User login
Rotating Hinge Distal Femur Replacement: A Turn for the Worse
Preoperatively periprosthetic joint infection with a postoperative complication of 180° rotation of the press-fit femoral component is a rare event, and knowledge of this possible complication is important for arthroplasty surgeons.
The use of a rotating hinge distal femur replacement (DFR) for significant bone and soft tissue defects in the setting of total knee arthroplasty (TKA) revision has become increasingly more common. Although significant advancements have been made in modern DFR components, complications and failure rates remain high. The unanticipated early failure presented serves as the first reported case in the literature to our knowledge of a 180° rotation of a press-fit DFR.
Originally, DFRs were used primarily for oncology patients with substantial bone loss following large mass excisions. The utility of DFRs has grown to include massive bone loss in the setting of TKA revision, periprosthetic fractures, and periprosthetic joint infections.1-3 DFRs help restore the joint line in the setting of significant bone loss and contain a rotating hinge mechanism that provides functional movement despite the loss of soft tissue constraints around the knee.1-3
DFRs have been associated with early postoperative mobilization and decreased need for ambulatory devices at 1 year in revision TKA and periprosthetic and geriatric distal femur fractures.4-6 Advances in prosthetic design, biomechanics, and fixation technique have led to improved survival rates.3 Despite these improvements, the overall complication rate remains high at 30 to 40%.3-7 Commonly reported complications after DFR include infection, aseptic loosening, soft tissue failure, and structural failure.3,4,7 Recent case studies also have reported on dislocation or disengagement of the rotating hinge.8-11
In this case report, we present a patient who had a DFR as the second stage of a 2-stage TKA revision due to a periprosthetic joint infection with a postoperative complication of 180° rotation of the press-fit femoral component. Although this is a rare event, knowledge of this possible complication is important for arthroplasty surgeons.
Case Presentation
A patient with a history of hypertension, osteopenia, and rheumatoid arthritis underwent a primary right TKA in 2007. Ten weeks postoperatively, the patient had a ground-level fall that resulted in a right periprosthetic supracondylar distal femur fracture that was treated with a distal femur locking plate. The patient healed, however, with a significant golf club deformity (Figure 1). The patient did well for more than a decade but in 2019 was admitted with pelvic inflammatory disease and adnexal abscess that was treated with broad-spectrum IV antibiotics. Shortly after this admission, the patient developed a right knee periprosthetic infection with cultures positive for Ureaplasma parvum.
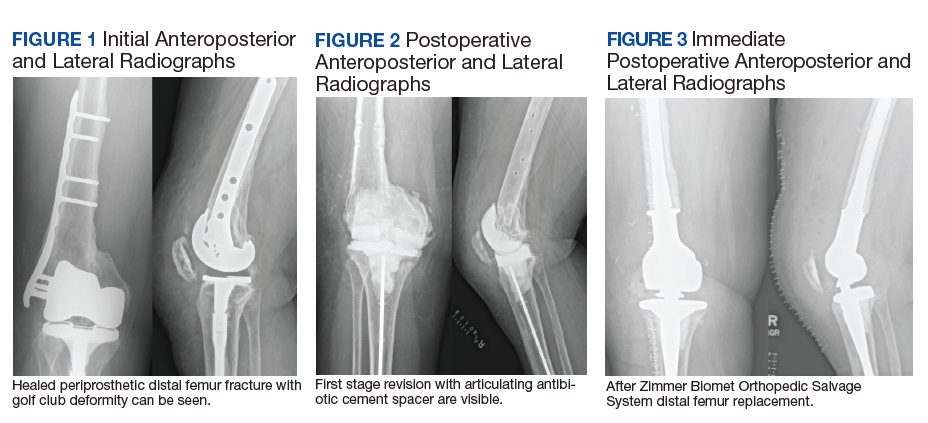
The patient then underwent a 2-stage revision of the infected TKA. Stage 1 consisted of explant of the TKA components as well as removal of the distal femur plate and screws and placement of an articulating antibiotic cement spacer (Figure 2). The patient completed 6 weeks of IV antibiotics. Following completion of the antibiotic course, we obtained a serum erythrocyte sedimentation rate, C-reactive protein level, and white blood cell count, which were all within normal limits. A knee aspiration was performed and did not show signs of residual infection. Frozen histopathology was sent during the second stage of the revision and did not show infection. After the results of the frozen histopathology returned, the antibiotic spacer was removed, and the femoral canal was thoroughly debrided. Cement and fibrous tissue in the femoral canal were carefully removed. In the setting of significant bone loss and soft tissue compromise due to the previous infection and distal femur fracture, the Zimmer Biomet Orthopedic Salvage System (OSS) with porous coated press-fit elliptical femoral stem was utilized.
The femoral canal was reamed until good cortical chatter was obtained at 16 mm. Per the Biomet OSS guide, “For bowed (curved) long and short press-fit stems, the final flexible reamer shaft diameter may need to be larger than the definitive trial and implant diameter.” After trialing, size 15.5 mm was selected for implantation. Intraoperatively the final stem was noted to have good interference fit after insertion and was stable throughout knee range of motion and varus/valgus stress testing. The patient did well with mobilization while in the hospital postoperatively and was discharged home (Figure 3).
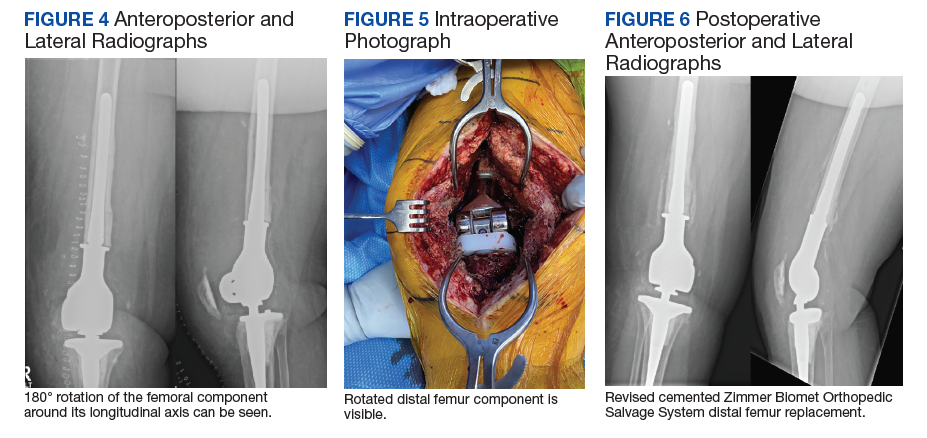
Five days after discharge, the patient kicked the repaired knee onto a chair for rest and elevation and experienced extreme pain and was unable to flex the knee. On presentation to the emergency department, the X-rays showed 180° rotation around the longitudinal axis of the femoral component without any other obvious component failure or fracture (Figure 4). The patient was taken back to surgery the following day. Intraoperatively, the femoral stem was found to be loose and rotated 180° (Figure 5). No failure or dislocation of the tibial or rotating hinge components were identified. The press-fit femoral stem was removed and replaced with a cemented stem (Figure 6).
The postoperative course after the revision surgery was uneventful, and the patient is doing well clinically with no pain, functional range of motion of 5 to 105°, and has returned to regular activities without difficulty.
Discussion
Despite advancements in DFRs and increasing use in the setting of revision TKA, the procedure remains high risk with respect to postoperative complications.3-7 Vertesich and colleagues demonstrated that 43.3% of patients who underwent DFR for failed TKA developed at least 1 postoperative complication that required a return to the operating room.7Physicians need to be aware of the high rate of complications and counsel patients appropriately preoperatively.
Complications after DFR include infection, aseptic loosening, soft tissue failure, and structural failure.4,7 Soft tissue failures include insufficiency or rupture of the extensor mechanism and patella dislocation.4,7 Structural failures include fracture of the hinge mechanism, dissociation of the component from the stem, rotating hinge-bushing failure, and dislocation of the hinge.4,7 In the acute postoperative period, the most common complications are infection and rotating-hinge dislocation/failure.3,12 There are various component options available for DFRs, including straight vs curved, cemented vs cementless/press-fit, and long vs short stems.13 Studies have sought to elucidate the ideal implant to decrease the rate of complications. Lu and colleagues demonstrated that curved press-fit short stems provided a stable interface without loosening over the short term (2 years) in 42 patients.13 No implant failures or incidences of aseptic loosening occurred in their study.13
The implant used in this case was a curved press-fit short-stem DFR. It was thought that this patient was young and with good enough bone quality that a press-fit short stem would be best in preserving bone stock. Both the technique guide and literature support reaming 0 to 2 mm greater than the planned stem size to accommodate the implant curvature.13 In this case, the intramedullary canal was reamed 0.5 mm larger than the curved stem that was implanted (16 mm and 15.5 mm, respectively). Intraoperatively during the index DFR, the component was stable and seemed to have a good press-fit interface. Despite this, obvious loosening of the component occurred with a relatively low-energy mechanism when the patient kicked the leg onto a chair, causing just enough force and femoral rotation to result in 180° rotation of the component.
Conclusions
We present this case report to make surgeons aware of this rare but serious complication. Although the final implant is a porous and curved stem, careful attention should be made during trialing to use the best-fitting implant to prevent this complication. If an adequate interference fit cannot be obtained, cementing the component may be required to prevent its loosening and catastrophic failure.
1. Sculco PK, Abdel MP, Hanssen AD, Lewallen DG. The management of bone loss in revision total knee arthroplasty: rebuild, reinforce, and augment. Bone Joint J. 2016;98-B(1 suppl A):120-124. doi:10.1302/0301-620X.98B1.36345
2. Harrison RJ Jr, Thacker MM, Pitcher JD, Temple HT, Scully SP. Distal femur replacement is useful in complex total knee arthroplasty revisions. Clin Orthop Relat Res. 2006;446:113-120. doi:10.1097/01.blo.0000214433.64774.1b
3. Smith EL, Shah A, Son SJ, et al. Survivorship of megaprostheses in revision hip and knee arthroplasty for septic and aseptic indications: a retrospective, multicenter study with minimum 2-year follow-up. Arthroplast Today. 2020;6(3):475-479. Published 2020 Jun 29. doi:10.1016/j.artd.2020.05.004
4. Wyles CC, Tibbo ME, Yuan BJ, Trousdale RT, Berry DJ, Abdel MP. Long-term results of total knee arthroplasty with contemporary distal femoral replacement. J Bone Joint Surg Am. 2020;102(1):45-51. doi:10.2106/JBJS.19.00489
5. Haidukewych GJ. Role of distal femoral replacement for periprosthetic fractures above a total knee arthroplasty: when and how?, J Orthop Trauma. 2019;33(suppl 6):S33-S35. doi:10.1097/BOT.0000000000001566
6. Hart GP, Kneisl JS, Springer BD, Patt JC, Karunakar MA. Open reduction vs distal femoral replacement arthroplasty for comminuted distal femur fractures in the patients 70 years and older: J Arthroplasty. 2017;32(1):202-206. doi:10.1016/j.arth.2016.06.006
7. Vertesich K, Puchner SE, Staats K, et al. Distal femoral reconstruction following failed total knee arthroplasty is accompanied with risk for complication and reduced joint function. BMC Musculoskelet Disord. 2019 Jan 31;20(1):47-54. doi:10.1186/s12891-019-2432-4
8. Biswas D, Haughom B, Mayle RE Jr, Della Valle CJ. Case report: Failure of rotating-hinge total knee prosthesis by disengagement of the hinge-post extension. Clin Orthop Relat Res. 2013;471(4):1389-1392. doi:10.1007/s11999-012-2736-2
9. Ward WG, Haight D, Ritchie P, Gordon S, Eckardt JJ. Dislocation of rotating hinge knee prostheses. A report of four cases. J Bone Joint Surg Am. 2005;87(5):1108-1112. doi:10.2106/JBJS.00837pp
10. Pacha-Vicente D, Malik A, Castellet-Feliu E, Nardi-Vilardaga J. Dislocation of rotating-hinge knee prostheses with antidislocation mechanism. J Arthroplasty. 2008;23(2):299-303. doi:10.1016/j.arth.2006.11.020
11. Manzano G, Schwarzkopf R. Posterior dislocation of the hinge-post extension in a rotating hinge total knee prosthesis. Case Rep Orthop. 2013;2013:756538. doi:10.1155/2013/756538
12. Vaishya R., Thapa, SS, Vaish A. Non-neoplastic indications and outcomes of the proximal and distal femur megaprosthesis: a critical review. Knee Surg Relat Res. 2020;32(1):18. Published 2020 Apr 9. doi:10.1186/s43019-020-00034-7
13. Lu M, Wang J, Xiao C, et al. Uncemented, curved, short endoprosthesis stem for distal femoral reconstruction: early follow-up outcomes. World J Surg Onc. 2018;16(1):183. doi:10.1186/s12957-018-1486-3
Preoperatively periprosthetic joint infection with a postoperative complication of 180° rotation of the press-fit femoral component is a rare event, and knowledge of this possible complication is important for arthroplasty surgeons.
Preoperatively periprosthetic joint infection with a postoperative complication of 180° rotation of the press-fit femoral component is a rare event, and knowledge of this possible complication is important for arthroplasty surgeons.
The use of a rotating hinge distal femur replacement (DFR) for significant bone and soft tissue defects in the setting of total knee arthroplasty (TKA) revision has become increasingly more common. Although significant advancements have been made in modern DFR components, complications and failure rates remain high. The unanticipated early failure presented serves as the first reported case in the literature to our knowledge of a 180° rotation of a press-fit DFR.
Originally, DFRs were used primarily for oncology patients with substantial bone loss following large mass excisions. The utility of DFRs has grown to include massive bone loss in the setting of TKA revision, periprosthetic fractures, and periprosthetic joint infections.1-3 DFRs help restore the joint line in the setting of significant bone loss and contain a rotating hinge mechanism that provides functional movement despite the loss of soft tissue constraints around the knee.1-3
DFRs have been associated with early postoperative mobilization and decreased need for ambulatory devices at 1 year in revision TKA and periprosthetic and geriatric distal femur fractures.4-6 Advances in prosthetic design, biomechanics, and fixation technique have led to improved survival rates.3 Despite these improvements, the overall complication rate remains high at 30 to 40%.3-7 Commonly reported complications after DFR include infection, aseptic loosening, soft tissue failure, and structural failure.3,4,7 Recent case studies also have reported on dislocation or disengagement of the rotating hinge.8-11
In this case report, we present a patient who had a DFR as the second stage of a 2-stage TKA revision due to a periprosthetic joint infection with a postoperative complication of 180° rotation of the press-fit femoral component. Although this is a rare event, knowledge of this possible complication is important for arthroplasty surgeons.
Case Presentation
A patient with a history of hypertension, osteopenia, and rheumatoid arthritis underwent a primary right TKA in 2007. Ten weeks postoperatively, the patient had a ground-level fall that resulted in a right periprosthetic supracondylar distal femur fracture that was treated with a distal femur locking plate. The patient healed, however, with a significant golf club deformity (Figure 1). The patient did well for more than a decade but in 2019 was admitted with pelvic inflammatory disease and adnexal abscess that was treated with broad-spectrum IV antibiotics. Shortly after this admission, the patient developed a right knee periprosthetic infection with cultures positive for Ureaplasma parvum.
The patient then underwent a 2-stage revision of the infected TKA. Stage 1 consisted of explant of the TKA components as well as removal of the distal femur plate and screws and placement of an articulating antibiotic cement spacer (Figure 2). The patient completed 6 weeks of IV antibiotics. Following completion of the antibiotic course, we obtained a serum erythrocyte sedimentation rate, C-reactive protein level, and white blood cell count, which were all within normal limits. A knee aspiration was performed and did not show signs of residual infection. Frozen histopathology was sent during the second stage of the revision and did not show infection. After the results of the frozen histopathology returned, the antibiotic spacer was removed, and the femoral canal was thoroughly debrided. Cement and fibrous tissue in the femoral canal were carefully removed. In the setting of significant bone loss and soft tissue compromise due to the previous infection and distal femur fracture, the Zimmer Biomet Orthopedic Salvage System (OSS) with porous coated press-fit elliptical femoral stem was utilized.
The femoral canal was reamed until good cortical chatter was obtained at 16 mm. Per the Biomet OSS guide, “For bowed (curved) long and short press-fit stems, the final flexible reamer shaft diameter may need to be larger than the definitive trial and implant diameter.” After trialing, size 15.5 mm was selected for implantation. Intraoperatively the final stem was noted to have good interference fit after insertion and was stable throughout knee range of motion and varus/valgus stress testing. The patient did well with mobilization while in the hospital postoperatively and was discharged home (Figure 3).
Five days after discharge, the patient kicked the repaired knee onto a chair for rest and elevation and experienced extreme pain and was unable to flex the knee. On presentation to the emergency department, the X-rays showed 180° rotation around the longitudinal axis of the femoral component without any other obvious component failure or fracture (Figure 4). The patient was taken back to surgery the following day. Intraoperatively, the femoral stem was found to be loose and rotated 180° (Figure 5). No failure or dislocation of the tibial or rotating hinge components were identified. The press-fit femoral stem was removed and replaced with a cemented stem (Figure 6).
The postoperative course after the revision surgery was uneventful, and the patient is doing well clinically with no pain, functional range of motion of 5 to 105°, and has returned to regular activities without difficulty.
Discussion
Despite advancements in DFRs and increasing use in the setting of revision TKA, the procedure remains high risk with respect to postoperative complications.3-7 Vertesich and colleagues demonstrated that 43.3% of patients who underwent DFR for failed TKA developed at least 1 postoperative complication that required a return to the operating room.7Physicians need to be aware of the high rate of complications and counsel patients appropriately preoperatively.
Complications after DFR include infection, aseptic loosening, soft tissue failure, and structural failure.4,7 Soft tissue failures include insufficiency or rupture of the extensor mechanism and patella dislocation.4,7 Structural failures include fracture of the hinge mechanism, dissociation of the component from the stem, rotating hinge-bushing failure, and dislocation of the hinge.4,7 In the acute postoperative period, the most common complications are infection and rotating-hinge dislocation/failure.3,12 There are various component options available for DFRs, including straight vs curved, cemented vs cementless/press-fit, and long vs short stems.13 Studies have sought to elucidate the ideal implant to decrease the rate of complications. Lu and colleagues demonstrated that curved press-fit short stems provided a stable interface without loosening over the short term (2 years) in 42 patients.13 No implant failures or incidences of aseptic loosening occurred in their study.13
The implant used in this case was a curved press-fit short-stem DFR. It was thought that this patient was young and with good enough bone quality that a press-fit short stem would be best in preserving bone stock. Both the technique guide and literature support reaming 0 to 2 mm greater than the planned stem size to accommodate the implant curvature.13 In this case, the intramedullary canal was reamed 0.5 mm larger than the curved stem that was implanted (16 mm and 15.5 mm, respectively). Intraoperatively during the index DFR, the component was stable and seemed to have a good press-fit interface. Despite this, obvious loosening of the component occurred with a relatively low-energy mechanism when the patient kicked the leg onto a chair, causing just enough force and femoral rotation to result in 180° rotation of the component.
Conclusions
We present this case report to make surgeons aware of this rare but serious complication. Although the final implant is a porous and curved stem, careful attention should be made during trialing to use the best-fitting implant to prevent this complication. If an adequate interference fit cannot be obtained, cementing the component may be required to prevent its loosening and catastrophic failure.
The use of a rotating hinge distal femur replacement (DFR) for significant bone and soft tissue defects in the setting of total knee arthroplasty (TKA) revision has become increasingly more common. Although significant advancements have been made in modern DFR components, complications and failure rates remain high. The unanticipated early failure presented serves as the first reported case in the literature to our knowledge of a 180° rotation of a press-fit DFR.
Originally, DFRs were used primarily for oncology patients with substantial bone loss following large mass excisions. The utility of DFRs has grown to include massive bone loss in the setting of TKA revision, periprosthetic fractures, and periprosthetic joint infections.1-3 DFRs help restore the joint line in the setting of significant bone loss and contain a rotating hinge mechanism that provides functional movement despite the loss of soft tissue constraints around the knee.1-3
DFRs have been associated with early postoperative mobilization and decreased need for ambulatory devices at 1 year in revision TKA and periprosthetic and geriatric distal femur fractures.4-6 Advances in prosthetic design, biomechanics, and fixation technique have led to improved survival rates.3 Despite these improvements, the overall complication rate remains high at 30 to 40%.3-7 Commonly reported complications after DFR include infection, aseptic loosening, soft tissue failure, and structural failure.3,4,7 Recent case studies also have reported on dislocation or disengagement of the rotating hinge.8-11
In this case report, we present a patient who had a DFR as the second stage of a 2-stage TKA revision due to a periprosthetic joint infection with a postoperative complication of 180° rotation of the press-fit femoral component. Although this is a rare event, knowledge of this possible complication is important for arthroplasty surgeons.
Case Presentation
A patient with a history of hypertension, osteopenia, and rheumatoid arthritis underwent a primary right TKA in 2007. Ten weeks postoperatively, the patient had a ground-level fall that resulted in a right periprosthetic supracondylar distal femur fracture that was treated with a distal femur locking plate. The patient healed, however, with a significant golf club deformity (Figure 1). The patient did well for more than a decade but in 2019 was admitted with pelvic inflammatory disease and adnexal abscess that was treated with broad-spectrum IV antibiotics. Shortly after this admission, the patient developed a right knee periprosthetic infection with cultures positive for Ureaplasma parvum.
The patient then underwent a 2-stage revision of the infected TKA. Stage 1 consisted of explant of the TKA components as well as removal of the distal femur plate and screws and placement of an articulating antibiotic cement spacer (Figure 2). The patient completed 6 weeks of IV antibiotics. Following completion of the antibiotic course, we obtained a serum erythrocyte sedimentation rate, C-reactive protein level, and white blood cell count, which were all within normal limits. A knee aspiration was performed and did not show signs of residual infection. Frozen histopathology was sent during the second stage of the revision and did not show infection. After the results of the frozen histopathology returned, the antibiotic spacer was removed, and the femoral canal was thoroughly debrided. Cement and fibrous tissue in the femoral canal were carefully removed. In the setting of significant bone loss and soft tissue compromise due to the previous infection and distal femur fracture, the Zimmer Biomet Orthopedic Salvage System (OSS) with porous coated press-fit elliptical femoral stem was utilized.
The femoral canal was reamed until good cortical chatter was obtained at 16 mm. Per the Biomet OSS guide, “For bowed (curved) long and short press-fit stems, the final flexible reamer shaft diameter may need to be larger than the definitive trial and implant diameter.” After trialing, size 15.5 mm was selected for implantation. Intraoperatively the final stem was noted to have good interference fit after insertion and was stable throughout knee range of motion and varus/valgus stress testing. The patient did well with mobilization while in the hospital postoperatively and was discharged home (Figure 3).
Five days after discharge, the patient kicked the repaired knee onto a chair for rest and elevation and experienced extreme pain and was unable to flex the knee. On presentation to the emergency department, the X-rays showed 180° rotation around the longitudinal axis of the femoral component without any other obvious component failure or fracture (Figure 4). The patient was taken back to surgery the following day. Intraoperatively, the femoral stem was found to be loose and rotated 180° (Figure 5). No failure or dislocation of the tibial or rotating hinge components were identified. The press-fit femoral stem was removed and replaced with a cemented stem (Figure 6).
The postoperative course after the revision surgery was uneventful, and the patient is doing well clinically with no pain, functional range of motion of 5 to 105°, and has returned to regular activities without difficulty.
Discussion
Despite advancements in DFRs and increasing use in the setting of revision TKA, the procedure remains high risk with respect to postoperative complications.3-7 Vertesich and colleagues demonstrated that 43.3% of patients who underwent DFR for failed TKA developed at least 1 postoperative complication that required a return to the operating room.7Physicians need to be aware of the high rate of complications and counsel patients appropriately preoperatively.
Complications after DFR include infection, aseptic loosening, soft tissue failure, and structural failure.4,7 Soft tissue failures include insufficiency or rupture of the extensor mechanism and patella dislocation.4,7 Structural failures include fracture of the hinge mechanism, dissociation of the component from the stem, rotating hinge-bushing failure, and dislocation of the hinge.4,7 In the acute postoperative period, the most common complications are infection and rotating-hinge dislocation/failure.3,12 There are various component options available for DFRs, including straight vs curved, cemented vs cementless/press-fit, and long vs short stems.13 Studies have sought to elucidate the ideal implant to decrease the rate of complications. Lu and colleagues demonstrated that curved press-fit short stems provided a stable interface without loosening over the short term (2 years) in 42 patients.13 No implant failures or incidences of aseptic loosening occurred in their study.13
The implant used in this case was a curved press-fit short-stem DFR. It was thought that this patient was young and with good enough bone quality that a press-fit short stem would be best in preserving bone stock. Both the technique guide and literature support reaming 0 to 2 mm greater than the planned stem size to accommodate the implant curvature.13 In this case, the intramedullary canal was reamed 0.5 mm larger than the curved stem that was implanted (16 mm and 15.5 mm, respectively). Intraoperatively during the index DFR, the component was stable and seemed to have a good press-fit interface. Despite this, obvious loosening of the component occurred with a relatively low-energy mechanism when the patient kicked the leg onto a chair, causing just enough force and femoral rotation to result in 180° rotation of the component.
