User login
People with cancer should be wary of taking dietary supplements
Cancer dietitian Lisa Cianciotta often finds herself sitting across from a patient who suddenly fishes a bottle of antioxidant supplements from their bag and says, “My friend told me this works really well,” or “I read on the Internet that this is supposed to be really good for cancer.”
Although taking an antioxidant pill sounds harmless, Ms. Cianciotta, a clinical dietitian who works with cancer patients at New York–Presbyterian Hospital, knows well that this popular dietary supplement can interfere with a patient’s radiation or chemotherapy.
But many patients with cancer believe these over-the-counter vitamins, minerals, or herbal remedies will help them, and most use at least one dietary supplement alongside their cancer treatment.
And that leaves Ms. Cianciotta with a delicate conversation ahead of her.
. Popular dietary supplements may, for instance, cancel the effects of a cancer treatment, making it less effective, or increase serious side effects, such as liver toxicity. But in other cases, supplementation, such as vitamin D for patients who lack the vitamin, may be beneficial, Ms. Cianciotta said.
These drug-supplement interactions can be hard to pinpoint, given that more than two-thirds of doctors don’t know their patients are using supplements.
Here’s what patients need to know about the potential risks of supplement use during treatment, and how oncologists can address this thorny, often poorly understood topic with patients.
The complex drug-supplement landscape
The list of dietary supplements and how they can interact with different treatments and cancer types is long and nuanced.
But certain supplements appear to affect cancer treatments regardless of other things and should be avoided. Any supplement that strongly alters the body’s levels of the protein cytochromes P450 is one example. This group of enzymes plays a key role in metabolizing drugs, including chemotherapy and immunotherapy agents.
Certain supplements – most notably St. John’s wort extract – may decrease or increase the activity of cytochrome P450, which can then affect the concentrations of anticancer drugs in the blood, said William Figg, PharmD, an associate director of the Center for Cancer Research at the National Cancer Institute in Bethesda, Md. Studies show, for instance, that this common herbal supplement can increase the activity of cytochrome P450, resulting in lower levels of cancer drugs.
Outside of drug metabolism, patients with hormone-related cancers, such as breast and prostate cancers, should steer clear of dietary supplements that can alter levels of testosterone or estrogen, Dr. Figg said. The evergreen shrub ashwagandha, for example, is marketed to reduce stress and fatigue, but can also increase testosterone levels – a potential problem for those with prostate cancer receiving androgen deprivation therapy, which lowers testosterone levels.
Many oncologists counsel patients against using antioxidant-based dietary supplements – particularly turmeric and green tea extract – while they have radiation therapy and certain chemotherapies. These therapies work by creating an abundance of highly reactive molecules called free radicals in tumor cells, which increase stress within these cells, ultimately killing them off. Antioxidants, in theory, can neutralize this effect, said Skyler Johnson, MD, a radiation oncologist at Huntsman Cancer Institute at the University of Utah, Salt Lake City. Some studies suggest that antioxidant supplements may lessen the effects of radiation and chemotherapy, although the evidence is mixed.
Some dietary supplements, including high-dose green tea extract and vitamin A, can cause kidney or liver toxicity, and “many cancer patients already have compromised kidney or liver function,” said Jun J. Mao, MD, chief of integrative medicine at Memorial Sloan Kettering Cancer Center in New York. Even herbs that don’t interfere with how well a cancer drug works, such as stevia, can increase treatment-related side effects, such as nausea and vomiting.
Another potential problem with dietary supplements: It’s nearly impossible to know exactly what’s in them. For instance, just last year, the Food and Drug Administration sent nearly 50 warning letters to companies marketing dietary supplements. The issue is that federal regulations governing production are less strict for supplements than for medications. As a result, some supplements contain ingredients not listed on the label.
One historical example was the supplement PC-SPES, a mix of eight herbs, marketed to men with prostate cancer. The supplement was recalled in 2002 after certain batches were found to contain traces of prescription drugs, including diethylstilbestrol, ethinyl estradiol, warfarin, and alprazolam.
To further complicate matters, some dietary supplements can be helpful. Most patients with cancer “are malnourished and missing out on nutrients they could be getting from food,” said Ms. Cianciotta.
Patients are tested routinely for vitamin deficiencies and receive supplements as needed, she said. Vitamin D and folic acid are two of the most common deficiencies in this patient population. Vitamin D supplementation can improve outcomes in patients who have received a stem cell transplant by aiding engraftment and rebuilding the immune system, while folic acid supplementation can help to raise low red blood cell counts and hemoglobin levels.
Although she rarely sees vitamin toxicity, Ms. Cianciotta stressed that more is not always better and supplement use, even when it seems safe or warranted due to a deficiency, should be taken under supervision, and monitored carefully by the patient’s care team.
Bringing supplement use into the light
Too often, providers are unaware of a patient’s supplement use.
A core reason: Dietary supplements are often touted as natural, which many patients equate with safety, said Samantha Heller, a senior clinical nutritionist at New York (N.Y.) University Langone Health.
That means patients may not know a supplement can act like a drug and interfere with their cancer treatment, and thus may not see the importance of telling their doctors.
Still, the promise of herbs, vitamins, and minerals can be alluring, and there are many reasons patients decide to partake. One major appeal: Dietary supplements can help some patients feel empowered.
“Cancer is a disease that takes away a lot of control from the individual. Taking supplements or herbs is a way to regain some sense of control,” said Dr. Mao.
The phenomenon can also be cultural, he said. Some people grow up taking herbs and supplements to stay healthy or combat health woes.
Pressure or advice from family or friends who may think they are helping a loved one with their dietary recommendations may play a role as well. Friends and family “cannot prescribe chemo, but they can buy herbs and supplements,” Dr. Mao said.
Patients seeking greater control over their health or who feel high levels of anxiety may be more likely to take suggestions from friends and family or more likely to believe false or misleading claims about the efficacy or safety of supplements, explained medical oncologist William Dahut, MD, chief scientific officer for the American Cancer Society.
Plus, social media often amplifies and normalizes this misinformation, noted Dr. Johnson. In a 2021 study published in the Journal of the National Cancer Institute, he and colleagues found that one-third of the most popular articles on cancer treatment posted to social media in 2018 and 2019 contained false, inaccurate, or misleading information that was often harmful.
Some of the false claims centered on unproven, potentially unsafe herbal remedies, according to Dr. Johnson. These included “lung cancer can be cured with cannabis oil” and “golden berries cure and prevent cancer.”
Given exaggerated claims of “cures,” some patients may keep their supplement use under the radar out of fear they will be judged or criticized.
“Clinicians should avoid making patients feel judged or telling people not to go online to do their own research,” Dr. Johnson said.
Guiding patients, instead, to accurate sources of online information may be one way to help patients feel empowered, he said. Cancer.gov and the Memorial Sloan Kettering Cancer Center’s About Herbs database provide accessible and accurate information on dietary supplements and cancer treatment for both health care professionals and patients, he noted.
If a particular supplement is not safe during treatment, providers should be able to explain why, said Ms. Cianciotta. In a recent study, 80% of health care providers surveyed believed that interactions between herbals and medications could be problematic, but only 15% could explain why.
“Being able to explain why we are discouraging a particular supplement right now tends to be much better received than just telling a patient not to take something, because it is bad,” she said.
Another key is listening closely to patients to understand why they may be taking a particular supplement. Does the patient feel out of control? Is nausea a problem?
“Allowing patients to tell you why they are using a particular supplement will often reveal unmet needs or psychosocial challenges,” Dr. Mao said. This information can allow providers to suggest an evidence-based alternative, such as mindfulness meditation or acupuncture, to manage stress.
And if a patient has received a dietary supplement from well-meaning family and friends?
“Simply telling a patient that a given supplement is useless or harmful could create family tension,” said Dr. Mao.
Instead, he recommends reframing the issue.
“We want to have a better understanding of how patients are tolerating chemo or immunotherapy before throwing other things on top of it. Let them know that now may just not be the right time to add a supplement to the mix,” Dr. Mao said.
The bottom line: “Patients want to play an active role in their own care, and we want to help them do that in a safe way,” he said.
A version of this article first appeared on WebMD.com.
Cancer dietitian Lisa Cianciotta often finds herself sitting across from a patient who suddenly fishes a bottle of antioxidant supplements from their bag and says, “My friend told me this works really well,” or “I read on the Internet that this is supposed to be really good for cancer.”
Although taking an antioxidant pill sounds harmless, Ms. Cianciotta, a clinical dietitian who works with cancer patients at New York–Presbyterian Hospital, knows well that this popular dietary supplement can interfere with a patient’s radiation or chemotherapy.
But many patients with cancer believe these over-the-counter vitamins, minerals, or herbal remedies will help them, and most use at least one dietary supplement alongside their cancer treatment.
And that leaves Ms. Cianciotta with a delicate conversation ahead of her.
. Popular dietary supplements may, for instance, cancel the effects of a cancer treatment, making it less effective, or increase serious side effects, such as liver toxicity. But in other cases, supplementation, such as vitamin D for patients who lack the vitamin, may be beneficial, Ms. Cianciotta said.
These drug-supplement interactions can be hard to pinpoint, given that more than two-thirds of doctors don’t know their patients are using supplements.
Here’s what patients need to know about the potential risks of supplement use during treatment, and how oncologists can address this thorny, often poorly understood topic with patients.
The complex drug-supplement landscape
The list of dietary supplements and how they can interact with different treatments and cancer types is long and nuanced.
But certain supplements appear to affect cancer treatments regardless of other things and should be avoided. Any supplement that strongly alters the body’s levels of the protein cytochromes P450 is one example. This group of enzymes plays a key role in metabolizing drugs, including chemotherapy and immunotherapy agents.
Certain supplements – most notably St. John’s wort extract – may decrease or increase the activity of cytochrome P450, which can then affect the concentrations of anticancer drugs in the blood, said William Figg, PharmD, an associate director of the Center for Cancer Research at the National Cancer Institute in Bethesda, Md. Studies show, for instance, that this common herbal supplement can increase the activity of cytochrome P450, resulting in lower levels of cancer drugs.
Outside of drug metabolism, patients with hormone-related cancers, such as breast and prostate cancers, should steer clear of dietary supplements that can alter levels of testosterone or estrogen, Dr. Figg said. The evergreen shrub ashwagandha, for example, is marketed to reduce stress and fatigue, but can also increase testosterone levels – a potential problem for those with prostate cancer receiving androgen deprivation therapy, which lowers testosterone levels.
Many oncologists counsel patients against using antioxidant-based dietary supplements – particularly turmeric and green tea extract – while they have radiation therapy and certain chemotherapies. These therapies work by creating an abundance of highly reactive molecules called free radicals in tumor cells, which increase stress within these cells, ultimately killing them off. Antioxidants, in theory, can neutralize this effect, said Skyler Johnson, MD, a radiation oncologist at Huntsman Cancer Institute at the University of Utah, Salt Lake City. Some studies suggest that antioxidant supplements may lessen the effects of radiation and chemotherapy, although the evidence is mixed.
Some dietary supplements, including high-dose green tea extract and vitamin A, can cause kidney or liver toxicity, and “many cancer patients already have compromised kidney or liver function,” said Jun J. Mao, MD, chief of integrative medicine at Memorial Sloan Kettering Cancer Center in New York. Even herbs that don’t interfere with how well a cancer drug works, such as stevia, can increase treatment-related side effects, such as nausea and vomiting.
Another potential problem with dietary supplements: It’s nearly impossible to know exactly what’s in them. For instance, just last year, the Food and Drug Administration sent nearly 50 warning letters to companies marketing dietary supplements. The issue is that federal regulations governing production are less strict for supplements than for medications. As a result, some supplements contain ingredients not listed on the label.
One historical example was the supplement PC-SPES, a mix of eight herbs, marketed to men with prostate cancer. The supplement was recalled in 2002 after certain batches were found to contain traces of prescription drugs, including diethylstilbestrol, ethinyl estradiol, warfarin, and alprazolam.
To further complicate matters, some dietary supplements can be helpful. Most patients with cancer “are malnourished and missing out on nutrients they could be getting from food,” said Ms. Cianciotta.
Patients are tested routinely for vitamin deficiencies and receive supplements as needed, she said. Vitamin D and folic acid are two of the most common deficiencies in this patient population. Vitamin D supplementation can improve outcomes in patients who have received a stem cell transplant by aiding engraftment and rebuilding the immune system, while folic acid supplementation can help to raise low red blood cell counts and hemoglobin levels.
Although she rarely sees vitamin toxicity, Ms. Cianciotta stressed that more is not always better and supplement use, even when it seems safe or warranted due to a deficiency, should be taken under supervision, and monitored carefully by the patient’s care team.
Bringing supplement use into the light
Too often, providers are unaware of a patient’s supplement use.
A core reason: Dietary supplements are often touted as natural, which many patients equate with safety, said Samantha Heller, a senior clinical nutritionist at New York (N.Y.) University Langone Health.
That means patients may not know a supplement can act like a drug and interfere with their cancer treatment, and thus may not see the importance of telling their doctors.
Still, the promise of herbs, vitamins, and minerals can be alluring, and there are many reasons patients decide to partake. One major appeal: Dietary supplements can help some patients feel empowered.
“Cancer is a disease that takes away a lot of control from the individual. Taking supplements or herbs is a way to regain some sense of control,” said Dr. Mao.
The phenomenon can also be cultural, he said. Some people grow up taking herbs and supplements to stay healthy or combat health woes.
Pressure or advice from family or friends who may think they are helping a loved one with their dietary recommendations may play a role as well. Friends and family “cannot prescribe chemo, but they can buy herbs and supplements,” Dr. Mao said.
Patients seeking greater control over their health or who feel high levels of anxiety may be more likely to take suggestions from friends and family or more likely to believe false or misleading claims about the efficacy or safety of supplements, explained medical oncologist William Dahut, MD, chief scientific officer for the American Cancer Society.
Plus, social media often amplifies and normalizes this misinformation, noted Dr. Johnson. In a 2021 study published in the Journal of the National Cancer Institute, he and colleagues found that one-third of the most popular articles on cancer treatment posted to social media in 2018 and 2019 contained false, inaccurate, or misleading information that was often harmful.
Some of the false claims centered on unproven, potentially unsafe herbal remedies, according to Dr. Johnson. These included “lung cancer can be cured with cannabis oil” and “golden berries cure and prevent cancer.”
Given exaggerated claims of “cures,” some patients may keep their supplement use under the radar out of fear they will be judged or criticized.
“Clinicians should avoid making patients feel judged or telling people not to go online to do their own research,” Dr. Johnson said.
Guiding patients, instead, to accurate sources of online information may be one way to help patients feel empowered, he said. Cancer.gov and the Memorial Sloan Kettering Cancer Center’s About Herbs database provide accessible and accurate information on dietary supplements and cancer treatment for both health care professionals and patients, he noted.
If a particular supplement is not safe during treatment, providers should be able to explain why, said Ms. Cianciotta. In a recent study, 80% of health care providers surveyed believed that interactions between herbals and medications could be problematic, but only 15% could explain why.
“Being able to explain why we are discouraging a particular supplement right now tends to be much better received than just telling a patient not to take something, because it is bad,” she said.
Another key is listening closely to patients to understand why they may be taking a particular supplement. Does the patient feel out of control? Is nausea a problem?
“Allowing patients to tell you why they are using a particular supplement will often reveal unmet needs or psychosocial challenges,” Dr. Mao said. This information can allow providers to suggest an evidence-based alternative, such as mindfulness meditation or acupuncture, to manage stress.
And if a patient has received a dietary supplement from well-meaning family and friends?
“Simply telling a patient that a given supplement is useless or harmful could create family tension,” said Dr. Mao.
Instead, he recommends reframing the issue.
“We want to have a better understanding of how patients are tolerating chemo or immunotherapy before throwing other things on top of it. Let them know that now may just not be the right time to add a supplement to the mix,” Dr. Mao said.
The bottom line: “Patients want to play an active role in their own care, and we want to help them do that in a safe way,” he said.
A version of this article first appeared on WebMD.com.
Cancer dietitian Lisa Cianciotta often finds herself sitting across from a patient who suddenly fishes a bottle of antioxidant supplements from their bag and says, “My friend told me this works really well,” or “I read on the Internet that this is supposed to be really good for cancer.”
Although taking an antioxidant pill sounds harmless, Ms. Cianciotta, a clinical dietitian who works with cancer patients at New York–Presbyterian Hospital, knows well that this popular dietary supplement can interfere with a patient’s radiation or chemotherapy.
But many patients with cancer believe these over-the-counter vitamins, minerals, or herbal remedies will help them, and most use at least one dietary supplement alongside their cancer treatment.
And that leaves Ms. Cianciotta with a delicate conversation ahead of her.
. Popular dietary supplements may, for instance, cancel the effects of a cancer treatment, making it less effective, or increase serious side effects, such as liver toxicity. But in other cases, supplementation, such as vitamin D for patients who lack the vitamin, may be beneficial, Ms. Cianciotta said.
These drug-supplement interactions can be hard to pinpoint, given that more than two-thirds of doctors don’t know their patients are using supplements.
Here’s what patients need to know about the potential risks of supplement use during treatment, and how oncologists can address this thorny, often poorly understood topic with patients.
The complex drug-supplement landscape
The list of dietary supplements and how they can interact with different treatments and cancer types is long and nuanced.
But certain supplements appear to affect cancer treatments regardless of other things and should be avoided. Any supplement that strongly alters the body’s levels of the protein cytochromes P450 is one example. This group of enzymes plays a key role in metabolizing drugs, including chemotherapy and immunotherapy agents.
Certain supplements – most notably St. John’s wort extract – may decrease or increase the activity of cytochrome P450, which can then affect the concentrations of anticancer drugs in the blood, said William Figg, PharmD, an associate director of the Center for Cancer Research at the National Cancer Institute in Bethesda, Md. Studies show, for instance, that this common herbal supplement can increase the activity of cytochrome P450, resulting in lower levels of cancer drugs.
Outside of drug metabolism, patients with hormone-related cancers, such as breast and prostate cancers, should steer clear of dietary supplements that can alter levels of testosterone or estrogen, Dr. Figg said. The evergreen shrub ashwagandha, for example, is marketed to reduce stress and fatigue, but can also increase testosterone levels – a potential problem for those with prostate cancer receiving androgen deprivation therapy, which lowers testosterone levels.
Many oncologists counsel patients against using antioxidant-based dietary supplements – particularly turmeric and green tea extract – while they have radiation therapy and certain chemotherapies. These therapies work by creating an abundance of highly reactive molecules called free radicals in tumor cells, which increase stress within these cells, ultimately killing them off. Antioxidants, in theory, can neutralize this effect, said Skyler Johnson, MD, a radiation oncologist at Huntsman Cancer Institute at the University of Utah, Salt Lake City. Some studies suggest that antioxidant supplements may lessen the effects of radiation and chemotherapy, although the evidence is mixed.
Some dietary supplements, including high-dose green tea extract and vitamin A, can cause kidney or liver toxicity, and “many cancer patients already have compromised kidney or liver function,” said Jun J. Mao, MD, chief of integrative medicine at Memorial Sloan Kettering Cancer Center in New York. Even herbs that don’t interfere with how well a cancer drug works, such as stevia, can increase treatment-related side effects, such as nausea and vomiting.
Another potential problem with dietary supplements: It’s nearly impossible to know exactly what’s in them. For instance, just last year, the Food and Drug Administration sent nearly 50 warning letters to companies marketing dietary supplements. The issue is that federal regulations governing production are less strict for supplements than for medications. As a result, some supplements contain ingredients not listed on the label.
One historical example was the supplement PC-SPES, a mix of eight herbs, marketed to men with prostate cancer. The supplement was recalled in 2002 after certain batches were found to contain traces of prescription drugs, including diethylstilbestrol, ethinyl estradiol, warfarin, and alprazolam.
To further complicate matters, some dietary supplements can be helpful. Most patients with cancer “are malnourished and missing out on nutrients they could be getting from food,” said Ms. Cianciotta.
Patients are tested routinely for vitamin deficiencies and receive supplements as needed, she said. Vitamin D and folic acid are two of the most common deficiencies in this patient population. Vitamin D supplementation can improve outcomes in patients who have received a stem cell transplant by aiding engraftment and rebuilding the immune system, while folic acid supplementation can help to raise low red blood cell counts and hemoglobin levels.
Although she rarely sees vitamin toxicity, Ms. Cianciotta stressed that more is not always better and supplement use, even when it seems safe or warranted due to a deficiency, should be taken under supervision, and monitored carefully by the patient’s care team.
Bringing supplement use into the light
Too often, providers are unaware of a patient’s supplement use.
A core reason: Dietary supplements are often touted as natural, which many patients equate with safety, said Samantha Heller, a senior clinical nutritionist at New York (N.Y.) University Langone Health.
