User login
An infant with a tender bump on her ear
A biopsy of the lesion was performed that showed a well-defined nodulocystic tumor composed of nests of basaloid cells that are undergoing trichilemmal keratinization. Shadow cells are seen as well as small areas of calcification. There is also a histiocytic infiltrate with multinucleated giant cells. The histologic diagnosis is of a pilomatrixoma.
Pilomatrixoma, also known as calcifying epithelioma of Malherbe, was first described in 1880, as a tumor of sebaceous gland origin. Later, in 1961, Robert Forbis Jr, MD, and Elson B. Helwig, MD, coined the term pilomatrixoma to describe the hair follicle matrix as the source of the tumor. Pilomatrixomas are commonly seen in the pediatric population, usually in children between 8 and 13 years of age. Our patient is one of the youngest described. The lesions are commonly seen on the face and neck in about 70% of the cases followed by the upper extremities, back, and legs. Clinically, the lesions appear as a firm dermal papule or nodule, which moves freely and may have associated erythema on the skin surface or a blueish gray hue on the underlying skin.

Most pilomatrixomas that have been studied have shown a mutation in Exon 3 of the beta-catenin gene (CTNNB1). The beta-catenin molecule is a subunit of the cadherin protein, which is part of an important pathway in the terminal hair follicle differentiation. Beta-catenin also plays an important role in the Wnt pathway, which regulates cell fate as well as early embryonic patterning. Beta-catenin is responsible for forming adhesion junctions among cells. There have also been immunohistochemical studies that have shown a BCL2 proto-oncogene overexpression to pilomatrixoma.
There are several genetic syndromes that have been associated with the presence of pilomatrixomas: Turner syndrome (XO chromosome abnormality associated with short stature and cardiac defects), Gardner syndrome (polyposis coli and colon and rectal cancer), myotonic dystrophy, Rubinstein-Taybi syndrome (characterized by broad thumbs and toes, short stature, distinctive facial features, and varying degrees of intellectual disability), and trisomy 9. On physical examination our patient didn’t present with any of the typical features or history that could suggest any of these syndromes. A close follow-up and evaluation by a geneticist was recommended because after the initial visit she developed a second lesion on the forehead.
The differential diagnosis for this lesion includes other cysts that may occur on the ear such as epidermal inclusion cyst or dermoid cysts, though these lesions do not tend to be as firm as pilomatrixomas are, which can help with the diagnosis. Dermoid cysts are made of dermal and epidermal components. They are usually present at birth and are commonly seen on the scalp and the periorbital face.
Keloids are rubbery nodules of scar tissue that can form on sites of trauma, and although the lesion occurred after she had her ears pierced, the consistency and rapid growth of the lesion as well as the pathological description made this benign fibrous growth less likely.
When pilomatrixomas are inflamed they can be confused with vascular growths: in this particular case, a hemangioma or another vascular tumor such as a tufted angioma or kaposiform hemangioendothelioma. An ultrasound of the lesion could have helped in the differential diagnosis of the lesion.
Pilomatrixomas can grow significantly and in some cases get inflamed or infected. Surgical management of pilomatrixomas is often required because the lesions do not regress spontaneously.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Forbis R Jr and Helwig EB. Arch Dermatol 1961;83:606-18.
Schwarz Y et al. Int J Pediatr Otorhinolaryngol. 2016 Jun;85:148-53.
A biopsy of the lesion was performed that showed a well-defined nodulocystic tumor composed of nests of basaloid cells that are undergoing trichilemmal keratinization. Shadow cells are seen as well as small areas of calcification. There is also a histiocytic infiltrate with multinucleated giant cells. The histologic diagnosis is of a pilomatrixoma.
Pilomatrixoma, also known as calcifying epithelioma of Malherbe, was first described in 1880, as a tumor of sebaceous gland origin. Later, in 1961, Robert Forbis Jr, MD, and Elson B. Helwig, MD, coined the term pilomatrixoma to describe the hair follicle matrix as the source of the tumor. Pilomatrixomas are commonly seen in the pediatric population, usually in children between 8 and 13 years of age. Our patient is one of the youngest described. The lesions are commonly seen on the face and neck in about 70% of the cases followed by the upper extremities, back, and legs. Clinically, the lesions appear as a firm dermal papule or nodule, which moves freely and may have associated erythema on the skin surface or a blueish gray hue on the underlying skin.
Most pilomatrixomas that have been studied have shown a mutation in Exon 3 of the beta-catenin gene (CTNNB1). The beta-catenin molecule is a subunit of the cadherin protein, which is part of an important pathway in the terminal hair follicle differentiation. Beta-catenin also plays an important role in the Wnt pathway, which regulates cell fate as well as early embryonic patterning. Beta-catenin is responsible for forming adhesion junctions among cells. There have also been immunohistochemical studies that have shown a BCL2 proto-oncogene overexpression to pilomatrixoma.
There are several genetic syndromes that have been associated with the presence of pilomatrixomas: Turner syndrome (XO chromosome abnormality associated with short stature and cardiac defects), Gardner syndrome (polyposis coli and colon and rectal cancer), myotonic dystrophy, Rubinstein-Taybi syndrome (characterized by broad thumbs and toes, short stature, distinctive facial features, and varying degrees of intellectual disability), and trisomy 9. On physical examination our patient didn’t present with any of the typical features or history that could suggest any of these syndromes. A close follow-up and evaluation by a geneticist was recommended because after the initial visit she developed a second lesion on the forehead.
The differential diagnosis for this lesion includes other cysts that may occur on the ear such as epidermal inclusion cyst or dermoid cysts, though these lesions do not tend to be as firm as pilomatrixomas are, which can help with the diagnosis. Dermoid cysts are made of dermal and epidermal components. They are usually present at birth and are commonly seen on the scalp and the periorbital face.
Keloids are rubbery nodules of scar tissue that can form on sites of trauma, and although the lesion occurred after she had her ears pierced, the consistency and rapid growth of the lesion as well as the pathological description made this benign fibrous growth less likely.
When pilomatrixomas are inflamed they can be confused with vascular growths: in this particular case, a hemangioma or another vascular tumor such as a tufted angioma or kaposiform hemangioendothelioma. An ultrasound of the lesion could have helped in the differential diagnosis of the lesion.
Pilomatrixomas can grow significantly and in some cases get inflamed or infected. Surgical management of pilomatrixomas is often required because the lesions do not regress spontaneously.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Forbis R Jr and Helwig EB. Arch Dermatol 1961;83:606-18.
Schwarz Y et al. Int J Pediatr Otorhinolaryngol. 2016 Jun;85:148-53.
A biopsy of the lesion was performed that showed a well-defined nodulocystic tumor composed of nests of basaloid cells that are undergoing trichilemmal keratinization. Shadow cells are seen as well as small areas of calcification. There is also a histiocytic infiltrate with multinucleated giant cells. The histologic diagnosis is of a pilomatrixoma.
Pilomatrixoma, also known as calcifying epithelioma of Malherbe, was first described in 1880, as a tumor of sebaceous gland origin. Later, in 1961, Robert Forbis Jr, MD, and Elson B. Helwig, MD, coined the term pilomatrixoma to describe the hair follicle matrix as the source of the tumor. Pilomatrixomas are commonly seen in the pediatric population, usually in children between 8 and 13 years of age. Our patient is one of the youngest described. The lesions are commonly seen on the face and neck in about 70% of the cases followed by the upper extremities, back, and legs. Clinically, the lesions appear as a firm dermal papule or nodule, which moves freely and may have associated erythema on the skin surface or a blueish gray hue on the underlying skin.
Most pilomatrixomas that have been studied have shown a mutation in Exon 3 of the beta-catenin gene (CTNNB1). The beta-catenin molecule is a subunit of the cadherin protein, which is part of an important pathway in the terminal hair follicle differentiation. Beta-catenin also plays an important role in the Wnt pathway, which regulates cell fate as well as early embryonic patterning. Beta-catenin is responsible for forming adhesion junctions among cells. There have also been immunohistochemical studies that have shown a BCL2 proto-oncogene overexpression to pilomatrixoma.
There are several genetic syndromes that have been associated with the presence of pilomatrixomas: Turner syndrome (XO chromosome abnormality associated with short stature and cardiac defects), Gardner syndrome (polyposis coli and colon and rectal cancer), myotonic dystrophy, Rubinstein-Taybi syndrome (characterized by broad thumbs and toes, short stature, distinctive facial features, and varying degrees of intellectual disability), and trisomy 9. On physical examination our patient didn’t present with any of the typical features or history that could suggest any of these syndromes. A close follow-up and evaluation by a geneticist was recommended because after the initial visit she developed a second lesion on the forehead.
The differential diagnosis for this lesion includes other cysts that may occur on the ear such as epidermal inclusion cyst or dermoid cysts, though these lesions do not tend to be as firm as pilomatrixomas are, which can help with the diagnosis. Dermoid cysts are made of dermal and epidermal components. They are usually present at birth and are commonly seen on the scalp and the periorbital face.
Keloids are rubbery nodules of scar tissue that can form on sites of trauma, and although the lesion occurred after she had her ears pierced, the consistency and rapid growth of the lesion as well as the pathological description made this benign fibrous growth less likely.
When pilomatrixomas are inflamed they can be confused with vascular growths: in this particular case, a hemangioma or another vascular tumor such as a tufted angioma or kaposiform hemangioendothelioma. An ultrasound of the lesion could have helped in the differential diagnosis of the lesion.
Pilomatrixomas can grow significantly and in some cases get inflamed or infected. Surgical management of pilomatrixomas is often required because the lesions do not regress spontaneously.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Forbis R Jr and Helwig EB. Arch Dermatol 1961;83:606-18.
Schwarz Y et al. Int J Pediatr Otorhinolaryngol. 2016 Jun;85:148-53.
A 4-month-old female was referred to our clinic for evaluation of a bump on the right ear. The lesion was first noted at 2 months of age as a little pimple. She was evaluated by her pediatrician and was treated with topical and oral antibiotics without resolution of the lesion. The bump continued to grow and seemed tender to palpation, so she was referred to dermatology for evaluation.
She was born via normal vaginal delivery at 40 weeks. Her mother has no medical conditions and the pregnancy was uneventful. She has been growing and developing well. She takes vitamin D and is currently breast fed.
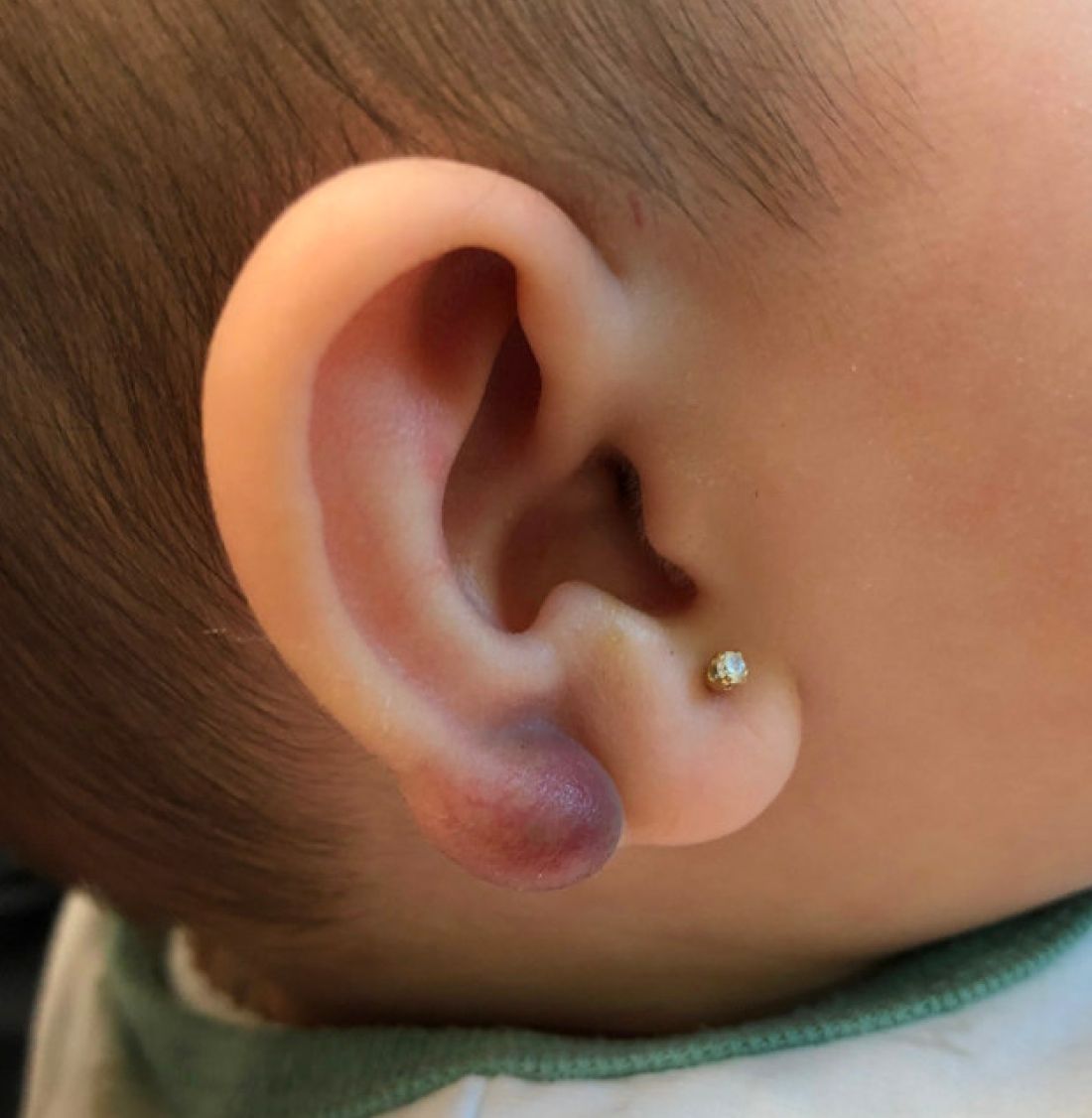
There have been no other family members with similar lesions. She had her ears pierced at a month of age without any complications.
On skin examination she has a firm red nodule on the right ear that appears slightly tender to touch. She has no other skin lesions of concern. She has normal muscle tone and there are no other abnormalities noted on the physical exam. She has no hepatomegaly, splenomegaly, or lymphadenopathy.
Nonblanching Rash on the Legs and Chest
The Diagnosis: Leukemia Cutis
Hematoxylin and eosin staining revealed an infiltration of monomorphic atypical myeloid cells with cleaved nuclei within the dermis, with a relatively uninvolved epidermis (Figure, A). The cells formed aggregates in single-file lines along dermal collagen bundles. Occasional Auer rods, which are crystal aggregates of the enzyme myeloperoxidase, a marker unique to cells of the myeloid lineage (Figure, B) were appreciated.
.")
Immunohistochemical staining for myeloperoxidase was weakly positive; however, flow cytometric evaluation of the bone marrow aspirate revealed that approximately 20% of all CD45+ cells were myeloid blasts. These findings confirmed the diagnosis of recurrent acute myeloid leukemia (AML). The diagnosis of AML can be confirmed with a bone marrow biopsy demonstrating more than 20% of the total cells in blast form as well as evidence that the cells are of myeloid origin, which can be inferred by the presence of Auer rods, positive myeloperoxidase staining, or immunophenotyping. In our patient, the Auer rods, myeloperoxidase staining, and atypical myeloid cells on skin biopsy, in conjunction with the bone marrow biopsy results, confirmed leukemia cutis.
Leukemia cutis is the infiltration of neoplastic proliferating leukocytes in the epidermis, dermis, or subcutis from a primary or more commonly metastatic malignancy. Leukemic cutaneous involvement is seen in up to 13% of leukemia patients and most commonly is seen in monocytic or myelomonocytic forms of AML.1 It may present anywhere on the body but mostly is found on the back, trunk, and head. It also may have a predilection for areas with a history of trauma or inflammation. The lesions most often are firm, erythematous to violaceous papules and nodules, though leukemia cutis can present with hemorrhagic ulcers, purpura, or other cutaneous manifestations of concomitant thrombocytopenia such as petechiae and ecchymoses.2 Involvement of the lower extremities mimicking venous stasis dermatitis has been described.3,4
Treatment of leukemia cutis requires targeting the underlying leukemia2 under the guidance of hematology and oncology as well as the use of chemotherapeutic agents.5 The presence of leukemia cutis is a poor prognostic sign, and a discussion regarding goals of care often is appropriate. Our patient initially responded to FLAG (fludarabine, cytarabine, filgrastim) chemotherapy induction and consolidation, which was followed by midostaurin maintenance. However, she ultimately regressed, requiring decitabine and gilteritinib treatment, and died 9 months later from the course of the disease.
Although typically asymptomatic and presenting on the lower limbs, capillaritis (also known as the pigmented purpuric dermatoses) consists of a set of cutaneous conditions that often are chronic and relapsing in nature, as opposed to our patient’s subacute presentation. These benign conditions have several distinct morphologies; some are characterized by pigmented macules or pinpoint red-brown petechiae that most often are found on the legs but also are seen on the trunk and upper extremities.6 Of the various clinical presentations of capillaritis, our patient’s skin findings may be most consistent with pigmented purpuric lichenoid dermatitis of Gougerot and Blum, in which purpuric red-brown papules coalesce into plaques, though her lesions were not raised. The other pigmented purpuric dermatoses can present with cayenne pepper–colored petechiae, golden-brown macules, pruritic purpuric patches, or red-brown annular patches,6 which were not seen in our patient.
Venous stasis dermatitis also favors the lower extremities7; however, it classically includes the medial malleolus and often presents with scaling and hyperpigmentation from hemosiderin deposition.8 It often is associated with pruritus, as opposed to the nonpruritic nonpainful lesions in leukemia cutis. Other signs of venous insufficiency also may be appreciated, including edema or varicose veins,7 which were not evident in our patient.
Leukocytoclastic vasculitis, a small vessel vasculitis, also appears as palpable or macular purpura, which classically is asymptomatic and erupts on the shins approximately 1 week after an inciting exposure,9 such as medications, pathogens, or autoimmune diseases. One of the least distinctive vasculitides is polyarteritis nodosa, a form of medium vessel vasculitis, which presents most often with palpable purpura or painful nodules on the lower extremities and may be accompanied by livedo reticularis or digital necrosis.9 Acute leukemia may be accompanied by inflammatory paraneoplastic conditions including vasculitis, which is thought to be due to leukemic cells infiltrating and damaging blood vessels.10
Pretibial myxedema is closely associated with Graves disease and shares some features seen in the presentation of our patient’s leukemia cutis. It is asymptomatic, classically affects the pretibial regions, and most commonly affects older adults and women.11,12 Pretibial myxedema presents with thick indurated plaques rather than patches. Our patient did not demonstrate ophthalmopathy, which nearly always precedes pretibial myxedema.12 The most common form of pretibial myxedema is nonpitting, though nodular, plaquelike, polypoid, and elephantiasic forms also exist.11 Pretibial myxedema classically favors the shins; however, it also can affect the ankles, dorsal aspects of the feet, and toes. The characteristic induration of the skin is believed to be the result of excess fibroblast production of glycosaminoglycans in the dermis and subcutis likely triggered by stimulation of fibroblast thyroid stimulating hormone receptors.11
- Bakst RL, Tallman MS, Douer D, et al. How I treat extramedullary acute myeloid leukemia. Blood. 2011;118:3785-3793.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Other lymphoproliferative and myeloproliferative diseases. In: Bolognia JL, Schaffer JV, Duncan KO, et al, eds. Dermatology Essentials. 2nd ed. Elsevier; 2014:973-977.
- Papadavid E, Panayiotides I, Katoulis A, et al. Stasis dermatitis-like leukaemic infiltration in a patient with myelodysplastic syndrome. Clin Exp Dermatol. 2008;33:298-300.
- Chang HY, Wong KM, Bosenberg M, et al. Myelogenous leukemia cutis resembling stasis dermatitis. J Am Acad Dermatol. 2003;49:128-129.
- Aguilera SB, Zarraga M, Rosen L. Leukemia cutis in a patient with acute myelogenous leukemia: a case report and review of the literature. Cutis. 2010;85:31-36.
- Kim DH, Seo SH, Ahn HH, et al. Characteristics and clinical manifestations of pigmented purpuric dermatosis. Ann Dermatol. 2015;27:404-410.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Other eczematous eruptions. In: Bolognia JL, Schaffer JV, Duncan KO, et al, eds. Dermatology Essentials. 2nd ed. Elsevier; 2014:103-108.
- Krooks JA, Weatherall AG. Leukemia cutis in acute myeloid leukemia signifies a poor prognosis. Cutis. 2018;102:266, 271-272.
- Wetter DA, Dutz JP, Shinkai K, et al. Cutaneous vasculitis. In: Bolognia JL, Schaffer JV, Lorenzo C, eds. Dermatology. 4th ed. Elsevier; 2018:409-439.
- Jones D, Dorfman DM, Barnhill RL, et al. Leukemic vasculitis: a feature of leukemia cutis in some patients. Am J Clin Pathol. 1997;107:637-642.
- Fatourechi V. Pretibial myxedema: pathophysiology and treatment options. Am J Clin Dermatol. 2005;6:295-309.
- Fatourechi V, Pajouhi M, Fransway AF. Dermopathy of Graves disease (pretibial myxedema). review of 150 cases. Medicine (Baltimore). 1994;73:1-7.
The Diagnosis: Leukemia Cutis
Hematoxylin and eosin staining revealed an infiltration of monomorphic atypical myeloid cells with cleaved nuclei within the dermis, with a relatively uninvolved epidermis (Figure, A). The cells formed aggregates in single-file lines along dermal collagen bundles. Occasional Auer rods, which are crystal aggregates of the enzyme myeloperoxidase, a marker unique to cells of the myeloid lineage (Figure, B) were appreciated.
Immunohistochemical staining for myeloperoxidase was weakly positive; however, flow cytometric evaluation of the bone marrow aspirate revealed that approximately 20% of all CD45+ cells were myeloid blasts. These findings confirmed the diagnosis of recurrent acute myeloid leukemia (AML). The diagnosis of AML can be confirmed with a bone marrow biopsy demonstrating more than 20% of the total cells in blast form as well as evidence that the cells are of myeloid origin, which can be inferred by the presence of Auer rods, positive myeloperoxidase staining, or immunophenotyping. In our patient, the Auer rods, myeloperoxidase staining, and atypical myeloid cells on skin biopsy, in conjunction with the bone marrow biopsy results, confirmed leukemia cutis.
Leukemia cutis is the infiltration of neoplastic proliferating leukocytes in the epidermis, dermis, or subcutis from a primary or more commonly metastatic malignancy. Leukemic cutaneous involvement is seen in up to 13% of leukemia patients and most commonly is seen in monocytic or myelomonocytic forms of AML.1 It may present anywhere on the body but mostly is found on the back, trunk, and head. It also may have a predilection for areas with a history of trauma or inflammation. The lesions most often are firm, erythematous to violaceous papules and nodules, though leukemia cutis can present with hemorrhagic ulcers, purpura, or other cutaneous manifestations of concomitant thrombocytopenia such as petechiae and ecchymoses.2 Involvement of the lower extremities mimicking venous stasis dermatitis has been described.3,4
Treatment of leukemia cutis requires targeting the underlying leukemia2 under the guidance of hematology and oncology as well as the use of chemotherapeutic agents.5 The presence of leukemia cutis is a poor prognostic sign, and a discussion regarding goals of care often is appropriate. Our patient initially responded to FLAG (fludarabine, cytarabine, filgrastim) chemotherapy induction and consolidation, which was followed by midostaurin maintenance. However, she ultimately regressed, requiring decitabine and gilteritinib treatment, and died 9 months later from the course of the disease.
Although typically asymptomatic and presenting on the lower limbs, capillaritis (also known as the pigmented purpuric dermatoses) consists of a set of cutaneous conditions that often are chronic and relapsing in nature, as opposed to our patient’s subacute presentation. These benign conditions have several distinct morphologies; some are characterized by pigmented macules or pinpoint red-brown petechiae that most often are found on the legs but also are seen on the trunk and upper extremities.6 Of the various clinical presentations of capillaritis, our patient’s skin findings may be most consistent with pigmented purpuric lichenoid dermatitis of Gougerot and Blum, in which purpuric red-brown papules coalesce into plaques, though her lesions were not raised. The other pigmented purpuric dermatoses can present with cayenne pepper–colored petechiae, golden-brown macules, pruritic purpuric patches, or red-brown annular patches,6 which were not seen in our patient.
Venous stasis dermatitis also favors the lower extremities7; however, it classically includes the medial malleolus and often presents with scaling and hyperpigmentation from hemosiderin deposition.8 It often is associated with pruritus, as opposed to the nonpruritic nonpainful lesions in leukemia cutis. Other signs of venous insufficiency also may be appreciated, including edema or varicose veins,7 which were not evident in our patient.
Leukocytoclastic vasculitis, a small vessel vasculitis, also appears as palpable or macular purpura, which classically is asymptomatic and erupts on the shins approximately 1 week after an inciting exposure,9 such as medications, pathogens, or autoimmune diseases. One of the least distinctive vasculitides is polyarteritis nodosa, a form of medium vessel vasculitis, which presents most often with palpable purpura or painful nodules on the lower extremities and may be accompanied by livedo reticularis or digital necrosis.9 Acute leukemia may be accompanied by inflammatory paraneoplastic conditions including vasculitis, which is thought to be due to leukemic cells infiltrating and damaging blood vessels.10
Pretibial myxedema is closely associated with Graves disease and shares some features seen in the presentation of our patient’s leukemia cutis. It is asymptomatic, classically affects the pretibial regions, and most commonly affects older adults and women.11,12 Pretibial myxedema presents with thick indurated plaques rather than patches. Our patient did not demonstrate ophthalmopathy, which nearly always precedes pretibial myxedema.12 The most common form of pretibial myxedema is nonpitting, though nodular, plaquelike, polypoid, and elephantiasic forms also exist.11 Pretibial myxedema classically favors the shins; however, it also can affect the ankles, dorsal aspects of the feet, and toes. The characteristic induration of the skin is believed to be the result of excess fibroblast production of glycosaminoglycans in the dermis and subcutis likely triggered by stimulation of fibroblast thyroid stimulating hormone receptors.11
The Diagnosis: Leukemia Cutis
Hematoxylin and eosin staining revealed an infiltration of monomorphic atypical myeloid cells with cleaved nuclei within the dermis, with a relatively uninvolved epidermis (Figure, A). The cells formed aggregates in single-file lines along dermal collagen bundles. Occasional Auer rods, which are crystal aggregates of the enzyme myeloperoxidase, a marker unique to cells of the myeloid lineage (Figure, B) were appreciated.
Immunohistochemical staining for myeloperoxidase was weakly positive; however, flow cytometric evaluation of the bone marrow aspirate revealed that approximately 20% of all CD45+ cells were myeloid blasts. These findings confirmed the diagnosis of recurrent acute myeloid leukemia (AML). The diagnosis of AML can be confirmed with a bone marrow biopsy demonstrating more than 20% of the total cells in blast form as well as evidence that the cells are of myeloid origin, which can be inferred by the presence of Auer rods, positive myeloperoxidase staining, or immunophenotyping. In our patient, the Auer rods, myeloperoxidase staining, and atypical myeloid cells on skin biopsy, in conjunction with the bone marrow biopsy results, confirmed leukemia cutis.
Leukemia cutis is the infiltration of neoplastic proliferating leukocytes in the epidermis, dermis, or subcutis from a primary or more commonly metastatic malignancy. Leukemic cutaneous involvement is seen in up to 13% of leukemia patients and most commonly is seen in monocytic or myelomonocytic forms of AML.1 It may present anywhere on the body but mostly is found on the back, trunk, and head. It also may have a predilection for areas with a history of trauma or inflammation. The lesions most often are firm, erythematous to violaceous papules and nodules, though leukemia cutis can present with hemorrhagic ulcers, purpura, or other cutaneous manifestations of concomitant thrombocytopenia such as petechiae and ecchymoses.2 Involvement of the lower extremities mimicking venous stasis dermatitis has been described.3,4
Treatment of leukemia cutis requires targeting the underlying leukemia2 under the guidance of hematology and oncology as well as the use of chemotherapeutic agents.5 The presence of leukemia cutis is a poor prognostic sign, and a discussion regarding goals of care often is appropriate. Our patient initially responded to FLAG (fludarabine, cytarabine, filgrastim) chemotherapy induction and consolidation, which was followed by midostaurin maintenance. However, she ultimately regressed, requiring decitabine and gilteritinib treatment, and died 9 months later from the course of the disease.
Although typically asymptomatic and presenting on the lower limbs, capillaritis (also known as the pigmented purpuric dermatoses) consists of a set of cutaneous conditions that often are chronic and relapsing in nature, as opposed to our patient’s subacute presentation. These benign conditions have several distinct morphologies; some are characterized by pigmented macules or pinpoint red-brown petechiae that most often are found on the legs but also are seen on the trunk and upper extremities.6 Of the various clinical presentations of capillaritis, our patient’s skin findings may be most consistent with pigmented purpuric lichenoid dermatitis of Gougerot and Blum, in which purpuric red-brown papules coalesce into plaques, though her lesions were not raised. The other pigmented purpuric dermatoses can present with cayenne pepper–colored petechiae, golden-brown macules, pruritic purpuric patches, or red-brown annular patches,6 which were not seen in our patient.
Venous stasis dermatitis also favors the lower extremities7; however, it classically includes the medial malleolus and often presents with scaling and hyperpigmentation from hemosiderin deposition.8 It often is associated with pruritus, as opposed to the nonpruritic nonpainful lesions in leukemia cutis. Other signs of venous insufficiency also may be appreciated, including edema or varicose veins,7 which were not evident in our patient.
Leukocytoclastic vasculitis, a small vessel vasculitis, also appears as palpable or macular purpura, which classically is asymptomatic and erupts on the shins approximately 1 week after an inciting exposure,9 such as medications, pathogens, or autoimmune diseases. One of the least distinctive vasculitides is polyarteritis nodosa, a form of medium vessel vasculitis, which presents most often with palpable purpura or painful nodules on the lower extremities and may be accompanied by livedo reticularis or digital necrosis.9 Acute leukemia may be accompanied by inflammatory paraneoplastic conditions including vasculitis, which is thought to be due to leukemic cells infiltrating and damaging blood vessels.10
Pretibial myxedema is closely associated with Graves disease and shares some features seen in the presentation of our patient’s leukemia cutis. It is asymptomatic, classically affects the pretibial regions, and most commonly affects older adults and women.11,12 Pretibial myxedema presents with thick indurated plaques rather than patches. Our patient did not demonstrate ophthalmopathy, which nearly always precedes pretibial myxedema.12 The most common form of pretibial myxedema is nonpitting, though nodular, plaquelike, polypoid, and elephantiasic forms also exist.11 Pretibial myxedema classically favors the shins; however, it also can affect the ankles, dorsal aspects of the feet, and toes. The characteristic induration of the skin is believed to be the result of excess fibroblast production of glycosaminoglycans in the dermis and subcutis likely triggered by stimulation of fibroblast thyroid stimulating hormone receptors.11
- Bakst RL, Tallman MS, Douer D, et al. How I treat extramedullary acute myeloid leukemia. Blood. 2011;118:3785-3793.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Other lymphoproliferative and myeloproliferative diseases. In: Bolognia JL, Schaffer JV, Duncan KO, et al, eds. Dermatology Essentials. 2nd ed. Elsevier; 2014:973-977.
- Papadavid E, Panayiotides I, Katoulis A, et al. Stasis dermatitis-like leukaemic infiltration in a patient with myelodysplastic syndrome. Clin Exp Dermatol. 2008;33:298-300.
- Chang HY, Wong KM, Bosenberg M, et al. Myelogenous leukemia cutis resembling stasis dermatitis. J Am Acad Dermatol. 2003;49:128-129.
- Aguilera SB, Zarraga M, Rosen L. Leukemia cutis in a patient with acute myelogenous leukemia: a case report and review of the literature. Cutis. 2010;85:31-36.
- Kim DH, Seo SH, Ahn HH, et al. Characteristics and clinical manifestations of pigmented purpuric dermatosis. Ann Dermatol. 2015;27:404-410.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Other eczematous eruptions. In: Bolognia JL, Schaffer JV, Duncan KO, et al, eds. Dermatology Essentials. 2nd ed. Elsevier; 2014:103-108.
- Krooks JA, Weatherall AG. Leukemia cutis in acute myeloid leukemia signifies a poor prognosis. Cutis. 2018;102:266, 271-272.
- Wetter DA, Dutz JP, Shinkai K, et al. Cutaneous vasculitis. In: Bolognia JL, Schaffer JV, Lorenzo C, eds. Dermatology. 4th ed. Elsevier; 2018:409-439.
- Jones D, Dorfman DM, Barnhill RL, et al. Leukemic vasculitis: a feature of leukemia cutis in some patients. Am J Clin Pathol. 1997;107:637-642.
- Fatourechi V. Pretibial myxedema: pathophysiology and treatment options. Am J Clin Dermatol. 2005;6:295-309.
- Fatourechi V, Pajouhi M, Fransway AF. Dermopathy of Graves disease (pretibial myxedema). review of 150 cases. Medicine (Baltimore). 1994;73:1-7.
- Bakst RL, Tallman MS, Douer D, et al. How I treat extramedullary acute myeloid leukemia. Blood. 2011;118:3785-3793.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Other lymphoproliferative and myeloproliferative diseases. In: Bolognia JL, Schaffer JV, Duncan KO, et al, eds. Dermatology Essentials. 2nd ed. Elsevier; 2014:973-977.
- Papadavid E, Panayiotides I, Katoulis A, et al. Stasis dermatitis-like leukaemic infiltration in a patient with myelodysplastic syndrome. Clin Exp Dermatol. 2008;33:298-300.
- Chang HY, Wong KM, Bosenberg M, et al. Myelogenous leukemia cutis resembling stasis dermatitis. J Am Acad Dermatol. 2003;49:128-129.
- Aguilera SB, Zarraga M, Rosen L. Leukemia cutis in a patient with acute myelogenous leukemia: a case report and review of the literature. Cutis. 2010;85:31-36.
- Kim DH, Seo SH, Ahn HH, et al. Characteristics and clinical manifestations of pigmented purpuric dermatosis. Ann Dermatol. 2015;27:404-410.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Other eczematous eruptions. In: Bolognia JL, Schaffer JV, Duncan KO, et al, eds. Dermatology Essentials. 2nd ed. Elsevier; 2014:103-108.
- Krooks JA, Weatherall AG. Leukemia cutis in acute myeloid leukemia signifies a poor prognosis. Cutis. 2018;102:266, 271-272.
- Wetter DA, Dutz JP, Shinkai K, et al. Cutaneous vasculitis. In: Bolognia JL, Schaffer JV, Lorenzo C, eds. Dermatology. 4th ed. Elsevier; 2018:409-439.
- Jones D, Dorfman DM, Barnhill RL, et al. Leukemic vasculitis: a feature of leukemia cutis in some patients. Am J Clin Pathol. 1997;107:637-642.
- Fatourechi V. Pretibial myxedema: pathophysiology and treatment options. Am J Clin Dermatol. 2005;6:295-309.
- Fatourechi V, Pajouhi M, Fransway AF. Dermopathy of Graves disease (pretibial myxedema). review of 150 cases. Medicine (Baltimore). 1994;73:1-7.
A 67-year-old woman with history of atrial fibrillation and leukemia presented with a nonpruritic nonpainful rash of 10 days' duration that began on the distal lower extremities (top) and then spread superiorly. She reported having a sore throat and mouth, cough, night sweats, unintentional weight loss, and lymphadenopathy. Physical examination revealed pink-purple nonblanching macules and patches on the lower extremities extending from the ankles to the knees. She also had firm pink papules on the chest (bottom) and back. Punch biopsies of the skin on the chest and leg were obtained for histologic examination and immunohistochemical staining.


