User login
Concurrent Atopic Dermatitis and Psoriasis Vulgaris: Implications for Targeted Biologic Therapy
Psoriasis vulgaris is a chronic inflammatory skin condition associated with notable elevation in helper T cell (TH) production of TH1/TH17-mediated inflammatory cytokines, including IL-17A.1 Upon binding of IL-17A to IL-17 receptors in the skin, an inflammatory cascade is triggered, resulting in the classic clinical appearance of psoriasis. Moderate to severe psoriasis often is managed by suppressing TH1/TH17-mediated inflammation using targeted immune therapy such as secukinumab, an IL-17A inhibitor.2 Atopic dermatitis (AD), another chronic inflammatory dermatosis, is associated with substantial elevation in TH2-mediated inflammatory cytokines, such as IL-4.3 Dupilumab, which interacts with IL-4R, disrupts the IL-4 and IL-13 signaling pathways and demonstrates considerable efficacy in the treatment of moderate to severe AD.4
A case series has shown that suppression of the TH1/TH17-mediated inflammation of psoriasis may paradoxically result in the development of TH2-mediated AD.5 Similarly, a recent case report described a patient who developed psoriasis following treatment of AD with dupilumab.6 Herein, we describe a patient with a history of psoriasis that was well controlled with secukinumab who developed severe refractory erythrodermic AD that resolved with dupilumab treatment. Following clearance of AD with dupilumab, he exhibited psoriasis recurrence.
Case Report
A 39-year-old man with a lifelong history of psoriasis was admitted to the hospital for management of severe erythroderma. Four years prior, secukinumab was initiated for treatment of psoriasis, resulting in excellent clinical response. He discontinued secukinumab after 2 years of treatment because of insurance coverage issues and managed his condition with only topical corticosteroids. He restarted secukinumab 10 months before admission because of a psoriasis flare. Shortly after resuming secukinumab, he developed a severe exfoliative erythroderma that was not responsive to corticosteroids, etanercept, methotrexate, or ustekinumab.

On initial presentation, physical examination revealed diffuse erythema and scaling with associated edema of the face, trunk, and extremities (Figure 1). A biopsy from the patient’s right arm demonstrated a superficial perivascular inflammatory infiltrate composed of lymphocytes, histiocytes, and scattered eosinophils consistent with spongiotic dermatitis (Figure 2). Cyclosporine 225 mg twice daily and topical corticosteroids were started.

Over the next several months, the patient had several admissions secondary to recurrent skin abscesses in the setting of refractory erythroderma. He underwent trials of infliximab, corticosteroids, intravenous immunoglobulin, guselkumab, and acitretin with minimal improvement. He underwent an extensive laboratory and radiologic workup, which was notable for cyclical peripheral eosinophilia and elevated IgE levels correlating with the erythroderma flares. A second biopsy was obtained and continued to demonstrate changes consistent with AD.

Four months after the initial hospitalization, all psoriasis medications were stopped, and the patient was started on dupilumab 300 mg/2 mL every 2 weeks and an 8-week oral prednisone taper. This combination led to notable clinical improvement and resolution of peripheral eosinophilia. Several months after disease remission, he began to develop worsening erythema and pruritus on the trunk and extremities, followed by the development of new psoriatic lesions (Figure 3) with a biopsy consistent with psoriasis (Figure 4). The patient was continued on dupilumab, but cyclosporine was added. The patient self-discontinued dupilumab owing to injection-site discomfort and has been slowly weaning off oral cyclosporine with 1 to 2 remaining eczematous plaques and 1 to 2 psoriatic plaques managed by topical corticosteroids.

Comment
We present a patient with psoriasis that was well controlled on secukinumab who developed severe AD following treatment with secukinumab. The AD resolved following treatment with dupilumab and a tapering dose of prednisone. However, after several months of treatment with dupilumab alone, he began to develop psoriatic lesions again. This case supports findings in a case series describing the development of AD in patients with psoriasis treated with IL-17 inhibitors5 and a recent case report describing a patient with AD who developed psoriasis following treatment with an IL-4/IL-13 inhibitor.6
Recognized adverse effects demonstrate biologic medications’ contributions to both normal as well as aberrant immunologic responses. For example, IL-17 plays an essential role in innate and adaptive immune responses against infections at mucosal and cutaneous interfaces, as demonstrated by chronic mucocutaneous candidiasis in patients with genetic defects in IL-17–related pathways.7 Similarly, in patients taking IL-17 antagonists, an increase in the incidence of Candida infections has been observed.8 In patients with concurrent psoriasis and inflammatory bowel disease (IBD), treatment with IL-17 inhibitors is contraindicated due to the risk of exacerbating the IBD. This observation is somewhat paradoxical, as increased IL-17 release by TH17 cells is implicated in the pathogenesis of IBD.9 Interestingly, it is now thought that IL-17 may play a protective role in T-cell–driven intestinal inflammation through induction of protective intestinal epithelial gene expression and increased mucosal defense against gut microbes, explaining the worsening of IBD in patients on IL-17 inhibitors.10 These adverse effects illustrate the complicated and varied roles biologic medications play in immunologic response.
Given that TH1 and TH2 exert opposing immune mechanisms, it is uncommon for psoriasis and AD to coexist in a single patient. However, patients who exhibit concurrent findings may represent a unique population in which psoriasis and AD coexist, perhaps because of an underlying genetic predisposition. Moreover, targeted treatment of pathways unique to these disease processes may result in paradoxical flaring of the nontargeted pathway. It also is possible that inhibition of a specific T-cell pathway in a subset of patients will result in an immunologic imbalance, favoring increased activity of the opposing pathway in the absence of coexisting disease. In the case presented here, the findings may be explained by secukinumab’s inhibition of TH1/TH17-mediated inflammation, which resulted in a shift to a TH2-mediated inflammatory response manifesting as AD, as well as dupilumab’s inhibition of TH2-mediated inflammation, which caused a shift back to TH1-mediated inflammatory pathways. Additionally, for patients with changing morphologies exacerbated by biologic medications, alternative diagnoses, such as cutaneous T-cell lymphoma, may be considered.
Conclusion
We report an unusual case of secukinumab-induced AD in a patient with psoriasis that resolved following several months of treatment with dupilumab and a tapering dose of prednisone. Subsequently, this same patient developed re-emergence of psoriatic lesions with continued use of dupilumab, which was eventually discontinued by the patient despite appropriate disease control. In addition to illustrating the underlying pathophysiologic mechanisms of 2 common inflammatory dermatologic conditions, this case highlights how pharmacologic interventions targeted at specific immunologic pathways may have unintended consequences. Further investigation into the effects of targeted biologics on the TH1/TH2 immune axis is warranted to better understand the mechanism and possible implications of the phenotypic switching presented in this case.
- Diani M, Altomare G, Reali E. T helper cell subsets in clinical manifestations of psoriasis. J Immunol Res. 2016;2016:7692024.
- Langley RG, Elewski BE, Lebwohl M, et al. Secukinumab in plaque psoriasis—results of two phase 3 trials. N Engl J Med. 2014;371:326-338.
- van der Heijden FL, Wierenga EA, Bos JD, et al. High frequency of IL-4-producing CD4+ allergen-specific T lymphocytes in atopic dermatitis lesional skin. J Invest Dermatol. 1991;97:389-394.
- Beck LA, Thaçi D, Hamilton JD, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med. 2014;371:130-139.
- Lai FYX, Higgins E, Smith CH, et al. Morphologic switch from psoriasiform to eczematous dermatitis after anti-IL-17 therapy: a case series. JAMA Dermatol. 2019;155:1082-1084.
- Varma A, Levitt J. Dupilumab-induced phenotype switching from atopic dermatitis to psoriasis. JAAD Case Rep. 2020;6:217-218.
- Ling Y, Puel A. IL-17 and infections. Actas Dermosifiliogr. 2014;105(suppl 1):34-40.
- Saunte DM, Mrowietz U, Puig L, et al. Candida infections in patients with psoriasis and psoriatic arthritis treated with interleukin-17 inhibitors and their practical management. Br J Dermatol. 2017;177:47-62.
- Hölttä V, Klemetti P, Sipponen T, et al. IL-23/IL-17 immunity as a hallmark of Crohn’s disease. Inflamm Bowel Dis. 2008;14:1175-1184.
- Smith MK, Pai J, Panaccione R, et al. Crohn’s-like disease in a patient exposed to anti-interleukin-17 blockade (ixekizumab) for the treatment of chronic plaque psoriasis: a case report. BMC Gastroenterol. 2019;19:162.
Psoriasis vulgaris is a chronic inflammatory skin condition associated with notable elevation in helper T cell (TH) production of TH1/TH17-mediated inflammatory cytokines, including IL-17A.1 Upon binding of IL-17A to IL-17 receptors in the skin, an inflammatory cascade is triggered, resulting in the classic clinical appearance of psoriasis. Moderate to severe psoriasis often is managed by suppressing TH1/TH17-mediated inflammation using targeted immune therapy such as secukinumab, an IL-17A inhibitor.2 Atopic dermatitis (AD), another chronic inflammatory dermatosis, is associated with substantial elevation in TH2-mediated inflammatory cytokines, such as IL-4.3 Dupilumab, which interacts with IL-4R, disrupts the IL-4 and IL-13 signaling pathways and demonstrates considerable efficacy in the treatment of moderate to severe AD.4
A case series has shown that suppression of the TH1/TH17-mediated inflammation of psoriasis may paradoxically result in the development of TH2-mediated AD.5 Similarly, a recent case report described a patient who developed psoriasis following treatment of AD with dupilumab.6 Herein, we describe a patient with a history of psoriasis that was well controlled with secukinumab who developed severe refractory erythrodermic AD that resolved with dupilumab treatment. Following clearance of AD with dupilumab, he exhibited psoriasis recurrence.
Case Report
A 39-year-old man with a lifelong history of psoriasis was admitted to the hospital for management of severe erythroderma. Four years prior, secukinumab was initiated for treatment of psoriasis, resulting in excellent clinical response. He discontinued secukinumab after 2 years of treatment because of insurance coverage issues and managed his condition with only topical corticosteroids. He restarted secukinumab 10 months before admission because of a psoriasis flare. Shortly after resuming secukinumab, he developed a severe exfoliative erythroderma that was not responsive to corticosteroids, etanercept, methotrexate, or ustekinumab.
On initial presentation, physical examination revealed diffuse erythema and scaling with associated edema of the face, trunk, and extremities (Figure 1). A biopsy from the patient’s right arm demonstrated a superficial perivascular inflammatory infiltrate composed of lymphocytes, histiocytes, and scattered eosinophils consistent with spongiotic dermatitis (Figure 2). Cyclosporine 225 mg twice daily and topical corticosteroids were started.
Over the next several months, the patient had several admissions secondary to recurrent skin abscesses in the setting of refractory erythroderma. He underwent trials of infliximab, corticosteroids, intravenous immunoglobulin, guselkumab, and acitretin with minimal improvement. He underwent an extensive laboratory and radiologic workup, which was notable for cyclical peripheral eosinophilia and elevated IgE levels correlating with the erythroderma flares. A second biopsy was obtained and continued to demonstrate changes consistent with AD.
Four months after the initial hospitalization, all psoriasis medications were stopped, and the patient was started on dupilumab 300 mg/2 mL every 2 weeks and an 8-week oral prednisone taper. This combination led to notable clinical improvement and resolution of peripheral eosinophilia. Several months after disease remission, he began to develop worsening erythema and pruritus on the trunk and extremities, followed by the development of new psoriatic lesions (Figure 3) with a biopsy consistent with psoriasis (Figure 4). The patient was continued on dupilumab, but cyclosporine was added. The patient self-discontinued dupilumab owing to injection-site discomfort and has been slowly weaning off oral cyclosporine with 1 to 2 remaining eczematous plaques and 1 to 2 psoriatic plaques managed by topical corticosteroids.
Comment
We present a patient with psoriasis that was well controlled on secukinumab who developed severe AD following treatment with secukinumab. The AD resolved following treatment with dupilumab and a tapering dose of prednisone. However, after several months of treatment with dupilumab alone, he began to develop psoriatic lesions again. This case supports findings in a case series describing the development of AD in patients with psoriasis treated with IL-17 inhibitors5 and a recent case report describing a patient with AD who developed psoriasis following treatment with an IL-4/IL-13 inhibitor.6
Recognized adverse effects demonstrate biologic medications’ contributions to both normal as well as aberrant immunologic responses. For example, IL-17 plays an essential role in innate and adaptive immune responses against infections at mucosal and cutaneous interfaces, as demonstrated by chronic mucocutaneous candidiasis in patients with genetic defects in IL-17–related pathways.7 Similarly, in patients taking IL-17 antagonists, an increase in the incidence of Candida infections has been observed.8 In patients with concurrent psoriasis and inflammatory bowel disease (IBD), treatment with IL-17 inhibitors is contraindicated due to the risk of exacerbating the IBD. This observation is somewhat paradoxical, as increased IL-17 release by TH17 cells is implicated in the pathogenesis of IBD.9 Interestingly, it is now thought that IL-17 may play a protective role in T-cell–driven intestinal inflammation through induction of protective intestinal epithelial gene expression and increased mucosal defense against gut microbes, explaining the worsening of IBD in patients on IL-17 inhibitors.10 These adverse effects illustrate the complicated and varied roles biologic medications play in immunologic response.
Given that TH1 and TH2 exert opposing immune mechanisms, it is uncommon for psoriasis and AD to coexist in a single patient. However, patients who exhibit concurrent findings may represent a unique population in which psoriasis and AD coexist, perhaps because of an underlying genetic predisposition. Moreover, targeted treatment of pathways unique to these disease processes may result in paradoxical flaring of the nontargeted pathway. It also is possible that inhibition of a specific T-cell pathway in a subset of patients will result in an immunologic imbalance, favoring increased activity of the opposing pathway in the absence of coexisting disease. In the case presented here, the findings may be explained by secukinumab’s inhibition of TH1/TH17-mediated inflammation, which resulted in a shift to a TH2-mediated inflammatory response manifesting as AD, as well as dupilumab’s inhibition of TH2-mediated inflammation, which caused a shift back to TH1-mediated inflammatory pathways. Additionally, for patients with changing morphologies exacerbated by biologic medications, alternative diagnoses, such as cutaneous T-cell lymphoma, may be considered.
Conclusion
We report an unusual case of secukinumab-induced AD in a patient with psoriasis that resolved following several months of treatment with dupilumab and a tapering dose of prednisone. Subsequently, this same patient developed re-emergence of psoriatic lesions with continued use of dupilumab, which was eventually discontinued by the patient despite appropriate disease control. In addition to illustrating the underlying pathophysiologic mechanisms of 2 common inflammatory dermatologic conditions, this case highlights how pharmacologic interventions targeted at specific immunologic pathways may have unintended consequences. Further investigation into the effects of targeted biologics on the TH1/TH2 immune axis is warranted to better understand the mechanism and possible implications of the phenotypic switching presented in this case.
Psoriasis vulgaris is a chronic inflammatory skin condition associated with notable elevation in helper T cell (TH) production of TH1/TH17-mediated inflammatory cytokines, including IL-17A.1 Upon binding of IL-17A to IL-17 receptors in the skin, an inflammatory cascade is triggered, resulting in the classic clinical appearance of psoriasis. Moderate to severe psoriasis often is managed by suppressing TH1/TH17-mediated inflammation using targeted immune therapy such as secukinumab, an IL-17A inhibitor.2 Atopic dermatitis (AD), another chronic inflammatory dermatosis, is associated with substantial elevation in TH2-mediated inflammatory cytokines, such as IL-4.3 Dupilumab, which interacts with IL-4R, disrupts the IL-4 and IL-13 signaling pathways and demonstrates considerable efficacy in the treatment of moderate to severe AD.4
A case series has shown that suppression of the TH1/TH17-mediated inflammation of psoriasis may paradoxically result in the development of TH2-mediated AD.5 Similarly, a recent case report described a patient who developed psoriasis following treatment of AD with dupilumab.6 Herein, we describe a patient with a history of psoriasis that was well controlled with secukinumab who developed severe refractory erythrodermic AD that resolved with dupilumab treatment. Following clearance of AD with dupilumab, he exhibited psoriasis recurrence.
Case Report
A 39-year-old man with a lifelong history of psoriasis was admitted to the hospital for management of severe erythroderma. Four years prior, secukinumab was initiated for treatment of psoriasis, resulting in excellent clinical response. He discontinued secukinumab after 2 years of treatment because of insurance coverage issues and managed his condition with only topical corticosteroids. He restarted secukinumab 10 months before admission because of a psoriasis flare. Shortly after resuming secukinumab, he developed a severe exfoliative erythroderma that was not responsive to corticosteroids, etanercept, methotrexate, or ustekinumab.
On initial presentation, physical examination revealed diffuse erythema and scaling with associated edema of the face, trunk, and extremities (Figure 1). A biopsy from the patient’s right arm demonstrated a superficial perivascular inflammatory infiltrate composed of lymphocytes, histiocytes, and scattered eosinophils consistent with spongiotic dermatitis (Figure 2). Cyclosporine 225 mg twice daily and topical corticosteroids were started.
Over the next several months, the patient had several admissions secondary to recurrent skin abscesses in the setting of refractory erythroderma. He underwent trials of infliximab, corticosteroids, intravenous immunoglobulin, guselkumab, and acitretin with minimal improvement. He underwent an extensive laboratory and radiologic workup, which was notable for cyclical peripheral eosinophilia and elevated IgE levels correlating with the erythroderma flares. A second biopsy was obtained and continued to demonstrate changes consistent with AD.
Four months after the initial hospitalization, all psoriasis medications were stopped, and the patient was started on dupilumab 300 mg/2 mL every 2 weeks and an 8-week oral prednisone taper. This combination led to notable clinical improvement and resolution of peripheral eosinophilia. Several months after disease remission, he began to develop worsening erythema and pruritus on the trunk and extremities, followed by the development of new psoriatic lesions (Figure 3) with a biopsy consistent with psoriasis (Figure 4). The patient was continued on dupilumab, but cyclosporine was added. The patient self-discontinued dupilumab owing to injection-site discomfort and has been slowly weaning off oral cyclosporine with 1 to 2 remaining eczematous plaques and 1 to 2 psoriatic plaques managed by topical corticosteroids.
Comment
We present a patient with psoriasis that was well controlled on secukinumab who developed severe AD following treatment with secukinumab. The AD resolved following treatment with dupilumab and a tapering dose of prednisone. However, after several months of treatment with dupilumab alone, he began to develop psoriatic lesions again. This case supports findings in a case series describing the development of AD in patients with psoriasis treated with IL-17 inhibitors5 and a recent case report describing a patient with AD who developed psoriasis following treatment with an IL-4/IL-13 inhibitor.6
Recognized adverse effects demonstrate biologic medications’ contributions to both normal as well as aberrant immunologic responses. For example, IL-17 plays an essential role in innate and adaptive immune responses against infections at mucosal and cutaneous interfaces, as demonstrated by chronic mucocutaneous candidiasis in patients with genetic defects in IL-17–related pathways.7 Similarly, in patients taking IL-17 antagonists, an increase in the incidence of Candida infections has been observed.8 In patients with concurrent psoriasis and inflammatory bowel disease (IBD), treatment with IL-17 inhibitors is contraindicated due to the risk of exacerbating the IBD. This observation is somewhat paradoxical, as increased IL-17 release by TH17 cells is implicated in the pathogenesis of IBD.9 Interestingly, it is now thought that IL-17 may play a protective role in T-cell–driven intestinal inflammation through induction of protective intestinal epithelial gene expression and increased mucosal defense against gut microbes, explaining the worsening of IBD in patients on IL-17 inhibitors.10 These adverse effects illustrate the complicated and varied roles biologic medications play in immunologic response.
Given that TH1 and TH2 exert opposing immune mechanisms, it is uncommon for psoriasis and AD to coexist in a single patient. However, patients who exhibit concurrent findings may represent a unique population in which psoriasis and AD coexist, perhaps because of an underlying genetic predisposition. Moreover, targeted treatment of pathways unique to these disease processes may result in paradoxical flaring of the nontargeted pathway. It also is possible that inhibition of a specific T-cell pathway in a subset of patients will result in an immunologic imbalance, favoring increased activity of the opposing pathway in the absence of coexisting disease. In the case presented here, the findings may be explained by secukinumab’s inhibition of TH1/TH17-mediated inflammation, which resulted in a shift to a TH2-mediated inflammatory response manifesting as AD, as well as dupilumab’s inhibition of TH2-mediated inflammation, which caused a shift back to TH1-mediated inflammatory pathways. Additionally, for patients with changing morphologies exacerbated by biologic medications, alternative diagnoses, such as cutaneous T-cell lymphoma, may be considered.
Conclusion
We report an unusual case of secukinumab-induced AD in a patient with psoriasis that resolved following several months of treatment with dupilumab and a tapering dose of prednisone. Subsequently, this same patient developed re-emergence of psoriatic lesions with continued use of dupilumab, which was eventually discontinued by the patient despite appropriate disease control. In addition to illustrating the underlying pathophysiologic mechanisms of 2 common inflammatory dermatologic conditions, this case highlights how pharmacologic interventions targeted at specific immunologic pathways may have unintended consequences. Further investigation into the effects of targeted biologics on the TH1/TH2 immune axis is warranted to better understand the mechanism and possible implications of the phenotypic switching presented in this case.
- Diani M, Altomare G, Reali E. T helper cell subsets in clinical manifestations of psoriasis. J Immunol Res. 2016;2016:7692024.
- Langley RG, Elewski BE, Lebwohl M, et al. Secukinumab in plaque psoriasis—results of two phase 3 trials. N Engl J Med. 2014;371:326-338.
- van der Heijden FL, Wierenga EA, Bos JD, et al. High frequency of IL-4-producing CD4+ allergen-specific T lymphocytes in atopic dermatitis lesional skin. J Invest Dermatol. 1991;97:389-394.
- Beck LA, Thaçi D, Hamilton JD, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med. 2014;371:130-139.
- Lai FYX, Higgins E, Smith CH, et al. Morphologic switch from psoriasiform to eczematous dermatitis after anti-IL-17 therapy: a case series. JAMA Dermatol. 2019;155:1082-1084.
- Varma A, Levitt J. Dupilumab-induced phenotype switching from atopic dermatitis to psoriasis. JAAD Case Rep. 2020;6:217-218.
- Ling Y, Puel A. IL-17 and infections. Actas Dermosifiliogr. 2014;105(suppl 1):34-40.
- Saunte DM, Mrowietz U, Puig L, et al. Candida infections in patients with psoriasis and psoriatic arthritis treated with interleukin-17 inhibitors and their practical management. Br J Dermatol. 2017;177:47-62.
- Hölttä V, Klemetti P, Sipponen T, et al. IL-23/IL-17 immunity as a hallmark of Crohn’s disease. Inflamm Bowel Dis. 2008;14:1175-1184.
- Smith MK, Pai J, Panaccione R, et al. Crohn’s-like disease in a patient exposed to anti-interleukin-17 blockade (ixekizumab) for the treatment of chronic plaque psoriasis: a case report. BMC Gastroenterol. 2019;19:162.
- Diani M, Altomare G, Reali E. T helper cell subsets in clinical manifestations of psoriasis. J Immunol Res. 2016;2016:7692024.
- Langley RG, Elewski BE, Lebwohl M, et al. Secukinumab in plaque psoriasis—results of two phase 3 trials. N Engl J Med. 2014;371:326-338.
- van der Heijden FL, Wierenga EA, Bos JD, et al. High frequency of IL-4-producing CD4+ allergen-specific T lymphocytes in atopic dermatitis lesional skin. J Invest Dermatol. 1991;97:389-394.
- Beck LA, Thaçi D, Hamilton JD, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med. 2014;371:130-139.
- Lai FYX, Higgins E, Smith CH, et al. Morphologic switch from psoriasiform to eczematous dermatitis after anti-IL-17 therapy: a case series. JAMA Dermatol. 2019;155:1082-1084.
- Varma A, Levitt J. Dupilumab-induced phenotype switching from atopic dermatitis to psoriasis. JAAD Case Rep. 2020;6:217-218.
- Ling Y, Puel A. IL-17 and infections. Actas Dermosifiliogr. 2014;105(suppl 1):34-40.
- Saunte DM, Mrowietz U, Puig L, et al. Candida infections in patients with psoriasis and psoriatic arthritis treated with interleukin-17 inhibitors and their practical management. Br J Dermatol. 2017;177:47-62.
- Hölttä V, Klemetti P, Sipponen T, et al. IL-23/IL-17 immunity as a hallmark of Crohn’s disease. Inflamm Bowel Dis. 2008;14:1175-1184.
- Smith MK, Pai J, Panaccione R, et al. Crohn’s-like disease in a patient exposed to anti-interleukin-17 blockade (ixekizumab) for the treatment of chronic plaque psoriasis: a case report. BMC Gastroenterol. 2019;19:162.
Practice Points
- Treatment of psoriasis vulgaris, a helper T cell TH1/TH17-mediated skin condition, with secukinumab may result in phenotypic switching to TH2-mediated atopic dermatitis.
- Atopic dermatitis responds well to dupilumab but may result in phenotypic switching to psoriasis.
- Biologic therapies targeted at specific immunologic pathways may have unintended consequences on the TH1/TH2 immune axis.

