User login
75-year-old woman • right-side rib pain • radiating shoulder pain • history of hypertension & hypercholesterolemia • Dx?
THE CASE
A 75-year-old woman presented to the primary care clinic with right-side rib pain. The patient said the pain started 1 week earlier, after she ate fried chicken for dinner, and had since been exacerbated by rich meals, lying supine, and taking a deep inspiratory breath. She also said that prior to coming to the clinic that day, the pain had been radiating to her right shoulder.
The patient denied experiencing associated fevers, chills, shortness of breath, chest pain, nausea, vomiting, constipation, diarrhea, or changes in stool color. She had a history of hypertension, for which she was taking lisinopril 20 mg/d, and hypercholesterolemia, for which she was on simvastatin 10 mg/d. She was additionally using timolol ophthalmic solution for her glaucoma.
During the examination, the patient’s vital signs were stable, with a pulse of 80 beats/min, a respiratory rate of 16 breaths/min, and an oxygen saturation of 98% on room air. The patient had no abdominal tenderness upon palpation, and the physical exam revealed no abnormalities. An in-office electrocardiogram was performed, with normal results. Additionally, a comprehensive metabolic panel, lipase test, and
THE DIAGNOSIS
Based on the lab results, a stat computed tomography pulmonary angiogram (CTPA) was ordered and showed a right segmental and subsegmental pulmonary embolism (PE; FIGURE 1).

DISCUSSION
PE shares pathophysiologic mechanisms with deep vein thrombosis (DVT), and together these comprise venous thromboembolism (VTE). Risk factors for VTE include hypercoagulable disorders, use of estrogens, active malignancy, and immobilization.1 Unprovoked VTE occurs in the absence of identifiable risk factors and carries a higher risk of recurrence.2,3 While PE is classically thought to occur in the setting of a DVT, there is increasing literature describing de novo PE that can occur independent of a DVT.4
Common symptoms of PE include tachycardia, tachypnea, and pleuritic chest pain.5 Abdominal pain is a rare symptom described in some case reports.6,7 Thus, a high clinical suspicion is needed for diagnosis of PE.
The Wells criteria is an established model for risk stratifying patients presenting with possible VTE (TABLE).8 For patients with low pretest probability, as in this case, a

Continue to: Length of treatment depends on gender and etiology
Length of treatment depends on gender and etiology
The cornerstone treatment for stable patients with VTE is therapeutic anticoagulation. The new oral anticoagulants, which directly inhibit factor Xa or thrombin, have become increasingly popular for management of VTE, in part because they don’t require INR testing and monitoring.2
The duration of anticoagulation, particularly in unprovoked PE, is debatable. As noted earlier, patients with an unprovoked PE are at higher risk of recurrence than those with a reversible cause, so the question becomes whether these patients should have indefinite anticoagulation.2,3 Studies examining risk stratification of patients with a first, unprovoked VTE have found that men have the highest risk of recurrence, followed by women who were not taking estrogen during the index VTE, and lastly women who were taking estrogen therapy during the index VTE and subsequently discontinued it.2,3,10
Thus, it is reasonable to give women the option to discontinue anticoagulation in the setting of a negative
Our patient was directed to the emergency department for further monitoring following CT confirmation. She was discharged home after being deemed stable and prescribed apixaban 10 mg/d. A venous duplex ultrasound performed 12 days later for knee pain revealed no venous thrombosis. A CT of the abdomen performed 3 months later for other reasons revealed a normal gallbladder with no visible stones.
Apixaban was continued for 3 months and discontinued after discussion of risks and benefits of therapy cessation in the setting of a normal
Continue to: THE TAKEAWAY
THE TAKEAWAY
PE carries a significantly high mortality rate and can manifest with nonspecific and masquerading signs. A high index of suspicion is required to place PE on the differential diagnosis and carry out appropriate testing. Our patient presented with a history consistent with biliary colic but with pleuritic chest pain that warranted consideration of a PE.
CORRESPONDENCE
Alyssa Anderson, MD, 1 Continental Drive, Elizabethtown, PA 17022; [email protected]
1. Israel HL, Goldstein F. The varied clinical manifestations of pulmonary embolism. Ann Intern Med. 1957;47:202-226. doi: 10.7326/0003-4819-47-2-202
2. Rehman H, John E, Parikh P. Pulmonary embolism presenting as abdominal pain: an atypical presentation of a common diagnosis. Case Rep Emerg Med. 2016;2016:1-3. doi: 10.1155/2016/7832895
3. Park ES, Cho JY, Seo J-H, et al. Pulmonary embolism presenting with acute abdominal pain in a girl with stable ankle fracture and inherited antithrombin deficiency. Blood Res. 2018;53:81-83. doi: 10.5045/br.2018.53.1.81
4. Tapson VF. Acute pulmonary embolism. N Engl J Med. 2008;358:1037-1052. doi: 10.1056/NEJMra072753
5. Agrawal V, Kim ESH. Risk of recurrent venous thromboembolism after an initial episode: risk stratification and implications for long-term treatment. Curr Cardiol Rep. 2019;21:24. doi: 10.1007/s11886-019-1111-2
6. Kearon C, Parpia S, Spencer FA, et al. Long‐term risk of recurrence in patients with a first unprovoked venous thromboembolism managed according to d‐dimer results; A cohort study. J Thromb Haemost. 2019;17:1144-1152. doi: 10.1111/jth.14458
7. Van Gent J-M, Zander AL, Olson EJ, et al. Pulmonary embolism without deep venous thrombosis. J Trauma Acute Care Surg. 2014;76:1270-1274. doi: 10.1097/TA.0000000000000233
8. Wells PS, Anderson DR, Rodger M, et al. Excluding pulmonary embolism at the bedside without diagnostic imaging: management of patients with suspected pulmonary embolism presenting to the emergency department by using a simple clinical model and d-dimer. Ann Intern Med. 2001;135:98-107. doi: 10.7326/0003-4819-135-2-200107170-00010
9. Kline JA. Diagnosis and exclusion of pulmonary embolism. Thromb Res. 2018;163:207-220. doi: 10.1016/j.thromres.2017.06.002
10. Kearon C, Akl EA, Ornelas J, et al. Antithrombotic therapy for VTE disease. Chest. 2016;149:315-352. doi: 10.1016/j.chest.2015.11.026
THE CASE
A 75-year-old woman presented to the primary care clinic with right-side rib pain. The patient said the pain started 1 week earlier, after she ate fried chicken for dinner, and had since been exacerbated by rich meals, lying supine, and taking a deep inspiratory breath. She also said that prior to coming to the clinic that day, the pain had been radiating to her right shoulder.
The patient denied experiencing associated fevers, chills, shortness of breath, chest pain, nausea, vomiting, constipation, diarrhea, or changes in stool color. She had a history of hypertension, for which she was taking lisinopril 20 mg/d, and hypercholesterolemia, for which she was on simvastatin 10 mg/d. She was additionally using timolol ophthalmic solution for her glaucoma.
During the examination, the patient’s vital signs were stable, with a pulse of 80 beats/min, a respiratory rate of 16 breaths/min, and an oxygen saturation of 98% on room air. The patient had no abdominal tenderness upon palpation, and the physical exam revealed no abnormalities. An in-office electrocardiogram was performed, with normal results. Additionally, a comprehensive metabolic panel, lipase test, and
THE DIAGNOSIS
Based on the lab results, a stat computed tomography pulmonary angiogram (CTPA) was ordered and showed a right segmental and subsegmental pulmonary embolism (PE; FIGURE 1).
DISCUSSION
PE shares pathophysiologic mechanisms with deep vein thrombosis (DVT), and together these comprise venous thromboembolism (VTE). Risk factors for VTE include hypercoagulable disorders, use of estrogens, active malignancy, and immobilization.1 Unprovoked VTE occurs in the absence of identifiable risk factors and carries a higher risk of recurrence.2,3 While PE is classically thought to occur in the setting of a DVT, there is increasing literature describing de novo PE that can occur independent of a DVT.4
Common symptoms of PE include tachycardia, tachypnea, and pleuritic chest pain.5 Abdominal pain is a rare symptom described in some case reports.6,7 Thus, a high clinical suspicion is needed for diagnosis of PE.
The Wells criteria is an established model for risk stratifying patients presenting with possible VTE (TABLE).8 For patients with low pretest probability, as in this case, a
Continue to: Length of treatment depends on gender and etiology
Length of treatment depends on gender and etiology
The cornerstone treatment for stable patients with VTE is therapeutic anticoagulation. The new oral anticoagulants, which directly inhibit factor Xa or thrombin, have become increasingly popular for management of VTE, in part because they don’t require INR testing and monitoring.2
The duration of anticoagulation, particularly in unprovoked PE, is debatable. As noted earlier, patients with an unprovoked PE are at higher risk of recurrence than those with a reversible cause, so the question becomes whether these patients should have indefinite anticoagulation.2,3 Studies examining risk stratification of patients with a first, unprovoked VTE have found that men have the highest risk of recurrence, followed by women who were not taking estrogen during the index VTE, and lastly women who were taking estrogen therapy during the index VTE and subsequently discontinued it.2,3,10
Thus, it is reasonable to give women the option to discontinue anticoagulation in the setting of a negative
Our patient was directed to the emergency department for further monitoring following CT confirmation. She was discharged home after being deemed stable and prescribed apixaban 10 mg/d. A venous duplex ultrasound performed 12 days later for knee pain revealed no venous thrombosis. A CT of the abdomen performed 3 months later for other reasons revealed a normal gallbladder with no visible stones.
Apixaban was continued for 3 months and discontinued after discussion of risks and benefits of therapy cessation in the setting of a normal
Continue to: THE TAKEAWAY
THE TAKEAWAY
PE carries a significantly high mortality rate and can manifest with nonspecific and masquerading signs. A high index of suspicion is required to place PE on the differential diagnosis and carry out appropriate testing. Our patient presented with a history consistent with biliary colic but with pleuritic chest pain that warranted consideration of a PE.
CORRESPONDENCE
Alyssa Anderson, MD, 1 Continental Drive, Elizabethtown, PA 17022; [email protected]
THE CASE
A 75-year-old woman presented to the primary care clinic with right-side rib pain. The patient said the pain started 1 week earlier, after she ate fried chicken for dinner, and had since been exacerbated by rich meals, lying supine, and taking a deep inspiratory breath. She also said that prior to coming to the clinic that day, the pain had been radiating to her right shoulder.
The patient denied experiencing associated fevers, chills, shortness of breath, chest pain, nausea, vomiting, constipation, diarrhea, or changes in stool color. She had a history of hypertension, for which she was taking lisinopril 20 mg/d, and hypercholesterolemia, for which she was on simvastatin 10 mg/d. She was additionally using timolol ophthalmic solution for her glaucoma.
During the examination, the patient’s vital signs were stable, with a pulse of 80 beats/min, a respiratory rate of 16 breaths/min, and an oxygen saturation of 98% on room air. The patient had no abdominal tenderness upon palpation, and the physical exam revealed no abnormalities. An in-office electrocardiogram was performed, with normal results. Additionally, a comprehensive metabolic panel, lipase test, and
THE DIAGNOSIS
Based on the lab results, a stat computed tomography pulmonary angiogram (CTPA) was ordered and showed a right segmental and subsegmental pulmonary embolism (PE; FIGURE 1).
DISCUSSION
PE shares pathophysiologic mechanisms with deep vein thrombosis (DVT), and together these comprise venous thromboembolism (VTE). Risk factors for VTE include hypercoagulable disorders, use of estrogens, active malignancy, and immobilization.1 Unprovoked VTE occurs in the absence of identifiable risk factors and carries a higher risk of recurrence.2,3 While PE is classically thought to occur in the setting of a DVT, there is increasing literature describing de novo PE that can occur independent of a DVT.4
Common symptoms of PE include tachycardia, tachypnea, and pleuritic chest pain.5 Abdominal pain is a rare symptom described in some case reports.6,7 Thus, a high clinical suspicion is needed for diagnosis of PE.
The Wells criteria is an established model for risk stratifying patients presenting with possible VTE (TABLE).8 For patients with low pretest probability, as in this case, a
Continue to: Length of treatment depends on gender and etiology
Length of treatment depends on gender and etiology
The cornerstone treatment for stable patients with VTE is therapeutic anticoagulation. The new oral anticoagulants, which directly inhibit factor Xa or thrombin, have become increasingly popular for management of VTE, in part because they don’t require INR testing and monitoring.2
The duration of anticoagulation, particularly in unprovoked PE, is debatable. As noted earlier, patients with an unprovoked PE are at higher risk of recurrence than those with a reversible cause, so the question becomes whether these patients should have indefinite anticoagulation.2,3 Studies examining risk stratification of patients with a first, unprovoked VTE have found that men have the highest risk of recurrence, followed by women who were not taking estrogen during the index VTE, and lastly women who were taking estrogen therapy during the index VTE and subsequently discontinued it.2,3,10
Thus, it is reasonable to give women the option to discontinue anticoagulation in the setting of a negative
Our patient was directed to the emergency department for further monitoring following CT confirmation. She was discharged home after being deemed stable and prescribed apixaban 10 mg/d. A venous duplex ultrasound performed 12 days later for knee pain revealed no venous thrombosis. A CT of the abdomen performed 3 months later for other reasons revealed a normal gallbladder with no visible stones.
Apixaban was continued for 3 months and discontinued after discussion of risks and benefits of therapy cessation in the setting of a normal
Continue to: THE TAKEAWAY
THE TAKEAWAY
PE carries a significantly high mortality rate and can manifest with nonspecific and masquerading signs. A high index of suspicion is required to place PE on the differential diagnosis and carry out appropriate testing. Our patient presented with a history consistent with biliary colic but with pleuritic chest pain that warranted consideration of a PE.
CORRESPONDENCE
Alyssa Anderson, MD, 1 Continental Drive, Elizabethtown, PA 17022; [email protected]
1. Israel HL, Goldstein F. The varied clinical manifestations of pulmonary embolism. Ann Intern Med. 1957;47:202-226. doi: 10.7326/0003-4819-47-2-202
2. Rehman H, John E, Parikh P. Pulmonary embolism presenting as abdominal pain: an atypical presentation of a common diagnosis. Case Rep Emerg Med. 2016;2016:1-3. doi: 10.1155/2016/7832895
3. Park ES, Cho JY, Seo J-H, et al. Pulmonary embolism presenting with acute abdominal pain in a girl with stable ankle fracture and inherited antithrombin deficiency. Blood Res. 2018;53:81-83. doi: 10.5045/br.2018.53.1.81
4. Tapson VF. Acute pulmonary embolism. N Engl J Med. 2008;358:1037-1052. doi: 10.1056/NEJMra072753
5. Agrawal V, Kim ESH. Risk of recurrent venous thromboembolism after an initial episode: risk stratification and implications for long-term treatment. Curr Cardiol Rep. 2019;21:24. doi: 10.1007/s11886-019-1111-2
6. Kearon C, Parpia S, Spencer FA, et al. Long‐term risk of recurrence in patients with a first unprovoked venous thromboembolism managed according to d‐dimer results; A cohort study. J Thromb Haemost. 2019;17:1144-1152. doi: 10.1111/jth.14458
7. Van Gent J-M, Zander AL, Olson EJ, et al. Pulmonary embolism without deep venous thrombosis. J Trauma Acute Care Surg. 2014;76:1270-1274. doi: 10.1097/TA.0000000000000233
8. Wells PS, Anderson DR, Rodger M, et al. Excluding pulmonary embolism at the bedside without diagnostic imaging: management of patients with suspected pulmonary embolism presenting to the emergency department by using a simple clinical model and d-dimer. Ann Intern Med. 2001;135:98-107. doi: 10.7326/0003-4819-135-2-200107170-00010
9. Kline JA. Diagnosis and exclusion of pulmonary embolism. Thromb Res. 2018;163:207-220. doi: 10.1016/j.thromres.2017.06.002
10. Kearon C, Akl EA, Ornelas J, et al. Antithrombotic therapy for VTE disease. Chest. 2016;149:315-352. doi: 10.1016/j.chest.2015.11.026
1. Israel HL, Goldstein F. The varied clinical manifestations of pulmonary embolism. Ann Intern Med. 1957;47:202-226. doi: 10.7326/0003-4819-47-2-202
2. Rehman H, John E, Parikh P. Pulmonary embolism presenting as abdominal pain: an atypical presentation of a common diagnosis. Case Rep Emerg Med. 2016;2016:1-3. doi: 10.1155/2016/7832895
3. Park ES, Cho JY, Seo J-H, et al. Pulmonary embolism presenting with acute abdominal pain in a girl with stable ankle fracture and inherited antithrombin deficiency. Blood Res. 2018;53:81-83. doi: 10.5045/br.2018.53.1.81
4. Tapson VF. Acute pulmonary embolism. N Engl J Med. 2008;358:1037-1052. doi: 10.1056/NEJMra072753
5. Agrawal V, Kim ESH. Risk of recurrent venous thromboembolism after an initial episode: risk stratification and implications for long-term treatment. Curr Cardiol Rep. 2019;21:24. doi: 10.1007/s11886-019-1111-2
6. Kearon C, Parpia S, Spencer FA, et al. Long‐term risk of recurrence in patients with a first unprovoked venous thromboembolism managed according to d‐dimer results; A cohort study. J Thromb Haemost. 2019;17:1144-1152. doi: 10.1111/jth.14458
7. Van Gent J-M, Zander AL, Olson EJ, et al. Pulmonary embolism without deep venous thrombosis. J Trauma Acute Care Surg. 2014;76:1270-1274. doi: 10.1097/TA.0000000000000233
8. Wells PS, Anderson DR, Rodger M, et al. Excluding pulmonary embolism at the bedside without diagnostic imaging: management of patients with suspected pulmonary embolism presenting to the emergency department by using a simple clinical model and d-dimer. Ann Intern Med. 2001;135:98-107. doi: 10.7326/0003-4819-135-2-200107170-00010
9. Kline JA. Diagnosis and exclusion of pulmonary embolism. Thromb Res. 2018;163:207-220. doi: 10.1016/j.thromres.2017.06.002
10. Kearon C, Akl EA, Ornelas J, et al. Antithrombotic therapy for VTE disease. Chest. 2016;149:315-352. doi: 10.1016/j.chest.2015.11.026

Acyclovir-Resistant Cutaneous Herpes Simplex Virus in DOCK8 Deficiency
Dedicator of cytokinesis 8 (DOCK8 ) deficiency is the major cause of autosomal-recessive hyper-IgEsyndrome. 1 Characteristic clinical features including eosinophilia, eczema, and recurrent Staphylococcus aureus cutaneous and respiratory tract infections are common in DOCK8 deficiency, similar to the autosomal-dominant form of hyper-IgE syndrome that is due to defi c iency of signal transducer and activation of transcription 3 (STAT-3 ). 1 In addition, patients with DOCK8 deficiency are particularly susceptible to asthma; food allergies; lymphomas; and severe cutaneous viral infections, including herpes simplex virus (HSV), molluscum contagiosum, varicella-zoster virus, and human papillomavirus. Since the discovery of the DOCK8 gene in 2009, various studies have sought to elucidate the mechanistic contribution of DOCK8 to the dermatologic immune environment. 2 Although cutaneous viral infections such as those caused by HSV typically are short lived and self-limiting in immunocompetent hosts, they have proven to be severe and recalcitrant in the setting of DOCK8 deficiency. 1 Herein, we report the case of a 32-month-old girl with homozygous DOCK8 deficiency who developed acyclovir-resistant cutaneous HSV.
Case Report
A 32-month-old girl presented with an approximately 2-cm linear erosion along the left posterior auricular sulcus at month 9 of a hospital stay for recurrent infections. Her medical history was notable for multiple upper respiratory tract infections, diffuse eczema, and food allergies. She had presented to an outside hospital at 14 months of age with herpetic gingivostomatitis and eczema herpeticum that was successfully treated with acyclovir. She was readmitted at 20 months of age due to Pneumocystis jiroveci pneumonia, pancytopenia, and disseminated histoplasmosis. Prophylactic oral acyclovir (20 mg/kg twice daily) was started, given her history of HSV infection. Because of recurrent infections, she underwent an immunodeficiency workup. Whole exome sequencing analysis revealed a homozygous deletion c.(528+1_529−1)_(1516+1_1517−1)del in DOCK8 gene–affecting exons 5 to 13. The patient was transferred to our hospital for continued care and as a potential candidate for bone marrow transplant following resolution of the disseminated histoplasmosis infection.
During her hospitalization at the current presentation, she was noted to have a 2-cm linear erosion along the left posterior auricular sulcus. Initial wound care with bacitracin ointment was applied to the area while specimens were obtained and empiric oral acyclovir therapy was initiated (20 mg/kg 4 times daily [QID]), given a clinical impression consistent with cutaneous HSV infection despite acyclovir prophylaxis. Direct immunofluorescence and viral cultures were positive for HSV-1, while bacterial cultures grew methicillin-susceptible S aureus. Cephalexin and mupirocin ointment were started, and acyclovir was continued. After 2 weeks of therapy, there was no visible change in the wound; cultures were repeated, again showing the wound contained HSV. Bacterial cultures this time grew Pseudomonas putida, and the antibiotic regimen was transitioned to cefepime.
After no response to the continued course of therapeutic acyclovir, HSV cultures were sent to the Centers for Disease Control and Prevention for resistance testing, and biopsy of the lesion was performed by the otolaryngology service to rule out malignancy and potential alternative diagnoses. Histopathology showed only reactive inflammation without visible microorganisms on tissue HSV-1/HSV-2 immunostain; however, tissue viral culture was positive for HSV-1. The patient was transitioned back to acyclovir (intravenous [IV] 20 mg/kg QID) with the addition of empiric foscarnet (IV 40 mg/kg 3 times daily) given the worsening appearance of the lesion. The HSV acyclovir resistance test results from the Centers for Disease Control and Prevention returned soon after and were positive for resistance (median infectious dose, 3.29 µg/L [reference interval, sensitive <2.00 µg/L; resistant >1.90 µg/L]). The patient completed a 21-day course of combination foscarnet and acyclovir therapy, during which time the lesion showed notable improvement and healing. The patient was continued on prophylactic acyclovir (IV 20 mg/kg QID). Unfortunately, the patient eventually died due to complications related to pneumonia.
Comment
Infection in Patients With DOCK8 Deficiency—The gene DOCK8 has emerged as playing a central role in both innate and adaptive immunity, as it is expressed primarily in immune cells and serves as a mediator of numerous processes, including immune synapse formation, cell signaling and trafficking, antibody and cytokine production, and lymphocyte memory.3 Cells that are critical for combating cutaneous viral infections, including skin-resident memory T cells and natural killer cells, are defective, which leads to a severely immunocompromised state in DOCK8-deficient patients with a particular susceptibility to infectious and inflammatory dermatologic disease.4
Herpes simplex virus infection commonly is seen in DOCK8 deficiency, with retrospective analysis of a DOCK8-deficient cohort revealing HSV infection in approximately 38% of patients.5 Prophylactic acyclovir is essential for DOCK8-deficient individuals with a history of HSV infection given the tendency of the virus to reactivate.6 However, despite prophylaxis, our patient developed an HSV-positive posterior auricular erosion that continued to progress even after increase of the acyclovir dose. Acyclovir resistance testing of the HSV isolated from the wound was positive, confirming the clinical suspicion of the presence of acyclovir-resistant HSV infection.
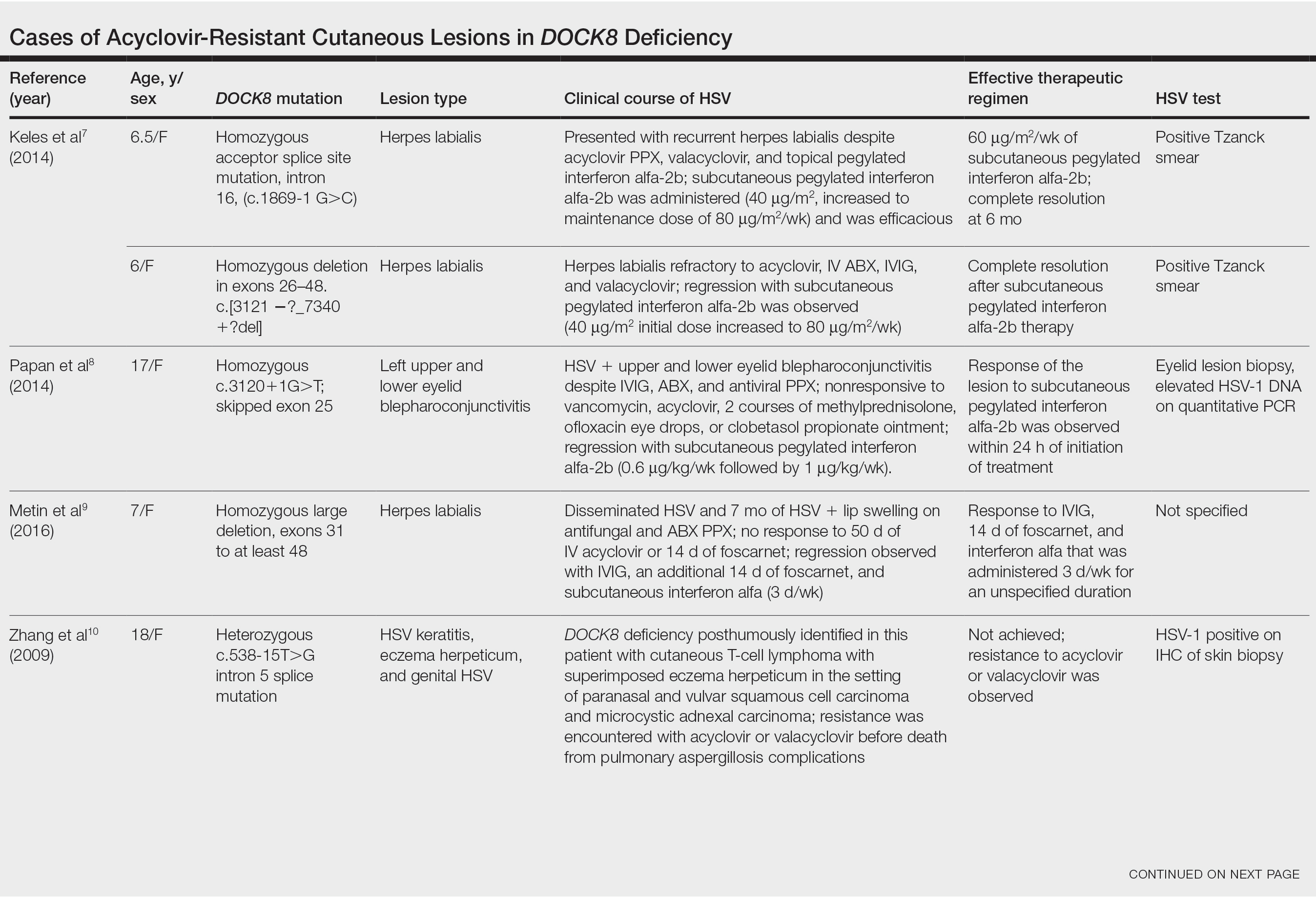
Acyclovir-Resistant HSV—Acyclovir-resistant HSV in immunosuppressed individuals was first noted in 1982, and most cases since then have occurred in the setting of AIDS and in organ transplant recipients.6 Few reports of acyclovir-resistant HSV in DOCK8 deficiency exist, and to our knowledge, our patient is the youngest DOCK8-deficient individual to be documented with acyclovir-resistant HSV infection.1,7-15 We identified relevant cases from the PubMed and EMBASE databases using the search terms DOCK8 deficiency and acyclovir and DOCK8 deficiency and herpes. The eTable lists other reported cases of acyclovir-resistant HSV in DOCK8-deficient patients. The majority of cases involved school-aged females. Lesion types varied and included herpes labialis, eczema herpeticum, and blepharoconjunctivitis. Escalation of therapy and resolution of the lesion was seen in some cases with administration of subcutaneous pegylated interferon alfa-2b.
Treatment Alternatives—Acyclovir competitively inhibits viral DNA polymerase by incorporating into elongating viral DNA strands and halting chain synthesis. Acyclovir requires triphosphorylation for activation, and viral thymidine kinase is responsible for the first phosphorylation event. Ninety-five percent of cases of acyclovir resistance are secondary to mutations in viral thymidine kinase. Foscarnet also inhibits viral DNA polymerase but does so directly without the need to be phosphorylated first.6 For this reason, foscarnet often is the drug of choice in the treatment of acyclovir-resistant HSV, as evidenced in our patient. However, foscarnet-resistant HSV strains may develop from mutations in the DNA polymerase gene.
Cidofovir is a nucleotide analogue that requires phosphorylation by host, as opposed to viral, kinases for antiviral activity. Intravenous and topical formulations of cidofovir have proven effective in the treatment of acyclovir- and foscarnet-resistant HSV lesions.6 Cidofovir also can be applied intralesionally, a method that provides targeted therapy and minimizes cidofovir-associated nephrotoxicity.12 Reports of systemic interferon alfa therapy for acyclovir-resistant HSV also exist. A study found IFN-⍺ production by peripheral blood mononuclear cells in DOCK8-deficient individuals to be significantly reduced relative to controls (P<.05).7 There has been complete resolution of acyclovir-resistant HSV lesions with subcutaneous pegylated interferon alfa-2b injections in several DOCK8-deficient patients.7-9
The need for escalating therapy in DOCK8-deficient individuals with acyclovir-resistant HSV infection underscores the essential role of DOCK8 in dermatologic immunity. Our case demonstrates that a high degree of suspicion for cutaneous HSV infection should be adopted in DOCK8-deficient patients of any age, regardless of acyclovir prophylaxis. Viral culture in addition to bacterial cultures should be performed early in patients with cutaneous erosions, and the threshold for HSV resistance testing should be low to minimize morbidity associated with these infections. Early resistance testing in our case could have prevented prolongation of infection and likely eliminated the need for a biopsy.
Conclusion
DOCK8 deficiency presents a unique challenge to dermatologists and other health care providers given the susceptibility of affected individuals to developing a reservoir of severe and potentially resistant viral cutaneous infections. Prophylactic acyclovir may not be sufficient for HSV suppression, even in the youngest of patients, and suspicion for resistance should be high to avoid delays in adequate treatment.
- Chu EY, Freeman AF, Jing H, et al. Cutaneous manifestations of DOCK8 deficiency syndrome. Arch Dermatol. 2012;148:79-84. doi:10.1001/archdermatol.2011.262
- Aydin SE, Kilic SS, Aytekin C, et al. DOCK8 deficiency: clinical and immunological phenotype and treatment options—a review of 136 patients. J Clin Immunol. 2015;35:189-198. doi:10.1007/s10875-014-0126-0
- Kearney CJ, Randall KL, Oliaro J. DOCK8 regulates signal transduction events to control immunity. Cell Mol Immunol. 2017;14:406-411. doi:10.1038/cmi.2017.9
- Zhang Q, Dove CG, Hor JL, et al. DOCK8 regulates lymphocyte shape integrity for skin antiviral immunity. J Exp Med. 2014;211:2549-2566. doi:10.1084/jem.20141307
- Engelhardt KR, Gertz EM, Keles S, et al. The extended clinical phenotype of 64 patients with DOCK8 deficiency. J Allergy Clin Immunol. 2015;136:402-412. doi:10.1016/j.jaci.2014.12.1945
- Chilukuri S, Rosen T. Management of acyclovir-resistant herpes simplex virus. Dermatol Clin. 2003;21:311-320. doi:10.1016/S0733-8635(02)00093-1
- Keles S, Jabara HH, Reisli I, et al. Plasmacytoid dendritic cell depletion in DOCK8 deficiency: rescue of severe herpetic infections with interferon alpha-2b therapy. J Allergy Clin Immunol. 2014;133:1753-1755.e3. doi:10.1016/j.jaci.2014.03.032
- Papan C, Hagl B, Heinz V, et al Beneficial IFN-α treatment of tumorous herpes simplex blepharoconjunctivitis in dedicator of cytokinesis 8 deficiency. J Allergy Clin Immunol. 2014;133:1456-1458. doi:10.1016/j.jaci.2014.02.008
- Metin A, Kanik-Yuksek S, Ozkaya-Parlakay A, et al. Giant herpes labialis in a child with DOCK8-deficient hyper-IgE syndrome. Pediatr Neonatol. 2016;57:79-80. doi:10.1016/j.pedneo.2015.04.011
- Zhang Q, Davis JC, Lamborn IT, et al. Combined immunodeficiency associated with DOCK8 mutations. N Engl J Med. 2009;361:2046-2055. doi:10.1056/NEJMoa0905506
- Lei JY, Wang Y, Jaffe ES, et al. Microcystic adnexal carcinoma associated with primary immunodeficiency, recurrent diffuse herpes simplex virus infection, and cutaneous T-cell lymphoma. Am J Dermatopathol. 2000;22:524-529. doi:10.1097/00000372-200012000-00008
- Castelo-Soccio L, Bernardin R, Stern J, et al. Successful treatment of acyclovir-resistant herpes simplex virus with intralesional cidofovir. Arch Dermatol. 2010;146:124-126. doi:10.1001/archdermatol.2009.363
- Shah NN, Freeman AF, Hickstein DD. Addendum to: haploidentical related donor hematopoietic stem cell transplantation for DOCK8 deficiency using post-transplantation cyclophosphamide. Biol Blood Marrow Transplant. 2019;25:E65-E67. doi:10.1016/j.bbmt.2018.11.014
- Freeman AF, Yazigi N, Shah NN, et al. Tandem orthotopic living donor liver transplantation followed by same donor haploidentical hematopoietic stem cell transplantation for DOCK8 deficiency. Transplantation. 2019;103:2144-2149. doi:10.1097/TP.0000000000002649
- Casto AM, Stout SC, Selvarangan R, et al. Evaluation of genotypic antiviral resistance testing as an alternative to phenotypic testing in a patient with DOCK8 deficiency and severe HSV-1 disease. J Infect Dis. 2020;221:2035-2042. doi:10.1093/infdis/jiaa020
Dedicator of cytokinesis 8 (DOCK8 ) deficiency is the major cause of autosomal-recessive hyper-IgEsyndrome. 1 Characteristic clinical features including eosinophilia, eczema, and recurrent Staphylococcus aureus cutaneous and respiratory tract infections are common in DOCK8 deficiency, similar to the autosomal-dominant form of hyper-IgE syndrome that is due to defi c iency of signal transducer and activation of transcription 3 (STAT-3 ). 1 In addition, patients with DOCK8 deficiency are particularly susceptible to asthma; food allergies; lymphomas; and severe cutaneous viral infections, including herpes simplex virus (HSV), molluscum contagiosum, varicella-zoster virus, and human papillomavirus. Since the discovery of the DOCK8 gene in 2009, various studies have sought to elucidate the mechanistic contribution of DOCK8 to the dermatologic immune environment. 2 Although cutaneous viral infections such as those caused by HSV typically are short lived and self-limiting in immunocompetent hosts, they have proven to be severe and recalcitrant in the setting of DOCK8 deficiency. 1 Herein, we report the case of a 32-month-old girl with homozygous DOCK8 deficiency who developed acyclovir-resistant cutaneous HSV.
Case Report
A 32-month-old girl presented with an approximately 2-cm linear erosion along the left posterior auricular sulcus at month 9 of a hospital stay for recurrent infections. Her medical history was notable for multiple upper respiratory tract infections, diffuse eczema, and food allergies. She had presented to an outside hospital at 14 months of age with herpetic gingivostomatitis and eczema herpeticum that was successfully treated with acyclovir. She was readmitted at 20 months of age due to Pneumocystis jiroveci pneumonia, pancytopenia, and disseminated histoplasmosis. Prophylactic oral acyclovir (20 mg/kg twice daily) was started, given her history of HSV infection. Because of recurrent infections, she underwent an immunodeficiency workup. Whole exome sequencing analysis revealed a homozygous deletion c.(528+1_529−1)_(1516+1_1517−1)del in DOCK8 gene–affecting exons 5 to 13. The patient was transferred to our hospital for continued care and as a potential candidate for bone marrow transplant following resolution of the disseminated histoplasmosis infection.
During her hospitalization at the current presentation, she was noted to have a 2-cm linear erosion along the left posterior auricular sulcus. Initial wound care with bacitracin ointment was applied to the area while specimens were obtained and empiric oral acyclovir therapy was initiated (20 mg/kg 4 times daily [QID]), given a clinical impression consistent with cutaneous HSV infection despite acyclovir prophylaxis. Direct immunofluorescence and viral cultures were positive for HSV-1, while bacterial cultures grew methicillin-susceptible S aureus. Cephalexin and mupirocin ointment were started, and acyclovir was continued. After 2 weeks of therapy, there was no visible change in the wound; cultures were repeated, again showing the wound contained HSV. Bacterial cultures this time grew Pseudomonas putida, and the antibiotic regimen was transitioned to cefepime.
