User login
Linearly Curved, Blackish Macule on the Wrist
Linear Basal Cell Carcinoma
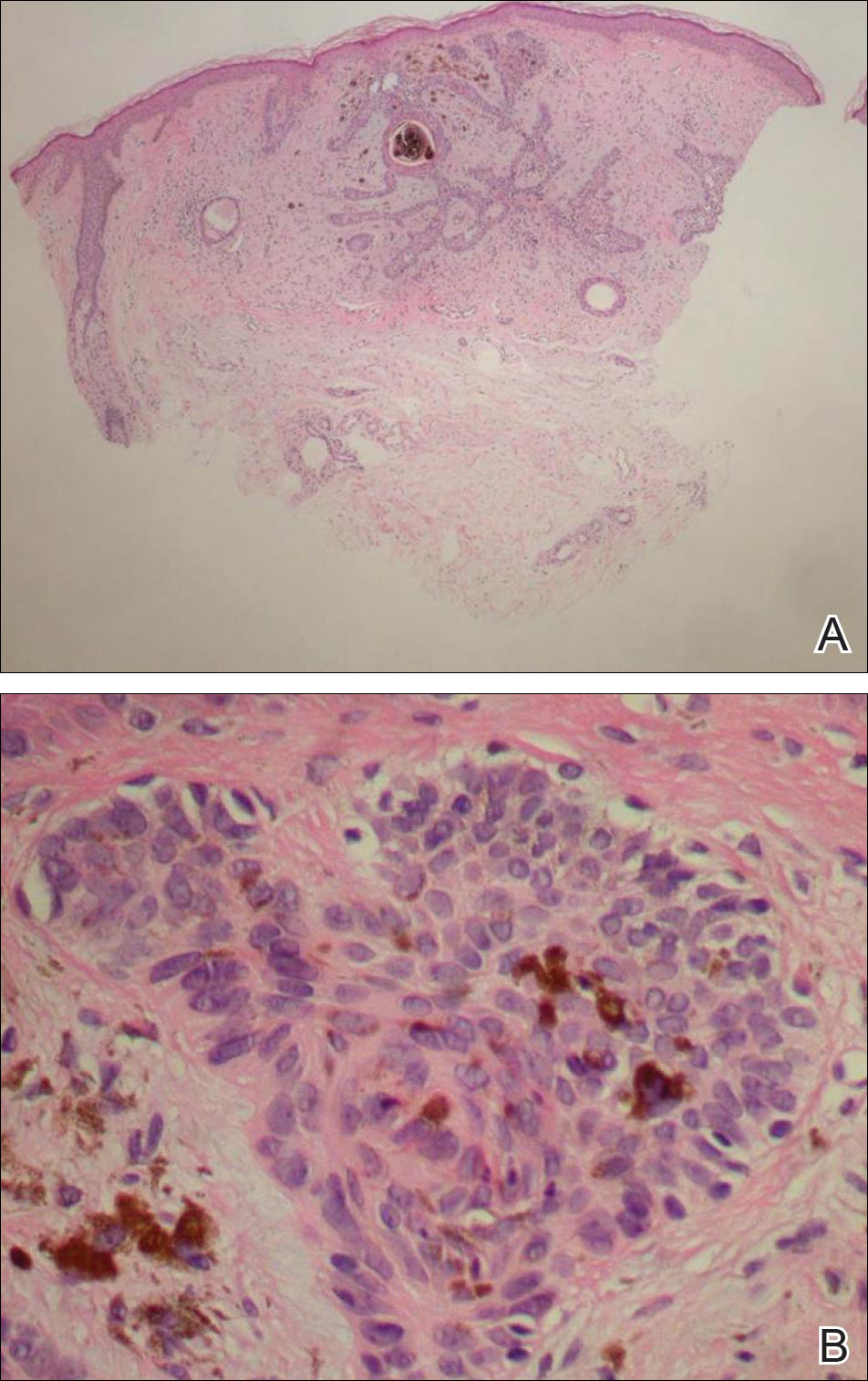
On examination, the lesion was suspected to be a nevocellular nevus, foreign body granuloma, or venous lake; however, a skin biopsy specimen from the lesion on the left wrist revealed a tumor mass of basaloid cells, peripheral palisading arrangement, and scattered pigment granules (Figure 1). Tumor cells were negative for S-100 protein staining. These findings were consistent with a diagnosis of linear basal cell carcinoma (BCC). The lesion was removed by simple excision with primary closure of the wound. The surgical margins were free of tumor cells. The lesion had not recurred at 6-month follow-up. The patient was subsequently lost to follow-up.
Basal cell carcinoma presents with diverse clinical features, and several morphologic and histologic variants have been reported.1 Linear BCC was described as a distinct clinical entity in 1985 by Lewis2 in a 73-year-old man with a 20-mm linear pigmented lesion on the left cheek. Linear BCC often is not recognized or categorized as such by clinicians, as some may think that linear BCC is not a distinct entity but rather is one of the diverse clinical features of BCC.3 Linear BCC is believed to have specific clinical and histologic features and can be regarded as a distinct entity.4 Mavrikakis et al5 objectively defined linear BCC as a lesion that appeared to extend preferentially in one direction, resulting in a lesion with relatively straight borders and a length much greater than the width (3:1 ratio). Our patient presented with a linearly curved lesion, which is a rare feature of BCC.
Linear BCC occurs in equal proportions in men and women aged 40 to 87 years. More than 92% of reported patients were older than 60 years.6 The most common site for linear BCC is the periocular area, with the majority of lesions occurring on the cheek or lower eyelid. The second most common site is the neck, followed by the trunk, lower face, and inguinal skin fold.3,5
The mechanism of linearity has been speculated. The majority of the reported cases of linear BCC have no history of trauma.7 However, focal trauma has been assumed to be a risk factor for the development of linear BCC, so the possibility that the Köbner phenomenon may be related to its linear pattern has been proposed.8 The Köbner phenomenon can be implicated in our case, as there was a history of surgery, which resulted in a scar.
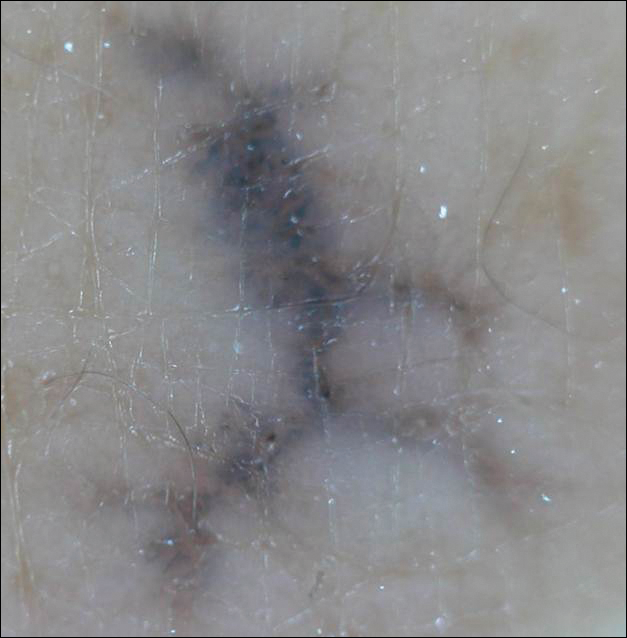
Menzies9 described dermoscopic features of pigmented BCC and stated that the diagnosis of pigmented BCC required the presence of 1 or more of the following 6 positive features: large blue-gray ovoid nests; multiple blue-gray globules; maple leaf–like areas; spoke wheel areas; ulceration; and arborizing treelike vessels. In our case, there were multiple blue-gray globules and a streak that resembled ginseng (Figure 2).
Linear BCC is an uncommon morphological variant that requires clinical recognition. Our case was unique because of the ginsenglike streak on dermoscopy and possible association with a prior trauma.
- Sexton M, Jones DB, Maloney ME. Histologic pattern analysis of basal cell carcinoma. study of a series of 1,039 consecutive neoplasms. J Am Acad Dermatol. 1990;23(6, pt 1):1118-1126.
- Lewis JE. Linear basal cell epithelioma. Int J Dermatol. 1985;24:124-125.
- Mavrikakis I, Malhotra R, Selva D, et al. Linear basal cell carcinoma: a distinct clinical entity. J Plast Reconstr Aesthet Surg. 2006;59:419-423.
- Jellouli A, Triki S, Zghal M, et al. Linear basal cell carcinoma. Actas Dermosifiliogr. 2010;101:648-650.
- Mavrikakis I, Malhotra R, Barlow R, et al. Linear basal cell carcinoma: a distinct clinical entity in the periocular region [published online January 10, 2006]. Ophthalmology. 2006;113:338-342.
- Lim KK, Randle HW, Roenigk RK, et al. Linear basal cell carcinoma: report of seventeen cases and review of the presentation and treatment. Dermatol Surg. 1999;25:63-67.
- Iga N, Sakurai K, Fujii H, et al. Linear basal cell carcinoma at the external genitalia. J Dermatol. 2014;41:275-276.
- Peschen M, Lo JS, Snow SN, et al. Linear basal cell carcinoma. Cutis. 1993;51:287-289.
- Menzies SW. Dermoscopy of pigmented basal cell carcinoma. Clin Dermatol. 2002;20:268-269.
Linear Basal Cell Carcinoma
On examination, the lesion was suspected to be a nevocellular nevus, foreign body granuloma, or venous lake; however, a skin biopsy specimen from the lesion on the left wrist revealed a tumor mass of basaloid cells, peripheral palisading arrangement, and scattered pigment granules (Figure 1). Tumor cells were negative for S-100 protein staining. These findings were consistent with a diagnosis of linear basal cell carcinoma (BCC). The lesion was removed by simple excision with primary closure of the wound. The surgical margins were free of tumor cells. The lesion had not recurred at 6-month follow-up. The patient was subsequently lost to follow-up.
Basal cell carcinoma presents with diverse clinical features, and several morphologic and histologic variants have been reported.1 Linear BCC was described as a distinct clinical entity in 1985 by Lewis2 in a 73-year-old man with a 20-mm linear pigmented lesion on the left cheek. Linear BCC often is not recognized or categorized as such by clinicians, as some may think that linear BCC is not a distinct entity but rather is one of the diverse clinical features of BCC.3 Linear BCC is believed to have specific clinical and histologic features and can be regarded as a distinct entity.4 Mavrikakis et al5 objectively defined linear BCC as a lesion that appeared to extend preferentially in one direction, resulting in a lesion with relatively straight borders and a length much greater than the width (3:1 ratio). Our patient presented with a linearly curved lesion, which is a rare feature of BCC.
Linear BCC occurs in equal proportions in men and women aged 40 to 87 years. More than 92% of reported patients were older than 60 years.6 The most common site for linear BCC is the periocular area, with the majority of lesions occurring on the cheek or lower eyelid. The second most common site is the neck, followed by the trunk, lower face, and inguinal skin fold.3,5
The mechanism of linearity has been speculated. The majority of the reported cases of linear BCC have no history of trauma.7 However, focal trauma has been assumed to be a risk factor for the development of linear BCC, so the possibility that the Köbner phenomenon may be related to its linear pattern has been proposed.8 The Köbner phenomenon can be implicated in our case, as there was a history of surgery, which resulted in a scar.
Menzies9 described dermoscopic features of pigmented BCC and stated that the diagnosis of pigmented BCC required the presence of 1 or more of the following 6 positive features: large blue-gray ovoid nests; multiple blue-gray globules; maple leaf–like areas; spoke wheel areas; ulceration; and arborizing treelike vessels. In our case, there were multiple blue-gray globules and a streak that resembled ginseng (Figure 2).
Linear BCC is an uncommon morphological variant that requires clinical recognition. Our case was unique because of the ginsenglike streak on dermoscopy and possible association with a prior trauma.
Linear Basal Cell Carcinoma
On examination, the lesion was suspected to be a nevocellular nevus, foreign body granuloma, or venous lake; however, a skin biopsy specimen from the lesion on the left wrist revealed a tumor mass of basaloid cells, peripheral palisading arrangement, and scattered pigment granules (Figure 1). Tumor cells were negative for S-100 protein staining. These findings were consistent with a diagnosis of linear basal cell carcinoma (BCC). The lesion was removed by simple excision with primary closure of the wound. The surgical margins were free of tumor cells. The lesion had not recurred at 6-month follow-up. The patient was subsequently lost to follow-up.
Basal cell carcinoma presents with diverse clinical features, and several morphologic and histologic variants have been reported.1 Linear BCC was described as a distinct clinical entity in 1985 by Lewis2 in a 73-year-old man with a 20-mm linear pigmented lesion on the left cheek. Linear BCC often is not recognized or categorized as such by clinicians, as some may think that linear BCC is not a distinct entity but rather is one of the diverse clinical features of BCC.3 Linear BCC is believed to have specific clinical and histologic features and can be regarded as a distinct entity.4 Mavrikakis et al5 objectively defined linear BCC as a lesion that appeared to extend preferentially in one direction, resulting in a lesion with relatively straight borders and a length much greater than the width (3:1 ratio). Our patient presented with a linearly curved lesion, which is a rare feature of BCC.
Linear BCC occurs in equal proportions in men and women aged 40 to 87 years. More than 92% of reported patients were older than 60 years.6 The most common site for linear BCC is the periocular area, with the majority of lesions occurring on the cheek or lower eyelid. The second most common site is the neck, followed by the trunk, lower face, and inguinal skin fold.3,5
The mechanism of linearity has been speculated. The majority of the reported cases of linear BCC have no history of trauma.7 However, focal trauma has been assumed to be a risk factor for the development of linear BCC, so the possibility that the Köbner phenomenon may be related to its linear pattern has been proposed.8 The Köbner phenomenon can be implicated in our case, as there was a history of surgery, which resulted in a scar.
Menzies9 described dermoscopic features of pigmented BCC and stated that the diagnosis of pigmented BCC required the presence of 1 or more of the following 6 positive features: large blue-gray ovoid nests; multiple blue-gray globules; maple leaf–like areas; spoke wheel areas; ulceration; and arborizing treelike vessels. In our case, there were multiple blue-gray globules and a streak that resembled ginseng (Figure 2).
Linear BCC is an uncommon morphological variant that requires clinical recognition. Our case was unique because of the ginsenglike streak on dermoscopy and possible association with a prior trauma.
- Sexton M, Jones DB, Maloney ME. Histologic pattern analysis of basal cell carcinoma. study of a series of 1,039 consecutive neoplasms. J Am Acad Dermatol. 1990;23(6, pt 1):1118-1126.
- Lewis JE. Linear basal cell epithelioma. Int J Dermatol. 1985;24:124-125.
- Mavrikakis I, Malhotra R, Selva D, et al. Linear basal cell carcinoma: a distinct clinical entity. J Plast Reconstr Aesthet Surg. 2006;59:419-423.
- Jellouli A, Triki S, Zghal M, et al. Linear basal cell carcinoma. Actas Dermosifiliogr. 2010;101:648-650.
- Mavrikakis I, Malhotra R, Barlow R, et al. Linear basal cell carcinoma: a distinct clinical entity in the periocular region [published online January 10, 2006]. Ophthalmology. 2006;113:338-342.
- Lim KK, Randle HW, Roenigk RK, et al. Linear basal cell carcinoma: report of seventeen cases and review of the presentation and treatment. Dermatol Surg. 1999;25:63-67.
- Iga N, Sakurai K, Fujii H, et al. Linear basal cell carcinoma at the external genitalia. J Dermatol. 2014;41:275-276.
- Peschen M, Lo JS, Snow SN, et al. Linear basal cell carcinoma. Cutis. 1993;51:287-289.
- Menzies SW. Dermoscopy of pigmented basal cell carcinoma. Clin Dermatol. 2002;20:268-269.
- Sexton M, Jones DB, Maloney ME. Histologic pattern analysis of basal cell carcinoma. study of a series of 1,039 consecutive neoplasms. J Am Acad Dermatol. 1990;23(6, pt 1):1118-1126.
- Lewis JE. Linear basal cell epithelioma. Int J Dermatol. 1985;24:124-125.
- Mavrikakis I, Malhotra R, Selva D, et al. Linear basal cell carcinoma: a distinct clinical entity. J Plast Reconstr Aesthet Surg. 2006;59:419-423.
- Jellouli A, Triki S, Zghal M, et al. Linear basal cell carcinoma. Actas Dermosifiliogr. 2010;101:648-650.
- Mavrikakis I, Malhotra R, Barlow R, et al. Linear basal cell carcinoma: a distinct clinical entity in the periocular region [published online January 10, 2006]. Ophthalmology. 2006;113:338-342.
- Lim KK, Randle HW, Roenigk RK, et al. Linear basal cell carcinoma: report of seventeen cases and review of the presentation and treatment. Dermatol Surg. 1999;25:63-67.
- Iga N, Sakurai K, Fujii H, et al. Linear basal cell carcinoma at the external genitalia. J Dermatol. 2014;41:275-276.
- Peschen M, Lo JS, Snow SN, et al. Linear basal cell carcinoma. Cutis. 1993;51:287-289.
- Menzies SW. Dermoscopy of pigmented basal cell carcinoma. Clin Dermatol. 2002;20:268-269.

Spontaneous Repigmentation of Silvery Hair in an Infant With Congenital Hydrops Fetalis and Hypoproteinemia
Silvery hair is characteristic of 3 rare autosomal-recessive disorders—Chédiak-Higashi syndrome (CHS), Elejalde syndrome (ES), and Griscelli syndrome (GS)—which are associated with mutations in various genes that encode several proteins involved in the intracellular processing and movement of melanosomes. We report the case of a 2-month-old male infant with transient silvery hair and generalized hypopigmentation of the skin and eyes who did not have any genetic mutations associated with the classic syndromes that usually are characterized by transient silvery hair.
Case Report
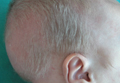
A 2-month-old male infant presented to the dermatology department for evaluation of silvery hair with generalized hypopigmentation of the skin and eyes (Figure 1) that had developed at 1 month of age. His parents were healthy, nonconsanguineous, and reported no family history of silvery hair. The patient was delivered by cesarean section at 35 weeks’ gestation. His medical history was remarkable for congenital hydrops fetalis with pleuropericardial effusion, ascites, soft-tissue edema, and hydrocele with no signs of any congenital infection. Both the patient and his mother were O Rh +.
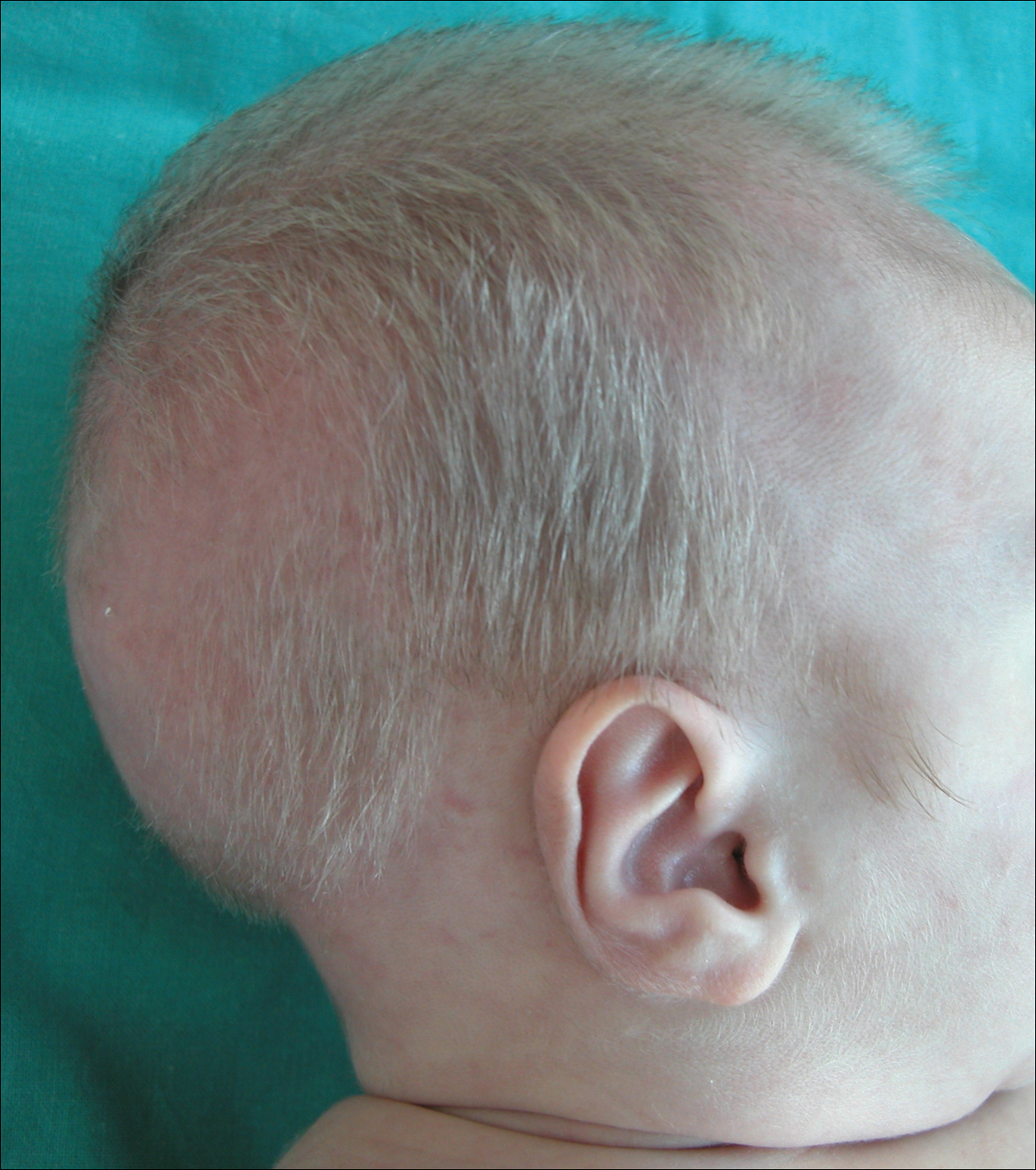
Several studies were performed following delivery. A direct Coombs test was negative. Blood studies revealed hypothyroidism and hypoalbuminemia secondary to protein loss associated with fetal hydrops. Cerebral, abdominal, and renal ultrasound; echocardiogram; thoracic and abdominal computed tomography; and cerebral magnetic resonance imaging revealed no abnormalities.
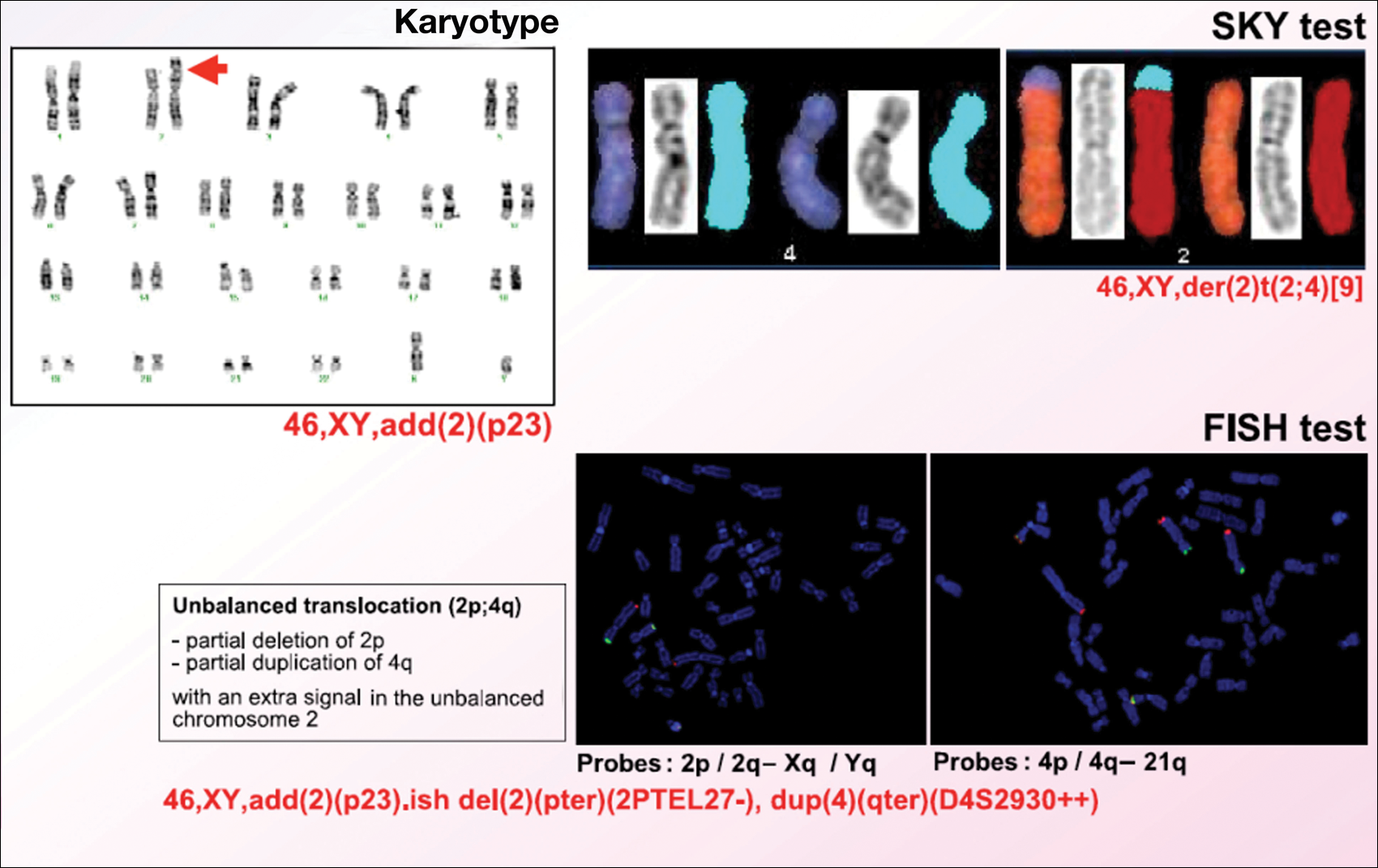
Karyotype results showed 46,XY,add(2)(p23), and subsequent spectral karyotyping and fluorescence in situ hybridization tests identified a chromosomal abnormality (46,XY,add[2][p23].ish del[2][pter][2PTEL27‒], dup[4][qter][D4S2930++])(Figure 2). Parental karyotypes were normal.
After birth, the infant was admitted to the neonatal intensive care unit for 50 days and received pleural and peritoneal drainages, mechanical ventilation, vasoactive drugs, parenteral nutrition with resolution of the hypoalbuminemia, levothyroxine, and intravenous antibiotics for central venous catheter infection. No drugs known to be associated with hypopigmentation of the hair, skin, or eyes were administered.
Two weeks after discharge from the neonatal intensive care unit, the patient was referred to our department. Physical examination revealed silvery hair on the scalp, eyebrows, and eyelashes, along with generalized hypopigmentation of the skin and eyes. Abdominal, cardiovascular, respiratory, and neurologic examination revealed no abnormalities, and no hepatosplenomegaly, lymphadenopathy, nystagmus, or strabismus was noted.
Light microscopy of the hair revealed small and regular aggregates of melanin along the hair shaft, predominantly in the medulla (Figure 3). Light microscopy of a skin biopsy specimen showed normal pigmentation in the melanocytes and no giant melanosomes. The melanocyte count was within reference range. A peripheral blood smear showed no giant granules in the granulocytes. No treatment was administered and the patient was followed closely every month. When the patient returned for follow-up at 9 months of age, physical examination revealed brown hair on the head, eyebrows, and eyelashes, as well as normal pigmentation of the skin and eyes (Figure 4). Thyroid function was normal and no recurrent infections of any type were noted. At follow-up at the age of 4 years, he showed normal neurological and psychological development with brown hair, no recurrent infections, and normal thyroid function. Given that CHS, ES, and GS had been ruled out, the clinical presentation and the genetic mutation detected may indicate that this case represents a new entity characterized by transient silvery hair.


Comment
Silvery hair is a known feature of CHS, ES, and GS (Table). The characteristic hypopigmentation associated with these autosomal-recessive disorders is the result of impaired melanosome transport leading to failed transfer of melanin to keratinocytes. These disorders differ from oculocutaneous albinism in that melanin synthesis is unaffected.
