User login
60-year-old man • chronic cough • history of GERD & dyslipidemia • throat tickle • Dx?
THE CASE
A 60-year-old man with a past medical history of gastroesophageal reflux disease (GERD) and dyslipidemia presented to his family physician for evaluation of chronic cough. Five years prior, the patient had developed a high fever and respiratory symptoms, including a cough, and was believed to have had severe otitis media. He was treated with multiple courses of antibiotics and corticosteroids for persistent otitis media. Although the condition eventually resolved, his cough continued.
The persistent cough prompted the patient to consult a succession of specialists. First, he saw a gastroenterologist; following an esophagogastroduodenoscopy, he was prescribed pantoprazole. Despite the proton-pump inhibitor (PPI) therapy, the cough remained. Next, he had multiple visits with an otolaryngologist but that yielded no specific diagnosis for the cough. He also saw an allergist-immunologist, who identified a ragweed allergy, gave him a diagnosis of cough-variant asthma, and prescribed antihistamines and mometasone furoate and formoterol fumarate dihydrate. Neither was helpful.
After 5 years of frustration, the patient complained to his family physician that he still had a cough and “a tickle” in his throat that was worsened by speaking and drinking cold beverages. He denied fever, shortness of breath, nausea, vomiting, or any other associated symptoms.
THE DIAGNOSIS
The failed treatment attempts with antihistamines, corticosteroids, bronchodilators, and PPI therapy excluded multiple etiologies for the cough. The throat discomfort and feeling of a “tickle” prompted us to consider a nerve-related disorder on the differential. The diagnosis of laryngeal sensory neuropathy (LSN) was considered.
DISCUSSION
LSN is a relatively uncommon cause of chronic refractory cough that can also manifest with throat discomfort, dysphagia, and dysphonia.1 It is thought to result from some type of insult to the recurrent laryngeal nerve or superior laryngeal nerve via viral infections, metabolic changes, or mechanical trauma, leading to a change in the firing threshold.2 The hypothesis of nerve damage is supported by the increased incidence of LSN in patients with goiters and those with type 2 diabetes.3,4 When there is a decrease in the laryngeal sensory threshold, dysfunctional laryngeal behavior results, leading to symptoms such as persistent cough and throat clearing.
Diagnosis. LSN is often diagnosed clinically, after GERD, allergies, asthma, angiotensin-converting enzyme inhibitor intake, and psychogenic disorders have been ruled out.1 Our patient had a prior diagnosis or investigation of nearly all of these conditions. Other clues pointing to an LSN diagnosis include a cough lasting 8 weeks or more, recurrent sensory disturbances (such as a tickle) of instantaneous onset before each cough episode, triggers that can include talking or a change in air temperature, daily coughing episodes numbering in the 10s to 100s, and a nonproductive cough.5,6
Beyond clinical clues, laryngeal electromyography, which evaluates the neuromuscular system in the larynx by recording action potentials generated in the laryngeal muscles during contraction, can be used for diagnosis.4 Videostroboscopy, which allows for an enlarged and slow motion view of the vocal cords, can also be used.
Continue to: Treatment
Treatment. To both confirm the diagnosis and treat the patient in a rapid, practical fashion, a trial of a neuromodulating agent such as pregabalin or gabapentin can be employed.6-9 A study identifying 28 LSN patients found symptomatic relief in 68% of patients taking gabapentin 100 to 900 mg/d.2 In another study, 12 LSN patients given pregabalin found relief after a 1-month regimen.1 Another study of 12 patients showed amitriptyline hydrochloride and gabapentin provided a positive response in 2 months, and the addition of reflux precautions and acid-suppression therapy was helpful.9 Finally, a group of 32 patients trialed on 3 different medications (amitriptyline, desipramine, and gabapentin) found similar efficacy among the 3.6
Another option. Aside from medications, botulinum toxin type A has been shown in a case series to directly decrease laryngeal hypertonicity and possibly reduce neurogenic inflammation and neuropeptide-mediated cough.10 Another study found that 18 patients with neurogenic cough who received superior laryngeal nerve blocks had cough severity index scores decrease from an average of 26.8 pretreatment to 14.6 posttreatment (P < .0001).11
Our patient agreed to a trial of gabapentin 300 mg once a day, with titration up to a maximum of 900 mg tid. When the patient returned to the clinic 4 months later, he reported that when he reached 300 mg bid, the cough completely resolved.
THE TAKEAWAY
A persistent cough with minimal identifiable triggers is a huge disruption to a patient’s life; having to visit multiple specialists before receiving a diagnosis compounds that. In our patient’s case, the process took 5 years, which underscores how important it is that LSN be considered in the differential diagnosis. Since this is generally a diagnosis of exclusion, it is important to take a careful history of a patient with a chronic cough. If LSN seems likely, trialing a patient on neuromodulating medication is the next best step, with dose titration if necessary.
Selena R. Pasadyn, 675 West 130th Street, Hinckley, OH, 44233; [email protected]
1. Halum SL, Sycamore DL, McRae BR. A new treatment option for laryngeal sensory neuropathy. Laryngoscope. 2009;119:1844-1847.
2. Lee B, Woo P. Chronic cough as a sign of laryngeal sensory neuropathy: diagnosis and treatment. Ann Otol Rhinol Laryngol. 2005;114:253-257.
3. Hamdan AL, Jabour J, Azar ST. Goiter and laryngeal sensory neuropathy. Int J Otolaryngol. 2013;2013:765265.
4. Hamdan AL, Dowli A, Barazi R, et al. Laryngeal sensory neuropathy in patients with diabetes mellitus. J Laryngol Otol. 2014;128:725-729.
5. Bastian RW, Vaidya AM, Delsupehe KG. Sensory neuropathic cough: a common and treatable cause of chronic cough. Otolaryngol Head Neck Surg. 2006;135:17-21.
6. Bastian ZJ, Bastian RW. The use of neuralgia medications to treat sensory neuropathic cough: our experience in a retrospective cohort of thirty-two patients. PeerJ. 2015;3:e816.
7. Van de Kerkhove C, Goeminne PC, Van Bleyenbergh P, et al. A cohort description and analysis of the effect of gabapentin on idiopathic cough. Cough. 2012;8:9.
8. Mishriki YY. Laryngeal neuropathy as a cause of chronic intractable cough. Am J Med. 2007;120:e5.
9. Norris BK, Schweinfurth JM. Management of recurrent laryngeal sensory neuropathic symptoms. Ann Otol Rhinol Laryngol. 2010;119:188-191.
10. Chu MW, Lieser JD, Sinacori JT. Use of botulinum toxin type a for chronic cough: a neuropathic model. Arch Otolaryngol Head Neck Surg. 2010;136:447.
11. Simpson CB, Tibbetts KM, Loochtan MJ, et al. Treatment of chronic neurogenic cough with in-office superior laryngeal nerve block. Laryngoscope. 2018;128:1898-1903.
THE CASE
A 60-year-old man with a past medical history of gastroesophageal reflux disease (GERD) and dyslipidemia presented to his family physician for evaluation of chronic cough. Five years prior, the patient had developed a high fever and respiratory symptoms, including a cough, and was believed to have had severe otitis media. He was treated with multiple courses of antibiotics and corticosteroids for persistent otitis media. Although the condition eventually resolved, his cough continued.
The persistent cough prompted the patient to consult a succession of specialists. First, he saw a gastroenterologist; following an esophagogastroduodenoscopy, he was prescribed pantoprazole. Despite the proton-pump inhibitor (PPI) therapy, the cough remained. Next, he had multiple visits with an otolaryngologist but that yielded no specific diagnosis for the cough. He also saw an allergist-immunologist, who identified a ragweed allergy, gave him a diagnosis of cough-variant asthma, and prescribed antihistamines and mometasone furoate and formoterol fumarate dihydrate. Neither was helpful.
After 5 years of frustration, the patient complained to his family physician that he still had a cough and “a tickle” in his throat that was worsened by speaking and drinking cold beverages. He denied fever, shortness of breath, nausea, vomiting, or any other associated symptoms.
THE DIAGNOSIS
The failed treatment attempts with antihistamines, corticosteroids, bronchodilators, and PPI therapy excluded multiple etiologies for the cough. The throat discomfort and feeling of a “tickle” prompted us to consider a nerve-related disorder on the differential. The diagnosis of laryngeal sensory neuropathy (LSN) was considered.
DISCUSSION
LSN is a relatively uncommon cause of chronic refractory cough that can also manifest with throat discomfort, dysphagia, and dysphonia.1 It is thought to result from some type of insult to the recurrent laryngeal nerve or superior laryngeal nerve via viral infections, metabolic changes, or mechanical trauma, leading to a change in the firing threshold.2 The hypothesis of nerve damage is supported by the increased incidence of LSN in patients with goiters and those with type 2 diabetes.3,4 When there is a decrease in the laryngeal sensory threshold, dysfunctional laryngeal behavior results, leading to symptoms such as persistent cough and throat clearing.
Diagnosis. LSN is often diagnosed clinically, after GERD, allergies, asthma, angiotensin-converting enzyme inhibitor intake, and psychogenic disorders have been ruled out.1 Our patient had a prior diagnosis or investigation of nearly all of these conditions. Other clues pointing to an LSN diagnosis include a cough lasting 8 weeks or more, recurrent sensory disturbances (such as a tickle) of instantaneous onset before each cough episode, triggers that can include talking or a change in air temperature, daily coughing episodes numbering in the 10s to 100s, and a nonproductive cough.5,6
Beyond clinical clues, laryngeal electromyography, which evaluates the neuromuscular system in the larynx by recording action potentials generated in the laryngeal muscles during contraction, can be used for diagnosis.4 Videostroboscopy, which allows for an enlarged and slow motion view of the vocal cords, can also be used.
Continue to: Treatment
Treatment. To both confirm the diagnosis and treat the patient in a rapid, practical fashion, a trial of a neuromodulating agent such as pregabalin or gabapentin can be employed.6-9 A study identifying 28 LSN patients found symptomatic relief in 68% of patients taking gabapentin 100 to 900 mg/d.2 In another study, 12 LSN patients given pregabalin found relief after a 1-month regimen.1 Another study of 12 patients showed amitriptyline hydrochloride and gabapentin provided a positive response in 2 months, and the addition of reflux precautions and acid-suppression therapy was helpful.9 Finally, a group of 32 patients trialed on 3 different medications (amitriptyline, desipramine, and gabapentin) found similar efficacy among the 3.6
Another option. Aside from medications, botulinum toxin type A has been shown in a case series to directly decrease laryngeal hypertonicity and possibly reduce neurogenic inflammation and neuropeptide-mediated cough.10 Another study found that 18 patients with neurogenic cough who received superior laryngeal nerve blocks had cough severity index scores decrease from an average of 26.8 pretreatment to 14.6 posttreatment (P < .0001).11
Our patient agreed to a trial of gabapentin 300 mg once a day, with titration up to a maximum of 900 mg tid. When the patient returned to the clinic 4 months later, he reported that when he reached 300 mg bid, the cough completely resolved.
THE TAKEAWAY
A persistent cough with minimal identifiable triggers is a huge disruption to a patient’s life; having to visit multiple specialists before receiving a diagnosis compounds that. In our patient’s case, the process took 5 years, which underscores how important it is that LSN be considered in the differential diagnosis. Since this is generally a diagnosis of exclusion, it is important to take a careful history of a patient with a chronic cough. If LSN seems likely, trialing a patient on neuromodulating medication is the next best step, with dose titration if necessary.
Selena R. Pasadyn, 675 West 130th Street, Hinckley, OH, 44233; [email protected]
THE CASE
A 60-year-old man with a past medical history of gastroesophageal reflux disease (GERD) and dyslipidemia presented to his family physician for evaluation of chronic cough. Five years prior, the patient had developed a high fever and respiratory symptoms, including a cough, and was believed to have had severe otitis media. He was treated with multiple courses of antibiotics and corticosteroids for persistent otitis media. Although the condition eventually resolved, his cough continued.
The persistent cough prompted the patient to consult a succession of specialists. First, he saw a gastroenterologist; following an esophagogastroduodenoscopy, he was prescribed pantoprazole. Despite the proton-pump inhibitor (PPI) therapy, the cough remained. Next, he had multiple visits with an otolaryngologist but that yielded no specific diagnosis for the cough. He also saw an allergist-immunologist, who identified a ragweed allergy, gave him a diagnosis of cough-variant asthma, and prescribed antihistamines and mometasone furoate and formoterol fumarate dihydrate. Neither was helpful.
After 5 years of frustration, the patient complained to his family physician that he still had a cough and “a tickle” in his throat that was worsened by speaking and drinking cold beverages. He denied fever, shortness of breath, nausea, vomiting, or any other associated symptoms.
THE DIAGNOSIS
The failed treatment attempts with antihistamines, corticosteroids, bronchodilators, and PPI therapy excluded multiple etiologies for the cough. The throat discomfort and feeling of a “tickle” prompted us to consider a nerve-related disorder on the differential. The diagnosis of laryngeal sensory neuropathy (LSN) was considered.
DISCUSSION
LSN is a relatively uncommon cause of chronic refractory cough that can also manifest with throat discomfort, dysphagia, and dysphonia.1 It is thought to result from some type of insult to the recurrent laryngeal nerve or superior laryngeal nerve via viral infections, metabolic changes, or mechanical trauma, leading to a change in the firing threshold.2 The hypothesis of nerve damage is supported by the increased incidence of LSN in patients with goiters and those with type 2 diabetes.3,4 When there is a decrease in the laryngeal sensory threshold, dysfunctional laryngeal behavior results, leading to symptoms such as persistent cough and throat clearing.
Diagnosis. LSN is often diagnosed clinically, after GERD, allergies, asthma, angiotensin-converting enzyme inhibitor intake, and psychogenic disorders have been ruled out.1 Our patient had a prior diagnosis or investigation of nearly all of these conditions. Other clues pointing to an LSN diagnosis include a cough lasting 8 weeks or more, recurrent sensory disturbances (such as a tickle) of instantaneous onset before each cough episode, triggers that can include talking or a change in air temperature, daily coughing episodes numbering in the 10s to 100s, and a nonproductive cough.5,6
Beyond clinical clues, laryngeal electromyography, which evaluates the neuromuscular system in the larynx by recording action potentials generated in the laryngeal muscles during contraction, can be used for diagnosis.4 Videostroboscopy, which allows for an enlarged and slow motion view of the vocal cords, can also be used.
Continue to: Treatment
Treatment. To both confirm the diagnosis and treat the patient in a rapid, practical fashion, a trial of a neuromodulating agent such as pregabalin or gabapentin can be employed.6-9 A study identifying 28 LSN patients found symptomatic relief in 68% of patients taking gabapentin 100 to 900 mg/d.2 In another study, 12 LSN patients given pregabalin found relief after a 1-month regimen.1 Another study of 12 patients showed amitriptyline hydrochloride and gabapentin provided a positive response in 2 months, and the addition of reflux precautions and acid-suppression therapy was helpful.9 Finally, a group of 32 patients trialed on 3 different medications (amitriptyline, desipramine, and gabapentin) found similar efficacy among the 3.6
Another option. Aside from medications, botulinum toxin type A has been shown in a case series to directly decrease laryngeal hypertonicity and possibly reduce neurogenic inflammation and neuropeptide-mediated cough.10 Another study found that 18 patients with neurogenic cough who received superior laryngeal nerve blocks had cough severity index scores decrease from an average of 26.8 pretreatment to 14.6 posttreatment (P < .0001).11
Our patient agreed to a trial of gabapentin 300 mg once a day, with titration up to a maximum of 900 mg tid. When the patient returned to the clinic 4 months later, he reported that when he reached 300 mg bid, the cough completely resolved.
THE TAKEAWAY
A persistent cough with minimal identifiable triggers is a huge disruption to a patient’s life; having to visit multiple specialists before receiving a diagnosis compounds that. In our patient’s case, the process took 5 years, which underscores how important it is that LSN be considered in the differential diagnosis. Since this is generally a diagnosis of exclusion, it is important to take a careful history of a patient with a chronic cough. If LSN seems likely, trialing a patient on neuromodulating medication is the next best step, with dose titration if necessary.
Selena R. Pasadyn, 675 West 130th Street, Hinckley, OH, 44233; [email protected]
1. Halum SL, Sycamore DL, McRae BR. A new treatment option for laryngeal sensory neuropathy. Laryngoscope. 2009;119:1844-1847.
2. Lee B, Woo P. Chronic cough as a sign of laryngeal sensory neuropathy: diagnosis and treatment. Ann Otol Rhinol Laryngol. 2005;114:253-257.
3. Hamdan AL, Jabour J, Azar ST. Goiter and laryngeal sensory neuropathy. Int J Otolaryngol. 2013;2013:765265.
4. Hamdan AL, Dowli A, Barazi R, et al. Laryngeal sensory neuropathy in patients with diabetes mellitus. J Laryngol Otol. 2014;128:725-729.
5. Bastian RW, Vaidya AM, Delsupehe KG. Sensory neuropathic cough: a common and treatable cause of chronic cough. Otolaryngol Head Neck Surg. 2006;135:17-21.
6. Bastian ZJ, Bastian RW. The use of neuralgia medications to treat sensory neuropathic cough: our experience in a retrospective cohort of thirty-two patients. PeerJ. 2015;3:e816.
7. Van de Kerkhove C, Goeminne PC, Van Bleyenbergh P, et al. A cohort description and analysis of the effect of gabapentin on idiopathic cough. Cough. 2012;8:9.
8. Mishriki YY. Laryngeal neuropathy as a cause of chronic intractable cough. Am J Med. 2007;120:e5.
9. Norris BK, Schweinfurth JM. Management of recurrent laryngeal sensory neuropathic symptoms. Ann Otol Rhinol Laryngol. 2010;119:188-191.
10. Chu MW, Lieser JD, Sinacori JT. Use of botulinum toxin type a for chronic cough: a neuropathic model. Arch Otolaryngol Head Neck Surg. 2010;136:447.
11. Simpson CB, Tibbetts KM, Loochtan MJ, et al. Treatment of chronic neurogenic cough with in-office superior laryngeal nerve block. Laryngoscope. 2018;128:1898-1903.
1. Halum SL, Sycamore DL, McRae BR. A new treatment option for laryngeal sensory neuropathy. Laryngoscope. 2009;119:1844-1847.
2. Lee B, Woo P. Chronic cough as a sign of laryngeal sensory neuropathy: diagnosis and treatment. Ann Otol Rhinol Laryngol. 2005;114:253-257.
3. Hamdan AL, Jabour J, Azar ST. Goiter and laryngeal sensory neuropathy. Int J Otolaryngol. 2013;2013:765265.
4. Hamdan AL, Dowli A, Barazi R, et al. Laryngeal sensory neuropathy in patients with diabetes mellitus. J Laryngol Otol. 2014;128:725-729.
5. Bastian RW, Vaidya AM, Delsupehe KG. Sensory neuropathic cough: a common and treatable cause of chronic cough. Otolaryngol Head Neck Surg. 2006;135:17-21.
6. Bastian ZJ, Bastian RW. The use of neuralgia medications to treat sensory neuropathic cough: our experience in a retrospective cohort of thirty-two patients. PeerJ. 2015;3:e816.
7. Van de Kerkhove C, Goeminne PC, Van Bleyenbergh P, et al. A cohort description and analysis of the effect of gabapentin on idiopathic cough. Cough. 2012;8:9.
8. Mishriki YY. Laryngeal neuropathy as a cause of chronic intractable cough. Am J Med. 2007;120:e5.
9. Norris BK, Schweinfurth JM. Management of recurrent laryngeal sensory neuropathic symptoms. Ann Otol Rhinol Laryngol. 2010;119:188-191.
10. Chu MW, Lieser JD, Sinacori JT. Use of botulinum toxin type a for chronic cough: a neuropathic model. Arch Otolaryngol Head Neck Surg. 2010;136:447.
11. Simpson CB, Tibbetts KM, Loochtan MJ, et al. Treatment of chronic neurogenic cough with in-office superior laryngeal nerve block. Laryngoscope. 2018;128:1898-1903.

20-year-old man • sudden-onset chest pain • worsening pain with cough and exertion • Dx?
THE CASE
A 20-year-old man presented to our clinic with a 3-day history of nonradiating chest pain located at the center of his chest. Past medical history included idiopathic neonatal giant-cell hepatitis and subsequent liver transplant at 1 month of age; he had been followed by the transplant team without rejection or infection and was in otherwise good health prior to the chest pain.
On the day of symptom onset, he was walking inside his house and fell to his knees with a chest pain described as “a punch” to the center of the chest that lasted for a few seconds. He was able to continue his daily activities without limitation despite a constant, squeezing, centrally located chest pain. The pain worsened with cough and exertion.
A few hours later, he went to an urgent care center for evaluation. There, he reported, his chest radiograph and electrocardiogram (EKG) results were normal and he was given a diagnosis of musculoskeletal chest pain. Over the next 3 days, his chest pain persisted but did not worsen. He was taking 500 mg of naproxen every 8 hours with no improvement. No other acute or chronic medications were being taken. He had no significant family history. A review of systems was otherwise negative.
On physical exam, his vital statistics included a height of 6’4”; weight, 261 lb; body mass index, 31.8; temperature, 98.7 °F; blood pressure, 134/77 mm Hg; heart rate, 92 beats/min; respiratory rate, 18 breaths/min; and oxygen saturation, 96%. Throughout the exam, he demonstrated no acute distress, appeared well, and was talkative; however, he reported having a “constant, squeezing” chest pain that did not worsen with palpation of the chest. The rest of his physical exam was unremarkable.
Although he reported that his EKG and chest radiograph were normal 3 days prior, repeat chest radiograph and EKG were ordered due to his unexplained, active chest pain and the lack of immediate access to the prior results.
THE DIAGNOSIS
The chest radiograph (FIGURE 1A) showed a “mildly ectatic ascending thoracic aorta” that had increased since a chest radiograph from 6 years prior (FIGURE 1B) and “was concerning for an aneurysm.” Computed tomography (CT) angiography (FIGURE 2) then confirmed a 7-cm aneurysm of the ascending aorta, with findings suggestive of a retrograde ascending aortic dissection.

DISCUSSION
The average age of a patient with acute aortic dissection (AAD) is 63 years; only 7% occur in people younger than 40.1 AAD is often accompanied by a predisposing risk factor such as a connective tissue disease, bicuspid aortic valve, longstanding hypertension, trauma, or larger aortic dimensions.2,3 Younger patients are more likely to have predisposing risk factors of Marfan syndrome, prior aortic surgery, or a bicuspid aortic valve.3

Continue to: A literature review did not reveal...
A literature review did not reveal any known correlation between the patient’s history of giant-cell hepatitis or antirejection therapy with thoracic aortic dissection. Furthermore, liver transplant is not known to be a specific risk factor for AAD in pediatric patients or outside the immediate postoperative period. Therefore, there were no known predisposing risk factors for AAD in our patient.
The most common clinical feature of AAD is chest pain, which occurs in 75% of patients.1 Other clinical symptoms include hypertension and diaphoresis.2,4 However, classic clinical findings are not always displayed, making the diagnosis difficult.2,4 The classical description of “tearing pain” is seen in only 51% of patients, and 5% to 15% of patients present without any pain.1
Commonly missed or misdiagnosed. The diagnosis of AAD has been missed during the initial exam in 38% of patients.4 As seen in our case, symptoms may be initially diagnosed as musculoskeletal chest pain. Based on symptoms, AAD can be incorrectly diagnosed as an acute myocardial infarction or vascular embolization.2,4
Every hour after symptom onset, the mortality rate of untreated AAD increases 1% to 2%,with no difference based on age.3,4 Different reports have shown mortality rates between 7% and 30%.4
Effective imaging is crucial to the diagnosis and treatment of AAD, given the occurrence of atypical presentation, missed diagnosis, and high mortality rate.4 A chest radiograph will show a widened mediastinum, but the preferred diagnostic tests are a CT or transthoracic echocardiogram.2,4 Once the diagnosis of AAD is confirmed, an aortic angiogram is the preferred test to determine the extent of the dissection prior to surgical treatment.2
Continue to: Classification dictates treatment
Classification dictates treatment. AAD is classified based on where the dissection of the aorta occurs. If the dissection involves the ascending aorta, it is classified as a type A AAD and should immediately be treated with emergent surgery in order to prevent complications including myocardial infarction, cardiac tamponade, and aortic rupture.2,4,5 If the dissection is limited to the descending aorta, it is classified as a type B AAD and can be medically managed by controlling pain and lowering blood pressure; if symptoms persist, surgical management may be required.2 After hospital discharge, AAD patients are followed closely with medical therapy, serial imaging, and reoperation if necessary.4
Our patient underwent emergent surgery for aortic root/ascending aortic replacement with a mechanical valve. He tolerated the procedure well. Surgical tissue pathology of the aortic segment showed a wall of elastic vessel with medial degeneration and dissection, and the tissue pathology of the aorta leaflets showed valvular tissue with myxoid degeneration.
THE TAKEAWAY
It is critical to keep AAD in the differential diagnosis of a patient presenting with acute onset of chest pain, as AAD often has an atypical presentation and can easily be misdiagnosed. Effective imaging is crucial to diagnosis, and immediate treatment is essential to patient survival.
CORRESPONDENCE
Rachel A. Reedy, PA, University of Florida, Department of General Pediatrics, 7046 SW Archer Road, Gainesville, FL 32608; [email protected]
1. Pineault J, Ouimet D, Pichette V, Vallée M. A case of aortic dissection in a young adult: a refresher of the literature of this “great masquerader.” Int J Gen Med. 2011;4:889-893.
2. Agabegi SS, Agabegi ElD, Ring AC. Diseases of the cardiovascular system. In: Jackson A, ed. Step-up to Medicine. 3rd ed. Lippincott Williams & Wilkins; 2012:54-55.
3. Januzzi JL, Isselbacher EM, Fattori R, et al. Characterizing the young patient with aortic dissection: results from the International Registry of Aortic Dissection (IRAD). J Am Coll Cardiol. 2004;43:665-669.
4. Tsai TT, Trimarchi S, Nienaber CA. Acute aortic dissection: perspectives from the International Registry of Acute Aortic Dissection (IRAD). Eur J Vasc Endovasc Surg. 2009;37:149-159.
5. Trimarchi S, Eagle KA, Nienaber CA, et al. Role of age in acute type A aortic dissection outcome: Report from the International Registry of Acute Aortic Dissection (IRAD). J Thorac Cardiovasc Surg. 2010;140:784-789.
THE CASE
A 20-year-old man presented to our clinic with a 3-day history of nonradiating chest pain located at the center of his chest. Past medical history included idiopathic neonatal giant-cell hepatitis and subsequent liver transplant at 1 month of age; he had been followed by the transplant team without rejection or infection and was in otherwise good health prior to the chest pain.
On the day of symptom onset, he was walking inside his house and fell to his knees with a chest pain described as “a punch” to the center of the chest that lasted for a few seconds. He was able to continue his daily activities without limitation despite a constant, squeezing, centrally located chest pain. The pain worsened with cough and exertion.
A few hours later, he went to an urgent care center for evaluation. There, he reported, his chest radiograph and electrocardiogram (EKG) results were normal and he was given a diagnosis of musculoskeletal chest pain. Over the next 3 days, his chest pain persisted but did not worsen. He was taking 500 mg of naproxen every 8 hours with no improvement. No other acute or chronic medications were being taken. He had no significant family history. A review of systems was otherwise negative.
On physical exam, his vital statistics included a height of 6’4”; weight, 261 lb; body mass index, 31.8; temperature, 98.7 °F; blood pressure, 134/77 mm Hg; heart rate, 92 beats/min; respiratory rate, 18 breaths/min; and oxygen saturation, 96%. Throughout the exam, he demonstrated no acute distress, appeared well, and was talkative; however, he reported having a “constant, squeezing” chest pain that did not worsen with palpation of the chest. The rest of his physical exam was unremarkable.
Although he reported that his EKG and chest radiograph were normal 3 days prior, repeat chest radiograph and EKG were ordered due to his unexplained, active chest pain and the lack of immediate access to the prior results.
THE DIAGNOSIS
The chest radiograph (FIGURE 1A) showed a “mildly ectatic ascending thoracic aorta” that had increased since a chest radiograph from 6 years prior (FIGURE 1B) and “was concerning for an aneurysm.” Computed tomography (CT) angiography (FIGURE 2) then confirmed a 7-cm aneurysm of the ascending aorta, with findings suggestive of a retrograde ascending aortic dissection.
DISCUSSION
The average age of a patient with acute aortic dissection (AAD) is 63 years; only 7% occur in people younger than 40.1 AAD is often accompanied by a predisposing risk factor such as a connective tissue disease, bicuspid aortic valve, longstanding hypertension, trauma, or larger aortic dimensions.2,3 Younger patients are more likely to have predisposing risk factors of Marfan syndrome, prior aortic surgery, or a bicuspid aortic valve.3
Continue to: A literature review did not reveal...
A literature review did not reveal any known correlation between the patient’s history of giant-cell hepatitis or antirejection therapy with thoracic aortic dissection. Furthermore, liver transplant is not known to be a specific risk factor for AAD in pediatric patients or outside the immediate postoperative period. Therefore, there were no known predisposing risk factors for AAD in our patient.
