User login
IVIG proves effective for dermatomyositis in phase 3 trial
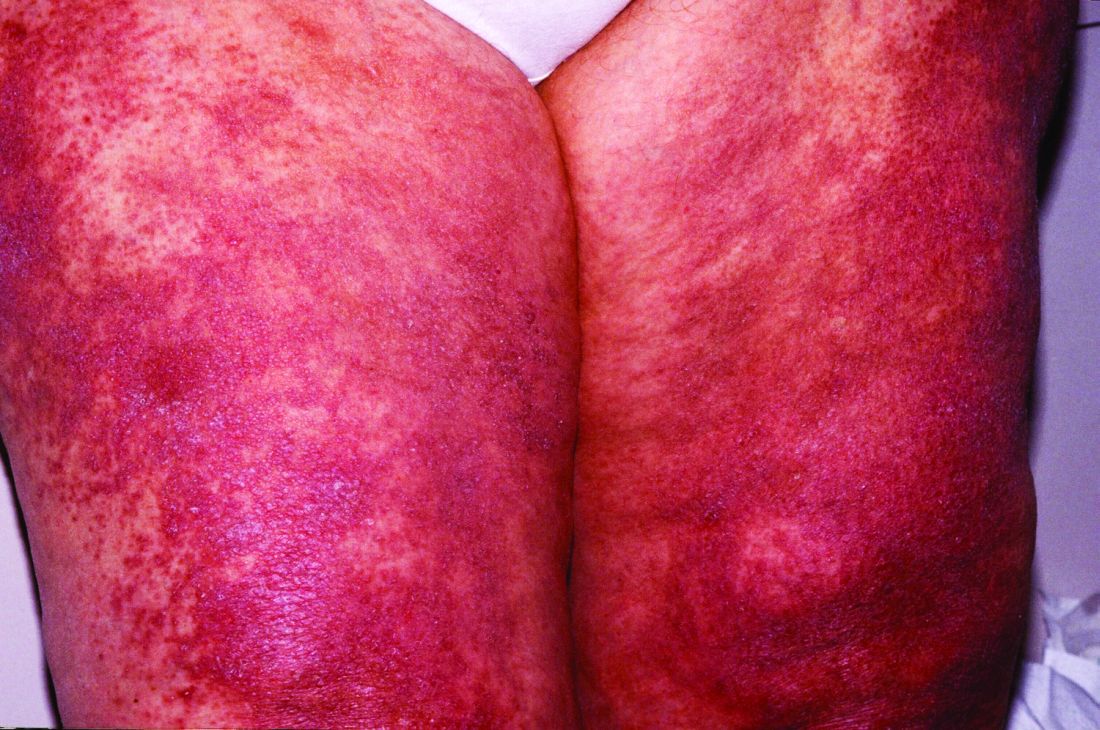
With use of intravenous immunoglobulin for the treatment of adults with dermatomyositis, a significantly higher percentage of patients experienced at least minimal improvement in disease activity in comparison with placebo in the first-ever phase 3 trial of the blood-product therapy for the condition.
Until this trial, published in the New England Journal of Medicine, there had not been an extensive evaluation of IVIG for the treatment of dermatomyositis, the study’s authors noted.
Glucocorticoids are typically offered as first-line therapy, followed by various immunosuppressants. IVIG is composed of purified liquid IgG concentrates from human plasma. It has been prescribed off label as second- or third-line therapy for dermatomyositis, usually along with immunosuppressive drugs. In European guidelines, it has been recommended as a glucocorticoid-sparing agent for patients with this condition.

“The study provides support that IVIG is effective in treating the signs and symptoms of patients with dermatomyositis, at least in the short term,” said David Fiorentino, MD, PhD, professor of dermatology and associate residency program director at Stanford Health Care, Stanford, California, who was not involved in the study.
“IVIG appears to be effective for patients with any severity level and works relatively quickly [within 1 month of therapy],” he added. “IVIG is effective in treating both the muscle symptoms as well as the rash of dermatomyositis, which is important, as both organ systems can cause significant patient morbidity in this disease.”
Time to improvement was shorter with IVIG than with placebo (a median of 35 days vs. 115 days), said Kathryn H. Dao, MD, associate professor in the division of rheumatic diseases at the University of Texas Southwestern Medical Center, Dallas, who was not involved in the study.
The study’s greatest strengths are its international, multicenter, randomized, placebo-controlled design, Dr. Dao said. In addition, “these patients were permitted to be on background medicines that we typically use in real-world situations.”
Study methodology
Researchers led by Rohit Aggarwal, MD, of the division of rheumatology and clinical immunology at the University of Pittsburgh, recruited patients aged 18-80 years with active dermatomyositis. Individuals were randomly assigned in a 1:1 ratio to receive either IVIG at a dose of 2.0 g/kg of body weight or placebo (0.9% sodium chloride) every 4 weeks for 16 weeks.
Those who were administered placebo and those who did not experience confirmed clinical deterioration while receiving IVIG could participate in an open-label extension phase for another 24 weeks.
The primary endpoint was a response, defined as a Total Improvement Score (TIS) of at least 20 (indicating at least minimal improvement) at week 16 and no confirmed deterioration up to week 16. The TIS is a weighted composite score that reflects the change in a core set of six measures of myositis activity over time. Scores span from 0 to 100, with higher scores indicating more significant improvement.
Secondary endpoints
Key secondary endpoints included moderate improvement (TIS ≥ 40) and major improvement (TIS ≥ 60) and change in score on the Cutaneous Dermatomyositis Disease Area and Severity Index.
A total of 95 patients underwent randomization; 47 patients received IVIG and 48 received placebo. At 16 weeks, a TIS of at least 20 occurred in 37 of 47 (79%) patients who received IVIG and in 21 of 48 (44%) patients with placebo (difference, 35%; 95% confidence interval, 17%-53%; P < .001).
The results with respect to the secondary endpoints, including at least moderate improvement and major improvement, were generally in the same direction as the results of the primary endpoint analysis, except for change in creatine kinase (CK) level (an individual core measure of the TIS), which did not differ meaningfully between the two groups.
Adverse events
Over the course of 40 weeks, 282 treatment-related adverse events were documented among patients who received IVIG. Headache was experienced by 42%, pyrexia by 19%, and nausea by 16%. Nine serious adverse events occurred and were believed to be associated with IVIG, including six thromboembolic events.
Despite the favorable outcome observed with IVIG, in an editorial that accompanied the study, Anthony A. Amato, MD, of Brigham and Women’s Hospital and Harvard Medical School, Boston, noted that “most of the core components of the TIS are subjective. Because of the high percentage of patients who had a response with placebo, large numbers of patients will be needed in future trials to show a significant difference between trial groups, or the primary endpoint would need to be set higher (e.g., a TIS of ≥40).”
Dr. Dao thought it was significant that the study proactively assessed patients for venous thrombotic events (VTEs) after each infusion. There were eight events in six patients who received IVIG. “Of interest and possibly practice changing is the finding that slowing the IVIG infusion rate from 0.12 to 0.04 mL/kg per minute reduced the incidence of VTEs from 1.54/100 patient-months to 0.54/100 patient-months,” she said. “This is important, as it informs clinicians that IVIG infusion rates should be slower for patients with active dermatomyositis to reduce the risk for blood clots.”
Study weaknesses
A considerable proportion of patients with dermatomyositis do not have clinical muscle involvement but do have rash and do not substantially differ in any other ways from those with classic dermatomyositis, Dr. Fiorentino said.
“These patients were not eligible to enter the trial, and so we have no data on the efficacy of IVIG in this population,” he said. “Unfortunately, these patients might now be denied insurance reimbursement for IVIG therapy, given that they are not part of the indicated patient population in the label.”
In addition, there is limited information about Black, Asian, or Hispanic patients because few of those patients participated in the study. That is also the case for patients younger than 18, which for this disease is relevant because incidence peaks in younger patients (juvenile dermatomyositis), Dr. Fiorentino noted.
Among the study’s weaknesses, Dr. Dao noted that more than 70% of participants were women. The study was short in duration, fewer than half of patients underwent muscle biopsy to confirm myositis, and only two thirds of patients underwent electromyography/nerve conduction studies to show evidence of myositis. There was a high placebo response (44%), the CK values were not high at the start of the trial, and they did not change with treatment.
No analysis was performed to evaluate the efficacy of IVIG across dermatomyositis subgroups – defined by autoantibodies – but the study likely was not powered to do so. These subgroups might respond differently to IVIG, yielding important information, Fiorentino said.
The study provided efficacy data for only one formulation of IVIG, Octagam 10%, which was approved for dermatomyositis by the Food and Drug Administration in 2021 on the basis of this trial. However, in the United States, patients with dermatomyositis are treated with multiple brands of IVIG. “The decision around IVIG brand is largely determined by third-party payers, and for the most part, the different brands are used interchangeably from the standpoint of the treating provider,” Dr. Fiorentino said. “This will likely continue to be the case, as the results of this study are generally being extrapolated to all brands of IVIG.”
Multiple IVIG brands that have been used for immune-mediated diseases differ in concentration, content of IgA, sugar concentration, additives, and preparations (for example, the need for reconstitution vs. being ready to use), Dr. Dao said. Octagam 10% is the only brand approved by the FDA for adult dermatomyositis; hence, cost can be an issue for patients if other brands are used off label. The typical cost of IVIG is $100-$400 per gram; a typical course of treatment is estimated to be $30,000-$40,000 per month. “However, if Octagam is not available or a patient has a reaction to it, clinicians may use other IVIG brands as deemed medically necessary to treat their patients,” she said.
Dr. Aggarwal has financial relationships with more than 15 pharmaceutical companies, including Octapharma, which provided financial support for this trial. Some of the coauthors were employees of Octapharma or had financial relationships with the company. Dr. Dao disclosed no relevant financial relationships. Dr. Fiorentino has conducted sponsored research for Pfizer and Argenyx, has received research funding from Serono, and is a paid adviser to Bristol-Myers Squibb, Janssen, Acelyrin, and Corbus.
A version of this article first appeared on Medscape.com.
With use of intravenous immunoglobulin for the treatment of adults with dermatomyositis, a significantly higher percentage of patients experienced at least minimal improvement in disease activity in comparison with placebo in the first-ever phase 3 trial of the blood-product therapy for the condition.
Until this trial, published in the New England Journal of Medicine, there had not been an extensive evaluation of IVIG for the treatment of dermatomyositis, the study’s authors noted.
Glucocorticoids are typically offered as first-line therapy, followed by various immunosuppressants. IVIG is composed of purified liquid IgG concentrates from human plasma. It has been prescribed off label as second- or third-line therapy for dermatomyositis, usually along with immunosuppressive drugs. In European guidelines, it has been recommended as a glucocorticoid-sparing agent for patients with this condition.
“The study provides support that IVIG is effective in treating the signs and symptoms of patients with dermatomyositis, at least in the short term,” said David Fiorentino, MD, PhD, professor of dermatology and associate residency program director at Stanford Health Care, Stanford, California, who was not involved in the study.
“IVIG appears to be effective for patients with any severity level and works relatively quickly [within 1 month of therapy],” he added. “IVIG is effective in treating both the muscle symptoms as well as the rash of dermatomyositis, which is important, as both organ systems can cause significant patient morbidity in this disease.”
Time to improvement was shorter with IVIG than with placebo (a median of 35 days vs. 115 days), said Kathryn H. Dao, MD, associate professor in the division of rheumatic diseases at the University of Texas Southwestern Medical Center, Dallas, who was not involved in the study.
The study’s greatest strengths are its international, multicenter, randomized, placebo-controlled design, Dr. Dao said. In addition, “these patients were permitted to be on background medicines that we typically use in real-world situations.”
Study methodology
Researchers led by Rohit Aggarwal, MD, of the division of rheumatology and clinical immunology at the University of Pittsburgh, recruited patients aged 18-80 years with active dermatomyositis. Individuals were randomly assigned in a 1:1 ratio to receive either IVIG at a dose of 2.0 g/kg of body weight or placebo (0.9% sodium chloride) every 4 weeks for 16 weeks.
Those who were administered placebo and those who did not experience confirmed clinical deterioration while receiving IVIG could participate in an open-label extension phase for another 24 weeks.
The primary endpoint was a response, defined as a Total Improvement Score (TIS) of at least 20 (indicating at least minimal improvement) at week 16 and no confirmed deterioration up to week 16. The TIS is a weighted composite score that reflects the change in a core set of six measures of myositis activity over time. Scores span from 0 to 100, with higher scores indicating more significant improvement.
Secondary endpoints
Key secondary endpoints included moderate improvement (TIS ≥ 40) and major improvement (TIS ≥ 60) and change in score on the Cutaneous Dermatomyositis Disease Area and Severity Index.
A total of 95 patients underwent randomization; 47 patients received IVIG and 48 received placebo. At 16 weeks, a TIS of at least 20 occurred in 37 of 47 (79%) patients who received IVIG and in 21 of 48 (44%) patients with placebo (difference, 35%; 95% confidence interval, 17%-53%; P < .001).
The results with respect to the secondary endpoints, including at least moderate improvement and major improvement, were generally in the same direction as the results of the primary endpoint analysis, except for change in creatine kinase (CK) level (an individual core measure of the TIS), which did not differ meaningfully between the two groups.
Adverse events
Over the course of 40 weeks, 282 treatment-related adverse events were documented among patients who received IVIG. Headache was experienced by 42%, pyrexia by 19%, and nausea by 16%. Nine serious adverse events occurred and were believed to be associated with IVIG, including six thromboembolic events.
Despite the favorable outcome observed with IVIG, in an editorial that accompanied the study, Anthony A. Amato, MD, of Brigham and Women’s Hospital and Harvard Medical School, Boston, noted that “most of the core components of the TIS are subjective. Because of the high percentage of patients who had a response with placebo, large numbers of patients will be needed in future trials to show a significant difference between trial groups, or the primary endpoint would need to be set higher (e.g., a TIS of ≥40).”
Dr. Dao thought it was significant that the study proactively assessed patients for venous thrombotic events (VTEs) after each infusion. There were eight events in six patients who received IVIG. “Of interest and possibly practice changing is the finding that slowing the IVIG infusion rate from 0.12 to 0.04 mL/kg per minute reduced the incidence of VTEs from 1.54/100 patient-months to 0.54/100 patient-months,” she said. “This is important, as it informs clinicians that IVIG infusion rates should be slower for patients with active dermatomyositis to reduce the risk for blood clots.”
Study weaknesses
A considerable proportion of patients with dermatomyositis do not have clinical muscle involvement but do have rash and do not substantially differ in any other ways from those with classic dermatomyositis, Dr. Fiorentino said.
“These patients were not eligible to enter the trial, and so we have no data on the efficacy of IVIG in this population,” he said. “Unfortunately, these patients might now be denied insurance reimbursement for IVIG therapy, given that they are not part of the indicated patient population in the label.”
In addition, there is limited information about Black, Asian, or Hispanic patients because few of those patients participated in the study. That is also the case for patients younger than 18, which for this disease is relevant because incidence peaks in younger patients (juvenile dermatomyositis), Dr. Fiorentino noted.
Among the study’s weaknesses, Dr. Dao noted that more than 70% of participants were women. The study was short in duration, fewer than half of patients underwent muscle biopsy to confirm myositis, and only two thirds of patients underwent electromyography/nerve conduction studies to show evidence of myositis. There was a high placebo response (44%), the CK values were not high at the start of the trial, and they did not change with treatment.
No analysis was performed to evaluate the efficacy of IVIG across dermatomyositis subgroups – defined by autoantibodies – but the study likely was not powered to do so. These subgroups might respond differently to IVIG, yielding important information, Fiorentino said.
The study provided efficacy data for only one formulation of IVIG, Octagam 10%, which was approved for dermatomyositis by the Food and Drug Administration in 2021 on the basis of this trial. However, in the United States, patients with dermatomyositis are treated with multiple brands of IVIG. “The decision around IVIG brand is largely determined by third-party payers, and for the most part, the different brands are used interchangeably from the standpoint of the treating provider,” Dr. Fiorentino said. “This will likely continue to be the case, as the results of this study are generally being extrapolated to all brands of IVIG.”
Multiple IVIG brands that have been used for immune-mediated diseases differ in concentration, content of IgA, sugar concentration, additives, and preparations (for example, the need for reconstitution vs. being ready to use), Dr. Dao said. Octagam 10% is the only brand approved by the FDA for adult dermatomyositis; hence, cost can be an issue for patients if other brands are used off label. The typical cost of IVIG is $100-$400 per gram; a typical course of treatment is estimated to be $30,000-$40,000 per month. “However, if Octagam is not available or a patient has a reaction to it, clinicians may use other IVIG brands as deemed medically necessary to treat their patients,” she said.
Dr. Aggarwal has financial relationships with more than 15 pharmaceutical companies, including Octapharma, which provided financial support for this trial. Some of the coauthors were employees of Octapharma or had financial relationships with the company. Dr. Dao disclosed no relevant financial relationships. Dr. Fiorentino has conducted sponsored research for Pfizer and Argenyx, has received research funding from Serono, and is a paid adviser to Bristol-Myers Squibb, Janssen, Acelyrin, and Corbus.
A version of this article first appeared on Medscape.com.
With use of intravenous immunoglobulin for the treatment of adults with dermatomyositis, a significantly higher percentage of patients experienced at least minimal improvement in disease activity in comparison with placebo in the first-ever phase 3 trial of the blood-product therapy for the condition.
Until this trial, published in the New England Journal of Medicine, there had not been an extensive evaluation of IVIG for the treatment of dermatomyositis, the study’s authors noted.
Glucocorticoids are typically offered as first-line therapy, followed by various immunosuppressants. IVIG is composed of purified liquid IgG concentrates from human plasma. It has been prescribed off label as second- or third-line therapy for dermatomyositis, usually along with immunosuppressive drugs. In European guidelines, it has been recommended as a glucocorticoid-sparing agent for patients with this condition.
“The study provides support that IVIG is effective in treating the signs and symptoms of patients with dermatomyositis, at least in the short term,” said David Fiorentino, MD, PhD, professor of dermatology and associate residency program director at Stanford Health Care, Stanford, California, who was not involved in the study.
“IVIG appears to be effective for patients with any severity level and works relatively quickly [within 1 month of therapy],” he added. “IVIG is effective in treating both the muscle symptoms as well as the rash of dermatomyositis, which is important, as both organ systems can cause significant patient morbidity in this disease.”
Time to improvement was shorter with IVIG than with placebo (a median of 35 days vs. 115 days), said Kathryn H. Dao, MD, associate professor in the division of rheumatic diseases at the University of Texas Southwestern Medical Center, Dallas, who was not involved in the study.
The study’s greatest strengths are its international, multicenter, randomized, placebo-controlled design, Dr. Dao said. In addition, “these patients were permitted to be on background medicines that we typically use in real-world situations.”
Study methodology
Researchers led by Rohit Aggarwal, MD, of the division of rheumatology and clinical immunology at the University of Pittsburgh, recruited patients aged 18-80 years with active dermatomyositis. Individuals were randomly assigned in a 1:1 ratio to receive either IVIG at a dose of 2.0 g/kg of body weight or placebo (0.9% sodium chloride) every 4 weeks for 16 weeks.
Those who were administered placebo and those who did not experience confirmed clinical deterioration while receiving IVIG could participate in an open-label extension phase for another 24 weeks.
The primary endpoint was a response, defined as a Total Improvement Score (TIS) of at least 20 (indicating at least minimal improvement) at week 16 and no confirmed deterioration up to week 16. The TIS is a weighted composite score that reflects the change in a core set of six measures of myositis activity over time. Scores span from 0 to 100, with higher scores indicating more significant improvement.
Secondary endpoints
Key secondary endpoints included moderate improvement (TIS ≥ 40) and major improvement (TIS ≥ 60) and change in score on the Cutaneous Dermatomyositis Disease Area and Severity Index.
A total of 95 patients underwent randomization; 47 patients received IVIG and 48 received placebo. At 16 weeks, a TIS of at least 20 occurred in 37 of 47 (79%) patients who received IVIG and in 21 of 48 (44%) patients with placebo (difference, 35%; 95% confidence interval, 17%-53%; P < .001).
The results with respect to the secondary endpoints, including at least moderate improvement and major improvement, were generally in the same direction as the results of the primary endpoint analysis, except for change in creatine kinase (CK) level (an individual core measure of the TIS), which did not differ meaningfully between the two groups.
Adverse events
Over the course of 40 weeks, 282 treatment-related adverse events were documented among patients who received IVIG. Headache was experienced by 42%, pyrexia by 19%, and nausea by 16%. Nine serious adverse events occurred and were believed to be associated with IVIG, including six thromboembolic events.
Despite the favorable outcome observed with IVIG, in an editorial that accompanied the study, Anthony A. Amato, MD, of Brigham and Women’s Hospital and Harvard Medical School, Boston, noted that “most of the core components of the TIS are subjective. Because of the high percentage of patients who had a response with placebo, large numbers of patients will be needed in future trials to show a significant difference between trial groups, or the primary endpoint would need to be set higher (e.g., a TIS of ≥40).”
Dr. Dao thought it was significant that the study proactively assessed patients for venous thrombotic events (VTEs) after each infusion. There were eight events in six patients who received IVIG. “Of interest and possibly practice changing is the finding that slowing the IVIG infusion rate from 0.12 to 0.04 mL/kg per minute reduced the incidence of VTEs from 1.54/100 patient-months to 0.54/100 patient-months,” she said. “This is important, as it informs clinicians that IVIG infusion rates should be slower for patients with active dermatomyositis to reduce the risk for blood clots.”
