User login
FDA-approved peanut immunotherapy protocol comes with a cost
Peanut allergy immunotherapy now comes with approval from the US Food and Drug Administration (FDA), but it also comes with protocols, standards, and paperwork. Whether it will be widely adopted has yet to be determined.
A few dozen allergists around the world have been offering food allergy immunotherapy for many years, having developed their own measuring techniques using store-bought food.
But the vast majority of allergists are not interested in developing home-grown treatments, not only because it involves research and development, but also because it comes with legal risks.
“Finally we have another treatment option,” said Edwin Kim, MD, from the UNC Allergy and Immunology Clinic in Chapel Hill, N.C. “This is what we were waiting for. It’s not cowboy stuff; this works.”
In January, the FDA approved peanut allergen powder (Palforzia) for patients 4-17 years of age, as reported by Medscape Medical News.
The pill contains measured doses of peanut flour and comes with a protocol that will allow allergists to bring patients to a peanut tolerance of 300 mg (about one peanut) and a black-box warning about anaphylaxis risk.
And before allergists can prescribe it, they must take a Risk Evaluation and Mitigation Strategy course to learn about dosing and the allergic reaction protocol.
“That may scare some away,” said Dr. Kim, who discussed the FDA-approved option during his presentation at the American College of Allergy, Asthma & Immunology 2020 Annual Scientific Meeting.
Allergic reaction, including the potential for anaphylaxis, has always been an issue with immunotherapy.
“People make the argument that there is a difference” between an expected allergic reaction – such as one that occurs after the administration of immunotherapy – and an unexpected reaction, he said. Because an expected reaction can be treated quickly, “some feel these expected reactions don’t matter so much.”
“Others say a reaction is a reaction” and argue that if, a treatment causes reaction, then it doesn’t make sense, he explained.
It comes down to patients – they must be willing to take a risk to develop tolerance and improve their quality of life – and the allergists willing to treat them.
The peanut powder involves paperwork, preauthorization forms, denials of care, a higher price tag, regimented procedures, and a prerequisite number of visits with patients. “Not everyone will want to do this,” said Dr. Kim.
The regimen involves three phases. During initial dose escalation, five doses are administered in the office on day 1. Then, over the next 6 months, updoses are administered during 11 in-office sessions and a 300-mg tolerance is achieved. Finally, to maintain tolerance to one peanut, daily doses are administered at home.
The drug cost alone is about $4,200 a year, according to Institute for Clinical and Economic Review. Peanut flour from the grocery store is cheaper, but comes with the risk of bacteria or other contamination.
“This product offers some reassurance, and that matters,” Dr. Kim said.
It’s good to have more options for food allergy treatment. “We need a more proactive way to treat food allergy; avoidance is not good enough,” he explained. “And presumably, at some point, the patient will be able to eat a grocery-store peanut instead of buying the pills.”
The art of medicine
But not all allergists will be able to make the protocol work. And it’s not clear whether there is room to alter treatment and offer patients with a higher tolerance a higher starting dose. What we do know, though, is that “the product leaves little room for ‘the art of medicine,’ ” Kim said.
That art is practiced by Arnon Elizur, MD, from the Shamir Medical Center in Tzrifin, Israel, but it’s backed by a rigid home-grown protocol.
Since 2010, he has treated 1,800 patients for peanut allergy, updosing slowly to a tolerance of 3,000 mg of peanut, the equivalent of 10 peanuts. He keeps the maintenance dose at four peanuts (1,200 mg). His center takes a personalized approach, starting patients on the highest dose they can tolerate and working up, with daily patient check-ins from home and a staff available around the clock to answer questions and deal with reactions.
“We aim for full sensitization,” Dr. Elizur said in an interview.
The peanut pill is “a big step forward” for immunotherapy, he said. It is “a standardized product, checked for bacteria and allergen content, which is available to a wide community of physicians.”
But, he pointed out, “it’s expensive.” And it’s only for peanut. “There are millions of food-allergic patients around the world dying from adverse reactions to many different kinds of food. We don’t want to wait for years for a product for all of them. We can use the actual food.”
He questions the lifelong maintenance protocol with a daily 300-mg pill. “If you can’t eat a peanut, why would you buy a drug that’s a peanut?” he asked.
He also said he’s disappointed that the product is not indicated for adults.
At the Shamir clinic, reactions are closely monitored. “Some are mild, others we treat with autoinjectors, epinephrine,” he reported. “Those are the most undesirable.”
Data from his center show that reactions occur in about 15% of patients. But his treatment success rates are good. In an average of 8 months, he is able to get 80% of his adult patients to full sensitization.
But it’s not for all patients or for all clinics, he acknowledged. “We continue to look at this balance in quality of life throughout the process. Our goal is to improve the quality of life threshold.”
Treatment that involves “native food” is “a lot of work” and requires “a lot of investment,” Dr. Elizur said. His center uses a web reporting system to maintain a 24/7 dialogue with patients, “and we look at the reports every day.” They also have a physician on call at all times. “Not everyone can commit to providing care throughout the day and night.”
His center charges the equivalent of $US3,000 per food allergy treated. “That’s whether it takes 6 months or 2 years,” he said.
There are more than 1,000 people on his 3-year waiting list.
“This is the first year that the American College of Allergy, Asthma, and Immunology is not hosting a pro–con debate on oral immunotherapy,” Dr. Kim pointed out. “We have a therapy now.”
However, the pandemic has slowed treatment uptake. “Immunotherapy is not easy to do, whether it’s FDA approved or not,” he explained. With at least 11 doctor visits in the first 6 months – each visit is between 30 minutes and 2-3 hours long – it hasn’t been possible to set up this year. “It’s not ideal.”
It will be interesting to see “how this will roll out and how it will be adopted,” Dr. Kim said. “From a food allergy point of view, the next 12 months are going to be very interesting.”
Dr. Kim reports receiving consulting honorarium from Aimmune, the maker of Palforzia; being on the clinical medical advisory board for DBV Technologies; and consulting for Aimmune, Allakos, Allergenis, DBV, Duke Clinical Research Institute, Ukko Incorporated, Vibrant America, and Kenota Health. Dr. Elizur has disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
Peanut allergy immunotherapy now comes with approval from the US Food and Drug Administration (FDA), but it also comes with protocols, standards, and paperwork. Whether it will be widely adopted has yet to be determined.
A few dozen allergists around the world have been offering food allergy immunotherapy for many years, having developed their own measuring techniques using store-bought food.
But the vast majority of allergists are not interested in developing home-grown treatments, not only because it involves research and development, but also because it comes with legal risks.
“Finally we have another treatment option,” said Edwin Kim, MD, from the UNC Allergy and Immunology Clinic in Chapel Hill, N.C. “This is what we were waiting for. It’s not cowboy stuff; this works.”
In January, the FDA approved peanut allergen powder (Palforzia) for patients 4-17 years of age, as reported by Medscape Medical News.
The pill contains measured doses of peanut flour and comes with a protocol that will allow allergists to bring patients to a peanut tolerance of 300 mg (about one peanut) and a black-box warning about anaphylaxis risk.
And before allergists can prescribe it, they must take a Risk Evaluation and Mitigation Strategy course to learn about dosing and the allergic reaction protocol.
“That may scare some away,” said Dr. Kim, who discussed the FDA-approved option during his presentation at the American College of Allergy, Asthma & Immunology 2020 Annual Scientific Meeting.
Allergic reaction, including the potential for anaphylaxis, has always been an issue with immunotherapy.
“People make the argument that there is a difference” between an expected allergic reaction – such as one that occurs after the administration of immunotherapy – and an unexpected reaction, he said. Because an expected reaction can be treated quickly, “some feel these expected reactions don’t matter so much.”
“Others say a reaction is a reaction” and argue that if, a treatment causes reaction, then it doesn’t make sense, he explained.
It comes down to patients – they must be willing to take a risk to develop tolerance and improve their quality of life – and the allergists willing to treat them.
The peanut powder involves paperwork, preauthorization forms, denials of care, a higher price tag, regimented procedures, and a prerequisite number of visits with patients. “Not everyone will want to do this,” said Dr. Kim.
The regimen involves three phases. During initial dose escalation, five doses are administered in the office on day 1. Then, over the next 6 months, updoses are administered during 11 in-office sessions and a 300-mg tolerance is achieved. Finally, to maintain tolerance to one peanut, daily doses are administered at home.
The drug cost alone is about $4,200 a year, according to Institute for Clinical and Economic Review. Peanut flour from the grocery store is cheaper, but comes with the risk of bacteria or other contamination.
“This product offers some reassurance, and that matters,” Dr. Kim said.
It’s good to have more options for food allergy treatment. “We need a more proactive way to treat food allergy; avoidance is not good enough,” he explained. “And presumably, at some point, the patient will be able to eat a grocery-store peanut instead of buying the pills.”
The art of medicine
But not all allergists will be able to make the protocol work. And it’s not clear whether there is room to alter treatment and offer patients with a higher tolerance a higher starting dose. What we do know, though, is that “the product leaves little room for ‘the art of medicine,’ ” Kim said.
That art is practiced by Arnon Elizur, MD, from the Shamir Medical Center in Tzrifin, Israel, but it’s backed by a rigid home-grown protocol.
Since 2010, he has treated 1,800 patients for peanut allergy, updosing slowly to a tolerance of 3,000 mg of peanut, the equivalent of 10 peanuts. He keeps the maintenance dose at four peanuts (1,200 mg). His center takes a personalized approach, starting patients on the highest dose they can tolerate and working up, with daily patient check-ins from home and a staff available around the clock to answer questions and deal with reactions.
“We aim for full sensitization,” Dr. Elizur said in an interview.
The peanut pill is “a big step forward” for immunotherapy, he said. It is “a standardized product, checked for bacteria and allergen content, which is available to a wide community of physicians.”
But, he pointed out, “it’s expensive.” And it’s only for peanut. “There are millions of food-allergic patients around the world dying from adverse reactions to many different kinds of food. We don’t want to wait for years for a product for all of them. We can use the actual food.”
He questions the lifelong maintenance protocol with a daily 300-mg pill. “If you can’t eat a peanut, why would you buy a drug that’s a peanut?” he asked.
He also said he’s disappointed that the product is not indicated for adults.
At the Shamir clinic, reactions are closely monitored. “Some are mild, others we treat with autoinjectors, epinephrine,” he reported. “Those are the most undesirable.”
Data from his center show that reactions occur in about 15% of patients. But his treatment success rates are good. In an average of 8 months, he is able to get 80% of his adult patients to full sensitization.
But it’s not for all patients or for all clinics, he acknowledged. “We continue to look at this balance in quality of life throughout the process. Our goal is to improve the quality of life threshold.”
Treatment that involves “native food” is “a lot of work” and requires “a lot of investment,” Dr. Elizur said. His center uses a web reporting system to maintain a 24/7 dialogue with patients, “and we look at the reports every day.” They also have a physician on call at all times. “Not everyone can commit to providing care throughout the day and night.”
His center charges the equivalent of $US3,000 per food allergy treated. “That’s whether it takes 6 months or 2 years,” he said.
There are more than 1,000 people on his 3-year waiting list.
“This is the first year that the American College of Allergy, Asthma, and Immunology is not hosting a pro–con debate on oral immunotherapy,” Dr. Kim pointed out. “We have a therapy now.”
However, the pandemic has slowed treatment uptake. “Immunotherapy is not easy to do, whether it’s FDA approved or not,” he explained. With at least 11 doctor visits in the first 6 months – each visit is between 30 minutes and 2-3 hours long – it hasn’t been possible to set up this year. “It’s not ideal.”
It will be interesting to see “how this will roll out and how it will be adopted,” Dr. Kim said. “From a food allergy point of view, the next 12 months are going to be very interesting.”
Dr. Kim reports receiving consulting honorarium from Aimmune, the maker of Palforzia; being on the clinical medical advisory board for DBV Technologies; and consulting for Aimmune, Allakos, Allergenis, DBV, Duke Clinical Research Institute, Ukko Incorporated, Vibrant America, and Kenota Health. Dr. Elizur has disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
Peanut allergy immunotherapy now comes with approval from the US Food and Drug Administration (FDA), but it also comes with protocols, standards, and paperwork. Whether it will be widely adopted has yet to be determined.
A few dozen allergists around the world have been offering food allergy immunotherapy for many years, having developed their own measuring techniques using store-bought food.
But the vast majority of allergists are not interested in developing home-grown treatments, not only because it involves research and development, but also because it comes with legal risks.
“Finally we have another treatment option,” said Edwin Kim, MD, from the UNC Allergy and Immunology Clinic in Chapel Hill, N.C. “This is what we were waiting for. It’s not cowboy stuff; this works.”
In January, the FDA approved peanut allergen powder (Palforzia) for patients 4-17 years of age, as reported by Medscape Medical News.
The pill contains measured doses of peanut flour and comes with a protocol that will allow allergists to bring patients to a peanut tolerance of 300 mg (about one peanut) and a black-box warning about anaphylaxis risk.
And before allergists can prescribe it, they must take a Risk Evaluation and Mitigation Strategy course to learn about dosing and the allergic reaction protocol.
“That may scare some away,” said Dr. Kim, who discussed the FDA-approved option during his presentation at the American College of Allergy, Asthma & Immunology 2020 Annual Scientific Meeting.
Allergic reaction, including the potential for anaphylaxis, has always been an issue with immunotherapy.
“People make the argument that there is a difference” between an expected allergic reaction – such as one that occurs after the administration of immunotherapy – and an unexpected reaction, he said. Because an expected reaction can be treated quickly, “some feel these expected reactions don’t matter so much.”
“Others say a reaction is a reaction” and argue that if, a treatment causes reaction, then it doesn’t make sense, he explained.
It comes down to patients – they must be willing to take a risk to develop tolerance and improve their quality of life – and the allergists willing to treat them.
The peanut powder involves paperwork, preauthorization forms, denials of care, a higher price tag, regimented procedures, and a prerequisite number of visits with patients. “Not everyone will want to do this,” said Dr. Kim.
The regimen involves three phases. During initial dose escalation, five doses are administered in the office on day 1. Then, over the next 6 months, updoses are administered during 11 in-office sessions and a 300-mg tolerance is achieved. Finally, to maintain tolerance to one peanut, daily doses are administered at home.
The drug cost alone is about $4,200 a year, according to Institute for Clinical and Economic Review. Peanut flour from the grocery store is cheaper, but comes with the risk of bacteria or other contamination.
“This product offers some reassurance, and that matters,” Dr. Kim said.
It’s good to have more options for food allergy treatment. “We need a more proactive way to treat food allergy; avoidance is not good enough,” he explained. “And presumably, at some point, the patient will be able to eat a grocery-store peanut instead of buying the pills.”
The art of medicine
But not all allergists will be able to make the protocol work. And it’s not clear whether there is room to alter treatment and offer patients with a higher tolerance a higher starting dose. What we do know, though, is that “the product leaves little room for ‘the art of medicine,’ ” Kim said.
That art is practiced by Arnon Elizur, MD, from the Shamir Medical Center in Tzrifin, Israel, but it’s backed by a rigid home-grown protocol.
Since 2010, he has treated 1,800 patients for peanut allergy, updosing slowly to a tolerance of 3,000 mg of peanut, the equivalent of 10 peanuts. He keeps the maintenance dose at four peanuts (1,200 mg). His center takes a personalized approach, starting patients on the highest dose they can tolerate and working up, with daily patient check-ins from home and a staff available around the clock to answer questions and deal with reactions.
“We aim for full sensitization,” Dr. Elizur said in an interview.
The peanut pill is “a big step forward” for immunotherapy, he said. It is “a standardized product, checked for bacteria and allergen content, which is available to a wide community of physicians.”
But, he pointed out, “it’s expensive.” And it’s only for peanut. “There are millions of food-allergic patients around the world dying from adverse reactions to many different kinds of food. We don’t want to wait for years for a product for all of them. We can use the actual food.”
He questions the lifelong maintenance protocol with a daily 300-mg pill. “If you can’t eat a peanut, why would you buy a drug that’s a peanut?” he asked.
He also said he’s disappointed that the product is not indicated for adults.
At the Shamir clinic, reactions are closely monitored. “Some are mild, others we treat with autoinjectors, epinephrine,” he reported. “Those are the most undesirable.”
