User login
Pigmented Papules on the Face, Neck, and Chest
The Diagnosis: Syringoma
Syringomas are benign adnexal tumors with distinct histopathologic features, including the characteristic comma- or tadpole-shaped tail comprised of dilated cystic eccrine ducts. Clinically, syringomas typically present predominantly in the periorbital region in adolescent girls. They may present as solitary or multiple lesions, and sites such as the genital area, palms, scalp, and chest rarely can be involved.1 Eruptive syringoma is a clinical subtype of syringoma that is seen on the face, neck, chest, and axillae that predominantly occurs in females with skin of color in countries such as Asia and Africa before or during puberty.2,3 Lesions appear as small, flesh-colored or slightly pigmented, flat-topped papules.3 The condition can be cosmetically disfiguring and difficult to treat, especially in patients with darker skin.
. B, Glittering yellow-whitish round structures over a fading pink-brown background")
In our patient, dermoscopic evaluation revealed reticular light brown lines, structureless light brown areas, clustered brown dots, globules, and reticular vessels on a faint background (Figure 1A). Glittering yellow-whitish round structures over a fading pink-brown background also were seen at some sites (Figure 1B). Histologic examination of a neck lesion revealed an epidermis with focal acanthosis; the upper dermis had tumor islands and ducts with cells with round to vesicular nuclei and eosinophilic cytoplasm. A well-circumscribed tumor in the dermis composed of tubules of varying sizes lined by cuboidal cells was seen, consistent with syringoma (Figure 2).

Dermoscopic features of syringomas have not been widely studied. Hayashi et al4 reported the dermoscopic features of unilateral linear syringomas as a delicate and faint reticular pigmentation network and multiple hypopigmented areas. Sakiyama et al5 also defined an incomplete pigment network with faint erythema in 2 eruptive syringoma cases.
Treatment of this condition is for cosmetic reasons only, and there are no reports of long-term morbidity associated with the disease.6,7 Multiple therapeutic options are available but are associated with complications such as hyperpigmentation and sclerosis in patients with skin of color due to the dermal location of these syringomas. Management of syringomas includes topical and surgical methods, including topical retinoids such as tretinoin and atropine solution 1%; surgical methods include dermabrasion, excision, cryotherapy, electrocautery, electrofulguration, laser therapy, and chemical cautery. However, there is a substantial risk for recurrence with these treatment options. In a case series of 5 patients with periorbital syringomas, treatment using radiofrequency and a CO2 laser was performed with favorable outcomes, highlighting the use of combination therapies for treatment.8 Seo et al9 reported a retrospective case series of 92 patients with periorbital syringomas in which they treated one group with CO2 laser and the other with botulinum toxin A injection; CO2 laser combined with botulinum toxin A showed a greater effect than laser treatment alone. The differential diagnosis includes pigmented plane warts, sebaceous hyperplasia, eruptive xanthomas, and hidrocystomas. Pigmented plane warts characteristically present as flat-topped papules with small hemorrhagic dots or tiny pinpoint vessels on dermoscopy. In sebaceous hyperplasia, yellowish umbilicated papular lesions are seen with crown vessels on dermoscopy. Eruptive xanthomas usually are erythematous to yellow, dome-shaped papules that appear mainly over the extensor aspects of the extremities. Hidrocystoma presents as a solitary translucent larger syringomalike lesion commonly seen in the periorbital region and/or on the cheeks.
We report a case of widespread syringomas with multiple close mimickers such as pigmented plane warts; however, dermoscopy of the lesions helped to arrive at the diagnosis. Dermatologists should be aware of this condition and its benign nature to ensure correct diagnosis and appropriate treatment.
- Williams K, Shinkai K. Evaluation and management of the patient with multiple syringomas: a systematic review of the literature. J Am Acad Dermatol. 2016;74:1234.e9-1240.e9.
- Tsunemi Y, Ihn H, Saeki H, et al. Generalized eruptive syringoma. Pediatr Dermatol. 2005;22:492-493.
- Singh S, Tewari R, Gupta S. An unusual case of generalised eruptive syringoma in an adult male. Med J Armed Forces India. 2014;70:389-391.
- Hayashi Y, Tanaka M, Nakajima S, et al. Unilateral linear syringoma in a Japanese female: dermoscopic differentiation from lichen lanus linearis. Dermatol Rep. 2011;3:E42.
- Sakiyama M, Maeda M, Fujimoto N, et al. Eruptive syringoma localized in intertriginous areas. J Dtsch Dermatol Ges. 2014;12:72-73.
- Wang JI, Roenigk HH Jr. Treatment of multiple facial syringomas with the carbon dioxide (CO2) laser. Dermatol Surg. 1999;25:136-139.
- Tsunemi Y, Ihn H, Saeki H, et al. Generalized eruptive syringoma. Pediatr Dermatol. 2005;22:492-493.
- Hasson A, Farias MM, Nicklas C, et al. Periorbital syringoma treated with radiofrequency and carbon dioxide (CO2) laser in 5 patients. J Drugs Dermatol. 2012;11:879-880.
- Seo HM, Choi JY, Min J, et al. Carbon dioxide laser combined with botulinum toxin A for patients with periorbital syringomas [published online March 31, 2016]. J Cosmet Laser Ther. 2016;18:149-153.
The Diagnosis: Syringoma
Syringomas are benign adnexal tumors with distinct histopathologic features, including the characteristic comma- or tadpole-shaped tail comprised of dilated cystic eccrine ducts. Clinically, syringomas typically present predominantly in the periorbital region in adolescent girls. They may present as solitary or multiple lesions, and sites such as the genital area, palms, scalp, and chest rarely can be involved.1 Eruptive syringoma is a clinical subtype of syringoma that is seen on the face, neck, chest, and axillae that predominantly occurs in females with skin of color in countries such as Asia and Africa before or during puberty.2,3 Lesions appear as small, flesh-colored or slightly pigmented, flat-topped papules.3 The condition can be cosmetically disfiguring and difficult to treat, especially in patients with darker skin.
In our patient, dermoscopic evaluation revealed reticular light brown lines, structureless light brown areas, clustered brown dots, globules, and reticular vessels on a faint background (Figure 1A). Glittering yellow-whitish round structures over a fading pink-brown background also were seen at some sites (Figure 1B). Histologic examination of a neck lesion revealed an epidermis with focal acanthosis; the upper dermis had tumor islands and ducts with cells with round to vesicular nuclei and eosinophilic cytoplasm. A well-circumscribed tumor in the dermis composed of tubules of varying sizes lined by cuboidal cells was seen, consistent with syringoma (Figure 2).
Dermoscopic features of syringomas have not been widely studied. Hayashi et al4 reported the dermoscopic features of unilateral linear syringomas as a delicate and faint reticular pigmentation network and multiple hypopigmented areas. Sakiyama et al5 also defined an incomplete pigment network with faint erythema in 2 eruptive syringoma cases.
Treatment of this condition is for cosmetic reasons only, and there are no reports of long-term morbidity associated with the disease.6,7 Multiple therapeutic options are available but are associated with complications such as hyperpigmentation and sclerosis in patients with skin of color due to the dermal location of these syringomas. Management of syringomas includes topical and surgical methods, including topical retinoids such as tretinoin and atropine solution 1%; surgical methods include dermabrasion, excision, cryotherapy, electrocautery, electrofulguration, laser therapy, and chemical cautery. However, there is a substantial risk for recurrence with these treatment options. In a case series of 5 patients with periorbital syringomas, treatment using radiofrequency and a CO2 laser was performed with favorable outcomes, highlighting the use of combination therapies for treatment.8 Seo et al9 reported a retrospective case series of 92 patients with periorbital syringomas in which they treated one group with CO2 laser and the other with botulinum toxin A injection; CO2 laser combined with botulinum toxin A showed a greater effect than laser treatment alone. The differential diagnosis includes pigmented plane warts, sebaceous hyperplasia, eruptive xanthomas, and hidrocystomas. Pigmented plane warts characteristically present as flat-topped papules with small hemorrhagic dots or tiny pinpoint vessels on dermoscopy. In sebaceous hyperplasia, yellowish umbilicated papular lesions are seen with crown vessels on dermoscopy. Eruptive xanthomas usually are erythematous to yellow, dome-shaped papules that appear mainly over the extensor aspects of the extremities. Hidrocystoma presents as a solitary translucent larger syringomalike lesion commonly seen in the periorbital region and/or on the cheeks.
We report a case of widespread syringomas with multiple close mimickers such as pigmented plane warts; however, dermoscopy of the lesions helped to arrive at the diagnosis. Dermatologists should be aware of this condition and its benign nature to ensure correct diagnosis and appropriate treatment.
The Diagnosis: Syringoma
Syringomas are benign adnexal tumors with distinct histopathologic features, including the characteristic comma- or tadpole-shaped tail comprised of dilated cystic eccrine ducts. Clinically, syringomas typically present predominantly in the periorbital region in adolescent girls. They may present as solitary or multiple lesions, and sites such as the genital area, palms, scalp, and chest rarely can be involved.1 Eruptive syringoma is a clinical subtype of syringoma that is seen on the face, neck, chest, and axillae that predominantly occurs in females with skin of color in countries such as Asia and Africa before or during puberty.2,3 Lesions appear as small, flesh-colored or slightly pigmented, flat-topped papules.3 The condition can be cosmetically disfiguring and difficult to treat, especially in patients with darker skin.
In our patient, dermoscopic evaluation revealed reticular light brown lines, structureless light brown areas, clustered brown dots, globules, and reticular vessels on a faint background (Figure 1A). Glittering yellow-whitish round structures over a fading pink-brown background also were seen at some sites (Figure 1B). Histologic examination of a neck lesion revealed an epidermis with focal acanthosis; the upper dermis had tumor islands and ducts with cells with round to vesicular nuclei and eosinophilic cytoplasm. A well-circumscribed tumor in the dermis composed of tubules of varying sizes lined by cuboidal cells was seen, consistent with syringoma (Figure 2).
Dermoscopic features of syringomas have not been widely studied. Hayashi et al4 reported the dermoscopic features of unilateral linear syringomas as a delicate and faint reticular pigmentation network and multiple hypopigmented areas. Sakiyama et al5 also defined an incomplete pigment network with faint erythema in 2 eruptive syringoma cases.
Treatment of this condition is for cosmetic reasons only, and there are no reports of long-term morbidity associated with the disease.6,7 Multiple therapeutic options are available but are associated with complications such as hyperpigmentation and sclerosis in patients with skin of color due to the dermal location of these syringomas. Management of syringomas includes topical and surgical methods, including topical retinoids such as tretinoin and atropine solution 1%; surgical methods include dermabrasion, excision, cryotherapy, electrocautery, electrofulguration, laser therapy, and chemical cautery. However, there is a substantial risk for recurrence with these treatment options. In a case series of 5 patients with periorbital syringomas, treatment using radiofrequency and a CO2 laser was performed with favorable outcomes, highlighting the use of combination therapies for treatment.8 Seo et al9 reported a retrospective case series of 92 patients with periorbital syringomas in which they treated one group with CO2 laser and the other with botulinum toxin A injection; CO2 laser combined with botulinum toxin A showed a greater effect than laser treatment alone. The differential diagnosis includes pigmented plane warts, sebaceous hyperplasia, eruptive xanthomas, and hidrocystomas. Pigmented plane warts characteristically present as flat-topped papules with small hemorrhagic dots or tiny pinpoint vessels on dermoscopy. In sebaceous hyperplasia, yellowish umbilicated papular lesions are seen with crown vessels on dermoscopy. Eruptive xanthomas usually are erythematous to yellow, dome-shaped papules that appear mainly over the extensor aspects of the extremities. Hidrocystoma presents as a solitary translucent larger syringomalike lesion commonly seen in the periorbital region and/or on the cheeks.
We report a case of widespread syringomas with multiple close mimickers such as pigmented plane warts; however, dermoscopy of the lesions helped to arrive at the diagnosis. Dermatologists should be aware of this condition and its benign nature to ensure correct diagnosis and appropriate treatment.
- Williams K, Shinkai K. Evaluation and management of the patient with multiple syringomas: a systematic review of the literature. J Am Acad Dermatol. 2016;74:1234.e9-1240.e9.
- Tsunemi Y, Ihn H, Saeki H, et al. Generalized eruptive syringoma. Pediatr Dermatol. 2005;22:492-493.
- Singh S, Tewari R, Gupta S. An unusual case of generalised eruptive syringoma in an adult male. Med J Armed Forces India. 2014;70:389-391.
- Hayashi Y, Tanaka M, Nakajima S, et al. Unilateral linear syringoma in a Japanese female: dermoscopic differentiation from lichen lanus linearis. Dermatol Rep. 2011;3:E42.
- Sakiyama M, Maeda M, Fujimoto N, et al. Eruptive syringoma localized in intertriginous areas. J Dtsch Dermatol Ges. 2014;12:72-73.
- Wang JI, Roenigk HH Jr. Treatment of multiple facial syringomas with the carbon dioxide (CO2) laser. Dermatol Surg. 1999;25:136-139.
- Tsunemi Y, Ihn H, Saeki H, et al. Generalized eruptive syringoma. Pediatr Dermatol. 2005;22:492-493.
- Hasson A, Farias MM, Nicklas C, et al. Periorbital syringoma treated with radiofrequency and carbon dioxide (CO2) laser in 5 patients. J Drugs Dermatol. 2012;11:879-880.
- Seo HM, Choi JY, Min J, et al. Carbon dioxide laser combined with botulinum toxin A for patients with periorbital syringomas [published online March 31, 2016]. J Cosmet Laser Ther. 2016;18:149-153.
- Williams K, Shinkai K. Evaluation and management of the patient with multiple syringomas: a systematic review of the literature. J Am Acad Dermatol. 2016;74:1234.e9-1240.e9.
- Tsunemi Y, Ihn H, Saeki H, et al. Generalized eruptive syringoma. Pediatr Dermatol. 2005;22:492-493.
- Singh S, Tewari R, Gupta S. An unusual case of generalised eruptive syringoma in an adult male. Med J Armed Forces India. 2014;70:389-391.
- Hayashi Y, Tanaka M, Nakajima S, et al. Unilateral linear syringoma in a Japanese female: dermoscopic differentiation from lichen lanus linearis. Dermatol Rep. 2011;3:E42.
- Sakiyama M, Maeda M, Fujimoto N, et al. Eruptive syringoma localized in intertriginous areas. J Dtsch Dermatol Ges. 2014;12:72-73.
- Wang JI, Roenigk HH Jr. Treatment of multiple facial syringomas with the carbon dioxide (CO2) laser. Dermatol Surg. 1999;25:136-139.
- Tsunemi Y, Ihn H, Saeki H, et al. Generalized eruptive syringoma. Pediatr Dermatol. 2005;22:492-493.
- Hasson A, Farias MM, Nicklas C, et al. Periorbital syringoma treated with radiofrequency and carbon dioxide (CO2) laser in 5 patients. J Drugs Dermatol. 2012;11:879-880.
- Seo HM, Choi JY, Min J, et al. Carbon dioxide laser combined with botulinum toxin A for patients with periorbital syringomas [published online March 31, 2016]. J Cosmet Laser Ther. 2016;18:149-153.
A 46-year-old woman presented with multiple asymptomatic, flesh-colored, hyperpigmented papules on the face of 5 to 6 months’ duration that were progressively increasing in number. The lesions first appeared near the eyebrows and cheeks (top) and subsequently spread to involve the neck. She had no notable medical history. Cutaneous examination revealed multiple tan to brown papules over the periorbital, malar, and neck regions ranging in size from 1 to 5 mm. The lesions over the periorbital region were arranged in a linear pattern (bottom). Similar lesions also were present on the chest and arms. No other sites were involved, and systemic examination was normal.


Solitary Pink Plaque on the Neck
The Diagnosis: Plaque-type Syringoma
A biopsy demonstrated multiple basaloid islands of tumor cells in the reticular dermis with ductal differentiation, some with a commalike tail. The ducts were lined by 2 to 3 layers of small uniform cuboidal cells without atypia and contained inspissated secretions within the lumina of scattered ducts. There was an associated fibrotic collagenous stroma. There was no evidence of perineural invasion and no deep dermal or subcutaneous extension (Figure 1). Additional cytokeratin immunohistochemical staining highlighted the adnexal proliferation (Figure 2). A diagnosis of plaque-type syringoma (PTS) was made.

