User login
Study: Pretreatment ECG not always needed in babies with hemangiomas
Routine ECG screening in infants before they receive propranolol for hemangiomas is “not likely to be an effective screening tool in patients with otherwise normal physical examination and family history” and may even cause harmful delays in treatment, study authors concluded.
“As previously published guidelines suggest, it is likely that an indication-driven ECG strategy is a better approach, because there is a low incidence of ECG abnormalities that would limit propranolol use in children,” wrote Kevin B. Yarbrough, MD, a dermatologist at Phoenix Children’s Hospital, and his associates. The results “support published guidelines for propranolol initiation and are congruent with findings from other investigators” (Pediatr Dermatol. 2016 Nov;33[6]:615-20).
In the retrospective study, Dr. Yarbrough and his associates tracked 162 patients (median age, 5.2 months) who underwent routine ECG screening at several clinics before propranolol treatment for hemangiomas from 2008 to 2013. The ECGs were read as abnormal in 69 cases (43%); the most common abnormality was left ventricular hypertrophy (16 patients), followed by right ventricular hypertrophy (8), sinus bradycardia (6), and sinus tachycardia (5).
Cardiologists cleared all 69 patients for propranolol treatment, which they received. “No patients in our cohort experienced an adverse effect during treatment that could have been predicted or prevented by ECG before initiation of the propranolol,” the authors wrote.
“Routine ECG adds to the cost of treating hemangiomas and leads to unnecessary consultations and testing. Even more importantly, abnormalities detected on ECG can lead to delays in treatment initiation, which in turn can lead to greater patient morbidity, as seen in the case of our patient whose hemangioma ulcerated while awaiting cardiology consultation,” they added.
Still, they noted that ECG tests should still be performed on “infants with bradycardia or cardiac arrhythmia found during initial physical examination, a family history of congenital heart disease or arrhythmias, and a maternal history of connective tissue disease.”
Study funding information was not provided. One of the study authors reported that he was a clinical investigator for Pierre Fabre Dermatologie, the manufacturer of the oral propranolol product Hemangeol.
Routine ECG screening in infants before they receive propranolol for hemangiomas is “not likely to be an effective screening tool in patients with otherwise normal physical examination and family history” and may even cause harmful delays in treatment, study authors concluded.
“As previously published guidelines suggest, it is likely that an indication-driven ECG strategy is a better approach, because there is a low incidence of ECG abnormalities that would limit propranolol use in children,” wrote Kevin B. Yarbrough, MD, a dermatologist at Phoenix Children’s Hospital, and his associates. The results “support published guidelines for propranolol initiation and are congruent with findings from other investigators” (Pediatr Dermatol. 2016 Nov;33[6]:615-20).
In the retrospective study, Dr. Yarbrough and his associates tracked 162 patients (median age, 5.2 months) who underwent routine ECG screening at several clinics before propranolol treatment for hemangiomas from 2008 to 2013. The ECGs were read as abnormal in 69 cases (43%); the most common abnormality was left ventricular hypertrophy (16 patients), followed by right ventricular hypertrophy (8), sinus bradycardia (6), and sinus tachycardia (5).
Cardiologists cleared all 69 patients for propranolol treatment, which they received. “No patients in our cohort experienced an adverse effect during treatment that could have been predicted or prevented by ECG before initiation of the propranolol,” the authors wrote.
“Routine ECG adds to the cost of treating hemangiomas and leads to unnecessary consultations and testing. Even more importantly, abnormalities detected on ECG can lead to delays in treatment initiation, which in turn can lead to greater patient morbidity, as seen in the case of our patient whose hemangioma ulcerated while awaiting cardiology consultation,” they added.
Still, they noted that ECG tests should still be performed on “infants with bradycardia or cardiac arrhythmia found during initial physical examination, a family history of congenital heart disease or arrhythmias, and a maternal history of connective tissue disease.”
Study funding information was not provided. One of the study authors reported that he was a clinical investigator for Pierre Fabre Dermatologie, the manufacturer of the oral propranolol product Hemangeol.
Routine ECG screening in infants before they receive propranolol for hemangiomas is “not likely to be an effective screening tool in patients with otherwise normal physical examination and family history” and may even cause harmful delays in treatment, study authors concluded.
“As previously published guidelines suggest, it is likely that an indication-driven ECG strategy is a better approach, because there is a low incidence of ECG abnormalities that would limit propranolol use in children,” wrote Kevin B. Yarbrough, MD, a dermatologist at Phoenix Children’s Hospital, and his associates. The results “support published guidelines for propranolol initiation and are congruent with findings from other investigators” (Pediatr Dermatol. 2016 Nov;33[6]:615-20).
In the retrospective study, Dr. Yarbrough and his associates tracked 162 patients (median age, 5.2 months) who underwent routine ECG screening at several clinics before propranolol treatment for hemangiomas from 2008 to 2013. The ECGs were read as abnormal in 69 cases (43%); the most common abnormality was left ventricular hypertrophy (16 patients), followed by right ventricular hypertrophy (8), sinus bradycardia (6), and sinus tachycardia (5).
Cardiologists cleared all 69 patients for propranolol treatment, which they received. “No patients in our cohort experienced an adverse effect during treatment that could have been predicted or prevented by ECG before initiation of the propranolol,” the authors wrote.
“Routine ECG adds to the cost of treating hemangiomas and leads to unnecessary consultations and testing. Even more importantly, abnormalities detected on ECG can lead to delays in treatment initiation, which in turn can lead to greater patient morbidity, as seen in the case of our patient whose hemangioma ulcerated while awaiting cardiology consultation,” they added.
Still, they noted that ECG tests should still be performed on “infants with bradycardia or cardiac arrhythmia found during initial physical examination, a family history of congenital heart disease or arrhythmias, and a maternal history of connective tissue disease.”
Study funding information was not provided. One of the study authors reported that he was a clinical investigator for Pierre Fabre Dermatologie, the manufacturer of the oral propranolol product Hemangeol.
FROM PEDIATRIC DERMATOLOGY
Key clinical point: While it’s appropriate in some cases, routine ECG screening appears to be unnecessary before administering propranolol to infants to treat hemangiomas.
Major finding: All 69 infants whose screening ECGs turned up abnormalities were subsequently cleared by cardiologists.
Data source: A retrospective analysis of 162 patients with infantile hemangiomas seen at various clinics from 2008 to 2013.
Disclosures: Study funding information was not provided. One of the study authors, Alfons L. Krol, MD, reported being a clinical investigator for Pierre Fabre Dermatologie, the manufacturer of the oral propranolol product Hemangeol.
Nasal infantile hemangiomas develop most complications
Infantile hemangiomas of the nose develop more complications than those at all other body sites combined, according to a report published in Pediatric Dermatology.
In what they described as the largest study to date to assess nasal infantile hemangiomas, researchers assessed which traits are associated with complications and predict residual skin changes at the age of 5 years. “Nasal infantile hemangiomas pose an immediate risk of airway obstruction because infants are obligate nasal breathers, and may have long-term functional and psychosocial consequences if involution is incomplete or development of surrounding structures, such as nasal cartilage, is compromised,” said Maria S. Kryatova of the departments of pediatrics and dermatology, Johns Hopkins University, Baltimore, and her associates.
The investigators identified all patients younger than 18 years who had been treated at their academic referral center for nasal infantile hemangiomas between 2001 and 2014. They performed retrospective chart reviews, which included photographs, for 89 participants. The parents of 63 of these children were interviewed when the participants reached a median age of 5 years and provided comparison photographs taken at their entry into kindergarten.
Thirty-five children (39%) developed one or more complications at some time during follow-up, including airway compromise, compression, or functional impairment; ulceration; visual obstruction or ocular compression; and infection. In comparison, the Hemangioma Investigator Group has previously reported a 24% overall rate of complications at all body sites. Similarly, the proportion of study participants who received at least one type of treatment (propranolol, oral steroids, pulsed dye laser, surgery, topical timolol, intralesional corticosteroids, yttrium-aluminum-garnet laser, carbon dioxide laser, or fraxel laser) was markedly higher (80%) than that reported previously by the Hemangioma Investigator Group for all body sites (38%).
“Our study is the first to report a significant association between [the hemangioma’s location on the nose] and depth. Lesions on the nasal dorsum are unlikely to be deep, whereas nasal tip lesions are unlikely to be superficial. Deep vertical growth may be limited by underlying nasal bone in the dorsum but less so by the soft tissue of the nasal tip.” Alternatively, as suggested by other investigators, an embryologic explanation is also possible – “the fusion lines between neural crest–derived mesenchyme and ectoderm-derived nasal placodes may have different properties in the vicinity of the nasal dorsum and nasal tip that predispose them to the development of superficial and deep hemangiomas, respectively,” Ms. Kryatova and her associates reported (Ped Dermatol. 2016;33[6]:652-8).
Segmental- and indeterminate-type lesions were more likely than focal-type lesions to develop ulceration, compression, or functional obstruction, and mixed-depth hemangiomas were more likely than deep or superficial hemangiomas to ulcerate. Overall, the lesions had involuted by kindergarten age in 70% of the study participants but persisted in 30%, and most of the children with involution showed residual skin changes such as telangiectasia (14 children), fibrofatty tissue (11 children), and scarring (9 children).
These findings show that a multicenter study to expand on these conclusions and to determine the best treatment algorithm for nasal infantile hemangiomas is warranted, the investigators added.
Infantile hemangiomas of the nose develop more complications than those at all other body sites combined, according to a report published in Pediatric Dermatology.
In what they described as the largest study to date to assess nasal infantile hemangiomas, researchers assessed which traits are associated with complications and predict residual skin changes at the age of 5 years. “Nasal infantile hemangiomas pose an immediate risk of airway obstruction because infants are obligate nasal breathers, and may have long-term functional and psychosocial consequences if involution is incomplete or development of surrounding structures, such as nasal cartilage, is compromised,” said Maria S. Kryatova of the departments of pediatrics and dermatology, Johns Hopkins University, Baltimore, and her associates.
The investigators identified all patients younger than 18 years who had been treated at their academic referral center for nasal infantile hemangiomas between 2001 and 2014. They performed retrospective chart reviews, which included photographs, for 89 participants. The parents of 63 of these children were interviewed when the participants reached a median age of 5 years and provided comparison photographs taken at their entry into kindergarten.
Thirty-five children (39%) developed one or more complications at some time during follow-up, including airway compromise, compression, or functional impairment; ulceration; visual obstruction or ocular compression; and infection. In comparison, the Hemangioma Investigator Group has previously reported a 24% overall rate of complications at all body sites. Similarly, the proportion of study participants who received at least one type of treatment (propranolol, oral steroids, pulsed dye laser, surgery, topical timolol, intralesional corticosteroids, yttrium-aluminum-garnet laser, carbon dioxide laser, or fraxel laser) was markedly higher (80%) than that reported previously by the Hemangioma Investigator Group for all body sites (38%).
“Our study is the first to report a significant association between [the hemangioma’s location on the nose] and depth. Lesions on the nasal dorsum are unlikely to be deep, whereas nasal tip lesions are unlikely to be superficial. Deep vertical growth may be limited by underlying nasal bone in the dorsum but less so by the soft tissue of the nasal tip.” Alternatively, as suggested by other investigators, an embryologic explanation is also possible – “the fusion lines between neural crest–derived mesenchyme and ectoderm-derived nasal placodes may have different properties in the vicinity of the nasal dorsum and nasal tip that predispose them to the development of superficial and deep hemangiomas, respectively,” Ms. Kryatova and her associates reported (Ped Dermatol. 2016;33[6]:652-8).
Segmental- and indeterminate-type lesions were more likely than focal-type lesions to develop ulceration, compression, or functional obstruction, and mixed-depth hemangiomas were more likely than deep or superficial hemangiomas to ulcerate. Overall, the lesions had involuted by kindergarten age in 70% of the study participants but persisted in 30%, and most of the children with involution showed residual skin changes such as telangiectasia (14 children), fibrofatty tissue (11 children), and scarring (9 children).
These findings show that a multicenter study to expand on these conclusions and to determine the best treatment algorithm for nasal infantile hemangiomas is warranted, the investigators added.
Infantile hemangiomas of the nose develop more complications than those at all other body sites combined, according to a report published in Pediatric Dermatology.
In what they described as the largest study to date to assess nasal infantile hemangiomas, researchers assessed which traits are associated with complications and predict residual skin changes at the age of 5 years. “Nasal infantile hemangiomas pose an immediate risk of airway obstruction because infants are obligate nasal breathers, and may have long-term functional and psychosocial consequences if involution is incomplete or development of surrounding structures, such as nasal cartilage, is compromised,” said Maria S. Kryatova of the departments of pediatrics and dermatology, Johns Hopkins University, Baltimore, and her associates.
The investigators identified all patients younger than 18 years who had been treated at their academic referral center for nasal infantile hemangiomas between 2001 and 2014. They performed retrospective chart reviews, which included photographs, for 89 participants. The parents of 63 of these children were interviewed when the participants reached a median age of 5 years and provided comparison photographs taken at their entry into kindergarten.
Thirty-five children (39%) developed one or more complications at some time during follow-up, including airway compromise, compression, or functional impairment; ulceration; visual obstruction or ocular compression; and infection. In comparison, the Hemangioma Investigator Group has previously reported a 24% overall rate of complications at all body sites. Similarly, the proportion of study participants who received at least one type of treatment (propranolol, oral steroids, pulsed dye laser, surgery, topical timolol, intralesional corticosteroids, yttrium-aluminum-garnet laser, carbon dioxide laser, or fraxel laser) was markedly higher (80%) than that reported previously by the Hemangioma Investigator Group for all body sites (38%).
“Our study is the first to report a significant association between [the hemangioma’s location on the nose] and depth. Lesions on the nasal dorsum are unlikely to be deep, whereas nasal tip lesions are unlikely to be superficial. Deep vertical growth may be limited by underlying nasal bone in the dorsum but less so by the soft tissue of the nasal tip.” Alternatively, as suggested by other investigators, an embryologic explanation is also possible – “the fusion lines between neural crest–derived mesenchyme and ectoderm-derived nasal placodes may have different properties in the vicinity of the nasal dorsum and nasal tip that predispose them to the development of superficial and deep hemangiomas, respectively,” Ms. Kryatova and her associates reported (Ped Dermatol. 2016;33[6]:652-8).
Segmental- and indeterminate-type lesions were more likely than focal-type lesions to develop ulceration, compression, or functional obstruction, and mixed-depth hemangiomas were more likely than deep or superficial hemangiomas to ulcerate. Overall, the lesions had involuted by kindergarten age in 70% of the study participants but persisted in 30%, and most of the children with involution showed residual skin changes such as telangiectasia (14 children), fibrofatty tissue (11 children), and scarring (9 children).
These findings show that a multicenter study to expand on these conclusions and to determine the best treatment algorithm for nasal infantile hemangiomas is warranted, the investigators added.
Key clinical point: Infantile hemangiomas of the nose develop more complications than those at all other sites combined.
Major finding: Thirty-five children (39%) developed one or more complications at some time during follow-up, including airway compromise, compression, or functional impairment; lesion ulceration; visual obstruction or ocular compression; and infection.
Data source: A retrospective chart review involving 89 patients with nasal infantile hemangiomas who were followed up at 5 years of age.
Disclosures: No sponsor was cited for this study, and the authors didn’t report their financial disclosures.
Vitiligo Patients Experience Barriers in Accessing Care
Vitiligo is a disorder typified by loss of pigmentation. Worldwide estimates of disease demonstrate 0.4% to 2% prevalence.1 Vitiligo generally is felt to be an autoimmune disorder with a complex multifactorial inheritance.2 Therapeutic options for vitiligo are largely off label and include topical corticosteroids, topical calcineurin inhibitors, narrowband UVB (NB-UVB) light phototherapy, and excimer (308 nm) laser therapy.3,4 Therapies for vitiligo are time consuming, as most topical therapies require twice-daily application. Additionally, many patients require 2 or more topical therapies due to involvement of both the head and neck as well as other body sites.3,4 Generalized disease often is treated with NB-UVB therapy 3 times weekly in-office visits, while excimer laser therapy is used for limited disease resistant to topical agents.3,4
Many barriers to good outcomes and care exist for patients with vitiligo.5 Patients may experience reduced quality of life and/or sexual dysfunction because of vitiligo lesions. The purpose of this pilot study was to identify barriers to access of care in vitiligo patients.
Methods
A survey was designed and then reviewed for unclear wording by members of the local vitiligo support group at Mount Sinai St. Luke’s-Roosevelt Hospital and Beth Israel Medical Centers (New York, New York). Linguistic revision and clarifications were added to the survey to correct identified communication problems. The survey was then posted using an Internet-based survey software. Links to the survey were sent via email to 107 individuals in a LISTSERV comprising Vitiligo Support International members who participated in a New York City support group (led by C.G. and N.B.S.). Only 1 email was used per household and only individuals 18 years or older could participate. These individuals were asked to complete a deidentified, 82-question, institutional review board–reviewed and exempted survey addressing issues affecting delivery and receipt of medical care for vitiligo.
Data were analyzed using the χ2 test, analysis of variance, or Student t test depending on the type of variable (categorical vs continuous). Fisher exact or Wilcoxon-Mann-Whitney tests were used when distributional assumptions were not met. A type I error rate (α=.05) was used to determine statistical significance. All analyses were performed using SAS 9.3 software.
Results
Respondents
The survey was completed by 81% (n=87) of individuals. The mean (SD) age of the treated patients about whom the respondents communicated was 33 (16) years and 71% (n=62) were women. The majority of respondents (64 [74%]) reported their race as white, followed by African American/black (12 [14%]), Hispanic (7 [8%]), and Asian (4 [5%]). Twenty-nine percent (22/76) of respondents reported a family income of less than $50,000 per year, 34% (26/76) reported an income of $50,000 to $100,000, and 37% (28/76) reported an income greater than $100,000, while 11 respondents did not report income.
Number of Physicians Seen
Respondents had reportedly seen an average (SD) number of 2 (1) physicians in the past/present before being offered any therapy for vitiligo and only 37% (32/87) of respondents reported being offered therapy by the first physician they saw. The number of physicians seen did not have a statistical relationship with years with vitiligo (ie, disease duration), sex, race, age of onset, income level, or number of sites affected.
Number of Sites Affected
The survey identified the following 23 sites affected by vitiligo: scalp, forehead, eyelids, lips, nose, cheeks, chin, neck, chest, stomach, back, upper arms, forearms, hands, wrists, fingers, genitalia, buttocks, thighs, calves/shins, ankles, feet, and toes. The average (SD) number of sites affected was 12 (6). The number of sites affected was correlated to the recommendation for phototherapy, while the recommendation for excimer laser therapy was inversely associated with the number of sites affected. The median number of sites affected for those who were not prescribed phototherapy was 10 (interquartile range [IQR]=9; P=.05); the median number of sites affected for those who were prescribed phototherapy was 15 (IQR=11). The association between the number of sites affected and whether the patient proceeded with phototherapy was not statistically significant. The need for phototherapy was not related to years with vitiligo (ie, disease duration), sex, or race.
Excimer laser therapy was prescribed more often to patients with fewer sites affected (median of 9 [IQR=3] vs median of 15 [IQR=9]; P=.04). Respondents who had fewer sites affected were on average more likely to proceed with excimer laser therapy (median of 8 [IQR=4] vs median of 11 [IQR=5]). The association between the number of sites affected and whether the patient proceeded with excimer laser therapy was not statistically significant.
Access to Topical Medications
Forty-one percent (36/87) of respondents reported difficulty accessing 1 or more topical therapies. Of 52 respondents who were prescribed a topical corticosteroid, 12 (23%) reported difficulty accessing therapy. Of 67 respondents who were prescribed a topical calcineurin inhibitor, 27 (40%) reported difficulty accessing medication (tacrolimus, n=17; pimecrolimus, n=10). Calcipotriene prescription coverage was not specifically addressed in this survey, as it usually is a second-line or adjunctive medication. Difficulty getting topical tacrolimus but not topical corticosteroids was associated with female sex (P=.03) but was not associated with race, income level, or level of education. Difficulty obtaining medication was not related to race, sex, level of education, or income level.
Consequences of Phototherapy
Twenty-three of 34 respondents (68%) who were told they required phototherapy actually received phototherapy and reported paying $38 weekly (IQR=$75). The majority of patients who proceeded with phototherapy lived (17/23 [74%]) or worked (16/23 [70%]) within 20 minutes of the therapy center. Self-reported response to phototherapy was good to very good in 65% (15/23) of respondents and no response in 30% (7/23); only 1 respondent reported worsening vitiligo. Sixty percent (15/25) of respondents said they were not satisfied with phototherapy. Respondents who were satisfied with the outcome of phototherapy had on average fewer sites affected by vitiligo (mean [SD], 10 [8]; P=.05). The association with other demographic and economic parameters (eg, sex, race, level of education, income level) was not statistically significant. Proceeding with phototherapy was not related to race, sex, level of education, or income level.