Data from his center show that reactions occur in about 15% of patients. But his treatment success rates are good. In an average of 8 months, he is able to get 80% of his adult patients to full sensitization.
But it’s not for all patients or for all clinics, he acknowledged. “We continue to look at this balance in quality of life throughout the process. Our goal is to improve the quality of life threshold.”
Treatment that involves “native food” is “a lot of work” and requires “a lot of investment,” Dr. Elizur said. His center uses a web reporting system to maintain a 24/7 dialogue with patients, “and we look at the reports every day.” They also have a physician on call at all times. “Not everyone can commit to providing care throughout the day and night.”
His center charges the equivalent of $US3,000 per food allergy treated. “That’s whether it takes 6 months or 2 years,” he said.
There are more than 1,000 people on his 3-year waiting list.
“This is the first year that the American College of Allergy, Asthma, and Immunology is not hosting a pro–con debate on oral immunotherapy,” Dr. Kim pointed out. “We have a therapy now.”
However, the pandemic has slowed treatment uptake. “Immunotherapy is not easy to do, whether it’s FDA approved or not,” he explained. With at least 11 doctor visits in the first 6 months – each visit is between 30 minutes and 2-3 hours long – it hasn’t been possible to set up this year. “It’s not ideal.”
It will be interesting to see “how this will roll out and how it will be adopted,” Dr. Kim said. “From a food allergy point of view, the next 12 months are going to be very interesting.”
Dr. Kim reports receiving consulting honorarium from Aimmune, the maker of Palforzia; being on the clinical medical advisory board for DBV Technologies; and consulting for Aimmune, Allakos, Allergenis, DBV, Duke Clinical Research Institute, Ukko Incorporated, Vibrant America, and Kenota Health. Dr. Elizur has disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
Poverty raises depression risk in patients with cystic fibrosis
Poor people with chronic illness have greater difficulty managing their disease than do their better-off counterparts, and a new study confirms this reality for patients with cystic fibrosis.

and anxiety symptoms, according to a new cross-sectional study. The data were drawn from the Cystic Fibrosis Foundation’s Success with Therapies Research Consortium.
“Assessing the special challenges that individuals with lower SES face, including financial barriers, is essential to understand how we can address the unique combinations of adherence barriers. In other chronic disorders, financial barriers or lower socioeconomic status is associated with nonadherence, but this relationship has not been well established in cystic fibrosis,” said Kimberly Dickinson, MD, MPH, of Johns Hopkins University, Baltimore, during her presentation of the results at the virtual North American Cystic Fibrosis Conference.

“I’ve always thought that my patients in the poorer population were doing worse, and I think this demonstrates that that’s true,” said Robert Giusti, MD, in an interview. Dr. Giusti is a clinical professor of pediatrics at the New York University and director of the Pediatric Cystic Fibrosis Center in New York. He was not involved in the study.
“These are very pertinent issues, especially if you think about the pandemic, and some of the issues related to mental health. It just highlights the importance of socioeconomic status and screening for some of the known risk factors so that we can develop interventions or programs to provide equitable care to all of our cystic fibrosis patients,” said Ryan Perkins, MD, who moderated the session where the study was presented. He is a pediatric and adult pulmonary fellow at Boston Children’s Hospital and Brigham and Women’s Hospital, also in Boston.
The researchers looked retrospectively at 1 year’s worth of pharmacy refill receipts and number of times prescriptions were refilled versus the number of times prescribed, then calculated medicinal possession ratios. This was cross-referenced with annual household income and insurance status of patients with CF at 12 pediatric and 9 adult CF care centers, for a total of 376 patients (128 pediatric and 248 adult).
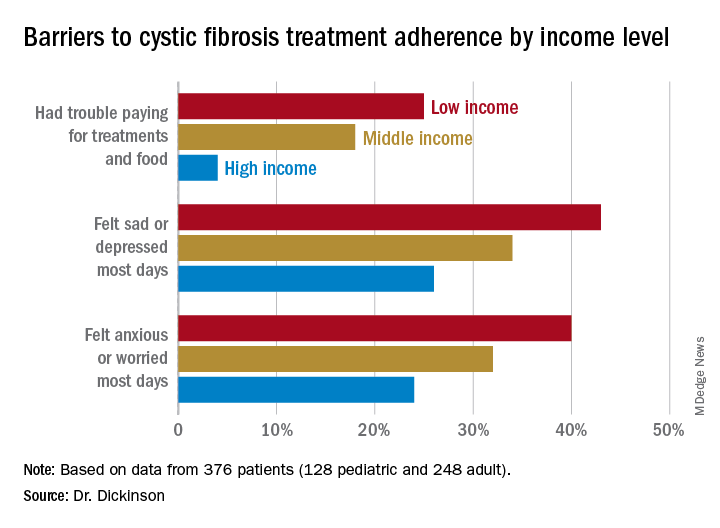
In this population, 32% of participants had public or no insurance, 68% had private or military insurance. The public/no insurance group was more likely than the private/military insurance group to report having trouble paying for treatments, food, or critical expenses related to CF care (23.3% vs. 12.1%, respectively); feeling symptoms on most days of depression (42.5% vs. 31.3%) or anxiety (40.0% vs. 28.5%); and experiencing conflict or stress with loved ones over treatments (30.0% vs. 20.3%) (P < .05 for all).
In all, 35% had a household income less than $40,000 per year, 33% between $44,000 and $100,000, and 32% higher than $100,000. The low-income group had a lower composite medication possession ratio (0.41) than the middle- (0.44) or high-income (0.52) groups, were more likely to have trouble paying for treatments, food, or treatment-related expenses (25%, 18%, 4%, respectively); were more likely most days to report symptoms of depression (43%, 34%, 26%) or anxiety (40%, 32%, 24%), and to have concerns about whether treatments were effective (42%, 27%, 29%). They were more likely to not be able to maintain a daily schedule or routine for treatments (28%, 22%, 14%).
The study showed that adherence barriers and suboptimal adherence are issues that cross all socioeconomic categories, though they were more problematic in the lowest bracket. Greater anxiety and depression among lower income individuals and those with private or no insurance was a key finding, according to Dr. Dickinson. “It highlights the importance of screening for mental health comorbidities that may impact non-adherence,” she said.
The study received funding from the Cystic Fibrosis Foundation. Dr. Dickinson, Dr. Giusti, and Dr. Perkins have no relevant financial disclosures.
Poor people with chronic illness have greater difficulty managing their disease than do their better-off counterparts, and a new study confirms this reality for patients with cystic fibrosis.
and anxiety symptoms, according to a new cross-sectional study. The data were drawn from the Cystic Fibrosis Foundation’s Success with Therapies Research Consortium.
“Assessing the special challenges that individuals with lower SES face, including financial barriers, is essential to understand how we can address the unique combinations of adherence barriers. In other chronic disorders, financial barriers or lower socioeconomic status is associated with nonadherence, but this relationship has not been well established in cystic fibrosis,” said Kimberly Dickinson, MD, MPH, of Johns Hopkins University, Baltimore, during her presentation of the results at the virtual North American Cystic Fibrosis Conference.
“I’ve always thought that my patients in the poorer population were doing worse, and I think this demonstrates that that’s true,” said Robert Giusti, MD, in an interview. Dr. Giusti is a clinical professor of pediatrics at the New York University and director of the Pediatric Cystic Fibrosis Center in New York. He was not involved in the study.
“These are very pertinent issues, especially if you think about the pandemic, and some of the issues related to mental health. It just highlights the importance of socioeconomic status and screening for some of the known risk factors so that we can develop interventions or programs to provide equitable care to all of our cystic fibrosis patients,” said Ryan Perkins, MD, who moderated the session where the study was presented. He is a pediatric and adult pulmonary fellow at Boston Children’s Hospital and Brigham and Women’s Hospital, also in Boston.
The researchers looked retrospectively at 1 year’s worth of pharmacy refill receipts and number of times prescriptions were refilled versus the number of times prescribed, then calculated medicinal possession ratios. This was cross-referenced with annual household income and insurance status of patients with CF at 12 pediatric and 9 adult CF care centers, for a total of 376 patients (128 pediatric and 248 adult).
In this population, 32% of participants had public or no insurance, 68% had private or military insurance. The public/no insurance group was more likely than the private/military insurance group to report having trouble paying for treatments, food, or critical expenses related to CF care (23.3% vs. 12.1%, respectively); feeling symptoms on most days of depression (42.5% vs. 31.3%) or anxiety (40.0% vs. 28.5%); and experiencing conflict or stress with loved ones over treatments (30.0% vs. 20.3%) (P < .05 for all).
In all, 35% had a household income less than $40,000 per year, 33% between $44,000 and $100,000, and 32% higher than $100,000. The low-income group had a lower composite medication possession ratio (0.41) than the middle- (0.44) or high-income (0.52) groups, were more likely to have trouble paying for treatments, food, or treatment-related expenses (25%, 18%, 4%, respectively); were more likely most days to report symptoms of depression (43%, 34%, 26%) or anxiety (40%, 32%, 24%), and to have concerns about whether treatments were effective (42%, 27%, 29%). They were more likely to not be able to maintain a daily schedule or routine for treatments (28%, 22%, 14%).
The study showed that adherence barriers and suboptimal adherence are issues that cross all socioeconomic categories, though they were more problematic in the lowest bracket. Greater anxiety and depression among lower income individuals and those with private or no insurance was a key finding, according to Dr. Dickinson. “It highlights the importance of screening for mental health comorbidities that may impact non-adherence,” she said.
The study received funding from the Cystic Fibrosis Foundation. Dr. Dickinson, Dr. Giusti, and Dr. Perkins have no relevant financial disclosures.
Poor people with chronic illness have greater difficulty managing their disease than do their better-off counterparts, and a new study confirms this reality for patients with cystic fibrosis.
and anxiety symptoms, according to a new cross-sectional study. The data were drawn from the Cystic Fibrosis Foundation’s Success with Therapies Research Consortium.
“Assessing the special challenges that individuals with lower SES face, including financial barriers, is essential to understand how we can address the unique combinations of adherence barriers. In other chronic disorders, financial barriers or lower socioeconomic status is associated with nonadherence, but this relationship has not been well established in cystic fibrosis,” said Kimberly Dickinson, MD, MPH, of Johns Hopkins University, Baltimore, during her presentation of the results at the virtual North American Cystic Fibrosis Conference.
“I’ve always thought that my patients in the poorer population were doing worse, and I think this demonstrates that that’s true,” said Robert Giusti, MD, in an interview. Dr. Giusti is a clinical professor of pediatrics at the New York University and director of the Pediatric Cystic Fibrosis Center in New York. He was not involved in the study.
“These are very pertinent issues, especially if you think about the pandemic, and some of the issues related to mental health. It just highlights the importance of socioeconomic status and screening for some of the known risk factors so that we can develop interventions or programs to provide equitable care to all of our cystic fibrosis patients,” said Ryan Perkins, MD, who moderated the session where the study was presented. He is a pediatric and adult pulmonary fellow at Boston Children’s Hospital and Brigham and Women’s Hospital, also in Boston.
The researchers looked retrospectively at 1 year’s worth of pharmacy refill receipts and number of times prescriptions were refilled versus the number of times prescribed, then calculated medicinal possession ratios. This was cross-referenced with annual household income and insurance status of patients with CF at 12 pediatric and 9 adult CF care centers, for a total of 376 patients (128 pediatric and 248 adult).
In this population, 32% of participants had public or no insurance, 68% had private or military insurance. The public/no insurance group was more likely than the private/military insurance group to report having trouble paying for treatments, food, or critical expenses related to CF care (23.3% vs. 12.1%, respectively); feeling symptoms on most days of depression (42.5% vs. 31.3%) or anxiety (40.0% vs. 28.5%); and experiencing conflict or stress with loved ones over treatments (30.0% vs. 20.3%) (P < .05 for all).
In all, 35% had a household income less than $40,000 per year, 33% between $44,000 and $100,000, and 32% higher than $100,000. The low-income group had a lower composite medication possession ratio (0.41) than the middle- (0.44) or high-income (0.52) groups, were more likely to have trouble paying for treatments, food, or treatment-related expenses (25%, 18%, 4%, respectively); were more likely most days to report symptoms of depression (43%, 34%, 26%) or anxiety (40%, 32%, 24%), and to have concerns about whether treatments were effective (42%, 27%, 29%). They were more likely to not be able to maintain a daily schedule or routine for treatments (28%, 22%, 14%).
The study showed that adherence barriers and suboptimal adherence are issues that cross all socioeconomic categories, though they were more problematic in the lowest bracket. Greater anxiety and depression among lower income individuals and those with private or no insurance was a key finding, according to Dr. Dickinson. “It highlights the importance of screening for mental health comorbidities that may impact non-adherence,” she said.
The study received funding from the Cystic Fibrosis Foundation. Dr. Dickinson, Dr. Giusti, and Dr. Perkins have no relevant financial disclosures.
FROM NACFC 2020
.
Dripping, dabbing, and bongs: Can’t tell the players without a scorecard
E-cigarettes may be synonymous with vaping to most physicians, but there are other ways for patients to inhale nicotine or tetrahydrocannabinol-containing aerosols, according to investigators at the Cleveland Clinic. 
Humberto Choi, MD, and associates wrote in the Annals of the American Thoracic Society.
These “alternate aerosol inhalation methods” have been poorly described thus far, so little is known about their scope of use and potential health impact, they noted.
Dripping involves an e-cigarette modified to expose the heating coil. The e-cigarette liquid is dripped directly onto the hot coil, which produces immediate aerosolization and results in a thicker cloud.
Dripping “may expose users to higher levels of nicotine compared to e-cigarette inhalation” and lead to “increased release of volatile aldehydes as a result of the higher heating potential of direct atomizer exposure,” the investigators suggested.
Water pipes, or bongs, produce both smoke and vapor, although an electronic vaporizer can be attached to create a “vape bong.” About 21% of daily cannabis users report using a bong, but tobacco inhalation is less common. Cases of severe pulmonary infections have been associated with bong use, along with a couple of tuberculosis clusters, Dr. Choi and associates said.
Dabbing uses butane-extracted, concentrated cannabis oil inhaled through a modified water pipe or bong or a smaller device called a “dab pen.” A small amount, or “dab,” of the product is placed on the “nail,” which replaces the bowl of the water pipe, heated with a blowtorch, and inhaled through the pipe, the researchers explained.
The prevalence of dabbing is unknown, but “the most recent Monitoring the Future survey of high school seniors shows that 11.9% of students have used a marijuana vaporizer at some point in their life,” they said.
Besides the fire risks involved in creating the material needed for dabbing – use of heating plates, ovens, and devices for removing butane vapors – inhalation of residual butane vapors could lead to vomiting, cardiac arrhythmias, acute encephalopathy, and respiratory depression, Dr. Choi and associates said.
Nicotine dependence is also a concern, as is the possibility of withdrawal symptoms. “Patients presenting with prolonged and severe vomiting, psychotic symptoms, or other acute neuropsychiatric symptoms should raise the suspicion of [tetrahydrocannabinol]-containing products especially synthetic cannabinoids,” they wrote.
SOURCE: Choi H et al. Ann Am Thorac Soc. 2020 Oct 14. doi: 10.1513/AnnalsATS.202005-511CME.
E-cigarettes may be synonymous with vaping to most physicians, but there are other ways for patients to inhale nicotine or tetrahydrocannabinol-containing aerosols, according to investigators at the Cleveland Clinic.
Humberto Choi, MD, and associates wrote in the Annals of the American Thoracic Society.
These “alternate aerosol inhalation methods” have been poorly described thus far, so little is known about their scope of use and potential health impact, they noted.
Dripping involves an e-cigarette modified to expose the heating coil. The e-cigarette liquid is dripped directly onto the hot coil, which produces immediate aerosolization and results in a thicker cloud.
Dripping “may expose users to higher levels of nicotine compared to e-cigarette inhalation” and lead to “increased release of volatile aldehydes as a result of the higher heating potential of direct atomizer exposure,” the investigators suggested.
Water pipes, or bongs, produce both smoke and vapor, although an electronic vaporizer can be attached to create a “vape bong.” About 21% of daily cannabis users report using a bong, but tobacco inhalation is less common. Cases of severe pulmonary infections have been associated with bong use, along with a couple of tuberculosis clusters, Dr. Choi and associates said.
