User login
Dupilumab-Induced Facial Flushing After Alcohol Consumption
Dupilumab is a fully humanized monoclonal antibody to the α subunit of the IL-4 receptor that inhibits the action of helper T cell (TH2)–type cytokines IL-4 and IL-13. Dupilumab was approved by the US Food and Drug Administration (FDA) in 2017 for the treatment of moderate to severe atopic dermatitis (AD). We report 2 patients with AD who were treated with dupilumab and subsequently developed facial flushing after consuming alcohol.
Case Report
Patient 1
A 24-year-old woman presented to the dermatology clinic with a lifelong history of moderate to severe AD. She had a medical history of asthma and seasonal allergies, which were treated with fexofenadine and an inhaler, as needed. The patient had an affected body surface area of approximately 70% and had achieved only partial relief with topical corticosteroids and topical calcineurin inhibitors.
Because her disease was severe, the patient was started on dupilumab at FDA-approved dosing for AD: a 600-mg subcutaneous (SC) loading dose, followed by 300 mg SC every 2 weeks. She reported rapid skin clearance within 2 weeks of the start of treatment. Her course was complicated by mild head and neck dermatitis.
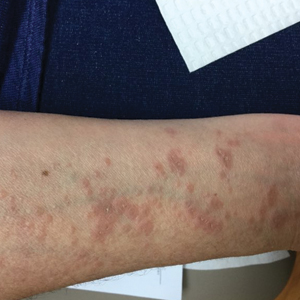
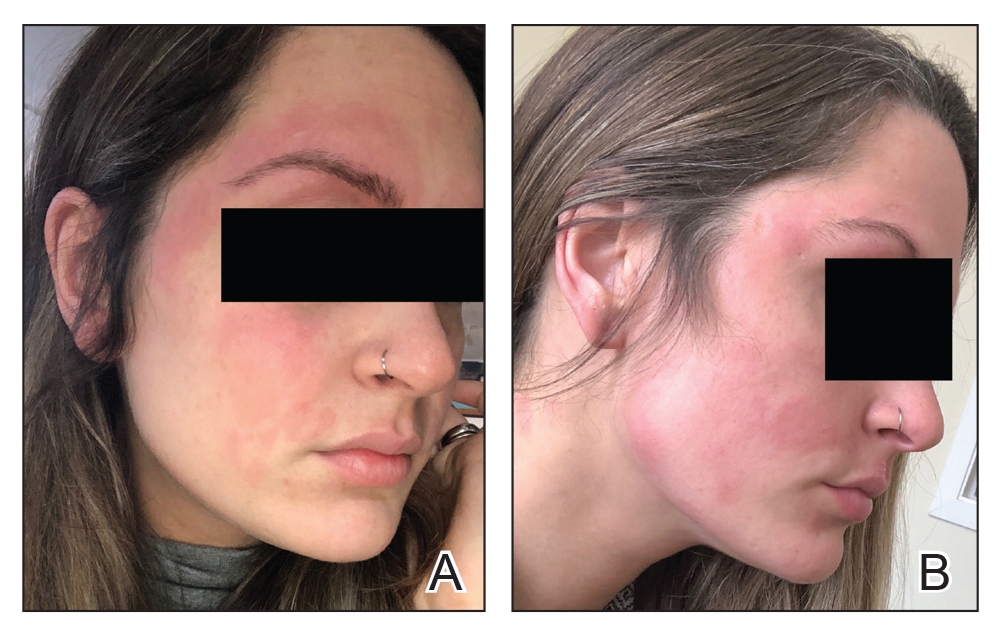
Seven months after starting treatment, the patient began to acutely experience erythema and warmth over the entire face that was triggered by drinking alcohol (Figure). Before starting dupilumab, she had consumed alcohol on multiple occasions without a flushing effect. This new finding was distinguishable from her facial dermatitis. Onset was within a few minutes after drinking alcohol; flushing self-resolved in 15 to 30 minutes. Although diffuse, erythema and warmth were concentrated around the jawline, eyebrows, and ears and occurred every time the patient drank alcohol. Moreover, she reported that consumption of hard (ie, distilled) liquor, specifically tequila, caused a more severe presentation. She denied other symptoms associated with dupilumab.
Patient 2
A 32-year-old man presented to the dermatology clinic with a 10-year history of moderate to severe AD. He had a medical history of asthma (treated with albuterol, montelukast, and fluticasone); allergic rhinitis; and severe environmental allergies, including sensitivity to dust mites, dogs, trees, and grass.
For AD, the patient had been treated with topical corticosteroids and the Goeckerman regimen (a combination of phototherapy and crude coal tar). He experienced only partial relief with topical corticosteroids; the Goeckerman regimen cleared his skin, but he had quick recurrence after approximately 1 month. Given his work schedule, the patient was unable to resume phototherapy.
Because of symptoms related to the patient’s severe allergies, his allergist prescribed dupilumab: a 600-mg SC loading dose, followed by 300 mg SC every 2 weeks. The patient reported near-complete resolution of AD symptoms approximately 2 months after initiating treatment. He reported a few episodes of mild conjunctivitis that self-resolved after the first month of treatment.
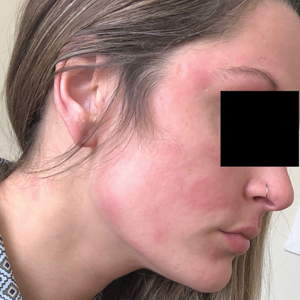
Three weeks after initiating dupilumab, the patient noticed new-onset facial flushing in response to consuming alcohol. He described flushing as sudden immediate redness and warmth concentrated around the forehead, eyes, and cheeks. He reported that flushing was worse with hard liquor than with beer. Flushing would slowly subside over approximately 30 minutes despite continued alcohol consumption.
Comment
Two other single-patient case reports have discussed similar findings of alcohol-induced flushing associated with dupilumab.1,2 Both of those patients—a 19-year-old woman and a 26-year-old woman—had not experienced flushing before beginning treatment with dupilumab for AD. Both experienced onset of facial flushing months after beginning dupilumab even though both had consumed alcohol before starting dupilumab, similar to the cases presented here. One patient had a history of asthma; the other had a history of seasonal and environmental allergies.
Possible Mechanism of Action
Acute alcohol ingestion causes dermal vasodilation of the skin (ie, flushing).3 A proposed mechanism is that flushing results from direct action on central vascular-control mechanisms. This theory results from observations that individuals with quadriplegia lack notable ethanol-induced vasodilation, suggesting that ethanol has a central neural site of action.Although some research has indicated that ethanol might induce these effects by altering the action of certain hormones (eg, angiotensin, vasopressin, and catecholamines), the precise mechanism by which ethanol alters vascular function in humans remains unexplained.3
Deficiencies in alcohol dehydrogenase (ADH), aldehyde dehydrogenase 2, and certain cytochrome P450 enzymes also might contribute to facial flushing. People of Asian, especially East Asian, descent often respond to an acute dose of ethanol with symptoms of facial flushing—predominantly the result of an elevated blood level of acetaldehyde caused by an inherited deficiency of aldehyde dehydrogenase 2,4 which is downstream from ADH in the metabolic pathway of alcohol. The major enzyme system responsible for metabolism of ethanol is ADH; however, the cytochrome P450–dependent ethanol-oxidizing system—including major CYP450 isoforms CYP3A, CYP2C19, CYP2C9, CYP1A2, and CYP2D6, as well as minor CYP450 isoforms, such as CYP2E1— also are involved, to a lesser extent.5
A Role for Dupilumab?
A recent pharmacokinetic study found that dupilumab appears to have little effect on the activity of the major CYP450 isoforms. However, the drug’s effect on ADH and minor CYP450 minor isoforms is unknown. Prior drug-drug interaction studies have shown that certain cytokines and cytokine modulators can markedly influence the expression, stability, and activity of specific CYP450 enzymes.6 For example, IL-6 causes a reduction in messenger RNA for CYP3A4 and, to a lesser extent, for other isoforms.7 Whether dupilumab influences enzymes involved in processing alcohol requires further study.
Conclusion
We describe 2 cases of dupilumab-induced facial flushing after alcohol consumption. The mechanism of this dupilumab-associated flushing is unknown and requires further research.
- Herz S, Petri M, Sondermann W. New alcohol flushing in a patient with atopic dermatitis under therapy with dupilumab. Dermatol Ther. 2019;32:e12762. doi:10.1111/dth.12762
- Igelman SJ, Na C, Simpson EL. Alcohol-induced facial flushing in a patient with atopic dermatitis treated with dupilumab. JAAD Case Rep. 2020;6:139-140. doi:10.1016/j.jdcr.2019.12.002
- Malpas SC, Robinson BJ, Maling TJ. Mechanism of ethanol-induced vasodilation. J Appl Physiol (1985). 1990;68:731-734. doi:10.1152/jappl.1990.68.2.731
- Brooks PJ, Enoch M-A, Goldman D, et al. The alcohol flushing response: an unrecognized risk factor for esophageal cancer from alcohol consumption. PLoS Med. 2009;6:e50. doi:10.1371/journal.pmed.1000050
- Cederbaum AI. Alcohol metabolism. Clin Liver Dis. 2012;16:667-685. doi:10.1016/j.cld.2012.08.002
- Davis JD, Bansal A, Hassman D, et al. Evaluation of potential disease-mediated drug-drug interaction in patients with moderate-to-severe atopic dermatitis receiving dupilumab. Clin Pharmacol Ther. 2018;104:1146-1154. doi:10.1002/cpt.1058
- Mimura H, Kobayashi K, Xu L, et al. Effects of cytokines on CYP3A4 expression and reversal of the effects by anti-cytokine agents in the three-dimensionally cultured human hepatoma cell line FLC-4. Drug Metab Pharmacokinet. 2015;30:105-110. doi:10.1016/j.dmpk.2014.09.004
Dupilumab is a fully humanized monoclonal antibody to the α subunit of the IL-4 receptor that inhibits the action of helper T cell (TH2)–type cytokines IL-4 and IL-13. Dupilumab was approved by the US Food and Drug Administration (FDA) in 2017 for the treatment of moderate to severe atopic dermatitis (AD). We report 2 patients with AD who were treated with dupilumab and subsequently developed facial flushing after consuming alcohol.
Case Report
Patient 1
A 24-year-old woman presented to the dermatology clinic with a lifelong history of moderate to severe AD. She had a medical history of asthma and seasonal allergies, which were treated with fexofenadine and an inhaler, as needed. The patient had an affected body surface area of approximately 70% and had achieved only partial relief with topical corticosteroids and topical calcineurin inhibitors.
Because her disease was severe, the patient was started on dupilumab at FDA-approved dosing for AD: a 600-mg subcutaneous (SC) loading dose, followed by 300 mg SC every 2 weeks. She reported rapid skin clearance within 2 weeks of the start of treatment. Her course was complicated by mild head and neck dermatitis.
Seven months after starting treatment, the patient began to acutely experience erythema and warmth over the entire face that was triggered by drinking alcohol (Figure). Before starting dupilumab, she had consumed alcohol on multiple occasions without a flushing effect. This new finding was distinguishable from her facial dermatitis. Onset was within a few minutes after drinking alcohol; flushing self-resolved in 15 to 30 minutes. Although diffuse, erythema and warmth were concentrated around the jawline, eyebrows, and ears and occurred every time the patient drank alcohol. Moreover, she reported that consumption of hard (ie, distilled) liquor, specifically tequila, caused a more severe presentation. She denied other symptoms associated with dupilumab.
Patient 2
A 32-year-old man presented to the dermatology clinic with a 10-year history of moderate to severe AD. He had a medical history of asthma (treated with albuterol, montelukast, and fluticasone); allergic rhinitis; and severe environmental allergies, including sensitivity to dust mites, dogs, trees, and grass.
For AD, the patient had been treated with topical corticosteroids and the Goeckerman regimen (a combination of phototherapy and crude coal tar). He experienced only partial relief with topical corticosteroids; the Goeckerman regimen cleared his skin, but he had quick recurrence after approximately 1 month. Given his work schedule, the patient was unable to resume phototherapy.
Because of symptoms related to the patient’s severe allergies, his allergist prescribed dupilumab: a 600-mg SC loading dose, followed by 300 mg SC every 2 weeks. The patient reported near-complete resolution of AD symptoms approximately 2 months after initiating treatment. He reported a few episodes of mild conjunctivitis that self-resolved after the first month of treatment.
Three weeks after initiating dupilumab, the patient noticed new-onset facial flushing in response to consuming alcohol. He described flushing as sudden immediate redness and warmth concentrated around the forehead, eyes, and cheeks. He reported that flushing was worse with hard liquor than with beer. Flushing would slowly subside over approximately 30 minutes despite continued alcohol consumption.
Comment
Two other single-patient case reports have discussed similar findings of alcohol-induced flushing associated with dupilumab.1,2 Both of those patients—a 19-year-old woman and a 26-year-old woman—had not experienced flushing before beginning treatment with dupilumab for AD. Both experienced onset of facial flushing months after beginning dupilumab even though both had consumed alcohol before starting dupilumab, similar to the cases presented here. One patient had a history of asthma; the other had a history of seasonal and environmental allergies.
Possible Mechanism of Action
Acute alcohol ingestion causes dermal vasodilation of the skin (ie, flushing).3 A proposed mechanism is that flushing results from direct action on central vascular-control mechanisms. This theory results from observations that individuals with quadriplegia lack notable ethanol-induced vasodilation, suggesting that ethanol has a central neural site of action.Although some research has indicated that ethanol might induce these effects by altering the action of certain hormones (eg, angiotensin, vasopressin, and catecholamines), the precise mechanism by which ethanol alters vascular function in humans remains unexplained.3
Deficiencies in alcohol dehydrogenase (ADH), aldehyde dehydrogenase 2, and certain cytochrome P450 enzymes also might contribute to facial flushing. People of Asian, especially East Asian, descent often respond to an acute dose of ethanol with symptoms of facial flushing—predominantly the result of an elevated blood level of acetaldehyde caused by an inherited deficiency of aldehyde dehydrogenase 2,4 which is downstream from ADH in the metabolic pathway of alcohol. The major enzyme system responsible for metabolism of ethanol is ADH; however, the cytochrome P450–dependent ethanol-oxidizing system—including major CYP450 isoforms CYP3A, CYP2C19, CYP2C9, CYP1A2, and CYP2D6, as well as minor CYP450 isoforms, such as CYP2E1— also are involved, to a lesser extent.5
A Role for Dupilumab?
A recent pharmacokinetic study found that dupilumab appears to have little effect on the activity of the major CYP450 isoforms. However, the drug’s effect on ADH and minor CYP450 minor isoforms is unknown. Prior drug-drug interaction studies have shown that certain cytokines and cytokine modulators can markedly influence the expression, stability, and activity of specific CYP450 enzymes.6 For example, IL-6 causes a reduction in messenger RNA for CYP3A4 and, to a lesser extent, for other isoforms.7 Whether dupilumab influences enzymes involved in processing alcohol requires further study.
Conclusion
We describe 2 cases of dupilumab-induced facial flushing after alcohol consumption. The mechanism of this dupilumab-associated flushing is unknown and requires further research.
Dupilumab is a fully humanized monoclonal antibody to the α subunit of the IL-4 receptor that inhibits the action of helper T cell (TH2)–type cytokines IL-4 and IL-13. Dupilumab was approved by the US Food and Drug Administration (FDA) in 2017 for the treatment of moderate to severe atopic dermatitis (AD). We report 2 patients with AD who were treated with dupilumab and subsequently developed facial flushing after consuming alcohol.
Case Report
Patient 1
A 24-year-old woman presented to the dermatology clinic with a lifelong history of moderate to severe AD. She had a medical history of asthma and seasonal allergies, which were treated with fexofenadine and an inhaler, as needed. The patient had an affected body surface area of approximately 70% and had achieved only partial relief with topical corticosteroids and topical calcineurin inhibitors.
Because her disease was severe, the patient was started on dupilumab at FDA-approved dosing for AD: a 600-mg subcutaneous (SC) loading dose, followed by 300 mg SC every 2 weeks. She reported rapid skin clearance within 2 weeks of the start of treatment. Her course was complicated by mild head and neck dermatitis.
Seven months after starting treatment, the patient began to acutely experience erythema and warmth over the entire face that was triggered by drinking alcohol (Figure). Before starting dupilumab, she had consumed alcohol on multiple occasions without a flushing effect. This new finding was distinguishable from her facial dermatitis. Onset was within a few minutes after drinking alcohol; flushing self-resolved in 15 to 30 minutes. Although diffuse, erythema and warmth were concentrated around the jawline, eyebrows, and ears and occurred every time the patient drank alcohol. Moreover, she reported that consumption of hard (ie, distilled) liquor, specifically tequila, caused a more severe presentation. She denied other symptoms associated with dupilumab.
Patient 2
A 32-year-old man presented to the dermatology clinic with a 10-year history of moderate to severe AD. He had a medical history of asthma (treated with albuterol, montelukast, and fluticasone); allergic rhinitis; and severe environmental allergies, including sensitivity to dust mites, dogs, trees, and grass.
For AD, the patient had been treated with topical corticosteroids and the Goeckerman regimen (a combination of phototherapy and crude coal tar). He experienced only partial relief with topical corticosteroids; the Goeckerman regimen cleared his skin, but he had quick recurrence after approximately 1 month. Given his work schedule, the patient was unable to resume phototherapy.
Because of symptoms related to the patient’s severe allergies, his allergist prescribed dupilumab: a 600-mg SC loading dose, followed by 300 mg SC every 2 weeks. The patient reported near-complete resolution of AD symptoms approximately 2 months after initiating treatment. He reported a few episodes of mild conjunctivitis that self-resolved after the first month of treatment.
Three weeks after initiating dupilumab, the patient noticed new-onset facial flushing in response to consuming alcohol. He described flushing as sudden immediate redness and warmth concentrated around the forehead, eyes, and cheeks. He reported that flushing was worse with hard liquor than with beer. Flushing would slowly subside over approximately 30 minutes despite continued alcohol consumption.
Comment
Two other single-patient case reports have discussed similar findings of alcohol-induced flushing associated with dupilumab.1,2 Both of those patients—a 19-year-old woman and a 26-year-old woman—had not experienced flushing before beginning treatment with dupilumab for AD. Both experienced onset of facial flushing months after beginning dupilumab even though both had consumed alcohol before starting dupilumab, similar to the cases presented here. One patient had a history of asthma; the other had a history of seasonal and environmental allergies.
Possible Mechanism of Action
Acute alcohol ingestion causes dermal vasodilation of the skin (ie, flushing).3 A proposed mechanism is that flushing results from direct action on central vascular-control mechanisms. This theory results from observations that individuals with quadriplegia lack notable ethanol-induced vasodilation, suggesting that ethanol has a central neural site of action.Although some research has indicated that ethanol might induce these effects by altering the action of certain hormones (eg, angiotensin, vasopressin, and catecholamines), the precise mechanism by which ethanol alters vascular function in humans remains unexplained.3
Deficiencies in alcohol dehydrogenase (ADH), aldehyde dehydrogenase 2, and certain cytochrome P450 enzymes also might contribute to facial flushing. People of Asian, especially East Asian, descent often respond to an acute dose of ethanol with symptoms of facial flushing—predominantly the result of an elevated blood level of acetaldehyde caused by an inherited deficiency of aldehyde dehydrogenase 2,4 which is downstream from ADH in the metabolic pathway of alcohol. The major enzyme system responsible for metabolism of ethanol is ADH; however, the cytochrome P450–dependent ethanol-oxidizing system—including major CYP450 isoforms CYP3A, CYP2C19, CYP2C9, CYP1A2, and CYP2D6, as well as minor CYP450 isoforms, such as CYP2E1— also are involved, to a lesser extent.5
A Role for Dupilumab?
A recent pharmacokinetic study found that dupilumab appears to have little effect on the activity of the major CYP450 isoforms. However, the drug’s effect on ADH and minor CYP450 minor isoforms is unknown. Prior drug-drug interaction studies have shown that certain cytokines and cytokine modulators can markedly influence the expression, stability, and activity of specific CYP450 enzymes.6 For example, IL-6 causes a reduction in messenger RNA for CYP3A4 and, to a lesser extent, for other isoforms.7 Whether dupilumab influences enzymes involved in processing alcohol requires further study.
Conclusion
We describe 2 cases of dupilumab-induced facial flushing after alcohol consumption. The mechanism of this dupilumab-associated flushing is unknown and requires further research.
- Herz S, Petri M, Sondermann W. New alcohol flushing in a patient with atopic dermatitis under therapy with dupilumab. Dermatol Ther. 2019;32:e12762. doi:10.1111/dth.12762
- Igelman SJ, Na C, Simpson EL. Alcohol-induced facial flushing in a patient with atopic dermatitis treated with dupilumab. JAAD Case Rep. 2020;6:139-140. doi:10.1016/j.jdcr.2019.12.002
- Malpas SC, Robinson BJ, Maling TJ. Mechanism of ethanol-induced vasodilation. J Appl Physiol (1985). 1990;68:731-734. doi:10.1152/jappl.1990.68.2.731
- Brooks PJ, Enoch M-A, Goldman D, et al. The alcohol flushing response: an unrecognized risk factor for esophageal cancer from alcohol consumption. PLoS Med. 2009;6:e50. doi:10.1371/journal.pmed.1000050
- Cederbaum AI. Alcohol metabolism. Clin Liver Dis. 2012;16:667-685. doi:10.1016/j.cld.2012.08.002
- Davis JD, Bansal A, Hassman D, et al. Evaluation of potential disease-mediated drug-drug interaction in patients with moderate-to-severe atopic dermatitis receiving dupilumab. Clin Pharmacol Ther. 2018;104:1146-1154. doi:10.1002/cpt.1058
- Mimura H, Kobayashi K, Xu L, et al. Effects of cytokines on CYP3A4 expression and reversal of the effects by anti-cytokine agents in the three-dimensionally cultured human hepatoma cell line FLC-4. Drug Metab Pharmacokinet. 2015;30:105-110. doi:10.1016/j.dmpk.2014.09.004
- Herz S, Petri M, Sondermann W. New alcohol flushing in a patient with atopic dermatitis under therapy with dupilumab. Dermatol Ther. 2019;32:e12762. doi:10.1111/dth.12762
- Igelman SJ, Na C, Simpson EL. Alcohol-induced facial flushing in a patient with atopic dermatitis treated with dupilumab. JAAD Case Rep. 2020;6:139-140. doi:10.1016/j.jdcr.2019.12.002
- Malpas SC, Robinson BJ, Maling TJ. Mechanism of ethanol-induced vasodilation. J Appl Physiol (1985). 1990;68:731-734. doi:10.1152/jappl.1990.68.2.731
- Brooks PJ, Enoch M-A, Goldman D, et al. The alcohol flushing response: an unrecognized risk factor for esophageal cancer from alcohol consumption. PLoS Med. 2009;6:e50. doi:10.1371/journal.pmed.1000050
- Cederbaum AI. Alcohol metabolism. Clin Liver Dis. 2012;16:667-685. doi:10.1016/j.cld.2012.08.002
- Davis JD, Bansal A, Hassman D, et al. Evaluation of potential disease-mediated drug-drug interaction in patients with moderate-to-severe atopic dermatitis receiving dupilumab. Clin Pharmacol Ther. 2018;104:1146-1154. doi:10.1002/cpt.1058
- Mimura H, Kobayashi K, Xu L, et al. Effects of cytokines on CYP3A4 expression and reversal of the effects by anti-cytokine agents in the three-dimensionally cultured human hepatoma cell line FLC-4. Drug Metab Pharmacokinet. 2015;30:105-110. doi:10.1016/j.dmpk.2014.09.004
Practice Points
- Dupilumab is a fully humanized monoclonal antibody that inhibits the action of IL-4 and IL-13. It was approved by the US Food and Drug Administration in 2017 for treatment of moderate to severe atopic dermatitis.
- Facial flushing after alcohol consumption may be an emerging side effect of dupilumab.
- Whether dupilumab influences enzymes involved in processing alcohol requires further study.
Myasthenic Crisis After Recurrent COVID-19 Infection
A patient with myasthenia gravis who survived 2 COVID-19 infections required plasmapheresis to recover from an acute crisis.
COVID-19 is still in the early stages of understanding, although it is known to be complicated by individual patient comorbidities. The management and treatment of COVID-19 continues to quickly evolve as more is discovered regarding the virus. Multiple treatments have been preliminarily tested and used under a Food and Drug Administration emergency use authorization (EUA) determination. The long-term success of these therapies, however, is yet to be determined. Additionally, if a patient has a second clinical presentation for COVID-19, it is not known whether this represents latency with subsequent reactivation from the previous infection or a second de novo infection. The uncertainty calls into question the duration of immunity, if any, following a primary infection.
COVID-19 management becomes more complicated when patients have complex medical conditions, such as myasthenia gravis (MG). This autoimmune neuromuscular disorder can present with varying weakness, and many patients are on immunomodulator medications. The weakness can worsen into a myasthenic crisis (MC), resulting in profound weakness of the respiratory muscles. Therefore, patients with MG are at increased risk for COVID-19 and may have a more complicated course when infected.
Our patient with MG presented for severe COVID-19 symptoms twice and later developed MC. He received 2 treatment modalities available under an EUA (remdesivir and convalescent plasma) for COVID-19, resulting in symptom resolution and a negative polymerize chain reaction (PCR) test result for the virus. However, after receiving his typical maintenance therapy of IV immunoglobulin (IVIG) for his MG, he again developed symptoms consistent with COVID-19 and tested positive. After recovering from the second episode of COVID-19, the patient went into MC requiring plasmapheresis.
Case Presentation
A 56-year-old male, US Army veteran presented to Carl R. Darnall Army Medical Center emergency department (ED) 6 days after testing positive for COVID-19, with worsening sputum, cough, congestion, dyspnea, and fever. Due to his MG, the patient had a home oxygen monitor and reported that his oxygenation saturation dropped below 90% with minimal exertion. His medical history was significant for MG, status postthymectomy and radiation treatment, left hemidiaphragm paralysis secondary to phrenic nerve injury, and corticosteroid-induced insulin-dependent diabetes mellitus. His current home medications included pyridostigmine 60 mg 3 times a day, mycophenolate (MMF) 1500 mg twice daily, IV immunoglobulin (IVIG) every 3 weeks, insulin aspart up to 16 U per meal, insulin glargine 30 U twice a day, dulaglutide 0.75 mg every week, and metformin 1000 mg twice daily.
On initial examination, the patient’s heart rate (HR) was 111 beats/min, respiratory rate (RR), 22 breaths/min, blood pressure (BP), 138/88 mm Hg, temperature, 100.9 oF, and his initial pulse oximetry, 91% on room air. On physical examination, the patient was tachypneic, though without other signs of respiratory distress. Lung auscultation revealed no adventitial lung sounds. His cardiac examination was notable only for tachycardia. His neurologic examination demonstrated intact cranial nerves, with 5 out of 5 (scale 1 to 5) strength throughout the upper and lower extremities, sensation was intact to light touch, and he had normal cerebellar function. The rest of the examination was normal.
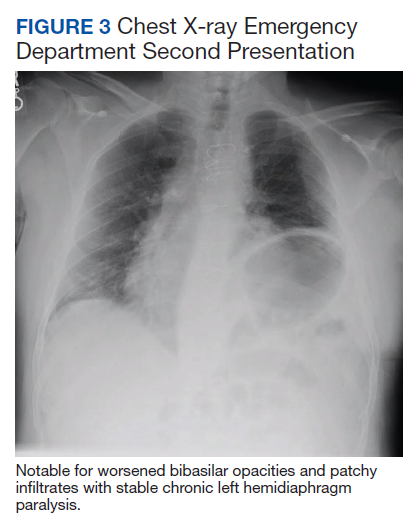
Initial laboratory investigation was notable for a white blood cell count of 14.15x103 cells/mcL with 84% neutrophils, and 6% lymphocytes. Additional tests revealed a C-reactive protein (CRP) level, 17.97 mg/dL (reference range, 0-0.5 mg/dL), ferritin level, 647 ng/mL (reference range, 22-274 ng/mL), d-dimer, 0.64 mcg/mL (reference range, 0-0.47mcg/mL), and a repeated positive COVID-19 PCR test. A portable chest X-ray showed bibasilar opacities (Figure 1).
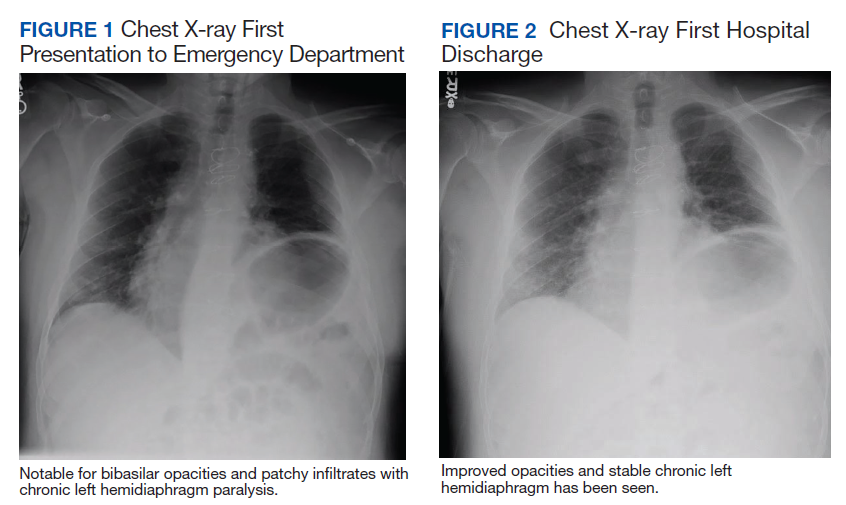
The patient was diagnosed with COVID-19 and admitted to the intensive care unit (ICU). In the ICU, the patient received 1 U of convalescent plasma (CP) and started on a course of IV remdesivir 100 mg/d consistent with the EUA. He also received a 5-day course of ceftriaxone and azithromycin for possible community acquired pneumonia (CAP). As part of the patient’s MG maintenance medications, he received IVIG 4 g while in the ICU. Throughout his ICU stay, he required supplemental nasal cannula oxygenation to maintain his oxygen saturation > 93%. After 8 days in the ICU, his oxygen requirements decreased, and the patient was transferred out of the ICU and remdesivir was discontinued. On hospital day 10, a repeat COVID-19 PCR test was negative, inflammatory markers returned to within normal limits, and a repeat chest X-ray showed improvement from admission (Figure 2). Having recovered significantly, he was discharged home.
Three weeks later, the patient again presented to the MTF with 3 days of dyspnea, cough, fever, nausea, and vomiting. One day before symptom onset, he had received his maintenance IVIG infusion. The patient reported that his home oxygen saturation was 82% with minimal exertion. On ED presentation his HR was 107 beats/min, RR, 28 breaths/min, temperature, 98.1 oF, BP 118/71 mm Hg, and oxygen saturation, 92% on 2L nasal cannula. His examination was most notable for tachypnea with accessory muscle use. At this time, his neurologic examination was unchanged from prior admission with grossly intact cranial nerves and symmetric 5 of 5 motor strength in all extremities.
