion was mostly asymptomatic; however, if it was irritated, the child would cry. He was otherwise healthy and had no history of skin conditions. There was no family history of skin conditions, birthmarks, or vascular malformations. On physical examination, we observed an isolated, approximately 3-cm, well-circumscribed, mobile, flesh-colored and violaceous nodule on the mid frontal scalp (Figure 1). At that time our differential diagnosis included a complex hemangioma or other vascular proliferation, nevus lipomatosis, or even a soft-tissue malignancy such as a sarcoma. Prior to biopsy, we ordered magnetic resonance imaging (MRI) to evaluate for intracranial extension of the lesion. The MRI revealed a 3.2-cm frontal midline scalp mass with complex imaging characteristics, and the radiologist gave a differential diagnosis of hemangioma, teratoma, or less likely liposarcoma (Figure 2). Fortunately, there was no evidence of central nervous system or intracranial invasion. We then proceeded with excisional biopsy, which grossly revealed a nodular, well-circumscribed, yellow mass (Figure 3). The wound was closed with primary closure. Histologically, there was a lobulated tumor with thin, well-vascularized connective tissue septa within a myxoid stroma (Figure 4A). The tumor was composed of lipocytes in varying stages of maturity without obvious nuclear atypia (Figures 4B and 4C), leading to a diagnosis of lipoblastoma.
Lipoblastoma (also known as an embryonic lipoma) is a rare variant of lipoma. It is a benign neoplasm of immature white fat cells primarily seen in children younger than 3 years. It is reportedly twice as common in boys versus girls. Lipoblastomas present as enlarging, soft, mobile, painless nodules, usually 3 to 5 cm in diameter. The extremities are the favored location, but they also have been described on the head, neck, and trunk. Additionally, mediastinal and retroperitoneal lipoblastomas have been documented.1 The tumors may be symptomatic, particularly when involving the neck or mediastinum. In rare instances, they may present with respiratory distress.2 Multiple cases of head and neck lipoblastomas have been published in the English-language literature.3 Growing evidence supports that a chromosomal breakpoint abnormality at 8q11-q13 may be implicated in the pathogenesis.4
mass without intracranial extension.
Most lipoblastomas are well circumscribed, encapsulated, and limited to the subcutis. However, lipoblastomatosis is the diffuse counterpart to lipoblastoma, affecting deeper soft tissue and often infiltrating adjacent skeletal muscle.1
Diagnosis is made by histologic evaluation. Lipoblastoma appears as immature fat cells in varying stages of maturity with septa separating them into lobules. There should not be nuclear atypia.5 The histologic differential diagnosis includes other adipose tumors, most chiefly myxoid liposarcoma. These tumors have a lobular pattern without prominent septae and contain nuclear atypia with atypical mitotic figures. Myxoid liposarcoma has an infiltrating pattern similar to lipoblastomatosis and has a metastatic rate up to 60%.6 Imaging studies such as MRI are helpful in diagnosis, particularly in head and neck or visceral cases.3 The treatment of choice of lipoblastoma is wide excision. With complete removal, tumors rarely recur. Recurrences are more common in lipoblastomatosis or with incompletely excised primary lesions.3 A 14% to 24% recurrence rate has been recognized. Cytogenic analysis of lipomatous tumors has begun to reveal translocations in chromosome 8q11-13 region breakpoint abnormalities and translocations, specifically involving the pleomorphic adenoma gene 1, PLAG1, as the oncogenic target in lipoblastoma.6 Identification of these molecular mutations may provide aid in differentiating histologically similar-appearing tumors in the future.
This case illustrates a rare benign childhood tumor that can be difficult to diagnose prior to histologic examination. Our patient did not fit the typical description of a lipoblastoma, as his tumor was axially located as opposed to the more common peripheral presentation. We aim to raise awareness of this diagnosis as more cases are being recognized.
- Kaddu S, Kohler S. Muscle, adipose, and cartilage neoplasms. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. St. Louis, MO: Mosby; 2003:1988-1989.