When questioned how many aspects of daily life (eg, work, home, school) were affected by phototherapy, 40% (35/87) of respondents reported that more than one life parameter was disturbed. Thirty-five percent (8/23) of respondents who received phototherapy reported that it affected their daily life “quite a bit” or “severely.” More respondents were likely to report that the therapy interfered with their life “somewhat,” “quite a bit,” or “severely” (76% [19/25]; 95% confidence interval, 55%-92%; P=.01) rather than “not at all” or “a little.”
Excimer Laser
Nine of 17 respondents (53%) who were recommended to undergo excimer laser therapy actually received therapy and reported paying $100 weekly (IQR=$60).
There was a trend toward significance of excimer usage being associated with lower age quartile (0–20 years)(P=.0553) and income more than $100,000 (P=.0788), neither of which reached statistical significance.
Insurance Coverage
Respondents were offered 7 answer options regarding the reason for noncoverage of topical calcineurin inhibitors. They were allowed to pick more than one reason where appropriate. For individuals who were prescribed topical tacrolimus but did not receive drug (n=17), the following reasons were cited: “no insurance coverage for the medication” (59% [10/17]), “your deductible was too high” (24% [4/17]), “prior authorization failed to produce coverage of the medication” (24% [4/17]), “your copay was prohibitively expensive” (24% [4/17]), “you were uncomfortable with the medication’s side effects” (18% [3/17]), “the tube was too small to cover your skin affected areas” (12% [2/17]), and “other” (29% [5/17]). Three patients selected 3 or more reasons, 8 patients selected 2 reasons, and 5 patients selected one reason.
Comment
It has been reported that patients with vitiligo may have difficulty related to treatment compliance for a variety of reasons.5 We identified notable barriers that arise for some, if not all, patients with vitiligo in the United States at some point in their care, including interference with other aspects of daily life, lack of coverage by current health insurance provider, and high out-of-pocket expenses, in addition to the negative effects of vitiligo on quality of life that have already been reported.6,7 These barriers are not a function of race/ethnicity, income level, or age of onset, but they may be impacted, as in the case of tacrolimus, by female sex. It is clear that, based on this study’s numbers, many patients will be unable to receive and/or comply with recommended treatment plans.
A limitation of this analysis is the study population, a select group of patients who had not been prescribed all the therapies in question. The sample size may not be large enough to demonstrate differences between level of education, race, or income level; however, even with a sample size of 87 respondents, the barriers to access of care are prominent. Larger population-based surveys would potentially tease out patterns of barriers not apparent with a smaller sample. No data were generated specific to calcipotriene, and this medication was not specified as a write-in agent on open question by any respondents; therefore, access to topical calcipotriene cannot be projected from this study. Phototherapy was queried as a nonspecific term and the breakdown of NB-UVB versus psoralen plus UVA was not available for this survey. Data suggesting a burden of socioeconomic barriers have been reported for atopic dermatitis8 and psoriasis,9 which corroborate the need for greater research in the field of access to care in dermatology.
Despite some advancement in the care of vitiligo, patients often are unable to access preferred or recommended treatment modalities. Standard recommendations for care are initial usage of calcineurin inhibitors for facial involvement and topical high-potency corticosteroids for involvement of the body.3,4 Based on this survey, it would seem that many patients are not able to receive the standard of care. Similarly, NB-UVB phototherapy and excimer laser therapy are recommended for widespread vitiligo and lesions unresponsive to topical care. It would seem that almost half of our respondents did not have access to one or more of the recommended therapies. Barriers to care may have substantial clinical and psychological outcomes, which were not evaluated in this study but merit future research.
- Krüger C, Schallreuter KU. A review of the worldwide prevalence of vitiligo in children/adolescents and adults. Int J Dermatol. 2012;51:1206-1212.
- Jin Y, Birlea SA, Fain PR, et al. Genome-wide association analyses identify 13 new susceptibility loci for generalized vitiligo. Nat Genet. 2012;44:676-680.
- Silverberg NB. Pediatric vitiligo. Pediatr Clin North Am. 2014;61:347-366.
- Taieb A, Alomar A, Böhm M, et al, Vitiligo European Task Force (VETF); European Academy of Dermatology and Venereology (EADV); Union Europénne des Médecins Spécialistes (UEMS). Guidelines for the management of vitiligo: the European Dermatology Forum consensus. Br J Dermatol. 2013;168:5-19.
- Abraham S, Raghavan P. Myths and facts about vitiligo: an epidemiological study. Indian J Pharm Sci. 2015;77:8-13.
- Silverberg JI, Silverberg NB. Quality of life impairment in children and adolescents with vitiligo. Pediatr Dermatol. 2014;31:309-318.
- Silverberg JI, Silverberg NB. Association between vitiligo extent and distribution and quality-of-life impairment. JAMA Dermatol. 2013;149:159-164.
- Silverberg JI, Hanifin JM. Adult eczema prevalence and associations with asthma and other health and demographic factors: a US population-based study. J Allergy Clin Immunol. 2013;132:1132-1138.
- Hamilton MP, Ntais D, Griffiths CE, et al. Psoriasis treatment and management—a systematic review of full economic evaluations. Br J Dermatol. 2015;172:574-583.
Vitiligo is a disorder typified by loss of pigmentation. Worldwide estimates of disease demonstrate 0.4% to 2% prevalence.1 Vitiligo generally is felt to be an autoimmune disorder with a complex multifactorial inheritance.2 Therapeutic options for vitiligo are largely off label and include topical corticosteroids, topical calcineurin inhibitors, narrowband UVB (NB-UVB) light phototherapy, and excimer (308 nm) laser therapy.3,4 Therapies for vitiligo are time consuming, as most topical therapies require twice-daily application. Additionally, many patients require 2 or more topical therapies due to involvement of both the head and neck as well as other body sites.3,4 Generalized disease often is treated with NB-UVB therapy 3 times weekly in-office visits, while excimer laser therapy is used for limited disease resistant to topical agents.3,4
Many barriers to good outcomes and care exist for patients with vitiligo.5 Patients may experience reduced quality of life and/or sexual dysfunction because of vitiligo lesions. The purpose of this pilot study was to identify barriers to access of care in vitiligo patients.
Methods
A survey was designed and then reviewed for unclear wording by members of the local vitiligo support group at Mount Sinai St. Luke’s-Roosevelt Hospital and Beth Israel Medical Centers (New York, New York). Linguistic revision and clarifications were added to the survey to correct identified communication problems. The survey was then posted using an Internet-based survey software. Links to the survey were sent via email to 107 individuals in a LISTSERV comprising Vitiligo Support International members who participated in a New York City support group (led by C.G. and N.B.S.). Only 1 email was used per household and only individuals 18 years or older could participate. These individuals were asked to complete a deidentified, 82-question, institutional review board–reviewed and exempted survey addressing issues affecting delivery and receipt of medical care for vitiligo.
Data were analyzed using the χ2 test, analysis of variance, or Student t test depending on the type of variable (categorical vs continuous). Fisher exact or Wilcoxon-Mann-Whitney tests were used when distributional assumptions were not met. A type I error rate (α=.05) was used to determine statistical significance. All analyses were performed using SAS 9.3 software.
Results
Respondents
The survey was completed by 81% (n=87) of individuals. The mean (SD) age of the treated patients about whom the respondents communicated was 33 (16) years and 71% (n=62) were women. The majority of respondents (64 [74%]) reported their race as white, followed by African American/black (12 [14%]), Hispanic (7 [8%]), and Asian (4 [5%]). Twenty-nine percent (22/76) of respondents reported a family income of less than $50,000 per year, 34% (26/76) reported an income of $50,000 to $100,000, and 37% (28/76) reported an income greater than $100,000, while 11 respondents did not report income.
Number of Physicians Seen
Respondents had reportedly seen an average (SD) number of 2 (1) physicians in the past/present before being offered any therapy for vitiligo and only 37% (32/87) of respondents reported being offered therapy by the first physician they saw. The number of physicians seen did not have a statistical relationship with years with vitiligo (ie, disease duration), sex, race, age of onset, income level, or number of sites affected.
Number of Sites Affected
The survey identified the following 23 sites affected by vitiligo: scalp, forehead, eyelids, lips, nose, cheeks, chin, neck, chest, stomach, back, upper arms, forearms, hands, wrists, fingers, genitalia, buttocks, thighs, calves/shins, ankles, feet, and toes. The average (SD) number of sites affected was 12 (6). The number of sites affected was correlated to the recommendation for phototherapy, while the recommendation for excimer laser therapy was inversely associated with the number of sites affected. The median number of sites affected for those who were not prescribed phototherapy was 10 (interquartile range [IQR]=9; P=.05); the median number of sites affected for those who were prescribed phototherapy was 15 (IQR=11). The association between the number of sites affected and whether the patient proceeded with phototherapy was not statistically significant. The need for phototherapy was not related to years with vitiligo (ie, disease duration), sex, or race.
Excimer laser therapy was prescribed more often to patients with fewer sites affected (median of 9 [IQR=3] vs median of 15 [IQR=9]; P=.04). Respondents who had fewer sites affected were on average more likely to proceed with excimer laser therapy (median of 8 [IQR=4] vs median of 11 [IQR=5]). The association between the number of sites affected and whether the patient proceeded with excimer laser therapy was not statistically significant.
Access to Topical Medications
Forty-one percent (36/87) of respondents reported difficulty accessing 1 or more topical therapies. Of 52 respondents who were prescribed a topical corticosteroid, 12 (23%) reported difficulty accessing therapy. Of 67 respondents who were prescribed a topical calcineurin inhibitor, 27 (40%) reported difficulty accessing medication (tacrolimus, n=17; pimecrolimus, n=10). Calcipotriene prescription coverage was not specifically addressed in this survey, as it usually is a second-line or adjunctive medication. Difficulty getting topical tacrolimus but not topical corticosteroids was associated with female sex (P=.03) but was not associated with race, income level, or level of education. Difficulty obtaining medication was not related to race, sex, level of education, or income level.
Consequences of Phototherapy
Twenty-three of 34 respondents (68%) who were told they required phototherapy actually received phototherapy and reported paying $38 weekly (IQR=$75). The majority of patients who proceeded with phototherapy lived (17/23 [74%]) or worked (16/23 [70%]) within 20 minutes of the therapy center. Self-reported response to phototherapy was good to very good in 65% (15/23) of respondents and no response in 30% (7/23); only 1 respondent reported worsening vitiligo. Sixty percent (15/25) of respondents said they were not satisfied with phototherapy. Respondents who were satisfied with the outcome of phototherapy had on average fewer sites affected by vitiligo (mean [SD], 10 [8]; P=.05). The association with other demographic and economic parameters (eg, sex, race, level of education, income level) was not statistically significant. Proceeding with phototherapy was not related to race, sex, level of education, or income level.
When questioned how many aspects of daily life (eg, work, home, school) were affected by phototherapy, 40% (35/87) of respondents reported that more than one life parameter was disturbed. Thirty-five percent (8/23) of respondents who received phototherapy reported that it affected their daily life “quite a bit” or “severely.” More respondents were likely to report that the therapy interfered with their life “somewhat,” “quite a bit,” or “severely” (76% [19/25]; 95% confidence interval, 55%-92%; P=.01) rather than “not at all” or “a little.”
Excimer Laser
Nine of 17 respondents (53%) who were recommended to undergo excimer laser therapy actually received therapy and reported paying $100 weekly (IQR=$60).
There was a trend toward significance of excimer usage being associated with lower age quartile (0–20 years)(P=.0553) and income more than $100,000 (P=.0788), neither of which reached statistical significance.
Insurance Coverage
Respondents were offered 7 answer options regarding the reason for noncoverage of topical calcineurin inhibitors. They were allowed to pick more than one reason where appropriate. For individuals who were prescribed topical tacrolimus but did not receive drug (n=17), the following reasons were cited: “no insurance coverage for the medication” (59% [10/17]), “your deductible was too high” (24% [4/17]), “prior authorization failed to produce coverage of the medication” (24% [4/17]), “your copay was prohibitively expensive” (24% [4/17]), “you were uncomfortable with the medication’s side effects” (18% [3/17]), “the tube was too small to cover your skin affected areas” (12% [2/17]), and “other” (29% [5/17]). Three patients selected 3 or more reasons, 8 patients selected 2 reasons, and 5 patients selected one reason.
Comment
It has been reported that patients with vitiligo may have difficulty related to treatment compliance for a variety of reasons.5 We identified notable barriers that arise for some, if not all, patients with vitiligo in the United States at some point in their care, including interference with other aspects of daily life, lack of coverage by current health insurance provider, and high out-of-pocket expenses, in addition to the negative effects of vitiligo on quality of life that have already been reported.6,7 These barriers are not a function of race/ethnicity, income level, or age of onset, but they may be impacted, as in the case of tacrolimus, by female sex. It is clear that, based on this study’s numbers, many patients will be unable to receive and/or comply with recommended treatment plans.
A limitation of this analysis is the study population, a select group of patients who had not been prescribed all the therapies in question. The sample size may not be large enough to demonstrate differences between level of education, race, or income level; however, even with a sample size of 87 respondents, the barriers to access of care are prominent. Larger population-based surveys would potentially tease out patterns of barriers not apparent with a smaller sample. No data were generated specific to calcipotriene, and this medication was not specified as a write-in agent on open question by any respondents; therefore, access to topical calcipotriene cannot be projected from this study. Phototherapy was queried as a nonspecific term and the breakdown of NB-UVB versus psoralen plus UVA was not available for this survey. Data suggesting a burden of socioeconomic barriers have been reported for atopic dermatitis8 and psoriasis,9 which corroborate the need for greater research in the field of access to care in dermatology.
Despite some advancement in the care of vitiligo, patients often are unable to access preferred or recommended treatment modalities. Standard recommendations for care are initial usage of calcineurin inhibitors for facial involvement and topical high-potency corticosteroids for involvement of the body.3,4 Based on this survey, it would seem that many patients are not able to receive the standard of care. Similarly, NB-UVB phototherapy and excimer laser therapy are recommended for widespread vitiligo and lesions unresponsive to topical care. It would seem that almost half of our respondents did not have access to one or more of the recommended therapies. Barriers to care may have substantial clinical and psychological outcomes, which were not evaluated in this study but merit future research.
Vitiligo is a disorder typified by loss of pigmentation. Worldwide estimates of disease demonstrate 0.4% to 2% prevalence.1 Vitiligo generally is felt to be an autoimmune disorder with a complex multifactorial inheritance.2 Therapeutic options for vitiligo are largely off label and include topical corticosteroids, topical calcineurin inhibitors, narrowband UVB (NB-UVB) light phototherapy, and excimer (308 nm) laser therapy.3,4 Therapies for vitiligo are time consuming, as most topical therapies require twice-daily application. Additionally, many patients require 2 or more topical therapies due to involvement of both the head and neck as well as other body sites.3,4 Generalized disease often is treated with NB-UVB therapy 3 times weekly in-office visits, while excimer laser therapy is used for limited disease resistant to topical agents.3,4
Many barriers to good outcomes and care exist for patients with vitiligo.5 Patients may experience reduced quality of life and/or sexual dysfunction because of vitiligo lesions. The purpose of this pilot study was to identify barriers to access of care in vitiligo patients.
Methods
A survey was designed and then reviewed for unclear wording by members of the local vitiligo support group at Mount Sinai St. Luke’s-Roosevelt Hospital and Beth Israel Medical Centers (New York, New York). Linguistic revision and clarifications were added to the survey to correct identified communication problems. The survey was then posted using an Internet-based survey software. Links to the survey were sent via email to 107 individuals in a LISTSERV comprising Vitiligo Support International members who participated in a New York City support group (led by C.G. and N.B.S.). Only 1 email was used per household and only individuals 18 years or older could participate. These individuals were asked to complete a deidentified, 82-question, institutional review board–reviewed and exempted survey addressing issues affecting delivery and receipt of medical care for vitiligo.
Data were analyzed using the χ2 test, analysis of variance, or Student t test depending on the type of variable (categorical vs continuous). Fisher exact or Wilcoxon-Mann-Whitney tests were used when distributional assumptions were not met. A type I error rate (α=.05) was used to determine statistical significance. All analyses were performed using SAS 9.3 software.
Results
Respondents
The survey was completed by 81% (n=87) of individuals. The mean (SD) age of the treated patients about whom the respondents communicated was 33 (16) years and 71% (n=62) were women. The majority of respondents (64 [74%]) reported their race as white, followed by African American/black (12 [14%]), Hispanic (7 [8%]), and Asian (4 [5%]). Twenty-nine percent (22/76) of respondents reported a family income of less than $50,000 per year, 34% (26/76) reported an income of $50,000 to $100,000, and 37% (28/76) reported an income greater than $100,000, while 11 respondents did not report income.
Number of Physicians Seen
Respondents had reportedly seen an average (SD) number of 2 (1) physicians in the past/present before being offered any therapy for vitiligo and only 37% (32/87) of respondents reported being offered therapy by the first physician they saw. The number of physicians seen did not have a statistical relationship with years with vitiligo (ie, disease duration), sex, race, age of onset, income level, or number of sites affected.
Number of Sites Affected
The survey identified the following 23 sites affected by vitiligo: scalp, forehead, eyelids, lips, nose, cheeks, chin, neck, chest, stomach, back, upper arms, forearms, hands, wrists, fingers, genitalia, buttocks, thighs, calves/shins, ankles, feet, and toes. The average (SD) number of sites affected was 12 (6). The number of sites affected was correlated to the recommendation for phototherapy, while the recommendation for excimer laser therapy was inversely associated with the number of sites affected. The median number of sites affected for those who were not prescribed phototherapy was 10 (interquartile range [IQR]=9; P=.05); the median number of sites affected for those who were prescribed phototherapy was 15 (IQR=11). The association between the number of sites affected and whether the patient proceeded with phototherapy was not statistically significant. The need for phototherapy was not related to years with vitiligo (ie, disease duration), sex, or race.
Excimer laser therapy was prescribed more often to patients with fewer sites affected (median of 9 [IQR=3] vs median of 15 [IQR=9]; P=.04). Respondents who had fewer sites affected were on average more likely to proceed with excimer laser therapy (median of 8 [IQR=4] vs median of 11 [IQR=5]). The association between the number of sites affected and whether the patient proceeded with excimer laser therapy was not statistically significant.
Access to Topical Medications
Forty-one percent (36/87) of respondents reported difficulty accessing 1 or more topical therapies. Of 52 respondents who were prescribed a topical corticosteroid, 12 (23%) reported difficulty accessing therapy. Of 67 respondents who were prescribed a topical calcineurin inhibitor, 27 (40%) reported difficulty accessing medication (tacrolimus, n=17; pimecrolimus, n=10). Calcipotriene prescription coverage was not specifically addressed in this survey, as it usually is a second-line or adjunctive medication. Difficulty getting topical tacrolimus but not topical corticosteroids was associated with female sex (P=.03) but was not associated with race, income level, or level of education. Difficulty obtaining medication was not related to race, sex, level of education, or income level.
Consequences of Phototherapy
Twenty-three of 34 respondents (68%) who were told they required phototherapy actually received phototherapy and reported paying $38 weekly (IQR=$75). The majority of patients who proceeded with phototherapy lived (17/23 [74%]) or worked (16/23 [70%]) within 20 minutes of the therapy center. Self-reported response to phototherapy was good to very good in 65% (15/23) of respondents and no response in 30% (7/23); only 1 respondent reported worsening vitiligo. Sixty percent (15/25) of respondents said they were not satisfied with phototherapy. Respondents who were satisfied with the outcome of phototherapy had on average fewer sites affected by vitiligo (mean [SD], 10 [8]; P=.05). The association with other demographic and economic parameters (eg, sex, race, level of education, income level) was not statistically significant. Proceeding with phototherapy was not related to race, sex, level of education, or income level.
When questioned how many aspects of daily life (eg, work, home, school) were affected by phototherapy, 40% (35/87) of respondents reported that more than one life parameter was disturbed. Thirty-five percent (8/23) of respondents who received phototherapy reported that it affected their daily life “quite a bit” or “severely.” More respondents were likely to report that the therapy interfered with their life “somewhat,” “quite a bit,” or “severely” (76% [19/25]; 95% confidence interval, 55%-92%; P=.01) rather than “not at all” or “a little.”
Excimer Laser
Nine of 17 respondents (53%) who were recommended to undergo excimer laser therapy actually received therapy and reported paying $100 weekly (IQR=$60).
There was a trend toward significance of excimer usage being associated with lower age quartile (0–20 years)(P=.0553) and income more than $100,000 (P=.0788), neither of which reached statistical significance.