Syringomas are benign dermal sweat gland tumors that typically present as flesh-colored papules on the cheeks or periorbital area of young females. Plaque-type tumors as well as papulonodular, eruptive, disseminated, urticaria pigmentosa–like, lichen planus–like, or milialike syringomas also have been reported. Syringomas may be associated with certain medical conditions such as Down syndrome, Nicolau-Balus syndrome, and both scarring and nonscarring alopecias.1 The clear cell variant of syringoma often is associated with diabetes mellitus.2 Kikuchi et al3 first described PTS in 1979. Plaque-type syringomas rarely are reported in the literature, and sites of involvement include the head and neck region, upper lip, chest, upper extremities, vulva, penis, and scrotum.4-6
.")
Histologically, syringomatous lesions are composed of multiple small ducts lined by 2 to 3 layers of cuboidal epithelium. The ducts may be arranged in nests or strands of basaloid cells surrounded by a dense fibrotic stroma. Occasionally, the ducts will form a comma- or teardropshaped tail; however, this also may be observed in desmoplastic trichoepithelioma (DTE).7 Perineural invasion is absent in syringomas. Syringomas exhibit a lateral growth pattern that typically is limited to the upper half of the reticular dermis and spares the underlying subcutis, muscle, and bone. The growth pattern may be discontinuous with proliferations juxtaposed by normal-appearing skin.8 Syringomas usually express progesterone receptors and are known to proliferate at puberty, suggesting that these neoplasms are under hormonal control.9 Although syringomas are benign, various treatment options that may be pursued for cosmetic purposes include radiofrequency, staged excision, laser ablation, and oral isotretinoin.8,10 If only a superficial biopsy is obtained, syringomas may display features of other adnexal neoplasms, including microcystic adnexal carcinoma (MAC), DTE, morpheaform basal cell carcinoma (BCC), and inflammatory linear verrucous epidermal nevus (ILVEN).
Microcystic adnexal carcinoma is a locally aggressive neoplasm first described by Goldstein et al11 in 1982 an indurated, ill-defined plaque or nodule on the face with a predilection for the upper and lower lip. Prior radiation therapy and immunosuppression are risk factors for the development of MAC.12 Histologically, the superficial portion displays small cornifying cysts interspersed with islands of basaloid cells and may mimic a syringoma. However, the deeper portions demonstrate ducts lined by a single layer of cells with a background of hyalinized and sclerotic stroma. The tumor cells may occupy the deep dermis and underlying subcutis, muscle, or bone and demonstrate an infiltrative growth pattern and perineural invasion. Treatment includes Mohs micrographic surgery.
Desmoplastic trichoepitheliomas most commonly present as solitary white to yellowish annular papules or plaques with a central dell located on sun-exposed areas of the face, cheeks, or chin. This benign neoplasm has a bimodal age distribution, primarily affecting females either in childhood or adulthood.13 Histologically, strands and nests of basaloid epithelial cells proliferate in a dense eosinophilic desmoplastic stroma. The basaloid islands are narrow and cordlike with growth parallel to the surface epidermis and do not dive deeply into the deep dermis or subcutis. Ductal differentiation with associated secretions typically is not seen in DTE.1 Calcifications and foreign body granulomatous infiltrates may be present. Merkel cells also are present in this tumor and may be highlighted by immunohistochemistry with cytokeratin 20.14 Rarely, desmoplastic trichoepitheliomas may transform into trichoblastic carcinomas. Treatment may consist of surgical excision or Mohs micrographic surgery.
Morpheaform BCC also is included in the clinical and histopathologic differential diagnosis of infiltrative basaloid neoplasms. It is one of the more aggressive variants of BCC. The use of immunohistochemical staining may aid in differentiating between these sclerosing adnexal neoplasms.15 For example, pleckstrin homologylike domain family A member 1 (PHLDA1) is a stem cell marker that is heavily expressed in DTE as a specific follicular bulge marker but is not present in a morpheaform BCC. This highlights the follicular nature of DTEs at the molecular level. BerEP4 is a monoclonal antibody that serves as an epithelial marker for 2 glycopolypeptides: 34 and 39 kDa. This antibody may demonstrate positivity in morpheaform BCC but does not stain cells of interest in MAC.
Inflammatory linear verrucous epidermal nevus clinically presents with erythematous and warty papules in a linear distribution following the Blaschko lines. The papules often are reported to be intensely pruritic and usually are localized to one extremity.16 Although adultonset forms of ILVEN have been described,17 it most commonly is diagnosed in young children. Histologically, ILVEN consists of psoriasiform epidermal hyperplasia with alternating areas of parakeratosis and orthokeratosis with underlying agranulosis and hypergranulosis, respectively.18 The upper dermis contains a perivascular lymphocytic infiltrate. Treatment with laser therapy and surgical excision has led to both symptomatic and clinical improvement of ILVEN.16
Plaque-type syringomas are a rare variant of syringomas that clinically may mimic other common inflammatory and neoplastic conditions. An adequate biopsy is imperative to differentiate between adnexal neoplasms, as a small superficial biopsy of a syringoma may demonstrate features observed in other malignant or locally aggressive neoplasms. In our patient, the small ducts lined by cuboidal epithelium with no cellular atypia and no deep dermal growth or perineural invasion allowed for the diagnosis of PTS. Therapeutic options were reviewed with our patient, including oral isotretinoin, laser therapy, and staged excision. Ultimately, our patient elected not to pursue treatment, and she is being monitored clinically for any changes in appearance or symptoms.
- Suwattee P, McClelland MC, Huiras EE, et al. Plaque-type syringoma: two cases misdiagnosed as microcystic adnexal carcinoma [published online November 12, 2007]. J Cutan Pathol. 2008;35:570-574.
- Furue M, Hori Y, Nakabayashi Y. Clear-cell syringoma. association with diabetes mellitus. Am J Dermatopathol. 1984;6:131-138.
- Kikuchi I, Idemori M, Okazaki M. Plaque type syringoma. J Dermatol. 1979;6:329-331.
- Kavala M, Can B, Zindanci I, et al. Vulvar pruritus caused by syringoma of the vulva. Int J Dermatol. 2008;47:831-832.
- Cohen PR, Tschen JA, Rapini RP. Penile syringoma: reports and review of patients with syringoma located on the penis. J Clin Aesthet Dermatol. 2013;6:38-42.
- Okuda H, Tei N, Shimizu K, et al. Chondroid syringoma of the scrotum. Int J Urol. 2008;15:944-945.
- Wallace JS, Bond JS, Seidel GD, et al. An important mimicker: plaquetype syringoma mistakenly diagnosed as microcystic adnexal carcinoma. Dermatol Surg. 2014;40:810-812.
- Clark M, Duprey C, Sutton A, et al. Plaque-type syringoma masquerading as microcystic adnexal carcinoma: review of the literature and description of a novel technique that emphasizes lesion architecture to help make the diagnosis. Am J Dermatopathol. 2019;41:E98-E101.
- Wallace ML, Smoller BR. Progesterone receptor positivity supports hormonal control of syringomas. J Cutan Pathol. 1995;22:442-445.
- Mainitz M, Schmidt JB, Gebhart W. Response of multiple syringomas to isotretinoin. Acta Derm Venereol. 1986;66:51-55.
- Goldstein DJ, Barr RJ, Santa Cruz DJ. Microcystic adnexal carcinoma: a distinct clinicopathologic entity. Cancer. 1982;50:566-572.
- Pujol RM, LeBoit PE, Su WP. Microcystic adnexal carcinoma with extensive sebaceous differentiation. Am J Dermatopathol. 1997;19:358-362.
- Rahman J, Tahir M, Arekemase H, et al. Desmoplastic trichoepithelioma: histopathologic and immunohistochemical criteria for differentiation of a rare benign hair follicle tumor from other cutaneous adnexal tumors. Cureus. 2020;12:E9703.
- Abesamis-Cubillan E, El-Shabrawi-Caelen L, LeBoit PE. Merkel cells and sclerosing epithelial neoplasms. Am J Dermatopathol. 2000;22:311-315.
- Sellheyer K, Nelson P, Kutzner H, et al. The immunohistochemical differential diagnosis of microcystic adnexal carcinoma, desmoplastic trichoepithelioma and morpheaform basal cell carcinoma using BerEP4 and stem cell markers. J Cutan Pathol. 2013;40:363-370.
- Gianfaldoni S, Tchernev G, Gianfaldoni R, et al. A case of “inflammatory linear verrucous epidermal nevus” (ILVEN) treated with CO2 laser ablation. Open Access Maced J Med Sci. 2017;5:454-457.
- Kawaguchi H, Takeuchi M, Ono H, et al. Adult onset of inflammatory linear verrucous epidermal nevus [published online October 27, 1999]. J Dermatol. 1999;26:599-602.
- Patterson JW, Hosler GA, Prenshaw KL, et al. The psoriasiform reaction pattern. In: Patterson JW. Weedon’s Skin Pathology. 5th ed. Elsevier; 2021:99-120.
The Diagnosis: Plaque-type Syringoma
A biopsy demonstrated multiple basaloid islands of tumor cells in the reticular dermis with ductal differentiation, some with a commalike tail. The ducts were lined by 2 to 3 layers of small uniform cuboidal cells without atypia and contained inspissated secretions within the lumina of scattered ducts. There was an associated fibrotic collagenous stroma. There was no evidence of perineural invasion and no deep dermal or subcutaneous extension (Figure 1). Additional cytokeratin immunohistochemical staining highlighted the adnexal proliferation (Figure 2). A diagnosis of plaque-type syringoma (PTS) was made.
Syringomas are benign dermal sweat gland tumors that typically present as flesh-colored papules on the cheeks or periorbital area of young females. Plaque-type tumors as well as papulonodular, eruptive, disseminated, urticaria pigmentosa–like, lichen planus–like, or milialike syringomas also have been reported. Syringomas may be associated with certain medical conditions such as Down syndrome, Nicolau-Balus syndrome, and both scarring and nonscarring alopecias.1 The clear cell variant of syringoma often is associated with diabetes mellitus.2 Kikuchi et al3 first described PTS in 1979. Plaque-type syringomas rarely are reported in the literature, and sites of involvement include the head and neck region, upper lip, chest, upper extremities, vulva, penis, and scrotum.4-6
Histologically, syringomatous lesions are composed of multiple small ducts lined by 2 to 3 layers of cuboidal epithelium. The ducts may be arranged in nests or strands of basaloid cells surrounded by a dense fibrotic stroma. Occasionally, the ducts will form a comma- or teardropshaped tail; however, this also may be observed in desmoplastic trichoepithelioma (DTE).7 Perineural invasion is absent in syringomas. Syringomas exhibit a lateral growth pattern that typically is limited to the upper half of the reticular dermis and spares the underlying subcutis, muscle, and bone. The growth pattern may be discontinuous with proliferations juxtaposed by normal-appearing skin.8 Syringomas usually express progesterone receptors and are known to proliferate at puberty, suggesting that these neoplasms are under hormonal control.9 Although syringomas are benign, various treatment options that may be pursued for cosmetic purposes include radiofrequency, staged excision, laser ablation, and oral isotretinoin.8,10 If only a superficial biopsy is obtained, syringomas may display features of other adnexal neoplasms, including microcystic adnexal carcinoma (MAC), DTE, morpheaform basal cell carcinoma (BCC), and inflammatory linear verrucous epidermal nevus (ILVEN).
Microcystic adnexal carcinoma is a locally aggressive neoplasm first described by Goldstein et al11 in 1982 an indurated, ill-defined plaque or nodule on the face with a predilection for the upper and lower lip. Prior radiation therapy and immunosuppression are risk factors for the development of MAC.12 Histologically, the superficial portion displays small cornifying cysts interspersed with islands of basaloid cells and may mimic a syringoma. However, the deeper portions demonstrate ducts lined by a single layer of cells with a background of hyalinized and sclerotic stroma. The tumor cells may occupy the deep dermis and underlying subcutis, muscle, or bone and demonstrate an infiltrative growth pattern and perineural invasion. Treatment includes Mohs micrographic surgery.
Desmoplastic trichoepitheliomas most commonly present as solitary white to yellowish annular papules or plaques with a central dell located on sun-exposed areas of the face, cheeks, or chin. This benign neoplasm has a bimodal age distribution, primarily affecting females either in childhood or adulthood.13 Histologically, strands and nests of basaloid epithelial cells proliferate in a dense eosinophilic desmoplastic stroma. The basaloid islands are narrow and cordlike with growth parallel to the surface epidermis and do not dive deeply into the deep dermis or subcutis. Ductal differentiation with associated secretions typically is not seen in DTE.1 Calcifications and foreign body granulomatous infiltrates may be present. Merkel cells also are present in this tumor and may be highlighted by immunohistochemistry with cytokeratin 20.14 Rarely, desmoplastic trichoepitheliomas may transform into trichoblastic carcinomas. Treatment may consist of surgical excision or Mohs micrographic surgery.
Morpheaform BCC also is included in the clinical and histopathologic differential diagnosis of infiltrative basaloid neoplasms. It is one of the more aggressive variants of BCC. The use of immunohistochemical staining may aid in differentiating between these sclerosing adnexal neoplasms.15 For example, pleckstrin homologylike domain family A member 1 (PHLDA1) is a stem cell marker that is heavily expressed in DTE as a specific follicular bulge marker but is not present in a morpheaform BCC. This highlights the follicular nature of DTEs at the molecular level. BerEP4 is a monoclonal antibody that serves as an epithelial marker for 2 glycopolypeptides: 34 and 39 kDa. This antibody may demonstrate positivity in morpheaform BCC but does not stain cells of interest in MAC.
Inflammatory linear verrucous epidermal nevus clinically presents with erythematous and warty papules in a linear distribution following the Blaschko lines. The papules often are reported to be intensely pruritic and usually are localized to one extremity.16 Although adultonset forms of ILVEN have been described,17 it most commonly is diagnosed in young children. Histologically, ILVEN consists of psoriasiform epidermal hyperplasia with alternating areas of parakeratosis and orthokeratosis with underlying agranulosis and hypergranulosis, respectively.18 The upper dermis contains a perivascular lymphocytic infiltrate. Treatment with laser therapy and surgical excision has led to both symptomatic and clinical improvement of ILVEN.16
Plaque-type syringomas are a rare variant of syringomas that clinically may mimic other common inflammatory and neoplastic conditions. An adequate biopsy is imperative to differentiate between adnexal neoplasms, as a small superficial biopsy of a syringoma may demonstrate features observed in other malignant or locally aggressive neoplasms. In our patient, the small ducts lined by cuboidal epithelium with no cellular atypia and no deep dermal growth or perineural invasion allowed for the diagnosis of PTS. Therapeutic options were reviewed with our patient, including oral isotretinoin, laser therapy, and staged excision. Ultimately, our patient elected not to pursue treatment, and she is being monitored clinically for any changes in appearance or symptoms.
The Diagnosis: Plaque-type Syringoma
A biopsy demonstrated multiple basaloid islands of tumor cells in the reticular dermis with ductal differentiation, some with a commalike tail. The ducts were lined by 2 to 3 layers of small uniform cuboidal cells without atypia and contained inspissated secretions within the lumina of scattered ducts. There was an associated fibrotic collagenous stroma. There was no evidence of perineural invasion and no deep dermal or subcutaneous extension (Figure 1). Additional cytokeratin immunohistochemical staining highlighted the adnexal proliferation (Figure 2). A diagnosis of plaque-type syringoma (PTS) was made.
Syringomas are benign dermal sweat gland tumors that typically present as flesh-colored papules on the cheeks or periorbital area of young females. Plaque-type tumors as well as papulonodular, eruptive, disseminated, urticaria pigmentosa–like, lichen planus–like, or milialike syringomas also have been reported. Syringomas may be associated with certain medical conditions such as Down syndrome, Nicolau-Balus syndrome, and both scarring and nonscarring alopecias.1 The clear cell variant of syringoma often is associated with diabetes mellitus.2 Kikuchi et al3 first described PTS in 1979. Plaque-type syringomas rarely are reported in the literature, and sites of involvement include the head and neck region, upper lip, chest, upper extremities, vulva, penis, and scrotum.4-6
Histologically, syringomatous lesions are composed of multiple small ducts lined by 2 to 3 layers of cuboidal epithelium. The ducts may be arranged in nests or strands of basaloid cells surrounded by a dense fibrotic stroma. Occasionally, the ducts will form a comma- or teardropshaped tail; however, this also may be observed in desmoplastic trichoepithelioma (DTE).7 Perineural invasion is absent in syringomas. Syringomas exhibit a lateral growth pattern that typically is limited to the upper half of the reticular dermis and spares the underlying subcutis, muscle, and bone. The growth pattern may be discontinuous with proliferations juxtaposed by normal-appearing skin.8 Syringomas usually express progesterone receptors and are known to proliferate at puberty, suggesting that these neoplasms are under hormonal control.9 Although syringomas are benign, various treatment options that may be pursued for cosmetic purposes include radiofrequency, staged excision, laser ablation, and oral isotretinoin.8,10 If only a superficial biopsy is obtained, syringomas may display features of other adnexal neoplasms, including microcystic adnexal carcinoma (MAC), DTE, morpheaform basal cell carcinoma (BCC), and inflammatory linear verrucous epidermal nevus (ILVEN).
Microcystic adnexal carcinoma is a locally aggressive neoplasm first described by Goldstein et al11 in 1982 an indurated, ill-defined plaque or nodule on the face with a predilection for the upper and lower lip. Prior radiation therapy and immunosuppression are risk factors for the development of MAC.12 Histologically, the superficial portion displays small cornifying cysts interspersed with islands of basaloid cells and may mimic a syringoma. However, the deeper portions demonstrate ducts lined by a single layer of cells with a background of hyalinized and sclerotic stroma. The tumor cells may occupy the deep dermis and underlying subcutis, muscle, or bone and demonstrate an infiltrative growth pattern and perineural invasion. Treatment includes Mohs micrographic surgery.
Desmoplastic trichoepitheliomas most commonly present as solitary white to yellowish annular papules or plaques with a central dell located on sun-exposed areas of the face, cheeks, or chin. This benign neoplasm has a bimodal age distribution, primarily affecting females either in childhood or adulthood.13 Histologically, strands and nests of basaloid epithelial cells proliferate in a dense eosinophilic desmoplastic stroma. The basaloid islands are narrow and cordlike with growth parallel to the surface epidermis and do not dive deeply into the deep dermis or subcutis. Ductal differentiation with associated secretions typically is not seen in DTE.1 Calcifications and foreign body granulomatous infiltrates may be present. Merkel cells also are present in this tumor and may be highlighted by immunohistochemistry with cytokeratin 20.14 Rarely, desmoplastic trichoepitheliomas may transform into trichoblastic carcinomas. Treatment may consist of surgical excision or Mohs micrographic surgery.
Morpheaform BCC also is included in the clinical and histopathologic differential diagnosis of infiltrative basaloid neoplasms. It is one of the more aggressive variants of BCC. The use of immunohistochemical staining may aid in differentiating between these sclerosing adnexal neoplasms.15 For example, pleckstrin homologylike domain family A member 1 (PHLDA1) is a stem cell marker that is heavily expressed in DTE as a specific follicular bulge marker but is not present in a morpheaform BCC. This highlights the follicular nature of DTEs at the molecular level. BerEP4 is a monoclonal antibody that serves as an epithelial marker for 2 glycopolypeptides: 34 and 39 kDa. This antibody may demonstrate positivity in morpheaform BCC but does not stain cells of interest in MAC.
Inflammatory linear verrucous epidermal nevus clinically presents with erythematous and warty papules in a linear distribution following the Blaschko lines. The papules often are reported to be intensely pruritic and usually are localized to one extremity.16 Although adultonset forms of ILVEN have been described,17 it most commonly is diagnosed in young children. Histologically, ILVEN consists of psoriasiform epidermal hyperplasia with alternating areas of parakeratosis and orthokeratosis with underlying agranulosis and hypergranulosis, respectively.18 The upper dermis contains a perivascular lymphocytic infiltrate. Treatment with laser therapy and surgical excision has led to both symptomatic and clinical improvement of ILVEN.16
Plaque-type syringomas are a rare variant of syringomas that clinically may mimic other common inflammatory and neoplastic conditions. An adequate biopsy is imperative to differentiate between adnexal neoplasms, as a small superficial biopsy of a syringoma may demonstrate features observed in other malignant or locally aggressive neoplasms. In our patient, the small ducts lined by cuboidal epithelium with no cellular atypia and no deep dermal growth or perineural invasion allowed for the diagnosis of PTS. Therapeutic options were reviewed with our patient, including oral isotretinoin, laser therapy, and staged excision. Ultimately, our patient elected not to pursue treatment, and she is being monitored clinically for any changes in appearance or symptoms.
- Suwattee P, McClelland MC, Huiras EE, et al. Plaque-type syringoma: two cases misdiagnosed as microcystic adnexal carcinoma [published online November 12, 2007]. J Cutan Pathol. 2008;35:570-574.
- Furue M, Hori Y, Nakabayashi Y. Clear-cell syringoma. association with diabetes mellitus. Am J Dermatopathol. 1984;6:131-138.
- Kikuchi I, Idemori M, Okazaki M. Plaque type syringoma. J Dermatol. 1979;6:329-331.
- Kavala M, Can B, Zindanci I, et al. Vulvar pruritus caused by syringoma of the vulva. Int J Dermatol. 2008;47:831-832.
- Cohen PR, Tschen JA, Rapini RP. Penile syringoma: reports and review of patients with syringoma located on the penis. J Clin Aesthet Dermatol. 2013;6:38-42.
- Okuda H, Tei N, Shimizu K, et al. Chondroid syringoma of the scrotum. Int J Urol. 2008;15:944-945.
- Wallace JS, Bond JS, Seidel GD, et al. An important mimicker: plaquetype syringoma mistakenly diagnosed as microcystic adnexal carcinoma. Dermatol Surg. 2014;40:810-812.
- Clark M, Duprey C, Sutton A, et al. Plaque-type syringoma masquerading as microcystic adnexal carcinoma: review of the literature and description of a novel technique that emphasizes lesion architecture to help make the diagnosis. Am J Dermatopathol. 2019;41:E98-E101.
- Wallace ML, Smoller BR. Progesterone receptor positivity supports hormonal control of syringomas. J Cutan Pathol. 1995;22:442-445.
- Mainitz M, Schmidt JB, Gebhart W. Response of multiple syringomas to isotretinoin. Acta Derm Venereol. 1986;66:51-55.
- Goldstein DJ, Barr RJ, Santa Cruz DJ. Microcystic adnexal carcinoma: a distinct clinicopathologic entity. Cancer. 1982;50:566-572.
- Pujol RM, LeBoit PE, Su WP. Microcystic adnexal carcinoma with extensive sebaceous differentiation. Am J Dermatopathol. 1997;19:358-362.
- Rahman J, Tahir M, Arekemase H, et al. Desmoplastic trichoepithelioma: histopathologic and immunohistochemical criteria for differentiation of a rare benign hair follicle tumor from other cutaneous adnexal tumors. Cureus. 2020;12:E9703.
- Abesamis-Cubillan E, El-Shabrawi-Caelen L, LeBoit PE. Merkel cells and sclerosing epithelial neoplasms. Am J Dermatopathol. 2000;22:311-315.
- Sellheyer K, Nelson P, Kutzner H, et al. The immunohistochemical differential diagnosis of microcystic adnexal carcinoma, desmoplastic trichoepithelioma and morpheaform basal cell carcinoma using BerEP4 and stem cell markers. J Cutan Pathol. 2013;40:363-370.
- Gianfaldoni S, Tchernev G, Gianfaldoni R, et al. A case of “inflammatory linear verrucous epidermal nevus” (ILVEN) treated with CO2 laser ablation. Open Access Maced J Med Sci. 2017;5:454-457.
- Kawaguchi H, Takeuchi M, Ono H, et al. Adult onset of inflammatory linear verrucous epidermal nevus [published online October 27, 1999]. J Dermatol. 1999;26:599-602.
- Patterson JW, Hosler GA, Prenshaw KL, et al. The psoriasiform reaction pattern. In: Patterson JW. Weedon’s Skin Pathology. 5th ed. Elsevier; 2021:99-120.
- Suwattee P, McClelland MC, Huiras EE, et al. Plaque-type syringoma: two cases misdiagnosed as microcystic adnexal carcinoma [published online November 12, 2007]. J Cutan Pathol. 2008;35:570-574.
- Furue M, Hori Y, Nakabayashi Y. Clear-cell syringoma. association with diabetes mellitus. Am J Dermatopathol. 1984;6:131-138.
- Kikuchi I, Idemori M, Okazaki M. Plaque type syringoma. J Dermatol. 1979;6:329-331.
- Kavala M, Can B, Zindanci I, et al. Vulvar pruritus caused by syringoma of the vulva. Int J Dermatol. 2008;47:831-832.
- Cohen PR, Tschen JA, Rapini RP. Penile syringoma: reports and review of patients with syringoma located on the penis. J Clin Aesthet Dermatol. 2013;6:38-42.
- Okuda H, Tei N, Shimizu K, et al. Chondroid syringoma of the scrotum. Int J Urol. 2008;15:944-945.
- Wallace JS, Bond JS, Seidel GD, et al. An important mimicker: plaquetype syringoma mistakenly diagnosed as microcystic adnexal carcinoma. Dermatol Surg. 2014;40:810-812.
- Clark M, Duprey C, Sutton A, et al. Plaque-type syringoma masquerading as microcystic adnexal carcinoma: review of the literature and description of a novel technique that emphasizes lesion architecture to help make the diagnosis. Am J Dermatopathol. 2019;41:E98-E101.
- Wallace ML, Smoller BR. Progesterone receptor positivity supports hormonal control of syringomas. J Cutan Pathol. 1995;22:442-445.
- Mainitz M, Schmidt JB, Gebhart W. Response of multiple syringomas to isotretinoin. Acta Derm Venereol. 1986;66:51-55.
- Goldstein DJ, Barr RJ, Santa Cruz DJ. Microcystic adnexal carcinoma: a distinct clinicopathologic entity. Cancer. 1982;50:566-572.
- Pujol RM, LeBoit PE, Su WP. Microcystic adnexal carcinoma with extensive sebaceous differentiation. Am J Dermatopathol. 1997;19:358-362.
- Rahman J, Tahir M, Arekemase H, et al. Desmoplastic trichoepithelioma: histopathologic and immunohistochemical criteria for differentiation of a rare benign hair follicle tumor from other cutaneous adnexal tumors. Cureus. 2020;12:E9703.
- Abesamis-Cubillan E, El-Shabrawi-Caelen L, LeBoit PE. Merkel cells and sclerosing epithelial neoplasms. Am J Dermatopathol. 2000;22:311-315.
- Sellheyer K, Nelson P, Kutzner H, et al. The immunohistochemical differential diagnosis of microcystic adnexal carcinoma, desmoplastic trichoepithelioma and morpheaform basal cell carcinoma using BerEP4 and stem cell markers. J Cutan Pathol. 2013;40:363-370.
- Gianfaldoni S, Tchernev G, Gianfaldoni R, et al. A case of “inflammatory linear verrucous epidermal nevus” (ILVEN) treated with CO2 laser ablation. Open Access Maced J Med Sci. 2017;5:454-457.
- Kawaguchi H, Takeuchi M, Ono H, et al. Adult onset of inflammatory linear verrucous epidermal nevus [published online October 27, 1999]. J Dermatol. 1999;26:599-602.
- Patterson JW, Hosler GA, Prenshaw KL, et al. The psoriasiform reaction pattern. In: Patterson JW. Weedon’s Skin Pathology. 5th ed. Elsevier; 2021:99-120.
A 17-year-old adolescent girl presented with a solitary, 8-cm, pink plaque on the anterior aspect of the neck of 5 years’ duration. No similar skin findings were present elsewhere on the body. The rash was not painful or pruritic, and she denied prior trauma to the site. The patient previously had tried a salicylic acid bodywash as well as mupirocin cream 2% and mometasone ointment with no improvement. Her medical history was unremarkable, and she had no known allergies. There was no family history of a similar rash. Physical examination revealed no palpable subcutaneous lumps or masses and no lymphadenopathy of the head or neck. An incisional biopsy was performed.