The most common clinical feature of AAD is chest pain, which occurs in 75% of patients.1 Other clinical symptoms include hypertension and diaphoresis.2,4 However, classic clinical findings are not always displayed, making the diagnosis difficult.2,4 The classical description of “tearing pain” is seen in only 51% of patients, and 5% to 15% of patients present without any pain.1
Commonly missed or misdiagnosed. The diagnosis of AAD has been missed during the initial exam in 38% of patients.4 As seen in our case, symptoms may be initially diagnosed as musculoskeletal chest pain. Based on symptoms, AAD can be incorrectly diagnosed as an acute myocardial infarction or vascular embolization.2,4
Every hour after symptom onset, the mortality rate of untreated AAD increases 1% to 2%,with no difference based on age.3,4 Different reports have shown mortality rates between 7% and 30%.4
Effective imaging is crucial to the diagnosis and treatment of AAD, given the occurrence of atypical presentation, missed diagnosis, and high mortality rate.4 A chest radiograph will show a widened mediastinum, but the preferred diagnostic tests are a CT or transthoracic echocardiogram.2,4 Once the diagnosis of AAD is confirmed, an aortic angiogram is the preferred test to determine the extent of the dissection prior to surgical treatment.2
Continue to: Classification dictates treatment
Classification dictates treatment. AAD is classified based on where the dissection of the aorta occurs. If the dissection involves the ascending aorta, it is classified as a type A AAD and should immediately be treated with emergent surgery in order to prevent complications including myocardial infarction, cardiac tamponade, and aortic rupture.2,4,5 If the dissection is limited to the descending aorta, it is classified as a type B AAD and can be medically managed by controlling pain and lowering blood pressure; if symptoms persist, surgical management may be required.2 After hospital discharge, AAD patients are followed closely with medical therapy, serial imaging, and reoperation if necessary.4
Our patient underwent emergent surgery for aortic root/ascending aortic replacement with a mechanical valve. He tolerated the procedure well. Surgical tissue pathology of the aortic segment showed a wall of elastic vessel with medial degeneration and dissection, and the tissue pathology of the aorta leaflets showed valvular tissue with myxoid degeneration.
THE TAKEAWAY
It is critical to keep AAD in the differential diagnosis of a patient presenting with acute onset of chest pain, as AAD often has an atypical presentation and can easily be misdiagnosed. Effective imaging is crucial to diagnosis, and immediate treatment is essential to patient survival.
CORRESPONDENCE
Rachel A. Reedy, PA, University of Florida, Department of General Pediatrics, 7046 SW Archer Road, Gainesville, FL 32608; [email protected]
THE CASE
A 20-year-old man presented to our clinic with a 3-day history of nonradiating chest pain located at the center of his chest. Past medical history included idiopathic neonatal giant-cell hepatitis and subsequent liver transplant at 1 month of age; he had been followed by the transplant team without rejection or infection and was in otherwise good health prior to the chest pain.
On the day of symptom onset, he was walking inside his house and fell to his knees with a chest pain described as “a punch” to the center of the chest that lasted for a few seconds. He was able to continue his daily activities without limitation despite a constant, squeezing, centrally located chest pain. The pain worsened with cough and exertion.
A few hours later, he went to an urgent care center for evaluation. There, he reported, his chest radiograph and electrocardiogram (EKG) results were normal and he was given a diagnosis of musculoskeletal chest pain. Over the next 3 days, his chest pain persisted but did not worsen. He was taking 500 mg of naproxen every 8 hours with no improvement. No other acute or chronic medications were being taken. He had no significant family history. A review of systems was otherwise negative.
On physical exam, his vital statistics included a height of 6’4”; weight, 261 lb; body mass index, 31.8; temperature, 98.7 °F; blood pressure, 134/77 mm Hg; heart rate, 92 beats/min; respiratory rate, 18 breaths/min; and oxygen saturation, 96%. Throughout the exam, he demonstrated no acute distress, appeared well, and was talkative; however, he reported having a “constant, squeezing” chest pain that did not worsen with palpation of the chest. The rest of his physical exam was unremarkable.
Although he reported that his EKG and chest radiograph were normal 3 days prior, repeat chest radiograph and EKG were ordered due to his unexplained, active chest pain and the lack of immediate access to the prior results.
THE DIAGNOSIS
The chest radiograph (FIGURE 1A) showed a “mildly ectatic ascending thoracic aorta” that had increased since a chest radiograph from 6 years prior (FIGURE 1B) and “was concerning for an aneurysm.” Computed tomography (CT) angiography (FIGURE 2) then confirmed a 7-cm aneurysm of the ascending aorta, with findings suggestive of a retrograde ascending aortic dissection.
DISCUSSION
The average age of a patient with acute aortic dissection (AAD) is 63 years; only 7% occur in people younger than 40.1 AAD is often accompanied by a predisposing risk factor such as a connective tissue disease, bicuspid aortic valve, longstanding hypertension, trauma, or larger aortic dimensions.2,3 Younger patients are more likely to have predisposing risk factors of Marfan syndrome, prior aortic surgery, or a bicuspid aortic valve.3
Continue to: A literature review did not reveal...
A literature review did not reveal any known correlation between the patient’s history of giant-cell hepatitis or antirejection therapy with thoracic aortic dissection. Furthermore, liver transplant is not known to be a specific risk factor for AAD in pediatric patients or outside the immediate postoperative period. Therefore, there were no known predisposing risk factors for AAD in our patient.
The most common clinical feature of AAD is chest pain, which occurs in 75% of patients.1 Other clinical symptoms include hypertension and diaphoresis.2,4 However, classic clinical findings are not always displayed, making the diagnosis difficult.2,4 The classical description of “tearing pain” is seen in only 51% of patients, and 5% to 15% of patients present without any pain.1
Commonly missed or misdiagnosed. The diagnosis of AAD has been missed during the initial exam in 38% of patients.4 As seen in our case, symptoms may be initially diagnosed as musculoskeletal chest pain. Based on symptoms, AAD can be incorrectly diagnosed as an acute myocardial infarction or vascular embolization.2,4
Every hour after symptom onset, the mortality rate of untreated AAD increases 1% to 2%,with no difference based on age.3,4 Different reports have shown mortality rates between 7% and 30%.4
Effective imaging is crucial to the diagnosis and treatment of AAD, given the occurrence of atypical presentation, missed diagnosis, and high mortality rate.4 A chest radiograph will show a widened mediastinum, but the preferred diagnostic tests are a CT or transthoracic echocardiogram.2,4 Once the diagnosis of AAD is confirmed, an aortic angiogram is the preferred test to determine the extent of the dissection prior to surgical treatment.2
Continue to: Classification dictates treatment
Classification dictates treatment. AAD is classified based on where the dissection of the aorta occurs. If the dissection involves the ascending aorta, it is classified as a type A AAD and should immediately be treated with emergent surgery in order to prevent complications including myocardial infarction, cardiac tamponade, and aortic rupture.2,4,5 If the dissection is limited to the descending aorta, it is classified as a type B AAD and can be medically managed by controlling pain and lowering blood pressure; if symptoms persist, surgical management may be required.2 After hospital discharge, AAD patients are followed closely with medical therapy, serial imaging, and reoperation if necessary.4
Our patient underwent emergent surgery for aortic root/ascending aortic replacement with a mechanical valve. He tolerated the procedure well. Surgical tissue pathology of the aortic segment showed a wall of elastic vessel with medial degeneration and dissection, and the tissue pathology of the aorta leaflets showed valvular tissue with myxoid degeneration.
THE TAKEAWAY
It is critical to keep AAD in the differential diagnosis of a patient presenting with acute onset of chest pain, as AAD often has an atypical presentation and can easily be misdiagnosed. Effective imaging is crucial to diagnosis, and immediate treatment is essential to patient survival.
CORRESPONDENCE
Rachel A. Reedy, PA, University of Florida, Department of General Pediatrics, 7046 SW Archer Road, Gainesville, FL 32608; [email protected]
1. Pineault J, Ouimet D, Pichette V, Vallée M. A case of aortic dissection in a young adult: a refresher of the literature of this “great masquerader.” Int J Gen Med. 2011;4:889-893.
2. Agabegi SS, Agabegi ElD, Ring AC. Diseases of the cardiovascular system. In: Jackson A, ed. Step-up to Medicine. 3rd ed. Lippincott Williams & Wilkins; 2012:54-55.
3. Januzzi JL, Isselbacher EM, Fattori R, et al. Characterizing the young patient with aortic dissection: results from the International Registry of Aortic Dissection (IRAD). J Am Coll Cardiol. 2004;43:665-669.
4. Tsai TT, Trimarchi S, Nienaber CA. Acute aortic dissection: perspectives from the International Registry of Acute Aortic Dissection (IRAD). Eur J Vasc Endovasc Surg. 2009;37:149-159.
5. Trimarchi S, Eagle KA, Nienaber CA, et al. Role of age in acute type A aortic dissection outcome: Report from the International Registry of Acute Aortic Dissection (IRAD). J Thorac Cardiovasc Surg. 2010;140:784-789.
1. Pineault J, Ouimet D, Pichette V, Vallée M. A case of aortic dissection in a young adult: a refresher of the literature of this “great masquerader.” Int J Gen Med. 2011;4:889-893.
2. Agabegi SS, Agabegi ElD, Ring AC. Diseases of the cardiovascular system. In: Jackson A, ed. Step-up to Medicine. 3rd ed. Lippincott Williams & Wilkins; 2012:54-55.
3. Januzzi JL, Isselbacher EM, Fattori R, et al. Characterizing the young patient with aortic dissection: results from the International Registry of Aortic Dissection (IRAD). J Am Coll Cardiol. 2004;43:665-669.
4. Tsai TT, Trimarchi S, Nienaber CA. Acute aortic dissection: perspectives from the International Registry of Acute Aortic Dissection (IRAD). Eur J Vasc Endovasc Surg. 2009;37:149-159.
5. Trimarchi S, Eagle KA, Nienaber CA, et al. Role of age in acute type A aortic dissection outcome: Report from the International Registry of Acute Aortic Dissection (IRAD). J Thorac Cardiovasc Surg. 2010;140:784-789.

Cutaneous Insulin-Derived Amyloidosis Presenting as Hyperkeratotic Nodules
Amyloidosis consists of approximately 30 protein-folding disorders sharing the common feature of abnormal extracellular amyloid deposition. In each condition, a specific soluble precursor protein aggregates to form the insoluble fibrils of amyloid, characterized by the beta-pleated sheet structure.1 Amyloidosis occurs as either a systemic or localized process. Insulin-derived (AIns) amyloidosis, a localized process occurring at insulin injection sites, was first reported in 1983.2 There were fewer than 20 reported cases until 2014, when 57 additional cases were reported by just 2 institutions,3,4 indicating that AIns amyloidosis may be more common than previously thought.3,5
Despite the increasing prevalence of diabetes mellitus and insulin use, there is a paucity of published cases of AIns amyloidosis. The lack of awareness of this condition among both dermatologists and general practitioners may be in part due to its variable clinical manifestations. We describe 2 patients with unique presentations of localized amyloidosis at repeated insulin injection sites.
Case Reports
Patient 1
A 39-year-old man with a history of type 1 diabetes mellitus presented with 4 asymptomatic nodules on the lateral thighs in areas of previous insulin injection. He first noticed the lesions 9 months prior to presentation and subsequently switched the injection site to the abdomen without development of new nodules. Despite being compliant with his insulin regimen, he had a long history of irregular glucose control, including frequent hypoglycemic episodes. The patient was using regular and neutral protamine hagedorn insulin.
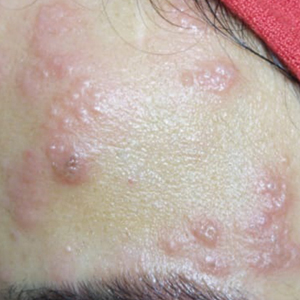
On physical examination, 2 soft, nontender, exophytic nodules were noted on each upper thigh with surrounding hyperpigmented and hyperkeratotic collarettes (Figure 1). The nodules ranged in size from 2 to 3.5 cm in diameter.
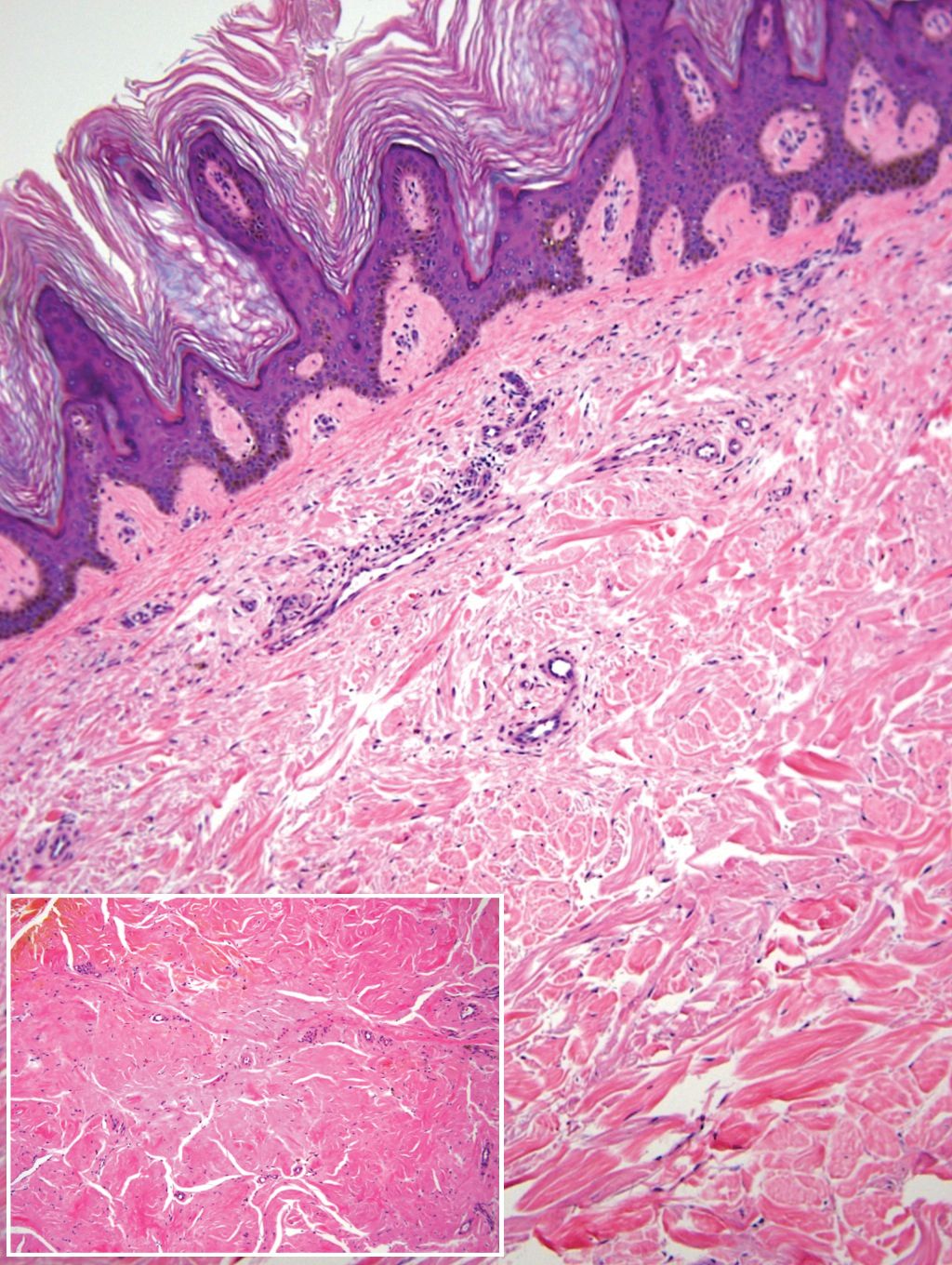
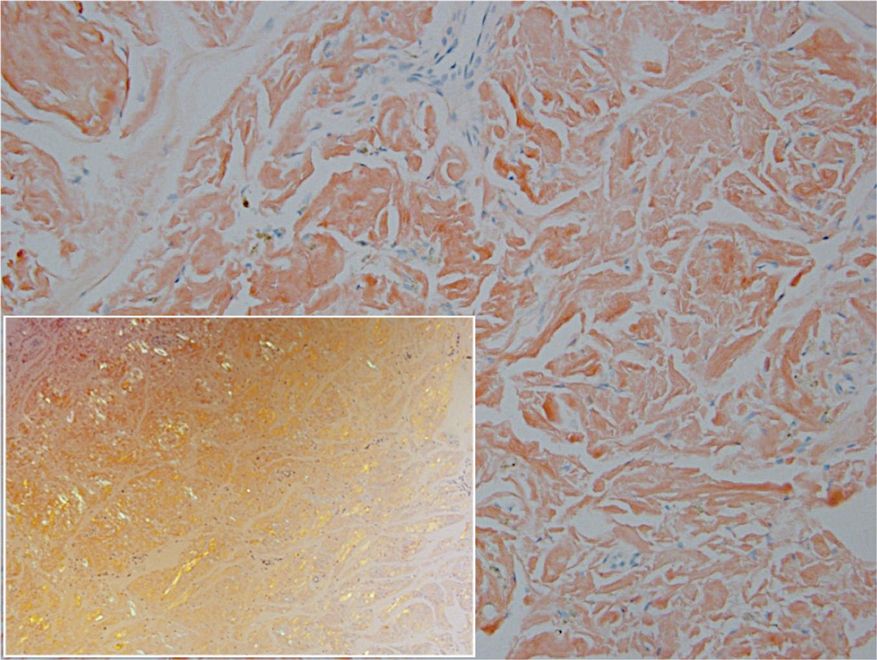
Remarkable laboratory data included a fasting glucose level of 207 mg/dL (reference range, 70–110 mg/dL) and a glycohemoglobin of 8.8% (reference range, <5.7%). Serum protein electrophoresis and immunofixation were normal. Histopathology of the lesions demonstrated diffuse deposition of pink amorphous material associated with prominent papillomatosis, hyperkeratosis, and acanthosis (Figure 2). Congo red staining was positive with green birefringence under polarized light, indicative of amyloid deposits (Figure 3). Liquid chromatography–tandem mass spectrometry of the specimens was consistent with deposition of AIns amyloidosis.
Due to the size and persistent nature of the lesions, the nodules were removed by tangential excision. In addition, the patient was advised to continue rotating injection sites frequently. His blood glucose levels are now well controlled, and he has not developed any new nodules.
Patient 2
A 53-year-old woman with a history of type 2 diabetes mellitus presented with painful subcutaneous nodules on the lower abdomen at sites of previous insulin injections. The nodules developed approximately 1 month after she started treatment with neutral protamine hagedorn insulin and had been slowly enlarging over the past year. She tried switching injection sites after noticing the lesions, but the nodules persisted. The patient had a long history of poor glucose control with chronically elevated glycohemoglobin and blood glucose levels.
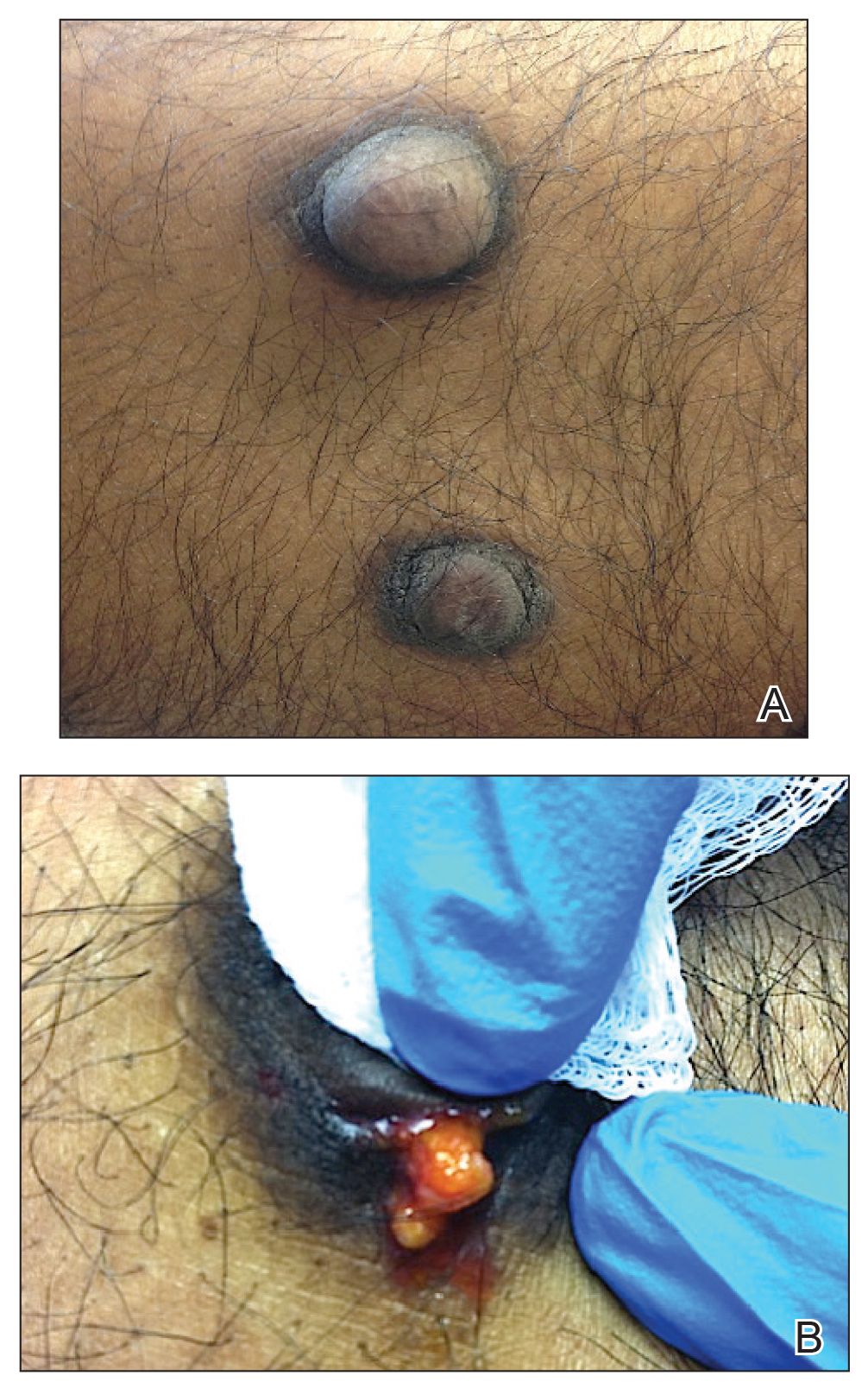
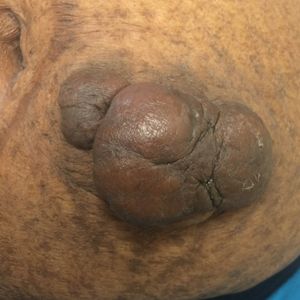
On physical examination, 2 hyperpigmented, exophytic, smooth nodules were noted on the right and left lower abdomen, ranging in size from 2.5 to 5.5 cm in diameter (Figure 4).
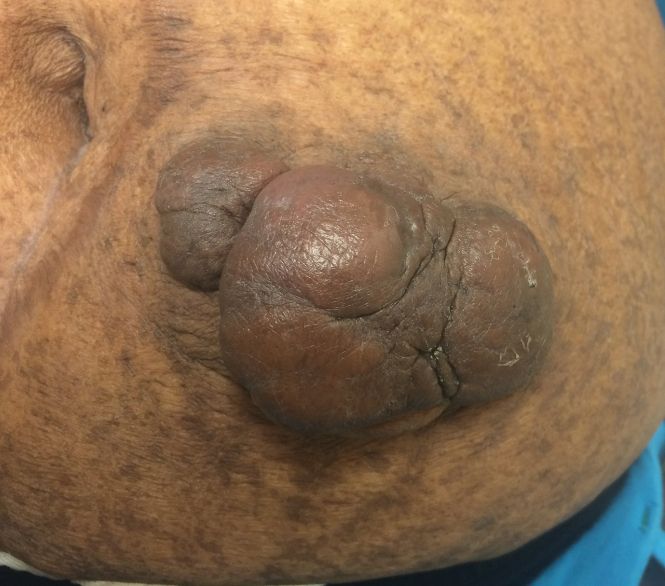
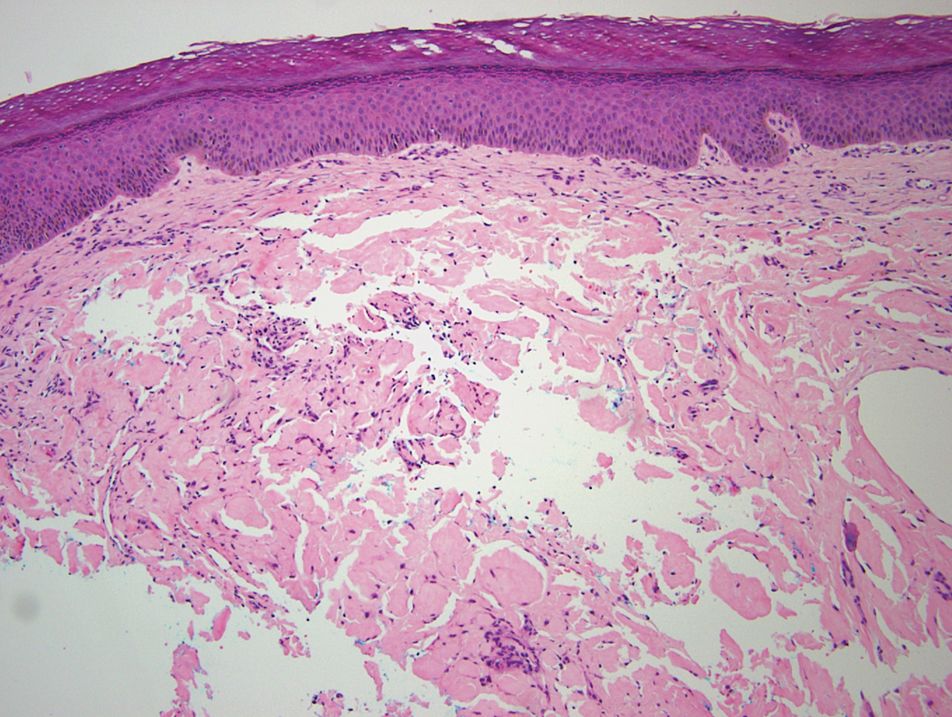
Relevant laboratory data included a fasting glucose level of 197 mg/dL and a glycohemoglobin of 9.3%. A biopsy of the lesion on the left lower abdomen revealed eosinophilic amorphous deposits with fissuring in the dermis (Figure 5). Congo red stain was positive with green birefringence under polarized light. Liquid chromatography–tandem mass spectrometry of the specimen showed deposition of AIns amyloid. The patient began injecting away from the amyloid nodules without development of any new lesions. The original nodules have persisted, and surgical excision is planned.
Comment
Insulin is the suspected precursor protein in AIns amyloidosis, but the exact pathogenesis is unknown. The protein that is derived from insulin in these tumors is now identified as AIns amyloidosis.5,6 It is hypothesized that insulin accumulates locally and is converted to amyloid by an unknown mechanism.7 Other potential contributory factors include chronic inflammation and foreign body reactions developing around amyloid deposits, as well as repeated trauma from injections into a single site.4,5 It appears that lesions may derive from a wide range of insulin types and occur after variable time periods.
A majority of cases of iatrogenic amyloid have been described as single, firm, subcutaneous masses at an injection site that commonly are misdiagnosed as lipomas or lipohypertrophy.7-11 To our knowledge, none of the reported cases resembled the multiple, discrete, exophytic nodules seen in our patients.3,4 The surrounding hyperkeratosis noted in patient 1 is another uncommon feature of AIns amyloidosis (Figures 1 and 2). Only 3 AIns amyloidosis cases described lesions with acanthosis nigricans–like changes, only 1 of which provided a clinical image.6,7,12The mechanism for the acanthosis nigricans–like changes may have been due to the high levels of insulin at the injection site. It has been suggested that the activation of insulinlike growth factor receptor by insulin leads to the proliferation of keratinocytes and fibroblasts.6 Histologic examination of AIns amyloidosis lesions generally demonstrates deposition of homogenous eosinophilic material consistent with amyloid, as well as positive Congo red staining with green birefringence by polarization. Immunohistologic staining with insulin antibody with or without proteomic analysis of the amyloid deposits can confirm the diagnosis. In both of our patients’ specimens, liquid chromatography–tandem mass spectrometry was performed for proteomic analysis, and results were consistent with AIns amyloidosis.
Reports in the literature have suggested that the deposition of amyloid at insulin injection sites has the potential to interfere with insulin absorption, leading to poor glucose control.4,11,13 Hence, injection site rotation is a crucial aspect of treatment and prevention of AIns amyloidosis. In their study of 4 patients, Nagase et al4 compared serum insulin levels after insulin injection into amyloid nodules vs insulin levels after injection into normal skin. Insulin absorption at the amyloid sites was 34% of that at normal sites. Given these results, patients should be instructed to inject away from the amyloid deposit once it is identified.6 Glucose levels should be monitored closely when patients first inject away from the amyloid mass, as injection of the same dosage to an area of normal skin can lead to increased insulin absorption and hypoglycemia.4,6 It is possible that the frequent hypoglycemic episodes noted in patient 1 were due to increased insulin sensitivity after switching to injection sites away from amyloid lesions.
Conclusion
Our patients demonstrate unique presentations of localized cutaneous amyloidosis at repeated insulin injection sites. We report these cases to complement the current data of iatrogenic amyloidosis and provide insight into this likely underreported phenomenon.
- Hazenberg BPC. Amyloidosis: a clinical overview. Rheum Dis Clin North Am. 2013;39:323-345.
- Storkel S, Schneider HM, Muntefering H, et al. Iatrogenic, insulin-dependent, local amyloidosis. Lab Invest. 1983;48:108-111.
- D’souza A, Theis JD, Vrana JA, et al. Pharmaceutical amyloidosis associated with subcutaneous insulin and enfuvirtide administration. Amyloid. 2014;21:71-75.
- Nagase T, Iwaya K, Iwaki Y, et al. Insulin-derived amyloidosis and poor glycemic control: a case series. Am J Med. 2014;127:450-454.
- Gupta Y, Singla G, Singla R. Insulin-derived amyloidosis. Indian J Endocrinol Metab. 2015;19:174-177.