At this second ED visit, laboratory results demonstrated a CRP of 3.44 mg/dL, ferritin 2019 ng/mL, d-dimer, 3.39 mcg/mL, and a positive COVID-19 PCR result. His chest X-ray demonstrated new peripheral opacities compared with the X-ray at discharge (Figure 3). He required ICU admission again for his COVID-19 symptoms.
During his ICU course he continued to require supplemental oxygen by nasal cannula, though never required intubation. This second admission, he was again treated empirically for CAP with levofloxacin 750 mg daily for 5 days. He was discharged after 14 days with symptom resolution and down trending of inflammatory markers, though he was not retested for COVID-19.
Four days after his second discharge, he presented to the ED for a third time with diffuse weakness, dysphagia, and dysarthria of 1 day. His HR was 87/beats/min; RR, 17 breaths/min; temperature, 98.7 oF; BP, 144/81 mm Hg; and oxygen saturation, 98% on room air. His examination was significant for slurred speech, bilateral ptosis, 3 of 5 strength in bilateral finger flexion/abduction, wrist extension, knee and ankle flexion/extension; 4 of 5 strength in bilateral proximal muscle testing of deltoid, and hip; normal sensation, cerebellar function and reflexes. His negative inspiratory force (NIF) maximal effort was −30 cmH2O. He was determined to be in MC without evidence of COIVD-19 symptoms, and laboratory results were within normal limits, including a negative COVID-19 PCR. As he received IVIG as maintenance therapy, plasmapheresis was recommended to treat his MC, which required transfer to an outside civilian facility.
At the outside hospital, the patient underwent 5 rounds of plasmapheresis over 10 days. By the third treatment his strength had returned with resolution of the bulbar symptoms and no supplemental oxygen requirements. The patient was discharged and continued his original dosages of MMF and pyridostigmine. At 3 months, he remained asymptomatic from a COVID-19 standpoint and stable from a MG standpoint.
Discussion
Reinfection with the COVID-19 has been continuously debated with alternative explanations suggested for a positive test after a previous negative PCR test in the setting of symptom resolution.1,2 Proposed causes include dynamic PCR results due to prolonged viral shedding and inaccurate or poorly sensitive tests. The repeat positive cases in these scenarios, however, occurred in asymptomatic patients.1,2 COVID-19 shedding averages 20 to 22 days after symptom onset but has been seen up to 36 days after symptom resolution.2,3 This would suggest that fluctuating results during the immediate postsymptom period may be due to variations in viral shedding load and or sampling error—especially in asymptomatic patients. On the other hand, patients who experience return of symptoms days to weeks after previous convalescence leave clinicians wondering whether this represents clinical latency with reactivation or COVID-19 reinfection. A separate case of initial COVID-19 in a patient that had subsequent clinical recovery with a negative PCR developed recurrent respiratory symptoms and had a positive PCR test only 10 days later, further highlighting the reinfection vs reactivation issue of COVID-19.2 Further understanding of this issue may have implications on the extent of natural immunity following primary infection; potential vaccine dosage schedules; and global public health policies.
Although reactivation may be plausible given his immunomodulatory therapy, our patient’s second COVID-19 symptoms started 40 days after the initial symptoms, and 26 days after the initial course resolution; previous cases of return of severe symptoms occurred between 3 and 6 days.1 Given our patient’s time course between resolution and return of symptoms, if latency is the mechanism at play, this case demonstrates an exceptionally longer latency period than the ones that have been reported. Additionally, if latency is an issue in COVID-19, using remdesivir as a treatment further complicates the understanding of this disease.
Remdesivir, a nucleoside analogue antiviral, was shown to benefit recovery in patients with severe symptoms in the Adaptive COVID-19 Treatment Trial-1 study.4 Our patient had originally been placed on a 10-day course; however, on treatment day 8, his symptoms resolved and the remdesivir was discontinued. This is a similar finding to half the patients in the 10-day arm of the study by McCreary and colleagues.5 Although our patient was asymptomatic 4 weeks after the start of remdesivir, consistent with the majority of patients in the McCreary 10-day study arm, further comparison of the presented patient is limited due to study length and follow-up considerations.5 No previous data exist on reactivation, reinfection, or long-term mortality after being treated with remdesivir for COVID-19 infection.
IVIG is being studied in the treatment of COVID-19 and bears consideration as it relates to our patient. There is no evidence that IVIG used in the treatment of autoimmune diseases increases the risk of infection compared with that of other medications used in the treatment of such diseases. Furthermore, the current guidance from the MG expert panel does not suggest that IVIG increases the risk of contracting COVID-19 aside from the risks of exposure to hospital infrastructure.6 Yet the guidance does not discuss the use of IVIG for MG in patients who are already symptomatic from COVID-19 or for patients recovering from the clinical disease or does it discuss a possible compounding risk of thromboembolic events associated with IVIG and COVID-19.6,7 Our patient received his maintenance IVIG during his first admission without any worsening of symptoms or increased oxygen requirements. The day following our patient’s next scheduled IVIG infusion—while asymptomatic—he again developed respiratory symptoms; this could suggest that IVIG did not contribute to his second clinical course nor protect against.
CP is a treatment modality that has been used and studied in previous infectious outbreaks such as the first severe acute respiratory syndrome, and the H1N1 influenza virus.8 Current data on CP for COVID-19 are limited, but early descriptive studies have shown a benefit in improvement of symptoms 5 days sooner in those requiring supplemental oxygen, but no benefit for those requiring mechanical ventilation.9 Like patients that benefitted in these studies, our patient received CP early, 6 days after first testing positive and onset of symptoms. This patient’s reinfection or return of symptoms draws into question the hindrance or even prevention of long-term immunity from administration of CP.
COVID-19 presents many challenges when managing this patient’s coexisting MG, especially as the patient was already being treated with immunosuppressing therapies. The guidance does recommend continuation of standard MG therapies during hospitalizations, including immunosuppression medications such as MMF.6 Immunosuppression is associated with worsened severity of COVID-19 symptoms, although no relation exists to degree of immunosuppression and severity.7,10 To the best of our knowledge there has been no case report of reinfection or reactivation of COVID-19 associated with immunosuppressive agents used in the treatment of MG.
Our patient also was taking pyridostigmine for the treatment of his MG. There is no evidence this medication increases the risk of infection; but the cholinergic activity can increase bronchial secretions, which could theoretically worsen the COVID-19 respiratory symptoms.6,11 During both ICU admissions, our patient continued pyridostigmine use, observing complete return to baseline after discharge. Given the possible association with worsened respiratory outcomes after the second ICU admission, the balance between managing MG symptoms and COVID-19 symptoms needs further examination.
The patient was in MC during his third presentation to the ED. Although respiratory symptoms may be difficult to differentiate from COVID-19, the additional neurologic symptoms seen in this patient allowed for quick determination of the need for MC treatment. There are many potential etiologies contributing to the development of the MC presented here, and it was likely due to multifactorial precipitants. A common cause of MC is viral upper respiratory infections, further challenging the care of these patients during this pandemic.12 Many medications have been cited as causing a MC, 2 of which our patient received during admission for COVID-19: azithromycin and levoquin.12 Although the patient did not receive hydroxychloroquine, which was still being considered as an appropriate COVID-19 treatment at the time, it also is a drug known for precipitating MC and its use scrutinized in patients with MG.12
A key aspect to diagnosing and guiding therapies in myasthenic crisis in addition to the clinical symptoms of acute weakness is respiratory assessment through the nonaerosolizing NIF test.12 Our patient’s NIF measured < 30 cmH2O when in MC, while the reference range is < 75 cmH2O, and for mechanical ventilation is recommended at 20 cmH2O. Although the patient was maintaining O2 saturation > 95%, his NIF value was concerning, and preparations were made in case of precipitous decline. Compounding the NIF assessment in this patient is his history of left phrenic nerve palsy. Without a documented baseline NIF, results were limited in determining his diaphragm strength.13 Treatment for MC includes IVIG or plasmapheresis, since this patient had failed his maintenance therapy IVIG, plasmapheresis was coordinated for definitive therapy.
Conclusions
Federal facilities have seen an increase in the amount of respiratory complaints over the past months. Although COVID-19 is a concerning diagnosis, it is crucial to consider comorbidities in the diagnostic workup of each, even with a previous recent diagnosis of COVID-19. As treatment recommendations for COVID-19 continue to fluctuate coupled with the limitations and difficulties associated with MG patients, so too treatment and evaluation must be carefully considered at each presentation.
1. Gousseff M, Penot P, Gallay L, et al. Clinical recurrences of COVID-19 symptoms after recovery: viral relapse, reinfection or inflammatory rebound? J Infect. 2020;81(5):816-846. doi:10.1016/j.jinf.2020.06.073
2. Duggan NM, Ludy SM, Shannon BC, Reisner AT, Wilcox SR. Is novel coronavirus 2019 reinfection possible? Interpreting dynamic SARS-CoV-2 test results. Am J Emerg Med. 2021;39:256.e1-256.e3. doi:10.1016/j.ajem.2020.06.079
3. Li J, Zhang L, Liu B, Song D. Case report: viral shedding for 60 days in a woman with COVID-19. Am J Trop Med Hyg. 2020;102(6):1210-1213. doi:10.4269/ajtmh.20-0275
4. Beigel JH, Tomashek KM, Dodd LE. Remdesivir for the treatment of Covid-19 - preliminary report. Reply. N Engl J Med. 2020;383(10):994. doi:10.1056/NEJMc2022236
5. McCreary EK, Angus DC. Efficacy of remdesivir in COVID-19. JAMA. 2020;324(11):1041-1042. doi:10.1001/jama.2020.16337
6. International MG/COVID-19 Working Group; Jacob S, Muppidi S, Gordon A, et al. Guidance for the management of myasthenia gravis (MG) and Lambert-Eaton myasthenic syndrome (LEMS) during the COVID-19 pandemic. J Neurol Sci. 2020;412:116803. doi:10.1016/j.jns.2020.116803
7. Anand P, Slama MCC, Kaku M, et al. COVID-19 in patients with myasthenia gravis. Muscle Nerve. 2020;62(2):254-258. doi:10.1002/mus.26918
8. Wooding DJ, Bach H. Treatment of COVID-19 with convalescent plasma: lessons from past coronavirus outbreaks. Clin Microbiol Infect. 2020;26(10):1436-1446. doi:10.1016/j.cmi.2020.08.005
9. Salazar E, Perez KK, Ashraf M, et al. Treatment of coronavirus disease 2019 (covid-19) patients with convalescent plasma. Am J Pathol. 2020;190(8):1680-1690. doi:10.1016/j.ajpath.2020.05.014
10. Ryan C, Minc A, Caceres J, et al. Predicting severe outcomes in Covid-19 related illness using only patient demographics, comorbidities and symptoms [published online ahead of print, 2020 Sep 9]. Am J Emerg Med. 2020;S0735-6757(20)30809-3. doi:10.1016/j.ajem.2020.09.017
11. Singh S, Govindarajan R. COVID-19 and generalized myasthenia gravis exacerbation: a case report. Clin Neurol Neurosurg. 2020;196:106045. doi:10.1016/j.clineuro.2020.106045
12. Wendell LC, Levine JM. Myasthenic crisis. Neurohospitalist. 2011;1(1):16-22. doi:10.1177/1941875210382918
13. Dubé BP, Dres M. Diaphragm dysfunction: diagnostic approaches and management strategies. J Clin Med. 2016;5(12):113. Published 2016 Dec 5. doi:10.3390/jcm5120113
A patient with myasthenia gravis who survived 2 COVID-19 infections required plasmapheresis to recover from an acute crisis.
A patient with myasthenia gravis who survived 2 COVID-19 infections required plasmapheresis to recover from an acute crisis.
COVID-19 is still in the early stages of understanding, although it is known to be complicated by individual patient comorbidities. The management and treatment of COVID-19 continues to quickly evolve as more is discovered regarding the virus. Multiple treatments have been preliminarily tested and used under a Food and Drug Administration emergency use authorization (EUA) determination. The long-term success of these therapies, however, is yet to be determined. Additionally, if a patient has a second clinical presentation for COVID-19, it is not known whether this represents latency with subsequent reactivation from the previous infection or a second de novo infection. The uncertainty calls into question the duration of immunity, if any, following a primary infection.
COVID-19 management becomes more complicated when patients have complex medical conditions, such as myasthenia gravis (MG). This autoimmune neuromuscular disorder can present with varying weakness, and many patients are on immunomodulator medications. The weakness can worsen into a myasthenic crisis (MC), resulting in profound weakness of the respiratory muscles. Therefore, patients with MG are at increased risk for COVID-19 and may have a more complicated course when infected.
Our patient with MG presented for severe COVID-19 symptoms twice and later developed MC. He received 2 treatment modalities available under an EUA (remdesivir and convalescent plasma) for COVID-19, resulting in symptom resolution and a negative polymerize chain reaction (PCR) test result for the virus. However, after receiving his typical maintenance therapy of IV immunoglobulin (IVIG) for his MG, he again developed symptoms consistent with COVID-19 and tested positive. After recovering from the second episode of COVID-19, the patient went into MC requiring plasmapheresis.
Case Presentation
A 56-year-old male, US Army veteran presented to Carl R. Darnall Army Medical Center emergency department (ED) 6 days after testing positive for COVID-19, with worsening sputum, cough, congestion, dyspnea, and fever. Due to his MG, the patient had a home oxygen monitor and reported that his oxygenation saturation dropped below 90% with minimal exertion. His medical history was significant for MG, status postthymectomy and radiation treatment, left hemidiaphragm paralysis secondary to phrenic nerve injury, and corticosteroid-induced insulin-dependent diabetes mellitus. His current home medications included pyridostigmine 60 mg 3 times a day, mycophenolate (MMF) 1500 mg twice daily, IV immunoglobulin (IVIG) every 3 weeks, insulin aspart up to 16 U per meal, insulin glargine 30 U twice a day, dulaglutide 0.75 mg every week, and metformin 1000 mg twice daily.
On initial examination, the patient’s heart rate (HR) was 111 beats/min, respiratory rate (RR), 22 breaths/min, blood pressure (BP), 138/88 mm Hg, temperature, 100.9 oF, and his initial pulse oximetry, 91% on room air. On physical examination, the patient was tachypneic, though without other signs of respiratory distress. Lung auscultation revealed no adventitial lung sounds. His cardiac examination was notable only for tachycardia. His neurologic examination demonstrated intact cranial nerves, with 5 out of 5 (scale 1 to 5) strength throughout the upper and lower extremities, sensation was intact to light touch, and he had normal cerebellar function. The rest of the examination was normal.
Initial laboratory investigation was notable for a white blood cell count of 14.15x103 cells/mcL with 84% neutrophils, and 6% lymphocytes. Additional tests revealed a C-reactive protein (CRP) level, 17.97 mg/dL (reference range, 0-0.5 mg/dL), ferritin level, 647 ng/mL (reference range, 22-274 ng/mL), d-dimer, 0.64 mcg/mL (reference range, 0-0.47mcg/mL), and a repeated positive COVID-19 PCR test. A portable chest X-ray showed bibasilar opacities (Figure 1).
The patient was diagnosed with COVID-19 and admitted to the intensive care unit (ICU). In the ICU, the patient received 1 U of convalescent plasma (CP) and started on a course of IV remdesivir 100 mg/d consistent with the EUA. He also received a 5-day course of ceftriaxone and azithromycin for possible community acquired pneumonia (CAP). As part of the patient’s MG maintenance medications, he received IVIG 4 g while in the ICU. Throughout his ICU stay, he required supplemental nasal cannula oxygenation to maintain his oxygen saturation > 93%. After 8 days in the ICU, his oxygen requirements decreased, and the patient was transferred out of the ICU and remdesivir was discontinued. On hospital day 10, a repeat COVID-19 PCR test was negative, inflammatory markers returned to within normal limits, and a repeat chest X-ray showed improvement from admission (Figure 2). Having recovered significantly, he was discharged home.
Three weeks later, the patient again presented to the MTF with 3 days of dyspnea, cough, fever, nausea, and vomiting. One day before symptom onset, he had received his maintenance IVIG infusion. The patient reported that his home oxygen saturation was 82% with minimal exertion. On ED presentation his HR was 107 beats/min, RR, 28 breaths/min, temperature, 98.1 oF, BP 118/71 mm Hg, and oxygen saturation, 92% on 2L nasal cannula. His examination was most notable for tachypnea with accessory muscle use. At this time, his neurologic examination was unchanged from prior admission with grossly intact cranial nerves and symmetric 5 of 5 motor strength in all extremities.
At this second ED visit, laboratory results demonstrated a CRP of 3.44 mg/dL, ferritin 2019 ng/mL, d-dimer, 3.39 mcg/mL, and a positive COVID-19 PCR result. His chest X-ray demonstrated new peripheral opacities compared with the X-ray at discharge (Figure 3). He required ICU admission again for his COVID-19 symptoms.
During his ICU course he continued to require supplemental oxygen by nasal cannula, though never required intubation. This second admission, he was again treated empirically for CAP with levofloxacin 750 mg daily for 5 days. He was discharged after 14 days with symptom resolution and down trending of inflammatory markers, though he was not retested for COVID-19.
Four days after his second discharge, he presented to the ED for a third time with diffuse weakness, dysphagia, and dysarthria of 1 day. His HR was 87/beats/min; RR, 17 breaths/min; temperature, 98.7 oF; BP, 144/81 mm Hg; and oxygen saturation, 98% on room air. His examination was significant for slurred speech, bilateral ptosis, 3 of 5 strength in bilateral finger flexion/abduction, wrist extension, knee and ankle flexion/extension; 4 of 5 strength in bilateral proximal muscle testing of deltoid, and hip; normal sensation, cerebellar function and reflexes. His negative inspiratory force (NIF) maximal effort was −30 cmH2O. He was determined to be in MC without evidence of COIVD-19 symptoms, and laboratory results were within normal limits, including a negative COVID-19 PCR. As he received IVIG as maintenance therapy, plasmapheresis was recommended to treat his MC, which required transfer to an outside civilian facility.
At the outside hospital, the patient underwent 5 rounds of plasmapheresis over 10 days. By the third treatment his strength had returned with resolution of the bulbar symptoms and no supplemental oxygen requirements. The patient was discharged and continued his original dosages of MMF and pyridostigmine. At 3 months, he remained asymptomatic from a COVID-19 standpoint and stable from a MG standpoint.
Discussion
Reinfection with the COVID-19 has been continuously debated with alternative explanations suggested for a positive test after a previous negative PCR test in the setting of symptom resolution.1,2 Proposed causes include dynamic PCR results due to prolonged viral shedding and inaccurate or poorly sensitive tests. The repeat positive cases in these scenarios, however, occurred in asymptomatic patients.1,2 COVID-19 shedding averages 20 to 22 days after symptom onset but has been seen up to 36 days after symptom resolution.2,3 This would suggest that fluctuating results during the immediate postsymptom period may be due to variations in viral shedding load and or sampling error—especially in asymptomatic patients. On the other hand, patients who experience return of symptoms days to weeks after previous convalescence leave clinicians wondering whether this represents clinical latency with reactivation or COVID-19 reinfection. A separate case of initial COVID-19 in a patient that had subsequent clinical recovery with a negative PCR developed recurrent respiratory symptoms and had a positive PCR test only 10 days later, further highlighting the reinfection vs reactivation issue of COVID-19.2 Further understanding of this issue may have implications on the extent of natural immunity following primary infection; potential vaccine dosage schedules; and global public health policies.
Although reactivation may be plausible given his immunomodulatory therapy, our patient’s second COVID-19 symptoms started 40 days after the initial symptoms, and 26 days after the initial course resolution; previous cases of return of severe symptoms occurred between 3 and 6 days.1 Given our patient’s time course between resolution and return of symptoms, if latency is the mechanism at play, this case demonstrates an exceptionally longer latency period than the ones that have been reported. Additionally, if latency is an issue in COVID-19, using remdesivir as a treatment further complicates the understanding of this disease.
Remdesivir, a nucleoside analogue antiviral, was shown to benefit recovery in patients with severe symptoms in the Adaptive COVID-19 Treatment Trial-1 study.4 Our patient had originally been placed on a 10-day course; however, on treatment day 8, his symptoms resolved and the remdesivir was discontinued. This is a similar finding to half the patients in the 10-day arm of the study by McCreary and colleagues.5 Although our patient was asymptomatic 4 weeks after the start of remdesivir, consistent with the majority of patients in the McCreary 10-day study arm, further comparison of the presented patient is limited due to study length and follow-up considerations.5 No previous data exist on reactivation, reinfection, or long-term mortality after being treated with remdesivir for COVID-19 infection.
IVIG is being studied in the treatment of COVID-19 and bears consideration as it relates to our patient. There is no evidence that IVIG used in the treatment of autoimmune diseases increases the risk of infection compared with that of other medications used in the treatment of such diseases. Furthermore, the current guidance from the MG expert panel does not suggest that IVIG increases the risk of contracting COVID-19 aside from the risks of exposure to hospital infrastructure.6 Yet the guidance does not discuss the use of IVIG for MG in patients who are already symptomatic from COVID-19 or for patients recovering from the clinical disease or does it discuss a possible compounding risk of thromboembolic events associated with IVIG and COVID-19.6,7 Our patient received his maintenance IVIG during his first admission without any worsening of symptoms or increased oxygen requirements. The day following our patient’s next scheduled IVIG infusion—while asymptomatic—he again developed respiratory symptoms; this could suggest that IVIG did not contribute to his second clinical course nor protect against.
CP is a treatment modality that has been used and studied in previous infectious outbreaks such as the first severe acute respiratory syndrome, and the H1N1 influenza virus.8 Current data on CP for COVID-19 are limited, but early descriptive studies have shown a benefit in improvement of symptoms 5 days sooner in those requiring supplemental oxygen, but no benefit for those requiring mechanical ventilation.9 Like patients that benefitted in these studies, our patient received CP early, 6 days after first testing positive and onset of symptoms. This patient’s reinfection or return of symptoms draws into question the hindrance or even prevention of long-term immunity from administration of CP.
COVID-19 presents many challenges when managing this patient’s coexisting MG, especially as the patient was already being treated with immunosuppressing therapies. The guidance does recommend continuation of standard MG therapies during hospitalizations, including immunosuppression medications such as MMF.6 Immunosuppression is associated with worsened severity of COVID-19 symptoms, although no relation exists to degree of immunosuppression and severity.7,10 To the best of our knowledge there has been no case report of reinfection or reactivation of COVID-19 associated with immunosuppressive agents used in the treatment of MG.
Our patient also was taking pyridostigmine for the treatment of his MG. There is no evidence this medication increases the risk of infection; but the cholinergic activity can increase bronchial secretions, which could theoretically worsen the COVID-19 respiratory symptoms.6,11 During both ICU admissions, our patient continued pyridostigmine use, observing complete return to baseline after discharge. Given the possible association with worsened respiratory outcomes after the second ICU admission, the balance between managing MG symptoms and COVID-19 symptoms needs further examination.
The patient was in MC during his third presentation to the ED. Although respiratory symptoms may be difficult to differentiate from COVID-19, the additional neurologic symptoms seen in this patient allowed for quick determination of the need for MC treatment. There are many potential etiologies contributing to the development of the MC presented here, and it was likely due to multifactorial precipitants. A common cause of MC is viral upper respiratory infections, further challenging the care of these patients during this pandemic.12 Many medications have been cited as causing a MC, 2 of which our patient received during admission for COVID-19: azithromycin and levoquin.12 Although the patient did not receive hydroxychloroquine, which was still being considered as an appropriate COVID-19 treatment at the time, it also is a drug known for precipitating MC and its use scrutinized in patients with MG.12
A key aspect to diagnosing and guiding therapies in myasthenic crisis in addition to the clinical symptoms of acute weakness is respiratory assessment through the nonaerosolizing NIF test.12 Our patient’s NIF measured < 30 cmH2O when in MC, while the reference range is < 75 cmH2O, and for mechanical ventilation is recommended at 20 cmH2O. Although the patient was maintaining O2 saturation > 95%, his NIF value was concerning, and preparations were made in case of precipitous decline. Compounding the NIF assessment in this patient is his history of left phrenic nerve palsy. Without a documented baseline NIF, results were limited in determining his diaphragm strength.13 Treatment for MC includes IVIG or plasmapheresis, since this patient had failed his maintenance therapy IVIG, plasmapheresis was coordinated for definitive therapy.
Conclusions
Federal facilities have seen an increase in the amount of respiratory complaints over the past months. Although COVID-19 is a concerning diagnosis, it is crucial to consider comorbidities in the diagnostic workup of each, even with a previous recent diagnosis of COVID-19. As treatment recommendations for COVID-19 continue to fluctuate coupled with the limitations and difficulties associated with MG patients, so too treatment and evaluation must be carefully considered at each presentation.
COVID-19 is still in the early stages of understanding, although it is known to be complicated by individual patient comorbidities. The management and treatment of COVID-19 continues to quickly evolve as more is discovered regarding the virus. Multiple treatments have been preliminarily tested and used under a Food and Drug Administration emergency use authorization (EUA) determination. The long-term success of these therapies, however, is yet to be determined. Additionally, if a patient has a second clinical presentation for COVID-19, it is not known whether this represents latency with subsequent reactivation from the previous infection or a second de novo infection. The uncertainty calls into question the duration of immunity, if any, following a primary infection.
COVID-19 management becomes more complicated when patients have complex medical conditions, such as myasthenia gravis (MG). This autoimmune neuromuscular disorder can present with varying weakness, and many patients are on immunomodulator medications. The weakness can worsen into a myasthenic crisis (MC), resulting in profound weakness of the respiratory muscles. Therefore, patients with MG are at increased risk for COVID-19 and may have a more complicated course when infected.
Our patient with MG presented for severe COVID-19 symptoms twice and later developed MC. He received 2 treatment modalities available under an EUA (remdesivir and convalescent plasma) for COVID-19, resulting in symptom resolution and a negative polymerize chain reaction (PCR) test result for the virus. However, after receiving his typical maintenance therapy of IV immunoglobulin (IVIG) for his MG, he again developed symptoms consistent with COVID-19 and tested positive. After recovering from the second episode of COVID-19, the patient went into MC requiring plasmapheresis.
Case Presentation
A 56-year-old male, US Army veteran presented to Carl R. Darnall Army Medical Center emergency department (ED) 6 days after testing positive for COVID-19, with worsening sputum, cough, congestion, dyspnea, and fever. Due to his MG, the patient had a home oxygen monitor and reported that his oxygenation saturation dropped below 90% with minimal exertion. His medical history was significant for MG, status postthymectomy and radiation treatment, left hemidiaphragm paralysis secondary to phrenic nerve injury, and corticosteroid-induced insulin-dependent diabetes mellitus. His current home medications included pyridostigmine 60 mg 3 times a day, mycophenolate (MMF) 1500 mg twice daily, IV immunoglobulin (IVIG) every 3 weeks, insulin aspart up to 16 U per meal, insulin glargine 30 U twice a day, dulaglutide 0.75 mg every week, and metformin 1000 mg twice daily.
On initial examination, the patient’s heart rate (HR) was 111 beats/min, respiratory rate (RR), 22 breaths/min, blood pressure (BP), 138/88 mm Hg, temperature, 100.9 oF, and his initial pulse oximetry, 91% on room air. On physical examination, the patient was tachypneic, though without other signs of respiratory distress. Lung auscultation revealed no adventitial lung sounds. His cardiac examination was notable only for tachycardia. His neurologic examination demonstrated intact cranial nerves, with 5 out of 5 (scale 1 to 5) strength throughout the upper and lower extremities, sensation was intact to light touch, and he had normal cerebellar function. The rest of the examination was normal.
Initial laboratory investigation was notable for a white blood cell count of 14.15x103 cells/mcL with 84% neutrophils, and 6% lymphocytes. Additional tests revealed a C-reactive protein (CRP) level, 17.97 mg/dL (reference range, 0-0.5 mg/dL), ferritin level, 647 ng/mL (reference range, 22-274 ng/mL), d-dimer, 0.64 mcg/mL (reference range, 0-0.47mcg/mL), and a repeated positive COVID-19 PCR test. A portable chest X-ray showed bibasilar opacities (Figure 1).
The patient was diagnosed with COVID-19 and admitted to the intensive care unit (ICU). In the ICU, the patient received 1 U of convalescent plasma (CP) and started on a course of IV remdesivir 100 mg/d consistent with the EUA. He also received a 5-day course of ceftriaxone and azithromycin for possible community acquired pneumonia (CAP). As part of the patient’s MG maintenance medications, he received IVIG 4 g while in the ICU. Throughout his ICU stay, he required supplemental nasal cannula oxygenation to maintain his oxygen saturation > 93%. After 8 days in the ICU, his oxygen requirements decreased, and the patient was transferred out of the ICU and remdesivir was discontinued. On hospital day 10, a repeat COVID-19 PCR test was negative, inflammatory markers returned to within normal limits, and a repeat chest X-ray showed improvement from admission (Figure 2). Having recovered significantly, he was discharged home.
Three weeks later, the patient again presented to the MTF with 3 days of dyspnea, cough, fever, nausea, and vomiting. One day before symptom onset, he had received his maintenance IVIG infusion. The patient reported that his home oxygen saturation was 82% with minimal exertion. On ED presentation his HR was 107 beats/min, RR, 28 breaths/min, temperature, 98.1 oF, BP 118/71 mm Hg, and oxygen saturation, 92% on 2L nasal cannula. His examination was most notable for tachypnea with accessory muscle use. At this time, his neurologic examination was unchanged from prior admission with grossly intact cranial nerves and symmetric 5 of 5 motor strength in all extremities.
At this second ED visit, laboratory results demonstrated a CRP of 3.44 mg/dL, ferritin 2019 ng/mL, d-dimer, 3.39 mcg/mL, and a positive COVID-19 PCR result. His chest X-ray demonstrated new peripheral opacities compared with the X-ray at discharge (Figure 3). He required ICU admission again for his COVID-19 symptoms.
During his ICU course he continued to require supplemental oxygen by nasal cannula, though never required intubation. This second admission, he was again treated empirically for CAP with levofloxacin 750 mg daily for 5 days. He was discharged after 14 days with symptom resolution and down trending of inflammatory markers, though he was not retested for COVID-19.
Four days after his second discharge, he presented to the ED for a third time with diffuse weakness, dysphagia, and dysarthria of 1 day. His HR was 87/beats/min; RR, 17 breaths/min; temperature, 98.7 oF; BP, 144/81 mm Hg; and oxygen saturation, 98% on room air. His examination was significant for slurred speech, bilateral ptosis, 3 of 5 strength in bilateral finger flexion/abduction, wrist extension, knee and ankle flexion/extension; 4 of 5 strength in bilateral proximal muscle testing of deltoid, and hip; normal sensation, cerebellar function and reflexes. His negative inspiratory force (NIF) maximal effort was −30 cmH2O. He was determined to be in MC without evidence of COIVD-19 symptoms, and laboratory results were within normal limits, including a negative COVID-19 PCR. As he received IVIG as maintenance therapy, plasmapheresis was recommended to treat his MC, which required transfer to an outside civilian facility.