After no response to the continued course of therapeutic acyclovir, HSV cultures were sent to the Centers for Disease Control and Prevention for resistance testing, and biopsy of the lesion was performed by the otolaryngology service to rule out malignancy and potential alternative diagnoses. Histopathology showed only reactive inflammation without visible microorganisms on tissue HSV-1/HSV-2 immunostain; however, tissue viral culture was positive for HSV-1. The patient was transitioned back to acyclovir (intravenous [IV] 20 mg/kg QID) with the addition of empiric foscarnet (IV 40 mg/kg 3 times daily) given the worsening appearance of the lesion. The HSV acyclovir resistance test results from the Centers for Disease Control and Prevention returned soon after and were positive for resistance (median infectious dose, 3.29 µg/L [reference interval, sensitive <2.00 µg/L; resistant >1.90 µg/L]). The patient completed a 21-day course of combination foscarnet and acyclovir therapy, during which time the lesion showed notable improvement and healing. The patient was continued on prophylactic acyclovir (IV 20 mg/kg QID). Unfortunately, the patient eventually died due to complications related to pneumonia.
Comment
Infection in Patients With DOCK8 Deficiency—The gene DOCK8 has emerged as playing a central role in both innate and adaptive immunity, as it is expressed primarily in immune cells and serves as a mediator of numerous processes, including immune synapse formation, cell signaling and trafficking, antibody and cytokine production, and lymphocyte memory.3 Cells that are critical for combating cutaneous viral infections, including skin-resident memory T cells and natural killer cells, are defective, which leads to a severely immunocompromised state in DOCK8-deficient patients with a particular susceptibility to infectious and inflammatory dermatologic disease.4
Herpes simplex virus infection commonly is seen in DOCK8 deficiency, with retrospective analysis of a DOCK8-deficient cohort revealing HSV infection in approximately 38% of patients.5 Prophylactic acyclovir is essential for DOCK8-deficient individuals with a history of HSV infection given the tendency of the virus to reactivate.6 However, despite prophylaxis, our patient developed an HSV-positive posterior auricular erosion that continued to progress even after increase of the acyclovir dose. Acyclovir resistance testing of the HSV isolated from the wound was positive, confirming the clinical suspicion of the presence of acyclovir-resistant HSV infection.
Acyclovir-Resistant HSV—Acyclovir-resistant HSV in immunosuppressed individuals was first noted in 1982, and most cases since then have occurred in the setting of AIDS and in organ transplant recipients.6 Few reports of acyclovir-resistant HSV in DOCK8 deficiency exist, and to our knowledge, our patient is the youngest DOCK8-deficient individual to be documented with acyclovir-resistant HSV infection.1,7-15 We identified relevant cases from the PubMed and EMBASE databases using the search terms DOCK8 deficiency and acyclovir and DOCK8 deficiency and herpes. The eTable lists other reported cases of acyclovir-resistant HSV in DOCK8-deficient patients. The majority of cases involved school-aged females. Lesion types varied and included herpes labialis, eczema herpeticum, and blepharoconjunctivitis. Escalation of therapy and resolution of the lesion was seen in some cases with administration of subcutaneous pegylated interferon alfa-2b.
Treatment Alternatives—Acyclovir competitively inhibits viral DNA polymerase by incorporating into elongating viral DNA strands and halting chain synthesis. Acyclovir requires triphosphorylation for activation, and viral thymidine kinase is responsible for the first phosphorylation event. Ninety-five percent of cases of acyclovir resistance are secondary to mutations in viral thymidine kinase. Foscarnet also inhibits viral DNA polymerase but does so directly without the need to be phosphorylated first.6 For this reason, foscarnet often is the drug of choice in the treatment of acyclovir-resistant HSV, as evidenced in our patient. However, foscarnet-resistant HSV strains may develop from mutations in the DNA polymerase gene.
Cidofovir is a nucleotide analogue that requires phosphorylation by host, as opposed to viral, kinases for antiviral activity. Intravenous and topical formulations of cidofovir have proven effective in the treatment of acyclovir- and foscarnet-resistant HSV lesions.6 Cidofovir also can be applied intralesionally, a method that provides targeted therapy and minimizes cidofovir-associated nephrotoxicity.12 Reports of systemic interferon alfa therapy for acyclovir-resistant HSV also exist. A study found IFN-⍺ production by peripheral blood mononuclear cells in DOCK8-deficient individuals to be significantly reduced relative to controls (P<.05).7 There has been complete resolution of acyclovir-resistant HSV lesions with subcutaneous pegylated interferon alfa-2b injections in several DOCK8-deficient patients.7-9
The need for escalating therapy in DOCK8-deficient individuals with acyclovir-resistant HSV infection underscores the essential role of DOCK8 in dermatologic immunity. Our case demonstrates that a high degree of suspicion for cutaneous HSV infection should be adopted in DOCK8-deficient patients of any age, regardless of acyclovir prophylaxis. Viral culture in addition to bacterial cultures should be performed early in patients with cutaneous erosions, and the threshold for HSV resistance testing should be low to minimize morbidity associated with these infections. Early resistance testing in our case could have prevented prolongation of infection and likely eliminated the need for a biopsy.
Conclusion
DOCK8 deficiency presents a unique challenge to dermatologists and other health care providers given the susceptibility of affected individuals to developing a reservoir of severe and potentially resistant viral cutaneous infections. Prophylactic acyclovir may not be sufficient for HSV suppression, even in the youngest of patients, and suspicion for resistance should be high to avoid delays in adequate treatment.
Dedicator of cytokinesis 8 (DOCK8 ) deficiency is the major cause of autosomal-recessive hyper-IgEsyndrome. 1 Characteristic clinical features including eosinophilia, eczema, and recurrent Staphylococcus aureus cutaneous and respiratory tract infections are common in DOCK8 deficiency, similar to the autosomal-dominant form of hyper-IgE syndrome that is due to defi c iency of signal transducer and activation of transcription 3 (STAT-3 ). 1 In addition, patients with DOCK8 deficiency are particularly susceptible to asthma; food allergies; lymphomas; and severe cutaneous viral infections, including herpes simplex virus (HSV), molluscum contagiosum, varicella-zoster virus, and human papillomavirus. Since the discovery of the DOCK8 gene in 2009, various studies have sought to elucidate the mechanistic contribution of DOCK8 to the dermatologic immune environment. 2 Although cutaneous viral infections such as those caused by HSV typically are short lived and self-limiting in immunocompetent hosts, they have proven to be severe and recalcitrant in the setting of DOCK8 deficiency. 1 Herein, we report the case of a 32-month-old girl with homozygous DOCK8 deficiency who developed acyclovir-resistant cutaneous HSV.
Case Report
A 32-month-old girl presented with an approximately 2-cm linear erosion along the left posterior auricular sulcus at month 9 of a hospital stay for recurrent infections. Her medical history was notable for multiple upper respiratory tract infections, diffuse eczema, and food allergies. She had presented to an outside hospital at 14 months of age with herpetic gingivostomatitis and eczema herpeticum that was successfully treated with acyclovir. She was readmitted at 20 months of age due to Pneumocystis jiroveci pneumonia, pancytopenia, and disseminated histoplasmosis. Prophylactic oral acyclovir (20 mg/kg twice daily) was started, given her history of HSV infection. Because of recurrent infections, she underwent an immunodeficiency workup. Whole exome sequencing analysis revealed a homozygous deletion c.(528+1_529−1)_(1516+1_1517−1)del in DOCK8 gene–affecting exons 5 to 13. The patient was transferred to our hospital for continued care and as a potential candidate for bone marrow transplant following resolution of the disseminated histoplasmosis infection.
During her hospitalization at the current presentation, she was noted to have a 2-cm linear erosion along the left posterior auricular sulcus. Initial wound care with bacitracin ointment was applied to the area while specimens were obtained and empiric oral acyclovir therapy was initiated (20 mg/kg 4 times daily [QID]), given a clinical impression consistent with cutaneous HSV infection despite acyclovir prophylaxis. Direct immunofluorescence and viral cultures were positive for HSV-1, while bacterial cultures grew methicillin-susceptible S aureus. Cephalexin and mupirocin ointment were started, and acyclovir was continued. After 2 weeks of therapy, there was no visible change in the wound; cultures were repeated, again showing the wound contained HSV. Bacterial cultures this time grew Pseudomonas putida, and the antibiotic regimen was transitioned to cefepime.
After no response to the continued course of therapeutic acyclovir, HSV cultures were sent to the Centers for Disease Control and Prevention for resistance testing, and biopsy of the lesion was performed by the otolaryngology service to rule out malignancy and potential alternative diagnoses. Histopathology showed only reactive inflammation without visible microorganisms on tissue HSV-1/HSV-2 immunostain; however, tissue viral culture was positive for HSV-1. The patient was transitioned back to acyclovir (intravenous [IV] 20 mg/kg QID) with the addition of empiric foscarnet (IV 40 mg/kg 3 times daily) given the worsening appearance of the lesion. The HSV acyclovir resistance test results from the Centers for Disease Control and Prevention returned soon after and were positive for resistance (median infectious dose, 3.29 µg/L [reference interval, sensitive <2.00 µg/L; resistant >1.90 µg/L]). The patient completed a 21-day course of combination foscarnet and acyclovir therapy, during which time the lesion showed notable improvement and healing. The patient was continued on prophylactic acyclovir (IV 20 mg/kg QID). Unfortunately, the patient eventually died due to complications related to pneumonia.
Comment
Infection in Patients With DOCK8 Deficiency—The gene DOCK8 has emerged as playing a central role in both innate and adaptive immunity, as it is expressed primarily in immune cells and serves as a mediator of numerous processes, including immune synapse formation, cell signaling and trafficking, antibody and cytokine production, and lymphocyte memory.3 Cells that are critical for combating cutaneous viral infections, including skin-resident memory T cells and natural killer cells, are defective, which leads to a severely immunocompromised state in DOCK8-deficient patients with a particular susceptibility to infectious and inflammatory dermatologic disease.4
Herpes simplex virus infection commonly is seen in DOCK8 deficiency, with retrospective analysis of a DOCK8-deficient cohort revealing HSV infection in approximately 38% of patients.5 Prophylactic acyclovir is essential for DOCK8-deficient individuals with a history of HSV infection given the tendency of the virus to reactivate.6 However, despite prophylaxis, our patient developed an HSV-positive posterior auricular erosion that continued to progress even after increase of the acyclovir dose. Acyclovir resistance testing of the HSV isolated from the wound was positive, confirming the clinical suspicion of the presence of acyclovir-resistant HSV infection.
Acyclovir-Resistant HSV—Acyclovir-resistant HSV in immunosuppressed individuals was first noted in 1982, and most cases since then have occurred in the setting of AIDS and in organ transplant recipients.6 Few reports of acyclovir-resistant HSV in DOCK8 deficiency exist, and to our knowledge, our patient is the youngest DOCK8-deficient individual to be documented with acyclovir-resistant HSV infection.1,7-15 We identified relevant cases from the PubMed and EMBASE databases using the search terms DOCK8 deficiency and acyclovir and DOCK8 deficiency and herpes. The eTable lists other reported cases of acyclovir-resistant HSV in DOCK8-deficient patients. The majority of cases involved school-aged females. Lesion types varied and included herpes labialis, eczema herpeticum, and blepharoconjunctivitis. Escalation of therapy and resolution of the lesion was seen in some cases with administration of subcutaneous pegylated interferon alfa-2b.
Treatment Alternatives—Acyclovir competitively inhibits viral DNA polymerase by incorporating into elongating viral DNA strands and halting chain synthesis. Acyclovir requires triphosphorylation for activation, and viral thymidine kinase is responsible for the first phosphorylation event. Ninety-five percent of cases of acyclovir resistance are secondary to mutations in viral thymidine kinase. Foscarnet also inhibits viral DNA polymerase but does so directly without the need to be phosphorylated first.6 For this reason, foscarnet often is the drug of choice in the treatment of acyclovir-resistant HSV, as evidenced in our patient. However, foscarnet-resistant HSV strains may develop from mutations in the DNA polymerase gene.
Cidofovir is a nucleotide analogue that requires phosphorylation by host, as opposed to viral, kinases for antiviral activity. Intravenous and topical formulations of cidofovir have proven effective in the treatment of acyclovir- and foscarnet-resistant HSV lesions.6 Cidofovir also can be applied intralesionally, a method that provides targeted therapy and minimizes cidofovir-associated nephrotoxicity.12 Reports of systemic interferon alfa therapy for acyclovir-resistant HSV also exist. A study found IFN-⍺ production by peripheral blood mononuclear cells in DOCK8-deficient individuals to be significantly reduced relative to controls (P<.05).7 There has been complete resolution of acyclovir-resistant HSV lesions with subcutaneous pegylated interferon alfa-2b injections in several DOCK8-deficient patients.7-9
The need for escalating therapy in DOCK8-deficient individuals with acyclovir-resistant HSV infection underscores the essential role of DOCK8 in dermatologic immunity. Our case demonstrates that a high degree of suspicion for cutaneous HSV infection should be adopted in DOCK8-deficient patients of any age, regardless of acyclovir prophylaxis. Viral culture in addition to bacterial cultures should be performed early in patients with cutaneous erosions, and the threshold for HSV resistance testing should be low to minimize morbidity associated with these infections. Early resistance testing in our case could have prevented prolongation of infection and likely eliminated the need for a biopsy.
Conclusion
DOCK8 deficiency presents a unique challenge to dermatologists and other health care providers given the susceptibility of affected individuals to developing a reservoir of severe and potentially resistant viral cutaneous infections. Prophylactic acyclovir may not be sufficient for HSV suppression, even in the youngest of patients, and suspicion for resistance should be high to avoid delays in adequate treatment.
- Chu EY, Freeman AF, Jing H, et al. Cutaneous manifestations of DOCK8 deficiency syndrome. Arch Dermatol. 2012;148:79-84. doi:10.1001/archdermatol.2011.262
- Aydin SE, Kilic SS, Aytekin C, et al. DOCK8 deficiency: clinical and immunological phenotype and treatment options—a review of 136 patients. J Clin Immunol. 2015;35:189-198. doi:10.1007/s10875-014-0126-0
- Kearney CJ, Randall KL, Oliaro J. DOCK8 regulates signal transduction events to control immunity. Cell Mol Immunol. 2017;14:406-411. doi:10.1038/cmi.2017.9
- Zhang Q, Dove CG, Hor JL, et al. DOCK8 regulates lymphocyte shape integrity for skin antiviral immunity. J Exp Med. 2014;211:2549-2566. doi:10.1084/jem.20141307
- Engelhardt KR, Gertz EM, Keles S, et al. The extended clinical phenotype of 64 patients with DOCK8 deficiency. J Allergy Clin Immunol. 2015;136:402-412. doi:10.1016/j.jaci.2014.12.1945
- Chilukuri S, Rosen T. Management of acyclovir-resistant herpes simplex virus. Dermatol Clin. 2003;21:311-320. doi:10.1016/S0733-8635(02)00093-1
- Keles S, Jabara HH, Reisli I, et al. Plasmacytoid dendritic cell depletion in DOCK8 deficiency: rescue of severe herpetic infections with interferon alpha-2b therapy. J Allergy Clin Immunol. 2014;133:1753-1755.e3. doi:10.1016/j.jaci.2014.03.032
- Papan C, Hagl B, Heinz V, et al Beneficial IFN-α treatment of tumorous herpes simplex blepharoconjunctivitis in dedicator of cytokinesis 8 deficiency. J Allergy Clin Immunol. 2014;133:1456-1458. doi:10.1016/j.jaci.2014.02.008
- Metin A, Kanik-Yuksek S, Ozkaya-Parlakay A, et al. Giant herpes labialis in a child with DOCK8-deficient hyper-IgE syndrome. Pediatr Neonatol. 2016;57:79-80. doi:10.1016/j.pedneo.2015.04.011
- Zhang Q, Davis JC, Lamborn IT, et al. Combined immunodeficiency associated with DOCK8 mutations. N Engl J Med. 2009;361:2046-2055. doi:10.1056/NEJMoa0905506
- Lei JY, Wang Y, Jaffe ES, et al. Microcystic adnexal carcinoma associated with primary immunodeficiency, recurrent diffuse herpes simplex virus infection, and cutaneous T-cell lymphoma. Am J Dermatopathol. 2000;22:524-529. doi:10.1097/00000372-200012000-00008
- Castelo-Soccio L, Bernardin R, Stern J, et al. Successful treatment of acyclovir-resistant herpes simplex virus with intralesional cidofovir. Arch Dermatol. 2010;146:124-126. doi:10.1001/archdermatol.2009.363
- Shah NN, Freeman AF, Hickstein DD. Addendum to: haploidentical related donor hematopoietic stem cell transplantation for DOCK8 deficiency using post-transplantation cyclophosphamide. Biol Blood Marrow Transplant. 2019;25:E65-E67. doi:10.1016/j.bbmt.2018.11.014
- Freeman AF, Yazigi N, Shah NN, et al. Tandem orthotopic living donor liver transplantation followed by same donor haploidentical hematopoietic stem cell transplantation for DOCK8 deficiency. Transplantation. 2019;103:2144-2149. doi:10.1097/TP.0000000000002649
- Casto AM, Stout SC, Selvarangan R, et al. Evaluation of genotypic antiviral resistance testing as an alternative to phenotypic testing in a patient with DOCK8 deficiency and severe HSV-1 disease. J Infect Dis. 2020;221:2035-2042. doi:10.1093/infdis/jiaa020
- Chu EY, Freeman AF, Jing H, et al. Cutaneous manifestations of DOCK8 deficiency syndrome. Arch Dermatol. 2012;148:79-84. doi:10.1001/archdermatol.2011.262
- Aydin SE, Kilic SS, Aytekin C, et al. DOCK8 deficiency: clinical and immunological phenotype and treatment options—a review of 136 patients. J Clin Immunol. 2015;35:189-198. doi:10.1007/s10875-014-0126-0
- Kearney CJ, Randall KL, Oliaro J. DOCK8 regulates signal transduction events to control immunity. Cell Mol Immunol. 2017;14:406-411. doi:10.1038/cmi.2017.9
- Zhang Q, Dove CG, Hor JL, et al. DOCK8 regulates lymphocyte shape integrity for skin antiviral immunity. J Exp Med. 2014;211:2549-2566. doi:10.1084/jem.20141307
- Engelhardt KR, Gertz EM, Keles S, et al. The extended clinical phenotype of 64 patients with DOCK8 deficiency. J Allergy Clin Immunol. 2015;136:402-412. doi:10.1016/j.jaci.2014.12.1945
- Chilukuri S, Rosen T. Management of acyclovir-resistant herpes simplex virus. Dermatol Clin. 2003;21:311-320. doi:10.1016/S0733-8635(02)00093-1
- Keles S, Jabara HH, Reisli I, et al. Plasmacytoid dendritic cell depletion in DOCK8 deficiency: rescue of severe herpetic infections with interferon alpha-2b therapy. J Allergy Clin Immunol. 2014;133:1753-1755.e3. doi:10.1016/j.jaci.2014.03.032
- Papan C, Hagl B, Heinz V, et al Beneficial IFN-α treatment of tumorous herpes simplex blepharoconjunctivitis in dedicator of cytokinesis 8 deficiency. J Allergy Clin Immunol. 2014;133:1456-1458. doi:10.1016/j.jaci.2014.02.008
- Metin A, Kanik-Yuksek S, Ozkaya-Parlakay A, et al. Giant herpes labialis in a child with DOCK8-deficient hyper-IgE syndrome. Pediatr Neonatol. 2016;57:79-80. doi:10.1016/j.pedneo.2015.04.011
- Zhang Q, Davis JC, Lamborn IT, et al. Combined immunodeficiency associated with DOCK8 mutations. N Engl J Med. 2009;361:2046-2055. doi:10.1056/NEJMoa0905506
- Lei JY, Wang Y, Jaffe ES, et al. Microcystic adnexal carcinoma associated with primary immunodeficiency, recurrent diffuse herpes simplex virus infection, and cutaneous T-cell lymphoma. Am J Dermatopathol. 2000;22:524-529. doi:10.1097/00000372-200012000-00008
- Castelo-Soccio L, Bernardin R, Stern J, et al. Successful treatment of acyclovir-resistant herpes simplex virus with intralesional cidofovir. Arch Dermatol. 2010;146:124-126. doi:10.1001/archdermatol.2009.363
- Shah NN, Freeman AF, Hickstein DD. Addendum to: haploidentical related donor hematopoietic stem cell transplantation for DOCK8 deficiency using post-transplantation cyclophosphamide. Biol Blood Marrow Transplant. 2019;25:E65-E67. doi:10.1016/j.bbmt.2018.11.014
- Freeman AF, Yazigi N, Shah NN, et al. Tandem orthotopic living donor liver transplantation followed by same donor haploidentical hematopoietic stem cell transplantation for DOCK8 deficiency. Transplantation. 2019;103:2144-2149. doi:10.1097/TP.0000000000002649
- Casto AM, Stout SC, Selvarangan R, et al. Evaluation of genotypic antiviral resistance testing as an alternative to phenotypic testing in a patient with DOCK8 deficiency and severe HSV-1 disease. J Infect Dis. 2020;221:2035-2042. doi:10.1093/infdis/jiaa020
Practice Points
- Patients with dedicator of cytokinesis 8 ( DOCK 8 ) deficiency are susceptible to development of severe recalcitrant viral cutaneous infections, including herpes simplex virus (HSV).
- Dermatologists should be aware that prophylactic acyclovir may not be sufficient for HSV suppression in the setting of severe immunodeficiency.
- Acyclovir-resistant cutaneous HSV lesions require escalation of therapy, which may include addition of foscarnet, cidofovir, or subcutaneous pegylated interferon alfa-2b to the therapeutic regimen.
- Viral culture should be performed on suspicious lesions in DOCK 8 -deficient patients despite acyclovir prophylaxis, and the threshold for HSV resistance testing should be low.
Bullous Amyloidosis Masquerading as Pseudoporphyria
Cutaneous amyloidosis encompasses a variety of clinical presentations. Primary localized cutaneous amyloidosis comprises lichen amyloidosis, macular amyloidosis, and nodular amyloidosis.1 Macular and lichen amyloidosis result from keratin deposits, while nodular amyloidosis results from cutaneous infiltration of plasma cells.2 Primary systemic amyloidosis is due to a plasma cell dyscrasia, particularly multiple myeloma, while secondary systemic amyloidosis occurs in the setting of restrictive cardiomyopathy, congestive heart failure, renal dysfunction, or chronic inflammation, as seen with rheumatoid arthritis, tuberculosis, and various autoinflammatory disorders.2 Plasma cell proliferative disorders are associated with various skin disorders, which may result from aggregated misfolded monoclonal immunoglobulins, indicating light chain–related systemic amyloidosis. Mucocutaneous lesions can occur in 30% to 40% of cases of primary systemic amyloidosis and may present as purpura, ecchymoses, waxy thickening, plaques, subcutaneous nodules, and/or bullae.3,4 When blistering is present, the differential diagnosis is broad and includes autoimmune bullous disease, drug eruptions, enoxaparin-induced bullous hemorrhagic dermatosis, deposition diseases, allergic contact dermatitis, bullous cellulitis, bullous bite reactions, neutrophilic dermatosis, and bullous lichen sclerosus.5 Herein, we present a case of a woman with a bullous skin eruption who eventually was diagnosed with bullous amyloidosis subsequent to a diagnosis of multiple myeloma.
Case Report
A 70-year-old woman presented to our dermatology clinic for evaluation of well-demarcated, hemorrhagic, flaccid vesicles and focal erosions with a rim of erythema on the distal forearms and hands. A shave biopsy from the right forearm showed cell-poor subepidermal vesicular dermatitis. Enzyme-linked immunosorbent assays for bullous pemphigoid antigens 1 and 2 as well as urinary porphyrins were negative. Direct immunofluorescence showed granular IgM at the basement membrane zone around vessels and cytoid bodies. At this time, a preliminary diagnosis of pseudoporphyria was suspected, though no classic medications (eg, nonsteroidal anti-inflammatory drugs, furosemide, antibiotics) or exogenous trigger factors (eg, UV light exposure, dialysis) were temporally related. Three months later, the patient presented with a large hemorrhagic bulla on the distal left forearm (Figure 1) and healing erosions on the dorsal fingers and upper back. Clobetasol ointment was initiated, as an autoimmune bullous dermatosis was suspected.

Approximately 1 year after she was first seen in our outpatient clinic, the patient was hospitalized for induction of chemotherapy—cyclophosphamide, bortezomib, and dexamethasone—for a new diagnosis of stage III multiple myeloma. A workup for back pain revealed multiple compression fractures and a plasma cell neoplasm with elevated λ light chains, which was confirmed with a bone marrow biopsy. During an inpatient dermatology consultation, we noted the development of intraoral hemorrhagic vesicles and worsening generalization of the hemorrhagic bullae, with healing erosions and intact hemorrhagic bullae on the dorsal hands, fingers (Figure 2), and upper back.
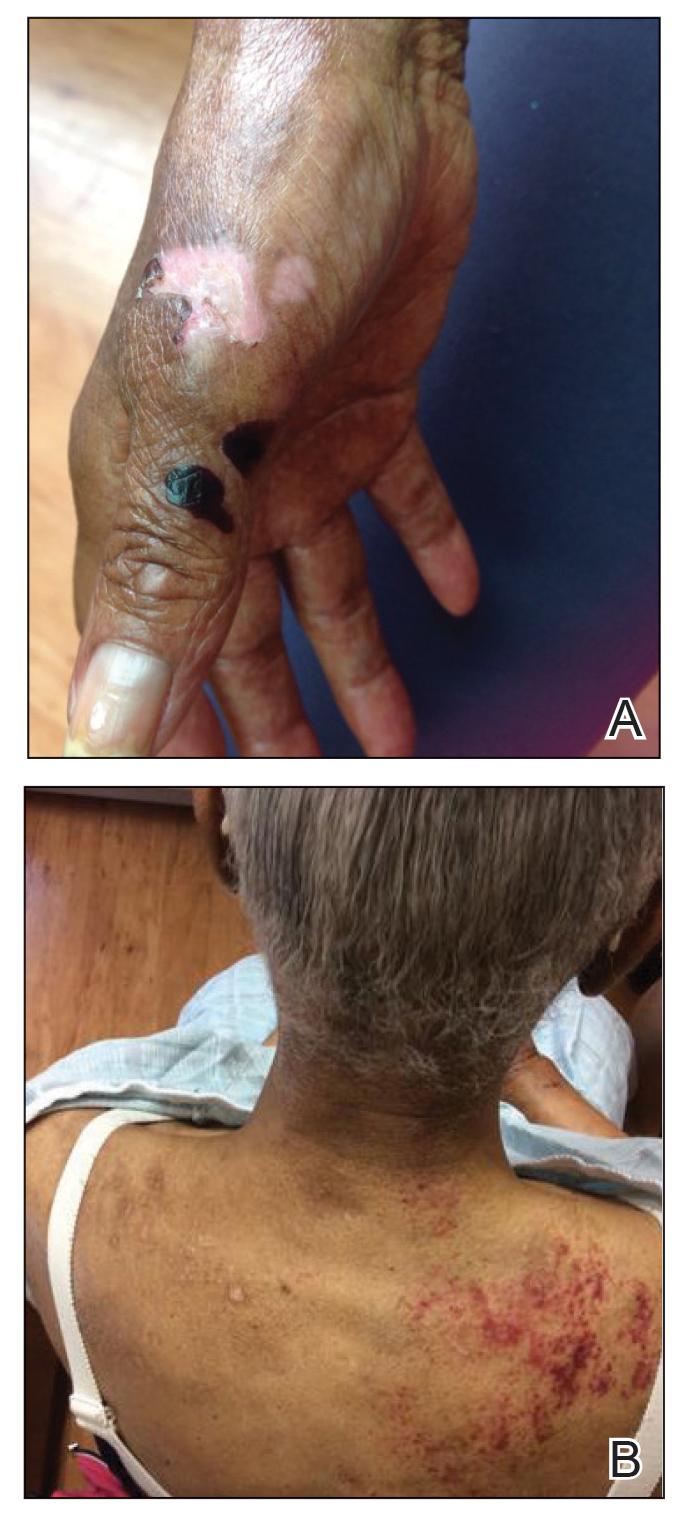
A repeat biopsy displayed bullous amyloidosis. Histopathologic examination revealed an ulcerated subepidermal blister with fibrin deposition at the ulcer base. A periadnexal, scant, eosinophilic deposition with extravasated red blood cells was appreciated. Amorphous eosinophilic deposits were found within the detached fragment of the epidermis and inflammatory infiltrate. A Congo red stain highlighted these areas with a salmon pink–colored material. Congo red staining showed a moderate amount of pale, apple green, birefringent deposit within these areas on polarized light examination.
A few months later, the patient was re-admitted, and the amount of skin detachment prompted the primary team to ask for another consultation. Although the extensive skin sloughing resembled toxic epidermal necrolysis, a repeat biopsy confirmed bullous amyloidosis.
Comment
Amyloidosis Histopathology—Amyloidoses represent a wide array of disorders with deposition of β-pleated sheets or amyloid fibrils, often with cutaneous manifestations.2,3 Primary systemic amyloidosis has been associated with underlying dyscrasia or multiple myeloma.6 In such cases, the skin lesions of multiple myeloma may result from a collection of misfolded monoclonal immunoglobulins or their fragments, as in light chain–related systemic amyloidosis.3 Histopathologically, both systemic and cutaneous amyloidosis appear similar and display deposition of amorphous, eosinophilic, fissured amyloid material in the dermis. Congo red stains the material orange-red and will display a characteristic apple green birefringence under polarized light.4 Although bullous amyloid lesions are rare, the cutaneous forms of these lesions can be an important sign of plasma cell dyscrasia.7
Presentation of Bullous Amyloidosis—Bullous manifestations rarely have been noted in the primary cutaneous forms of amyloidosis.5,8,9 Importantly, cutaneous blistering more often is linked to systemic forms of amyloidosis with multiorgan involvement, including primary systemic and myeloma-associated amyloidosis.5,10 However, patients with localized bullous cutaneous amyloidosis without systemic involvement also have been seen.10,11 Bullae may occur at any time, with contents that frequently are hemorrhagic due to capillary fragility.12,13 Bullous manifestations raise the differential diagnoses of bullous pemphigoid, epidermolysis bullosa acquisita, linear IgA disease, porphyria cutanea tarda, pseudoporphyria, bullous drug eruption, bullous eruption of renal dialysis, or bullous lupus erythematosus.5,13-17
In our patient, the acral distribution of bullae, presence of hemorrhage, chronicity of symptoms, and negative enzyme-linked immunosorbent assay initially suggested a diagnosis of pseudoporphyria. However, the presence of intraoral hemorrhagic vesicles and subsequent confirmatory pathology aided in differentiating bullous amyloidosis from pseudoporphyria. Nodular localized primary cutaneous amyloidosis, a rare form of skin-restricted amyloidoses, can coexist with bullous lesions. Of note, reported cases of nodular localized primary cutaneous amyloidosis did not result in development of multiple myeloma.5,10
Bullae are located either subepidermally or intradermally, and bullous lesions of cutaneous amyloidosis typically demonstrate subepidermal or superficial intradermal clefting on light microscopy.5,10,12 Histopathology of bullous amyloidosis shows intradermal or subepidermal blister formation and amorphous eosinophilic material showing apple green birefringence with Congo red staining deposited in the dermis and/or around the adipocytes and blood vessel walls.12,18-20 In prior cases, direct immunofluorescence of bullous amyloidosis revealed absent immunoglobulin (IgG, IgA, IgM) or complement (C3 and C9) deposits in the basement membrane zone or dermis.13,21,22 In these cases, electron microscopy was useful in diagnosis, as it showed the presence of amyloid deposits.21,22
Cause of Bullae—Various mechanisms are thought to trigger the blister formation in amyloidosis. Bullae created from trauma or friction often present as tense painful blisters that commonly are hemorrhagic.10,23 Amyloid deposits in the walls of blood vessels and the affinity of dermal amyloid in blood vessel walls to surrounding collagen likely leads to increased fragility of capillaries and the dermal matrix, hemorrhagic tendency, and infrapapillary blisters, thus creating hemorrhagic bullous eruptions.24,25 Specifically, close proximity of immunoglobulin-derived amyloid oligomers to epidermal keratinocytes may be toxic and therefore could trigger subepidermal bullous change.5 Additionally, alteration in the physicochemical properties of the amyloidal protein might explain bullous eruption.9 Trauma or rubbing of the hands and feet may precipitate the acral blister formation in bullous amyloidosis.5,11
Due to deposition of these amyloid fibrils, skin bleeding in these patients is called amyloid or pinch purpura. Vessel wall fragility and damage by amyloid are the principal causes of periorbital and gastrointestinal tract bleeding.26 Destruction of the lamina densa and widening of the intercellular space between keratinocytes by amyloid globules induce skin fragility.11
Although uncommon, various cases of bullous amyloidosis have been reported in the literature. Multiple myeloma patients represent the majority of those reported to have bullous amyloidosis.6,7,13,24,27-30 Plasmacytoma-associated bullous amyloid purpura and paraproteinemia also have been noted.25 Multiple myeloma with secondary AL amyloidosis has been seen with amyloid purpura and atraumatic ecchymoses of the face, highlighting the hemorrhage noted in these patients.26
Management of Amyloidosis—Various treatment options have been attempted for primary cutaneous amyloidosis, including oral retinoids, corticosteroids, cyclophosphamide, cyclosporine, amitriptyline, colchicine, cepharanthin, tacrolimus, dimethyl sulfoxide, vitamin D3 analogs, capsaicin, menthol, hydrocolloid dressings, surgical modalities, laser treatment, and phototherapy.1 There is no clear consensus for therapeutic modalities except for treating the underlying plasma cell dyscrasia in primary systemic amyloidosis.
Conclusion
We report the case of a patient displaying signs of pseudoporphyria that ultimately proved to be bullous amyloidosis, or what we termed pseudopseudoporphyria. Bullous amyloidosis should be considered in the differential diagnoses of hemorrhagic bullous skin eruptions. Particular attention should be given to a systemic workup for multiple myeloma when hemorrhagic vesicles/bullae are chronic and coexist with purpura, angina bullosa hemorrhagica, fatigue/weight loss, and/or macroglossia.
- Weidner T, Illing T, Elsner P. Primary localized cutaneous amyloidosis: a systematic treatment review. Am J Clin Dermatol. 2017;18:629-642.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Amyloidosis. Dermatology Essentials. Elsevier Saunders; 2014:341-345.
- Bhutani M, Shahid Z, Schnebelen A, et al. Cutaneous manifestations of multiple myeloma and other plasma cell proliferative disorders. Semin Oncol. 2016;43:395-400.
- Terushkin V, Boyd KP, Patel RR, et al. Primary localized cutaneous amyloidosis. Dermatol Online J. 2013;19:20711.
- LaChance A, Phelps A, Finch J, et al. Nodular localized primary cutaneous amyloidosis: a bullous variant. Clin Exp Dermatol. 2014;39:344-347.
- Gonzalez-Ramos J, Garrido-Gutiérrez C, González-Silva Y, et al. Relapsing bullous amyloidosis of the oral mucosa and acquired cutis laxa in a patient with multiple myeloma: a rare triple association. Clin Exp Dermatol. 2017;42:410-412.
- Kanoh T. Bullous amyloidosis [in Japanese]. Rinsho Ketsueki. 1993;34:1050-1052.
- Johnson TM, Rapini RP, Hebert AA, et al. Bullous amyloidosis. Cutis. 1989;43:346-352.
- Houman MH, Smiti KM, Ben Ghorbel I, et al. Bullous amyloidosis. Ann Dermatol Venereol. 2002;129:299-302.
- Sanusi T, Li Y, Qian Y, et al. Primary localized cutaneous nodular amyloidosis with bullous lesions. Indian J Dermatol Venereol Leprol. 2015;81:400-402.
- Ochiai T, Morishima T, Hao T, et al. Bullous amyloidosis: the mechanism of blister formation revealed by electron microscopy. J Cutan Pathol. 2001;28:407-411.
- Chu CH, Chan JY, Hsieh SW, et al. Diffuse ecchymoses and blisters on a yellowish waxy base: a case of bullous amyloidosis. J Dermatol. 2016;43:713-714.
- Wang XD, Shen H, Liu ZH. Diffuse haemorrhagic bullous amyloidosis with multiple myeloma. Clin Exp Dermatol. 2008;33:94-96.
- Biswas P, Aggarwal I, Sen D, et al. Bullous pemphigoid clinically presenting as lichen amyloidosis. Indian J Dermatol Venereol Leprol. 2014;80:544-546.
- Bluhm JF 3rd. Bullous dermatosis vs amyloidosis. Arch Dermatol. 1981;117:252.