- Kudo-Watanuki S, Kurihara E, Yamamoto K, et al. Coexistence of insulin-derived amyloidosis and an overlying acanthosis nigricans-like lesion at the site of insulin injection. Clin Exp Dermatol. 2013;38:25-29.
- Yumlu S, Barany R, Eriksson M, et al. Localized insulin-derived amyloidosis in patients with diabetes mellitus: a case report. Hum Pathol. 2009;40:1655-1660.
- Okamura S, Hayashino Y, Kore-Eda S, et al. Localized amyloidosis at the site of repeated insulin injection in a patient with type 2 diabetes. Diabetes Care. 2013;36:E200.
- Dische FE, Wernstedt C, Westermark GT, et al. Insulin as an amyloid-fibril protein at sites of repeated insulin injections in a diabetic patient. Diabetologia. 1988;31:158-161.
- Swift B, Hawkins PN, Richards C, et al. Examination of insulin injection sites: an unexpected finding of localized amyloidosis. Diabetic Med. 2002;19:881-882.
- Albert SG, Obadiah J, Parseghian SA, et al. Severe insulin resistance associated with subcutaneous amyloid deposition. Diabetes Res Clin Pract. 2007;75:374-376.
- Nandeesh BN, Rajalakshmi T, Shubha B. Cutaneous amyloidosis and insulin with coexistence of acanthosis nigricans. Indian J Pathol Microbiol. 2014;57:127-129.
- Endo JO, Rocken C, Lamb S, et al. Nodular amyloidosis in a diabetic patient with frequent hypoglycemia: sequelae of repeatedly injecting insulin without site rotation. J Am Acad Dermatol. 2010;63:E113-E114.
Amyloidosis consists of approximately 30 protein-folding disorders sharing the common feature of abnormal extracellular amyloid deposition. In each condition, a specific soluble precursor protein aggregates to form the insoluble fibrils of amyloid, characterized by the beta-pleated sheet structure.1 Amyloidosis occurs as either a systemic or localized process. Insulin-derived (AIns) amyloidosis, a localized process occurring at insulin injection sites, was first reported in 1983.2 There were fewer than 20 reported cases until 2014, when 57 additional cases were reported by just 2 institutions,3,4 indicating that AIns amyloidosis may be more common than previously thought.3,5
Despite the increasing prevalence of diabetes mellitus and insulin use, there is a paucity of published cases of AIns amyloidosis. The lack of awareness of this condition among both dermatologists and general practitioners may be in part due to its variable clinical manifestations. We describe 2 patients with unique presentations of localized amyloidosis at repeated insulin injection sites.
Case Reports
Patient 1
A 39-year-old man with a history of type 1 diabetes mellitus presented with 4 asymptomatic nodules on the lateral thighs in areas of previous insulin injection. He first noticed the lesions 9 months prior to presentation and subsequently switched the injection site to the abdomen without development of new nodules. Despite being compliant with his insulin regimen, he had a long history of irregular glucose control, including frequent hypoglycemic episodes. The patient was using regular and neutral protamine hagedorn insulin.
On physical examination, 2 soft, nontender, exophytic nodules were noted on each upper thigh with surrounding hyperpigmented and hyperkeratotic collarettes (Figure 1). The nodules ranged in size from 2 to 3.5 cm in diameter.
Remarkable laboratory data included a fasting glucose level of 207 mg/dL (reference range, 70–110 mg/dL) and a glycohemoglobin of 8.8% (reference range, <5.7%). Serum protein electrophoresis and immunofixation were normal. Histopathology of the lesions demonstrated diffuse deposition of pink amorphous material associated with prominent papillomatosis, hyperkeratosis, and acanthosis (Figure 2). Congo red staining was positive with green birefringence under polarized light, indicative of amyloid deposits (Figure 3). Liquid chromatography–tandem mass spectrometry of the specimens was consistent with deposition of AIns amyloidosis.
Due to the size and persistent nature of the lesions, the nodules were removed by tangential excision. In addition, the patient was advised to continue rotating injection sites frequently. His blood glucose levels are now well controlled, and he has not developed any new nodules.
Patient 2
A 53-year-old woman with a history of type 2 diabetes mellitus presented with painful subcutaneous nodules on the lower abdomen at sites of previous insulin injections. The nodules developed approximately 1 month after she started treatment with neutral protamine hagedorn insulin and had been slowly enlarging over the past year. She tried switching injection sites after noticing the lesions, but the nodules persisted. The patient had a long history of poor glucose control with chronically elevated glycohemoglobin and blood glucose levels.
On physical examination, 2 hyperpigmented, exophytic, smooth nodules were noted on the right and left lower abdomen, ranging in size from 2.5 to 5.5 cm in diameter (Figure 4).
Relevant laboratory data included a fasting glucose level of 197 mg/dL and a glycohemoglobin of 9.3%. A biopsy of the lesion on the left lower abdomen revealed eosinophilic amorphous deposits with fissuring in the dermis (Figure 5). Congo red stain was positive with green birefringence under polarized light. Liquid chromatography–tandem mass spectrometry of the specimen showed deposition of AIns amyloid. The patient began injecting away from the amyloid nodules without development of any new lesions. The original nodules have persisted, and surgical excision is planned.
Comment
Insulin is the suspected precursor protein in AIns amyloidosis, but the exact pathogenesis is unknown. The protein that is derived from insulin in these tumors is now identified as AIns amyloidosis.5,6 It is hypothesized that insulin accumulates locally and is converted to amyloid by an unknown mechanism.7 Other potential contributory factors include chronic inflammation and foreign body reactions developing around amyloid deposits, as well as repeated trauma from injections into a single site.4,5 It appears that lesions may derive from a wide range of insulin types and occur after variable time periods.
A majority of cases of iatrogenic amyloid have been described as single, firm, subcutaneous masses at an injection site that commonly are misdiagnosed as lipomas or lipohypertrophy.7-11 To our knowledge, none of the reported cases resembled the multiple, discrete, exophytic nodules seen in our patients.3,4 The surrounding hyperkeratosis noted in patient 1 is another uncommon feature of AIns amyloidosis (Figures 1 and 2). Only 3 AIns amyloidosis cases described lesions with acanthosis nigricans–like changes, only 1 of which provided a clinical image.6,7,12The mechanism for the acanthosis nigricans–like changes may have been due to the high levels of insulin at the injection site. It has been suggested that the activation of insulinlike growth factor receptor by insulin leads to the proliferation of keratinocytes and fibroblasts.6 Histologic examination of AIns amyloidosis lesions generally demonstrates deposition of homogenous eosinophilic material consistent with amyloid, as well as positive Congo red staining with green birefringence by polarization. Immunohistologic staining with insulin antibody with or without proteomic analysis of the amyloid deposits can confirm the diagnosis. In both of our patients’ specimens, liquid chromatography–tandem mass spectrometry was performed for proteomic analysis, and results were consistent with AIns amyloidosis.
Reports in the literature have suggested that the deposition of amyloid at insulin injection sites has the potential to interfere with insulin absorption, leading to poor glucose control.4,11,13 Hence, injection site rotation is a crucial aspect of treatment and prevention of AIns amyloidosis. In their study of 4 patients, Nagase et al4 compared serum insulin levels after insulin injection into amyloid nodules vs insulin levels after injection into normal skin. Insulin absorption at the amyloid sites was 34% of that at normal sites. Given these results, patients should be instructed to inject away from the amyloid deposit once it is identified.6 Glucose levels should be monitored closely when patients first inject away from the amyloid mass, as injection of the same dosage to an area of normal skin can lead to increased insulin absorption and hypoglycemia.4,6 It is possible that the frequent hypoglycemic episodes noted in patient 1 were due to increased insulin sensitivity after switching to injection sites away from amyloid lesions.
Conclusion
Our patients demonstrate unique presentations of localized cutaneous amyloidosis at repeated insulin injection sites. We report these cases to complement the current data of iatrogenic amyloidosis and provide insight into this likely underreported phenomenon.
Amyloidosis consists of approximately 30 protein-folding disorders sharing the common feature of abnormal extracellular amyloid deposition. In each condition, a specific soluble precursor protein aggregates to form the insoluble fibrils of amyloid, characterized by the beta-pleated sheet structure.1 Amyloidosis occurs as either a systemic or localized process. Insulin-derived (AIns) amyloidosis, a localized process occurring at insulin injection sites, was first reported in 1983.2 There were fewer than 20 reported cases until 2014, when 57 additional cases were reported by just 2 institutions,3,4 indicating that AIns amyloidosis may be more common than previously thought.3,5
Despite the increasing prevalence of diabetes mellitus and insulin use, there is a paucity of published cases of AIns amyloidosis. The lack of awareness of this condition among both dermatologists and general practitioners may be in part due to its variable clinical manifestations. We describe 2 patients with unique presentations of localized amyloidosis at repeated insulin injection sites.
Case Reports
Patient 1
A 39-year-old man with a history of type 1 diabetes mellitus presented with 4 asymptomatic nodules on the lateral thighs in areas of previous insulin injection. He first noticed the lesions 9 months prior to presentation and subsequently switched the injection site to the abdomen without development of new nodules. Despite being compliant with his insulin regimen, he had a long history of irregular glucose control, including frequent hypoglycemic episodes. The patient was using regular and neutral protamine hagedorn insulin.
On physical examination, 2 soft, nontender, exophytic nodules were noted on each upper thigh with surrounding hyperpigmented and hyperkeratotic collarettes (Figure 1). The nodules ranged in size from 2 to 3.5 cm in diameter.
Remarkable laboratory data included a fasting glucose level of 207 mg/dL (reference range, 70–110 mg/dL) and a glycohemoglobin of 8.8% (reference range, <5.7%). Serum protein electrophoresis and immunofixation were normal. Histopathology of the lesions demonstrated diffuse deposition of pink amorphous material associated with prominent papillomatosis, hyperkeratosis, and acanthosis (Figure 2). Congo red staining was positive with green birefringence under polarized light, indicative of amyloid deposits (Figure 3). Liquid chromatography–tandem mass spectrometry of the specimens was consistent with deposition of AIns amyloidosis.
Due to the size and persistent nature of the lesions, the nodules were removed by tangential excision. In addition, the patient was advised to continue rotating injection sites frequently. His blood glucose levels are now well controlled, and he has not developed any new nodules.
Patient 2
A 53-year-old woman with a history of type 2 diabetes mellitus presented with painful subcutaneous nodules on the lower abdomen at sites of previous insulin injections. The nodules developed approximately 1 month after she started treatment with neutral protamine hagedorn insulin and had been slowly enlarging over the past year. She tried switching injection sites after noticing the lesions, but the nodules persisted. The patient had a long history of poor glucose control with chronically elevated glycohemoglobin and blood glucose levels.
On physical examination, 2 hyperpigmented, exophytic, smooth nodules were noted on the right and left lower abdomen, ranging in size from 2.5 to 5.5 cm in diameter (Figure 4).
Relevant laboratory data included a fasting glucose level of 197 mg/dL and a glycohemoglobin of 9.3%. A biopsy of the lesion on the left lower abdomen revealed eosinophilic amorphous deposits with fissuring in the dermis (Figure 5). Congo red stain was positive with green birefringence under polarized light. Liquid chromatography–tandem mass spectrometry of the specimen showed deposition of AIns amyloid. The patient began injecting away from the amyloid nodules without development of any new lesions. The original nodules have persisted, and surgical excision is planned.
Comment
Insulin is the suspected precursor protein in AIns amyloidosis, but the exact pathogenesis is unknown. The protein that is derived from insulin in these tumors is now identified as AIns amyloidosis.5,6 It is hypothesized that insulin accumulates locally and is converted to amyloid by an unknown mechanism.7 Other potential contributory factors include chronic inflammation and foreign body reactions developing around amyloid deposits, as well as repeated trauma from injections into a single site.4,5 It appears that lesions may derive from a wide range of insulin types and occur after variable time periods.
A majority of cases of iatrogenic amyloid have been described as single, firm, subcutaneous masses at an injection site that commonly are misdiagnosed as lipomas or lipohypertrophy.7-11 To our knowledge, none of the reported cases resembled the multiple, discrete, exophytic nodules seen in our patients.3,4 The surrounding hyperkeratosis noted in patient 1 is another uncommon feature of AIns amyloidosis (Figures 1 and 2). Only 3 AIns amyloidosis cases described lesions with acanthosis nigricans–like changes, only 1 of which provided a clinical image.6,7,12The mechanism for the acanthosis nigricans–like changes may have been due to the high levels of insulin at the injection site. It has been suggested that the activation of insulinlike growth factor receptor by insulin leads to the proliferation of keratinocytes and fibroblasts.6 Histologic examination of AIns amyloidosis lesions generally demonstrates deposition of homogenous eosinophilic material consistent with amyloid, as well as positive Congo red staining with green birefringence by polarization. Immunohistologic staining with insulin antibody with or without proteomic analysis of the amyloid deposits can confirm the diagnosis. In both of our patients’ specimens, liquid chromatography–tandem mass spectrometry was performed for proteomic analysis, and results were consistent with AIns amyloidosis.
Reports in the literature have suggested that the deposition of amyloid at insulin injection sites has the potential to interfere with insulin absorption, leading to poor glucose control.4,11,13 Hence, injection site rotation is a crucial aspect of treatment and prevention of AIns amyloidosis. In their study of 4 patients, Nagase et al4 compared serum insulin levels after insulin injection into amyloid nodules vs insulin levels after injection into normal skin. Insulin absorption at the amyloid sites was 34% of that at normal sites. Given these results, patients should be instructed to inject away from the amyloid deposit once it is identified.6 Glucose levels should be monitored closely when patients first inject away from the amyloid mass, as injection of the same dosage to an area of normal skin can lead to increased insulin absorption and hypoglycemia.4,6 It is possible that the frequent hypoglycemic episodes noted in patient 1 were due to increased insulin sensitivity after switching to injection sites away from amyloid lesions.
Conclusion
Our patients demonstrate unique presentations of localized cutaneous amyloidosis at repeated insulin injection sites. We report these cases to complement the current data of iatrogenic amyloidosis and provide insight into this likely underreported phenomenon.
- Hazenberg BPC. Amyloidosis: a clinical overview. Rheum Dis Clin North Am. 2013;39:323-345.
- Storkel S, Schneider HM, Muntefering H, et al. Iatrogenic, insulin-dependent, local amyloidosis. Lab Invest. 1983;48:108-111.
- D’souza A, Theis JD, Vrana JA, et al. Pharmaceutical amyloidosis associated with subcutaneous insulin and enfuvirtide administration. Amyloid. 2014;21:71-75.
- Nagase T, Iwaya K, Iwaki Y, et al. Insulin-derived amyloidosis and poor glycemic control: a case series. Am J Med. 2014;127:450-454.
- Gupta Y, Singla G, Singla R. Insulin-derived amyloidosis. Indian J Endocrinol Metab. 2015;19:174-177.
- Kudo-Watanuki S, Kurihara E, Yamamoto K, et al. Coexistence of insulin-derived amyloidosis and an overlying acanthosis nigricans-like lesion at the site of insulin injection. Clin Exp Dermatol. 2013;38:25-29.
- Yumlu S, Barany R, Eriksson M, et al. Localized insulin-derived amyloidosis in patients with diabetes mellitus: a case report. Hum Pathol. 2009;40:1655-1660.
- Okamura S, Hayashino Y, Kore-Eda S, et al. Localized amyloidosis at the site of repeated insulin injection in a patient with type 2 diabetes. Diabetes Care. 2013;36:E200.
- Dische FE, Wernstedt C, Westermark GT, et al. Insulin as an amyloid-fibril protein at sites of repeated insulin injections in a diabetic patient. Diabetologia. 1988;31:158-161.
- Swift B, Hawkins PN, Richards C, et al. Examination of insulin injection sites: an unexpected finding of localized amyloidosis. Diabetic Med. 2002;19:881-882.
- Albert SG, Obadiah J, Parseghian SA, et al. Severe insulin resistance associated with subcutaneous amyloid deposition. Diabetes Res Clin Pract. 2007;75:374-376.
- Nandeesh BN, Rajalakshmi T, Shubha B. Cutaneous amyloidosis and insulin with coexistence of acanthosis nigricans. Indian J Pathol Microbiol. 2014;57:127-129.
- Endo JO, Rocken C, Lamb S, et al. Nodular amyloidosis in a diabetic patient with frequent hypoglycemia: sequelae of repeatedly injecting insulin without site rotation. J Am Acad Dermatol. 2010;63:E113-E114.
- Hazenberg BPC. Amyloidosis: a clinical overview. Rheum Dis Clin North Am. 2013;39:323-345.
- Storkel S, Schneider HM, Muntefering H, et al. Iatrogenic, insulin-dependent, local amyloidosis. Lab Invest. 1983;48:108-111.
- D’souza A, Theis JD, Vrana JA, et al. Pharmaceutical amyloidosis associated with subcutaneous insulin and enfuvirtide administration. Amyloid. 2014;21:71-75.
- Nagase T, Iwaya K, Iwaki Y, et al. Insulin-derived amyloidosis and poor glycemic control: a case series. Am J Med. 2014;127:450-454.
- Gupta Y, Singla G, Singla R. Insulin-derived amyloidosis. Indian J Endocrinol Metab. 2015;19:174-177.
- Kudo-Watanuki S, Kurihara E, Yamamoto K, et al. Coexistence of insulin-derived amyloidosis and an overlying acanthosis nigricans-like lesion at the site of insulin injection. Clin Exp Dermatol. 2013;38:25-29.
- Yumlu S, Barany R, Eriksson M, et al. Localized insulin-derived amyloidosis in patients with diabetes mellitus: a case report. Hum Pathol. 2009;40:1655-1660.
- Okamura S, Hayashino Y, Kore-Eda S, et al. Localized amyloidosis at the site of repeated insulin injection in a patient with type 2 diabetes. Diabetes Care. 2013;36:E200.
- Dische FE, Wernstedt C, Westermark GT, et al. Insulin as an amyloid-fibril protein at sites of repeated insulin injections in a diabetic patient. Diabetologia. 1988;31:158-161.
- Swift B, Hawkins PN, Richards C, et al. Examination of insulin injection sites: an unexpected finding of localized amyloidosis. Diabetic Med. 2002;19:881-882.
- Albert SG, Obadiah J, Parseghian SA, et al. Severe insulin resistance associated with subcutaneous amyloid deposition. Diabetes Res Clin Pract. 2007;75:374-376.
- Nandeesh BN, Rajalakshmi T, Shubha B. Cutaneous amyloidosis and insulin with coexistence of acanthosis nigricans. Indian J Pathol Microbiol. 2014;57:127-129.
- Endo JO, Rocken C, Lamb S, et al. Nodular amyloidosis in a diabetic patient with frequent hypoglycemia: sequelae of repeatedly injecting insulin without site rotation. J Am Acad Dermatol. 2010;63:E113-E114.
Practice Points
- Deposition of amyloid at insulin injection sites has the potential to interfere with insulin absorption, leading to poor glucose control.
- Patients with insulin-derived (AIns) amyloidosis may initially present after noticing nodular deposits.
- Insulin injection site rotation is a crucial aspect of treatment and prevention of AIns amyloidosis.
Sequential Targeted Treatment for a Geriatric Patient with Acute Myeloid Leukemia with Concurrent FLT3-TKD and IDH1 Mutations
Nearly 20,000 patients are diagnosed with acute myeloid leukemia (AML) in the US annually.1 Despite the use of aggressive chemotherapeutic agents, the prognosis remains poor, with a mean 5-year survival of 28.3%.2 Fortunately, with the refinement of next-generation sequencing (NGS) hematology panels and development of systemic targeted therapies, the treatment landscape for eligible patients has improved, both in frontline and relapsed or refractory (R/R) patients.
Specifically, investigations into alterations within the FMS-like tyrosine kinase (FLT3) and isocitrate dehydrogenase (IDH) genes have led to the discovery of a number of targeted treatments. Midostaurin is US Food and Drug Administration (FDA)-approved for use in combination with induction chemotherapy for patients with internal tandem duplication of the FLT3 (FLT3-ITD) gene or mutations within the tyrosine kinase domain (FLT3-TKD).3 Ivosidenib is indicated for frontline treatment for those who are poor candidates for induction chemotherapy, and R/R patients who have an R132H mutation in IDH1.4,5 Enasidenib is FDA-approved for R/R patients with R140Q, R172S, and R172K mutations in IDH2.6
The optimal treatment for patients with AML with ≥ 2 clinically actionable mutations has not been established. In this article we describe a geriatric patient who initially was diagnosed with AML with concurrent FLT3-TKD and IDH1 mutations and received targeted, sequential management. We detail changes in disease phenotype and mutational status by repeating an NGS hematology panel and cytogenetic studies after each stage of therapy. Lastly, we discuss the clonal evolution apparent within leukemic cells with use of ≥ 1 or more targeted agents.
Case Presentation
A 68-year-old man presented to the Emergency Department at The Durham Veterans Affairs Medical Center in North Carolina with fatigue and light-headedness. Because of his symptoms and pancytopenia, a bone marrow aspiration and trephine biopsy were performed, which showed 57% myeloblasts, 12% promyelocytes/myelocytes, and 2% metamyelocytes in 20 to 30% cellular bone marrow. Flow cytometry confirmed a blast population consistent with AML. A LeukoVantage (Quest Diagnostics) hematologic NGS panel revealed the presence of FLT3-TKD, IDH1, RUNX1, BCOR-E1477, and SF3B1 mutations (Table). Initial fluorescence in situ hybridization (FISH) results showed a normal pattern of hybridization with no translocations. His disease was deemed to be intermediate-high risk because of the presence of FLT3-TKD and RUNX1 mutations, despite the normal cytogenetic profile and absence of additional clinical features.
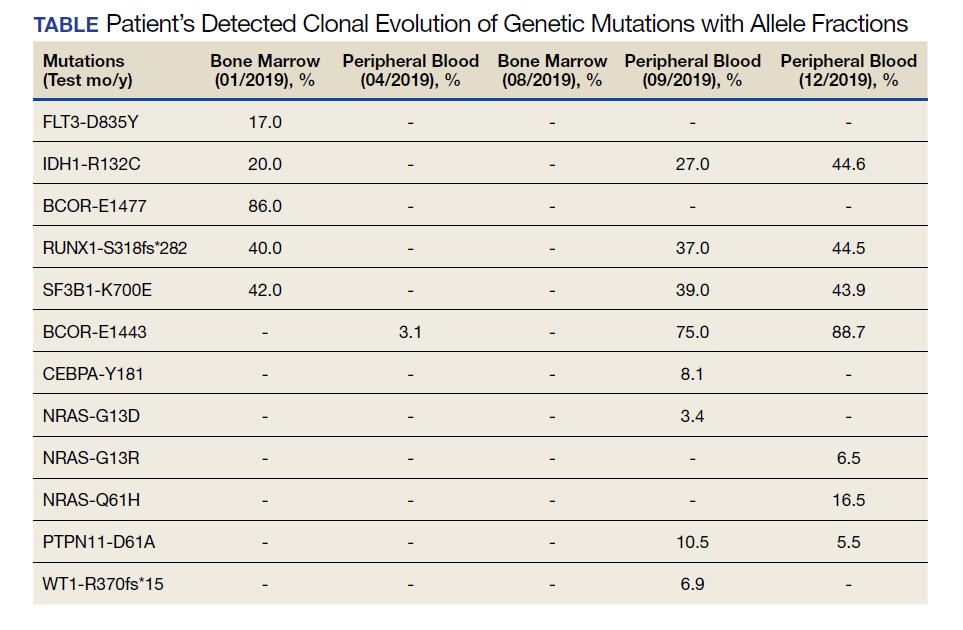
Induction chemotherapy was started with idarubicin, 12 mg/m2, on days 1 to 3 and cytarabine, 200 mg/m2, on days 1 to 7. Because of the presence of a FLT3-TKD mutation, midostaurin was planned for days 8 to 21. After induction chemotherapy, a bone marrow biopsy on day 14 revealed an acellular marrow with no observed myeloblasts. A bone marrow biopsy conducted before initiating consolidation therapy, revealed 30% cellularity with morphologic remission. However, flow cytometry found 5% myeloblasts expressing CD34, CD117, CD13, CD38, and HLA-DR, consistent with measurable residual disease. He received 2 cycles of consolidation therapy with high-dose cytarabine combined with midostaurin. After the patient's second cycle of consolidation, he continued to experience transfusion-dependent cytopenias. Another bone marrow evaluation demonstrated 10% cellularity with nearly all cells appearing to be myeloblasts. A repeat LeukoVantage NGS panel demonstrated undetectable FLT3-TKD mutation and persistent IDH1-R123C mutation. FISH studies revealed a complex karyotype with monosomy of chromosomes 5 and 7 and trisomy of chromosome 8.
We discussed with the patient and his family the options available, which included initiating targeted therapy for his IDH1 mutation, administering hypomethylation therapy with or without venetoclax, or pursuing palliative measures. We collectively decided to pursue therapy with single-agent oral ivosidenib, 500 mg daily. After 1 month of treatment, our patient developed worsening fatigue. His white blood cell count had increased to > 43 k/cm2, raising concern for differentiation syndrome.
A review of the peripheral smear showed a wide-spectrum of maturing granulocytes, with a large percentage of blasts. Peripheral flow cytometry confirmed a blast population of 15%. After a short period of symptom improvement with steroids, the patient developed worsening confusion. Brain imaging identified 2 subdural hemorrhages. Because of a significant peripheral blast population and the development of these hemorrhages, palliative measures were pursued, and the patient was discharged to an inpatient hospice facility. A final NGS panel performed from peripheral blood detected mutations in IDH1, RUNX1, PTPN11, NRAS, BCOR-E1443, and SF3B1 genes.
Discussion
To our knowledge, this is the first reported case of a patient who sequentially received targeted treatments directed against both FLT3 and IDH1 mutations. Initial management with midostaurin and cytarabine resulted in sustained remission of his FLT3-TKD mutation. However, despite receiving prompt standard of care with combination induction chemotherapy and targeted therapy, the patient experienced unfavorable clonal evolution based upon his molecular and cytogenetic testing. Addition of ivosidenib as a second targeting agent for his IDH1 mutation did not achieve a second remission.
Clonal evolution is a well-described phenomenon in hematology. Indolent conditions, such as clonal hematopoiesis of intermediate potential, or malignancies, such as myelodysplastic syndromes and myeloproliferative neoplasms, could transform into acute leukemia through the accumulation of driver mutations and/or cytogenetic abnormalities. Clonal evolution often is viewed as the culprit in patients with AML whose disease relapses after remission with initial chemotherapy.7-10 With the increasing availability of commercial NGS panels designed to assess mutations among patients experiencing hematologic malignancies, patterns of relapse, and, models of clonal evolution could be observed closely in patients with AML.
We were able to monitor molecular changes within our patient’s predominant clonal populations by repeating peripheral comprehensive NGS panels after lines of targeted therapies. The repeated sequencing revealed that clones with FLT3-TKD mutations responded to midostaurin with first-line chemotherapy whereas it was unclear whether clones with IDH1 mutation responded to ivosidenib. Development of complex cytogenetic findings along with the clonal expansion of BCOR mutation-harboring cells likely contributed to our patient’s acutely worsening condition. Several studies have found that the presence of a BCOR mutation in adults with AML leads to lower overall survival and relapse-free survival.11,12 As of now, there are no treatments specifically targeting BCOR mutations.
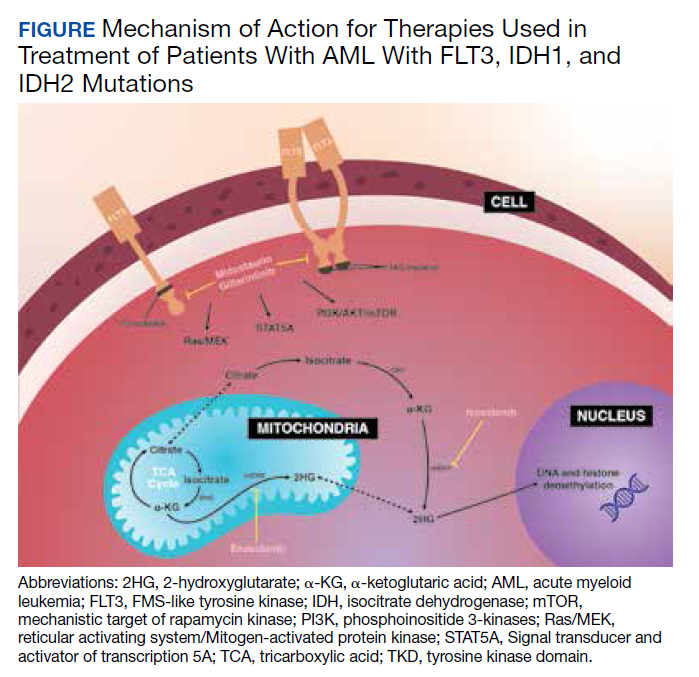
Although there are novel targeting agents with proven efficacy for both FLT3 and IDH1 mutations (Figure), it is difficult to determine which pathogenic mutation drives disease onset. No evidence suggests that these drugs could be administered in tandem. At the present time, interest is directed towards targeting all AML subclones simultaneously, which could reduce the likelihood of evolution among founder clones.7,10,13 In their comparison between molecular profiles and outcomes of patients with AML, Papaemmanuil and colleagues observed that > 80% of patients with AML harbor ≥ 2 driver mutations concurrently.14 Moreover, FLT3-ITD and IDH1 mutations tend to co-occur in approximately 9 to 27% of AML cases.15-18 Available targeted agents for AML are relatively new and hematologists’ familiarity with these drugs is continuing to grow. As the number of novel agents increases, investigations directed toward assessing the safety profile and efficacy of combining targeted agents will be beneficial for patients with AML with ≥ 1 driver mutation.