At the outside hospital, the patient underwent 5 rounds of plasmapheresis over 10 days. By the third treatment his strength had returned with resolution of the bulbar symptoms and no supplemental oxygen requirements. The patient was discharged and continued his original dosages of MMF and pyridostigmine. At 3 months, he remained asymptomatic from a COVID-19 standpoint and stable from a MG standpoint.
Discussion
Reinfection with the COVID-19 has been continuously debated with alternative explanations suggested for a positive test after a previous negative PCR test in the setting of symptom resolution.1,2 Proposed causes include dynamic PCR results due to prolonged viral shedding and inaccurate or poorly sensitive tests. The repeat positive cases in these scenarios, however, occurred in asymptomatic patients.1,2 COVID-19 shedding averages 20 to 22 days after symptom onset but has been seen up to 36 days after symptom resolution.2,3 This would suggest that fluctuating results during the immediate postsymptom period may be due to variations in viral shedding load and or sampling error—especially in asymptomatic patients. On the other hand, patients who experience return of symptoms days to weeks after previous convalescence leave clinicians wondering whether this represents clinical latency with reactivation or COVID-19 reinfection. A separate case of initial COVID-19 in a patient that had subsequent clinical recovery with a negative PCR developed recurrent respiratory symptoms and had a positive PCR test only 10 days later, further highlighting the reinfection vs reactivation issue of COVID-19.2 Further understanding of this issue may have implications on the extent of natural immunity following primary infection; potential vaccine dosage schedules; and global public health policies.
Although reactivation may be plausible given his immunomodulatory therapy, our patient’s second COVID-19 symptoms started 40 days after the initial symptoms, and 26 days after the initial course resolution; previous cases of return of severe symptoms occurred between 3 and 6 days.1 Given our patient’s time course between resolution and return of symptoms, if latency is the mechanism at play, this case demonstrates an exceptionally longer latency period than the ones that have been reported. Additionally, if latency is an issue in COVID-19, using remdesivir as a treatment further complicates the understanding of this disease.
Remdesivir, a nucleoside analogue antiviral, was shown to benefit recovery in patients with severe symptoms in the Adaptive COVID-19 Treatment Trial-1 study.4 Our patient had originally been placed on a 10-day course; however, on treatment day 8, his symptoms resolved and the remdesivir was discontinued. This is a similar finding to half the patients in the 10-day arm of the study by McCreary and colleagues.5 Although our patient was asymptomatic 4 weeks after the start of remdesivir, consistent with the majority of patients in the McCreary 10-day study arm, further comparison of the presented patient is limited due to study length and follow-up considerations.5 No previous data exist on reactivation, reinfection, or long-term mortality after being treated with remdesivir for COVID-19 infection.
IVIG is being studied in the treatment of COVID-19 and bears consideration as it relates to our patient. There is no evidence that IVIG used in the treatment of autoimmune diseases increases the risk of infection compared with that of other medications used in the treatment of such diseases. Furthermore, the current guidance from the MG expert panel does not suggest that IVIG increases the risk of contracting COVID-19 aside from the risks of exposure to hospital infrastructure.6 Yet the guidance does not discuss the use of IVIG for MG in patients who are already symptomatic from COVID-19 or for patients recovering from the clinical disease or does it discuss a possible compounding risk of thromboembolic events associated with IVIG and COVID-19.6,7 Our patient received his maintenance IVIG during his first admission without any worsening of symptoms or increased oxygen requirements. The day following our patient’s next scheduled IVIG infusion—while asymptomatic—he again developed respiratory symptoms; this could suggest that IVIG did not contribute to his second clinical course nor protect against.
CP is a treatment modality that has been used and studied in previous infectious outbreaks such as the first severe acute respiratory syndrome, and the H1N1 influenza virus.8 Current data on CP for COVID-19 are limited, but early descriptive studies have shown a benefit in improvement of symptoms 5 days sooner in those requiring supplemental oxygen, but no benefit for those requiring mechanical ventilation.9 Like patients that benefitted in these studies, our patient received CP early, 6 days after first testing positive and onset of symptoms. This patient’s reinfection or return of symptoms draws into question the hindrance or even prevention of long-term immunity from administration of CP.
COVID-19 presents many challenges when managing this patient’s coexisting MG, especially as the patient was already being treated with immunosuppressing therapies. The guidance does recommend continuation of standard MG therapies during hospitalizations, including immunosuppression medications such as MMF.6 Immunosuppression is associated with worsened severity of COVID-19 symptoms, although no relation exists to degree of immunosuppression and severity.7,10 To the best of our knowledge there has been no case report of reinfection or reactivation of COVID-19 associated with immunosuppressive agents used in the treatment of MG.
Our patient also was taking pyridostigmine for the treatment of his MG. There is no evidence this medication increases the risk of infection; but the cholinergic activity can increase bronchial secretions, which could theoretically worsen the COVID-19 respiratory symptoms.6,11 During both ICU admissions, our patient continued pyridostigmine use, observing complete return to baseline after discharge. Given the possible association with worsened respiratory outcomes after the second ICU admission, the balance between managing MG symptoms and COVID-19 symptoms needs further examination.
The patient was in MC during his third presentation to the ED. Although respiratory symptoms may be difficult to differentiate from COVID-19, the additional neurologic symptoms seen in this patient allowed for quick determination of the need for MC treatment. There are many potential etiologies contributing to the development of the MC presented here, and it was likely due to multifactorial precipitants. A common cause of MC is viral upper respiratory infections, further challenging the care of these patients during this pandemic.12 Many medications have been cited as causing a MC, 2 of which our patient received during admission for COVID-19: azithromycin and levoquin.12 Although the patient did not receive hydroxychloroquine, which was still being considered as an appropriate COVID-19 treatment at the time, it also is a drug known for precipitating MC and its use scrutinized in patients with MG.12
A key aspect to diagnosing and guiding therapies in myasthenic crisis in addition to the clinical symptoms of acute weakness is respiratory assessment through the nonaerosolizing NIF test.12 Our patient’s NIF measured < 30 cmH2O when in MC, while the reference range is < 75 cmH2O, and for mechanical ventilation is recommended at 20 cmH2O. Although the patient was maintaining O2 saturation > 95%, his NIF value was concerning, and preparations were made in case of precipitous decline. Compounding the NIF assessment in this patient is his history of left phrenic nerve palsy. Without a documented baseline NIF, results were limited in determining his diaphragm strength.13 Treatment for MC includes IVIG or plasmapheresis, since this patient had failed his maintenance therapy IVIG, plasmapheresis was coordinated for definitive therapy.
Conclusions
Federal facilities have seen an increase in the amount of respiratory complaints over the past months. Although COVID-19 is a concerning diagnosis, it is crucial to consider comorbidities in the diagnostic workup of each, even with a previous recent diagnosis of COVID-19. As treatment recommendations for COVID-19 continue to fluctuate coupled with the limitations and difficulties associated with MG patients, so too treatment and evaluation must be carefully considered at each presentation.
1. Gousseff M, Penot P, Gallay L, et al. Clinical recurrences of COVID-19 symptoms after recovery: viral relapse, reinfection or inflammatory rebound? J Infect. 2020;81(5):816-846. doi:10.1016/j.jinf.2020.06.073
2. Duggan NM, Ludy SM, Shannon BC, Reisner AT, Wilcox SR. Is novel coronavirus 2019 reinfection possible? Interpreting dynamic SARS-CoV-2 test results. Am J Emerg Med. 2021;39:256.e1-256.e3. doi:10.1016/j.ajem.2020.06.079
3. Li J, Zhang L, Liu B, Song D. Case report: viral shedding for 60 days in a woman with COVID-19. Am J Trop Med Hyg. 2020;102(6):1210-1213. doi:10.4269/ajtmh.20-0275
4. Beigel JH, Tomashek KM, Dodd LE. Remdesivir for the treatment of Covid-19 - preliminary report. Reply. N Engl J Med. 2020;383(10):994. doi:10.1056/NEJMc2022236
5. McCreary EK, Angus DC. Efficacy of remdesivir in COVID-19. JAMA. 2020;324(11):1041-1042. doi:10.1001/jama.2020.16337
6. International MG/COVID-19 Working Group; Jacob S, Muppidi S, Gordon A, et al. Guidance for the management of myasthenia gravis (MG) and Lambert-Eaton myasthenic syndrome (LEMS) during the COVID-19 pandemic. J Neurol Sci. 2020;412:116803. doi:10.1016/j.jns.2020.116803
7. Anand P, Slama MCC, Kaku M, et al. COVID-19 in patients with myasthenia gravis. Muscle Nerve. 2020;62(2):254-258. doi:10.1002/mus.26918
8. Wooding DJ, Bach H. Treatment of COVID-19 with convalescent plasma: lessons from past coronavirus outbreaks. Clin Microbiol Infect. 2020;26(10):1436-1446. doi:10.1016/j.cmi.2020.08.005
9. Salazar E, Perez KK, Ashraf M, et al. Treatment of coronavirus disease 2019 (covid-19) patients with convalescent plasma. Am J Pathol. 2020;190(8):1680-1690. doi:10.1016/j.ajpath.2020.05.014
10. Ryan C, Minc A, Caceres J, et al. Predicting severe outcomes in Covid-19 related illness using only patient demographics, comorbidities and symptoms [published online ahead of print, 2020 Sep 9]. Am J Emerg Med. 2020;S0735-6757(20)30809-3. doi:10.1016/j.ajem.2020.09.017
11. Singh S, Govindarajan R. COVID-19 and generalized myasthenia gravis exacerbation: a case report. Clin Neurol Neurosurg. 2020;196:106045. doi:10.1016/j.clineuro.2020.106045
12. Wendell LC, Levine JM. Myasthenic crisis. Neurohospitalist. 2011;1(1):16-22. doi:10.1177/1941875210382918
13. Dubé BP, Dres M. Diaphragm dysfunction: diagnostic approaches and management strategies. J Clin Med. 2016;5(12):113. Published 2016 Dec 5. doi:10.3390/jcm5120113
1. Gousseff M, Penot P, Gallay L, et al. Clinical recurrences of COVID-19 symptoms after recovery: viral relapse, reinfection or inflammatory rebound? J Infect. 2020;81(5):816-846. doi:10.1016/j.jinf.2020.06.073
2. Duggan NM, Ludy SM, Shannon BC, Reisner AT, Wilcox SR. Is novel coronavirus 2019 reinfection possible? Interpreting dynamic SARS-CoV-2 test results. Am J Emerg Med. 2021;39:256.e1-256.e3. doi:10.1016/j.ajem.2020.06.079
3. Li J, Zhang L, Liu B, Song D. Case report: viral shedding for 60 days in a woman with COVID-19. Am J Trop Med Hyg. 2020;102(6):1210-1213. doi:10.4269/ajtmh.20-0275
4. Beigel JH, Tomashek KM, Dodd LE. Remdesivir for the treatment of Covid-19 - preliminary report. Reply. N Engl J Med. 2020;383(10):994. doi:10.1056/NEJMc2022236
5. McCreary EK, Angus DC. Efficacy of remdesivir in COVID-19. JAMA. 2020;324(11):1041-1042. doi:10.1001/jama.2020.16337
6. International MG/COVID-19 Working Group; Jacob S, Muppidi S, Gordon A, et al. Guidance for the management of myasthenia gravis (MG) and Lambert-Eaton myasthenic syndrome (LEMS) during the COVID-19 pandemic. J Neurol Sci. 2020;412:116803. doi:10.1016/j.jns.2020.116803
7. Anand P, Slama MCC, Kaku M, et al. COVID-19 in patients with myasthenia gravis. Muscle Nerve. 2020;62(2):254-258. doi:10.1002/mus.26918
8. Wooding DJ, Bach H. Treatment of COVID-19 with convalescent plasma: lessons from past coronavirus outbreaks. Clin Microbiol Infect. 2020;26(10):1436-1446. doi:10.1016/j.cmi.2020.08.005
9. Salazar E, Perez KK, Ashraf M, et al. Treatment of coronavirus disease 2019 (covid-19) patients with convalescent plasma. Am J Pathol. 2020;190(8):1680-1690. doi:10.1016/j.ajpath.2020.05.014
10. Ryan C, Minc A, Caceres J, et al. Predicting severe outcomes in Covid-19 related illness using only patient demographics, comorbidities and symptoms [published online ahead of print, 2020 Sep 9]. Am J Emerg Med. 2020;S0735-6757(20)30809-3. doi:10.1016/j.ajem.2020.09.017
11. Singh S, Govindarajan R. COVID-19 and generalized myasthenia gravis exacerbation: a case report. Clin Neurol Neurosurg. 2020;196:106045. doi:10.1016/j.clineuro.2020.106045
12. Wendell LC, Levine JM. Myasthenic crisis. Neurohospitalist. 2011;1(1):16-22. doi:10.1177/1941875210382918
13. Dubé BP, Dres M. Diaphragm dysfunction: diagnostic approaches and management strategies. J Clin Med. 2016;5(12):113. Published 2016 Dec 5. doi:10.3390/jcm5120113
27-year-old woman • postpartum seizures • PTSD • history of depression • Dx?
THE CASE
A 27-year-old woman presented to the family medicine clinic to establish care for a recent onset of seizures, for which she had previously been admitted, 4 months after delivering her first child. Her pregnancy was complicated by type 1 diabetes and poor glycemic control. Labor was induced at 37 weeks; however, vaginal delivery was impeded by arrest of dilation. An emergency cesarean section was performed under general anesthesia, resulting in a healthy newborn male.
Six weeks after giving birth, the patient was started on sertraline 50 mg/d for postpartum depression. Her history was significant for depression 8 years prior that was controlled with psychotherapy, and treated prior to coming to our clinic. She had not experienced any depressive symptoms during pregnancy.
Three months postpartum, she was hospitalized for recurrent syncopal episodes. They lasted about 2 minutes, with prodromal generalized weakness followed by loss of consciousness. There was no post-event confusion, tongue-biting, or incontinence. Physical exam, electroencephalogram (EEG), echocardiogram, and magnetic resonance imaging of the head and neck demonstrated no acute findings.
These episodes escalated in frequency weeks after they began, involving as many as 40 daily attacks, some of which lasted up to 45 minutes. During these events, the patient was nonresponsive but reported reliving the delivery of her child. Upon initial consultation with Neurology, no cause was found, and she was advised to wear a helmet, stop driving, and refrain from carrying her son. No antiepileptic medications were initiated because there were no EEG findings that supported seizure, and her mood had not improved, despite an increase in sertraline dosage, a switch to citalopram, and the addition of bupropion. She described anxiety, nightmares, and intrusive thoughts during psychotherapy sessions. Her psychiatrist gave her an additional diagnosis of posttraumatic stress disorder (PTSD) secondary to her delivery. The family medicine clinic assisted the patient and her family throughout her care by functioning as a home base for her.
Eight months following initial symptoms, repeat evaluation with a video-EEG revealed no evidence of EEG changes during seizure-like activity.
THE DIAGNOSIS
The patient was given a diagnosis of
DISCUSSION
With a prevalence of 5% to 10% and 20% to 40% in outpatient and inpatient epilepsy clinics respectively, PNES events have become of increasing interest to physicians.2 There are few cases of PNES in women during pregnancy reported in the literature.3,4 This is the first case report of PNES with postpartum onset.
Continue to: Epilepsy vs psychogenic nonepileptic seizures
Epilepsy vs psychogenic nonepileptic seizures
PNES episodes appear similar to epileptic seizures, but without a definitive neurobiologic source.2,3 However, recent literature suggests the root cause may be found in abnormalities in neurologic networks, such as dysfunction of frontal and parietal lobe connectivity and increased communication from emotional centers of the brain.2,5 There are no typical pathognomonic symptoms of PNES, leading to diagnostic difficulty.2 A definitive diagnosis may be made when a patient experiences seizures without EEG abnormalities.2 Further diagnostic brain imaging is unnecessary.
Trauma may be the underlying cause
A predominance of PNES in both women and young adults, with no definitive associated factors, has been reported in the literature.2 Studies suggest childhood sexual abuse, physical abuse, traumatic brain injury, and health-related trauma, such as distressing medical experiences and surgeries, may be risk factors, while depression, misdiagnosis, and mistreatment can heighten seizure activity.2,3
Treatment requires a multidisciplinary team
Effective management of PNES requires collaboration between the primary care physician, neurologist, psychiatrist, and psychotherapist, with an emphasis on evaluation and control of the underlying trigger(s).3 Randomized controlled trials have demonstrated the efficacy of cognitive behavioral therapy (CBT), supportive care, and patient education in reducing seizure frequency at the 6-month follow-up.3,6 Additional studies have reported the best prognostic factor in PNES management is patient employment of an internal locus of control—the patient’s belief that they control life events.7,8 Case series suggest electroconvulsive therapy (ECT) is an effective alternative mood stabilization and seizure reduction therapy when tolerated.9
Our patient tried several combinations of treatment to manage PNES and comorbid psychiatric conditions, including CBT, antidepressants, and anxiolytics. After about 5 treatment failures, she pursued ECT for treatment-resistant depression and PNES frequency reduction but failed to tolerate therapy. Currently, her PNES has been reduced to 1 to 2 weekly episodes with a 200 mg/d dose of lamotrigine as a mood stabilizer combined with CBT.
THE TAKEAWAY
Providers should investigate a patient’s history and psychologic disposition when the patient presents with seizure-like behavior without a neurobiologic source or with a negative video-EEG study. A history of depression, traumatic experience, PTSD, or other psychosocial triggers must be noted early to prevent a delay in treatment when PNES is part of the differential. Due to a delayed diagnosis of PNES in our patient, she went without full treatment for almost 12 months and experienced worsening episodes. The primary care physician plays an integral role in early identification and intervention through anticipatory guidance, initial work-up, and support for patients with suspected PNES (TABLE).

CORRESPONDENCE
Karim Hanna, MD, 13330 USF Laurel Drive, Tampa, FL; [email protected]
1. LaFrance WC Jr, Baker GA, Duncan R, et al. Minimum requirements for the diagnosis of psychogenic nonepileptic seizures: a staged approach: a report from the International League Against Epilepsy Nonepileptic Seizures Task Force. Epilepsia. 2013;54:2005-2018. doi: 10.1111/epi.12356
2. Asadi-Pooya AA, Sperling MR. Epidemiology of psychogenic nonepileptic seizures. Epilepsy Behav. 2015;46:60-65. doi: 10.1016/j.yebeh.2015.03.015
3. Devireddy VK, Sharma A. A case of psychogenic non-epileptic seizures, unresponsive type, in pregnancy. Prim Care Companion CNS Disord. 2014;16:PCC.13l01574. doi: 10.4088/PCC.13l01574
4. DeToledo JC, Lowe MR, Puig A. Nonepileptic seizures in pregnancy. Neurology. 2000;55:120-121. doi: 10.1212/wnl.55.1.120
5. Ding J-R, An D, Liao W, et al. Altered functional and structural connectivity networks in psychogenic non-epileptic seizures. PLoS One. 2013;8:e63850. doi: 10.1371/journal.pone.0063850
6. Goldstein LH, Chalder T, Chigwedere C, et al. Cognitive-behavioral therapy for psychogenic nonepileptic seizures: a pilot RCT. Neurology. 2010;74:1986-1994. doi: 0.1212/WNL.0b013e3181e39658
7. McLaughlin DP, Pachana NA, McFarland K. The impact of depression, seizure variables and locus of control on health related quality of life in a community dwelling sample of older adults. Seizure. 2010;19:232-236. doi: 10.1016/j.seizure.2010.02.008
8. Duncan R, Anderson J, Cullen B, et al. Predictors of 6-month and 3-year outcomes after psychological intervention for psychogenic non epileptic seizures. Seizure. 2016;36:22-26. doi: 10.1016/j.seizure.2015.12.016
9. Blumer D, Rice S, Adamolekun B. Electroconvulsive treatment for nonepileptic seizure disorders. Epilepsy Behav. 2009;15:382-387. doi: 10.1016/j.yebeh.2009.05.004
THE CASE
A 27-year-old woman presented to the family medicine clinic to establish care for a recent onset of seizures, for which she had previously been admitted, 4 months after delivering her first child. Her pregnancy was complicated by type 1 diabetes and poor glycemic control. Labor was induced at 37 weeks; however, vaginal delivery was impeded by arrest of dilation. An emergency cesarean section was performed under general anesthesia, resulting in a healthy newborn male.
Six weeks after giving birth, the patient was started on sertraline 50 mg/d for postpartum depression. Her history was significant for depression 8 years prior that was controlled with psychotherapy, and treated prior to coming to our clinic. She had not experienced any depressive symptoms during pregnancy.
Three months postpartum, she was hospitalized for recurrent syncopal episodes. They lasted about 2 minutes, with prodromal generalized weakness followed by loss of consciousness. There was no post-event confusion, tongue-biting, or incontinence. Physical exam, electroencephalogram (EEG), echocardiogram, and magnetic resonance imaging of the head and neck demonstrated no acute findings.
These episodes escalated in frequency weeks after they began, involving as many as 40 daily attacks, some of which lasted up to 45 minutes. During these events, the patient was nonresponsive but reported reliving the delivery of her child. Upon initial consultation with Neurology, no cause was found, and she was advised to wear a helmet, stop driving, and refrain from carrying her son. No antiepileptic medications were initiated because there were no EEG findings that supported seizure, and her mood had not improved, despite an increase in sertraline dosage, a switch to citalopram, and the addition of bupropion. She described anxiety, nightmares, and intrusive thoughts during psychotherapy sessions. Her psychiatrist gave her an additional diagnosis of posttraumatic stress disorder (PTSD) secondary to her delivery. The family medicine clinic assisted the patient and her family throughout her care by functioning as a home base for her.
Eight months following initial symptoms, repeat evaluation with a video-EEG revealed no evidence of EEG changes during seizure-like activity.
THE DIAGNOSIS
The patient was given a diagnosis of
DISCUSSION
With a prevalence of 5% to 10% and 20% to 40% in outpatient and inpatient epilepsy clinics respectively, PNES events have become of increasing interest to physicians.2 There are few cases of PNES in women during pregnancy reported in the literature.3,4 This is the first case report of PNES with postpartum onset.
Continue to: Epilepsy vs psychogenic nonepileptic seizures
Epilepsy vs psychogenic nonepileptic seizures
PNES episodes appear similar to epileptic seizures, but without a definitive neurobiologic source.2,3 However, recent literature suggests the root cause may be found in abnormalities in neurologic networks, such as dysfunction of frontal and parietal lobe connectivity and increased communication from emotional centers of the brain.2,5 There are no typical pathognomonic symptoms of PNES, leading to diagnostic difficulty.2 A definitive diagnosis may be made when a patient experiences seizures without EEG abnormalities.2 Further diagnostic brain imaging is unnecessary.
Trauma may be the underlying cause
A predominance of PNES in both women and young adults, with no definitive associated factors, has been reported in the literature.2 Studies suggest childhood sexual abuse, physical abuse, traumatic brain injury, and health-related trauma, such as distressing medical experiences and surgeries, may be risk factors, while depression, misdiagnosis, and mistreatment can heighten seizure activity.2,3
Treatment requires a multidisciplinary team
Effective management of PNES requires collaboration between the primary care physician, neurologist, psychiatrist, and psychotherapist, with an emphasis on evaluation and control of the underlying trigger(s).3 Randomized controlled trials have demonstrated the efficacy of cognitive behavioral therapy (CBT), supportive care, and patient education in reducing seizure frequency at the 6-month follow-up.3,6 Additional studies have reported the best prognostic factor in PNES management is patient employment of an internal locus of control—the patient’s belief that they control life events.7,8 Case series suggest electroconvulsive therapy (ECT) is an effective alternative mood stabilization and seizure reduction therapy when tolerated.9
Our patient tried several combinations of treatment to manage PNES and comorbid psychiatric conditions, including CBT, antidepressants, and anxiolytics. After about 5 treatment failures, she pursued ECT for treatment-resistant depression and PNES frequency reduction but failed to tolerate therapy. Currently, her PNES has been reduced to 1 to 2 weekly episodes with a 200 mg/d dose of lamotrigine as a mood stabilizer combined with CBT.
THE TAKEAWAY
Providers should investigate a patient’s history and psychologic disposition when the patient presents with seizure-like behavior without a neurobiologic source or with a negative video-EEG study. A history of depression, traumatic experience, PTSD, or other psychosocial triggers must be noted early to prevent a delay in treatment when PNES is part of the differential. Due to a delayed diagnosis of PNES in our patient, she went without full treatment for almost 12 months and experienced worsening episodes. The primary care physician plays an integral role in early identification and intervention through anticipatory guidance, initial work-up, and support for patients with suspected PNES (TABLE).
CORRESPONDENCE
Karim Hanna, MD, 13330 USF Laurel Drive, Tampa, FL; [email protected]
THE CASE
A 27-year-old woman presented to the family medicine clinic to establish care for a recent onset of seizures, for which she had previously been admitted, 4 months after delivering her first child. Her pregnancy was complicated by type 1 diabetes and poor glycemic control. Labor was induced at 37 weeks; however, vaginal delivery was impeded by arrest of dilation. An emergency cesarean section was performed under general anesthesia, resulting in a healthy newborn male.
Six weeks after giving birth, the patient was started on sertraline 50 mg/d for postpartum depression. Her history was significant for depression 8 years prior that was controlled with psychotherapy, and treated prior to coming to our clinic. She had not experienced any depressive symptoms during pregnancy.
Three months postpartum, she was hospitalized for recurrent syncopal episodes. They lasted about 2 minutes, with prodromal generalized weakness followed by loss of consciousness. There was no post-event confusion, tongue-biting, or incontinence. Physical exam, electroencephalogram (EEG), echocardiogram, and magnetic resonance imaging of the head and neck demonstrated no acute findings.
These episodes escalated in frequency weeks after they began, involving as many as 40 daily attacks, some of which lasted up to 45 minutes. During these events, the patient was nonresponsive but reported reliving the delivery of her child. Upon initial consultation with Neurology, no cause was found, and she was advised to wear a helmet, stop driving, and refrain from carrying her son. No antiepileptic medications were initiated because there were no EEG findings that supported seizure, and her mood had not improved, despite an increase in sertraline dosage, a switch to citalopram, and the addition of bupropion. She described anxiety, nightmares, and intrusive thoughts during psychotherapy sessions. Her psychiatrist gave her an additional diagnosis of posttraumatic stress disorder (PTSD) secondary to her delivery. The family medicine clinic assisted the patient and her family throughout her care by functioning as a home base for her.
Eight months following initial symptoms, repeat evaluation with a video-EEG revealed no evidence of EEG changes during seizure-like activity.
THE DIAGNOSIS
The patient was given a diagnosis of
DISCUSSION
With a prevalence of 5% to 10% and 20% to 40% in outpatient and inpatient epilepsy clinics respectively, PNES events have become of increasing interest to physicians.2 There are few cases of PNES in women during pregnancy reported in the literature.3,4 This is the first case report of PNES with postpartum onset.
Continue to: Epilepsy vs psychogenic nonepileptic seizures
Epilepsy vs psychogenic nonepileptic seizures
PNES episodes appear similar to epileptic seizures, but without a definitive neurobiologic source.2,3 However, recent literature suggests the root cause may be found in abnormalities in neurologic networks, such as dysfunction of frontal and parietal lobe connectivity and increased communication from emotional centers of the brain.2,5 There are no typical pathognomonic symptoms of PNES, leading to diagnostic difficulty.2 A definitive diagnosis may be made when a patient experiences seizures without EEG abnormalities.2 Further diagnostic brain imaging is unnecessary.
Trauma may be the underlying cause
A predominance of PNES in both women and young adults, with no definitive associated factors, has been reported in the literature.2 Studies suggest childhood sexual abuse, physical abuse, traumatic brain injury, and health-related trauma, such as distressing medical experiences and surgeries, may be risk factors, while depression, misdiagnosis, and mistreatment can heighten seizure activity.2,3
Treatment requires a multidisciplinary team
Effective management of PNES requires collaboration between the primary care physician, neurologist, psychiatrist, and psychotherapist, with an emphasis on evaluation and control of the underlying trigger(s).3 Randomized controlled trials have demonstrated the efficacy of cognitive behavioral therapy (CBT), supportive care, and patient education in reducing seizure frequency at the 6-month follow-up.3,6 Additional studies have reported the best prognostic factor in PNES management is patient employment of an internal locus of control—the patient’s belief that they control life events.7,8 Case series suggest electroconvulsive therapy (ECT) is an effective alternative mood stabilization and seizure reduction therapy when tolerated.9
Our patient tried several combinations of treatment to manage PNES and comorbid psychiatric conditions, including CBT, antidepressants, and anxiolytics. After about 5 treatment failures, she pursued ECT for treatment-resistant depression and PNES frequency reduction but failed to tolerate therapy. Currently, her PNES has been reduced to 1 to 2 weekly episodes with a 200 mg/d dose of lamotrigine as a mood stabilizer combined with CBT.
THE TAKEAWAY
Providers should investigate a patient’s history and psychologic disposition when the patient presents with seizure-like behavior without a neurobiologic source or with a negative video-EEG study. A history of depression, traumatic experience, PTSD, or other psychosocial triggers must be noted early to prevent a delay in treatment when PNES is part of the differential. Due to a delayed diagnosis of PNES in our patient, she went without full treatment for almost 12 months and experienced worsening episodes. The primary care physician plays an integral role in early identification and intervention through anticipatory guidance, initial work-up, and support for patients with suspected PNES (TABLE).