Firm Mobile Nodule on the Scalp
The Diagnosis: Metastatic Carcinoid Tumor
Carcinoid tumors are derived from neuroendocrine cell compartments and generally arise in the gastrointestinal tract, with a quarter of carcinoids arising in the small bowel.1 Carcinoid tumors have an incidence of approximately 2 to 5 per 100,000 patients.2 Metastasis of carcinoids is approximately 31.2% to 46.7%.1 Metastasis to the skin is uncommon; we present a rare case of a carcinoid tumor of the terminal ileum with metastasis to the scalp.
Unlike our patient, most patients with carcinoid tumors have an indolent clinical course. The most common cutaneous symptom is flushing, which occurs in 75% of patients.3 Secreted vasoactive peptides such as serotonin may cause other symptoms such as tachycardia, diarrhea, and bronchospasm; together, these symptoms comprise carcinoid syndrome. Carcinoid syndrome requires metastasis of the tumor to the liver or a site outside of the gastrointestinal tract because the liver will metabolize the secreted serotonin. However, even in patients with liver metastasis, carcinoid syndrome only occurs in approximately 10% of patients.4 Common skin findings of carcinoid syndrome include pellagralike dermatitis, flushing, and sclerodermalike changes.5 Our patient experienced several episodes of presyncope with symptoms of dyspnea, lightheadedness, and flushing but did not have bronchospasm or recurrent diarrhea. Intramuscular octreotide improved some symptoms.
The scalp accounts for approximately 15% of cutaneous metastases, the most common being from the lung, renal, and breast cancers.6 Cutaneous metastases of carcinoid tumors are rare. A PubMed search of articles indexed for MEDLINE using the terms metastatic AND [carcinoid OR neuroendocrine] tumors AND [skin OR cutaneous] revealed 47 cases.7-11 Similar to other skin metastases, cutaneous metastases of carcinoid tumors commonly present as firm erythematous nodules of varying sizes that may be asymptomatic, tender, or pruritic (Figure 1). Cases of carcinoid tumors with cutaneous metastasis as the initial and only presenting sign are exceedingly rare.12

Histology of carcinoid tumors reveals a dermal neoplasm composed of loosely cohesive, mildly atypical, polygonal cells with salt-and-pepper chromatin and eosinophilic cytoplasm, which are similar findings to the primary tumor. The cells may grow in the typical trabecular or organoid neuroendocrine pattern or exhibit a pseudoglandular growth pattern with prominent vessels (quiz image, top).12 Positive chromogranin and synaptophysin immunostaining are the most common and reliable markers used for the diagnosis of carcinoid tumors.
.")
An important histopathologic differential diagnosis is the aggressive Merkel cell carcinoma, which also demonstrates homogenous salt-and-pepper chromatin but exhibits a higher mitotic rate and positive cytokeratin 20 staining (Figure 2).13 Basal cell carcinoma (BCC) also may display similar features, including a blue tumor at scanning magnification and nodular or infiltrative growth patterns. The cell morphology of BCC is characterized by islands of basaloid cells with minimal cytoplasm and frequent apoptosis, connecting to the epidermis with peripheral palisading, retraction artifact, and a myxoid stroma; BCC lacks the salt-and-pepper chromatin commonly seen in carcinoid tumors (Figure 3). Basal cell carcinoma is characterized by positive BerEP4 (epithelial cell adhesion molecule immunostain), cytokeratin 5/6, and cytokeratin 14 uptake. Cytokeratin 20, often used to diagnose Merkel cell carcinoma, is negative in BCC. Chromogranin and synaptophysin occasionally may be positive in BCC.14
.")
The superficial Ewing sarcoma family of tumors also may be included in the differential diagnosis of small round cell tumors of the skin, but they are very rare. These tumors possess strong positive membranous staining of cytokeratin 99 and also can stain positively for synaptophysin and chromogranin.15 Epithelial membrane antigen, which is negative in Ewing sarcomas, is positive in carcinoid tumors.16 Neuroendocrine tumors of all sites share similar basic morphologic patterns, and multiple primary tumors should be considered, including small cell lung carcinoma (Figure 4).17,18 Red granulations and true glandular lumina typically are not seen in the lungs but are common in gastrointestinal carcinoids.18 Regarding immunohistochemistry, TTF-1 is negative and CDX2 is positive in gastroenteropancreatic carcinoids, suggesting that these 2 markers can help distinguish carcinoids of unknown primary origin.19