Dabbing uses butane-extracted, concentrated cannabis oil inhaled through a modified water pipe or bong or a smaller device called a “dab pen.” A small amount, or “dab,” of the product is placed on the “nail,” which replaces the bowl of the water pipe, heated with a blowtorch, and inhaled through the pipe, the researchers explained.
The prevalence of dabbing is unknown, but “the most recent Monitoring the Future survey of high school seniors shows that 11.9% of students have used a marijuana vaporizer at some point in their life,” they said.
Besides the fire risks involved in creating the material needed for dabbing – use of heating plates, ovens, and devices for removing butane vapors – inhalation of residual butane vapors could lead to vomiting, cardiac arrhythmias, acute encephalopathy, and respiratory depression, Dr. Choi and associates said.
Nicotine dependence is also a concern, as is the possibility of withdrawal symptoms. “Patients presenting with prolonged and severe vomiting, psychotic symptoms, or other acute neuropsychiatric symptoms should raise the suspicion of [tetrahydrocannabinol]-containing products especially synthetic cannabinoids,” they wrote.
SOURCE: Choi H et al. Ann Am Thorac Soc. 2020 Oct 14. doi: 10.1513/AnnalsATS.202005-511CME.
E-cigarettes may be synonymous with vaping to most physicians, but there are other ways for patients to inhale nicotine or tetrahydrocannabinol-containing aerosols, according to investigators at the Cleveland Clinic.
Humberto Choi, MD, and associates wrote in the Annals of the American Thoracic Society.
These “alternate aerosol inhalation methods” have been poorly described thus far, so little is known about their scope of use and potential health impact, they noted.
Dripping involves an e-cigarette modified to expose the heating coil. The e-cigarette liquid is dripped directly onto the hot coil, which produces immediate aerosolization and results in a thicker cloud.
Dripping “may expose users to higher levels of nicotine compared to e-cigarette inhalation” and lead to “increased release of volatile aldehydes as a result of the higher heating potential of direct atomizer exposure,” the investigators suggested.
Water pipes, or bongs, produce both smoke and vapor, although an electronic vaporizer can be attached to create a “vape bong.” About 21% of daily cannabis users report using a bong, but tobacco inhalation is less common. Cases of severe pulmonary infections have been associated with bong use, along with a couple of tuberculosis clusters, Dr. Choi and associates said.
Dabbing uses butane-extracted, concentrated cannabis oil inhaled through a modified water pipe or bong or a smaller device called a “dab pen.” A small amount, or “dab,” of the product is placed on the “nail,” which replaces the bowl of the water pipe, heated with a blowtorch, and inhaled through the pipe, the researchers explained.
The prevalence of dabbing is unknown, but “the most recent Monitoring the Future survey of high school seniors shows that 11.9% of students have used a marijuana vaporizer at some point in their life,” they said.
Besides the fire risks involved in creating the material needed for dabbing – use of heating plates, ovens, and devices for removing butane vapors – inhalation of residual butane vapors could lead to vomiting, cardiac arrhythmias, acute encephalopathy, and respiratory depression, Dr. Choi and associates said.
Nicotine dependence is also a concern, as is the possibility of withdrawal symptoms. “Patients presenting with prolonged and severe vomiting, psychotic symptoms, or other acute neuropsychiatric symptoms should raise the suspicion of [tetrahydrocannabinol]-containing products especially synthetic cannabinoids,” they wrote.
SOURCE: Choi H et al. Ann Am Thorac Soc. 2020 Oct 14. doi: 10.1513/AnnalsATS.202005-511CME.
FROM ANNALS OF THE AMERICAN THORACIC SOCIETY
Triple combination therapy for cystic fibrosis linked to plunging hospitalizations
.
The triple combination therapy elexacaftor/tezacaftor/ivacaftor was associated with a near elimination of hospital stays in one hospital in Oregon, according to a new report. The hospital savings still weren’t nearly enough to pay for the cost of therapy, but the study underscores what many institutions have observed and adds a new layer to the view of quality of life improvements that the new therapy brings.
“After we started prescribing it, we noticed pretty quickly that hospitalizations appeared to be declining after patients started triple combination therapy, and we were hearing [similar reports] from other centers as well. We wanted to quantify this,” Eric C. Walter, MD, a pulmonologist at the Kaiser Permanente Cystic Fibrosis Clinic in Portland, Ore., said during a presentation of the results at the virtual North American Cystic Fibrosis Conference.
“We’re seeing that across the board in real practice, the number of cystic fibrosis patients that have to be hospitalized since starting this triple combination has gone down,” Robert Giusti, MD, said in an interview. “When they’ve had pulmonary exacerbations in the past, it was frequently because they failed outpatient antibiotics, but I think with triple combination therapy, if they do get sick, the likelihood is they will respond to oral antibiotics, so they may not need that prolonged IV course in the hospital.” Dr. Giusti is clinical professor of pediatrics at New York University and director of the Pediatric Cystic Fibrosis Center. He was not involved in the study.
The therapy gained Food and Drug Administration approval in 2019 for the treatment of individuals with CF who are aged 12 years and older, and who have at least one copy of the F508del mutation. Its cost is about $317,000 per year within the Kaiser Permanente system, according to Dr. Walter. His group compared hospitalization days for CF-related diagnoses from Jan. 1 through Aug. 31, 2020, before and after initiation of triple combination therapy.
Of 47 eligible patients, 32 initiated therapy during the study period; 38% had severe lung disease, defined by forced expiratory volume in 1 second (FEV1) value less than 40%. In 2020, before initiation of therapy, there were an average of 27 hospital days per month, all among patients with severe lung disease.
Among the therapy group, there were no hospitalizations after initiation of therapy through Aug. 31. Dr. Walter noted that the first hospitalization of a patient on triple combination therapy didn’t occur until early October.
At an average daily cost of $6,700, the researchers calculated that triple combination therapy saved about $189,000 per month in this group of patients. Comparing numbers to previous years, in which some patients with FEV1 greater than 40% were hospitalized, the researchers calculated that the therapy saved about $151,000 per month among individuals with severe lung disease: Patients with severe lung disease contributed about 80% to total hospital costs.
The drug itself for the whole group cost $845,000, dwarfing the $189,000 savings overall. But among patients with severe disease, hospitalization savings were about $151,000 per month, while the drug cost in this group was $316,800 per month.
Cost savings are important, but the improvement in quality of life for a patient – avoiding hospitalization, fewer impacts on work and education – should not be overlooked, according to Ryan Perkins, MD, a pediatric and adult pulmonary fellow at Boston Children’s Hospital and Brigham and Women’s Hospital, who moderated the session. “Some of these aren’t things people typically quantify and assign a price tag to,” Dr. Perkins said in an interview.
A big limitation of the work is that it was conducted during the COVID-19 pandemic, which may have reduced hospitalizations. “We did have patients that called in, told us they were sick, that they needed to be treated for an exacerbation but didn’t want to go to the hospital,” said Dr. Walter. To help adjust for this, Dr. Walter’s team plans to compare intravenous antibiotic exposure before and after triple combination therapy, reasoning that it could help clarify the pandemic’s impact on hospitalizations.
Dr. Walter, Dr. Giusti, and Dr. Perkins have no relevant financial disclosures.
SOURCE: Walter E et al. NACFC 2020. Abstract 795.
.
The triple combination therapy elexacaftor/tezacaftor/ivacaftor was associated with a near elimination of hospital stays in one hospital in Oregon, according to a new report. The hospital savings still weren’t nearly enough to pay for the cost of therapy, but the study underscores what many institutions have observed and adds a new layer to the view of quality of life improvements that the new therapy brings.
“After we started prescribing it, we noticed pretty quickly that hospitalizations appeared to be declining after patients started triple combination therapy, and we were hearing [similar reports] from other centers as well. We wanted to quantify this,” Eric C. Walter, MD, a pulmonologist at the Kaiser Permanente Cystic Fibrosis Clinic in Portland, Ore., said during a presentation of the results at the virtual North American Cystic Fibrosis Conference.
“We’re seeing that across the board in real practice, the number of cystic fibrosis patients that have to be hospitalized since starting this triple combination has gone down,” Robert Giusti, MD, said in an interview. “When they’ve had pulmonary exacerbations in the past, it was frequently because they failed outpatient antibiotics, but I think with triple combination therapy, if they do get sick, the likelihood is they will respond to oral antibiotics, so they may not need that prolonged IV course in the hospital.” Dr. Giusti is clinical professor of pediatrics at New York University and director of the Pediatric Cystic Fibrosis Center. He was not involved in the study.
The therapy gained Food and Drug Administration approval in 2019 for the treatment of individuals with CF who are aged 12 years and older, and who have at least one copy of the F508del mutation. Its cost is about $317,000 per year within the Kaiser Permanente system, according to Dr. Walter. His group compared hospitalization days for CF-related diagnoses from Jan. 1 through Aug. 31, 2020, before and after initiation of triple combination therapy.
Of 47 eligible patients, 32 initiated therapy during the study period; 38% had severe lung disease, defined by forced expiratory volume in 1 second (FEV1) value less than 40%. In 2020, before initiation of therapy, there were an average of 27 hospital days per month, all among patients with severe lung disease.
Among the therapy group, there were no hospitalizations after initiation of therapy through Aug. 31. Dr. Walter noted that the first hospitalization of a patient on triple combination therapy didn’t occur until early October.
At an average daily cost of $6,700, the researchers calculated that triple combination therapy saved about $189,000 per month in this group of patients. Comparing numbers to previous years, in which some patients with FEV1 greater than 40% were hospitalized, the researchers calculated that the therapy saved about $151,000 per month among individuals with severe lung disease: Patients with severe lung disease contributed about 80% to total hospital costs.
The drug itself for the whole group cost $845,000, dwarfing the $189,000 savings overall. But among patients with severe disease, hospitalization savings were about $151,000 per month, while the drug cost in this group was $316,800 per month.
Cost savings are important, but the improvement in quality of life for a patient – avoiding hospitalization, fewer impacts on work and education – should not be overlooked, according to Ryan Perkins, MD, a pediatric and adult pulmonary fellow at Boston Children’s Hospital and Brigham and Women’s Hospital, who moderated the session. “Some of these aren’t things people typically quantify and assign a price tag to,” Dr. Perkins said in an interview.
A big limitation of the work is that it was conducted during the COVID-19 pandemic, which may have reduced hospitalizations. “We did have patients that called in, told us they were sick, that they needed to be treated for an exacerbation but didn’t want to go to the hospital,” said Dr. Walter. To help adjust for this, Dr. Walter’s team plans to compare intravenous antibiotic exposure before and after triple combination therapy, reasoning that it could help clarify the pandemic’s impact on hospitalizations.
Dr. Walter, Dr. Giusti, and Dr. Perkins have no relevant financial disclosures.
SOURCE: Walter E et al. NACFC 2020. Abstract 795.
.
The triple combination therapy elexacaftor/tezacaftor/ivacaftor was associated with a near elimination of hospital stays in one hospital in Oregon, according to a new report. The hospital savings still weren’t nearly enough to pay for the cost of therapy, but the study underscores what many institutions have observed and adds a new layer to the view of quality of life improvements that the new therapy brings.
“After we started prescribing it, we noticed pretty quickly that hospitalizations appeared to be declining after patients started triple combination therapy, and we were hearing [similar reports] from other centers as well. We wanted to quantify this,” Eric C. Walter, MD, a pulmonologist at the Kaiser Permanente Cystic Fibrosis Clinic in Portland, Ore., said during a presentation of the results at the virtual North American Cystic Fibrosis Conference.
“We’re seeing that across the board in real practice, the number of cystic fibrosis patients that have to be hospitalized since starting this triple combination has gone down,” Robert Giusti, MD, said in an interview. “When they’ve had pulmonary exacerbations in the past, it was frequently because they failed outpatient antibiotics, but I think with triple combination therapy, if they do get sick, the likelihood is they will respond to oral antibiotics, so they may not need that prolonged IV course in the hospital.” Dr. Giusti is clinical professor of pediatrics at New York University and director of the Pediatric Cystic Fibrosis Center. He was not involved in the study.
The therapy gained Food and Drug Administration approval in 2019 for the treatment of individuals with CF who are aged 12 years and older, and who have at least one copy of the F508del mutation. Its cost is about $317,000 per year within the Kaiser Permanente system, according to Dr. Walter. His group compared hospitalization days for CF-related diagnoses from Jan. 1 through Aug. 31, 2020, before and after initiation of triple combination therapy.
Of 47 eligible patients, 32 initiated therapy during the study period; 38% had severe lung disease, defined by forced expiratory volume in 1 second (FEV1) value less than 40%. In 2020, before initiation of therapy, there were an average of 27 hospital days per month, all among patients with severe lung disease.
Among the therapy group, there were no hospitalizations after initiation of therapy through Aug. 31. Dr. Walter noted that the first hospitalization of a patient on triple combination therapy didn’t occur until early October.
At an average daily cost of $6,700, the researchers calculated that triple combination therapy saved about $189,000 per month in this group of patients. Comparing numbers to previous years, in which some patients with FEV1 greater than 40% were hospitalized, the researchers calculated that the therapy saved about $151,000 per month among individuals with severe lung disease: Patients with severe lung disease contributed about 80% to total hospital costs.
The drug itself for the whole group cost $845,000, dwarfing the $189,000 savings overall. But among patients with severe disease, hospitalization savings were about $151,000 per month, while the drug cost in this group was $316,800 per month.
Cost savings are important, but the improvement in quality of life for a patient – avoiding hospitalization, fewer impacts on work and education – should not be overlooked, according to Ryan Perkins, MD, a pediatric and adult pulmonary fellow at Boston Children’s Hospital and Brigham and Women’s Hospital, who moderated the session. “Some of these aren’t things people typically quantify and assign a price tag to,” Dr. Perkins said in an interview.
A big limitation of the work is that it was conducted during the COVID-19 pandemic, which may have reduced hospitalizations. “We did have patients that called in, told us they were sick, that they needed to be treated for an exacerbation but didn’t want to go to the hospital,” said Dr. Walter. To help adjust for this, Dr. Walter’s team plans to compare intravenous antibiotic exposure before and after triple combination therapy, reasoning that it could help clarify the pandemic’s impact on hospitalizations.
Dr. Walter, Dr. Giusti, and Dr. Perkins have no relevant financial disclosures.
SOURCE: Walter E et al. NACFC 2020. Abstract 795.
FROM NACFC 2020
Cystic fibrosis patients’ vulnerability to COVID-19 infection: Preliminary data ease fears
But early results suggest that social distance measures and perhaps the younger average age of individuals with CF have prevented a severe impact on this patient population.
Not all of the news is good. Some research suggests that posttransplant individuals may be at greater risk of severe outcomes. However, researchers warned that the data are too sparse to draw firm conclusions, and ongoing analyses of patient registries and other sources should lend greater insight into the burden of COVID-19 among individuals with CF. Those were some of the conclusions presented at a session of the virtual North American Cystic Fibrosis Conference.
D.B. Sanders, MD, who is a pediatric pulmonologist at Riley Hospital for Children and the Indiana University, both in Indianapolis, presented data from the Cystic Fibrosis Foundation’s Patient Registry, which includes patients in the United States. As in other populations, he showed that health care use has gone down among individuals with CF. From April to September 2019, 81% of clinical encounters were in the clinic and 12% in the hospital. Over the same period in 2020, those numbers dropped to 35% and 4%, respectively, with 30% by phone or computer. In-person health care use rebounded somewhat between July 1 and Sept. 16, with 53% of encounters at the clinic, 5% at the hospital, and 28% conducted virtually. There were also dips in forced expiratory volume in one second (FEV1) and microbiology testing, from about 90% occurring during health encounters at the end of 2019 to fewer than 10% of encounters by April.
As of Aug. 17, Dr. Sanders reported that 3,048 individuals with CF had been tested for COVID-19, with 174 positive results.
Racial and ethnic disparities in positive test results seen in other populations were also observable among individuals with CF. Several groups made up a higher proportion of COVID-19–positive CF patients than the general CF population, including Hispanics (18% vs. 9%), Blacks (7% vs. 5%), and individuals with FEV1 value less than 40% predicted (14% vs. 8%).