- Bluhm JF 3rd. Bullous amyloidosis vs epidermolysis bullosa acquisita. JAMA. 1981;245:32.
- Murphy GM, Wright J, Nicholls DS, et al. Sunbed-induced pseudoporphyria. Br J Dermatol. 1989;120:555-562.
- Pramatarov K, Lazarova A, Mateev G, et al. Bullous hemorrhagic primary systemic amyloidosis. Int J Dermatol. 1990;29:211-213.
- Bieber T, Ruzicka T, Linke RP, et al. Hemorrhagic bullous amyloidosis. a histologic, immunocytochemical, and ultrastructural study of two patients. Arch Dermatol. 1988;124:1683-1686.
- Khoo BP, Tay YK. Lichen amyloidosis: a bullous variant. Ann Acad Med Singapore. 2000;29:105-107.
- Asahina A, Hasegawa K, Ishiyama M, et al. Bullous amyloidosis mimicking bullous pemphigoid: usefulness of electron microscopic examination. Acta Derm Venereol. 2010;90:427-428.
- Schmutz JL, Barbaud A, Cuny JF, et al. Bullous amyloidosis [in French]. Ann Dermatol Venereol. 1988;115:295-301.
- Lachmann HJ, Hawkins PN. Amyloidosis of the skin. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. McGraw-Hill; 2012:1574-1583.
- Grundmann JU, Bonnekoh B, Gollnick H. Extensive haemorrhagic-bullous skin manifestation of systemic AA-amyloidosis associated with IgG lambda-myeloma. Eur J Dermatol. 2000;10:139-142.
- Hödl S, Turek TD, Kerl H. Plasmocytoma-associated bullous hemorrhagic amyloidosis of the skin [in German]. Hautarzt. 1982;33:556-558.
- Colucci G, Alberio L, Demarmels Biasiutti F, et al. Bilateral periorbital ecchymoses. an often missed sign of amyloid purpura. Hamostaseologie. 2014;34:249-252.
- Behera B, Pattnaik M, Sahu B, et al. Cutaneous manifestations of multiple myeloma. Indian J Dermatol. 2016;61:668-671.
- Fujita Y, Tsuji-Abe Y, Sato-Matsumura KC, et al. Nail dystrophy and blisters as sole manifestations in myeloma-associated amyloidosis. J Am Acad Dermatol. 2006;54:712-714.
- Chang SL, Lai PC, Cheng CJ, et al. Bullous amyloidosis in a hemodialysis patient is myeloma-associated rather than hemodialysis-associated amyloidosis. Amyloid. 2007;14:153-156.
- Winzer M, Ruppert M, Baretton G, et al. Bullous poikilodermatitic amyloidosis of the skin with junctional bulla development in IgG light chain plasmacytoma of the lambda type. histology, immunohistology and electron microscopy [in German]. Hautarzt. 1992;43:199-204.
Cutaneous amyloidosis encompasses a variety of clinical presentations. Primary localized cutaneous amyloidosis comprises lichen amyloidosis, macular amyloidosis, and nodular amyloidosis.1 Macular and lichen amyloidosis result from keratin deposits, while nodular amyloidosis results from cutaneous infiltration of plasma cells.2 Primary systemic amyloidosis is due to a plasma cell dyscrasia, particularly multiple myeloma, while secondary systemic amyloidosis occurs in the setting of restrictive cardiomyopathy, congestive heart failure, renal dysfunction, or chronic inflammation, as seen with rheumatoid arthritis, tuberculosis, and various autoinflammatory disorders.2 Plasma cell proliferative disorders are associated with various skin disorders, which may result from aggregated misfolded monoclonal immunoglobulins, indicating light chain–related systemic amyloidosis. Mucocutaneous lesions can occur in 30% to 40% of cases of primary systemic amyloidosis and may present as purpura, ecchymoses, waxy thickening, plaques, subcutaneous nodules, and/or bullae.3,4 When blistering is present, the differential diagnosis is broad and includes autoimmune bullous disease, drug eruptions, enoxaparin-induced bullous hemorrhagic dermatosis, deposition diseases, allergic contact dermatitis, bullous cellulitis, bullous bite reactions, neutrophilic dermatosis, and bullous lichen sclerosus.5 Herein, we present a case of a woman with a bullous skin eruption who eventually was diagnosed with bullous amyloidosis subsequent to a diagnosis of multiple myeloma.
Case Report
A 70-year-old woman presented to our dermatology clinic for evaluation of well-demarcated, hemorrhagic, flaccid vesicles and focal erosions with a rim of erythema on the distal forearms and hands. A shave biopsy from the right forearm showed cell-poor subepidermal vesicular dermatitis. Enzyme-linked immunosorbent assays for bullous pemphigoid antigens 1 and 2 as well as urinary porphyrins were negative. Direct immunofluorescence showed granular IgM at the basement membrane zone around vessels and cytoid bodies. At this time, a preliminary diagnosis of pseudoporphyria was suspected, though no classic medications (eg, nonsteroidal anti-inflammatory drugs, furosemide, antibiotics) or exogenous trigger factors (eg, UV light exposure, dialysis) were temporally related. Three months later, the patient presented with a large hemorrhagic bulla on the distal left forearm (Figure 1) and healing erosions on the dorsal fingers and upper back. Clobetasol ointment was initiated, as an autoimmune bullous dermatosis was suspected.
Approximately 1 year after she was first seen in our outpatient clinic, the patient was hospitalized for induction of chemotherapy—cyclophosphamide, bortezomib, and dexamethasone—for a new diagnosis of stage III multiple myeloma. A workup for back pain revealed multiple compression fractures and a plasma cell neoplasm with elevated λ light chains, which was confirmed with a bone marrow biopsy. During an inpatient dermatology consultation, we noted the development of intraoral hemorrhagic vesicles and worsening generalization of the hemorrhagic bullae, with healing erosions and intact hemorrhagic bullae on the dorsal hands, fingers (Figure 2), and upper back.
A repeat biopsy displayed bullous amyloidosis. Histopathologic examination revealed an ulcerated subepidermal blister with fibrin deposition at the ulcer base. A periadnexal, scant, eosinophilic deposition with extravasated red blood cells was appreciated. Amorphous eosinophilic deposits were found within the detached fragment of the epidermis and inflammatory infiltrate. A Congo red stain highlighted these areas with a salmon pink–colored material. Congo red staining showed a moderate amount of pale, apple green, birefringent deposit within these areas on polarized light examination.
A few months later, the patient was re-admitted, and the amount of skin detachment prompted the primary team to ask for another consultation. Although the extensive skin sloughing resembled toxic epidermal necrolysis, a repeat biopsy confirmed bullous amyloidosis.
Comment
Amyloidosis Histopathology—Amyloidoses represent a wide array of disorders with deposition of β-pleated sheets or amyloid fibrils, often with cutaneous manifestations.2,3 Primary systemic amyloidosis has been associated with underlying dyscrasia or multiple myeloma.6 In such cases, the skin lesions of multiple myeloma may result from a collection of misfolded monoclonal immunoglobulins or their fragments, as in light chain–related systemic amyloidosis.3 Histopathologically, both systemic and cutaneous amyloidosis appear similar and display deposition of amorphous, eosinophilic, fissured amyloid material in the dermis. Congo red stains the material orange-red and will display a characteristic apple green birefringence under polarized light.4 Although bullous amyloid lesions are rare, the cutaneous forms of these lesions can be an important sign of plasma cell dyscrasia.7
Presentation of Bullous Amyloidosis—Bullous manifestations rarely have been noted in the primary cutaneous forms of amyloidosis.5,8,9 Importantly, cutaneous blistering more often is linked to systemic forms of amyloidosis with multiorgan involvement, including primary systemic and myeloma-associated amyloidosis.5,10 However, patients with localized bullous cutaneous amyloidosis without systemic involvement also have been seen.10,11 Bullae may occur at any time, with contents that frequently are hemorrhagic due to capillary fragility.12,13 Bullous manifestations raise the differential diagnoses of bullous pemphigoid, epidermolysis bullosa acquisita, linear IgA disease, porphyria cutanea tarda, pseudoporphyria, bullous drug eruption, bullous eruption of renal dialysis, or bullous lupus erythematosus.5,13-17
In our patient, the acral distribution of bullae, presence of hemorrhage, chronicity of symptoms, and negative enzyme-linked immunosorbent assay initially suggested a diagnosis of pseudoporphyria. However, the presence of intraoral hemorrhagic vesicles and subsequent confirmatory pathology aided in differentiating bullous amyloidosis from pseudoporphyria. Nodular localized primary cutaneous amyloidosis, a rare form of skin-restricted amyloidoses, can coexist with bullous lesions. Of note, reported cases of nodular localized primary cutaneous amyloidosis did not result in development of multiple myeloma.5,10
Bullae are located either subepidermally or intradermally, and bullous lesions of cutaneous amyloidosis typically demonstrate subepidermal or superficial intradermal clefting on light microscopy.5,10,12 Histopathology of bullous amyloidosis shows intradermal or subepidermal blister formation and amorphous eosinophilic material showing apple green birefringence with Congo red staining deposited in the dermis and/or around the adipocytes and blood vessel walls.12,18-20 In prior cases, direct immunofluorescence of bullous amyloidosis revealed absent immunoglobulin (IgG, IgA, IgM) or complement (C3 and C9) deposits in the basement membrane zone or dermis.13,21,22 In these cases, electron microscopy was useful in diagnosis, as it showed the presence of amyloid deposits.21,22
Cause of Bullae—Various mechanisms are thought to trigger the blister formation in amyloidosis. Bullae created from trauma or friction often present as tense painful blisters that commonly are hemorrhagic.10,23 Amyloid deposits in the walls of blood vessels and the affinity of dermal amyloid in blood vessel walls to surrounding collagen likely leads to increased fragility of capillaries and the dermal matrix, hemorrhagic tendency, and infrapapillary blisters, thus creating hemorrhagic bullous eruptions.24,25 Specifically, close proximity of immunoglobulin-derived amyloid oligomers to epidermal keratinocytes may be toxic and therefore could trigger subepidermal bullous change.5 Additionally, alteration in the physicochemical properties of the amyloidal protein might explain bullous eruption.9 Trauma or rubbing of the hands and feet may precipitate the acral blister formation in bullous amyloidosis.5,11
Due to deposition of these amyloid fibrils, skin bleeding in these patients is called amyloid or pinch purpura. Vessel wall fragility and damage by amyloid are the principal causes of periorbital and gastrointestinal tract bleeding.26 Destruction of the lamina densa and widening of the intercellular space between keratinocytes by amyloid globules induce skin fragility.11
Although uncommon, various cases of bullous amyloidosis have been reported in the literature. Multiple myeloma patients represent the majority of those reported to have bullous amyloidosis.6,7,13,24,27-30 Plasmacytoma-associated bullous amyloid purpura and paraproteinemia also have been noted.25 Multiple myeloma with secondary AL amyloidosis has been seen with amyloid purpura and atraumatic ecchymoses of the face, highlighting the hemorrhage noted in these patients.26
Management of Amyloidosis—Various treatment options have been attempted for primary cutaneous amyloidosis, including oral retinoids, corticosteroids, cyclophosphamide, cyclosporine, amitriptyline, colchicine, cepharanthin, tacrolimus, dimethyl sulfoxide, vitamin D3 analogs, capsaicin, menthol, hydrocolloid dressings, surgical modalities, laser treatment, and phototherapy.1 There is no clear consensus for therapeutic modalities except for treating the underlying plasma cell dyscrasia in primary systemic amyloidosis.
Conclusion
We report the case of a patient displaying signs of pseudoporphyria that ultimately proved to be bullous amyloidosis, or what we termed pseudopseudoporphyria. Bullous amyloidosis should be considered in the differential diagnoses of hemorrhagic bullous skin eruptions. Particular attention should be given to a systemic workup for multiple myeloma when hemorrhagic vesicles/bullae are chronic and coexist with purpura, angina bullosa hemorrhagica, fatigue/weight loss, and/or macroglossia.
Cutaneous amyloidosis encompasses a variety of clinical presentations. Primary localized cutaneous amyloidosis comprises lichen amyloidosis, macular amyloidosis, and nodular amyloidosis.1 Macular and lichen amyloidosis result from keratin deposits, while nodular amyloidosis results from cutaneous infiltration of plasma cells.2 Primary systemic amyloidosis is due to a plasma cell dyscrasia, particularly multiple myeloma, while secondary systemic amyloidosis occurs in the setting of restrictive cardiomyopathy, congestive heart failure, renal dysfunction, or chronic inflammation, as seen with rheumatoid arthritis, tuberculosis, and various autoinflammatory disorders.2 Plasma cell proliferative disorders are associated with various skin disorders, which may result from aggregated misfolded monoclonal immunoglobulins, indicating light chain–related systemic amyloidosis. Mucocutaneous lesions can occur in 30% to 40% of cases of primary systemic amyloidosis and may present as purpura, ecchymoses, waxy thickening, plaques, subcutaneous nodules, and/or bullae.3,4 When blistering is present, the differential diagnosis is broad and includes autoimmune bullous disease, drug eruptions, enoxaparin-induced bullous hemorrhagic dermatosis, deposition diseases, allergic contact dermatitis, bullous cellulitis, bullous bite reactions, neutrophilic dermatosis, and bullous lichen sclerosus.5 Herein, we present a case of a woman with a bullous skin eruption who eventually was diagnosed with bullous amyloidosis subsequent to a diagnosis of multiple myeloma.
Case Report
A 70-year-old woman presented to our dermatology clinic for evaluation of well-demarcated, hemorrhagic, flaccid vesicles and focal erosions with a rim of erythema on the distal forearms and hands. A shave biopsy from the right forearm showed cell-poor subepidermal vesicular dermatitis. Enzyme-linked immunosorbent assays for bullous pemphigoid antigens 1 and 2 as well as urinary porphyrins were negative. Direct immunofluorescence showed granular IgM at the basement membrane zone around vessels and cytoid bodies. At this time, a preliminary diagnosis of pseudoporphyria was suspected, though no classic medications (eg, nonsteroidal anti-inflammatory drugs, furosemide, antibiotics) or exogenous trigger factors (eg, UV light exposure, dialysis) were temporally related. Three months later, the patient presented with a large hemorrhagic bulla on the distal left forearm (Figure 1) and healing erosions on the dorsal fingers and upper back. Clobetasol ointment was initiated, as an autoimmune bullous dermatosis was suspected.
Approximately 1 year after she was first seen in our outpatient clinic, the patient was hospitalized for induction of chemotherapy—cyclophosphamide, bortezomib, and dexamethasone—for a new diagnosis of stage III multiple myeloma. A workup for back pain revealed multiple compression fractures and a plasma cell neoplasm with elevated λ light chains, which was confirmed with a bone marrow biopsy. During an inpatient dermatology consultation, we noted the development of intraoral hemorrhagic vesicles and worsening generalization of the hemorrhagic bullae, with healing erosions and intact hemorrhagic bullae on the dorsal hands, fingers (Figure 2), and upper back.
A repeat biopsy displayed bullous amyloidosis. Histopathologic examination revealed an ulcerated subepidermal blister with fibrin deposition at the ulcer base. A periadnexal, scant, eosinophilic deposition with extravasated red blood cells was appreciated. Amorphous eosinophilic deposits were found within the detached fragment of the epidermis and inflammatory infiltrate. A Congo red stain highlighted these areas with a salmon pink–colored material. Congo red staining showed a moderate amount of pale, apple green, birefringent deposit within these areas on polarized light examination.
A few months later, the patient was re-admitted, and the amount of skin detachment prompted the primary team to ask for another consultation. Although the extensive skin sloughing resembled toxic epidermal necrolysis, a repeat biopsy confirmed bullous amyloidosis.
Comment
Amyloidosis Histopathology—Amyloidoses represent a wide array of disorders with deposition of β-pleated sheets or amyloid fibrils, often with cutaneous manifestations.2,3 Primary systemic amyloidosis has been associated with underlying dyscrasia or multiple myeloma.6 In such cases, the skin lesions of multiple myeloma may result from a collection of misfolded monoclonal immunoglobulins or their fragments, as in light chain–related systemic amyloidosis.3 Histopathologically, both systemic and cutaneous amyloidosis appear similar and display deposition of amorphous, eosinophilic, fissured amyloid material in the dermis. Congo red stains the material orange-red and will display a characteristic apple green birefringence under polarized light.4 Although bullous amyloid lesions are rare, the cutaneous forms of these lesions can be an important sign of plasma cell dyscrasia.7
Presentation of Bullous Amyloidosis—Bullous manifestations rarely have been noted in the primary cutaneous forms of amyloidosis.5,8,9 Importantly, cutaneous blistering more often is linked to systemic forms of amyloidosis with multiorgan involvement, including primary systemic and myeloma-associated amyloidosis.5,10 However, patients with localized bullous cutaneous amyloidosis without systemic involvement also have been seen.10,11 Bullae may occur at any time, with contents that frequently are hemorrhagic due to capillary fragility.12,13 Bullous manifestations raise the differential diagnoses of bullous pemphigoid, epidermolysis bullosa acquisita, linear IgA disease, porphyria cutanea tarda, pseudoporphyria, bullous drug eruption, bullous eruption of renal dialysis, or bullous lupus erythematosus.5,13-17
In our patient, the acral distribution of bullae, presence of hemorrhage, chronicity of symptoms, and negative enzyme-linked immunosorbent assay initially suggested a diagnosis of pseudoporphyria. However, the presence of intraoral hemorrhagic vesicles and subsequent confirmatory pathology aided in differentiating bullous amyloidosis from pseudoporphyria. Nodular localized primary cutaneous amyloidosis, a rare form of skin-restricted amyloidoses, can coexist with bullous lesions. Of note, reported cases of nodular localized primary cutaneous amyloidosis did not result in development of multiple myeloma.5,10
Bullae are located either subepidermally or intradermally, and bullous lesions of cutaneous amyloidosis typically demonstrate subepidermal or superficial intradermal clefting on light microscopy.5,10,12 Histopathology of bullous amyloidosis shows intradermal or subepidermal blister formation and amorphous eosinophilic material showing apple green birefringence with Congo red staining deposited in the dermis and/or around the adipocytes and blood vessel walls.12,18-20 In prior cases, direct immunofluorescence of bullous amyloidosis revealed absent immunoglobulin (IgG, IgA, IgM) or complement (C3 and C9) deposits in the basement membrane zone or dermis.13,21,22 In these cases, electron microscopy was useful in diagnosis, as it showed the presence of amyloid deposits.21,22
Cause of Bullae—Various mechanisms are thought to trigger the blister formation in amyloidosis. Bullae created from trauma or friction often present as tense painful blisters that commonly are hemorrhagic.10,23 Amyloid deposits in the walls of blood vessels and the affinity of dermal amyloid in blood vessel walls to surrounding collagen likely leads to increased fragility of capillaries and the dermal matrix, hemorrhagic tendency, and infrapapillary blisters, thus creating hemorrhagic bullous eruptions.24,25 Specifically, close proximity of immunoglobulin-derived amyloid oligomers to epidermal keratinocytes may be toxic and therefore could trigger subepidermal bullous change.5 Additionally, alteration in the physicochemical properties of the amyloidal protein might explain bullous eruption.9 Trauma or rubbing of the hands and feet may precipitate the acral blister formation in bullous amyloidosis.5,11
Due to deposition of these amyloid fibrils, skin bleeding in these patients is called amyloid or pinch purpura. Vessel wall fragility and damage by amyloid are the principal causes of periorbital and gastrointestinal tract bleeding.26 Destruction of the lamina densa and widening of the intercellular space between keratinocytes by amyloid globules induce skin fragility.11
Although uncommon, various cases of bullous amyloidosis have been reported in the literature. Multiple myeloma patients represent the majority of those reported to have bullous amyloidosis.6,7,13,24,27-30 Plasmacytoma-associated bullous amyloid purpura and paraproteinemia also have been noted.25 Multiple myeloma with secondary AL amyloidosis has been seen with amyloid purpura and atraumatic ecchymoses of the face, highlighting the hemorrhage noted in these patients.26
Management of Amyloidosis—Various treatment options have been attempted for primary cutaneous amyloidosis, including oral retinoids, corticosteroids, cyclophosphamide, cyclosporine, amitriptyline, colchicine, cepharanthin, tacrolimus, dimethyl sulfoxide, vitamin D3 analogs, capsaicin, menthol, hydrocolloid dressings, surgical modalities, laser treatment, and phototherapy.1 There is no clear consensus for therapeutic modalities except for treating the underlying plasma cell dyscrasia in primary systemic amyloidosis.
Conclusion
We report the case of a patient displaying signs of pseudoporphyria that ultimately proved to be bullous amyloidosis, or what we termed pseudopseudoporphyria. Bullous amyloidosis should be considered in the differential diagnoses of hemorrhagic bullous skin eruptions. Particular attention should be given to a systemic workup for multiple myeloma when hemorrhagic vesicles/bullae are chronic and coexist with purpura, angina bullosa hemorrhagica, fatigue/weight loss, and/or macroglossia.
- Weidner T, Illing T, Elsner P. Primary localized cutaneous amyloidosis: a systematic treatment review. Am J Clin Dermatol. 2017;18:629-642.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Amyloidosis. Dermatology Essentials. Elsevier Saunders; 2014:341-345.
- Bhutani M, Shahid Z, Schnebelen A, et al. Cutaneous manifestations of multiple myeloma and other plasma cell proliferative disorders. Semin Oncol. 2016;43:395-400.
- Terushkin V, Boyd KP, Patel RR, et al. Primary localized cutaneous amyloidosis. Dermatol Online J. 2013;19:20711.
- LaChance A, Phelps A, Finch J, et al. Nodular localized primary cutaneous amyloidosis: a bullous variant. Clin Exp Dermatol. 2014;39:344-347.
- Gonzalez-Ramos J, Garrido-Gutiérrez C, González-Silva Y, et al. Relapsing bullous amyloidosis of the oral mucosa and acquired cutis laxa in a patient with multiple myeloma: a rare triple association. Clin Exp Dermatol. 2017;42:410-412.
- Kanoh T. Bullous amyloidosis [in Japanese]. Rinsho Ketsueki. 1993;34:1050-1052.
- Johnson TM, Rapini RP, Hebert AA, et al. Bullous amyloidosis. Cutis. 1989;43:346-352.
- Houman MH, Smiti KM, Ben Ghorbel I, et al. Bullous amyloidosis. Ann Dermatol Venereol. 2002;129:299-302.
- Sanusi T, Li Y, Qian Y, et al. Primary localized cutaneous nodular amyloidosis with bullous lesions. Indian J Dermatol Venereol Leprol. 2015;81:400-402.
- Ochiai T, Morishima T, Hao T, et al. Bullous amyloidosis: the mechanism of blister formation revealed by electron microscopy. J Cutan Pathol. 2001;28:407-411.
- Chu CH, Chan JY, Hsieh SW, et al. Diffuse ecchymoses and blisters on a yellowish waxy base: a case of bullous amyloidosis. J Dermatol. 2016;43:713-714.
- Wang XD, Shen H, Liu ZH. Diffuse haemorrhagic bullous amyloidosis with multiple myeloma. Clin Exp Dermatol. 2008;33:94-96.
- Biswas P, Aggarwal I, Sen D, et al. Bullous pemphigoid clinically presenting as lichen amyloidosis. Indian J Dermatol Venereol Leprol. 2014;80:544-546.
- Bluhm JF 3rd. Bullous dermatosis vs amyloidosis. Arch Dermatol. 1981;117:252.
- Bluhm JF 3rd. Bullous amyloidosis vs epidermolysis bullosa acquisita. JAMA. 1981;245:32.
- Murphy GM, Wright J, Nicholls DS, et al. Sunbed-induced pseudoporphyria. Br J Dermatol. 1989;120:555-562.
- Pramatarov K, Lazarova A, Mateev G, et al. Bullous hemorrhagic primary systemic amyloidosis. Int J Dermatol. 1990;29:211-213.
- Bieber T, Ruzicka T, Linke RP, et al. Hemorrhagic bullous amyloidosis. a histologic, immunocytochemical, and ultrastructural study of two patients. Arch Dermatol. 1988;124:1683-1686.
- Khoo BP, Tay YK. Lichen amyloidosis: a bullous variant. Ann Acad Med Singapore. 2000;29:105-107.
- Asahina A, Hasegawa K, Ishiyama M, et al. Bullous amyloidosis mimicking bullous pemphigoid: usefulness of electron microscopic examination. Acta Derm Venereol. 2010;90:427-428.
- Schmutz JL, Barbaud A, Cuny JF, et al. Bullous amyloidosis [in French]. Ann Dermatol Venereol. 1988;115:295-301.
- Lachmann HJ, Hawkins PN. Amyloidosis of the skin. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. McGraw-Hill; 2012:1574-1583.
- Grundmann JU, Bonnekoh B, Gollnick H. Extensive haemorrhagic-bullous skin manifestation of systemic AA-amyloidosis associated with IgG lambda-myeloma. Eur J Dermatol. 2000;10:139-142.
- Hödl S, Turek TD, Kerl H. Plasmocytoma-associated bullous hemorrhagic amyloidosis of the skin [in German]. Hautarzt. 1982;33:556-558.
- Colucci G, Alberio L, Demarmels Biasiutti F, et al. Bilateral periorbital ecchymoses. an often missed sign of amyloid purpura. Hamostaseologie. 2014;34:249-252.
- Behera B, Pattnaik M, Sahu B, et al. Cutaneous manifestations of multiple myeloma. Indian J Dermatol. 2016;61:668-671.
- Fujita Y, Tsuji-Abe Y, Sato-Matsumura KC, et al. Nail dystrophy and blisters as sole manifestations in myeloma-associated amyloidosis. J Am Acad Dermatol. 2006;54:712-714.
- Chang SL, Lai PC, Cheng CJ, et al. Bullous amyloidosis in a hemodialysis patient is myeloma-associated rather than hemodialysis-associated amyloidosis. Amyloid. 2007;14:153-156.
- Winzer M, Ruppert M, Baretton G, et al. Bullous poikilodermatitic amyloidosis of the skin with junctional bulla development in IgG light chain plasmacytoma of the lambda type. histology, immunohistology and electron microscopy [in German]. Hautarzt. 1992;43:199-204.
- Weidner T, Illing T, Elsner P. Primary localized cutaneous amyloidosis: a systematic treatment review. Am J Clin Dermatol. 2017;18:629-642.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Amyloidosis. Dermatology Essentials. Elsevier Saunders; 2014:341-345.
- Bhutani M, Shahid Z, Schnebelen A, et al. Cutaneous manifestations of multiple myeloma and other plasma cell proliferative disorders. Semin Oncol. 2016;43:395-400.
- Terushkin V, Boyd KP, Patel RR, et al. Primary localized cutaneous amyloidosis. Dermatol Online J. 2013;19:20711.
- LaChance A, Phelps A, Finch J, et al. Nodular localized primary cutaneous amyloidosis: a bullous variant. Clin Exp Dermatol. 2014;39:344-347.
- Gonzalez-Ramos J, Garrido-Gutiérrez C, González-Silva Y, et al. Relapsing bullous amyloidosis of the oral mucosa and acquired cutis laxa in a patient with multiple myeloma: a rare triple association. Clin Exp Dermatol. 2017;42:410-412.
- Kanoh T. Bullous amyloidosis [in Japanese]. Rinsho Ketsueki. 1993;34:1050-1052.
- Johnson TM, Rapini RP, Hebert AA, et al. Bullous amyloidosis. Cutis. 1989;43:346-352.
- Houman MH, Smiti KM, Ben Ghorbel I, et al. Bullous amyloidosis. Ann Dermatol Venereol. 2002;129:299-302.
- Sanusi T, Li Y, Qian Y, et al. Primary localized cutaneous nodular amyloidosis with bullous lesions. Indian J Dermatol Venereol Leprol. 2015;81:400-402.
- Ochiai T, Morishima T, Hao T, et al. Bullous amyloidosis: the mechanism of blister formation revealed by electron microscopy. J Cutan Pathol. 2001;28:407-411.
- Chu CH, Chan JY, Hsieh SW, et al. Diffuse ecchymoses and blisters on a yellowish waxy base: a case of bullous amyloidosis. J Dermatol. 2016;43:713-714.
- Wang XD, Shen H, Liu ZH. Diffuse haemorrhagic bullous amyloidosis with multiple myeloma. Clin Exp Dermatol. 2008;33:94-96.
- Biswas P, Aggarwal I, Sen D, et al. Bullous pemphigoid clinically presenting as lichen amyloidosis. Indian J Dermatol Venereol Leprol. 2014;80:544-546.
- Bluhm JF 3rd. Bullous dermatosis vs amyloidosis. Arch Dermatol. 1981;117:252.
- Bluhm JF 3rd. Bullous amyloidosis vs epidermolysis bullosa acquisita. JAMA. 1981;245:32.
- Murphy GM, Wright J, Nicholls DS, et al. Sunbed-induced pseudoporphyria. Br J Dermatol. 1989;120:555-562.
- Pramatarov K, Lazarova A, Mateev G, et al. Bullous hemorrhagic primary systemic amyloidosis. Int J Dermatol. 1990;29:211-213.
- Bieber T, Ruzicka T, Linke RP, et al. Hemorrhagic bullous amyloidosis. a histologic, immunocytochemical, and ultrastructural study of two patients. Arch Dermatol. 1988;124:1683-1686.
- Khoo BP, Tay YK. Lichen amyloidosis: a bullous variant. Ann Acad Med Singapore. 2000;29:105-107.
- Asahina A, Hasegawa K, Ishiyama M, et al. Bullous amyloidosis mimicking bullous pemphigoid: usefulness of electron microscopic examination. Acta Derm Venereol. 2010;90:427-428.
- Schmutz JL, Barbaud A, Cuny JF, et al. Bullous amyloidosis [in French]. Ann Dermatol Venereol. 1988;115:295-301.
- Lachmann HJ, Hawkins PN. Amyloidosis of the skin. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. McGraw-Hill; 2012:1574-1583.
- Grundmann JU, Bonnekoh B, Gollnick H. Extensive haemorrhagic-bullous skin manifestation of systemic AA-amyloidosis associated with IgG lambda-myeloma. Eur J Dermatol. 2000;10:139-142.
- Hödl S, Turek TD, Kerl H. Plasmocytoma-associated bullous hemorrhagic amyloidosis of the skin [in German]. Hautarzt. 1982;33:556-558.
- Colucci G, Alberio L, Demarmels Biasiutti F, et al. Bilateral periorbital ecchymoses. an often missed sign of amyloid purpura. Hamostaseologie. 2014;34:249-252.
- Behera B, Pattnaik M, Sahu B, et al. Cutaneous manifestations of multiple myeloma. Indian J Dermatol. 2016;61:668-671.
- Fujita Y, Tsuji-Abe Y, Sato-Matsumura KC, et al. Nail dystrophy and blisters as sole manifestations in myeloma-associated amyloidosis. J Am Acad Dermatol. 2006;54:712-714.
- Chang SL, Lai PC, Cheng CJ, et al. Bullous amyloidosis in a hemodialysis patient is myeloma-associated rather than hemodialysis-associated amyloidosis. Amyloid. 2007;14:153-156.
- Winzer M, Ruppert M, Baretton G, et al. Bullous poikilodermatitic amyloidosis of the skin with junctional bulla development in IgG light chain plasmacytoma of the lambda type. histology, immunohistology and electron microscopy [in German]. Hautarzt. 1992;43:199-204.
Practice Points
- Primary systemic amyloidosis, including the rare cutaneous bullous amyloidosis, often is difficult to diagnose and has been associated with underlying plasma cell dyscrasia or multiple myeloma.
- When evaluating patients with initially convincing signs of pseudoporphyria, it is imperative to consider the diagnosis of bullous amyloidosis, which additionally can present with intraoral hemorrhagic vesicles and have confirmatory histopathologic features.
- Further investigation for multiple myeloma is warranted when patients with a chronic hemorrhagic bullous condition also present with symptoms of purpura, angina bullosa hemorrhagica, fatigue, weight loss, and/or macroglossia. Accurate diagnosis of bullous amyloidosis and timely treatment of its underlying cause will contribute to better, more proactive patient care.
75-year-old man • fatigue • unintentional weight loss • anemia • Dx?
THE CASE
A 75-year-old man with a history of osteoarthritis presented to our clinic with worsening weakness over the previous month. His signs and symptoms included profound fatigue, subjective fevers, a 10-pound weight loss, ankle swelling, myalgias in his legs and back, shortness of breath, and a persistent cough. The patient was otherwise previously healthy.
The patient’s heart and lung exams were normal. Initial outpatient labs showed significantly elevated inflammatory markers, with an erythrocyte sedimentation rate (ESR) of 102 mm/h (normal range for men ≥ 50 years, 0-20 mm/h) and a C-reactive protein (CRP) level of 11.1 mg/L (normal range, < 3 mg/L). The patient also had an elevated white blood cell count of 12,000/mcL (normal range, 4500-11,000/mcL). His hemoglobin was low (11 g/dL; normal range, 13.5-17.5 g/dL) and so was his albumin level (2.9 g/dL; normal range, 3.4-5.4 g/dL). The results of his prostate-specific antigen and brain natriuretic peptide tests were both normal. The results of a computed tomography scan of his thorax, abdomen, and pelvis were negative for malignancy.
The patient returned to our clinic 3 days later with severe weakness, which inhibited him from walking. He complained of a severe spasmodic pain between his shoulder blades. He denied joint stiffness, headaches, vision changes, or jaw claudication. The patient’s son had noted an overall increase in his father’s baseline heart rate, with readings increasing from the 50 beats/min range to the 70 beats/min range; this raised concern for a catecholamine-secreting tumor. There was also concern for occult infection and malignancy, or an autoimmune process, such as polymyalgia rheumatica. Due to his extreme weakness, the patient was directly admitted to the hospital for further work-up.
THE DIAGNOSIS
Concern for a smoldering infection prompted an order for a transthoracic echocardiogram. Images revealed a large mass on the mitral valve (FIGURE 1). Blood cultures quickly grew Streptococcus sanguinis. Additional work-up with a transesophageal echocardiogram (TEE) showed a “windsock” deformity (thinning and ballooning of the mitral valve), a known sequela of infective endocarditis (FIGURE 2).1 Further history obtained after the TEE revealed the patient had had a routine dental cleaning the month before his symptoms began. A murmur was then also detected.

DISCUSSION
Infective endocarditis (IE) is uncommon and difficult to diagnose; it has a high early-mortality rate of 30%.2 TEE is the recommended imaging study for IE, because it is more sensitive than a transthoracic echocardiogram for identifying vegetations on the valves and it is more cost effective.3

The modified Duke Criteria provide guidance for diagnosis of endocarditis. Major criteria focus on positive blood cultures and evidence of endocardial involvement. Minor criteria include predisposing heart conditions, intravenous drug use (IVDU), fever, and vascular and immunologic phenomena. As many as 90% of patients have a fever and often experience weight loss.4 Murmurs are auscultated in up to 85% of patients, and embolic features are present in up to 25% of patients at the time of diagnosis.4 In the developed world, Janeway lesions, Osler nodes, and splinter hemorrhages are increasingly rare, as patients usually present earlier in the disease course.4 While ESR and CRP are generally elevated in cases of IE, they are not part of the Duke Criteria.4
A closer look at risk factors
In 2007, guidelines for the prevention, treatment, and management of endocarditis were given significant categorical revision by the American Heart Association for the first time in 50 years.5 Recommendations for antibiotic prophylaxis prior to dental procedures became more restrictive, to include only 4 groups of high-risk patients: those with prosthetic cardiac valves, those with a history of IE, those with congenital heart disease, and cardiac transplant recipients.4 The rationale for these restrictions included the small risk for anaphylaxis and potential increase in risk for bacterial resistance associated with antibiotic prophylaxis.4 A review published in 2021 noted no increase in the frequency of, nor the morbidity and mortality from, viridans group streptococcal IE since the guideline updates.5
Continue to: There is an emerging consensus...
There is an emerging consensus that poor oral hygiene and gingival bleeding after tooth brushing promote a chronic low-grade bacteremia that may be more strongly associated with IE than an isolated dental extraction.6 Poor dental hygiene, defined as dental plaque and calculus, is especially common in the elderly, who are known to let their dental hygiene lapse.6 In our patient’s case, his generally poor oral hygiene was more likely the cause of his IE than his routine dental cleaning.
Other risk factors include IV drug use. At our tertiary care hospital in western North Carolina, 48% of patients with endocarditis had an additional diagnosis of opiate or narcotic dependence (Ryan Tilton, PharmD, email communication, June 7, 2018). Interestingly, though, only 16% of patients in North America with endocarditis were found to be currently using IV drugs.7
Our patient was treated with IV antibiotics for 4 weeks and underwent rehabilitation at a skilled nursing facility. Four weeks after diagnosis, he underwent an endoscopic porcine mitral valve replacement. Two months after that, he returned to his previously active lifestyle and began riding his stationary bike. The patient also began taking a daily aspirin. Consistent with current guidelines, he now gets antibiotic prophylaxis prior to dental procedures.