Conclusions
We present this case report to make surgeons aware of this rare but serious complication. Although the final implant is a porous and curved stem, careful attention should be made during trialing to use the best-fitting implant to prevent this complication. If an adequate interference fit cannot be obtained, cementing the component may be required to prevent its loosening and catastrophic failure.
1. Sculco PK, Abdel MP, Hanssen AD, Lewallen DG. The management of bone loss in revision total knee arthroplasty: rebuild, reinforce, and augment. Bone Joint J. 2016;98-B(1 suppl A):120-124. doi:10.1302/0301-620X.98B1.36345
2. Harrison RJ Jr, Thacker MM, Pitcher JD, Temple HT, Scully SP. Distal femur replacement is useful in complex total knee arthroplasty revisions. Clin Orthop Relat Res. 2006;446:113-120. doi:10.1097/01.blo.0000214433.64774.1b
3. Smith EL, Shah A, Son SJ, et al. Survivorship of megaprostheses in revision hip and knee arthroplasty for septic and aseptic indications: a retrospective, multicenter study with minimum 2-year follow-up. Arthroplast Today. 2020;6(3):475-479. Published 2020 Jun 29. doi:10.1016/j.artd.2020.05.004
4. Wyles CC, Tibbo ME, Yuan BJ, Trousdale RT, Berry DJ, Abdel MP. Long-term results of total knee arthroplasty with contemporary distal femoral replacement. J Bone Joint Surg Am. 2020;102(1):45-51. doi:10.2106/JBJS.19.00489
5. Haidukewych GJ. Role of distal femoral replacement for periprosthetic fractures above a total knee arthroplasty: when and how?, J Orthop Trauma. 2019;33(suppl 6):S33-S35. doi:10.1097/BOT.0000000000001566
6. Hart GP, Kneisl JS, Springer BD, Patt JC, Karunakar MA. Open reduction vs distal femoral replacement arthroplasty for comminuted distal femur fractures in the patients 70 years and older: J Arthroplasty. 2017;32(1):202-206. doi:10.1016/j.arth.2016.06.006
7. Vertesich K, Puchner SE, Staats K, et al. Distal femoral reconstruction following failed total knee arthroplasty is accompanied with risk for complication and reduced joint function. BMC Musculoskelet Disord. 2019 Jan 31;20(1):47-54. doi:10.1186/s12891-019-2432-4
8. Biswas D, Haughom B, Mayle RE Jr, Della Valle CJ. Case report: Failure of rotating-hinge total knee prosthesis by disengagement of the hinge-post extension. Clin Orthop Relat Res. 2013;471(4):1389-1392. doi:10.1007/s11999-012-2736-2
9. Ward WG, Haight D, Ritchie P, Gordon S, Eckardt JJ. Dislocation of rotating hinge knee prostheses. A report of four cases. J Bone Joint Surg Am. 2005;87(5):1108-1112. doi:10.2106/JBJS.00837pp
10. Pacha-Vicente D, Malik A, Castellet-Feliu E, Nardi-Vilardaga J. Dislocation of rotating-hinge knee prostheses with antidislocation mechanism. J Arthroplasty. 2008;23(2):299-303. doi:10.1016/j.arth.2006.11.020
11. Manzano G, Schwarzkopf R. Posterior dislocation of the hinge-post extension in a rotating hinge total knee prosthesis. Case Rep Orthop. 2013;2013:756538. doi:10.1155/2013/756538
12. Vaishya R., Thapa, SS, Vaish A. Non-neoplastic indications and outcomes of the proximal and distal femur megaprosthesis: a critical review. Knee Surg Relat Res. 2020;32(1):18. Published 2020 Apr 9. doi:10.1186/s43019-020-00034-7
13. Lu M, Wang J, Xiao C, et al. Uncemented, curved, short endoprosthesis stem for distal femoral reconstruction: early follow-up outcomes. World J Surg Onc. 2018;16(1):183. doi:10.1186/s12957-018-1486-3
1. Sculco PK, Abdel MP, Hanssen AD, Lewallen DG. The management of bone loss in revision total knee arthroplasty: rebuild, reinforce, and augment. Bone Joint J. 2016;98-B(1 suppl A):120-124. doi:10.1302/0301-620X.98B1.36345
2. Harrison RJ Jr, Thacker MM, Pitcher JD, Temple HT, Scully SP. Distal femur replacement is useful in complex total knee arthroplasty revisions. Clin Orthop Relat Res. 2006;446:113-120. doi:10.1097/01.blo.0000214433.64774.1b
3. Smith EL, Shah A, Son SJ, et al. Survivorship of megaprostheses in revision hip and knee arthroplasty for septic and aseptic indications: a retrospective, multicenter study with minimum 2-year follow-up. Arthroplast Today. 2020;6(3):475-479. Published 2020 Jun 29. doi:10.1016/j.artd.2020.05.004
4. Wyles CC, Tibbo ME, Yuan BJ, Trousdale RT, Berry DJ, Abdel MP. Long-term results of total knee arthroplasty with contemporary distal femoral replacement. J Bone Joint Surg Am. 2020;102(1):45-51. doi:10.2106/JBJS.19.00489
5. Haidukewych GJ. Role of distal femoral replacement for periprosthetic fractures above a total knee arthroplasty: when and how?, J Orthop Trauma. 2019;33(suppl 6):S33-S35. doi:10.1097/BOT.0000000000001566
6. Hart GP, Kneisl JS, Springer BD, Patt JC, Karunakar MA. Open reduction vs distal femoral replacement arthroplasty for comminuted distal femur fractures in the patients 70 years and older: J Arthroplasty. 2017;32(1):202-206. doi:10.1016/j.arth.2016.06.006
7. Vertesich K, Puchner SE, Staats K, et al. Distal femoral reconstruction following failed total knee arthroplasty is accompanied with risk for complication and reduced joint function. BMC Musculoskelet Disord. 2019 Jan 31;20(1):47-54. doi:10.1186/s12891-019-2432-4
8. Biswas D, Haughom B, Mayle RE Jr, Della Valle CJ. Case report: Failure of rotating-hinge total knee prosthesis by disengagement of the hinge-post extension. Clin Orthop Relat Res. 2013;471(4):1389-1392. doi:10.1007/s11999-012-2736-2
9. Ward WG, Haight D, Ritchie P, Gordon S, Eckardt JJ. Dislocation of rotating hinge knee prostheses. A report of four cases. J Bone Joint Surg Am. 2005;87(5):1108-1112. doi:10.2106/JBJS.00837pp
10. Pacha-Vicente D, Malik A, Castellet-Feliu E, Nardi-Vilardaga J. Dislocation of rotating-hinge knee prostheses with antidislocation mechanism. J Arthroplasty. 2008;23(2):299-303. doi:10.1016/j.arth.2006.11.020
11. Manzano G, Schwarzkopf R. Posterior dislocation of the hinge-post extension in a rotating hinge total knee prosthesis. Case Rep Orthop. 2013;2013:756538. doi:10.1155/2013/756538
12. Vaishya R., Thapa, SS, Vaish A. Non-neoplastic indications and outcomes of the proximal and distal femur megaprosthesis: a critical review. Knee Surg Relat Res. 2020;32(1):18. Published 2020 Apr 9. doi:10.1186/s43019-020-00034-7
13. Lu M, Wang J, Xiao C, et al. Uncemented, curved, short endoprosthesis stem for distal femoral reconstruction: early follow-up outcomes. World J Surg Onc. 2018;16(1):183. doi:10.1186/s12957-018-1486-3
Herpes Zoster Following a Nucleoside-Modified Messenger RNA COVID-19 Vaccine
Since the end of 2019, COVID-19 infection caused by SARS-CoV-2 has spread in a worldwide pandemic. The first cutaneous manifestations possibly linked to COVID-19 were reported in spring 2020.1 Herpes zoster (HZ) was suspected as a predictive cutaneous manifestation of COVID-19 with a debated prognostic significance.2 The end of 2020 was marked with the beginning of vaccination against COVID-19, and safety studies reported few side effects after vaccination with nucleoside-modified messenger RNA (mRNA) COVID-19 vaccines.3 Real-life use of vaccines could lead to the occurrence of potential side effects (or fortuitous medical events) that were not observed in these studies. We report a series of 5 cases of HZ occurring after vaccination with a nucleoside-modified mRNA COVID-19 vaccine extracted from a declarative cohort of cutaneous reactions in our vaccination center.
Case Series
We identified 2 men and 3 women (Table) who experienced HZ after vaccination with a nucleoside-modified mRNA COVID-19 vaccine (Comirnaty, Pfizer-BioNTech). Patients fulfilled French governmental criteria for vaccination at the time of the report—older than 75 years or a health care professional—and they were vaccinated at the vaccination center of a French university hospital. The median age of the patients was 56 years (interquartile range [IQR], 51–82 years). One patient was diagnosed with COVID-19 in February 2020. A medical history of HZ was found in 1 patient. No medical history of immunosuppression was noted. Herpes zoster was observed on the same side of the body as the vaccination site in 4 patients. The median delay before the onset of symptoms was 6 days (IQR, 1–15 days) after injection. The median duration of the symptoms was 13 days (IQR, 11.5–16.5 days). Clinical signs of HZ were mild with few vesicles in 4 patients, and we observed a notably long delay between the onset of pain and the eruption of vesicles in 2 cases (4 and 10 days, respectively). The clinical diagnosis of HZ was confirmed by a dermatologist for all patients (Figures 1 and 2). Polymerase chain reaction assays for the detection of the varicella-zoster virus were performed in 2 cases and were positive. A complete blood cell count was performed in 1 patient, and we observed isolated lymphopenia (500/mm3 [reference range, 1000–4000/mm3]). Herpes zoster occurred after the first dose of vaccine in 4 patients and after the second dose for 1 patient. Three patients were treated with antiviral therapy (acyclovir) for 7 days. Three patients recovered from symptoms within 2 weeks and 2 patients within 1 week.

Comment
We report a series of HZ cases occurring after vaccination with a nucleoside-modified mRNA COVID-19 vaccine. We did not observe complicated HZ, and most of the time, HZ lesions were located on the same side of the body as the vaccine injection. One case of HZ after COVID-19 vaccination was reported by Bostan and Yalici-Armagan,4 but it followed injection with an inactivated vaccine, which is different from our series. Herpes zoster remains rarely reported, mainly following mRNA COVID-19 vaccination.5

Cases of HZ after vaccination have been reported after the live attenuated zoster or yellow fever vaccines, but HZ should not appear as a concomitant effect after any type of vaccines.6,7 Kawai et al8 reported that the incidence rate of HZ ranged from 3 to 5 cases per 1000 person-years in North America, Europe, and Asia-Pacific. The risk for recurrence of HZ ranged from 1% to 6% depending on the type of study design, age distribution of studied populations, and definition.8 In another retrospective database analysis in Israel, the incidence density rate of HZ was 3.46 cases per 1000 person-years in the total population and 12.8 cases per 1000 person-years in immunocompromised patients, therefore the immunocompromised status is important to consider.9

In our declarative cohort of skin eruptions before vaccination, we recorded 11 cases of HZ among 148 skin eruptions (7.43%) at the time of the study, but the design of the study did not allow us to estimate the exact incidence of HZ in the global COVID-19–vaccinated population because our study was not based on a systematic and prospective analysis of all vaccinated patients. The comparison between the prevalence of HZ in the COVID-19–vaccinated population and the nonvaccinated population is difficult owing to the lack of data about HZ in the nonvaccinated population at the time of our analysis. Furthermore, we did not include all vaccinated patients in a prospective follow-up. We highlight the importance of medical history of patients that differed between vaccinated patients (at the time of our analysis) and the global population due to French governmental access criteria to vaccination. The link to prior SARS-CoV-2 infection was uncertain because a medical history of COVID-19 was found in only 1 patient. Only 1 patient had a history of HZ, which is not a contraindication of COVID-19 vaccination.
Postinjection pains are frequent with COVID-19 vaccines, but clinical signs such as extension of pain, burning sensation, and eruption of vesicles should lead the physician to consider the diagnosis of HZ, regardless of the delay between the injection and the symptoms. Indeed, the onset of symptoms could be late, and the clinical presentation initially may be mistaken for an injection-site reaction, which is a frequent known side effect of vaccines. These new cases do not prove causality between COVID-19 vaccination and HZ. Varicella-zoster virus remains latent in dorsal-root or ganglia after primary infection, and HZ caused by reactivation of varicella-zoster virus may occur spontaneously or be triggered. In our series, we did not observe medical history of immunosuppression, and no other known risk factors of HZ (eg, radiation therapy, physical trauma, fever after vaccination) were recorded. The pathophysiologic mechanism remains elusive, but local vaccine-induced immunomodulation or an inflammatory state may be involved.
Conclusion
Our case series highlights that clinicians must remain vigilant to diagnose HZ early to prevent potential complications, such as postherpetic neuralgia. Also, vaccination should not be contraindicated in patients with medical history of HZ; the occurrence of HZ does not justify avoiding the second injection of the vaccine due to the benefit of vaccination.
- Recalcati S. Cutaneous manifestations in COVID-19: a first perspective. J Eur Acad Dermatol Venereol. 2020;34:E212-E213.
- Elsaie ML, Youssef EA, Nada HA. Herpes zoster might be an indicator for latent COVID 19 infection. Dermatol Ther. 2020;33:e13666.
- Polack FP, Thomas SJ, Kitchin N, et al. Safety and efficacy of the BNT162b2 mRNA COVID-19 vaccine. N Engl J Med. 2020;383:2603-2615.
- Bostan E, Yalici-Armagan B. Herpes zoster following inactivated COVID-19 vaccine: a coexistence or coincidence? J Cosmet Dermatol. 2021;20:1566-1567.
- Desai HD, Sharma K, Shah A, et al. Can SARS-CoV-2 vaccine increase the risk of reactivation of varicella zoster? a systematic review. J Cosmet Dermatol. 2021;20:3350-3361.
- Fahlbusch M, Wesselmann U, Lehmann P. Herpes zoster after varicella-zoster vaccination [in German]. Hautarzt. 2013;64:107-109.
- Bayas JM, González-Alvarez R, Guinovart C. Herpes zoster after yellow fever vaccination. J Travel Med. 2007;14:65-66.
- Kawai K, Gebremeskel BG, Acosta CJ. Systematic review of incidence and complications of herpes zoster: towards a global perspective. BMJ Open. 2014;10;4:E004833.
- Weitzman D, Shavit O, Stein M, et al. A population based study of the epidemiology of herpes zoster and its complications. J Infect. 2013;67:463-469.
Since the end of 2019, COVID-19 infection caused by SARS-CoV-2 has spread in a worldwide pandemic. The first cutaneous manifestations possibly linked to COVID-19 were reported in spring 2020.1 Herpes zoster (HZ) was suspected as a predictive cutaneous manifestation of COVID-19 with a debated prognostic significance.2 The end of 2020 was marked with the beginning of vaccination against COVID-19, and safety studies reported few side effects after vaccination with nucleoside-modified messenger RNA (mRNA) COVID-19 vaccines.3 Real-life use of vaccines could lead to the occurrence of potential side effects (or fortuitous medical events) that were not observed in these studies. We report a series of 5 cases of HZ occurring after vaccination with a nucleoside-modified mRNA COVID-19 vaccine extracted from a declarative cohort of cutaneous reactions in our vaccination center.
Case Series
We identified 2 men and 3 women (Table) who experienced HZ after vaccination with a nucleoside-modified mRNA COVID-19 vaccine (Comirnaty, Pfizer-BioNTech). Patients fulfilled French governmental criteria for vaccination at the time of the report—older than 75 years or a health care professional—and they were vaccinated at the vaccination center of a French university hospital. The median age of the patients was 56 years (interquartile range [IQR], 51–82 years). One patient was diagnosed with COVID-19 in February 2020. A medical history of HZ was found in 1 patient. No medical history of immunosuppression was noted. Herpes zoster was observed on the same side of the body as the vaccination site in 4 patients. The median delay before the onset of symptoms was 6 days (IQR, 1–15 days) after injection. The median duration of the symptoms was 13 days (IQR, 11.5–16.5 days). Clinical signs of HZ were mild with few vesicles in 4 patients, and we observed a notably long delay between the onset of pain and the eruption of vesicles in 2 cases (4 and 10 days, respectively). The clinical diagnosis of HZ was confirmed by a dermatologist for all patients (Figures 1 and 2). Polymerase chain reaction assays for the detection of the varicella-zoster virus were performed in 2 cases and were positive. A complete blood cell count was performed in 1 patient, and we observed isolated lymphopenia (500/mm3 [reference range, 1000–4000/mm3]). Herpes zoster occurred after the first dose of vaccine in 4 patients and after the second dose for 1 patient. Three patients were treated with antiviral therapy (acyclovir) for 7 days. Three patients recovered from symptoms within 2 weeks and 2 patients within 1 week.
Comment
We report a series of HZ cases occurring after vaccination with a nucleoside-modified mRNA COVID-19 vaccine. We did not observe complicated HZ, and most of the time, HZ lesions were located on the same side of the body as the vaccine injection. One case of HZ after COVID-19 vaccination was reported by Bostan and Yalici-Armagan,4 but it followed injection with an inactivated vaccine, which is different from our series. Herpes zoster remains rarely reported, mainly following mRNA COVID-19 vaccination.5
Cases of HZ after vaccination have been reported after the live attenuated zoster or yellow fever vaccines, but HZ should not appear as a concomitant effect after any type of vaccines.6,7 Kawai et al8 reported that the incidence rate of HZ ranged from 3 to 5 cases per 1000 person-years in North America, Europe, and Asia-Pacific. The risk for recurrence of HZ ranged from 1% to 6% depending on the type of study design, age distribution of studied populations, and definition.8 In another retrospective database analysis in Israel, the incidence density rate of HZ was 3.46 cases per 1000 person-years in the total population and 12.8 cases per 1000 person-years in immunocompromised patients, therefore the immunocompromised status is important to consider.9
In our declarative cohort of skin eruptions before vaccination, we recorded 11 cases of HZ among 148 skin eruptions (7.43%) at the time of the study, but the design of the study did not allow us to estimate the exact incidence of HZ in the global COVID-19–vaccinated population because our study was not based on a systematic and prospective analysis of all vaccinated patients. The comparison between the prevalence of HZ in the COVID-19–vaccinated population and the nonvaccinated population is difficult owing to the lack of data about HZ in the nonvaccinated population at the time of our analysis. Furthermore, we did not include all vaccinated patients in a prospective follow-up. We highlight the importance of medical history of patients that differed between vaccinated patients (at the time of our analysis) and the global population due to French governmental access criteria to vaccination. The link to prior SARS-CoV-2 infection was uncertain because a medical history of COVID-19 was found in only 1 patient. Only 1 patient had a history of HZ, which is not a contraindication of COVID-19 vaccination.
Postinjection pains are frequent with COVID-19 vaccines, but clinical signs such as extension of pain, burning sensation, and eruption of vesicles should lead the physician to consider the diagnosis of HZ, regardless of the delay between the injection and the symptoms. Indeed, the onset of symptoms could be late, and the clinical presentation initially may be mistaken for an injection-site reaction, which is a frequent known side effect of vaccines. These new cases do not prove causality between COVID-19 vaccination and HZ. Varicella-zoster virus remains latent in dorsal-root or ganglia after primary infection, and HZ caused by reactivation of varicella-zoster virus may occur spontaneously or be triggered. In our series, we did not observe medical history of immunosuppression, and no other known risk factors of HZ (eg, radiation therapy, physical trauma, fever after vaccination) were recorded. The pathophysiologic mechanism remains elusive, but local vaccine-induced immunomodulation or an inflammatory state may be involved.
Conclusion
Our case series highlights that clinicians must remain vigilant to diagnose HZ early to prevent potential complications, such as postherpetic neuralgia. Also, vaccination should not be contraindicated in patients with medical history of HZ; the occurrence of HZ does not justify avoiding the second injection of the vaccine due to the benefit of vaccination.
Since the end of 2019, COVID-19 infection caused by SARS-CoV-2 has spread in a worldwide pandemic. The first cutaneous manifestations possibly linked to COVID-19 were reported in spring 2020.1 Herpes zoster (HZ) was suspected as a predictive cutaneous manifestation of COVID-19 with a debated prognostic significance.2 The end of 2020 was marked with the beginning of vaccination against COVID-19, and safety studies reported few side effects after vaccination with nucleoside-modified messenger RNA (mRNA) COVID-19 vaccines.3 Real-life use of vaccines could lead to the occurrence of potential side effects (or fortuitous medical events) that were not observed in these studies. We report a series of 5 cases of HZ occurring after vaccination with a nucleoside-modified mRNA COVID-19 vaccine extracted from a declarative cohort of cutaneous reactions in our vaccination center.
Case Series
We identified 2 men and 3 women (Table) who experienced HZ after vaccination with a nucleoside-modified mRNA COVID-19 vaccine (Comirnaty, Pfizer-BioNTech). Patients fulfilled French governmental criteria for vaccination at the time of the report—older than 75 years or a health care professional—and they were vaccinated at the vaccination center of a French university hospital. The median age of the patients was 56 years (interquartile range [IQR], 51–82 years). One patient was diagnosed with COVID-19 in February 2020. A medical history of HZ was found in 1 patient. No medical history of immunosuppression was noted. Herpes zoster was observed on the same side of the body as the vaccination site in 4 patients. The median delay before the onset of symptoms was 6 days (IQR, 1–15 days) after injection. The median duration of the symptoms was 13 days (IQR, 11.5–16.5 days). Clinical signs of HZ were mild with few vesicles in 4 patients, and we observed a notably long delay between the onset of pain and the eruption of vesicles in 2 cases (4 and 10 days, respectively). The clinical diagnosis of HZ was confirmed by a dermatologist for all patients (Figures 1 and 2). Polymerase chain reaction assays for the detection of the varicella-zoster virus were performed in 2 cases and were positive. A complete blood cell count was performed in 1 patient, and we observed isolated lymphopenia (500/mm3 [reference range, 1000–4000/mm3]). Herpes zoster occurred after the first dose of vaccine in 4 patients and after the second dose for 1 patient. Three patients were treated with antiviral therapy (acyclovir) for 7 days. Three patients recovered from symptoms within 2 weeks and 2 patients within 1 week.
Comment
We report a series of HZ cases occurring after vaccination with a nucleoside-modified mRNA COVID-19 vaccine. We did not observe complicated HZ, and most of the time, HZ lesions were located on the same side of the body as the vaccine injection. One case of HZ after COVID-19 vaccination was reported by Bostan and Yalici-Armagan,4 but it followed injection with an inactivated vaccine, which is different from our series. Herpes zoster remains rarely reported, mainly following mRNA COVID-19 vaccination.5
Cases of HZ after vaccination have been reported after the live attenuated zoster or yellow fever vaccines, but HZ should not appear as a concomitant effect after any type of vaccines.6,7 Kawai et al8 reported that the incidence rate of HZ ranged from 3 to 5 cases per 1000 person-years in North America, Europe, and Asia-Pacific. The risk for recurrence of HZ ranged from 1% to 6% depending on the type of study design, age distribution of studied populations, and definition.8 In another retrospective database analysis in Israel, the incidence density rate of HZ was 3.46 cases per 1000 person-years in the total population and 12.8 cases per 1000 person-years in immunocompromised patients, therefore the immunocompromised status is important to consider.9
In our declarative cohort of skin eruptions before vaccination, we recorded 11 cases of HZ among 148 skin eruptions (7.43%) at the time of the study, but the design of the study did not allow us to estimate the exact incidence of HZ in the global COVID-19–vaccinated population because our study was not based on a systematic and prospective analysis of all vaccinated patients. The comparison between the prevalence of HZ in the COVID-19–vaccinated population and the nonvaccinated population is difficult owing to the lack of data about HZ in the nonvaccinated population at the time of our analysis. Furthermore, we did not include all vaccinated patients in a prospective follow-up. We highlight the importance of medical history of patients that differed between vaccinated patients (at the time of our analysis) and the global population due to French governmental access criteria to vaccination. The link to prior SARS-CoV-2 infection was uncertain because a medical history of COVID-19 was found in only 1 patient. Only 1 patient had a history of HZ, which is not a contraindication of COVID-19 vaccination.
Postinjection pains are frequent with COVID-19 vaccines, but clinical signs such as extension of pain, burning sensation, and eruption of vesicles should lead the physician to consider the diagnosis of HZ, regardless of the delay between the injection and the symptoms. Indeed, the onset of symptoms could be late, and the clinical presentation initially may be mistaken for an injection-site reaction, which is a frequent known side effect of vaccines. These new cases do not prove causality between COVID-19 vaccination and HZ. Varicella-zoster virus remains latent in dorsal-root or ganglia after primary infection, and HZ caused by reactivation of varicella-zoster virus may occur spontaneously or be triggered. In our series, we did not observe medical history of immunosuppression, and no other known risk factors of HZ (eg, radiation therapy, physical trauma, fever after vaccination) were recorded. The pathophysiologic mechanism remains elusive, but local vaccine-induced immunomodulation or an inflammatory state may be involved.