As of Sept. 17, there had been 51 hospitalizations and two deaths in the United States among 212 individuals with CF who tested positive for COVID-19, with increasing numbers that mirror trends in the U.S. population. One death occurred in a patient with advanced lung disease, the other in a post–lung transplant patient. “Thankfully [the numbers are] not higher, but this is being followed very closely,” said Dr. Sanders during his presentation.
One encouraging bit of news was that hospitalizations among individuals with CF have dropped since the start of the pandemic. “I think this shows how good our families are at socially distancing, wearing masks, and now that they not being exposed to viruses, I think we’re seeing the fruits of this with fewer hospitalizations,” said Dr. Sanders. He noted that it’s possible some of the decline could have been to reluctance to go to the hospital, and the introduction of triple combination cystic fibrosis transmembrane conductance regulator modulator therapy has also likely contributed. “We were already seeing fewer hospitalizations even before the pandemic hit,” he said.
At the session, Rebecca Cosgriff, director of data and quality improvement at the Cystic Fibrosis Trust in the United Kingdom, presented an international perspective on COVID-19 cases among individuals with CF. At the beginning of the pandemic, the Cystic Fibrosis Global Registry Harmonization Group recruited country coordinators to collect anonymized data on infections, hospitalizations, and other outcomes. In April, the group published its initial findings from 40 cases in eight countries, which concluded that these cases generally resembled the broader population in clinical course, which assuaged initial fears.
Ms. Cosgriff reported on results from a second round of data collection with a cutoff date of June 19, which expanded to 19 countries and included many from South America and more in Europe. The network encompassed about 85,000 individuals with CF, and tallied 181 cases of COVID-19. A total of 149 cases were nontransplant, and 32 were posttransplant (28 lung only). Fully 15% of the nontransplant group were over age 40 years, compared with 41% in the transplant group. Homozygous F508del mutations were more common in the posttransplant group (59% vs. 36%). However, lung function, as estimated by the best FEV1 measured in the previous year prior to infection, differed between the nontransplant (73%) and posttransplant (80%) COVID-19 patients.
Across all age groups, hospitalizations were more common in patients with best FEV1 percentage predicted values less than 70% (P = .001). Ms. Cosgriff also expressed concern about the posttransplant group. “Across all outcomes that might be indicative of infection severity – hospitalization, ICU admission, new supplementary oxygen, and non-invasive ventilation – the proportion of the posttransplant group was higher across the board,” she said during her presentation.
There were seven deaths. Ms. Cosgriff noted that there were too few deaths to analyze trends, but she presented a slide showing characteristics of deceased patients. “Factors like being post–lung transplant, being male, having less FEV1 than predicted, being over 40, or having CF-related diabetes, all appear pretty frequently amongst the cohort of people who died,” she said.
Overall, the results of these surveys are encouraging, suggesting that early fears that COVID-19 cases could be more severe among individuals with CF may not have been borne out so far. Dr. Sanders noted in his talk that there aren’t enough cases in the U.S. cohort to show links to risk factors with statistical significance. “But thankfully we’re not seeing a host of negative outcomes,” he said.
Dr. Sanders and Ms Cosgriff have no relevant financial disclosures.
But early results suggest that social distance measures and perhaps the younger average age of individuals with CF have prevented a severe impact on this patient population.
Not all of the news is good. Some research suggests that posttransplant individuals may be at greater risk of severe outcomes. However, researchers warned that the data are too sparse to draw firm conclusions, and ongoing analyses of patient registries and other sources should lend greater insight into the burden of COVID-19 among individuals with CF. Those were some of the conclusions presented at a session of the virtual North American Cystic Fibrosis Conference.
D.B. Sanders, MD, who is a pediatric pulmonologist at Riley Hospital for Children and the Indiana University, both in Indianapolis, presented data from the Cystic Fibrosis Foundation’s Patient Registry, which includes patients in the United States. As in other populations, he showed that health care use has gone down among individuals with CF. From April to September 2019, 81% of clinical encounters were in the clinic and 12% in the hospital. Over the same period in 2020, those numbers dropped to 35% and 4%, respectively, with 30% by phone or computer. In-person health care use rebounded somewhat between July 1 and Sept. 16, with 53% of encounters at the clinic, 5% at the hospital, and 28% conducted virtually. There were also dips in forced expiratory volume in one second (FEV1) and microbiology testing, from about 90% occurring during health encounters at the end of 2019 to fewer than 10% of encounters by April.
As of Aug. 17, Dr. Sanders reported that 3,048 individuals with CF had been tested for COVID-19, with 174 positive results.
Racial and ethnic disparities in positive test results seen in other populations were also observable among individuals with CF. Several groups made up a higher proportion of COVID-19–positive CF patients than the general CF population, including Hispanics (18% vs. 9%), Blacks (7% vs. 5%), and individuals with FEV1 value less than 40% predicted (14% vs. 8%).
As of Sept. 17, there had been 51 hospitalizations and two deaths in the United States among 212 individuals with CF who tested positive for COVID-19, with increasing numbers that mirror trends in the U.S. population. One death occurred in a patient with advanced lung disease, the other in a post–lung transplant patient. “Thankfully [the numbers are] not higher, but this is being followed very closely,” said Dr. Sanders during his presentation.
One encouraging bit of news was that hospitalizations among individuals with CF have dropped since the start of the pandemic. “I think this shows how good our families are at socially distancing, wearing masks, and now that they not being exposed to viruses, I think we’re seeing the fruits of this with fewer hospitalizations,” said Dr. Sanders. He noted that it’s possible some of the decline could have been to reluctance to go to the hospital, and the introduction of triple combination cystic fibrosis transmembrane conductance regulator modulator therapy has also likely contributed. “We were already seeing fewer hospitalizations even before the pandemic hit,” he said.
At the session, Rebecca Cosgriff, director of data and quality improvement at the Cystic Fibrosis Trust in the United Kingdom, presented an international perspective on COVID-19 cases among individuals with CF. At the beginning of the pandemic, the Cystic Fibrosis Global Registry Harmonization Group recruited country coordinators to collect anonymized data on infections, hospitalizations, and other outcomes. In April, the group published its initial findings from 40 cases in eight countries, which concluded that these cases generally resembled the broader population in clinical course, which assuaged initial fears.
Ms. Cosgriff reported on results from a second round of data collection with a cutoff date of June 19, which expanded to 19 countries and included many from South America and more in Europe. The network encompassed about 85,000 individuals with CF, and tallied 181 cases of COVID-19. A total of 149 cases were nontransplant, and 32 were posttransplant (28 lung only). Fully 15% of the nontransplant group were over age 40 years, compared with 41% in the transplant group. Homozygous F508del mutations were more common in the posttransplant group (59% vs. 36%). However, lung function, as estimated by the best FEV1 measured in the previous year prior to infection, differed between the nontransplant (73%) and posttransplant (80%) COVID-19 patients.
Across all age groups, hospitalizations were more common in patients with best FEV1 percentage predicted values less than 70% (P = .001). Ms. Cosgriff also expressed concern about the posttransplant group. “Across all outcomes that might be indicative of infection severity – hospitalization, ICU admission, new supplementary oxygen, and non-invasive ventilation – the proportion of the posttransplant group was higher across the board,” she said during her presentation.
There were seven deaths. Ms. Cosgriff noted that there were too few deaths to analyze trends, but she presented a slide showing characteristics of deceased patients. “Factors like being post–lung transplant, being male, having less FEV1 than predicted, being over 40, or having CF-related diabetes, all appear pretty frequently amongst the cohort of people who died,” she said.
Overall, the results of these surveys are encouraging, suggesting that early fears that COVID-19 cases could be more severe among individuals with CF may not have been borne out so far. Dr. Sanders noted in his talk that there aren’t enough cases in the U.S. cohort to show links to risk factors with statistical significance. “But thankfully we’re not seeing a host of negative outcomes,” he said.
Dr. Sanders and Ms Cosgriff have no relevant financial disclosures.
But early results suggest that social distance measures and perhaps the younger average age of individuals with CF have prevented a severe impact on this patient population.
Not all of the news is good. Some research suggests that posttransplant individuals may be at greater risk of severe outcomes. However, researchers warned that the data are too sparse to draw firm conclusions, and ongoing analyses of patient registries and other sources should lend greater insight into the burden of COVID-19 among individuals with CF. Those were some of the conclusions presented at a session of the virtual North American Cystic Fibrosis Conference.
D.B. Sanders, MD, who is a pediatric pulmonologist at Riley Hospital for Children and the Indiana University, both in Indianapolis, presented data from the Cystic Fibrosis Foundation’s Patient Registry, which includes patients in the United States. As in other populations, he showed that health care use has gone down among individuals with CF. From April to September 2019, 81% of clinical encounters were in the clinic and 12% in the hospital. Over the same period in 2020, those numbers dropped to 35% and 4%, respectively, with 30% by phone or computer. In-person health care use rebounded somewhat between July 1 and Sept. 16, with 53% of encounters at the clinic, 5% at the hospital, and 28% conducted virtually. There were also dips in forced expiratory volume in one second (FEV1) and microbiology testing, from about 90% occurring during health encounters at the end of 2019 to fewer than 10% of encounters by April.
As of Aug. 17, Dr. Sanders reported that 3,048 individuals with CF had been tested for COVID-19, with 174 positive results.
Racial and ethnic disparities in positive test results seen in other populations were also observable among individuals with CF. Several groups made up a higher proportion of COVID-19–positive CF patients than the general CF population, including Hispanics (18% vs. 9%), Blacks (7% vs. 5%), and individuals with FEV1 value less than 40% predicted (14% vs. 8%).
As of Sept. 17, there had been 51 hospitalizations and two deaths in the United States among 212 individuals with CF who tested positive for COVID-19, with increasing numbers that mirror trends in the U.S. population. One death occurred in a patient with advanced lung disease, the other in a post–lung transplant patient. “Thankfully [the numbers are] not higher, but this is being followed very closely,” said Dr. Sanders during his presentation.
One encouraging bit of news was that hospitalizations among individuals with CF have dropped since the start of the pandemic. “I think this shows how good our families are at socially distancing, wearing masks, and now that they not being exposed to viruses, I think we’re seeing the fruits of this with fewer hospitalizations,” said Dr. Sanders. He noted that it’s possible some of the decline could have been to reluctance to go to the hospital, and the introduction of triple combination cystic fibrosis transmembrane conductance regulator modulator therapy has also likely contributed. “We were already seeing fewer hospitalizations even before the pandemic hit,” he said.
At the session, Rebecca Cosgriff, director of data and quality improvement at the Cystic Fibrosis Trust in the United Kingdom, presented an international perspective on COVID-19 cases among individuals with CF. At the beginning of the pandemic, the Cystic Fibrosis Global Registry Harmonization Group recruited country coordinators to collect anonymized data on infections, hospitalizations, and other outcomes. In April, the group published its initial findings from 40 cases in eight countries, which concluded that these cases generally resembled the broader population in clinical course, which assuaged initial fears.
Ms. Cosgriff reported on results from a second round of data collection with a cutoff date of June 19, which expanded to 19 countries and included many from South America and more in Europe. The network encompassed about 85,000 individuals with CF, and tallied 181 cases of COVID-19. A total of 149 cases were nontransplant, and 32 were posttransplant (28 lung only). Fully 15% of the nontransplant group were over age 40 years, compared with 41% in the transplant group. Homozygous F508del mutations were more common in the posttransplant group (59% vs. 36%). However, lung function, as estimated by the best FEV1 measured in the previous year prior to infection, differed between the nontransplant (73%) and posttransplant (80%) COVID-19 patients.
Across all age groups, hospitalizations were more common in patients with best FEV1 percentage predicted values less than 70% (P = .001). Ms. Cosgriff also expressed concern about the posttransplant group. “Across all outcomes that might be indicative of infection severity – hospitalization, ICU admission, new supplementary oxygen, and non-invasive ventilation – the proportion of the posttransplant group was higher across the board,” she said during her presentation.
There were seven deaths. Ms. Cosgriff noted that there were too few deaths to analyze trends, but she presented a slide showing characteristics of deceased patients. “Factors like being post–lung transplant, being male, having less FEV1 than predicted, being over 40, or having CF-related diabetes, all appear pretty frequently amongst the cohort of people who died,” she said.
Overall, the results of these surveys are encouraging, suggesting that early fears that COVID-19 cases could be more severe among individuals with CF may not have been borne out so far. Dr. Sanders noted in his talk that there aren’t enough cases in the U.S. cohort to show links to risk factors with statistical significance. “But thankfully we’re not seeing a host of negative outcomes,” he said.
Dr. Sanders and Ms Cosgriff have no relevant financial disclosures.
FROM NACFC 2020
Pregnancy can be safe with interstitial lung disease
Pregnant women with interstitial lung disease (ILD) related to autoimmune disease may not need to terminate their pregnancies if they have close monitoring before, during, and after pregnancy with a multidisciplinary team of physicians, new research suggests.
Senior author Megan Clowse, MD, MPH, associate professor of medicine in the division of rheumatology at Duke University, Durham, N.C., explained during a press conference at the virtual annual meeting of the American College of Rheumatology that women with ILD are often advised by obstetricians or rheumatologists to avoid conception or terminate their pregnancies, though evidence for that has been based on small studies of 9-15 patients that have had mixed results.
“Many of these pregnancies were delivered 20-30 years ago, definitely with different rheumatic and obstetric care than we can provide now,” she said. “It’s really time to rethink our approach to interstitial lung disease and pregnancy.”
This study showed that while adverse pregnancy outcomes are common in these women, overall maternal morbidity and mortality are low.
ILD may be a secondary disease in people who have scleroderma, lupus, and sarcoidosis.
Largest study to date
This Pfizer-sponsored retrospective study of 67 pregnant women is the largest to date, and it analyzed 94 pregnancies (including five sets of twins).
Sarah Rae Easter, MD, maternal-fetal medicine doctor in the department of obstetrics and gynecology at Brigham and Women’s Hospital, Boston, called the work “exciting” as the researchers were able to look back at a large number of cases for a rare condition for more than 20 years.
“Their data provides much-needed evidence to provide some reassurance for women affected by this type of pulmonary disease regarding the relative safety of pregnancy,” she said in an interview.
Study spanned 23 years
The researchers reviewed pregnancy records in patients diagnosed with ILD secondary to autoimmune disease at Duke University Health System from January 1996 to July 2019.
They classified the severity of ILD based on two standard breathing tests – forced vital capacity and diffusion capacity for carbon monoxide.
Overall, 69% of the women were diagnosed with sarcoidosis and the remaining 31% had a connective tissue disease associated with ILD (CTD-ILD). Of those measured for ILD severity, 11% were severe, 25% were moderate, 50% were mild, and 14% were normal. Their average maternal age was 32.1 and 83% were Black.
While 70% of the pregnancies resulted in live births, 9% were terminated. The remainder resulted in miscarriage or stillbirth.
Researchers reported a 15% rate of preeclampsia, a 34% rate of the composite measure PROMISSE-Adverse Pregnancy Outcome (APO), and a 15% rate of PROMISSE-APO SEVERE. Patients with severe disease had the highest rates of PROMISSE-APO (P = .03 across groups).
(PROMISSE stands for the Predictors of Pregnancy Outcome: Biomarkers in Antiphospholipid Antibody Syndrome and Systemic Lupus Erythematosus study.)
None of the women died
Dr. Clowse said it was a pleasant surprise to find that none of the women died, though patients with severe ILD had more adverse outcomes. Only 2.1% were treated in an intensive care unit during or soon after delivery. In 4.2%, ILD patients had significant shortness of breath due to fluid volume overload around the time of delivery.
For the women who had normal-to-moderate lung disease, Dr. Clowse said, “they really had remarkably good outcomes, really pretty comparable to the general population. About 15% delivered preterm and about 20% suffered a pregnancy loss.”
Dr. Easter, who was not involved with the study, noted the large number of Black women in the cohort.
“Focusing in on improving outcomes for Black and Brown women related to pregnancy in our country is a much-needed undertaking,” Dr. Easter said.
Being able to quote percentages from this research, based on a good-sized study “at least gives people a benchmark about what kind of risk they are willing to assume for themselves,” she said.
For providers, being able to place this rare disease within the spectrum of other diseases where there is more data is also very helpful, she said.