Metastases in carcinoid tumors are common, with one study noting that the highest frequency of small intestinal metastases was from the ileal subset.20 At the time of diagnosis, 58% to 64% of patients with small intestine carcinoid tumors already had nonlocalized disease, with frequent sites being the lymph nodes (89.8%), liver (44.1%), lungs (13.6%), and peritoneum (13.6%). Regional and distant metastases are associated with substantially worse prognoses, with survival rates of 71.7% and 38.5%, respectively.1 Treatment of symptomatic unresectable disease focuses on symptomatic management with somatostatin analogs that also control tumor growth.21
We present a rare case of scalp metastasis of a carcinoid tumor of the terminal ileum. Distant metastasis is associated with poorer prognosis and should be considered in patients with a known history of a carcinoid tumor.
Acknowledgment—We would like to acknowledge the Research Histology and Tissue Imaging Core at University of Illinois Chicago Research Resources Center for the immunohistochemistry studies.
- Modlin IM, Lye KD, Kidd M. A 5-decade analysis of 13,715 carcinoid tumors. Cancer. 2003;97:934-959.
- Lawrence B, Gustafsson BI, Chan A, et al. The epidemiology of gastroenteropancreatic neuroendocrine tumors. Endocrinol Metab Clin North Am. 2011;40:1-18, vii.
- Sabir S, James WD, Schuchter LM. Cutaneous manifestations of cancer. Curr Opin Oncol. 1999;11:139-144.
- Tomassetti P. Clinical aspects of carcinoid tumours. Italian J Gastroenterol Hepatol. 1999;31(suppl 2):S143-S146.
- Bell HK, Poston GJ, Vora J, et al. Cutaneous manifestations of the malignant carcinoid syndrome. Br J Dermatol. 2005;152:71-75.
- Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29(2 pt 1):228-236.
- Garcia A, Mays S, Silapunt S. Metastatic neuroendocrine carcinoma in the skin. Dermatol Online J. 2017;23:13030/qt9052w9x1.
- Ciliberti MP, Carbonara R, Grillo A, et al. Unexpected response to palliative radiotherapy for subcutaneous metastases of an advanced small cell pancreatic neuroendocrine carcinoma: a case report of two different radiation schedules. BMC Cancer. 2020;20:311.
- Devnani B, Kumar R, Pathy S, et al. Cutaneous metastases from neuroendocrine carcinoma of the cervix: an unusual metastatic lesion from an uncommon malignancy. Curr Probl Cancer. 2018; 42:527-533.
- Falto-Aizpurua L, Seyfer S, Krishnan B, et al. Cutaneous metastasis of a pulmonary carcinoid tumor. Cutis. 2017;99:E13-E15.
- Dhingra R, Tse JY, Saif MW. Cutaneous metastasis of gastroenteropancreatic neuroendocrine tumors (GEP-Nets)[published online September 8, 2018]. JOP. 2018;19.
- Jedrych J, Busam K, Klimstra DS, et al. Cutaneous metastases as an initial manifestation of visceral well-differentiated neuroendocrine tumor: a report of four cases and a review of literature. J Cutan Pathol. 2014;41:113-122.
- Lloyd RV. Practical markers used in the diagnosis of neuroendocrine tumors. Endocr Pathol. 2003;14:293-301.
- Stanoszek LM, Wang GY, Harms PW. Histologic mimics of basal cell carcinoma. Arch Pathol Lab Med. 2017;141:1490-1502.
- Machado I, Llombart B, Calabuig-Fariñas S, et al. Superficial Ewing’s sarcoma family of tumors: a clinicopathological study with differential diagnoses. J Cutan Pathol. 2011;38:636-643.
- D’Cruze L, Dutta R, Rao S, et al. The role of immunohistochemistry in the analysis of the spectrum of small round cell tumours at a tertiary care centre. J Clin Diagn Res. 2013;7:1377-1382.
- Chirila DN, Turdeanu NA, Constantea NA, et al. Multiple malignant tumors. Chirurgia (Bucur). 2013;108:498-502.
- Rekhtman N. Neuroendocrine tumors of the lung: an update. Arch Pathol Lab Med. 2010;134:1628-1638.
- Lin X, Saad RS, Luckasevic TM, et al. Diagnostic value of CDX-2 and TTF-1 expressions in separating metastatic neuroendocrine neoplasms of unknown origin. Appl Immunohistochem Mol Morphol. 2007;15:407-414.
- Olney JR, Urdaneta LF, Al-Jurf AS, et al. Carcinoid tumors of the gastrointestinal tract. Am Surg. 1985;51:37-41.
- Strosberg JR, Halfdanarson TR, Bellizzi AM, et al. The North American Neuroendocrine Tumor Society consensus guidelines for surveillance and medical management of midgut neuroendocrine tumors. Pancreas. 2017;46:707-714.
The Diagnosis: Metastatic Carcinoid Tumor
Carcinoid tumors are derived from neuroendocrine cell compartments and generally arise in the gastrointestinal tract, with a quarter of carcinoids arising in the small bowel.1 Carcinoid tumors have an incidence of approximately 2 to 5 per 100,000 patients.2 Metastasis of carcinoids is approximately 31.2% to 46.7%.1 Metastasis to the skin is uncommon; we present a rare case of a carcinoid tumor of the terminal ileum with metastasis to the scalp.
Unlike our patient, most patients with carcinoid tumors have an indolent clinical course. The most common cutaneous symptom is flushing, which occurs in 75% of patients.3 Secreted vasoactive peptides such as serotonin may cause other symptoms such as tachycardia, diarrhea, and bronchospasm; together, these symptoms comprise carcinoid syndrome. Carcinoid syndrome requires metastasis of the tumor to the liver or a site outside of the gastrointestinal tract because the liver will metabolize the secreted serotonin. However, even in patients with liver metastasis, carcinoid syndrome only occurs in approximately 10% of patients.4 Common skin findings of carcinoid syndrome include pellagralike dermatitis, flushing, and sclerodermalike changes.5 Our patient experienced several episodes of presyncope with symptoms of dyspnea, lightheadedness, and flushing but did not have bronchospasm or recurrent diarrhea. Intramuscular octreotide improved some symptoms.
The scalp accounts for approximately 15% of cutaneous metastases, the most common being from the lung, renal, and breast cancers.6 Cutaneous metastases of carcinoid tumors are rare. A PubMed search of articles indexed for MEDLINE using the terms metastatic AND [carcinoid OR neuroendocrine] tumors AND [skin OR cutaneous] revealed 47 cases.7-11 Similar to other skin metastases, cutaneous metastases of carcinoid tumors commonly present as firm erythematous nodules of varying sizes that may be asymptomatic, tender, or pruritic (Figure 1). Cases of carcinoid tumors with cutaneous metastasis as the initial and only presenting sign are exceedingly rare.12
Histology of carcinoid tumors reveals a dermal neoplasm composed of loosely cohesive, mildly atypical, polygonal cells with salt-and-pepper chromatin and eosinophilic cytoplasm, which are similar findings to the primary tumor. The cells may grow in the typical trabecular or organoid neuroendocrine pattern or exhibit a pseudoglandular growth pattern with prominent vessels (quiz image, top).12 Positive chromogranin and synaptophysin immunostaining are the most common and reliable markers used for the diagnosis of carcinoid tumors.
An important histopathologic differential diagnosis is the aggressive Merkel cell carcinoma, which also demonstrates homogenous salt-and-pepper chromatin but exhibits a higher mitotic rate and positive cytokeratin 20 staining (Figure 2).13 Basal cell carcinoma (BCC) also may display similar features, including a blue tumor at scanning magnification and nodular or infiltrative growth patterns. The cell morphology of BCC is characterized by islands of basaloid cells with minimal cytoplasm and frequent apoptosis, connecting to the epidermis with peripheral palisading, retraction artifact, and a myxoid stroma; BCC lacks the salt-and-pepper chromatin commonly seen in carcinoid tumors (Figure 3). Basal cell carcinoma is characterized by positive BerEP4 (epithelial cell adhesion molecule immunostain), cytokeratin 5/6, and cytokeratin 14 uptake. Cytokeratin 20, often used to diagnose Merkel cell carcinoma, is negative in BCC. Chromogranin and synaptophysin occasionally may be positive in BCC.14
The superficial Ewing sarcoma family of tumors also may be included in the differential diagnosis of small round cell tumors of the skin, but they are very rare. These tumors possess strong positive membranous staining of cytokeratin 99 and also can stain positively for synaptophysin and chromogranin.15 Epithelial membrane antigen, which is negative in Ewing sarcomas, is positive in carcinoid tumors.16 Neuroendocrine tumors of all sites share similar basic morphologic patterns, and multiple primary tumors should be considered, including small cell lung carcinoma (Figure 4).17,18 Red granulations and true glandular lumina typically are not seen in the lungs but are common in gastrointestinal carcinoids.18 Regarding immunohistochemistry, TTF-1 is negative and CDX2 is positive in gastroenteropancreatic carcinoids, suggesting that these 2 markers can help distinguish carcinoids of unknown primary origin.19
Metastases in carcinoid tumors are common, with one study noting that the highest frequency of small intestinal metastases was from the ileal subset.20 At the time of diagnosis, 58% to 64% of patients with small intestine carcinoid tumors already had nonlocalized disease, with frequent sites being the lymph nodes (89.8%), liver (44.1%), lungs (13.6%), and peritoneum (13.6%). Regional and distant metastases are associated with substantially worse prognoses, with survival rates of 71.7% and 38.5%, respectively.1 Treatment of symptomatic unresectable disease focuses on symptomatic management with somatostatin analogs that also control tumor growth.21
We present a rare case of scalp metastasis of a carcinoid tumor of the terminal ileum. Distant metastasis is associated with poorer prognosis and should be considered in patients with a known history of a carcinoid tumor.
Acknowledgment—We would like to acknowledge the Research Histology and Tissue Imaging Core at University of Illinois Chicago Research Resources Center for the immunohistochemistry studies.
The Diagnosis: Metastatic Carcinoid Tumor
Carcinoid tumors are derived from neuroendocrine cell compartments and generally arise in the gastrointestinal tract, with a quarter of carcinoids arising in the small bowel.1 Carcinoid tumors have an incidence of approximately 2 to 5 per 100,000 patients.2 Metastasis of carcinoids is approximately 31.2% to 46.7%.1 Metastasis to the skin is uncommon; we present a rare case of a carcinoid tumor of the terminal ileum with metastasis to the scalp.
Unlike our patient, most patients with carcinoid tumors have an indolent clinical course. The most common cutaneous symptom is flushing, which occurs in 75% of patients.3 Secreted vasoactive peptides such as serotonin may cause other symptoms such as tachycardia, diarrhea, and bronchospasm; together, these symptoms comprise carcinoid syndrome. Carcinoid syndrome requires metastasis of the tumor to the liver or a site outside of the gastrointestinal tract because the liver will metabolize the secreted serotonin. However, even in patients with liver metastasis, carcinoid syndrome only occurs in approximately 10% of patients.4 Common skin findings of carcinoid syndrome include pellagralike dermatitis, flushing, and sclerodermalike changes.5 Our patient experienced several episodes of presyncope with symptoms of dyspnea, lightheadedness, and flushing but did not have bronchospasm or recurrent diarrhea. Intramuscular octreotide improved some symptoms.
The scalp accounts for approximately 15% of cutaneous metastases, the most common being from the lung, renal, and breast cancers.6 Cutaneous metastases of carcinoid tumors are rare. A PubMed search of articles indexed for MEDLINE using the terms metastatic AND [carcinoid OR neuroendocrine] tumors AND [skin OR cutaneous] revealed 47 cases.7-11 Similar to other skin metastases, cutaneous metastases of carcinoid tumors commonly present as firm erythematous nodules of varying sizes that may be asymptomatic, tender, or pruritic (Figure 1). Cases of carcinoid tumors with cutaneous metastasis as the initial and only presenting sign are exceedingly rare.12
Histology of carcinoid tumors reveals a dermal neoplasm composed of loosely cohesive, mildly atypical, polygonal cells with salt-and-pepper chromatin and eosinophilic cytoplasm, which are similar findings to the primary tumor. The cells may grow in the typical trabecular or organoid neuroendocrine pattern or exhibit a pseudoglandular growth pattern with prominent vessels (quiz image, top).12 Positive chromogranin and synaptophysin immunostaining are the most common and reliable markers used for the diagnosis of carcinoid tumors.
An important histopathologic differential diagnosis is the aggressive Merkel cell carcinoma, which also demonstrates homogenous salt-and-pepper chromatin but exhibits a higher mitotic rate and positive cytokeratin 20 staining (Figure 2).13 Basal cell carcinoma (BCC) also may display similar features, including a blue tumor at scanning magnification and nodular or infiltrative growth patterns. The cell morphology of BCC is characterized by islands of basaloid cells with minimal cytoplasm and frequent apoptosis, connecting to the epidermis with peripheral palisading, retraction artifact, and a myxoid stroma; BCC lacks the salt-and-pepper chromatin commonly seen in carcinoid tumors (Figure 3). Basal cell carcinoma is characterized by positive BerEP4 (epithelial cell adhesion molecule immunostain), cytokeratin 5/6, and cytokeratin 14 uptake. Cytokeratin 20, often used to diagnose Merkel cell carcinoma, is negative in BCC. Chromogranin and synaptophysin occasionally may be positive in BCC.14
The superficial Ewing sarcoma family of tumors also may be included in the differential diagnosis of small round cell tumors of the skin, but they are very rare. These tumors possess strong positive membranous staining of cytokeratin 99 and also can stain positively for synaptophysin and chromogranin.15 Epithelial membrane antigen, which is negative in Ewing sarcomas, is positive in carcinoid tumors.16 Neuroendocrine tumors of all sites share similar basic morphologic patterns, and multiple primary tumors should be considered, including small cell lung carcinoma (Figure 4).17,18 Red granulations and true glandular lumina typically are not seen in the lungs but are common in gastrointestinal carcinoids.18 Regarding immunohistochemistry, TTF-1 is negative and CDX2 is positive in gastroenteropancreatic carcinoids, suggesting that these 2 markers can help distinguish carcinoids of unknown primary origin.19
Metastases in carcinoid tumors are common, with one study noting that the highest frequency of small intestinal metastases was from the ileal subset.20 At the time of diagnosis, 58% to 64% of patients with small intestine carcinoid tumors already had nonlocalized disease, with frequent sites being the lymph nodes (89.8%), liver (44.1%), lungs (13.6%), and peritoneum (13.6%). Regional and distant metastases are associated with substantially worse prognoses, with survival rates of 71.7% and 38.5%, respectively.1 Treatment of symptomatic unresectable disease focuses on symptomatic management with somatostatin analogs that also control tumor growth.21
We present a rare case of scalp metastasis of a carcinoid tumor of the terminal ileum. Distant metastasis is associated with poorer prognosis and should be considered in patients with a known history of a carcinoid tumor.
Acknowledgment—We would like to acknowledge the Research Histology and Tissue Imaging Core at University of Illinois Chicago Research Resources Center for the immunohistochemistry studies.
- Modlin IM, Lye KD, Kidd M. A 5-decade analysis of 13,715 carcinoid tumors. Cancer. 2003;97:934-959.
- Lawrence B, Gustafsson BI, Chan A, et al. The epidemiology of gastroenteropancreatic neuroendocrine tumors. Endocrinol Metab Clin North Am. 2011;40:1-18, vii.
- Sabir S, James WD, Schuchter LM. Cutaneous manifestations of cancer. Curr Opin Oncol. 1999;11:139-144.
- Tomassetti P. Clinical aspects of carcinoid tumours. Italian J Gastroenterol Hepatol. 1999;31(suppl 2):S143-S146.
- Bell HK, Poston GJ, Vora J, et al. Cutaneous manifestations of the malignant carcinoid syndrome. Br J Dermatol. 2005;152:71-75.
- Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29(2 pt 1):228-236.
- Garcia A, Mays S, Silapunt S. Metastatic neuroendocrine carcinoma in the skin. Dermatol Online J. 2017;23:13030/qt9052w9x1.
- Ciliberti MP, Carbonara R, Grillo A, et al. Unexpected response to palliative radiotherapy for subcutaneous metastases of an advanced small cell pancreatic neuroendocrine carcinoma: a case report of two different radiation schedules. BMC Cancer. 2020;20:311.
- Devnani B, Kumar R, Pathy S, et al. Cutaneous metastases from neuroendocrine carcinoma of the cervix: an unusual metastatic lesion from an uncommon malignancy. Curr Probl Cancer. 2018; 42:527-533.
- Falto-Aizpurua L, Seyfer S, Krishnan B, et al. Cutaneous metastasis of a pulmonary carcinoid tumor. Cutis. 2017;99:E13-E15.
- Dhingra R, Tse JY, Saif MW. Cutaneous metastasis of gastroenteropancreatic neuroendocrine tumors (GEP-Nets)[published online September 8, 2018]. JOP. 2018;19.
- Jedrych J, Busam K, Klimstra DS, et al. Cutaneous metastases as an initial manifestation of visceral well-differentiated neuroendocrine tumor: a report of four cases and a review of literature. J Cutan Pathol. 2014;41:113-122.
- Lloyd RV. Practical markers used in the diagnosis of neuroendocrine tumors. Endocr Pathol. 2003;14:293-301.
- Stanoszek LM, Wang GY, Harms PW. Histologic mimics of basal cell carcinoma. Arch Pathol Lab Med. 2017;141:1490-1502.
- Machado I, Llombart B, Calabuig-Fariñas S, et al. Superficial Ewing’s sarcoma family of tumors: a clinicopathological study with differential diagnoses. J Cutan Pathol. 2011;38:636-643.
- D’Cruze L, Dutta R, Rao S, et al. The role of immunohistochemistry in the analysis of the spectrum of small round cell tumours at a tertiary care centre. J Clin Diagn Res. 2013;7:1377-1382.
- Chirila DN, Turdeanu NA, Constantea NA, et al. Multiple malignant tumors. Chirurgia (Bucur). 2013;108:498-502.
- Rekhtman N. Neuroendocrine tumors of the lung: an update. Arch Pathol Lab Med. 2010;134:1628-1638.
- Lin X, Saad RS, Luckasevic TM, et al. Diagnostic value of CDX-2 and TTF-1 expressions in separating metastatic neuroendocrine neoplasms of unknown origin. Appl Immunohistochem Mol Morphol. 2007;15:407-414.
- Olney JR, Urdaneta LF, Al-Jurf AS, et al. Carcinoid tumors of the gastrointestinal tract. Am Surg. 1985;51:37-41.
- Strosberg JR, Halfdanarson TR, Bellizzi AM, et al. The North American Neuroendocrine Tumor Society consensus guidelines for surveillance and medical management of midgut neuroendocrine tumors. Pancreas. 2017;46:707-714.
- Modlin IM, Lye KD, Kidd M. A 5-decade analysis of 13,715 carcinoid tumors. Cancer. 2003;97:934-959.
- Lawrence B, Gustafsson BI, Chan A, et al. The epidemiology of gastroenteropancreatic neuroendocrine tumors. Endocrinol Metab Clin North Am. 2011;40:1-18, vii.
- Sabir S, James WD, Schuchter LM. Cutaneous manifestations of cancer. Curr Opin Oncol. 1999;11:139-144.
- Tomassetti P. Clinical aspects of carcinoid tumours. Italian J Gastroenterol Hepatol. 1999;31(suppl 2):S143-S146.
- Bell HK, Poston GJ, Vora J, et al. Cutaneous manifestations of the malignant carcinoid syndrome. Br J Dermatol. 2005;152:71-75.
- Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29(2 pt 1):228-236.
- Garcia A, Mays S, Silapunt S. Metastatic neuroendocrine carcinoma in the skin. Dermatol Online J. 2017;23:13030/qt9052w9x1.
- Ciliberti MP, Carbonara R, Grillo A, et al. Unexpected response to palliative radiotherapy for subcutaneous metastases of an advanced small cell pancreatic neuroendocrine carcinoma: a case report of two different radiation schedules. BMC Cancer. 2020;20:311.
- Devnani B, Kumar R, Pathy S, et al. Cutaneous metastases from neuroendocrine carcinoma of the cervix: an unusual metastatic lesion from an uncommon malignancy. Curr Probl Cancer. 2018; 42:527-533.
- Falto-Aizpurua L, Seyfer S, Krishnan B, et al. Cutaneous metastasis of a pulmonary carcinoid tumor. Cutis. 2017;99:E13-E15.
- Dhingra R, Tse JY, Saif MW. Cutaneous metastasis of gastroenteropancreatic neuroendocrine tumors (GEP-Nets)[published online September 8, 2018]. JOP. 2018;19.
- Jedrych J, Busam K, Klimstra DS, et al. Cutaneous metastases as an initial manifestation of visceral well-differentiated neuroendocrine tumor: a report of four cases and a review of literature. J Cutan Pathol. 2014;41:113-122.
- Lloyd RV. Practical markers used in the diagnosis of neuroendocrine tumors. Endocr Pathol. 2003;14:293-301.
- Stanoszek LM, Wang GY, Harms PW. Histologic mimics of basal cell carcinoma. Arch Pathol Lab Med. 2017;141:1490-1502.
- Machado I, Llombart B, Calabuig-Fariñas S, et al. Superficial Ewing’s sarcoma family of tumors: a clinicopathological study with differential diagnoses. J Cutan Pathol. 2011;38:636-643.
- D’Cruze L, Dutta R, Rao S, et al. The role of immunohistochemistry in the analysis of the spectrum of small round cell tumours at a tertiary care centre. J Clin Diagn Res. 2013;7:1377-1382.
- Chirila DN, Turdeanu NA, Constantea NA, et al. Multiple malignant tumors. Chirurgia (Bucur). 2013;108:498-502.
- Rekhtman N. Neuroendocrine tumors of the lung: an update. Arch Pathol Lab Med. 2010;134:1628-1638.
- Lin X, Saad RS, Luckasevic TM, et al. Diagnostic value of CDX-2 and TTF-1 expressions in separating metastatic neuroendocrine neoplasms of unknown origin. Appl Immunohistochem Mol Morphol. 2007;15:407-414.
- Olney JR, Urdaneta LF, Al-Jurf AS, et al. Carcinoid tumors of the gastrointestinal tract. Am Surg. 1985;51:37-41.
- Strosberg JR, Halfdanarson TR, Bellizzi AM, et al. The North American Neuroendocrine Tumor Society consensus guidelines for surveillance and medical management of midgut neuroendocrine tumors. Pancreas. 2017;46:707-714.
A 47-year-old woman was admitted to the hospital with abdominal pain and flushing. She had a history of a midgut carcinoid that originated in the ileum with metastasis to the colon, liver, and pancreas. Dermatologic examination revealed a firm, nontender, mobile, 7-mm scalp nodule with a pink-purple overlying epidermis. The lesion was associated with a slight decrease in hair density. A 4-mm punch biopsy was performed.



Tender Nonhealing Lesion on the Leg
The Diagnosis: Calciphylaxis
Calciphylaxis is a rare life-threatening condition that most often is seen in patients with end-stage renal disease at a rate of 35 per 10,000 chronic dialysis patients.1 It less commonly has been described in nonuremic patients. The exact incidence of nonuremic calciphylaxis is unknown, but multiple risk factors have been identified, such as alcoholic liver disease, primary hyperparathyroidism, connective tissue diseases, and underlying malignancies. Other less common risk factors include type 2 diabetes mellitus, hypercoagulable disorders, obesity, hypoalbuminemia, and warfarin/ corticosteroid use.2 However, most often no obvious triggers are identified.1
Regardless of the etiology, calciphylaxis is characterized by the calcification of blood vessels and connective tissues, leading to vessel injury, intimal fibrosis, and thrombosis, followed by ischemic necrosis of the skin and soft tissue. It is postulated that microvascular calcification occurs as an active cell-mediated process that depends on the balance between the promoters and inhibitors of calcification.1 In our patient, liver disease likely predisposed formation of calcification through the creation of an environment susceptible to vascular injury via decreased synthesis of proteins C and S.3 Synthesis of fetuin-A, a protein that acts as a circulating inhibitor of vascular ossification/calcification, also is decreased in calcification. Another inhibitor of calcification, matrix Gla protein, is unable to undergo activation through vitamin K–dependent carboxylation secondary to liver disease–induced vitamin K deficiency.3 Microvascular calcification without calciphylaxis may occur in other conditions such as type 2 diabetes mellitus. Therefore, clinicopathologic correlation is important in determining the diagnosis.
Calciphylaxis has a variety of clinical presentations depending on the stage of disease. It begins as a fixed, indurated, livedo reticularis–like plaque. The lesions become increasingly violaceous with intermixed areas of light blanched skin secondary to ischemia and then develop retiform pupura.4 Eventually, affected sites can become bullous and ulcerate or form a necrotic eschar. Severe pain is a cardinal feature throughout all stages.4 Lesions in nonuremic calciphylaxis most commonly are located in the central and/or proximal areas of the body.2