Multiple Fingerlike Projections on the Leg
The Diagnosis: Elephantiasis Nostras Verrucosa
Histopathology revealed a benign fibroepithelial polyp demonstrating areas of hyperkeratosis, acanthosis, and focal papillomatosis (Figure, A). Increased superficial vessels with dilated lymphatics, stellate fibroblasts, edematous stroma, and plasmolymphocytosis also were noted (Figure, B). Clinical and histopathological findings led to a diagnosis of lymphedema papules in the setting of elephantiasis nostra verrucosa (ENV).
. B, Dilated lymphatics, stellate fibroblasts, edematous stroma, and superficial plasmolymphocytosis")
Elephantiasis nostras verrucosa is a complication of long-standing nonfilarial obstruction of lymphatic drainage leading to grotesque enlargement of the affected areas. Common cutaneous manifestations of ENV include nonpitting edema, dermal fibrosis, and extensive hyperkeratosis with verrucous and papillomatous lesions.1 In the beginning stages of ENV, the skin has a cobblestonelike appearance. As the disease progresses, the verrucous lesions continue to enlarge, giving the affected area a mossy appearance. Although less common, groupings of large papillomas similar to our patient’s presentation also can form.2 Ulcer formation is more likely to occur in advanced disease states, increasing the risk for bacterial and fungal colonization. Elephantiasis nostras verrucosa classically affects the legs; however, this condition can develop in any area with chronic lymphedema. Cases of ENV involving the arms, abdomen, scrotum, and ear have been documented.3-5
The pathogenesis of ENV involves the proliferation of fibroblasts and fibrosis secondary to lymphostasis and inflammation.6 When interstitial fluid builds up in the affected region, the protein-rich fluid is believed to trigger fibrogenesis and increase macrophage, keratinocyte, and adipocyte activity.7 Because of this inflammatory process, dilation and fibrosis of the lymphatic channels develop. Lymphatic obstruction can have several etiologies, most notably infection and malignancy. Staphylococcal lymphangitis and erysipelas create fibrosis of the lymphatic system and are the main infectious causes of ENV.6 Large tumors or lymphomas are insidious causes of lymphatic obstruction and should be ruled out when investigating for ENV. Other risk factors include obesity, chronic venous insufficiency, surgery, trauma, radiation, and uncontrolled congestive heart failure.1,6,8
An ENV diagnosis is clinicopathologic, involving a comprehensive metabolic panel and complete blood cell count with differential. A biopsy is needed for pathologic confirmation and to rule out malignancy. Histologically, ENV is characterized by pseudoepitheliomatous hyperplasia, dermal fibrosis, hyperkeratosis of the epidermis, and dilated lymphatic vessels.6,8 Additional studies for diagnosis include wound and lymph node culture, Wood lamp examination, and lymphoscintigraphy.
Given the chronic and progressive nature of the disease, ENV is difficult to treat. There currently is no standard of treatment, but the mainstay of management involves reducing peripheral edema. Lifestyle changes including weight loss, extremity elevation, and increased ambulation are helpful first-line therapies.3 Compression of the affected extremity using stockings or intermittent pneumatic compression devices has proven to be beneficial with long-term use.7 Patients should be followed for wound care to prevent the infection of ulcers.2 Pharmacologic treatments include systemic retinoids, which have been shown to reduce the appearance of hyperkeratosis, verrucous lesions, and papillomatous nodules.6 Prophylactic antibiotics are reserved for advanced stages of disease or in patients with recurrent infections.2,7 In severe cases of ENV that are unresponsive to medical management, surgical intervention such as lymphatic anastomosis and debulking may be considered.9,10
Other diagnoses to consider for ENV include pretibial myxedema, lymphatic filariasis, Stewart-Treves syndrome, and papillomatosis cutis carcinoides. Pretibial myxedema is an uncommon dermatologic manifestation of Graves disease. It is a local autoimmune reaction in the cutaneous tissue characterized by hyperpigmentation, nonpitting edema, and nodules on the anterior leg. Histopathology shows increased hyaluronic acid and chondroitin as well as compression of dermal lymphatics.11
Filariasis is a parasitic infection caused by Wuchereria bancrofti, Brugia malayi or Brugia timori, and Onchocerca volvulus.6 This condition presents with elephantiasis of the affected extremities but should be considered in areas endemic for filarial parasites such as tropical and subtropical countries.12 Eosinophilia and identification of microfilaria in a peripheral blood smear would indicate parasitic infection. Stewart-Treves syndrome is a rare angiosarcoma that arises in areas of chronic lymphedema. This condition classically is seen on the upper extremities following a mastectomy with lymphadenectomy, lymph node irradiation, or both.
Stewart-Treves syndrome presents with coalescing purpuric macules and nodules that eventually coalesce into cutaneous masses. Histopathology reveals proliferating vascular channels that split apart dermal collagen with hyperchromatism and pleomorphism in the tumor endothelial cells that line these channels.13
Papillomatosis cutis carcinoides is a low-grade squamous cell carcinoma that occurs secondary to human papillomavirus commonly affecting the mouth, anogenital area, and the plantar surfaces of the feet. It presents with exophytic growths and ulcerated tumors that are unilateral and asymmetrical. The presence of blunt-shaped tumor projections extending deep into the dermis to form sinuses and keratin-filled cysts is characteristic of papillomatosis cutis carcinoides.14
- Dean SM, Zirwas MJ, Horst AV. Elephantiasis nostras verrucosa: an institutional analysis of 21 cases. J Am Acad Dermatol. 2011;64: 1104-1110. doi:10.1016/j.jaad.2010.04.047
- Fife CE, Farrow W, Hebert AA, et al. Skin and wound care in lymphedema patients: a taxonomy, primer, and literature review. Adv Skin Wound Care. 2017;30:305-318. doi:10.1097/01.ASW.0000520501.23702.82
- Boyd J, Sloan S, Meffert J. Elephantiasis nostrum verrucosa of the abdomen: clinical results with tazarotene. J Drugs Dermatol. 2004; 3:446-448.
- Nakai K, Taoka R, Sugimoto M, et al. Genital elephantiasis possibly caused by chronic inguinal eczema with streptococcal infection. J Dermatol. 2019;46:E196-E198. doi:10.1111/1346-8138.14746
- Carlson JA, Mazza J, Kircher K, et al. Otophyma: a case report and review of the literature of lymphedema (elephantiasis) of the ear. Am J Dermatopathol. 2008;30:67-72. doi:10.1097/DAD.0b013e31815cd937
- Sisto K, Khachemoune A. Elephantiasis nostras verrucosa: a review. Am J Clin Dermatol. 2008;9:141-146. doi:10.2165/00128071-200809030-00001
- Yoho RM, Budny AM, Pea AS. Elephantiasis nostras verrucosa. J Am Podiatr Med Assoc. 2006;96:442-444. doi:10.7547/0960442
- Yosipovitch G, DeVore A, Dawn A. Obesity and the skin: skin physiology and skin manifestations of obesity. J Am Acad Dermatol. 2007;56:901-920. doi:10.1016/j.jaad.2006.12.004
- Iwao F, Sato-Matsumura KC, Sawamura D, et al. Elephantiasis nostras verrucosa successfully treated by surgical debridement. Dermatol Surg. 2004;30:939-941. doi:10.1111/j.1524-4725.2004.30267.x
- Tiwari A, Cheng KS, Button M, et al. Differential diagnosis, investigation, and current treatment of lower limb lymphedema. Arch Surg. 2003;138:152-161. doi:10.1001/archsurg.138.2.152
- Fatourechi V. Pretibial myxedema: pathophysiology and treatment options. Am J Clin Dermatol. 2005;6:295-309. doi:10.2165 /00128071-200506050-00003
- Addiss DG, Brady MA. Morbidity management in the Global Programme to Eliminate Lymphatic Filariasis: a review of the scientific literature. Filaria J. 2007;6:2. doi:10.1186/1475-2883-6-2
- Bernia E, Rios-Viñuela E, Requena C. Stewart-Treves syndrome. JAMA Dermatol. 2021;157:721. doi:10.1001/jamadermatol.2021.0341
- Schwartz RA. Verrucous carcinoma of the skin and mucosa. J Am Acad Dermatol. 1995;32:1-24. doi:10.1016/0190-9622(95)90177-9
The Diagnosis: Elephantiasis Nostras Verrucosa
Histopathology revealed a benign fibroepithelial polyp demonstrating areas of hyperkeratosis, acanthosis, and focal papillomatosis (Figure, A). Increased superficial vessels with dilated lymphatics, stellate fibroblasts, edematous stroma, and plasmolymphocytosis also were noted (Figure, B). Clinical and histopathological findings led to a diagnosis of lymphedema papules in the setting of elephantiasis nostra verrucosa (ENV).
Elephantiasis nostras verrucosa is a complication of long-standing nonfilarial obstruction of lymphatic drainage leading to grotesque enlargement of the affected areas. Common cutaneous manifestations of ENV include nonpitting edema, dermal fibrosis, and extensive hyperkeratosis with verrucous and papillomatous lesions.1 In the beginning stages of ENV, the skin has a cobblestonelike appearance. As the disease progresses, the verrucous lesions continue to enlarge, giving the affected area a mossy appearance. Although less common, groupings of large papillomas similar to our patient’s presentation also can form.2 Ulcer formation is more likely to occur in advanced disease states, increasing the risk for bacterial and fungal colonization. Elephantiasis nostras verrucosa classically affects the legs; however, this condition can develop in any area with chronic lymphedema. Cases of ENV involving the arms, abdomen, scrotum, and ear have been documented.3-5
The pathogenesis of ENV involves the proliferation of fibroblasts and fibrosis secondary to lymphostasis and inflammation.6 When interstitial fluid builds up in the affected region, the protein-rich fluid is believed to trigger fibrogenesis and increase macrophage, keratinocyte, and adipocyte activity.7 Because of this inflammatory process, dilation and fibrosis of the lymphatic channels develop. Lymphatic obstruction can have several etiologies, most notably infection and malignancy. Staphylococcal lymphangitis and erysipelas create fibrosis of the lymphatic system and are the main infectious causes of ENV.6 Large tumors or lymphomas are insidious causes of lymphatic obstruction and should be ruled out when investigating for ENV. Other risk factors include obesity, chronic venous insufficiency, surgery, trauma, radiation, and uncontrolled congestive heart failure.1,6,8
An ENV diagnosis is clinicopathologic, involving a comprehensive metabolic panel and complete blood cell count with differential. A biopsy is needed for pathologic confirmation and to rule out malignancy. Histologically, ENV is characterized by pseudoepitheliomatous hyperplasia, dermal fibrosis, hyperkeratosis of the epidermis, and dilated lymphatic vessels.6,8 Additional studies for diagnosis include wound and lymph node culture, Wood lamp examination, and lymphoscintigraphy.
Given the chronic and progressive nature of the disease, ENV is difficult to treat. There currently is no standard of treatment, but the mainstay of management involves reducing peripheral edema. Lifestyle changes including weight loss, extremity elevation, and increased ambulation are helpful first-line therapies.3 Compression of the affected extremity using stockings or intermittent pneumatic compression devices has proven to be beneficial with long-term use.7 Patients should be followed for wound care to prevent the infection of ulcers.2 Pharmacologic treatments include systemic retinoids, which have been shown to reduce the appearance of hyperkeratosis, verrucous lesions, and papillomatous nodules.6 Prophylactic antibiotics are reserved for advanced stages of disease or in patients with recurrent infections.2,7 In severe cases of ENV that are unresponsive to medical management, surgical intervention such as lymphatic anastomosis and debulking may be considered.9,10
Other diagnoses to consider for ENV include pretibial myxedema, lymphatic filariasis, Stewart-Treves syndrome, and papillomatosis cutis carcinoides. Pretibial myxedema is an uncommon dermatologic manifestation of Graves disease. It is a local autoimmune reaction in the cutaneous tissue characterized by hyperpigmentation, nonpitting edema, and nodules on the anterior leg. Histopathology shows increased hyaluronic acid and chondroitin as well as compression of dermal lymphatics.11
Filariasis is a parasitic infection caused by Wuchereria bancrofti, Brugia malayi or Brugia timori, and Onchocerca volvulus.6 This condition presents with elephantiasis of the affected extremities but should be considered in areas endemic for filarial parasites such as tropical and subtropical countries.12 Eosinophilia and identification of microfilaria in a peripheral blood smear would indicate parasitic infection. Stewart-Treves syndrome is a rare angiosarcoma that arises in areas of chronic lymphedema. This condition classically is seen on the upper extremities following a mastectomy with lymphadenectomy, lymph node irradiation, or both.
Stewart-Treves syndrome presents with coalescing purpuric macules and nodules that eventually coalesce into cutaneous masses. Histopathology reveals proliferating vascular channels that split apart dermal collagen with hyperchromatism and pleomorphism in the tumor endothelial cells that line these channels.13
Papillomatosis cutis carcinoides is a low-grade squamous cell carcinoma that occurs secondary to human papillomavirus commonly affecting the mouth, anogenital area, and the plantar surfaces of the feet. It presents with exophytic growths and ulcerated tumors that are unilateral and asymmetrical. The presence of blunt-shaped tumor projections extending deep into the dermis to form sinuses and keratin-filled cysts is characteristic of papillomatosis cutis carcinoides.14
The Diagnosis: Elephantiasis Nostras Verrucosa
Histopathology revealed a benign fibroepithelial polyp demonstrating areas of hyperkeratosis, acanthosis, and focal papillomatosis (Figure, A). Increased superficial vessels with dilated lymphatics, stellate fibroblasts, edematous stroma, and plasmolymphocytosis also were noted (Figure, B). Clinical and histopathological findings led to a diagnosis of lymphedema papules in the setting of elephantiasis nostra verrucosa (ENV).
Elephantiasis nostras verrucosa is a complication of long-standing nonfilarial obstruction of lymphatic drainage leading to grotesque enlargement of the affected areas. Common cutaneous manifestations of ENV include nonpitting edema, dermal fibrosis, and extensive hyperkeratosis with verrucous and papillomatous lesions.1 In the beginning stages of ENV, the skin has a cobblestonelike appearance. As the disease progresses, the verrucous lesions continue to enlarge, giving the affected area a mossy appearance. Although less common, groupings of large papillomas similar to our patient’s presentation also can form.2 Ulcer formation is more likely to occur in advanced disease states, increasing the risk for bacterial and fungal colonization. Elephantiasis nostras verrucosa classically affects the legs; however, this condition can develop in any area with chronic lymphedema. Cases of ENV involving the arms, abdomen, scrotum, and ear have been documented.3-5
The pathogenesis of ENV involves the proliferation of fibroblasts and fibrosis secondary to lymphostasis and inflammation.6 When interstitial fluid builds up in the affected region, the protein-rich fluid is believed to trigger fibrogenesis and increase macrophage, keratinocyte, and adipocyte activity.7 Because of this inflammatory process, dilation and fibrosis of the lymphatic channels develop. Lymphatic obstruction can have several etiologies, most notably infection and malignancy. Staphylococcal lymphangitis and erysipelas create fibrosis of the lymphatic system and are the main infectious causes of ENV.6 Large tumors or lymphomas are insidious causes of lymphatic obstruction and should be ruled out when investigating for ENV. Other risk factors include obesity, chronic venous insufficiency, surgery, trauma, radiation, and uncontrolled congestive heart failure.1,6,8
An ENV diagnosis is clinicopathologic, involving a comprehensive metabolic panel and complete blood cell count with differential. A biopsy is needed for pathologic confirmation and to rule out malignancy. Histologically, ENV is characterized by pseudoepitheliomatous hyperplasia, dermal fibrosis, hyperkeratosis of the epidermis, and dilated lymphatic vessels.6,8 Additional studies for diagnosis include wound and lymph node culture, Wood lamp examination, and lymphoscintigraphy.
Given the chronic and progressive nature of the disease, ENV is difficult to treat. There currently is no standard of treatment, but the mainstay of management involves reducing peripheral edema. Lifestyle changes including weight loss, extremity elevation, and increased ambulation are helpful first-line therapies.3 Compression of the affected extremity using stockings or intermittent pneumatic compression devices has proven to be beneficial with long-term use.7 Patients should be followed for wound care to prevent the infection of ulcers.2 Pharmacologic treatments include systemic retinoids, which have been shown to reduce the appearance of hyperkeratosis, verrucous lesions, and papillomatous nodules.6 Prophylactic antibiotics are reserved for advanced stages of disease or in patients with recurrent infections.2,7 In severe cases of ENV that are unresponsive to medical management, surgical intervention such as lymphatic anastomosis and debulking may be considered.9,10
Other diagnoses to consider for ENV include pretibial myxedema, lymphatic filariasis, Stewart-Treves syndrome, and papillomatosis cutis carcinoides. Pretibial myxedema is an uncommon dermatologic manifestation of Graves disease. It is a local autoimmune reaction in the cutaneous tissue characterized by hyperpigmentation, nonpitting edema, and nodules on the anterior leg. Histopathology shows increased hyaluronic acid and chondroitin as well as compression of dermal lymphatics.11
Filariasis is a parasitic infection caused by Wuchereria bancrofti, Brugia malayi or Brugia timori, and Onchocerca volvulus.6 This condition presents with elephantiasis of the affected extremities but should be considered in areas endemic for filarial parasites such as tropical and subtropical countries.12 Eosinophilia and identification of microfilaria in a peripheral blood smear would indicate parasitic infection. Stewart-Treves syndrome is a rare angiosarcoma that arises in areas of chronic lymphedema. This condition classically is seen on the upper extremities following a mastectomy with lymphadenectomy, lymph node irradiation, or both.
Stewart-Treves syndrome presents with coalescing purpuric macules and nodules that eventually coalesce into cutaneous masses. Histopathology reveals proliferating vascular channels that split apart dermal collagen with hyperchromatism and pleomorphism in the tumor endothelial cells that line these channels.13
Papillomatosis cutis carcinoides is a low-grade squamous cell carcinoma that occurs secondary to human papillomavirus commonly affecting the mouth, anogenital area, and the plantar surfaces of the feet. It presents with exophytic growths and ulcerated tumors that are unilateral and asymmetrical. The presence of blunt-shaped tumor projections extending deep into the dermis to form sinuses and keratin-filled cysts is characteristic of papillomatosis cutis carcinoides.14
- Dean SM, Zirwas MJ, Horst AV. Elephantiasis nostras verrucosa: an institutional analysis of 21 cases. J Am Acad Dermatol. 2011;64: 1104-1110. doi:10.1016/j.jaad.2010.04.047
- Fife CE, Farrow W, Hebert AA, et al. Skin and wound care in lymphedema patients: a taxonomy, primer, and literature review. Adv Skin Wound Care. 2017;30:305-318. doi:10.1097/01.ASW.0000520501.23702.82
- Boyd J, Sloan S, Meffert J. Elephantiasis nostrum verrucosa of the abdomen: clinical results with tazarotene. J Drugs Dermatol. 2004; 3:446-448.
- Nakai K, Taoka R, Sugimoto M, et al. Genital elephantiasis possibly caused by chronic inguinal eczema with streptococcal infection. J Dermatol. 2019;46:E196-E198. doi:10.1111/1346-8138.14746
- Carlson JA, Mazza J, Kircher K, et al. Otophyma: a case report and review of the literature of lymphedema (elephantiasis) of the ear. Am J Dermatopathol. 2008;30:67-72. doi:10.1097/DAD.0b013e31815cd937
- Sisto K, Khachemoune A. Elephantiasis nostras verrucosa: a review. Am J Clin Dermatol. 2008;9:141-146. doi:10.2165/00128071-200809030-00001
- Yoho RM, Budny AM, Pea AS. Elephantiasis nostras verrucosa. J Am Podiatr Med Assoc. 2006;96:442-444. doi:10.7547/0960442
- Yosipovitch G, DeVore A, Dawn A. Obesity and the skin: skin physiology and skin manifestations of obesity. J Am Acad Dermatol. 2007;56:901-920. doi:10.1016/j.jaad.2006.12.004
- Iwao F, Sato-Matsumura KC, Sawamura D, et al. Elephantiasis nostras verrucosa successfully treated by surgical debridement. Dermatol Surg. 2004;30:939-941. doi:10.1111/j.1524-4725.2004.30267.x
- Tiwari A, Cheng KS, Button M, et al. Differential diagnosis, investigation, and current treatment of lower limb lymphedema. Arch Surg. 2003;138:152-161. doi:10.1001/archsurg.138.2.152
- Fatourechi V. Pretibial myxedema: pathophysiology and treatment options. Am J Clin Dermatol. 2005;6:295-309. doi:10.2165 /00128071-200506050-00003
- Addiss DG, Brady MA. Morbidity management in the Global Programme to Eliminate Lymphatic Filariasis: a review of the scientific literature. Filaria J. 2007;6:2. doi:10.1186/1475-2883-6-2
- Bernia E, Rios-Viñuela E, Requena C. Stewart-Treves syndrome. JAMA Dermatol. 2021;157:721. doi:10.1001/jamadermatol.2021.0341
- Schwartz RA. Verrucous carcinoma of the skin and mucosa. J Am Acad Dermatol. 1995;32:1-24. doi:10.1016/0190-9622(95)90177-9
- Dean SM, Zirwas MJ, Horst AV. Elephantiasis nostras verrucosa: an institutional analysis of 21 cases. J Am Acad Dermatol. 2011;64: 1104-1110. doi:10.1016/j.jaad.2010.04.047
- Fife CE, Farrow W, Hebert AA, et al. Skin and wound care in lymphedema patients: a taxonomy, primer, and literature review. Adv Skin Wound Care. 2017;30:305-318. doi:10.1097/01.ASW.0000520501.23702.82
- Boyd J, Sloan S, Meffert J. Elephantiasis nostrum verrucosa of the abdomen: clinical results with tazarotene. J Drugs Dermatol. 2004; 3:446-448.
- Nakai K, Taoka R, Sugimoto M, et al. Genital elephantiasis possibly caused by chronic inguinal eczema with streptococcal infection. J Dermatol. 2019;46:E196-E198. doi:10.1111/1346-8138.14746
- Carlson JA, Mazza J, Kircher K, et al. Otophyma: a case report and review of the literature of lymphedema (elephantiasis) of the ear. Am J Dermatopathol. 2008;30:67-72. doi:10.1097/DAD.0b013e31815cd937
- Sisto K, Khachemoune A. Elephantiasis nostras verrucosa: a review. Am J Clin Dermatol. 2008;9:141-146. doi:10.2165/00128071-200809030-00001
- Yoho RM, Budny AM, Pea AS. Elephantiasis nostras verrucosa. J Am Podiatr Med Assoc. 2006;96:442-444. doi:10.7547/0960442
- Yosipovitch G, DeVore A, Dawn A. Obesity and the skin: skin physiology and skin manifestations of obesity. J Am Acad Dermatol. 2007;56:901-920. doi:10.1016/j.jaad.2006.12.004
- Iwao F, Sato-Matsumura KC, Sawamura D, et al. Elephantiasis nostras verrucosa successfully treated by surgical debridement. Dermatol Surg. 2004;30:939-941. doi:10.1111/j.1524-4725.2004.30267.x
- Tiwari A, Cheng KS, Button M, et al. Differential diagnosis, investigation, and current treatment of lower limb lymphedema. Arch Surg. 2003;138:152-161. doi:10.1001/archsurg.138.2.152
- Fatourechi V. Pretibial myxedema: pathophysiology and treatment options. Am J Clin Dermatol. 2005;6:295-309. doi:10.2165 /00128071-200506050-00003
- Addiss DG, Brady MA. Morbidity management in the Global Programme to Eliminate Lymphatic Filariasis: a review of the scientific literature. Filaria J. 2007;6:2. doi:10.1186/1475-2883-6-2
- Bernia E, Rios-Viñuela E, Requena C. Stewart-Treves syndrome. JAMA Dermatol. 2021;157:721. doi:10.1001/jamadermatol.2021.0341
- Schwartz RA. Verrucous carcinoma of the skin and mucosa. J Am Acad Dermatol. 1995;32:1-24. doi:10.1016/0190-9622(95)90177-9
A 61-year-old man presented with painful skin growths on the right pretibial region of several months’ duration. The patient reported pain due to friction between the lesions and underlying skin, leading to erosions. His medical history was remarkable for morbid obesity (body mass index of 62), chronic venous stasis, and chronic lymphedema. The patient was followed for wound care of venous stasis ulcers. Dermatologic examination revealed multiple 5- to 30-mm, flesh-colored, fingerlike projections on the right tibial region. A biopsy was obtained and submitted for histopathologic analysis.


Focal Palmoplantar Keratoderma and Gingival Keratosis Caused by a KRT16 Mutation
To the Editor:
Focal palmoplantar keratoderma and gingival keratosis (FPGK)(Online Mendelian Inheritance in Man [OMIM] 148730) is a rare autosomal-dominant syndrome featuring focal, pressure-related, painful palmoplantar keratoderma and gingival hyperkeratosis presenting as leukokeratosis. Focal palmoplantar keratoderma and gingival keratosis was first defined by Gorlin1 in 1976. Since then, only a few cases have been reported, but no causative mutations have been identified.2
Focal pressure-related palmoplantar keratoderma (PPK) and oral hyperkeratosis also are seen in pachyonychia congenita (PC)(OMIM 167200, 615726, 615728, 167210), a rare autosomal-dominant disorder of keratinization characterized by PPK and nail dystrophy. Patients with PC often present with plantar pain; more variable features include oral leukokeratosis, follicular hyperkeratosis, pilosebaceous and epidermal inclusion cysts, hoarseness, hyperhidrosis, and natal teeth. Pachyonychia congenita is caused by mutation in keratin genes KRT6A, KRT6B, KRT16, or KRT17.
Focal palmoplantar keratoderma and gingival keratosis as well as PC are distinct from other forms of PPK with gingival involvement such as
Despite the common features of FPGK and PC, they are considered distinct disorders due to absence of nail changes in FPGK and no prior evidence of a common genetic cause. We present a patient with familial FPGK found by whole exome sequencing to be caused by a mutation in KRT16.
 showing focal palmoplantar keratoderma and gingival keratosis in those heterozygous for KRT16 mutation p.R127H")
The proband was a 57-year-old man born to unrelated parents (Figure 1). He had no skin problems at birth, and his development was normal. He had painful focal keratoderma since childhood that were most prominent at pressure points on the soles and toes (Figure 2A), in addition to gingival hyperkeratosis and oral leukokeratosis (Figure 2B). He had no associated abnormalities of the skin, hair, or teeth and no nail findings (Figure 2C). He reported that his father and 2 of his 3 sisters were affected with similar symptoms. A punch biopsy of the right fifth toe was consistent with verrucous epidermal hyperplasia with perinuclear keratinization in the spinous layer (Figure 3A). A gingival biopsy showed perinuclear eosinophilic globules and basophilic stranding in the cytoplasm (Figure 3B). His older sister had more severe and painful focal keratoderma of the soles, punctate keratoderma of the palms, gingival hyperkeratosis, and leukokeratosis of the tongue.

Whole exome sequencing of the proband revealed a heterozygous missense mutation in KRT16 (c.380G>A, p.R127H, rs57424749). Sanger sequencing confirmed this mutation and showed that it was heterozygous in both of his affected sisters and absent in his unaffected niece (Figure 1). The patient was treated with topical and systemic retinoids, keratolytics, and mechanical removal to moderate effect, with noted improvement in the appearance and associated pain of the plantar keratoderma.