Dr. Clowse said in an interview that the preponderance of Black women in the study was a surprise but may be explained by two factors: Sarcoidosis is seen more frequently in Black women and in the study area in North Carolina there is a large population of Black women.
“Also, our patients with more severe lupus, the ones who are more likely to have interstitial lung disease, are often Black and that’s likely contributing as well,” she said.
Multidisciplinary teams advised
Dr. Clowse emphasized that women with ILD need multidisciplinary teams in pregnancy and should be managed at tertiary care centers where there is a full complement of obstetric and internal medicine experts.
“We do recommend evaluating the severity of their lungs and their heart disease around the time of pregnancy and during pregnancy if they have shortness of breath,” she said.
“We currently recommend that these patients with moderate or severe disease stay in the hospital for up to a week, just for monitoring,” she said.
Dr. Easter said having that kind of access to a large academic healthcare center should be an important part of the decision-making.
Patients need to think about whether they would have access to care similar to what the researchers are describing when they are making the decision to pursue or continue pregnancy, she said.
The study was sponsored by Pfizer Inc. Dr. Clowse reported relationships with UCB, GlaxoSmithKline, AstraZeneca, and Pfizer. Dr. Easter has disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
Pregnant women with interstitial lung disease (ILD) related to autoimmune disease may not need to terminate their pregnancies if they have close monitoring before, during, and after pregnancy with a multidisciplinary team of physicians, new research suggests.
Senior author Megan Clowse, MD, MPH, associate professor of medicine in the division of rheumatology at Duke University, Durham, N.C., explained during a press conference at the virtual annual meeting of the American College of Rheumatology that women with ILD are often advised by obstetricians or rheumatologists to avoid conception or terminate their pregnancies, though evidence for that has been based on small studies of 9-15 patients that have had mixed results.
“Many of these pregnancies were delivered 20-30 years ago, definitely with different rheumatic and obstetric care than we can provide now,” she said. “It’s really time to rethink our approach to interstitial lung disease and pregnancy.”
This study showed that while adverse pregnancy outcomes are common in these women, overall maternal morbidity and mortality are low.
ILD may be a secondary disease in people who have scleroderma, lupus, and sarcoidosis.
Largest study to date
This Pfizer-sponsored retrospective study of 67 pregnant women is the largest to date, and it analyzed 94 pregnancies (including five sets of twins).
Sarah Rae Easter, MD, maternal-fetal medicine doctor in the department of obstetrics and gynecology at Brigham and Women’s Hospital, Boston, called the work “exciting” as the researchers were able to look back at a large number of cases for a rare condition for more than 20 years.
“Their data provides much-needed evidence to provide some reassurance for women affected by this type of pulmonary disease regarding the relative safety of pregnancy,” she said in an interview.
Study spanned 23 years
The researchers reviewed pregnancy records in patients diagnosed with ILD secondary to autoimmune disease at Duke University Health System from January 1996 to July 2019.
They classified the severity of ILD based on two standard breathing tests – forced vital capacity and diffusion capacity for carbon monoxide.
Overall, 69% of the women were diagnosed with sarcoidosis and the remaining 31% had a connective tissue disease associated with ILD (CTD-ILD). Of those measured for ILD severity, 11% were severe, 25% were moderate, 50% were mild, and 14% were normal. Their average maternal age was 32.1 and 83% were Black.
While 70% of the pregnancies resulted in live births, 9% were terminated. The remainder resulted in miscarriage or stillbirth.
Researchers reported a 15% rate of preeclampsia, a 34% rate of the composite measure PROMISSE-Adverse Pregnancy Outcome (APO), and a 15% rate of PROMISSE-APO SEVERE. Patients with severe disease had the highest rates of PROMISSE-APO (P = .03 across groups).
(PROMISSE stands for the Predictors of Pregnancy Outcome: Biomarkers in Antiphospholipid Antibody Syndrome and Systemic Lupus Erythematosus study.)
None of the women died
Dr. Clowse said it was a pleasant surprise to find that none of the women died, though patients with severe ILD had more adverse outcomes. Only 2.1% were treated in an intensive care unit during or soon after delivery. In 4.2%, ILD patients had significant shortness of breath due to fluid volume overload around the time of delivery.
For the women who had normal-to-moderate lung disease, Dr. Clowse said, “they really had remarkably good outcomes, really pretty comparable to the general population. About 15% delivered preterm and about 20% suffered a pregnancy loss.”
Dr. Easter, who was not involved with the study, noted the large number of Black women in the cohort.
“Focusing in on improving outcomes for Black and Brown women related to pregnancy in our country is a much-needed undertaking,” Dr. Easter said.
Being able to quote percentages from this research, based on a good-sized study “at least gives people a benchmark about what kind of risk they are willing to assume for themselves,” she said.
For providers, being able to place this rare disease within the spectrum of other diseases where there is more data is also very helpful, she said.
Dr. Clowse said in an interview that the preponderance of Black women in the study was a surprise but may be explained by two factors: Sarcoidosis is seen more frequently in Black women and in the study area in North Carolina there is a large population of Black women.
“Also, our patients with more severe lupus, the ones who are more likely to have interstitial lung disease, are often Black and that’s likely contributing as well,” she said.
Multidisciplinary teams advised
Dr. Clowse emphasized that women with ILD need multidisciplinary teams in pregnancy and should be managed at tertiary care centers where there is a full complement of obstetric and internal medicine experts.
“We do recommend evaluating the severity of their lungs and their heart disease around the time of pregnancy and during pregnancy if they have shortness of breath,” she said.
“We currently recommend that these patients with moderate or severe disease stay in the hospital for up to a week, just for monitoring,” she said.
Dr. Easter said having that kind of access to a large academic healthcare center should be an important part of the decision-making.
Patients need to think about whether they would have access to care similar to what the researchers are describing when they are making the decision to pursue or continue pregnancy, she said.
The study was sponsored by Pfizer Inc. Dr. Clowse reported relationships with UCB, GlaxoSmithKline, AstraZeneca, and Pfizer. Dr. Easter has disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
Pregnant women with interstitial lung disease (ILD) related to autoimmune disease may not need to terminate their pregnancies if they have close monitoring before, during, and after pregnancy with a multidisciplinary team of physicians, new research suggests.
Senior author Megan Clowse, MD, MPH, associate professor of medicine in the division of rheumatology at Duke University, Durham, N.C., explained during a press conference at the virtual annual meeting of the American College of Rheumatology that women with ILD are often advised by obstetricians or rheumatologists to avoid conception or terminate their pregnancies, though evidence for that has been based on small studies of 9-15 patients that have had mixed results.
“Many of these pregnancies were delivered 20-30 years ago, definitely with different rheumatic and obstetric care than we can provide now,” she said. “It’s really time to rethink our approach to interstitial lung disease and pregnancy.”
This study showed that while adverse pregnancy outcomes are common in these women, overall maternal morbidity and mortality are low.
ILD may be a secondary disease in people who have scleroderma, lupus, and sarcoidosis.
Largest study to date
This Pfizer-sponsored retrospective study of 67 pregnant women is the largest to date, and it analyzed 94 pregnancies (including five sets of twins).
Sarah Rae Easter, MD, maternal-fetal medicine doctor in the department of obstetrics and gynecology at Brigham and Women’s Hospital, Boston, called the work “exciting” as the researchers were able to look back at a large number of cases for a rare condition for more than 20 years.
“Their data provides much-needed evidence to provide some reassurance for women affected by this type of pulmonary disease regarding the relative safety of pregnancy,” she said in an interview.
Study spanned 23 years
The researchers reviewed pregnancy records in patients diagnosed with ILD secondary to autoimmune disease at Duke University Health System from January 1996 to July 2019.
They classified the severity of ILD based on two standard breathing tests – forced vital capacity and diffusion capacity for carbon monoxide.
Overall, 69% of the women were diagnosed with sarcoidosis and the remaining 31% had a connective tissue disease associated with ILD (CTD-ILD). Of those measured for ILD severity, 11% were severe, 25% were moderate, 50% were mild, and 14% were normal. Their average maternal age was 32.1 and 83% were Black.
While 70% of the pregnancies resulted in live births, 9% were terminated. The remainder resulted in miscarriage or stillbirth.
Researchers reported a 15% rate of preeclampsia, a 34% rate of the composite measure PROMISSE-Adverse Pregnancy Outcome (APO), and a 15% rate of PROMISSE-APO SEVERE. Patients with severe disease had the highest rates of PROMISSE-APO (P = .03 across groups).
(PROMISSE stands for the Predictors of Pregnancy Outcome: Biomarkers in Antiphospholipid Antibody Syndrome and Systemic Lupus Erythematosus study.)
None of the women died
Dr. Clowse said it was a pleasant surprise to find that none of the women died, though patients with severe ILD had more adverse outcomes. Only 2.1% were treated in an intensive care unit during or soon after delivery. In 4.2%, ILD patients had significant shortness of breath due to fluid volume overload around the time of delivery.
For the women who had normal-to-moderate lung disease, Dr. Clowse said, “they really had remarkably good outcomes, really pretty comparable to the general population. About 15% delivered preterm and about 20% suffered a pregnancy loss.”
Dr. Easter, who was not involved with the study, noted the large number of Black women in the cohort.
“Focusing in on improving outcomes for Black and Brown women related to pregnancy in our country is a much-needed undertaking,” Dr. Easter said.
Being able to quote percentages from this research, based on a good-sized study “at least gives people a benchmark about what kind of risk they are willing to assume for themselves,” she said.
For providers, being able to place this rare disease within the spectrum of other diseases where there is more data is also very helpful, she said.
Dr. Clowse said in an interview that the preponderance of Black women in the study was a surprise but may be explained by two factors: Sarcoidosis is seen more frequently in Black women and in the study area in North Carolina there is a large population of Black women.
“Also, our patients with more severe lupus, the ones who are more likely to have interstitial lung disease, are often Black and that’s likely contributing as well,” she said.
Multidisciplinary teams advised
Dr. Clowse emphasized that women with ILD need multidisciplinary teams in pregnancy and should be managed at tertiary care centers where there is a full complement of obstetric and internal medicine experts.
“We do recommend evaluating the severity of their lungs and their heart disease around the time of pregnancy and during pregnancy if they have shortness of breath,” she said.
“We currently recommend that these patients with moderate or severe disease stay in the hospital for up to a week, just for monitoring,” she said.
Dr. Easter said having that kind of access to a large academic healthcare center should be an important part of the decision-making.
Patients need to think about whether they would have access to care similar to what the researchers are describing when they are making the decision to pursue or continue pregnancy, she said.
The study was sponsored by Pfizer Inc. Dr. Clowse reported relationships with UCB, GlaxoSmithKline, AstraZeneca, and Pfizer. Dr. Easter has disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
Medication adherence challenges and helpers
For most chronic diseases, up to 20%-30% of the pills that are prescribed are not taken. In the case of inhalers for asthma and COPD, patients miss over half of the prescribed doses.
There are many things that contribute to the problem of poor adherence, but people often just simply forget. Thankfully, there are tools designed to help remind patients of what they need to take and when. A survey of apps developed to help patients remember to take their medicines found more than 700 available in Apple and Android app stores.1 Most apps focus on medication alerts, reminders, and medication logs.2 A recent review showed that apps have some – yet limited – effectiveness in increasing adherence, with patient self-reported improvements of 7%-40%.3

Another perhaps more promising area of improving adherence involves high-tech advances in the way medications can be taken. Inhalers are a primary target as they are complicated devices. A patient has to breathe in at the correct time after the inhaler is actuated, and the inhaler works optimally only if the rate of inhalation is sufficient to carry the medication into the lungs.
A number of companies have developed attachments for inhalers (and even inhalers themselves) that can record when the medication is taken through a Bluetooth connection to a patient’s smartphone. These can also assess inspiratory flow. Reminders to take the medication are built into the app, and those reminders disappear if the medication is taken. Patients can receive feedback about the quality of their timing and inspiratory rate to maximize medication delivery to the lungs.4
We learned long ago that it is difficult to take medications three to four times a day, so extended-release tablets were developed to reduce the frequency to once or twice a day. A great deal of work is now being done behind the scenes to develop medications that decrease the need for patients to remember to take their medications. The best examples of this are the long-acting reversible contraception (LARC) devices, specifically IUDs and Nexplanon. Compared with traditional oral contraceptives that need to be taken daily, LARCs reduce the rate of pregnancy by five- to tenfold.
We also now have medications for osteoporosis that can be taken monthly, or even annually. When bisphosphonates were first developed for osteoporosis prevention, they needed to be taken daily. Then a weekly bisphosphonate was developed. Now there is a once-monthly oral bisphosphonate, Ibandronate, and even a once yearly IV bisphosphonate.
Exciting developments have also occurred in the management of diabetes. We may be tempted to take for granted how once-daily long-acting insulin, which releases insulin slowly over the course of a day, has revolutionized the diabetic treatment since its Food and Drug Administration approval in 2000. Yet progress did not end there. The first GLP-1 receptor agonist for diabetes was approved in 2005 and was a twice-a-day medicine. Shortly afterward, a daily GLP-1 was approved, and now there are three once-weekly GLP-1 receptor agonists.
Several pharmaceutical manufacturers are now working on a once-weekly insulin,5 as well as an implantable GLP-1 receptor agonist that will need to be replaced every 6-12 months.6 Imagine your patient coming in once a year to replace his or her potent glucose lowering medication – one that offers a low incidence of hypoglycemia, maintains glucose control all year long, and requires no adherence to a complicated medication regimen.
Similar technology is being used to develop a once-yearly anti-HIV prophylactic medication delivery system.7 This could help prevent the spread of HIV in areas of the world where it may be difficult for people to take daily medications.7
The many technological advances we have described may help us reduce our likelihood of missing a dose of a medication. We are hopeful that progress in this area will continue, and that one day medication adherence will require even less effort from patients than it does today.
Dr. Notte is a family physician and chief medical officer of Abington (Pa.) Hospital–Jefferson Health. Follow him on Twitter (@doctornotte). Dr. Skolnik is professor of family and community medicine at Sidney Kimmel Medical College, Philadelphia, and associate director of the family medicine residency program at Abington Hospital–Jefferson Health. They have no conflicts related to the content of this piece.
References
1. Tabi K et al. Mobile apps for medication management: Review and analysis. JMIR Mhealth Uhealth. 2019 Sep 7(9):13608.
2. Park JYE et al. Mobile phone apps targeting medication adherence: Quality assessment and content analysis of user reviews. JMIR Mhealth Uhealth. 2019 Jan 31;7(1):e11919.
3. Pérez-Jover V et al. Mobile apps for increasing treatment adherence: Systematic review. J Med Internet Res. 2019;21(6):e12505. doi: 10.2196/12505.
4. 4 Smart inhalers that could be lifesaving for people living with asthma & COPD. MyTherapy, July 11, 2019.
5. Rosenstock J et al. Once-weekly insulin for type 2 diabetes without previous insulin treatment. N Engl J Med. 2020 Sep 22. doi: 10.1056/NEJMoa2022474.
6. GLP-1 agonists: From 2 daily injections to 1 per week and beyond. DiaTribe, Jan. 10, 2018.
7. Long-acting HIV prevention tools. Hiv.gov, July 20, 2019.
For most chronic diseases, up to 20%-30% of the pills that are prescribed are not taken. In the case of inhalers for asthma and COPD, patients miss over half of the prescribed doses.
There are many things that contribute to the problem of poor adherence, but people often just simply forget. Thankfully, there are tools designed to help remind patients of what they need to take and when. A survey of apps developed to help patients remember to take their medicines found more than 700 available in Apple and Android app stores.1 Most apps focus on medication alerts, reminders, and medication logs.2 A recent review showed that apps have some – yet limited – effectiveness in increasing adherence, with patient self-reported improvements of 7%-40%.3
Another perhaps more promising area of improving adherence involves high-tech advances in the way medications can be taken. Inhalers are a primary target as they are complicated devices. A patient has to breathe in at the correct time after the inhaler is actuated, and the inhaler works optimally only if the rate of inhalation is sufficient to carry the medication into the lungs.