CORRESPONDENCE
Karim Hanna, MD, 13330 USF Laurel Drive, Tampa, FL; [email protected]
1. LaFrance WC Jr, Baker GA, Duncan R, et al. Minimum requirements for the diagnosis of psychogenic nonepileptic seizures: a staged approach: a report from the International League Against Epilepsy Nonepileptic Seizures Task Force. Epilepsia. 2013;54:2005-2018. doi: 10.1111/epi.12356
2. Asadi-Pooya AA, Sperling MR. Epidemiology of psychogenic nonepileptic seizures. Epilepsy Behav. 2015;46:60-65. doi: 10.1016/j.yebeh.2015.03.015
3. Devireddy VK, Sharma A. A case of psychogenic non-epileptic seizures, unresponsive type, in pregnancy. Prim Care Companion CNS Disord. 2014;16:PCC.13l01574. doi: 10.4088/PCC.13l01574
4. DeToledo JC, Lowe MR, Puig A. Nonepileptic seizures in pregnancy. Neurology. 2000;55:120-121. doi: 10.1212/wnl.55.1.120
5. Ding J-R, An D, Liao W, et al. Altered functional and structural connectivity networks in psychogenic non-epileptic seizures. PLoS One. 2013;8:e63850. doi: 10.1371/journal.pone.0063850
6. Goldstein LH, Chalder T, Chigwedere C, et al. Cognitive-behavioral therapy for psychogenic nonepileptic seizures: a pilot RCT. Neurology. 2010;74:1986-1994. doi: 0.1212/WNL.0b013e3181e39658
7. McLaughlin DP, Pachana NA, McFarland K. The impact of depression, seizure variables and locus of control on health related quality of life in a community dwelling sample of older adults. Seizure. 2010;19:232-236. doi: 10.1016/j.seizure.2010.02.008
8. Duncan R, Anderson J, Cullen B, et al. Predictors of 6-month and 3-year outcomes after psychological intervention for psychogenic non epileptic seizures. Seizure. 2016;36:22-26. doi: 10.1016/j.seizure.2015.12.016
9. Blumer D, Rice S, Adamolekun B. Electroconvulsive treatment for nonepileptic seizure disorders. Epilepsy Behav. 2009;15:382-387. doi: 10.1016/j.yebeh.2009.05.004
1. LaFrance WC Jr, Baker GA, Duncan R, et al. Minimum requirements for the diagnosis of psychogenic nonepileptic seizures: a staged approach: a report from the International League Against Epilepsy Nonepileptic Seizures Task Force. Epilepsia. 2013;54:2005-2018. doi: 10.1111/epi.12356
2. Asadi-Pooya AA, Sperling MR. Epidemiology of psychogenic nonepileptic seizures. Epilepsy Behav. 2015;46:60-65. doi: 10.1016/j.yebeh.2015.03.015
3. Devireddy VK, Sharma A. A case of psychogenic non-epileptic seizures, unresponsive type, in pregnancy. Prim Care Companion CNS Disord. 2014;16:PCC.13l01574. doi: 10.4088/PCC.13l01574
4. DeToledo JC, Lowe MR, Puig A. Nonepileptic seizures in pregnancy. Neurology. 2000;55:120-121. doi: 10.1212/wnl.55.1.120
5. Ding J-R, An D, Liao W, et al. Altered functional and structural connectivity networks in psychogenic non-epileptic seizures. PLoS One. 2013;8:e63850. doi: 10.1371/journal.pone.0063850
6. Goldstein LH, Chalder T, Chigwedere C, et al. Cognitive-behavioral therapy for psychogenic nonepileptic seizures: a pilot RCT. Neurology. 2010;74:1986-1994. doi: 0.1212/WNL.0b013e3181e39658
7. McLaughlin DP, Pachana NA, McFarland K. The impact of depression, seizure variables and locus of control on health related quality of life in a community dwelling sample of older adults. Seizure. 2010;19:232-236. doi: 10.1016/j.seizure.2010.02.008
8. Duncan R, Anderson J, Cullen B, et al. Predictors of 6-month and 3-year outcomes after psychological intervention for psychogenic non epileptic seizures. Seizure. 2016;36:22-26. doi: 10.1016/j.seizure.2015.12.016
9. Blumer D, Rice S, Adamolekun B. Electroconvulsive treatment for nonepileptic seizure disorders. Epilepsy Behav. 2009;15:382-387. doi: 10.1016/j.yebeh.2009.05.004

Unexpected Complications: A Case of Rosacea Fulminans in Pregnancy
Rosacea fulminans (RF) is a rare facial dermatosis characterized by its fulminating course. 1 It presents with superficial and deep-seated papules, pustules, and nodules combined with an intense reddish or cyanotic erythema localized to the face. Furthermore, there is an absence of comedones and involvement of the chest or back. 2 Rosacea fulminans primarily affects women and often is, but not always, proceeded by seborrhea, chronic acne vulgaris, or rosacea. Although the etiology of RF remains unknown, immunologic, hormonal, and vascular factors have been implicated. 3 We report a case of RF in a pregnant patient with a history of mild acne as a teenager that was long ago resolved.
Case Report
A 32-year-old pregnant woman (10 weeks’ gestation) presented with a rapidly progressing inflammatory disorder of the face of 1 month’s duration. The lesions developed 3 weeks after beginning progesterone therapy (200 mg vaginal suppository) for infertility due to polycystic ovary syndrome. Despite discontinuing progesterone for the last month, the patient’s lesions had dramatically worsened (Figure 1). Empiric cephalosporin treatment prescribed by her primary care physician yielded no improvement. Physical examination at the current presentation revealed erythematous nodules and pustules all over the face, coalescing into large thick plaques on the patient’s right cheek and chin. Submental nodes were palpable and tender. Based on the initial clinical findings, acne conglobata secondary to progesterone therapy was considered. The patient was given intralesional triamcinolone (2.5 mg/cc) injections to all larger nodules and several blue light treatments.
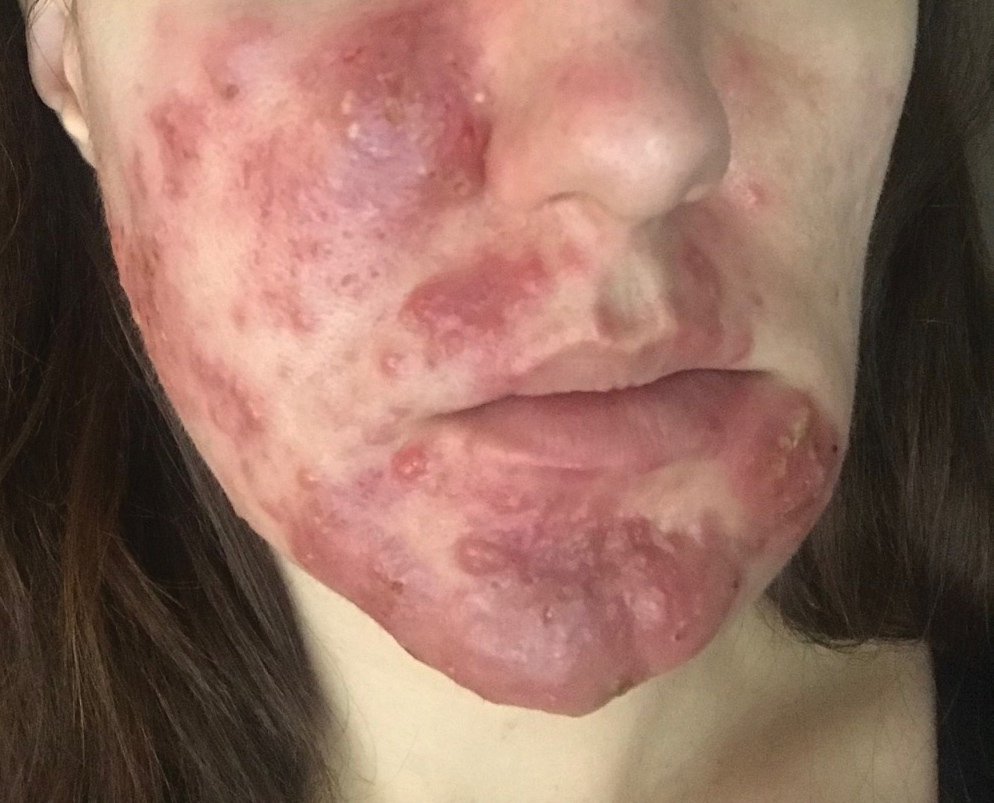
The injected areas had improved 5 days after the initial visit; however, the chin and right paranasal cheek developed even more nodules and papules coalescing into large plaques. After consulting the patient’s obstetrician, prednisone (20 mg once daily) was initiated. Three weeks later, the patient’s nodular lesions had improved, but there was a showering of more than 100 pustules and increased general erythema of the entire face (Figure 2). Crotamiton cream 10% (every day before noon), ivermectin cream 1% (every night at bedtime), and sodium sulfacetamide cleanser 10% once daily were added to the treatment plan.

At 16 weeks’ gestation, there was slight improvement; however, there was still erythema on the entire face with scattered pustules and multiple papules and nodules. Many small ice-pick scars were seen on the cheeks and forehead. No comedones were observed. A punch biopsy of an intact papule showed a prominent inflammatory infiltrate with granulomatous reaction and numerous neutrophils predominantly affecting hair follicles. Based on the clinical presentation and histopathology, a diagnosis of RF was made. Azithromycin (250 mg once daily) and metronidazole cream 0.75% twice daily were added. Two weeks later there were fewer nodules but many papules, edema, and intense erythema. The prednisone dosage was increased to 40 mg once daily. Two weeks later, the patient showed improvement with fewer lesions, less edema, and less erythema. The patient was instructed to finish the azithromycin course and discontinue use. At 28 weeks’ gestation, a prednisone taper was started with the intention to reduce the daily dose by delivery.
The patient delivered a healthy girl (birth weight, 1.985 kg) prematurely at 34 weeks’ gestation. At 2 months postpartum, the patient’s existing lesions continued to spontaneously improve; however, she still had numerous nodules and papules and continued to develop new lesions and form additional scars. Isotretinoin was instituted at 3 months postpartum upon cessation of nursing. Three months later (40 mg/d isotretinoin), the patient was nearly clear. At 8 months postpartum, isotretinoin was discontinued after a course of 150 mg/kg.
Comment
Rosacea fulminans initially was called pyoderma faciale but was later regarded as a severe form of rosacea and was renamed rosacea fulminans.2 According to a PubMed search of articles indexed for MEDLINE using the terms pregnancy and rosacea fulminans or pyoderma faciale, we identified 12 publications reporting 20 cases of RF associated with pregnancy (Table). Although there is no substantial evidence regarding the exact mechanism, these cases indicate that pregnancy can be an exacerbating or causative factor in the pathogenesis of RF.
In addition to pregnancy, RF has been associated with inflammatory bowel disease, thyroid and liver disease, erythema nodosum, and severe emotional trauma. However, no organism has been consistently isolated, and no evidence of family history has been reported.1 Histopathologic findings are dependent on the stage of disease. Massive infiltrates of neutrophils may be observed in early stages. In older lesions, infiltrates take the form of epithelioid cell granulomas.2
Treatment of RF during pregnancy is challenging. Early and aggressive treatment with retinoids, tetracycline antibiotics, antiandrogenic contraceptives, and dapsone is recommended in patients who are not pregnant; these therapies are all contraindicated in pregnancy. Topical steroids can be safely used; however, systemic steroids usually are required to control RF. The use of systemic steroids can only be justified if the risks for intrauterine growth retardation, maternal diabetes mellitus, and hypertension outweigh the benefits of treating this severe disfiguring skin condition.10 A study by Bakar et al13 indicated that azithromycin is an effective and safe alternative in the treatment of RF. It has a superior pharmacokinetic profile compared to other macrolides and does not pose increased risks for congenital malformation or miscarriage. Because of the concomitant use of both azithromycin and prednisone, it is not possible to determine which had the larger role in the patient’s improvement.
Isotretinoin therapy in our patient led to substantial improvement of RF. Time will tell if the response will be durable. Also unknown is the risk for recurrence with subsequent pregnancies, which has not been reported in the literature. Although it is difficult to confidently say that pregnancy was the inciting factor in this patient’s RF, this case certainly provides more evidence for a link between pregnancy and RF.
- Jarrett R, Gonsalves R, Anstey AV. Differing obstetric outcomes of rosacea fulminans in pregnancy: report of three cases with review of pathogenesis and management. Clin Exp Dermatol. 2010;35:888-891. doi:10.1111/j.1365-2230.2010.03846.x
- Ferahbas A, Utas S, Mistik S, et al. Rosacea fulminans in pregnancy: case report and review of the literature. Am J Clin Dermatol. 2006;7:141-144. doi:10.2165/00128071-200607020-00007
- Fuentelsaz V, Ara M, Corredera C, et al. Rosacea fulminans in pregnancy: successful treatment with azithromycin. Clin Exp Dermatol. 2011;36:674-676. doi:10.1111/j.1365-2230.2011.04042.x
- Garayar Cantero M, Garabito Solovera E, Aguado García Á, et al. Use of permethrin in the treatment of rosacea fulminans during pregnancy: one case report. Dermatol Ther. 2020;33:E13436. doi:10.1111/dth.13436
- Demir O, Tas IS, Gunay B, et al. A rare dermatologic disease in pregnancy: rosacea fulminans—case report and review of the literature. Open Access Maced J Med Sci. 2018;6:1438-1441. doi:10.3889/oamjms.2018.267
- Markou AG, Alessandrini V, Muray JM, et al. Rosacea fulminans during pregnancy. Clin Exp Obstet Gynecol. 2017;44:157-159.
- Haenen CCP, Kouwenhoven STP, van Doorn R. Rosacea fulminans in pregnancy [in Dutch]. Ned Tijdschr Geneeskd. 2015;159:A8334.
- de Morais e Silva FA, Bonassi M, Steiner D, et al. Rosacea fulminans in pregnancy with ocular perforation. J Dtsch Dermatol Ges. 2011;9:542-543. doi:10.1111/j.1610-0387.2011.07616.x
- Cisse M, Maruani A, Bré C. Rosacea fulminans in the early course of a pregnancy by in vitro fertilization with embryo transfer [in French]. Ann Dermatol Venereol. 2008;135:675-678. doi:10.1016/j.annder.2008.04.015
- Lewis VJ, Holme SA, Wright A, et al. Rosacea fulminans in pregnancy. Br J Dermatol. 2004;151:917-919. doi:10.1111/j.1365-2133.2004.06190.x
- Plewig G, Jansen T, Kligman AM. Pyoderma faciale. a review and report of 20 additional cases: is it rosacea? Arch Dermatol. 1992;128:1611-1617. doi:10.1001/archderm.128.12.1611
- Massa MC, Su WP. Pyoderma faciale: a clinical study of twenty-nine patients. J Am Acad Dermatol. 1982;6:84-91. doi:10.1016/s0190-9622(82)70008-8
- Bakar O, Demirçay Z, Gürbüz O. Therapeutic potential of azithromycin in rosacea. Int J Dermatol. 2004;43:151-154. doi:10.1111/j.1365-4632.2004.01958.x
Rosacea fulminans (RF) is a rare facial dermatosis characterized by its fulminating course. 1 It presents with superficial and deep-seated papules, pustules, and nodules combined with an intense reddish or cyanotic erythema localized to the face. Furthermore, there is an absence of comedones and involvement of the chest or back. 2 Rosacea fulminans primarily affects women and often is, but not always, proceeded by seborrhea, chronic acne vulgaris, or rosacea. Although the etiology of RF remains unknown, immunologic, hormonal, and vascular factors have been implicated. 3 We report a case of RF in a pregnant patient with a history of mild acne as a teenager that was long ago resolved.
Case Report
A 32-year-old pregnant woman (10 weeks’ gestation) presented with a rapidly progressing inflammatory disorder of the face of 1 month’s duration. The lesions developed 3 weeks after beginning progesterone therapy (200 mg vaginal suppository) for infertility due to polycystic ovary syndrome. Despite discontinuing progesterone for the last month, the patient’s lesions had dramatically worsened (Figure 1). Empiric cephalosporin treatment prescribed by her primary care physician yielded no improvement. Physical examination at the current presentation revealed erythematous nodules and pustules all over the face, coalescing into large thick plaques on the patient’s right cheek and chin. Submental nodes were palpable and tender. Based on the initial clinical findings, acne conglobata secondary to progesterone therapy was considered. The patient was given intralesional triamcinolone (2.5 mg/cc) injections to all larger nodules and several blue light treatments.
The injected areas had improved 5 days after the initial visit; however, the chin and right paranasal cheek developed even more nodules and papules coalescing into large plaques. After consulting the patient’s obstetrician, prednisone (20 mg once daily) was initiated. Three weeks later, the patient’s nodular lesions had improved, but there was a showering of more than 100 pustules and increased general erythema of the entire face (Figure 2). Crotamiton cream 10% (every day before noon), ivermectin cream 1% (every night at bedtime), and sodium sulfacetamide cleanser 10% once daily were added to the treatment plan.
At 16 weeks’ gestation, there was slight improvement; however, there was still erythema on the entire face with scattered pustules and multiple papules and nodules. Many small ice-pick scars were seen on the cheeks and forehead. No comedones were observed. A punch biopsy of an intact papule showed a prominent inflammatory infiltrate with granulomatous reaction and numerous neutrophils predominantly affecting hair follicles. Based on the clinical presentation and histopathology, a diagnosis of RF was made. Azithromycin (250 mg once daily) and metronidazole cream 0.75% twice daily were added. Two weeks later there were fewer nodules but many papules, edema, and intense erythema. The prednisone dosage was increased to 40 mg once daily. Two weeks later, the patient showed improvement with fewer lesions, less edema, and less erythema. The patient was instructed to finish the azithromycin course and discontinue use. At 28 weeks’ gestation, a prednisone taper was started with the intention to reduce the daily dose by delivery.
The patient delivered a healthy girl (birth weight, 1.985 kg) prematurely at 34 weeks’ gestation. At 2 months postpartum, the patient’s existing lesions continued to spontaneously improve; however, she still had numerous nodules and papules and continued to develop new lesions and form additional scars. Isotretinoin was instituted at 3 months postpartum upon cessation of nursing. Three months later (40 mg/d isotretinoin), the patient was nearly clear. At 8 months postpartum, isotretinoin was discontinued after a course of 150 mg/kg.
Comment
Rosacea fulminans initially was called pyoderma faciale but was later regarded as a severe form of rosacea and was renamed rosacea fulminans.2 According to a PubMed search of articles indexed for MEDLINE using the terms pregnancy and rosacea fulminans or pyoderma faciale, we identified 12 publications reporting 20 cases of RF associated with pregnancy (Table). Although there is no substantial evidence regarding the exact mechanism, these cases indicate that pregnancy can be an exacerbating or causative factor in the pathogenesis of RF.
In addition to pregnancy, RF has been associated with inflammatory bowel disease, thyroid and liver disease, erythema nodosum, and severe emotional trauma. However, no organism has been consistently isolated, and no evidence of family history has been reported.1 Histopathologic findings are dependent on the stage of disease. Massive infiltrates of neutrophils may be observed in early stages. In older lesions, infiltrates take the form of epithelioid cell granulomas.2
Treatment of RF during pregnancy is challenging. Early and aggressive treatment with retinoids, tetracycline antibiotics, antiandrogenic contraceptives, and dapsone is recommended in patients who are not pregnant; these therapies are all contraindicated in pregnancy. Topical steroids can be safely used; however, systemic steroids usually are required to control RF. The use of systemic steroids can only be justified if the risks for intrauterine growth retardation, maternal diabetes mellitus, and hypertension outweigh the benefits of treating this severe disfiguring skin condition.10 A study by Bakar et al13 indicated that azithromycin is an effective and safe alternative in the treatment of RF. It has a superior pharmacokinetic profile compared to other macrolides and does not pose increased risks for congenital malformation or miscarriage. Because of the concomitant use of both azithromycin and prednisone, it is not possible to determine which had the larger role in the patient’s improvement.
Isotretinoin therapy in our patient led to substantial improvement of RF. Time will tell if the response will be durable. Also unknown is the risk for recurrence with subsequent pregnancies, which has not been reported in the literature. Although it is difficult to confidently say that pregnancy was the inciting factor in this patient’s RF, this case certainly provides more evidence for a link between pregnancy and RF.
Rosacea fulminans (RF) is a rare facial dermatosis characterized by its fulminating course. 1 It presents with superficial and deep-seated papules, pustules, and nodules combined with an intense reddish or cyanotic erythema localized to the face. Furthermore, there is an absence of comedones and involvement of the chest or back. 2 Rosacea fulminans primarily affects women and often is, but not always, proceeded by seborrhea, chronic acne vulgaris, or rosacea. Although the etiology of RF remains unknown, immunologic, hormonal, and vascular factors have been implicated. 3 We report a case of RF in a pregnant patient with a history of mild acne as a teenager that was long ago resolved.
Case Report
A 32-year-old pregnant woman (10 weeks’ gestation) presented with a rapidly progressing inflammatory disorder of the face of 1 month’s duration. The lesions developed 3 weeks after beginning progesterone therapy (200 mg vaginal suppository) for infertility due to polycystic ovary syndrome. Despite discontinuing progesterone for the last month, the patient’s lesions had dramatically worsened (Figure 1). Empiric cephalosporin treatment prescribed by her primary care physician yielded no improvement. Physical examination at the current presentation revealed erythematous nodules and pustules all over the face, coalescing into large thick plaques on the patient’s right cheek and chin. Submental nodes were palpable and tender. Based on the initial clinical findings, acne conglobata secondary to progesterone therapy was considered. The patient was given intralesional triamcinolone (2.5 mg/cc) injections to all larger nodules and several blue light treatments.
The injected areas had improved 5 days after the initial visit; however, the chin and right paranasal cheek developed even more nodules and papules coalescing into large plaques. After consulting the patient’s obstetrician, prednisone (20 mg once daily) was initiated. Three weeks later, the patient’s nodular lesions had improved, but there was a showering of more than 100 pustules and increased general erythema of the entire face (Figure 2). Crotamiton cream 10% (every day before noon), ivermectin cream 1% (every night at bedtime), and sodium sulfacetamide cleanser 10% once daily were added to the treatment plan.
At 16 weeks’ gestation, there was slight improvement; however, there was still erythema on the entire face with scattered pustules and multiple papules and nodules. Many small ice-pick scars were seen on the cheeks and forehead. No comedones were observed. A punch biopsy of an intact papule showed a prominent inflammatory infiltrate with granulomatous reaction and numerous neutrophils predominantly affecting hair follicles. Based on the clinical presentation and histopathology, a diagnosis of RF was made. Azithromycin (250 mg once daily) and metronidazole cream 0.75% twice daily were added. Two weeks later there were fewer nodules but many papules, edema, and intense erythema. The prednisone dosage was increased to 40 mg once daily. Two weeks later, the patient showed improvement with fewer lesions, less edema, and less erythema. The patient was instructed to finish the azithromycin course and discontinue use. At 28 weeks’ gestation, a prednisone taper was started with the intention to reduce the daily dose by delivery.
The patient delivered a healthy girl (birth weight, 1.985 kg) prematurely at 34 weeks’ gestation. At 2 months postpartum, the patient’s existing lesions continued to spontaneously improve; however, she still had numerous nodules and papules and continued to develop new lesions and form additional scars. Isotretinoin was instituted at 3 months postpartum upon cessation of nursing. Three months later (40 mg/d isotretinoin), the patient was nearly clear. At 8 months postpartum, isotretinoin was discontinued after a course of 150 mg/kg.
Comment
Rosacea fulminans initially was called pyoderma faciale but was later regarded as a severe form of rosacea and was renamed rosacea fulminans.2 According to a PubMed search of articles indexed for MEDLINE using the terms pregnancy and rosacea fulminans or pyoderma faciale, we identified 12 publications reporting 20 cases of RF associated with pregnancy (Table). Although there is no substantial evidence regarding the exact mechanism, these cases indicate that pregnancy can be an exacerbating or causative factor in the pathogenesis of RF.
In addition to pregnancy, RF has been associated with inflammatory bowel disease, thyroid and liver disease, erythema nodosum, and severe emotional trauma. However, no organism has been consistently isolated, and no evidence of family history has been reported.1 Histopathologic findings are dependent on the stage of disease. Massive infiltrates of neutrophils may be observed in early stages. In older lesions, infiltrates take the form of epithelioid cell granulomas.2
Treatment of RF during pregnancy is challenging. Early and aggressive treatment with retinoids, tetracycline antibiotics, antiandrogenic contraceptives, and dapsone is recommended in patients who are not pregnant; these therapies are all contraindicated in pregnancy. Topical steroids can be safely used; however, systemic steroids usually are required to control RF. The use of systemic steroids can only be justified if the risks for intrauterine growth retardation, maternal diabetes mellitus, and hypertension outweigh the benefits of treating this severe disfiguring skin condition.10 A study by Bakar et al13 indicated that azithromycin is an effective and safe alternative in the treatment of RF. It has a superior pharmacokinetic profile compared to other macrolides and does not pose increased risks for congenital malformation or miscarriage. Because of the concomitant use of both azithromycin and prednisone, it is not possible to determine which had the larger role in the patient’s improvement.
Isotretinoin therapy in our patient led to substantial improvement of RF. Time will tell if the response will be durable. Also unknown is the risk for recurrence with subsequent pregnancies, which has not been reported in the literature. Although it is difficult to confidently say that pregnancy was the inciting factor in this patient’s RF, this case certainly provides more evidence for a link between pregnancy and RF.
- Jarrett R, Gonsalves R, Anstey AV. Differing obstetric outcomes of rosacea fulminans in pregnancy: report of three cases with review of pathogenesis and management. Clin Exp Dermatol. 2010;35:888-891. doi:10.1111/j.1365-2230.2010.03846.x
- Ferahbas A, Utas S, Mistik S, et al. Rosacea fulminans in pregnancy: case report and review of the literature. Am J Clin Dermatol. 2006;7:141-144. doi:10.2165/00128071-200607020-00007
- Fuentelsaz V, Ara M, Corredera C, et al. Rosacea fulminans in pregnancy: successful treatment with azithromycin. Clin Exp Dermatol. 2011;36:674-676. doi:10.1111/j.1365-2230.2011.04042.x
- Garayar Cantero M, Garabito Solovera E, Aguado García Á, et al. Use of permethrin in the treatment of rosacea fulminans during pregnancy: one case report. Dermatol Ther. 2020;33:E13436. doi:10.1111/dth.13436
- Demir O, Tas IS, Gunay B, et al. A rare dermatologic disease in pregnancy: rosacea fulminans—case report and review of the literature. Open Access Maced J Med Sci. 2018;6:1438-1441. doi:10.3889/oamjms.2018.267
- Markou AG, Alessandrini V, Muray JM, et al. Rosacea fulminans during pregnancy. Clin Exp Obstet Gynecol. 2017;44:157-159.
- Haenen CCP, Kouwenhoven STP, van Doorn R. Rosacea fulminans in pregnancy [in Dutch]. Ned Tijdschr Geneeskd. 2015;159:A8334.
- de Morais e Silva FA, Bonassi M, Steiner D, et al. Rosacea fulminans in pregnancy with ocular perforation. J Dtsch Dermatol Ges. 2011;9:542-543. doi:10.1111/j.1610-0387.2011.07616.x
- Cisse M, Maruani A, Bré C. Rosacea fulminans in the early course of a pregnancy by in vitro fertilization with embryo transfer [in French]. Ann Dermatol Venereol. 2008;135:675-678. doi:10.1016/j.annder.2008.04.015
- Lewis VJ, Holme SA, Wright A, et al. Rosacea fulminans in pregnancy. Br J Dermatol. 2004;151:917-919. doi:10.1111/j.1365-2133.2004.06190.x
- Plewig G, Jansen T, Kligman AM. Pyoderma faciale. a review and report of 20 additional cases: is it rosacea? Arch Dermatol. 1992;128:1611-1617. doi:10.1001/archderm.128.12.1611
- Massa MC, Su WP. Pyoderma faciale: a clinical study of twenty-nine patients. J Am Acad Dermatol. 1982;6:84-91. doi:10.1016/s0190-9622(82)70008-8
- Bakar O, Demirçay Z, Gürbüz O. Therapeutic potential of azithromycin in rosacea. Int J Dermatol. 2004;43:151-154. doi:10.1111/j.1365-4632.2004.01958.x
- Jarrett R, Gonsalves R, Anstey AV. Differing obstetric outcomes of rosacea fulminans in pregnancy: report of three cases with review of pathogenesis and management. Clin Exp Dermatol. 2010;35:888-891. doi:10.1111/j.1365-2230.2010.03846.x
- Ferahbas A, Utas S, Mistik S, et al. Rosacea fulminans in pregnancy: case report and review of the literature. Am J Clin Dermatol. 2006;7:141-144. doi:10.2165/00128071-200607020-00007
- Fuentelsaz V, Ara M, Corredera C, et al. Rosacea fulminans in pregnancy: successful treatment with azithromycin. Clin Exp Dermatol. 2011;36:674-676. doi:10.1111/j.1365-2230.2011.04042.x
- Garayar Cantero M, Garabito Solovera E, Aguado García Á, et al. Use of permethrin in the treatment of rosacea fulminans during pregnancy: one case report. Dermatol Ther. 2020;33:E13436. doi:10.1111/dth.13436
- Demir O, Tas IS, Gunay B, et al. A rare dermatologic disease in pregnancy: rosacea fulminans—case report and review of the literature. Open Access Maced J Med Sci. 2018;6:1438-1441. doi:10.3889/oamjms.2018.267
- Markou AG, Alessandrini V, Muray JM, et al. Rosacea fulminans during pregnancy. Clin Exp Obstet Gynecol. 2017;44:157-159.
- Haenen CCP, Kouwenhoven STP, van Doorn R. Rosacea fulminans in pregnancy [in Dutch]. Ned Tijdschr Geneeskd. 2015;159:A8334.
- de Morais e Silva FA, Bonassi M, Steiner D, et al. Rosacea fulminans in pregnancy with ocular perforation. J Dtsch Dermatol Ges. 2011;9:542-543. doi:10.1111/j.1610-0387.2011.07616.x
- Cisse M, Maruani A, Bré C. Rosacea fulminans in the early course of a pregnancy by in vitro fertilization with embryo transfer [in French]. Ann Dermatol Venereol. 2008;135:675-678. doi:10.1016/j.annder.2008.04.015
- Lewis VJ, Holme SA, Wright A, et al. Rosacea fulminans in pregnancy. Br J Dermatol. 2004;151:917-919. doi:10.1111/j.1365-2133.2004.06190.x
- Plewig G, Jansen T, Kligman AM. Pyoderma faciale. a review and report of 20 additional cases: is it rosacea? Arch Dermatol. 1992;128:1611-1617. doi:10.1001/archderm.128.12.1611
- Massa MC, Su WP. Pyoderma faciale: a clinical study of twenty-nine patients. J Am Acad Dermatol. 1982;6:84-91. doi:10.1016/s0190-9622(82)70008-8
- Bakar O, Demirçay Z, Gürbüz O. Therapeutic potential of azithromycin in rosacea. Int J Dermatol. 2004;43:151-154. doi:10.1111/j.1365-4632.2004.01958.x
Practice Points
- Rosacea fulminans (RF) is a rare facial dermatosis that can present in pregnant patients.
- Treatment of RF in a pregnant patient requires special considerations because typical therapies are contraindicated in pregnancy.

A Pilot With Electrical Pain in the Face
An intracranial epidermoid cyst is an unusual but treatable cause of trigeminal neuralgia.
A 25-year-old male student pilot presented to his flight surgeon in Corpus Christi, Texas, with a 1-year history of episodic left-sided facial pain. He described the pain as electric-like with subsequent tingling sensation. These symptoms were always located on the left side of his tongue and lower lip. They were provoked by chewing, touching, or brushing his teeth. The most recent episode had lasted for 3 days before resolving. He noted 2 similar episodes a few months earlier that he related to periods of high stress.
On physical examination, the student pilot was well appearing with unremarkable vital signs. There were no skin lesions of the head or neck region. His tongue was midline without cutaneous lesions or atrophy. There was no facial numbness or weakness of the mastication muscles. There were no oropharyngeal mucosal or anatomic abnormalities. He had no lymphadenopathy. The remainder of the physical examination was unremarkable. He was seen by both dental medicine and oral surgery providers who did not identify an underlying cause for his symptoms.