Conclusions
For our patient with AML, sequential targeted management of FLT3-TKD and IDH1 mutations was not beneficial. Higher-risk disease features, such as the development of a complex karyotype, likely contributed to our patient’s poor response to second-line ivosidenib. The sequential NGS malignant hematology panels allowed us to closely monitor changes to the molecular structure of our patient’s AML after each line of targeted therapy. Future investigations of combining targeted agents for patients with AML with concurrent actionable mutations would provide insight into outcomes of treating multiple clonal populations simultaneously.
1. De Kouchkovsky I, Abdul-Hay M. Acute myeloid leukemia: a comprehensive review and 2016 update. Blood Cancer J. 2016;6(7):e441. doi:10.1038/bcj.2016.50.
2. National Cancer Institute. Cancer Stat Facts: Leukemia — acute myeloid leukemia (AML). Accessed November 4, 2020. https://seer.cancer.gov/statfacts/html/amyl.html
3. Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med. 2017;377(5):454-464. doi:10.1056/NEJMoa1614359.
4. DiNardo CD, Stein EM, de Botton S, et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med. 2018;378(25):2386-2398. doi:10.1056/NEJMoa1716984.
5. Roboz, GJ, DiNardo, CD, Stein, EM, et al. Ivosidenib induces deep durable remissions in patients with newly diagnosed IDH1-mutant acute myeloid leukemia. Blood. 2019;135(7), 463-471. doi: 10.1182/blood.2019002140
6. Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722-731. doi:10.1182/blood-2017-04-779405.
7. Jan M, Majeti R. Clonal evolution of acute leukemia genomes. Oncogene. 2013;32(2):135-140. doi:10.1038/onc.2012.48.
8. Grove CS, Vassiliou GS. Acute myeloid leukaemia: a paradigm for the clonal evolution of cancer? Dis Model Mech. 2014;7(8):941-951. doi:10.1242/dmm.015974.
9. Anderson K, Lutz C, van Delft FW, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature. 2011;469(7330):356-561. doi: 10.1038/nature09650.
10. Ding L, Ley TJ, Larson DE, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481(7382):506-510. doi:10.1038/nature10738.
11. Terada K, Yamaguchi H, Ueki T, et al. Usefulness of BCOR gene mutation as a prognostic factor in acute myeloid leukemia with intermediate cytogenetic prognosis. Genes Chromosomes Cancer. 2018;57(8):401-408. doi:10.1002/gcc.22542.
12. Grossmann V, Tiacci E, Holmes AB, et al. Whole-exome sequencing identifies somatic mutations of BCOR in acute myeloid leukemia with normal karyotype. Blood. 2011;118(23):6153-6163. doi:10.1182/blood-2011-07-365320.
13. Parkin B, Ouillette P, Li Y, et al. Clonal evolution and devolution after chemotherapy in adult acute myelogenous leukemia. Blood. 2013;121(2):369-377. doi:10.1182/blood-2012-04-427039.
14. Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209-2221. doi:10.1056/NEJMoa1516192.
15. DiNardo CD, Ravandi F, Agresta S, et al. Characteristics, clinical outcome, and prognostic significance of IDH mutations in AML. Am J Hematol. 2015;90(8):732-736. doi:10.1002/ajh.24072.
16. Rakheja D, Konoplev S, Medeiros LJ, Chen W. IDH mutations in acute myeloid leukemia. Hum Pathol. 2012;43 (10):1541-1551. doi:10.1016/j.humpath.2012.05.003.
17. Lai C, Doucette K, Norsworthy K. Recent drug approvals for acute myeloid leukemia. J H Oncol. 2019;12(1):100. doi:10.1186/s13045-019-0774-x.
18. Boddu P, Takahashi K, Pemmaraju N, et al. Influence of IDH on FLT3-ITD status in newly diagnosed AML. Leukemia. 2017;31(11):2526-2529. doi:10.1038/leu.2017.244.
Nearly 20,000 patients are diagnosed with acute myeloid leukemia (AML) in the US annually.1 Despite the use of aggressive chemotherapeutic agents, the prognosis remains poor, with a mean 5-year survival of 28.3%.2 Fortunately, with the refinement of next-generation sequencing (NGS) hematology panels and development of systemic targeted therapies, the treatment landscape for eligible patients has improved, both in frontline and relapsed or refractory (R/R) patients.
Specifically, investigations into alterations within the FMS-like tyrosine kinase (FLT3) and isocitrate dehydrogenase (IDH) genes have led to the discovery of a number of targeted treatments. Midostaurin is US Food and Drug Administration (FDA)-approved for use in combination with induction chemotherapy for patients with internal tandem duplication of the FLT3 (FLT3-ITD) gene or mutations within the tyrosine kinase domain (FLT3-TKD).3 Ivosidenib is indicated for frontline treatment for those who are poor candidates for induction chemotherapy, and R/R patients who have an R132H mutation in IDH1.4,5 Enasidenib is FDA-approved for R/R patients with R140Q, R172S, and R172K mutations in IDH2.6
The optimal treatment for patients with AML with ≥ 2 clinically actionable mutations has not been established. In this article we describe a geriatric patient who initially was diagnosed with AML with concurrent FLT3-TKD and IDH1 mutations and received targeted, sequential management. We detail changes in disease phenotype and mutational status by repeating an NGS hematology panel and cytogenetic studies after each stage of therapy. Lastly, we discuss the clonal evolution apparent within leukemic cells with use of ≥ 1 or more targeted agents.
Case Presentation
A 68-year-old man presented to the Emergency Department at The Durham Veterans Affairs Medical Center in North Carolina with fatigue and light-headedness. Because of his symptoms and pancytopenia, a bone marrow aspiration and trephine biopsy were performed, which showed 57% myeloblasts, 12% promyelocytes/myelocytes, and 2% metamyelocytes in 20 to 30% cellular bone marrow. Flow cytometry confirmed a blast population consistent with AML. A LeukoVantage (Quest Diagnostics) hematologic NGS panel revealed the presence of FLT3-TKD, IDH1, RUNX1, BCOR-E1477, and SF3B1 mutations (Table). Initial fluorescence in situ hybridization (FISH) results showed a normal pattern of hybridization with no translocations. His disease was deemed to be intermediate-high risk because of the presence of FLT3-TKD and RUNX1 mutations, despite the normal cytogenetic profile and absence of additional clinical features.
Induction chemotherapy was started with idarubicin, 12 mg/m2, on days 1 to 3 and cytarabine, 200 mg/m2, on days 1 to 7. Because of the presence of a FLT3-TKD mutation, midostaurin was planned for days 8 to 21. After induction chemotherapy, a bone marrow biopsy on day 14 revealed an acellular marrow with no observed myeloblasts. A bone marrow biopsy conducted before initiating consolidation therapy, revealed 30% cellularity with morphologic remission. However, flow cytometry found 5% myeloblasts expressing CD34, CD117, CD13, CD38, and HLA-DR, consistent with measurable residual disease. He received 2 cycles of consolidation therapy with high-dose cytarabine combined with midostaurin. After the patient's second cycle of consolidation, he continued to experience transfusion-dependent cytopenias. Another bone marrow evaluation demonstrated 10% cellularity with nearly all cells appearing to be myeloblasts. A repeat LeukoVantage NGS panel demonstrated undetectable FLT3-TKD mutation and persistent IDH1-R123C mutation. FISH studies revealed a complex karyotype with monosomy of chromosomes 5 and 7 and trisomy of chromosome 8.
We discussed with the patient and his family the options available, which included initiating targeted therapy for his IDH1 mutation, administering hypomethylation therapy with or without venetoclax, or pursuing palliative measures. We collectively decided to pursue therapy with single-agent oral ivosidenib, 500 mg daily. After 1 month of treatment, our patient developed worsening fatigue. His white blood cell count had increased to > 43 k/cm2, raising concern for differentiation syndrome.
A review of the peripheral smear showed a wide-spectrum of maturing granulocytes, with a large percentage of blasts. Peripheral flow cytometry confirmed a blast population of 15%. After a short period of symptom improvement with steroids, the patient developed worsening confusion. Brain imaging identified 2 subdural hemorrhages. Because of a significant peripheral blast population and the development of these hemorrhages, palliative measures were pursued, and the patient was discharged to an inpatient hospice facility. A final NGS panel performed from peripheral blood detected mutations in IDH1, RUNX1, PTPN11, NRAS, BCOR-E1443, and SF3B1 genes.
Discussion
To our knowledge, this is the first reported case of a patient who sequentially received targeted treatments directed against both FLT3 and IDH1 mutations. Initial management with midostaurin and cytarabine resulted in sustained remission of his FLT3-TKD mutation. However, despite receiving prompt standard of care with combination induction chemotherapy and targeted therapy, the patient experienced unfavorable clonal evolution based upon his molecular and cytogenetic testing. Addition of ivosidenib as a second targeting agent for his IDH1 mutation did not achieve a second remission.
Clonal evolution is a well-described phenomenon in hematology. Indolent conditions, such as clonal hematopoiesis of intermediate potential, or malignancies, such as myelodysplastic syndromes and myeloproliferative neoplasms, could transform into acute leukemia through the accumulation of driver mutations and/or cytogenetic abnormalities. Clonal evolution often is viewed as the culprit in patients with AML whose disease relapses after remission with initial chemotherapy.7-10 With the increasing availability of commercial NGS panels designed to assess mutations among patients experiencing hematologic malignancies, patterns of relapse, and, models of clonal evolution could be observed closely in patients with AML.
We were able to monitor molecular changes within our patient’s predominant clonal populations by repeating peripheral comprehensive NGS panels after lines of targeted therapies. The repeated sequencing revealed that clones with FLT3-TKD mutations responded to midostaurin with first-line chemotherapy whereas it was unclear whether clones with IDH1 mutation responded to ivosidenib. Development of complex cytogenetic findings along with the clonal expansion of BCOR mutation-harboring cells likely contributed to our patient’s acutely worsening condition. Several studies have found that the presence of a BCOR mutation in adults with AML leads to lower overall survival and relapse-free survival.11,12 As of now, there are no treatments specifically targeting BCOR mutations.
Although there are novel targeting agents with proven efficacy for both FLT3 and IDH1 mutations (Figure), it is difficult to determine which pathogenic mutation drives disease onset. No evidence suggests that these drugs could be administered in tandem. At the present time, interest is directed towards targeting all AML subclones simultaneously, which could reduce the likelihood of evolution among founder clones.7,10,13 In their comparison between molecular profiles and outcomes of patients with AML, Papaemmanuil and colleagues observed that > 80% of patients with AML harbor ≥ 2 driver mutations concurrently.14 Moreover, FLT3-ITD and IDH1 mutations tend to co-occur in approximately 9 to 27% of AML cases.15-18 Available targeted agents for AML are relatively new and hematologists’ familiarity with these drugs is continuing to grow. As the number of novel agents increases, investigations directed toward assessing the safety profile and efficacy of combining targeted agents will be beneficial for patients with AML with ≥ 1 driver mutation.
Conclusions
For our patient with AML, sequential targeted management of FLT3-TKD and IDH1 mutations was not beneficial. Higher-risk disease features, such as the development of a complex karyotype, likely contributed to our patient’s poor response to second-line ivosidenib. The sequential NGS malignant hematology panels allowed us to closely monitor changes to the molecular structure of our patient’s AML after each line of targeted therapy. Future investigations of combining targeted agents for patients with AML with concurrent actionable mutations would provide insight into outcomes of treating multiple clonal populations simultaneously.
Nearly 20,000 patients are diagnosed with acute myeloid leukemia (AML) in the US annually.1 Despite the use of aggressive chemotherapeutic agents, the prognosis remains poor, with a mean 5-year survival of 28.3%.2 Fortunately, with the refinement of next-generation sequencing (NGS) hematology panels and development of systemic targeted therapies, the treatment landscape for eligible patients has improved, both in frontline and relapsed or refractory (R/R) patients.
Specifically, investigations into alterations within the FMS-like tyrosine kinase (FLT3) and isocitrate dehydrogenase (IDH) genes have led to the discovery of a number of targeted treatments. Midostaurin is US Food and Drug Administration (FDA)-approved for use in combination with induction chemotherapy for patients with internal tandem duplication of the FLT3 (FLT3-ITD) gene or mutations within the tyrosine kinase domain (FLT3-TKD).3 Ivosidenib is indicated for frontline treatment for those who are poor candidates for induction chemotherapy, and R/R patients who have an R132H mutation in IDH1.4,5 Enasidenib is FDA-approved for R/R patients with R140Q, R172S, and R172K mutations in IDH2.6
The optimal treatment for patients with AML with ≥ 2 clinically actionable mutations has not been established. In this article we describe a geriatric patient who initially was diagnosed with AML with concurrent FLT3-TKD and IDH1 mutations and received targeted, sequential management. We detail changes in disease phenotype and mutational status by repeating an NGS hematology panel and cytogenetic studies after each stage of therapy. Lastly, we discuss the clonal evolution apparent within leukemic cells with use of ≥ 1 or more targeted agents.
Case Presentation
A 68-year-old man presented to the Emergency Department at The Durham Veterans Affairs Medical Center in North Carolina with fatigue and light-headedness. Because of his symptoms and pancytopenia, a bone marrow aspiration and trephine biopsy were performed, which showed 57% myeloblasts, 12% promyelocytes/myelocytes, and 2% metamyelocytes in 20 to 30% cellular bone marrow. Flow cytometry confirmed a blast population consistent with AML. A LeukoVantage (Quest Diagnostics) hematologic NGS panel revealed the presence of FLT3-TKD, IDH1, RUNX1, BCOR-E1477, and SF3B1 mutations (Table). Initial fluorescence in situ hybridization (FISH) results showed a normal pattern of hybridization with no translocations. His disease was deemed to be intermediate-high risk because of the presence of FLT3-TKD and RUNX1 mutations, despite the normal cytogenetic profile and absence of additional clinical features.
Induction chemotherapy was started with idarubicin, 12 mg/m2, on days 1 to 3 and cytarabine, 200 mg/m2, on days 1 to 7. Because of the presence of a FLT3-TKD mutation, midostaurin was planned for days 8 to 21. After induction chemotherapy, a bone marrow biopsy on day 14 revealed an acellular marrow with no observed myeloblasts. A bone marrow biopsy conducted before initiating consolidation therapy, revealed 30% cellularity with morphologic remission. However, flow cytometry found 5% myeloblasts expressing CD34, CD117, CD13, CD38, and HLA-DR, consistent with measurable residual disease. He received 2 cycles of consolidation therapy with high-dose cytarabine combined with midostaurin. After the patient's second cycle of consolidation, he continued to experience transfusion-dependent cytopenias. Another bone marrow evaluation demonstrated 10% cellularity with nearly all cells appearing to be myeloblasts. A repeat LeukoVantage NGS panel demonstrated undetectable FLT3-TKD mutation and persistent IDH1-R123C mutation. FISH studies revealed a complex karyotype with monosomy of chromosomes 5 and 7 and trisomy of chromosome 8.
We discussed with the patient and his family the options available, which included initiating targeted therapy for his IDH1 mutation, administering hypomethylation therapy with or without venetoclax, or pursuing palliative measures. We collectively decided to pursue therapy with single-agent oral ivosidenib, 500 mg daily. After 1 month of treatment, our patient developed worsening fatigue. His white blood cell count had increased to > 43 k/cm2, raising concern for differentiation syndrome.
A review of the peripheral smear showed a wide-spectrum of maturing granulocytes, with a large percentage of blasts. Peripheral flow cytometry confirmed a blast population of 15%. After a short period of symptom improvement with steroids, the patient developed worsening confusion. Brain imaging identified 2 subdural hemorrhages. Because of a significant peripheral blast population and the development of these hemorrhages, palliative measures were pursued, and the patient was discharged to an inpatient hospice facility. A final NGS panel performed from peripheral blood detected mutations in IDH1, RUNX1, PTPN11, NRAS, BCOR-E1443, and SF3B1 genes.
Discussion
To our knowledge, this is the first reported case of a patient who sequentially received targeted treatments directed against both FLT3 and IDH1 mutations. Initial management with midostaurin and cytarabine resulted in sustained remission of his FLT3-TKD mutation. However, despite receiving prompt standard of care with combination induction chemotherapy and targeted therapy, the patient experienced unfavorable clonal evolution based upon his molecular and cytogenetic testing. Addition of ivosidenib as a second targeting agent for his IDH1 mutation did not achieve a second remission.
Clonal evolution is a well-described phenomenon in hematology. Indolent conditions, such as clonal hematopoiesis of intermediate potential, or malignancies, such as myelodysplastic syndromes and myeloproliferative neoplasms, could transform into acute leukemia through the accumulation of driver mutations and/or cytogenetic abnormalities. Clonal evolution often is viewed as the culprit in patients with AML whose disease relapses after remission with initial chemotherapy.7-10 With the increasing availability of commercial NGS panels designed to assess mutations among patients experiencing hematologic malignancies, patterns of relapse, and, models of clonal evolution could be observed closely in patients with AML.
We were able to monitor molecular changes within our patient’s predominant clonal populations by repeating peripheral comprehensive NGS panels after lines of targeted therapies. The repeated sequencing revealed that clones with FLT3-TKD mutations responded to midostaurin with first-line chemotherapy whereas it was unclear whether clones with IDH1 mutation responded to ivosidenib. Development of complex cytogenetic findings along with the clonal expansion of BCOR mutation-harboring cells likely contributed to our patient’s acutely worsening condition. Several studies have found that the presence of a BCOR mutation in adults with AML leads to lower overall survival and relapse-free survival.11,12 As of now, there are no treatments specifically targeting BCOR mutations.
Although there are novel targeting agents with proven efficacy for both FLT3 and IDH1 mutations (Figure), it is difficult to determine which pathogenic mutation drives disease onset. No evidence suggests that these drugs could be administered in tandem. At the present time, interest is directed towards targeting all AML subclones simultaneously, which could reduce the likelihood of evolution among founder clones.7,10,13 In their comparison between molecular profiles and outcomes of patients with AML, Papaemmanuil and colleagues observed that > 80% of patients with AML harbor ≥ 2 driver mutations concurrently.14 Moreover, FLT3-ITD and IDH1 mutations tend to co-occur in approximately 9 to 27% of AML cases.15-18 Available targeted agents for AML are relatively new and hematologists’ familiarity with these drugs is continuing to grow. As the number of novel agents increases, investigations directed toward assessing the safety profile and efficacy of combining targeted agents will be beneficial for patients with AML with ≥ 1 driver mutation.
Conclusions
For our patient with AML, sequential targeted management of FLT3-TKD and IDH1 mutations was not beneficial. Higher-risk disease features, such as the development of a complex karyotype, likely contributed to our patient’s poor response to second-line ivosidenib. The sequential NGS malignant hematology panels allowed us to closely monitor changes to the molecular structure of our patient’s AML after each line of targeted therapy. Future investigations of combining targeted agents for patients with AML with concurrent actionable mutations would provide insight into outcomes of treating multiple clonal populations simultaneously.
1. De Kouchkovsky I, Abdul-Hay M. Acute myeloid leukemia: a comprehensive review and 2016 update. Blood Cancer J. 2016;6(7):e441. doi:10.1038/bcj.2016.50.
2. National Cancer Institute. Cancer Stat Facts: Leukemia — acute myeloid leukemia (AML). Accessed November 4, 2020. https://seer.cancer.gov/statfacts/html/amyl.html
3. Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med. 2017;377(5):454-464. doi:10.1056/NEJMoa1614359.
4. DiNardo CD, Stein EM, de Botton S, et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med. 2018;378(25):2386-2398. doi:10.1056/NEJMoa1716984.
5. Roboz, GJ, DiNardo, CD, Stein, EM, et al. Ivosidenib induces deep durable remissions in patients with newly diagnosed IDH1-mutant acute myeloid leukemia. Blood. 2019;135(7), 463-471. doi: 10.1182/blood.2019002140
6. Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722-731. doi:10.1182/blood-2017-04-779405.
7. Jan M, Majeti R. Clonal evolution of acute leukemia genomes. Oncogene. 2013;32(2):135-140. doi:10.1038/onc.2012.48.
8. Grove CS, Vassiliou GS. Acute myeloid leukaemia: a paradigm for the clonal evolution of cancer? Dis Model Mech. 2014;7(8):941-951. doi:10.1242/dmm.015974.
9. Anderson K, Lutz C, van Delft FW, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature. 2011;469(7330):356-561. doi: 10.1038/nature09650.
10. Ding L, Ley TJ, Larson DE, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481(7382):506-510. doi:10.1038/nature10738.
11. Terada K, Yamaguchi H, Ueki T, et al. Usefulness of BCOR gene mutation as a prognostic factor in acute myeloid leukemia with intermediate cytogenetic prognosis. Genes Chromosomes Cancer. 2018;57(8):401-408. doi:10.1002/gcc.22542.
12. Grossmann V, Tiacci E, Holmes AB, et al. Whole-exome sequencing identifies somatic mutations of BCOR in acute myeloid leukemia with normal karyotype. Blood. 2011;118(23):6153-6163. doi:10.1182/blood-2011-07-365320.
13. Parkin B, Ouillette P, Li Y, et al. Clonal evolution and devolution after chemotherapy in adult acute myelogenous leukemia. Blood. 2013;121(2):369-377. doi:10.1182/blood-2012-04-427039.
14. Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209-2221. doi:10.1056/NEJMoa1516192.
15. DiNardo CD, Ravandi F, Agresta S, et al. Characteristics, clinical outcome, and prognostic significance of IDH mutations in AML. Am J Hematol. 2015;90(8):732-736. doi:10.1002/ajh.24072.
16. Rakheja D, Konoplev S, Medeiros LJ, Chen W. IDH mutations in acute myeloid leukemia. Hum Pathol. 2012;43 (10):1541-1551. doi:10.1016/j.humpath.2012.05.003.
17. Lai C, Doucette K, Norsworthy K. Recent drug approvals for acute myeloid leukemia. J H Oncol. 2019;12(1):100. doi:10.1186/s13045-019-0774-x.
18. Boddu P, Takahashi K, Pemmaraju N, et al. Influence of IDH on FLT3-ITD status in newly diagnosed AML. Leukemia. 2017;31(11):2526-2529. doi:10.1038/leu.2017.244.
1. De Kouchkovsky I, Abdul-Hay M. Acute myeloid leukemia: a comprehensive review and 2016 update. Blood Cancer J. 2016;6(7):e441. doi:10.1038/bcj.2016.50.
2. National Cancer Institute. Cancer Stat Facts: Leukemia — acute myeloid leukemia (AML). Accessed November 4, 2020. https://seer.cancer.gov/statfacts/html/amyl.html
3. Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med. 2017;377(5):454-464. doi:10.1056/NEJMoa1614359.
4. DiNardo CD, Stein EM, de Botton S, et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med. 2018;378(25):2386-2398. doi:10.1056/NEJMoa1716984.
5. Roboz, GJ, DiNardo, CD, Stein, EM, et al. Ivosidenib induces deep durable remissions in patients with newly diagnosed IDH1-mutant acute myeloid leukemia. Blood. 2019;135(7), 463-471. doi: 10.1182/blood.2019002140
6. Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722-731. doi:10.1182/blood-2017-04-779405.
7. Jan M, Majeti R. Clonal evolution of acute leukemia genomes. Oncogene. 2013;32(2):135-140. doi:10.1038/onc.2012.48.
8. Grove CS, Vassiliou GS. Acute myeloid leukaemia: a paradigm for the clonal evolution of cancer? Dis Model Mech. 2014;7(8):941-951. doi:10.1242/dmm.015974.
9. Anderson K, Lutz C, van Delft FW, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature. 2011;469(7330):356-561. doi: 10.1038/nature09650.
10. Ding L, Ley TJ, Larson DE, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481(7382):506-510. doi:10.1038/nature10738.
11. Terada K, Yamaguchi H, Ueki T, et al. Usefulness of BCOR gene mutation as a prognostic factor in acute myeloid leukemia with intermediate cytogenetic prognosis. Genes Chromosomes Cancer. 2018;57(8):401-408. doi:10.1002/gcc.22542.
12. Grossmann V, Tiacci E, Holmes AB, et al. Whole-exome sequencing identifies somatic mutations of BCOR in acute myeloid leukemia with normal karyotype. Blood. 2011;118(23):6153-6163. doi:10.1182/blood-2011-07-365320.
13. Parkin B, Ouillette P, Li Y, et al. Clonal evolution and devolution after chemotherapy in adult acute myelogenous leukemia. Blood. 2013;121(2):369-377. doi:10.1182/blood-2012-04-427039.
14. Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209-2221. doi:10.1056/NEJMoa1516192.
15. DiNardo CD, Ravandi F, Agresta S, et al. Characteristics, clinical outcome, and prognostic significance of IDH mutations in AML. Am J Hematol. 2015;90(8):732-736. doi:10.1002/ajh.24072.
16. Rakheja D, Konoplev S, Medeiros LJ, Chen W. IDH mutations in acute myeloid leukemia. Hum Pathol. 2012;43 (10):1541-1551. doi:10.1016/j.humpath.2012.05.003.
17. Lai C, Doucette K, Norsworthy K. Recent drug approvals for acute myeloid leukemia. J H Oncol. 2019;12(1):100. doi:10.1186/s13045-019-0774-x.
18. Boddu P, Takahashi K, Pemmaraju N, et al. Influence of IDH on FLT3-ITD status in newly diagnosed AML. Leukemia. 2017;31(11):2526-2529. doi:10.1038/leu.2017.244.
Long-Term Successful Treatment of Indolent Systemic Mastocytosis With Omalizumab
This case study suggests that omalizumab may help prevent anaphylaxis and reduce disease burden associated with systemic mastocytosis, but further studies and formal clinical trials are needed to confirm these findings.
Mastocytosis is a rare disease that causes allergic and anaphylactic symptoms due to chronic or episodic, excessive mast cell degranulation as well as mast cell infiltration of the skin or other organs.1 Mast cells aid in innate immunity by generation of a vasodilatory and inflammatory response and are significant contributors to allergic reactions. Cutaneous mastocytosis is defined by isolated skin involvement. Systemic mastocytosis (SM) is characterized by mast cell infiltration of extracutaneous organs, most often bone marrow.2

Background
SM is divided into distinct subtypes (Table 1). Nonadvanced SM subtypes include indolent SM and smoldering SM. These are the most common forms and tend to have more slowly progressing courses without evidence of organ tissue dysfunction, a myelodysplastic syndrome, or of a myeloproliferative disorder.3 Advanced SM is less common and is associated with organ tissue dysfunction. It also may be associated with myeloproliferative, myelodysplastic, or lymphoproliferative hematologic neoplasms, and subtypes include aggressive SM, SM with an associated hematologic neoplasm, and mast cell leukemia (Table 2).4

Treatment options approved by the US Food and Drug Administration (FDA) for advanced SM include disease-altering medications, such as tyrosine kinase inhibitors (eg, imatinib), but the approved treatment options for nonadvanced SM are generally aimed at managing only symptoms (Table 3). Although not approved by the FDA for the treatment of SM, omalizumab may aid in the prevention of anaphylaxis, the reduction of disease burden, and the improvement in quality of life for patients with SM.5 Omalizumab is a humanized monoclonal antibody against the Fc portion of immunoglobulin E (IgE). It is approved by the FDA for treatment of asthma as well as chronic idiopathic urticaria.6
Case Presentation
A 32-year-old female initially presented to Womack Army Medical Center at Fort Bragg, North Carolina, for evaluation due to recurrent episodes of anaphylaxis occurring 1 to 2 times per month as well as chronic skin rashes that progressed over the previous 5 years (Figure). She initially was diagnosed with idiopathic anaphylaxis and subsequently had multiple emergency department (ED) and clinic visits for vasovagal syncope, unexplained allergic reactions, dizziness, giddiness, and shortness of breath. More recently, she was diagnosed with idiopathic urticaria.
The patient reported at least 12 episodes in the previous year involving facial flushing that proceeded inferiorly, chest tightness, shortness of breath, labored breathing, crampy abdominal pain, and nausea without urticaria or significant pruritus. These bouts often were accompanied by mild facial angioedema, acute sinus pressure, vomiting, tachycardia, and lightheadedness. She reported experiencing brief losses of consciousness with at least 4 of these episodes. Home and ED blood pressure measurements revealed hypotension on several occasions with systolic readings in the 80s. She also developed nonpruritic freckles on her upper chest initially with subsequent increase in number and spread to involve her entire trunk, proximal extremities, and eventually distal extremities.
The patient had received intramuscular epinephrine several times, which led to rapid resolution of her symptoms. Intensive care unit admission for observation overnight was deemed necessary following one of her first episodes, but she did not require intubation or vasopressor support. Eventually, she began treating most episodes at home with diphenhydramine, ranitidine, and occasionally an epinephrine auto-injector, only presenting to the ED for severe dyspnea or loss of consciousness. Some episodes awoke her from sleeping but no triggers were identified (eg, foods, alcohol, supplements, medications, insect stings, latex exposure, exercise, strong emotions, or menstrual cycle).
Examination revealed hyperpigmented macules and papules scattered on the trunk and extremities, with a positive Darier sign. Punch biopsy of one of the macules revealed focal basal cell hyperpigmentation and sheets of benign-appearing mast cells in the superficial dermis, highlighted by CD117 immunohistochemical stain. A serum tryptase level was obtained and found to be significantly elevated (134 mcg/L). The patient was diagnosed with maculopapular cutaneous mastocytosis (urticaria pigmentosa).
A bone marrow biopsy revealed multiple prominent infiltrates of monomorphic, spindled, CD117-positive, CD2-positive, and CD25-positive mast cells arranged interstitially and paratrabecularly, with associated reticulin fibrosis. Indolent SM was diagnosed according to the World Health Organization classification system with multifocal, dense aggregates of mast cells (> 25%) in the bone marrow and with persistently elevated serum tryptase levels (134, 134, 151, and 159 ng/mL) without laboratory evidence of an associated clonal myeloid disorder or findings consistent with infiltrating bone lesions on full body magnetic resonance imaging scan.4
Despite maximal antihistamine and antileukotriene therapy with ranitidine (150 mg twice daily), cetirizine (10 mg twice daily), montelukast (10 mg daily), and cromolyn sodium (200 mg daily), the patient continued to experience recurrent episodes of anaphylaxis requiring subcutaneous epinephrine and systemic corticosteroids. In May 2016, the patient began a trial of off-label therapy with omalizumab injections (300 mg subcutaneous every 4 weeks). She has continued on therapy for more than 4 years and experienced only 1 anaphylactic episode. She also has had significant improvement in cutaneous symptoms.