That means patients may not know a supplement can act like a drug and interfere with their cancer treatment, and thus may not see the importance of telling their doctors.
Still, the promise of herbs, vitamins, and minerals can be alluring, and there are many reasons patients decide to partake. One major appeal: Dietary supplements can help some patients feel empowered.
“Cancer is a disease that takes away a lot of control from the individual. Taking supplements or herbs is a way to regain some sense of control,” said Dr. Mao.
The phenomenon can also be cultural, he said. Some people grow up taking herbs and supplements to stay healthy or combat health woes.
Pressure or advice from family or friends who may think they are helping a loved one with their dietary recommendations may play a role as well. Friends and family “cannot prescribe chemo, but they can buy herbs and supplements,” Dr. Mao said.
Patients seeking greater control over their health or who feel high levels of anxiety may be more likely to take suggestions from friends and family or more likely to believe false or misleading claims about the efficacy or safety of supplements, explained medical oncologist William Dahut, MD, chief scientific officer for the American Cancer Society.
Plus, social media often amplifies and normalizes this misinformation, noted Dr. Johnson. In a 2021 study published in the Journal of the National Cancer Institute, he and colleagues found that one-third of the most popular articles on cancer treatment posted to social media in 2018 and 2019 contained false, inaccurate, or misleading information that was often harmful.
Some of the false claims centered on unproven, potentially unsafe herbal remedies, according to Dr. Johnson. These included “lung cancer can be cured with cannabis oil” and “golden berries cure and prevent cancer.”
Given exaggerated claims of “cures,” some patients may keep their supplement use under the radar out of fear they will be judged or criticized.
“Clinicians should avoid making patients feel judged or telling people not to go online to do their own research,” Dr. Johnson said.
Guiding patients, instead, to accurate sources of online information may be one way to help patients feel empowered, he said. Cancer.gov and the Memorial Sloan Kettering Cancer Center’s About Herbs database provide accessible and accurate information on dietary supplements and cancer treatment for both health care professionals and patients, he noted.
If a particular supplement is not safe during treatment, providers should be able to explain why, said Ms. Cianciotta. In a recent study, 80% of health care providers surveyed believed that interactions between herbals and medications could be problematic, but only 15% could explain why.
“Being able to explain why we are discouraging a particular supplement right now tends to be much better received than just telling a patient not to take something, because it is bad,” she said.
Another key is listening closely to patients to understand why they may be taking a particular supplement. Does the patient feel out of control? Is nausea a problem?
“Allowing patients to tell you why they are using a particular supplement will often reveal unmet needs or psychosocial challenges,” Dr. Mao said. This information can allow providers to suggest an evidence-based alternative, such as mindfulness meditation or acupuncture, to manage stress.
And if a patient has received a dietary supplement from well-meaning family and friends?
“Simply telling a patient that a given supplement is useless or harmful could create family tension,” said Dr. Mao.
Instead, he recommends reframing the issue.
“We want to have a better understanding of how patients are tolerating chemo or immunotherapy before throwing other things on top of it. Let them know that now may just not be the right time to add a supplement to the mix,” Dr. Mao said.
The bottom line: “Patients want to play an active role in their own care, and we want to help them do that in a safe way,” he said.
A version of this article first appeared on WebMD.com.
Tucatinib plus trastuzumab approved for HER2+ colorectal cancer
The U.S. Food and Drug Administration has granted accelerated approval to tucatinib (Tukysa) in combination with trastuzumab for use in RAS wild-type, HER2-positive unresectable or metastatic colorectal cancer that has progressed after fluoropyrimidine, oxaliplatin, and irinotecan-based chemotherapy.
This is the first FDA-approved treatment for HER2-positive metastatic colorectal cancer, maker Seagen said in a Jan. 19 press release.
“Historically, patients with HER2-positive metastatic colorectal cancer who have progressed following frontline therapy have had poor outcomes. The FDA approval of a chemotherapy-free combination regimen that specifically targets HER2 is great news for these patients,” John Strickler, MD, associate professor of medicine at Duke University Medical Center, Durham, N.C., said in the press release.
Dr. Strickler was the lead investigator on the approval trial, dubbed MOUNTAINEER, which involved 84 patients who met the treatment criteria and who had also been treated with an anti-VEGF antibody. Participants whose tumors were deficient in mismatch repair proteins or were microsatellite instability–high must also have received a PD-1 inhibitor. Patients who received prior anti-HER2 therapy were excluded, the FDA explained in its own press release.
Participants were treated with tucatinib 300 mg orally twice daily– the recommended dose in product labeling – with trastuzumab administered at a loading dose of 8 mg/kg intravenously on day 1 of cycle 1 followed by a maintenance dose of trastuzumab 6 mg/kg on day 1 of each subsequent 21-day cycle.
Overall response rate was 38%, and median duration of response was 12.4 months.
The most common adverse events, occurring in at least 20% of study participants, were diarrhea, fatigue, rash, nausea, abdominal pain, infusion related reactions, and pyrexia. The most common laboratory abnormalities were increased creatinine, decreased lymphocytes, increased alanine aminotransferase, and decreased hemoglobin, among others.
Serious adverse reactions occurred in 22% of patients. The most common (occurring in ≥ 2% of patients) were intestinal obstruction (7%); urinary tract infection (3.5%); and pneumonia, abdominal pain, and rectal perforation (2.3% each). Adverse reactions leading to permanent discontinuation occurred in 6% of patients, including increased alanine aminotransferase in 2.3%.
Continued approval for the indication may be contingent upon verification and description of clinical benefit in confirmatory trials, the company said.
A global, randomized phase 3 clinical trial (MOUNTAINEER-03) is ongoing and is comparing tucatinib in combination with trastuzumab and mFOLFOX6 with standard of care and is intended to serve as a confirmatory trial, the company said.
Tucatinib is already approved in combination with trastuzumab and capecitabine for use in the treatment of advanced unresectable or metastatic HER2-positive breast cancer.
A version of this article first appeared on Medscape.com.
The U.S. Food and Drug Administration has granted accelerated approval to tucatinib (Tukysa) in combination with trastuzumab for use in RAS wild-type, HER2-positive unresectable or metastatic colorectal cancer that has progressed after fluoropyrimidine, oxaliplatin, and irinotecan-based chemotherapy.
This is the first FDA-approved treatment for HER2-positive metastatic colorectal cancer, maker Seagen said in a Jan. 19 press release.
“Historically, patients with HER2-positive metastatic colorectal cancer who have progressed following frontline therapy have had poor outcomes. The FDA approval of a chemotherapy-free combination regimen that specifically targets HER2 is great news for these patients,” John Strickler, MD, associate professor of medicine at Duke University Medical Center, Durham, N.C., said in the press release.
Dr. Strickler was the lead investigator on the approval trial, dubbed MOUNTAINEER, which involved 84 patients who met the treatment criteria and who had also been treated with an anti-VEGF antibody. Participants whose tumors were deficient in mismatch repair proteins or were microsatellite instability–high must also have received a PD-1 inhibitor. Patients who received prior anti-HER2 therapy were excluded, the FDA explained in its own press release.
Participants were treated with tucatinib 300 mg orally twice daily– the recommended dose in product labeling – with trastuzumab administered at a loading dose of 8 mg/kg intravenously on day 1 of cycle 1 followed by a maintenance dose of trastuzumab 6 mg/kg on day 1 of each subsequent 21-day cycle.
Overall response rate was 38%, and median duration of response was 12.4 months.
The most common adverse events, occurring in at least 20% of study participants, were diarrhea, fatigue, rash, nausea, abdominal pain, infusion related reactions, and pyrexia. The most common laboratory abnormalities were increased creatinine, decreased lymphocytes, increased alanine aminotransferase, and decreased hemoglobin, among others.
Serious adverse reactions occurred in 22% of patients. The most common (occurring in ≥ 2% of patients) were intestinal obstruction (7%); urinary tract infection (3.5%); and pneumonia, abdominal pain, and rectal perforation (2.3% each). Adverse reactions leading to permanent discontinuation occurred in 6% of patients, including increased alanine aminotransferase in 2.3%.
Continued approval for the indication may be contingent upon verification and description of clinical benefit in confirmatory trials, the company said.
A global, randomized phase 3 clinical trial (MOUNTAINEER-03) is ongoing and is comparing tucatinib in combination with trastuzumab and mFOLFOX6 with standard of care and is intended to serve as a confirmatory trial, the company said.
Tucatinib is already approved in combination with trastuzumab and capecitabine for use in the treatment of advanced unresectable or metastatic HER2-positive breast cancer.
A version of this article first appeared on Medscape.com.
The U.S. Food and Drug Administration has granted accelerated approval to tucatinib (Tukysa) in combination with trastuzumab for use in RAS wild-type, HER2-positive unresectable or metastatic colorectal cancer that has progressed after fluoropyrimidine, oxaliplatin, and irinotecan-based chemotherapy.
This is the first FDA-approved treatment for HER2-positive metastatic colorectal cancer, maker Seagen said in a Jan. 19 press release.
“Historically, patients with HER2-positive metastatic colorectal cancer who have progressed following frontline therapy have had poor outcomes. The FDA approval of a chemotherapy-free combination regimen that specifically targets HER2 is great news for these patients,” John Strickler, MD, associate professor of medicine at Duke University Medical Center, Durham, N.C., said in the press release.
Dr. Strickler was the lead investigator on the approval trial, dubbed MOUNTAINEER, which involved 84 patients who met the treatment criteria and who had also been treated with an anti-VEGF antibody. Participants whose tumors were deficient in mismatch repair proteins or were microsatellite instability–high must also have received a PD-1 inhibitor. Patients who received prior anti-HER2 therapy were excluded, the FDA explained in its own press release.
Participants were treated with tucatinib 300 mg orally twice daily– the recommended dose in product labeling – with trastuzumab administered at a loading dose of 8 mg/kg intravenously on day 1 of cycle 1 followed by a maintenance dose of trastuzumab 6 mg/kg on day 1 of each subsequent 21-day cycle.
Overall response rate was 38%, and median duration of response was 12.4 months.
The most common adverse events, occurring in at least 20% of study participants, were diarrhea, fatigue, rash, nausea, abdominal pain, infusion related reactions, and pyrexia. The most common laboratory abnormalities were increased creatinine, decreased lymphocytes, increased alanine aminotransferase, and decreased hemoglobin, among others.
Serious adverse reactions occurred in 22% of patients. The most common (occurring in ≥ 2% of patients) were intestinal obstruction (7%); urinary tract infection (3.5%); and pneumonia, abdominal pain, and rectal perforation (2.3% each). Adverse reactions leading to permanent discontinuation occurred in 6% of patients, including increased alanine aminotransferase in 2.3%.
Continued approval for the indication may be contingent upon verification and description of clinical benefit in confirmatory trials, the company said.
A global, randomized phase 3 clinical trial (MOUNTAINEER-03) is ongoing and is comparing tucatinib in combination with trastuzumab and mFOLFOX6 with standard of care and is intended to serve as a confirmatory trial, the company said.
Tucatinib is already approved in combination with trastuzumab and capecitabine for use in the treatment of advanced unresectable or metastatic HER2-positive breast cancer.
A version of this article first appeared on Medscape.com.
Highly anticipated HIV vaccine fails in large trial
officials announced Wednesday.
The vaccine had been in development since 2019 and was given to 3,900 study participants through October 2022, but data shows it does not protect against HIV compared with a placebo, according to developer Janssen Pharmaceutical.
Experts estimate the failure means there won’t be another potential vaccine on the horizon for 3 to 5 years, the New York Times reported.
“It’s obviously disappointing,” Anthony Fauci, MD, former head of the National Institute of Allergy and Infectious Diseases, told MSNBC, noting that other areas of HIV treatment research are promising. “I don’t think that people should give up on the field of the HIV vaccine.”
No safety issues had been identified with the vaccine during the trial, which studied the experimental treatment in men who have sex with men or with transgender people.
There is no cure for HIV, but disease progression can be managed with existing treatments. HIV attacks the body’s immune system and destroys white blood cells, increasing the risk of other infections. More than 1.5 million people worldwide were infected with HIV in 2021 and 38.4 million people are living with the virus, according to UNAIDS.
A version of this article first appeared on WebMD.com.
officials announced Wednesday.
The vaccine had been in development since 2019 and was given to 3,900 study participants through October 2022, but data shows it does not protect against HIV compared with a placebo, according to developer Janssen Pharmaceutical.
Experts estimate the failure means there won’t be another potential vaccine on the horizon for 3 to 5 years, the New York Times reported.
“It’s obviously disappointing,” Anthony Fauci, MD, former head of the National Institute of Allergy and Infectious Diseases, told MSNBC, noting that other areas of HIV treatment research are promising. “I don’t think that people should give up on the field of the HIV vaccine.”
No safety issues had been identified with the vaccine during the trial, which studied the experimental treatment in men who have sex with men or with transgender people.
There is no cure for HIV, but disease progression can be managed with existing treatments. HIV attacks the body’s immune system and destroys white blood cells, increasing the risk of other infections. More than 1.5 million people worldwide were infected with HIV in 2021 and 38.4 million people are living with the virus, according to UNAIDS.
A version of this article first appeared on WebMD.com.
officials announced Wednesday.
The vaccine had been in development since 2019 and was given to 3,900 study participants through October 2022, but data shows it does not protect against HIV compared with a placebo, according to developer Janssen Pharmaceutical.
Experts estimate the failure means there won’t be another potential vaccine on the horizon for 3 to 5 years, the New York Times reported.
“It’s obviously disappointing,” Anthony Fauci, MD, former head of the National Institute of Allergy and Infectious Diseases, told MSNBC, noting that other areas of HIV treatment research are promising. “I don’t think that people should give up on the field of the HIV vaccine.”
No safety issues had been identified with the vaccine during the trial, which studied the experimental treatment in men who have sex with men or with transgender people.
There is no cure for HIV, but disease progression can be managed with existing treatments. HIV attacks the body’s immune system and destroys white blood cells, increasing the risk of other infections. More than 1.5 million people worldwide were infected with HIV in 2021 and 38.4 million people are living with the virus, according to UNAIDS.
A version of this article first appeared on WebMD.com.
Best estimates made for hydroxychloroquine retinopathy risk
A new study likely makes the best estimate yet of the degree of retinopathy risk that patients who take the antimalarial drug hydroxychloroquine (HCQ) can expect, deriving mainly from the cumulative dose taken during the first 5 years of use, according to a study published in Annals of Internal Medicine.
HCQ works to decrease activity in a patient’s immune system, which is effective in many cases of systemic lupus erythematosus, one of the most common indications for the drug. However, an adverse outcome of treatment can be HCQ retinopathy, a progressive form of vision loss in patients taking HCQ over an extended period (mostly for longer than 5 years). The disease is often asymptomatic, although some patients do present a paracentral scotoma and a decrease in color vision. Patients may also notice flashing shapes in their vision and find that they have difficulty reading. Eventually, HCQ retinopathy can lead to loss of visual acuity, loss of peripheral vision, and loss of night vision.
Researchers from Kaiser Permanente Northern California and Harvard Medical School analyzed 3,325 persons who received HCQ for 5 or more years between 2004 and 2020. Their goal was to both characterize the long-term risk for incident HCQ retinopathy and examine the degree to which average HCQ dose within the first 5 years of treatment serves as a prediction of the risk.
The researchers then estimated the risk for developing retinopathy after 15 years, according to patients’ average dosing levels during the first 5 years of therapy. Overall, 81 participants developed HCQ retinopathy with overall cumulative incidences of 2.5% after 10 years and 8.6% after 15 years; the risk was greater for those given a higher dose during the first 5 years of treatment.
The mechanism of how HCQ toxicity may occur is still not completely known. There is evidence that toxicity happens because HCQ binds to melanin in both the retinal pigment epithelium and uvea in high concentrations. HCQ can interfere with lysosomal function, leading to oxidation and accumulation of lysosomes, which can cause dysfunction of the retinal pigment epithelium.
Progressive retinopathy can continue even after the drug is stopped. “It’s thought to be a very mild but important risk,” said Nilanjana Bose, MD, MBA, a rheumatologist with Memorial Hermann Health System in Houston. “Patients taking HCQ must be screened for retinal issues, most certainly elderly patients and patients with any kind of comorbidities.”

A 2021 joint position statement from the American College of Rheumatology, American Academy of Dermatology, the Rheumatologic Dermatology Society, and the American Academy of Ophthalmology recommends a baseline eye exam within a few months after starting therapy, then additional screening at 5 years on HCQ and annually thereafter.
“Early detection of retinopathy is important in overall visual prognosis, because toxicity can continue even after discontinuation of the medication,” said Rukhsana G. Mirza, MD, professor of ophthalmology and medical education at Northwestern University in Chicago.

“Examination alone is not sufficient to evaluate early changes, and specialized testing must be done. These include color photos, visual field tests, optical coherence tomography, fundus autofluorescence and in some cases, multifocal electroretinogram. Also, the AAO [American Academy of Ophthalmology] has specific recommendations related to Asian patients as they may have a different pattern of retinopathy that must also be considered.”
More accurate risk measurements
This news organization asked study coauthor April Jorge, MD, assistant professor of medicine in the division of rheumatology, allergy, and immunology at Massachusetts General Hospital and Harvard Medical School, Boston, to discuss the study, how it correlates to past research, and what it adds that’s new and useful to rheumatologists and ophthalmologists:
Question: Your research found that a higher dose of HCQ in the first 5 years of treatment led to a greater risk of retinopathy. Is there any indication that a lower dose given more frequently, either within that 5-year period or longer, would pose a similar risk?
Answer: In our study, we assessed the HCQ dose in the first 5 years of use but followed patients who continued the medication longer than 5 years, through up to 15 years of use. Therefore, we compared the risk of HCQ retinopathy associated with different HCQ dosages but for the same duration of use. We found that for any dose of HCQ, the risk of retinopathy increases the longer the medication is used. However, patients who used a higher dose of HCQ had a higher risk of developing retinopathy over time.

Although current guidelines recommend avoiding any HCQ dose over 5 mg/kg per day to reduce the risk of retinopathy, we found a higher risk of retinopathy associated with dosing over 6 mg/kg per day than between 5 and 6 mg/kg per day and the lowest risk with dosing under 5 mg/kg per day.
Q: How does your study align with and/or expand upon previous research regarding HCQ risk?
A: An important prior study of hydroxychloroquine retinopathy was the 2014 study by Ronald B. Melles, MD, and Michael F. Marmor, MD, published in JAMA Ophthalmology. Prior to our present study, that was the largest study to use the modern screening method (optical coherence tomography) to detect HCQ retinopathy. That screening tool is more sensitive than older methods, so it can detect early/mild cases of retinopathy that are typically asymptomatic. Compared to older studies, that 2014 study found a much higher risk of HCQ retinopathy than was previously appreciated.
However, that 2014 study did have some key limitations that could affect the risk estimates, such as using prevalent cases. A key feature of our present study is that we took several important steps to generate more accurate risk estimates. This included using an incident user cohort and detecting incident retinopathy cases through serial review of optical coherence tomography (screening) studies.
To achieve a high degree of methodologic rigor in correctly identifying retinopathy outcomes, we had expert ophthalmologists perform masked adjudication of all screening studies, and we assessed the intra-rater reliability of these study interpretations. Therefore, our study adds to the literature more accurate estimates of retinopathy risk. We found a lower cumulative incidence of retinopathy than was identified in the 2014 study, but the risk is still noteworthy.
Also unique to our study, we graded the severity of HCQ retinopathy outcomes. This was important, as we found that the majority of retinopathy cases detected through routine screening are mild and presumed to be asymptomatic. This will likely be reassuring news for patients that we can screen for this adverse event to detect it early and prevent vision loss.
Another important difference was that we assessed the risk of retinopathy associated with using over 6 mg/kg per day, between 5 and 6 mg/kg per day, and less than 5 mg/kg per day, whereas the highest dosing group assessed in the 2014 study included all patients using over 5 mg/kg per day. The risk was considerably higher in the > 6 mg/kg per day group than in the 5-6 mg/kg per day group.
Q: How can rheumatologists and ophthalmologists use this new information specifically to better treat their patients?
A: Our study provides more accurate estimates of the risk of HCQ retinopathy than in prior studies. These risk estimates can be used when rheumatologists (and other clinicians who prescribe HCQ) consider the risks and benefits of this otherwise important and well-tolerated medication. The risk associated with different dose ranges could also inform dosing decisions, since dosing over 6 mg/kg per day may be more of a concern than using doses in the 5-6 mg/kg range. Ophthalmologists can also use these new risk estimates to counsel patients of the importance of HCQ retinopathy screening and can also hopefully provide some reassurance to patients that the risk of severe retinopathy is low as long as they are being monitored.