Rapid Desensitization after a Type I Hypersensitivity Reaction to Ceftazidime/Avibactam
Cerebral palsy (CP) embodies a collection of disorders involving permanent but nonprogressive motor dysfunction secondary to one of a variety of abnormal disturbances that can occur in the developing fetal or infantile brain.1 The motor impairment of CP classically leads to irregularities in muscle tone, posture, and/or movement, resulting in limitations of functional abilities that vary in severity.1,2 Patients with CP commonly experience dysphagia, gastroesophageal reflux disease, impaired airway clearance, chest wall and spine deformities, restrictive lung disease, and/or recurrent aspiration.1 Consequently, pulmonary disease is the leading cause of morbidity and mortality in patients with severe CP, characterized by recurrent bacterial infections.3,4
Frequent antibiotic use increases the risk of multidrug-resistant pathogen formation and hypersensitivity to antibiotics. Life-threatening allergic reactions in a patient population with impaired lung function significantly complicates patient management, often leading to suboptimal treatment with second-line agents.5 This case study describes a previously penicillin-tolerant patient with CP who developed a type I hypersensitivity reaction to ceftazidime/avibactam and was treated successfully with the antibiotic after rapid induction of temporary tolerance.
Case Presentation
A 34-year-old male with a complex medical history of severe spastic CP and atonic seizures was recently diagnosed with adenocarcinoma of the colon and admitted for ileostomy and sigmoidectomy. The surgery was complicated by spillage of intestinal contents into the peritoneal cavity 3 days postoperation. The patient was urgently taken to the operating room for exploratory laparotomy, culminating in remaining colectomy, complete abdominal washout, and wound vacuum placement. He continued to deteriorate clinically over the next few weeks, beginning with the development of feculent peritonitis and septic shock. Respiratory distress ensued, and the patient required a tracheostomy with mechanical ventilation. A computed tomography of the chest was consistent with multifocal pneumonia, and a respiratory culture of bronchioalveolar lavage fluid cultivated Klebsiella pneumoniae, a carbapenem-resistant Enterobacteriaceae.
The infectious disease service was consulted and recommended ceftazidime/avibactam as the only acceptable antibiotic to treat this organism. The patient had no history of drug hypersensitivities. However, he developed diffuse, generalized urticaria and predominately right-sided flushing immediately following the onset of the antibiotic infusion. The urticaria was pruritic. The patient did not have angioedema, and he did not experience any adverse respiratory, cardiac, gastrointestinal, or neurologic symptoms. The infusion was ceased immediately, and the patient was treated with a combination of diphenhydramine 50 mg IV and ranitidine 50 mg IV. Resolution of his hypersensitivity symptoms occurred within an hour of treatment, and vital signs remained stable with no resurgence of symptoms. At the time of his reaction, the patient also was taking pantoprazole, valproate, metoprolol, risperidone, and oxycodone as needed for pain. A tryptase level was not measured.
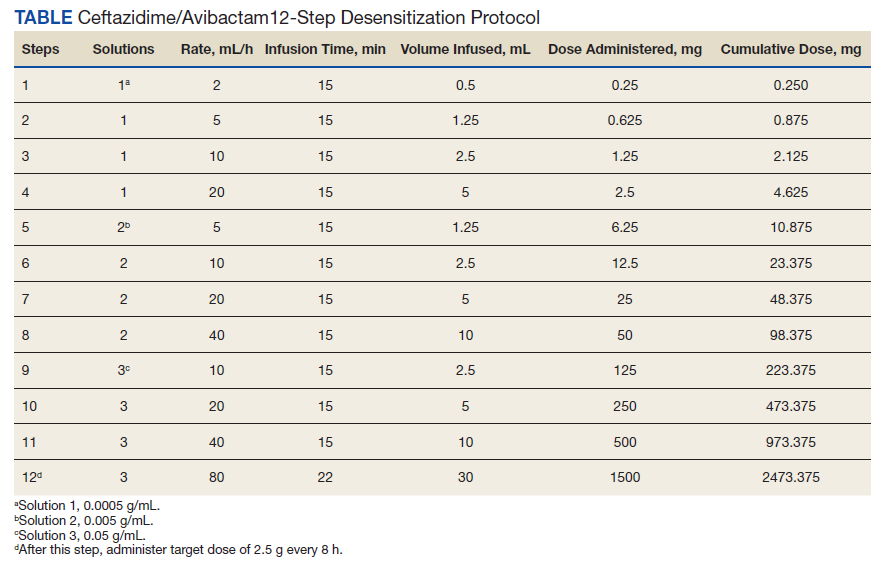
The allergy and immunology service was consulted for rapid desensitization to ceftazidime/avibactam as the culture and sensitivity test demonstrated the bacterium to be resistant to alternative antibiotics. Skin testing to ceftazidime/avibactam was deferred at the time due to the patient’s critical illness. The patient was premedicated with diphenhydramine and ranitidine 50 mg IV. Rapid IV desensitization was performed using a standard 12-step protocol developed for chemotherapeutic agents but demonstrated as safe and effective when applied to antibiotics in patients with cystic fibrosis.5 The antibiotic was administered in sequential 15-minute intervals for a total of 12 progressively doubled doses with continuous monitoring for the appearance of allergic reactions (Table). The target dose of 2.5 g was successfully achieved, and the patient tolerated a complete 14-day treatment regimen with no further adverse reactions to the medication. During the remainder of his hospital admission, the patient improved significantly without further complications.
Discussion
This is the first reported case in the literature to describe a type I hypersensitivity reaction with rapid IV induction of tolerance to ceftazidime/avibactam. We describe his reaction as type I hypersensitivity because the patient developed immediate generalized urticaria and flushing. Use of a safe desensitization protocol, demonstrated in this case report, is paramount to optimal management of infections in patient populations with severely decreased lung function, such as CP.5-7 It provides a safe and effective technique to maintain patients on first line, preferred therapy, despite their increased risk of potentially life-threatening allergic reactions.
Interestingly, this patient previously tolerated penicillins and cephalosporins without adverse reactions, suggesting the possibility of a non–IgE-mediated vs an IgE-mediated mechanism to the hypersensitivity reaction. The patient also was receiving oxycodone at the time of his reaction, and oxycodone can cause nonspecific mast cell degranulation. Additional information from skin testing to ceftazidime/avibactam could help determine whether the patient had an IgE-mediated hypersensitivity reaction. This information could help clarify the culprit agent and guide further avoidance recommendations.
Unfortunately, because the patient was critically ill, skin testing was not performed, and he underwent an urgent antibiotic desensitization with success. It was recommended that the patient follow up in the allergy and immunology clinic for further evaluation with skin testing to ceftazidime/avibactam as well as other β-lactams to determine his future risk of reaction. Unfortunately, he was lost to follow-up.
Frequent IV antibiotic use is a risk factor for the development of antibiotic allergies.8,9 This patient had received many prior courses of IV antibiotics, and this factor most likely contributed to his immediate hypersensitivity reaction to ceftazidime/avibactam. Fortunately, he tolerated a rapid induction of tolerance.
As life expectancies for patients with chronic medical conditions that involve recurrent infections increase, the associated emergence of multidrug-resistant pathogens and necessity for use of novel combination antibiotics should prompt further investigation of nonirritating doses of these drugs for skin testing in the case of drug hypersensitivities. This information would be essential for skin prick testing and determination of whether patients have a true IgE-mediated reaction to these antibiotics.
Conclusions
This is the first case report demonstrating a successful rapid induction of tolerance for the antibiotic ceftazidime/avibactam. Fortunately, the patient tolerated the desensitization procedure without further adverse reactions, and he had a resolution of his infection.
1. Rosenbaum P, Paneth N, Leviton A, et al. A report: the definition and classification of cerebral palsy April 2006. Dev Med Child Neurol. 2007;109:8-14.
2. Haak P, Lenski M, Hidecker MJ, et al. Cerebral palsy and aging. Dev Med Child Neurol. 2009;51(suppl 4):16-23. doi:10.1111/j.1469-8749.2009.03428.x
3. Duruflé-Tapin A, Colin A, Nicolas B, Lebreton C, Dauvergne F, Gallien P. Analysis of the medical causes of death in cerebral palsy. Ann Phys Rehabil Med. 2014;57(1):24-37. doi:10.1016/j.rehab.2013.11.002
4. Boel L, Pernet K, Toussaint M, et al. Respiratory morbidity in children with cerebral palsy: an overview. Dev Med Child Neurol. 2019;61(6):646-653. doi:10.1111/dmcn.14060
5. Legere HJ 3rd, Palis RI, Rodriguez Bouza T, Uluer AZ, Castells MC. A safe protocol for rapid desensitization in patients with cystic fibrosis and antibiotic hypersensitivity. J Cyst Fibros. 2009;8(6):418-424. doi:10.1016/j.jcf.2009.08.002
6. Castells M. Rapid desensitization for hypersensitivity reactions to medications. Immunol Allergy Clin North Am. 2009;29(3):585-606. doi:10.1016/j.iac.2009.04.012
7. Liu A, Fanning L, Chong H, et al. Desensitization regimens for drug allergy: state of the art in the 21st century. Clin Exp Allergy. 2011;41(12):1679-1689. doi:10.1111/j.1365-2222.2011.03825.x
8. Thong BY, Tan TC. Epidemiology and risk factors for drug allergy. Br J Clin Pharmacol. 2011;71(5):684-700. doi:10.1111/j.1365-2125.2010.03774.x
9. Adkinson NF Jr. Risk factors for drug allergy. J Allergy Clin Immunol. 1984;74(4, pt 2):567-572. doi:10.1016/0091-6749(84)90108-8
Cerebral palsy (CP) embodies a collection of disorders involving permanent but nonprogressive motor dysfunction secondary to one of a variety of abnormal disturbances that can occur in the developing fetal or infantile brain.1 The motor impairment of CP classically leads to irregularities in muscle tone, posture, and/or movement, resulting in limitations of functional abilities that vary in severity.1,2 Patients with CP commonly experience dysphagia, gastroesophageal reflux disease, impaired airway clearance, chest wall and spine deformities, restrictive lung disease, and/or recurrent aspiration.1 Consequently, pulmonary disease is the leading cause of morbidity and mortality in patients with severe CP, characterized by recurrent bacterial infections.3,4
Frequent antibiotic use increases the risk of multidrug-resistant pathogen formation and hypersensitivity to antibiotics. Life-threatening allergic reactions in a patient population with impaired lung function significantly complicates patient management, often leading to suboptimal treatment with second-line agents.5 This case study describes a previously penicillin-tolerant patient with CP who developed a type I hypersensitivity reaction to ceftazidime/avibactam and was treated successfully with the antibiotic after rapid induction of temporary tolerance.
Case Presentation
A 34-year-old male with a complex medical history of severe spastic CP and atonic seizures was recently diagnosed with adenocarcinoma of the colon and admitted for ileostomy and sigmoidectomy. The surgery was complicated by spillage of intestinal contents into the peritoneal cavity 3 days postoperation. The patient was urgently taken to the operating room for exploratory laparotomy, culminating in remaining colectomy, complete abdominal washout, and wound vacuum placement. He continued to deteriorate clinically over the next few weeks, beginning with the development of feculent peritonitis and septic shock. Respiratory distress ensued, and the patient required a tracheostomy with mechanical ventilation. A computed tomography of the chest was consistent with multifocal pneumonia, and a respiratory culture of bronchioalveolar lavage fluid cultivated Klebsiella pneumoniae, a carbapenem-resistant Enterobacteriaceae.
The infectious disease service was consulted and recommended ceftazidime/avibactam as the only acceptable antibiotic to treat this organism. The patient had no history of drug hypersensitivities. However, he developed diffuse, generalized urticaria and predominately right-sided flushing immediately following the onset of the antibiotic infusion. The urticaria was pruritic. The patient did not have angioedema, and he did not experience any adverse respiratory, cardiac, gastrointestinal, or neurologic symptoms. The infusion was ceased immediately, and the patient was treated with a combination of diphenhydramine 50 mg IV and ranitidine 50 mg IV. Resolution of his hypersensitivity symptoms occurred within an hour of treatment, and vital signs remained stable with no resurgence of symptoms. At the time of his reaction, the patient also was taking pantoprazole, valproate, metoprolol, risperidone, and oxycodone as needed for pain. A tryptase level was not measured.
The allergy and immunology service was consulted for rapid desensitization to ceftazidime/avibactam as the culture and sensitivity test demonstrated the bacterium to be resistant to alternative antibiotics. Skin testing to ceftazidime/avibactam was deferred at the time due to the patient’s critical illness. The patient was premedicated with diphenhydramine and ranitidine 50 mg IV. Rapid IV desensitization was performed using a standard 12-step protocol developed for chemotherapeutic agents but demonstrated as safe and effective when applied to antibiotics in patients with cystic fibrosis.5 The antibiotic was administered in sequential 15-minute intervals for a total of 12 progressively doubled doses with continuous monitoring for the appearance of allergic reactions (Table). The target dose of 2.5 g was successfully achieved, and the patient tolerated a complete 14-day treatment regimen with no further adverse reactions to the medication. During the remainder of his hospital admission, the patient improved significantly without further complications.
Discussion
This is the first reported case in the literature to describe a type I hypersensitivity reaction with rapid IV induction of tolerance to ceftazidime/avibactam. We describe his reaction as type I hypersensitivity because the patient developed immediate generalized urticaria and flushing. Use of a safe desensitization protocol, demonstrated in this case report, is paramount to optimal management of infections in patient populations with severely decreased lung function, such as CP.5-7 It provides a safe and effective technique to maintain patients on first line, preferred therapy, despite their increased risk of potentially life-threatening allergic reactions.
Interestingly, this patient previously tolerated penicillins and cephalosporins without adverse reactions, suggesting the possibility of a non–IgE-mediated vs an IgE-mediated mechanism to the hypersensitivity reaction. The patient also was receiving oxycodone at the time of his reaction, and oxycodone can cause nonspecific mast cell degranulation. Additional information from skin testing to ceftazidime/avibactam could help determine whether the patient had an IgE-mediated hypersensitivity reaction. This information could help clarify the culprit agent and guide further avoidance recommendations.
Unfortunately, because the patient was critically ill, skin testing was not performed, and he underwent an urgent antibiotic desensitization with success. It was recommended that the patient follow up in the allergy and immunology clinic for further evaluation with skin testing to ceftazidime/avibactam as well as other β-lactams to determine his future risk of reaction. Unfortunately, he was lost to follow-up.
Frequent IV antibiotic use is a risk factor for the development of antibiotic allergies.8,9 This patient had received many prior courses of IV antibiotics, and this factor most likely contributed to his immediate hypersensitivity reaction to ceftazidime/avibactam. Fortunately, he tolerated a rapid induction of tolerance.
As life expectancies for patients with chronic medical conditions that involve recurrent infections increase, the associated emergence of multidrug-resistant pathogens and necessity for use of novel combination antibiotics should prompt further investigation of nonirritating doses of these drugs for skin testing in the case of drug hypersensitivities. This information would be essential for skin prick testing and determination of whether patients have a true IgE-mediated reaction to these antibiotics.
Conclusions
This is the first case report demonstrating a successful rapid induction of tolerance for the antibiotic ceftazidime/avibactam. Fortunately, the patient tolerated the desensitization procedure without further adverse reactions, and he had a resolution of his infection.
Cerebral palsy (CP) embodies a collection of disorders involving permanent but nonprogressive motor dysfunction secondary to one of a variety of abnormal disturbances that can occur in the developing fetal or infantile brain.1 The motor impairment of CP classically leads to irregularities in muscle tone, posture, and/or movement, resulting in limitations of functional abilities that vary in severity.1,2 Patients with CP commonly experience dysphagia, gastroesophageal reflux disease, impaired airway clearance, chest wall and spine deformities, restrictive lung disease, and/or recurrent aspiration.1 Consequently, pulmonary disease is the leading cause of morbidity and mortality in patients with severe CP, characterized by recurrent bacterial infections.3,4
Frequent antibiotic use increases the risk of multidrug-resistant pathogen formation and hypersensitivity to antibiotics. Life-threatening allergic reactions in a patient population with impaired lung function significantly complicates patient management, often leading to suboptimal treatment with second-line agents.5 This case study describes a previously penicillin-tolerant patient with CP who developed a type I hypersensitivity reaction to ceftazidime/avibactam and was treated successfully with the antibiotic after rapid induction of temporary tolerance.
Case Presentation
A 34-year-old male with a complex medical history of severe spastic CP and atonic seizures was recently diagnosed with adenocarcinoma of the colon and admitted for ileostomy and sigmoidectomy. The surgery was complicated by spillage of intestinal contents into the peritoneal cavity 3 days postoperation. The patient was urgently taken to the operating room for exploratory laparotomy, culminating in remaining colectomy, complete abdominal washout, and wound vacuum placement. He continued to deteriorate clinically over the next few weeks, beginning with the development of feculent peritonitis and septic shock. Respiratory distress ensued, and the patient required a tracheostomy with mechanical ventilation. A computed tomography of the chest was consistent with multifocal pneumonia, and a respiratory culture of bronchioalveolar lavage fluid cultivated Klebsiella pneumoniae, a carbapenem-resistant Enterobacteriaceae.
The infectious disease service was consulted and recommended ceftazidime/avibactam as the only acceptable antibiotic to treat this organism. The patient had no history of drug hypersensitivities. However, he developed diffuse, generalized urticaria and predominately right-sided flushing immediately following the onset of the antibiotic infusion. The urticaria was pruritic. The patient did not have angioedema, and he did not experience any adverse respiratory, cardiac, gastrointestinal, or neurologic symptoms. The infusion was ceased immediately, and the patient was treated with a combination of diphenhydramine 50 mg IV and ranitidine 50 mg IV. Resolution of his hypersensitivity symptoms occurred within an hour of treatment, and vital signs remained stable with no resurgence of symptoms. At the time of his reaction, the patient also was taking pantoprazole, valproate, metoprolol, risperidone, and oxycodone as needed for pain. A tryptase level was not measured.
The allergy and immunology service was consulted for rapid desensitization to ceftazidime/avibactam as the culture and sensitivity test demonstrated the bacterium to be resistant to alternative antibiotics. Skin testing to ceftazidime/avibactam was deferred at the time due to the patient’s critical illness. The patient was premedicated with diphenhydramine and ranitidine 50 mg IV. Rapid IV desensitization was performed using a standard 12-step protocol developed for chemotherapeutic agents but demonstrated as safe and effective when applied to antibiotics in patients with cystic fibrosis.5 The antibiotic was administered in sequential 15-minute intervals for a total of 12 progressively doubled doses with continuous monitoring for the appearance of allergic reactions (Table). The target dose of 2.5 g was successfully achieved, and the patient tolerated a complete 14-day treatment regimen with no further adverse reactions to the medication. During the remainder of his hospital admission, the patient improved significantly without further complications.
Discussion
This is the first reported case in the literature to describe a type I hypersensitivity reaction with rapid IV induction of tolerance to ceftazidime/avibactam. We describe his reaction as type I hypersensitivity because the patient developed immediate generalized urticaria and flushing. Use of a safe desensitization protocol, demonstrated in this case report, is paramount to optimal management of infections in patient populations with severely decreased lung function, such as CP.5-7 It provides a safe and effective technique to maintain patients on first line, preferred therapy, despite their increased risk of potentially life-threatening allergic reactions.
Interestingly, this patient previously tolerated penicillins and cephalosporins without adverse reactions, suggesting the possibility of a non–IgE-mediated vs an IgE-mediated mechanism to the hypersensitivity reaction. The patient also was receiving oxycodone at the time of his reaction, and oxycodone can cause nonspecific mast cell degranulation. Additional information from skin testing to ceftazidime/avibactam could help determine whether the patient had an IgE-mediated hypersensitivity reaction. This information could help clarify the culprit agent and guide further avoidance recommendations.
Unfortunately, because the patient was critically ill, skin testing was not performed, and he underwent an urgent antibiotic desensitization with success. It was recommended that the patient follow up in the allergy and immunology clinic for further evaluation with skin testing to ceftazidime/avibactam as well as other β-lactams to determine his future risk of reaction. Unfortunately, he was lost to follow-up.
Frequent IV antibiotic use is a risk factor for the development of antibiotic allergies.8,9 This patient had received many prior courses of IV antibiotics, and this factor most likely contributed to his immediate hypersensitivity reaction to ceftazidime/avibactam. Fortunately, he tolerated a rapid induction of tolerance.
As life expectancies for patients with chronic medical conditions that involve recurrent infections increase, the associated emergence of multidrug-resistant pathogens and necessity for use of novel combination antibiotics should prompt further investigation of nonirritating doses of these drugs for skin testing in the case of drug hypersensitivities. This information would be essential for skin prick testing and determination of whether patients have a true IgE-mediated reaction to these antibiotics.
Conclusions
This is the first case report demonstrating a successful rapid induction of tolerance for the antibiotic ceftazidime/avibactam. Fortunately, the patient tolerated the desensitization procedure without further adverse reactions, and he had a resolution of his infection.
1. Rosenbaum P, Paneth N, Leviton A, et al. A report: the definition and classification of cerebral palsy April 2006. Dev Med Child Neurol. 2007;109:8-14.
2. Haak P, Lenski M, Hidecker MJ, et al. Cerebral palsy and aging. Dev Med Child Neurol. 2009;51(suppl 4):16-23. doi:10.1111/j.1469-8749.2009.03428.x
3. Duruflé-Tapin A, Colin A, Nicolas B, Lebreton C, Dauvergne F, Gallien P. Analysis of the medical causes of death in cerebral palsy. Ann Phys Rehabil Med. 2014;57(1):24-37. doi:10.1016/j.rehab.2013.11.002
4. Boel L, Pernet K, Toussaint M, et al. Respiratory morbidity in children with cerebral palsy: an overview. Dev Med Child Neurol. 2019;61(6):646-653. doi:10.1111/dmcn.14060
5. Legere HJ 3rd, Palis RI, Rodriguez Bouza T, Uluer AZ, Castells MC. A safe protocol for rapid desensitization in patients with cystic fibrosis and antibiotic hypersensitivity. J Cyst Fibros. 2009;8(6):418-424. doi:10.1016/j.jcf.2009.08.002
6. Castells M. Rapid desensitization for hypersensitivity reactions to medications. Immunol Allergy Clin North Am. 2009;29(3):585-606. doi:10.1016/j.iac.2009.04.012
7. Liu A, Fanning L, Chong H, et al. Desensitization regimens for drug allergy: state of the art in the 21st century. Clin Exp Allergy. 2011;41(12):1679-1689. doi:10.1111/j.1365-2222.2011.03825.x
8. Thong BY, Tan TC. Epidemiology and risk factors for drug allergy. Br J Clin Pharmacol. 2011;71(5):684-700. doi:10.1111/j.1365-2125.2010.03774.x
9. Adkinson NF Jr. Risk factors for drug allergy. J Allergy Clin Immunol. 1984;74(4, pt 2):567-572. doi:10.1016/0091-6749(84)90108-8
1. Rosenbaum P, Paneth N, Leviton A, et al. A report: the definition and classification of cerebral palsy April 2006. Dev Med Child Neurol. 2007;109:8-14.
2. Haak P, Lenski M, Hidecker MJ, et al. Cerebral palsy and aging. Dev Med Child Neurol. 2009;51(suppl 4):16-23. doi:10.1111/j.1469-8749.2009.03428.x
3. Duruflé-Tapin A, Colin A, Nicolas B, Lebreton C, Dauvergne F, Gallien P. Analysis of the medical causes of death in cerebral palsy. Ann Phys Rehabil Med. 2014;57(1):24-37. doi:10.1016/j.rehab.2013.11.002
4. Boel L, Pernet K, Toussaint M, et al. Respiratory morbidity in children with cerebral palsy: an overview. Dev Med Child Neurol. 2019;61(6):646-653. doi:10.1111/dmcn.14060
5. Legere HJ 3rd, Palis RI, Rodriguez Bouza T, Uluer AZ, Castells MC. A safe protocol for rapid desensitization in patients with cystic fibrosis and antibiotic hypersensitivity. J Cyst Fibros. 2009;8(6):418-424. doi:10.1016/j.jcf.2009.08.002
6. Castells M. Rapid desensitization for hypersensitivity reactions to medications. Immunol Allergy Clin North Am. 2009;29(3):585-606. doi:10.1016/j.iac.2009.04.012
7. Liu A, Fanning L, Chong H, et al. Desensitization regimens for drug allergy: state of the art in the 21st century. Clin Exp Allergy. 2011;41(12):1679-1689. doi:10.1111/j.1365-2222.2011.03825.x
8. Thong BY, Tan TC. Epidemiology and risk factors for drug allergy. Br J Clin Pharmacol. 2011;71(5):684-700. doi:10.1111/j.1365-2125.2010.03774.x
9. Adkinson NF Jr. Risk factors for drug allergy. J Allergy Clin Immunol. 1984;74(4, pt 2):567-572. doi:10.1016/0091-6749(84)90108-8
The Balance of Truth-Telling and Respect for Confidentiality: The Ethics of Case Reports
Medical case reports are as old as the healing profession itself.1 These ancient medical stories have a modern definition: “A case report is a narrative that describes, for medical, scientific or educational purposes, a medical problem experienced by one or more patients.”2 Case report experts describe the 3-fold purposes of this type of research: as a mainstay of education; a harbinger of emerging illnesses; and an appraiser of new interventions. Case-based education has long been a pillar of health professions education: Nurses, doctors, and allied health professionals are taught and learn through reading and discussing with their teachers and each other about cases of their own patients and of those in the literature.3 Case reports also have helped identify and raise awareness of new diseases and rare conditions, such as HIV.4 Finally, case reports have alerted regulatory agencies and the medical community about medication adverse effects, such as birth defects from thalidomide.5
Case reports also have been criticized on both scientific and ethical grounds. Critics argue that many case reports often lack the rigor and consistency of other types of research.6 Three recent trends in medical publication have strengthened the validity of these criticisms: the increase in the popularity of case reports; the corresponding increase in submissions to journals, including Federal Practitioner; and the rise of predatory publishers.7,8
The ethical scrutiny of case reports discussed in this column focuses on the tension between providing readers with adequate, accurate information to fulfil the goals of case reports while also protecting patient confidentiality. The latter issue during most of the history of medicine was not considered by health care professionals when the prevailing paternalism supported a professional-oriented approach to health care. The rise of bioethics in the 1960s and 1970s began the shift toward patient autonomy in medical decision making and patient rights to control their protected health information that rendered case reports ethically problematic.
To address both changes in ethical standards and scientific limitations, a committee of clinicians, researchers, and journal editors formed the Case Report (CARE) group.2,8 The group undertook an effort to improve the quality of case reports. From 2011 to 2012, they developed the CARE guidelines for clinical case reporting. The guidance took the form of a Statement and Checklist presented at the 2013 International Congress on Peer Review and Biomedical Publication. Since their presentation, multiple prestigious medical journals in many countries have implemented these recommendations.
As part of an overall effort to raise the ethical caliber of our own journal, Federal Practitioner will begin to implement the CARE guidelines for case reports for all future submissions. Use of the CARE recommendations will help prospective authors enhance the scientific value and ethical caliber of case reports submitted to the journal as well as assist the Federal Practitioner editorial team, editorial board, and peer reviewers to evaluate submissions more judiciously.
An essential part of the CARE guidelines is that the patient who is the subject of the case report provide informed consent for the publication of their personal narrative. The CARE group considers this an “ethical duty” of authors and editors alike. In “exceptional circumstances” such as if the patient is a minor or permanently incapacitated, a guardian or relative may grant consent. In the rare event that even with exhaustive attempts, if informed consent cannot be obtained from a patient or their representative, then the authors of the case report must submit a statement to this effect.4 Some journals may require that the authors obtain the approval of an institutional review board or the permission of an ethics or other institutional committee or a privacy officer.2
Requesting the patient’s consent is an extension of the shared decision making that is now a best practice in clinical care into the arena of research, making the patient or their representative a partner in the work. Ethicists have recommended inviting patients or relatives to read a draft of the case report and agree to its publication or request specific modifications to the manuscript. The CARE group rightly points out that with the rise of open notes in medical documentation, patients increasingly have access to their charts in near or real time.2 Gone are the days of Sir William Osler when only doctors read medical journals and all of these technical developments as well as standards of research and social changes in the practitioner-patient relationship make it imperative that writers and editors join together to make case reports more transparent, accurate, and consistent.7
An additional step to protect patient privacy is the requirement that authors either de-identify potentially identifiable health information, such as age, birth, death, admission, and discharge dates, or in some instances obtain separate consent for the release of that protected data.8 These restrictions constitute a challenge to case report authors who in some instances may consider these same facts critical to the integrity of the case presentation that have made some scholars doubt their continued viability. After all, the contribution of the case to the medical literature often lies in its very particularity. Conversely, no matter how frustrated we might become during writing a case report, we would not want to see our own protected health information or that of our family on a website or in print without our knowledge or approval. Indeed, the International Committee of Medical Journal Editors states that “If identifying characteristics are de-identified, authors should provide assurance, and editors should so note, that such changes do not distort scientific meaning.”9
However, the exponential growth of the internet, the spread of social media, and the ubiquity of a plethora of electronic devices, which prior generations of writers and readers could not even imagine, make these limitations necessary to protect patient privacy and the public’s trust in health care professionals. The CARE guidelines can help authors of case reports hone the art of anonymizing the protected health information of subjects of case reports, such as ethnicity and occupation, while accurately conveying the clinical specifics of the case that make it valuable to students and colleagues.
We at Federal Practitioner recognize there is a real tension between truth-telling in case report publication and respect for patient confidentiality that will never be perfectly achieved, but is one that is important for medical knowledge, making it worthy of the continuous efforts of authors and editors to negotiate.
1. Nissen T, Wynn R. The history of the case report: a selective review. JRSM Open. 2014;5(4):2054270414523410. Published 2014 Mar 12. doi:10.1177/2054270414523410
2. Gagnier JJ, Kienle G, Altman DG, et al. The CARE guidelines: consensus-based clinical case reporting guideline development. BMJ Case Rep. 2013;2013:bcr2013201554. Published 2013 Oct 23. doi:10.1136/bcr-2013-201554
3. McLean SF. Case-based learning and its application in medical and health-care fields: a review of worldwide literature. J Med Educ Curric Dev. 2016;3:JMECD.S20377. Published 2016 Apr 27. doi:10.4137/JMECD.S20377
4. Centers for Disease Control (CDC). Pneumocystis pneumonia—Los Angeles. MMWR Morb Mortal Wkly Rep. 1981;30(21):250-252.
5. McBride WG. Thalidomide and congenital abnormalities. Lancet 1961;278(7216):1358. doi:10.1016/S0140-6736(61)90927-8
6. Vandenbroucke JP. In defense of case reports and case series. Ann Intern Med. 2001;134(4):330-334. doi:10.7326/0003-4819-134-4-200102200-00017
7. Rosoff PM. Can the case report withstand ethical scrutiny? Hastings Cent Rep. 2019;49(6):17-21. doi:10.1002/hast.1065
8. Riley DS, Barber MS, Kienle GS, et al. CARE guidelines for case reports: explanation and elaboration document. J Clin Epidemiol. 2017;89:218-235. doi:10.1016/j.jclinepi.2017.04.026
9. International Committee of Medical Journal Editors. Recommendations for the conduct, reporting, editing, and publication of scholarly work in medical journals. Updated December 2021. Accessed January 31, 2022. http://www.icmje.org/news-and-editorials/new_journal_dec2021.html
Medical case reports are as old as the healing profession itself.1 These ancient medical stories have a modern definition: “A case report is a narrative that describes, for medical, scientific or educational purposes, a medical problem experienced by one or more patients.”2 Case report experts describe the 3-fold purposes of this type of research: as a mainstay of education; a harbinger of emerging illnesses; and an appraiser of new interventions. Case-based education has long been a pillar of health professions education: Nurses, doctors, and allied health professionals are taught and learn through reading and discussing with their teachers and each other about cases of their own patients and of those in the literature.3 Case reports also have helped identify and raise awareness of new diseases and rare conditions, such as HIV.4 Finally, case reports have alerted regulatory agencies and the medical community about medication adverse effects, such as birth defects from thalidomide.5
Case reports also have been criticized on both scientific and ethical grounds. Critics argue that many case reports often lack the rigor and consistency of other types of research.6 Three recent trends in medical publication have strengthened the validity of these criticisms: the increase in the popularity of case reports; the corresponding increase in submissions to journals, including Federal Practitioner; and the rise of predatory publishers.7,8
The ethical scrutiny of case reports discussed in this column focuses on the tension between providing readers with adequate, accurate information to fulfil the goals of case reports while also protecting patient confidentiality. The latter issue during most of the history of medicine was not considered by health care professionals when the prevailing paternalism supported a professional-oriented approach to health care. The rise of bioethics in the 1960s and 1970s began the shift toward patient autonomy in medical decision making and patient rights to control their protected health information that rendered case reports ethically problematic.
To address both changes in ethical standards and scientific limitations, a committee of clinicians, researchers, and journal editors formed the Case Report (CARE) group.2,8 The group undertook an effort to improve the quality of case reports. From 2011 to 2012, they developed the CARE guidelines for clinical case reporting. The guidance took the form of a Statement and Checklist presented at the 2013 International Congress on Peer Review and Biomedical Publication. Since their presentation, multiple prestigious medical journals in many countries have implemented these recommendations.
As part of an overall effort to raise the ethical caliber of our own journal, Federal Practitioner will begin to implement the CARE guidelines for case reports for all future submissions. Use of the CARE recommendations will help prospective authors enhance the scientific value and ethical caliber of case reports submitted to the journal as well as assist the Federal Practitioner editorial team, editorial board, and peer reviewers to evaluate submissions more judiciously.
An essential part of the CARE guidelines is that the patient who is the subject of the case report provide informed consent for the publication of their personal narrative. The CARE group considers this an “ethical duty” of authors and editors alike. In “exceptional circumstances” such as if the patient is a minor or permanently incapacitated, a guardian or relative may grant consent. In the rare event that even with exhaustive attempts, if informed consent cannot be obtained from a patient or their representative, then the authors of the case report must submit a statement to this effect.4 Some journals may require that the authors obtain the approval of an institutional review board or the permission of an ethics or other institutional committee or a privacy officer.2
Requesting the patient’s consent is an extension of the shared decision making that is now a best practice in clinical care into the arena of research, making the patient or their representative a partner in the work. Ethicists have recommended inviting patients or relatives to read a draft of the case report and agree to its publication or request specific modifications to the manuscript. The CARE group rightly points out that with the rise of open notes in medical documentation, patients increasingly have access to their charts in near or real time.2 Gone are the days of Sir William Osler when only doctors read medical journals and all of these technical developments as well as standards of research and social changes in the practitioner-patient relationship make it imperative that writers and editors join together to make case reports more transparent, accurate, and consistent.7
An additional step to protect patient privacy is the requirement that authors either de-identify potentially identifiable health information, such as age, birth, death, admission, and discharge dates, or in some instances obtain separate consent for the release of that protected data.8 These restrictions constitute a challenge to case report authors who in some instances may consider these same facts critical to the integrity of the case presentation that have made some scholars doubt their continued viability. After all, the contribution of the case to the medical literature often lies in its very particularity. Conversely, no matter how frustrated we might become during writing a case report, we would not want to see our own protected health information or that of our family on a website or in print without our knowledge or approval. Indeed, the International Committee of Medical Journal Editors states that “If identifying characteristics are de-identified, authors should provide assurance, and editors should so note, that such changes do not distort scientific meaning.”9
However, the exponential growth of the internet, the spread of social media, and the ubiquity of a plethora of electronic devices, which prior generations of writers and readers could not even imagine, make these limitations necessary to protect patient privacy and the public’s trust in health care professionals. The CARE guidelines can help authors of case reports hone the art of anonymizing the protected health information of subjects of case reports, such as ethnicity and occupation, while accurately conveying the clinical specifics of the case that make it valuable to students and colleagues.
We at Federal Practitioner recognize there is a real tension between truth-telling in case report publication and respect for patient confidentiality that will never be perfectly achieved, but is one that is important for medical knowledge, making it worthy of the continuous efforts of authors and editors to negotiate.
Medical case reports are as old as the healing profession itself.1 These ancient medical stories have a modern definition: “A case report is a narrative that describes, for medical, scientific or educational purposes, a medical problem experienced by one or more patients.”2 Case report experts describe the 3-fold purposes of this type of research: as a mainstay of education; a harbinger of emerging illnesses; and an appraiser of new interventions. Case-based education has long been a pillar of health professions education: Nurses, doctors, and allied health professionals are taught and learn through reading and discussing with their teachers and each other about cases of their own patients and of those in the literature.3 Case reports also have helped identify and raise awareness of new diseases and rare conditions, such as HIV.4 Finally, case reports have alerted regulatory agencies and the medical community about medication adverse effects, such as birth defects from thalidomide.5
Case reports also have been criticized on both scientific and ethical grounds. Critics argue that many case reports often lack the rigor and consistency of other types of research.6 Three recent trends in medical publication have strengthened the validity of these criticisms: the increase in the popularity of case reports; the corresponding increase in submissions to journals, including Federal Practitioner; and the rise of predatory publishers.7,8
The ethical scrutiny of case reports discussed in this column focuses on the tension between providing readers with adequate, accurate information to fulfil the goals of case reports while also protecting patient confidentiality. The latter issue during most of the history of medicine was not considered by health care professionals when the prevailing paternalism supported a professional-oriented approach to health care. The rise of bioethics in the 1960s and 1970s began the shift toward patient autonomy in medical decision making and patient rights to control their protected health information that rendered case reports ethically problematic.
To address both changes in ethical standards and scientific limitations, a committee of clinicians, researchers, and journal editors formed the Case Report (CARE) group.2,8 The group undertook an effort to improve the quality of case reports. From 2011 to 2012, they developed the CARE guidelines for clinical case reporting. The guidance took the form of a Statement and Checklist presented at the 2013 International Congress on Peer Review and Biomedical Publication. Since their presentation, multiple prestigious medical journals in many countries have implemented these recommendations.
As part of an overall effort to raise the ethical caliber of our own journal, Federal Practitioner will begin to implement the CARE guidelines for case reports for all future submissions. Use of the CARE recommendations will help prospective authors enhance the scientific value and ethical caliber of case reports submitted to the journal as well as assist the Federal Practitioner editorial team, editorial board, and peer reviewers to evaluate submissions more judiciously.
An essential part of the CARE guidelines is that the patient who is the subject of the case report provide informed consent for the publication of their personal narrative. The CARE group considers this an “ethical duty” of authors and editors alike. In “exceptional circumstances” such as if the patient is a minor or permanently incapacitated, a guardian or relative may grant consent. In the rare event that even with exhaustive attempts, if informed consent cannot be obtained from a patient or their representative, then the authors of the case report must submit a statement to this effect.4 Some journals may require that the authors obtain the approval of an institutional review board or the permission of an ethics or other institutional committee or a privacy officer.2
Requesting the patient’s consent is an extension of the shared decision making that is now a best practice in clinical care into the arena of research, making the patient or their representative a partner in the work. Ethicists have recommended inviting patients or relatives to read a draft of the case report and agree to its publication or request specific modifications to the manuscript. The CARE group rightly points out that with the rise of open notes in medical documentation, patients increasingly have access to their charts in near or real time.2 Gone are the days of Sir William Osler when only doctors read medical journals and all of these technical developments as well as standards of research and social changes in the practitioner-patient relationship make it imperative that writers and editors join together to make case reports more transparent, accurate, and consistent.7
An additional step to protect patient privacy is the requirement that authors either de-identify potentially identifiable health information, such as age, birth, death, admission, and discharge dates, or in some instances obtain separate consent for the release of that protected data.8 These restrictions constitute a challenge to case report authors who in some instances may consider these same facts critical to the integrity of the case presentation that have made some scholars doubt their continued viability. After all, the contribution of the case to the medical literature often lies in its very particularity. Conversely, no matter how frustrated we might become during writing a case report, we would not want to see our own protected health information or that of our family on a website or in print without our knowledge or approval. Indeed, the International Committee of Medical Journal Editors states that “If identifying characteristics are de-identified, authors should provide assurance, and editors should so note, that such changes do not distort scientific meaning.”9
However, the exponential growth of the internet, the spread of social media, and the ubiquity of a plethora of electronic devices, which prior generations of writers and readers could not even imagine, make these limitations necessary to protect patient privacy and the public’s trust in health care professionals. The CARE guidelines can help authors of case reports hone the art of anonymizing the protected health information of subjects of case reports, such as ethnicity and occupation, while accurately conveying the clinical specifics of the case that make it valuable to students and colleagues.
We at Federal Practitioner recognize there is a real tension between truth-telling in case report publication and respect for patient confidentiality that will never be perfectly achieved, but is one that is important for medical knowledge, making it worthy of the continuous efforts of authors and editors to negotiate.
1. Nissen T, Wynn R. The history of the case report: a selective review. JRSM Open. 2014;5(4):2054270414523410. Published 2014 Mar 12. doi:10.1177/2054270414523410
2. Gagnier JJ, Kienle G, Altman DG, et al. The CARE guidelines: consensus-based clinical case reporting guideline development. BMJ Case Rep. 2013;2013:bcr2013201554. Published 2013 Oct 23. doi:10.1136/bcr-2013-201554
3. McLean SF. Case-based learning and its application in medical and health-care fields: a review of worldwide literature. J Med Educ Curric Dev. 2016;3:JMECD.S20377. Published 2016 Apr 27. doi:10.4137/JMECD.S20377
4. Centers for Disease Control (CDC). Pneumocystis pneumonia—Los Angeles. MMWR Morb Mortal Wkly Rep. 1981;30(21):250-252.
5. McBride WG. Thalidomide and congenital abnormalities. Lancet 1961;278(7216):1358. doi:10.1016/S0140-6736(61)90927-8
6. Vandenbroucke JP. In defense of case reports and case series. Ann Intern Med. 2001;134(4):330-334. doi:10.7326/0003-4819-134-4-200102200-00017
7. Rosoff PM. Can the case report withstand ethical scrutiny? Hastings Cent Rep. 2019;49(6):17-21. doi:10.1002/hast.1065
8. Riley DS, Barber MS, Kienle GS, et al. CARE guidelines for case reports: explanation and elaboration document. J Clin Epidemiol. 2017;89:218-235. doi:10.1016/j.jclinepi.2017.04.026
9. International Committee of Medical Journal Editors. Recommendations for the conduct, reporting, editing, and publication of scholarly work in medical journals. Updated December 2021. Accessed January 31, 2022. http://www.icmje.org/news-and-editorials/new_journal_dec2021.html
1. Nissen T, Wynn R. The history of the case report: a selective review. JRSM Open. 2014;5(4):2054270414523410. Published 2014 Mar 12. doi:10.1177/2054270414523410
2. Gagnier JJ, Kienle G, Altman DG, et al. The CARE guidelines: consensus-based clinical case reporting guideline development. BMJ Case Rep. 2013;2013:bcr2013201554. Published 2013 Oct 23. doi:10.1136/bcr-2013-201554
3. McLean SF. Case-based learning and its application in medical and health-care fields: a review of worldwide literature. J Med Educ Curric Dev. 2016;3:JMECD.S20377. Published 2016 Apr 27. doi:10.4137/JMECD.S20377
4. Centers for Disease Control (CDC). Pneumocystis pneumonia—Los Angeles. MMWR Morb Mortal Wkly Rep. 1981;30(21):250-252.
5. McBride WG. Thalidomide and congenital abnormalities. Lancet 1961;278(7216):1358. doi:10.1016/S0140-6736(61)90927-8
6. Vandenbroucke JP. In defense of case reports and case series. Ann Intern Med. 2001;134(4):330-334. doi:10.7326/0003-4819-134-4-200102200-00017
7. Rosoff PM. Can the case report withstand ethical scrutiny? Hastings Cent Rep. 2019;49(6):17-21. doi:10.1002/hast.1065
8. Riley DS, Barber MS, Kienle GS, et al. CARE guidelines for case reports: explanation and elaboration document. J Clin Epidemiol. 2017;89:218-235. doi:10.1016/j.jclinepi.2017.04.026
9. International Committee of Medical Journal Editors. Recommendations for the conduct, reporting, editing, and publication of scholarly work in medical journals. Updated December 2021. Accessed January 31, 2022. http://www.icmje.org/news-and-editorials/new_journal_dec2021.html
Guttate Psoriasis Following COVID-19 Infection
Psoriasis is an inflammatory skin condition affecting 1% to 5% of the world population. 1 Guttate psoriasis is a subgroup of psoriasis that most commonly presents as raindroplike, erythematous, silvery, scaly papules. There have been limited reports of guttate psoriasis caused by rhinovirus and COVID-19 infection, but a PubMed search of articles indexed for MEDLINE using the term COVID-19 guttate psoriasis yielded only 3 documented cases of a psoriatic flare secondary to SARS-CoV-2 infection. 1-4 Herein, we detail a case in which a patient with mild SARS-CoV-2 infection who did not have a personal or family history of psoriasis experienced a moderate psoriatic flare 3 weeks after diagnosis of COVID-19.
Case Report
A 55-year-old woman was diagnosed with COVID-19 after SARS-CoV-2 RNA was detected from a nasopharyngeal swab. She reported moderate fatigue but no other symptoms. At the time of infection, she was not taking medications and reported neither a personal nor family history of psoriasis.
Three weeks after the COVID-19 diagnosis, she reported erythematous scaly papules only on the trunk and backs of the legs. Two months after the COVID-19 diagnosis, she was evaluated in our practice and diagnosed with guttate psoriasis. The patient refused biopsy. Physical examination revealed that the affected body surface area had increased to 5%; erythematous, silvery, scaly papules were found on the trunk, anterior and posterior legs, and lateral thighs (Figure). At the time of evaluation, she did not report joint pain or nail changes.

