User login
Vesicular Eruption Secondary to Bites by Larval Amblyomma americanum
Case Report
A 58-year-old woman presented to the dermatology office with a widespread pruritic eruption of 3 days’ duration that started in the groin and spread to the rest of the body. No treatments had been attempted. She had no notable medical history, and she denied any recent illness, change in personal care products, or new medications or supplements. She reported a camping trip 2 weeks prior to presentation on the east end of Long Island, New York. She later learned that others on the same trip developed a similar, albeit less widespread, eruption.

Physical examination revealed clear vesicles on the arms, legs, trunk, and pubic area (Figure 1). Dermoscopy revealed a small lone star tick larva in the center of one of the vesicles (Figure 2). The type of tick larva was identified using resources from the Centers for Disease Control and Prevention (Figure 3).1 Careful inspection revealed dark marks on various vesicles, mostly in the perineum, yielding nearly 20 larvae, which were removed with forceps. The patient was counseled to cover herself in petrolatum for 2 to 3 hours with the hope of smothering any remaining tick larvae. She was given triamcinolone cream and was encouraged to take a nonsedating antihistamine for itch. The patient was seen back in clinic 2 weeks later and the eruption had resolved.

Comment
Spread of Tick-Borne Disease—Ticks and tick-borne disease are increasing major health concerns for humans, domesticated animals, and livestock. Reported cases of bacterial and protozoan tick-borne disease doubled in the United States between 2004 and 2016. Ninety percent of the nearly 60,000 cases of nationally notifiable vector-borne diseases reported in 2017 were linked to ticks.2 Geographic ranges of multiple tick species continue to expand, which is thought to be secondary to rising global temperatures, ecologic changes, reforestation, and increases in commerce and travel (Figure 4).3 Not only have warming temperatures contributed to geographic range expansion, they also may extend ticks’ active season. The lone star tick (Amblyomma americanum) is widely distributed throughout much of the eastern United States.4 The range of A americanum has expanded north in recent years from its prior core range in the southeastern United States.2 One study found that from 2006 to 2016, the vector tick species most commonly collected from humans and submitted to a tick surveillance system in New Jersey shifted from Ixodes scapularis to A americanum.5

Bites by Amblyomma Ticks—As with most hard ticks, the life cycle of A americanum lasts 2 years and includes the egg, the 6-legged larva or “seed tick,” the 8-legged immature nymph, and the 8-legged reproductively mature adult (Figure 3). Amblyomma americanum can lay several thousand eggs.2 Because our patient had numerous bites, it is plausible that she came into contact with a nest of newly hatched tick larvae. Morphogenesis from larva to nymph, then nymph to adult, requires a blood meal.6,7 The larvae emerge from eggs deposited on the ground and then crawl up low vegetation where they can easily attach to passing hosts. The tick clings to hair or clothing and waits until the host is at rest before moving to a favorable location and then bites.8 When attaching, ticks inject an anesthetic akin to lidocaine, making the bite painless. A tick may spend up to 24 hours on the host prior to biting and then feed for 2 hours to 7 days before releasing.9 For the majority of tick-borne illnesses, the tick must remain attached for 24 to 48 hours before disease is transmitted.10
 in the United States.")
All stages of
Even when the ticks do not transmit disease, tick bites can cause impressive local reactions. Uncomplicated bites can be painful and leave a puncture wound that can take 1 to 2 weeks to heal.13 Rarely, bites can cause a delayed hypersensitivity reaction including fever, pruritus, and urticaria. Granulomas can develop if a tick is improperly removed.9 Other reports describe prurigo lesions, skin hemorrhage, papular urticaria, diffuse papules, vesicles and bullae, necrotic ulcers, and patchy alopecia.14,15 A 2015 systematic controlled study of human bite reactions from A americanum demonstrated the development of itchy erythematous papules and vesicles within 48 hours of larval tick attachment to research participants. The study found tissue damage from A americanum mouthparts, and degranulating mast cells may be evident in as little as 15 minutes.16 The severity of individual skin reaction is hypothesized to depend on several variables, such as the duration of feeding, size of mouthparts, type of tick secretions, changes in secretions during feeding, and prior exposures of the host.14
Tick Removal—If patients present to clinic with ticks attached, removal can be challenging. Removal recommendations call for use of blunt forceps or tweezers. Ticks should be grasped near the skin with consistent pressure, and the tick should be pulled straight out, perpendicular to the skin. Twisting motions can cause the head to separate from the body and remain in the bite wound. Immediately following removal, the area should be cleansed with a disinfectant.10,17 After the tick is removed, some studies recommend storing the tick at −20 °C; should the patient develop disease, the tick could be sent for evaluation.6,17 If there is no clinical or serologic evidence of infection, testing for the presence of antibodies against tick-borne bacteria at presentation and at 3 and 6 weeks is not recommended due to low sensitivity, low positive predictive value, and cost. Clinicians must only observe and treat if disease occurs.17
Prevention of Tick Bites—Tick bites are best prevented by avoiding tick-infested areas; when these areas are unavoidable, tick bites may be prevented by wearing long pants with the pant legs tucked into boots. In addition, applying topical DEET (N,N-diethyl-m-toluamide) repellent to exposed skin and treating clothing with permethrin can be helpful.17 When used alone, DEET provides greater than 90% protection for up to 2.7 hours against A americanum.18 Permethrin-treated clothing alone is 79% to 100% effective at killing A americanum ticks or disabling them for several hours.19
Conclusion
Tick-borne illness is an increasingly important cause of human infectious disease. In addition to their role as a disease vector, ticks can produce primary skin disorders. This case posed a diagnostic challenge because of the unusually large number and wide distribution of bites as well as the subsequent vesicular reaction that ensued. It is important to keep tick larvae or adult tick bites in the differential when evaluating a patient to expedite tick removal and begin clinical monitoring. Recognition of A americanum larvae as a potential cause of pruritic papules may be helpful in similar cases. In addition, it is important for dermatologists to be aware of the tick species in their area.
- Centers for Disease Control and Prevention. Tick ID. Accessed February 21, 2022. https://www.cdc.gov/ticks/tickbornediseases/tickID.html
- Molaei G, Little EAH, Williams SC, et al. Bracing for the worst—range expansion of the lone star tick in the northeastern United States. N Engl J Med. 2019;381:2189-2192.
- Centers for Disease Control and Prevention, Division of Vector-Borne Diseases. Lone star tick (Amblyomma americanum). Accessed March 23, 2022. https://www.cdc.gov/ticks/maps/lone_star_tick.pdf
- Reynolds HH, Elston DM. What’s eating you? lone star tick (Amblyomma americanum). Cutis. 2017;99:111-114.
- Jordan RA, Egizi A. The growing importance of lone star ticks in a Lyme disease endemic county: passive tick surveillance in Monmouth County, NJ, 2006–2016. PLoS One. 2019;14:E0211778.
- Singh-Behl D, La Rosa SP, Tomecki KJ. Tick-borne infections. Dermatol Clin. 2003;21:237-244, v.
- Spach DH, Liles WC, Campbell GL, et al. Tick-borne diseases in the United States. N Engl J Med. 1993;329:936-947.
- Duckworth PF Jr, Hayden GF, Reed CN. Human infestation by Amblyomma americanum larvae (“seed ticks”). South Med J. 1985;78:751-753.
- Middleton DB. Tick-borne infections. what starts as a tiny bite may have a serious outcome. Postgrad Med. 1994;95:131-139.
- Moody EK, Barker RW, White JL, et al. Ticks and tick-borne diseases in Oklahoma. J Okla State Med Assoc. 1998;91:438-445.
- Jones BE. Human ‘seed tick’ infestation. Amblyomma americanum larvae. Arch Dermatol. 1981;117:812-814.
- Centers for Disease Control and Prevention. Tick bite prophylaxis. Accessed February 21, 2022. https://www.cdc.gov/ticks/tickbornediseases/tick-bite-prophylaxis.html
- Fisher EJ, Mo J, Lucky AW. Multiple pruritic papules from lone star tick larvae bites. Arch Dermatol. 2006;142:491-494.
- Krinsky WL. Dermatoses associated with the bites of mites and ticks (Arthropoda: Acari). Int J Dermatol. 1983;22:75-91.
- Yesudian P, Thambiah AS. Persistent papules after tick-bites. Dermatologica. 1973;147:214-218.
- Goddard J, Portugal JS. Cutaneous lesions due to bites by larval Amblyomma americanum ticks. JAMA Dermatol. 2015;151:1373-1375.
- Parola P, Raoult D. Ticks and tickborne bacterial diseases in humans: an emerging infectious threat. Clin Infect Dis. 2001;32:897-928.
- Solberg VB, Klein TA, McPherson KR, et al. Field evaluation of DEET and a piperidine repellent (AI3-37220) against Amblyomma americanum (Acari: Ixodidae). J Med Entomol. 1995;32:870-875.
- Evans SR, Korch GW Jr, Lawson MA. Comparative field evaluation of permethrin and DEET-treated military uniforms for personal protection against ticks (Acari). J Med Entomol. 1990;27:829-834.
Case Report
A 58-year-old woman presented to the dermatology office with a widespread pruritic eruption of 3 days’ duration that started in the groin and spread to the rest of the body. No treatments had been attempted. She had no notable medical history, and she denied any recent illness, change in personal care products, or new medications or supplements. She reported a camping trip 2 weeks prior to presentation on the east end of Long Island, New York. She later learned that others on the same trip developed a similar, albeit less widespread, eruption.
Physical examination revealed clear vesicles on the arms, legs, trunk, and pubic area (Figure 1). Dermoscopy revealed a small lone star tick larva in the center of one of the vesicles (Figure 2). The type of tick larva was identified using resources from the Centers for Disease Control and Prevention (Figure 3).1 Careful inspection revealed dark marks on various vesicles, mostly in the perineum, yielding nearly 20 larvae, which were removed with forceps. The patient was counseled to cover herself in petrolatum for 2 to 3 hours with the hope of smothering any remaining tick larvae. She was given triamcinolone cream and was encouraged to take a nonsedating antihistamine for itch. The patient was seen back in clinic 2 weeks later and the eruption had resolved.
Comment
Spread of Tick-Borne Disease—Ticks and tick-borne disease are increasing major health concerns for humans, domesticated animals, and livestock. Reported cases of bacterial and protozoan tick-borne disease doubled in the United States between 2004 and 2016. Ninety percent of the nearly 60,000 cases of nationally notifiable vector-borne diseases reported in 2017 were linked to ticks.2 Geographic ranges of multiple tick species continue to expand, which is thought to be secondary to rising global temperatures, ecologic changes, reforestation, and increases in commerce and travel (Figure 4).3 Not only have warming temperatures contributed to geographic range expansion, they also may extend ticks’ active season. The lone star tick (Amblyomma americanum) is widely distributed throughout much of the eastern United States.4 The range of A americanum has expanded north in recent years from its prior core range in the southeastern United States.2 One study found that from 2006 to 2016, the vector tick species most commonly collected from humans and submitted to a tick surveillance system in New Jersey shifted from Ixodes scapularis to A americanum.5
Bites by Amblyomma Ticks—As with most hard ticks, the life cycle of A americanum lasts 2 years and includes the egg, the 6-legged larva or “seed tick,” the 8-legged immature nymph, and the 8-legged reproductively mature adult (Figure 3). Amblyomma americanum can lay several thousand eggs.2 Because our patient had numerous bites, it is plausible that she came into contact with a nest of newly hatched tick larvae. Morphogenesis from larva to nymph, then nymph to adult, requires a blood meal.6,7 The larvae emerge from eggs deposited on the ground and then crawl up low vegetation where they can easily attach to passing hosts. The tick clings to hair or clothing and waits until the host is at rest before moving to a favorable location and then bites.8 When attaching, ticks inject an anesthetic akin to lidocaine, making the bite painless. A tick may spend up to 24 hours on the host prior to biting and then feed for 2 hours to 7 days before releasing.9 For the majority of tick-borne illnesses, the tick must remain attached for 24 to 48 hours before disease is transmitted.10
All stages of
Even when the ticks do not transmit disease, tick bites can cause impressive local reactions. Uncomplicated bites can be painful and leave a puncture wound that can take 1 to 2 weeks to heal.13 Rarely, bites can cause a delayed hypersensitivity reaction including fever, pruritus, and urticaria. Granulomas can develop if a tick is improperly removed.9 Other reports describe prurigo lesions, skin hemorrhage, papular urticaria, diffuse papules, vesicles and bullae, necrotic ulcers, and patchy alopecia.14,15 A 2015 systematic controlled study of human bite reactions from A americanum demonstrated the development of itchy erythematous papules and vesicles within 48 hours of larval tick attachment to research participants. The study found tissue damage from A americanum mouthparts, and degranulating mast cells may be evident in as little as 15 minutes.16 The severity of individual skin reaction is hypothesized to depend on several variables, such as the duration of feeding, size of mouthparts, type of tick secretions, changes in secretions during feeding, and prior exposures of the host.14
Tick Removal—If patients present to clinic with ticks attached, removal can be challenging. Removal recommendations call for use of blunt forceps or tweezers. Ticks should be grasped near the skin with consistent pressure, and the tick should be pulled straight out, perpendicular to the skin. Twisting motions can cause the head to separate from the body and remain in the bite wound. Immediately following removal, the area should be cleansed with a disinfectant.10,17 After the tick is removed, some studies recommend storing the tick at −20 °C; should the patient develop disease, the tick could be sent for evaluation.6,17 If there is no clinical or serologic evidence of infection, testing for the presence of antibodies against tick-borne bacteria at presentation and at 3 and 6 weeks is not recommended due to low sensitivity, low positive predictive value, and cost. Clinicians must only observe and treat if disease occurs.17
Prevention of Tick Bites—Tick bites are best prevented by avoiding tick-infested areas; when these areas are unavoidable, tick bites may be prevented by wearing long pants with the pant legs tucked into boots. In addition, applying topical DEET (N,N-diethyl-m-toluamide) repellent to exposed skin and treating clothing with permethrin can be helpful.17 When used alone, DEET provides greater than 90% protection for up to 2.7 hours against A americanum.18 Permethrin-treated clothing alone is 79% to 100% effective at killing A americanum ticks or disabling them for several hours.19
Conclusion
Tick-borne illness is an increasingly important cause of human infectious disease. In addition to their role as a disease vector, ticks can produce primary skin disorders. This case posed a diagnostic challenge because of the unusually large number and wide distribution of bites as well as the subsequent vesicular reaction that ensued. It is important to keep tick larvae or adult tick bites in the differential when evaluating a patient to expedite tick removal and begin clinical monitoring. Recognition of A americanum larvae as a potential cause of pruritic papules may be helpful in similar cases. In addition, it is important for dermatologists to be aware of the tick species in their area.
Case Report
A 58-year-old woman presented to the dermatology office with a widespread pruritic eruption of 3 days’ duration that started in the groin and spread to the rest of the body. No treatments had been attempted. She had no notable medical history, and she denied any recent illness, change in personal care products, or new medications or supplements. She reported a camping trip 2 weeks prior to presentation on the east end of Long Island, New York. She later learned that others on the same trip developed a similar, albeit less widespread, eruption.
Physical examination revealed clear vesicles on the arms, legs, trunk, and pubic area (Figure 1). Dermoscopy revealed a small lone star tick larva in the center of one of the vesicles (Figure 2). The type of tick larva was identified using resources from the Centers for Disease Control and Prevention (Figure 3).1 Careful inspection revealed dark marks on various vesicles, mostly in the perineum, yielding nearly 20 larvae, which were removed with forceps. The patient was counseled to cover herself in petrolatum for 2 to 3 hours with the hope of smothering any remaining tick larvae. She was given triamcinolone cream and was encouraged to take a nonsedating antihistamine for itch. The patient was seen back in clinic 2 weeks later and the eruption had resolved.
Comment
Spread of Tick-Borne Disease—Ticks and tick-borne disease are increasing major health concerns for humans, domesticated animals, and livestock. Reported cases of bacterial and protozoan tick-borne disease doubled in the United States between 2004 and 2016. Ninety percent of the nearly 60,000 cases of nationally notifiable vector-borne diseases reported in 2017 were linked to ticks.2 Geographic ranges of multiple tick species continue to expand, which is thought to be secondary to rising global temperatures, ecologic changes, reforestation, and increases in commerce and travel (Figure 4).3 Not only have warming temperatures contributed to geographic range expansion, they also may extend ticks’ active season. The lone star tick (Amblyomma americanum) is widely distributed throughout much of the eastern United States.4 The range of A americanum has expanded north in recent years from its prior core range in the southeastern United States.2 One study found that from 2006 to 2016, the vector tick species most commonly collected from humans and submitted to a tick surveillance system in New Jersey shifted from Ixodes scapularis to A americanum.5
Bites by Amblyomma Ticks—As with most hard ticks, the life cycle of A americanum lasts 2 years and includes the egg, the 6-legged larva or “seed tick,” the 8-legged immature nymph, and the 8-legged reproductively mature adult (Figure 3). Amblyomma americanum can lay several thousand eggs.2 Because our patient had numerous bites, it is plausible that she came into contact with a nest of newly hatched tick larvae. Morphogenesis from larva to nymph, then nymph to adult, requires a blood meal.6,7 The larvae emerge from eggs deposited on the ground and then crawl up low vegetation where they can easily attach to passing hosts. The tick clings to hair or clothing and waits until the host is at rest before moving to a favorable location and then bites.8 When attaching, ticks inject an anesthetic akin to lidocaine, making the bite painless. A tick may spend up to 24 hours on the host prior to biting and then feed for 2 hours to 7 days before releasing.9 For the majority of tick-borne illnesses, the tick must remain attached for 24 to 48 hours before disease is transmitted.10
All stages of
Even when the ticks do not transmit disease, tick bites can cause impressive local reactions. Uncomplicated bites can be painful and leave a puncture wound that can take 1 to 2 weeks to heal.13 Rarely, bites can cause a delayed hypersensitivity reaction including fever, pruritus, and urticaria. Granulomas can develop if a tick is improperly removed.9 Other reports describe prurigo lesions, skin hemorrhage, papular urticaria, diffuse papules, vesicles and bullae, necrotic ulcers, and patchy alopecia.14,15 A 2015 systematic controlled study of human bite reactions from A americanum demonstrated the development of itchy erythematous papules and vesicles within 48 hours of larval tick attachment to research participants. The study found tissue damage from A americanum mouthparts, and degranulating mast cells may be evident in as little as 15 minutes.16 The severity of individual skin reaction is hypothesized to depend on several variables, such as the duration of feeding, size of mouthparts, type of tick secretions, changes in secretions during feeding, and prior exposures of the host.14
Tick Removal—If patients present to clinic with ticks attached, removal can be challenging. Removal recommendations call for use of blunt forceps or tweezers. Ticks should be grasped near the skin with consistent pressure, and the tick should be pulled straight out, perpendicular to the skin. Twisting motions can cause the head to separate from the body and remain in the bite wound. Immediately following removal, the area should be cleansed with a disinfectant.10,17 After the tick is removed, some studies recommend storing the tick at −20 °C; should the patient develop disease, the tick could be sent for evaluation.6,17 If there is no clinical or serologic evidence of infection, testing for the presence of antibodies against tick-borne bacteria at presentation and at 3 and 6 weeks is not recommended due to low sensitivity, low positive predictive value, and cost. Clinicians must only observe and treat if disease occurs.17
Prevention of Tick Bites—Tick bites are best prevented by avoiding tick-infested areas; when these areas are unavoidable, tick bites may be prevented by wearing long pants with the pant legs tucked into boots. In addition, applying topical DEET (N,N-diethyl-m-toluamide) repellent to exposed skin and treating clothing with permethrin can be helpful.17 When used alone, DEET provides greater than 90% protection for up to 2.7 hours against A americanum.18 Permethrin-treated clothing alone is 79% to 100% effective at killing A americanum ticks or disabling them for several hours.19
Conclusion
Tick-borne illness is an increasingly important cause of human infectious disease. In addition to their role as a disease vector, ticks can produce primary skin disorders. This case posed a diagnostic challenge because of the unusually large number and wide distribution of bites as well as the subsequent vesicular reaction that ensued. It is important to keep tick larvae or adult tick bites in the differential when evaluating a patient to expedite tick removal and begin clinical monitoring. Recognition of A americanum larvae as a potential cause of pruritic papules may be helpful in similar cases. In addition, it is important for dermatologists to be aware of the tick species in their area.
- Centers for Disease Control and Prevention. Tick ID. Accessed February 21, 2022. https://www.cdc.gov/ticks/tickbornediseases/tickID.html
- Molaei G, Little EAH, Williams SC, et al. Bracing for the worst—range expansion of the lone star tick in the northeastern United States. N Engl J Med. 2019;381:2189-2192.
- Centers for Disease Control and Prevention, Division of Vector-Borne Diseases. Lone star tick (Amblyomma americanum). Accessed March 23, 2022. https://www.cdc.gov/ticks/maps/lone_star_tick.pdf
- Reynolds HH, Elston DM. What’s eating you? lone star tick (Amblyomma americanum). Cutis. 2017;99:111-114.
- Jordan RA, Egizi A. The growing importance of lone star ticks in a Lyme disease endemic county: passive tick surveillance in Monmouth County, NJ, 2006–2016. PLoS One. 2019;14:E0211778.
- Singh-Behl D, La Rosa SP, Tomecki KJ. Tick-borne infections. Dermatol Clin. 2003;21:237-244, v.
- Spach DH, Liles WC, Campbell GL, et al. Tick-borne diseases in the United States. N Engl J Med. 1993;329:936-947.
- Duckworth PF Jr, Hayden GF, Reed CN. Human infestation by Amblyomma americanum larvae (“seed ticks”). South Med J. 1985;78:751-753.
- Middleton DB. Tick-borne infections. what starts as a tiny bite may have a serious outcome. Postgrad Med. 1994;95:131-139.
- Moody EK, Barker RW, White JL, et al. Ticks and tick-borne diseases in Oklahoma. J Okla State Med Assoc. 1998;91:438-445.
- Jones BE. Human ‘seed tick’ infestation. Amblyomma americanum larvae. Arch Dermatol. 1981;117:812-814.
- Centers for Disease Control and Prevention. Tick bite prophylaxis. Accessed February 21, 2022. https://www.cdc.gov/ticks/tickbornediseases/tick-bite-prophylaxis.html
- Fisher EJ, Mo J, Lucky AW. Multiple pruritic papules from lone star tick larvae bites. Arch Dermatol. 2006;142:491-494.
- Krinsky WL. Dermatoses associated with the bites of mites and ticks (Arthropoda: Acari). Int J Dermatol. 1983;22:75-91.
- Yesudian P, Thambiah AS. Persistent papules after tick-bites. Dermatologica. 1973;147:214-218.
- Goddard J, Portugal JS. Cutaneous lesions due to bites by larval Amblyomma americanum ticks. JAMA Dermatol. 2015;151:1373-1375.
- Parola P, Raoult D. Ticks and tickborne bacterial diseases in humans: an emerging infectious threat. Clin Infect Dis. 2001;32:897-928.
- Solberg VB, Klein TA, McPherson KR, et al. Field evaluation of DEET and a piperidine repellent (AI3-37220) against Amblyomma americanum (Acari: Ixodidae). J Med Entomol. 1995;32:870-875.
- Evans SR, Korch GW Jr, Lawson MA. Comparative field evaluation of permethrin and DEET-treated military uniforms for personal protection against ticks (Acari). J Med Entomol. 1990;27:829-834.
- Centers for Disease Control and Prevention. Tick ID. Accessed February 21, 2022. https://www.cdc.gov/ticks/tickbornediseases/tickID.html
- Molaei G, Little EAH, Williams SC, et al. Bracing for the worst—range expansion of the lone star tick in the northeastern United States. N Engl J Med. 2019;381:2189-2192.
- Centers for Disease Control and Prevention, Division of Vector-Borne Diseases. Lone star tick (Amblyomma americanum). Accessed March 23, 2022. https://www.cdc.gov/ticks/maps/lone_star_tick.pdf
- Reynolds HH, Elston DM. What’s eating you? lone star tick (Amblyomma americanum). Cutis. 2017;99:111-114.
- Jordan RA, Egizi A. The growing importance of lone star ticks in a Lyme disease endemic county: passive tick surveillance in Monmouth County, NJ, 2006–2016. PLoS One. 2019;14:E0211778.
- Singh-Behl D, La Rosa SP, Tomecki KJ. Tick-borne infections. Dermatol Clin. 2003;21:237-244, v.
- Spach DH, Liles WC, Campbell GL, et al. Tick-borne diseases in the United States. N Engl J Med. 1993;329:936-947.
- Duckworth PF Jr, Hayden GF, Reed CN. Human infestation by Amblyomma americanum larvae (“seed ticks”). South Med J. 1985;78:751-753.
- Middleton DB. Tick-borne infections. what starts as a tiny bite may have a serious outcome. Postgrad Med. 1994;95:131-139.
- Moody EK, Barker RW, White JL, et al. Ticks and tick-borne diseases in Oklahoma. J Okla State Med Assoc. 1998;91:438-445.
- Jones BE. Human ‘seed tick’ infestation. Amblyomma americanum larvae. Arch Dermatol. 1981;117:812-814.
- Centers for Disease Control and Prevention. Tick bite prophylaxis. Accessed February 21, 2022. https://www.cdc.gov/ticks/tickbornediseases/tick-bite-prophylaxis.html
- Fisher EJ, Mo J, Lucky AW. Multiple pruritic papules from lone star tick larvae bites. Arch Dermatol. 2006;142:491-494.
- Krinsky WL. Dermatoses associated with the bites of mites and ticks (Arthropoda: Acari). Int J Dermatol. 1983;22:75-91.
- Yesudian P, Thambiah AS. Persistent papules after tick-bites. Dermatologica. 1973;147:214-218.
- Goddard J, Portugal JS. Cutaneous lesions due to bites by larval Amblyomma americanum ticks. JAMA Dermatol. 2015;151:1373-1375.
- Parola P, Raoult D. Ticks and tickborne bacterial diseases in humans: an emerging infectious threat. Clin Infect Dis. 2001;32:897-928.
- Solberg VB, Klein TA, McPherson KR, et al. Field evaluation of DEET and a piperidine repellent (AI3-37220) against Amblyomma americanum (Acari: Ixodidae). J Med Entomol. 1995;32:870-875.
- Evans SR, Korch GW Jr, Lawson MA. Comparative field evaluation of permethrin and DEET-treated military uniforms for personal protection against ticks (Acari). J Med Entomol. 1990;27:829-834.
Practice Points
- The range of Amblyomma americanum has expanded north in recent years from its core range in the southeastern United States. Warming temperatures also have increased the duration of the ticks’ active season.
- Amblyomma americanum can lay several thousand eggs. A person happening upon a newly hatched nest of larval ticks could sustain a widespread vesicular eruption secondary to tick bites.
- It is important to keep larval tick infestation in the differential when evaluating a patient with a new widespread vesicular eruption to expedite prompt removal of the offending ticks and to begin clinical monitoring.

Angioimmunoblastic T-cell Lymphoma Mimicking DRESS Syndrome
Angioimmunoblastic T-cell lymphoma (AITL) is a rare and aggressive lymphoma arising from follicular T-helper cells that predominantly affects older adults and carries a 5-year overall survival rate of 32%.1 Notably, as many as 50% of AITL patients present with a skin rash in addition to the more common but nonspecific acute-onset generalized lymphadenopathy, hepatosplenomegaly, and anemia.2 At presentation, most AITL patients are already at an advanced (III/IV) stage of disease.
Formerly known as angioimmunoblastic lymphadenopathy with dysproteinemia, AITL was once considered a benign entity that carried a risk for malignant transformation. As more cases have been identified and explored, this entity has been recategorized as a frank lymphoma.3 Therefore, it is critical that AITL be diagnosed and treated as early as possible.
We present the case of a 65-year-old man with clinical features that resembled drug reaction with eosinophilia and systemic symptoms (DRESS syndrome). After extensive workup, he was found to have AITL. This atypical case highlights the importance of maintaining a flexible differential diagnosis in patients with a persistent rash that does not improve with appropriate drug withdrawal and therapy.
Case Report
A 65-year-old Filipino man whose medical history was notable for hepatitis B that had been treated with entecavir for years without issue was admitted to the internal medicine service with fever of unknown origin and malaise of approximately 6 weeks’ duration. Six days prior to admission and 5 days after completing courses of the antiviral oseltamivir phosphate and amoxicillin for an upper respiratory tract infection and sinusitis, he developed worsening of an intermittently pruritic rash of approximately 1 month's duration. The dermatology department was consulted the day of hospital admission for evaluation of the rash. Chronic home medications included entecavir, lisinopril/hydrochlorothiazide, amlodipine, atorvastatin, metformin, salsalate, and over-the-counter nonsteroidal anti-inflammatory drugs (NSAIDs) as needed.
Physical examination was notable for mild erythema and scale distributed across the entire face; mild facial edema; and a blanchable, nonconfluent, macular erythema distributed across the trunk and upper and proximal lower extremities (Figure). In addition, the patient displayed conjunctival injection, pitting edema of the hands, and bilateral cervical and inguinal lymphadenopathy.

Laboratory tests revealed mild leukocytosis (11.6×109/L, [reference range, 4.0–10.5×109/L]), anemia (hemoglobin, 125 g/L (reference range, 138–170 g/L); hematocrit, 36.9%, [reference range, 40.0%–50.0%)], eosinophilia (1.07×109/L [reference range, 0.00–0.70×109/L)], hyponatremia, hypokalemia, and a mildly elevated creatinine level. Computed tomography and full-body positron-emission tomography (PET) scans during admission demonstrated diffuse lymphadenopathy. A skin biopsy from the left chest and a left inguinal lymph node biopsy also were performed.
Despite the lack of a clear medication trigger within the usual timeline for severe cutaneous drug-induced hypersensitivity reactions, DRESS syndrome was high on the differential diagnosis at the time of the initial presentation given the diffuse morbilliform eruption with pruritus, facial edema, eosinophilia, and lymphadenopathy.
Home medications were discontinued except for amlodipine, atorvastatin, and entecavir. The patient was treated symptomatically with topical steroids because it was believed that, if the clinical presentation represented DRESS syndrome, it was a mild variant that could be treated topically.4 His case was considered mild because of a lack of confirmed organ dysfunction and a mild protracted course.
After discharge following a 3-day inpatient stay, the patient was followed in the clinic weekly for 3 weeks without considerable change in the skin or laboratory findings. Discontinuation of entecavir was discussed and approved by his hepatologist.
Posthospitalization analysis of the punch biopsy specimen from the chest performed during the patient’s hospital stay revealed a superficial and deep dermal lymphoid infiltrate comprising CD3-, CD5-, and programmed cell death protein 1–positive cells with cytologic atypia in a perivascular distribution. Analysis of the lymph node biopsy specimen performed during the hospitalization showed effacement of the nodal architecture, a polymorphous lymphoid cell population with irregular nuclear contour, and abundant clear cytoplasm associated with high endothelial venules (HEVs). Cells of interest were positive for CD3, CD4, CD2, CD5, and CD7, with a subset staining positive for programmed cell death protein 1, inducible costimulator, CD10, and chemokine (C-X-C motif) ligand (CXCL) 13. CD21 demonstrated an expanded follicular dendritic cell meshwork in association with HEVs. Polymerase chain reaction revealed a clonal T-cell population. These findings of the skin and lymph node biopsies were consistent with AITL. Subsequent bone marrow biopsy with flow cytometry showed a normal CD4:CD8 ratio in T cells and no increase in natural killer cells.
Cyclophosphamide–hydroxydaunorubicin–Oncovin–prednisone (CHOP) chemotherapy was initiated; the patient completed a total of 6 cycles. He has had near resolution of the skin findings and is considered in remission based on a PET scan performed approximately 7 months after the initial presentation.
Comment
Angioimmunoblastic T-cell lymphoma is a rare peripheral T-cell lymphoma, part of a group of aggressive neoplasms that constitute approximately 15% of peripheral T-cell lymphomas and approximately 2% of non-Hodgkin lymphomas in adults worldwide.5 Cutaneous involvement occurs in approximately half of AITL cases and can be the first manifestation of disease.2 Skin findings are largely nonspecific, ranging from simple morbilliform rashes to erythroderma, at times manifesting with purpura.
Given this variability in the presentation of AITL, early diagnosis is challenging in the absence of more specific signs and symptoms.2 It can conceivably be mistaken for common entities such as viral exanthems or drug eruptions, depending on the history and context. DRESS syndrome, a T cell-mediated, delayed type-IV hypersensitivity drug reaction can present in a manner highly similar to that of AITL, with cutaneous involvement (diffuse morbilliform rash, fever, facial edema, and generalized lymphadenopathy) and variable systemic involvement. Laboratory findings of eosinophilia, atypical lymphocytes, and thrombocytopenia also might be seen in both entities.6 Furthermore, the AITL in our patient was accompanied by electrolyte disturbances that were concerning for syndrome of inappropriate antidiuretic hormone secretion, a rare complication of patients with DRESS syndrome complicated by encephalitis.7,8
Our patient met 4 RegiSCAR criteria for DRESS syndrome, warranting high clinical suspicion for an offending drug.9 DRESS syndrome can be caused by numerous medications—most commonly anticonvulsants, sulfonamides, antibiotics, allopurinol, and NSAIDs. A review of our patient’s medication list identified NSAIDs (including salsalate), entecavir, and amoxicillin, as possible culpable medications. Notably, the only new addition to the patient’s regimen was amoxicillin, which did not fit the typical 2- to 8-week timeline for a DRESS syndrome nidus.10 Our patient’s fever began well before the antibiotic was initiated, and skin findings appeared within 1 week after the course of amoxicillin was completed. Although there is documented variability in the latency of onset of DRESS syndrome following administration of a culprit medication,11 it is critical to maintain a broad differential diagnosis to allow for further diagnostic information to be obtained, especially when the medication timeline does not align with the clinical presentation.
DRESS syndrome is far more common than AITL. Similarities in their clinical presentation pose a substantial challenge and often cause a delay in the diagnosis of AITL, which is made by excisional tissue biopsy, most commonly of a lymph node, with assessment of morphology and immunophenotyping. Histologic assessment of tissue reveals a polymorphous infiltrate of variably sized atypical lymphocytes with prominent arborizing HEVs as well as expanded populations of follicular dendritic cells that can be detected by CD21 staining. Cells express CD3 and CD4, variably express BCL6 (B-cell lymphoma 6 antigen) and CD10, and also may have partial or complete loss of expression of a subset of pan T-cell antigens (CD2, CD3, CD5, and CD7).12-18
The treatment approach to AITL mirrors that of other nodal peripheral T-cell lymphomas, including chemotherapy and consideration of autologous stem-cell transplantation. Recent prospective trials of CHOP and CHOP-like chemotherapy have reported 3-year event-free survival and overall survival rates of 50% and 68%, respectively.19 Novel chemotherapeutic targets and gene-expression profiling are being investigated as potential therapeutic avenues.20
Conclusion
DRESS syndrome and AITL can have near-identical presentations. Clinicians should maintain a high index of suspicion for AITL in patients with presumed DRESS syndrome whose rash does not improve with appropriate drug withdrawal and steroid therapy or who lack a strong offending medication history. In such cases, skin and lymph node biopsies should be performed as early as possible to evaluate for AITL and so that appropriate therapy can be initiated.
- Federico M, Rudiger T, Bellei M, et al. Clinicopathologic characteristics of angioimmunoblastic T-cell lymphoma: analysis of the international peripheral T-cell lymphoma project. J Clin Oncol. 2013;31:240-246. doi:10.1200/JCO.2011.37.3647
- Botros N, Cerroni L, Shawwa A, et al. Cutaneous manifestations of angioimmunoblastic T-cell lymphoma: clinical and pathological characteristics. Am J Dermatopathol. 2015;37:274-283. doi:10.1097/DAD.0000000000000144
- Sachsida-Colombo E, Barbosa Mariano LC, Bastos FQ, et al. A difficult case of angioimmunoblastic T-cell lymphoma to diagnose. Rev Bras Hematol Hemoter. 2016;38:82-85. doi:10.1016/j.bjhh.2015.11.002
- Funck-Brentano E, Duong T-A, Bouvresse S, et al. Therapeutic management of DRESS: a retrospective study of 38 cases. J Am Acad Dermatol. 2015;72:246-252. doi:10.1016/j.jaad.2014.10.032
- Lunning MA, Vose JM. Angioimmunoblastic T-cell lymphoma: the many-faced lymphoma. Blood. 2017;129:1095-1102. doi:10.1182/blood-2016-09-692541
- Sato R, Itoh M, Suzuki H, et al. Pathological findings of lymphadenopathy in drug-induced hypersensitivity syndrome (DIHS)/drug reaction with eosinophilia and systemic syndrome (DRESS): similarities with angioimmunoblastic T-cell lymphoma. Eur J Dermatol. 2017;27:201-202. doi:10.1684/ejd.2016.2954
- Osizik L, Tanriover MD, Saka E. Autoimmune limbic encephalitis and syndrome of inappropriate antidiuretic hormone secretion associated with lamotrigine-induced drug rash with eosinophilia and systemic symptoms (DRESS) syndrome. Intern Med. 2015;55:1393-1396. doi:10.2169/internalmedicine.55.6035
- Sakuma K, Kano Y, Fukuhara M, et al. Syndrome of inappropriate secretion of antidiuretic hormone associated with limbic encephalitis in a patient with drug-induced hypersensitivity syndrome. Clin Exp Dermatol. 2008;33:287-290. doi:10.1111/j.1365-2230.2007.02645.x
- Pannu AK, Saroch A. Diagnostic criteria for drug rash and eosinophilia with systemic symptoms. J Family Med Prim Care. 2017;6:693-694. doi:10.4103/2249-4863.222050
- Kardaun SH, Sekula P, Valeyrie-Allanore L, et al; RegiSCAR study group. Drug reaction with eosinophilia and systemic symptoms (DRESS): an original multisystem adverse drug reaction. results from the prospective RegiSCAR study. Br J Dermatol. 2013;169:1071-1080. doi:10.1111/bjd.12501
- Soria A, Bernier C, Veyrac G, et al. Drug reaction with eosinophilia and systemic symptoms may occur within 2 weeks of drug exposure: a retrospective study. J Am Acad Dermatol. 2020;82:606.
- Loghavi S, Wang SA, Medeiros LJ, et al. Immunophenotypic and diagnostic characterization of angioimmunoblastic T-cell lymphoma by advanced flow cytometric technology. Leuk Lymphoma. 2016;57:2804-2812. doi:10.3109/10428194.2016.1170827
- Lee S-S, R, Odenwald T, et al. Angioimmunoblastic T cell lymphoma is derived from mature T-helper cells with varying expression and loss of detectable CD4. Int J Cancer. 2003;103:12-20. doi:10.1002/ijc.10758
- Feller AC, Griesser H, Schilling CV, et al. Clonal gene rearrangement patterns correlate with immunophenotype and clinical parameters in patients with angioimmunoblastic lymphadenopathy. Am J Pathol. 1988;133:549-556.
- Swerdlow SH, Campo E, Harris NL, et al, eds. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues. IARC Press; 2008.
- Attygalle A, Al-Jehani R, Diss TC, et al. Neoplastic T cells in angioimmunoblastic T-cell lymphoma express CD10. Blood. 2002;99:627-633. doi:10.1182/blood.v99.2.627
- Mourad N, Mounier N, J, et al; Groupe d’Etude des Lymphomes de l’Adulte. Clinical, biologic, and pathologic features in 157 patients with angioimmunoblastic T-cell lymphoma treated within the Groupe d’Etude des Lymphomes de l’Adulte (GELA) trials. Blood. 2008;111:4463-4470. doi:10.1182/blood-2007-08-105759
- Marafioti T, Paterson JC, Ballabio E, et al. The inducible T-cell co-stimulator molecule is expressed on subsets of T cells and is a new marker of lymphomas of T follicular helper cell-derivation. Haematologica. 2010;95:432-439. doi:10.3324/haematol.2009.010991
- Schmitz N, L, Ziepert M, et al. Treatment and prognosis of mature T-cell and NK-cell lymphoma: an analysis of patients withT-cell lymphoma treated in studies of the German High-Grade Non-Hodgkin Lymphoma Study Group. Blood. 2010;116:3418-3425. doi:10.1182/blood-2010-02-270785
- Moskowitz AJ. Practical treatment approach for angioimmunoblastic T-cell lymphoma. J Oncol Pract. 2019;15:137-143. doi:10.1200/JOP.18.00511
Angioimmunoblastic T-cell lymphoma (AITL) is a rare and aggressive lymphoma arising from follicular T-helper cells that predominantly affects older adults and carries a 5-year overall survival rate of 32%.1 Notably, as many as 50% of AITL patients present with a skin rash in addition to the more common but nonspecific acute-onset generalized lymphadenopathy, hepatosplenomegaly, and anemia.2 At presentation, most AITL patients are already at an advanced (III/IV) stage of disease.