The study authors were supported by grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases and the Rheumatology Research Foundation. The authors report no relevant financial relationships. Dr. Bose and Dr. Mirza had no relevant disclosures.
A version of this article first appeared on Medscape.com.
A new study likely makes the best estimate yet of the degree of retinopathy risk that patients who take the antimalarial drug hydroxychloroquine (HCQ) can expect, deriving mainly from the cumulative dose taken during the first 5 years of use, according to a study published in Annals of Internal Medicine.
HCQ works to decrease activity in a patient’s immune system, which is effective in many cases of systemic lupus erythematosus, one of the most common indications for the drug. However, an adverse outcome of treatment can be HCQ retinopathy, a progressive form of vision loss in patients taking HCQ over an extended period (mostly for longer than 5 years). The disease is often asymptomatic, although some patients do present a paracentral scotoma and a decrease in color vision. Patients may also notice flashing shapes in their vision and find that they have difficulty reading. Eventually, HCQ retinopathy can lead to loss of visual acuity, loss of peripheral vision, and loss of night vision.
Researchers from Kaiser Permanente Northern California and Harvard Medical School analyzed 3,325 persons who received HCQ for 5 or more years between 2004 and 2020. Their goal was to both characterize the long-term risk for incident HCQ retinopathy and examine the degree to which average HCQ dose within the first 5 years of treatment serves as a prediction of the risk.
The researchers then estimated the risk for developing retinopathy after 15 years, according to patients’ average dosing levels during the first 5 years of therapy. Overall, 81 participants developed HCQ retinopathy with overall cumulative incidences of 2.5% after 10 years and 8.6% after 15 years; the risk was greater for those given a higher dose during the first 5 years of treatment.
The mechanism of how HCQ toxicity may occur is still not completely known. There is evidence that toxicity happens because HCQ binds to melanin in both the retinal pigment epithelium and uvea in high concentrations. HCQ can interfere with lysosomal function, leading to oxidation and accumulation of lysosomes, which can cause dysfunction of the retinal pigment epithelium.
Progressive retinopathy can continue even after the drug is stopped. “It’s thought to be a very mild but important risk,” said Nilanjana Bose, MD, MBA, a rheumatologist with Memorial Hermann Health System in Houston. “Patients taking HCQ must be screened for retinal issues, most certainly elderly patients and patients with any kind of comorbidities.”
A 2021 joint position statement from the American College of Rheumatology, American Academy of Dermatology, the Rheumatologic Dermatology Society, and the American Academy of Ophthalmology recommends a baseline eye exam within a few months after starting therapy, then additional screening at 5 years on HCQ and annually thereafter.
“Early detection of retinopathy is important in overall visual prognosis, because toxicity can continue even after discontinuation of the medication,” said Rukhsana G. Mirza, MD, professor of ophthalmology and medical education at Northwestern University in Chicago.
“Examination alone is not sufficient to evaluate early changes, and specialized testing must be done. These include color photos, visual field tests, optical coherence tomography, fundus autofluorescence and in some cases, multifocal electroretinogram. Also, the AAO [American Academy of Ophthalmology] has specific recommendations related to Asian patients as they may have a different pattern of retinopathy that must also be considered.”
More accurate risk measurements
This news organization asked study coauthor April Jorge, MD, assistant professor of medicine in the division of rheumatology, allergy, and immunology at Massachusetts General Hospital and Harvard Medical School, Boston, to discuss the study, how it correlates to past research, and what it adds that’s new and useful to rheumatologists and ophthalmologists:
Question: Your research found that a higher dose of HCQ in the first 5 years of treatment led to a greater risk of retinopathy. Is there any indication that a lower dose given more frequently, either within that 5-year period or longer, would pose a similar risk?
Answer: In our study, we assessed the HCQ dose in the first 5 years of use but followed patients who continued the medication longer than 5 years, through up to 15 years of use. Therefore, we compared the risk of HCQ retinopathy associated with different HCQ dosages but for the same duration of use. We found that for any dose of HCQ, the risk of retinopathy increases the longer the medication is used. However, patients who used a higher dose of HCQ had a higher risk of developing retinopathy over time.
Although current guidelines recommend avoiding any HCQ dose over 5 mg/kg per day to reduce the risk of retinopathy, we found a higher risk of retinopathy associated with dosing over 6 mg/kg per day than between 5 and 6 mg/kg per day and the lowest risk with dosing under 5 mg/kg per day.
Q: How does your study align with and/or expand upon previous research regarding HCQ risk?
A: An important prior study of hydroxychloroquine retinopathy was the 2014 study by Ronald B. Melles, MD, and Michael F. Marmor, MD, published in JAMA Ophthalmology. Prior to our present study, that was the largest study to use the modern screening method (optical coherence tomography) to detect HCQ retinopathy. That screening tool is more sensitive than older methods, so it can detect early/mild cases of retinopathy that are typically asymptomatic. Compared to older studies, that 2014 study found a much higher risk of HCQ retinopathy than was previously appreciated.
However, that 2014 study did have some key limitations that could affect the risk estimates, such as using prevalent cases. A key feature of our present study is that we took several important steps to generate more accurate risk estimates. This included using an incident user cohort and detecting incident retinopathy cases through serial review of optical coherence tomography (screening) studies.
To achieve a high degree of methodologic rigor in correctly identifying retinopathy outcomes, we had expert ophthalmologists perform masked adjudication of all screening studies, and we assessed the intra-rater reliability of these study interpretations. Therefore, our study adds to the literature more accurate estimates of retinopathy risk. We found a lower cumulative incidence of retinopathy than was identified in the 2014 study, but the risk is still noteworthy.
Also unique to our study, we graded the severity of HCQ retinopathy outcomes. This was important, as we found that the majority of retinopathy cases detected through routine screening are mild and presumed to be asymptomatic. This will likely be reassuring news for patients that we can screen for this adverse event to detect it early and prevent vision loss.
Another important difference was that we assessed the risk of retinopathy associated with using over 6 mg/kg per day, between 5 and 6 mg/kg per day, and less than 5 mg/kg per day, whereas the highest dosing group assessed in the 2014 study included all patients using over 5 mg/kg per day. The risk was considerably higher in the > 6 mg/kg per day group than in the 5-6 mg/kg per day group.
Q: How can rheumatologists and ophthalmologists use this new information specifically to better treat their patients?
A: Our study provides more accurate estimates of the risk of HCQ retinopathy than in prior studies. These risk estimates can be used when rheumatologists (and other clinicians who prescribe HCQ) consider the risks and benefits of this otherwise important and well-tolerated medication. The risk associated with different dose ranges could also inform dosing decisions, since dosing over 6 mg/kg per day may be more of a concern than using doses in the 5-6 mg/kg range. Ophthalmologists can also use these new risk estimates to counsel patients of the importance of HCQ retinopathy screening and can also hopefully provide some reassurance to patients that the risk of severe retinopathy is low as long as they are being monitored.
The study authors were supported by grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases and the Rheumatology Research Foundation. The authors report no relevant financial relationships. Dr. Bose and Dr. Mirza had no relevant disclosures.
A version of this article first appeared on Medscape.com.
A new study likely makes the best estimate yet of the degree of retinopathy risk that patients who take the antimalarial drug hydroxychloroquine (HCQ) can expect, deriving mainly from the cumulative dose taken during the first 5 years of use, according to a study published in Annals of Internal Medicine.
HCQ works to decrease activity in a patient’s immune system, which is effective in many cases of systemic lupus erythematosus, one of the most common indications for the drug. However, an adverse outcome of treatment can be HCQ retinopathy, a progressive form of vision loss in patients taking HCQ over an extended period (mostly for longer than 5 years). The disease is often asymptomatic, although some patients do present a paracentral scotoma and a decrease in color vision. Patients may also notice flashing shapes in their vision and find that they have difficulty reading. Eventually, HCQ retinopathy can lead to loss of visual acuity, loss of peripheral vision, and loss of night vision.
Researchers from Kaiser Permanente Northern California and Harvard Medical School analyzed 3,325 persons who received HCQ for 5 or more years between 2004 and 2020. Their goal was to both characterize the long-term risk for incident HCQ retinopathy and examine the degree to which average HCQ dose within the first 5 years of treatment serves as a prediction of the risk.
The researchers then estimated the risk for developing retinopathy after 15 years, according to patients’ average dosing levels during the first 5 years of therapy. Overall, 81 participants developed HCQ retinopathy with overall cumulative incidences of 2.5% after 10 years and 8.6% after 15 years; the risk was greater for those given a higher dose during the first 5 years of treatment.
The mechanism of how HCQ toxicity may occur is still not completely known. There is evidence that toxicity happens because HCQ binds to melanin in both the retinal pigment epithelium and uvea in high concentrations. HCQ can interfere with lysosomal function, leading to oxidation and accumulation of lysosomes, which can cause dysfunction of the retinal pigment epithelium.
Progressive retinopathy can continue even after the drug is stopped. “It’s thought to be a very mild but important risk,” said Nilanjana Bose, MD, MBA, a rheumatologist with Memorial Hermann Health System in Houston. “Patients taking HCQ must be screened for retinal issues, most certainly elderly patients and patients with any kind of comorbidities.”
A 2021 joint position statement from the American College of Rheumatology, American Academy of Dermatology, the Rheumatologic Dermatology Society, and the American Academy of Ophthalmology recommends a baseline eye exam within a few months after starting therapy, then additional screening at 5 years on HCQ and annually thereafter.
“Early detection of retinopathy is important in overall visual prognosis, because toxicity can continue even after discontinuation of the medication,” said Rukhsana G. Mirza, MD, professor of ophthalmology and medical education at Northwestern University in Chicago.
“Examination alone is not sufficient to evaluate early changes, and specialized testing must be done. These include color photos, visual field tests, optical coherence tomography, fundus autofluorescence and in some cases, multifocal electroretinogram. Also, the AAO [American Academy of Ophthalmology] has specific recommendations related to Asian patients as they may have a different pattern of retinopathy that must also be considered.”
More accurate risk measurements
This news organization asked study coauthor April Jorge, MD, assistant professor of medicine in the division of rheumatology, allergy, and immunology at Massachusetts General Hospital and Harvard Medical School, Boston, to discuss the study, how it correlates to past research, and what it adds that’s new and useful to rheumatologists and ophthalmologists:
Question: Your research found that a higher dose of HCQ in the first 5 years of treatment led to a greater risk of retinopathy. Is there any indication that a lower dose given more frequently, either within that 5-year period or longer, would pose a similar risk?
Answer: In our study, we assessed the HCQ dose in the first 5 years of use but followed patients who continued the medication longer than 5 years, through up to 15 years of use. Therefore, we compared the risk of HCQ retinopathy associated with different HCQ dosages but for the same duration of use. We found that for any dose of HCQ, the risk of retinopathy increases the longer the medication is used. However, patients who used a higher dose of HCQ had a higher risk of developing retinopathy over time.
Although current guidelines recommend avoiding any HCQ dose over 5 mg/kg per day to reduce the risk of retinopathy, we found a higher risk of retinopathy associated with dosing over 6 mg/kg per day than between 5 and 6 mg/kg per day and the lowest risk with dosing under 5 mg/kg per day.
Q: How does your study align with and/or expand upon previous research regarding HCQ risk?
A: An important prior study of hydroxychloroquine retinopathy was the 2014 study by Ronald B. Melles, MD, and Michael F. Marmor, MD, published in JAMA Ophthalmology. Prior to our present study, that was the largest study to use the modern screening method (optical coherence tomography) to detect HCQ retinopathy. That screening tool is more sensitive than older methods, so it can detect early/mild cases of retinopathy that are typically asymptomatic. Compared to older studies, that 2014 study found a much higher risk of HCQ retinopathy than was previously appreciated.
However, that 2014 study did have some key limitations that could affect the risk estimates, such as using prevalent cases. A key feature of our present study is that we took several important steps to generate more accurate risk estimates. This included using an incident user cohort and detecting incident retinopathy cases through serial review of optical coherence tomography (screening) studies.
To achieve a high degree of methodologic rigor in correctly identifying retinopathy outcomes, we had expert ophthalmologists perform masked adjudication of all screening studies, and we assessed the intra-rater reliability of these study interpretations. Therefore, our study adds to the literature more accurate estimates of retinopathy risk. We found a lower cumulative incidence of retinopathy than was identified in the 2014 study, but the risk is still noteworthy.
Also unique to our study, we graded the severity of HCQ retinopathy outcomes. This was important, as we found that the majority of retinopathy cases detected through routine screening are mild and presumed to be asymptomatic. This will likely be reassuring news for patients that we can screen for this adverse event to detect it early and prevent vision loss.
Another important difference was that we assessed the risk of retinopathy associated with using over 6 mg/kg per day, between 5 and 6 mg/kg per day, and less than 5 mg/kg per day, whereas the highest dosing group assessed in the 2014 study included all patients using over 5 mg/kg per day. The risk was considerably higher in the > 6 mg/kg per day group than in the 5-6 mg/kg per day group.
Q: How can rheumatologists and ophthalmologists use this new information specifically to better treat their patients?
A: Our study provides more accurate estimates of the risk of HCQ retinopathy than in prior studies. These risk estimates can be used when rheumatologists (and other clinicians who prescribe HCQ) consider the risks and benefits of this otherwise important and well-tolerated medication. The risk associated with different dose ranges could also inform dosing decisions, since dosing over 6 mg/kg per day may be more of a concern than using doses in the 5-6 mg/kg range. Ophthalmologists can also use these new risk estimates to counsel patients of the importance of HCQ retinopathy screening and can also hopefully provide some reassurance to patients that the risk of severe retinopathy is low as long as they are being monitored.
The study authors were supported by grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases and the Rheumatology Research Foundation. The authors report no relevant financial relationships. Dr. Bose and Dr. Mirza had no relevant disclosures.
A version of this article first appeared on Medscape.com.
FROM ANNALS OF INTERNAL MEDICINE
HRT may prevent Alzheimer’s in high-risk women
new research suggests.
Results from a cohort study of almost 1,200 women showed that use of HRT was associated with higher delayed memory scores and larger entorhinal and hippocampal brain volumes – areas that are affected early by Alzheimer’s disease (AD) pathology.
HRT was also found to be most effective, as seen by larger hippocampal volume, when introduced during early perimenopause.
“Clinicians are very much aware of the susceptibility of women to cognitive disturbances during menopause,” lead author Rasha Saleh, MD, senior research associate, University of East Anglia (England), said in an interview.
“Identifying the at-risk APOE4 women and early HRT introduction can be of benefit. Confirming our findings in a clinical trial would be the next step forward,” Dr. Saleh said.
The findings were published online in Alzheimer’s Research and Therapy.
Personalized approaches
Dr. Saleh noted that estrogen receptors are localized in various areas of the brain, including cognition-related areas. Estrogen regulates such things as neuroinflammatory status, glucose utilization, and lipid metabolism.
“The decline of estrogen during menopause can lead to disturbance in these functions, which can accelerate AD-related pathology,” she said.
HRT during the menopausal transition and afterward is “being considered as a strategy to mitigate cognitive decline,” the investigators wrote. Early observational studies have suggested that oral estrogen “may be protective against dementia,” but results of clinical trials have been inconsistent, and some have even shown “harmful effects.”
The current researchers were “interested in the personalized approaches in the prevention of AD,” Dr. Saleh said. Preclinical and pilot data from her group have shown that women with APOE4 have “better cognitive test scores with nutritional and hormonal interventions.”
This led Dr. Saleh to hypothesize that HRT would be of more cognitive benefit for those with versus without APOE4, particularly when introduced early during the menopausal transition.
To investigate this hypothesis, the researchers analyzed baseline data from participants in the European Prevention of Alzheimer’s Dementia (EPAD) cohort. This project was initiated in 2015 with the aim of developing longitudinal models over the entire course of AD prior to dementia clinical diagnosis.
Participants were recruited from 10 European countries. All were required to be at least 50 years old, to have not been diagnosed with dementia at baseline, and to have no medical or psychiatric illness that could potentially exclude them from further research.
The current study included 1,178 women (mean age, 65.1 years), who were divided by genotype into non-APOE4 and APOE4 groups. HRT treatment for current or previous users included estrogen alone or estrogen plus progestogens via oral or transdermal administration routes, and at different doses.
The four tests used to assess cognition were the Mini-Mental State Examination dot counting to evaluate verbal working memory, the Repeatable Battery for the Assessment of Neuropsychological Status (RBANS) total score, the Four Mountain Test, and the supermarket trolley virtual reality test.
Brain MRI data were collected. The researchers focused on the medial temporal lobe as the “main brain region regulating cognition and memory processing.” This lobe includes the hippocampus, the parahippocampus, the entorhinal cortex, and the amygdala.
‘Critical window’
The researchers found a “trend” toward an APOE-HRT interaction (P-interaction = .097) for the total RBANS score. In particular, it was significant for the RBANS delayed memory index, where scores were consistently higher for women with APOE4 who had received HRT, compared with all other groups (P-interaction = .009).
Within-genotype group comparisons showed that HRT users had a higher RBANS total scale score and delayed memory index (P = .045 and P = .002, respectively), but only among APOE4 carriers. Effect size analyses showed a large effect of HRT use on the Four Mountain Test score and the supermarket trolley virtual reality test score (Cohen’s d = 0.988 and 1.2, respectively).
“This large effect was found only in APOE4 carriers,” the investigators noted.
Similarly, a moderate to large effect of HRT on the left entorhinal volume was observed in APOE4 carriers (Cohen’s d = 0.63).
In members of the APOE4 group who received HRT, the left entorhinal and left and right amygdala volumes were larger, compared with both no-APOE4 and non-HRT users (P-interaction = .002, .003, and .005, respectively). Similar trends were observed for the right entorhinal volume (P = .074).
In addition, among HRT users, the left entorhinal volume was larger (P = .03); the right and left anterior cingulate gyrus volumes were smaller (P = .003 and .062, respectively); and the left superior frontal gyrus volume was larger (P = .009) in comparison with women who did not receive HRT, independently of their APOE genotype.
Early use of HRT among APOE4 carriers was associated with larger right and left hippocampal volume (P = .035 and P = .028, respectively) – an association not found in non-APOE4 carriers. The association was also not significant when participants were not stratified by APOE genotype.
“The key important point here is the timing, or the ‘critical window,’ when HRT can be of most benefit,” Dr. Saleh said. “This is most beneficial when introduced early, before the neuropathology becomes irreversible.”
Study limitations include its cross-sectional design, which precludes the establishment of a causal relationship, and the fact that information regarding the type and dose of estrogen was not available for all participants.
HRT is not without risk, Dr. Saleh noted. She recommended that clinicians “carry out various screening tests to make sure that a woman is eligible for HRT and not at risk of hypercoagulability, for instance.”
Risk-benefit ratio
In a comment, Howard Fillit, MD, cofounder and chief science officer at the Alzheimer’s Drug Discovery Foundation, called the study “exactly the kind of work that needs to be done.”
Dr. Fillit, who was not involved with the current research, is a clinical professor of geriatric medicine, palliative care medicine, and neuroscience at Mount Sinai Hospital, New York.
He compared the process with that of osteoporosis. “We know that if women are treated [with HRT] at the time of the menopause, you can prevent the rapid bone loss that occurs with rapid estrogen loss. But if you wait 5, 10 years out, once the bone loss has occurred, the HRT doesn’t really have any impact on osteoporosis risk because the horse is already out of the barn,” he said.
Although HRT carries risks, “they can clearly be managed; and if it’s proven that estrogen or hormone replacement around the time of the menopause can be protective [against AD], the risk-benefit ratio of HRT could be in favor of treatment,” Dr. Fillit added.
The study was conducted as part of the Medical Research Council NuBrain Consortium. The investigators and Dr. Fillit reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
new research suggests.
Results from a cohort study of almost 1,200 women showed that use of HRT was associated with higher delayed memory scores and larger entorhinal and hippocampal brain volumes – areas that are affected early by Alzheimer’s disease (AD) pathology.
HRT was also found to be most effective, as seen by larger hippocampal volume, when introduced during early perimenopause.
“Clinicians are very much aware of the susceptibility of women to cognitive disturbances during menopause,” lead author Rasha Saleh, MD, senior research associate, University of East Anglia (England), said in an interview.
“Identifying the at-risk APOE4 women and early HRT introduction can be of benefit. Confirming our findings in a clinical trial would be the next step forward,” Dr. Saleh said.
The findings were published online in Alzheimer’s Research and Therapy.
Personalized approaches
Dr. Saleh noted that estrogen receptors are localized in various areas of the brain, including cognition-related areas. Estrogen regulates such things as neuroinflammatory status, glucose utilization, and lipid metabolism.
“The decline of estrogen during menopause can lead to disturbance in these functions, which can accelerate AD-related pathology,” she said.