The patient was treated with triamcinolone acetonide cream 0.1% twice daily for 2 to 4 weeks. The guttate psoriasis resolved.
Comment
A sudden psoriatic flare can be linked to dysregulation of the innate immune response. Guttate psoriasis and generalized plaque-type psoriasis are postulated to have similar pathogenetic mechanisms, but guttate psoriasis is the only type of psoriasis that originates from viral infection. Initially, viral RNA will stimulate the toll-like receptor 3 protein, leading to increased production of the pathogenic cytokine IL-36γ and pathogenic chemokine CXCL8 (also known as IL-8), both of which are biomarkers for psoriasis.1 Specifically, IL-36γ and CXCL8 are known to further stimulate the proinflammatory cascade during the innate immune response displayed in guttate psoriasis.5,6
Our patient had a mild case of COVID-19, and she first reported the erythematous and scaly papules 3 weeks after infection. Dysregulation of proinflammatory cytokines must have started in the initial stages—within 7 days—of the viral infection. Guttate psoriasis arises within 3 weeks of infection with other viral and bacterial triggers, most commonly with streptococcal infections.1
Rodríguez et al7 described a phenomenon in which both SARS-CoV-2 and Middle East respiratory syndrome, both caused by a coronavirus, can lead to a reduction of type I interferon, which in turn leads to failure of control of viral replication during initial stages of a viral infection. This triggers an increase in proinflammatory cytokines and chemokines, including IL‐36γ and CXCL8. This pathologic mechanism might apply to SARS-CoV-2, as demonstrated in our patient’s sudden psoriatic flare 3 weeks after the COVID-19 diagnosis. However, further investigation and quantification of the putatively involved cytokines is necessary for confirmation.
Conclusion
Psoriasis, a chronic inflammatory skin condition, has been linked predominantly to genetic and environmental factors. Guttate psoriasis as a secondary reaction after streptococcal tonsillar and respiratory infections has been reported.1
Our case is the fourth documented case of guttate psoriasis secondary to COVID-19 infection.2-4 However, it is the second documented case of a patient with a diagnosis of guttate psoriasis secondary to COVID-19 infection who had neither a personal nor family history of psoriasis.
Because SARS-CoV-2 is a novel virus, the long-term effects of COVID-19 remain unclear. We report this case and its findings to introduce a novel clinical manifestation of SARS-CoV-2 infection.
- Sbidian E, Madrange M, Viguier M, et al. Respiratory virus infection triggers acute psoriasis flares across different clinical subtypes and genetic backgrounds. Br J Dermatol. 2019;181:1304-1306. doi:10.1111/bjd.18203
- Gananandan K, Sacks B, Ewing I. Guttate psoriasis secondary to COVID-19. BMJ Case Rep. 2020;13:e237367. doi:10.1136/bcr-2020-237367
- Rouai M, Rabhi F, Mansouri N, et al. New-onset guttate psoriasis secondary to COVID-19. Clin Case Rep. 2021;9:e04542. doi:10.1002/ccr3.4542
- Agarwal A, Tripathy T, Kar BR. Guttate flare in a patient with chronic plaque psoriasis following COVID-19 infection: a case report. J Cosmet Dermatol. 2021;20:3064-3065. doi:10.1111/jocd.14396
- Madonna S, Girolomoni G, Dinarello CA, et al. The significance of IL-36 hyperactivation and IL-36R targeting in psoriasis. Int J Mol Sci. 2019;20:3318. doi:10.3390/ijms20133318
- Nedoszytko B, Sokołowska-Wojdyło M, Ruckemann-Dziurdzin´ska K, et al. Chemokines and cytokines network in the pathogenesis of the inflammatory skin diseases: atopic dermatitis, psoriasis and skin mastocytosis. Postepy Dermatol Alergol. 2014;31:84-91. doi:10.5114/pdia.2014.40920
- Rodríguez Y, Novelli L, Rojas M, et al. Autoinflammatory and autoimmune conditions at the crossroad of COVID-19. J Autoimmun. 2020;114:102506. doi:10.1016/j.jaut.2020.102506
Psoriasis is an inflammatory skin condition affecting 1% to 5% of the world population. 1 Guttate psoriasis is a subgroup of psoriasis that most commonly presents as raindroplike, erythematous, silvery, scaly papules. There have been limited reports of guttate psoriasis caused by rhinovirus and COVID-19 infection, but a PubMed search of articles indexed for MEDLINE using the term COVID-19 guttate psoriasis yielded only 3 documented cases of a psoriatic flare secondary to SARS-CoV-2 infection. 1-4 Herein, we detail a case in which a patient with mild SARS-CoV-2 infection who did not have a personal or family history of psoriasis experienced a moderate psoriatic flare 3 weeks after diagnosis of COVID-19.
Case Report
A 55-year-old woman was diagnosed with COVID-19 after SARS-CoV-2 RNA was detected from a nasopharyngeal swab. She reported moderate fatigue but no other symptoms. At the time of infection, she was not taking medications and reported neither a personal nor family history of psoriasis.
Three weeks after the COVID-19 diagnosis, she reported erythematous scaly papules only on the trunk and backs of the legs. Two months after the COVID-19 diagnosis, she was evaluated in our practice and diagnosed with guttate psoriasis. The patient refused biopsy. Physical examination revealed that the affected body surface area had increased to 5%; erythematous, silvery, scaly papules were found on the trunk, anterior and posterior legs, and lateral thighs (Figure). At the time of evaluation, she did not report joint pain or nail changes.
The patient was treated with triamcinolone acetonide cream 0.1% twice daily for 2 to 4 weeks. The guttate psoriasis resolved.
Comment
A sudden psoriatic flare can be linked to dysregulation of the innate immune response. Guttate psoriasis and generalized plaque-type psoriasis are postulated to have similar pathogenetic mechanisms, but guttate psoriasis is the only type of psoriasis that originates from viral infection. Initially, viral RNA will stimulate the toll-like receptor 3 protein, leading to increased production of the pathogenic cytokine IL-36γ and pathogenic chemokine CXCL8 (also known as IL-8), both of which are biomarkers for psoriasis.1 Specifically, IL-36γ and CXCL8 are known to further stimulate the proinflammatory cascade during the innate immune response displayed in guttate psoriasis.5,6
Our patient had a mild case of COVID-19, and she first reported the erythematous and scaly papules 3 weeks after infection. Dysregulation of proinflammatory cytokines must have started in the initial stages—within 7 days—of the viral infection. Guttate psoriasis arises within 3 weeks of infection with other viral and bacterial triggers, most commonly with streptococcal infections.1
Rodríguez et al7 described a phenomenon in which both SARS-CoV-2 and Middle East respiratory syndrome, both caused by a coronavirus, can lead to a reduction of type I interferon, which in turn leads to failure of control of viral replication during initial stages of a viral infection. This triggers an increase in proinflammatory cytokines and chemokines, including IL‐36γ and CXCL8. This pathologic mechanism might apply to SARS-CoV-2, as demonstrated in our patient’s sudden psoriatic flare 3 weeks after the COVID-19 diagnosis. However, further investigation and quantification of the putatively involved cytokines is necessary for confirmation.
Conclusion
Psoriasis, a chronic inflammatory skin condition, has been linked predominantly to genetic and environmental factors. Guttate psoriasis as a secondary reaction after streptococcal tonsillar and respiratory infections has been reported.1
Our case is the fourth documented case of guttate psoriasis secondary to COVID-19 infection.2-4 However, it is the second documented case of a patient with a diagnosis of guttate psoriasis secondary to COVID-19 infection who had neither a personal nor family history of psoriasis.
Because SARS-CoV-2 is a novel virus, the long-term effects of COVID-19 remain unclear. We report this case and its findings to introduce a novel clinical manifestation of SARS-CoV-2 infection.
Psoriasis is an inflammatory skin condition affecting 1% to 5% of the world population. 1 Guttate psoriasis is a subgroup of psoriasis that most commonly presents as raindroplike, erythematous, silvery, scaly papules. There have been limited reports of guttate psoriasis caused by rhinovirus and COVID-19 infection, but a PubMed search of articles indexed for MEDLINE using the term COVID-19 guttate psoriasis yielded only 3 documented cases of a psoriatic flare secondary to SARS-CoV-2 infection. 1-4 Herein, we detail a case in which a patient with mild SARS-CoV-2 infection who did not have a personal or family history of psoriasis experienced a moderate psoriatic flare 3 weeks after diagnosis of COVID-19.
Case Report
A 55-year-old woman was diagnosed with COVID-19 after SARS-CoV-2 RNA was detected from a nasopharyngeal swab. She reported moderate fatigue but no other symptoms. At the time of infection, she was not taking medications and reported neither a personal nor family history of psoriasis.
Three weeks after the COVID-19 diagnosis, she reported erythematous scaly papules only on the trunk and backs of the legs. Two months after the COVID-19 diagnosis, she was evaluated in our practice and diagnosed with guttate psoriasis. The patient refused biopsy. Physical examination revealed that the affected body surface area had increased to 5%; erythematous, silvery, scaly papules were found on the trunk, anterior and posterior legs, and lateral thighs (Figure). At the time of evaluation, she did not report joint pain or nail changes.
The patient was treated with triamcinolone acetonide cream 0.1% twice daily for 2 to 4 weeks. The guttate psoriasis resolved.
Comment
A sudden psoriatic flare can be linked to dysregulation of the innate immune response. Guttate psoriasis and generalized plaque-type psoriasis are postulated to have similar pathogenetic mechanisms, but guttate psoriasis is the only type of psoriasis that originates from viral infection. Initially, viral RNA will stimulate the toll-like receptor 3 protein, leading to increased production of the pathogenic cytokine IL-36γ and pathogenic chemokine CXCL8 (also known as IL-8), both of which are biomarkers for psoriasis.1 Specifically, IL-36γ and CXCL8 are known to further stimulate the proinflammatory cascade during the innate immune response displayed in guttate psoriasis.5,6
Our patient had a mild case of COVID-19, and she first reported the erythematous and scaly papules 3 weeks after infection. Dysregulation of proinflammatory cytokines must have started in the initial stages—within 7 days—of the viral infection. Guttate psoriasis arises within 3 weeks of infection with other viral and bacterial triggers, most commonly with streptococcal infections.1
Rodríguez et al7 described a phenomenon in which both SARS-CoV-2 and Middle East respiratory syndrome, both caused by a coronavirus, can lead to a reduction of type I interferon, which in turn leads to failure of control of viral replication during initial stages of a viral infection. This triggers an increase in proinflammatory cytokines and chemokines, including IL‐36γ and CXCL8. This pathologic mechanism might apply to SARS-CoV-2, as demonstrated in our patient’s sudden psoriatic flare 3 weeks after the COVID-19 diagnosis. However, further investigation and quantification of the putatively involved cytokines is necessary for confirmation.
Conclusion
Psoriasis, a chronic inflammatory skin condition, has been linked predominantly to genetic and environmental factors. Guttate psoriasis as a secondary reaction after streptococcal tonsillar and respiratory infections has been reported.1
Our case is the fourth documented case of guttate psoriasis secondary to COVID-19 infection.2-4 However, it is the second documented case of a patient with a diagnosis of guttate psoriasis secondary to COVID-19 infection who had neither a personal nor family history of psoriasis.
Because SARS-CoV-2 is a novel virus, the long-term effects of COVID-19 remain unclear. We report this case and its findings to introduce a novel clinical manifestation of SARS-CoV-2 infection.
- Sbidian E, Madrange M, Viguier M, et al. Respiratory virus infection triggers acute psoriasis flares across different clinical subtypes and genetic backgrounds. Br J Dermatol. 2019;181:1304-1306. doi:10.1111/bjd.18203
- Gananandan K, Sacks B, Ewing I. Guttate psoriasis secondary to COVID-19. BMJ Case Rep. 2020;13:e237367. doi:10.1136/bcr-2020-237367
- Rouai M, Rabhi F, Mansouri N, et al. New-onset guttate psoriasis secondary to COVID-19. Clin Case Rep. 2021;9:e04542. doi:10.1002/ccr3.4542
- Agarwal A, Tripathy T, Kar BR. Guttate flare in a patient with chronic plaque psoriasis following COVID-19 infection: a case report. J Cosmet Dermatol. 2021;20:3064-3065. doi:10.1111/jocd.14396
- Madonna S, Girolomoni G, Dinarello CA, et al. The significance of IL-36 hyperactivation and IL-36R targeting in psoriasis. Int J Mol Sci. 2019;20:3318. doi:10.3390/ijms20133318
- Nedoszytko B, Sokołowska-Wojdyło M, Ruckemann-Dziurdzin´ska K, et al. Chemokines and cytokines network in the pathogenesis of the inflammatory skin diseases: atopic dermatitis, psoriasis and skin mastocytosis. Postepy Dermatol Alergol. 2014;31:84-91. doi:10.5114/pdia.2014.40920
- Rodríguez Y, Novelli L, Rojas M, et al. Autoinflammatory and autoimmune conditions at the crossroad of COVID-19. J Autoimmun. 2020;114:102506. doi:10.1016/j.jaut.2020.102506
- Sbidian E, Madrange M, Viguier M, et al. Respiratory virus infection triggers acute psoriasis flares across different clinical subtypes and genetic backgrounds. Br J Dermatol. 2019;181:1304-1306. doi:10.1111/bjd.18203
- Gananandan K, Sacks B, Ewing I. Guttate psoriasis secondary to COVID-19. BMJ Case Rep. 2020;13:e237367. doi:10.1136/bcr-2020-237367
- Rouai M, Rabhi F, Mansouri N, et al. New-onset guttate psoriasis secondary to COVID-19. Clin Case Rep. 2021;9:e04542. doi:10.1002/ccr3.4542
- Agarwal A, Tripathy T, Kar BR. Guttate flare in a patient with chronic plaque psoriasis following COVID-19 infection: a case report. J Cosmet Dermatol. 2021;20:3064-3065. doi:10.1111/jocd.14396
- Madonna S, Girolomoni G, Dinarello CA, et al. The significance of IL-36 hyperactivation and IL-36R targeting in psoriasis. Int J Mol Sci. 2019;20:3318. doi:10.3390/ijms20133318
- Nedoszytko B, Sokołowska-Wojdyło M, Ruckemann-Dziurdzin´ska K, et al. Chemokines and cytokines network in the pathogenesis of the inflammatory skin diseases: atopic dermatitis, psoriasis and skin mastocytosis. Postepy Dermatol Alergol. 2014;31:84-91. doi:10.5114/pdia.2014.40920
- Rodríguez Y, Novelli L, Rojas M, et al. Autoinflammatory and autoimmune conditions at the crossroad of COVID-19. J Autoimmun. 2020;114:102506. doi:10.1016/j.jaut.2020.102506
Practice Points
- Guttate psoriasis is the only type of psoriasis that originates from viral infection.
- Dysregulation of proinflammatory cytokines during COVID-19 infection in our patient led to development of guttate psoriasis 3 weeks later.