Conclusion
Our case series highlights that clinicians must remain vigilant to diagnose HZ early to prevent potential complications, such as postherpetic neuralgia. Also, vaccination should not be contraindicated in patients with medical history of HZ; the occurrence of HZ does not justify avoiding the second injection of the vaccine due to the benefit of vaccination.
- Recalcati S. Cutaneous manifestations in COVID-19: a first perspective. J Eur Acad Dermatol Venereol. 2020;34:E212-E213.
- Elsaie ML, Youssef EA, Nada HA. Herpes zoster might be an indicator for latent COVID 19 infection. Dermatol Ther. 2020;33:e13666.
- Polack FP, Thomas SJ, Kitchin N, et al. Safety and efficacy of the BNT162b2 mRNA COVID-19 vaccine. N Engl J Med. 2020;383:2603-2615.
- Bostan E, Yalici-Armagan B. Herpes zoster following inactivated COVID-19 vaccine: a coexistence or coincidence? J Cosmet Dermatol. 2021;20:1566-1567.
- Desai HD, Sharma K, Shah A, et al. Can SARS-CoV-2 vaccine increase the risk of reactivation of varicella zoster? a systematic review. J Cosmet Dermatol. 2021;20:3350-3361.
- Fahlbusch M, Wesselmann U, Lehmann P. Herpes zoster after varicella-zoster vaccination [in German]. Hautarzt. 2013;64:107-109.
- Bayas JM, González-Alvarez R, Guinovart C. Herpes zoster after yellow fever vaccination. J Travel Med. 2007;14:65-66.
- Kawai K, Gebremeskel BG, Acosta CJ. Systematic review of incidence and complications of herpes zoster: towards a global perspective. BMJ Open. 2014;10;4:E004833.
- Weitzman D, Shavit O, Stein M, et al. A population based study of the epidemiology of herpes zoster and its complications. J Infect. 2013;67:463-469.
- Recalcati S. Cutaneous manifestations in COVID-19: a first perspective. J Eur Acad Dermatol Venereol. 2020;34:E212-E213.
- Elsaie ML, Youssef EA, Nada HA. Herpes zoster might be an indicator for latent COVID 19 infection. Dermatol Ther. 2020;33:e13666.
- Polack FP, Thomas SJ, Kitchin N, et al. Safety and efficacy of the BNT162b2 mRNA COVID-19 vaccine. N Engl J Med. 2020;383:2603-2615.
- Bostan E, Yalici-Armagan B. Herpes zoster following inactivated COVID-19 vaccine: a coexistence or coincidence? J Cosmet Dermatol. 2021;20:1566-1567.
- Desai HD, Sharma K, Shah A, et al. Can SARS-CoV-2 vaccine increase the risk of reactivation of varicella zoster? a systematic review. J Cosmet Dermatol. 2021;20:3350-3361.
- Fahlbusch M, Wesselmann U, Lehmann P. Herpes zoster after varicella-zoster vaccination [in German]. Hautarzt. 2013;64:107-109.
- Bayas JM, González-Alvarez R, Guinovart C. Herpes zoster after yellow fever vaccination. J Travel Med. 2007;14:65-66.
- Kawai K, Gebremeskel BG, Acosta CJ. Systematic review of incidence and complications of herpes zoster: towards a global perspective. BMJ Open. 2014;10;4:E004833.
- Weitzman D, Shavit O, Stein M, et al. A population based study of the epidemiology of herpes zoster and its complications. J Infect. 2013;67:463-469.
Practice Points
- Herpes zoster (HZ) has been reported following COVID-19 vaccination.
- Postinjection pain is common with COVID-19 vaccination, but clinical signs such as extension of pain, burning sensation, and eruption of vesicles should lead the physician to consider the diagnosis of HZ, regardless of the delay in onset between the injection and the symptoms.
- When indicated, the second vaccine dose should not be avoided in patients who are diagnosed with HZ.

Secretan Syndrome: A Fluctuating Case of Factitious Lymphedema
Secretan syndrome (SS) represents a recurrent or chronic form of factitious lymphedema, usually affecting the dorsal aspect of the hand.1-3 It is accepted as a subtype of Munchausen syndrome whereby the patient self-inflicts and simulates lymphedema.1,2 Historically, many of the cases reported with the term Charcot’s oedème bleu are now believed to represent clinical variants of SS.4-6
Case Report
A 38-year-old Turkish woman presented with progressive swelling of the right hand of 2 years’ duration that had caused difficulty in manual work and reduction in manual dexterity. She previously had sought medical treatment for this condition by visiting several hospitals. According to her medical record, the following laboratory or radiologic tests had revealed negative or normal findings, except for obvious soft-tissue edema: bacterial and fungal cultures, plain radiography, Doppler ultrasonography, lymphoscintigraphy, magnetic resonance imaging, fine needle aspiration, and punch biopsy. Reflex sympathetic dystrophy, compartment syndrome, filariasis, tuberculosis, and lymphatic and venous obstruction were all excluded by appropriate testing. Our patient was in good health prior to onset of this disorder, and her medical history was unremarkable. There was no family history of a similar condition.
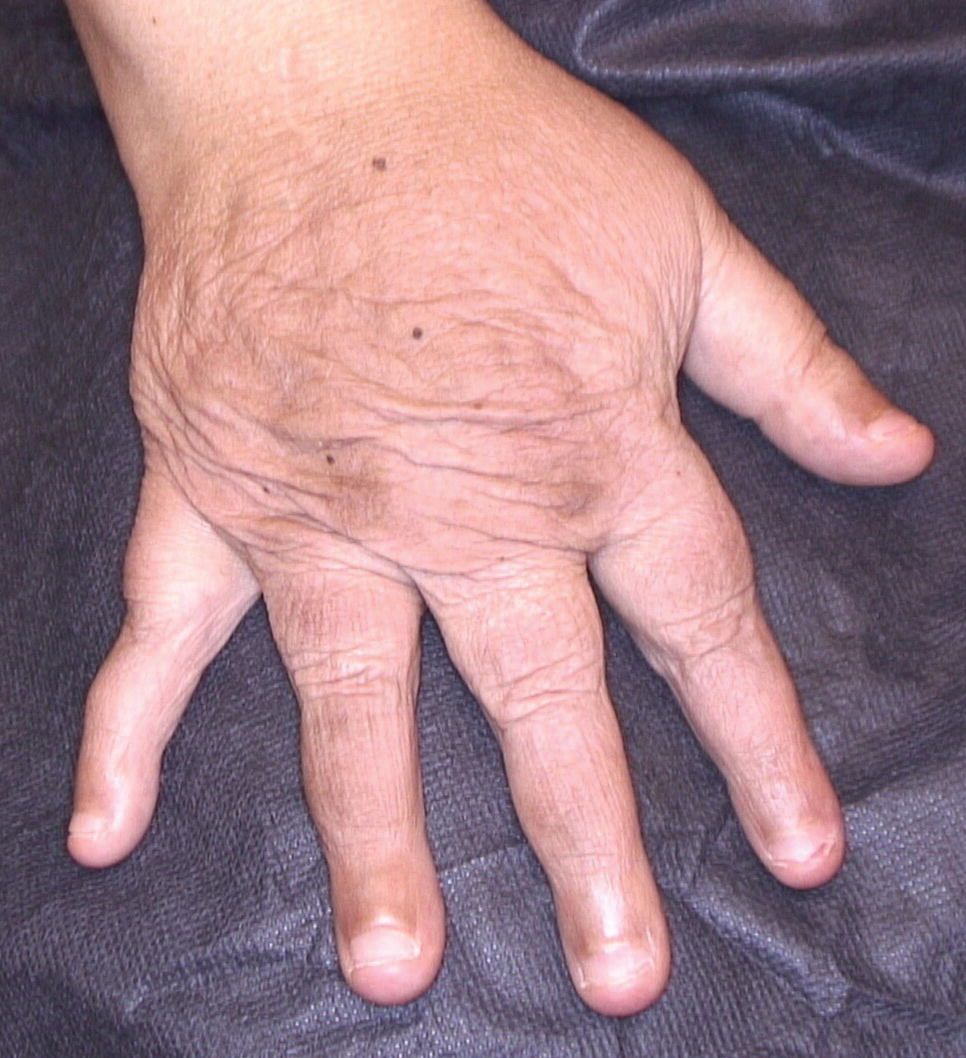
Dermatologic examination revealed brawny, soft, pitting edema; erythema; and crusts affecting the dorsal aspect of the right hand and proximal parts of the fingers (Figure 1). The yellow discoloration of the skin and nails was attributed to potassium permanganate wet dressings. Under an elastic bandage at the wrist, which the patient unrolled herself, a sharp line of demarcation was evident, separating the lymphedematous and normal parts of the arm. There was no axillary lymphadenopathy.

The patient’s affect was discordant to the manifestation of the cutaneous findings. She wanted to show every physician in the department how swollen her hand was and seemed to be happy with this condition. Although she displayed no signs of disturbance when the affected extremity was touched or handled, she reported severe pain and tenderness as well as difficulty in housework. She noted that she normally resided in a city and that the swelling had started at the time she had relocated to a rural village to take care of her bedridden mother-in-law. She was under an intensive workload in the village, and the condition of the hand was impeding manual work.
Factitious lymphedema was considered, and hospitalization was recommended. The patient was then lost to follow-up; however, one of her relatives noted that the patient had returned to the city. When she presented again 1 year later, almost all physical signs had disappeared (Figure 2), and a psychiatric referral was recommended. A Minnesota Multiphasic Personality Inventory test yielded an invalid result due to the patient’s exaggeration of her preexisting physical symptoms. Further psychiatric workup was rejected by the patient.
Almost a year after the psychiatric referral, the patient’s follow-up photographs revealed that the lymphedema recurred when she went to visit her mother-in-law in the rural village and that it was completely ameliorated when she returned to the city. Thus, a positive “mother-in-law provocation test” was accepted as final proof of the self-inflicted nature of the condition.
Comment
In 1901, Henri Francois Secretan, a Swiss physician, reported workmen who had persistent hard swellings on the dorsal aspect of the hands after minor work-related trauma for which they had compensation claims.7 In his original report, Secretan did not suggest self-inflicted trauma in the etiology of this disorder.5,8,9 In 1890, Jean Martin Charcot, a French neurologist, described oedème bleu, a term that is now believed to denote a condition similar to SS.4-6 Currently, SS is attributed to self-inflicted injury and is considered a form of factitious lymphedema.9 As in dermatitis artefacta, most patients with SS are young women, and male patients with the condition tend to be older.3,8
The mechanism used to provoke this factitious lymphedema might be of traumatic or obstructive nature. Secretan syndrome either is induced by intermittent or constant application of a tourniquet, ligature, cord, elastic bandage, scarf, kerchief, rubber band, or compress around the affected extremity, or by repetitive blunt trauma, force, or skin irritation.1,4,5,8-10 There was an underlying psychopathology in all reported cases.1,8,11 Factitious lymphedema is unconsciously motivated and consciously produced.4,12 The affected patient often is experiencing a serious emotional conflict and is unlikely to be a malingerer, although exaggeration of symptoms may occur, as in our patient.12 Psychiatric evaluation in SS may uncover neurosis, hysteria, frank psychosis, schizophrenia, masochism, depression, or an abnormal personality disorder.1,12
Patients with SS present with recurrent or chronic lymphedema, usually affecting the dominant hand.1 Involvement usually is unilateral; bilateral cases are rare.3,6 Secretan syndrome is not solely limited to the hands; it also may involve the upper and lower extremities, including the feet.3,11 There may be a clear line of demarcation, a ring, sulcus, distinct circumferential linear bands of erythema, discoloration, or ecchymoses, separating the normal and lymphedematous parts of the extremity.1,4,6,8-10,12 Patients usually attempt to hide the constricted areas from sight.1 Over time, flexion contractures may develop due to peritendinous fibrosis.6 Histopathology displays a hematoma with adhesions to the extensor tendons; a hematoma surrounded by a thickened scar; or changes similar to ganglion tissue with cystic areas of mucin, fibrosis, and myxoid degeneration.4,6
Factitious lymphedema can only be definitively diagnosed when the patient confesses or is caught self-inflicting the injury. Nevertheless, a diagnosis by exclusion is possible.4 Lymphangiography, lymphoscintigraphy, vascular Doppler ultrasonography, and magnetic resonance imaging may be helpful in excluding congenital and acquired causes of lymphedema and venous obstruction.1,3,9,11 Magnetic resonance imaging may show soft tissue and tendon edema as well as diffuse peritendinous fibrosis extending to the fascia of the dorsal interosseous muscles.3,4
Factitious lymphedema should be suspected in all patients with recurrent or chronic unilateral lymphedema without an explicable or apparent predisposing factor.4,11,12 Patients with SS typically visit several hospitals or institutions; see many physicians; and willingly accept, request, and undergo unnecessary extensive, invasive, and costly diagnostic and therapeutic procedures and prolonged hospitalizations.1,2,5,12 The disorder promptly responds to immobilization and elevation of the limb.2,4 Plaster casts may prove useful in prevention of compression and thus amelioration of the lymphedema.1,4,6 Once the diagnosis is confirmed, direct confrontation should be avoided and ideally the patient should be referred for psychiatric evaluation.1,2,4,5,8,12 If the patient admits self-inflicting behavior, psychotherapy and/or behavior modification therapy along with psychotropic medications may be helpful to relieve emotional and behavioral symptoms.12 Unfortunately, if the patient denies a self-inflicting role in the occurrence of lymphedema and persists in self-injurious behavior, psychotherapy or psychotropic medications will be futile.9
1. Miyamoto Y, Hamanaka T, Yokoyama S, et al. Factitious lymphedema of the upper limb. Kawasaki Med J. 1979;5:39-45.
2. de Oliveira RK, Bayer LR, Lauxen D, et al. Factitious lesions of the hand. Rev Bras Ortop. 2013;48:381-386.
3. Hahm MH, Yi JH. A case report of Secretan’s disease in both hands. J Korean Soc Radiol. 2013;68:511-514.
4. Eldridge MP, Grunert BK, Matloub HS. Streamlined classification of psychopathological hand disorders: a literature review. Hand (NY). 2008;3:118-128.
5. Ostlere LS, Harris D, Denton C, et al. Boxing-glove hand: an unusual presentation of dermatitis artefacta. J Am Acad Dermatol. 1993;28:120-122.
6. Winkelmann RK, Barker SM. Factitial traumatic panniculitis. J Am Acad Dermatol. 1985;13:988-994.
7. Secretan H. Oederne dur et hyperplasie traumatique du metacarpe dorsal. RevMed Suisse Romande. 1901;21:409-416.
8. Barth JH, Pegum JS. The case of the speckled band: acquired lymphedema due to constriction bands. J Am Acad Dermatol. 1986;15:296-297.
9. Birman MV, Lee DH. Factitious disorders of the upper extremity. J Am Acad Orthop Surg. 2012;20:78-85.
10. Nwaejike N, Archbold H, Wilson DS. Factitious lymphoedema as a psychiatric condition mimicking reflex sympathetic dystrophy: a case report. J Med Case Rep. 2008;2:216.
11. De Fátima Guerreiro Godoy M, Pereira De Godoy JM. Factitious lymphedema of the arm: case report and review of publications. Eur J Phys Rehabil Med. 2015;51:337-339.
12. Abhari SAA, Alimalayeri N, Abhari SSA, et al. Factitious lymphedema of the hand. Iran J Psychiatry. 2006;1:166-168.
Secretan syndrome (SS) represents a recurrent or chronic form of factitious lymphedema, usually affecting the dorsal aspect of the hand.1-3 It is accepted as a subtype of Munchausen syndrome whereby the patient self-inflicts and simulates lymphedema.1,2 Historically, many of the cases reported with the term Charcot’s oedème bleu are now believed to represent clinical variants of SS.4-6
Case Report
A 38-year-old Turkish woman presented with progressive swelling of the right hand of 2 years’ duration that had caused difficulty in manual work and reduction in manual dexterity. She previously had sought medical treatment for this condition by visiting several hospitals. According to her medical record, the following laboratory or radiologic tests had revealed negative or normal findings, except for obvious soft-tissue edema: bacterial and fungal cultures, plain radiography, Doppler ultrasonography, lymphoscintigraphy, magnetic resonance imaging, fine needle aspiration, and punch biopsy. Reflex sympathetic dystrophy, compartment syndrome, filariasis, tuberculosis, and lymphatic and venous obstruction were all excluded by appropriate testing. Our patient was in good health prior to onset of this disorder, and her medical history was unremarkable. There was no family history of a similar condition.
Dermatologic examination revealed brawny, soft, pitting edema; erythema; and crusts affecting the dorsal aspect of the right hand and proximal parts of the fingers (Figure 1). The yellow discoloration of the skin and nails was attributed to potassium permanganate wet dressings. Under an elastic bandage at the wrist, which the patient unrolled herself, a sharp line of demarcation was evident, separating the lymphedematous and normal parts of the arm. There was no axillary lymphadenopathy.
The patient’s affect was discordant to the manifestation of the cutaneous findings. She wanted to show every physician in the department how swollen her hand was and seemed to be happy with this condition. Although she displayed no signs of disturbance when the affected extremity was touched or handled, she reported severe pain and tenderness as well as difficulty in housework. She noted that she normally resided in a city and that the swelling had started at the time she had relocated to a rural village to take care of her bedridden mother-in-law. She was under an intensive workload in the village, and the condition of the hand was impeding manual work.
Factitious lymphedema was considered, and hospitalization was recommended. The patient was then lost to follow-up; however, one of her relatives noted that the patient had returned to the city. When she presented again 1 year later, almost all physical signs had disappeared (Figure 2), and a psychiatric referral was recommended. A Minnesota Multiphasic Personality Inventory test yielded an invalid result due to the patient’s exaggeration of her preexisting physical symptoms. Further psychiatric workup was rejected by the patient.
Almost a year after the psychiatric referral, the patient’s follow-up photographs revealed that the lymphedema recurred when she went to visit her mother-in-law in the rural village and that it was completely ameliorated when she returned to the city. Thus, a positive “mother-in-law provocation test” was accepted as final proof of the self-inflicted nature of the condition.
Comment
In 1901, Henri Francois Secretan, a Swiss physician, reported workmen who had persistent hard swellings on the dorsal aspect of the hands after minor work-related trauma for which they had compensation claims.7 In his original report, Secretan did not suggest self-inflicted trauma in the etiology of this disorder.5,8,9 In 1890, Jean Martin Charcot, a French neurologist, described oedème bleu, a term that is now believed to denote a condition similar to SS.4-6 Currently, SS is attributed to self-inflicted injury and is considered a form of factitious lymphedema.9 As in dermatitis artefacta, most patients with SS are young women, and male patients with the condition tend to be older.3,8
The mechanism used to provoke this factitious lymphedema might be of traumatic or obstructive nature. Secretan syndrome either is induced by intermittent or constant application of a tourniquet, ligature, cord, elastic bandage, scarf, kerchief, rubber band, or compress around the affected extremity, or by repetitive blunt trauma, force, or skin irritation.1,4,5,8-10 There was an underlying psychopathology in all reported cases.1,8,11 Factitious lymphedema is unconsciously motivated and consciously produced.4,12 The affected patient often is experiencing a serious emotional conflict and is unlikely to be a malingerer, although exaggeration of symptoms may occur, as in our patient.12 Psychiatric evaluation in SS may uncover neurosis, hysteria, frank psychosis, schizophrenia, masochism, depression, or an abnormal personality disorder.1,12
Patients with SS present with recurrent or chronic lymphedema, usually affecting the dominant hand.1 Involvement usually is unilateral; bilateral cases are rare.3,6 Secretan syndrome is not solely limited to the hands; it also may involve the upper and lower extremities, including the feet.3,11 There may be a clear line of demarcation, a ring, sulcus, distinct circumferential linear bands of erythema, discoloration, or ecchymoses, separating the normal and lymphedematous parts of the extremity.1,4,6,8-10,12 Patients usually attempt to hide the constricted areas from sight.1 Over time, flexion contractures may develop due to peritendinous fibrosis.6 Histopathology displays a hematoma with adhesions to the extensor tendons; a hematoma surrounded by a thickened scar; or changes similar to ganglion tissue with cystic areas of mucin, fibrosis, and myxoid degeneration.4,6
Factitious lymphedema can only be definitively diagnosed when the patient confesses or is caught self-inflicting the injury. Nevertheless, a diagnosis by exclusion is possible.4 Lymphangiography, lymphoscintigraphy, vascular Doppler ultrasonography, and magnetic resonance imaging may be helpful in excluding congenital and acquired causes of lymphedema and venous obstruction.1,3,9,11 Magnetic resonance imaging may show soft tissue and tendon edema as well as diffuse peritendinous fibrosis extending to the fascia of the dorsal interosseous muscles.3,4
Factitious lymphedema should be suspected in all patients with recurrent or chronic unilateral lymphedema without an explicable or apparent predisposing factor.4,11,12 Patients with SS typically visit several hospitals or institutions; see many physicians; and willingly accept, request, and undergo unnecessary extensive, invasive, and costly diagnostic and therapeutic procedures and prolonged hospitalizations.1,2,5,12 The disorder promptly responds to immobilization and elevation of the limb.2,4 Plaster casts may prove useful in prevention of compression and thus amelioration of the lymphedema.1,4,6 Once the diagnosis is confirmed, direct confrontation should be avoided and ideally the patient should be referred for psychiatric evaluation.1,2,4,5,8,12 If the patient admits self-inflicting behavior, psychotherapy and/or behavior modification therapy along with psychotropic medications may be helpful to relieve emotional and behavioral symptoms.12 Unfortunately, if the patient denies a self-inflicting role in the occurrence of lymphedema and persists in self-injurious behavior, psychotherapy or psychotropic medications will be futile.9
Secretan syndrome (SS) represents a recurrent or chronic form of factitious lymphedema, usually affecting the dorsal aspect of the hand.1-3 It is accepted as a subtype of Munchausen syndrome whereby the patient self-inflicts and simulates lymphedema.1,2 Historically, many of the cases reported with the term Charcot’s oedème bleu are now believed to represent clinical variants of SS.4-6
Case Report
A 38-year-old Turkish woman presented with progressive swelling of the right hand of 2 years’ duration that had caused difficulty in manual work and reduction in manual dexterity. She previously had sought medical treatment for this condition by visiting several hospitals. According to her medical record, the following laboratory or radiologic tests had revealed negative or normal findings, except for obvious soft-tissue edema: bacterial and fungal cultures, plain radiography, Doppler ultrasonography, lymphoscintigraphy, magnetic resonance imaging, fine needle aspiration, and punch biopsy. Reflex sympathetic dystrophy, compartment syndrome, filariasis, tuberculosis, and lymphatic and venous obstruction were all excluded by appropriate testing. Our patient was in good health prior to onset of this disorder, and her medical history was unremarkable. There was no family history of a similar condition.
Dermatologic examination revealed brawny, soft, pitting edema; erythema; and crusts affecting the dorsal aspect of the right hand and proximal parts of the fingers (Figure 1). The yellow discoloration of the skin and nails was attributed to potassium permanganate wet dressings. Under an elastic bandage at the wrist, which the patient unrolled herself, a sharp line of demarcation was evident, separating the lymphedematous and normal parts of the arm. There was no axillary lymphadenopathy.
The patient’s affect was discordant to the manifestation of the cutaneous findings. She wanted to show every physician in the department how swollen her hand was and seemed to be happy with this condition. Although she displayed no signs of disturbance when the affected extremity was touched or handled, she reported severe pain and tenderness as well as difficulty in housework. She noted that she normally resided in a city and that the swelling had started at the time she had relocated to a rural village to take care of her bedridden mother-in-law. She was under an intensive workload in the village, and the condition of the hand was impeding manual work.
Factitious lymphedema was considered, and hospitalization was recommended. The patient was then lost to follow-up; however, one of her relatives noted that the patient had returned to the city. When she presented again 1 year later, almost all physical signs had disappeared (Figure 2), and a psychiatric referral was recommended. A Minnesota Multiphasic Personality Inventory test yielded an invalid result due to the patient’s exaggeration of her preexisting physical symptoms. Further psychiatric workup was rejected by the patient.
Almost a year after the psychiatric referral, the patient’s follow-up photographs revealed that the lymphedema recurred when she went to visit her mother-in-law in the rural village and that it was completely ameliorated when she returned to the city. Thus, a positive “mother-in-law provocation test” was accepted as final proof of the self-inflicted nature of the condition.