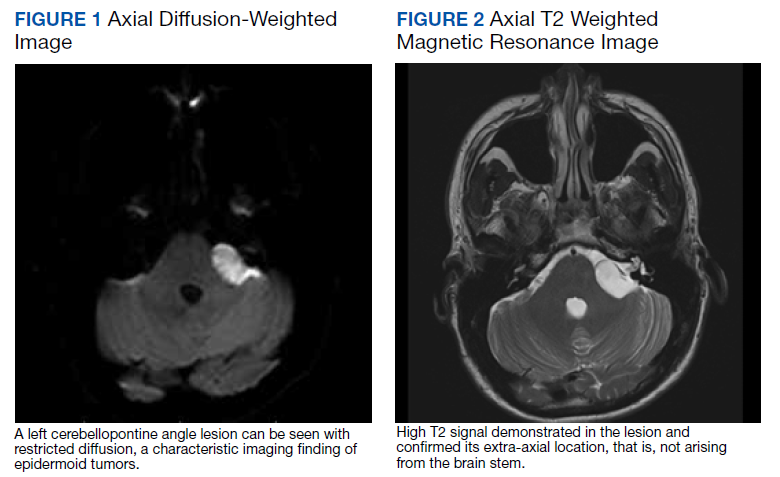
When symptoms recurred a fourth time, the student was referred for a magnetic resonance imaging (MRI) of the brain with and without contrast. The MRI demonstrated a left-sided extra-axial mass, involving the cerebellopontine angle (CPA), with imaging features most consistent with an epidermoid cyst (Figures 1, 2, and 3). An audiogram performed at the time of diagnosis revealed no sensorineural hearing loss.
Discussion
Epidermoid cysts are extra-axial tumors that are benign and slow growing. They constitute about 1% of all intracranial tumors.1 They most commonly occur at the CPA but can also arise in the fourth ventricle and suprasellar regions.2 Epidermoid cysts constitute about 5 to 7% of all CPA tumors.3,4 The 2 most common presenting symptoms of these tumors are headache and cranial nerve dysfunction.1 Other presenting symptoms may include ataxia, hemiparesis, and tinnitus.
On computed tomography (CT), epidermoid cysts can be identical in density to cerebrospinal fluid, making early detection difficult. On MRI, the lesion is easily seen on diffusion-weighted imaging, due to hyperintensity and restricted diffusion. The cysts rarely enhance, unlike the more common tumors in this region, vestibular schwannomas and meningiomas.5
Total neurosurgical resection of the epidermoid cyst is the optimal treatment and is possible in most cases.6 Management of these cysts may prove difficult because of their close proximity to the cranial nerves and brain stem. A near-total excision may be necessary for those tumors that have strong adhesions to neurovascular structures.7 Literature reports that recurrence after surgery is rare in cases of subtotal removal.8,9 Reported postoperative complications may include aseptic meningitis and cranial nerve dysfunction.10
Management
The patient was informed of the presumed diagnosis of brain epidermoid cyst and sent for neurosurgery evaluation. Surgery was indicated and via a retrosigmoid craniotomy, the tumor was removed in total with no complications. On 3-month postoperative follow-up, MRI showed no evidence of residual epidermoid (Figure 4). On physical examination at the follow-up, he was alert and oriented. His surgical incision was well healed. He was neurologically intact with a normal gait. He was released without restrictions from neurosurgical care.
The patient wished to continue flying after successful resection of his cyst. The neurosurgical procedure for removal of the epidermoid cyst medically disqualified him for military aviation.11 As the patient had no neurologic deficits, a waiver was submitted on his behalf to the Naval Aerospace Medical Institute. The waiver was granted for flying duties, and the patient returned to training. He has had no return of symptoms to date.
Conclusions
An intracranial epidermoid cyst is an unusual but treatable cause of trigeminal neuralgia. Gross total removal, without cranial nerve or cerebellar deficits, resulted in the patient’s complete return to health and training as a pilot.
1. Farhoud A, Khedr W, Aboul-Enein H. Surgical resection of cerebellopontine epidermoid cysts: limitations and outcome. J Neurol Surg B Skull Base. 2018;79(2):167‐172. doi:10.1055/s-0037-1606220
2. Hung LC, Wu CS, Lin CC, Fang WK, Hsu YC. Epidermoid cyst presenting as isolated trigeminal neuralgia - two case reports. Acta Neurol Taiwan. 2013;22(3):133‐137.
3. Feng R, Gu X, Hu J, et al. Surgical treatment and radiotherapy of epidermoid cyst with malignant transformation in cerebellopontine angle. Int J Clin Exp Med. 2014;7(1):312‐315.
4. Friedmann DR, Grobelny B, Golfinos JG, Roland JT. Nonschwannoma tumors of the cerebellopontine angle. Otolaryngol Clin North Am. 2015;48(3):461-475. doi:10.1016/j.ote.2015.02.006
5. CPA-IAC In: Harnsberger HR, Glastonbury CM, Michel MA, Koch BL Branstetter BF IV. Diagnostic Imaging: Head and Neck, 2nd ed. Amirsys, Inc; 2011:VI(8):6-9 6. Hasegawa M, Nouri M, Nagahisa S, et al. Cerebellopontine angle epidermoid cysts: clinical presentations and surgical outcome. Neurosurg Rev. 2016;39(2):259‐267. doi:10.1007/s10143-015-0684-5
7. Safavi-Abbasi S, Di Rocco F, Bambakidis N, et al. Has management of epidermoid tumors of the cerebellopontine angle improved? A surgical synopsis of the past and present. Skull Base. 2008;18(2):85‐98. doi:10.1055/s-2007-991108
8. Son DW, Choi CH, Cha SH. Epidermoid tumors in the cerebellopontine angle presenting with trigeminal neuralgia. J Korean Neurosurg Soc. 2010;47(4):271‐277. doi:10.3340/jkns.2010.47.4.271
9. Schiefer TK, Link MJ. Epidermoids of the cerebellopontine angle: a 20-year experience. Surg Neurol. 2008;70(6):584-590; discussion 590. doi:10.1016/j.surneu.2007.12.021
10. Meng L, Yuguang L, Feng L, Wandong S, Shugan Z, Chengyuan W. Cerebellopontine angle epidermoids presenting with trigeminal neuralgia. J Clin Neurosci. 2005;12(7):784‐786. doi:10.1016/j.jocn.2004.09.023
11. Naval Aerospace Medical Institute. US Navy Aeromedical reference and waiver guide. Updated March 31, 2021. Accessed June 17, 2021. https://www.med.navy.mil/sites/nmotc/nami/arwg/Documents/WaiverGuide/Complete_Waiver_Guide.pdf
An intracranial epidermoid cyst is an unusual but treatable cause of trigeminal neuralgia.
An intracranial epidermoid cyst is an unusual but treatable cause of trigeminal neuralgia.
A 25-year-old male student pilot presented to his flight surgeon in Corpus Christi, Texas, with a 1-year history of episodic left-sided facial pain. He described the pain as electric-like with subsequent tingling sensation. These symptoms were always located on the left side of his tongue and lower lip. They were provoked by chewing, touching, or brushing his teeth. The most recent episode had lasted for 3 days before resolving. He noted 2 similar episodes a few months earlier that he related to periods of high stress.
On physical examination, the student pilot was well appearing with unremarkable vital signs. There were no skin lesions of the head or neck region. His tongue was midline without cutaneous lesions or atrophy. There was no facial numbness or weakness of the mastication muscles. There were no oropharyngeal mucosal or anatomic abnormalities. He had no lymphadenopathy. The remainder of the physical examination was unremarkable. He was seen by both dental medicine and oral surgery providers who did not identify an underlying cause for his symptoms.
When symptoms recurred a fourth time, the student was referred for a magnetic resonance imaging (MRI) of the brain with and without contrast. The MRI demonstrated a left-sided extra-axial mass, involving the cerebellopontine angle (CPA), with imaging features most consistent with an epidermoid cyst (Figures 1, 2, and 3). An audiogram performed at the time of diagnosis revealed no sensorineural hearing loss.
Discussion
Epidermoid cysts are extra-axial tumors that are benign and slow growing. They constitute about 1% of all intracranial tumors.1 They most commonly occur at the CPA but can also arise in the fourth ventricle and suprasellar regions.2 Epidermoid cysts constitute about 5 to 7% of all CPA tumors.3,4 The 2 most common presenting symptoms of these tumors are headache and cranial nerve dysfunction.1 Other presenting symptoms may include ataxia, hemiparesis, and tinnitus.
On computed tomography (CT), epidermoid cysts can be identical in density to cerebrospinal fluid, making early detection difficult. On MRI, the lesion is easily seen on diffusion-weighted imaging, due to hyperintensity and restricted diffusion. The cysts rarely enhance, unlike the more common tumors in this region, vestibular schwannomas and meningiomas.5
Total neurosurgical resection of the epidermoid cyst is the optimal treatment and is possible in most cases.6 Management of these cysts may prove difficult because of their close proximity to the cranial nerves and brain stem. A near-total excision may be necessary for those tumors that have strong adhesions to neurovascular structures.7 Literature reports that recurrence after surgery is rare in cases of subtotal removal.8,9 Reported postoperative complications may include aseptic meningitis and cranial nerve dysfunction.10
Management
The patient was informed of the presumed diagnosis of brain epidermoid cyst and sent for neurosurgery evaluation. Surgery was indicated and via a retrosigmoid craniotomy, the tumor was removed in total with no complications. On 3-month postoperative follow-up, MRI showed no evidence of residual epidermoid (Figure 4). On physical examination at the follow-up, he was alert and oriented. His surgical incision was well healed. He was neurologically intact with a normal gait. He was released without restrictions from neurosurgical care.
The patient wished to continue flying after successful resection of his cyst. The neurosurgical procedure for removal of the epidermoid cyst medically disqualified him for military aviation.11 As the patient had no neurologic deficits, a waiver was submitted on his behalf to the Naval Aerospace Medical Institute. The waiver was granted for flying duties, and the patient returned to training. He has had no return of symptoms to date.
Conclusions
An intracranial epidermoid cyst is an unusual but treatable cause of trigeminal neuralgia. Gross total removal, without cranial nerve or cerebellar deficits, resulted in the patient’s complete return to health and training as a pilot.
A 25-year-old male student pilot presented to his flight surgeon in Corpus Christi, Texas, with a 1-year history of episodic left-sided facial pain. He described the pain as electric-like with subsequent tingling sensation. These symptoms were always located on the left side of his tongue and lower lip. They were provoked by chewing, touching, or brushing his teeth. The most recent episode had lasted for 3 days before resolving. He noted 2 similar episodes a few months earlier that he related to periods of high stress.
On physical examination, the student pilot was well appearing with unremarkable vital signs. There were no skin lesions of the head or neck region. His tongue was midline without cutaneous lesions or atrophy. There was no facial numbness or weakness of the mastication muscles. There were no oropharyngeal mucosal or anatomic abnormalities. He had no lymphadenopathy. The remainder of the physical examination was unremarkable. He was seen by both dental medicine and oral surgery providers who did not identify an underlying cause for his symptoms.
When symptoms recurred a fourth time, the student was referred for a magnetic resonance imaging (MRI) of the brain with and without contrast. The MRI demonstrated a left-sided extra-axial mass, involving the cerebellopontine angle (CPA), with imaging features most consistent with an epidermoid cyst (Figures 1, 2, and 3). An audiogram performed at the time of diagnosis revealed no sensorineural hearing loss.
Discussion
Epidermoid cysts are extra-axial tumors that are benign and slow growing. They constitute about 1% of all intracranial tumors.1 They most commonly occur at the CPA but can also arise in the fourth ventricle and suprasellar regions.2 Epidermoid cysts constitute about 5 to 7% of all CPA tumors.3,4 The 2 most common presenting symptoms of these tumors are headache and cranial nerve dysfunction.1 Other presenting symptoms may include ataxia, hemiparesis, and tinnitus.
On computed tomography (CT), epidermoid cysts can be identical in density to cerebrospinal fluid, making early detection difficult. On MRI, the lesion is easily seen on diffusion-weighted imaging, due to hyperintensity and restricted diffusion. The cysts rarely enhance, unlike the more common tumors in this region, vestibular schwannomas and meningiomas.5
Total neurosurgical resection of the epidermoid cyst is the optimal treatment and is possible in most cases.6 Management of these cysts may prove difficult because of their close proximity to the cranial nerves and brain stem. A near-total excision may be necessary for those tumors that have strong adhesions to neurovascular structures.7 Literature reports that recurrence after surgery is rare in cases of subtotal removal.8,9 Reported postoperative complications may include aseptic meningitis and cranial nerve dysfunction.10
Management
The patient was informed of the presumed diagnosis of brain epidermoid cyst and sent for neurosurgery evaluation. Surgery was indicated and via a retrosigmoid craniotomy, the tumor was removed in total with no complications. On 3-month postoperative follow-up, MRI showed no evidence of residual epidermoid (Figure 4). On physical examination at the follow-up, he was alert and oriented. His surgical incision was well healed. He was neurologically intact with a normal gait. He was released without restrictions from neurosurgical care.
The patient wished to continue flying after successful resection of his cyst. The neurosurgical procedure for removal of the epidermoid cyst medically disqualified him for military aviation.11 As the patient had no neurologic deficits, a waiver was submitted on his behalf to the Naval Aerospace Medical Institute. The waiver was granted for flying duties, and the patient returned to training. He has had no return of symptoms to date.
Conclusions
An intracranial epidermoid cyst is an unusual but treatable cause of trigeminal neuralgia. Gross total removal, without cranial nerve or cerebellar deficits, resulted in the patient’s complete return to health and training as a pilot.
1. Farhoud A, Khedr W, Aboul-Enein H. Surgical resection of cerebellopontine epidermoid cysts: limitations and outcome. J Neurol Surg B Skull Base. 2018;79(2):167‐172. doi:10.1055/s-0037-1606220
2. Hung LC, Wu CS, Lin CC, Fang WK, Hsu YC. Epidermoid cyst presenting as isolated trigeminal neuralgia - two case reports. Acta Neurol Taiwan. 2013;22(3):133‐137.
3. Feng R, Gu X, Hu J, et al. Surgical treatment and radiotherapy of epidermoid cyst with malignant transformation in cerebellopontine angle. Int J Clin Exp Med. 2014;7(1):312‐315.
4. Friedmann DR, Grobelny B, Golfinos JG, Roland JT. Nonschwannoma tumors of the cerebellopontine angle. Otolaryngol Clin North Am. 2015;48(3):461-475. doi:10.1016/j.ote.2015.02.006
5. CPA-IAC In: Harnsberger HR, Glastonbury CM, Michel MA, Koch BL Branstetter BF IV. Diagnostic Imaging: Head and Neck, 2nd ed. Amirsys, Inc; 2011:VI(8):6-9 6. Hasegawa M, Nouri M, Nagahisa S, et al. Cerebellopontine angle epidermoid cysts: clinical presentations and surgical outcome. Neurosurg Rev. 2016;39(2):259‐267. doi:10.1007/s10143-015-0684-5
7. Safavi-Abbasi S, Di Rocco F, Bambakidis N, et al. Has management of epidermoid tumors of the cerebellopontine angle improved? A surgical synopsis of the past and present. Skull Base. 2008;18(2):85‐98. doi:10.1055/s-2007-991108
8. Son DW, Choi CH, Cha SH. Epidermoid tumors in the cerebellopontine angle presenting with trigeminal neuralgia. J Korean Neurosurg Soc. 2010;47(4):271‐277. doi:10.3340/jkns.2010.47.4.271
9. Schiefer TK, Link MJ. Epidermoids of the cerebellopontine angle: a 20-year experience. Surg Neurol. 2008;70(6):584-590; discussion 590. doi:10.1016/j.surneu.2007.12.021
10. Meng L, Yuguang L, Feng L, Wandong S, Shugan Z, Chengyuan W. Cerebellopontine angle epidermoids presenting with trigeminal neuralgia. J Clin Neurosci. 2005;12(7):784‐786. doi:10.1016/j.jocn.2004.09.023
11. Naval Aerospace Medical Institute. US Navy Aeromedical reference and waiver guide. Updated March 31, 2021. Accessed June 17, 2021. https://www.med.navy.mil/sites/nmotc/nami/arwg/Documents/WaiverGuide/Complete_Waiver_Guide.pdf
1. Farhoud A, Khedr W, Aboul-Enein H. Surgical resection of cerebellopontine epidermoid cysts: limitations and outcome. J Neurol Surg B Skull Base. 2018;79(2):167‐172. doi:10.1055/s-0037-1606220
2. Hung LC, Wu CS, Lin CC, Fang WK, Hsu YC. Epidermoid cyst presenting as isolated trigeminal neuralgia - two case reports. Acta Neurol Taiwan. 2013;22(3):133‐137.
3. Feng R, Gu X, Hu J, et al. Surgical treatment and radiotherapy of epidermoid cyst with malignant transformation in cerebellopontine angle. Int J Clin Exp Med. 2014;7(1):312‐315.
4. Friedmann DR, Grobelny B, Golfinos JG, Roland JT. Nonschwannoma tumors of the cerebellopontine angle. Otolaryngol Clin North Am. 2015;48(3):461-475. doi:10.1016/j.ote.2015.02.006
5. CPA-IAC In: Harnsberger HR, Glastonbury CM, Michel MA, Koch BL Branstetter BF IV. Diagnostic Imaging: Head and Neck, 2nd ed. Amirsys, Inc; 2011:VI(8):6-9 6. Hasegawa M, Nouri M, Nagahisa S, et al. Cerebellopontine angle epidermoid cysts: clinical presentations and surgical outcome. Neurosurg Rev. 2016;39(2):259‐267. doi:10.1007/s10143-015-0684-5
7. Safavi-Abbasi S, Di Rocco F, Bambakidis N, et al. Has management of epidermoid tumors of the cerebellopontine angle improved? A surgical synopsis of the past and present. Skull Base. 2008;18(2):85‐98. doi:10.1055/s-2007-991108
8. Son DW, Choi CH, Cha SH. Epidermoid tumors in the cerebellopontine angle presenting with trigeminal neuralgia. J Korean Neurosurg Soc. 2010;47(4):271‐277. doi:10.3340/jkns.2010.47.4.271
9. Schiefer TK, Link MJ. Epidermoids of the cerebellopontine angle: a 20-year experience. Surg Neurol. 2008;70(6):584-590; discussion 590. doi:10.1016/j.surneu.2007.12.021
10. Meng L, Yuguang L, Feng L, Wandong S, Shugan Z, Chengyuan W. Cerebellopontine angle epidermoids presenting with trigeminal neuralgia. J Clin Neurosci. 2005;12(7):784‐786. doi:10.1016/j.jocn.2004.09.023
11. Naval Aerospace Medical Institute. US Navy Aeromedical reference and waiver guide. Updated March 31, 2021. Accessed June 17, 2021. https://www.med.navy.mil/sites/nmotc/nami/arwg/Documents/WaiverGuide/Complete_Waiver_Guide.pdf
Results of Laboratory Monitoring in Patients Taking Isotretinoin for Acne
Introduced in 1982, isotretinoin is a retinoid derivative that has been widely used to treat various dermatologic conditions such as acne vulgaris, rosacea, hidradenitis suppurativa, and hair folliculitis. 1 It remains one of the most effective drugs for the treatment of all forms of acne vulgaris, especially the nodulocystic type, and exerts its effects via different mechanisms that affect the major domains involved in the pathogenesis of acne. 2 One month after treatment initiation, isotretinoin suppresses sebum production by decreasing the size and activity of sebaceous glands. In addition, it notably stabilizes keratinization of the skin and decreases the number of Propionibacterium acnes, which will minimize the inflammation associated with acne. 3,4 Despite its beneficial effects, isotretinoin therapy has been associated with several complications. The most commonly reported adverse effects include fissured lips, dry skin, eczema, epistaxis, dry eyes, gastrointestinal tract upset, angular stomatitis, and back pain. Less frequent systemic adverse effects have been reported and relate mainly to teratogenicity, pancreatitis, drug-induced hepatotoxicity, leukopenia, and thrombocytopenia. 5
Isotretinoin use has been associated with alterations in hepatic and lipid profiles; elevations of serum liver enzymes and triglycerides (TGs) following isotretinoin treatment have been reported.4 Consequently, different protocols for laboratory monitoring during isotretinoin therapy have been established and utilized by various health care institutes.6 Despite the time and economic investment involved, certain protocols recommend repetition of liver function tests and several other laboratory parameters following a baseline test.7 The aim of this study was to determine the prevalence of laboratory changes in alanine aminotransferase (ALT), aspartate aminotransferase (AST), cholesterol, and TGs among patients with acne receiving isotretinoin therapy, as well as to link the initial and second laboratory readings of the aforementioned parameters following initiation of isotretinoin treatment.
Materials and Methods
This retrospective cohort design study obtained patient data, including laboratory test results, from the Electronic System for Integrated Health Information at King Khalid University Hospital (KKUH)(Riyadh, Saudi Arabia). All patients older than 16 years who presented with acne vulgaris to the dermatology department at KKUH; who received a course of isotretinoin for at least 4 weeks between 2011 and 2016; and who had available baseline readings of ALT, AST, cholesterol, and TGs, as well as 2 concurrent follow-up readings after isotretinoin treatment initiation, were included in this study. Patients with only 1 reading following treatment initiation and those receiving isotretinoin treatment for reasons other than acne were excluded. This study was approved by the institutional review board of the College of Medicine at King Saud University (Riyadh, Saudi Arabia)(E-18-3310).
Statistical Analysis
Data were entered into a Microsoft Excel document, and statistical analysis was performed using SPSS (version 22.0). Data were represented as numbers and percentages. Repeated measures analysis was performed using the Cochran Q test to compare proportions of abnormal laboratory values among 3 groups: baseline, first reading, and second reading. When test results were significant, a post hoc test was used to compare proportions between any 2 groups. Moreover, a Spearman rank correlation was performed to investigate the association between the daily isotretinoin dose and the laboratory parameters. Results with P<.05 were considered statistically significant.
Results
During the study period, treatment with oral isotretinoin was undertaken by 386 patients at KKUH. Several of these patients were excluded due to incomplete medical records. The age of the studied patients ranged from 17 to 60 years, with a median age of 24 years (interquartile range, 20−28 years). The daily administered dose ranged from 10 to 80 mg, with a median dose of 30 mg (interquartile range, 20−40 mg), as illustrated in the Table. Repeated-measures analysis of liver enzymes (AST and ALT), total cholesterol, and TGs is detailed in eTable 1. Eight (2.2%) of 371 patients showed abnormal baseline AST levels. The first follow-up measurements of AST revealed high levels in 7 (1.9%) patients. This figure doubled (14 [3.8%] patients) at the second follow-up, with no statistically significant differences (P>.05). Likewise, ALT showed abnormally high levels at baseline and at both the first and second follow-ups (47/371 [12.7%], 49/371 [13.2%], and 37/371 [10.0%], respectively) with no significant differences (P>.05). Furthermore, the proportions of high cholesterol levels at baseline and at both the first and second follow-ups (40/331 [12.1%], 72/331 [21.8%], and 62/331 [18.7%], respectively) showed a statistically significant difference (P=.001). The proportions of high cholesterol levels in both the first and second follow-ups were significantly higher than the baseline proportions (P=.001 and P=.002, respectively). However, the percentages of high cholesterol were reduced at the second reading relative to the first but with no significant differences. Regarding TGs, there was a statistically significant difference in the proportions of high levels over time (5/320 [1.6%], 12/320 [3.8%], and 14/320 [4.4%] at baseline and at the first and second readings, respectively). Moreover, pairwise comparison among the 3 readings revealed a significant difference between the second follow-up and the baseline levels (P=.048). eTable 2 demonstrates statistically significant positive weak associations between the daily administered isotretinoin dose and each of the cholesterol and TG levels, both at the first and second follow-up readings (P<.05).
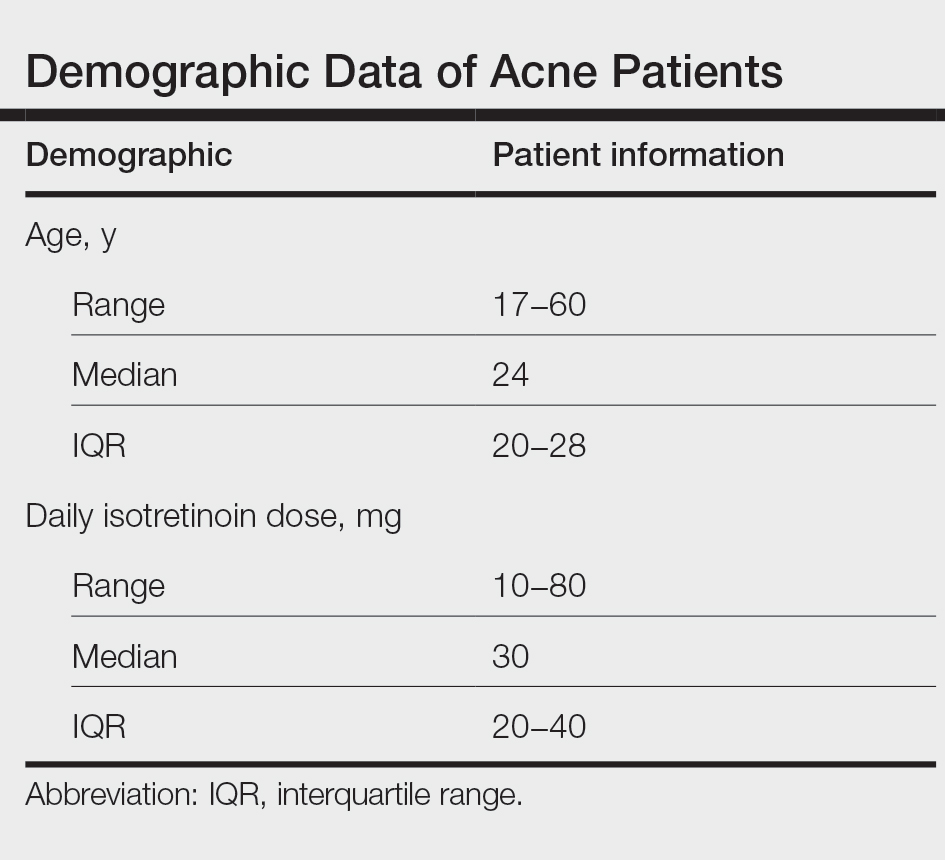

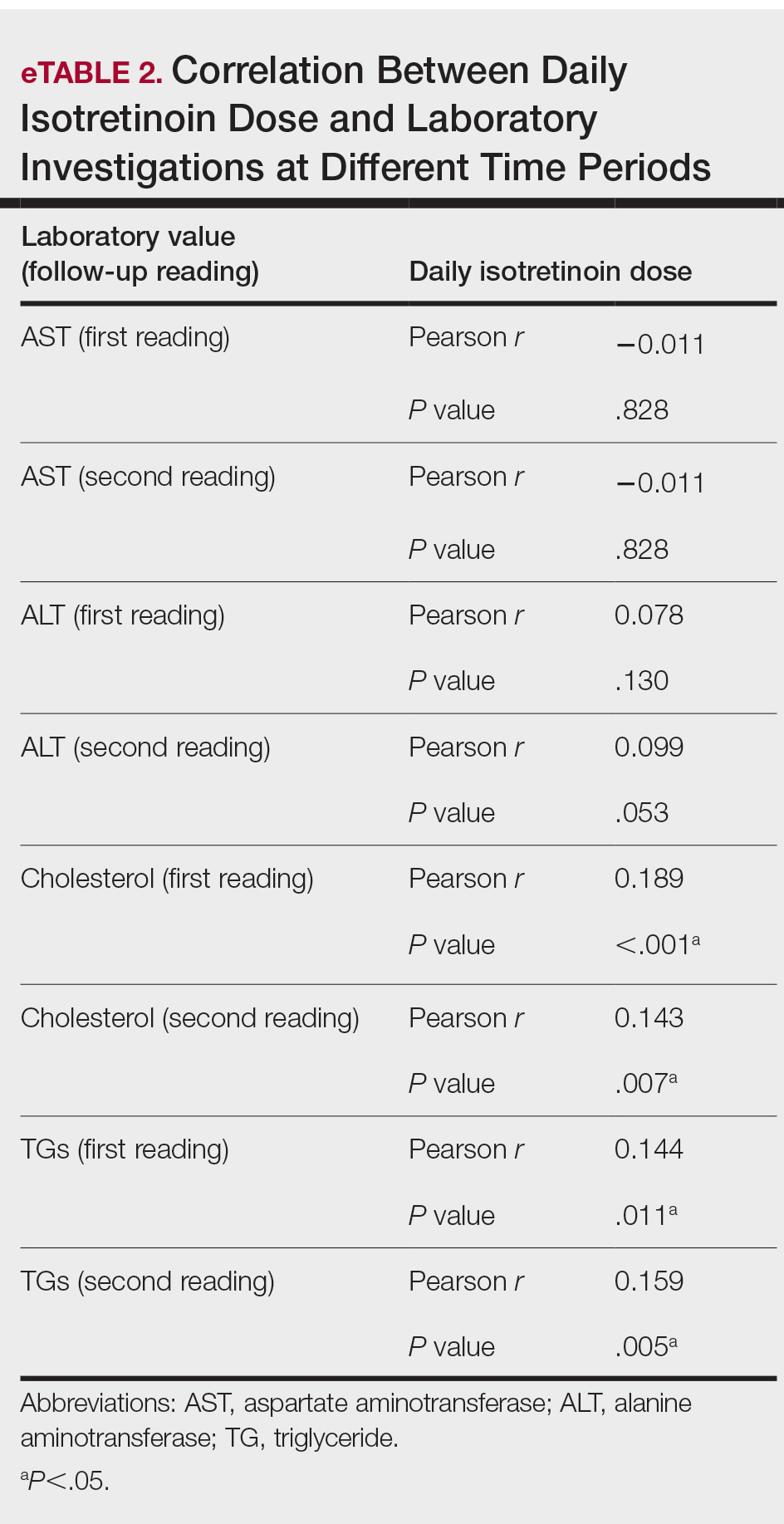
Comment
Evaluation of the effects of isotretinoin on liver enzymes and lipids has suggested that oral isotretinoin may cause alterations in liver aminotransferases (AST and ALT) and lipid profiles to various degrees.8 Furthermore, there are controversies regarding the routine laboratory monitoring of these patients. Some studies have reported severe alterations in serum liver transaminase and lipid levels, and they support the need for careful monitoring when treating patients with isotretinoin. However, other studies have reported that adverse effects are minimal, with no need for costly laboratory monitoring.9
Our study explored the profile of changes in liver aminotransferases (AST and ALT), cholesterol, and TGs in patients with acne who had been treated with oral isotretinoin. The cholesterol levels showed a nonprogressive increase, with a prevalence rate of 21.8% and 18.7% at the first and second follow-ups, respectively. Likewise, the frequency of high TG levels was 3.8% and 4.4%, respectively, with significant differences from the baseline levels (P=.041). However, liver enzymes were less affected by isotretinoin therapy than lipid profiles. Both AST and ALT showed nonsignificant minimal elevations during follow-up of the patients.