Severe Acute Systemic Reaction After the First Injections of Ixekizumab
Case Report
A 39-year-old woman who was otherwise healthy presented with fatigue, malaise, a resolving rash, focal lymphadenopathy, increasing distal arthritis, dactylitis, resolving ecchymoses, and acute onycholysis of 1 week’s duration that developed 13 days after initiating ixekizumab. The patient had a history of psoriasis and psoriatic arthritis for more than 10 years. She had been successfully treated in the past for psoriasis with adalimumab for several years; however, adalimumab was discontinued after an episode of Clostridium difficile colitis. The patient had a negative purified protein derivative (tuberculin) test prior to starting biologics as she works in the health care field. Routine follow-up purified protein derivative (tuberculin) test was positive. She discontinued all therapy for psoriasis and psoriatic arthritis prior to being appropriately treated for 6 months under the care of infectious disease physicians. She then had several pregnancies and chose to restart biologic treatment after weaning her third child from breastfeeding, as her skin and joint disease were notably flaring.
Ustekinumab was chosen to shift treatment away from tumor necrosis factor (TNF) α inhibitors. The patient's condition was under relatively good control for 1 year; however, she experienced notable gastrointestinal tract upset (ie, intermittent diarrhea and constipation), despite multiple negative tests for C difficile. The patient was referred to see a gastroenterologist but never followed up. Due to long-term low-grade gastrointestinal problems, ustekinumab was discontinued, and the gastrointestinal symptoms resolved without treatment.
Given the side effects noted with TNF-α and IL-12/23 inhibitors and the fact that the patient’s cutaneous and joint disease were notable, the decision was made to start the IL-17A inhibitor ixekizumab. The patient administered 2 injections, one in each thigh. Within 12 hours, she experienced severe injection-site pain. The pain was so severe that it woke her from sleep the night of the first injections. She then developed severe pain in the right axilla that limited upper extremity mobility. Within 48 hours, she developed an erythematous, nonpruritic, nonscaly, mottled rash on the right breast that began to resolve within 24 hours without treatment. In addition, 3 days after the injections, she developed ecchymoses on the trunk and extremities without any identifiable trauma, severe acute onycholysis in several fingernails (Figure 1) and toenails, dactylitis such that she could not wear her wedding ring, and a flare of psoriatic arthritis in the fingers and ankles.

At the current presentation (2 weeks after the injections), the patient reported malaise, flulike symptoms, and low-grade intermittent fevers. Results from a hematology panel displayed leukopenia at 2.69×103/μL (reference range, 3.54–9.06×103/μL) and thrombocytopenia at 114×103/μL (reference range, 165–415×103/μL).1 Her most recent laboratory results before the ixekizumab injections displayed a white blood cell count level at 4.6×103/μL and platelet count at 159×103/μL. C-reactive protein and erythrocyte sedimentation rate were within reference range. A shave biopsy of an erythematous nodule on the proximal interphalangeal joint of the fourth finger on the right hand displayed spongiotic dermatitis with eosinophils (Figure 2).

Interestingly, the psoriatic plaques on the scalp, trunk, and extremities had nearly completely resolved after only the first 2 injections. However, given the side effects, the second dose of ixekizumab was held, repeat laboratory tests were ordered to ensure normalization of cytopenia, and the patient was transitioned to pulse-dose topical steroids to control the remaining psoriatic plaques.
One week after presentation (3 weeks after the initial injections), the patient’s systemic symptoms had almost completely resolved, and she denied any further concerns. Her fingernails and toenails, however, continued to show the changes of onycholysis noted at the visit.
Comment
Ixekizumab is a human IgG4 monoclonal antibody that binds to IL-17A, one of the cytokines involved in the pathogenesis of psoriasis. The monoclonal antibody prevents its attachment to the IL-17 receptor, which inhibits the release of further cytokines and chemokines, decreasing the inflammatory and immune response.2
Ixekizumab was approved by the US Food and Drug Administration for plaque psoriasis after 3 clinical trials—UNCOVER-1, UNCOVER-2, and UNCOVER-3—were performed. In UNCOVER-3, the most common side effects that occurred—nasopharyngitis, upper respiratory tract infection, injection-site reaction, arthralgia, headache, and infections (specifically candidiasis)—generally were well tolerated. More serious adverse events included cardiovascular and cerebrovascular events, inflammatory bowel disease, and nonmelanoma skin cancer.3
Notable laboratory abnormalities that have been documented from ixekizumab include elevated liver function tests (eg, alanine aminotransferase, aspartate aminotransferase, bilirubin, and alkaline phosphatase), as well as leukopenia, neutropenia, and thrombocytopenia.4 Although short-term thrombocytopenia, as described in our patient, provides an explanation for the bruising noted on observation, it is unusual to note such notable ecchymoses within days of the first injection.
Onycholysis has not been documented as a side effect of ixekizumab; however, it has been reported as an adverse event from other biologic medications. Sfikakis et al5 reported 5 patients who developed psoriatic skin lesions after treatment with 3 different anti-TNF biologics—infliximab, adalimumab, or etanercept—fo
The exact pathophysiology of these adverse events has not been clearly understood, but it has been proposed that anti-TNF biologics may initiate an autoimmune reaction in the skin and nails, leading to paradoxical psoriasis and nail changes such as onycholysis. Tumor necrosis factor may have a regulatory role in the skin that prevents autoreactive T cells, such as cutaneous lymphocyte antigen–expressing T cells that promote the formation of psoriasiform lesions. By inhibiting TNF, there can be an underlying activation of autoreactive T cells that leads to tissue destruction in the skin and nails.6 Anti-TNF biologics also could increase CXCR3, a chemokine receptor that allows autoreactive T cells to enter the skin and cause pathology.7
IL-17A and IL-17F also have been shown to upregulate the expression of TNF receptor II in synoviocytes,8 which demonstrates that IL-17 works in synergy with TNF-α to promote an inflammatory reaction.9 Due to the inhibitory effects of ixekizumab, psoriatic arthritis should theoretically improve. However, if there is an alteration in the inflammatory sequence, then the regulatory role of TNF could be suppressed and psoriatic arthritis could become exacerbated. Additionally, its associated symptoms, such as dactylitis, could develop, as seen in our patient.4 Because psoriatic arthritis is closely associated with nail changes of psoriasis, it is conceivable that acute arthritic flares and acute onycholysis are both induced by the same cytokine dysregulation. Further studies and a larger patient population need to be evaluated to determine the exact cause of the acute exacerbation of psoriatic arthritis with concomitant nail changes as noted in our patient.
Acute onycholysis (within 72 hours) is a rare side effect of ixekizumab. It can be postulated that our patient’s severe acute onycholysis associated with a flare of psoriatic arthritis could be due to idiosyncratic immune dysregulation, promoting the activity of autoreactive T cells. The pharmacologic effects of ixekizumab occur through the inhibition of IL-17. We propose that by inhibiting IL-17 with associated TNF alterations, an altered inflammatory cascade could promote an autoimmune reaction leading to the described pathology.
- Kratz A, Pesce MA, Basner RC, et al. Laboratory values of clinical importance. In: Kasper D, Fauci A, Hauser S, et al, eds. Harrison’s Principles of Internal Medicine. 19th ed. McGraw-Hill; 2014.
- Ixekizumab. Package insert. Eli Lilly & Co; 2017.
- Gordon KB, Blauvelt A, Papp KA, et al. Phase 3 trials of ixekizumab in moderate-to-severe plaque psoriasis. N Engl J Med. 2016;375:345-356.
- Leonardi C, Matheson R, Zachariae C, et al. Anti-interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N Engl J Med. 2012;366:1190-1199.
- Sfikakis PP, Iliopoulos A, Elezoglou A, et al. Psoriasis induced by anti-tumor necrosis factor therapy: a paradoxical adverse reaction. Arthritis Rheum. 2005;52:2513-2518.
- Berg EL, Yoshino T, Rott LS, et al. The cutaneous lymphocyte antigen is a skin lymphocyte homing receptor for the vascular lectin endothelial cell-leukocyte adhesion molecule 1. J Exp Med. 1991;174:1461-1466.
- Flier J, Boorsma DM, van Beek PJ, et al. Differential expression of CXCR3 targeting chemokines CXCL10, CXCL9, and CXCL11 in different types of skin inflammation. J Pathol. 2001;194:398-405.
- Zrioual S, Ecochard R, Tournadre A, et al. Genome-wide comparison between IL-17A- and IL-17F-induced effects in human rheumatoid arthritis synoviocytes. J Immunol. 2009;182:3112-3120.
- Gaffen SL. The role of interleukin-17 in the pathogenesis of rheumatoid arthritis. Curr Rheumatol Rep. 2009;11:365-370.
Case Report
A 39-year-old woman who was otherwise healthy presented with fatigue, malaise, a resolving rash, focal lymphadenopathy, increasing distal arthritis, dactylitis, resolving ecchymoses, and acute onycholysis of 1 week’s duration that developed 13 days after initiating ixekizumab. The patient had a history of psoriasis and psoriatic arthritis for more than 10 years. She had been successfully treated in the past for psoriasis with adalimumab for several years; however, adalimumab was discontinued after an episode of Clostridium difficile colitis. The patient had a negative purified protein derivative (tuberculin) test prior to starting biologics as she works in the health care field. Routine follow-up purified protein derivative (tuberculin) test was positive. She discontinued all therapy for psoriasis and psoriatic arthritis prior to being appropriately treated for 6 months under the care of infectious disease physicians. She then had several pregnancies and chose to restart biologic treatment after weaning her third child from breastfeeding, as her skin and joint disease were notably flaring.
Ustekinumab was chosen to shift treatment away from tumor necrosis factor (TNF) α inhibitors. The patient's condition was under relatively good control for 1 year; however, she experienced notable gastrointestinal tract upset (ie, intermittent diarrhea and constipation), despite multiple negative tests for C difficile. The patient was referred to see a gastroenterologist but never followed up. Due to long-term low-grade gastrointestinal problems, ustekinumab was discontinued, and the gastrointestinal symptoms resolved without treatment.
Given the side effects noted with TNF-α and IL-12/23 inhibitors and the fact that the patient’s cutaneous and joint disease were notable, the decision was made to start the IL-17A inhibitor ixekizumab. The patient administered 2 injections, one in each thigh. Within 12 hours, she experienced severe injection-site pain. The pain was so severe that it woke her from sleep the night of the first injections. She then developed severe pain in the right axilla that limited upper extremity mobility. Within 48 hours, she developed an erythematous, nonpruritic, nonscaly, mottled rash on the right breast that began to resolve within 24 hours without treatment. In addition, 3 days after the injections, she developed ecchymoses on the trunk and extremities without any identifiable trauma, severe acute onycholysis in several fingernails (Figure 1) and toenails, dactylitis such that she could not wear her wedding ring, and a flare of psoriatic arthritis in the fingers and ankles.
At the current presentation (2 weeks after the injections), the patient reported malaise, flulike symptoms, and low-grade intermittent fevers. Results from a hematology panel displayed leukopenia at 2.69×103/μL (reference range, 3.54–9.06×103/μL) and thrombocytopenia at 114×103/μL (reference range, 165–415×103/μL).1 Her most recent laboratory results before the ixekizumab injections displayed a white blood cell count level at 4.6×103/μL and platelet count at 159×103/μL. C-reactive protein and erythrocyte sedimentation rate were within reference range. A shave biopsy of an erythematous nodule on the proximal interphalangeal joint of the fourth finger on the right hand displayed spongiotic dermatitis with eosinophils (Figure 2).
Interestingly, the psoriatic plaques on the scalp, trunk, and extremities had nearly completely resolved after only the first 2 injections. However, given the side effects, the second dose of ixekizumab was held, repeat laboratory tests were ordered to ensure normalization of cytopenia, and the patient was transitioned to pulse-dose topical steroids to control the remaining psoriatic plaques.
One week after presentation (3 weeks after the initial injections), the patient’s systemic symptoms had almost completely resolved, and she denied any further concerns. Her fingernails and toenails, however, continued to show the changes of onycholysis noted at the visit.
Comment
Ixekizumab is a human IgG4 monoclonal antibody that binds to IL-17A, one of the cytokines involved in the pathogenesis of psoriasis. The monoclonal antibody prevents its attachment to the IL-17 receptor, which inhibits the release of further cytokines and chemokines, decreasing the inflammatory and immune response.2
Ixekizumab was approved by the US Food and Drug Administration for plaque psoriasis after 3 clinical trials—UNCOVER-1, UNCOVER-2, and UNCOVER-3—were performed. In UNCOVER-3, the most common side effects that occurred—nasopharyngitis, upper respiratory tract infection, injection-site reaction, arthralgia, headache, and infections (specifically candidiasis)—generally were well tolerated. More serious adverse events included cardiovascular and cerebrovascular events, inflammatory bowel disease, and nonmelanoma skin cancer.3
Notable laboratory abnormalities that have been documented from ixekizumab include elevated liver function tests (eg, alanine aminotransferase, aspartate aminotransferase, bilirubin, and alkaline phosphatase), as well as leukopenia, neutropenia, and thrombocytopenia.4 Although short-term thrombocytopenia, as described in our patient, provides an explanation for the bruising noted on observation, it is unusual to note such notable ecchymoses within days of the first injection.
Onycholysis has not been documented as a side effect of ixekizumab; however, it has been reported as an adverse event from other biologic medications. Sfikakis et al5 reported 5 patients who developed psoriatic skin lesions after treatment with 3 different anti-TNF biologics—infliximab, adalimumab, or etanercept—fo
The exact pathophysiology of these adverse events has not been clearly understood, but it has been proposed that anti-TNF biologics may initiate an autoimmune reaction in the skin and nails, leading to paradoxical psoriasis and nail changes such as onycholysis. Tumor necrosis factor may have a regulatory role in the skin that prevents autoreactive T cells, such as cutaneous lymphocyte antigen–expressing T cells that promote the formation of psoriasiform lesions. By inhibiting TNF, there can be an underlying activation of autoreactive T cells that leads to tissue destruction in the skin and nails.6 Anti-TNF biologics also could increase CXCR3, a chemokine receptor that allows autoreactive T cells to enter the skin and cause pathology.7
IL-17A and IL-17F also have been shown to upregulate the expression of TNF receptor II in synoviocytes,8 which demonstrates that IL-17 works in synergy with TNF-α to promote an inflammatory reaction.9 Due to the inhibitory effects of ixekizumab, psoriatic arthritis should theoretically improve. However, if there is an alteration in the inflammatory sequence, then the regulatory role of TNF could be suppressed and psoriatic arthritis could become exacerbated. Additionally, its associated symptoms, such as dactylitis, could develop, as seen in our patient.4 Because psoriatic arthritis is closely associated with nail changes of psoriasis, it is conceivable that acute arthritic flares and acute onycholysis are both induced by the same cytokine dysregulation. Further studies and a larger patient population need to be evaluated to determine the exact cause of the acute exacerbation of psoriatic arthritis with concomitant nail changes as noted in our patient.
Acute onycholysis (within 72 hours) is a rare side effect of ixekizumab. It can be postulated that our patient’s severe acute onycholysis associated with a flare of psoriatic arthritis could be due to idiosyncratic immune dysregulation, promoting the activity of autoreactive T cells. The pharmacologic effects of ixekizumab occur through the inhibition of IL-17. We propose that by inhibiting IL-17 with associated TNF alterations, an altered inflammatory cascade could promote an autoimmune reaction leading to the described pathology.
Case Report
A 39-year-old woman who was otherwise healthy presented with fatigue, malaise, a resolving rash, focal lymphadenopathy, increasing distal arthritis, dactylitis, resolving ecchymoses, and acute onycholysis of 1 week’s duration that developed 13 days after initiating ixekizumab. The patient had a history of psoriasis and psoriatic arthritis for more than 10 years. She had been successfully treated in the past for psoriasis with adalimumab for several years; however, adalimumab was discontinued after an episode of Clostridium difficile colitis. The patient had a negative purified protein derivative (tuberculin) test prior to starting biologics as she works in the health care field. Routine follow-up purified protein derivative (tuberculin) test was positive. She discontinued all therapy for psoriasis and psoriatic arthritis prior to being appropriately treated for 6 months under the care of infectious disease physicians. She then had several pregnancies and chose to restart biologic treatment after weaning her third child from breastfeeding, as her skin and joint disease were notably flaring.
Ustekinumab was chosen to shift treatment away from tumor necrosis factor (TNF) α inhibitors. The patient's condition was under relatively good control for 1 year; however, she experienced notable gastrointestinal tract upset (ie, intermittent diarrhea and constipation), despite multiple negative tests for C difficile. The patient was referred to see a gastroenterologist but never followed up. Due to long-term low-grade gastrointestinal problems, ustekinumab was discontinued, and the gastrointestinal symptoms resolved without treatment.
Given the side effects noted with TNF-α and IL-12/23 inhibitors and the fact that the patient’s cutaneous and joint disease were notable, the decision was made to start the IL-17A inhibitor ixekizumab. The patient administered 2 injections, one in each thigh. Within 12 hours, she experienced severe injection-site pain. The pain was so severe that it woke her from sleep the night of the first injections. She then developed severe pain in the right axilla that limited upper extremity mobility. Within 48 hours, she developed an erythematous, nonpruritic, nonscaly, mottled rash on the right breast that began to resolve within 24 hours without treatment. In addition, 3 days after the injections, she developed ecchymoses on the trunk and extremities without any identifiable trauma, severe acute onycholysis in several fingernails (Figure 1) and toenails, dactylitis such that she could not wear her wedding ring, and a flare of psoriatic arthritis in the fingers and ankles.
At the current presentation (2 weeks after the injections), the patient reported malaise, flulike symptoms, and low-grade intermittent fevers. Results from a hematology panel displayed leukopenia at 2.69×103/μL (reference range, 3.54–9.06×103/μL) and thrombocytopenia at 114×103/μL (reference range, 165–415×103/μL).1 Her most recent laboratory results before the ixekizumab injections displayed a white blood cell count level at 4.6×103/μL and platelet count at 159×103/μL. C-reactive protein and erythrocyte sedimentation rate were within reference range. A shave biopsy of an erythematous nodule on the proximal interphalangeal joint of the fourth finger on the right hand displayed spongiotic dermatitis with eosinophils (Figure 2).
Interestingly, the psoriatic plaques on the scalp, trunk, and extremities had nearly completely resolved after only the first 2 injections. However, given the side effects, the second dose of ixekizumab was held, repeat laboratory tests were ordered to ensure normalization of cytopenia, and the patient was transitioned to pulse-dose topical steroids to control the remaining psoriatic plaques.
One week after presentation (3 weeks after the initial injections), the patient’s systemic symptoms had almost completely resolved, and she denied any further concerns. Her fingernails and toenails, however, continued to show the changes of onycholysis noted at the visit.
Comment
Ixekizumab is a human IgG4 monoclonal antibody that binds to IL-17A, one of the cytokines involved in the pathogenesis of psoriasis. The monoclonal antibody prevents its attachment to the IL-17 receptor, which inhibits the release of further cytokines and chemokines, decreasing the inflammatory and immune response.2
Ixekizumab was approved by the US Food and Drug Administration for plaque psoriasis after 3 clinical trials—UNCOVER-1, UNCOVER-2, and UNCOVER-3—were performed. In UNCOVER-3, the most common side effects that occurred—nasopharyngitis, upper respiratory tract infection, injection-site reaction, arthralgia, headache, and infections (specifically candidiasis)—generally were well tolerated. More serious adverse events included cardiovascular and cerebrovascular events, inflammatory bowel disease, and nonmelanoma skin cancer.3
Notable laboratory abnormalities that have been documented from ixekizumab include elevated liver function tests (eg, alanine aminotransferase, aspartate aminotransferase, bilirubin, and alkaline phosphatase), as well as leukopenia, neutropenia, and thrombocytopenia.4 Although short-term thrombocytopenia, as described in our patient, provides an explanation for the bruising noted on observation, it is unusual to note such notable ecchymoses within days of the first injection.
Onycholysis has not been documented as a side effect of ixekizumab; however, it has been reported as an adverse event from other biologic medications. Sfikakis et al5 reported 5 patients who developed psoriatic skin lesions after treatment with 3 different anti-TNF biologics—infliximab, adalimumab, or etanercept—fo
The exact pathophysiology of these adverse events has not been clearly understood, but it has been proposed that anti-TNF biologics may initiate an autoimmune reaction in the skin and nails, leading to paradoxical psoriasis and nail changes such as onycholysis. Tumor necrosis factor may have a regulatory role in the skin that prevents autoreactive T cells, such as cutaneous lymphocyte antigen–expressing T cells that promote the formation of psoriasiform lesions. By inhibiting TNF, there can be an underlying activation of autoreactive T cells that leads to tissue destruction in the skin and nails.6 Anti-TNF biologics also could increase CXCR3, a chemokine receptor that allows autoreactive T cells to enter the skin and cause pathology.7
IL-17A and IL-17F also have been shown to upregulate the expression of TNF receptor II in synoviocytes,8 which demonstrates that IL-17 works in synergy with TNF-α to promote an inflammatory reaction.9 Due to the inhibitory effects of ixekizumab, psoriatic arthritis should theoretically improve. However, if there is an alteration in the inflammatory sequence, then the regulatory role of TNF could be suppressed and psoriatic arthritis could become exacerbated. Additionally, its associated symptoms, such as dactylitis, could develop, as seen in our patient.4 Because psoriatic arthritis is closely associated with nail changes of psoriasis, it is conceivable that acute arthritic flares and acute onycholysis are both induced by the same cytokine dysregulation. Further studies and a larger patient population need to be evaluated to determine the exact cause of the acute exacerbation of psoriatic arthritis with concomitant nail changes as noted in our patient.
Acute onycholysis (within 72 hours) is a rare side effect of ixekizumab. It can be postulated that our patient’s severe acute onycholysis associated with a flare of psoriatic arthritis could be due to idiosyncratic immune dysregulation, promoting the activity of autoreactive T cells. The pharmacologic effects of ixekizumab occur through the inhibition of IL-17. We propose that by inhibiting IL-17 with associated TNF alterations, an altered inflammatory cascade could promote an autoimmune reaction leading to the described pathology.
- Kratz A, Pesce MA, Basner RC, et al. Laboratory values of clinical importance. In: Kasper D, Fauci A, Hauser S, et al, eds. Harrison’s Principles of Internal Medicine. 19th ed. McGraw-Hill; 2014.
- Ixekizumab. Package insert. Eli Lilly & Co; 2017.
- Gordon KB, Blauvelt A, Papp KA, et al. Phase 3 trials of ixekizumab in moderate-to-severe plaque psoriasis. N Engl J Med. 2016;375:345-356.
- Leonardi C, Matheson R, Zachariae C, et al. Anti-interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N Engl J Med. 2012;366:1190-1199.
- Sfikakis PP, Iliopoulos A, Elezoglou A, et al. Psoriasis induced by anti-tumor necrosis factor therapy: a paradoxical adverse reaction. Arthritis Rheum. 2005;52:2513-2518.
- Berg EL, Yoshino T, Rott LS, et al. The cutaneous lymphocyte antigen is a skin lymphocyte homing receptor for the vascular lectin endothelial cell-leukocyte adhesion molecule 1. J Exp Med. 1991;174:1461-1466.
- Flier J, Boorsma DM, van Beek PJ, et al. Differential expression of CXCR3 targeting chemokines CXCL10, CXCL9, and CXCL11 in different types of skin inflammation. J Pathol. 2001;194:398-405.
- Zrioual S, Ecochard R, Tournadre A, et al. Genome-wide comparison between IL-17A- and IL-17F-induced effects in human rheumatoid arthritis synoviocytes. J Immunol. 2009;182:3112-3120.
- Gaffen SL. The role of interleukin-17 in the pathogenesis of rheumatoid arthritis. Curr Rheumatol Rep. 2009;11:365-370.
- Kratz A, Pesce MA, Basner RC, et al. Laboratory values of clinical importance. In: Kasper D, Fauci A, Hauser S, et al, eds. Harrison’s Principles of Internal Medicine. 19th ed. McGraw-Hill; 2014.
- Ixekizumab. Package insert. Eli Lilly & Co; 2017.
- Gordon KB, Blauvelt A, Papp KA, et al. Phase 3 trials of ixekizumab in moderate-to-severe plaque psoriasis. N Engl J Med. 2016;375:345-356.
- Leonardi C, Matheson R, Zachariae C, et al. Anti-interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N Engl J Med. 2012;366:1190-1199.
- Sfikakis PP, Iliopoulos A, Elezoglou A, et al. Psoriasis induced by anti-tumor necrosis factor therapy: a paradoxical adverse reaction. Arthritis Rheum. 2005;52:2513-2518.
- Berg EL, Yoshino T, Rott LS, et al. The cutaneous lymphocyte antigen is a skin lymphocyte homing receptor for the vascular lectin endothelial cell-leukocyte adhesion molecule 1. J Exp Med. 1991;174:1461-1466.
- Flier J, Boorsma DM, van Beek PJ, et al. Differential expression of CXCR3 targeting chemokines CXCL10, CXCL9, and CXCL11 in different types of skin inflammation. J Pathol. 2001;194:398-405.
- Zrioual S, Ecochard R, Tournadre A, et al. Genome-wide comparison between IL-17A- and IL-17F-induced effects in human rheumatoid arthritis synoviocytes. J Immunol. 2009;182:3112-3120.
- Gaffen SL. The role of interleukin-17 in the pathogenesis of rheumatoid arthritis. Curr Rheumatol Rep. 2009;11:365-370.
Practice Points
- Psoriasis is an autoimmune disorder with a predominance of CD4+ and CD8+ T cells that release cytokines, such as tumor necrosis factor 11α and interleukins, which promote inflammation in the skin and joints and is associated with systemic inflammation predisposing patients to cardiovascular disease.
- Common adverse effects of most biologic medications for psoriasis include injection-site pain and rash, fever, malaise, back pain, urticaria and flushing, edema, dyspnea, and nausea.
- Ixekizumab is a humanized IL-17A antagonist intended for adults with moderate to severe psoriasis. Certain rare side effects specific to ixekizumab include inflammatory bowel disease, thrombocytopenia, severe injection-site reactions, and candidiasis.
- Acute onycholysis and acute exacerbation of arthritis/dactylitis are rare side effects of ixekizumab therapy.