HRT during the menopausal transition and afterward is “being considered as a strategy to mitigate cognitive decline,” the investigators wrote. Early observational studies have suggested that oral estrogen “may be protective against dementia,” but results of clinical trials have been inconsistent, and some have even shown “harmful effects.”
The current researchers were “interested in the personalized approaches in the prevention of AD,” Dr. Saleh said. Preclinical and pilot data from her group have shown that women with APOE4 have “better cognitive test scores with nutritional and hormonal interventions.”
This led Dr. Saleh to hypothesize that HRT would be of more cognitive benefit for those with versus without APOE4, particularly when introduced early during the menopausal transition.
To investigate this hypothesis, the researchers analyzed baseline data from participants in the European Prevention of Alzheimer’s Dementia (EPAD) cohort. This project was initiated in 2015 with the aim of developing longitudinal models over the entire course of AD prior to dementia clinical diagnosis.
Participants were recruited from 10 European countries. All were required to be at least 50 years old, to have not been diagnosed with dementia at baseline, and to have no medical or psychiatric illness that could potentially exclude them from further research.
The current study included 1,178 women (mean age, 65.1 years), who were divided by genotype into non-APOE4 and APOE4 groups. HRT treatment for current or previous users included estrogen alone or estrogen plus progestogens via oral or transdermal administration routes, and at different doses.
The four tests used to assess cognition were the Mini-Mental State Examination dot counting to evaluate verbal working memory, the Repeatable Battery for the Assessment of Neuropsychological Status (RBANS) total score, the Four Mountain Test, and the supermarket trolley virtual reality test.
Brain MRI data were collected. The researchers focused on the medial temporal lobe as the “main brain region regulating cognition and memory processing.” This lobe includes the hippocampus, the parahippocampus, the entorhinal cortex, and the amygdala.
‘Critical window’
The researchers found a “trend” toward an APOE-HRT interaction (P-interaction = .097) for the total RBANS score. In particular, it was significant for the RBANS delayed memory index, where scores were consistently higher for women with APOE4 who had received HRT, compared with all other groups (P-interaction = .009).
Within-genotype group comparisons showed that HRT users had a higher RBANS total scale score and delayed memory index (P = .045 and P = .002, respectively), but only among APOE4 carriers. Effect size analyses showed a large effect of HRT use on the Four Mountain Test score and the supermarket trolley virtual reality test score (Cohen’s d = 0.988 and 1.2, respectively).
“This large effect was found only in APOE4 carriers,” the investigators noted.
Similarly, a moderate to large effect of HRT on the left entorhinal volume was observed in APOE4 carriers (Cohen’s d = 0.63).
In members of the APOE4 group who received HRT, the left entorhinal and left and right amygdala volumes were larger, compared with both no-APOE4 and non-HRT users (P-interaction = .002, .003, and .005, respectively). Similar trends were observed for the right entorhinal volume (P = .074).
In addition, among HRT users, the left entorhinal volume was larger (P = .03); the right and left anterior cingulate gyrus volumes were smaller (P = .003 and .062, respectively); and the left superior frontal gyrus volume was larger (P = .009) in comparison with women who did not receive HRT, independently of their APOE genotype.
Early use of HRT among APOE4 carriers was associated with larger right and left hippocampal volume (P = .035 and P = .028, respectively) – an association not found in non-APOE4 carriers. The association was also not significant when participants were not stratified by APOE genotype.
“The key important point here is the timing, or the ‘critical window,’ when HRT can be of most benefit,” Dr. Saleh said. “This is most beneficial when introduced early, before the neuropathology becomes irreversible.”
Study limitations include its cross-sectional design, which precludes the establishment of a causal relationship, and the fact that information regarding the type and dose of estrogen was not available for all participants.
HRT is not without risk, Dr. Saleh noted. She recommended that clinicians “carry out various screening tests to make sure that a woman is eligible for HRT and not at risk of hypercoagulability, for instance.”
Risk-benefit ratio
In a comment, Howard Fillit, MD, cofounder and chief science officer at the Alzheimer’s Drug Discovery Foundation, called the study “exactly the kind of work that needs to be done.”
Dr. Fillit, who was not involved with the current research, is a clinical professor of geriatric medicine, palliative care medicine, and neuroscience at Mount Sinai Hospital, New York.
He compared the process with that of osteoporosis. “We know that if women are treated [with HRT] at the time of the menopause, you can prevent the rapid bone loss that occurs with rapid estrogen loss. But if you wait 5, 10 years out, once the bone loss has occurred, the HRT doesn’t really have any impact on osteoporosis risk because the horse is already out of the barn,” he said.
Although HRT carries risks, “they can clearly be managed; and if it’s proven that estrogen or hormone replacement around the time of the menopause can be protective [against AD], the risk-benefit ratio of HRT could be in favor of treatment,” Dr. Fillit added.
The study was conducted as part of the Medical Research Council NuBrain Consortium. The investigators and Dr. Fillit reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
new research suggests.
Results from a cohort study of almost 1,200 women showed that use of HRT was associated with higher delayed memory scores and larger entorhinal and hippocampal brain volumes – areas that are affected early by Alzheimer’s disease (AD) pathology.
HRT was also found to be most effective, as seen by larger hippocampal volume, when introduced during early perimenopause.
“Clinicians are very much aware of the susceptibility of women to cognitive disturbances during menopause,” lead author Rasha Saleh, MD, senior research associate, University of East Anglia (England), said in an interview.
“Identifying the at-risk APOE4 women and early HRT introduction can be of benefit. Confirming our findings in a clinical trial would be the next step forward,” Dr. Saleh said.
The findings were published online in Alzheimer’s Research and Therapy.
Personalized approaches
Dr. Saleh noted that estrogen receptors are localized in various areas of the brain, including cognition-related areas. Estrogen regulates such things as neuroinflammatory status, glucose utilization, and lipid metabolism.
“The decline of estrogen during menopause can lead to disturbance in these functions, which can accelerate AD-related pathology,” she said.
HRT during the menopausal transition and afterward is “being considered as a strategy to mitigate cognitive decline,” the investigators wrote. Early observational studies have suggested that oral estrogen “may be protective against dementia,” but results of clinical trials have been inconsistent, and some have even shown “harmful effects.”
The current researchers were “interested in the personalized approaches in the prevention of AD,” Dr. Saleh said. Preclinical and pilot data from her group have shown that women with APOE4 have “better cognitive test scores with nutritional and hormonal interventions.”
This led Dr. Saleh to hypothesize that HRT would be of more cognitive benefit for those with versus without APOE4, particularly when introduced early during the menopausal transition.
To investigate this hypothesis, the researchers analyzed baseline data from participants in the European Prevention of Alzheimer’s Dementia (EPAD) cohort. This project was initiated in 2015 with the aim of developing longitudinal models over the entire course of AD prior to dementia clinical diagnosis.
Participants were recruited from 10 European countries. All were required to be at least 50 years old, to have not been diagnosed with dementia at baseline, and to have no medical or psychiatric illness that could potentially exclude them from further research.
The current study included 1,178 women (mean age, 65.1 years), who were divided by genotype into non-APOE4 and APOE4 groups. HRT treatment for current or previous users included estrogen alone or estrogen plus progestogens via oral or transdermal administration routes, and at different doses.
The four tests used to assess cognition were the Mini-Mental State Examination dot counting to evaluate verbal working memory, the Repeatable Battery for the Assessment of Neuropsychological Status (RBANS) total score, the Four Mountain Test, and the supermarket trolley virtual reality test.
Brain MRI data were collected. The researchers focused on the medial temporal lobe as the “main brain region regulating cognition and memory processing.” This lobe includes the hippocampus, the parahippocampus, the entorhinal cortex, and the amygdala.
‘Critical window’
The researchers found a “trend” toward an APOE-HRT interaction (P-interaction = .097) for the total RBANS score. In particular, it was significant for the RBANS delayed memory index, where scores were consistently higher for women with APOE4 who had received HRT, compared with all other groups (P-interaction = .009).
Within-genotype group comparisons showed that HRT users had a higher RBANS total scale score and delayed memory index (P = .045 and P = .002, respectively), but only among APOE4 carriers. Effect size analyses showed a large effect of HRT use on the Four Mountain Test score and the supermarket trolley virtual reality test score (Cohen’s d = 0.988 and 1.2, respectively).
“This large effect was found only in APOE4 carriers,” the investigators noted.
Similarly, a moderate to large effect of HRT on the left entorhinal volume was observed in APOE4 carriers (Cohen’s d = 0.63).
In members of the APOE4 group who received HRT, the left entorhinal and left and right amygdala volumes were larger, compared with both no-APOE4 and non-HRT users (P-interaction = .002, .003, and .005, respectively). Similar trends were observed for the right entorhinal volume (P = .074).
In addition, among HRT users, the left entorhinal volume was larger (P = .03); the right and left anterior cingulate gyrus volumes were smaller (P = .003 and .062, respectively); and the left superior frontal gyrus volume was larger (P = .009) in comparison with women who did not receive HRT, independently of their APOE genotype.
Early use of HRT among APOE4 carriers was associated with larger right and left hippocampal volume (P = .035 and P = .028, respectively) – an association not found in non-APOE4 carriers. The association was also not significant when participants were not stratified by APOE genotype.
“The key important point here is the timing, or the ‘critical window,’ when HRT can be of most benefit,” Dr. Saleh said. “This is most beneficial when introduced early, before the neuropathology becomes irreversible.”
Study limitations include its cross-sectional design, which precludes the establishment of a causal relationship, and the fact that information regarding the type and dose of estrogen was not available for all participants.
HRT is not without risk, Dr. Saleh noted. She recommended that clinicians “carry out various screening tests to make sure that a woman is eligible for HRT and not at risk of hypercoagulability, for instance.”
Risk-benefit ratio
In a comment, Howard Fillit, MD, cofounder and chief science officer at the Alzheimer’s Drug Discovery Foundation, called the study “exactly the kind of work that needs to be done.”
Dr. Fillit, who was not involved with the current research, is a clinical professor of geriatric medicine, palliative care medicine, and neuroscience at Mount Sinai Hospital, New York.
He compared the process with that of osteoporosis. “We know that if women are treated [with HRT] at the time of the menopause, you can prevent the rapid bone loss that occurs with rapid estrogen loss. But if you wait 5, 10 years out, once the bone loss has occurred, the HRT doesn’t really have any impact on osteoporosis risk because the horse is already out of the barn,” he said.
Although HRT carries risks, “they can clearly be managed; and if it’s proven that estrogen or hormone replacement around the time of the menopause can be protective [against AD], the risk-benefit ratio of HRT could be in favor of treatment,” Dr. Fillit added.
The study was conducted as part of the Medical Research Council NuBrain Consortium. The investigators and Dr. Fillit reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM ALZHEIMER’S RESEARCH AND THERAPY
Transition to Tenecteplase From t-PA for Acute Ischemic Stroke at Walter Reed National Military Medical Center
Tissue plasminogen activator (t-PA) has been the standard IV thrombolytic used in acute ischemic stroke treatment since its US Food and Drug Administration (FDA) approval in 1995. Trials have established this drug’s efficacy in the treatment of acute ischemic stroke and the appropriate patient population for therapy.1-3 Published guidelines and experiences have made clear that a written protocol with extensive personnel training is important to deliver this care properly.4
Tenecteplase has been available for use in the treatment of acute myocardial infarction (MI) and studied in acute ischemic strokes since 2000. Recent large multicenter trials have suggested tenecteplase may work better than t-PA in the recanalization of large vessel occlusions (LVOs) and have provided guidance on proper dosing in acute ischemic stroke victims.5-8 Compared with t-PA, tenecteplase has a longer half-life, is more fibrin specific (causing less coagulopathy), and is more resistant to endogenous plasminogen activator inhibitor.9,10 Using tenecteplase for acute ischemic stroke is simpler as a single dose bolus rather than a bolus followed by a 1-hour infusion with t-PA. Immediate mechanical thrombectomy for LVO is less complicated without the 1-hour t-PA infusion.5,6 Tenecteplase use also allows for nonthrombectomy hospitals to accelerate transfer times for patients who need thrombectomy following thrombolysis by eliminating the need for critical care nurse–staffed ambulances for interfacility transfer.11 Tenecteplase also is cheaper: Tenecteplase costs $3748 per vial, whereas t-PA costs $5800 per vial equating to roughly a $2000 savings per patient.12,13 Finally, the pharmacy formulary is simplified by using a single thrombolytic agent for both cardiac and neurologic emergencies.
Tenecteplase does have some drawbacks to consider. Currently, tenecteplase is not approved by the FDA for the indication of acute ischemic stroke, though the drug is endorsed by the American Heart Association stroke guidelines of 2019 as an alternative to t-PA.14 There is no stroke-specific preparation of the drug, leading to potential dosing errors. Therefore, a systematic process to safely transition from t-PA to tenecteplase for acute ischemic stroke was undertaken at Walter Reed National Military Medical Center (WRNMMC) in Bethesda, Maryland. Here, we report the process required in making a complex switch in thrombolytic medication along with the potential benefits of making this transition.
OBSERVATIONS
The process to implement tenecteplase required extensive training and education for staff physicians, nurses, pharmacists, radiologists, trainees, and the rapid response team. Our institution administered IV thrombolytic drugs up to 25 times annually to acute ischemic stroke victims, meaning we had to train personnel extensively and repeatedly.
In preparation for the transition to tenecteplase, hospital leadership gathered staff for multidisciplinary administrative meetings that included neurology, emergency medicine, intensive care, pharmacy, radiology, and nursing departments. The purpose of these meetings was to establish a standard operating procedure (SOP) to ensure a safe transition. This process began in May 2020 and involved regular meetings to draft and revise our SOP. Additionally, several leadership and training sessions were held over a 6-month period. Stroke boxes were developed that contained the required evaluation tools, consent forms, medications (tenecteplase and treatments for known complications), dosing cards, and instructions. Final approval of the updated acute ischemic stroke hospital policy was obtained in November 2020 and signed by the above departments.
All inclusion and exclusion criteria were determined to be the same for tenecteplase as they were for t-PA with the notable exception that the WAKE-UP trial protocol would not be supported until further evidence became available.9 The results of the WAKE-UP trial had previously been used at WRNMMC to justify administration of t-PA in patients who awoke with symptoms of acute ischemic stroke, the last known well was unclear or > 4.5 hours, and for whom a magnetic resonance imaging (MRI) of the brain could be obtained rapidly. Based on the WAKE-UP trial, if the MRI scan of the brain in these patients demonstrated restricted diffusion without fluid attenuated inversion recovery (FLAIR) signal changes (diffusion-weighted [DWI]-FLAIR mismatch sign), this indicated that the stroke had likely occurred recently, and it was safe to administer t-PA. This allowed for administration of t-PA outside the standard treatment window of 4.5 hours from last known well, especially in the cases of patients who awoke with symptoms.
Since safety data are not yet available for the use of tenecteplase in this fashion, the WAKE-UP trial protocol was not used as an inclusion criterion. The informed consent form was modified, and the following scenarios were outlined: (1) If the patient or surrogate is immediately available to consent, paper consent will be documented with the additional note that tenecteplase is being used off-label; and (2) If the patient cannot consent and a surrogate is not immediately available, the medicine will be used emergently as long as the neurology resident and attending physicians agree.15
Risk mitigation was considered carefully. The stroke box described above is stocked and maintained by the pharmacy as we have transitioned to using designated pharmacists for the storage and preparation of tenecteplase. We highly recommend the use of designated pharmacists or emergency department pharmacists in this manner to avoid dosing errors.7,16 Since the current pharmacy-provided tenecteplase bottle contains twice the maximum dose indicated for ischemic stroke, only a 5 mL syringe is included in the stroke box to ensure a maximum dose of 25 mg is drawn up after reconstitution. Dosing card charts were made like existing dosing card charts for t-PA to quickly calculate the 0.25 mg/kg dose. In training, the difference in dosing in ischemic stroke was emphasized. Finally, pharmacy has taken responsibility for dosing the medication during stroke codes.
Any medical personnel at WRNMMC can initiate a stroke code by sending a page to the neurology consult service (Figure). 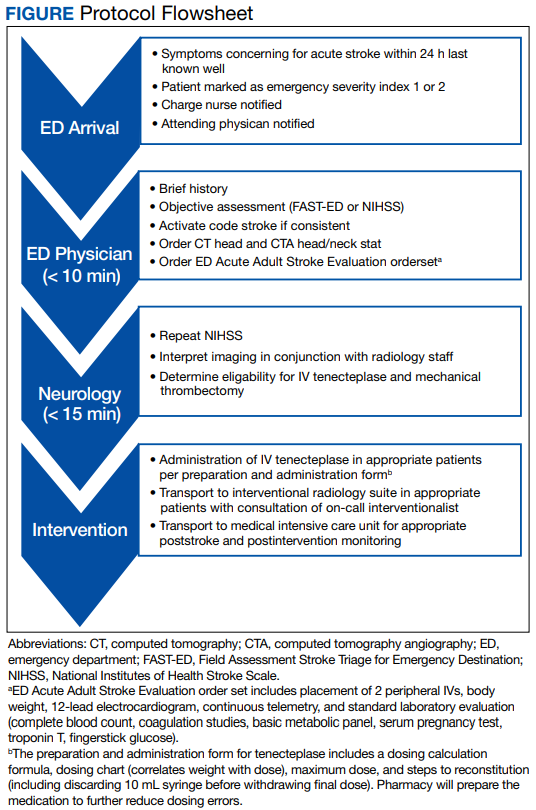
TRANSITION AND RESULTS
From November 2020 to December 2021, 10 patients have been treated in total at WRNMMC (Table). 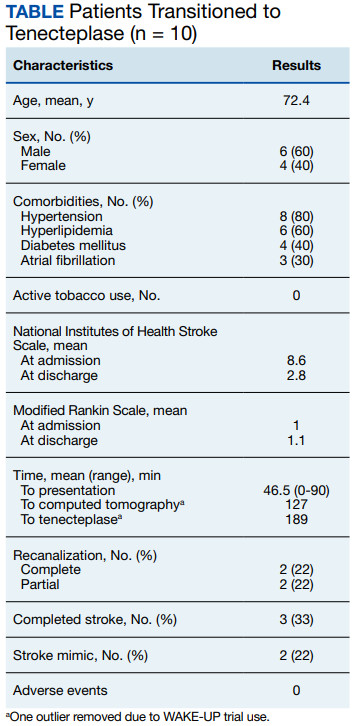
CONCLUSIONS
The available evidence supports the transition from t-PA to tenecteplase for acute ischemic stroke. The successful transition required months of preparation involving multidisciplinary meetings between neurology, nursing, pharmacy, radiology, rapid response teams, critical care, and emergency medicine departments. Safeguards must be implemented to avoid a tenecteplase dosing error that can lead to potentially life-threatening adverse effects. The results at WRNMMC thus far are promising for safety and efficacy. Several process improvements are planned: a hospital-wide overhead page will accompany the direct page to neurology; other team members, including radiology and pharmacy, will be included on the acute stroke alert; and a stroke-specific paging application will be implemented to better track real-time stroke metrics and improve flow. These measures mirror processes that are occurring in institutions that treat acute stroke patients.