A number of companies have developed attachments for inhalers (and even inhalers themselves) that can record when the medication is taken through a Bluetooth connection to a patient’s smartphone. These can also assess inspiratory flow. Reminders to take the medication are built into the app, and those reminders disappear if the medication is taken. Patients can receive feedback about the quality of their timing and inspiratory rate to maximize medication delivery to the lungs.4
We learned long ago that it is difficult to take medications three to four times a day, so extended-release tablets were developed to reduce the frequency to once or twice a day. A great deal of work is now being done behind the scenes to develop medications that decrease the need for patients to remember to take their medications. The best examples of this are the long-acting reversible contraception (LARC) devices, specifically IUDs and Nexplanon. Compared with traditional oral contraceptives that need to be taken daily, LARCs reduce the rate of pregnancy by five- to tenfold.
We also now have medications for osteoporosis that can be taken monthly, or even annually. When bisphosphonates were first developed for osteoporosis prevention, they needed to be taken daily. Then a weekly bisphosphonate was developed. Now there is a once-monthly oral bisphosphonate, Ibandronate, and even a once yearly IV bisphosphonate.
Exciting developments have also occurred in the management of diabetes. We may be tempted to take for granted how once-daily long-acting insulin, which releases insulin slowly over the course of a day, has revolutionized the diabetic treatment since its Food and Drug Administration approval in 2000. Yet progress did not end there. The first GLP-1 receptor agonist for diabetes was approved in 2005 and was a twice-a-day medicine. Shortly afterward, a daily GLP-1 was approved, and now there are three once-weekly GLP-1 receptor agonists.
Several pharmaceutical manufacturers are now working on a once-weekly insulin,5 as well as an implantable GLP-1 receptor agonist that will need to be replaced every 6-12 months.6 Imagine your patient coming in once a year to replace his or her potent glucose lowering medication – one that offers a low incidence of hypoglycemia, maintains glucose control all year long, and requires no adherence to a complicated medication regimen.
Similar technology is being used to develop a once-yearly anti-HIV prophylactic medication delivery system.7 This could help prevent the spread of HIV in areas of the world where it may be difficult for people to take daily medications.7
The many technological advances we have described may help us reduce our likelihood of missing a dose of a medication. We are hopeful that progress in this area will continue, and that one day medication adherence will require even less effort from patients than it does today.
Dr. Notte is a family physician and chief medical officer of Abington (Pa.) Hospital–Jefferson Health. Follow him on Twitter (@doctornotte). Dr. Skolnik is professor of family and community medicine at Sidney Kimmel Medical College, Philadelphia, and associate director of the family medicine residency program at Abington Hospital–Jefferson Health. They have no conflicts related to the content of this piece.
References
1. Tabi K et al. Mobile apps for medication management: Review and analysis. JMIR Mhealth Uhealth. 2019 Sep 7(9):13608.
2. Park JYE et al. Mobile phone apps targeting medication adherence: Quality assessment and content analysis of user reviews. JMIR Mhealth Uhealth. 2019 Jan 31;7(1):e11919.
3. Pérez-Jover V et al. Mobile apps for increasing treatment adherence: Systematic review. J Med Internet Res. 2019;21(6):e12505. doi: 10.2196/12505.
4. 4 Smart inhalers that could be lifesaving for people living with asthma & COPD. MyTherapy, July 11, 2019.
5. Rosenstock J et al. Once-weekly insulin for type 2 diabetes without previous insulin treatment. N Engl J Med. 2020 Sep 22. doi: 10.1056/NEJMoa2022474.
6. GLP-1 agonists: From 2 daily injections to 1 per week and beyond. DiaTribe, Jan. 10, 2018.
7. Long-acting HIV prevention tools. Hiv.gov, July 20, 2019.
For most chronic diseases, up to 20%-30% of the pills that are prescribed are not taken. In the case of inhalers for asthma and COPD, patients miss over half of the prescribed doses.
There are many things that contribute to the problem of poor adherence, but people often just simply forget. Thankfully, there are tools designed to help remind patients of what they need to take and when. A survey of apps developed to help patients remember to take their medicines found more than 700 available in Apple and Android app stores.1 Most apps focus on medication alerts, reminders, and medication logs.2 A recent review showed that apps have some – yet limited – effectiveness in increasing adherence, with patient self-reported improvements of 7%-40%.3
Another perhaps more promising area of improving adherence involves high-tech advances in the way medications can be taken. Inhalers are a primary target as they are complicated devices. A patient has to breathe in at the correct time after the inhaler is actuated, and the inhaler works optimally only if the rate of inhalation is sufficient to carry the medication into the lungs.
A number of companies have developed attachments for inhalers (and even inhalers themselves) that can record when the medication is taken through a Bluetooth connection to a patient’s smartphone. These can also assess inspiratory flow. Reminders to take the medication are built into the app, and those reminders disappear if the medication is taken. Patients can receive feedback about the quality of their timing and inspiratory rate to maximize medication delivery to the lungs.4
We learned long ago that it is difficult to take medications three to four times a day, so extended-release tablets were developed to reduce the frequency to once or twice a day. A great deal of work is now being done behind the scenes to develop medications that decrease the need for patients to remember to take their medications. The best examples of this are the long-acting reversible contraception (LARC) devices, specifically IUDs and Nexplanon. Compared with traditional oral contraceptives that need to be taken daily, LARCs reduce the rate of pregnancy by five- to tenfold.
We also now have medications for osteoporosis that can be taken monthly, or even annually. When bisphosphonates were first developed for osteoporosis prevention, they needed to be taken daily. Then a weekly bisphosphonate was developed. Now there is a once-monthly oral bisphosphonate, Ibandronate, and even a once yearly IV bisphosphonate.
Exciting developments have also occurred in the management of diabetes. We may be tempted to take for granted how once-daily long-acting insulin, which releases insulin slowly over the course of a day, has revolutionized the diabetic treatment since its Food and Drug Administration approval in 2000. Yet progress did not end there. The first GLP-1 receptor agonist for diabetes was approved in 2005 and was a twice-a-day medicine. Shortly afterward, a daily GLP-1 was approved, and now there are three once-weekly GLP-1 receptor agonists.
Several pharmaceutical manufacturers are now working on a once-weekly insulin,5 as well as an implantable GLP-1 receptor agonist that will need to be replaced every 6-12 months.6 Imagine your patient coming in once a year to replace his or her potent glucose lowering medication – one that offers a low incidence of hypoglycemia, maintains glucose control all year long, and requires no adherence to a complicated medication regimen.
Similar technology is being used to develop a once-yearly anti-HIV prophylactic medication delivery system.7 This could help prevent the spread of HIV in areas of the world where it may be difficult for people to take daily medications.7
The many technological advances we have described may help us reduce our likelihood of missing a dose of a medication. We are hopeful that progress in this area will continue, and that one day medication adherence will require even less effort from patients than it does today.
Dr. Notte is a family physician and chief medical officer of Abington (Pa.) Hospital–Jefferson Health. Follow him on Twitter (@doctornotte). Dr. Skolnik is professor of family and community medicine at Sidney Kimmel Medical College, Philadelphia, and associate director of the family medicine residency program at Abington Hospital–Jefferson Health. They have no conflicts related to the content of this piece.
References
1. Tabi K et al. Mobile apps for medication management: Review and analysis. JMIR Mhealth Uhealth. 2019 Sep 7(9):13608.
2. Park JYE et al. Mobile phone apps targeting medication adherence: Quality assessment and content analysis of user reviews. JMIR Mhealth Uhealth. 2019 Jan 31;7(1):e11919.
3. Pérez-Jover V et al. Mobile apps for increasing treatment adherence: Systematic review. J Med Internet Res. 2019;21(6):e12505. doi: 10.2196/12505.
4. 4 Smart inhalers that could be lifesaving for people living with asthma & COPD. MyTherapy, July 11, 2019.
5. Rosenstock J et al. Once-weekly insulin for type 2 diabetes without previous insulin treatment. N Engl J Med. 2020 Sep 22. doi: 10.1056/NEJMoa2022474.
6. GLP-1 agonists: From 2 daily injections to 1 per week and beyond. DiaTribe, Jan. 10, 2018.
7. Long-acting HIV prevention tools. Hiv.gov, July 20, 2019.
Home spirometry improved monitoring of cystic fibrosis patients during COVID-19 pandemic
Home spirometry has become increasingly used among cystic fibrosis patients during the COVID-19 pandemic, and new research suggests that home devices perform reasonably well. Forced expiratory volume in 1 second (FEV1) values were a bit lower than values seen in clinical spirometry performed in the same patient at a nearby time point, but the procedure reliably picked up decreases in FEV1, potentially helping patients and clinicians spot exacerbations early.
“Home spirometry was sort of a curiosity that was slowly working its way into cystic fibrosis research in 2019, and then all of a sudden in 2020 it became front and center as the only way to continue with clinical monitoring and research in many cases,” Alexander Paynter, MS, a biostatistician at the Cystic Fibrosis Foundation’s Therapeutic Development Network Coordinating Center, said during a talk at the virtual North American Cystic Fibrosis Conference.
To better determine how closely home spirometry matches clinical spirometry, Mr. Paynter and his colleagues analyzed data from the eICE study, which included 267 cystic fibrosis patients aged 14 and over at 14 cystic fibrosis centers. They were randomized to use home spirometry as an early intervention to detect exacerbations, or to continue usual clinic care with visits to the clinic every 3 months. The dataset includes twice-weekly home spirometry values, with a full-year of follow-up data. The researchers compared the home spirometry data to the clinical data closest in time to it. Clinic spirometry data with no corresponding home data within 7 days were discarded.
There was an estimated difference of –2.01 mL between home and clinic tests, with home spirometry producing lower values (95% confidence interval, –3.56 to –0.45). “There is actually a bias in home spirometry as compared to clinic spirometry,” concluded Mr. Paynter.
One explanation for lower values in home spirometry is that users are inexperienced with the device. If that’s true, then agreement should improve over time, but the researchers didn’t see strong evidence of that. Among 44 patients who completed five clinical visits, there was a difference of –2.97 (standard deviation [SD], 10.51) at baseline, –1.66 at 3 months (SD, 13.49), –3.7 at 6 months (SD, 12.44), –0.86 at 9 months (SD, 13.73), and –0.53 at 12 months (SD, 13.35). Though there was improvement over time, “we don’t find a lot of evidence that this bias completely resolves,” said Mr. Paynter.
In fact, a more likely explanation is the presence of coaching by a technician during clinical spirometry, according to Robert J. Giusti, MD, clinical professor of pediatrics and director of the Pediatric Cystic Fibrosis Center at New York University. “When they’re doing it at home, they don’t do it with the same effort, so I think that coaching through telemedicine during the home spirometry would make that difference disappear,” he said when asked to comment on the study.
The researchers found that change-based endpoints were similar between clinic and at-home spirometry. Compared to baseline, the two showed similar declines over time. “The clinic and home observations tend to track each other pretty well. At 6 months, for instance, it’s about a change of three points decrease (in both). But the bad news is that the variability is much greater in home devices,” said Mr. Paynter, noting larger confidence intervals and standard deviation values associated with home spirometry. That could influence future clinical designs that may rely on home spirometry, since a larger confidence interval means reduced power, which could double or even quadruple the number of participants needed to achieve the required power, he said.
But from a clinical standpoint, the ability of home spirometry to consistently detect a change from baseline could be quite valuable to future patient management, according to Dr. Giusti. “It looks like home spirometry will show that kind of a decrease, so that it’s still sensitive to pick up the concern that a patient is getting worse at home,” he said.
That could be useful even after the COVID-19 pandemic passes, as patients continue to embrace home monitoring. Physicians could keep track of patients and keep them focused on their care and treatment through frequent telemedicine visits combined with home spirometry. “I really think home spirometry will keep us more focused on how the patients are doing and make for better outcomes,” said Dr. Giusti.
Mr. Paynter and Dr. Giusti have no relevant financial disclosures.
SOURCE: Alex Paynter et al. NACFC 2020. Poster 643.
Home spirometry has become increasingly used among cystic fibrosis patients during the COVID-19 pandemic, and new research suggests that home devices perform reasonably well. Forced expiratory volume in 1 second (FEV1) values were a bit lower than values seen in clinical spirometry performed in the same patient at a nearby time point, but the procedure reliably picked up decreases in FEV1, potentially helping patients and clinicians spot exacerbations early.
“Home spirometry was sort of a curiosity that was slowly working its way into cystic fibrosis research in 2019, and then all of a sudden in 2020 it became front and center as the only way to continue with clinical monitoring and research in many cases,” Alexander Paynter, MS, a biostatistician at the Cystic Fibrosis Foundation’s Therapeutic Development Network Coordinating Center, said during a talk at the virtual North American Cystic Fibrosis Conference.
To better determine how closely home spirometry matches clinical spirometry, Mr. Paynter and his colleagues analyzed data from the eICE study, which included 267 cystic fibrosis patients aged 14 and over at 14 cystic fibrosis centers. They were randomized to use home spirometry as an early intervention to detect exacerbations, or to continue usual clinic care with visits to the clinic every 3 months. The dataset includes twice-weekly home spirometry values, with a full-year of follow-up data. The researchers compared the home spirometry data to the clinical data closest in time to it. Clinic spirometry data with no corresponding home data within 7 days were discarded.
There was an estimated difference of –2.01 mL between home and clinic tests, with home spirometry producing lower values (95% confidence interval, –3.56 to –0.45). “There is actually a bias in home spirometry as compared to clinic spirometry,” concluded Mr. Paynter.
One explanation for lower values in home spirometry is that users are inexperienced with the device. If that’s true, then agreement should improve over time, but the researchers didn’t see strong evidence of that. Among 44 patients who completed five clinical visits, there was a difference of –2.97 (standard deviation [SD], 10.51) at baseline, –1.66 at 3 months (SD, 13.49), –3.7 at 6 months (SD, 12.44), –0.86 at 9 months (SD, 13.73), and –0.53 at 12 months (SD, 13.35). Though there was improvement over time, “we don’t find a lot of evidence that this bias completely resolves,” said Mr. Paynter.
In fact, a more likely explanation is the presence of coaching by a technician during clinical spirometry, according to Robert J. Giusti, MD, clinical professor of pediatrics and director of the Pediatric Cystic Fibrosis Center at New York University. “When they’re doing it at home, they don’t do it with the same effort, so I think that coaching through telemedicine during the home spirometry would make that difference disappear,” he said when asked to comment on the study.
The researchers found that change-based endpoints were similar between clinic and at-home spirometry. Compared to baseline, the two showed similar declines over time. “The clinic and home observations tend to track each other pretty well. At 6 months, for instance, it’s about a change of three points decrease (in both). But the bad news is that the variability is much greater in home devices,” said Mr. Paynter, noting larger confidence intervals and standard deviation values associated with home spirometry. That could influence future clinical designs that may rely on home spirometry, since a larger confidence interval means reduced power, which could double or even quadruple the number of participants needed to achieve the required power, he said.
But from a clinical standpoint, the ability of home spirometry to consistently detect a change from baseline could be quite valuable to future patient management, according to Dr. Giusti. “It looks like home spirometry will show that kind of a decrease, so that it’s still sensitive to pick up the concern that a patient is getting worse at home,” he said.
That could be useful even after the COVID-19 pandemic passes, as patients continue to embrace home monitoring. Physicians could keep track of patients and keep them focused on their care and treatment through frequent telemedicine visits combined with home spirometry. “I really think home spirometry will keep us more focused on how the patients are doing and make for better outcomes,” said Dr. Giusti.
Mr. Paynter and Dr. Giusti have no relevant financial disclosures.
SOURCE: Alex Paynter et al. NACFC 2020. Poster 643.
Home spirometry has become increasingly used among cystic fibrosis patients during the COVID-19 pandemic, and new research suggests that home devices perform reasonably well. Forced expiratory volume in 1 second (FEV1) values were a bit lower than values seen in clinical spirometry performed in the same patient at a nearby time point, but the procedure reliably picked up decreases in FEV1, potentially helping patients and clinicians spot exacerbations early.
“Home spirometry was sort of a curiosity that was slowly working its way into cystic fibrosis research in 2019, and then all of a sudden in 2020 it became front and center as the only way to continue with clinical monitoring and research in many cases,” Alexander Paynter, MS, a biostatistician at the Cystic Fibrosis Foundation’s Therapeutic Development Network Coordinating Center, said during a talk at the virtual North American Cystic Fibrosis Conference.