Formerly known as angioimmunoblastic lymphadenopathy with dysproteinemia, AITL was once considered a benign entity that carried a risk for malignant transformation. As more cases have been identified and explored, this entity has been recategorized as a frank lymphoma.3 Therefore, it is critical that AITL be diagnosed and treated as early as possible.
We present the case of a 65-year-old man with clinical features that resembled drug reaction with eosinophilia and systemic symptoms (DRESS syndrome). After extensive workup, he was found to have AITL. This atypical case highlights the importance of maintaining a flexible differential diagnosis in patients with a persistent rash that does not improve with appropriate drug withdrawal and therapy.
Case Report
A 65-year-old Filipino man whose medical history was notable for hepatitis B that had been treated with entecavir for years without issue was admitted to the internal medicine service with fever of unknown origin and malaise of approximately 6 weeks’ duration. Six days prior to admission and 5 days after completing courses of the antiviral oseltamivir phosphate and amoxicillin for an upper respiratory tract infection and sinusitis, he developed worsening of an intermittently pruritic rash of approximately 1 month's duration. The dermatology department was consulted the day of hospital admission for evaluation of the rash. Chronic home medications included entecavir, lisinopril/hydrochlorothiazide, amlodipine, atorvastatin, metformin, salsalate, and over-the-counter nonsteroidal anti-inflammatory drugs (NSAIDs) as needed.
Physical examination was notable for mild erythema and scale distributed across the entire face; mild facial edema; and a blanchable, nonconfluent, macular erythema distributed across the trunk and upper and proximal lower extremities (Figure). In addition, the patient displayed conjunctival injection, pitting edema of the hands, and bilateral cervical and inguinal lymphadenopathy.
Laboratory tests revealed mild leukocytosis (11.6×109/L, [reference range, 4.0–10.5×109/L]), anemia (hemoglobin, 125 g/L (reference range, 138–170 g/L); hematocrit, 36.9%, [reference range, 40.0%–50.0%)], eosinophilia (1.07×109/L [reference range, 0.00–0.70×109/L)], hyponatremia, hypokalemia, and a mildly elevated creatinine level. Computed tomography and full-body positron-emission tomography (PET) scans during admission demonstrated diffuse lymphadenopathy. A skin biopsy from the left chest and a left inguinal lymph node biopsy also were performed.
Despite the lack of a clear medication trigger within the usual timeline for severe cutaneous drug-induced hypersensitivity reactions, DRESS syndrome was high on the differential diagnosis at the time of the initial presentation given the diffuse morbilliform eruption with pruritus, facial edema, eosinophilia, and lymphadenopathy.
Home medications were discontinued except for amlodipine, atorvastatin, and entecavir. The patient was treated symptomatically with topical steroids because it was believed that, if the clinical presentation represented DRESS syndrome, it was a mild variant that could be treated topically.4 His case was considered mild because of a lack of confirmed organ dysfunction and a mild protracted course.
After discharge following a 3-day inpatient stay, the patient was followed in the clinic weekly for 3 weeks without considerable change in the skin or laboratory findings. Discontinuation of entecavir was discussed and approved by his hepatologist.
Posthospitalization analysis of the punch biopsy specimen from the chest performed during the patient’s hospital stay revealed a superficial and deep dermal lymphoid infiltrate comprising CD3-, CD5-, and programmed cell death protein 1–positive cells with cytologic atypia in a perivascular distribution. Analysis of the lymph node biopsy specimen performed during the hospitalization showed effacement of the nodal architecture, a polymorphous lymphoid cell population with irregular nuclear contour, and abundant clear cytoplasm associated with high endothelial venules (HEVs). Cells of interest were positive for CD3, CD4, CD2, CD5, and CD7, with a subset staining positive for programmed cell death protein 1, inducible costimulator, CD10, and chemokine (C-X-C motif) ligand (CXCL) 13. CD21 demonstrated an expanded follicular dendritic cell meshwork in association with HEVs. Polymerase chain reaction revealed a clonal T-cell population. These findings of the skin and lymph node biopsies were consistent with AITL. Subsequent bone marrow biopsy with flow cytometry showed a normal CD4:CD8 ratio in T cells and no increase in natural killer cells.
Cyclophosphamide–hydroxydaunorubicin–Oncovin–prednisone (CHOP) chemotherapy was initiated; the patient completed a total of 6 cycles. He has had near resolution of the skin findings and is considered in remission based on a PET scan performed approximately 7 months after the initial presentation.
Comment
Angioimmunoblastic T-cell lymphoma is a rare peripheral T-cell lymphoma, part of a group of aggressive neoplasms that constitute approximately 15% of peripheral T-cell lymphomas and approximately 2% of non-Hodgkin lymphomas in adults worldwide.5 Cutaneous involvement occurs in approximately half of AITL cases and can be the first manifestation of disease.2 Skin findings are largely nonspecific, ranging from simple morbilliform rashes to erythroderma, at times manifesting with purpura.
Given this variability in the presentation of AITL, early diagnosis is challenging in the absence of more specific signs and symptoms.2 It can conceivably be mistaken for common entities such as viral exanthems or drug eruptions, depending on the history and context. DRESS syndrome, a T cell-mediated, delayed type-IV hypersensitivity drug reaction can present in a manner highly similar to that of AITL, with cutaneous involvement (diffuse morbilliform rash, fever, facial edema, and generalized lymphadenopathy) and variable systemic involvement. Laboratory findings of eosinophilia, atypical lymphocytes, and thrombocytopenia also might be seen in both entities.6 Furthermore, the AITL in our patient was accompanied by electrolyte disturbances that were concerning for syndrome of inappropriate antidiuretic hormone secretion, a rare complication of patients with DRESS syndrome complicated by encephalitis.7,8
Our patient met 4 RegiSCAR criteria for DRESS syndrome, warranting high clinical suspicion for an offending drug.9 DRESS syndrome can be caused by numerous medications—most commonly anticonvulsants, sulfonamides, antibiotics, allopurinol, and NSAIDs. A review of our patient’s medication list identified NSAIDs (including salsalate), entecavir, and amoxicillin, as possible culpable medications. Notably, the only new addition to the patient’s regimen was amoxicillin, which did not fit the typical 2- to 8-week timeline for a DRESS syndrome nidus.10 Our patient’s fever began well before the antibiotic was initiated, and skin findings appeared within 1 week after the course of amoxicillin was completed. Although there is documented variability in the latency of onset of DRESS syndrome following administration of a culprit medication,11 it is critical to maintain a broad differential diagnosis to allow for further diagnostic information to be obtained, especially when the medication timeline does not align with the clinical presentation.
DRESS syndrome is far more common than AITL. Similarities in their clinical presentation pose a substantial challenge and often cause a delay in the diagnosis of AITL, which is made by excisional tissue biopsy, most commonly of a lymph node, with assessment of morphology and immunophenotyping. Histologic assessment of tissue reveals a polymorphous infiltrate of variably sized atypical lymphocytes with prominent arborizing HEVs as well as expanded populations of follicular dendritic cells that can be detected by CD21 staining. Cells express CD3 and CD4, variably express BCL6 (B-cell lymphoma 6 antigen) and CD10, and also may have partial or complete loss of expression of a subset of pan T-cell antigens (CD2, CD3, CD5, and CD7).12-18
The treatment approach to AITL mirrors that of other nodal peripheral T-cell lymphomas, including chemotherapy and consideration of autologous stem-cell transplantation. Recent prospective trials of CHOP and CHOP-like chemotherapy have reported 3-year event-free survival and overall survival rates of 50% and 68%, respectively.19 Novel chemotherapeutic targets and gene-expression profiling are being investigated as potential therapeutic avenues.20
Conclusion
DRESS syndrome and AITL can have near-identical presentations. Clinicians should maintain a high index of suspicion for AITL in patients with presumed DRESS syndrome whose rash does not improve with appropriate drug withdrawal and steroid therapy or who lack a strong offending medication history. In such cases, skin and lymph node biopsies should be performed as early as possible to evaluate for AITL and so that appropriate therapy can be initiated.
Angioimmunoblastic T-cell lymphoma (AITL) is a rare and aggressive lymphoma arising from follicular T-helper cells that predominantly affects older adults and carries a 5-year overall survival rate of 32%.1 Notably, as many as 50% of AITL patients present with a skin rash in addition to the more common but nonspecific acute-onset generalized lymphadenopathy, hepatosplenomegaly, and anemia.2 At presentation, most AITL patients are already at an advanced (III/IV) stage of disease.
Formerly known as angioimmunoblastic lymphadenopathy with dysproteinemia, AITL was once considered a benign entity that carried a risk for malignant transformation. As more cases have been identified and explored, this entity has been recategorized as a frank lymphoma.3 Therefore, it is critical that AITL be diagnosed and treated as early as possible.
We present the case of a 65-year-old man with clinical features that resembled drug reaction with eosinophilia and systemic symptoms (DRESS syndrome). After extensive workup, he was found to have AITL. This atypical case highlights the importance of maintaining a flexible differential diagnosis in patients with a persistent rash that does not improve with appropriate drug withdrawal and therapy.
Case Report
A 65-year-old Filipino man whose medical history was notable for hepatitis B that had been treated with entecavir for years without issue was admitted to the internal medicine service with fever of unknown origin and malaise of approximately 6 weeks’ duration. Six days prior to admission and 5 days after completing courses of the antiviral oseltamivir phosphate and amoxicillin for an upper respiratory tract infection and sinusitis, he developed worsening of an intermittently pruritic rash of approximately 1 month's duration. The dermatology department was consulted the day of hospital admission for evaluation of the rash. Chronic home medications included entecavir, lisinopril/hydrochlorothiazide, amlodipine, atorvastatin, metformin, salsalate, and over-the-counter nonsteroidal anti-inflammatory drugs (NSAIDs) as needed.
Physical examination was notable for mild erythema and scale distributed across the entire face; mild facial edema; and a blanchable, nonconfluent, macular erythema distributed across the trunk and upper and proximal lower extremities (Figure). In addition, the patient displayed conjunctival injection, pitting edema of the hands, and bilateral cervical and inguinal lymphadenopathy.
Laboratory tests revealed mild leukocytosis (11.6×109/L, [reference range, 4.0–10.5×109/L]), anemia (hemoglobin, 125 g/L (reference range, 138–170 g/L); hematocrit, 36.9%, [reference range, 40.0%–50.0%)], eosinophilia (1.07×109/L [reference range, 0.00–0.70×109/L)], hyponatremia, hypokalemia, and a mildly elevated creatinine level. Computed tomography and full-body positron-emission tomography (PET) scans during admission demonstrated diffuse lymphadenopathy. A skin biopsy from the left chest and a left inguinal lymph node biopsy also were performed.
Despite the lack of a clear medication trigger within the usual timeline for severe cutaneous drug-induced hypersensitivity reactions, DRESS syndrome was high on the differential diagnosis at the time of the initial presentation given the diffuse morbilliform eruption with pruritus, facial edema, eosinophilia, and lymphadenopathy.
Home medications were discontinued except for amlodipine, atorvastatin, and entecavir. The patient was treated symptomatically with topical steroids because it was believed that, if the clinical presentation represented DRESS syndrome, it was a mild variant that could be treated topically.4 His case was considered mild because of a lack of confirmed organ dysfunction and a mild protracted course.
After discharge following a 3-day inpatient stay, the patient was followed in the clinic weekly for 3 weeks without considerable change in the skin or laboratory findings. Discontinuation of entecavir was discussed and approved by his hepatologist.
Posthospitalization analysis of the punch biopsy specimen from the chest performed during the patient’s hospital stay revealed a superficial and deep dermal lymphoid infiltrate comprising CD3-, CD5-, and programmed cell death protein 1–positive cells with cytologic atypia in a perivascular distribution. Analysis of the lymph node biopsy specimen performed during the hospitalization showed effacement of the nodal architecture, a polymorphous lymphoid cell population with irregular nuclear contour, and abundant clear cytoplasm associated with high endothelial venules (HEVs). Cells of interest were positive for CD3, CD4, CD2, CD5, and CD7, with a subset staining positive for programmed cell death protein 1, inducible costimulator, CD10, and chemokine (C-X-C motif) ligand (CXCL) 13. CD21 demonstrated an expanded follicular dendritic cell meshwork in association with HEVs. Polymerase chain reaction revealed a clonal T-cell population. These findings of the skin and lymph node biopsies were consistent with AITL. Subsequent bone marrow biopsy with flow cytometry showed a normal CD4:CD8 ratio in T cells and no increase in natural killer cells.
Cyclophosphamide–hydroxydaunorubicin–Oncovin–prednisone (CHOP) chemotherapy was initiated; the patient completed a total of 6 cycles. He has had near resolution of the skin findings and is considered in remission based on a PET scan performed approximately 7 months after the initial presentation.
Comment
Angioimmunoblastic T-cell lymphoma is a rare peripheral T-cell lymphoma, part of a group of aggressive neoplasms that constitute approximately 15% of peripheral T-cell lymphomas and approximately 2% of non-Hodgkin lymphomas in adults worldwide.5 Cutaneous involvement occurs in approximately half of AITL cases and can be the first manifestation of disease.2 Skin findings are largely nonspecific, ranging from simple morbilliform rashes to erythroderma, at times manifesting with purpura.
Given this variability in the presentation of AITL, early diagnosis is challenging in the absence of more specific signs and symptoms.2 It can conceivably be mistaken for common entities such as viral exanthems or drug eruptions, depending on the history and context. DRESS syndrome, a T cell-mediated, delayed type-IV hypersensitivity drug reaction can present in a manner highly similar to that of AITL, with cutaneous involvement (diffuse morbilliform rash, fever, facial edema, and generalized lymphadenopathy) and variable systemic involvement. Laboratory findings of eosinophilia, atypical lymphocytes, and thrombocytopenia also might be seen in both entities.6 Furthermore, the AITL in our patient was accompanied by electrolyte disturbances that were concerning for syndrome of inappropriate antidiuretic hormone secretion, a rare complication of patients with DRESS syndrome complicated by encephalitis.7,8
Our patient met 4 RegiSCAR criteria for DRESS syndrome, warranting high clinical suspicion for an offending drug.9 DRESS syndrome can be caused by numerous medications—most commonly anticonvulsants, sulfonamides, antibiotics, allopurinol, and NSAIDs. A review of our patient’s medication list identified NSAIDs (including salsalate), entecavir, and amoxicillin, as possible culpable medications. Notably, the only new addition to the patient’s regimen was amoxicillin, which did not fit the typical 2- to 8-week timeline for a DRESS syndrome nidus.10 Our patient’s fever began well before the antibiotic was initiated, and skin findings appeared within 1 week after the course of amoxicillin was completed. Although there is documented variability in the latency of onset of DRESS syndrome following administration of a culprit medication,11 it is critical to maintain a broad differential diagnosis to allow for further diagnostic information to be obtained, especially when the medication timeline does not align with the clinical presentation.
DRESS syndrome is far more common than AITL. Similarities in their clinical presentation pose a substantial challenge and often cause a delay in the diagnosis of AITL, which is made by excisional tissue biopsy, most commonly of a lymph node, with assessment of morphology and immunophenotyping. Histologic assessment of tissue reveals a polymorphous infiltrate of variably sized atypical lymphocytes with prominent arborizing HEVs as well as expanded populations of follicular dendritic cells that can be detected by CD21 staining. Cells express CD3 and CD4, variably express BCL6 (B-cell lymphoma 6 antigen) and CD10, and also may have partial or complete loss of expression of a subset of pan T-cell antigens (CD2, CD3, CD5, and CD7).12-18
The treatment approach to AITL mirrors that of other nodal peripheral T-cell lymphomas, including chemotherapy and consideration of autologous stem-cell transplantation. Recent prospective trials of CHOP and CHOP-like chemotherapy have reported 3-year event-free survival and overall survival rates of 50% and 68%, respectively.19 Novel chemotherapeutic targets and gene-expression profiling are being investigated as potential therapeutic avenues.20
Conclusion
DRESS syndrome and AITL can have near-identical presentations. Clinicians should maintain a high index of suspicion for AITL in patients with presumed DRESS syndrome whose rash does not improve with appropriate drug withdrawal and steroid therapy or who lack a strong offending medication history. In such cases, skin and lymph node biopsies should be performed as early as possible to evaluate for AITL and so that appropriate therapy can be initiated.
- Federico M, Rudiger T, Bellei M, et al. Clinicopathologic characteristics of angioimmunoblastic T-cell lymphoma: analysis of the international peripheral T-cell lymphoma project. J Clin Oncol. 2013;31:240-246. doi:10.1200/JCO.2011.37.3647
- Botros N, Cerroni L, Shawwa A, et al. Cutaneous manifestations of angioimmunoblastic T-cell lymphoma: clinical and pathological characteristics. Am J Dermatopathol. 2015;37:274-283. doi:10.1097/DAD.0000000000000144
- Sachsida-Colombo E, Barbosa Mariano LC, Bastos FQ, et al. A difficult case of angioimmunoblastic T-cell lymphoma to diagnose. Rev Bras Hematol Hemoter. 2016;38:82-85. doi:10.1016/j.bjhh.2015.11.002
- Funck-Brentano E, Duong T-A, Bouvresse S, et al. Therapeutic management of DRESS: a retrospective study of 38 cases. J Am Acad Dermatol. 2015;72:246-252. doi:10.1016/j.jaad.2014.10.032
- Lunning MA, Vose JM. Angioimmunoblastic T-cell lymphoma: the many-faced lymphoma. Blood. 2017;129:1095-1102. doi:10.1182/blood-2016-09-692541
- Sato R, Itoh M, Suzuki H, et al. Pathological findings of lymphadenopathy in drug-induced hypersensitivity syndrome (DIHS)/drug reaction with eosinophilia and systemic syndrome (DRESS): similarities with angioimmunoblastic T-cell lymphoma. Eur J Dermatol. 2017;27:201-202. doi:10.1684/ejd.2016.2954
- Osizik L, Tanriover MD, Saka E. Autoimmune limbic encephalitis and syndrome of inappropriate antidiuretic hormone secretion associated with lamotrigine-induced drug rash with eosinophilia and systemic symptoms (DRESS) syndrome. Intern Med. 2015;55:1393-1396. doi:10.2169/internalmedicine.55.6035
- Sakuma K, Kano Y, Fukuhara M, et al. Syndrome of inappropriate secretion of antidiuretic hormone associated with limbic encephalitis in a patient with drug-induced hypersensitivity syndrome. Clin Exp Dermatol. 2008;33:287-290. doi:10.1111/j.1365-2230.2007.02645.x
- Pannu AK, Saroch A. Diagnostic criteria for drug rash and eosinophilia with systemic symptoms. J Family Med Prim Care. 2017;6:693-694. doi:10.4103/2249-4863.222050
- Kardaun SH, Sekula P, Valeyrie-Allanore L, et al; RegiSCAR study group. Drug reaction with eosinophilia and systemic symptoms (DRESS): an original multisystem adverse drug reaction. results from the prospective RegiSCAR study. Br J Dermatol. 2013;169:1071-1080. doi:10.1111/bjd.12501
- Soria A, Bernier C, Veyrac G, et al. Drug reaction with eosinophilia and systemic symptoms may occur within 2 weeks of drug exposure: a retrospective study. J Am Acad Dermatol. 2020;82:606.
- Loghavi S, Wang SA, Medeiros LJ, et al. Immunophenotypic and diagnostic characterization of angioimmunoblastic T-cell lymphoma by advanced flow cytometric technology. Leuk Lymphoma. 2016;57:2804-2812. doi:10.3109/10428194.2016.1170827
- Lee S-S, R, Odenwald T, et al. Angioimmunoblastic T cell lymphoma is derived from mature T-helper cells with varying expression and loss of detectable CD4. Int J Cancer. 2003;103:12-20. doi:10.1002/ijc.10758
- Feller AC, Griesser H, Schilling CV, et al. Clonal gene rearrangement patterns correlate with immunophenotype and clinical parameters in patients with angioimmunoblastic lymphadenopathy. Am J Pathol. 1988;133:549-556.
- Swerdlow SH, Campo E, Harris NL, et al, eds. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues. IARC Press; 2008.
- Attygalle A, Al-Jehani R, Diss TC, et al. Neoplastic T cells in angioimmunoblastic T-cell lymphoma express CD10. Blood. 2002;99:627-633. doi:10.1182/blood.v99.2.627
- Mourad N, Mounier N, J, et al; Groupe d’Etude des Lymphomes de l’Adulte. Clinical, biologic, and pathologic features in 157 patients with angioimmunoblastic T-cell lymphoma treated within the Groupe d’Etude des Lymphomes de l’Adulte (GELA) trials. Blood. 2008;111:4463-4470. doi:10.1182/blood-2007-08-105759
- Marafioti T, Paterson JC, Ballabio E, et al. The inducible T-cell co-stimulator molecule is expressed on subsets of T cells and is a new marker of lymphomas of T follicular helper cell-derivation. Haematologica. 2010;95:432-439. doi:10.3324/haematol.2009.010991
- Schmitz N, L, Ziepert M, et al. Treatment and prognosis of mature T-cell and NK-cell lymphoma: an analysis of patients withT-cell lymphoma treated in studies of the German High-Grade Non-Hodgkin Lymphoma Study Group. Blood. 2010;116:3418-3425. doi:10.1182/blood-2010-02-270785
- Moskowitz AJ. Practical treatment approach for angioimmunoblastic T-cell lymphoma. J Oncol Pract. 2019;15:137-143. doi:10.1200/JOP.18.00511
- Federico M, Rudiger T, Bellei M, et al. Clinicopathologic characteristics of angioimmunoblastic T-cell lymphoma: analysis of the international peripheral T-cell lymphoma project. J Clin Oncol. 2013;31:240-246. doi:10.1200/JCO.2011.37.3647
- Botros N, Cerroni L, Shawwa A, et al. Cutaneous manifestations of angioimmunoblastic T-cell lymphoma: clinical and pathological characteristics. Am J Dermatopathol. 2015;37:274-283. doi:10.1097/DAD.0000000000000144
- Sachsida-Colombo E, Barbosa Mariano LC, Bastos FQ, et al. A difficult case of angioimmunoblastic T-cell lymphoma to diagnose. Rev Bras Hematol Hemoter. 2016;38:82-85. doi:10.1016/j.bjhh.2015.11.002
- Funck-Brentano E, Duong T-A, Bouvresse S, et al. Therapeutic management of DRESS: a retrospective study of 38 cases. J Am Acad Dermatol. 2015;72:246-252. doi:10.1016/j.jaad.2014.10.032
- Lunning MA, Vose JM. Angioimmunoblastic T-cell lymphoma: the many-faced lymphoma. Blood. 2017;129:1095-1102. doi:10.1182/blood-2016-09-692541
- Sato R, Itoh M, Suzuki H, et al. Pathological findings of lymphadenopathy in drug-induced hypersensitivity syndrome (DIHS)/drug reaction with eosinophilia and systemic syndrome (DRESS): similarities with angioimmunoblastic T-cell lymphoma. Eur J Dermatol. 2017;27:201-202. doi:10.1684/ejd.2016.2954
- Osizik L, Tanriover MD, Saka E. Autoimmune limbic encephalitis and syndrome of inappropriate antidiuretic hormone secretion associated with lamotrigine-induced drug rash with eosinophilia and systemic symptoms (DRESS) syndrome. Intern Med. 2015;55:1393-1396. doi:10.2169/internalmedicine.55.6035
- Sakuma K, Kano Y, Fukuhara M, et al. Syndrome of inappropriate secretion of antidiuretic hormone associated with limbic encephalitis in a patient with drug-induced hypersensitivity syndrome. Clin Exp Dermatol. 2008;33:287-290. doi:10.1111/j.1365-2230.2007.02645.x
- Pannu AK, Saroch A. Diagnostic criteria for drug rash and eosinophilia with systemic symptoms. J Family Med Prim Care. 2017;6:693-694. doi:10.4103/2249-4863.222050
- Kardaun SH, Sekula P, Valeyrie-Allanore L, et al; RegiSCAR study group. Drug reaction with eosinophilia and systemic symptoms (DRESS): an original multisystem adverse drug reaction. results from the prospective RegiSCAR study. Br J Dermatol. 2013;169:1071-1080. doi:10.1111/bjd.12501
- Soria A, Bernier C, Veyrac G, et al. Drug reaction with eosinophilia and systemic symptoms may occur within 2 weeks of drug exposure: a retrospective study. J Am Acad Dermatol. 2020;82:606.
- Loghavi S, Wang SA, Medeiros LJ, et al. Immunophenotypic and diagnostic characterization of angioimmunoblastic T-cell lymphoma by advanced flow cytometric technology. Leuk Lymphoma. 2016;57:2804-2812. doi:10.3109/10428194.2016.1170827
- Lee S-S, R, Odenwald T, et al. Angioimmunoblastic T cell lymphoma is derived from mature T-helper cells with varying expression and loss of detectable CD4. Int J Cancer. 2003;103:12-20. doi:10.1002/ijc.10758
- Feller AC, Griesser H, Schilling CV, et al. Clonal gene rearrangement patterns correlate with immunophenotype and clinical parameters in patients with angioimmunoblastic lymphadenopathy. Am J Pathol. 1988;133:549-556.
- Swerdlow SH, Campo E, Harris NL, et al, eds. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues. IARC Press; 2008.
- Attygalle A, Al-Jehani R, Diss TC, et al. Neoplastic T cells in angioimmunoblastic T-cell lymphoma express CD10. Blood. 2002;99:627-633. doi:10.1182/blood.v99.2.627
- Mourad N, Mounier N, J, et al; Groupe d’Etude des Lymphomes de l’Adulte. Clinical, biologic, and pathologic features in 157 patients with angioimmunoblastic T-cell lymphoma treated within the Groupe d’Etude des Lymphomes de l’Adulte (GELA) trials. Blood. 2008;111:4463-4470. doi:10.1182/blood-2007-08-105759
- Marafioti T, Paterson JC, Ballabio E, et al. The inducible T-cell co-stimulator molecule is expressed on subsets of T cells and is a new marker of lymphomas of T follicular helper cell-derivation. Haematologica. 2010;95:432-439. doi:10.3324/haematol.2009.010991
- Schmitz N, L, Ziepert M, et al. Treatment and prognosis of mature T-cell and NK-cell lymphoma: an analysis of patients withT-cell lymphoma treated in studies of the German High-Grade Non-Hodgkin Lymphoma Study Group. Blood. 2010;116:3418-3425. doi:10.1182/blood-2010-02-270785
- Moskowitz AJ. Practical treatment approach for angioimmunoblastic T-cell lymphoma. J Oncol Pract. 2019;15:137-143. doi:10.1200/JOP.18.00511
Practice Points
- It is important to maintain a high index of suspicion for angioimmunoblastic T-cell lymphoma in older patients with a longstanding rash and no clear culprit for drug reaction with eosinophilia and systemic symptoms (DRESS syndrome).
- Consider performing a lymph node biopsy early in the course of disease in patients with presumed DRESS syndrome who do not improve with drug withdrawal and steroid therapy.

Clinical Presentation of Subacute Combined Degeneration in a Patient With Chronic B12 Deficiency
Subacute combined degeneration (SCD) is an acquired neurologic complication of vitamin B12 (cobalamin) or, rarely, vitamin B9 (folate) deficiency. SCD is characterized by progressive demyelination of the dorsal and lateral spinal cord, resulting in peripheral neuropathy; gait ataxia; impaired proprioception, vibration, and fine touch; optic neuropathy; and cognitive impairment.1 In addition to SCD, other neurologic manifestations of B12 deficiency include dementia, depression, visual symptoms due to optic atrophy, and behavioral changes.2 The prevalence of SCD in the US has not been well documented, but B12 deficiency is reported at 6% in those aged < 60 years and 20% in those > 60 years.3
Causes of B12 and B9 deficiency include advanced age, low nutritional intake (eg, vegan diet), impaired absorption (eg, inflammatory bowel disease, autoimmune pernicious anemia, gastrectomy, pancreatic disease), alcohol use, tapeworm infection, medications, and high metabolic states.2,4 Impaired B12 absorption is common in patients taking medications, such as metformin and proton pump inhibitors (PPI), due to suppression of ileal membrane transport and intrinsic factor activity.5-7 B-vitamin deficiency can be exacerbated by states of increased cellular turnover, such as polycythemia vera, due to elevated DNA synthesis.
Patients may experience permanent neurologic damage when the diagnosis and treatment of SCD are missed or delayed. Early diagnosis of SCD can be challenging due to lack of specific hematologic markers. In addition, many other conditions such as diabetic neuropathy, malnutrition, toxic neuropathy, sarcoidosis, HIV, multiple sclerosis, polycythemia vera, and iron deficiency anemia have similar presentations and clinical findings.8 Anemia and/or macrocytosis are not specific to B12 deficiency.4 In addition, patients with B12 deficiency may have a normal complete blood count (CBC); those with concomitant iron deficiency may have minimal or no mean corpuscular volume (MCV) elevation.4 In patients suspected to have B12 deficiency based on clinical presentation or laboratory findings of macrocytosis, serum methylmalonic acid (MMA) can serve as a direct measure of B12 activity, with levels > 0.75 μmol/L almost always indicating cobalamin deficiency. 9 On the other hand, plasma total homocysteine (tHcy) is a sensitive marker for B12 deficiency. The active form of B12, holotranscobalamin, has also emerged as a specific measure of B12 deficiency.9 However, in patients with SCD, measurement of these markers may be unnecessary due to the severity of their clinical symptoms.
The diagnosis of SCD is further complicated because not all individuals who develop B12 or B9 deficiency will develop SCD. It is difficult to determine which patients will develop SCD because the minimum level of serum B12 required for normal function is unknown, and recent studies indicate that SCD may occur even at low-normal B12 and B9 levels.2,4,10 Commonly, a serum B12 level of < 200 pg/mL is considered deficient, while a level between 200 and 300 pg/mL is considered borderline.4 The goal level of serum B12 is > 300 pg/mL, which is considered normal.4 While serologic findings of B-vitamin deficiency are only moderately specific, radiographic findings are highly sensitive and specific for SCD. According to Briani and colleagues, the most consistent finding in SCD on magnetic resonance imaging (MRI) is a “symmetrical, abnormally increased T2 signal intensity, commonly confined to posterior or posterior and lateral columns in the cervical and thoracic spinal cord.”2
We present a case of SCD in a patient with low-normal vitamin B12 levels who presented with progressive sensorimotor deficits and vision loss. The patient was subsequently diagnosed with SCD by radiologic workup. His course was complicated by worsening neurologic deficits despite B12 replacement. The progression of his clinical symptoms demonstrates the need for prompt, aggressive B12 replacement in patients diagnosed with SCD.
Case Presentation
A 63-year-old man presented for neurologic evaluation of progressive gait disturbance, paresthesia, blurred vision, and increasing falls despite use of a walker. Pertinent medical history included polycythemia vera requiring phlebotomy for approximately 9 years, alcohol use disorder (18 servings weekly), type 2 diabetes mellitus, and a remote episode of transient ischemic attack (TIA). The patient reported a 5-year history of burning pain in all extremities. A prior physician diagnosis attributed the symptoms to polyneuropathy secondary to iron deficiency anemia in the setting of chronic phlebotomy for polycythemia vera and high erythrogenesis. He was prescribed gabapentin 600 mg 3 times daily for pain control. B12 deficiency was considered an unlikely etiology due to a low-normal serum level of 305 pg/mL (reference range, 190-950 pg/mL) and normocytosis, with MCV of 88 fL (reference range, 80-100 fL). The patient also reported a 3-year history of blurred vision, which was initially attributed to be secondary to diabetic retinopathy. One week prior to presenting to our clinic, he was evaluated by ophthalmology for new-onset, bilateral central visual field defects, and he was diagnosed with nutritional optic neuropathy.
Ophthalmology suspected B12 deficiency. Notable findings included reduced deep tendon reflexes (DTRs) in the upper extremities and absent DTRs in the lower extremities, reduced sensation to light touch in all extremities, absent sensation to pinprick, vibration, and temperature in the lower extremities, positive Romberg sign, and a wide-based antalgic gait with the ankles externally rotated bilaterally (Table 1)
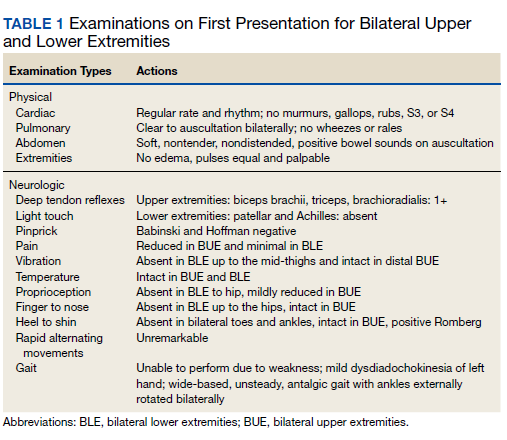
Previous cardiac evaluation failed to provide a diagnosis for syncopal episodes. MRI of the brain revealed nonspecific white matter changes consistent with chronic microvascular ischemic disease. Electromyography was limited due to pain but showed severe peripheral neuropathy. Laboratory results showed megalocytosis, low-normal serum B12 levels, and low serum folate levels (Table 2). The patient was diagnosed with polyneuropathy and was given intramuscular (IM) vitamin B12 1000 mcg once and a daily multivitamin (containing 25 mcg of B12). He was counseled on alcohol abstinence and medication adherence and was scheduled for follow-up in 3 months. He continued outpatient phlebotomy every 6 weeks for polycythemia.
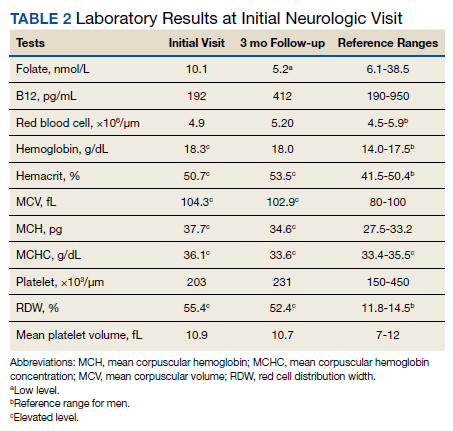
At 3-month follow-up, the patient reported medication adherence, continued alcohol use, and worsening of symptoms. Falls, which now occurred 2 to 3 times weekly despite proper use of a walker, were described as sudden loss of bilateral lower extremity strength without loss of consciousness, palpitations, or other prodrome. Laboratory results showed minimal changes. Physical examination of the patient demonstrated similar deficits as on initial presentation. The patient received one additional B12 1000 mcg IM. Gabapentin was replaced with pregabalin 75 mg twice daily due to persistent uncontrolled pain and paresthesia. The patient was scheduled for a 3-month followup (6 months from initial visit) and repeat serology.
At 6-month follow-up, the patient showed continued progression of disease with significant difficulty using the walker, worsening falls, and wheelchair use required. Physical examination showed decreased sensation bilaterally up to the knees, absent bilateral patellar and Achilles reflexes, and unsteady gait. Laboratory results showed persistent subclinical B12 deficiency. MRI of the brain and spine showed high T2 signaling in a pattern highly specific for SCD. A formal diagnosis of SCD was made. The patient received an additional B12 1000 mcg IM once. Follow-up phone call with the patient 1 month later revealed no progression or improvement of symptoms.
Radiographic Findings
MRI of the cervical and thoracic spine demonstrated abnormal high T2 signal starting from C2 and extending along the course of the cervical and thoracic spinal cord (Figure). MRI in SCD classically shows symmetric, bilateral high T2 signal within the dorsal columns; on axial images, there is typically an inverted “V” sign.2,4 There can also be abnormal cerebral white matter change; however, MRI of the brain in this patient did not show any abnormalities.2 The imaging differential for this appearance includes other metabolic deficiencies/toxicities: copper deficiency; vitamin E deficiency; methotrexateinduced myelopathy, and infectious causes: HIV vacuolar myelopathy; and neurosyphilis (tabes dorsalis).4
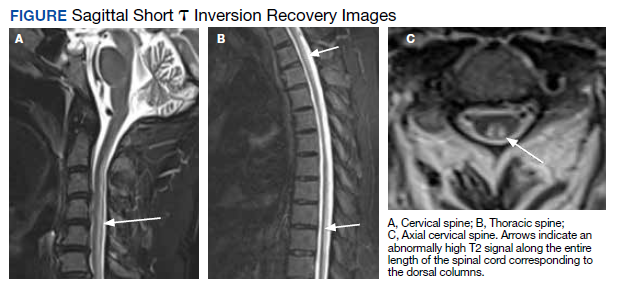
Discussion
This case demonstrates the clinical and radiographic findings of SCD and underscores the need for high-intensity dosing of B12 replacement in patients with SCD to prevent progression of the disease and development of morbidities.
Symptoms of SCD may manifest even when the vitamin levels are in low-normal levels. Its presentation is often nonspecific, thus radiologic workup is beneficial to elucidate the clinical picture. We support the use of spinal MRI in patients with clinical suspicion of SCD to help rule out other causes of myelopathy. However, an MRI is not indicated in all patients with B12 deficiency, especially those without myelopathic symptoms. Additionally, follow-up spinal MRIs are useful in monitoring the progression or improvement of SCD after B12 replacement.2 It is important to note that the MRI findings in SCD are not specific to B12 deficiency; other causes may present with similar radiographic findings.4 Therefore, radiologic findings must be correlated with a patient’s clinical presentation.
B12 replacement improves and may resolve clinical symptoms and abnormal radiographic findings of SCD. The treatment duration of B12 deficiency depends on the underlying etiology. Reversible causes, such as metformin use > 4 months, PPI use > 12 months, and dietary deficiency, require treatment until appropriate levels are reached and symptoms are resolved.4,11 The need for chronic metformin and PPI use should also be reassessed regularly. In patients who require long-term metformin use, IM administration of B12 1000 mcg annually should be considered, which will ensure adequate storage for more than 1 year.12,13 In patients who require long-term PPI use, the risk and benefits of continued use should be measured, and if needed, the lowest possible effective PPI dose is recommended.14 Irreversible causes of B12 deficiency, such as advanced age, prior gastrectomy, chronic pancreatitis, or autoimmune pernicious anemia, require lifelong supplementation of B12.4,11
In general, oral vitamin B12 replacement at 1000 to 2000 mcg daily may be as effective as parenteral replacement in patients with mild to moderate deficiency or neurologic symptoms.11 On the other hand, patients with SCD often require parenteral replacement of B12 due to the severity of their deficiency or neurologic symptoms, need for more rapid improvement in symptoms, and prevention of irreversible neurological deficits. 4,11 Appropriate B12 replacement in SCD requires intensive initial therapy which may involve IM B12 1000 mcg every other day for 2 weeks and additional IM supplementation every 2 to 3 months afterward until resolution of deficiency.4,14 IM replacement may also be considered in patients who are nonadherent to oral replacement or have an underlying gastrointestinal condition that impairs enteral absorption.4,11
B12 deficiency is frequently undertreated and can lead to progression of disease with significant morbidity. The need for highintensity dosing of B12 replacement is crucial in patients with SCD. Failure to respond to treatment, as shown from the lack of improvement of serum markers or symptoms, likely suggests undertreatment, treatment nonadherence, iron deficiency anemia, an unidentified malabsorption syndrome, or other diagnoses. In our case, significant undertreatment, compounded by his suspected iron deficiency anemia secondary to his polycythemia vera and chronic phlebotomies, are the most likel etiologies for his lack of clinical improvement.
Multiple factors may affect the prognosis of SCD. Males aged < 50 years with absence of anemia, spinal cord atrophy, Romberg sign, Babinski sign, or sensory deficits on examination have increased likelihood of eventual recovery of signs and symptoms of SCD; those with less spinal cord involvement (< 7 cord segments), contrast enhancement, and spinal cord edema also have improved outcomes.4,15
Conclusion
SCD is a rare but serious complication of chronic vitamin B12 deficiency that presents with a variety of neurological findings and may be easily confused with other illnesses. The condition is easily overlooked or misdiagnosed; thus, it is crucial to differentiate B12 deficiency from other common causes of neurologic symptoms. Specific findings on MRI are useful to support the clinical diagnosis of SCD and guide clinical decisions. Given the prevalence of B12 deficiency in the older adult population, clinicians should remain alert to the possibility of these conditions in patients who present with progressive neuropathy. Once a patient is diagnosed with SCD secondary to a B12 deficiency, appropriate B12 replacement is critical. Appropriate B12 replacement is aggressive and involves IM B12 1000 mcg every other day for 2 to 3 weeks, followed by additional IM administration every 2 months before transitioning to oral therapy. As seen in this case, failure to adequately replenish B12 can lead to progression or lack of resolution of SCD symptoms.