THE TAKEAWAY
This patient, without any history of IVDU or cardiac valvular abnormalities, presented with symptoms classic for a developing malignancy or possible rheumatologic condition. Subacute IE may manifest similarly, with vague symptoms such as myalgias, fatigue, chills, and/or anemia. In non-drug users, suspicion for endocarditis should be highest in men older than age 60. Also, it’s important to auscultate for a new heart murmur. In our patient’s case, no murmur was auscultated until after his TEE. JFP
CORRESPONDENCE
Ginger Poulton, MD, 123 Hendersonville Road, Asheville, NC 28803; [email protected]
1. Paruchuru PK, Adluri K, Patel RL. Windsock deformity of the mitral valve—a late presentation of endocarditis. Eur J Cardiothorac Surg. 2002;21:88. doi: 10.1016/s1010-7940(01)01038-7
2. Toyoda N, Chikwe J, Itagaki S, et al. Trends in infective endocarditis in California and New York State, 1998-2013. JAMA. 2017;317:1652-1660. doi: 10.1001/jama.2017.4287
3. Baddour LM, Wilson WR, Bayer AS, et al. Infective endocarditis in adults: diagnosis, antimicrobial therapy, and management of complications: a scientific statement for healthcare professionals from the American Heart Association. Circulation. 2015;132:1435-1486. doi: 10.1161/CIR.0000000000000296
4. Habib G, Lancellotti P, Antunes MJ, et al. 2015 ESC Guidelines for the management of infective endocarditis: The Task Force for the Management of Infective Endocarditis of the European Society of Cardiology (ESC). Endorsed by: European Association for Cardio-Thoracic Surgery (EACTS), the European Association of Nuclear Medicine (EANM). Eur Heart J. 2015;36:3075-3128. doi: 10.1093/eurheartj/ehv319
5. Wilson, WR, Gewitz, M, Lockhart PB et al. Prevention of Viridans Group Streptococcal Infective Endocarditis. A Scientific Statement from the American Heart Association. Circulation. 2021; 143e963-e978.
6. Lockhart PB, Brennan MT, Thornhill M, et al. Poor oral hygiene as a risk factor for infective endocarditis-related bacteremia. J Am Dent Assoc. 2009;140:1238-1244. doi: 10.14219/jada.archive.2009.0046
7. Murdoch DR, Corey GR, Hoen B, et al. Clinical presentation, etiology, and outcome of infective endocarditis in the 21st century: the International Collaboration on Endocarditis-Prospective Cohort Study. Arch Intern Med. 2009;169:463-473. doi: 10.1001/archinternmed.2008.603
THE CASE
A 75-year-old man with a history of osteoarthritis presented to our clinic with worsening weakness over the previous month. His signs and symptoms included profound fatigue, subjective fevers, a 10-pound weight loss, ankle swelling, myalgias in his legs and back, shortness of breath, and a persistent cough. The patient was otherwise previously healthy.
The patient’s heart and lung exams were normal. Initial outpatient labs showed significantly elevated inflammatory markers, with an erythrocyte sedimentation rate (ESR) of 102 mm/h (normal range for men ≥ 50 years, 0-20 mm/h) and a C-reactive protein (CRP) level of 11.1 mg/L (normal range, < 3 mg/L). The patient also had an elevated white blood cell count of 12,000/mcL (normal range, 4500-11,000/mcL). His hemoglobin was low (11 g/dL; normal range, 13.5-17.5 g/dL) and so was his albumin level (2.9 g/dL; normal range, 3.4-5.4 g/dL). The results of his prostate-specific antigen and brain natriuretic peptide tests were both normal. The results of a computed tomography scan of his thorax, abdomen, and pelvis were negative for malignancy.
The patient returned to our clinic 3 days later with severe weakness, which inhibited him from walking. He complained of a severe spasmodic pain between his shoulder blades. He denied joint stiffness, headaches, vision changes, or jaw claudication. The patient’s son had noted an overall increase in his father’s baseline heart rate, with readings increasing from the 50 beats/min range to the 70 beats/min range; this raised concern for a catecholamine-secreting tumor. There was also concern for occult infection and malignancy, or an autoimmune process, such as polymyalgia rheumatica. Due to his extreme weakness, the patient was directly admitted to the hospital for further work-up.
THE DIAGNOSIS
Concern for a smoldering infection prompted an order for a transthoracic echocardiogram. Images revealed a large mass on the mitral valve (FIGURE 1). Blood cultures quickly grew Streptococcus sanguinis. Additional work-up with a transesophageal echocardiogram (TEE) showed a “windsock” deformity (thinning and ballooning of the mitral valve), a known sequela of infective endocarditis (FIGURE 2).1 Further history obtained after the TEE revealed the patient had had a routine dental cleaning the month before his symptoms began. A murmur was then also detected.
DISCUSSION
Infective endocarditis (IE) is uncommon and difficult to diagnose; it has a high early-mortality rate of 30%.2 TEE is the recommended imaging study for IE, because it is more sensitive than a transthoracic echocardiogram for identifying vegetations on the valves and it is more cost effective.3
The modified Duke Criteria provide guidance for diagnosis of endocarditis. Major criteria focus on positive blood cultures and evidence of endocardial involvement. Minor criteria include predisposing heart conditions, intravenous drug use (IVDU), fever, and vascular and immunologic phenomena. As many as 90% of patients have a fever and often experience weight loss.4 Murmurs are auscultated in up to 85% of patients, and embolic features are present in up to 25% of patients at the time of diagnosis.4 In the developed world, Janeway lesions, Osler nodes, and splinter hemorrhages are increasingly rare, as patients usually present earlier in the disease course.4 While ESR and CRP are generally elevated in cases of IE, they are not part of the Duke Criteria.4
A closer look at risk factors
In 2007, guidelines for the prevention, treatment, and management of endocarditis were given significant categorical revision by the American Heart Association for the first time in 50 years.5 Recommendations for antibiotic prophylaxis prior to dental procedures became more restrictive, to include only 4 groups of high-risk patients: those with prosthetic cardiac valves, those with a history of IE, those with congenital heart disease, and cardiac transplant recipients.4 The rationale for these restrictions included the small risk for anaphylaxis and potential increase in risk for bacterial resistance associated with antibiotic prophylaxis.4 A review published in 2021 noted no increase in the frequency of, nor the morbidity and mortality from, viridans group streptococcal IE since the guideline updates.5
Continue to: There is an emerging consensus...
There is an emerging consensus that poor oral hygiene and gingival bleeding after tooth brushing promote a chronic low-grade bacteremia that may be more strongly associated with IE than an isolated dental extraction.6 Poor dental hygiene, defined as dental plaque and calculus, is especially common in the elderly, who are known to let their dental hygiene lapse.6 In our patient’s case, his generally poor oral hygiene was more likely the cause of his IE than his routine dental cleaning.
Other risk factors include IV drug use. At our tertiary care hospital in western North Carolina, 48% of patients with endocarditis had an additional diagnosis of opiate or narcotic dependence (Ryan Tilton, PharmD, email communication, June 7, 2018). Interestingly, though, only 16% of patients in North America with endocarditis were found to be currently using IV drugs.7
Our patient was treated with IV antibiotics for 4 weeks and underwent rehabilitation at a skilled nursing facility. Four weeks after diagnosis, he underwent an endoscopic porcine mitral valve replacement. Two months after that, he returned to his previously active lifestyle and began riding his stationary bike. The patient also began taking a daily aspirin. Consistent with current guidelines, he now gets antibiotic prophylaxis prior to dental procedures.
THE TAKEAWAY
This patient, without any history of IVDU or cardiac valvular abnormalities, presented with symptoms classic for a developing malignancy or possible rheumatologic condition. Subacute IE may manifest similarly, with vague symptoms such as myalgias, fatigue, chills, and/or anemia. In non-drug users, suspicion for endocarditis should be highest in men older than age 60. Also, it’s important to auscultate for a new heart murmur. In our patient’s case, no murmur was auscultated until after his TEE. JFP
CORRESPONDENCE
Ginger Poulton, MD, 123 Hendersonville Road, Asheville, NC 28803; [email protected]
THE CASE
A 75-year-old man with a history of osteoarthritis presented to our clinic with worsening weakness over the previous month. His signs and symptoms included profound fatigue, subjective fevers, a 10-pound weight loss, ankle swelling, myalgias in his legs and back, shortness of breath, and a persistent cough. The patient was otherwise previously healthy.
The patient’s heart and lung exams were normal. Initial outpatient labs showed significantly elevated inflammatory markers, with an erythrocyte sedimentation rate (ESR) of 102 mm/h (normal range for men ≥ 50 years, 0-20 mm/h) and a C-reactive protein (CRP) level of 11.1 mg/L (normal range, < 3 mg/L). The patient also had an elevated white blood cell count of 12,000/mcL (normal range, 4500-11,000/mcL). His hemoglobin was low (11 g/dL; normal range, 13.5-17.5 g/dL) and so was his albumin level (2.9 g/dL; normal range, 3.4-5.4 g/dL). The results of his prostate-specific antigen and brain natriuretic peptide tests were both normal. The results of a computed tomography scan of his thorax, abdomen, and pelvis were negative for malignancy.
The patient returned to our clinic 3 days later with severe weakness, which inhibited him from walking. He complained of a severe spasmodic pain between his shoulder blades. He denied joint stiffness, headaches, vision changes, or jaw claudication. The patient’s son had noted an overall increase in his father’s baseline heart rate, with readings increasing from the 50 beats/min range to the 70 beats/min range; this raised concern for a catecholamine-secreting tumor. There was also concern for occult infection and malignancy, or an autoimmune process, such as polymyalgia rheumatica. Due to his extreme weakness, the patient was directly admitted to the hospital for further work-up.
THE DIAGNOSIS
Concern for a smoldering infection prompted an order for a transthoracic echocardiogram. Images revealed a large mass on the mitral valve (FIGURE 1). Blood cultures quickly grew Streptococcus sanguinis. Additional work-up with a transesophageal echocardiogram (TEE) showed a “windsock” deformity (thinning and ballooning of the mitral valve), a known sequela of infective endocarditis (FIGURE 2).1 Further history obtained after the TEE revealed the patient had had a routine dental cleaning the month before his symptoms began. A murmur was then also detected.
DISCUSSION
Infective endocarditis (IE) is uncommon and difficult to diagnose; it has a high early-mortality rate of 30%.2 TEE is the recommended imaging study for IE, because it is more sensitive than a transthoracic echocardiogram for identifying vegetations on the valves and it is more cost effective.3
The modified Duke Criteria provide guidance for diagnosis of endocarditis. Major criteria focus on positive blood cultures and evidence of endocardial involvement. Minor criteria include predisposing heart conditions, intravenous drug use (IVDU), fever, and vascular and immunologic phenomena. As many as 90% of patients have a fever and often experience weight loss.4 Murmurs are auscultated in up to 85% of patients, and embolic features are present in up to 25% of patients at the time of diagnosis.4 In the developed world, Janeway lesions, Osler nodes, and splinter hemorrhages are increasingly rare, as patients usually present earlier in the disease course.4 While ESR and CRP are generally elevated in cases of IE, they are not part of the Duke Criteria.4
A closer look at risk factors
In 2007, guidelines for the prevention, treatment, and management of endocarditis were given significant categorical revision by the American Heart Association for the first time in 50 years.5 Recommendations for antibiotic prophylaxis prior to dental procedures became more restrictive, to include only 4 groups of high-risk patients: those with prosthetic cardiac valves, those with a history of IE, those with congenital heart disease, and cardiac transplant recipients.4 The rationale for these restrictions included the small risk for anaphylaxis and potential increase in risk for bacterial resistance associated with antibiotic prophylaxis.4 A review published in 2021 noted no increase in the frequency of, nor the morbidity and mortality from, viridans group streptococcal IE since the guideline updates.5
Continue to: There is an emerging consensus...
There is an emerging consensus that poor oral hygiene and gingival bleeding after tooth brushing promote a chronic low-grade bacteremia that may be more strongly associated with IE than an isolated dental extraction.6 Poor dental hygiene, defined as dental plaque and calculus, is especially common in the elderly, who are known to let their dental hygiene lapse.6 In our patient’s case, his generally poor oral hygiene was more likely the cause of his IE than his routine dental cleaning.
Other risk factors include IV drug use. At our tertiary care hospital in western North Carolina, 48% of patients with endocarditis had an additional diagnosis of opiate or narcotic dependence (Ryan Tilton, PharmD, email communication, June 7, 2018). Interestingly, though, only 16% of patients in North America with endocarditis were found to be currently using IV drugs.7
Our patient was treated with IV antibiotics for 4 weeks and underwent rehabilitation at a skilled nursing facility. Four weeks after diagnosis, he underwent an endoscopic porcine mitral valve replacement. Two months after that, he returned to his previously active lifestyle and began riding his stationary bike. The patient also began taking a daily aspirin. Consistent with current guidelines, he now gets antibiotic prophylaxis prior to dental procedures.
THE TAKEAWAY
This patient, without any history of IVDU or cardiac valvular abnormalities, presented with symptoms classic for a developing malignancy or possible rheumatologic condition. Subacute IE may manifest similarly, with vague symptoms such as myalgias, fatigue, chills, and/or anemia. In non-drug users, suspicion for endocarditis should be highest in men older than age 60. Also, it’s important to auscultate for a new heart murmur. In our patient’s case, no murmur was auscultated until after his TEE. JFP
CORRESPONDENCE
Ginger Poulton, MD, 123 Hendersonville Road, Asheville, NC 28803; [email protected]
1. Paruchuru PK, Adluri K, Patel RL. Windsock deformity of the mitral valve—a late presentation of endocarditis. Eur J Cardiothorac Surg. 2002;21:88. doi: 10.1016/s1010-7940(01)01038-7
2. Toyoda N, Chikwe J, Itagaki S, et al. Trends in infective endocarditis in California and New York State, 1998-2013. JAMA. 2017;317:1652-1660. doi: 10.1001/jama.2017.4287
3. Baddour LM, Wilson WR, Bayer AS, et al. Infective endocarditis in adults: diagnosis, antimicrobial therapy, and management of complications: a scientific statement for healthcare professionals from the American Heart Association. Circulation. 2015;132:1435-1486. doi: 10.1161/CIR.0000000000000296
4. Habib G, Lancellotti P, Antunes MJ, et al. 2015 ESC Guidelines for the management of infective endocarditis: The Task Force for the Management of Infective Endocarditis of the European Society of Cardiology (ESC). Endorsed by: European Association for Cardio-Thoracic Surgery (EACTS), the European Association of Nuclear Medicine (EANM). Eur Heart J. 2015;36:3075-3128. doi: 10.1093/eurheartj/ehv319
5. Wilson, WR, Gewitz, M, Lockhart PB et al. Prevention of Viridans Group Streptococcal Infective Endocarditis. A Scientific Statement from the American Heart Association. Circulation. 2021; 143e963-e978.
6. Lockhart PB, Brennan MT, Thornhill M, et al. Poor oral hygiene as a risk factor for infective endocarditis-related bacteremia. J Am Dent Assoc. 2009;140:1238-1244. doi: 10.14219/jada.archive.2009.0046
7. Murdoch DR, Corey GR, Hoen B, et al. Clinical presentation, etiology, and outcome of infective endocarditis in the 21st century: the International Collaboration on Endocarditis-Prospective Cohort Study. Arch Intern Med. 2009;169:463-473. doi: 10.1001/archinternmed.2008.603
1. Paruchuru PK, Adluri K, Patel RL. Windsock deformity of the mitral valve—a late presentation of endocarditis. Eur J Cardiothorac Surg. 2002;21:88. doi: 10.1016/s1010-7940(01)01038-7
2. Toyoda N, Chikwe J, Itagaki S, et al. Trends in infective endocarditis in California and New York State, 1998-2013. JAMA. 2017;317:1652-1660. doi: 10.1001/jama.2017.4287
3. Baddour LM, Wilson WR, Bayer AS, et al. Infective endocarditis in adults: diagnosis, antimicrobial therapy, and management of complications: a scientific statement for healthcare professionals from the American Heart Association. Circulation. 2015;132:1435-1486. doi: 10.1161/CIR.0000000000000296
4. Habib G, Lancellotti P, Antunes MJ, et al. 2015 ESC Guidelines for the management of infective endocarditis: The Task Force for the Management of Infective Endocarditis of the European Society of Cardiology (ESC). Endorsed by: European Association for Cardio-Thoracic Surgery (EACTS), the European Association of Nuclear Medicine (EANM). Eur Heart J. 2015;36:3075-3128. doi: 10.1093/eurheartj/ehv319
5. Wilson, WR, Gewitz, M, Lockhart PB et al. Prevention of Viridans Group Streptococcal Infective Endocarditis. A Scientific Statement from the American Heart Association. Circulation. 2021; 143e963-e978.
6. Lockhart PB, Brennan MT, Thornhill M, et al. Poor oral hygiene as a risk factor for infective endocarditis-related bacteremia. J Am Dent Assoc. 2009;140:1238-1244. doi: 10.14219/jada.archive.2009.0046
7. Murdoch DR, Corey GR, Hoen B, et al. Clinical presentation, etiology, and outcome of infective endocarditis in the 21st century: the International Collaboration on Endocarditis-Prospective Cohort Study. Arch Intern Med. 2009;169:463-473. doi: 10.1001/archinternmed.2008.603

78-year-old man • tail bone pain • unintended weight loss • history of diabetes and hypertension • Dx?
THE CASE
A 78-year-old man with a history of diabetes and hypertension was referred to the outpatient surgical office with a chief complaint of “tail bone pain” that had started after a fall a year earlier. The patient complained that the pain was worse when sitting and at nighttime. He also admitted to a 7-lb weight loss over the past 2 months without change in diet or appetite. He denied symptoms of incontinence, urinary retention, sharp stabbing pains in the lower extremities, night sweats, or anorexia.
The patient first visited an urgent care facility on the day after the fall because he was experiencing pain in his “tail bone” region while riding his lawn mower. A pelvic x-ray was performed at that time and showed no coccyx fracture. He received a steroid injection in the right sacroiliac joint, which provided some relief for a month. Throughout the course of the year, he was given 6 steroid injections into his sacroiliac joint by his primary care provider (PCP) and clinicians at his local urgent care facility. One year after the fall, the patient’s PCP ordered a computed tomography (CT) scan of the abdomen and pelvis, which revealed a 4.6 x 7.5–cm soft-tissue mass with bony destruction of the lower sacrum and coccyx that extended into the sacral and coccygeal canal (FIGURE 1).
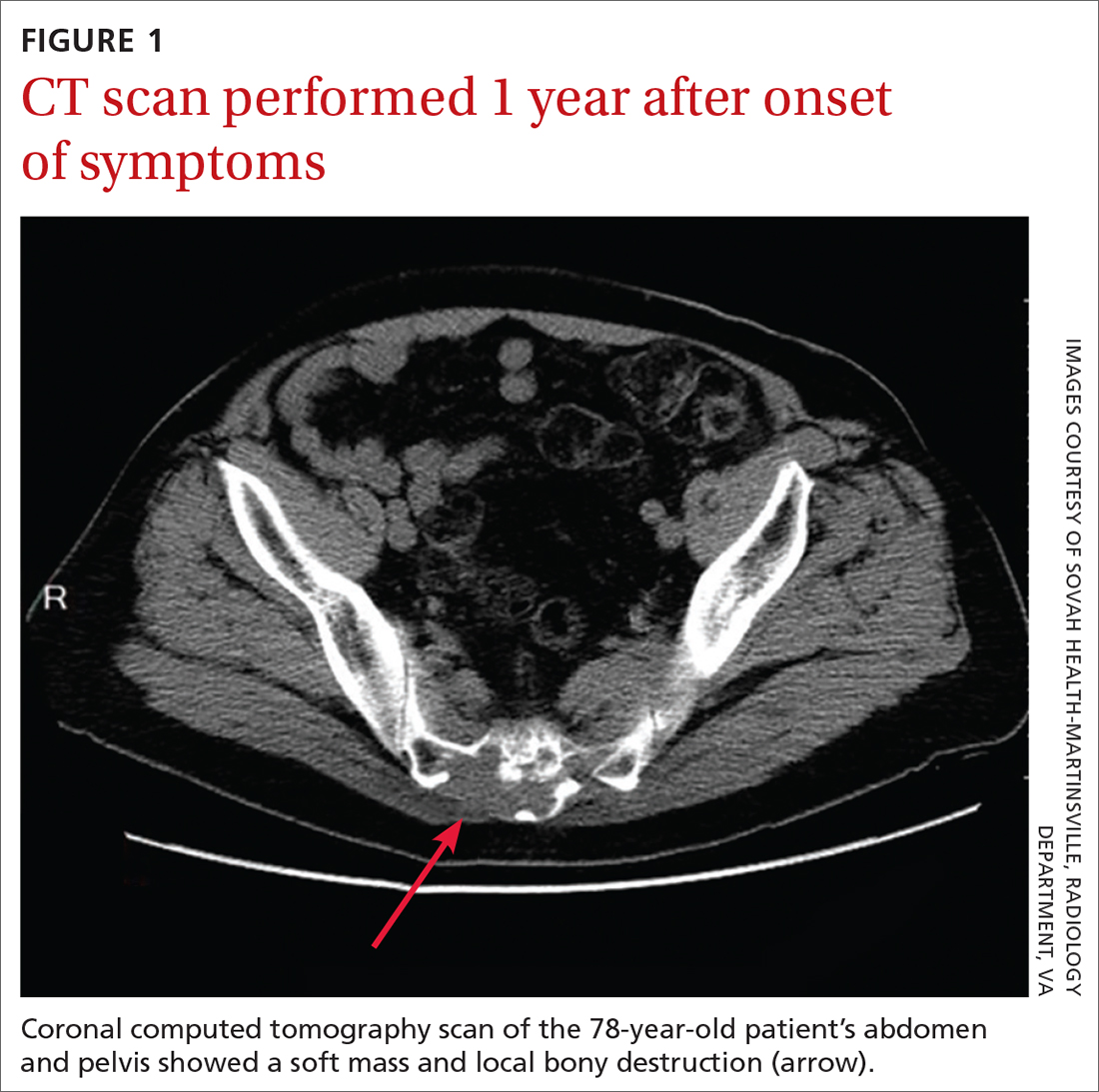
On exam in our surgical office, the patient was found to be alert and oriented. His neurologic exam was unremarkable, with an intact motor and sensory exam and no symptoms of cauda equina syndrome. During palpation over the lower sacrum and coccyx, both tenderness and a boggy, soft mass were observed. Nerve impingement was most likely caused by the size of the mass.
THE DIAGNOSIS
Biopsy revealed a large tan-gray, gelatinous, soft-tissue mass that was necrotizing through the lower sacrum. The diagnosis of a sacral chordoma was confirmed with magnetic resonance imaging of the pelvis, which demonstrated a 4.6 × 8.1–cm destructive expansile sacrococcygeal tumor with an exophytic soft-tissue component (FIGURE 2). The tumor also involved the piriformis and gluteus maximus muscles bilaterally.
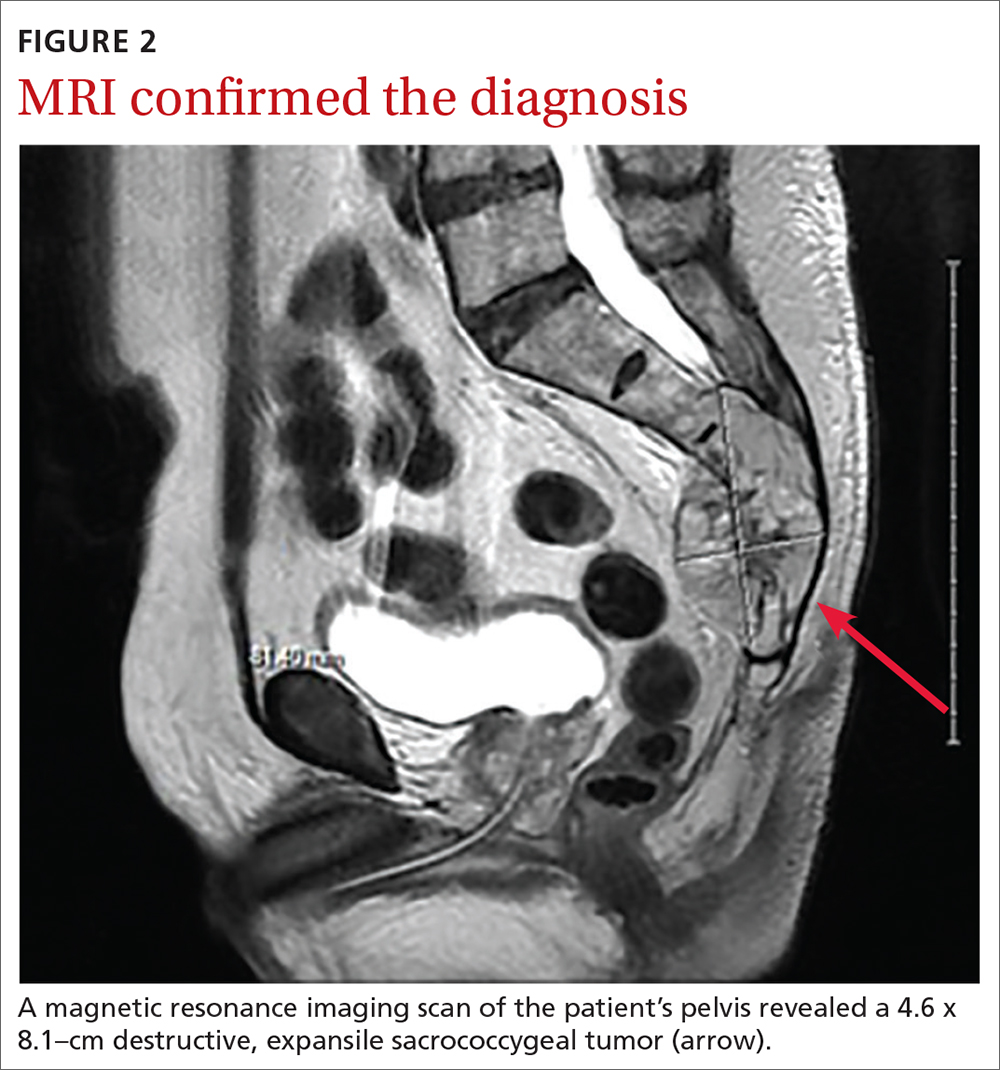
DISCUSSION
Chordomas are rare, malignant bone tumors that grow slowly and originate from embryonic remnants of the notochord.1 They are most commonly seen in the sacrococcygeal segment (50%) but are also seen in the spheno-occipital synchondrosis (30%-35%) and other spinal segments such as C2 and lumbar spine.2 Chordomas are typically seen in middle-aged patients, with sacral chordomas occurring predominantly in men compared to women (3:1).2
Slow to grow, slow to diagnose
The difficulty with diagnosing sacral chordomas lies in the tendency for these tumors to grow extremely slowly, making detection challenging due to a lack of symptoms in the early clinical course. Once the tumors cause noticeable symptoms, they are usually large and extensively locally invasive. As a result, most patients experience delayed diagnosis, with an average symptom duration of 2.3 years prior to diagnosis.3
Reexamining a common problem as a symptom of a rare condition
The most commonly manifesting symptom of sacral chordomas is lower back pain that is typically dull and worse with sitting.3,4 Since lower back pain is the leading cause of disability, it is difficult to determine when back pain is simply a benign consequence of aging or muscular pain and when it is, in fact, pathologic.5 A thorough history and physical are crucial in making the distinction.
Continue to: Clinical red flags...
Clinical red flags include pain with neurologic symptoms (including paresthesia, urinary or bowel disturbances, and weakness in the lower limbs), pain in the lower back with or without coccyx pain that persists and gradually worsens over time, and pain that fails to resolve.3 These symptoms are collectively strong indicators of underlying sacral pathology and should warrant further investigation, including a CT and MRI of the involved area.
Survival rate is improved by surgery
The gold standard for treatment of sacral chordomas is surgical resection with adequate margins, as these tumors are both radio- and chemo-insensitive.6 It is generally accepted that achieving a wide surgical margin is the most important predictor of survival and of reducing local recurrence in patients with sacrococcygeal chordoma.7-9
The survival rate varies after a posterior-only surgical approach; some studies cite the 5-year survival rate as 100% and others state the 7-year survival rate as 5%.4 The wide variation is likely due to small trial size, a lack of evidence, and how invasive the disease is at the time of surgery.
The recurrence rate 5 years after surgery is approximately 20%.4 The rate of urinary and fecal incontinence after surgery using a posterior-only approach is between 20% and 100%; some of this variation may be due to which spinal level is involved.4 If S3 is affected, there is almost always perineal anesthesia along with bowel and bladder incontinence.4
This patient was referred to Neurosurgery and underwent resection. He recovered well from surgery but suffered from some residual urinary incontinence. The patient did not receive chemotherapy or radiation, and further work-up revealed no evidence of metastasis.
Continue to: THE TAKEAWAY
THE TAKEAWAY
The diagnosis of sacral chordoma remains challenging. A history of clinical red flags, especially persistent lower back pain with neuropathy, should prompt an aggressive investigation to rule out underlying pathology. Other signs on physical exam could include urinary or bowel disturbances, weakness in the lower limbs, saddle anesthesia, new foot drop, and/or laxity of the anal sphincter.5 Early detection and surgical intervention are crucial for these patients to experience a better prognosis and preserve maximum function.
CORRESPONDENCE
Ginger Poulton, MD, 123 Hendersonville Road, Asheville, NC 28803; [email protected]
1. Zabel-du Bois A, Nikoghosyan A, Schwahofer A, et al. Intensity modulated radiotherapy in the management of sacral chordoma in primary versus recurrent disease. Radiother Oncol. 2010;97:408-412. doi: 10.1016/j.radonc.2010.10.008
2. Murphey MD, Andrews CL, Flemming DJ, et al. Primary tumors of the spine: radiologic pathologic correlation. Radiographics. 1996;1131-1158. doi: 10.1148/radiographics.16.5.8888395
3. Jeys L, Gibbins R, Evans G, et al. Sacral chordoma: a diagnosis not to be sat on? Int Orthopaedics. 2008;32:269-272. doi: 10.1007/s00264-006-0296-3
4. Pillai S, Govender, S. Sacral chordoma: a review of literature. J Orthop. 2018;15:679-684. doi: 10.1016/j.jor.2018.04.001
5. Traeger A, Buchbinder R, Harris I, et al. Diagnosis and management of low-back pain in primary care. CMAJ. 2017;189:E1386-E1395. doi: 10.1503/cmaj.170527
6. Walcott BP, Nahed BV, Mohyeldin A, et al. Chordoma: current concepts, management, and future directions. Lancet Oncol. 2012;13:e69-76. doi: 10.1016/S1470-2045(11)70337-0
7. Bergh P, Kindblom LG, Gunterberg B, et al. Prognostic factors in chordoma of the sacrum and mobile spine: a study of 39 patients. Cancer. 2000;88:2122-2134. doi: 10.1002/(sici)1097-0142(20000501)88:9<2122::aid-cncr19>3.0.co;2-1
8. Boriani S, Bandiera S, Biagini R, et al. Chordoma of the mobile spine: fifty years of experience. Spine. 2006;31:493-503. doi: 10.1097/01.brs.0000200038.30869.27
9. Hanna SA, Aston WJ, Briggs TW, et al. Sacral chordoma: can local recurrence after sacrectomy be predicted? Clin Orthop Relat Res. 2008;466:2217-2223. doi: 10.1007/s11999-008-0356-7
THE CASE
A 78-year-old man with a history of diabetes and hypertension was referred to the outpatient surgical office with a chief complaint of “tail bone pain” that had started after a fall a year earlier. The patient complained that the pain was worse when sitting and at nighttime. He also admitted to a 7-lb weight loss over the past 2 months without change in diet or appetite. He denied symptoms of incontinence, urinary retention, sharp stabbing pains in the lower extremities, night sweats, or anorexia.
The patient first visited an urgent care facility on the day after the fall because he was experiencing pain in his “tail bone” region while riding his lawn mower. A pelvic x-ray was performed at that time and showed no coccyx fracture. He received a steroid injection in the right sacroiliac joint, which provided some relief for a month. Throughout the course of the year, he was given 6 steroid injections into his sacroiliac joint by his primary care provider (PCP) and clinicians at his local urgent care facility. One year after the fall, the patient’s PCP ordered a computed tomography (CT) scan of the abdomen and pelvis, which revealed a 4.6 x 7.5–cm soft-tissue mass with bony destruction of the lower sacrum and coccyx that extended into the sacral and coccygeal canal (FIGURE 1).
On exam in our surgical office, the patient was found to be alert and oriented. His neurologic exam was unremarkable, with an intact motor and sensory exam and no symptoms of cauda equina syndrome. During palpation over the lower sacrum and coccyx, both tenderness and a boggy, soft mass were observed. Nerve impingement was most likely caused by the size of the mass.
THE DIAGNOSIS
Biopsy revealed a large tan-gray, gelatinous, soft-tissue mass that was necrotizing through the lower sacrum. The diagnosis of a sacral chordoma was confirmed with magnetic resonance imaging of the pelvis, which demonstrated a 4.6 × 8.1–cm destructive expansile sacrococcygeal tumor with an exophytic soft-tissue component (FIGURE 2). The tumor also involved the piriformis and gluteus maximus muscles bilaterally.
DISCUSSION
Chordomas are rare, malignant bone tumors that grow slowly and originate from embryonic remnants of the notochord.1 They are most commonly seen in the sacrococcygeal segment (50%) but are also seen in the spheno-occipital synchondrosis (30%-35%) and other spinal segments such as C2 and lumbar spine.2 Chordomas are typically seen in middle-aged patients, with sacral chordomas occurring predominantly in men compared to women (3:1).2
Slow to grow, slow to diagnose
The difficulty with diagnosing sacral chordomas lies in the tendency for these tumors to grow extremely slowly, making detection challenging due to a lack of symptoms in the early clinical course. Once the tumors cause noticeable symptoms, they are usually large and extensively locally invasive. As a result, most patients experience delayed diagnosis, with an average symptom duration of 2.3 years prior to diagnosis.3
Reexamining a common problem as a symptom of a rare condition
The most commonly manifesting symptom of sacral chordomas is lower back pain that is typically dull and worse with sitting.3,4 Since lower back pain is the leading cause of disability, it is difficult to determine when back pain is simply a benign consequence of aging or muscular pain and when it is, in fact, pathologic.5 A thorough history and physical are crucial in making the distinction.
Continue to: Clinical red flags...
Clinical red flags include pain with neurologic symptoms (including paresthesia, urinary or bowel disturbances, and weakness in the lower limbs), pain in the lower back with or without coccyx pain that persists and gradually worsens over time, and pain that fails to resolve.3 These symptoms are collectively strong indicators of underlying sacral pathology and should warrant further investigation, including a CT and MRI of the involved area.
Survival rate is improved by surgery
The gold standard for treatment of sacral chordomas is surgical resection with adequate margins, as these tumors are both radio- and chemo-insensitive.6 It is generally accepted that achieving a wide surgical margin is the most important predictor of survival and of reducing local recurrence in patients with sacrococcygeal chordoma.7-9
The survival rate varies after a posterior-only surgical approach; some studies cite the 5-year survival rate as 100% and others state the 7-year survival rate as 5%.4 The wide variation is likely due to small trial size, a lack of evidence, and how invasive the disease is at the time of surgery.
The recurrence rate 5 years after surgery is approximately 20%.4 The rate of urinary and fecal incontinence after surgery using a posterior-only approach is between 20% and 100%; some of this variation may be due to which spinal level is involved.4 If S3 is affected, there is almost always perineal anesthesia along with bowel and bladder incontinence.4
This patient was referred to Neurosurgery and underwent resection. He recovered well from surgery but suffered from some residual urinary incontinence. The patient did not receive chemotherapy or radiation, and further work-up revealed no evidence of metastasis.
Continue to: THE TAKEAWAY
THE TAKEAWAY
The diagnosis of sacral chordoma remains challenging. A history of clinical red flags, especially persistent lower back pain with neuropathy, should prompt an aggressive investigation to rule out underlying pathology. Other signs on physical exam could include urinary or bowel disturbances, weakness in the lower limbs, saddle anesthesia, new foot drop, and/or laxity of the anal sphincter.5 Early detection and surgical intervention are crucial for these patients to experience a better prognosis and preserve maximum function.