Phenotypic heterogeneity is common in PC, though PC due to KRT6A mutations demonstrates more severe nail disease with oral lesions, cysts, and follicular hyperkeratosis, while PC caused by KRT16 mutations generally presents with more extensive and painful PPK.4KRT16 mutations affecting p.R127 are frequent causes of PC, and genotype-phenotype correlations have been observed. Individuals with p.R127P mutations exhibit more severe disease with earlier age of onset, more extensive nail involvement and oral leukokeratosis, and greater impact on daily quality of life than in individuals with p.R127C mutations.5 Cases of PC with KRT16 p.R127S and p.R127G mutations also have been observed. The KRT16 c.380G>A, p.R127H mutation we documented has been reported in one kindred with PC who presented with PPK, oral leukokeratosis, toenail thickening, and pilosebaceous and follicular hyperkeratosis.6
Although patients with FPGK lack the thickening of fingernails and/or toenails considered a defining feature of PC, the disorders otherwise are phenotypically similar, suggesting the possibility of common pathogenesis. One linkage study of familial FPGK excluded genetic intervals containing type I and type II keratins but was limited to a single small kindred.2 This study and our data together suggest that, similar to PC, there are multiple genes in which mutations cause FPGK.
Murine Krt16 knockouts show distinct phenotypes depending on the mouse strain in which they are propagated, ranging from perinatal lethality to differences in the severity of oral and PPK lesions.7 These observations provide evidence that additional genetic variants contribute to Krt16 phenotypes in mice and suggest the same could be true for humans.
We propose that some cases of FPGK are due to mutations in KRT16 and thus share a genetic pathogenesis with PC, underscoring the utility of whole exome sequencing in providing genetic diagnoses for disorders that are genetically and clinically heterogeneous. Further biologic investigation of phenotypes caused by KRT16 mutation may reveal respective contributions of additional genetic variation and environmental effects to the variable clinical presentations.
- Gorlin RJ. Focal palmoplantar and marginal gingival hyperkeratosis—a syndrome. Birth Defects Orig Artic Ser. 1976;12:239-242.
- Kolde G, Hennies HC, Bethke G, et al. Focal palmoplantar and gingival keratosis: a distinct palmoplantar ectodermal dysplasia with epidermolytic alterations but lack of mutations in known keratins. J Am Acad Dermatol. 2005;52(3 pt 1):403-409.
- Duchatelet S, Hovnanian A. Olmsted syndrome: clinical, molecular and therapeutic aspects. Orphanet J Rare Dis. 2015;10:33.
- Spaunhurst KM, Hogendorf AM, Smith FJ, et al. Pachyonychia congenita patients with mutations in KRT6A have more extensive disease compared with patients who have mutations in KRT16. Br J Dermatol. 2012;166:875-878.
- Fu T, Leachman SA, Wilson NJ, et al. Genotype-phenotype correlations among pachyonychia congenita patients with K16 mutations. J Invest Dermatol. 2011;131:1025-1028.
- Wilson NJ, O’Toole EA, Milstone LM, et al. The molecular genetic analysis of the expanding pachyonychia congenita case collection. Br J Dermatol. 2014;171:343-355.
- Zieman A, Coulombe PA. The keratin 16 null phenotype is modestly impacted by genetic strain background in mice. Exp Dermatol. 2018;27:672-674.
To the Editor:
Focal palmoplantar keratoderma and gingival keratosis (FPGK)(Online Mendelian Inheritance in Man [OMIM] 148730) is a rare autosomal-dominant syndrome featuring focal, pressure-related, painful palmoplantar keratoderma and gingival hyperkeratosis presenting as leukokeratosis. Focal palmoplantar keratoderma and gingival keratosis was first defined by Gorlin1 in 1976. Since then, only a few cases have been reported, but no causative mutations have been identified.2
Focal pressure-related palmoplantar keratoderma (PPK) and oral hyperkeratosis also are seen in pachyonychia congenita (PC)(OMIM 167200, 615726, 615728, 167210), a rare autosomal-dominant disorder of keratinization characterized by PPK and nail dystrophy. Patients with PC often present with plantar pain; more variable features include oral leukokeratosis, follicular hyperkeratosis, pilosebaceous and epidermal inclusion cysts, hoarseness, hyperhidrosis, and natal teeth. Pachyonychia congenita is caused by mutation in keratin genes KRT6A, KRT6B, KRT16, or KRT17.
Focal palmoplantar keratoderma and gingival keratosis as well as PC are distinct from other forms of PPK with gingival involvement such as
Despite the common features of FPGK and PC, they are considered distinct disorders due to absence of nail changes in FPGK and no prior evidence of a common genetic cause. We present a patient with familial FPGK found by whole exome sequencing to be caused by a mutation in KRT16.
The proband was a 57-year-old man born to unrelated parents (Figure 1). He had no skin problems at birth, and his development was normal. He had painful focal keratoderma since childhood that were most prominent at pressure points on the soles and toes (Figure 2A), in addition to gingival hyperkeratosis and oral leukokeratosis (Figure 2B). He had no associated abnormalities of the skin, hair, or teeth and no nail findings (Figure 2C). He reported that his father and 2 of his 3 sisters were affected with similar symptoms. A punch biopsy of the right fifth toe was consistent with verrucous epidermal hyperplasia with perinuclear keratinization in the spinous layer (Figure 3A). A gingival biopsy showed perinuclear eosinophilic globules and basophilic stranding in the cytoplasm (Figure 3B). His older sister had more severe and painful focal keratoderma of the soles, punctate keratoderma of the palms, gingival hyperkeratosis, and leukokeratosis of the tongue.
Whole exome sequencing of the proband revealed a heterozygous missense mutation in KRT16 (c.380G>A, p.R127H, rs57424749). Sanger sequencing confirmed this mutation and showed that it was heterozygous in both of his affected sisters and absent in his unaffected niece (Figure 1). The patient was treated with topical and systemic retinoids, keratolytics, and mechanical removal to moderate effect, with noted improvement in the appearance and associated pain of the plantar keratoderma.
Phenotypic heterogeneity is common in PC, though PC due to KRT6A mutations demonstrates more severe nail disease with oral lesions, cysts, and follicular hyperkeratosis, while PC caused by KRT16 mutations generally presents with more extensive and painful PPK.4KRT16 mutations affecting p.R127 are frequent causes of PC, and genotype-phenotype correlations have been observed. Individuals with p.R127P mutations exhibit more severe disease with earlier age of onset, more extensive nail involvement and oral leukokeratosis, and greater impact on daily quality of life than in individuals with p.R127C mutations.5 Cases of PC with KRT16 p.R127S and p.R127G mutations also have been observed. The KRT16 c.380G>A, p.R127H mutation we documented has been reported in one kindred with PC who presented with PPK, oral leukokeratosis, toenail thickening, and pilosebaceous and follicular hyperkeratosis.6
Although patients with FPGK lack the thickening of fingernails and/or toenails considered a defining feature of PC, the disorders otherwise are phenotypically similar, suggesting the possibility of common pathogenesis. One linkage study of familial FPGK excluded genetic intervals containing type I and type II keratins but was limited to a single small kindred.2 This study and our data together suggest that, similar to PC, there are multiple genes in which mutations cause FPGK.
Murine Krt16 knockouts show distinct phenotypes depending on the mouse strain in which they are propagated, ranging from perinatal lethality to differences in the severity of oral and PPK lesions.7 These observations provide evidence that additional genetic variants contribute to Krt16 phenotypes in mice and suggest the same could be true for humans.
We propose that some cases of FPGK are due to mutations in KRT16 and thus share a genetic pathogenesis with PC, underscoring the utility of whole exome sequencing in providing genetic diagnoses for disorders that are genetically and clinically heterogeneous. Further biologic investigation of phenotypes caused by KRT16 mutation may reveal respective contributions of additional genetic variation and environmental effects to the variable clinical presentations.
To the Editor:
Focal palmoplantar keratoderma and gingival keratosis (FPGK)(Online Mendelian Inheritance in Man [OMIM] 148730) is a rare autosomal-dominant syndrome featuring focal, pressure-related, painful palmoplantar keratoderma and gingival hyperkeratosis presenting as leukokeratosis. Focal palmoplantar keratoderma and gingival keratosis was first defined by Gorlin1 in 1976. Since then, only a few cases have been reported, but no causative mutations have been identified.2
Focal pressure-related palmoplantar keratoderma (PPK) and oral hyperkeratosis also are seen in pachyonychia congenita (PC)(OMIM 167200, 615726, 615728, 167210), a rare autosomal-dominant disorder of keratinization characterized by PPK and nail dystrophy. Patients with PC often present with plantar pain; more variable features include oral leukokeratosis, follicular hyperkeratosis, pilosebaceous and epidermal inclusion cysts, hoarseness, hyperhidrosis, and natal teeth. Pachyonychia congenita is caused by mutation in keratin genes KRT6A, KRT6B, KRT16, or KRT17.
Focal palmoplantar keratoderma and gingival keratosis as well as PC are distinct from other forms of PPK with gingival involvement such as
Despite the common features of FPGK and PC, they are considered distinct disorders due to absence of nail changes in FPGK and no prior evidence of a common genetic cause. We present a patient with familial FPGK found by whole exome sequencing to be caused by a mutation in KRT16.
The proband was a 57-year-old man born to unrelated parents (Figure 1). He had no skin problems at birth, and his development was normal. He had painful focal keratoderma since childhood that were most prominent at pressure points on the soles and toes (Figure 2A), in addition to gingival hyperkeratosis and oral leukokeratosis (Figure 2B). He had no associated abnormalities of the skin, hair, or teeth and no nail findings (Figure 2C). He reported that his father and 2 of his 3 sisters were affected with similar symptoms. A punch biopsy of the right fifth toe was consistent with verrucous epidermal hyperplasia with perinuclear keratinization in the spinous layer (Figure 3A). A gingival biopsy showed perinuclear eosinophilic globules and basophilic stranding in the cytoplasm (Figure 3B). His older sister had more severe and painful focal keratoderma of the soles, punctate keratoderma of the palms, gingival hyperkeratosis, and leukokeratosis of the tongue.
Whole exome sequencing of the proband revealed a heterozygous missense mutation in KRT16 (c.380G>A, p.R127H, rs57424749). Sanger sequencing confirmed this mutation and showed that it was heterozygous in both of his affected sisters and absent in his unaffected niece (Figure 1). The patient was treated with topical and systemic retinoids, keratolytics, and mechanical removal to moderate effect, with noted improvement in the appearance and associated pain of the plantar keratoderma.
Phenotypic heterogeneity is common in PC, though PC due to KRT6A mutations demonstrates more severe nail disease with oral lesions, cysts, and follicular hyperkeratosis, while PC caused by KRT16 mutations generally presents with more extensive and painful PPK.4KRT16 mutations affecting p.R127 are frequent causes of PC, and genotype-phenotype correlations have been observed. Individuals with p.R127P mutations exhibit more severe disease with earlier age of onset, more extensive nail involvement and oral leukokeratosis, and greater impact on daily quality of life than in individuals with p.R127C mutations.5 Cases of PC with KRT16 p.R127S and p.R127G mutations also have been observed. The KRT16 c.380G>A, p.R127H mutation we documented has been reported in one kindred with PC who presented with PPK, oral leukokeratosis, toenail thickening, and pilosebaceous and follicular hyperkeratosis.6
Although patients with FPGK lack the thickening of fingernails and/or toenails considered a defining feature of PC, the disorders otherwise are phenotypically similar, suggesting the possibility of common pathogenesis. One linkage study of familial FPGK excluded genetic intervals containing type I and type II keratins but was limited to a single small kindred.2 This study and our data together suggest that, similar to PC, there are multiple genes in which mutations cause FPGK.
Murine Krt16 knockouts show distinct phenotypes depending on the mouse strain in which they are propagated, ranging from perinatal lethality to differences in the severity of oral and PPK lesions.7 These observations provide evidence that additional genetic variants contribute to Krt16 phenotypes in mice and suggest the same could be true for humans.
We propose that some cases of FPGK are due to mutations in KRT16 and thus share a genetic pathogenesis with PC, underscoring the utility of whole exome sequencing in providing genetic diagnoses for disorders that are genetically and clinically heterogeneous. Further biologic investigation of phenotypes caused by KRT16 mutation may reveal respective contributions of additional genetic variation and environmental effects to the variable clinical presentations.
- Gorlin RJ. Focal palmoplantar and marginal gingival hyperkeratosis—a syndrome. Birth Defects Orig Artic Ser. 1976;12:239-242.
- Kolde G, Hennies HC, Bethke G, et al. Focal palmoplantar and gingival keratosis: a distinct palmoplantar ectodermal dysplasia with epidermolytic alterations but lack of mutations in known keratins. J Am Acad Dermatol. 2005;52(3 pt 1):403-409.
- Duchatelet S, Hovnanian A. Olmsted syndrome: clinical, molecular and therapeutic aspects. Orphanet J Rare Dis. 2015;10:33.
- Spaunhurst KM, Hogendorf AM, Smith FJ, et al. Pachyonychia congenita patients with mutations in KRT6A have more extensive disease compared with patients who have mutations in KRT16. Br J Dermatol. 2012;166:875-878.
- Fu T, Leachman SA, Wilson NJ, et al. Genotype-phenotype correlations among pachyonychia congenita patients with K16 mutations. J Invest Dermatol. 2011;131:1025-1028.
- Wilson NJ, O’Toole EA, Milstone LM, et al. The molecular genetic analysis of the expanding pachyonychia congenita case collection. Br J Dermatol. 2014;171:343-355.
- Zieman A, Coulombe PA. The keratin 16 null phenotype is modestly impacted by genetic strain background in mice. Exp Dermatol. 2018;27:672-674.
- Gorlin RJ. Focal palmoplantar and marginal gingival hyperkeratosis—a syndrome. Birth Defects Orig Artic Ser. 1976;12:239-242.
- Kolde G, Hennies HC, Bethke G, et al. Focal palmoplantar and gingival keratosis: a distinct palmoplantar ectodermal dysplasia with epidermolytic alterations but lack of mutations in known keratins. J Am Acad Dermatol. 2005;52(3 pt 1):403-409.
- Duchatelet S, Hovnanian A. Olmsted syndrome: clinical, molecular and therapeutic aspects. Orphanet J Rare Dis. 2015;10:33.
- Spaunhurst KM, Hogendorf AM, Smith FJ, et al. Pachyonychia congenita patients with mutations in KRT6A have more extensive disease compared with patients who have mutations in KRT16. Br J Dermatol. 2012;166:875-878.
- Fu T, Leachman SA, Wilson NJ, et al. Genotype-phenotype correlations among pachyonychia congenita patients with K16 mutations. J Invest Dermatol. 2011;131:1025-1028.
- Wilson NJ, O’Toole EA, Milstone LM, et al. The molecular genetic analysis of the expanding pachyonychia congenita case collection. Br J Dermatol. 2014;171:343-355.
- Zieman A, Coulombe PA. The keratin 16 null phenotype is modestly impacted by genetic strain background in mice. Exp Dermatol. 2018;27:672-674.
Practice Points
- Focal palmoplantar keratoderma and gingival keratosis (FPGK) is a rare autosomal-dominant syndrome featuring focal, pressure-related, painful palmoplantar keratoderma (PPK) and gingival hyperkeratosis presenting as leukokeratosis.
- Focal pressure-related PPK and oral hyperkeratosis also are seen in pachyonychia congenita (PC), which is caused by mutations in keratin genes and is distinguished from FPGK by characteristic nail changes.
- A shared causative gene suggests that FPGK should be considered part of the PC spectrum.

Fungated Eroded Plaque on the Arm
The Diagnosis: Cutaneous Blastomycosis
A skin biopsy and fungal cultures confirmed the diagnosis of cutaneous blastomycosis. Grocott- Gomori methenamine-silver staining highlighted fungal organisms with refractile walls and broad-based budding consistent with cutaneous blastomycosis (Figure 1). The histopathologic specimen also demonstrated marked pseudoepitheliomatous hyperplasia (Figure 2A) with neutrophilic microabscesses (Figure 2B). Acid-fast bacillus and Fite staining were negative for bacterial organisms. A fungal culture was positive for Blastomyces dermatitidis. Urine and serum blastomycosis antigen were positive. Although Histoplasma serum antigen also was positive, this likely was from cross-reactivity. Chest radiography was negative for lung involvement, and the patient displayed no neurologic symptoms. He was started on oral itraconazole therapy for the treatment of cutaneous blastomycosis.

Blastomyces dermatitidis, the causative organism of blastomycosis, is endemic to the Ohio and Mississippi River valleys, Great Lakes region, and southeastern United States. It is a thermally dimorphic fungus found in soils that grows as a mold at 25 °C and yeast at 37 °C. Primary infection of the lungs—blastomycosis pneumonia—often is the only clinical manifestation1; however, subsequent hematogenous dissemination to extrapulmonary sites such as the skin, bones, and genitourinary system can occur. Cutaneous blastomycosis, the most common extrapulmonary manifestation, typically follows pulmonary infection. In rare cases, it can occur from direct inoculation.2,3 Skin lesions can occur anywhere but frequently are found on exposed surfaces of the head, neck, and extremities. Lesions classically present as verrucous crusting plaques with draining microabscesses. Violaceous nodules, ulcers, and pustules also may occur.1

Diagnosis involves obtaining a thorough history of possible environmental exposures such as the patient’s geographic area of residence, occupation, and outdoor activities involving soil or decaying wood. Because blastomycosis can remain latent, remote exposures are relevant. Definitive diagnosis of cutaneous blastomycosis involves skin biopsy of the lesion with fungal culture, but the yeast’s distinctive thick wall and broad-based budding seen with periodic acid–Schiff or Grocott-Gomori methenamine-silver staining provides a rapid presumptive diagnosis.3 Pseudoepitheliomatous hyperplasia and microabscesses also are characteristic features.2 Urine antigen testing for a component of the polysaccharide cell wall has a sensitivity of 93% but a lower specificity of 79% due to cross-reactivity with histoplasmosis.4 Treatment consists of itraconazole for mild to moderate blastomycosis or amphotericin B for those with severe disease or central nervous system involvement or those who are immunosuppressed.1
The differential diagnosis for our patient’s lesion included infectious vs neoplastic etiologies. Histoplasma capsulatum, the dimorphic fungus that causes histoplasmosis, also is endemic to the Ohio and Mississippi River valleys. It is found in soil and droppings of some bats and birds such as chickens and pigeons. Similar to blastomycosis, the primary infection site most commonly is the lungs. It subsequently may disseminate to the skin or less commonly via direct inoculation of injured skin. It can present as papules, plaques, ulcers, purpura, or abscesses. Unlike blastomycosis, tissue biopsy of a cutaneous lesion reveals granuloma formation and distinctive oval, narrow-based budding yeast.5 Atypical mycobacteria are another source of infection to consider. For example, cutaneous Mycobacterium kansasii may present as papules and pustules forming verrucous or granulomatous plaques and ulceration. Histopathologic findings distinguishing mycobacterial infection from blastomycosis include granulomas and acid-fast bacilli in histiocytes.6
Noninfectious etiologies in the differential may include cutaneous squamous cell carcinoma or pemphigus vegetans. Squamous cell carcinoma may present with a broad range of clinical features—papules, plaques, or nodules with smooth, scaly, verrucous, or ulcerative secondary features all are possible presentations.7 Fairskinned individuals, such as our patient, would be at a higher risk in sun-damaged skin. Histologically, cutaneous squamous cell carcinoma is defined as an invasion of the dermis by neoplastic squamous epithelial cells in the form of cords, sheets, individual cells, nodules, or cystic structures.7 Pemphigus vegetans is the rarest variant of a group of autoimmune vesiculobullous diseases known as pemphigus. It can be differentiated from the most common variant—pemphigus vulgaris—by the presence of vegetative plaques in intertriginous areas. However, these verrucous vegetations can be misleading and make clinical diagnosis difficult. Histopathologic findings of hyperkeratosis, pseudoepitheliomatous hyperplasia, papillomatosis, and acantholysis with a suprabasal cleft would confirm the diagnosis.8
In summary, cutaneous blastomycosis classically presents as verrucous crusting plaques, as seen in our patient. It is important to conduct a thorough history for environmental exposures, but definitive diagnosis of cutaneous blastomycosis involves skin biopsy with fungal culture. Treatment depends on the severity of disease and organ involvement. Itraconazole would be appropriate for mild to moderate blastomycosis.
- Miceli A, Krishnamurthy K. Blastomycosis. StatPearls. StatPearls Publishing; 2022. Accessed June 21, 2022. https://www.ncbi.nlm.nih.gov/books/NBK441987/
- Gray NA, Baddour LM. Cutaneous inoculation blastomycosis. Clin Infect Dis. 2002;34:E44-E49.
- Schwartz IS, Kauffman CA. Blastomycosis. Semin Respir Crit Care Med. 2020;41:31-41. doi:10.1055/s-0039-3400281
- Castillo CG, Kauffman CA, Miceli MH. Blastomycosis. Infect Dis Clin North Am. 2016;30:247-264. doi:10.1016/j.idc.2015.10.002
- Raggio B. Primary cutaneous histoplasmosis. Ear Nose Throat J. 2018;97:346-348.
- Bhambri S, Bhambri A, Del Rosso JQ. Atypical mycobacterial cutaneous infections. Dermatol Clin. 2009;27:63-73. doi:10.1016/j.det.2008.07.009
- Parekh V, Seykora JT. Cutaneous squamous cell carcinoma. Clin Lab Med. 2017;37:503-525. doi:10.1016/j.cll.2017.06.003
- Messersmith L, Krauland K. Pemphigus vegetans. StatPearls. StatPearls Publishing; 2022. Accessed June 21, 2022. https://www.ncbi.nlm.nih.gov/books/NBK545229/
The Diagnosis: Cutaneous Blastomycosis
A skin biopsy and fungal cultures confirmed the diagnosis of cutaneous blastomycosis. Grocott- Gomori methenamine-silver staining highlighted fungal organisms with refractile walls and broad-based budding consistent with cutaneous blastomycosis (Figure 1). The histopathologic specimen also demonstrated marked pseudoepitheliomatous hyperplasia (Figure 2A) with neutrophilic microabscesses (Figure 2B). Acid-fast bacillus and Fite staining were negative for bacterial organisms. A fungal culture was positive for Blastomyces dermatitidis. Urine and serum blastomycosis antigen were positive. Although Histoplasma serum antigen also was positive, this likely was from cross-reactivity. Chest radiography was negative for lung involvement, and the patient displayed no neurologic symptoms. He was started on oral itraconazole therapy for the treatment of cutaneous blastomycosis.
Blastomyces dermatitidis, the causative organism of blastomycosis, is endemic to the Ohio and Mississippi River valleys, Great Lakes region, and southeastern United States. It is a thermally dimorphic fungus found in soils that grows as a mold at 25 °C and yeast at 37 °C. Primary infection of the lungs—blastomycosis pneumonia—often is the only clinical manifestation1; however, subsequent hematogenous dissemination to extrapulmonary sites such as the skin, bones, and genitourinary system can occur. Cutaneous blastomycosis, the most common extrapulmonary manifestation, typically follows pulmonary infection. In rare cases, it can occur from direct inoculation.2,3 Skin lesions can occur anywhere but frequently are found on exposed surfaces of the head, neck, and extremities. Lesions classically present as verrucous crusting plaques with draining microabscesses. Violaceous nodules, ulcers, and pustules also may occur.1
Diagnosis involves obtaining a thorough history of possible environmental exposures such as the patient’s geographic area of residence, occupation, and outdoor activities involving soil or decaying wood. Because blastomycosis can remain latent, remote exposures are relevant. Definitive diagnosis of cutaneous blastomycosis involves skin biopsy of the lesion with fungal culture, but the yeast’s distinctive thick wall and broad-based budding seen with periodic acid–Schiff or Grocott-Gomori methenamine-silver staining provides a rapid presumptive diagnosis.3 Pseudoepitheliomatous hyperplasia and microabscesses also are characteristic features.2 Urine antigen testing for a component of the polysaccharide cell wall has a sensitivity of 93% but a lower specificity of 79% due to cross-reactivity with histoplasmosis.4 Treatment consists of itraconazole for mild to moderate blastomycosis or amphotericin B for those with severe disease or central nervous system involvement or those who are immunosuppressed.1
The differential diagnosis for our patient’s lesion included infectious vs neoplastic etiologies. Histoplasma capsulatum, the dimorphic fungus that causes histoplasmosis, also is endemic to the Ohio and Mississippi River valleys. It is found in soil and droppings of some bats and birds such as chickens and pigeons. Similar to blastomycosis, the primary infection site most commonly is the lungs. It subsequently may disseminate to the skin or less commonly via direct inoculation of injured skin. It can present as papules, plaques, ulcers, purpura, or abscesses. Unlike blastomycosis, tissue biopsy of a cutaneous lesion reveals granuloma formation and distinctive oval, narrow-based budding yeast.5 Atypical mycobacteria are another source of infection to consider. For example, cutaneous Mycobacterium kansasii may present as papules and pustules forming verrucous or granulomatous plaques and ulceration. Histopathologic findings distinguishing mycobacterial infection from blastomycosis include granulomas and acid-fast bacilli in histiocytes.6
Noninfectious etiologies in the differential may include cutaneous squamous cell carcinoma or pemphigus vegetans. Squamous cell carcinoma may present with a broad range of clinical features—papules, plaques, or nodules with smooth, scaly, verrucous, or ulcerative secondary features all are possible presentations.7 Fairskinned individuals, such as our patient, would be at a higher risk in sun-damaged skin. Histologically, cutaneous squamous cell carcinoma is defined as an invasion of the dermis by neoplastic squamous epithelial cells in the form of cords, sheets, individual cells, nodules, or cystic structures.7 Pemphigus vegetans is the rarest variant of a group of autoimmune vesiculobullous diseases known as pemphigus. It can be differentiated from the most common variant—pemphigus vulgaris—by the presence of vegetative plaques in intertriginous areas. However, these verrucous vegetations can be misleading and make clinical diagnosis difficult. Histopathologic findings of hyperkeratosis, pseudoepitheliomatous hyperplasia, papillomatosis, and acantholysis with a suprabasal cleft would confirm the diagnosis.8
In summary, cutaneous blastomycosis classically presents as verrucous crusting plaques, as seen in our patient. It is important to conduct a thorough history for environmental exposures, but definitive diagnosis of cutaneous blastomycosis involves skin biopsy with fungal culture. Treatment depends on the severity of disease and organ involvement. Itraconazole would be appropriate for mild to moderate blastomycosis.
The Diagnosis: Cutaneous Blastomycosis
A skin biopsy and fungal cultures confirmed the diagnosis of cutaneous blastomycosis. Grocott- Gomori methenamine-silver staining highlighted fungal organisms with refractile walls and broad-based budding consistent with cutaneous blastomycosis (Figure 1). The histopathologic specimen also demonstrated marked pseudoepitheliomatous hyperplasia (Figure 2A) with neutrophilic microabscesses (Figure 2B). Acid-fast bacillus and Fite staining were negative for bacterial organisms. A fungal culture was positive for Blastomyces dermatitidis. Urine and serum blastomycosis antigen were positive. Although Histoplasma serum antigen also was positive, this likely was from cross-reactivity. Chest radiography was negative for lung involvement, and the patient displayed no neurologic symptoms. He was started on oral itraconazole therapy for the treatment of cutaneous blastomycosis.
Blastomyces dermatitidis, the causative organism of blastomycosis, is endemic to the Ohio and Mississippi River valleys, Great Lakes region, and southeastern United States. It is a thermally dimorphic fungus found in soils that grows as a mold at 25 °C and yeast at 37 °C. Primary infection of the lungs—blastomycosis pneumonia—often is the only clinical manifestation1; however, subsequent hematogenous dissemination to extrapulmonary sites such as the skin, bones, and genitourinary system can occur. Cutaneous blastomycosis, the most common extrapulmonary manifestation, typically follows pulmonary infection. In rare cases, it can occur from direct inoculation.2,3 Skin lesions can occur anywhere but frequently are found on exposed surfaces of the head, neck, and extremities. Lesions classically present as verrucous crusting plaques with draining microabscesses. Violaceous nodules, ulcers, and pustules also may occur.1
Diagnosis involves obtaining a thorough history of possible environmental exposures such as the patient’s geographic area of residence, occupation, and outdoor activities involving soil or decaying wood. Because blastomycosis can remain latent, remote exposures are relevant. Definitive diagnosis of cutaneous blastomycosis involves skin biopsy of the lesion with fungal culture, but the yeast’s distinctive thick wall and broad-based budding seen with periodic acid–Schiff or Grocott-Gomori methenamine-silver staining provides a rapid presumptive diagnosis.3 Pseudoepitheliomatous hyperplasia and microabscesses also are characteristic features.2 Urine antigen testing for a component of the polysaccharide cell wall has a sensitivity of 93% but a lower specificity of 79% due to cross-reactivity with histoplasmosis.4 Treatment consists of itraconazole for mild to moderate blastomycosis or amphotericin B for those with severe disease or central nervous system involvement or those who are immunosuppressed.1
The differential diagnosis for our patient’s lesion included infectious vs neoplastic etiologies. Histoplasma capsulatum, the dimorphic fungus that causes histoplasmosis, also is endemic to the Ohio and Mississippi River valleys. It is found in soil and droppings of some bats and birds such as chickens and pigeons. Similar to blastomycosis, the primary infection site most commonly is the lungs. It subsequently may disseminate to the skin or less commonly via direct inoculation of injured skin. It can present as papules, plaques, ulcers, purpura, or abscesses. Unlike blastomycosis, tissue biopsy of a cutaneous lesion reveals granuloma formation and distinctive oval, narrow-based budding yeast.5 Atypical mycobacteria are another source of infection to consider. For example, cutaneous Mycobacterium kansasii may present as papules and pustules forming verrucous or granulomatous plaques and ulceration. Histopathologic findings distinguishing mycobacterial infection from blastomycosis include granulomas and acid-fast bacilli in histiocytes.6
Noninfectious etiologies in the differential may include cutaneous squamous cell carcinoma or pemphigus vegetans. Squamous cell carcinoma may present with a broad range of clinical features—papules, plaques, or nodules with smooth, scaly, verrucous, or ulcerative secondary features all are possible presentations.7 Fairskinned individuals, such as our patient, would be at a higher risk in sun-damaged skin. Histologically, cutaneous squamous cell carcinoma is defined as an invasion of the dermis by neoplastic squamous epithelial cells in the form of cords, sheets, individual cells, nodules, or cystic structures.7 Pemphigus vegetans is the rarest variant of a group of autoimmune vesiculobullous diseases known as pemphigus. It can be differentiated from the most common variant—pemphigus vulgaris—by the presence of vegetative plaques in intertriginous areas. However, these verrucous vegetations can be misleading and make clinical diagnosis difficult. Histopathologic findings of hyperkeratosis, pseudoepitheliomatous hyperplasia, papillomatosis, and acantholysis with a suprabasal cleft would confirm the diagnosis.8
In summary, cutaneous blastomycosis classically presents as verrucous crusting plaques, as seen in our patient. It is important to conduct a thorough history for environmental exposures, but definitive diagnosis of cutaneous blastomycosis involves skin biopsy with fungal culture. Treatment depends on the severity of disease and organ involvement. Itraconazole would be appropriate for mild to moderate blastomycosis.
- Miceli A, Krishnamurthy K. Blastomycosis. StatPearls. StatPearls Publishing; 2022. Accessed June 21, 2022. https://www.ncbi.nlm.nih.gov/books/NBK441987/
- Gray NA, Baddour LM. Cutaneous inoculation blastomycosis. Clin Infect Dis. 2002;34:E44-E49.
- Schwartz IS, Kauffman CA. Blastomycosis. Semin Respir Crit Care Med. 2020;41:31-41. doi:10.1055/s-0039-3400281
- Castillo CG, Kauffman CA, Miceli MH. Blastomycosis. Infect Dis Clin North Am. 2016;30:247-264. doi:10.1016/j.idc.2015.10.002
- Raggio B. Primary cutaneous histoplasmosis. Ear Nose Throat J. 2018;97:346-348.
- Bhambri S, Bhambri A, Del Rosso JQ. Atypical mycobacterial cutaneous infections. Dermatol Clin. 2009;27:63-73. doi:10.1016/j.det.2008.07.009
- Parekh V, Seykora JT. Cutaneous squamous cell carcinoma. Clin Lab Med. 2017;37:503-525. doi:10.1016/j.cll.2017.06.003
- Messersmith L, Krauland K. Pemphigus vegetans. StatPearls. StatPearls Publishing; 2022. Accessed June 21, 2022. https://www.ncbi.nlm.nih.gov/books/NBK545229/
- Miceli A, Krishnamurthy K. Blastomycosis. StatPearls. StatPearls Publishing; 2022. Accessed June 21, 2022. https://www.ncbi.nlm.nih.gov/books/NBK441987/
- Gray NA, Baddour LM. Cutaneous inoculation blastomycosis. Clin Infect Dis. 2002;34:E44-E49.
- Schwartz IS, Kauffman CA. Blastomycosis. Semin Respir Crit Care Med. 2020;41:31-41. doi:10.1055/s-0039-3400281
- Castillo CG, Kauffman CA, Miceli MH. Blastomycosis. Infect Dis Clin North Am. 2016;30:247-264. doi:10.1016/j.idc.2015.10.002
- Raggio B. Primary cutaneous histoplasmosis. Ear Nose Throat J. 2018;97:346-348.
- Bhambri S, Bhambri A, Del Rosso JQ. Atypical mycobacterial cutaneous infections. Dermatol Clin. 2009;27:63-73. doi:10.1016/j.det.2008.07.009
- Parekh V, Seykora JT. Cutaneous squamous cell carcinoma. Clin Lab Med. 2017;37:503-525. doi:10.1016/j.cll.2017.06.003
- Messersmith L, Krauland K. Pemphigus vegetans. StatPearls. StatPearls Publishing; 2022. Accessed June 21, 2022. https://www.ncbi.nlm.nih.gov/books/NBK545229/
A 39-year-old man from Ohio presented with a tender, 10×6-cm, fungated, eroded plaque on the right medial upper arm that developed over the last 4 years. He initially noticed a firm lump under the right arm 4 years prior that was diagnosed as possible cellulitis at an outside clinic and treated with trimethoprim-sulfamethoxazole. The lesion then began to erode and became a chronic nonhealing wound. Approximately 1 year prior to the current presentation, the patient recalled unloading a truckload of soil around the same time the wound began to enlarge in diameter and depth. He denied any prior or current respiratory or systemic symptoms including fevers, chills, or weight loss.