1. Lees KR, Bluhmki E, von Kummer R, et al. Time to treatment with intravenous alteplase and outcome in stroke: an updated pooled analysis of ECASS, ATLANTIS, NINDS, and EPITHET trials. Lancet. 2010;375(9727):1695-1703. doi:10.1016/S0140-6736(10)60491-6
2. National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. Tissue plasminogen activator for acute ischemic stroke. N Engl J Med. 1995;333(24):1581- 1587. doi:10.1056/NEJM199512143332401
3. Hacke W, Donnan G, Fieschi C, et al. Association of outcome with early stroke treatment: pooled analysis of ATLANTIS, ECASS, and NINDS rt-PA stroke trials. Lancet. 2004;363(9411):768-774. doi:10.1016/S0140-6736(04)15692-4
4. Jauch EC, Saver JL, Adams HP Jr, et al. Guidelines for the early management of patients with acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2013;44(3):870-947. doi:10.1161/STR.0b013e318284056a
5. Campbell B, Mitchell P, Churilov L, et al. Tenecteplase versus alteplase before thrombectomy for ischemic stroke. N Engl J Med. 2018;378(17):1573-1582. doi:10.1056/nejmoa1716405
6. Yang P, Zhang Y, Zhang L, et al. Endovascular thrombectomy with or without intravenous alteplase in acute stroke. N Engl J Med. 2020;382(21):1981-1993. doi:10.1056/NEJMoa2001123
7. Menon BK, Buck BH, Singh N, et al. Intravenous tenecteplase compared with alteplase for acute ischaemic stroke in Canada (AcT): a pragmatic, multicentre, open-label, registry-linked, randomised, controlled, noninferiority trial. Lancet. 2022;400(10347):161-169. doi:10.1016/S0140-6736(22)01054-6
8. Campbell BCV, Mitchell PJ, Churilov L, et al. Effect of intravenous tenecteplase dose on cerebral reperfusion before thrombectomy in patients with large vessel occlusion ischemic stroke: the EXTEND-IA TNK part 2 randomized clinical trial. JAMA. 2020;323(13):1257- 1265. doi:10.1001/jama.2020.1511
9. Warach SJ, Dula AN, Milling TJ Jr. Tenecteplase thrombolysis for acute ischemic stroke. Stroke. 2020;51(11):3440- 3451. doi:10.1161/STROKEAHA.120.029749
10. Huang X, Moreton FC, Kalladka D, et al. Coagulation and fibrinolytic activity of tenecteplase and alteplase in acute ischemic stroke. Stroke. 2015;46(12):3543-3546. doi:10.1161/STROKEAHA.115.011290
11. Burgos AM, Saver JL. Evidence that tenecteplase is noninferior to alteplase for acute ischemic stroke: meta-analysis of 5 randomized trials. Stroke. 2019;50(8):2156-2162. doi:10.1161/STROKEAHA.119.025080
12. Potla N, Ganti L. Tenecteplase vs. alteplase for acute ischemic stroke: a systematic review. Int J Emerg Med. 2022;15(1). doi:10.1186/s12245-021-00399-w
13. Warach SJ, Winegar A, Ottenbacher A, Miller C, Gibson D. Abstract WMP52: reduced hospital costs for ischemic stroke treated with tenecteplase. Stroke. 2022;53(suppl 1):AWMP52. doi:10.1161/str.53.suppl_1.WMP52
14. Powers WJ, Rabinstein AA, Ackerson T, et al. Guidelines for the Early Management of Patients With Acute Ischemic Stroke: 2019 Update to the 2018 Guidelines for the Early Management of Acute Ischemic Stroke: A Guideline for Healthcare Professionals From the American Heart Association/American Stroke Association. Stroke. 2019;50(12):e344-e418. doi:10.1161/str.0000000000000211
15. Faris H, Dewar B, Dowlatshahi D, et al. Ethical justification for deferral of consent in the AcT trial for acute ischemic stroke. Stroke. 2022;53(7):2420-2423. doi:10.1161/strokeaha.122.038760
16. Kvistad CE, Næss H, Helleberg BH, et al. Tenecteplase versus alteplase for the management of acute ischaemic stroke in Norway (NOR-TEST 2, part A): a phase 3, randomised, open-label, blinded endpoint, non-inferiority trial. Lancet Neurol. 2022;21(6):511-519. doi:10.1016/S1474-4422(22)00124-7
Tissue plasminogen activator (t-PA) has been the standard IV thrombolytic used in acute ischemic stroke treatment since its US Food and Drug Administration (FDA) approval in 1995. Trials have established this drug’s efficacy in the treatment of acute ischemic stroke and the appropriate patient population for therapy.1-3 Published guidelines and experiences have made clear that a written protocol with extensive personnel training is important to deliver this care properly.4
Tenecteplase has been available for use in the treatment of acute myocardial infarction (MI) and studied in acute ischemic strokes since 2000. Recent large multicenter trials have suggested tenecteplase may work better than t-PA in the recanalization of large vessel occlusions (LVOs) and have provided guidance on proper dosing in acute ischemic stroke victims.5-8 Compared with t-PA, tenecteplase has a longer half-life, is more fibrin specific (causing less coagulopathy), and is more resistant to endogenous plasminogen activator inhibitor.9,10 Using tenecteplase for acute ischemic stroke is simpler as a single dose bolus rather than a bolus followed by a 1-hour infusion with t-PA. Immediate mechanical thrombectomy for LVO is less complicated without the 1-hour t-PA infusion.5,6 Tenecteplase use also allows for nonthrombectomy hospitals to accelerate transfer times for patients who need thrombectomy following thrombolysis by eliminating the need for critical care nurse–staffed ambulances for interfacility transfer.11 Tenecteplase also is cheaper: Tenecteplase costs $3748 per vial, whereas t-PA costs $5800 per vial equating to roughly a $2000 savings per patient.12,13 Finally, the pharmacy formulary is simplified by using a single thrombolytic agent for both cardiac and neurologic emergencies.
Tenecteplase does have some drawbacks to consider. Currently, tenecteplase is not approved by the FDA for the indication of acute ischemic stroke, though the drug is endorsed by the American Heart Association stroke guidelines of 2019 as an alternative to t-PA.14 There is no stroke-specific preparation of the drug, leading to potential dosing errors. Therefore, a systematic process to safely transition from t-PA to tenecteplase for acute ischemic stroke was undertaken at Walter Reed National Military Medical Center (WRNMMC) in Bethesda, Maryland. Here, we report the process required in making a complex switch in thrombolytic medication along with the potential benefits of making this transition.
OBSERVATIONS
The process to implement tenecteplase required extensive training and education for staff physicians, nurses, pharmacists, radiologists, trainees, and the rapid response team. Our institution administered IV thrombolytic drugs up to 25 times annually to acute ischemic stroke victims, meaning we had to train personnel extensively and repeatedly.
In preparation for the transition to tenecteplase, hospital leadership gathered staff for multidisciplinary administrative meetings that included neurology, emergency medicine, intensive care, pharmacy, radiology, and nursing departments. The purpose of these meetings was to establish a standard operating procedure (SOP) to ensure a safe transition. This process began in May 2020 and involved regular meetings to draft and revise our SOP. Additionally, several leadership and training sessions were held over a 6-month period. Stroke boxes were developed that contained the required evaluation tools, consent forms, medications (tenecteplase and treatments for known complications), dosing cards, and instructions. Final approval of the updated acute ischemic stroke hospital policy was obtained in November 2020 and signed by the above departments.
All inclusion and exclusion criteria were determined to be the same for tenecteplase as they were for t-PA with the notable exception that the WAKE-UP trial protocol would not be supported until further evidence became available.9 The results of the WAKE-UP trial had previously been used at WRNMMC to justify administration of t-PA in patients who awoke with symptoms of acute ischemic stroke, the last known well was unclear or > 4.5 hours, and for whom a magnetic resonance imaging (MRI) of the brain could be obtained rapidly. Based on the WAKE-UP trial, if the MRI scan of the brain in these patients demonstrated restricted diffusion without fluid attenuated inversion recovery (FLAIR) signal changes (diffusion-weighted [DWI]-FLAIR mismatch sign), this indicated that the stroke had likely occurred recently, and it was safe to administer t-PA. This allowed for administration of t-PA outside the standard treatment window of 4.5 hours from last known well, especially in the cases of patients who awoke with symptoms.
Since safety data are not yet available for the use of tenecteplase in this fashion, the WAKE-UP trial protocol was not used as an inclusion criterion. The informed consent form was modified, and the following scenarios were outlined: (1) If the patient or surrogate is immediately available to consent, paper consent will be documented with the additional note that tenecteplase is being used off-label; and (2) If the patient cannot consent and a surrogate is not immediately available, the medicine will be used emergently as long as the neurology resident and attending physicians agree.15
Risk mitigation was considered carefully. The stroke box described above is stocked and maintained by the pharmacy as we have transitioned to using designated pharmacists for the storage and preparation of tenecteplase. We highly recommend the use of designated pharmacists or emergency department pharmacists in this manner to avoid dosing errors.7,16 Since the current pharmacy-provided tenecteplase bottle contains twice the maximum dose indicated for ischemic stroke, only a 5 mL syringe is included in the stroke box to ensure a maximum dose of 25 mg is drawn up after reconstitution. Dosing card charts were made like existing dosing card charts for t-PA to quickly calculate the 0.25 mg/kg dose. In training, the difference in dosing in ischemic stroke was emphasized. Finally, pharmacy has taken responsibility for dosing the medication during stroke codes.
Any medical personnel at WRNMMC can initiate a stroke code by sending a page to the neurology consult service (Figure).
TRANSITION AND RESULTS
From November 2020 to December 2021, 10 patients have been treated in total at WRNMMC (Table).
CONCLUSIONS
The available evidence supports the transition from t-PA to tenecteplase for acute ischemic stroke. The successful transition required months of preparation involving multidisciplinary meetings between neurology, nursing, pharmacy, radiology, rapid response teams, critical care, and emergency medicine departments. Safeguards must be implemented to avoid a tenecteplase dosing error that can lead to potentially life-threatening adverse effects. The results at WRNMMC thus far are promising for safety and efficacy. Several process improvements are planned: a hospital-wide overhead page will accompany the direct page to neurology; other team members, including radiology and pharmacy, will be included on the acute stroke alert; and a stroke-specific paging application will be implemented to better track real-time stroke metrics and improve flow. These measures mirror processes that are occurring in institutions that treat acute stroke patients.
Tissue plasminogen activator (t-PA) has been the standard IV thrombolytic used in acute ischemic stroke treatment since its US Food and Drug Administration (FDA) approval in 1995. Trials have established this drug’s efficacy in the treatment of acute ischemic stroke and the appropriate patient population for therapy.1-3 Published guidelines and experiences have made clear that a written protocol with extensive personnel training is important to deliver this care properly.4
Tenecteplase has been available for use in the treatment of acute myocardial infarction (MI) and studied in acute ischemic strokes since 2000. Recent large multicenter trials have suggested tenecteplase may work better than t-PA in the recanalization of large vessel occlusions (LVOs) and have provided guidance on proper dosing in acute ischemic stroke victims.5-8 Compared with t-PA, tenecteplase has a longer half-life, is more fibrin specific (causing less coagulopathy), and is more resistant to endogenous plasminogen activator inhibitor.9,10 Using tenecteplase for acute ischemic stroke is simpler as a single dose bolus rather than a bolus followed by a 1-hour infusion with t-PA. Immediate mechanical thrombectomy for LVO is less complicated without the 1-hour t-PA infusion.5,6 Tenecteplase use also allows for nonthrombectomy hospitals to accelerate transfer times for patients who need thrombectomy following thrombolysis by eliminating the need for critical care nurse–staffed ambulances for interfacility transfer.11 Tenecteplase also is cheaper: Tenecteplase costs $3748 per vial, whereas t-PA costs $5800 per vial equating to roughly a $2000 savings per patient.12,13 Finally, the pharmacy formulary is simplified by using a single thrombolytic agent for both cardiac and neurologic emergencies.
Tenecteplase does have some drawbacks to consider. Currently, tenecteplase is not approved by the FDA for the indication of acute ischemic stroke, though the drug is endorsed by the American Heart Association stroke guidelines of 2019 as an alternative to t-PA.14 There is no stroke-specific preparation of the drug, leading to potential dosing errors. Therefore, a systematic process to safely transition from t-PA to tenecteplase for acute ischemic stroke was undertaken at Walter Reed National Military Medical Center (WRNMMC) in Bethesda, Maryland. Here, we report the process required in making a complex switch in thrombolytic medication along with the potential benefits of making this transition.
OBSERVATIONS
The process to implement tenecteplase required extensive training and education for staff physicians, nurses, pharmacists, radiologists, trainees, and the rapid response team. Our institution administered IV thrombolytic drugs up to 25 times annually to acute ischemic stroke victims, meaning we had to train personnel extensively and repeatedly.
In preparation for the transition to tenecteplase, hospital leadership gathered staff for multidisciplinary administrative meetings that included neurology, emergency medicine, intensive care, pharmacy, radiology, and nursing departments. The purpose of these meetings was to establish a standard operating procedure (SOP) to ensure a safe transition. This process began in May 2020 and involved regular meetings to draft and revise our SOP. Additionally, several leadership and training sessions were held over a 6-month period. Stroke boxes were developed that contained the required evaluation tools, consent forms, medications (tenecteplase and treatments for known complications), dosing cards, and instructions. Final approval of the updated acute ischemic stroke hospital policy was obtained in November 2020 and signed by the above departments.
All inclusion and exclusion criteria were determined to be the same for tenecteplase as they were for t-PA with the notable exception that the WAKE-UP trial protocol would not be supported until further evidence became available.9 The results of the WAKE-UP trial had previously been used at WRNMMC to justify administration of t-PA in patients who awoke with symptoms of acute ischemic stroke, the last known well was unclear or > 4.5 hours, and for whom a magnetic resonance imaging (MRI) of the brain could be obtained rapidly. Based on the WAKE-UP trial, if the MRI scan of the brain in these patients demonstrated restricted diffusion without fluid attenuated inversion recovery (FLAIR) signal changes (diffusion-weighted [DWI]-FLAIR mismatch sign), this indicated that the stroke had likely occurred recently, and it was safe to administer t-PA. This allowed for administration of t-PA outside the standard treatment window of 4.5 hours from last known well, especially in the cases of patients who awoke with symptoms.
Since safety data are not yet available for the use of tenecteplase in this fashion, the WAKE-UP trial protocol was not used as an inclusion criterion. The informed consent form was modified, and the following scenarios were outlined: (1) If the patient or surrogate is immediately available to consent, paper consent will be documented with the additional note that tenecteplase is being used off-label; and (2) If the patient cannot consent and a surrogate is not immediately available, the medicine will be used emergently as long as the neurology resident and attending physicians agree.15
Risk mitigation was considered carefully. The stroke box described above is stocked and maintained by the pharmacy as we have transitioned to using designated pharmacists for the storage and preparation of tenecteplase. We highly recommend the use of designated pharmacists or emergency department pharmacists in this manner to avoid dosing errors.7,16 Since the current pharmacy-provided tenecteplase bottle contains twice the maximum dose indicated for ischemic stroke, only a 5 mL syringe is included in the stroke box to ensure a maximum dose of 25 mg is drawn up after reconstitution. Dosing card charts were made like existing dosing card charts for t-PA to quickly calculate the 0.25 mg/kg dose. In training, the difference in dosing in ischemic stroke was emphasized. Finally, pharmacy has taken responsibility for dosing the medication during stroke codes.
Any medical personnel at WRNMMC can initiate a stroke code by sending a page to the neurology consult service (Figure).
TRANSITION AND RESULTS
From November 2020 to December 2021, 10 patients have been treated in total at WRNMMC (Table).
CONCLUSIONS
The available evidence supports the transition from t-PA to tenecteplase for acute ischemic stroke. The successful transition required months of preparation involving multidisciplinary meetings between neurology, nursing, pharmacy, radiology, rapid response teams, critical care, and emergency medicine departments. Safeguards must be implemented to avoid a tenecteplase dosing error that can lead to potentially life-threatening adverse effects. The results at WRNMMC thus far are promising for safety and efficacy. Several process improvements are planned: a hospital-wide overhead page will accompany the direct page to neurology; other team members, including radiology and pharmacy, will be included on the acute stroke alert; and a stroke-specific paging application will be implemented to better track real-time stroke metrics and improve flow. These measures mirror processes that are occurring in institutions that treat acute stroke patients.
1. Lees KR, Bluhmki E, von Kummer R, et al. Time to treatment with intravenous alteplase and outcome in stroke: an updated pooled analysis of ECASS, ATLANTIS, NINDS, and EPITHET trials. Lancet. 2010;375(9727):1695-1703. doi:10.1016/S0140-6736(10)60491-6
2. National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. Tissue plasminogen activator for acute ischemic stroke. N Engl J Med. 1995;333(24):1581- 1587. doi:10.1056/NEJM199512143332401
3. Hacke W, Donnan G, Fieschi C, et al. Association of outcome with early stroke treatment: pooled analysis of ATLANTIS, ECASS, and NINDS rt-PA stroke trials. Lancet. 2004;363(9411):768-774. doi:10.1016/S0140-6736(04)15692-4
4. Jauch EC, Saver JL, Adams HP Jr, et al. Guidelines for the early management of patients with acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2013;44(3):870-947. doi:10.1161/STR.0b013e318284056a
5. Campbell B, Mitchell P, Churilov L, et al. Tenecteplase versus alteplase before thrombectomy for ischemic stroke. N Engl J Med. 2018;378(17):1573-1582. doi:10.1056/nejmoa1716405
6. Yang P, Zhang Y, Zhang L, et al. Endovascular thrombectomy with or without intravenous alteplase in acute stroke. N Engl J Med. 2020;382(21):1981-1993. doi:10.1056/NEJMoa2001123
7. Menon BK, Buck BH, Singh N, et al. Intravenous tenecteplase compared with alteplase for acute ischaemic stroke in Canada (AcT): a pragmatic, multicentre, open-label, registry-linked, randomised, controlled, noninferiority trial. Lancet. 2022;400(10347):161-169. doi:10.1016/S0140-6736(22)01054-6
8. Campbell BCV, Mitchell PJ, Churilov L, et al. Effect of intravenous tenecteplase dose on cerebral reperfusion before thrombectomy in patients with large vessel occlusion ischemic stroke: the EXTEND-IA TNK part 2 randomized clinical trial. JAMA. 2020;323(13):1257- 1265. doi:10.1001/jama.2020.1511
9. Warach SJ, Dula AN, Milling TJ Jr. Tenecteplase thrombolysis for acute ischemic stroke. Stroke. 2020;51(11):3440- 3451. doi:10.1161/STROKEAHA.120.029749
10. Huang X, Moreton FC, Kalladka D, et al. Coagulation and fibrinolytic activity of tenecteplase and alteplase in acute ischemic stroke. Stroke. 2015;46(12):3543-3546. doi:10.1161/STROKEAHA.115.011290
11. Burgos AM, Saver JL. Evidence that tenecteplase is noninferior to alteplase for acute ischemic stroke: meta-analysis of 5 randomized trials. Stroke. 2019;50(8):2156-2162. doi:10.1161/STROKEAHA.119.025080
12. Potla N, Ganti L. Tenecteplase vs. alteplase for acute ischemic stroke: a systematic review. Int J Emerg Med. 2022;15(1). doi:10.1186/s12245-021-00399-w
13. Warach SJ, Winegar A, Ottenbacher A, Miller C, Gibson D. Abstract WMP52: reduced hospital costs for ischemic stroke treated with tenecteplase. Stroke. 2022;53(suppl 1):AWMP52. doi:10.1161/str.53.suppl_1.WMP52
14. Powers WJ, Rabinstein AA, Ackerson T, et al. Guidelines for the Early Management of Patients With Acute Ischemic Stroke: 2019 Update to the 2018 Guidelines for the Early Management of Acute Ischemic Stroke: A Guideline for Healthcare Professionals From the American Heart Association/American Stroke Association. Stroke. 2019;50(12):e344-e418. doi:10.1161/str.0000000000000211
15. Faris H, Dewar B, Dowlatshahi D, et al. Ethical justification for deferral of consent in the AcT trial for acute ischemic stroke. Stroke. 2022;53(7):2420-2423. doi:10.1161/strokeaha.122.038760
16. Kvistad CE, Næss H, Helleberg BH, et al. Tenecteplase versus alteplase for the management of acute ischaemic stroke in Norway (NOR-TEST 2, part A): a phase 3, randomised, open-label, blinded endpoint, non-inferiority trial. Lancet Neurol. 2022;21(6):511-519. doi:10.1016/S1474-4422(22)00124-7
1. Lees KR, Bluhmki E, von Kummer R, et al. Time to treatment with intravenous alteplase and outcome in stroke: an updated pooled analysis of ECASS, ATLANTIS, NINDS, and EPITHET trials. Lancet. 2010;375(9727):1695-1703. doi:10.1016/S0140-6736(10)60491-6
2. National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. Tissue plasminogen activator for acute ischemic stroke. N Engl J Med. 1995;333(24):1581- 1587. doi:10.1056/NEJM199512143332401
3. Hacke W, Donnan G, Fieschi C, et al. Association of outcome with early stroke treatment: pooled analysis of ATLANTIS, ECASS, and NINDS rt-PA stroke trials. Lancet. 2004;363(9411):768-774. doi:10.1016/S0140-6736(04)15692-4
4. Jauch EC, Saver JL, Adams HP Jr, et al. Guidelines for the early management of patients with acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2013;44(3):870-947. doi:10.1161/STR.0b013e318284056a
5. Campbell B, Mitchell P, Churilov L, et al. Tenecteplase versus alteplase before thrombectomy for ischemic stroke. N Engl J Med. 2018;378(17):1573-1582. doi:10.1056/nejmoa1716405
6. Yang P, Zhang Y, Zhang L, et al. Endovascular thrombectomy with or without intravenous alteplase in acute stroke. N Engl J Med. 2020;382(21):1981-1993. doi:10.1056/NEJMoa2001123
7. Menon BK, Buck BH, Singh N, et al. Intravenous tenecteplase compared with alteplase for acute ischaemic stroke in Canada (AcT): a pragmatic, multicentre, open-label, registry-linked, randomised, controlled, noninferiority trial. Lancet. 2022;400(10347):161-169. doi:10.1016/S0140-6736(22)01054-6
8. Campbell BCV, Mitchell PJ, Churilov L, et al. Effect of intravenous tenecteplase dose on cerebral reperfusion before thrombectomy in patients with large vessel occlusion ischemic stroke: the EXTEND-IA TNK part 2 randomized clinical trial. JAMA. 2020;323(13):1257- 1265. doi:10.1001/jama.2020.1511
9. Warach SJ, Dula AN, Milling TJ Jr. Tenecteplase thrombolysis for acute ischemic stroke. Stroke. 2020;51(11):3440- 3451. doi:10.1161/STROKEAHA.120.029749
10. Huang X, Moreton FC, Kalladka D, et al. Coagulation and fibrinolytic activity of tenecteplase and alteplase in acute ischemic stroke. Stroke. 2015;46(12):3543-3546. doi:10.1161/STROKEAHA.115.011290
11. Burgos AM, Saver JL. Evidence that tenecteplase is noninferior to alteplase for acute ischemic stroke: meta-analysis of 5 randomized trials. Stroke. 2019;50(8):2156-2162. doi:10.1161/STROKEAHA.119.025080
12. Potla N, Ganti L. Tenecteplase vs. alteplase for acute ischemic stroke: a systematic review. Int J Emerg Med. 2022;15(1). doi:10.1186/s12245-021-00399-w
13. Warach SJ, Winegar A, Ottenbacher A, Miller C, Gibson D. Abstract WMP52: reduced hospital costs for ischemic stroke treated with tenecteplase. Stroke. 2022;53(suppl 1):AWMP52. doi:10.1161/str.53.suppl_1.WMP52
14. Powers WJ, Rabinstein AA, Ackerson T, et al. Guidelines for the Early Management of Patients With Acute Ischemic Stroke: 2019 Update to the 2018 Guidelines for the Early Management of Acute Ischemic Stroke: A Guideline for Healthcare Professionals From the American Heart Association/American Stroke Association. Stroke. 2019;50(12):e344-e418. doi:10.1161/str.0000000000000211
15. Faris H, Dewar B, Dowlatshahi D, et al. Ethical justification for deferral of consent in the AcT trial for acute ischemic stroke. Stroke. 2022;53(7):2420-2423. doi:10.1161/strokeaha.122.038760
16. Kvistad CE, Næss H, Helleberg BH, et al. Tenecteplase versus alteplase for the management of acute ischaemic stroke in Norway (NOR-TEST 2, part A): a phase 3, randomised, open-label, blinded endpoint, non-inferiority trial. Lancet Neurol. 2022;21(6):511-519. doi:10.1016/S1474-4422(22)00124-7
PPI use in type 2 diabetes links with cardiovascular events
Among people with type 2 diabetes who self-reported regularly using a proton pump inhibitor (PPI), the incidence of cardiovascular disease (CVD) events as well as all-cause death was significantly increased in a study of more than 19,000 people with type 2 diabetes in a prospective U.K. database.