Insurance Coverage
Respondents were offered 7 answer options regarding the reason for noncoverage of topical calcineurin inhibitors. They were allowed to pick more than one reason where appropriate. For individuals who were prescribed topical tacrolimus but did not receive drug (n=17), the following reasons were cited: “no insurance coverage for the medication” (59% [10/17]), “your deductible was too high” (24% [4/17]), “prior authorization failed to produce coverage of the medication” (24% [4/17]), “your copay was prohibitively expensive” (24% [4/17]), “you were uncomfortable with the medication’s side effects” (18% [3/17]), “the tube was too small to cover your skin affected areas” (12% [2/17]), and “other” (29% [5/17]). Three patients selected 3 or more reasons, 8 patients selected 2 reasons, and 5 patients selected one reason.
Comment
It has been reported that patients with vitiligo may have difficulty related to treatment compliance for a variety of reasons.5 We identified notable barriers that arise for some, if not all, patients with vitiligo in the United States at some point in their care, including interference with other aspects of daily life, lack of coverage by current health insurance provider, and high out-of-pocket expenses, in addition to the negative effects of vitiligo on quality of life that have already been reported.6,7 These barriers are not a function of race/ethnicity, income level, or age of onset, but they may be impacted, as in the case of tacrolimus, by female sex. It is clear that, based on this study’s numbers, many patients will be unable to receive and/or comply with recommended treatment plans.
A limitation of this analysis is the study population, a select group of patients who had not been prescribed all the therapies in question. The sample size may not be large enough to demonstrate differences between level of education, race, or income level; however, even with a sample size of 87 respondents, the barriers to access of care are prominent. Larger population-based surveys would potentially tease out patterns of barriers not apparent with a smaller sample. No data were generated specific to calcipotriene, and this medication was not specified as a write-in agent on open question by any respondents; therefore, access to topical calcipotriene cannot be projected from this study. Phototherapy was queried as a nonspecific term and the breakdown of NB-UVB versus psoralen plus UVA was not available for this survey. Data suggesting a burden of socioeconomic barriers have been reported for atopic dermatitis8 and psoriasis,9 which corroborate the need for greater research in the field of access to care in dermatology.
Despite some advancement in the care of vitiligo, patients often are unable to access preferred or recommended treatment modalities. Standard recommendations for care are initial usage of calcineurin inhibitors for facial involvement and topical high-potency corticosteroids for involvement of the body.3,4 Based on this survey, it would seem that many patients are not able to receive the standard of care. Similarly, NB-UVB phototherapy and excimer laser therapy are recommended for widespread vitiligo and lesions unresponsive to topical care. It would seem that almost half of our respondents did not have access to one or more of the recommended therapies. Barriers to care may have substantial clinical and psychological outcomes, which were not evaluated in this study but merit future research.
- Krüger C, Schallreuter KU. A review of the worldwide prevalence of vitiligo in children/adolescents and adults. Int J Dermatol. 2012;51:1206-1212.
- Jin Y, Birlea SA, Fain PR, et al. Genome-wide association analyses identify 13 new susceptibility loci for generalized vitiligo. Nat Genet. 2012;44:676-680.
- Silverberg NB. Pediatric vitiligo. Pediatr Clin North Am. 2014;61:347-366.
- Taieb A, Alomar A, Böhm M, et al, Vitiligo European Task Force (VETF); European Academy of Dermatology and Venereology (EADV); Union Europénne des Médecins Spécialistes (UEMS). Guidelines for the management of vitiligo: the European Dermatology Forum consensus. Br J Dermatol. 2013;168:5-19.
- Abraham S, Raghavan P. Myths and facts about vitiligo: an epidemiological study. Indian J Pharm Sci. 2015;77:8-13.
- Silverberg JI, Silverberg NB. Quality of life impairment in children and adolescents with vitiligo. Pediatr Dermatol. 2014;31:309-318.
- Silverberg JI, Silverberg NB. Association between vitiligo extent and distribution and quality-of-life impairment. JAMA Dermatol. 2013;149:159-164.
- Silverberg JI, Hanifin JM. Adult eczema prevalence and associations with asthma and other health and demographic factors: a US population-based study. J Allergy Clin Immunol. 2013;132:1132-1138.
- Hamilton MP, Ntais D, Griffiths CE, et al. Psoriasis treatment and management—a systematic review of full economic evaluations. Br J Dermatol. 2015;172:574-583.
- Krüger C, Schallreuter KU. A review of the worldwide prevalence of vitiligo in children/adolescents and adults. Int J Dermatol. 2012;51:1206-1212.
- Jin Y, Birlea SA, Fain PR, et al. Genome-wide association analyses identify 13 new susceptibility loci for generalized vitiligo. Nat Genet. 2012;44:676-680.
- Silverberg NB. Pediatric vitiligo. Pediatr Clin North Am. 2014;61:347-366.
- Taieb A, Alomar A, Böhm M, et al, Vitiligo European Task Force (VETF); European Academy of Dermatology and Venereology (EADV); Union Europénne des Médecins Spécialistes (UEMS). Guidelines for the management of vitiligo: the European Dermatology Forum consensus. Br J Dermatol. 2013;168:5-19.
- Abraham S, Raghavan P. Myths and facts about vitiligo: an epidemiological study. Indian J Pharm Sci. 2015;77:8-13.
- Silverberg JI, Silverberg NB. Quality of life impairment in children and adolescents with vitiligo. Pediatr Dermatol. 2014;31:309-318.
- Silverberg JI, Silverberg NB. Association between vitiligo extent and distribution and quality-of-life impairment. JAMA Dermatol. 2013;149:159-164.
- Silverberg JI, Hanifin JM. Adult eczema prevalence and associations with asthma and other health and demographic factors: a US population-based study. J Allergy Clin Immunol. 2013;132:1132-1138.
- Hamilton MP, Ntais D, Griffiths CE, et al. Psoriasis treatment and management—a systematic review of full economic evaluations. Br J Dermatol. 2015;172:574-583.
Practice Points
- Patients with vitiligo may experience difficulty receiving the care prescribed to them.
- It is best to identify barriers such as work schedule or distance before recommending a treatment plan.
Accuracy and Sources of Images From Direct Google Image Searches for Common Dermatology Terms
To the Editor:
Prior studies have assessed the quality of text-based dermatology information on the Internet using traditional search engine queries.1 However, little is understood about the sources, accuracy, and quality of online dermatology images derived from direct image searches. Previous work has shown that direct search engine image queries were largely accurate for 3 pediatric dermatology diagnosis searches: atopic dermatitis, lichen striatus, and subcutaneous fat necrosis.2 We assessed images obtained for common dermatologic conditions from a Google image search (GIS) compared to a traditional text-based Google web search (GWS).
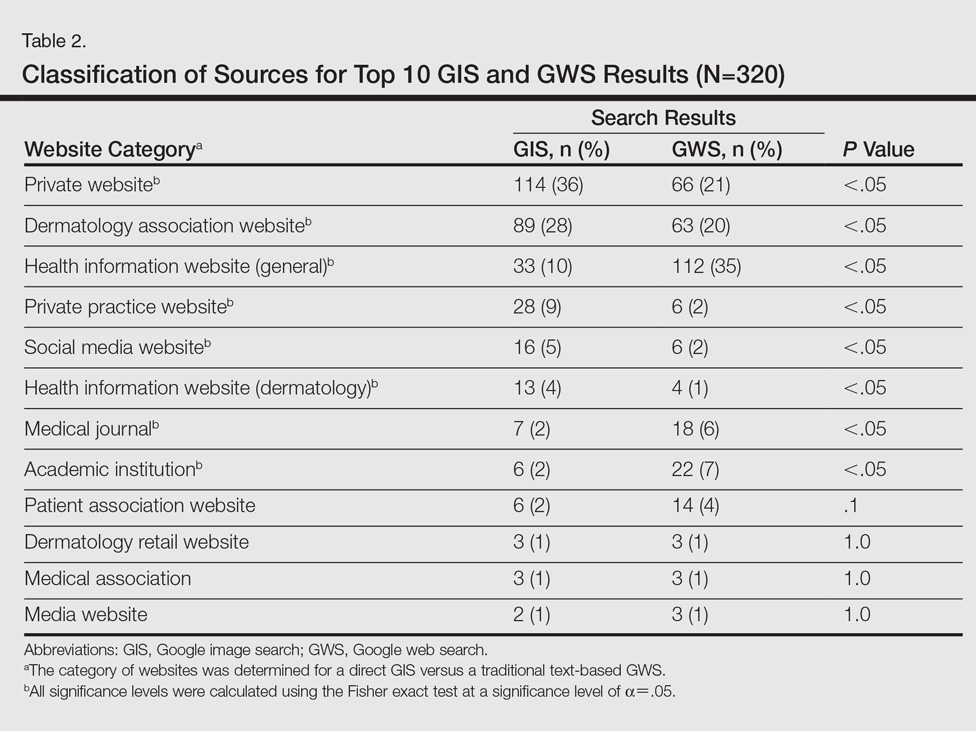
Image results for 32 unique dermatologic search terms were analyzed (Table 1). These search terms were selected using the results of a prior study that identified the most common dermatologic diagnoses that led users to the 2 most popular dermatology-specific websites worldwide: the American Academy of Dermatology (www.aad.org) and DermNet New Zealand (www.dermnetnz.org).3 The Alexa directory (www.alexa.com), a large publicly available Internet analytics resource, was used to determine the most common dermatology search terms that led a user to either www.dermnetnz.org or www.aad.org. In addition, searches for the 3 most common types of skin cancer—melanoma, squamous cell carcinoma, and basal cell carcinoma—were included. Each term was entered into a GIS and a GWS. The first 10 results, which represent 92% of the websites ultimately visited by users,4 were analyzed. The source, diagnostic accuracy, and Fitzpatrick skin type of the images was determined. Website sources were organized into 11 categories. All data collection occurred within a 1-week period in August 2015.
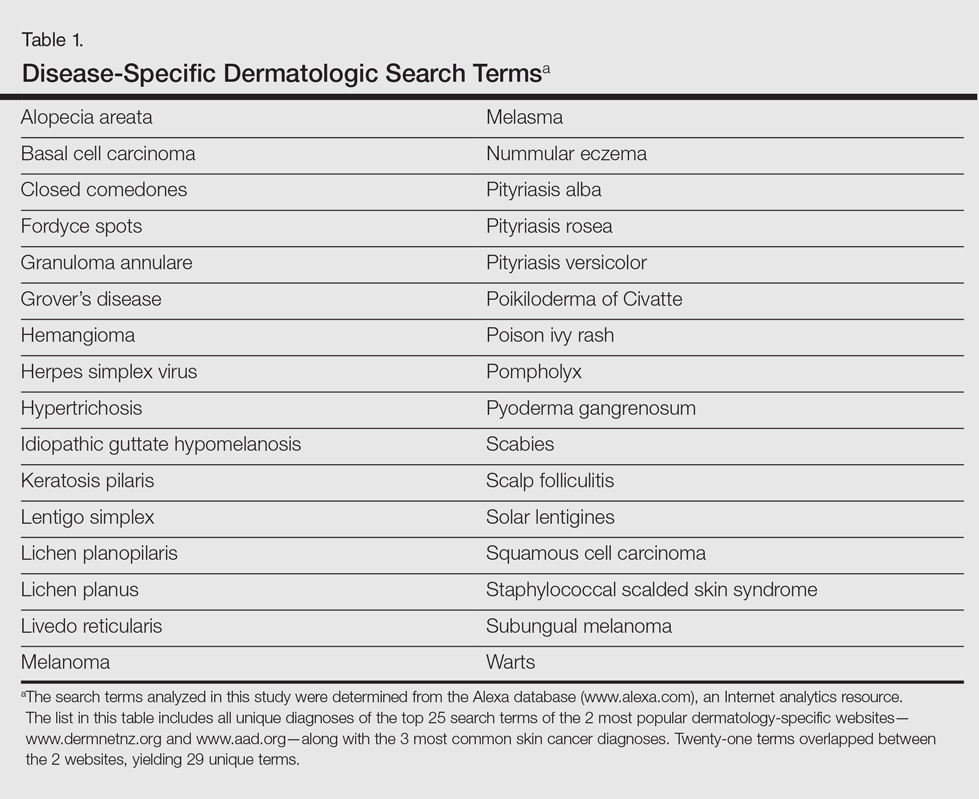
A total of 320 images were analyzed. In the GIS, private websites (36%), dermatology association websites (28%), and general health information websites (10%) were the 3 most common sources. In the GWS, health information websites (35%), private websites (21%), and dermatology association websites (20%) accounted for the most common sources (Table 2). The majority of images were of Fitzpatrick skin types I and II (89%) and nearly all images were diagnostically accurate (98%). There was no statistically significant difference in accuracy of diagnosis between physician-associated websites (100% accuracy) versus nonphysician-associated sites (98% accuracy, P=.25).
Our results showed high diagnostic accuracy among the top GIS results for common dermatology search terms. Diagnostic accuracy did not vary between websites that were physician associated versus those that were not. Our results are comparable to the reported accuracy of online dermatologic health information.1 In GIS results, the majority of images were provided by private websites, whereas the top websites in GWS results were health information websites.
Only 1% of images were of Fitzpatrick skin types VI and VII. Presentation of skin diseases is remarkably different based on the patient’s skin type.5 The shortage of readily accessible images of skin of color is in line with the lack of familiarity physicians and trainees have with dermatologic conditions in ethnic skin.6
Based on the results from this analysis, providers and patients searching for dermatologic conditions via a direct GIS should be cognizant of several considerations. Although our results showed that GIS was accurate, the searcher should note that image-based searches are not accompanied by relevant text that can help confirm relevancy and accuracy. Image searches depend on textual tags added by the source website. Websites that represent dermatological associations and academic centers can add an additional layer of confidence for users. Patients and clinicians also should be aware that the consideration of a patient’s Fitzpatrick skin type is critical when assessing the relevancy of a GIS result. In conclusion, search results via GIS queries are accurate for the dermatological diagnoses tested but may be lacking in skin of color variations, suggesting a potential unmet need based on our growing ethnic skin population.
- Jensen JD, Dunnick CA, Arbuckle HA, et al. Dermatology information on the Internet: an appraisal by dermatologists and dermatology residents. J Am Acad Dermatol. 2010;63:1101-1103.
- Cutrone M, Grimalt R. Dermatological image search engines on the Internet: do they work? J Eur Acad Dermatol Venereol. 2007;21:175-177.
- Xu S, Nault A, Bhatia A. Search and engagement analysis of association websites representing dermatologists—implications and opportunities for web visibility and patient education: website rankings of dermatology associations. Pract Dermatol. In press.
- comScore releases July 2015 U.S. desktop search engine rankings [press release]. Reston, VA: comScore, Inc; August 14, 2015. http://www.comscore.com/Insights/Market-Rankings/comScore-Releases-July-2015-U.S.-Desktop-Search-Engine-Rankings. Accessed October 18, 2016.
- Kundu RV, Patterson S. Dermatologic conditions in skin of color: part I. special considerations for common skin disorders. Am Fam Physician. 2013;87:850-856.
- Nijhawan RI, Jacob SE, Woolery-Lloyd H. Skin of color education in dermatology residency programs: does residency training reflect the changing demographics of the United States? J Am Acad Dermatol. 2008;59:615-618.
To the Editor:
Prior studies have assessed the quality of text-based dermatology information on the Internet using traditional search engine queries.1 However, little is understood about the sources, accuracy, and quality of online dermatology images derived from direct image searches. Previous work has shown that direct search engine image queries were largely accurate for 3 pediatric dermatology diagnosis searches: atopic dermatitis, lichen striatus, and subcutaneous fat necrosis.2 We assessed images obtained for common dermatologic conditions from a Google image search (GIS) compared to a traditional text-based Google web search (GWS).
Image results for 32 unique dermatologic search terms were analyzed (Table 1). These search terms were selected using the results of a prior study that identified the most common dermatologic diagnoses that led users to the 2 most popular dermatology-specific websites worldwide: the American Academy of Dermatology (www.aad.org) and DermNet New Zealand (www.dermnetnz.org).3 The Alexa directory (www.alexa.com), a large publicly available Internet analytics resource, was used to determine the most common dermatology search terms that led a user to either www.dermnetnz.org or www.aad.org. In addition, searches for the 3 most common types of skin cancer—melanoma, squamous cell carcinoma, and basal cell carcinoma—were included. Each term was entered into a GIS and a GWS. The first 10 results, which represent 92% of the websites ultimately visited by users,4 were analyzed. The source, diagnostic accuracy, and Fitzpatrick skin type of the images was determined. Website sources were organized into 11 categories. All data collection occurred within a 1-week period in August 2015.
A total of 320 images were analyzed. In the GIS, private websites (36%), dermatology association websites (28%), and general health information websites (10%) were the 3 most common sources. In the GWS, health information websites (35%), private websites (21%), and dermatology association websites (20%) accounted for the most common sources (Table 2). The majority of images were of Fitzpatrick skin types I and II (89%) and nearly all images were diagnostically accurate (98%). There was no statistically significant difference in accuracy of diagnosis between physician-associated websites (100% accuracy) versus nonphysician-associated sites (98% accuracy, P=.25).
Our results showed high diagnostic accuracy among the top GIS results for common dermatology search terms. Diagnostic accuracy did not vary between websites that were physician associated versus those that were not. Our results are comparable to the reported accuracy of online dermatologic health information.1 In GIS results, the majority of images were provided by private websites, whereas the top websites in GWS results were health information websites.
Only 1% of images were of Fitzpatrick skin types VI and VII. Presentation of skin diseases is remarkably different based on the patient’s skin type.5 The shortage of readily accessible images of skin of color is in line with the lack of familiarity physicians and trainees have with dermatologic conditions in ethnic skin.6
Based on the results from this analysis, providers and patients searching for dermatologic conditions via a direct GIS should be cognizant of several considerations. Although our results showed that GIS was accurate, the searcher should note that image-based searches are not accompanied by relevant text that can help confirm relevancy and accuracy. Image searches depend on textual tags added by the source website. Websites that represent dermatological associations and academic centers can add an additional layer of confidence for users. Patients and clinicians also should be aware that the consideration of a patient’s Fitzpatrick skin type is critical when assessing the relevancy of a GIS result. In conclusion, search results via GIS queries are accurate for the dermatological diagnoses tested but may be lacking in skin of color variations, suggesting a potential unmet need based on our growing ethnic skin population.
To the Editor:
Prior studies have assessed the quality of text-based dermatology information on the Internet using traditional search engine queries.1 However, little is understood about the sources, accuracy, and quality of online dermatology images derived from direct image searches. Previous work has shown that direct search engine image queries were largely accurate for 3 pediatric dermatology diagnosis searches: atopic dermatitis, lichen striatus, and subcutaneous fat necrosis.2 We assessed images obtained for common dermatologic conditions from a Google image search (GIS) compared to a traditional text-based Google web search (GWS).
Image results for 32 unique dermatologic search terms were analyzed (Table 1). These search terms were selected using the results of a prior study that identified the most common dermatologic diagnoses that led users to the 2 most popular dermatology-specific websites worldwide: the American Academy of Dermatology (www.aad.org) and DermNet New Zealand (www.dermnetnz.org).3 The Alexa directory (www.alexa.com), a large publicly available Internet analytics resource, was used to determine the most common dermatology search terms that led a user to either www.dermnetnz.org or www.aad.org. In addition, searches for the 3 most common types of skin cancer—melanoma, squamous cell carcinoma, and basal cell carcinoma—were included. Each term was entered into a GIS and a GWS. The first 10 results, which represent 92% of the websites ultimately visited by users,4 were analyzed. The source, diagnostic accuracy, and Fitzpatrick skin type of the images was determined. Website sources were organized into 11 categories. All data collection occurred within a 1-week period in August 2015.
A total of 320 images were analyzed. In the GIS, private websites (36%), dermatology association websites (28%), and general health information websites (10%) were the 3 most common sources. In the GWS, health information websites (35%), private websites (21%), and dermatology association websites (20%) accounted for the most common sources (Table 2). The majority of images were of Fitzpatrick skin types I and II (89%) and nearly all images were diagnostically accurate (98%). There was no statistically significant difference in accuracy of diagnosis between physician-associated websites (100% accuracy) versus nonphysician-associated sites (98% accuracy, P=.25).
Our results showed high diagnostic accuracy among the top GIS results for common dermatology search terms. Diagnostic accuracy did not vary between websites that were physician associated versus those that were not. Our results are comparable to the reported accuracy of online dermatologic health information.1 In GIS results, the majority of images were provided by private websites, whereas the top websites in GWS results were health information websites.