Study weaknesses
A considerable proportion of patients with dermatomyositis do not have clinical muscle involvement but do have rash and do not substantially differ in any other ways from those with classic dermatomyositis, Dr. Fiorentino said.
“These patients were not eligible to enter the trial, and so we have no data on the efficacy of IVIG in this population,” he said. “Unfortunately, these patients might now be denied insurance reimbursement for IVIG therapy, given that they are not part of the indicated patient population in the label.”
In addition, there is limited information about Black, Asian, or Hispanic patients because few of those patients participated in the study. That is also the case for patients younger than 18, which for this disease is relevant because incidence peaks in younger patients (juvenile dermatomyositis), Dr. Fiorentino noted.
Among the study’s weaknesses, Dr. Dao noted that more than 70% of participants were women. The study was short in duration, fewer than half of patients underwent muscle biopsy to confirm myositis, and only two thirds of patients underwent electromyography/nerve conduction studies to show evidence of myositis. There was a high placebo response (44%), the CK values were not high at the start of the trial, and they did not change with treatment.
No analysis was performed to evaluate the efficacy of IVIG across dermatomyositis subgroups – defined by autoantibodies – but the study likely was not powered to do so. These subgroups might respond differently to IVIG, yielding important information, Fiorentino said.
The study provided efficacy data for only one formulation of IVIG, Octagam 10%, which was approved for dermatomyositis by the Food and Drug Administration in 2021 on the basis of this trial. However, in the United States, patients with dermatomyositis are treated with multiple brands of IVIG. “The decision around IVIG brand is largely determined by third-party payers, and for the most part, the different brands are used interchangeably from the standpoint of the treating provider,” Dr. Fiorentino said. “This will likely continue to be the case, as the results of this study are generally being extrapolated to all brands of IVIG.”
Multiple IVIG brands that have been used for immune-mediated diseases differ in concentration, content of IgA, sugar concentration, additives, and preparations (for example, the need for reconstitution vs. being ready to use), Dr. Dao said. Octagam 10% is the only brand approved by the FDA for adult dermatomyositis; hence, cost can be an issue for patients if other brands are used off label. The typical cost of IVIG is $100-$400 per gram; a typical course of treatment is estimated to be $30,000-$40,000 per month. “However, if Octagam is not available or a patient has a reaction to it, clinicians may use other IVIG brands as deemed medically necessary to treat their patients,” she said.
Dr. Aggarwal has financial relationships with more than 15 pharmaceutical companies, including Octapharma, which provided financial support for this trial. Some of the coauthors were employees of Octapharma or had financial relationships with the company. Dr. Dao disclosed no relevant financial relationships. Dr. Fiorentino has conducted sponsored research for Pfizer and Argenyx, has received research funding from Serono, and is a paid adviser to Bristol-Myers Squibb, Janssen, Acelyrin, and Corbus.
A version of this article first appeared on Medscape.com.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Lasker awardee pioneered prenatal DNA testing
For Yuk Ming Dennis Lo, BM BCh, DPhil, a 1996 paper showing the detection of tumor DNA in blood plasma would prove a turning point.
Since the 1960s, clinicians had been searching for a way to glimpse into a fetus’ genetic makeup without disturbing the pregnancy – a fascination Dr. Lo, at the time a graduate student in the United Kingdom, shared.
But the article triggered a thought. If cancer cells could release their DNA into blood plasma, then maybe fetuses could, too. “I had the strange thought that the cancer growing in the patients is a little bit like the placenta that has implanted into the uterus,” he told The New York Times.
The answer was yes. In 1997, having returned home to Hong Kong, Dr. Lo published a seminal article showing that cell-free fetal DNA could be detected in maternal blood.
He went on to devise methods to detect markers for Down syndrome, creating a noninvasive test that is more than 99% accurate for ruling out the disorder, along with screenings for trisomy 18 (Edwards syndrome), trisomy 13 (Patau syndrome), and other chromosome abnormalities.
With the commercial launch of noninvasive prenatal testing (NIPT) in 2011, health care centers around the world quickly embraced the technology as a safe alternative to more invasive methods, such as amniocentesis, for identifying fetal abnormalities. NIPT is now available in over 60 countries and is widely used by clinicians, according to the Lasker Foundation, which granted him the 2022 Lasker DeBakey Clinical Medical Research Award, along with a $250,000 prize.
“I am pleased that since its launch, noninvasive prenatal testing has become a standard of care,” Dr. Lo, chair of the department of chemical pathology at The Chinese University of Hong Kong, said in a video on the Lasker website. “It has also stimulated a global interest in the diagnostic applications of plasma DNA, especially in the area of cancer liquid biopsies and transplantation monitoring. I look forward to seeing these and other yet to be developed applications improving health care worldwide.”
Dr. Lo’s work has inspired clinical advances and applications, including Rh factor assessments, innovations in cancer technology, transplantation, and beyond, according to Lasker.
Iris Krishna, MD, MPH, director of Perinatal Quality in the Emory Perinatal Center at Emory University School of Medicine, Atlanta, said Dr. Lo’s work has also provided opportunities to screen for other genetic disorders, such as microdeletion syndromes and single gene disorders
“As we continue to learn about the possibilities of this technology, it is imperative for the clinician to be knowledgeable of the benefits and limitations of cell-free DNA screening to be able to counsel their patients appropriately,” Dr. Krishna said.
A COVID clearinghouse
Lauren Gardner, PhD, professor in the department of civil and systems engineering at Johns Hopkins University, Baltimore, received the Lasker Bloomberg Public Service Award for her work on the Johns Hopkins’ COVID-19 dashboard, a critical tool for the dissemination of public health data in real time.
According to the Los Angeles Times, Ensheng Dong, Dr. Gardner’s graduate student, approached her about tracking cases of the emerging infection in his home country of China. Mr. Dong mined Chinese websites for early cases of COVID-19 and created online maps using the information. At Dr. Gardner’s suggestion, he expanded the database to include global data.
At the time, according to Lasker, no other institution was providing this information. The World Health Organization created summaries of daily COVID-19 counts, but the data were not as accessible. Dr. Gardner said timely and obtainable information was crucial to craft nimble and rational strategies for combating the pandemic.
“Given the amount of misinformation in circulation and the highly politicized nature of the COVID-19 public health crisis, our work enabled individuals to access the information they needed to make informed decisions to protect themselves, which was especially critical in those locations with delayed or nonexistent policies in place,” Dr. Gardner said in a statement.
Dr. Gardner said she was excited to pursue additional data-centric projects. “I am optimistic that in the future, timely public health information will become increasingly available, especially in times of crisis,” she said. “Moving forward, I am excited to build on our learnings from COVID-19 and transfer that knowledge to address other problems facing societies.”
New knowledge of cells, immunology, and disease
The 2022 Albert Lasker Basic Research Award honored three scientists who helped identify a family of proteins that connect cells and assist the immune system in attaching to its targets. The proteins, called integrins, are needed for cells to interact with each other to build complex structures in the body. They are also key to the process T cells undergo to recognize and attack cancer cells.
Awardees Richard O. Hynes, MA, PhD, distinguished professor of cancer research at Massachusetts Institute of Technology; Erkki Ruoslahti, MD, PhD, distinguished professor emeritus at Sanford Burnham Prebys Medical Discovery Institute, La Jolla, California; and Timothy A. Springer, PhD, professor of biological chemistry and molecular biology at Boston Children’s Hospital and Harvard Medical School, independently identified a cell-surface–associated protein that helps cells attach to the extracellular matrix.
“Many of the mysteries of how integrins work are only being discovered today,” Dr. Springer said in his acceptance remarks online.
The discoveries related to integrins have led to several clinical advances, including the development of drugs like the eyedrops lifitegrast, the biologic agent vedolizumab (made using integrins Springer discovered), and tirofiban, a medication used to hamper clotting in cardiovascular diseases.
A version of this article first appeared on Medscape.com.
For Yuk Ming Dennis Lo, BM BCh, DPhil, a 1996 paper showing the detection of tumor DNA in blood plasma would prove a turning point.
Since the 1960s, clinicians had been searching for a way to glimpse into a fetus’ genetic makeup without disturbing the pregnancy – a fascination Dr. Lo, at the time a graduate student in the United Kingdom, shared.
But the article triggered a thought. If cancer cells could release their DNA into blood plasma, then maybe fetuses could, too. “I had the strange thought that the cancer growing in the patients is a little bit like the placenta that has implanted into the uterus,” he told The New York Times.
The answer was yes. In 1997, having returned home to Hong Kong, Dr. Lo published a seminal article showing that cell-free fetal DNA could be detected in maternal blood.
He went on to devise methods to detect markers for Down syndrome, creating a noninvasive test that is more than 99% accurate for ruling out the disorder, along with screenings for trisomy 18 (Edwards syndrome), trisomy 13 (Patau syndrome), and other chromosome abnormalities.
With the commercial launch of noninvasive prenatal testing (NIPT) in 2011, health care centers around the world quickly embraced the technology as a safe alternative to more invasive methods, such as amniocentesis, for identifying fetal abnormalities. NIPT is now available in over 60 countries and is widely used by clinicians, according to the Lasker Foundation, which granted him the 2022 Lasker DeBakey Clinical Medical Research Award, along with a $250,000 prize.
“I am pleased that since its launch, noninvasive prenatal testing has become a standard of care,” Dr. Lo, chair of the department of chemical pathology at The Chinese University of Hong Kong, said in a video on the Lasker website. “It has also stimulated a global interest in the diagnostic applications of plasma DNA, especially in the area of cancer liquid biopsies and transplantation monitoring. I look forward to seeing these and other yet to be developed applications improving health care worldwide.”
Dr. Lo’s work has inspired clinical advances and applications, including Rh factor assessments, innovations in cancer technology, transplantation, and beyond, according to Lasker.
Iris Krishna, MD, MPH, director of Perinatal Quality in the Emory Perinatal Center at Emory University School of Medicine, Atlanta, said Dr. Lo’s work has also provided opportunities to screen for other genetic disorders, such as microdeletion syndromes and single gene disorders
“As we continue to learn about the possibilities of this technology, it is imperative for the clinician to be knowledgeable of the benefits and limitations of cell-free DNA screening to be able to counsel their patients appropriately,” Dr. Krishna said.
A COVID clearinghouse
Lauren Gardner, PhD, professor in the department of civil and systems engineering at Johns Hopkins University, Baltimore, received the Lasker Bloomberg Public Service Award for her work on the Johns Hopkins’ COVID-19 dashboard, a critical tool for the dissemination of public health data in real time.
According to the Los Angeles Times, Ensheng Dong, Dr. Gardner’s graduate student, approached her about tracking cases of the emerging infection in his home country of China. Mr. Dong mined Chinese websites for early cases of COVID-19 and created online maps using the information. At Dr. Gardner’s suggestion, he expanded the database to include global data.
At the time, according to Lasker, no other institution was providing this information. The World Health Organization created summaries of daily COVID-19 counts, but the data were not as accessible. Dr. Gardner said timely and obtainable information was crucial to craft nimble and rational strategies for combating the pandemic.
“Given the amount of misinformation in circulation and the highly politicized nature of the COVID-19 public health crisis, our work enabled individuals to access the information they needed to make informed decisions to protect themselves, which was especially critical in those locations with delayed or nonexistent policies in place,” Dr. Gardner said in a statement.
Dr. Gardner said she was excited to pursue additional data-centric projects. “I am optimistic that in the future, timely public health information will become increasingly available, especially in times of crisis,” she said. “Moving forward, I am excited to build on our learnings from COVID-19 and transfer that knowledge to address other problems facing societies.”
New knowledge of cells, immunology, and disease
The 2022 Albert Lasker Basic Research Award honored three scientists who helped identify a family of proteins that connect cells and assist the immune system in attaching to its targets. The proteins, called integrins, are needed for cells to interact with each other to build complex structures in the body. They are also key to the process T cells undergo to recognize and attack cancer cells.
Awardees Richard O. Hynes, MA, PhD, distinguished professor of cancer research at Massachusetts Institute of Technology; Erkki Ruoslahti, MD, PhD, distinguished professor emeritus at Sanford Burnham Prebys Medical Discovery Institute, La Jolla, California; and Timothy A. Springer, PhD, professor of biological chemistry and molecular biology at Boston Children’s Hospital and Harvard Medical School, independently identified a cell-surface–associated protein that helps cells attach to the extracellular matrix.
“Many of the mysteries of how integrins work are only being discovered today,” Dr. Springer said in his acceptance remarks online.
The discoveries related to integrins have led to several clinical advances, including the development of drugs like the eyedrops lifitegrast, the biologic agent vedolizumab (made using integrins Springer discovered), and tirofiban, a medication used to hamper clotting in cardiovascular diseases.
A version of this article first appeared on Medscape.com.
For Yuk Ming Dennis Lo, BM BCh, DPhil, a 1996 paper showing the detection of tumor DNA in blood plasma would prove a turning point.
Since the 1960s, clinicians had been searching for a way to glimpse into a fetus’ genetic makeup without disturbing the pregnancy – a fascination Dr. Lo, at the time a graduate student in the United Kingdom, shared.
But the article triggered a thought. If cancer cells could release their DNA into blood plasma, then maybe fetuses could, too. “I had the strange thought that the cancer growing in the patients is a little bit like the placenta that has implanted into the uterus,” he told The New York Times.
The answer was yes. In 1997, having returned home to Hong Kong, Dr. Lo published a seminal article showing that cell-free fetal DNA could be detected in maternal blood.
He went on to devise methods to detect markers for Down syndrome, creating a noninvasive test that is more than 99% accurate for ruling out the disorder, along with screenings for trisomy 18 (Edwards syndrome), trisomy 13 (Patau syndrome), and other chromosome abnormalities.
With the commercial launch of noninvasive prenatal testing (NIPT) in 2011, health care centers around the world quickly embraced the technology as a safe alternative to more invasive methods, such as amniocentesis, for identifying fetal abnormalities. NIPT is now available in over 60 countries and is widely used by clinicians, according to the Lasker Foundation, which granted him the 2022 Lasker DeBakey Clinical Medical Research Award, along with a $250,000 prize.
“I am pleased that since its launch, noninvasive prenatal testing has become a standard of care,” Dr. Lo, chair of the department of chemical pathology at The Chinese University of Hong Kong, said in a video on the Lasker website. “It has also stimulated a global interest in the diagnostic applications of plasma DNA, especially in the area of cancer liquid biopsies and transplantation monitoring. I look forward to seeing these and other yet to be developed applications improving health care worldwide.”
Dr. Lo’s work has inspired clinical advances and applications, including Rh factor assessments, innovations in cancer technology, transplantation, and beyond, according to Lasker.
Iris Krishna, MD, MPH, director of Perinatal Quality in the Emory Perinatal Center at Emory University School of Medicine, Atlanta, said Dr. Lo’s work has also provided opportunities to screen for other genetic disorders, such as microdeletion syndromes and single gene disorders
“As we continue to learn about the possibilities of this technology, it is imperative for the clinician to be knowledgeable of the benefits and limitations of cell-free DNA screening to be able to counsel their patients appropriately,” Dr. Krishna said.
A COVID clearinghouse
Lauren Gardner, PhD, professor in the department of civil and systems engineering at Johns Hopkins University, Baltimore, received the Lasker Bloomberg Public Service Award for her work on the Johns Hopkins’ COVID-19 dashboard, a critical tool for the dissemination of public health data in real time.
According to the Los Angeles Times, Ensheng Dong, Dr. Gardner’s graduate student, approached her about tracking cases of the emerging infection in his home country of China. Mr. Dong mined Chinese websites for early cases of COVID-19 and created online maps using the information. At Dr. Gardner’s suggestion, he expanded the database to include global data.
At the time, according to Lasker, no other institution was providing this information. The World Health Organization created summaries of daily COVID-19 counts, but the data were not as accessible. Dr. Gardner said timely and obtainable information was crucial to craft nimble and rational strategies for combating the pandemic.
“Given the amount of misinformation in circulation and the highly politicized nature of the COVID-19 public health crisis, our work enabled individuals to access the information they needed to make informed decisions to protect themselves, which was especially critical in those locations with delayed or nonexistent policies in place,” Dr. Gardner said in a statement.
Dr. Gardner said she was excited to pursue additional data-centric projects. “I am optimistic that in the future, timely public health information will become increasingly available, especially in times of crisis,” she said. “Moving forward, I am excited to build on our learnings from COVID-19 and transfer that knowledge to address other problems facing societies.”
New knowledge of cells, immunology, and disease
The 2022 Albert Lasker Basic Research Award honored three scientists who helped identify a family of proteins that connect cells and assist the immune system in attaching to its targets. The proteins, called integrins, are needed for cells to interact with each other to build complex structures in the body. They are also key to the process T cells undergo to recognize and attack cancer cells.
Awardees Richard O. Hynes, MA, PhD, distinguished professor of cancer research at Massachusetts Institute of Technology; Erkki Ruoslahti, MD, PhD, distinguished professor emeritus at Sanford Burnham Prebys Medical Discovery Institute, La Jolla, California; and Timothy A. Springer, PhD, professor of biological chemistry and molecular biology at Boston Children’s Hospital and Harvard Medical School, independently identified a cell-surface–associated protein that helps cells attach to the extracellular matrix.
“Many of the mysteries of how integrins work are only being discovered today,” Dr. Springer said in his acceptance remarks online.
The discoveries related to integrins have led to several clinical advances, including the development of drugs like the eyedrops lifitegrast, the biologic agent vedolizumab (made using integrins Springer discovered), and tirofiban, a medication used to hamper clotting in cardiovascular diseases.
A version of this article first appeared on Medscape.com.
Cancer as a full contact sport
John worked as a handyman and lived on a small sailboat in a marina. When he was diagnosed with metastatic kidney cancer at age 48, he quickly fell through the cracks. He failed to show to appointments and took oral anticancer treatments, but just sporadically. He had Medicaid, so insurance wasn’t the issue. It was everything else.
John was behind on his slip fees; he hadn’t been able to work for some time because of his progressive weakness and pain. He was chronically in danger of getting kicked out of his makeshift home aboard the boat. He had no reliable transportation to the clinic and so he didn’t come to appointments regularly. The specialty pharmacy refused to deliver his expensive oral chemotherapy to his address at the marina. He went days without eating full meals because he was too weak to cook for himself. Plus, he was estranged from his family who were unaware of his illness. His oncologist was overwhelmed trying to take care of him. He had a reasonable chance of achieving disease control on first-line oral therapy, but his problems seemed to hinder these chances at every turn. She was distraught – what could she do?

Enter the team approach. John’s oncologist reached out to our palliative care program for help. We recognized that this was a job too big for us alone so we connected John with the Extensivist Medicine program at UCLA Health, a high-intensity primary care program led by a physician specializing in primary care for high-risk individuals. The program provides wraparound outpatient services for chronically and seriously ill patients, like John, who are at risk for falling through the cracks. John went from receiving very little support to now having an entire team of caring professionals focused on helping him achieve his best possible outcome despite the seriousness of his disease.
He now had the support of a high-functioning team with clearly defined roles. Social work connected him with housing, food, and transportation resources. A nurse called him every day to check in and make sure he was taking medications and reminded him about his upcoming appointments. Case management helped him get needed equipment, such as grab bars and a walker. As his palliative care nurse practitioner, I counseled him on understanding his prognosis and planning ahead for medical emergencies. Our psycho-oncology clinicians helped John reconcile with his family, who were more than willing to take him in once they realized how ill he was. Once these social factors were addressed, John could more easily stay current with his oral chemotherapy, giving him the best chance possible to achieve a robust treatment response that could buy him more time.
And, John did get that time – he got 6 months of improved quality of life, during which he reconnected with his family, including his children, and rebuilt these important relationships. Eventually treatment failed him. His disease, already widely metastatic, became more active and painful. He accepted hospice care at his sister’s house and we transitioned him from our team to the hospice team. He died peacefully surrounded by family.
Interprofessional teamwork is fundamental to treat ‘total pain’
None of this would have been possible without the work of high-functioning teams. It is a commonly held belief that interprofessional teamwork is fundamental to the care of patients and families living with serious illness. But why? How did this idea come about? And what evidence is there to support teamwork?
Dame Cicely Saunders, who founded the modern hospice movement in mid-20th century England, embodied the interdisciplinary team by working first as a nurse, then a social worker, and finally as a physician. She wrote about patients’ “total pain,” the crisis of physical, spiritual, social, and emotional distress that many people have at the end of life. She understood that no single health care discipline was adequate to the task of addressing each of these domains equally well. Thus, hospice became synonymous with care provided by a quartet of specialists – physicians, nurses, social workers, and chaplains. Nowadays, there are other specialists that are added to the mix – home health aides, pharmacists, physical and occupational therapists, music and pet therapists, and so on.
But in medicine, like all areas of science, convention and tradition only go so far. What evidence is there to support the work of an interdisciplinary team in managing the distress of patients and families living with advanced illnesses? It turns out that there is good evidence to support the use of high-functioning interdisciplinary teams in the care of the seriously ill. Palliative care is associated with improved patient outcomes, including improvements in symptom control, quality of life, and end of life care, when it is delivered by an interdisciplinary team rather than by a solo practitioner.