Comment
In 1901, Henri Francois Secretan, a Swiss physician, reported workmen who had persistent hard swellings on the dorsal aspect of the hands after minor work-related trauma for which they had compensation claims.7 In his original report, Secretan did not suggest self-inflicted trauma in the etiology of this disorder.5,8,9 In 1890, Jean Martin Charcot, a French neurologist, described oedème bleu, a term that is now believed to denote a condition similar to SS.4-6 Currently, SS is attributed to self-inflicted injury and is considered a form of factitious lymphedema.9 As in dermatitis artefacta, most patients with SS are young women, and male patients with the condition tend to be older.3,8
The mechanism used to provoke this factitious lymphedema might be of traumatic or obstructive nature. Secretan syndrome either is induced by intermittent or constant application of a tourniquet, ligature, cord, elastic bandage, scarf, kerchief, rubber band, or compress around the affected extremity, or by repetitive blunt trauma, force, or skin irritation.1,4,5,8-10 There was an underlying psychopathology in all reported cases.1,8,11 Factitious lymphedema is unconsciously motivated and consciously produced.4,12 The affected patient often is experiencing a serious emotional conflict and is unlikely to be a malingerer, although exaggeration of symptoms may occur, as in our patient.12 Psychiatric evaluation in SS may uncover neurosis, hysteria, frank psychosis, schizophrenia, masochism, depression, or an abnormal personality disorder.1,12
Patients with SS present with recurrent or chronic lymphedema, usually affecting the dominant hand.1 Involvement usually is unilateral; bilateral cases are rare.3,6 Secretan syndrome is not solely limited to the hands; it also may involve the upper and lower extremities, including the feet.3,11 There may be a clear line of demarcation, a ring, sulcus, distinct circumferential linear bands of erythema, discoloration, or ecchymoses, separating the normal and lymphedematous parts of the extremity.1,4,6,8-10,12 Patients usually attempt to hide the constricted areas from sight.1 Over time, flexion contractures may develop due to peritendinous fibrosis.6 Histopathology displays a hematoma with adhesions to the extensor tendons; a hematoma surrounded by a thickened scar; or changes similar to ganglion tissue with cystic areas of mucin, fibrosis, and myxoid degeneration.4,6
Factitious lymphedema can only be definitively diagnosed when the patient confesses or is caught self-inflicting the injury. Nevertheless, a diagnosis by exclusion is possible.4 Lymphangiography, lymphoscintigraphy, vascular Doppler ultrasonography, and magnetic resonance imaging may be helpful in excluding congenital and acquired causes of lymphedema and venous obstruction.1,3,9,11 Magnetic resonance imaging may show soft tissue and tendon edema as well as diffuse peritendinous fibrosis extending to the fascia of the dorsal interosseous muscles.3,4
Factitious lymphedema should be suspected in all patients with recurrent or chronic unilateral lymphedema without an explicable or apparent predisposing factor.4,11,12 Patients with SS typically visit several hospitals or institutions; see many physicians; and willingly accept, request, and undergo unnecessary extensive, invasive, and costly diagnostic and therapeutic procedures and prolonged hospitalizations.1,2,5,12 The disorder promptly responds to immobilization and elevation of the limb.2,4 Plaster casts may prove useful in prevention of compression and thus amelioration of the lymphedema.1,4,6 Once the diagnosis is confirmed, direct confrontation should be avoided and ideally the patient should be referred for psychiatric evaluation.1,2,4,5,8,12 If the patient admits self-inflicting behavior, psychotherapy and/or behavior modification therapy along with psychotropic medications may be helpful to relieve emotional and behavioral symptoms.12 Unfortunately, if the patient denies a self-inflicting role in the occurrence of lymphedema and persists in self-injurious behavior, psychotherapy or psychotropic medications will be futile.9
1. Miyamoto Y, Hamanaka T, Yokoyama S, et al. Factitious lymphedema of the upper limb. Kawasaki Med J. 1979;5:39-45.
2. de Oliveira RK, Bayer LR, Lauxen D, et al. Factitious lesions of the hand. Rev Bras Ortop. 2013;48:381-386.
3. Hahm MH, Yi JH. A case report of Secretan’s disease in both hands. J Korean Soc Radiol. 2013;68:511-514.
4. Eldridge MP, Grunert BK, Matloub HS. Streamlined classification of psychopathological hand disorders: a literature review. Hand (NY). 2008;3:118-128.
5. Ostlere LS, Harris D, Denton C, et al. Boxing-glove hand: an unusual presentation of dermatitis artefacta. J Am Acad Dermatol. 1993;28:120-122.
6. Winkelmann RK, Barker SM. Factitial traumatic panniculitis. J Am Acad Dermatol. 1985;13:988-994.
7. Secretan H. Oederne dur et hyperplasie traumatique du metacarpe dorsal. RevMed Suisse Romande. 1901;21:409-416.
8. Barth JH, Pegum JS. The case of the speckled band: acquired lymphedema due to constriction bands. J Am Acad Dermatol. 1986;15:296-297.
9. Birman MV, Lee DH. Factitious disorders of the upper extremity. J Am Acad Orthop Surg. 2012;20:78-85.
10. Nwaejike N, Archbold H, Wilson DS. Factitious lymphoedema as a psychiatric condition mimicking reflex sympathetic dystrophy: a case report. J Med Case Rep. 2008;2:216.
11. De Fátima Guerreiro Godoy M, Pereira De Godoy JM. Factitious lymphedema of the arm: case report and review of publications. Eur J Phys Rehabil Med. 2015;51:337-339.
12. Abhari SAA, Alimalayeri N, Abhari SSA, et al. Factitious lymphedema of the hand. Iran J Psychiatry. 2006;1:166-168.
1. Miyamoto Y, Hamanaka T, Yokoyama S, et al. Factitious lymphedema of the upper limb. Kawasaki Med J. 1979;5:39-45.
2. de Oliveira RK, Bayer LR, Lauxen D, et al. Factitious lesions of the hand. Rev Bras Ortop. 2013;48:381-386.
3. Hahm MH, Yi JH. A case report of Secretan’s disease in both hands. J Korean Soc Radiol. 2013;68:511-514.
4. Eldridge MP, Grunert BK, Matloub HS. Streamlined classification of psychopathological hand disorders: a literature review. Hand (NY). 2008;3:118-128.
5. Ostlere LS, Harris D, Denton C, et al. Boxing-glove hand: an unusual presentation of dermatitis artefacta. J Am Acad Dermatol. 1993;28:120-122.
6. Winkelmann RK, Barker SM. Factitial traumatic panniculitis. J Am Acad Dermatol. 1985;13:988-994.
7. Secretan H. Oederne dur et hyperplasie traumatique du metacarpe dorsal. RevMed Suisse Romande. 1901;21:409-416.
8. Barth JH, Pegum JS. The case of the speckled band: acquired lymphedema due to constriction bands. J Am Acad Dermatol. 1986;15:296-297.
9. Birman MV, Lee DH. Factitious disorders of the upper extremity. J Am Acad Orthop Surg. 2012;20:78-85.
10. Nwaejike N, Archbold H, Wilson DS. Factitious lymphoedema as a psychiatric condition mimicking reflex sympathetic dystrophy: a case report. J Med Case Rep. 2008;2:216.
11. De Fátima Guerreiro Godoy M, Pereira De Godoy JM. Factitious lymphedema of the arm: case report and review of publications. Eur J Phys Rehabil Med. 2015;51:337-339.
12. Abhari SAA, Alimalayeri N, Abhari SSA, et al. Factitious lymphedema of the hand. Iran J Psychiatry. 2006;1:166-168.
Practice Points
- Secretan syndrome is a recurrent or chronic form of factitious lymphedema that usually affects the dorsal aspect of the hand; it is accepted as a subtype of Munchausen syndrome.
- Secretan syndrome usually is induced by compression of the extremity by tourniquets, ligatures, cords, or similar equipment.
- This unconsciously motivated and consciously produced lymphedema is an expression of underlying psychiatric disease.
Not All Pulmonary Nodules in Smokers are Lung Cancer
Identification of pulmonary nodules in older adults who smoke immediately brings concern for malignancy in the mind of clinicians. This is particularly the case in patients with significant smoking history. According to the National Cancer Institute in 2019, 12.9% of all new cancer cases were lung cancers.1 Screening for lung cancer, especially in patients with increased risk from smoking, is imperative to early detection and treatment. However, 20% of patients will be overdiagnosed by lung cancer-screening techniques.2 The rate of malignancy noted on a patient’s first screening computed tomography (CT) scan was between 3.7% and 5.5%.3
Rheumatoid arthritis (RA) is an autoimmune inflammatory condition that mainly affects the joints. Extraarticular manifestations can arise in various locations throughout the body, however. These manifestations are commonly observed in the skin, heart, and lungs.4 Prevalence of pulmonary rheumatoid nodules ranges from < 0.4% in radiologic studies to 32% in lung biopsies of patients with RA and nodules.5
Furthermore, there is a strong association between the risk of rheumatoid nodules in patients with positive serum rheumatoid factor (RF) and smoking history.6 Solitary pulmonary nodules in patients with RA can coexist with bronchogenic carcinoma, making their diagnosis more important.7
Case Presentation
A 54-year-old woman with a 30 pack-year smoking history and history of RA initially presented to the emergency department for cough and dyspnea for 5-day duration. Her initial diagnosis was bronchitis based on presenting symptom profile. A chest CT demonstrated 3 cavitary pulmonary nodules, 1 measuring 2.4 x 2.0 cm in the right middle lobe, and 2 additional nodules, measuring 1.8 x 1.4 and 1.5 x 1.4 in the left upper lobe (Figure). She had no improvement of symptoms after a 7-day course of doxycycline. The patient was taking methotrexate 15 mg weekly and golimumab 50 mg subcutaneously every 4 weeks as treatment for RA, prescribed by her rheumatologist.
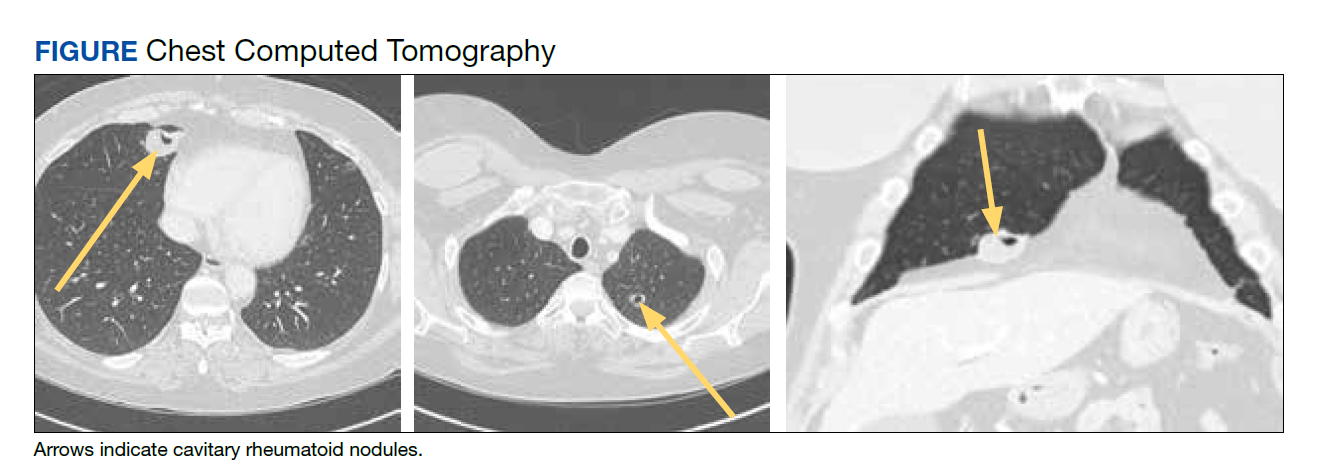
Pulmonology was consulted and a positron emission tomography-CT (PET-CT) confirmed several cavitary pulmonary nodules involving both lungs with no suspicious fluorodeoxyglucose (FDG) uptake. The largest lesion was in the right middle lobe with FDG uptake of 1.9. Additional nodules were found in the left upper lobe, measuring 1.8 x 1.4 cm with FDG of 4.01, and in the left lung apex, measuring 1.5 x 1.4 cm with uptake of 3.53. CTguided percutaneous fine needle aspiration (PFNA) of the right middle lobe lung nodule demonstrated granuloma with central inflammatory debris. Grocott methenamine silver (GMS) stain was negative for fungal organism, acid-fast bacteria (AFB) stain was negative for acid-fast bacilli, and CD20 and CD3 immunostaining demonstrated mixed B- and T-cell populations. There was no evidence of atypia or malignancy. The biopsy demonstrated granuloma with central inflammatory debris on a background of densely fibrotic tissue and lympho-plasmatic inflammation. This finding confirmed the diagnosis of RA with pulmonary involvement.
Outpatient follow-up was established with a pulmonologist and rheumatologist. Methotrexate 15 mg weekly and golimumab subcutaneously 50 mg every 4 weeks were prescribed for the patient. The nodules are being monitored based on Fleischer guidelines with CT imaging 3 to 6 months following initial presentation. Further imaging will be considered at 18 to 24 months as well to further assess stability of the nodules and monitor for changes in size, shape, and necrosis. The patient also was encouraged to quit smoking. Her clinical course since the diagnosis has been stable.
Discussion
The differential diagnosis for new multiple pulmonary nodules on imaging studies is broad and includes infectious processes, such as tuberculosis, as well as other mycobacterial, fungal, and bacterial infections. Noninfectious causes of lung disease are an even broader category of consideration. Noninfectious pulmonary nodules differential includes sarcoidosis, granulomatous with polyangiitis, hypersensitivity pneumonitis, methotrexate drug reaction, pulmonary manifestations of systemic conditions, such as RA chronic granulomatous disease and malignancy.8 Bronchogenic carcinoma was suspected in this patient due to her smoking history. Squamous cell carcinoma was also considered as the lesion was cavitary. AFB and GMS stains were negative for fungi. Langerhans cell histiocytosis were considered but ruled out as these lesions contain larger numbers of eosinophils than described in the pathology report. Histoplasma and coccidiosis laboratory tests were obtained as the patient lived in a region endemic to both these fungi but were negative (Table). A diagnosis of rheumatoid nodule was made based on the clinical setting, typical radiographic, histopathology features, and negative cultures.
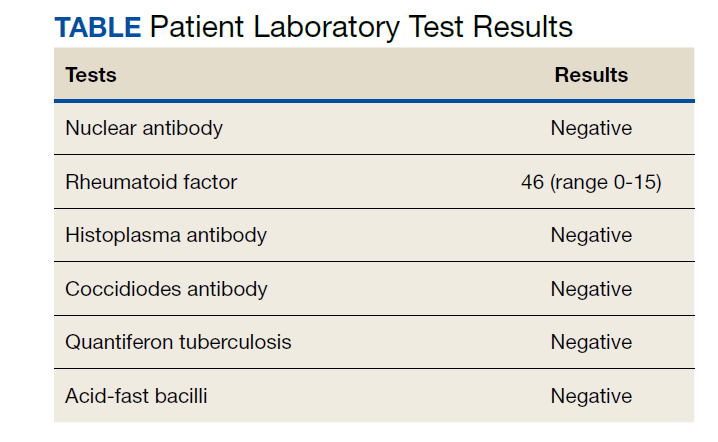
This case is unique due to the quality and location of the rheumatoid nodules within the lungs. Pulmonary manifestations of RA are usually subcutaneous or subpleural, solid, and peripherally located.9 This patient’s nodules were necrobiotic and located within the lung parenchyma. There was significant cavitation. These factors are atypical features of pulmonary RA.
Pulmonary RA can have many associated symptoms and remains an important factor in patient mortality. Estimates demonstrate that 10 to 20% of RA-related deaths are secondary to pulmonary manifestations.10 There are a wide array of symptoms and presentations to be aware of clinically. These symptoms are often nondescript, widely sensitive to many disease processes, and nonspecific to pulmonary RA. These symptoms include dyspnea, wheezing, and nonproductive cough.10 Bronchiectasis is a common symptom as well as small airway obstruction.10 Consolidated necrobiotic lesions are present in up to 20% of pulmonary RA cases.10 Generally these lesions are asymptomatic but can also be associated with pneumothorax, hemoptysis, and airway obstruction.10 Awareness of these symptoms is important for diagnosis and monitoring clinical improvement in patients.
Further workup is necessary to differentiate malignancy-related pulmonary nodules and other causes; if the index of suspicion is high for malignancy as in our case, the workup should be more aggressive. Biopsy is mandatory in such cases to rule out infections and malignancy, as it is highly sensitive and specific. The main problem hindering management is when a clinician fails to include this in their differential diagnosis. This further elucidates the importance of awareness of this diagnosis. Suspicious lesions in a proper clinical setting should be followed up by imaging studies and confirmatory histopathological diagnosis. Typical follow-up is 3 months after initial presentation to assess stability and possibly 18 to 24 months as well based on Fleischer guidelines.
Various treatment modalities have been tried as per literature, including tocilizumab and rituximab. 11,12 Our patient is currently being treated with golimumab based on outpatient rheumatologist recommendations.
Conclusions
This case demonstrates the importance of a careful workup to narrow a broad differential. Medical diagnosis of pulmonary nodules requires an in-depth workup, including clinical evaluation, laboratory and pulmonary functions tests, as well as various imaging studies.
1. Lung and Bronchus Cancer - Cancer Stat Facts. SEER. Accessed February 2, 2020. https://seer.cancer.gov /statfacts/html/lungb.html
2. Shaughnessy AF. One in Five Patients Overdiagnosed with Lung Cancer Screening. Am Fam Physician. 2014 Jul 15;90(2):112.
3. McWilliams A, Tammemagi MC, Mayo JR, et al. Probability of cancer in pulmonary nodules detected on first screening CT. N Engl J Med. 2013;369;910-919. doi:10.1056/NEJMoa1214726
4. Stamp LK, Cleland LG. Rheumatoid arthritis. In: Thompson LU, Ward WE, eds. Optimizing Women’s Health through Nutrition. CRC Press; 2008; 279-320.
5. Yousem SA, Colby TV, Carrington CB. Lung biopsy in rheumatoid arthritis. Am Rev Respir Dis. 1985;131(5):770-777. doi:10.1164/arrd.1985.131.5.770
6. Nyhäll-Wåhlin BM, Jacobsson LT, Petersson IF, Turesson C; BARFOT study group. Smoking is a strong risk factor for rheumatoid nodules in early rheumatoid arthritis. Ann Rheum Dis. 2006;65(5):601-606. doi:10.1136/ard.2005.039172
7. Shenberger KN, Schned AR, Taylor TH. Rheumatoid disease and bronchogenic carcinoma—case report and review of the literature. J Rheumatol. 1984;11:226–228.
8. Mukhopadhyay S, Wilcox BE, Myers JL, et al. Pulmonary necrotizing granulomas of unknown cause clinical and pathologic analysis of 131 patients with completely resected nodules. Chest. 2013;144(3):813-824. doi:10.1378/chest.12-2113
9. Ohshimo S, Guzman J, Costabel U, Bonella F. Differential diagnosis of granulomatous lung disease: clues and pitfalls: Number 4 in the Series “Pathology for the clinician.” Edited by Peter Dorfmüller and Alberto Cavazza. Eur Respir Rev. 2017;26(145):170012. Published 2017 Aug 9. doi:10.1183/16000617.0012-2017
10. Brown KK. Rheumatoid lung disease. Proc Am Thorac Soc. 2007;4(5):443-448. doi:10.1513/pats.200703-045MS
11. Braun MG, Wagener P. Regression von peripheren und pulmonalen Rheumaknoten unter Rituximab-Therapie [Regression of peripheral and pulmonary rheumatoid nodules under therapy with rituximab]. Z Rheumatol. 2013;72(2):166-171. doi:10.1007/s00393-012-1054-0
12. Andres M, Vela P, Romera C. Marked improvement of lung rheumatoid nodules after treatment with tocilizumab. Rheumatology (Oxford). 2012;51(6):1132-1134. doi:10.1093/rheumatology/ker455
Identification of pulmonary nodules in older adults who smoke immediately brings concern for malignancy in the mind of clinicians. This is particularly the case in patients with significant smoking history. According to the National Cancer Institute in 2019, 12.9% of all new cancer cases were lung cancers.1 Screening for lung cancer, especially in patients with increased risk from smoking, is imperative to early detection and treatment. However, 20% of patients will be overdiagnosed by lung cancer-screening techniques.2 The rate of malignancy noted on a patient’s first screening computed tomography (CT) scan was between 3.7% and 5.5%.3
Rheumatoid arthritis (RA) is an autoimmune inflammatory condition that mainly affects the joints. Extraarticular manifestations can arise in various locations throughout the body, however. These manifestations are commonly observed in the skin, heart, and lungs.4 Prevalence of pulmonary rheumatoid nodules ranges from < 0.4% in radiologic studies to 32% in lung biopsies of patients with RA and nodules.5
Furthermore, there is a strong association between the risk of rheumatoid nodules in patients with positive serum rheumatoid factor (RF) and smoking history.6 Solitary pulmonary nodules in patients with RA can coexist with bronchogenic carcinoma, making their diagnosis more important.7
Case Presentation
A 54-year-old woman with a 30 pack-year smoking history and history of RA initially presented to the emergency department for cough and dyspnea for 5-day duration. Her initial diagnosis was bronchitis based on presenting symptom profile. A chest CT demonstrated 3 cavitary pulmonary nodules, 1 measuring 2.4 x 2.0 cm in the right middle lobe, and 2 additional nodules, measuring 1.8 x 1.4 and 1.5 x 1.4 in the left upper lobe (Figure). She had no improvement of symptoms after a 7-day course of doxycycline. The patient was taking methotrexate 15 mg weekly and golimumab 50 mg subcutaneously every 4 weeks as treatment for RA, prescribed by her rheumatologist.
Pulmonology was consulted and a positron emission tomography-CT (PET-CT) confirmed several cavitary pulmonary nodules involving both lungs with no suspicious fluorodeoxyglucose (FDG) uptake. The largest lesion was in the right middle lobe with FDG uptake of 1.9. Additional nodules were found in the left upper lobe, measuring 1.8 x 1.4 cm with FDG of 4.01, and in the left lung apex, measuring 1.5 x 1.4 cm with uptake of 3.53. CTguided percutaneous fine needle aspiration (PFNA) of the right middle lobe lung nodule demonstrated granuloma with central inflammatory debris. Grocott methenamine silver (GMS) stain was negative for fungal organism, acid-fast bacteria (AFB) stain was negative for acid-fast bacilli, and CD20 and CD3 immunostaining demonstrated mixed B- and T-cell populations. There was no evidence of atypia or malignancy. The biopsy demonstrated granuloma with central inflammatory debris on a background of densely fibrotic tissue and lympho-plasmatic inflammation. This finding confirmed the diagnosis of RA with pulmonary involvement.
Outpatient follow-up was established with a pulmonologist and rheumatologist. Methotrexate 15 mg weekly and golimumab subcutaneously 50 mg every 4 weeks were prescribed for the patient. The nodules are being monitored based on Fleischer guidelines with CT imaging 3 to 6 months following initial presentation. Further imaging will be considered at 18 to 24 months as well to further assess stability of the nodules and monitor for changes in size, shape, and necrosis. The patient also was encouraged to quit smoking. Her clinical course since the diagnosis has been stable.
Discussion
The differential diagnosis for new multiple pulmonary nodules on imaging studies is broad and includes infectious processes, such as tuberculosis, as well as other mycobacterial, fungal, and bacterial infections. Noninfectious causes of lung disease are an even broader category of consideration. Noninfectious pulmonary nodules differential includes sarcoidosis, granulomatous with polyangiitis, hypersensitivity pneumonitis, methotrexate drug reaction, pulmonary manifestations of systemic conditions, such as RA chronic granulomatous disease and malignancy.8 Bronchogenic carcinoma was suspected in this patient due to her smoking history. Squamous cell carcinoma was also considered as the lesion was cavitary. AFB and GMS stains were negative for fungi. Langerhans cell histiocytosis were considered but ruled out as these lesions contain larger numbers of eosinophils than described in the pathology report. Histoplasma and coccidiosis laboratory tests were obtained as the patient lived in a region endemic to both these fungi but were negative (Table). A diagnosis of rheumatoid nodule was made based on the clinical setting, typical radiographic, histopathology features, and negative cultures.
This case is unique due to the quality and location of the rheumatoid nodules within the lungs. Pulmonary manifestations of RA are usually subcutaneous or subpleural, solid, and peripherally located.9 This patient’s nodules were necrobiotic and located within the lung parenchyma. There was significant cavitation. These factors are atypical features of pulmonary RA.
Pulmonary RA can have many associated symptoms and remains an important factor in patient mortality. Estimates demonstrate that 10 to 20% of RA-related deaths are secondary to pulmonary manifestations.10 There are a wide array of symptoms and presentations to be aware of clinically. These symptoms are often nondescript, widely sensitive to many disease processes, and nonspecific to pulmonary RA. These symptoms include dyspnea, wheezing, and nonproductive cough.10 Bronchiectasis is a common symptom as well as small airway obstruction.10 Consolidated necrobiotic lesions are present in up to 20% of pulmonary RA cases.10 Generally these lesions are asymptomatic but can also be associated with pneumothorax, hemoptysis, and airway obstruction.10 Awareness of these symptoms is important for diagnosis and monitoring clinical improvement in patients.
Further workup is necessary to differentiate malignancy-related pulmonary nodules and other causes; if the index of suspicion is high for malignancy as in our case, the workup should be more aggressive. Biopsy is mandatory in such cases to rule out infections and malignancy, as it is highly sensitive and specific. The main problem hindering management is when a clinician fails to include this in their differential diagnosis. This further elucidates the importance of awareness of this diagnosis. Suspicious lesions in a proper clinical setting should be followed up by imaging studies and confirmatory histopathological diagnosis. Typical follow-up is 3 months after initial presentation to assess stability and possibly 18 to 24 months as well based on Fleischer guidelines.
Various treatment modalities have been tried as per literature, including tocilizumab and rituximab. 11,12 Our patient is currently being treated with golimumab based on outpatient rheumatologist recommendations.