- Benato C, Falezza G, Lonardoni A, et al. Acute respiratory distress caused by a giant mediastinal lipoblastoma in a 16-month-old boy. Ann Thorac Surg. 2011;92:119-120.
- Pham NS, Poirier B, Fuller SC, et al. Pediatric lipoblastoma in the head and neck: a systematic review of 48 reported cases. Int J Pediatr Otorhinolaryngol. 2010;74:723-728.
- Chen Z, Coffin CM, Scott S, et al. Evidence by spectral karyotyping that 8q11.2 is nonrandomly involved in lipoblastoma. J Mol Diagn. 2000;2:73-77.
- Weedon, D. Weedon’s Skin Pathology. 3rd ed. China: Churchill Livingstone; 2010.
- Hicks J, Dilley A, Patal D. Lipoblastoma and lipoblastomatosis in infancy and childhood: histopathologic, ultrastructural, and cytogenetic features. Ultrastruct Pathol. 2001;25:321-333.
- Kaddu S, Kohler S. Muscle, adipose, and cartilage neoplasms. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. St. Louis, MO: Mosby; 2003:1988-1989.
- Benato C, Falezza G, Lonardoni A, et al. Acute respiratory distress caused by a giant mediastinal lipoblastoma in a 16-month-old boy. Ann Thorac Surg. 2011;92:119-120.
- Pham NS, Poirier B, Fuller SC, et al. Pediatric lipoblastoma in the head and neck: a systematic review of 48 reported cases. Int J Pediatr Otorhinolaryngol. 2010;74:723-728.
- Chen Z, Coffin CM, Scott S, et al. Evidence by spectral karyotyping that 8q11.2 is nonrandomly involved in lipoblastoma. J Mol Diagn. 2000;2:73-77.
- Weedon, D. Weedon’s Skin Pathology. 3rd ed. China: Churchill Livingstone; 2010.
- Hicks J, Dilley A, Patal D. Lipoblastoma and lipoblastomatosis in infancy and childhood: histopathologic, ultrastructural, and cytogenetic features. Ultrastruct Pathol. 2001;25:321-333.
Practice Points
- Lipoblastoma is a benign neoplasm of immature white fat cells primarily seen in children younger than 3 years.
- Lipoblastomas often present as painless nodules located on the extremities.
- Histologically, lipoblastoma reveals immature adipose cells in varying stages of maturity arranged into lobules separated by septae.
- Consider magnetic resonance imaging if visceral extension is a concern; otherwise, surgical excision is curative in most cases.

Verrucous Coalescing Dry Papules and Plaques on the Hip and Flank
The Diagnosis: Blaschkitis
A punch biopsy from the right lateral hip was performed. Histopathologic examination revealed orthokeratosis overlying mild psoriasiform epidermal hyperplasia associated with a lichenoid infiltrate composed almost entirely of lymphocytes (Figure). The infiltrate did not entirely obscure the dermoepidermal junction and spared the adnexal structures. The clinical presentation along with histopathologic analysis confirmed a diagnosis of blaschkitis. The lesions were treated with triamcinolone ointment twice daily as needed, and the patient reported symptomatic and clinical improvement with this intervention at 4-week follow-up.