58-year-old man • bilateral shoulder pain • history of prostate cancer • limited shoulder range of motion • Dx?
THE CASE
A 58-year-old African American man with a past medical history of prostate cancer, hypertension, hyperlipidemia, osteoarthritis, and gastroesophageal reflux disease presented to our office to establish care with a new provider. He complained of bilateral shoulder pain, that was worse on the right side, for the past year. He denied any previous falls, trauma, or injury. He reported that lifting his grandkids was becoming increasingly difficult due to the pain but denied any weakness or neurologic symptoms. He had been using over-the-counter nonsteroidal anti-inflammatory drugs (NSAIDs), which provided minimal relief.
On physical examination, the overlying skin was normal and there was no tenderness to palpation. His shoulder range of motion was limited with complete flexion, but otherwise intact. Muscle strength was 5 out of 5 bilaterally, and neurovascular and sensory examinations were normal. On the right side, the Empty Can Test was positive, but the Neer and Apley tests were negative. All testing was negative on the left side.
The patient was referred for 10 sessions of physical therapy, which he completed. His pain persisted, and an x-ray of his right shoulder was performed. The x-ray indicated a high-riding humeral head, and magnetic resonance imaging (MRI) of the right shoulder was recommended due to possible rotator cuff tendinopathy.
The MRI demonstrated a full-thickness tear of the distal supraspinatus tendon along with “metastatic lesions” (FIGURE). As a result, a bone scan was obtained and revealed activity in the proximal right humerus; however, it was nonconclusive for osteoblastic metastasis. A positron emission tomography (PET) scan was ordered, which revealed findings suggestive of bony metastasis in the proximal left tibia, distal shaft of the right tibia, and the right and left humeral heads. The patient was then scheduled for a bone biopsy; a chest, abdomen, and pelvis computed tomography (CT) scan with IV and oral contrast was also ordered.

THE DIAGNOSIS
A bone biopsy of the left tibia indicated prominent non-necrotizing granulomatous inflammation and stains were negative for microorganisms. The CT scan demonstrated peribronchial vascular reticulonodular opacities in the upper lung zones compatible with sarcoidosis; no metastatic lesions were identified. Laboratory studies were obtained and demonstrated an elevated angiotensin-converting enzyme (ACE) level consistent with sarcoidosis. The cumulative test results pointed to a diagnosis of osseous sarcoidosis.
DISCUSSION
Osseous sarcoidosis is a rare manifestation of larger systemic disease. It is estimated that bony lesions occur in only 3% to 13% of patients with sarcoidosis.1 Bone involvement is most common in African Americans and occurs primarily in the hands and feet.1-3
Osseous lesions are comprised of noncaseating granulomatous inflammation.4,5 They are often asymptomatic but can be painful and associated with overlying skin disease and soft-tissue swelling.1,4 Although it’s not typical, patients may present with symptoms such as pain, stiffness, or fractures. On CT imaging and MRI (as in this case), osseous lesions can be confused with metastatic bone disease, and biopsy may be required for diagnosis.4
Continue to: There are multiple patterns of bone involvement
There are multiple patterns of bone involvement in osseous sarcoidosis, ranging from large cystic lesions that can lead to stress fractures to “tunnels” or “lace-like” reticulated patterns found in the bones of the hands and feet. 2,3,5,6 Long bone involvement is typically limited to the proximal and distal thirds of the bone.6 Sarcoidosis is also known to involve the axial skeleton, and less commonly, the cranial vault.6 Although multiple variations may manifest over time, skin changes usually precede bone lesions3,6; however, that was not the case with this patient.
Treatment entails pain management
Up to 50% of patients with bone lesions are symptomatic and may require treatment.3,5 Treatment is reserved for these symptomatic patients, with the goal of pain reduction.2,3,7
Low- to moderate-dose corticosteroids have been shown to relieve soft-tissue swelling and decrease pain.2,3,7 A prolonged course of steroids is not recommended, due to the risk of osteoporosis and fractures, and does not normalize bone structure.3,7
Other options. NSAIDs, such as colchicine and indomethacin, have also been found to be effective in pain management.7 Treatments such as methotrexate and hydroxychloroquine may be considered for those cases that are refractory to steroids.2
Given the extent of our patient’s disease, he was referred to multiple specialists to rule out further organ involvement. He was found to have neurosarcoidosis on brain imaging and was subsequently treated with prednisone 10 mg/d. The patient is being routinely monitored for active disease at various intervals or as symptoms arise.
THE TAKEAWAY
Consideration for systemic diseases (eg, sarcoidosis) should be given to patients presenting with musculoskeletal complaints without a significant history of trauma or injury. In those with risk factors associated with a higher incidence of sarcoidosis, such as age and race, a work-up should include imaging and biopsy. Treatment (eg, corticosteroids, NSAIDs) is provided to those patients who are symptomatic, with the goal of symptom relief.3
1. Rao DA, Dellaripa PF. Extrapulmonary manifestations of sarcoidosis. Rheum Dis Clin North Am. 2013;39:277-297. doi: 10.1016/j.rdc.2013.02.007
2. Kobak S. Sarcoidosis: a rheumatologist’s perspective. Ther Adv Musculoskelet Dis. 2015;7:196-205. doi: 10.1177/1759720X15591310
3. Bechman K, Christidis D, Walsh S, et al. A review of the musculoskeletal manifestations of sarcoidosis. Rheumatology (Oxford). 2018;57:777-783. doi: 10.1093/rheumatology/kex317
4. Iannuzzi MC, Rybicki BA, Teirstein AS. Sarcoidosis. N Engl J Med. 2007;357:2153-2165. doi: 10.1056/NEJMra071714
5. Yachoui R, Parker BJ, Nguyen TT. Bone and bone marrow involvement in sarcoidosis. Rheumatol Int. 2015;35:1917-1924. doi: 10.1007/s00296-015-3341-y
6. Aptel S, Lecocq-Teixeira S, Olivier P, et al. Multimodality evaluation of musculoskeletal sarcoidosis: Imaging findings and literature review. Diagn Interv Imaging. 2016;97:5-18. doi: 10.1016/j.diii.2014.11.038
7. Wilcox A, Bharadwaj P, Sharma OP. Bone sarcoidosis. Curr Opin Rheumatol. 2000;12:321-330. doi: 10.1097/00002281-200007000-00016
THE CASE
A 58-year-old African American man with a past medical history of prostate cancer, hypertension, hyperlipidemia, osteoarthritis, and gastroesophageal reflux disease presented to our office to establish care with a new provider. He complained of bilateral shoulder pain, that was worse on the right side, for the past year. He denied any previous falls, trauma, or injury. He reported that lifting his grandkids was becoming increasingly difficult due to the pain but denied any weakness or neurologic symptoms. He had been using over-the-counter nonsteroidal anti-inflammatory drugs (NSAIDs), which provided minimal relief.
On physical examination, the overlying skin was normal and there was no tenderness to palpation. His shoulder range of motion was limited with complete flexion, but otherwise intact. Muscle strength was 5 out of 5 bilaterally, and neurovascular and sensory examinations were normal. On the right side, the Empty Can Test was positive, but the Neer and Apley tests were negative. All testing was negative on the left side.
The patient was referred for 10 sessions of physical therapy, which he completed. His pain persisted, and an x-ray of his right shoulder was performed. The x-ray indicated a high-riding humeral head, and magnetic resonance imaging (MRI) of the right shoulder was recommended due to possible rotator cuff tendinopathy.
The MRI demonstrated a full-thickness tear of the distal supraspinatus tendon along with “metastatic lesions” (FIGURE). As a result, a bone scan was obtained and revealed activity in the proximal right humerus; however, it was nonconclusive for osteoblastic metastasis. A positron emission tomography (PET) scan was ordered, which revealed findings suggestive of bony metastasis in the proximal left tibia, distal shaft of the right tibia, and the right and left humeral heads. The patient was then scheduled for a bone biopsy; a chest, abdomen, and pelvis computed tomography (CT) scan with IV and oral contrast was also ordered.
THE DIAGNOSIS
A bone biopsy of the left tibia indicated prominent non-necrotizing granulomatous inflammation and stains were negative for microorganisms. The CT scan demonstrated peribronchial vascular reticulonodular opacities in the upper lung zones compatible with sarcoidosis; no metastatic lesions were identified. Laboratory studies were obtained and demonstrated an elevated angiotensin-converting enzyme (ACE) level consistent with sarcoidosis. The cumulative test results pointed to a diagnosis of osseous sarcoidosis.
DISCUSSION
Osseous sarcoidosis is a rare manifestation of larger systemic disease. It is estimated that bony lesions occur in only 3% to 13% of patients with sarcoidosis.1 Bone involvement is most common in African Americans and occurs primarily in the hands and feet.1-3
Osseous lesions are comprised of noncaseating granulomatous inflammation.4,5 They are often asymptomatic but can be painful and associated with overlying skin disease and soft-tissue swelling.1,4 Although it’s not typical, patients may present with symptoms such as pain, stiffness, or fractures. On CT imaging and MRI (as in this case), osseous lesions can be confused with metastatic bone disease, and biopsy may be required for diagnosis.4
Continue to: There are multiple patterns of bone involvement
There are multiple patterns of bone involvement in osseous sarcoidosis, ranging from large cystic lesions that can lead to stress fractures to “tunnels” or “lace-like” reticulated patterns found in the bones of the hands and feet. 2,3,5,6 Long bone involvement is typically limited to the proximal and distal thirds of the bone.6 Sarcoidosis is also known to involve the axial skeleton, and less commonly, the cranial vault.6 Although multiple variations may manifest over time, skin changes usually precede bone lesions3,6; however, that was not the case with this patient.
Treatment entails pain management
Up to 50% of patients with bone lesions are symptomatic and may require treatment.3,5 Treatment is reserved for these symptomatic patients, with the goal of pain reduction.2,3,7
Low- to moderate-dose corticosteroids have been shown to relieve soft-tissue swelling and decrease pain.2,3,7 A prolonged course of steroids is not recommended, due to the risk of osteoporosis and fractures, and does not normalize bone structure.3,7
Other options. NSAIDs, such as colchicine and indomethacin, have also been found to be effective in pain management.7 Treatments such as methotrexate and hydroxychloroquine may be considered for those cases that are refractory to steroids.2
Given the extent of our patient’s disease, he was referred to multiple specialists to rule out further organ involvement. He was found to have neurosarcoidosis on brain imaging and was subsequently treated with prednisone 10 mg/d. The patient is being routinely monitored for active disease at various intervals or as symptoms arise.
THE TAKEAWAY
Consideration for systemic diseases (eg, sarcoidosis) should be given to patients presenting with musculoskeletal complaints without a significant history of trauma or injury. In those with risk factors associated with a higher incidence of sarcoidosis, such as age and race, a work-up should include imaging and biopsy. Treatment (eg, corticosteroids, NSAIDs) is provided to those patients who are symptomatic, with the goal of symptom relief.3
THE CASE
A 58-year-old African American man with a past medical history of prostate cancer, hypertension, hyperlipidemia, osteoarthritis, and gastroesophageal reflux disease presented to our office to establish care with a new provider. He complained of bilateral shoulder pain, that was worse on the right side, for the past year. He denied any previous falls, trauma, or injury. He reported that lifting his grandkids was becoming increasingly difficult due to the pain but denied any weakness or neurologic symptoms. He had been using over-the-counter nonsteroidal anti-inflammatory drugs (NSAIDs), which provided minimal relief.
On physical examination, the overlying skin was normal and there was no tenderness to palpation. His shoulder range of motion was limited with complete flexion, but otherwise intact. Muscle strength was 5 out of 5 bilaterally, and neurovascular and sensory examinations were normal. On the right side, the Empty Can Test was positive, but the Neer and Apley tests were negative. All testing was negative on the left side.
The patient was referred for 10 sessions of physical therapy, which he completed. His pain persisted, and an x-ray of his right shoulder was performed. The x-ray indicated a high-riding humeral head, and magnetic resonance imaging (MRI) of the right shoulder was recommended due to possible rotator cuff tendinopathy.
The MRI demonstrated a full-thickness tear of the distal supraspinatus tendon along with “metastatic lesions” (FIGURE). As a result, a bone scan was obtained and revealed activity in the proximal right humerus; however, it was nonconclusive for osteoblastic metastasis. A positron emission tomography (PET) scan was ordered, which revealed findings suggestive of bony metastasis in the proximal left tibia, distal shaft of the right tibia, and the right and left humeral heads. The patient was then scheduled for a bone biopsy; a chest, abdomen, and pelvis computed tomography (CT) scan with IV and oral contrast was also ordered.
THE DIAGNOSIS
A bone biopsy of the left tibia indicated prominent non-necrotizing granulomatous inflammation and stains were negative for microorganisms. The CT scan demonstrated peribronchial vascular reticulonodular opacities in the upper lung zones compatible with sarcoidosis; no metastatic lesions were identified. Laboratory studies were obtained and demonstrated an elevated angiotensin-converting enzyme (ACE) level consistent with sarcoidosis. The cumulative test results pointed to a diagnosis of osseous sarcoidosis.
DISCUSSION
Osseous sarcoidosis is a rare manifestation of larger systemic disease. It is estimated that bony lesions occur in only 3% to 13% of patients with sarcoidosis.1 Bone involvement is most common in African Americans and occurs primarily in the hands and feet.1-3
Osseous lesions are comprised of noncaseating granulomatous inflammation.4,5 They are often asymptomatic but can be painful and associated with overlying skin disease and soft-tissue swelling.1,4 Although it’s not typical, patients may present with symptoms such as pain, stiffness, or fractures. On CT imaging and MRI (as in this case), osseous lesions can be confused with metastatic bone disease, and biopsy may be required for diagnosis.4
Continue to: There are multiple patterns of bone involvement
There are multiple patterns of bone involvement in osseous sarcoidosis, ranging from large cystic lesions that can lead to stress fractures to “tunnels” or “lace-like” reticulated patterns found in the bones of the hands and feet. 2,3,5,6 Long bone involvement is typically limited to the proximal and distal thirds of the bone.6 Sarcoidosis is also known to involve the axial skeleton, and less commonly, the cranial vault.6 Although multiple variations may manifest over time, skin changes usually precede bone lesions3,6; however, that was not the case with this patient.
Treatment entails pain management
Up to 50% of patients with bone lesions are symptomatic and may require treatment.3,5 Treatment is reserved for these symptomatic patients, with the goal of pain reduction.2,3,7
Low- to moderate-dose corticosteroids have been shown to relieve soft-tissue swelling and decrease pain.2,3,7 A prolonged course of steroids is not recommended, due to the risk of osteoporosis and fractures, and does not normalize bone structure.3,7
Other options. NSAIDs, such as colchicine and indomethacin, have also been found to be effective in pain management.7 Treatments such as methotrexate and hydroxychloroquine may be considered for those cases that are refractory to steroids.2
Given the extent of our patient’s disease, he was referred to multiple specialists to rule out further organ involvement. He was found to have neurosarcoidosis on brain imaging and was subsequently treated with prednisone 10 mg/d. The patient is being routinely monitored for active disease at various intervals or as symptoms arise.
THE TAKEAWAY
Consideration for systemic diseases (eg, sarcoidosis) should be given to patients presenting with musculoskeletal complaints without a significant history of trauma or injury. In those with risk factors associated with a higher incidence of sarcoidosis, such as age and race, a work-up should include imaging and biopsy. Treatment (eg, corticosteroids, NSAIDs) is provided to those patients who are symptomatic, with the goal of symptom relief.3
1. Rao DA, Dellaripa PF. Extrapulmonary manifestations of sarcoidosis. Rheum Dis Clin North Am. 2013;39:277-297. doi: 10.1016/j.rdc.2013.02.007
2. Kobak S. Sarcoidosis: a rheumatologist’s perspective. Ther Adv Musculoskelet Dis. 2015;7:196-205. doi: 10.1177/1759720X15591310
3. Bechman K, Christidis D, Walsh S, et al. A review of the musculoskeletal manifestations of sarcoidosis. Rheumatology (Oxford). 2018;57:777-783. doi: 10.1093/rheumatology/kex317
4. Iannuzzi MC, Rybicki BA, Teirstein AS. Sarcoidosis. N Engl J Med. 2007;357:2153-2165. doi: 10.1056/NEJMra071714
5. Yachoui R, Parker BJ, Nguyen TT. Bone and bone marrow involvement in sarcoidosis. Rheumatol Int. 2015;35:1917-1924. doi: 10.1007/s00296-015-3341-y
6. Aptel S, Lecocq-Teixeira S, Olivier P, et al. Multimodality evaluation of musculoskeletal sarcoidosis: Imaging findings and literature review. Diagn Interv Imaging. 2016;97:5-18. doi: 10.1016/j.diii.2014.11.038
7. Wilcox A, Bharadwaj P, Sharma OP. Bone sarcoidosis. Curr Opin Rheumatol. 2000;12:321-330. doi: 10.1097/00002281-200007000-00016
1. Rao DA, Dellaripa PF. Extrapulmonary manifestations of sarcoidosis. Rheum Dis Clin North Am. 2013;39:277-297. doi: 10.1016/j.rdc.2013.02.007
2. Kobak S. Sarcoidosis: a rheumatologist’s perspective. Ther Adv Musculoskelet Dis. 2015;7:196-205. doi: 10.1177/1759720X15591310
3. Bechman K, Christidis D, Walsh S, et al. A review of the musculoskeletal manifestations of sarcoidosis. Rheumatology (Oxford). 2018;57:777-783. doi: 10.1093/rheumatology/kex317
4. Iannuzzi MC, Rybicki BA, Teirstein AS. Sarcoidosis. N Engl J Med. 2007;357:2153-2165. doi: 10.1056/NEJMra071714
5. Yachoui R, Parker BJ, Nguyen TT. Bone and bone marrow involvement in sarcoidosis. Rheumatol Int. 2015;35:1917-1924. doi: 10.1007/s00296-015-3341-y
6. Aptel S, Lecocq-Teixeira S, Olivier P, et al. Multimodality evaluation of musculoskeletal sarcoidosis: Imaging findings and literature review. Diagn Interv Imaging. 2016;97:5-18. doi: 10.1016/j.diii.2014.11.038
7. Wilcox A, Bharadwaj P, Sharma OP. Bone sarcoidosis. Curr Opin Rheumatol. 2000;12:321-330. doi: 10.1097/00002281-200007000-00016

52-year-old man • syncopal episode • chest pain • mild lightheadedness • Dx?
THE CASE
A 52-year-old man with a history of hypertension and gastroesophageal reflux disease (GERD) presented to the emergency department (ED) after an episode of syncope. He reported that the syncope occurred soon after he stood up to go to the kitchen to make dinner but was without prodrome or associated symptoms. He recalled little of the event, and the episode was unwitnessed. He had a few bruises on his arms but no significant injuries.
On questioning, he reported occasional palpitations but no changes in his normal exercise tolerance. His only medication was lisinopril 10 mg/d.
In the ED, his vital signs, physical exam (including orthostatic vital signs), basic labs (including troponin I), and a 12-lead EKG were normal. After a cardiology consultation, he was discharged home with a 30-day ambulatory rhythm monitor.
A few days later, while walking up and down some hills, he experienced about 15 seconds of chest pain accompanied by mild lightheadedness. Thinking it might be related to his GERD, he took some over-the-counter antacids when he returned home, since these had been effective for him in the past.
However, the rhythm monitoring company contacted the EKG lab to transmit a concerning strip (FIGURE). They also reported that the patient had been contacted and reported no further symptoms.