Clinical suspicion is essential for diagnosis. Skin biopsy is the standard method for confirmation in unclear cases. The classic histologic features include intravascular and extravascular calcification, microthrombosis, and fibrointimal hyperplasia of the small dermal and subcutaneous arteries and arterioles, leading to ischemia and intense septal panniculitis.1 Von Kossa immunostaining is used to increase the detection of calcium deposits (Figure 1).1 In addition to the classic changes, our case demonstrated a rare histologic variant with pseudoxanthoma elasticum (PXE)–like changes (Figure 2), which are thought to occur secondary to pathologic elastin fibrogenesis or increased proteolytic activity resulting in abnormal remodeling of the extracellular matrix in the setting of increased calcification of elastin fibers.5 Detection of PXE-like changes may be a helpful clue when specimens lack other characteristic signs.
.")
Wound care, pain control, and addressing underlying causes are mainstays of therapy. Sodium thiosulfate, an antioxidant with vasodilatory properties that also inhibits adipocyte calcification and blocks the ability of adipocytes to induce calcification of vascular smooth-muscle cells, also is useful. Antibiotic prophylaxis is not indicated.1
.")
Even with treatment, both uremic and nonuremic calciphylaxis have a dismal prognosis; 1-year mortality is approximately 50% to 60% and rises to 80% at 2 years.4 Lesion location affects prognosis, and more proximal lesions portend worse outcomes. In patients with both proximal and distal lesions, there is a 90% mortality rate within 1 year. Ulceration also portends worse outcomes, as the wounds often are resistant to healing and act as nidi for infection.4 Septicemia is the most common cause of death.1
Ecthyma gangrenosum is a cutaneous manifestation secondary to an infection most commonly associated with Pseudomonas aeruginosa.6 It often presents in immunocompromised patients with an underlying gramnegative septicemia.7 The clinical presentation initially begins with painless macules that rapidly progress into necrotic ulcers, usually accompanied by associated systemic symptoms such as fever, chills, and hypotension. Histopathology reveals numerous gram-negative rods around necrotic vessels.7
Idiopathic purpura fulminans is the rarest form of purpura fulminans. It is caused by autoantibody formation against protein S, resulting in protein S depletion and subsequent hypercoagulability.8 It usually occurs 7 to 10 days after the onset of a precipitating infection. Lesions begin as erythematous macules that progress within hours to painful, sharply defined areas of purpura and hemorrhagic cutaneous necrosis that may extend to deeper tissues.8 Secondary infection of gangrenous tissue may occur. Distribution usually is diffuse and signs of septic shock and disseminated intravascular coagulation usually are present.
Hughes syndrome, also known as antiphospholipid syndrome, is an acquired autoimmune disorder that manifests clinically as recurrent arterial or venous thrombosis.9 Cutaneous manifestations consist of livedo reticularis, arterial and venous ulcers, and superficial thrombophlebitis.10 Laboratory testing for antiphospholipid antibodies and obtaining a detailed history of the patient’s cardiovascular health are crucial for diagnosis.9
Necrotizing fasciitis typically begins as an inconspicuous superficial cutaneous infection that rapidly is transmitted to the fascia. Infection can spread along fascial planes for several days without affecting the overlying skin, leading to delayed diagnosis.11 The first signs to appear are disproportionate pain and a change in skin color to reddish-purple or bluish-gray. Next, the skin will become indurated, swollen, shiny, and more painful.11 Skin breakdown will begin in 3 to 5 days and is accompanied by bullae and cutaneous gangrene. The involved area becomes painless due to thrombosis of the small vessels that supply the superficial nerves.12 Septic shock ultimately will develop if untreated.
We present a rare case of nonuremic calciphylaxis. We encourage dermatologists to include calciphylaxis in the differential when evaluating any patient with a painful retiform rash or ulcerated eschar, even in the absence of renal disease.
- Nigwekar SU, Thadhani R, Brandenburg VM. Calciphylaxis. N Engl J Med. 2018;378:1704-1714.
- Nigwekar SU, Wolf M, Sterns RH, et al. Calciphylaxis from nonuremic causes: a systematic review. Clin J Am Soc Nephrol. 2008;3:1139-1143.
- Sammour YM, Saleh HM, Gad MM, et al. Non-uremic calciphylaxis associated with alcoholic hepatitis: a case report. World J Hepatol. 2019;11:127-132.
- James WD, Elston DM, Treat J, et al, eds. Cutaneous vascular diseases. Andrews’ Diseases of the Skin: Clinical Dermatology. Elsevier; 2020:813-861.
- Nathoo RK, Harb JN, Auerbach J, et al. Pseudoxanthoma elasticum-like changes in nonuremic calciphylaxis: case series and brief review of a helpful diagnostic clue. J Cutan Pathol. 2017;44:1064-1069.
- Vaiman M, Lazarovitch T, Heller L, et al. Ecthyma gangrenosum and ecthyma-like lesions: review article. Eur J Clin Microbiol Infect Dis Off Publ Eur Soc Clin Microbiol. 2015;34:633-639.
- Greene SL, Su WP, Muller SA. Ecthyma gangrenosum: report of clinical, histopathologic, and bacteriologic aspects of eight cases. J Am Acad Dermatol. 1984;11(5 pt 1):781-787.
- Levin M, Eley BS, Louis J, et al. Postinfectious purpura fulminans caused by an autoantibody directed against protein S. J Pediatr. 1995;127:355-363.
- Hughes G. Hughes syndrome: the antiphospholipid syndrome—a clinical overview. Clin Rev Allergy Immunol. 2007;32:3-12.
- Chang Y, Dabiri G, Damstetter E, et al. Coagulation disorders and their cutaneous presentations: pathophysiology. J Am Acad Dermatol. 2016;74:783-792; quiz 793-794.
- Fais P, Viero A, Viel G, et al. Necrotizing fasciitis: case series and review of the literature on clinical and medico-legal diagnostic challenges. Int J Legal Med. 2018;132:1357-1366.
- Brook I. Microbiology and management of soft tissue and muscle infections. Int J Surg Lond Engl. 2008;6:328-338.
The Diagnosis: Calciphylaxis
Calciphylaxis is a rare life-threatening condition that most often is seen in patients with end-stage renal disease at a rate of 35 per 10,000 chronic dialysis patients.1 It less commonly has been described in nonuremic patients. The exact incidence of nonuremic calciphylaxis is unknown, but multiple risk factors have been identified, such as alcoholic liver disease, primary hyperparathyroidism, connective tissue diseases, and underlying malignancies. Other less common risk factors include type 2 diabetes mellitus, hypercoagulable disorders, obesity, hypoalbuminemia, and warfarin/ corticosteroid use.2 However, most often no obvious triggers are identified.1
Regardless of the etiology, calciphylaxis is characterized by the calcification of blood vessels and connective tissues, leading to vessel injury, intimal fibrosis, and thrombosis, followed by ischemic necrosis of the skin and soft tissue. It is postulated that microvascular calcification occurs as an active cell-mediated process that depends on the balance between the promoters and inhibitors of calcification.1 In our patient, liver disease likely predisposed formation of calcification through the creation of an environment susceptible to vascular injury via decreased synthesis of proteins C and S.3 Synthesis of fetuin-A, a protein that acts as a circulating inhibitor of vascular ossification/calcification, also is decreased in calcification. Another inhibitor of calcification, matrix Gla protein, is unable to undergo activation through vitamin K–dependent carboxylation secondary to liver disease–induced vitamin K deficiency.3 Microvascular calcification without calciphylaxis may occur in other conditions such as type 2 diabetes mellitus. Therefore, clinicopathologic correlation is important in determining the diagnosis.
Calciphylaxis has a variety of clinical presentations depending on the stage of disease. It begins as a fixed, indurated, livedo reticularis–like plaque. The lesions become increasingly violaceous with intermixed areas of light blanched skin secondary to ischemia and then develop retiform pupura.4 Eventually, affected sites can become bullous and ulcerate or form a necrotic eschar. Severe pain is a cardinal feature throughout all stages.4 Lesions in nonuremic calciphylaxis most commonly are located in the central and/or proximal areas of the body.2
Clinical suspicion is essential for diagnosis. Skin biopsy is the standard method for confirmation in unclear cases. The classic histologic features include intravascular and extravascular calcification, microthrombosis, and fibrointimal hyperplasia of the small dermal and subcutaneous arteries and arterioles, leading to ischemia and intense septal panniculitis.1 Von Kossa immunostaining is used to increase the detection of calcium deposits (Figure 1).1 In addition to the classic changes, our case demonstrated a rare histologic variant with pseudoxanthoma elasticum (PXE)–like changes (Figure 2), which are thought to occur secondary to pathologic elastin fibrogenesis or increased proteolytic activity resulting in abnormal remodeling of the extracellular matrix in the setting of increased calcification of elastin fibers.5 Detection of PXE-like changes may be a helpful clue when specimens lack other characteristic signs.
Wound care, pain control, and addressing underlying causes are mainstays of therapy. Sodium thiosulfate, an antioxidant with vasodilatory properties that also inhibits adipocyte calcification and blocks the ability of adipocytes to induce calcification of vascular smooth-muscle cells, also is useful. Antibiotic prophylaxis is not indicated.1
Even with treatment, both uremic and nonuremic calciphylaxis have a dismal prognosis; 1-year mortality is approximately 50% to 60% and rises to 80% at 2 years.4 Lesion location affects prognosis, and more proximal lesions portend worse outcomes. In patients with both proximal and distal lesions, there is a 90% mortality rate within 1 year. Ulceration also portends worse outcomes, as the wounds often are resistant to healing and act as nidi for infection.4 Septicemia is the most common cause of death.1
Ecthyma gangrenosum is a cutaneous manifestation secondary to an infection most commonly associated with Pseudomonas aeruginosa.6 It often presents in immunocompromised patients with an underlying gramnegative septicemia.7 The clinical presentation initially begins with painless macules that rapidly progress into necrotic ulcers, usually accompanied by associated systemic symptoms such as fever, chills, and hypotension. Histopathology reveals numerous gram-negative rods around necrotic vessels.7
Idiopathic purpura fulminans is the rarest form of purpura fulminans. It is caused by autoantibody formation against protein S, resulting in protein S depletion and subsequent hypercoagulability.8 It usually occurs 7 to 10 days after the onset of a precipitating infection. Lesions begin as erythematous macules that progress within hours to painful, sharply defined areas of purpura and hemorrhagic cutaneous necrosis that may extend to deeper tissues.8 Secondary infection of gangrenous tissue may occur. Distribution usually is diffuse and signs of septic shock and disseminated intravascular coagulation usually are present.
Hughes syndrome, also known as antiphospholipid syndrome, is an acquired autoimmune disorder that manifests clinically as recurrent arterial or venous thrombosis.9 Cutaneous manifestations consist of livedo reticularis, arterial and venous ulcers, and superficial thrombophlebitis.10 Laboratory testing for antiphospholipid antibodies and obtaining a detailed history of the patient’s cardiovascular health are crucial for diagnosis.9
Necrotizing fasciitis typically begins as an inconspicuous superficial cutaneous infection that rapidly is transmitted to the fascia. Infection can spread along fascial planes for several days without affecting the overlying skin, leading to delayed diagnosis.11 The first signs to appear are disproportionate pain and a change in skin color to reddish-purple or bluish-gray. Next, the skin will become indurated, swollen, shiny, and more painful.11 Skin breakdown will begin in 3 to 5 days and is accompanied by bullae and cutaneous gangrene. The involved area becomes painless due to thrombosis of the small vessels that supply the superficial nerves.12 Septic shock ultimately will develop if untreated.
We present a rare case of nonuremic calciphylaxis. We encourage dermatologists to include calciphylaxis in the differential when evaluating any patient with a painful retiform rash or ulcerated eschar, even in the absence of renal disease.
The Diagnosis: Calciphylaxis
Calciphylaxis is a rare life-threatening condition that most often is seen in patients with end-stage renal disease at a rate of 35 per 10,000 chronic dialysis patients.1 It less commonly has been described in nonuremic patients. The exact incidence of nonuremic calciphylaxis is unknown, but multiple risk factors have been identified, such as alcoholic liver disease, primary hyperparathyroidism, connective tissue diseases, and underlying malignancies. Other less common risk factors include type 2 diabetes mellitus, hypercoagulable disorders, obesity, hypoalbuminemia, and warfarin/ corticosteroid use.2 However, most often no obvious triggers are identified.1
Regardless of the etiology, calciphylaxis is characterized by the calcification of blood vessels and connective tissues, leading to vessel injury, intimal fibrosis, and thrombosis, followed by ischemic necrosis of the skin and soft tissue. It is postulated that microvascular calcification occurs as an active cell-mediated process that depends on the balance between the promoters and inhibitors of calcification.1 In our patient, liver disease likely predisposed formation of calcification through the creation of an environment susceptible to vascular injury via decreased synthesis of proteins C and S.3 Synthesis of fetuin-A, a protein that acts as a circulating inhibitor of vascular ossification/calcification, also is decreased in calcification. Another inhibitor of calcification, matrix Gla protein, is unable to undergo activation through vitamin K–dependent carboxylation secondary to liver disease–induced vitamin K deficiency.3 Microvascular calcification without calciphylaxis may occur in other conditions such as type 2 diabetes mellitus. Therefore, clinicopathologic correlation is important in determining the diagnosis.
Calciphylaxis has a variety of clinical presentations depending on the stage of disease. It begins as a fixed, indurated, livedo reticularis–like plaque. The lesions become increasingly violaceous with intermixed areas of light blanched skin secondary to ischemia and then develop retiform pupura.4 Eventually, affected sites can become bullous and ulcerate or form a necrotic eschar. Severe pain is a cardinal feature throughout all stages.4 Lesions in nonuremic calciphylaxis most commonly are located in the central and/or proximal areas of the body.2
Clinical suspicion is essential for diagnosis. Skin biopsy is the standard method for confirmation in unclear cases. The classic histologic features include intravascular and extravascular calcification, microthrombosis, and fibrointimal hyperplasia of the small dermal and subcutaneous arteries and arterioles, leading to ischemia and intense septal panniculitis.1 Von Kossa immunostaining is used to increase the detection of calcium deposits (Figure 1).1 In addition to the classic changes, our case demonstrated a rare histologic variant with pseudoxanthoma elasticum (PXE)–like changes (Figure 2), which are thought to occur secondary to pathologic elastin fibrogenesis or increased proteolytic activity resulting in abnormal remodeling of the extracellular matrix in the setting of increased calcification of elastin fibers.5 Detection of PXE-like changes may be a helpful clue when specimens lack other characteristic signs.
Wound care, pain control, and addressing underlying causes are mainstays of therapy. Sodium thiosulfate, an antioxidant with vasodilatory properties that also inhibits adipocyte calcification and blocks the ability of adipocytes to induce calcification of vascular smooth-muscle cells, also is useful. Antibiotic prophylaxis is not indicated.1
Even with treatment, both uremic and nonuremic calciphylaxis have a dismal prognosis; 1-year mortality is approximately 50% to 60% and rises to 80% at 2 years.4 Lesion location affects prognosis, and more proximal lesions portend worse outcomes. In patients with both proximal and distal lesions, there is a 90% mortality rate within 1 year. Ulceration also portends worse outcomes, as the wounds often are resistant to healing and act as nidi for infection.4 Septicemia is the most common cause of death.1
Ecthyma gangrenosum is a cutaneous manifestation secondary to an infection most commonly associated with Pseudomonas aeruginosa.6 It often presents in immunocompromised patients with an underlying gramnegative septicemia.7 The clinical presentation initially begins with painless macules that rapidly progress into necrotic ulcers, usually accompanied by associated systemic symptoms such as fever, chills, and hypotension. Histopathology reveals numerous gram-negative rods around necrotic vessels.7
Idiopathic purpura fulminans is the rarest form of purpura fulminans. It is caused by autoantibody formation against protein S, resulting in protein S depletion and subsequent hypercoagulability.8 It usually occurs 7 to 10 days after the onset of a precipitating infection. Lesions begin as erythematous macules that progress within hours to painful, sharply defined areas of purpura and hemorrhagic cutaneous necrosis that may extend to deeper tissues.8 Secondary infection of gangrenous tissue may occur. Distribution usually is diffuse and signs of septic shock and disseminated intravascular coagulation usually are present.
Hughes syndrome, also known as antiphospholipid syndrome, is an acquired autoimmune disorder that manifests clinically as recurrent arterial or venous thrombosis.9 Cutaneous manifestations consist of livedo reticularis, arterial and venous ulcers, and superficial thrombophlebitis.10 Laboratory testing for antiphospholipid antibodies and obtaining a detailed history of the patient’s cardiovascular health are crucial for diagnosis.9
Necrotizing fasciitis typically begins as an inconspicuous superficial cutaneous infection that rapidly is transmitted to the fascia. Infection can spread along fascial planes for several days without affecting the overlying skin, leading to delayed diagnosis.11 The first signs to appear are disproportionate pain and a change in skin color to reddish-purple or bluish-gray. Next, the skin will become indurated, swollen, shiny, and more painful.11 Skin breakdown will begin in 3 to 5 days and is accompanied by bullae and cutaneous gangrene. The involved area becomes painless due to thrombosis of the small vessels that supply the superficial nerves.12 Septic shock ultimately will develop if untreated.
We present a rare case of nonuremic calciphylaxis. We encourage dermatologists to include calciphylaxis in the differential when evaluating any patient with a painful retiform rash or ulcerated eschar, even in the absence of renal disease.
- Nigwekar SU, Thadhani R, Brandenburg VM. Calciphylaxis. N Engl J Med. 2018;378:1704-1714.
- Nigwekar SU, Wolf M, Sterns RH, et al. Calciphylaxis from nonuremic causes: a systematic review. Clin J Am Soc Nephrol. 2008;3:1139-1143.
- Sammour YM, Saleh HM, Gad MM, et al. Non-uremic calciphylaxis associated with alcoholic hepatitis: a case report. World J Hepatol. 2019;11:127-132.
- James WD, Elston DM, Treat J, et al, eds. Cutaneous vascular diseases. Andrews’ Diseases of the Skin: Clinical Dermatology. Elsevier; 2020:813-861.
- Nathoo RK, Harb JN, Auerbach J, et al. Pseudoxanthoma elasticum-like changes in nonuremic calciphylaxis: case series and brief review of a helpful diagnostic clue. J Cutan Pathol. 2017;44:1064-1069.
- Vaiman M, Lazarovitch T, Heller L, et al. Ecthyma gangrenosum and ecthyma-like lesions: review article. Eur J Clin Microbiol Infect Dis Off Publ Eur Soc Clin Microbiol. 2015;34:633-639.
- Greene SL, Su WP, Muller SA. Ecthyma gangrenosum: report of clinical, histopathologic, and bacteriologic aspects of eight cases. J Am Acad Dermatol. 1984;11(5 pt 1):781-787.
- Levin M, Eley BS, Louis J, et al. Postinfectious purpura fulminans caused by an autoantibody directed against protein S. J Pediatr. 1995;127:355-363.
- Hughes G. Hughes syndrome: the antiphospholipid syndrome—a clinical overview. Clin Rev Allergy Immunol. 2007;32:3-12.
- Chang Y, Dabiri G, Damstetter E, et al. Coagulation disorders and their cutaneous presentations: pathophysiology. J Am Acad Dermatol. 2016;74:783-792; quiz 793-794.
- Fais P, Viero A, Viel G, et al. Necrotizing fasciitis: case series and review of the literature on clinical and medico-legal diagnostic challenges. Int J Legal Med. 2018;132:1357-1366.
- Brook I. Microbiology and management of soft tissue and muscle infections. Int J Surg Lond Engl. 2008;6:328-338.
- Nigwekar SU, Thadhani R, Brandenburg VM. Calciphylaxis. N Engl J Med. 2018;378:1704-1714.
- Nigwekar SU, Wolf M, Sterns RH, et al. Calciphylaxis from nonuremic causes: a systematic review. Clin J Am Soc Nephrol. 2008;3:1139-1143.
- Sammour YM, Saleh HM, Gad MM, et al. Non-uremic calciphylaxis associated with alcoholic hepatitis: a case report. World J Hepatol. 2019;11:127-132.
- James WD, Elston DM, Treat J, et al, eds. Cutaneous vascular diseases. Andrews’ Diseases of the Skin: Clinical Dermatology. Elsevier; 2020:813-861.
- Nathoo RK, Harb JN, Auerbach J, et al. Pseudoxanthoma elasticum-like changes in nonuremic calciphylaxis: case series and brief review of a helpful diagnostic clue. J Cutan Pathol. 2017;44:1064-1069.
- Vaiman M, Lazarovitch T, Heller L, et al. Ecthyma gangrenosum and ecthyma-like lesions: review article. Eur J Clin Microbiol Infect Dis Off Publ Eur Soc Clin Microbiol. 2015;34:633-639.
- Greene SL, Su WP, Muller SA. Ecthyma gangrenosum: report of clinical, histopathologic, and bacteriologic aspects of eight cases. J Am Acad Dermatol. 1984;11(5 pt 1):781-787.
- Levin M, Eley BS, Louis J, et al. Postinfectious purpura fulminans caused by an autoantibody directed against protein S. J Pediatr. 1995;127:355-363.
- Hughes G. Hughes syndrome: the antiphospholipid syndrome—a clinical overview. Clin Rev Allergy Immunol. 2007;32:3-12.
- Chang Y, Dabiri G, Damstetter E, et al. Coagulation disorders and their cutaneous presentations: pathophysiology. J Am Acad Dermatol. 2016;74:783-792; quiz 793-794.
- Fais P, Viero A, Viel G, et al. Necrotizing fasciitis: case series and review of the literature on clinical and medico-legal diagnostic challenges. Int J Legal Med. 2018;132:1357-1366.
- Brook I. Microbiology and management of soft tissue and muscle infections. Int J Surg Lond Engl. 2008;6:328-338.
A 50-year-old woman presented to our dermatology clinic with an exquisitely tender, nonhealing lesion on the left leg of 2 weeks’ duration that began as a small red-purplish spot. She applied a triple antibiotic ointment and wrapped the area with gauze daily but reported that it continued to enlarge and darken in color before forming a “scab.” She noted occasional seropurulent discharge and denied any trauma or new exposures to the area. She was seen at a local emergency department 3 days prior to presentation and was prescribed oral clindamycin for suspected cellulitis, but she denied any improvement with the initiation of antibiotics. Her medical history was notable for obesity, depression, hypothyroidism, and liver disease secondary to alcohol use disorder. She reported that she drank a pint of vodka daily. Her medications included pantoprazole, spironolactone, bumetanide, citalopram, levothyroxine, naltrexone, tramadol, and a multivitamin. Physical examination revealed violaceous mottling with areas of superficial erythema and ulceration with necrotic eschars on the proximal left thigh that were extremely painful. A biopsy was obtained for confirmation of diagnosis, but the patient died before the results were returned.