Discussion
Mast cell overactivation and degranulation in mastocytosis is largely driven by the IgE antibody, which plays a significant role in atopic conditions, immediate hypersensitivity reactions, and anaphylaxis, as well as in the immunologic response to parasitic infections. The severity of atopic disease seems to be associated with serum IgE levels in many patients.7 IgE binding to surface receptors on mast cells and eosinophils prompts the release of toxic mediators, incites inflammation, and induces allergic symptoms.8 Activation of mast cells is classically elicited by IgE binding to the high-affinity Fcε RI receptor, the expression of which correlates with IgE levels.9
The anti-IgE, recombinant, humanized immunoglobulin G monoclonal antibody, omalizumab, decreases mastocytic and eosinophilic symptoms by binding and inhibiting IgE. This diminishes free IgE levels, inhibits IgE binding to the Fcε RI receptor, and affects downregulation of this high-affinity receptor on mast cells and basophils.6 Omalizumab is currently FDA approved only for the treatment of moderate-to-severe, persistent, allergic asthma that is not controlled by inhaled corticosteroids in patients aged ≥ 6 years, and for chronic idiopathic urticaria not controlled by H1 antihistamine therapy in patients aged ≥ 12 years.10 However, it stands to reason that this therapy also should be effective in the treatment of other poorly controlled atopic conditions, especially mastocytosis, the symptoms of which are driven by excessive mast cell degranulation and tissue infiltration.
As early as 2007, preliminary data showed that treatment with omalizumab could decrease the frequency of episodes of anaphylaxis.11 A National Institutes of Health case report followed 2 patients, one for 5 months and the other for 24 months. Both patients experienced a decrease in frequency of anaphylaxis following initiation of omalizumab. In 2010, a second case report described the treatment of an Australian patient with recurrent idiopathic anaphylaxis also diagnosed with SM. After initiation of treatment with omalizumab, she, too, experienced decreased frequency of episodes of anaphylaxis over 14 months.12 A review of patients treated at the Mastocytosis Centre Odense University Hospital in Denmark was published in 2017. Of 13 patients with SM treated with omalizumab, 5 experienced what was considered a complete response to the medication, with 3 each experiencing major and partial responses.5 The median treatment time in these patients was 27 months. Each of these cases showed significant promise in the use of omalizumab to treat SM, informing the decision to attempt this treatment in our patient.
The potential positive effects of omalizumab in reducing symptom severity in patients with SM was further supported by a 2017 meta-analysis. This review included several individual case reports noting that omalizumab could decrease frequency of pulmonary and gastrointestinal manifestations of SM.13 A small randomized control trial of omalizumab for treatment of mild symptoms of SM found improvement in disease severity, although neither primary nor secondary endpoints reached statistical significance.14
This case demonstrates a substantial, long-term, clinical benefit and quality of life improvement with omalizumab therapy in a patient with indolent SM that was not adequately controlled by conventional therapies. This is evidenced by an impressive decline in the frequency of mastocytic anaphylactic episodes as well as diminished patient-endorsed cutaneous symptoms.
This case provides further evidence of the efficacy of this therapy in diminishing disease burden for patients with SM who are otherwise limited to treatments aimed at transient symptomatic relief without significant alteration of the underlying cause of symptoms. At the time this article was written, our patient had now 52 months of continuous treatment without any adverse reactions noted, suggesting the treatment's long-term efficacy. It also adds to a small but growing body of literature that supports the use of anti-IgE therapy as a treatment option for improved management of this distressing, life-altering illness. Even in the time that our patient has been receiving omalizumab for SM, another small case series of 2 patients has been published showing sustained treatment effect at 12 years of therapy.15 This adds further insight that omalizumab can offer long-term, safe treatment for this limiting condition.
Omalizumab therapy is not without risk, but for patients afflicted by unrestrained mastocytic disease, the benefits may outweigh the risks. The most common significant risk with this medication is anaphylaxis, occurring in 1 to 2 per 1,000 patients, usually within 2 hours of an injection.16 This may correlate to the underlying degree of atopy in patients receiving omalizumab, and the risk of anaphylaxis is relatively low compared with that of many other biologic medications.17 Additionally, early data from initial phases of clinical trials indicated a potentially elevated malignancy risk with omalizumab. However, subsequent pooled analysis of larger numbers of patients has decreased suspicion that a causal relationship exists.18
Conclusions
Omalizumab has proven value in the treatment of atopic conditions, such as asthma and idiopathic urticaria, for which it has been approved for use by the FDA. Its effectiveness in significantly decreasing free serum IgE levels, and inhibiting IgE activation of mast cells makes it a possible treatment option for patients with SM who are not sufficiently controlled with conventional therapy. The findings in this case suggest that omalizumab may be effective in the prevention of anaphylaxis and in the reduction of disease burden associated with SM. Further studies and formal clinical trials are needed to confirm these findings. Patients should be counseled appropriately concerning the risks, benefits, and off-label status of this treatment option.
1. Theoharides TC, Valent P, Akin C. Mast cells, mastocytosis, and related disorders. N Engl J Med. 2015;373(2):163-172. doi:10.1056/NEJMra1409760
2. Valent P, Sperr WR, Schwartz LB, Horny H-P. Diagnosis and classification of mast cell proliferative disorders: delineation from immunologic diseases and non-mast cell hematopoietic neoplasms. J Allergy Clin Immunol. 2004;114(1):3-11. doi:10.1016/j.jaci.2004.02.045
3. Valent P, Sotlar K, Sperr WR, et al. Refined diagnostic criteria and classification of mast cell leukemia (MCL) and myelomastocytic leukemia (MML): a consensus proposal. Ann Oncol. 2014;25(9):1691-1700. doi:10.1093/annonc/mdu047
4. Valent P, Akin C, Metcalfe DD. Mastocytosis: 2016 updated WHO classification and novel emerging treatment concepts. Blood. 2017;129(11):1420-1427. doi:10.1182/blood-2016-09-731893
5. Broesby-Olsen S, Vestergaard H, Mortz CG, et al. Omalizumab prevents anaphylaxis and improves symptoms in systemic mastocytosis: Efficacy and safety observations. 2018;73(1):230-238. doi:10.1111/all.13237
6. Kaplan AP, Giménez-Arnau AM, Saini SS.Mechanisms of action that contribute to efficacy of omalizumab in chronic spontaneous urticaria. Allergy. 2017;72(4):519-533. doi:10.1111/all.13083
7. Borish L, Chipps B, Deniz Y, Gujrathi S, Zheng B, Dolan C; TENOR Study Group. Total serum IgE levels in a large cohort of patients with severe or difficult-to-treat asthma. Ann Allergy Asthma Immunol. 2005;95(3):247-253. doi:10.1016/S1081-1206(10)61221-5
8. Corry DB, Kheradmand F. Induction and regulation of the IgE response. Nature. 1999;402(suppl 6760):18-23. doi:10.1038/35037014
9. MacGlashan D, McKenzie-White J, Chichester K, et al. In vitro regulation of FcRIα expression on human basophils by IgE antibody. Blood. 1998;91(5):1633-1643.
10. XOLAIR [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation. Revised 2019. Accessed November 11, 2020. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/103976s5234lbl.pdf
11. Carter MC, Robyn JA, Bressler PB, Walker JC, Shapiro GC, and Metcalfe DD. Omalizumab for the treatment of unprovoked anaphylaxis in patients with systemic mastocytosis. J Allergy Clin Immunol. 2007;119(6):1550-1551. doi:10.1016/j.jaci.2007.03.032
12. Douglass JA, Carroll K, Voskamp A, Bourke P, Wei A, O’Hehir RE. Omalizumab is effective in treating systemic mastocytosis in a nonatopic patient. Allergy. 2010; 65(7):926-927. doi:10.1111/j.1398-9995.2009.02259.x
13. Le M, Miedzybrodzki B, Olynych T, Chapdelaine H, Ben-Shoshan M. Natural history and treatment of cutaneous and systemic mastocytosis. Postgrad Med. 2017;129(8):896-901. doi:10.1080/00325481.2017.1364124
14. Distler M, Maul J-T, Steiner T, et al. Efficacy of omalizumab in mastocytosis: allusive indication obtained from a prospective, double-blind, multicenter study (XOLMA Study) [published online ahead of print January 20, 2020]. Dermatology. doi:10.1159/000504842
15. Constantine G, Bressler P, Petroni D, Metcalfe D, Carter M. Twelve-year follow-up of omalizumab for anaphylaxis in 2 patients with systemic mastocytosis. J Allergy Clin Immunol Pract. 2019;7(4)1314-1316. doi:10.1016/j.jaip.2018.07.041
16. Fanta CH. Asthma. N Engl J Med. 2009;360(10):1002-1014. doi:10.1056/NEJMra0804579
17. Baldo BA. Adverse events to monoclonal antibodies used for cancer therapy: focus on hypersensitivity responses. Oncoimmunology. 2013;2(10):e26333. doi:10.4161/onci.26333
18. Busse W, Buhl R, Fernandez Vidaurre C, et al. Omalizumab and the risk of malignancy: results from a pooled analysis. J Allergy Clin Immunol. 2012;129(4):983-989.e6. doi:10.1016/j.jaci.2012.01.033.
19. Castells M, Akin C. Mastocytosis (cutaneous and systemic): epidemiology, pathogenesis, and clinical manifestations. Accessed December 8, 2020. Updated June 12, 2018. https://www.uptodate.com/contents/mastocytosis-cutaneous-and-systemic-epidemiology-pathogenesis-and-clinical-manifestations
20. Czarny J, Lange M, Lugowska-Umer H, Nowicki R. Cutaneous mastocytosis treatment: strategies, limitations, and perspectives. Postepy Dermatol Alergol. 2018;35(6):541-545. doi:10.5114/ada.2018.77605
This case study suggests that omalizumab may help prevent anaphylaxis and reduce disease burden associated with systemic mastocytosis, but further studies and formal clinical trials are needed to confirm these findings.
This case study suggests that omalizumab may help prevent anaphylaxis and reduce disease burden associated with systemic mastocytosis, but further studies and formal clinical trials are needed to confirm these findings.
Mastocytosis is a rare disease that causes allergic and anaphylactic symptoms due to chronic or episodic, excessive mast cell degranulation as well as mast cell infiltration of the skin or other organs.1 Mast cells aid in innate immunity by generation of a vasodilatory and inflammatory response and are significant contributors to allergic reactions. Cutaneous mastocytosis is defined by isolated skin involvement. Systemic mastocytosis (SM) is characterized by mast cell infiltration of extracutaneous organs, most often bone marrow.2
Background
SM is divided into distinct subtypes (Table 1). Nonadvanced SM subtypes include indolent SM and smoldering SM. These are the most common forms and tend to have more slowly progressing courses without evidence of organ tissue dysfunction, a myelodysplastic syndrome, or of a myeloproliferative disorder.3 Advanced SM is less common and is associated with organ tissue dysfunction. It also may be associated with myeloproliferative, myelodysplastic, or lymphoproliferative hematologic neoplasms, and subtypes include aggressive SM, SM with an associated hematologic neoplasm, and mast cell leukemia (Table 2).4
Treatment options approved by the US Food and Drug Administration (FDA) for advanced SM include disease-altering medications, such as tyrosine kinase inhibitors (eg, imatinib), but the approved treatment options for nonadvanced SM are generally aimed at managing only symptoms (Table 3). Although not approved by the FDA for the treatment of SM, omalizumab may aid in the prevention of anaphylaxis, the reduction of disease burden, and the improvement in quality of life for patients with SM.5 Omalizumab is a humanized monoclonal antibody against the Fc portion of immunoglobulin E (IgE). It is approved by the FDA for treatment of asthma as well as chronic idiopathic urticaria.6
Case Presentation
A 32-year-old female initially presented to Womack Army Medical Center at Fort Bragg, North Carolina, for evaluation due to recurrent episodes of anaphylaxis occurring 1 to 2 times per month as well as chronic skin rashes that progressed over the previous 5 years (Figure). She initially was diagnosed with idiopathic anaphylaxis and subsequently had multiple emergency department (ED) and clinic visits for vasovagal syncope, unexplained allergic reactions, dizziness, giddiness, and shortness of breath. More recently, she was diagnosed with idiopathic urticaria.
The patient reported at least 12 episodes in the previous year involving facial flushing that proceeded inferiorly, chest tightness, shortness of breath, labored breathing, crampy abdominal pain, and nausea without urticaria or significant pruritus. These bouts often were accompanied by mild facial angioedema, acute sinus pressure, vomiting, tachycardia, and lightheadedness. She reported experiencing brief losses of consciousness with at least 4 of these episodes. Home and ED blood pressure measurements revealed hypotension on several occasions with systolic readings in the 80s. She also developed nonpruritic freckles on her upper chest initially with subsequent increase in number and spread to involve her entire trunk, proximal extremities, and eventually distal extremities.
The patient had received intramuscular epinephrine several times, which led to rapid resolution of her symptoms. Intensive care unit admission for observation overnight was deemed necessary following one of her first episodes, but she did not require intubation or vasopressor support. Eventually, she began treating most episodes at home with diphenhydramine, ranitidine, and occasionally an epinephrine auto-injector, only presenting to the ED for severe dyspnea or loss of consciousness. Some episodes awoke her from sleeping but no triggers were identified (eg, foods, alcohol, supplements, medications, insect stings, latex exposure, exercise, strong emotions, or menstrual cycle).
Examination revealed hyperpigmented macules and papules scattered on the trunk and extremities, with a positive Darier sign. Punch biopsy of one of the macules revealed focal basal cell hyperpigmentation and sheets of benign-appearing mast cells in the superficial dermis, highlighted by CD117 immunohistochemical stain. A serum tryptase level was obtained and found to be significantly elevated (134 mcg/L). The patient was diagnosed with maculopapular cutaneous mastocytosis (urticaria pigmentosa).
A bone marrow biopsy revealed multiple prominent infiltrates of monomorphic, spindled, CD117-positive, CD2-positive, and CD25-positive mast cells arranged interstitially and paratrabecularly, with associated reticulin fibrosis. Indolent SM was diagnosed according to the World Health Organization classification system with multifocal, dense aggregates of mast cells (> 25%) in the bone marrow and with persistently elevated serum tryptase levels (134, 134, 151, and 159 ng/mL) without laboratory evidence of an associated clonal myeloid disorder or findings consistent with infiltrating bone lesions on full body magnetic resonance imaging scan.4
Despite maximal antihistamine and antileukotriene therapy with ranitidine (150 mg twice daily), cetirizine (10 mg twice daily), montelukast (10 mg daily), and cromolyn sodium (200 mg daily), the patient continued to experience recurrent episodes of anaphylaxis requiring subcutaneous epinephrine and systemic corticosteroids. In May 2016, the patient began a trial of off-label therapy with omalizumab injections (300 mg subcutaneous every 4 weeks). She has continued on therapy for more than 4 years and experienced only 1 anaphylactic episode. She also has had significant improvement in cutaneous symptoms.
Discussion
Mast cell overactivation and degranulation in mastocytosis is largely driven by the IgE antibody, which plays a significant role in atopic conditions, immediate hypersensitivity reactions, and anaphylaxis, as well as in the immunologic response to parasitic infections. The severity of atopic disease seems to be associated with serum IgE levels in many patients.7 IgE binding to surface receptors on mast cells and eosinophils prompts the release of toxic mediators, incites inflammation, and induces allergic symptoms.8 Activation of mast cells is classically elicited by IgE binding to the high-affinity Fcε RI receptor, the expression of which correlates with IgE levels.9
The anti-IgE, recombinant, humanized immunoglobulin G monoclonal antibody, omalizumab, decreases mastocytic and eosinophilic symptoms by binding and inhibiting IgE. This diminishes free IgE levels, inhibits IgE binding to the Fcε RI receptor, and affects downregulation of this high-affinity receptor on mast cells and basophils.6 Omalizumab is currently FDA approved only for the treatment of moderate-to-severe, persistent, allergic asthma that is not controlled by inhaled corticosteroids in patients aged ≥ 6 years, and for chronic idiopathic urticaria not controlled by H1 antihistamine therapy in patients aged ≥ 12 years.10 However, it stands to reason that this therapy also should be effective in the treatment of other poorly controlled atopic conditions, especially mastocytosis, the symptoms of which are driven by excessive mast cell degranulation and tissue infiltration.
As early as 2007, preliminary data showed that treatment with omalizumab could decrease the frequency of episodes of anaphylaxis.11 A National Institutes of Health case report followed 2 patients, one for 5 months and the other for 24 months. Both patients experienced a decrease in frequency of anaphylaxis following initiation of omalizumab. In 2010, a second case report described the treatment of an Australian patient with recurrent idiopathic anaphylaxis also diagnosed with SM. After initiation of treatment with omalizumab, she, too, experienced decreased frequency of episodes of anaphylaxis over 14 months.12 A review of patients treated at the Mastocytosis Centre Odense University Hospital in Denmark was published in 2017. Of 13 patients with SM treated with omalizumab, 5 experienced what was considered a complete response to the medication, with 3 each experiencing major and partial responses.5 The median treatment time in these patients was 27 months. Each of these cases showed significant promise in the use of omalizumab to treat SM, informing the decision to attempt this treatment in our patient.
The potential positive effects of omalizumab in reducing symptom severity in patients with SM was further supported by a 2017 meta-analysis. This review included several individual case reports noting that omalizumab could decrease frequency of pulmonary and gastrointestinal manifestations of SM.13 A small randomized control trial of omalizumab for treatment of mild symptoms of SM found improvement in disease severity, although neither primary nor secondary endpoints reached statistical significance.14
This case demonstrates a substantial, long-term, clinical benefit and quality of life improvement with omalizumab therapy in a patient with indolent SM that was not adequately controlled by conventional therapies. This is evidenced by an impressive decline in the frequency of mastocytic anaphylactic episodes as well as diminished patient-endorsed cutaneous symptoms.
This case provides further evidence of the efficacy of this therapy in diminishing disease burden for patients with SM who are otherwise limited to treatments aimed at transient symptomatic relief without significant alteration of the underlying cause of symptoms. At the time this article was written, our patient had now 52 months of continuous treatment without any adverse reactions noted, suggesting the treatment's long-term efficacy. It also adds to a small but growing body of literature that supports the use of anti-IgE therapy as a treatment option for improved management of this distressing, life-altering illness. Even in the time that our patient has been receiving omalizumab for SM, another small case series of 2 patients has been published showing sustained treatment effect at 12 years of therapy.15 This adds further insight that omalizumab can offer long-term, safe treatment for this limiting condition.
Omalizumab therapy is not without risk, but for patients afflicted by unrestrained mastocytic disease, the benefits may outweigh the risks. The most common significant risk with this medication is anaphylaxis, occurring in 1 to 2 per 1,000 patients, usually within 2 hours of an injection.16 This may correlate to the underlying degree of atopy in patients receiving omalizumab, and the risk of anaphylaxis is relatively low compared with that of many other biologic medications.17 Additionally, early data from initial phases of clinical trials indicated a potentially elevated malignancy risk with omalizumab. However, subsequent pooled analysis of larger numbers of patients has decreased suspicion that a causal relationship exists.18
Conclusions
Omalizumab has proven value in the treatment of atopic conditions, such as asthma and idiopathic urticaria, for which it has been approved for use by the FDA. Its effectiveness in significantly decreasing free serum IgE levels, and inhibiting IgE activation of mast cells makes it a possible treatment option for patients with SM who are not sufficiently controlled with conventional therapy. The findings in this case suggest that omalizumab may be effective in the prevention of anaphylaxis and in the reduction of disease burden associated with SM. Further studies and formal clinical trials are needed to confirm these findings. Patients should be counseled appropriately concerning the risks, benefits, and off-label status of this treatment option.
Mastocytosis is a rare disease that causes allergic and anaphylactic symptoms due to chronic or episodic, excessive mast cell degranulation as well as mast cell infiltration of the skin or other organs.1 Mast cells aid in innate immunity by generation of a vasodilatory and inflammatory response and are significant contributors to allergic reactions. Cutaneous mastocytosis is defined by isolated skin involvement. Systemic mastocytosis (SM) is characterized by mast cell infiltration of extracutaneous organs, most often bone marrow.2
Background
SM is divided into distinct subtypes (Table 1). Nonadvanced SM subtypes include indolent SM and smoldering SM. These are the most common forms and tend to have more slowly progressing courses without evidence of organ tissue dysfunction, a myelodysplastic syndrome, or of a myeloproliferative disorder.3 Advanced SM is less common and is associated with organ tissue dysfunction. It also may be associated with myeloproliferative, myelodysplastic, or lymphoproliferative hematologic neoplasms, and subtypes include aggressive SM, SM with an associated hematologic neoplasm, and mast cell leukemia (Table 2).4
Treatment options approved by the US Food and Drug Administration (FDA) for advanced SM include disease-altering medications, such as tyrosine kinase inhibitors (eg, imatinib), but the approved treatment options for nonadvanced SM are generally aimed at managing only symptoms (Table 3). Although not approved by the FDA for the treatment of SM, omalizumab may aid in the prevention of anaphylaxis, the reduction of disease burden, and the improvement in quality of life for patients with SM.5 Omalizumab is a humanized monoclonal antibody against the Fc portion of immunoglobulin E (IgE). It is approved by the FDA for treatment of asthma as well as chronic idiopathic urticaria.6
Case Presentation
A 32-year-old female initially presented to Womack Army Medical Center at Fort Bragg, North Carolina, for evaluation due to recurrent episodes of anaphylaxis occurring 1 to 2 times per month as well as chronic skin rashes that progressed over the previous 5 years (Figure). She initially was diagnosed with idiopathic anaphylaxis and subsequently had multiple emergency department (ED) and clinic visits for vasovagal syncope, unexplained allergic reactions, dizziness, giddiness, and shortness of breath. More recently, she was diagnosed with idiopathic urticaria.
The patient reported at least 12 episodes in the previous year involving facial flushing that proceeded inferiorly, chest tightness, shortness of breath, labored breathing, crampy abdominal pain, and nausea without urticaria or significant pruritus. These bouts often were accompanied by mild facial angioedema, acute sinus pressure, vomiting, tachycardia, and lightheadedness. She reported experiencing brief losses of consciousness with at least 4 of these episodes. Home and ED blood pressure measurements revealed hypotension on several occasions with systolic readings in the 80s. She also developed nonpruritic freckles on her upper chest initially with subsequent increase in number and spread to involve her entire trunk, proximal extremities, and eventually distal extremities.
The patient had received intramuscular epinephrine several times, which led to rapid resolution of her symptoms. Intensive care unit admission for observation overnight was deemed necessary following one of her first episodes, but she did not require intubation or vasopressor support. Eventually, she began treating most episodes at home with diphenhydramine, ranitidine, and occasionally an epinephrine auto-injector, only presenting to the ED for severe dyspnea or loss of consciousness. Some episodes awoke her from sleeping but no triggers were identified (eg, foods, alcohol, supplements, medications, insect stings, latex exposure, exercise, strong emotions, or menstrual cycle).
Examination revealed hyperpigmented macules and papules scattered on the trunk and extremities, with a positive Darier sign. Punch biopsy of one of the macules revealed focal basal cell hyperpigmentation and sheets of benign-appearing mast cells in the superficial dermis, highlighted by CD117 immunohistochemical stain. A serum tryptase level was obtained and found to be significantly elevated (134 mcg/L). The patient was diagnosed with maculopapular cutaneous mastocytosis (urticaria pigmentosa).
A bone marrow biopsy revealed multiple prominent infiltrates of monomorphic, spindled, CD117-positive, CD2-positive, and CD25-positive mast cells arranged interstitially and paratrabecularly, with associated reticulin fibrosis. Indolent SM was diagnosed according to the World Health Organization classification system with multifocal, dense aggregates of mast cells (> 25%) in the bone marrow and with persistently elevated serum tryptase levels (134, 134, 151, and 159 ng/mL) without laboratory evidence of an associated clonal myeloid disorder or findings consistent with infiltrating bone lesions on full body magnetic resonance imaging scan.4
Despite maximal antihistamine and antileukotriene therapy with ranitidine (150 mg twice daily), cetirizine (10 mg twice daily), montelukast (10 mg daily), and cromolyn sodium (200 mg daily), the patient continued to experience recurrent episodes of anaphylaxis requiring subcutaneous epinephrine and systemic corticosteroids. In May 2016, the patient began a trial of off-label therapy with omalizumab injections (300 mg subcutaneous every 4 weeks). She has continued on therapy for more than 4 years and experienced only 1 anaphylactic episode. She also has had significant improvement in cutaneous symptoms.
Discussion
Mast cell overactivation and degranulation in mastocytosis is largely driven by the IgE antibody, which plays a significant role in atopic conditions, immediate hypersensitivity reactions, and anaphylaxis, as well as in the immunologic response to parasitic infections. The severity of atopic disease seems to be associated with serum IgE levels in many patients.7 IgE binding to surface receptors on mast cells and eosinophils prompts the release of toxic mediators, incites inflammation, and induces allergic symptoms.8 Activation of mast cells is classically elicited by IgE binding to the high-affinity Fcε RI receptor, the expression of which correlates with IgE levels.9
The anti-IgE, recombinant, humanized immunoglobulin G monoclonal antibody, omalizumab, decreases mastocytic and eosinophilic symptoms by binding and inhibiting IgE. This diminishes free IgE levels, inhibits IgE binding to the Fcε RI receptor, and affects downregulation of this high-affinity receptor on mast cells and basophils.6 Omalizumab is currently FDA approved only for the treatment of moderate-to-severe, persistent, allergic asthma that is not controlled by inhaled corticosteroids in patients aged ≥ 6 years, and for chronic idiopathic urticaria not controlled by H1 antihistamine therapy in patients aged ≥ 12 years.10 However, it stands to reason that this therapy also should be effective in the treatment of other poorly controlled atopic conditions, especially mastocytosis, the symptoms of which are driven by excessive mast cell degranulation and tissue infiltration.
As early as 2007, preliminary data showed that treatment with omalizumab could decrease the frequency of episodes of anaphylaxis.11 A National Institutes of Health case report followed 2 patients, one for 5 months and the other for 24 months. Both patients experienced a decrease in frequency of anaphylaxis following initiation of omalizumab. In 2010, a second case report described the treatment of an Australian patient with recurrent idiopathic anaphylaxis also diagnosed with SM. After initiation of treatment with omalizumab, she, too, experienced decreased frequency of episodes of anaphylaxis over 14 months.12 A review of patients treated at the Mastocytosis Centre Odense University Hospital in Denmark was published in 2017. Of 13 patients with SM treated with omalizumab, 5 experienced what was considered a complete response to the medication, with 3 each experiencing major and partial responses.5 The median treatment time in these patients was 27 months. Each of these cases showed significant promise in the use of omalizumab to treat SM, informing the decision to attempt this treatment in our patient.
The potential positive effects of omalizumab in reducing symptom severity in patients with SM was further supported by a 2017 meta-analysis. This review included several individual case reports noting that omalizumab could decrease frequency of pulmonary and gastrointestinal manifestations of SM.13 A small randomized control trial of omalizumab for treatment of mild symptoms of SM found improvement in disease severity, although neither primary nor secondary endpoints reached statistical significance.14
This case demonstrates a substantial, long-term, clinical benefit and quality of life improvement with omalizumab therapy in a patient with indolent SM that was not adequately controlled by conventional therapies. This is evidenced by an impressive decline in the frequency of mastocytic anaphylactic episodes as well as diminished patient-endorsed cutaneous symptoms.
This case provides further evidence of the efficacy of this therapy in diminishing disease burden for patients with SM who are otherwise limited to treatments aimed at transient symptomatic relief without significant alteration of the underlying cause of symptoms. At the time this article was written, our patient had now 52 months of continuous treatment without any adverse reactions noted, suggesting the treatment's long-term efficacy. It also adds to a small but growing body of literature that supports the use of anti-IgE therapy as a treatment option for improved management of this distressing, life-altering illness. Even in the time that our patient has been receiving omalizumab for SM, another small case series of 2 patients has been published showing sustained treatment effect at 12 years of therapy.15 This adds further insight that omalizumab can offer long-term, safe treatment for this limiting condition.
Omalizumab therapy is not without risk, but for patients afflicted by unrestrained mastocytic disease, the benefits may outweigh the risks. The most common significant risk with this medication is anaphylaxis, occurring in 1 to 2 per 1,000 patients, usually within 2 hours of an injection.16 This may correlate to the underlying degree of atopy in patients receiving omalizumab, and the risk of anaphylaxis is relatively low compared with that of many other biologic medications.17 Additionally, early data from initial phases of clinical trials indicated a potentially elevated malignancy risk with omalizumab. However, subsequent pooled analysis of larger numbers of patients has decreased suspicion that a causal relationship exists.18
Conclusions
Omalizumab has proven value in the treatment of atopic conditions, such as asthma and idiopathic urticaria, for which it has been approved for use by the FDA. Its effectiveness in significantly decreasing free serum IgE levels, and inhibiting IgE activation of mast cells makes it a possible treatment option for patients with SM who are not sufficiently controlled with conventional therapy. The findings in this case suggest that omalizumab may be effective in the prevention of anaphylaxis and in the reduction of disease burden associated with SM. Further studies and formal clinical trials are needed to confirm these findings. Patients should be counseled appropriately concerning the risks, benefits, and off-label status of this treatment option.