To better determine how closely home spirometry matches clinical spirometry, Mr. Paynter and his colleagues analyzed data from the eICE study, which included 267 cystic fibrosis patients aged 14 and over at 14 cystic fibrosis centers. They were randomized to use home spirometry as an early intervention to detect exacerbations, or to continue usual clinic care with visits to the clinic every 3 months. The dataset includes twice-weekly home spirometry values, with a full-year of follow-up data. The researchers compared the home spirometry data to the clinical data closest in time to it. Clinic spirometry data with no corresponding home data within 7 days were discarded.
There was an estimated difference of –2.01 mL between home and clinic tests, with home spirometry producing lower values (95% confidence interval, –3.56 to –0.45). “There is actually a bias in home spirometry as compared to clinic spirometry,” concluded Mr. Paynter.
One explanation for lower values in home spirometry is that users are inexperienced with the device. If that’s true, then agreement should improve over time, but the researchers didn’t see strong evidence of that. Among 44 patients who completed five clinical visits, there was a difference of –2.97 (standard deviation [SD], 10.51) at baseline, –1.66 at 3 months (SD, 13.49), –3.7 at 6 months (SD, 12.44), –0.86 at 9 months (SD, 13.73), and –0.53 at 12 months (SD, 13.35). Though there was improvement over time, “we don’t find a lot of evidence that this bias completely resolves,” said Mr. Paynter.
In fact, a more likely explanation is the presence of coaching by a technician during clinical spirometry, according to Robert J. Giusti, MD, clinical professor of pediatrics and director of the Pediatric Cystic Fibrosis Center at New York University. “When they’re doing it at home, they don’t do it with the same effort, so I think that coaching through telemedicine during the home spirometry would make that difference disappear,” he said when asked to comment on the study.
The researchers found that change-based endpoints were similar between clinic and at-home spirometry. Compared to baseline, the two showed similar declines over time. “The clinic and home observations tend to track each other pretty well. At 6 months, for instance, it’s about a change of three points decrease (in both). But the bad news is that the variability is much greater in home devices,” said Mr. Paynter, noting larger confidence intervals and standard deviation values associated with home spirometry. That could influence future clinical designs that may rely on home spirometry, since a larger confidence interval means reduced power, which could double or even quadruple the number of participants needed to achieve the required power, he said.
But from a clinical standpoint, the ability of home spirometry to consistently detect a change from baseline could be quite valuable to future patient management, according to Dr. Giusti. “It looks like home spirometry will show that kind of a decrease, so that it’s still sensitive to pick up the concern that a patient is getting worse at home,” he said.
That could be useful even after the COVID-19 pandemic passes, as patients continue to embrace home monitoring. Physicians could keep track of patients and keep them focused on their care and treatment through frequent telemedicine visits combined with home spirometry. “I really think home spirometry will keep us more focused on how the patients are doing and make for better outcomes,” said Dr. Giusti.
Mr. Paynter and Dr. Giusti have no relevant financial disclosures.
SOURCE: Alex Paynter et al. NACFC 2020. Poster 643.
FROM NACFC 2020
Cystic fibrosis treatment: Triple combination benefits patients with advanced disease
New CFTR [cystic fibrosis transmembrane conductance regulator] modulator therapies can offer life-altering benefits to some patients with cystic fibrosis, even those with advanced disease.
, according to a multicenter analysis of patients taking elexacaftor, tezacaftor, and ivacaftor.
The study participants had a percent predicted forced expiratory volume in 1 second (ppFEV1) of 40% or below, or other high-risk factors. Researchers compared them to control patients who were genetically ineligible for triple combination therapy.
Previous studies of such patients on individual drugs or previous combinations showed increases in lung function in patients with advanced disease, though the magnitude of improvement varied across regimens. “With this improvement, it’s unclear how CFTR modulators should affect lung transplant referral timing,” Brent Bermingham, MD, said during a presentation of the study at the virtual North American Cystic Fibrosis Conference.
“The rationale for our study was that despite patients with advanced lung disease being excluded from phase III trials (of elexacaftor, tezacaftor, and ivacaftor), they are receiving a therapy with an unknown clinical efficacy and safety profile,” said Dr. Bermingham, a pulmonary and critical care fellow at the Medical University of South Carolina, Charleston.
Lung transplant referral guidelines recommend that physicians initiate discussions about the potential benefit of lung transplant when FEV1 drops below 50% of the predicted value. Patients should be referred for a transplant when the value is below 50% and rapidly declining (>20% decline in the past 12 months), when it drops below 40% with accompanying predictors of shortened survival, or when it drops below 30%. The guidelines were published before approval of triple combination therapy.
The researchers conducted an open-label retrospective analysis of 60 patients started on triple combination therapy between September 2019 and February 2020 at three centers in the Southeast. They compared percent predicted ppFEV1 values prior to initiation of therapy to ppFEV1 values obtained 2-12 weeks after the start of therapy. Patients on therapy were compared with 10 genetically ineligible controls. The two groups were generally similar aside from genetic status, though 100% of the therapy group had pancreatic insufficiency, compared with 90% of controls (P = .013).
The therapeutic group experienced a 7.8% increase in ppFEV1 after starting therapy (P < .001), compared with a 0.5% decrease in controls (P = .65). Before initiation of therapy, 33% of the therapy group met the criteria for initiating a transplant discussion, while 67% had been recommended for transplant. After therapy, 55% met the criteria for discussion, 33% were recommended for transplant, and 12% no longer met the criteria for discussion of transplantation. Fifty percent of controls were in discussion, and this dropped to 40%, while 50% were referred for transplantation, and this increased to 60%. On therapy, transplant referral candidates had an increase of forced vital capacity from 48.9 to 59.16 (P < .001).
Adverse events were rare, with only one discontinuation that occurred following a lung transplant and was not believed to be treatment related.
“Our study had a large number of patients taken from multiple centers, which suggests generalizabilty and real-world experience,” said Dr. Bermingham.
The results are encouraging, said Robert J. Giusti, MD, clinical professor of pediatrics at the New York University and director of the Pediatric Cystic Fibrosis Center.
“We’re all remarking how wonderful patients feel these days. It’s really a disease-altering treatment. But for the high-risk group, whose FEV1 is less than 40%, those are the patients we’re more concerned about because we thought maybe they had too much lung disease, and that they wouldn’t benefit from triple combination. But they seem to be improving, so that’s very reassuring,” said Dr. Giusti, who was not involved in the study.
The study received funding from the Cystic Fibrosis Foundation and Dartmouth College. Dr. Bermingham and Dr. Giusti have no relevant financial disclosures.
SOURCE: Bermingham B et al. NACFC 2020, Abstract 645.
New CFTR [cystic fibrosis transmembrane conductance regulator] modulator therapies can offer life-altering benefits to some patients with cystic fibrosis, even those with advanced disease.
, according to a multicenter analysis of patients taking elexacaftor, tezacaftor, and ivacaftor.
The study participants had a percent predicted forced expiratory volume in 1 second (ppFEV1) of 40% or below, or other high-risk factors. Researchers compared them to control patients who were genetically ineligible for triple combination therapy.
Previous studies of such patients on individual drugs or previous combinations showed increases in lung function in patients with advanced disease, though the magnitude of improvement varied across regimens. “With this improvement, it’s unclear how CFTR modulators should affect lung transplant referral timing,” Brent Bermingham, MD, said during a presentation of the study at the virtual North American Cystic Fibrosis Conference.
“The rationale for our study was that despite patients with advanced lung disease being excluded from phase III trials (of elexacaftor, tezacaftor, and ivacaftor), they are receiving a therapy with an unknown clinical efficacy and safety profile,” said Dr. Bermingham, a pulmonary and critical care fellow at the Medical University of South Carolina, Charleston.
Lung transplant referral guidelines recommend that physicians initiate discussions about the potential benefit of lung transplant when FEV1 drops below 50% of the predicted value. Patients should be referred for a transplant when the value is below 50% and rapidly declining (>20% decline in the past 12 months), when it drops below 40% with accompanying predictors of shortened survival, or when it drops below 30%. The guidelines were published before approval of triple combination therapy.
The researchers conducted an open-label retrospective analysis of 60 patients started on triple combination therapy between September 2019 and February 2020 at three centers in the Southeast. They compared percent predicted ppFEV1 values prior to initiation of therapy to ppFEV1 values obtained 2-12 weeks after the start of therapy. Patients on therapy were compared with 10 genetically ineligible controls. The two groups were generally similar aside from genetic status, though 100% of the therapy group had pancreatic insufficiency, compared with 90% of controls (P = .013).
The therapeutic group experienced a 7.8% increase in ppFEV1 after starting therapy (P < .001), compared with a 0.5% decrease in controls (P = .65). Before initiation of therapy, 33% of the therapy group met the criteria for initiating a transplant discussion, while 67% had been recommended for transplant. After therapy, 55% met the criteria for discussion, 33% were recommended for transplant, and 12% no longer met the criteria for discussion of transplantation. Fifty percent of controls were in discussion, and this dropped to 40%, while 50% were referred for transplantation, and this increased to 60%. On therapy, transplant referral candidates had an increase of forced vital capacity from 48.9 to 59.16 (P < .001).
Adverse events were rare, with only one discontinuation that occurred following a lung transplant and was not believed to be treatment related.
“Our study had a large number of patients taken from multiple centers, which suggests generalizabilty and real-world experience,” said Dr. Bermingham.
The results are encouraging, said Robert J. Giusti, MD, clinical professor of pediatrics at the New York University and director of the Pediatric Cystic Fibrosis Center.
“We’re all remarking how wonderful patients feel these days. It’s really a disease-altering treatment. But for the high-risk group, whose FEV1 is less than 40%, those are the patients we’re more concerned about because we thought maybe they had too much lung disease, and that they wouldn’t benefit from triple combination. But they seem to be improving, so that’s very reassuring,” said Dr. Giusti, who was not involved in the study.
The study received funding from the Cystic Fibrosis Foundation and Dartmouth College. Dr. Bermingham and Dr. Giusti have no relevant financial disclosures.
SOURCE: Bermingham B et al. NACFC 2020, Abstract 645.
New CFTR [cystic fibrosis transmembrane conductance regulator] modulator therapies can offer life-altering benefits to some patients with cystic fibrosis, even those with advanced disease.
, according to a multicenter analysis of patients taking elexacaftor, tezacaftor, and ivacaftor.
The study participants had a percent predicted forced expiratory volume in 1 second (ppFEV1) of 40% or below, or other high-risk factors. Researchers compared them to control patients who were genetically ineligible for triple combination therapy.
Previous studies of such patients on individual drugs or previous combinations showed increases in lung function in patients with advanced disease, though the magnitude of improvement varied across regimens. “With this improvement, it’s unclear how CFTR modulators should affect lung transplant referral timing,” Brent Bermingham, MD, said during a presentation of the study at the virtual North American Cystic Fibrosis Conference.
“The rationale for our study was that despite patients with advanced lung disease being excluded from phase III trials (of elexacaftor, tezacaftor, and ivacaftor), they are receiving a therapy with an unknown clinical efficacy and safety profile,” said Dr. Bermingham, a pulmonary and critical care fellow at the Medical University of South Carolina, Charleston.
Lung transplant referral guidelines recommend that physicians initiate discussions about the potential benefit of lung transplant when FEV1 drops below 50% of the predicted value. Patients should be referred for a transplant when the value is below 50% and rapidly declining (>20% decline in the past 12 months), when it drops below 40% with accompanying predictors of shortened survival, or when it drops below 30%. The guidelines were published before approval of triple combination therapy.
The researchers conducted an open-label retrospective analysis of 60 patients started on triple combination therapy between September 2019 and February 2020 at three centers in the Southeast. They compared percent predicted ppFEV1 values prior to initiation of therapy to ppFEV1 values obtained 2-12 weeks after the start of therapy. Patients on therapy were compared with 10 genetically ineligible controls. The two groups were generally similar aside from genetic status, though 100% of the therapy group had pancreatic insufficiency, compared with 90% of controls (P = .013).
The therapeutic group experienced a 7.8% increase in ppFEV1 after starting therapy (P < .001), compared with a 0.5% decrease in controls (P = .65). Before initiation of therapy, 33% of the therapy group met the criteria for initiating a transplant discussion, while 67% had been recommended for transplant. After therapy, 55% met the criteria for discussion, 33% were recommended for transplant, and 12% no longer met the criteria for discussion of transplantation. Fifty percent of controls were in discussion, and this dropped to 40%, while 50% were referred for transplantation, and this increased to 60%. On therapy, transplant referral candidates had an increase of forced vital capacity from 48.9 to 59.16 (P < .001).
Adverse events were rare, with only one discontinuation that occurred following a lung transplant and was not believed to be treatment related.
“Our study had a large number of patients taken from multiple centers, which suggests generalizabilty and real-world experience,” said Dr. Bermingham.
The results are encouraging, said Robert J. Giusti, MD, clinical professor of pediatrics at the New York University and director of the Pediatric Cystic Fibrosis Center.
“We’re all remarking how wonderful patients feel these days. It’s really a disease-altering treatment. But for the high-risk group, whose FEV1 is less than 40%, those are the patients we’re more concerned about because we thought maybe they had too much lung disease, and that they wouldn’t benefit from triple combination. But they seem to be improving, so that’s very reassuring,” said Dr. Giusti, who was not involved in the study.
The study received funding from the Cystic Fibrosis Foundation and Dartmouth College. Dr. Bermingham and Dr. Giusti have no relevant financial disclosures.
SOURCE: Bermingham B et al. NACFC 2020, Abstract 645.
FROM NACFC 2020
Biometric changes on fitness trackers, smartwatches detect COVID-19
A smartphone app that combines passively collected physiologic data from wearable devices, such as fitness trackers, and self-reported symptoms can discriminate between COVID-19–positive and –negative individuals among those who report symptoms, new data suggest.

After analyzing data from more than 30,000 participants, researchers from the Digital Engagement and Tracking for Early Control and Treatment (DETECT) study concluded that adding individual changes in sensor data improves models based on symptoms alone for differentiating symptomatic persons who are COVID-19 positive and symptomatic persons who are COVID-19 negative.
The combination can potentially identify infection clusters before wider community spread occurs, Giorgio Quer, PhD, and colleagues report in an article published online Oct. 29 in Nature Medicine. DETECT investigators note that marrying participant-reported symptoms with personal sensor data, such as deviation from normal sleep duration and resting heart rate, resulted in an area under the curve (AUC) of 0.80 (interquartile range [IQR], 0.73-0.86) for differentiating between symptomatic individuals who were positive and those who were negative for COVID-19.
“By better characterizing each individual’s unique baseline, you can then identify changes that may indicate that someone has a viral illness,” said Dr. Quer, director of artificial intelligence at Scripps Research Translational Institute in La Jolla, Calif. “In previous research, we found that the proportion of individuals with elevated resting heart rate and sleep duration compared with their normal could significantly improve real-time detection of influenza-like illness rates at the state level,” he said in an interview.
Thus, continuous passively captured data may be a useful adjunct to bricks-and-mortar site testing, which is generally a one-off or infrequent sampling assay and is not always easily accessible, he added. Furthermore, traditional screening with temperature and symptom reporting is inadequate. An elevation in temperature is not as common as frequently believed for people who test positive for COVID-19, Dr. Quer continued. “Early identification via sensor variables of those who are presymptomatic or even asymptomatic would be especially valuable, as people may potentially be infectious during this period, and early detection is the ultimate goal,” Dr. Quer said.
According to his group, adding these physiologic changes from baseline values significantly outperformed detection (P < .01) using a British model described in an earlier study by by Cristina Menni, PhD, and associates. That method, in which symptoms were considered alone, yielded an AUC of 0.71 (IQR, 0.63-0.79).
According to Dr. Quer, one in five Americans currently wear an electronic device. “If we could enroll even a small percentage of these individuals, we’d be able to potentially identify clusters before they have the opportunity to spread,” he said.
DETECT study details
During the period March 15 to June 7, 2020, the study enrolled 30,529 participants from all 50 states. They ranged in age from younger than 35 years (23.1%) to older than 65 years (12.8%); the majority (63.5%) were aged 35-65 years, and 62% were women. Sensor devices in use by the cohort included Fitbit activity trackers (78.4%) and Apple HealthKit (31.2%).