Conclusions
This case demonstrates the importance of a careful workup to narrow a broad differential. Medical diagnosis of pulmonary nodules requires an in-depth workup, including clinical evaluation, laboratory and pulmonary functions tests, as well as various imaging studies.
Identification of pulmonary nodules in older adults who smoke immediately brings concern for malignancy in the mind of clinicians. This is particularly the case in patients with significant smoking history. According to the National Cancer Institute in 2019, 12.9% of all new cancer cases were lung cancers.1 Screening for lung cancer, especially in patients with increased risk from smoking, is imperative to early detection and treatment. However, 20% of patients will be overdiagnosed by lung cancer-screening techniques.2 The rate of malignancy noted on a patient’s first screening computed tomography (CT) scan was between 3.7% and 5.5%.3
Rheumatoid arthritis (RA) is an autoimmune inflammatory condition that mainly affects the joints. Extraarticular manifestations can arise in various locations throughout the body, however. These manifestations are commonly observed in the skin, heart, and lungs.4 Prevalence of pulmonary rheumatoid nodules ranges from < 0.4% in radiologic studies to 32% in lung biopsies of patients with RA and nodules.5
Furthermore, there is a strong association between the risk of rheumatoid nodules in patients with positive serum rheumatoid factor (RF) and smoking history.6 Solitary pulmonary nodules in patients with RA can coexist with bronchogenic carcinoma, making their diagnosis more important.7
Case Presentation
A 54-year-old woman with a 30 pack-year smoking history and history of RA initially presented to the emergency department for cough and dyspnea for 5-day duration. Her initial diagnosis was bronchitis based on presenting symptom profile. A chest CT demonstrated 3 cavitary pulmonary nodules, 1 measuring 2.4 x 2.0 cm in the right middle lobe, and 2 additional nodules, measuring 1.8 x 1.4 and 1.5 x 1.4 in the left upper lobe (Figure). She had no improvement of symptoms after a 7-day course of doxycycline. The patient was taking methotrexate 15 mg weekly and golimumab 50 mg subcutaneously every 4 weeks as treatment for RA, prescribed by her rheumatologist.
Pulmonology was consulted and a positron emission tomography-CT (PET-CT) confirmed several cavitary pulmonary nodules involving both lungs with no suspicious fluorodeoxyglucose (FDG) uptake. The largest lesion was in the right middle lobe with FDG uptake of 1.9. Additional nodules were found in the left upper lobe, measuring 1.8 x 1.4 cm with FDG of 4.01, and in the left lung apex, measuring 1.5 x 1.4 cm with uptake of 3.53. CTguided percutaneous fine needle aspiration (PFNA) of the right middle lobe lung nodule demonstrated granuloma with central inflammatory debris. Grocott methenamine silver (GMS) stain was negative for fungal organism, acid-fast bacteria (AFB) stain was negative for acid-fast bacilli, and CD20 and CD3 immunostaining demonstrated mixed B- and T-cell populations. There was no evidence of atypia or malignancy. The biopsy demonstrated granuloma with central inflammatory debris on a background of densely fibrotic tissue and lympho-plasmatic inflammation. This finding confirmed the diagnosis of RA with pulmonary involvement.
Outpatient follow-up was established with a pulmonologist and rheumatologist. Methotrexate 15 mg weekly and golimumab subcutaneously 50 mg every 4 weeks were prescribed for the patient. The nodules are being monitored based on Fleischer guidelines with CT imaging 3 to 6 months following initial presentation. Further imaging will be considered at 18 to 24 months as well to further assess stability of the nodules and monitor for changes in size, shape, and necrosis. The patient also was encouraged to quit smoking. Her clinical course since the diagnosis has been stable.
Discussion
The differential diagnosis for new multiple pulmonary nodules on imaging studies is broad and includes infectious processes, such as tuberculosis, as well as other mycobacterial, fungal, and bacterial infections. Noninfectious causes of lung disease are an even broader category of consideration. Noninfectious pulmonary nodules differential includes sarcoidosis, granulomatous with polyangiitis, hypersensitivity pneumonitis, methotrexate drug reaction, pulmonary manifestations of systemic conditions, such as RA chronic granulomatous disease and malignancy.8 Bronchogenic carcinoma was suspected in this patient due to her smoking history. Squamous cell carcinoma was also considered as the lesion was cavitary. AFB and GMS stains were negative for fungi. Langerhans cell histiocytosis were considered but ruled out as these lesions contain larger numbers of eosinophils than described in the pathology report. Histoplasma and coccidiosis laboratory tests were obtained as the patient lived in a region endemic to both these fungi but were negative (Table). A diagnosis of rheumatoid nodule was made based on the clinical setting, typical radiographic, histopathology features, and negative cultures.
This case is unique due to the quality and location of the rheumatoid nodules within the lungs. Pulmonary manifestations of RA are usually subcutaneous or subpleural, solid, and peripherally located.9 This patient’s nodules were necrobiotic and located within the lung parenchyma. There was significant cavitation. These factors are atypical features of pulmonary RA.
Pulmonary RA can have many associated symptoms and remains an important factor in patient mortality. Estimates demonstrate that 10 to 20% of RA-related deaths are secondary to pulmonary manifestations.10 There are a wide array of symptoms and presentations to be aware of clinically. These symptoms are often nondescript, widely sensitive to many disease processes, and nonspecific to pulmonary RA. These symptoms include dyspnea, wheezing, and nonproductive cough.10 Bronchiectasis is a common symptom as well as small airway obstruction.10 Consolidated necrobiotic lesions are present in up to 20% of pulmonary RA cases.10 Generally these lesions are asymptomatic but can also be associated with pneumothorax, hemoptysis, and airway obstruction.10 Awareness of these symptoms is important for diagnosis and monitoring clinical improvement in patients.
Further workup is necessary to differentiate malignancy-related pulmonary nodules and other causes; if the index of suspicion is high for malignancy as in our case, the workup should be more aggressive. Biopsy is mandatory in such cases to rule out infections and malignancy, as it is highly sensitive and specific. The main problem hindering management is when a clinician fails to include this in their differential diagnosis. This further elucidates the importance of awareness of this diagnosis. Suspicious lesions in a proper clinical setting should be followed up by imaging studies and confirmatory histopathological diagnosis. Typical follow-up is 3 months after initial presentation to assess stability and possibly 18 to 24 months as well based on Fleischer guidelines.
Various treatment modalities have been tried as per literature, including tocilizumab and rituximab. 11,12 Our patient is currently being treated with golimumab based on outpatient rheumatologist recommendations.
Conclusions
This case demonstrates the importance of a careful workup to narrow a broad differential. Medical diagnosis of pulmonary nodules requires an in-depth workup, including clinical evaluation, laboratory and pulmonary functions tests, as well as various imaging studies.
1. Lung and Bronchus Cancer - Cancer Stat Facts. SEER. Accessed February 2, 2020. https://seer.cancer.gov /statfacts/html/lungb.html
2. Shaughnessy AF. One in Five Patients Overdiagnosed with Lung Cancer Screening. Am Fam Physician. 2014 Jul 15;90(2):112.
3. McWilliams A, Tammemagi MC, Mayo JR, et al. Probability of cancer in pulmonary nodules detected on first screening CT. N Engl J Med. 2013;369;910-919. doi:10.1056/NEJMoa1214726
4. Stamp LK, Cleland LG. Rheumatoid arthritis. In: Thompson LU, Ward WE, eds. Optimizing Women’s Health through Nutrition. CRC Press; 2008; 279-320.
5. Yousem SA, Colby TV, Carrington CB. Lung biopsy in rheumatoid arthritis. Am Rev Respir Dis. 1985;131(5):770-777. doi:10.1164/arrd.1985.131.5.770
6. Nyhäll-Wåhlin BM, Jacobsson LT, Petersson IF, Turesson C; BARFOT study group. Smoking is a strong risk factor for rheumatoid nodules in early rheumatoid arthritis. Ann Rheum Dis. 2006;65(5):601-606. doi:10.1136/ard.2005.039172
7. Shenberger KN, Schned AR, Taylor TH. Rheumatoid disease and bronchogenic carcinoma—case report and review of the literature. J Rheumatol. 1984;11:226–228.
8. Mukhopadhyay S, Wilcox BE, Myers JL, et al. Pulmonary necrotizing granulomas of unknown cause clinical and pathologic analysis of 131 patients with completely resected nodules. Chest. 2013;144(3):813-824. doi:10.1378/chest.12-2113
9. Ohshimo S, Guzman J, Costabel U, Bonella F. Differential diagnosis of granulomatous lung disease: clues and pitfalls: Number 4 in the Series “Pathology for the clinician.” Edited by Peter Dorfmüller and Alberto Cavazza. Eur Respir Rev. 2017;26(145):170012. Published 2017 Aug 9. doi:10.1183/16000617.0012-2017
10. Brown KK. Rheumatoid lung disease. Proc Am Thorac Soc. 2007;4(5):443-448. doi:10.1513/pats.200703-045MS
11. Braun MG, Wagener P. Regression von peripheren und pulmonalen Rheumaknoten unter Rituximab-Therapie [Regression of peripheral and pulmonary rheumatoid nodules under therapy with rituximab]. Z Rheumatol. 2013;72(2):166-171. doi:10.1007/s00393-012-1054-0
12. Andres M, Vela P, Romera C. Marked improvement of lung rheumatoid nodules after treatment with tocilizumab. Rheumatology (Oxford). 2012;51(6):1132-1134. doi:10.1093/rheumatology/ker455
1. Lung and Bronchus Cancer - Cancer Stat Facts. SEER. Accessed February 2, 2020. https://seer.cancer.gov /statfacts/html/lungb.html
2. Shaughnessy AF. One in Five Patients Overdiagnosed with Lung Cancer Screening. Am Fam Physician. 2014 Jul 15;90(2):112.
3. McWilliams A, Tammemagi MC, Mayo JR, et al. Probability of cancer in pulmonary nodules detected on first screening CT. N Engl J Med. 2013;369;910-919. doi:10.1056/NEJMoa1214726
4. Stamp LK, Cleland LG. Rheumatoid arthritis. In: Thompson LU, Ward WE, eds. Optimizing Women’s Health through Nutrition. CRC Press; 2008; 279-320.
5. Yousem SA, Colby TV, Carrington CB. Lung biopsy in rheumatoid arthritis. Am Rev Respir Dis. 1985;131(5):770-777. doi:10.1164/arrd.1985.131.5.770
6. Nyhäll-Wåhlin BM, Jacobsson LT, Petersson IF, Turesson C; BARFOT study group. Smoking is a strong risk factor for rheumatoid nodules in early rheumatoid arthritis. Ann Rheum Dis. 2006;65(5):601-606. doi:10.1136/ard.2005.039172
7. Shenberger KN, Schned AR, Taylor TH. Rheumatoid disease and bronchogenic carcinoma—case report and review of the literature. J Rheumatol. 1984;11:226–228.
8. Mukhopadhyay S, Wilcox BE, Myers JL, et al. Pulmonary necrotizing granulomas of unknown cause clinical and pathologic analysis of 131 patients with completely resected nodules. Chest. 2013;144(3):813-824. doi:10.1378/chest.12-2113
9. Ohshimo S, Guzman J, Costabel U, Bonella F. Differential diagnosis of granulomatous lung disease: clues and pitfalls: Number 4 in the Series “Pathology for the clinician.” Edited by Peter Dorfmüller and Alberto Cavazza. Eur Respir Rev. 2017;26(145):170012. Published 2017 Aug 9. doi:10.1183/16000617.0012-2017
10. Brown KK. Rheumatoid lung disease. Proc Am Thorac Soc. 2007;4(5):443-448. doi:10.1513/pats.200703-045MS
11. Braun MG, Wagener P. Regression von peripheren und pulmonalen Rheumaknoten unter Rituximab-Therapie [Regression of peripheral and pulmonary rheumatoid nodules under therapy with rituximab]. Z Rheumatol. 2013;72(2):166-171. doi:10.1007/s00393-012-1054-0
12. Andres M, Vela P, Romera C. Marked improvement of lung rheumatoid nodules after treatment with tocilizumab. Rheumatology (Oxford). 2012;51(6):1132-1134. doi:10.1093/rheumatology/ker455
Pheochromocytoma: An Incidental Finding in an Asymptomatic Older Adult With Renal Oncocytoma
A high index of suspicion for pheochromocytoma is necessary during the workup of secondary hypertension as untreated pheochromocytoma may lead to significant morbidity and mortality, especially in patients who require any surgical treatment.
Pheochromocytoma is a rare catecholamine-secreting tumor of chromaffin cells of the adrenal medulla or sympathetic ganglia, occurring in about 0.2 to 0.5% of patients with hypertension.1-3 However, in a review of 54 autopsy-proven cases of pheochromocytoma, about 50% of the patients with hypertension were not clinically suspected for pheochromocytoma.4
Pheochromocytoma is usually diagnosed based on symptoms of hyperadrenergic spells, resistant hypertension, especially in the young, with a pressor response to the anesthesia stress test and adrenal incidentaloma.
The classic triad of symptoms associated with pheochromocytoma includes episodic headache (90%), sweating (60-70%), and palpitations (70%).2,5 Sustained or paroxysmal hypertension is the most common symptom reported in about 95% of patients with pheochromocytoma. Other symptoms include pallor, tremors, dyspnea, generalized weakness, orthostatic hypotension, cardiomyopathy, or hyperglycemia.6 However, about 10% of patients with pheochromocytoma are asymptomatic or mildly symptomatic.7 Secondary causes of hypertension are usually suspected in multidrug resistant or sudden early onset of hypertension.8
Approximately 10% of catecholamine-secreting tumors are malignant.9-11 Benign and malignant pheochromocytoma have a similar biochemical and histologic presentation and are differentiated based on local invasion into the surrounding tissues and organs (eg, kidney, liver) or distant metastasis.
A typical workup of a suspected patient with pheochromocytoma includes biochemical tests, including measurements of urinary and fractionated plasma metanephrines and catecholamine. Patients with positive biochemical tests should undergo localization of the tumor with an imaging study either with an adrenal/abdominal magnetic resonance imaging (MRI) or computed tomography (CT) scan. If a patient has paraganglioma or an adrenal mass > 10 cm or negative abdominal imaging with a positive biochemical test, further imaging with an iobenguane I-123 scan is needed (Figure 1).

In this article, we present an unusual case of asymptomatic pheochromocytoma in a patient with right-sided renal oncocytoma who underwent an uneventful nephrectomy and adrenalectomy.
Case Presentation
A 72-year-old male with a medical history of diabetes, hypertension, sensory neuropathy, benign prostatic hypertrophy (BPH) status posttransurethral resection of the prostate, and chronic renal failure presented to establish care with the Arizona Kidney Disease and Hypertension Center. His medications included losartan 50 mg by mouth daily, diltiazem 180 mg extended-release by mouth daily, carvedilol 6.25 mg by mouth twice a day, and tamsulosin 0.4 mg by mouth daily. His presenting vitals were blood pressure (BP), 112/74 left arm sitting, pulse, 63/beats per min, and body mass index, 34. On physical examination, the patient was alert and oriented, and the chest was clear to auscultation without wheeze or rhonchi. On cardiac examination, heart rate and rhythm were regular; S1 and S2 were normal with no added murmurs, rubs or gallops, and no jugular venous distension. The abdomen was soft, nontender, with no palpable mass. His laboratory results showed sodium, 142 mmol/L; potassium, 5.3 mmol/L; chloride, 101 mmol/L; carbon dioxide, 24 mmol/L; albumin, 4.3 g/dL; creatinine, 1.89 mg/dL; blood urea nitrogen, 29 mg/dL; estimated glomerular filtration rate non-African American, 35 mL/min/1.73; 24-h urine creatinine clearance, 105 mL/min; protein, 1306 mg/24 h (Table).
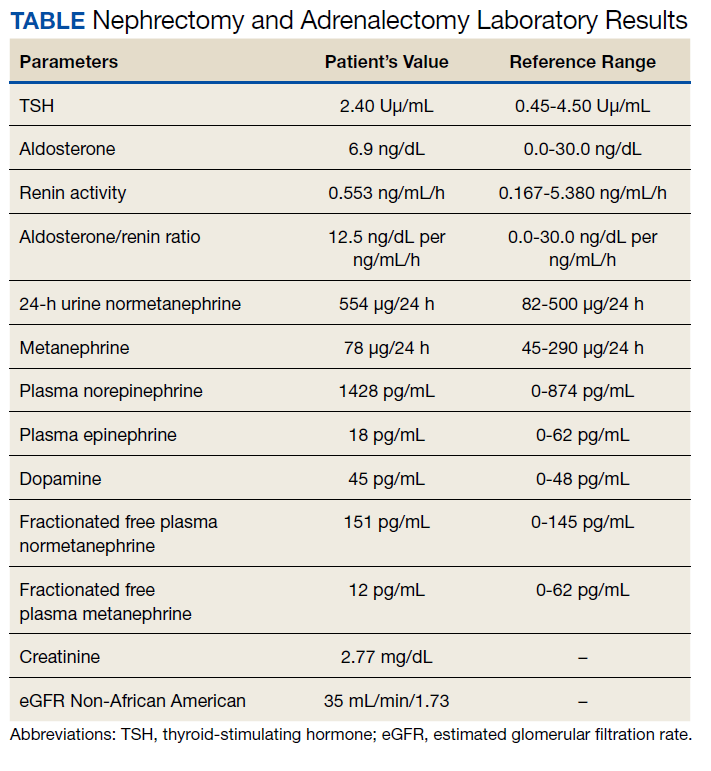
His renal ultrasound showed an exophytic isoechoic mass or complex cyst at the lateral aspect of the lower pole of the right kidney, measuring 45 mm in diameter. An MRI of the abdomen with and without contrast showed a solid partially exophytic mass of the posterolateral interpolar cortex of the right kidney, measuring 5.9 cm in the greatest dimension (Figure 2). No definite involvement of Gerota fascia was noted, a 1-cm metastasis to the right adrenal gland was present, renal veins were patent, and there was no upper retroperitoneal lymphadenopathy.
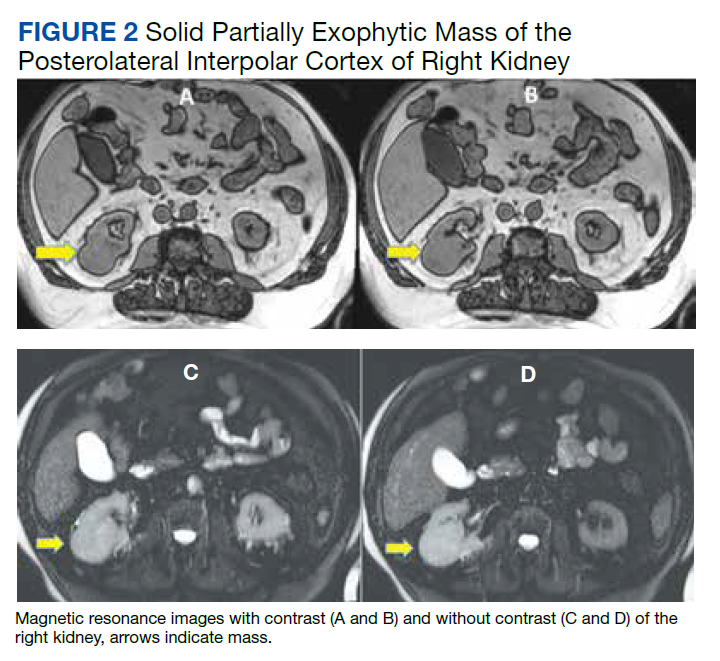
Treatment and Follow-up
The patient underwent right-hand-assisted lap-aroscopic radical nephrectomy and right adre-nalectomy without any complications. However, the surgical pathology report showed oncocytoma of the kidney (5.7 cm), pheochromocytoma of the adrenal gland (1.4 cm), and papillary adenoma of the kidney (0.7 cm). Right kidney nephrectomy showed non-neoplastic renal parenchyma, diabetic glomerulosclerosis (Renal Pathology Society 2010 diabetic nephropathy class IIb), severe mesangial expansion, moderate interstitial fibrosis, moderate arteriosclerosis, and mild arteriolosclerosis.
A fluorodeoxyglucose-positron emission tomography (FDG-PET) scan was significant for right nephrectomy and adrenalectomy and showed no significant evidence of residual neoplasm or local or distant metastases. A nuclear medicine (iobenguane I-123) tumor and single positron emission computed tomography (SPECT) scan showed normal activity throughout the body and no evidence of abnormal activity (Figure 3).
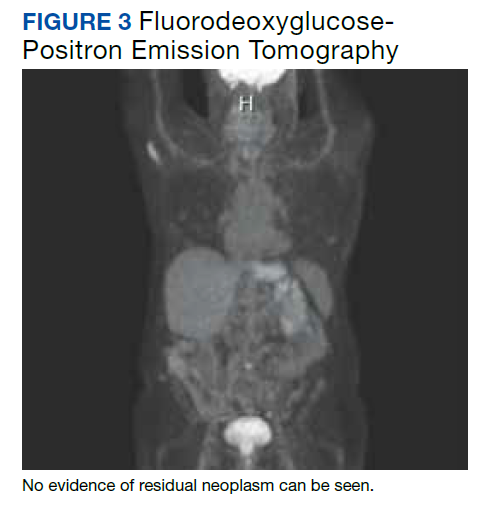
Discussion
Pheochromocytoma is a rare cause of secondary hypertension. However, the real numbers are thought to be > 0.2 to 0.5%.1,2,4 Patients with pheochromocytoma should undergo surgical adrenal resection after appropriate medical preparation. Patients with pheochromocytoma who are not diagnosed preoperatively have increased surgical mortality rates due to fatal hypertensive crises, malignant arrhythmia, and multiorgan failure as a result of hypertensive crisis.15 Anesthetic drugs during surgery also can exacerbate the cardiotoxic effects of catecholamines. Short-acting anesthetic agents, such as fentanyl, are used in patients with pheochromocytoma.16
This case of pheochromocytoma illustrated no classic symptoms of episodic headache, sweating, and tachycardia, and the patient was otherwise asymptomatic. BP was well controlled with losartan, diltiazem, and a β-blocker with α-blocking activity (carvedilol). As the patient was not known to have pheochromocytoma, he did not undergo preoperative medical therapy. Figure 4 illustrates the receptors stimulate catecholamines, and the drugs blocking these receptors prevent hypertensive crisis during surgery. However, the surgery was without potential complications (ie, hypertensive crisis, malignant arrhythmia, or multiorgan failure). The patient was diagnosed incidentally on histopathology after right radical nephrectomy and adrenalectomy due to solid partially exophytic right renal mass (5.9 cm) with right adrenal metastasis. About 10% of patients are asymptomatic or mildly symptomatic.7 Sometimes, the symptoms may be ignored because of the episodic nature. Other possible reasons can be small, nonfunctional tumors or the use of antihypertensive medications suppressing the symptoms.7
The adrenal mass that was initially thought to be a metastasis of right kidney mass was later confirmed as pheochromocytoma. One possible explanation for uneventful surgery could be the use of β-blocker with α-blocking activity (carvedilol), α-1 adrenergic blocker (tamsulosin) along with nondihydropyridine calcium channel blocker (diltiazem) as part of the patient’s antihypertensive and BPH medication regimen. Another possible explanation could be silent or episodically secreting pheochromocytoma with a small functional portion.
Subsequent workup after adrenalectomy, including urinary and fractionated plasma metanephrines and catecholamines, were not consistent with catecholamine hypersecretion. A 24-hour urine fractionated metanephrines test has about 98% sensitivity and 98% specificity. Elevated plasma norepinephrine was thought to be due to renal failure because it was < 3-fold the upper limit of normal, which is considered to be a possible indication of pheochromocytoma.17,18 The nuclear medicine (iobenguane I-123) tumor, SPECT, and FDG-PET CT studies were negative for residual pheochromocytoma. Other imaging studies to consider in patients with suspected catecholamine-secreting tumor with positive biochemical test and negative abdominal imaging are a whole-body MRI scan, 68-Ga DOTATATE (gallium 68 1,4,7,10-tetraazacyclododecane-1,4,7,10 tetraacetic acid-octreotate) or FDG-PET scan.19
In a review of 54 autopsy-proven pheochromocytoma cases by Sutton and colleagues in 1981, 74% of the patients were not clinically suspected for pheochromocytoma in their life.4 Similarly, in a retrospective study of hospital autopsies by McNeil and colleagues, one incidental pheochromocytoma was detected in every 2031 autopsies (0.05%).20 In another case series of 41 patients with pheochromocytoma-related adrenalectomy, almost 50% of the pheochromocytomas were detected incidentally on imaging studies.21 Although the number of incidental findings are decreasing due to advances in screening techniques, a significant number of patients remain undiagnosed. Multiple cases of diagnosis of pheochromocytoma on autopsy of patients who died of hemodynamic instability (ie, hypertensive crisis, hypotension crisis precipitated by surgery for adrenal or nonadrenal conditions) are reported.3 To the best of our knowledge, there are no case reports published on the diagnosis of pheochromocytoma after adrenalectomy in an asymptomatic patient without intraoperative complications.