1. Theoharides TC, Valent P, Akin C. Mast cells, mastocytosis, and related disorders. N Engl J Med. 2015;373(2):163-172. doi:10.1056/NEJMra1409760
2. Valent P, Sperr WR, Schwartz LB, Horny H-P. Diagnosis and classification of mast cell proliferative disorders: delineation from immunologic diseases and non-mast cell hematopoietic neoplasms. J Allergy Clin Immunol. 2004;114(1):3-11. doi:10.1016/j.jaci.2004.02.045
3. Valent P, Sotlar K, Sperr WR, et al. Refined diagnostic criteria and classification of mast cell leukemia (MCL) and myelomastocytic leukemia (MML): a consensus proposal. Ann Oncol. 2014;25(9):1691-1700. doi:10.1093/annonc/mdu047
4. Valent P, Akin C, Metcalfe DD. Mastocytosis: 2016 updated WHO classification and novel emerging treatment concepts. Blood. 2017;129(11):1420-1427. doi:10.1182/blood-2016-09-731893
5. Broesby-Olsen S, Vestergaard H, Mortz CG, et al. Omalizumab prevents anaphylaxis and improves symptoms in systemic mastocytosis: Efficacy and safety observations. 2018;73(1):230-238. doi:10.1111/all.13237
6. Kaplan AP, Giménez-Arnau AM, Saini SS.Mechanisms of action that contribute to efficacy of omalizumab in chronic spontaneous urticaria. Allergy. 2017;72(4):519-533. doi:10.1111/all.13083
7. Borish L, Chipps B, Deniz Y, Gujrathi S, Zheng B, Dolan C; TENOR Study Group. Total serum IgE levels in a large cohort of patients with severe or difficult-to-treat asthma. Ann Allergy Asthma Immunol. 2005;95(3):247-253. doi:10.1016/S1081-1206(10)61221-5
8. Corry DB, Kheradmand F. Induction and regulation of the IgE response. Nature. 1999;402(suppl 6760):18-23. doi:10.1038/35037014
9. MacGlashan D, McKenzie-White J, Chichester K, et al. In vitro regulation of FcRIα expression on human basophils by IgE antibody. Blood. 1998;91(5):1633-1643.
10. XOLAIR [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation. Revised 2019. Accessed November 11, 2020. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/103976s5234lbl.pdf
11. Carter MC, Robyn JA, Bressler PB, Walker JC, Shapiro GC, and Metcalfe DD. Omalizumab for the treatment of unprovoked anaphylaxis in patients with systemic mastocytosis. J Allergy Clin Immunol. 2007;119(6):1550-1551. doi:10.1016/j.jaci.2007.03.032
12. Douglass JA, Carroll K, Voskamp A, Bourke P, Wei A, O’Hehir RE. Omalizumab is effective in treating systemic mastocytosis in a nonatopic patient. Allergy. 2010; 65(7):926-927. doi:10.1111/j.1398-9995.2009.02259.x
13. Le M, Miedzybrodzki B, Olynych T, Chapdelaine H, Ben-Shoshan M. Natural history and treatment of cutaneous and systemic mastocytosis. Postgrad Med. 2017;129(8):896-901. doi:10.1080/00325481.2017.1364124
14. Distler M, Maul J-T, Steiner T, et al. Efficacy of omalizumab in mastocytosis: allusive indication obtained from a prospective, double-blind, multicenter study (XOLMA Study) [published online ahead of print January 20, 2020]. Dermatology. doi:10.1159/000504842
15. Constantine G, Bressler P, Petroni D, Metcalfe D, Carter M. Twelve-year follow-up of omalizumab for anaphylaxis in 2 patients with systemic mastocytosis. J Allergy Clin Immunol Pract. 2019;7(4)1314-1316. doi:10.1016/j.jaip.2018.07.041
16. Fanta CH. Asthma. N Engl J Med. 2009;360(10):1002-1014. doi:10.1056/NEJMra0804579
17. Baldo BA. Adverse events to monoclonal antibodies used for cancer therapy: focus on hypersensitivity responses. Oncoimmunology. 2013;2(10):e26333. doi:10.4161/onci.26333
18. Busse W, Buhl R, Fernandez Vidaurre C, et al. Omalizumab and the risk of malignancy: results from a pooled analysis. J Allergy Clin Immunol. 2012;129(4):983-989.e6. doi:10.1016/j.jaci.2012.01.033.
19. Castells M, Akin C. Mastocytosis (cutaneous and systemic): epidemiology, pathogenesis, and clinical manifestations. Accessed December 8, 2020. Updated June 12, 2018. https://www.uptodate.com/contents/mastocytosis-cutaneous-and-systemic-epidemiology-pathogenesis-and-clinical-manifestations
20. Czarny J, Lange M, Lugowska-Umer H, Nowicki R. Cutaneous mastocytosis treatment: strategies, limitations, and perspectives. Postepy Dermatol Alergol. 2018;35(6):541-545. doi:10.5114/ada.2018.77605
1. Theoharides TC, Valent P, Akin C. Mast cells, mastocytosis, and related disorders. N Engl J Med. 2015;373(2):163-172. doi:10.1056/NEJMra1409760
2. Valent P, Sperr WR, Schwartz LB, Horny H-P. Diagnosis and classification of mast cell proliferative disorders: delineation from immunologic diseases and non-mast cell hematopoietic neoplasms. J Allergy Clin Immunol. 2004;114(1):3-11. doi:10.1016/j.jaci.2004.02.045
3. Valent P, Sotlar K, Sperr WR, et al. Refined diagnostic criteria and classification of mast cell leukemia (MCL) and myelomastocytic leukemia (MML): a consensus proposal. Ann Oncol. 2014;25(9):1691-1700. doi:10.1093/annonc/mdu047
4. Valent P, Akin C, Metcalfe DD. Mastocytosis: 2016 updated WHO classification and novel emerging treatment concepts. Blood. 2017;129(11):1420-1427. doi:10.1182/blood-2016-09-731893
5. Broesby-Olsen S, Vestergaard H, Mortz CG, et al. Omalizumab prevents anaphylaxis and improves symptoms in systemic mastocytosis: Efficacy and safety observations. 2018;73(1):230-238. doi:10.1111/all.13237
6. Kaplan AP, Giménez-Arnau AM, Saini SS.Mechanisms of action that contribute to efficacy of omalizumab in chronic spontaneous urticaria. Allergy. 2017;72(4):519-533. doi:10.1111/all.13083
7. Borish L, Chipps B, Deniz Y, Gujrathi S, Zheng B, Dolan C; TENOR Study Group. Total serum IgE levels in a large cohort of patients with severe or difficult-to-treat asthma. Ann Allergy Asthma Immunol. 2005;95(3):247-253. doi:10.1016/S1081-1206(10)61221-5
8. Corry DB, Kheradmand F. Induction and regulation of the IgE response. Nature. 1999;402(suppl 6760):18-23. doi:10.1038/35037014
9. MacGlashan D, McKenzie-White J, Chichester K, et al. In vitro regulation of FcRIα expression on human basophils by IgE antibody. Blood. 1998;91(5):1633-1643.
10. XOLAIR [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation. Revised 2019. Accessed November 11, 2020. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/103976s5234lbl.pdf
11. Carter MC, Robyn JA, Bressler PB, Walker JC, Shapiro GC, and Metcalfe DD. Omalizumab for the treatment of unprovoked anaphylaxis in patients with systemic mastocytosis. J Allergy Clin Immunol. 2007;119(6):1550-1551. doi:10.1016/j.jaci.2007.03.032
12. Douglass JA, Carroll K, Voskamp A, Bourke P, Wei A, O’Hehir RE. Omalizumab is effective in treating systemic mastocytosis in a nonatopic patient. Allergy. 2010; 65(7):926-927. doi:10.1111/j.1398-9995.2009.02259.x
13. Le M, Miedzybrodzki B, Olynych T, Chapdelaine H, Ben-Shoshan M. Natural history and treatment of cutaneous and systemic mastocytosis. Postgrad Med. 2017;129(8):896-901. doi:10.1080/00325481.2017.1364124
14. Distler M, Maul J-T, Steiner T, et al. Efficacy of omalizumab in mastocytosis: allusive indication obtained from a prospective, double-blind, multicenter study (XOLMA Study) [published online ahead of print January 20, 2020]. Dermatology. doi:10.1159/000504842
15. Constantine G, Bressler P, Petroni D, Metcalfe D, Carter M. Twelve-year follow-up of omalizumab for anaphylaxis in 2 patients with systemic mastocytosis. J Allergy Clin Immunol Pract. 2019;7(4)1314-1316. doi:10.1016/j.jaip.2018.07.041
16. Fanta CH. Asthma. N Engl J Med. 2009;360(10):1002-1014. doi:10.1056/NEJMra0804579
17. Baldo BA. Adverse events to monoclonal antibodies used for cancer therapy: focus on hypersensitivity responses. Oncoimmunology. 2013;2(10):e26333. doi:10.4161/onci.26333
18. Busse W, Buhl R, Fernandez Vidaurre C, et al. Omalizumab and the risk of malignancy: results from a pooled analysis. J Allergy Clin Immunol. 2012;129(4):983-989.e6. doi:10.1016/j.jaci.2012.01.033.
19. Castells M, Akin C. Mastocytosis (cutaneous and systemic): epidemiology, pathogenesis, and clinical manifestations. Accessed December 8, 2020. Updated June 12, 2018. https://www.uptodate.com/contents/mastocytosis-cutaneous-and-systemic-epidemiology-pathogenesis-and-clinical-manifestations
20. Czarny J, Lange M, Lugowska-Umer H, Nowicki R. Cutaneous mastocytosis treatment: strategies, limitations, and perspectives. Postepy Dermatol Alergol. 2018;35(6):541-545. doi:10.5114/ada.2018.77605
Erythema Ab Igne and Malignant Transformation to Squamous Cell Carcinoma
Case Report
A 67-year-old Black woman presented with a long-standing history of pruritus and “scaly thick bumps” on the lower extremities. Upon further questioning, she reported a 30-year history of placing her feet by an electric space heater and daily baths in “very hot” water. A review of systems and medical history were unremarkable, and the patient was not on any medications. Initial physical examination of the lower extremities demonstrated lichenified plaques and scattered, firm, ulcerated nodules surrounded by mottled postinflammatory hyperpigmentation with sharp demarcation at the midcalf bilaterally (Figure 1).
Subsequently, the patient was shown to have multiple actinic keratoses and SCCs, both in situ and invasive, within the areas of EAI (Figure 2). The patient had no actinic keratoses or other cutaneous malignant neoplasms elsewhere on the skin. Management of actinic keratoses, SCC in situ, and invasive SCC on the lower extremities included numerous excisions, treatment with liquid nitrogen, and topical 5-fluorouracil under occlusion. The patient continues to be monitored frequently.
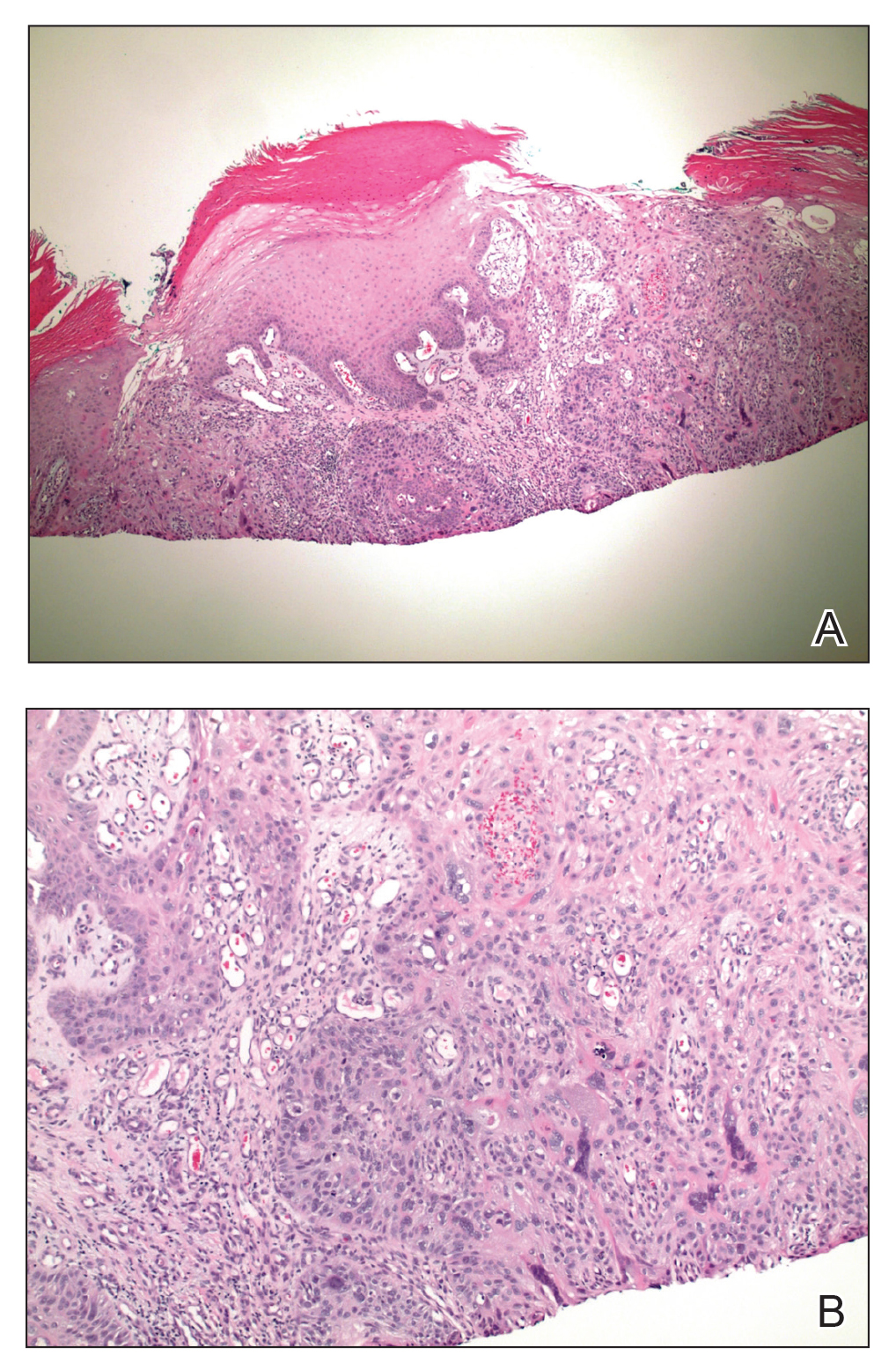
Comment
Presentation of EAI
Erythema ab igne is a cutaneous reaction resulting from prolonged exposure to an infrared heat source at temperatures insufficient to cause a burn (37 °F to 11
Histopathology of EAI
Histologically, later stages of EAI can demonstrate focal hyperkeratosis with dyskeratosis and increased dermal elastosis, similar to actinic damage, with a predisposition to develop SCC.2 Notably, early reports document various heat-induced carcinomas, including kangri-burn cancers among Kashmiris, kang thermal cancers in China, and kairo cancers in Japan.2,4,5 More recent reports identify cutaneous carcinomas arising specifically in the setting of EAI, most commonly SCC3; Merkel cell carcinoma and cutaneous marginal zone lymphoma are less commonly reported malignancies.6,7 Given the frequency of malignant transformation within sites of thermal exposure, chronic heat exposure may share a common pathophysiology with SCC and other neoplasms, including Merkel cell carcinoma and cutaneous marginal zone lymphoma.
SCC in Black Individuals
Squamous cell carcinoma is the most common skin cancer in Black individuals, with a notably higher incidence in high-risk subpopulations (immunosuppressed patients). Unlike White individuals, SCCs frequently occur in non–sun-exposed areas in Black individuals and are associated with unique risk factors, such as human papillomavirus, as demonstrated in Black transplant patients.8 A retrospective study examining the characteristics of SCC on the legs of Black individuals documented atypical hyperkeratotic neoplasms surrounded by abnormal pigmentation and mottling of surrounding skin.9 Morphologic skin changes could be the result of chronic thermal damage: Numerous patients reported a history of leg warming from an open heat source. Other patients had an actual diagnosis of EAI. The predilection for less-exposed skin suggests UV radiation (UVR) might be a less important predisposing risk factor for this racial group, and the increased mortality associated with SCC in Black individuals might represent a more aggressive nature to this subset of SCCs.9 Furthermore, infrared radiation (IRR), such as fires and coal stoves, might have the potential to stimulate skin changes similar to those associated with UVR and ultimately malignant changes.
Infrared Radiation
Compared to UVR, little is known about the biological effects of IRR (wavelength, 760 nm to 1 mm), to which human skin is constantly exposed from natural and artificial light sources. Early studies have demonstrated the carcinogenic potential of IRR, observing an augmentation of UVR-induced tumorigenesis in the presence of heat. More recently, IRR was observed to stimulate increased collagenase production from dermal fibroblasts and influence pathways (extracellular signal-related kinases 1/2 and p38 mitogen-activated protein kinases) in a similar fashion to UVB and UVA.10,11 Therefore, IRR might be capable of eliciting molecular responses comparable to those caused by UVR.
Conclusion
Although SCC in association with EAI is uncommon, historical reports of thermal cancers and scientific observations of IRR-induced biological and molecular effects support EAI as a predisposing risk factor for SCC and the important need for close monitoring by physicians. Studies are needed to further elucidate the pathologic effects of IRR, with more promotion of caution relating to thermal exposure.
- Milchak M, Smucker J, Chung CG, et al. Erythema ab igne due to heating pad use: a case report and review of clinical presentation, prevention, and complications. Case Rep Med. 2016;2016:1862480.
- Miller K, Hunt R, Chu J, et al. Erythema ab igne. Dermatol Online J. 2011;17:28. Accessed December 10, 2020. https://escholarship.org/uc/item/47z4v01z
- Wharton JB, Sheehan DJ, Lesher JL Jr. Squamous cell carcinoma in situ arising in the setting of erythema ab igne. J Drugs Dermatol. 2008;7:488-489.
- Neve EF. Kangri-burn cancer. Br Med J. 1923;2:1255-1256.
- Laycock HT. The kang cancer of North-West China. Br Med J. 1948;1:982.
- Wharton J, Roffwarg D, Miller J, et al. Cutaneous marginal zone lymphoma arising in the setting of erythema ab igne. J Am Acad Dermatol. 2010;62:1080-1081.
- Jones CS, Tyring SK, Lee PC, et al. Development of neuroendocrine (Merkel cell) carcinoma mixed with squamous cell carcinoma in erythema ab igne. Arch Dermatol. 1988;124:110-113.
- Pritchett EN, Doyle A, Shaver CM, et al. Nonmelanoma skin cancer in nonwhite organ transplant recipients. JAMA Dermatol. 2016;152:1348-1353.
- McCall CO, Chen SC. Squamous cell carcinoma of the legs in African Americans. J Am Acad Dermatol. 2002;47:524-529.
- Freeman RG, Knox JM. Influence of temperature on ultraviolet injury. Arch Dermatol. 1964;89:858-864.
- Schieke SM, Schroeder P, Krutmann J. Cutaneous effects of infrared radiation: from clinical observations to molecular response mechanisms. Photodermatol Photoimmunol Photomed. 2003;19:228-234.
Case Report
A 67-year-old Black woman presented with a long-standing history of pruritus and “scaly thick bumps” on the lower extremities. Upon further questioning, she reported a 30-year history of placing her feet by an electric space heater and daily baths in “very hot” water. A review of systems and medical history were unremarkable, and the patient was not on any medications. Initial physical examination of the lower extremities demonstrated lichenified plaques and scattered, firm, ulcerated nodules surrounded by mottled postinflammatory hyperpigmentation with sharp demarcation at the midcalf bilaterally (Figure 1).
Subsequently, the patient was shown to have multiple actinic keratoses and SCCs, both in situ and invasive, within the areas of EAI (Figure 2). The patient had no actinic keratoses or other cutaneous malignant neoplasms elsewhere on the skin. Management of actinic keratoses, SCC in situ, and invasive SCC on the lower extremities included numerous excisions, treatment with liquid nitrogen, and topical 5-fluorouracil under occlusion. The patient continues to be monitored frequently.
Comment
Presentation of EAI
Erythema ab igne is a cutaneous reaction resulting from prolonged exposure to an infrared heat source at temperatures insufficient to cause a burn (37 °F to 11
Histopathology of EAI
Histologically, later stages of EAI can demonstrate focal hyperkeratosis with dyskeratosis and increased dermal elastosis, similar to actinic damage, with a predisposition to develop SCC.2 Notably, early reports document various heat-induced carcinomas, including kangri-burn cancers among Kashmiris, kang thermal cancers in China, and kairo cancers in Japan.2,4,5 More recent reports identify cutaneous carcinomas arising specifically in the setting of EAI, most commonly SCC3; Merkel cell carcinoma and cutaneous marginal zone lymphoma are less commonly reported malignancies.6,7 Given the frequency of malignant transformation within sites of thermal exposure, chronic heat exposure may share a common pathophysiology with SCC and other neoplasms, including Merkel cell carcinoma and cutaneous marginal zone lymphoma.
SCC in Black Individuals
Squamous cell carcinoma is the most common skin cancer in Black individuals, with a notably higher incidence in high-risk subpopulations (immunosuppressed patients). Unlike White individuals, SCCs frequently occur in non–sun-exposed areas in Black individuals and are associated with unique risk factors, such as human papillomavirus, as demonstrated in Black transplant patients.8 A retrospective study examining the characteristics of SCC on the legs of Black individuals documented atypical hyperkeratotic neoplasms surrounded by abnormal pigmentation and mottling of surrounding skin.9 Morphologic skin changes could be the result of chronic thermal damage: Numerous patients reported a history of leg warming from an open heat source. Other patients had an actual diagnosis of EAI. The predilection for less-exposed skin suggests UV radiation (UVR) might be a less important predisposing risk factor for this racial group, and the increased mortality associated with SCC in Black individuals might represent a more aggressive nature to this subset of SCCs.9 Furthermore, infrared radiation (IRR), such as fires and coal stoves, might have the potential to stimulate skin changes similar to those associated with UVR and ultimately malignant changes.
Infrared Radiation
Compared to UVR, little is known about the biological effects of IRR (wavelength, 760 nm to 1 mm), to which human skin is constantly exposed from natural and artificial light sources. Early studies have demonstrated the carcinogenic potential of IRR, observing an augmentation of UVR-induced tumorigenesis in the presence of heat. More recently, IRR was observed to stimulate increased collagenase production from dermal fibroblasts and influence pathways (extracellular signal-related kinases 1/2 and p38 mitogen-activated protein kinases) in a similar fashion to UVB and UVA.10,11 Therefore, IRR might be capable of eliciting molecular responses comparable to those caused by UVR.
Conclusion
Although SCC in association with EAI is uncommon, historical reports of thermal cancers and scientific observations of IRR-induced biological and molecular effects support EAI as a predisposing risk factor for SCC and the important need for close monitoring by physicians. Studies are needed to further elucidate the pathologic effects of IRR, with more promotion of caution relating to thermal exposure.
Case Report
A 67-year-old Black woman presented with a long-standing history of pruritus and “scaly thick bumps” on the lower extremities. Upon further questioning, she reported a 30-year history of placing her feet by an electric space heater and daily baths in “very hot” water. A review of systems and medical history were unremarkable, and the patient was not on any medications. Initial physical examination of the lower extremities demonstrated lichenified plaques and scattered, firm, ulcerated nodules surrounded by mottled postinflammatory hyperpigmentation with sharp demarcation at the midcalf bilaterally (Figure 1).
Subsequently, the patient was shown to have multiple actinic keratoses and SCCs, both in situ and invasive, within the areas of EAI (Figure 2). The patient had no actinic keratoses or other cutaneous malignant neoplasms elsewhere on the skin. Management of actinic keratoses, SCC in situ, and invasive SCC on the lower extremities included numerous excisions, treatment with liquid nitrogen, and topical 5-fluorouracil under occlusion. The patient continues to be monitored frequently.
Comment
Presentation of EAI
Erythema ab igne is a cutaneous reaction resulting from prolonged exposure to an infrared heat source at temperatures insufficient to cause a burn (37 °F to 11
Histopathology of EAI
Histologically, later stages of EAI can demonstrate focal hyperkeratosis with dyskeratosis and increased dermal elastosis, similar to actinic damage, with a predisposition to develop SCC.2 Notably, early reports document various heat-induced carcinomas, including kangri-burn cancers among Kashmiris, kang thermal cancers in China, and kairo cancers in Japan.2,4,5 More recent reports identify cutaneous carcinomas arising specifically in the setting of EAI, most commonly SCC3; Merkel cell carcinoma and cutaneous marginal zone lymphoma are less commonly reported malignancies.6,7 Given the frequency of malignant transformation within sites of thermal exposure, chronic heat exposure may share a common pathophysiology with SCC and other neoplasms, including Merkel cell carcinoma and cutaneous marginal zone lymphoma.
SCC in Black Individuals
Squamous cell carcinoma is the most common skin cancer in Black individuals, with a notably higher incidence in high-risk subpopulations (immunosuppressed patients). Unlike White individuals, SCCs frequently occur in non–sun-exposed areas in Black individuals and are associated with unique risk factors, such as human papillomavirus, as demonstrated in Black transplant patients.8 A retrospective study examining the characteristics of SCC on the legs of Black individuals documented atypical hyperkeratotic neoplasms surrounded by abnormal pigmentation and mottling of surrounding skin.9 Morphologic skin changes could be the result of chronic thermal damage: Numerous patients reported a history of leg warming from an open heat source. Other patients had an actual diagnosis of EAI. The predilection for less-exposed skin suggests UV radiation (UVR) might be a less important predisposing risk factor for this racial group, and the increased mortality associated with SCC in Black individuals might represent a more aggressive nature to this subset of SCCs.9 Furthermore, infrared radiation (IRR), such as fires and coal stoves, might have the potential to stimulate skin changes similar to those associated with UVR and ultimately malignant changes.
Infrared Radiation
Compared to UVR, little is known about the biological effects of IRR (wavelength, 760 nm to 1 mm), to which human skin is constantly exposed from natural and artificial light sources. Early studies have demonstrated the carcinogenic potential of IRR, observing an augmentation of UVR-induced tumorigenesis in the presence of heat. More recently, IRR was observed to stimulate increased collagenase production from dermal fibroblasts and influence pathways (extracellular signal-related kinases 1/2 and p38 mitogen-activated protein kinases) in a similar fashion to UVB and UVA.10,11 Therefore, IRR might be capable of eliciting molecular responses comparable to those caused by UVR.
Conclusion
Although SCC in association with EAI is uncommon, historical reports of thermal cancers and scientific observations of IRR-induced biological and molecular effects support EAI as a predisposing risk factor for SCC and the important need for close monitoring by physicians. Studies are needed to further elucidate the pathologic effects of IRR, with more promotion of caution relating to thermal exposure.
- Milchak M, Smucker J, Chung CG, et al. Erythema ab igne due to heating pad use: a case report and review of clinical presentation, prevention, and complications. Case Rep Med. 2016;2016:1862480.
- Miller K, Hunt R, Chu J, et al. Erythema ab igne. Dermatol Online J. 2011;17:28. Accessed December 10, 2020. https://escholarship.org/uc/item/47z4v01z
- Wharton JB, Sheehan DJ, Lesher JL Jr. Squamous cell carcinoma in situ arising in the setting of erythema ab igne. J Drugs Dermatol. 2008;7:488-489.
- Neve EF. Kangri-burn cancer. Br Med J. 1923;2:1255-1256.
- Laycock HT. The kang cancer of North-West China. Br Med J. 1948;1:982.
- Wharton J, Roffwarg D, Miller J, et al. Cutaneous marginal zone lymphoma arising in the setting of erythema ab igne. J Am Acad Dermatol. 2010;62:1080-1081.
- Jones CS, Tyring SK, Lee PC, et al. Development of neuroendocrine (Merkel cell) carcinoma mixed with squamous cell carcinoma in erythema ab igne. Arch Dermatol. 1988;124:110-113.
- Pritchett EN, Doyle A, Shaver CM, et al. Nonmelanoma skin cancer in nonwhite organ transplant recipients. JAMA Dermatol. 2016;152:1348-1353.
- McCall CO, Chen SC. Squamous cell carcinoma of the legs in African Americans. J Am Acad Dermatol. 2002;47:524-529.
- Freeman RG, Knox JM. Influence of temperature on ultraviolet injury. Arch Dermatol. 1964;89:858-864.
- Schieke SM, Schroeder P, Krutmann J. Cutaneous effects of infrared radiation: from clinical observations to molecular response mechanisms. Photodermatol Photoimmunol Photomed. 2003;19:228-234.
- Milchak M, Smucker J, Chung CG, et al. Erythema ab igne due to heating pad use: a case report and review of clinical presentation, prevention, and complications. Case Rep Med. 2016;2016:1862480.
- Miller K, Hunt R, Chu J, et al. Erythema ab igne. Dermatol Online J. 2011;17:28. Accessed December 10, 2020. https://escholarship.org/uc/item/47z4v01z
- Wharton JB, Sheehan DJ, Lesher JL Jr. Squamous cell carcinoma in situ arising in the setting of erythema ab igne. J Drugs Dermatol. 2008;7:488-489.
- Neve EF. Kangri-burn cancer. Br Med J. 1923;2:1255-1256.
- Laycock HT. The kang cancer of North-West China. Br Med J. 1948;1:982.
- Wharton J, Roffwarg D, Miller J, et al. Cutaneous marginal zone lymphoma arising in the setting of erythema ab igne. J Am Acad Dermatol. 2010;62:1080-1081.
- Jones CS, Tyring SK, Lee PC, et al. Development of neuroendocrine (Merkel cell) carcinoma mixed with squamous cell carcinoma in erythema ab igne. Arch Dermatol. 1988;124:110-113.
- Pritchett EN, Doyle A, Shaver CM, et al. Nonmelanoma skin cancer in nonwhite organ transplant recipients. JAMA Dermatol. 2016;152:1348-1353.