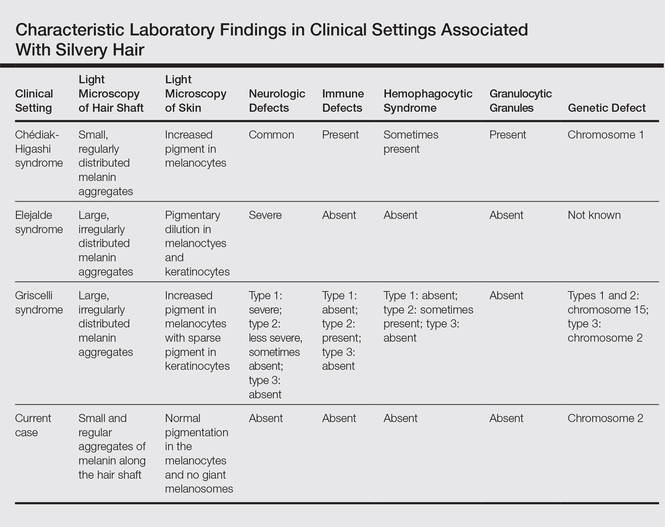
Chédiak-Higashi syndrome is characterized by generalized hypopigmentation of the skin and eyes, silvery hair, neurologic and immune dysfunction, lymphoproliferative disorders, and large granules in granulocytes and other cell types.1-3 A common complication of CHS is hemophagocytic lymphohistiocytosis, which is characterized by fever, jaundice, lymphadenopathy, hepatosplenomegaly, and pancytopenia.4 Pigmentary dilution of the irises also may be present, along with photophobia, strabismus, nystagmus, and impaired visual acuity. Chédiak-Higashi syndrome is the result of a genetic defect in the lysosomal trafficking regulator gene, also known as CHS1 (located on chromosome 1q42.1‒q42.2).5 Melanin in the hair shaft is distributed uniformly in multiple small aggregates. Light microscopy of the skin typically shows giant melanosomes in melanocytes and aberrant keratinocyte maturation.
Elejalde syndrome is characterized by silvery hair (eyelashes and eyebrows), neurologic defects, and normal immunologic function.6,7 The underlying molecular basis remains unknown. It appears related to or allelic to GS type 1 and thus associated with mutations in MYO5A (myosin VA); however, the gene mutation responsible has yet to be defined.8 Light microscopy of the hair shaft usually shows an irregular distribution of large melanin aggregates, primarily in the medulla.9,10 Skin biopsy generally shows irregular distribution and irregular size of melanin granules in the basal layer.11 Leukocytes usually show no abnormal cytoplasmic granules. Ocular involvement is common and may present as nystagmus, diplopia, hypopigmented retinas, and/or papilledema.
In GS, hair microscopy generally reveals large aggregates of melanin pigment distributed irregularly along the hair shaft. Granulocytes typically show no giant granules. Light microscopy of the skin usually shows increased pigment in melanocytes with sparse pigment in keratinocytes. Griscelli syndrome is classified into 3 types.12 In GS type 1, patients have silvery gray hair, light-colored skin, severe neurologic defects,13 and normal immune status. This variant is caused by a mutation in the MYO5A gene located on chromosome 15q21. In GS type 2, patients have silvery gray hair, pyogenic infections, an accelerated phase of hemophagocytic lymphohistiocytosis, and variable neurologic defects in the absence of primary neurologic disease.14,15 This variant is caused by a mutation in the RAB27A (member RAS oncogene family) gene located on chromosome 15q21. In GS type 3, patients exhibit generalized hypopigmentation of the skin and hair with no abnormalities of the nervous or immune systems. There are 2 different mutations associated with GS type 3: the first is located on chromosome 2q37.3, causing a mutation in MLPH (melanophilin), and the second is caused by an F-exon deletion in the MYO5A gene.14
Our patient had silvery hair, generalized hypopigmentation of the skin and eyes, and normal central nervous system function with no other ocular involvement and no evidence of recurrent infections of any kind. Light microscopy showed small and regular melanin pigment aggregates in the hair shaft, which differs from the irregular pigment aggregates in GS and ES.
The regular melanin pigment aggregates observed along the hair shaft were consistent with CHS, but other manifestations of this syndrome were absent: ocular, neurologic, hematologic, and immunologic abnormalities with presence of giant intracytoplasmic granules in leukocytes, and giant melanosomes in melanocytes. In our patient, the absence of these features along with the spontaneous repigmentation of the silvery hair, improvement of thyroid function, reversal of hypoalbuminemia, and the chromosomopathy detected make a diagnosis of CHS highly improbable.
We concluded that the silvery hair noted in our patient resulted from the 46,XY,add(2)(p23) chromosomal abnormality. This mutation could affect some of the genes that control the trafficking of melanosomes or could induce hypothyroidism and hypoproteinemia associated with congenital hydrops fetalis (Figure 5).
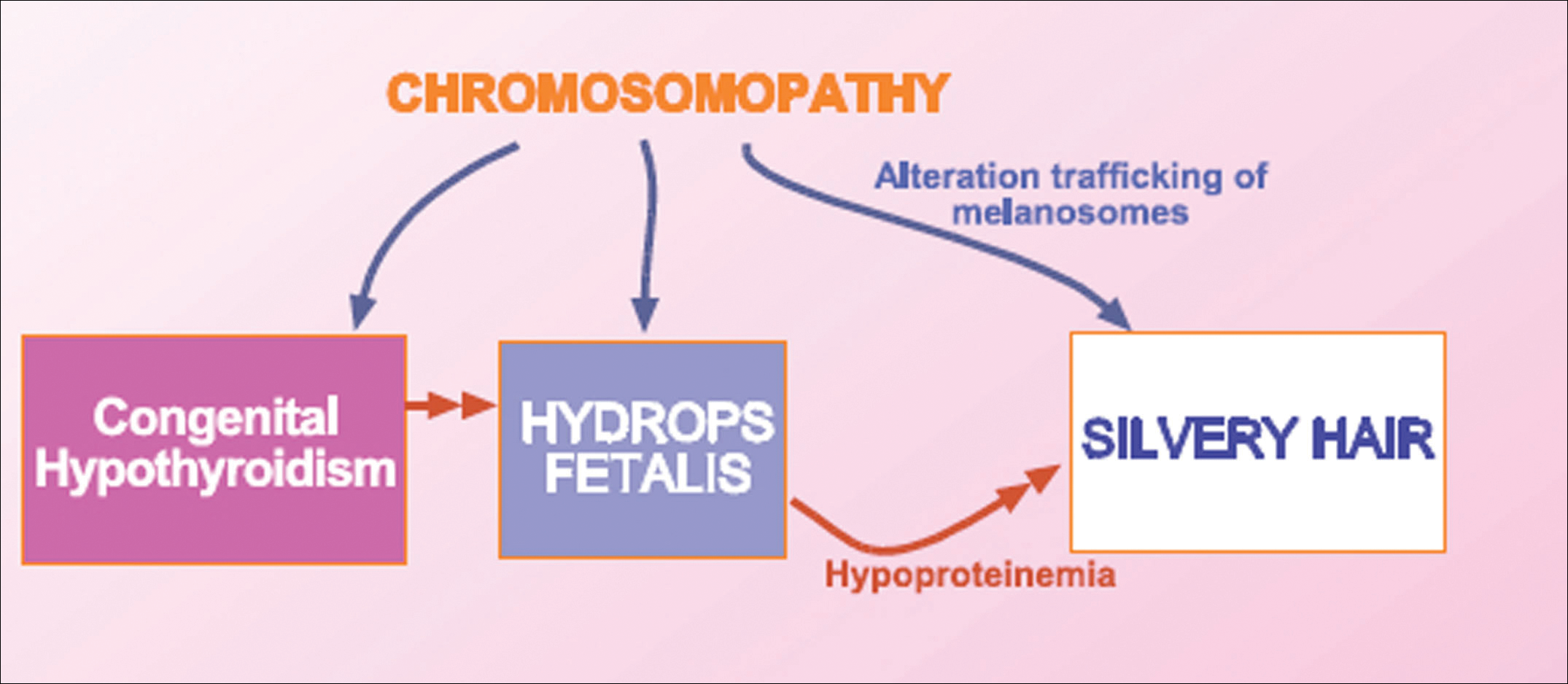
Hydrops fetalis is a potentially fatal condition characterized by severe edema (swelling) in a fetus or neonate. There are 2 types of hydrops fetalis: immune and nonimmune. Immune hydrops fetalis may develop in an Rh+ fetus with an Rh– mother, as the mother’s immune cells begin to break down the red blood cells of the fetus, resulting in anemia in the fetus with subsequent fetal heart failure, leading to an accumulation of large amounts of fluid in the tissues and organs. Nonimmune hydrops fetalis can occur secondary to diseases that interfere with the fetus’s ability to manage fluid (eg, severe anemia; congenital infections; urinary, lymphatic, heart, or thoracic defects; inborn errors of metabolism; chromosomal abnormalities). Case studies have suggested that congenital hypothyroidism could be a cause of nonimmune hydrops fetalis.16,17 Thyroid hormone deficiency reduces stimulation of adrenergic receptors in the lymphatic system and lungs, thereby decreasing lymph flow and protein efflux to the lymphatic system and decreasing clearance of liquid from the lungs. The final result is lymph vessel engorgement and subsequent leakage of lymphatic fluid to pleural spaces, causing hydrops fetalis and chylothorax.
The 46,XY,add(2)(p23) chromosomal abnormality has not been commonly associated with hypothyroidism and hydrops fetalis. The silvery hair in our patient was transient and spontaneously repigmented to brown over the course of follow-up in conjunction with improved physiologic changes. We concluded that the silvery hair in our patient was induced by his hypoproteinemic status secondary to hydrops fetalis and hypothyroidism.
Conclusion
In addition to CHS, ES, and GS, the differential diagnosis for silvery hair with abnormal skin pigmentation in children should include 46,XY,add(2)(p23) mutation, as was detected in our patient. Evaluation should include light microscopy of the hair shaft, skin biopsy, assessment of immune function, peripheral blood smear, and neurologic and eye examinations.
- White JG. The Chédiak-Higashi syndrome: a possible lysosomal disease. Blood. 1966;28:143-156.
- Introne W, Boissy RE, Gahl WA. Clinical, molecular, and cell biological aspects of Chédiak-Higashi syndrome. Mol Genet Metab. 1999;68:283-303.
- Kaplan J, De Domenico I, Ward DM. Chédiak-Higashi syndrome. Curr Opin Hematol. 2008;15:22-29.
- Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis [published online December 7, 2006]. Eur J Pediatr. 2007;166:95-109.
- Morrone K, Wang Y, Huizing M, et al. Two novel mutations identified in an African-American child with Chédiak-Higashi syndrome [published online March 24, 2010]. Case Report Med. 2010;2010:967535.
- Ivanovich J, Mallory S, Storer T, et al. 12-year-old male with Elejalde syndrome (neuroectodermal melanolysosomal disease). Am J Med Genet. 2001;98:313-316.
- Cahali JB, Fernandez SA, Oliveira ZN, et al. Elejalde syndrome: report of a case and review of the literature. Pediatr Dermatol. 2004;21:479-482.
- Bahadoran P, Ortonne JP, Ballotti R, et al. Comment on Elejalde syndrome and relationship with Griscelli syndrome. Am J Med Genet. 2003;116:408-409.
- Duran-McKinster C, Rodriguez-Jurado R, Ridaura C, et al. Elejalde syndrome—a melanolysosomal neurocutaneous syndrome: clinical and morphological findings in 7 patients. Arch Dermatol. 1999;135:182-186.
- Happle R. Neurocutaneous diseases. In: Freedberg IM, Eisen AZ, Wolff K, et al, eds. Dermatology in General Medicine. 5th ed. New York, NY: McGraw-Hill; 1999:2131-2148.
- Sanal O, Yel L, Kucukali T, et al. An allelic variant of Griscelli disease: presentation with severe hypotonia, mental-motor retardation, and hypopigmentation consistent with Elejalde syndrome (neuroectodermal melanolysosomal disorder). J Neurol. 2000;247:570-572.
- Malhotra AK, Bhaskar G, Nanda M, et al. Griscelli syndrome. J Am Acad Dermatol. 2006;55:337-340.
- Al-Idrissi E, ElGhazali G, Alzahrani M, et al. Premature birth, respiratory distress, intracerebral hemorrhage, and silvery-gray hair: differential diagnosis of the 3 types of Griscelli syndrome. J Pediatr Hematol Oncol. 2010;32:494-496.
- Ménasché G, Ho CH, Sanal O, et al. Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1). J Clin Invest. 2003;112:450-456.
- Griscelli C, Durandy A, Guy-Grand D, et al. A syndrome associating partial albinism and immunodeficiency. Am J Med. 1978;65:691-702.
- Narchi H. Congenital hypothyroidism and nonimmune hydrops fetalis: associated? Pediatrics. 1999;104:1416-1417.
- Kessel I, Makhoul IR, Sujov P. Congenital hypothyroidism and nonimmune hydrops fetalis: associated? Pediatrics. 1999;103:E9.
Silvery hair is characteristic of 3 rare autosomal-recessive disorders—Chédiak-Higashi syndrome (CHS), Elejalde syndrome (ES), and Griscelli syndrome (GS)—which are associated with mutations in various genes that encode several proteins involved in the intracellular processing and movement of melanosomes. We report the case of a 2-month-old male infant with transient silvery hair and generalized hypopigmentation of the skin and eyes who did not have any genetic mutations associated with the classic syndromes that usually are characterized by transient silvery hair.
Case Report
A 2-month-old male infant presented to the dermatology department for evaluation of silvery hair with generalized hypopigmentation of the skin and eyes (Figure 1) that had developed at 1 month of age. His parents were healthy, nonconsanguineous, and reported no family history of silvery hair. The patient was delivered by cesarean section at 35 weeks’ gestation. His medical history was remarkable for congenital hydrops fetalis with pleuropericardial effusion, ascites, soft-tissue edema, and hydrocele with no signs of any congenital infection. Both the patient and his mother were O Rh +.
Several studies were performed following delivery. A direct Coombs test was negative. Blood studies revealed hypothyroidism and hypoalbuminemia secondary to protein loss associated with fetal hydrops. Cerebral, abdominal, and renal ultrasound; echocardiogram; thoracic and abdominal computed tomography; and cerebral magnetic resonance imaging revealed no abnormalities.
Karyotype results showed 46,XY,add(2)(p23), and subsequent spectral karyotyping and fluorescence in situ hybridization tests identified a chromosomal abnormality (46,XY,add[2][p23].ish del[2][pter][2PTEL27‒], dup[4][qter][D4S2930++])(Figure 2). Parental karyotypes were normal.
After birth, the infant was admitted to the neonatal intensive care unit for 50 days and received pleural and peritoneal drainages, mechanical ventilation, vasoactive drugs, parenteral nutrition with resolution of the hypoalbuminemia, levothyroxine, and intravenous antibiotics for central venous catheter infection. No drugs known to be associated with hypopigmentation of the hair, skin, or eyes were administered.
Two weeks after discharge from the neonatal intensive care unit, the patient was referred to our department. Physical examination revealed silvery hair on the scalp, eyebrows, and eyelashes, along with generalized hypopigmentation of the skin and eyes. Abdominal, cardiovascular, respiratory, and neurologic examination revealed no abnormalities, and no hepatosplenomegaly, lymphadenopathy, nystagmus, or strabismus was noted.
Light microscopy of the hair revealed small and regular aggregates of melanin along the hair shaft, predominantly in the medulla (Figure 3). Light microscopy of a skin biopsy specimen showed normal pigmentation in the melanocytes and no giant melanosomes. The melanocyte count was within reference range. A peripheral blood smear showed no giant granules in the granulocytes. No treatment was administered and the patient was followed closely every month. When the patient returned for follow-up at 9 months of age, physical examination revealed brown hair on the head, eyebrows, and eyelashes, as well as normal pigmentation of the skin and eyes (Figure 4). Thyroid function was normal and no recurrent infections of any type were noted. At follow-up at the age of 4 years, he showed normal neurological and psychological development with brown hair, no recurrent infections, and normal thyroid function. Given that CHS, ES, and GS had been ruled out, the clinical presentation and the genetic mutation detected may indicate that this case represents a new entity characterized by transient silvery hair.
Comment
Silvery hair is a known feature of CHS, ES, and GS (Table). The characteristic hypopigmentation associated with these autosomal-recessive disorders is the result of impaired melanosome transport leading to failed transfer of melanin to keratinocytes. These disorders differ from oculocutaneous albinism in that melanin synthesis is unaffected.
Chédiak-Higashi syndrome is characterized by generalized hypopigmentation of the skin and eyes, silvery hair, neurologic and immune dysfunction, lymphoproliferative disorders, and large granules in granulocytes and other cell types.1-3 A common complication of CHS is hemophagocytic lymphohistiocytosis, which is characterized by fever, jaundice, lymphadenopathy, hepatosplenomegaly, and pancytopenia.4 Pigmentary dilution of the irises also may be present, along with photophobia, strabismus, nystagmus, and impaired visual acuity. Chédiak-Higashi syndrome is the result of a genetic defect in the lysosomal trafficking regulator gene, also known as CHS1 (located on chromosome 1q42.1‒q42.2).5 Melanin in the hair shaft is distributed uniformly in multiple small aggregates. Light microscopy of the skin typically shows giant melanosomes in melanocytes and aberrant keratinocyte maturation.
Elejalde syndrome is characterized by silvery hair (eyelashes and eyebrows), neurologic defects, and normal immunologic function.6,7 The underlying molecular basis remains unknown. It appears related to or allelic to GS type 1 and thus associated with mutations in MYO5A (myosin VA); however, the gene mutation responsible has yet to be defined.8 Light microscopy of the hair shaft usually shows an irregular distribution of large melanin aggregates, primarily in the medulla.9,10 Skin biopsy generally shows irregular distribution and irregular size of melanin granules in the basal layer.11 Leukocytes usually show no abnormal cytoplasmic granules. Ocular involvement is common and may present as nystagmus, diplopia, hypopigmented retinas, and/or papilledema.
In GS, hair microscopy generally reveals large aggregates of melanin pigment distributed irregularly along the hair shaft. Granulocytes typically show no giant granules. Light microscopy of the skin usually shows increased pigment in melanocytes with sparse pigment in keratinocytes. Griscelli syndrome is classified into 3 types.12 In GS type 1, patients have silvery gray hair, light-colored skin, severe neurologic defects,13 and normal immune status. This variant is caused by a mutation in the MYO5A gene located on chromosome 15q21. In GS type 2, patients have silvery gray hair, pyogenic infections, an accelerated phase of hemophagocytic lymphohistiocytosis, and variable neurologic defects in the absence of primary neurologic disease.14,15 This variant is caused by a mutation in the RAB27A (member RAS oncogene family) gene located on chromosome 15q21. In GS type 3, patients exhibit generalized hypopigmentation of the skin and hair with no abnormalities of the nervous or immune systems. There are 2 different mutations associated with GS type 3: the first is located on chromosome 2q37.3, causing a mutation in MLPH (melanophilin), and the second is caused by an F-exon deletion in the MYO5A gene.14
Our patient had silvery hair, generalized hypopigmentation of the skin and eyes, and normal central nervous system function with no other ocular involvement and no evidence of recurrent infections of any kind. Light microscopy showed small and regular melanin pigment aggregates in the hair shaft, which differs from the irregular pigment aggregates in GS and ES.
The regular melanin pigment aggregates observed along the hair shaft were consistent with CHS, but other manifestations of this syndrome were absent: ocular, neurologic, hematologic, and immunologic abnormalities with presence of giant intracytoplasmic granules in leukocytes, and giant melanosomes in melanocytes. In our patient, the absence of these features along with the spontaneous repigmentation of the silvery hair, improvement of thyroid function, reversal of hypoalbuminemia, and the chromosomopathy detected make a diagnosis of CHS highly improbable.
We concluded that the silvery hair noted in our patient resulted from the 46,XY,add(2)(p23) chromosomal abnormality. This mutation could affect some of the genes that control the trafficking of melanosomes or could induce hypothyroidism and hypoproteinemia associated with congenital hydrops fetalis (Figure 5).
Hydrops fetalis is a potentially fatal condition characterized by severe edema (swelling) in a fetus or neonate. There are 2 types of hydrops fetalis: immune and nonimmune. Immune hydrops fetalis may develop in an Rh+ fetus with an Rh– mother, as the mother’s immune cells begin to break down the red blood cells of the fetus, resulting in anemia in the fetus with subsequent fetal heart failure, leading to an accumulation of large amounts of fluid in the tissues and organs. Nonimmune hydrops fetalis can occur secondary to diseases that interfere with the fetus’s ability to manage fluid (eg, severe anemia; congenital infections; urinary, lymphatic, heart, or thoracic defects; inborn errors of metabolism; chromosomal abnormalities). Case studies have suggested that congenital hypothyroidism could be a cause of nonimmune hydrops fetalis.16,17 Thyroid hormone deficiency reduces stimulation of adrenergic receptors in the lymphatic system and lungs, thereby decreasing lymph flow and protein efflux to the lymphatic system and decreasing clearance of liquid from the lungs. The final result is lymph vessel engorgement and subsequent leakage of lymphatic fluid to pleural spaces, causing hydrops fetalis and chylothorax.
The 46,XY,add(2)(p23) chromosomal abnormality has not been commonly associated with hypothyroidism and hydrops fetalis. The silvery hair in our patient was transient and spontaneously repigmented to brown over the course of follow-up in conjunction with improved physiologic changes. We concluded that the silvery hair in our patient was induced by his hypoproteinemic status secondary to hydrops fetalis and hypothyroidism.
Conclusion
In addition to CHS, ES, and GS, the differential diagnosis for silvery hair with abnormal skin pigmentation in children should include 46,XY,add(2)(p23) mutation, as was detected in our patient. Evaluation should include light microscopy of the hair shaft, skin biopsy, assessment of immune function, peripheral blood smear, and neurologic and eye examinations.
Silvery hair is characteristic of 3 rare autosomal-recessive disorders—Chédiak-Higashi syndrome (CHS), Elejalde syndrome (ES), and Griscelli syndrome (GS)—which are associated with mutations in various genes that encode several proteins involved in the intracellular processing and movement of melanosomes. We report the case of a 2-month-old male infant with transient silvery hair and generalized hypopigmentation of the skin and eyes who did not have any genetic mutations associated with the classic syndromes that usually are characterized by transient silvery hair.
Case Report
A 2-month-old male infant presented to the dermatology department for evaluation of silvery hair with generalized hypopigmentation of the skin and eyes (Figure 1) that had developed at 1 month of age. His parents were healthy, nonconsanguineous, and reported no family history of silvery hair. The patient was delivered by cesarean section at 35 weeks’ gestation. His medical history was remarkable for congenital hydrops fetalis with pleuropericardial effusion, ascites, soft-tissue edema, and hydrocele with no signs of any congenital infection. Both the patient and his mother were O Rh +.
Several studies were performed following delivery. A direct Coombs test was negative. Blood studies revealed hypothyroidism and hypoalbuminemia secondary to protein loss associated with fetal hydrops. Cerebral, abdominal, and renal ultrasound; echocardiogram; thoracic and abdominal computed tomography; and cerebral magnetic resonance imaging revealed no abnormalities.
Karyotype results showed 46,XY,add(2)(p23), and subsequent spectral karyotyping and fluorescence in situ hybridization tests identified a chromosomal abnormality (46,XY,add[2][p23].ish del[2][pter][2PTEL27‒], dup[4][qter][D4S2930++])(Figure 2). Parental karyotypes were normal.
After birth, the infant was admitted to the neonatal intensive care unit for 50 days and received pleural and peritoneal drainages, mechanical ventilation, vasoactive drugs, parenteral nutrition with resolution of the hypoalbuminemia, levothyroxine, and intravenous antibiotics for central venous catheter infection. No drugs known to be associated with hypopigmentation of the hair, skin, or eyes were administered.
Two weeks after discharge from the neonatal intensive care unit, the patient was referred to our department. Physical examination revealed silvery hair on the scalp, eyebrows, and eyelashes, along with generalized hypopigmentation of the skin and eyes. Abdominal, cardiovascular, respiratory, and neurologic examination revealed no abnormalities, and no hepatosplenomegaly, lymphadenopathy, nystagmus, or strabismus was noted.
Light microscopy of the hair revealed small and regular aggregates of melanin along the hair shaft, predominantly in the medulla (Figure 3). Light microscopy of a skin biopsy specimen showed normal pigmentation in the melanocytes and no giant melanosomes. The melanocyte count was within reference range. A peripheral blood smear showed no giant granules in the granulocytes. No treatment was administered and the patient was followed closely every month. When the patient returned for follow-up at 9 months of age, physical examination revealed brown hair on the head, eyebrows, and eyelashes, as well as normal pigmentation of the skin and eyes (Figure 4). Thyroid function was normal and no recurrent infections of any type were noted. At follow-up at the age of 4 years, he showed normal neurological and psychological development with brown hair, no recurrent infections, and normal thyroid function. Given that CHS, ES, and GS had been ruled out, the clinical presentation and the genetic mutation detected may indicate that this case represents a new entity characterized by transient silvery hair.
Comment
Silvery hair is a known feature of CHS, ES, and GS (Table). The characteristic hypopigmentation associated with these autosomal-recessive disorders is the result of impaired melanosome transport leading to failed transfer of melanin to keratinocytes. These disorders differ from oculocutaneous albinism in that melanin synthesis is unaffected.
Chédiak-Higashi syndrome is characterized by generalized hypopigmentation of the skin and eyes, silvery hair, neurologic and immune dysfunction, lymphoproliferative disorders, and large granules in granulocytes and other cell types.1-3 A common complication of CHS is hemophagocytic lymphohistiocytosis, which is characterized by fever, jaundice, lymphadenopathy, hepatosplenomegaly, and pancytopenia.4 Pigmentary dilution of the irises also may be present, along with photophobia, strabismus, nystagmus, and impaired visual acuity. Chédiak-Higashi syndrome is the result of a genetic defect in the lysosomal trafficking regulator gene, also known as CHS1 (located on chromosome 1q42.1‒q42.2).5 Melanin in the hair shaft is distributed uniformly in multiple small aggregates. Light microscopy of the skin typically shows giant melanosomes in melanocytes and aberrant keratinocyte maturation.
Elejalde syndrome is characterized by silvery hair (eyelashes and eyebrows), neurologic defects, and normal immunologic function.6,7 The underlying molecular basis remains unknown. It appears related to or allelic to GS type 1 and thus associated with mutations in MYO5A (myosin VA); however, the gene mutation responsible has yet to be defined.8 Light microscopy of the hair shaft usually shows an irregular distribution of large melanin aggregates, primarily in the medulla.9,10 Skin biopsy generally shows irregular distribution and irregular size of melanin granules in the basal layer.11 Leukocytes usually show no abnormal cytoplasmic granules. Ocular involvement is common and may present as nystagmus, diplopia, hypopigmented retinas, and/or papilledema.
In GS, hair microscopy generally reveals large aggregates of melanin pigment distributed irregularly along the hair shaft. Granulocytes typically show no giant granules. Light microscopy of the skin usually shows increased pigment in melanocytes with sparse pigment in keratinocytes. Griscelli syndrome is classified into 3 types.12 In GS type 1, patients have silvery gray hair, light-colored skin, severe neurologic defects,13 and normal immune status. This variant is caused by a mutation in the MYO5A gene located on chromosome 15q21. In GS type 2, patients have silvery gray hair, pyogenic infections, an accelerated phase of hemophagocytic lymphohistiocytosis, and variable neurologic defects in the absence of primary neurologic disease.14,15 This variant is caused by a mutation in the RAB27A (member RAS oncogene family) gene located on chromosome 15q21. In GS type 3, patients exhibit generalized hypopigmentation of the skin and hair with no abnormalities of the nervous or immune systems. There are 2 different mutations associated with GS type 3: the first is located on chromosome 2q37.3, causing a mutation in MLPH (melanophilin), and the second is caused by an F-exon deletion in the MYO5A gene.14
Our patient had silvery hair, generalized hypopigmentation of the skin and eyes, and normal central nervous system function with no other ocular involvement and no evidence of recurrent infections of any kind. Light microscopy showed small and regular melanin pigment aggregates in the hair shaft, which differs from the irregular pigment aggregates in GS and ES.