You may think that teamwork is most useful for patients like John who have seemingly intractable social barriers. But it is also true that for even patients with many more social advantages teamwork improves quality of life. I got to see this up close recently in my own life.
Teamwork improves quality of life
My father recently passed away after a 9-month battle with advanced cancer. He had every advantage possible – financial stability, high health literacy, an incredibly devoted spouse who happens to be an RN, good insurance, and access to top-notch medical care. Yet, even he benefited from a team approach. It started small, with the oncologist and oncology NP providing excellent, patient-centered care. Then it grew to include myself as the daughter/palliative care nurse practitioner who made recommendations for treating his nausea and ensured that his advance directive was completed and uploaded to his chart. When my dad needed physical therapy, the home health agency sent a wonderful physical therapist, who brought all sorts of equipment that kept him more functional than he would have been otherwise. Other family members helped out – my sisters helped connect my dad with a priest who came to the home to provide spiritual care, which was crucial to ensuring that he was at peace. And, in his final days, my dad had the hospice team to help manage his symptoms and his family members to provide hands-on care.
The complexity of cancer care has long necessitated a team approach to planning cancer treatment – known as a tumor board – with medical oncology, radiation oncology, surgery, and pathology all weighing in. It makes sense that patients and their families would also need a team of clinicians representing different specialty areas to assist with the wide array of physical, psychosocial, practical, and spiritual concerns that arise throughout the cancer disease trajectory.
Ms. D’Ambruoso is a hospice and palliative care nurse practitioner for UCLA Health Cancer Care, Santa Monica, Calif.
John worked as a handyman and lived on a small sailboat in a marina. When he was diagnosed with metastatic kidney cancer at age 48, he quickly fell through the cracks. He failed to show to appointments and took oral anticancer treatments, but just sporadically. He had Medicaid, so insurance wasn’t the issue. It was everything else.
John was behind on his slip fees; he hadn’t been able to work for some time because of his progressive weakness and pain. He was chronically in danger of getting kicked out of his makeshift home aboard the boat. He had no reliable transportation to the clinic and so he didn’t come to appointments regularly. The specialty pharmacy refused to deliver his expensive oral chemotherapy to his address at the marina. He went days without eating full meals because he was too weak to cook for himself. Plus, he was estranged from his family who were unaware of his illness. His oncologist was overwhelmed trying to take care of him. He had a reasonable chance of achieving disease control on first-line oral therapy, but his problems seemed to hinder these chances at every turn. She was distraught – what could she do?
Enter the team approach. John’s oncologist reached out to our palliative care program for help. We recognized that this was a job too big for us alone so we connected John with the Extensivist Medicine program at UCLA Health, a high-intensity primary care program led by a physician specializing in primary care for high-risk individuals. The program provides wraparound outpatient services for chronically and seriously ill patients, like John, who are at risk for falling through the cracks. John went from receiving very little support to now having an entire team of caring professionals focused on helping him achieve his best possible outcome despite the seriousness of his disease.
He now had the support of a high-functioning team with clearly defined roles. Social work connected him with housing, food, and transportation resources. A nurse called him every day to check in and make sure he was taking medications and reminded him about his upcoming appointments. Case management helped him get needed equipment, such as grab bars and a walker. As his palliative care nurse practitioner, I counseled him on understanding his prognosis and planning ahead for medical emergencies. Our psycho-oncology clinicians helped John reconcile with his family, who were more than willing to take him in once they realized how ill he was. Once these social factors were addressed, John could more easily stay current with his oral chemotherapy, giving him the best chance possible to achieve a robust treatment response that could buy him more time.
And, John did get that time – he got 6 months of improved quality of life, during which he reconnected with his family, including his children, and rebuilt these important relationships. Eventually treatment failed him. His disease, already widely metastatic, became more active and painful. He accepted hospice care at his sister’s house and we transitioned him from our team to the hospice team. He died peacefully surrounded by family.
Interprofessional teamwork is fundamental to treat ‘total pain’
None of this would have been possible without the work of high-functioning teams. It is a commonly held belief that interprofessional teamwork is fundamental to the care of patients and families living with serious illness. But why? How did this idea come about? And what evidence is there to support teamwork?
Dame Cicely Saunders, who founded the modern hospice movement in mid-20th century England, embodied the interdisciplinary team by working first as a nurse, then a social worker, and finally as a physician. She wrote about patients’ “total pain,” the crisis of physical, spiritual, social, and emotional distress that many people have at the end of life. She understood that no single health care discipline was adequate to the task of addressing each of these domains equally well. Thus, hospice became synonymous with care provided by a quartet of specialists – physicians, nurses, social workers, and chaplains. Nowadays, there are other specialists that are added to the mix – home health aides, pharmacists, physical and occupational therapists, music and pet therapists, and so on.
But in medicine, like all areas of science, convention and tradition only go so far. What evidence is there to support the work of an interdisciplinary team in managing the distress of patients and families living with advanced illnesses? It turns out that there is good evidence to support the use of high-functioning interdisciplinary teams in the care of the seriously ill. Palliative care is associated with improved patient outcomes, including improvements in symptom control, quality of life, and end of life care, when it is delivered by an interdisciplinary team rather than by a solo practitioner.
You may think that teamwork is most useful for patients like John who have seemingly intractable social barriers. But it is also true that for even patients with many more social advantages teamwork improves quality of life. I got to see this up close recently in my own life.
Teamwork improves quality of life
My father recently passed away after a 9-month battle with advanced cancer. He had every advantage possible – financial stability, high health literacy, an incredibly devoted spouse who happens to be an RN, good insurance, and access to top-notch medical care. Yet, even he benefited from a team approach. It started small, with the oncologist and oncology NP providing excellent, patient-centered care. Then it grew to include myself as the daughter/palliative care nurse practitioner who made recommendations for treating his nausea and ensured that his advance directive was completed and uploaded to his chart. When my dad needed physical therapy, the home health agency sent a wonderful physical therapist, who brought all sorts of equipment that kept him more functional than he would have been otherwise. Other family members helped out – my sisters helped connect my dad with a priest who came to the home to provide spiritual care, which was crucial to ensuring that he was at peace. And, in his final days, my dad had the hospice team to help manage his symptoms and his family members to provide hands-on care.
The complexity of cancer care has long necessitated a team approach to planning cancer treatment – known as a tumor board – with medical oncology, radiation oncology, surgery, and pathology all weighing in. It makes sense that patients and their families would also need a team of clinicians representing different specialty areas to assist with the wide array of physical, psychosocial, practical, and spiritual concerns that arise throughout the cancer disease trajectory.
Ms. D’Ambruoso is a hospice and palliative care nurse practitioner for UCLA Health Cancer Care, Santa Monica, Calif.
John worked as a handyman and lived on a small sailboat in a marina. When he was diagnosed with metastatic kidney cancer at age 48, he quickly fell through the cracks. He failed to show to appointments and took oral anticancer treatments, but just sporadically. He had Medicaid, so insurance wasn’t the issue. It was everything else.
John was behind on his slip fees; he hadn’t been able to work for some time because of his progressive weakness and pain. He was chronically in danger of getting kicked out of his makeshift home aboard the boat. He had no reliable transportation to the clinic and so he didn’t come to appointments regularly. The specialty pharmacy refused to deliver his expensive oral chemotherapy to his address at the marina. He went days without eating full meals because he was too weak to cook for himself. Plus, he was estranged from his family who were unaware of his illness. His oncologist was overwhelmed trying to take care of him. He had a reasonable chance of achieving disease control on first-line oral therapy, but his problems seemed to hinder these chances at every turn. She was distraught – what could she do?
Enter the team approach. John’s oncologist reached out to our palliative care program for help. We recognized that this was a job too big for us alone so we connected John with the Extensivist Medicine program at UCLA Health, a high-intensity primary care program led by a physician specializing in primary care for high-risk individuals. The program provides wraparound outpatient services for chronically and seriously ill patients, like John, who are at risk for falling through the cracks. John went from receiving very little support to now having an entire team of caring professionals focused on helping him achieve his best possible outcome despite the seriousness of his disease.
He now had the support of a high-functioning team with clearly defined roles. Social work connected him with housing, food, and transportation resources. A nurse called him every day to check in and make sure he was taking medications and reminded him about his upcoming appointments. Case management helped him get needed equipment, such as grab bars and a walker. As his palliative care nurse practitioner, I counseled him on understanding his prognosis and planning ahead for medical emergencies. Our psycho-oncology clinicians helped John reconcile with his family, who were more than willing to take him in once they realized how ill he was. Once these social factors were addressed, John could more easily stay current with his oral chemotherapy, giving him the best chance possible to achieve a robust treatment response that could buy him more time.
And, John did get that time – he got 6 months of improved quality of life, during which he reconnected with his family, including his children, and rebuilt these important relationships. Eventually treatment failed him. His disease, already widely metastatic, became more active and painful. He accepted hospice care at his sister’s house and we transitioned him from our team to the hospice team. He died peacefully surrounded by family.
Interprofessional teamwork is fundamental to treat ‘total pain’
None of this would have been possible without the work of high-functioning teams. It is a commonly held belief that interprofessional teamwork is fundamental to the care of patients and families living with serious illness. But why? How did this idea come about? And what evidence is there to support teamwork?
Dame Cicely Saunders, who founded the modern hospice movement in mid-20th century England, embodied the interdisciplinary team by working first as a nurse, then a social worker, and finally as a physician. She wrote about patients’ “total pain,” the crisis of physical, spiritual, social, and emotional distress that many people have at the end of life. She understood that no single health care discipline was adequate to the task of addressing each of these domains equally well. Thus, hospice became synonymous with care provided by a quartet of specialists – physicians, nurses, social workers, and chaplains. Nowadays, there are other specialists that are added to the mix – home health aides, pharmacists, physical and occupational therapists, music and pet therapists, and so on.
But in medicine, like all areas of science, convention and tradition only go so far. What evidence is there to support the work of an interdisciplinary team in managing the distress of patients and families living with advanced illnesses? It turns out that there is good evidence to support the use of high-functioning interdisciplinary teams in the care of the seriously ill. Palliative care is associated with improved patient outcomes, including improvements in symptom control, quality of life, and end of life care, when it is delivered by an interdisciplinary team rather than by a solo practitioner.
You may think that teamwork is most useful for patients like John who have seemingly intractable social barriers. But it is also true that for even patients with many more social advantages teamwork improves quality of life. I got to see this up close recently in my own life.
Teamwork improves quality of life
My father recently passed away after a 9-month battle with advanced cancer. He had every advantage possible – financial stability, high health literacy, an incredibly devoted spouse who happens to be an RN, good insurance, and access to top-notch medical care. Yet, even he benefited from a team approach. It started small, with the oncologist and oncology NP providing excellent, patient-centered care. Then it grew to include myself as the daughter/palliative care nurse practitioner who made recommendations for treating his nausea and ensured that his advance directive was completed and uploaded to his chart. When my dad needed physical therapy, the home health agency sent a wonderful physical therapist, who brought all sorts of equipment that kept him more functional than he would have been otherwise. Other family members helped out – my sisters helped connect my dad with a priest who came to the home to provide spiritual care, which was crucial to ensuring that he was at peace. And, in his final days, my dad had the hospice team to help manage his symptoms and his family members to provide hands-on care.
The complexity of cancer care has long necessitated a team approach to planning cancer treatment – known as a tumor board – with medical oncology, radiation oncology, surgery, and pathology all weighing in. It makes sense that patients and their families would also need a team of clinicians representing different specialty areas to assist with the wide array of physical, psychosocial, practical, and spiritual concerns that arise throughout the cancer disease trajectory.
Ms. D’Ambruoso is a hospice and palliative care nurse practitioner for UCLA Health Cancer Care, Santa Monica, Calif.
ALS drug gets FDA panel thumbs-up after rare second look
In a rare second review of a new drug application,
By a vote of 7-2, the FDA Peripheral and Central Nervous System Drugs Advisory Committee reversed course on AMX0035 (Amylyx Pharmaceuticals), a combination of sodium phenylbutyrate and taurursodiol.
The panel previously voted 6-4 to reject the drug, ruling that data provided by Amylyx had failed to demonstrate that the survival benefit reported in the only clinical trial of AMX0035 so far was a direct result of the drug.
This time, two panelists who previously voted no were swayed by the drug maker’s new analysis of previously presented research, more than 1,300 public comments in support of the drug, supportive testimony from ALS patients and clinicians, and assurances from company executives that Amylyx would pull the drug from the market if results of an ongoing phase 3 clinical trial show the drug doesn’t work.
“As in March, today we have to have an internal dialogue between our scientific scrutiny and clinical compassion,” said Liana G. Apostolova, MD, from Indiana University, Indianapolis, who originally voted against the application.
“Today I also saw additional confirmatory evidence that was not unequivocally persuasive but was nonetheless reassuring,” Dr. Apostolova said. “Because of that I am voting in support of AMX0035.”
A rare second chance
ALS (Lou Gehrig’s disease) is a progressive, fatal neurodegenerative disease affecting nerve cells in the brain and spinal cord that causes loss of motor control. It is rare, affecting about 30,000 people in the United States with another 5,000 new cases diagnosed each year. Most people with the disease die within 2 years of diagnosis.
The FDA has approved two therapies for ALS, but both have limited efficacy.
Typically, FDA approval requires two large studies or one study with a “very persuasive” effect on survival.
Amylyx’s application is based on a single study, the multicenter, two-phase CENTAUR trial. In that trial, 137 people with ALS received AMX0035 or placebo for 24 weeks.
Researchers found that patients receiving AMX0035 had a 25% slower decline in function, compared with the those taking placebo. A change of 20% or more is considered clinically meaningful.
The investigators also found a statistically significant median difference of 4.8 months in time to death, first hospitalization, or tracheostomy/permanent assisted ventilation in the group originally assigned to receive AMX0035 compared with the group originally assigned to receive placebo (hazard ratio, 0.62; P = .023).
In the panel’s previous vote against the drug application, members cited several issues with the study, concluding that it did not offer persuasive or robust evidence of efficacy. They also cited missing data assumptions in the primary analysis, issues of randomization and imbalances in concomitant use of riluzole and edaravone, the two FDA-approved drugs for ALS.
The FDA later requested additional information from Amylyx, delayed its final ruling on the new drug application to Sept. 29, and called for a second review meeting – a virtually unheard-of move.
An FDA review posted in advance of the meeting Sept. 29 had hinted at a different outcome. In that report, regulators said new data from Amylyx were not “sufficiently independent or persuasive” to establish effectiveness.
However, FDA officials in the meeting stressed the importance of considering unmet medical need in ALS in the panel’s decision-making process.
“Recognizing the substantial unmet medical need in ALS, we feel that it is important that the committee is afforded the opportunity to consider this new information, along with the information presented at the prior meeting, in that context,” Billy Dunn, MD, director of the FDA Office of Neuroscience, said during the meeting.
Panelists heard additional data that Amylyx claims confirms the results of the CENTAUR study, including new analyses of the previously submitted survival data and new data from that study and an open-label extension.
They also provided new information on a biomarker data from a phase 2 study of AMX0035 to treat Alzheimer’s disease.
“I think we note the limitations of the analyses, but we still haven’t taken it off the table that they could be considered as confirmatory evidence and that’s why we’re here today,” said Teresa Buracchio, MD, director of the division of neurology for the FDA.
Two members of the panel who voted no in March stuck with that position at the Sept. 29 meeting.
“Unfortunately, I don’t believe the new evidence we’ve reviewed, while promising, combined with that prior evidence, constitutes substantial evidence of effectiveness,” said panelist Caleb Alexander, MD, a professor of epidemiology and medicine at the Johns Hopkins University Center for Drug Safety and Effectiveness, Baltimore.
Dr. Alexander, who also voted no in March, said that post hoc data presented at the meeting were not enough to assuage concerns that led him and others to reject the drug in March.
A challenging situation
Amylyx is currently leading the 48-week international, phase 3, placebo-controlled PHOENIX clinical trial of AMX0035. The study has enrolled about half of its 600-patient target.
“Undoubtedly, the results of the phase 3 study would be highly informative for a regulatory decision on the current ... review for AMX0035,” said Emily Freilich, MD, of the FDA.
However, results aren’t expected until late 2023 or early 2024, which “places the agency in a challenging situation of potentially making a regulatory decision that may not be subsequently confirmed by the results of the ongoing study.”
In June, Amylyx received conditional approval in Canada for the drug, but final approval depends on the outcome of the PHOENIX trial. The FDA does not offer a conditional approval track.
“If AMX0035 is not approved now, the FDA anticipated decision will likely happen in 2025, underscoring the critical importance of today’s outcome,” said Tammy Sarnelli, MPAHC, global head of Regulatory Affairs for Amylyx Pharmaceuticals.
If the FDA were to approve AMX0035 and results from the PHOENIX trial ultimately fail to prove efficacy, Justin Klee, co-CEO and cofounder of Amylyx Pharmaceuticals, said the company would withdraw the drug.
“To be clear, if PHOENIX is not successful, we will do what is right for patients, which includes voluntarily removing the product from the market,” Mr. Klee said.
Regardless of the company’s decision, FDA officials noted that the agency does have the ability to recall a drug from the market if studies show that it no longer meets requirements for approval.
“The FDA, with all due respect, significantly understates the complexity and likelihood of their pulling a product from the market,” Dr. Alexander said. “Whether or not they can ultimately pull a product from the market is no substitute for the evidentiary thresholds that are required for market access.”
A version of this article first appeared on Medscape.com.
In a rare second review of a new drug application,
By a vote of 7-2, the FDA Peripheral and Central Nervous System Drugs Advisory Committee reversed course on AMX0035 (Amylyx Pharmaceuticals), a combination of sodium phenylbutyrate and taurursodiol.
The panel previously voted 6-4 to reject the drug, ruling that data provided by Amylyx had failed to demonstrate that the survival benefit reported in the only clinical trial of AMX0035 so far was a direct result of the drug.
This time, two panelists who previously voted no were swayed by the drug maker’s new analysis of previously presented research, more than 1,300 public comments in support of the drug, supportive testimony from ALS patients and clinicians, and assurances from company executives that Amylyx would pull the drug from the market if results of an ongoing phase 3 clinical trial show the drug doesn’t work.
“As in March, today we have to have an internal dialogue between our scientific scrutiny and clinical compassion,” said Liana G. Apostolova, MD, from Indiana University, Indianapolis, who originally voted against the application.
“Today I also saw additional confirmatory evidence that was not unequivocally persuasive but was nonetheless reassuring,” Dr. Apostolova said. “Because of that I am voting in support of AMX0035.”
A rare second chance
ALS (Lou Gehrig’s disease) is a progressive, fatal neurodegenerative disease affecting nerve cells in the brain and spinal cord that causes loss of motor control. It is rare, affecting about 30,000 people in the United States with another 5,000 new cases diagnosed each year. Most people with the disease die within 2 years of diagnosis.
The FDA has approved two therapies for ALS, but both have limited efficacy.
Typically, FDA approval requires two large studies or one study with a “very persuasive” effect on survival.
Amylyx’s application is based on a single study, the multicenter, two-phase CENTAUR trial. In that trial, 137 people with ALS received AMX0035 or placebo for 24 weeks.
Researchers found that patients receiving AMX0035 had a 25% slower decline in function, compared with the those taking placebo. A change of 20% or more is considered clinically meaningful.
The investigators also found a statistically significant median difference of 4.8 months in time to death, first hospitalization, or tracheostomy/permanent assisted ventilation in the group originally assigned to receive AMX0035 compared with the group originally assigned to receive placebo (hazard ratio, 0.62; P = .023).
In the panel’s previous vote against the drug application, members cited several issues with the study, concluding that it did not offer persuasive or robust evidence of efficacy. They also cited missing data assumptions in the primary analysis, issues of randomization and imbalances in concomitant use of riluzole and edaravone, the two FDA-approved drugs for ALS.
The FDA later requested additional information from Amylyx, delayed its final ruling on the new drug application to Sept. 29, and called for a second review meeting – a virtually unheard-of move.
An FDA review posted in advance of the meeting Sept. 29 had hinted at a different outcome. In that report, regulators said new data from Amylyx were not “sufficiently independent or persuasive” to establish effectiveness.
However, FDA officials in the meeting stressed the importance of considering unmet medical need in ALS in the panel’s decision-making process.
“Recognizing the substantial unmet medical need in ALS, we feel that it is important that the committee is afforded the opportunity to consider this new information, along with the information presented at the prior meeting, in that context,” Billy Dunn, MD, director of the FDA Office of Neuroscience, said during the meeting.
Panelists heard additional data that Amylyx claims confirms the results of the CENTAUR study, including new analyses of the previously submitted survival data and new data from that study and an open-label extension.
They also provided new information on a biomarker data from a phase 2 study of AMX0035 to treat Alzheimer’s disease.
“I think we note the limitations of the analyses, but we still haven’t taken it off the table that they could be considered as confirmatory evidence and that’s why we’re here today,” said Teresa Buracchio, MD, director of the division of neurology for the FDA.
Two members of the panel who voted no in March stuck with that position at the Sept. 29 meeting.
“Unfortunately, I don’t believe the new evidence we’ve reviewed, while promising, combined with that prior evidence, constitutes substantial evidence of effectiveness,” said panelist Caleb Alexander, MD, a professor of epidemiology and medicine at the Johns Hopkins University Center for Drug Safety and Effectiveness, Baltimore.
Dr. Alexander, who also voted no in March, said that post hoc data presented at the meeting were not enough to assuage concerns that led him and others to reject the drug in March.