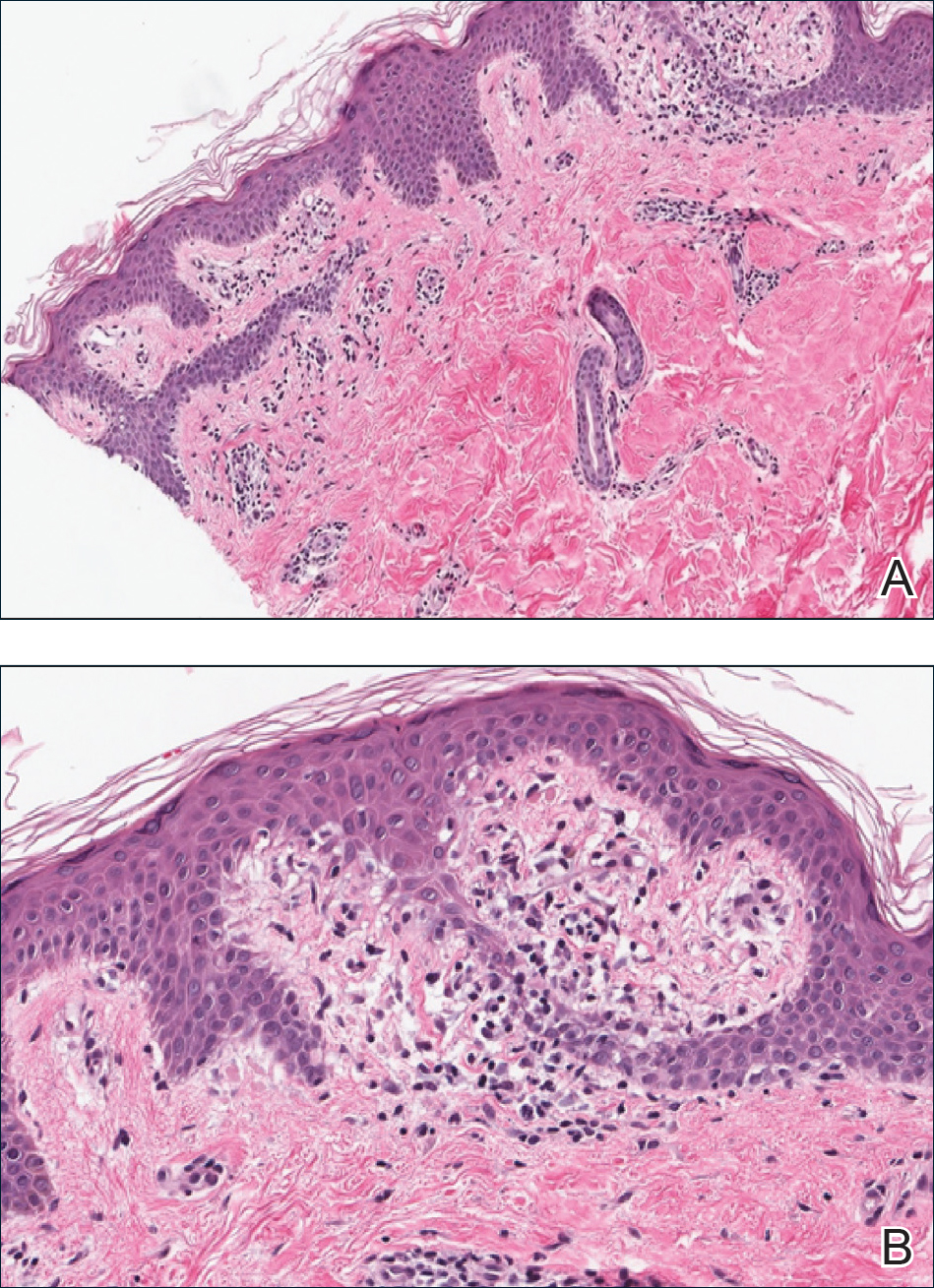
Described by Grosshans and Marot1 in 1990, blaschkitis is an acquired inflammatory dermatosis following the lines of Blaschko. It predominantly is seen on the trunk and typically presents with pruritic papules and vesicles. It frequently has a relapsing course and is more commonly found in adults. Blaschkitis is considered a form of cutaneous mosaicism representing somatic mutations affecting epidermal cell growth and migration during embryogenesis. It has been proposed that these aberrant cells are not clinically apparent at birth; however, viral infection and drug or other environmental triggers can induce antigen presentation of the clone cells activating a T cell–mediated inflammatory response.2-4
The differential diagnosis includes other acquired Blaschko-linear dermatoses such as lichen striatus, inflammatory linear verrucous epidermal nevus, unilateral lichen planus, linear lichen sclerosus, linear psoriasis, linear fixed drug reaction, lichen nitidus, and others.1,4 Given the overlap in clinical and histopathological presentations of the entities in the differential, it often is difficult to discern the etiology of the papular and vesicular eruption in question. Discrimination of one etiology from the others is further confounded by the fact that these lesions can all be pruritic and initially are treated with topical corticosteroids. A degree of clinical suspicion for blaschkitis coupled with prior understanding of lesional manifestations is helpful in this circumstance. Although classic lichen planus often affects the arms, legs, flexor surfaces, and occasionally the oral mucosa, blaschkitis generally is limited to the trunk. The lesions of lichen planus generally are violaceous, flattopped, polygonal papules that tend to coalesce. They often have a thin, transparent, and adherent scale overlying the papular lesions, and they occasionally demonstrate Wickham striae, which are faint white reticulated networks typically seen in oral mucosal lesions. In the case of lichen nitidus, lesions often follow a geometric line due to the Köbner response, whereas physical trauma from scratching or injury causes lesions to form along the line of insult. Assessing patients for any newly initiated medications can help eliminate lichenoid drug eruptions. Lichen striatus perhaps has the most overlap with blaschkitis, having been described as also following the lines of Blaschko but occurring in children rather than adults. Inflammatory linear verrucous epidermal nevi also can be distinguished from blaschkitis on this premise, as these lesions arise during the first 5 years of life and generally affect the lower extremities.4,5