During median follow-up of about 11 years, regular use of a PPI by people with type 2 diabetes was significantly linked with a 27% relative increase in the incidence of coronary artery disease, compared with nonuse of a PPI, after full adjustment for potential confounding variables.
The results also show PPI use was significantly linked after full adjustment with a 34% relative increase in MI, a 35% relative increase in heart failure, and a 30% relative increase in all-cause death, say a team of Chinese researchers in a recent report in the Journal of Clinical Endocrinology and Metabolism.
PPIs are a medication class widely used in both over-the-counter and prescription formulations to reduce acid production in the stomach and to treat gastroesophageal reflux disease and other acid-related disorders. The PPI class includes such widely used agents as esomeprazole (Nexium), lansoprazole (Prevacid), and omeprazole (Prilosec).
The analyses in this report, which used data collected in the UK Biobank, are “rigorous,” and the findings of “a modest elevation of CVD risk are consistent with a growing number of observational studies in populations with and without diabetes,” commented Mary R. Rooney, PhD, an epidemiologist at Johns Hopkins University, Baltimore, who focuses on diabetes and cardiovascular diseases.
Prior observational reports
For example, a report from a prospective, observational study of more than 4300 U.S. residents published in 2021 that Dr. Rooney coauthored documented that cumulative PPI exposure for more than 5 years was significantly linked with a twofold increase in the rate of CVD events, compared with people who did not use a PPI. (This analysis did not examine a possible effect of diabetes status.)
And in a separate prospective, observational study of more than 1,000 Australians with type 2 diabetes, initiation of PPI treatment was significantly linked with a 3.6-fold increased incidence of CVD events, compared with PPI nonuse.
However, Dr. Rooney cautioned that the role of PPI use in raising CVD events “is still an unresolved question. It is too soon to tell if PPI use in people with diabetes should trigger additional caution.” Findings are needed from prospective, randomized trials to determine more definitively whether PPIs play a causal role in the incidence of CVD events, she said in an interview.
U.S. practice often results in unwarranted prolongation of PPI treatment, said the authors of an editorial that accompanied the 2021 report by Dr. Rooney and coauthors.
Long-term PPI use threatens harm
“The practice of initiating stress ulcer prophylaxis [by administering a PPI] in critical care is common,” wrote the authors of the 2021 editorial, Nitin Malik, MD, and William S. Weintraub, MD. “Although it is data driven and well intentioned, the possibility of causing harm – if it is continued on a long-term basis after resolution of the acute illness – is palpable.”
The new analyses using UK Biobank data included 19,229 adults with type 2 diabetes and no preexisting coronary artery disease, MI, heart failure, or stroke. The cohort included 15,954 people (83%) who did not report using a PPI and 3,275 who currently used PPIs regularly. Study limitations include self-report as the only verification of PPI use and lack of information on type of PPI, dose size, or use duration.
The findings remained consistent in several sensitivity analyses, including a propensity score–matched analysis and after further adjustment for use of histamine2 receptor antagonists, a drug class with indications similar to those for PPIs.
The authors of the report speculated that mechanisms that might link PPI use and increased CVD and mortality risk could include changes to the gut microbiota and possible interactions between PPIs and antiplatelet agents.
The study received no commercial funding. The authors and Dr. Rooney disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Among people with type 2 diabetes who self-reported regularly using a proton pump inhibitor (PPI), the incidence of cardiovascular disease (CVD) events as well as all-cause death was significantly increased in a study of more than 19,000 people with type 2 diabetes in a prospective U.K. database.
During median follow-up of about 11 years, regular use of a PPI by people with type 2 diabetes was significantly linked with a 27% relative increase in the incidence of coronary artery disease, compared with nonuse of a PPI, after full adjustment for potential confounding variables.
The results also show PPI use was significantly linked after full adjustment with a 34% relative increase in MI, a 35% relative increase in heart failure, and a 30% relative increase in all-cause death, say a team of Chinese researchers in a recent report in the Journal of Clinical Endocrinology and Metabolism.
PPIs are a medication class widely used in both over-the-counter and prescription formulations to reduce acid production in the stomach and to treat gastroesophageal reflux disease and other acid-related disorders. The PPI class includes such widely used agents as esomeprazole (Nexium), lansoprazole (Prevacid), and omeprazole (Prilosec).
The analyses in this report, which used data collected in the UK Biobank, are “rigorous,” and the findings of “a modest elevation of CVD risk are consistent with a growing number of observational studies in populations with and without diabetes,” commented Mary R. Rooney, PhD, an epidemiologist at Johns Hopkins University, Baltimore, who focuses on diabetes and cardiovascular diseases.
Prior observational reports
For example, a report from a prospective, observational study of more than 4300 U.S. residents published in 2021 that Dr. Rooney coauthored documented that cumulative PPI exposure for more than 5 years was significantly linked with a twofold increase in the rate of CVD events, compared with people who did not use a PPI. (This analysis did not examine a possible effect of diabetes status.)
And in a separate prospective, observational study of more than 1,000 Australians with type 2 diabetes, initiation of PPI treatment was significantly linked with a 3.6-fold increased incidence of CVD events, compared with PPI nonuse.
However, Dr. Rooney cautioned that the role of PPI use in raising CVD events “is still an unresolved question. It is too soon to tell if PPI use in people with diabetes should trigger additional caution.” Findings are needed from prospective, randomized trials to determine more definitively whether PPIs play a causal role in the incidence of CVD events, she said in an interview.
U.S. practice often results in unwarranted prolongation of PPI treatment, said the authors of an editorial that accompanied the 2021 report by Dr. Rooney and coauthors.
Long-term PPI use threatens harm
“The practice of initiating stress ulcer prophylaxis [by administering a PPI] in critical care is common,” wrote the authors of the 2021 editorial, Nitin Malik, MD, and William S. Weintraub, MD. “Although it is data driven and well intentioned, the possibility of causing harm – if it is continued on a long-term basis after resolution of the acute illness – is palpable.”
The new analyses using UK Biobank data included 19,229 adults with type 2 diabetes and no preexisting coronary artery disease, MI, heart failure, or stroke. The cohort included 15,954 people (83%) who did not report using a PPI and 3,275 who currently used PPIs regularly. Study limitations include self-report as the only verification of PPI use and lack of information on type of PPI, dose size, or use duration.
The findings remained consistent in several sensitivity analyses, including a propensity score–matched analysis and after further adjustment for use of histamine2 receptor antagonists, a drug class with indications similar to those for PPIs.
The authors of the report speculated that mechanisms that might link PPI use and increased CVD and mortality risk could include changes to the gut microbiota and possible interactions between PPIs and antiplatelet agents.
The study received no commercial funding. The authors and Dr. Rooney disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Among people with type 2 diabetes who self-reported regularly using a proton pump inhibitor (PPI), the incidence of cardiovascular disease (CVD) events as well as all-cause death was significantly increased in a study of more than 19,000 people with type 2 diabetes in a prospective U.K. database.
During median follow-up of about 11 years, regular use of a PPI by people with type 2 diabetes was significantly linked with a 27% relative increase in the incidence of coronary artery disease, compared with nonuse of a PPI, after full adjustment for potential confounding variables.
The results also show PPI use was significantly linked after full adjustment with a 34% relative increase in MI, a 35% relative increase in heart failure, and a 30% relative increase in all-cause death, say a team of Chinese researchers in a recent report in the Journal of Clinical Endocrinology and Metabolism.
PPIs are a medication class widely used in both over-the-counter and prescription formulations to reduce acid production in the stomach and to treat gastroesophageal reflux disease and other acid-related disorders. The PPI class includes such widely used agents as esomeprazole (Nexium), lansoprazole (Prevacid), and omeprazole (Prilosec).
The analyses in this report, which used data collected in the UK Biobank, are “rigorous,” and the findings of “a modest elevation of CVD risk are consistent with a growing number of observational studies in populations with and without diabetes,” commented Mary R. Rooney, PhD, an epidemiologist at Johns Hopkins University, Baltimore, who focuses on diabetes and cardiovascular diseases.
Prior observational reports
For example, a report from a prospective, observational study of more than 4300 U.S. residents published in 2021 that Dr. Rooney coauthored documented that cumulative PPI exposure for more than 5 years was significantly linked with a twofold increase in the rate of CVD events, compared with people who did not use a PPI. (This analysis did not examine a possible effect of diabetes status.)
And in a separate prospective, observational study of more than 1,000 Australians with type 2 diabetes, initiation of PPI treatment was significantly linked with a 3.6-fold increased incidence of CVD events, compared with PPI nonuse.
However, Dr. Rooney cautioned that the role of PPI use in raising CVD events “is still an unresolved question. It is too soon to tell if PPI use in people with diabetes should trigger additional caution.” Findings are needed from prospective, randomized trials to determine more definitively whether PPIs play a causal role in the incidence of CVD events, she said in an interview.
U.S. practice often results in unwarranted prolongation of PPI treatment, said the authors of an editorial that accompanied the 2021 report by Dr. Rooney and coauthors.
Long-term PPI use threatens harm
“The practice of initiating stress ulcer prophylaxis [by administering a PPI] in critical care is common,” wrote the authors of the 2021 editorial, Nitin Malik, MD, and William S. Weintraub, MD. “Although it is data driven and well intentioned, the possibility of causing harm – if it is continued on a long-term basis after resolution of the acute illness – is palpable.”
The new analyses using UK Biobank data included 19,229 adults with type 2 diabetes and no preexisting coronary artery disease, MI, heart failure, or stroke. The cohort included 15,954 people (83%) who did not report using a PPI and 3,275 who currently used PPIs regularly. Study limitations include self-report as the only verification of PPI use and lack of information on type of PPI, dose size, or use duration.
The findings remained consistent in several sensitivity analyses, including a propensity score–matched analysis and after further adjustment for use of histamine2 receptor antagonists, a drug class with indications similar to those for PPIs.
The authors of the report speculated that mechanisms that might link PPI use and increased CVD and mortality risk could include changes to the gut microbiota and possible interactions between PPIs and antiplatelet agents.
The study received no commercial funding. The authors and Dr. Rooney disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM THE JOURNAL OF CLINICAL ENDOCRINOLOGY AND METABOLISM
Some BP meds tied to significantly lower risk for dementia, Alzheimer’s
Antihypertensive medications that stimulate rather than inhibit type 2 and 4 angiotensin II receptors can lower the rate of dementia among new users of these medications, new research suggests.
Results from a cohort study of more than 57,000 older Medicare beneficiaries showed that the initiation of antihypertensives that stimulate the receptors was linked to a 16% lower risk for incident Alzheimer’s disease and related dementia (ADRD) and an 18% lower risk for vascular dementia compared with those that inhibit the receptors.
“Achieving appropriate blood pressure control is essential for maximizing brain health, and this promising research suggests certain antihypertensives could yield brain benefit compared to others,” lead study author Zachary A. Marcum, PharmD, PhD, associate professor, University of Washington School of Pharmacy, Seattle, told this news organization.
The findings were published online in JAMA Network Open.
Medicare beneficiaries
Previous observational studies showed that antihypertensive medications that stimulate type 2 and 4 angiotensin II receptors, in comparison with those that don’t, were associated with lower rates of dementia. However, those studies included individuals with prevalent hypertension and were relatively small.
The new retrospective cohort study included a random sample of 57,773 Medicare beneficiaries aged at least 65 years with new-onset hypertension. The mean age of participants was 73.8 years, 62.9% were women, and 86.9% were White.
Over the course of the study, some participants filled at least one prescription for a stimulating angiotensin II receptor type 2 and 4, such as angiotensin II receptor type 1 blockers, dihydropyridine calcium channel blockers, and thiazide diuretics.
Others participants filled a prescription for an inhibiting type 2 and 4 angiotensin II receptors, including angiotensin-converting enzyme (ACE) inhibitors, beta-blockers, and nondihydropyridine calcium channel blockers.
“All these medications lower blood pressure, but they do it in different ways,” said Dr. Marcum.
The researchers were interested in the varying activity of these drugs at the type 2 and 4 angiotensin II receptors.
For each 30-day interval, they categorized beneficiaries into four groups: a stimulating medication group (n = 4,879) consisting of individuals mostly taking stimulating antihypertensives; an inhibiting medication group (n = 10,303) that mostly included individuals prescribed this type of antihypertensive; a mixed group (n = 2,179) that included a combination of the first two classifications; and a nonuser group (n = 40,413) of individuals who were not using either type of drug.
The primary outcome was time to first occurrence of ADRD. The secondary outcome was time to first occurrence of vascular dementia.
Researchers controlled for cardiovascular risk factors and sociodemographic characteristics, such as age, sex, race/ethnicity, and receipt of low-income subsidy.
Unanswered questions
After adjustments, results showed that initiation of an antihypertensive medication regimen that exclusively stimulates, rather than inhibits, type 2 and 4 angiotensin II receptors was associated with a 16% lower risk for incident ADRD over a follow-up of just under 7 years (hazard ratio, 0.84; 95% confidence interval, 0.79-0.90; P < .001).
The mixed regimen was also associated with statistically significant (P = .001) reduced odds of ADRD compared with the inhibiting medications.
As for vascular dementia, use of stimulating vs. inhibiting medications was associated with an 18% lower risk (HR, 0.82; 95% CI, 0.69-0.96; P = .02).
Again, use of the mixed regimen was associated with reduced risk of vascular dementia compared with the inhibiting medications (P = .03).
A variety of potential mechanisms might explain the superiority of stimulating agents when it comes to dementia risk, said Dr. Marcum. These could include, for example, increased blood flow to the brain and reduced amyloid.
“But more mechanistic work is needed as well as evaluation of dose responses, because that’s not something we looked at in this study,” Dr. Marcum said. “There are still a lot of unanswered questions.”
Stimulators instead of inhibitors?
The results of the current analysis come on the heels of some previous work showing the benefits of lowering blood pressure. For example, the Systolic Blood Pressure Intervention Trial (SPRINT) showed that targeting a systolic blood pressure below 120 mm Hg significantly reduces risk for heart disease, stroke, and death from these diseases.
But in contrast to previous research, the current study included only beneficiaries with incident hypertension and new use of antihypertensive medications, and it adjusted for time-varying confounding.
Prescribing stimulating instead of inhibiting treatments could make a difference at the population level, Dr. Marcum noted.
“If we could shift the prescribing a little bit from inhibiting to stimulating, that could possibly reduce dementia risk,” he said.
However, “we’re not suggesting [that all patients] have their regimen switched,” he added.
That’s because inhibiting medications still have an important place in the antihypertensive treatment armamentarium, Dr. Marcum noted. As an example, beta-blockers are used post heart attack.
As well, factors such as cost and side effects should be taken into consideration when prescribing an antihypertensive drug.
The new results could be used to set up a comparison in a future randomized controlled trial that would provide the strongest evidence for estimating causal effects of treatments, said Dr. Marcum.
‘More convincing’
Carlos G. Santos-Gallego, MD, Icahn School of Medicine at Mount Sinai, New York, said the study is “more convincing” than previous related research, as it has a larger sample size and a longer follow-up.

“And the exquisite statistical analysis gives more robustness, more solidity, to the hypothesis that drugs that stimulate type 2 and 4 angiotensin II receptors might be protective for dementia,” said Dr. Santos-Gallego, who was not involved with the research.
However, he noted that the retrospective study had some limitations, including the underdiagnosis of dementia. “The diagnosis of dementia is, honestly, very poorly done in the clinical setting,” he said.
As well, the study could be subject to “confounding by indication,” Dr. Santos-Gallego said. “There could be a third variable, another confounding factor, that’s responsible both for the dementia and for the prescription of these drugs,” he added.
For example, he noted that comorbidities such as atrial fibrillation, myocardial infarction, and heart failure might increase the risk of dementia.
He agreed with the investigators that a randomized clinical trial would address these limitations. “All comorbidities would be equally shared” in the randomized groups, and all participants would be given “a specific test for dementia at the same time,” Dr. Santos-Gallego said.
Still, he noted that the new results are in keeping with hypertension guidelines that recommend stimulating drugs.
“This trial definitely shows that the current hypertension guidelines are good treatment for our patients, not only to control blood pressure and not only to prevent infarction to prevent stroke but also to prevent dementia,” said Dr. Santos-Gallego.
Also commenting for this news organization, Heather Snyder, PhD, vice president of medical and scientific relations at the Alzheimer’s Association, said the new data provide “clarity” on why previous research had differing results on the effect of antihypertensives on cognition.
Among the caveats of this new analysis is that “it’s unclear if the demographics in this study are fully representative of Medicare beneficiaries,” said Dr. Snyder.
She, too, said a clinical trial is important “to understand if there is a preventative and/or treatment potential in the medications that stimulate type 2 and 4 angiotensin II receptors.”
The study received funding from the National Institute on Aging. Dr. Marcum and Dr. Santos-Gallego have reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Antihypertensive medications that stimulate rather than inhibit type 2 and 4 angiotensin II receptors can lower the rate of dementia among new users of these medications, new research suggests.
Results from a cohort study of more than 57,000 older Medicare beneficiaries showed that the initiation of antihypertensives that stimulate the receptors was linked to a 16% lower risk for incident Alzheimer’s disease and related dementia (ADRD) and an 18% lower risk for vascular dementia compared with those that inhibit the receptors.
“Achieving appropriate blood pressure control is essential for maximizing brain health, and this promising research suggests certain antihypertensives could yield brain benefit compared to others,” lead study author Zachary A. Marcum, PharmD, PhD, associate professor, University of Washington School of Pharmacy, Seattle, told this news organization.
The findings were published online in JAMA Network Open.
Medicare beneficiaries
Previous observational studies showed that antihypertensive medications that stimulate type 2 and 4 angiotensin II receptors, in comparison with those that don’t, were associated with lower rates of dementia. However, those studies included individuals with prevalent hypertension and were relatively small.