Only 1% of images were of Fitzpatrick skin types VI and VII. Presentation of skin diseases is remarkably different based on the patient’s skin type.5 The shortage of readily accessible images of skin of color is in line with the lack of familiarity physicians and trainees have with dermatologic conditions in ethnic skin.6
Based on the results from this analysis, providers and patients searching for dermatologic conditions via a direct GIS should be cognizant of several considerations. Although our results showed that GIS was accurate, the searcher should note that image-based searches are not accompanied by relevant text that can help confirm relevancy and accuracy. Image searches depend on textual tags added by the source website. Websites that represent dermatological associations and academic centers can add an additional layer of confidence for users. Patients and clinicians also should be aware that the consideration of a patient’s Fitzpatrick skin type is critical when assessing the relevancy of a GIS result. In conclusion, search results via GIS queries are accurate for the dermatological diagnoses tested but may be lacking in skin of color variations, suggesting a potential unmet need based on our growing ethnic skin population.
- Jensen JD, Dunnick CA, Arbuckle HA, et al. Dermatology information on the Internet: an appraisal by dermatologists and dermatology residents. J Am Acad Dermatol. 2010;63:1101-1103.
- Cutrone M, Grimalt R. Dermatological image search engines on the Internet: do they work? J Eur Acad Dermatol Venereol. 2007;21:175-177.
- Xu S, Nault A, Bhatia A. Search and engagement analysis of association websites representing dermatologists—implications and opportunities for web visibility and patient education: website rankings of dermatology associations. Pract Dermatol. In press.
- comScore releases July 2015 U.S. desktop search engine rankings [press release]. Reston, VA: comScore, Inc; August 14, 2015. http://www.comscore.com/Insights/Market-Rankings/comScore-Releases-July-2015-U.S.-Desktop-Search-Engine-Rankings. Accessed October 18, 2016.
- Kundu RV, Patterson S. Dermatologic conditions in skin of color: part I. special considerations for common skin disorders. Am Fam Physician. 2013;87:850-856.
- Nijhawan RI, Jacob SE, Woolery-Lloyd H. Skin of color education in dermatology residency programs: does residency training reflect the changing demographics of the United States? J Am Acad Dermatol. 2008;59:615-618.
- Jensen JD, Dunnick CA, Arbuckle HA, et al. Dermatology information on the Internet: an appraisal by dermatologists and dermatology residents. J Am Acad Dermatol. 2010;63:1101-1103.
- Cutrone M, Grimalt R. Dermatological image search engines on the Internet: do they work? J Eur Acad Dermatol Venereol. 2007;21:175-177.
- Xu S, Nault A, Bhatia A. Search and engagement analysis of association websites representing dermatologists—implications and opportunities for web visibility and patient education: website rankings of dermatology associations. Pract Dermatol. In press.
- comScore releases July 2015 U.S. desktop search engine rankings [press release]. Reston, VA: comScore, Inc; August 14, 2015. http://www.comscore.com/Insights/Market-Rankings/comScore-Releases-July-2015-U.S.-Desktop-Search-Engine-Rankings. Accessed October 18, 2016.
- Kundu RV, Patterson S. Dermatologic conditions in skin of color: part I. special considerations for common skin disorders. Am Fam Physician. 2013;87:850-856.
- Nijhawan RI, Jacob SE, Woolery-Lloyd H. Skin of color education in dermatology residency programs: does residency training reflect the changing demographics of the United States? J Am Acad Dermatol. 2008;59:615-618.
Practice Points
- Direct Google image searches largely deliver accurate results for common dermatological diagnoses.
- Greater effort should be made to include more publicly available images for dermatological diseases in darker skin types.
Novel De Novo Heterozygous Frameshift Mutation of the ADAR1 Gene in Heavy Dyschromatosis Symmetrica Hereditaria
To the Editor:
Dyschromatosis symmetrica hereditaria (DSH)(Online Mendelian Inheritance in Man 127400), also called reticulate acropigmentation of Dohi, is a pigmentary genodermatosis characterized by a mixture of hyperpigmented and hypopigmented macules of various sizes on the dorsal aspects of the hands and feet. Linkage analysis has revealed that the DSH gene locus resides on chromosome 1q11-q21,1 and the adenosine deaminase RNA specific gene, ADAR1 (also called DSRAD), in this region has been identified as being responsible for the development of DSH.2 We report a sporadic case of severe DSH with the ADAR1 gene detected in a mutation analysis.
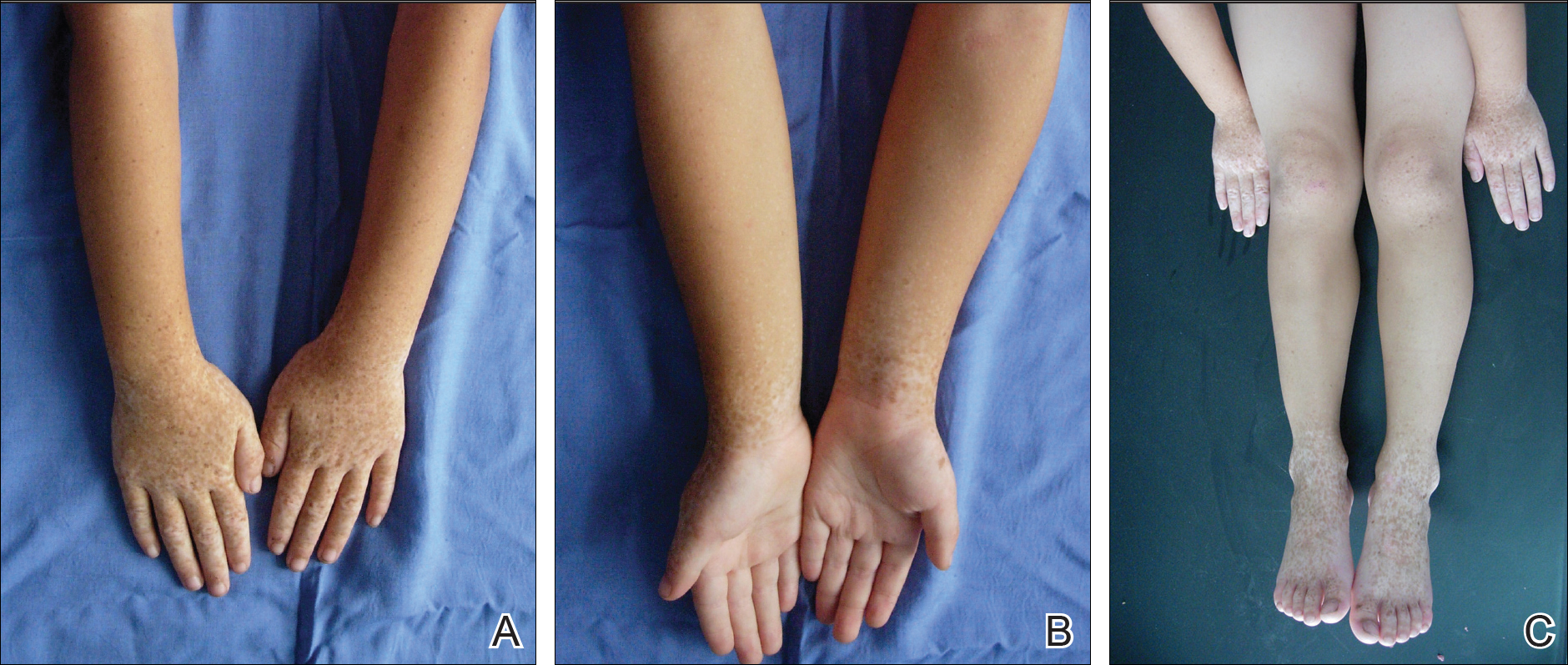
A 6-year-old girl presented with a mixture of hyperpigmented and hypopigmented macules on the dorsal aspects of the hands and feet and the curved side of the wrists, heels, and knees, as well as scattered frecklelike and depigmented spots on the face, ears, neck, arms, and upper back (Figure 1). Her parents noted that hyperpigmented and hypopigmented macules on the dorsal aspects of the hands developed at 5 months of age. Exacerbation after exposure to sunlight resulted in the eruption becoming remarkable in summer and fainter in winter. The skin lesions gradually became more progressive. Physical examination revealed that the patient generally was healthy.
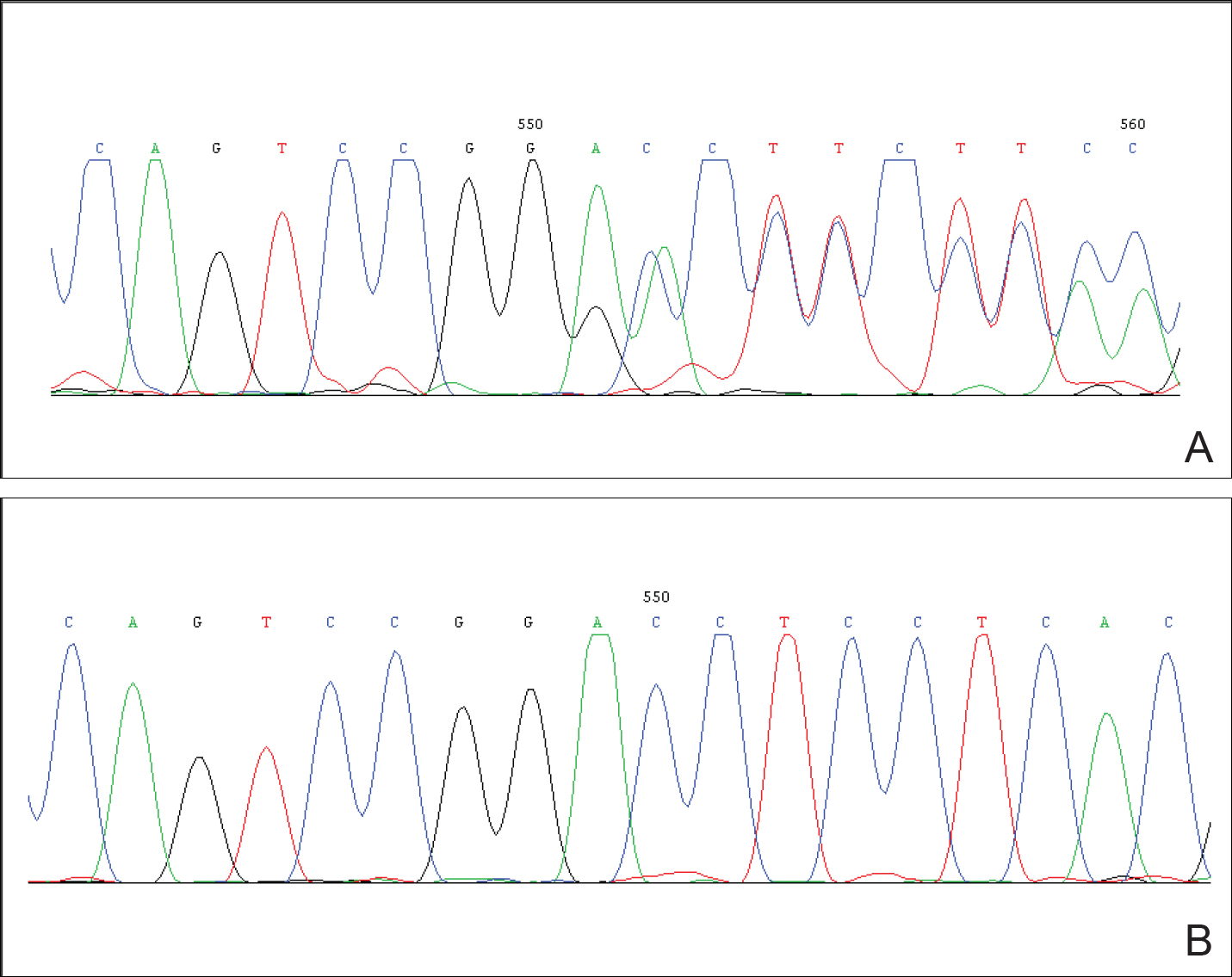
After obtaining informed consent, we performed a mutation analysis of the ADAR1 gene in our patient and her parents. We used a kit to extract genomic DNA from peripheral blood, which was then used to amplify the exons of the ADAR1 gene with intronic flanking sequences by polymerase chain reaction with the primer.3 After amplification, polymerase chain reaction products were purified. We sequenced the ADAR1 gene. Sequence comparisons and analysis found that the patient (proband) carried a heterozygous insertional mutation c.2253insG in exon 6 of the ADAR1 gene. This mutation was not detected in the proband’s healthy parents and 100 normal individuals (Figure 2).
Dyschromatosis symmetrica hereditaria is acquired by autosomal-dominant inheritance and is mainly reported in Asians, especially in Japan and China. Oyama et al4 reviewed 185 cases of DSH in Japan and found the onset of this disease usually was during infancy or childhood; 73% of patients developed the skin lesions before 6 years of age. Suzuki et al5 reported 10 unrelated Japanese patients and found the onset of disease ranged from 1 year of age to childhood. Zhang et al1,6 investigated 78 Chinese patients with DSH including 8 multigenerational families and 2 sporadic patients and found the age of disease onset ranged from 6 months to 15 years of age. The age of onset in our patient (5 months) was younger than these prior reports.
Patients with DSH have a characteristic appearance including a mixture of hyperpigmented and hypopigmented macules of various sizes on the dorsal aspects of the hands and feet. Few patients have similar lesions on the knees and elbows. Many patients have frecklelike macules on the face and arms.1-6 One patient has been described with scattered depigmented spots on the face and chest.1 Our patient had a characteristic appearance as well as some special manifestations including skin lesions on the curved side of the wrist, ears, neck, and upper back.
The human ADAR1 gene spans 30 kilobase and contains 15 exons. It encodes RNA-specific adenosine deaminase composed of 1226 amino acid residues. This enzyme is important for various functions such as site-specific RNA editing and nuclear translation. This enzyme has 2 Z-alpha domains, 3 double-stranded RNA–binding domains, and the putative deaminase domain corresponding to exon 2, exons 2 to 7, and exons 9 to 14 of ADAR1, respectively.6
Mutation analysis of the ADAR1 gene in this case showed heterozygous insertion mutation c.2253insG in exon 6 of the ADAR1 gene, which changed the reading frame, and 475 amino acid residues in C-terminus are replaced by 90 amino acid residues (TSSRAQVRLPSKSWGSLVPSRLRTQQEA RQAGSSRCGSPCLDWGEREGRTHGFHRG NPSDRGQSQKNYAPPLKVPRSTAKT DTPSHWQHLP). This mutation was not detected in the proband’s healthy parents and the 100 control individuals, which indicated that it was a de novo mutation and the pathogenic mutation of DSH rather than a common polymorphism.
In conclusion, we report a novel mutation of the ADAR1 gene with a heavy clinical phenotype in DSH. This study expands the spectrum of clinical manifestations and demonstrates the ADAR1 mutation in DSH.
Acknowledgments
We are most grateful to the patient and her family for taking part in our study.
- Zhang XJ, Gao M, Li M, et al. Identification of a locus for dyschromatosis symmetrica hereditaria at chromosome 1q11-1q21. J Invest Dermatol. 2003;120:776-780.
- Miyamura Y, Suzuki T, Kono M, et al. Mutations of the RNA-specific adenosine deaminase gene (DSRAD) are involved in dyschromatosis symmetrica hereditaria [published online August 11, 2003]. Am J Hum Genet. 2003;73:693-699.
- Li M, Li C, Hua H, et al. Identification of two novel mutations in Chinese patients with dyschromatosis symmetrica hereditaria [published online October 8, 2005]. Arch Dermatol Res. 2005;297:196-200.
- Oyama M, Shimizu H, Ohata Y, et al. Dyschromatosis symmetrica hereditaria (reticulate acropigmentation of Dohi): report of a Japanese family with the condition and a literature review of 185 cases. Br J Dermatol. 1999;140:491-496.
- Suzuki N, Suzuki T, Inagaki K, et al. Ten novel mutations of the ADAR1 gene in Japanese patients with dyschromatosis symmetrica hereditaria [published online August 17, 2006]. J Invest Dermatol. 2007;127:309-311.
- Zhang XJ, He PP, Li M, et al. Seven novel mutations of the ADAR gene in Chinese families and sporadic patients with dyschromatosis symmetrica hereditaria (DSH). Hum Mutat. 2004;23:629-630.
To the Editor:
Dyschromatosis symmetrica hereditaria (DSH)(Online Mendelian Inheritance in Man 127400), also called reticulate acropigmentation of Dohi, is a pigmentary genodermatosis characterized by a mixture of hyperpigmented and hypopigmented macules of various sizes on the dorsal aspects of the hands and feet. Linkage analysis has revealed that the DSH gene locus resides on chromosome 1q11-q21,1 and the adenosine deaminase RNA specific gene, ADAR1 (also called DSRAD), in this region has been identified as being responsible for the development of DSH.2 We report a sporadic case of severe DSH with the ADAR1 gene detected in a mutation analysis.
A 6-year-old girl presented with a mixture of hyperpigmented and hypopigmented macules on the dorsal aspects of the hands and feet and the curved side of the wrists, heels, and knees, as well as scattered frecklelike and depigmented spots on the face, ears, neck, arms, and upper back (Figure 1). Her parents noted that hyperpigmented and hypopigmented macules on the dorsal aspects of the hands developed at 5 months of age. Exacerbation after exposure to sunlight resulted in the eruption becoming remarkable in summer and fainter in winter. The skin lesions gradually became more progressive. Physical examination revealed that the patient generally was healthy.
After obtaining informed consent, we performed a mutation analysis of the ADAR1 gene in our patient and her parents. We used a kit to extract genomic DNA from peripheral blood, which was then used to amplify the exons of the ADAR1 gene with intronic flanking sequences by polymerase chain reaction with the primer.3 After amplification, polymerase chain reaction products were purified. We sequenced the ADAR1 gene. Sequence comparisons and analysis found that the patient (proband) carried a heterozygous insertional mutation c.2253insG in exon 6 of the ADAR1 gene. This mutation was not detected in the proband’s healthy parents and 100 normal individuals (Figure 2).
Dyschromatosis symmetrica hereditaria is acquired by autosomal-dominant inheritance and is mainly reported in Asians, especially in Japan and China. Oyama et al4 reviewed 185 cases of DSH in Japan and found the onset of this disease usually was during infancy or childhood; 73% of patients developed the skin lesions before 6 years of age. Suzuki et al5 reported 10 unrelated Japanese patients and found the onset of disease ranged from 1 year of age to childhood. Zhang et al1,6 investigated 78 Chinese patients with DSH including 8 multigenerational families and 2 sporadic patients and found the age of disease onset ranged from 6 months to 15 years of age. The age of onset in our patient (5 months) was younger than these prior reports.
Patients with DSH have a characteristic appearance including a mixture of hyperpigmented and hypopigmented macules of various sizes on the dorsal aspects of the hands and feet. Few patients have similar lesions on the knees and elbows. Many patients have frecklelike macules on the face and arms.1-6 One patient has been described with scattered depigmented spots on the face and chest.1 Our patient had a characteristic appearance as well as some special manifestations including skin lesions on the curved side of the wrist, ears, neck, and upper back.
The human ADAR1 gene spans 30 kilobase and contains 15 exons. It encodes RNA-specific adenosine deaminase composed of 1226 amino acid residues. This enzyme is important for various functions such as site-specific RNA editing and nuclear translation. This enzyme has 2 Z-alpha domains, 3 double-stranded RNA–binding domains, and the putative deaminase domain corresponding to exon 2, exons 2 to 7, and exons 9 to 14 of ADAR1, respectively.6
Mutation analysis of the ADAR1 gene in this case showed heterozygous insertion mutation c.2253insG in exon 6 of the ADAR1 gene, which changed the reading frame, and 475 amino acid residues in C-terminus are replaced by 90 amino acid residues (TSSRAQVRLPSKSWGSLVPSRLRTQQEA RQAGSSRCGSPCLDWGEREGRTHGFHRG NPSDRGQSQKNYAPPLKVPRSTAKT DTPSHWQHLP). This mutation was not detected in the proband’s healthy parents and the 100 control individuals, which indicated that it was a de novo mutation and the pathogenic mutation of DSH rather than a common polymorphism.
In conclusion, we report a novel mutation of the ADAR1 gene with a heavy clinical phenotype in DSH. This study expands the spectrum of clinical manifestations and demonstrates the ADAR1 mutation in DSH.
Acknowledgments
We are most grateful to the patient and her family for taking part in our study.
To the Editor:
Dyschromatosis symmetrica hereditaria (DSH)(Online Mendelian Inheritance in Man 127400), also called reticulate acropigmentation of Dohi, is a pigmentary genodermatosis characterized by a mixture of hyperpigmented and hypopigmented macules of various sizes on the dorsal aspects of the hands and feet. Linkage analysis has revealed that the DSH gene locus resides on chromosome 1q11-q21,1 and the adenosine deaminase RNA specific gene, ADAR1 (also called DSRAD), in this region has been identified as being responsible for the development of DSH.2 We report a sporadic case of severe DSH with the ADAR1 gene detected in a mutation analysis.