Histopathology is somewhat variable but generally includes spongiotic dermatitis with concomitant interface
dermatitis characterized by T-cell infiltration. Spongiosis is a feature less commonly seen in lichen striatus. Lesions can progress over time from spongiotic dermatitis to spongiotic psoriasiform dermatitis and later to spongiotic psoriasiform lichenoid dermatitis.4 Treatment of blaschkitis should begin with reassurance of the benign nature of the dermatosis. Pruritic symptoms can be managed with a course of topical steroids.
Blaschkitis is a rare and self-limiting acquired dermatosis that should be incorporated into the differential diagnosis of Blaschko-linear dermatoses. Further investigation is needed to determine if blaschkitis and lichen striatus represent the ends of a disease spectrum or completely distinct entities.
- Grosshans E, Marot L. Blaschkitis in adults. Ann Dermatol Venereol. 1990;117:9-15.
- Müller CS, Schmaltz R, Vogt T, et al. Lichen striatus and blaschkitis: reappraisal of the concept of blaschkolinear dermatoses [published online November 29, 2010]. Br J Dermatol. 2011;164:257-262.
- Sun BK, Tsao H. X-chromosome inactivation and skin disease. J Invest Dermatol. 2008;128:2753-2759.
- Keegan BR, Kamino H, Fangman W, et al. “Pediatric blaschkitis”: expanding spectrum of childhood acquired Blaschko-linear dermatoses. Pediatr Dermatol. 2007;24:261-267.
- Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012.
The Diagnosis: Blaschkitis
A punch biopsy from the right lateral hip was performed. Histopathologic examination revealed orthokeratosis overlying mild psoriasiform epidermal hyperplasia associated with a lichenoid infiltrate composed almost entirely of lymphocytes (Figure). The infiltrate did not entirely obscure the dermoepidermal junction and spared the adnexal structures. The clinical presentation along with histopathologic analysis confirmed a diagnosis of blaschkitis. The lesions were treated with triamcinolone ointment twice daily as needed, and the patient reported symptomatic and clinical improvement with this intervention at 4-week follow-up.