- McCall CO, Chen SC. Squamous cell carcinoma of the legs in African Americans. J Am Acad Dermatol. 2002;47:524-529.
- Freeman RG, Knox JM. Influence of temperature on ultraviolet injury. Arch Dermatol. 1964;89:858-864.
- Schieke SM, Schroeder P, Krutmann J. Cutaneous effects of infrared radiation: from clinical observations to molecular response mechanisms. Photodermatol Photoimmunol Photomed. 2003;19:228-234.
Practice Points
- Erythema ab igne (EAI) is a cutaneous reaction in response to prolonged exposure to infrared heat sources at temperatures insufficient to induce a burn.
- Common infrared heat sources include open fires, coal stoves, heating pads, laptop computers, and electric space heaters.
- Although considered a chronic pigmentary disorder, EAI rarely can progress to malignant transformation, including squamous cell carcinoma. Patients with EAI should be monitored long-term for malignant transformation.
17-year-old girl • abdominal pain • lower-leg itching • dark urine and yellow eyes • Dx?
THE CASE
A 17-year-old White girl with no known past medical history presented to the emergency department (ED) with complaints of abdominal pain and pruritus. The abdominal pain had started 9 days prior and lasted for 3 days. One day after resolution, she developed bilateral lower extremity itching, which was not relieved with loratadine.
Review of systems included dark urine and yellow eyes noted for several days. The patient denied nausea, vomiting, diarrhea, constipation, fevers, chills, arthralgias, recent illness, travel, or sick contacts. Immunizations were up to date. The patient had no history of surgery or liver disease and no pertinent family history. Her current medications included ethinyl estradiol/norethindrone acetate for birth control and minocycline for acne vulgaris. She had been taking the latter medication for 2 years. No additional medications were noted, including vitamins, over-the-counter medications, or supplements. She denied smoking and alcohol or recreational drug use.
In the ED, the patient had normal vital signs. Physical exam findings included bilateral scleral icterus and scattered skin excoriations on the hands, arms, back of the neck, and feet. At the time of hospital admission, the patient’s minocycline and birth control were held under the initial presumption that one or both might be contributing to her presentation.
Pertinent laboratory findings included aspartate transaminase (AST), 828 U/L (normal range, 2-40 U/L); alanine aminotransferase (ALT), 784 U/L (normal range, 3-30 U/L); lactic acid dehydrogenase, 520 U/L (normal range, 140-280 U/L); alkaline phosphatase, 119 U/L (normal range, 44-147 U/L); total bilirubin, 1.9 µmol/L (normal range, 2-18 µmol/L); and direct bilirubin, 1.3 µmol/L (normal range, 0-4 µmol/L). Baseline liver function test results (prior to admission) were unknown. Results of a coagulation panel, complete blood count, basic metabolic panel, amylase, lipase, urine toxicology, and urinalysis all were within normal limits.
Ultrasound of the abdomen revealed a normal abdomen, liver, pancreas, gallbladder, and common bile duct. This imaging study was negative for other obstructive pathologies.
THE DIAGNOSIS
During hospital admission, a noninvasive liver work-up was pursued by Gastroenterology. A hepatitis panel, Epstein-Barr virus testing, and levels of ceruloplasmin and acetaminophen were all found to be within normal limits, excluding additional causes of liver disease. Serum antinuclear antibody (ANA) testing was significantly positive, with a titer of 1:640 (range, < 1:20) and, as noted above, liver transaminases were severely elevated, leading to a presumptive diagnosis of drug-induced liver pathology.
Continue to: During outpatient follow-up...
During outpatient follow-up with Gastroenterology 2 days after discharge, the patient’s liver transaminases and bilirubin continued to trend upward (to a maximum ALT of 871 U/L; AST, 1097 U/L; alkaline phosphatase, 122 U/L; and bilirubin, 2.9 µmol/L). Immunoglobulin G was 1342 mg/mL (normal range, 694-1618 mg/mL).
An ultrasound-guided liver biopsy was performed; it demonstrated lobular, portal, and periportal hepatitis with focal bridging necrosis, consistent with a diagnosis of autoimmune hepatitis. Mild-to-moderate focal cholestasis was demonstrated, consistent with cholestatic hepatitis.
DISCUSSION
Autoimmune hepatitis is characterized by inflammation of the liver, secondary to the presence of circulating antibodies or hypergammaglobulinemia. The pathogenesis is thought to involve a T-cell–mediated immune attack on the liver. Based on case reports,the use of minocycline is associated with risk for liver injury, although the incidence is rare.1-4 Use of this medication may be associated with autoimmune disease in patients who are predisposed to autoimmune tendencies or who have genetic predeterminants.
Diagnosis is typically made based on abnormalities in aminotransferases (AST, ALT), elevation in serum immunoglobulins, and positive auto-antibody titers including ANA, smooth muscle antibodies, and anti-liver kidney microsomal type 1 antibodies. Although clinical presentations tend to differ, the confirmatory diagnosis is typically made histologically, with the presence of lobular and perivenular necro-inflammatory changes and plasma cell infiltration.5
Other infectious and metabolic causes of hepatitis should be excluded. Many medications and herbal agents have been noted to cause autoimmune hepatitis or similar syndromes that mimic the condition.
Medication history. Review of the case patient’s medication list identified ethinyl estradiol/norethindrone acetate and minocycline as potential culprits. Ethinyl estradiol/norethindrone acetate is a low-dose combination oral contraceptive pill (OCP). Although earlier formulations of OCPs were associated with hepatobiliary complications, these adverse effects are noted to be rare in the absence of predisposing conditions.6 In some cases, OCPs have been linked to cholestasis, chronic hepatocellular carcinoma, or hepatic adenomas, but studies have shown that these medications do not affect the course of acute liver failure.7
Continue to: Minocycline...
Minocycline is a second-generation tetracycline commonly used to treat acne vulgaris. Long-term treatment with minocycline has been associated with severe adverse effects, including autoimmune and hypersensitivity reactions.8 Minocycline-associated hepatotoxicity can be due to a systemic hypersensitivity reaction, occurring within a few weeks of therapy initiation, whereas autoimmune hepatitis manifests after a year or more of exposure to the medication (as in this case). Patients may present acutely several months after starting the medication, with symptoms of jaundice, fatigue, and/or joint aches. The acute liver injury is typically self-limited and often resolves with cessation of the drug. However, patients may require corticosteroids and immunosuppressive therapy.
Which is it? Histologically, drug-induced autoimmune hepatitis is indistinguishable from idiopathic autoimmune hepatitis.3 The estimated incidence of idiopathic autoimmune liver disease ranges from 0.7 to 2 out of 100,000 population.9 A systematic review of the literature identified 65 reported cases of liver damage associated with minocycline specifically.1
In this case, given the patient’s 2-year history of minocycline use, it is possible that she developed an acute presentation of autoimmune hepatitis. With drug-induced autoimmune liver injury, complete resolution occurs after withdrawal of the offending medication, and a response to corticosteroid therapy supports the diagnosis. Recurrence of signs or symptoms following corticosteroid cessation may indicate idiopathic autoimmune hepatitis as opposed to a drug-induced form.2
Our patient was started on steroid and immunomodulator therapy, with prednisone 40 mg/d and mycophenolate 250 mg bid. At follow-up with Gastroenterology, the patient’s symptoms and liver function test results had improved significantly (AST, 27 U/L; ALT, 14 U/L; alkaline phosphatase, 51 U/L; and total bilirubin, 0.4 µmol/L). The patient was continued on a prednisone taper while simultaneously titrating mycophenolate. The ultimate plan of care included continuing mycophenolate for a total of 4 to 5 years.
THE TAKEAWAY
During evaluation of a patient with new-onset liver disease, it is important to inquire about prescription medications, drugs, vitamins, and herbal supplements as possible contributors to the disease process. This case highlights the importance of monitoring patients while on minocycline and of weighing the risks vs benefits of long-term therapy. It has been suggested that liver enzymes be tested before therapy initiation and about every 3 months during long-term antibiotic treatment.4 Careful consideration and caution should be taken prior to the initiation of medications that have been linked to rare, but important, adverse reactions.
ACKNOWLEDGEMENT
The authors would like to thank Frank Bauer, MD, and Eva Sotil, MD, for their contributions to this case presentation.
CORRESPONDENCE
Andrea Gillis, DO, Asylum Hill Family Medicine Center, 99 Woodland Street, Hartford, CT 06105; andrea.gillis@ trinityhealthofne.org
1. Lawrenson RA, Seaman HE, Sundström A, et al. Liver damage associated with minocycline use in acne: a systematic review of the published literature and pharmacovigilance data. Drug Saf. 2000;23:333-349.
2. Teitelbaum JE, Perez-Atayde AR, Cohen M, et al. Minocycline-related autoimmune hepatitis case series and literature review. Arch Pediatr Adolesc Med. 1998;152:1132-1136.
3. Goldstein NS, Bayati N, Silverman AL, et al. Minocycline as a cause of drug induced autoimmune hepatitis: report of four cases and comparison with autoimmune hepatitis. Am J Clinic Pathol. 2000;114:591-598.
4. Ramakrishna J, Johnson AR, Banner BF. Long-term minocycline use for acne in healthy adolescents can cause severe autoimmune hepatitis. J Clin Gastroenterol. 2009;43:787-790.
5. Nguyen Canh H, Harada K, Ouchi H, et al. Acute presentation of autoimmune hepatitis: a multicentre study with detailed histological evaluation in a large cohort of patients. J Clin Pathol. 2017;70:961-969.
6. Lindberg MC. Hepatobiliary complications of oral contraceptives. J Gen Intern Med. 1992; 7:199-209.
7. Kapp N, Tilley IB, Curtis KM. The effects of hormonal contraceptive use among women with viral hepatitis or cirrhosis of the liver: a systematic review. Contraception. 2009;80:381-386.
8. DeLemos AS, Foureau DM, Jacobs C, et al. Drug-induced liver injury with autoimmune features. Semin Liver Dis. 2014;34:194-204.
9. Jepsen P, Gronbaek L, Vilstrup H. Worldwide incidence of autoimmune liver disease. Dig Dis. 2015;33(suppl 2):2-12.
THE CASE
A 17-year-old White girl with no known past medical history presented to the emergency department (ED) with complaints of abdominal pain and pruritus. The abdominal pain had started 9 days prior and lasted for 3 days. One day after resolution, she developed bilateral lower extremity itching, which was not relieved with loratadine.
Review of systems included dark urine and yellow eyes noted for several days. The patient denied nausea, vomiting, diarrhea, constipation, fevers, chills, arthralgias, recent illness, travel, or sick contacts. Immunizations were up to date. The patient had no history of surgery or liver disease and no pertinent family history. Her current medications included ethinyl estradiol/norethindrone acetate for birth control and minocycline for acne vulgaris. She had been taking the latter medication for 2 years. No additional medications were noted, including vitamins, over-the-counter medications, or supplements. She denied smoking and alcohol or recreational drug use.
In the ED, the patient had normal vital signs. Physical exam findings included bilateral scleral icterus and scattered skin excoriations on the hands, arms, back of the neck, and feet. At the time of hospital admission, the patient’s minocycline and birth control were held under the initial presumption that one or both might be contributing to her presentation.
Pertinent laboratory findings included aspartate transaminase (AST), 828 U/L (normal range, 2-40 U/L); alanine aminotransferase (ALT), 784 U/L (normal range, 3-30 U/L); lactic acid dehydrogenase, 520 U/L (normal range, 140-280 U/L); alkaline phosphatase, 119 U/L (normal range, 44-147 U/L); total bilirubin, 1.9 µmol/L (normal range, 2-18 µmol/L); and direct bilirubin, 1.3 µmol/L (normal range, 0-4 µmol/L). Baseline liver function test results (prior to admission) were unknown. Results of a coagulation panel, complete blood count, basic metabolic panel, amylase, lipase, urine toxicology, and urinalysis all were within normal limits.
Ultrasound of the abdomen revealed a normal abdomen, liver, pancreas, gallbladder, and common bile duct. This imaging study was negative for other obstructive pathologies.
THE DIAGNOSIS
During hospital admission, a noninvasive liver work-up was pursued by Gastroenterology. A hepatitis panel, Epstein-Barr virus testing, and levels of ceruloplasmin and acetaminophen were all found to be within normal limits, excluding additional causes of liver disease. Serum antinuclear antibody (ANA) testing was significantly positive, with a titer of 1:640 (range, < 1:20) and, as noted above, liver transaminases were severely elevated, leading to a presumptive diagnosis of drug-induced liver pathology.
Continue to: During outpatient follow-up...
During outpatient follow-up with Gastroenterology 2 days after discharge, the patient’s liver transaminases and bilirubin continued to trend upward (to a maximum ALT of 871 U/L; AST, 1097 U/L; alkaline phosphatase, 122 U/L; and bilirubin, 2.9 µmol/L). Immunoglobulin G was 1342 mg/mL (normal range, 694-1618 mg/mL).
An ultrasound-guided liver biopsy was performed; it demonstrated lobular, portal, and periportal hepatitis with focal bridging necrosis, consistent with a diagnosis of autoimmune hepatitis. Mild-to-moderate focal cholestasis was demonstrated, consistent with cholestatic hepatitis.
DISCUSSION
Autoimmune hepatitis is characterized by inflammation of the liver, secondary to the presence of circulating antibodies or hypergammaglobulinemia. The pathogenesis is thought to involve a T-cell–mediated immune attack on the liver. Based on case reports,the use of minocycline is associated with risk for liver injury, although the incidence is rare.1-4 Use of this medication may be associated with autoimmune disease in patients who are predisposed to autoimmune tendencies or who have genetic predeterminants.
Diagnosis is typically made based on abnormalities in aminotransferases (AST, ALT), elevation in serum immunoglobulins, and positive auto-antibody titers including ANA, smooth muscle antibodies, and anti-liver kidney microsomal type 1 antibodies. Although clinical presentations tend to differ, the confirmatory diagnosis is typically made histologically, with the presence of lobular and perivenular necro-inflammatory changes and plasma cell infiltration.5
Other infectious and metabolic causes of hepatitis should be excluded. Many medications and herbal agents have been noted to cause autoimmune hepatitis or similar syndromes that mimic the condition.
Medication history. Review of the case patient’s medication list identified ethinyl estradiol/norethindrone acetate and minocycline as potential culprits. Ethinyl estradiol/norethindrone acetate is a low-dose combination oral contraceptive pill (OCP). Although earlier formulations of OCPs were associated with hepatobiliary complications, these adverse effects are noted to be rare in the absence of predisposing conditions.6 In some cases, OCPs have been linked to cholestasis, chronic hepatocellular carcinoma, or hepatic adenomas, but studies have shown that these medications do not affect the course of acute liver failure.7
Continue to: Minocycline...
Minocycline is a second-generation tetracycline commonly used to treat acne vulgaris. Long-term treatment with minocycline has been associated with severe adverse effects, including autoimmune and hypersensitivity reactions.8 Minocycline-associated hepatotoxicity can be due to a systemic hypersensitivity reaction, occurring within a few weeks of therapy initiation, whereas autoimmune hepatitis manifests after a year or more of exposure to the medication (as in this case). Patients may present acutely several months after starting the medication, with symptoms of jaundice, fatigue, and/or joint aches. The acute liver injury is typically self-limited and often resolves with cessation of the drug. However, patients may require corticosteroids and immunosuppressive therapy.
Which is it? Histologically, drug-induced autoimmune hepatitis is indistinguishable from idiopathic autoimmune hepatitis.3 The estimated incidence of idiopathic autoimmune liver disease ranges from 0.7 to 2 out of 100,000 population.9 A systematic review of the literature identified 65 reported cases of liver damage associated with minocycline specifically.1
In this case, given the patient’s 2-year history of minocycline use, it is possible that she developed an acute presentation of autoimmune hepatitis. With drug-induced autoimmune liver injury, complete resolution occurs after withdrawal of the offending medication, and a response to corticosteroid therapy supports the diagnosis. Recurrence of signs or symptoms following corticosteroid cessation may indicate idiopathic autoimmune hepatitis as opposed to a drug-induced form.2
Our patient was started on steroid and immunomodulator therapy, with prednisone 40 mg/d and mycophenolate 250 mg bid. At follow-up with Gastroenterology, the patient’s symptoms and liver function test results had improved significantly (AST, 27 U/L; ALT, 14 U/L; alkaline phosphatase, 51 U/L; and total bilirubin, 0.4 µmol/L). The patient was continued on a prednisone taper while simultaneously titrating mycophenolate. The ultimate plan of care included continuing mycophenolate for a total of 4 to 5 years.
THE TAKEAWAY
During evaluation of a patient with new-onset liver disease, it is important to inquire about prescription medications, drugs, vitamins, and herbal supplements as possible contributors to the disease process. This case highlights the importance of monitoring patients while on minocycline and of weighing the risks vs benefits of long-term therapy. It has been suggested that liver enzymes be tested before therapy initiation and about every 3 months during long-term antibiotic treatment.4 Careful consideration and caution should be taken prior to the initiation of medications that have been linked to rare, but important, adverse reactions.
ACKNOWLEDGEMENT
The authors would like to thank Frank Bauer, MD, and Eva Sotil, MD, for their contributions to this case presentation.
CORRESPONDENCE
Andrea Gillis, DO, Asylum Hill Family Medicine Center, 99 Woodland Street, Hartford, CT 06105; andrea.gillis@ trinityhealthofne.org
THE CASE
A 17-year-old White girl with no known past medical history presented to the emergency department (ED) with complaints of abdominal pain and pruritus. The abdominal pain had started 9 days prior and lasted for 3 days. One day after resolution, she developed bilateral lower extremity itching, which was not relieved with loratadine.
Review of systems included dark urine and yellow eyes noted for several days. The patient denied nausea, vomiting, diarrhea, constipation, fevers, chills, arthralgias, recent illness, travel, or sick contacts. Immunizations were up to date. The patient had no history of surgery or liver disease and no pertinent family history. Her current medications included ethinyl estradiol/norethindrone acetate for birth control and minocycline for acne vulgaris. She had been taking the latter medication for 2 years. No additional medications were noted, including vitamins, over-the-counter medications, or supplements. She denied smoking and alcohol or recreational drug use.
In the ED, the patient had normal vital signs. Physical exam findings included bilateral scleral icterus and scattered skin excoriations on the hands, arms, back of the neck, and feet. At the time of hospital admission, the patient’s minocycline and birth control were held under the initial presumption that one or both might be contributing to her presentation.
Pertinent laboratory findings included aspartate transaminase (AST), 828 U/L (normal range, 2-40 U/L); alanine aminotransferase (ALT), 784 U/L (normal range, 3-30 U/L); lactic acid dehydrogenase, 520 U/L (normal range, 140-280 U/L); alkaline phosphatase, 119 U/L (normal range, 44-147 U/L); total bilirubin, 1.9 µmol/L (normal range, 2-18 µmol/L); and direct bilirubin, 1.3 µmol/L (normal range, 0-4 µmol/L). Baseline liver function test results (prior to admission) were unknown. Results of a coagulation panel, complete blood count, basic metabolic panel, amylase, lipase, urine toxicology, and urinalysis all were within normal limits.
Ultrasound of the abdomen revealed a normal abdomen, liver, pancreas, gallbladder, and common bile duct. This imaging study was negative for other obstructive pathologies.
THE DIAGNOSIS
During hospital admission, a noninvasive liver work-up was pursued by Gastroenterology. A hepatitis panel, Epstein-Barr virus testing, and levels of ceruloplasmin and acetaminophen were all found to be within normal limits, excluding additional causes of liver disease. Serum antinuclear antibody (ANA) testing was significantly positive, with a titer of 1:640 (range, < 1:20) and, as noted above, liver transaminases were severely elevated, leading to a presumptive diagnosis of drug-induced liver pathology.
Continue to: During outpatient follow-up...
During outpatient follow-up with Gastroenterology 2 days after discharge, the patient’s liver transaminases and bilirubin continued to trend upward (to a maximum ALT of 871 U/L; AST, 1097 U/L; alkaline phosphatase, 122 U/L; and bilirubin, 2.9 µmol/L). Immunoglobulin G was 1342 mg/mL (normal range, 694-1618 mg/mL).
An ultrasound-guided liver biopsy was performed; it demonstrated lobular, portal, and periportal hepatitis with focal bridging necrosis, consistent with a diagnosis of autoimmune hepatitis. Mild-to-moderate focal cholestasis was demonstrated, consistent with cholestatic hepatitis.
DISCUSSION
Autoimmune hepatitis is characterized by inflammation of the liver, secondary to the presence of circulating antibodies or hypergammaglobulinemia. The pathogenesis is thought to involve a T-cell–mediated immune attack on the liver. Based on case reports,the use of minocycline is associated with risk for liver injury, although the incidence is rare.1-4 Use of this medication may be associated with autoimmune disease in patients who are predisposed to autoimmune tendencies or who have genetic predeterminants.
Diagnosis is typically made based on abnormalities in aminotransferases (AST, ALT), elevation in serum immunoglobulins, and positive auto-antibody titers including ANA, smooth muscle antibodies, and anti-liver kidney microsomal type 1 antibodies. Although clinical presentations tend to differ, the confirmatory diagnosis is typically made histologically, with the presence of lobular and perivenular necro-inflammatory changes and plasma cell infiltration.5
Other infectious and metabolic causes of hepatitis should be excluded. Many medications and herbal agents have been noted to cause autoimmune hepatitis or similar syndromes that mimic the condition.
Medication history. Review of the case patient’s medication list identified ethinyl estradiol/norethindrone acetate and minocycline as potential culprits. Ethinyl estradiol/norethindrone acetate is a low-dose combination oral contraceptive pill (OCP). Although earlier formulations of OCPs were associated with hepatobiliary complications, these adverse effects are noted to be rare in the absence of predisposing conditions.6 In some cases, OCPs have been linked to cholestasis, chronic hepatocellular carcinoma, or hepatic adenomas, but studies have shown that these medications do not affect the course of acute liver failure.7
Continue to: Minocycline...
Minocycline is a second-generation tetracycline commonly used to treat acne vulgaris. Long-term treatment with minocycline has been associated with severe adverse effects, including autoimmune and hypersensitivity reactions.8 Minocycline-associated hepatotoxicity can be due to a systemic hypersensitivity reaction, occurring within a few weeks of therapy initiation, whereas autoimmune hepatitis manifests after a year or more of exposure to the medication (as in this case). Patients may present acutely several months after starting the medication, with symptoms of jaundice, fatigue, and/or joint aches. The acute liver injury is typically self-limited and often resolves with cessation of the drug. However, patients may require corticosteroids and immunosuppressive therapy.
Which is it? Histologically, drug-induced autoimmune hepatitis is indistinguishable from idiopathic autoimmune hepatitis.3 The estimated incidence of idiopathic autoimmune liver disease ranges from 0.7 to 2 out of 100,000 population.9 A systematic review of the literature identified 65 reported cases of liver damage associated with minocycline specifically.1
In this case, given the patient’s 2-year history of minocycline use, it is possible that she developed an acute presentation of autoimmune hepatitis. With drug-induced autoimmune liver injury, complete resolution occurs after withdrawal of the offending medication, and a response to corticosteroid therapy supports the diagnosis. Recurrence of signs or symptoms following corticosteroid cessation may indicate idiopathic autoimmune hepatitis as opposed to a drug-induced form.2
Our patient was started on steroid and immunomodulator therapy, with prednisone 40 mg/d and mycophenolate 250 mg bid. At follow-up with Gastroenterology, the patient’s symptoms and liver function test results had improved significantly (AST, 27 U/L; ALT, 14 U/L; alkaline phosphatase, 51 U/L; and total bilirubin, 0.4 µmol/L). The patient was continued on a prednisone taper while simultaneously titrating mycophenolate. The ultimate plan of care included continuing mycophenolate for a total of 4 to 5 years.
THE TAKEAWAY
During evaluation of a patient with new-onset liver disease, it is important to inquire about prescription medications, drugs, vitamins, and herbal supplements as possible contributors to the disease process. This case highlights the importance of monitoring patients while on minocycline and of weighing the risks vs benefits of long-term therapy. It has been suggested that liver enzymes be tested before therapy initiation and about every 3 months during long-term antibiotic treatment.4 Careful consideration and caution should be taken prior to the initiation of medications that have been linked to rare, but important, adverse reactions.
ACKNOWLEDGEMENT
The authors would like to thank Frank Bauer, MD, and Eva Sotil, MD, for their contributions to this case presentation.
CORRESPONDENCE
Andrea Gillis, DO, Asylum Hill Family Medicine Center, 99 Woodland Street, Hartford, CT 06105; andrea.gillis@ trinityhealthofne.org
1. Lawrenson RA, Seaman HE, Sundström A, et al. Liver damage associated with minocycline use in acne: a systematic review of the published literature and pharmacovigilance data. Drug Saf. 2000;23:333-349.
2. Teitelbaum JE, Perez-Atayde AR, Cohen M, et al. Minocycline-related autoimmune hepatitis case series and literature review. Arch Pediatr Adolesc Med. 1998;152:1132-1136.
3. Goldstein NS, Bayati N, Silverman AL, et al. Minocycline as a cause of drug induced autoimmune hepatitis: report of four cases and comparison with autoimmune hepatitis. Am J Clinic Pathol. 2000;114:591-598.
4. Ramakrishna J, Johnson AR, Banner BF. Long-term minocycline use for acne in healthy adolescents can cause severe autoimmune hepatitis. J Clin Gastroenterol. 2009;43:787-790.
5. Nguyen Canh H, Harada K, Ouchi H, et al. Acute presentation of autoimmune hepatitis: a multicentre study with detailed histological evaluation in a large cohort of patients. J Clin Pathol. 2017;70:961-969.
6. Lindberg MC. Hepatobiliary complications of oral contraceptives. J Gen Intern Med. 1992; 7:199-209.
7. Kapp N, Tilley IB, Curtis KM. The effects of hormonal contraceptive use among women with viral hepatitis or cirrhosis of the liver: a systematic review. Contraception. 2009;80:381-386.
8. DeLemos AS, Foureau DM, Jacobs C, et al. Drug-induced liver injury with autoimmune features. Semin Liver Dis. 2014;34:194-204.
9. Jepsen P, Gronbaek L, Vilstrup H. Worldwide incidence of autoimmune liver disease. Dig Dis. 2015;33(suppl 2):2-12.
1. Lawrenson RA, Seaman HE, Sundström A, et al. Liver damage associated with minocycline use in acne: a systematic review of the published literature and pharmacovigilance data. Drug Saf. 2000;23:333-349.
2. Teitelbaum JE, Perez-Atayde AR, Cohen M, et al. Minocycline-related autoimmune hepatitis case series and literature review. Arch Pediatr Adolesc Med. 1998;152:1132-1136.
3. Goldstein NS, Bayati N, Silverman AL, et al. Minocycline as a cause of drug induced autoimmune hepatitis: report of four cases and comparison with autoimmune hepatitis. Am J Clinic Pathol. 2000;114:591-598.
4. Ramakrishna J, Johnson AR, Banner BF. Long-term minocycline use for acne in healthy adolescents can cause severe autoimmune hepatitis. J Clin Gastroenterol. 2009;43:787-790.
5. Nguyen Canh H, Harada K, Ouchi H, et al. Acute presentation of autoimmune hepatitis: a multicentre study with detailed histological evaluation in a large cohort of patients. J Clin Pathol. 2017;70:961-969.
6. Lindberg MC. Hepatobiliary complications of oral contraceptives. J Gen Intern Med. 1992; 7:199-209.
7. Kapp N, Tilley IB, Curtis KM. The effects of hormonal contraceptive use among women with viral hepatitis or cirrhosis of the liver: a systematic review. Contraception. 2009;80:381-386.
8. DeLemos AS, Foureau DM, Jacobs C, et al. Drug-induced liver injury with autoimmune features. Semin Liver Dis. 2014;34:194-204.
9. Jepsen P, Gronbaek L, Vilstrup H. Worldwide incidence of autoimmune liver disease. Dig Dis. 2015;33(suppl 2):2-12.

Herpes Zoster May Be a Marker for COVID-19 Infection During Pregnancy
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the most recently identified member of the zoonotic pathogens of coronaviruses. It caused an outbreak of pneumonia in December 2019 in Wuhan, China.1 Among all related acute respiratory syndromes (SARS-CoV, Middle East respiratory syndrome coronavirus), SARS-CoV-2 remains to be the most infectious, has the highest potential for human transmission, and can eventually result in acute respiratory distress syndrome.2,3
Only 15% of coronavirus disease 2019 (COVID-19) cases progress to pneumonia, and approximately 5% of these cases develop acute respiratory distress syndrome, septic shock, and/or multiple organ failure. The majority of cases only exhibit mild to moderate symptoms.4,5 A wide array of skin manifestations in COVID-19 infection have been reported, including maculopapular eruptions, morbilliform rashes, urticaria, chickenpoxlike lesions, livedo reticularis, COVID toes, erythema multiforme, pityriasis rosea, and several other patterns.6 We report a case of herpes zoster (HZ) complication in a COVID-19–positive woman who was 27 weeks pregnant.