Similar to our findings, Zane et al6 at the University of California, San Francisco, studied 13,772 patients with acne who underwent oral isotretinoin therapy between 1995 and 2002. They reported a cumulative incidence of new abnormalities in patients with normal values at baseline at a frequency of 44% for TG levels, 31% for total cholesterol levels, and 11% for transaminase levels. Moreover, they suggested that these abnormalities generally were transient and reversible.6 Another retrospective study in Brazil included 130 patients who were treated with isotretinoin for 3 months and reported that TG levels had increased beyond the normal range in 11% of patients, whereas 8.6% had elevated AST levels and 7.3% had elevated ALT levels.8 Comparable to our findings, Kizilyel et al10 concluded that isotretinoin appeared to have a greater effect on lipids than on liver enzymes, and they recommended its use with careful monitoring.
The transient effects of isotretinoin therapy on lipid profiles were highlighted in an earlier study. It has been reported that the changes in low-density lipoprotein and TGs returned to baseline levels 2 months following termination of treatment.11 Although many studies have reported alterations in serum transaminase and lipid levels, other studies fail to report any such effects. Alcalay et al7 investigated 907 patients who completed a treatment course lasting 5 to 9 months. They reported that only 1.5% of patients had serum TG levels above 400 mg. Additionally, serum levels of liver enzymes were not elevated to a degree necessitating discontinuation of treatment. They concluded that isotretinoin is a safe therapeutic drug and suggested that there is no need for routine laboratory follow-up in young healthy patients apart from a pregnancy test for females.7 In addition, Brito et al12 conducted a prospective clinical and laboratory evaluation of 150 patients being treated with oral isotretinoin prior to the start of therapy, 1 month after therapy initiation, and every 3 months thereafter until the completion of treatment. They found no statistically significant changes in liver transaminase, TG, or cholesterol levels.12 In another study of 30 participants, Baxter et al13 also reported no significant changes in TG or cholesterol levels measured at baseline or during treatment with isotretinoin. Furthermore, a systematic review and meta-analysis has estimated the laboratory changes that occur during isotretinoin therapy of acne vulgaris.14 The evidence revealed in this study does not support monthly laboratory testing for use of standard doses of oral isotretinoin for the typical patient with acne.
Conclusion
In our study, liver enzymes were less affected than lipids in patients who were treated with isotretinoin. Additionally, laboratory alterations in lipid profiles were nonprogressive and nonsevere. Consequently, isotretinoin may be administered with minimal concern for changes in serum transaminase and lipid profile. However, physicians should exercise caution when administering isotretinoin in patients with a history of abnormal findings.
- Kaymak Y, Ilter N. The results and side effects of systemic isotretinoin treatment in 100 patients with acne vulgaris. Dermatol Nurs. 2006;18:576-580.
- Al-Mutairi N, Manchanda Y, Nour-Eldin O, et al. Isotretinoin in acne vulgaris: a prospective analysis of 160 cases from Kuwait. J Drugs Dermatol. 2005;4:369-373.
- Agarwal US, Besarwal RK, Bhola K. Oral isotretinoin in different dose regimens for acne vulgaris: a randomized comparative trial. Indian J Dermatol Venereol Leprol. 2011;77:688-694.
- Hansen TJ, Lucking S, Miller JJ, et al. Standardized laboratory monitoring with use of isotretinoin in acne. J Am Acad Dermatol. 2016;75:323-328.
- Strauss JS, Rapini RP, Shalita AR, et al. Isotretinoin therapy for acne: results of a multicenter dose-response study. J Am Acad Dermatol. 1984;10:490-496.
- Zane LT, Leyden WA, Marqueling AL, et al. A population-based analysis of laboratory abnormalities during isotretinoin therapy for acne vulgaris. Arch Dermatol. 2006;142:1016-1022.
- Alcalay J, Landau M, Zucker A. Analysis of laboratory data in acne patients treated with isotretinoin: is there really a need to perform routine laboratory tests? J Dermatolog Treat. 2001;12:9-12.
- Vieira AS, Beijamini V, Melchiors AC. The effect of isotretinoin on triglycerides and liver aminotransferases. An Bras Dermatol. 2012;87:382-387.
- Bauer LB, Ornelas JN, Elston DM, et al. Isotretinoin: controversies, facts, and recommendations. Expert Rev Clin Pharmacol. 2016;9:1435-1442.
- Kizilyel O, Metin MS, Elmas ÖF, et al. Effects of oral isotretinoin on lipids and liver enzymes in acne patients. Cutis. 2014;94:234-238.
- Bershad S, Rubinstein A, Paterniti JR, et al. Changes in plasma lipids and lipoproteins during isotretinoin therapy for acne. N Engl J Med. 1985;313:981-985.
- Brito MDFDM, Sant’Anna IP, Galindo JCS, et al. Evaluation of clinical adverse effects and laboratory alterations in patients with acne vulgaris treated with oral isotretinoin. An Bras Dermatol. 2010;85:331-337.
- Baxter KF, Ling TC, Barth JH, et al. Retrospective survey of serum lipids in patients receiving more than three courses of isotretinoin. J Dermatolog Treat. 2004;14:216-218.
- Lee YH, Scharnitz TP, Muscat J, et al. Laboratory monitoring during isotretinoin therapy for acne: a systematic review and meta-analysis. JAMA Dermatol. 2016;152:35-44.
Introduced in 1982, isotretinoin is a retinoid derivative that has been widely used to treat various dermatologic conditions such as acne vulgaris, rosacea, hidradenitis suppurativa, and hair folliculitis. 1 It remains one of the most effective drugs for the treatment of all forms of acne vulgaris, especially the nodulocystic type, and exerts its effects via different mechanisms that affect the major domains involved in the pathogenesis of acne. 2 One month after treatment initiation, isotretinoin suppresses sebum production by decreasing the size and activity of sebaceous glands. In addition, it notably stabilizes keratinization of the skin and decreases the number of Propionibacterium acnes, which will minimize the inflammation associated with acne. 3,4 Despite its beneficial effects, isotretinoin therapy has been associated with several complications. The most commonly reported adverse effects include fissured lips, dry skin, eczema, epistaxis, dry eyes, gastrointestinal tract upset, angular stomatitis, and back pain. Less frequent systemic adverse effects have been reported and relate mainly to teratogenicity, pancreatitis, drug-induced hepatotoxicity, leukopenia, and thrombocytopenia. 5
Isotretinoin use has been associated with alterations in hepatic and lipid profiles; elevations of serum liver enzymes and triglycerides (TGs) following isotretinoin treatment have been reported.4 Consequently, different protocols for laboratory monitoring during isotretinoin therapy have been established and utilized by various health care institutes.6 Despite the time and economic investment involved, certain protocols recommend repetition of liver function tests and several other laboratory parameters following a baseline test.7 The aim of this study was to determine the prevalence of laboratory changes in alanine aminotransferase (ALT), aspartate aminotransferase (AST), cholesterol, and TGs among patients with acne receiving isotretinoin therapy, as well as to link the initial and second laboratory readings of the aforementioned parameters following initiation of isotretinoin treatment.
Materials and Methods
This retrospective cohort design study obtained patient data, including laboratory test results, from the Electronic System for Integrated Health Information at King Khalid University Hospital (KKUH)(Riyadh, Saudi Arabia). All patients older than 16 years who presented with acne vulgaris to the dermatology department at KKUH; who received a course of isotretinoin for at least 4 weeks between 2011 and 2016; and who had available baseline readings of ALT, AST, cholesterol, and TGs, as well as 2 concurrent follow-up readings after isotretinoin treatment initiation, were included in this study. Patients with only 1 reading following treatment initiation and those receiving isotretinoin treatment for reasons other than acne were excluded. This study was approved by the institutional review board of the College of Medicine at King Saud University (Riyadh, Saudi Arabia)(E-18-3310).
Statistical Analysis
Data were entered into a Microsoft Excel document, and statistical analysis was performed using SPSS (version 22.0). Data were represented as numbers and percentages. Repeated measures analysis was performed using the Cochran Q test to compare proportions of abnormal laboratory values among 3 groups: baseline, first reading, and second reading. When test results were significant, a post hoc test was used to compare proportions between any 2 groups. Moreover, a Spearman rank correlation was performed to investigate the association between the daily isotretinoin dose and the laboratory parameters. Results with P<.05 were considered statistically significant.
Results
During the study period, treatment with oral isotretinoin was undertaken by 386 patients at KKUH. Several of these patients were excluded due to incomplete medical records. The age of the studied patients ranged from 17 to 60 years, with a median age of 24 years (interquartile range, 20−28 years). The daily administered dose ranged from 10 to 80 mg, with a median dose of 30 mg (interquartile range, 20−40 mg), as illustrated in the Table. Repeated-measures analysis of liver enzymes (AST and ALT), total cholesterol, and TGs is detailed in eTable 1. Eight (2.2%) of 371 patients showed abnormal baseline AST levels. The first follow-up measurements of AST revealed high levels in 7 (1.9%) patients. This figure doubled (14 [3.8%] patients) at the second follow-up, with no statistically significant differences (P>.05). Likewise, ALT showed abnormally high levels at baseline and at both the first and second follow-ups (47/371 [12.7%], 49/371 [13.2%], and 37/371 [10.0%], respectively) with no significant differences (P>.05). Furthermore, the proportions of high cholesterol levels at baseline and at both the first and second follow-ups (40/331 [12.1%], 72/331 [21.8%], and 62/331 [18.7%], respectively) showed a statistically significant difference (P=.001). The proportions of high cholesterol levels in both the first and second follow-ups were significantly higher than the baseline proportions (P=.001 and P=.002, respectively). However, the percentages of high cholesterol were reduced at the second reading relative to the first but with no significant differences. Regarding TGs, there was a statistically significant difference in the proportions of high levels over time (5/320 [1.6%], 12/320 [3.8%], and 14/320 [4.4%] at baseline and at the first and second readings, respectively). Moreover, pairwise comparison among the 3 readings revealed a significant difference between the second follow-up and the baseline levels (P=.048). eTable 2 demonstrates statistically significant positive weak associations between the daily administered isotretinoin dose and each of the cholesterol and TG levels, both at the first and second follow-up readings (P<.05).
Comment
Evaluation of the effects of isotretinoin on liver enzymes and lipids has suggested that oral isotretinoin may cause alterations in liver aminotransferases (AST and ALT) and lipid profiles to various degrees.8 Furthermore, there are controversies regarding the routine laboratory monitoring of these patients. Some studies have reported severe alterations in serum liver transaminase and lipid levels, and they support the need for careful monitoring when treating patients with isotretinoin. However, other studies have reported that adverse effects are minimal, with no need for costly laboratory monitoring.9
Our study explored the profile of changes in liver aminotransferases (AST and ALT), cholesterol, and TGs in patients with acne who had been treated with oral isotretinoin. The cholesterol levels showed a nonprogressive increase, with a prevalence rate of 21.8% and 18.7% at the first and second follow-ups, respectively. Likewise, the frequency of high TG levels was 3.8% and 4.4%, respectively, with significant differences from the baseline levels (P=.041). However, liver enzymes were less affected by isotretinoin therapy than lipid profiles. Both AST and ALT showed nonsignificant minimal elevations during follow-up of the patients.
Similar to our findings, Zane et al6 at the University of California, San Francisco, studied 13,772 patients with acne who underwent oral isotretinoin therapy between 1995 and 2002. They reported a cumulative incidence of new abnormalities in patients with normal values at baseline at a frequency of 44% for TG levels, 31% for total cholesterol levels, and 11% for transaminase levels. Moreover, they suggested that these abnormalities generally were transient and reversible.6 Another retrospective study in Brazil included 130 patients who were treated with isotretinoin for 3 months and reported that TG levels had increased beyond the normal range in 11% of patients, whereas 8.6% had elevated AST levels and 7.3% had elevated ALT levels.8 Comparable to our findings, Kizilyel et al10 concluded that isotretinoin appeared to have a greater effect on lipids than on liver enzymes, and they recommended its use with careful monitoring.
The transient effects of isotretinoin therapy on lipid profiles were highlighted in an earlier study. It has been reported that the changes in low-density lipoprotein and TGs returned to baseline levels 2 months following termination of treatment.11 Although many studies have reported alterations in serum transaminase and lipid levels, other studies fail to report any such effects. Alcalay et al7 investigated 907 patients who completed a treatment course lasting 5 to 9 months. They reported that only 1.5% of patients had serum TG levels above 400 mg. Additionally, serum levels of liver enzymes were not elevated to a degree necessitating discontinuation of treatment. They concluded that isotretinoin is a safe therapeutic drug and suggested that there is no need for routine laboratory follow-up in young healthy patients apart from a pregnancy test for females.7 In addition, Brito et al12 conducted a prospective clinical and laboratory evaluation of 150 patients being treated with oral isotretinoin prior to the start of therapy, 1 month after therapy initiation, and every 3 months thereafter until the completion of treatment. They found no statistically significant changes in liver transaminase, TG, or cholesterol levels.12 In another study of 30 participants, Baxter et al13 also reported no significant changes in TG or cholesterol levels measured at baseline or during treatment with isotretinoin. Furthermore, a systematic review and meta-analysis has estimated the laboratory changes that occur during isotretinoin therapy of acne vulgaris.14 The evidence revealed in this study does not support monthly laboratory testing for use of standard doses of oral isotretinoin for the typical patient with acne.
Conclusion
In our study, liver enzymes were less affected than lipids in patients who were treated with isotretinoin. Additionally, laboratory alterations in lipid profiles were nonprogressive and nonsevere. Consequently, isotretinoin may be administered with minimal concern for changes in serum transaminase and lipid profile. However, physicians should exercise caution when administering isotretinoin in patients with a history of abnormal findings.
Introduced in 1982, isotretinoin is a retinoid derivative that has been widely used to treat various dermatologic conditions such as acne vulgaris, rosacea, hidradenitis suppurativa, and hair folliculitis. 1 It remains one of the most effective drugs for the treatment of all forms of acne vulgaris, especially the nodulocystic type, and exerts its effects via different mechanisms that affect the major domains involved in the pathogenesis of acne. 2 One month after treatment initiation, isotretinoin suppresses sebum production by decreasing the size and activity of sebaceous glands. In addition, it notably stabilizes keratinization of the skin and decreases the number of Propionibacterium acnes, which will minimize the inflammation associated with acne. 3,4 Despite its beneficial effects, isotretinoin therapy has been associated with several complications. The most commonly reported adverse effects include fissured lips, dry skin, eczema, epistaxis, dry eyes, gastrointestinal tract upset, angular stomatitis, and back pain. Less frequent systemic adverse effects have been reported and relate mainly to teratogenicity, pancreatitis, drug-induced hepatotoxicity, leukopenia, and thrombocytopenia. 5
Isotretinoin use has been associated with alterations in hepatic and lipid profiles; elevations of serum liver enzymes and triglycerides (TGs) following isotretinoin treatment have been reported.4 Consequently, different protocols for laboratory monitoring during isotretinoin therapy have been established and utilized by various health care institutes.6 Despite the time and economic investment involved, certain protocols recommend repetition of liver function tests and several other laboratory parameters following a baseline test.7 The aim of this study was to determine the prevalence of laboratory changes in alanine aminotransferase (ALT), aspartate aminotransferase (AST), cholesterol, and TGs among patients with acne receiving isotretinoin therapy, as well as to link the initial and second laboratory readings of the aforementioned parameters following initiation of isotretinoin treatment.
Materials and Methods
This retrospective cohort design study obtained patient data, including laboratory test results, from the Electronic System for Integrated Health Information at King Khalid University Hospital (KKUH)(Riyadh, Saudi Arabia). All patients older than 16 years who presented with acne vulgaris to the dermatology department at KKUH; who received a course of isotretinoin for at least 4 weeks between 2011 and 2016; and who had available baseline readings of ALT, AST, cholesterol, and TGs, as well as 2 concurrent follow-up readings after isotretinoin treatment initiation, were included in this study. Patients with only 1 reading following treatment initiation and those receiving isotretinoin treatment for reasons other than acne were excluded. This study was approved by the institutional review board of the College of Medicine at King Saud University (Riyadh, Saudi Arabia)(E-18-3310).
Statistical Analysis
Data were entered into a Microsoft Excel document, and statistical analysis was performed using SPSS (version 22.0). Data were represented as numbers and percentages. Repeated measures analysis was performed using the Cochran Q test to compare proportions of abnormal laboratory values among 3 groups: baseline, first reading, and second reading. When test results were significant, a post hoc test was used to compare proportions between any 2 groups. Moreover, a Spearman rank correlation was performed to investigate the association between the daily isotretinoin dose and the laboratory parameters. Results with P<.05 were considered statistically significant.
Results
During the study period, treatment with oral isotretinoin was undertaken by 386 patients at KKUH. Several of these patients were excluded due to incomplete medical records. The age of the studied patients ranged from 17 to 60 years, with a median age of 24 years (interquartile range, 20−28 years). The daily administered dose ranged from 10 to 80 mg, with a median dose of 30 mg (interquartile range, 20−40 mg), as illustrated in the Table. Repeated-measures analysis of liver enzymes (AST and ALT), total cholesterol, and TGs is detailed in eTable 1. Eight (2.2%) of 371 patients showed abnormal baseline AST levels. The first follow-up measurements of AST revealed high levels in 7 (1.9%) patients. This figure doubled (14 [3.8%] patients) at the second follow-up, with no statistically significant differences (P>.05). Likewise, ALT showed abnormally high levels at baseline and at both the first and second follow-ups (47/371 [12.7%], 49/371 [13.2%], and 37/371 [10.0%], respectively) with no significant differences (P>.05). Furthermore, the proportions of high cholesterol levels at baseline and at both the first and second follow-ups (40/331 [12.1%], 72/331 [21.8%], and 62/331 [18.7%], respectively) showed a statistically significant difference (P=.001). The proportions of high cholesterol levels in both the first and second follow-ups were significantly higher than the baseline proportions (P=.001 and P=.002, respectively). However, the percentages of high cholesterol were reduced at the second reading relative to the first but with no significant differences. Regarding TGs, there was a statistically significant difference in the proportions of high levels over time (5/320 [1.6%], 12/320 [3.8%], and 14/320 [4.4%] at baseline and at the first and second readings, respectively). Moreover, pairwise comparison among the 3 readings revealed a significant difference between the second follow-up and the baseline levels (P=.048). eTable 2 demonstrates statistically significant positive weak associations between the daily administered isotretinoin dose and each of the cholesterol and TG levels, both at the first and second follow-up readings (P<.05).
Comment
Evaluation of the effects of isotretinoin on liver enzymes and lipids has suggested that oral isotretinoin may cause alterations in liver aminotransferases (AST and ALT) and lipid profiles to various degrees.8 Furthermore, there are controversies regarding the routine laboratory monitoring of these patients. Some studies have reported severe alterations in serum liver transaminase and lipid levels, and they support the need for careful monitoring when treating patients with isotretinoin. However, other studies have reported that adverse effects are minimal, with no need for costly laboratory monitoring.9
Our study explored the profile of changes in liver aminotransferases (AST and ALT), cholesterol, and TGs in patients with acne who had been treated with oral isotretinoin. The cholesterol levels showed a nonprogressive increase, with a prevalence rate of 21.8% and 18.7% at the first and second follow-ups, respectively. Likewise, the frequency of high TG levels was 3.8% and 4.4%, respectively, with significant differences from the baseline levels (P=.041). However, liver enzymes were less affected by isotretinoin therapy than lipid profiles. Both AST and ALT showed nonsignificant minimal elevations during follow-up of the patients.
Similar to our findings, Zane et al6 at the University of California, San Francisco, studied 13,772 patients with acne who underwent oral isotretinoin therapy between 1995 and 2002. They reported a cumulative incidence of new abnormalities in patients with normal values at baseline at a frequency of 44% for TG levels, 31% for total cholesterol levels, and 11% for transaminase levels. Moreover, they suggested that these abnormalities generally were transient and reversible.6 Another retrospective study in Brazil included 130 patients who were treated with isotretinoin for 3 months and reported that TG levels had increased beyond the normal range in 11% of patients, whereas 8.6% had elevated AST levels and 7.3% had elevated ALT levels.8 Comparable to our findings, Kizilyel et al10 concluded that isotretinoin appeared to have a greater effect on lipids than on liver enzymes, and they recommended its use with careful monitoring.
The transient effects of isotretinoin therapy on lipid profiles were highlighted in an earlier study. It has been reported that the changes in low-density lipoprotein and TGs returned to baseline levels 2 months following termination of treatment.11 Although many studies have reported alterations in serum transaminase and lipid levels, other studies fail to report any such effects. Alcalay et al7 investigated 907 patients who completed a treatment course lasting 5 to 9 months. They reported that only 1.5% of patients had serum TG levels above 400 mg. Additionally, serum levels of liver enzymes were not elevated to a degree necessitating discontinuation of treatment. They concluded that isotretinoin is a safe therapeutic drug and suggested that there is no need for routine laboratory follow-up in young healthy patients apart from a pregnancy test for females.7 In addition, Brito et al12 conducted a prospective clinical and laboratory evaluation of 150 patients being treated with oral isotretinoin prior to the start of therapy, 1 month after therapy initiation, and every 3 months thereafter until the completion of treatment. They found no statistically significant changes in liver transaminase, TG, or cholesterol levels.12 In another study of 30 participants, Baxter et al13 also reported no significant changes in TG or cholesterol levels measured at baseline or during treatment with isotretinoin. Furthermore, a systematic review and meta-analysis has estimated the laboratory changes that occur during isotretinoin therapy of acne vulgaris.14 The evidence revealed in this study does not support monthly laboratory testing for use of standard doses of oral isotretinoin for the typical patient with acne.
Conclusion
In our study, liver enzymes were less affected than lipids in patients who were treated with isotretinoin. Additionally, laboratory alterations in lipid profiles were nonprogressive and nonsevere. Consequently, isotretinoin may be administered with minimal concern for changes in serum transaminase and lipid profile. However, physicians should exercise caution when administering isotretinoin in patients with a history of abnormal findings.
- Kaymak Y, Ilter N. The results and side effects of systemic isotretinoin treatment in 100 patients with acne vulgaris. Dermatol Nurs. 2006;18:576-580.
- Al-Mutairi N, Manchanda Y, Nour-Eldin O, et al. Isotretinoin in acne vulgaris: a prospective analysis of 160 cases from Kuwait. J Drugs Dermatol. 2005;4:369-373.
- Agarwal US, Besarwal RK, Bhola K. Oral isotretinoin in different dose regimens for acne vulgaris: a randomized comparative trial. Indian J Dermatol Venereol Leprol. 2011;77:688-694.
- Hansen TJ, Lucking S, Miller JJ, et al. Standardized laboratory monitoring with use of isotretinoin in acne. J Am Acad Dermatol. 2016;75:323-328.
- Strauss JS, Rapini RP, Shalita AR, et al. Isotretinoin therapy for acne: results of a multicenter dose-response study. J Am Acad Dermatol. 1984;10:490-496.
- Zane LT, Leyden WA, Marqueling AL, et al. A population-based analysis of laboratory abnormalities during isotretinoin therapy for acne vulgaris. Arch Dermatol. 2006;142:1016-1022.
- Alcalay J, Landau M, Zucker A. Analysis of laboratory data in acne patients treated with isotretinoin: is there really a need to perform routine laboratory tests? J Dermatolog Treat. 2001;12:9-12.
- Vieira AS, Beijamini V, Melchiors AC. The effect of isotretinoin on triglycerides and liver aminotransferases. An Bras Dermatol. 2012;87:382-387.
- Bauer LB, Ornelas JN, Elston DM, et al. Isotretinoin: controversies, facts, and recommendations. Expert Rev Clin Pharmacol. 2016;9:1435-1442.
- Kizilyel O, Metin MS, Elmas ÖF, et al. Effects of oral isotretinoin on lipids and liver enzymes in acne patients. Cutis. 2014;94:234-238.
- Bershad S, Rubinstein A, Paterniti JR, et al. Changes in plasma lipids and lipoproteins during isotretinoin therapy for acne. N Engl J Med. 1985;313:981-985.
- Brito MDFDM, Sant’Anna IP, Galindo JCS, et al. Evaluation of clinical adverse effects and laboratory alterations in patients with acne vulgaris treated with oral isotretinoin. An Bras Dermatol. 2010;85:331-337.
- Baxter KF, Ling TC, Barth JH, et al. Retrospective survey of serum lipids in patients receiving more than three courses of isotretinoin. J Dermatolog Treat. 2004;14:216-218.
- Lee YH, Scharnitz TP, Muscat J, et al. Laboratory monitoring during isotretinoin therapy for acne: a systematic review and meta-analysis. JAMA Dermatol. 2016;152:35-44.
- Kaymak Y, Ilter N. The results and side effects of systemic isotretinoin treatment in 100 patients with acne vulgaris. Dermatol Nurs. 2006;18:576-580.
- Al-Mutairi N, Manchanda Y, Nour-Eldin O, et al. Isotretinoin in acne vulgaris: a prospective analysis of 160 cases from Kuwait. J Drugs Dermatol. 2005;4:369-373.
- Agarwal US, Besarwal RK, Bhola K. Oral isotretinoin in different dose regimens for acne vulgaris: a randomized comparative trial. Indian J Dermatol Venereol Leprol. 2011;77:688-694.
- Hansen TJ, Lucking S, Miller JJ, et al. Standardized laboratory monitoring with use of isotretinoin in acne. J Am Acad Dermatol. 2016;75:323-328.
- Strauss JS, Rapini RP, Shalita AR, et al. Isotretinoin therapy for acne: results of a multicenter dose-response study. J Am Acad Dermatol. 1984;10:490-496.
- Zane LT, Leyden WA, Marqueling AL, et al. A population-based analysis of laboratory abnormalities during isotretinoin therapy for acne vulgaris. Arch Dermatol. 2006;142:1016-1022.
- Alcalay J, Landau M, Zucker A. Analysis of laboratory data in acne patients treated with isotretinoin: is there really a need to perform routine laboratory tests? J Dermatolog Treat. 2001;12:9-12.
- Vieira AS, Beijamini V, Melchiors AC. The effect of isotretinoin on triglycerides and liver aminotransferases. An Bras Dermatol. 2012;87:382-387.
- Bauer LB, Ornelas JN, Elston DM, et al. Isotretinoin: controversies, facts, and recommendations. Expert Rev Clin Pharmacol. 2016;9:1435-1442.
- Kizilyel O, Metin MS, Elmas ÖF, et al. Effects of oral isotretinoin on lipids and liver enzymes in acne patients. Cutis. 2014;94:234-238.
- Bershad S, Rubinstein A, Paterniti JR, et al. Changes in plasma lipids and lipoproteins during isotretinoin therapy for acne. N Engl J Med. 1985;313:981-985.
- Brito MDFDM, Sant’Anna IP, Galindo JCS, et al. Evaluation of clinical adverse effects and laboratory alterations in patients with acne vulgaris treated with oral isotretinoin. An Bras Dermatol. 2010;85:331-337.
- Baxter KF, Ling TC, Barth JH, et al. Retrospective survey of serum lipids in patients receiving more than three courses of isotretinoin. J Dermatolog Treat. 2004;14:216-218.
- Lee YH, Scharnitz TP, Muscat J, et al. Laboratory monitoring during isotretinoin therapy for acne: a systematic review and meta-analysis. JAMA Dermatol. 2016;152:35-44.
Practice Points
- Isotretinoin is the mainstay treatment for severe acne.
- Cost and convenience to patients should always be considered.
- Frequent monitoring for laboratory changes during isotretinoin treatment is not warranted.
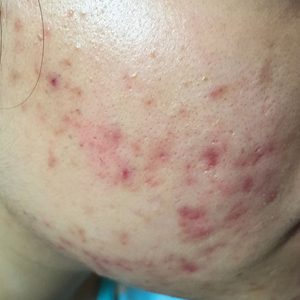
5-year-old boy • calf pain • fever • cough & rhinitis • Dx?
THE CASE
A 5-year-old previously healthy white boy presented to clinic with bilateral calf pain and refusal to bear weight since awakening that morning. Associated symptoms included a 3-day history of generalized fatigue, subjective fevers, cough, congestion, and rhinitis. The night prior to presentation, he showed no symptoms of gait abnormalities, muscle pain, or weakness. There was no history of similar symptoms, trauma, overexertion, foreign travel, or family history of musculoskeletal disease. He was fully immunized, except for the annual influenza vaccine. He was not taking any medications. This case occurred before the onset of the COVID-19 pandemic.
Objective findings included fever of 101 °F, refusal to bear weight, and symmetrical bilateral tenderness to palpation of the gastrocnemius-soleus complex. Pain was elicited with passive dorsiflexion. There was no erythema, edema, or sensory deficits, and the distal leg compartments were soft. There was normal range of motion of the hips, knees, and ankles. Dorsalis pedis pulses were 2+, and patella reflexes were 2/4 bilaterally.
Lab results included a white blood cell count of 2500/μL (normal range, 4500 to 11,000/μL);absolute neutrophil count, 900/μL (1500 to 8000/μL); platelet count, 131,000/μL (150,000 to 450,000/μL); creatine kinase level, 869 IU/L (22 to 198 U/L); and aspartate aminotransferase level, 116 U/L (8 to 33 U/L). A rapid influenza swab was positive for influenza B. Plain films of the bilateral hips and lower extremities were unremarkable. C-reactive protein (CRP) level, urinalysis, and renal function tests were within normal limits. Creatine kinase (CK) level peaked (1935 U/L; normal range, 22 to 198 U/L) within the first 24 hours of presentation and then trended down.
The Diagnosis
The patient’s sudden onset of symmetrical bilateral calf pain in the setting of an upper respiratory tract infection was extremely suspicious for benign acute childhood myositis (BACM). Lab work and radiologic evaluation were performed to rule out more ominous causes of refusal to bear weight. The suspicion of BACM was further validated by influenza B serology, an elevated CK, and a normal CRP.
Discussion
BACM was first described by Lundberg in 1957.1 The overall incidence and prevalence are unclear.2 A viral prodrome involving rhinorrhea, low-grade fever, sore throat, cough, and malaise typically precedes bilateral calf pain by 3 days.2-4 Myositis symptoms typically last for 4 days.3 While several infectious etiologies have been linked to this condition, influenza B has the greatest association.5,6
❚ Patient population. BACM occurs predominately in school-aged children (6-8 years old) and has a male-to-female ratio of 2:1.3,5,6 In a retrospective study of 219 children, BACM was strongly associated with male gender and ages 6 to 9 years.3 In another retrospective study of 54 children,80% of patients were male, and the mean age was 7.3 years.5
❚ Key symptoms and differential. The distinguishing feature of BACM is bilateral symmetric gastrocnemius-soleus tenderness.2,4 Additionally, the lack of neurologic symptoms is an important differentiator, as long as refusal to bear weight is not mistaken for weakness.6 These features help to distinguish BACM from other items in the differential, including trauma, Guillain-Barre syndrome, osteomyelitis, malignancy, deep vein thrombosis, and inherited musculoskeletal disorders.2
Continue to: Labratory evaluation...