CORRESPONDENCE
Ginger Poulton, MD, 123 Hendersonville Road, Asheville, NC 28803; [email protected]
THE CASE
A 78-year-old man with a history of diabetes and hypertension was referred to the outpatient surgical office with a chief complaint of “tail bone pain” that had started after a fall a year earlier. The patient complained that the pain was worse when sitting and at nighttime. He also admitted to a 7-lb weight loss over the past 2 months without change in diet or appetite. He denied symptoms of incontinence, urinary retention, sharp stabbing pains in the lower extremities, night sweats, or anorexia.
The patient first visited an urgent care facility on the day after the fall because he was experiencing pain in his “tail bone” region while riding his lawn mower. A pelvic x-ray was performed at that time and showed no coccyx fracture. He received a steroid injection in the right sacroiliac joint, which provided some relief for a month. Throughout the course of the year, he was given 6 steroid injections into his sacroiliac joint by his primary care provider (PCP) and clinicians at his local urgent care facility. One year after the fall, the patient’s PCP ordered a computed tomography (CT) scan of the abdomen and pelvis, which revealed a 4.6 x 7.5–cm soft-tissue mass with bony destruction of the lower sacrum and coccyx that extended into the sacral and coccygeal canal (FIGURE 1).
On exam in our surgical office, the patient was found to be alert and oriented. His neurologic exam was unremarkable, with an intact motor and sensory exam and no symptoms of cauda equina syndrome. During palpation over the lower sacrum and coccyx, both tenderness and a boggy, soft mass were observed. Nerve impingement was most likely caused by the size of the mass.
THE DIAGNOSIS
Biopsy revealed a large tan-gray, gelatinous, soft-tissue mass that was necrotizing through the lower sacrum. The diagnosis of a sacral chordoma was confirmed with magnetic resonance imaging of the pelvis, which demonstrated a 4.6 × 8.1–cm destructive expansile sacrococcygeal tumor with an exophytic soft-tissue component (FIGURE 2). The tumor also involved the piriformis and gluteus maximus muscles bilaterally.
DISCUSSION
Chordomas are rare, malignant bone tumors that grow slowly and originate from embryonic remnants of the notochord.1 They are most commonly seen in the sacrococcygeal segment (50%) but are also seen in the spheno-occipital synchondrosis (30%-35%) and other spinal segments such as C2 and lumbar spine.2 Chordomas are typically seen in middle-aged patients, with sacral chordomas occurring predominantly in men compared to women (3:1).2
Slow to grow, slow to diagnose
The difficulty with diagnosing sacral chordomas lies in the tendency for these tumors to grow extremely slowly, making detection challenging due to a lack of symptoms in the early clinical course. Once the tumors cause noticeable symptoms, they are usually large and extensively locally invasive. As a result, most patients experience delayed diagnosis, with an average symptom duration of 2.3 years prior to diagnosis.3
Reexamining a common problem as a symptom of a rare condition
The most commonly manifesting symptom of sacral chordomas is lower back pain that is typically dull and worse with sitting.3,4 Since lower back pain is the leading cause of disability, it is difficult to determine when back pain is simply a benign consequence of aging or muscular pain and when it is, in fact, pathologic.5 A thorough history and physical are crucial in making the distinction.
Continue to: Clinical red flags...
Clinical red flags include pain with neurologic symptoms (including paresthesia, urinary or bowel disturbances, and weakness in the lower limbs), pain in the lower back with or without coccyx pain that persists and gradually worsens over time, and pain that fails to resolve.3 These symptoms are collectively strong indicators of underlying sacral pathology and should warrant further investigation, including a CT and MRI of the involved area.
Survival rate is improved by surgery
The gold standard for treatment of sacral chordomas is surgical resection with adequate margins, as these tumors are both radio- and chemo-insensitive.6 It is generally accepted that achieving a wide surgical margin is the most important predictor of survival and of reducing local recurrence in patients with sacrococcygeal chordoma.7-9
The survival rate varies after a posterior-only surgical approach; some studies cite the 5-year survival rate as 100% and others state the 7-year survival rate as 5%.4 The wide variation is likely due to small trial size, a lack of evidence, and how invasive the disease is at the time of surgery.
The recurrence rate 5 years after surgery is approximately 20%.4 The rate of urinary and fecal incontinence after surgery using a posterior-only approach is between 20% and 100%; some of this variation may be due to which spinal level is involved.4 If S3 is affected, there is almost always perineal anesthesia along with bowel and bladder incontinence.4
This patient was referred to Neurosurgery and underwent resection. He recovered well from surgery but suffered from some residual urinary incontinence. The patient did not receive chemotherapy or radiation, and further work-up revealed no evidence of metastasis.
Continue to: THE TAKEAWAY
THE TAKEAWAY
The diagnosis of sacral chordoma remains challenging. A history of clinical red flags, especially persistent lower back pain with neuropathy, should prompt an aggressive investigation to rule out underlying pathology. Other signs on physical exam could include urinary or bowel disturbances, weakness in the lower limbs, saddle anesthesia, new foot drop, and/or laxity of the anal sphincter.5 Early detection and surgical intervention are crucial for these patients to experience a better prognosis and preserve maximum function.
CORRESPONDENCE
Ginger Poulton, MD, 123 Hendersonville Road, Asheville, NC 28803; [email protected]
1. Zabel-du Bois A, Nikoghosyan A, Schwahofer A, et al. Intensity modulated radiotherapy in the management of sacral chordoma in primary versus recurrent disease. Radiother Oncol. 2010;97:408-412. doi: 10.1016/j.radonc.2010.10.008
2. Murphey MD, Andrews CL, Flemming DJ, et al. Primary tumors of the spine: radiologic pathologic correlation. Radiographics. 1996;1131-1158. doi: 10.1148/radiographics.16.5.8888395
3. Jeys L, Gibbins R, Evans G, et al. Sacral chordoma: a diagnosis not to be sat on? Int Orthopaedics. 2008;32:269-272. doi: 10.1007/s00264-006-0296-3
4. Pillai S, Govender, S. Sacral chordoma: a review of literature. J Orthop. 2018;15:679-684. doi: 10.1016/j.jor.2018.04.001
5. Traeger A, Buchbinder R, Harris I, et al. Diagnosis and management of low-back pain in primary care. CMAJ. 2017;189:E1386-E1395. doi: 10.1503/cmaj.170527
6. Walcott BP, Nahed BV, Mohyeldin A, et al. Chordoma: current concepts, management, and future directions. Lancet Oncol. 2012;13:e69-76. doi: 10.1016/S1470-2045(11)70337-0
7. Bergh P, Kindblom LG, Gunterberg B, et al. Prognostic factors in chordoma of the sacrum and mobile spine: a study of 39 patients. Cancer. 2000;88:2122-2134. doi: 10.1002/(sici)1097-0142(20000501)88:9<2122::aid-cncr19>3.0.co;2-1
8. Boriani S, Bandiera S, Biagini R, et al. Chordoma of the mobile spine: fifty years of experience. Spine. 2006;31:493-503. doi: 10.1097/01.brs.0000200038.30869.27
9. Hanna SA, Aston WJ, Briggs TW, et al. Sacral chordoma: can local recurrence after sacrectomy be predicted? Clin Orthop Relat Res. 2008;466:2217-2223. doi: 10.1007/s11999-008-0356-7
1. Zabel-du Bois A, Nikoghosyan A, Schwahofer A, et al. Intensity modulated radiotherapy in the management of sacral chordoma in primary versus recurrent disease. Radiother Oncol. 2010;97:408-412. doi: 10.1016/j.radonc.2010.10.008
2. Murphey MD, Andrews CL, Flemming DJ, et al. Primary tumors of the spine: radiologic pathologic correlation. Radiographics. 1996;1131-1158. doi: 10.1148/radiographics.16.5.8888395
3. Jeys L, Gibbins R, Evans G, et al. Sacral chordoma: a diagnosis not to be sat on? Int Orthopaedics. 2008;32:269-272. doi: 10.1007/s00264-006-0296-3
4. Pillai S, Govender, S. Sacral chordoma: a review of literature. J Orthop. 2018;15:679-684. doi: 10.1016/j.jor.2018.04.001
5. Traeger A, Buchbinder R, Harris I, et al. Diagnosis and management of low-back pain in primary care. CMAJ. 2017;189:E1386-E1395. doi: 10.1503/cmaj.170527
6. Walcott BP, Nahed BV, Mohyeldin A, et al. Chordoma: current concepts, management, and future directions. Lancet Oncol. 2012;13:e69-76. doi: 10.1016/S1470-2045(11)70337-0
7. Bergh P, Kindblom LG, Gunterberg B, et al. Prognostic factors in chordoma of the sacrum and mobile spine: a study of 39 patients. Cancer. 2000;88:2122-2134. doi: 10.1002/(sici)1097-0142(20000501)88:9<2122::aid-cncr19>3.0.co;2-1
8. Boriani S, Bandiera S, Biagini R, et al. Chordoma of the mobile spine: fifty years of experience. Spine. 2006;31:493-503. doi: 10.1097/01.brs.0000200038.30869.27
9. Hanna SA, Aston WJ, Briggs TW, et al. Sacral chordoma: can local recurrence after sacrectomy be predicted? Clin Orthop Relat Res. 2008;466:2217-2223. doi: 10.1007/s11999-008-0356-7

Orbital Varix Masquerading as an Intraorbital Lymphoma
Clinical context was paramount to the diagnosis and management of a patient with periorbital pain and a history of systemic lymphoma.
We present a case of an orbital varix masquerading as an orbital lymphoma. Our case underscores the importance of clinical correlation and thorough study of the ordered films by the ordering health care provider.
Case Presentation
An 84-year-old female veteran presented to the Bay Pines Veterans Affairs Healthcare System emergency department. She had a past ocular history of nonproliferative diabetic retinopathy in both eyes (OU) and senile cataracts OU. She had a complicated medical history most notable for congestive heart failure and Stage IV B cell follicular lymphoma, having received 6 rounds of chemotherapy, and has since been on rituximab maintenance therapy for the past few years.
The patient reported dyspnea on exertion, 30-pound weight gain, and ocular pain in her right eye (OD), more so than her left eye (OS) that was severe enough to wake her from sleep. She endorsed an associated headache but reported no visual loss or any other ocular symptoms other than conjunctival injection. On examination, the patient demonstrated jugular venous distension. X-ray imaging obtained in the emergency department demonstrated bilateral pleural effusions. Our patient was admitted subsequently for an exacerbation of congestive heart failure. She was monitored for euvolemia and discharged 4 days later.
During admission, imaging of the orbits was obtained. Computed tomography (CT) of the head without contrast demonstrated at least 4 intraorbital masses in the right orbit, measuring up to 22 mm in maximum diameter and at least 3 intraorbital masses in the left orbit, measuring up to 16 mm in diameter (Figure 1). Magnetic resonance imaging (MRI) with contrast of the brain and orbits was ordered, which demonstrated multiple bilateral uniformly enhancing, primarily extraconal masses present in both orbits, the largest of which occupied the superomedial aspect of the right orbit and measured 12 x 18 x 20 mm. Further, the ophthalmic veins were noted to be engorged. The cavernous did not demonstrate any thrombosis. No other ocular structures were compromised, although there was compression of the extraocular muscles in both orbits (Figures 2, 3, 4, 5, and 6). At that time, the reading radiologist suggested the most likely diagnosis was metastatic orbital lymphoma given the clinical history, which became the working diagnosis.
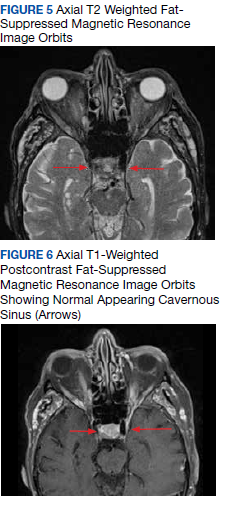
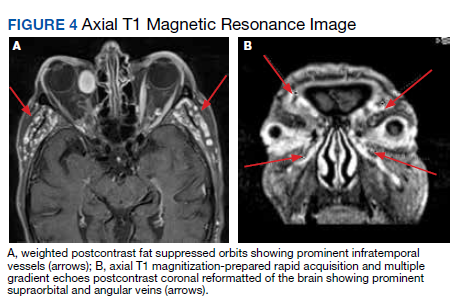
A few days after admission, the patient received an ophthalmic evaluation at the eye clinic. Visual acuity (VA) at this time was 20/200 that pinholed (PH) 20/70 OD and 20/30 without pinhole improvement OS. Refraction was -2.50 + 1.50 × 120 OD and -0.25 + 0.50 × 065 OS, which yielded visual acuities of 20/60 and 20/30, respectively. There was no afferent pupillary defect and pupils were symmetric. Goldmann tonometry demonstrated pressures of 11 mm of mercury OU at 1630. Slit-lamp and dilated fundus examinations were within normal limits except for 2+ nuclear sclerotic cataracts, large cups of 0.6 OD and 0.7 OS, and a mild epiretinal membrane OD. The decision was made to refer the patient to oculoplastic service for biopsy of the lesion to rule out a metastatic lymphoid solid tumor. At this juncture, the working diagnosis continued to be metastatic orbital lymphoma.
The patient underwent right anterior orbitotomy. Intraoperatively, after dissection to the lesion was accomplished, it was noted that the mass displayed a blue to purple hue consistent with a vascular malformation. It was decided to continue careful dissection instead of obtaining a biopsy. Continued dissection further corroborated a vascular lesion. Meticulous hemostasis was maintained during the dissection; however, dissection was halted after about 35-mm depth into the orbit, given concern for damaging the optic nerve. The feeding vessel to the lesion was tied off with two 5-0 vicryl sutures, and the specimen was cut distal to the ligation. During the procedure, pupillary function was continually checked. The rest of the surgery proceeded without any difficulty, and the specimen was sent off to pathology.
Pathology returned as an orbital varix with no thrombosis or malignant tissue. Surgery to remove lesions of the left orbit was deferred given radiologic findings consistent with vascular lesions, similar to the removed lesion from the right orbit. The patient is currently without residual periorbital pain after diuresis, and the patient’s oncological management continues to be maintenance rituximab. The remaining lesions will be monitored with yearly serial imaging.
Discussion
In a study of 242 patients, Bacorn and colleagues found that a clinician’s preoperative assessment correlated with histopathologic diagnosis in 75.7% of cases, whereas the radiology report was correct in only 52.4% of cases.1 Retrospective analysis identified clues that could have been used to more rapidly elucidate the true diagnosis for our patient.
In regard to symptomatology, orbital varices present with intermittent proptosis, vision loss, and rarely, periorbital pain unless thrombosed.2,3 The severity of periorbital pain experienced by our patient is atypical of an orbital varix especially in the absence of a phlebolith. A specific feature of orbital varix is enlargement with the Valsalva maneuver.3 Although the patient did not report the notedsymptoms, more pointed questioning may have helped elucidate our patient’s true diagnosis sooner.
Radiologically, the presence of a partial flow void (decreased signal on T2) is useful for confirming the vascular nature of a lesion as was present in our case. Specific to the radiologic evaluation of orbital varices, it is recommended to obtain imaging with and without the Valsalva maneuver.4 Ultrasound is a superb tool in our armamentarium to image orbital lesions. B-scan ultrasound with and without Valsalva should be able to demonstrate variation in size when standing (minimal distension) vs lying flat with Valsalva (maximal distension).4 Further, Doppler ultrasound would be able to demonstrate changes in flow within the lesion when comparing previously mentioned maneuvers.4 Orbital lymphoma would not demonstrate this variation.
The size change of an orbital varix lesion may be further demonstrated on head CT with contrast. On CT, an orbital varix will demonstrate isodensity to other venous structures, whereas orbital lymphomas will be hyperdense when compared to extraocular muscles.4,5 Further, a head CT without contrast may demonstrate phleboliths within an orbital varix.4 MRI should be performed with the Valsalva maneuver. On T1 and T2 studies, orbital varices demonstrate hypointensity when compared to extraocular muscles (EOMs).4 Lymphomas demonstrate a very specific radiologic pattern on MRI. On T1, they demonstrate isointensity to hypointensity when compared to EOMS, and on T2, they demonstrate iso- to hyperintensity when compared to EOMs.5 With respect to fluorodeoxyglucose (FDG) positron emission tomography (PET), our patient’s orbital lesion did not demonstrate FDG uptake. In patients where lymphoma previously demonstrated FDG PET uptake, the absence of such uptake strongly argues against malignant nature of the lesion (Figure 7).
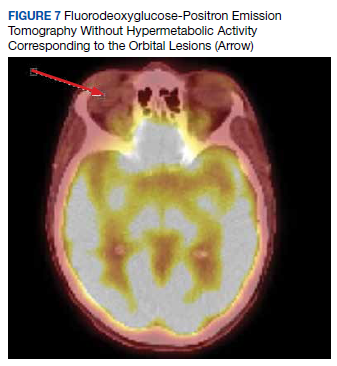
Prominently enhancing lesions are more likely to represent varices, aneurysms, or other highly or completely vascular lesions. Any intraorbital intervention should be conducted as though a vascular lesion is within the differential, and appropriate care should be taken even if not specifically enunciated in the radiologic report.
Management of orbital varices is not standardized; however, these lesions tend to be observed if no significant proptosis, pain, thrombosis, diplopia, or compression of the optic nerve is present. In such cases, surgical intervention is performed; however, the lesions may recur. Our patient’s presentation coincided with her heart failure exacerbation most likely secondary to flow disruption and fluid overload in the venous system, thereby exacerbating her orbital varices. The resolution of our patient’s orbital pain in the left orbit was likely due to improved distension after achieving euvolemia after diuresis. In cases where varices are secondary to a correctable etiologies, treatment of these etiologies are in order. Chen and colleagues reported a case of pulsatile proptosis associated with fluid overload in a newly diagnosed case of heart failure secondary to mitral regurgitation.6 Thus, orbital pain due to worsened orbital varices may represent a symptom of fluid overload and the provider may look for etiologies of this disease process.
Conclusions
We present a case of an orbital varix masquerading as an orbital lymphoma. While the ruling out of a diagnosis that might portend a poor prognosis is always of paramount importance, proper use of investigative studies and a thorough history could have helped elucidate the true diagnosis sooner: In this case an orbital varix masquerading as an orbital lymphoma. Mainly, the use of the Valsalva maneuver during the physical examination (resulting in proptosis) and during radiologic studies might have obviated the need for formal biopsy. Furthermore, orbital pain may be a presenting symptom of fluid overload in patients with a history of orbital varices.
1. Bacorn C, Gokoffski KK, Lin LK. Clinical correlation recommended: accuracy of clinician versus radiologic interpretation of the imaging of orbital lesions. Orbit. 2021;40(2):133-137. doi:10.1080/01676830.2020.1752742
2. Shams PN, Cugati S, Wells T, Huilgol S, Selva D. Orbital varix thrombosis and review of orbital vascular anomalies in blue rubber bleb nevus syndrome. Ophthalmic Plast Reconstr Surg. 2015;31(4):e82-e86. doi:10.1097/IOP.0000000000000107
3. Islam N, Mireskandari K, Rose GE. Orbital varices and orbital wall defects. Br J Ophthalmol. 2004;88(8):1092-1093.
4. Smoker WR, Gentry LR, Yee NK, Reede DL, Nerad JA. Vascular lesions of the orbit: more than meets the eye. Radiographics. 2008;28(1):185-325. doi:10.1148/rg.281075040
5. Karcioglu ZA, ed. Orbital Tumors. New York; 2005. Chap 13:133-140.
6. Chen Z, Jones H. A case of tricuspid regurgitation and congestive cardiac failure presenting with orbital pulsation. JRSM Cardiovasc Dis. 2012;1(1):cvd.2012.012005. Published 2012 Apr 5. doi:10.1258/cvd.2012.012005
Clinical context was paramount to the diagnosis and management of a patient with periorbital pain and a history of systemic lymphoma.
Clinical context was paramount to the diagnosis and management of a patient with periorbital pain and a history of systemic lymphoma.
We present a case of an orbital varix masquerading as an orbital lymphoma. Our case underscores the importance of clinical correlation and thorough study of the ordered films by the ordering health care provider.
Case Presentation
An 84-year-old female veteran presented to the Bay Pines Veterans Affairs Healthcare System emergency department. She had a past ocular history of nonproliferative diabetic retinopathy in both eyes (OU) and senile cataracts OU. She had a complicated medical history most notable for congestive heart failure and Stage IV B cell follicular lymphoma, having received 6 rounds of chemotherapy, and has since been on rituximab maintenance therapy for the past few years.
The patient reported dyspnea on exertion, 30-pound weight gain, and ocular pain in her right eye (OD), more so than her left eye (OS) that was severe enough to wake her from sleep. She endorsed an associated headache but reported no visual loss or any other ocular symptoms other than conjunctival injection. On examination, the patient demonstrated jugular venous distension. X-ray imaging obtained in the emergency department demonstrated bilateral pleural effusions. Our patient was admitted subsequently for an exacerbation of congestive heart failure. She was monitored for euvolemia and discharged 4 days later.
During admission, imaging of the orbits was obtained. Computed tomography (CT) of the head without contrast demonstrated at least 4 intraorbital masses in the right orbit, measuring up to 22 mm in maximum diameter and at least 3 intraorbital masses in the left orbit, measuring up to 16 mm in diameter (Figure 1). Magnetic resonance imaging (MRI) with contrast of the brain and orbits was ordered, which demonstrated multiple bilateral uniformly enhancing, primarily extraconal masses present in both orbits, the largest of which occupied the superomedial aspect of the right orbit and measured 12 x 18 x 20 mm. Further, the ophthalmic veins were noted to be engorged. The cavernous did not demonstrate any thrombosis. No other ocular structures were compromised, although there was compression of the extraocular muscles in both orbits (Figures 2, 3, 4, 5, and 6). At that time, the reading radiologist suggested the most likely diagnosis was metastatic orbital lymphoma given the clinical history, which became the working diagnosis.
A few days after admission, the patient received an ophthalmic evaluation at the eye clinic. Visual acuity (VA) at this time was 20/200 that pinholed (PH) 20/70 OD and 20/30 without pinhole improvement OS. Refraction was -2.50 + 1.50 × 120 OD and -0.25 + 0.50 × 065 OS, which yielded visual acuities of 20/60 and 20/30, respectively. There was no afferent pupillary defect and pupils were symmetric. Goldmann tonometry demonstrated pressures of 11 mm of mercury OU at 1630. Slit-lamp and dilated fundus examinations were within normal limits except for 2+ nuclear sclerotic cataracts, large cups of 0.6 OD and 0.7 OS, and a mild epiretinal membrane OD. The decision was made to refer the patient to oculoplastic service for biopsy of the lesion to rule out a metastatic lymphoid solid tumor. At this juncture, the working diagnosis continued to be metastatic orbital lymphoma.
The patient underwent right anterior orbitotomy. Intraoperatively, after dissection to the lesion was accomplished, it was noted that the mass displayed a blue to purple hue consistent with a vascular malformation. It was decided to continue careful dissection instead of obtaining a biopsy. Continued dissection further corroborated a vascular lesion. Meticulous hemostasis was maintained during the dissection; however, dissection was halted after about 35-mm depth into the orbit, given concern for damaging the optic nerve. The feeding vessel to the lesion was tied off with two 5-0 vicryl sutures, and the specimen was cut distal to the ligation. During the procedure, pupillary function was continually checked. The rest of the surgery proceeded without any difficulty, and the specimen was sent off to pathology.
Pathology returned as an orbital varix with no thrombosis or malignant tissue. Surgery to remove lesions of the left orbit was deferred given radiologic findings consistent with vascular lesions, similar to the removed lesion from the right orbit. The patient is currently without residual periorbital pain after diuresis, and the patient’s oncological management continues to be maintenance rituximab. The remaining lesions will be monitored with yearly serial imaging.
Discussion
In a study of 242 patients, Bacorn and colleagues found that a clinician’s preoperative assessment correlated with histopathologic diagnosis in 75.7% of cases, whereas the radiology report was correct in only 52.4% of cases.1 Retrospective analysis identified clues that could have been used to more rapidly elucidate the true diagnosis for our patient.
In regard to symptomatology, orbital varices present with intermittent proptosis, vision loss, and rarely, periorbital pain unless thrombosed.2,3 The severity of periorbital pain experienced by our patient is atypical of an orbital varix especially in the absence of a phlebolith. A specific feature of orbital varix is enlargement with the Valsalva maneuver.3 Although the patient did not report the notedsymptoms, more pointed questioning may have helped elucidate our patient’s true diagnosis sooner.
Radiologically, the presence of a partial flow void (decreased signal on T2) is useful for confirming the vascular nature of a lesion as was present in our case. Specific to the radiologic evaluation of orbital varices, it is recommended to obtain imaging with and without the Valsalva maneuver.4 Ultrasound is a superb tool in our armamentarium to image orbital lesions. B-scan ultrasound with and without Valsalva should be able to demonstrate variation in size when standing (minimal distension) vs lying flat with Valsalva (maximal distension).4 Further, Doppler ultrasound would be able to demonstrate changes in flow within the lesion when comparing previously mentioned maneuvers.4 Orbital lymphoma would not demonstrate this variation.
The size change of an orbital varix lesion may be further demonstrated on head CT with contrast. On CT, an orbital varix will demonstrate isodensity to other venous structures, whereas orbital lymphomas will be hyperdense when compared to extraocular muscles.4,5 Further, a head CT without contrast may demonstrate phleboliths within an orbital varix.4 MRI should be performed with the Valsalva maneuver. On T1 and T2 studies, orbital varices demonstrate hypointensity when compared to extraocular muscles (EOMs).4 Lymphomas demonstrate a very specific radiologic pattern on MRI. On T1, they demonstrate isointensity to hypointensity when compared to EOMS, and on T2, they demonstrate iso- to hyperintensity when compared to EOMs.5 With respect to fluorodeoxyglucose (FDG) positron emission tomography (PET), our patient’s orbital lesion did not demonstrate FDG uptake. In patients where lymphoma previously demonstrated FDG PET uptake, the absence of such uptake strongly argues against malignant nature of the lesion (Figure 7).
Prominently enhancing lesions are more likely to represent varices, aneurysms, or other highly or completely vascular lesions. Any intraorbital intervention should be conducted as though a vascular lesion is within the differential, and appropriate care should be taken even if not specifically enunciated in the radiologic report.
Management of orbital varices is not standardized; however, these lesions tend to be observed if no significant proptosis, pain, thrombosis, diplopia, or compression of the optic nerve is present. In such cases, surgical intervention is performed; however, the lesions may recur. Our patient’s presentation coincided with her heart failure exacerbation most likely secondary to flow disruption and fluid overload in the venous system, thereby exacerbating her orbital varices. The resolution of our patient’s orbital pain in the left orbit was likely due to improved distension after achieving euvolemia after diuresis. In cases where varices are secondary to a correctable etiologies, treatment of these etiologies are in order. Chen and colleagues reported a case of pulsatile proptosis associated with fluid overload in a newly diagnosed case of heart failure secondary to mitral regurgitation.6 Thus, orbital pain due to worsened orbital varices may represent a symptom of fluid overload and the provider may look for etiologies of this disease process.
Conclusions
We present a case of an orbital varix masquerading as an orbital lymphoma. While the ruling out of a diagnosis that might portend a poor prognosis is always of paramount importance, proper use of investigative studies and a thorough history could have helped elucidate the true diagnosis sooner: In this case an orbital varix masquerading as an orbital lymphoma. Mainly, the use of the Valsalva maneuver during the physical examination (resulting in proptosis) and during radiologic studies might have obviated the need for formal biopsy. Furthermore, orbital pain may be a presenting symptom of fluid overload in patients with a history of orbital varices.
We present a case of an orbital varix masquerading as an orbital lymphoma. Our case underscores the importance of clinical correlation and thorough study of the ordered films by the ordering health care provider.
Case Presentation
An 84-year-old female veteran presented to the Bay Pines Veterans Affairs Healthcare System emergency department. She had a past ocular history of nonproliferative diabetic retinopathy in both eyes (OU) and senile cataracts OU. She had a complicated medical history most notable for congestive heart failure and Stage IV B cell follicular lymphoma, having received 6 rounds of chemotherapy, and has since been on rituximab maintenance therapy for the past few years.
The patient reported dyspnea on exertion, 30-pound weight gain, and ocular pain in her right eye (OD), more so than her left eye (OS) that was severe enough to wake her from sleep. She endorsed an associated headache but reported no visual loss or any other ocular symptoms other than conjunctival injection. On examination, the patient demonstrated jugular venous distension. X-ray imaging obtained in the emergency department demonstrated bilateral pleural effusions. Our patient was admitted subsequently for an exacerbation of congestive heart failure. She was monitored for euvolemia and discharged 4 days later.
During admission, imaging of the orbits was obtained. Computed tomography (CT) of the head without contrast demonstrated at least 4 intraorbital masses in the right orbit, measuring up to 22 mm in maximum diameter and at least 3 intraorbital masses in the left orbit, measuring up to 16 mm in diameter (Figure 1). Magnetic resonance imaging (MRI) with contrast of the brain and orbits was ordered, which demonstrated multiple bilateral uniformly enhancing, primarily extraconal masses present in both orbits, the largest of which occupied the superomedial aspect of the right orbit and measured 12 x 18 x 20 mm. Further, the ophthalmic veins were noted to be engorged. The cavernous did not demonstrate any thrombosis. No other ocular structures were compromised, although there was compression of the extraocular muscles in both orbits (Figures 2, 3, 4, 5, and 6). At that time, the reading radiologist suggested the most likely diagnosis was metastatic orbital lymphoma given the clinical history, which became the working diagnosis.
A few days after admission, the patient received an ophthalmic evaluation at the eye clinic. Visual acuity (VA) at this time was 20/200 that pinholed (PH) 20/70 OD and 20/30 without pinhole improvement OS. Refraction was -2.50 + 1.50 × 120 OD and -0.25 + 0.50 × 065 OS, which yielded visual acuities of 20/60 and 20/30, respectively. There was no afferent pupillary defect and pupils were symmetric. Goldmann tonometry demonstrated pressures of 11 mm of mercury OU at 1630. Slit-lamp and dilated fundus examinations were within normal limits except for 2+ nuclear sclerotic cataracts, large cups of 0.6 OD and 0.7 OS, and a mild epiretinal membrane OD. The decision was made to refer the patient to oculoplastic service for biopsy of the lesion to rule out a metastatic lymphoid solid tumor. At this juncture, the working diagnosis continued to be metastatic orbital lymphoma.
The patient underwent right anterior orbitotomy. Intraoperatively, after dissection to the lesion was accomplished, it was noted that the mass displayed a blue to purple hue consistent with a vascular malformation. It was decided to continue careful dissection instead of obtaining a biopsy. Continued dissection further corroborated a vascular lesion. Meticulous hemostasis was maintained during the dissection; however, dissection was halted after about 35-mm depth into the orbit, given concern for damaging the optic nerve. The feeding vessel to the lesion was tied off with two 5-0 vicryl sutures, and the specimen was cut distal to the ligation. During the procedure, pupillary function was continually checked. The rest of the surgery proceeded without any difficulty, and the specimen was sent off to pathology.
Pathology returned as an orbital varix with no thrombosis or malignant tissue. Surgery to remove lesions of the left orbit was deferred given radiologic findings consistent with vascular lesions, similar to the removed lesion from the right orbit. The patient is currently without residual periorbital pain after diuresis, and the patient’s oncological management continues to be maintenance rituximab. The remaining lesions will be monitored with yearly serial imaging.
Discussion
In a study of 242 patients, Bacorn and colleagues found that a clinician’s preoperative assessment correlated with histopathologic diagnosis in 75.7% of cases, whereas the radiology report was correct in only 52.4% of cases.1 Retrospective analysis identified clues that could have been used to more rapidly elucidate the true diagnosis for our patient.
In regard to symptomatology, orbital varices present with intermittent proptosis, vision loss, and rarely, periorbital pain unless thrombosed.2,3 The severity of periorbital pain experienced by our patient is atypical of an orbital varix especially in the absence of a phlebolith. A specific feature of orbital varix is enlargement with the Valsalva maneuver.3 Although the patient did not report the notedsymptoms, more pointed questioning may have helped elucidate our patient’s true diagnosis sooner.
Radiologically, the presence of a partial flow void (decreased signal on T2) is useful for confirming the vascular nature of a lesion as was present in our case. Specific to the radiologic evaluation of orbital varices, it is recommended to obtain imaging with and without the Valsalva maneuver.4 Ultrasound is a superb tool in our armamentarium to image orbital lesions. B-scan ultrasound with and without Valsalva should be able to demonstrate variation in size when standing (minimal distension) vs lying flat with Valsalva (maximal distension).4 Further, Doppler ultrasound would be able to demonstrate changes in flow within the lesion when comparing previously mentioned maneuvers.4 Orbital lymphoma would not demonstrate this variation.
The size change of an orbital varix lesion may be further demonstrated on head CT with contrast. On CT, an orbital varix will demonstrate isodensity to other venous structures, whereas orbital lymphomas will be hyperdense when compared to extraocular muscles.4,5 Further, a head CT without contrast may demonstrate phleboliths within an orbital varix.4 MRI should be performed with the Valsalva maneuver. On T1 and T2 studies, orbital varices demonstrate hypointensity when compared to extraocular muscles (EOMs).4 Lymphomas demonstrate a very specific radiologic pattern on MRI. On T1, they demonstrate isointensity to hypointensity when compared to EOMS, and on T2, they demonstrate iso- to hyperintensity when compared to EOMs.5 With respect to fluorodeoxyglucose (FDG) positron emission tomography (PET), our patient’s orbital lesion did not demonstrate FDG uptake. In patients where lymphoma previously demonstrated FDG PET uptake, the absence of such uptake strongly argues against malignant nature of the lesion (Figure 7).
Prominently enhancing lesions are more likely to represent varices, aneurysms, or other highly or completely vascular lesions. Any intraorbital intervention should be conducted as though a vascular lesion is within the differential, and appropriate care should be taken even if not specifically enunciated in the radiologic report.
Management of orbital varices is not standardized; however, these lesions tend to be observed if no significant proptosis, pain, thrombosis, diplopia, or compression of the optic nerve is present. In such cases, surgical intervention is performed; however, the lesions may recur. Our patient’s presentation coincided with her heart failure exacerbation most likely secondary to flow disruption and fluid overload in the venous system, thereby exacerbating her orbital varices. The resolution of our patient’s orbital pain in the left orbit was likely due to improved distension after achieving euvolemia after diuresis. In cases where varices are secondary to a correctable etiologies, treatment of these etiologies are in order. Chen and colleagues reported a case of pulsatile proptosis associated with fluid overload in a newly diagnosed case of heart failure secondary to mitral regurgitation.6 Thus, orbital pain due to worsened orbital varices may represent a symptom of fluid overload and the provider may look for etiologies of this disease process.
Conclusions
We present a case of an orbital varix masquerading as an orbital lymphoma. While the ruling out of a diagnosis that might portend a poor prognosis is always of paramount importance, proper use of investigative studies and a thorough history could have helped elucidate the true diagnosis sooner: In this case an orbital varix masquerading as an orbital lymphoma. Mainly, the use of the Valsalva maneuver during the physical examination (resulting in proptosis) and during radiologic studies might have obviated the need for formal biopsy. Furthermore, orbital pain may be a presenting symptom of fluid overload in patients with a history of orbital varices.
1. Bacorn C, Gokoffski KK, Lin LK. Clinical correlation recommended: accuracy of clinician versus radiologic interpretation of the imaging of orbital lesions. Orbit. 2021;40(2):133-137. doi:10.1080/01676830.2020.1752742
2. Shams PN, Cugati S, Wells T, Huilgol S, Selva D. Orbital varix thrombosis and review of orbital vascular anomalies in blue rubber bleb nevus syndrome. Ophthalmic Plast Reconstr Surg. 2015;31(4):e82-e86. doi:10.1097/IOP.0000000000000107
3. Islam N, Mireskandari K, Rose GE. Orbital varices and orbital wall defects. Br J Ophthalmol. 2004;88(8):1092-1093.
4. Smoker WR, Gentry LR, Yee NK, Reede DL, Nerad JA. Vascular lesions of the orbit: more than meets the eye. Radiographics. 2008;28(1):185-325. doi:10.1148/rg.281075040
5. Karcioglu ZA, ed. Orbital Tumors. New York; 2005. Chap 13:133-140.
6. Chen Z, Jones H. A case of tricuspid regurgitation and congestive cardiac failure presenting with orbital pulsation. JRSM Cardiovasc Dis. 2012;1(1):cvd.2012.012005. Published 2012 Apr 5. doi:10.1258/cvd.2012.012005
1. Bacorn C, Gokoffski KK, Lin LK. Clinical correlation recommended: accuracy of clinician versus radiologic interpretation of the imaging of orbital lesions. Orbit. 2021;40(2):133-137. doi:10.1080/01676830.2020.1752742
2. Shams PN, Cugati S, Wells T, Huilgol S, Selva D. Orbital varix thrombosis and review of orbital vascular anomalies in blue rubber bleb nevus syndrome. Ophthalmic Plast Reconstr Surg. 2015;31(4):e82-e86. doi:10.1097/IOP.0000000000000107
3. Islam N, Mireskandari K, Rose GE. Orbital varices and orbital wall defects. Br J Ophthalmol. 2004;88(8):1092-1093.
4. Smoker WR, Gentry LR, Yee NK, Reede DL, Nerad JA. Vascular lesions of the orbit: more than meets the eye. Radiographics. 2008;28(1):185-325. doi:10.1148/rg.281075040
5. Karcioglu ZA, ed. Orbital Tumors. New York; 2005. Chap 13:133-140.
6. Chen Z, Jones H. A case of tricuspid regurgitation and congestive cardiac failure presenting with orbital pulsation. JRSM Cardiovasc Dis. 2012;1(1):cvd.2012.012005. Published 2012 Apr 5. doi:10.1258/cvd.2012.012005
Hemophagocytic Lymphohistiocytosis: Early Treatment Leading to an Excellent Outcome
HLH is a rare and deadly disease increasingly more present in adults, but following treatment protocol may yield favorable results.