A challenging situation
Amylyx is currently leading the 48-week international, phase 3, placebo-controlled PHOENIX clinical trial of AMX0035. The study has enrolled about half of its 600-patient target.
“Undoubtedly, the results of the phase 3 study would be highly informative for a regulatory decision on the current ... review for AMX0035,” said Emily Freilich, MD, of the FDA.
However, results aren’t expected until late 2023 or early 2024, which “places the agency in a challenging situation of potentially making a regulatory decision that may not be subsequently confirmed by the results of the ongoing study.”
In June, Amylyx received conditional approval in Canada for the drug, but final approval depends on the outcome of the PHOENIX trial. The FDA does not offer a conditional approval track.
“If AMX0035 is not approved now, the FDA anticipated decision will likely happen in 2025, underscoring the critical importance of today’s outcome,” said Tammy Sarnelli, MPAHC, global head of Regulatory Affairs for Amylyx Pharmaceuticals.
If the FDA were to approve AMX0035 and results from the PHOENIX trial ultimately fail to prove efficacy, Justin Klee, co-CEO and cofounder of Amylyx Pharmaceuticals, said the company would withdraw the drug.
“To be clear, if PHOENIX is not successful, we will do what is right for patients, which includes voluntarily removing the product from the market,” Mr. Klee said.
Regardless of the company’s decision, FDA officials noted that the agency does have the ability to recall a drug from the market if studies show that it no longer meets requirements for approval.
“The FDA, with all due respect, significantly understates the complexity and likelihood of their pulling a product from the market,” Dr. Alexander said. “Whether or not they can ultimately pull a product from the market is no substitute for the evidentiary thresholds that are required for market access.”
A version of this article first appeared on Medscape.com.
In a rare second review of a new drug application,
By a vote of 7-2, the FDA Peripheral and Central Nervous System Drugs Advisory Committee reversed course on AMX0035 (Amylyx Pharmaceuticals), a combination of sodium phenylbutyrate and taurursodiol.
The panel previously voted 6-4 to reject the drug, ruling that data provided by Amylyx had failed to demonstrate that the survival benefit reported in the only clinical trial of AMX0035 so far was a direct result of the drug.
This time, two panelists who previously voted no were swayed by the drug maker’s new analysis of previously presented research, more than 1,300 public comments in support of the drug, supportive testimony from ALS patients and clinicians, and assurances from company executives that Amylyx would pull the drug from the market if results of an ongoing phase 3 clinical trial show the drug doesn’t work.
“As in March, today we have to have an internal dialogue between our scientific scrutiny and clinical compassion,” said Liana G. Apostolova, MD, from Indiana University, Indianapolis, who originally voted against the application.
“Today I also saw additional confirmatory evidence that was not unequivocally persuasive but was nonetheless reassuring,” Dr. Apostolova said. “Because of that I am voting in support of AMX0035.”
A rare second chance
ALS (Lou Gehrig’s disease) is a progressive, fatal neurodegenerative disease affecting nerve cells in the brain and spinal cord that causes loss of motor control. It is rare, affecting about 30,000 people in the United States with another 5,000 new cases diagnosed each year. Most people with the disease die within 2 years of diagnosis.
The FDA has approved two therapies for ALS, but both have limited efficacy.
Typically, FDA approval requires two large studies or one study with a “very persuasive” effect on survival.
Amylyx’s application is based on a single study, the multicenter, two-phase CENTAUR trial. In that trial, 137 people with ALS received AMX0035 or placebo for 24 weeks.
Researchers found that patients receiving AMX0035 had a 25% slower decline in function, compared with the those taking placebo. A change of 20% or more is considered clinically meaningful.
The investigators also found a statistically significant median difference of 4.8 months in time to death, first hospitalization, or tracheostomy/permanent assisted ventilation in the group originally assigned to receive AMX0035 compared with the group originally assigned to receive placebo (hazard ratio, 0.62; P = .023).
In the panel’s previous vote against the drug application, members cited several issues with the study, concluding that it did not offer persuasive or robust evidence of efficacy. They also cited missing data assumptions in the primary analysis, issues of randomization and imbalances in concomitant use of riluzole and edaravone, the two FDA-approved drugs for ALS.
The FDA later requested additional information from Amylyx, delayed its final ruling on the new drug application to Sept. 29, and called for a second review meeting – a virtually unheard-of move.
An FDA review posted in advance of the meeting Sept. 29 had hinted at a different outcome. In that report, regulators said new data from Amylyx were not “sufficiently independent or persuasive” to establish effectiveness.
However, FDA officials in the meeting stressed the importance of considering unmet medical need in ALS in the panel’s decision-making process.
“Recognizing the substantial unmet medical need in ALS, we feel that it is important that the committee is afforded the opportunity to consider this new information, along with the information presented at the prior meeting, in that context,” Billy Dunn, MD, director of the FDA Office of Neuroscience, said during the meeting.
Panelists heard additional data that Amylyx claims confirms the results of the CENTAUR study, including new analyses of the previously submitted survival data and new data from that study and an open-label extension.
They also provided new information on a biomarker data from a phase 2 study of AMX0035 to treat Alzheimer’s disease.
“I think we note the limitations of the analyses, but we still haven’t taken it off the table that they could be considered as confirmatory evidence and that’s why we’re here today,” said Teresa Buracchio, MD, director of the division of neurology for the FDA.
Two members of the panel who voted no in March stuck with that position at the Sept. 29 meeting.
“Unfortunately, I don’t believe the new evidence we’ve reviewed, while promising, combined with that prior evidence, constitutes substantial evidence of effectiveness,” said panelist Caleb Alexander, MD, a professor of epidemiology and medicine at the Johns Hopkins University Center for Drug Safety and Effectiveness, Baltimore.
Dr. Alexander, who also voted no in March, said that post hoc data presented at the meeting were not enough to assuage concerns that led him and others to reject the drug in March.
A challenging situation
Amylyx is currently leading the 48-week international, phase 3, placebo-controlled PHOENIX clinical trial of AMX0035. The study has enrolled about half of its 600-patient target.
“Undoubtedly, the results of the phase 3 study would be highly informative for a regulatory decision on the current ... review for AMX0035,” said Emily Freilich, MD, of the FDA.
However, results aren’t expected until late 2023 or early 2024, which “places the agency in a challenging situation of potentially making a regulatory decision that may not be subsequently confirmed by the results of the ongoing study.”
In June, Amylyx received conditional approval in Canada for the drug, but final approval depends on the outcome of the PHOENIX trial. The FDA does not offer a conditional approval track.
“If AMX0035 is not approved now, the FDA anticipated decision will likely happen in 2025, underscoring the critical importance of today’s outcome,” said Tammy Sarnelli, MPAHC, global head of Regulatory Affairs for Amylyx Pharmaceuticals.
If the FDA were to approve AMX0035 and results from the PHOENIX trial ultimately fail to prove efficacy, Justin Klee, co-CEO and cofounder of Amylyx Pharmaceuticals, said the company would withdraw the drug.
“To be clear, if PHOENIX is not successful, we will do what is right for patients, which includes voluntarily removing the product from the market,” Mr. Klee said.
Regardless of the company’s decision, FDA officials noted that the agency does have the ability to recall a drug from the market if studies show that it no longer meets requirements for approval.
“The FDA, with all due respect, significantly understates the complexity and likelihood of their pulling a product from the market,” Dr. Alexander said. “Whether or not they can ultimately pull a product from the market is no substitute for the evidentiary thresholds that are required for market access.”
A version of this article first appeared on Medscape.com.
Increasing primary care doctors’ knowledge of IPF could speed up diagnoses, suggests white paper
The nonspecific nature of the symptoms of idiopathic pulmonary fibrosis (IPF) especially in early stages, and the relative rarity of IPF compared with other conditions that have similar symptoms, may contribute to a delay in diagnosis in the primary care setting, wrote Daniel F. Dilling, MD, of Loyola University Chicago, Maywood, Ill., and colleagues in Chest: Clinical Perspectives (Dilling et al. State of Practice: Factors Driving Diagnostic Delays in Idiopathic Pulmonary Fibrosis. Chest. 2022).
“We have learned over and over again through research, and also through talking with our own patients with IPF, that there is often a long lag between the first signs of the disease and a diagnosis of IPF,” corresponding author Dr. Dilling said in an interview.
“Even some pulmonary specialists can be uncertain about how to approach the diagnosis when a CT scan or other test first suggests the possibility; this can cost a patient precious time, as being on drug therapy earlier can result in preservation of lung function,” he said. “By sounding the alarm bell with this paper, we hope to promote awareness and education/training within the primary care community as well as the pulmonary community, and also to make all of them aware of the possibility of referral to specialty ILD [interstitial lung disease] centers when desired and possible,” he added.
The researchers conducted a pair of online surveys to inform the development of improving education on IPF among primary care providers.
In the white paper, which can be accessed online, the authors reported results of the surveys. One included 100 general pulmonologists and the other included 306 primary care physicians (156 practiced family physicians and 150 practiced general internal medicine). The data were collected between April 11, 2022, and May 16, 2022. Participants were asked to respond to a patient case scenario of a 55-year-old woman with nonspecific symptoms such as shortness of breath on moderate exertion, cough, exhaustion, and trouble sleeping.
The PCPs were most likely to evaluate the patient for a cardiac condition (46%), 25% would evaluate for chronic obstructive pulmonary disease (COPD), and 23% for asthma. More than half (58%) ranked progressive fibrosing ILD as one of their bottom two diagnoses.
A total of 87% of PCPs said they would begin a diagnostic workup to evaluate symptoms if the patient had no preexisting respiratory disease, compared with 61% for patients with a respiratory diagnosis.
Although 93% of PCPs cited a chest x-ray as part of the initial patient workup, fewer than half said they would order an echocardiogram, spirometry, or pulmonary function test (PFT), and 11% said they would include diffusion capacity testing in the initial workup.
In addition, PCPs were less likely to ask patients about issues that might prompt an IPF diagnosis, such as exposures to agents through work, hobbies, the environment, or comorbidities.
In the pulmonology survey, more than 75% of respondents cited patient history, high-resolution tomography scan, serologic testing, and review for autoimmune disease symptoms as first steps in a diagnostic response to patients with suspected IPF.
Differences between PCPs’ and pulmonolgists’ responses
Both PCPs and pulmonologists responded to several questions to assess knowledge and opinion gaps related to IPF. Overall, pulmonologists were more likely than PCPs to cite both imaging and testing issues and waiting 6-8 weeks after symptom onset before imaging as contributing factors to diagnostic delays.
PCPs more often expressed beliefs that delayed diagnosis had little impact on a patient with IPF, and that the treatments may be worse than the disease.
Dr. Dilling said he was not surprised by the survey findings, as similar clues about the underdiagnosis of IPF have surfaced in prior studies.
“We need to get the word out to primary care physicians, to pulmonary physicians, and even to the public, that idiopathic pulmonary fibrosis and other forms of interstitial lung disease are out there and prevalent, and that making the right diagnosis in a timely way can lead to better outcomes for patients,” he said.
The take-home message for primary care is to think outside the COPD box, said Dr. Dilling. “Just because someone has shortness of breath or cough and used to smoke does not automatically mean that they have COPD,” he emphasized. “Listen carefully for crackles (rales) on exam. Get spirometry or PFTs before you secure the diagnosis of COPD, or else you will be missing all of your cases of pulmonary fibrosis; think of pulmonary fibrosis and use imaging to help guide your diagnosis,” he said.
The authors suggested several education goals for PCPs, including establishing the importance of early evaluation, outlining the correct approach to a patient workup, encouraging prompt referral, and empowering PCPs as part of the team approach to IPF patients’ care. For pulmonologists, only 11% of those surveyed said they were aware of the latest developments in antifibrotic research, and education efforts might include information about drug pipelines and clinical trials, as well as technology.
Looking ahead, “We need to better understand how to find the pulmonary fibrosis in the community,” Dr. Dilling said. This understanding may come in part from greater education and awareness, he noted. However, eventually there may be ways to enhance the reading of PFTs and of CT scans through artificial intelligence technologies that would not only prompt clinicians to recognize what they are seeing, but would prompt them to refer and send the patient on the correct diagnostic path as soon as possible, he added.
Key message: Include ILD in differential diagnosis of patients with shortness of breath and/or cough
Advances in diagnostics and therapies for interstitial lung disease can take time to be absorbed and adopted, and patients with ILD and pulmonologists caring for ILD, specifically IPF, continue to report delays in diagnosis and therapy, said Krishna Thavarajah, MD, a pulmonologist at Henry Ford Hospital, Detroit, Mich., in an interview.
The current study findings of the time to diagnosis and the approach to patient workups echo her own clinical experience, Dr. Thavarajah said. “There is a delay in IPF diagnosis as physicians look to more common diagnoses, such as cardiac disease or chronic obstructive pulmonary disease, prior to pursuit of additional workup, and the attitude toward treatment has, in some ways, lagged behind advances in therapy, including timing and feasibility of therapy for IPF,” she said.
The key message for primary care physicians is to include ILD in the differential diagnosis of patients with shortness of breath and/or cough, especially if the initial cardiac and pulmonary test (meaning at least a chest x-ray and pulmonary function tests, including a diffusion capacity) are not pointing to an alternative cause within 3 months of presentation, Dr. Thavarajah said.
Once IPF is diagnosed, primary care clinicians should know that there are FDA-approved therapies that improve survival, said Dr. Thavarajah. “There are identifiable and treatable comorbid conditions,” she added. “The statement of ‘time lost is lung lost’ sums up the care of an IPF patient; partnerships between primary care clinicians, pulmonologists, and referral centers can provide the patient multiple levels of support with quality-of-life interventions, treatments, and also clinical trials, delivered by a team of providers,” she said.
In the wake of the current study, more research is needed with outcome studies regarding educational interventions targeting primary care and pulmonologists on appropriate workup, timing of workup, and current therapy for IPF patients, she added.
The white paper received no outside funding. The authors and Dr. Thavarajah had no financial conflicts to disclose.
The nonspecific nature of the symptoms of idiopathic pulmonary fibrosis (IPF) especially in early stages, and the relative rarity of IPF compared with other conditions that have similar symptoms, may contribute to a delay in diagnosis in the primary care setting, wrote Daniel F. Dilling, MD, of Loyola University Chicago, Maywood, Ill., and colleagues in Chest: Clinical Perspectives (Dilling et al. State of Practice: Factors Driving Diagnostic Delays in Idiopathic Pulmonary Fibrosis. Chest. 2022).
“We have learned over and over again through research, and also through talking with our own patients with IPF, that there is often a long lag between the first signs of the disease and a diagnosis of IPF,” corresponding author Dr. Dilling said in an interview.
“Even some pulmonary specialists can be uncertain about how to approach the diagnosis when a CT scan or other test first suggests the possibility; this can cost a patient precious time, as being on drug therapy earlier can result in preservation of lung function,” he said. “By sounding the alarm bell with this paper, we hope to promote awareness and education/training within the primary care community as well as the pulmonary community, and also to make all of them aware of the possibility of referral to specialty ILD [interstitial lung disease] centers when desired and possible,” he added.
The researchers conducted a pair of online surveys to inform the development of improving education on IPF among primary care providers.
In the white paper, which can be accessed online, the authors reported results of the surveys. One included 100 general pulmonologists and the other included 306 primary care physicians (156 practiced family physicians and 150 practiced general internal medicine). The data were collected between April 11, 2022, and May 16, 2022. Participants were asked to respond to a patient case scenario of a 55-year-old woman with nonspecific symptoms such as shortness of breath on moderate exertion, cough, exhaustion, and trouble sleeping.
The PCPs were most likely to evaluate the patient for a cardiac condition (46%), 25% would evaluate for chronic obstructive pulmonary disease (COPD), and 23% for asthma. More than half (58%) ranked progressive fibrosing ILD as one of their bottom two diagnoses.
A total of 87% of PCPs said they would begin a diagnostic workup to evaluate symptoms if the patient had no preexisting respiratory disease, compared with 61% for patients with a respiratory diagnosis.
Although 93% of PCPs cited a chest x-ray as part of the initial patient workup, fewer than half said they would order an echocardiogram, spirometry, or pulmonary function test (PFT), and 11% said they would include diffusion capacity testing in the initial workup.
In addition, PCPs were less likely to ask patients about issues that might prompt an IPF diagnosis, such as exposures to agents through work, hobbies, the environment, or comorbidities.
In the pulmonology survey, more than 75% of respondents cited patient history, high-resolution tomography scan, serologic testing, and review for autoimmune disease symptoms as first steps in a diagnostic response to patients with suspected IPF.
Differences between PCPs’ and pulmonolgists’ responses
Both PCPs and pulmonologists responded to several questions to assess knowledge and opinion gaps related to IPF. Overall, pulmonologists were more likely than PCPs to cite both imaging and testing issues and waiting 6-8 weeks after symptom onset before imaging as contributing factors to diagnostic delays.
PCPs more often expressed beliefs that delayed diagnosis had little impact on a patient with IPF, and that the treatments may be worse than the disease.
Dr. Dilling said he was not surprised by the survey findings, as similar clues about the underdiagnosis of IPF have surfaced in prior studies.
“We need to get the word out to primary care physicians, to pulmonary physicians, and even to the public, that idiopathic pulmonary fibrosis and other forms of interstitial lung disease are out there and prevalent, and that making the right diagnosis in a timely way can lead to better outcomes for patients,” he said.
The take-home message for primary care is to think outside the COPD box, said Dr. Dilling. “Just because someone has shortness of breath or cough and used to smoke does not automatically mean that they have COPD,” he emphasized. “Listen carefully for crackles (rales) on exam. Get spirometry or PFTs before you secure the diagnosis of COPD, or else you will be missing all of your cases of pulmonary fibrosis; think of pulmonary fibrosis and use imaging to help guide your diagnosis,” he said.
The authors suggested several education goals for PCPs, including establishing the importance of early evaluation, outlining the correct approach to a patient workup, encouraging prompt referral, and empowering PCPs as part of the team approach to IPF patients’ care. For pulmonologists, only 11% of those surveyed said they were aware of the latest developments in antifibrotic research, and education efforts might include information about drug pipelines and clinical trials, as well as technology.
Looking ahead, “We need to better understand how to find the pulmonary fibrosis in the community,” Dr. Dilling said. This understanding may come in part from greater education and awareness, he noted. However, eventually there may be ways to enhance the reading of PFTs and of CT scans through artificial intelligence technologies that would not only prompt clinicians to recognize what they are seeing, but would prompt them to refer and send the patient on the correct diagnostic path as soon as possible, he added.
Key message: Include ILD in differential diagnosis of patients with shortness of breath and/or cough
Advances in diagnostics and therapies for interstitial lung disease can take time to be absorbed and adopted, and patients with ILD and pulmonologists caring for ILD, specifically IPF, continue to report delays in diagnosis and therapy, said Krishna Thavarajah, MD, a pulmonologist at Henry Ford Hospital, Detroit, Mich., in an interview.
The current study findings of the time to diagnosis and the approach to patient workups echo her own clinical experience, Dr. Thavarajah said. “There is a delay in IPF diagnosis as physicians look to more common diagnoses, such as cardiac disease or chronic obstructive pulmonary disease, prior to pursuit of additional workup, and the attitude toward treatment has, in some ways, lagged behind advances in therapy, including timing and feasibility of therapy for IPF,” she said.
The key message for primary care physicians is to include ILD in the differential diagnosis of patients with shortness of breath and/or cough, especially if the initial cardiac and pulmonary test (meaning at least a chest x-ray and pulmonary function tests, including a diffusion capacity) are not pointing to an alternative cause within 3 months of presentation, Dr. Thavarajah said.
Once IPF is diagnosed, primary care clinicians should know that there are FDA-approved therapies that improve survival, said Dr. Thavarajah. “There are identifiable and treatable comorbid conditions,” she added. “The statement of ‘time lost is lung lost’ sums up the care of an IPF patient; partnerships between primary care clinicians, pulmonologists, and referral centers can provide the patient multiple levels of support with quality-of-life interventions, treatments, and also clinical trials, delivered by a team of providers,” she said.
In the wake of the current study, more research is needed with outcome studies regarding educational interventions targeting primary care and pulmonologists on appropriate workup, timing of workup, and current therapy for IPF patients, she added.
The white paper received no outside funding. The authors and Dr. Thavarajah had no financial conflicts to disclose.
The nonspecific nature of the symptoms of idiopathic pulmonary fibrosis (IPF) especially in early stages, and the relative rarity of IPF compared with other conditions that have similar symptoms, may contribute to a delay in diagnosis in the primary care setting, wrote Daniel F. Dilling, MD, of Loyola University Chicago, Maywood, Ill., and colleagues in Chest: Clinical Perspectives (Dilling et al. State of Practice: Factors Driving Diagnostic Delays in Idiopathic Pulmonary Fibrosis. Chest. 2022).
“We have learned over and over again through research, and also through talking with our own patients with IPF, that there is often a long lag between the first signs of the disease and a diagnosis of IPF,” corresponding author Dr. Dilling said in an interview.
“Even some pulmonary specialists can be uncertain about how to approach the diagnosis when a CT scan or other test first suggests the possibility; this can cost a patient precious time, as being on drug therapy earlier can result in preservation of lung function,” he said. “By sounding the alarm bell with this paper, we hope to promote awareness and education/training within the primary care community as well as the pulmonary community, and also to make all of them aware of the possibility of referral to specialty ILD [interstitial lung disease] centers when desired and possible,” he added.