Erythematous Papules on the Ears
The Diagnosis: Borrelial Lymphocytoma (Lymphocytoma Cutis)
A punch biopsy revealed an atypical lobular lymphoid infiltrate within the dermis and subcutaneous tissue with a mixed composition of CD3+ T cells and CD20+ B cells (quiz image, bottom). Immunohistochemical studies revealed a normal CD4:CD8 ratio with preservation of CD5 and CD7. CD30 was largely negative. CD21 failed to detect follicular dendritic cell networks, and κ/λ light chain staining confirmed a preserved ratio of polytypic plasma cells. There was limited staining with B-cell lymphoma (Bcl-2 and Bcl-6). Polymerase chain reaction studies for both T- and B-cell receptors were negative (polyclonal).
Lyme disease is the most frequently reported vectorborne infectious disease in the United States, and borrelial lymphocytoma (BL) is a rare clinical sequela. Borrelial lymphocytoma is a variant of lymphocytoma cutis (also known as benign reactive lymphoid hyperplasia), which is an inflammatory lesion that can mimic malignant lymphoma clinically and histologically. Lymphocytoma cutis is considered the prototypical example of cutaneous B-cell pseudolymphoma.1 Due to suspicion for lymphocytoma cutis based on the histologic findings and characteristic location of the lesions in our patient, Lyme serologies were ordered and were positive for IgM antibodies against p23, p39, and p41 antigens in high titers. Our patient was treated with doxycycline 100 mg twice daily for 3 weeks with complete resolution of the lesions at 3-month follow-up.
Clinically, BL appears as erythematous papules, plaques, or nodules commonly on the lobules of the ears (quiz image, top). Most cases of lymphocytoma cutis are idiopathic but may be triggered by identifiable associated etiologies including Borrelia burgdorferi, Leishmania donovani, molluscum contagiosum, herpes zoster virus, vaccinations, tattoos, insect bites, and drugs. The main differential diagnosis of lymphocytoma cutis is cutaneous B-cell lymphoma. Pseudolymphoma of the skin can mimic nearly all immunohistochemical staining patterns of true B-cell lymphomas.2
Primary cutaneous follicle center lymphoma frequently occurs on the head and neck. This true lymphoma of the skin can demonstrate prominent follicle centers with centrocytes and fragmented germinal centers (Figure 1) or show a diffuse pattern.3 Most cases show conspicuous Bcl-6 staining, and IgH gene rearrangements can detect a clonal B-cell population in more than 50% of cases.4
Diffuse large B-cell lymphoma can occur as a primary cutaneous malignancy or as a manifestation of systemic disease.4 When arising in the skin, lesions tend to affect the extremities, and the disease is classified as diffuse large B-cell lymphoma, leg type. Histologically, sheets of large atypical lymphocytes with numerous mitoses are seen (Figure 2). These cells stain positively with Bcl-2 and frequently demonstrate Bcl-6 and MUM-1, none of which were seen in our case.4 Lymphomatoid papulosis (LyP) tends to present with relapsing erythematous papules. Patients occasionally develop LyP in association with mycosis fungoides or other lymphomas. Both LyP and primary cutaneous anaplastic large cell lymphoma demonstrate conspicuous CD30+ large cells that can be multinucleated or resemble the Reed-Sternberg cells seen in Hodgkin lymphoma (Figure 3).4 Arthropod bite reactions are common but may be confused with lymphomas and pseudolymphomas. The perivascular lymphocytic infiltrate seen in arthropod bite reactions may be dense and usually is associated with numerous eosinophils (Figure 4). Occasional plasma cells also can be seen, and if the infiltrate closely adheres to vascular structures, a diagnosis of erythema chronicum migrans also can be considered. Patients with chronic lymphocytic leukemia/lymphoma may demonstrate exaggerated or persistent arthropod bite reactions, and atypical lymphocytes can be detected admixed with the otherwise reactive infiltrate.4
Borrelia burgdorferi is primarily endemic to North America and Europe. It is a spirochete bacterium spread by the Ixodes tick that was first recognized as the etiologic agent in 1975 in Old Lyme, Connecticut, where it received its name.5 Most reported cases of Lyme disease occur in the northeastern United States, which correlates with this case given our patient’s place of residence.6 Borrelial lymphocytoma cutis occurs in areas endemic for the Ixodes tick in Europe and North America.7 When describing the genotyping of Borrelia seen in BL, the strain B burgdorferi previously was grouped with Borrelia afzelii and Borrelia garinii.2 In the contemporary literature, however, B burgdorferi is referred to as sensu stricto when specifically talking about the strain B burgdorferi, and the term sensu lato is used when referencing the combination of strains (B burgdorferi, B afzelii, B garinii).
A 2016 study by Maraspin et al8 comprising 144 patients diagnosed with BL showed that the lesions mainly were located on the breast (106 patients [73.6%]) and the earlobe (27 patients [18.8%]), with the remaining cases occurring elsewhere on the body (11 patients [7.6%]). The Borrelia strains isolated from the BL lesions included B afzelii, Borrelia bissettii, and B garinii, with B afzelii being the most commonly identified (84.6% [11/13]).8
Borrelial lymphocytoma usually is categorized as a form of early disseminated Lyme disease and is treated as such. The treatment of choice for early disseminated Lyme disease is doxycycline 100 mg twice daily for 14 to 21 days. Ceftriaxone and azithromycin are reasonable treatment options for patients who have tetracycline allergies or who are pregnant.9
In conclusion, the presentation of red papules or nodules on the ears should prompt clinical suspicion of Lyme disease, particularly in endemic areas. Differentiating pseudolymphomas from true lymphomas and other reactive conditions can be challenging.
- Mitteldorf C, Kempf W. Cutaneous pseudolymphoma. Surg Pathol Clin. 2017;10:455-476. doi:10.1016/j.path.2017.01.002
- Colli C, Leinweber B, Müllegger R, et al. Borrelia burgdorferiassociated lymphocytoma cutis: clinicopathologic, immunophenotypic, and molecular study of 106 cases. J Cutan Pathol. 2004;31:232-240. doi:10.1111/j.0303-6987.2003.00167.x
- Wehbe AM, Neppalli V, Syrbu S, et al. Diffuse follicle centre lymphoma presents with high frequency of extranodal disease. J Clin Oncol. 2008;26(15 suppl):19511. doi:10.1200/jco.2008.26.15_suppl.19511
- Patterson JW, Hosler GA. Cutaneous infiltrates—lymphomatous and leukemic. In: Patterson JW, ed. Weedon’s Skin Pathology. 4th ed. Elsevier; 2016:1171-1217.
- Cardenas-de la Garza JA, De la Cruz-Valadez E, Ocampo -Candiani J, et al. Clinical spectrum of Lyme disease. Eur J Clin Microbiol Infect Dis. 2019;38:201-208. doi:10.1007/s10096-018-3417-1
- Shapiro ED, Gerber MA. Lyme disease. Clin Infect Dis. 2000;31:533-542. doi:10.1086/313982
- Kandhari R, Kandhari S, Jain S. Borrelial lymphocytoma cutis: a diagnostic dilemma. Indian J Dermatol. 2014;59:595-597. doi:10.4103/0019-5154.143530
- Maraspin V, Nahtigal Klevišar M, Ružic´-Sabljic´ E, et al. Borrelial lymphocytoma in adult patients. Clin Infect Dis. 2016;63:914-921. doi:10.1093/cid/ciw417
- Wormser GP, Dattwyler RJ, Shapiro ED, et al. The clinical assessment, treatment, and prevention of Lyme disease, human granulocytic anaplasmosis, and babesiosis: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis. 2006; 43:1089-1134. doi:10.1086/508667
The Diagnosis: Borrelial Lymphocytoma (Lymphocytoma Cutis)
A punch biopsy revealed an atypical lobular lymphoid infiltrate within the dermis and subcutaneous tissue with a mixed composition of CD3+ T cells and CD20+ B cells (quiz image, bottom). Immunohistochemical studies revealed a normal CD4:CD8 ratio with preservation of CD5 and CD7. CD30 was largely negative. CD21 failed to detect follicular dendritic cell networks, and κ/λ light chain staining confirmed a preserved ratio of polytypic plasma cells. There was limited staining with B-cell lymphoma (Bcl-2 and Bcl-6). Polymerase chain reaction studies for both T- and B-cell receptors were negative (polyclonal).
Lyme disease is the most frequently reported vectorborne infectious disease in the United States, and borrelial lymphocytoma (BL) is a rare clinical sequela. Borrelial lymphocytoma is a variant of lymphocytoma cutis (also known as benign reactive lymphoid hyperplasia), which is an inflammatory lesion that can mimic malignant lymphoma clinically and histologically. Lymphocytoma cutis is considered the prototypical example of cutaneous B-cell pseudolymphoma.1 Due to suspicion for lymphocytoma cutis based on the histologic findings and characteristic location of the lesions in our patient, Lyme serologies were ordered and were positive for IgM antibodies against p23, p39, and p41 antigens in high titers. Our patient was treated with doxycycline 100 mg twice daily for 3 weeks with complete resolution of the lesions at 3-month follow-up.
Clinically, BL appears as erythematous papules, plaques, or nodules commonly on the lobules of the ears (quiz image, top). Most cases of lymphocytoma cutis are idiopathic but may be triggered by identifiable associated etiologies including Borrelia burgdorferi, Leishmania donovani, molluscum contagiosum, herpes zoster virus, vaccinations, tattoos, insect bites, and drugs. The main differential diagnosis of lymphocytoma cutis is cutaneous B-cell lymphoma. Pseudolymphoma of the skin can mimic nearly all immunohistochemical staining patterns of true B-cell lymphomas.2
Primary cutaneous follicle center lymphoma frequently occurs on the head and neck. This true lymphoma of the skin can demonstrate prominent follicle centers with centrocytes and fragmented germinal centers (Figure 1) or show a diffuse pattern.3 Most cases show conspicuous Bcl-6 staining, and IgH gene rearrangements can detect a clonal B-cell population in more than 50% of cases.4
Diffuse large B-cell lymphoma can occur as a primary cutaneous malignancy or as a manifestation of systemic disease.4 When arising in the skin, lesions tend to affect the extremities, and the disease is classified as diffuse large B-cell lymphoma, leg type. Histologically, sheets of large atypical lymphocytes with numerous mitoses are seen (Figure 2). These cells stain positively with Bcl-2 and frequently demonstrate Bcl-6 and MUM-1, none of which were seen in our case.4 Lymphomatoid papulosis (LyP) tends to present with relapsing erythematous papules. Patients occasionally develop LyP in association with mycosis fungoides or other lymphomas. Both LyP and primary cutaneous anaplastic large cell lymphoma demonstrate conspicuous CD30+ large cells that can be multinucleated or resemble the Reed-Sternberg cells seen in Hodgkin lymphoma (Figure 3).4 Arthropod bite reactions are common but may be confused with lymphomas and pseudolymphomas. The perivascular lymphocytic infiltrate seen in arthropod bite reactions may be dense and usually is associated with numerous eosinophils (Figure 4). Occasional plasma cells also can be seen, and if the infiltrate closely adheres to vascular structures, a diagnosis of erythema chronicum migrans also can be considered. Patients with chronic lymphocytic leukemia/lymphoma may demonstrate exaggerated or persistent arthropod bite reactions, and atypical lymphocytes can be detected admixed with the otherwise reactive infiltrate.4
Borrelia burgdorferi is primarily endemic to North America and Europe. It is a spirochete bacterium spread by the Ixodes tick that was first recognized as the etiologic agent in 1975 in Old Lyme, Connecticut, where it received its name.5 Most reported cases of Lyme disease occur in the northeastern United States, which correlates with this case given our patient’s place of residence.6 Borrelial lymphocytoma cutis occurs in areas endemic for the Ixodes tick in Europe and North America.7 When describing the genotyping of Borrelia seen in BL, the strain B burgdorferi previously was grouped with Borrelia afzelii and Borrelia garinii.2 In the contemporary literature, however, B burgdorferi is referred to as sensu stricto when specifically talking about the strain B burgdorferi, and the term sensu lato is used when referencing the combination of strains (B burgdorferi, B afzelii, B garinii).
A 2016 study by Maraspin et al8 comprising 144 patients diagnosed with BL showed that the lesions mainly were located on the breast (106 patients [73.6%]) and the earlobe (27 patients [18.8%]), with the remaining cases occurring elsewhere on the body (11 patients [7.6%]). The Borrelia strains isolated from the BL lesions included B afzelii, Borrelia bissettii, and B garinii, with B afzelii being the most commonly identified (84.6% [11/13]).8
Borrelial lymphocytoma usually is categorized as a form of early disseminated Lyme disease and is treated as such. The treatment of choice for early disseminated Lyme disease is doxycycline 100 mg twice daily for 14 to 21 days. Ceftriaxone and azithromycin are reasonable treatment options for patients who have tetracycline allergies or who are pregnant.9
In conclusion, the presentation of red papules or nodules on the ears should prompt clinical suspicion of Lyme disease, particularly in endemic areas. Differentiating pseudolymphomas from true lymphomas and other reactive conditions can be challenging.
The Diagnosis: Borrelial Lymphocytoma (Lymphocytoma Cutis)
A punch biopsy revealed an atypical lobular lymphoid infiltrate within the dermis and subcutaneous tissue with a mixed composition of CD3+ T cells and CD20+ B cells (quiz image, bottom). Immunohistochemical studies revealed a normal CD4:CD8 ratio with preservation of CD5 and CD7. CD30 was largely negative. CD21 failed to detect follicular dendritic cell networks, and κ/λ light chain staining confirmed a preserved ratio of polytypic plasma cells. There was limited staining with B-cell lymphoma (Bcl-2 and Bcl-6). Polymerase chain reaction studies for both T- and B-cell receptors were negative (polyclonal).
Lyme disease is the most frequently reported vectorborne infectious disease in the United States, and borrelial lymphocytoma (BL) is a rare clinical sequela. Borrelial lymphocytoma is a variant of lymphocytoma cutis (also known as benign reactive lymphoid hyperplasia), which is an inflammatory lesion that can mimic malignant lymphoma clinically and histologically. Lymphocytoma cutis is considered the prototypical example of cutaneous B-cell pseudolymphoma.1 Due to suspicion for lymphocytoma cutis based on the histologic findings and characteristic location of the lesions in our patient, Lyme serologies were ordered and were positive for IgM antibodies against p23, p39, and p41 antigens in high titers. Our patient was treated with doxycycline 100 mg twice daily for 3 weeks with complete resolution of the lesions at 3-month follow-up.
Clinically, BL appears as erythematous papules, plaques, or nodules commonly on the lobules of the ears (quiz image, top). Most cases of lymphocytoma cutis are idiopathic but may be triggered by identifiable associated etiologies including Borrelia burgdorferi, Leishmania donovani, molluscum contagiosum, herpes zoster virus, vaccinations, tattoos, insect bites, and drugs. The main differential diagnosis of lymphocytoma cutis is cutaneous B-cell lymphoma. Pseudolymphoma of the skin can mimic nearly all immunohistochemical staining patterns of true B-cell lymphomas.2
Primary cutaneous follicle center lymphoma frequently occurs on the head and neck. This true lymphoma of the skin can demonstrate prominent follicle centers with centrocytes and fragmented germinal centers (Figure 1) or show a diffuse pattern.3 Most cases show conspicuous Bcl-6 staining, and IgH gene rearrangements can detect a clonal B-cell population in more than 50% of cases.4
Diffuse large B-cell lymphoma can occur as a primary cutaneous malignancy or as a manifestation of systemic disease.4 When arising in the skin, lesions tend to affect the extremities, and the disease is classified as diffuse large B-cell lymphoma, leg type. Histologically, sheets of large atypical lymphocytes with numerous mitoses are seen (Figure 2). These cells stain positively with Bcl-2 and frequently demonstrate Bcl-6 and MUM-1, none of which were seen in our case.4 Lymphomatoid papulosis (LyP) tends to present with relapsing erythematous papules. Patients occasionally develop LyP in association with mycosis fungoides or other lymphomas. Both LyP and primary cutaneous anaplastic large cell lymphoma demonstrate conspicuous CD30+ large cells that can be multinucleated or resemble the Reed-Sternberg cells seen in Hodgkin lymphoma (Figure 3).4 Arthropod bite reactions are common but may be confused with lymphomas and pseudolymphomas. The perivascular lymphocytic infiltrate seen in arthropod bite reactions may be dense and usually is associated with numerous eosinophils (Figure 4). Occasional plasma cells also can be seen, and if the infiltrate closely adheres to vascular structures, a diagnosis of erythema chronicum migrans also can be considered. Patients with chronic lymphocytic leukemia/lymphoma may demonstrate exaggerated or persistent arthropod bite reactions, and atypical lymphocytes can be detected admixed with the otherwise reactive infiltrate.4
Borrelia burgdorferi is primarily endemic to North America and Europe. It is a spirochete bacterium spread by the Ixodes tick that was first recognized as the etiologic agent in 1975 in Old Lyme, Connecticut, where it received its name.5 Most reported cases of Lyme disease occur in the northeastern United States, which correlates with this case given our patient’s place of residence.6 Borrelial lymphocytoma cutis occurs in areas endemic for the Ixodes tick in Europe and North America.7 When describing the genotyping of Borrelia seen in BL, the strain B burgdorferi previously was grouped with Borrelia afzelii and Borrelia garinii.2 In the contemporary literature, however, B burgdorferi is referred to as sensu stricto when specifically talking about the strain B burgdorferi, and the term sensu lato is used when referencing the combination of strains (B burgdorferi, B afzelii, B garinii).
A 2016 study by Maraspin et al8 comprising 144 patients diagnosed with BL showed that the lesions mainly were located on the breast (106 patients [73.6%]) and the earlobe (27 patients [18.8%]), with the remaining cases occurring elsewhere on the body (11 patients [7.6%]). The Borrelia strains isolated from the BL lesions included B afzelii, Borrelia bissettii, and B garinii, with B afzelii being the most commonly identified (84.6% [11/13]).8
Borrelial lymphocytoma usually is categorized as a form of early disseminated Lyme disease and is treated as such. The treatment of choice for early disseminated Lyme disease is doxycycline 100 mg twice daily for 14 to 21 days. Ceftriaxone and azithromycin are reasonable treatment options for patients who have tetracycline allergies or who are pregnant.9
In conclusion, the presentation of red papules or nodules on the ears should prompt clinical suspicion of Lyme disease, particularly in endemic areas. Differentiating pseudolymphomas from true lymphomas and other reactive conditions can be challenging.
- Mitteldorf C, Kempf W. Cutaneous pseudolymphoma. Surg Pathol Clin. 2017;10:455-476. doi:10.1016/j.path.2017.01.002
- Colli C, Leinweber B, Müllegger R, et al. Borrelia burgdorferiassociated lymphocytoma cutis: clinicopathologic, immunophenotypic, and molecular study of 106 cases. J Cutan Pathol. 2004;31:232-240. doi:10.1111/j.0303-6987.2003.00167.x
- Wehbe AM, Neppalli V, Syrbu S, et al. Diffuse follicle centre lymphoma presents with high frequency of extranodal disease. J Clin Oncol. 2008;26(15 suppl):19511. doi:10.1200/jco.2008.26.15_suppl.19511
- Patterson JW, Hosler GA. Cutaneous infiltrates—lymphomatous and leukemic. In: Patterson JW, ed. Weedon’s Skin Pathology. 4th ed. Elsevier; 2016:1171-1217.
- Cardenas-de la Garza JA, De la Cruz-Valadez E, Ocampo -Candiani J, et al. Clinical spectrum of Lyme disease. Eur J Clin Microbiol Infect Dis. 2019;38:201-208. doi:10.1007/s10096-018-3417-1
- Shapiro ED, Gerber MA. Lyme disease. Clin Infect Dis. 2000;31:533-542. doi:10.1086/313982
- Kandhari R, Kandhari S, Jain S. Borrelial lymphocytoma cutis: a diagnostic dilemma. Indian J Dermatol. 2014;59:595-597. doi:10.4103/0019-5154.143530
- Maraspin V, Nahtigal Klevišar M, Ružic´-Sabljic´ E, et al. Borrelial lymphocytoma in adult patients. Clin Infect Dis. 2016;63:914-921. doi:10.1093/cid/ciw417
- Wormser GP, Dattwyler RJ, Shapiro ED, et al. The clinical assessment, treatment, and prevention of Lyme disease, human granulocytic anaplasmosis, and babesiosis: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis. 2006; 43:1089-1134. doi:10.1086/508667
- Mitteldorf C, Kempf W. Cutaneous pseudolymphoma. Surg Pathol Clin. 2017;10:455-476. doi:10.1016/j.path.2017.01.002
- Colli C, Leinweber B, Müllegger R, et al. Borrelia burgdorferiassociated lymphocytoma cutis: clinicopathologic, immunophenotypic, and molecular study of 106 cases. J Cutan Pathol. 2004;31:232-240. doi:10.1111/j.0303-6987.2003.00167.x
- Wehbe AM, Neppalli V, Syrbu S, et al. Diffuse follicle centre lymphoma presents with high frequency of extranodal disease. J Clin Oncol. 2008;26(15 suppl):19511. doi:10.1200/jco.2008.26.15_suppl.19511
- Patterson JW, Hosler GA. Cutaneous infiltrates—lymphomatous and leukemic. In: Patterson JW, ed. Weedon’s Skin Pathology. 4th ed. Elsevier; 2016:1171-1217.
- Cardenas-de la Garza JA, De la Cruz-Valadez E, Ocampo -Candiani J, et al. Clinical spectrum of Lyme disease. Eur J Clin Microbiol Infect Dis. 2019;38:201-208. doi:10.1007/s10096-018-3417-1
- Shapiro ED, Gerber MA. Lyme disease. Clin Infect Dis. 2000;31:533-542. doi:10.1086/313982
- Kandhari R, Kandhari S, Jain S. Borrelial lymphocytoma cutis: a diagnostic dilemma. Indian J Dermatol. 2014;59:595-597. doi:10.4103/0019-5154.143530
- Maraspin V, Nahtigal Klevišar M, Ružic´-Sabljic´ E, et al. Borrelial lymphocytoma in adult patients. Clin Infect Dis. 2016;63:914-921. doi:10.1093/cid/ciw417
- Wormser GP, Dattwyler RJ, Shapiro ED, et al. The clinical assessment, treatment, and prevention of Lyme disease, human granulocytic anaplasmosis, and babesiosis: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis. 2006; 43:1089-1134. doi:10.1086/508667
A 53-year-old man with a history of atopic dermatitis presented with pain and redness of the lobules of both ears of 9 months’ duration. He had no known allergies and took no medications. He lived in suburban Virginia and had not recently traveled outside of the region. Physical examination revealed tender erythematous and edematous nodules on the lobules of both ears (top). There was no evidence of arthritis or neurologic deficits. A punch biopsy was performed (bottom).