The goal of preoperative medical therapy includes BP control, prevention of tachycardia, and volume expansion. The preoperative medications regimens are combined α- and β-adrenergic blockade, calcium channel blockers, and metyrosine. According to clinical practice guidelines of the Endocrine Society in 2014, the α-adrenergic blockers should be started first at least 7 days before surgery to control BP and to cause vasodilation. Early use of α-blockers is required to prevent cardiotoxicity. The β-adrenergic blockers should be started after the adequate α-adrenergic blockade, typically 2 to 3 days before surgery, as early use can cause vasoconstriction in patients with pheochromocytoma. The α-adrenergic blockers include phenoxybenzamine (nonselective long-acting nonspecific α-adrenergic blocking agent), and selective α-1 adrenergic blockers (doxazosin, prazosin, terazosin). The β-adrenergic blocker (ie, propranolol, metoprolol) should be started cautiously with a low dose and slowly titrated to control heart rate. A high sodium diet and increased fluid intake also are recommended 7 to 14 days before surgery. A sudden drop in catecholamines can cause hypotension during an operation. Continuous fluid infusions are given to prevent hypotension.22 Similarly, anesthetic agents also should be modified to prevent cardiotoxic effects. Rocuronium and vecuronium are less cardiotoxic compared with other sympathomimetic muscle relaxants. Short-acting anesthetic agents, such as fentanyl, are preferred. α-blockers are continued throughout the operation. Biochemical testing with fractionated metanephrines is performed about 1 to 2 weeks postoperatively to look for recurrence of the disease.23
Secondary causes of hypertension are suspected in multidrug resistant or sudden early onset of hypertension before aged 40 years. Pheochromocytoma is a rare cause of secondary hypertension, and older adult patients are rarely diagnosed with pheochromocytoma.24 In this report, pheochromocytoma was detected in a 72-year-old hypertensive patient. Therefore, a pheochromocytoma diagnosis should not be ignored in the older adult patient with adrenal mass and hypertension treated with more than one drug. The authors recommend any patient undergoing surgery with adrenal lesion should be considered for the screening of possible pheochromocytoma and prepared preoperatively, especially any patient with renal cell carcinoma with adrenal metastasis.
Conclusions
Asymptomatic pheochromocytoma is an unusual but serious condition, especially for patients undergoing a surgical procedure. An adrenal mass may be ignored in asymptomatic or mildly symptomatic older adult patients and is mostly considered as adrenal metastasis when present with other malignancies. Fortunately, the nephrectomy and adrenalectomy in our case of asymptomatic pheochromocytoma was uneventful, but pheochromocytoma should be ruled out before a surgical procedure, as an absence of medical pretreatment can lead to serious consequences. Therefore, we suggest a more careful screening of pheochromocytoma in patients with an adrenal mass (primary or metastatic) and hypertension treated with multiple antihypertensive drugs, even in older adult patients.
1. Omura M, Saito J, Yamaguchi K, Kakuta Y, Nishikawa T. Prospective study on the prevalence of secondary hypertension among hypertensive patients visiting a general outpatient clinic in Japan. Hypertens Res. 2004;27(3):193-202. doi:10.1291/hypres.27.193
2. Stein PP, Black HR. A simplified diagnostic approach to pheochromocytoma: a review of the literature and report of one institution’s experience. Medicine (Baltimore). 1991;70(1):46-66. doi:10.1097/00005792-199101000-00004
3. Beard CM, Sheps SG, Kurland LT, Carney JA, Lie JT. Occurrence of pheochromocytoma in Rochester, Minnesota, 1950 through 1979. Mayo Clin Proc. 1983;58(12):802-804.
4. Sutton MG, Sheps SG, Lie JT. Prevalence of clinically unsuspected pheochromocytoma: review of a 50-year autopsy series. Mayo Clin Proc. 1981;56(6):354-360.
5. Manger WM, Gifford RW Jr. Pheochromocytoma. J Clin Hypertens (Greenwich). 2002;4(1):62-72. doi:10.1111/j.1524-6175.2002.01452.x
6. Kassim TA, Clarke DD, Mai VQ, Clyde PW, Mohamed Shakir KM. Catecholamine-induced cardiomyopathy. Endocr Pract. 2008;14(9):1137-1149. doi:10.4158/EP.14.9.1137
7. Kudva YC, Young WF, Thompson GB, Grant CS, Van Heerden JA. Adrenal incidentaloma: an important component of the clinical presentation spectrum of benign sporadic adrenal pheochromocytoma. The Endocrinologist. 1999;9(2):77-80. doi:10.1097/00019616-199903000-00002
8. Puar TH, Mok Y, Debajyoti R, Khoo J, How CH, Ng AK. Secondary hypertension in adults. Singapore Med J. 2016;57(5):228-232. doi:10.11622/smedj.2016087
9. Bravo EL. Pheochromocytoma: new concepts and future trends. Kidney Int. 1991;40(3):544-556. doi:10.1038/ki.1991.244
10. Plouin PF, Chatellier G, Fofol I, Corvol P. Tumor recurrence and hypertension persistence after successful pheochromocytoma operation. Hypertension. 1997;29(5):1133-1139. doi:10.1161/01.hyp.29.5.1133
11. Hamidi O, Young WF Jr, Iñiguez-Ariza NM, et al. Malignant pheochromocytoma and paraganglioma: 272 patients over 55 years. J Clin Endocrinol Metab. 2017;102(9):3296-3305. doi:10.1210/jc.2017-00992
12. Kenny L, Rizzo V, Trevis J, Assimakopoulou E, Timon D. The unexpected diagnosis of phaeochromocytoma in the anaesthetic room. Ann Card Anaesth. 2018;21(3):307-310. doi:10.4103/aca.ACA_206_17
13. Johnston PC, Silversides JA, Wallace H, et al. Phaeochromocytoma crisis: two cases of undiagnosed phaeochromocytoma presenting after elective nonrelated surgical procedures. Case Rep Anesthesiol. 2013;2013:514714. doi:10.1155/2013/514714
14. Shen SJ, Cheng HM, Chiu AW, Chou CW, Chen JY. Perioperative hypertensive crisis in clinically silent pheochromocytomas: report of four cases. Chang Gung Med J. 2005;28(1):44-50.
15. Lo CY, Lam KY, Wat MS, Lam KS. Adrenal pheochromocytoma remains a frequently overlooked diagnosis. Am J Surg. 2000;179(3):212-215. doi:10.1016/s0002-9610(00)00296-8
16. Myklejord DJ. Undiagnosed pheochromocytoma: the anesthesiologist nightmare. Clin Med Res. 2004;2(1):59-62. doi:10.3121/cmr.2.1.59
17. Stumvoll M, Radjaipour M, Seif F. Diagnostic considerations in pheochromocytoma and chronic hemodialysis: case report and review of the literature. Am J Nephrol. 1995;15(2):147-151. doi:10.1159/000168820
18. Morioka M, Yuihama S, Nakajima T, et al. Incidentally discovered pheochromocytoma in long-term hemodialysis patients. Int J Urol. 2002;9(12):700-703. doi:10.1046/j.1442-2042.2002.00553.x
19. ˇCtvrtlík F, Koranda P, Schovánek J, Škarda J, Hartmann I, Tüdös Z. Current diagnostic imaging of pheochromocytomas and implications for therapeutic strategy. Exp Ther Med. 2018;15(4):3151-3160. doi:10.3892/etm.2018.5871
20. McNeil AR, Blok BH, Koelmeyer TD, Burke MP, Hilton JM. Phaeochromocytomas discovered during coronial autopsies in Sydney, Melbourne and Auckland. Aust N Z J Med. 2000;30(6):648-652. doi:10.1111/j.1445-5994.2000.tb04358.x
21. Baguet JP, Hammer L, Mazzuco TL, et al. Circumstances of discovery of phaeochromocytoma: a retrospective study of 41 consecutive patients. Eur J Endocrinol. 2004;150(5):681-686. doi:10.1530/eje.0.1500681
22. Lenders JW, Duh QY, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014;99(6):1915-1942. doi:10.1210/jc.2014-1498
23. Dortzbach K, Gainsburg DM, Frost EA. Variants of pheochromocytoma and their anesthetic implications--a case report and literature review. Middle East J Anaesthesiol. 2010;20(6):897-905.
24. Januszewicz W, Chodakowska J, Styczy´nski G. Secondary hypertension in the elderly. J Hum Hypertens. 1998;12(9):603-606. doi:10.1038/sj.jhh.1000673
A high index of suspicion for pheochromocytoma is necessary during the workup of secondary hypertension as untreated pheochromocytoma may lead to significant morbidity and mortality, especially in patients who require any surgical treatment.
A high index of suspicion for pheochromocytoma is necessary during the workup of secondary hypertension as untreated pheochromocytoma may lead to significant morbidity and mortality, especially in patients who require any surgical treatment.
Pheochromocytoma is a rare catecholamine-secreting tumor of chromaffin cells of the adrenal medulla or sympathetic ganglia, occurring in about 0.2 to 0.5% of patients with hypertension.1-3 However, in a review of 54 autopsy-proven cases of pheochromocytoma, about 50% of the patients with hypertension were not clinically suspected for pheochromocytoma.4
Pheochromocytoma is usually diagnosed based on symptoms of hyperadrenergic spells, resistant hypertension, especially in the young, with a pressor response to the anesthesia stress test and adrenal incidentaloma.
The classic triad of symptoms associated with pheochromocytoma includes episodic headache (90%), sweating (60-70%), and palpitations (70%).2,5 Sustained or paroxysmal hypertension is the most common symptom reported in about 95% of patients with pheochromocytoma. Other symptoms include pallor, tremors, dyspnea, generalized weakness, orthostatic hypotension, cardiomyopathy, or hyperglycemia.6 However, about 10% of patients with pheochromocytoma are asymptomatic or mildly symptomatic.7 Secondary causes of hypertension are usually suspected in multidrug resistant or sudden early onset of hypertension.8
Approximately 10% of catecholamine-secreting tumors are malignant.9-11 Benign and malignant pheochromocytoma have a similar biochemical and histologic presentation and are differentiated based on local invasion into the surrounding tissues and organs (eg, kidney, liver) or distant metastasis.
A typical workup of a suspected patient with pheochromocytoma includes biochemical tests, including measurements of urinary and fractionated plasma metanephrines and catecholamine. Patients with positive biochemical tests should undergo localization of the tumor with an imaging study either with an adrenal/abdominal magnetic resonance imaging (MRI) or computed tomography (CT) scan. If a patient has paraganglioma or an adrenal mass > 10 cm or negative abdominal imaging with a positive biochemical test, further imaging with an iobenguane I-123 scan is needed (Figure 1).
In this article, we present an unusual case of asymptomatic pheochromocytoma in a patient with right-sided renal oncocytoma who underwent an uneventful nephrectomy and adrenalectomy.
Case Presentation
A 72-year-old male with a medical history of diabetes, hypertension, sensory neuropathy, benign prostatic hypertrophy (BPH) status posttransurethral resection of the prostate, and chronic renal failure presented to establish care with the Arizona Kidney Disease and Hypertension Center. His medications included losartan 50 mg by mouth daily, diltiazem 180 mg extended-release by mouth daily, carvedilol 6.25 mg by mouth twice a day, and tamsulosin 0.4 mg by mouth daily. His presenting vitals were blood pressure (BP), 112/74 left arm sitting, pulse, 63/beats per min, and body mass index, 34. On physical examination, the patient was alert and oriented, and the chest was clear to auscultation without wheeze or rhonchi. On cardiac examination, heart rate and rhythm were regular; S1 and S2 were normal with no added murmurs, rubs or gallops, and no jugular venous distension. The abdomen was soft, nontender, with no palpable mass. His laboratory results showed sodium, 142 mmol/L; potassium, 5.3 mmol/L; chloride, 101 mmol/L; carbon dioxide, 24 mmol/L; albumin, 4.3 g/dL; creatinine, 1.89 mg/dL; blood urea nitrogen, 29 mg/dL; estimated glomerular filtration rate non-African American, 35 mL/min/1.73; 24-h urine creatinine clearance, 105 mL/min; protein, 1306 mg/24 h (Table).
His renal ultrasound showed an exophytic isoechoic mass or complex cyst at the lateral aspect of the lower pole of the right kidney, measuring 45 mm in diameter. An MRI of the abdomen with and without contrast showed a solid partially exophytic mass of the posterolateral interpolar cortex of the right kidney, measuring 5.9 cm in the greatest dimension (Figure 2). No definite involvement of Gerota fascia was noted, a 1-cm metastasis to the right adrenal gland was present, renal veins were patent, and there was no upper retroperitoneal lymphadenopathy.
Treatment and Follow-up
The patient underwent right-hand-assisted lap-aroscopic radical nephrectomy and right adre-nalectomy without any complications. However, the surgical pathology report showed oncocytoma of the kidney (5.7 cm), pheochromocytoma of the adrenal gland (1.4 cm), and papillary adenoma of the kidney (0.7 cm). Right kidney nephrectomy showed non-neoplastic renal parenchyma, diabetic glomerulosclerosis (Renal Pathology Society 2010 diabetic nephropathy class IIb), severe mesangial expansion, moderate interstitial fibrosis, moderate arteriosclerosis, and mild arteriolosclerosis.
A fluorodeoxyglucose-positron emission tomography (FDG-PET) scan was significant for right nephrectomy and adrenalectomy and showed no significant evidence of residual neoplasm or local or distant metastases. A nuclear medicine (iobenguane I-123) tumor and single positron emission computed tomography (SPECT) scan showed normal activity throughout the body and no evidence of abnormal activity (Figure 3).
Discussion
Pheochromocytoma is a rare cause of secondary hypertension. However, the real numbers are thought to be > 0.2 to 0.5%.1,2,4 Patients with pheochromocytoma should undergo surgical adrenal resection after appropriate medical preparation. Patients with pheochromocytoma who are not diagnosed preoperatively have increased surgical mortality rates due to fatal hypertensive crises, malignant arrhythmia, and multiorgan failure as a result of hypertensive crisis.15 Anesthetic drugs during surgery also can exacerbate the cardiotoxic effects of catecholamines. Short-acting anesthetic agents, such as fentanyl, are used in patients with pheochromocytoma.16
This case of pheochromocytoma illustrated no classic symptoms of episodic headache, sweating, and tachycardia, and the patient was otherwise asymptomatic. BP was well controlled with losartan, diltiazem, and a β-blocker with α-blocking activity (carvedilol). As the patient was not known to have pheochromocytoma, he did not undergo preoperative medical therapy. Figure 4 illustrates the receptors stimulate catecholamines, and the drugs blocking these receptors prevent hypertensive crisis during surgery. However, the surgery was without potential complications (ie, hypertensive crisis, malignant arrhythmia, or multiorgan failure). The patient was diagnosed incidentally on histopathology after right radical nephrectomy and adrenalectomy due to solid partially exophytic right renal mass (5.9 cm) with right adrenal metastasis. About 10% of patients are asymptomatic or mildly symptomatic.7 Sometimes, the symptoms may be ignored because of the episodic nature. Other possible reasons can be small, nonfunctional tumors or the use of antihypertensive medications suppressing the symptoms.7
The adrenal mass that was initially thought to be a metastasis of right kidney mass was later confirmed as pheochromocytoma. One possible explanation for uneventful surgery could be the use of β-blocker with α-blocking activity (carvedilol), α-1 adrenergic blocker (tamsulosin) along with nondihydropyridine calcium channel blocker (diltiazem) as part of the patient’s antihypertensive and BPH medication regimen. Another possible explanation could be silent or episodically secreting pheochromocytoma with a small functional portion.
Subsequent workup after adrenalectomy, including urinary and fractionated plasma metanephrines and catecholamines, were not consistent with catecholamine hypersecretion. A 24-hour urine fractionated metanephrines test has about 98% sensitivity and 98% specificity. Elevated plasma norepinephrine was thought to be due to renal failure because it was < 3-fold the upper limit of normal, which is considered to be a possible indication of pheochromocytoma.17,18 The nuclear medicine (iobenguane I-123) tumor, SPECT, and FDG-PET CT studies were negative for residual pheochromocytoma. Other imaging studies to consider in patients with suspected catecholamine-secreting tumor with positive biochemical test and negative abdominal imaging are a whole-body MRI scan, 68-Ga DOTATATE (gallium 68 1,4,7,10-tetraazacyclododecane-1,4,7,10 tetraacetic acid-octreotate) or FDG-PET scan.19
In a review of 54 autopsy-proven pheochromocytoma cases by Sutton and colleagues in 1981, 74% of the patients were not clinically suspected for pheochromocytoma in their life.4 Similarly, in a retrospective study of hospital autopsies by McNeil and colleagues, one incidental pheochromocytoma was detected in every 2031 autopsies (0.05%).20 In another case series of 41 patients with pheochromocytoma-related adrenalectomy, almost 50% of the pheochromocytomas were detected incidentally on imaging studies.21 Although the number of incidental findings are decreasing due to advances in screening techniques, a significant number of patients remain undiagnosed. Multiple cases of diagnosis of pheochromocytoma on autopsy of patients who died of hemodynamic instability (ie, hypertensive crisis, hypotension crisis precipitated by surgery for adrenal or nonadrenal conditions) are reported.3 To the best of our knowledge, there are no case reports published on the diagnosis of pheochromocytoma after adrenalectomy in an asymptomatic patient without intraoperative complications.
The goal of preoperative medical therapy includes BP control, prevention of tachycardia, and volume expansion. The preoperative medications regimens are combined α- and β-adrenergic blockade, calcium channel blockers, and metyrosine. According to clinical practice guidelines of the Endocrine Society in 2014, the α-adrenergic blockers should be started first at least 7 days before surgery to control BP and to cause vasodilation. Early use of α-blockers is required to prevent cardiotoxicity. The β-adrenergic blockers should be started after the adequate α-adrenergic blockade, typically 2 to 3 days before surgery, as early use can cause vasoconstriction in patients with pheochromocytoma. The α-adrenergic blockers include phenoxybenzamine (nonselective long-acting nonspecific α-adrenergic blocking agent), and selective α-1 adrenergic blockers (doxazosin, prazosin, terazosin). The β-adrenergic blocker (ie, propranolol, metoprolol) should be started cautiously with a low dose and slowly titrated to control heart rate. A high sodium diet and increased fluid intake also are recommended 7 to 14 days before surgery. A sudden drop in catecholamines can cause hypotension during an operation. Continuous fluid infusions are given to prevent hypotension.22 Similarly, anesthetic agents also should be modified to prevent cardiotoxic effects. Rocuronium and vecuronium are less cardiotoxic compared with other sympathomimetic muscle relaxants. Short-acting anesthetic agents, such as fentanyl, are preferred. α-blockers are continued throughout the operation. Biochemical testing with fractionated metanephrines is performed about 1 to 2 weeks postoperatively to look for recurrence of the disease.23
Secondary causes of hypertension are suspected in multidrug resistant or sudden early onset of hypertension before aged 40 years. Pheochromocytoma is a rare cause of secondary hypertension, and older adult patients are rarely diagnosed with pheochromocytoma.24 In this report, pheochromocytoma was detected in a 72-year-old hypertensive patient. Therefore, a pheochromocytoma diagnosis should not be ignored in the older adult patient with adrenal mass and hypertension treated with more than one drug. The authors recommend any patient undergoing surgery with adrenal lesion should be considered for the screening of possible pheochromocytoma and prepared preoperatively, especially any patient with renal cell carcinoma with adrenal metastasis.
Conclusions
Asymptomatic pheochromocytoma is an unusual but serious condition, especially for patients undergoing a surgical procedure. An adrenal mass may be ignored in asymptomatic or mildly symptomatic older adult patients and is mostly considered as adrenal metastasis when present with other malignancies. Fortunately, the nephrectomy and adrenalectomy in our case of asymptomatic pheochromocytoma was uneventful, but pheochromocytoma should be ruled out before a surgical procedure, as an absence of medical pretreatment can lead to serious consequences. Therefore, we suggest a more careful screening of pheochromocytoma in patients with an adrenal mass (primary or metastatic) and hypertension treated with multiple antihypertensive drugs, even in older adult patients.
Pheochromocytoma is a rare catecholamine-secreting tumor of chromaffin cells of the adrenal medulla or sympathetic ganglia, occurring in about 0.2 to 0.5% of patients with hypertension.1-3 However, in a review of 54 autopsy-proven cases of pheochromocytoma, about 50% of the patients with hypertension were not clinically suspected for pheochromocytoma.4
Pheochromocytoma is usually diagnosed based on symptoms of hyperadrenergic spells, resistant hypertension, especially in the young, with a pressor response to the anesthesia stress test and adrenal incidentaloma.
The classic triad of symptoms associated with pheochromocytoma includes episodic headache (90%), sweating (60-70%), and palpitations (70%).2,5 Sustained or paroxysmal hypertension is the most common symptom reported in about 95% of patients with pheochromocytoma. Other symptoms include pallor, tremors, dyspnea, generalized weakness, orthostatic hypotension, cardiomyopathy, or hyperglycemia.6 However, about 10% of patients with pheochromocytoma are asymptomatic or mildly symptomatic.7 Secondary causes of hypertension are usually suspected in multidrug resistant or sudden early onset of hypertension.8
Approximately 10% of catecholamine-secreting tumors are malignant.9-11 Benign and malignant pheochromocytoma have a similar biochemical and histologic presentation and are differentiated based on local invasion into the surrounding tissues and organs (eg, kidney, liver) or distant metastasis.
A typical workup of a suspected patient with pheochromocytoma includes biochemical tests, including measurements of urinary and fractionated plasma metanephrines and catecholamine. Patients with positive biochemical tests should undergo localization of the tumor with an imaging study either with an adrenal/abdominal magnetic resonance imaging (MRI) or computed tomography (CT) scan. If a patient has paraganglioma or an adrenal mass > 10 cm or negative abdominal imaging with a positive biochemical test, further imaging with an iobenguane I-123 scan is needed (Figure 1).
In this article, we present an unusual case of asymptomatic pheochromocytoma in a patient with right-sided renal oncocytoma who underwent an uneventful nephrectomy and adrenalectomy.
Case Presentation
A 72-year-old male with a medical history of diabetes, hypertension, sensory neuropathy, benign prostatic hypertrophy (BPH) status posttransurethral resection of the prostate, and chronic renal failure presented to establish care with the Arizona Kidney Disease and Hypertension Center. His medications included losartan 50 mg by mouth daily, diltiazem 180 mg extended-release by mouth daily, carvedilol 6.25 mg by mouth twice a day, and tamsulosin 0.4 mg by mouth daily. His presenting vitals were blood pressure (BP), 112/74 left arm sitting, pulse, 63/beats per min, and body mass index, 34. On physical examination, the patient was alert and oriented, and the chest was clear to auscultation without wheeze or rhonchi. On cardiac examination, heart rate and rhythm were regular; S1 and S2 were normal with no added murmurs, rubs or gallops, and no jugular venous distension. The abdomen was soft, nontender, with no palpable mass. His laboratory results showed sodium, 142 mmol/L; potassium, 5.3 mmol/L; chloride, 101 mmol/L; carbon dioxide, 24 mmol/L; albumin, 4.3 g/dL; creatinine, 1.89 mg/dL; blood urea nitrogen, 29 mg/dL; estimated glomerular filtration rate non-African American, 35 mL/min/1.73; 24-h urine creatinine clearance, 105 mL/min; protein, 1306 mg/24 h (Table).
His renal ultrasound showed an exophytic isoechoic mass or complex cyst at the lateral aspect of the lower pole of the right kidney, measuring 45 mm in diameter. An MRI of the abdomen with and without contrast showed a solid partially exophytic mass of the posterolateral interpolar cortex of the right kidney, measuring 5.9 cm in the greatest dimension (Figure 2). No definite involvement of Gerota fascia was noted, a 1-cm metastasis to the right adrenal gland was present, renal veins were patent, and there was no upper retroperitoneal lymphadenopathy.
Treatment and Follow-up
The patient underwent right-hand-assisted lap-aroscopic radical nephrectomy and right adre-nalectomy without any complications. However, the surgical pathology report showed oncocytoma of the kidney (5.7 cm), pheochromocytoma of the adrenal gland (1.4 cm), and papillary adenoma of the kidney (0.7 cm). Right kidney nephrectomy showed non-neoplastic renal parenchyma, diabetic glomerulosclerosis (Renal Pathology Society 2010 diabetic nephropathy class IIb), severe mesangial expansion, moderate interstitial fibrosis, moderate arteriosclerosis, and mild arteriolosclerosis.
A fluorodeoxyglucose-positron emission tomography (FDG-PET) scan was significant for right nephrectomy and adrenalectomy and showed no significant evidence of residual neoplasm or local or distant metastases. A nuclear medicine (iobenguane I-123) tumor and single positron emission computed tomography (SPECT) scan showed normal activity throughout the body and no evidence of abnormal activity (Figure 3).