Participants downloaded an app called MyDataHelps, which collects smartwatch and activity tracker information, including self-reported symptoms and diagnostic testing results. The app also monitors changes from baseline in resting heart rate, sleep duration, and physical activity, as measured by steps.
Overall, 3,811 participants reported having at least one symptom of some kind (e.g., fatigue, cough, dyspnea, loss of taste or smell). Of these, 54 reported testing positive for COVID-19, and 279 reported testing negative.
Sleep and activity were significantly different for the positive and negative groups, with an AUC of 0.68 (IQR, 0.57-0.79) for the sleep metric and 0.69 (IQR, 0.61-0.77) for the activity metric, suggesting that these parameters were more affected in COVID-19–positive participants.
When the investigators combined resting heart rate, sleep, and activity into a single metric, predictive performance improved to an AUC of 0.72 (IQR, 0.64-0.80).
The next step, Dr. Quer said, is to include an alert to notify users of possible infection.
Alerting users to possible COVID-19 infection
In a similar study, an alert feature was already incorporated. The study, led by Michael P. Snyder, PhD, director of the Center for Genomics and Personalized Medicine at Stanford (Calif.) University, will soon be published online in Nature Biomedical Engineering. In that study, presymptomatic detection of COVID-19 was achieved in more than 80% of participants using resting heart rate.
“The median is 4 days prior to symptom formation,” Dr. Snyder said in an interview. “We have an alarm system to notify people when their heart rate is elevated. So a positive signal from a smartwatch can be used to follow up by polymerase chain reaction [testing].”
Dr. Snyder said these approaches offer a roadmap to containing widespread infections. “Public health authorities need to be open to these technologies and begin incorporating them into their tracking,” he said. “Right now, people do temperature checks, which are of limited value. Resting heart rate is much better information.”
Although the DETECT researchers have not yet received feedback on their results, they believe public health authorities could recommend the use of such apps. “These are devices that people routinely wear for tracking their fitness and sleep, so it would be relatively easy to use the data for viral illness tracking,” said co–lead author Jennifer Radin, PhD, an epidemiologist at Scripps. “Testing resources are still limited and don’t allow for routine serial testing of individuals who may be asymptomatic or presymptomatic. Wearables can offer a different way to routinely monitor and screen people for changes in their data that may indicate COVID-19.”
The marshaling of data through consumer digital platforms to fight the coronavirus is gaining ground. New York State and New Jersey are already embracing smartphone apps to alert individuals to possible exposure to the virus.
More than 710,000 New Yorkers have downloaded the COVID NY Alert app, launched in October to help protect individuals and communities from COVID-19 by sending alerts without compromising privacy or personal information. “Upon receiving a notification about a potential exposure, users are then able to self-quarantine, get tested, and reduce the potential exposure risk to family, friends, coworkers, and others,” Jonah Bruno, a spokesperson for the New York State Department of Health, said in an interview.
And recently the Mayo Clinic and Safe Health Systems launched a platform to store COVID-19 testing and vaccination data.
Both the Scripps and Stanford platforms are part of a global technologic response to the COVID-19 pandemic. Prospective studies, led by device manufacturers and academic institutions, allow individuals to voluntarily share sensor and clinical data to address the crisis. Similar approaches have been used to track COVID-19 in large populations in Germany via the Corona Data Donation app.
The study by Dr. Quer and colleagues was funded by a grant from the National Center for Advancing Translational Sciences at the National Institutes of Health. One coauthor reported grants from Janssen and personal fees from Otsuka and Livongo outside of the submitted work. The other authors have disclosed no relevant financial relationships. Dr. Snyder has ties to Personalis, Qbio, January, SensOmics, Protos, Mirvie, and Oralome.
A version of this article originally appeared on Medscape.com.
A smartphone app that combines passively collected physiologic data from wearable devices, such as fitness trackers, and self-reported symptoms can discriminate between COVID-19–positive and –negative individuals among those who report symptoms, new data suggest.
After analyzing data from more than 30,000 participants, researchers from the Digital Engagement and Tracking for Early Control and Treatment (DETECT) study concluded that adding individual changes in sensor data improves models based on symptoms alone for differentiating symptomatic persons who are COVID-19 positive and symptomatic persons who are COVID-19 negative.
The combination can potentially identify infection clusters before wider community spread occurs, Giorgio Quer, PhD, and colleagues report in an article published online Oct. 29 in Nature Medicine. DETECT investigators note that marrying participant-reported symptoms with personal sensor data, such as deviation from normal sleep duration and resting heart rate, resulted in an area under the curve (AUC) of 0.80 (interquartile range [IQR], 0.73-0.86) for differentiating between symptomatic individuals who were positive and those who were negative for COVID-19.
“By better characterizing each individual’s unique baseline, you can then identify changes that may indicate that someone has a viral illness,” said Dr. Quer, director of artificial intelligence at Scripps Research Translational Institute in La Jolla, Calif. “In previous research, we found that the proportion of individuals with elevated resting heart rate and sleep duration compared with their normal could significantly improve real-time detection of influenza-like illness rates at the state level,” he said in an interview.
Thus, continuous passively captured data may be a useful adjunct to bricks-and-mortar site testing, which is generally a one-off or infrequent sampling assay and is not always easily accessible, he added. Furthermore, traditional screening with temperature and symptom reporting is inadequate. An elevation in temperature is not as common as frequently believed for people who test positive for COVID-19, Dr. Quer continued. “Early identification via sensor variables of those who are presymptomatic or even asymptomatic would be especially valuable, as people may potentially be infectious during this period, and early detection is the ultimate goal,” Dr. Quer said.
According to his group, adding these physiologic changes from baseline values significantly outperformed detection (P < .01) using a British model described in an earlier study by by Cristina Menni, PhD, and associates. That method, in which symptoms were considered alone, yielded an AUC of 0.71 (IQR, 0.63-0.79).
According to Dr. Quer, one in five Americans currently wear an electronic device. “If we could enroll even a small percentage of these individuals, we’d be able to potentially identify clusters before they have the opportunity to spread,” he said.
DETECT study details
During the period March 15 to June 7, 2020, the study enrolled 30,529 participants from all 50 states. They ranged in age from younger than 35 years (23.1%) to older than 65 years (12.8%); the majority (63.5%) were aged 35-65 years, and 62% were women. Sensor devices in use by the cohort included Fitbit activity trackers (78.4%) and Apple HealthKit (31.2%).
Participants downloaded an app called MyDataHelps, which collects smartwatch and activity tracker information, including self-reported symptoms and diagnostic testing results. The app also monitors changes from baseline in resting heart rate, sleep duration, and physical activity, as measured by steps.
Overall, 3,811 participants reported having at least one symptom of some kind (e.g., fatigue, cough, dyspnea, loss of taste or smell). Of these, 54 reported testing positive for COVID-19, and 279 reported testing negative.
Sleep and activity were significantly different for the positive and negative groups, with an AUC of 0.68 (IQR, 0.57-0.79) for the sleep metric and 0.69 (IQR, 0.61-0.77) for the activity metric, suggesting that these parameters were more affected in COVID-19–positive participants.
When the investigators combined resting heart rate, sleep, and activity into a single metric, predictive performance improved to an AUC of 0.72 (IQR, 0.64-0.80).
The next step, Dr. Quer said, is to include an alert to notify users of possible infection.
Alerting users to possible COVID-19 infection
In a similar study, an alert feature was already incorporated. The study, led by Michael P. Snyder, PhD, director of the Center for Genomics and Personalized Medicine at Stanford (Calif.) University, will soon be published online in Nature Biomedical Engineering. In that study, presymptomatic detection of COVID-19 was achieved in more than 80% of participants using resting heart rate.
“The median is 4 days prior to symptom formation,” Dr. Snyder said in an interview. “We have an alarm system to notify people when their heart rate is elevated. So a positive signal from a smartwatch can be used to follow up by polymerase chain reaction [testing].”
Dr. Snyder said these approaches offer a roadmap to containing widespread infections. “Public health authorities need to be open to these technologies and begin incorporating them into their tracking,” he said. “Right now, people do temperature checks, which are of limited value. Resting heart rate is much better information.”
Although the DETECT researchers have not yet received feedback on their results, they believe public health authorities could recommend the use of such apps. “These are devices that people routinely wear for tracking their fitness and sleep, so it would be relatively easy to use the data for viral illness tracking,” said co–lead author Jennifer Radin, PhD, an epidemiologist at Scripps. “Testing resources are still limited and don’t allow for routine serial testing of individuals who may be asymptomatic or presymptomatic. Wearables can offer a different way to routinely monitor and screen people for changes in their data that may indicate COVID-19.”
The marshaling of data through consumer digital platforms to fight the coronavirus is gaining ground. New York State and New Jersey are already embracing smartphone apps to alert individuals to possible exposure to the virus.
More than 710,000 New Yorkers have downloaded the COVID NY Alert app, launched in October to help protect individuals and communities from COVID-19 by sending alerts without compromising privacy or personal information. “Upon receiving a notification about a potential exposure, users are then able to self-quarantine, get tested, and reduce the potential exposure risk to family, friends, coworkers, and others,” Jonah Bruno, a spokesperson for the New York State Department of Health, said in an interview.
And recently the Mayo Clinic and Safe Health Systems launched a platform to store COVID-19 testing and vaccination data.
Both the Scripps and Stanford platforms are part of a global technologic response to the COVID-19 pandemic. Prospective studies, led by device manufacturers and academic institutions, allow individuals to voluntarily share sensor and clinical data to address the crisis. Similar approaches have been used to track COVID-19 in large populations in Germany via the Corona Data Donation app.
The study by Dr. Quer and colleagues was funded by a grant from the National Center for Advancing Translational Sciences at the National Institutes of Health. One coauthor reported grants from Janssen and personal fees from Otsuka and Livongo outside of the submitted work. The other authors have disclosed no relevant financial relationships. Dr. Snyder has ties to Personalis, Qbio, January, SensOmics, Protos, Mirvie, and Oralome.
A version of this article originally appeared on Medscape.com.
A smartphone app that combines passively collected physiologic data from wearable devices, such as fitness trackers, and self-reported symptoms can discriminate between COVID-19–positive and –negative individuals among those who report symptoms, new data suggest.
After analyzing data from more than 30,000 participants, researchers from the Digital Engagement and Tracking for Early Control and Treatment (DETECT) study concluded that adding individual changes in sensor data improves models based on symptoms alone for differentiating symptomatic persons who are COVID-19 positive and symptomatic persons who are COVID-19 negative.
The combination can potentially identify infection clusters before wider community spread occurs, Giorgio Quer, PhD, and colleagues report in an article published online Oct. 29 in Nature Medicine. DETECT investigators note that marrying participant-reported symptoms with personal sensor data, such as deviation from normal sleep duration and resting heart rate, resulted in an area under the curve (AUC) of 0.80 (interquartile range [IQR], 0.73-0.86) for differentiating between symptomatic individuals who were positive and those who were negative for COVID-19.
“By better characterizing each individual’s unique baseline, you can then identify changes that may indicate that someone has a viral illness,” said Dr. Quer, director of artificial intelligence at Scripps Research Translational Institute in La Jolla, Calif. “In previous research, we found that the proportion of individuals with elevated resting heart rate and sleep duration compared with their normal could significantly improve real-time detection of influenza-like illness rates at the state level,” he said in an interview.
Thus, continuous passively captured data may be a useful adjunct to bricks-and-mortar site testing, which is generally a one-off or infrequent sampling assay and is not always easily accessible, he added. Furthermore, traditional screening with temperature and symptom reporting is inadequate. An elevation in temperature is not as common as frequently believed for people who test positive for COVID-19, Dr. Quer continued. “Early identification via sensor variables of those who are presymptomatic or even asymptomatic would be especially valuable, as people may potentially be infectious during this period, and early detection is the ultimate goal,” Dr. Quer said.
According to his group, adding these physiologic changes from baseline values significantly outperformed detection (P < .01) using a British model described in an earlier study by by Cristina Menni, PhD, and associates. That method, in which symptoms were considered alone, yielded an AUC of 0.71 (IQR, 0.63-0.79).
According to Dr. Quer, one in five Americans currently wear an electronic device. “If we could enroll even a small percentage of these individuals, we’d be able to potentially identify clusters before they have the opportunity to spread,” he said.
DETECT study details
During the period March 15 to June 7, 2020, the study enrolled 30,529 participants from all 50 states. They ranged in age from younger than 35 years (23.1%) to older than 65 years (12.8%); the majority (63.5%) were aged 35-65 years, and 62% were women. Sensor devices in use by the cohort included Fitbit activity trackers (78.4%) and Apple HealthKit (31.2%).
Participants downloaded an app called MyDataHelps, which collects smartwatch and activity tracker information, including self-reported symptoms and diagnostic testing results. The app also monitors changes from baseline in resting heart rate, sleep duration, and physical activity, as measured by steps.
Overall, 3,811 participants reported having at least one symptom of some kind (e.g., fatigue, cough, dyspnea, loss of taste or smell). Of these, 54 reported testing positive for COVID-19, and 279 reported testing negative.
Sleep and activity were significantly different for the positive and negative groups, with an AUC of 0.68 (IQR, 0.57-0.79) for the sleep metric and 0.69 (IQR, 0.61-0.77) for the activity metric, suggesting that these parameters were more affected in COVID-19–positive participants.
When the investigators combined resting heart rate, sleep, and activity into a single metric, predictive performance improved to an AUC of 0.72 (IQR, 0.64-0.80).
The next step, Dr. Quer said, is to include an alert to notify users of possible infection.
Alerting users to possible COVID-19 infection
In a similar study, an alert feature was already incorporated. The study, led by Michael P. Snyder, PhD, director of the Center for Genomics and Personalized Medicine at Stanford (Calif.) University, will soon be published online in Nature Biomedical Engineering. In that study, presymptomatic detection of COVID-19 was achieved in more than 80% of participants using resting heart rate.
“The median is 4 days prior to symptom formation,” Dr. Snyder said in an interview. “We have an alarm system to notify people when their heart rate is elevated. So a positive signal from a smartwatch can be used to follow up by polymerase chain reaction [testing].”
Dr. Snyder said these approaches offer a roadmap to containing widespread infections. “Public health authorities need to be open to these technologies and begin incorporating them into their tracking,” he said. “Right now, people do temperature checks, which are of limited value. Resting heart rate is much better information.”
Although the DETECT researchers have not yet received feedback on their results, they believe public health authorities could recommend the use of such apps. “These are devices that people routinely wear for tracking their fitness and sleep, so it would be relatively easy to use the data for viral illness tracking,” said co–lead author Jennifer Radin, PhD, an epidemiologist at Scripps. “Testing resources are still limited and don’t allow for routine serial testing of individuals who may be asymptomatic or presymptomatic. Wearables can offer a different way to routinely monitor and screen people for changes in their data that may indicate COVID-19.”
The marshaling of data through consumer digital platforms to fight the coronavirus is gaining ground. New York State and New Jersey are already embracing smartphone apps to alert individuals to possible exposure to the virus.
More than 710,000 New Yorkers have downloaded the COVID NY Alert app, launched in October to help protect individuals and communities from COVID-19 by sending alerts without compromising privacy or personal information. “Upon receiving a notification about a potential exposure, users are then able to self-quarantine, get tested, and reduce the potential exposure risk to family, friends, coworkers, and others,” Jonah Bruno, a spokesperson for the New York State Department of Health, said in an interview.
And recently the Mayo Clinic and Safe Health Systems launched a platform to store COVID-19 testing and vaccination data.
Both the Scripps and Stanford platforms are part of a global technologic response to the COVID-19 pandemic. Prospective studies, led by device manufacturers and academic institutions, allow individuals to voluntarily share sensor and clinical data to address the crisis. Similar approaches have been used to track COVID-19 in large populations in Germany via the Corona Data Donation app.
The study by Dr. Quer and colleagues was funded by a grant from the National Center for Advancing Translational Sciences at the National Institutes of Health. One coauthor reported grants from Janssen and personal fees from Otsuka and Livongo outside of the submitted work. The other authors have disclosed no relevant financial relationships. Dr. Snyder has ties to Personalis, Qbio, January, SensOmics, Protos, Mirvie, and Oralome.
A version of this article originally appeared on Medscape.com.