The researchers conducted a pair of online surveys to inform the development of improving education on IPF among primary care providers.
In the white paper, which can be accessed online, the authors reported results of the surveys. One included 100 general pulmonologists and the other included 306 primary care physicians (156 practiced family physicians and 150 practiced general internal medicine). The data were collected between April 11, 2022, and May 16, 2022. Participants were asked to respond to a patient case scenario of a 55-year-old woman with nonspecific symptoms such as shortness of breath on moderate exertion, cough, exhaustion, and trouble sleeping.
The PCPs were most likely to evaluate the patient for a cardiac condition (46%), 25% would evaluate for chronic obstructive pulmonary disease (COPD), and 23% for asthma. More than half (58%) ranked progressive fibrosing ILD as one of their bottom two diagnoses.
A total of 87% of PCPs said they would begin a diagnostic workup to evaluate symptoms if the patient had no preexisting respiratory disease, compared with 61% for patients with a respiratory diagnosis.
Although 93% of PCPs cited a chest x-ray as part of the initial patient workup, fewer than half said they would order an echocardiogram, spirometry, or pulmonary function test (PFT), and 11% said they would include diffusion capacity testing in the initial workup.
In addition, PCPs were less likely to ask patients about issues that might prompt an IPF diagnosis, such as exposures to agents through work, hobbies, the environment, or comorbidities.
In the pulmonology survey, more than 75% of respondents cited patient history, high-resolution tomography scan, serologic testing, and review for autoimmune disease symptoms as first steps in a diagnostic response to patients with suspected IPF.
Differences between PCPs’ and pulmonolgists’ responses
Both PCPs and pulmonologists responded to several questions to assess knowledge and opinion gaps related to IPF. Overall, pulmonologists were more likely than PCPs to cite both imaging and testing issues and waiting 6-8 weeks after symptom onset before imaging as contributing factors to diagnostic delays.
PCPs more often expressed beliefs that delayed diagnosis had little impact on a patient with IPF, and that the treatments may be worse than the disease.
Dr. Dilling said he was not surprised by the survey findings, as similar clues about the underdiagnosis of IPF have surfaced in prior studies.
“We need to get the word out to primary care physicians, to pulmonary physicians, and even to the public, that idiopathic pulmonary fibrosis and other forms of interstitial lung disease are out there and prevalent, and that making the right diagnosis in a timely way can lead to better outcomes for patients,” he said.
The take-home message for primary care is to think outside the COPD box, said Dr. Dilling. “Just because someone has shortness of breath or cough and used to smoke does not automatically mean that they have COPD,” he emphasized. “Listen carefully for crackles (rales) on exam. Get spirometry or PFTs before you secure the diagnosis of COPD, or else you will be missing all of your cases of pulmonary fibrosis; think of pulmonary fibrosis and use imaging to help guide your diagnosis,” he said.
The authors suggested several education goals for PCPs, including establishing the importance of early evaluation, outlining the correct approach to a patient workup, encouraging prompt referral, and empowering PCPs as part of the team approach to IPF patients’ care. For pulmonologists, only 11% of those surveyed said they were aware of the latest developments in antifibrotic research, and education efforts might include information about drug pipelines and clinical trials, as well as technology.
Looking ahead, “We need to better understand how to find the pulmonary fibrosis in the community,” Dr. Dilling said. This understanding may come in part from greater education and awareness, he noted. However, eventually there may be ways to enhance the reading of PFTs and of CT scans through artificial intelligence technologies that would not only prompt clinicians to recognize what they are seeing, but would prompt them to refer and send the patient on the correct diagnostic path as soon as possible, he added.
Key message: Include ILD in differential diagnosis of patients with shortness of breath and/or cough
Advances in diagnostics and therapies for interstitial lung disease can take time to be absorbed and adopted, and patients with ILD and pulmonologists caring for ILD, specifically IPF, continue to report delays in diagnosis and therapy, said Krishna Thavarajah, MD, a pulmonologist at Henry Ford Hospital, Detroit, Mich., in an interview.
The current study findings of the time to diagnosis and the approach to patient workups echo her own clinical experience, Dr. Thavarajah said. “There is a delay in IPF diagnosis as physicians look to more common diagnoses, such as cardiac disease or chronic obstructive pulmonary disease, prior to pursuit of additional workup, and the attitude toward treatment has, in some ways, lagged behind advances in therapy, including timing and feasibility of therapy for IPF,” she said.
The key message for primary care physicians is to include ILD in the differential diagnosis of patients with shortness of breath and/or cough, especially if the initial cardiac and pulmonary test (meaning at least a chest x-ray and pulmonary function tests, including a diffusion capacity) are not pointing to an alternative cause within 3 months of presentation, Dr. Thavarajah said.
Once IPF is diagnosed, primary care clinicians should know that there are FDA-approved therapies that improve survival, said Dr. Thavarajah. “There are identifiable and treatable comorbid conditions,” she added. “The statement of ‘time lost is lung lost’ sums up the care of an IPF patient; partnerships between primary care clinicians, pulmonologists, and referral centers can provide the patient multiple levels of support with quality-of-life interventions, treatments, and also clinical trials, delivered by a team of providers,” she said.
In the wake of the current study, more research is needed with outcome studies regarding educational interventions targeting primary care and pulmonologists on appropriate workup, timing of workup, and current therapy for IPF patients, she added.
The white paper received no outside funding. The authors and Dr. Thavarajah had no financial conflicts to disclose.
FROM CHEST CLINICAL PERSPECTIVES
Shift in child hospice care is a lifeline for parents seeking a measure of comfort and hope
POMONA, CALIF. – When you first meet 17-month-old Aaron Martinez, it’s not obvious that something is catastrophically wrong.
What you see is a beautiful little boy with smooth, lustrous skin, an abundance of glossy brown hair, and a disarming smile. What you hear are coos and cries that don’t immediately signal anything is horribly awry.
But his parents, Adriana Pinedo and Hector Martinez, know the truth painfully well.
Although Ms. Pinedo’s doctors and midwife had described the pregnancy as “perfect” for all 9 months, Aaron was born with most of his brain cells dead, the result of two strokes and a massive bleed he sustained while in utero.
Doctors aren’t sure what caused the anomalies that left Aaron with virtually no cognitive function or physical mobility. His voluminous hair hides a head whose circumference is too small for his age. He has epilepsy that triggers multiple seizures each day, and his smile is not always what it seems. “It could be a smile; it could be a seizure,” Ms. Pinedo said.
Shortly after Aaron was born, doctors told Ms. Pinedo, 34, and Mr. Martinez, 35, there was no hope and they should “let nature take its course.” They would learn months later that the doctors had not expected the boy to live more than 5 days. It was on Day 5 that his parents put him in home hospice care, an arrangement that has continued into his second year of life.
The family gets weekly visits from hospice nurses, therapists, social workers, and a chaplain in the cramped one-bedroom apartment they rent from the people who live in the main house on the same lot on a quiet residential street in this Inland Empire city.
One of the main criteria for hospice care, established by Medicare largely for seniors but also applied to children, is a diagnosis of 6 months or less to live. Yet over the course of 17 months, Aaron’s medical team has repeatedly recertified his hospice eligibility.
Under a provision of the 2010 Affordable Care Act, children enrolled in Medicaid or the Children’s Health Insurance Program are allowed, unlike adults, to be in hospice while continuing to receive curative or life-extending care. Commercial insurers are not required to cover this “concurrent care,” but many now do.
More than a decade since its inception, concurrent care is widely credited with improving the quality of life for many terminally ill children, easing stress on the family and, in some cases, sustaining hope for a cure. But the arrangement can contribute to a painful dilemma for parents like Ms. Pinedo and Mr. Martinez, who are torn between their fierce commitment to their son and the futility of knowing that his condition leaves him with no future worth hoping for.
“We could lose a life, but if he continues to live this way, we’ll lose three,” said Ms. Pinedo. “There’s no quality of life for him or for us.”
Aaron’s doctors now say he could conceivably live for years. His body hasn’t stopped growing since he was born. He’s in the 96th percentile for height for his age, and his weight is about average.
His parents have talked about “graduating” him from hospice. But he is never stable for long, and they welcome the visits from their hospice team. The seizures, sometimes 30 a day, are a persistent assault on his brain and, as he grows, the medications intended to control them must be changed or the doses recalibrated. He is at continual risk of gastrointestinal problems and potentially deadly fluid buildup in his lungs.
Ms. Pinedo, who works from home for a nonprofit public health organization, spends much of her time with Aaron, while Mr. Martinez works as a landscaper. She has chosen to live in the moment, she said, because otherwise her mind wanders to a future in which either “he could die – or he won’t, and I’ll end up changing the diapers of a 40-year-old man.” Either of those “are going to suck.”
While cancer is one of the major illnesses afflicting children in hospice, many others, like Aaron, have rare congenital defects, severe neurological impairments, or uncommon metabolic deficiencies.
“We have diseases that families tell us are 1 of 10 cases in the world,” said Glen Komatsu, MD, medical director of Torrance, Calif.–based TrinityKids Care, which provides home hospice services to Aaron and more than 70 other kids in Los Angeles and Orange counties.
In the years leading up to the ACA’s implementation, pediatric health advocates lobbied hard for the concurrent care provision. Without the possibility of life-extending care or hope for a cure, many parents refused to put their terminally ill kids in hospice, thinking it was tantamount to giving up on them. That meant the whole family missed out on the support hospice can provide, not just pain relief and comfort for the dying child, but emotional and spiritual care for parents and siblings under extreme duress.
TrinityKids Care, run by the large national Catholic health system Providence, doesn’t just send nurses, social workers, and chaplains into homes. For patients able to participate, and their siblings, it also offers art and science projects, exercise classes, movies, and music. During the pandemic, these activities have been conducted via Zoom, and volunteers deliver needed supplies to the children’s homes.
The ability to get treatments that prolong their lives is a major reason children in concurrent care are more likely than adults to outlive the 6-months-to-live diagnosis required for hospice.
“Concurrent care, by its very intention, very clearly is going to extend their lives, and by extending their lives they’re no longer going to be hospice-eligible if you use the 6-month life expectancy criteria,” said David Steinhorn, MD, a pediatric intensive care physician in Virginia, who has helped develop numerous children’s hospice programs across the United States.
Another factor is that kids, even sick ones, are simply more robust than many older people.
“Sick kids are often otherwise healthy, except for one organ,” said Debra Lotstein, MD, chief of the division of comfort and palliative care at Children’s Hospital Los Angeles. “They may have cancer in their body, but their hearts are good and their lungs are good, compared to a 90-year-old who at baseline is just not as resilient.”
All of Aaron Martinez’s vital organs, except for his brain, seem to be working. “There have been times when we’ve brought him in, and the nurse looks at the chart and looks at him, and she can’t believe it’s that child,” said Mr. Martinez.
When kids live past the 6-month life expectancy, they must be recertified to stay in hospice. In many cases, Dr. Steinhorn said, he is willing to recertify his pediatric patients indefinitely.
Even with doctors advocating for them, it’s not always easy for children to get into hospice care. Most hospices care primarily for adults and are reluctant to take kids.
“The hospice will say: ‘We don’t have the capacity to treat children. Our nurses aren’t trained. It’s different. We just can’t do it,’ ” said Lori Butterworth, cofounder of the Children’s Hospice and Palliative Care Coalition of California in Watsonville. “The other reason is not wanting to, because it’s existentially devastating and sad and hard.”
Finances also play a role. Home hospice care is paid at a per diem rate set by Medicare – slightly over $200 a day for the first 2 months, about $161 a day after that – and it is typically the same for kids and adults. Children, particularly those with rare conditions, often require more intensive and innovative care, so the per diem doesn’t stretch as far.
The concurrent care provision has made taking pediatric patients more viable for hospice organizations, Dr. Steinhorn and others said. Under the ACA, many of the expenses for certain medications and medical services can be shifted to the patient’s primary insurance, leaving hospices responsible for pain relief and comfort care.
Even so, the relatively small number of kids who die each year from protracted ailments hardly makes pediatric hospice an appealing line of business in an industry craving growth, especially one in which private equity investors are active and seeking a big payday.
In California, only 21 of 1,336 hospices reported having a specialized pediatric hospice program, and 59 said they served at least one patient under age 21, according to an analysis of 2020 state data by Cordt Kassner, CEO of Hospice Analytics in Colorado Springs.
Hospice providers that do cater to children often face a more basic challenge: Even with the possibility of concurrent care, many parents still equate hospice with acceptance of death. That was the case initially for Matt and Reese Sonnen, Los Angeles residents whose daughter, Layla, was born with a seizure disorder that had no name: Her brain had simply failed to develop in the womb, and an MRI showed “fluid taking up space where the brain wasn’t,” her mother said.
When Layla’s team first mentioned hospice, “I was in the car on my phone, and I almost crashed the car,” Mrs. Sonnen recalled. “The first thought that came to mind was: ‘It is just the end,’ but we felt she was nowhere near it, because she was strong, she was mighty. She was my little girl. She was going to get through this.”
About 3 months later, as Layla’s nervous system deteriorated, causing her to writhe in pain, her parents agreed to enroll her in hospice with TrinityKids Care. She died weeks later, not long after her second birthday. She was in her mother’s arms, with Mr. Sonnen close by.
“All of a sudden, Layla breathed out a big rush of air. The nurse looked at me and said: ‘That was her last breath.’ I was literally breathing in her last breath,” Mrs. Sonnen recounted. “I never wanted to breathe again, because now I felt I had her in my lungs. Don’t make me laugh, don’t make me exhale.”
Layla’s parents have no regrets about their decision to put her in hospice. “It was the absolute right decision, and in hindsight we should have done it sooner,” Mr. Sonnen said. “She was suffering, and we had blinders on.”
Ms. Pinedo said she is “infinitely grateful” for hospice, despite the heartache of Aaron’s condition. Sometimes the social worker will stop by, she said, just to say hello and drop off a latte, a small gesture that can feel very uplifting. “They’ve been our lifeline,” she said.
Ms. Pinedo talks about a friend of hers with a healthy baby, also named Aaron, who is pregnant with her second child. “All the stuff that was on our list, they’re living. And I love them dearly. But it’s almost hard to look, because it’s like looking at the stuff that you didn’t get. It’s like Christmas Day, staring through the window at the neighbor’s house, and you’re sitting there in the cold.”
Yet she seems palpably torn between that bleak remorse and the unconditional love parents feel toward their children. At one point, Ms. Pinedo interrupted herself midsentence and turned to her son, who was in Mr. Martinez’s arms: “Yes, Papi, you are so stinking cute, and you are still my dream come true.”
This story was produced by KHN, which publishes California Healthline, an editorially independent service of the California Health Care Foundation. KHN (Kaiser Health News) is a national newsroom that produces in-depth journalism about health issues. Together with Policy Analysis and Polling, KHN is one of the three major operating programs at KFF (Kaiser Family Foundation). KFF is an endowed nonprofit organization providing information on health issues to the nation.
POMONA, CALIF. – When you first meet 17-month-old Aaron Martinez, it’s not obvious that something is catastrophically wrong.
What you see is a beautiful little boy with smooth, lustrous skin, an abundance of glossy brown hair, and a disarming smile. What you hear are coos and cries that don’t immediately signal anything is horribly awry.
But his parents, Adriana Pinedo and Hector Martinez, know the truth painfully well.
Although Ms. Pinedo’s doctors and midwife had described the pregnancy as “perfect” for all 9 months, Aaron was born with most of his brain cells dead, the result of two strokes and a massive bleed he sustained while in utero.
Doctors aren’t sure what caused the anomalies that left Aaron with virtually no cognitive function or physical mobility. His voluminous hair hides a head whose circumference is too small for his age. He has epilepsy that triggers multiple seizures each day, and his smile is not always what it seems. “It could be a smile; it could be a seizure,” Ms. Pinedo said.
Shortly after Aaron was born, doctors told Ms. Pinedo, 34, and Mr. Martinez, 35, there was no hope and they should “let nature take its course.” They would learn months later that the doctors had not expected the boy to live more than 5 days. It was on Day 5 that his parents put him in home hospice care, an arrangement that has continued into his second year of life.
The family gets weekly visits from hospice nurses, therapists, social workers, and a chaplain in the cramped one-bedroom apartment they rent from the people who live in the main house on the same lot on a quiet residential street in this Inland Empire city.
One of the main criteria for hospice care, established by Medicare largely for seniors but also applied to children, is a diagnosis of 6 months or less to live. Yet over the course of 17 months, Aaron’s medical team has repeatedly recertified his hospice eligibility.
Under a provision of the 2010 Affordable Care Act, children enrolled in Medicaid or the Children’s Health Insurance Program are allowed, unlike adults, to be in hospice while continuing to receive curative or life-extending care. Commercial insurers are not required to cover this “concurrent care,” but many now do.
More than a decade since its inception, concurrent care is widely credited with improving the quality of life for many terminally ill children, easing stress on the family and, in some cases, sustaining hope for a cure. But the arrangement can contribute to a painful dilemma for parents like Ms. Pinedo and Mr. Martinez, who are torn between their fierce commitment to their son and the futility of knowing that his condition leaves him with no future worth hoping for.
“We could lose a life, but if he continues to live this way, we’ll lose three,” said Ms. Pinedo. “There’s no quality of life for him or for us.”
Aaron’s doctors now say he could conceivably live for years. His body hasn’t stopped growing since he was born. He’s in the 96th percentile for height for his age, and his weight is about average.
His parents have talked about “graduating” him from hospice. But he is never stable for long, and they welcome the visits from their hospice team. The seizures, sometimes 30 a day, are a persistent assault on his brain and, as he grows, the medications intended to control them must be changed or the doses recalibrated. He is at continual risk of gastrointestinal problems and potentially deadly fluid buildup in his lungs.
Ms. Pinedo, who works from home for a nonprofit public health organization, spends much of her time with Aaron, while Mr. Martinez works as a landscaper. She has chosen to live in the moment, she said, because otherwise her mind wanders to a future in which either “he could die – or he won’t, and I’ll end up changing the diapers of a 40-year-old man.” Either of those “are going to suck.”
While cancer is one of the major illnesses afflicting children in hospice, many others, like Aaron, have rare congenital defects, severe neurological impairments, or uncommon metabolic deficiencies.
“We have diseases that families tell us are 1 of 10 cases in the world,” said Glen Komatsu, MD, medical director of Torrance, Calif.–based TrinityKids Care, which provides home hospice services to Aaron and more than 70 other kids in Los Angeles and Orange counties.
In the years leading up to the ACA’s implementation, pediatric health advocates lobbied hard for the concurrent care provision. Without the possibility of life-extending care or hope for a cure, many parents refused to put their terminally ill kids in hospice, thinking it was tantamount to giving up on them. That meant the whole family missed out on the support hospice can provide, not just pain relief and comfort for the dying child, but emotional and spiritual care for parents and siblings under extreme duress.
TrinityKids Care, run by the large national Catholic health system Providence, doesn’t just send nurses, social workers, and chaplains into homes. For patients able to participate, and their siblings, it also offers art and science projects, exercise classes, movies, and music. During the pandemic, these activities have been conducted via Zoom, and volunteers deliver needed supplies to the children’s homes.
The ability to get treatments that prolong their lives is a major reason children in concurrent care are more likely than adults to outlive the 6-months-to-live diagnosis required for hospice.
“Concurrent care, by its very intention, very clearly is going to extend their lives, and by extending their lives they’re no longer going to be hospice-eligible if you use the 6-month life expectancy criteria,” said David Steinhorn, MD, a pediatric intensive care physician in Virginia, who has helped develop numerous children’s hospice programs across the United States.
Another factor is that kids, even sick ones, are simply more robust than many older people.
“Sick kids are often otherwise healthy, except for one organ,” said Debra Lotstein, MD, chief of the division of comfort and palliative care at Children’s Hospital Los Angeles. “They may have cancer in their body, but their hearts are good and their lungs are good, compared to a 90-year-old who at baseline is just not as resilient.”
All of Aaron Martinez’s vital organs, except for his brain, seem to be working. “There have been times when we’ve brought him in, and the nurse looks at the chart and looks at him, and she can’t believe it’s that child,” said Mr. Martinez.
When kids live past the 6-month life expectancy, they must be recertified to stay in hospice. In many cases, Dr. Steinhorn said, he is willing to recertify his pediatric patients indefinitely.
Even with doctors advocating for them, it’s not always easy for children to get into hospice care. Most hospices care primarily for adults and are reluctant to take kids.
“The hospice will say: ‘We don’t have the capacity to treat children. Our nurses aren’t trained. It’s different. We just can’t do it,’ ” said Lori Butterworth, cofounder of the Children’s Hospice and Palliative Care Coalition of California in Watsonville. “The other reason is not wanting to, because it’s existentially devastating and sad and hard.”
Finances also play a role. Home hospice care is paid at a per diem rate set by Medicare – slightly over $200 a day for the first 2 months, about $161 a day after that – and it is typically the same for kids and adults. Children, particularly those with rare conditions, often require more intensive and innovative care, so the per diem doesn’t stretch as far.
The concurrent care provision has made taking pediatric patients more viable for hospice organizations, Dr. Steinhorn and others said. Under the ACA, many of the expenses for certain medications and medical services can be shifted to the patient’s primary insurance, leaving hospices responsible for pain relief and comfort care.