Discussion
Pheochromocytoma is a rare cause of secondary hypertension. However, the real numbers are thought to be > 0.2 to 0.5%.1,2,4 Patients with pheochromocytoma should undergo surgical adrenal resection after appropriate medical preparation. Patients with pheochromocytoma who are not diagnosed preoperatively have increased surgical mortality rates due to fatal hypertensive crises, malignant arrhythmia, and multiorgan failure as a result of hypertensive crisis.15 Anesthetic drugs during surgery also can exacerbate the cardiotoxic effects of catecholamines. Short-acting anesthetic agents, such as fentanyl, are used in patients with pheochromocytoma.16
This case of pheochromocytoma illustrated no classic symptoms of episodic headache, sweating, and tachycardia, and the patient was otherwise asymptomatic. BP was well controlled with losartan, diltiazem, and a β-blocker with α-blocking activity (carvedilol). As the patient was not known to have pheochromocytoma, he did not undergo preoperative medical therapy. Figure 4 illustrates the receptors stimulate catecholamines, and the drugs blocking these receptors prevent hypertensive crisis during surgery. However, the surgery was without potential complications (ie, hypertensive crisis, malignant arrhythmia, or multiorgan failure). The patient was diagnosed incidentally on histopathology after right radical nephrectomy and adrenalectomy due to solid partially exophytic right renal mass (5.9 cm) with right adrenal metastasis. About 10% of patients are asymptomatic or mildly symptomatic.7 Sometimes, the symptoms may be ignored because of the episodic nature. Other possible reasons can be small, nonfunctional tumors or the use of antihypertensive medications suppressing the symptoms.7
The adrenal mass that was initially thought to be a metastasis of right kidney mass was later confirmed as pheochromocytoma. One possible explanation for uneventful surgery could be the use of β-blocker with α-blocking activity (carvedilol), α-1 adrenergic blocker (tamsulosin) along with nondihydropyridine calcium channel blocker (diltiazem) as part of the patient’s antihypertensive and BPH medication regimen. Another possible explanation could be silent or episodically secreting pheochromocytoma with a small functional portion.
Subsequent workup after adrenalectomy, including urinary and fractionated plasma metanephrines and catecholamines, were not consistent with catecholamine hypersecretion. A 24-hour urine fractionated metanephrines test has about 98% sensitivity and 98% specificity. Elevated plasma norepinephrine was thought to be due to renal failure because it was < 3-fold the upper limit of normal, which is considered to be a possible indication of pheochromocytoma.17,18 The nuclear medicine (iobenguane I-123) tumor, SPECT, and FDG-PET CT studies were negative for residual pheochromocytoma. Other imaging studies to consider in patients with suspected catecholamine-secreting tumor with positive biochemical test and negative abdominal imaging are a whole-body MRI scan, 68-Ga DOTATATE (gallium 68 1,4,7,10-tetraazacyclododecane-1,4,7,10 tetraacetic acid-octreotate) or FDG-PET scan.19
In a review of 54 autopsy-proven pheochromocytoma cases by Sutton and colleagues in 1981, 74% of the patients were not clinically suspected for pheochromocytoma in their life.4 Similarly, in a retrospective study of hospital autopsies by McNeil and colleagues, one incidental pheochromocytoma was detected in every 2031 autopsies (0.05%).20 In another case series of 41 patients with pheochromocytoma-related adrenalectomy, almost 50% of the pheochromocytomas were detected incidentally on imaging studies.21 Although the number of incidental findings are decreasing due to advances in screening techniques, a significant number of patients remain undiagnosed. Multiple cases of diagnosis of pheochromocytoma on autopsy of patients who died of hemodynamic instability (ie, hypertensive crisis, hypotension crisis precipitated by surgery for adrenal or nonadrenal conditions) are reported.3 To the best of our knowledge, there are no case reports published on the diagnosis of pheochromocytoma after adrenalectomy in an asymptomatic patient without intraoperative complications.
The goal of preoperative medical therapy includes BP control, prevention of tachycardia, and volume expansion. The preoperative medications regimens are combined α- and β-adrenergic blockade, calcium channel blockers, and metyrosine. According to clinical practice guidelines of the Endocrine Society in 2014, the α-adrenergic blockers should be started first at least 7 days before surgery to control BP and to cause vasodilation. Early use of α-blockers is required to prevent cardiotoxicity. The β-adrenergic blockers should be started after the adequate α-adrenergic blockade, typically 2 to 3 days before surgery, as early use can cause vasoconstriction in patients with pheochromocytoma. The α-adrenergic blockers include phenoxybenzamine (nonselective long-acting nonspecific α-adrenergic blocking agent), and selective α-1 adrenergic blockers (doxazosin, prazosin, terazosin). The β-adrenergic blocker (ie, propranolol, metoprolol) should be started cautiously with a low dose and slowly titrated to control heart rate. A high sodium diet and increased fluid intake also are recommended 7 to 14 days before surgery. A sudden drop in catecholamines can cause hypotension during an operation. Continuous fluid infusions are given to prevent hypotension.22 Similarly, anesthetic agents also should be modified to prevent cardiotoxic effects. Rocuronium and vecuronium are less cardiotoxic compared with other sympathomimetic muscle relaxants. Short-acting anesthetic agents, such as fentanyl, are preferred. α-blockers are continued throughout the operation. Biochemical testing with fractionated metanephrines is performed about 1 to 2 weeks postoperatively to look for recurrence of the disease.23
Secondary causes of hypertension are suspected in multidrug resistant or sudden early onset of hypertension before aged 40 years. Pheochromocytoma is a rare cause of secondary hypertension, and older adult patients are rarely diagnosed with pheochromocytoma.24 In this report, pheochromocytoma was detected in a 72-year-old hypertensive patient. Therefore, a pheochromocytoma diagnosis should not be ignored in the older adult patient with adrenal mass and hypertension treated with more than one drug. The authors recommend any patient undergoing surgery with adrenal lesion should be considered for the screening of possible pheochromocytoma and prepared preoperatively, especially any patient with renal cell carcinoma with adrenal metastasis.
Conclusions
Asymptomatic pheochromocytoma is an unusual but serious condition, especially for patients undergoing a surgical procedure. An adrenal mass may be ignored in asymptomatic or mildly symptomatic older adult patients and is mostly considered as adrenal metastasis when present with other malignancies. Fortunately, the nephrectomy and adrenalectomy in our case of asymptomatic pheochromocytoma was uneventful, but pheochromocytoma should be ruled out before a surgical procedure, as an absence of medical pretreatment can lead to serious consequences. Therefore, we suggest a more careful screening of pheochromocytoma in patients with an adrenal mass (primary or metastatic) and hypertension treated with multiple antihypertensive drugs, even in older adult patients.
1. Omura M, Saito J, Yamaguchi K, Kakuta Y, Nishikawa T. Prospective study on the prevalence of secondary hypertension among hypertensive patients visiting a general outpatient clinic in Japan. Hypertens Res. 2004;27(3):193-202. doi:10.1291/hypres.27.193
2. Stein PP, Black HR. A simplified diagnostic approach to pheochromocytoma: a review of the literature and report of one institution’s experience. Medicine (Baltimore). 1991;70(1):46-66. doi:10.1097/00005792-199101000-00004
3. Beard CM, Sheps SG, Kurland LT, Carney JA, Lie JT. Occurrence of pheochromocytoma in Rochester, Minnesota, 1950 through 1979. Mayo Clin Proc. 1983;58(12):802-804.
4. Sutton MG, Sheps SG, Lie JT. Prevalence of clinically unsuspected pheochromocytoma: review of a 50-year autopsy series. Mayo Clin Proc. 1981;56(6):354-360.
5. Manger WM, Gifford RW Jr. Pheochromocytoma. J Clin Hypertens (Greenwich). 2002;4(1):62-72. doi:10.1111/j.1524-6175.2002.01452.x
6. Kassim TA, Clarke DD, Mai VQ, Clyde PW, Mohamed Shakir KM. Catecholamine-induced cardiomyopathy. Endocr Pract. 2008;14(9):1137-1149. doi:10.4158/EP.14.9.1137
7. Kudva YC, Young WF, Thompson GB, Grant CS, Van Heerden JA. Adrenal incidentaloma: an important component of the clinical presentation spectrum of benign sporadic adrenal pheochromocytoma. The Endocrinologist. 1999;9(2):77-80. doi:10.1097/00019616-199903000-00002
8. Puar TH, Mok Y, Debajyoti R, Khoo J, How CH, Ng AK. Secondary hypertension in adults. Singapore Med J. 2016;57(5):228-232. doi:10.11622/smedj.2016087
9. Bravo EL. Pheochromocytoma: new concepts and future trends. Kidney Int. 1991;40(3):544-556. doi:10.1038/ki.1991.244
10. Plouin PF, Chatellier G, Fofol I, Corvol P. Tumor recurrence and hypertension persistence after successful pheochromocytoma operation. Hypertension. 1997;29(5):1133-1139. doi:10.1161/01.hyp.29.5.1133
11. Hamidi O, Young WF Jr, Iñiguez-Ariza NM, et al. Malignant pheochromocytoma and paraganglioma: 272 patients over 55 years. J Clin Endocrinol Metab. 2017;102(9):3296-3305. doi:10.1210/jc.2017-00992
12. Kenny L, Rizzo V, Trevis J, Assimakopoulou E, Timon D. The unexpected diagnosis of phaeochromocytoma in the anaesthetic room. Ann Card Anaesth. 2018;21(3):307-310. doi:10.4103/aca.ACA_206_17
13. Johnston PC, Silversides JA, Wallace H, et al. Phaeochromocytoma crisis: two cases of undiagnosed phaeochromocytoma presenting after elective nonrelated surgical procedures. Case Rep Anesthesiol. 2013;2013:514714. doi:10.1155/2013/514714
14. Shen SJ, Cheng HM, Chiu AW, Chou CW, Chen JY. Perioperative hypertensive crisis in clinically silent pheochromocytomas: report of four cases. Chang Gung Med J. 2005;28(1):44-50.
15. Lo CY, Lam KY, Wat MS, Lam KS. Adrenal pheochromocytoma remains a frequently overlooked diagnosis. Am J Surg. 2000;179(3):212-215. doi:10.1016/s0002-9610(00)00296-8
16. Myklejord DJ. Undiagnosed pheochromocytoma: the anesthesiologist nightmare. Clin Med Res. 2004;2(1):59-62. doi:10.3121/cmr.2.1.59
17. Stumvoll M, Radjaipour M, Seif F. Diagnostic considerations in pheochromocytoma and chronic hemodialysis: case report and review of the literature. Am J Nephrol. 1995;15(2):147-151. doi:10.1159/000168820
18. Morioka M, Yuihama S, Nakajima T, et al. Incidentally discovered pheochromocytoma in long-term hemodialysis patients. Int J Urol. 2002;9(12):700-703. doi:10.1046/j.1442-2042.2002.00553.x
19. ˇCtvrtlík F, Koranda P, Schovánek J, Škarda J, Hartmann I, Tüdös Z. Current diagnostic imaging of pheochromocytomas and implications for therapeutic strategy. Exp Ther Med. 2018;15(4):3151-3160. doi:10.3892/etm.2018.5871
20. McNeil AR, Blok BH, Koelmeyer TD, Burke MP, Hilton JM. Phaeochromocytomas discovered during coronial autopsies in Sydney, Melbourne and Auckland. Aust N Z J Med. 2000;30(6):648-652. doi:10.1111/j.1445-5994.2000.tb04358.x
21. Baguet JP, Hammer L, Mazzuco TL, et al. Circumstances of discovery of phaeochromocytoma: a retrospective study of 41 consecutive patients. Eur J Endocrinol. 2004;150(5):681-686. doi:10.1530/eje.0.1500681
22. Lenders JW, Duh QY, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014;99(6):1915-1942. doi:10.1210/jc.2014-1498
23. Dortzbach K, Gainsburg DM, Frost EA. Variants of pheochromocytoma and their anesthetic implications--a case report and literature review. Middle East J Anaesthesiol. 2010;20(6):897-905.
24. Januszewicz W, Chodakowska J, Styczy´nski G. Secondary hypertension in the elderly. J Hum Hypertens. 1998;12(9):603-606. doi:10.1038/sj.jhh.1000673
1. Omura M, Saito J, Yamaguchi K, Kakuta Y, Nishikawa T. Prospective study on the prevalence of secondary hypertension among hypertensive patients visiting a general outpatient clinic in Japan. Hypertens Res. 2004;27(3):193-202. doi:10.1291/hypres.27.193
2. Stein PP, Black HR. A simplified diagnostic approach to pheochromocytoma: a review of the literature and report of one institution’s experience. Medicine (Baltimore). 1991;70(1):46-66. doi:10.1097/00005792-199101000-00004
3. Beard CM, Sheps SG, Kurland LT, Carney JA, Lie JT. Occurrence of pheochromocytoma in Rochester, Minnesota, 1950 through 1979. Mayo Clin Proc. 1983;58(12):802-804.
4. Sutton MG, Sheps SG, Lie JT. Prevalence of clinically unsuspected pheochromocytoma: review of a 50-year autopsy series. Mayo Clin Proc. 1981;56(6):354-360.
5. Manger WM, Gifford RW Jr. Pheochromocytoma. J Clin Hypertens (Greenwich). 2002;4(1):62-72. doi:10.1111/j.1524-6175.2002.01452.x
6. Kassim TA, Clarke DD, Mai VQ, Clyde PW, Mohamed Shakir KM. Catecholamine-induced cardiomyopathy. Endocr Pract. 2008;14(9):1137-1149. doi:10.4158/EP.14.9.1137
7. Kudva YC, Young WF, Thompson GB, Grant CS, Van Heerden JA. Adrenal incidentaloma: an important component of the clinical presentation spectrum of benign sporadic adrenal pheochromocytoma. The Endocrinologist. 1999;9(2):77-80. doi:10.1097/00019616-199903000-00002
8. Puar TH, Mok Y, Debajyoti R, Khoo J, How CH, Ng AK. Secondary hypertension in adults. Singapore Med J. 2016;57(5):228-232. doi:10.11622/smedj.2016087
9. Bravo EL. Pheochromocytoma: new concepts and future trends. Kidney Int. 1991;40(3):544-556. doi:10.1038/ki.1991.244
10. Plouin PF, Chatellier G, Fofol I, Corvol P. Tumor recurrence and hypertension persistence after successful pheochromocytoma operation. Hypertension. 1997;29(5):1133-1139. doi:10.1161/01.hyp.29.5.1133
11. Hamidi O, Young WF Jr, Iñiguez-Ariza NM, et al. Malignant pheochromocytoma and paraganglioma: 272 patients over 55 years. J Clin Endocrinol Metab. 2017;102(9):3296-3305. doi:10.1210/jc.2017-00992
12. Kenny L, Rizzo V, Trevis J, Assimakopoulou E, Timon D. The unexpected diagnosis of phaeochromocytoma in the anaesthetic room. Ann Card Anaesth. 2018;21(3):307-310. doi:10.4103/aca.ACA_206_17
13. Johnston PC, Silversides JA, Wallace H, et al. Phaeochromocytoma crisis: two cases of undiagnosed phaeochromocytoma presenting after elective nonrelated surgical procedures. Case Rep Anesthesiol. 2013;2013:514714. doi:10.1155/2013/514714
14. Shen SJ, Cheng HM, Chiu AW, Chou CW, Chen JY. Perioperative hypertensive crisis in clinically silent pheochromocytomas: report of four cases. Chang Gung Med J. 2005;28(1):44-50.
15. Lo CY, Lam KY, Wat MS, Lam KS. Adrenal pheochromocytoma remains a frequently overlooked diagnosis. Am J Surg. 2000;179(3):212-215. doi:10.1016/s0002-9610(00)00296-8
16. Myklejord DJ. Undiagnosed pheochromocytoma: the anesthesiologist nightmare. Clin Med Res. 2004;2(1):59-62. doi:10.3121/cmr.2.1.59
17. Stumvoll M, Radjaipour M, Seif F. Diagnostic considerations in pheochromocytoma and chronic hemodialysis: case report and review of the literature. Am J Nephrol. 1995;15(2):147-151. doi:10.1159/000168820
18. Morioka M, Yuihama S, Nakajima T, et al. Incidentally discovered pheochromocytoma in long-term hemodialysis patients. Int J Urol. 2002;9(12):700-703. doi:10.1046/j.1442-2042.2002.00553.x
19. ˇCtvrtlík F, Koranda P, Schovánek J, Škarda J, Hartmann I, Tüdös Z. Current diagnostic imaging of pheochromocytomas and implications for therapeutic strategy. Exp Ther Med. 2018;15(4):3151-3160. doi:10.3892/etm.2018.5871
20. McNeil AR, Blok BH, Koelmeyer TD, Burke MP, Hilton JM. Phaeochromocytomas discovered during coronial autopsies in Sydney, Melbourne and Auckland. Aust N Z J Med. 2000;30(6):648-652. doi:10.1111/j.1445-5994.2000.tb04358.x
21. Baguet JP, Hammer L, Mazzuco TL, et al. Circumstances of discovery of phaeochromocytoma: a retrospective study of 41 consecutive patients. Eur J Endocrinol. 2004;150(5):681-686. doi:10.1530/eje.0.1500681
22. Lenders JW, Duh QY, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014;99(6):1915-1942. doi:10.1210/jc.2014-1498
23. Dortzbach K, Gainsburg DM, Frost EA. Variants of pheochromocytoma and their anesthetic implications--a case report and literature review. Middle East J Anaesthesiol. 2010;20(6):897-905.
24. Januszewicz W, Chodakowska J, Styczy´nski G. Secondary hypertension in the elderly. J Hum Hypertens. 1998;12(9):603-606. doi:10.1038/sj.jhh.1000673
Widespread Necrotizing Purpura and Lucio Phenomenon as the First Diagnostic Presentation of Diffuse Nonnodular Lepromatous Leprosy
Case Report
A 70-year-old man living in Esna, Luxor, Egypt presented to the Department of Rheumatology and Rehabilitation with widespread gangrenous skin lesions associated with ulcers of 2 weeks’ duration. One year prior, the patient had an insidious onset of nocturnal fever, bilateral leg edema, and numbness and a tingling sensation in both hands. He presented some laboratory and radiologic investigations that were performed at another hospital prior to the current presentation, which revealed thrombocytopenia, mild splenomegaly, and generalized lymphadenopathy. An excisional left axillary lymph node biopsy was performed at another hospital prior to the current presentation, and the pathology report provided by the patient described a reactive, foamy, histiocyte-rich lesion, suggesting a diagnosis of hemophagocytic lymphohistiocytosis. The patient had no diabetes or hypertension and no history of deep vein thrombosis, stroke, or unintentional weight loss. No medications were taken prior to the onset of the skin lesions, and his family history was irrelevant.
General examination at the current presentation revealed a fever (temperature, 101.3 °F [38.5 °C]), a normal heart rate (90 beats per minute), normal blood pressure (120/80 mmHg), normal respiratory rate (14 breaths per minute), accentuated heart sounds, and normal vesicular breathing without adventitious sounds. He had saddle nose, loss of the outer third of the eyebrows, and marked reduction in the density of the eyelashes (madarosis). Bilateral pitting edema of the legs also was present. Neurologic examination revealed hypoesthesia in a glove-and-stocking pattern, thickened peripheral nerves, and trophic changes over both hands; however, he had normal muscle power and deep reflexes. Joint examination revealed no abnormalities. Skin examination revealed widespread, reticulated, necrotizing, purpuric lesions on the arms, legs, abdomen, and ears, some associated with gangrenous ulcerations and hemorrhagic blisters. Scattered vasculitic ulcers and gangrenous patches were seen on the fingers. A gangrenous ulcer mimicking Fournier gangrene was seen involving the scrotal skin in addition to a gangrenous lesion on the glans penis (Figure 1–3). Unaffected skin appeared smooth, shiny, and edematous and showed no nodular lesions. Peripheral pulsations were intact.
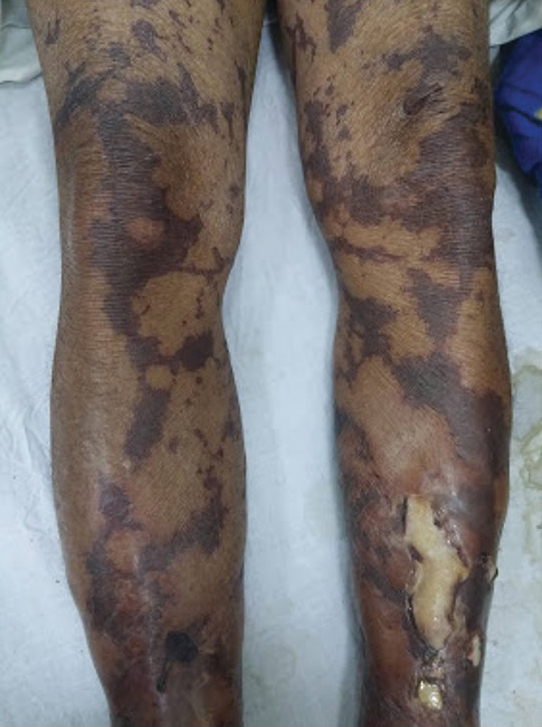
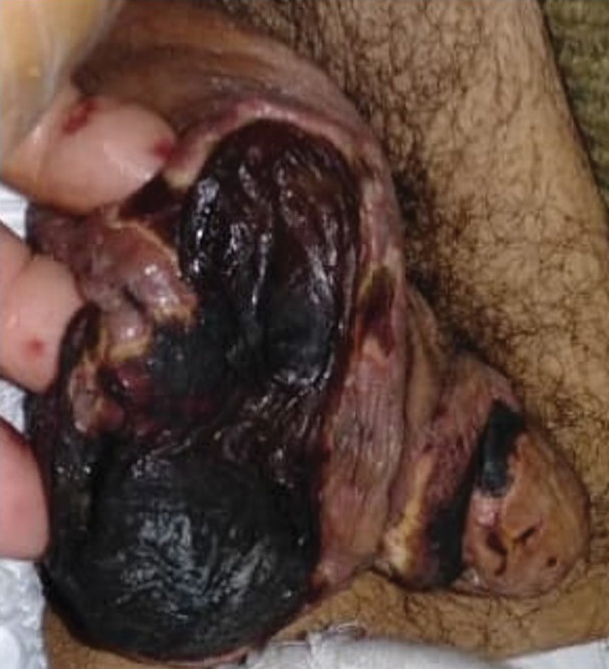
Positive findings from a wide panel of laboratory investigations included an elevated erythrocyte sedimentation rate (103 mm for the first hour [reference range, 0–22 mm]), high C-reactive protein (50.7 mg/L [reference range, up to 6 mg/L]), anemia (hemoglobin count, 7.3 g/dL [reference range, 13.5–17.5 g/dL]), thrombocytopenia (45×103/mm3 [reference range, 150×103/mm3), low serum albumin (2.3 g/dL [reference range, 3.4–5.4 g/dL]), elevated IgG and IgM anticardiolipin antibodies (IgG, 21.4 IgG phospholipid [GPL] units [reference range, <10 IgG phospholipid (GPL) units]; IgM, 59.4 IgM phospholipid (MPL) units [reference range, <7 IgM phospholipid (MPL) units]), positive lupus anticoagulant panel test, elevated anti-β2 glycoprotein antibodies (IgG, 17.5
Nerve conduction velocity showed axonal sensory polyneuropathy. Motor nerve conduction studies for median and ulnar nerves were within normal range. Lower-limb nerves assessment was limited by the ulcerated areas and marked edema. Echocardiography was unremarkable. Arterial Doppler studies were only available for the upper limbs and were unremarkable.
A punch biopsy was taken from one of the necrotizing purpuric lesions on the legs, and histopathologic examination revealed foci of epidermal necrosis and subepidermal separation and superficial and deep perivascular and periadnexal infiltrates extending into the fat lobules. The infiltrates were mainly made up of foamy macrophages, and some contained globi (lepra cells), in addition to lymphocytes and many neutrophils with nuclear dust. Blood vessels in the superficial and deep dermis and in the subcutaneous fat showed fibrinoid necrosis in their walls with neutrophils infiltrating the walls and thrombi in the lumens (Figure 4). Modified Ziehl-Neelsen staining revealed clumps of acid-fast lepra bacilli inside vascular lumina and endothelial cell lining and within the foamy macrophages (Figure 5). Slit-skin smear examination was performed twice and yielded negative results. The slide and paraffin block of the already performed lymph node biopsy were retrieved. Examination revealed aggregates of foamy histiocytes surrounded by lymphocytes and plasma cells replacing normal lymphoid follicles. Modified Ziehl-Neelsen stain was performed, and clusters of acid-fast bacilli were detected within the foamy histiocytic infiltrate (Figure 6).
According to the results of the skin biopsy, the revised result of the lymph node biopsy, and the pattern of neurologic deficit together with clinical and laboratory correlation, the patient was diagnosed with diffuse nonnodular lepromatous leprosy presenting with Lucio phenomenon (Lucio leprosy) and associated with lepromatous lymphadenitis.
The patient received the following treatment: methylprednisolone 500 mg (intravenous pulse therapy) followed by daily oral administration of prednisolone 10 mg, rifampicin 300 mg, dapsone 100 mg, clofazimine 100 mg, acetylsalicylic acid 150 mg, and enoxaparin sodium 80 mg. In addition, the scrotal Fournier gangrene–like lesion was treated by surgical debridement followed by vacuum therapy. By the second week after treatment, the gangrenous lesions of the fingers developed a line of demarcation, and the skin infarctions started to recede.