The new retrospective cohort study included a random sample of 57,773 Medicare beneficiaries aged at least 65 years with new-onset hypertension. The mean age of participants was 73.8 years, 62.9% were women, and 86.9% were White.
Over the course of the study, some participants filled at least one prescription for a stimulating angiotensin II receptor type 2 and 4, such as angiotensin II receptor type 1 blockers, dihydropyridine calcium channel blockers, and thiazide diuretics.
Others participants filled a prescription for an inhibiting type 2 and 4 angiotensin II receptors, including angiotensin-converting enzyme (ACE) inhibitors, beta-blockers, and nondihydropyridine calcium channel blockers.
“All these medications lower blood pressure, but they do it in different ways,” said Dr. Marcum.
The researchers were interested in the varying activity of these drugs at the type 2 and 4 angiotensin II receptors.
For each 30-day interval, they categorized beneficiaries into four groups: a stimulating medication group (n = 4,879) consisting of individuals mostly taking stimulating antihypertensives; an inhibiting medication group (n = 10,303) that mostly included individuals prescribed this type of antihypertensive; a mixed group (n = 2,179) that included a combination of the first two classifications; and a nonuser group (n = 40,413) of individuals who were not using either type of drug.
The primary outcome was time to first occurrence of ADRD. The secondary outcome was time to first occurrence of vascular dementia.
Researchers controlled for cardiovascular risk factors and sociodemographic characteristics, such as age, sex, race/ethnicity, and receipt of low-income subsidy.
Unanswered questions
After adjustments, results showed that initiation of an antihypertensive medication regimen that exclusively stimulates, rather than inhibits, type 2 and 4 angiotensin II receptors was associated with a 16% lower risk for incident ADRD over a follow-up of just under 7 years (hazard ratio, 0.84; 95% confidence interval, 0.79-0.90; P < .001).
The mixed regimen was also associated with statistically significant (P = .001) reduced odds of ADRD compared with the inhibiting medications.
As for vascular dementia, use of stimulating vs. inhibiting medications was associated with an 18% lower risk (HR, 0.82; 95% CI, 0.69-0.96; P = .02).
Again, use of the mixed regimen was associated with reduced risk of vascular dementia compared with the inhibiting medications (P = .03).
A variety of potential mechanisms might explain the superiority of stimulating agents when it comes to dementia risk, said Dr. Marcum. These could include, for example, increased blood flow to the brain and reduced amyloid.
“But more mechanistic work is needed as well as evaluation of dose responses, because that’s not something we looked at in this study,” Dr. Marcum said. “There are still a lot of unanswered questions.”
Stimulators instead of inhibitors?
The results of the current analysis come on the heels of some previous work showing the benefits of lowering blood pressure. For example, the Systolic Blood Pressure Intervention Trial (SPRINT) showed that targeting a systolic blood pressure below 120 mm Hg significantly reduces risk for heart disease, stroke, and death from these diseases.
But in contrast to previous research, the current study included only beneficiaries with incident hypertension and new use of antihypertensive medications, and it adjusted for time-varying confounding.
Prescribing stimulating instead of inhibiting treatments could make a difference at the population level, Dr. Marcum noted.
“If we could shift the prescribing a little bit from inhibiting to stimulating, that could possibly reduce dementia risk,” he said.
However, “we’re not suggesting [that all patients] have their regimen switched,” he added.
That’s because inhibiting medications still have an important place in the antihypertensive treatment armamentarium, Dr. Marcum noted. As an example, beta-blockers are used post heart attack.
As well, factors such as cost and side effects should be taken into consideration when prescribing an antihypertensive drug.
The new results could be used to set up a comparison in a future randomized controlled trial that would provide the strongest evidence for estimating causal effects of treatments, said Dr. Marcum.
‘More convincing’
Carlos G. Santos-Gallego, MD, Icahn School of Medicine at Mount Sinai, New York, said the study is “more convincing” than previous related research, as it has a larger sample size and a longer follow-up.
“And the exquisite statistical analysis gives more robustness, more solidity, to the hypothesis that drugs that stimulate type 2 and 4 angiotensin II receptors might be protective for dementia,” said Dr. Santos-Gallego, who was not involved with the research.
However, he noted that the retrospective study had some limitations, including the underdiagnosis of dementia. “The diagnosis of dementia is, honestly, very poorly done in the clinical setting,” he said.
As well, the study could be subject to “confounding by indication,” Dr. Santos-Gallego said. “There could be a third variable, another confounding factor, that’s responsible both for the dementia and for the prescription of these drugs,” he added.
For example, he noted that comorbidities such as atrial fibrillation, myocardial infarction, and heart failure might increase the risk of dementia.
He agreed with the investigators that a randomized clinical trial would address these limitations. “All comorbidities would be equally shared” in the randomized groups, and all participants would be given “a specific test for dementia at the same time,” Dr. Santos-Gallego said.
Still, he noted that the new results are in keeping with hypertension guidelines that recommend stimulating drugs.
“This trial definitely shows that the current hypertension guidelines are good treatment for our patients, not only to control blood pressure and not only to prevent infarction to prevent stroke but also to prevent dementia,” said Dr. Santos-Gallego.
Also commenting for this news organization, Heather Snyder, PhD, vice president of medical and scientific relations at the Alzheimer’s Association, said the new data provide “clarity” on why previous research had differing results on the effect of antihypertensives on cognition.
Among the caveats of this new analysis is that “it’s unclear if the demographics in this study are fully representative of Medicare beneficiaries,” said Dr. Snyder.
She, too, said a clinical trial is important “to understand if there is a preventative and/or treatment potential in the medications that stimulate type 2 and 4 angiotensin II receptors.”
The study received funding from the National Institute on Aging. Dr. Marcum and Dr. Santos-Gallego have reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Antihypertensive medications that stimulate rather than inhibit type 2 and 4 angiotensin II receptors can lower the rate of dementia among new users of these medications, new research suggests.
Results from a cohort study of more than 57,000 older Medicare beneficiaries showed that the initiation of antihypertensives that stimulate the receptors was linked to a 16% lower risk for incident Alzheimer’s disease and related dementia (ADRD) and an 18% lower risk for vascular dementia compared with those that inhibit the receptors.
“Achieving appropriate blood pressure control is essential for maximizing brain health, and this promising research suggests certain antihypertensives could yield brain benefit compared to others,” lead study author Zachary A. Marcum, PharmD, PhD, associate professor, University of Washington School of Pharmacy, Seattle, told this news organization.
The findings were published online in JAMA Network Open.
Medicare beneficiaries
Previous observational studies showed that antihypertensive medications that stimulate type 2 and 4 angiotensin II receptors, in comparison with those that don’t, were associated with lower rates of dementia. However, those studies included individuals with prevalent hypertension and were relatively small.
The new retrospective cohort study included a random sample of 57,773 Medicare beneficiaries aged at least 65 years with new-onset hypertension. The mean age of participants was 73.8 years, 62.9% were women, and 86.9% were White.
Over the course of the study, some participants filled at least one prescription for a stimulating angiotensin II receptor type 2 and 4, such as angiotensin II receptor type 1 blockers, dihydropyridine calcium channel blockers, and thiazide diuretics.
Others participants filled a prescription for an inhibiting type 2 and 4 angiotensin II receptors, including angiotensin-converting enzyme (ACE) inhibitors, beta-blockers, and nondihydropyridine calcium channel blockers.
“All these medications lower blood pressure, but they do it in different ways,” said Dr. Marcum.
The researchers were interested in the varying activity of these drugs at the type 2 and 4 angiotensin II receptors.
For each 30-day interval, they categorized beneficiaries into four groups: a stimulating medication group (n = 4,879) consisting of individuals mostly taking stimulating antihypertensives; an inhibiting medication group (n = 10,303) that mostly included individuals prescribed this type of antihypertensive; a mixed group (n = 2,179) that included a combination of the first two classifications; and a nonuser group (n = 40,413) of individuals who were not using either type of drug.
The primary outcome was time to first occurrence of ADRD. The secondary outcome was time to first occurrence of vascular dementia.
Researchers controlled for cardiovascular risk factors and sociodemographic characteristics, such as age, sex, race/ethnicity, and receipt of low-income subsidy.
Unanswered questions
After adjustments, results showed that initiation of an antihypertensive medication regimen that exclusively stimulates, rather than inhibits, type 2 and 4 angiotensin II receptors was associated with a 16% lower risk for incident ADRD over a follow-up of just under 7 years (hazard ratio, 0.84; 95% confidence interval, 0.79-0.90; P < .001).
The mixed regimen was also associated with statistically significant (P = .001) reduced odds of ADRD compared with the inhibiting medications.
As for vascular dementia, use of stimulating vs. inhibiting medications was associated with an 18% lower risk (HR, 0.82; 95% CI, 0.69-0.96; P = .02).
Again, use of the mixed regimen was associated with reduced risk of vascular dementia compared with the inhibiting medications (P = .03).
A variety of potential mechanisms might explain the superiority of stimulating agents when it comes to dementia risk, said Dr. Marcum. These could include, for example, increased blood flow to the brain and reduced amyloid.
“But more mechanistic work is needed as well as evaluation of dose responses, because that’s not something we looked at in this study,” Dr. Marcum said. “There are still a lot of unanswered questions.”
Stimulators instead of inhibitors?
The results of the current analysis come on the heels of some previous work showing the benefits of lowering blood pressure. For example, the Systolic Blood Pressure Intervention Trial (SPRINT) showed that targeting a systolic blood pressure below 120 mm Hg significantly reduces risk for heart disease, stroke, and death from these diseases.
But in contrast to previous research, the current study included only beneficiaries with incident hypertension and new use of antihypertensive medications, and it adjusted for time-varying confounding.
Prescribing stimulating instead of inhibiting treatments could make a difference at the population level, Dr. Marcum noted.
“If we could shift the prescribing a little bit from inhibiting to stimulating, that could possibly reduce dementia risk,” he said.
However, “we’re not suggesting [that all patients] have their regimen switched,” he added.
That’s because inhibiting medications still have an important place in the antihypertensive treatment armamentarium, Dr. Marcum noted. As an example, beta-blockers are used post heart attack.
As well, factors such as cost and side effects should be taken into consideration when prescribing an antihypertensive drug.
The new results could be used to set up a comparison in a future randomized controlled trial that would provide the strongest evidence for estimating causal effects of treatments, said Dr. Marcum.
‘More convincing’
Carlos G. Santos-Gallego, MD, Icahn School of Medicine at Mount Sinai, New York, said the study is “more convincing” than previous related research, as it has a larger sample size and a longer follow-up.
“And the exquisite statistical analysis gives more robustness, more solidity, to the hypothesis that drugs that stimulate type 2 and 4 angiotensin II receptors might be protective for dementia,” said Dr. Santos-Gallego, who was not involved with the research.
However, he noted that the retrospective study had some limitations, including the underdiagnosis of dementia. “The diagnosis of dementia is, honestly, very poorly done in the clinical setting,” he said.
As well, the study could be subject to “confounding by indication,” Dr. Santos-Gallego said. “There could be a third variable, another confounding factor, that’s responsible both for the dementia and for the prescription of these drugs,” he added.
For example, he noted that comorbidities such as atrial fibrillation, myocardial infarction, and heart failure might increase the risk of dementia.
He agreed with the investigators that a randomized clinical trial would address these limitations. “All comorbidities would be equally shared” in the randomized groups, and all participants would be given “a specific test for dementia at the same time,” Dr. Santos-Gallego said.
Still, he noted that the new results are in keeping with hypertension guidelines that recommend stimulating drugs.
“This trial definitely shows that the current hypertension guidelines are good treatment for our patients, not only to control blood pressure and not only to prevent infarction to prevent stroke but also to prevent dementia,” said Dr. Santos-Gallego.
Also commenting for this news organization, Heather Snyder, PhD, vice president of medical and scientific relations at the Alzheimer’s Association, said the new data provide “clarity” on why previous research had differing results on the effect of antihypertensives on cognition.
Among the caveats of this new analysis is that “it’s unclear if the demographics in this study are fully representative of Medicare beneficiaries,” said Dr. Snyder.
She, too, said a clinical trial is important “to understand if there is a preventative and/or treatment potential in the medications that stimulate type 2 and 4 angiotensin II receptors.”
The study received funding from the National Institute on Aging. Dr. Marcum and Dr. Santos-Gallego have reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FDA approves second antiamyloid for Alzheimer’s disease
Like its controversial cousin aducanumab (Aduhelm, Biogen/Eisai), lecanemab was approved under the FDA’s accelerated approval pathway, which can be used to fast-track a drug that provides a meaningful therapeutic advantage over existing treatments for a serious or life-threatening illness.
Unlike aducanumab, however, there was no formal FDA advisory committee meeting on lecanemab prior to approval.
“Alzheimer’s disease immeasurably incapacitates the lives of those who suffer from it and has devastating effects on their loved ones,” Billy Dunn, MD, director of the Office of Neuroscience in the FDA’s Center for Drug Evaluation and Research, said in a press release.
“This treatment option is the latest therapy to target and affect the underlying disease process of Alzheimer’s, instead of only treating the symptoms of the disease,” Dr. Dunn added.
Eisai has reported that lecanemab will cost $26,500 a year.
Modest benefit, adverse events
The FDA noted, “The labeling states that treatment with Leqembi should be initiated in patients with mild cognitive impairment or mild dementia stage of disease, the population in which treatment was studied in clinical trials.”
The agency approved the treatment on the basis of findings from the CLARITY AD trial, which showed modest cognitive benefit for patients with early AD – but at a cost of increased risk for amyloid-related edema and effusions.
The trial enrolled 1,795 adults with mild cognitive impairment or early Alzheimer’s disease in whom amyloid pathology in the brain had been confirmed. Treatment consisted of lecanemab 10 mg/kg biweekly or matching placebo.
After 18 months of treatment, lecanemab slowed cognitive decline by 27%, compared with placebo, as measured by the Clinical Dementia Rating–Sum of Boxes (CDR-SB). This was an absolute difference of 0.45 points (change from baseline, 1.21 for lecanemab vs. 1.66 with placebo; P < .001).
While the results are “welcome news,” a 0.45-point difference on the CDR-SB might not be clinically meaningful, authors of a recent editorial in The Lancet cautioned.
Amyloid-related imaging abnormalities that manifest as edema or microhemorrhages also occurred in one in five patients taking lecanemab.
In addition, a newly published case report in The New England Journal of Medicine describes a patient with Alzheimer’s disease who was taking lecanemab and who died after experiencing numerous intracerebral hemorrhages during treatment with tissue plasminogen activator (tPA) for acute ischemic stroke.
“The findings raise the possibility of cerebral hemorrhages and necrotizing vasculopathy associated with tPA infusion in a patient with cerebrovascular amyloid who had received lecanemab,” the authors wrote.
Alzheimer’s Association reaction
Still, in anticipation of accelerated approval of lecanemab and the antiamyloid drug donanemab (Eli Lilly), which the FDA has also fast-tracked, the Alzheimer’s Association filed a formal request last month with the Centers for Medicare & Medicaid Services asking that it provide full and unrestricted coverage for FDA-approved Alzheimer’s disease treatments.
In a letter addressed to CMS administrator Chiquita Brooks-LaSure, the association asked the agency to remove the requirements for “coverage with evidence development” in its national coverage determination for FDA-approved antiamyloid monoclonal antibodies.
“Each day matters when it comes to slowing the progression of this disease,” Joanne Pike, DrPH, president and CEO for the Alzheimer’s Association, noted in a news release at the time.
“The current CMS policy to severely limit access to these treatments eliminates people’s options, is resulting in continued irreversible disease progression, and contributes to greater health inequities. That’s not acceptable,” Dr. Pike added.
After news of today’s approval was released, Dr. Pike noted in a new release, “The Alzheimer’s Association welcomes and celebrates this action by the FDA. We now have a second approved treatment that changes the course of Alzheimer’s disease in a meaningful way for people in the early stages of the disease.”
Maria C. Carrillo, PhD, chief science officer at the Alzheimer’s Association, called today’s approval “a milestone achievement.”
“The progress we’ve seen in not only this class of treatments but also in the diversification of treatment types and targets over the past few years is exciting and provides real hope to those impacted by this devastating disease,” Dr. Carrillo said.
Critical issues
Commenting on the approval, Alvaro Pascual-Leone, MD, PhD, professor of neurology at Harvard Medical School, Boston, and chief medical officer at Linus Health, said FDA approval of lecanemab and its adoption in the clinic represent a “very exciting development and prospect; but arguably some critical issues need to be considered.”
He noted that the health care system “is not currently prepared to cope with the challenges and demands of lecanemab,” as well as future pharmacologic agents.
“First, we need better workflows to identify suitable patients who can most benefit from this treatment,” said Dr. Pascual-Leone. He added that beyond identification of cognitive difficulties, amyloid status will need to be determined.
“Presently, this requires expensive and invasive tests,” such as positron-emission tomography scans or lumbar punctures for cerebrospinal fluid analysis. However, these are not fully covered by insurance companies and would be challenging to fully scale, he noted.
“In addition to screening, health systems will need to resolve the logistics challenges around the administration of lecanemab with twice-monthly infusions and the need for careful longitudinal evaluations for potential side effects,” said Dr. Pascual-Leone.
“While lecanemab may represent the first disease-modifying therapy widely available for early Alzheimer’s disease, the likely more promising approach is the addition of other therapies to lecanemab as part of a multi-intervention strategy combining pharmacologic and nonpharmacologic interventions,” he added.
Dr. Pascual-Leone has served as a paid member on scientific advisory boards for Neuroelectrics, Magstim, TetraNeuron, Skin2Neuron, MedRhythms, and Hearts Radiant and is a cofounder of TI Solutions and Linus Health.
A version of this article first appeared on Medscape.com.
This article was updated 1/9/23.
Like its controversial cousin aducanumab (Aduhelm, Biogen/Eisai), lecanemab was approved under the FDA’s accelerated approval pathway, which can be used to fast-track a drug that provides a meaningful therapeutic advantage over existing treatments for a serious or life-threatening illness.
Unlike aducanumab, however, there was no formal FDA advisory committee meeting on lecanemab prior to approval.
“Alzheimer’s disease immeasurably incapacitates the lives of those who suffer from it and has devastating effects on their loved ones,” Billy Dunn, MD, director of the Office of Neuroscience in the FDA’s Center for Drug Evaluation and Research, said in a press release.
“This treatment option is the latest therapy to target and affect the underlying disease process of Alzheimer’s, instead of only treating the symptoms of the disease,” Dr. Dunn added.
Eisai has reported that lecanemab will cost $26,500 a year.
Modest benefit, adverse events
The FDA noted, “The labeling states that treatment with Leqembi should be initiated in patients with mild cognitive impairment or mild dementia stage of disease, the population in which treatment was studied in clinical trials.”
The agency approved the treatment on the basis of findings from the CLARITY AD trial, which showed modest cognitive benefit for patients with early AD – but at a cost of increased risk for amyloid-related edema and effusions.
The trial enrolled 1,795 adults with mild cognitive impairment or early Alzheimer’s disease in whom amyloid pathology in the brain had been confirmed. Treatment consisted of lecanemab 10 mg/kg biweekly or matching placebo.
After 18 months of treatment, lecanemab slowed cognitive decline by 27%, compared with placebo, as measured by the Clinical Dementia Rating–Sum of Boxes (CDR-SB). This was an absolute difference of 0.45 points (change from baseline, 1.21 for lecanemab vs. 1.66 with placebo; P < .001).
While the results are “welcome news,” a 0.45-point difference on the CDR-SB might not be clinically meaningful, authors of a recent editorial in The Lancet cautioned.
Amyloid-related imaging abnormalities that manifest as edema or microhemorrhages also occurred in one in five patients taking lecanemab.
In addition, a newly published case report in The New England Journal of Medicine describes a patient with Alzheimer’s disease who was taking lecanemab and who died after experiencing numerous intracerebral hemorrhages during treatment with tissue plasminogen activator (tPA) for acute ischemic stroke.
“The findings raise the possibility of cerebral hemorrhages and necrotizing vasculopathy associated with tPA infusion in a patient with cerebrovascular amyloid who had received lecanemab,” the authors wrote.
Alzheimer’s Association reaction
Still, in anticipation of accelerated approval of lecanemab and the antiamyloid drug donanemab (Eli Lilly), which the FDA has also fast-tracked, the Alzheimer’s Association filed a formal request last month with the Centers for Medicare & Medicaid Services asking that it provide full and unrestricted coverage for FDA-approved Alzheimer’s disease treatments.
In a letter addressed to CMS administrator Chiquita Brooks-LaSure, the association asked the agency to remove the requirements for “coverage with evidence development” in its national coverage determination for FDA-approved antiamyloid monoclonal antibodies.
“Each day matters when it comes to slowing the progression of this disease,” Joanne Pike, DrPH, president and CEO for the Alzheimer’s Association, noted in a news release at the time.