Described by Grosshans and Marot1 in 1990, blaschkitis is an acquired inflammatory dermatosis following the lines of Blaschko. It predominantly is seen on the trunk and typically presents with pruritic papules and vesicles. It frequently has a relapsing course and is more commonly found in adults. Blaschkitis is considered a form of cutaneous mosaicism representing somatic mutations affecting epidermal cell growth and migration during embryogenesis. It has been proposed that these aberrant cells are not clinically apparent at birth; however, viral infection and drug or other environmental triggers can induce antigen presentation of the clone cells activating a T cell–mediated inflammatory response.2-4
The differential diagnosis includes other acquired Blaschko-linear dermatoses such as lichen striatus, inflammatory linear verrucous epidermal nevus, unilateral lichen planus, linear lichen sclerosus, linear psoriasis, linear fixed drug reaction, lichen nitidus, and others.1,4 Given the overlap in clinical and histopathological presentations of the entities in the differential, it often is difficult to discern the etiology of the papular and vesicular eruption in question. Discrimination of one etiology from the others is further confounded by the fact that these lesions can all be pruritic and initially are treated with topical corticosteroids. A degree of clinical suspicion for blaschkitis coupled with prior understanding of lesional manifestations is helpful in this circumstance. Although classic lichen planus often affects the arms, legs, flexor surfaces, and occasionally the oral mucosa, blaschkitis generally is limited to the trunk. The lesions of lichen planus generally are violaceous, flattopped, polygonal papules that tend to coalesce. They often have a thin, transparent, and adherent scale overlying the papular lesions, and they occasionally demonstrate Wickham striae, which are faint white reticulated networks typically seen in oral mucosal lesions. In the case of lichen nitidus, lesions often follow a geometric line due to the Köbner response, whereas physical trauma from scratching or injury causes lesions to form along the line of insult. Assessing patients for any newly initiated medications can help eliminate lichenoid drug eruptions. Lichen striatus perhaps has the most overlap with blaschkitis, having been described as also following the lines of Blaschko but occurring in children rather than adults. Inflammatory linear verrucous epidermal nevi also can be distinguished from blaschkitis on this premise, as these lesions arise during the first 5 years of life and generally affect the lower extremities.4,5
Histopathology is somewhat variable but generally includes spongiotic dermatitis with concomitant interface
dermatitis characterized by T-cell infiltration. Spongiosis is a feature less commonly seen in lichen striatus. Lesions can progress over time from spongiotic dermatitis to spongiotic psoriasiform dermatitis and later to spongiotic psoriasiform lichenoid dermatitis.4 Treatment of blaschkitis should begin with reassurance of the benign nature of the dermatosis. Pruritic symptoms can be managed with a course of topical steroids.
Blaschkitis is a rare and self-limiting acquired dermatosis that should be incorporated into the differential diagnosis of Blaschko-linear dermatoses. Further investigation is needed to determine if blaschkitis and lichen striatus represent the ends of a disease spectrum or completely distinct entities.
The Diagnosis: Blaschkitis
A punch biopsy from the right lateral hip was performed. Histopathologic examination revealed orthokeratosis overlying mild psoriasiform epidermal hyperplasia associated with a lichenoid infiltrate composed almost entirely of lymphocytes (Figure). The infiltrate did not entirely obscure the dermoepidermal junction and spared the adnexal structures. The clinical presentation along with histopathologic analysis confirmed a diagnosis of blaschkitis. The lesions were treated with triamcinolone ointment twice daily as needed, and the patient reported symptomatic and clinical improvement with this intervention at 4-week follow-up.