A 6-year-old girl presented with a mixture of hyperpigmented and hypopigmented macules on the dorsal aspects of the hands and feet and the curved side of the wrists, heels, and knees, as well as scattered frecklelike and depigmented spots on the face, ears, neck, arms, and upper back (Figure 1). Her parents noted that hyperpigmented and hypopigmented macules on the dorsal aspects of the hands developed at 5 months of age. Exacerbation after exposure to sunlight resulted in the eruption becoming remarkable in summer and fainter in winter. The skin lesions gradually became more progressive. Physical examination revealed that the patient generally was healthy.
After obtaining informed consent, we performed a mutation analysis of the ADAR1 gene in our patient and her parents. We used a kit to extract genomic DNA from peripheral blood, which was then used to amplify the exons of the ADAR1 gene with intronic flanking sequences by polymerase chain reaction with the primer.3 After amplification, polymerase chain reaction products were purified. We sequenced the ADAR1 gene. Sequence comparisons and analysis found that the patient (proband) carried a heterozygous insertional mutation c.2253insG in exon 6 of the ADAR1 gene. This mutation was not detected in the proband’s healthy parents and 100 normal individuals (Figure 2).
Dyschromatosis symmetrica hereditaria is acquired by autosomal-dominant inheritance and is mainly reported in Asians, especially in Japan and China. Oyama et al4 reviewed 185 cases of DSH in Japan and found the onset of this disease usually was during infancy or childhood; 73% of patients developed the skin lesions before 6 years of age. Suzuki et al5 reported 10 unrelated Japanese patients and found the onset of disease ranged from 1 year of age to childhood. Zhang et al1,6 investigated 78 Chinese patients with DSH including 8 multigenerational families and 2 sporadic patients and found the age of disease onset ranged from 6 months to 15 years of age. The age of onset in our patient (5 months) was younger than these prior reports.
Patients with DSH have a characteristic appearance including a mixture of hyperpigmented and hypopigmented macules of various sizes on the dorsal aspects of the hands and feet. Few patients have similar lesions on the knees and elbows. Many patients have frecklelike macules on the face and arms.1-6 One patient has been described with scattered depigmented spots on the face and chest.1 Our patient had a characteristic appearance as well as some special manifestations including skin lesions on the curved side of the wrist, ears, neck, and upper back.
The human ADAR1 gene spans 30 kilobase and contains 15 exons. It encodes RNA-specific adenosine deaminase composed of 1226 amino acid residues. This enzyme is important for various functions such as site-specific RNA editing and nuclear translation. This enzyme has 2 Z-alpha domains, 3 double-stranded RNA–binding domains, and the putative deaminase domain corresponding to exon 2, exons 2 to 7, and exons 9 to 14 of ADAR1, respectively.6
Mutation analysis of the ADAR1 gene in this case showed heterozygous insertion mutation c.2253insG in exon 6 of the ADAR1 gene, which changed the reading frame, and 475 amino acid residues in C-terminus are replaced by 90 amino acid residues (TSSRAQVRLPSKSWGSLVPSRLRTQQEA RQAGSSRCGSPCLDWGEREGRTHGFHRG NPSDRGQSQKNYAPPLKVPRSTAKT DTPSHWQHLP). This mutation was not detected in the proband’s healthy parents and the 100 control individuals, which indicated that it was a de novo mutation and the pathogenic mutation of DSH rather than a common polymorphism.
In conclusion, we report a novel mutation of the ADAR1 gene with a heavy clinical phenotype in DSH. This study expands the spectrum of clinical manifestations and demonstrates the ADAR1 mutation in DSH.
Acknowledgments
We are most grateful to the patient and her family for taking part in our study.
- Zhang XJ, Gao M, Li M, et al. Identification of a locus for dyschromatosis symmetrica hereditaria at chromosome 1q11-1q21. J Invest Dermatol. 2003;120:776-780.
- Miyamura Y, Suzuki T, Kono M, et al. Mutations of the RNA-specific adenosine deaminase gene (DSRAD) are involved in dyschromatosis symmetrica hereditaria [published online August 11, 2003]. Am J Hum Genet. 2003;73:693-699.
- Li M, Li C, Hua H, et al. Identification of two novel mutations in Chinese patients with dyschromatosis symmetrica hereditaria [published online October 8, 2005]. Arch Dermatol Res. 2005;297:196-200.
- Oyama M, Shimizu H, Ohata Y, et al. Dyschromatosis symmetrica hereditaria (reticulate acropigmentation of Dohi): report of a Japanese family with the condition and a literature review of 185 cases. Br J Dermatol. 1999;140:491-496.
- Suzuki N, Suzuki T, Inagaki K, et al. Ten novel mutations of the ADAR1 gene in Japanese patients with dyschromatosis symmetrica hereditaria [published online August 17, 2006]. J Invest Dermatol. 2007;127:309-311.
- Zhang XJ, He PP, Li M, et al. Seven novel mutations of the ADAR gene in Chinese families and sporadic patients with dyschromatosis symmetrica hereditaria (DSH). Hum Mutat. 2004;23:629-630.
- Zhang XJ, Gao M, Li M, et al. Identification of a locus for dyschromatosis symmetrica hereditaria at chromosome 1q11-1q21. J Invest Dermatol. 2003;120:776-780.
- Miyamura Y, Suzuki T, Kono M, et al. Mutations of the RNA-specific adenosine deaminase gene (DSRAD) are involved in dyschromatosis symmetrica hereditaria [published online August 11, 2003]. Am J Hum Genet. 2003;73:693-699.
- Li M, Li C, Hua H, et al. Identification of two novel mutations in Chinese patients with dyschromatosis symmetrica hereditaria [published online October 8, 2005]. Arch Dermatol Res. 2005;297:196-200.
- Oyama M, Shimizu H, Ohata Y, et al. Dyschromatosis symmetrica hereditaria (reticulate acropigmentation of Dohi): report of a Japanese family with the condition and a literature review of 185 cases. Br J Dermatol. 1999;140:491-496.
- Suzuki N, Suzuki T, Inagaki K, et al. Ten novel mutations of the ADAR1 gene in Japanese patients with dyschromatosis symmetrica hereditaria [published online August 17, 2006]. J Invest Dermatol. 2007;127:309-311.
- Zhang XJ, He PP, Li M, et al. Seven novel mutations of the ADAR gene in Chinese families and sporadic patients with dyschromatosis symmetrica hereditaria (DSH). Hum Mutat. 2004;23:629-630.
Practice Points
- The adenosine deaminase RNA specific gene, ADAR1, has been identified as being responsible for the development of dyschromatosis symmetrica hereditaria (DSH).
- The characteristic appearance of DSH is a mixture of hyperpigmented and hypopigmented macules of various sizes on the dorsal aspects of the hands and feet.
Desmoplastic Hairless Hypopigmented Nevus
To the Editor:
We report 2 cases of desmoplastic hairless hypopigmented nevi (DHHN), which are giant congenital melanocytic nevi (GCMN) that show sclerosis with progressive loss of pigment and hair. These changes in GCMN could be considered signs of regression.
A 6-year-old boy presented in the dermatology department with an asymptomatic skin lesion on the right buttock since birth. The parents claimed that the lesion was darkly pigmented at birth and gradually increased in size, with progressive reduction in color in the last 2 years. Physical examination revealed a 10×6-cm, well-defined, raised plaque on the upper medial side of the right buttock (Figure 1). The plaque was firm with a shiny smooth surface and was devoid of hair. The surface was flesh colored with scattered pigmented spots. A punch biopsy of the lesion showed increased melanin content in the basal cell layer. The upper dermis showed small nests of epithelioid nevus cells, most of them containing melanin pigment (Figure 2). In the lower two-thirds of the dermis, nevus cells were both epithelioid and spindle shaped and were arranged in between thick sclerotic collagen bundles with an increased number of fibroblasts. There was a marked reduction in the number of hair follicles. Immunohistochemical staining results were S-100 positive and CD34 negative.
A 5-year-old boy presented in the dermatology department with a large hairy GCMN covering most of the trunk since birth. In the last 1.5 years the parents noted gradual fading of color, decreased hair density, and increased induration of the nevus. Physical examination revealed a large plaque covering the anterior aspect of the trunk (Figure 3) and the back extending down to the buttocks. The lesion formed large skin folds that were more pronounced on the back. The nevus was darkly pigmented with large areas of lighter color that were indurated, devoid of hair, and showed small spots of dark pigmentation. A punch biopsy from the lesion showed small nests of nevus cells in the upper part of the reticular dermis. In the lower part of the dermis, nevus cells were arranged in single units in between thick collagen bundles.
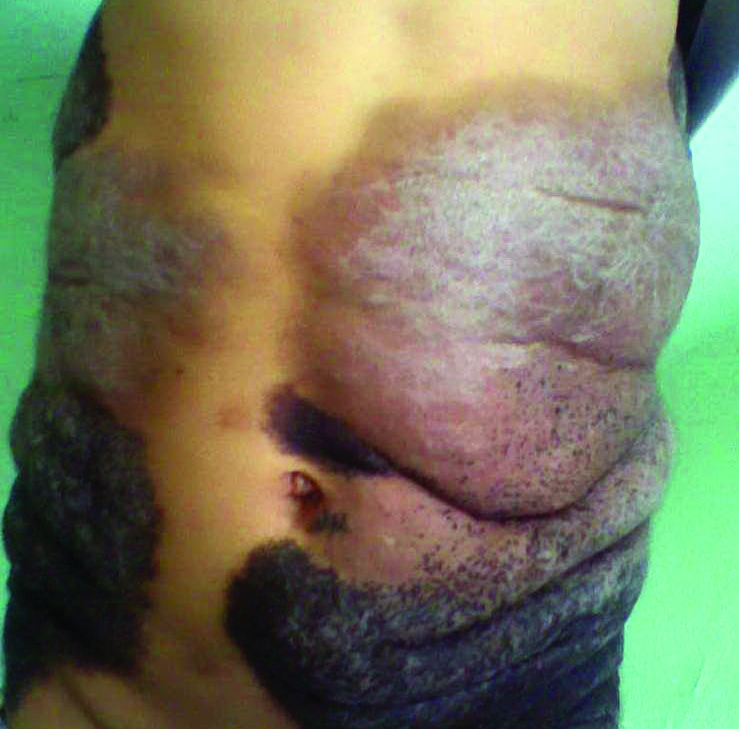
In 2003, Ruiz-Maldonado et al1 described 4 cases of GCMN that showed progressive loss of pigmentation, sclerosis, and hair loss. They proposed the term desmoplastic hairless hypopigmented nevus for their cases and considered it as a variant of GCMN.1 Prior to these reported cases, 2 similar cases were described. The first was a report by Hogan et al2 in 1988 of a 7-month-old girl with a GCMN involving the occipital area and the upper back that became indurated and ulcerated with progressive involution that led to complete disappearance of the nevus. The second was a report by Pattee et al3 in 2001 of a newborn with a GCMN located on the trunk with progressive sclerodermiform reaction. After surgical excision of the nevus, the sclerotic margin disappeared.3
Following the report by Ruiz-Maldonaldo et al,1 5 more cases of DHHN were described.4-8 All cases of DHHN share the same clinical and histopathological features. The clinical features include a GCMN present since birth with progressive sclerosis over time and loss of both pigmentation and hair. Histologically, DHHN shows the typical changes of a congenital melanocytic nevus with decreased numbers of nevus cells, thick sclerotic collagen bundles of the reticular dermis, increased number of fibroblasts, and decreased number of hair follicles. The progressive reduction in the number of nevus cells in melanocytic nevi is considered a sign of regression. Spontaneous regression was rarely described in GCMN, and all the reported cases of regression were associated with desmoplasia.4 Desmoplasia is thought to be induced by either melanocytes that function as adaptive fibroblasts or by fibroblasts themselves, as fibroblasts can show multifunctional differentiation capabilities.9 The direct correlation between the increased induration of DHHN and pigment depletion supports the former hypothesis. The absence of inflammatory cells within the sections of DHHN lesions is against the possibility of an immune-mediated reaction as a cause for the clinical and histological changes seen in this rare form of GCMN. The progressive hair loss in DHHN may be explained by the progressive fibrotic changes in the reticular dermis that affect the blood supply to follicles, leading to atrophy or even absence of the follicles. The progressive reduction in the number of nevus cells in DHHN reduces the potential for malignant transformation and hence following a watchful waiting strategy is a reasonable way to manage these nevi.
We present 2 patients with DHHN, which is a rare form of GCMN that shows signs of regression. The cause of these changes is still unclear.
- Ruiz-Maldonado R, Orozco-Covarrubias L, Ridaura-Sanz C, et al. Desmoplastic hairless hypopigmented naevus: a variant of giant congenital melanocytic naevus. Br J Dermatol. 2003;148:1253-1257.
- Hogan DJ, Murphy F, Bremner RM. Spontaneous resolution of a giant congenital melanocytic nevus. Pediatr Dermatol. 1988;5:170-172.
- Pattee SF, Hansen RC, Bangert JL, et al. Giant congenital nevus with progressive sclerodermoid reaction in a newborn. Pediatr Dermatol. 2001;18:321-324.
- Boente MC, Asial RA. Desmoplastic hairless hypopigmented nevus (DHHN). a distinct variant of giant melanocytic nevus. Eur J Dermatol. 2005;15:451-453.
- Bushby SA, Rajan NJ, Shehade SA. Spontaneous resolution of a giant melanocytic naevus involving a desmoplastic process. Br J Dermatol. 2005;153(suppl 1):13-19.
- Martin JM, Jorda E, Monteagudo C, et al. Desmoplastic giant congenital nevus with progressive depigmentation. J Am Acad Dermatol. 2007;56(suppl 2):S10-S14.
- Hermandez-Martin A, Torrelo A, Echevarria C, et al. Ulcerated sclerotic giant congenital melanocytic naevus: case report and review of the literature. Clin Exp Dermatol. 2007;32:529-532.
- Werner B, Carvalho VO, Nacif SB, et al. Desmoplastic hypopigmented hairless nevus: a variant with progressive depigmentation, induration and overgrowth [published online May 16, 2011]. Pediatr Dermatol. 2012;29:336-340.
- Fearns C, Dowdle EB. The desmoplastic response: induction of collagen synthesis by melanoma cells in vitro. Int J Cancer. 1992;50:621-627.
To the Editor:
We report 2 cases of desmoplastic hairless hypopigmented nevi (DHHN), which are giant congenital melanocytic nevi (GCMN) that show sclerosis with progressive loss of pigment and hair. These changes in GCMN could be considered signs of regression.
A 6-year-old boy presented in the dermatology department with an asymptomatic skin lesion on the right buttock since birth. The parents claimed that the lesion was darkly pigmented at birth and gradually increased in size, with progressive reduction in color in the last 2 years. Physical examination revealed a 10×6-cm, well-defined, raised plaque on the upper medial side of the right buttock (Figure 1). The plaque was firm with a shiny smooth surface and was devoid of hair. The surface was flesh colored with scattered pigmented spots. A punch biopsy of the lesion showed increased melanin content in the basal cell layer. The upper dermis showed small nests of epithelioid nevus cells, most of them containing melanin pigment (Figure 2). In the lower two-thirds of the dermis, nevus cells were both epithelioid and spindle shaped and were arranged in between thick sclerotic collagen bundles with an increased number of fibroblasts. There was a marked reduction in the number of hair follicles. Immunohistochemical staining results were S-100 positive and CD34 negative.
A 5-year-old boy presented in the dermatology department with a large hairy GCMN covering most of the trunk since birth. In the last 1.5 years the parents noted gradual fading of color, decreased hair density, and increased induration of the nevus. Physical examination revealed a large plaque covering the anterior aspect of the trunk (Figure 3) and the back extending down to the buttocks. The lesion formed large skin folds that were more pronounced on the back. The nevus was darkly pigmented with large areas of lighter color that were indurated, devoid of hair, and showed small spots of dark pigmentation. A punch biopsy from the lesion showed small nests of nevus cells in the upper part of the reticular dermis. In the lower part of the dermis, nevus cells were arranged in single units in between thick collagen bundles.
In 2003, Ruiz-Maldonado et al1 described 4 cases of GCMN that showed progressive loss of pigmentation, sclerosis, and hair loss. They proposed the term desmoplastic hairless hypopigmented nevus for their cases and considered it as a variant of GCMN.1 Prior to these reported cases, 2 similar cases were described. The first was a report by Hogan et al2 in 1988 of a 7-month-old girl with a GCMN involving the occipital area and the upper back that became indurated and ulcerated with progressive involution that led to complete disappearance of the nevus. The second was a report by Pattee et al3 in 2001 of a newborn with a GCMN located on the trunk with progressive sclerodermiform reaction. After surgical excision of the nevus, the sclerotic margin disappeared.3
Following the report by Ruiz-Maldonaldo et al,1 5 more cases of DHHN were described.4-8 All cases of DHHN share the same clinical and histopathological features. The clinical features include a GCMN present since birth with progressive sclerosis over time and loss of both pigmentation and hair. Histologically, DHHN shows the typical changes of a congenital melanocytic nevus with decreased numbers of nevus cells, thick sclerotic collagen bundles of the reticular dermis, increased number of fibroblasts, and decreased number of hair follicles. The progressive reduction in the number of nevus cells in melanocytic nevi is considered a sign of regression. Spontaneous regression was rarely described in GCMN, and all the reported cases of regression were associated with desmoplasia.4 Desmoplasia is thought to be induced by either melanocytes that function as adaptive fibroblasts or by fibroblasts themselves, as fibroblasts can show multifunctional differentiation capabilities.9 The direct correlation between the increased induration of DHHN and pigment depletion supports the former hypothesis. The absence of inflammatory cells within the sections of DHHN lesions is against the possibility of an immune-mediated reaction as a cause for the clinical and histological changes seen in this rare form of GCMN. The progressive hair loss in DHHN may be explained by the progressive fibrotic changes in the reticular dermis that affect the blood supply to follicles, leading to atrophy or even absence of the follicles. The progressive reduction in the number of nevus cells in DHHN reduces the potential for malignant transformation and hence following a watchful waiting strategy is a reasonable way to manage these nevi.
We present 2 patients with DHHN, which is a rare form of GCMN that shows signs of regression. The cause of these changes is still unclear.
To the Editor:
We report 2 cases of desmoplastic hairless hypopigmented nevi (DHHN), which are giant congenital melanocytic nevi (GCMN) that show sclerosis with progressive loss of pigment and hair. These changes in GCMN could be considered signs of regression.
A 6-year-old boy presented in the dermatology department with an asymptomatic skin lesion on the right buttock since birth. The parents claimed that the lesion was darkly pigmented at birth and gradually increased in size, with progressive reduction in color in the last 2 years. Physical examination revealed a 10×6-cm, well-defined, raised plaque on the upper medial side of the right buttock (Figure 1). The plaque was firm with a shiny smooth surface and was devoid of hair. The surface was flesh colored with scattered pigmented spots. A punch biopsy of the lesion showed increased melanin content in the basal cell layer. The upper dermis showed small nests of epithelioid nevus cells, most of them containing melanin pigment (Figure 2). In the lower two-thirds of the dermis, nevus cells were both epithelioid and spindle shaped and were arranged in between thick sclerotic collagen bundles with an increased number of fibroblasts. There was a marked reduction in the number of hair follicles. Immunohistochemical staining results were S-100 positive and CD34 negative.
A 5-year-old boy presented in the dermatology department with a large hairy GCMN covering most of the trunk since birth. In the last 1.5 years the parents noted gradual fading of color, decreased hair density, and increased induration of the nevus. Physical examination revealed a large plaque covering the anterior aspect of the trunk (Figure 3) and the back extending down to the buttocks. The lesion formed large skin folds that were more pronounced on the back. The nevus was darkly pigmented with large areas of lighter color that were indurated, devoid of hair, and showed small spots of dark pigmentation. A punch biopsy from the lesion showed small nests of nevus cells in the upper part of the reticular dermis. In the lower part of the dermis, nevus cells were arranged in single units in between thick collagen bundles.