“The current CMS policy to severely limit access to these treatments eliminates people’s options, is resulting in continued irreversible disease progression, and contributes to greater health inequities. That’s not acceptable,” Dr. Pike added.
After news of today’s approval was released, Dr. Pike noted in a new release, “The Alzheimer’s Association welcomes and celebrates this action by the FDA. We now have a second approved treatment that changes the course of Alzheimer’s disease in a meaningful way for people in the early stages of the disease.”
Maria C. Carrillo, PhD, chief science officer at the Alzheimer’s Association, called today’s approval “a milestone achievement.”
“The progress we’ve seen in not only this class of treatments but also in the diversification of treatment types and targets over the past few years is exciting and provides real hope to those impacted by this devastating disease,” Dr. Carrillo said.
Critical issues
Commenting on the approval, Alvaro Pascual-Leone, MD, PhD, professor of neurology at Harvard Medical School, Boston, and chief medical officer at Linus Health, said FDA approval of lecanemab and its adoption in the clinic represent a “very exciting development and prospect; but arguably some critical issues need to be considered.”
He noted that the health care system “is not currently prepared to cope with the challenges and demands of lecanemab,” as well as future pharmacologic agents.
“First, we need better workflows to identify suitable patients who can most benefit from this treatment,” said Dr. Pascual-Leone. He added that beyond identification of cognitive difficulties, amyloid status will need to be determined.
“Presently, this requires expensive and invasive tests,” such as positron-emission tomography scans or lumbar punctures for cerebrospinal fluid analysis. However, these are not fully covered by insurance companies and would be challenging to fully scale, he noted.
“In addition to screening, health systems will need to resolve the logistics challenges around the administration of lecanemab with twice-monthly infusions and the need for careful longitudinal evaluations for potential side effects,” said Dr. Pascual-Leone.
“While lecanemab may represent the first disease-modifying therapy widely available for early Alzheimer’s disease, the likely more promising approach is the addition of other therapies to lecanemab as part of a multi-intervention strategy combining pharmacologic and nonpharmacologic interventions,” he added.
Dr. Pascual-Leone has served as a paid member on scientific advisory boards for Neuroelectrics, Magstim, TetraNeuron, Skin2Neuron, MedRhythms, and Hearts Radiant and is a cofounder of TI Solutions and Linus Health.
A version of this article first appeared on Medscape.com.
This article was updated 1/9/23.
Like its controversial cousin aducanumab (Aduhelm, Biogen/Eisai), lecanemab was approved under the FDA’s accelerated approval pathway, which can be used to fast-track a drug that provides a meaningful therapeutic advantage over existing treatments for a serious or life-threatening illness.
Unlike aducanumab, however, there was no formal FDA advisory committee meeting on lecanemab prior to approval.
“Alzheimer’s disease immeasurably incapacitates the lives of those who suffer from it and has devastating effects on their loved ones,” Billy Dunn, MD, director of the Office of Neuroscience in the FDA’s Center for Drug Evaluation and Research, said in a press release.
“This treatment option is the latest therapy to target and affect the underlying disease process of Alzheimer’s, instead of only treating the symptoms of the disease,” Dr. Dunn added.
Eisai has reported that lecanemab will cost $26,500 a year.
Modest benefit, adverse events
The FDA noted, “The labeling states that treatment with Leqembi should be initiated in patients with mild cognitive impairment or mild dementia stage of disease, the population in which treatment was studied in clinical trials.”
The agency approved the treatment on the basis of findings from the CLARITY AD trial, which showed modest cognitive benefit for patients with early AD – but at a cost of increased risk for amyloid-related edema and effusions.
The trial enrolled 1,795 adults with mild cognitive impairment or early Alzheimer’s disease in whom amyloid pathology in the brain had been confirmed. Treatment consisted of lecanemab 10 mg/kg biweekly or matching placebo.
After 18 months of treatment, lecanemab slowed cognitive decline by 27%, compared with placebo, as measured by the Clinical Dementia Rating–Sum of Boxes (CDR-SB). This was an absolute difference of 0.45 points (change from baseline, 1.21 for lecanemab vs. 1.66 with placebo; P < .001).
While the results are “welcome news,” a 0.45-point difference on the CDR-SB might not be clinically meaningful, authors of a recent editorial in The Lancet cautioned.
Amyloid-related imaging abnormalities that manifest as edema or microhemorrhages also occurred in one in five patients taking lecanemab.
In addition, a newly published case report in The New England Journal of Medicine describes a patient with Alzheimer’s disease who was taking lecanemab and who died after experiencing numerous intracerebral hemorrhages during treatment with tissue plasminogen activator (tPA) for acute ischemic stroke.
“The findings raise the possibility of cerebral hemorrhages and necrotizing vasculopathy associated with tPA infusion in a patient with cerebrovascular amyloid who had received lecanemab,” the authors wrote.
Alzheimer’s Association reaction
Still, in anticipation of accelerated approval of lecanemab and the antiamyloid drug donanemab (Eli Lilly), which the FDA has also fast-tracked, the Alzheimer’s Association filed a formal request last month with the Centers for Medicare & Medicaid Services asking that it provide full and unrestricted coverage for FDA-approved Alzheimer’s disease treatments.
In a letter addressed to CMS administrator Chiquita Brooks-LaSure, the association asked the agency to remove the requirements for “coverage with evidence development” in its national coverage determination for FDA-approved antiamyloid monoclonal antibodies.
“Each day matters when it comes to slowing the progression of this disease,” Joanne Pike, DrPH, president and CEO for the Alzheimer’s Association, noted in a news release at the time.
“The current CMS policy to severely limit access to these treatments eliminates people’s options, is resulting in continued irreversible disease progression, and contributes to greater health inequities. That’s not acceptable,” Dr. Pike added.
After news of today’s approval was released, Dr. Pike noted in a new release, “The Alzheimer’s Association welcomes and celebrates this action by the FDA. We now have a second approved treatment that changes the course of Alzheimer’s disease in a meaningful way for people in the early stages of the disease.”
Maria C. Carrillo, PhD, chief science officer at the Alzheimer’s Association, called today’s approval “a milestone achievement.”
“The progress we’ve seen in not only this class of treatments but also in the diversification of treatment types and targets over the past few years is exciting and provides real hope to those impacted by this devastating disease,” Dr. Carrillo said.
Critical issues
Commenting on the approval, Alvaro Pascual-Leone, MD, PhD, professor of neurology at Harvard Medical School, Boston, and chief medical officer at Linus Health, said FDA approval of lecanemab and its adoption in the clinic represent a “very exciting development and prospect; but arguably some critical issues need to be considered.”
He noted that the health care system “is not currently prepared to cope with the challenges and demands of lecanemab,” as well as future pharmacologic agents.
“First, we need better workflows to identify suitable patients who can most benefit from this treatment,” said Dr. Pascual-Leone. He added that beyond identification of cognitive difficulties, amyloid status will need to be determined.
“Presently, this requires expensive and invasive tests,” such as positron-emission tomography scans or lumbar punctures for cerebrospinal fluid analysis. However, these are not fully covered by insurance companies and would be challenging to fully scale, he noted.
“In addition to screening, health systems will need to resolve the logistics challenges around the administration of lecanemab with twice-monthly infusions and the need for careful longitudinal evaluations for potential side effects,” said Dr. Pascual-Leone.
“While lecanemab may represent the first disease-modifying therapy widely available for early Alzheimer’s disease, the likely more promising approach is the addition of other therapies to lecanemab as part of a multi-intervention strategy combining pharmacologic and nonpharmacologic interventions,” he added.
Dr. Pascual-Leone has served as a paid member on scientific advisory boards for Neuroelectrics, Magstim, TetraNeuron, Skin2Neuron, MedRhythms, and Hearts Radiant and is a cofounder of TI Solutions and Linus Health.
A version of this article first appeared on Medscape.com.
This article was updated 1/9/23.
IV ketamine a promising option for resistant depression in older adults
Results showed nearly 50% of participants responded to ketamine and 25% achieved complete remission from TRD, as measured by scores on the Montgomery-Asberg Depression Rating Scale (MADRS).

“Our pilot study suggests that IV ketamine is well-tolerated, safe, and associated with improvement in late-life TRD,” co-investigator Marie Anne Gebara, MD, assistant professor of psychiatry at the University of Pittsburgh, told this news organization.
Dr. Gebara pointed out the treatment “may not be appropriate for all patients with TRD,” such as those with a history of psychotic symptoms or uncontrolled hypertension; but “it appears to be a promising option.”
The findings were published online in the American Journal of Geriatric Psychiatry.
Lack of data in seniors
Although ketamine has been shown in prior research to rapidly reduce suicidal ideation in adults, there has been a lack of data on its efficacy and safety in older adults, the current investigators note.
“Almost 50% of older adults suffering from depression have TRD, which is a leading cause of disability, excess mortality from suicide, and dementia,” Dr. Gebara said.
She added that after two failed trials of antidepressants, “older adults have few evidence-based choices: aripiprazole or bupropion augmentation, transcranial magnetic stimulation, or electroconvulsive therapy. Novel treatments with rapid benefit are needed as long-term outcomes are poor and recurrence rates are high.”
Dr. Gebara and colleagues at five sites (Columbia University, New York State Psychiatric Institute, University of Toronto, University of Pittsburgh, and Washington University in St. Louis) each enrolled five participants aged 60 and older into the pilot study between October 2020 and November 2021, for a total of 25 participants (mean age, 71 years).
Each participant was recruited from patient registries or referred by behavioral health or primary care providers and diagnosed with TRD, which was defined as an episode of major depressive disorder without psychotic features that persisted despite two or more trials of antidepressants including at least one evidence-based second-line treatment.
Participants had to take an oral antidepressant dosage for at least 1 month prior to the start of the IV ketamine infusions, and continue their antidepressant for the length of the trial.
They received IV ketamine twice weekly for 4 weeks. The dosage was weight-dependent.
At the end of the 4 weeks, participants who achieved a MADRS total score of less than 10 or had a 30% or greater reduction from their baseline MADRS score entered another 4-week phase of the trial. This phase consisted of once-weekly administration of IV ketamine.
Larger plans
Results showed 15 of the 25 participants (60%) experienced a 30% or higher reduction in MADRS scores in the first phase of the study. The mean change in MADRS total score from the beginning to the end of the first phase was a decrease of 9.4 points (P < .01).
At the end of the continuation phase, half (48%) met criteria for response and 27% met criteria for remission.
After ketamine administration, the researchers also found an improvement in Fluid Cognition Composite Score (Cohen’s d value = .61), indicating a medium to large effect size, and in three measures of executive function.
Overall, adverse events were rare and did not keep patients from participating in the study, the investigators note. Five of the 25 participants reported infusion-induced hypertension that was transient.
Study limitations cited include the small sample size and the absence of randomization and placebo control or comparison treatment.
“We were very pleased with these findings because they establish the safety of this novel intervention in older adults,” Dr. Gebara said.
“After establishing safety and tolerability, we can plan for larger, randomized controlled trials that will allow us to determine the effectiveness of IV ketamine for older adults with TRD,” she added.
Multiple mechanisms
In a comment, Gerard Sanacora, MD, PhD, professor of psychiatry at Yale University and director of the Yale Depression Research Program, New Haven, Conn., noted multiple mechanisms likely contribute to the antidepressant effects of ketamine.

Dr. Sanacora has independently researched the effects of ketamine but was not involved with the current study.
“Much of the work to date has focused on the drug’s proximal effects on the glutamatergic neurotransmitter system and the resulting enhancement of adaptive neuroplasticity in several brain regions,” he said.
“However, there is also evidence to suggest other neurotransmitter systems and possibly even neuroinflammatory regulators are also contributing to the effect,” Dr. Sanacora added.
He noted that these mechanisms are also likely amplified by the “hope, optimism, expectations, and improved medical management overall that are known to be associated with treatments that require close monitoring and follow-up with health care providers.”
Dr. Gebara noted that “internal/department funds at each site” were used to support the study. She also reported receiving support from Otsuka US. Disclosures for the other investigators are listed in the original article. Dr. Sanacora has reported having “no major direct conflicts” with the study.
A version of this article first appeared on Medscape.com.
Results showed nearly 50% of participants responded to ketamine and 25% achieved complete remission from TRD, as measured by scores on the Montgomery-Asberg Depression Rating Scale (MADRS).
“Our pilot study suggests that IV ketamine is well-tolerated, safe, and associated with improvement in late-life TRD,” co-investigator Marie Anne Gebara, MD, assistant professor of psychiatry at the University of Pittsburgh, told this news organization.
Dr. Gebara pointed out the treatment “may not be appropriate for all patients with TRD,” such as those with a history of psychotic symptoms or uncontrolled hypertension; but “it appears to be a promising option.”
The findings were published online in the American Journal of Geriatric Psychiatry.
Lack of data in seniors
Although ketamine has been shown in prior research to rapidly reduce suicidal ideation in adults, there has been a lack of data on its efficacy and safety in older adults, the current investigators note.
“Almost 50% of older adults suffering from depression have TRD, which is a leading cause of disability, excess mortality from suicide, and dementia,” Dr. Gebara said.
She added that after two failed trials of antidepressants, “older adults have few evidence-based choices: aripiprazole or bupropion augmentation, transcranial magnetic stimulation, or electroconvulsive therapy. Novel treatments with rapid benefit are needed as long-term outcomes are poor and recurrence rates are high.”
Dr. Gebara and colleagues at five sites (Columbia University, New York State Psychiatric Institute, University of Toronto, University of Pittsburgh, and Washington University in St. Louis) each enrolled five participants aged 60 and older into the pilot study between October 2020 and November 2021, for a total of 25 participants (mean age, 71 years).
Each participant was recruited from patient registries or referred by behavioral health or primary care providers and diagnosed with TRD, which was defined as an episode of major depressive disorder without psychotic features that persisted despite two or more trials of antidepressants including at least one evidence-based second-line treatment.
Participants had to take an oral antidepressant dosage for at least 1 month prior to the start of the IV ketamine infusions, and continue their antidepressant for the length of the trial.
They received IV ketamine twice weekly for 4 weeks. The dosage was weight-dependent.
At the end of the 4 weeks, participants who achieved a MADRS total score of less than 10 or had a 30% or greater reduction from their baseline MADRS score entered another 4-week phase of the trial. This phase consisted of once-weekly administration of IV ketamine.
Larger plans
Results showed 15 of the 25 participants (60%) experienced a 30% or higher reduction in MADRS scores in the first phase of the study. The mean change in MADRS total score from the beginning to the end of the first phase was a decrease of 9.4 points (P < .01).
At the end of the continuation phase, half (48%) met criteria for response and 27% met criteria for remission.
After ketamine administration, the researchers also found an improvement in Fluid Cognition Composite Score (Cohen’s d value = .61), indicating a medium to large effect size, and in three measures of executive function.
Overall, adverse events were rare and did not keep patients from participating in the study, the investigators note. Five of the 25 participants reported infusion-induced hypertension that was transient.
Study limitations cited include the small sample size and the absence of randomization and placebo control or comparison treatment.
“We were very pleased with these findings because they establish the safety of this novel intervention in older adults,” Dr. Gebara said.
“After establishing safety and tolerability, we can plan for larger, randomized controlled trials that will allow us to determine the effectiveness of IV ketamine for older adults with TRD,” she added.
Multiple mechanisms
In a comment, Gerard Sanacora, MD, PhD, professor of psychiatry at Yale University and director of the Yale Depression Research Program, New Haven, Conn., noted multiple mechanisms likely contribute to the antidepressant effects of ketamine.
Dr. Sanacora has independently researched the effects of ketamine but was not involved with the current study.
“Much of the work to date has focused on the drug’s proximal effects on the glutamatergic neurotransmitter system and the resulting enhancement of adaptive neuroplasticity in several brain regions,” he said.
“However, there is also evidence to suggest other neurotransmitter systems and possibly even neuroinflammatory regulators are also contributing to the effect,” Dr. Sanacora added.
He noted that these mechanisms are also likely amplified by the “hope, optimism, expectations, and improved medical management overall that are known to be associated with treatments that require close monitoring and follow-up with health care providers.”
Dr. Gebara noted that “internal/department funds at each site” were used to support the study. She also reported receiving support from Otsuka US. Disclosures for the other investigators are listed in the original article. Dr. Sanacora has reported having “no major direct conflicts” with the study.
A version of this article first appeared on Medscape.com.
Results showed nearly 50% of participants responded to ketamine and 25% achieved complete remission from TRD, as measured by scores on the Montgomery-Asberg Depression Rating Scale (MADRS).
“Our pilot study suggests that IV ketamine is well-tolerated, safe, and associated with improvement in late-life TRD,” co-investigator Marie Anne Gebara, MD, assistant professor of psychiatry at the University of Pittsburgh, told this news organization.
Dr. Gebara pointed out the treatment “may not be appropriate for all patients with TRD,” such as those with a history of psychotic symptoms or uncontrolled hypertension; but “it appears to be a promising option.”
The findings were published online in the American Journal of Geriatric Psychiatry.
Lack of data in seniors
Although ketamine has been shown in prior research to rapidly reduce suicidal ideation in adults, there has been a lack of data on its efficacy and safety in older adults, the current investigators note.
“Almost 50% of older adults suffering from depression have TRD, which is a leading cause of disability, excess mortality from suicide, and dementia,” Dr. Gebara said.
She added that after two failed trials of antidepressants, “older adults have few evidence-based choices: aripiprazole or bupropion augmentation, transcranial magnetic stimulation, or electroconvulsive therapy. Novel treatments with rapid benefit are needed as long-term outcomes are poor and recurrence rates are high.”
Dr. Gebara and colleagues at five sites (Columbia University, New York State Psychiatric Institute, University of Toronto, University of Pittsburgh, and Washington University in St. Louis) each enrolled five participants aged 60 and older into the pilot study between October 2020 and November 2021, for a total of 25 participants (mean age, 71 years).
Each participant was recruited from patient registries or referred by behavioral health or primary care providers and diagnosed with TRD, which was defined as an episode of major depressive disorder without psychotic features that persisted despite two or more trials of antidepressants including at least one evidence-based second-line treatment.
Participants had to take an oral antidepressant dosage for at least 1 month prior to the start of the IV ketamine infusions, and continue their antidepressant for the length of the trial.
They received IV ketamine twice weekly for 4 weeks. The dosage was weight-dependent.
At the end of the 4 weeks, participants who achieved a MADRS total score of less than 10 or had a 30% or greater reduction from their baseline MADRS score entered another 4-week phase of the trial. This phase consisted of once-weekly administration of IV ketamine.
Larger plans
Results showed 15 of the 25 participants (60%) experienced a 30% or higher reduction in MADRS scores in the first phase of the study. The mean change in MADRS total score from the beginning to the end of the first phase was a decrease of 9.4 points (P < .01).
At the end of the continuation phase, half (48%) met criteria for response and 27% met criteria for remission.
After ketamine administration, the researchers also found an improvement in Fluid Cognition Composite Score (Cohen’s d value = .61), indicating a medium to large effect size, and in three measures of executive function.
Overall, adverse events were rare and did not keep patients from participating in the study, the investigators note. Five of the 25 participants reported infusion-induced hypertension that was transient.
Study limitations cited include the small sample size and the absence of randomization and placebo control or comparison treatment.
“We were very pleased with these findings because they establish the safety of this novel intervention in older adults,” Dr. Gebara said.
“After establishing safety and tolerability, we can plan for larger, randomized controlled trials that will allow us to determine the effectiveness of IV ketamine for older adults with TRD,” she added.
Multiple mechanisms
In a comment, Gerard Sanacora, MD, PhD, professor of psychiatry at Yale University and director of the Yale Depression Research Program, New Haven, Conn., noted multiple mechanisms likely contribute to the antidepressant effects of ketamine.
Dr. Sanacora has independently researched the effects of ketamine but was not involved with the current study.
“Much of the work to date has focused on the drug’s proximal effects on the glutamatergic neurotransmitter system and the resulting enhancement of adaptive neuroplasticity in several brain regions,” he said.
“However, there is also evidence to suggest other neurotransmitter systems and possibly even neuroinflammatory regulators are also contributing to the effect,” Dr. Sanacora added.
He noted that these mechanisms are also likely amplified by the “hope, optimism, expectations, and improved medical management overall that are known to be associated with treatments that require close monitoring and follow-up with health care providers.”
Dr. Gebara noted that “internal/department funds at each site” were used to support the study. She also reported receiving support from Otsuka US. Disclosures for the other investigators are listed in the original article. Dr. Sanacora has reported having “no major direct conflicts” with the study.
A version of this article first appeared on Medscape.com.
FROM THE AMERICAN JOURNAL OF GERIATRIC PSYCHIATRY