1. Gürsoy AE, Kolukısa M, Babacan-Yıldız G, Celebi A. Subacute Combined Degeneration of the Spinal Cord due to Different Etiologies and Improvement of MRI Findings. Case Rep Neurol Med. 2013;2013:159649. doi:10.1155/2013/159649
2. Briani C, Dalla Torre C, Citton V, et al. Cobalamin deficiency: clinical picture and radiological findings. Nutrients. 2013;5(11):4521-4539. Published 2013 Nov 15. doi:10.3390/nu5114521
3. Hunt A, Harrington D, Robinson S. Vitamin B12 deficiency. BMJ. 2014;349:g5226. Published 2014 Sep 4. doi:10.1136/bmj.g5226
4. Qudsiya Z, De Jesus O. Subacute combined degeneration of the spinal cord. [Updated 2021 Feb 7]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing. Updated August 30, 2021. Accessed January 5, 2022. https://www.ncbi.nlm.nih.gov/books /NBK559316/
5. de Jager J, Kooy A, Lehert P, et al. Long term treatment with metformin in patients with type 2 diabetes and risk of vitamin B-12 deficiency: randomised placebo controlled trial. BMJ. 2010;340:c2181. Published 2010 May 20. doi:10.1136/bmj.c2181
6. Aroda VR, Edelstein SL, Goldberg RB, et al. Longterm Metformin Use and Vitamin B12 Deficiency in the Diabetes Prevention Program Outcomes Study. J Clin Endocrinol Metab. 2016;101(4):1754-1761. doi:10.1210/jc.2015-3754
7. Lam JR, Schneider JL, Zhao W, Corley DA. Proton pump inhibitor and histamine 2 receptor antagonist use and vitamin B12 deficiency. JAMA. 2013;310(22):2435-2442. doi:10.1001/jama.2013.280490
8. Mihalj M, Titlic´ M, Bonacin D, Dogaš Z. Sensomotor axonal peripheral neuropathy as a first complication of polycythemia rubra vera: A report of 3 cases. Am J Case Rep. 2013;14:385-387. Published 2013 Sep 25. doi:10.12659/AJCR.884016
9. Devalia V, Hamilton MS, Molloy AM; British Committee for Standards in Haematology. Guidelines for the diagnosis and treatment of cobalamin and folate disorders. Br J Haematol. 2014;166(4):496-513. doi:10.1111/bjh.12959
10. Cao J, Xu S, Liu C. Is serum vitamin B12 decrease a necessity for the diagnosis of subacute combined degeneration?: A meta-analysis. Medicine (Baltimore). 2020;99(14):e19700.doi:10.1097/MD.0000000000019700
11. Langan RC, Goodbred AJ. Vitamin B12 Deficiency: Recognition and Management. Am Fam Physician. 2017;96(6):384-389.
12. Mazokopakis EE, Starakis IK. Recommendations for diagnosis and management of metformin-induced vitamin B12 (Cbl) deficiency. Diabetes Res Clin Pract. 2012;97(3):359-367. doi:10.1016/j.diabres.2012.06.001
13. Mahajan R, Gupta K. Revisiting Metformin: Annual Vitamin B12 Supplementation may become Mandatory with Long-Term Metformin Use. J Young Pharm. 2010;2(4):428-429. doi:10.4103/0975-1483.71621
14. Parks NE. Metabolic and Toxic Myelopathies. Continuum (Minneap Minn). 2021;27(1):143-162. doi:10.1212/CON.0000000000000963
15. Vasconcelos OM, Poehm EH, McCarter RJ, Campbell WW, Quezado ZM. Potential outcome factors in subacute combined degeneration: review of observational studies. J Gen Intern Med. 2006;21(10):1063-1068. doi:10.1111/j.1525-1497.2006.00525.x
Subacute combined degeneration (SCD) is an acquired neurologic complication of vitamin B12 (cobalamin) or, rarely, vitamin B9 (folate) deficiency. SCD is characterized by progressive demyelination of the dorsal and lateral spinal cord, resulting in peripheral neuropathy; gait ataxia; impaired proprioception, vibration, and fine touch; optic neuropathy; and cognitive impairment.1 In addition to SCD, other neurologic manifestations of B12 deficiency include dementia, depression, visual symptoms due to optic atrophy, and behavioral changes.2 The prevalence of SCD in the US has not been well documented, but B12 deficiency is reported at 6% in those aged < 60 years and 20% in those > 60 years.3
Causes of B12 and B9 deficiency include advanced age, low nutritional intake (eg, vegan diet), impaired absorption (eg, inflammatory bowel disease, autoimmune pernicious anemia, gastrectomy, pancreatic disease), alcohol use, tapeworm infection, medications, and high metabolic states.2,4 Impaired B12 absorption is common in patients taking medications, such as metformin and proton pump inhibitors (PPI), due to suppression of ileal membrane transport and intrinsic factor activity.5-7 B-vitamin deficiency can be exacerbated by states of increased cellular turnover, such as polycythemia vera, due to elevated DNA synthesis.
Patients may experience permanent neurologic damage when the diagnosis and treatment of SCD are missed or delayed. Early diagnosis of SCD can be challenging due to lack of specific hematologic markers. In addition, many other conditions such as diabetic neuropathy, malnutrition, toxic neuropathy, sarcoidosis, HIV, multiple sclerosis, polycythemia vera, and iron deficiency anemia have similar presentations and clinical findings.8 Anemia and/or macrocytosis are not specific to B12 deficiency.4 In addition, patients with B12 deficiency may have a normal complete blood count (CBC); those with concomitant iron deficiency may have minimal or no mean corpuscular volume (MCV) elevation.4 In patients suspected to have B12 deficiency based on clinical presentation or laboratory findings of macrocytosis, serum methylmalonic acid (MMA) can serve as a direct measure of B12 activity, with levels > 0.75 μmol/L almost always indicating cobalamin deficiency. 9 On the other hand, plasma total homocysteine (tHcy) is a sensitive marker for B12 deficiency. The active form of B12, holotranscobalamin, has also emerged as a specific measure of B12 deficiency.9 However, in patients with SCD, measurement of these markers may be unnecessary due to the severity of their clinical symptoms.
The diagnosis of SCD is further complicated because not all individuals who develop B12 or B9 deficiency will develop SCD. It is difficult to determine which patients will develop SCD because the minimum level of serum B12 required for normal function is unknown, and recent studies indicate that SCD may occur even at low-normal B12 and B9 levels.2,4,10 Commonly, a serum B12 level of < 200 pg/mL is considered deficient, while a level between 200 and 300 pg/mL is considered borderline.4 The goal level of serum B12 is > 300 pg/mL, which is considered normal.4 While serologic findings of B-vitamin deficiency are only moderately specific, radiographic findings are highly sensitive and specific for SCD. According to Briani and colleagues, the most consistent finding in SCD on magnetic resonance imaging (MRI) is a “symmetrical, abnormally increased T2 signal intensity, commonly confined to posterior or posterior and lateral columns in the cervical and thoracic spinal cord.”2
We present a case of SCD in a patient with low-normal vitamin B12 levels who presented with progressive sensorimotor deficits and vision loss. The patient was subsequently diagnosed with SCD by radiologic workup. His course was complicated by worsening neurologic deficits despite B12 replacement. The progression of his clinical symptoms demonstrates the need for prompt, aggressive B12 replacement in patients diagnosed with SCD.
Case Presentation
A 63-year-old man presented for neurologic evaluation of progressive gait disturbance, paresthesia, blurred vision, and increasing falls despite use of a walker. Pertinent medical history included polycythemia vera requiring phlebotomy for approximately 9 years, alcohol use disorder (18 servings weekly), type 2 diabetes mellitus, and a remote episode of transient ischemic attack (TIA). The patient reported a 5-year history of burning pain in all extremities. A prior physician diagnosis attributed the symptoms to polyneuropathy secondary to iron deficiency anemia in the setting of chronic phlebotomy for polycythemia vera and high erythrogenesis. He was prescribed gabapentin 600 mg 3 times daily for pain control. B12 deficiency was considered an unlikely etiology due to a low-normal serum level of 305 pg/mL (reference range, 190-950 pg/mL) and normocytosis, with MCV of 88 fL (reference range, 80-100 fL). The patient also reported a 3-year history of blurred vision, which was initially attributed to be secondary to diabetic retinopathy. One week prior to presenting to our clinic, he was evaluated by ophthalmology for new-onset, bilateral central visual field defects, and he was diagnosed with nutritional optic neuropathy.
Ophthalmology suspected B12 deficiency. Notable findings included reduced deep tendon reflexes (DTRs) in the upper extremities and absent DTRs in the lower extremities, reduced sensation to light touch in all extremities, absent sensation to pinprick, vibration, and temperature in the lower extremities, positive Romberg sign, and a wide-based antalgic gait with the ankles externally rotated bilaterally (Table 1)
Previous cardiac evaluation failed to provide a diagnosis for syncopal episodes. MRI of the brain revealed nonspecific white matter changes consistent with chronic microvascular ischemic disease. Electromyography was limited due to pain but showed severe peripheral neuropathy. Laboratory results showed megalocytosis, low-normal serum B12 levels, and low serum folate levels (Table 2). The patient was diagnosed with polyneuropathy and was given intramuscular (IM) vitamin B12 1000 mcg once and a daily multivitamin (containing 25 mcg of B12). He was counseled on alcohol abstinence and medication adherence and was scheduled for follow-up in 3 months. He continued outpatient phlebotomy every 6 weeks for polycythemia.
At 3-month follow-up, the patient reported medication adherence, continued alcohol use, and worsening of symptoms. Falls, which now occurred 2 to 3 times weekly despite proper use of a walker, were described as sudden loss of bilateral lower extremity strength without loss of consciousness, palpitations, or other prodrome. Laboratory results showed minimal changes. Physical examination of the patient demonstrated similar deficits as on initial presentation. The patient received one additional B12 1000 mcg IM. Gabapentin was replaced with pregabalin 75 mg twice daily due to persistent uncontrolled pain and paresthesia. The patient was scheduled for a 3-month followup (6 months from initial visit) and repeat serology.
At 6-month follow-up, the patient showed continued progression of disease with significant difficulty using the walker, worsening falls, and wheelchair use required. Physical examination showed decreased sensation bilaterally up to the knees, absent bilateral patellar and Achilles reflexes, and unsteady gait. Laboratory results showed persistent subclinical B12 deficiency. MRI of the brain and spine showed high T2 signaling in a pattern highly specific for SCD. A formal diagnosis of SCD was made. The patient received an additional B12 1000 mcg IM once. Follow-up phone call with the patient 1 month later revealed no progression or improvement of symptoms.
Radiographic Findings
MRI of the cervical and thoracic spine demonstrated abnormal high T2 signal starting from C2 and extending along the course of the cervical and thoracic spinal cord (Figure). MRI in SCD classically shows symmetric, bilateral high T2 signal within the dorsal columns; on axial images, there is typically an inverted “V” sign.2,4 There can also be abnormal cerebral white matter change; however, MRI of the brain in this patient did not show any abnormalities.2 The imaging differential for this appearance includes other metabolic deficiencies/toxicities: copper deficiency; vitamin E deficiency; methotrexateinduced myelopathy, and infectious causes: HIV vacuolar myelopathy; and neurosyphilis (tabes dorsalis).4
Discussion
This case demonstrates the clinical and radiographic findings of SCD and underscores the need for high-intensity dosing of B12 replacement in patients with SCD to prevent progression of the disease and development of morbidities.
Symptoms of SCD may manifest even when the vitamin levels are in low-normal levels. Its presentation is often nonspecific, thus radiologic workup is beneficial to elucidate the clinical picture. We support the use of spinal MRI in patients with clinical suspicion of SCD to help rule out other causes of myelopathy. However, an MRI is not indicated in all patients with B12 deficiency, especially those without myelopathic symptoms. Additionally, follow-up spinal MRIs are useful in monitoring the progression or improvement of SCD after B12 replacement.2 It is important to note that the MRI findings in SCD are not specific to B12 deficiency; other causes may present with similar radiographic findings.4 Therefore, radiologic findings must be correlated with a patient’s clinical presentation.
B12 replacement improves and may resolve clinical symptoms and abnormal radiographic findings of SCD. The treatment duration of B12 deficiency depends on the underlying etiology. Reversible causes, such as metformin use > 4 months, PPI use > 12 months, and dietary deficiency, require treatment until appropriate levels are reached and symptoms are resolved.4,11 The need for chronic metformin and PPI use should also be reassessed regularly. In patients who require long-term metformin use, IM administration of B12 1000 mcg annually should be considered, which will ensure adequate storage for more than 1 year.12,13 In patients who require long-term PPI use, the risk and benefits of continued use should be measured, and if needed, the lowest possible effective PPI dose is recommended.14 Irreversible causes of B12 deficiency, such as advanced age, prior gastrectomy, chronic pancreatitis, or autoimmune pernicious anemia, require lifelong supplementation of B12.4,11
In general, oral vitamin B12 replacement at 1000 to 2000 mcg daily may be as effective as parenteral replacement in patients with mild to moderate deficiency or neurologic symptoms.11 On the other hand, patients with SCD often require parenteral replacement of B12 due to the severity of their deficiency or neurologic symptoms, need for more rapid improvement in symptoms, and prevention of irreversible neurological deficits. 4,11 Appropriate B12 replacement in SCD requires intensive initial therapy which may involve IM B12 1000 mcg every other day for 2 weeks and additional IM supplementation every 2 to 3 months afterward until resolution of deficiency.4,14 IM replacement may also be considered in patients who are nonadherent to oral replacement or have an underlying gastrointestinal condition that impairs enteral absorption.4,11
B12 deficiency is frequently undertreated and can lead to progression of disease with significant morbidity. The need for highintensity dosing of B12 replacement is crucial in patients with SCD. Failure to respond to treatment, as shown from the lack of improvement of serum markers or symptoms, likely suggests undertreatment, treatment nonadherence, iron deficiency anemia, an unidentified malabsorption syndrome, or other diagnoses. In our case, significant undertreatment, compounded by his suspected iron deficiency anemia secondary to his polycythemia vera and chronic phlebotomies, are the most likel etiologies for his lack of clinical improvement.
Multiple factors may affect the prognosis of SCD. Males aged < 50 years with absence of anemia, spinal cord atrophy, Romberg sign, Babinski sign, or sensory deficits on examination have increased likelihood of eventual recovery of signs and symptoms of SCD; those with less spinal cord involvement (< 7 cord segments), contrast enhancement, and spinal cord edema also have improved outcomes.4,15
Conclusion
SCD is a rare but serious complication of chronic vitamin B12 deficiency that presents with a variety of neurological findings and may be easily confused with other illnesses. The condition is easily overlooked or misdiagnosed; thus, it is crucial to differentiate B12 deficiency from other common causes of neurologic symptoms. Specific findings on MRI are useful to support the clinical diagnosis of SCD and guide clinical decisions. Given the prevalence of B12 deficiency in the older adult population, clinicians should remain alert to the possibility of these conditions in patients who present with progressive neuropathy. Once a patient is diagnosed with SCD secondary to a B12 deficiency, appropriate B12 replacement is critical. Appropriate B12 replacement is aggressive and involves IM B12 1000 mcg every other day for 2 to 3 weeks, followed by additional IM administration every 2 months before transitioning to oral therapy. As seen in this case, failure to adequately replenish B12 can lead to progression or lack of resolution of SCD symptoms.
Subacute combined degeneration (SCD) is an acquired neurologic complication of vitamin B12 (cobalamin) or, rarely, vitamin B9 (folate) deficiency. SCD is characterized by progressive demyelination of the dorsal and lateral spinal cord, resulting in peripheral neuropathy; gait ataxia; impaired proprioception, vibration, and fine touch; optic neuropathy; and cognitive impairment.1 In addition to SCD, other neurologic manifestations of B12 deficiency include dementia, depression, visual symptoms due to optic atrophy, and behavioral changes.2 The prevalence of SCD in the US has not been well documented, but B12 deficiency is reported at 6% in those aged < 60 years and 20% in those > 60 years.3
Causes of B12 and B9 deficiency include advanced age, low nutritional intake (eg, vegan diet), impaired absorption (eg, inflammatory bowel disease, autoimmune pernicious anemia, gastrectomy, pancreatic disease), alcohol use, tapeworm infection, medications, and high metabolic states.2,4 Impaired B12 absorption is common in patients taking medications, such as metformin and proton pump inhibitors (PPI), due to suppression of ileal membrane transport and intrinsic factor activity.5-7 B-vitamin deficiency can be exacerbated by states of increased cellular turnover, such as polycythemia vera, due to elevated DNA synthesis.
Patients may experience permanent neurologic damage when the diagnosis and treatment of SCD are missed or delayed. Early diagnosis of SCD can be challenging due to lack of specific hematologic markers. In addition, many other conditions such as diabetic neuropathy, malnutrition, toxic neuropathy, sarcoidosis, HIV, multiple sclerosis, polycythemia vera, and iron deficiency anemia have similar presentations and clinical findings.8 Anemia and/or macrocytosis are not specific to B12 deficiency.4 In addition, patients with B12 deficiency may have a normal complete blood count (CBC); those with concomitant iron deficiency may have minimal or no mean corpuscular volume (MCV) elevation.4 In patients suspected to have B12 deficiency based on clinical presentation or laboratory findings of macrocytosis, serum methylmalonic acid (MMA) can serve as a direct measure of B12 activity, with levels > 0.75 μmol/L almost always indicating cobalamin deficiency. 9 On the other hand, plasma total homocysteine (tHcy) is a sensitive marker for B12 deficiency. The active form of B12, holotranscobalamin, has also emerged as a specific measure of B12 deficiency.9 However, in patients with SCD, measurement of these markers may be unnecessary due to the severity of their clinical symptoms.
The diagnosis of SCD is further complicated because not all individuals who develop B12 or B9 deficiency will develop SCD. It is difficult to determine which patients will develop SCD because the minimum level of serum B12 required for normal function is unknown, and recent studies indicate that SCD may occur even at low-normal B12 and B9 levels.2,4,10 Commonly, a serum B12 level of < 200 pg/mL is considered deficient, while a level between 200 and 300 pg/mL is considered borderline.4 The goal level of serum B12 is > 300 pg/mL, which is considered normal.4 While serologic findings of B-vitamin deficiency are only moderately specific, radiographic findings are highly sensitive and specific for SCD. According to Briani and colleagues, the most consistent finding in SCD on magnetic resonance imaging (MRI) is a “symmetrical, abnormally increased T2 signal intensity, commonly confined to posterior or posterior and lateral columns in the cervical and thoracic spinal cord.”2
We present a case of SCD in a patient with low-normal vitamin B12 levels who presented with progressive sensorimotor deficits and vision loss. The patient was subsequently diagnosed with SCD by radiologic workup. His course was complicated by worsening neurologic deficits despite B12 replacement. The progression of his clinical symptoms demonstrates the need for prompt, aggressive B12 replacement in patients diagnosed with SCD.
Case Presentation
A 63-year-old man presented for neurologic evaluation of progressive gait disturbance, paresthesia, blurred vision, and increasing falls despite use of a walker. Pertinent medical history included polycythemia vera requiring phlebotomy for approximately 9 years, alcohol use disorder (18 servings weekly), type 2 diabetes mellitus, and a remote episode of transient ischemic attack (TIA). The patient reported a 5-year history of burning pain in all extremities. A prior physician diagnosis attributed the symptoms to polyneuropathy secondary to iron deficiency anemia in the setting of chronic phlebotomy for polycythemia vera and high erythrogenesis. He was prescribed gabapentin 600 mg 3 times daily for pain control. B12 deficiency was considered an unlikely etiology due to a low-normal serum level of 305 pg/mL (reference range, 190-950 pg/mL) and normocytosis, with MCV of 88 fL (reference range, 80-100 fL). The patient also reported a 3-year history of blurred vision, which was initially attributed to be secondary to diabetic retinopathy. One week prior to presenting to our clinic, he was evaluated by ophthalmology for new-onset, bilateral central visual field defects, and he was diagnosed with nutritional optic neuropathy.
Ophthalmology suspected B12 deficiency. Notable findings included reduced deep tendon reflexes (DTRs) in the upper extremities and absent DTRs in the lower extremities, reduced sensation to light touch in all extremities, absent sensation to pinprick, vibration, and temperature in the lower extremities, positive Romberg sign, and a wide-based antalgic gait with the ankles externally rotated bilaterally (Table 1)
Previous cardiac evaluation failed to provide a diagnosis for syncopal episodes. MRI of the brain revealed nonspecific white matter changes consistent with chronic microvascular ischemic disease. Electromyography was limited due to pain but showed severe peripheral neuropathy. Laboratory results showed megalocytosis, low-normal serum B12 levels, and low serum folate levels (Table 2). The patient was diagnosed with polyneuropathy and was given intramuscular (IM) vitamin B12 1000 mcg once and a daily multivitamin (containing 25 mcg of B12). He was counseled on alcohol abstinence and medication adherence and was scheduled for follow-up in 3 months. He continued outpatient phlebotomy every 6 weeks for polycythemia.
At 3-month follow-up, the patient reported medication adherence, continued alcohol use, and worsening of symptoms. Falls, which now occurred 2 to 3 times weekly despite proper use of a walker, were described as sudden loss of bilateral lower extremity strength without loss of consciousness, palpitations, or other prodrome. Laboratory results showed minimal changes. Physical examination of the patient demonstrated similar deficits as on initial presentation. The patient received one additional B12 1000 mcg IM. Gabapentin was replaced with pregabalin 75 mg twice daily due to persistent uncontrolled pain and paresthesia. The patient was scheduled for a 3-month followup (6 months from initial visit) and repeat serology.
At 6-month follow-up, the patient showed continued progression of disease with significant difficulty using the walker, worsening falls, and wheelchair use required. Physical examination showed decreased sensation bilaterally up to the knees, absent bilateral patellar and Achilles reflexes, and unsteady gait. Laboratory results showed persistent subclinical B12 deficiency. MRI of the brain and spine showed high T2 signaling in a pattern highly specific for SCD. A formal diagnosis of SCD was made. The patient received an additional B12 1000 mcg IM once. Follow-up phone call with the patient 1 month later revealed no progression or improvement of symptoms.
Radiographic Findings
MRI of the cervical and thoracic spine demonstrated abnormal high T2 signal starting from C2 and extending along the course of the cervical and thoracic spinal cord (Figure). MRI in SCD classically shows symmetric, bilateral high T2 signal within the dorsal columns; on axial images, there is typically an inverted “V” sign.2,4 There can also be abnormal cerebral white matter change; however, MRI of the brain in this patient did not show any abnormalities.2 The imaging differential for this appearance includes other metabolic deficiencies/toxicities: copper deficiency; vitamin E deficiency; methotrexateinduced myelopathy, and infectious causes: HIV vacuolar myelopathy; and neurosyphilis (tabes dorsalis).4
Discussion
This case demonstrates the clinical and radiographic findings of SCD and underscores the need for high-intensity dosing of B12 replacement in patients with SCD to prevent progression of the disease and development of morbidities.
Symptoms of SCD may manifest even when the vitamin levels are in low-normal levels. Its presentation is often nonspecific, thus radiologic workup is beneficial to elucidate the clinical picture. We support the use of spinal MRI in patients with clinical suspicion of SCD to help rule out other causes of myelopathy. However, an MRI is not indicated in all patients with B12 deficiency, especially those without myelopathic symptoms. Additionally, follow-up spinal MRIs are useful in monitoring the progression or improvement of SCD after B12 replacement.2 It is important to note that the MRI findings in SCD are not specific to B12 deficiency; other causes may present with similar radiographic findings.4 Therefore, radiologic findings must be correlated with a patient’s clinical presentation.
B12 replacement improves and may resolve clinical symptoms and abnormal radiographic findings of SCD. The treatment duration of B12 deficiency depends on the underlying etiology. Reversible causes, such as metformin use > 4 months, PPI use > 12 months, and dietary deficiency, require treatment until appropriate levels are reached and symptoms are resolved.4,11 The need for chronic metformin and PPI use should also be reassessed regularly. In patients who require long-term metformin use, IM administration of B12 1000 mcg annually should be considered, which will ensure adequate storage for more than 1 year.12,13 In patients who require long-term PPI use, the risk and benefits of continued use should be measured, and if needed, the lowest possible effective PPI dose is recommended.14 Irreversible causes of B12 deficiency, such as advanced age, prior gastrectomy, chronic pancreatitis, or autoimmune pernicious anemia, require lifelong supplementation of B12.4,11
In general, oral vitamin B12 replacement at 1000 to 2000 mcg daily may be as effective as parenteral replacement in patients with mild to moderate deficiency or neurologic symptoms.11 On the other hand, patients with SCD often require parenteral replacement of B12 due to the severity of their deficiency or neurologic symptoms, need for more rapid improvement in symptoms, and prevention of irreversible neurological deficits. 4,11 Appropriate B12 replacement in SCD requires intensive initial therapy which may involve IM B12 1000 mcg every other day for 2 weeks and additional IM supplementation every 2 to 3 months afterward until resolution of deficiency.4,14 IM replacement may also be considered in patients who are nonadherent to oral replacement or have an underlying gastrointestinal condition that impairs enteral absorption.4,11
B12 deficiency is frequently undertreated and can lead to progression of disease with significant morbidity. The need for highintensity dosing of B12 replacement is crucial in patients with SCD. Failure to respond to treatment, as shown from the lack of improvement of serum markers or symptoms, likely suggests undertreatment, treatment nonadherence, iron deficiency anemia, an unidentified malabsorption syndrome, or other diagnoses. In our case, significant undertreatment, compounded by his suspected iron deficiency anemia secondary to his polycythemia vera and chronic phlebotomies, are the most likel etiologies for his lack of clinical improvement.
Multiple factors may affect the prognosis of SCD. Males aged < 50 years with absence of anemia, spinal cord atrophy, Romberg sign, Babinski sign, or sensory deficits on examination have increased likelihood of eventual recovery of signs and symptoms of SCD; those with less spinal cord involvement (< 7 cord segments), contrast enhancement, and spinal cord edema also have improved outcomes.4,15
Conclusion
SCD is a rare but serious complication of chronic vitamin B12 deficiency that presents with a variety of neurological findings and may be easily confused with other illnesses. The condition is easily overlooked or misdiagnosed; thus, it is crucial to differentiate B12 deficiency from other common causes of neurologic symptoms. Specific findings on MRI are useful to support the clinical diagnosis of SCD and guide clinical decisions. Given the prevalence of B12 deficiency in the older adult population, clinicians should remain alert to the possibility of these conditions in patients who present with progressive neuropathy. Once a patient is diagnosed with SCD secondary to a B12 deficiency, appropriate B12 replacement is critical. Appropriate B12 replacement is aggressive and involves IM B12 1000 mcg every other day for 2 to 3 weeks, followed by additional IM administration every 2 months before transitioning to oral therapy. As seen in this case, failure to adequately replenish B12 can lead to progression or lack of resolution of SCD symptoms.
1. Gürsoy AE, Kolukısa M, Babacan-Yıldız G, Celebi A. Subacute Combined Degeneration of the Spinal Cord due to Different Etiologies and Improvement of MRI Findings. Case Rep Neurol Med. 2013;2013:159649. doi:10.1155/2013/159649
2. Briani C, Dalla Torre C, Citton V, et al. Cobalamin deficiency: clinical picture and radiological findings. Nutrients. 2013;5(11):4521-4539. Published 2013 Nov 15. doi:10.3390/nu5114521
3. Hunt A, Harrington D, Robinson S. Vitamin B12 deficiency. BMJ. 2014;349:g5226. Published 2014 Sep 4. doi:10.1136/bmj.g5226
4. Qudsiya Z, De Jesus O. Subacute combined degeneration of the spinal cord. [Updated 2021 Feb 7]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing. Updated August 30, 2021. Accessed January 5, 2022. https://www.ncbi.nlm.nih.gov/books /NBK559316/
5. de Jager J, Kooy A, Lehert P, et al. Long term treatment with metformin in patients with type 2 diabetes and risk of vitamin B-12 deficiency: randomised placebo controlled trial. BMJ. 2010;340:c2181. Published 2010 May 20. doi:10.1136/bmj.c2181
6. Aroda VR, Edelstein SL, Goldberg RB, et al. Longterm Metformin Use and Vitamin B12 Deficiency in the Diabetes Prevention Program Outcomes Study. J Clin Endocrinol Metab. 2016;101(4):1754-1761. doi:10.1210/jc.2015-3754
7. Lam JR, Schneider JL, Zhao W, Corley DA. Proton pump inhibitor and histamine 2 receptor antagonist use and vitamin B12 deficiency. JAMA. 2013;310(22):2435-2442. doi:10.1001/jama.2013.280490
8. Mihalj M, Titlic´ M, Bonacin D, Dogaš Z. Sensomotor axonal peripheral neuropathy as a first complication of polycythemia rubra vera: A report of 3 cases. Am J Case Rep. 2013;14:385-387. Published 2013 Sep 25. doi:10.12659/AJCR.884016
9. Devalia V, Hamilton MS, Molloy AM; British Committee for Standards in Haematology. Guidelines for the diagnosis and treatment of cobalamin and folate disorders. Br J Haematol. 2014;166(4):496-513. doi:10.1111/bjh.12959
10. Cao J, Xu S, Liu C. Is serum vitamin B12 decrease a necessity for the diagnosis of subacute combined degeneration?: A meta-analysis. Medicine (Baltimore). 2020;99(14):e19700.doi:10.1097/MD.0000000000019700
11. Langan RC, Goodbred AJ. Vitamin B12 Deficiency: Recognition and Management. Am Fam Physician. 2017;96(6):384-389.
12. Mazokopakis EE, Starakis IK. Recommendations for diagnosis and management of metformin-induced vitamin B12 (Cbl) deficiency. Diabetes Res Clin Pract. 2012;97(3):359-367. doi:10.1016/j.diabres.2012.06.001
13. Mahajan R, Gupta K. Revisiting Metformin: Annual Vitamin B12 Supplementation may become Mandatory with Long-Term Metformin Use. J Young Pharm. 2010;2(4):428-429. doi:10.4103/0975-1483.71621
14. Parks NE. Metabolic and Toxic Myelopathies. Continuum (Minneap Minn). 2021;27(1):143-162. doi:10.1212/CON.0000000000000963
15. Vasconcelos OM, Poehm EH, McCarter RJ, Campbell WW, Quezado ZM. Potential outcome factors in subacute combined degeneration: review of observational studies. J Gen Intern Med. 2006;21(10):1063-1068. doi:10.1111/j.1525-1497.2006.00525.x
1. Gürsoy AE, Kolukısa M, Babacan-Yıldız G, Celebi A. Subacute Combined Degeneration of the Spinal Cord due to Different Etiologies and Improvement of MRI Findings. Case Rep Neurol Med. 2013;2013:159649. doi:10.1155/2013/159649
2. Briani C, Dalla Torre C, Citton V, et al. Cobalamin deficiency: clinical picture and radiological findings. Nutrients. 2013;5(11):4521-4539. Published 2013 Nov 15. doi:10.3390/nu5114521
3. Hunt A, Harrington D, Robinson S. Vitamin B12 deficiency. BMJ. 2014;349:g5226. Published 2014 Sep 4. doi:10.1136/bmj.g5226
4. Qudsiya Z, De Jesus O. Subacute combined degeneration of the spinal cord. [Updated 2021 Feb 7]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing. Updated August 30, 2021. Accessed January 5, 2022. https://www.ncbi.nlm.nih.gov/books /NBK559316/
5. de Jager J, Kooy A, Lehert P, et al. Long term treatment with metformin in patients with type 2 diabetes and risk of vitamin B-12 deficiency: randomised placebo controlled trial. BMJ. 2010;340:c2181. Published 2010 May 20. doi:10.1136/bmj.c2181
6. Aroda VR, Edelstein SL, Goldberg RB, et al. Longterm Metformin Use and Vitamin B12 Deficiency in the Diabetes Prevention Program Outcomes Study. J Clin Endocrinol Metab. 2016;101(4):1754-1761. doi:10.1210/jc.2015-3754
7. Lam JR, Schneider JL, Zhao W, Corley DA. Proton pump inhibitor and histamine 2 receptor antagonist use and vitamin B12 deficiency. JAMA. 2013;310(22):2435-2442. doi:10.1001/jama.2013.280490
8. Mihalj M, Titlic´ M, Bonacin D, Dogaš Z. Sensomotor axonal peripheral neuropathy as a first complication of polycythemia rubra vera: A report of 3 cases. Am J Case Rep. 2013;14:385-387. Published 2013 Sep 25. doi:10.12659/AJCR.884016
9. Devalia V, Hamilton MS, Molloy AM; British Committee for Standards in Haematology. Guidelines for the diagnosis and treatment of cobalamin and folate disorders. Br J Haematol. 2014;166(4):496-513. doi:10.1111/bjh.12959
10. Cao J, Xu S, Liu C. Is serum vitamin B12 decrease a necessity for the diagnosis of subacute combined degeneration?: A meta-analysis. Medicine (Baltimore). 2020;99(14):e19700.doi:10.1097/MD.0000000000019700
11. Langan RC, Goodbred AJ. Vitamin B12 Deficiency: Recognition and Management. Am Fam Physician. 2017;96(6):384-389.
12. Mazokopakis EE, Starakis IK. Recommendations for diagnosis and management of metformin-induced vitamin B12 (Cbl) deficiency. Diabetes Res Clin Pract. 2012;97(3):359-367. doi:10.1016/j.diabres.2012.06.001
13. Mahajan R, Gupta K. Revisiting Metformin: Annual Vitamin B12 Supplementation may become Mandatory with Long-Term Metformin Use. J Young Pharm. 2010;2(4):428-429. doi:10.4103/0975-1483.71621
14. Parks NE. Metabolic and Toxic Myelopathies. Continuum (Minneap Minn). 2021;27(1):143-162. doi:10.1212/CON.0000000000000963
15. Vasconcelos OM, Poehm EH, McCarter RJ, Campbell WW, Quezado ZM. Potential outcome factors in subacute combined degeneration: review of observational studies. J Gen Intern Med. 2006;21(10):1063-1068. doi:10.1111/j.1525-1497.2006.00525.x
Reactivation of a BCG Vaccination Scar Following the First Dose of the Moderna COVID-19 Vaccine
The COVID-19 pandemic has resulted in notable morbidity and mortality worldwide. In December 2020, the US Food and Drug Administration issued an Emergency Use Authorization for 2 messenger RNA (mRNA) vaccines—produced by Pfizer-BioNTech and Moderna—for the prevention of COVID-19. Phase 3 trials of the vaccine developed by Moderna showed 94.1% efficacy at preventing COVID-19 after 2 doses.1
Common cutaneous adverse effects of the Moderna COVID-19 Vaccine include injection-site reactions, such as pain, induration, and erythema. Less frequently reported dermatologic adverse effects include diffuse bullous rash and hypersensitivity reactions.1 We report a case of reactivation of a BCG vaccination scar after the first dose of the Moderna COVID-19 Vaccine.
Case Report
A 48-year-old Asian man who was otherwise healthy presented with erythema, induration, and mild pruritus on the deltoid muscle of the left arm, near the scar from an earlier BCG vaccine, which he received at approximately 5 years of age when living in Taiwan. The patient received the first dose of the Moderna COVID-19 Vaccine approximately 5 to 7 cm distant from the BCG vaccination scar. One to 2 days after inoculation, the patient endorsed tenderness at the site of COVID-19 vaccination but denied systemic symptoms. He had never been given a diagnosis of COVID-19. His SARS-CoV-2 antibody status was unknown.
Eight days later, the patient noticed a well-defined, erythematous, indurated plaque with mild itchiness overlying and around the BCG vaccination scar that did not involve the COVID-19 vaccination site. The following day, the redness and induration became worse (Figure).

The patient was otherwise well. Vital signs were normal; there was no lymphadenopathy. The rash resolved without treatment over the next 4 days.
Comment
The BCG vaccine is an intradermal live attenuated virus vaccine used to prevent certain forms of tuberculosis and potentially other Mycobacterium infections. Although the vaccine is not routinely administered in the United States, it is part of the vaccination schedule in most countries, administered most often to newborns and infants. Administration of the BCG vaccine commonly results in mild localized erythema, swelling, and pain at the injection site. Most inoculated patients also develop an ulcer that heals with the characteristic BCG vaccination scar.2,3
There is evidence that the BCG vaccine can enhance the innate immune system response and might decrease the rate of infection by unrelated pathogens, including viruses.4 Several epidemiologic studies have suggested that the BCG vaccine might offer some protection against COVID-19, possibly due to a resemblance of the amino acid sequences of BCG and SARS-CoV-2, which might provoke cross-reactive T cells.5,6 Further studies are underway to determine whether the BCG vaccine is truly protective against COVID-19.
BCG vaccination scar reactivation presents as redness, swelling, or ulceration at the BCG injection site months to years after inoculation. Although erythema and induration of the BCG scar are not included in the diagnostic criteria of Kawasaki disease, likely due to variable vaccine requirements in different countries, these findings are largely recognized as specific for Kawasaki disease and present in approximately half of affected patients who received the BCG vaccine.2
Heat Shock Proteins—Heat shock proteins (HSPs) are produced by cells in response to stressors. The proposed mechanism of BCG vaccination scar reactivation is a cross-reaction between human homologue HSP 63 and Mycobacterium HSP 65, leading to hyperactivity of the immune system against BCG.7 There also are reports of reactivation of a BCG vaccination scar from measles infection and influenza vaccination.2,8,9 Most prior reports of BCG vaccination scar reactivation have been in pediatric patients; our patient is an adult who received the BCG vaccine more than 40 years ago.
Mechanism of Reactivation—The mechanism of BCG vaccination scar reactivation in our patient, who received the Moderna COVID-19 Vaccine, is unclear. Possible mechanisms include (1) release of HSP mediated by the COVID-19 vaccine, leading to an immune response at the BCG vaccine scar, or (2) another immune-mediated cross-reaction between BCG and the Moderna COVID-19 Vaccine mRNA nanoparticle or encoded spike protein antigen. It has been hypothesized that the BCG vaccine might offer some protection against COVID-19; this remains uncertain and is under further investigation.10 A recent retrospective cohort study showed that a BCG vaccination booster may decrease COVID-19 infection rates in higher-risk populations.11
Conclusion
We present a case of BCG vaccine scar reactivation occurring after a dose of the Moderna COVID-19 Vaccine, a likely underreported, self-limiting, cutaneous adverse effect of this mRNA vaccine.
- Baden LR, El Sahly HM, Essink B, et al; COVE Study Group. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N Engl J Med. 2020;384:403-416. doi:10.1056/NEJMoa2035389
- Muthuvelu S, Lim KS, Huang L-Y, et al. Measles infection causing bacillus Calmette-Guérin reactivation: a case report. BMC Pediatr. 2019;19:251. doi:10.1186/s12887-019-1635-z
- Fatima S, Kumari A, Das G, et al. Tuberculosis vaccine: a journey from BCG to present. Life Sci. 2020;252:117594. doi:10.1016/j.lfs.2020.117594
- O’Neill LAJ, Netea MG. BCG-induced trained immunity: can it offer protection against COVID-19? Nat Rev Immunol. 2020;20:335-337. doi:10.1038/s41577-020-0337-y
- Brooks NA, Puri A, Garg S, et al. The association of coronavirus disease-19 mortality and prior bacille Calmette-Guérin vaccination: a robust ecological analysis using unsupervised machine learning. Sci Rep. 2021;11:774. doi:10.1038/s41598-020-80787-z
- Tomita Y, Sato R, Ikeda T, et al. BCG vaccine may generate cross-reactive T-cells against SARS-CoV-2: in silico analyses and a hypothesis. Vaccine. 2020;38:6352-6356. doi:10.1016/j.vaccine.2020.08.045
- Lim KYY, Chua MC, Tan NWH, et al. Reactivation of BCG inoculation site in a child with febrile exanthema of 3 days duration: an early indicator of incomplete Kawasaki disease. BMJ Case Rep. 2020;13:E239648. doi:10.1136/bcr-2020-239648
- Kondo M, Goto H, Yamamoto S. First case of redness and erosion at bacillus Calmette-Guérin inoculation site after vaccination against influenza. J Dermatol. 2016;43:1229-1231. doi:10.1111/1346-8138.13365
- Chavarri-Guerra Y, Soto-Pérez-de-Celis E. Erythema at the bacillus Calmette-Guerin scar after influenza vaccination. Rev Soc Bras Med Trop. 2019;53:E20190390. doi:10.1590/0037-8682-0390-2019
- Fu W, Ho P-C, Liu C-L, et al. Reconcile the debate over protective effects of BCG vaccine against COVID-19. Sci Rep. 2021;11:8356. doi:10.1038/s41598-021-87731-9
- Amirlak L, Haddad R, Hardy JD, et al. Effectiveness of booster BCG vaccination in preventing COVID-19 infection. Hum Vaccin Immunother. 2021;17:3913-3915. doi:10.1080/21645515.2021.1956228
The COVID-19 pandemic has resulted in notable morbidity and mortality worldwide. In December 2020, the US Food and Drug Administration issued an Emergency Use Authorization for 2 messenger RNA (mRNA) vaccines—produced by Pfizer-BioNTech and Moderna—for the prevention of COVID-19. Phase 3 trials of the vaccine developed by Moderna showed 94.1% efficacy at preventing COVID-19 after 2 doses.1
Common cutaneous adverse effects of the Moderna COVID-19 Vaccine include injection-site reactions, such as pain, induration, and erythema. Less frequently reported dermatologic adverse effects include diffuse bullous rash and hypersensitivity reactions.1 We report a case of reactivation of a BCG vaccination scar after the first dose of the Moderna COVID-19 Vaccine.