In 2003, Ruiz-Maldonado et al1 described 4 cases of GCMN that showed progressive loss of pigmentation, sclerosis, and hair loss. They proposed the term desmoplastic hairless hypopigmented nevus for their cases and considered it as a variant of GCMN.1 Prior to these reported cases, 2 similar cases were described. The first was a report by Hogan et al2 in 1988 of a 7-month-old girl with a GCMN involving the occipital area and the upper back that became indurated and ulcerated with progressive involution that led to complete disappearance of the nevus. The second was a report by Pattee et al3 in 2001 of a newborn with a GCMN located on the trunk with progressive sclerodermiform reaction. After surgical excision of the nevus, the sclerotic margin disappeared.3
Following the report by Ruiz-Maldonaldo et al,1 5 more cases of DHHN were described.4-8 All cases of DHHN share the same clinical and histopathological features. The clinical features include a GCMN present since birth with progressive sclerosis over time and loss of both pigmentation and hair. Histologically, DHHN shows the typical changes of a congenital melanocytic nevus with decreased numbers of nevus cells, thick sclerotic collagen bundles of the reticular dermis, increased number of fibroblasts, and decreased number of hair follicles. The progressive reduction in the number of nevus cells in melanocytic nevi is considered a sign of regression. Spontaneous regression was rarely described in GCMN, and all the reported cases of regression were associated with desmoplasia.4 Desmoplasia is thought to be induced by either melanocytes that function as adaptive fibroblasts or by fibroblasts themselves, as fibroblasts can show multifunctional differentiation capabilities.9 The direct correlation between the increased induration of DHHN and pigment depletion supports the former hypothesis. The absence of inflammatory cells within the sections of DHHN lesions is against the possibility of an immune-mediated reaction as a cause for the clinical and histological changes seen in this rare form of GCMN. The progressive hair loss in DHHN may be explained by the progressive fibrotic changes in the reticular dermis that affect the blood supply to follicles, leading to atrophy or even absence of the follicles. The progressive reduction in the number of nevus cells in DHHN reduces the potential for malignant transformation and hence following a watchful waiting strategy is a reasonable way to manage these nevi.
We present 2 patients with DHHN, which is a rare form of GCMN that shows signs of regression. The cause of these changes is still unclear.
- Ruiz-Maldonado R, Orozco-Covarrubias L, Ridaura-Sanz C, et al. Desmoplastic hairless hypopigmented naevus: a variant of giant congenital melanocytic naevus. Br J Dermatol. 2003;148:1253-1257.
- Hogan DJ, Murphy F, Bremner RM. Spontaneous resolution of a giant congenital melanocytic nevus. Pediatr Dermatol. 1988;5:170-172.
- Pattee SF, Hansen RC, Bangert JL, et al. Giant congenital nevus with progressive sclerodermoid reaction in a newborn. Pediatr Dermatol. 2001;18:321-324.
- Boente MC, Asial RA. Desmoplastic hairless hypopigmented nevus (DHHN). a distinct variant of giant melanocytic nevus. Eur J Dermatol. 2005;15:451-453.
- Bushby SA, Rajan NJ, Shehade SA. Spontaneous resolution of a giant melanocytic naevus involving a desmoplastic process. Br J Dermatol. 2005;153(suppl 1):13-19.
- Martin JM, Jorda E, Monteagudo C, et al. Desmoplastic giant congenital nevus with progressive depigmentation. J Am Acad Dermatol. 2007;56(suppl 2):S10-S14.
- Hermandez-Martin A, Torrelo A, Echevarria C, et al. Ulcerated sclerotic giant congenital melanocytic naevus: case report and review of the literature. Clin Exp Dermatol. 2007;32:529-532.
- Werner B, Carvalho VO, Nacif SB, et al. Desmoplastic hypopigmented hairless nevus: a variant with progressive depigmentation, induration and overgrowth [published online May 16, 2011]. Pediatr Dermatol. 2012;29:336-340.
- Fearns C, Dowdle EB. The desmoplastic response: induction of collagen synthesis by melanoma cells in vitro. Int J Cancer. 1992;50:621-627.
- Ruiz-Maldonado R, Orozco-Covarrubias L, Ridaura-Sanz C, et al. Desmoplastic hairless hypopigmented naevus: a variant of giant congenital melanocytic naevus. Br J Dermatol. 2003;148:1253-1257.
- Hogan DJ, Murphy F, Bremner RM. Spontaneous resolution of a giant congenital melanocytic nevus. Pediatr Dermatol. 1988;5:170-172.
- Pattee SF, Hansen RC, Bangert JL, et al. Giant congenital nevus with progressive sclerodermoid reaction in a newborn. Pediatr Dermatol. 2001;18:321-324.
- Boente MC, Asial RA. Desmoplastic hairless hypopigmented nevus (DHHN). a distinct variant of giant melanocytic nevus. Eur J Dermatol. 2005;15:451-453.
- Bushby SA, Rajan NJ, Shehade SA. Spontaneous resolution of a giant melanocytic naevus involving a desmoplastic process. Br J Dermatol. 2005;153(suppl 1):13-19.
- Martin JM, Jorda E, Monteagudo C, et al. Desmoplastic giant congenital nevus with progressive depigmentation. J Am Acad Dermatol. 2007;56(suppl 2):S10-S14.
- Hermandez-Martin A, Torrelo A, Echevarria C, et al. Ulcerated sclerotic giant congenital melanocytic naevus: case report and review of the literature. Clin Exp Dermatol. 2007;32:529-532.
- Werner B, Carvalho VO, Nacif SB, et al. Desmoplastic hypopigmented hairless nevus: a variant with progressive depigmentation, induration and overgrowth [published online May 16, 2011]. Pediatr Dermatol. 2012;29:336-340.
- Fearns C, Dowdle EB. The desmoplastic response: induction of collagen synthesis by melanoma cells in vitro. Int J Cancer. 1992;50:621-627.
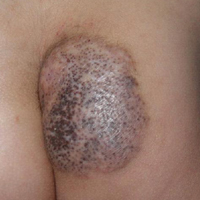
Nevus Spilus: Is the Presence of Hair Associated With an Increased Risk for Melanoma?
The term nevus spilus (NS), also known as speckled lentiginous nevus, was first used in the 19th century to describe lesions with background café au lait–like lentiginous melanocytic hyperplasia speckled with small, 1- to 3-mm, darker foci. The dark spots reflect lentigines; junctional, compound, and intradermal nevus cell nests; and more rarely Spitz and blue nevi. Both macular and papular subtypes have been described.1 This birthmark is quite common, occurring in 1.3% to 2.3% of the adult population worldwide.2 Hypertrichosis has been described in NS.3-9 Two subsequent cases of malignant melanoma in hairy NS suggested that lesions may be particularly prone to malignant degeneration.4,8 We report an additional case of hairy NS that was not associated with melanoma and consider whether dermatologists should warn their patients about this association.
Case Report
A 26-year-old woman presented with a stable 7×8-cm, tan-brown, macular, pigmented birthmark studded with darker 1- to 2-mm, irregular, brown-black and blue, confettilike macules on the left proximal lateral thigh that had been present since birth (Figure 1). Dark terminal hairs were present, arising from both the darker and lighter pigmented areas but not the surrounding normal skin.
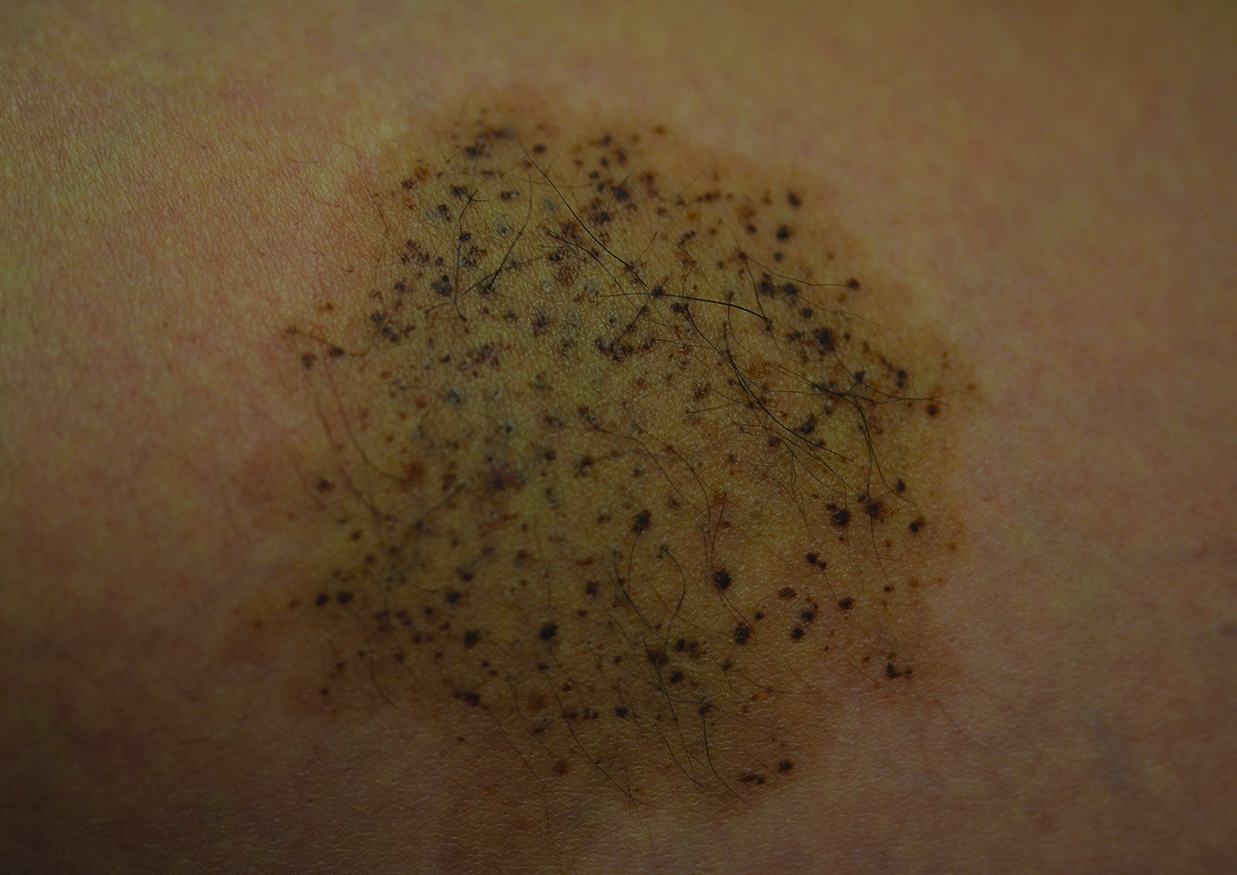
A 4-mm punch biopsy from one of the dark blue macules demonstrated uniform lentiginous melanocytic hyperplasia and nevus cell nests adjacent to the sweat glands extending into the mid dermis (Figure 2). No clinical evidence of malignant degeneration was present.
Comment
The risk for melanoma is increased in classic nonspeckled congenital nevi and the risk correlates with the size of the lesion and most probably the number of nevus cells in the lesion that increase the risk for a random mutation.8,10,11 It is likely that NS with or without hair presages a small increased risk for melanoma,6,9,12 which is not surprising because NS is a subtype of congenital melanocytic nevus (CMN), a condition that is present at birth and results from a proliferation of melanocytes.6 Nevus spilus, however, appears to have a notably lower risk for malignant degeneration than other classic CMN of the same size. The following support for this hypothesis is offered: First, CMN have nevus cells broadly filling the dermis that extend more deeply into the dermis than NS (Figure 2A).10 In our estimation, CMN have at least 100 times the number of nevus cells per square centimeter compared to NS. The potential for malignant degeneration of any one melanocyte is greater when more are present. Second, although some NS lesions evolve, classic CMN are universally more proliferative than NS.10,13 The involved skin in CMN thickens over time with increased numbers of melanocytes and marked overgrowth of adjacent tissue. Melanocytes in a proliferative phase may be more likely to undergo malignant degeneration.10
A PubMed search of articles indexed for MEDLINE using the search term nevus spilus and melanoma yielded 2 cases4,8 of melanoma arising among 15 cases of hairy NS in the literature, which led to the suggestion that the presence of hair could be associated with an increased risk for malignant degeneration in NS (Table). This apparent high incidence of melanoma most likely reflects referral/publication bias rather than a statistically significant association. In fact, the clinical lesion most clinically similar to hairy NS is Becker nevus, with tan macules demonstrating lentiginous melanocytic hyperplasia associated with numerous coarse terminal hairs. There is no indication that Becker nevi have a considerable premalignant potential, though one case of melanoma arising in a Becker nevus has been reported.9 There is no evidence to suggest that classic CMN with hypertrichosis has a greater premalignant potential than similar lesions without hypertrichosis.

We noticed the presence of hair in our patient’s lesion only after reports in the literature caused us to look for this phenomenon.9 This occurrence may actually be quite common. We do not recommend prophylactic excision of NS and believe the risk for malignant degeneration is low in NS with or without hair, though larger NS (>4 cm), especially giant, zosteriform, or segmental lesions, may have a greater risk.1,6,9,10 It is prudent for physicians to carefully examine NS and sample suspicious foci, especially when patients describe a lesion as changing.
- Vidaurri-de la Cruz H, Happle R. Two distinct types of speckled lentiginous nevi characterized by macular versus papular speckles. Dermatology. 2006;212:53-58.
- Ly L, Christie M, Swain S, et al. Melanoma(s) arising in large segmental speckled lentiginous nevi: a case series. J Am Acad Dermatol. 2011;64:1190-1193.
- Prose NS, Heilman E, Felman YM, et al. Multiple benign juvenile melanoma. J Am Acad Dermatol. 1983;9:236-242.
- Grinspan D, Casala A, Abulafia J, et al. Melanoma on dysplastic nevus spilus. Int J Dermatol. 1997;36:499-502 .
- Langenbach N, Pfau A, Landthaler M, et al. Naevi spili, café-au-lait spots and melanocytic naevi aggregated alongside Blaschko’s lines, with a review of segmental melanocytic lesions. Acta Derm Venereol. 1998;78:378-380.
- Schaffer JV, Orlow SJ, Lazova R, et al. Speckled lentiginous nevus: within the spectrum of congenital melanocytic nevi. Arch Dermatol. 2001;137:172-178.
- Saraswat A, Dogra S, Bansali A, et al. Phakomatosis pigmentokeratotica associated with hypophosphataemic vitamin D–resistant rickets: improvement in phosphate homeostasis after partial laser ablation. Br J Dermatol. 2003;148:1074-1076.
- Zeren-Bilgin
i , Gür S, Aydın O, et al. Melanoma arising in a hairy nevus spilus. Int J Dermatol. 2006;45:1362-1364. - Singh S, Jain N, Khanna N, et al. Hairy nevus spilus: a case series. Pediatr Dermatol. 2013;30:100-104.
- Price HN, Schaffer JV. Congenital melanocytic nevi—when to worry and how to treat: facts and controversies. Clin Dermatol. 2010;28:293-302.
- Alikhan Ali, Ibrahimi OA, Eisen DB. Congenital melanocytic nevi: where are we now? J Am Acad Dermatol. 2012;67:495.e1-495.e17.
- Haenssle HA, Kaune KM, Buhl T, et al. Melanoma arising in segmental nevus spilus: detection by sequential digital dermatoscopy. J Am Acad Dermatol. 2009;61:337-341.
- Cohen LM. Nevus spilus: congenital or acquired? Arch Dermatol. 2001;137:215-216.
The term nevus spilus (NS), also known as speckled lentiginous nevus, was first used in the 19th century to describe lesions with background café au lait–like lentiginous melanocytic hyperplasia speckled with small, 1- to 3-mm, darker foci. The dark spots reflect lentigines; junctional, compound, and intradermal nevus cell nests; and more rarely Spitz and blue nevi. Both macular and papular subtypes have been described.1 This birthmark is quite common, occurring in 1.3% to 2.3% of the adult population worldwide.2 Hypertrichosis has been described in NS.3-9 Two subsequent cases of malignant melanoma in hairy NS suggested that lesions may be particularly prone to malignant degeneration.4,8 We report an additional case of hairy NS that was not associated with melanoma and consider whether dermatologists should warn their patients about this association.
Case Report
A 26-year-old woman presented with a stable 7×8-cm, tan-brown, macular, pigmented birthmark studded with darker 1- to 2-mm, irregular, brown-black and blue, confettilike macules on the left proximal lateral thigh that had been present since birth (Figure 1). Dark terminal hairs were present, arising from both the darker and lighter pigmented areas but not the surrounding normal skin.
A 4-mm punch biopsy from one of the dark blue macules demonstrated uniform lentiginous melanocytic hyperplasia and nevus cell nests adjacent to the sweat glands extending into the mid dermis (Figure 2). No clinical evidence of malignant degeneration was present.
Comment
The risk for melanoma is increased in classic nonspeckled congenital nevi and the risk correlates with the size of the lesion and most probably the number of nevus cells in the lesion that increase the risk for a random mutation.8,10,11 It is likely that NS with or without hair presages a small increased risk for melanoma,6,9,12 which is not surprising because NS is a subtype of congenital melanocytic nevus (CMN), a condition that is present at birth and results from a proliferation of melanocytes.6 Nevus spilus, however, appears to have a notably lower risk for malignant degeneration than other classic CMN of the same size. The following support for this hypothesis is offered: First, CMN have nevus cells broadly filling the dermis that extend more deeply into the dermis than NS (Figure 2A).10 In our estimation, CMN have at least 100 times the number of nevus cells per square centimeter compared to NS. The potential for malignant degeneration of any one melanocyte is greater when more are present. Second, although some NS lesions evolve, classic CMN are universally more proliferative than NS.10,13 The involved skin in CMN thickens over time with increased numbers of melanocytes and marked overgrowth of adjacent tissue. Melanocytes in a proliferative phase may be more likely to undergo malignant degeneration.10
A PubMed search of articles indexed for MEDLINE using the search term nevus spilus and melanoma yielded 2 cases4,8 of melanoma arising among 15 cases of hairy NS in the literature, which led to the suggestion that the presence of hair could be associated with an increased risk for malignant degeneration in NS (Table). This apparent high incidence of melanoma most likely reflects referral/publication bias rather than a statistically significant association. In fact, the clinical lesion most clinically similar to hairy NS is Becker nevus, with tan macules demonstrating lentiginous melanocytic hyperplasia associated with numerous coarse terminal hairs. There is no indication that Becker nevi have a considerable premalignant potential, though one case of melanoma arising in a Becker nevus has been reported.9 There is no evidence to suggest that classic CMN with hypertrichosis has a greater premalignant potential than similar lesions without hypertrichosis.
We noticed the presence of hair in our patient’s lesion only after reports in the literature caused us to look for this phenomenon.9 This occurrence may actually be quite common. We do not recommend prophylactic excision of NS and believe the risk for malignant degeneration is low in NS with or without hair, though larger NS (>4 cm), especially giant, zosteriform, or segmental lesions, may have a greater risk.1,6,9,10 It is prudent for physicians to carefully examine NS and sample suspicious foci, especially when patients describe a lesion as changing.
The term nevus spilus (NS), also known as speckled lentiginous nevus, was first used in the 19th century to describe lesions with background café au lait–like lentiginous melanocytic hyperplasia speckled with small, 1- to 3-mm, darker foci. The dark spots reflect lentigines; junctional, compound, and intradermal nevus cell nests; and more rarely Spitz and blue nevi. Both macular and papular subtypes have been described.1 This birthmark is quite common, occurring in 1.3% to 2.3% of the adult population worldwide.2 Hypertrichosis has been described in NS.3-9 Two subsequent cases of malignant melanoma in hairy NS suggested that lesions may be particularly prone to malignant degeneration.4,8 We report an additional case of hairy NS that was not associated with melanoma and consider whether dermatologists should warn their patients about this association.
Case Report
A 26-year-old woman presented with a stable 7×8-cm, tan-brown, macular, pigmented birthmark studded with darker 1- to 2-mm, irregular, brown-black and blue, confettilike macules on the left proximal lateral thigh that had been present since birth (Figure 1). Dark terminal hairs were present, arising from both the darker and lighter pigmented areas but not the surrounding normal skin.
A 4-mm punch biopsy from one of the dark blue macules demonstrated uniform lentiginous melanocytic hyperplasia and nevus cell nests adjacent to the sweat glands extending into the mid dermis (Figure 2). No clinical evidence of malignant degeneration was present.