❚ Laboratory evaluation will often show mild neutropenia, thrombocytopenia, and mild elevation in CK.7,8 CRP is typically normal.4,7,9 In a retrospective study of 28 admissions for BACM from 2001 to 2012, common findings included leukopenia (35%), neutropenia (25%), and thrombocytopenia (21%). The median CK value was 4181 U/L.4 In another analysis of BACM cases, mean CK was 1872 U/L.5
❚ Biopsy is unnecessary; however, calf muscle samples from 11 of 12 children with suspected BACM due to influenza B infection were consistent with patchy necrosis without significant myositis.10
❚ Complications. Rhabdomyolysis, although rare, has been reported with BACM. In 1 analysis, 10 of 316 patients with influenza-associated myositis developed rhabdomyolysis; 8 experienced renal failure. Rhabdomyolysis was 4 times more likely to occur in girls, and 86% of cases were associated with influenza A.6 Common manifestations of rhabdomyolysis associated with influenza include diffuse myopathy, gross hematuria, and myoglobinuria.6
❚ Treatment is mainly supportive.4,8,9 Antivirals typically are not indicated, as the bilateral calf pain manifests during the recovery phase of the illness.4,9,11 BACM is self-limited and should resolve within 3 days of myositis manifestation.2 Patients should follow up in 2 to 3 weeks to verify symptom resolution.2
If muscle pain, swelling, and tenderness worsen, further work-up is indicated. In more severe cases, including those involving renal failure, intensive care management and even dialysis may be necessary.4,6
❚ Our patient was hospitalized due to fever in the setting of neutropenia. Ultimately, he was treated with acetaminophen and intravenous fluids for mild dehydration and elevated CK levels. He was discharged home after 3 days, at which time he had complete resolution of pain and was able to resume normal activities.
The Takeaway
Benign acute childhood myositis is a self-limited disorder with an excellent prognosis. It has a typical presentation and therefore should be a clinical diagnosis; however, investigative studies may be warranted to rule out more ominous causes. Reassurance to family that the condition should self-resolve in a few days is important. Close follow-up should be scheduled to ensure resolution of symptoms.
CORRESPONDENCE
Nicholas A. Rathjen, DO, William Beaumont Army Medical Center, Department of Soldier and Family Care, 11335 SSG Sims Street, Fort Bliss, TX 79918; nicholas.a.rathjen@gmail. com
- Lundberg A. Myalgia cruris epidemica. Acta Paediatr. 1957;46:18-31. doi: 10.1111/j.1651-2227.1957.tb08627.x
- Magee H, Goldman RD. Viral myositis in children. Can Fam Physician. 2017;63:365-368.
- Mall S, Buchholz U, Tibussek D, et al. A large outbreak of influenza B-associated benign acute childhood myositis in Germany, 2007/2008. Pediatr Infect Dis J. 2011;30:e142-e146. doi: 10.1097/INF.0b013e318217e356
- Santos JA, Albuquerque C, Lito D, et al. Benign acute childhood myositis: an alarming condition with an excellent prognosis! Am J Emerg Med. 2014;32:1418-1419. doi: 10.1016/j.ajem.2014.08.022
- Rosenberg T, Heitner S, Scolnik D, et al. Outcome of benign acute childhood myositis: the experience of 2 large tertiary care pediatric hospitals. Pediatr Emerg Care. 2018;34:400-402. doi: 10.1097/PEC.0000000000000830
- Agyeman P, Duppenthaler A, Heininger U, et al. Influenza-associated myositis in children. Infection. 2004;32:199-203. doi: 10.1007/s15010-004-4003-2
- Mackay MT, Kornberg AJ, Shield LK, et al. Benign acute childhood myositis: laboratory and clinical features. Neurology. 1999;53:2127-2131. doi: 10.1212/wnl.53.9.2127
- Neocleous C, Spanou C, Mpampalis E, et al. Unnecessary diagnostic investigations in benign acute childhood myositis: a case series report. Scott Med J. 2012;57:182. doi: 10.1258/smj.2012.012023
- Felipe Cavagnaro SM, Alejandra Aird G, Ingrid Harwardt R, et al. Benign acute childhood myositis: clinical series and literature review. Rev Chil Pediatr. 2017;88:268-274. doi: 10.1016/j.rchipe.2016.07.002
- Bove KE, Hilton PK, Partin J, et al. Morphology of acute myopathy associated with influenza B infection. Pediatric Pathology. 1983;1:51-66. https://doi.org/10.3109/15513818309048284
- Koliou M, Hadjiloizou S, Ourani S, et al. A case of benign acute childhood myositis associated with influenza A (HINI) virus infection. Clin Microbiol Infect. 2010;16:193-195. doi: 10.1111/j.1469-0691.2009.03064.x
THE CASE
A 5-year-old previously healthy white boy presented to clinic with bilateral calf pain and refusal to bear weight since awakening that morning. Associated symptoms included a 3-day history of generalized fatigue, subjective fevers, cough, congestion, and rhinitis. The night prior to presentation, he showed no symptoms of gait abnormalities, muscle pain, or weakness. There was no history of similar symptoms, trauma, overexertion, foreign travel, or family history of musculoskeletal disease. He was fully immunized, except for the annual influenza vaccine. He was not taking any medications. This case occurred before the onset of the COVID-19 pandemic.
Objective findings included fever of 101 °F, refusal to bear weight, and symmetrical bilateral tenderness to palpation of the gastrocnemius-soleus complex. Pain was elicited with passive dorsiflexion. There was no erythema, edema, or sensory deficits, and the distal leg compartments were soft. There was normal range of motion of the hips, knees, and ankles. Dorsalis pedis pulses were 2+, and patella reflexes were 2/4 bilaterally.
Lab results included a white blood cell count of 2500/μL (normal range, 4500 to 11,000/μL);absolute neutrophil count, 900/μL (1500 to 8000/μL); platelet count, 131,000/μL (150,000 to 450,000/μL); creatine kinase level, 869 IU/L (22 to 198 U/L); and aspartate aminotransferase level, 116 U/L (8 to 33 U/L). A rapid influenza swab was positive for influenza B. Plain films of the bilateral hips and lower extremities were unremarkable. C-reactive protein (CRP) level, urinalysis, and renal function tests were within normal limits. Creatine kinase (CK) level peaked (1935 U/L; normal range, 22 to 198 U/L) within the first 24 hours of presentation and then trended down.
The Diagnosis
The patient’s sudden onset of symmetrical bilateral calf pain in the setting of an upper respiratory tract infection was extremely suspicious for benign acute childhood myositis (BACM). Lab work and radiologic evaluation were performed to rule out more ominous causes of refusal to bear weight. The suspicion of BACM was further validated by influenza B serology, an elevated CK, and a normal CRP.
Discussion
BACM was first described by Lundberg in 1957.1 The overall incidence and prevalence are unclear.2 A viral prodrome involving rhinorrhea, low-grade fever, sore throat, cough, and malaise typically precedes bilateral calf pain by 3 days.2-4 Myositis symptoms typically last for 4 days.3 While several infectious etiologies have been linked to this condition, influenza B has the greatest association.5,6
❚ Patient population. BACM occurs predominately in school-aged children (6-8 years old) and has a male-to-female ratio of 2:1.3,5,6 In a retrospective study of 219 children, BACM was strongly associated with male gender and ages 6 to 9 years.3 In another retrospective study of 54 children,80% of patients were male, and the mean age was 7.3 years.5
❚ Key symptoms and differential. The distinguishing feature of BACM is bilateral symmetric gastrocnemius-soleus tenderness.2,4 Additionally, the lack of neurologic symptoms is an important differentiator, as long as refusal to bear weight is not mistaken for weakness.6 These features help to distinguish BACM from other items in the differential, including trauma, Guillain-Barre syndrome, osteomyelitis, malignancy, deep vein thrombosis, and inherited musculoskeletal disorders.2
Continue to: Labratory evaluation...
❚ Laboratory evaluation will often show mild neutropenia, thrombocytopenia, and mild elevation in CK.7,8 CRP is typically normal.4,7,9 In a retrospective study of 28 admissions for BACM from 2001 to 2012, common findings included leukopenia (35%), neutropenia (25%), and thrombocytopenia (21%). The median CK value was 4181 U/L.4 In another analysis of BACM cases, mean CK was 1872 U/L.5
❚ Biopsy is unnecessary; however, calf muscle samples from 11 of 12 children with suspected BACM due to influenza B infection were consistent with patchy necrosis without significant myositis.10
❚ Complications. Rhabdomyolysis, although rare, has been reported with BACM. In 1 analysis, 10 of 316 patients with influenza-associated myositis developed rhabdomyolysis; 8 experienced renal failure. Rhabdomyolysis was 4 times more likely to occur in girls, and 86% of cases were associated with influenza A.6 Common manifestations of rhabdomyolysis associated with influenza include diffuse myopathy, gross hematuria, and myoglobinuria.6
❚ Treatment is mainly supportive.4,8,9 Antivirals typically are not indicated, as the bilateral calf pain manifests during the recovery phase of the illness.4,9,11 BACM is self-limited and should resolve within 3 days of myositis manifestation.2 Patients should follow up in 2 to 3 weeks to verify symptom resolution.2
If muscle pain, swelling, and tenderness worsen, further work-up is indicated. In more severe cases, including those involving renal failure, intensive care management and even dialysis may be necessary.4,6
❚ Our patient was hospitalized due to fever in the setting of neutropenia. Ultimately, he was treated with acetaminophen and intravenous fluids for mild dehydration and elevated CK levels. He was discharged home after 3 days, at which time he had complete resolution of pain and was able to resume normal activities.
The Takeaway
Benign acute childhood myositis is a self-limited disorder with an excellent prognosis. It has a typical presentation and therefore should be a clinical diagnosis; however, investigative studies may be warranted to rule out more ominous causes. Reassurance to family that the condition should self-resolve in a few days is important. Close follow-up should be scheduled to ensure resolution of symptoms.
CORRESPONDENCE
Nicholas A. Rathjen, DO, William Beaumont Army Medical Center, Department of Soldier and Family Care, 11335 SSG Sims Street, Fort Bliss, TX 79918; nicholas.a.rathjen@gmail. com
THE CASE
A 5-year-old previously healthy white boy presented to clinic with bilateral calf pain and refusal to bear weight since awakening that morning. Associated symptoms included a 3-day history of generalized fatigue, subjective fevers, cough, congestion, and rhinitis. The night prior to presentation, he showed no symptoms of gait abnormalities, muscle pain, or weakness. There was no history of similar symptoms, trauma, overexertion, foreign travel, or family history of musculoskeletal disease. He was fully immunized, except for the annual influenza vaccine. He was not taking any medications. This case occurred before the onset of the COVID-19 pandemic.
Objective findings included fever of 101 °F, refusal to bear weight, and symmetrical bilateral tenderness to palpation of the gastrocnemius-soleus complex. Pain was elicited with passive dorsiflexion. There was no erythema, edema, or sensory deficits, and the distal leg compartments were soft. There was normal range of motion of the hips, knees, and ankles. Dorsalis pedis pulses were 2+, and patella reflexes were 2/4 bilaterally.
Lab results included a white blood cell count of 2500/μL (normal range, 4500 to 11,000/μL);absolute neutrophil count, 900/μL (1500 to 8000/μL); platelet count, 131,000/μL (150,000 to 450,000/μL); creatine kinase level, 869 IU/L (22 to 198 U/L); and aspartate aminotransferase level, 116 U/L (8 to 33 U/L). A rapid influenza swab was positive for influenza B. Plain films of the bilateral hips and lower extremities were unremarkable. C-reactive protein (CRP) level, urinalysis, and renal function tests were within normal limits. Creatine kinase (CK) level peaked (1935 U/L; normal range, 22 to 198 U/L) within the first 24 hours of presentation and then trended down.
The Diagnosis
The patient’s sudden onset of symmetrical bilateral calf pain in the setting of an upper respiratory tract infection was extremely suspicious for benign acute childhood myositis (BACM). Lab work and radiologic evaluation were performed to rule out more ominous causes of refusal to bear weight. The suspicion of BACM was further validated by influenza B serology, an elevated CK, and a normal CRP.
Discussion
BACM was first described by Lundberg in 1957.1 The overall incidence and prevalence are unclear.2 A viral prodrome involving rhinorrhea, low-grade fever, sore throat, cough, and malaise typically precedes bilateral calf pain by 3 days.2-4 Myositis symptoms typically last for 4 days.3 While several infectious etiologies have been linked to this condition, influenza B has the greatest association.5,6
❚ Patient population. BACM occurs predominately in school-aged children (6-8 years old) and has a male-to-female ratio of 2:1.3,5,6 In a retrospective study of 219 children, BACM was strongly associated with male gender and ages 6 to 9 years.3 In another retrospective study of 54 children,80% of patients were male, and the mean age was 7.3 years.5
❚ Key symptoms and differential. The distinguishing feature of BACM is bilateral symmetric gastrocnemius-soleus tenderness.2,4 Additionally, the lack of neurologic symptoms is an important differentiator, as long as refusal to bear weight is not mistaken for weakness.6 These features help to distinguish BACM from other items in the differential, including trauma, Guillain-Barre syndrome, osteomyelitis, malignancy, deep vein thrombosis, and inherited musculoskeletal disorders.2
Continue to: Labratory evaluation...
❚ Laboratory evaluation will often show mild neutropenia, thrombocytopenia, and mild elevation in CK.7,8 CRP is typically normal.4,7,9 In a retrospective study of 28 admissions for BACM from 2001 to 2012, common findings included leukopenia (35%), neutropenia (25%), and thrombocytopenia (21%). The median CK value was 4181 U/L.4 In another analysis of BACM cases, mean CK was 1872 U/L.5
❚ Biopsy is unnecessary; however, calf muscle samples from 11 of 12 children with suspected BACM due to influenza B infection were consistent with patchy necrosis without significant myositis.10
❚ Complications. Rhabdomyolysis, although rare, has been reported with BACM. In 1 analysis, 10 of 316 patients with influenza-associated myositis developed rhabdomyolysis; 8 experienced renal failure. Rhabdomyolysis was 4 times more likely to occur in girls, and 86% of cases were associated with influenza A.6 Common manifestations of rhabdomyolysis associated with influenza include diffuse myopathy, gross hematuria, and myoglobinuria.6
❚ Treatment is mainly supportive.4,8,9 Antivirals typically are not indicated, as the bilateral calf pain manifests during the recovery phase of the illness.4,9,11 BACM is self-limited and should resolve within 3 days of myositis manifestation.2 Patients should follow up in 2 to 3 weeks to verify symptom resolution.2
If muscle pain, swelling, and tenderness worsen, further work-up is indicated. In more severe cases, including those involving renal failure, intensive care management and even dialysis may be necessary.4,6
❚ Our patient was hospitalized due to fever in the setting of neutropenia. Ultimately, he was treated with acetaminophen and intravenous fluids for mild dehydration and elevated CK levels. He was discharged home after 3 days, at which time he had complete resolution of pain and was able to resume normal activities.
The Takeaway
Benign acute childhood myositis is a self-limited disorder with an excellent prognosis. It has a typical presentation and therefore should be a clinical diagnosis; however, investigative studies may be warranted to rule out more ominous causes. Reassurance to family that the condition should self-resolve in a few days is important. Close follow-up should be scheduled to ensure resolution of symptoms.
CORRESPONDENCE
Nicholas A. Rathjen, DO, William Beaumont Army Medical Center, Department of Soldier and Family Care, 11335 SSG Sims Street, Fort Bliss, TX 79918; nicholas.a.rathjen@gmail. com
- Lundberg A. Myalgia cruris epidemica. Acta Paediatr. 1957;46:18-31. doi: 10.1111/j.1651-2227.1957.tb08627.x
- Magee H, Goldman RD. Viral myositis in children. Can Fam Physician. 2017;63:365-368.
- Mall S, Buchholz U, Tibussek D, et al. A large outbreak of influenza B-associated benign acute childhood myositis in Germany, 2007/2008. Pediatr Infect Dis J. 2011;30:e142-e146. doi: 10.1097/INF.0b013e318217e356
- Santos JA, Albuquerque C, Lito D, et al. Benign acute childhood myositis: an alarming condition with an excellent prognosis! Am J Emerg Med. 2014;32:1418-1419. doi: 10.1016/j.ajem.2014.08.022
- Rosenberg T, Heitner S, Scolnik D, et al. Outcome of benign acute childhood myositis: the experience of 2 large tertiary care pediatric hospitals. Pediatr Emerg Care. 2018;34:400-402. doi: 10.1097/PEC.0000000000000830
- Agyeman P, Duppenthaler A, Heininger U, et al. Influenza-associated myositis in children. Infection. 2004;32:199-203. doi: 10.1007/s15010-004-4003-2
- Mackay MT, Kornberg AJ, Shield LK, et al. Benign acute childhood myositis: laboratory and clinical features. Neurology. 1999;53:2127-2131. doi: 10.1212/wnl.53.9.2127
- Neocleous C, Spanou C, Mpampalis E, et al. Unnecessary diagnostic investigations in benign acute childhood myositis: a case series report. Scott Med J. 2012;57:182. doi: 10.1258/smj.2012.012023
- Felipe Cavagnaro SM, Alejandra Aird G, Ingrid Harwardt R, et al. Benign acute childhood myositis: clinical series and literature review. Rev Chil Pediatr. 2017;88:268-274. doi: 10.1016/j.rchipe.2016.07.002
- Bove KE, Hilton PK, Partin J, et al. Morphology of acute myopathy associated with influenza B infection. Pediatric Pathology. 1983;1:51-66. https://doi.org/10.3109/15513818309048284
- Koliou M, Hadjiloizou S, Ourani S, et al. A case of benign acute childhood myositis associated with influenza A (HINI) virus infection. Clin Microbiol Infect. 2010;16:193-195. doi: 10.1111/j.1469-0691.2009.03064.x
- Lundberg A. Myalgia cruris epidemica. Acta Paediatr. 1957;46:18-31. doi: 10.1111/j.1651-2227.1957.tb08627.x
- Magee H, Goldman RD. Viral myositis in children. Can Fam Physician. 2017;63:365-368.
- Mall S, Buchholz U, Tibussek D, et al. A large outbreak of influenza B-associated benign acute childhood myositis in Germany, 2007/2008. Pediatr Infect Dis J. 2011;30:e142-e146. doi: 10.1097/INF.0b013e318217e356
- Santos JA, Albuquerque C, Lito D, et al. Benign acute childhood myositis: an alarming condition with an excellent prognosis! Am J Emerg Med. 2014;32:1418-1419. doi: 10.1016/j.ajem.2014.08.022
- Rosenberg T, Heitner S, Scolnik D, et al. Outcome of benign acute childhood myositis: the experience of 2 large tertiary care pediatric hospitals. Pediatr Emerg Care. 2018;34:400-402. doi: 10.1097/PEC.0000000000000830
- Agyeman P, Duppenthaler A, Heininger U, et al. Influenza-associated myositis in children. Infection. 2004;32:199-203. doi: 10.1007/s15010-004-4003-2
- Mackay MT, Kornberg AJ, Shield LK, et al. Benign acute childhood myositis: laboratory and clinical features. Neurology. 1999;53:2127-2131. doi: 10.1212/wnl.53.9.2127
- Neocleous C, Spanou C, Mpampalis E, et al. Unnecessary diagnostic investigations in benign acute childhood myositis: a case series report. Scott Med J. 2012;57:182. doi: 10.1258/smj.2012.012023
- Felipe Cavagnaro SM, Alejandra Aird G, Ingrid Harwardt R, et al. Benign acute childhood myositis: clinical series and literature review. Rev Chil Pediatr. 2017;88:268-274. doi: 10.1016/j.rchipe.2016.07.002
- Bove KE, Hilton PK, Partin J, et al. Morphology of acute myopathy associated with influenza B infection. Pediatric Pathology. 1983;1:51-66. https://doi.org/10.3109/15513818309048284
- Koliou M, Hadjiloizou S, Ourani S, et al. A case of benign acute childhood myositis associated with influenza A (HINI) virus infection. Clin Microbiol Infect. 2010;16:193-195. doi: 10.1111/j.1469-0691.2009.03064.x
Stopping Empagliflozin Unmasks Heart Failure
SGLT2 inhibitors have been shown to have a role in the management of heart failure in patients with type 2 diabetes mellitus, but there is a risk of exacerbation when discontinued.
About 40% of patients with heart failure (HF) also have type 2 diabetes mellitus (T2DM).1 Certain sodium-glucose cotransporter-2 (SGLT2) inhibitors have benefited patients with HF.2 We report a case of a patient with T2DM who had signs and symptoms of hypervolemia after discontinuing the SGLT2 inhibitor empagliflozin. The patient was found to have previously undiagnosed HF.
Case Presentation
A 58-year-old male presented for care at Malcolm Randall Veterans Affairs Medical Center in Gainseville, Florida, diabetes clinic. The patient was diagnosed with T2DM at age 32 years. At 36 years, he was started on subcutaneous insulin injections, and was switched to insulin pump therapy in his early 40s. At the time of evaluation, the T2DM was managed using an insulin pump, metformin, and acarbose. He had been prescribed empagliflozin 10 mg several months before presentation, but the medication ran out about 1 month prior to evaluation, and additional refills were unavailable.
The patient reported a 1-month history of worsening exertional shortness of breath, decreased exercise tolerance, and lower extremity swelling. Vitals signs, including respiratory rate and oxygen saturation were within normal limits. Bibasilar crackles and bilateral 2+ pitting pedal edema were noted. The remaining examination was unrevealing. His most recent glycated hemoglobin A1c level from 1 month prior to the presentation was 6.4%.
Given the patient’s shortness of breath and evidence of fluid overload on examination, brain natriuretic peptide was obtained and was significantly elevated at 5,895 pg/mL. A transthoracic echocardiogram revealed left ventricular ejection fraction < 20%. The patient was started on furosemide 40 mg, pending receipt of empagliflozin. A cardiology evaluation also was recommended.
Cardiac catheterization identified significant obstructions to the left anterior descending and left circumflex arteries. The patient underwent successful percutaneous coronary intervention to these areas. Following initiation of medications and coronary revascularization, the patient reported significant symptom improvement. At the follow-up evaluation 8 weeks later, he was symptom free, and his physical examination was consistent with euvolemia.
Discussion
T2DM has been associated with adverse cardiovascular outcomes, including atherosclerotic heart disease and HF. There are several theories about the relationship between T2DM and HF, though the exact pathophysiology of this relationship is unknown.3,4 One theory suggests diabetic cardiomyopathy as the cause. In patients with diabetic cardiomyopathy, there is early development of diastolic dysfunction, which eventually progresses to ventricular dysfunction. There is continued stimulation of the renin-angiotensin-aldosterone system that leads to death of cardiomyocytes, fibrosis, and remodeling, which worsens pump failure.5
SGLT2 inhibitors decrease hyperglycemia and hyperinsulinemia, potentially reducing HF risk. SGLT2 inhibitors decrease blood glucose levels by inhibiting SGLT2 in the proximal tubule, leading to a decrease of glucose reabsorption and an increase in excretion.6,7 The EMPA-REG OUTCOME trial looked at cardiovascular outcomes in patients with T2DM at high risk for adverse cardiac events. There was a significant risk reduction in deaths and hospitalizations for HF in patients treated with empagliflozin.8
The EMPRISE study specifically examined empagliflozin and its effects on hospitalization for HF.2 When compared with patients treated with sitagliptin, there was a statistically significant decrease in hospitalization for HF in patients with T2DM, both with and without preexisting cardiovascular disease.
This case highlights the relationship between T2DM and HF. We also show how the use of empagliflozin may have helped manage the patient’s undiagnosed HF and how its discontinuation luckily unmasked it. Routine evaluation for HF in patients with T2DM is not done, but likely there are patients who would benefit, especially given the strong, albeit less known, association between these 2 conditions.
Further studies are needed to determine the type of patients who would benefit most from HF screening. For now, the best practice is to obtain a complete medical history that includes current and recently discontinued medications as well a thorough physical examination for signs of fluid overload and cardiovascular compromise. Patients who may have signs concerning for HF can have appropriate testing and intervention.
Conclusions
SGLT2 inhibitors have been shown to have a role in the management of HF in patients with T2DM. There is a risk of exacerbation or unmasking of HF when discontinuing SGLT2 inhibitors. To our knowledge, this is the first paper describing the discovery of HF following interruption of SGLT2 inhibitor treatment. The clinician and patient should monitor for signs and symptoms of fluid overload when stopping therapy. Further research into the benefits of a more comprehensive evaluation is needed.
1. Thomas MC. Type 2 diabetes and heart failure: challenges and solutions. Curr Cardiol Rev. 2016;12(3):249-255. doi:10.2174/1573403X12666160606120254
2. Patorno E, Pawar A, Franklin J, et al. Empagliflozin and the risk of heart failure hospitalization in routine clinical care: a first analysis from the EMPRISE study. Circulation. 2019;139(25):2822-2830. doi:10.1161/CIRCULATIONAHA.118.039177
3. Packer M. Heart failure: the most important, preventable, and treatable cardiovascular complication of type 2 diabetes. Diabetes Care. 2018;41(1):11-13. doi:10.2337/dci17-0052
4. Thrainsdottir I, Aspelund T, Thorheirsson G, et al. The association between glucose abnormalities and heart failure in the population-based Reykjavík study. Diabetes Care. 2005;28(3):612-616. doi:10.2337/diacare.28.3.612
5. Bell D, Goncalves E. Heart failure in the patient with diabetes: epidemiology, aetiology, prognosis, therapy and the effect of glucose-lowering medications. Diabetes Obes Metab. 2019;21(6):1277-1290. doi:10.1111/dom.13652
6. Nair S, Wilding JPH. Sodium glucose cotransporter 2 Inhibitors as a new treatment for diabetes mellitus. J Clin Endocrinol Metab. 2010;95(1):34-42. doi:10.1210/jc.2009-0473
7. Ali A, Bain S, Hicks D, et al; Improving Diabetes Steering Committee. SGLT2 inhibitors: cardiovascular benefits beyond HbA1c- translating evidence into practice. Diabetes Ther. 2019;10(5):1595-1622. doi:10.1007/s13300-019-0657-8
8. Zinman B, Wanner C, Lachin J, et al; EMPA-REG OUTCOME Investigators. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373(22):2117-2128. doi:10.1056/NEJMoa1504720
SGLT2 inhibitors have been shown to have a role in the management of heart failure in patients with type 2 diabetes mellitus, but there is a risk of exacerbation when discontinued.
SGLT2 inhibitors have been shown to have a role in the management of heart failure in patients with type 2 diabetes mellitus, but there is a risk of exacerbation when discontinued.
About 40% of patients with heart failure (HF) also have type 2 diabetes mellitus (T2DM).1 Certain sodium-glucose cotransporter-2 (SGLT2) inhibitors have benefited patients with HF.2 We report a case of a patient with T2DM who had signs and symptoms of hypervolemia after discontinuing the SGLT2 inhibitor empagliflozin. The patient was found to have previously undiagnosed HF.
Case Presentation
A 58-year-old male presented for care at Malcolm Randall Veterans Affairs Medical Center in Gainseville, Florida, diabetes clinic. The patient was diagnosed with T2DM at age 32 years. At 36 years, he was started on subcutaneous insulin injections, and was switched to insulin pump therapy in his early 40s. At the time of evaluation, the T2DM was managed using an insulin pump, metformin, and acarbose. He had been prescribed empagliflozin 10 mg several months before presentation, but the medication ran out about 1 month prior to evaluation, and additional refills were unavailable.
The patient reported a 1-month history of worsening exertional shortness of breath, decreased exercise tolerance, and lower extremity swelling. Vitals signs, including respiratory rate and oxygen saturation were within normal limits. Bibasilar crackles and bilateral 2+ pitting pedal edema were noted. The remaining examination was unrevealing. His most recent glycated hemoglobin A1c level from 1 month prior to the presentation was 6.4%.
Given the patient’s shortness of breath and evidence of fluid overload on examination, brain natriuretic peptide was obtained and was significantly elevated at 5,895 pg/mL. A transthoracic echocardiogram revealed left ventricular ejection fraction < 20%. The patient was started on furosemide 40 mg, pending receipt of empagliflozin. A cardiology evaluation also was recommended.
Cardiac catheterization identified significant obstructions to the left anterior descending and left circumflex arteries. The patient underwent successful percutaneous coronary intervention to these areas. Following initiation of medications and coronary revascularization, the patient reported significant symptom improvement. At the follow-up evaluation 8 weeks later, he was symptom free, and his physical examination was consistent with euvolemia.
Discussion
T2DM has been associated with adverse cardiovascular outcomes, including atherosclerotic heart disease and HF. There are several theories about the relationship between T2DM and HF, though the exact pathophysiology of this relationship is unknown.3,4 One theory suggests diabetic cardiomyopathy as the cause. In patients with diabetic cardiomyopathy, there is early development of diastolic dysfunction, which eventually progresses to ventricular dysfunction. There is continued stimulation of the renin-angiotensin-aldosterone system that leads to death of cardiomyocytes, fibrosis, and remodeling, which worsens pump failure.5
SGLT2 inhibitors decrease hyperglycemia and hyperinsulinemia, potentially reducing HF risk. SGLT2 inhibitors decrease blood glucose levels by inhibiting SGLT2 in the proximal tubule, leading to a decrease of glucose reabsorption and an increase in excretion.6,7 The EMPA-REG OUTCOME trial looked at cardiovascular outcomes in patients with T2DM at high risk for adverse cardiac events. There was a significant risk reduction in deaths and hospitalizations for HF in patients treated with empagliflozin.8
The EMPRISE study specifically examined empagliflozin and its effects on hospitalization for HF.2 When compared with patients treated with sitagliptin, there was a statistically significant decrease in hospitalization for HF in patients with T2DM, both with and without preexisting cardiovascular disease.
This case highlights the relationship between T2DM and HF. We also show how the use of empagliflozin may have helped manage the patient’s undiagnosed HF and how its discontinuation luckily unmasked it. Routine evaluation for HF in patients with T2DM is not done, but likely there are patients who would benefit, especially given the strong, albeit less known, association between these 2 conditions.
Further studies are needed to determine the type of patients who would benefit most from HF screening. For now, the best practice is to obtain a complete medical history that includes current and recently discontinued medications as well a thorough physical examination for signs of fluid overload and cardiovascular compromise. Patients who may have signs concerning for HF can have appropriate testing and intervention.
Conclusions
SGLT2 inhibitors have been shown to have a role in the management of HF in patients with T2DM. There is a risk of exacerbation or unmasking of HF when discontinuing SGLT2 inhibitors. To our knowledge, this is the first paper describing the discovery of HF following interruption of SGLT2 inhibitor treatment. The clinician and patient should monitor for signs and symptoms of fluid overload when stopping therapy. Further research into the benefits of a more comprehensive evaluation is needed.