Described by Grosshans and Marot1 in 1990, blaschkitis is an acquired inflammatory dermatosis following the lines of Blaschko. It predominantly is seen on the trunk and typically presents with pruritic papules and vesicles. It frequently has a relapsing course and is more commonly found in adults. Blaschkitis is considered a form of cutaneous mosaicism representing somatic mutations affecting epidermal cell growth and migration during embryogenesis. It has been proposed that these aberrant cells are not clinically apparent at birth; however, viral infection and drug or other environmental triggers can induce antigen presentation of the clone cells activating a T cell–mediated inflammatory response.2-4
The differential diagnosis includes other acquired Blaschko-linear dermatoses such as lichen striatus, inflammatory linear verrucous epidermal nevus, unilateral lichen planus, linear lichen sclerosus, linear psoriasis, linear fixed drug reaction, lichen nitidus, and others.1,4 Given the overlap in clinical and histopathological presentations of the entities in the differential, it often is difficult to discern the etiology of the papular and vesicular eruption in question. Discrimination of one etiology from the others is further confounded by the fact that these lesions can all be pruritic and initially are treated with topical corticosteroids. A degree of clinical suspicion for blaschkitis coupled with prior understanding of lesional manifestations is helpful in this circumstance. Although classic lichen planus often affects the arms, legs, flexor surfaces, and occasionally the oral mucosa, blaschkitis generally is limited to the trunk. The lesions of lichen planus generally are violaceous, flattopped, polygonal papules that tend to coalesce. They often have a thin, transparent, and adherent scale overlying the papular lesions, and they occasionally demonstrate Wickham striae, which are faint white reticulated networks typically seen in oral mucosal lesions. In the case of lichen nitidus, lesions often follow a geometric line due to the Köbner response, whereas physical trauma from scratching or injury causes lesions to form along the line of insult. Assessing patients for any newly initiated medications can help eliminate lichenoid drug eruptions. Lichen striatus perhaps has the most overlap with blaschkitis, having been described as also following the lines of Blaschko but occurring in children rather than adults. Inflammatory linear verrucous epidermal nevi also can be distinguished from blaschkitis on this premise, as these lesions arise during the first 5 years of life and generally affect the lower extremities.4,5
Histopathology is somewhat variable but generally includes spongiotic dermatitis with concomitant interface
dermatitis characterized by T-cell infiltration. Spongiosis is a feature less commonly seen in lichen striatus. Lesions can progress over time from spongiotic dermatitis to spongiotic psoriasiform dermatitis and later to spongiotic psoriasiform lichenoid dermatitis.4 Treatment of blaschkitis should begin with reassurance of the benign nature of the dermatosis. Pruritic symptoms can be managed with a course of topical steroids.
Blaschkitis is a rare and self-limiting acquired dermatosis that should be incorporated into the differential diagnosis of Blaschko-linear dermatoses. Further investigation is needed to determine if blaschkitis and lichen striatus represent the ends of a disease spectrum or completely distinct entities.
- Grosshans E, Marot L. Blaschkitis in adults. Ann Dermatol Venereol. 1990;117:9-15.
- Müller CS, Schmaltz R, Vogt T, et al. Lichen striatus and blaschkitis: reappraisal of the concept of blaschkolinear dermatoses [published online November 29, 2010]. Br J Dermatol. 2011;164:257-262.
- Sun BK, Tsao H. X-chromosome inactivation and skin disease. J Invest Dermatol. 2008;128:2753-2759.
- Keegan BR, Kamino H, Fangman W, et al. “Pediatric blaschkitis”: expanding spectrum of childhood acquired Blaschko-linear dermatoses. Pediatr Dermatol. 2007;24:261-267.
- Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012.
- Grosshans E, Marot L. Blaschkitis in adults. Ann Dermatol Venereol. 1990;117:9-15.
- Müller CS, Schmaltz R, Vogt T, et al. Lichen striatus and blaschkitis: reappraisal of the concept of blaschkolinear dermatoses [published online November 29, 2010]. Br J Dermatol. 2011;164:257-262.
- Sun BK, Tsao H. X-chromosome inactivation and skin disease. J Invest Dermatol. 2008;128:2753-2759.
- Keegan BR, Kamino H, Fangman W, et al. “Pediatric blaschkitis”: expanding spectrum of childhood acquired Blaschko-linear dermatoses. Pediatr Dermatol. 2007;24:261-267.
- Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012.