When is an allergic reaction to raw plant food due to tree pollen?
A new guideline aims to help primary care doctors differentiate pollen food syndrome (PFS) – a cross-reactive allergic reaction to certain raw, but not cooked, plant foods – from other food allergies.
The guideline from the British Society of Allergy and Clinical Immunology (BSACI) focuses on birch tree pollen, the major sensitizing PFS allergen in Northern Europe. Providers may be able to diagnose PFS related to birch pollen from clinical history alone, including the foods involved and the rapidity of symptom onset, write lead author Isabel J. Skypala, PhD, RD, of Imperial College London, and her colleagues.
The new BSACI guideline for diagnosis and management of PFS was published in Clinical & Experimental Allergy.
PFS is common and increasingly prevalent
PFS – also called oral allergy syndrome and pollen food allergy syndrome – is common and increasingly prevalent. PFS can begin at any age but usually starts in pollen-sensitized school-age children and adults with seasonal allergic rhinitis.
Symptoms from similar proteins in food
Mild to moderate allergic symptoms develop quickly when people sensitized to birch pollen eat raw plant foods that contain proteins similar to those in the pollen, such as pathogenesis-related protein PR-10. The allergens are broken down by cooking or processing.
Symptoms usually occur immediately or within 15 minutes of eating. Patients may have tingling; itching or soreness in the mouth, throat, or ears; mild lip and oral mucosa angioedema; itchy hands, sneezing, or eye symptoms; tongue or pharynx angioedema; perioral rash; cough; abdominal pain; nausea; and/or worsening of eczema. In children, itch and rash may predominate.
Triggers depend on pollen type
PFS triggers vary depending on a person’s pollen sensitization, which is affected by their geographic area and local dietary habits. In the United Kingdom, almost 70% of birch-allergic adults and more than 40% of birch-allergic children have PFS, the authors write.
Typical triggers include eating apples, stone fruits, kiwis, carrots, celery, hazelnuts, almonds, walnuts, soymilk, and peanuts, as well as peeling potatoes or other root vegetables. Freshly prepared vegetable or fruit smoothies or juices, celery, soymilk, raw nuts, large quantities of roasted nuts, and concentrated nut products can cause more severe reactions.
Diagnostic clinical history
If a patient answers yes to these questions, they almost certainly have PFS, the authors write:
- Are symptoms caused by raw fruits, nuts, carrots, or celery?
- Are the same trigger foods tolerated when they’re cooked well or roasted?
- Do symptoms come immediately or within a few minutes of eating?
- Do symptoms occur in the oropharynx and include tingling, itching, or swelling?
- Does the patient have seasonal allergic rhinitis or sensitization to pollen?
Testing needed for some cases
Allergy tests may be needed for people who report atypical or severe reactions or who also react to cooked or processed plant foods, such as roasted nuts, nuts in foods, fruits or vegetables in juices and smoothies, and soy products other than milk. Tests may also be needed for people who react to foods that are not linked with PFS, such as cashews, pistachios, macadamias, sesame seeds, beans, lentils, and chickpeas.
Whether PFS reactions also occur to roasted hazelnuts, almonds, walnuts, Brazil nuts, or peanuts, either alone or in composite foods such as chocolates, spreads, desserts, and snacks, is unclear.
An oral food challenge to confirm PFS is needed only if the history and diagnostic tests are inconclusive or if the patient is avoiding multiple foods.
Dietary management
PFS is managed by excluding known trigger foods. This becomes challenging for patients with preexisting food allergies and for vegetarians and vegans.
Personalized dietary advice is needed to avoid nutritional imbalance, minimize anxiety and unnecessary food restrictions, and improve quality of life. Reactions after accidental exposure often resolve without medication, and if antihistamines are needed, they rarely require self-injectable devices.
Guideline helpful beyond the United Kingdom and birch pollen
Allyson S. Larkin, MD, associate professor of pediatrics at the University of Pittsburgh School of Medicine, told this news organization in an email that the guideline summarizes in great detail the pathophysiology behind PFS and highlights how component testing may help diagnose patients and manage the condition.
“Patients worry very much about the progression and severity of allergic reactions,” said Dr. Larkin, who was not involved in the guideline development.
“As the authors note, recognizing the nutritional consequences of dietary restrictions is important, and nutrition consults and suitable alternative suggestions are very helpful for these patients, especially for those with food allergy or who are vegetarian or vegan.”
Jill A. Poole, MD, professor of medicine and chief of the Division of Allergy and Immunology at the University of Nebraska College of Medicine, Omaha, noted that PFS, although common, is underrecognized by the public and by health care providers.
“People are not allergic to the specific food, but they are allergic to a seasonal allergen, such as birch tree, that cross-reacts with the food protein, which is typically changed with cooking,” she explained in an email.
“This differs from reactions by those who have moderate to severe allergic food-specific reactions that may include systemic reactions like anaphylaxis from eating certain foods,” she said.
“Importantly, the number of cross-reacting foods with seasonal pollens continues to grow, and the extent of testing has expanded in recent years,” advised Dr. Poole, who also was not involved in the guideline development.
The authors recommend further related research into food immunotherapy and other novel PFS treatments. They also want to raise awareness of factors affecting PFS prevalence, such as increased spread and allergenicity of pollen due to climate change, pollution, the global consumption of previously local traditional foods, and the increase in vegetarian and vegan diets.
The authors, Dr. Larkin, and Dr. Poole report no relevant financial relationships involving this guideline. The guideline was not funded.
A version of this article first appeared on Medscape.com.
A new guideline aims to help primary care doctors differentiate pollen food syndrome (PFS) – a cross-reactive allergic reaction to certain raw, but not cooked, plant foods – from other food allergies.
The guideline from the British Society of Allergy and Clinical Immunology (BSACI) focuses on birch tree pollen, the major sensitizing PFS allergen in Northern Europe. Providers may be able to diagnose PFS related to birch pollen from clinical history alone, including the foods involved and the rapidity of symptom onset, write lead author Isabel J. Skypala, PhD, RD, of Imperial College London, and her colleagues.
The new BSACI guideline for diagnosis and management of PFS was published in Clinical & Experimental Allergy.
PFS is common and increasingly prevalent
PFS – also called oral allergy syndrome and pollen food allergy syndrome – is common and increasingly prevalent. PFS can begin at any age but usually starts in pollen-sensitized school-age children and adults with seasonal allergic rhinitis.
Symptoms from similar proteins in food
Mild to moderate allergic symptoms develop quickly when people sensitized to birch pollen eat raw plant foods that contain proteins similar to those in the pollen, such as pathogenesis-related protein PR-10. The allergens are broken down by cooking or processing.
Symptoms usually occur immediately or within 15 minutes of eating. Patients may have tingling; itching or soreness in the mouth, throat, or ears; mild lip and oral mucosa angioedema; itchy hands, sneezing, or eye symptoms; tongue or pharynx angioedema; perioral rash; cough; abdominal pain; nausea; and/or worsening of eczema. In children, itch and rash may predominate.
Triggers depend on pollen type
PFS triggers vary depending on a person’s pollen sensitization, which is affected by their geographic area and local dietary habits. In the United Kingdom, almost 70% of birch-allergic adults and more than 40% of birch-allergic children have PFS, the authors write.
Typical triggers include eating apples, stone fruits, kiwis, carrots, celery, hazelnuts, almonds, walnuts, soymilk, and peanuts, as well as peeling potatoes or other root vegetables. Freshly prepared vegetable or fruit smoothies or juices, celery, soymilk, raw nuts, large quantities of roasted nuts, and concentrated nut products can cause more severe reactions.
Diagnostic clinical history
If a patient answers yes to these questions, they almost certainly have PFS, the authors write:
- Are symptoms caused by raw fruits, nuts, carrots, or celery?
- Are the same trigger foods tolerated when they’re cooked well or roasted?
- Do symptoms come immediately or within a few minutes of eating?
- Do symptoms occur in the oropharynx and include tingling, itching, or swelling?
- Does the patient have seasonal allergic rhinitis or sensitization to pollen?
Testing needed for some cases
Allergy tests may be needed for people who report atypical or severe reactions or who also react to cooked or processed plant foods, such as roasted nuts, nuts in foods, fruits or vegetables in juices and smoothies, and soy products other than milk. Tests may also be needed for people who react to foods that are not linked with PFS, such as cashews, pistachios, macadamias, sesame seeds, beans, lentils, and chickpeas.
Whether PFS reactions also occur to roasted hazelnuts, almonds, walnuts, Brazil nuts, or peanuts, either alone or in composite foods such as chocolates, spreads, desserts, and snacks, is unclear.
An oral food challenge to confirm PFS is needed only if the history and diagnostic tests are inconclusive or if the patient is avoiding multiple foods.
Dietary management
PFS is managed by excluding known trigger foods. This becomes challenging for patients with preexisting food allergies and for vegetarians and vegans.
Personalized dietary advice is needed to avoid nutritional imbalance, minimize anxiety and unnecessary food restrictions, and improve quality of life. Reactions after accidental exposure often resolve without medication, and if antihistamines are needed, they rarely require self-injectable devices.
Guideline helpful beyond the United Kingdom and birch pollen
Allyson S. Larkin, MD, associate professor of pediatrics at the University of Pittsburgh School of Medicine, told this news organization in an email that the guideline summarizes in great detail the pathophysiology behind PFS and highlights how component testing may help diagnose patients and manage the condition.
“Patients worry very much about the progression and severity of allergic reactions,” said Dr. Larkin, who was not involved in the guideline development.
“As the authors note, recognizing the nutritional consequences of dietary restrictions is important, and nutrition consults and suitable alternative suggestions are very helpful for these patients, especially for those with food allergy or who are vegetarian or vegan.”
Jill A. Poole, MD, professor of medicine and chief of the Division of Allergy and Immunology at the University of Nebraska College of Medicine, Omaha, noted that PFS, although common, is underrecognized by the public and by health care providers.
“People are not allergic to the specific food, but they are allergic to a seasonal allergen, such as birch tree, that cross-reacts with the food protein, which is typically changed with cooking,” she explained in an email.
“This differs from reactions by those who have moderate to severe allergic food-specific reactions that may include systemic reactions like anaphylaxis from eating certain foods,” she said.
“Importantly, the number of cross-reacting foods with seasonal pollens continues to grow, and the extent of testing has expanded in recent years,” advised Dr. Poole, who also was not involved in the guideline development.
The authors recommend further related research into food immunotherapy and other novel PFS treatments. They also want to raise awareness of factors affecting PFS prevalence, such as increased spread and allergenicity of pollen due to climate change, pollution, the global consumption of previously local traditional foods, and the increase in vegetarian and vegan diets.
The authors, Dr. Larkin, and Dr. Poole report no relevant financial relationships involving this guideline. The guideline was not funded.
A version of this article first appeared on Medscape.com.
A new guideline aims to help primary care doctors differentiate pollen food syndrome (PFS) – a cross-reactive allergic reaction to certain raw, but not cooked, plant foods – from other food allergies.
The guideline from the British Society of Allergy and Clinical Immunology (BSACI) focuses on birch tree pollen, the major sensitizing PFS allergen in Northern Europe. Providers may be able to diagnose PFS related to birch pollen from clinical history alone, including the foods involved and the rapidity of symptom onset, write lead author Isabel J. Skypala, PhD, RD, of Imperial College London, and her colleagues.
The new BSACI guideline for diagnosis and management of PFS was published in Clinical & Experimental Allergy.
PFS is common and increasingly prevalent
PFS – also called oral allergy syndrome and pollen food allergy syndrome – is common and increasingly prevalent. PFS can begin at any age but usually starts in pollen-sensitized school-age children and adults with seasonal allergic rhinitis.
Symptoms from similar proteins in food
Mild to moderate allergic symptoms develop quickly when people sensitized to birch pollen eat raw plant foods that contain proteins similar to those in the pollen, such as pathogenesis-related protein PR-10. The allergens are broken down by cooking or processing.
Symptoms usually occur immediately or within 15 minutes of eating. Patients may have tingling; itching or soreness in the mouth, throat, or ears; mild lip and oral mucosa angioedema; itchy hands, sneezing, or eye symptoms; tongue or pharynx angioedema; perioral rash; cough; abdominal pain; nausea; and/or worsening of eczema. In children, itch and rash may predominate.
Triggers depend on pollen type
PFS triggers vary depending on a person’s pollen sensitization, which is affected by their geographic area and local dietary habits. In the United Kingdom, almost 70% of birch-allergic adults and more than 40% of birch-allergic children have PFS, the authors write.
Typical triggers include eating apples, stone fruits, kiwis, carrots, celery, hazelnuts, almonds, walnuts, soymilk, and peanuts, as well as peeling potatoes or other root vegetables. Freshly prepared vegetable or fruit smoothies or juices, celery, soymilk, raw nuts, large quantities of roasted nuts, and concentrated nut products can cause more severe reactions.
Diagnostic clinical history
If a patient answers yes to these questions, they almost certainly have PFS, the authors write:
- Are symptoms caused by raw fruits, nuts, carrots, or celery?
- Are the same trigger foods tolerated when they’re cooked well or roasted?
- Do symptoms come immediately or within a few minutes of eating?
- Do symptoms occur in the oropharynx and include tingling, itching, or swelling?
- Does the patient have seasonal allergic rhinitis or sensitization to pollen?
Testing needed for some cases
Allergy tests may be needed for people who report atypical or severe reactions or who also react to cooked or processed plant foods, such as roasted nuts, nuts in foods, fruits or vegetables in juices and smoothies, and soy products other than milk. Tests may also be needed for people who react to foods that are not linked with PFS, such as cashews, pistachios, macadamias, sesame seeds, beans, lentils, and chickpeas.
Whether PFS reactions also occur to roasted hazelnuts, almonds, walnuts, Brazil nuts, or peanuts, either alone or in composite foods such as chocolates, spreads, desserts, and snacks, is unclear.
An oral food challenge to confirm PFS is needed only if the history and diagnostic tests are inconclusive or if the patient is avoiding multiple foods.
Dietary management
PFS is managed by excluding known trigger foods. This becomes challenging for patients with preexisting food allergies and for vegetarians and vegans.
Personalized dietary advice is needed to avoid nutritional imbalance, minimize anxiety and unnecessary food restrictions, and improve quality of life. Reactions after accidental exposure often resolve without medication, and if antihistamines are needed, they rarely require self-injectable devices.
Guideline helpful beyond the United Kingdom and birch pollen
Allyson S. Larkin, MD, associate professor of pediatrics at the University of Pittsburgh School of Medicine, told this news organization in an email that the guideline summarizes in great detail the pathophysiology behind PFS and highlights how component testing may help diagnose patients and manage the condition.
“Patients worry very much about the progression and severity of allergic reactions,” said Dr. Larkin, who was not involved in the guideline development.
“As the authors note, recognizing the nutritional consequences of dietary restrictions is important, and nutrition consults and suitable alternative suggestions are very helpful for these patients, especially for those with food allergy or who are vegetarian or vegan.”
Jill A. Poole, MD, professor of medicine and chief of the Division of Allergy and Immunology at the University of Nebraska College of Medicine, Omaha, noted that PFS, although common, is underrecognized by the public and by health care providers.
“People are not allergic to the specific food, but they are allergic to a seasonal allergen, such as birch tree, that cross-reacts with the food protein, which is typically changed with cooking,” she explained in an email.
“This differs from reactions by those who have moderate to severe allergic food-specific reactions that may include systemic reactions like anaphylaxis from eating certain foods,” she said.
“Importantly, the number of cross-reacting foods with seasonal pollens continues to grow, and the extent of testing has expanded in recent years,” advised Dr. Poole, who also was not involved in the guideline development.
The authors recommend further related research into food immunotherapy and other novel PFS treatments. They also want to raise awareness of factors affecting PFS prevalence, such as increased spread and allergenicity of pollen due to climate change, pollution, the global consumption of previously local traditional foods, and the increase in vegetarian and vegan diets.
The authors, Dr. Larkin, and Dr. Poole report no relevant financial relationships involving this guideline. The guideline was not funded.
A version of this article first appeared on Medscape.com.
A 10-year-old with a red bump on her lower lip
The patient’s history and examination are consistent with a diagnosis of pyogenic granuloma. Specifically, the history of rapid growth, friable nature, associated bleeding, and hemorrhagic crusting point to pyogenic granuloma as the most likely diagnosis.
Pyogenic granuloma is an acquired benign vascular growth of the skin or mucous membranes.1 It most frequently occurs in children and young adults and most commonly affects the skin of the head, trunk, and extremities.2 Common mucosal sites include the gingiva, lips, and tongue.2 The etiology of pyogenic granuloma is unknown, though it is thought to be a process akin to the overgrowth of granulation tissue.3,4 Expression of angiogenic factors and subsequent vascular hyperplasia are also implicated as key players in the pathogenesis of pyogenic granuloma.1,4 In addition, several associated factors and inciting triggers have been proposed including trauma, infections, and hormonal fluctuations.3-5 However, the majority of patients do not report predisposing factors or a history of prior trauma at the site.3,6