Case Report
A 48-year-old Asian man who was otherwise healthy presented with erythema, induration, and mild pruritus on the deltoid muscle of the left arm, near the scar from an earlier BCG vaccine, which he received at approximately 5 years of age when living in Taiwan. The patient received the first dose of the Moderna COVID-19 Vaccine approximately 5 to 7 cm distant from the BCG vaccination scar. One to 2 days after inoculation, the patient endorsed tenderness at the site of COVID-19 vaccination but denied systemic symptoms. He had never been given a diagnosis of COVID-19. His SARS-CoV-2 antibody status was unknown.
Eight days later, the patient noticed a well-defined, erythematous, indurated plaque with mild itchiness overlying and around the BCG vaccination scar that did not involve the COVID-19 vaccination site. The following day, the redness and induration became worse (Figure).
The patient was otherwise well. Vital signs were normal; there was no lymphadenopathy. The rash resolved without treatment over the next 4 days.
Comment
The BCG vaccine is an intradermal live attenuated virus vaccine used to prevent certain forms of tuberculosis and potentially other Mycobacterium infections. Although the vaccine is not routinely administered in the United States, it is part of the vaccination schedule in most countries, administered most often to newborns and infants. Administration of the BCG vaccine commonly results in mild localized erythema, swelling, and pain at the injection site. Most inoculated patients also develop an ulcer that heals with the characteristic BCG vaccination scar.2,3
There is evidence that the BCG vaccine can enhance the innate immune system response and might decrease the rate of infection by unrelated pathogens, including viruses.4 Several epidemiologic studies have suggested that the BCG vaccine might offer some protection against COVID-19, possibly due to a resemblance of the amino acid sequences of BCG and SARS-CoV-2, which might provoke cross-reactive T cells.5,6 Further studies are underway to determine whether the BCG vaccine is truly protective against COVID-19.
BCG vaccination scar reactivation presents as redness, swelling, or ulceration at the BCG injection site months to years after inoculation. Although erythema and induration of the BCG scar are not included in the diagnostic criteria of Kawasaki disease, likely due to variable vaccine requirements in different countries, these findings are largely recognized as specific for Kawasaki disease and present in approximately half of affected patients who received the BCG vaccine.2
Heat Shock Proteins—Heat shock proteins (HSPs) are produced by cells in response to stressors. The proposed mechanism of BCG vaccination scar reactivation is a cross-reaction between human homologue HSP 63 and Mycobacterium HSP 65, leading to hyperactivity of the immune system against BCG.7 There also are reports of reactivation of a BCG vaccination scar from measles infection and influenza vaccination.2,8,9 Most prior reports of BCG vaccination scar reactivation have been in pediatric patients; our patient is an adult who received the BCG vaccine more than 40 years ago.
Mechanism of Reactivation—The mechanism of BCG vaccination scar reactivation in our patient, who received the Moderna COVID-19 Vaccine, is unclear. Possible mechanisms include (1) release of HSP mediated by the COVID-19 vaccine, leading to an immune response at the BCG vaccine scar, or (2) another immune-mediated cross-reaction between BCG and the Moderna COVID-19 Vaccine mRNA nanoparticle or encoded spike protein antigen. It has been hypothesized that the BCG vaccine might offer some protection against COVID-19; this remains uncertain and is under further investigation.10 A recent retrospective cohort study showed that a BCG vaccination booster may decrease COVID-19 infection rates in higher-risk populations.11
Conclusion
We present a case of BCG vaccine scar reactivation occurring after a dose of the Moderna COVID-19 Vaccine, a likely underreported, self-limiting, cutaneous adverse effect of this mRNA vaccine.
The COVID-19 pandemic has resulted in notable morbidity and mortality worldwide. In December 2020, the US Food and Drug Administration issued an Emergency Use Authorization for 2 messenger RNA (mRNA) vaccines—produced by Pfizer-BioNTech and Moderna—for the prevention of COVID-19. Phase 3 trials of the vaccine developed by Moderna showed 94.1% efficacy at preventing COVID-19 after 2 doses.1
Common cutaneous adverse effects of the Moderna COVID-19 Vaccine include injection-site reactions, such as pain, induration, and erythema. Less frequently reported dermatologic adverse effects include diffuse bullous rash and hypersensitivity reactions.1 We report a case of reactivation of a BCG vaccination scar after the first dose of the Moderna COVID-19 Vaccine.
Case Report
A 48-year-old Asian man who was otherwise healthy presented with erythema, induration, and mild pruritus on the deltoid muscle of the left arm, near the scar from an earlier BCG vaccine, which he received at approximately 5 years of age when living in Taiwan. The patient received the first dose of the Moderna COVID-19 Vaccine approximately 5 to 7 cm distant from the BCG vaccination scar. One to 2 days after inoculation, the patient endorsed tenderness at the site of COVID-19 vaccination but denied systemic symptoms. He had never been given a diagnosis of COVID-19. His SARS-CoV-2 antibody status was unknown.
Eight days later, the patient noticed a well-defined, erythematous, indurated plaque with mild itchiness overlying and around the BCG vaccination scar that did not involve the COVID-19 vaccination site. The following day, the redness and induration became worse (Figure).
The patient was otherwise well. Vital signs were normal; there was no lymphadenopathy. The rash resolved without treatment over the next 4 days.
Comment
The BCG vaccine is an intradermal live attenuated virus vaccine used to prevent certain forms of tuberculosis and potentially other Mycobacterium infections. Although the vaccine is not routinely administered in the United States, it is part of the vaccination schedule in most countries, administered most often to newborns and infants. Administration of the BCG vaccine commonly results in mild localized erythema, swelling, and pain at the injection site. Most inoculated patients also develop an ulcer that heals with the characteristic BCG vaccination scar.2,3
There is evidence that the BCG vaccine can enhance the innate immune system response and might decrease the rate of infection by unrelated pathogens, including viruses.4 Several epidemiologic studies have suggested that the BCG vaccine might offer some protection against COVID-19, possibly due to a resemblance of the amino acid sequences of BCG and SARS-CoV-2, which might provoke cross-reactive T cells.5,6 Further studies are underway to determine whether the BCG vaccine is truly protective against COVID-19.
BCG vaccination scar reactivation presents as redness, swelling, or ulceration at the BCG injection site months to years after inoculation. Although erythema and induration of the BCG scar are not included in the diagnostic criteria of Kawasaki disease, likely due to variable vaccine requirements in different countries, these findings are largely recognized as specific for Kawasaki disease and present in approximately half of affected patients who received the BCG vaccine.2
Heat Shock Proteins—Heat shock proteins (HSPs) are produced by cells in response to stressors. The proposed mechanism of BCG vaccination scar reactivation is a cross-reaction between human homologue HSP 63 and Mycobacterium HSP 65, leading to hyperactivity of the immune system against BCG.7 There also are reports of reactivation of a BCG vaccination scar from measles infection and influenza vaccination.2,8,9 Most prior reports of BCG vaccination scar reactivation have been in pediatric patients; our patient is an adult who received the BCG vaccine more than 40 years ago.
Mechanism of Reactivation—The mechanism of BCG vaccination scar reactivation in our patient, who received the Moderna COVID-19 Vaccine, is unclear. Possible mechanisms include (1) release of HSP mediated by the COVID-19 vaccine, leading to an immune response at the BCG vaccine scar, or (2) another immune-mediated cross-reaction between BCG and the Moderna COVID-19 Vaccine mRNA nanoparticle or encoded spike protein antigen. It has been hypothesized that the BCG vaccine might offer some protection against COVID-19; this remains uncertain and is under further investigation.10 A recent retrospective cohort study showed that a BCG vaccination booster may decrease COVID-19 infection rates in higher-risk populations.11
Conclusion
We present a case of BCG vaccine scar reactivation occurring after a dose of the Moderna COVID-19 Vaccine, a likely underreported, self-limiting, cutaneous adverse effect of this mRNA vaccine.
- Baden LR, El Sahly HM, Essink B, et al; COVE Study Group. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N Engl J Med. 2020;384:403-416. doi:10.1056/NEJMoa2035389
- Muthuvelu S, Lim KS, Huang L-Y, et al. Measles infection causing bacillus Calmette-Guérin reactivation: a case report. BMC Pediatr. 2019;19:251. doi:10.1186/s12887-019-1635-z
- Fatima S, Kumari A, Das G, et al. Tuberculosis vaccine: a journey from BCG to present. Life Sci. 2020;252:117594. doi:10.1016/j.lfs.2020.117594
- O’Neill LAJ, Netea MG. BCG-induced trained immunity: can it offer protection against COVID-19? Nat Rev Immunol. 2020;20:335-337. doi:10.1038/s41577-020-0337-y
- Brooks NA, Puri A, Garg S, et al. The association of coronavirus disease-19 mortality and prior bacille Calmette-Guérin vaccination: a robust ecological analysis using unsupervised machine learning. Sci Rep. 2021;11:774. doi:10.1038/s41598-020-80787-z
- Tomita Y, Sato R, Ikeda T, et al. BCG vaccine may generate cross-reactive T-cells against SARS-CoV-2: in silico analyses and a hypothesis. Vaccine. 2020;38:6352-6356. doi:10.1016/j.vaccine.2020.08.045
- Lim KYY, Chua MC, Tan NWH, et al. Reactivation of BCG inoculation site in a child with febrile exanthema of 3 days duration: an early indicator of incomplete Kawasaki disease. BMJ Case Rep. 2020;13:E239648. doi:10.1136/bcr-2020-239648
- Kondo M, Goto H, Yamamoto S. First case of redness and erosion at bacillus Calmette-Guérin inoculation site after vaccination against influenza. J Dermatol. 2016;43:1229-1231. doi:10.1111/1346-8138.13365
- Chavarri-Guerra Y, Soto-Pérez-de-Celis E. Erythema at the bacillus Calmette-Guerin scar after influenza vaccination. Rev Soc Bras Med Trop. 2019;53:E20190390. doi:10.1590/0037-8682-0390-2019
- Fu W, Ho P-C, Liu C-L, et al. Reconcile the debate over protective effects of BCG vaccine against COVID-19. Sci Rep. 2021;11:8356. doi:10.1038/s41598-021-87731-9
- Amirlak L, Haddad R, Hardy JD, et al. Effectiveness of booster BCG vaccination in preventing COVID-19 infection. Hum Vaccin Immunother. 2021;17:3913-3915. doi:10.1080/21645515.2021.1956228
- Baden LR, El Sahly HM, Essink B, et al; COVE Study Group. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N Engl J Med. 2020;384:403-416. doi:10.1056/NEJMoa2035389
- Muthuvelu S, Lim KS, Huang L-Y, et al. Measles infection causing bacillus Calmette-Guérin reactivation: a case report. BMC Pediatr. 2019;19:251. doi:10.1186/s12887-019-1635-z
- Fatima S, Kumari A, Das G, et al. Tuberculosis vaccine: a journey from BCG to present. Life Sci. 2020;252:117594. doi:10.1016/j.lfs.2020.117594
- O’Neill LAJ, Netea MG. BCG-induced trained immunity: can it offer protection against COVID-19? Nat Rev Immunol. 2020;20:335-337. doi:10.1038/s41577-020-0337-y
- Brooks NA, Puri A, Garg S, et al. The association of coronavirus disease-19 mortality and prior bacille Calmette-Guérin vaccination: a robust ecological analysis using unsupervised machine learning. Sci Rep. 2021;11:774. doi:10.1038/s41598-020-80787-z
- Tomita Y, Sato R, Ikeda T, et al. BCG vaccine may generate cross-reactive T-cells against SARS-CoV-2: in silico analyses and a hypothesis. Vaccine. 2020;38:6352-6356. doi:10.1016/j.vaccine.2020.08.045
- Lim KYY, Chua MC, Tan NWH, et al. Reactivation of BCG inoculation site in a child with febrile exanthema of 3 days duration: an early indicator of incomplete Kawasaki disease. BMJ Case Rep. 2020;13:E239648. doi:10.1136/bcr-2020-239648
- Kondo M, Goto H, Yamamoto S. First case of redness and erosion at bacillus Calmette-Guérin inoculation site after vaccination against influenza. J Dermatol. 2016;43:1229-1231. doi:10.1111/1346-8138.13365
- Chavarri-Guerra Y, Soto-Pérez-de-Celis E. Erythema at the bacillus Calmette-Guerin scar after influenza vaccination. Rev Soc Bras Med Trop. 2019;53:E20190390. doi:10.1590/0037-8682-0390-2019
- Fu W, Ho P-C, Liu C-L, et al. Reconcile the debate over protective effects of BCG vaccine against COVID-19. Sci Rep. 2021;11:8356. doi:10.1038/s41598-021-87731-9
- Amirlak L, Haddad R, Hardy JD, et al. Effectiveness of booster BCG vaccination in preventing COVID-19 infection. Hum Vaccin Immunother. 2021;17:3913-3915. doi:10.1080/21645515.2021.1956228
Practice Points
- BCG vaccination scar reactivation is a potential benign, self-limited reaction in patients who receive the Moderna COVID-19 Vaccine.
- Symptoms of BCG vaccination scar reactivation, which is seen more commonly in children with Kawasaki disease, include redness, swelling, and ulceration.

3-year-old girl • fever • cervical lymphadenopathy • leukocytosis • Dx?
THE CASE
A previously healthy 3-year-old girl presented to the emergency department with 4 days of fever and 2 days of right-side neck pain. The maximum temperature at home was 103 °F. The patient was irritable and vomited once. There were no other apparent or reported symptoms.
The neck exam was notable for nonfluctuant, swollen, and tender lymph nodes on the right side. Her sclera and conjunctiva were clear, and her oropharynx was unremarkable. Lab work revealed leukocytosis, with a white blood cell (WBC) count of 15.5 × 103/µL (normal range, 4.0-10.0 × 103/µL). She was given one 20 cc/kg normal saline bolus, started on intravenous clindamycin for presumed cervical lymphadenitis, and admitted to the hospital.
On Day 2, the patient developed a fine maculopapular rash on her chest, abdomen, and back. She had spiking fevers—as high as 102.2 °F—despite being on antibiotics for more than 24 hours. The erythrocyte sedimentation rate (ESR) was 39 mm/h (0-20 mm/h), and C-reactive protein (CRP) was 71.4 mg/L (0.0-4.9 mg/L). Due to concern for abscess, a neck ultrasound was performed; it showed a chain of enlarged lymph nodes in the right neck (largest, 2.3 × 1.1 × 1.4 cm) and no abscess.
On Day 3, clindamycin was switched to intravenous ampicillin/sulbactam because a nasal swab for methicillin-resistant Staphylococcus aureus was negative. A swab for respiratory viral infections was also negative. The patient then developed notable facial swelling, bilateral bulbar conjunctival injection with limbic sparing, and swelling of her hands and feet.
THE DIAGNOSIS
By the end of Day 3, the patient’s lab studies demonstrated microcytic anemia and low albumin (2.5 g/dL), but no transaminitis, thrombocytosis, or sterile pyuria. An electrocardiogram was unremarkable. A pediatric echocardiogram revealed hyperemic coronaries, indicating inflammation. The coronary arteries were measured in the upper limits of normal, and the patient’s Z-scores were < 2.5. (A Z-score < 2 indicates no involvement, 2 to < 2.5 indicates dilation, and ≥ 2.5 indicates aneurysm abnormality.1) An ultrasound of the right upper quadrant revealed an enlarged/elongated gallbladder. The patient therefore met clinical criteria for Kawasaki disease.
DISCUSSION
Kawasaki disease is a self-limited vasculitis of childhood and the leading cause of acquired heart disease in children in developed countries.1 The annual incidence of Kawasaki disease in North America is about 25 cases per 100,000 children < 5 years of age.1 In the United States, incidence is highest in Asian and Pacific Islander populations (30 per 100,000) and is particularly high among those of Japanese ancestry (~200 per 100,000).2 Disease prevalence is also noteworthy in Non-Hispanic African American (17 per 100,000) and Hispanic (16 per 100,000) populations.2
Diagnosis of Kawasaki disease requires presence of fever lasting at least 5 days and at least 4 of the following: bilateral bulbar conjunctival injection, oral mucous membrane changes (erythematous or cracked lips, erythematous pharynx, strawberry tongue), peripheral extremity changes (erythema of palms or soles, edema of hands or feet, and/or periungual desquamation), diffuse maculopapular rash, and cervical lymphadenopathy (≥ 1.5 cm, often unilateral). If ≥ 4 criteria are met, Kawasaki disease can be diagnosed on the fourth day of illness.1
Continue to: Laboratory findings suggesting...
Laboratory findings suggesting Kawasaki disease include a WBC count ≥ 15,000/mcL, normocytic, normochromic anemia, platelets ≥ 450,000/mcL after 7 days of illness, sterile pyuria (≥ 10 WBCs/high-power field), serum alanine aminotransferase level > 50 U/L, and serum albumin ≤ 3 g/dL.
Cardiac abnormalities are not included in the diagnostic criteria for Kawasaki disease but provide evidence in cases of incomplete Kawasaki disease if ≥ 4 criteria are not met and there is strong clinical suspicion.1 Incomplete Kawasaki disease should be considered in a patient with a CRP level ≥ 3 mg/dL and/or ESR ≥ 40 mm/h, ≥ 3 supplemental laboratory criteria, or a positive echocardiogram.1
Ultrasound imaging may reveal cervical lymph nodes resembling a “cluster of grapes.”3 The case patient’s imaging showed a “chain of enlarged lymph nodes.” She likely had gallbladder “hydrops” due to its increased longitudinal and horizontal diameter and lack of other anatomic changes.4
Prompt treatment is essential
Treatment for complete and incomplete Kawasaki disease is a single high dose of intravenous immunoglobulin (IVIG) along with aspirin. Patients meeting criteria should be treated as soon as the diagnosis is established.5 A single high dose of IVIG (2 g/kg), administered over 10 to 12 hours, should be initiated within 5 to 10 days of disease onset. Administering IVIG in the acute phase of Kawasaki disease reduces the prevalence of coronary artery abnormalities.6 Corticosteroids may be used as adjunctive therapy for patients with high risk of IVIG resistance.1,7-9
Our patient was not deemed to be at high risk for IVIG resistance (Non-Japanese patient, age at fever onset > 6 months, absence of coronary artery aneurysm9) and was administered IVIG on Day 4. She was also given moderate-dose aspirin, then later transitioned to low-dose aspirin. The patient’s fevers improved, she was less irritable, and she had periods of playfulness. Physical exam then showed erythematous and cracked lips with peeling skin.
Continue to: The patient was discharged...
The patient was discharged home on Day 8, after her fever resolved, with instructions to continue low-dose aspirin and to obtain a repeat echocardiogram, gallbladder ultrasound, and lab work in 2 weeks. At her follow-up appointment, periungual desquamation was noted, and ultrasound showed continued enlarged/elongated gallbladder. A repeat echocardiogram was not available. Overall, the patient recovered from Kawasaki disease after therapeutic intervention.
THE TAKEAWAY
Kawasaki disease can be relatively rare in North American populations, but it is important for physicians to be able to recognize and treat it. Untreated children have a 25% chance of developing coronary artery aneurysms.1,10,11 Early treatment with IVIG can decrease risk to 5%, resulting in an excellent medium- to long-term prognosis for patients.10 Thorough physical examination and an appropriate degree of clinical suspicion was key in this case of Kawasaki disease.
Taisha Doo, MD, 1401 Madison Street, Suite #100, Seattle, WA 98104; [email protected]
1. McCrindle BW, Rowley AH, Newburger JW, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a scientific statement for health professionals from the American Heart Association. Circulation. 2017;135:e927-e999. doi: 10.1161/CIR.0000000000000484
2. Holman RC, Belay ED, Christensen KY, et al. Hospitalizations for Kawasaki syndrome among children in the United States, 1997-2007. Pediatr Infect Dis. 2010;29:483-488. doi: 10.1097/INF.0b013e3181cf8705
3. Tashiro N, Matsubara T, Uchida M, et al. Ultrasonographic evaluation of cervical lymph nodes in Kawasaki disease. Pediatrics. 2002;109:e77. doi: 10.1542/peds.109.5.e77
4. Chen CJ, Huang FC, Taio MM, et al. Sonographic gallbladder abnormality is associated with intravenous immunoglobulin resistance in Kawasaki disease. Scientific World J. 2012;2012:485758. doi: 10.1100/2012/485758
5. Dominguez SR, Anderson MS, El-Adawy M, et al. Preventing coronary artery abnormalities: a need for earlier diagnosis and treatment of Kawasaki disease. Pediatr Infect Dis J. 2012;31:1217-1220. doi: 10.1097/INF.0b013e318266bcf9
6. Kuo HC. Preventing coronary artery lesions in Kawasaki disease. Biomed J. 2017;40:141-146. doi: 10.1016/j.bj.2017.04.002
7. Chen S, Dong Y, Yin Y, et al. Intravenous immunoglobulin plus corticosteroid to prevent coronary artery abnormalities in Kawasaki disease: a meta-analysis. Heart. 2013;99:76-82. doi: 10.1136/heartjnl-2012-302126
8. Chantasiriwan N, Silvilairat S, Makonkawkeyoon K, et al. Predictors of intravenous immunoglobulin resistance and coronary artery aneurysm in patients with Kawasaki disease, Paediatr Int Child Health. 2018;38:209-212. doi: 10.1080/20469047.2018.1471381
9. Son MBF, Gauvreau K, Tremoulet AH, et al. Risk model development and validation for prediction of coronary artery aneurysms in Kawasaki disease in a North American population. J Am Heart Assoc. 2019;8:e011319. doi: 10.1161/JAHA.118.011319
10. de La Harpe M, di Bernardo S, Hofer M, et al. Thirty years of Kawasaki disease: a single-center study at the University Hospital of Lausanne. Front Pediatr. 2019;7:11. doi: 10.3389/fped.2019.00011
11. Newburger JW, Takahashi M, Gerber MA, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Circulation. 2004;110:2747-2771. doi: 10.1161/01.CIR.0000145143.19711.78
THE CASE
A previously healthy 3-year-old girl presented to the emergency department with 4 days of fever and 2 days of right-side neck pain. The maximum temperature at home was 103 °F. The patient was irritable and vomited once. There were no other apparent or reported symptoms.
The neck exam was notable for nonfluctuant, swollen, and tender lymph nodes on the right side. Her sclera and conjunctiva were clear, and her oropharynx was unremarkable. Lab work revealed leukocytosis, with a white blood cell (WBC) count of 15.5 × 103/µL (normal range, 4.0-10.0 × 103/µL). She was given one 20 cc/kg normal saline bolus, started on intravenous clindamycin for presumed cervical lymphadenitis, and admitted to the hospital.
On Day 2, the patient developed a fine maculopapular rash on her chest, abdomen, and back. She had spiking fevers—as high as 102.2 °F—despite being on antibiotics for more than 24 hours. The erythrocyte sedimentation rate (ESR) was 39 mm/h (0-20 mm/h), and C-reactive protein (CRP) was 71.4 mg/L (0.0-4.9 mg/L). Due to concern for abscess, a neck ultrasound was performed; it showed a chain of enlarged lymph nodes in the right neck (largest, 2.3 × 1.1 × 1.4 cm) and no abscess.
On Day 3, clindamycin was switched to intravenous ampicillin/sulbactam because a nasal swab for methicillin-resistant Staphylococcus aureus was negative. A swab for respiratory viral infections was also negative. The patient then developed notable facial swelling, bilateral bulbar conjunctival injection with limbic sparing, and swelling of her hands and feet.
THE DIAGNOSIS
By the end of Day 3, the patient’s lab studies demonstrated microcytic anemia and low albumin (2.5 g/dL), but no transaminitis, thrombocytosis, or sterile pyuria. An electrocardiogram was unremarkable. A pediatric echocardiogram revealed hyperemic coronaries, indicating inflammation. The coronary arteries were measured in the upper limits of normal, and the patient’s Z-scores were < 2.5. (A Z-score < 2 indicates no involvement, 2 to < 2.5 indicates dilation, and ≥ 2.5 indicates aneurysm abnormality.1) An ultrasound of the right upper quadrant revealed an enlarged/elongated gallbladder. The patient therefore met clinical criteria for Kawasaki disease.
DISCUSSION
Kawasaki disease is a self-limited vasculitis of childhood and the leading cause of acquired heart disease in children in developed countries.1 The annual incidence of Kawasaki disease in North America is about 25 cases per 100,000 children < 5 years of age.1 In the United States, incidence is highest in Asian and Pacific Islander populations (30 per 100,000) and is particularly high among those of Japanese ancestry (~200 per 100,000).2 Disease prevalence is also noteworthy in Non-Hispanic African American (17 per 100,000) and Hispanic (16 per 100,000) populations.2
Diagnosis of Kawasaki disease requires presence of fever lasting at least 5 days and at least 4 of the following: bilateral bulbar conjunctival injection, oral mucous membrane changes (erythematous or cracked lips, erythematous pharynx, strawberry tongue), peripheral extremity changes (erythema of palms or soles, edema of hands or feet, and/or periungual desquamation), diffuse maculopapular rash, and cervical lymphadenopathy (≥ 1.5 cm, often unilateral). If ≥ 4 criteria are met, Kawasaki disease can be diagnosed on the fourth day of illness.1
Continue to: Laboratory findings suggesting...
Laboratory findings suggesting Kawasaki disease include a WBC count ≥ 15,000/mcL, normocytic, normochromic anemia, platelets ≥ 450,000/mcL after 7 days of illness, sterile pyuria (≥ 10 WBCs/high-power field), serum alanine aminotransferase level > 50 U/L, and serum albumin ≤ 3 g/dL.
Cardiac abnormalities are not included in the diagnostic criteria for Kawasaki disease but provide evidence in cases of incomplete Kawasaki disease if ≥ 4 criteria are not met and there is strong clinical suspicion.1 Incomplete Kawasaki disease should be considered in a patient with a CRP level ≥ 3 mg/dL and/or ESR ≥ 40 mm/h, ≥ 3 supplemental laboratory criteria, or a positive echocardiogram.1
Ultrasound imaging may reveal cervical lymph nodes resembling a “cluster of grapes.”3 The case patient’s imaging showed a “chain of enlarged lymph nodes.” She likely had gallbladder “hydrops” due to its increased longitudinal and horizontal diameter and lack of other anatomic changes.4
Prompt treatment is essential
Treatment for complete and incomplete Kawasaki disease is a single high dose of intravenous immunoglobulin (IVIG) along with aspirin. Patients meeting criteria should be treated as soon as the diagnosis is established.5 A single high dose of IVIG (2 g/kg), administered over 10 to 12 hours, should be initiated within 5 to 10 days of disease onset. Administering IVIG in the acute phase of Kawasaki disease reduces the prevalence of coronary artery abnormalities.6 Corticosteroids may be used as adjunctive therapy for patients with high risk of IVIG resistance.1,7-9
Our patient was not deemed to be at high risk for IVIG resistance (Non-Japanese patient, age at fever onset > 6 months, absence of coronary artery aneurysm9) and was administered IVIG on Day 4. She was also given moderate-dose aspirin, then later transitioned to low-dose aspirin. The patient’s fevers improved, she was less irritable, and she had periods of playfulness. Physical exam then showed erythematous and cracked lips with peeling skin.
Continue to: The patient was discharged...
The patient was discharged home on Day 8, after her fever resolved, with instructions to continue low-dose aspirin and to obtain a repeat echocardiogram, gallbladder ultrasound, and lab work in 2 weeks. At her follow-up appointment, periungual desquamation was noted, and ultrasound showed continued enlarged/elongated gallbladder. A repeat echocardiogram was not available. Overall, the patient recovered from Kawasaki disease after therapeutic intervention.
THE TAKEAWAY
Kawasaki disease can be relatively rare in North American populations, but it is important for physicians to be able to recognize and treat it. Untreated children have a 25% chance of developing coronary artery aneurysms.1,10,11 Early treatment with IVIG can decrease risk to 5%, resulting in an excellent medium- to long-term prognosis for patients.10 Thorough physical examination and an appropriate degree of clinical suspicion was key in this case of Kawasaki disease.
Taisha Doo, MD, 1401 Madison Street, Suite #100, Seattle, WA 98104; [email protected]
THE CASE
A previously healthy 3-year-old girl presented to the emergency department with 4 days of fever and 2 days of right-side neck pain. The maximum temperature at home was 103 °F. The patient was irritable and vomited once. There were no other apparent or reported symptoms.
The neck exam was notable for nonfluctuant, swollen, and tender lymph nodes on the right side. Her sclera and conjunctiva were clear, and her oropharynx was unremarkable. Lab work revealed leukocytosis, with a white blood cell (WBC) count of 15.5 × 103/µL (normal range, 4.0-10.0 × 103/µL). She was given one 20 cc/kg normal saline bolus, started on intravenous clindamycin for presumed cervical lymphadenitis, and admitted to the hospital.
On Day 2, the patient developed a fine maculopapular rash on her chest, abdomen, and back. She had spiking fevers—as high as 102.2 °F—despite being on antibiotics for more than 24 hours. The erythrocyte sedimentation rate (ESR) was 39 mm/h (0-20 mm/h), and C-reactive protein (CRP) was 71.4 mg/L (0.0-4.9 mg/L). Due to concern for abscess, a neck ultrasound was performed; it showed a chain of enlarged lymph nodes in the right neck (largest, 2.3 × 1.1 × 1.4 cm) and no abscess.
On Day 3, clindamycin was switched to intravenous ampicillin/sulbactam because a nasal swab for methicillin-resistant Staphylococcus aureus was negative. A swab for respiratory viral infections was also negative. The patient then developed notable facial swelling, bilateral bulbar conjunctival injection with limbic sparing, and swelling of her hands and feet.
THE DIAGNOSIS
By the end of Day 3, the patient’s lab studies demonstrated microcytic anemia and low albumin (2.5 g/dL), but no transaminitis, thrombocytosis, or sterile pyuria. An electrocardiogram was unremarkable. A pediatric echocardiogram revealed hyperemic coronaries, indicating inflammation. The coronary arteries were measured in the upper limits of normal, and the patient’s Z-scores were < 2.5. (A Z-score < 2 indicates no involvement, 2 to < 2.5 indicates dilation, and ≥ 2.5 indicates aneurysm abnormality.1) An ultrasound of the right upper quadrant revealed an enlarged/elongated gallbladder. The patient therefore met clinical criteria for Kawasaki disease.
DISCUSSION
Kawasaki disease is a self-limited vasculitis of childhood and the leading cause of acquired heart disease in children in developed countries.1 The annual incidence of Kawasaki disease in North America is about 25 cases per 100,000 children < 5 years of age.1 In the United States, incidence is highest in Asian and Pacific Islander populations (30 per 100,000) and is particularly high among those of Japanese ancestry (~200 per 100,000).2 Disease prevalence is also noteworthy in Non-Hispanic African American (17 per 100,000) and Hispanic (16 per 100,000) populations.2
Diagnosis of Kawasaki disease requires presence of fever lasting at least 5 days and at least 4 of the following: bilateral bulbar conjunctival injection, oral mucous membrane changes (erythematous or cracked lips, erythematous pharynx, strawberry tongue), peripheral extremity changes (erythema of palms or soles, edema of hands or feet, and/or periungual desquamation), diffuse maculopapular rash, and cervical lymphadenopathy (≥ 1.5 cm, often unilateral). If ≥ 4 criteria are met, Kawasaki disease can be diagnosed on the fourth day of illness.1
Continue to: Laboratory findings suggesting...
Laboratory findings suggesting Kawasaki disease include a WBC count ≥ 15,000/mcL, normocytic, normochromic anemia, platelets ≥ 450,000/mcL after 7 days of illness, sterile pyuria (≥ 10 WBCs/high-power field), serum alanine aminotransferase level > 50 U/L, and serum albumin ≤ 3 g/dL.
Cardiac abnormalities are not included in the diagnostic criteria for Kawasaki disease but provide evidence in cases of incomplete Kawasaki disease if ≥ 4 criteria are not met and there is strong clinical suspicion.1 Incomplete Kawasaki disease should be considered in a patient with a CRP level ≥ 3 mg/dL and/or ESR ≥ 40 mm/h, ≥ 3 supplemental laboratory criteria, or a positive echocardiogram.1
Ultrasound imaging may reveal cervical lymph nodes resembling a “cluster of grapes.”3 The case patient’s imaging showed a “chain of enlarged lymph nodes.” She likely had gallbladder “hydrops” due to its increased longitudinal and horizontal diameter and lack of other anatomic changes.4
Prompt treatment is essential
Treatment for complete and incomplete Kawasaki disease is a single high dose of intravenous immunoglobulin (IVIG) along with aspirin. Patients meeting criteria should be treated as soon as the diagnosis is established.5 A single high dose of IVIG (2 g/kg), administered over 10 to 12 hours, should be initiated within 5 to 10 days of disease onset. Administering IVIG in the acute phase of Kawasaki disease reduces the prevalence of coronary artery abnormalities.6 Corticosteroids may be used as adjunctive therapy for patients with high risk of IVIG resistance.1,7-9
Our patient was not deemed to be at high risk for IVIG resistance (Non-Japanese patient, age at fever onset > 6 months, absence of coronary artery aneurysm9) and was administered IVIG on Day 4. She was also given moderate-dose aspirin, then later transitioned to low-dose aspirin. The patient’s fevers improved, she was less irritable, and she had periods of playfulness. Physical exam then showed erythematous and cracked lips with peeling skin.
Continue to: The patient was discharged...
The patient was discharged home on Day 8, after her fever resolved, with instructions to continue low-dose aspirin and to obtain a repeat echocardiogram, gallbladder ultrasound, and lab work in 2 weeks. At her follow-up appointment, periungual desquamation was noted, and ultrasound showed continued enlarged/elongated gallbladder. A repeat echocardiogram was not available. Overall, the patient recovered from Kawasaki disease after therapeutic intervention.
THE TAKEAWAY
Kawasaki disease can be relatively rare in North American populations, but it is important for physicians to be able to recognize and treat it. Untreated children have a 25% chance of developing coronary artery aneurysms.1,10,11 Early treatment with IVIG can decrease risk to 5%, resulting in an excellent medium- to long-term prognosis for patients.10 Thorough physical examination and an appropriate degree of clinical suspicion was key in this case of Kawasaki disease.
Taisha Doo, MD, 1401 Madison Street, Suite #100, Seattle, WA 98104; [email protected]
1. McCrindle BW, Rowley AH, Newburger JW, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a scientific statement for health professionals from the American Heart Association. Circulation. 2017;135:e927-e999. doi: 10.1161/CIR.0000000000000484
2. Holman RC, Belay ED, Christensen KY, et al. Hospitalizations for Kawasaki syndrome among children in the United States, 1997-2007. Pediatr Infect Dis. 2010;29:483-488. doi: 10.1097/INF.0b013e3181cf8705
3. Tashiro N, Matsubara T, Uchida M, et al. Ultrasonographic evaluation of cervical lymph nodes in Kawasaki disease. Pediatrics. 2002;109:e77. doi: 10.1542/peds.109.5.e77
4. Chen CJ, Huang FC, Taio MM, et al. Sonographic gallbladder abnormality is associated with intravenous immunoglobulin resistance in Kawasaki disease. Scientific World J. 2012;2012:485758. doi: 10.1100/2012/485758
5. Dominguez SR, Anderson MS, El-Adawy M, et al. Preventing coronary artery abnormalities: a need for earlier diagnosis and treatment of Kawasaki disease. Pediatr Infect Dis J. 2012;31:1217-1220. doi: 10.1097/INF.0b013e318266bcf9
6. Kuo HC. Preventing coronary artery lesions in Kawasaki disease. Biomed J. 2017;40:141-146. doi: 10.1016/j.bj.2017.04.002
7. Chen S, Dong Y, Yin Y, et al. Intravenous immunoglobulin plus corticosteroid to prevent coronary artery abnormalities in Kawasaki disease: a meta-analysis. Heart. 2013;99:76-82. doi: 10.1136/heartjnl-2012-302126
8. Chantasiriwan N, Silvilairat S, Makonkawkeyoon K, et al. Predictors of intravenous immunoglobulin resistance and coronary artery aneurysm in patients with Kawasaki disease, Paediatr Int Child Health. 2018;38:209-212. doi: 10.1080/20469047.2018.1471381
9. Son MBF, Gauvreau K, Tremoulet AH, et al. Risk model development and validation for prediction of coronary artery aneurysms in Kawasaki disease in a North American population. J Am Heart Assoc. 2019;8:e011319. doi: 10.1161/JAHA.118.011319
10. de La Harpe M, di Bernardo S, Hofer M, et al. Thirty years of Kawasaki disease: a single-center study at the University Hospital of Lausanne. Front Pediatr. 2019;7:11. doi: 10.3389/fped.2019.00011
11. Newburger JW, Takahashi M, Gerber MA, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Circulation. 2004;110:2747-2771. doi: 10.1161/01.CIR.0000145143.19711.78
1. McCrindle BW, Rowley AH, Newburger JW, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a scientific statement for health professionals from the American Heart Association. Circulation. 2017;135:e927-e999. doi: 10.1161/CIR.0000000000000484
2. Holman RC, Belay ED, Christensen KY, et al. Hospitalizations for Kawasaki syndrome among children in the United States, 1997-2007. Pediatr Infect Dis. 2010;29:483-488. doi: 10.1097/INF.0b013e3181cf8705
3. Tashiro N, Matsubara T, Uchida M, et al. Ultrasonographic evaluation of cervical lymph nodes in Kawasaki disease. Pediatrics. 2002;109:e77. doi: 10.1542/peds.109.5.e77
4. Chen CJ, Huang FC, Taio MM, et al. Sonographic gallbladder abnormality is associated with intravenous immunoglobulin resistance in Kawasaki disease. Scientific World J. 2012;2012:485758. doi: 10.1100/2012/485758
5. Dominguez SR, Anderson MS, El-Adawy M, et al. Preventing coronary artery abnormalities: a need for earlier diagnosis and treatment of Kawasaki disease. Pediatr Infect Dis J. 2012;31:1217-1220. doi: 10.1097/INF.0b013e318266bcf9
6. Kuo HC. Preventing coronary artery lesions in Kawasaki disease. Biomed J. 2017;40:141-146. doi: 10.1016/j.bj.2017.04.002
7. Chen S, Dong Y, Yin Y, et al. Intravenous immunoglobulin plus corticosteroid to prevent coronary artery abnormalities in Kawasaki disease: a meta-analysis. Heart. 2013;99:76-82. doi: 10.1136/heartjnl-2012-302126
8. Chantasiriwan N, Silvilairat S, Makonkawkeyoon K, et al. Predictors of intravenous immunoglobulin resistance and coronary artery aneurysm in patients with Kawasaki disease, Paediatr Int Child Health. 2018;38:209-212. doi: 10.1080/20469047.2018.1471381
9. Son MBF, Gauvreau K, Tremoulet AH, et al. Risk model development and validation for prediction of coronary artery aneurysms in Kawasaki disease in a North American population. J Am Heart Assoc. 2019;8:e011319. doi: 10.1161/JAHA.118.011319
10. de La Harpe M, di Bernardo S, Hofer M, et al. Thirty years of Kawasaki disease: a single-center study at the University Hospital of Lausanne. Front Pediatr. 2019;7:11. doi: 10.3389/fped.2019.00011
11. Newburger JW, Takahashi M, Gerber MA, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Circulation. 2004;110:2747-2771. doi: 10.1161/01.CIR.0000145143.19711.78

Telescoping Stents to Maintain a 3-Way Patency of the Airway
There are several malignant and nonmalignant conditions that can lead to central airway obstruction (CAO) resulting in lobar collapse. The clinical consequences range from significant dyspnea to respiratory failure. Airway stenting has been used to maintain patency of obstructed airways and relieve symptoms. Before lung cancer screening became more common, approximately 10% of lung cancers at presentation had evidence of CAO.1
On occasion, an endobronchial malignancy involves the right mainstem (RMS) bronchus near the orifice of the right upper lobe (RUL).2 Such strategically located lesions pose a challenge to relieve the RMS obstruction through stenting, securing airway patency into the bronchus intermedius (BI) while avoiding obstruction of the RUL bronchus. The use of endobronchial silicone stents, hybrid covered stents, as well as self-expanding metal stents (SEMS) is an established mode of relieving CAO due to malignant disease.3 We reviewed the literature for approaches that were available before and after the date of the index case reported here.