About 40% of patients with heart failure (HF) also have type 2 diabetes mellitus (T2DM).1 Certain sodium-glucose cotransporter-2 (SGLT2) inhibitors have benefited patients with HF.2 We report a case of a patient with T2DM who had signs and symptoms of hypervolemia after discontinuing the SGLT2 inhibitor empagliflozin. The patient was found to have previously undiagnosed HF.
Case Presentation
A 58-year-old male presented for care at Malcolm Randall Veterans Affairs Medical Center in Gainseville, Florida, diabetes clinic. The patient was diagnosed with T2DM at age 32 years. At 36 years, he was started on subcutaneous insulin injections, and was switched to insulin pump therapy in his early 40s. At the time of evaluation, the T2DM was managed using an insulin pump, metformin, and acarbose. He had been prescribed empagliflozin 10 mg several months before presentation, but the medication ran out about 1 month prior to evaluation, and additional refills were unavailable.
The patient reported a 1-month history of worsening exertional shortness of breath, decreased exercise tolerance, and lower extremity swelling. Vitals signs, including respiratory rate and oxygen saturation were within normal limits. Bibasilar crackles and bilateral 2+ pitting pedal edema were noted. The remaining examination was unrevealing. His most recent glycated hemoglobin A1c level from 1 month prior to the presentation was 6.4%.
Given the patient’s shortness of breath and evidence of fluid overload on examination, brain natriuretic peptide was obtained and was significantly elevated at 5,895 pg/mL. A transthoracic echocardiogram revealed left ventricular ejection fraction < 20%. The patient was started on furosemide 40 mg, pending receipt of empagliflozin. A cardiology evaluation also was recommended.
Cardiac catheterization identified significant obstructions to the left anterior descending and left circumflex arteries. The patient underwent successful percutaneous coronary intervention to these areas. Following initiation of medications and coronary revascularization, the patient reported significant symptom improvement. At the follow-up evaluation 8 weeks later, he was symptom free, and his physical examination was consistent with euvolemia.
Discussion
T2DM has been associated with adverse cardiovascular outcomes, including atherosclerotic heart disease and HF. There are several theories about the relationship between T2DM and HF, though the exact pathophysiology of this relationship is unknown.3,4 One theory suggests diabetic cardiomyopathy as the cause. In patients with diabetic cardiomyopathy, there is early development of diastolic dysfunction, which eventually progresses to ventricular dysfunction. There is continued stimulation of the renin-angiotensin-aldosterone system that leads to death of cardiomyocytes, fibrosis, and remodeling, which worsens pump failure.5
SGLT2 inhibitors decrease hyperglycemia and hyperinsulinemia, potentially reducing HF risk. SGLT2 inhibitors decrease blood glucose levels by inhibiting SGLT2 in the proximal tubule, leading to a decrease of glucose reabsorption and an increase in excretion.6,7 The EMPA-REG OUTCOME trial looked at cardiovascular outcomes in patients with T2DM at high risk for adverse cardiac events. There was a significant risk reduction in deaths and hospitalizations for HF in patients treated with empagliflozin.8
The EMPRISE study specifically examined empagliflozin and its effects on hospitalization for HF.2 When compared with patients treated with sitagliptin, there was a statistically significant decrease in hospitalization for HF in patients with T2DM, both with and without preexisting cardiovascular disease.
This case highlights the relationship between T2DM and HF. We also show how the use of empagliflozin may have helped manage the patient’s undiagnosed HF and how its discontinuation luckily unmasked it. Routine evaluation for HF in patients with T2DM is not done, but likely there are patients who would benefit, especially given the strong, albeit less known, association between these 2 conditions.
Further studies are needed to determine the type of patients who would benefit most from HF screening. For now, the best practice is to obtain a complete medical history that includes current and recently discontinued medications as well a thorough physical examination for signs of fluid overload and cardiovascular compromise. Patients who may have signs concerning for HF can have appropriate testing and intervention.
Conclusions
SGLT2 inhibitors have been shown to have a role in the management of HF in patients with T2DM. There is a risk of exacerbation or unmasking of HF when discontinuing SGLT2 inhibitors. To our knowledge, this is the first paper describing the discovery of HF following interruption of SGLT2 inhibitor treatment. The clinician and patient should monitor for signs and symptoms of fluid overload when stopping therapy. Further research into the benefits of a more comprehensive evaluation is needed.
1. Thomas MC. Type 2 diabetes and heart failure: challenges and solutions. Curr Cardiol Rev. 2016;12(3):249-255. doi:10.2174/1573403X12666160606120254
2. Patorno E, Pawar A, Franklin J, et al. Empagliflozin and the risk of heart failure hospitalization in routine clinical care: a first analysis from the EMPRISE study. Circulation. 2019;139(25):2822-2830. doi:10.1161/CIRCULATIONAHA.118.039177
3. Packer M. Heart failure: the most important, preventable, and treatable cardiovascular complication of type 2 diabetes. Diabetes Care. 2018;41(1):11-13. doi:10.2337/dci17-0052
4. Thrainsdottir I, Aspelund T, Thorheirsson G, et al. The association between glucose abnormalities and heart failure in the population-based Reykjavík study. Diabetes Care. 2005;28(3):612-616. doi:10.2337/diacare.28.3.612
5. Bell D, Goncalves E. Heart failure in the patient with diabetes: epidemiology, aetiology, prognosis, therapy and the effect of glucose-lowering medications. Diabetes Obes Metab. 2019;21(6):1277-1290. doi:10.1111/dom.13652
6. Nair S, Wilding JPH. Sodium glucose cotransporter 2 Inhibitors as a new treatment for diabetes mellitus. J Clin Endocrinol Metab. 2010;95(1):34-42. doi:10.1210/jc.2009-0473
7. Ali A, Bain S, Hicks D, et al; Improving Diabetes Steering Committee. SGLT2 inhibitors: cardiovascular benefits beyond HbA1c- translating evidence into practice. Diabetes Ther. 2019;10(5):1595-1622. doi:10.1007/s13300-019-0657-8
8. Zinman B, Wanner C, Lachin J, et al; EMPA-REG OUTCOME Investigators. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373(22):2117-2128. doi:10.1056/NEJMoa1504720
1. Thomas MC. Type 2 diabetes and heart failure: challenges and solutions. Curr Cardiol Rev. 2016;12(3):249-255. doi:10.2174/1573403X12666160606120254
2. Patorno E, Pawar A, Franklin J, et al. Empagliflozin and the risk of heart failure hospitalization in routine clinical care: a first analysis from the EMPRISE study. Circulation. 2019;139(25):2822-2830. doi:10.1161/CIRCULATIONAHA.118.039177
3. Packer M. Heart failure: the most important, preventable, and treatable cardiovascular complication of type 2 diabetes. Diabetes Care. 2018;41(1):11-13. doi:10.2337/dci17-0052
4. Thrainsdottir I, Aspelund T, Thorheirsson G, et al. The association between glucose abnormalities and heart failure in the population-based Reykjavík study. Diabetes Care. 2005;28(3):612-616. doi:10.2337/diacare.28.3.612
5. Bell D, Goncalves E. Heart failure in the patient with diabetes: epidemiology, aetiology, prognosis, therapy and the effect of glucose-lowering medications. Diabetes Obes Metab. 2019;21(6):1277-1290. doi:10.1111/dom.13652
6. Nair S, Wilding JPH. Sodium glucose cotransporter 2 Inhibitors as a new treatment for diabetes mellitus. J Clin Endocrinol Metab. 2010;95(1):34-42. doi:10.1210/jc.2009-0473
7. Ali A, Bain S, Hicks D, et al; Improving Diabetes Steering Committee. SGLT2 inhibitors: cardiovascular benefits beyond HbA1c- translating evidence into practice. Diabetes Ther. 2019;10(5):1595-1622. doi:10.1007/s13300-019-0657-8
8. Zinman B, Wanner C, Lachin J, et al; EMPA-REG OUTCOME Investigators. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373(22):2117-2128. doi:10.1056/NEJMoa1504720
Nivolumab-Induced Granuloma Annulare
Granuloma annulare (GA) is a benign, cutaneous, granulomatous disease of unclear etiology. Typically, GA presents in young adults as asymptomatic, annular, flesh-colored to pink papules and plaques, commonly on the upper and lower extremities. Histologically, GA is characterized by mucin deposition, palisading or an interstitial granulomatous pattern, and collagen and elastic fiber degeneration.1
Granuloma annulare has been associated with various medications and medical conditions, including diabetes mellitus, hyperlipidemia, thyroid disease, and HIV.1 More recently, immune-checkpoint inhibitors (ICIs) have been reported to trigger GA.2 We report a case of nivolumab-induced GA in a 54-year-old woman.
Case Report
A 54-year-old woman presented with an itchy rash on the upper extremities, face, and chest of 4 months’ duration. The patient noted that the rash started on the hands and progressed to include the arms, face, and chest. She also reported associated mild tenderness. She had a history of stage IV non–small-cell lung carcinoma with metastases to the ribs and adrenal glands. She had been started on biweekly intravenous infusions of the ICI nivolumab by her oncologist approximately 1 year prior to the current presentation after failing a course of conventional chemotherapy. The most recent positron emission tomography–computed tomography scan 1 month prior to presentation showed a stable lung mass with radiologic disappearance of metastases, indicating a favorable response to nivolumab. The patient also had a history of hypothyroidism and depression, which were treated with oral levothyroxine 75 μg once daily and oral sertraline 50 mg once daily, respectively, both for longer than 5 years.
Physical examination revealed annular, erythematous, flat-topped papules, some with surmounting fine scale, coalescing into larger plaques along the dorsal surface of the hands and arms (Figure 1) as well as the forehead and chest. A biopsy of a papule on the dorsal aspect of the left hand revealed nodules of histiocytes admixed with Langerhans giant cells within the dermis; mucin was noted centrally within some nodules (Figure 2). Periodic acid–Schiff staining was negative for fungal elements compared to control. Polarization of the specimen was negative for foreign bodies. The biopsy findings therefore were consistent with a diagnosis of GA.
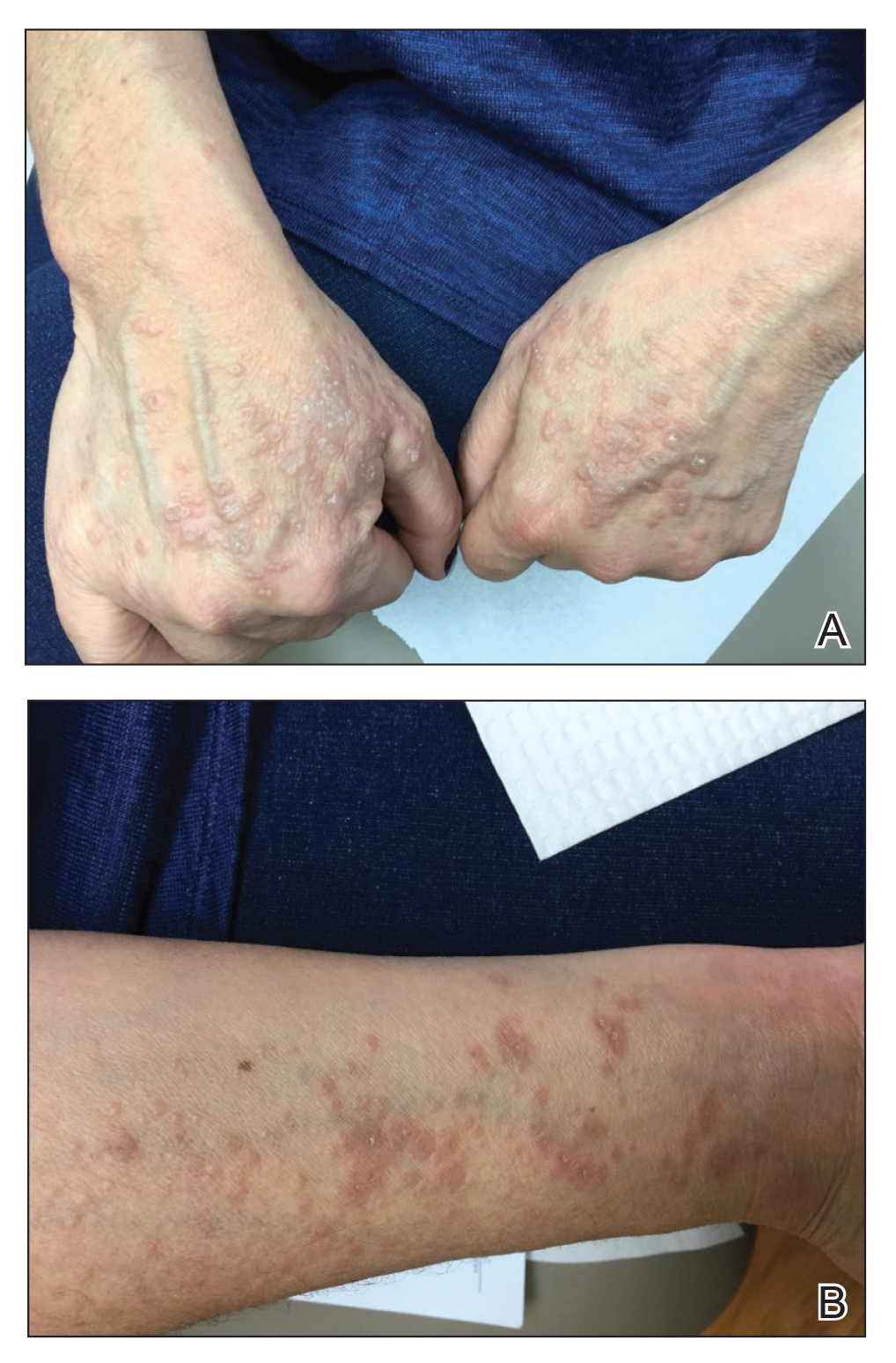
A 3-month treatment course of betamethasone dipropionate 0.05% cream twice daily failed. Narrowband UVB phototherapy was then initiated at 3 sessions weekly. The eruption of GA improved after 3 months of phototherapy. Subsequently, the patient was lost to follow-up.
Comment
Discovery of specific immune checkpoints in tumor-induced immunosuppression revolutionized oncologic therapy. An example is the programmed cell-death protein 1 (PD-1) receptor that is expressed on activated immune cells, including T cells and macrophages.3,4 Upon binding to the PD-1 ligand (PD-L1), T-cell proliferation is inhibited, resulting in downregulation of the immune response. As a result, tumor cells have evolved to overexpress PD-L1 to evade immunologic detection.3 Nivolumab, a fully human IgG4 antibody to PD-1, has emerged along with other ICIs as effective treatments for numerous cancers, including melanoma and non–small-cell lung cancer. By disrupting downregulation of T cells, ICIs improve immune-mediated antitumor activity.3
However, the resulting immunologic disturbance by ICIs has been reported to induce various cutaneous and systemic immune-mediated adverse reactions, including granulomatous reactions such as sarcoidosis, GA, and a cutaneous sarcoidlike granulomatous reaction.1,2,5,6 Our patient represents a rare case of nivolumab-induced GA.
Recent evidence suggests that GA might be caused in part by a cell-mediated hypersensitivity reaction that is regulated by a helper T cell subset 1 inflammatory reaction. Through release of cytokines by activated CD4+ T cells, macrophages are recruited, forming the granulomatous pattern and secreting enzymes that can degrade connective tissue. Nivolumab and other ICIs can thus trigger this reaction because their blockade of PD-1 enhances T cell–mediated immune reactions.2 In addition, because macrophages themselves also express PD-1, ICIs can directly enhance macrophage recruitment and proliferation, further increasing the risk of a granulomatous reaction.4
Interestingly, cutaneous adverse reactions to nivolumab have been associated with improved survival in melanoma patients.7 The nature of this association with granulomatous reactions in general and with GA specifically remains to be determined.
Conclusion
Since the approval of the first PD-1 inhibitors, pembrolizumab and nivolumab, in 2014, other ICIs targeting the immune checkpoint pathway have been developed. Newer agents targeting PD-L1 (avelumab, atezolizumab, and durvalumab) were recently approved. Additionally, cemiplimab, another PD-1 inhibitor, was approved by the US Food and Drug Administration in 2018 for the treatment of advanced cutaneous squamous cell carcinoma.8 Indications for all ICIs also have expanded considerably.3 Therefore, the incidence of immune-mediated adverse reactions, including GA, is bound to increase. Physicians should be cognizant of this association to accurately diagnose and effectively treat adverse reactions in patients who are taking ICIs.
- Piette EW, Rosenbach M. Granuloma annulare: pathogenesis, disease associations and triggers, and therapeutic options. J Am Acad Dermatol. 2016;75:467-479. doi:10.1016/j.jaad.2015.03.055
- Wu J, Kwong BY, Martires KJ, et al. Granuloma annulare associated with immune checkpoint inhibitors. J Eur Acad Dermatol. 2018;32:E124-E126. doi:10.1111/jdv.14617
- Gong J, Chehrazi-Raffle A, Reddi S, et al. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations. J Immunother Cancer. 2018;6:8. doi:10.1186/s40425-018-0316-z
- Gordon SR, Maute RL, Dulken BW, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545:495-499. doi:10.1038/nature22396
- Birnbaum MR, Ma MW, Fleisig S, et al. Nivolumab-related cutaneous sarcoidosis in a patient with lung adenocarcinoma. JAAD Case Rep. 2017;3:208-211. doi:10.1016/j.jdcr.2017.02.015
- Danlos F-X, Pagès C, Baroudjian B, et al. Nivolumab-induced sarcoid-like granulomatous reaction in a patient with advanced melanoma. Chest. 2016;149:E133-E136. doi:10.1016/j.chest.2015.10.082
- Freeman-Keller M, Kim Y, Cronin H, et al. Nivolumab in resected and unresectable metastatic melanoma: characteristics of immune-related adverse events and association with outcomes. Clin Cancer Res. 2016;22:886-894. doi:10.1158/1078-0432.CCR-15-1136
- Migden MR, Rischin D, Schmults CD, et al. PD-1 blockade with cemiplimab in advanced cutaneous squamous-cell carcinoma. N Engl J Med. 2018;379:341-351. doi:10.1056/NEJMoa1805131
Granuloma annulare (GA) is a benign, cutaneous, granulomatous disease of unclear etiology. Typically, GA presents in young adults as asymptomatic, annular, flesh-colored to pink papules and plaques, commonly on the upper and lower extremities. Histologically, GA is characterized by mucin deposition, palisading or an interstitial granulomatous pattern, and collagen and elastic fiber degeneration.1
Granuloma annulare has been associated with various medications and medical conditions, including diabetes mellitus, hyperlipidemia, thyroid disease, and HIV.1 More recently, immune-checkpoint inhibitors (ICIs) have been reported to trigger GA.2 We report a case of nivolumab-induced GA in a 54-year-old woman.
Case Report
A 54-year-old woman presented with an itchy rash on the upper extremities, face, and chest of 4 months’ duration. The patient noted that the rash started on the hands and progressed to include the arms, face, and chest. She also reported associated mild tenderness. She had a history of stage IV non–small-cell lung carcinoma with metastases to the ribs and adrenal glands. She had been started on biweekly intravenous infusions of the ICI nivolumab by her oncologist approximately 1 year prior to the current presentation after failing a course of conventional chemotherapy. The most recent positron emission tomography–computed tomography scan 1 month prior to presentation showed a stable lung mass with radiologic disappearance of metastases, indicating a favorable response to nivolumab. The patient also had a history of hypothyroidism and depression, which were treated with oral levothyroxine 75 μg once daily and oral sertraline 50 mg once daily, respectively, both for longer than 5 years.
Physical examination revealed annular, erythematous, flat-topped papules, some with surmounting fine scale, coalescing into larger plaques along the dorsal surface of the hands and arms (Figure 1) as well as the forehead and chest. A biopsy of a papule on the dorsal aspect of the left hand revealed nodules of histiocytes admixed with Langerhans giant cells within the dermis; mucin was noted centrally within some nodules (Figure 2). Periodic acid–Schiff staining was negative for fungal elements compared to control. Polarization of the specimen was negative for foreign bodies. The biopsy findings therefore were consistent with a diagnosis of GA.
A 3-month treatment course of betamethasone dipropionate 0.05% cream twice daily failed. Narrowband UVB phototherapy was then initiated at 3 sessions weekly. The eruption of GA improved after 3 months of phototherapy. Subsequently, the patient was lost to follow-up.
Comment
Discovery of specific immune checkpoints in tumor-induced immunosuppression revolutionized oncologic therapy. An example is the programmed cell-death protein 1 (PD-1) receptor that is expressed on activated immune cells, including T cells and macrophages.3,4 Upon binding to the PD-1 ligand (PD-L1), T-cell proliferation is inhibited, resulting in downregulation of the immune response. As a result, tumor cells have evolved to overexpress PD-L1 to evade immunologic detection.3 Nivolumab, a fully human IgG4 antibody to PD-1, has emerged along with other ICIs as effective treatments for numerous cancers, including melanoma and non–small-cell lung cancer. By disrupting downregulation of T cells, ICIs improve immune-mediated antitumor activity.3
However, the resulting immunologic disturbance by ICIs has been reported to induce various cutaneous and systemic immune-mediated adverse reactions, including granulomatous reactions such as sarcoidosis, GA, and a cutaneous sarcoidlike granulomatous reaction.1,2,5,6 Our patient represents a rare case of nivolumab-induced GA.
Recent evidence suggests that GA might be caused in part by a cell-mediated hypersensitivity reaction that is regulated by a helper T cell subset 1 inflammatory reaction. Through release of cytokines by activated CD4+ T cells, macrophages are recruited, forming the granulomatous pattern and secreting enzymes that can degrade connective tissue. Nivolumab and other ICIs can thus trigger this reaction because their blockade of PD-1 enhances T cell–mediated immune reactions.2 In addition, because macrophages themselves also express PD-1, ICIs can directly enhance macrophage recruitment and proliferation, further increasing the risk of a granulomatous reaction.4
Interestingly, cutaneous adverse reactions to nivolumab have been associated with improved survival in melanoma patients.7 The nature of this association with granulomatous reactions in general and with GA specifically remains to be determined.
Conclusion
Since the approval of the first PD-1 inhibitors, pembrolizumab and nivolumab, in 2014, other ICIs targeting the immune checkpoint pathway have been developed. Newer agents targeting PD-L1 (avelumab, atezolizumab, and durvalumab) were recently approved. Additionally, cemiplimab, another PD-1 inhibitor, was approved by the US Food and Drug Administration in 2018 for the treatment of advanced cutaneous squamous cell carcinoma.8 Indications for all ICIs also have expanded considerably.3 Therefore, the incidence of immune-mediated adverse reactions, including GA, is bound to increase. Physicians should be cognizant of this association to accurately diagnose and effectively treat adverse reactions in patients who are taking ICIs.
Granuloma annulare (GA) is a benign, cutaneous, granulomatous disease of unclear etiology. Typically, GA presents in young adults as asymptomatic, annular, flesh-colored to pink papules and plaques, commonly on the upper and lower extremities. Histologically, GA is characterized by mucin deposition, palisading or an interstitial granulomatous pattern, and collagen and elastic fiber degeneration.1
Granuloma annulare has been associated with various medications and medical conditions, including diabetes mellitus, hyperlipidemia, thyroid disease, and HIV.1 More recently, immune-checkpoint inhibitors (ICIs) have been reported to trigger GA.2 We report a case of nivolumab-induced GA in a 54-year-old woman.
Case Report
A 54-year-old woman presented with an itchy rash on the upper extremities, face, and chest of 4 months’ duration. The patient noted that the rash started on the hands and progressed to include the arms, face, and chest. She also reported associated mild tenderness. She had a history of stage IV non–small-cell lung carcinoma with metastases to the ribs and adrenal glands. She had been started on biweekly intravenous infusions of the ICI nivolumab by her oncologist approximately 1 year prior to the current presentation after failing a course of conventional chemotherapy. The most recent positron emission tomography–computed tomography scan 1 month prior to presentation showed a stable lung mass with radiologic disappearance of metastases, indicating a favorable response to nivolumab. The patient also had a history of hypothyroidism and depression, which were treated with oral levothyroxine 75 μg once daily and oral sertraline 50 mg once daily, respectively, both for longer than 5 years.
Physical examination revealed annular, erythematous, flat-topped papules, some with surmounting fine scale, coalescing into larger plaques along the dorsal surface of the hands and arms (Figure 1) as well as the forehead and chest. A biopsy of a papule on the dorsal aspect of the left hand revealed nodules of histiocytes admixed with Langerhans giant cells within the dermis; mucin was noted centrally within some nodules (Figure 2). Periodic acid–Schiff staining was negative for fungal elements compared to control. Polarization of the specimen was negative for foreign bodies. The biopsy findings therefore were consistent with a diagnosis of GA.
A 3-month treatment course of betamethasone dipropionate 0.05% cream twice daily failed. Narrowband UVB phototherapy was then initiated at 3 sessions weekly. The eruption of GA improved after 3 months of phototherapy. Subsequently, the patient was lost to follow-up.
Comment
Discovery of specific immune checkpoints in tumor-induced immunosuppression revolutionized oncologic therapy. An example is the programmed cell-death protein 1 (PD-1) receptor that is expressed on activated immune cells, including T cells and macrophages.3,4 Upon binding to the PD-1 ligand (PD-L1), T-cell proliferation is inhibited, resulting in downregulation of the immune response. As a result, tumor cells have evolved to overexpress PD-L1 to evade immunologic detection.3 Nivolumab, a fully human IgG4 antibody to PD-1, has emerged along with other ICIs as effective treatments for numerous cancers, including melanoma and non–small-cell lung cancer. By disrupting downregulation of T cells, ICIs improve immune-mediated antitumor activity.3
However, the resulting immunologic disturbance by ICIs has been reported to induce various cutaneous and systemic immune-mediated adverse reactions, including granulomatous reactions such as sarcoidosis, GA, and a cutaneous sarcoidlike granulomatous reaction.1,2,5,6 Our patient represents a rare case of nivolumab-induced GA.
Recent evidence suggests that GA might be caused in part by a cell-mediated hypersensitivity reaction that is regulated by a helper T cell subset 1 inflammatory reaction. Through release of cytokines by activated CD4+ T cells, macrophages are recruited, forming the granulomatous pattern and secreting enzymes that can degrade connective tissue. Nivolumab and other ICIs can thus trigger this reaction because their blockade of PD-1 enhances T cell–mediated immune reactions.2 In addition, because macrophages themselves also express PD-1, ICIs can directly enhance macrophage recruitment and proliferation, further increasing the risk of a granulomatous reaction.4
Interestingly, cutaneous adverse reactions to nivolumab have been associated with improved survival in melanoma patients.7 The nature of this association with granulomatous reactions in general and with GA specifically remains to be determined.
Conclusion
Since the approval of the first PD-1 inhibitors, pembrolizumab and nivolumab, in 2014, other ICIs targeting the immune checkpoint pathway have been developed. Newer agents targeting PD-L1 (avelumab, atezolizumab, and durvalumab) were recently approved. Additionally, cemiplimab, another PD-1 inhibitor, was approved by the US Food and Drug Administration in 2018 for the treatment of advanced cutaneous squamous cell carcinoma.8 Indications for all ICIs also have expanded considerably.3 Therefore, the incidence of immune-mediated adverse reactions, including GA, is bound to increase. Physicians should be cognizant of this association to accurately diagnose and effectively treat adverse reactions in patients who are taking ICIs.
- Piette EW, Rosenbach M. Granuloma annulare: pathogenesis, disease associations and triggers, and therapeutic options. J Am Acad Dermatol. 2016;75:467-479. doi:10.1016/j.jaad.2015.03.055
- Wu J, Kwong BY, Martires KJ, et al. Granuloma annulare associated with immune checkpoint inhibitors. J Eur Acad Dermatol. 2018;32:E124-E126. doi:10.1111/jdv.14617
- Gong J, Chehrazi-Raffle A, Reddi S, et al. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations. J Immunother Cancer. 2018;6:8. doi:10.1186/s40425-018-0316-z
- Gordon SR, Maute RL, Dulken BW, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545:495-499. doi:10.1038/nature22396
- Birnbaum MR, Ma MW, Fleisig S, et al. Nivolumab-related cutaneous sarcoidosis in a patient with lung adenocarcinoma. JAAD Case Rep. 2017;3:208-211. doi:10.1016/j.jdcr.2017.02.015
- Danlos F-X, Pagès C, Baroudjian B, et al. Nivolumab-induced sarcoid-like granulomatous reaction in a patient with advanced melanoma. Chest. 2016;149:E133-E136. doi:10.1016/j.chest.2015.10.082
- Freeman-Keller M, Kim Y, Cronin H, et al. Nivolumab in resected and unresectable metastatic melanoma: characteristics of immune-related adverse events and association with outcomes. Clin Cancer Res. 2016;22:886-894. doi:10.1158/1078-0432.CCR-15-1136
- Migden MR, Rischin D, Schmults CD, et al. PD-1 blockade with cemiplimab in advanced cutaneous squamous-cell carcinoma. N Engl J Med. 2018;379:341-351. doi:10.1056/NEJMoa1805131
- Piette EW, Rosenbach M. Granuloma annulare: pathogenesis, disease associations and triggers, and therapeutic options. J Am Acad Dermatol. 2016;75:467-479. doi:10.1016/j.jaad.2015.03.055
- Wu J, Kwong BY, Martires KJ, et al. Granuloma annulare associated with immune checkpoint inhibitors. J Eur Acad Dermatol. 2018;32:E124-E126. doi:10.1111/jdv.14617
- Gong J, Chehrazi-Raffle A, Reddi S, et al. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations. J Immunother Cancer. 2018;6:8. doi:10.1186/s40425-018-0316-z
- Gordon SR, Maute RL, Dulken BW, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545:495-499. doi:10.1038/nature22396
- Birnbaum MR, Ma MW, Fleisig S, et al. Nivolumab-related cutaneous sarcoidosis in a patient with lung adenocarcinoma. JAAD Case Rep. 2017;3:208-211. doi:10.1016/j.jdcr.2017.02.015
- Danlos F-X, Pagès C, Baroudjian B, et al. Nivolumab-induced sarcoid-like granulomatous reaction in a patient with advanced melanoma. Chest. 2016;149:E133-E136. doi:10.1016/j.chest.2015.10.082
- Freeman-Keller M, Kim Y, Cronin H, et al. Nivolumab in resected and unresectable metastatic melanoma: characteristics of immune-related adverse events and association with outcomes. Clin Cancer Res. 2016;22:886-894. doi:10.1158/1078-0432.CCR-15-1136
- Migden MR, Rischin D, Schmults CD, et al. PD-1 blockade with cemiplimab in advanced cutaneous squamous-cell carcinoma. N Engl J Med. 2018;379:341-351. doi:10.1056/NEJMoa1805131
Practice Points
- Immune-related adverse events (irAEs) frequently occur in patients on immunotherapy, with the skin representing the most common site of involvement.
- Although rare, granulomatous reactions such as granuloma annulare increasingly are recognized as potential irAEs.
- Clinicians should be aware of this novel association to accurately diagnose and effectively treat adverse reactions in patients receiving immunotherapy.