.")
.")
Acute Generalized Exanthematous Pustulosis Induced by the Second-Generation Antipsychotic Cariprazine
To the Editor:
A 57-year-old woman presented to an outpatient clinic with severe pruritus and burning of the skin as well as subjective fevers and chills. She had been discharged from a psychiatric hospital for attempted suicide 1 day prior. There were no recent changes in the medication regimen, which consisted of linaclotide, fluoxetine, lorazepam, and gabapentin. While admitted, the patient was started on the atypical antipsychotic cariprazine. Within 24 hours of the first dose, she developed severe facial erythema that progressed to diffuse erythema over more than 60% of the body surface area. The attending psychiatrist promptly discontinued cariprazine. During the next 24 hours, there were no reports of fever, leukocytosis, or signs of systemic organ involvement. Given the patient’s mental and medical stability, she was discharged with instructions to follow up with the outpatient dermatology clinic.
At the current presentation, physical examination revealed innumerable 1- to 4-mm pustules coalescing to lakes of pus on an erythematous base over more than 60% of the body surface area (Figure 1). The mucous membranes were clear of lesions, the Nikolsky sign was negative, and the patient’s temperature was 99.6 °F in the office. Complete blood cell count and complete metabolic panel results were within reference range.

A 4-mm abdominal punch biopsy showed subcorneal neutrophilic pustules, papillary dermal edema, and superficial dermal lymphohistiocytic inflammation with numerous neutrophils, eosinophils, and extravasated red blood cells, consistent with acute generalized exanthematous pustulosis (AGEP)(Figure 2). The patient was started on wet wraps with triamcinolone cream 0.1%.

Two days later, physical examination revealed the erythema noted on initial examination had notably decreased, and the patient no longer reported burning or pruritus. One week after initial presentation to the clinic, the patient’s rash had resolved, and only a few small areas of desquamation remained.
Acute generalized exanthematous pustulosis is a severe cutaneous adverse reaction characterized by the development of numerous nonfollicular sterile pustules on an edematous and erythematous base. In almost 90% of reported cases, the cause is related to use of antibiotics, antifungals, antimalarials, or diltiazem (a calcium channel blocker). This rare cutaneous reaction occurs in 1 to 5 patients per million per year1; it carries a 1% to 2% mortality rate with proper supportive treatment.
The clinical symptoms of AGEP typically present 24 to 48 hours after drug initiation with the rapid development of dozens to thousands of 1- to 4-mm pustules, typically localized to the flexor surfaces and face. In the setting of AGEP, acute onset of fever and leukocytosis typically occur at the time of the cutaneous eruption. These features were absent in this patient. The eruption usually starts on the face and then migrates to the trunk and extremities, sparing the palms and soles. Systemic involvement most commonly presents as hepatic, renal, or pulmonary insufficiency, which has been seen in 20% of cases.2
The immunologic response associated with the reaction has been studied in vitro. Drug-specific CD8 T cells use perforin/granzyme B and Fas ligand mechanisms to induce apoptosis of the keratinocytes within the epidermis, leading to vesicle formation.3 During the very first stages of formation, vesicles mainly comprise CD8 T cells and keratinocytes. These cells then begin producing CXC-18, a potent neutrophil chemokine, leading to extensive chemotaxis of neutrophils into vesicles, which then rapidly transform to pustules.3 This rapid transformation leads to the lakes of pustules, a description often associated with AGEP.
Treatment of AGEP is mainly supportive and consists of discontinuing use of the causative agent. Topical corticosteroids can be used during the pustular phase for symptom management. There is no evidence that systemic steroids reduce the duration of the disease.2 Other supportive measures such as application of wet wraps can be used to provide comfort.
Cutaneous adverse drug reactions commonly are associated with psychiatric pharmacotherapy, but first-and second-generation antipsychotics rarely are associated with these types of reactions. In this patient, the causative agent of the AGEP was cariprazine, an atypical antipsychotic that had no reported association with AGEP or cutaneous adverse drug reactions prior to this presentation.
- Fernando SL. Acute generalised exanthematous pustulosis. Australas J Dermatol. 2012;53:87-92.
- Feldmeyer L, Heidemeyer K, Yawalkar N. Acute generalized exanthematous pustulosis: pathogenesis, genetic background, clinical variants and therapy. Int J Mol Sci. 2016;17:1214.
- Szatkowski J, Schwartz RA. Acute generalized exanthematous pustulosis (AGEP): a review and update. J Am Acad Dermatol. 2015;73:843-848.
To the Editor:
A 57-year-old woman presented to an outpatient clinic with severe pruritus and burning of the skin as well as subjective fevers and chills. She had been discharged from a psychiatric hospital for attempted suicide 1 day prior. There were no recent changes in the medication regimen, which consisted of linaclotide, fluoxetine, lorazepam, and gabapentin. While admitted, the patient was started on the atypical antipsychotic cariprazine. Within 24 hours of the first dose, she developed severe facial erythema that progressed to diffuse erythema over more than 60% of the body surface area. The attending psychiatrist promptly discontinued cariprazine. During the next 24 hours, there were no reports of fever, leukocytosis, or signs of systemic organ involvement. Given the patient’s mental and medical stability, she was discharged with instructions to follow up with the outpatient dermatology clinic.
At the current presentation, physical examination revealed innumerable 1- to 4-mm pustules coalescing to lakes of pus on an erythematous base over more than 60% of the body surface area (Figure 1). The mucous membranes were clear of lesions, the Nikolsky sign was negative, and the patient’s temperature was 99.6 °F in the office. Complete blood cell count and complete metabolic panel results were within reference range.
A 4-mm abdominal punch biopsy showed subcorneal neutrophilic pustules, papillary dermal edema, and superficial dermal lymphohistiocytic inflammation with numerous neutrophils, eosinophils, and extravasated red blood cells, consistent with acute generalized exanthematous pustulosis (AGEP)(Figure 2). The patient was started on wet wraps with triamcinolone cream 0.1%.
Two days later, physical examination revealed the erythema noted on initial examination had notably decreased, and the patient no longer reported burning or pruritus. One week after initial presentation to the clinic, the patient’s rash had resolved, and only a few small areas of desquamation remained.
Acute generalized exanthematous pustulosis is a severe cutaneous adverse reaction characterized by the development of numerous nonfollicular sterile pustules on an edematous and erythematous base. In almost 90% of reported cases, the cause is related to use of antibiotics, antifungals, antimalarials, or diltiazem (a calcium channel blocker). This rare cutaneous reaction occurs in 1 to 5 patients per million per year1; it carries a 1% to 2% mortality rate with proper supportive treatment.
The clinical symptoms of AGEP typically present 24 to 48 hours after drug initiation with the rapid development of dozens to thousands of 1- to 4-mm pustules, typically localized to the flexor surfaces and face. In the setting of AGEP, acute onset of fever and leukocytosis typically occur at the time of the cutaneous eruption. These features were absent in this patient. The eruption usually starts on the face and then migrates to the trunk and extremities, sparing the palms and soles. Systemic involvement most commonly presents as hepatic, renal, or pulmonary insufficiency, which has been seen in 20% of cases.2
The immunologic response associated with the reaction has been studied in vitro. Drug-specific CD8 T cells use perforin/granzyme B and Fas ligand mechanisms to induce apoptosis of the keratinocytes within the epidermis, leading to vesicle formation.3 During the very first stages of formation, vesicles mainly comprise CD8 T cells and keratinocytes. These cells then begin producing CXC-18, a potent neutrophil chemokine, leading to extensive chemotaxis of neutrophils into vesicles, which then rapidly transform to pustules.3 This rapid transformation leads to the lakes of pustules, a description often associated with AGEP.
Treatment of AGEP is mainly supportive and consists of discontinuing use of the causative agent. Topical corticosteroids can be used during the pustular phase for symptom management. There is no evidence that systemic steroids reduce the duration of the disease.2 Other supportive measures such as application of wet wraps can be used to provide comfort.
Cutaneous adverse drug reactions commonly are associated with psychiatric pharmacotherapy, but first-and second-generation antipsychotics rarely are associated with these types of reactions. In this patient, the causative agent of the AGEP was cariprazine, an atypical antipsychotic that had no reported association with AGEP or cutaneous adverse drug reactions prior to this presentation.
To the Editor:
A 57-year-old woman presented to an outpatient clinic with severe pruritus and burning of the skin as well as subjective fevers and chills. She had been discharged from a psychiatric hospital for attempted suicide 1 day prior. There were no recent changes in the medication regimen, which consisted of linaclotide, fluoxetine, lorazepam, and gabapentin. While admitted, the patient was started on the atypical antipsychotic cariprazine. Within 24 hours of the first dose, she developed severe facial erythema that progressed to diffuse erythema over more than 60% of the body surface area. The attending psychiatrist promptly discontinued cariprazine. During the next 24 hours, there were no reports of fever, leukocytosis, or signs of systemic organ involvement. Given the patient’s mental and medical stability, she was discharged with instructions to follow up with the outpatient dermatology clinic.
At the current presentation, physical examination revealed innumerable 1- to 4-mm pustules coalescing to lakes of pus on an erythematous base over more than 60% of the body surface area (Figure 1). The mucous membranes were clear of lesions, the Nikolsky sign was negative, and the patient’s temperature was 99.6 °F in the office. Complete blood cell count and complete metabolic panel results were within reference range.
A 4-mm abdominal punch biopsy showed subcorneal neutrophilic pustules, papillary dermal edema, and superficial dermal lymphohistiocytic inflammation with numerous neutrophils, eosinophils, and extravasated red blood cells, consistent with acute generalized exanthematous pustulosis (AGEP)(Figure 2). The patient was started on wet wraps with triamcinolone cream 0.1%.
Two days later, physical examination revealed the erythema noted on initial examination had notably decreased, and the patient no longer reported burning or pruritus. One week after initial presentation to the clinic, the patient’s rash had resolved, and only a few small areas of desquamation remained.
Acute generalized exanthematous pustulosis is a severe cutaneous adverse reaction characterized by the development of numerous nonfollicular sterile pustules on an edematous and erythematous base. In almost 90% of reported cases, the cause is related to use of antibiotics, antifungals, antimalarials, or diltiazem (a calcium channel blocker). This rare cutaneous reaction occurs in 1 to 5 patients per million per year1; it carries a 1% to 2% mortality rate with proper supportive treatment.
The clinical symptoms of AGEP typically present 24 to 48 hours after drug initiation with the rapid development of dozens to thousands of 1- to 4-mm pustules, typically localized to the flexor surfaces and face. In the setting of AGEP, acute onset of fever and leukocytosis typically occur at the time of the cutaneous eruption. These features were absent in this patient. The eruption usually starts on the face and then migrates to the trunk and extremities, sparing the palms and soles. Systemic involvement most commonly presents as hepatic, renal, or pulmonary insufficiency, which has been seen in 20% of cases.2
The immunologic response associated with the reaction has been studied in vitro. Drug-specific CD8 T cells use perforin/granzyme B and Fas ligand mechanisms to induce apoptosis of the keratinocytes within the epidermis, leading to vesicle formation.3 During the very first stages of formation, vesicles mainly comprise CD8 T cells and keratinocytes. These cells then begin producing CXC-18, a potent neutrophil chemokine, leading to extensive chemotaxis of neutrophils into vesicles, which then rapidly transform to pustules.3 This rapid transformation leads to the lakes of pustules, a description often associated with AGEP.
Treatment of AGEP is mainly supportive and consists of discontinuing use of the causative agent. Topical corticosteroids can be used during the pustular phase for symptom management. There is no evidence that systemic steroids reduce the duration of the disease.2 Other supportive measures such as application of wet wraps can be used to provide comfort.
Cutaneous adverse drug reactions commonly are associated with psychiatric pharmacotherapy, but first-and second-generation antipsychotics rarely are associated with these types of reactions. In this patient, the causative agent of the AGEP was cariprazine, an atypical antipsychotic that had no reported association with AGEP or cutaneous adverse drug reactions prior to this presentation.
- Fernando SL. Acute generalised exanthematous pustulosis. Australas J Dermatol. 2012;53:87-92.
- Feldmeyer L, Heidemeyer K, Yawalkar N. Acute generalized exanthematous pustulosis: pathogenesis, genetic background, clinical variants and therapy. Int J Mol Sci. 2016;17:1214.
- Szatkowski J, Schwartz RA. Acute generalized exanthematous pustulosis (AGEP): a review and update. J Am Acad Dermatol. 2015;73:843-848.
- Fernando SL. Acute generalised exanthematous pustulosis. Australas J Dermatol. 2012;53:87-92.
- Feldmeyer L, Heidemeyer K, Yawalkar N. Acute generalized exanthematous pustulosis: pathogenesis, genetic background, clinical variants and therapy. Int J Mol Sci. 2016;17:1214.
- Szatkowski J, Schwartz RA. Acute generalized exanthematous pustulosis (AGEP): a review and update. J Am Acad Dermatol. 2015;73:843-848.
Practice Points
- The second-generation antipsychotic cariprazine has been shown to be a potential causative agent in acute generalized exanthematous pustulosis (AGEP).
- Treatment of AGEP is mainly supportive and consists of discontinuation of the causative agent as well as symptom control using cold compresses and topical corticosteroids.
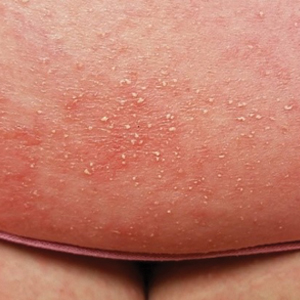
Itchy Vesicular Rash
The Diagnosis: Tinea Corporis Bullosa
At the time of presentation, a potassium hydroxide (KOH) preparation, fungal culture, and punch biopsy of the right ventral wrist was performed. The KOH preparation was positive for fungal hyphae characteristic of dermatophyte infections. Histologically, the biopsy showed intraepidermal and subepidermal blisters with neutrophil- and lymphocyte-rich contents (Figure 1). Fungal hyphae and spores were present within the stratum corneum and superficial epidermis (Figure 2), and fungal cultures grew Microsporum canis. The extent of the rash (upper and lower extremities, chest, and back), positive fungal culture, and KOH preparation all supported the diagnosis of tinea corporis bullosa, which was confirmed with biopsy. Oral prednisone use was discouraged and triamcinolone ointment was discontinued given that inappropriate treatment with steroids in the setting of fungal infection suppresses an inflammatory response and alters clinical appearance, obviating the persistent underlying infection.

Tinea corporis bullosa is a rare superficial dermatophyte fungal infection that often is acquired by close personto- person contact or contact with domestic animals. The infection begins as a circular pruritic plaque, generally with raised borders, which may be erythematous or hyperpigmented. By definition, tinea corporis occurs in sites other than the face, feet, hands, or groin area. Bullae formation is thought to be secondary to a delayed hypersensitivity reaction provoked by the presence of a dermatophyte antigen.1

Linear IgA bullous dermatosis is an immunemediated disease characterized by IgA deposition at the dermoepidermal junction. Linear IgA bullous dermatosis classically presents as widespread tense vesicles in an arciform or annular pattern. Mucosal involvement is common and typically presents with erosions, ulcerations, and scarring.2 Given the absence of mucosal involvement in our patient and a positive KOH preparation, linear IgA bullous dermatosis was an unlikely diagnosis.
Benign inoculation lymphoreticulosis, more commonly known as cat scratch disease (CSD), is a Bartonella henselae infection that results from a cat scratch or bite. Cat scratch disease can present as localized cutaneous and nodal involvement (lymphadenopathy) near the site of inoculation, or it may present as disseminated disease. Cutaneous lesions generally progress through vesicular, erythematous, and papular phases. Regional lymphadenopathy proximal to the inoculation site is the hallmark of CSD.3 Given the absence of lymphadenopathy in our patient as well as the sporadic distribution of lesions, CSD was an unlikely diagnosis.
Dermatitis herpetiformis (DH) is an autoimmune disorder with cutaneous manifestations of gluten sensitivity. Dermatitis herpetiformis presents as extremely pruritic papules and vesicles arranged in groups on areas such as the elbows, dorsal aspects of the forearms, knees, scalp, back, and buttocks. Most patients with DH have celiac disease or small bowel disease related to gluten sensitivity.4 Given our patient’s acute presentation in adulthood and lack of gluten sensitivity, DH was an unlikely diagnosis.
Bullous fixed drug reaction is a cutaneous eruption that typically presents in the setting of exposure to an offending drug/agent. Drug reactions can have various cutaneous presentations, with the most common being pigmented macules that progress into plaques.5 Given the isolated nature of our patient’s episode and apparent lack of association with medication, bullous fixed drug reaction was an unlikely diagnosis.
Tinea corporis bullosa is a rare clinical variant of tinea corporis that has only been reported in patients with a history of contact with different animals. There are many causative organisms related to tinea corporis; Trichophyton rubrum is the most common etiology of tinea corporis, while tinea corporis due to close contact with domesticated animals often is caused by M canis.6 The immunoinhibitory properties of the mannans in the fungal cell wall allow the organisms to adhere to the skin prior to invasion. Cutaneous invasion into dead cornified layers of the skin is credited to the proteases, subtilisinlike proteases (subtilases), and keratinases produced by the fungus.1 There are many different clinical presentations of tinea corporis due to the variability of causative organisms. An annular (ring-shaped) lesion with a central plaque and advancing border is the most typical presentation. Tinea corporis bullosa is characterized by the presence of bullae or vesicles in the borders of the scaly plaque. Rupture of the bullae subsequently leads to erosions and crusts over the plaque.
The diagnosis of tinea corporis bullosa often is clinical if the lesion is typical and can be confirmed using KOH preparation and fungal culture. Once the diagnosis is confirmed, topical antifungals are the standard treatment approach for localized superficial tinea corporis. Systemic antifungal treatment can be initiated if the lesion is extensive, recurrent, chronic, or unresponsive to topical treatment.1 Given our patient’s characteristic presentation, she was managed with an over-the-counter topical antifungal (terbinafine). The patient’s lesions dramatically improved, rendering oral therapy unnecessary. At 1-month follow-up, the rash had nearly resolved.
- Leung AK, Lam JM, Leong KF, et al. Tinea corporis: an updated review [published online July 20, 2020]. Drugs Context. doi:10.7573/dic.2020-5-6
- Guide SV, Marinkovich MP. Linear IgA bullous dermatosis. Clin Dermatol. 2001;19:719-727.
- Lamps LW, Scott MA. Cat-scratch disease: historic, clinical, and pathologic perspectives. Pathology Patterns Reviews. 2004;121(suppl):S71-S80.
- Caproni M, Antiga E, Melani L, et al. Guidelines for the diagnosis and treatment of dermatitis herpetiformis. J Eur Acad Dermatol Venereol. 2009;23:633-638.
- Patel S, John AM, Handler MZ, et al. Fixed drug eruptions: an update, emphasizing the potentially lethal generalized bullous fixed drug eruption. Am J Clin Dermatol. 2020;21:393-399.
- Ziemer M, Seyfarth F, Elsner P, et al. Atypical manifestations of tinea corporis. Mycoses. 2007;50:31-35.
The Diagnosis: Tinea Corporis Bullosa
At the time of presentation, a potassium hydroxide (KOH) preparation, fungal culture, and punch biopsy of the right ventral wrist was performed. The KOH preparation was positive for fungal hyphae characteristic of dermatophyte infections. Histologically, the biopsy showed intraepidermal and subepidermal blisters with neutrophil- and lymphocyte-rich contents (Figure 1). Fungal hyphae and spores were present within the stratum corneum and superficial epidermis (Figure 2), and fungal cultures grew Microsporum canis. The extent of the rash (upper and lower extremities, chest, and back), positive fungal culture, and KOH preparation all supported the diagnosis of tinea corporis bullosa, which was confirmed with biopsy. Oral prednisone use was discouraged and triamcinolone ointment was discontinued given that inappropriate treatment with steroids in the setting of fungal infection suppresses an inflammatory response and alters clinical appearance, obviating the persistent underlying infection.
Tinea corporis bullosa is a rare superficial dermatophyte fungal infection that often is acquired by close personto- person contact or contact with domestic animals. The infection begins as a circular pruritic plaque, generally with raised borders, which may be erythematous or hyperpigmented. By definition, tinea corporis occurs in sites other than the face, feet, hands, or groin area. Bullae formation is thought to be secondary to a delayed hypersensitivity reaction provoked by the presence of a dermatophyte antigen.1
Linear IgA bullous dermatosis is an immunemediated disease characterized by IgA deposition at the dermoepidermal junction. Linear IgA bullous dermatosis classically presents as widespread tense vesicles in an arciform or annular pattern. Mucosal involvement is common and typically presents with erosions, ulcerations, and scarring.2 Given the absence of mucosal involvement in our patient and a positive KOH preparation, linear IgA bullous dermatosis was an unlikely diagnosis.
Benign inoculation lymphoreticulosis, more commonly known as cat scratch disease (CSD), is a Bartonella henselae infection that results from a cat scratch or bite. Cat scratch disease can present as localized cutaneous and nodal involvement (lymphadenopathy) near the site of inoculation, or it may present as disseminated disease. Cutaneous lesions generally progress through vesicular, erythematous, and papular phases. Regional lymphadenopathy proximal to the inoculation site is the hallmark of CSD.3 Given the absence of lymphadenopathy in our patient as well as the sporadic distribution of lesions, CSD was an unlikely diagnosis.
Dermatitis herpetiformis (DH) is an autoimmune disorder with cutaneous manifestations of gluten sensitivity. Dermatitis herpetiformis presents as extremely pruritic papules and vesicles arranged in groups on areas such as the elbows, dorsal aspects of the forearms, knees, scalp, back, and buttocks. Most patients with DH have celiac disease or small bowel disease related to gluten sensitivity.4 Given our patient’s acute presentation in adulthood and lack of gluten sensitivity, DH was an unlikely diagnosis.
Bullous fixed drug reaction is a cutaneous eruption that typically presents in the setting of exposure to an offending drug/agent. Drug reactions can have various cutaneous presentations, with the most common being pigmented macules that progress into plaques.5 Given the isolated nature of our patient’s episode and apparent lack of association with medication, bullous fixed drug reaction was an unlikely diagnosis.
Tinea corporis bullosa is a rare clinical variant of tinea corporis that has only been reported in patients with a history of contact with different animals. There are many causative organisms related to tinea corporis; Trichophyton rubrum is the most common etiology of tinea corporis, while tinea corporis due to close contact with domesticated animals often is caused by M canis.6 The immunoinhibitory properties of the mannans in the fungal cell wall allow the organisms to adhere to the skin prior to invasion. Cutaneous invasion into dead cornified layers of the skin is credited to the proteases, subtilisinlike proteases (subtilases), and keratinases produced by the fungus.1 There are many different clinical presentations of tinea corporis due to the variability of causative organisms. An annular (ring-shaped) lesion with a central plaque and advancing border is the most typical presentation. Tinea corporis bullosa is characterized by the presence of bullae or vesicles in the borders of the scaly plaque. Rupture of the bullae subsequently leads to erosions and crusts over the plaque.
The diagnosis of tinea corporis bullosa often is clinical if the lesion is typical and can be confirmed using KOH preparation and fungal culture. Once the diagnosis is confirmed, topical antifungals are the standard treatment approach for localized superficial tinea corporis. Systemic antifungal treatment can be initiated if the lesion is extensive, recurrent, chronic, or unresponsive to topical treatment.1 Given our patient’s characteristic presentation, she was managed with an over-the-counter topical antifungal (terbinafine). The patient’s lesions dramatically improved, rendering oral therapy unnecessary. At 1-month follow-up, the rash had nearly resolved.
The Diagnosis: Tinea Corporis Bullosa
At the time of presentation, a potassium hydroxide (KOH) preparation, fungal culture, and punch biopsy of the right ventral wrist was performed. The KOH preparation was positive for fungal hyphae characteristic of dermatophyte infections. Histologically, the biopsy showed intraepidermal and subepidermal blisters with neutrophil- and lymphocyte-rich contents (Figure 1). Fungal hyphae and spores were present within the stratum corneum and superficial epidermis (Figure 2), and fungal cultures grew Microsporum canis. The extent of the rash (upper and lower extremities, chest, and back), positive fungal culture, and KOH preparation all supported the diagnosis of tinea corporis bullosa, which was confirmed with biopsy. Oral prednisone use was discouraged and triamcinolone ointment was discontinued given that inappropriate treatment with steroids in the setting of fungal infection suppresses an inflammatory response and alters clinical appearance, obviating the persistent underlying infection.
Tinea corporis bullosa is a rare superficial dermatophyte fungal infection that often is acquired by close personto- person contact or contact with domestic animals. The infection begins as a circular pruritic plaque, generally with raised borders, which may be erythematous or hyperpigmented. By definition, tinea corporis occurs in sites other than the face, feet, hands, or groin area. Bullae formation is thought to be secondary to a delayed hypersensitivity reaction provoked by the presence of a dermatophyte antigen.1
Linear IgA bullous dermatosis is an immunemediated disease characterized by IgA deposition at the dermoepidermal junction. Linear IgA bullous dermatosis classically presents as widespread tense vesicles in an arciform or annular pattern. Mucosal involvement is common and typically presents with erosions, ulcerations, and scarring.2 Given the absence of mucosal involvement in our patient and a positive KOH preparation, linear IgA bullous dermatosis was an unlikely diagnosis.
Benign inoculation lymphoreticulosis, more commonly known as cat scratch disease (CSD), is a Bartonella henselae infection that results from a cat scratch or bite. Cat scratch disease can present as localized cutaneous and nodal involvement (lymphadenopathy) near the site of inoculation, or it may present as disseminated disease. Cutaneous lesions generally progress through vesicular, erythematous, and papular phases. Regional lymphadenopathy proximal to the inoculation site is the hallmark of CSD.3 Given the absence of lymphadenopathy in our patient as well as the sporadic distribution of lesions, CSD was an unlikely diagnosis.
Dermatitis herpetiformis (DH) is an autoimmune disorder with cutaneous manifestations of gluten sensitivity. Dermatitis herpetiformis presents as extremely pruritic papules and vesicles arranged in groups on areas such as the elbows, dorsal aspects of the forearms, knees, scalp, back, and buttocks. Most patients with DH have celiac disease or small bowel disease related to gluten sensitivity.4 Given our patient’s acute presentation in adulthood and lack of gluten sensitivity, DH was an unlikely diagnosis.
Bullous fixed drug reaction is a cutaneous eruption that typically presents in the setting of exposure to an offending drug/agent. Drug reactions can have various cutaneous presentations, with the most common being pigmented macules that progress into plaques.5 Given the isolated nature of our patient’s episode and apparent lack of association with medication, bullous fixed drug reaction was an unlikely diagnosis.
Tinea corporis bullosa is a rare clinical variant of tinea corporis that has only been reported in patients with a history of contact with different animals. There are many causative organisms related to tinea corporis; Trichophyton rubrum is the most common etiology of tinea corporis, while tinea corporis due to close contact with domesticated animals often is caused by M canis.6 The immunoinhibitory properties of the mannans in the fungal cell wall allow the organisms to adhere to the skin prior to invasion. Cutaneous invasion into dead cornified layers of the skin is credited to the proteases, subtilisinlike proteases (subtilases), and keratinases produced by the fungus.1 There are many different clinical presentations of tinea corporis due to the variability of causative organisms. An annular (ring-shaped) lesion with a central plaque and advancing border is the most typical presentation. Tinea corporis bullosa is characterized by the presence of bullae or vesicles in the borders of the scaly plaque. Rupture of the bullae subsequently leads to erosions and crusts over the plaque.
The diagnosis of tinea corporis bullosa often is clinical if the lesion is typical and can be confirmed using KOH preparation and fungal culture. Once the diagnosis is confirmed, topical antifungals are the standard treatment approach for localized superficial tinea corporis. Systemic antifungal treatment can be initiated if the lesion is extensive, recurrent, chronic, or unresponsive to topical treatment.1 Given our patient’s characteristic presentation, she was managed with an over-the-counter topical antifungal (terbinafine). The patient’s lesions dramatically improved, rendering oral therapy unnecessary. At 1-month follow-up, the rash had nearly resolved.
- Leung AK, Lam JM, Leong KF, et al. Tinea corporis: an updated review [published online July 20, 2020]. Drugs Context. doi:10.7573/dic.2020-5-6
- Guide SV, Marinkovich MP. Linear IgA bullous dermatosis. Clin Dermatol. 2001;19:719-727.
- Lamps LW, Scott MA. Cat-scratch disease: historic, clinical, and pathologic perspectives. Pathology Patterns Reviews. 2004;121(suppl):S71-S80.
- Caproni M, Antiga E, Melani L, et al. Guidelines for the diagnosis and treatment of dermatitis herpetiformis. J Eur Acad Dermatol Venereol. 2009;23:633-638.
- Patel S, John AM, Handler MZ, et al. Fixed drug eruptions: an update, emphasizing the potentially lethal generalized bullous fixed drug eruption. Am J Clin Dermatol. 2020;21:393-399.
- Ziemer M, Seyfarth F, Elsner P, et al. Atypical manifestations of tinea corporis. Mycoses. 2007;50:31-35.
- Leung AK, Lam JM, Leong KF, et al. Tinea corporis: an updated review [published online July 20, 2020]. Drugs Context. doi:10.7573/dic.2020-5-6
- Guide SV, Marinkovich MP. Linear IgA bullous dermatosis. Clin Dermatol. 2001;19:719-727.
- Lamps LW, Scott MA. Cat-scratch disease: historic, clinical, and pathologic perspectives. Pathology Patterns Reviews. 2004;121(suppl):S71-S80.
- Caproni M, Antiga E, Melani L, et al. Guidelines for the diagnosis and treatment of dermatitis herpetiformis. J Eur Acad Dermatol Venereol. 2009;23:633-638.
- Patel S, John AM, Handler MZ, et al. Fixed drug eruptions: an update, emphasizing the potentially lethal generalized bullous fixed drug eruption. Am J Clin Dermatol. 2020;21:393-399.
- Ziemer M, Seyfarth F, Elsner P, et al. Atypical manifestations of tinea corporis. Mycoses. 2007;50:31-35.
A 38-year-old woman presented with a rash of 5 days’ duration that initially appeared on the wrists after playing with her kitten, with subsequent involvement of the chest, back, abdomen, and upper and lower extremities. Physical examination revealed multiple annular plaques with raised erythematous borders, rare peripheral vesicles, and superficial central scaling. Extreme pruritus accompanied the plaques, both of which developed after playing with her kitten. The patient noted that all lesions on the upper extremities evolved in areas subject to deep puncture while more superficially excoriated areas were unaffected. She denied any other prior skin conditions and had received a 5-day course of azithromycin without improvement prior to presentation; triamcinolone ointment 0.1% had provided only temporary relief. Primary care providers prescribed a short course of oral prednisone; however, she did not start it prior to presentation.