The regular melanin pigment aggregates observed along the hair shaft were consistent with CHS, but other manifestations of this syndrome were absent: ocular, neurologic, hematologic, and immunologic abnormalities with presence of giant intracytoplasmic granules in leukocytes, and giant melanosomes in melanocytes. In our patient, the absence of these features along with the spontaneous repigmentation of the silvery hair, improvement of thyroid function, reversal of hypoalbuminemia, and the chromosomopathy detected make a diagnosis of CHS highly improbable.
We concluded that the silvery hair noted in our patient resulted from the 46,XY,add(2)(p23) chromosomal abnormality. This mutation could affect some of the genes that control the trafficking of melanosomes or could induce hypothyroidism and hypoproteinemia associated with congenital hydrops fetalis (Figure 5).
Hydrops fetalis is a potentially fatal condition characterized by severe edema (swelling) in a fetus or neonate. There are 2 types of hydrops fetalis: immune and nonimmune. Immune hydrops fetalis may develop in an Rh+ fetus with an Rh– mother, as the mother’s immune cells begin to break down the red blood cells of the fetus, resulting in anemia in the fetus with subsequent fetal heart failure, leading to an accumulation of large amounts of fluid in the tissues and organs. Nonimmune hydrops fetalis can occur secondary to diseases that interfere with the fetus’s ability to manage fluid (eg, severe anemia; congenital infections; urinary, lymphatic, heart, or thoracic defects; inborn errors of metabolism; chromosomal abnormalities). Case studies have suggested that congenital hypothyroidism could be a cause of nonimmune hydrops fetalis.16,17 Thyroid hormone deficiency reduces stimulation of adrenergic receptors in the lymphatic system and lungs, thereby decreasing lymph flow and protein efflux to the lymphatic system and decreasing clearance of liquid from the lungs. The final result is lymph vessel engorgement and subsequent leakage of lymphatic fluid to pleural spaces, causing hydrops fetalis and chylothorax.
The 46,XY,add(2)(p23) chromosomal abnormality has not been commonly associated with hypothyroidism and hydrops fetalis. The silvery hair in our patient was transient and spontaneously repigmented to brown over the course of follow-up in conjunction with improved physiologic changes. We concluded that the silvery hair in our patient was induced by his hypoproteinemic status secondary to hydrops fetalis and hypothyroidism.
Conclusion
In addition to CHS, ES, and GS, the differential diagnosis for silvery hair with abnormal skin pigmentation in children should include 46,XY,add(2)(p23) mutation, as was detected in our patient. Evaluation should include light microscopy of the hair shaft, skin biopsy, assessment of immune function, peripheral blood smear, and neurologic and eye examinations.
- White JG. The Chédiak-Higashi syndrome: a possible lysosomal disease. Blood. 1966;28:143-156.
- Introne W, Boissy RE, Gahl WA. Clinical, molecular, and cell biological aspects of Chédiak-Higashi syndrome. Mol Genet Metab. 1999;68:283-303.
- Kaplan J, De Domenico I, Ward DM. Chédiak-Higashi syndrome. Curr Opin Hematol. 2008;15:22-29.
- Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis [published online December 7, 2006]. Eur J Pediatr. 2007;166:95-109.
- Morrone K, Wang Y, Huizing M, et al. Two novel mutations identified in an African-American child with Chédiak-Higashi syndrome [published online March 24, 2010]. Case Report Med. 2010;2010:967535.
- Ivanovich J, Mallory S, Storer T, et al. 12-year-old male with Elejalde syndrome (neuroectodermal melanolysosomal disease). Am J Med Genet. 2001;98:313-316.
- Cahali JB, Fernandez SA, Oliveira ZN, et al. Elejalde syndrome: report of a case and review of the literature. Pediatr Dermatol. 2004;21:479-482.
- Bahadoran P, Ortonne JP, Ballotti R, et al. Comment on Elejalde syndrome and relationship with Griscelli syndrome. Am J Med Genet. 2003;116:408-409.
- Duran-McKinster C, Rodriguez-Jurado R, Ridaura C, et al. Elejalde syndrome—a melanolysosomal neurocutaneous syndrome: clinical and morphological findings in 7 patients. Arch Dermatol. 1999;135:182-186.
- Happle R. Neurocutaneous diseases. In: Freedberg IM, Eisen AZ, Wolff K, et al, eds. Dermatology in General Medicine. 5th ed. New York, NY: McGraw-Hill; 1999:2131-2148.
- Sanal O, Yel L, Kucukali T, et al. An allelic variant of Griscelli disease: presentation with severe hypotonia, mental-motor retardation, and hypopigmentation consistent with Elejalde syndrome (neuroectodermal melanolysosomal disorder). J Neurol. 2000;247:570-572.
- Malhotra AK, Bhaskar G, Nanda M, et al. Griscelli syndrome. J Am Acad Dermatol. 2006;55:337-340.
- Al-Idrissi E, ElGhazali G, Alzahrani M, et al. Premature birth, respiratory distress, intracerebral hemorrhage, and silvery-gray hair: differential diagnosis of the 3 types of Griscelli syndrome. J Pediatr Hematol Oncol. 2010;32:494-496.
- Ménasché G, Ho CH, Sanal O, et al. Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1). J Clin Invest. 2003;112:450-456.
- Griscelli C, Durandy A, Guy-Grand D, et al. A syndrome associating partial albinism and immunodeficiency. Am J Med. 1978;65:691-702.
- Narchi H. Congenital hypothyroidism and nonimmune hydrops fetalis: associated? Pediatrics. 1999;104:1416-1417.
- Kessel I, Makhoul IR, Sujov P. Congenital hypothyroidism and nonimmune hydrops fetalis: associated? Pediatrics. 1999;103:E9.
- White JG. The Chédiak-Higashi syndrome: a possible lysosomal disease. Blood. 1966;28:143-156.
- Introne W, Boissy RE, Gahl WA. Clinical, molecular, and cell biological aspects of Chédiak-Higashi syndrome. Mol Genet Metab. 1999;68:283-303.
- Kaplan J, De Domenico I, Ward DM. Chédiak-Higashi syndrome. Curr Opin Hematol. 2008;15:22-29.
- Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis [published online December 7, 2006]. Eur J Pediatr. 2007;166:95-109.
- Morrone K, Wang Y, Huizing M, et al. Two novel mutations identified in an African-American child with Chédiak-Higashi syndrome [published online March 24, 2010]. Case Report Med. 2010;2010:967535.
- Ivanovich J, Mallory S, Storer T, et al. 12-year-old male with Elejalde syndrome (neuroectodermal melanolysosomal disease). Am J Med Genet. 2001;98:313-316.
- Cahali JB, Fernandez SA, Oliveira ZN, et al. Elejalde syndrome: report of a case and review of the literature. Pediatr Dermatol. 2004;21:479-482.
- Bahadoran P, Ortonne JP, Ballotti R, et al. Comment on Elejalde syndrome and relationship with Griscelli syndrome. Am J Med Genet. 2003;116:408-409.
- Duran-McKinster C, Rodriguez-Jurado R, Ridaura C, et al. Elejalde syndrome—a melanolysosomal neurocutaneous syndrome: clinical and morphological findings in 7 patients. Arch Dermatol. 1999;135:182-186.
- Happle R. Neurocutaneous diseases. In: Freedberg IM, Eisen AZ, Wolff K, et al, eds. Dermatology in General Medicine. 5th ed. New York, NY: McGraw-Hill; 1999:2131-2148.
- Sanal O, Yel L, Kucukali T, et al. An allelic variant of Griscelli disease: presentation with severe hypotonia, mental-motor retardation, and hypopigmentation consistent with Elejalde syndrome (neuroectodermal melanolysosomal disorder). J Neurol. 2000;247:570-572.
- Malhotra AK, Bhaskar G, Nanda M, et al. Griscelli syndrome. J Am Acad Dermatol. 2006;55:337-340.
- Al-Idrissi E, ElGhazali G, Alzahrani M, et al. Premature birth, respiratory distress, intracerebral hemorrhage, and silvery-gray hair: differential diagnosis of the 3 types of Griscelli syndrome. J Pediatr Hematol Oncol. 2010;32:494-496.
- Ménasché G, Ho CH, Sanal O, et al. Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1). J Clin Invest. 2003;112:450-456.
- Griscelli C, Durandy A, Guy-Grand D, et al. A syndrome associating partial albinism and immunodeficiency. Am J Med. 1978;65:691-702.
- Narchi H. Congenital hypothyroidism and nonimmune hydrops fetalis: associated? Pediatrics. 1999;104:1416-1417.
- Kessel I, Makhoul IR, Sujov P. Congenital hypothyroidism and nonimmune hydrops fetalis: associated? Pediatrics. 1999;103:E9.
Practice Points
- Silvery hair is characteristic of 3 rare autosomal-recessive disorders: Chédiak-Higashi syndrome, Elejalde syndrome, and Griscelli syndrome.
- Hypopigmentation is the result of impaired melanosome transport leading to failed transfer of melanin to keratinocytes.
- Evaluation should include light microscopy of the hair shaft, skin biopsy, assessment of immune function, peripheral blood smear, and neurologic and eye examinations.
Management of Vitiligo Patients With Surgical Interventions
Vitiligo is a common, asymptomatic, acquired depigmentation disorder that is caused by an unknown etiology. Lesions appear as sharply demarcated, depigmented macules and patches that are scattered symmetrically or unsymmetrically over the body. The presentation can be delineated based on the segmental or nonsegmental nature of the disease. According to the revised classification/nomenclature of vitiligo,1 the disorder can be classified as nonsegmental, segmental, mixed, or unclassified. The pathogenesis of the vitiligo disease process is due to multiple modalities that contribute to melanocyte loss. Theories for melanocyte destruction include but are not limited to autoimmunity, biochemicals, epidermal cytokines, increased hydrogen peroxide and free radicals, and humoral and cellular immune alteration.2,3
Despite its long history, the most frustrating aspect of the vitiligo disease process remains its treatment due to limited efficacy, frequent application of topicals, and the need for high-potency steroids. Medical therapies usually are the first line of treatment and are most effective with few side effects for bilateral nonsegmental or evolving vitiligo.2 Some of the primary therapies with the highest efficacies appear to be calcipotriene and psoralen plus UVA, psoralen plus UVA as monotherapy, excimer laser, narrowband UVB, oral steroids, 8-methoxypsoralen, tacrolimus, and topical steroids.4 The theory is that these treatments would be successful if the patient had active melanocytes in the external root sheath that would be able to repigment a patch of vitiligo.5 Hence, it would be more difficult to treat areas such as the dorsal aspect of the fingers and toes because they lack hair-bearing areas with melanocytes.6 The alternative approach to treating vitiligo patches would be surgical intervention techniques, as they provide melanocytic cells to a previously depigmented area.3,5 The focus of this article is to evaluate the efficacy and appropriate use of some of the surgical procedures that can be used in the treatment of vitiligo patients.
Candidate Selection
First, vitiligo patients for whom first-line treatment with medical therapies has failed are candidates for surgical techniques. The second vital component is to clinically confirm the diagnosis of vitiligo as opposed to other genetic, infectious, or autoimmune causes of pigment loss. Lastly, the vitiligo patch should be stable. A stable vitiligo patch does not continue to progress and is no longer responsive to topical medications that are meant to repigment for a discernible period of time.7
Classification of Disease Stage
To classify the stage of vitiligo prior to surgical intervention, Gauthier8 created a basic grading system: grade I, with partial depletion of epidermal melanocytes in a vitiligo patch that responds to repigmentation in a follicular pattern evenly such as on the face and neck; grade II, with complete depletion of epidermal melanocytes with a usual follicular pattern of repigmentation; and grade III, indicating complete depletion of follicular melanocytes with no hope of response to medical therapy. According to Rusfianti and Wirohadidjodjo,2 the surgical techniques that have developed over the years for treatment of grade III vitiligo patients include split-thickness skin grafting, suction blister grafting, miniature punch grafting, and cultured melanocyte transplantation.
Surgical Techniques
Split-thickness skin grafting is an older procedure that entails the use of a harvesting graft site with no pigment loss and dermabrasion of the recipient area to allow interaction with the wound bed.9 With proper care and minimal movement or wrinkling of the graft site, patients can have repigmentation without skip areas.
Suction blister grafting is another tried and tested surgical intervention. Hasegawa et al10 conducted a study of 15 patients (13 males, 2 females; age range, 16–38 years) diagnosed with segmental vitiligo who were treated using the suction blister grafting technique with CO2 laser resurfacing. Patients were recruited 1 month prior to initiating therapy and no other treatments were used during the month or in conjunction with the surgical intervention. Suction blisters were harvested from the left thigh and transferred in saline to the recipient site, which was abraded with 1 pass of the short-pulse CO2 laser system. The recipient sites were then closed with 7-0 nylon sutures and covered tightly with tie-over dressings for at least 1 week. Within 6 months of the procedure, a treatment response of 100% was seen in 15 patients, making it an effective method for treatment-resistant vitiligo patients.10
Miniature punch grafting is another possible treatment option for resistant cases of vitiligo. Mapar et al11 conducted a study in 25 patients (21 women, 4 men; age range, 20–47 years) who had been diagnosed with stable vitiligo (ie, no progression in the last 2 years) and were treated with single hair follicle transplant versus miniature punch grafting. The theory behind the study was to use the melanocytic reservoir noted in the normal hair follicle to repigment the vitiligo patch. With follow-up of both methods of treatment, there was no statistical difference in treatment results.11 A similar study was conducted by Malakar and Lahiri12 in patients with lip leukoderma (a variant of vitiligo). One hundred eight patients (41 males, 67 females; age range, 14–62 years) who had been diagnosed with stable lip leukoderma (ie, stable vitiligo for at least 6 months) underwent treatment via autologous miniature punch grafting. Punch biopsies were performed in donor sites of the buttocks and upper thighs with 72% of patients noting complete repigmentation. Complications noted were herpes labialis–induced lip leukoderma, which ultimately led to rejection of the graft site.12 Overall, however, miniature punch grafting is a viable surgical option in stable vitiligo patients.
Cultured melanocyte transplantation, or a noncultured epidermal suspension, was first initiated in 1992.13 Silpa-Archa et al14 conducted an open, split-comparison study of 6 vitiligo patients (5 women, 1 man; age range, 20–65 years) with stable lesions. Fifty percent of patients received autologous pigmented skin cellular suspension, which was applied to vitiligo-affected skin that was treated with a fractionated CO2 laser, and 50% received dermabrasion. Composite dressing was placed overlying the site with dressing removal in 1 week. The degree of repigmentation was based on a modified vitiligo area scoring index scale of poor (0%–25%), fair (26%–50%), good (51%–75%), very good (76%–90%), or excellent (91%–100%). Overall repigmentation was very good to excellent in all 6 patients.14 Potentially, this method can far improve the surgical treatment options for future vitiligo patients.
Final Thoughts
Overall, when evaluating surgical interventions for the treatment of vitiligo, careful consideration of the patient’s disease progression, failed therapies, outcome expectations, and repigmentation is warranted prior to initiating any procedure. For appropriate candidates, a range of surgical methodologies has proven to be effective in treatment of stable vitiligo patients.
- Taïeb A, Picardo M; VETF members. The definition and assessment of vitiligo: a consensus report of the Vitiligo European Task Force. Pigment Cell Res. 2007;20:27-35. Cited by: Ezzedine K, Lim HW, Suzuki T, et al; Vitiligo Global Issue Consensus Conference Panelists. Revised classification/nomenclature of vitiligo and related issues: the Vitiligo Global Issues Consensus Conference. Pigment Cell Melanoma Res. 2012;25:E1-E13.
- Rusfianti M, Wirohadidjodjo YW. Dermatosurgical techniques for repigmentation of vitiligo. Int J Dermatol. 2006;45:411-417.
- Falabella R. Surgical therapies for vitiligo. Clin Dermatol. 1997;15:927-939.
- Whitton ME, Pinart M, Batchelor J, et al. Interventions for vitiligo. Cochrane Database Syst Rev. 2015;2:CD003263.
- Mulekar SV, Isedeh P. Surgical interventions for vitiligo: an evidence-based review. Br J Dermatol. 2013;169(suppl 3):57-66.
- Dutta AK, Mandal SB. A clinical study of 650 vitiligo cases and their classification. Indian J Dermatol. 1969;14:103-111.
- Falabella R, Arrunategui A, Barona MI, et al. The minigrafting test for vitiligo: detection of stable lesions for melanocyte transplantation. J Am Acad Dermatol. 1995;32:228-232.
- Gauthier Y. Le vitiligo. Gaz Med. 1994;101:8-12.
- Malakar S, Malakar RS. Surgical pearl: composite film and graft unit for the recipient area dressing after split-thickness skin grafting in vitiligo. J Am Acad Dermatol. 2001;44:856-858.
- Hasegawa T, Suga Y, Ikejima A, et al. Suction blister grafting with CO2 laser resurfacing of the graft recipient site for vitiligo. J Dermatol. 2007;34:490-492.
- Mapar MA, Safarpour M, Mapar M, et al. A comparative study of the mini-punch grafting and hair follicle transplantation in the treatment of refractory and stable vitiligo. J Am Acad Dermatol. 2014;70:743-747.
- Malakar S, Lahiri K. Punch grafting for lip leukoderma. Dermatology. 2004;208:125-128.
- Gauthier Y, Surleve-Bazeille JE. Autologous grafting with noncultured melanocytes: a simplified method for treatment of depigmented lesions. J Am Acad Dermatol. 1992;26(2, pt 1):191-194.
- Silpa-Archa N, Griffith JL, Williams MS, et al. Prospective comparison of recipient-site preparation with fractional carbon dioxide laser versus dermabrasion and recipient-site dressing composition in melanocyte-keratinocyte transplantation procedure in vitiligo: a preliminary study [published online January 24, 2016]. Br J Dermatol. 2016;174:895-897.
Vitiligo is a common, asymptomatic, acquired depigmentation disorder that is caused by an unknown etiology. Lesions appear as sharply demarcated, depigmented macules and patches that are scattered symmetrically or unsymmetrically over the body. The presentation can be delineated based on the segmental or nonsegmental nature of the disease. According to the revised classification/nomenclature of vitiligo,1 the disorder can be classified as nonsegmental, segmental, mixed, or unclassified. The pathogenesis of the vitiligo disease process is due to multiple modalities that contribute to melanocyte loss. Theories for melanocyte destruction include but are not limited to autoimmunity, biochemicals, epidermal cytokines, increased hydrogen peroxide and free radicals, and humoral and cellular immune alteration.2,3
Despite its long history, the most frustrating aspect of the vitiligo disease process remains its treatment due to limited efficacy, frequent application of topicals, and the need for high-potency steroids. Medical therapies usually are the first line of treatment and are most effective with few side effects for bilateral nonsegmental or evolving vitiligo.2 Some of the primary therapies with the highest efficacies appear to be calcipotriene and psoralen plus UVA, psoralen plus UVA as monotherapy, excimer laser, narrowband UVB, oral steroids, 8-methoxypsoralen, tacrolimus, and topical steroids.4 The theory is that these treatments would be successful if the patient had active melanocytes in the external root sheath that would be able to repigment a patch of vitiligo.5 Hence, it would be more difficult to treat areas such as the dorsal aspect of the fingers and toes because they lack hair-bearing areas with melanocytes.6 The alternative approach to treating vitiligo patches would be surgical intervention techniques, as they provide melanocytic cells to a previously depigmented area.3,5 The focus of this article is to evaluate the efficacy and appropriate use of some of the surgical procedures that can be used in the treatment of vitiligo patients.
Candidate Selection
First, vitiligo patients for whom first-line treatment with medical therapies has failed are candidates for surgical techniques. The second vital component is to clinically confirm the diagnosis of vitiligo as opposed to other genetic, infectious, or autoimmune causes of pigment loss. Lastly, the vitiligo patch should be stable. A stable vitiligo patch does not continue to progress and is no longer responsive to topical medications that are meant to repigment for a discernible period of time.7
Classification of Disease Stage
To classify the stage of vitiligo prior to surgical intervention, Gauthier8 created a basic grading system: grade I, with partial depletion of epidermal melanocytes in a vitiligo patch that responds to repigmentation in a follicular pattern evenly such as on the face and neck; grade II, with complete depletion of epidermal melanocytes with a usual follicular pattern of repigmentation; and grade III, indicating complete depletion of follicular melanocytes with no hope of response to medical therapy. According to Rusfianti and Wirohadidjodjo,2 the surgical techniques that have developed over the years for treatment of grade III vitiligo patients include split-thickness skin grafting, suction blister grafting, miniature punch grafting, and cultured melanocyte transplantation.
Surgical Techniques
Split-thickness skin grafting is an older procedure that entails the use of a harvesting graft site with no pigment loss and dermabrasion of the recipient area to allow interaction with the wound bed.9 With proper care and minimal movement or wrinkling of the graft site, patients can have repigmentation without skip areas.
Suction blister grafting is another tried and tested surgical intervention. Hasegawa et al10 conducted a study of 15 patients (13 males, 2 females; age range, 16–38 years) diagnosed with segmental vitiligo who were treated using the suction blister grafting technique with CO2 laser resurfacing. Patients were recruited 1 month prior to initiating therapy and no other treatments were used during the month or in conjunction with the surgical intervention. Suction blisters were harvested from the left thigh and transferred in saline to the recipient site, which was abraded with 1 pass of the short-pulse CO2 laser system. The recipient sites were then closed with 7-0 nylon sutures and covered tightly with tie-over dressings for at least 1 week. Within 6 months of the procedure, a treatment response of 100% was seen in 15 patients, making it an effective method for treatment-resistant vitiligo patients.10
Miniature punch grafting is another possible treatment option for resistant cases of vitiligo. Mapar et al11 conducted a study in 25 patients (21 women, 4 men; age range, 20–47 years) who had been diagnosed with stable vitiligo (ie, no progression in the last 2 years) and were treated with single hair follicle transplant versus miniature punch grafting. The theory behind the study was to use the melanocytic reservoir noted in the normal hair follicle to repigment the vitiligo patch. With follow-up of both methods of treatment, there was no statistical difference in treatment results.11 A similar study was conducted by Malakar and Lahiri12 in patients with lip leukoderma (a variant of vitiligo). One hundred eight patients (41 males, 67 females; age range, 14–62 years) who had been diagnosed with stable lip leukoderma (ie, stable vitiligo for at least 6 months) underwent treatment via autologous miniature punch grafting. Punch biopsies were performed in donor sites of the buttocks and upper thighs with 72% of patients noting complete repigmentation. Complications noted were herpes labialis–induced lip leukoderma, which ultimately led to rejection of the graft site.12 Overall, however, miniature punch grafting is a viable surgical option in stable vitiligo patients.
Cultured melanocyte transplantation, or a noncultured epidermal suspension, was first initiated in 1992.13 Silpa-Archa et al14 conducted an open, split-comparison study of 6 vitiligo patients (5 women, 1 man; age range, 20–65 years) with stable lesions. Fifty percent of patients received autologous pigmented skin cellular suspension, which was applied to vitiligo-affected skin that was treated with a fractionated CO2 laser, and 50% received dermabrasion. Composite dressing was placed overlying the site with dressing removal in 1 week. The degree of repigmentation was based on a modified vitiligo area scoring index scale of poor (0%–25%), fair (26%–50%), good (51%–75%), very good (76%–90%), or excellent (91%–100%). Overall repigmentation was very good to excellent in all 6 patients.14 Potentially, this method can far improve the surgical treatment options for future vitiligo patients.
Final Thoughts
Overall, when evaluating surgical interventions for the treatment of vitiligo, careful consideration of the patient’s disease progression, failed therapies, outcome expectations, and repigmentation is warranted prior to initiating any procedure. For appropriate candidates, a range of surgical methodologies has proven to be effective in treatment of stable vitiligo patients.
Vitiligo is a common, asymptomatic, acquired depigmentation disorder that is caused by an unknown etiology. Lesions appear as sharply demarcated, depigmented macules and patches that are scattered symmetrically or unsymmetrically over the body. The presentation can be delineated based on the segmental or nonsegmental nature of the disease. According to the revised classification/nomenclature of vitiligo,1 the disorder can be classified as nonsegmental, segmental, mixed, or unclassified. The pathogenesis of the vitiligo disease process is due to multiple modalities that contribute to melanocyte loss. Theories for melanocyte destruction include but are not limited to autoimmunity, biochemicals, epidermal cytokines, increased hydrogen peroxide and free radicals, and humoral and cellular immune alteration.2,3
Despite its long history, the most frustrating aspect of the vitiligo disease process remains its treatment due to limited efficacy, frequent application of topicals, and the need for high-potency steroids. Medical therapies usually are the first line of treatment and are most effective with few side effects for bilateral nonsegmental or evolving vitiligo.2 Some of the primary therapies with the highest efficacies appear to be calcipotriene and psoralen plus UVA, psoralen plus UVA as monotherapy, excimer laser, narrowband UVB, oral steroids, 8-methoxypsoralen, tacrolimus, and topical steroids.4 The theory is that these treatments would be successful if the patient had active melanocytes in the external root sheath that would be able to repigment a patch of vitiligo.5 Hence, it would be more difficult to treat areas such as the dorsal aspect of the fingers and toes because they lack hair-bearing areas with melanocytes.6 The alternative approach to treating vitiligo patches would be surgical intervention techniques, as they provide melanocytic cells to a previously depigmented area.3,5 The focus of this article is to evaluate the efficacy and appropriate use of some of the surgical procedures that can be used in the treatment of vitiligo patients.
Candidate Selection
First, vitiligo patients for whom first-line treatment with medical therapies has failed are candidates for surgical techniques. The second vital component is to clinically confirm the diagnosis of vitiligo as opposed to other genetic, infectious, or autoimmune causes of pigment loss. Lastly, the vitiligo patch should be stable. A stable vitiligo patch does not continue to progress and is no longer responsive to topical medications that are meant to repigment for a discernible period of time.7
Classification of Disease Stage
To classify the stage of vitiligo prior to surgical intervention, Gauthier8 created a basic grading system: grade I, with partial depletion of epidermal melanocytes in a vitiligo patch that responds to repigmentation in a follicular pattern evenly such as on the face and neck; grade II, with complete depletion of epidermal melanocytes with a usual follicular pattern of repigmentation; and grade III, indicating complete depletion of follicular melanocytes with no hope of response to medical therapy. According to Rusfianti and Wirohadidjodjo,2 the surgical techniques that have developed over the years for treatment of grade III vitiligo patients include split-thickness skin grafting, suction blister grafting, miniature punch grafting, and cultured melanocyte transplantation.
Surgical Techniques
Split-thickness skin grafting is an older procedure that entails the use of a harvesting graft site with no pigment loss and dermabrasion of the recipient area to allow interaction with the wound bed.9 With proper care and minimal movement or wrinkling of the graft site, patients can have repigmentation without skip areas.
Suction blister grafting is another tried and tested surgical intervention. Hasegawa et al10 conducted a study of 15 patients (13 males, 2 females; age range, 16–38 years) diagnosed with segmental vitiligo who were treated using the suction blister grafting technique with CO2 laser resurfacing. Patients were recruited 1 month prior to initiating therapy and no other treatments were used during the month or in conjunction with the surgical intervention. Suction blisters were harvested from the left thigh and transferred in saline to the recipient site, which was abraded with 1 pass of the short-pulse CO2 laser system. The recipient sites were then closed with 7-0 nylon sutures and covered tightly with tie-over dressings for at least 1 week. Within 6 months of the procedure, a treatment response of 100% was seen in 15 patients, making it an effective method for treatment-resistant vitiligo patients.10
Miniature punch grafting is another possible treatment option for resistant cases of vitiligo. Mapar et al11 conducted a study in 25 patients (21 women, 4 men; age range, 20–47 years) who had been diagnosed with stable vitiligo (ie, no progression in the last 2 years) and were treated with single hair follicle transplant versus miniature punch grafting. The theory behind the study was to use the melanocytic reservoir noted in the normal hair follicle to repigment the vitiligo patch. With follow-up of both methods of treatment, there was no statistical difference in treatment results.11 A similar study was conducted by Malakar and Lahiri12 in patients with lip leukoderma (a variant of vitiligo). One hundred eight patients (41 males, 67 females; age range, 14–62 years) who had been diagnosed with stable lip leukoderma (ie, stable vitiligo for at least 6 months) underwent treatment via autologous miniature punch grafting. Punch biopsies were performed in donor sites of the buttocks and upper thighs with 72% of patients noting complete repigmentation. Complications noted were herpes labialis–induced lip leukoderma, which ultimately led to rejection of the graft site.12 Overall, however, miniature punch grafting is a viable surgical option in stable vitiligo patients.