Hemophagocytic lymphohistiocytosis (HLH) is a rare and deadly disease in which unregulated proliferation of histiocytes and T-cell infiltration takes place. It is known as a pediatric disease in which gene defects result in impaired cytotoxic NK- and T-cell function. It has been associated with autosomal recessive inheritance pattern. Without therapy, survival for these patients with active familial HLH is approximately 2 months.
Recognition of the disease has increased over the years, and as a result the diagnosis of HLH in adults also has increased. An acquired form can be triggered by viruses like Epstein-Barr virus, influenza, HIV, lymphoid malignancies, rheumatologic disorders, or immunodeficiency disorders. Survival rates for untreated HLH have been reported at < 5%.1 Despite early recognition and adequate treatment, HLH carries an overall mortality of 50% in the initial presentation, 90% die in the first 8 weeks of treatment due to uncontrolled disease.2
Case Presentation
A 56-year-old man with no active medical issues except for a remote history of non-Hodgkin lymphoma treated with chemotherapy and splenectomy in 1990 presented to the Veterans Affairs Caribbean Healthcare System in San Juan, Puerto Rico. He was admitted to the medicine ward due to community acquired pneumonia. Three days into admission his clinical status deteriorated, and the patient was transferred to the intensive care unit (ICU) due to acute respiratory failure and sepsis secondary to worsening pneumonia. Chest imaging demonstrated rapidly progressing diffuse bilateral infiltrates. Due to the severity of the chest imaging, a diagnostic bronchoscopy was performed.
The patient’s antibiotics regimen was empirically escalated to vancomycin 1500 mg IV every 12 hours and meropenem 2 g IV every 8 hours. Despite optimization of therapy, the patient did not show clinical signs of improvement. Febrile episodes persisted, pulmonary infiltrates and hypoxemia worsened, and the patient required a neuromuscular blockade. Since the bronchoscopy was nondiagnostic and deterioration persistent, the differential diagnosis was broadened. This led to the ordering of inflammatory markers. Laboratory testing showed ferritin levels > 16,000 ng/mL, pointing to HLH as a possible diagnosis. Further workup was remarkable for triglycerides of 1234 mg/dL and a fibrinogen of 0.77 g/L. In the setting of bicytopenia and persistent fever, HLH-94 regimen was started with dexamethasone 40 mg daily and etoposide 100 mg/m2. CD25 levels of 154,701 pg/mL were demonstrated as well as a decreased immunoglobulin (Ig) G levels with absent IgM and IgA. Bone marrow biopsy was consistent with hemophagocytosis. The patient eventually was extubated and sent to the oncology ward to continue chemotherapy.
Discussion
A high clinical suspicion is warranted for rapid diagnosis and treatment as HLH evolves in most cases to multiorgan failure and death. The diagnostic criteria for HLH was developed by the Histiocyte Society in 1991 and then restructured in 2004.3,4 In the first diagnostic tool developed in 1991, diagnosis was based on 5 criteria (fever, splenomegaly, bicytopenia, hypertriglyceridemia and/or hypofibrinogenemia, and hemophagocytosis). Three additional laboratory findings were also described as part of HLH diagnosis since 2004: low or absent NK-cell-activity, hyperferritinemia of > 500 ng/dL, and high-soluble interleukin-2-receptor levels (CD25) > 2400 U/mL. Overall, 5 of 8 criteria are needed for the HLH diagnosis.
Despite the common use of these diagnostic criteria, they were developed for the pediatric population but have not been validated for adult patients.5 For adult patients, the HScore was developed in 2014. It has 9 variables: 3 are based on clinical findings (known underlying immunosuppression, high temperature, and organomegaly; 5 are based on laboratory values (ferritin, serum glutamic oxaloacetic transaminase, cytopenia, triglycerides, and fibrinogen levels); the last variable uses cytologic findings in the bone marrow. In the initial study, probability of having HLH ranged from < 1% with an HScore of ≤ 90% to > 99% with an HScore of ≥ 250 in noncritically ill adults.5 A recently published retrospective study demonstrated the diagnostic reliability of both the HLH-2004 criteria and HScore in critically ill adult patients. This study concluded that the best prediction accuracy of HLH diagnosis for a cutoff of 4 fulfilled HLH-2004 criteria had a 95.0% sensitivity and 93.6% specificity and HScore cutoff of 168 reached a 100% sensitivity and 94.1% specificity.6
The early negative bronchoscopy lowered the possibility of an infection as the etiology of the clinical presentation and narrowed the hyperferritinemia differential diagnosis. Hyperferritinemia has a sensitivity and specificity of > 90% for diagnosis when above 10,000 ng/dL in the pediatric population.7 This is not the case in adults. Hyperferritinemia is a marker of different inflammatory responses, such as histoplasmosis infection, malignancy, or iron overload rather than an isolated diagnostic tool for HLH.8 It has been reported that CD25 levels less than the diagnostic threshold of 2400 U/mL have a 100% sensitivity for the diagnosis and therefore can rule out the diagnosis. When this is taken into consideration, it can be concluded that CD25 level is a better diagnostic tool when compared with ferritin, but its main limitation is its lack of widespread availability.9 Still, there is a limited number of pathologies that are associated with marked hyperferritinemia, specifically using thresholds of more than 6000 ng/dL.10 Taking into consideration the high mortality of untreated HLH, isolated hyperferritinemia still warrants HLH workup to aggressively pursue the diagnosis and improve outcomes.
The goal of therapy in HLH is prompt inactivation of the dysregulated inflammation with aggressive immunosuppression. In our deteriorating patient, the treatment was started with only 4 of the 8 HLH-2004 diagnostic criteria being met. As per the 2018 Histiocyte Society consensus statement, the decision to start the HLH-94 treatment relies on not only the HLH-2004 diagnostic criteria, but also the patient’s clinical evolution.11 In 1994 the Histiocyte Society also published a treatment protocol termed HLH-94. A Korean retrospective study demonstrated that this protocol led to a 5-year survival rate of 60 to 80% depending on the HLH trigger and response to initial treatment.12 The protocol consists of etoposide at 150 mg/m2, 2 weekly doses in the first 2 weeks and then 1 dose weekly for the next 6 weeks. Dexamethasone is the steroid of choice as it readily crosses the blood-brain barrier. Its dosage consists of 10 mg/m2 for the first 2 weeks and then it is halved every 2 weeks until the eighth week of treatment. A slow taper follows to avoid adrenal insufficiency. Once 8 weeks of treatment have been completed, cyclosporine is added to a goal trough of 200 mcg/dL. If there is central nervous system (CNS) involvement, early aggressive treatment with intrathecal methotrexate is indicated if no improvement is noted during initial therapy.11
In 2004 the Histiocyte Society restructured the HLH-94 treatment protocol with the aim of presenting a more aggressive treatment strategy. The protocol added cyclosporine to the initial induction therapy, rather than later in the ninth week as HLH-94. Neither the use of cyclosporine nor the HLH-2004 have been demonstrated to be superior to the use of etoposide and dexamethasone alone or in the HLH-94 protocol, respectively.13 Cyclosporine is associated with adverse effects (AEs) and may have many contraindications in the acute phase of the disease. Therefore, the HLH-94 protocol is still the recommended regimen.11
To assess adequate clinical response, several clinical and laboratory parameters are followed. Clinically, resolution of fever, improvement in hepatosplenomegaly, lymphadenopathy, and mental status can be useful. Laboratories can be used to assess improvement from organ specific damage such as hepatic involvement or cytopenia. The limitation of these diagnostic studies is that they could falsely suggest an inadequate response to treatment due to concomitant infection or medication AEs. Other markers such as ferritin levels, CD25, and NK cell activity levels are more specific to HLH. Out of them, a decreasing ferritin level has the needed specificity and widespread availability for repeated assessment. On the other hand, both CD25 and NK cell activity are readily available only in specialized centers. An initial high ferritin level is a marker for a poor prognosis, and the rate of decline correlates with mortality. Studies have demonstrated that persistently elevated ferritin levels after treatment initiation are associated with worse outcomes.14,15
Several salvage treatments have been identified in recalcitrant or relapsing disease. In general, chemotherapy needs to be intensified, either by returning to the initial high dosage if recurrence occurs in the weaning phase of treatment or adding other agents if no response was initially achieved. Emapalumab, an interferon γ antibody, was approved by the US Food and Drug Administration for the treatment of intractable HLH after it demonstrated that when added to dexamethasone, it lead to treatment response in 17 out of 27 pediatric patients, with a relatively safe AE profile.16 The goal of intensifying chemotherapy is to have the patient tolerate allogenic stem cell transplant, which is clinically indicated in familial HLH, malignancy induced HLH, and recalcitrant cases. In patients who undergo hematopoietic cell transplantation (HCT) there is a tendency to increase survival to 66% at 5 years.12
Conclusions
HLH is a rare and deadly disease increasingly more present in adults. Our patient who initially presented with a sepsis diagnosis was suspected of having a hematologic etiology for his clinical findings due to markedly elevated ferritin levels. In our patient, the HLH-94 treatment protocol was used, yielding favorable results. Given the lack of specific scientific data backing updated protocols such as HLH-2004 and a comparatively favorable safety profile, current guidelines still recommend using the HLH-94 treatment protocol. Decreasing ferritin levels may be used in conjunction with clinical improvement to demonstrate therapeutic response. Persistence of disease despite standard treatment may warrant novel therapies, such as emapalumab or HCT. Physicians need to be wary of an HLH diagnosis as early identification and treatment may improve its otherwise grim prognosis.
1. Chen TY, Hsu MH, Kuo HC, Sheen JM, Cheng MC, Lin YJ. Outcome analysis of pediatric hemophagocytic lymphohistiocytosis. J Formos Med Assoc. 2021;120(1, pt 1):172-179. doi:10.1016/j.jfma.2020.03.025
2. Henter JI, Samuelsson-Horne A, Aricò M, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002;100(7):2367-2373. doi:10.1182/blood-2002-01-0172
3. Henter JI, Elinder G, Ost A. Diagnostic guidelines for hemophagocytic lymphohistiocytosis. The FHL Study Group of the Histiocyte Society. Semin Oncol. 1991;18(1):29-33.
4. Henter JI, Horne A, Aricó M, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124-131. doi:10.1002/pbc.21039
5. Knaak C, Nyvlt P, Schuster FS, et al. Hemophagocytic lymphohistiocytosis in critically ill patients: diagnostic reliability of HLH-2004 criteria and HScore. Crit Care. 2020;24(1):244. Published 2020 May 24. doi:10.1186/s13054-020-02941-3
6. Fardet L, Galicier L, Lambotte O, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014;66(9):2613-2620. doi:10.1002/art.38690
7. La Rosée P, Horne A, Hines M, et al. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood. 2019;133(23):2465-2477. doi:10.1182/blood.2018894618
8. Schaffner M, Rosenstein L, Ballas Z, Suneja M. Significance of Hyperferritinemia in Hospitalized Adults. Am J Med Sci. 2017;354(2):152-158. doi:10.1016/j.amjms.2017.04.016
9. Hayden A, Lin M, Park S, et al. Soluble interleukin-2 receptor is a sensitive diagnostic test in adult HLH. Blood Adv. 2017;1(26):2529-2534. Published 2017 Dec 6. doi:10.1182/bloodadvances.2017012310
10. Belfeki N, Strazzulla A, Picque M, Diamantis S. Extreme hyperferritinemia: etiological spectrum and impact on prognosis. Reumatismo. 2020;71(4):199-202. Published 2020 Jan 28. doi:10.4081/reumatismo.2019.1221
11. Ehl S, Astigarraga I, von Bahr Greenwood T, et al. Recommendations for the use of etoposide-based therapy and bone marrow transplantation for the treatment of HLH: consensus statements by the HLH Steering Committee of the Histiocyte Society. J Allergy Clin Immunol Pract. 2018;6(5):1508-1517. doi:10.1016/j.jaip.2018.05.031
12. Yoon JH, Park SS, Jeon YW, et al. Treatment outcomes and prognostic factors in adult patients with secondary hemophagocytic lymphohistiocytosis not associated with malignancy. Haematologica. 2019;104(2):269-276. doi:10.3324/haematol.2018.198655
13. Bergsten E, Horne A, Aricó M, et al. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: long-term results of the cooperative HLH-2004 study. Blood. 2017;130(25):2728-2738. doi:10.1182/blood-2017-06-788349
14. Lin TF, Ferlic-Stark LL, Allen CE, Kozinetz CA, McClain KL. Rate of decline of ferritin in patients with hemophagocytic lymphohistiocytosis as a prognostic variable for mortality. Pediatr Blood Cancer. 2011;56(1):154-155. doi:10.1002/pbc.22774
15. Zhou J, Zhou J, Shen DT, Goyal H, Wu ZQ, Xu HG. Development and validation of the prognostic value of ferritin in adult patients with Hemophagocytic Lymphohistiocytosis. Orphanet J Rare Dis. 2020;15(1):71. Published 2020 Mar 12. doi:10.1186/s13023-020-1336-616. Locatelli F, Jordan MB, Allen CE, et al. Safety and efficacy of emapalumab in pediatric patients with primary hemophagocytic lymphohistiocytosis. Presented at: American Society of Hematology Annual Meeting, November 29, 2018. Blood. 2018;132(suppl 1):LBA-6. doi:10.1182/blood-2018-120810
HLH is a rare and deadly disease increasingly more present in adults, but following treatment protocol may yield favorable results.
HLH is a rare and deadly disease increasingly more present in adults, but following treatment protocol may yield favorable results.
Hemophagocytic lymphohistiocytosis (HLH) is a rare and deadly disease in which unregulated proliferation of histiocytes and T-cell infiltration takes place. It is known as a pediatric disease in which gene defects result in impaired cytotoxic NK- and T-cell function. It has been associated with autosomal recessive inheritance pattern. Without therapy, survival for these patients with active familial HLH is approximately 2 months.
Recognition of the disease has increased over the years, and as a result the diagnosis of HLH in adults also has increased. An acquired form can be triggered by viruses like Epstein-Barr virus, influenza, HIV, lymphoid malignancies, rheumatologic disorders, or immunodeficiency disorders. Survival rates for untreated HLH have been reported at < 5%.1 Despite early recognition and adequate treatment, HLH carries an overall mortality of 50% in the initial presentation, 90% die in the first 8 weeks of treatment due to uncontrolled disease.2
Case Presentation
A 56-year-old man with no active medical issues except for a remote history of non-Hodgkin lymphoma treated with chemotherapy and splenectomy in 1990 presented to the Veterans Affairs Caribbean Healthcare System in San Juan, Puerto Rico. He was admitted to the medicine ward due to community acquired pneumonia. Three days into admission his clinical status deteriorated, and the patient was transferred to the intensive care unit (ICU) due to acute respiratory failure and sepsis secondary to worsening pneumonia. Chest imaging demonstrated rapidly progressing diffuse bilateral infiltrates. Due to the severity of the chest imaging, a diagnostic bronchoscopy was performed.
The patient’s antibiotics regimen was empirically escalated to vancomycin 1500 mg IV every 12 hours and meropenem 2 g IV every 8 hours. Despite optimization of therapy, the patient did not show clinical signs of improvement. Febrile episodes persisted, pulmonary infiltrates and hypoxemia worsened, and the patient required a neuromuscular blockade. Since the bronchoscopy was nondiagnostic and deterioration persistent, the differential diagnosis was broadened. This led to the ordering of inflammatory markers. Laboratory testing showed ferritin levels > 16,000 ng/mL, pointing to HLH as a possible diagnosis. Further workup was remarkable for triglycerides of 1234 mg/dL and a fibrinogen of 0.77 g/L. In the setting of bicytopenia and persistent fever, HLH-94 regimen was started with dexamethasone 40 mg daily and etoposide 100 mg/m2. CD25 levels of 154,701 pg/mL were demonstrated as well as a decreased immunoglobulin (Ig) G levels with absent IgM and IgA. Bone marrow biopsy was consistent with hemophagocytosis. The patient eventually was extubated and sent to the oncology ward to continue chemotherapy.
Discussion
A high clinical suspicion is warranted for rapid diagnosis and treatment as HLH evolves in most cases to multiorgan failure and death. The diagnostic criteria for HLH was developed by the Histiocyte Society in 1991 and then restructured in 2004.3,4 In the first diagnostic tool developed in 1991, diagnosis was based on 5 criteria (fever, splenomegaly, bicytopenia, hypertriglyceridemia and/or hypofibrinogenemia, and hemophagocytosis). Three additional laboratory findings were also described as part of HLH diagnosis since 2004: low or absent NK-cell-activity, hyperferritinemia of > 500 ng/dL, and high-soluble interleukin-2-receptor levels (CD25) > 2400 U/mL. Overall, 5 of 8 criteria are needed for the HLH diagnosis.
Despite the common use of these diagnostic criteria, they were developed for the pediatric population but have not been validated for adult patients.5 For adult patients, the HScore was developed in 2014. It has 9 variables: 3 are based on clinical findings (known underlying immunosuppression, high temperature, and organomegaly; 5 are based on laboratory values (ferritin, serum glutamic oxaloacetic transaminase, cytopenia, triglycerides, and fibrinogen levels); the last variable uses cytologic findings in the bone marrow. In the initial study, probability of having HLH ranged from < 1% with an HScore of ≤ 90% to > 99% with an HScore of ≥ 250 in noncritically ill adults.5 A recently published retrospective study demonstrated the diagnostic reliability of both the HLH-2004 criteria and HScore in critically ill adult patients. This study concluded that the best prediction accuracy of HLH diagnosis for a cutoff of 4 fulfilled HLH-2004 criteria had a 95.0% sensitivity and 93.6% specificity and HScore cutoff of 168 reached a 100% sensitivity and 94.1% specificity.6
The early negative bronchoscopy lowered the possibility of an infection as the etiology of the clinical presentation and narrowed the hyperferritinemia differential diagnosis. Hyperferritinemia has a sensitivity and specificity of > 90% for diagnosis when above 10,000 ng/dL in the pediatric population.7 This is not the case in adults. Hyperferritinemia is a marker of different inflammatory responses, such as histoplasmosis infection, malignancy, or iron overload rather than an isolated diagnostic tool for HLH.8 It has been reported that CD25 levels less than the diagnostic threshold of 2400 U/mL have a 100% sensitivity for the diagnosis and therefore can rule out the diagnosis. When this is taken into consideration, it can be concluded that CD25 level is a better diagnostic tool when compared with ferritin, but its main limitation is its lack of widespread availability.9 Still, there is a limited number of pathologies that are associated with marked hyperferritinemia, specifically using thresholds of more than 6000 ng/dL.10 Taking into consideration the high mortality of untreated HLH, isolated hyperferritinemia still warrants HLH workup to aggressively pursue the diagnosis and improve outcomes.
The goal of therapy in HLH is prompt inactivation of the dysregulated inflammation with aggressive immunosuppression. In our deteriorating patient, the treatment was started with only 4 of the 8 HLH-2004 diagnostic criteria being met. As per the 2018 Histiocyte Society consensus statement, the decision to start the HLH-94 treatment relies on not only the HLH-2004 diagnostic criteria, but also the patient’s clinical evolution.11 In 1994 the Histiocyte Society also published a treatment protocol termed HLH-94. A Korean retrospective study demonstrated that this protocol led to a 5-year survival rate of 60 to 80% depending on the HLH trigger and response to initial treatment.12 The protocol consists of etoposide at 150 mg/m2, 2 weekly doses in the first 2 weeks and then 1 dose weekly for the next 6 weeks. Dexamethasone is the steroid of choice as it readily crosses the blood-brain barrier. Its dosage consists of 10 mg/m2 for the first 2 weeks and then it is halved every 2 weeks until the eighth week of treatment. A slow taper follows to avoid adrenal insufficiency. Once 8 weeks of treatment have been completed, cyclosporine is added to a goal trough of 200 mcg/dL. If there is central nervous system (CNS) involvement, early aggressive treatment with intrathecal methotrexate is indicated if no improvement is noted during initial therapy.11
In 2004 the Histiocyte Society restructured the HLH-94 treatment protocol with the aim of presenting a more aggressive treatment strategy. The protocol added cyclosporine to the initial induction therapy, rather than later in the ninth week as HLH-94. Neither the use of cyclosporine nor the HLH-2004 have been demonstrated to be superior to the use of etoposide and dexamethasone alone or in the HLH-94 protocol, respectively.13 Cyclosporine is associated with adverse effects (AEs) and may have many contraindications in the acute phase of the disease. Therefore, the HLH-94 protocol is still the recommended regimen.11
To assess adequate clinical response, several clinical and laboratory parameters are followed. Clinically, resolution of fever, improvement in hepatosplenomegaly, lymphadenopathy, and mental status can be useful. Laboratories can be used to assess improvement from organ specific damage such as hepatic involvement or cytopenia. The limitation of these diagnostic studies is that they could falsely suggest an inadequate response to treatment due to concomitant infection or medication AEs. Other markers such as ferritin levels, CD25, and NK cell activity levels are more specific to HLH. Out of them, a decreasing ferritin level has the needed specificity and widespread availability for repeated assessment. On the other hand, both CD25 and NK cell activity are readily available only in specialized centers. An initial high ferritin level is a marker for a poor prognosis, and the rate of decline correlates with mortality. Studies have demonstrated that persistently elevated ferritin levels after treatment initiation are associated with worse outcomes.14,15
Several salvage treatments have been identified in recalcitrant or relapsing disease. In general, chemotherapy needs to be intensified, either by returning to the initial high dosage if recurrence occurs in the weaning phase of treatment or adding other agents if no response was initially achieved. Emapalumab, an interferon γ antibody, was approved by the US Food and Drug Administration for the treatment of intractable HLH after it demonstrated that when added to dexamethasone, it lead to treatment response in 17 out of 27 pediatric patients, with a relatively safe AE profile.16 The goal of intensifying chemotherapy is to have the patient tolerate allogenic stem cell transplant, which is clinically indicated in familial HLH, malignancy induced HLH, and recalcitrant cases. In patients who undergo hematopoietic cell transplantation (HCT) there is a tendency to increase survival to 66% at 5 years.12
Conclusions
HLH is a rare and deadly disease increasingly more present in adults. Our patient who initially presented with a sepsis diagnosis was suspected of having a hematologic etiology for his clinical findings due to markedly elevated ferritin levels. In our patient, the HLH-94 treatment protocol was used, yielding favorable results. Given the lack of specific scientific data backing updated protocols such as HLH-2004 and a comparatively favorable safety profile, current guidelines still recommend using the HLH-94 treatment protocol. Decreasing ferritin levels may be used in conjunction with clinical improvement to demonstrate therapeutic response. Persistence of disease despite standard treatment may warrant novel therapies, such as emapalumab or HCT. Physicians need to be wary of an HLH diagnosis as early identification and treatment may improve its otherwise grim prognosis.
Hemophagocytic lymphohistiocytosis (HLH) is a rare and deadly disease in which unregulated proliferation of histiocytes and T-cell infiltration takes place. It is known as a pediatric disease in which gene defects result in impaired cytotoxic NK- and T-cell function. It has been associated with autosomal recessive inheritance pattern. Without therapy, survival for these patients with active familial HLH is approximately 2 months.
Recognition of the disease has increased over the years, and as a result the diagnosis of HLH in adults also has increased. An acquired form can be triggered by viruses like Epstein-Barr virus, influenza, HIV, lymphoid malignancies, rheumatologic disorders, or immunodeficiency disorders. Survival rates for untreated HLH have been reported at < 5%.1 Despite early recognition and adequate treatment, HLH carries an overall mortality of 50% in the initial presentation, 90% die in the first 8 weeks of treatment due to uncontrolled disease.2
Case Presentation
A 56-year-old man with no active medical issues except for a remote history of non-Hodgkin lymphoma treated with chemotherapy and splenectomy in 1990 presented to the Veterans Affairs Caribbean Healthcare System in San Juan, Puerto Rico. He was admitted to the medicine ward due to community acquired pneumonia. Three days into admission his clinical status deteriorated, and the patient was transferred to the intensive care unit (ICU) due to acute respiratory failure and sepsis secondary to worsening pneumonia. Chest imaging demonstrated rapidly progressing diffuse bilateral infiltrates. Due to the severity of the chest imaging, a diagnostic bronchoscopy was performed.
The patient’s antibiotics regimen was empirically escalated to vancomycin 1500 mg IV every 12 hours and meropenem 2 g IV every 8 hours. Despite optimization of therapy, the patient did not show clinical signs of improvement. Febrile episodes persisted, pulmonary infiltrates and hypoxemia worsened, and the patient required a neuromuscular blockade. Since the bronchoscopy was nondiagnostic and deterioration persistent, the differential diagnosis was broadened. This led to the ordering of inflammatory markers. Laboratory testing showed ferritin levels > 16,000 ng/mL, pointing to HLH as a possible diagnosis. Further workup was remarkable for triglycerides of 1234 mg/dL and a fibrinogen of 0.77 g/L. In the setting of bicytopenia and persistent fever, HLH-94 regimen was started with dexamethasone 40 mg daily and etoposide 100 mg/m2. CD25 levels of 154,701 pg/mL were demonstrated as well as a decreased immunoglobulin (Ig) G levels with absent IgM and IgA. Bone marrow biopsy was consistent with hemophagocytosis. The patient eventually was extubated and sent to the oncology ward to continue chemotherapy.
Discussion
A high clinical suspicion is warranted for rapid diagnosis and treatment as HLH evolves in most cases to multiorgan failure and death. The diagnostic criteria for HLH was developed by the Histiocyte Society in 1991 and then restructured in 2004.3,4 In the first diagnostic tool developed in 1991, diagnosis was based on 5 criteria (fever, splenomegaly, bicytopenia, hypertriglyceridemia and/or hypofibrinogenemia, and hemophagocytosis). Three additional laboratory findings were also described as part of HLH diagnosis since 2004: low or absent NK-cell-activity, hyperferritinemia of > 500 ng/dL, and high-soluble interleukin-2-receptor levels (CD25) > 2400 U/mL. Overall, 5 of 8 criteria are needed for the HLH diagnosis.
Despite the common use of these diagnostic criteria, they were developed for the pediatric population but have not been validated for adult patients.5 For adult patients, the HScore was developed in 2014. It has 9 variables: 3 are based on clinical findings (known underlying immunosuppression, high temperature, and organomegaly; 5 are based on laboratory values (ferritin, serum glutamic oxaloacetic transaminase, cytopenia, triglycerides, and fibrinogen levels); the last variable uses cytologic findings in the bone marrow. In the initial study, probability of having HLH ranged from < 1% with an HScore of ≤ 90% to > 99% with an HScore of ≥ 250 in noncritically ill adults.5 A recently published retrospective study demonstrated the diagnostic reliability of both the HLH-2004 criteria and HScore in critically ill adult patients. This study concluded that the best prediction accuracy of HLH diagnosis for a cutoff of 4 fulfilled HLH-2004 criteria had a 95.0% sensitivity and 93.6% specificity and HScore cutoff of 168 reached a 100% sensitivity and 94.1% specificity.6
The early negative bronchoscopy lowered the possibility of an infection as the etiology of the clinical presentation and narrowed the hyperferritinemia differential diagnosis. Hyperferritinemia has a sensitivity and specificity of > 90% for diagnosis when above 10,000 ng/dL in the pediatric population.7 This is not the case in adults. Hyperferritinemia is a marker of different inflammatory responses, such as histoplasmosis infection, malignancy, or iron overload rather than an isolated diagnostic tool for HLH.8 It has been reported that CD25 levels less than the diagnostic threshold of 2400 U/mL have a 100% sensitivity for the diagnosis and therefore can rule out the diagnosis. When this is taken into consideration, it can be concluded that CD25 level is a better diagnostic tool when compared with ferritin, but its main limitation is its lack of widespread availability.9 Still, there is a limited number of pathologies that are associated with marked hyperferritinemia, specifically using thresholds of more than 6000 ng/dL.10 Taking into consideration the high mortality of untreated HLH, isolated hyperferritinemia still warrants HLH workup to aggressively pursue the diagnosis and improve outcomes.
The goal of therapy in HLH is prompt inactivation of the dysregulated inflammation with aggressive immunosuppression. In our deteriorating patient, the treatment was started with only 4 of the 8 HLH-2004 diagnostic criteria being met. As per the 2018 Histiocyte Society consensus statement, the decision to start the HLH-94 treatment relies on not only the HLH-2004 diagnostic criteria, but also the patient’s clinical evolution.11 In 1994 the Histiocyte Society also published a treatment protocol termed HLH-94. A Korean retrospective study demonstrated that this protocol led to a 5-year survival rate of 60 to 80% depending on the HLH trigger and response to initial treatment.12 The protocol consists of etoposide at 150 mg/m2, 2 weekly doses in the first 2 weeks and then 1 dose weekly for the next 6 weeks. Dexamethasone is the steroid of choice as it readily crosses the blood-brain barrier. Its dosage consists of 10 mg/m2 for the first 2 weeks and then it is halved every 2 weeks until the eighth week of treatment. A slow taper follows to avoid adrenal insufficiency. Once 8 weeks of treatment have been completed, cyclosporine is added to a goal trough of 200 mcg/dL. If there is central nervous system (CNS) involvement, early aggressive treatment with intrathecal methotrexate is indicated if no improvement is noted during initial therapy.11
In 2004 the Histiocyte Society restructured the HLH-94 treatment protocol with the aim of presenting a more aggressive treatment strategy. The protocol added cyclosporine to the initial induction therapy, rather than later in the ninth week as HLH-94. Neither the use of cyclosporine nor the HLH-2004 have been demonstrated to be superior to the use of etoposide and dexamethasone alone or in the HLH-94 protocol, respectively.13 Cyclosporine is associated with adverse effects (AEs) and may have many contraindications in the acute phase of the disease. Therefore, the HLH-94 protocol is still the recommended regimen.11
To assess adequate clinical response, several clinical and laboratory parameters are followed. Clinically, resolution of fever, improvement in hepatosplenomegaly, lymphadenopathy, and mental status can be useful. Laboratories can be used to assess improvement from organ specific damage such as hepatic involvement or cytopenia. The limitation of these diagnostic studies is that they could falsely suggest an inadequate response to treatment due to concomitant infection or medication AEs. Other markers such as ferritin levels, CD25, and NK cell activity levels are more specific to HLH. Out of them, a decreasing ferritin level has the needed specificity and widespread availability for repeated assessment. On the other hand, both CD25 and NK cell activity are readily available only in specialized centers. An initial high ferritin level is a marker for a poor prognosis, and the rate of decline correlates with mortality. Studies have demonstrated that persistently elevated ferritin levels after treatment initiation are associated with worse outcomes.14,15
Several salvage treatments have been identified in recalcitrant or relapsing disease. In general, chemotherapy needs to be intensified, either by returning to the initial high dosage if recurrence occurs in the weaning phase of treatment or adding other agents if no response was initially achieved. Emapalumab, an interferon γ antibody, was approved by the US Food and Drug Administration for the treatment of intractable HLH after it demonstrated that when added to dexamethasone, it lead to treatment response in 17 out of 27 pediatric patients, with a relatively safe AE profile.16 The goal of intensifying chemotherapy is to have the patient tolerate allogenic stem cell transplant, which is clinically indicated in familial HLH, malignancy induced HLH, and recalcitrant cases. In patients who undergo hematopoietic cell transplantation (HCT) there is a tendency to increase survival to 66% at 5 years.12
Conclusions
HLH is a rare and deadly disease increasingly more present in adults. Our patient who initially presented with a sepsis diagnosis was suspected of having a hematologic etiology for his clinical findings due to markedly elevated ferritin levels. In our patient, the HLH-94 treatment protocol was used, yielding favorable results. Given the lack of specific scientific data backing updated protocols such as HLH-2004 and a comparatively favorable safety profile, current guidelines still recommend using the HLH-94 treatment protocol. Decreasing ferritin levels may be used in conjunction with clinical improvement to demonstrate therapeutic response. Persistence of disease despite standard treatment may warrant novel therapies, such as emapalumab or HCT. Physicians need to be wary of an HLH diagnosis as early identification and treatment may improve its otherwise grim prognosis.
1. Chen TY, Hsu MH, Kuo HC, Sheen JM, Cheng MC, Lin YJ. Outcome analysis of pediatric hemophagocytic lymphohistiocytosis. J Formos Med Assoc. 2021;120(1, pt 1):172-179. doi:10.1016/j.jfma.2020.03.025
2. Henter JI, Samuelsson-Horne A, Aricò M, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002;100(7):2367-2373. doi:10.1182/blood-2002-01-0172
3. Henter JI, Elinder G, Ost A. Diagnostic guidelines for hemophagocytic lymphohistiocytosis. The FHL Study Group of the Histiocyte Society. Semin Oncol. 1991;18(1):29-33.
4. Henter JI, Horne A, Aricó M, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124-131. doi:10.1002/pbc.21039
5. Knaak C, Nyvlt P, Schuster FS, et al. Hemophagocytic lymphohistiocytosis in critically ill patients: diagnostic reliability of HLH-2004 criteria and HScore. Crit Care. 2020;24(1):244. Published 2020 May 24. doi:10.1186/s13054-020-02941-3
6. Fardet L, Galicier L, Lambotte O, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014;66(9):2613-2620. doi:10.1002/art.38690
7. La Rosée P, Horne A, Hines M, et al. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood. 2019;133(23):2465-2477. doi:10.1182/blood.2018894618
8. Schaffner M, Rosenstein L, Ballas Z, Suneja M. Significance of Hyperferritinemia in Hospitalized Adults. Am J Med Sci. 2017;354(2):152-158. doi:10.1016/j.amjms.2017.04.016
9. Hayden A, Lin M, Park S, et al. Soluble interleukin-2 receptor is a sensitive diagnostic test in adult HLH. Blood Adv. 2017;1(26):2529-2534. Published 2017 Dec 6. doi:10.1182/bloodadvances.2017012310
10. Belfeki N, Strazzulla A, Picque M, Diamantis S. Extreme hyperferritinemia: etiological spectrum and impact on prognosis. Reumatismo. 2020;71(4):199-202. Published 2020 Jan 28. doi:10.4081/reumatismo.2019.1221
11. Ehl S, Astigarraga I, von Bahr Greenwood T, et al. Recommendations for the use of etoposide-based therapy and bone marrow transplantation for the treatment of HLH: consensus statements by the HLH Steering Committee of the Histiocyte Society. J Allergy Clin Immunol Pract. 2018;6(5):1508-1517. doi:10.1016/j.jaip.2018.05.031
12. Yoon JH, Park SS, Jeon YW, et al. Treatment outcomes and prognostic factors in adult patients with secondary hemophagocytic lymphohistiocytosis not associated with malignancy. Haematologica. 2019;104(2):269-276. doi:10.3324/haematol.2018.198655
13. Bergsten E, Horne A, Aricó M, et al. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: long-term results of the cooperative HLH-2004 study. Blood. 2017;130(25):2728-2738. doi:10.1182/blood-2017-06-788349
14. Lin TF, Ferlic-Stark LL, Allen CE, Kozinetz CA, McClain KL. Rate of decline of ferritin in patients with hemophagocytic lymphohistiocytosis as a prognostic variable for mortality. Pediatr Blood Cancer. 2011;56(1):154-155. doi:10.1002/pbc.22774
15. Zhou J, Zhou J, Shen DT, Goyal H, Wu ZQ, Xu HG. Development and validation of the prognostic value of ferritin in adult patients with Hemophagocytic Lymphohistiocytosis. Orphanet J Rare Dis. 2020;15(1):71. Published 2020 Mar 12. doi:10.1186/s13023-020-1336-616. Locatelli F, Jordan MB, Allen CE, et al. Safety and efficacy of emapalumab in pediatric patients with primary hemophagocytic lymphohistiocytosis. Presented at: American Society of Hematology Annual Meeting, November 29, 2018. Blood. 2018;132(suppl 1):LBA-6. doi:10.1182/blood-2018-120810
1. Chen TY, Hsu MH, Kuo HC, Sheen JM, Cheng MC, Lin YJ. Outcome analysis of pediatric hemophagocytic lymphohistiocytosis. J Formos Med Assoc. 2021;120(1, pt 1):172-179. doi:10.1016/j.jfma.2020.03.025
2. Henter JI, Samuelsson-Horne A, Aricò M, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002;100(7):2367-2373. doi:10.1182/blood-2002-01-0172
3. Henter JI, Elinder G, Ost A. Diagnostic guidelines for hemophagocytic lymphohistiocytosis. The FHL Study Group of the Histiocyte Society. Semin Oncol. 1991;18(1):29-33.