Case Presentation
A 65-year-old veteran with a history of smoking presented to a US Department of Veterans Affairs Medical Center (VAMC) in 2011, with hemoptysis of 2-week duration. Computed tomography (CT) of the chest revealed a 5.3 × 4.2 × 6.5 cm right mediastinal mass and a 3.0 × 2.8 × 3 cm right hilar mass. Flexible bronchoscopy revealed > 80% occlusion of the RMS and BI due to a medially located mass sparing the RUL orifice, which was patent (Figure 1). Airways distal to the BI were free of disease. Endobronchial biopsies revealed poorly differentiated non-small cell carcinoma of the lung. The patient was referred to the interventional pulmonary service for further airway management.
Under general anesthesia and through a size-9 endotracheal tube, piecemeal debulking of the mass using a cryoprobe was performed. Argon photocoagulation (APC) was used to control bleeding. Balloon bronchoplasty was performed next with pulmonary Boston Scientific CRE balloon at the BI and the RMS bronchus. Under fluoroscopic guidance, a 12 × 30 mm self-expanding hybrid Merit Medical AERO stent was placed distally into the BI. Next, a 14 × 30 mm AERO stent was placed proximally in the RMS bronchus with its distal end telescoped into the smaller distal stent for a distance of 3 to 4 mm at a slanted angle. The overlap was deliberately performed at the level of RUL takeoff. Forcing the distal end of the proximal larger stent into a smaller stent created mechanical stress. The angled alignment channeled this mechanical stress so that the distal end of the proximal stent flared open laterally into the RUL orifice to allow for ventilation (Figure 2). On follow-up 6 months later, all 3 airways remained patent with stents in place (Figure 3).
The patient returned to the VAMC and underwent chemotherapy with carboplatin and paclitaxel cycles that were completed in May 2012, as well as completing 6300 centigray (cGy) of radiation to the area. This led to regression of the tumor permitting removal of the proximal stent in October 2012. Unfortunately, upon follow-up in July 2013, a hypermetabolic lesion in the right upper posterior chest was noted to be eroding the third rib. Biopsy proved it to be poorly differentiated non-small cell lung cancer. Palliative external beam radiation was used to treat this lesion with a total of 3780 cGy completed by the end of August 2013.
Sadly, the patient was admitted later in 2013 with worsening cough and shortness of breath. Chest and abdominal CTs showed an increase in the size of the right apical mass, and mediastinal lymphadenopathy, as well as innumerable nodules in the left lung. The mass had recurred and extended distal to the stent into the lower and middle lobes. New liver nodule and lytic lesion within left ischial tuberosity, T12, L1, and S1 vertebral bodies were noted. The pulmonary service reached out to us via email and we recommended either additional chemoradiotherapy or palliative care. At that point the tumor was widespread and resistant to therapy. It extended beyond the central airways making airway debulking futile. Stents are palliative in nature and we believed that the initial stenting allowed the patient to get chemoradiation by improving functional status through preventing collapse of the right lung. As a result, the patient had about 19 months of a remission period with quality of life. The patient ultimately died under the care of palliative care in inpatient hospice setting.
Literature Review
A literature review revealed multiple approaches to preserving a 3-way patent airway at the takeoff of the RUL (Table). One approach to alleviating such an obstruction favors placing a straight silicone stent from the RMS into the BI, closing off the orifice of the RUL (Figure 4A).4 However, this entails sacrificing ventilation of the RUL. An alternative suggested by Peled and colleagues was carried out successfully in 3 patients. After placing a stent to relieve the obstruction, a Nd:YAG laser is used to create a window in the stent in proximity to the RUL orifice, which allows preservation or ventilations to the RUL (Figure 4B).5
A third effective approach utilizes silicone Y stents, which are usually employed for relief of obstruction at the level of the main carina.6,7 Instead of deploying them at the main carina, they would be deployed at the secondary carina, which the RUL makes with the BI, often with customized cutting for adjustment of the stent limbs to the appropriate size of the RUL and BI (Figure 4C). This approach has been successfully used to maintain RUL ventilation.2
A fourth technique involves using an Oki stent, a dedicated bifurcated silicone stent, which was first described in 2013. It is designed for the RMS bronchus around the RUL and BI bifurcation, enabling the stent to maintain airway patency in the right lung without affecting the trachea and carina (Figure 4D). The arm located in the RUL prevents migration.8 A fifth technique involves deploying a precisely selected Oki stent specially modified based on a printed 3-dimensional (3D) model of the airways after computer-aided simulation.9A sixth technique employs de novo custom printing stents based on 3D models of the tracheobronchial tree constructed based on CT imaging. This approach creates more accurately fitting stents.1

Discussion
The RUL contributes roughly 5 to 10% of the total oxygenation capacity of the lung.10 In patients with lung cancer and limited pulmonary reserve, preserving ventilation to the RUL can be clinically important. The chosen method to relieve endobronchial obstruction depends on several variables, including expertise, ability of the patient to undergo general anesthesia for rigid or flexible bronchoscopy, stent availability, and airway anatomy.
This case illustrates a new method to deal with lesions close to the RUL orifice. This maneuver may not be possible with all types of stents. AERO stents are fully covered (Figure 4E). In contrast, stents that are uncovered at both distal ends, such as a Boston Scientific Ultraflex stent, may not be adequate for such a maneuver. Intercalating uncovered ends of SEMS may allow for tumor in-growth through the uncovered metal mesh near the RUL orifice and may paradoxically compromise both the RUL and BI. The diameter of AERO stents is slightly larger at its ends.11 This helps prevent migration, which in this case maintained the crucial overlap of the stents. On the other hand, use of AERO stents may be associated with a higher risk of infection.12 Precise measurements of the airway diameter are essential given the difference in internal and external stent diameter with silicone stents.
Silicone stents migrate more readily than SEMS and may not be well suited for the procedure we performed. In our case, we wished to maintain ventilation for the RUL; hence, we elected not to bypass it with a silicone stent. We did not have access to a YAG. Moreover, laser carries more energy than APC. Nd:YAG laser has been reported to cause airway fire when used with silicone stents.13 Several authors have reported the use of silicone Y stents at the primary or secondary carina to preserve luminal patency.6,7 Airway anatomy and the angle of the Y may require modification of these stents prior to their use. Cutting stents may compromise their integrity. The bifurcating limb prevents migration which can be a significant concern with the tubular silicone stents. An important consideration for patients in advanced stages of malignancy is that placement of such stent requires undergoing general anesthesia and rigid bronchoscopy, unlike with AERO and metal stents that can be deployed with fiberoptic bronchoscopy under moderate sedation. As such, we did not elect to use a silicone Y stent. Accumulation of secretions or formation of granulation tissue at the orifices can result in recurrence of obstruction.14
Advances in 3D printing seem to be the future of customized airway stenting. This could help clinicians overcome the challenges of improperly sized stents and distorted airway anatomy. Cases have reported successful use of 3D-printed patient-specific airway prostheses.15,16 However, their use is not common practice, as there is a limited amount of materials that are flexible, biocompatible, and approved by the US Food and Drug Administration (FDA) for medical use. Infection control is another layer of consideration in such stents. Standardization of materials and regulation of personalized devices and their cleansing protocols is neccesary.17 At the time of this case, Oki stents and 3D printing were not available in the market. This report provides a viable alternative to use AERO stents for this maneuver.
Conclusions
Patients presenting with malignant CAO near the RUL require a personalized approach to treatment, considering their overall health, functional status, nature and location of CAO, and degree of symptoms. Once a decision is made to stent the airway, careful assessment of airway anatomy, delineation of obstruction, available expertise, and types of stents available needs to be made to preserve ventilation to the nondiseased RUL. Airway stents are expensive and need to be used wisely for palliation and allowing for a quality life while the patient receives more definitive targeted therapy.
Acknowledgments
The authors would like to gratefully acknowledge Dr Jenny Kim, who referred the patient to the interventional service and helped obtain consent for publishing the case.
1. Criner GJ, Eberhardt R, Fernandez-Bussy S, et al. Interventional bronchoscopy. Am J Respir Crit Care Med. 2020;202(1):29-50. doi:10.1164/rccm.201907-1292SO
2. Oki M, Saka H, Kitagawa C, Kogure Y. Silicone y-stent placement on the carina between bronchus to the right upper lobe and bronchus intermedius. Ann Thorac Surg. 2009;87(3):971-974. doi:10.1016/j.athoracsur.2008.06.049
3. Ernst A, Feller-Kopman D, Becker HD, Mehta AC. Central airway obstruction. Am J Respir Crit Care Med. 2004;169(12):1278-1297. doi:10.1164/rccm.200210-1181SO
4. Liu Y-H, Wu Y-C, Hsieh M-J, Ko P-J. Straight bronchial stent placement across the right upper lobe bronchus: A simple alternative for the management of airway obstruction around the carina and right main bronchus. J Thorac Cardiovasc Surg. 2011;141(1):303-305.e1.doi:10.1016/j.jtcvs.2010.06.015
5. Peled N, Shitrit D, Bendayan D, Kramer MR. Right upper lobe ‘window’ in right main bronchus stenting. Eur J Cardiothorac Surg. 2006;30(4):680-682. doi:10.1016/j.ejcts.2006.07.020
6. Dumon J-F, Dumon MC. Dumon-Novatech Y-stents: a four-year experience with 50 tracheobronchial tumors involving the carina. J Bronchol. 2000;7(1):26-32 doi:10.1097/00128594-200007000-00005
7. Dutau H, Toutblanc B, Lamb C, Seijo L. Use of the Dumon Y-stent in the management of malignant disease involving the carina: a retrospective review of 86 patients. Chest. 2004;126(3):951-958. doi:10.1378/chest.126.3.951
8. Dalar L, Abul Y. Safety and efficacy of Oki stenting used to treat obstructions in the right mainstem bronchus. J Bronchol Interv Pulmonol. 2018;25(3):212-217. doi:10.1097/LBR.0000000000000486
9. Guibert N, Moreno B, Plat G, Didier A, Mazieres J, Hermant C. Stenting of complex malignant central-airway obstruction guided by a three-dimensional printed model of the airways. Ann Thorac Surg. 2017;103(4):e357-e359. doi:10.1016/j.athoracsur.2016.09.082
10. Win T, Tasker AD, Groves AM, et al. Ventilation-perfusion scintigraphy to predict postoperative pulmonary function in lung cancer patients undergoing pneumonectomy. AJR Am J Roentgenol. 2006;187(5):1260-1265. doi:10.2214/AJR.04.1973
11. Mehta AC. AERO self-expanding hybrid stent for airway stenosis. Expert Rev Med Devices. 2008;5(5):553-557. doi:10.1586/17434440.5.5.553
12. Ost DE, Shah AM, Lei X, et al. Respiratory infections increase the risk of granulation tissue formation following airway stenting in patients with malignant airway obstruction. Chest. 2012;141(6):1473-1481. doi:10.1378/chest.11-2005
13. Scherer TA. Nd-YAG laser ignition of silicone endobronchial stents. Chest. 2000;117(5):1449-1454. doi:10.1378/chest.117.5.1449
14. Folch E, Keyes C. Airway stents. Ann Cardiothorac Surg. 2018;7(2):273-283. doi:10.21037/acs.2018.03.08
15. Cheng GZ, Folch E, Brik R, et al. Three-dimensional modeled T-tube design and insertion in a patient with tracheal dehiscence. Chest. 2015;148(4):e106-e108. doi:10.1378/chest.15-0240
16. Tam MD, Laycock SD, Jayne D, Babar J, Noble B. 3-D printouts of the tracheobronchial tree generated from CT images as an aid to management in a case of tracheobronchial chondromalacia caused by relapsing polychondritis. J Radiol Case Rep. 2013;7(8):34-43. Published 2013 Aug 1. doi:10.3941/jrcr.v7i8.1390
17. Alraiyes AH, Avasarala SK, Machuzak MS, Gildea TR. 3D printing for airway disease. AME Med J. 2019;4:14. doi:10.21037/amj.2019.01.05
There are several malignant and nonmalignant conditions that can lead to central airway obstruction (CAO) resulting in lobar collapse. The clinical consequences range from significant dyspnea to respiratory failure. Airway stenting has been used to maintain patency of obstructed airways and relieve symptoms. Before lung cancer screening became more common, approximately 10% of lung cancers at presentation had evidence of CAO.1
On occasion, an endobronchial malignancy involves the right mainstem (RMS) bronchus near the orifice of the right upper lobe (RUL).2 Such strategically located lesions pose a challenge to relieve the RMS obstruction through stenting, securing airway patency into the bronchus intermedius (BI) while avoiding obstruction of the RUL bronchus. The use of endobronchial silicone stents, hybrid covered stents, as well as self-expanding metal stents (SEMS) is an established mode of relieving CAO due to malignant disease.3 We reviewed the literature for approaches that were available before and after the date of the index case reported here.
Case Presentation
A 65-year-old veteran with a history of smoking presented to a US Department of Veterans Affairs Medical Center (VAMC) in 2011, with hemoptysis of 2-week duration. Computed tomography (CT) of the chest revealed a 5.3 × 4.2 × 6.5 cm right mediastinal mass and a 3.0 × 2.8 × 3 cm right hilar mass. Flexible bronchoscopy revealed > 80% occlusion of the RMS and BI due to a medially located mass sparing the RUL orifice, which was patent (Figure 1). Airways distal to the BI were free of disease. Endobronchial biopsies revealed poorly differentiated non-small cell carcinoma of the lung. The patient was referred to the interventional pulmonary service for further airway management.
Under general anesthesia and through a size-9 endotracheal tube, piecemeal debulking of the mass using a cryoprobe was performed. Argon photocoagulation (APC) was used to control bleeding. Balloon bronchoplasty was performed next with pulmonary Boston Scientific CRE balloon at the BI and the RMS bronchus. Under fluoroscopic guidance, a 12 × 30 mm self-expanding hybrid Merit Medical AERO stent was placed distally into the BI. Next, a 14 × 30 mm AERO stent was placed proximally in the RMS bronchus with its distal end telescoped into the smaller distal stent for a distance of 3 to 4 mm at a slanted angle. The overlap was deliberately performed at the level of RUL takeoff. Forcing the distal end of the proximal larger stent into a smaller stent created mechanical stress. The angled alignment channeled this mechanical stress so that the distal end of the proximal stent flared open laterally into the RUL orifice to allow for ventilation (Figure 2). On follow-up 6 months later, all 3 airways remained patent with stents in place (Figure 3).
The patient returned to the VAMC and underwent chemotherapy with carboplatin and paclitaxel cycles that were completed in May 2012, as well as completing 6300 centigray (cGy) of radiation to the area. This led to regression of the tumor permitting removal of the proximal stent in October 2012. Unfortunately, upon follow-up in July 2013, a hypermetabolic lesion in the right upper posterior chest was noted to be eroding the third rib. Biopsy proved it to be poorly differentiated non-small cell lung cancer. Palliative external beam radiation was used to treat this lesion with a total of 3780 cGy completed by the end of August 2013.
Sadly, the patient was admitted later in 2013 with worsening cough and shortness of breath. Chest and abdominal CTs showed an increase in the size of the right apical mass, and mediastinal lymphadenopathy, as well as innumerable nodules in the left lung. The mass had recurred and extended distal to the stent into the lower and middle lobes. New liver nodule and lytic lesion within left ischial tuberosity, T12, L1, and S1 vertebral bodies were noted. The pulmonary service reached out to us via email and we recommended either additional chemoradiotherapy or palliative care. At that point the tumor was widespread and resistant to therapy. It extended beyond the central airways making airway debulking futile. Stents are palliative in nature and we believed that the initial stenting allowed the patient to get chemoradiation by improving functional status through preventing collapse of the right lung. As a result, the patient had about 19 months of a remission period with quality of life. The patient ultimately died under the care of palliative care in inpatient hospice setting.
Literature Review
A literature review revealed multiple approaches to preserving a 3-way patent airway at the takeoff of the RUL (Table). One approach to alleviating such an obstruction favors placing a straight silicone stent from the RMS into the BI, closing off the orifice of the RUL (Figure 4A).4 However, this entails sacrificing ventilation of the RUL. An alternative suggested by Peled and colleagues was carried out successfully in 3 patients. After placing a stent to relieve the obstruction, a Nd:YAG laser is used to create a window in the stent in proximity to the RUL orifice, which allows preservation or ventilations to the RUL (Figure 4B).5
A third effective approach utilizes silicone Y stents, which are usually employed for relief of obstruction at the level of the main carina.6,7 Instead of deploying them at the main carina, they would be deployed at the secondary carina, which the RUL makes with the BI, often with customized cutting for adjustment of the stent limbs to the appropriate size of the RUL and BI (Figure 4C). This approach has been successfully used to maintain RUL ventilation.2
A fourth technique involves using an Oki stent, a dedicated bifurcated silicone stent, which was first described in 2013. It is designed for the RMS bronchus around the RUL and BI bifurcation, enabling the stent to maintain airway patency in the right lung without affecting the trachea and carina (Figure 4D). The arm located in the RUL prevents migration.8 A fifth technique involves deploying a precisely selected Oki stent specially modified based on a printed 3-dimensional (3D) model of the airways after computer-aided simulation.9A sixth technique employs de novo custom printing stents based on 3D models of the tracheobronchial tree constructed based on CT imaging. This approach creates more accurately fitting stents.1
Discussion
The RUL contributes roughly 5 to 10% of the total oxygenation capacity of the lung.10 In patients with lung cancer and limited pulmonary reserve, preserving ventilation to the RUL can be clinically important. The chosen method to relieve endobronchial obstruction depends on several variables, including expertise, ability of the patient to undergo general anesthesia for rigid or flexible bronchoscopy, stent availability, and airway anatomy.
This case illustrates a new method to deal with lesions close to the RUL orifice. This maneuver may not be possible with all types of stents. AERO stents are fully covered (Figure 4E). In contrast, stents that are uncovered at both distal ends, such as a Boston Scientific Ultraflex stent, may not be adequate for such a maneuver. Intercalating uncovered ends of SEMS may allow for tumor in-growth through the uncovered metal mesh near the RUL orifice and may paradoxically compromise both the RUL and BI. The diameter of AERO stents is slightly larger at its ends.11 This helps prevent migration, which in this case maintained the crucial overlap of the stents. On the other hand, use of AERO stents may be associated with a higher risk of infection.12 Precise measurements of the airway diameter are essential given the difference in internal and external stent diameter with silicone stents.
Silicone stents migrate more readily than SEMS and may not be well suited for the procedure we performed. In our case, we wished to maintain ventilation for the RUL; hence, we elected not to bypass it with a silicone stent. We did not have access to a YAG. Moreover, laser carries more energy than APC. Nd:YAG laser has been reported to cause airway fire when used with silicone stents.13 Several authors have reported the use of silicone Y stents at the primary or secondary carina to preserve luminal patency.6,7 Airway anatomy and the angle of the Y may require modification of these stents prior to their use. Cutting stents may compromise their integrity. The bifurcating limb prevents migration which can be a significant concern with the tubular silicone stents. An important consideration for patients in advanced stages of malignancy is that placement of such stent requires undergoing general anesthesia and rigid bronchoscopy, unlike with AERO and metal stents that can be deployed with fiberoptic bronchoscopy under moderate sedation. As such, we did not elect to use a silicone Y stent. Accumulation of secretions or formation of granulation tissue at the orifices can result in recurrence of obstruction.14
Advances in 3D printing seem to be the future of customized airway stenting. This could help clinicians overcome the challenges of improperly sized stents and distorted airway anatomy. Cases have reported successful use of 3D-printed patient-specific airway prostheses.15,16 However, their use is not common practice, as there is a limited amount of materials that are flexible, biocompatible, and approved by the US Food and Drug Administration (FDA) for medical use. Infection control is another layer of consideration in such stents. Standardization of materials and regulation of personalized devices and their cleansing protocols is neccesary.17 At the time of this case, Oki stents and 3D printing were not available in the market. This report provides a viable alternative to use AERO stents for this maneuver.
Conclusions
Patients presenting with malignant CAO near the RUL require a personalized approach to treatment, considering their overall health, functional status, nature and location of CAO, and degree of symptoms. Once a decision is made to stent the airway, careful assessment of airway anatomy, delineation of obstruction, available expertise, and types of stents available needs to be made to preserve ventilation to the nondiseased RUL. Airway stents are expensive and need to be used wisely for palliation and allowing for a quality life while the patient receives more definitive targeted therapy.
Acknowledgments
The authors would like to gratefully acknowledge Dr Jenny Kim, who referred the patient to the interventional service and helped obtain consent for publishing the case.
There are several malignant and nonmalignant conditions that can lead to central airway obstruction (CAO) resulting in lobar collapse. The clinical consequences range from significant dyspnea to respiratory failure. Airway stenting has been used to maintain patency of obstructed airways and relieve symptoms. Before lung cancer screening became more common, approximately 10% of lung cancers at presentation had evidence of CAO.1
On occasion, an endobronchial malignancy involves the right mainstem (RMS) bronchus near the orifice of the right upper lobe (RUL).2 Such strategically located lesions pose a challenge to relieve the RMS obstruction through stenting, securing airway patency into the bronchus intermedius (BI) while avoiding obstruction of the RUL bronchus. The use of endobronchial silicone stents, hybrid covered stents, as well as self-expanding metal stents (SEMS) is an established mode of relieving CAO due to malignant disease.3 We reviewed the literature for approaches that were available before and after the date of the index case reported here.
Case Presentation
A 65-year-old veteran with a history of smoking presented to a US Department of Veterans Affairs Medical Center (VAMC) in 2011, with hemoptysis of 2-week duration. Computed tomography (CT) of the chest revealed a 5.3 × 4.2 × 6.5 cm right mediastinal mass and a 3.0 × 2.8 × 3 cm right hilar mass. Flexible bronchoscopy revealed > 80% occlusion of the RMS and BI due to a medially located mass sparing the RUL orifice, which was patent (Figure 1). Airways distal to the BI were free of disease. Endobronchial biopsies revealed poorly differentiated non-small cell carcinoma of the lung. The patient was referred to the interventional pulmonary service for further airway management.
Under general anesthesia and through a size-9 endotracheal tube, piecemeal debulking of the mass using a cryoprobe was performed. Argon photocoagulation (APC) was used to control bleeding. Balloon bronchoplasty was performed next with pulmonary Boston Scientific CRE balloon at the BI and the RMS bronchus. Under fluoroscopic guidance, a 12 × 30 mm self-expanding hybrid Merit Medical AERO stent was placed distally into the BI. Next, a 14 × 30 mm AERO stent was placed proximally in the RMS bronchus with its distal end telescoped into the smaller distal stent for a distance of 3 to 4 mm at a slanted angle. The overlap was deliberately performed at the level of RUL takeoff. Forcing the distal end of the proximal larger stent into a smaller stent created mechanical stress. The angled alignment channeled this mechanical stress so that the distal end of the proximal stent flared open laterally into the RUL orifice to allow for ventilation (Figure 2). On follow-up 6 months later, all 3 airways remained patent with stents in place (Figure 3).
The patient returned to the VAMC and underwent chemotherapy with carboplatin and paclitaxel cycles that were completed in May 2012, as well as completing 6300 centigray (cGy) of radiation to the area. This led to regression of the tumor permitting removal of the proximal stent in October 2012. Unfortunately, upon follow-up in July 2013, a hypermetabolic lesion in the right upper posterior chest was noted to be eroding the third rib. Biopsy proved it to be poorly differentiated non-small cell lung cancer. Palliative external beam radiation was used to treat this lesion with a total of 3780 cGy completed by the end of August 2013.
Sadly, the patient was admitted later in 2013 with worsening cough and shortness of breath. Chest and abdominal CTs showed an increase in the size of the right apical mass, and mediastinal lymphadenopathy, as well as innumerable nodules in the left lung. The mass had recurred and extended distal to the stent into the lower and middle lobes. New liver nodule and lytic lesion within left ischial tuberosity, T12, L1, and S1 vertebral bodies were noted. The pulmonary service reached out to us via email and we recommended either additional chemoradiotherapy or palliative care. At that point the tumor was widespread and resistant to therapy. It extended beyond the central airways making airway debulking futile. Stents are palliative in nature and we believed that the initial stenting allowed the patient to get chemoradiation by improving functional status through preventing collapse of the right lung. As a result, the patient had about 19 months of a remission period with quality of life. The patient ultimately died under the care of palliative care in inpatient hospice setting.
Literature Review
A literature review revealed multiple approaches to preserving a 3-way patent airway at the takeoff of the RUL (Table). One approach to alleviating such an obstruction favors placing a straight silicone stent from the RMS into the BI, closing off the orifice of the RUL (Figure 4A).4 However, this entails sacrificing ventilation of the RUL. An alternative suggested by Peled and colleagues was carried out successfully in 3 patients. After placing a stent to relieve the obstruction, a Nd:YAG laser is used to create a window in the stent in proximity to the RUL orifice, which allows preservation or ventilations to the RUL (Figure 4B).5
A third effective approach utilizes silicone Y stents, which are usually employed for relief of obstruction at the level of the main carina.6,7 Instead of deploying them at the main carina, they would be deployed at the secondary carina, which the RUL makes with the BI, often with customized cutting for adjustment of the stent limbs to the appropriate size of the RUL and BI (Figure 4C). This approach has been successfully used to maintain RUL ventilation.2
A fourth technique involves using an Oki stent, a dedicated bifurcated silicone stent, which was first described in 2013. It is designed for the RMS bronchus around the RUL and BI bifurcation, enabling the stent to maintain airway patency in the right lung without affecting the trachea and carina (Figure 4D). The arm located in the RUL prevents migration.8 A fifth technique involves deploying a precisely selected Oki stent specially modified based on a printed 3-dimensional (3D) model of the airways after computer-aided simulation.9A sixth technique employs de novo custom printing stents based on 3D models of the tracheobronchial tree constructed based on CT imaging. This approach creates more accurately fitting stents.1
Discussion
The RUL contributes roughly 5 to 10% of the total oxygenation capacity of the lung.10 In patients with lung cancer and limited pulmonary reserve, preserving ventilation to the RUL can be clinically important. The chosen method to relieve endobronchial obstruction depends on several variables, including expertise, ability of the patient to undergo general anesthesia for rigid or flexible bronchoscopy, stent availability, and airway anatomy.
This case illustrates a new method to deal with lesions close to the RUL orifice. This maneuver may not be possible with all types of stents. AERO stents are fully covered (Figure 4E). In contrast, stents that are uncovered at both distal ends, such as a Boston Scientific Ultraflex stent, may not be adequate for such a maneuver. Intercalating uncovered ends of SEMS may allow for tumor in-growth through the uncovered metal mesh near the RUL orifice and may paradoxically compromise both the RUL and BI. The diameter of AERO stents is slightly larger at its ends.11 This helps prevent migration, which in this case maintained the crucial overlap of the stents. On the other hand, use of AERO stents may be associated with a higher risk of infection.12 Precise measurements of the airway diameter are essential given the difference in internal and external stent diameter with silicone stents.
Silicone stents migrate more readily than SEMS and may not be well suited for the procedure we performed. In our case, we wished to maintain ventilation for the RUL; hence, we elected not to bypass it with a silicone stent. We did not have access to a YAG. Moreover, laser carries more energy than APC. Nd:YAG laser has been reported to cause airway fire when used with silicone stents.13 Several authors have reported the use of silicone Y stents at the primary or secondary carina to preserve luminal patency.6,7 Airway anatomy and the angle of the Y may require modification of these stents prior to their use. Cutting stents may compromise their integrity. The bifurcating limb prevents migration which can be a significant concern with the tubular silicone stents. An important consideration for patients in advanced stages of malignancy is that placement of such stent requires undergoing general anesthesia and rigid bronchoscopy, unlike with AERO and metal stents that can be deployed with fiberoptic bronchoscopy under moderate sedation. As such, we did not elect to use a silicone Y stent. Accumulation of secretions or formation of granulation tissue at the orifices can result in recurrence of obstruction.14
Advances in 3D printing seem to be the future of customized airway stenting. This could help clinicians overcome the challenges of improperly sized stents and distorted airway anatomy. Cases have reported successful use of 3D-printed patient-specific airway prostheses.15,16 However, their use is not common practice, as there is a limited amount of materials that are flexible, biocompatible, and approved by the US Food and Drug Administration (FDA) for medical use. Infection control is another layer of consideration in such stents. Standardization of materials and regulation of personalized devices and their cleansing protocols is neccesary.17 At the time of this case, Oki stents and 3D printing were not available in the market. This report provides a viable alternative to use AERO stents for this maneuver.
Conclusions
Patients presenting with malignant CAO near the RUL require a personalized approach to treatment, considering their overall health, functional status, nature and location of CAO, and degree of symptoms. Once a decision is made to stent the airway, careful assessment of airway anatomy, delineation of obstruction, available expertise, and types of stents available needs to be made to preserve ventilation to the nondiseased RUL. Airway stents are expensive and need to be used wisely for palliation and allowing for a quality life while the patient receives more definitive targeted therapy.
Acknowledgments
The authors would like to gratefully acknowledge Dr Jenny Kim, who referred the patient to the interventional service and helped obtain consent for publishing the case.
1. Criner GJ, Eberhardt R, Fernandez-Bussy S, et al. Interventional bronchoscopy. Am J Respir Crit Care Med. 2020;202(1):29-50. doi:10.1164/rccm.201907-1292SO
2. Oki M, Saka H, Kitagawa C, Kogure Y. Silicone y-stent placement on the carina between bronchus to the right upper lobe and bronchus intermedius. Ann Thorac Surg. 2009;87(3):971-974. doi:10.1016/j.athoracsur.2008.06.049
3. Ernst A, Feller-Kopman D, Becker HD, Mehta AC. Central airway obstruction. Am J Respir Crit Care Med. 2004;169(12):1278-1297. doi:10.1164/rccm.200210-1181SO
4. Liu Y-H, Wu Y-C, Hsieh M-J, Ko P-J. Straight bronchial stent placement across the right upper lobe bronchus: A simple alternative for the management of airway obstruction around the carina and right main bronchus. J Thorac Cardiovasc Surg. 2011;141(1):303-305.e1.doi:10.1016/j.jtcvs.2010.06.015
5. Peled N, Shitrit D, Bendayan D, Kramer MR. Right upper lobe ‘window’ in right main bronchus stenting. Eur J Cardiothorac Surg. 2006;30(4):680-682. doi:10.1016/j.ejcts.2006.07.020
6. Dumon J-F, Dumon MC. Dumon-Novatech Y-stents: a four-year experience with 50 tracheobronchial tumors involving the carina. J Bronchol. 2000;7(1):26-32 doi:10.1097/00128594-200007000-00005
7. Dutau H, Toutblanc B, Lamb C, Seijo L. Use of the Dumon Y-stent in the management of malignant disease involving the carina: a retrospective review of 86 patients. Chest. 2004;126(3):951-958. doi:10.1378/chest.126.3.951
8. Dalar L, Abul Y. Safety and efficacy of Oki stenting used to treat obstructions in the right mainstem bronchus. J Bronchol Interv Pulmonol. 2018;25(3):212-217. doi:10.1097/LBR.0000000000000486
9. Guibert N, Moreno B, Plat G, Didier A, Mazieres J, Hermant C. Stenting of complex malignant central-airway obstruction guided by a three-dimensional printed model of the airways. Ann Thorac Surg. 2017;103(4):e357-e359. doi:10.1016/j.athoracsur.2016.09.082
10. Win T, Tasker AD, Groves AM, et al. Ventilation-perfusion scintigraphy to predict postoperative pulmonary function in lung cancer patients undergoing pneumonectomy. AJR Am J Roentgenol. 2006;187(5):1260-1265. doi:10.2214/AJR.04.1973
11. Mehta AC. AERO self-expanding hybrid stent for airway stenosis. Expert Rev Med Devices. 2008;5(5):553-557. doi:10.1586/17434440.5.5.553
12. Ost DE, Shah AM, Lei X, et al. Respiratory infections increase the risk of granulation tissue formation following airway stenting in patients with malignant airway obstruction. Chest. 2012;141(6):1473-1481. doi:10.1378/chest.11-2005
13. Scherer TA. Nd-YAG laser ignition of silicone endobronchial stents. Chest. 2000;117(5):1449-1454. doi:10.1378/chest.117.5.1449
14. Folch E, Keyes C. Airway stents. Ann Cardiothorac Surg. 2018;7(2):273-283. doi:10.21037/acs.2018.03.08
15. Cheng GZ, Folch E, Brik R, et al. Three-dimensional modeled T-tube design and insertion in a patient with tracheal dehiscence. Chest. 2015;148(4):e106-e108. doi:10.1378/chest.15-0240
16. Tam MD, Laycock SD, Jayne D, Babar J, Noble B. 3-D printouts of the tracheobronchial tree generated from CT images as an aid to management in a case of tracheobronchial chondromalacia caused by relapsing polychondritis. J Radiol Case Rep. 2013;7(8):34-43. Published 2013 Aug 1. doi:10.3941/jrcr.v7i8.1390
17. Alraiyes AH, Avasarala SK, Machuzak MS, Gildea TR. 3D printing for airway disease. AME Med J. 2019;4:14. doi:10.21037/amj.2019.01.05
1. Criner GJ, Eberhardt R, Fernandez-Bussy S, et al. Interventional bronchoscopy. Am J Respir Crit Care Med. 2020;202(1):29-50. doi:10.1164/rccm.201907-1292SO
2. Oki M, Saka H, Kitagawa C, Kogure Y. Silicone y-stent placement on the carina between bronchus to the right upper lobe and bronchus intermedius. Ann Thorac Surg. 2009;87(3):971-974. doi:10.1016/j.athoracsur.2008.06.049
3. Ernst A, Feller-Kopman D, Becker HD, Mehta AC. Central airway obstruction. Am J Respir Crit Care Med. 2004;169(12):1278-1297. doi:10.1164/rccm.200210-1181SO
4. Liu Y-H, Wu Y-C, Hsieh M-J, Ko P-J. Straight bronchial stent placement across the right upper lobe bronchus: A simple alternative for the management of airway obstruction around the carina and right main bronchus. J Thorac Cardiovasc Surg. 2011;141(1):303-305.e1.doi:10.1016/j.jtcvs.2010.06.015
5. Peled N, Shitrit D, Bendayan D, Kramer MR. Right upper lobe ‘window’ in right main bronchus stenting. Eur J Cardiothorac Surg. 2006;30(4):680-682. doi:10.1016/j.ejcts.2006.07.020
6. Dumon J-F, Dumon MC. Dumon-Novatech Y-stents: a four-year experience with 50 tracheobronchial tumors involving the carina. J Bronchol. 2000;7(1):26-32 doi:10.1097/00128594-200007000-00005
7. Dutau H, Toutblanc B, Lamb C, Seijo L. Use of the Dumon Y-stent in the management of malignant disease involving the carina: a retrospective review of 86 patients. Chest. 2004;126(3):951-958. doi:10.1378/chest.126.3.951
8. Dalar L, Abul Y. Safety and efficacy of Oki stenting used to treat obstructions in the right mainstem bronchus. J Bronchol Interv Pulmonol. 2018;25(3):212-217. doi:10.1097/LBR.0000000000000486
9. Guibert N, Moreno B, Plat G, Didier A, Mazieres J, Hermant C. Stenting of complex malignant central-airway obstruction guided by a three-dimensional printed model of the airways. Ann Thorac Surg. 2017;103(4):e357-e359. doi:10.1016/j.athoracsur.2016.09.082
10. Win T, Tasker AD, Groves AM, et al. Ventilation-perfusion scintigraphy to predict postoperative pulmonary function in lung cancer patients undergoing pneumonectomy. AJR Am J Roentgenol. 2006;187(5):1260-1265. doi:10.2214/AJR.04.1973
11. Mehta AC. AERO self-expanding hybrid stent for airway stenosis. Expert Rev Med Devices. 2008;5(5):553-557. doi:10.1586/17434440.5.5.553
12. Ost DE, Shah AM, Lei X, et al. Respiratory infections increase the risk of granulation tissue formation following airway stenting in patients with malignant airway obstruction. Chest. 2012;141(6):1473-1481. doi:10.1378/chest.11-2005
13. Scherer TA. Nd-YAG laser ignition of silicone endobronchial stents. Chest. 2000;117(5):1449-1454. doi:10.1378/chest.117.5.1449
14. Folch E, Keyes C. Airway stents. Ann Cardiothorac Surg. 2018;7(2):273-283. doi:10.21037/acs.2018.03.08
15. Cheng GZ, Folch E, Brik R, et al. Three-dimensional modeled T-tube design and insertion in a patient with tracheal dehiscence. Chest. 2015;148(4):e106-e108. doi:10.1378/chest.15-0240
16. Tam MD, Laycock SD, Jayne D, Babar J, Noble B. 3-D printouts of the tracheobronchial tree generated from CT images as an aid to management in a case of tracheobronchial chondromalacia caused by relapsing polychondritis. J Radiol Case Rep. 2013;7(8):34-43. Published 2013 Aug 1. doi:10.3941/jrcr.v7i8.1390
17. Alraiyes AH, Avasarala SK, Machuzak MS, Gildea TR. 3D printing for airway disease. AME Med J. 2019;4:14. doi:10.21037/amj.2019.01.05
Interstitial Granulomatous Dermatitis as an Adverse Reaction to Vedolizumab
The number of monoclonal antibodies developed for therapeutic use has rapidly expanded over the last decade due to their generally favorable adverse effect (AE) profiles and efficacy.1 Tumor necrosis factor α inhibitors and general integrin antagonists are well-known examples of such monoclonal antibodies. Common conditions utilizing immunotherapy include inflammatory bowel diseases (IBDs), such as Crohn disease and ulcerative colitis (UC).2
The monoclonal antibody vedolizumab, approved in 2014 for moderate to severe UC and Crohn disease, selectively antagonizes α4β7 integrin to target a specific population of gastrointestinal T lymphocytes, preventing their mobilization to areas of inflammation.3 Adverse effects in patients treated with vedolizumab occur at a rate comparable to placebo and largely are considered nonserious4,5; the most commonly reported AE is disease exacerbation (13%–17% of patients).5,6 Published reports of cutaneous AEs at administration of vedolizumab include urticaria during infusion, appearance of cutaneous manifestations characteristic of IBD, psoriasis, Henoch-Schönlein purpura, and Sweet syndrome.7-10
We present the case of a 61-year-old woman with UC who developed reactive granulomatous dermatitis (RGD), interstitial granulomatous dermatitis (IGD) type secondary to vedolizumab. This adverse reaction has not, to our knowledge, been previously reported.
Case Report
A 61-year-old woman with a medical history of UC treated with vedolizumab and myelodysplastic syndrome treated with intravenous immunoglobulin (due to hypogammaglobulinemia following allogeneic stem cell transplantation 14 months prior) presented with a concern of a rash. The patient had been in a baseline state of health until 1 week after receiving her second dose of vedolizumab, at which time she developed a mildly pruritic maculopapular rash on the back and chest. Triamcinolone ointment and hydroxyzine were recommended during an initial telehealth consultation with an oncologist with minimal improvement. The rash continued to spread distally with worsening pruritus.
The patient returned to her oncologist for a routine follow-up appointment 5 days after initial teleconsultation. She reported poor oral intake due to oropharyngeal pain and a worsening rash; her husband added a report of recent onset of somnolence. She was admitted to the hospital, and intravenous fluids were administered.
At admission, the patient was hypotensive; vital signs were otherwise normal. Physical examination revealed the oropharynx was erythematous. Pink lichenoid papules coalescing into plaques were present diffusely across the trunk, arms, and legs; the hands, feet, and face were spared (Figure 1).