Cultured melanocyte transplantation, or a noncultured epidermal suspension, was first initiated in 1992.13 Silpa-Archa et al14 conducted an open, split-comparison study of 6 vitiligo patients (5 women, 1 man; age range, 20–65 years) with stable lesions. Fifty percent of patients received autologous pigmented skin cellular suspension, which was applied to vitiligo-affected skin that was treated with a fractionated CO2 laser, and 50% received dermabrasion. Composite dressing was placed overlying the site with dressing removal in 1 week. The degree of repigmentation was based on a modified vitiligo area scoring index scale of poor (0%–25%), fair (26%–50%), good (51%–75%), very good (76%–90%), or excellent (91%–100%). Overall repigmentation was very good to excellent in all 6 patients.14 Potentially, this method can far improve the surgical treatment options for future vitiligo patients.
Final Thoughts
Overall, when evaluating surgical interventions for the treatment of vitiligo, careful consideration of the patient’s disease progression, failed therapies, outcome expectations, and repigmentation is warranted prior to initiating any procedure. For appropriate candidates, a range of surgical methodologies has proven to be effective in treatment of stable vitiligo patients.
- Taïeb A, Picardo M; VETF members. The definition and assessment of vitiligo: a consensus report of the Vitiligo European Task Force. Pigment Cell Res. 2007;20:27-35. Cited by: Ezzedine K, Lim HW, Suzuki T, et al; Vitiligo Global Issue Consensus Conference Panelists. Revised classification/nomenclature of vitiligo and related issues: the Vitiligo Global Issues Consensus Conference. Pigment Cell Melanoma Res. 2012;25:E1-E13.
- Rusfianti M, Wirohadidjodjo YW. Dermatosurgical techniques for repigmentation of vitiligo. Int J Dermatol. 2006;45:411-417.
- Falabella R. Surgical therapies for vitiligo. Clin Dermatol. 1997;15:927-939.
- Whitton ME, Pinart M, Batchelor J, et al. Interventions for vitiligo. Cochrane Database Syst Rev. 2015;2:CD003263.
- Mulekar SV, Isedeh P. Surgical interventions for vitiligo: an evidence-based review. Br J Dermatol. 2013;169(suppl 3):57-66.
- Dutta AK, Mandal SB. A clinical study of 650 vitiligo cases and their classification. Indian J Dermatol. 1969;14:103-111.
- Falabella R, Arrunategui A, Barona MI, et al. The minigrafting test for vitiligo: detection of stable lesions for melanocyte transplantation. J Am Acad Dermatol. 1995;32:228-232.
- Gauthier Y. Le vitiligo. Gaz Med. 1994;101:8-12.
- Malakar S, Malakar RS. Surgical pearl: composite film and graft unit for the recipient area dressing after split-thickness skin grafting in vitiligo. J Am Acad Dermatol. 2001;44:856-858.
- Hasegawa T, Suga Y, Ikejima A, et al. Suction blister grafting with CO2 laser resurfacing of the graft recipient site for vitiligo. J Dermatol. 2007;34:490-492.
- Mapar MA, Safarpour M, Mapar M, et al. A comparative study of the mini-punch grafting and hair follicle transplantation in the treatment of refractory and stable vitiligo. J Am Acad Dermatol. 2014;70:743-747.
- Malakar S, Lahiri K. Punch grafting for lip leukoderma. Dermatology. 2004;208:125-128.
- Gauthier Y, Surleve-Bazeille JE. Autologous grafting with noncultured melanocytes: a simplified method for treatment of depigmented lesions. J Am Acad Dermatol. 1992;26(2, pt 1):191-194.
- Silpa-Archa N, Griffith JL, Williams MS, et al. Prospective comparison of recipient-site preparation with fractional carbon dioxide laser versus dermabrasion and recipient-site dressing composition in melanocyte-keratinocyte transplantation procedure in vitiligo: a preliminary study [published online January 24, 2016]. Br J Dermatol. 2016;174:895-897.
- Taïeb A, Picardo M; VETF members. The definition and assessment of vitiligo: a consensus report of the Vitiligo European Task Force. Pigment Cell Res. 2007;20:27-35. Cited by: Ezzedine K, Lim HW, Suzuki T, et al; Vitiligo Global Issue Consensus Conference Panelists. Revised classification/nomenclature of vitiligo and related issues: the Vitiligo Global Issues Consensus Conference. Pigment Cell Melanoma Res. 2012;25:E1-E13.
- Rusfianti M, Wirohadidjodjo YW. Dermatosurgical techniques for repigmentation of vitiligo. Int J Dermatol. 2006;45:411-417.
- Falabella R. Surgical therapies for vitiligo. Clin Dermatol. 1997;15:927-939.
- Whitton ME, Pinart M, Batchelor J, et al. Interventions for vitiligo. Cochrane Database Syst Rev. 2015;2:CD003263.
- Mulekar SV, Isedeh P. Surgical interventions for vitiligo: an evidence-based review. Br J Dermatol. 2013;169(suppl 3):57-66.
- Dutta AK, Mandal SB. A clinical study of 650 vitiligo cases and their classification. Indian J Dermatol. 1969;14:103-111.
- Falabella R, Arrunategui A, Barona MI, et al. The minigrafting test for vitiligo: detection of stable lesions for melanocyte transplantation. J Am Acad Dermatol. 1995;32:228-232.
- Gauthier Y. Le vitiligo. Gaz Med. 1994;101:8-12.
- Malakar S, Malakar RS. Surgical pearl: composite film and graft unit for the recipient area dressing after split-thickness skin grafting in vitiligo. J Am Acad Dermatol. 2001;44:856-858.
- Hasegawa T, Suga Y, Ikejima A, et al. Suction blister grafting with CO2 laser resurfacing of the graft recipient site for vitiligo. J Dermatol. 2007;34:490-492.
- Mapar MA, Safarpour M, Mapar M, et al. A comparative study of the mini-punch grafting and hair follicle transplantation in the treatment of refractory and stable vitiligo. J Am Acad Dermatol. 2014;70:743-747.
- Malakar S, Lahiri K. Punch grafting for lip leukoderma. Dermatology. 2004;208:125-128.
- Gauthier Y, Surleve-Bazeille JE. Autologous grafting with noncultured melanocytes: a simplified method for treatment of depigmented lesions. J Am Acad Dermatol. 1992;26(2, pt 1):191-194.
- Silpa-Archa N, Griffith JL, Williams MS, et al. Prospective comparison of recipient-site preparation with fractional carbon dioxide laser versus dermabrasion and recipient-site dressing composition in melanocyte-keratinocyte transplantation procedure in vitiligo: a preliminary study [published online January 24, 2016]. Br J Dermatol. 2016;174:895-897.

Laser Best Practices for Darker Skin Types

What does your patient need to know at the first visit? Does it apply to patients of all genders and ages?
Before performing laser procedures on patients with richly pigmented skin (Fitzpatrick skin types IV–VI), patients need to be informed of the higher risk for pigmentary alterations as potential complications of the procedure. Specifically, hyperpigmentation or hypopigmentation can occur postprocedure, depending on the type of device used, the treatment settings, the technique, the underlying skin disorder being treated, and the patient’s individual response to treatment. Fortunately, these pigment alterations are in most cases self-limited but can last for weeks to months depending on the severity and the nature of the dyspigmentation.
What are your go-to treatments? What are the side effects?
Notwithstanding the higher risks for pigmentary alterations, lasers can be extremely useful for the management of numerous dermatologic concerns in patients with Fitzpatrick skin types IV to VI including laser hair removal for pseudofolliculitis barbae or nonablative fractional laser resurfacing for acne scarring and pigmentary disorders. My go-to treatments include the following: long-pulsed 1064-nm Nd:YAG laser for hair removal in Fitzpatrick skin types V to VI, 808-nm diode laser with linear scanning of hair removal in Fitzpatrick skin type IV or less, 1550-nm erbium-doped nonablative fractional laser for acne scarring in Fitzpatrick skin types IV to VI, and low-power diode 1927-nm fractional laser for melasma and postinflammatory hyperpigmentation in Fitzpatrick skin types IV to VI.
All of these procedures are performed with conservative treatment settings such as low fluences and longer pulse durations for laser hair removal and low treatment densities for fractional laser procedures. Prior to laser resurfacing, I recommend hydroquinone cream 4% twice daily starting 2 weeks before the first session and for 4 weeks posttreatment. These recommendations are based on published evidence (see Suggested Readings) as well as anecdotal experience.
How do you keep patients compliant with treatment?
Emphasizing the need for broad-spectrum sunscreen and avoidance of intense sun exposure before and after laser treatments is important during the initial consultation and prior to each treatment. I warn my patients of the higher risk for hyperpigmentation if the skin is tanned or has recently had intense sun exposure.
What do you do if they refuse treatment?
If patients refuse laser treatment or recommended precautions, then I will consider nonlaser treatment options.
What resources do you recommend to patients for more information?
I recommend patients visit the Skin of Color Society website (www.skinofcolorsociety.org).
Suggested Readings
- Alexis AF. Fractional laser resurfacing of acne scarring in patients with Fitzpatrick skin types IV-VI. J Drugs Dermatol. 2011;10(12 suppl):s6-s7.
- Alexis AF. Lasers and light-based therapies in ethnic skin: treatment options and recommendations for Fitzpatrick skin types V and VI. Br J Dermatol. 2013;169(suppl 3):91-97.
- Alexis AF, Coley MK, Nijhawan RI, et al. Nonablative fractional laser resurfacing for acne scarring in patients with Fitzpatrick skin phototypes IV-VI. Dermatol Surg. 2016;42:392-402.
- Battle EF, Hobbs LM. Laser-assisted hair removal for darker skin types. Dermatol Ther. 2004;17:177-183.
- Clark CM, Silverberg JI, Alexis AF. A retrospective chart review to assess the safety of nonablative fractional laser resurfacing in Fitzpatrick skin types IV to VI. J Drugs Dermatol. 2013;12:428-431.
- Ross EV, Cooke LM, Timko AL, et al. Treatment of pseudofolliculitis barbae in skin types IV, V, and VI with a long-pulsed neodymium:yttrium aluminum garnet laser. J Am Acad Dermatol. 2002;47:263-270.
What does your patient need to know at the first visit? Does it apply to patients of all genders and ages?
Before performing laser procedures on patients with richly pigmented skin (Fitzpatrick skin types IV–VI), patients need to be informed of the higher risk for pigmentary alterations as potential complications of the procedure. Specifically, hyperpigmentation or hypopigmentation can occur postprocedure, depending on the type of device used, the treatment settings, the technique, the underlying skin disorder being treated, and the patient’s individual response to treatment. Fortunately, these pigment alterations are in most cases self-limited but can last for weeks to months depending on the severity and the nature of the dyspigmentation.
What are your go-to treatments? What are the side effects?
Notwithstanding the higher risks for pigmentary alterations, lasers can be extremely useful for the management of numerous dermatologic concerns in patients with Fitzpatrick skin types IV to VI including laser hair removal for pseudofolliculitis barbae or nonablative fractional laser resurfacing for acne scarring and pigmentary disorders. My go-to treatments include the following: long-pulsed 1064-nm Nd:YAG laser for hair removal in Fitzpatrick skin types V to VI, 808-nm diode laser with linear scanning of hair removal in Fitzpatrick skin type IV or less, 1550-nm erbium-doped nonablative fractional laser for acne scarring in Fitzpatrick skin types IV to VI, and low-power diode 1927-nm fractional laser for melasma and postinflammatory hyperpigmentation in Fitzpatrick skin types IV to VI.
All of these procedures are performed with conservative treatment settings such as low fluences and longer pulse durations for laser hair removal and low treatment densities for fractional laser procedures. Prior to laser resurfacing, I recommend hydroquinone cream 4% twice daily starting 2 weeks before the first session and for 4 weeks posttreatment. These recommendations are based on published evidence (see Suggested Readings) as well as anecdotal experience.
How do you keep patients compliant with treatment?
Emphasizing the need for broad-spectrum sunscreen and avoidance of intense sun exposure before and after laser treatments is important during the initial consultation and prior to each treatment. I warn my patients of the higher risk for hyperpigmentation if the skin is tanned or has recently had intense sun exposure.
What do you do if they refuse treatment?
If patients refuse laser treatment or recommended precautions, then I will consider nonlaser treatment options.
What resources do you recommend to patients for more information?
I recommend patients visit the Skin of Color Society website (www.skinofcolorsociety.org).
Suggested Readings
- Alexis AF. Fractional laser resurfacing of acne scarring in patients with Fitzpatrick skin types IV-VI. J Drugs Dermatol. 2011;10(12 suppl):s6-s7.
- Alexis AF. Lasers and light-based therapies in ethnic skin: treatment options and recommendations for Fitzpatrick skin types V and VI. Br J Dermatol. 2013;169(suppl 3):91-97.
- Alexis AF, Coley MK, Nijhawan RI, et al. Nonablative fractional laser resurfacing for acne scarring in patients with Fitzpatrick skin phototypes IV-VI. Dermatol Surg. 2016;42:392-402.
- Battle EF, Hobbs LM. Laser-assisted hair removal for darker skin types. Dermatol Ther. 2004;17:177-183.
- Clark CM, Silverberg JI, Alexis AF. A retrospective chart review to assess the safety of nonablative fractional laser resurfacing in Fitzpatrick skin types IV to VI. J Drugs Dermatol. 2013;12:428-431.
- Ross EV, Cooke LM, Timko AL, et al. Treatment of pseudofolliculitis barbae in skin types IV, V, and VI with a long-pulsed neodymium:yttrium aluminum garnet laser. J Am Acad Dermatol. 2002;47:263-270.
What does your patient need to know at the first visit? Does it apply to patients of all genders and ages?
Before performing laser procedures on patients with richly pigmented skin (Fitzpatrick skin types IV–VI), patients need to be informed of the higher risk for pigmentary alterations as potential complications of the procedure. Specifically, hyperpigmentation or hypopigmentation can occur postprocedure, depending on the type of device used, the treatment settings, the technique, the underlying skin disorder being treated, and the patient’s individual response to treatment. Fortunately, these pigment alterations are in most cases self-limited but can last for weeks to months depending on the severity and the nature of the dyspigmentation.
What are your go-to treatments? What are the side effects?
Notwithstanding the higher risks for pigmentary alterations, lasers can be extremely useful for the management of numerous dermatologic concerns in patients with Fitzpatrick skin types IV to VI including laser hair removal for pseudofolliculitis barbae or nonablative fractional laser resurfacing for acne scarring and pigmentary disorders. My go-to treatments include the following: long-pulsed 1064-nm Nd:YAG laser for hair removal in Fitzpatrick skin types V to VI, 808-nm diode laser with linear scanning of hair removal in Fitzpatrick skin type IV or less, 1550-nm erbium-doped nonablative fractional laser for acne scarring in Fitzpatrick skin types IV to VI, and low-power diode 1927-nm fractional laser for melasma and postinflammatory hyperpigmentation in Fitzpatrick skin types IV to VI.
All of these procedures are performed with conservative treatment settings such as low fluences and longer pulse durations for laser hair removal and low treatment densities for fractional laser procedures. Prior to laser resurfacing, I recommend hydroquinone cream 4% twice daily starting 2 weeks before the first session and for 4 weeks posttreatment. These recommendations are based on published evidence (see Suggested Readings) as well as anecdotal experience.
How do you keep patients compliant with treatment?
Emphasizing the need for broad-spectrum sunscreen and avoidance of intense sun exposure before and after laser treatments is important during the initial consultation and prior to each treatment. I warn my patients of the higher risk for hyperpigmentation if the skin is tanned or has recently had intense sun exposure.
What do you do if they refuse treatment?
If patients refuse laser treatment or recommended precautions, then I will consider nonlaser treatment options.
What resources do you recommend to patients for more information?
I recommend patients visit the Skin of Color Society website (www.skinofcolorsociety.org).
Suggested Readings
- Alexis AF. Fractional laser resurfacing of acne scarring in patients with Fitzpatrick skin types IV-VI. J Drugs Dermatol. 2011;10(12 suppl):s6-s7.
- Alexis AF. Lasers and light-based therapies in ethnic skin: treatment options and recommendations for Fitzpatrick skin types V and VI. Br J Dermatol. 2013;169(suppl 3):91-97.
- Alexis AF, Coley MK, Nijhawan RI, et al. Nonablative fractional laser resurfacing for acne scarring in patients with Fitzpatrick skin phototypes IV-VI. Dermatol Surg. 2016;42:392-402.
- Battle EF, Hobbs LM. Laser-assisted hair removal for darker skin types. Dermatol Ther. 2004;17:177-183.
- Clark CM, Silverberg JI, Alexis AF. A retrospective chart review to assess the safety of nonablative fractional laser resurfacing in Fitzpatrick skin types IV to VI. J Drugs Dermatol. 2013;12:428-431.
- Ross EV, Cooke LM, Timko AL, et al. Treatment of pseudofolliculitis barbae in skin types IV, V, and VI with a long-pulsed neodymium:yttrium aluminum garnet laser. J Am Acad Dermatol. 2002;47:263-270.

Exophytic Scalp Tumor
The Diagnosis: Primary Cutaneous Carcinosarcoma
A generous shave biopsy and debulking performed on the initial visit revealed an infiltrating tumor consisting of malignant epithelial and stromal components (Figure). The basaloid and squamoid epithelial cells were keratin positive. The stromal cells demonstrated positivity for CD10 but were keratin negative. The epithelial portion of the tumor was composed mostly of basaloid islands of cells with nuclear pleomorphism, scattered mitoses, and focal sebaceous differentiation. The mesenchymal portion of the tumor displayed florid pleomorphism and polymorphism, with many large atypical cells and proliferation. A diagnosis of primary cutaneous carcinosarcoma (PCC) was rendered. Head and neck computed tomography showed tumor penetration of less than 1 cm into scalp soft tissues with no involvement of the underlying bone. There was some evidence of swelling of the supragaleal soft tissues without indication of perineural spread. An 11-mm hyperlucent lower cervical lymph node on the left side that likely represented an incidental finding was noted. Surgical excision with margin evaluation was recommended, but the patient declined. He instead received radiation therapy to the left side of the posterior scalp with a total dose of 30 Gy at 6 Gy per fraction and 1 fraction daily. The patient was found to have a well-healed scar with no evidence of recurrence at 4-week follow-up and again at 5 months after radiation therapy.
|
|
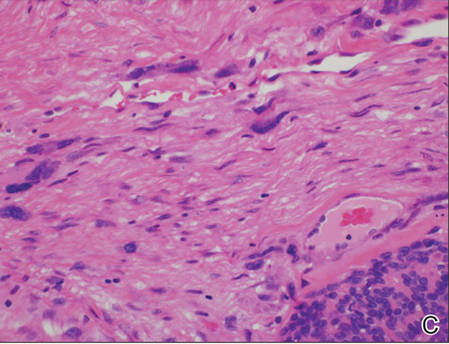
|
| A generous shave biopsy and debulking performed on the initial visit revealed an inflitrating tumor consisting on malignant epithelial and stromal components (A-C)(H&E; original magnifications ×10, ×20, and ×40, respectively). |
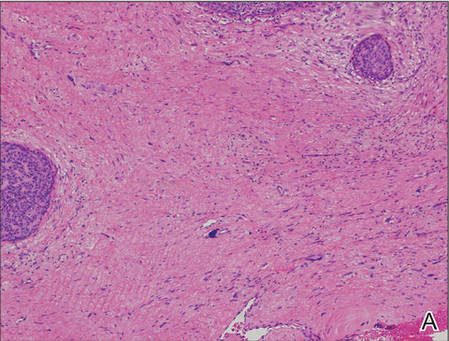
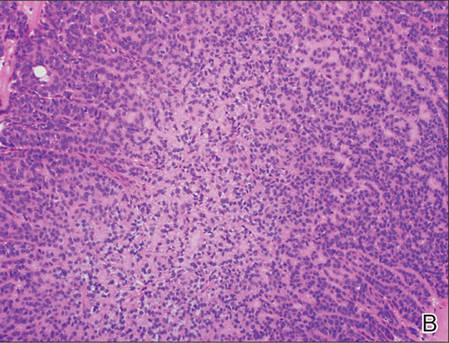
Primary cutaneous carcinosarcoma is a rare biphasic neoplasm of unknown etiology that is characterized by the presence of both malignant epithelial and mesenchymal components.1 Carcinosarcomas have been reported in both the male and female reproductive tracts, urinary tract, gastrointestinal tract, lungs, breasts, larynx, thymus, and thyroid but is uncommon as a primary neoplasm of the skin.2 Epidermal PCC occurs with greater frequency in males than in females and typically presents in the eighth or ninth decades of life.3 These tumors tend to arise in sun-exposed regions, most commonly on the face and scalp.2
Morphologically, PCCs typically are exophytic growths that often feature surface ulceration and may or may not bleed upon palpation.4 Primary cutaneous carcinosarcomas may present as long-standing lesions that have undergone rapid transformation in the weeks preceding presentation.4 It is not uncommon for PCC lesions to carry the clinical diagnosis of squamous cell carcinoma, which suggests notable morphologic overlap between these entities. Histopathologically, PCC shows a basal cell carcinoma and/or a squamous cell carcinoma epithelial component intimately admixed with a sarcomatous component.5 The mesenchymal component of PCC typically resembles a superficial malignant fibrous histiocytoma characterized by pleomorphic nuclei and cytoplasm, necrosis, and an increased number of mitotic figures.2 Immunohistochemistry can be beneficial in the diagnosis of PCC. A combination of p63 and AE1/AE3 stains can be used to confirm cells of epithelial origin. Staining with vimentin, CD10, or caldesmon can help to delineate the mesenchymal component of PCC.
Epidermal PCC most commonly affects elderly individuals with a history of extensive sun exposure. It has been suggested that p53 mutations due to UV damage are key in tumor formation for both epithelial and mesenchymal elements.5 Literature supports a monoclonal origin for the epithelial and mesenchymal components of this tumor; however, there is insufficient evidence.6 Surgical excision is the primary treatment modality for epidermal PCC, but adjuvant or substitutive radiotherapy has been used in some cases.4 The prognosis of PCC is notably better than its visceral counterpart due to early diagnosis and treatment of easily visible lesions. Epidermal PCC has a 70% 5-year disease-free survival rate, while adnexal PCC tends to occur in younger patients and has a 25% 5-year disease-free survival rate.3 Due to the rarity of reported cases and limited follow-up, the long-term prognosis for PCC remains unclear.
We report an unusual case of PCC on the scalp that was successfully treated with radiation therapy alone. This modality should be considered in patients with large tumors who refuse surgery or are not good surgical candidates.
1. El Harroudi T, Ech-Charif S, Amrani M, et al. Primary carcinosarcoma of the skin. J Hand Microsurg. 2010;2:79-81.
2. Patel NK, McKee PH, Smith NP. Primary metaplastic carcinoma (carcinosarcoma) of the skin: a clinicopathologic study of four cases and review of the literature. Am J Dermatopathol. 1997;19:363-372.
3. Hong SH, Hong SJ, Lee Y, et al. Primary cutaneous carcinosarcoma of the shoulder: case report with literature review. Dermatol Surg. 2013;39:338-340.
4. Syme-Grant J, Syme-Grant NJ, Motta L, et al. Are primary cutaneous carcinosarcomas underdiagnosed? five cases and a review of the literature. J Plast Reconstr Aesthet Surg. 2006;59:1402-1408.
5. Tran TA, Muller S, Chaudahri PJ, et al. Cutaneous carcinosarcoma: adnexal vs. epidermal types define high- and low-risk tumors. results of a meta-analysis. J Cutan Pathol. 2005;32:2-11.
6. Paniz Mondolfi AE, Jour G, Johnson M, et al. Primary cutaneous carcinosarcoma: insights into its clonal origin and mutational pattern expression analysis through next-generation sequencing. Hum Pathol. 2013;44:2853-2860.
The Diagnosis: Primary Cutaneous Carcinosarcoma
A generous shave biopsy and debulking performed on the initial visit revealed an infiltrating tumor consisting of malignant epithelial and stromal components (Figure). The basaloid and squamoid epithelial cells were keratin positive. The stromal cells demonstrated positivity for CD10 but were keratin negative. The epithelial portion of the tumor was composed mostly of basaloid islands of cells with nuclear pleomorphism, scattered mitoses, and focal sebaceous differentiation. The mesenchymal portion of the tumor displayed florid pleomorphism and polymorphism, with many large atypical cells and proliferation. A diagnosis of primary cutaneous carcinosarcoma (PCC) was rendered. Head and neck computed tomography showed tumor penetration of less than 1 cm into scalp soft tissues with no involvement of the underlying bone. There was some evidence of swelling of the supragaleal soft tissues without indication of perineural spread. An 11-mm hyperlucent lower cervical lymph node on the left side that likely represented an incidental finding was noted. Surgical excision with margin evaluation was recommended, but the patient declined. He instead received radiation therapy to the left side of the posterior scalp with a total dose of 30 Gy at 6 Gy per fraction and 1 fraction daily. The patient was found to have a well-healed scar with no evidence of recurrence at 4-week follow-up and again at 5 months after radiation therapy.
|
|
|
|
| A generous shave biopsy and debulking performed on the initial visit revealed an inflitrating tumor consisting on malignant epithelial and stromal components (A-C)(H&E; original magnifications ×10, ×20, and ×40, respectively). |
Primary cutaneous carcinosarcoma is a rare biphasic neoplasm of unknown etiology that is characterized by the presence of both malignant epithelial and mesenchymal components.1 Carcinosarcomas have been reported in both the male and female reproductive tracts, urinary tract, gastrointestinal tract, lungs, breasts, larynx, thymus, and thyroid but is uncommon as a primary neoplasm of the skin.2 Epidermal PCC occurs with greater frequency in males than in females and typically presents in the eighth or ninth decades of life.3 These tumors tend to arise in sun-exposed regions, most commonly on the face and scalp.2
Morphologically, PCCs typically are exophytic growths that often feature surface ulceration and may or may not bleed upon palpation.4 Primary cutaneous carcinosarcomas may present as long-standing lesions that have undergone rapid transformation in the weeks preceding presentation.4 It is not uncommon for PCC lesions to carry the clinical diagnosis of squamous cell carcinoma, which suggests notable morphologic overlap between these entities. Histopathologically, PCC shows a basal cell carcinoma and/or a squamous cell carcinoma epithelial component intimately admixed with a sarcomatous component.5 The mesenchymal component of PCC typically resembles a superficial malignant fibrous histiocytoma characterized by pleomorphic nuclei and cytoplasm, necrosis, and an increased number of mitotic figures.2 Immunohistochemistry can be beneficial in the diagnosis of PCC. A combination of p63 and AE1/AE3 stains can be used to confirm cells of epithelial origin. Staining with vimentin, CD10, or caldesmon can help to delineate the mesenchymal component of PCC.
Epidermal PCC most commonly affects elderly individuals with a history of extensive sun exposure. It has been suggested that p53 mutations due to UV damage are key in tumor formation for both epithelial and mesenchymal elements.5 Literature supports a monoclonal origin for the epithelial and mesenchymal components of this tumor; however, there is insufficient evidence.6 Surgical excision is the primary treatment modality for epidermal PCC, but adjuvant or substitutive radiotherapy has been used in some cases.4 The prognosis of PCC is notably better than its visceral counterpart due to early diagnosis and treatment of easily visible lesions. Epidermal PCC has a 70% 5-year disease-free survival rate, while adnexal PCC tends to occur in younger patients and has a 25% 5-year disease-free survival rate.3 Due to the rarity of reported cases and limited follow-up, the long-term prognosis for PCC remains unclear.