Clinically, pyogenic granuloma usually presents as a painless, erythematous, dome-shaped friable papule or nodule that easily bleeds and may ulcerate. It typically undergoes a period of growth over weeks to months followed by stabilization. Occasionally, pyogenic granulomas will spontaneously involute, though most do not.7 Pyogenic granuloma may occur within an existing capillary malformation, such as a port wine stain, spontaneously or as a sequela of laser treatment.8,9 Diagnosis of pyogenic granuloma can typically be made clinically on the basis of history and exam. Dermoscopic evaluation of pyogenic granuloma will reveal a homogeneous papule with a surrounding white-brown collarette, and potentially white intersecting lines.10 Histopathologic evaluation may be necessary to differentiate lesions from conditions that may mimic pyogenic granuloma.
What’s on the differential?
The differential diagnosis for pyogenic granuloma consists of Spitz nevus, cherry hemangioma, amelanotic melanoma, and glomus tumor.
Spitz nevus
Spitz nevus (spindle and epithelial cell nevus) is a benign melanocytic lesion that classically appears as a sharply circumscribed, smooth, dome-shaped, pink-red, or brown papule or plaque. There is typically a history of rapid growth over several months followed by stabilization. It usually presents in childhood or adolescence and is most commonly located on the face and extremities. While there are similarities in the appearance of Spitz nevi and pyogenic granuloma, Spitz nevi are not usually friable nor associated with bleeding as in our patient. Furthermore, on dermoscopy, Spitz nevus typically exhibits a starburst pattern with regularly distributed dotted vessels, or a peripheral globular pattern with reticular depigmentation. The definitive diagnosis of Spitz nevi relies on histopathologic evaluation, which is critical for discriminating Spitz nevi from melanoma.
Cherry hemangioma

Cherry angiomas are the most common type of acquired benign vascular proliferation. They present as small, bright red or violaceous macules or papules. However, they typically appear in early to midadulthood and increase in number with age. The age of our patient and solitary presentation of the lesion make this diagnosis unlikely. In addition, cherry angiomas are not usually associated with bleeding. It is important to note that, depending on the age of the patient, pyogenic granuloma may also be confused with infantile hemangioma. Infantile hemangiomas may become bright red papules, nodules, or plaques that appear in early infancy. They characteristically involute, which does not typically happen with pyogenic granuloma.
Amelanotic melanoma
Amelanotic melanoma is an uncommon variant of melanoma with little to no pigmentation. It may appear as a skin-colored to light-brown, pink, or red macule, papule, or nodule. The lesion may be asymmetric with irregular and well-defined borders. The variable and uncharacteristic appearance of this melanoma variant makes it diagnostically challenging and it is often confused with benign lesions including pyogenic granuloma. Dermoscopy can help distinguish amelanotic melanoma from other benign conditions, and will reveal areas of pink to white, polymorphous vessels and crystalline structures. However, ultimately biopsy and histopathological evaluation is necessary for accurate diagnosis.
Glomus tumor
Glomus tumors are rare, benign neoplasms originating from cells of the glomus body that presents as a red-purple, vascular papule or nodule. They are usually found in areas rich in glomus bodies, such as the subungual regions, fingertips, palms, wrists, and forearms. Glomus tumors are typically associated with tenderness, paroxysmal pain, and cold sensitivity. They do not bleed or ulcerate. While pyogenic granuloma may be confused for glomus tumor when present on the fingers or extremities, the location of the lesion in our patient is not consistent with a diagnosis of glomus tumor.
Management and disease course
Management with procedural or topical interventions is usually pursued for pyogenic granuloma because of frequent bleeding and ulceration of lesions. The most common approach is simple excision by a scoop or shave technique, with or without curettage and most commonly with electrocautery of the base. Other options include full-thickness excision, destruction with laser therapy, cryotherapy, or topical treatments (for example, timolol).11 Lesion recurrence can occur with both surgical and nonsurgical management.11 Regardless of management technique, it is useful to obtain histopathologic evaluation of tissue for accurate diagnosis.
Our patient underwent surgical destruction of her lower-lip lesion with shave excision followed by electrocautery. The surgical specimen was sent for pathology, which confirmed the diagnosis of pyogenic granuloma. The patient experienced no complications from the procedure and did not have recurrence of the lesion.
Ms. Sui is a research associate in the department of dermatology, division of pediatric and adolescent dermatology, University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Neither Ms. Sui nor Dr. Eichenfield have any relevant financial disclosures.
References
1. Lin RL and Janniger CK. Cutis. 2004 Oct;74(4):229-33.
2. Harris MN et al. J Am Acad Dermatol. 2000 Jun;42(6):1012-6.
3. Pagliai KA and Cohen BA. Pediatr Dermatol. 2004 Jan-Feb;21(1):10-3.
4. Kamal R et al. J Oral Maxillofac Pathol. 2012 Jan;16(1):79-82.
5. Requena L and Sangueza OP. J Am Acad Dermatol. 1997 Dec;37(6):887-919.
6. Patrice SJ et al. Pediatr Dermatol. 1991 Dec;8(4):267-76.
7. Luba MC et al. Am Fam Physician. 2003 Feb 15;67(4):729-38.
8. Swerlick RA and Cooper PH. J Am Acad Dermatol. 1983 May;8(5):627-30.
9. Sheehan DJ and Lesher JL Jr. Cutis. 2004 Mar;73(3):175-80.
10. Zaballos P et al. Br J Dermatol. 2006 Jun;154(6):1108-11.
11. Lee J et al. J Plast Reconstr Aesthet Surg. 2011 Sep;64(9):1216-20. .
The patient’s history and examination are consistent with a diagnosis of pyogenic granuloma. Specifically, the history of rapid growth, friable nature, associated bleeding, and hemorrhagic crusting point to pyogenic granuloma as the most likely diagnosis.
Pyogenic granuloma is an acquired benign vascular growth of the skin or mucous membranes.1 It most frequently occurs in children and young adults and most commonly affects the skin of the head, trunk, and extremities.2 Common mucosal sites include the gingiva, lips, and tongue.2 The etiology of pyogenic granuloma is unknown, though it is thought to be a process akin to the overgrowth of granulation tissue.3,4 Expression of angiogenic factors and subsequent vascular hyperplasia are also implicated as key players in the pathogenesis of pyogenic granuloma.1,4 In addition, several associated factors and inciting triggers have been proposed including trauma, infections, and hormonal fluctuations.3-5 However, the majority of patients do not report predisposing factors or a history of prior trauma at the site.3,6
Clinically, pyogenic granuloma usually presents as a painless, erythematous, dome-shaped friable papule or nodule that easily bleeds and may ulcerate. It typically undergoes a period of growth over weeks to months followed by stabilization. Occasionally, pyogenic granulomas will spontaneously involute, though most do not.7 Pyogenic granuloma may occur within an existing capillary malformation, such as a port wine stain, spontaneously or as a sequela of laser treatment.8,9 Diagnosis of pyogenic granuloma can typically be made clinically on the basis of history and exam. Dermoscopic evaluation of pyogenic granuloma will reveal a homogeneous papule with a surrounding white-brown collarette, and potentially white intersecting lines.10 Histopathologic evaluation may be necessary to differentiate lesions from conditions that may mimic pyogenic granuloma.
What’s on the differential?
The differential diagnosis for pyogenic granuloma consists of Spitz nevus, cherry hemangioma, amelanotic melanoma, and glomus tumor.
Spitz nevus
Spitz nevus (spindle and epithelial cell nevus) is a benign melanocytic lesion that classically appears as a sharply circumscribed, smooth, dome-shaped, pink-red, or brown papule or plaque. There is typically a history of rapid growth over several months followed by stabilization. It usually presents in childhood or adolescence and is most commonly located on the face and extremities. While there are similarities in the appearance of Spitz nevi and pyogenic granuloma, Spitz nevi are not usually friable nor associated with bleeding as in our patient. Furthermore, on dermoscopy, Spitz nevus typically exhibits a starburst pattern with regularly distributed dotted vessels, or a peripheral globular pattern with reticular depigmentation. The definitive diagnosis of Spitz nevi relies on histopathologic evaluation, which is critical for discriminating Spitz nevi from melanoma.
Cherry hemangioma
Cherry angiomas are the most common type of acquired benign vascular proliferation. They present as small, bright red or violaceous macules or papules. However, they typically appear in early to midadulthood and increase in number with age. The age of our patient and solitary presentation of the lesion make this diagnosis unlikely. In addition, cherry angiomas are not usually associated with bleeding. It is important to note that, depending on the age of the patient, pyogenic granuloma may also be confused with infantile hemangioma. Infantile hemangiomas may become bright red papules, nodules, or plaques that appear in early infancy. They characteristically involute, which does not typically happen with pyogenic granuloma.
Amelanotic melanoma
Amelanotic melanoma is an uncommon variant of melanoma with little to no pigmentation. It may appear as a skin-colored to light-brown, pink, or red macule, papule, or nodule. The lesion may be asymmetric with irregular and well-defined borders. The variable and uncharacteristic appearance of this melanoma variant makes it diagnostically challenging and it is often confused with benign lesions including pyogenic granuloma. Dermoscopy can help distinguish amelanotic melanoma from other benign conditions, and will reveal areas of pink to white, polymorphous vessels and crystalline structures. However, ultimately biopsy and histopathological evaluation is necessary for accurate diagnosis.
Glomus tumor
Glomus tumors are rare, benign neoplasms originating from cells of the glomus body that presents as a red-purple, vascular papule or nodule. They are usually found in areas rich in glomus bodies, such as the subungual regions, fingertips, palms, wrists, and forearms. Glomus tumors are typically associated with tenderness, paroxysmal pain, and cold sensitivity. They do not bleed or ulcerate. While pyogenic granuloma may be confused for glomus tumor when present on the fingers or extremities, the location of the lesion in our patient is not consistent with a diagnosis of glomus tumor.
Management and disease course
Management with procedural or topical interventions is usually pursued for pyogenic granuloma because of frequent bleeding and ulceration of lesions. The most common approach is simple excision by a scoop or shave technique, with or without curettage and most commonly with electrocautery of the base. Other options include full-thickness excision, destruction with laser therapy, cryotherapy, or topical treatments (for example, timolol).11 Lesion recurrence can occur with both surgical and nonsurgical management.11 Regardless of management technique, it is useful to obtain histopathologic evaluation of tissue for accurate diagnosis.
Our patient underwent surgical destruction of her lower-lip lesion with shave excision followed by electrocautery. The surgical specimen was sent for pathology, which confirmed the diagnosis of pyogenic granuloma. The patient experienced no complications from the procedure and did not have recurrence of the lesion.
Ms. Sui is a research associate in the department of dermatology, division of pediatric and adolescent dermatology, University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Neither Ms. Sui nor Dr. Eichenfield have any relevant financial disclosures.
References
1. Lin RL and Janniger CK. Cutis. 2004 Oct;74(4):229-33.
2. Harris MN et al. J Am Acad Dermatol. 2000 Jun;42(6):1012-6.
3. Pagliai KA and Cohen BA. Pediatr Dermatol. 2004 Jan-Feb;21(1):10-3.
4. Kamal R et al. J Oral Maxillofac Pathol. 2012 Jan;16(1):79-82.
5. Requena L and Sangueza OP. J Am Acad Dermatol. 1997 Dec;37(6):887-919.
6. Patrice SJ et al. Pediatr Dermatol. 1991 Dec;8(4):267-76.
7. Luba MC et al. Am Fam Physician. 2003 Feb 15;67(4):729-38.
8. Swerlick RA and Cooper PH. J Am Acad Dermatol. 1983 May;8(5):627-30.
9. Sheehan DJ and Lesher JL Jr. Cutis. 2004 Mar;73(3):175-80.
10. Zaballos P et al. Br J Dermatol. 2006 Jun;154(6):1108-11.
11. Lee J et al. J Plast Reconstr Aesthet Surg. 2011 Sep;64(9):1216-20. .
The patient’s history and examination are consistent with a diagnosis of pyogenic granuloma. Specifically, the history of rapid growth, friable nature, associated bleeding, and hemorrhagic crusting point to pyogenic granuloma as the most likely diagnosis.
Pyogenic granuloma is an acquired benign vascular growth of the skin or mucous membranes.1 It most frequently occurs in children and young adults and most commonly affects the skin of the head, trunk, and extremities.2 Common mucosal sites include the gingiva, lips, and tongue.2 The etiology of pyogenic granuloma is unknown, though it is thought to be a process akin to the overgrowth of granulation tissue.3,4 Expression of angiogenic factors and subsequent vascular hyperplasia are also implicated as key players in the pathogenesis of pyogenic granuloma.1,4 In addition, several associated factors and inciting triggers have been proposed including trauma, infections, and hormonal fluctuations.3-5 However, the majority of patients do not report predisposing factors or a history of prior trauma at the site.3,6
Clinically, pyogenic granuloma usually presents as a painless, erythematous, dome-shaped friable papule or nodule that easily bleeds and may ulcerate. It typically undergoes a period of growth over weeks to months followed by stabilization. Occasionally, pyogenic granulomas will spontaneously involute, though most do not.7 Pyogenic granuloma may occur within an existing capillary malformation, such as a port wine stain, spontaneously or as a sequela of laser treatment.8,9 Diagnosis of pyogenic granuloma can typically be made clinically on the basis of history and exam. Dermoscopic evaluation of pyogenic granuloma will reveal a homogeneous papule with a surrounding white-brown collarette, and potentially white intersecting lines.10 Histopathologic evaluation may be necessary to differentiate lesions from conditions that may mimic pyogenic granuloma.
What’s on the differential?
The differential diagnosis for pyogenic granuloma consists of Spitz nevus, cherry hemangioma, amelanotic melanoma, and glomus tumor.
Spitz nevus
Spitz nevus (spindle and epithelial cell nevus) is a benign melanocytic lesion that classically appears as a sharply circumscribed, smooth, dome-shaped, pink-red, or brown papule or plaque. There is typically a history of rapid growth over several months followed by stabilization. It usually presents in childhood or adolescence and is most commonly located on the face and extremities. While there are similarities in the appearance of Spitz nevi and pyogenic granuloma, Spitz nevi are not usually friable nor associated with bleeding as in our patient. Furthermore, on dermoscopy, Spitz nevus typically exhibits a starburst pattern with regularly distributed dotted vessels, or a peripheral globular pattern with reticular depigmentation. The definitive diagnosis of Spitz nevi relies on histopathologic evaluation, which is critical for discriminating Spitz nevi from melanoma.
Cherry hemangioma
Cherry angiomas are the most common type of acquired benign vascular proliferation. They present as small, bright red or violaceous macules or papules. However, they typically appear in early to midadulthood and increase in number with age. The age of our patient and solitary presentation of the lesion make this diagnosis unlikely. In addition, cherry angiomas are not usually associated with bleeding. It is important to note that, depending on the age of the patient, pyogenic granuloma may also be confused with infantile hemangioma. Infantile hemangiomas may become bright red papules, nodules, or plaques that appear in early infancy. They characteristically involute, which does not typically happen with pyogenic granuloma.
Amelanotic melanoma
Amelanotic melanoma is an uncommon variant of melanoma with little to no pigmentation. It may appear as a skin-colored to light-brown, pink, or red macule, papule, or nodule. The lesion may be asymmetric with irregular and well-defined borders. The variable and uncharacteristic appearance of this melanoma variant makes it diagnostically challenging and it is often confused with benign lesions including pyogenic granuloma. Dermoscopy can help distinguish amelanotic melanoma from other benign conditions, and will reveal areas of pink to white, polymorphous vessels and crystalline structures. However, ultimately biopsy and histopathological evaluation is necessary for accurate diagnosis.
Glomus tumor
Glomus tumors are rare, benign neoplasms originating from cells of the glomus body that presents as a red-purple, vascular papule or nodule. They are usually found in areas rich in glomus bodies, such as the subungual regions, fingertips, palms, wrists, and forearms. Glomus tumors are typically associated with tenderness, paroxysmal pain, and cold sensitivity. They do not bleed or ulcerate. While pyogenic granuloma may be confused for glomus tumor when present on the fingers or extremities, the location of the lesion in our patient is not consistent with a diagnosis of glomus tumor.
Management and disease course
Management with procedural or topical interventions is usually pursued for pyogenic granuloma because of frequent bleeding and ulceration of lesions. The most common approach is simple excision by a scoop or shave technique, with or without curettage and most commonly with electrocautery of the base. Other options include full-thickness excision, destruction with laser therapy, cryotherapy, or topical treatments (for example, timolol).11 Lesion recurrence can occur with both surgical and nonsurgical management.11 Regardless of management technique, it is useful to obtain histopathologic evaluation of tissue for accurate diagnosis.
Our patient underwent surgical destruction of her lower-lip lesion with shave excision followed by electrocautery. The surgical specimen was sent for pathology, which confirmed the diagnosis of pyogenic granuloma. The patient experienced no complications from the procedure and did not have recurrence of the lesion.
Ms. Sui is a research associate in the department of dermatology, division of pediatric and adolescent dermatology, University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Neither Ms. Sui nor Dr. Eichenfield have any relevant financial disclosures.
References
1. Lin RL and Janniger CK. Cutis. 2004 Oct;74(4):229-33.
2. Harris MN et al. J Am Acad Dermatol. 2000 Jun;42(6):1012-6.
3. Pagliai KA and Cohen BA. Pediatr Dermatol. 2004 Jan-Feb;21(1):10-3.
4. Kamal R et al. J Oral Maxillofac Pathol. 2012 Jan;16(1):79-82.
5. Requena L and Sangueza OP. J Am Acad Dermatol. 1997 Dec;37(6):887-919.
6. Patrice SJ et al. Pediatr Dermatol. 1991 Dec;8(4):267-76.
7. Luba MC et al. Am Fam Physician. 2003 Feb 15;67(4):729-38.
8. Swerlick RA and Cooper PH. J Am Acad Dermatol. 1983 May;8(5):627-30.
9. Sheehan DJ and Lesher JL Jr. Cutis. 2004 Mar;73(3):175-80.
10. Zaballos P et al. Br J Dermatol. 2006 Jun;154(6):1108-11.
11. Lee J et al. J Plast Reconstr Aesthet Surg. 2011 Sep;64(9):1216-20. .
Scattered Flesh-Colored Papules in a Linear Array in the Setting of Diffuse Skin Thickening
The Diagnosis: Scleromyxedema
A punch biopsy of the upper back performed at an outside institution revealed increased histiocytes and abundant interstitial mucin confined to the papillary dermis (Figures 1 and 2), consistent with the lichen myxedematosus (LM) papules that may be seen in scleromyxedema. Serum protein electrophoresis revealed the presence of a protein of restricted mobility on the gamma region that occupied 5.3% of the total protein (0.3 g/dL). Urine protein electrophoresis showed free kappa light chain monoclonal protein in the gamma region. Immunofixation electrophoresis revealed the presence of IgG kappa monoclonal protein in the gamma region with 10% monotype kappa cells. The presence of Raynaud phenomenon and positive antinuclear antibody (1:320, speckled) was noted. Laboratory studies for thyroid-stimulating hormone, C-reactive protein, Scl-70 antibody, myositis panel, ribonucleoprotein antibody, Smith antibody, Sjögren syndrome–related antigens A and B antibodies, rheumatoid factor, and RNA polymerase III antibody all were within reference range. Our patient was treated with monthly intravenous immunoglobulin (IVIG), and he noted substantial improvement in skin findings after 3 months of IVIG.
.")
Localized lichen myxedematosus is a rare idiopathic cutaneous disease that clinically is characterized by waxy indurated papules and histologically is characterized by diffuse mucin deposition and fibroblast proliferation in the upper dermis.1 Scleromyxedema is a diffuse variant of LM in which the papules and plaques of LM are associated with skin thickening involving almost the entire body and associated systemic disease. The exact mechanism of this disease is unknown, but the most widely accepted hypothesis is that immunoglobulins and cytokines contribute to the synthesis of glycosaminoglycans and thereby the deposition of mucin in the dermis.2 Scleromyxedema has a chronic course and generally responds poorly to existing treatments.1 Partial improvement has been demonstrated in treatment with topical calcineurin inhibitors and topical steroids.2
.")
The differential diagnosis in our patient included scleromyxedema, scleredema, scleroderma, LM, and reticular erythematosus mucinosis. He was diagnosed with scleromyxedema with kappa monoclonal gammopathy. Scleromyxedema is a rare disorder involving the deposition of mucinous material in the papillary dermis that causes the formation of infiltrative skin lesions.3 The etiology is unknown, but the presence of a monoclonal protein is an important characteristic of this disorder. It is important to rule out thyroid disease as a possible etiology before concluding that the disease process is driven by the monoclonal gammopathy; this will help determine appropriate therapies.4,5 Usually the monoclonal protein is associated with the IgG lambda subtype. Intravenous immunoglobulin often is considered as a first-line treatment of scleromyxedema and usually is administered at a dosage of 2 g/kg divided over 2 to 5 consecutive days per month.3 Previously, our patient had been treated with IVIG for 3 years for chronic inflammatory demyelinating polyneuropathy and had stopped 1 to 2 years before his cutaneous symptoms started. Generally, scleromyxedema patients must stay on IVIG long-term to prevent relapse, typically every 6 to 8 weeks. Second-line treatments for scleromyxedema include systemic corticosteroids and thalidomide.6 Scleromyxedema and LM have several clinical and histopathologic features in common. Our patient’s biopsy revealed increased mucin deposition associated with fibroblast proliferation confined to the superficial dermis. These histologic changes can be seen in the setting of either LM or scleromyxedema. Our patient’s diffuse skin thickening and monoclonal gammopathy were more characteristic of scleromyxedema. In contrast, LM is a localized eruption with no internal organ manifestations and no associated systemic disease, such as monoclonal gammopathy and thyroid disease.
Scleredema adultorum of Buschke (also referred to as scleredema) is a rare idiopathic dermatologic condition characterized by thickening and tightening of the skin that leads to firm, nonpitting, woody edema that initially involves the upper back and neck but can spread to the face, scalp, and shoulders; importantly, scleredema spares the hands and feet.7 Scleredema has been associated with type 2 diabetes mellitus, streptococcal upper respiratory tract infections, and monoclonal gammopathy.8 Although our patient did have a monoclonal gammopathy, he also experienced prominent hand involvement with diffuse skin thickening, which is not typical of scleredema. Additionally, biopsy of scleredema would show increased mucin but would not show the proliferation of fibroblasts that was seen in our patient’s biopsy. Furthermore, scleredema has more profound diffuse superficial and deep mucin deposition compared to scleromyxedema. Scleroderma is an autoimmune cutaneous condition that is divided into 2 categories: localized scleroderma and systemic sclerosis (SSc).9 Localized scleroderma (also called morphea) often is characterized by indurated hyperpigmented or hypopigmented lesions. There is an absence of Raynaud phenomenon, telangiectasia, and systemic disease.9 Systemic sclerosis is further divided into 2 categories—limited cutaneous and diffuse cutaneous—which are differentiated by the extent of organ system involvement. Limited cutaneous SSc involves calcinosis, Raynaud phenomenon, esophageal dysmotility, skin sclerosis distal to the elbows and knees, and telangiectasia.9 Diffuse cutaneous SSc is characterized by Raynaud phenomenon; cutaneous sclerosis proximal to the elbows and knees; and fibrosis of the gastrointestinal, pulmonary, renal, and cardiac systems.9 Scl-70 antibodies are specific for diffuse cutaneous SSc, and centromere antibodies are specific for limited cutaneous SSc. Scleromyxedema shares many of the same clinical symptoms as scleroderma; therefore, histopathologic examination is important for differentiating these disorders. Histologically, scleroderma is characterized by thickened collagen bundles associated with a variable degree of perivascular and interstitial lymphoplasmacytic inflammation. No increased dermal mucin is present.9 Our patient did not have the clinical cutaneous features of localized scleroderma and lacked the signs of internal organ involvement that typically are found in SSc. He did have Raynaud phenomenon but did not have matlike telangiectases or Scl-70 or centromere antibodies.
Reticular erythematosus mucinosis (REM) is a rare inflammatory cutaneous disease that is characterized by diffuse reticular erythematous macules or papules that may be asymptomatic or associated with pruritus.10 Reticular erythematosus mucinosis most frequently affects middle-aged women and appears on the trunk.9 Our patient was not part of the demographic group most frequently affected by REM. More importantly, our patient’s lesions were not erythematous or reticular in appearance, making the diagnosis of REM unlikely. Furthermore, REM has no associated cutaneous sclerosis or induration.
- Nofal A, Amer H, Alakad R, et al. Lichen myxedematosus: diagnostic criteria, classification, and severity grading. Int J Dermatol. 2017;56:284-290.
- Christman MP, Sukhdeo K, Kim RH, et al. Papular mucinosis, or localized lichen myxedematosus (LM)(discrete papular type). Dermatol Online J. 2017;23:8.
- Haber R, Bachour J, El Gemayel M. Scleromyxedema treatment: a systematic review and update. Int J Dermatol. 2020;59:1191-1201.
- Hazan E, Griffin TD Jr, Jabbour SA, et al. Scleromyxedema in a patient with thyroid disease: an atypical case or a case for revised criteria? Cutis. 2020;105:E6-E10.
- Shenoy A, Steixner J, Beltrani V, et al. Discrete papular lichen myxedematosus and scleromyxedema with hypothyroidism: a report of two cases. Case Rep Dermatol. 2019;11:64-70.
- Hoffman JHO, Enk AH. Scleromyxedema. J Dtsch Dermatol Ges. 2020;18:1449-1467.
- Beers WH, Ince AI, Moore TL. Scleredema adultorum of Buschke: a case report and review of the literature. Semin Arthritis Rheum. 2006;35:355-359.
- Miguel D, Schliemann S, Elsner P. Treatment of scleroderma adultorum Buschke: a systematic review. Acta Derm Venereol. 2018;98:305-309.
- Rongioletti F, Ferreli C, Atzori L, et al. Scleroderma with an update about clinicopathological correlation. G Ital Dermatol Venereol. 2018;153:208-215.
- Ocanha-Xavier JP, Cola-Senra CO, Xavier-Junior JCC. Reticular erythematous mucinosis: literature review and case report of a 24-year-old patient with systemic erythematosus lupus. Lupus. 2021;30:325-335.
The Diagnosis: Scleromyxedema
A punch biopsy of the upper back performed at an outside institution revealed increased histiocytes and abundant interstitial mucin confined to the papillary dermis (Figures 1 and 2), consistent with the lichen myxedematosus (LM) papules that may be seen in scleromyxedema. Serum protein electrophoresis revealed the presence of a protein of restricted mobility on the gamma region that occupied 5.3% of the total protein (0.3 g/dL). Urine protein electrophoresis showed free kappa light chain monoclonal protein in the gamma region. Immunofixation electrophoresis revealed the presence of IgG kappa monoclonal protein in the gamma region with 10% monotype kappa cells. The presence of Raynaud phenomenon and positive antinuclear antibody (1:320, speckled) was noted. Laboratory studies for thyroid-stimulating hormone, C-reactive protein, Scl-70 antibody, myositis panel, ribonucleoprotein antibody, Smith antibody, Sjögren syndrome–related antigens A and B antibodies, rheumatoid factor, and RNA polymerase III antibody all were within reference range. Our patient was treated with monthly intravenous immunoglobulin (IVIG), and he noted substantial improvement in skin findings after 3 months of IVIG.
Localized lichen myxedematosus is a rare idiopathic cutaneous disease that clinically is characterized by waxy indurated papules and histologically is characterized by diffuse mucin deposition and fibroblast proliferation in the upper dermis.1 Scleromyxedema is a diffuse variant of LM in which the papules and plaques of LM are associated with skin thickening involving almost the entire body and associated systemic disease. The exact mechanism of this disease is unknown, but the most widely accepted hypothesis is that immunoglobulins and cytokines contribute to the synthesis of glycosaminoglycans and thereby the deposition of mucin in the dermis.2 Scleromyxedema has a chronic course and generally responds poorly to existing treatments.1 Partial improvement has been demonstrated in treatment with topical calcineurin inhibitors and topical steroids.2
The differential diagnosis in our patient included scleromyxedema, scleredema, scleroderma, LM, and reticular erythematosus mucinosis. He was diagnosed with scleromyxedema with kappa monoclonal gammopathy. Scleromyxedema is a rare disorder involving the deposition of mucinous material in the papillary dermis that causes the formation of infiltrative skin lesions.3 The etiology is unknown, but the presence of a monoclonal protein is an important characteristic of this disorder. It is important to rule out thyroid disease as a possible etiology before concluding that the disease process is driven by the monoclonal gammopathy; this will help determine appropriate therapies.4,5 Usually the monoclonal protein is associated with the IgG lambda subtype. Intravenous immunoglobulin often is considered as a first-line treatment of scleromyxedema and usually is administered at a dosage of 2 g/kg divided over 2 to 5 consecutive days per month.3 Previously, our patient had been treated with IVIG for 3 years for chronic inflammatory demyelinating polyneuropathy and had stopped 1 to 2 years before his cutaneous symptoms started. Generally, scleromyxedema patients must stay on IVIG long-term to prevent relapse, typically every 6 to 8 weeks. Second-line treatments for scleromyxedema include systemic corticosteroids and thalidomide.6 Scleromyxedema and LM have several clinical and histopathologic features in common. Our patient’s biopsy revealed increased mucin deposition associated with fibroblast proliferation confined to the superficial dermis. These histologic changes can be seen in the setting of either LM or scleromyxedema. Our patient’s diffuse skin thickening and monoclonal gammopathy were more characteristic of scleromyxedema. In contrast, LM is a localized eruption with no internal organ manifestations and no associated systemic disease, such as monoclonal gammopathy and thyroid disease.
Scleredema adultorum of Buschke (also referred to as scleredema) is a rare idiopathic dermatologic condition characterized by thickening and tightening of the skin that leads to firm, nonpitting, woody edema that initially involves the upper back and neck but can spread to the face, scalp, and shoulders; importantly, scleredema spares the hands and feet.7 Scleredema has been associated with type 2 diabetes mellitus, streptococcal upper respiratory tract infections, and monoclonal gammopathy.8 Although our patient did have a monoclonal gammopathy, he also experienced prominent hand involvement with diffuse skin thickening, which is not typical of scleredema. Additionally, biopsy of scleredema would show increased mucin but would not show the proliferation of fibroblasts that was seen in our patient’s biopsy. Furthermore, scleredema has more profound diffuse superficial and deep mucin deposition compared to scleromyxedema. Scleroderma is an autoimmune cutaneous condition that is divided into 2 categories: localized scleroderma and systemic sclerosis (SSc).9 Localized scleroderma (also called morphea) often is characterized by indurated hyperpigmented or hypopigmented lesions. There is an absence of Raynaud phenomenon, telangiectasia, and systemic disease.9 Systemic sclerosis is further divided into 2 categories—limited cutaneous and diffuse cutaneous—which are differentiated by the extent of organ system involvement. Limited cutaneous SSc involves calcinosis, Raynaud phenomenon, esophageal dysmotility, skin sclerosis distal to the elbows and knees, and telangiectasia.9 Diffuse cutaneous SSc is characterized by Raynaud phenomenon; cutaneous sclerosis proximal to the elbows and knees; and fibrosis of the gastrointestinal, pulmonary, renal, and cardiac systems.9 Scl-70 antibodies are specific for diffuse cutaneous SSc, and centromere antibodies are specific for limited cutaneous SSc. Scleromyxedema shares many of the same clinical symptoms as scleroderma; therefore, histopathologic examination is important for differentiating these disorders. Histologically, scleroderma is characterized by thickened collagen bundles associated with a variable degree of perivascular and interstitial lymphoplasmacytic inflammation. No increased dermal mucin is present.9 Our patient did not have the clinical cutaneous features of localized scleroderma and lacked the signs of internal organ involvement that typically are found in SSc. He did have Raynaud phenomenon but did not have matlike telangiectases or Scl-70 or centromere antibodies.
Reticular erythematosus mucinosis (REM) is a rare inflammatory cutaneous disease that is characterized by diffuse reticular erythematous macules or papules that may be asymptomatic or associated with pruritus.10 Reticular erythematosus mucinosis most frequently affects middle-aged women and appears on the trunk.9 Our patient was not part of the demographic group most frequently affected by REM. More importantly, our patient’s lesions were not erythematous or reticular in appearance, making the diagnosis of REM unlikely. Furthermore, REM has no associated cutaneous sclerosis or induration.
The Diagnosis: Scleromyxedema
A punch biopsy of the upper back performed at an outside institution revealed increased histiocytes and abundant interstitial mucin confined to the papillary dermis (Figures 1 and 2), consistent with the lichen myxedematosus (LM) papules that may be seen in scleromyxedema. Serum protein electrophoresis revealed the presence of a protein of restricted mobility on the gamma region that occupied 5.3% of the total protein (0.3 g/dL). Urine protein electrophoresis showed free kappa light chain monoclonal protein in the gamma region. Immunofixation electrophoresis revealed the presence of IgG kappa monoclonal protein in the gamma region with 10% monotype kappa cells. The presence of Raynaud phenomenon and positive antinuclear antibody (1:320, speckled) was noted. Laboratory studies for thyroid-stimulating hormone, C-reactive protein, Scl-70 antibody, myositis panel, ribonucleoprotein antibody, Smith antibody, Sjögren syndrome–related antigens A and B antibodies, rheumatoid factor, and RNA polymerase III antibody all were within reference range. Our patient was treated with monthly intravenous immunoglobulin (IVIG), and he noted substantial improvement in skin findings after 3 months of IVIG.
Localized lichen myxedematosus is a rare idiopathic cutaneous disease that clinically is characterized by waxy indurated papules and histologically is characterized by diffuse mucin deposition and fibroblast proliferation in the upper dermis.1 Scleromyxedema is a diffuse variant of LM in which the papules and plaques of LM are associated with skin thickening involving almost the entire body and associated systemic disease. The exact mechanism of this disease is unknown, but the most widely accepted hypothesis is that immunoglobulins and cytokines contribute to the synthesis of glycosaminoglycans and thereby the deposition of mucin in the dermis.2 Scleromyxedema has a chronic course and generally responds poorly to existing treatments.1 Partial improvement has been demonstrated in treatment with topical calcineurin inhibitors and topical steroids.2
The differential diagnosis in our patient included scleromyxedema, scleredema, scleroderma, LM, and reticular erythematosus mucinosis. He was diagnosed with scleromyxedema with kappa monoclonal gammopathy. Scleromyxedema is a rare disorder involving the deposition of mucinous material in the papillary dermis that causes the formation of infiltrative skin lesions.3 The etiology is unknown, but the presence of a monoclonal protein is an important characteristic of this disorder. It is important to rule out thyroid disease as a possible etiology before concluding that the disease process is driven by the monoclonal gammopathy; this will help determine appropriate therapies.4,5 Usually the monoclonal protein is associated with the IgG lambda subtype. Intravenous immunoglobulin often is considered as a first-line treatment of scleromyxedema and usually is administered at a dosage of 2 g/kg divided over 2 to 5 consecutive days per month.3 Previously, our patient had been treated with IVIG for 3 years for chronic inflammatory demyelinating polyneuropathy and had stopped 1 to 2 years before his cutaneous symptoms started. Generally, scleromyxedema patients must stay on IVIG long-term to prevent relapse, typically every 6 to 8 weeks. Second-line treatments for scleromyxedema include systemic corticosteroids and thalidomide.6 Scleromyxedema and LM have several clinical and histopathologic features in common. Our patient’s biopsy revealed increased mucin deposition associated with fibroblast proliferation confined to the superficial dermis. These histologic changes can be seen in the setting of either LM or scleromyxedema. Our patient’s diffuse skin thickening and monoclonal gammopathy were more characteristic of scleromyxedema. In contrast, LM is a localized eruption with no internal organ manifestations and no associated systemic disease, such as monoclonal gammopathy and thyroid disease.
Scleredema adultorum of Buschke (also referred to as scleredema) is a rare idiopathic dermatologic condition characterized by thickening and tightening of the skin that leads to firm, nonpitting, woody edema that initially involves the upper back and neck but can spread to the face, scalp, and shoulders; importantly, scleredema spares the hands and feet.7 Scleredema has been associated with type 2 diabetes mellitus, streptococcal upper respiratory tract infections, and monoclonal gammopathy.8 Although our patient did have a monoclonal gammopathy, he also experienced prominent hand involvement with diffuse skin thickening, which is not typical of scleredema. Additionally, biopsy of scleredema would show increased mucin but would not show the proliferation of fibroblasts that was seen in our patient’s biopsy. Furthermore, scleredema has more profound diffuse superficial and deep mucin deposition compared to scleromyxedema. Scleroderma is an autoimmune cutaneous condition that is divided into 2 categories: localized scleroderma and systemic sclerosis (SSc).9 Localized scleroderma (also called morphea) often is characterized by indurated hyperpigmented or hypopigmented lesions. There is an absence of Raynaud phenomenon, telangiectasia, and systemic disease.9 Systemic sclerosis is further divided into 2 categories—limited cutaneous and diffuse cutaneous—which are differentiated by the extent of organ system involvement. Limited cutaneous SSc involves calcinosis, Raynaud phenomenon, esophageal dysmotility, skin sclerosis distal to the elbows and knees, and telangiectasia.9 Diffuse cutaneous SSc is characterized by Raynaud phenomenon; cutaneous sclerosis proximal to the elbows and knees; and fibrosis of the gastrointestinal, pulmonary, renal, and cardiac systems.9 Scl-70 antibodies are specific for diffuse cutaneous SSc, and centromere antibodies are specific for limited cutaneous SSc. Scleromyxedema shares many of the same clinical symptoms as scleroderma; therefore, histopathologic examination is important for differentiating these disorders. Histologically, scleroderma is characterized by thickened collagen bundles associated with a variable degree of perivascular and interstitial lymphoplasmacytic inflammation. No increased dermal mucin is present.9 Our patient did not have the clinical cutaneous features of localized scleroderma and lacked the signs of internal organ involvement that typically are found in SSc. He did have Raynaud phenomenon but did not have matlike telangiectases or Scl-70 or centromere antibodies.
Reticular erythematosus mucinosis (REM) is a rare inflammatory cutaneous disease that is characterized by diffuse reticular erythematous macules or papules that may be asymptomatic or associated with pruritus.10 Reticular erythematosus mucinosis most frequently affects middle-aged women and appears on the trunk.9 Our patient was not part of the demographic group most frequently affected by REM. More importantly, our patient’s lesions were not erythematous or reticular in appearance, making the diagnosis of REM unlikely. Furthermore, REM has no associated cutaneous sclerosis or induration.
- Nofal A, Amer H, Alakad R, et al. Lichen myxedematosus: diagnostic criteria, classification, and severity grading. Int J Dermatol. 2017;56:284-290.
- Christman MP, Sukhdeo K, Kim RH, et al. Papular mucinosis, or localized lichen myxedematosus (LM)(discrete papular type). Dermatol Online J. 2017;23:8.
- Haber R, Bachour J, El Gemayel M. Scleromyxedema treatment: a systematic review and update. Int J Dermatol. 2020;59:1191-1201.
- Hazan E, Griffin TD Jr, Jabbour SA, et al. Scleromyxedema in a patient with thyroid disease: an atypical case or a case for revised criteria? Cutis. 2020;105:E6-E10.
- Shenoy A, Steixner J, Beltrani V, et al. Discrete papular lichen myxedematosus and scleromyxedema with hypothyroidism: a report of two cases. Case Rep Dermatol. 2019;11:64-70.
- Hoffman JHO, Enk AH. Scleromyxedema. J Dtsch Dermatol Ges. 2020;18:1449-1467.
- Beers WH, Ince AI, Moore TL. Scleredema adultorum of Buschke: a case report and review of the literature. Semin Arthritis Rheum. 2006;35:355-359.
- Miguel D, Schliemann S, Elsner P. Treatment of scleroderma adultorum Buschke: a systematic review. Acta Derm Venereol. 2018;98:305-309.
- Rongioletti F, Ferreli C, Atzori L, et al. Scleroderma with an update about clinicopathological correlation. G Ital Dermatol Venereol. 2018;153:208-215.
- Ocanha-Xavier JP, Cola-Senra CO, Xavier-Junior JCC. Reticular erythematous mucinosis: literature review and case report of a 24-year-old patient with systemic erythematosus lupus. Lupus. 2021;30:325-335.
- Nofal A, Amer H, Alakad R, et al. Lichen myxedematosus: diagnostic criteria, classification, and severity grading. Int J Dermatol. 2017;56:284-290.
- Christman MP, Sukhdeo K, Kim RH, et al. Papular mucinosis, or localized lichen myxedematosus (LM)(discrete papular type). Dermatol Online J. 2017;23:8.
- Haber R, Bachour J, El Gemayel M. Scleromyxedema treatment: a systematic review and update. Int J Dermatol. 2020;59:1191-1201.
- Hazan E, Griffin TD Jr, Jabbour SA, et al. Scleromyxedema in a patient with thyroid disease: an atypical case or a case for revised criteria? Cutis. 2020;105:E6-E10.
- Shenoy A, Steixner J, Beltrani V, et al. Discrete papular lichen myxedematosus and scleromyxedema with hypothyroidism: a report of two cases. Case Rep Dermatol. 2019;11:64-70.
- Hoffman JHO, Enk AH. Scleromyxedema. J Dtsch Dermatol Ges. 2020;18:1449-1467.
- Beers WH, Ince AI, Moore TL. Scleredema adultorum of Buschke: a case report and review of the literature. Semin Arthritis Rheum. 2006;35:355-359.
- Miguel D, Schliemann S, Elsner P. Treatment of scleroderma adultorum Buschke: a systematic review. Acta Derm Venereol. 2018;98:305-309.
- Rongioletti F, Ferreli C, Atzori L, et al. Scleroderma with an update about clinicopathological correlation. G Ital Dermatol Venereol. 2018;153:208-215.
- Ocanha-Xavier JP, Cola-Senra CO, Xavier-Junior JCC. Reticular erythematous mucinosis: literature review and case report of a 24-year-old patient with systemic erythematosus lupus. Lupus. 2021;30:325-335.
A 76-year-old man presented to our clinic with diffusely thickened and tightened skin that worsened over the course of 1 year, as well as numerous scattered small, firm, flesh-colored papules arranged in a linear pattern over the face, ears, neck, chest, abdomen, arms, hands, and knees. His symptoms progressed to include substantial skin thickening initially over the thighs followed by the arms, chest, back (top), and face. He developed confluent cobblestonelike plaques over the elbows and hands (bottom) and eventually developed decreased oral aperture limiting oral intake as well as decreased range of motion in the hands. The patient had a deep furrowed appearance of the brow accompanied by discrete, scattered, flesh-colored papules on the forehead and behind the ears. Deep furrows also were present on the back. When the proximal interphalangeal joints of the hands were extended, elevated rings with central depression were seen instead of horizontal folds.