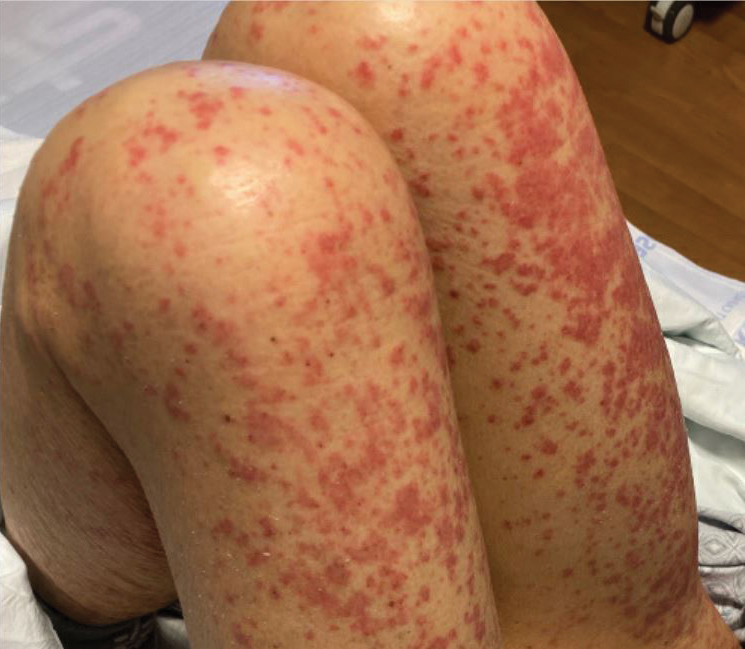
A complete blood cell count and comprehensive metabolic panel were unremarkable. A lumbar puncture, chest radiograph, blood cultures, urinalysis, and urine cultures did not identify a clear infectious cause for the rash, though the workup for infection did raise concern about active cytomegalovirus (CMV) infection with colitis and pneumonitis. Computed tomography of the head showed no acute hemorrhage.
Dermatology was consulted and determined that the appearance of the rash was most consistent with a lichenoid drug eruption, likely secondary to vedolizumab that was administered 1 week before the rash onset. Analysis of a skin biopsy revealed a dense dermal histiocytic and lymphocytic infiltrate in close approximation to blood vessels, confirmed by immunohistochemical staining for CD45, CD43, CD68, CD34, c-KIT, and myeloperoxidase (Figures 2A and 2B). Colloidal iron staining of the specimen revealed no mucin (Figure 2C).
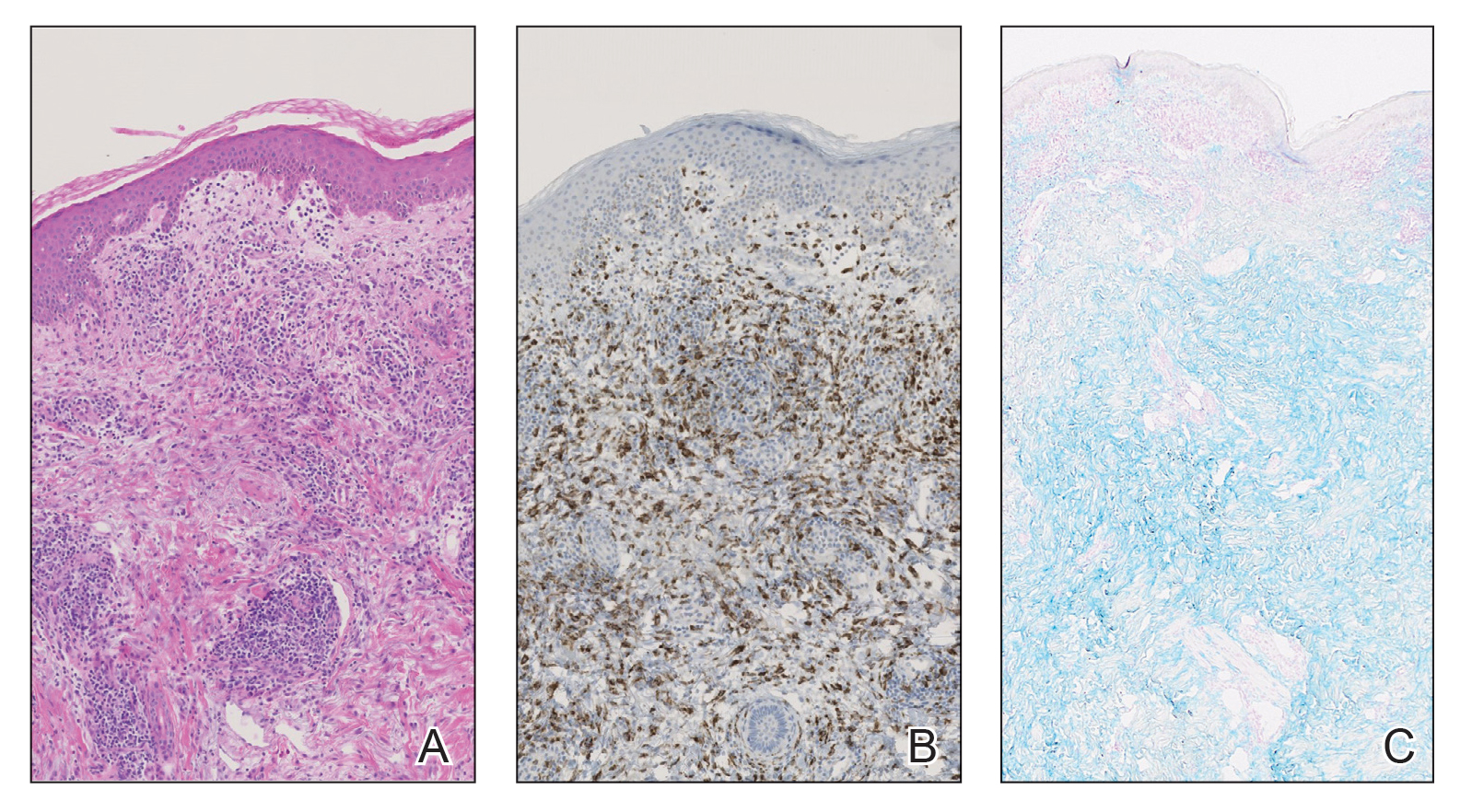
Taken together, the clinical presentation and histopathologic findings were determined to be most consistent with RGD, IGD type, with secondary vasculitis due to vedolizumab. The patient was treated with triamcinolone ointment and low-dose prednisone. Vedolizumab was discontinued. The rash resolved several weeks after cessation of vedolizumab.
Comment
This case describes the development of RGD, IGD type, as an AE of vedolizumab for the treatment of IBD. Reactive granulomatous dermatitis encompasses a spectrum of cutaneous reactions that includes the diagnosis formerly distinctly identified as IGD.11 This variety of RGD is characterized by histologic findings of heavy histiocytic inflammation in the reticular layer of the dermis with interstitial and perivascular neutrophils, lymphocytes, and histiocytes, as well as the absence of mucin. Interstitial granulomatous dermatitis–type reactions commonly are associated with autoimmune conditions and medications, with accumulating examples occurring in the setting of other biologic therapies, including the IL-6 receptor inhibitor tocilizumab; the programmed death receptor-1 inhibitor nivolumab; and the tumor necrosis factor α inhibitors infliximab, etanercept, and adalimumab.12-15
Although our patient represents CMV infection while being treated with vedolizumab, the relationship between the two is unclear. Development of CMV infection while receiving vedolizumab has been reported in the literature in a patient who was concurrently immunosuppressed with azathioprine.16 In contrast, vedolizumab administration has been utilized as a treatment of CMV infection in IBD patients, either alone or in combination with antiviral agents, with successful resolution of infection.17,18 Additional observations of the interaction between CMV infection and vedolizumab would be required to determine if the onset of CMV infection in this patient represents an additional risk of the medication.
Identifying a relationship between a monoclonal antibody therapy, such as vedolizumab, and RGD, IGD type, might be difficult in clinical practice, particularly if this type of reaction has not been previously associated with the culprit medication. In our patient, onset of cutaneous findings in relation to dosing of vedolizumab and exclusion of other possible causes of the rash supported the decision to stop vedolizumab. However, this decision often is challenging in patients with multiple concurrent medical conditions and those whose therapeutic options are limited.
Conclusion
Ulcerative colitis is not an uncommon condition; utilization of targeted monoclonal antibodies as a treatment strategy is expanding.2,19 As implementation of vedolizumab as a targeted biologic therapy for this disease increases, additional cases of IGD might emerge with greater frequency. Because IBD and autoimmune conditions have a tendency to coincide, awareness of the reaction presented here might be particularly important for dermatologists managing cutaneous manifestations of autoimmune conditions, as patients might present with a clinical picture complicated by preexisting skin findings.20 Furthermore, as reports of RGD, IGD type, in response to several monoclonal antibodies accumulate, it is prudent for all physicians to be aware of this potential complication of this class of medication so that they can make educated decisions about continuing monoclonal antibody therapy.
- Grilo AL, Mantalaris A. The increasingly human and profitable monoclonal antibody market. Trends Biotechnol. 2019;37:9-16. doi:10.1016/j.tibtech.2018.05.014
- Yu H, MacIsaac D, Wong JJ, et al. Market share and costs of biologic therapies for inflammatory bowel disease in the USA. Aliment Pharmacol Ther. 2018;47:364-370. doi:10.1111/apt.14430
- Wyant T, Fedyk E, Abhyankar B. An overview of the mechanism of action of the monoclonal antibody vedolizumab. J Crohns Colitis. 2016;10:1437-1444. doi:10.1093/ecco-jcc/jjw092
- Mosli MH, MacDonald JK, Bickston SJ, et al. Vedolizumab for induction and maintenance of remission in ulcerative colitis: a Cochrane systematic review and meta-analysis. Inflamm Bowel Dis. 2015;21:1151-1159. doi:10.1097/MIB.0000000000000396
- Cohen RD, Bhayat F, Blake A, et al. The safety profile of vedolizumab in ulcerative colitis and Crohn’s disease: 4 years of global post-marketing data. J Crohns Colitis. 2020;14:192-204. doi:10.1093/ecco-jcc/jjz137
- Sands BE, Feagan BG, Rutgeerts P, et al. Effects of vedolizumab induction therapy for patients with Crohn’s disease in whom tumor necrosis factor antagonist treatment failed. Gastroenterology. 2014;147:618-627.e3. doi:10.1053/j.gastro.2014.05.008
- Tadbiri S, Peyrin-Biroulet L, Serrero M, et al; . Impact of vedolizumab therapy on extra-intestinal manifestations in patients with inflammatory bowel disease: a multicentre cohort study nested in the OBSERV-IBD cohort. Aliment Pharmacol Ther. 2018;47:485-493. doi:10.1111/apt.14419
- Pereira Guedes T, Pedroto I, Lago P. Vedolizumab-associated psoriasis: until where does gut selectivity go? Rev Esp Enferm Dig. 2020;112:580-581. doi:10.17235/reed.2020.6817/2019
- Gold SL, Magro C, Scherl E. A unique infusion reaction to vedolizumab in a patient with Crohn’s disease. Gastroenterology. 2018;155:981-982. doi:10.1053/j.gastro.2018.03.048
- Martínez Andrés B, Sastre Lozano V, Sánchez Melgarejo JF. Sweet syndrome after treatment with vedolizumab in a patient with Crohn’s disease. Rev Esp Enferm Dig. 2018;110:530. doi:10.17235/reed.2018.5603/2018
- Rosenbach M, English JC 3rd. Reactive granulomatous dermatitis: a review of palisaded neutrophilic and granulomatous dermatitis, interstitial granulomatous dermatitis, interstitial granulomatous drug reaction, and a proposed reclassification. Dermatol Clin. 2015;33:373-387. doi:10.1016/j.det.2015.03.005
- Crowson AN, Magro C. Interstitial granulomatous dermatitis with arthritis. Hum Pathol. 2004;35:779-780. doi:10.1016/j.humpath.2004.05.001
- Altemir A, Iglesias-Sancho M, Sola-Casas MdeLA, et al. Interstitial granulomatous dermatitis following tocilizumab, a paradoxical reaction? Dermatol Ther. 2020;33:e14207. doi:10.1111/dth.14207
- Singh P, Wolfe SP, Alloo A, et al. Interstitial granulomatous dermatitis and granulomatous arteritis in the setting of PD-1 inhibitor therapy for metastatic melanoma. J Cutan Pathol. 2020;47:65-69. doi:10.1111/cup.13562
- Deng A, Harvey V, Sina B, et al. Interstitial granulomatous dermatitis associated with the use of tumor necrosis factor alpha inhibitors. Arch Dermatol. 2006;142:198-202. doi:10.1001/archderm.142.2.198
- Bonfanti E, Bracco C, Biancheri P, et al. Fever during anti-integrin therapy: new immunodeficiency. Eur J Case Rep Intern Med. 2020;7:001288. doi:10.12890/2020_001288
- A, Lenarcik M, E. Resolution of CMV infection in the bowel on vedolizumab therapy. J Crohns Colitis. 2019;13:1234-1235. doi:10.1093/ecco-jcc/jjz033
- Hommel C, Pillet S, Rahier J-F. Comment on: ‘Resolution of CMV infection in the bowel on vedolizumab therapy’. J Crohns Colitis. 2020;14:148-149. doi:10.1093/ecco-jcc/jjz108
- Ng SC, Shi HY, Hamidi N, et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet. 2017;390:2769-2778. doi:10.1016/S0140-6736(17)32448-0
- Halling ML, Kjeldsen J, Knudsen T, et al. Patients with inflammatory bowel disease have increased risk of autoimmune and inflammatory diseases. World J Gastroenterol. 2017;23:6137-6146. doi:10.3748/wjg.v23.i33.6137
The number of monoclonal antibodies developed for therapeutic use has rapidly expanded over the last decade due to their generally favorable adverse effect (AE) profiles and efficacy.1 Tumor necrosis factor α inhibitors and general integrin antagonists are well-known examples of such monoclonal antibodies. Common conditions utilizing immunotherapy include inflammatory bowel diseases (IBDs), such as Crohn disease and ulcerative colitis (UC).2
The monoclonal antibody vedolizumab, approved in 2014 for moderate to severe UC and Crohn disease, selectively antagonizes α4β7 integrin to target a specific population of gastrointestinal T lymphocytes, preventing their mobilization to areas of inflammation.3 Adverse effects in patients treated with vedolizumab occur at a rate comparable to placebo and largely are considered nonserious4,5; the most commonly reported AE is disease exacerbation (13%–17% of patients).5,6 Published reports of cutaneous AEs at administration of vedolizumab include urticaria during infusion, appearance of cutaneous manifestations characteristic of IBD, psoriasis, Henoch-Schönlein purpura, and Sweet syndrome.7-10
We present the case of a 61-year-old woman with UC who developed reactive granulomatous dermatitis (RGD), interstitial granulomatous dermatitis (IGD) type secondary to vedolizumab. This adverse reaction has not, to our knowledge, been previously reported.
Case Report
A 61-year-old woman with a medical history of UC treated with vedolizumab and myelodysplastic syndrome treated with intravenous immunoglobulin (due to hypogammaglobulinemia following allogeneic stem cell transplantation 14 months prior) presented with a concern of a rash. The patient had been in a baseline state of health until 1 week after receiving her second dose of vedolizumab, at which time she developed a mildly pruritic maculopapular rash on the back and chest. Triamcinolone ointment and hydroxyzine were recommended during an initial telehealth consultation with an oncologist with minimal improvement. The rash continued to spread distally with worsening pruritus.
The patient returned to her oncologist for a routine follow-up appointment 5 days after initial teleconsultation. She reported poor oral intake due to oropharyngeal pain and a worsening rash; her husband added a report of recent onset of somnolence. She was admitted to the hospital, and intravenous fluids were administered.
At admission, the patient was hypotensive; vital signs were otherwise normal. Physical examination revealed the oropharynx was erythematous. Pink lichenoid papules coalescing into plaques were present diffusely across the trunk, arms, and legs; the hands, feet, and face were spared (Figure 1).
A complete blood cell count and comprehensive metabolic panel were unremarkable. A lumbar puncture, chest radiograph, blood cultures, urinalysis, and urine cultures did not identify a clear infectious cause for the rash, though the workup for infection did raise concern about active cytomegalovirus (CMV) infection with colitis and pneumonitis. Computed tomography of the head showed no acute hemorrhage.
Dermatology was consulted and determined that the appearance of the rash was most consistent with a lichenoid drug eruption, likely secondary to vedolizumab that was administered 1 week before the rash onset. Analysis of a skin biopsy revealed a dense dermal histiocytic and lymphocytic infiltrate in close approximation to blood vessels, confirmed by immunohistochemical staining for CD45, CD43, CD68, CD34, c-KIT, and myeloperoxidase (Figures 2A and 2B). Colloidal iron staining of the specimen revealed no mucin (Figure 2C).
Taken together, the clinical presentation and histopathologic findings were determined to be most consistent with RGD, IGD type, with secondary vasculitis due to vedolizumab. The patient was treated with triamcinolone ointment and low-dose prednisone. Vedolizumab was discontinued. The rash resolved several weeks after cessation of vedolizumab.
Comment
This case describes the development of RGD, IGD type, as an AE of vedolizumab for the treatment of IBD. Reactive granulomatous dermatitis encompasses a spectrum of cutaneous reactions that includes the diagnosis formerly distinctly identified as IGD.11 This variety of RGD is characterized by histologic findings of heavy histiocytic inflammation in the reticular layer of the dermis with interstitial and perivascular neutrophils, lymphocytes, and histiocytes, as well as the absence of mucin. Interstitial granulomatous dermatitis–type reactions commonly are associated with autoimmune conditions and medications, with accumulating examples occurring in the setting of other biologic therapies, including the IL-6 receptor inhibitor tocilizumab; the programmed death receptor-1 inhibitor nivolumab; and the tumor necrosis factor α inhibitors infliximab, etanercept, and adalimumab.12-15
Although our patient represents CMV infection while being treated with vedolizumab, the relationship between the two is unclear. Development of CMV infection while receiving vedolizumab has been reported in the literature in a patient who was concurrently immunosuppressed with azathioprine.16 In contrast, vedolizumab administration has been utilized as a treatment of CMV infection in IBD patients, either alone or in combination with antiviral agents, with successful resolution of infection.17,18 Additional observations of the interaction between CMV infection and vedolizumab would be required to determine if the onset of CMV infection in this patient represents an additional risk of the medication.
Identifying a relationship between a monoclonal antibody therapy, such as vedolizumab, and RGD, IGD type, might be difficult in clinical practice, particularly if this type of reaction has not been previously associated with the culprit medication. In our patient, onset of cutaneous findings in relation to dosing of vedolizumab and exclusion of other possible causes of the rash supported the decision to stop vedolizumab. However, this decision often is challenging in patients with multiple concurrent medical conditions and those whose therapeutic options are limited.
Conclusion
Ulcerative colitis is not an uncommon condition; utilization of targeted monoclonal antibodies as a treatment strategy is expanding.2,19 As implementation of vedolizumab as a targeted biologic therapy for this disease increases, additional cases of IGD might emerge with greater frequency. Because IBD and autoimmune conditions have a tendency to coincide, awareness of the reaction presented here might be particularly important for dermatologists managing cutaneous manifestations of autoimmune conditions, as patients might present with a clinical picture complicated by preexisting skin findings.20 Furthermore, as reports of RGD, IGD type, in response to several monoclonal antibodies accumulate, it is prudent for all physicians to be aware of this potential complication of this class of medication so that they can make educated decisions about continuing monoclonal antibody therapy.
The number of monoclonal antibodies developed for therapeutic use has rapidly expanded over the last decade due to their generally favorable adverse effect (AE) profiles and efficacy.1 Tumor necrosis factor α inhibitors and general integrin antagonists are well-known examples of such monoclonal antibodies. Common conditions utilizing immunotherapy include inflammatory bowel diseases (IBDs), such as Crohn disease and ulcerative colitis (UC).2
The monoclonal antibody vedolizumab, approved in 2014 for moderate to severe UC and Crohn disease, selectively antagonizes α4β7 integrin to target a specific population of gastrointestinal T lymphocytes, preventing their mobilization to areas of inflammation.3 Adverse effects in patients treated with vedolizumab occur at a rate comparable to placebo and largely are considered nonserious4,5; the most commonly reported AE is disease exacerbation (13%–17% of patients).5,6 Published reports of cutaneous AEs at administration of vedolizumab include urticaria during infusion, appearance of cutaneous manifestations characteristic of IBD, psoriasis, Henoch-Schönlein purpura, and Sweet syndrome.7-10
We present the case of a 61-year-old woman with UC who developed reactive granulomatous dermatitis (RGD), interstitial granulomatous dermatitis (IGD) type secondary to vedolizumab. This adverse reaction has not, to our knowledge, been previously reported.
Case Report
A 61-year-old woman with a medical history of UC treated with vedolizumab and myelodysplastic syndrome treated with intravenous immunoglobulin (due to hypogammaglobulinemia following allogeneic stem cell transplantation 14 months prior) presented with a concern of a rash. The patient had been in a baseline state of health until 1 week after receiving her second dose of vedolizumab, at which time she developed a mildly pruritic maculopapular rash on the back and chest. Triamcinolone ointment and hydroxyzine were recommended during an initial telehealth consultation with an oncologist with minimal improvement. The rash continued to spread distally with worsening pruritus.
The patient returned to her oncologist for a routine follow-up appointment 5 days after initial teleconsultation. She reported poor oral intake due to oropharyngeal pain and a worsening rash; her husband added a report of recent onset of somnolence. She was admitted to the hospital, and intravenous fluids were administered.
At admission, the patient was hypotensive; vital signs were otherwise normal. Physical examination revealed the oropharynx was erythematous. Pink lichenoid papules coalescing into plaques were present diffusely across the trunk, arms, and legs; the hands, feet, and face were spared (Figure 1).
A complete blood cell count and comprehensive metabolic panel were unremarkable. A lumbar puncture, chest radiograph, blood cultures, urinalysis, and urine cultures did not identify a clear infectious cause for the rash, though the workup for infection did raise concern about active cytomegalovirus (CMV) infection with colitis and pneumonitis. Computed tomography of the head showed no acute hemorrhage.
Dermatology was consulted and determined that the appearance of the rash was most consistent with a lichenoid drug eruption, likely secondary to vedolizumab that was administered 1 week before the rash onset. Analysis of a skin biopsy revealed a dense dermal histiocytic and lymphocytic infiltrate in close approximation to blood vessels, confirmed by immunohistochemical staining for CD45, CD43, CD68, CD34, c-KIT, and myeloperoxidase (Figures 2A and 2B). Colloidal iron staining of the specimen revealed no mucin (Figure 2C).
Taken together, the clinical presentation and histopathologic findings were determined to be most consistent with RGD, IGD type, with secondary vasculitis due to vedolizumab. The patient was treated with triamcinolone ointment and low-dose prednisone. Vedolizumab was discontinued. The rash resolved several weeks after cessation of vedolizumab.
Comment
This case describes the development of RGD, IGD type, as an AE of vedolizumab for the treatment of IBD. Reactive granulomatous dermatitis encompasses a spectrum of cutaneous reactions that includes the diagnosis formerly distinctly identified as IGD.11 This variety of RGD is characterized by histologic findings of heavy histiocytic inflammation in the reticular layer of the dermis with interstitial and perivascular neutrophils, lymphocytes, and histiocytes, as well as the absence of mucin. Interstitial granulomatous dermatitis–type reactions commonly are associated with autoimmune conditions and medications, with accumulating examples occurring in the setting of other biologic therapies, including the IL-6 receptor inhibitor tocilizumab; the programmed death receptor-1 inhibitor nivolumab; and the tumor necrosis factor α inhibitors infliximab, etanercept, and adalimumab.12-15
Although our patient represents CMV infection while being treated with vedolizumab, the relationship between the two is unclear. Development of CMV infection while receiving vedolizumab has been reported in the literature in a patient who was concurrently immunosuppressed with azathioprine.16 In contrast, vedolizumab administration has been utilized as a treatment of CMV infection in IBD patients, either alone or in combination with antiviral agents, with successful resolution of infection.17,18 Additional observations of the interaction between CMV infection and vedolizumab would be required to determine if the onset of CMV infection in this patient represents an additional risk of the medication.
Identifying a relationship between a monoclonal antibody therapy, such as vedolizumab, and RGD, IGD type, might be difficult in clinical practice, particularly if this type of reaction has not been previously associated with the culprit medication. In our patient, onset of cutaneous findings in relation to dosing of vedolizumab and exclusion of other possible causes of the rash supported the decision to stop vedolizumab. However, this decision often is challenging in patients with multiple concurrent medical conditions and those whose therapeutic options are limited.
Conclusion
Ulcerative colitis is not an uncommon condition; utilization of targeted monoclonal antibodies as a treatment strategy is expanding.2,19 As implementation of vedolizumab as a targeted biologic therapy for this disease increases, additional cases of IGD might emerge with greater frequency. Because IBD and autoimmune conditions have a tendency to coincide, awareness of the reaction presented here might be particularly important for dermatologists managing cutaneous manifestations of autoimmune conditions, as patients might present with a clinical picture complicated by preexisting skin findings.20 Furthermore, as reports of RGD, IGD type, in response to several monoclonal antibodies accumulate, it is prudent for all physicians to be aware of this potential complication of this class of medication so that they can make educated decisions about continuing monoclonal antibody therapy.
- Grilo AL, Mantalaris A. The increasingly human and profitable monoclonal antibody market. Trends Biotechnol. 2019;37:9-16. doi:10.1016/j.tibtech.2018.05.014
- Yu H, MacIsaac D, Wong JJ, et al. Market share and costs of biologic therapies for inflammatory bowel disease in the USA. Aliment Pharmacol Ther. 2018;47:364-370. doi:10.1111/apt.14430
- Wyant T, Fedyk E, Abhyankar B. An overview of the mechanism of action of the monoclonal antibody vedolizumab. J Crohns Colitis. 2016;10:1437-1444. doi:10.1093/ecco-jcc/jjw092
- Mosli MH, MacDonald JK, Bickston SJ, et al. Vedolizumab for induction and maintenance of remission in ulcerative colitis: a Cochrane systematic review and meta-analysis. Inflamm Bowel Dis. 2015;21:1151-1159. doi:10.1097/MIB.0000000000000396
- Cohen RD, Bhayat F, Blake A, et al. The safety profile of vedolizumab in ulcerative colitis and Crohn’s disease: 4 years of global post-marketing data. J Crohns Colitis. 2020;14:192-204. doi:10.1093/ecco-jcc/jjz137
- Sands BE, Feagan BG, Rutgeerts P, et al. Effects of vedolizumab induction therapy for patients with Crohn’s disease in whom tumor necrosis factor antagonist treatment failed. Gastroenterology. 2014;147:618-627.e3. doi:10.1053/j.gastro.2014.05.008
- Tadbiri S, Peyrin-Biroulet L, Serrero M, et al; . Impact of vedolizumab therapy on extra-intestinal manifestations in patients with inflammatory bowel disease: a multicentre cohort study nested in the OBSERV-IBD cohort. Aliment Pharmacol Ther. 2018;47:485-493. doi:10.1111/apt.14419
- Pereira Guedes T, Pedroto I, Lago P. Vedolizumab-associated psoriasis: until where does gut selectivity go? Rev Esp Enferm Dig. 2020;112:580-581. doi:10.17235/reed.2020.6817/2019
- Gold SL, Magro C, Scherl E. A unique infusion reaction to vedolizumab in a patient with Crohn’s disease. Gastroenterology. 2018;155:981-982. doi:10.1053/j.gastro.2018.03.048
- Martínez Andrés B, Sastre Lozano V, Sánchez Melgarejo JF. Sweet syndrome after treatment with vedolizumab in a patient with Crohn’s disease. Rev Esp Enferm Dig. 2018;110:530. doi:10.17235/reed.2018.5603/2018
- Rosenbach M, English JC 3rd. Reactive granulomatous dermatitis: a review of palisaded neutrophilic and granulomatous dermatitis, interstitial granulomatous dermatitis, interstitial granulomatous drug reaction, and a proposed reclassification. Dermatol Clin. 2015;33:373-387. doi:10.1016/j.det.2015.03.005
- Crowson AN, Magro C. Interstitial granulomatous dermatitis with arthritis. Hum Pathol. 2004;35:779-780. doi:10.1016/j.humpath.2004.05.001
- Altemir A, Iglesias-Sancho M, Sola-Casas MdeLA, et al. Interstitial granulomatous dermatitis following tocilizumab, a paradoxical reaction? Dermatol Ther. 2020;33:e14207. doi:10.1111/dth.14207
- Singh P, Wolfe SP, Alloo A, et al. Interstitial granulomatous dermatitis and granulomatous arteritis in the setting of PD-1 inhibitor therapy for metastatic melanoma. J Cutan Pathol. 2020;47:65-69. doi:10.1111/cup.13562
- Deng A, Harvey V, Sina B, et al. Interstitial granulomatous dermatitis associated with the use of tumor necrosis factor alpha inhibitors. Arch Dermatol. 2006;142:198-202. doi:10.1001/archderm.142.2.198
- Bonfanti E, Bracco C, Biancheri P, et al. Fever during anti-integrin therapy: new immunodeficiency. Eur J Case Rep Intern Med. 2020;7:001288. doi:10.12890/2020_001288
- A, Lenarcik M, E. Resolution of CMV infection in the bowel on vedolizumab therapy. J Crohns Colitis. 2019;13:1234-1235. doi:10.1093/ecco-jcc/jjz033
- Hommel C, Pillet S, Rahier J-F. Comment on: ‘Resolution of CMV infection in the bowel on vedolizumab therapy’. J Crohns Colitis. 2020;14:148-149. doi:10.1093/ecco-jcc/jjz108
- Ng SC, Shi HY, Hamidi N, et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet. 2017;390:2769-2778. doi:10.1016/S0140-6736(17)32448-0
- Halling ML, Kjeldsen J, Knudsen T, et al. Patients with inflammatory bowel disease have increased risk of autoimmune and inflammatory diseases. World J Gastroenterol. 2017;23:6137-6146. doi:10.3748/wjg.v23.i33.6137
- Grilo AL, Mantalaris A. The increasingly human and profitable monoclonal antibody market. Trends Biotechnol. 2019;37:9-16. doi:10.1016/j.tibtech.2018.05.014
- Yu H, MacIsaac D, Wong JJ, et al. Market share and costs of biologic therapies for inflammatory bowel disease in the USA. Aliment Pharmacol Ther. 2018;47:364-370. doi:10.1111/apt.14430
- Wyant T, Fedyk E, Abhyankar B. An overview of the mechanism of action of the monoclonal antibody vedolizumab. J Crohns Colitis. 2016;10:1437-1444. doi:10.1093/ecco-jcc/jjw092
- Mosli MH, MacDonald JK, Bickston SJ, et al. Vedolizumab for induction and maintenance of remission in ulcerative colitis: a Cochrane systematic review and meta-analysis. Inflamm Bowel Dis. 2015;21:1151-1159. doi:10.1097/MIB.0000000000000396
- Cohen RD, Bhayat F, Blake A, et al. The safety profile of vedolizumab in ulcerative colitis and Crohn’s disease: 4 years of global post-marketing data. J Crohns Colitis. 2020;14:192-204. doi:10.1093/ecco-jcc/jjz137
- Sands BE, Feagan BG, Rutgeerts P, et al. Effects of vedolizumab induction therapy for patients with Crohn’s disease in whom tumor necrosis factor antagonist treatment failed. Gastroenterology. 2014;147:618-627.e3. doi:10.1053/j.gastro.2014.05.008
- Tadbiri S, Peyrin-Biroulet L, Serrero M, et al; . Impact of vedolizumab therapy on extra-intestinal manifestations in patients with inflammatory bowel disease: a multicentre cohort study nested in the OBSERV-IBD cohort. Aliment Pharmacol Ther. 2018;47:485-493. doi:10.1111/apt.14419
- Pereira Guedes T, Pedroto I, Lago P. Vedolizumab-associated psoriasis: until where does gut selectivity go? Rev Esp Enferm Dig. 2020;112:580-581. doi:10.17235/reed.2020.6817/2019
- Gold SL, Magro C, Scherl E. A unique infusion reaction to vedolizumab in a patient with Crohn’s disease. Gastroenterology. 2018;155:981-982. doi:10.1053/j.gastro.2018.03.048
- Martínez Andrés B, Sastre Lozano V, Sánchez Melgarejo JF. Sweet syndrome after treatment with vedolizumab in a patient with Crohn’s disease. Rev Esp Enferm Dig. 2018;110:530. doi:10.17235/reed.2018.5603/2018
- Rosenbach M, English JC 3rd. Reactive granulomatous dermatitis: a review of palisaded neutrophilic and granulomatous dermatitis, interstitial granulomatous dermatitis, interstitial granulomatous drug reaction, and a proposed reclassification. Dermatol Clin. 2015;33:373-387. doi:10.1016/j.det.2015.03.005
- Crowson AN, Magro C. Interstitial granulomatous dermatitis with arthritis. Hum Pathol. 2004;35:779-780. doi:10.1016/j.humpath.2004.05.001
- Altemir A, Iglesias-Sancho M, Sola-Casas MdeLA, et al. Interstitial granulomatous dermatitis following tocilizumab, a paradoxical reaction? Dermatol Ther. 2020;33:e14207. doi:10.1111/dth.14207
- Singh P, Wolfe SP, Alloo A, et al. Interstitial granulomatous dermatitis and granulomatous arteritis in the setting of PD-1 inhibitor therapy for metastatic melanoma. J Cutan Pathol. 2020;47:65-69. doi:10.1111/cup.13562
- Deng A, Harvey V, Sina B, et al. Interstitial granulomatous dermatitis associated with the use of tumor necrosis factor alpha inhibitors. Arch Dermatol. 2006;142:198-202. doi:10.1001/archderm.142.2.198
- Bonfanti E, Bracco C, Biancheri P, et al. Fever during anti-integrin therapy: new immunodeficiency. Eur J Case Rep Intern Med. 2020;7:001288. doi:10.12890/2020_001288
- A, Lenarcik M, E. Resolution of CMV infection in the bowel on vedolizumab therapy. J Crohns Colitis. 2019;13:1234-1235. doi:10.1093/ecco-jcc/jjz033
- Hommel C, Pillet S, Rahier J-F. Comment on: ‘Resolution of CMV infection in the bowel on vedolizumab therapy’. J Crohns Colitis. 2020;14:148-149. doi:10.1093/ecco-jcc/jjz108
- Ng SC, Shi HY, Hamidi N, et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet. 2017;390:2769-2778. doi:10.1016/S0140-6736(17)32448-0
- Halling ML, Kjeldsen J, Knudsen T, et al. Patients with inflammatory bowel disease have increased risk of autoimmune and inflammatory diseases. World J Gastroenterol. 2017;23:6137-6146. doi:10.3748/wjg.v23.i33.6137
Practice Points
- Reactive granulomatous dermatitis, interstitial granulomatous dermatitis (IGD) type, can occur as an adverse reaction to vedolizumab despite the minimal adverse effect profile of the medication.
- Evidence of IGD type reactions to monoclonal antibodies is accumulating; this disorder can be considered in the differential diagnosis for patients who develop a new rash when treated with an agent of this therapeutic class.

Mild Grisel Syndrome: Expanding the Differential for Posttonsillectomy Adenoidectomy Symptoms
Tonsillectomy with or without adenoidectomy (T&A) is the second most common pediatric surgical procedure in the United States.1 It is most often performed during childhood between 5 and 8 years of age with a second peak observed between 17 and 21 years of age in the adolescent and young adult populations.2 While recurrent tonsillitis has been traditionally associated with tonsillectomy, sleep disordered breathing with obstructive sleep apnea is now the primary indication for the procedure.1
Up to 97% of T&As are performed as an outpatient same-day surgery not requiring inpatient admission.2 Although largely a safe and routinely performed surgery, several complications have been described. Due to the outpatient nature of the procedure, the complications are often encountered in the emergency department (ED) and sometimes in primary care settings. Common complications (outside of the perioperative time frame) include nausea, vomiting, otalgia, odynophagia, infection of the throat (broadly), and hemorrhage; uncommon complications include subcutaneous emphysema, taste disorders, and Eagle syndrome. Some complications are rarer still and carry significant morbidity and even mortality, including mediastinitis, cervical osteomyelitis, and Grisel syndrome.3 The following case encourages the clinician to expand the differential for a patient presenting after T&A.
Case Presentation
A child aged < 3 years was brought to the ED by their mother. She reported neck pain and stiffness 10 days after T&A with concurrent tympanostomy tube placement at an outside pediatric hospital. At triage, their heart rate was 94 bpm, temperature was 98.2 °F, respiratory rate, 22 breaths per minute, and oxygen saturation, 97% on room air. The mother of the patient (MOP) had been giving the prescribed oral liquid formulations of ibuprofen and acetaminophen with hydrocodone as directed. No drug allergies were reported, and immunizations were up to date for age. Other medical and surgical history included eczema and remote cutaneous hemangioma resection. The patient lived at home with 2 parents and was not exposed to smoke; their family history was noncontributory.
Since the surgery, the MOP had noticed constant and increasing neck stiffness, specifically with looking up and down but not side to side. She also had noticed swelling behind both ears. She reported no substantial decrease in intake by mouth or decrease in urine or bowel frequency. On review of systems, she reported no fever, vomiting, difficulty breathing, bleeding from the mouth or nose, eye or ear drainage, or rash.
On physical examination, the patient was alert and in no acute distress; active and playful on an electronic device but was notably not moving their head, which was held in a forward-looking position without any signs of trauma. When asked, the child would not flex or extend their neck but would rotate a few degrees from neutral to both sides. Even with moving the electronic device up and down in space, no active neck extension or flexion could be elicited. The examination of the head, eyes, ears, nose, and throat was otherwise only remarkable for palpable and mildly tender postauricular lymph nodes and diffuse erythema in the posterior pharynx. Cardiopulmonary, abdominal, skin, and extremity examinations were unremarkable.
With concern for an infectious process, the physician ordered blood chemistry and hematology tests along with neck radiography. While awaiting the results, the patient was given a weight-based bolus of normal saline, and the home pain regimen was administered. An attempt was made to passively flex and extend the neck as the child slept in their mother’s arms, but the patient immediately awoke and began to cry.
All values of the comprehensive metabolic panel were within normal limits except for a slight elevation in the blood urea nitrogen to 21 mg/dL and glucose to 159 mg/dL. The complete blood count was unrevealing. The computed tomography (CT) scan with contrast of the soft tissues of the neck was limited by motion artifact but showed a head held in axial rotation with soft tissue irregularity in the anterior aspect of the adenoids (Figure 1). There was what appeared to be normal lymphadenopathy in the hypopharynx, but the soft tissues were otherwise unremarkable.
The on-call pediatric otolaryngologist at the hospital where the procedure was performed was paged. On hearing the details of the case, the specialist was concerned for Grisel syndrome and requested to see the patient in their facility. No additional recommendations for care were provided; the mother was updated and agreed to transfer. The patient was comfortable and stable with repeat vitals as follows: heart rate, 86 beats per minute, blood pressure, 99/62, temperature, 98.3 °F, respiratory rate, 20 breaths per minute, and oxygen saturation, 99% on room air.
On arrival at the receiving facility, the emergency team performed a history and physical that revealed no significant changes from the initial evaluation. They then facilitated evaluation by the pediatric otolaryngologist who conducted a more directed physical examination. Decreased active and passive range of motion (ROM) of the neck without rotatory restriction was again noted. They also observed scant fibrinous exudate within the oropharynx and tonsillar fossa, which was normal in the setting of the recent surgery. They recommended additional analgesia with intramuscular ketorolac, weight-based dosing at 1 mg/kg.
With repeat examination after this additional analgesic, ROM of the neck first passive then active had improved. The patient was then discharged to follow up in the coming days with instructions to continue the pain and anti-inflammatory regimen. They were not started on an antibiotic at that time nor were they placed in a cervical collar. At the follow-up, the MOP reported persistence of neck stiffness for a few days initially but then observed slow improvement. By postoperative day 18, the stiffness had resolved. No other follow-up or referrals related to this issue were readily apparent in review of the patient’s health record.