We report an unusual case of PCC on the scalp that was successfully treated with radiation therapy alone. This modality should be considered in patients with large tumors who refuse surgery or are not good surgical candidates.
The Diagnosis: Primary Cutaneous Carcinosarcoma
A generous shave biopsy and debulking performed on the initial visit revealed an infiltrating tumor consisting of malignant epithelial and stromal components (Figure). The basaloid and squamoid epithelial cells were keratin positive. The stromal cells demonstrated positivity for CD10 but were keratin negative. The epithelial portion of the tumor was composed mostly of basaloid islands of cells with nuclear pleomorphism, scattered mitoses, and focal sebaceous differentiation. The mesenchymal portion of the tumor displayed florid pleomorphism and polymorphism, with many large atypical cells and proliferation. A diagnosis of primary cutaneous carcinosarcoma (PCC) was rendered. Head and neck computed tomography showed tumor penetration of less than 1 cm into scalp soft tissues with no involvement of the underlying bone. There was some evidence of swelling of the supragaleal soft tissues without indication of perineural spread. An 11-mm hyperlucent lower cervical lymph node on the left side that likely represented an incidental finding was noted. Surgical excision with margin evaluation was recommended, but the patient declined. He instead received radiation therapy to the left side of the posterior scalp with a total dose of 30 Gy at 6 Gy per fraction and 1 fraction daily. The patient was found to have a well-healed scar with no evidence of recurrence at 4-week follow-up and again at 5 months after radiation therapy.
|
|
|
|
| A generous shave biopsy and debulking performed on the initial visit revealed an inflitrating tumor consisting on malignant epithelial and stromal components (A-C)(H&E; original magnifications ×10, ×20, and ×40, respectively). |
Primary cutaneous carcinosarcoma is a rare biphasic neoplasm of unknown etiology that is characterized by the presence of both malignant epithelial and mesenchymal components.1 Carcinosarcomas have been reported in both the male and female reproductive tracts, urinary tract, gastrointestinal tract, lungs, breasts, larynx, thymus, and thyroid but is uncommon as a primary neoplasm of the skin.2 Epidermal PCC occurs with greater frequency in males than in females and typically presents in the eighth or ninth decades of life.3 These tumors tend to arise in sun-exposed regions, most commonly on the face and scalp.2
Morphologically, PCCs typically are exophytic growths that often feature surface ulceration and may or may not bleed upon palpation.4 Primary cutaneous carcinosarcomas may present as long-standing lesions that have undergone rapid transformation in the weeks preceding presentation.4 It is not uncommon for PCC lesions to carry the clinical diagnosis of squamous cell carcinoma, which suggests notable morphologic overlap between these entities. Histopathologically, PCC shows a basal cell carcinoma and/or a squamous cell carcinoma epithelial component intimately admixed with a sarcomatous component.5 The mesenchymal component of PCC typically resembles a superficial malignant fibrous histiocytoma characterized by pleomorphic nuclei and cytoplasm, necrosis, and an increased number of mitotic figures.2 Immunohistochemistry can be beneficial in the diagnosis of PCC. A combination of p63 and AE1/AE3 stains can be used to confirm cells of epithelial origin. Staining with vimentin, CD10, or caldesmon can help to delineate the mesenchymal component of PCC.
Epidermal PCC most commonly affects elderly individuals with a history of extensive sun exposure. It has been suggested that p53 mutations due to UV damage are key in tumor formation for both epithelial and mesenchymal elements.5 Literature supports a monoclonal origin for the epithelial and mesenchymal components of this tumor; however, there is insufficient evidence.6 Surgical excision is the primary treatment modality for epidermal PCC, but adjuvant or substitutive radiotherapy has been used in some cases.4 The prognosis of PCC is notably better than its visceral counterpart due to early diagnosis and treatment of easily visible lesions. Epidermal PCC has a 70% 5-year disease-free survival rate, while adnexal PCC tends to occur in younger patients and has a 25% 5-year disease-free survival rate.3 Due to the rarity of reported cases and limited follow-up, the long-term prognosis for PCC remains unclear.
We report an unusual case of PCC on the scalp that was successfully treated with radiation therapy alone. This modality should be considered in patients with large tumors who refuse surgery or are not good surgical candidates.
1. El Harroudi T, Ech-Charif S, Amrani M, et al. Primary carcinosarcoma of the skin. J Hand Microsurg. 2010;2:79-81.
2. Patel NK, McKee PH, Smith NP. Primary metaplastic carcinoma (carcinosarcoma) of the skin: a clinicopathologic study of four cases and review of the literature. Am J Dermatopathol. 1997;19:363-372.
3. Hong SH, Hong SJ, Lee Y, et al. Primary cutaneous carcinosarcoma of the shoulder: case report with literature review. Dermatol Surg. 2013;39:338-340.
4. Syme-Grant J, Syme-Grant NJ, Motta L, et al. Are primary cutaneous carcinosarcomas underdiagnosed? five cases and a review of the literature. J Plast Reconstr Aesthet Surg. 2006;59:1402-1408.
5. Tran TA, Muller S, Chaudahri PJ, et al. Cutaneous carcinosarcoma: adnexal vs. epidermal types define high- and low-risk tumors. results of a meta-analysis. J Cutan Pathol. 2005;32:2-11.
6. Paniz Mondolfi AE, Jour G, Johnson M, et al. Primary cutaneous carcinosarcoma: insights into its clonal origin and mutational pattern expression analysis through next-generation sequencing. Hum Pathol. 2013;44:2853-2860.
1. El Harroudi T, Ech-Charif S, Amrani M, et al. Primary carcinosarcoma of the skin. J Hand Microsurg. 2010;2:79-81.
2. Patel NK, McKee PH, Smith NP. Primary metaplastic carcinoma (carcinosarcoma) of the skin: a clinicopathologic study of four cases and review of the literature. Am J Dermatopathol. 1997;19:363-372.
3. Hong SH, Hong SJ, Lee Y, et al. Primary cutaneous carcinosarcoma of the shoulder: case report with literature review. Dermatol Surg. 2013;39:338-340.
4. Syme-Grant J, Syme-Grant NJ, Motta L, et al. Are primary cutaneous carcinosarcomas underdiagnosed? five cases and a review of the literature. J Plast Reconstr Aesthet Surg. 2006;59:1402-1408.
5. Tran TA, Muller S, Chaudahri PJ, et al. Cutaneous carcinosarcoma: adnexal vs. epidermal types define high- and low-risk tumors. results of a meta-analysis. J Cutan Pathol. 2005;32:2-11.
6. Paniz Mondolfi AE, Jour G, Johnson M, et al. Primary cutaneous carcinosarcoma: insights into its clonal origin and mutational pattern expression analysis through next-generation sequencing. Hum Pathol. 2013;44:2853-2860.
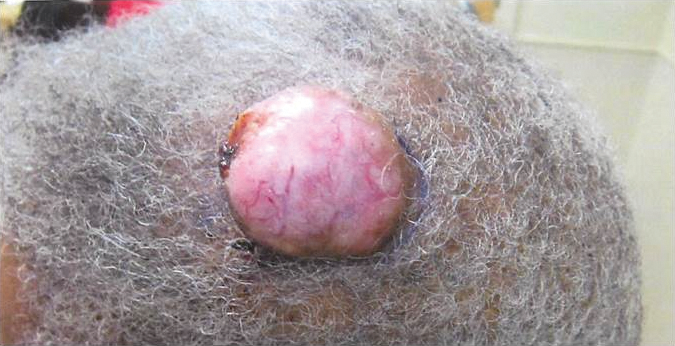
An 81-year-old man presented with a 3.5×3.0-cm pink exophytic tumor with an eroded surface and prominent vascularity on the left side of the parietal scalp. The patient reported that the tumor had been present for more than 30 years but recently had grown larger in size. He denied pain or pruritus in association with the lesion and did not report any systemic symptoms. He had received no prior treatments for the tumor.

Prevalence of Glaucoma in Patients With Vitiligo
Vitiligo is an acquired idiopathic disease of unknown etiology. Characterized by depigmented maculae and melanocytic destruction, it usually presents in childhood or young adulthood. The incidence of vitiligo ranges from 0.5% to 2% globally and there is no racial or gender predilection.1
Patients with vitiligo may exhibit pigmentary abnormalities of the iris and retina.2 Noninflammatory depigmented lesions of the ocular fundus observed in vitiligo indicate a local loss of melanocytes.1 The fact that melanocytes are present not only in the skin and roots of the hair but also in the uvea and stria vascularis of the inner ear may explain the ophthalmologic disorders that accompany vitiligo.3 The term glaucoma refers to a large number of diseases that share a common feature: a distinctive and progressive optic neuropathy that may derive from various risks and is associated with a gradual loss of the visual field. If the disorder is not diagnosed and treated properly it could cause blindness.
Glaucoma is classified on the basis of the underlying abnormality that causes intraocular pressure (IOP) to rise. Glaucoma is first divided into open-angle and angle-closure glaucoma; glaucoma associated with developmental anomalies is then subdivided according to specific alterations.4
A PubMed search of articles indexed for MEDLINE using the terms vitiligo and glaucoma revealed only 1 study examining the incidence of glaucoma in patients with vitiligo.5 In the study reported here, we determined the presence of and possible risk factors for glaucoma in patients with vitiligo who had presented to the dermatology polyclinic.
Methods
We registered 49 patients diagnosed with vitiligo by clinical and Wood light examination and 20 age- and sex-matched healthy controls. Patients who were using topical corticosteroid treatments for vitiligo lesions located on the face were excluded from the study due to the glaucoma-inducing effects of corticosteroids. Similarly, patients who received drugs with sympathetic and parasympathetic action that can cause glaucoma were excluded.
The patients received a comprehensive ophthalmologic examination that included visual acuity testing, refraction, IOP measurement, gonioscopy, and fundus examination. All patients and controls underwent visual field tests and optic nerve head analyses using a confocal scanning laser ophthalmoscope. Glaucoma was diagnosed based on fundus examination, IOP measurement, field of vision evaluation, and optic nerve head analysis.
Informed consent was obtained from all participants. The research protocol was approved by the university hospital ethics committee.
Results
The study registered a total of 49 patients with vitiligo (28 female; 21 male) and 20 healthy controls (10 female; 10 male) with a variety of demographic and clinical characteristics (Table 1).
Mean (SD) IOP values were 13.83 (2.84) mm Hg for the right eye and 13.89 (2.60) mm Hg for the left eye in the vitiligo group. Values were 14.35 (2.56) mm Hg and 14.95 (2.92) mm Hg, respectively, in the control group. The IOP differences between the 2 groups were not statistically significant (P>.05).
Nine patients (18.4%) in the vitiligo group were found to have signs of normal-tension glaucoma (NTG). Optic nerve damage and vision loss occurs in the presence of normal IOP in NTG. There were no signs of NTG in the control group. Normal-tension glaucoma was diagnosed in the vitiligo group based on glaucomatous optic disc appearance, visual field defects, and structural analysis of the entire optic nerve head in confocal scanning laser ophthalmoscope. The NTG difference between the vitiligo and control groups was statistically significant (P=.04).
In the vitiligo group, of the 9 patients who had NTG, 6 had periorbital vitiligo lesions; the remaining 3 had none. Although patients who had periorbital lesions had a higher rate of glaucoma relative to the patients without periorbital lesions, the difference was not statistically significant (P>.05).
No statistically significant differences (P>.05) were found between patients with vitiligo with and without glaucoma in terms of age, sex, disease duration, family history of vitiligo, presence or absence of periorbital involvement, manner of involvement, percentage of the involved body areas, and IOP (Table 1).
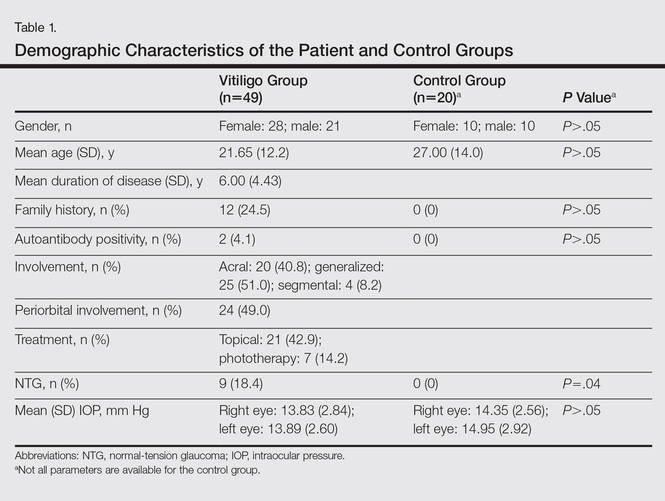
Comment
Glaucoma is characterized by increased IOP, visual field loss, and changes in the optic nerve head. Although elevated IOP is common in ocular hypertension as well as in glaucoma, there is no glaucomatous visual field loss in ocular hypertension. In NTG, on the other hand, glaucomatous visual field loss and optic nerve head changes occur without an increase in IOP.6 Normal-tension glaucoma is a particular type of open-angle glaucoma. It is believed that NTG and high-tension glaucoma induce optic nerve head damage through different means.7 Alternative theories have been put forth to account for the glaucomatous damage to the optic nerve head that occurs in NTG, despite normal or close to normal IOP. These theories include vascular disorders (eg, ischemia, which interrupts the orthograde or retrograde axonal transport), excessive accumulation of free radicals, triggering of apoptosis, and low resistance of lamina cribrosa.8
Although there are various studies exploring ocular symptoms in patients with vitiligo,9-15 only 1 study has examined the incidence of glaucoma in this group of patients.5 Biswas et al11 examined ocular signs in 100 patients with vitiligo and found that 23% of patients had hypopigmented foci in the iris, 18% had pigmentation in the anterior chamber, 11% had chorioretinal degeneration, 9% had hypopigmentation of the retinal pigment epithelium, 5% had uveitis, and 34% were evaluated as normal. In this study, the authors concluded that there was a strong relationship between vitiligo and eye diseases.11 When Gopal et al9 compared the eye examinations of 150 vitiligo patients and 100 healthy controls, they found uveitis, iris, and retinal pigmentary abnormalities in 16% of the vitiligo patients (P<.001).
Rogosić et al5 examined the incidence of glaucoma in 42 patients with vitiligo and found primary open-angle glaucoma in 24 (57%) patients. The patients had a mean age of 56 years, mean disease duration of 13 years, and mean IOP of 18 mm Hg for the right eye and 17.5 mm Hg for the left eye. The incidence of glaucoma was significantly higher in patients with vitiligo (P<.001) and increased with disease duration.5
Similar studies, however, have failed to show a relationship between vitiligo and glaucoma. In a study that evaluated the retinal pigment epithelium and the optic nerve in patients with vitiligo, Perossini et al10 found that the fundus examination of the patients was perfectly normal.
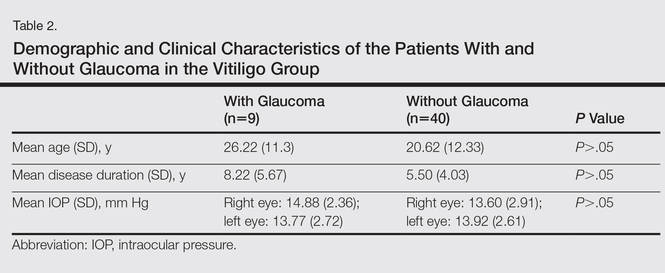
In our study, we detected NTG in 18.4% of patients with vitiligo. We did not find a significant statistical difference between patients with and without glaucoma (Table 2). Rogosić et al5 found a significant relationship between age and glaucoma incidence, but we did not find such a relationship, which we believe is because the mean age of our patients was lower than the prior study.
In vitiligo, melanocytes are destroyed through an unknown mechanism. Although the cellular and molecular mechanisms causing melanocytic destruction have not yet been determined, various hypotheses have been put forward to explain the etiopathogenesis of vitiligo. Among these, the most commonly held hypotheses are the neural, self-destruction, and autoimmune hypotheses.16
Based on the observation that stress and serious trauma could precipitate or trigger the onset of vitiligo,16 the neural hypothesis holds that neurochemical mediators released from the edges of the nerve endings exert toxic effects on melanocytes. The fact that both melanocytes and choroidal pigment cells originate from the mesenchyme and dermatomal spreading of segmental vitiligo are arguments propounded in favor of this hypothesis.17
The self-destruction hypothesis suggests that the intrinsic protective mechanisms that normally enable melanocytes to eliminate toxic intermediate products or metabolites on the melanogenesis path have been impaired in patients with vitiligo.18,19 There is evidence of increased oxidative stress over the whole epidermis of patients with vitiligo.20 Thus, free radicals affect melanin and cause membrane damage via lipid peroxidation reactions.21
The autoimmune hypothesis proposes a clinical relationship between vitiligo and several diseases believed to be autoimmune. Because the macrophage infiltration observed in vitiligo lesions is more pronounced on the perilesional skin, this hypothesis holds that macrophages may play a role in melanocyte removal.21 The Koebner phenomenon observed in vitiligo lends support to the critical role of trauma in the etiopathogenesis of the disease.
Although we could not explain the co-presence of vitiligo and glaucoma, we believe that it may result from the fact that both diseases are observed in tissues that have the same embryologic origin and etiology, perhaps vascular or neural disorders, excessive accumulation of free radicals, or the triggering of apoptosis. Dermatologists should be alert to the presence of glaucoma in patients with vitiligo because glaucoma is an eye disease that progresses slowly and may lead to vision loss.
1. Ortonne JP. Vitiligo and other disorders of hypopigmentation. In: Bolognia JB, Jorizzo JL, Rapini RP, eds. Dermatology. 1st ed. New York, NY: Mosby; 2003:947-973.
2. Ortonne JP, Bahadoran P, Fitzpatrick TB, et al. Hypomelanoses and hypermelanoses. In: Freedberg IM, Eisen AZ, Wolff K, eds. Fitzpatrick’s Dermatology in General Medicine. 6th ed. New York, NY: McGraw-Hill; 2003:836-881.
3. van den Wijngaard R, Wijngaard R, Wankowiczs-Kalinsa A, et al. Autoimmune melanocyte destruction in vitiligo. Lab Invest. 2001;81:1061-1067.
4. Shields MB, Ritch R, Krupin T. Classification of the glaucomas. In: Ritch R, Shields MB, Krupin T, eds. The Glaucomas. St Louis, MO: C.V. Mosby Co; 1996:717-725.
5. Rogosić V, Bojić L, Puizina-Ivić N, et al. Vitiligo and glaucoma–an association or a coincidence? a pilot study. Acta Dermatovenerol Croat. 2010;18:21-26.
6. Anderson DR. Normal-tension glaucoma (low-tension glaucoma). Indian J Ophthalmol. 2011;59(suppl 59):S97-S101.
7. Iwata K. Primary open angle glaucoma and low tension glaucoma–pathogenesis and mechanism of optic nerve damage [in Japanese]. Nippon Ganka Gakkai Zasshi. 1992;96:1501-1531.
8. Hitchings RA, Anderton SA. A comparative study of visual field defects seen in patients with low-tension glaucoma and chronic simple glaucoma. Br J Ophthalmol. 1983;67:818-821.
9. Gopal KV, Rama Rao GR, Kumar YH, et al. Vitiligo: a part of a systemic autoimmune process. Indian J Dermatol Venereol Leprol. 2007;73:162-165.
10. Perossini M, Turio E, Perossini T, et al. Vitiligo: ocular and electrophysiological findings. G Ital Dermatol Venereol. 2010;145:141-149.
11. Biswas G, Barbhuiya JN, Biswas MC, et al. Clinical pattern of ocular manifestations in vitiligo. J Indian Med Assoc. 2003;101:478-480.
12. Park S, Albert DM, Bolognia JL. Ocular manifestations of pigmentary disorders. Dermatol Clin. 1992;10:609-622.
13. Albert DM, Nordlund JJ, Lerner AB. Ocular abnormalities occurring with vitiligo. Ophthalmology. 1979;86:1145-1160.
14. Wagoner MD, Albert DM, Lerner AB, et al. New observations on vitiligo and ocular disease. Am J Ophthalmol. 1983;96:16-26.
15. Cowan CL Jr, Halder RM, Grimes PE, et al. Ocular disturbances in vitiligo. J Am Acad Dermatol. 1986;15:17-24.
16. Orecchia GE. Neural pathogenesis. In: Hann SK, Nordlund JJ. Vitiligo. Oxford, England: Blackwell Science Ltd; 2000:142-150.
17. Braun-Falco O, Plewig G, Wolf HH, et al. Disorders of melanin pigmentation. In: Bartels V, ed. Dermatology. Berlin, Germany: Springer; 2000:1013-1042.
18. Tüzün Y, Kotoğyan A, Aydemir EH, et al. Pigmentasyon bozuklukları. In: Baransü O. Dermatoloji. 2nd ed. Istanbul: Nobel Tıp Kitabevi; 1994:557-559.
19. Odom RB, James WD, Berger TG. Disturbances of pigmentation. In: Odom RB, James WD, Berger TG. Andrews’ Diseases of the Skin. 9th ed. Philadelphia, PA: W.B. Saunders Company; 2000:1065-1068.
20. Schallreuter KU. Biochemical theory of vitiligo: a role of pteridines in pigmentation. In: Hann SK, Nordlund JJ. Vitiligo. London, England: Blackwell Science Ltd; 2000:151-159.
21. van den Wijngaard R, Wankowicz-Kalinska A, Le Poole C, et al. Local immune response in skin of generalized vitiligo patients. destruction of melanocytes is associated with the predominent presence of CLA+T cells at the perilesional site. Lab Invest. 2000;80:1299-1309.
Vitiligo is an acquired idiopathic disease of unknown etiology. Characterized by depigmented maculae and melanocytic destruction, it usually presents in childhood or young adulthood. The incidence of vitiligo ranges from 0.5% to 2% globally and there is no racial or gender predilection.1
Patients with vitiligo may exhibit pigmentary abnormalities of the iris and retina.2 Noninflammatory depigmented lesions of the ocular fundus observed in vitiligo indicate a local loss of melanocytes.1 The fact that melanocytes are present not only in the skin and roots of the hair but also in the uvea and stria vascularis of the inner ear may explain the ophthalmologic disorders that accompany vitiligo.3 The term glaucoma refers to a large number of diseases that share a common feature: a distinctive and progressive optic neuropathy that may derive from various risks and is associated with a gradual loss of the visual field. If the disorder is not diagnosed and treated properly it could cause blindness.
Glaucoma is classified on the basis of the underlying abnormality that causes intraocular pressure (IOP) to rise. Glaucoma is first divided into open-angle and angle-closure glaucoma; glaucoma associated with developmental anomalies is then subdivided according to specific alterations.4
A PubMed search of articles indexed for MEDLINE using the terms vitiligo and glaucoma revealed only 1 study examining the incidence of glaucoma in patients with vitiligo.5 In the study reported here, we determined the presence of and possible risk factors for glaucoma in patients with vitiligo who had presented to the dermatology polyclinic.
Methods
We registered 49 patients diagnosed with vitiligo by clinical and Wood light examination and 20 age- and sex-matched healthy controls. Patients who were using topical corticosteroid treatments for vitiligo lesions located on the face were excluded from the study due to the glaucoma-inducing effects of corticosteroids. Similarly, patients who received drugs with sympathetic and parasympathetic action that can cause glaucoma were excluded.
The patients received a comprehensive ophthalmologic examination that included visual acuity testing, refraction, IOP measurement, gonioscopy, and fundus examination. All patients and controls underwent visual field tests and optic nerve head analyses using a confocal scanning laser ophthalmoscope. Glaucoma was diagnosed based on fundus examination, IOP measurement, field of vision evaluation, and optic nerve head analysis.
Informed consent was obtained from all participants. The research protocol was approved by the university hospital ethics committee.
Results
The study registered a total of 49 patients with vitiligo (28 female; 21 male) and 20 healthy controls (10 female; 10 male) with a variety of demographic and clinical characteristics (Table 1).
Mean (SD) IOP values were 13.83 (2.84) mm Hg for the right eye and 13.89 (2.60) mm Hg for the left eye in the vitiligo group. Values were 14.35 (2.56) mm Hg and 14.95 (2.92) mm Hg, respectively, in the control group. The IOP differences between the 2 groups were not statistically significant (P>.05).
Nine patients (18.4%) in the vitiligo group were found to have signs of normal-tension glaucoma (NTG). Optic nerve damage and vision loss occurs in the presence of normal IOP in NTG. There were no signs of NTG in the control group. Normal-tension glaucoma was diagnosed in the vitiligo group based on glaucomatous optic disc appearance, visual field defects, and structural analysis of the entire optic nerve head in confocal scanning laser ophthalmoscope. The NTG difference between the vitiligo and control groups was statistically significant (P=.04).
In the vitiligo group, of the 9 patients who had NTG, 6 had periorbital vitiligo lesions; the remaining 3 had none. Although patients who had periorbital lesions had a higher rate of glaucoma relative to the patients without periorbital lesions, the difference was not statistically significant (P>.05).
No statistically significant differences (P>.05) were found between patients with vitiligo with and without glaucoma in terms of age, sex, disease duration, family history of vitiligo, presence or absence of periorbital involvement, manner of involvement, percentage of the involved body areas, and IOP (Table 1).
Comment
Glaucoma is characterized by increased IOP, visual field loss, and changes in the optic nerve head. Although elevated IOP is common in ocular hypertension as well as in glaucoma, there is no glaucomatous visual field loss in ocular hypertension. In NTG, on the other hand, glaucomatous visual field loss and optic nerve head changes occur without an increase in IOP.6 Normal-tension glaucoma is a particular type of open-angle glaucoma. It is believed that NTG and high-tension glaucoma induce optic nerve head damage through different means.7 Alternative theories have been put forth to account for the glaucomatous damage to the optic nerve head that occurs in NTG, despite normal or close to normal IOP. These theories include vascular disorders (eg, ischemia, which interrupts the orthograde or retrograde axonal transport), excessive accumulation of free radicals, triggering of apoptosis, and low resistance of lamina cribrosa.8
Although there are various studies exploring ocular symptoms in patients with vitiligo,9-15 only 1 study has examined the incidence of glaucoma in this group of patients.5 Biswas et al11 examined ocular signs in 100 patients with vitiligo and found that 23% of patients had hypopigmented foci in the iris, 18% had pigmentation in the anterior chamber, 11% had chorioretinal degeneration, 9% had hypopigmentation of the retinal pigment epithelium, 5% had uveitis, and 34% were evaluated as normal. In this study, the authors concluded that there was a strong relationship between vitiligo and eye diseases.11 When Gopal et al9 compared the eye examinations of 150 vitiligo patients and 100 healthy controls, they found uveitis, iris, and retinal pigmentary abnormalities in 16% of the vitiligo patients (P<.001).
Rogosić et al5 examined the incidence of glaucoma in 42 patients with vitiligo and found primary open-angle glaucoma in 24 (57%) patients. The patients had a mean age of 56 years, mean disease duration of 13 years, and mean IOP of 18 mm Hg for the right eye and 17.5 mm Hg for the left eye. The incidence of glaucoma was significantly higher in patients with vitiligo (P<.001) and increased with disease duration.5
Similar studies, however, have failed to show a relationship between vitiligo and glaucoma. In a study that evaluated the retinal pigment epithelium and the optic nerve in patients with vitiligo, Perossini et al10 found that the fundus examination of the patients was perfectly normal.
In our study, we detected NTG in 18.4% of patients with vitiligo. We did not find a significant statistical difference between patients with and without glaucoma (Table 2). Rogosić et al5 found a significant relationship between age and glaucoma incidence, but we did not find such a relationship, which we believe is because the mean age of our patients was lower than the prior study.