THE DIAGNOSIS
Most notable on the patient’s rhythm strip was a continuously varying QRS complex, which was indicative of polymorphic ventricular tachycardia and consistent with the patient’s syncope and other symptoms. Less obvious at first glance was an ST-segment elevation in the preceding beats. Comparison to a post-episode tracing (FIGURE) highlights the abnormality. Polymorphic ventricular tachycardia resolves in 1 of 2 ways: It will either stop on its own (causing syncope if it lasts more than a few seconds) or it will devolve into ventricular fibrillation, causing cardiac arrest.1
The combination of these findings and the clinical scenario prompted a recommendation that the patient report to the ED for admission (his wife drove him). He was admitted to the intensive care unit (ICU) for continuous telemetry monitoring, and a cardiac catheterization was ordered. The procedure revealed a 99% thrombotic mid-right coronary artery lesion, for which aspiration thrombectomy and uncomplicated stenting were performed.
Continue to: DISCUSSION
DISCUSSION
Guidelines from the American College of Cardiology/American Heart Association/Heart Rhythm Society recommend a detailed history and physical exam, as well as an EKG, for the initial evaluation of syncope.2 If this does not point to a diagnosis (and depending on the presentation and other factors), an ambulatory rhythm monitor can be considered. Other possible testing modalities include stress testing, resting transthoracic echocardiography, electrophysiologic testing, and cardiac magnetic resonance imaging or computed tomography.
Is the cause cardiac? The guidelines suggest that a cardiac cause of syncope is more likely if several of the following factors are present: age > 60 years; male sex; presence of known heart disease (acquired or congenital); brief prodrome (eg, palpitations) or no prodrome; exertional or supine syncope; 1 to 2 episodes; an abnormal cardiac exam; and a family history of premature sudden death.2 A noncardiac cause is suggested by other factors: younger age; no known cardiac disease; standing or a position change from supine to sitting/standing; prodrome; specific triggers (eg, dehydration, pain); and frequent and prolonged stereotypic episodes.2
While the guidelines do not specify the number of factors or endorse a specific scoring system, such tools have been developed. For example, the EGSYS (Evaluation of Guidelines in Syncope Study) Score assigns 1 point for each of 6 factors: palpitations; heart disease and/or abnormal EKG; effort syncope; supine syncope; precipitating or predisposing factors; and autonomic prodromes. A score ≥ 3 identified cardiac syncope with a sensitivity of 95%, but with a specificity of only 61%. In the derivation study, patients with a score ≥ 3 had higher mortality than those with a lower score (17 vs 3%; P < .001).3
Myocardial ischemia can trigger ventricular arrhythmias. In the GUSTO-1 trial of fibrinolytic therapy in patients with acute ST-segment elevation myocardial infarction (n = 40,895), the incidence of ventricular tachycardia or ventricular fibrillation was 10.2%.4 In a pooled analysis (4 trials; n = 26,416) of patients who were treated for non–ST-segment elevation or unstable angina-type acute coronary syndromes, the rate of these arrhythmias was markedly lower (2.1%).5 The risk of ventricular arrhythmia is one reason close monitoring (eg, continuous telemetry, ICU admission) is the standard of care for patients with acute coronary syndromes.
Our patient experienced syncope upon standing, which suggested a noncardiac cause (usually orthostatic hypotension). However, the history of palpitations increased the suspicion for a cardiac cause, and thus the rhythm monitor was ordered.
THE TAKEAWAY
This case was unusual in that ambulatory monitoring captured electrocardiographic evidence of myocardial ischemia leading directly to a ventricular arrhythmia. In the evaluation of syncope, a detailed history, physical exam, and a baseline 12-lead EKG can sometimes give clues to an arrhythmic cause of syncope (eg, Brugada syndrome, prior infarct pattern, prolonged QTc, bradycardia, heart block, arrhythmogenic right ventricular cardiomyopathy)—but prolonged rhythm monitoring is sometimes needed to identify a cause.
Michael A. Chen, MD, PhD, Harborview Medical Center, University of Washington School of Medicine, 325 9th Avenue, Box 359748 (Cardiology), Seattle, WA 98104; [email protected]
1. Viskin S, Chorin E, Viskin D, et al. Polymorphic ventricular tachycardia: terminology, mechanism, diagnosis, and emergency therapy. Circulation. 2021;144:823-839. doi: 10.1161/CIRCULATIONAHA.121.055783
2. Shen W-K, Sheldon RS, Benditt DG, et al. 2017 ACC/AHA/HRS guideline for the evaluation and management of patients with syncope: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol. 2017;70:620-663. doi: 10.1016/j.jacc.2017.03.002
3. Del Rosso A, Ungar A, Maggi R, et al. Clinical predictors of cardiac syncope at initial evaluation in patients referred urgently to a general hospital: the EGSYS score. Heart. 2008;94:1528-1529. doi: 10.1136/hrt.2008.143123
4. Newby KH, Thompson T, Stebbins A, et al. Sustained ventricular arrhythmias in patients receiving thrombolytic therapy: incidence and outcomes. The GUSTO Investigators. Circulation. 1998;98:2567-2573. doi: 10.1161/01.cir.98.23.2567
5. Al-Khatib SM, Granger CB, Huang Y, et al. Sustained ventricular arrhythmias among patients with acute coronary syndromes with no ST-segment elevation: incidence, predictors, and outcomes. Circulation. 2002;106:309-12. doi: 10.1161/01.cir.0000022692.49934.e3
THE CASE
A 52-year-old man with a history of hypertension and gastroesophageal reflux disease (GERD) presented to the emergency department (ED) after an episode of syncope. He reported that the syncope occurred soon after he stood up to go to the kitchen to make dinner but was without prodrome or associated symptoms. He recalled little of the event, and the episode was unwitnessed. He had a few bruises on his arms but no significant injuries.
On questioning, he reported occasional palpitations but no changes in his normal exercise tolerance. His only medication was lisinopril 10 mg/d.
In the ED, his vital signs, physical exam (including orthostatic vital signs), basic labs (including troponin I), and a 12-lead EKG were normal. After a cardiology consultation, he was discharged home with a 30-day ambulatory rhythm monitor.
A few days later, while walking up and down some hills, he experienced about 15 seconds of chest pain accompanied by mild lightheadedness. Thinking it might be related to his GERD, he took some over-the-counter antacids when he returned home, since these had been effective for him in the past.
However, the rhythm monitoring company contacted the EKG lab to transmit a concerning strip (FIGURE). They also reported that the patient had been contacted and reported no further symptoms.
THE DIAGNOSIS
Most notable on the patient’s rhythm strip was a continuously varying QRS complex, which was indicative of polymorphic ventricular tachycardia and consistent with the patient’s syncope and other symptoms. Less obvious at first glance was an ST-segment elevation in the preceding beats. Comparison to a post-episode tracing (FIGURE) highlights the abnormality. Polymorphic ventricular tachycardia resolves in 1 of 2 ways: It will either stop on its own (causing syncope if it lasts more than a few seconds) or it will devolve into ventricular fibrillation, causing cardiac arrest.1
The combination of these findings and the clinical scenario prompted a recommendation that the patient report to the ED for admission (his wife drove him). He was admitted to the intensive care unit (ICU) for continuous telemetry monitoring, and a cardiac catheterization was ordered. The procedure revealed a 99% thrombotic mid-right coronary artery lesion, for which aspiration thrombectomy and uncomplicated stenting were performed.
Continue to: DISCUSSION
DISCUSSION
Guidelines from the American College of Cardiology/American Heart Association/Heart Rhythm Society recommend a detailed history and physical exam, as well as an EKG, for the initial evaluation of syncope.2 If this does not point to a diagnosis (and depending on the presentation and other factors), an ambulatory rhythm monitor can be considered. Other possible testing modalities include stress testing, resting transthoracic echocardiography, electrophysiologic testing, and cardiac magnetic resonance imaging or computed tomography.
Is the cause cardiac? The guidelines suggest that a cardiac cause of syncope is more likely if several of the following factors are present: age > 60 years; male sex; presence of known heart disease (acquired or congenital); brief prodrome (eg, palpitations) or no prodrome; exertional or supine syncope; 1 to 2 episodes; an abnormal cardiac exam; and a family history of premature sudden death.2 A noncardiac cause is suggested by other factors: younger age; no known cardiac disease; standing or a position change from supine to sitting/standing; prodrome; specific triggers (eg, dehydration, pain); and frequent and prolonged stereotypic episodes.2
While the guidelines do not specify the number of factors or endorse a specific scoring system, such tools have been developed. For example, the EGSYS (Evaluation of Guidelines in Syncope Study) Score assigns 1 point for each of 6 factors: palpitations; heart disease and/or abnormal EKG; effort syncope; supine syncope; precipitating or predisposing factors; and autonomic prodromes. A score ≥ 3 identified cardiac syncope with a sensitivity of 95%, but with a specificity of only 61%. In the derivation study, patients with a score ≥ 3 had higher mortality than those with a lower score (17 vs 3%; P < .001).3
Myocardial ischemia can trigger ventricular arrhythmias. In the GUSTO-1 trial of fibrinolytic therapy in patients with acute ST-segment elevation myocardial infarction (n = 40,895), the incidence of ventricular tachycardia or ventricular fibrillation was 10.2%.4 In a pooled analysis (4 trials; n = 26,416) of patients who were treated for non–ST-segment elevation or unstable angina-type acute coronary syndromes, the rate of these arrhythmias was markedly lower (2.1%).5 The risk of ventricular arrhythmia is one reason close monitoring (eg, continuous telemetry, ICU admission) is the standard of care for patients with acute coronary syndromes.
Our patient experienced syncope upon standing, which suggested a noncardiac cause (usually orthostatic hypotension). However, the history of palpitations increased the suspicion for a cardiac cause, and thus the rhythm monitor was ordered.
THE TAKEAWAY
This case was unusual in that ambulatory monitoring captured electrocardiographic evidence of myocardial ischemia leading directly to a ventricular arrhythmia. In the evaluation of syncope, a detailed history, physical exam, and a baseline 12-lead EKG can sometimes give clues to an arrhythmic cause of syncope (eg, Brugada syndrome, prior infarct pattern, prolonged QTc, bradycardia, heart block, arrhythmogenic right ventricular cardiomyopathy)—but prolonged rhythm monitoring is sometimes needed to identify a cause.
Michael A. Chen, MD, PhD, Harborview Medical Center, University of Washington School of Medicine, 325 9th Avenue, Box 359748 (Cardiology), Seattle, WA 98104; [email protected]
THE CASE
A 52-year-old man with a history of hypertension and gastroesophageal reflux disease (GERD) presented to the emergency department (ED) after an episode of syncope. He reported that the syncope occurred soon after he stood up to go to the kitchen to make dinner but was without prodrome or associated symptoms. He recalled little of the event, and the episode was unwitnessed. He had a few bruises on his arms but no significant injuries.
On questioning, he reported occasional palpitations but no changes in his normal exercise tolerance. His only medication was lisinopril 10 mg/d.
In the ED, his vital signs, physical exam (including orthostatic vital signs), basic labs (including troponin I), and a 12-lead EKG were normal. After a cardiology consultation, he was discharged home with a 30-day ambulatory rhythm monitor.
A few days later, while walking up and down some hills, he experienced about 15 seconds of chest pain accompanied by mild lightheadedness. Thinking it might be related to his GERD, he took some over-the-counter antacids when he returned home, since these had been effective for him in the past.
However, the rhythm monitoring company contacted the EKG lab to transmit a concerning strip (FIGURE). They also reported that the patient had been contacted and reported no further symptoms.
THE DIAGNOSIS
Most notable on the patient’s rhythm strip was a continuously varying QRS complex, which was indicative of polymorphic ventricular tachycardia and consistent with the patient’s syncope and other symptoms. Less obvious at first glance was an ST-segment elevation in the preceding beats. Comparison to a post-episode tracing (FIGURE) highlights the abnormality. Polymorphic ventricular tachycardia resolves in 1 of 2 ways: It will either stop on its own (causing syncope if it lasts more than a few seconds) or it will devolve into ventricular fibrillation, causing cardiac arrest.1
The combination of these findings and the clinical scenario prompted a recommendation that the patient report to the ED for admission (his wife drove him). He was admitted to the intensive care unit (ICU) for continuous telemetry monitoring, and a cardiac catheterization was ordered. The procedure revealed a 99% thrombotic mid-right coronary artery lesion, for which aspiration thrombectomy and uncomplicated stenting were performed.
Continue to: DISCUSSION
DISCUSSION
Guidelines from the American College of Cardiology/American Heart Association/Heart Rhythm Society recommend a detailed history and physical exam, as well as an EKG, for the initial evaluation of syncope.2 If this does not point to a diagnosis (and depending on the presentation and other factors), an ambulatory rhythm monitor can be considered. Other possible testing modalities include stress testing, resting transthoracic echocardiography, electrophysiologic testing, and cardiac magnetic resonance imaging or computed tomography.
Is the cause cardiac? The guidelines suggest that a cardiac cause of syncope is more likely if several of the following factors are present: age > 60 years; male sex; presence of known heart disease (acquired or congenital); brief prodrome (eg, palpitations) or no prodrome; exertional or supine syncope; 1 to 2 episodes; an abnormal cardiac exam; and a family history of premature sudden death.2 A noncardiac cause is suggested by other factors: younger age; no known cardiac disease; standing or a position change from supine to sitting/standing; prodrome; specific triggers (eg, dehydration, pain); and frequent and prolonged stereotypic episodes.2
While the guidelines do not specify the number of factors or endorse a specific scoring system, such tools have been developed. For example, the EGSYS (Evaluation of Guidelines in Syncope Study) Score assigns 1 point for each of 6 factors: palpitations; heart disease and/or abnormal EKG; effort syncope; supine syncope; precipitating or predisposing factors; and autonomic prodromes. A score ≥ 3 identified cardiac syncope with a sensitivity of 95%, but with a specificity of only 61%. In the derivation study, patients with a score ≥ 3 had higher mortality than those with a lower score (17 vs 3%; P < .001).3
Myocardial ischemia can trigger ventricular arrhythmias. In the GUSTO-1 trial of fibrinolytic therapy in patients with acute ST-segment elevation myocardial infarction (n = 40,895), the incidence of ventricular tachycardia or ventricular fibrillation was 10.2%.4 In a pooled analysis (4 trials; n = 26,416) of patients who were treated for non–ST-segment elevation or unstable angina-type acute coronary syndromes, the rate of these arrhythmias was markedly lower (2.1%).5 The risk of ventricular arrhythmia is one reason close monitoring (eg, continuous telemetry, ICU admission) is the standard of care for patients with acute coronary syndromes.
Our patient experienced syncope upon standing, which suggested a noncardiac cause (usually orthostatic hypotension). However, the history of palpitations increased the suspicion for a cardiac cause, and thus the rhythm monitor was ordered.
THE TAKEAWAY
This case was unusual in that ambulatory monitoring captured electrocardiographic evidence of myocardial ischemia leading directly to a ventricular arrhythmia. In the evaluation of syncope, a detailed history, physical exam, and a baseline 12-lead EKG can sometimes give clues to an arrhythmic cause of syncope (eg, Brugada syndrome, prior infarct pattern, prolonged QTc, bradycardia, heart block, arrhythmogenic right ventricular cardiomyopathy)—but prolonged rhythm monitoring is sometimes needed to identify a cause.
Michael A. Chen, MD, PhD, Harborview Medical Center, University of Washington School of Medicine, 325 9th Avenue, Box 359748 (Cardiology), Seattle, WA 98104; [email protected]
1. Viskin S, Chorin E, Viskin D, et al. Polymorphic ventricular tachycardia: terminology, mechanism, diagnosis, and emergency therapy. Circulation. 2021;144:823-839. doi: 10.1161/CIRCULATIONAHA.121.055783
2. Shen W-K, Sheldon RS, Benditt DG, et al. 2017 ACC/AHA/HRS guideline for the evaluation and management of patients with syncope: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol. 2017;70:620-663. doi: 10.1016/j.jacc.2017.03.002
3. Del Rosso A, Ungar A, Maggi R, et al. Clinical predictors of cardiac syncope at initial evaluation in patients referred urgently to a general hospital: the EGSYS score. Heart. 2008;94:1528-1529. doi: 10.1136/hrt.2008.143123
4. Newby KH, Thompson T, Stebbins A, et al. Sustained ventricular arrhythmias in patients receiving thrombolytic therapy: incidence and outcomes. The GUSTO Investigators. Circulation. 1998;98:2567-2573. doi: 10.1161/01.cir.98.23.2567
5. Al-Khatib SM, Granger CB, Huang Y, et al. Sustained ventricular arrhythmias among patients with acute coronary syndromes with no ST-segment elevation: incidence, predictors, and outcomes. Circulation. 2002;106:309-12. doi: 10.1161/01.cir.0000022692.49934.e3
1. Viskin S, Chorin E, Viskin D, et al. Polymorphic ventricular tachycardia: terminology, mechanism, diagnosis, and emergency therapy. Circulation. 2021;144:823-839. doi: 10.1161/CIRCULATIONAHA.121.055783
2. Shen W-K, Sheldon RS, Benditt DG, et al. 2017 ACC/AHA/HRS guideline for the evaluation and management of patients with syncope: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol. 2017;70:620-663. doi: 10.1016/j.jacc.2017.03.002
3. Del Rosso A, Ungar A, Maggi R, et al. Clinical predictors of cardiac syncope at initial evaluation in patients referred urgently to a general hospital: the EGSYS score. Heart. 2008;94:1528-1529. doi: 10.1136/hrt.2008.143123
4. Newby KH, Thompson T, Stebbins A, et al. Sustained ventricular arrhythmias in patients receiving thrombolytic therapy: incidence and outcomes. The GUSTO Investigators. Circulation. 1998;98:2567-2573. doi: 10.1161/01.cir.98.23.2567
5. Al-Khatib SM, Granger CB, Huang Y, et al. Sustained ventricular arrhythmias among patients with acute coronary syndromes with no ST-segment elevation: incidence, predictors, and outcomes. Circulation. 2002;106:309-12. doi: 10.1161/01.cir.0000022692.49934.e3