Comment
The risk for melanoma is increased in classic nonspeckled congenital nevi and the risk correlates with the size of the lesion and most probably the number of nevus cells in the lesion that increase the risk for a random mutation.8,10,11 It is likely that NS with or without hair presages a small increased risk for melanoma,6,9,12 which is not surprising because NS is a subtype of congenital melanocytic nevus (CMN), a condition that is present at birth and results from a proliferation of melanocytes.6 Nevus spilus, however, appears to have a notably lower risk for malignant degeneration than other classic CMN of the same size. The following support for this hypothesis is offered: First, CMN have nevus cells broadly filling the dermis that extend more deeply into the dermis than NS (Figure 2A).10 In our estimation, CMN have at least 100 times the number of nevus cells per square centimeter compared to NS. The potential for malignant degeneration of any one melanocyte is greater when more are present. Second, although some NS lesions evolve, classic CMN are universally more proliferative than NS.10,13 The involved skin in CMN thickens over time with increased numbers of melanocytes and marked overgrowth of adjacent tissue. Melanocytes in a proliferative phase may be more likely to undergo malignant degeneration.10
A PubMed search of articles indexed for MEDLINE using the search term nevus spilus and melanoma yielded 2 cases4,8 of melanoma arising among 15 cases of hairy NS in the literature, which led to the suggestion that the presence of hair could be associated with an increased risk for malignant degeneration in NS (Table). This apparent high incidence of melanoma most likely reflects referral/publication bias rather than a statistically significant association. In fact, the clinical lesion most clinically similar to hairy NS is Becker nevus, with tan macules demonstrating lentiginous melanocytic hyperplasia associated with numerous coarse terminal hairs. There is no indication that Becker nevi have a considerable premalignant potential, though one case of melanoma arising in a Becker nevus has been reported.9 There is no evidence to suggest that classic CMN with hypertrichosis has a greater premalignant potential than similar lesions without hypertrichosis.
We noticed the presence of hair in our patient’s lesion only after reports in the literature caused us to look for this phenomenon.9 This occurrence may actually be quite common. We do not recommend prophylactic excision of NS and believe the risk for malignant degeneration is low in NS with or without hair, though larger NS (>4 cm), especially giant, zosteriform, or segmental lesions, may have a greater risk.1,6,9,10 It is prudent for physicians to carefully examine NS and sample suspicious foci, especially when patients describe a lesion as changing.
- Vidaurri-de la Cruz H, Happle R. Two distinct types of speckled lentiginous nevi characterized by macular versus papular speckles. Dermatology. 2006;212:53-58.
- Ly L, Christie M, Swain S, et al. Melanoma(s) arising in large segmental speckled lentiginous nevi: a case series. J Am Acad Dermatol. 2011;64:1190-1193.
- Prose NS, Heilman E, Felman YM, et al. Multiple benign juvenile melanoma. J Am Acad Dermatol. 1983;9:236-242.
- Grinspan D, Casala A, Abulafia J, et al. Melanoma on dysplastic nevus spilus. Int J Dermatol. 1997;36:499-502 .
- Langenbach N, Pfau A, Landthaler M, et al. Naevi spili, café-au-lait spots and melanocytic naevi aggregated alongside Blaschko’s lines, with a review of segmental melanocytic lesions. Acta Derm Venereol. 1998;78:378-380.
- Schaffer JV, Orlow SJ, Lazova R, et al. Speckled lentiginous nevus: within the spectrum of congenital melanocytic nevi. Arch Dermatol. 2001;137:172-178.
- Saraswat A, Dogra S, Bansali A, et al. Phakomatosis pigmentokeratotica associated with hypophosphataemic vitamin D–resistant rickets: improvement in phosphate homeostasis after partial laser ablation. Br J Dermatol. 2003;148:1074-1076.
- Zeren-Bilgin
i , Gür S, Aydın O, et al. Melanoma arising in a hairy nevus spilus. Int J Dermatol. 2006;45:1362-1364. - Singh S, Jain N, Khanna N, et al. Hairy nevus spilus: a case series. Pediatr Dermatol. 2013;30:100-104.
- Price HN, Schaffer JV. Congenital melanocytic nevi—when to worry and how to treat: facts and controversies. Clin Dermatol. 2010;28:293-302.
- Alikhan Ali, Ibrahimi OA, Eisen DB. Congenital melanocytic nevi: where are we now? J Am Acad Dermatol. 2012;67:495.e1-495.e17.
- Haenssle HA, Kaune KM, Buhl T, et al. Melanoma arising in segmental nevus spilus: detection by sequential digital dermatoscopy. J Am Acad Dermatol. 2009;61:337-341.
- Cohen LM. Nevus spilus: congenital or acquired? Arch Dermatol. 2001;137:215-216.
- Vidaurri-de la Cruz H, Happle R. Two distinct types of speckled lentiginous nevi characterized by macular versus papular speckles. Dermatology. 2006;212:53-58.
- Ly L, Christie M, Swain S, et al. Melanoma(s) arising in large segmental speckled lentiginous nevi: a case series. J Am Acad Dermatol. 2011;64:1190-1193.
- Prose NS, Heilman E, Felman YM, et al. Multiple benign juvenile melanoma. J Am Acad Dermatol. 1983;9:236-242.
- Grinspan D, Casala A, Abulafia J, et al. Melanoma on dysplastic nevus spilus. Int J Dermatol. 1997;36:499-502 .
- Langenbach N, Pfau A, Landthaler M, et al. Naevi spili, café-au-lait spots and melanocytic naevi aggregated alongside Blaschko’s lines, with a review of segmental melanocytic lesions. Acta Derm Venereol. 1998;78:378-380.
- Schaffer JV, Orlow SJ, Lazova R, et al. Speckled lentiginous nevus: within the spectrum of congenital melanocytic nevi. Arch Dermatol. 2001;137:172-178.
- Saraswat A, Dogra S, Bansali A, et al. Phakomatosis pigmentokeratotica associated with hypophosphataemic vitamin D–resistant rickets: improvement in phosphate homeostasis after partial laser ablation. Br J Dermatol. 2003;148:1074-1076.
- Zeren-Bilgin
i , Gür S, Aydın O, et al. Melanoma arising in a hairy nevus spilus. Int J Dermatol. 2006;45:1362-1364. - Singh S, Jain N, Khanna N, et al. Hairy nevus spilus: a case series. Pediatr Dermatol. 2013;30:100-104.
- Price HN, Schaffer JV. Congenital melanocytic nevi—when to worry and how to treat: facts and controversies. Clin Dermatol. 2010;28:293-302.
- Alikhan Ali, Ibrahimi OA, Eisen DB. Congenital melanocytic nevi: where are we now? J Am Acad Dermatol. 2012;67:495.e1-495.e17.
- Haenssle HA, Kaune KM, Buhl T, et al. Melanoma arising in segmental nevus spilus: detection by sequential digital dermatoscopy. J Am Acad Dermatol. 2009;61:337-341.
- Cohen LM. Nevus spilus: congenital or acquired? Arch Dermatol. 2001;137:215-216.
Practice Points
- Nevus spilus (NS) appears as a café au lait macule studded with darker brown “moles.”
- Although melanoma has been described in NS, it is rare.
- There is no evidence that hairy NS are predisposed to melanoma.
Painful Ulcerations Above the Malleoli
The Diagnosis: Livedoid Vasculopathy
Livedoid vasculopathy (LV) is a rare cutaneous disorder that most commonly affects the lower legs. It has an estimated incidence of 1 case per 100,000 per year and predominantly affects women.1 The disease pathogenesis is not fully understood but is thought to involve thrombosis and occlusion of dermal vessels resulting in tissue hypoxia.2 Both inherited and acquired thrombophilic conditions frequently are seen in patients with LV.3,4 Livedoid vasculopathy also has been described as idiopathic5 and is associated with immune complex deposition.6 However, the number of cases of idiopathic LV may be overestimated; as technological advancements to detect coagulation abnormalities improve, it is hypothesized that this entity will be identified less often.2,4
Livedoid vasculopathy has been described in the literature using the term PPURPLE (painful purpuric ulcers with reticular pattern of lower extremities).7 The triad of livedo racemosa, recurrent painful ulcerations, and residual healing with atrophie blanche characterizes the clinical manifestations of LV; however, all 3 characteristics do not need to appear simultaneously for a diagnosis to be made. The condition has a chronic course with spontaneous remissions and exacerbations. Episodic ulcerations occur, especially in the summertime, and heal slowly, leaving behind atrophic, porcelain white, stellate-shaped scars called atrophie blanche. Livedo racemosa also may be seen in Sneddon syndrome; however, these patients experience neurologic symptoms secondary to cerebrovascular occlusion. In contrast to livedo racemosa, acquired livedo reticularis represents a physiologic hypoperfusion pattern that occurs in response to cold exposure.8 A localized sharp pain, known as angina cutis, typically precedes the clinical symptom of painful ulcerations.9 Atrophie blanche once was thought to be specific to LV but has been seen in other diseases such as systemic lupus erythematosus and chronic venous insufficiency.2
The diagnosis of LV is based on identification of characteristic clinical features and skin biopsy. In almost all biopsy specimens, histopathology reveals fibrinoid occlusion of vessels in the superficial and mid dermis.4 Other findings may include epidermal necrosis and vessel wall hyalinization and infarction2 (Figure). Because LV is commonly misdiagnosed as vasculitis, the absence of hallmark features of vasculitis such as neutrophilic infiltrate of blood vessel walls and fibrinoid necrosis suggest the diagnosis. Extensive laboratory evaluation for inherited and acquired coagulation abnormalities should be performed.
Treatment of LV is difficult, as there is currently no consensus on optimal therapy. The mainstay of therapy is to reduce pain, prevent infection, and reduce ulceration and development of atrophie blanche. Underlying causes should be identified and appropriately treated. Because the primary pathogenesis of LV is considered to be a hypercoagulable state, first-line treatment often includes therapies to enhance blood flow and prevent thrombosis such as smoking cessation, antiplatelet therapy, and pentoxifylline. Vasodilating agents, anti-inflammatory agents, anticoagulation, and fibrinolytic therapy also have been used with varying degrees of success.7
- Fritsch P, Zelger B. Livedo vasculitis [in German]. Hautarzt. 1995;46:215-224; quiz 222-223.
- Kerk N, Goerge T. Livedoid vasculopathy—a thrombotic disease. Vasa. 2013;42:317-322.
- Stevanovic DV. Atrophie blanche. a sign of dermal blood occlusion. Arch Dermatol. 1974;109:858-862.
- Hairston BR, Davis MD, Pittelkow MR, et al. Livedoid vasculopathy: further evidence for procoagulant pathogenesis. Arch Dermatol. 2006;142:1413-1418.
- Shornick JK, Nicholes BK, Bergstresser PR, et al. Idiopathic atrophie blanche. J Am Acad Dermatol. 1983;8:792-798.
- Feldaker M, Hines EA Jr, Kierland RR. Livedo reticularis with ulcerations. Circulation. 1956;13:196-216.
- Callen JP. Livedoid vasculopathy: what it is and how the patient should be evaluated and treated. Arch Dermatol. 2006;142:1481-1482.
- Copeman PW. Livedo reticularis. signs in the skin of disturbance of blood viscosity and of blood flow. Br J Dermatol. 1975;93:519-529.
- Goerge T. Livedoid vasculopathy. pathogenesis, diagnosis and treatment of cutaneous infarction [in German]. Hautarzt. 2011;62:627-634; quiz 635.
The Diagnosis: Livedoid Vasculopathy
Livedoid vasculopathy (LV) is a rare cutaneous disorder that most commonly affects the lower legs. It has an estimated incidence of 1 case per 100,000 per year and predominantly affects women.1 The disease pathogenesis is not fully understood but is thought to involve thrombosis and occlusion of dermal vessels resulting in tissue hypoxia.2 Both inherited and acquired thrombophilic conditions frequently are seen in patients with LV.3,4 Livedoid vasculopathy also has been described as idiopathic5 and is associated with immune complex deposition.6 However, the number of cases of idiopathic LV may be overestimated; as technological advancements to detect coagulation abnormalities improve, it is hypothesized that this entity will be identified less often.2,4
Livedoid vasculopathy has been described in the literature using the term PPURPLE (painful purpuric ulcers with reticular pattern of lower extremities).7 The triad of livedo racemosa, recurrent painful ulcerations, and residual healing with atrophie blanche characterizes the clinical manifestations of LV; however, all 3 characteristics do not need to appear simultaneously for a diagnosis to be made. The condition has a chronic course with spontaneous remissions and exacerbations. Episodic ulcerations occur, especially in the summertime, and heal slowly, leaving behind atrophic, porcelain white, stellate-shaped scars called atrophie blanche. Livedo racemosa also may be seen in Sneddon syndrome; however, these patients experience neurologic symptoms secondary to cerebrovascular occlusion. In contrast to livedo racemosa, acquired livedo reticularis represents a physiologic hypoperfusion pattern that occurs in response to cold exposure.8 A localized sharp pain, known as angina cutis, typically precedes the clinical symptom of painful ulcerations.9 Atrophie blanche once was thought to be specific to LV but has been seen in other diseases such as systemic lupus erythematosus and chronic venous insufficiency.2
The diagnosis of LV is based on identification of characteristic clinical features and skin biopsy. In almost all biopsy specimens, histopathology reveals fibrinoid occlusion of vessels in the superficial and mid dermis.4 Other findings may include epidermal necrosis and vessel wall hyalinization and infarction2 (Figure). Because LV is commonly misdiagnosed as vasculitis, the absence of hallmark features of vasculitis such as neutrophilic infiltrate of blood vessel walls and fibrinoid necrosis suggest the diagnosis. Extensive laboratory evaluation for inherited and acquired coagulation abnormalities should be performed.
Treatment of LV is difficult, as there is currently no consensus on optimal therapy. The mainstay of therapy is to reduce pain, prevent infection, and reduce ulceration and development of atrophie blanche. Underlying causes should be identified and appropriately treated. Because the primary pathogenesis of LV is considered to be a hypercoagulable state, first-line treatment often includes therapies to enhance blood flow and prevent thrombosis such as smoking cessation, antiplatelet therapy, and pentoxifylline. Vasodilating agents, anti-inflammatory agents, anticoagulation, and fibrinolytic therapy also have been used with varying degrees of success.7
The Diagnosis: Livedoid Vasculopathy
Livedoid vasculopathy (LV) is a rare cutaneous disorder that most commonly affects the lower legs. It has an estimated incidence of 1 case per 100,000 per year and predominantly affects women.1 The disease pathogenesis is not fully understood but is thought to involve thrombosis and occlusion of dermal vessels resulting in tissue hypoxia.2 Both inherited and acquired thrombophilic conditions frequently are seen in patients with LV.3,4 Livedoid vasculopathy also has been described as idiopathic5 and is associated with immune complex deposition.6 However, the number of cases of idiopathic LV may be overestimated; as technological advancements to detect coagulation abnormalities improve, it is hypothesized that this entity will be identified less often.2,4
Livedoid vasculopathy has been described in the literature using the term PPURPLE (painful purpuric ulcers with reticular pattern of lower extremities).7 The triad of livedo racemosa, recurrent painful ulcerations, and residual healing with atrophie blanche characterizes the clinical manifestations of LV; however, all 3 characteristics do not need to appear simultaneously for a diagnosis to be made. The condition has a chronic course with spontaneous remissions and exacerbations. Episodic ulcerations occur, especially in the summertime, and heal slowly, leaving behind atrophic, porcelain white, stellate-shaped scars called atrophie blanche. Livedo racemosa also may be seen in Sneddon syndrome; however, these patients experience neurologic symptoms secondary to cerebrovascular occlusion. In contrast to livedo racemosa, acquired livedo reticularis represents a physiologic hypoperfusion pattern that occurs in response to cold exposure.8 A localized sharp pain, known as angina cutis, typically precedes the clinical symptom of painful ulcerations.9 Atrophie blanche once was thought to be specific to LV but has been seen in other diseases such as systemic lupus erythematosus and chronic venous insufficiency.2
The diagnosis of LV is based on identification of characteristic clinical features and skin biopsy. In almost all biopsy specimens, histopathology reveals fibrinoid occlusion of vessels in the superficial and mid dermis.4 Other findings may include epidermal necrosis and vessel wall hyalinization and infarction2 (Figure). Because LV is commonly misdiagnosed as vasculitis, the absence of hallmark features of vasculitis such as neutrophilic infiltrate of blood vessel walls and fibrinoid necrosis suggest the diagnosis. Extensive laboratory evaluation for inherited and acquired coagulation abnormalities should be performed.
Treatment of LV is difficult, as there is currently no consensus on optimal therapy. The mainstay of therapy is to reduce pain, prevent infection, and reduce ulceration and development of atrophie blanche. Underlying causes should be identified and appropriately treated. Because the primary pathogenesis of LV is considered to be a hypercoagulable state, first-line treatment often includes therapies to enhance blood flow and prevent thrombosis such as smoking cessation, antiplatelet therapy, and pentoxifylline. Vasodilating agents, anti-inflammatory agents, anticoagulation, and fibrinolytic therapy also have been used with varying degrees of success.7
- Fritsch P, Zelger B. Livedo vasculitis [in German]. Hautarzt. 1995;46:215-224; quiz 222-223.
- Kerk N, Goerge T. Livedoid vasculopathy—a thrombotic disease. Vasa. 2013;42:317-322.
- Stevanovic DV. Atrophie blanche. a sign of dermal blood occlusion. Arch Dermatol. 1974;109:858-862.
- Hairston BR, Davis MD, Pittelkow MR, et al. Livedoid vasculopathy: further evidence for procoagulant pathogenesis. Arch Dermatol. 2006;142:1413-1418.
- Shornick JK, Nicholes BK, Bergstresser PR, et al. Idiopathic atrophie blanche. J Am Acad Dermatol. 1983;8:792-798.
- Feldaker M, Hines EA Jr, Kierland RR. Livedo reticularis with ulcerations. Circulation. 1956;13:196-216.
- Callen JP. Livedoid vasculopathy: what it is and how the patient should be evaluated and treated. Arch Dermatol. 2006;142:1481-1482.
- Copeman PW. Livedo reticularis. signs in the skin of disturbance of blood viscosity and of blood flow. Br J Dermatol. 1975;93:519-529.
- Goerge T. Livedoid vasculopathy. pathogenesis, diagnosis and treatment of cutaneous infarction [in German]. Hautarzt. 2011;62:627-634; quiz 635.
- Fritsch P, Zelger B. Livedo vasculitis [in German]. Hautarzt. 1995;46:215-224; quiz 222-223.
- Kerk N, Goerge T. Livedoid vasculopathy—a thrombotic disease. Vasa. 2013;42:317-322.
- Stevanovic DV. Atrophie blanche. a sign of dermal blood occlusion. Arch Dermatol. 1974;109:858-862.
- Hairston BR, Davis MD, Pittelkow MR, et al. Livedoid vasculopathy: further evidence for procoagulant pathogenesis. Arch Dermatol. 2006;142:1413-1418.
- Shornick JK, Nicholes BK, Bergstresser PR, et al. Idiopathic atrophie blanche. J Am Acad Dermatol. 1983;8:792-798.
- Feldaker M, Hines EA Jr, Kierland RR. Livedo reticularis with ulcerations. Circulation. 1956;13:196-216.
- Callen JP. Livedoid vasculopathy: what it is and how the patient should be evaluated and treated. Arch Dermatol. 2006;142:1481-1482.
- Copeman PW. Livedo reticularis. signs in the skin of disturbance of blood viscosity and of blood flow. Br J Dermatol. 1975;93:519-529.
- Goerge T. Livedoid vasculopathy. pathogenesis, diagnosis and treatment of cutaneous infarction [in German]. Hautarzt. 2011;62:627-634; quiz 635.
A 58-year-old woman presented in the summertime with skin discoloration of the bilateral lower legs and painful ulcerations above the medial and lateral malleoli of 15 years’ duration. She denied any recent trauma to the area or change in skin lesion appearance with cold exposure. Extensive laboratory evaluation for inherited and acquired coagulation abnormalities was negative. A punch biopsy specimen obtained from the left anterior lower leg revealed vascular thrombi with extravasated erythrocytes and a sparse perivascular inflammatory cell infiltrate.
Subungual Onycholemmal Cyst of the Toenail Mimicking Subungual Melanoma
Case Report
A 23-year-old woman presented with a horizontal split along the midline of the right great toenail associated with some tenderness of 2 to 3 months’ duration. Approximately 5 years prior, she noticed a bluish-colored area under the nail that had been steadily increasing in size. She denied a history of trauma, drainage, or bleeding. There was no history of other nail abnormalities. Her medications and personal, family, and social history were noncontributory.
Physical examination of the right great toenail revealed a horizontal split of the nail plate with a bluish hue visible under the nail plate (Figure 1A). The remaining toenails and fingernails were normal. A punch biopsy of the nail bed was performed with a presumptive clinical diagnosis of subungual melanoma versus melanocytic nevus versus cyst (Figure 1B). Nail plate avulsion revealed a blackened nail bed dotted with areas of bluish color and a red friable nodule present focally. Upon further inspection, extension was apparent into the distal matrix.
Histopathologic examination revealed a cystic structure with an epithelial lining mostly reminiscent of an isthmus catagen cyst admixed with the presence of both an intermittent focal granular layer and an eosinophilic cuticle surrounding pink laminated keratin, most consistent with a diagnosis of subungual onycholemmal cyst (SOC)(Figure 2). A reexcision was performed with removal of half of the nail bed, including a portion of the distal matrix extending inferiorly to the bone. Variably sized, epithelium-lined, keratin-filled cystic structures emanated from the nail bed epithelium. There were foci of hemorrhage and granulation tissue secondary to cyst rupture (Figure 3). The defect healed by secondary intention. No clinical evidence of recurrence was seen at 6-month follow-up.