Even so, the relatively small number of kids who die each year from protracted ailments hardly makes pediatric hospice an appealing line of business in an industry craving growth, especially one in which private equity investors are active and seeking a big payday.
In California, only 21 of 1,336 hospices reported having a specialized pediatric hospice program, and 59 said they served at least one patient under age 21, according to an analysis of 2020 state data by Cordt Kassner, CEO of Hospice Analytics in Colorado Springs.
Hospice providers that do cater to children often face a more basic challenge: Even with the possibility of concurrent care, many parents still equate hospice with acceptance of death. That was the case initially for Matt and Reese Sonnen, Los Angeles residents whose daughter, Layla, was born with a seizure disorder that had no name: Her brain had simply failed to develop in the womb, and an MRI showed “fluid taking up space where the brain wasn’t,” her mother said.
When Layla’s team first mentioned hospice, “I was in the car on my phone, and I almost crashed the car,” Mrs. Sonnen recalled. “The first thought that came to mind was: ‘It is just the end,’ but we felt she was nowhere near it, because she was strong, she was mighty. She was my little girl. She was going to get through this.”
About 3 months later, as Layla’s nervous system deteriorated, causing her to writhe in pain, her parents agreed to enroll her in hospice with TrinityKids Care. She died weeks later, not long after her second birthday. She was in her mother’s arms, with Mr. Sonnen close by.
“All of a sudden, Layla breathed out a big rush of air. The nurse looked at me and said: ‘That was her last breath.’ I was literally breathing in her last breath,” Mrs. Sonnen recounted. “I never wanted to breathe again, because now I felt I had her in my lungs. Don’t make me laugh, don’t make me exhale.”
Layla’s parents have no regrets about their decision to put her in hospice. “It was the absolute right decision, and in hindsight we should have done it sooner,” Mr. Sonnen said. “She was suffering, and we had blinders on.”
Ms. Pinedo said she is “infinitely grateful” for hospice, despite the heartache of Aaron’s condition. Sometimes the social worker will stop by, she said, just to say hello and drop off a latte, a small gesture that can feel very uplifting. “They’ve been our lifeline,” she said.
Ms. Pinedo talks about a friend of hers with a healthy baby, also named Aaron, who is pregnant with her second child. “All the stuff that was on our list, they’re living. And I love them dearly. But it’s almost hard to look, because it’s like looking at the stuff that you didn’t get. It’s like Christmas Day, staring through the window at the neighbor’s house, and you’re sitting there in the cold.”
Yet she seems palpably torn between that bleak remorse and the unconditional love parents feel toward their children. At one point, Ms. Pinedo interrupted herself midsentence and turned to her son, who was in Mr. Martinez’s arms: “Yes, Papi, you are so stinking cute, and you are still my dream come true.”
This story was produced by KHN, which publishes California Healthline, an editorially independent service of the California Health Care Foundation. KHN (Kaiser Health News) is a national newsroom that produces in-depth journalism about health issues. Together with Policy Analysis and Polling, KHN is one of the three major operating programs at KFF (Kaiser Family Foundation). KFF is an endowed nonprofit organization providing information on health issues to the nation.
POMONA, CALIF. – When you first meet 17-month-old Aaron Martinez, it’s not obvious that something is catastrophically wrong.
What you see is a beautiful little boy with smooth, lustrous skin, an abundance of glossy brown hair, and a disarming smile. What you hear are coos and cries that don’t immediately signal anything is horribly awry.
But his parents, Adriana Pinedo and Hector Martinez, know the truth painfully well.
Although Ms. Pinedo’s doctors and midwife had described the pregnancy as “perfect” for all 9 months, Aaron was born with most of his brain cells dead, the result of two strokes and a massive bleed he sustained while in utero.
Doctors aren’t sure what caused the anomalies that left Aaron with virtually no cognitive function or physical mobility. His voluminous hair hides a head whose circumference is too small for his age. He has epilepsy that triggers multiple seizures each day, and his smile is not always what it seems. “It could be a smile; it could be a seizure,” Ms. Pinedo said.
Shortly after Aaron was born, doctors told Ms. Pinedo, 34, and Mr. Martinez, 35, there was no hope and they should “let nature take its course.” They would learn months later that the doctors had not expected the boy to live more than 5 days. It was on Day 5 that his parents put him in home hospice care, an arrangement that has continued into his second year of life.
The family gets weekly visits from hospice nurses, therapists, social workers, and a chaplain in the cramped one-bedroom apartment they rent from the people who live in the main house on the same lot on a quiet residential street in this Inland Empire city.
One of the main criteria for hospice care, established by Medicare largely for seniors but also applied to children, is a diagnosis of 6 months or less to live. Yet over the course of 17 months, Aaron’s medical team has repeatedly recertified his hospice eligibility.
Under a provision of the 2010 Affordable Care Act, children enrolled in Medicaid or the Children’s Health Insurance Program are allowed, unlike adults, to be in hospice while continuing to receive curative or life-extending care. Commercial insurers are not required to cover this “concurrent care,” but many now do.
More than a decade since its inception, concurrent care is widely credited with improving the quality of life for many terminally ill children, easing stress on the family and, in some cases, sustaining hope for a cure. But the arrangement can contribute to a painful dilemma for parents like Ms. Pinedo and Mr. Martinez, who are torn between their fierce commitment to their son and the futility of knowing that his condition leaves him with no future worth hoping for.
“We could lose a life, but if he continues to live this way, we’ll lose three,” said Ms. Pinedo. “There’s no quality of life for him or for us.”
Aaron’s doctors now say he could conceivably live for years. His body hasn’t stopped growing since he was born. He’s in the 96th percentile for height for his age, and his weight is about average.
His parents have talked about “graduating” him from hospice. But he is never stable for long, and they welcome the visits from their hospice team. The seizures, sometimes 30 a day, are a persistent assault on his brain and, as he grows, the medications intended to control them must be changed or the doses recalibrated. He is at continual risk of gastrointestinal problems and potentially deadly fluid buildup in his lungs.
Ms. Pinedo, who works from home for a nonprofit public health organization, spends much of her time with Aaron, while Mr. Martinez works as a landscaper. She has chosen to live in the moment, she said, because otherwise her mind wanders to a future in which either “he could die – or he won’t, and I’ll end up changing the diapers of a 40-year-old man.” Either of those “are going to suck.”
While cancer is one of the major illnesses afflicting children in hospice, many others, like Aaron, have rare congenital defects, severe neurological impairments, or uncommon metabolic deficiencies.
“We have diseases that families tell us are 1 of 10 cases in the world,” said Glen Komatsu, MD, medical director of Torrance, Calif.–based TrinityKids Care, which provides home hospice services to Aaron and more than 70 other kids in Los Angeles and Orange counties.
In the years leading up to the ACA’s implementation, pediatric health advocates lobbied hard for the concurrent care provision. Without the possibility of life-extending care or hope for a cure, many parents refused to put their terminally ill kids in hospice, thinking it was tantamount to giving up on them. That meant the whole family missed out on the support hospice can provide, not just pain relief and comfort for the dying child, but emotional and spiritual care for parents and siblings under extreme duress.
TrinityKids Care, run by the large national Catholic health system Providence, doesn’t just send nurses, social workers, and chaplains into homes. For patients able to participate, and their siblings, it also offers art and science projects, exercise classes, movies, and music. During the pandemic, these activities have been conducted via Zoom, and volunteers deliver needed supplies to the children’s homes.
The ability to get treatments that prolong their lives is a major reason children in concurrent care are more likely than adults to outlive the 6-months-to-live diagnosis required for hospice.
“Concurrent care, by its very intention, very clearly is going to extend their lives, and by extending their lives they’re no longer going to be hospice-eligible if you use the 6-month life expectancy criteria,” said David Steinhorn, MD, a pediatric intensive care physician in Virginia, who has helped develop numerous children’s hospice programs across the United States.
Another factor is that kids, even sick ones, are simply more robust than many older people.
“Sick kids are often otherwise healthy, except for one organ,” said Debra Lotstein, MD, chief of the division of comfort and palliative care at Children’s Hospital Los Angeles. “They may have cancer in their body, but their hearts are good and their lungs are good, compared to a 90-year-old who at baseline is just not as resilient.”
All of Aaron Martinez’s vital organs, except for his brain, seem to be working. “There have been times when we’ve brought him in, and the nurse looks at the chart and looks at him, and she can’t believe it’s that child,” said Mr. Martinez.
When kids live past the 6-month life expectancy, they must be recertified to stay in hospice. In many cases, Dr. Steinhorn said, he is willing to recertify his pediatric patients indefinitely.
Even with doctors advocating for them, it’s not always easy for children to get into hospice care. Most hospices care primarily for adults and are reluctant to take kids.
“The hospice will say: ‘We don’t have the capacity to treat children. Our nurses aren’t trained. It’s different. We just can’t do it,’ ” said Lori Butterworth, cofounder of the Children’s Hospice and Palliative Care Coalition of California in Watsonville. “The other reason is not wanting to, because it’s existentially devastating and sad and hard.”
Finances also play a role. Home hospice care is paid at a per diem rate set by Medicare – slightly over $200 a day for the first 2 months, about $161 a day after that – and it is typically the same for kids and adults. Children, particularly those with rare conditions, often require more intensive and innovative care, so the per diem doesn’t stretch as far.
The concurrent care provision has made taking pediatric patients more viable for hospice organizations, Dr. Steinhorn and others said. Under the ACA, many of the expenses for certain medications and medical services can be shifted to the patient’s primary insurance, leaving hospices responsible for pain relief and comfort care.
Even so, the relatively small number of kids who die each year from protracted ailments hardly makes pediatric hospice an appealing line of business in an industry craving growth, especially one in which private equity investors are active and seeking a big payday.
In California, only 21 of 1,336 hospices reported having a specialized pediatric hospice program, and 59 said they served at least one patient under age 21, according to an analysis of 2020 state data by Cordt Kassner, CEO of Hospice Analytics in Colorado Springs.
Hospice providers that do cater to children often face a more basic challenge: Even with the possibility of concurrent care, many parents still equate hospice with acceptance of death. That was the case initially for Matt and Reese Sonnen, Los Angeles residents whose daughter, Layla, was born with a seizure disorder that had no name: Her brain had simply failed to develop in the womb, and an MRI showed “fluid taking up space where the brain wasn’t,” her mother said.
When Layla’s team first mentioned hospice, “I was in the car on my phone, and I almost crashed the car,” Mrs. Sonnen recalled. “The first thought that came to mind was: ‘It is just the end,’ but we felt she was nowhere near it, because she was strong, she was mighty. She was my little girl. She was going to get through this.”
About 3 months later, as Layla’s nervous system deteriorated, causing her to writhe in pain, her parents agreed to enroll her in hospice with TrinityKids Care. She died weeks later, not long after her second birthday. She was in her mother’s arms, with Mr. Sonnen close by.
“All of a sudden, Layla breathed out a big rush of air. The nurse looked at me and said: ‘That was her last breath.’ I was literally breathing in her last breath,” Mrs. Sonnen recounted. “I never wanted to breathe again, because now I felt I had her in my lungs. Don’t make me laugh, don’t make me exhale.”
Layla’s parents have no regrets about their decision to put her in hospice. “It was the absolute right decision, and in hindsight we should have done it sooner,” Mr. Sonnen said. “She was suffering, and we had blinders on.”
Ms. Pinedo said she is “infinitely grateful” for hospice, despite the heartache of Aaron’s condition. Sometimes the social worker will stop by, she said, just to say hello and drop off a latte, a small gesture that can feel very uplifting. “They’ve been our lifeline,” she said.
Ms. Pinedo talks about a friend of hers with a healthy baby, also named Aaron, who is pregnant with her second child. “All the stuff that was on our list, they’re living. And I love them dearly. But it’s almost hard to look, because it’s like looking at the stuff that you didn’t get. It’s like Christmas Day, staring through the window at the neighbor’s house, and you’re sitting there in the cold.”
Yet she seems palpably torn between that bleak remorse and the unconditional love parents feel toward their children. At one point, Ms. Pinedo interrupted herself midsentence and turned to her son, who was in Mr. Martinez’s arms: “Yes, Papi, you are so stinking cute, and you are still my dream come true.”
This story was produced by KHN, which publishes California Healthline, an editorially independent service of the California Health Care Foundation. KHN (Kaiser Health News) is a national newsroom that produces in-depth journalism about health issues. Together with Policy Analysis and Polling, KHN is one of the three major operating programs at KFF (Kaiser Family Foundation). KFF is an endowed nonprofit organization providing information on health issues to the nation.
Uncombable hair syndrome: One gene, variants responsible for many cases
that manifests during infancy, investigators have reported.
The findings are from a cohort study published in JAMA Dermatology, which involved 107 unrelated children and adults suspected of having UHS, as well as family members, all of whom were recruited from January 2013 to December 2021. Genetic analyses were conducted in Germany from January 2014 to December 2021 with exome sequencing.
Study builds on prior research
Senior author Regina C. Betz, MD, professor of dermatogenetics at the Institute of Human Genetics, University Hospital Bonn, Germany, said that in 2016, she and her coinvestigators authored a study on the molecular genetics of UHS. That study, which involved 18 people with UHS, identified variants in three genes – PADI3, TCHH, and TGM3 – that encode proteins that play a role in the formation of the hair shaft. The investigators described how a deficiency in the shaping and mechanical strengthening of the hair shaft occurs in the UHS phenotype, which is characterized by dry, frizzy, and wiry hair that cannot be combed flat.
As a result of that previous work, “we base the assignment or confirmation of a clinical diagnosis of UHS on molecular genetic diagnostics,” the authors write in the new study, rather than on the clinical appearance of the hair and the physical examination of the patient, with confirmation on microscopical examination of the hair shaft.
Social media as instrument in finding study participants
Following the 2016 study, Dr. Betz and colleagues were contacted by many clinicians and by the public through Facebook and other social media platforms with details about possible cases of UHS, an autosomal recessive disorder. Through these contacts, blood samples, saliva, or DNA was sent to the investigators’ laboratory from 89 unrelated index patients (69 female patients, 20 male patients) suspected of having UHS. This resulted in the identification of pathogenic variants in 69 cases, the investigators write.
“In the first study, we had 18 patients, and then we tried to collect as many as possible” to determine the main mechanism behind UHS, Dr. Betz said. One question is whether there are additional genes responsible for UHS, she noted. “Even now, we are not sure, because in 25% [of cases in the new study], we didn’t find any mutation in the three known genes.”
The current study resulted in the discovery of eight novel pathogenic variants in PADI3, which are responsible for 71.0% (76) of the 107 cases. Of those, “6 were single observations and 2 were observed in 3 and 2 individuals, respectively,” the investigators write.
Children can grow out of this disorder, but it can also persist into adulthood, Dr. Betz noted. Communication that investigators had with parents of the children with UHS revealed that these children are often the targets of bullying by other children, she added.
She and her and colleagues will continue this research and are currently studying adults who have UHS.
Research leads to possible treatment pathways
Jeff Donovan, MD, FRCPC, FAAD, a dermatologist and medical director of the Donovan Hair Clinic in Whistler, British Columbia, described these findings as fundamental to understanding UHS and creating pathways to possible treatments.
The study “identifies more about the genetic basis of this challenging condition,” said Dr. Donovan, who is also clinical instructor in the department of dermatology at the University of British Columbia, Vancouver, and president of the Canadian Hair Loss Foundation. “We really need this type of information in order to have any sort of clue in terms of how to treat it,” he told this news organization.
“In the hair loss world, it’s pretty clear that if you can understand the genetic basis of things, or the basic science of a condition, whether it’s the basic genetics or the basic immunology, you give yourself the best chance to develop good treatments,” said Dr. Donovan.
The article provides advanced genetic information of the condition, such that geneticists can test for at least three markers if they are suspecting UHS, Dr. Donovan observed.
Condition can lead to bullying
Dr. Donovan also commented that UHS can have a detrimental impact on children with regard to socializing with their peers. “Having hair that sticks out and is very full like this is challenging because kids do get teased,” he said.
“It is often the parents who are the most affected” when a child aged 2-5 years has a hair condition such as UHS. But at age 5-9, “children are developing self-identity and an understanding of various aspects of self-esteem and what they look like and what others look like. And that’s where the teasing really starts. And that’s where it does become troublesome.”
Dr. Betz and Dr. Donovan have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
that manifests during infancy, investigators have reported.
The findings are from a cohort study published in JAMA Dermatology, which involved 107 unrelated children and adults suspected of having UHS, as well as family members, all of whom were recruited from January 2013 to December 2021. Genetic analyses were conducted in Germany from January 2014 to December 2021 with exome sequencing.
Study builds on prior research
Senior author Regina C. Betz, MD, professor of dermatogenetics at the Institute of Human Genetics, University Hospital Bonn, Germany, said that in 2016, she and her coinvestigators authored a study on the molecular genetics of UHS. That study, which involved 18 people with UHS, identified variants in three genes – PADI3, TCHH, and TGM3 – that encode proteins that play a role in the formation of the hair shaft. The investigators described how a deficiency in the shaping and mechanical strengthening of the hair shaft occurs in the UHS phenotype, which is characterized by dry, frizzy, and wiry hair that cannot be combed flat.
As a result of that previous work, “we base the assignment or confirmation of a clinical diagnosis of UHS on molecular genetic diagnostics,” the authors write in the new study, rather than on the clinical appearance of the hair and the physical examination of the patient, with confirmation on microscopical examination of the hair shaft.
Social media as instrument in finding study participants
Following the 2016 study, Dr. Betz and colleagues were contacted by many clinicians and by the public through Facebook and other social media platforms with details about possible cases of UHS, an autosomal recessive disorder. Through these contacts, blood samples, saliva, or DNA was sent to the investigators’ laboratory from 89 unrelated index patients (69 female patients, 20 male patients) suspected of having UHS. This resulted in the identification of pathogenic variants in 69 cases, the investigators write.
“In the first study, we had 18 patients, and then we tried to collect as many as possible” to determine the main mechanism behind UHS, Dr. Betz said. One question is whether there are additional genes responsible for UHS, she noted. “Even now, we are not sure, because in 25% [of cases in the new study], we didn’t find any mutation in the three known genes.”
The current study resulted in the discovery of eight novel pathogenic variants in PADI3, which are responsible for 71.0% (76) of the 107 cases. Of those, “6 were single observations and 2 were observed in 3 and 2 individuals, respectively,” the investigators write.
Children can grow out of this disorder, but it can also persist into adulthood, Dr. Betz noted. Communication that investigators had with parents of the children with UHS revealed that these children are often the targets of bullying by other children, she added.
She and her and colleagues will continue this research and are currently studying adults who have UHS.
Research leads to possible treatment pathways
Jeff Donovan, MD, FRCPC, FAAD, a dermatologist and medical director of the Donovan Hair Clinic in Whistler, British Columbia, described these findings as fundamental to understanding UHS and creating pathways to possible treatments.
The study “identifies more about the genetic basis of this challenging condition,” said Dr. Donovan, who is also clinical instructor in the department of dermatology at the University of British Columbia, Vancouver, and president of the Canadian Hair Loss Foundation. “We really need this type of information in order to have any sort of clue in terms of how to treat it,” he told this news organization.
“In the hair loss world, it’s pretty clear that if you can understand the genetic basis of things, or the basic science of a condition, whether it’s the basic genetics or the basic immunology, you give yourself the best chance to develop good treatments,” said Dr. Donovan.
The article provides advanced genetic information of the condition, such that geneticists can test for at least three markers if they are suspecting UHS, Dr. Donovan observed.
Condition can lead to bullying
Dr. Donovan also commented that UHS can have a detrimental impact on children with regard to socializing with their peers. “Having hair that sticks out and is very full like this is challenging because kids do get teased,” he said.
“It is often the parents who are the most affected” when a child aged 2-5 years has a hair condition such as UHS. But at age 5-9, “children are developing self-identity and an understanding of various aspects of self-esteem and what they look like and what others look like. And that’s where the teasing really starts. And that’s where it does become troublesome.”
Dr. Betz and Dr. Donovan have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
that manifests during infancy, investigators have reported.
The findings are from a cohort study published in JAMA Dermatology, which involved 107 unrelated children and adults suspected of having UHS, as well as family members, all of whom were recruited from January 2013 to December 2021. Genetic analyses were conducted in Germany from January 2014 to December 2021 with exome sequencing.
Study builds on prior research
Senior author Regina C. Betz, MD, professor of dermatogenetics at the Institute of Human Genetics, University Hospital Bonn, Germany, said that in 2016, she and her coinvestigators authored a study on the molecular genetics of UHS. That study, which involved 18 people with UHS, identified variants in three genes – PADI3, TCHH, and TGM3 – that encode proteins that play a role in the formation of the hair shaft. The investigators described how a deficiency in the shaping and mechanical strengthening of the hair shaft occurs in the UHS phenotype, which is characterized by dry, frizzy, and wiry hair that cannot be combed flat.
As a result of that previous work, “we base the assignment or confirmation of a clinical diagnosis of UHS on molecular genetic diagnostics,” the authors write in the new study, rather than on the clinical appearance of the hair and the physical examination of the patient, with confirmation on microscopical examination of the hair shaft.
Social media as instrument in finding study participants
Following the 2016 study, Dr. Betz and colleagues were contacted by many clinicians and by the public through Facebook and other social media platforms with details about possible cases of UHS, an autosomal recessive disorder. Through these contacts, blood samples, saliva, or DNA was sent to the investigators’ laboratory from 89 unrelated index patients (69 female patients, 20 male patients) suspected of having UHS. This resulted in the identification of pathogenic variants in 69 cases, the investigators write.