4. Henter JI, Horne A, Aricó M, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124-131. doi:10.1002/pbc.21039
5. Knaak C, Nyvlt P, Schuster FS, et al. Hemophagocytic lymphohistiocytosis in critically ill patients: diagnostic reliability of HLH-2004 criteria and HScore. Crit Care. 2020;24(1):244. Published 2020 May 24. doi:10.1186/s13054-020-02941-3
6. Fardet L, Galicier L, Lambotte O, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014;66(9):2613-2620. doi:10.1002/art.38690
7. La Rosée P, Horne A, Hines M, et al. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood. 2019;133(23):2465-2477. doi:10.1182/blood.2018894618
8. Schaffner M, Rosenstein L, Ballas Z, Suneja M. Significance of Hyperferritinemia in Hospitalized Adults. Am J Med Sci. 2017;354(2):152-158. doi:10.1016/j.amjms.2017.04.016
9. Hayden A, Lin M, Park S, et al. Soluble interleukin-2 receptor is a sensitive diagnostic test in adult HLH. Blood Adv. 2017;1(26):2529-2534. Published 2017 Dec 6. doi:10.1182/bloodadvances.2017012310
10. Belfeki N, Strazzulla A, Picque M, Diamantis S. Extreme hyperferritinemia: etiological spectrum and impact on prognosis. Reumatismo. 2020;71(4):199-202. Published 2020 Jan 28. doi:10.4081/reumatismo.2019.1221
11. Ehl S, Astigarraga I, von Bahr Greenwood T, et al. Recommendations for the use of etoposide-based therapy and bone marrow transplantation for the treatment of HLH: consensus statements by the HLH Steering Committee of the Histiocyte Society. J Allergy Clin Immunol Pract. 2018;6(5):1508-1517. doi:10.1016/j.jaip.2018.05.031
12. Yoon JH, Park SS, Jeon YW, et al. Treatment outcomes and prognostic factors in adult patients with secondary hemophagocytic lymphohistiocytosis not associated with malignancy. Haematologica. 2019;104(2):269-276. doi:10.3324/haematol.2018.198655
13. Bergsten E, Horne A, Aricó M, et al. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: long-term results of the cooperative HLH-2004 study. Blood. 2017;130(25):2728-2738. doi:10.1182/blood-2017-06-788349
14. Lin TF, Ferlic-Stark LL, Allen CE, Kozinetz CA, McClain KL. Rate of decline of ferritin in patients with hemophagocytic lymphohistiocytosis as a prognostic variable for mortality. Pediatr Blood Cancer. 2011;56(1):154-155. doi:10.1002/pbc.22774
15. Zhou J, Zhou J, Shen DT, Goyal H, Wu ZQ, Xu HG. Development and validation of the prognostic value of ferritin in adult patients with Hemophagocytic Lymphohistiocytosis. Orphanet J Rare Dis. 2020;15(1):71. Published 2020 Mar 12. doi:10.1186/s13023-020-1336-616. Locatelli F, Jordan MB, Allen CE, et al. Safety and efficacy of emapalumab in pediatric patients with primary hemophagocytic lymphohistiocytosis. Presented at: American Society of Hematology Annual Meeting, November 29, 2018. Blood. 2018;132(suppl 1):LBA-6. doi:10.1182/blood-2018-120810
Three Primary Cancers in a Veteran With Agent Orange and Agent Blue Exposures
A Vietnam War veteran’s exposures likely contributed to his cancer diagnoses, but these associations are confounded by his substance use, particularly cigarette smoking.
Known as the “6 rainbow herbicides,” based on their identifying color on storage containers, the United States widely deployed the herbicides agents orange, green, pink, purple, white, and blue during the Vietnam War to deny the enemy cover and destroy crops.1 Unfortunately, all these herbicides were found to have contained some form of carcinogen. Agent Blue’s active ingredient consisted of sodium cacodylate trihydrate (C2H6AsNaO2), a compound that is metabolized into the organic form of the carcinogen arsenic before eventually converting into its relatively less toxic inorganic form.2 Agent Orange’s defoliating agent is 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). All rainbow herbicides except Agent Blue were unintentionally contaminated with carcinogenic dioxins. Agent Blue contained the carcinogen cacodylic acid, an organoarsenic acid. Today, herbicides no longer contain polychlorinated dibenzo-p-dioxins such as TCDD or arsenic due to strict manufacturing restrictions.2,3 In the treatment of veteran populations, knowledge of the 6 rainbow herbicides’ carcinogenic potential is important.
Between 1962 and 1971, the United States sprayed more than 45 million liters of Agent Orange on Vietnam and at least 366 kg of TCDD on South Vietnam.1,4 However, because Agent Orange was not a known carcinogen during the Vietnam War, records of exposure are poor. Additionally, individuals in Vietnam during this period were not the only ones exposed to this carcinogen as Agent Orange also was sprayed in Thailand and Korea.5 Even today there are still locations in Vietnam where Agent Orange concentrations exceed internationally acceptable levels. The Da Nang, Bien Hoa, and Phu Cat airports in Vietnam have been found to have dioxin levels exceeding 1000 ppt (parts of dioxin per trillion parts of lipid) toxicity equivalence in the soil. Although the Vietnam government is working toward decontaminating these and many other dioxin hotspots, residents in these locations are exposed to higher than internationally acceptable levels of dioxin.6
Despite receiving less media attention, Vietnam War veterans and Vietnamese soldiers and civilians were exposed to significant amounts of arsenic-based Agent Blue. Arsenic is a compound which has no environmental half-life and is carcinogenic humans if inhaled or ingested.2 Between 1962 and 1971, the United States distributed 7.8 million liters of Agent Blue containing 1,232,400 kg of arsenic across 300,000 hectares of rice paddies, 100,000 hectares of forest, and perimeters of all military bases during the Vietnam War.2,5 According to a review by Saha and colleagues, lower levels of arsenic exposure are associated with acute and chronic diseases, including cancers, of all organ systems.7
The following case presentation involves a Vietnam War veteran aged 70 years who was exposed to Agent Orange and developed 3 primary cancers, including cutaneous large B-cell non-Hodgkin lymphoma (NHL), high-grade urothelial carcinoma, and anal carcinoma in situ. Epidemiologically, this is an uncommon occurrence as only 8% of cancer survivors in the United States have been diagnosed with > 1 cancer.8
With no family history of cancer, the development of multiple malignancies raises concern for a history of toxin exposure. This report of a Vietnam War veteran with multiple conditions found to be associated with Agent Orange exposure provides an opportunity to discuss the role this exposure may have on the development of a comprehensive list of medical conditions as described by the literature. Additionally, the potential contributions of other confounding toxin exposures such as cigarette smoking, excessive alcohol use, and potential Agent Blue exposure on our patients’ health will be discussed.
Case Presentation
A male aged 70 years with Stage IV primary cutaneous large B-cell NHL, incompletely resected high-grade urothelial cancer, carcinoma in situ of the anal canal, and peripheral arterial disease (PAD) presented to the primary care clinic at the Washington DC Veterans Affairs Medical Center (DCVAMC) with concern for left leg ischemia. He also reported 2 large telangiectasias on his back for 6 months accompanied by lymphadenopathy and intermittent night sweats.
He was last seen at the DCVAMC 15 months prior after his twelfth dose of rituximab treatment for NHL. However, the patient failed to return for completion of his treatment due to frustration with the lengthy chemotherapy and follow-up process. Additionally, the patient's history included 3 failed arterial stents with complete nonadherence to the prescribed clopidogrel, resulting in the failure of 3 more subsequent graft placements. On presentation, the patient continued to report nonadherence with the clopidogrel.
The patient’s medical history included coronary artery disease (CAD) status after 2 stents in the left anterior descending artery and 1 stent in the proximal circumflex artery placed 4 years prior. He also had a history of hypertension, type 2 diabetes mellitus (T2DM), amyloid light-chain (AL) amyloidosis, aortic aneurysm, cataracts, obesity, treated hepatitis B and C, and posttraumatic stress disorder. He had no family history of cancer or AL amyloidosis; however, he noted that he was estranged from his family.
His social history was notable for active cigarette smoking up to 3 packs per day for 40 years and consuming large quantities of alcohol—at one point as many as 20 beers per day over a period of 4.5 years. He had a distant history of cocaine use but no current use, which was supported with negative urinary toxicology screens for illicit drugs over the past year.
Our patient also reported a history of Agent Orange exposure. As an artilleryman in the US Army III Corps, he was deployed for about 1 year in the most heavily sprayed regions of Vietnam, including Bien Hua, Long Binh, Xuan Loc, and Camp Zion for about 2 to 4 months at each location.
Hospital Course
The patient was treated on an inpatient basis for expedited workup and treatment for his urothelial carcinoma, NHL, and ischemic limb. His urothelial carcinoma was successfully resected, and the telangiectasias on his back were biopsied and found to be consistent with his known cutaneous large B-cell NHL, for which plans to resume outpatient chemotherapy were made. The patient’s 3 arterial grafts in his left leg were confirmed to have failed, and the patient was counseled that he would soon likely require an amputation of his ischemic leg.
Discussion
We must rely on our patient’s historical recall as there are no widely available laboratory tests or physical examination findings to confirm and/or determine the magnitude of TCDD or arsenic exposure.9-11
Exposures
The patient was stationed in Bien Hoa, the second highest dioxin-contaminated air base in Vietnam (Figure).6 Dioxin also is known to be a particularly persistent environmental pollutant, such that in January 2018, Bien Hoa was found to still have dioxin levels higher than what is considered internationally acceptable. In fact, these levels were deemed significant enough to lead the United States and Vietnamese government to sign a memorandum of intent to begin cleanup of this airport.6 TCDD is known to have a half-life of about 7.6 years, and its long half-life is mainly attributed to its slow elimination process from its stores within the liver and fat, consisting of passive excretion through the gut wall and slow metabolism by the liver.12,13 Thus, as an artilleryman mainly operating 105 howitzers within the foliage of Vietnam, our patient was exposed not only to high levels of this persistent environmental pollutant on a daily basis, but this toxin likely remained within his system for many years after his return from Vietnam.
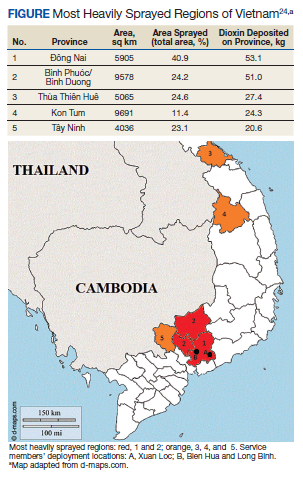
Our patient also had a convincing history for potential Agent Blue exposure through both inhalation and ingestion of contaminated food and water. Additionally, his description of deforestations occurring within a matter of days increased the level of suspicion for Agent Blue exposure. This is because Agent Blue was the herbicide of choice for missions requiring rapid deforestation, achieving defoliation as quickly as 1 to 2 days.14 Additionally, our patient was stationed within cities in southern Vietnam near Agent Blue hot spots, such as Da Nang and Saigon, and Agent Blue was sprayed along the perimeter of all military bases.2
Levels of Evidence
Using the Veterans and Agent Orange Update in 2018 as our guide, we reviewed the quality of evidence suggesting an association between many of our patient’s comorbidities to Agent Orange exposure.5 This publication categorizes the level of evidence for association between health conditions and Agent Orange exposure in 4 main categories (Table 1).
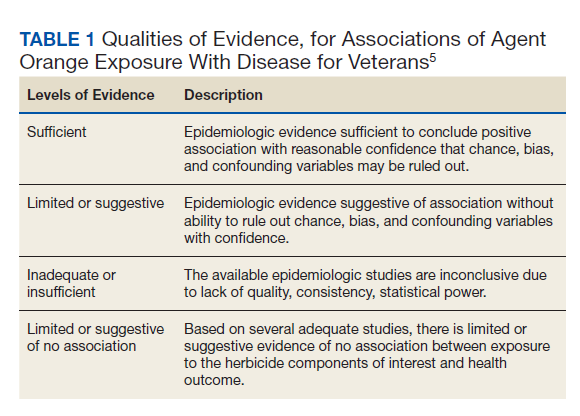
In the Veterans and Agent Orange Update, NHL notably has a sufficient level of evidence of association with Agent Orange exposure.5 Although our patient’s extensive history of polysubstance use confounds the effect Agent Orange may have had on his health, cutaneous large B-cell NHL is an interesting exception as literature does not support even a correlative link between smoking and excessive alcohol use with primary cutaneous large B-cell NHL. Several case-control studies have found little to no association with cigarette smoking and the large B-cell subtype of NHL.15,16 Moreover, several studies have found that moderate- to-heavy alcohol use, especially beer, may have a protective effect against the development of NHL.17 Of note, our patient’s alcoholic beverage of choice was beer. Regarding our patient’s distant history of cocaine use, it has been reported that cocaine use, in the absence of an HIV infection, has not been found to increase the risk of developing NHL.18 Similarly, arsenic exposure has not been associated with NHL in the literature.19,20
The 2018 update also upgraded bladder carcinoma from having inadequate or insufficient to a limited or suggestive level of evidence for association.5 However, our patient’s most significant risk factor for bladder cancer was smoking, with a meta-analysis of 430,000 patients reporting a risk ratio (RR) of 3.14 for current cigarette smokers.21 The patient’s arsenic exposure from Agent Blue also increased his risk of developing bladder cancer. Several studies suggest a strong association between environmental arsenic exposure and bladder cancer.22-26 A 30-year meta-analysis of 40 studies by Saint-Jacques and colleagues reported that the incidence of bladder cancer was found to increase in a dose-dependent manner, with higher concentrations of arsenic contaminated wate, with incidence rising from 2.7 to 5.8 times as the amount of arsenic contamination water increased from 10 to 150 mg/L.
Our patient’s history is concerning for higher than average Agent Blue exposure compared with that of most Vietnam War veterans. Given the dose-dependent effect of arsenic on bladder cancer risk, both our patient’s history of smoking and Agent Blue exposure are risk factors in the development of his bladder cancer.22 These likely played a more significant role in his development of bladder cancer than did his Agent Orange exposure.
Finally, smoking is the most significant risk factor in our patient’s development of anal carcinoma in situ. The 2018 Agent Orange update does report limited/suggested evidence of no association between Agent Orange and anal carcinoma.5 It also is unknown whether Agent Blue exposure is a contributing cause to his development of anal carcinoma in situ.27 However, current smokers are at significant risk of developing anal cancer independent of age.28-30 Given our patient’s extensive smoking history, this is the most likely contributing factor.
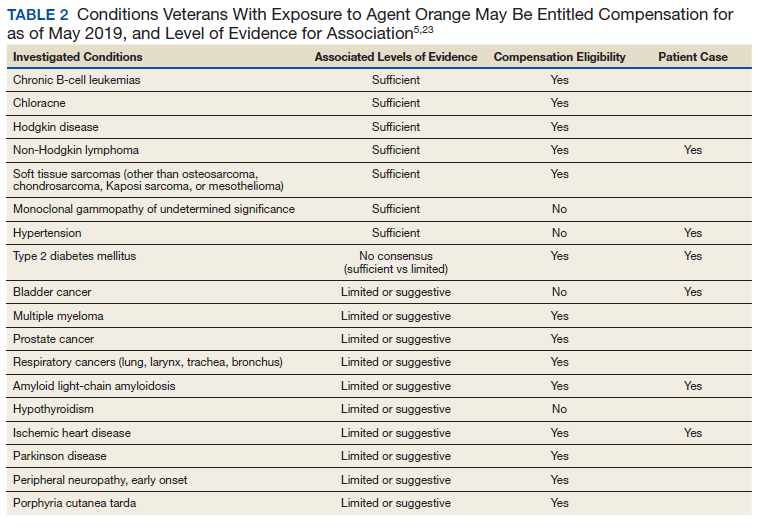
Our patient also had several noncancer-related comorbidities with correlative associations with Agent Orange exposure of varying degrees (Table 2). Somewhat surprising, the development of our patient’s hypertension and T2DM may be associated in some way with his history of Agent Orange exposure. Hypertension had been recategorized from having limited or suggestive evidence to sufficient evidence in this committee’s most recent publication, and the committee is undecided on whether T2DM has a sufficient vs limited level of evidence for association with Agent Orange exposure.5 On the other hand, the committee continues to classify both ischemic heart disease and AL amyloidosis as having a limited or suggestive level of evidence that links Agent Orange exposure to these conditions.5
Arsenic may be another risk factor for our patient’s development of CAD and arterial insufficiency. Arsenic exposure is theorized to cause a direct toxic effect on coronary arteries, and arsenic exposure has been linked to PAD, CAD, and hypertension.31-34 Other significant and compelling risk factors for cardiovascular disease in our patient included his extensive history of heavy cigarette smoking, poorly controlled T2DM, obesity, and hypertension.35-37 AL amyloidosis is a rare disorder with an incidence of only 9 to 14 cases per million person-years.38,39 This disorder has not been linked to smoking or arsenic exposure in the literature. As our patient does not have a history of plasma dyscrasias or a family history of AL amyloidosis, the only known risk factors for AL amyloidosis that apply to our patient included NHL and Agent Orange exposure—NHL being a condition that is noted to be strongly correlated with Agent Orange exposure as discussed previously.5,36,40,41
Conclusions
This case describes a Vietnam War veteran with significant exposure to rainbow herbicides and considerable polysubstance who developed 3 primary cancers and several chronic medical conditions. His exposure to Agents Orange and Blue likely contributed to his medical problems, but these associations are confounded by his substance use, particularly cigarette smoking. Of all his comorbidities, our patient’s NHL is the condition most likely to be associated with his history of Agent Orange exposure. His Agent Blue exposure also increased his risk for developing bladder cancer, cardiovascular disease, and PAD.
This case also highlights the importance of evaluating Vietnam War veterans for rainbow herbicide exposure and the complexity associated with attributing diseases to these exposures. All veterans who served in the inland waterways of Vietnam between 1962 and 1975; in the Korean Demilitarized Zone between April 1, 1968 and August 31, 1971; or in Thailand between February 28, 1961 and May 7, 1975 were at risk of rainbow herbicide exposure. These veterans may not only be eligible for disability compensation but also should be screened for associated comorbidities as outlined by current research.42 We hope that this report will serve as an aid in achieving this mission.
1. Stellman JM, Stellman SD, Christian R, Weber T, Tomasallo C. The extent and patterns of usage of Agent Orange and other herbicides in Vietnam. Nature. 2003;422(6933):681-687. doi:10.1038/nature01537
2. Olson K, Cihacek L. The fate of Agent Blue, the arsenic based herbicide, used in South Vietnam during the Vietnam War. Open J Soil Sci. 2020;10:518-577. doi:10.4236/ojss.2020.1011027
3. Lee Chang A, Dym AA, Venegas-Borsellino C, et al. Comparison between simulation-based training and lecture-based education in teaching situation awareness. a randomized controlled study. Ann Am Thorac Soc. 2017;14(4):529-535. doi:10.1513/AnnalsATS.201612-950OC
4. Stellman SD. Agent Orange during the Vietnam War: the lingering issue of its civilian and military health impact. Am J Public Health. 2018;108(6):726-728. doi:10.2105/AJPH.2018.304426
5. National Academies of Sciences, Engineering, and Medicine. Veterans and Agent Orange: Update 11 (2018). The National Academies Press; 2018. doi:10.17226/25137
6. Martin MF. US Agent Orange/dioxin assistance to Vietnam. Updated January 15, 2021. Accessed June 17, 2021. https://fas.org/sgp/crs/row/R44268.pdf
7. Saha JC, Dikshit AK, Bandyopadhyay M, Saha KC. A review of arsenic poisoning and its effects on human health. Crit Rev Environ Sci Technol. 1999;29(3):281-313. doi:10.1080/10643389991259227
8. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7-34. doi:10.3322/caac.21551
9. American Cancer Society. Agent Orange and cancer risk. Updated June 9, 2020. Accessed June 17, 2021. https://www.cancer.org/cancer/cancer-causes/agent-orange-and-cancer.html
10. US Department of Veterans Affairs. Veterans’ diseases associated with Agent Orange. Updated June 16, 2021. Accessed June 17, 2021. https://www.publichealth.va.gov/exposures/agentorange/conditions/index.asp
11. Katz SA. On the use of hair analysis for assessing arsenic intoxication. Int J Environ Res Public Health. 2019;16(6):977. Published 2019 Mar 18. doi:10.3390/ijerph16060977
12. Chang ET, Boffetta P, Adami HO, Mandel JS. A critical review of the epidemiology of Agent Orange or 2,3,7,8-tetrachlorodibenzo-p-dioxin and lymphoid malignancies. Ann Epidemiol. 2015;25(4):275-292.e30. doi:10.1016/j.annepidem.2015.01.002
13. Kramárová E, Kogevinas M, Anh CT, et al. Exposure to Agent Orange and occurrence of soft-tissue sarcomas or non-Hodgkin lymphomas: an ongoing study in Vietnam. Environ Health Perspect. 1998;106 Suppl 2(suppl 2):671-678. doi:10.1289/ehp.106-1533419
14. Institute of Medicine (US) Committee to Review the Health Effects in Vietnam Veterans of Exposure to Herbicides. Veterans and Agent Orange: Health Effects of Herbicides Used in Vietnam. National Academies Press US; 1994.
15. Morton LM, Hartge P, Holford TR, et al. Cigarette smoking and risk of non-Hodgkin lymphoma: a pooled analysis from the International Lymphoma Epidemiology Consortium (interlymph). Cancer Epidemiol Biomarkers Prev. 2005;14(4):925-933. doi:10.1158/1055-9965.EPI-04-0693
16. Schöllkopf C, Smedby KE, Hjalgrim H, et al. Cigarette smoking and risk of non-Hodgkin’s lymphoma--a population-based case-control study. Cancer Epidemiol Biomarkers Prev. 2005;14(7):1791-1796. doi:10.1158/1055-9965.EPI-05-0077
17. Psaltopoulou T, Sergentanis TN, Ntanasis-Stathopoulos I, Tzanninis IG, Tsilimigras DI, Dimopoulos MA. Alcohol consumption and risk of hematological malignancies: a meta-analysis of prospective studies. Int J Cancer. 2018;143(3):486-495. doi:10.1002/ijc.31330
18. Aujla AS, Lee SH. Association between cocaine use and hematological malignancies. J Clin Oncol. 2016;34(15_suppl):e19072-e19072. doi:10.1200/JCO.2016.34.15_suppl.e19072
19. Mao Y, Hu J, Ugnat AM, White K. Non-Hodgkin’s lymphoma and occupational exposure to chemicals in Canada. Canadian Cancer Registries Epidemiology Research Group. Ann Oncol. 2000;11 (suppl 1):69-73. doi:10.1093/annonc/11.suppl_1.S69
20. Kelekci KH, Bilgin I, Ermete M. Arsenical keratoses and non-Hodgkin’s lymphoma: arsenic-induced or coincidental conditions? J Pakistan Assoc Dermatol. 2012;22(4):366-369.
21. Antoni S, Ferlay J, Soerjomataram I, Znaor A, Jemal A, Bray F. Bladder cancer incidence and mortality: a global overview and recent trends. Eur Urol. 2017;71(1):96-108. doi:10.1016/j.eururo.2016.06.010
22. Saint-Jacques N, Parker L, Brown P, Dummer TJ. Arsenic in drinking water and urinary tract cancers: a systematic review of 30 years of epidemiological evidence. Environ Health. 2014;13:44. Published 2014 Jun 2. doi:10.1186/1476-069X-13-44
23. Radosavljevic′ V, Jakovljevic′ B. Arsenic and bladder cancer: observations and suggestions. J Environ Health. 2008;71(3):40-42.
24. Marsit CJ, Karagas MR, Schned A, Kelsey KT. Carcinogen exposure and epigenetic silencing in bladder cancer. Ann N Y Acad Sci. 2006;1076(1):810-821. doi:10.1196/annals.1371.031
25. Mendez WM Jr, Eftim S, Cohen J, et al. Relationships between arsenic concentrations in drinking water and lung and bladder cancer incidence in U.S. counties. J Expo Sci Environ Epidemiol. 2017;27(3):235-243. doi:10.1038/jes.2016.58
26. Pal DK, Agrawal A, Ghosh S, Ghosh A. Association of arsenic with recurrence of urinary bladder cancer. Trop Doct. 2020;50(4):325-330. doi:10.1177/0049475520930155
27. García-Esquinas E, Pollán M, Umans JG, et al. Arsenic exposure and cancer mortality in a US-based prospective cohort: the strong heart study [published correction appears in Cancer Epidemiol Biomarkers Prev. 2013;22(8):1479]. Cancer Epidemiol Biomarkers Prev. 2013;22(11):1944-1953. doi:10.1158/1055-9965.EPI-13-0234-T
28. Daling JR, Madeleine MM, Johnson LG, et al. Human papillomavirus, smoking, and sexual practices in the etiology of anal cancer. Cancer. 2004;101(2):270-280. doi:10.1002/cncr.20365
29. Bertisch B, Franceschi S, Lise M, et al; Swiss HIV Cohort Study Investigators. Risk factors for anal cancer in persons infected with HIV: a nested case-control study in the Swiss HIV Cohort Study. Am J Epidemiol. 2013;178(6):877-884. doi:10.1093/aje/kwt153
30. Rabkin CS, Biggar RJ, Melbye M, Curtis RE. Second primary cancers following anal and cervical carcinoma: evidence of shared etiologic factors. Am J Epidemiol. 1992;136(1):54-58. doi:10.1093/oxfordjournals.aje.a116420

31. Newman JD, Navas-Acien A, Kuo CC, et al. Peripheral arterial disease and its association with arsenic exposure and metabolism in the Strong Heart Study. Am J Epidemiol. 2016;184(11):806-817. doi:10.1093/aje/kww002
32. Moon KA, Guallar E, Umans JG, et al. Association between exposure to low to moderate arsenic levels and incident cardiovascular disease. A prospective cohort study. Ann Intern Med. 2013;159(10):649-659. doi:10.7326/0003-4819-159-10-201311190-00719
33. Moon K, Guallar E, Navas-Acien A. Arsenic exposure and cardiovascular disease: an updated systematic review. Curr Atheroscler Rep. 2012;14(6):542-555. doi:10.1007/s11883-012-0280-x
34. Stea F, Bianchi F, Cori L, Sicari R. Cardiovascular effects of arsenic: clinical and epidemiological findings. Environ Sci Pollut Res Int. 2014;21(1):244-251. doi:10.1007/s11356-013-2113-z
35. Burns DM. Epidemiology of smoking-induced cardiovascular disease. Prog Cardiovasc Dis. 2003;46(1):11-29. doi:10.1016/s0033-0620(03)00079-3
36. Merlini G, Dispenzieri A, Sanchorawala V, et al. Systemic immunoglobulin light chain amyloidosis. Nat Rev Dis Primers. 2018;4(1):38. Published 2018 Oct 25. doi:10.1038/s41572-018-0034-3
37. Dokken BB. The pathophysiology of cardiovascular disease and diabetes: beyond blood pressure and lipids. Diabetes Spectrum. 2008;21(3):160-165. doi:10.2337/diaspect.21.3.160
38. Vaxman I, Gertz M. Recent advances in the diagnosis, risk stratification, and management of systemic light-chain amyloidosis. Acta Haematol. 2019;141(2):93-106. doi:10.1159/000495455
39. Quock TP, Yan T, Chang E, Guthrie S, Broder MS. Epidemiology of AL amyloidosis: a real-world study using US claims data. Blood Adv. 2018;2(10):1046-1053. doi:10.1182/bloodadvances.2018016402
40. Basset M, Defrancesco I, Milani P, et al. Nonlymphoplasmacytic lymphomas associated with light-chain amyloidosis. Blood. 2020;135(4):293-296. doi:10.1182/blood.2019002762
41. Kyle RA, Therneau TM, Rajkumar SV, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med. 2002;346(8):564-569. doi:10.1056/NEJMoa01133202
42. US Department of Veterans Affairs. Agent Orange registry health exam for veterans. Updated May 28, 2021. Accessed June 17, 2021. https://www.publichealth.va.gov/exposures/agentorange/benefits/registry-exam.asp
A Vietnam War veteran’s exposures likely contributed to his cancer diagnoses, but these associations are confounded by his substance use, particularly cigarette smoking.
A Vietnam War veteran’s exposures likely contributed to his cancer diagnoses, but these associations are confounded by his substance use, particularly cigarette smoking.
Known as the “6 rainbow herbicides,” based on their identifying color on storage containers, the United States widely deployed the herbicides agents orange, green, pink, purple, white, and blue during the Vietnam War to deny the enemy cover and destroy crops.1 Unfortunately, all these herbicides were found to have contained some form of carcinogen. Agent Blue’s active ingredient consisted of sodium cacodylate trihydrate (C2H6AsNaO2), a compound that is metabolized into the organic form of the carcinogen arsenic before eventually converting into its relatively less toxic inorganic form.2 Agent Orange’s defoliating agent is 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). All rainbow herbicides except Agent Blue were unintentionally contaminated with carcinogenic dioxins. Agent Blue contained the carcinogen cacodylic acid, an organoarsenic acid. Today, herbicides no longer contain polychlorinated dibenzo-p-dioxins such as TCDD or arsenic due to strict manufacturing restrictions.2,3 In the treatment of veteran populations, knowledge of the 6 rainbow herbicides’ carcinogenic potential is important.
Between 1962 and 1971, the United States sprayed more than 45 million liters of Agent Orange on Vietnam and at least 366 kg of TCDD on South Vietnam.1,4 However, because Agent Orange was not a known carcinogen during the Vietnam War, records of exposure are poor. Additionally, individuals in Vietnam during this period were not the only ones exposed to this carcinogen as Agent Orange also was sprayed in Thailand and Korea.5 Even today there are still locations in Vietnam where Agent Orange concentrations exceed internationally acceptable levels. The Da Nang, Bien Hoa, and Phu Cat airports in Vietnam have been found to have dioxin levels exceeding 1000 ppt (parts of dioxin per trillion parts of lipid) toxicity equivalence in the soil. Although the Vietnam government is working toward decontaminating these and many other dioxin hotspots, residents in these locations are exposed to higher than internationally acceptable levels of dioxin.6
Despite receiving less media attention, Vietnam War veterans and Vietnamese soldiers and civilians were exposed to significant amounts of arsenic-based Agent Blue. Arsenic is a compound which has no environmental half-life and is carcinogenic humans if inhaled or ingested.2 Between 1962 and 1971, the United States distributed 7.8 million liters of Agent Blue containing 1,232,400 kg of arsenic across 300,000 hectares of rice paddies, 100,000 hectares of forest, and perimeters of all military bases during the Vietnam War.2,5 According to a review by Saha and colleagues, lower levels of arsenic exposure are associated with acute and chronic diseases, including cancers, of all organ systems.7
The following case presentation involves a Vietnam War veteran aged 70 years who was exposed to Agent Orange and developed 3 primary cancers, including cutaneous large B-cell non-Hodgkin lymphoma (NHL), high-grade urothelial carcinoma, and anal carcinoma in situ. Epidemiologically, this is an uncommon occurrence as only 8% of cancer survivors in the United States have been diagnosed with > 1 cancer.8
With no family history of cancer, the development of multiple malignancies raises concern for a history of toxin exposure. This report of a Vietnam War veteran with multiple conditions found to be associated with Agent Orange exposure provides an opportunity to discuss the role this exposure may have on the development of a comprehensive list of medical conditions as described by the literature. Additionally, the potential contributions of other confounding toxin exposures such as cigarette smoking, excessive alcohol use, and potential Agent Blue exposure on our patients’ health will be discussed.
Case Presentation
A male aged 70 years with Stage IV primary cutaneous large B-cell NHL, incompletely resected high-grade urothelial cancer, carcinoma in situ of the anal canal, and peripheral arterial disease (PAD) presented to the primary care clinic at the Washington DC Veterans Affairs Medical Center (DCVAMC) with concern for left leg ischemia. He also reported 2 large telangiectasias on his back for 6 months accompanied by lymphadenopathy and intermittent night sweats.
He was last seen at the DCVAMC 15 months prior after his twelfth dose of rituximab treatment for NHL. However, the patient failed to return for completion of his treatment due to frustration with the lengthy chemotherapy and follow-up process. Additionally, the patient's history included 3 failed arterial stents with complete nonadherence to the prescribed clopidogrel, resulting in the failure of 3 more subsequent graft placements. On presentation, the patient continued to report nonadherence with the clopidogrel.
The patient’s medical history included coronary artery disease (CAD) status after 2 stents in the left anterior descending artery and 1 stent in the proximal circumflex artery placed 4 years prior. He also had a history of hypertension, type 2 diabetes mellitus (T2DM), amyloid light-chain (AL) amyloidosis, aortic aneurysm, cataracts, obesity, treated hepatitis B and C, and posttraumatic stress disorder. He had no family history of cancer or AL amyloidosis; however, he noted that he was estranged from his family.
His social history was notable for active cigarette smoking up to 3 packs per day for 40 years and consuming large quantities of alcohol—at one point as many as 20 beers per day over a period of 4.5 years. He had a distant history of cocaine use but no current use, which was supported with negative urinary toxicology screens for illicit drugs over the past year.
Our patient also reported a history of Agent Orange exposure. As an artilleryman in the US Army III Corps, he was deployed for about 1 year in the most heavily sprayed regions of Vietnam, including Bien Hua, Long Binh, Xuan Loc, and Camp Zion for about 2 to 4 months at each location.
Hospital Course
The patient was treated on an inpatient basis for expedited workup and treatment for his urothelial carcinoma, NHL, and ischemic limb. His urothelial carcinoma was successfully resected, and the telangiectasias on his back were biopsied and found to be consistent with his known cutaneous large B-cell NHL, for which plans to resume outpatient chemotherapy were made. The patient’s 3 arterial grafts in his left leg were confirmed to have failed, and the patient was counseled that he would soon likely require an amputation of his ischemic leg.
Discussion
We must rely on our patient’s historical recall as there are no widely available laboratory tests or physical examination findings to confirm and/or determine the magnitude of TCDD or arsenic exposure.9-11
Exposures
The patient was stationed in Bien Hoa, the second highest dioxin-contaminated air base in Vietnam (Figure).6 Dioxin also is known to be a particularly persistent environmental pollutant, such that in January 2018, Bien Hoa was found to still have dioxin levels higher than what is considered internationally acceptable. In fact, these levels were deemed significant enough to lead the United States and Vietnamese government to sign a memorandum of intent to begin cleanup of this airport.6 TCDD is known to have a half-life of about 7.6 years, and its long half-life is mainly attributed to its slow elimination process from its stores within the liver and fat, consisting of passive excretion through the gut wall and slow metabolism by the liver.12,13 Thus, as an artilleryman mainly operating 105 howitzers within the foliage of Vietnam, our patient was exposed not only to high levels of this persistent environmental pollutant on a daily basis, but this toxin likely remained within his system for many years after his return from Vietnam.
Our patient also had a convincing history for potential Agent Blue exposure through both inhalation and ingestion of contaminated food and water. Additionally, his description of deforestations occurring within a matter of days increased the level of suspicion for Agent Blue exposure. This is because Agent Blue was the herbicide of choice for missions requiring rapid deforestation, achieving defoliation as quickly as 1 to 2 days.14 Additionally, our patient was stationed within cities in southern Vietnam near Agent Blue hot spots, such as Da Nang and Saigon, and Agent Blue was sprayed along the perimeter of all military bases.2
Levels of Evidence
Using the Veterans and Agent Orange Update in 2018 as our guide, we reviewed the quality of evidence suggesting an association between many of our patient’s comorbidities to Agent Orange exposure.5 This publication categorizes the level of evidence for association between health conditions and Agent Orange exposure in 4 main categories (Table 1).