Discussion
Grisel syndrome is the atraumatic rotary subluxation of the atlantoaxial joint, specifically, the atlas (C1 vertebra) rotates to a fixed, nonanatomic position while the axis (C2 vertebra) remains in normal alignment in relation to the remainder of the spinal column. The subluxation occurs in the absence of ligamentous injury but is associated with an increase in ligamentous laxity.4 The atlas is a ring-shaped vertebra with 2 lateral masses connected by anterior and posterior arches; it lacks a spinous process unlike other vertebrae. It articulates with the skull by means of the 2 articular facets on the superior aspect of the lateral masses. Articulation with the axis occurs at 3 sites: 2 articular facets on the inferior portion of the lateral masses of the atlas and a facet for the dens on the posterior portion of the anterior arch. The dens projects superiorly from the body of the axis and is bound posteriorly by the transverse ligament of the atlas.5
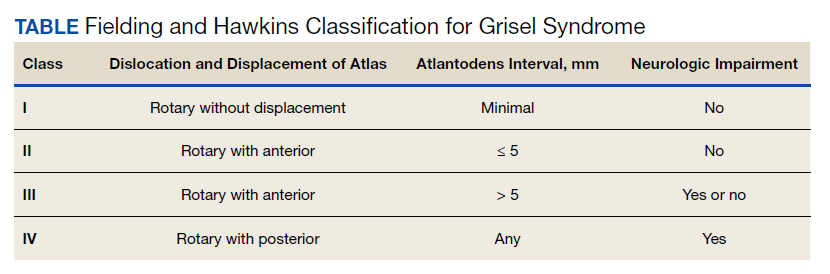
The degree of subluxation seen in Grisel syndrome correlates to the disease severity and is classified by the Fielding and Hawkins (FH) system (Table). This system accounts for the distance from the atlas to the dens (atlantodens interval) and the relative asymmetry of the atlantoaxial joint.6 In a normal adult, the upper limit of normal for the atlantodens interval is 3 mm, whereas this distance increases to 4.5 mm for the pediatric population.7 Type I (FH-I) involves rotary subluxation alone without any increase in the atlantodens interval; in FH-II, that interval has increased from normal but to no more than 5 mm. FH-I and FH-II are the most encountered and are not associated with neurologic impairment. In FH-III, neurologic deficits can be present, and the atlantodens interval is increased to > 5 mm. Different from FH-II and FH-III in which anterior dislocation of the atlas with reference to the dens is observed, FH-IV involves a rotary movement of the atlas with concurrent posterior displacement and often involves spinal cord compression.6
Subluxation and displacement without trauma are key components of Grisel syndrome. The 2-hit hypothesis is often used to explain how this can occur, ie, 2 anomalies must be present simultaneously for this condition to develop. First, the laxity of the transverse ligament, the posterior wall of the dens, and other atlantoaxial ligaments must be increased. Second, an asymmetric contraction of the deep erector muscles of the neck either abruptly or more insidiously rotate and dislocate the atlas.8 The pathophysiology is not exactly understood, but the most commonly held hypothesis describes contiguous spread of infection or inflammatory mediators from the pharynx to the ligaments and muscles described.6
Spread could occur via the venous system. The posterior superior pharyngeal region is drained by the periodontoidal venous plexus; the connections here with the pharyngovertebral veins allow for the embolization of infectious or other proinflammatory material to the prevertebral fascia. These emboli induce fasciitis and subsequent aberrant relaxation of the ligaments. In reaction to the inflammation or increased laxity, contiguous muscles of the deep neck contract and freeze the joint out of anatomic alignment.4
The abnormal alignment is apparent grossly as torticollis. Most broadly, torticollis describes an anomalous head posture due to involuntary muscle contractions of neck muscles and specifically describes chin deviation to the side. The antecollis and retrocollis subtypes of torticollis describe forward flexion and backward extension of the neck, respectively.7 Torticollis (broadly) is the most frequently reported condition of those found to have Grisel syndrome (90.7%); other common presenting conditions include neck pain (81.5%) and neck stiffness (31.5%). Fever is found in only 27.8% of cases. Pediatric patients (aged ≤ 12 years) are the most commonly affected, accounting for 87% of cases with an observed 4:1 male to female predominance.7,8 Symptoms begin most often within the first week from the inciting event in 85% of the cases.8 Head and neck surgery precedes up to 67% of cases, and infectious etiologies largely account for the remaining cases.7 Of the postsurgical cases, 55.6% had undergone T&A.8
Although anomalous head posture or neck stiffness following T&A would be of great clinic concern for Grisel syndrome, radiographic studies play a confirmatory role. CT scan is used to evaluate the bony structures, with 3D reconstruction of the cervical spine being most useful to determine the presence and degree of subluxation.8 Magnetic resonance imaging also aids in diagnosis to evaluate ligamentous structures in the area of concern as well as in the evaluation of spinal cord compression.6 Laboratory tests are largely unhelpful in making or excluding the diagnosis.8
If Grisel syndrome is suspected, both the original surgeon (if preceded by surgery) and the neurosurgical team should be consulted. Although no widely adopted guidelines exist for the management of this rare disease, general practice patterns have emerged with the degree of intervention predictably correlating to disease severity. FH-I is usually treated with nonsteroidal anti-inflammatory drugs and muscle relaxants with or without a soft cervical collar. For FH-II, closed reduction and immobilization in a stiff cervical collar is recommended. If no neurologic defect is present, FH-III is treated with bed rest, a period of inline cervical traction, and subsequent immobilization. FH-III with neurologic sequelae and all FH-IV necessitate emergent neurosurgical consultation.4 Surgical intervention is a last resort but is required in up to 24.1% of cases.8
Antibiotic therapy is not routinely given unless clear infectious etiology is identified. No standard antibiotic regimen exists, but coverage for typical upper respiratory pathogens likely suffices. Empiric antibiotic therapy is not recommended for all causes of Grisel syndrome, ie, when the underlying cause is not yet elucidated.6 One case of Grisel syndrome occurring in the setting of cervical osteomyelitis has been described, though, and required prolonged IV antibiotics.3 Physical therapy is recommended as adjunct with no limitations for range of motion save for that of the patient’s individual pain threshold.4
Possibly attributable to waxing and waning ligamentous laxity and strength of the neck muscle contraction, the atlantodens interval and the degree of subluxation can change, making Grisel syndrome dynamic. As such, the FH classification can change, necessitating more or less aggressive therapy. A neurologic evaluation is recommended at least every 2 weeks after the diagnosis is made. If initial identification or recognition of known disease progression is delayed, serious complications can develop. Acutely, spinal cord compression can lead to quadriplegia and death; more insidious complications include reduced neck mobility, dysphonia, and dysphagia.4 As serious, life-threatening complications can arise from Grisel syndrome while good functional outcomes can be achieved with timely and appropriate treatment, the clinician should be inspired to have a high clinical suspicion for this syndrome given the right context.
Conclusions
The patient experienced a desirable outcome with minimal, conservative treatment. As such, the pathology in this case was likely attributed to the mildest form of Grisel syndrome (FH-I). The follow-up was reassuring as well, revealing no worsening or progression of symptoms. The initial evaluation in this case was limited by the inadequacy of the CT scan. Motion artifact in the pharynx prevented the definite exclusion of deep space infection, while the rotation of the head in combination with motion artifact in the cranial-most portions of the vertebral column made determining alignment difficult. One clear axial image, though, does show rotation of the atlas (Figure 2). The uncertainty at the end of our workup prompted surgical consultation, not, admittedly, concern for Grisel syndrome. Awareness of this disease entity is nevertheless important and clinically relevant. Early identification and treatment is associated with decreased morbidity and improvement in long-term functional outcomes.6 Despite its rarity, the clinician should consider Grisel syndrome in any pediatric patient presenting with neck stiffness following the commonly performed T&A.
1. Ramos SD, Mukerji S, Pine HS. Tonsillectomy and adenoidectomy. Pediatr Clin North Am. 2013;60(4):793-807. doi:10.1016/j.pcl.2013.04.015
2. Stoner MJ, Dulaurier M. Pediatric ENT emergencies. Emerg Med Clin North Am. 2013;31(3):795-808. doi:10.1016/j.emc.2013.04.005
3. Leong SC, Karoos PD, Papouliakos SM, et al. Unusual complications of tonsillectomy: a systematic review. Am J Otolaryngol. 2007;28(6):419-422. doi:10.1016/j.amjoto.2006.10.016
4. Fath L, Cebula H, Santin MN, Cocab A, Debrya C, Proustb F. The Grisel’s syndrome: a non-traumatic subluxation of the atlantoaxial joint. Neurochirurgie. 2018;64(4):327-330. doi:10.1016/j.neuchi.2018.02.001
5. Moore K, Agur A, Dalley A. Essential Clinical Anatomy. 5th ed. Baltimore: Lippincott, Williams, and Wilkins; 2015:282-287.
6. Spennato P, Nicosia G, Rapanà A, et al. Grisel syndrome following adenoidectomy: surgical management in a case with delayed diagnosis. World Neurosurg. 2015;84(5):1494.e7-e12.
7. Anania P, Pavone P, Pacetti M, et al. Grisel syndrome in pediatric age: a single-center Italian experience and review of the literature. World Neurosurg. 2019;125:374-382. doi:10.1016/j.wneu.2019.02.035
8. Aldriweesh T, Altheyab F, Alenezi M, et al. Grisel’s syndrome post otolaryngology procedures: a systematic review. Int J Pediatr Otorhinolaryngol. 2020;137:110-125. doi:10.1016/j.ijporl.2020.110225
Tonsillectomy with or without adenoidectomy (T&A) is the second most common pediatric surgical procedure in the United States.1 It is most often performed during childhood between 5 and 8 years of age with a second peak observed between 17 and 21 years of age in the adolescent and young adult populations.2 While recurrent tonsillitis has been traditionally associated with tonsillectomy, sleep disordered breathing with obstructive sleep apnea is now the primary indication for the procedure.1
Up to 97% of T&As are performed as an outpatient same-day surgery not requiring inpatient admission.2 Although largely a safe and routinely performed surgery, several complications have been described. Due to the outpatient nature of the procedure, the complications are often encountered in the emergency department (ED) and sometimes in primary care settings. Common complications (outside of the perioperative time frame) include nausea, vomiting, otalgia, odynophagia, infection of the throat (broadly), and hemorrhage; uncommon complications include subcutaneous emphysema, taste disorders, and Eagle syndrome. Some complications are rarer still and carry significant morbidity and even mortality, including mediastinitis, cervical osteomyelitis, and Grisel syndrome.3 The following case encourages the clinician to expand the differential for a patient presenting after T&A.
Case Presentation
A child aged < 3 years was brought to the ED by their mother. She reported neck pain and stiffness 10 days after T&A with concurrent tympanostomy tube placement at an outside pediatric hospital. At triage, their heart rate was 94 bpm, temperature was 98.2 °F, respiratory rate, 22 breaths per minute, and oxygen saturation, 97% on room air. The mother of the patient (MOP) had been giving the prescribed oral liquid formulations of ibuprofen and acetaminophen with hydrocodone as directed. No drug allergies were reported, and immunizations were up to date for age. Other medical and surgical history included eczema and remote cutaneous hemangioma resection. The patient lived at home with 2 parents and was not exposed to smoke; their family history was noncontributory.
Since the surgery, the MOP had noticed constant and increasing neck stiffness, specifically with looking up and down but not side to side. She also had noticed swelling behind both ears. She reported no substantial decrease in intake by mouth or decrease in urine or bowel frequency. On review of systems, she reported no fever, vomiting, difficulty breathing, bleeding from the mouth or nose, eye or ear drainage, or rash.
On physical examination, the patient was alert and in no acute distress; active and playful on an electronic device but was notably not moving their head, which was held in a forward-looking position without any signs of trauma. When asked, the child would not flex or extend their neck but would rotate a few degrees from neutral to both sides. Even with moving the electronic device up and down in space, no active neck extension or flexion could be elicited. The examination of the head, eyes, ears, nose, and throat was otherwise only remarkable for palpable and mildly tender postauricular lymph nodes and diffuse erythema in the posterior pharynx. Cardiopulmonary, abdominal, skin, and extremity examinations were unremarkable.
With concern for an infectious process, the physician ordered blood chemistry and hematology tests along with neck radiography. While awaiting the results, the patient was given a weight-based bolus of normal saline, and the home pain regimen was administered. An attempt was made to passively flex and extend the neck as the child slept in their mother’s arms, but the patient immediately awoke and began to cry.
All values of the comprehensive metabolic panel were within normal limits except for a slight elevation in the blood urea nitrogen to 21 mg/dL and glucose to 159 mg/dL. The complete blood count was unrevealing. The computed tomography (CT) scan with contrast of the soft tissues of the neck was limited by motion artifact but showed a head held in axial rotation with soft tissue irregularity in the anterior aspect of the adenoids (Figure 1). There was what appeared to be normal lymphadenopathy in the hypopharynx, but the soft tissues were otherwise unremarkable.
The on-call pediatric otolaryngologist at the hospital where the procedure was performed was paged. On hearing the details of the case, the specialist was concerned for Grisel syndrome and requested to see the patient in their facility. No additional recommendations for care were provided; the mother was updated and agreed to transfer. The patient was comfortable and stable with repeat vitals as follows: heart rate, 86 beats per minute, blood pressure, 99/62, temperature, 98.3 °F, respiratory rate, 20 breaths per minute, and oxygen saturation, 99% on room air.
On arrival at the receiving facility, the emergency team performed a history and physical that revealed no significant changes from the initial evaluation. They then facilitated evaluation by the pediatric otolaryngologist who conducted a more directed physical examination. Decreased active and passive range of motion (ROM) of the neck without rotatory restriction was again noted. They also observed scant fibrinous exudate within the oropharynx and tonsillar fossa, which was normal in the setting of the recent surgery. They recommended additional analgesia with intramuscular ketorolac, weight-based dosing at 1 mg/kg.
With repeat examination after this additional analgesic, ROM of the neck first passive then active had improved. The patient was then discharged to follow up in the coming days with instructions to continue the pain and anti-inflammatory regimen. They were not started on an antibiotic at that time nor were they placed in a cervical collar. At the follow-up, the MOP reported persistence of neck stiffness for a few days initially but then observed slow improvement. By postoperative day 18, the stiffness had resolved. No other follow-up or referrals related to this issue were readily apparent in review of the patient’s health record.
Discussion
Grisel syndrome is the atraumatic rotary subluxation of the atlantoaxial joint, specifically, the atlas (C1 vertebra) rotates to a fixed, nonanatomic position while the axis (C2 vertebra) remains in normal alignment in relation to the remainder of the spinal column. The subluxation occurs in the absence of ligamentous injury but is associated with an increase in ligamentous laxity.4 The atlas is a ring-shaped vertebra with 2 lateral masses connected by anterior and posterior arches; it lacks a spinous process unlike other vertebrae. It articulates with the skull by means of the 2 articular facets on the superior aspect of the lateral masses. Articulation with the axis occurs at 3 sites: 2 articular facets on the inferior portion of the lateral masses of the atlas and a facet for the dens on the posterior portion of the anterior arch. The dens projects superiorly from the body of the axis and is bound posteriorly by the transverse ligament of the atlas.5
The degree of subluxation seen in Grisel syndrome correlates to the disease severity and is classified by the Fielding and Hawkins (FH) system (Table). This system accounts for the distance from the atlas to the dens (atlantodens interval) and the relative asymmetry of the atlantoaxial joint.6 In a normal adult, the upper limit of normal for the atlantodens interval is 3 mm, whereas this distance increases to 4.5 mm for the pediatric population.7 Type I (FH-I) involves rotary subluxation alone without any increase in the atlantodens interval; in FH-II, that interval has increased from normal but to no more than 5 mm. FH-I and FH-II are the most encountered and are not associated with neurologic impairment. In FH-III, neurologic deficits can be present, and the atlantodens interval is increased to > 5 mm. Different from FH-II and FH-III in which anterior dislocation of the atlas with reference to the dens is observed, FH-IV involves a rotary movement of the atlas with concurrent posterior displacement and often involves spinal cord compression.6
Subluxation and displacement without trauma are key components of Grisel syndrome. The 2-hit hypothesis is often used to explain how this can occur, ie, 2 anomalies must be present simultaneously for this condition to develop. First, the laxity of the transverse ligament, the posterior wall of the dens, and other atlantoaxial ligaments must be increased. Second, an asymmetric contraction of the deep erector muscles of the neck either abruptly or more insidiously rotate and dislocate the atlas.8 The pathophysiology is not exactly understood, but the most commonly held hypothesis describes contiguous spread of infection or inflammatory mediators from the pharynx to the ligaments and muscles described.6
Spread could occur via the venous system. The posterior superior pharyngeal region is drained by the periodontoidal venous plexus; the connections here with the pharyngovertebral veins allow for the embolization of infectious or other proinflammatory material to the prevertebral fascia. These emboli induce fasciitis and subsequent aberrant relaxation of the ligaments. In reaction to the inflammation or increased laxity, contiguous muscles of the deep neck contract and freeze the joint out of anatomic alignment.4
The abnormal alignment is apparent grossly as torticollis. Most broadly, torticollis describes an anomalous head posture due to involuntary muscle contractions of neck muscles and specifically describes chin deviation to the side. The antecollis and retrocollis subtypes of torticollis describe forward flexion and backward extension of the neck, respectively.7 Torticollis (broadly) is the most frequently reported condition of those found to have Grisel syndrome (90.7%); other common presenting conditions include neck pain (81.5%) and neck stiffness (31.5%). Fever is found in only 27.8% of cases. Pediatric patients (aged ≤ 12 years) are the most commonly affected, accounting for 87% of cases with an observed 4:1 male to female predominance.7,8 Symptoms begin most often within the first week from the inciting event in 85% of the cases.8 Head and neck surgery precedes up to 67% of cases, and infectious etiologies largely account for the remaining cases.7 Of the postsurgical cases, 55.6% had undergone T&A.8
Although anomalous head posture or neck stiffness following T&A would be of great clinic concern for Grisel syndrome, radiographic studies play a confirmatory role. CT scan is used to evaluate the bony structures, with 3D reconstruction of the cervical spine being most useful to determine the presence and degree of subluxation.8 Magnetic resonance imaging also aids in diagnosis to evaluate ligamentous structures in the area of concern as well as in the evaluation of spinal cord compression.6 Laboratory tests are largely unhelpful in making or excluding the diagnosis.8
If Grisel syndrome is suspected, both the original surgeon (if preceded by surgery) and the neurosurgical team should be consulted. Although no widely adopted guidelines exist for the management of this rare disease, general practice patterns have emerged with the degree of intervention predictably correlating to disease severity. FH-I is usually treated with nonsteroidal anti-inflammatory drugs and muscle relaxants with or without a soft cervical collar. For FH-II, closed reduction and immobilization in a stiff cervical collar is recommended. If no neurologic defect is present, FH-III is treated with bed rest, a period of inline cervical traction, and subsequent immobilization. FH-III with neurologic sequelae and all FH-IV necessitate emergent neurosurgical consultation.4 Surgical intervention is a last resort but is required in up to 24.1% of cases.8
Antibiotic therapy is not routinely given unless clear infectious etiology is identified. No standard antibiotic regimen exists, but coverage for typical upper respiratory pathogens likely suffices. Empiric antibiotic therapy is not recommended for all causes of Grisel syndrome, ie, when the underlying cause is not yet elucidated.6 One case of Grisel syndrome occurring in the setting of cervical osteomyelitis has been described, though, and required prolonged IV antibiotics.3 Physical therapy is recommended as adjunct with no limitations for range of motion save for that of the patient’s individual pain threshold.4
Possibly attributable to waxing and waning ligamentous laxity and strength of the neck muscle contraction, the atlantodens interval and the degree of subluxation can change, making Grisel syndrome dynamic. As such, the FH classification can change, necessitating more or less aggressive therapy. A neurologic evaluation is recommended at least every 2 weeks after the diagnosis is made. If initial identification or recognition of known disease progression is delayed, serious complications can develop. Acutely, spinal cord compression can lead to quadriplegia and death; more insidious complications include reduced neck mobility, dysphonia, and dysphagia.4 As serious, life-threatening complications can arise from Grisel syndrome while good functional outcomes can be achieved with timely and appropriate treatment, the clinician should be inspired to have a high clinical suspicion for this syndrome given the right context.
Conclusions
The patient experienced a desirable outcome with minimal, conservative treatment. As such, the pathology in this case was likely attributed to the mildest form of Grisel syndrome (FH-I). The follow-up was reassuring as well, revealing no worsening or progression of symptoms. The initial evaluation in this case was limited by the inadequacy of the CT scan. Motion artifact in the pharynx prevented the definite exclusion of deep space infection, while the rotation of the head in combination with motion artifact in the cranial-most portions of the vertebral column made determining alignment difficult. One clear axial image, though, does show rotation of the atlas (Figure 2). The uncertainty at the end of our workup prompted surgical consultation, not, admittedly, concern for Grisel syndrome. Awareness of this disease entity is nevertheless important and clinically relevant. Early identification and treatment is associated with decreased morbidity and improvement in long-term functional outcomes.6 Despite its rarity, the clinician should consider Grisel syndrome in any pediatric patient presenting with neck stiffness following the commonly performed T&A.
Tonsillectomy with or without adenoidectomy (T&A) is the second most common pediatric surgical procedure in the United States.1 It is most often performed during childhood between 5 and 8 years of age with a second peak observed between 17 and 21 years of age in the adolescent and young adult populations.2 While recurrent tonsillitis has been traditionally associated with tonsillectomy, sleep disordered breathing with obstructive sleep apnea is now the primary indication for the procedure.1
Up to 97% of T&As are performed as an outpatient same-day surgery not requiring inpatient admission.2 Although largely a safe and routinely performed surgery, several complications have been described. Due to the outpatient nature of the procedure, the complications are often encountered in the emergency department (ED) and sometimes in primary care settings. Common complications (outside of the perioperative time frame) include nausea, vomiting, otalgia, odynophagia, infection of the throat (broadly), and hemorrhage; uncommon complications include subcutaneous emphysema, taste disorders, and Eagle syndrome. Some complications are rarer still and carry significant morbidity and even mortality, including mediastinitis, cervical osteomyelitis, and Grisel syndrome.3 The following case encourages the clinician to expand the differential for a patient presenting after T&A.
Case Presentation
A child aged < 3 years was brought to the ED by their mother. She reported neck pain and stiffness 10 days after T&A with concurrent tympanostomy tube placement at an outside pediatric hospital. At triage, their heart rate was 94 bpm, temperature was 98.2 °F, respiratory rate, 22 breaths per minute, and oxygen saturation, 97% on room air. The mother of the patient (MOP) had been giving the prescribed oral liquid formulations of ibuprofen and acetaminophen with hydrocodone as directed. No drug allergies were reported, and immunizations were up to date for age. Other medical and surgical history included eczema and remote cutaneous hemangioma resection. The patient lived at home with 2 parents and was not exposed to smoke; their family history was noncontributory.
Since the surgery, the MOP had noticed constant and increasing neck stiffness, specifically with looking up and down but not side to side. She also had noticed swelling behind both ears. She reported no substantial decrease in intake by mouth or decrease in urine or bowel frequency. On review of systems, she reported no fever, vomiting, difficulty breathing, bleeding from the mouth or nose, eye or ear drainage, or rash.
On physical examination, the patient was alert and in no acute distress; active and playful on an electronic device but was notably not moving their head, which was held in a forward-looking position without any signs of trauma. When asked, the child would not flex or extend their neck but would rotate a few degrees from neutral to both sides. Even with moving the electronic device up and down in space, no active neck extension or flexion could be elicited. The examination of the head, eyes, ears, nose, and throat was otherwise only remarkable for palpable and mildly tender postauricular lymph nodes and diffuse erythema in the posterior pharynx. Cardiopulmonary, abdominal, skin, and extremity examinations were unremarkable.
With concern for an infectious process, the physician ordered blood chemistry and hematology tests along with neck radiography. While awaiting the results, the patient was given a weight-based bolus of normal saline, and the home pain regimen was administered. An attempt was made to passively flex and extend the neck as the child slept in their mother’s arms, but the patient immediately awoke and began to cry.
All values of the comprehensive metabolic panel were within normal limits except for a slight elevation in the blood urea nitrogen to 21 mg/dL and glucose to 159 mg/dL. The complete blood count was unrevealing. The computed tomography (CT) scan with contrast of the soft tissues of the neck was limited by motion artifact but showed a head held in axial rotation with soft tissue irregularity in the anterior aspect of the adenoids (Figure 1). There was what appeared to be normal lymphadenopathy in the hypopharynx, but the soft tissues were otherwise unremarkable.
The on-call pediatric otolaryngologist at the hospital where the procedure was performed was paged. On hearing the details of the case, the specialist was concerned for Grisel syndrome and requested to see the patient in their facility. No additional recommendations for care were provided; the mother was updated and agreed to transfer. The patient was comfortable and stable with repeat vitals as follows: heart rate, 86 beats per minute, blood pressure, 99/62, temperature, 98.3 °F, respiratory rate, 20 breaths per minute, and oxygen saturation, 99% on room air.
On arrival at the receiving facility, the emergency team performed a history and physical that revealed no significant changes from the initial evaluation. They then facilitated evaluation by the pediatric otolaryngologist who conducted a more directed physical examination. Decreased active and passive range of motion (ROM) of the neck without rotatory restriction was again noted. They also observed scant fibrinous exudate within the oropharynx and tonsillar fossa, which was normal in the setting of the recent surgery. They recommended additional analgesia with intramuscular ketorolac, weight-based dosing at 1 mg/kg.
With repeat examination after this additional analgesic, ROM of the neck first passive then active had improved. The patient was then discharged to follow up in the coming days with instructions to continue the pain and anti-inflammatory regimen. They were not started on an antibiotic at that time nor were they placed in a cervical collar. At the follow-up, the MOP reported persistence of neck stiffness for a few days initially but then observed slow improvement. By postoperative day 18, the stiffness had resolved. No other follow-up or referrals related to this issue were readily apparent in review of the patient’s health record.
Discussion
Grisel syndrome is the atraumatic rotary subluxation of the atlantoaxial joint, specifically, the atlas (C1 vertebra) rotates to a fixed, nonanatomic position while the axis (C2 vertebra) remains in normal alignment in relation to the remainder of the spinal column. The subluxation occurs in the absence of ligamentous injury but is associated with an increase in ligamentous laxity.4 The atlas is a ring-shaped vertebra with 2 lateral masses connected by anterior and posterior arches; it lacks a spinous process unlike other vertebrae. It articulates with the skull by means of the 2 articular facets on the superior aspect of the lateral masses. Articulation with the axis occurs at 3 sites: 2 articular facets on the inferior portion of the lateral masses of the atlas and a facet for the dens on the posterior portion of the anterior arch. The dens projects superiorly from the body of the axis and is bound posteriorly by the transverse ligament of the atlas.5
The degree of subluxation seen in Grisel syndrome correlates to the disease severity and is classified by the Fielding and Hawkins (FH) system (Table). This system accounts for the distance from the atlas to the dens (atlantodens interval) and the relative asymmetry of the atlantoaxial joint.6 In a normal adult, the upper limit of normal for the atlantodens interval is 3 mm, whereas this distance increases to 4.5 mm for the pediatric population.7 Type I (FH-I) involves rotary subluxation alone without any increase in the atlantodens interval; in FH-II, that interval has increased from normal but to no more than 5 mm. FH-I and FH-II are the most encountered and are not associated with neurologic impairment. In FH-III, neurologic deficits can be present, and the atlantodens interval is increased to > 5 mm. Different from FH-II and FH-III in which anterior dislocation of the atlas with reference to the dens is observed, FH-IV involves a rotary movement of the atlas with concurrent posterior displacement and often involves spinal cord compression.6
Subluxation and displacement without trauma are key components of Grisel syndrome. The 2-hit hypothesis is often used to explain how this can occur, ie, 2 anomalies must be present simultaneously for this condition to develop. First, the laxity of the transverse ligament, the posterior wall of the dens, and other atlantoaxial ligaments must be increased. Second, an asymmetric contraction of the deep erector muscles of the neck either abruptly or more insidiously rotate and dislocate the atlas.8 The pathophysiology is not exactly understood, but the most commonly held hypothesis describes contiguous spread of infection or inflammatory mediators from the pharynx to the ligaments and muscles described.6
Spread could occur via the venous system. The posterior superior pharyngeal region is drained by the periodontoidal venous plexus; the connections here with the pharyngovertebral veins allow for the embolization of infectious or other proinflammatory material to the prevertebral fascia. These emboli induce fasciitis and subsequent aberrant relaxation of the ligaments. In reaction to the inflammation or increased laxity, contiguous muscles of the deep neck contract and freeze the joint out of anatomic alignment.4
The abnormal alignment is apparent grossly as torticollis. Most broadly, torticollis describes an anomalous head posture due to involuntary muscle contractions of neck muscles and specifically describes chin deviation to the side. The antecollis and retrocollis subtypes of torticollis describe forward flexion and backward extension of the neck, respectively.7 Torticollis (broadly) is the most frequently reported condition of those found to have Grisel syndrome (90.7%); other common presenting conditions include neck pain (81.5%) and neck stiffness (31.5%). Fever is found in only 27.8% of cases. Pediatric patients (aged ≤ 12 years) are the most commonly affected, accounting for 87% of cases with an observed 4:1 male to female predominance.7,8 Symptoms begin most often within the first week from the inciting event in 85% of the cases.8 Head and neck surgery precedes up to 67% of cases, and infectious etiologies largely account for the remaining cases.7 Of the postsurgical cases, 55.6% had undergone T&A.8
Although anomalous head posture or neck stiffness following T&A would be of great clinic concern for Grisel syndrome, radiographic studies play a confirmatory role. CT scan is used to evaluate the bony structures, with 3D reconstruction of the cervical spine being most useful to determine the presence and degree of subluxation.8 Magnetic resonance imaging also aids in diagnosis to evaluate ligamentous structures in the area of concern as well as in the evaluation of spinal cord compression.6 Laboratory tests are largely unhelpful in making or excluding the diagnosis.8
If Grisel syndrome is suspected, both the original surgeon (if preceded by surgery) and the neurosurgical team should be consulted. Although no widely adopted guidelines exist for the management of this rare disease, general practice patterns have emerged with the degree of intervention predictably correlating to disease severity. FH-I is usually treated with nonsteroidal anti-inflammatory drugs and muscle relaxants with or without a soft cervical collar. For FH-II, closed reduction and immobilization in a stiff cervical collar is recommended. If no neurologic defect is present, FH-III is treated with bed rest, a period of inline cervical traction, and subsequent immobilization. FH-III with neurologic sequelae and all FH-IV necessitate emergent neurosurgical consultation.4 Surgical intervention is a last resort but is required in up to 24.1% of cases.8
Antibiotic therapy is not routinely given unless clear infectious etiology is identified. No standard antibiotic regimen exists, but coverage for typical upper respiratory pathogens likely suffices. Empiric antibiotic therapy is not recommended for all causes of Grisel syndrome, ie, when the underlying cause is not yet elucidated.6 One case of Grisel syndrome occurring in the setting of cervical osteomyelitis has been described, though, and required prolonged IV antibiotics.3 Physical therapy is recommended as adjunct with no limitations for range of motion save for that of the patient’s individual pain threshold.4
Possibly attributable to waxing and waning ligamentous laxity and strength of the neck muscle contraction, the atlantodens interval and the degree of subluxation can change, making Grisel syndrome dynamic. As such, the FH classification can change, necessitating more or less aggressive therapy. A neurologic evaluation is recommended at least every 2 weeks after the diagnosis is made. If initial identification or recognition of known disease progression is delayed, serious complications can develop. Acutely, spinal cord compression can lead to quadriplegia and death; more insidious complications include reduced neck mobility, dysphonia, and dysphagia.4 As serious, life-threatening complications can arise from Grisel syndrome while good functional outcomes can be achieved with timely and appropriate treatment, the clinician should be inspired to have a high clinical suspicion for this syndrome given the right context.
Conclusions
The patient experienced a desirable outcome with minimal, conservative treatment. As such, the pathology in this case was likely attributed to the mildest form of Grisel syndrome (FH-I). The follow-up was reassuring as well, revealing no worsening or progression of symptoms. The initial evaluation in this case was limited by the inadequacy of the CT scan. Motion artifact in the pharynx prevented the definite exclusion of deep space infection, while the rotation of the head in combination with motion artifact in the cranial-most portions of the vertebral column made determining alignment difficult. One clear axial image, though, does show rotation of the atlas (Figure 2). The uncertainty at the end of our workup prompted surgical consultation, not, admittedly, concern for Grisel syndrome. Awareness of this disease entity is nevertheless important and clinically relevant. Early identification and treatment is associated with decreased morbidity and improvement in long-term functional outcomes.6 Despite its rarity, the clinician should consider Grisel syndrome in any pediatric patient presenting with neck stiffness following the commonly performed T&A.
1. Ramos SD, Mukerji S, Pine HS. Tonsillectomy and adenoidectomy. Pediatr Clin North Am. 2013;60(4):793-807. doi:10.1016/j.pcl.2013.04.015
2. Stoner MJ, Dulaurier M. Pediatric ENT emergencies. Emerg Med Clin North Am. 2013;31(3):795-808. doi:10.1016/j.emc.2013.04.005
3. Leong SC, Karoos PD, Papouliakos SM, et al. Unusual complications of tonsillectomy: a systematic review. Am J Otolaryngol. 2007;28(6):419-422. doi:10.1016/j.amjoto.2006.10.016
4. Fath L, Cebula H, Santin MN, Cocab A, Debrya C, Proustb F. The Grisel’s syndrome: a non-traumatic subluxation of the atlantoaxial joint. Neurochirurgie. 2018;64(4):327-330. doi:10.1016/j.neuchi.2018.02.001
5. Moore K, Agur A, Dalley A. Essential Clinical Anatomy. 5th ed. Baltimore: Lippincott, Williams, and Wilkins; 2015:282-287.
6. Spennato P, Nicosia G, Rapanà A, et al. Grisel syndrome following adenoidectomy: surgical management in a case with delayed diagnosis. World Neurosurg. 2015;84(5):1494.e7-e12.
7. Anania P, Pavone P, Pacetti M, et al. Grisel syndrome in pediatric age: a single-center Italian experience and review of the literature. World Neurosurg. 2019;125:374-382. doi:10.1016/j.wneu.2019.02.035
8. Aldriweesh T, Altheyab F, Alenezi M, et al. Grisel’s syndrome post otolaryngology procedures: a systematic review. Int J Pediatr Otorhinolaryngol. 2020;137:110-125. doi:10.1016/j.ijporl.2020.110225
1. Ramos SD, Mukerji S, Pine HS. Tonsillectomy and adenoidectomy. Pediatr Clin North Am. 2013;60(4):793-807. doi:10.1016/j.pcl.2013.04.015
2. Stoner MJ, Dulaurier M. Pediatric ENT emergencies. Emerg Med Clin North Am. 2013;31(3):795-808. doi:10.1016/j.emc.2013.04.005
3. Leong SC, Karoos PD, Papouliakos SM, et al. Unusual complications of tonsillectomy: a systematic review. Am J Otolaryngol. 2007;28(6):419-422. doi:10.1016/j.amjoto.2006.10.016
4. Fath L, Cebula H, Santin MN, Cocab A, Debrya C, Proustb F. The Grisel’s syndrome: a non-traumatic subluxation of the atlantoaxial joint. Neurochirurgie. 2018;64(4):327-330. doi:10.1016/j.neuchi.2018.02.001
5. Moore K, Agur A, Dalley A. Essential Clinical Anatomy. 5th ed. Baltimore: Lippincott, Williams, and Wilkins; 2015:282-287.
6. Spennato P, Nicosia G, Rapanà A, et al. Grisel syndrome following adenoidectomy: surgical management in a case with delayed diagnosis. World Neurosurg. 2015;84(5):1494.e7-e12.
7. Anania P, Pavone P, Pacetti M, et al. Grisel syndrome in pediatric age: a single-center Italian experience and review of the literature. World Neurosurg. 2019;125:374-382. doi:10.1016/j.wneu.2019.02.035
8. Aldriweesh T, Altheyab F, Alenezi M, et al. Grisel’s syndrome post otolaryngology procedures: a systematic review. Int J Pediatr Otorhinolaryngol. 2020;137:110-125. doi:10.1016/j.ijporl.2020.110225
Repeat Laparoscopic Cholecystectomy for Duplicated Gallbladder After 16-Year Interval
Gallbladder duplication is a congenital abnormality of the hepatobiliary system and often is not considered in the evaluation of a patient with right upper quadrant pain. Accuracy of the most commonly used imaging study to assess for biliary disease, abdominal ultrasound, is highly dependent on the skills of the ultrasonographer, and given its relative rarity, this condition is often not considered prior to planned cholecystectomy.1 Small case reviews found that < 50% of gallbladder duplications are diagnosed preoperatively despite use of ultrasound or computed tomography (CT) scan.2-4 Failure to recognize duplicate gallbladder anatomy in symptomatic patients may result in incomplete surgical management, an increase in perioperative complications, and years of morbidity due to unresolved symptoms. Once a patient has had a cholecystectomy, symptoms are presumed to be due to a nonbiliary etiology and an extensive, often repetitive, workup is pursued before “repeat cholecystectomy” is considered.5
Case Presentation
A 63-year-old man was referred to gastroenterology for recurrent episodic right upper quadrant pain. He reported intermittent both right and left upper abdominal pain that was variable in quality. At times it was associated with an empty stomach prior to meals; at other times, onset was 30 to 60 minutes after meals. The patient also reported significant flatulence and bloating and intermittent loose stools. Sixteen years before, he underwent a laparoscopic cholecystectomy. He reported that the pain he experienced before the cholecystectomy never resolved after surgery but occurred less frequently. For the next 16 years, the patient did not seek evaluation of his ongoing but infrequent symptoms until his pain became a daily occurrence. The patient’s surgical history included a remote open vagotomy and antrectomy for peptic ulcer disease, laparoscopic appendectomy, and a laparoscopic cholecystectomy for reported biliary colic.
The gastroenterology evaluation included a colonoscopy and esophagogastroduodenoscopy (EGD); both were benign and without findings specific to identify the etiology for the patient’s pain. The patient was given a course of rifaximin 1200 mg daily for 7 days for possible bacterial overgrowth and placed on a proton pump inhibitor twice daily. Neither of these interventions helped resolve the patient’s symptoms. Further workup was pursued by gastroenterology to include a right upper quadrant ultrasound that showed a structure most consistent with a small gallbladder containing a small polyp vs stone. Magnetic resonance cholangiopancreatography (MRCP) also was performed and showed the presence of a small gallbladder with a small 2-mm filling defect and an otherwise benign biliary tree. MRCP images and EGD documented a Billroth 1 reconstruction at the time of his remote antrectomy and vagotomy (Figure 1).

The patient was referred to general surgery for consideration of a repeat cholecystectomy. He confirmed the history of intermittent upper abdominal pain for the past 16 years, which was similar to the symptoms he had experienced before his original laparoscopic cholecystectomy. On examination, the patient had a body mass index of 38, had a large upper midline incision from his prior antrectomy and vagotomy procedure, and several scars presumed to be port incision scars to the right lateral abdominal wall. Hospital records were obtained from the patient’s prior workup for biliary colic and cholecystectomy 16 years before. The preoperative abdominal ultrasound examination showed a mildly distended gallbladder but was notably described as “quite limited due to patient’s body habitus and liver is not well seen.” No additional imaging was documented in his presurgical evaluation notes and imaging records.
The operative report described a gallbladder that was densely adherent to adjacent fat and omental tissue with significant adhesions secondary to the prior vagotomy and antrectomy procedure. The cystic duct and artery were dissected free at the level of their junction with the gallbladder infundibulum. The cystic artery was divided with a harmonic scalpel. Following this the gallbladder body was dissected free from the liver bed in top-down fashion. A 0 Vicryl Endoloop suture was placed over the gallbladder and secured just past the origin of the cystic duct on the gallbladder infundibulum and the cystic duct divided above this suture. No surgical clips were used, which corresponded with the lack of surgical clips seen in imaging in his recent gastroenterology workup. No documentation of an intraoperative cholangiogram existed or was considered in the operative report.
The pathology report from this first cholecystectomy procedure noted the removed specimen to be an unopened 6-cm gallbladder containing 2 small yellow stones that otherwise were benign. At the time of this patient’s re-presentation to general surgery, there was suspicion that the patient’s prior surgical procedure had not been a cholecystectomy but rather a subtotal cholecystectomy. However, after appropriate workup and review of prior records, the patient had, indeed, previously undergone cholecystectomy and represented a rare case of gallbladder duplication resulting in abdominal pain for 16 years after his index operation.