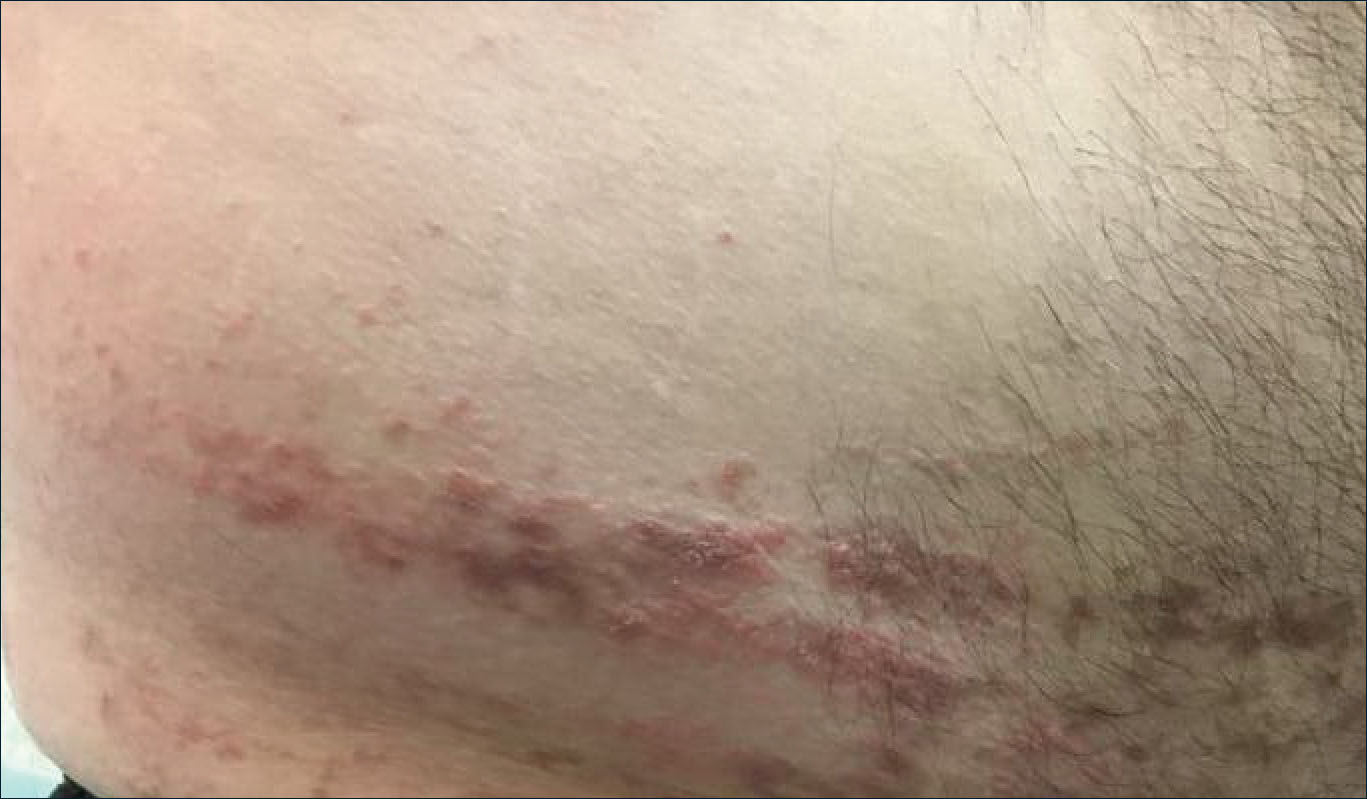
A 31-year-old man presented with a recurring pruritic rash on the right lateral flank and hip of 2 years’ duration. Physical examination revealed erythematous, verrucous, dry papules and plaques coalescing into larger plaques on the right flank and hip in dermatomal distributions involving the T10 and T11 dermatomes. A few papules were scattered in a linear eruption along the right flank and right upper thigh. Some postinflammatory changes were noted. No rash was noted over any other area of the body. Physical examination was otherwise unremarkable.