“In the first study, we had 18 patients, and then we tried to collect as many as possible” to determine the main mechanism behind UHS, Dr. Betz said. One question is whether there are additional genes responsible for UHS, she noted. “Even now, we are not sure, because in 25% [of cases in the new study], we didn’t find any mutation in the three known genes.”
The current study resulted in the discovery of eight novel pathogenic variants in PADI3, which are responsible for 71.0% (76) of the 107 cases. Of those, “6 were single observations and 2 were observed in 3 and 2 individuals, respectively,” the investigators write.
Children can grow out of this disorder, but it can also persist into adulthood, Dr. Betz noted. Communication that investigators had with parents of the children with UHS revealed that these children are often the targets of bullying by other children, she added.
She and her and colleagues will continue this research and are currently studying adults who have UHS.
Research leads to possible treatment pathways
Jeff Donovan, MD, FRCPC, FAAD, a dermatologist and medical director of the Donovan Hair Clinic in Whistler, British Columbia, described these findings as fundamental to understanding UHS and creating pathways to possible treatments.
The study “identifies more about the genetic basis of this challenging condition,” said Dr. Donovan, who is also clinical instructor in the department of dermatology at the University of British Columbia, Vancouver, and president of the Canadian Hair Loss Foundation. “We really need this type of information in order to have any sort of clue in terms of how to treat it,” he told this news organization.
“In the hair loss world, it’s pretty clear that if you can understand the genetic basis of things, or the basic science of a condition, whether it’s the basic genetics or the basic immunology, you give yourself the best chance to develop good treatments,” said Dr. Donovan.
The article provides advanced genetic information of the condition, such that geneticists can test for at least three markers if they are suspecting UHS, Dr. Donovan observed.
Condition can lead to bullying
Dr. Donovan also commented that UHS can have a detrimental impact on children with regard to socializing with their peers. “Having hair that sticks out and is very full like this is challenging because kids do get teased,” he said.
“It is often the parents who are the most affected” when a child aged 2-5 years has a hair condition such as UHS. But at age 5-9, “children are developing self-identity and an understanding of various aspects of self-esteem and what they look like and what others look like. And that’s where the teasing really starts. And that’s where it does become troublesome.”
Dr. Betz and Dr. Donovan have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM JAMA DERMATOLOGY
AAP guidance helps distinguish bleeding disorders from abuse
In some cases, bruising or bleeding from bleeding disorders may look like signs of child abuse, but new guidance may help clinicians distinguish one from the other.
On Sept. 19 the American Academy of Pediatrics published two reports – a clinical report and a technical report – in the October 2022 issue of Pediatrics on evaluating for bleeding disorders when child abuse is suspected.
The reports were written by the AAP Section on Hematology/Oncology and the AAP Council on Child Abuse and Neglect.
One doesn’t rule out the other
The reports emphasize that laboratory testing of bleeding cannot always rule out abuse, just as a history of trauma (accidental or nonaccidental) may not rule out a bleeding disorder or other medical condition.
In the clinical report, led by James Anderst, MD, MSCI, with the division of child adversity and resilience, Children’s Mercy Hospital, University of Missouri–Kansas City, the researchers note that infants are at especially high risk of abusive bruising/bleeding, but bleeding disorders may also present in infancy.
The authors give an example of a situation when taking a thorough history won’t necessarily rule out a bleeding disorder: Male infants who have been circumcised with no significant bleeding issues may still have a bleeding disorder. Therefore, laboratory evaluations are often needed to detect disordered bleeding.
Children’s medications should be documented, the authors note, because certain drugs, such as nonsteroidal anti-inflammatory drugs, some antibiotics, antiepileptics, and herbal supplements, can affect tests that might be used to detect bleeding disorders.
Likewise, asking about restrictive or unusual diets or alternative therapies is important as some could increase the likelihood of bleeding/bruising.
Signs that bleeding disorder is not likely
The authors advise that, if a child has any of the following, an evaluation for a bleeding disorder is generally not needed:
- Caregivers’ description of trauma sufficiently explains the bruising.
- The child or an independent witness can provide a history of abuse or nonabusive trauma that explains the bruising.
- The outline of the bruising follows an object or hand pattern.
- The location of the bruising is on the ears, neck, or genitals.
“Bruising to the ears, neck, or genitals is rarely seen in either accidental injuries or in children with bleeding disorders,” the authors write.
Specification of which locations for injuries are more indicative of abuse in both mobile and immobile children was among the most important information from the paper, Seattle pediatrician Timothy Joos, MD, said in an interview.
Also very helpful, he said, was the listing of which tests should be done if bruising looks like potential abuse.
The authors write that if bruising is concerning for abuse that necessitates evaluation for bleeding disorders, the following tests should be done: PT (prothrombin time); aPTT (activated partial thromboplastin time); von Willebrand Factor (VWF) activity (Ristocetin cofactor); factor VIII activity level; factor IX activity level; and a complete blood count, including platelets.
“I think that’s what a lot of us suspected, but there’s not a lot of summary evidence regarding that until now,” Dr. Joos said.
Case-by-case decisions on when to test
The decision on whether to evaluate for a bleeding disorder may be made case by case.
If there is no obvious known trauma or intracranial hemorrhage (ICH), particularly subdural hematoma (SDH) in a nonmobile child, abuse should be suspected, the authors write.
They acknowledge that children can have ICH, such as a small SDH or an epidural hematoma, under the point of impact from a short fall.
“However,” the authors write, “short falls rarely result in significant brain injury.”
Conditions may affect screening tests
Screening tests for bleeding disorders can be falsely positive or falsely negative, the authors caution in the technical report, led by Shannon Carpenter, MD, MS, with the department of pediatrics, University of Missouri–Kansas City.
- If coagulation laboratory test specimens sit in a hot metal box all day, for instance, factor levels may be falsely low, the authors explain.
- Conversely, factors such as VWF and factor VIII are acute-phase reactants and factor levels will be deceptively high if blood specimens are taken in a stressful time.
- Patients who have a traumatic brain injury often show temporary coagulopathy that does not signal a congenital disorder.
Vitamin K deficiency
The technical report explains that if an infant, typically younger than 6 months, presents with bleeding/bruising that raises flags for abuse and has a long PT, clinicians should confirm vitamin K was provided at birth and/or testing for vitamin K deficiency should be performed.
Not all states require vitamin K to be administered at birth and some parents refuse it. Deficiency can lead to bleeding in the skin or from mucosal surfaces from circumcision, generalized ecchymoses, and large intramuscular hemorrhages or ICH.
When infants don’t get vitamin K at birth, vitamin K deficiency bleeding (VKDB) is seen most often in the first days of life, the technical report states. It can also occur 1-3 months after birth.
“Late VKDB occurs from the first month to 3 months after birth,” the authors write. “This deficiency is more prevalent in breast-fed babies, because human milk contains less vitamin K than does cow milk.”
Overall, the authors write, extensive lab tests are usually not necessary, given the rarity of most bleeding disorders and specific clinical factors that decrease the odds that a bleeding disorder caused the child’s findings.
Dr. Joos said the decisions described in this paper are the kind that can keep pediatricians up at night.
“Any kind of guidance is helpful in these difficult cases,” he said. “These are scenarios that can often happen in the middle of the night, and you’re often struggling with evidence or past experience that can help you make some of these decisions.”
Authors of the reports and Dr. Joos declared no relevant financial relationships.
In some cases, bruising or bleeding from bleeding disorders may look like signs of child abuse, but new guidance may help clinicians distinguish one from the other.
On Sept. 19 the American Academy of Pediatrics published two reports – a clinical report and a technical report – in the October 2022 issue of Pediatrics on evaluating for bleeding disorders when child abuse is suspected.
The reports were written by the AAP Section on Hematology/Oncology and the AAP Council on Child Abuse and Neglect.
One doesn’t rule out the other
The reports emphasize that laboratory testing of bleeding cannot always rule out abuse, just as a history of trauma (accidental or nonaccidental) may not rule out a bleeding disorder or other medical condition.
In the clinical report, led by James Anderst, MD, MSCI, with the division of child adversity and resilience, Children’s Mercy Hospital, University of Missouri–Kansas City, the researchers note that infants are at especially high risk of abusive bruising/bleeding, but bleeding disorders may also present in infancy.
The authors give an example of a situation when taking a thorough history won’t necessarily rule out a bleeding disorder: Male infants who have been circumcised with no significant bleeding issues may still have a bleeding disorder. Therefore, laboratory evaluations are often needed to detect disordered bleeding.
Children’s medications should be documented, the authors note, because certain drugs, such as nonsteroidal anti-inflammatory drugs, some antibiotics, antiepileptics, and herbal supplements, can affect tests that might be used to detect bleeding disorders.
Likewise, asking about restrictive or unusual diets or alternative therapies is important as some could increase the likelihood of bleeding/bruising.
Signs that bleeding disorder is not likely
The authors advise that, if a child has any of the following, an evaluation for a bleeding disorder is generally not needed:
- Caregivers’ description of trauma sufficiently explains the bruising.
- The child or an independent witness can provide a history of abuse or nonabusive trauma that explains the bruising.
- The outline of the bruising follows an object or hand pattern.
- The location of the bruising is on the ears, neck, or genitals.
“Bruising to the ears, neck, or genitals is rarely seen in either accidental injuries or in children with bleeding disorders,” the authors write.
Specification of which locations for injuries are more indicative of abuse in both mobile and immobile children was among the most important information from the paper, Seattle pediatrician Timothy Joos, MD, said in an interview.
Also very helpful, he said, was the listing of which tests should be done if bruising looks like potential abuse.
The authors write that if bruising is concerning for abuse that necessitates evaluation for bleeding disorders, the following tests should be done: PT (prothrombin time); aPTT (activated partial thromboplastin time); von Willebrand Factor (VWF) activity (Ristocetin cofactor); factor VIII activity level; factor IX activity level; and a complete blood count, including platelets.
“I think that’s what a lot of us suspected, but there’s not a lot of summary evidence regarding that until now,” Dr. Joos said.
Case-by-case decisions on when to test
The decision on whether to evaluate for a bleeding disorder may be made case by case.
If there is no obvious known trauma or intracranial hemorrhage (ICH), particularly subdural hematoma (SDH) in a nonmobile child, abuse should be suspected, the authors write.
They acknowledge that children can have ICH, such as a small SDH or an epidural hematoma, under the point of impact from a short fall.
“However,” the authors write, “short falls rarely result in significant brain injury.”
Conditions may affect screening tests
Screening tests for bleeding disorders can be falsely positive or falsely negative, the authors caution in the technical report, led by Shannon Carpenter, MD, MS, with the department of pediatrics, University of Missouri–Kansas City.
- If coagulation laboratory test specimens sit in a hot metal box all day, for instance, factor levels may be falsely low, the authors explain.
- Conversely, factors such as VWF and factor VIII are acute-phase reactants and factor levels will be deceptively high if blood specimens are taken in a stressful time.
- Patients who have a traumatic brain injury often show temporary coagulopathy that does not signal a congenital disorder.
Vitamin K deficiency
The technical report explains that if an infant, typically younger than 6 months, presents with bleeding/bruising that raises flags for abuse and has a long PT, clinicians should confirm vitamin K was provided at birth and/or testing for vitamin K deficiency should be performed.
Not all states require vitamin K to be administered at birth and some parents refuse it. Deficiency can lead to bleeding in the skin or from mucosal surfaces from circumcision, generalized ecchymoses, and large intramuscular hemorrhages or ICH.
When infants don’t get vitamin K at birth, vitamin K deficiency bleeding (VKDB) is seen most often in the first days of life, the technical report states. It can also occur 1-3 months after birth.
“Late VKDB occurs from the first month to 3 months after birth,” the authors write. “This deficiency is more prevalent in breast-fed babies, because human milk contains less vitamin K than does cow milk.”
Overall, the authors write, extensive lab tests are usually not necessary, given the rarity of most bleeding disorders and specific clinical factors that decrease the odds that a bleeding disorder caused the child’s findings.
Dr. Joos said the decisions described in this paper are the kind that can keep pediatricians up at night.
“Any kind of guidance is helpful in these difficult cases,” he said. “These are scenarios that can often happen in the middle of the night, and you’re often struggling with evidence or past experience that can help you make some of these decisions.”
Authors of the reports and Dr. Joos declared no relevant financial relationships.
In some cases, bruising or bleeding from bleeding disorders may look like signs of child abuse, but new guidance may help clinicians distinguish one from the other.
On Sept. 19 the American Academy of Pediatrics published two reports – a clinical report and a technical report – in the October 2022 issue of Pediatrics on evaluating for bleeding disorders when child abuse is suspected.
The reports were written by the AAP Section on Hematology/Oncology and the AAP Council on Child Abuse and Neglect.
One doesn’t rule out the other
The reports emphasize that laboratory testing of bleeding cannot always rule out abuse, just as a history of trauma (accidental or nonaccidental) may not rule out a bleeding disorder or other medical condition.
In the clinical report, led by James Anderst, MD, MSCI, with the division of child adversity and resilience, Children’s Mercy Hospital, University of Missouri–Kansas City, the researchers note that infants are at especially high risk of abusive bruising/bleeding, but bleeding disorders may also present in infancy.
The authors give an example of a situation when taking a thorough history won’t necessarily rule out a bleeding disorder: Male infants who have been circumcised with no significant bleeding issues may still have a bleeding disorder. Therefore, laboratory evaluations are often needed to detect disordered bleeding.
Children’s medications should be documented, the authors note, because certain drugs, such as nonsteroidal anti-inflammatory drugs, some antibiotics, antiepileptics, and herbal supplements, can affect tests that might be used to detect bleeding disorders.
Likewise, asking about restrictive or unusual diets or alternative therapies is important as some could increase the likelihood of bleeding/bruising.
Signs that bleeding disorder is not likely
The authors advise that, if a child has any of the following, an evaluation for a bleeding disorder is generally not needed:
- Caregivers’ description of trauma sufficiently explains the bruising.
- The child or an independent witness can provide a history of abuse or nonabusive trauma that explains the bruising.
- The outline of the bruising follows an object or hand pattern.
- The location of the bruising is on the ears, neck, or genitals.
“Bruising to the ears, neck, or genitals is rarely seen in either accidental injuries or in children with bleeding disorders,” the authors write.
Specification of which locations for injuries are more indicative of abuse in both mobile and immobile children was among the most important information from the paper, Seattle pediatrician Timothy Joos, MD, said in an interview.
Also very helpful, he said, was the listing of which tests should be done if bruising looks like potential abuse.
The authors write that if bruising is concerning for abuse that necessitates evaluation for bleeding disorders, the following tests should be done: PT (prothrombin time); aPTT (activated partial thromboplastin time); von Willebrand Factor (VWF) activity (Ristocetin cofactor); factor VIII activity level; factor IX activity level; and a complete blood count, including platelets.
“I think that’s what a lot of us suspected, but there’s not a lot of summary evidence regarding that until now,” Dr. Joos said.
Case-by-case decisions on when to test
The decision on whether to evaluate for a bleeding disorder may be made case by case.
If there is no obvious known trauma or intracranial hemorrhage (ICH), particularly subdural hematoma (SDH) in a nonmobile child, abuse should be suspected, the authors write.
They acknowledge that children can have ICH, such as a small SDH or an epidural hematoma, under the point of impact from a short fall.
“However,” the authors write, “short falls rarely result in significant brain injury.”
Conditions may affect screening tests
Screening tests for bleeding disorders can be falsely positive or falsely negative, the authors caution in the technical report, led by Shannon Carpenter, MD, MS, with the department of pediatrics, University of Missouri–Kansas City.
- If coagulation laboratory test specimens sit in a hot metal box all day, for instance, factor levels may be falsely low, the authors explain.
- Conversely, factors such as VWF and factor VIII are acute-phase reactants and factor levels will be deceptively high if blood specimens are taken in a stressful time.
- Patients who have a traumatic brain injury often show temporary coagulopathy that does not signal a congenital disorder.
Vitamin K deficiency
The technical report explains that if an infant, typically younger than 6 months, presents with bleeding/bruising that raises flags for abuse and has a long PT, clinicians should confirm vitamin K was provided at birth and/or testing for vitamin K deficiency should be performed.
Not all states require vitamin K to be administered at birth and some parents refuse it. Deficiency can lead to bleeding in the skin or from mucosal surfaces from circumcision, generalized ecchymoses, and large intramuscular hemorrhages or ICH.
When infants don’t get vitamin K at birth, vitamin K deficiency bleeding (VKDB) is seen most often in the first days of life, the technical report states. It can also occur 1-3 months after birth.
“Late VKDB occurs from the first month to 3 months after birth,” the authors write. “This deficiency is more prevalent in breast-fed babies, because human milk contains less vitamin K than does cow milk.”
Overall, the authors write, extensive lab tests are usually not necessary, given the rarity of most bleeding disorders and specific clinical factors that decrease the odds that a bleeding disorder caused the child’s findings.
Dr. Joos said the decisions described in this paper are the kind that can keep pediatricians up at night.
“Any kind of guidance is helpful in these difficult cases,” he said. “These are scenarios that can often happen in the middle of the night, and you’re often struggling with evidence or past experience that can help you make some of these decisions.”
Authors of the reports and Dr. Joos declared no relevant financial relationships.
FROM PEDIATRICS
A farewell to arms? Drug approvals based on single-arm trials can be flawed
PARIS – with results that should only be used, under certain conditions, for accelerated approvals that should then be followed by confirmatory studies.
In fact, many drugs approved over the last decade based solely on data from single-arm trials have been subsequently withdrawn when put through the rigors of a head-to-head randomized controlled trial, according to Bishal Gyawali, MD, PhD, from the department of oncology at Queen’s University, Kingston, Ont.
“Single-arm trials are not meant to provide confirmatory evidence sufficient for approval; However, that ship has sailed, and we have several drugs that are approved on the basis of single-arm trials, but we need to make sure that those approvals are accelerated or conditional approvals, not regular approval,” he said in a presentation included in a special session on drug approvals at the European Society for Medical Oncology Congress.
“We should not allow premature regular approval based on single-arm trials, because once a drug gets conditional approval, access is not an issue. Patients will have access to the drug anyway, but we should ensure that robust evidence follows, and long-term follow-up data are needed to develop confidence in the efficacy outcomes that are seen in single-arm trials,” he said.
In many cases, single-arm trials are large enough or of long enough duration that investigators could have reasonably performed a randomized controlled trial (RCT) in the first place, Dr. Gyawali added.
Why do single-arm trials?
The term “single-arm registration trial” is something of an oxymoron, he said, noting that the purpose of such trials should be whether to take the drug to a phase 3, randomized trial. But as authors of a 2019 study in JAMA Network Open showed, of a sample of phase 3 RCTs, 42% did not have a prior phase 2 trial, and 28% had a negative phase 2 trial. Single-arm trials may be acceptable for conditional drug approvals if all of the following conditions are met:
- A RCT is not possible because the disease is rare or randomization would be unethical.
- The safety of the drug is established and its potential benefits outweigh its risks.
- The drug is associated with a high and durable overall or objective response rate.
- The mechanism of action is supported by a strong scientific rationale, and if the drug may meet an unmet medical need.
Survival endpoints won’t do
Efficacy endpoints typically used in RCTs, such as progression-free survival (PFS) and overall survival (OS) can be misleading because they may be a result of the natural history of the disease and not the drug being tested, whereas ORRs are almost certainly reflective of the action of the drug itself, because spontaneous tumor regression is a rare phenomenon, Dr. Gyawali said.
He cautioned, however, that the ORR of placebo is not zero percent. For example in a 2018 study of sorafenib (Nexavar) versus placebo for advanced or refractory desmoid tumors, the ORR with the active drug was 33%, and the ORR for placebo was 20%.
It’s also open to question, he said, what constitutes an acceptably high ORR and duration of response, pointing to Food and Drug Administration accelerated approval of an indication for nivolumab (Opdivo) for treatment of patients with hepatocellular carcinoma (HCC) that had progressed on sorafenib. In the single-arm trial used as the basis for approval, the ORRs as assessed by an independent central review committee blinded to the results was 14.3%.
“So, nivolumab in hepatocellular cancer was approved on the basis of a response rate lower than that of placebo, albeit in a different tumor. But the point I’m trying to show here is we don’t have a good definition of what is a good response rate,” he said.
In July 2021, Bristol-Myers Squibb voluntarily withdrew the HCC indication for nivolumab, following negative results of the CheckMate 459 trial and a 5-4 vote against continuing the accelerated approval.
On second thought ...
Citing data compiled by Nathan I. Cherny, MD, from Shaare Zedek Medical Center, Jerusalem, Dr. Gyawali noted that 58 of 161 FDA approvals from 2017 to 2021 of drugs for adult solid tumors were based on single-arm trials. Of the 58 drugs, 39 received accelerated approvals, and 19 received regular approvals; of the 39 that received accelerated approvals, 4 were subsequently withdrawn, 8 were converted to regular approvals, and the remainder continued as accelerated approvals.
Interestingly, the median response rate among all the drugs was 40%, and did not differ between the type of approval received, suggesting that response rates are not predictive of whether a drug will receive a conditional or full-fledged go-ahead.
What’s rare and safe?