Second opinions on melanocytic lesions swayed when first opinion is known
, diminishing the value and accuracy of an independent analysis.
In a novel effort to determine whether previous interpretations sway second opinions, 149 dermatopathologists were asked to read melanocytic skin biopsy specimens without access to the initial pathology report. A year or more later they read them again but now with access to the initial reading.

The study showed that the participants, independent of many variables, such as years of experience or frequency with which they offered second options, were more likely to upgrade or downgrade the severity of the specimens in accordance with the initial report even if their original reading was correct.
If the goal of a second dermatopathologist opinion is to obtain an independent diagnostic opinion, the message from this study is that they “should be blinded to first opinions,” according to the authors of this study, led by Joann G. Elmore, MD, professor of medicine, University of California, Los Angeles. The study was published online in JAMA Dermatology.
Two-phase study has 1-year washout
The study was conducted in two phases. In phase 1, a nationally representative sample of volunteer dermatopathologists performed 878 interpretations. In phase 2, conducted after a washout period of 12 months or more, the dermatopathologists read a random subset of the same cases evaluated in phase 1, but this time, unlike the first, they were first exposed to prior pathology reports.
Ultimately, “the dermatologists provided more than 5,000 interpretations of study cases, which was a big contribution of time,” Dr. Elmore said in an interview. Grateful for their critical contribution, she speculated that they were driven by the importance of the question being asked.
When categorized by the Melanocytic Pathology Assessment Tool (MPAT), which rates specimens from benign (class 1) to pT1b invasive melanoma (class 4), the influence of the prior report went in both directions, so that the likelihood of upgrading or downgrading went in accordance with the grading in the original dermatopathology report.
As a result, the risk of a less severe interpretation on the second relative to the first reading was 38% greater if the initial dermatopathology report had a lower grade (relative risk, 1.38; 95% confidence interval [CI], 1.19-1.59). The risk of upgrading the second report if the initial pathology report had a higher grade was increased by more than 50% (RR, 1.52; 95% CI, 1.34-1.73).
The greater likelihood of upgrading than downgrading is “understandable,” Dr. Elmore said. “I think this is consistent with the concern about missing something,” she explained.
According to Dr. Elmore, one of the greatest concerns regarding the bias imposed by the original pathology report is that the switch of opinions often went from one that was accurate to one that was inaccurate.
If the phase 1 diagnosis was accurate but upgraded in the phase 2 diagnosis, the risk of inaccuracy was almost doubled (RR, 1.96; 95% CI, 1.31-2.93). If the phase 1 report was inaccurate, the relative risk of changing the phase 2 diagnosis was still high but lower than if it was accurate (RR, 1.46; 95% CI, 1.27-1.68).
“That is, even when the phase 1 diagnoses agreed with the consensus reference diagnosis, they were swayed away from the correct diagnosis in phase 2 [when the initial pathology report characterized the specimen as higher grade],” Dr. Elmore reported.
Conversely, the risk of downgrading was about the same whether the phase 1 evaluation was accurate (RR, 1.37; 95% CI, 1.14-1.64) or inaccurate (RR 1.32; 95% CI, 1.07-1.64).
Downward and upward shifts in severity from an accurate diagnosis are concerning because of the likelihood they will lead to overtreatment or undertreatment. The problem, according to data from this study, is that dermatologists making a second opinion cannot judge their own susceptibility to being swayed by the original report.
Pathologists might be unaware of bias
At baseline, the participants were asked whether they thought they were influenced by the first interpretation when providing a second opinion. Although 69% acknowledged that they might be “somewhat influenced,” 31% maintained that they do not take initial reports into consideration. When the two groups were compared, the risk of downgrading was nearly identical. The risk of upgrading was lower in those claiming to disregard initial reports (RR, 1.29) relative to those who said they were “somewhat influenced” by a previous diagnosis (RR, 1.64), but the difference was not significant.
The actual risk of bias incurred by prior pathology reports might be greater than that captured in this study for several reasons, according to the investigators. They pointed out that all participants were experienced and board-certified and might therefore be expected to be more confident in their interpretations than an unselected group of dermatopathologists. In addition, participants might have been more careful in their interpretations knowing they were participating in a study.
“There are a lot of data to support the value of second opinions [in dermatopathology and other areas], but we need to consider the process of how they are being obtained,” Dr. Elmore said. “There needs to be a greater emphasis on providing an independent analysis.”
More than 60% of the dermatologists participating in this study reported that they agreed or strongly agreed with the premise that they prefer to have the original dermatopathology report when they offer a second opinion. Dr. Elmore said that the desire of those offering a second opinion to have as much information in front of them as possible is understandable, but the bias imposed by the original report weakens the value of the second opinion.
Blind reading of pathology reports needed
“These data suggest that seeing the original report sways opinions and that includes swaying opinions away from an accurate reading,” Dr. Elmore said. She thinks that for dermatopathologists to render a valuable and independent second opinion, the specimens should be examined “at least initially” without access to the first report.
The results of this study were not surprising to Vishal Anil Patel, MD, director of the Cutaneous Oncology Program, George Washington University Cancer Center, Washington. He made the point that physicians “are human first and foremost and not perfect machines.” As a result, he suggested bias and error are inevitable.
Although strategies to avoid bias are likely to offer some protection against inaccuracy, he said that diagnostic support tools such as artificial intelligence might be the right direction for improving inter- and intra-rater reliability.
Ruifeng Guo, MD, PhD, a consultant in the division of anatomic pathology at the Mayo Clinic, Rochester, Minn., agreed with the basic premise of the study, but he cautioned that restricting access to the initial pathology report might not always be the right approach.
It is true that “dermatopathologists providing a second opinion in diagnosing cutaneous melanoma are mostly unaware of the risk of bias if they read the initial pathology report,” said Dr. Guo, but restricting access comes with risks.
“There are also times critical information may be contained in the initial pathology report that needs to be considered when providing a second opinion consultation,” he noted. Ultimately, the decision to read or not read the initial report should be decided “on an individual basis.”
The study was funded by grants from the National Cancer Institute. Dr. Elmore, Dr. Patel, and Dr. Guo reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
, diminishing the value and accuracy of an independent analysis.
In a novel effort to determine whether previous interpretations sway second opinions, 149 dermatopathologists were asked to read melanocytic skin biopsy specimens without access to the initial pathology report. A year or more later they read them again but now with access to the initial reading.
The study showed that the participants, independent of many variables, such as years of experience or frequency with which they offered second options, were more likely to upgrade or downgrade the severity of the specimens in accordance with the initial report even if their original reading was correct.
If the goal of a second dermatopathologist opinion is to obtain an independent diagnostic opinion, the message from this study is that they “should be blinded to first opinions,” according to the authors of this study, led by Joann G. Elmore, MD, professor of medicine, University of California, Los Angeles. The study was published online in JAMA Dermatology.
Two-phase study has 1-year washout
The study was conducted in two phases. In phase 1, a nationally representative sample of volunteer dermatopathologists performed 878 interpretations. In phase 2, conducted after a washout period of 12 months or more, the dermatopathologists read a random subset of the same cases evaluated in phase 1, but this time, unlike the first, they were first exposed to prior pathology reports.
Ultimately, “the dermatologists provided more than 5,000 interpretations of study cases, which was a big contribution of time,” Dr. Elmore said in an interview. Grateful for their critical contribution, she speculated that they were driven by the importance of the question being asked.
When categorized by the Melanocytic Pathology Assessment Tool (MPAT), which rates specimens from benign (class 1) to pT1b invasive melanoma (class 4), the influence of the prior report went in both directions, so that the likelihood of upgrading or downgrading went in accordance with the grading in the original dermatopathology report.
As a result, the risk of a less severe interpretation on the second relative to the first reading was 38% greater if the initial dermatopathology report had a lower grade (relative risk, 1.38; 95% confidence interval [CI], 1.19-1.59). The risk of upgrading the second report if the initial pathology report had a higher grade was increased by more than 50% (RR, 1.52; 95% CI, 1.34-1.73).
The greater likelihood of upgrading than downgrading is “understandable,” Dr. Elmore said. “I think this is consistent with the concern about missing something,” she explained.
According to Dr. Elmore, one of the greatest concerns regarding the bias imposed by the original pathology report is that the switch of opinions often went from one that was accurate to one that was inaccurate.
If the phase 1 diagnosis was accurate but upgraded in the phase 2 diagnosis, the risk of inaccuracy was almost doubled (RR, 1.96; 95% CI, 1.31-2.93). If the phase 1 report was inaccurate, the relative risk of changing the phase 2 diagnosis was still high but lower than if it was accurate (RR, 1.46; 95% CI, 1.27-1.68).
“That is, even when the phase 1 diagnoses agreed with the consensus reference diagnosis, they were swayed away from the correct diagnosis in phase 2 [when the initial pathology report characterized the specimen as higher grade],” Dr. Elmore reported.
Conversely, the risk of downgrading was about the same whether the phase 1 evaluation was accurate (RR, 1.37; 95% CI, 1.14-1.64) or inaccurate (RR 1.32; 95% CI, 1.07-1.64).
Downward and upward shifts in severity from an accurate diagnosis are concerning because of the likelihood they will lead to overtreatment or undertreatment. The problem, according to data from this study, is that dermatologists making a second opinion cannot judge their own susceptibility to being swayed by the original report.
Pathologists might be unaware of bias
At baseline, the participants were asked whether they thought they were influenced by the first interpretation when providing a second opinion. Although 69% acknowledged that they might be “somewhat influenced,” 31% maintained that they do not take initial reports into consideration. When the two groups were compared, the risk of downgrading was nearly identical. The risk of upgrading was lower in those claiming to disregard initial reports (RR, 1.29) relative to those who said they were “somewhat influenced” by a previous diagnosis (RR, 1.64), but the difference was not significant.
The actual risk of bias incurred by prior pathology reports might be greater than that captured in this study for several reasons, according to the investigators. They pointed out that all participants were experienced and board-certified and might therefore be expected to be more confident in their interpretations than an unselected group of dermatopathologists. In addition, participants might have been more careful in their interpretations knowing they were participating in a study.
“There are a lot of data to support the value of second opinions [in dermatopathology and other areas], but we need to consider the process of how they are being obtained,” Dr. Elmore said. “There needs to be a greater emphasis on providing an independent analysis.”
More than 60% of the dermatologists participating in this study reported that they agreed or strongly agreed with the premise that they prefer to have the original dermatopathology report when they offer a second opinion. Dr. Elmore said that the desire of those offering a second opinion to have as much information in front of them as possible is understandable, but the bias imposed by the original report weakens the value of the second opinion.
Blind reading of pathology reports needed
“These data suggest that seeing the original report sways opinions and that includes swaying opinions away from an accurate reading,” Dr. Elmore said. She thinks that for dermatopathologists to render a valuable and independent second opinion, the specimens should be examined “at least initially” without access to the first report.
The results of this study were not surprising to Vishal Anil Patel, MD, director of the Cutaneous Oncology Program, George Washington University Cancer Center, Washington. He made the point that physicians “are human first and foremost and not perfect machines.” As a result, he suggested bias and error are inevitable.
Although strategies to avoid bias are likely to offer some protection against inaccuracy, he said that diagnostic support tools such as artificial intelligence might be the right direction for improving inter- and intra-rater reliability.
Ruifeng Guo, MD, PhD, a consultant in the division of anatomic pathology at the Mayo Clinic, Rochester, Minn., agreed with the basic premise of the study, but he cautioned that restricting access to the initial pathology report might not always be the right approach.
It is true that “dermatopathologists providing a second opinion in diagnosing cutaneous melanoma are mostly unaware of the risk of bias if they read the initial pathology report,” said Dr. Guo, but restricting access comes with risks.
“There are also times critical information may be contained in the initial pathology report that needs to be considered when providing a second opinion consultation,” he noted. Ultimately, the decision to read or not read the initial report should be decided “on an individual basis.”
The study was funded by grants from the National Cancer Institute. Dr. Elmore, Dr. Patel, and Dr. Guo reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
, diminishing the value and accuracy of an independent analysis.
In a novel effort to determine whether previous interpretations sway second opinions, 149 dermatopathologists were asked to read melanocytic skin biopsy specimens without access to the initial pathology report. A year or more later they read them again but now with access to the initial reading.
The study showed that the participants, independent of many variables, such as years of experience or frequency with which they offered second options, were more likely to upgrade or downgrade the severity of the specimens in accordance with the initial report even if their original reading was correct.
If the goal of a second dermatopathologist opinion is to obtain an independent diagnostic opinion, the message from this study is that they “should be blinded to first opinions,” according to the authors of this study, led by Joann G. Elmore, MD, professor of medicine, University of California, Los Angeles. The study was published online in JAMA Dermatology.
Two-phase study has 1-year washout
The study was conducted in two phases. In phase 1, a nationally representative sample of volunteer dermatopathologists performed 878 interpretations. In phase 2, conducted after a washout period of 12 months or more, the dermatopathologists read a random subset of the same cases evaluated in phase 1, but this time, unlike the first, they were first exposed to prior pathology reports.
Ultimately, “the dermatologists provided more than 5,000 interpretations of study cases, which was a big contribution of time,” Dr. Elmore said in an interview. Grateful for their critical contribution, she speculated that they were driven by the importance of the question being asked.
When categorized by the Melanocytic Pathology Assessment Tool (MPAT), which rates specimens from benign (class 1) to pT1b invasive melanoma (class 4), the influence of the prior report went in both directions, so that the likelihood of upgrading or downgrading went in accordance with the grading in the original dermatopathology report.
As a result, the risk of a less severe interpretation on the second relative to the first reading was 38% greater if the initial dermatopathology report had a lower grade (relative risk, 1.38; 95% confidence interval [CI], 1.19-1.59). The risk of upgrading the second report if the initial pathology report had a higher grade was increased by more than 50% (RR, 1.52; 95% CI, 1.34-1.73).
The greater likelihood of upgrading than downgrading is “understandable,” Dr. Elmore said. “I think this is consistent with the concern about missing something,” she explained.
According to Dr. Elmore, one of the greatest concerns regarding the bias imposed by the original pathology report is that the switch of opinions often went from one that was accurate to one that was inaccurate.
If the phase 1 diagnosis was accurate but upgraded in the phase 2 diagnosis, the risk of inaccuracy was almost doubled (RR, 1.96; 95% CI, 1.31-2.93). If the phase 1 report was inaccurate, the relative risk of changing the phase 2 diagnosis was still high but lower than if it was accurate (RR, 1.46; 95% CI, 1.27-1.68).
“That is, even when the phase 1 diagnoses agreed with the consensus reference diagnosis, they were swayed away from the correct diagnosis in phase 2 [when the initial pathology report characterized the specimen as higher grade],” Dr. Elmore reported.
Conversely, the risk of downgrading was about the same whether the phase 1 evaluation was accurate (RR, 1.37; 95% CI, 1.14-1.64) or inaccurate (RR 1.32; 95% CI, 1.07-1.64).
Downward and upward shifts in severity from an accurate diagnosis are concerning because of the likelihood they will lead to overtreatment or undertreatment. The problem, according to data from this study, is that dermatologists making a second opinion cannot judge their own susceptibility to being swayed by the original report.
Pathologists might be unaware of bias
At baseline, the participants were asked whether they thought they were influenced by the first interpretation when providing a second opinion. Although 69% acknowledged that they might be “somewhat influenced,” 31% maintained that they do not take initial reports into consideration. When the two groups were compared, the risk of downgrading was nearly identical. The risk of upgrading was lower in those claiming to disregard initial reports (RR, 1.29) relative to those who said they were “somewhat influenced” by a previous diagnosis (RR, 1.64), but the difference was not significant.
The actual risk of bias incurred by prior pathology reports might be greater than that captured in this study for several reasons, according to the investigators. They pointed out that all participants were experienced and board-certified and might therefore be expected to be more confident in their interpretations than an unselected group of dermatopathologists. In addition, participants might have been more careful in their interpretations knowing they were participating in a study.
“There are a lot of data to support the value of second opinions [in dermatopathology and other areas], but we need to consider the process of how they are being obtained,” Dr. Elmore said. “There needs to be a greater emphasis on providing an independent analysis.”
More than 60% of the dermatologists participating in this study reported that they agreed or strongly agreed with the premise that they prefer to have the original dermatopathology report when they offer a second opinion. Dr. Elmore said that the desire of those offering a second opinion to have as much information in front of them as possible is understandable, but the bias imposed by the original report weakens the value of the second opinion.
Blind reading of pathology reports needed
“These data suggest that seeing the original report sways opinions and that includes swaying opinions away from an accurate reading,” Dr. Elmore said. She thinks that for dermatopathologists to render a valuable and independent second opinion, the specimens should be examined “at least initially” without access to the first report.
The results of this study were not surprising to Vishal Anil Patel, MD, director of the Cutaneous Oncology Program, George Washington University Cancer Center, Washington. He made the point that physicians “are human first and foremost and not perfect machines.” As a result, he suggested bias and error are inevitable.
Although strategies to avoid bias are likely to offer some protection against inaccuracy, he said that diagnostic support tools such as artificial intelligence might be the right direction for improving inter- and intra-rater reliability.
Ruifeng Guo, MD, PhD, a consultant in the division of anatomic pathology at the Mayo Clinic, Rochester, Minn., agreed with the basic premise of the study, but he cautioned that restricting access to the initial pathology report might not always be the right approach.
It is true that “dermatopathologists providing a second opinion in diagnosing cutaneous melanoma are mostly unaware of the risk of bias if they read the initial pathology report,” said Dr. Guo, but restricting access comes with risks.
“There are also times critical information may be contained in the initial pathology report that needs to be considered when providing a second opinion consultation,” he noted. Ultimately, the decision to read or not read the initial report should be decided “on an individual basis.”
The study was funded by grants from the National Cancer Institute. Dr. Elmore, Dr. Patel, and Dr. Guo reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Exaggerated Facial Lines on the Forehead and Cheeks
The Diagnosis: Pachydermoperiostosis
Histopathology of the forehead punch biopsy demonstrated sebaceous hyperplasia with an occupation rate of greater than 40%, increased mucin, elastic fiber degeneration, and fibrosis. Pachydermia is graded from 0 to 3 depending on the degree of these changes; our patient met criteria for grade 3 pachydermia (Figure 1). Radiography revealed diffuse cortical thickening of the long bones that was most marked in the left femur (Figure 2); however, no other findings were demonstrative of Paget disease.
. B, Colloidal iron stain demonstrated increased mucin (original magnification ×4). C, Verhoeff-van Gieson stain showed elastic fiber")
Pachydermoperiostosis (PDP)(also known as Touraine-Solente-Golé syndrome or primary hypertrophic osteoarthropathy) is a rare genetic condition that affects both the dermatologic and skeletal systems. Clinical features of the disease include progressive thickening and furrowing of the skin on the scalp and face (known as pachydermia), digital clubbing, and periostosis. Other potential cutaneous features include seborrhea, acne, hyperhidrosis of the palms and soles, cutis verticis gyrata, eczema, and a burning sensation of the hands and feet. Myelofibrosis and gastrointestinal abnormalities also have been reported.1

The disease typically affects males (7:1 ratio); also, men typically display a more severe phenotype of the disease.2 It most commonly begins during puberty and follows a generally progressive course of 5 to 20 years before eventually stabilizing. Both autosomal-dominant with incomplete penetrance and recessive inheritance versions of PDP can occur. Prostaglandin E2 (PGE2) has been implicated in the pathogenesis of PDP; PGE2 is important in the inflammatory response and may evolve from disrupted protein degradation pathways.3 Sasaki et al4 additionally reported that the severity of pachydermia clinically and histologically appeared to correlate with the serum PGE2 levels in affected patients. Prostaglandin E2 causes a vasodilatory effect, perhaps explaining the clubbing observed in PDP, and also modifies the activity of osteoblasts and osteoclasts, causing the bone remodeling observed in the disease.4
In our patient, the initial differential diagnosis included PDP, as well as lepromatous leprosy, acromegaly, Paget disease of the bone, amyloidosis, scleromyxedema, and cutaneous T-cell lymphoma. However, the time course of the disease, lack of numerous symmetric thickened plaques and madarosis, and pathology argued against lepromatous leprosy. Acromegaly was ruled out due to lack of macroglossia as well as laboratory analysis within reference range including IGF-1 levels and thyroid function tests. Biopsy findings ultimately ruled out amyloidosis and cutaneous T-cell lymphoma. The bone scan revealed diffuse cortical thickening consistent with PDP, and there were no other radiologic findings suggestive of Paget disease. Pachydermoperiostosis is diagnosed using the Borochowitz criteria, which entails that 2 of the following 4 fulfillment criteria must be met: familial transmission, pachydermia, digital clubbing, and/or bony involvement with evidence of radiologic alterations or pain. Our patient met all 4 criteria. The clinical manifestations of PDP are variable with respect to skin and bone changes. The various clinical expressions include the complete form (ie, pachydermia, cutis verticis gyrata, periostosis), the incomplete form (ie, absence of cutis verticis gyrata), and forme fruste (ie, pachydermia with minimal or absent periostosis).5
Management for PDP involves surgical correction for cosmesis as well as for functional concerns if present. Symptoms secondary to periostosis should be managed with symptomatic treatment such as nonsteroidal antiinflammatory drugs. Patients managed with etoricoxib, a COX-2–selective nonsteroidal anti-inflammatory drug, have had normalized inflammatory markers that resulted in the lessening of forehead skin folds. Oral aescin has been shown to relieve joint pain due to its antiedematous effect.6 Our patient received treatment with nonsteroidal anti-inflammatory drugs for symptomatic management of the associated joint pain but unfortunately was lost to follow-up.
- Castori M, Sinibaldi L, Mingarelli R, et al. Pachydermoperiostosis: an update. Clin Genet. 2005;68:477-486.
- Reginato AJ, Shipachasse V, Guerrero R. Familial idiopathic hypertrophic osteoarthropathy and cranial suture defects in children. Skel Radiol. 1982;8:105-109.
- Coggins KG, Coffman TM, Koller BH. The Hippocratic finger points the blame at PGE2. Nat Genet. 2008;40:691-692.
- Sasaki T, Niizeki H, Shimizu A, et al. Identification of mutations in the prostaglandin transporter gene SLCO2A1 and its phenotype-genotype correlation in Japanese patients with pachydermoperiostosis. J Dermatol Sci. 2012;68:36-44.
- Bhaskaranand K, Shetty RR, Bhat AK. Pachydermoperiostosis: three case reports. J Orthop Surg (Hong Kong). 2001;9:61-66.
- Zhang H, Yang B. Successful treatment of pachydermoperiostosis patients with etoricoxib, aescin, and arthroscopic synovectomy: two case reports. Medicine (Baltimore). 2017;96:E8865.
The Diagnosis: Pachydermoperiostosis
Histopathology of the forehead punch biopsy demonstrated sebaceous hyperplasia with an occupation rate of greater than 40%, increased mucin, elastic fiber degeneration, and fibrosis. Pachydermia is graded from 0 to 3 depending on the degree of these changes; our patient met criteria for grade 3 pachydermia (Figure 1). Radiography revealed diffuse cortical thickening of the long bones that was most marked in the left femur (Figure 2); however, no other findings were demonstrative of Paget disease.
Pachydermoperiostosis (PDP)(also known as Touraine-Solente-Golé syndrome or primary hypertrophic osteoarthropathy) is a rare genetic condition that affects both the dermatologic and skeletal systems. Clinical features of the disease include progressive thickening and furrowing of the skin on the scalp and face (known as pachydermia), digital clubbing, and periostosis. Other potential cutaneous features include seborrhea, acne, hyperhidrosis of the palms and soles, cutis verticis gyrata, eczema, and a burning sensation of the hands and feet. Myelofibrosis and gastrointestinal abnormalities also have been reported.1
The disease typically affects males (7:1 ratio); also, men typically display a more severe phenotype of the disease.2 It most commonly begins during puberty and follows a generally progressive course of 5 to 20 years before eventually stabilizing. Both autosomal-dominant with incomplete penetrance and recessive inheritance versions of PDP can occur. Prostaglandin E2 (PGE2) has been implicated in the pathogenesis of PDP; PGE2 is important in the inflammatory response and may evolve from disrupted protein degradation pathways.3 Sasaki et al4 additionally reported that the severity of pachydermia clinically and histologically appeared to correlate with the serum PGE2 levels in affected patients. Prostaglandin E2 causes a vasodilatory effect, perhaps explaining the clubbing observed in PDP, and also modifies the activity of osteoblasts and osteoclasts, causing the bone remodeling observed in the disease.4
In our patient, the initial differential diagnosis included PDP, as well as lepromatous leprosy, acromegaly, Paget disease of the bone, amyloidosis, scleromyxedema, and cutaneous T-cell lymphoma. However, the time course of the disease, lack of numerous symmetric thickened plaques and madarosis, and pathology argued against lepromatous leprosy. Acromegaly was ruled out due to lack of macroglossia as well as laboratory analysis within reference range including IGF-1 levels and thyroid function tests. Biopsy findings ultimately ruled out amyloidosis and cutaneous T-cell lymphoma. The bone scan revealed diffuse cortical thickening consistent with PDP, and there were no other radiologic findings suggestive of Paget disease. Pachydermoperiostosis is diagnosed using the Borochowitz criteria, which entails that 2 of the following 4 fulfillment criteria must be met: familial transmission, pachydermia, digital clubbing, and/or bony involvement with evidence of radiologic alterations or pain. Our patient met all 4 criteria. The clinical manifestations of PDP are variable with respect to skin and bone changes. The various clinical expressions include the complete form (ie, pachydermia, cutis verticis gyrata, periostosis), the incomplete form (ie, absence of cutis verticis gyrata), and forme fruste (ie, pachydermia with minimal or absent periostosis).5
Management for PDP involves surgical correction for cosmesis as well as for functional concerns if present. Symptoms secondary to periostosis should be managed with symptomatic treatment such as nonsteroidal antiinflammatory drugs. Patients managed with etoricoxib, a COX-2–selective nonsteroidal anti-inflammatory drug, have had normalized inflammatory markers that resulted in the lessening of forehead skin folds. Oral aescin has been shown to relieve joint pain due to its antiedematous effect.6 Our patient received treatment with nonsteroidal anti-inflammatory drugs for symptomatic management of the associated joint pain but unfortunately was lost to follow-up.
The Diagnosis: Pachydermoperiostosis
Histopathology of the forehead punch biopsy demonstrated sebaceous hyperplasia with an occupation rate of greater than 40%, increased mucin, elastic fiber degeneration, and fibrosis. Pachydermia is graded from 0 to 3 depending on the degree of these changes; our patient met criteria for grade 3 pachydermia (Figure 1). Radiography revealed diffuse cortical thickening of the long bones that was most marked in the left femur (Figure 2); however, no other findings were demonstrative of Paget disease.
Pachydermoperiostosis (PDP)(also known as Touraine-Solente-Golé syndrome or primary hypertrophic osteoarthropathy) is a rare genetic condition that affects both the dermatologic and skeletal systems. Clinical features of the disease include progressive thickening and furrowing of the skin on the scalp and face (known as pachydermia), digital clubbing, and periostosis. Other potential cutaneous features include seborrhea, acne, hyperhidrosis of the palms and soles, cutis verticis gyrata, eczema, and a burning sensation of the hands and feet. Myelofibrosis and gastrointestinal abnormalities also have been reported.1
The disease typically affects males (7:1 ratio); also, men typically display a more severe phenotype of the disease.2 It most commonly begins during puberty and follows a generally progressive course of 5 to 20 years before eventually stabilizing. Both autosomal-dominant with incomplete penetrance and recessive inheritance versions of PDP can occur. Prostaglandin E2 (PGE2) has been implicated in the pathogenesis of PDP; PGE2 is important in the inflammatory response and may evolve from disrupted protein degradation pathways.3 Sasaki et al4 additionally reported that the severity of pachydermia clinically and histologically appeared to correlate with the serum PGE2 levels in affected patients. Prostaglandin E2 causes a vasodilatory effect, perhaps explaining the clubbing observed in PDP, and also modifies the activity of osteoblasts and osteoclasts, causing the bone remodeling observed in the disease.4
In our patient, the initial differential diagnosis included PDP, as well as lepromatous leprosy, acromegaly, Paget disease of the bone, amyloidosis, scleromyxedema, and cutaneous T-cell lymphoma. However, the time course of the disease, lack of numerous symmetric thickened plaques and madarosis, and pathology argued against lepromatous leprosy. Acromegaly was ruled out due to lack of macroglossia as well as laboratory analysis within reference range including IGF-1 levels and thyroid function tests. Biopsy findings ultimately ruled out amyloidosis and cutaneous T-cell lymphoma. The bone scan revealed diffuse cortical thickening consistent with PDP, and there were no other radiologic findings suggestive of Paget disease. Pachydermoperiostosis is diagnosed using the Borochowitz criteria, which entails that 2 of the following 4 fulfillment criteria must be met: familial transmission, pachydermia, digital clubbing, and/or bony involvement with evidence of radiologic alterations or pain. Our patient met all 4 criteria. The clinical manifestations of PDP are variable with respect to skin and bone changes. The various clinical expressions include the complete form (ie, pachydermia, cutis verticis gyrata, periostosis), the incomplete form (ie, absence of cutis verticis gyrata), and forme fruste (ie, pachydermia with minimal or absent periostosis).5
Management for PDP involves surgical correction for cosmesis as well as for functional concerns if present. Symptoms secondary to periostosis should be managed with symptomatic treatment such as nonsteroidal antiinflammatory drugs. Patients managed with etoricoxib, a COX-2–selective nonsteroidal anti-inflammatory drug, have had normalized inflammatory markers that resulted in the lessening of forehead skin folds. Oral aescin has been shown to relieve joint pain due to its antiedematous effect.6 Our patient received treatment with nonsteroidal anti-inflammatory drugs for symptomatic management of the associated joint pain but unfortunately was lost to follow-up.
- Castori M, Sinibaldi L, Mingarelli R, et al. Pachydermoperiostosis: an update. Clin Genet. 2005;68:477-486.
- Reginato AJ, Shipachasse V, Guerrero R. Familial idiopathic hypertrophic osteoarthropathy and cranial suture defects in children. Skel Radiol. 1982;8:105-109.
- Coggins KG, Coffman TM, Koller BH. The Hippocratic finger points the blame at PGE2. Nat Genet. 2008;40:691-692.
- Sasaki T, Niizeki H, Shimizu A, et al. Identification of mutations in the prostaglandin transporter gene SLCO2A1 and its phenotype-genotype correlation in Japanese patients with pachydermoperiostosis. J Dermatol Sci. 2012;68:36-44.
- Bhaskaranand K, Shetty RR, Bhat AK. Pachydermoperiostosis: three case reports. J Orthop Surg (Hong Kong). 2001;9:61-66.
- Zhang H, Yang B. Successful treatment of pachydermoperiostosis patients with etoricoxib, aescin, and arthroscopic synovectomy: two case reports. Medicine (Baltimore). 2017;96:E8865.
- Castori M, Sinibaldi L, Mingarelli R, et al. Pachydermoperiostosis: an update. Clin Genet. 2005;68:477-486.
- Reginato AJ, Shipachasse V, Guerrero R. Familial idiopathic hypertrophic osteoarthropathy and cranial suture defects in children. Skel Radiol. 1982;8:105-109.
- Coggins KG, Coffman TM, Koller BH. The Hippocratic finger points the blame at PGE2. Nat Genet. 2008;40:691-692.
- Sasaki T, Niizeki H, Shimizu A, et al. Identification of mutations in the prostaglandin transporter gene SLCO2A1 and its phenotype-genotype correlation in Japanese patients with pachydermoperiostosis. J Dermatol Sci. 2012;68:36-44.
- Bhaskaranand K, Shetty RR, Bhat AK. Pachydermoperiostosis: three case reports. J Orthop Surg (Hong Kong). 2001;9:61-66.
- Zhang H, Yang B. Successful treatment of pachydermoperiostosis patients with etoricoxib, aescin, and arthroscopic synovectomy: two case reports. Medicine (Baltimore). 2017;96:E8865.
A 36-year-old man presented to the emergency department with an olecranon fracture after falling from a tree. The patient had a medical history of type 2 diabetes mellitus and a surgical history of facial cosmetic surgery. He underwent internal fixation with orthopedic surgery for the olecranon fracture, and dermatology subsequently was consulted due to diffuse skin changes on the face. He reported that these dermatologic changes began around 17 years of age and had progressed to the current presentation. He denied itching, burning, pain, or contact with armadillos. A family history revealed the patient’s brother also had a similar appearance. Physical examination revealed exaggerated facial lines on the forehead (top) and cheeks. Digital clubbing and skin thickening were noted on the hands (bottom) and feet; examination of the back revealed multiple hypopigmented patches. Observation of the scalp showed multiple symmetric ridges and grooves with sparse overlying hair consistent with cutis verticis gyrata. A punch biopsy of the forehead was obtained as well as bone radiography taken previously by the primary team.