In vitiligo, melanocytes are destroyed through an unknown mechanism. Although the cellular and molecular mechanisms causing melanocytic destruction have not yet been determined, various hypotheses have been put forward to explain the etiopathogenesis of vitiligo. Among these, the most commonly held hypotheses are the neural, self-destruction, and autoimmune hypotheses.16
Based on the observation that stress and serious trauma could precipitate or trigger the onset of vitiligo,16 the neural hypothesis holds that neurochemical mediators released from the edges of the nerve endings exert toxic effects on melanocytes. The fact that both melanocytes and choroidal pigment cells originate from the mesenchyme and dermatomal spreading of segmental vitiligo are arguments propounded in favor of this hypothesis.17
The self-destruction hypothesis suggests that the intrinsic protective mechanisms that normally enable melanocytes to eliminate toxic intermediate products or metabolites on the melanogenesis path have been impaired in patients with vitiligo.18,19 There is evidence of increased oxidative stress over the whole epidermis of patients with vitiligo.20 Thus, free radicals affect melanin and cause membrane damage via lipid peroxidation reactions.21
The autoimmune hypothesis proposes a clinical relationship between vitiligo and several diseases believed to be autoimmune. Because the macrophage infiltration observed in vitiligo lesions is more pronounced on the perilesional skin, this hypothesis holds that macrophages may play a role in melanocyte removal.21 The Koebner phenomenon observed in vitiligo lends support to the critical role of trauma in the etiopathogenesis of the disease.
Although we could not explain the co-presence of vitiligo and glaucoma, we believe that it may result from the fact that both diseases are observed in tissues that have the same embryologic origin and etiology, perhaps vascular or neural disorders, excessive accumulation of free radicals, or the triggering of apoptosis. Dermatologists should be alert to the presence of glaucoma in patients with vitiligo because glaucoma is an eye disease that progresses slowly and may lead to vision loss.
Vitiligo is an acquired idiopathic disease of unknown etiology. Characterized by depigmented maculae and melanocytic destruction, it usually presents in childhood or young adulthood. The incidence of vitiligo ranges from 0.5% to 2% globally and there is no racial or gender predilection.1
Patients with vitiligo may exhibit pigmentary abnormalities of the iris and retina.2 Noninflammatory depigmented lesions of the ocular fundus observed in vitiligo indicate a local loss of melanocytes.1 The fact that melanocytes are present not only in the skin and roots of the hair but also in the uvea and stria vascularis of the inner ear may explain the ophthalmologic disorders that accompany vitiligo.3 The term glaucoma refers to a large number of diseases that share a common feature: a distinctive and progressive optic neuropathy that may derive from various risks and is associated with a gradual loss of the visual field. If the disorder is not diagnosed and treated properly it could cause blindness.
Glaucoma is classified on the basis of the underlying abnormality that causes intraocular pressure (IOP) to rise. Glaucoma is first divided into open-angle and angle-closure glaucoma; glaucoma associated with developmental anomalies is then subdivided according to specific alterations.4
A PubMed search of articles indexed for MEDLINE using the terms vitiligo and glaucoma revealed only 1 study examining the incidence of glaucoma in patients with vitiligo.5 In the study reported here, we determined the presence of and possible risk factors for glaucoma in patients with vitiligo who had presented to the dermatology polyclinic.
Methods
We registered 49 patients diagnosed with vitiligo by clinical and Wood light examination and 20 age- and sex-matched healthy controls. Patients who were using topical corticosteroid treatments for vitiligo lesions located on the face were excluded from the study due to the glaucoma-inducing effects of corticosteroids. Similarly, patients who received drugs with sympathetic and parasympathetic action that can cause glaucoma were excluded.
The patients received a comprehensive ophthalmologic examination that included visual acuity testing, refraction, IOP measurement, gonioscopy, and fundus examination. All patients and controls underwent visual field tests and optic nerve head analyses using a confocal scanning laser ophthalmoscope. Glaucoma was diagnosed based on fundus examination, IOP measurement, field of vision evaluation, and optic nerve head analysis.
Informed consent was obtained from all participants. The research protocol was approved by the university hospital ethics committee.
Results
The study registered a total of 49 patients with vitiligo (28 female; 21 male) and 20 healthy controls (10 female; 10 male) with a variety of demographic and clinical characteristics (Table 1).
Mean (SD) IOP values were 13.83 (2.84) mm Hg for the right eye and 13.89 (2.60) mm Hg for the left eye in the vitiligo group. Values were 14.35 (2.56) mm Hg and 14.95 (2.92) mm Hg, respectively, in the control group. The IOP differences between the 2 groups were not statistically significant (P>.05).
Nine patients (18.4%) in the vitiligo group were found to have signs of normal-tension glaucoma (NTG). Optic nerve damage and vision loss occurs in the presence of normal IOP in NTG. There were no signs of NTG in the control group. Normal-tension glaucoma was diagnosed in the vitiligo group based on glaucomatous optic disc appearance, visual field defects, and structural analysis of the entire optic nerve head in confocal scanning laser ophthalmoscope. The NTG difference between the vitiligo and control groups was statistically significant (P=.04).
In the vitiligo group, of the 9 patients who had NTG, 6 had periorbital vitiligo lesions; the remaining 3 had none. Although patients who had periorbital lesions had a higher rate of glaucoma relative to the patients without periorbital lesions, the difference was not statistically significant (P>.05).
No statistically significant differences (P>.05) were found between patients with vitiligo with and without glaucoma in terms of age, sex, disease duration, family history of vitiligo, presence or absence of periorbital involvement, manner of involvement, percentage of the involved body areas, and IOP (Table 1).
Comment
Glaucoma is characterized by increased IOP, visual field loss, and changes in the optic nerve head. Although elevated IOP is common in ocular hypertension as well as in glaucoma, there is no glaucomatous visual field loss in ocular hypertension. In NTG, on the other hand, glaucomatous visual field loss and optic nerve head changes occur without an increase in IOP.6 Normal-tension glaucoma is a particular type of open-angle glaucoma. It is believed that NTG and high-tension glaucoma induce optic nerve head damage through different means.7 Alternative theories have been put forth to account for the glaucomatous damage to the optic nerve head that occurs in NTG, despite normal or close to normal IOP. These theories include vascular disorders (eg, ischemia, which interrupts the orthograde or retrograde axonal transport), excessive accumulation of free radicals, triggering of apoptosis, and low resistance of lamina cribrosa.8
Although there are various studies exploring ocular symptoms in patients with vitiligo,9-15 only 1 study has examined the incidence of glaucoma in this group of patients.5 Biswas et al11 examined ocular signs in 100 patients with vitiligo and found that 23% of patients had hypopigmented foci in the iris, 18% had pigmentation in the anterior chamber, 11% had chorioretinal degeneration, 9% had hypopigmentation of the retinal pigment epithelium, 5% had uveitis, and 34% were evaluated as normal. In this study, the authors concluded that there was a strong relationship between vitiligo and eye diseases.11 When Gopal et al9 compared the eye examinations of 150 vitiligo patients and 100 healthy controls, they found uveitis, iris, and retinal pigmentary abnormalities in 16% of the vitiligo patients (P<.001).
Rogosić et al5 examined the incidence of glaucoma in 42 patients with vitiligo and found primary open-angle glaucoma in 24 (57%) patients. The patients had a mean age of 56 years, mean disease duration of 13 years, and mean IOP of 18 mm Hg for the right eye and 17.5 mm Hg for the left eye. The incidence of glaucoma was significantly higher in patients with vitiligo (P<.001) and increased with disease duration.5
Similar studies, however, have failed to show a relationship between vitiligo and glaucoma. In a study that evaluated the retinal pigment epithelium and the optic nerve in patients with vitiligo, Perossini et al10 found that the fundus examination of the patients was perfectly normal.
In our study, we detected NTG in 18.4% of patients with vitiligo. We did not find a significant statistical difference between patients with and without glaucoma (Table 2). Rogosić et al5 found a significant relationship between age and glaucoma incidence, but we did not find such a relationship, which we believe is because the mean age of our patients was lower than the prior study.
In vitiligo, melanocytes are destroyed through an unknown mechanism. Although the cellular and molecular mechanisms causing melanocytic destruction have not yet been determined, various hypotheses have been put forward to explain the etiopathogenesis of vitiligo. Among these, the most commonly held hypotheses are the neural, self-destruction, and autoimmune hypotheses.16
Based on the observation that stress and serious trauma could precipitate or trigger the onset of vitiligo,16 the neural hypothesis holds that neurochemical mediators released from the edges of the nerve endings exert toxic effects on melanocytes. The fact that both melanocytes and choroidal pigment cells originate from the mesenchyme and dermatomal spreading of segmental vitiligo are arguments propounded in favor of this hypothesis.17
The self-destruction hypothesis suggests that the intrinsic protective mechanisms that normally enable melanocytes to eliminate toxic intermediate products or metabolites on the melanogenesis path have been impaired in patients with vitiligo.18,19 There is evidence of increased oxidative stress over the whole epidermis of patients with vitiligo.20 Thus, free radicals affect melanin and cause membrane damage via lipid peroxidation reactions.21
The autoimmune hypothesis proposes a clinical relationship between vitiligo and several diseases believed to be autoimmune. Because the macrophage infiltration observed in vitiligo lesions is more pronounced on the perilesional skin, this hypothesis holds that macrophages may play a role in melanocyte removal.21 The Koebner phenomenon observed in vitiligo lends support to the critical role of trauma in the etiopathogenesis of the disease.
Although we could not explain the co-presence of vitiligo and glaucoma, we believe that it may result from the fact that both diseases are observed in tissues that have the same embryologic origin and etiology, perhaps vascular or neural disorders, excessive accumulation of free radicals, or the triggering of apoptosis. Dermatologists should be alert to the presence of glaucoma in patients with vitiligo because glaucoma is an eye disease that progresses slowly and may lead to vision loss.
1. Ortonne JP. Vitiligo and other disorders of hypopigmentation. In: Bolognia JB, Jorizzo JL, Rapini RP, eds. Dermatology. 1st ed. New York, NY: Mosby; 2003:947-973.
2. Ortonne JP, Bahadoran P, Fitzpatrick TB, et al. Hypomelanoses and hypermelanoses. In: Freedberg IM, Eisen AZ, Wolff K, eds. Fitzpatrick’s Dermatology in General Medicine. 6th ed. New York, NY: McGraw-Hill; 2003:836-881.
3. van den Wijngaard R, Wijngaard R, Wankowiczs-Kalinsa A, et al. Autoimmune melanocyte destruction in vitiligo. Lab Invest. 2001;81:1061-1067.
4. Shields MB, Ritch R, Krupin T. Classification of the glaucomas. In: Ritch R, Shields MB, Krupin T, eds. The Glaucomas. St Louis, MO: C.V. Mosby Co; 1996:717-725.
5. Rogosić V, Bojić L, Puizina-Ivić N, et al. Vitiligo and glaucoma–an association or a coincidence? a pilot study. Acta Dermatovenerol Croat. 2010;18:21-26.
6. Anderson DR. Normal-tension glaucoma (low-tension glaucoma). Indian J Ophthalmol. 2011;59(suppl 59):S97-S101.
7. Iwata K. Primary open angle glaucoma and low tension glaucoma–pathogenesis and mechanism of optic nerve damage [in Japanese]. Nippon Ganka Gakkai Zasshi. 1992;96:1501-1531.
8. Hitchings RA, Anderton SA. A comparative study of visual field defects seen in patients with low-tension glaucoma and chronic simple glaucoma. Br J Ophthalmol. 1983;67:818-821.
9. Gopal KV, Rama Rao GR, Kumar YH, et al. Vitiligo: a part of a systemic autoimmune process. Indian J Dermatol Venereol Leprol. 2007;73:162-165.
10. Perossini M, Turio E, Perossini T, et al. Vitiligo: ocular and electrophysiological findings. G Ital Dermatol Venereol. 2010;145:141-149.
11. Biswas G, Barbhuiya JN, Biswas MC, et al. Clinical pattern of ocular manifestations in vitiligo. J Indian Med Assoc. 2003;101:478-480.
12. Park S, Albert DM, Bolognia JL. Ocular manifestations of pigmentary disorders. Dermatol Clin. 1992;10:609-622.
13. Albert DM, Nordlund JJ, Lerner AB. Ocular abnormalities occurring with vitiligo. Ophthalmology. 1979;86:1145-1160.
14. Wagoner MD, Albert DM, Lerner AB, et al. New observations on vitiligo and ocular disease. Am J Ophthalmol. 1983;96:16-26.
15. Cowan CL Jr, Halder RM, Grimes PE, et al. Ocular disturbances in vitiligo. J Am Acad Dermatol. 1986;15:17-24.
16. Orecchia GE. Neural pathogenesis. In: Hann SK, Nordlund JJ. Vitiligo. Oxford, England: Blackwell Science Ltd; 2000:142-150.
17. Braun-Falco O, Plewig G, Wolf HH, et al. Disorders of melanin pigmentation. In: Bartels V, ed. Dermatology. Berlin, Germany: Springer; 2000:1013-1042.
18. Tüzün Y, Kotoğyan A, Aydemir EH, et al. Pigmentasyon bozuklukları. In: Baransü O. Dermatoloji. 2nd ed. Istanbul: Nobel Tıp Kitabevi; 1994:557-559.
19. Odom RB, James WD, Berger TG. Disturbances of pigmentation. In: Odom RB, James WD, Berger TG. Andrews’ Diseases of the Skin. 9th ed. Philadelphia, PA: W.B. Saunders Company; 2000:1065-1068.
20. Schallreuter KU. Biochemical theory of vitiligo: a role of pteridines in pigmentation. In: Hann SK, Nordlund JJ. Vitiligo. London, England: Blackwell Science Ltd; 2000:151-159.
21. van den Wijngaard R, Wankowicz-Kalinska A, Le Poole C, et al. Local immune response in skin of generalized vitiligo patients. destruction of melanocytes is associated with the predominent presence of CLA+T cells at the perilesional site. Lab Invest. 2000;80:1299-1309.
1. Ortonne JP. Vitiligo and other disorders of hypopigmentation. In: Bolognia JB, Jorizzo JL, Rapini RP, eds. Dermatology. 1st ed. New York, NY: Mosby; 2003:947-973.
2. Ortonne JP, Bahadoran P, Fitzpatrick TB, et al. Hypomelanoses and hypermelanoses. In: Freedberg IM, Eisen AZ, Wolff K, eds. Fitzpatrick’s Dermatology in General Medicine. 6th ed. New York, NY: McGraw-Hill; 2003:836-881.
3. van den Wijngaard R, Wijngaard R, Wankowiczs-Kalinsa A, et al. Autoimmune melanocyte destruction in vitiligo. Lab Invest. 2001;81:1061-1067.
4. Shields MB, Ritch R, Krupin T. Classification of the glaucomas. In: Ritch R, Shields MB, Krupin T, eds. The Glaucomas. St Louis, MO: C.V. Mosby Co; 1996:717-725.
5. Rogosić V, Bojić L, Puizina-Ivić N, et al. Vitiligo and glaucoma–an association or a coincidence? a pilot study. Acta Dermatovenerol Croat. 2010;18:21-26.
6. Anderson DR. Normal-tension glaucoma (low-tension glaucoma). Indian J Ophthalmol. 2011;59(suppl 59):S97-S101.
7. Iwata K. Primary open angle glaucoma and low tension glaucoma–pathogenesis and mechanism of optic nerve damage [in Japanese]. Nippon Ganka Gakkai Zasshi. 1992;96:1501-1531.
8. Hitchings RA, Anderton SA. A comparative study of visual field defects seen in patients with low-tension glaucoma and chronic simple glaucoma. Br J Ophthalmol. 1983;67:818-821.
9. Gopal KV, Rama Rao GR, Kumar YH, et al. Vitiligo: a part of a systemic autoimmune process. Indian J Dermatol Venereol Leprol. 2007;73:162-165.
10. Perossini M, Turio E, Perossini T, et al. Vitiligo: ocular and electrophysiological findings. G Ital Dermatol Venereol. 2010;145:141-149.
11. Biswas G, Barbhuiya JN, Biswas MC, et al. Clinical pattern of ocular manifestations in vitiligo. J Indian Med Assoc. 2003;101:478-480.
12. Park S, Albert DM, Bolognia JL. Ocular manifestations of pigmentary disorders. Dermatol Clin. 1992;10:609-622.
13. Albert DM, Nordlund JJ, Lerner AB. Ocular abnormalities occurring with vitiligo. Ophthalmology. 1979;86:1145-1160.
14. Wagoner MD, Albert DM, Lerner AB, et al. New observations on vitiligo and ocular disease. Am J Ophthalmol. 1983;96:16-26.
15. Cowan CL Jr, Halder RM, Grimes PE, et al. Ocular disturbances in vitiligo. J Am Acad Dermatol. 1986;15:17-24.
16. Orecchia GE. Neural pathogenesis. In: Hann SK, Nordlund JJ. Vitiligo. Oxford, England: Blackwell Science Ltd; 2000:142-150.
17. Braun-Falco O, Plewig G, Wolf HH, et al. Disorders of melanin pigmentation. In: Bartels V, ed. Dermatology. Berlin, Germany: Springer; 2000:1013-1042.
18. Tüzün Y, Kotoğyan A, Aydemir EH, et al. Pigmentasyon bozuklukları. In: Baransü O. Dermatoloji. 2nd ed. Istanbul: Nobel Tıp Kitabevi; 1994:557-559.
19. Odom RB, James WD, Berger TG. Disturbances of pigmentation. In: Odom RB, James WD, Berger TG. Andrews’ Diseases of the Skin. 9th ed. Philadelphia, PA: W.B. Saunders Company; 2000:1065-1068.
20. Schallreuter KU. Biochemical theory of vitiligo: a role of pteridines in pigmentation. In: Hann SK, Nordlund JJ. Vitiligo. London, England: Blackwell Science Ltd; 2000:151-159.
21. van den Wijngaard R, Wankowicz-Kalinska A, Le Poole C, et al. Local immune response in skin of generalized vitiligo patients. destruction of melanocytes is associated with the predominent presence of CLA+T cells at the perilesional site. Lab Invest. 2000;80:1299-1309.
Practice Points
- Patients with vitiligo may exhibit pigmentary abnormalities of the iris and retina.
- Normal-tension glaucoma may develop in patients with vitiligo.
- Glaucoma progresses slowly and may lead to vision loss; as a result, dermatologists should be alert to the presence of glaucoma in vitiligo patients.

Clinical Pearl: Increasing Utility of Isopropyl Alcohol for Cutaneous Dyschromia
Practice Gap
Conditions with dyschromia including terra firma-forme dermatosis (TFFD), confluent and reticulate papillomatosis (CARP), and acanthosis nigricans are difficult to distinguish from one another.
Diagnostic Tools
Since its development in 1920, dermatologists have utilized isopropyl alcohol in ways that exceed conventional antimicrobial purposes. If TFFP, CARP, and acanthosis nigricans are suspected, the first step in any algorithmic approach should be to rub the skin with an alcohol pad using firm continuous pressure in an attempt to remove pigmentation. Complete resolution of dyspigmentation strongly supports a diagnosis of TFFD1 and can be curative (Figure). Alcohol can similarly lighten CARP but to a lesser degree than TFFD.2 In contrast, acanthosis nigricans will display minimal to no improvement with isopropyl alcohol.
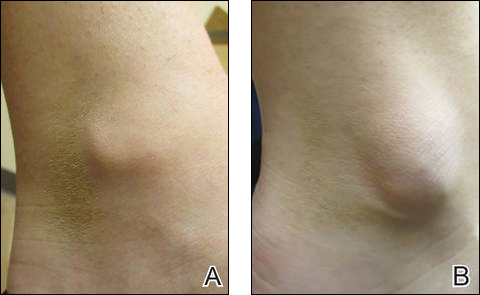
Practice Implications
Isopropyl alcohol has few side effects and each swab costs less than a dime. It is extremely cost effective compared to biopsy and subsequent pathology and laboratory costs. Patients appreciate a noninvasive initial approach, and it is rewarding to treat a cosmetically disturbing condition with ease.
Swabbing the skin with alcohol pads reflects light and improves visualization of veins that should be avoided during surgery. Alcohol-based gel inhibits bacterial colonization, reduces dermatoscope-related nosocomial infection, and enhances dermoscopic resolution.3 Alcohol swabs quickly remove gentian violet, which aids in porokeratosis diagnosis; the pathognomonic cornoid lamella of porokeratosis retains gentian violet.4 A solution of 70% isopropyl alcohol preserves myiasis larvae better than formalin, which causes larval tissue hardening. Alcohol also can be squeezed into the central punctum in myiasis as a form of treatment.5 In conclusion, alcohol represents a convenient, inexpensive, and helpful tool in the dermatologist’s armamentarium that should not be forgotten.
- Browning J, Rosen T. Terra firma-forme dermatosis revisited. Dermatol Online J. 2005;11:15.
- Berk DR. Confluent and reticulated papillomatosis response to 70% alcohol swabbing. Arch Dermatol. 2011;147:247-248.
- Kelly SC, Purcell SM. Prevention of nosocomial infection during dermoscopy? Dermatol Surg. 2006;32:552-555.
- Thomas CJ, Elston DM. Medical pearl: Gentian violet to highlight the cornoid lamella in disseminated superficial actinic porokeratosis. J Am Acad Dermatol. 2005;52(3, pt 1):513-514.
- Meinking TL, Burkhart CN, Burkhart CG. Changing paradigms in parasitic infections: common dermatological helminthic infections and cutaneous myiasis. Clin Dermatol. 2003;21:407-416.
Practice Gap
Conditions with dyschromia including terra firma-forme dermatosis (TFFD), confluent and reticulate papillomatosis (CARP), and acanthosis nigricans are difficult to distinguish from one another.
Diagnostic Tools
Since its development in 1920, dermatologists have utilized isopropyl alcohol in ways that exceed conventional antimicrobial purposes. If TFFP, CARP, and acanthosis nigricans are suspected, the first step in any algorithmic approach should be to rub the skin with an alcohol pad using firm continuous pressure in an attempt to remove pigmentation. Complete resolution of dyspigmentation strongly supports a diagnosis of TFFD1 and can be curative (Figure). Alcohol can similarly lighten CARP but to a lesser degree than TFFD.2 In contrast, acanthosis nigricans will display minimal to no improvement with isopropyl alcohol.
Practice Implications
Isopropyl alcohol has few side effects and each swab costs less than a dime. It is extremely cost effective compared to biopsy and subsequent pathology and laboratory costs. Patients appreciate a noninvasive initial approach, and it is rewarding to treat a cosmetically disturbing condition with ease.
Swabbing the skin with alcohol pads reflects light and improves visualization of veins that should be avoided during surgery. Alcohol-based gel inhibits bacterial colonization, reduces dermatoscope-related nosocomial infection, and enhances dermoscopic resolution.3 Alcohol swabs quickly remove gentian violet, which aids in porokeratosis diagnosis; the pathognomonic cornoid lamella of porokeratosis retains gentian violet.4 A solution of 70% isopropyl alcohol preserves myiasis larvae better than formalin, which causes larval tissue hardening. Alcohol also can be squeezed into the central punctum in myiasis as a form of treatment.5 In conclusion, alcohol represents a convenient, inexpensive, and helpful tool in the dermatologist’s armamentarium that should not be forgotten.
Practice Gap
Conditions with dyschromia including terra firma-forme dermatosis (TFFD), confluent and reticulate papillomatosis (CARP), and acanthosis nigricans are difficult to distinguish from one another.
Diagnostic Tools
Since its development in 1920, dermatologists have utilized isopropyl alcohol in ways that exceed conventional antimicrobial purposes. If TFFP, CARP, and acanthosis nigricans are suspected, the first step in any algorithmic approach should be to rub the skin with an alcohol pad using firm continuous pressure in an attempt to remove pigmentation. Complete resolution of dyspigmentation strongly supports a diagnosis of TFFD1 and can be curative (Figure). Alcohol can similarly lighten CARP but to a lesser degree than TFFD.2 In contrast, acanthosis nigricans will display minimal to no improvement with isopropyl alcohol.
Practice Implications
Isopropyl alcohol has few side effects and each swab costs less than a dime. It is extremely cost effective compared to biopsy and subsequent pathology and laboratory costs. Patients appreciate a noninvasive initial approach, and it is rewarding to treat a cosmetically disturbing condition with ease.
Swabbing the skin with alcohol pads reflects light and improves visualization of veins that should be avoided during surgery. Alcohol-based gel inhibits bacterial colonization, reduces dermatoscope-related nosocomial infection, and enhances dermoscopic resolution.3 Alcohol swabs quickly remove gentian violet, which aids in porokeratosis diagnosis; the pathognomonic cornoid lamella of porokeratosis retains gentian violet.4 A solution of 70% isopropyl alcohol preserves myiasis larvae better than formalin, which causes larval tissue hardening. Alcohol also can be squeezed into the central punctum in myiasis as a form of treatment.5 In conclusion, alcohol represents a convenient, inexpensive, and helpful tool in the dermatologist’s armamentarium that should not be forgotten.
- Browning J, Rosen T. Terra firma-forme dermatosis revisited. Dermatol Online J. 2005;11:15.
- Berk DR. Confluent and reticulated papillomatosis response to 70% alcohol swabbing. Arch Dermatol. 2011;147:247-248.
- Kelly SC, Purcell SM. Prevention of nosocomial infection during dermoscopy? Dermatol Surg. 2006;32:552-555.
- Thomas CJ, Elston DM. Medical pearl: Gentian violet to highlight the cornoid lamella in disseminated superficial actinic porokeratosis. J Am Acad Dermatol. 2005;52(3, pt 1):513-514.
- Meinking TL, Burkhart CN, Burkhart CG. Changing paradigms in parasitic infections: common dermatological helminthic infections and cutaneous myiasis. Clin Dermatol. 2003;21:407-416.
- Browning J, Rosen T. Terra firma-forme dermatosis revisited. Dermatol Online J. 2005;11:15.
- Berk DR. Confluent and reticulated papillomatosis response to 70% alcohol swabbing. Arch Dermatol. 2011;147:247-248.
- Kelly SC, Purcell SM. Prevention of nosocomial infection during dermoscopy? Dermatol Surg. 2006;32:552-555.
- Thomas CJ, Elston DM. Medical pearl: Gentian violet to highlight the cornoid lamella in disseminated superficial actinic porokeratosis. J Am Acad Dermatol. 2005;52(3, pt 1):513-514.
- Meinking TL, Burkhart CN, Burkhart CG. Changing paradigms in parasitic infections: common dermatological helminthic infections and cutaneous myiasis. Clin Dermatol. 2003;21:407-416.
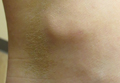
Diagnosing Porokeratosis of Mibelli Every Time: A Novel Biopsy Technique to Maximize Histopathologic Confirmation
Porokeratosis of Mibelli (PM) is a lesion characterized by a surrounding cornoid lamella with variable nonspecific findings (eg, atrophy, acanthosis, verrucous hyperplasia) in the center of the lesion that typically presents in infancy to early childhood.1 We report a case of PM in which a prior biopsy from the center of the lesion demonstrated papulosquamous dermatitis. We propose a 3-step technique to ensure proper orientation of a punch biopsy in cases of suspected PM.
Case Report
A 3-year-old girl presented with an erythematous, hypopigmented, scaling plaque on the posterior aspect of the left ankle surrounded by a hard rim. The plaque was first noted at 12 months of age and had slowly enlarged as the patient grew. Six months prior, a biopsy from the center of the lesion performed at another facility demonstrated a papulosquamous dermatitis.
Physical examination revealed a lesion that was 4.2-cm long, 2.2-cm wide at the superior pole, and 3.5-cm wide at the inferior pole (Figure 1). A line was drawn with a skin marker perpendicular to the rim of the lesion (Figure 2A) and a 6-mm punch biopsy was performed, centered at the intersection of the drawn line and the cornoid lamella (Figure 2B). The tissue was then bisected at the bedside along the skin marker line with a #15 blade (Figure 2C) and submitted in formalin for histologic processing. Histologic examination revealed an invagination of the epidermis producing a tier of parakeratotic cells with its apex pointed away from the center of the lesion. Dyskeratotic cells were noted at the base of the parakeratosis (Figure 3). Verrucous hyperplasia was present in the central portion of the specimen adjacent to the cornoid lamella. Based on these histopathologic findings, the correct diagnosis of PM was made.