The definition of a rare disease in the United States is one that affects fewer than 40,000 per year, and in Europe it’s an incidence rate of less than 6 per 100,000 population, Dr. Gyawali noted. But he argued that even non–small cell lung cancer, the most common form of cancer in the world, could be considered rare if it is broken down into subtypes that are treated according to specific mutations that may occur in a relatively small number of patients.
He also noted that a specific drug’s safety, one of the most important criteria for granting approval to a drug based on a single-arm trial, can be difficult to judge without adequate controls for comparison.
Cherry-picking patients
Winette van der Graaf, MD, president of the European Organization for the Research and Treatment of Cancer, who attended the session where Dr. Gyawali’s presentation was played, said in an interview that clinicians should cast a critical eye on how trials are designed and conducted, including patient selection and choice of endpoints.
“One of the most obvious things to be concerned about is that we’re still having patients with good performance status enrolled, mostly PS 0 or 1, so how representative are these clinical trials for the patients we see in front of us on a daily basis?” she said.
“The other question is radiological endpoints, which we focus on with OS and PFS are most important for patients, especially if you consider that if patients may have asymptomatic disease, and we are only treating them with potentially toxic medication, what are we doing for them? Median overall survival when you look at all of these trials is only 4 months, so we really need to take into account how we affect patients in clinical trials,” she added.
Dr. van der Graaf emphasized that clinical trial investigators need to more routinely incorporate quality of life measures and other patient-reported outcomes in clinical trial results to help regulators and clinicians in practice get a better sense of the true clinical benefit of a new drug.
Dr. Gyawali did not disclose a funding source for his presentation. He reported consulting fees from Vivio Health and research grants from the American Society of Clinical Oncology. Dr. van der Graaf reported no conflicts of interest.
PARIS – with results that should only be used, under certain conditions, for accelerated approvals that should then be followed by confirmatory studies.
In fact, many drugs approved over the last decade based solely on data from single-arm trials have been subsequently withdrawn when put through the rigors of a head-to-head randomized controlled trial, according to Bishal Gyawali, MD, PhD, from the department of oncology at Queen’s University, Kingston, Ont.
“Single-arm trials are not meant to provide confirmatory evidence sufficient for approval; However, that ship has sailed, and we have several drugs that are approved on the basis of single-arm trials, but we need to make sure that those approvals are accelerated or conditional approvals, not regular approval,” he said in a presentation included in a special session on drug approvals at the European Society for Medical Oncology Congress.
“We should not allow premature regular approval based on single-arm trials, because once a drug gets conditional approval, access is not an issue. Patients will have access to the drug anyway, but we should ensure that robust evidence follows, and long-term follow-up data are needed to develop confidence in the efficacy outcomes that are seen in single-arm trials,” he said.
In many cases, single-arm trials are large enough or of long enough duration that investigators could have reasonably performed a randomized controlled trial (RCT) in the first place, Dr. Gyawali added.
Why do single-arm trials?
The term “single-arm registration trial” is something of an oxymoron, he said, noting that the purpose of such trials should be whether to take the drug to a phase 3, randomized trial. But as authors of a 2019 study in JAMA Network Open showed, of a sample of phase 3 RCTs, 42% did not have a prior phase 2 trial, and 28% had a negative phase 2 trial. Single-arm trials may be acceptable for conditional drug approvals if all of the following conditions are met:
- A RCT is not possible because the disease is rare or randomization would be unethical.
- The safety of the drug is established and its potential benefits outweigh its risks.
- The drug is associated with a high and durable overall or objective response rate.
- The mechanism of action is supported by a strong scientific rationale, and if the drug may meet an unmet medical need.
Survival endpoints won’t do
Efficacy endpoints typically used in RCTs, such as progression-free survival (PFS) and overall survival (OS) can be misleading because they may be a result of the natural history of the disease and not the drug being tested, whereas ORRs are almost certainly reflective of the action of the drug itself, because spontaneous tumor regression is a rare phenomenon, Dr. Gyawali said.
He cautioned, however, that the ORR of placebo is not zero percent. For example in a 2018 study of sorafenib (Nexavar) versus placebo for advanced or refractory desmoid tumors, the ORR with the active drug was 33%, and the ORR for placebo was 20%.
It’s also open to question, he said, what constitutes an acceptably high ORR and duration of response, pointing to Food and Drug Administration accelerated approval of an indication for nivolumab (Opdivo) for treatment of patients with hepatocellular carcinoma (HCC) that had progressed on sorafenib. In the single-arm trial used as the basis for approval, the ORRs as assessed by an independent central review committee blinded to the results was 14.3%.
“So, nivolumab in hepatocellular cancer was approved on the basis of a response rate lower than that of placebo, albeit in a different tumor. But the point I’m trying to show here is we don’t have a good definition of what is a good response rate,” he said.
In July 2021, Bristol-Myers Squibb voluntarily withdrew the HCC indication for nivolumab, following negative results of the CheckMate 459 trial and a 5-4 vote against continuing the accelerated approval.
On second thought ...
Citing data compiled by Nathan I. Cherny, MD, from Shaare Zedek Medical Center, Jerusalem, Dr. Gyawali noted that 58 of 161 FDA approvals from 2017 to 2021 of drugs for adult solid tumors were based on single-arm trials. Of the 58 drugs, 39 received accelerated approvals, and 19 received regular approvals; of the 39 that received accelerated approvals, 4 were subsequently withdrawn, 8 were converted to regular approvals, and the remainder continued as accelerated approvals.
Interestingly, the median response rate among all the drugs was 40%, and did not differ between the type of approval received, suggesting that response rates are not predictive of whether a drug will receive a conditional or full-fledged go-ahead.
What’s rare and safe?
The definition of a rare disease in the United States is one that affects fewer than 40,000 per year, and in Europe it’s an incidence rate of less than 6 per 100,000 population, Dr. Gyawali noted. But he argued that even non–small cell lung cancer, the most common form of cancer in the world, could be considered rare if it is broken down into subtypes that are treated according to specific mutations that may occur in a relatively small number of patients.
He also noted that a specific drug’s safety, one of the most important criteria for granting approval to a drug based on a single-arm trial, can be difficult to judge without adequate controls for comparison.
Cherry-picking patients
Winette van der Graaf, MD, president of the European Organization for the Research and Treatment of Cancer, who attended the session where Dr. Gyawali’s presentation was played, said in an interview that clinicians should cast a critical eye on how trials are designed and conducted, including patient selection and choice of endpoints.
“One of the most obvious things to be concerned about is that we’re still having patients with good performance status enrolled, mostly PS 0 or 1, so how representative are these clinical trials for the patients we see in front of us on a daily basis?” she said.
“The other question is radiological endpoints, which we focus on with OS and PFS are most important for patients, especially if you consider that if patients may have asymptomatic disease, and we are only treating them with potentially toxic medication, what are we doing for them? Median overall survival when you look at all of these trials is only 4 months, so we really need to take into account how we affect patients in clinical trials,” she added.
Dr. van der Graaf emphasized that clinical trial investigators need to more routinely incorporate quality of life measures and other patient-reported outcomes in clinical trial results to help regulators and clinicians in practice get a better sense of the true clinical benefit of a new drug.
Dr. Gyawali did not disclose a funding source for his presentation. He reported consulting fees from Vivio Health and research grants from the American Society of Clinical Oncology. Dr. van der Graaf reported no conflicts of interest.
PARIS – with results that should only be used, under certain conditions, for accelerated approvals that should then be followed by confirmatory studies.
In fact, many drugs approved over the last decade based solely on data from single-arm trials have been subsequently withdrawn when put through the rigors of a head-to-head randomized controlled trial, according to Bishal Gyawali, MD, PhD, from the department of oncology at Queen’s University, Kingston, Ont.
“Single-arm trials are not meant to provide confirmatory evidence sufficient for approval; However, that ship has sailed, and we have several drugs that are approved on the basis of single-arm trials, but we need to make sure that those approvals are accelerated or conditional approvals, not regular approval,” he said in a presentation included in a special session on drug approvals at the European Society for Medical Oncology Congress.
“We should not allow premature regular approval based on single-arm trials, because once a drug gets conditional approval, access is not an issue. Patients will have access to the drug anyway, but we should ensure that robust evidence follows, and long-term follow-up data are needed to develop confidence in the efficacy outcomes that are seen in single-arm trials,” he said.
In many cases, single-arm trials are large enough or of long enough duration that investigators could have reasonably performed a randomized controlled trial (RCT) in the first place, Dr. Gyawali added.
Why do single-arm trials?
The term “single-arm registration trial” is something of an oxymoron, he said, noting that the purpose of such trials should be whether to take the drug to a phase 3, randomized trial. But as authors of a 2019 study in JAMA Network Open showed, of a sample of phase 3 RCTs, 42% did not have a prior phase 2 trial, and 28% had a negative phase 2 trial. Single-arm trials may be acceptable for conditional drug approvals if all of the following conditions are met:
- A RCT is not possible because the disease is rare or randomization would be unethical.
- The safety of the drug is established and its potential benefits outweigh its risks.
- The drug is associated with a high and durable overall or objective response rate.
- The mechanism of action is supported by a strong scientific rationale, and if the drug may meet an unmet medical need.
Survival endpoints won’t do
Efficacy endpoints typically used in RCTs, such as progression-free survival (PFS) and overall survival (OS) can be misleading because they may be a result of the natural history of the disease and not the drug being tested, whereas ORRs are almost certainly reflective of the action of the drug itself, because spontaneous tumor regression is a rare phenomenon, Dr. Gyawali said.
He cautioned, however, that the ORR of placebo is not zero percent. For example in a 2018 study of sorafenib (Nexavar) versus placebo for advanced or refractory desmoid tumors, the ORR with the active drug was 33%, and the ORR for placebo was 20%.
It’s also open to question, he said, what constitutes an acceptably high ORR and duration of response, pointing to Food and Drug Administration accelerated approval of an indication for nivolumab (Opdivo) for treatment of patients with hepatocellular carcinoma (HCC) that had progressed on sorafenib. In the single-arm trial used as the basis for approval, the ORRs as assessed by an independent central review committee blinded to the results was 14.3%.
“So, nivolumab in hepatocellular cancer was approved on the basis of a response rate lower than that of placebo, albeit in a different tumor. But the point I’m trying to show here is we don’t have a good definition of what is a good response rate,” he said.
In July 2021, Bristol-Myers Squibb voluntarily withdrew the HCC indication for nivolumab, following negative results of the CheckMate 459 trial and a 5-4 vote against continuing the accelerated approval.
On second thought ...
Citing data compiled by Nathan I. Cherny, MD, from Shaare Zedek Medical Center, Jerusalem, Dr. Gyawali noted that 58 of 161 FDA approvals from 2017 to 2021 of drugs for adult solid tumors were based on single-arm trials. Of the 58 drugs, 39 received accelerated approvals, and 19 received regular approvals; of the 39 that received accelerated approvals, 4 were subsequently withdrawn, 8 were converted to regular approvals, and the remainder continued as accelerated approvals.
Interestingly, the median response rate among all the drugs was 40%, and did not differ between the type of approval received, suggesting that response rates are not predictive of whether a drug will receive a conditional or full-fledged go-ahead.
What’s rare and safe?
The definition of a rare disease in the United States is one that affects fewer than 40,000 per year, and in Europe it’s an incidence rate of less than 6 per 100,000 population, Dr. Gyawali noted. But he argued that even non–small cell lung cancer, the most common form of cancer in the world, could be considered rare if it is broken down into subtypes that are treated according to specific mutations that may occur in a relatively small number of patients.
He also noted that a specific drug’s safety, one of the most important criteria for granting approval to a drug based on a single-arm trial, can be difficult to judge without adequate controls for comparison.
Cherry-picking patients
Winette van der Graaf, MD, president of the European Organization for the Research and Treatment of Cancer, who attended the session where Dr. Gyawali’s presentation was played, said in an interview that clinicians should cast a critical eye on how trials are designed and conducted, including patient selection and choice of endpoints.
“One of the most obvious things to be concerned about is that we’re still having patients with good performance status enrolled, mostly PS 0 or 1, so how representative are these clinical trials for the patients we see in front of us on a daily basis?” she said.
“The other question is radiological endpoints, which we focus on with OS and PFS are most important for patients, especially if you consider that if patients may have asymptomatic disease, and we are only treating them with potentially toxic medication, what are we doing for them? Median overall survival when you look at all of these trials is only 4 months, so we really need to take into account how we affect patients in clinical trials,” she added.
Dr. van der Graaf emphasized that clinical trial investigators need to more routinely incorporate quality of life measures and other patient-reported outcomes in clinical trial results to help regulators and clinicians in practice get a better sense of the true clinical benefit of a new drug.
Dr. Gyawali did not disclose a funding source for his presentation. He reported consulting fees from Vivio Health and research grants from the American Society of Clinical Oncology. Dr. van der Graaf reported no conflicts of interest.
AT ESMO CONGRESS 2022
Ketamine promising for rare condition linked to autism
Also known as Helsmoortel–Van Der Aa syndrome, ADNP syndrome is caused by mutations in the ADNP gene. Studies in animal models suggest that low-dose ketamine increases expression of ADNP and is neuroprotective.
Intrigued by the preclinical evidence, Alexander Kolevzon, MD, clinical director of the Seaver Autism Center at Mount Sinai, New York, and colleagues treated 10 children with ADNP syndrome with a single low dose of ketamine (0.5mg/kg) infused intravenously over 40 minutes. The children ranged in ages 6-12 years.
Using parent-report instruments to assess treatment effects, ketamine was associated with “nominally significant” improvement in a variety of domains, including social behavior, attention-deficit and hyperactivity, restricted and repetitive behaviors, and sensory sensitivities.
Parent reports of improvement in these domains aligned with clinician-rated assessments based on the Clinical Global Impressions–Improvement scale.
The results also highlight the potential utility of electrophysiological measurement of auditory steady-state response and eye-tracking to track change with ketamine treatment, the researchers say.
The study was published online in Human Genetics and Genomic (HGG) Advances.
Hypothesis-generating
Ketamine was generally well tolerated. There were no clinically significant abnormalities in laboratory or cardiac monitoring, and there were no serious adverse events (AEs).
Treatment emergent AEs were all mild to moderate and no child required any interventions.
The most common AEs were elation/silliness in five children (50%), all of whom had a history of similar symptoms. Drowsiness and fatigue occurred in four children (40%) and two of them had a history of drowsiness. Aggression was likewise relatively common, reported in four children (40%), all of whom had aggression at baseline.
Decreased appetite emerged as a new AE in three children (30%), increased anxiety occurred in three children (30%), and irritability, nausea/vomiting, and restlessness each occurred in two children (20%).
The researchers caution that the findings are intended to be “hypothesis generating.”
“We are encouraged by these findings, which provide preliminary support for ketamine to help reduce negative effects of this devastating syndrome,” Dr. Kolevzon said in a news release from Mount Sinai.
Ketamine might help ease symptoms of ADNP syndrome “by increasing expression of the ADNP gene or by promoting synaptic plasticity through glutamatergic pathways,” Dr. Kolevzon told this news organization.
The next step, he said, is to get “a larger, placebo-controlled study approved for funding using repeated dosing over a longer duration of time. We are working with the FDA to get the design approved for an investigational new drug application.”
Support for the study was provided by the ADNP Kids Foundation and the Foundation for Mood Disorders. Support for mediKanren was provided by the National Center for Advancing Translational Sciences, and National Institutes of Health through the Biomedical Data Translator Program. Dr. Kolevzon is on the scientific advisory board of Ovid Therapeutics, Ritrova Therapeutics, and Jaguar Therapeutics and consults to Acadia, Alkermes, GW Pharmaceuticals, Neuren Pharmaceuticals, Clinilabs Drug Development Corporation, and Scioto Biosciences.
A version of this article first appeared on Medscape.com.
Also known as Helsmoortel–Van Der Aa syndrome, ADNP syndrome is caused by mutations in the ADNP gene. Studies in animal models suggest that low-dose ketamine increases expression of ADNP and is neuroprotective.
Intrigued by the preclinical evidence, Alexander Kolevzon, MD, clinical director of the Seaver Autism Center at Mount Sinai, New York, and colleagues treated 10 children with ADNP syndrome with a single low dose of ketamine (0.5mg/kg) infused intravenously over 40 minutes. The children ranged in ages 6-12 years.
Using parent-report instruments to assess treatment effects, ketamine was associated with “nominally significant” improvement in a variety of domains, including social behavior, attention-deficit and hyperactivity, restricted and repetitive behaviors, and sensory sensitivities.
Parent reports of improvement in these domains aligned with clinician-rated assessments based on the Clinical Global Impressions–Improvement scale.
The results also highlight the potential utility of electrophysiological measurement of auditory steady-state response and eye-tracking to track change with ketamine treatment, the researchers say.
The study was published online in Human Genetics and Genomic (HGG) Advances.
Hypothesis-generating
Ketamine was generally well tolerated. There were no clinically significant abnormalities in laboratory or cardiac monitoring, and there were no serious adverse events (AEs).
Treatment emergent AEs were all mild to moderate and no child required any interventions.
The most common AEs were elation/silliness in five children (50%), all of whom had a history of similar symptoms. Drowsiness and fatigue occurred in four children (40%) and two of them had a history of drowsiness. Aggression was likewise relatively common, reported in four children (40%), all of whom had aggression at baseline.
Decreased appetite emerged as a new AE in three children (30%), increased anxiety occurred in three children (30%), and irritability, nausea/vomiting, and restlessness each occurred in two children (20%).
The researchers caution that the findings are intended to be “hypothesis generating.”
“We are encouraged by these findings, which provide preliminary support for ketamine to help reduce negative effects of this devastating syndrome,” Dr. Kolevzon said in a news release from Mount Sinai.
Ketamine might help ease symptoms of ADNP syndrome “by increasing expression of the ADNP gene or by promoting synaptic plasticity through glutamatergic pathways,” Dr. Kolevzon told this news organization.
The next step, he said, is to get “a larger, placebo-controlled study approved for funding using repeated dosing over a longer duration of time. We are working with the FDA to get the design approved for an investigational new drug application.”
Support for the study was provided by the ADNP Kids Foundation and the Foundation for Mood Disorders. Support for mediKanren was provided by the National Center for Advancing Translational Sciences, and National Institutes of Health through the Biomedical Data Translator Program. Dr. Kolevzon is on the scientific advisory board of Ovid Therapeutics, Ritrova Therapeutics, and Jaguar Therapeutics and consults to Acadia, Alkermes, GW Pharmaceuticals, Neuren Pharmaceuticals, Clinilabs Drug Development Corporation, and Scioto Biosciences.
A version of this article first appeared on Medscape.com.
Also known as Helsmoortel–Van Der Aa syndrome, ADNP syndrome is caused by mutations in the ADNP gene. Studies in animal models suggest that low-dose ketamine increases expression of ADNP and is neuroprotective.
Intrigued by the preclinical evidence, Alexander Kolevzon, MD, clinical director of the Seaver Autism Center at Mount Sinai, New York, and colleagues treated 10 children with ADNP syndrome with a single low dose of ketamine (0.5mg/kg) infused intravenously over 40 minutes. The children ranged in ages 6-12 years.
Using parent-report instruments to assess treatment effects, ketamine was associated with “nominally significant” improvement in a variety of domains, including social behavior, attention-deficit and hyperactivity, restricted and repetitive behaviors, and sensory sensitivities.
Parent reports of improvement in these domains aligned with clinician-rated assessments based on the Clinical Global Impressions–Improvement scale.
The results also highlight the potential utility of electrophysiological measurement of auditory steady-state response and eye-tracking to track change with ketamine treatment, the researchers say.
The study was published online in Human Genetics and Genomic (HGG) Advances.
Hypothesis-generating
Ketamine was generally well tolerated. There were no clinically significant abnormalities in laboratory or cardiac monitoring, and there were no serious adverse events (AEs).
Treatment emergent AEs were all mild to moderate and no child required any interventions.
The most common AEs were elation/silliness in five children (50%), all of whom had a history of similar symptoms. Drowsiness and fatigue occurred in four children (40%) and two of them had a history of drowsiness. Aggression was likewise relatively common, reported in four children (40%), all of whom had aggression at baseline.
Decreased appetite emerged as a new AE in three children (30%), increased anxiety occurred in three children (30%), and irritability, nausea/vomiting, and restlessness each occurred in two children (20%).
The researchers caution that the findings are intended to be “hypothesis generating.”
“We are encouraged by these findings, which provide preliminary support for ketamine to help reduce negative effects of this devastating syndrome,” Dr. Kolevzon said in a news release from Mount Sinai.
Ketamine might help ease symptoms of ADNP syndrome “by increasing expression of the ADNP gene or by promoting synaptic plasticity through glutamatergic pathways,” Dr. Kolevzon told this news organization.
The next step, he said, is to get “a larger, placebo-controlled study approved for funding using repeated dosing over a longer duration of time. We are working with the FDA to get the design approved for an investigational new drug application.”
Support for the study was provided by the ADNP Kids Foundation and the Foundation for Mood Disorders. Support for mediKanren was provided by the National Center for Advancing Translational Sciences, and National Institutes of Health through the Biomedical Data Translator Program. Dr. Kolevzon is on the scientific advisory board of Ovid Therapeutics, Ritrova Therapeutics, and Jaguar Therapeutics and consults to Acadia, Alkermes, GW Pharmaceuticals, Neuren Pharmaceuticals, Clinilabs Drug Development Corporation, and Scioto Biosciences.
A version of this article first appeared on Medscape.com.
From Human Genetics and Genomic Advances