Rapidly Evolving Papulonodular Eruption in the Axilla
The Diagnosis: Lymphomatoid Papulosis
At the time of the initial visit, a punch biopsy was performed on the posterior shoulder girdle. Histopathology revealed mild epidermal spongiosis and acanthosis with associated parakeratosis and a dermal lymphocytic infiltrate with extravasated erythrocytes consistent with pityriasis rosea (Figure 1). Two weeks after the biopsy, the patient returned for suture removal and to discuss the biopsy results. The patient reported more evolving lesions despite completing the prescribed course of dicloxacillin. At this time, physical examination revealed the persistence of several reddishbrown papules along with new nodular lesions on the arms and thighs, some with central ulceration and crusting (Figure 2). A second biopsy of a nodular lesion on the right distal forearm was performed at this visit along with a superficial tissue culture, which was negative for bacterial or fungal elements. The biopsy revealed an atypical CD30+ lymphoid proliferation (Figure 3). These cells were strongly PD-L1 positive and also positive for CD3, CD4, and granzyme-B. Ki67 showed a high proliferation rate, and T-cell gene rearrangement studies were positive. Given these histologic findings and the clinical context of rapidly evolving skin lesions from small papules to nodular skin tumors, a diagnosis of lymphomatoid papulosis (LyP) was established.

Because of the notable pathologic discordance between the 2 biopsy specimens, re-evaluation of the initial specimen was requested. The initial biopsy was subsequently found to be CD30+ with an identical peak on gene rearrangement studies as the second biopsy, further validating the diagnosis of LyP (Figure 4). Our patient was offered low-dose methotrexate therapy but declined the treatment plan, as the skin lesions had begun to resolve.

Lymphomatoid papulosis is a chronic CD30+ lymphoproliferative disorder with a characteristic recurrent and self-remitting disease course.1,2 Although it typically has a benign clinical course, it is histologically malignant and considered a low-grade variant of cutaneous T-cell lymphoma. 2,3 The classic clinical presentation of LyP involves the presence of reddish-brown papules and nodules typically measuring less than 2.0 cm, which may show evidence of central ulceration, hemorrhage, necrosis, and/or crust formation.1-5 It is characteristic that a patient may present with these skin lesions in different stages of evolution and that biopsies of these lesions may reflect different histologic features depending on the age of the lesion, making a definitive diagnosis more difficult to obtain if not clinically correlated.1,2 Any part of the body may be involved; however, there appears to be a predilection for the trunk and extremities in most cases.1-3,5 The skin eruptions usually are asymptomatic, but pruritus is a commonly associated concern.1,2,4,5
 and a CD30+ lymphocytic infiltrate (original magnification ×2)")
Lymphomatoid papulosis can have a localized, clustered, or generalized distribution pattern and typically will spontaneously regress without treatment within 3 to 12 weeks of symptom onset.2,3 Lymphomatoid papulosis has a slight male predominance with a male to female ratio of 1.5:1. It occurs most commonly between 35 and 45 years of age, though it can present at any age. The overall duration of the disease can range from months to decades.2,3 Lymphomatoid papulosis makes up approximately 15% of all cutaneous T-cell lymphomas.2,3 Although the overall prognosis is excellent, patients with LyP are at an increased risk of developing cutaneous or systemic lymphoma, most commonly mycosis fungoides, anaplastic large cell lymphoma, or Hodgkin lymphoma.1-3 This increased lifelong risk is the reason that patients with LyP must be followed long-term every 6 to 12 months for surveillance of emerging malignancy.1,2,6

The pathogenesis of LyP remains unknown. Some have hypothesized a possible viral trigger; however, there is insufficient data to support this theory.2,6 A diagnostic hallmark of LyP is its CD30 positivity, which is a known marker for T-cell activation.6 The spontaneous regression of skin lesions that is characteristic of LyP is believed to involve the interactions between CD30 and its ligand (CD30L), which may contribute to apoptosis of neoplastic T cells.2,3,6 With regards to the possible mechanisms contributing to tumor progression in LyP, a mutation in the transforming growth factor β receptor gene on CD30+ tumor cells within LyP lesions may allow for these cells to evade growth regulation and progress to lymphoma.2,6 A large percentage of LyP biopsy specimens show evidence of T-cell receptor gene monoclonal rearrangement, which can aid in establishing a diagnosis.1,2
The histologic features of LyP can vary greatly depending on the age of the lesion sampled.1,2 Histologic subtypes of LyP have been established, with type A being the most common (approximately 75% of cases), displaying a wedge-shaped infiltrate of scattered or clustered, large, atypical CD30+ T cells.1,2 Types B through E vary in histologic features, with the exception that all subtypes contain a CD30+ lymphocytic infiltrate.2,3
Treatment of LyP depends on the symptom/disease burden that the patient is experiencing. For patients with a limited number of nonscarring skin lesions in areas that are not cosmetically sensitive, observation is recommended. 1-3 For symptomatic patients with an extensive number of lesions, particularly those that may be scarring and/or in cosmetically sensitive areas, low-dose oral methotrexate therapy is considered first-line treatment.1-4 A methotrexate dose of 5 to 20 mg weekly can be effective in reducing the number and severity of lesions, with duration of treatment depending on clinical response.1,2 For patients who have contraindications to or who cannot tolerate oral methotrexate, phototherapy using psoralen plus UVA twice weekly for 6 to 8 weeks is another treatment option.1,2 Topical corticosteroids also can be used in children or for patients experiencing substantial pruritus.1,2,4 Oral or topical retinoids, topical carmustine or mechlorethamine, and brentuximab (an anti-CD30 monoclonal antibody) are all alternative therapies that have shown some beneficial effects.1,2 In the event that any of the skin lesions do not spontaneously regress within a 3- to 12-week time frame, surgical excision or radiotherapy can be performed on those lesions.2
Primary cutaneous anaplastic large cell lymphoma (C-ALCL) is another CD30+ lymphoproliferative disorder with overlapping clinical and histopathological features of LyP. Recurrent crops of multiple lesions favor a diagnosis of LyP, whereas solitary lesions favor C-ALCL; however, multifocal C-ALCL cases may occur.2 Mycosis fungoides is the most common type of cutaneous T-cell lymphoma that characteristically presents in a patch, plaque, tumor progression. Although mycosis fungoides eventually may transform into a CD30+ lymphoma, our patient did not display the characteristic clinical progression to suggest this diagnosis. Pityriasis lichenoides et varioliformis acuta and pityriasis lichenoides chronica also fall into the spectrum of clonal T-cell cutaneous disorders that more commonly affect the pediatric population. Pityriasis lichenoides et varioliformis acuta has a marked CD8+ lymphocyte infiltrate, whereas pityriasis lichenoides chronica has more CD4+ lymphocytes. These disorders typically do not stain positive for CD30.2
All patients with a diagnosis of LyP should maintain lifelong, regular, 6- to 12-month follow-up visits to monitor disease status and screen for any evidence of developing malignancy.1,2,6 A thorough review of clinical history, complete skin examination, and physical examination with a particular focus on detection of lymphadenopathy and hepatosplenomegaly should be included at every followup visit.1 Systemic symptoms such as fever, night sweats, or weight loss are not typical features of LyP; therefore, patients who begin to develop these symptoms should be promptly evaluated for systemic lymphoma.1
- Kadin ME. Lymphomatoid papulosis. UpToDate website. Accessed June 4, 2022. https://www.uptodate.com/contents/lymphomatoid-papulosis
- Willemze R. Cutaneous T-cell lymphoma. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. Vol 2. 4th ed. Elsevier Saunders; 2017:2141-2143.
- Wiznia LE, Cohen JM, Beasley JM, et al. Lymphomatoid papulosis. Dermatol Online J. 2018;24:13030/qt4xt046c9.
- Wieser I, Oh CW, Talpur R, et al. Lymphomatoid papulosis: treatment response and associated lymphomas in a study of 180 patients. J Am Acad Dermatol. 2016;74:59-67. doi:10.1016/j.jaad.2015.09.013
- Wolff K, Johnson RA, Saavedra AP, et al. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 8th ed. McGraw-Hill Education; 2017.
- Kunishige JH, McDonald H, Alvarez G, et al. Lymphomatoid papulosis and associated lymphomas: a retrospective case series of 84 patients. Clin Exp Dermatol. 2009;34:576-581. doi:10.1111 /j.1365-2230.2008.03024.x
The Diagnosis: Lymphomatoid Papulosis
At the time of the initial visit, a punch biopsy was performed on the posterior shoulder girdle. Histopathology revealed mild epidermal spongiosis and acanthosis with associated parakeratosis and a dermal lymphocytic infiltrate with extravasated erythrocytes consistent with pityriasis rosea (Figure 1). Two weeks after the biopsy, the patient returned for suture removal and to discuss the biopsy results. The patient reported more evolving lesions despite completing the prescribed course of dicloxacillin. At this time, physical examination revealed the persistence of several reddishbrown papules along with new nodular lesions on the arms and thighs, some with central ulceration and crusting (Figure 2). A second biopsy of a nodular lesion on the right distal forearm was performed at this visit along with a superficial tissue culture, which was negative for bacterial or fungal elements. The biopsy revealed an atypical CD30+ lymphoid proliferation (Figure 3). These cells were strongly PD-L1 positive and also positive for CD3, CD4, and granzyme-B. Ki67 showed a high proliferation rate, and T-cell gene rearrangement studies were positive. Given these histologic findings and the clinical context of rapidly evolving skin lesions from small papules to nodular skin tumors, a diagnosis of lymphomatoid papulosis (LyP) was established.
Because of the notable pathologic discordance between the 2 biopsy specimens, re-evaluation of the initial specimen was requested. The initial biopsy was subsequently found to be CD30+ with an identical peak on gene rearrangement studies as the second biopsy, further validating the diagnosis of LyP (Figure 4). Our patient was offered low-dose methotrexate therapy but declined the treatment plan, as the skin lesions had begun to resolve.
Lymphomatoid papulosis is a chronic CD30+ lymphoproliferative disorder with a characteristic recurrent and self-remitting disease course.1,2 Although it typically has a benign clinical course, it is histologically malignant and considered a low-grade variant of cutaneous T-cell lymphoma. 2,3 The classic clinical presentation of LyP involves the presence of reddish-brown papules and nodules typically measuring less than 2.0 cm, which may show evidence of central ulceration, hemorrhage, necrosis, and/or crust formation.1-5 It is characteristic that a patient may present with these skin lesions in different stages of evolution and that biopsies of these lesions may reflect different histologic features depending on the age of the lesion, making a definitive diagnosis more difficult to obtain if not clinically correlated.1,2 Any part of the body may be involved; however, there appears to be a predilection for the trunk and extremities in most cases.1-3,5 The skin eruptions usually are asymptomatic, but pruritus is a commonly associated concern.1,2,4,5
Lymphomatoid papulosis can have a localized, clustered, or generalized distribution pattern and typically will spontaneously regress without treatment within 3 to 12 weeks of symptom onset.2,3 Lymphomatoid papulosis has a slight male predominance with a male to female ratio of 1.5:1. It occurs most commonly between 35 and 45 years of age, though it can present at any age. The overall duration of the disease can range from months to decades.2,3 Lymphomatoid papulosis makes up approximately 15% of all cutaneous T-cell lymphomas.2,3 Although the overall prognosis is excellent, patients with LyP are at an increased risk of developing cutaneous or systemic lymphoma, most commonly mycosis fungoides, anaplastic large cell lymphoma, or Hodgkin lymphoma.1-3 This increased lifelong risk is the reason that patients with LyP must be followed long-term every 6 to 12 months for surveillance of emerging malignancy.1,2,6
The pathogenesis of LyP remains unknown. Some have hypothesized a possible viral trigger; however, there is insufficient data to support this theory.2,6 A diagnostic hallmark of LyP is its CD30 positivity, which is a known marker for T-cell activation.6 The spontaneous regression of skin lesions that is characteristic of LyP is believed to involve the interactions between CD30 and its ligand (CD30L), which may contribute to apoptosis of neoplastic T cells.2,3,6 With regards to the possible mechanisms contributing to tumor progression in LyP, a mutation in the transforming growth factor β receptor gene on CD30+ tumor cells within LyP lesions may allow for these cells to evade growth regulation and progress to lymphoma.2,6 A large percentage of LyP biopsy specimens show evidence of T-cell receptor gene monoclonal rearrangement, which can aid in establishing a diagnosis.1,2
The histologic features of LyP can vary greatly depending on the age of the lesion sampled.1,2 Histologic subtypes of LyP have been established, with type A being the most common (approximately 75% of cases), displaying a wedge-shaped infiltrate of scattered or clustered, large, atypical CD30+ T cells.1,2 Types B through E vary in histologic features, with the exception that all subtypes contain a CD30+ lymphocytic infiltrate.2,3
Treatment of LyP depends on the symptom/disease burden that the patient is experiencing. For patients with a limited number of nonscarring skin lesions in areas that are not cosmetically sensitive, observation is recommended. 1-3 For symptomatic patients with an extensive number of lesions, particularly those that may be scarring and/or in cosmetically sensitive areas, low-dose oral methotrexate therapy is considered first-line treatment.1-4 A methotrexate dose of 5 to 20 mg weekly can be effective in reducing the number and severity of lesions, with duration of treatment depending on clinical response.1,2 For patients who have contraindications to or who cannot tolerate oral methotrexate, phototherapy using psoralen plus UVA twice weekly for 6 to 8 weeks is another treatment option.1,2 Topical corticosteroids also can be used in children or for patients experiencing substantial pruritus.1,2,4 Oral or topical retinoids, topical carmustine or mechlorethamine, and brentuximab (an anti-CD30 monoclonal antibody) are all alternative therapies that have shown some beneficial effects.1,2 In the event that any of the skin lesions do not spontaneously regress within a 3- to 12-week time frame, surgical excision or radiotherapy can be performed on those lesions.2
Primary cutaneous anaplastic large cell lymphoma (C-ALCL) is another CD30+ lymphoproliferative disorder with overlapping clinical and histopathological features of LyP. Recurrent crops of multiple lesions favor a diagnosis of LyP, whereas solitary lesions favor C-ALCL; however, multifocal C-ALCL cases may occur.2 Mycosis fungoides is the most common type of cutaneous T-cell lymphoma that characteristically presents in a patch, plaque, tumor progression. Although mycosis fungoides eventually may transform into a CD30+ lymphoma, our patient did not display the characteristic clinical progression to suggest this diagnosis. Pityriasis lichenoides et varioliformis acuta and pityriasis lichenoides chronica also fall into the spectrum of clonal T-cell cutaneous disorders that more commonly affect the pediatric population. Pityriasis lichenoides et varioliformis acuta has a marked CD8+ lymphocyte infiltrate, whereas pityriasis lichenoides chronica has more CD4+ lymphocytes. These disorders typically do not stain positive for CD30.2
All patients with a diagnosis of LyP should maintain lifelong, regular, 6- to 12-month follow-up visits to monitor disease status and screen for any evidence of developing malignancy.1,2,6 A thorough review of clinical history, complete skin examination, and physical examination with a particular focus on detection of lymphadenopathy and hepatosplenomegaly should be included at every followup visit.1 Systemic symptoms such as fever, night sweats, or weight loss are not typical features of LyP; therefore, patients who begin to develop these symptoms should be promptly evaluated for systemic lymphoma.1
The Diagnosis: Lymphomatoid Papulosis
At the time of the initial visit, a punch biopsy was performed on the posterior shoulder girdle. Histopathology revealed mild epidermal spongiosis and acanthosis with associated parakeratosis and a dermal lymphocytic infiltrate with extravasated erythrocytes consistent with pityriasis rosea (Figure 1). Two weeks after the biopsy, the patient returned for suture removal and to discuss the biopsy results. The patient reported more evolving lesions despite completing the prescribed course of dicloxacillin. At this time, physical examination revealed the persistence of several reddishbrown papules along with new nodular lesions on the arms and thighs, some with central ulceration and crusting (Figure 2). A second biopsy of a nodular lesion on the right distal forearm was performed at this visit along with a superficial tissue culture, which was negative for bacterial or fungal elements. The biopsy revealed an atypical CD30+ lymphoid proliferation (Figure 3). These cells were strongly PD-L1 positive and also positive for CD3, CD4, and granzyme-B. Ki67 showed a high proliferation rate, and T-cell gene rearrangement studies were positive. Given these histologic findings and the clinical context of rapidly evolving skin lesions from small papules to nodular skin tumors, a diagnosis of lymphomatoid papulosis (LyP) was established.
Because of the notable pathologic discordance between the 2 biopsy specimens, re-evaluation of the initial specimen was requested. The initial biopsy was subsequently found to be CD30+ with an identical peak on gene rearrangement studies as the second biopsy, further validating the diagnosis of LyP (Figure 4). Our patient was offered low-dose methotrexate therapy but declined the treatment plan, as the skin lesions had begun to resolve.
Lymphomatoid papulosis is a chronic CD30+ lymphoproliferative disorder with a characteristic recurrent and self-remitting disease course.1,2 Although it typically has a benign clinical course, it is histologically malignant and considered a low-grade variant of cutaneous T-cell lymphoma. 2,3 The classic clinical presentation of LyP involves the presence of reddish-brown papules and nodules typically measuring less than 2.0 cm, which may show evidence of central ulceration, hemorrhage, necrosis, and/or crust formation.1-5 It is characteristic that a patient may present with these skin lesions in different stages of evolution and that biopsies of these lesions may reflect different histologic features depending on the age of the lesion, making a definitive diagnosis more difficult to obtain if not clinically correlated.1,2 Any part of the body may be involved; however, there appears to be a predilection for the trunk and extremities in most cases.1-3,5 The skin eruptions usually are asymptomatic, but pruritus is a commonly associated concern.1,2,4,5
Lymphomatoid papulosis can have a localized, clustered, or generalized distribution pattern and typically will spontaneously regress without treatment within 3 to 12 weeks of symptom onset.2,3 Lymphomatoid papulosis has a slight male predominance with a male to female ratio of 1.5:1. It occurs most commonly between 35 and 45 years of age, though it can present at any age. The overall duration of the disease can range from months to decades.2,3 Lymphomatoid papulosis makes up approximately 15% of all cutaneous T-cell lymphomas.2,3 Although the overall prognosis is excellent, patients with LyP are at an increased risk of developing cutaneous or systemic lymphoma, most commonly mycosis fungoides, anaplastic large cell lymphoma, or Hodgkin lymphoma.1-3 This increased lifelong risk is the reason that patients with LyP must be followed long-term every 6 to 12 months for surveillance of emerging malignancy.1,2,6
The pathogenesis of LyP remains unknown. Some have hypothesized a possible viral trigger; however, there is insufficient data to support this theory.2,6 A diagnostic hallmark of LyP is its CD30 positivity, which is a known marker for T-cell activation.6 The spontaneous regression of skin lesions that is characteristic of LyP is believed to involve the interactions between CD30 and its ligand (CD30L), which may contribute to apoptosis of neoplastic T cells.2,3,6 With regards to the possible mechanisms contributing to tumor progression in LyP, a mutation in the transforming growth factor β receptor gene on CD30+ tumor cells within LyP lesions may allow for these cells to evade growth regulation and progress to lymphoma.2,6 A large percentage of LyP biopsy specimens show evidence of T-cell receptor gene monoclonal rearrangement, which can aid in establishing a diagnosis.1,2
The histologic features of LyP can vary greatly depending on the age of the lesion sampled.1,2 Histologic subtypes of LyP have been established, with type A being the most common (approximately 75% of cases), displaying a wedge-shaped infiltrate of scattered or clustered, large, atypical CD30+ T cells.1,2 Types B through E vary in histologic features, with the exception that all subtypes contain a CD30+ lymphocytic infiltrate.2,3
Treatment of LyP depends on the symptom/disease burden that the patient is experiencing. For patients with a limited number of nonscarring skin lesions in areas that are not cosmetically sensitive, observation is recommended. 1-3 For symptomatic patients with an extensive number of lesions, particularly those that may be scarring and/or in cosmetically sensitive areas, low-dose oral methotrexate therapy is considered first-line treatment.1-4 A methotrexate dose of 5 to 20 mg weekly can be effective in reducing the number and severity of lesions, with duration of treatment depending on clinical response.1,2 For patients who have contraindications to or who cannot tolerate oral methotrexate, phototherapy using psoralen plus UVA twice weekly for 6 to 8 weeks is another treatment option.1,2 Topical corticosteroids also can be used in children or for patients experiencing substantial pruritus.1,2,4 Oral or topical retinoids, topical carmustine or mechlorethamine, and brentuximab (an anti-CD30 monoclonal antibody) are all alternative therapies that have shown some beneficial effects.1,2 In the event that any of the skin lesions do not spontaneously regress within a 3- to 12-week time frame, surgical excision or radiotherapy can be performed on those lesions.2
Primary cutaneous anaplastic large cell lymphoma (C-ALCL) is another CD30+ lymphoproliferative disorder with overlapping clinical and histopathological features of LyP. Recurrent crops of multiple lesions favor a diagnosis of LyP, whereas solitary lesions favor C-ALCL; however, multifocal C-ALCL cases may occur.2 Mycosis fungoides is the most common type of cutaneous T-cell lymphoma that characteristically presents in a patch, plaque, tumor progression. Although mycosis fungoides eventually may transform into a CD30+ lymphoma, our patient did not display the characteristic clinical progression to suggest this diagnosis. Pityriasis lichenoides et varioliformis acuta and pityriasis lichenoides chronica also fall into the spectrum of clonal T-cell cutaneous disorders that more commonly affect the pediatric population. Pityriasis lichenoides et varioliformis acuta has a marked CD8+ lymphocyte infiltrate, whereas pityriasis lichenoides chronica has more CD4+ lymphocytes. These disorders typically do not stain positive for CD30.2
All patients with a diagnosis of LyP should maintain lifelong, regular, 6- to 12-month follow-up visits to monitor disease status and screen for any evidence of developing malignancy.1,2,6 A thorough review of clinical history, complete skin examination, and physical examination with a particular focus on detection of lymphadenopathy and hepatosplenomegaly should be included at every followup visit.1 Systemic symptoms such as fever, night sweats, or weight loss are not typical features of LyP; therefore, patients who begin to develop these symptoms should be promptly evaluated for systemic lymphoma.1
- Kadin ME. Lymphomatoid papulosis. UpToDate website. Accessed June 4, 2022. https://www.uptodate.com/contents/lymphomatoid-papulosis
- Willemze R. Cutaneous T-cell lymphoma. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. Vol 2. 4th ed. Elsevier Saunders; 2017:2141-2143.
- Wiznia LE, Cohen JM, Beasley JM, et al. Lymphomatoid papulosis. Dermatol Online J. 2018;24:13030/qt4xt046c9.
- Wieser I, Oh CW, Talpur R, et al. Lymphomatoid papulosis: treatment response and associated lymphomas in a study of 180 patients. J Am Acad Dermatol. 2016;74:59-67. doi:10.1016/j.jaad.2015.09.013
- Wolff K, Johnson RA, Saavedra AP, et al. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 8th ed. McGraw-Hill Education; 2017.
- Kunishige JH, McDonald H, Alvarez G, et al. Lymphomatoid papulosis and associated lymphomas: a retrospective case series of 84 patients. Clin Exp Dermatol. 2009;34:576-581. doi:10.1111 /j.1365-2230.2008.03024.x
- Kadin ME. Lymphomatoid papulosis. UpToDate website. Accessed June 4, 2022. https://www.uptodate.com/contents/lymphomatoid-papulosis
- Willemze R. Cutaneous T-cell lymphoma. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. Vol 2. 4th ed. Elsevier Saunders; 2017:2141-2143.
- Wiznia LE, Cohen JM, Beasley JM, et al. Lymphomatoid papulosis. Dermatol Online J. 2018;24:13030/qt4xt046c9.
- Wieser I, Oh CW, Talpur R, et al. Lymphomatoid papulosis: treatment response and associated lymphomas in a study of 180 patients. J Am Acad Dermatol. 2016;74:59-67. doi:10.1016/j.jaad.2015.09.013
- Wolff K, Johnson RA, Saavedra AP, et al. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 8th ed. McGraw-Hill Education; 2017.
- Kunishige JH, McDonald H, Alvarez G, et al. Lymphomatoid papulosis and associated lymphomas: a retrospective case series of 84 patients. Clin Exp Dermatol. 2009;34:576-581. doi:10.1111 /j.1365-2230.2008.03024.x
A 37-year-old woman presented to our dermatology clinic with a pruritic erythematous eruption involving the trunk, axillae, and proximal extremities of 10 days’ duration. Her medical history was notable only for eczema, and she denied taking any medications. Physical examination revealed scattered erythematous papules and crusts involving the trunk bilaterally and the extremities. We initially made a clinical diagnosis of bullous impetigo, and the patient was prescribed mupirocin ointment and dicloxacillin. At 1-week follow-up, the patient reported persistent skin lesions that were evolving despite therapy. Physical examination at this visit revealed an evolving eruption of multiple reddish-brown scaly papules involving the axillae, arms, forearms, and thighs, as depicted here.