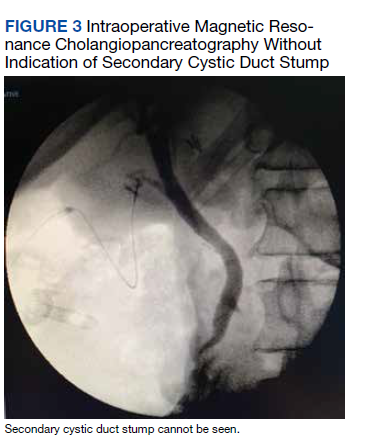
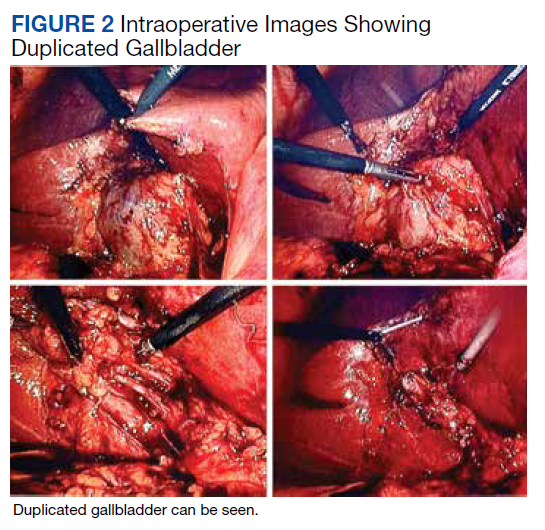
The patient was consented for repeat cholecystectomy and underwent a laparoscopic lysis of adhesions, cholecystectomy, and intraoperative cholangiogram. Significant scarring was found at the liver undersurface that would have been exposed during the original laparoscopic resection of the gallbladder from its liver bed. Deeper to this, a small saccular structure was identified as the duplicate gallbladder (Figure 2). Though the visualized gallbladder was small with a deep intrahepatic lie, the critical view of safety was achieved and was without additional variation. An intraoperative cholangiogram was performed to determine whether residual ductal stumps or other additional evidence of the previously removed gallbladder could be identified. The cholangiogram showed clear visualization of the cystic duct, common bile duct, right and left hepatic ducts, and contrast into the duodenum without abnormal variants. There was no visualized accessory or secondary cystic duct stump seen on the cholangiogram (Figure 3). Pathology of the repeat cholecystectomy specimen confirmed a 3-cm gallbladder with a distinct duct leading out of the gallbladder and the presence of several gallstones. The patient had an uneventful recovery after the repeat laparoscopic cholecystectomy with complete resolution of his upper abdominal pain.
Discussion
The first reported human case of gallbladder duplication was noted in a sacrificial victim of Emperor Augustus in 31 BCE. Sherren reported the first documented case of double accessory gallbladder in a living human in 1911.1,6 Though the exact incidence of gallbladder duplication is not fully known due to primary documentation from case reports, incidence is approximately 1 in 4000 to 5000 people. It was first formally classified by Boyden in 1926.7 Further anatomic classification based on morphology and embryogenesis was delineated by Harlaftis and colleagues in 1977, establishing type 1 and 2 structures of a duplicated gallbladder.8 Type 1 duplicated gallbladder anatomy shares a single cystic duct, whereas in type 2 each gallbladder has its own cystic duct. Later reports and studies identified triple gallbladders as well as trabecular variants with the most common classification used currently being the modified Harlaftis classification.9,10
The case presented here most likely represents either a Y-shaped type 1 primordial gallbladder or a type 2 accessory gallbladder based on historical data and intraoperative cholangiogram findings at the time of repeat cholecystectomy. Gallbladder duplication is clinically indistinguishable from regular gallbladder pathology preoperatively and can only be identified on imaging or intraoperatively.11 Prior case reports and studies have found that it is frequently missed on preoperative abdominal ultrasonography and CT in up to 50% of cases.12-14
The differential diagnosis of gallbladder duplication seen on preoperative imaging includes a gallbladder diverticulum, choledochal cyst, focal adenonomyomatosis, Phrygian cap, or folded gallbladder.1,2 Historically, the most definitive test for gallbladder duplication has been either intraoperative cholangiography, which can also clarify biliary anatomy, or endoscopic retrograde cholangiopancreatography with cholangiography.1,3 The debate over routine use of intraoperative cholangiography has been ongoing for the past several decades.15 Though intraoperative cholangiogram remains one of the most definitive tests for gallbladder duplication, given the overall low incidence of this variant, recommendation for routine intraoperative cholangiography solely to rule out gallbladder duplication cannot be definitively recommended based on our review of the literature. Currently, preoperative MRCP is the study of choice when there is concern from historical facts or from other imaging of gallbladder duplication as it is noninvasive and has a high degree of detail, particularly with 3D reconstructions.14,16 At the time of surgery, the most critical step to avoid inadvertent ductal injury is clear visualization of ductal anatomy and obtaining the critical view of safety.17 Though this will also assist in identifying some cases of gallbladder duplication, given the great variation of duplication, it will not prevent missing some variants. In our case, extensive local scarring from the patient’s prior antrectomy and vagotomy along with lack of the use of intraoperative cholangiography likely contributed to missing his duplication at the time of his index cholecystectomy.
Undiagnosed gallbladder duplication can lead to additional morbidity related to common entities associated with gallbladder pathology, such as biliary colic, cholecystitis, cholangitis, and pancreatitis. Additionally, case reports in the literature have documented more rare associations, such as empyema, carcinoma, cholecystoenteric fistula, and torsion, all associated with a duplicated gallbladder.18-21 Once identified pre- or intraoperatively, it is generally recommended that all gallbladders be removed in symptomatic patients and that intraoperative cholangiography be done to assure complete resection of the duplicated gallbladders and to avoid injury to the biliary trees.22-25
Conclusions
Gallbladder duplication and other congenital biliary anatomic variations should be considered before a biliary operation and included in the differential diagnosis when evaluating patients who have clinical symptoms consistent with biliary pathology. In addition, intraoperative cholangiogram should be performed during cholecystectomy if the inferior liver edge cannot be visualized well, as in the case of this patient where a prior foregut operation resulted in extensive adhesive disease. Intraoperative cholangiogram also should be considered in patients whose preoperative imaging does not visualize the right upper quadrant well due to patient habitus. Doing so may identify gallbladder duplication and allow for complete cholecystectomy as well as proper identification and management of cystic duct variants. Awareness and consideration of duplicated biliary variants can help prevent intraoperative complications related to biliary anomalies and avoid the morbidity related to recurrent biliary disease and the need for repeat operative procedures.
Acknowledgments
We extend our thanks to Veterans Affairs Puget Sound Healthcare System and the Departments of Surgery and Radiology for their support of this case report, and Lorrie Langdale, MD, and Roger Tatum, MD, for their mentorship of this project
1. Vezakis A, Pantiora E, Giannoulopoulos D, et al. A duplicated gallbladder in a patient presenting with acute cholangitis. A case study and a literature review. Ann Hepatol. 2019;18(1):240-245. doi:10.5604/01.3001.0012.7932
2. Barut Í, Tarhan ÖR, Dog^ru U, Bülbül M. Gallbladder duplication: diagnosed and treated by laparoscopy. Eur J Gen Med. 2006;3(3):142-145. doi:10.29333/ejgm/82396 3. Cozacov Y, Subhas G, Jacobs M, Parikh J. Total laparoscopic removal of accessory gallbladder: a case report and review of literature. World J Gastrointest Surg. 2015;7(12):398-402. doi:10.4240/wjgs.v7.i12.398
4. Musleh MG, Burnett H, Rajashanker B, Ammori BJ. Laparoscopic double cholecystectomy for duplicated gallbladder: a case report. Int J Surg Case Rep. 2017;41:502-504. Published 2017 Nov 27. doi:10.1016/j.ijscr.2017.11.046
5. Walbolt TD, Lalezarzadeh F. Laparoscopic management of a duplicated gallbladder: a case study and anatomic history. Surg Laparosc Endosc Percutan Tech. 2011;21(3):e156-e158. doi:10.1097/SLE.0b013e31821d47ce
6. Sherren J. A double gall-bladder removed by operation. Ann Surg. 1911;54(2):204-205. doi:10.1097/00000658-191108000-00009
7. Boyden EA. The accessory gall-bladder—an embryological and comparative study of aberrant biliary vesicles occurring in man and the domestic mammals. Am J Anat. 1926; 38(2):177-231. doi:10.1002/aja.1000380202
8. Harlaftis N, Gray SW, Skandalakis JE. Multiple gallbladders. Surg Gynecol Obstet. 1977;145(6):928-934.
9. Kim RD, Zendejas I, Velopulos C, et al. Duplicate gallbladder arising from the left hepatic duct: report of a case. Surg Today. 2009;39(6):536-539. doi:10.1007/s00595-008-3878-4
10. Causey MW, Miller S, Fernelius CA, Burgess JR, Brown TA, Newton C. Gallbladder duplication: evaluation, treatment, and classification. J Pediatr Surg. 2010;45(2):443-446. doi:10.1016/j.jpedsurg.2009.12.015
11. Apolo Romero EX, Gálvez Salazar PF, Estrada Chandi JA, et al. Gallbladder duplication and cholecystitis. J Surg Case Rep. 2018;2018(7):rjy158. Published 2018 Jul 3. doi:10.1093/jscr/rjy158
12. Gorecki PJ, Andrei VE, Musacchio T, Schein M. Double gallbladder originating from left hepatic duct: a case report and review of literature. JSLS. 1998;2(4):337-339.
13. Cueto García J, Weber A, Serrano Berry F, Tanur Tatz B. Double gallbladder treated successfully by laparoscopy. J Laparoendosc Surg. 1993;3(2):153-155. doi:10.1089/lps.1993.3.153
14. Fazio V, Damiano G, Palumbo VD, et al. An unexpected surprise at the end of a “quiet” cholecystectomy. A case report and review of the literature. Ann Ital Chir. 2012;83(3):265-267.
15. Flum DR, Dellinger EP, Cheadle A, Chan L, Koepsell T. Intraoperative cholangiography and risk of common bile duct injury during cholecystectomy. JAMA. 2003;289(13):1639-1644. doi:10.1001/jama.289.13.1639
16. Botsford A, McKay K, Hartery A, Hapgood C. MRCP imaging of duplicate gallbladder: a case report and review of the literature. Surg Radiol Anat. 2015;37(5):425-429. doi:10.1007/s00276-015-1456-1
17. Strasberg SM, Hertl M, Soper NJ. An analysis of the problem of biliary injury during laparoscopic cholecystectomy. J Am Coll Surg. 1995;180(1):101-125.
18. Raymond SW, Thrift CB. Carcinoma of a duplicated gall bladder. Ill Med J. 1956;110(5):239-240.
19. Cunningham JJ. Empyema of a duplicated gallbladder: echographic findings. J Clin Ultrasound. 1980;8(6):511-512. doi:10.1002/jcu.1870080612
20. Recht W. Torsion of a double gallbladder; a report of a case and a review of the literature. Br J Surg. 1952;39(156):342-344. doi:10.1002/bjs.18003915616
21. Ritchie AW, Crucioli V. Double gallbladder with cholecystocolic fistula: a case report. Br J Surg. 1980;67(2):145-146. doi:10.1002/bjs.1800670226
22. Shapiro T, Rennie W. Duplicate gallbladder cholecystitis after open cholecystectomy. Ann Emerg Med. 1999;33(5):584-587. doi:10.1016/s0196-0644(99)70348-3
23. Hobbs MS, Mai Q, Knuiman MW, Fletcher DR, Ridout SC. Surgeon experience and trends in intraoperative complications in laparoscopic cholecystectomy. Br J Surg. 2006;93(7):844-853. doi:10.1002/bjs.5333
24. Davidoff AM, Pappas TN, Murray EA, et al. Mechanisms of major biliary injury during laparoscopic cholecystectomy. Ann Surg. 1992;215(3):196-202. doi:10.1097/00000658-199203000-00002
25. Flowers JL, Zucker KA, Graham SM, Scovill WA, Imbembo AL, Bailey RW. Laparoscopic cholangiography. Results and indications. Ann Surg. 1992;215(3):209-216. doi:10.1097/00000658-199203000-00004
Gallbladder duplication is a congenital abnormality of the hepatobiliary system and often is not considered in the evaluation of a patient with right upper quadrant pain. Accuracy of the most commonly used imaging study to assess for biliary disease, abdominal ultrasound, is highly dependent on the skills of the ultrasonographer, and given its relative rarity, this condition is often not considered prior to planned cholecystectomy.1 Small case reviews found that < 50% of gallbladder duplications are diagnosed preoperatively despite use of ultrasound or computed tomography (CT) scan.2-4 Failure to recognize duplicate gallbladder anatomy in symptomatic patients may result in incomplete surgical management, an increase in perioperative complications, and years of morbidity due to unresolved symptoms. Once a patient has had a cholecystectomy, symptoms are presumed to be due to a nonbiliary etiology and an extensive, often repetitive, workup is pursued before “repeat cholecystectomy” is considered.5
Case Presentation
A 63-year-old man was referred to gastroenterology for recurrent episodic right upper quadrant pain. He reported intermittent both right and left upper abdominal pain that was variable in quality. At times it was associated with an empty stomach prior to meals; at other times, onset was 30 to 60 minutes after meals. The patient also reported significant flatulence and bloating and intermittent loose stools. Sixteen years before, he underwent a laparoscopic cholecystectomy. He reported that the pain he experienced before the cholecystectomy never resolved after surgery but occurred less frequently. For the next 16 years, the patient did not seek evaluation of his ongoing but infrequent symptoms until his pain became a daily occurrence. The patient’s surgical history included a remote open vagotomy and antrectomy for peptic ulcer disease, laparoscopic appendectomy, and a laparoscopic cholecystectomy for reported biliary colic.
The gastroenterology evaluation included a colonoscopy and esophagogastroduodenoscopy (EGD); both were benign and without findings specific to identify the etiology for the patient’s pain. The patient was given a course of rifaximin 1200 mg daily for 7 days for possible bacterial overgrowth and placed on a proton pump inhibitor twice daily. Neither of these interventions helped resolve the patient’s symptoms. Further workup was pursued by gastroenterology to include a right upper quadrant ultrasound that showed a structure most consistent with a small gallbladder containing a small polyp vs stone. Magnetic resonance cholangiopancreatography (MRCP) also was performed and showed the presence of a small gallbladder with a small 2-mm filling defect and an otherwise benign biliary tree. MRCP images and EGD documented a Billroth 1 reconstruction at the time of his remote antrectomy and vagotomy (Figure 1).
The patient was referred to general surgery for consideration of a repeat cholecystectomy. He confirmed the history of intermittent upper abdominal pain for the past 16 years, which was similar to the symptoms he had experienced before his original laparoscopic cholecystectomy. On examination, the patient had a body mass index of 38, had a large upper midline incision from his prior antrectomy and vagotomy procedure, and several scars presumed to be port incision scars to the right lateral abdominal wall. Hospital records were obtained from the patient’s prior workup for biliary colic and cholecystectomy 16 years before. The preoperative abdominal ultrasound examination showed a mildly distended gallbladder but was notably described as “quite limited due to patient’s body habitus and liver is not well seen.” No additional imaging was documented in his presurgical evaluation notes and imaging records.
The operative report described a gallbladder that was densely adherent to adjacent fat and omental tissue with significant adhesions secondary to the prior vagotomy and antrectomy procedure. The cystic duct and artery were dissected free at the level of their junction with the gallbladder infundibulum. The cystic artery was divided with a harmonic scalpel. Following this the gallbladder body was dissected free from the liver bed in top-down fashion. A 0 Vicryl Endoloop suture was placed over the gallbladder and secured just past the origin of the cystic duct on the gallbladder infundibulum and the cystic duct divided above this suture. No surgical clips were used, which corresponded with the lack of surgical clips seen in imaging in his recent gastroenterology workup. No documentation of an intraoperative cholangiogram existed or was considered in the operative report.
The pathology report from this first cholecystectomy procedure noted the removed specimen to be an unopened 6-cm gallbladder containing 2 small yellow stones that otherwise were benign. At the time of this patient’s re-presentation to general surgery, there was suspicion that the patient’s prior surgical procedure had not been a cholecystectomy but rather a subtotal cholecystectomy. However, after appropriate workup and review of prior records, the patient had, indeed, previously undergone cholecystectomy and represented a rare case of gallbladder duplication resulting in abdominal pain for 16 years after his index operation.
The patient was consented for repeat cholecystectomy and underwent a laparoscopic lysis of adhesions, cholecystectomy, and intraoperative cholangiogram. Significant scarring was found at the liver undersurface that would have been exposed during the original laparoscopic resection of the gallbladder from its liver bed. Deeper to this, a small saccular structure was identified as the duplicate gallbladder (Figure 2). Though the visualized gallbladder was small with a deep intrahepatic lie, the critical view of safety was achieved and was without additional variation. An intraoperative cholangiogram was performed to determine whether residual ductal stumps or other additional evidence of the previously removed gallbladder could be identified. The cholangiogram showed clear visualization of the cystic duct, common bile duct, right and left hepatic ducts, and contrast into the duodenum without abnormal variants. There was no visualized accessory or secondary cystic duct stump seen on the cholangiogram (Figure 3). Pathology of the repeat cholecystectomy specimen confirmed a 3-cm gallbladder with a distinct duct leading out of the gallbladder and the presence of several gallstones. The patient had an uneventful recovery after the repeat laparoscopic cholecystectomy with complete resolution of his upper abdominal pain.
Discussion
The first reported human case of gallbladder duplication was noted in a sacrificial victim of Emperor Augustus in 31 BCE. Sherren reported the first documented case of double accessory gallbladder in a living human in 1911.1,6 Though the exact incidence of gallbladder duplication is not fully known due to primary documentation from case reports, incidence is approximately 1 in 4000 to 5000 people. It was first formally classified by Boyden in 1926.7 Further anatomic classification based on morphology and embryogenesis was delineated by Harlaftis and colleagues in 1977, establishing type 1 and 2 structures of a duplicated gallbladder.8 Type 1 duplicated gallbladder anatomy shares a single cystic duct, whereas in type 2 each gallbladder has its own cystic duct. Later reports and studies identified triple gallbladders as well as trabecular variants with the most common classification used currently being the modified Harlaftis classification.9,10
The case presented here most likely represents either a Y-shaped type 1 primordial gallbladder or a type 2 accessory gallbladder based on historical data and intraoperative cholangiogram findings at the time of repeat cholecystectomy. Gallbladder duplication is clinically indistinguishable from regular gallbladder pathology preoperatively and can only be identified on imaging or intraoperatively.11 Prior case reports and studies have found that it is frequently missed on preoperative abdominal ultrasonography and CT in up to 50% of cases.12-14
The differential diagnosis of gallbladder duplication seen on preoperative imaging includes a gallbladder diverticulum, choledochal cyst, focal adenonomyomatosis, Phrygian cap, or folded gallbladder.1,2 Historically, the most definitive test for gallbladder duplication has been either intraoperative cholangiography, which can also clarify biliary anatomy, or endoscopic retrograde cholangiopancreatography with cholangiography.1,3 The debate over routine use of intraoperative cholangiography has been ongoing for the past several decades.15 Though intraoperative cholangiogram remains one of the most definitive tests for gallbladder duplication, given the overall low incidence of this variant, recommendation for routine intraoperative cholangiography solely to rule out gallbladder duplication cannot be definitively recommended based on our review of the literature. Currently, preoperative MRCP is the study of choice when there is concern from historical facts or from other imaging of gallbladder duplication as it is noninvasive and has a high degree of detail, particularly with 3D reconstructions.14,16 At the time of surgery, the most critical step to avoid inadvertent ductal injury is clear visualization of ductal anatomy and obtaining the critical view of safety.17 Though this will also assist in identifying some cases of gallbladder duplication, given the great variation of duplication, it will not prevent missing some variants. In our case, extensive local scarring from the patient’s prior antrectomy and vagotomy along with lack of the use of intraoperative cholangiography likely contributed to missing his duplication at the time of his index cholecystectomy.
Undiagnosed gallbladder duplication can lead to additional morbidity related to common entities associated with gallbladder pathology, such as biliary colic, cholecystitis, cholangitis, and pancreatitis. Additionally, case reports in the literature have documented more rare associations, such as empyema, carcinoma, cholecystoenteric fistula, and torsion, all associated with a duplicated gallbladder.18-21 Once identified pre- or intraoperatively, it is generally recommended that all gallbladders be removed in symptomatic patients and that intraoperative cholangiography be done to assure complete resection of the duplicated gallbladders and to avoid injury to the biliary trees.22-25
Conclusions
Gallbladder duplication and other congenital biliary anatomic variations should be considered before a biliary operation and included in the differential diagnosis when evaluating patients who have clinical symptoms consistent with biliary pathology. In addition, intraoperative cholangiogram should be performed during cholecystectomy if the inferior liver edge cannot be visualized well, as in the case of this patient where a prior foregut operation resulted in extensive adhesive disease. Intraoperative cholangiogram also should be considered in patients whose preoperative imaging does not visualize the right upper quadrant well due to patient habitus. Doing so may identify gallbladder duplication and allow for complete cholecystectomy as well as proper identification and management of cystic duct variants. Awareness and consideration of duplicated biliary variants can help prevent intraoperative complications related to biliary anomalies and avoid the morbidity related to recurrent biliary disease and the need for repeat operative procedures.
Acknowledgments
We extend our thanks to Veterans Affairs Puget Sound Healthcare System and the Departments of Surgery and Radiology for their support of this case report, and Lorrie Langdale, MD, and Roger Tatum, MD, for their mentorship of this project
Gallbladder duplication is a congenital abnormality of the hepatobiliary system and often is not considered in the evaluation of a patient with right upper quadrant pain. Accuracy of the most commonly used imaging study to assess for biliary disease, abdominal ultrasound, is highly dependent on the skills of the ultrasonographer, and given its relative rarity, this condition is often not considered prior to planned cholecystectomy.1 Small case reviews found that < 50% of gallbladder duplications are diagnosed preoperatively despite use of ultrasound or computed tomography (CT) scan.2-4 Failure to recognize duplicate gallbladder anatomy in symptomatic patients may result in incomplete surgical management, an increase in perioperative complications, and years of morbidity due to unresolved symptoms. Once a patient has had a cholecystectomy, symptoms are presumed to be due to a nonbiliary etiology and an extensive, often repetitive, workup is pursued before “repeat cholecystectomy” is considered.5
Case Presentation
A 63-year-old man was referred to gastroenterology for recurrent episodic right upper quadrant pain. He reported intermittent both right and left upper abdominal pain that was variable in quality. At times it was associated with an empty stomach prior to meals; at other times, onset was 30 to 60 minutes after meals. The patient also reported significant flatulence and bloating and intermittent loose stools. Sixteen years before, he underwent a laparoscopic cholecystectomy. He reported that the pain he experienced before the cholecystectomy never resolved after surgery but occurred less frequently. For the next 16 years, the patient did not seek evaluation of his ongoing but infrequent symptoms until his pain became a daily occurrence. The patient’s surgical history included a remote open vagotomy and antrectomy for peptic ulcer disease, laparoscopic appendectomy, and a laparoscopic cholecystectomy for reported biliary colic.
The gastroenterology evaluation included a colonoscopy and esophagogastroduodenoscopy (EGD); both were benign and without findings specific to identify the etiology for the patient’s pain. The patient was given a course of rifaximin 1200 mg daily for 7 days for possible bacterial overgrowth and placed on a proton pump inhibitor twice daily. Neither of these interventions helped resolve the patient’s symptoms. Further workup was pursued by gastroenterology to include a right upper quadrant ultrasound that showed a structure most consistent with a small gallbladder containing a small polyp vs stone. Magnetic resonance cholangiopancreatography (MRCP) also was performed and showed the presence of a small gallbladder with a small 2-mm filling defect and an otherwise benign biliary tree. MRCP images and EGD documented a Billroth 1 reconstruction at the time of his remote antrectomy and vagotomy (Figure 1).
The patient was referred to general surgery for consideration of a repeat cholecystectomy. He confirmed the history of intermittent upper abdominal pain for the past 16 years, which was similar to the symptoms he had experienced before his original laparoscopic cholecystectomy. On examination, the patient had a body mass index of 38, had a large upper midline incision from his prior antrectomy and vagotomy procedure, and several scars presumed to be port incision scars to the right lateral abdominal wall. Hospital records were obtained from the patient’s prior workup for biliary colic and cholecystectomy 16 years before. The preoperative abdominal ultrasound examination showed a mildly distended gallbladder but was notably described as “quite limited due to patient’s body habitus and liver is not well seen.” No additional imaging was documented in his presurgical evaluation notes and imaging records.
The operative report described a gallbladder that was densely adherent to adjacent fat and omental tissue with significant adhesions secondary to the prior vagotomy and antrectomy procedure. The cystic duct and artery were dissected free at the level of their junction with the gallbladder infundibulum. The cystic artery was divided with a harmonic scalpel. Following this the gallbladder body was dissected free from the liver bed in top-down fashion. A 0 Vicryl Endoloop suture was placed over the gallbladder and secured just past the origin of the cystic duct on the gallbladder infundibulum and the cystic duct divided above this suture. No surgical clips were used, which corresponded with the lack of surgical clips seen in imaging in his recent gastroenterology workup. No documentation of an intraoperative cholangiogram existed or was considered in the operative report.
The pathology report from this first cholecystectomy procedure noted the removed specimen to be an unopened 6-cm gallbladder containing 2 small yellow stones that otherwise were benign. At the time of this patient’s re-presentation to general surgery, there was suspicion that the patient’s prior surgical procedure had not been a cholecystectomy but rather a subtotal cholecystectomy. However, after appropriate workup and review of prior records, the patient had, indeed, previously undergone cholecystectomy and represented a rare case of gallbladder duplication resulting in abdominal pain for 16 years after his index operation.
The patient was consented for repeat cholecystectomy and underwent a laparoscopic lysis of adhesions, cholecystectomy, and intraoperative cholangiogram. Significant scarring was found at the liver undersurface that would have been exposed during the original laparoscopic resection of the gallbladder from its liver bed. Deeper to this, a small saccular structure was identified as the duplicate gallbladder (Figure 2). Though the visualized gallbladder was small with a deep intrahepatic lie, the critical view of safety was achieved and was without additional variation. An intraoperative cholangiogram was performed to determine whether residual ductal stumps or other additional evidence of the previously removed gallbladder could be identified. The cholangiogram showed clear visualization of the cystic duct, common bile duct, right and left hepatic ducts, and contrast into the duodenum without abnormal variants. There was no visualized accessory or secondary cystic duct stump seen on the cholangiogram (Figure 3). Pathology of the repeat cholecystectomy specimen confirmed a 3-cm gallbladder with a distinct duct leading out of the gallbladder and the presence of several gallstones. The patient had an uneventful recovery after the repeat laparoscopic cholecystectomy with complete resolution of his upper abdominal pain.
Discussion
The first reported human case of gallbladder duplication was noted in a sacrificial victim of Emperor Augustus in 31 BCE. Sherren reported the first documented case of double accessory gallbladder in a living human in 1911.1,6 Though the exact incidence of gallbladder duplication is not fully known due to primary documentation from case reports, incidence is approximately 1 in 4000 to 5000 people. It was first formally classified by Boyden in 1926.7 Further anatomic classification based on morphology and embryogenesis was delineated by Harlaftis and colleagues in 1977, establishing type 1 and 2 structures of a duplicated gallbladder.8 Type 1 duplicated gallbladder anatomy shares a single cystic duct, whereas in type 2 each gallbladder has its own cystic duct. Later reports and studies identified triple gallbladders as well as trabecular variants with the most common classification used currently being the modified Harlaftis classification.9,10
The case presented here most likely represents either a Y-shaped type 1 primordial gallbladder or a type 2 accessory gallbladder based on historical data and intraoperative cholangiogram findings at the time of repeat cholecystectomy. Gallbladder duplication is clinically indistinguishable from regular gallbladder pathology preoperatively and can only be identified on imaging or intraoperatively.11 Prior case reports and studies have found that it is frequently missed on preoperative abdominal ultrasonography and CT in up to 50% of cases.12-14
The differential diagnosis of gallbladder duplication seen on preoperative imaging includes a gallbladder diverticulum, choledochal cyst, focal adenonomyomatosis, Phrygian cap, or folded gallbladder.1,2 Historically, the most definitive test for gallbladder duplication has been either intraoperative cholangiography, which can also clarify biliary anatomy, or endoscopic retrograde cholangiopancreatography with cholangiography.1,3 The debate over routine use of intraoperative cholangiography has been ongoing for the past several decades.15 Though intraoperative cholangiogram remains one of the most definitive tests for gallbladder duplication, given the overall low incidence of this variant, recommendation for routine intraoperative cholangiography solely to rule out gallbladder duplication cannot be definitively recommended based on our review of the literature. Currently, preoperative MRCP is the study of choice when there is concern from historical facts or from other imaging of gallbladder duplication as it is noninvasive and has a high degree of detail, particularly with 3D reconstructions.14,16 At the time of surgery, the most critical step to avoid inadvertent ductal injury is clear visualization of ductal anatomy and obtaining the critical view of safety.17 Though this will also assist in identifying some cases of gallbladder duplication, given the great variation of duplication, it will not prevent missing some variants. In our case, extensive local scarring from the patient’s prior antrectomy and vagotomy along with lack of the use of intraoperative cholangiography likely contributed to missing his duplication at the time of his index cholecystectomy.
Undiagnosed gallbladder duplication can lead to additional morbidity related to common entities associated with gallbladder pathology, such as biliary colic, cholecystitis, cholangitis, and pancreatitis. Additionally, case reports in the literature have documented more rare associations, such as empyema, carcinoma, cholecystoenteric fistula, and torsion, all associated with a duplicated gallbladder.18-21 Once identified pre- or intraoperatively, it is generally recommended that all gallbladders be removed in symptomatic patients and that intraoperative cholangiography be done to assure complete resection of the duplicated gallbladders and to avoid injury to the biliary trees.22-25
Conclusions
Gallbladder duplication and other congenital biliary anatomic variations should be considered before a biliary operation and included in the differential diagnosis when evaluating patients who have clinical symptoms consistent with biliary pathology. In addition, intraoperative cholangiogram should be performed during cholecystectomy if the inferior liver edge cannot be visualized well, as in the case of this patient where a prior foregut operation resulted in extensive adhesive disease. Intraoperative cholangiogram also should be considered in patients whose preoperative imaging does not visualize the right upper quadrant well due to patient habitus. Doing so may identify gallbladder duplication and allow for complete cholecystectomy as well as proper identification and management of cystic duct variants. Awareness and consideration of duplicated biliary variants can help prevent intraoperative complications related to biliary anomalies and avoid the morbidity related to recurrent biliary disease and the need for repeat operative procedures.
Acknowledgments
We extend our thanks to Veterans Affairs Puget Sound Healthcare System and the Departments of Surgery and Radiology for their support of this case report, and Lorrie Langdale, MD, and Roger Tatum, MD, for their mentorship of this project
1. Vezakis A, Pantiora E, Giannoulopoulos D, et al. A duplicated gallbladder in a patient presenting with acute cholangitis. A case study and a literature review. Ann Hepatol. 2019;18(1):240-245. doi:10.5604/01.3001.0012.7932
2. Barut Í, Tarhan ÖR, Dog^ru U, Bülbül M. Gallbladder duplication: diagnosed and treated by laparoscopy. Eur J Gen Med. 2006;3(3):142-145. doi:10.29333/ejgm/82396 3. Cozacov Y, Subhas G, Jacobs M, Parikh J. Total laparoscopic removal of accessory gallbladder: a case report and review of literature. World J Gastrointest Surg. 2015;7(12):398-402. doi:10.4240/wjgs.v7.i12.398
4. Musleh MG, Burnett H, Rajashanker B, Ammori BJ. Laparoscopic double cholecystectomy for duplicated gallbladder: a case report. Int J Surg Case Rep. 2017;41:502-504. Published 2017 Nov 27. doi:10.1016/j.ijscr.2017.11.046
5. Walbolt TD, Lalezarzadeh F. Laparoscopic management of a duplicated gallbladder: a case study and anatomic history. Surg Laparosc Endosc Percutan Tech. 2011;21(3):e156-e158. doi:10.1097/SLE.0b013e31821d47ce
6. Sherren J. A double gall-bladder removed by operation. Ann Surg. 1911;54(2):204-205. doi:10.1097/00000658-191108000-00009
7. Boyden EA. The accessory gall-bladder—an embryological and comparative study of aberrant biliary vesicles occurring in man and the domestic mammals. Am J Anat. 1926; 38(2):177-231. doi:10.1002/aja.1000380202
8. Harlaftis N, Gray SW, Skandalakis JE. Multiple gallbladders. Surg Gynecol Obstet. 1977;145(6):928-934.
9. Kim RD, Zendejas I, Velopulos C, et al. Duplicate gallbladder arising from the left hepatic duct: report of a case. Surg Today. 2009;39(6):536-539. doi:10.1007/s00595-008-3878-4
10. Causey MW, Miller S, Fernelius CA, Burgess JR, Brown TA, Newton C. Gallbladder duplication: evaluation, treatment, and classification. J Pediatr Surg. 2010;45(2):443-446. doi:10.1016/j.jpedsurg.2009.12.015
11. Apolo Romero EX, Gálvez Salazar PF, Estrada Chandi JA, et al. Gallbladder duplication and cholecystitis. J Surg Case Rep. 2018;2018(7):rjy158. Published 2018 Jul 3. doi:10.1093/jscr/rjy158
12. Gorecki PJ, Andrei VE, Musacchio T, Schein M. Double gallbladder originating from left hepatic duct: a case report and review of literature. JSLS. 1998;2(4):337-339.
13. Cueto García J, Weber A, Serrano Berry F, Tanur Tatz B. Double gallbladder treated successfully by laparoscopy. J Laparoendosc Surg. 1993;3(2):153-155. doi:10.1089/lps.1993.3.153
14. Fazio V, Damiano G, Palumbo VD, et al. An unexpected surprise at the end of a “quiet” cholecystectomy. A case report and review of the literature. Ann Ital Chir. 2012;83(3):265-267.
15. Flum DR, Dellinger EP, Cheadle A, Chan L, Koepsell T. Intraoperative cholangiography and risk of common bile duct injury during cholecystectomy. JAMA. 2003;289(13):1639-1644. doi:10.1001/jama.289.13.1639
16. Botsford A, McKay K, Hartery A, Hapgood C. MRCP imaging of duplicate gallbladder: a case report and review of the literature. Surg Radiol Anat. 2015;37(5):425-429. doi:10.1007/s00276-015-1456-1
17. Strasberg SM, Hertl M, Soper NJ. An analysis of the problem of biliary injury during laparoscopic cholecystectomy. J Am Coll Surg. 1995;180(1):101-125.
18. Raymond SW, Thrift CB. Carcinoma of a duplicated gall bladder. Ill Med J. 1956;110(5):239-240.
19. Cunningham JJ. Empyema of a duplicated gallbladder: echographic findings. J Clin Ultrasound. 1980;8(6):511-512. doi:10.1002/jcu.1870080612
20. Recht W. Torsion of a double gallbladder; a report of a case and a review of the literature. Br J Surg. 1952;39(156):342-344. doi:10.1002/bjs.18003915616
21. Ritchie AW, Crucioli V. Double gallbladder with cholecystocolic fistula: a case report. Br J Surg. 1980;67(2):145-146. doi:10.1002/bjs.1800670226
22. Shapiro T, Rennie W. Duplicate gallbladder cholecystitis after open cholecystectomy. Ann Emerg Med. 1999;33(5):584-587. doi:10.1016/s0196-0644(99)70348-3
23. Hobbs MS, Mai Q, Knuiman MW, Fletcher DR, Ridout SC. Surgeon experience and trends in intraoperative complications in laparoscopic cholecystectomy. Br J Surg. 2006;93(7):844-853. doi:10.1002/bjs.5333
24. Davidoff AM, Pappas TN, Murray EA, et al. Mechanisms of major biliary injury during laparoscopic cholecystectomy. Ann Surg. 1992;215(3):196-202. doi:10.1097/00000658-199203000-00002
25. Flowers JL, Zucker KA, Graham SM, Scovill WA, Imbembo AL, Bailey RW. Laparoscopic cholangiography. Results and indications. Ann Surg. 1992;215(3):209-216. doi:10.1097/00000658-199203000-00004
1. Vezakis A, Pantiora E, Giannoulopoulos D, et al. A duplicated gallbladder in a patient presenting with acute cholangitis. A case study and a literature review. Ann Hepatol. 2019;18(1):240-245. doi:10.5604/01.3001.0012.7932
2. Barut Í, Tarhan ÖR, Dog^ru U, Bülbül M. Gallbladder duplication: diagnosed and treated by laparoscopy. Eur J Gen Med. 2006;3(3):142-145. doi:10.29333/ejgm/82396 3. Cozacov Y, Subhas G, Jacobs M, Parikh J. Total laparoscopic removal of accessory gallbladder: a case report and review of literature. World J Gastrointest Surg. 2015;7(12):398-402. doi:10.4240/wjgs.v7.i12.398
4. Musleh MG, Burnett H, Rajashanker B, Ammori BJ. Laparoscopic double cholecystectomy for duplicated gallbladder: a case report. Int J Surg Case Rep. 2017;41:502-504. Published 2017 Nov 27. doi:10.1016/j.ijscr.2017.11.046
5. Walbolt TD, Lalezarzadeh F. Laparoscopic management of a duplicated gallbladder: a case study and anatomic history. Surg Laparosc Endosc Percutan Tech. 2011;21(3):e156-e158. doi:10.1097/SLE.0b013e31821d47ce
6. Sherren J. A double gall-bladder removed by operation. Ann Surg. 1911;54(2):204-205. doi:10.1097/00000658-191108000-00009
7. Boyden EA. The accessory gall-bladder—an embryological and comparative study of aberrant biliary vesicles occurring in man and the domestic mammals. Am J Anat. 1926; 38(2):177-231. doi:10.1002/aja.1000380202
8. Harlaftis N, Gray SW, Skandalakis JE. Multiple gallbladders. Surg Gynecol Obstet. 1977;145(6):928-934.
9. Kim RD, Zendejas I, Velopulos C, et al. Duplicate gallbladder arising from the left hepatic duct: report of a case. Surg Today. 2009;39(6):536-539. doi:10.1007/s00595-008-3878-4
10. Causey MW, Miller S, Fernelius CA, Burgess JR, Brown TA, Newton C. Gallbladder duplication: evaluation, treatment, and classification. J Pediatr Surg. 2010;45(2):443-446. doi:10.1016/j.jpedsurg.2009.12.015
11. Apolo Romero EX, Gálvez Salazar PF, Estrada Chandi JA, et al. Gallbladder duplication and cholecystitis. J Surg Case Rep. 2018;2018(7):rjy158. Published 2018 Jul 3. doi:10.1093/jscr/rjy158
12. Gorecki PJ, Andrei VE, Musacchio T, Schein M. Double gallbladder originating from left hepatic duct: a case report and review of literature. JSLS. 1998;2(4):337-339.
13. Cueto García J, Weber A, Serrano Berry F, Tanur Tatz B. Double gallbladder treated successfully by laparoscopy. J Laparoendosc Surg. 1993;3(2):153-155. doi:10.1089/lps.1993.3.153
14. Fazio V, Damiano G, Palumbo VD, et al. An unexpected surprise at the end of a “quiet” cholecystectomy. A case report and review of the literature. Ann Ital Chir. 2012;83(3):265-267.
15. Flum DR, Dellinger EP, Cheadle A, Chan L, Koepsell T. Intraoperative cholangiography and risk of common bile duct injury during cholecystectomy. JAMA. 2003;289(13):1639-1644. doi:10.1001/jama.289.13.1639
16. Botsford A, McKay K, Hartery A, Hapgood C. MRCP imaging of duplicate gallbladder: a case report and review of the literature. Surg Radiol Anat. 2015;37(5):425-429. doi:10.1007/s00276-015-1456-1
17. Strasberg SM, Hertl M, Soper NJ. An analysis of the problem of biliary injury during laparoscopic cholecystectomy. J Am Coll Surg. 1995;180(1):101-125.
18. Raymond SW, Thrift CB. Carcinoma of a duplicated gall bladder. Ill Med J. 1956;110(5):239-240.
19. Cunningham JJ. Empyema of a duplicated gallbladder: echographic findings. J Clin Ultrasound. 1980;8(6):511-512. doi:10.1002/jcu.1870080612
20. Recht W. Torsion of a double gallbladder; a report of a case and a review of the literature. Br J Surg. 1952;39(156):342-344. doi:10.1002/bjs.18003915616
21. Ritchie AW, Crucioli V. Double gallbladder with cholecystocolic fistula: a case report. Br J Surg. 1980;67(2):145-146. doi:10.1002/bjs.1800670226
22. Shapiro T, Rennie W. Duplicate gallbladder cholecystitis after open cholecystectomy. Ann Emerg Med. 1999;33(5):584-587. doi:10.1016/s0196-0644(99)70348-3
23. Hobbs MS, Mai Q, Knuiman MW, Fletcher DR, Ridout SC. Surgeon experience and trends in intraoperative complications in laparoscopic cholecystectomy. Br J Surg. 2006;93(7):844-853. doi:10.1002/bjs.5333
24. Davidoff AM, Pappas TN, Murray EA, et al. Mechanisms of major biliary injury during laparoscopic cholecystectomy. Ann Surg. 1992;215(3):196-202. doi:10.1097/00000658-199203000-00002
25. Flowers JL, Zucker KA, Graham SM, Scovill WA, Imbembo AL, Bailey RW. Laparoscopic cholangiography. Results and indications. Ann Surg. 1992;215(3):209-216. doi:10.1097/00000658-199203000-00004