In the Veterans and Agent Orange Update, NHL notably has a sufficient level of evidence of association with Agent Orange exposure.5 Although our patient’s extensive history of polysubstance use confounds the effect Agent Orange may have had on his health, cutaneous large B-cell NHL is an interesting exception as literature does not support even a correlative link between smoking and excessive alcohol use with primary cutaneous large B-cell NHL. Several case-control studies have found little to no association with cigarette smoking and the large B-cell subtype of NHL.15,16 Moreover, several studies have found that moderate- to-heavy alcohol use, especially beer, may have a protective effect against the development of NHL.17 Of note, our patient’s alcoholic beverage of choice was beer. Regarding our patient’s distant history of cocaine use, it has been reported that cocaine use, in the absence of an HIV infection, has not been found to increase the risk of developing NHL.18 Similarly, arsenic exposure has not been associated with NHL in the literature.19,20
The 2018 update also upgraded bladder carcinoma from having inadequate or insufficient to a limited or suggestive level of evidence for association.5 However, our patient’s most significant risk factor for bladder cancer was smoking, with a meta-analysis of 430,000 patients reporting a risk ratio (RR) of 3.14 for current cigarette smokers.21 The patient’s arsenic exposure from Agent Blue also increased his risk of developing bladder cancer. Several studies suggest a strong association between environmental arsenic exposure and bladder cancer.22-26 A 30-year meta-analysis of 40 studies by Saint-Jacques and colleagues reported that the incidence of bladder cancer was found to increase in a dose-dependent manner, with higher concentrations of arsenic contaminated wate, with incidence rising from 2.7 to 5.8 times as the amount of arsenic contamination water increased from 10 to 150 mg/L.
Our patient’s history is concerning for higher than average Agent Blue exposure compared with that of most Vietnam War veterans. Given the dose-dependent effect of arsenic on bladder cancer risk, both our patient’s history of smoking and Agent Blue exposure are risk factors in the development of his bladder cancer.22 These likely played a more significant role in his development of bladder cancer than did his Agent Orange exposure.
Finally, smoking is the most significant risk factor in our patient’s development of anal carcinoma in situ. The 2018 Agent Orange update does report limited/suggested evidence of no association between Agent Orange and anal carcinoma.5 It also is unknown whether Agent Blue exposure is a contributing cause to his development of anal carcinoma in situ.27 However, current smokers are at significant risk of developing anal cancer independent of age.28-30 Given our patient’s extensive smoking history, this is the most likely contributing factor.
Our patient also had several noncancer-related comorbidities with correlative associations with Agent Orange exposure of varying degrees (Table 2). Somewhat surprising, the development of our patient’s hypertension and T2DM may be associated in some way with his history of Agent Orange exposure. Hypertension had been recategorized from having limited or suggestive evidence to sufficient evidence in this committee’s most recent publication, and the committee is undecided on whether T2DM has a sufficient vs limited level of evidence for association with Agent Orange exposure.5 On the other hand, the committee continues to classify both ischemic heart disease and AL amyloidosis as having a limited or suggestive level of evidence that links Agent Orange exposure to these conditions.5
Arsenic may be another risk factor for our patient’s development of CAD and arterial insufficiency. Arsenic exposure is theorized to cause a direct toxic effect on coronary arteries, and arsenic exposure has been linked to PAD, CAD, and hypertension.31-34 Other significant and compelling risk factors for cardiovascular disease in our patient included his extensive history of heavy cigarette smoking, poorly controlled T2DM, obesity, and hypertension.35-37 AL amyloidosis is a rare disorder with an incidence of only 9 to 14 cases per million person-years.38,39 This disorder has not been linked to smoking or arsenic exposure in the literature. As our patient does not have a history of plasma dyscrasias or a family history of AL amyloidosis, the only known risk factors for AL amyloidosis that apply to our patient included NHL and Agent Orange exposure—NHL being a condition that is noted to be strongly correlated with Agent Orange exposure as discussed previously.5,36,40,41
Conclusions
This case describes a Vietnam War veteran with significant exposure to rainbow herbicides and considerable polysubstance who developed 3 primary cancers and several chronic medical conditions. His exposure to Agents Orange and Blue likely contributed to his medical problems, but these associations are confounded by his substance use, particularly cigarette smoking. Of all his comorbidities, our patient’s NHL is the condition most likely to be associated with his history of Agent Orange exposure. His Agent Blue exposure also increased his risk for developing bladder cancer, cardiovascular disease, and PAD.
This case also highlights the importance of evaluating Vietnam War veterans for rainbow herbicide exposure and the complexity associated with attributing diseases to these exposures. All veterans who served in the inland waterways of Vietnam between 1962 and 1975; in the Korean Demilitarized Zone between April 1, 1968 and August 31, 1971; or in Thailand between February 28, 1961 and May 7, 1975 were at risk of rainbow herbicide exposure. These veterans may not only be eligible for disability compensation but also should be screened for associated comorbidities as outlined by current research.42 We hope that this report will serve as an aid in achieving this mission.
Known as the “6 rainbow herbicides,” based on their identifying color on storage containers, the United States widely deployed the herbicides agents orange, green, pink, purple, white, and blue during the Vietnam War to deny the enemy cover and destroy crops.1 Unfortunately, all these herbicides were found to have contained some form of carcinogen. Agent Blue’s active ingredient consisted of sodium cacodylate trihydrate (C2H6AsNaO2), a compound that is metabolized into the organic form of the carcinogen arsenic before eventually converting into its relatively less toxic inorganic form.2 Agent Orange’s defoliating agent is 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). All rainbow herbicides except Agent Blue were unintentionally contaminated with carcinogenic dioxins. Agent Blue contained the carcinogen cacodylic acid, an organoarsenic acid. Today, herbicides no longer contain polychlorinated dibenzo-p-dioxins such as TCDD or arsenic due to strict manufacturing restrictions.2,3 In the treatment of veteran populations, knowledge of the 6 rainbow herbicides’ carcinogenic potential is important.
Between 1962 and 1971, the United States sprayed more than 45 million liters of Agent Orange on Vietnam and at least 366 kg of TCDD on South Vietnam.1,4 However, because Agent Orange was not a known carcinogen during the Vietnam War, records of exposure are poor. Additionally, individuals in Vietnam during this period were not the only ones exposed to this carcinogen as Agent Orange also was sprayed in Thailand and Korea.5 Even today there are still locations in Vietnam where Agent Orange concentrations exceed internationally acceptable levels. The Da Nang, Bien Hoa, and Phu Cat airports in Vietnam have been found to have dioxin levels exceeding 1000 ppt (parts of dioxin per trillion parts of lipid) toxicity equivalence in the soil. Although the Vietnam government is working toward decontaminating these and many other dioxin hotspots, residents in these locations are exposed to higher than internationally acceptable levels of dioxin.6
Despite receiving less media attention, Vietnam War veterans and Vietnamese soldiers and civilians were exposed to significant amounts of arsenic-based Agent Blue. Arsenic is a compound which has no environmental half-life and is carcinogenic humans if inhaled or ingested.2 Between 1962 and 1971, the United States distributed 7.8 million liters of Agent Blue containing 1,232,400 kg of arsenic across 300,000 hectares of rice paddies, 100,000 hectares of forest, and perimeters of all military bases during the Vietnam War.2,5 According to a review by Saha and colleagues, lower levels of arsenic exposure are associated with acute and chronic diseases, including cancers, of all organ systems.7
The following case presentation involves a Vietnam War veteran aged 70 years who was exposed to Agent Orange and developed 3 primary cancers, including cutaneous large B-cell non-Hodgkin lymphoma (NHL), high-grade urothelial carcinoma, and anal carcinoma in situ. Epidemiologically, this is an uncommon occurrence as only 8% of cancer survivors in the United States have been diagnosed with > 1 cancer.8
With no family history of cancer, the development of multiple malignancies raises concern for a history of toxin exposure. This report of a Vietnam War veteran with multiple conditions found to be associated with Agent Orange exposure provides an opportunity to discuss the role this exposure may have on the development of a comprehensive list of medical conditions as described by the literature. Additionally, the potential contributions of other confounding toxin exposures such as cigarette smoking, excessive alcohol use, and potential Agent Blue exposure on our patients’ health will be discussed.
Case Presentation
A male aged 70 years with Stage IV primary cutaneous large B-cell NHL, incompletely resected high-grade urothelial cancer, carcinoma in situ of the anal canal, and peripheral arterial disease (PAD) presented to the primary care clinic at the Washington DC Veterans Affairs Medical Center (DCVAMC) with concern for left leg ischemia. He also reported 2 large telangiectasias on his back for 6 months accompanied by lymphadenopathy and intermittent night sweats.
He was last seen at the DCVAMC 15 months prior after his twelfth dose of rituximab treatment for NHL. However, the patient failed to return for completion of his treatment due to frustration with the lengthy chemotherapy and follow-up process. Additionally, the patient's history included 3 failed arterial stents with complete nonadherence to the prescribed clopidogrel, resulting in the failure of 3 more subsequent graft placements. On presentation, the patient continued to report nonadherence with the clopidogrel.
The patient’s medical history included coronary artery disease (CAD) status after 2 stents in the left anterior descending artery and 1 stent in the proximal circumflex artery placed 4 years prior. He also had a history of hypertension, type 2 diabetes mellitus (T2DM), amyloid light-chain (AL) amyloidosis, aortic aneurysm, cataracts, obesity, treated hepatitis B and C, and posttraumatic stress disorder. He had no family history of cancer or AL amyloidosis; however, he noted that he was estranged from his family.
His social history was notable for active cigarette smoking up to 3 packs per day for 40 years and consuming large quantities of alcohol—at one point as many as 20 beers per day over a period of 4.5 years. He had a distant history of cocaine use but no current use, which was supported with negative urinary toxicology screens for illicit drugs over the past year.
Our patient also reported a history of Agent Orange exposure. As an artilleryman in the US Army III Corps, he was deployed for about 1 year in the most heavily sprayed regions of Vietnam, including Bien Hua, Long Binh, Xuan Loc, and Camp Zion for about 2 to 4 months at each location.
Hospital Course
The patient was treated on an inpatient basis for expedited workup and treatment for his urothelial carcinoma, NHL, and ischemic limb. His urothelial carcinoma was successfully resected, and the telangiectasias on his back were biopsied and found to be consistent with his known cutaneous large B-cell NHL, for which plans to resume outpatient chemotherapy were made. The patient’s 3 arterial grafts in his left leg were confirmed to have failed, and the patient was counseled that he would soon likely require an amputation of his ischemic leg.
Discussion
We must rely on our patient’s historical recall as there are no widely available laboratory tests or physical examination findings to confirm and/or determine the magnitude of TCDD or arsenic exposure.9-11
Exposures
The patient was stationed in Bien Hoa, the second highest dioxin-contaminated air base in Vietnam (Figure).6 Dioxin also is known to be a particularly persistent environmental pollutant, such that in January 2018, Bien Hoa was found to still have dioxin levels higher than what is considered internationally acceptable. In fact, these levels were deemed significant enough to lead the United States and Vietnamese government to sign a memorandum of intent to begin cleanup of this airport.6 TCDD is known to have a half-life of about 7.6 years, and its long half-life is mainly attributed to its slow elimination process from its stores within the liver and fat, consisting of passive excretion through the gut wall and slow metabolism by the liver.12,13 Thus, as an artilleryman mainly operating 105 howitzers within the foliage of Vietnam, our patient was exposed not only to high levels of this persistent environmental pollutant on a daily basis, but this toxin likely remained within his system for many years after his return from Vietnam.
Our patient also had a convincing history for potential Agent Blue exposure through both inhalation and ingestion of contaminated food and water. Additionally, his description of deforestations occurring within a matter of days increased the level of suspicion for Agent Blue exposure. This is because Agent Blue was the herbicide of choice for missions requiring rapid deforestation, achieving defoliation as quickly as 1 to 2 days.14 Additionally, our patient was stationed within cities in southern Vietnam near Agent Blue hot spots, such as Da Nang and Saigon, and Agent Blue was sprayed along the perimeter of all military bases.2
Levels of Evidence
Using the Veterans and Agent Orange Update in 2018 as our guide, we reviewed the quality of evidence suggesting an association between many of our patient’s comorbidities to Agent Orange exposure.5 This publication categorizes the level of evidence for association between health conditions and Agent Orange exposure in 4 main categories (Table 1).
In the Veterans and Agent Orange Update, NHL notably has a sufficient level of evidence of association with Agent Orange exposure.5 Although our patient’s extensive history of polysubstance use confounds the effect Agent Orange may have had on his health, cutaneous large B-cell NHL is an interesting exception as literature does not support even a correlative link between smoking and excessive alcohol use with primary cutaneous large B-cell NHL. Several case-control studies have found little to no association with cigarette smoking and the large B-cell subtype of NHL.15,16 Moreover, several studies have found that moderate- to-heavy alcohol use, especially beer, may have a protective effect against the development of NHL.17 Of note, our patient’s alcoholic beverage of choice was beer. Regarding our patient’s distant history of cocaine use, it has been reported that cocaine use, in the absence of an HIV infection, has not been found to increase the risk of developing NHL.18 Similarly, arsenic exposure has not been associated with NHL in the literature.19,20
The 2018 update also upgraded bladder carcinoma from having inadequate or insufficient to a limited or suggestive level of evidence for association.5 However, our patient’s most significant risk factor for bladder cancer was smoking, with a meta-analysis of 430,000 patients reporting a risk ratio (RR) of 3.14 for current cigarette smokers.21 The patient’s arsenic exposure from Agent Blue also increased his risk of developing bladder cancer. Several studies suggest a strong association between environmental arsenic exposure and bladder cancer.22-26 A 30-year meta-analysis of 40 studies by Saint-Jacques and colleagues reported that the incidence of bladder cancer was found to increase in a dose-dependent manner, with higher concentrations of arsenic contaminated wate, with incidence rising from 2.7 to 5.8 times as the amount of arsenic contamination water increased from 10 to 150 mg/L.
Our patient’s history is concerning for higher than average Agent Blue exposure compared with that of most Vietnam War veterans. Given the dose-dependent effect of arsenic on bladder cancer risk, both our patient’s history of smoking and Agent Blue exposure are risk factors in the development of his bladder cancer.22 These likely played a more significant role in his development of bladder cancer than did his Agent Orange exposure.
Finally, smoking is the most significant risk factor in our patient’s development of anal carcinoma in situ. The 2018 Agent Orange update does report limited/suggested evidence of no association between Agent Orange and anal carcinoma.5 It also is unknown whether Agent Blue exposure is a contributing cause to his development of anal carcinoma in situ.27 However, current smokers are at significant risk of developing anal cancer independent of age.28-30 Given our patient’s extensive smoking history, this is the most likely contributing factor.
Our patient also had several noncancer-related comorbidities with correlative associations with Agent Orange exposure of varying degrees (Table 2). Somewhat surprising, the development of our patient’s hypertension and T2DM may be associated in some way with his history of Agent Orange exposure. Hypertension had been recategorized from having limited or suggestive evidence to sufficient evidence in this committee’s most recent publication, and the committee is undecided on whether T2DM has a sufficient vs limited level of evidence for association with Agent Orange exposure.5 On the other hand, the committee continues to classify both ischemic heart disease and AL amyloidosis as having a limited or suggestive level of evidence that links Agent Orange exposure to these conditions.5
Arsenic may be another risk factor for our patient’s development of CAD and arterial insufficiency. Arsenic exposure is theorized to cause a direct toxic effect on coronary arteries, and arsenic exposure has been linked to PAD, CAD, and hypertension.31-34 Other significant and compelling risk factors for cardiovascular disease in our patient included his extensive history of heavy cigarette smoking, poorly controlled T2DM, obesity, and hypertension.35-37 AL amyloidosis is a rare disorder with an incidence of only 9 to 14 cases per million person-years.38,39 This disorder has not been linked to smoking or arsenic exposure in the literature. As our patient does not have a history of plasma dyscrasias or a family history of AL amyloidosis, the only known risk factors for AL amyloidosis that apply to our patient included NHL and Agent Orange exposure—NHL being a condition that is noted to be strongly correlated with Agent Orange exposure as discussed previously.5,36,40,41
Conclusions
This case describes a Vietnam War veteran with significant exposure to rainbow herbicides and considerable polysubstance who developed 3 primary cancers and several chronic medical conditions. His exposure to Agents Orange and Blue likely contributed to his medical problems, but these associations are confounded by his substance use, particularly cigarette smoking. Of all his comorbidities, our patient’s NHL is the condition most likely to be associated with his history of Agent Orange exposure. His Agent Blue exposure also increased his risk for developing bladder cancer, cardiovascular disease, and PAD.
This case also highlights the importance of evaluating Vietnam War veterans for rainbow herbicide exposure and the complexity associated with attributing diseases to these exposures. All veterans who served in the inland waterways of Vietnam between 1962 and 1975; in the Korean Demilitarized Zone between April 1, 1968 and August 31, 1971; or in Thailand between February 28, 1961 and May 7, 1975 were at risk of rainbow herbicide exposure. These veterans may not only be eligible for disability compensation but also should be screened for associated comorbidities as outlined by current research.42 We hope that this report will serve as an aid in achieving this mission.
1. Stellman JM, Stellman SD, Christian R, Weber T, Tomasallo C. The extent and patterns of usage of Agent Orange and other herbicides in Vietnam. Nature. 2003;422(6933):681-687. doi:10.1038/nature01537
2. Olson K, Cihacek L. The fate of Agent Blue, the arsenic based herbicide, used in South Vietnam during the Vietnam War. Open J Soil Sci. 2020;10:518-577. doi:10.4236/ojss.2020.1011027
3. Lee Chang A, Dym AA, Venegas-Borsellino C, et al. Comparison between simulation-based training and lecture-based education in teaching situation awareness. a randomized controlled study. Ann Am Thorac Soc. 2017;14(4):529-535. doi:10.1513/AnnalsATS.201612-950OC
4. Stellman SD. Agent Orange during the Vietnam War: the lingering issue of its civilian and military health impact. Am J Public Health. 2018;108(6):726-728. doi:10.2105/AJPH.2018.304426
5. National Academies of Sciences, Engineering, and Medicine. Veterans and Agent Orange: Update 11 (2018). The National Academies Press; 2018. doi:10.17226/25137
6. Martin MF. US Agent Orange/dioxin assistance to Vietnam. Updated January 15, 2021. Accessed June 17, 2021. https://fas.org/sgp/crs/row/R44268.pdf
7. Saha JC, Dikshit AK, Bandyopadhyay M, Saha KC. A review of arsenic poisoning and its effects on human health. Crit Rev Environ Sci Technol. 1999;29(3):281-313. doi:10.1080/10643389991259227
8. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7-34. doi:10.3322/caac.21551
9. American Cancer Society. Agent Orange and cancer risk. Updated June 9, 2020. Accessed June 17, 2021. https://www.cancer.org/cancer/cancer-causes/agent-orange-and-cancer.html
10. US Department of Veterans Affairs. Veterans’ diseases associated with Agent Orange. Updated June 16, 2021. Accessed June 17, 2021. https://www.publichealth.va.gov/exposures/agentorange/conditions/index.asp
11. Katz SA. On the use of hair analysis for assessing arsenic intoxication. Int J Environ Res Public Health. 2019;16(6):977. Published 2019 Mar 18. doi:10.3390/ijerph16060977
12. Chang ET, Boffetta P, Adami HO, Mandel JS. A critical review of the epidemiology of Agent Orange or 2,3,7,8-tetrachlorodibenzo-p-dioxin and lymphoid malignancies. Ann Epidemiol. 2015;25(4):275-292.e30. doi:10.1016/j.annepidem.2015.01.002
13. Kramárová E, Kogevinas M, Anh CT, et al. Exposure to Agent Orange and occurrence of soft-tissue sarcomas or non-Hodgkin lymphomas: an ongoing study in Vietnam. Environ Health Perspect. 1998;106 Suppl 2(suppl 2):671-678. doi:10.1289/ehp.106-1533419
14. Institute of Medicine (US) Committee to Review the Health Effects in Vietnam Veterans of Exposure to Herbicides. Veterans and Agent Orange: Health Effects of Herbicides Used in Vietnam. National Academies Press US; 1994.
15. Morton LM, Hartge P, Holford TR, et al. Cigarette smoking and risk of non-Hodgkin lymphoma: a pooled analysis from the International Lymphoma Epidemiology Consortium (interlymph). Cancer Epidemiol Biomarkers Prev. 2005;14(4):925-933. doi:10.1158/1055-9965.EPI-04-0693
16. Schöllkopf C, Smedby KE, Hjalgrim H, et al. Cigarette smoking and risk of non-Hodgkin’s lymphoma--a population-based case-control study. Cancer Epidemiol Biomarkers Prev. 2005;14(7):1791-1796. doi:10.1158/1055-9965.EPI-05-0077
17. Psaltopoulou T, Sergentanis TN, Ntanasis-Stathopoulos I, Tzanninis IG, Tsilimigras DI, Dimopoulos MA. Alcohol consumption and risk of hematological malignancies: a meta-analysis of prospective studies. Int J Cancer. 2018;143(3):486-495. doi:10.1002/ijc.31330
18. Aujla AS, Lee SH. Association between cocaine use and hematological malignancies. J Clin Oncol. 2016;34(15_suppl):e19072-e19072. doi:10.1200/JCO.2016.34.15_suppl.e19072
19. Mao Y, Hu J, Ugnat AM, White K. Non-Hodgkin’s lymphoma and occupational exposure to chemicals in Canada. Canadian Cancer Registries Epidemiology Research Group. Ann Oncol. 2000;11 (suppl 1):69-73. doi:10.1093/annonc/11.suppl_1.S69
20. Kelekci KH, Bilgin I, Ermete M. Arsenical keratoses and non-Hodgkin’s lymphoma: arsenic-induced or coincidental conditions? J Pakistan Assoc Dermatol. 2012;22(4):366-369.
21. Antoni S, Ferlay J, Soerjomataram I, Znaor A, Jemal A, Bray F. Bladder cancer incidence and mortality: a global overview and recent trends. Eur Urol. 2017;71(1):96-108. doi:10.1016/j.eururo.2016.06.010
22. Saint-Jacques N, Parker L, Brown P, Dummer TJ. Arsenic in drinking water and urinary tract cancers: a systematic review of 30 years of epidemiological evidence. Environ Health. 2014;13:44. Published 2014 Jun 2. doi:10.1186/1476-069X-13-44
23. Radosavljevic′ V, Jakovljevic′ B. Arsenic and bladder cancer: observations and suggestions. J Environ Health. 2008;71(3):40-42.
24. Marsit CJ, Karagas MR, Schned A, Kelsey KT. Carcinogen exposure and epigenetic silencing in bladder cancer. Ann N Y Acad Sci. 2006;1076(1):810-821. doi:10.1196/annals.1371.031
25. Mendez WM Jr, Eftim S, Cohen J, et al. Relationships between arsenic concentrations in drinking water and lung and bladder cancer incidence in U.S. counties. J Expo Sci Environ Epidemiol. 2017;27(3):235-243. doi:10.1038/jes.2016.58
26. Pal DK, Agrawal A, Ghosh S, Ghosh A. Association of arsenic with recurrence of urinary bladder cancer. Trop Doct. 2020;50(4):325-330. doi:10.1177/0049475520930155
27. García-Esquinas E, Pollán M, Umans JG, et al. Arsenic exposure and cancer mortality in a US-based prospective cohort: the strong heart study [published correction appears in Cancer Epidemiol Biomarkers Prev. 2013;22(8):1479]. Cancer Epidemiol Biomarkers Prev. 2013;22(11):1944-1953. doi:10.1158/1055-9965.EPI-13-0234-T
28. Daling JR, Madeleine MM, Johnson LG, et al. Human papillomavirus, smoking, and sexual practices in the etiology of anal cancer. Cancer. 2004;101(2):270-280. doi:10.1002/cncr.20365
29. Bertisch B, Franceschi S, Lise M, et al; Swiss HIV Cohort Study Investigators. Risk factors for anal cancer in persons infected with HIV: a nested case-control study in the Swiss HIV Cohort Study. Am J Epidemiol. 2013;178(6):877-884. doi:10.1093/aje/kwt153
30. Rabkin CS, Biggar RJ, Melbye M, Curtis RE. Second primary cancers following anal and cervical carcinoma: evidence of shared etiologic factors. Am J Epidemiol. 1992;136(1):54-58. doi:10.1093/oxfordjournals.aje.a116420
31. Newman JD, Navas-Acien A, Kuo CC, et al. Peripheral arterial disease and its association with arsenic exposure and metabolism in the Strong Heart Study. Am J Epidemiol. 2016;184(11):806-817. doi:10.1093/aje/kww002
32. Moon KA, Guallar E, Umans JG, et al. Association between exposure to low to moderate arsenic levels and incident cardiovascular disease. A prospective cohort study. Ann Intern Med. 2013;159(10):649-659. doi:10.7326/0003-4819-159-10-201311190-00719
33. Moon K, Guallar E, Navas-Acien A. Arsenic exposure and cardiovascular disease: an updated systematic review. Curr Atheroscler Rep. 2012;14(6):542-555. doi:10.1007/s11883-012-0280-x
34. Stea F, Bianchi F, Cori L, Sicari R. Cardiovascular effects of arsenic: clinical and epidemiological findings. Environ Sci Pollut Res Int. 2014;21(1):244-251. doi:10.1007/s11356-013-2113-z
35. Burns DM. Epidemiology of smoking-induced cardiovascular disease. Prog Cardiovasc Dis. 2003;46(1):11-29. doi:10.1016/s0033-0620(03)00079-3
36. Merlini G, Dispenzieri A, Sanchorawala V, et al. Systemic immunoglobulin light chain amyloidosis. Nat Rev Dis Primers. 2018;4(1):38. Published 2018 Oct 25. doi:10.1038/s41572-018-0034-3
37. Dokken BB. The pathophysiology of cardiovascular disease and diabetes: beyond blood pressure and lipids. Diabetes Spectrum. 2008;21(3):160-165. doi:10.2337/diaspect.21.3.160
38. Vaxman I, Gertz M. Recent advances in the diagnosis, risk stratification, and management of systemic light-chain amyloidosis. Acta Haematol. 2019;141(2):93-106. doi:10.1159/000495455
39. Quock TP, Yan T, Chang E, Guthrie S, Broder MS. Epidemiology of AL amyloidosis: a real-world study using US claims data. Blood Adv. 2018;2(10):1046-1053. doi:10.1182/bloodadvances.2018016402
40. Basset M, Defrancesco I, Milani P, et al. Nonlymphoplasmacytic lymphomas associated with light-chain amyloidosis. Blood. 2020;135(4):293-296. doi:10.1182/blood.2019002762
41. Kyle RA, Therneau TM, Rajkumar SV, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med. 2002;346(8):564-569. doi:10.1056/NEJMoa01133202
42. US Department of Veterans Affairs. Agent Orange registry health exam for veterans. Updated May 28, 2021. Accessed June 17, 2021. https://www.publichealth.va.gov/exposures/agentorange/benefits/registry-exam.asp
1. Stellman JM, Stellman SD, Christian R, Weber T, Tomasallo C. The extent and patterns of usage of Agent Orange and other herbicides in Vietnam. Nature. 2003;422(6933):681-687. doi:10.1038/nature01537
2. Olson K, Cihacek L. The fate of Agent Blue, the arsenic based herbicide, used in South Vietnam during the Vietnam War. Open J Soil Sci. 2020;10:518-577. doi:10.4236/ojss.2020.1011027
3. Lee Chang A, Dym AA, Venegas-Borsellino C, et al. Comparison between simulation-based training and lecture-based education in teaching situation awareness. a randomized controlled study. Ann Am Thorac Soc. 2017;14(4):529-535. doi:10.1513/AnnalsATS.201612-950OC
4. Stellman SD. Agent Orange during the Vietnam War: the lingering issue of its civilian and military health impact. Am J Public Health. 2018;108(6):726-728. doi:10.2105/AJPH.2018.304426
5. National Academies of Sciences, Engineering, and Medicine. Veterans and Agent Orange: Update 11 (2018). The National Academies Press; 2018. doi:10.17226/25137
6. Martin MF. US Agent Orange/dioxin assistance to Vietnam. Updated January 15, 2021. Accessed June 17, 2021. https://fas.org/sgp/crs/row/R44268.pdf
7. Saha JC, Dikshit AK, Bandyopadhyay M, Saha KC. A review of arsenic poisoning and its effects on human health. Crit Rev Environ Sci Technol. 1999;29(3):281-313. doi:10.1080/10643389991259227
8. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7-34. doi:10.3322/caac.21551
9. American Cancer Society. Agent Orange and cancer risk. Updated June 9, 2020. Accessed June 17, 2021. https://www.cancer.org/cancer/cancer-causes/agent-orange-and-cancer.html
10. US Department of Veterans Affairs. Veterans’ diseases associated with Agent Orange. Updated June 16, 2021. Accessed June 17, 2021. https://www.publichealth.va.gov/exposures/agentorange/conditions/index.asp
11. Katz SA. On the use of hair analysis for assessing arsenic intoxication. Int J Environ Res Public Health. 2019;16(6):977. Published 2019 Mar 18. doi:10.3390/ijerph16060977
12. Chang ET, Boffetta P, Adami HO, Mandel JS. A critical review of the epidemiology of Agent Orange or 2,3,7,8-tetrachlorodibenzo-p-dioxin and lymphoid malignancies. Ann Epidemiol. 2015;25(4):275-292.e30. doi:10.1016/j.annepidem.2015.01.002
13. Kramárová E, Kogevinas M, Anh CT, et al. Exposure to Agent Orange and occurrence of soft-tissue sarcomas or non-Hodgkin lymphomas: an ongoing study in Vietnam. Environ Health Perspect. 1998;106 Suppl 2(suppl 2):671-678. doi:10.1289/ehp.106-1533419
14. Institute of Medicine (US) Committee to Review the Health Effects in Vietnam Veterans of Exposure to Herbicides. Veterans and Agent Orange: Health Effects of Herbicides Used in Vietnam. National Academies Press US; 1994.
15. Morton LM, Hartge P, Holford TR, et al. Cigarette smoking and risk of non-Hodgkin lymphoma: a pooled analysis from the International Lymphoma Epidemiology Consortium (interlymph). Cancer Epidemiol Biomarkers Prev. 2005;14(4):925-933. doi:10.1158/1055-9965.EPI-04-0693
16. Schöllkopf C, Smedby KE, Hjalgrim H, et al. Cigarette smoking and risk of non-Hodgkin’s lymphoma--a population-based case-control study. Cancer Epidemiol Biomarkers Prev. 2005;14(7):1791-1796. doi:10.1158/1055-9965.EPI-05-0077
17. Psaltopoulou T, Sergentanis TN, Ntanasis-Stathopoulos I, Tzanninis IG, Tsilimigras DI, Dimopoulos MA. Alcohol consumption and risk of hematological malignancies: a meta-analysis of prospective studies. Int J Cancer. 2018;143(3):486-495. doi:10.1002/ijc.31330
18. Aujla AS, Lee SH. Association between cocaine use and hematological malignancies. J Clin Oncol. 2016;34(15_suppl):e19072-e19072. doi:10.1200/JCO.2016.34.15_suppl.e19072
19. Mao Y, Hu J, Ugnat AM, White K. Non-Hodgkin’s lymphoma and occupational exposure to chemicals in Canada. Canadian Cancer Registries Epidemiology Research Group. Ann Oncol. 2000;11 (suppl 1):69-73. doi:10.1093/annonc/11.suppl_1.S69
20. Kelekci KH, Bilgin I, Ermete M. Arsenical keratoses and non-Hodgkin’s lymphoma: arsenic-induced or coincidental conditions? J Pakistan Assoc Dermatol. 2012;22(4):366-369.
21. Antoni S, Ferlay J, Soerjomataram I, Znaor A, Jemal A, Bray F. Bladder cancer incidence and mortality: a global overview and recent trends. Eur Urol. 2017;71(1):96-108. doi:10.1016/j.eururo.2016.06.010
22. Saint-Jacques N, Parker L, Brown P, Dummer TJ. Arsenic in drinking water and urinary tract cancers: a systematic review of 30 years of epidemiological evidence. Environ Health. 2014;13:44. Published 2014 Jun 2. doi:10.1186/1476-069X-13-44
23. Radosavljevic′ V, Jakovljevic′ B. Arsenic and bladder cancer: observations and suggestions. J Environ Health. 2008;71(3):40-42.
24. Marsit CJ, Karagas MR, Schned A, Kelsey KT. Carcinogen exposure and epigenetic silencing in bladder cancer. Ann N Y Acad Sci. 2006;1076(1):810-821. doi:10.1196/annals.1371.031
25. Mendez WM Jr, Eftim S, Cohen J, et al. Relationships between arsenic concentrations in drinking water and lung and bladder cancer incidence in U.S. counties. J Expo Sci Environ Epidemiol. 2017;27(3):235-243. doi:10.1038/jes.2016.58
26. Pal DK, Agrawal A, Ghosh S, Ghosh A. Association of arsenic with recurrence of urinary bladder cancer. Trop Doct. 2020;50(4):325-330. doi:10.1177/0049475520930155
27. García-Esquinas E, Pollán M, Umans JG, et al. Arsenic exposure and cancer mortality in a US-based prospective cohort: the strong heart study [published correction appears in Cancer Epidemiol Biomarkers Prev. 2013;22(8):1479]. Cancer Epidemiol Biomarkers Prev. 2013;22(11):1944-1953. doi:10.1158/1055-9965.EPI-13-0234-T
28. Daling JR, Madeleine MM, Johnson LG, et al. Human papillomavirus, smoking, and sexual practices in the etiology of anal cancer. Cancer. 2004;101(2):270-280. doi:10.1002/cncr.20365
29. Bertisch B, Franceschi S, Lise M, et al; Swiss HIV Cohort Study Investigators. Risk factors for anal cancer in persons infected with HIV: a nested case-control study in the Swiss HIV Cohort Study. Am J Epidemiol. 2013;178(6):877-884. doi:10.1093/aje/kwt153
30. Rabkin CS, Biggar RJ, Melbye M, Curtis RE. Second primary cancers following anal and cervical carcinoma: evidence of shared etiologic factors. Am J Epidemiol. 1992;136(1):54-58. doi:10.1093/oxfordjournals.aje.a116420
31. Newman JD, Navas-Acien A, Kuo CC, et al. Peripheral arterial disease and its association with arsenic exposure and metabolism in the Strong Heart Study. Am J Epidemiol. 2016;184(11):806-817. doi:10.1093/aje/kww002
32. Moon KA, Guallar E, Umans JG, et al. Association between exposure to low to moderate arsenic levels and incident cardiovascular disease. A prospective cohort study. Ann Intern Med. 2013;159(10):649-659. doi:10.7326/0003-4819-159-10-201311190-00719
33. Moon K, Guallar E, Navas-Acien A. Arsenic exposure and cardiovascular disease: an updated systematic review. Curr Atheroscler Rep. 2012;14(6):542-555. doi:10.1007/s11883-012-0280-x
34. Stea F, Bianchi F, Cori L, Sicari R. Cardiovascular effects of arsenic: clinical and epidemiological findings. Environ Sci Pollut Res Int. 2014;21(1):244-251. doi:10.1007/s11356-013-2113-z
35. Burns DM. Epidemiology of smoking-induced cardiovascular disease. Prog Cardiovasc Dis. 2003;46(1):11-29. doi:10.1016/s0033-0620(03)00079-3
36. Merlini G, Dispenzieri A, Sanchorawala V, et al. Systemic immunoglobulin light chain amyloidosis. Nat Rev Dis Primers. 2018;4(1):38. Published 2018 Oct 25. doi:10.1038/s41572-018-0034-3
37. Dokken BB. The pathophysiology of cardiovascular disease and diabetes: beyond blood pressure and lipids. Diabetes Spectrum. 2008;21(3):160-165. doi:10.2337/diaspect.21.3.160
38. Vaxman I, Gertz M. Recent advances in the diagnosis, risk stratification, and management of systemic light-chain amyloidosis. Acta Haematol. 2019;141(2):93-106. doi:10.1159/000495455
39. Quock TP, Yan T, Chang E, Guthrie S, Broder MS. Epidemiology of AL amyloidosis: a real-world study using US claims data. Blood Adv. 2018;2(10):1046-1053. doi:10.1182/bloodadvances.2018016402
40. Basset M, Defrancesco I, Milani P, et al. Nonlymphoplasmacytic lymphomas associated with light-chain amyloidosis. Blood. 2020;135(4):293-296. doi:10.1182/blood.2019002762
41. Kyle RA, Therneau TM, Rajkumar SV, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med. 2002;346(8):564-569. doi:10.1056/NEJMoa01133202
42. US Department of Veterans Affairs. Agent Orange registry health exam for veterans. Updated May 28, 2021. Accessed June 17, 2021. https://www.publichealth.va.gov/exposures/agentorange/benefits/registry-exam.asp