Case Report
A 36-year-old woman who was 27 weeks pregnant was referred by her obstetrician to the dermatology clinic. She presented with a low-grade fever and a vesicular painful rash. Physical examination revealed painful, itchy, dysesthetic papules and vesicles on the left side of the forehead along with mild edema of the left upper eyelid but no watering of the eye or photophobia. She reported episodes of fever (temperature, 38.9°C), fatigue, and myalgia over the last week. She had bouts of dyspnea and tachycardia that she thought were related to being in the late second trimester of pregnancy. The area surrounding the vesicular eruption was tender to touch. No dry cough or any gastrointestinal or urinary tract symptoms were noted. She reported a burning sensation when splashing water on the face or when exposed to air currents. One week following the initial symptoms, she experienced a painful vesicular rash along the upper left forehead (Figure) associated with eyelid edema. Oral and ocular mucosae were free of any presentations. She had no relevant history and had not experienced any complications during pregnancy. A diagnosis of HZ was made, and she was prescribed valacyclovir 1 g 3 times daily for 7 days, acetaminophen for the fever, and calamine lotion. We recommended COVID-19 testing based on her symptoms. A chest radiograph and a positive nasopharyngeal smear were consistent with COVID-19 infection. She reported via telephone follow-up 1 week after presentation that her skin condition had improved following the treatment course and that the vesicles eventually dried, leaving a crusting appearance after 5 to 7 days. Regarding her SARS-CoV-2 condition, her oxygen saturation was 95% at presentation; she self-quarantined at home; and she was treated with oseltamivir 75 mg orally every 12 hours for 5 days, azithromycin 500 mg orally daily, acetaminophen, and vitamin C. Electronic fetal heart rate monitoring and ultrasound examinations were performed to assess the condition of the fetus and were reported normal. At the time of writing this article, she was 32 weeks pregnant and tested negative to 2 consecutive nasopharyngeal swabs for COVID-19 and was in good general condition. She continued her pregnancy according to her obstetrician’s recommendations.
Comment
The incubation time of COVID-19 can be up to 14 days. Fever, dry cough, fatigue, and diarrhea have been speculated to be clinical symptoms; however, many cases may be asymptomatic. Aside from a medical or travel history at risk for COVID-19, diagnosis can be confirmed by detection of viral RNA by reverse transcriptase–polymerase chain reaction for nasopharyngeal swabs or bronchoalveolar fluid. Patients who are immunocompromised, older, or male or who have a history of cardiovascular conditions or debilitating chronic conditions are at an increased risk for severe disease and poor outcome compared to younger healthy individuals.7
The vesicular rash of COVID-19 has been reported to have different forms of presentation. A diffuse widespread pattern resembling hand-foot-and-mouth disease and a localized monomorphic pattern resembling chickenpox but with predilection to the trunk has been described.8
Physiologic changes in the immune and cardiopulmonary systems during pregnancy (eg, diaphragm elevation, increased oxygen consumption, edema of the respiratory tract mucosae) make pregnant women intolerant to hypoxia. The mortality rate of the 1918 influenza pandemic was 2.6% in the overall population but 37% among pregnant women.9 In 2009, pregnant women were reported to be at an increased risk for complications from the H1N1 influenza virus pandemic, with a higher estimated rate of hospital admission than the general population.10 In 2003, approximately 50% of pregnant women who received a diagnosis of SARS-CoV were admitted to the intensive care unit, approximately 33% of pregnant women with SARS-CoV required mechanical ventilation, and the mortality rate was as high as 25% for these women.11 To date, data on the effects of COVID-19 in pregnancy are limited to small case series.12-15
It was confirmed that COVID-19 infection is accompanied by a reduction in lymphocytes, monocytes, and eosinophils, along with a notable reduction of CD4/CD8 T cells, B cells, and natural killer cells. It was further revealed that nonsurvivor COVID-19 patients continued to show a decrease in lymphocyte counts along the course of their disease until death.16-18
Different mechanisms for lymphocyte depletion and deficiency were speculated among COVID-19 patients and include direct lymphocyte death through coronavirus angiotensin-converting enzyme 2–lymphocyte-expressed receptors; direct damage to lymphatic organs, such as the thymus and spleen, but this theory needs to be further investigated; direct lymphocyte apoptosis mediated by tumor necrosis factor α, IL-6, and other proinflammatory cytokines; and direct inhibition of lymphocytes by metabolic upset, such as acidosis.19,20
These causes may precipitate lymphopenia and impaired antiviral responses.21 It also has been postulated that the functional damage of CD4+ T cells may predispose patients with COVID-19 to severe disease.22 Such immune changes can render a patient more susceptible to developing shingles by reactivating varicella-zoster virus, which could be a sign of undiagnosed COVID-19 infection in younger age groups.
Two earlier reports discussed HZ among COVID-19–diagnosed patients. Shors23 presented a case of a patient who developed varicella-zoster virus reactivation of the V2 dermatome during the course of COVID-19 infection. In addition, the patient developed severe acute herpetic neuralgia despite the early initiation of antiviral therapy.23 Elsaie et al24 described 2 cases of patients during the pandemic who first presented with HZ before later being diagnosed with COVID-19 infection.
New information and cutaneous manifestations possibly related to COVID-19 are emerging every day. We report a pregnant female presenting with HZ during the course of COVID-19 infection, which suggests that the clinical presentation of HZ at the time of the current pandemic, especially if associated with other signs of COVID-19 infection, should be carefully monitored and reported for further assessment.
Acknowledgment
The authors would like to thank all the health care workers who have been fighting COVID-19 in Egypt and worldwide.
- Li Q, Guan X, Wu P, et al. Early transmission dynamics in Wuhan, China, of novel coronavirus-infected pneumonia. N Engl J Med. 2020;382:1199-1207.
- Zhang YZ, Holes EC. A genomic perspective on the origin and emergence of sars-cov-2. Cell. 2020;181:223-227.
- Prompetchara E, Ketloy C, Palaga T. Immune responses in COVID-19 and potential vaccines: lessons learned from SARS and MERS epidemic. Asian Pac J Allergy Immunol. 2020;38:1‐9.
- Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan0, China. Lancet. 2020;395:497-506.
- Xu Z, Shi L, Wang Y, et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med. 2020;8:420-422.
- Wollina U, Karadag˘ AS, Rowland-Payne C, et al. Cutaneous signs in COVID-19 patients: a review. Dermatol Ther. 2020;33:e13549.
- Lauer SA, Grantz KH, Bi Q, et al. The incubation period of coronavirus disease 2019 (COVID-19) from publicly reported confirmed cases: estimation and application. Ann Intern Med. 2020;172:577‐582.
- Fernandez-Nieto D, Ortega-Quijano D, Jimenez-Cauhe J, et al. Clinical and histological characterization of vesicular COVID-19 rashes: a prospective study in a tertiary care hospital. Clin Exp Dermatol. 2020;45:872-875.
- Gottfredsson M. The Spanish flu in Iceland 1918. Lessons in medicine and history [in Icelandic]. Laeknabladid. 2008;94:737-745.
- Jamieson D, Honein M, Rasmussen S, et al. H1N1 2009 influenza virus infection during pregnancy in the USA. Lancet. 2009;374:451-458.
- Ksiazek TG, Erdman D, Goldsmith CS. A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med. 2003;348:1953-1966.
- Chen H, Guo J, Wang C, et al. Clinical characteristics and intrauterine vertical transmission potential of COVID-19 infection in nine pregnant women: a retrospective review of medical records. Lancet. 2020;395:809‐815.
- Zhu H, Wang L, Fang C, et al. Clinical analysis of 10 neonates born to mothers with 2019-nCov pneumonia. Transl Pediatr. 2020;9:51-60.
- Liu Y, Chen H, Tang K, et al. Clinical manifestations and outcome of SARS-CoV-2 infection during pregnancy [published online March 4, 2020]. J Infect. doi:10.1016/j.jinf.2020.02.028.
- Zhang L, Jiang Y, Wei M, et al. Analysis of the pregnancy outcomes in pregnant women with COVID-19 in Hubei Province [in Chinese]. Zhonghua Fu Chan Ke Za Zhi. 2020;55:166-171.
- Henry BM, de Oliveira MHS, Benoit S, et al. Hematologic, biochemical and immune biomarker abnormalities associated with severe illness and mortality in coronavirus disease 2019 (COVID-19): a meta-analysis. Clin Chem Lab Med. 2020;58:1021-1028.
- Cai Q, Huang D, Ou P, et al. COVID-19 in a designated infectious diseases hospital outside Hubei Province, China. Allergy. 2020;75:1742-1752.
- Ruan Q, Yang K, Wang W, et al. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. 2020;46:846-884.
- Kumar A, Anil A, Sharma P, et al. Clinical features of COVID-19 and factors associated with severe clinical course: a systematic review and meta-analysis [preprint]. SSRN. doi:10.2139/ssrn.3566166.
- Xu H, Zhong L, Deng J, et al. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int J Oral Sci. 2020;12. https://doi.org/10.1038/s41368-020-0074-x.
- Li H, Liu L, Zhang D, et al. SARS-CoV-2 and viral sepsis: observations and hypotheses. Lancet. 2020;395:1517-1520.
- Zheng M, Gao Y, Wang G, et al. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell Mol Immunol. 2020;17:533-535.
- Shors AR. Herpes zoster and severe acute herpetic neuralgia as a complication of COVID-19 infection. JAAD Case Rep. 2020;6:656-657.
- Elsaie ML, Youssef EA, Nada HA. Herpes zoster might be an indicator for latent COVID 19 infection [published online May 23, 2020]. Dermatol Ther. doi:10.1111/dth.13666.
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the most recently identified member of the zoonotic pathogens of coronaviruses. It caused an outbreak of pneumonia in December 2019 in Wuhan, China.1 Among all related acute respiratory syndromes (SARS-CoV, Middle East respiratory syndrome coronavirus), SARS-CoV-2 remains to be the most infectious, has the highest potential for human transmission, and can eventually result in acute respiratory distress syndrome.2,3
Only 15% of coronavirus disease 2019 (COVID-19) cases progress to pneumonia, and approximately 5% of these cases develop acute respiratory distress syndrome, septic shock, and/or multiple organ failure. The majority of cases only exhibit mild to moderate symptoms.4,5 A wide array of skin manifestations in COVID-19 infection have been reported, including maculopapular eruptions, morbilliform rashes, urticaria, chickenpoxlike lesions, livedo reticularis, COVID toes, erythema multiforme, pityriasis rosea, and several other patterns.6 We report a case of herpes zoster (HZ) complication in a COVID-19–positive woman who was 27 weeks pregnant.
Case Report
A 36-year-old woman who was 27 weeks pregnant was referred by her obstetrician to the dermatology clinic. She presented with a low-grade fever and a vesicular painful rash. Physical examination revealed painful, itchy, dysesthetic papules and vesicles on the left side of the forehead along with mild edema of the left upper eyelid but no watering of the eye or photophobia. She reported episodes of fever (temperature, 38.9°C), fatigue, and myalgia over the last week. She had bouts of dyspnea and tachycardia that she thought were related to being in the late second trimester of pregnancy. The area surrounding the vesicular eruption was tender to touch. No dry cough or any gastrointestinal or urinary tract symptoms were noted. She reported a burning sensation when splashing water on the face or when exposed to air currents. One week following the initial symptoms, she experienced a painful vesicular rash along the upper left forehead (Figure) associated with eyelid edema. Oral and ocular mucosae were free of any presentations. She had no relevant history and had not experienced any complications during pregnancy. A diagnosis of HZ was made, and she was prescribed valacyclovir 1 g 3 times daily for 7 days, acetaminophen for the fever, and calamine lotion. We recommended COVID-19 testing based on her symptoms. A chest radiograph and a positive nasopharyngeal smear were consistent with COVID-19 infection. She reported via telephone follow-up 1 week after presentation that her skin condition had improved following the treatment course and that the vesicles eventually dried, leaving a crusting appearance after 5 to 7 days. Regarding her SARS-CoV-2 condition, her oxygen saturation was 95% at presentation; she self-quarantined at home; and she was treated with oseltamivir 75 mg orally every 12 hours for 5 days, azithromycin 500 mg orally daily, acetaminophen, and vitamin C. Electronic fetal heart rate monitoring and ultrasound examinations were performed to assess the condition of the fetus and were reported normal. At the time of writing this article, she was 32 weeks pregnant and tested negative to 2 consecutive nasopharyngeal swabs for COVID-19 and was in good general condition. She continued her pregnancy according to her obstetrician’s recommendations.
Comment
The incubation time of COVID-19 can be up to 14 days. Fever, dry cough, fatigue, and diarrhea have been speculated to be clinical symptoms; however, many cases may be asymptomatic. Aside from a medical or travel history at risk for COVID-19, diagnosis can be confirmed by detection of viral RNA by reverse transcriptase–polymerase chain reaction for nasopharyngeal swabs or bronchoalveolar fluid. Patients who are immunocompromised, older, or male or who have a history of cardiovascular conditions or debilitating chronic conditions are at an increased risk for severe disease and poor outcome compared to younger healthy individuals.7
The vesicular rash of COVID-19 has been reported to have different forms of presentation. A diffuse widespread pattern resembling hand-foot-and-mouth disease and a localized monomorphic pattern resembling chickenpox but with predilection to the trunk has been described.8
Physiologic changes in the immune and cardiopulmonary systems during pregnancy (eg, diaphragm elevation, increased oxygen consumption, edema of the respiratory tract mucosae) make pregnant women intolerant to hypoxia. The mortality rate of the 1918 influenza pandemic was 2.6% in the overall population but 37% among pregnant women.9 In 2009, pregnant women were reported to be at an increased risk for complications from the H1N1 influenza virus pandemic, with a higher estimated rate of hospital admission than the general population.10 In 2003, approximately 50% of pregnant women who received a diagnosis of SARS-CoV were admitted to the intensive care unit, approximately 33% of pregnant women with SARS-CoV required mechanical ventilation, and the mortality rate was as high as 25% for these women.11 To date, data on the effects of COVID-19 in pregnancy are limited to small case series.12-15
It was confirmed that COVID-19 infection is accompanied by a reduction in lymphocytes, monocytes, and eosinophils, along with a notable reduction of CD4/CD8 T cells, B cells, and natural killer cells. It was further revealed that nonsurvivor COVID-19 patients continued to show a decrease in lymphocyte counts along the course of their disease until death.16-18
Different mechanisms for lymphocyte depletion and deficiency were speculated among COVID-19 patients and include direct lymphocyte death through coronavirus angiotensin-converting enzyme 2–lymphocyte-expressed receptors; direct damage to lymphatic organs, such as the thymus and spleen, but this theory needs to be further investigated; direct lymphocyte apoptosis mediated by tumor necrosis factor α, IL-6, and other proinflammatory cytokines; and direct inhibition of lymphocytes by metabolic upset, such as acidosis.19,20
These causes may precipitate lymphopenia and impaired antiviral responses.21 It also has been postulated that the functional damage of CD4+ T cells may predispose patients with COVID-19 to severe disease.22 Such immune changes can render a patient more susceptible to developing shingles by reactivating varicella-zoster virus, which could be a sign of undiagnosed COVID-19 infection in younger age groups.
Two earlier reports discussed HZ among COVID-19–diagnosed patients. Shors23 presented a case of a patient who developed varicella-zoster virus reactivation of the V2 dermatome during the course of COVID-19 infection. In addition, the patient developed severe acute herpetic neuralgia despite the early initiation of antiviral therapy.23 Elsaie et al24 described 2 cases of patients during the pandemic who first presented with HZ before later being diagnosed with COVID-19 infection.
New information and cutaneous manifestations possibly related to COVID-19 are emerging every day. We report a pregnant female presenting with HZ during the course of COVID-19 infection, which suggests that the clinical presentation of HZ at the time of the current pandemic, especially if associated with other signs of COVID-19 infection, should be carefully monitored and reported for further assessment.
Acknowledgment
The authors would like to thank all the health care workers who have been fighting COVID-19 in Egypt and worldwide.
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the most recently identified member of the zoonotic pathogens of coronaviruses. It caused an outbreak of pneumonia in December 2019 in Wuhan, China.1 Among all related acute respiratory syndromes (SARS-CoV, Middle East respiratory syndrome coronavirus), SARS-CoV-2 remains to be the most infectious, has the highest potential for human transmission, and can eventually result in acute respiratory distress syndrome.2,3
Only 15% of coronavirus disease 2019 (COVID-19) cases progress to pneumonia, and approximately 5% of these cases develop acute respiratory distress syndrome, septic shock, and/or multiple organ failure. The majority of cases only exhibit mild to moderate symptoms.4,5 A wide array of skin manifestations in COVID-19 infection have been reported, including maculopapular eruptions, morbilliform rashes, urticaria, chickenpoxlike lesions, livedo reticularis, COVID toes, erythema multiforme, pityriasis rosea, and several other patterns.6 We report a case of herpes zoster (HZ) complication in a COVID-19–positive woman who was 27 weeks pregnant.
Case Report
A 36-year-old woman who was 27 weeks pregnant was referred by her obstetrician to the dermatology clinic. She presented with a low-grade fever and a vesicular painful rash. Physical examination revealed painful, itchy, dysesthetic papules and vesicles on the left side of the forehead along with mild edema of the left upper eyelid but no watering of the eye or photophobia. She reported episodes of fever (temperature, 38.9°C), fatigue, and myalgia over the last week. She had bouts of dyspnea and tachycardia that she thought were related to being in the late second trimester of pregnancy. The area surrounding the vesicular eruption was tender to touch. No dry cough or any gastrointestinal or urinary tract symptoms were noted. She reported a burning sensation when splashing water on the face or when exposed to air currents. One week following the initial symptoms, she experienced a painful vesicular rash along the upper left forehead (Figure) associated with eyelid edema. Oral and ocular mucosae were free of any presentations. She had no relevant history and had not experienced any complications during pregnancy. A diagnosis of HZ was made, and she was prescribed valacyclovir 1 g 3 times daily for 7 days, acetaminophen for the fever, and calamine lotion. We recommended COVID-19 testing based on her symptoms. A chest radiograph and a positive nasopharyngeal smear were consistent with COVID-19 infection. She reported via telephone follow-up 1 week after presentation that her skin condition had improved following the treatment course and that the vesicles eventually dried, leaving a crusting appearance after 5 to 7 days. Regarding her SARS-CoV-2 condition, her oxygen saturation was 95% at presentation; she self-quarantined at home; and she was treated with oseltamivir 75 mg orally every 12 hours for 5 days, azithromycin 500 mg orally daily, acetaminophen, and vitamin C. Electronic fetal heart rate monitoring and ultrasound examinations were performed to assess the condition of the fetus and were reported normal. At the time of writing this article, she was 32 weeks pregnant and tested negative to 2 consecutive nasopharyngeal swabs for COVID-19 and was in good general condition. She continued her pregnancy according to her obstetrician’s recommendations.
Comment
The incubation time of COVID-19 can be up to 14 days. Fever, dry cough, fatigue, and diarrhea have been speculated to be clinical symptoms; however, many cases may be asymptomatic. Aside from a medical or travel history at risk for COVID-19, diagnosis can be confirmed by detection of viral RNA by reverse transcriptase–polymerase chain reaction for nasopharyngeal swabs or bronchoalveolar fluid. Patients who are immunocompromised, older, or male or who have a history of cardiovascular conditions or debilitating chronic conditions are at an increased risk for severe disease and poor outcome compared to younger healthy individuals.7
The vesicular rash of COVID-19 has been reported to have different forms of presentation. A diffuse widespread pattern resembling hand-foot-and-mouth disease and a localized monomorphic pattern resembling chickenpox but with predilection to the trunk has been described.8
Physiologic changes in the immune and cardiopulmonary systems during pregnancy (eg, diaphragm elevation, increased oxygen consumption, edema of the respiratory tract mucosae) make pregnant women intolerant to hypoxia. The mortality rate of the 1918 influenza pandemic was 2.6% in the overall population but 37% among pregnant women.9 In 2009, pregnant women were reported to be at an increased risk for complications from the H1N1 influenza virus pandemic, with a higher estimated rate of hospital admission than the general population.10 In 2003, approximately 50% of pregnant women who received a diagnosis of SARS-CoV were admitted to the intensive care unit, approximately 33% of pregnant women with SARS-CoV required mechanical ventilation, and the mortality rate was as high as 25% for these women.11 To date, data on the effects of COVID-19 in pregnancy are limited to small case series.12-15
It was confirmed that COVID-19 infection is accompanied by a reduction in lymphocytes, monocytes, and eosinophils, along with a notable reduction of CD4/CD8 T cells, B cells, and natural killer cells. It was further revealed that nonsurvivor COVID-19 patients continued to show a decrease in lymphocyte counts along the course of their disease until death.16-18
Different mechanisms for lymphocyte depletion and deficiency were speculated among COVID-19 patients and include direct lymphocyte death through coronavirus angiotensin-converting enzyme 2–lymphocyte-expressed receptors; direct damage to lymphatic organs, such as the thymus and spleen, but this theory needs to be further investigated; direct lymphocyte apoptosis mediated by tumor necrosis factor α, IL-6, and other proinflammatory cytokines; and direct inhibition of lymphocytes by metabolic upset, such as acidosis.19,20
These causes may precipitate lymphopenia and impaired antiviral responses.21 It also has been postulated that the functional damage of CD4+ T cells may predispose patients with COVID-19 to severe disease.22 Such immune changes can render a patient more susceptible to developing shingles by reactivating varicella-zoster virus, which could be a sign of undiagnosed COVID-19 infection in younger age groups.
Two earlier reports discussed HZ among COVID-19–diagnosed patients. Shors23 presented a case of a patient who developed varicella-zoster virus reactivation of the V2 dermatome during the course of COVID-19 infection. In addition, the patient developed severe acute herpetic neuralgia despite the early initiation of antiviral therapy.23 Elsaie et al24 described 2 cases of patients during the pandemic who first presented with HZ before later being diagnosed with COVID-19 infection.
New information and cutaneous manifestations possibly related to COVID-19 are emerging every day. We report a pregnant female presenting with HZ during the course of COVID-19 infection, which suggests that the clinical presentation of HZ at the time of the current pandemic, especially if associated with other signs of COVID-19 infection, should be carefully monitored and reported for further assessment.
Acknowledgment
The authors would like to thank all the health care workers who have been fighting COVID-19 in Egypt and worldwide.
- Li Q, Guan X, Wu P, et al. Early transmission dynamics in Wuhan, China, of novel coronavirus-infected pneumonia. N Engl J Med. 2020;382:1199-1207.
- Zhang YZ, Holes EC. A genomic perspective on the origin and emergence of sars-cov-2. Cell. 2020;181:223-227.
- Prompetchara E, Ketloy C, Palaga T. Immune responses in COVID-19 and potential vaccines: lessons learned from SARS and MERS epidemic. Asian Pac J Allergy Immunol. 2020;38:1‐9.
- Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan0, China. Lancet. 2020;395:497-506.
- Xu Z, Shi L, Wang Y, et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med. 2020;8:420-422.
- Wollina U, Karadag˘ AS, Rowland-Payne C, et al. Cutaneous signs in COVID-19 patients: a review. Dermatol Ther. 2020;33:e13549.
- Lauer SA, Grantz KH, Bi Q, et al. The incubation period of coronavirus disease 2019 (COVID-19) from publicly reported confirmed cases: estimation and application. Ann Intern Med. 2020;172:577‐582.
- Fernandez-Nieto D, Ortega-Quijano D, Jimenez-Cauhe J, et al. Clinical and histological characterization of vesicular COVID-19 rashes: a prospective study in a tertiary care hospital. Clin Exp Dermatol. 2020;45:872-875.
- Gottfredsson M. The Spanish flu in Iceland 1918. Lessons in medicine and history [in Icelandic]. Laeknabladid. 2008;94:737-745.
- Jamieson D, Honein M, Rasmussen S, et al. H1N1 2009 influenza virus infection during pregnancy in the USA. Lancet. 2009;374:451-458.
- Ksiazek TG, Erdman D, Goldsmith CS. A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med. 2003;348:1953-1966.
- Chen H, Guo J, Wang C, et al. Clinical characteristics and intrauterine vertical transmission potential of COVID-19 infection in nine pregnant women: a retrospective review of medical records. Lancet. 2020;395:809‐815.
- Zhu H, Wang L, Fang C, et al. Clinical analysis of 10 neonates born to mothers with 2019-nCov pneumonia. Transl Pediatr. 2020;9:51-60.
- Liu Y, Chen H, Tang K, et al. Clinical manifestations and outcome of SARS-CoV-2 infection during pregnancy [published online March 4, 2020]. J Infect. doi:10.1016/j.jinf.2020.02.028.
- Zhang L, Jiang Y, Wei M, et al. Analysis of the pregnancy outcomes in pregnant women with COVID-19 in Hubei Province [in Chinese]. Zhonghua Fu Chan Ke Za Zhi. 2020;55:166-171.
- Henry BM, de Oliveira MHS, Benoit S, et al. Hematologic, biochemical and immune biomarker abnormalities associated with severe illness and mortality in coronavirus disease 2019 (COVID-19): a meta-analysis. Clin Chem Lab Med. 2020;58:1021-1028.
- Cai Q, Huang D, Ou P, et al. COVID-19 in a designated infectious diseases hospital outside Hubei Province, China. Allergy. 2020;75:1742-1752.
- Ruan Q, Yang K, Wang W, et al. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. 2020;46:846-884.
- Kumar A, Anil A, Sharma P, et al. Clinical features of COVID-19 and factors associated with severe clinical course: a systematic review and meta-analysis [preprint]. SSRN. doi:10.2139/ssrn.3566166.
- Xu H, Zhong L, Deng J, et al. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int J Oral Sci. 2020;12. https://doi.org/10.1038/s41368-020-0074-x.
- Li H, Liu L, Zhang D, et al. SARS-CoV-2 and viral sepsis: observations and hypotheses. Lancet. 2020;395:1517-1520.
- Zheng M, Gao Y, Wang G, et al. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell Mol Immunol. 2020;17:533-535.
- Shors AR. Herpes zoster and severe acute herpetic neuralgia as a complication of COVID-19 infection. JAAD Case Rep. 2020;6:656-657.
- Elsaie ML, Youssef EA, Nada HA. Herpes zoster might be an indicator for latent COVID 19 infection [published online May 23, 2020]. Dermatol Ther. doi:10.1111/dth.13666.
- Li Q, Guan X, Wu P, et al. Early transmission dynamics in Wuhan, China, of novel coronavirus-infected pneumonia. N Engl J Med. 2020;382:1199-1207.
- Zhang YZ, Holes EC. A genomic perspective on the origin and emergence of sars-cov-2. Cell. 2020;181:223-227.
- Prompetchara E, Ketloy C, Palaga T. Immune responses in COVID-19 and potential vaccines: lessons learned from SARS and MERS epidemic. Asian Pac J Allergy Immunol. 2020;38:1‐9.
- Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan0, China. Lancet. 2020;395:497-506.
- Xu Z, Shi L, Wang Y, et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med. 2020;8:420-422.
- Wollina U, Karadag˘ AS, Rowland-Payne C, et al. Cutaneous signs in COVID-19 patients: a review. Dermatol Ther. 2020;33:e13549.
- Lauer SA, Grantz KH, Bi Q, et al. The incubation period of coronavirus disease 2019 (COVID-19) from publicly reported confirmed cases: estimation and application. Ann Intern Med. 2020;172:577‐582.
- Fernandez-Nieto D, Ortega-Quijano D, Jimenez-Cauhe J, et al. Clinical and histological characterization of vesicular COVID-19 rashes: a prospective study in a tertiary care hospital. Clin Exp Dermatol. 2020;45:872-875.
- Gottfredsson M. The Spanish flu in Iceland 1918. Lessons in medicine and history [in Icelandic]. Laeknabladid. 2008;94:737-745.
- Jamieson D, Honein M, Rasmussen S, et al. H1N1 2009 influenza virus infection during pregnancy in the USA. Lancet. 2009;374:451-458.
- Ksiazek TG, Erdman D, Goldsmith CS. A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med. 2003;348:1953-1966.
- Chen H, Guo J, Wang C, et al. Clinical characteristics and intrauterine vertical transmission potential of COVID-19 infection in nine pregnant women: a retrospective review of medical records. Lancet. 2020;395:809‐815.
- Zhu H, Wang L, Fang C, et al. Clinical analysis of 10 neonates born to mothers with 2019-nCov pneumonia. Transl Pediatr. 2020;9:51-60.
- Liu Y, Chen H, Tang K, et al. Clinical manifestations and outcome of SARS-CoV-2 infection during pregnancy [published online March 4, 2020]. J Infect. doi:10.1016/j.jinf.2020.02.028.
- Zhang L, Jiang Y, Wei M, et al. Analysis of the pregnancy outcomes in pregnant women with COVID-19 in Hubei Province [in Chinese]. Zhonghua Fu Chan Ke Za Zhi. 2020;55:166-171.
- Henry BM, de Oliveira MHS, Benoit S, et al. Hematologic, biochemical and immune biomarker abnormalities associated with severe illness and mortality in coronavirus disease 2019 (COVID-19): a meta-analysis. Clin Chem Lab Med. 2020;58:1021-1028.
- Cai Q, Huang D, Ou P, et al. COVID-19 in a designated infectious diseases hospital outside Hubei Province, China. Allergy. 2020;75:1742-1752.
- Ruan Q, Yang K, Wang W, et al. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. 2020;46:846-884.
- Kumar A, Anil A, Sharma P, et al. Clinical features of COVID-19 and factors associated with severe clinical course: a systematic review and meta-analysis [preprint]. SSRN. doi:10.2139/ssrn.3566166.
- Xu H, Zhong L, Deng J, et al. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int J Oral Sci. 2020;12. https://doi.org/10.1038/s41368-020-0074-x.
- Li H, Liu L, Zhang D, et al. SARS-CoV-2 and viral sepsis: observations and hypotheses. Lancet. 2020;395:1517-1520.
- Zheng M, Gao Y, Wang G, et al. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell Mol Immunol. 2020;17:533-535.
- Shors AR. Herpes zoster and severe acute herpetic neuralgia as a complication of COVID-19 infection. JAAD Case Rep. 2020;6:656-657.
- Elsaie ML, Youssef EA, Nada HA. Herpes zoster might be an indicator for latent COVID 19 infection [published online May 23, 2020]. Dermatol Ther. doi:10.1111/dth.13666.
Practice Points
- The vesicular rash of coronavirus disease 2019 (COVID-19) has been reported to have different forms of presentation.
- Pregnant women appear to be at increased risk for complications from COVID-19 infection.
- The clinical presentation of herpes zoster should be carefully monitored and reported for further assessment, especially if associated with other signs of COVID-19 infection.