Scheduled Acetaminophen to Minimize Neuropsychiatric Symptoms in Wernicke-Korsakoff Syndrome
To manage the physical, cognitive, and emotional symptoms of a veteran hospitalized for Wernicke-Korsakoff syndrome secondary to chronic alcohol overuse, acetaminophen was administered in place of psychoactive medications.
Alcohol is the most common substance misused by veterans. 1 Veterans may m isuse alcohol as a result of mental illness or posttraumatic stress disorder (PTSD), having difficulties adjusting to civilian life, or because of heavy drinking habits acquired before leaving active duty. 2 One potential long-term effect of chronic alcohol misuse is Wernicke-Korsakoff syndrome (WKS), a neuropsychiatric condition secondary to a deficiency of thiamine. 3 The disease is characterized by altered mental status, oculomotor findings, and ataxia. 3 Patients with WKS may exhibit challenging behaviors, including aggression, disinhibition, and lack of awareness of their illness. 4 Due to long-standing cognitive and physical deficits, many patients require lifelong care with a focus on a palliative approach. 3
The mainstay of pharmacologic management for the neuropsychiatric symptoms of WKS continues to be psychoactive medications, such as antipsychotics, benzodiazepines, antidepressants, and anticonvulsant medications.4-6 Though atypical antipsychotic medications remain the most widely used, they have a high adverse effect (AE) profile.5,6 Among the potential AEs are metabolic syndrome, anticholinergic effects, QTc prolongation, orthostatic hypotension, extrapyramidal effects, sedation, and falls. There also is a US Food and Drug Administration boxed warning for increased risk of mortality.7 With the goal of improving and maintaining patient safety, pharmacologic interventions with lower AEs may be beneficial in the management of the neuropsychiatric symptoms of WKS.
This case describes a veteran who was initially hospitalized due to confusion, ataxia, and nystagmus secondary to chronic alcohol overuse. The aim of the case was to consider the use of acetaminophen in place of psychoactive medications as a way to manage neuropsychiatric symptoms of WKS even when pain was not present.
Case Presentation
A veteran presented to the local US Department of Veterans Affairs (VA) emergency department (ED) due to their spouse’s concern of acute onset confusion and ambulatory difficulties. The veteran’s medical history included extensive alcohol misuse, mild asthma, and diet-controlled hyperlipidemia. On initial evaluation, the veteran displayed symptoms of ataxia and confusion. When asked why the veteran was at the ED, the response was, “I just came to the hospital to find my sister.” Based on their medical history, clinical evaluation, and altered mental status, the veteran was admitted to the acute care medical service with a presumptive diagnosis of WKS.
On admission, the laboratory evaluation revealed normal alanine transaminase (ALT) and aspartate transaminase (AST) levels but markedly elevated γ-glutamyl transferase (GGT) consistent with alcohol toxicity. COVID-19 testing was negative. Magnetic resonance imaging (MRI) of the brain revealed evidence of alterations in the mammillary bodies and moderately severe cortical and cerebellar volume loss suggestive of long-standing alcohol use.
The veteran was hospitalized for 12 days and treated with high-dose IV thiamine, which resulted in improvement of their ophthalmic disorder (nystagmus) and ataxia. However, they continued to exhibit poor recall, confusion, and occasional agitation characterized by verbal outbursts and aggression toward the staff.
The veteran’s spouse worked full time and did not feel capable of providing the necessary follow-up care at home. The safest discharge plan found was to transfer the veteran to the local VA community living center (CLC) for physical therapy and further support of their marked cognitive decline and agitation.
Following admission to the CLC, the veteran was placed in a secured memory unit with staff trained specifically on management of veterans with cognitive impairment and behavioral concerns. As the veteran did not have decisional capacity on admission, the staff arranged a meeting with the spouse. Based on that conversation, the goals of care were to focus on a palliative approach and the hope that the veteran would one day be able to return home to their spouse.
At the CLC, the veteran was initially treated with thiamine 200 mg orally once daily and albuterol inhaler as needed. A clinical psychologist performed a comprehensive psychological evaluation on admission, which confirmed evidence of WKS with symptoms, including confusion, disorientation, and confabulation. There was no evidence of cultural diversity factors regarding the veteran’s delusional beliefs.
After the first full day in the CLC, the nursing staff observed anger and agitation that seemed to start midafternoon and continued until around dinnertime. The veteran displayed verbal outbursts, refusal to cooperate with the staff, and multiple attempts to leave the CLC. With the guidance of a geriatric psychiatrist, risperidone 1 mg once daily as needed was initiated, and staff continued with verbal redirection, both with limited efficacy. After 3 days, due to safety concerns for the veteran, other CLC patients, and CLC staff, risperidone dosing was increased to 1 mg twice daily, which had limited efficacy. Lorazepam 1 mg once daily also was added. A careful medication review was performed to minimize any potential AEs or interactions that might have contributed to the veteran’s behavior, but no pharmacologic interventions were found to fully abate their behavioral issues.
After 5 weeks of ongoing intermittent behavioral issues, the medical team again met to discuss new treatment options.A case reported by Husebo and colleagues used scheduled acetaminophen to help relieve neuropsychiatric symptoms of dementia in a patient who exhibited similar behavioral issues and did not respond well to antipsychotics or benzodiazepines.8 Although our veteran did not express or exhibit obvious pain, the medical team chose to trial this intervention, and the veteran was started on acetaminophen 650 mg orally 3 times daily. A comprehensive metabolic panel, including GGT and thyroid-stimulating hormone, was performed before starting acetaminophen; no abnormalities were noted. The clinical examination did not reveal physical abnormalities other than ataxia.
After 5 days of therapy with the scheduled acetaminophen, the veteran’s clinical behavior dramatically improved. The veteran exhibited infrequent agitated behavior and became cooperative with staff. Three days later, the scheduled lorazepam was discontinued, and eventually they were tapered off risperidone. One month after starting scheduled acetaminophen, the veteran had improved to a point where the staff determined a safe discharge plan could be initiated. The veteran’s nystagmus resolved and behavioral issues improved, although cognitive impairment persisted.
Due to COVID-19, a teleconference was scheduled with the veteran’s spouse to discuss a discharge plan. The spouse was pleased that the veteran had progressed adequately both functionally and behaviorally to make a safe discharge home possible. The spouse arranged to take family leave from their job to help support the veteran after discharge. The veteran was able to return home with a safe discharge plan 1 week later. The acetaminophen was continued with twice-daily dosing and was continued because there were no new behavioral issues. This was done to enhance postfacility adherence and minimize the risk of drug-drug interactions. Attempts to follow up with the veteran postdischarge were unfortunately unsuccessful as the family lived out of the local area.
Discussion
Alcohol misuse is a common finding in many US veterans, as well as in the general population.1,3 As a result, it is not uncommon to see patients with physical and psychological symptoms related to this abuse. Many of these patients will become verbally and physically abusive, thus having appropriate pharmacologic and nonpharmacologic interventions is important.
In this case study, the veteran was diagnosed with WKS and exhibited physical, cognitive, and emotional symptoms consistent with this disease. Although the physical symptoms improved with thiamine and abstinence from alcohol, their cognitive impairment, verbal outbursts, and aggressive demeanor persisted.
After using antipsychotic and anxiolytic medications with minimal clinical improvement, a trial of acetaminophen 650 mg 3 times daily was instituted. The patient’s behavior improved; demeanor became calmer, and they were easily redirected by the nursing staff. Psychological support was again employed, which enhanced and supported the veteran’s calmer demeanor. Although there is limited medical literature on the use of acetaminophen in clinical situations not related to pain, there has been research documenting its effect on social interaction.9,10
Acetaminophen is an analgesic medication that acts through central neural mechanisms. It has been hypothesized that social and physical pain rely on shared neurochemical underpinnings, and some of the regions of the brain involved in affective experience of physical pain also have been found to be involved in the experience of social pain.11 Acetaminophen may impact an individual’s social well-being as social pain processes.11 It has been shown to blunt reactivity to both physical pain as well as negative stimuli.11
Conclusions
A 2019 survey on alcohol and drug use found 5.6% of adults aged ≥ 18 have an alcohol use disorder.12 In severe cases, this can result in WKS. Although replacement of thiamine is critical for physical improvement, psychological deficits may persist. Small studies have advanced the concept of using scheduled acetaminophen even when the patient is not verbalizing or displaying pain.13 Although more research needs to be done on this topic, this palliative approach may be worth considering, especially if the risks of antipsychotics and anxiolytics outweigh the benefits.
1. National Institute on Drug Abuse. Substance use and military life drug facts. Published October 2019. Accessed November 10, 2021. https://www.drugabuse.gov/publications/drugfacts/substance-use-military-life
2. National Veterans Foundation. What statistics show about veteran substance abuse and why proper treatment is important. Published March 30, 2016. Accessed November 10, 2021. https://nvf.org/veteran-substance-abuse-statistics
3. National Center for Biotechnology Information. Korsakoff syndrome. Updated July 10, 2020. Accessed November 10, 2021. https://www.ncbi.nlm.nih.gov/books/NBK539854
4. Gerridzen IJ, Goossensen MA. Patients with Korsakoff syndrome in nursing homes: characteristics, comorbidity, and use of psychotropic drugs. Int Psychogeriatr. 2014;26(1):115-121. doi:10.1017/S1041610213001543
5. Press D, Alexander M. Management of neuropsychiatric symptoms of dementia. Updated October 2021. Accessed November 10, 2021. https://www.uptodate.com/contents/management-of-neuropsychiatric-symptoms-of-dementia
6. Steinberg M, Lyketsos CG. Atypical antipsychotic use in patients with dementia: Managing safety concerns. Am J Psychiatry. 2012;169(9):900-906. doi:10.1176/appi.ajp.2012.12030342
7. Jibson MD. Second-generation antipsychotic medications: pharmacology, administration, and side effects. https://www.uptodate.com/contents/second-generation-antipsychotic-medications-pharmacology-administration-and-side-effects
8. Husebo BS, Ballard C, Sandvik R, Nilsen OB, Aarsland D. Efficacy of treating pain to reduce behavioural disturbances in residents of nursing homes with dementia: cluster randomised clinical trial. BMJ. 2011;343:d4065. doi:10.1136/bmj.d4065
9. Fung K, Alden LE. Once hurt, twice shy: social pain contributes to social anxiety. Emotion. 2017;(2):231-239. doi:10.1037/emo0000223
10. Roberts ID, Krajbich I, Cheavens JS, Campo JV, Way BM. Acetaminophen Reduces Distrust in Individuals with Borderline Personality Disorder Features. Clin Psychol Sci. 2018;6(1):145-154. doi:10.1177/2167702617731374
11. Dewall CN, Macdonald G, Webster GD, et al. Acetaminophen reduces social pain: behavioral and neural evidence. Psychol Sci. 2010;21(7):931-937. doi:10.1177/0956797610374741
12. National Institute on Alcohol Abuse and Alcoholism. Alcohol facts and statistics. Updated June 2021. Accessed November 2, 202November 10, 2021. https://www.niaaa.nih.gov/publications/brochures-and-fact-sheets/alcohol-facts-and-statistics
13. Chibnall JT, Tait RC, Harman B, Luebbert RA. Effect of acetaminophen on behavior, well-being, and psychotropic medication use in nursing home residents with moderate-to-severe dementia. J Am Geriatrics Soc. 2005;53(11):1921-9. doi:10.1111/j.1532-5415.2005.53572.x
To manage the physical, cognitive, and emotional symptoms of a veteran hospitalized for Wernicke-Korsakoff syndrome secondary to chronic alcohol overuse, acetaminophen was administered in place of psychoactive medications.
To manage the physical, cognitive, and emotional symptoms of a veteran hospitalized for Wernicke-Korsakoff syndrome secondary to chronic alcohol overuse, acetaminophen was administered in place of psychoactive medications.
Alcohol is the most common substance misused by veterans. 1 Veterans may m isuse alcohol as a result of mental illness or posttraumatic stress disorder (PTSD), having difficulties adjusting to civilian life, or because of heavy drinking habits acquired before leaving active duty. 2 One potential long-term effect of chronic alcohol misuse is Wernicke-Korsakoff syndrome (WKS), a neuropsychiatric condition secondary to a deficiency of thiamine. 3 The disease is characterized by altered mental status, oculomotor findings, and ataxia. 3 Patients with WKS may exhibit challenging behaviors, including aggression, disinhibition, and lack of awareness of their illness. 4 Due to long-standing cognitive and physical deficits, many patients require lifelong care with a focus on a palliative approach. 3
The mainstay of pharmacologic management for the neuropsychiatric symptoms of WKS continues to be psychoactive medications, such as antipsychotics, benzodiazepines, antidepressants, and anticonvulsant medications.4-6 Though atypical antipsychotic medications remain the most widely used, they have a high adverse effect (AE) profile.5,6 Among the potential AEs are metabolic syndrome, anticholinergic effects, QTc prolongation, orthostatic hypotension, extrapyramidal effects, sedation, and falls. There also is a US Food and Drug Administration boxed warning for increased risk of mortality.7 With the goal of improving and maintaining patient safety, pharmacologic interventions with lower AEs may be beneficial in the management of the neuropsychiatric symptoms of WKS.
This case describes a veteran who was initially hospitalized due to confusion, ataxia, and nystagmus secondary to chronic alcohol overuse. The aim of the case was to consider the use of acetaminophen in place of psychoactive medications as a way to manage neuropsychiatric symptoms of WKS even when pain was not present.
Case Presentation
A veteran presented to the local US Department of Veterans Affairs (VA) emergency department (ED) due to their spouse’s concern of acute onset confusion and ambulatory difficulties. The veteran’s medical history included extensive alcohol misuse, mild asthma, and diet-controlled hyperlipidemia. On initial evaluation, the veteran displayed symptoms of ataxia and confusion. When asked why the veteran was at the ED, the response was, “I just came to the hospital to find my sister.” Based on their medical history, clinical evaluation, and altered mental status, the veteran was admitted to the acute care medical service with a presumptive diagnosis of WKS.
On admission, the laboratory evaluation revealed normal alanine transaminase (ALT) and aspartate transaminase (AST) levels but markedly elevated γ-glutamyl transferase (GGT) consistent with alcohol toxicity. COVID-19 testing was negative. Magnetic resonance imaging (MRI) of the brain revealed evidence of alterations in the mammillary bodies and moderately severe cortical and cerebellar volume loss suggestive of long-standing alcohol use.
The veteran was hospitalized for 12 days and treated with high-dose IV thiamine, which resulted in improvement of their ophthalmic disorder (nystagmus) and ataxia. However, they continued to exhibit poor recall, confusion, and occasional agitation characterized by verbal outbursts and aggression toward the staff.
The veteran’s spouse worked full time and did not feel capable of providing the necessary follow-up care at home. The safest discharge plan found was to transfer the veteran to the local VA community living center (CLC) for physical therapy and further support of their marked cognitive decline and agitation.
Following admission to the CLC, the veteran was placed in a secured memory unit with staff trained specifically on management of veterans with cognitive impairment and behavioral concerns. As the veteran did not have decisional capacity on admission, the staff arranged a meeting with the spouse. Based on that conversation, the goals of care were to focus on a palliative approach and the hope that the veteran would one day be able to return home to their spouse.
At the CLC, the veteran was initially treated with thiamine 200 mg orally once daily and albuterol inhaler as needed. A clinical psychologist performed a comprehensive psychological evaluation on admission, which confirmed evidence of WKS with symptoms, including confusion, disorientation, and confabulation. There was no evidence of cultural diversity factors regarding the veteran’s delusional beliefs.
After the first full day in the CLC, the nursing staff observed anger and agitation that seemed to start midafternoon and continued until around dinnertime. The veteran displayed verbal outbursts, refusal to cooperate with the staff, and multiple attempts to leave the CLC. With the guidance of a geriatric psychiatrist, risperidone 1 mg once daily as needed was initiated, and staff continued with verbal redirection, both with limited efficacy. After 3 days, due to safety concerns for the veteran, other CLC patients, and CLC staff, risperidone dosing was increased to 1 mg twice daily, which had limited efficacy. Lorazepam 1 mg once daily also was added. A careful medication review was performed to minimize any potential AEs or interactions that might have contributed to the veteran’s behavior, but no pharmacologic interventions were found to fully abate their behavioral issues.
After 5 weeks of ongoing intermittent behavioral issues, the medical team again met to discuss new treatment options.A case reported by Husebo and colleagues used scheduled acetaminophen to help relieve neuropsychiatric symptoms of dementia in a patient who exhibited similar behavioral issues and did not respond well to antipsychotics or benzodiazepines.8 Although our veteran did not express or exhibit obvious pain, the medical team chose to trial this intervention, and the veteran was started on acetaminophen 650 mg orally 3 times daily. A comprehensive metabolic panel, including GGT and thyroid-stimulating hormone, was performed before starting acetaminophen; no abnormalities were noted. The clinical examination did not reveal physical abnormalities other than ataxia.
After 5 days of therapy with the scheduled acetaminophen, the veteran’s clinical behavior dramatically improved. The veteran exhibited infrequent agitated behavior and became cooperative with staff. Three days later, the scheduled lorazepam was discontinued, and eventually they were tapered off risperidone. One month after starting scheduled acetaminophen, the veteran had improved to a point where the staff determined a safe discharge plan could be initiated. The veteran’s nystagmus resolved and behavioral issues improved, although cognitive impairment persisted.
Due to COVID-19, a teleconference was scheduled with the veteran’s spouse to discuss a discharge plan. The spouse was pleased that the veteran had progressed adequately both functionally and behaviorally to make a safe discharge home possible. The spouse arranged to take family leave from their job to help support the veteran after discharge. The veteran was able to return home with a safe discharge plan 1 week later. The acetaminophen was continued with twice-daily dosing and was continued because there were no new behavioral issues. This was done to enhance postfacility adherence and minimize the risk of drug-drug interactions. Attempts to follow up with the veteran postdischarge were unfortunately unsuccessful as the family lived out of the local area.
Discussion
Alcohol misuse is a common finding in many US veterans, as well as in the general population.1,3 As a result, it is not uncommon to see patients with physical and psychological symptoms related to this abuse. Many of these patients will become verbally and physically abusive, thus having appropriate pharmacologic and nonpharmacologic interventions is important.
In this case study, the veteran was diagnosed with WKS and exhibited physical, cognitive, and emotional symptoms consistent with this disease. Although the physical symptoms improved with thiamine and abstinence from alcohol, their cognitive impairment, verbal outbursts, and aggressive demeanor persisted.
After using antipsychotic and anxiolytic medications with minimal clinical improvement, a trial of acetaminophen 650 mg 3 times daily was instituted. The patient’s behavior improved; demeanor became calmer, and they were easily redirected by the nursing staff. Psychological support was again employed, which enhanced and supported the veteran’s calmer demeanor. Although there is limited medical literature on the use of acetaminophen in clinical situations not related to pain, there has been research documenting its effect on social interaction.9,10
Acetaminophen is an analgesic medication that acts through central neural mechanisms. It has been hypothesized that social and physical pain rely on shared neurochemical underpinnings, and some of the regions of the brain involved in affective experience of physical pain also have been found to be involved in the experience of social pain.11 Acetaminophen may impact an individual’s social well-being as social pain processes.11 It has been shown to blunt reactivity to both physical pain as well as negative stimuli.11
Conclusions
A 2019 survey on alcohol and drug use found 5.6% of adults aged ≥ 18 have an alcohol use disorder.12 In severe cases, this can result in WKS. Although replacement of thiamine is critical for physical improvement, psychological deficits may persist. Small studies have advanced the concept of using scheduled acetaminophen even when the patient is not verbalizing or displaying pain.13 Although more research needs to be done on this topic, this palliative approach may be worth considering, especially if the risks of antipsychotics and anxiolytics outweigh the benefits.
Alcohol is the most common substance misused by veterans. 1 Veterans may m isuse alcohol as a result of mental illness or posttraumatic stress disorder (PTSD), having difficulties adjusting to civilian life, or because of heavy drinking habits acquired before leaving active duty. 2 One potential long-term effect of chronic alcohol misuse is Wernicke-Korsakoff syndrome (WKS), a neuropsychiatric condition secondary to a deficiency of thiamine. 3 The disease is characterized by altered mental status, oculomotor findings, and ataxia. 3 Patients with WKS may exhibit challenging behaviors, including aggression, disinhibition, and lack of awareness of their illness. 4 Due to long-standing cognitive and physical deficits, many patients require lifelong care with a focus on a palliative approach. 3
The mainstay of pharmacologic management for the neuropsychiatric symptoms of WKS continues to be psychoactive medications, such as antipsychotics, benzodiazepines, antidepressants, and anticonvulsant medications.4-6 Though atypical antipsychotic medications remain the most widely used, they have a high adverse effect (AE) profile.5,6 Among the potential AEs are metabolic syndrome, anticholinergic effects, QTc prolongation, orthostatic hypotension, extrapyramidal effects, sedation, and falls. There also is a US Food and Drug Administration boxed warning for increased risk of mortality.7 With the goal of improving and maintaining patient safety, pharmacologic interventions with lower AEs may be beneficial in the management of the neuropsychiatric symptoms of WKS.
This case describes a veteran who was initially hospitalized due to confusion, ataxia, and nystagmus secondary to chronic alcohol overuse. The aim of the case was to consider the use of acetaminophen in place of psychoactive medications as a way to manage neuropsychiatric symptoms of WKS even when pain was not present.
Case Presentation
A veteran presented to the local US Department of Veterans Affairs (VA) emergency department (ED) due to their spouse’s concern of acute onset confusion and ambulatory difficulties. The veteran’s medical history included extensive alcohol misuse, mild asthma, and diet-controlled hyperlipidemia. On initial evaluation, the veteran displayed symptoms of ataxia and confusion. When asked why the veteran was at the ED, the response was, “I just came to the hospital to find my sister.” Based on their medical history, clinical evaluation, and altered mental status, the veteran was admitted to the acute care medical service with a presumptive diagnosis of WKS.
On admission, the laboratory evaluation revealed normal alanine transaminase (ALT) and aspartate transaminase (AST) levels but markedly elevated γ-glutamyl transferase (GGT) consistent with alcohol toxicity. COVID-19 testing was negative. Magnetic resonance imaging (MRI) of the brain revealed evidence of alterations in the mammillary bodies and moderately severe cortical and cerebellar volume loss suggestive of long-standing alcohol use.
The veteran was hospitalized for 12 days and treated with high-dose IV thiamine, which resulted in improvement of their ophthalmic disorder (nystagmus) and ataxia. However, they continued to exhibit poor recall, confusion, and occasional agitation characterized by verbal outbursts and aggression toward the staff.
The veteran’s spouse worked full time and did not feel capable of providing the necessary follow-up care at home. The safest discharge plan found was to transfer the veteran to the local VA community living center (CLC) for physical therapy and further support of their marked cognitive decline and agitation.
Following admission to the CLC, the veteran was placed in a secured memory unit with staff trained specifically on management of veterans with cognitive impairment and behavioral concerns. As the veteran did not have decisional capacity on admission, the staff arranged a meeting with the spouse. Based on that conversation, the goals of care were to focus on a palliative approach and the hope that the veteran would one day be able to return home to their spouse.
At the CLC, the veteran was initially treated with thiamine 200 mg orally once daily and albuterol inhaler as needed. A clinical psychologist performed a comprehensive psychological evaluation on admission, which confirmed evidence of WKS with symptoms, including confusion, disorientation, and confabulation. There was no evidence of cultural diversity factors regarding the veteran’s delusional beliefs.
After the first full day in the CLC, the nursing staff observed anger and agitation that seemed to start midafternoon and continued until around dinnertime. The veteran displayed verbal outbursts, refusal to cooperate with the staff, and multiple attempts to leave the CLC. With the guidance of a geriatric psychiatrist, risperidone 1 mg once daily as needed was initiated, and staff continued with verbal redirection, both with limited efficacy. After 3 days, due to safety concerns for the veteran, other CLC patients, and CLC staff, risperidone dosing was increased to 1 mg twice daily, which had limited efficacy. Lorazepam 1 mg once daily also was added. A careful medication review was performed to minimize any potential AEs or interactions that might have contributed to the veteran’s behavior, but no pharmacologic interventions were found to fully abate their behavioral issues.
After 5 weeks of ongoing intermittent behavioral issues, the medical team again met to discuss new treatment options.A case reported by Husebo and colleagues used scheduled acetaminophen to help relieve neuropsychiatric symptoms of dementia in a patient who exhibited similar behavioral issues and did not respond well to antipsychotics or benzodiazepines.8 Although our veteran did not express or exhibit obvious pain, the medical team chose to trial this intervention, and the veteran was started on acetaminophen 650 mg orally 3 times daily. A comprehensive metabolic panel, including GGT and thyroid-stimulating hormone, was performed before starting acetaminophen; no abnormalities were noted. The clinical examination did not reveal physical abnormalities other than ataxia.
After 5 days of therapy with the scheduled acetaminophen, the veteran’s clinical behavior dramatically improved. The veteran exhibited infrequent agitated behavior and became cooperative with staff. Three days later, the scheduled lorazepam was discontinued, and eventually they were tapered off risperidone. One month after starting scheduled acetaminophen, the veteran had improved to a point where the staff determined a safe discharge plan could be initiated. The veteran’s nystagmus resolved and behavioral issues improved, although cognitive impairment persisted.
Due to COVID-19, a teleconference was scheduled with the veteran’s spouse to discuss a discharge plan. The spouse was pleased that the veteran had progressed adequately both functionally and behaviorally to make a safe discharge home possible. The spouse arranged to take family leave from their job to help support the veteran after discharge. The veteran was able to return home with a safe discharge plan 1 week later. The acetaminophen was continued with twice-daily dosing and was continued because there were no new behavioral issues. This was done to enhance postfacility adherence and minimize the risk of drug-drug interactions. Attempts to follow up with the veteran postdischarge were unfortunately unsuccessful as the family lived out of the local area.
Discussion
Alcohol misuse is a common finding in many US veterans, as well as in the general population.1,3 As a result, it is not uncommon to see patients with physical and psychological symptoms related to this abuse. Many of these patients will become verbally and physically abusive, thus having appropriate pharmacologic and nonpharmacologic interventions is important.
In this case study, the veteran was diagnosed with WKS and exhibited physical, cognitive, and emotional symptoms consistent with this disease. Although the physical symptoms improved with thiamine and abstinence from alcohol, their cognitive impairment, verbal outbursts, and aggressive demeanor persisted.
After using antipsychotic and anxiolytic medications with minimal clinical improvement, a trial of acetaminophen 650 mg 3 times daily was instituted. The patient’s behavior improved; demeanor became calmer, and they were easily redirected by the nursing staff. Psychological support was again employed, which enhanced and supported the veteran’s calmer demeanor. Although there is limited medical literature on the use of acetaminophen in clinical situations not related to pain, there has been research documenting its effect on social interaction.9,10
Acetaminophen is an analgesic medication that acts through central neural mechanisms. It has been hypothesized that social and physical pain rely on shared neurochemical underpinnings, and some of the regions of the brain involved in affective experience of physical pain also have been found to be involved in the experience of social pain.11 Acetaminophen may impact an individual’s social well-being as social pain processes.11 It has been shown to blunt reactivity to both physical pain as well as negative stimuli.11
Conclusions
A 2019 survey on alcohol and drug use found 5.6% of adults aged ≥ 18 have an alcohol use disorder.12 In severe cases, this can result in WKS. Although replacement of thiamine is critical for physical improvement, psychological deficits may persist. Small studies have advanced the concept of using scheduled acetaminophen even when the patient is not verbalizing or displaying pain.13 Although more research needs to be done on this topic, this palliative approach may be worth considering, especially if the risks of antipsychotics and anxiolytics outweigh the benefits.
1. National Institute on Drug Abuse. Substance use and military life drug facts. Published October 2019. Accessed November 10, 2021. https://www.drugabuse.gov/publications/drugfacts/substance-use-military-life
2. National Veterans Foundation. What statistics show about veteran substance abuse and why proper treatment is important. Published March 30, 2016. Accessed November 10, 2021. https://nvf.org/veteran-substance-abuse-statistics
3. National Center for Biotechnology Information. Korsakoff syndrome. Updated July 10, 2020. Accessed November 10, 2021. https://www.ncbi.nlm.nih.gov/books/NBK539854
4. Gerridzen IJ, Goossensen MA. Patients with Korsakoff syndrome in nursing homes: characteristics, comorbidity, and use of psychotropic drugs. Int Psychogeriatr. 2014;26(1):115-121. doi:10.1017/S1041610213001543
5. Press D, Alexander M. Management of neuropsychiatric symptoms of dementia. Updated October 2021. Accessed November 10, 2021. https://www.uptodate.com/contents/management-of-neuropsychiatric-symptoms-of-dementia
6. Steinberg M, Lyketsos CG. Atypical antipsychotic use in patients with dementia: Managing safety concerns. Am J Psychiatry. 2012;169(9):900-906. doi:10.1176/appi.ajp.2012.12030342
7. Jibson MD. Second-generation antipsychotic medications: pharmacology, administration, and side effects. https://www.uptodate.com/contents/second-generation-antipsychotic-medications-pharmacology-administration-and-side-effects
8. Husebo BS, Ballard C, Sandvik R, Nilsen OB, Aarsland D. Efficacy of treating pain to reduce behavioural disturbances in residents of nursing homes with dementia: cluster randomised clinical trial. BMJ. 2011;343:d4065. doi:10.1136/bmj.d4065
9. Fung K, Alden LE. Once hurt, twice shy: social pain contributes to social anxiety. Emotion. 2017;(2):231-239. doi:10.1037/emo0000223
10. Roberts ID, Krajbich I, Cheavens JS, Campo JV, Way BM. Acetaminophen Reduces Distrust in Individuals with Borderline Personality Disorder Features. Clin Psychol Sci. 2018;6(1):145-154. doi:10.1177/2167702617731374
11. Dewall CN, Macdonald G, Webster GD, et al. Acetaminophen reduces social pain: behavioral and neural evidence. Psychol Sci. 2010;21(7):931-937. doi:10.1177/0956797610374741
12. National Institute on Alcohol Abuse and Alcoholism. Alcohol facts and statistics. Updated June 2021. Accessed November 2, 202November 10, 2021. https://www.niaaa.nih.gov/publications/brochures-and-fact-sheets/alcohol-facts-and-statistics
13. Chibnall JT, Tait RC, Harman B, Luebbert RA. Effect of acetaminophen on behavior, well-being, and psychotropic medication use in nursing home residents with moderate-to-severe dementia. J Am Geriatrics Soc. 2005;53(11):1921-9. doi:10.1111/j.1532-5415.2005.53572.x
1. National Institute on Drug Abuse. Substance use and military life drug facts. Published October 2019. Accessed November 10, 2021. https://www.drugabuse.gov/publications/drugfacts/substance-use-military-life
2. National Veterans Foundation. What statistics show about veteran substance abuse and why proper treatment is important. Published March 30, 2016. Accessed November 10, 2021. https://nvf.org/veteran-substance-abuse-statistics
3. National Center for Biotechnology Information. Korsakoff syndrome. Updated July 10, 2020. Accessed November 10, 2021. https://www.ncbi.nlm.nih.gov/books/NBK539854
4. Gerridzen IJ, Goossensen MA. Patients with Korsakoff syndrome in nursing homes: characteristics, comorbidity, and use of psychotropic drugs. Int Psychogeriatr. 2014;26(1):115-121. doi:10.1017/S1041610213001543
5. Press D, Alexander M. Management of neuropsychiatric symptoms of dementia. Updated October 2021. Accessed November 10, 2021. https://www.uptodate.com/contents/management-of-neuropsychiatric-symptoms-of-dementia
6. Steinberg M, Lyketsos CG. Atypical antipsychotic use in patients with dementia: Managing safety concerns. Am J Psychiatry. 2012;169(9):900-906. doi:10.1176/appi.ajp.2012.12030342
7. Jibson MD. Second-generation antipsychotic medications: pharmacology, administration, and side effects. https://www.uptodate.com/contents/second-generation-antipsychotic-medications-pharmacology-administration-and-side-effects
8. Husebo BS, Ballard C, Sandvik R, Nilsen OB, Aarsland D. Efficacy of treating pain to reduce behavioural disturbances in residents of nursing homes with dementia: cluster randomised clinical trial. BMJ. 2011;343:d4065. doi:10.1136/bmj.d4065
9. Fung K, Alden LE. Once hurt, twice shy: social pain contributes to social anxiety. Emotion. 2017;(2):231-239. doi:10.1037/emo0000223
10. Roberts ID, Krajbich I, Cheavens JS, Campo JV, Way BM. Acetaminophen Reduces Distrust in Individuals with Borderline Personality Disorder Features. Clin Psychol Sci. 2018;6(1):145-154. doi:10.1177/2167702617731374
11. Dewall CN, Macdonald G, Webster GD, et al. Acetaminophen reduces social pain: behavioral and neural evidence. Psychol Sci. 2010;21(7):931-937. doi:10.1177/0956797610374741
12. National Institute on Alcohol Abuse and Alcoholism. Alcohol facts and statistics. Updated June 2021. Accessed November 2, 202November 10, 2021. https://www.niaaa.nih.gov/publications/brochures-and-fact-sheets/alcohol-facts-and-statistics
13. Chibnall JT, Tait RC, Harman B, Luebbert RA. Effect of acetaminophen on behavior, well-being, and psychotropic medication use in nursing home residents with moderate-to-severe dementia. J Am Geriatrics Soc. 2005;53(11):1921-9. doi:10.1111/j.1532-5415.2005.53572.x
Using Telehealth Rehabilitation Therapy to Treat a Finger Flexor Tendon Repair During COVID-19
Telehealth-assisted finger rehabilitat ion therapy demonstrated good functional results following repair of a zone 2 flexor tendon laceration.
In 1948, Sterling Bunnell, MD, used the term no man’s land to describe the area between the A1 pulley at the volar aspect of the metacarpophalangeal joint and the insertion of the flexor digitorum superficialis tendons on the middle phalanx (zone 2).1 Bunnell’s description referenced the area of land in World War I between the trenches of opposing armies, and his goal was to emphasize the heightened risks of performing tendon repair in this area, as these repairs were notorious for poor outcomes. In lieu of tendon repair, Bunnell advocated treatment of tendon lacerations in this area with tendon excision and grafting.
It was not until the 1960s that researchers began to advocate for acute repair of tendons in this area.2,3 Since Verdan’s and Kleinart’s work, fastidious adherence to atraumatic technique and improvements in suture technique and rehabilitation protocols have allowed hand surgeons to repair tendons in this area with some level of success. Over the ensuing decades, acute repair of flexor tendon injuries within zone 2 has become the standard of care. The importance of meticulous technique during flexor tendon repair cannot be overemphasized; however, without appropriate hand therapy, even the most meticulous repair may fail.
COVID-19 has created significant barriers to patient care. Reducing travel and limiting face-to-face patient visits have been emphasized as methods that reduce spread of the virus, but these restrictions also prevent patients from easily accessing hand therapy. Recent adoption of telemedicine and videoconferencing technologies may help to reduce some of these barriers, but few previous studies have described the use of videoconferencing technology to supplant face-to-face hand therapy visits. This case describes the use of videoconferencing technology to provide hand therapy for a patient following repair of an acute flexor tendon laceration in zone 2.
Case Presentation
A patient aged < 50 years presented to a US Department of Veterans Affairs (VA) hand surgery clinic 2 days after sustaining a laceration to the flexor digitorum profundus (FDP) in zone 2 of the small finger while cleaning a knife. During the discussion of their treatment options and the recommended postoperative hand therapy protocol, the patient noted difficulty attending postoperative appointments due to COVID-19 as well as a lack of resources. Given these limitations and following discussion with our hand therapist, we discussed the potential for telehealth follow-up with videoconferencing. Four days following the injury, the patient underwent repair of the FDP. During surgery, the laceration was present at the level of the A3 pulley. The FDP was repaired using a 6-0 polypropylene synthetic suture for the epitendinous repair and 4-strand core suture repair using 3-0 Fiberwire suture in a modified cruciate fashion. The A2 and A4 pulleys were preserved, and venting of the pulleys was not required. At the time of surgery, the flexor digitorum superficialis and radial and ulnar digital neurovascular bundles were intact. Following surgical repair of the tendon, the patient was placed into a dorsal blocking splint with a plan for follow-up within 2 to 3 days.
The patient attended the first postoperative visit in person on postoperative day 2. During this visit, the postoperative splint and dressings were removed, and a forearm-based dorsal blocking orthosis was fabricated using thermoplastic. At this visit, the veteran relayed concerns regarding psychosocial and resource barriers in addition to concerns surrounding COVID-19 that would prevent travel to and from hand therapy appointments. Due to these concerns, a passive-motion protocol was initiated using the Indiana manual as a guide.4 The patient returned to the hand clinic at 2 weeks after surgery for evaluation by the operating surgeon and suture removal. All visits after the suture removal were conducted via either telehealth with videoconferencing or by telephone (Table 1).
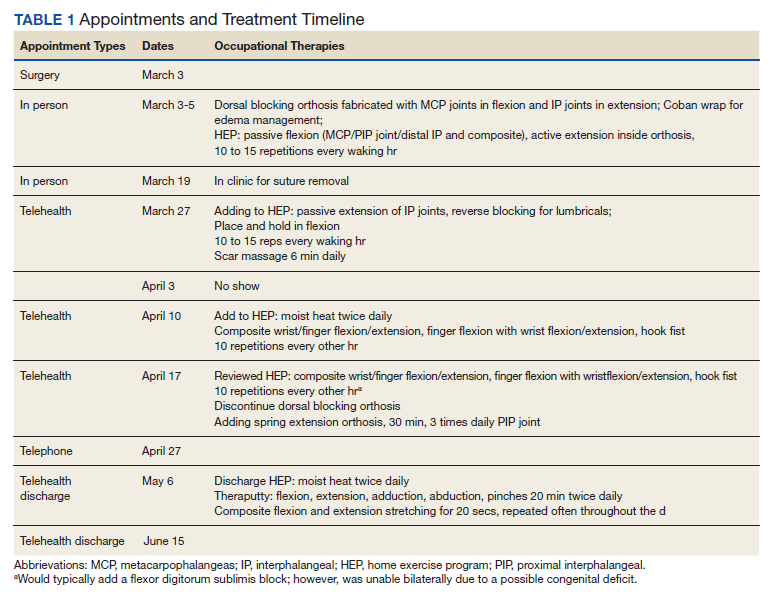
The operative team evaluated the patient 5 times following surgery. Only 2 of these visits were in-person. The patient attended 6 hand therapy sessions with 2 in-person visits to occupational therapy (Figure 1). The remaining 4 visits were conducted using videoconferencing. The patient received therapy supplies by mail as needed, and their use was reviewed in telerehabilitation sessions with videoconferencing as needed. During their postoperative course, the patient experienced little edema or scar tissue formation, and recovery was uncomplicated. The patient developed a mild extensor lag for which a proximal interphalangeal joint spring extension orthosis was provided via mail (Figure 2). The patient admitted only partial adherence with this orthosis, and at discharge, a 10-degree extensor lag remained. The patient was not concerned by this extension deficit and did not experience any associated functional deficits, demonstrated by scores on the Quick Disabilities of the Arm, Shoulder and Hand questionnaire and Patient Specific Functional Scale (Table 2).
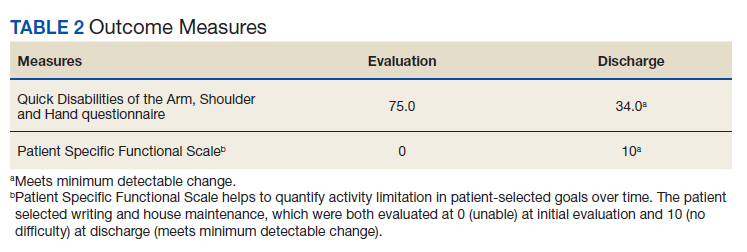