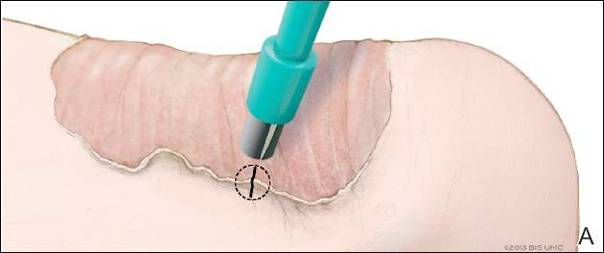
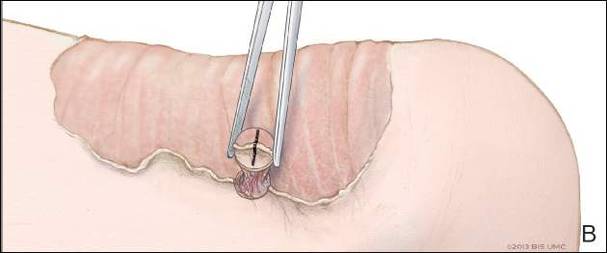
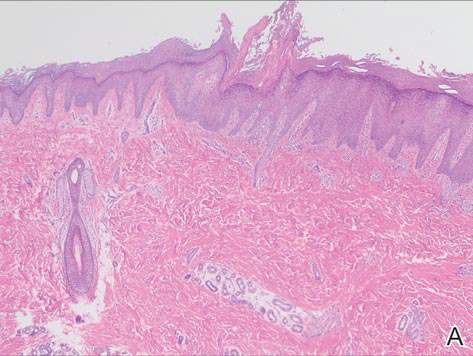
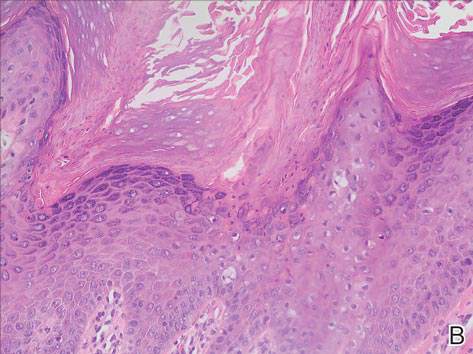
Comment
Porokeratosis of Mibelli is a rare condition that typically presents in infancy to early childhood.1 It may appear as small keratotic papules or larger plaques that reach several centimeters in diameter.2 There is a 7.5% risk for malignant transformation (eg, basal cell carcinoma, squamous cell carcinoma, Bowen disease).3 Variable nonspecific findings (eg, atrophy, acanthosis, verrucous hyperplasia) typically are present in the center of the lesion. In our case, a biopsy from the center of the plaque demonstrated verrucous hyperplasia. The incorrect diagnosis of PM as psoriasis also has been reported.4
We propose a 3-step technique to ensure proper orientation of a punch biopsy in cases of suspected PM. First, draw a line perpendicular to the rim of the lesion to mark the biopsy site (Figure 2A). Second, perform a punch biopsy centered at the intersection of the drawn line and the cornoid lamella (Figure 2B). Third, section the biopsied tissue with a #15 blade along the perpendicular line at the bedside (Figure 2C). The surgical pathology requisition should mention that the specimen has been transected and the cut edges should be placed down in the cassette, ensuring that the cornoid lamella will be present in cross-section on the slides.
If the punch biopsy specimen is not bisected, it can be difficult to orient it in the pathology laboratory, especially if the cornoid lamellae are not prominent. Furthermore, the technician processing the tissue may not be aware of the importance of sectioning the specimen perpendicular to the cornoid lamella. Following this procedure, diagnosis can be confirmed in virtually every case of PM.
- Richard G, Irvine A, Traupe H, et al. Ichthyosis and disorders of other conification. In: Schachner L, Hansen R, Krafchik B, et al, eds. Pediatric Dermatology. Philadelphia, PA: Elsevier Health Sciences; 2011:640-643.
- Pierson D, Bandel C, Ehrig, et al. Benign epidermal tumors and proliferations. In: Bolognia J, Jorizzo J, Rapini R, et al, eds. Dermatology. 1st ed. Vol 2. Edinburgh, Scotland: Elsevier; 2003:1707-1709.
- Cort DF, Abdel-Aziz AH. Epithelioma arising in porokeratosis of Mibelli. Br J Plast Surg. 1972;25:318-328.
- De Simone C, Paradisi A, Massi G, et al. Giant verrucous porokeratosis of Mibelli mimicking psoriasis in a patient with psoriasis. J Am Acad Dermatol. 2007;57:665-668.
Porokeratosis of Mibelli (PM) is a lesion characterized by a surrounding cornoid lamella with variable nonspecific findings (eg, atrophy, acanthosis, verrucous hyperplasia) in the center of the lesion that typically presents in infancy to early childhood.1 We report a case of PM in which a prior biopsy from the center of the lesion demonstrated papulosquamous dermatitis. We propose a 3-step technique to ensure proper orientation of a punch biopsy in cases of suspected PM.
Case Report
A 3-year-old girl presented with an erythematous, hypopigmented, scaling plaque on the posterior aspect of the left ankle surrounded by a hard rim. The plaque was first noted at 12 months of age and had slowly enlarged as the patient grew. Six months prior, a biopsy from the center of the lesion performed at another facility demonstrated a papulosquamous dermatitis.
Physical examination revealed a lesion that was 4.2-cm long, 2.2-cm wide at the superior pole, and 3.5-cm wide at the inferior pole (Figure 1). A line was drawn with a skin marker perpendicular to the rim of the lesion (Figure 2A) and a 6-mm punch biopsy was performed, centered at the intersection of the drawn line and the cornoid lamella (Figure 2B). The tissue was then bisected at the bedside along the skin marker line with a #15 blade (Figure 2C) and submitted in formalin for histologic processing. Histologic examination revealed an invagination of the epidermis producing a tier of parakeratotic cells with its apex pointed away from the center of the lesion. Dyskeratotic cells were noted at the base of the parakeratosis (Figure 3). Verrucous hyperplasia was present in the central portion of the specimen adjacent to the cornoid lamella. Based on these histopathologic findings, the correct diagnosis of PM was made.
Comment
Porokeratosis of Mibelli is a rare condition that typically presents in infancy to early childhood.1 It may appear as small keratotic papules or larger plaques that reach several centimeters in diameter.2 There is a 7.5% risk for malignant transformation (eg, basal cell carcinoma, squamous cell carcinoma, Bowen disease).3 Variable nonspecific findings (eg, atrophy, acanthosis, verrucous hyperplasia) typically are present in the center of the lesion. In our case, a biopsy from the center of the plaque demonstrated verrucous hyperplasia. The incorrect diagnosis of PM as psoriasis also has been reported.4
We propose a 3-step technique to ensure proper orientation of a punch biopsy in cases of suspected PM. First, draw a line perpendicular to the rim of the lesion to mark the biopsy site (Figure 2A). Second, perform a punch biopsy centered at the intersection of the drawn line and the cornoid lamella (Figure 2B). Third, section the biopsied tissue with a #15 blade along the perpendicular line at the bedside (Figure 2C). The surgical pathology requisition should mention that the specimen has been transected and the cut edges should be placed down in the cassette, ensuring that the cornoid lamella will be present in cross-section on the slides.
If the punch biopsy specimen is not bisected, it can be difficult to orient it in the pathology laboratory, especially if the cornoid lamellae are not prominent. Furthermore, the technician processing the tissue may not be aware of the importance of sectioning the specimen perpendicular to the cornoid lamella. Following this procedure, diagnosis can be confirmed in virtually every case of PM.
Porokeratosis of Mibelli (PM) is a lesion characterized by a surrounding cornoid lamella with variable nonspecific findings (eg, atrophy, acanthosis, verrucous hyperplasia) in the center of the lesion that typically presents in infancy to early childhood.1 We report a case of PM in which a prior biopsy from the center of the lesion demonstrated papulosquamous dermatitis. We propose a 3-step technique to ensure proper orientation of a punch biopsy in cases of suspected PM.
Case Report
A 3-year-old girl presented with an erythematous, hypopigmented, scaling plaque on the posterior aspect of the left ankle surrounded by a hard rim. The plaque was first noted at 12 months of age and had slowly enlarged as the patient grew. Six months prior, a biopsy from the center of the lesion performed at another facility demonstrated a papulosquamous dermatitis.
Physical examination revealed a lesion that was 4.2-cm long, 2.2-cm wide at the superior pole, and 3.5-cm wide at the inferior pole (Figure 1). A line was drawn with a skin marker perpendicular to the rim of the lesion (Figure 2A) and a 6-mm punch biopsy was performed, centered at the intersection of the drawn line and the cornoid lamella (Figure 2B). The tissue was then bisected at the bedside along the skin marker line with a #15 blade (Figure 2C) and submitted in formalin for histologic processing. Histologic examination revealed an invagination of the epidermis producing a tier of parakeratotic cells with its apex pointed away from the center of the lesion. Dyskeratotic cells were noted at the base of the parakeratosis (Figure 3). Verrucous hyperplasia was present in the central portion of the specimen adjacent to the cornoid lamella. Based on these histopathologic findings, the correct diagnosis of PM was made.
Comment
Porokeratosis of Mibelli is a rare condition that typically presents in infancy to early childhood.1 It may appear as small keratotic papules or larger plaques that reach several centimeters in diameter.2 There is a 7.5% risk for malignant transformation (eg, basal cell carcinoma, squamous cell carcinoma, Bowen disease).3 Variable nonspecific findings (eg, atrophy, acanthosis, verrucous hyperplasia) typically are present in the center of the lesion. In our case, a biopsy from the center of the plaque demonstrated verrucous hyperplasia. The incorrect diagnosis of PM as psoriasis also has been reported.4
We propose a 3-step technique to ensure proper orientation of a punch biopsy in cases of suspected PM. First, draw a line perpendicular to the rim of the lesion to mark the biopsy site (Figure 2A). Second, perform a punch biopsy centered at the intersection of the drawn line and the cornoid lamella (Figure 2B). Third, section the biopsied tissue with a #15 blade along the perpendicular line at the bedside (Figure 2C). The surgical pathology requisition should mention that the specimen has been transected and the cut edges should be placed down in the cassette, ensuring that the cornoid lamella will be present in cross-section on the slides.
If the punch biopsy specimen is not bisected, it can be difficult to orient it in the pathology laboratory, especially if the cornoid lamellae are not prominent. Furthermore, the technician processing the tissue may not be aware of the importance of sectioning the specimen perpendicular to the cornoid lamella. Following this procedure, diagnosis can be confirmed in virtually every case of PM.
- Richard G, Irvine A, Traupe H, et al. Ichthyosis and disorders of other conification. In: Schachner L, Hansen R, Krafchik B, et al, eds. Pediatric Dermatology. Philadelphia, PA: Elsevier Health Sciences; 2011:640-643.
- Pierson D, Bandel C, Ehrig, et al. Benign epidermal tumors and proliferations. In: Bolognia J, Jorizzo J, Rapini R, et al, eds. Dermatology. 1st ed. Vol 2. Edinburgh, Scotland: Elsevier; 2003:1707-1709.
- Cort DF, Abdel-Aziz AH. Epithelioma arising in porokeratosis of Mibelli. Br J Plast Surg. 1972;25:318-328.
- De Simone C, Paradisi A, Massi G, et al. Giant verrucous porokeratosis of Mibelli mimicking psoriasis in a patient with psoriasis. J Am Acad Dermatol. 2007;57:665-668.
- Richard G, Irvine A, Traupe H, et al. Ichthyosis and disorders of other conification. In: Schachner L, Hansen R, Krafchik B, et al, eds. Pediatric Dermatology. Philadelphia, PA: Elsevier Health Sciences; 2011:640-643.
- Pierson D, Bandel C, Ehrig, et al. Benign epidermal tumors and proliferations. In: Bolognia J, Jorizzo J, Rapini R, et al, eds. Dermatology. 1st ed. Vol 2. Edinburgh, Scotland: Elsevier; 2003:1707-1709.
- Cort DF, Abdel-Aziz AH. Epithelioma arising in porokeratosis of Mibelli. Br J Plast Surg. 1972;25:318-328.
- De Simone C, Paradisi A, Massi G, et al. Giant verrucous porokeratosis of Mibelli mimicking psoriasis in a patient with psoriasis. J Am Acad Dermatol. 2007;57:665-668.
Practice Points
- A biopsy from the center of a plaque of porokeratosis will produce nonspecific findings.
- Bisecting the punch specimen at the bedside along a line drawn perpendicular to the cornoid lamella guarantees proper orientation of the specimen.
MRSA incidence decreased in children as clindamycin resistance increased
The incidence of methicillin-resistant Staphylococcus aureus (MRSA) infections has decreased in children in recent years, but resistance to clindamycin has increased over the same period, a study showed.
“The epidemic of skin and soft tissue infections and invasive MRSA led to modifications of antimicrobial prescribing practices for suspected S. aureus infections,” reported Dr. Deena E. Sutter of the San Antonio Military Medical Center in Fort Sam Houston, Tex., and her associates. “Over the study period, erythromycin susceptibility among methicillin-susceptible S. aureus (MSSA) remained stable, suggesting that declining clindamycin susceptibility is a result of an increase in inducible resistance.”
The steady decline in clindamycin susceptibility “may lead to some concern about the continued reliance on clindamycin for the empirical treatment of presumptive S. aureus infections, although it is probably premature to abandon this effective antibiotic choice,” they wrote (Pediatrics. 2016 Mar. 1. doi: 10.1542/peds.2015-3099). “It is crucial that clinicians remain knowledgeable about local susceptibility rates as it would be prudent to consider [alternative] antimicrobial agents for empirical use when the local clindamycin susceptibility rate drops below 85%.”
The researchers retrospectively analyzed lab results from 39,209 patients under age 18 who were treated for S. aureus infections at one of the 266 U.S. facilities of the Military Health System from 2005 to 2014. The data included 41,745 S. aureus isolates, classified as MRSA if found resistant to cefoxitin, methicillin, or oxacillin and as methicillin susceptible (MSSA) if susceptible to those antimicrobials. The isolates had also been tested for susceptibility to ciprofloxacin, clindamycin, erythromycin, gentamicin, oxacillin, penicillin, rifampin, tetracycline, and trimethoprim/sulfamethoxazole (TMP/SMX).
During that decade, overall S. aureus susceptibility to clindamycin, ciprofloxacin, and TMP/SMX decreased – although susceptibility to TMP/SMX in 2014 stayed high at 98% – while overall susceptibility to erythromycin, gentamicin, and oxacillin increased. Specifically, 59% of S. aureus isolates were susceptible to oxacillin in 2005, which dropped briefly to 54% in 2007 before climbing to the 2014 rate of 68%.
Meanwhile, overall susceptibility to clindamycin dropped from 91% in 2005 to 86% in 2014, and MSSA susceptibility to clindamycin dropped from 91% in 2005 to 84% in 2014. “Erythromycin susceptibility remained stable among MSSA isolates throughout the study period at 63.5%, whereas MRSA susceptibility to erythromycin increased from 12.1% to 20.5%,” Dr. Sutter and her associates reported. “Ciprofloxacin susceptibility significantly decreased overall, although an initial decrease of 10.6% over the first 7 years of the study was subsequently followed by an increase of 6% between 2011 and 2014.”
Most of the isolates came from patients with skin and soft tissue infections, which were less likely to be susceptible to oxacillin than were other infections. Infections in children aged 1-5 years also were less likely to be susceptible to oxacillin, compared with infections in children of other age groups.
If the local clindamycin susceptibility rate falls below 85%, “beta-lactams, TMP/SMX, or tetracyclines may be used for less severe infections with intravenous vancomycin employed in severe cases,” the investigators said. “If overall MRSA rates continue to decline and clindamycin resistance among MSSA continues to increase, we may see a return to antistaphylococcal beta-lactam antimicrobial agents such as oxacillin or first-generation cephalosporins as preferred empirical therapy for presumed S. aureus infections.”
The research did not use external funding, and the authors reported no relevant financial disclosures.
Staphylococcus aureus is one of the most common organisms isolated from children with health care–associated infections, regardless of whether these infections had their onset in the community or were acquired in the hospital. Thus, the initial empiric treatment of a skin or soft tissue infection or invasive infection in a child almost always includes an antibiotic effective against S. aureus.
However, over the years, clindamycin susceptibility among S. aureus isolates has declined, likely related to the increased use of this agent for empiric as well as definitive treatment of community-acquired (CA) MRSA infections, encouraging the transmission of the genes associated with clindamycin resistance.
What are the implications of the findings from the report by Sutter et al. with respect to the selection of empiric antibiotics for children with suspected S. aureus infections? Currently, considering the still substantial MRSA resistance rates that exceed the 10%-15% level suggested by many experts as the threshold above which agents effective against CA-MRSA isolates should be administered for empiric treatment, changes in the selection of empiric antibiotics are not warranted. If rates of MRSA among S. aureus isolates from otherwise normal children are documented to drop below the 10%-15% threshold in different communities, a modification of current recommendations should be considered. It would also be important to understand why methicillin resistance is declining among S. aureus isolates from CA infections; this information may provide clues for preventing CA-MRSA infections with the use of vaccines or other means. The epidemiology of S. aureus infections in children has been changing over the past 2 decades, which is why it is critical to keep a very close eye on this common pathogen.
These comments were excerpted from an accompanying commentary by Dr. Sheldon L. Kaplan of the infectious disease service at Texas Children’s Hospital in Houston (Pediatrics. 2016 Mar 1. doi: 10.1542/peds.2016-0101). Dr. Kaplan has received research funds from Pfizer, Forest Laboratories, and Cubist.
Staphylococcus aureus is one of the most common organisms isolated from children with health care–associated infections, regardless of whether these infections had their onset in the community or were acquired in the hospital. Thus, the initial empiric treatment of a skin or soft tissue infection or invasive infection in a child almost always includes an antibiotic effective against S. aureus.
However, over the years, clindamycin susceptibility among S. aureus isolates has declined, likely related to the increased use of this agent for empiric as well as definitive treatment of community-acquired (CA) MRSA infections, encouraging the transmission of the genes associated with clindamycin resistance.
What are the implications of the findings from the report by Sutter et al. with respect to the selection of empiric antibiotics for children with suspected S. aureus infections? Currently, considering the still substantial MRSA resistance rates that exceed the 10%-15% level suggested by many experts as the threshold above which agents effective against CA-MRSA isolates should be administered for empiric treatment, changes in the selection of empiric antibiotics are not warranted. If rates of MRSA among S. aureus isolates from otherwise normal children are documented to drop below the 10%-15% threshold in different communities, a modification of current recommendations should be considered. It would also be important to understand why methicillin resistance is declining among S. aureus isolates from CA infections; this information may provide clues for preventing CA-MRSA infections with the use of vaccines or other means. The epidemiology of S. aureus infections in children has been changing over the past 2 decades, which is why it is critical to keep a very close eye on this common pathogen.
These comments were excerpted from an accompanying commentary by Dr. Sheldon L. Kaplan of the infectious disease service at Texas Children’s Hospital in Houston (Pediatrics. 2016 Mar 1. doi: 10.1542/peds.2016-0101). Dr. Kaplan has received research funds from Pfizer, Forest Laboratories, and Cubist.
Staphylococcus aureus is one of the most common organisms isolated from children with health care–associated infections, regardless of whether these infections had their onset in the community or were acquired in the hospital. Thus, the initial empiric treatment of a skin or soft tissue infection or invasive infection in a child almost always includes an antibiotic effective against S. aureus.
However, over the years, clindamycin susceptibility among S. aureus isolates has declined, likely related to the increased use of this agent for empiric as well as definitive treatment of community-acquired (CA) MRSA infections, encouraging the transmission of the genes associated with clindamycin resistance.
What are the implications of the findings from the report by Sutter et al. with respect to the selection of empiric antibiotics for children with suspected S. aureus infections? Currently, considering the still substantial MRSA resistance rates that exceed the 10%-15% level suggested by many experts as the threshold above which agents effective against CA-MRSA isolates should be administered for empiric treatment, changes in the selection of empiric antibiotics are not warranted. If rates of MRSA among S. aureus isolates from otherwise normal children are documented to drop below the 10%-15% threshold in different communities, a modification of current recommendations should be considered. It would also be important to understand why methicillin resistance is declining among S. aureus isolates from CA infections; this information may provide clues for preventing CA-MRSA infections with the use of vaccines or other means. The epidemiology of S. aureus infections in children has been changing over the past 2 decades, which is why it is critical to keep a very close eye on this common pathogen.
These comments were excerpted from an accompanying commentary by Dr. Sheldon L. Kaplan of the infectious disease service at Texas Children’s Hospital in Houston (Pediatrics. 2016 Mar 1. doi: 10.1542/peds.2016-0101). Dr. Kaplan has received research funds from Pfizer, Forest Laboratories, and Cubist.
The incidence of methicillin-resistant Staphylococcus aureus (MRSA) infections has decreased in children in recent years, but resistance to clindamycin has increased over the same period, a study showed.
“The epidemic of skin and soft tissue infections and invasive MRSA led to modifications of antimicrobial prescribing practices for suspected S. aureus infections,” reported Dr. Deena E. Sutter of the San Antonio Military Medical Center in Fort Sam Houston, Tex., and her associates. “Over the study period, erythromycin susceptibility among methicillin-susceptible S. aureus (MSSA) remained stable, suggesting that declining clindamycin susceptibility is a result of an increase in inducible resistance.”
The steady decline in clindamycin susceptibility “may lead to some concern about the continued reliance on clindamycin for the empirical treatment of presumptive S. aureus infections, although it is probably premature to abandon this effective antibiotic choice,” they wrote (Pediatrics. 2016 Mar. 1. doi: 10.1542/peds.2015-3099). “It is crucial that clinicians remain knowledgeable about local susceptibility rates as it would be prudent to consider [alternative] antimicrobial agents for empirical use when the local clindamycin susceptibility rate drops below 85%.”
The researchers retrospectively analyzed lab results from 39,209 patients under age 18 who were treated for S. aureus infections at one of the 266 U.S. facilities of the Military Health System from 2005 to 2014. The data included 41,745 S. aureus isolates, classified as MRSA if found resistant to cefoxitin, methicillin, or oxacillin and as methicillin susceptible (MSSA) if susceptible to those antimicrobials. The isolates had also been tested for susceptibility to ciprofloxacin, clindamycin, erythromycin, gentamicin, oxacillin, penicillin, rifampin, tetracycline, and trimethoprim/sulfamethoxazole (TMP/SMX).
During that decade, overall S. aureus susceptibility to clindamycin, ciprofloxacin, and TMP/SMX decreased – although susceptibility to TMP/SMX in 2014 stayed high at 98% – while overall susceptibility to erythromycin, gentamicin, and oxacillin increased. Specifically, 59% of S. aureus isolates were susceptible to oxacillin in 2005, which dropped briefly to 54% in 2007 before climbing to the 2014 rate of 68%.
Meanwhile, overall susceptibility to clindamycin dropped from 91% in 2005 to 86% in 2014, and MSSA susceptibility to clindamycin dropped from 91% in 2005 to 84% in 2014. “Erythromycin susceptibility remained stable among MSSA isolates throughout the study period at 63.5%, whereas MRSA susceptibility to erythromycin increased from 12.1% to 20.5%,” Dr. Sutter and her associates reported. “Ciprofloxacin susceptibility significantly decreased overall, although an initial decrease of 10.6% over the first 7 years of the study was subsequently followed by an increase of 6% between 2011 and 2014.”
Most of the isolates came from patients with skin and soft tissue infections, which were less likely to be susceptible to oxacillin than were other infections. Infections in children aged 1-5 years also were less likely to be susceptible to oxacillin, compared with infections in children of other age groups.
If the local clindamycin susceptibility rate falls below 85%, “beta-lactams, TMP/SMX, or tetracyclines may be used for less severe infections with intravenous vancomycin employed in severe cases,” the investigators said. “If overall MRSA rates continue to decline and clindamycin resistance among MSSA continues to increase, we may see a return to antistaphylococcal beta-lactam antimicrobial agents such as oxacillin or first-generation cephalosporins as preferred empirical therapy for presumed S. aureus infections.”
The research did not use external funding, and the authors reported no relevant financial disclosures.
The incidence of methicillin-resistant Staphylococcus aureus (MRSA) infections has decreased in children in recent years, but resistance to clindamycin has increased over the same period, a study showed.
“The epidemic of skin and soft tissue infections and invasive MRSA led to modifications of antimicrobial prescribing practices for suspected S. aureus infections,” reported Dr. Deena E. Sutter of the San Antonio Military Medical Center in Fort Sam Houston, Tex., and her associates. “Over the study period, erythromycin susceptibility among methicillin-susceptible S. aureus (MSSA) remained stable, suggesting that declining clindamycin susceptibility is a result of an increase in inducible resistance.”
The steady decline in clindamycin susceptibility “may lead to some concern about the continued reliance on clindamycin for the empirical treatment of presumptive S. aureus infections, although it is probably premature to abandon this effective antibiotic choice,” they wrote (Pediatrics. 2016 Mar. 1. doi: 10.1542/peds.2015-3099). “It is crucial that clinicians remain knowledgeable about local susceptibility rates as it would be prudent to consider [alternative] antimicrobial agents for empirical use when the local clindamycin susceptibility rate drops below 85%.”
The researchers retrospectively analyzed lab results from 39,209 patients under age 18 who were treated for S. aureus infections at one of the 266 U.S. facilities of the Military Health System from 2005 to 2014. The data included 41,745 S. aureus isolates, classified as MRSA if found resistant to cefoxitin, methicillin, or oxacillin and as methicillin susceptible (MSSA) if susceptible to those antimicrobials. The isolates had also been tested for susceptibility to ciprofloxacin, clindamycin, erythromycin, gentamicin, oxacillin, penicillin, rifampin, tetracycline, and trimethoprim/sulfamethoxazole (TMP/SMX).
During that decade, overall S. aureus susceptibility to clindamycin, ciprofloxacin, and TMP/SMX decreased – although susceptibility to TMP/SMX in 2014 stayed high at 98% – while overall susceptibility to erythromycin, gentamicin, and oxacillin increased. Specifically, 59% of S. aureus isolates were susceptible to oxacillin in 2005, which dropped briefly to 54% in 2007 before climbing to the 2014 rate of 68%.
Meanwhile, overall susceptibility to clindamycin dropped from 91% in 2005 to 86% in 2014, and MSSA susceptibility to clindamycin dropped from 91% in 2005 to 84% in 2014. “Erythromycin susceptibility remained stable among MSSA isolates throughout the study period at 63.5%, whereas MRSA susceptibility to erythromycin increased from 12.1% to 20.5%,” Dr. Sutter and her associates reported. “Ciprofloxacin susceptibility significantly decreased overall, although an initial decrease of 10.6% over the first 7 years of the study was subsequently followed by an increase of 6% between 2011 and 2014.”
Most of the isolates came from patients with skin and soft tissue infections, which were less likely to be susceptible to oxacillin than were other infections. Infections in children aged 1-5 years also were less likely to be susceptible to oxacillin, compared with infections in children of other age groups.
If the local clindamycin susceptibility rate falls below 85%, “beta-lactams, TMP/SMX, or tetracyclines may be used for less severe infections with intravenous vancomycin employed in severe cases,” the investigators said. “If overall MRSA rates continue to decline and clindamycin resistance among MSSA continues to increase, we may see a return to antistaphylococcal beta-lactam antimicrobial agents such as oxacillin or first-generation cephalosporins as preferred empirical therapy for presumed S. aureus infections.”
The research did not use external funding, and the authors reported no relevant financial disclosures.
FROM PEDIATRICS
Key clinical point: The incidence of methicillin-resistant Staphylococcus aureus infections has decreased in children in recent years while resistance to clindamycin has increased.
Major finding: MRSA susceptibility to oxacillin increased to 68.4% in 2014, and susceptibility dropped to 86% for clindamycin.
Data source: A retrospective analysis of 41,745 S. aureus isolates from 39,209 patients under age 18 years in the U.S. Military Health System between 2005 and 2014.
Disclosures: The research did not use external funding, and the authors reported no relevant financial disclosures.