Subungual onycholemmal cysts, also known as subungual epidermoid cysts or subungual epidermoid inclusions, are rare and distinctive nail abnormalities occurring in the dermis of the nail bed. We present a case of an SOC in a toenail mimicking subungual malignant melanoma.
Originally described by Samman1 in 1959, SOCs were attributed to trauma to the nail with resultant implantation of the epidermis into the deeper tissue. Lewin2,3 examined 90 postmortem fingernail and nail bed samples and found 8 subungual epidermoid cysts associated with clubbing of the fingernails. He postulated that the early pathogenesis of clubbing involved dermal fibroblast proliferation in the nail bed, leading to sequestration of nail bed epithelium into the dermis with resultant cyst formation. Microscopic subungual cysts also were identified in normal-appearing nails without evidence of trauma, thought to have arisen from the tips of the nail bed rete ridges by a process of bulbous proliferation rather than sequestration. These findings in normal nails suggest that SOCs may represent a more common entity than previously recognized.
It is imperative to recognize the presence of nail unit tumors early because of the risk for permanent nail plate dystrophy and the possibility of a malignant tumor.4,5 Subungual onycholemmal cysts may present with a wide spectrum of clinical findings including marked subungual hyperkeratosis, onychodystrophy, ridging, nail bed pigmentation, clubbing, thickening, or less often a normal-appearing nail. Based on reported cases, several trends are evident. Although nail dystrophy is most often asymptomatic, pain is not uncommon.5,6 It most commonly involves single digits, predominantly thumbs and great toenails.7,8 This predilection suggests that trauma or other local factors may be involved in its pathogenesis. Of note, trauma to the nail may occur years before the development of the lesions or it may not be recalled at all.
Diagnosis requires a degree of clinical suspicion and a nail bed biopsy with partial or total nail plate avulsion to visualize the pathologic portion of the nail bed. Because surgical intervention may lead to the implantation of epithelium, recurrences after nail biopsy or excision may occur.
In contrast to epidermal inclusion cysts arising in the skin, most SOCs do not have a granular layer.9 Hair and nails represent analogous differentiation products of the ectoderm. The nail matrix is homologous to portions of the hair matrix, while the nail bed epithelium is comparable to the outer root sheath of the hair follicle.7 Subungual onycholemmal cysts originate from the nail bed epithelium, which keratinizes in the absence of a granular layer, similar to the follicular isthmus outer root sheath. Thus, SOCs are comparable to the outer root sheath–derived isthmus-catagen cysts because of their abrupt central keratinization.8
Subungual onycholemmal cysts also must be distinguished from slowly growing malignant tumors of the nail bed epithelium, referred to as onycholemmal carcinomas by Alessi et al.10 This entity characteristically presents in elderly patients as a slowly growing, circumscribed, subungual discoloration that may ulcerate, destroying the nail apparatus and penetrating the phalangeal bone. On histopathology, it is characterized by small cysts filled with eosinophilic keratin devoid of a granular layer and lined by atypical squamous epithelium accompanied by solid nests and strands of atypical keratinocytes within the dermis.11 When a cystic component and clear cells predominate, the designation of malignant proliferating onycholemmal cyst has been applied. Its infiltrative growth pattern with destruction of the underlying bone makes it an important entity to exclude when considering the differential diagnosis of tumors of the nail bed.
Subungual melanomas comprise only 1% to 3% of malignant melanomas and 85% are initially misdiagnosed due to their rarity and nonspecific variable presentation. Aside from clinical evidence of Hutchinson sign in the early stages in almost all cases, accurate diagnosis of subungual melanoma and differentiation from SOCs relies on histopathology. A biopsy is necessary to make the diagnosis, but even microscopic findings may be nonspecific during the early stages.
Conclusion
We report a case of a 23-year-old woman with horizontal ridging and tenderness of the right great toenail associated with pigmentation of 5 years’ duration due to an SOC. The etiology of these subungual cysts, with or without nail abnormalities, still remains unclear. Its predilection for the thumbs and great toenails suggests that trauma or other local factors may be involved in its pathogenesis. Because of the rarity of this entity, there are no guidelines for surgical treatment. Subungual onycholemmal cysts may be an underrecognized and more common entity that must be considered when discussing tumors of the nail unit.
- Samman PD. The human toe nail. its genesis and blood supply. Br J Dermatol. 1959;71:296-302.
- Lewin K. The normal fingernail. Br J Dermatol. 1965;77:421-430.
- Lewin K. Subungual epidermoid inclusions. Br J Dermatol. 1969;81:671-675.
- Dominguez-Cherit J, Chanussot-Deprez C, Maria-Sarti H, et al. Nail unit tumors: a study of 234 patients in the dermatology department of the “Dr. Manuel Gea González” General Hospital in Mexico City. Dermatol Surg. 2008;34:1363-1371.
- Sáez-de-Ocariz MM, Domínguez-Cherit J, García-Corona C. Subungual epidermoid cysts. Int J Dermatol. 2001;40:524-526.
- Molly DO, Herbert K. Subungual epidermoid cyst. J Hand Surg Br. 2006;31:345.
- Telang GH, Jellinek N. Multiple calcified subungual epidermoid inclusions. J Am Acad Dermatol. 2007;56:336-339.
- Fanti PA, Tosti A. Subungual epidermoid inclusions: report of 8 cases. Dermatologica. 1989;178:209-212.
- Takiyoshi N, Nakano H, Matsuzaki T, et al. An eclipse in the subungual space: a diagnostic sign for a subungual epidermal cyst? Br J Dermatol. 2009;161:962-963.
- Alessi E, Coggi A, Gianotti R, et al. Onycholemmal carcinoma. Am J Dermatopathol. 2004;26:397-402.
- Inaoki M, Makino E, Adachi M, et al. Onycholemmal carcinoma. J Cutan Pathol. 2006;33:577-580.
Case Report
A 23-year-old woman presented with a horizontal split along the midline of the right great toenail associated with some tenderness of 2 to 3 months’ duration. Approximately 5 years prior, she noticed a bluish-colored area under the nail that had been steadily increasing in size. She denied a history of trauma, drainage, or bleeding. There was no history of other nail abnormalities. Her medications and personal, family, and social history were noncontributory.
Physical examination of the right great toenail revealed a horizontal split of the nail plate with a bluish hue visible under the nail plate (Figure 1A). The remaining toenails and fingernails were normal. A punch biopsy of the nail bed was performed with a presumptive clinical diagnosis of subungual melanoma versus melanocytic nevus versus cyst (Figure 1B). Nail plate avulsion revealed a blackened nail bed dotted with areas of bluish color and a red friable nodule present focally. Upon further inspection, extension was apparent into the distal matrix.
Histopathologic examination revealed a cystic structure with an epithelial lining mostly reminiscent of an isthmus catagen cyst admixed with the presence of both an intermittent focal granular layer and an eosinophilic cuticle surrounding pink laminated keratin, most consistent with a diagnosis of subungual onycholemmal cyst (SOC)(Figure 2). A reexcision was performed with removal of half of the nail bed, including a portion of the distal matrix extending inferiorly to the bone. Variably sized, epithelium-lined, keratin-filled cystic structures emanated from the nail bed epithelium. There were foci of hemorrhage and granulation tissue secondary to cyst rupture (Figure 3). The defect healed by secondary intention. No clinical evidence of recurrence was seen at 6-month follow-up.
Subungual onycholemmal cysts, also known as subungual epidermoid cysts or subungual epidermoid inclusions, are rare and distinctive nail abnormalities occurring in the dermis of the nail bed. We present a case of an SOC in a toenail mimicking subungual malignant melanoma.
Originally described by Samman1 in 1959, SOCs were attributed to trauma to the nail with resultant implantation of the epidermis into the deeper tissue. Lewin2,3 examined 90 postmortem fingernail and nail bed samples and found 8 subungual epidermoid cysts associated with clubbing of the fingernails. He postulated that the early pathogenesis of clubbing involved dermal fibroblast proliferation in the nail bed, leading to sequestration of nail bed epithelium into the dermis with resultant cyst formation. Microscopic subungual cysts also were identified in normal-appearing nails without evidence of trauma, thought to have arisen from the tips of the nail bed rete ridges by a process of bulbous proliferation rather than sequestration. These findings in normal nails suggest that SOCs may represent a more common entity than previously recognized.
It is imperative to recognize the presence of nail unit tumors early because of the risk for permanent nail plate dystrophy and the possibility of a malignant tumor.4,5 Subungual onycholemmal cysts may present with a wide spectrum of clinical findings including marked subungual hyperkeratosis, onychodystrophy, ridging, nail bed pigmentation, clubbing, thickening, or less often a normal-appearing nail. Based on reported cases, several trends are evident. Although nail dystrophy is most often asymptomatic, pain is not uncommon.5,6 It most commonly involves single digits, predominantly thumbs and great toenails.7,8 This predilection suggests that trauma or other local factors may be involved in its pathogenesis. Of note, trauma to the nail may occur years before the development of the lesions or it may not be recalled at all.
Diagnosis requires a degree of clinical suspicion and a nail bed biopsy with partial or total nail plate avulsion to visualize the pathologic portion of the nail bed. Because surgical intervention may lead to the implantation of epithelium, recurrences after nail biopsy or excision may occur.
In contrast to epidermal inclusion cysts arising in the skin, most SOCs do not have a granular layer.9 Hair and nails represent analogous differentiation products of the ectoderm. The nail matrix is homologous to portions of the hair matrix, while the nail bed epithelium is comparable to the outer root sheath of the hair follicle.7 Subungual onycholemmal cysts originate from the nail bed epithelium, which keratinizes in the absence of a granular layer, similar to the follicular isthmus outer root sheath. Thus, SOCs are comparable to the outer root sheath–derived isthmus-catagen cysts because of their abrupt central keratinization.8
Subungual onycholemmal cysts also must be distinguished from slowly growing malignant tumors of the nail bed epithelium, referred to as onycholemmal carcinomas by Alessi et al.10 This entity characteristically presents in elderly patients as a slowly growing, circumscribed, subungual discoloration that may ulcerate, destroying the nail apparatus and penetrating the phalangeal bone. On histopathology, it is characterized by small cysts filled with eosinophilic keratin devoid of a granular layer and lined by atypical squamous epithelium accompanied by solid nests and strands of atypical keratinocytes within the dermis.11 When a cystic component and clear cells predominate, the designation of malignant proliferating onycholemmal cyst has been applied. Its infiltrative growth pattern with destruction of the underlying bone makes it an important entity to exclude when considering the differential diagnosis of tumors of the nail bed.
Subungual melanomas comprise only 1% to 3% of malignant melanomas and 85% are initially misdiagnosed due to their rarity and nonspecific variable presentation. Aside from clinical evidence of Hutchinson sign in the early stages in almost all cases, accurate diagnosis of subungual melanoma and differentiation from SOCs relies on histopathology. A biopsy is necessary to make the diagnosis, but even microscopic findings may be nonspecific during the early stages.
Conclusion
We report a case of a 23-year-old woman with horizontal ridging and tenderness of the right great toenail associated with pigmentation of 5 years’ duration due to an SOC. The etiology of these subungual cysts, with or without nail abnormalities, still remains unclear. Its predilection for the thumbs and great toenails suggests that trauma or other local factors may be involved in its pathogenesis. Because of the rarity of this entity, there are no guidelines for surgical treatment. Subungual onycholemmal cysts may be an underrecognized and more common entity that must be considered when discussing tumors of the nail unit.
Case Report
A 23-year-old woman presented with a horizontal split along the midline of the right great toenail associated with some tenderness of 2 to 3 months’ duration. Approximately 5 years prior, she noticed a bluish-colored area under the nail that had been steadily increasing in size. She denied a history of trauma, drainage, or bleeding. There was no history of other nail abnormalities. Her medications and personal, family, and social history were noncontributory.
Physical examination of the right great toenail revealed a horizontal split of the nail plate with a bluish hue visible under the nail plate (Figure 1A). The remaining toenails and fingernails were normal. A punch biopsy of the nail bed was performed with a presumptive clinical diagnosis of subungual melanoma versus melanocytic nevus versus cyst (Figure 1B). Nail plate avulsion revealed a blackened nail bed dotted with areas of bluish color and a red friable nodule present focally. Upon further inspection, extension was apparent into the distal matrix.
Histopathologic examination revealed a cystic structure with an epithelial lining mostly reminiscent of an isthmus catagen cyst admixed with the presence of both an intermittent focal granular layer and an eosinophilic cuticle surrounding pink laminated keratin, most consistent with a diagnosis of subungual onycholemmal cyst (SOC)(Figure 2). A reexcision was performed with removal of half of the nail bed, including a portion of the distal matrix extending inferiorly to the bone. Variably sized, epithelium-lined, keratin-filled cystic structures emanated from the nail bed epithelium. There were foci of hemorrhage and granulation tissue secondary to cyst rupture (Figure 3). The defect healed by secondary intention. No clinical evidence of recurrence was seen at 6-month follow-up.
Subungual onycholemmal cysts, also known as subungual epidermoid cysts or subungual epidermoid inclusions, are rare and distinctive nail abnormalities occurring in the dermis of the nail bed. We present a case of an SOC in a toenail mimicking subungual malignant melanoma.
Originally described by Samman1 in 1959, SOCs were attributed to trauma to the nail with resultant implantation of the epidermis into the deeper tissue. Lewin2,3 examined 90 postmortem fingernail and nail bed samples and found 8 subungual epidermoid cysts associated with clubbing of the fingernails. He postulated that the early pathogenesis of clubbing involved dermal fibroblast proliferation in the nail bed, leading to sequestration of nail bed epithelium into the dermis with resultant cyst formation. Microscopic subungual cysts also were identified in normal-appearing nails without evidence of trauma, thought to have arisen from the tips of the nail bed rete ridges by a process of bulbous proliferation rather than sequestration. These findings in normal nails suggest that SOCs may represent a more common entity than previously recognized.
It is imperative to recognize the presence of nail unit tumors early because of the risk for permanent nail plate dystrophy and the possibility of a malignant tumor.4,5 Subungual onycholemmal cysts may present with a wide spectrum of clinical findings including marked subungual hyperkeratosis, onychodystrophy, ridging, nail bed pigmentation, clubbing, thickening, or less often a normal-appearing nail. Based on reported cases, several trends are evident. Although nail dystrophy is most often asymptomatic, pain is not uncommon.5,6 It most commonly involves single digits, predominantly thumbs and great toenails.7,8 This predilection suggests that trauma or other local factors may be involved in its pathogenesis. Of note, trauma to the nail may occur years before the development of the lesions or it may not be recalled at all.
Diagnosis requires a degree of clinical suspicion and a nail bed biopsy with partial or total nail plate avulsion to visualize the pathologic portion of the nail bed. Because surgical intervention may lead to the implantation of epithelium, recurrences after nail biopsy or excision may occur.
In contrast to epidermal inclusion cysts arising in the skin, most SOCs do not have a granular layer.9 Hair and nails represent analogous differentiation products of the ectoderm. The nail matrix is homologous to portions of the hair matrix, while the nail bed epithelium is comparable to the outer root sheath of the hair follicle.7 Subungual onycholemmal cysts originate from the nail bed epithelium, which keratinizes in the absence of a granular layer, similar to the follicular isthmus outer root sheath. Thus, SOCs are comparable to the outer root sheath–derived isthmus-catagen cysts because of their abrupt central keratinization.8
Subungual onycholemmal cysts also must be distinguished from slowly growing malignant tumors of the nail bed epithelium, referred to as onycholemmal carcinomas by Alessi et al.10 This entity characteristically presents in elderly patients as a slowly growing, circumscribed, subungual discoloration that may ulcerate, destroying the nail apparatus and penetrating the phalangeal bone. On histopathology, it is characterized by small cysts filled with eosinophilic keratin devoid of a granular layer and lined by atypical squamous epithelium accompanied by solid nests and strands of atypical keratinocytes within the dermis.11 When a cystic component and clear cells predominate, the designation of malignant proliferating onycholemmal cyst has been applied. Its infiltrative growth pattern with destruction of the underlying bone makes it an important entity to exclude when considering the differential diagnosis of tumors of the nail bed.
Subungual melanomas comprise only 1% to 3% of malignant melanomas and 85% are initially misdiagnosed due to their rarity and nonspecific variable presentation. Aside from clinical evidence of Hutchinson sign in the early stages in almost all cases, accurate diagnosis of subungual melanoma and differentiation from SOCs relies on histopathology. A biopsy is necessary to make the diagnosis, but even microscopic findings may be nonspecific during the early stages.
Conclusion
We report a case of a 23-year-old woman with horizontal ridging and tenderness of the right great toenail associated with pigmentation of 5 years’ duration due to an SOC. The etiology of these subungual cysts, with or without nail abnormalities, still remains unclear. Its predilection for the thumbs and great toenails suggests that trauma or other local factors may be involved in its pathogenesis. Because of the rarity of this entity, there are no guidelines for surgical treatment. Subungual onycholemmal cysts may be an underrecognized and more common entity that must be considered when discussing tumors of the nail unit.
- Samman PD. The human toe nail. its genesis and blood supply. Br J Dermatol. 1959;71:296-302.
- Lewin K. The normal fingernail. Br J Dermatol. 1965;77:421-430.
- Lewin K. Subungual epidermoid inclusions. Br J Dermatol. 1969;81:671-675.
- Dominguez-Cherit J, Chanussot-Deprez C, Maria-Sarti H, et al. Nail unit tumors: a study of 234 patients in the dermatology department of the “Dr. Manuel Gea González” General Hospital in Mexico City. Dermatol Surg. 2008;34:1363-1371.
- Sáez-de-Ocariz MM, Domínguez-Cherit J, García-Corona C. Subungual epidermoid cysts. Int J Dermatol. 2001;40:524-526.
- Molly DO, Herbert K. Subungual epidermoid cyst. J Hand Surg Br. 2006;31:345.
- Telang GH, Jellinek N. Multiple calcified subungual epidermoid inclusions. J Am Acad Dermatol. 2007;56:336-339.
- Fanti PA, Tosti A. Subungual epidermoid inclusions: report of 8 cases. Dermatologica. 1989;178:209-212.
- Takiyoshi N, Nakano H, Matsuzaki T, et al. An eclipse in the subungual space: a diagnostic sign for a subungual epidermal cyst? Br J Dermatol. 2009;161:962-963.
- Alessi E, Coggi A, Gianotti R, et al. Onycholemmal carcinoma. Am J Dermatopathol. 2004;26:397-402.
- Inaoki M, Makino E, Adachi M, et al. Onycholemmal carcinoma. J Cutan Pathol. 2006;33:577-580.
- Samman PD. The human toe nail. its genesis and blood supply. Br J Dermatol. 1959;71:296-302.
- Lewin K. The normal fingernail. Br J Dermatol. 1965;77:421-430.
- Lewin K. Subungual epidermoid inclusions. Br J Dermatol. 1969;81:671-675.
- Dominguez-Cherit J, Chanussot-Deprez C, Maria-Sarti H, et al. Nail unit tumors: a study of 234 patients in the dermatology department of the “Dr. Manuel Gea González” General Hospital in Mexico City. Dermatol Surg. 2008;34:1363-1371.
- Sáez-de-Ocariz MM, Domínguez-Cherit J, García-Corona C. Subungual epidermoid cysts. Int J Dermatol. 2001;40:524-526.
- Molly DO, Herbert K. Subungual epidermoid cyst. J Hand Surg Br. 2006;31:345.
- Telang GH, Jellinek N. Multiple calcified subungual epidermoid inclusions. J Am Acad Dermatol. 2007;56:336-339.
- Fanti PA, Tosti A. Subungual epidermoid inclusions: report of 8 cases. Dermatologica. 1989;178:209-212.
- Takiyoshi N, Nakano H, Matsuzaki T, et al. An eclipse in the subungual space: a diagnostic sign for a subungual epidermal cyst? Br J Dermatol. 2009;161:962-963.
- Alessi E, Coggi A, Gianotti R, et al. Onycholemmal carcinoma. Am J Dermatopathol. 2004;26:397-402.
- Inaoki M, Makino E, Adachi M, et al. Onycholemmal carcinoma. J Cutan Pathol. 2006;33:577-580.
Practice Points
- Trauma to the nail may occur years before the development of subungual onycholemmal cysts or it may not be recalled at all.
- Diagnosis requires a degree of clinical suspicion and a nail bed biopsy.
- Subungual onycholemmal cysts must be distinguished from slowly growing malignant tumors of the nail bed epithelium.