Discussion
Few studies have been published that address the efficacy of telerehabilitation after surgical management of traumatic injuries involving the upper extremity. One Australian study performed by Worboys and colleagues concluded that utilization of telehealth services for hand therapy visits may provide accurate patient assessment with favorable patient satisfaction.5 Another study performed in the UK by Gilbert and colleagues demonstrated that videoconferencing is well received by patients, as it may offer shorter wait times, improved convenience, and reduced travel cost.
The authors noted that although videoconferencing may not completely replace in-person therapy, it could act as an adjunct.6 While these in-person visits may be necessary, particularly to establish care, at least one study has demonstrated that patients may prefer follow-up via telehealth if provided the option.7 In a randomized, controlled study performed in Norway, patients were randomized to either an in-person or video consultation with an orthopedic outpatient clinic. Of patients randomized to the in-person clinic visit, 86% preferred to have follow-up via videoconferencing.7
Previous studies have demonstrated that telehealth may produce accurate patient assessment, with relatively high patient satisfaction. Given the COVID-19 pandemic and the limitations that this crisis has placed on in-person outpatient visits, clinics that previously may have been resistant to telehealth are adapting and using the technology to meet the needs of their population.8 The present case demonstrates that videoconferencing is feasible and may lead to successful results, even for cases requiring significant hand therapy follow-up, such as flexor tendon repairs.
Conclusions
Although in-person hand therapy remains the standard of care following flexor tendon repair of the hand, situations may exist in which hand therapy conducted via telehealth is better than no hand therapy at all. The present case study highlights the use of telehealth as an acceptable supplement to in-person postoperative visits.
In our case, use of a standardized protocol with an emphasis on hand function and patient satisfaction as opposed to strict range of motion measurements produced good results. Although a specific telehealth satisfaction measure was not used in this case, commonly used questionnaires may be integrated into future visits to improve telehealth implementation and patient experience. In this specific case, the veteran felt that hand function was regained and expressed general satisfaction with the telemedicine process at the conclusion of care. While telehealth was a useful adjunct in the treatment of the present patient, further study of videoconferencing should be conducted to determine whether hand therapy conducted via telehealth could be implemented more broadly following upper extremity surgery.
1. Hege JJ. History off-hand: Bunnell’s no-man’s land. Hand (NY). 2019;14(4):570-574. doi:10.1177/1558944717744337
2. Verdan C. Primary repair of flexor tendons. J Bone Joint Surg Am. 1960;42-A:647-657.
3. Kleinert HE, Kutz JE, Ashbell TS, et al. Primary repair of lacerated flexor tendon in no man’s land (abstract). J Bone Joint Surg. 1967;49A:577.
4. Cannon NM. Diagnosis and Treatment Manual for Physicians and Therapists: Upper Extremity Rehabilitation. 4th ed. Hand Rehabilitation Center of Indiana; 2001.
5. Worboys T, Brassington M, Ward EC, Cornwell PL. Delivering occupational therapy hand assessment and treatment sessions via telehealth. J Telemed Telecare. 2018;24(3):185-192. doi:10.1177/1357633X17691861
6. Gilbert AW, Jaggi A, May CR. What is the patient acceptability of real time 1:1 videoconferencing in an orthopaedics setting? A systematic review. Physiotherapy. 2018;104(2):178-186. doi:10.1016/j.physio.2017.11.217
7. Buvik A, Bugge E, Knutsen G, Smatresk A, Wilsgaard T. Patient reported outcomes with remote orthopaedic consultations by telemedicine: A randomised controlled trial. J Telemed Telecare. 2019;25(8):451-459. doi:10.1177/1357633X18783921
8. Loeb AE, Rao SS, Ficke JR, Morris CD, Riley LH 3rd, Levin AS. Departmental experience and lessons learned with accelerated introduction of telemedicine during the COVID-19 crisis. J Am Acad Orthop Surg. 2020;28(11):e469-e476. doi:10.5435/JAAOS-D-20-00380
Telehealth-assisted finger rehabilitat ion therapy demonstrated good functional results following repair of a zone 2 flexor tendon laceration.
Telehealth-assisted finger rehabilitat ion therapy demonstrated good functional results following repair of a zone 2 flexor tendon laceration.
In 1948, Sterling Bunnell, MD, used the term no man’s land to describe the area between the A1 pulley at the volar aspect of the metacarpophalangeal joint and the insertion of the flexor digitorum superficialis tendons on the middle phalanx (zone 2).1 Bunnell’s description referenced the area of land in World War I between the trenches of opposing armies, and his goal was to emphasize the heightened risks of performing tendon repair in this area, as these repairs were notorious for poor outcomes. In lieu of tendon repair, Bunnell advocated treatment of tendon lacerations in this area with tendon excision and grafting.
It was not until the 1960s that researchers began to advocate for acute repair of tendons in this area.2,3 Since Verdan’s and Kleinart’s work, fastidious adherence to atraumatic technique and improvements in suture technique and rehabilitation protocols have allowed hand surgeons to repair tendons in this area with some level of success. Over the ensuing decades, acute repair of flexor tendon injuries within zone 2 has become the standard of care. The importance of meticulous technique during flexor tendon repair cannot be overemphasized; however, without appropriate hand therapy, even the most meticulous repair may fail.
COVID-19 has created significant barriers to patient care. Reducing travel and limiting face-to-face patient visits have been emphasized as methods that reduce spread of the virus, but these restrictions also prevent patients from easily accessing hand therapy. Recent adoption of telemedicine and videoconferencing technologies may help to reduce some of these barriers, but few previous studies have described the use of videoconferencing technology to supplant face-to-face hand therapy visits. This case describes the use of videoconferencing technology to provide hand therapy for a patient following repair of an acute flexor tendon laceration in zone 2.
Case Presentation
A patient aged < 50 years presented to a US Department of Veterans Affairs (VA) hand surgery clinic 2 days after sustaining a laceration to the flexor digitorum profundus (FDP) in zone 2 of the small finger while cleaning a knife. During the discussion of their treatment options and the recommended postoperative hand therapy protocol, the patient noted difficulty attending postoperative appointments due to COVID-19 as well as a lack of resources. Given these limitations and following discussion with our hand therapist, we discussed the potential for telehealth follow-up with videoconferencing. Four days following the injury, the patient underwent repair of the FDP. During surgery, the laceration was present at the level of the A3 pulley. The FDP was repaired using a 6-0 polypropylene synthetic suture for the epitendinous repair and 4-strand core suture repair using 3-0 Fiberwire suture in a modified cruciate fashion. The A2 and A4 pulleys were preserved, and venting of the pulleys was not required. At the time of surgery, the flexor digitorum superficialis and radial and ulnar digital neurovascular bundles were intact. Following surgical repair of the tendon, the patient was placed into a dorsal blocking splint with a plan for follow-up within 2 to 3 days.
The patient attended the first postoperative visit in person on postoperative day 2. During this visit, the postoperative splint and dressings were removed, and a forearm-based dorsal blocking orthosis was fabricated using thermoplastic. At this visit, the veteran relayed concerns regarding psychosocial and resource barriers in addition to concerns surrounding COVID-19 that would prevent travel to and from hand therapy appointments. Due to these concerns, a passive-motion protocol was initiated using the Indiana manual as a guide.4 The patient returned to the hand clinic at 2 weeks after surgery for evaluation by the operating surgeon and suture removal. All visits after the suture removal were conducted via either telehealth with videoconferencing or by telephone (Table 1).
The operative team evaluated the patient 5 times following surgery. Only 2 of these visits were in-person. The patient attended 6 hand therapy sessions with 2 in-person visits to occupational therapy (Figure 1). The remaining 4 visits were conducted using videoconferencing. The patient received therapy supplies by mail as needed, and their use was reviewed in telerehabilitation sessions with videoconferencing as needed. During their postoperative course, the patient experienced little edema or scar tissue formation, and recovery was uncomplicated. The patient developed a mild extensor lag for which a proximal interphalangeal joint spring extension orthosis was provided via mail (Figure 2). The patient admitted only partial adherence with this orthosis, and at discharge, a 10-degree extensor lag remained. The patient was not concerned by this extension deficit and did not experience any associated functional deficits, demonstrated by scores on the Quick Disabilities of the Arm, Shoulder and Hand questionnaire and Patient Specific Functional Scale (Table 2).
Discussion
Few studies have been published that address the efficacy of telerehabilitation after surgical management of traumatic injuries involving the upper extremity. One Australian study performed by Worboys and colleagues concluded that utilization of telehealth services for hand therapy visits may provide accurate patient assessment with favorable patient satisfaction.5 Another study performed in the UK by Gilbert and colleagues demonstrated that videoconferencing is well received by patients, as it may offer shorter wait times, improved convenience, and reduced travel cost.
The authors noted that although videoconferencing may not completely replace in-person therapy, it could act as an adjunct.6 While these in-person visits may be necessary, particularly to establish care, at least one study has demonstrated that patients may prefer follow-up via telehealth if provided the option.7 In a randomized, controlled study performed in Norway, patients were randomized to either an in-person or video consultation with an orthopedic outpatient clinic. Of patients randomized to the in-person clinic visit, 86% preferred to have follow-up via videoconferencing.7
Previous studies have demonstrated that telehealth may produce accurate patient assessment, with relatively high patient satisfaction. Given the COVID-19 pandemic and the limitations that this crisis has placed on in-person outpatient visits, clinics that previously may have been resistant to telehealth are adapting and using the technology to meet the needs of their population.8 The present case demonstrates that videoconferencing is feasible and may lead to successful results, even for cases requiring significant hand therapy follow-up, such as flexor tendon repairs.
Conclusions
Although in-person hand therapy remains the standard of care following flexor tendon repair of the hand, situations may exist in which hand therapy conducted via telehealth is better than no hand therapy at all. The present case study highlights the use of telehealth as an acceptable supplement to in-person postoperative visits.
In our case, use of a standardized protocol with an emphasis on hand function and patient satisfaction as opposed to strict range of motion measurements produced good results. Although a specific telehealth satisfaction measure was not used in this case, commonly used questionnaires may be integrated into future visits to improve telehealth implementation and patient experience. In this specific case, the veteran felt that hand function was regained and expressed general satisfaction with the telemedicine process at the conclusion of care. While telehealth was a useful adjunct in the treatment of the present patient, further study of videoconferencing should be conducted to determine whether hand therapy conducted via telehealth could be implemented more broadly following upper extremity surgery.
In 1948, Sterling Bunnell, MD, used the term no man’s land to describe the area between the A1 pulley at the volar aspect of the metacarpophalangeal joint and the insertion of the flexor digitorum superficialis tendons on the middle phalanx (zone 2).1 Bunnell’s description referenced the area of land in World War I between the trenches of opposing armies, and his goal was to emphasize the heightened risks of performing tendon repair in this area, as these repairs were notorious for poor outcomes. In lieu of tendon repair, Bunnell advocated treatment of tendon lacerations in this area with tendon excision and grafting.
It was not until the 1960s that researchers began to advocate for acute repair of tendons in this area.2,3 Since Verdan’s and Kleinart’s work, fastidious adherence to atraumatic technique and improvements in suture technique and rehabilitation protocols have allowed hand surgeons to repair tendons in this area with some level of success. Over the ensuing decades, acute repair of flexor tendon injuries within zone 2 has become the standard of care. The importance of meticulous technique during flexor tendon repair cannot be overemphasized; however, without appropriate hand therapy, even the most meticulous repair may fail.
COVID-19 has created significant barriers to patient care. Reducing travel and limiting face-to-face patient visits have been emphasized as methods that reduce spread of the virus, but these restrictions also prevent patients from easily accessing hand therapy. Recent adoption of telemedicine and videoconferencing technologies may help to reduce some of these barriers, but few previous studies have described the use of videoconferencing technology to supplant face-to-face hand therapy visits. This case describes the use of videoconferencing technology to provide hand therapy for a patient following repair of an acute flexor tendon laceration in zone 2.
Case Presentation
A patient aged < 50 years presented to a US Department of Veterans Affairs (VA) hand surgery clinic 2 days after sustaining a laceration to the flexor digitorum profundus (FDP) in zone 2 of the small finger while cleaning a knife. During the discussion of their treatment options and the recommended postoperative hand therapy protocol, the patient noted difficulty attending postoperative appointments due to COVID-19 as well as a lack of resources. Given these limitations and following discussion with our hand therapist, we discussed the potential for telehealth follow-up with videoconferencing. Four days following the injury, the patient underwent repair of the FDP. During surgery, the laceration was present at the level of the A3 pulley. The FDP was repaired using a 6-0 polypropylene synthetic suture for the epitendinous repair and 4-strand core suture repair using 3-0 Fiberwire suture in a modified cruciate fashion. The A2 and A4 pulleys were preserved, and venting of the pulleys was not required. At the time of surgery, the flexor digitorum superficialis and radial and ulnar digital neurovascular bundles were intact. Following surgical repair of the tendon, the patient was placed into a dorsal blocking splint with a plan for follow-up within 2 to 3 days.
The patient attended the first postoperative visit in person on postoperative day 2. During this visit, the postoperative splint and dressings were removed, and a forearm-based dorsal blocking orthosis was fabricated using thermoplastic. At this visit, the veteran relayed concerns regarding psychosocial and resource barriers in addition to concerns surrounding COVID-19 that would prevent travel to and from hand therapy appointments. Due to these concerns, a passive-motion protocol was initiated using the Indiana manual as a guide.4 The patient returned to the hand clinic at 2 weeks after surgery for evaluation by the operating surgeon and suture removal. All visits after the suture removal were conducted via either telehealth with videoconferencing or by telephone (Table 1).
The operative team evaluated the patient 5 times following surgery. Only 2 of these visits were in-person. The patient attended 6 hand therapy sessions with 2 in-person visits to occupational therapy (Figure 1). The remaining 4 visits were conducted using videoconferencing. The patient received therapy supplies by mail as needed, and their use was reviewed in telerehabilitation sessions with videoconferencing as needed. During their postoperative course, the patient experienced little edema or scar tissue formation, and recovery was uncomplicated. The patient developed a mild extensor lag for which a proximal interphalangeal joint spring extension orthosis was provided via mail (Figure 2). The patient admitted only partial adherence with this orthosis, and at discharge, a 10-degree extensor lag remained. The patient was not concerned by this extension deficit and did not experience any associated functional deficits, demonstrated by scores on the Quick Disabilities of the Arm, Shoulder and Hand questionnaire and Patient Specific Functional Scale (Table 2).
Discussion
Few studies have been published that address the efficacy of telerehabilitation after surgical management of traumatic injuries involving the upper extremity. One Australian study performed by Worboys and colleagues concluded that utilization of telehealth services for hand therapy visits may provide accurate patient assessment with favorable patient satisfaction.5 Another study performed in the UK by Gilbert and colleagues demonstrated that videoconferencing is well received by patients, as it may offer shorter wait times, improved convenience, and reduced travel cost.
The authors noted that although videoconferencing may not completely replace in-person therapy, it could act as an adjunct.6 While these in-person visits may be necessary, particularly to establish care, at least one study has demonstrated that patients may prefer follow-up via telehealth if provided the option.7 In a randomized, controlled study performed in Norway, patients were randomized to either an in-person or video consultation with an orthopedic outpatient clinic. Of patients randomized to the in-person clinic visit, 86% preferred to have follow-up via videoconferencing.7
Previous studies have demonstrated that telehealth may produce accurate patient assessment, with relatively high patient satisfaction. Given the COVID-19 pandemic and the limitations that this crisis has placed on in-person outpatient visits, clinics that previously may have been resistant to telehealth are adapting and using the technology to meet the needs of their population.8 The present case demonstrates that videoconferencing is feasible and may lead to successful results, even for cases requiring significant hand therapy follow-up, such as flexor tendon repairs.
Conclusions
Although in-person hand therapy remains the standard of care following flexor tendon repair of the hand, situations may exist in which hand therapy conducted via telehealth is better than no hand therapy at all. The present case study highlights the use of telehealth as an acceptable supplement to in-person postoperative visits.
In our case, use of a standardized protocol with an emphasis on hand function and patient satisfaction as opposed to strict range of motion measurements produced good results. Although a specific telehealth satisfaction measure was not used in this case, commonly used questionnaires may be integrated into future visits to improve telehealth implementation and patient experience. In this specific case, the veteran felt that hand function was regained and expressed general satisfaction with the telemedicine process at the conclusion of care. While telehealth was a useful adjunct in the treatment of the present patient, further study of videoconferencing should be conducted to determine whether hand therapy conducted via telehealth could be implemented more broadly following upper extremity surgery.
1. Hege JJ. History off-hand: Bunnell’s no-man’s land. Hand (NY). 2019;14(4):570-574. doi:10.1177/1558944717744337
2. Verdan C. Primary repair of flexor tendons. J Bone Joint Surg Am. 1960;42-A:647-657.
3. Kleinert HE, Kutz JE, Ashbell TS, et al. Primary repair of lacerated flexor tendon in no man’s land (abstract). J Bone Joint Surg. 1967;49A:577.
4. Cannon NM. Diagnosis and Treatment Manual for Physicians and Therapists: Upper Extremity Rehabilitation. 4th ed. Hand Rehabilitation Center of Indiana; 2001.
5. Worboys T, Brassington M, Ward EC, Cornwell PL. Delivering occupational therapy hand assessment and treatment sessions via telehealth. J Telemed Telecare. 2018;24(3):185-192. doi:10.1177/1357633X17691861
6. Gilbert AW, Jaggi A, May CR. What is the patient acceptability of real time 1:1 videoconferencing in an orthopaedics setting? A systematic review. Physiotherapy. 2018;104(2):178-186. doi:10.1016/j.physio.2017.11.217
7. Buvik A, Bugge E, Knutsen G, Smatresk A, Wilsgaard T. Patient reported outcomes with remote orthopaedic consultations by telemedicine: A randomised controlled trial. J Telemed Telecare. 2019;25(8):451-459. doi:10.1177/1357633X18783921
8. Loeb AE, Rao SS, Ficke JR, Morris CD, Riley LH 3rd, Levin AS. Departmental experience and lessons learned with accelerated introduction of telemedicine during the COVID-19 crisis. J Am Acad Orthop Surg. 2020;28(11):e469-e476. doi:10.5435/JAAOS-D-20-00380
1. Hege JJ. History off-hand: Bunnell’s no-man’s land. Hand (NY). 2019;14(4):570-574. doi:10.1177/1558944717744337
2. Verdan C. Primary repair of flexor tendons. J Bone Joint Surg Am. 1960;42-A:647-657.
3. Kleinert HE, Kutz JE, Ashbell TS, et al. Primary repair of lacerated flexor tendon in no man’s land (abstract). J Bone Joint Surg. 1967;49A:577.
4. Cannon NM. Diagnosis and Treatment Manual for Physicians and Therapists: Upper Extremity Rehabilitation. 4th ed. Hand Rehabilitation Center of Indiana; 2001.
5. Worboys T, Brassington M, Ward EC, Cornwell PL. Delivering occupational therapy hand assessment and treatment sessions via telehealth. J Telemed Telecare. 2018;24(3):185-192. doi:10.1177/1357633X17691861
6. Gilbert AW, Jaggi A, May CR. What is the patient acceptability of real time 1:1 videoconferencing in an orthopaedics setting? A systematic review. Physiotherapy. 2018;104(2):178-186. doi:10.1016/j.physio.2017.11.217
7. Buvik A, Bugge E, Knutsen G, Smatresk A, Wilsgaard T. Patient reported outcomes with remote orthopaedic consultations by telemedicine: A randomised controlled trial. J Telemed Telecare. 2019;25(8):451-459. doi:10.1177/1357633X18783921
8. Loeb AE, Rao SS, Ficke JR, Morris CD, Riley LH 3rd, Levin AS. Departmental experience and lessons learned with accelerated introduction of telemedicine during the COVID-19 crisis. J Am Acad Orthop Surg. 2020;28(11):e469-e476. doi:10.5435/JAAOS-D-20-00380