Piebaldism in Children
Case Report
A 14-year-old adolescent girl presented with multiple asymptomatic light-colored patches on the forehead, bilateral arms, and legs that had been present since birth. The patient reported that the size of the patches had increased in proportion to her overall growth and that “brown spots” had gradually started to form within and around the patches. She noted that her father and paternal grandfather also had similar clinical findings. A review of systems was negative for hearing impairment, ocular abnormalities, and recurrent infections.
Physical examination revealed an otherwise healthy adolescent girl with Fitzpatrick skin type I and homogeneous blue eyes. Large symmetric depigmented patches were noted on the extensor surfaces of the mid legs and mid forearms (Figure). Macules of baseline pigment and hyperpigmentation were irregularly scattered within and at the periphery of the patches. A triangular hypopigmented patch at the hairline on the mid frontal scalp hairline was accompanied by depigmentation of terminal hairs in this region.
A clinical diagnosis of piebaldism was made and was discussed at length with the patient. Due to the benign nature of the condition and patient preference, no therapeutic intervention was pursued. It was recommended that she apply sunscreen daily for protection of the depigmented areas.
Comment
Piebaldism is a rare hereditary disorder of melanocyte development characterized clinically by the presence of congenital poliosis and leukoderma.1 The exact prevalence of piebaldism is unknown, but it has been estimated that less than 1 in 20,000 children are born with this condition.2 Poliosis circumscripta, traditionally known as white forelock, may be the only manifestation in 80% to 90% of cases and is present at birth.3 The white forelock typically appears in a triangular shape and the underlying skin of the scalp also is amelanotic. The eyebrows and eyelashes also may be involved.3
Characteristically, lesions of leukoderma are well-circumscribed, irregular, white patches that are often accompanied by hyperpigmented macules noted on both depigmented and unaffected adjacent skin.1 The lesions are classically distributed on the central forehead and anterior trunk, with extension to the flanks, anterior mid arms, and mid legs. Sparing of the dorsal midline, hands, feet, and periorificial area is characteristic.1
Depigmented patches typically are nonprogressive and persist into adulthood. Additional hyperpigmented macules may develop at or within the margins of the white patches. Partial or complete repigmentation may occur spontaneously or after trauma in some patients.2 Some children may develop café au lait lesions and may be misdiagnosed as concurrently having neurofibromatosis type I and piebaldism. If neurofibromatosis type I is suspected, patients should be thoroughly evaluated for other diagnostic criteria of this syndrome, as there may be cases of coexistence and overlap with piebaldism.4
Piebaldism is an autosomal-dominant inherited disorder and most commonly develops as a consequence of a mutation in the c-kit proto-oncogene (located on chromosome arm 14q12), which affects melanoblast migration, proliferation, differentiation, and survival.2 In piebaldism, the site of mutation within the gene correlates with the severity of the phenotype.5 Melanocytes are histologically and ultrastructurally absent or considerably reduced in depigmented patches but are normal in number in the hyperpigmented areas.2
Rare cases of piebaldism have been reported in association with other diseases, including congenital megacolon, congenital dyserythropoietic anemia type II, Diamond-Blackfan anemia, Grover disease (transient acantholytic dermatosis), and glycogen-storage disease type 1a.1,6 Poliosis alone may be the initial presentation of certain genetic syndromes, including Waardenburg syndrome (WS) and tuberous sclerosis; it also may be acquired in the setting of several conditions, including vitiligo, Vogt-Koyanagi-Harada syndrome, Alezzandrini syndrome, alopecia areata, and sarcoidosis.3
Notably, the diagnosis of piebaldism should alert the clinician to the possibility of WS, an autosomal-dominant disease characterized by a congenital white forelock, leukoderma in a piebaldlike distribution, lateral displacement of the medial canthi, a hypertrophic nasal root, heterochromia iridis, and progressive sensorineural hearing loss.7 Four clinical subtypes of WS have been described, with various gene mutations implicated: type 1 is the classic form, type 2 lacks dystopia canthorum and has a stonger association with deafness, type 3 is associated with limb abnormalities, and type 4 is associated with congenital megacolon. A case of WS type 1 has been described in association with facial nerve palsy and lingua plicata, 2 main features of Melkerson-Rosenthal syndrome.8 Depigmentation in WS is caused by the absence of melanocytes in the affected areas as well as failed migration of melanocytes to the ears and eyes.3 Waardenburg syndrome may be distinguished from piebaldism by characteristic facial features of the disease and should prompt a thorough ocular and auditory examination in affected patients.9
Although not a diagnostic criterion, poliosis rarely has been reported as one of the earliest associated findings of tuberous sclerosis.3,10 Major cutaneous features of this disease include facial angiofibromas, hypomelanotic macules, shagreen patches (connective tissue nevi), periungual fibromas, molluscum pendulum, and café au lait macules.
Vitiligo also may be considered in the differential diagnosis of piebaldism and can be distinguished by the presence of depigmented patches in a typical acral and periorificial distribution, lack of congential presentation, and relatively progressive course. Vitiligo is characterized by an acquired loss of epidermal melanocytes, leading to depigmented macules and patches.1,3
Vitiligo, poliosis, and alopecia areata usually are late clinical manifestations of Vogt-Koyanagi-Harada syndrome, a rare condition characterized by an autoimmune response to melanocyte-associated antigens. This condition initially presents with neurologic and ocular manifestations including headache, muscle weakness, tinnitus, uveitis, and choroiditis prior to dermatologic manifestations.11
Alezzandrini syndrome, a rare and closely related disorder, is distinctly characterized by whitening of scalp hair, eyebrows, and eyelashes, along with unilateral depigmentation of facial skin. This presentation is associated with ipsilateral visual changes and hearing abnormalities.12
The absence of abnormal ocular, auditory, and neurologic examinations, along with lack of characteristic cutaneous features indicating any of the aforementioned disorders, highly suggests a diagnosis of piebaldism.
Piebaldism is considered a relatively benign disorder but can be highly socially disabling, which presents a therapeutic challenge in affected children. Depigmented skin in piebaldism generally is considered unresponsive to medical or light therapy.1 Topical treatments with makeup or artificial pigmenting agents (eg, dihydroxyacetone [an ingredient used in sunless tanning products]) are useful but temporary. Sunscreen should be used judiciously to avoid sunburn and reduce carcinogenic potential.13
Several surgical techniques have been reported for treatment of leukoderma but with variable success. Of those reported, micropunch transplantation (minigrafting) using epidermal donor sites of 1 to 1.25 mm is a relatively inexpensive and effective method but is limited by scarring at the donor site.14 Autologous cultured epidermal cellular grafting with a controlled number of melanocytes is reported to achieve greater than 75% repigmentation. It requires fewer donor sites and, therefore, results in less scarring.15 Additionally, use of the erbium-doped:YAG laser aids in deepithelialization of the recipient site, allowing for treatment of large piebald lesions during a single operation.16 Despite these advances, additional studies are needed to improve quality of life in those affected.
- Janjua SA, Khachemoune A, Guldbakke KK. Piebaldism: a case report and a concise review of the literature. Cutis. 2007;80:411-414.
- Agarwal S, Ojha A. Piebaldism: a brief report and review of the literature. Indian Dermatol Online J. 2012;3:144-147.
- Sleiman R, Kurban M, Succaria F, et al. Poliosis circumscripta: overview and underlying causes. J Am Acad Dermatol. 2013;69:625-633.
- Oiso N, Fukai K, Kawada A, et al. Piebaldism. J Dermatol. 2013;40:330-355.
- López V, Jordá E. Piebaldism in a 2-year-old girl. Dermatol Online J. 2011;17:13.
- Ghoshal B, Sarkar N, Bhattacharjee M, et al. Glycogen storage disease 1a with piebaldism. Indian Pediatr. 2012;49:235-236.
- Salvatore S, Carnevale C, Infussi R, et al. Waardenburg syndrome: a review of literature and case reports. Clin Ter. 2012;163:e85-e94.
- Dourmishev AL, Dourmishev LA, Schwartz RA, et al. Waardenburg syndrome. Int J Dermatol. 1999;38:656-663.
- Fistarol SK, Itin PH. Disorders of pigmentation. J Dtsch Dermatol Ges. 2010;8:187-201.
- McWilliam RC, Stephenson JB. Depigmented hair. the earliest sign of tuberous sclerosis. Arch Dis Child. 1978;53:961-963.
- Chan EW, Sanjay S, Chang BC. Headache, red eyes, blurred vision and hearing loss. diagnosis: Vogt-Koyanagi-Harada syndrome. CMAJ. 2010;182:1205-1209.
- Andrade A, Pithon M. Alezzandrini syndrome: report of a sixth clinical case. Dermatology (Basel). 2011;222:8-9.
- Suga Y, Ikejima A, Matsuba S, et al. Medical pearl: DHA application for camouflaging segmental vitiligo and piebald lesions. J Am Acad Dermatol. 2002;47:436-438.
- Neves DR, Régis Júnior JR, Oliveira PJ, et al. Melanocyte transplant in piebaldism: case report. An Bras Dermatol. 2010;85:384-388.
- Van geel N, Wallaeys E, Goh BK, et al. Long-term results of noncultured epidermal cellular grafting in vitiligo, halo naevi, piebaldism and naevus depigmentosus. Br J Dermatol. 2010;163:1186-1193.
- Guerra L, Primavera G, Raskovic D, et al. Permanent repigmentation of piebaldism by erbium:YAG laser and autologous cultured epidermis. Br J Dermatol. 2004;150:715-721.
Case Report
A 14-year-old adolescent girl presented with multiple asymptomatic light-colored patches on the forehead, bilateral arms, and legs that had been present since birth. The patient reported that the size of the patches had increased in proportion to her overall growth and that “brown spots” had gradually started to form within and around the patches. She noted that her father and paternal grandfather also had similar clinical findings. A review of systems was negative for hearing impairment, ocular abnormalities, and recurrent infections.
Physical examination revealed an otherwise healthy adolescent girl with Fitzpatrick skin type I and homogeneous blue eyes. Large symmetric depigmented patches were noted on the extensor surfaces of the mid legs and mid forearms (Figure). Macules of baseline pigment and hyperpigmentation were irregularly scattered within and at the periphery of the patches. A triangular hypopigmented patch at the hairline on the mid frontal scalp hairline was accompanied by depigmentation of terminal hairs in this region.
A clinical diagnosis of piebaldism was made and was discussed at length with the patient. Due to the benign nature of the condition and patient preference, no therapeutic intervention was pursued. It was recommended that she apply sunscreen daily for protection of the depigmented areas.
Comment
Piebaldism is a rare hereditary disorder of melanocyte development characterized clinically by the presence of congenital poliosis and leukoderma.1 The exact prevalence of piebaldism is unknown, but it has been estimated that less than 1 in 20,000 children are born with this condition.2 Poliosis circumscripta, traditionally known as white forelock, may be the only manifestation in 80% to 90% of cases and is present at birth.3 The white forelock typically appears in a triangular shape and the underlying skin of the scalp also is amelanotic. The eyebrows and eyelashes also may be involved.3
Characteristically, lesions of leukoderma are well-circumscribed, irregular, white patches that are often accompanied by hyperpigmented macules noted on both depigmented and unaffected adjacent skin.1 The lesions are classically distributed on the central forehead and anterior trunk, with extension to the flanks, anterior mid arms, and mid legs. Sparing of the dorsal midline, hands, feet, and periorificial area is characteristic.1
Depigmented patches typically are nonprogressive and persist into adulthood. Additional hyperpigmented macules may develop at or within the margins of the white patches. Partial or complete repigmentation may occur spontaneously or after trauma in some patients.2 Some children may develop café au lait lesions and may be misdiagnosed as concurrently having neurofibromatosis type I and piebaldism. If neurofibromatosis type I is suspected, patients should be thoroughly evaluated for other diagnostic criteria of this syndrome, as there may be cases of coexistence and overlap with piebaldism.4
Piebaldism is an autosomal-dominant inherited disorder and most commonly develops as a consequence of a mutation in the c-kit proto-oncogene (located on chromosome arm 14q12), which affects melanoblast migration, proliferation, differentiation, and survival.2 In piebaldism, the site of mutation within the gene correlates with the severity of the phenotype.5 Melanocytes are histologically and ultrastructurally absent or considerably reduced in depigmented patches but are normal in number in the hyperpigmented areas.2
Rare cases of piebaldism have been reported in association with other diseases, including congenital megacolon, congenital dyserythropoietic anemia type II, Diamond-Blackfan anemia, Grover disease (transient acantholytic dermatosis), and glycogen-storage disease type 1a.1,6 Poliosis alone may be the initial presentation of certain genetic syndromes, including Waardenburg syndrome (WS) and tuberous sclerosis; it also may be acquired in the setting of several conditions, including vitiligo, Vogt-Koyanagi-Harada syndrome, Alezzandrini syndrome, alopecia areata, and sarcoidosis.3
Notably, the diagnosis of piebaldism should alert the clinician to the possibility of WS, an autosomal-dominant disease characterized by a congenital white forelock, leukoderma in a piebaldlike distribution, lateral displacement of the medial canthi, a hypertrophic nasal root, heterochromia iridis, and progressive sensorineural hearing loss.7 Four clinical subtypes of WS have been described, with various gene mutations implicated: type 1 is the classic form, type 2 lacks dystopia canthorum and has a stonger association with deafness, type 3 is associated with limb abnormalities, and type 4 is associated with congenital megacolon. A case of WS type 1 has been described in association with facial nerve palsy and lingua plicata, 2 main features of Melkerson-Rosenthal syndrome.8 Depigmentation in WS is caused by the absence of melanocytes in the affected areas as well as failed migration of melanocytes to the ears and eyes.3 Waardenburg syndrome may be distinguished from piebaldism by characteristic facial features of the disease and should prompt a thorough ocular and auditory examination in affected patients.9
Although not a diagnostic criterion, poliosis rarely has been reported as one of the earliest associated findings of tuberous sclerosis.3,10 Major cutaneous features of this disease include facial angiofibromas, hypomelanotic macules, shagreen patches (connective tissue nevi), periungual fibromas, molluscum pendulum, and café au lait macules.
Vitiligo also may be considered in the differential diagnosis of piebaldism and can be distinguished by the presence of depigmented patches in a typical acral and periorificial distribution, lack of congential presentation, and relatively progressive course. Vitiligo is characterized by an acquired loss of epidermal melanocytes, leading to depigmented macules and patches.1,3
Vitiligo, poliosis, and alopecia areata usually are late clinical manifestations of Vogt-Koyanagi-Harada syndrome, a rare condition characterized by an autoimmune response to melanocyte-associated antigens. This condition initially presents with neurologic and ocular manifestations including headache, muscle weakness, tinnitus, uveitis, and choroiditis prior to dermatologic manifestations.11
Alezzandrini syndrome, a rare and closely related disorder, is distinctly characterized by whitening of scalp hair, eyebrows, and eyelashes, along with unilateral depigmentation of facial skin. This presentation is associated with ipsilateral visual changes and hearing abnormalities.12
The absence of abnormal ocular, auditory, and neurologic examinations, along with lack of characteristic cutaneous features indicating any of the aforementioned disorders, highly suggests a diagnosis of piebaldism.
Piebaldism is considered a relatively benign disorder but can be highly socially disabling, which presents a therapeutic challenge in affected children. Depigmented skin in piebaldism generally is considered unresponsive to medical or light therapy.1 Topical treatments with makeup or artificial pigmenting agents (eg, dihydroxyacetone [an ingredient used in sunless tanning products]) are useful but temporary. Sunscreen should be used judiciously to avoid sunburn and reduce carcinogenic potential.13
Several surgical techniques have been reported for treatment of leukoderma but with variable success. Of those reported, micropunch transplantation (minigrafting) using epidermal donor sites of 1 to 1.25 mm is a relatively inexpensive and effective method but is limited by scarring at the donor site.14 Autologous cultured epidermal cellular grafting with a controlled number of melanocytes is reported to achieve greater than 75% repigmentation. It requires fewer donor sites and, therefore, results in less scarring.15 Additionally, use of the erbium-doped:YAG laser aids in deepithelialization of the recipient site, allowing for treatment of large piebald lesions during a single operation.16 Despite these advances, additional studies are needed to improve quality of life in those affected.
Case Report
A 14-year-old adolescent girl presented with multiple asymptomatic light-colored patches on the forehead, bilateral arms, and legs that had been present since birth. The patient reported that the size of the patches had increased in proportion to her overall growth and that “brown spots” had gradually started to form within and around the patches. She noted that her father and paternal grandfather also had similar clinical findings. A review of systems was negative for hearing impairment, ocular abnormalities, and recurrent infections.
Physical examination revealed an otherwise healthy adolescent girl with Fitzpatrick skin type I and homogeneous blue eyes. Large symmetric depigmented patches were noted on the extensor surfaces of the mid legs and mid forearms (Figure). Macules of baseline pigment and hyperpigmentation were irregularly scattered within and at the periphery of the patches. A triangular hypopigmented patch at the hairline on the mid frontal scalp hairline was accompanied by depigmentation of terminal hairs in this region.
A clinical diagnosis of piebaldism was made and was discussed at length with the patient. Due to the benign nature of the condition and patient preference, no therapeutic intervention was pursued. It was recommended that she apply sunscreen daily for protection of the depigmented areas.
Comment
Piebaldism is a rare hereditary disorder of melanocyte development characterized clinically by the presence of congenital poliosis and leukoderma.1 The exact prevalence of piebaldism is unknown, but it has been estimated that less than 1 in 20,000 children are born with this condition.2 Poliosis circumscripta, traditionally known as white forelock, may be the only manifestation in 80% to 90% of cases and is present at birth.3 The white forelock typically appears in a triangular shape and the underlying skin of the scalp also is amelanotic. The eyebrows and eyelashes also may be involved.3
Characteristically, lesions of leukoderma are well-circumscribed, irregular, white patches that are often accompanied by hyperpigmented macules noted on both depigmented and unaffected adjacent skin.1 The lesions are classically distributed on the central forehead and anterior trunk, with extension to the flanks, anterior mid arms, and mid legs. Sparing of the dorsal midline, hands, feet, and periorificial area is characteristic.1
Depigmented patches typically are nonprogressive and persist into adulthood. Additional hyperpigmented macules may develop at or within the margins of the white patches. Partial or complete repigmentation may occur spontaneously or after trauma in some patients.2 Some children may develop café au lait lesions and may be misdiagnosed as concurrently having neurofibromatosis type I and piebaldism. If neurofibromatosis type I is suspected, patients should be thoroughly evaluated for other diagnostic criteria of this syndrome, as there may be cases of coexistence and overlap with piebaldism.4
Piebaldism is an autosomal-dominant inherited disorder and most commonly develops as a consequence of a mutation in the c-kit proto-oncogene (located on chromosome arm 14q12), which affects melanoblast migration, proliferation, differentiation, and survival.2 In piebaldism, the site of mutation within the gene correlates with the severity of the phenotype.5 Melanocytes are histologically and ultrastructurally absent or considerably reduced in depigmented patches but are normal in number in the hyperpigmented areas.2
Rare cases of piebaldism have been reported in association with other diseases, including congenital megacolon, congenital dyserythropoietic anemia type II, Diamond-Blackfan anemia, Grover disease (transient acantholytic dermatosis), and glycogen-storage disease type 1a.1,6 Poliosis alone may be the initial presentation of certain genetic syndromes, including Waardenburg syndrome (WS) and tuberous sclerosis; it also may be acquired in the setting of several conditions, including vitiligo, Vogt-Koyanagi-Harada syndrome, Alezzandrini syndrome, alopecia areata, and sarcoidosis.3
Notably, the diagnosis of piebaldism should alert the clinician to the possibility of WS, an autosomal-dominant disease characterized by a congenital white forelock, leukoderma in a piebaldlike distribution, lateral displacement of the medial canthi, a hypertrophic nasal root, heterochromia iridis, and progressive sensorineural hearing loss.7 Four clinical subtypes of WS have been described, with various gene mutations implicated: type 1 is the classic form, type 2 lacks dystopia canthorum and has a stonger association with deafness, type 3 is associated with limb abnormalities, and type 4 is associated with congenital megacolon. A case of WS type 1 has been described in association with facial nerve palsy and lingua plicata, 2 main features of Melkerson-Rosenthal syndrome.8 Depigmentation in WS is caused by the absence of melanocytes in the affected areas as well as failed migration of melanocytes to the ears and eyes.3 Waardenburg syndrome may be distinguished from piebaldism by characteristic facial features of the disease and should prompt a thorough ocular and auditory examination in affected patients.9
Although not a diagnostic criterion, poliosis rarely has been reported as one of the earliest associated findings of tuberous sclerosis.3,10 Major cutaneous features of this disease include facial angiofibromas, hypomelanotic macules, shagreen patches (connective tissue nevi), periungual fibromas, molluscum pendulum, and café au lait macules.
Vitiligo also may be considered in the differential diagnosis of piebaldism and can be distinguished by the presence of depigmented patches in a typical acral and periorificial distribution, lack of congential presentation, and relatively progressive course. Vitiligo is characterized by an acquired loss of epidermal melanocytes, leading to depigmented macules and patches.1,3
Vitiligo, poliosis, and alopecia areata usually are late clinical manifestations of Vogt-Koyanagi-Harada syndrome, a rare condition characterized by an autoimmune response to melanocyte-associated antigens. This condition initially presents with neurologic and ocular manifestations including headache, muscle weakness, tinnitus, uveitis, and choroiditis prior to dermatologic manifestations.11
Alezzandrini syndrome, a rare and closely related disorder, is distinctly characterized by whitening of scalp hair, eyebrows, and eyelashes, along with unilateral depigmentation of facial skin. This presentation is associated with ipsilateral visual changes and hearing abnormalities.12
The absence of abnormal ocular, auditory, and neurologic examinations, along with lack of characteristic cutaneous features indicating any of the aforementioned disorders, highly suggests a diagnosis of piebaldism.
Piebaldism is considered a relatively benign disorder but can be highly socially disabling, which presents a therapeutic challenge in affected children. Depigmented skin in piebaldism generally is considered unresponsive to medical or light therapy.1 Topical treatments with makeup or artificial pigmenting agents (eg, dihydroxyacetone [an ingredient used in sunless tanning products]) are useful but temporary. Sunscreen should be used judiciously to avoid sunburn and reduce carcinogenic potential.13
Several surgical techniques have been reported for treatment of leukoderma but with variable success. Of those reported, micropunch transplantation (minigrafting) using epidermal donor sites of 1 to 1.25 mm is a relatively inexpensive and effective method but is limited by scarring at the donor site.14 Autologous cultured epidermal cellular grafting with a controlled number of melanocytes is reported to achieve greater than 75% repigmentation. It requires fewer donor sites and, therefore, results in less scarring.15 Additionally, use of the erbium-doped:YAG laser aids in deepithelialization of the recipient site, allowing for treatment of large piebald lesions during a single operation.16 Despite these advances, additional studies are needed to improve quality of life in those affected.
- Janjua SA, Khachemoune A, Guldbakke KK. Piebaldism: a case report and a concise review of the literature. Cutis. 2007;80:411-414.
- Agarwal S, Ojha A. Piebaldism: a brief report and review of the literature. Indian Dermatol Online J. 2012;3:144-147.
- Sleiman R, Kurban M, Succaria F, et al. Poliosis circumscripta: overview and underlying causes. J Am Acad Dermatol. 2013;69:625-633.
- Oiso N, Fukai K, Kawada A, et al. Piebaldism. J Dermatol. 2013;40:330-355.
- López V, Jordá E. Piebaldism in a 2-year-old girl. Dermatol Online J. 2011;17:13.
- Ghoshal B, Sarkar N, Bhattacharjee M, et al. Glycogen storage disease 1a with piebaldism. Indian Pediatr. 2012;49:235-236.
- Salvatore S, Carnevale C, Infussi R, et al. Waardenburg syndrome: a review of literature and case reports. Clin Ter. 2012;163:e85-e94.
- Dourmishev AL, Dourmishev LA, Schwartz RA, et al. Waardenburg syndrome. Int J Dermatol. 1999;38:656-663.
- Fistarol SK, Itin PH. Disorders of pigmentation. J Dtsch Dermatol Ges. 2010;8:187-201.
- McWilliam RC, Stephenson JB. Depigmented hair. the earliest sign of tuberous sclerosis. Arch Dis Child. 1978;53:961-963.
- Chan EW, Sanjay S, Chang BC. Headache, red eyes, blurred vision and hearing loss. diagnosis: Vogt-Koyanagi-Harada syndrome. CMAJ. 2010;182:1205-1209.
- Andrade A, Pithon M. Alezzandrini syndrome: report of a sixth clinical case. Dermatology (Basel). 2011;222:8-9.
- Suga Y, Ikejima A, Matsuba S, et al. Medical pearl: DHA application for camouflaging segmental vitiligo and piebald lesions. J Am Acad Dermatol. 2002;47:436-438.
- Neves DR, Régis Júnior JR, Oliveira PJ, et al. Melanocyte transplant in piebaldism: case report. An Bras Dermatol. 2010;85:384-388.
- Van geel N, Wallaeys E, Goh BK, et al. Long-term results of noncultured epidermal cellular grafting in vitiligo, halo naevi, piebaldism and naevus depigmentosus. Br J Dermatol. 2010;163:1186-1193.
- Guerra L, Primavera G, Raskovic D, et al. Permanent repigmentation of piebaldism by erbium:YAG laser and autologous cultured epidermis. Br J Dermatol. 2004;150:715-721.
- Janjua SA, Khachemoune A, Guldbakke KK. Piebaldism: a case report and a concise review of the literature. Cutis. 2007;80:411-414.
- Agarwal S, Ojha A. Piebaldism: a brief report and review of the literature. Indian Dermatol Online J. 2012;3:144-147.
- Sleiman R, Kurban M, Succaria F, et al. Poliosis circumscripta: overview and underlying causes. J Am Acad Dermatol. 2013;69:625-633.
- Oiso N, Fukai K, Kawada A, et al. Piebaldism. J Dermatol. 2013;40:330-355.
- López V, Jordá E. Piebaldism in a 2-year-old girl. Dermatol Online J. 2011;17:13.
- Ghoshal B, Sarkar N, Bhattacharjee M, et al. Glycogen storage disease 1a with piebaldism. Indian Pediatr. 2012;49:235-236.
- Salvatore S, Carnevale C, Infussi R, et al. Waardenburg syndrome: a review of literature and case reports. Clin Ter. 2012;163:e85-e94.
- Dourmishev AL, Dourmishev LA, Schwartz RA, et al. Waardenburg syndrome. Int J Dermatol. 1999;38:656-663.
- Fistarol SK, Itin PH. Disorders of pigmentation. J Dtsch Dermatol Ges. 2010;8:187-201.
- McWilliam RC, Stephenson JB. Depigmented hair. the earliest sign of tuberous sclerosis. Arch Dis Child. 1978;53:961-963.
- Chan EW, Sanjay S, Chang BC. Headache, red eyes, blurred vision and hearing loss. diagnosis: Vogt-Koyanagi-Harada syndrome. CMAJ. 2010;182:1205-1209.
- Andrade A, Pithon M. Alezzandrini syndrome: report of a sixth clinical case. Dermatology (Basel). 2011;222:8-9.
- Suga Y, Ikejima A, Matsuba S, et al. Medical pearl: DHA application for camouflaging segmental vitiligo and piebald lesions. J Am Acad Dermatol. 2002;47:436-438.
- Neves DR, Régis Júnior JR, Oliveira PJ, et al. Melanocyte transplant in piebaldism: case report. An Bras Dermatol. 2010;85:384-388.
- Van geel N, Wallaeys E, Goh BK, et al. Long-term results of noncultured epidermal cellular grafting in vitiligo, halo naevi, piebaldism and naevus depigmentosus. Br J Dermatol. 2010;163:1186-1193.
- Guerra L, Primavera G, Raskovic D, et al. Permanent repigmentation of piebaldism by erbium:YAG laser and autologous cultured epidermis. Br J Dermatol. 2004;150:715-721.
Practice Points
- Poliosis circumscripta (or white forelock) is commonly the only manifestation of piebaldism in children.
- Affected areas of leukoderma in piebaldism are classically distributed on the central forehead, anterior trunk, and mid extremities.
- The presence of congenital leukoderma should prompt a thorough skin examination and review of the patient’s medical history for evidence of ocular, auditory, and/or neurologic abnormalities.