Firm Exophytic Tumor on the Shin
The Diagnosis: Leiomyosarcoma
Cutaneous leiomyosarcomas are relatively rare neoplasms that favor the head, neck, and extremities of older adults.1 Dermal leiomyosarcomas originate from arrector pili and are locally aggressive, whereas subcutaneous leiomyosarcomas arise from vascular smooth muscle and metastasize in 30% to 60% of cases.2 Clinically, leiomyosarcomas present as solitary, firm, well-circumscribed nodules with possible ulceration and crusting.3 Histopathology of leiomyosarcoma shows fascicles of atypical spindle cells with blunt-ended nuclei and perinuclear glycogen vacuoles, variable atypia, and mitotic figures (quiz images). Definitive diagnosis is based on positive immunohistochemical staining for desmin and smooth muscle actin.4 Treatment entails complete removal via wide local excision or Mohs micrographic surgery.5
Atypical fibroxanthoma (AFX) is a malignant fibrohistiocytic neoplasm that arises in the dermis and preferentially affects the head and neck in older individuals.3 Atypical fibroxanthoma presents as a nonspecific, pinkred, sometimes ulcerated papule on sun-damaged skin that may clinically resemble a squamous cell carcinoma (SCC) or basal cell carcinoma.6 Histopathology shows pleomorphic spindle cells with hyperchromatic nuclei and abundant cytoplasm mixed with multinucleated giant cells and scattered mitotic figures (Figure 1). Immunohistochemistry is essential for distinguishing AFX from other spindle cell neoplasms. Atypical fibroxanthoma stains positively for vimentin, procollagen-1, CD10, and CD68 but is negative for S-100, human melanoma black 45, Melan-A, desmin, cytokeratin, p40, and p63.6 Treatment includes wide local excision or Mohs micrographic surgery.

Melanoma is an aggressive cancer with the propensity to metastasize. Both desmoplastic and spindle cell variants demonstrate atypical spindled melanocytes on histology, and desmoplasia is seen in the desmoplastic variant (Figure 2). In some cases, evaluation of the epidermis for melanoma in situ may aid in diagnosis.7 Clinical and prognostic features differ between the 2 variants. Desmoplastic melanomas usually present on the head and neck as scarlike nodules with a low rate of nodal involvement, while spindle cell melanomas can occur anywhere on the body, often are amelanotic, and are associated with widespread metastatic disease at the time of presentation.8 SOX10 (SRY-box transcription factor 10) and S-100 may be the only markers that are positive in desmoplastic melanoma.9,10 Treatment depends on the thickness of the lesion.11

Spindle cell SCC is a histologic variant of SCC characterized by spindled epithelial cells. Spindle cell SCC typically presents as an ulcerated or exophytic mass in sun-exposed areas or areas exposed to ionizing radiation, or in immunocompromised individuals. Histopathology shows spindled pleomorphic keratinocytes with elongated nuclei infiltrating the dermis and minimal keratinization (Figure 3).12 Immunohistochemistry is necessary to distinguish spindle cell SCC from other spindle cell tumors such as spindle cell melanoma, AFX, and leiomyosarcoma. Spindle cell SCC is positive for high-molecular-weight cytokeratin, p40, and p63. Mohs micrographic surgery provides the highest cure rate, and radiation therapy may be considered when clear surgical margins cannot be obtained.6

Undifferentiated pleomorphic sarcoma (UPS) (formerly known as malignant fibrous histiocytoma) describes tumors that resemble AFX but are more invasive. They commonly involve the soft tissue with a higher risk for both recurrence and metastasis than AFX.13 Histopathology shows marked cytologic pleomorphism, bizarre cellular forms, atypical mitoses, and ulceration (Figure 4).14 Diagnosis of UPS is by exclusion and is dependent on immunohistochemical studies. In contrast to AFX, UPS is more likely to be positive for LN-2 (CD74).6 Undifferentiated pleomorphic sarcoma has been treated with surgical excision in combination with chemical and radiation therapy, but due to limited data, optimal management is less clear compared to AFX.15 There is a substantial risk for local recurrence and metastasis, and the lungs are the most common sites of distant metastasis.13 In a study of 23 individuals with high-grade UPS, 5-year metastasis-free survival and local recurrence-free survival were 26% and 16%, respectively.10

- Massi D, Franchi A, Alos L, et al. Primary cutaneous leiomyosarcoma: clinicopathological analysis of 36 cases. Histopathology. 2010;56: 251-262. doi:10.1111/j.1365-2559.2009.03471.x
- Ciurea ME, Georgescu CV, Radu CC, et al. Cutaneous leiomyosarcoma—case report [published online June 25, 2014]. J Med Life. 2014;7:270-273.
- Fleury LFF, Sanches JA. Primary cutaneous sarcomas. An Bras Dermatol. 2006;81:207-221. doi:10.1590/s0365-05962006000300002
- Murback NDN, de Castro BC, Takita LC, et al. Cutaneous leiomyosarcoma on the face. An Bras Dermatol. 2018;93:262-264. doi:10.1590 /abd1806-4841.20186715
- Winchester DS, Hocker TL, Brewer JD, et al. Leiomyosarcoma of the skin: clinical, histopathologic, and prognostic factors that influence outcomes. J Am Acad Dermatol. 2014;71:919-925. doi:10.1016/j .jaad.2014.07.020
- Hollmig ST, Sachdev R, Cockerell CJ, et al. Spindle cell neoplasms encountered in dermatologic surgery: a review. Dermatol Surg. 2012;38:825-850. doi:10.1111/j.1524-4725.2012.02296.x
- De Almeida LS, Requena L, Rütten A, et al. Desmoplastic malignant melanoma: a clinicopathologic analysis of 113 cases. Am J Dermatopathol. 2008;30:207-215. doi:10.1097/DAD.0B013E3181716E6B
- Weissinger SE, Keil P, Silvers DN, et al. A diagnostic algorithm to distinguish desmoplastic from spindle cell melanoma. Mod Pathol. 2014;27:524-534. doi:10.1038/modpathol.2013.162
- Ohsie SJ, Sarantopoulos GP, Cochran AJ, et al. Immunohistochemical characteristics of melanoma. J Cutan Pathol. 2008;35:433-444. doi:10.1111/j.1600-0560.2007.00891.x
- Delisca GO, Mesko NW, Alamanda VK, et al. MFH and highgrade undifferentiated pleomorphic sarcoma—what’s in a name? [published online September 12, 2014]. J Surg Oncol. 2015;111:173-177. doi:10.1002/jso.23787
- Baron PL, Nguyen CL. Malignant of melanoma. In: Holzheimer RG, Mannick JA, eds. Surgical Treatment: Evidence-Based and Problem- Oriented. Zuckschwerdt; 2001. https://www.ncbi.nlm.nih.gov/books /NBK6877
- Wernheden E, Trøstrup H, Pedersen Pilt A. Unusual presentation of cutaneous spindle cell squamous cell carcinoma: a case report. Case Rep Dermatol. 2020;12:70-75. doi:10.1159/000507358
- Ramsey JK, Chen JL, Schoenfield L, et al. Undifferentiated pleomorphic sarcoma metastatic to the orbit. Ophthal Plast Reconstr Surg. 2018;34:E193-E195. doi:10.1097/IOP.0000000000001240
- Winchester D, Lehman J, Tello T, et al. Undifferentiated pleomorphic sarcoma: factors predictive of adverse outcomes. J Am Acad Dermatol. 2018;79:853-859. doi:10.1016/j.jaad.2018.05.022
- Soleymani T, Tyler Hollmig S. Conception and management of a poorly understood spectrum of dermatologic neoplasms: atypical fibroxanthoma, pleomorphic dermal sarcoma, and undifferentiated pleomorphic sarcoma. Curr Treat Options Oncol. 2017;18:50. doi:10.1007 /s11864-017-0489-6
The Diagnosis: Leiomyosarcoma
Cutaneous leiomyosarcomas are relatively rare neoplasms that favor the head, neck, and extremities of older adults.1 Dermal leiomyosarcomas originate from arrector pili and are locally aggressive, whereas subcutaneous leiomyosarcomas arise from vascular smooth muscle and metastasize in 30% to 60% of cases.2 Clinically, leiomyosarcomas present as solitary, firm, well-circumscribed nodules with possible ulceration and crusting.3 Histopathology of leiomyosarcoma shows fascicles of atypical spindle cells with blunt-ended nuclei and perinuclear glycogen vacuoles, variable atypia, and mitotic figures (quiz images). Definitive diagnosis is based on positive immunohistochemical staining for desmin and smooth muscle actin.4 Treatment entails complete removal via wide local excision or Mohs micrographic surgery.5
Atypical fibroxanthoma (AFX) is a malignant fibrohistiocytic neoplasm that arises in the dermis and preferentially affects the head and neck in older individuals.3 Atypical fibroxanthoma presents as a nonspecific, pinkred, sometimes ulcerated papule on sun-damaged skin that may clinically resemble a squamous cell carcinoma (SCC) or basal cell carcinoma.6 Histopathology shows pleomorphic spindle cells with hyperchromatic nuclei and abundant cytoplasm mixed with multinucleated giant cells and scattered mitotic figures (Figure 1). Immunohistochemistry is essential for distinguishing AFX from other spindle cell neoplasms. Atypical fibroxanthoma stains positively for vimentin, procollagen-1, CD10, and CD68 but is negative for S-100, human melanoma black 45, Melan-A, desmin, cytokeratin, p40, and p63.6 Treatment includes wide local excision or Mohs micrographic surgery.
Melanoma is an aggressive cancer with the propensity to metastasize. Both desmoplastic and spindle cell variants demonstrate atypical spindled melanocytes on histology, and desmoplasia is seen in the desmoplastic variant (Figure 2). In some cases, evaluation of the epidermis for melanoma in situ may aid in diagnosis.7 Clinical and prognostic features differ between the 2 variants. Desmoplastic melanomas usually present on the head and neck as scarlike nodules with a low rate of nodal involvement, while spindle cell melanomas can occur anywhere on the body, often are amelanotic, and are associated with widespread metastatic disease at the time of presentation.8 SOX10 (SRY-box transcription factor 10) and S-100 may be the only markers that are positive in desmoplastic melanoma.9,10 Treatment depends on the thickness of the lesion.11
Spindle cell SCC is a histologic variant of SCC characterized by spindled epithelial cells. Spindle cell SCC typically presents as an ulcerated or exophytic mass in sun-exposed areas or areas exposed to ionizing radiation, or in immunocompromised individuals. Histopathology shows spindled pleomorphic keratinocytes with elongated nuclei infiltrating the dermis and minimal keratinization (Figure 3).12 Immunohistochemistry is necessary to distinguish spindle cell SCC from other spindle cell tumors such as spindle cell melanoma, AFX, and leiomyosarcoma. Spindle cell SCC is positive for high-molecular-weight cytokeratin, p40, and p63. Mohs micrographic surgery provides the highest cure rate, and radiation therapy may be considered when clear surgical margins cannot be obtained.6
Undifferentiated pleomorphic sarcoma (UPS) (formerly known as malignant fibrous histiocytoma) describes tumors that resemble AFX but are more invasive. They commonly involve the soft tissue with a higher risk for both recurrence and metastasis than AFX.13 Histopathology shows marked cytologic pleomorphism, bizarre cellular forms, atypical mitoses, and ulceration (Figure 4).14 Diagnosis of UPS is by exclusion and is dependent on immunohistochemical studies. In contrast to AFX, UPS is more likely to be positive for LN-2 (CD74).6 Undifferentiated pleomorphic sarcoma has been treated with surgical excision in combination with chemical and radiation therapy, but due to limited data, optimal management is less clear compared to AFX.15 There is a substantial risk for local recurrence and metastasis, and the lungs are the most common sites of distant metastasis.13 In a study of 23 individuals with high-grade UPS, 5-year metastasis-free survival and local recurrence-free survival were 26% and 16%, respectively.10
The Diagnosis: Leiomyosarcoma
Cutaneous leiomyosarcomas are relatively rare neoplasms that favor the head, neck, and extremities of older adults.1 Dermal leiomyosarcomas originate from arrector pili and are locally aggressive, whereas subcutaneous leiomyosarcomas arise from vascular smooth muscle and metastasize in 30% to 60% of cases.2 Clinically, leiomyosarcomas present as solitary, firm, well-circumscribed nodules with possible ulceration and crusting.3 Histopathology of leiomyosarcoma shows fascicles of atypical spindle cells with blunt-ended nuclei and perinuclear glycogen vacuoles, variable atypia, and mitotic figures (quiz images). Definitive diagnosis is based on positive immunohistochemical staining for desmin and smooth muscle actin.4 Treatment entails complete removal via wide local excision or Mohs micrographic surgery.5
Atypical fibroxanthoma (AFX) is a malignant fibrohistiocytic neoplasm that arises in the dermis and preferentially affects the head and neck in older individuals.3 Atypical fibroxanthoma presents as a nonspecific, pinkred, sometimes ulcerated papule on sun-damaged skin that may clinically resemble a squamous cell carcinoma (SCC) or basal cell carcinoma.6 Histopathology shows pleomorphic spindle cells with hyperchromatic nuclei and abundant cytoplasm mixed with multinucleated giant cells and scattered mitotic figures (Figure 1). Immunohistochemistry is essential for distinguishing AFX from other spindle cell neoplasms. Atypical fibroxanthoma stains positively for vimentin, procollagen-1, CD10, and CD68 but is negative for S-100, human melanoma black 45, Melan-A, desmin, cytokeratin, p40, and p63.6 Treatment includes wide local excision or Mohs micrographic surgery.
Melanoma is an aggressive cancer with the propensity to metastasize. Both desmoplastic and spindle cell variants demonstrate atypical spindled melanocytes on histology, and desmoplasia is seen in the desmoplastic variant (Figure 2). In some cases, evaluation of the epidermis for melanoma in situ may aid in diagnosis.7 Clinical and prognostic features differ between the 2 variants. Desmoplastic melanomas usually present on the head and neck as scarlike nodules with a low rate of nodal involvement, while spindle cell melanomas can occur anywhere on the body, often are amelanotic, and are associated with widespread metastatic disease at the time of presentation.8 SOX10 (SRY-box transcription factor 10) and S-100 may be the only markers that are positive in desmoplastic melanoma.9,10 Treatment depends on the thickness of the lesion.11
Spindle cell SCC is a histologic variant of SCC characterized by spindled epithelial cells. Spindle cell SCC typically presents as an ulcerated or exophytic mass in sun-exposed areas or areas exposed to ionizing radiation, or in immunocompromised individuals. Histopathology shows spindled pleomorphic keratinocytes with elongated nuclei infiltrating the dermis and minimal keratinization (Figure 3).12 Immunohistochemistry is necessary to distinguish spindle cell SCC from other spindle cell tumors such as spindle cell melanoma, AFX, and leiomyosarcoma. Spindle cell SCC is positive for high-molecular-weight cytokeratin, p40, and p63. Mohs micrographic surgery provides the highest cure rate, and radiation therapy may be considered when clear surgical margins cannot be obtained.6
Undifferentiated pleomorphic sarcoma (UPS) (formerly known as malignant fibrous histiocytoma) describes tumors that resemble AFX but are more invasive. They commonly involve the soft tissue with a higher risk for both recurrence and metastasis than AFX.13 Histopathology shows marked cytologic pleomorphism, bizarre cellular forms, atypical mitoses, and ulceration (Figure 4).14 Diagnosis of UPS is by exclusion and is dependent on immunohistochemical studies. In contrast to AFX, UPS is more likely to be positive for LN-2 (CD74).6 Undifferentiated pleomorphic sarcoma has been treated with surgical excision in combination with chemical and radiation therapy, but due to limited data, optimal management is less clear compared to AFX.15 There is a substantial risk for local recurrence and metastasis, and the lungs are the most common sites of distant metastasis.13 In a study of 23 individuals with high-grade UPS, 5-year metastasis-free survival and local recurrence-free survival were 26% and 16%, respectively.10
- Massi D, Franchi A, Alos L, et al. Primary cutaneous leiomyosarcoma: clinicopathological analysis of 36 cases. Histopathology. 2010;56: 251-262. doi:10.1111/j.1365-2559.2009.03471.x
- Ciurea ME, Georgescu CV, Radu CC, et al. Cutaneous leiomyosarcoma—case report [published online June 25, 2014]. J Med Life. 2014;7:270-273.
- Fleury LFF, Sanches JA. Primary cutaneous sarcomas. An Bras Dermatol. 2006;81:207-221. doi:10.1590/s0365-05962006000300002
- Murback NDN, de Castro BC, Takita LC, et al. Cutaneous leiomyosarcoma on the face. An Bras Dermatol. 2018;93:262-264. doi:10.1590 /abd1806-4841.20186715
- Winchester DS, Hocker TL, Brewer JD, et al. Leiomyosarcoma of the skin: clinical, histopathologic, and prognostic factors that influence outcomes. J Am Acad Dermatol. 2014;71:919-925. doi:10.1016/j .jaad.2014.07.020
- Hollmig ST, Sachdev R, Cockerell CJ, et al. Spindle cell neoplasms encountered in dermatologic surgery: a review. Dermatol Surg. 2012;38:825-850. doi:10.1111/j.1524-4725.2012.02296.x
- De Almeida LS, Requena L, Rütten A, et al. Desmoplastic malignant melanoma: a clinicopathologic analysis of 113 cases. Am J Dermatopathol. 2008;30:207-215. doi:10.1097/DAD.0B013E3181716E6B
- Weissinger SE, Keil P, Silvers DN, et al. A diagnostic algorithm to distinguish desmoplastic from spindle cell melanoma. Mod Pathol. 2014;27:524-534. doi:10.1038/modpathol.2013.162
- Ohsie SJ, Sarantopoulos GP, Cochran AJ, et al. Immunohistochemical characteristics of melanoma. J Cutan Pathol. 2008;35:433-444. doi:10.1111/j.1600-0560.2007.00891.x
- Delisca GO, Mesko NW, Alamanda VK, et al. MFH and highgrade undifferentiated pleomorphic sarcoma—what’s in a name? [published online September 12, 2014]. J Surg Oncol. 2015;111:173-177. doi:10.1002/jso.23787
- Baron PL, Nguyen CL. Malignant of melanoma. In: Holzheimer RG, Mannick JA, eds. Surgical Treatment: Evidence-Based and Problem- Oriented. Zuckschwerdt; 2001. https://www.ncbi.nlm.nih.gov/books /NBK6877
- Wernheden E, Trøstrup H, Pedersen Pilt A. Unusual presentation of cutaneous spindle cell squamous cell carcinoma: a case report. Case Rep Dermatol. 2020;12:70-75. doi:10.1159/000507358
- Ramsey JK, Chen JL, Schoenfield L, et al. Undifferentiated pleomorphic sarcoma metastatic to the orbit. Ophthal Plast Reconstr Surg. 2018;34:E193-E195. doi:10.1097/IOP.0000000000001240
- Winchester D, Lehman J, Tello T, et al. Undifferentiated pleomorphic sarcoma: factors predictive of adverse outcomes. J Am Acad Dermatol. 2018;79:853-859. doi:10.1016/j.jaad.2018.05.022
- Soleymani T, Tyler Hollmig S. Conception and management of a poorly understood spectrum of dermatologic neoplasms: atypical fibroxanthoma, pleomorphic dermal sarcoma, and undifferentiated pleomorphic sarcoma. Curr Treat Options Oncol. 2017;18:50. doi:10.1007 /s11864-017-0489-6
- Massi D, Franchi A, Alos L, et al. Primary cutaneous leiomyosarcoma: clinicopathological analysis of 36 cases. Histopathology. 2010;56: 251-262. doi:10.1111/j.1365-2559.2009.03471.x
- Ciurea ME, Georgescu CV, Radu CC, et al. Cutaneous leiomyosarcoma—case report [published online June 25, 2014]. J Med Life. 2014;7:270-273.
- Fleury LFF, Sanches JA. Primary cutaneous sarcomas. An Bras Dermatol. 2006;81:207-221. doi:10.1590/s0365-05962006000300002
- Murback NDN, de Castro BC, Takita LC, et al. Cutaneous leiomyosarcoma on the face. An Bras Dermatol. 2018;93:262-264. doi:10.1590 /abd1806-4841.20186715
- Winchester DS, Hocker TL, Brewer JD, et al. Leiomyosarcoma of the skin: clinical, histopathologic, and prognostic factors that influence outcomes. J Am Acad Dermatol. 2014;71:919-925. doi:10.1016/j .jaad.2014.07.020
- Hollmig ST, Sachdev R, Cockerell CJ, et al. Spindle cell neoplasms encountered in dermatologic surgery: a review. Dermatol Surg. 2012;38:825-850. doi:10.1111/j.1524-4725.2012.02296.x
- De Almeida LS, Requena L, Rütten A, et al. Desmoplastic malignant melanoma: a clinicopathologic analysis of 113 cases. Am J Dermatopathol. 2008;30:207-215. doi:10.1097/DAD.0B013E3181716E6B
- Weissinger SE, Keil P, Silvers DN, et al. A diagnostic algorithm to distinguish desmoplastic from spindle cell melanoma. Mod Pathol. 2014;27:524-534. doi:10.1038/modpathol.2013.162
- Ohsie SJ, Sarantopoulos GP, Cochran AJ, et al. Immunohistochemical characteristics of melanoma. J Cutan Pathol. 2008;35:433-444. doi:10.1111/j.1600-0560.2007.00891.x
- Delisca GO, Mesko NW, Alamanda VK, et al. MFH and highgrade undifferentiated pleomorphic sarcoma—what’s in a name? [published online September 12, 2014]. J Surg Oncol. 2015;111:173-177. doi:10.1002/jso.23787
- Baron PL, Nguyen CL. Malignant of melanoma. In: Holzheimer RG, Mannick JA, eds. Surgical Treatment: Evidence-Based and Problem- Oriented. Zuckschwerdt; 2001. https://www.ncbi.nlm.nih.gov/books /NBK6877
- Wernheden E, Trøstrup H, Pedersen Pilt A. Unusual presentation of cutaneous spindle cell squamous cell carcinoma: a case report. Case Rep Dermatol. 2020;12:70-75. doi:10.1159/000507358
- Ramsey JK, Chen JL, Schoenfield L, et al. Undifferentiated pleomorphic sarcoma metastatic to the orbit. Ophthal Plast Reconstr Surg. 2018;34:E193-E195. doi:10.1097/IOP.0000000000001240
- Winchester D, Lehman J, Tello T, et al. Undifferentiated pleomorphic sarcoma: factors predictive of adverse outcomes. J Am Acad Dermatol. 2018;79:853-859. doi:10.1016/j.jaad.2018.05.022
- Soleymani T, Tyler Hollmig S. Conception and management of a poorly understood spectrum of dermatologic neoplasms: atypical fibroxanthoma, pleomorphic dermal sarcoma, and undifferentiated pleomorphic sarcoma. Curr Treat Options Oncol. 2017;18:50. doi:10.1007 /s11864-017-0489-6
A 62-year-old man presented with a firm, exophytic, 2.8×1.5-cm tumor on the left shin of 6 to 7 years’ duration. An excisional biopsy was obtained for histopathologic evaluation.