Chronic Retiform Purpura of the Abdomen and Thighs: A Fatal Case of Intravascular Large Cell Lymphoma
To the Editor:
Intravascular large cell lymphoma (ILCL) is a rare B-cell lymphoma that is defined by the presence of large neoplastic B cells in the lumen of blood vessels.1 At least 3 variants of ILCL have been described based on case reports and a small case series: classic, cutaneous, and hemophagocytic. The classic variant presents in elderly patients as nonspecific constitutional symptoms (fever or pain, or less frequently weight loss) or as signs of multiorgan failure (most commonly of the central nervous system). Skin involvement, which is present in nearly half of these patients, can take on multiple morphologies, including retiform purpura, ulcerated nodules, or pseudocellulitis. The cutaneous variant typically presents in middle-aged women with normal hematologic studies. Systemic involvement is less common in this variant of disease than the classic variant, which may partly explain why overall survival is superior in this variant. The hemophagocytic variant manifests as intravascular lymphoma accompanied by hemophagocytic syndrome (fever, hepatosplenomegaly, thrombocytopenia, and bone marrow involvement).

A 69-year-old man presented to the emergency department for failure to thrive and nonhealing wounds of 1 year’s duration. His medical history was notable for poorly controlled diabetes mellitus, progressive multifocal ischemic and hemorrhagic cerebral infarcts, and bilateral deep venous thromboses. Physical examination revealed large purpuric to brown plaques in a retiform configuration with central necrotic eschars on the thighs and abdomen (Figure 1). There was no palpable lymphadenopathy. Laboratory tests revealed normocytic anemia with a hemoglobin level of 10.5 g/dL (reference range, 12–18 g/dL), elevated lactate dehydrogenase level of 525 U/L (reference range, 118–242 U/L), elevated erythrocyte sedimentation rate of 73 mm/h (reference range, <20 mm/h), antinuclear antibody (ANA) titer of 1:2560 (reference range, <1:80), and polyclonal hypergammaglobulinemia. The patient’s white blood cell and platelet counts, creatinine level, and liver function tests were within reference range. Cryoglobulins, coagulation studies, and cardiolipin antibodies were negative. Chest and abdominal imaging also were negative. An incisional skin biopsy and skin punch biopsy showed thrombotic coagulopathy and dilated vessels. A bone marrow biopsy revealed a hypercellular marrow but no plasma cell neoplasm. A repeat incisional skin biopsy demonstrated large CD20+ and CD45+ atypical lymphocytes within the small capillaries of the deep dermis and subcutaneous fat (Figure 2), which confirmed ILCL. Too deconditioned to tolerate chemotherapy, the patient opted for palliative care and died 18 months after initial presentation.
. B, CD20 immunohistochemical staining highlighted atypical B cells")
The diagnosis of ILCL often is delayed for several reasons.2 Patients can present with a variety of signs and symptoms related to small vessel occlusion that can be misattributed to other conditions.3,4 In our case, the patient’s recurrent infarcts were thought to be due to his poorly controlled diabetes mellitus, which was diagnosed a few weeks prior, and a positive ANA, even though the workup for antiphospholipid syndrome was negative. Interestingly, a positive ANA (without signs or symptoms of lupus or other autoimmune conditions) has been reported in patients with lymphoma.3 A positive antineutrophil cytoplasmic antibody level (without symptoms or other signs of vasculitis) has been reported in patients with ILCL.4,5 Therefore, distractors are common.
Multiple incisional skin biopsies in the absence of clinical findings (ie, random skin biopsy) are moderately sensitive (77.8%) for the diagnosis of ILCL.2 In a study by Matsue et al,2 111 suspected cases of ILCL underwent 3 incisional biopsies of fat-containing areas of the skin, such as the thigh, abdomen, and upper arm. Intravascular large cell lymphoma was confirmed in 26 cases. Seven additional cases were diagnosed as ILCL, 2 by additional skin biopsies (1 by a second round and 1 by a third round) and 5 by internal organ biopsy (4 bone marrow and 1 adrenal gland). The remaining cases ultimately were found to be a diagnostic mimicker of ILCL, including non-ILCL.2 Although random skin biopsies are reasonably sensitive for ILCL, multiple biopsies are needed, and in some cases, sampling of an internal organ may be required to establish the diagnosis of ILCL.
The prognosis of ILCL is poor; the 3-year overall survival rate for classic and cutaneous variants is 22% and 56%, respectively.6 Anthracycline-based chemotherapy, such as CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone), is considered first-line treatment, and the addition of rituximab to the CHOP regimen may improve remission rates and survival.7
- Ponzoni M, Campo E, Nakamura S. Intravascular large B-cell lymphoma: a chameleon with multiple faces and many masks [published online August 15, 2018]. Blood. 2018;132:1561-1567. doi:10.1182/blood-2017-04-737445
- Matsue K, Abe Y, Kitadate A, et al. Sensitivity and specificity of incisional random skin biopsy for diagnosis of intravascular large B-cell lymphoma. Blood. 2019;133:1257-1259.
- Altintas A, Cil T, Pasa S, et al. Clinical significance of elevated antinuclear antibody test in patients with Hodgkin’s and non-Hodgkin’s lymphoma. Minerva Med. 2008;99:7-14.
- Shinkawa Y, Hatachi S, Yagita M. Intravascular large B-cell lymphoma with a high titer of proteinase-3-anti-neutrophil cytoplasmic antibody mimicking granulomatosis with polyangiitis. Mod Rheumatol. 2019;29:195-197.
- Sugiyama A, Kobayashi M, Daizo A, et al. Diffuse cerebral vasoconstriction in a intravascular lymphoma patient with a high serum MPO-ANCA level. Intern Med. 2017;56:1715-1718.
- Ferreri AJ, Campo E, Seymour JF, et al. Intravascular lymphoma: clinical presentation, natural history, management and prognostic factors in a series of 38 cases, with special emphasis on the ‘cutaneous variant.’ Br J Haematol. 2004;127:173-183.
- Ferreri AJM, Dognini GP, Bairey O, et al; International Extranodal Lyphoma Study Group. The addition of rituximab to anthracycline-based chemotherapy significantly improves outcome in ‘Western’ patients with intravascular large B-cell lymphoma [published online August 10, 2008]. Br J Haematol. 2008;143:253-257. doi:10.1111/j.1365-2141.2008.07338.x
To the Editor:
Intravascular large cell lymphoma (ILCL) is a rare B-cell lymphoma that is defined by the presence of large neoplastic B cells in the lumen of blood vessels.1 At least 3 variants of ILCL have been described based on case reports and a small case series: classic, cutaneous, and hemophagocytic. The classic variant presents in elderly patients as nonspecific constitutional symptoms (fever or pain, or less frequently weight loss) or as signs of multiorgan failure (most commonly of the central nervous system). Skin involvement, which is present in nearly half of these patients, can take on multiple morphologies, including retiform purpura, ulcerated nodules, or pseudocellulitis. The cutaneous variant typically presents in middle-aged women with normal hematologic studies. Systemic involvement is less common in this variant of disease than the classic variant, which may partly explain why overall survival is superior in this variant. The hemophagocytic variant manifests as intravascular lymphoma accompanied by hemophagocytic syndrome (fever, hepatosplenomegaly, thrombocytopenia, and bone marrow involvement).
A 69-year-old man presented to the emergency department for failure to thrive and nonhealing wounds of 1 year’s duration. His medical history was notable for poorly controlled diabetes mellitus, progressive multifocal ischemic and hemorrhagic cerebral infarcts, and bilateral deep venous thromboses. Physical examination revealed large purpuric to brown plaques in a retiform configuration with central necrotic eschars on the thighs and abdomen (Figure 1). There was no palpable lymphadenopathy. Laboratory tests revealed normocytic anemia with a hemoglobin level of 10.5 g/dL (reference range, 12–18 g/dL), elevated lactate dehydrogenase level of 525 U/L (reference range, 118–242 U/L), elevated erythrocyte sedimentation rate of 73 mm/h (reference range, <20 mm/h), antinuclear antibody (ANA) titer of 1:2560 (reference range, <1:80), and polyclonal hypergammaglobulinemia. The patient’s white blood cell and platelet counts, creatinine level, and liver function tests were within reference range. Cryoglobulins, coagulation studies, and cardiolipin antibodies were negative. Chest and abdominal imaging also were negative. An incisional skin biopsy and skin punch biopsy showed thrombotic coagulopathy and dilated vessels. A bone marrow biopsy revealed a hypercellular marrow but no plasma cell neoplasm. A repeat incisional skin biopsy demonstrated large CD20+ and CD45+ atypical lymphocytes within the small capillaries of the deep dermis and subcutaneous fat (Figure 2), which confirmed ILCL. Too deconditioned to tolerate chemotherapy, the patient opted for palliative care and died 18 months after initial presentation.
The diagnosis of ILCL often is delayed for several reasons.2 Patients can present with a variety of signs and symptoms related to small vessel occlusion that can be misattributed to other conditions.3,4 In our case, the patient’s recurrent infarcts were thought to be due to his poorly controlled diabetes mellitus, which was diagnosed a few weeks prior, and a positive ANA, even though the workup for antiphospholipid syndrome was negative. Interestingly, a positive ANA (without signs or symptoms of lupus or other autoimmune conditions) has been reported in patients with lymphoma.3 A positive antineutrophil cytoplasmic antibody level (without symptoms or other signs of vasculitis) has been reported in patients with ILCL.4,5 Therefore, distractors are common.
Multiple incisional skin biopsies in the absence of clinical findings (ie, random skin biopsy) are moderately sensitive (77.8%) for the diagnosis of ILCL.2 In a study by Matsue et al,2 111 suspected cases of ILCL underwent 3 incisional biopsies of fat-containing areas of the skin, such as the thigh, abdomen, and upper arm. Intravascular large cell lymphoma was confirmed in 26 cases. Seven additional cases were diagnosed as ILCL, 2 by additional skin biopsies (1 by a second round and 1 by a third round) and 5 by internal organ biopsy (4 bone marrow and 1 adrenal gland). The remaining cases ultimately were found to be a diagnostic mimicker of ILCL, including non-ILCL.2 Although random skin biopsies are reasonably sensitive for ILCL, multiple biopsies are needed, and in some cases, sampling of an internal organ may be required to establish the diagnosis of ILCL.
The prognosis of ILCL is poor; the 3-year overall survival rate for classic and cutaneous variants is 22% and 56%, respectively.6 Anthracycline-based chemotherapy, such as CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone), is considered first-line treatment, and the addition of rituximab to the CHOP regimen may improve remission rates and survival.7
To the Editor:
Intravascular large cell lymphoma (ILCL) is a rare B-cell lymphoma that is defined by the presence of large neoplastic B cells in the lumen of blood vessels.1 At least 3 variants of ILCL have been described based on case reports and a small case series: classic, cutaneous, and hemophagocytic. The classic variant presents in elderly patients as nonspecific constitutional symptoms (fever or pain, or less frequently weight loss) or as signs of multiorgan failure (most commonly of the central nervous system). Skin involvement, which is present in nearly half of these patients, can take on multiple morphologies, including retiform purpura, ulcerated nodules, or pseudocellulitis. The cutaneous variant typically presents in middle-aged women with normal hematologic studies. Systemic involvement is less common in this variant of disease than the classic variant, which may partly explain why overall survival is superior in this variant. The hemophagocytic variant manifests as intravascular lymphoma accompanied by hemophagocytic syndrome (fever, hepatosplenomegaly, thrombocytopenia, and bone marrow involvement).
A 69-year-old man presented to the emergency department for failure to thrive and nonhealing wounds of 1 year’s duration. His medical history was notable for poorly controlled diabetes mellitus, progressive multifocal ischemic and hemorrhagic cerebral infarcts, and bilateral deep venous thromboses. Physical examination revealed large purpuric to brown plaques in a retiform configuration with central necrotic eschars on the thighs and abdomen (Figure 1). There was no palpable lymphadenopathy. Laboratory tests revealed normocytic anemia with a hemoglobin level of 10.5 g/dL (reference range, 12–18 g/dL), elevated lactate dehydrogenase level of 525 U/L (reference range, 118–242 U/L), elevated erythrocyte sedimentation rate of 73 mm/h (reference range, <20 mm/h), antinuclear antibody (ANA) titer of 1:2560 (reference range, <1:80), and polyclonal hypergammaglobulinemia. The patient’s white blood cell and platelet counts, creatinine level, and liver function tests were within reference range. Cryoglobulins, coagulation studies, and cardiolipin antibodies were negative. Chest and abdominal imaging also were negative. An incisional skin biopsy and skin punch biopsy showed thrombotic coagulopathy and dilated vessels. A bone marrow biopsy revealed a hypercellular marrow but no plasma cell neoplasm. A repeat incisional skin biopsy demonstrated large CD20+ and CD45+ atypical lymphocytes within the small capillaries of the deep dermis and subcutaneous fat (Figure 2), which confirmed ILCL. Too deconditioned to tolerate chemotherapy, the patient opted for palliative care and died 18 months after initial presentation.
The diagnosis of ILCL often is delayed for several reasons.2 Patients can present with a variety of signs and symptoms related to small vessel occlusion that can be misattributed to other conditions.3,4 In our case, the patient’s recurrent infarcts were thought to be due to his poorly controlled diabetes mellitus, which was diagnosed a few weeks prior, and a positive ANA, even though the workup for antiphospholipid syndrome was negative. Interestingly, a positive ANA (without signs or symptoms of lupus or other autoimmune conditions) has been reported in patients with lymphoma.3 A positive antineutrophil cytoplasmic antibody level (without symptoms or other signs of vasculitis) has been reported in patients with ILCL.4,5 Therefore, distractors are common.
Multiple incisional skin biopsies in the absence of clinical findings (ie, random skin biopsy) are moderately sensitive (77.8%) for the diagnosis of ILCL.2 In a study by Matsue et al,2 111 suspected cases of ILCL underwent 3 incisional biopsies of fat-containing areas of the skin, such as the thigh, abdomen, and upper arm. Intravascular large cell lymphoma was confirmed in 26 cases. Seven additional cases were diagnosed as ILCL, 2 by additional skin biopsies (1 by a second round and 1 by a third round) and 5 by internal organ biopsy (4 bone marrow and 1 adrenal gland). The remaining cases ultimately were found to be a diagnostic mimicker of ILCL, including non-ILCL.2 Although random skin biopsies are reasonably sensitive for ILCL, multiple biopsies are needed, and in some cases, sampling of an internal organ may be required to establish the diagnosis of ILCL.
The prognosis of ILCL is poor; the 3-year overall survival rate for classic and cutaneous variants is 22% and 56%, respectively.6 Anthracycline-based chemotherapy, such as CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone), is considered first-line treatment, and the addition of rituximab to the CHOP regimen may improve remission rates and survival.7
- Ponzoni M, Campo E, Nakamura S. Intravascular large B-cell lymphoma: a chameleon with multiple faces and many masks [published online August 15, 2018]. Blood. 2018;132:1561-1567. doi:10.1182/blood-2017-04-737445
- Matsue K, Abe Y, Kitadate A, et al. Sensitivity and specificity of incisional random skin biopsy for diagnosis of intravascular large B-cell lymphoma. Blood. 2019;133:1257-1259.
- Altintas A, Cil T, Pasa S, et al. Clinical significance of elevated antinuclear antibody test in patients with Hodgkin’s and non-Hodgkin’s lymphoma. Minerva Med. 2008;99:7-14.
- Shinkawa Y, Hatachi S, Yagita M. Intravascular large B-cell lymphoma with a high titer of proteinase-3-anti-neutrophil cytoplasmic antibody mimicking granulomatosis with polyangiitis. Mod Rheumatol. 2019;29:195-197.
- Sugiyama A, Kobayashi M, Daizo A, et al. Diffuse cerebral vasoconstriction in a intravascular lymphoma patient with a high serum MPO-ANCA level. Intern Med. 2017;56:1715-1718.
- Ferreri AJ, Campo E, Seymour JF, et al. Intravascular lymphoma: clinical presentation, natural history, management and prognostic factors in a series of 38 cases, with special emphasis on the ‘cutaneous variant.’ Br J Haematol. 2004;127:173-183.
- Ferreri AJM, Dognini GP, Bairey O, et al; International Extranodal Lyphoma Study Group. The addition of rituximab to anthracycline-based chemotherapy significantly improves outcome in ‘Western’ patients with intravascular large B-cell lymphoma [published online August 10, 2008]. Br J Haematol. 2008;143:253-257. doi:10.1111/j.1365-2141.2008.07338.x
- Ponzoni M, Campo E, Nakamura S. Intravascular large B-cell lymphoma: a chameleon with multiple faces and many masks [published online August 15, 2018]. Blood. 2018;132:1561-1567. doi:10.1182/blood-2017-04-737445
- Matsue K, Abe Y, Kitadate A, et al. Sensitivity and specificity of incisional random skin biopsy for diagnosis of intravascular large B-cell lymphoma. Blood. 2019;133:1257-1259.
- Altintas A, Cil T, Pasa S, et al. Clinical significance of elevated antinuclear antibody test in patients with Hodgkin’s and non-Hodgkin’s lymphoma. Minerva Med. 2008;99:7-14.
- Shinkawa Y, Hatachi S, Yagita M. Intravascular large B-cell lymphoma with a high titer of proteinase-3-anti-neutrophil cytoplasmic antibody mimicking granulomatosis with polyangiitis. Mod Rheumatol. 2019;29:195-197.
- Sugiyama A, Kobayashi M, Daizo A, et al. Diffuse cerebral vasoconstriction in a intravascular lymphoma patient with a high serum MPO-ANCA level. Intern Med. 2017;56:1715-1718.
- Ferreri AJ, Campo E, Seymour JF, et al. Intravascular lymphoma: clinical presentation, natural history, management and prognostic factors in a series of 38 cases, with special emphasis on the ‘cutaneous variant.’ Br J Haematol. 2004;127:173-183.
- Ferreri AJM, Dognini GP, Bairey O, et al; International Extranodal Lyphoma Study Group. The addition of rituximab to anthracycline-based chemotherapy significantly improves outcome in ‘Western’ patients with intravascular large B-cell lymphoma [published online August 10, 2008]. Br J Haematol. 2008;143:253-257. doi:10.1111/j.1365-2141.2008.07338.x
Practice Points
- Intravascular large cell lymphoma (ILCL) is a life-threatening malignancy that can present with retiform purpura and other symptoms of vascular occlusion.
- The diagnosis of ILCL can be challenging because of the presence of distractors, and multiple biopsies may be required to establish pathology.