Comment
Despite a decrease in its prevalence through a World Health Organization (WHO)–empowered eradication program, leprosy still represents a health problem in endemic areas.1,2 It is characterized by a wide range of immune responses to Mycobacterium leprae, displaying a spectrum of clinical and histopathologic manifestations that vary from the tuberculoid or paucibacillary pole with a strong cell-mediated immune response and fewer organisms to the lepromatous or multibacillary pole with weaker cell-mediated immune response and higher loads of organisms.3 In addition to its well-known cutaneous and neurologic manifestations, leprosy can present with a variety of manifestations, including constitutional symptoms, musculoskeletal manifestations, and serologic abnormalities; thus, leprosy can mimic rheumatoid arthritis, spondyloarthritis, and vasculitis—a pitfall that may result in misdiagnosis as a rheumatologic disorder.3-7
The chronic course of leprosy can be disrupted by acute, immunologically mediated reactions known as lepra reactions, of which there are 3 types.8 Type I lepra reactions are cell mediated and occur mainly in patients with borderline disease, often representing an upgrade toward the tuberculoid pole; less often they represent a downgrade reaction. Nerves become painful and swollen with possible loss of function, and skin lesions become edematous and tender; sometimes arthritis develops.9 Type II lepra reactions, also known as erythema nodosum leprosum (ENL), occur in borderline lepromatous and lepromatous patients with a high bacillary load. They are characterized by fever, body aches, tender cutaneous/subcutaneous nodules that may ulcerate, possible bullous lesions, painful nerve swellings, swollen joints, iritis, lymphadenitis, glomerulonephritis, epididymo-orchitis, and hepatic affection. Both immune-complex and delayed hypersensitivity reactions play a role in ENL.8,10 The third reaction is a rare aggressive type known as Lucio phenomenon or Lucio leprosy, which presents with irregular-shaped, angulated, or stellar necrotizing purpuric lesions (hemorrhagic infacrtions) developing mainly on the extremities. The lesions evolve into ulcers that heal with atrophic scarring.2,11 Lucio phenomenon develops as a result of thrombotic vascular occlusion secondary to massive invasion of vascular endothelial cells by lepra bacilli.2,11-14 Involvement of the scrotal skin, such as in our patient, is rare.
Lucio phenomenon mainly is seen in Mexico and Central America, and few cases have been documented in Cuba, South America, the United States, India, Polynesia, South Africa, and Southeast Asia.15-17 It specifically occurs in patients with untreated, diffuse, nonnodular lepromatous leprosy (pure and primitive diffuse lepromatous leprosy (DLL)/diffuse leprosy of Lucio and Latapí). This type of leprosy was first described by Lucio and Alvarado18 in 1852 as a distinct form of lepromatous leprosy characterized by widespread and dense infiltration of the whole skin by lepra bacilli without the typical nodular lesions of leprosy, rendering its diagnosis challenging, especially in sporadic cases. Other manifestations of DLL include complete alopecia of the eyebrows and eyelashes, destructive rhinitis, and areas of anhidrosis and dyesthesia.2
Latapí and Chévez-Zomora19 defined Lucio phenomenon in 1948 as a form of histopathologic vasculitis restricted to patients with DLL. Histopathologically, in addition to the infiltration of the skin with acid-fast bacilli–laden foamy histiocytes, lesions of Lucio phenomenon show features of necrotizing (leukocytoclastic) vasculitis with fibrinoid necrosis20 or vascular thrombi with minimal perivascular lymphocytic infiltrate and no evidence of vasculitis.11 Medium to large vessels in the deep dermis and subcutaneous tissue show infiltration of their walls with a large number of macrophages laden with acid-fast bacilli.11 Cases with histopathologic features mimicking antiphospholipid syndrome with endothelial cell proliferation, thrombosis, and mild mononuclear cell infiltrate also may be seen.20 In all cases, ischemic epidermal necrosis is seen, as well as acid-fast bacilli, both singly and in clusters (globi) within endothelial cells and inside blood vessel lumina.
Although Lucio phenomenon initially was thought to be immune-complex mediated like ENL, it has been suggested that the main trigger is thrombotic vascular occlusion secondary to massive invasion of the vascular endothelial cells by the lepra bacilli, resulting in necrosis.14 Bacterial lipopolysaccharides promote the release of IL-1 and tumor necrosis factor α, which in turn stimulate the production of prostaglandins, IL-6, and coagulation factor III, leading to vascular thrombosis and tissue necrosis.21,22 Moreover, antiphospholipid antibodies, which have been found to be induced in response to certain infectious agents in genetically predisposed individuals,23 have been reported in patients with leprosy, mainly in association with lepromatous leprosy. The reported prevalence of anticardiolipin antibodies ranged from 37% to 98%, whereas anti-β2-glycoprotein I antibodies ranged from 3% to 19%, and antiprothrombin antibodies ranged from 6% to 45%.24,25 Antiphospholipid antibodies have been reported to play a role in the pathogenesis of Lucio phenomenon.11,13,15,26 Our case supports this hypothesis with positive anticardiolipin antibodies, anti-β2 glycoprotein antibodies, and positive lupus anticoagulant.
In accordance with Curi et al,2 who reported 5 cases of DLL with Lucio phenomenon, our patient showed a similar presentation with positive inflammatory markers in association with a negative autoimmune profile (ANA, ANCA-C&P) and negative venereal disease research laboratory test. It is important to mention that a positive autoimmune profile (ANA, ANCA-C&P) can be present in leprotic patients, causing possible diagnostic confusion with collagen diseases.27,28
An interesting finding in our case was the negative slit-skin smear results. Although the specificity of slit-skin smear is 100%, as it directly demonstrates the presence of acid-fast bacilli,29 its sensitivity is low and varies from 10% to 50%.30 The detection of acid-fast bacilli in tissue sections is reported to be a better method for confirming the diagnosis of leprosy.31
The provisional impression of hemophagocytic lymphohistiocytosis in the lymph node biopsy in our patient was excluded upon detection of acid-fast bacilli in the foamy histiocytes infiltrating the lymph node; moreover, the normal serum lipids and serum ferritin argued against this diagnosis.32 Leprosy tends to involve the lymph nodes, particularly in borderline, borderline lepromatous, and lepromatous forms.33 The incidence of lymph node involvement accompanied by skin lesions with the presence of acid-fast bacilli in the lymph nodes is 92.2%.34
Our patient showed an excellent response to antileprotic treatment, which was administered according to the WHO multidrug therapy guidelines for multibacillary leprosy,35 combined with low-dose prednisolone, acetylsalicylic acid, and anticoagulant treatment. Thalidomide and high-dose prednisolone (60 mg/d) combined with antileprotic treatment also have been reported to be successful in managing recurrent infarctions in leprosy.36 The Fournier-like gangrenous ulcer of the scrotum was managed by surgical debridement and vacuum therapy.
It is noteworthy that the WHO elimination goal for leprosy was to reduce the prevalence to less than 1 case per 10,000 population. Egypt is among the first countries in North Africa and the Middle East regions to achieve this target supervised by the National Leprosy Control Program as early as 1994; this was further reduced to 0.33 cases per 10,000 population in 2004, and reduced again in 2009; however, certain foci showed a prevalence rate more than the elimination target, particularly in the cities of Qena (1.12) and Sohag (2.47).37 Esna, where our patient is from, is an endemic area in Egypt.38
Conclusion
1. World Health Organization. World Health Statistics: 2011. World Health Organization; 2011. https://www.who.int/gho/publications/world_health_statistics/EN_WHS2011_Full.pdf
2. Curi PF, Villaroel JS, Migliore N, et al. Lucio’s phenomenon: report of five cases. Clin Rheumatol. 2016;35:1397-1401.
3. Shrestha B, Li YQ, Fu P. Leprosy mimics adult onset Still’s disease in a Chinese patient. Egypt Rheumatol. 2018;40:217-220.
4. Prasad S, Misra R, Aggarwal A, et al. Leprosy revealed in a rheumatology clinic: a case series. Int J Rheum Dis. 2013;16:129-133.
5. Chao G, Fang L, Lu C. Leprosy with ANA positive mistaken for connective tissue disease. Clin Rheumatol. 2013;32:645-648.
6. Chauhan S, Wakhlu A, Agarwal V. Arthritis in leprosy. Rheumatology. 2010;49:2237-2242.
7. Rath D, Bhargava S, Kundu BK. Leprosy mimicking common rheumatologic entities: a trial for the clinician in the era of biologics. Case Rep Rheumatol. 2014;2014:429698.
8. Cuevas J, Rodríguez-Peralto JL, Carrillo R, et al. Erythema nodosum leprosum: reactional leprosy. Semin Cutan Med Surg. 2007;26:126-130.
9. Henriques CC, Lopéz B, Mestre T, et al. Leprosy and rheumatoid arthritis: consequence or association? BMJ Case Rep. 2012;13:1-4.
10. Vázquez-Botet M, Sánchez JL. Erythema nodosum leprosum. Int J Dermatol. 1987;26:436-437.
11. Nunzie E, Ortega Cabrera LV, Macanchi Moncayo FM, et al. Lucio leprosy with Lucio’s phenomenon, digital gangrene and anticardiolipin antibodies. Lepr Rev. 2014;85:194-200.
12. Salvi S, Chopra A. Leprosy in a rheumatology setting: a challenging mimic to expose. Clin Rheumatol. 2013;32:1557-1563.
13. Azulay-Abulafia L, Pereira SL, Hardmann D, et al. Lucio phenomenon. vasculitis or occlusive vasculopathy? Hautarzt. 2006;57:1101-1105.
14. Benard G, Sakai-Valente NY, Bianconcini Trindade MA. Concomittant Lucio phenomenon and erythema nodosum in a leprosy patient: clues for their distinct pathogenesis. Am J Dermatopathol. 2009;31:288-292.
15. Rocha RH, Emerich PS, Diniz LM, et al. Lucio’s phenomenon: exuberant case report and review of Brazilian cases. An Bras Dermatol. 2016;91(suppl 5):S60-S63.
16. Costa IM, Kawano LB, Pereira CP, et al. Lucio’s phenomenon: a case report and review of the literature. Int J Dermatol. 2005;44:566-571.
17. Kumari R, Thappa DM, Basu D. A fatal case of Lucio phenomenon from India. Dermatol Online J. 2008;14:10.
18. Lucio R, Alvarado I. Opúsculo Sobre el Mal de San Lázaro o Elefantiasis de los Griegos. M. Murguía; 1852.
19. Latapí F, Chévez-Zamora A. The “spotted” leprosy of Lucio: an introduction to its clinical and histological study. Int J Lepr. 1948;16:421-437.
20. Vargas OF. Diffuse leprosy of Lucio and Latapí: a histologic study. Lepr Rev. 2007;78:248-260.
21. Latapí FR, Chevez-Zamora A. La lepra manchada de Lucio. Rev Dermatol Mex. 1978;22:102-107.
22. Monteiro R, Abreu MA, Tiezzi MG, et al. Fenômeno de Lúcio: mais um caso relatado no Brasil. An Bras Dermatol. 2012;87:296-300.
23. Gharavi EE, Chaimovich H, Cucucrull E, et al. Induction of antiphospholipid antibodies by immunization with synthetic bacterial & viral peptides. Lupus. 1999;8:449-455.
24. de Larrañaga GF, Forastiero RR, Martinuzzo ME, et al. High prevalence of antiphospholipid antibodies in leprosy: evaluation of antigen reactivity. Lupus. 2000;9:594-600.
25. Loizou S, Singh S, Wypkema E, et al. Anticardiolipin, anti-beta(2)-glycoprotein I and antiprothrombin antibodies in black South African patients with infectious disease. Ann Rheum Dis. 2003;62:1106-1111.
26. Akerkar SM, Bichile LS. Leprosy & gangrene: a rare association; role of antiphospholipid antibodies. BMC Infect Dis. 2005,5:74.
27. Horta-Baas G, Hernández-Cabrera MF, Barile-Fabris LA, et al. Multibacillary leprosy mimicking systemic lupus erythematosus: case report and literature review. Lupus. 2015;24:1095-1102.
28. Pradhan V, Badakere SS, Shankar KU. Increased incidence of cytoplasmic ANCA (cANCA) and other auto antibodies in leprosy patients from western India. Lepr Rev. 2004;75:50-56.
29. Oskam L. Diagnosis and classification of leprosy. Lepr Rev. 2002;73:17-26.
30. Rao PN. Recent advances in the control programs and therapy of leprosy. Indian J Dermatol Venereol Leprol. 2004;70:269-276.
31. Rao PN, Pratap D, Ramana Reddy AV, et al. Evaluation of leprosy patients with 1 to 5 skin lesions with relevance to their grouping into paucibacillary or multibacillary disease. Indian J Dermatol Venereol Leprol. 2006;72:207-210.
32. Rosado FGN, Kim AS. Hemophagocytic lymphohistiocytosis. an update on diagnosis and pathogenesis. Am J Clin Pathol. 2013;139:713-727.
33. Kar HK, Mohanty HC, Mohanty GN, et al. Clinicopathological study of lymph node involvement in leprosy. Lepr India. 1983;55:725-738.
34. Gupta JC, Panda PK, Shrivastava KK, et al. A histopathologic study of lymph nodes in 43 cases of leprosy. Lepr India. 1978;50:196-203.
35. WHO Expert Committee on Leprosy. Seventh Report. World Health Organization; 1998. https://apps.who.int/iris/bitstream/handle/10665/42060/WHO_TRS_874.pdf?sequence=1&isAllowed=y
36. Misra DP, Parida JR, Chowdhury AC, et al. Lepra reaction with Lucio phenomenon mimicking cutaneous vasculitis. Case Rep Immunol. 2014;2014:641989.
37. Amer A, Mansour A. Epidemiological study of leprosy in Egypt: 2005-2009. Egypt J Dermatol Venereol. 2014;34:70-73.
38. World Health Organization. Screening campaign aims to eliminate leprosy in Egypt. Published May 9, 2018. Accessed September 8, 2021. http://www.emro.who.int/egy/egypt-events/last-miless-activities-on-eliminating-leprosy-from-egypt.html
Case Report
A 70-year-old man living in Esna, Luxor, Egypt presented to the Department of Rheumatology and Rehabilitation with widespread gangrenous skin lesions associated with ulcers of 2 weeks’ duration. One year prior, the patient had an insidious onset of nocturnal fever, bilateral leg edema, and numbness and a tingling sensation in both hands. He presented some laboratory and radiologic investigations that were performed at another hospital prior to the current presentation, which revealed thrombocytopenia, mild splenomegaly, and generalized lymphadenopathy. An excisional left axillary lymph node biopsy was performed at another hospital prior to the current presentation, and the pathology report provided by the patient described a reactive, foamy, histiocyte-rich lesion, suggesting a diagnosis of hemophagocytic lymphohistiocytosis. The patient had no diabetes or hypertension and no history of deep vein thrombosis, stroke, or unintentional weight loss. No medications were taken prior to the onset of the skin lesions, and his family history was irrelevant.
General examination at the current presentation revealed a fever (temperature, 101.3 °F [38.5 °C]), a normal heart rate (90 beats per minute), normal blood pressure (120/80 mmHg), normal respiratory rate (14 breaths per minute), accentuated heart sounds, and normal vesicular breathing without adventitious sounds. He had saddle nose, loss of the outer third of the eyebrows, and marked reduction in the density of the eyelashes (madarosis). Bilateral pitting edema of the legs also was present. Neurologic examination revealed hypoesthesia in a glove-and-stocking pattern, thickened peripheral nerves, and trophic changes over both hands; however, he had normal muscle power and deep reflexes. Joint examination revealed no abnormalities. Skin examination revealed widespread, reticulated, necrotizing, purpuric lesions on the arms, legs, abdomen, and ears, some associated with gangrenous ulcerations and hemorrhagic blisters. Scattered vasculitic ulcers and gangrenous patches were seen on the fingers. A gangrenous ulcer mimicking Fournier gangrene was seen involving the scrotal skin in addition to a gangrenous lesion on the glans penis (Figure 1–3). Unaffected skin appeared smooth, shiny, and edematous and showed no nodular lesions. Peripheral pulsations were intact.
Positive findings from a wide panel of laboratory investigations included an elevated erythrocyte sedimentation rate (103 mm for the first hour [reference range, 0–22 mm]), high C-reactive protein (50.7 mg/L [reference range, up to 6 mg/L]), anemia (hemoglobin count, 7.3 g/dL [reference range, 13.5–17.5 g/dL]), thrombocytopenia (45×103/mm3 [reference range, 150×103/mm3), low serum albumin (2.3 g/dL [reference range, 3.4–5.4 g/dL]), elevated IgG and IgM anticardiolipin antibodies (IgG, 21.4 IgG phospholipid [GPL] units [reference range, <10 IgG phospholipid (GPL) units]; IgM, 59.4 IgM phospholipid (MPL) units [reference range, <7 IgM phospholipid (MPL) units]), positive lupus anticoagulant panel test, elevated anti-β2 glycoprotein antibodies (IgG, 17.5
Nerve conduction velocity showed axonal sensory polyneuropathy. Motor nerve conduction studies for median and ulnar nerves were within normal range. Lower-limb nerves assessment was limited by the ulcerated areas and marked edema. Echocardiography was unremarkable. Arterial Doppler studies were only available for the upper limbs and were unremarkable.
A punch biopsy was taken from one of the necrotizing purpuric lesions on the legs, and histopathologic examination revealed foci of epidermal necrosis and subepidermal separation and superficial and deep perivascular and periadnexal infiltrates extending into the fat lobules. The infiltrates were mainly made up of foamy macrophages, and some contained globi (lepra cells), in addition to lymphocytes and many neutrophils with nuclear dust. Blood vessels in the superficial and deep dermis and in the subcutaneous fat showed fibrinoid necrosis in their walls with neutrophils infiltrating the walls and thrombi in the lumens (Figure 4). Modified Ziehl-Neelsen staining revealed clumps of acid-fast lepra bacilli inside vascular lumina and endothelial cell lining and within the foamy macrophages (Figure 5). Slit-skin smear examination was performed twice and yielded negative results. The slide and paraffin block of the already performed lymph node biopsy were retrieved. Examination revealed aggregates of foamy histiocytes surrounded by lymphocytes and plasma cells replacing normal lymphoid follicles. Modified Ziehl-Neelsen stain was performed, and clusters of acid-fast bacilli were detected within the foamy histiocytic infiltrate (Figure 6).
According to the results of the skin biopsy, the revised result of the lymph node biopsy, and the pattern of neurologic deficit together with clinical and laboratory correlation, the patient was diagnosed with diffuse nonnodular lepromatous leprosy presenting with Lucio phenomenon (Lucio leprosy) and associated with lepromatous lymphadenitis.
The patient received the following treatment: methylprednisolone 500 mg (intravenous pulse therapy) followed by daily oral administration of prednisolone 10 mg, rifampicin 300 mg, dapsone 100 mg, clofazimine 100 mg, acetylsalicylic acid 150 mg, and enoxaparin sodium 80 mg. In addition, the scrotal Fournier gangrene–like lesion was treated by surgical debridement followed by vacuum therapy. By the second week after treatment, the gangrenous lesions of the fingers developed a line of demarcation, and the skin infarctions started to recede.
Comment
Despite a decrease in its prevalence through a World Health Organization (WHO)–empowered eradication program, leprosy still represents a health problem in endemic areas.1,2 It is characterized by a wide range of immune responses to Mycobacterium leprae, displaying a spectrum of clinical and histopathologic manifestations that vary from the tuberculoid or paucibacillary pole with a strong cell-mediated immune response and fewer organisms to the lepromatous or multibacillary pole with weaker cell-mediated immune response and higher loads of organisms.3 In addition to its well-known cutaneous and neurologic manifestations, leprosy can present with a variety of manifestations, including constitutional symptoms, musculoskeletal manifestations, and serologic abnormalities; thus, leprosy can mimic rheumatoid arthritis, spondyloarthritis, and vasculitis—a pitfall that may result in misdiagnosis as a rheumatologic disorder.3-7
The chronic course of leprosy can be disrupted by acute, immunologically mediated reactions known as lepra reactions, of which there are 3 types.8 Type I lepra reactions are cell mediated and occur mainly in patients with borderline disease, often representing an upgrade toward the tuberculoid pole; less often they represent a downgrade reaction. Nerves become painful and swollen with possible loss of function, and skin lesions become edematous and tender; sometimes arthritis develops.9 Type II lepra reactions, also known as erythema nodosum leprosum (ENL), occur in borderline lepromatous and lepromatous patients with a high bacillary load. They are characterized by fever, body aches, tender cutaneous/subcutaneous nodules that may ulcerate, possible bullous lesions, painful nerve swellings, swollen joints, iritis, lymphadenitis, glomerulonephritis, epididymo-orchitis, and hepatic affection. Both immune-complex and delayed hypersensitivity reactions play a role in ENL.8,10 The third reaction is a rare aggressive type known as Lucio phenomenon or Lucio leprosy, which presents with irregular-shaped, angulated, or stellar necrotizing purpuric lesions (hemorrhagic infacrtions) developing mainly on the extremities. The lesions evolve into ulcers that heal with atrophic scarring.2,11 Lucio phenomenon develops as a result of thrombotic vascular occlusion secondary to massive invasion of vascular endothelial cells by lepra bacilli.2,11-14 Involvement of the scrotal skin, such as in our patient, is rare.
Lucio phenomenon mainly is seen in Mexico and Central America, and few cases have been documented in Cuba, South America, the United States, India, Polynesia, South Africa, and Southeast Asia.15-17 It specifically occurs in patients with untreated, diffuse, nonnodular lepromatous leprosy (pure and primitive diffuse lepromatous leprosy (DLL)/diffuse leprosy of Lucio and Latapí). This type of leprosy was first described by Lucio and Alvarado18 in 1852 as a distinct form of lepromatous leprosy characterized by widespread and dense infiltration of the whole skin by lepra bacilli without the typical nodular lesions of leprosy, rendering its diagnosis challenging, especially in sporadic cases. Other manifestations of DLL include complete alopecia of the eyebrows and eyelashes, destructive rhinitis, and areas of anhidrosis and dyesthesia.2
Latapí and Chévez-Zomora19 defined Lucio phenomenon in 1948 as a form of histopathologic vasculitis restricted to patients with DLL. Histopathologically, in addition to the infiltration of the skin with acid-fast bacilli–laden foamy histiocytes, lesions of Lucio phenomenon show features of necrotizing (leukocytoclastic) vasculitis with fibrinoid necrosis20 or vascular thrombi with minimal perivascular lymphocytic infiltrate and no evidence of vasculitis.11 Medium to large vessels in the deep dermis and subcutaneous tissue show infiltration of their walls with a large number of macrophages laden with acid-fast bacilli.11 Cases with histopathologic features mimicking antiphospholipid syndrome with endothelial cell proliferation, thrombosis, and mild mononuclear cell infiltrate also may be seen.20 In all cases, ischemic epidermal necrosis is seen, as well as acid-fast bacilli, both singly and in clusters (globi) within endothelial cells and inside blood vessel lumina.
Although Lucio phenomenon initially was thought to be immune-complex mediated like ENL, it has been suggested that the main trigger is thrombotic vascular occlusion secondary to massive invasion of the vascular endothelial cells by the lepra bacilli, resulting in necrosis.14 Bacterial lipopolysaccharides promote the release of IL-1 and tumor necrosis factor α, which in turn stimulate the production of prostaglandins, IL-6, and coagulation factor III, leading to vascular thrombosis and tissue necrosis.21,22 Moreover, antiphospholipid antibodies, which have been found to be induced in response to certain infectious agents in genetically predisposed individuals,23 have been reported in patients with leprosy, mainly in association with lepromatous leprosy. The reported prevalence of anticardiolipin antibodies ranged from 37% to 98%, whereas anti-β2-glycoprotein I antibodies ranged from 3% to 19%, and antiprothrombin antibodies ranged from 6% to 45%.24,25 Antiphospholipid antibodies have been reported to play a role in the pathogenesis of Lucio phenomenon.11,13,15,26 Our case supports this hypothesis with positive anticardiolipin antibodies, anti-β2 glycoprotein antibodies, and positive lupus anticoagulant.
In accordance with Curi et al,2 who reported 5 cases of DLL with Lucio phenomenon, our patient showed a similar presentation with positive inflammatory markers in association with a negative autoimmune profile (ANA, ANCA-C&P) and negative venereal disease research laboratory test. It is important to mention that a positive autoimmune profile (ANA, ANCA-C&P) can be present in leprotic patients, causing possible diagnostic confusion with collagen diseases.27,28
An interesting finding in our case was the negative slit-skin smear results. Although the specificity of slit-skin smear is 100%, as it directly demonstrates the presence of acid-fast bacilli,29 its sensitivity is low and varies from 10% to 50%.30 The detection of acid-fast bacilli in tissue sections is reported to be a better method for confirming the diagnosis of leprosy.31
The provisional impression of hemophagocytic lymphohistiocytosis in the lymph node biopsy in our patient was excluded upon detection of acid-fast bacilli in the foamy histiocytes infiltrating the lymph node; moreover, the normal serum lipids and serum ferritin argued against this diagnosis.32 Leprosy tends to involve the lymph nodes, particularly in borderline, borderline lepromatous, and lepromatous forms.33 The incidence of lymph node involvement accompanied by skin lesions with the presence of acid-fast bacilli in the lymph nodes is 92.2%.34
Our patient showed an excellent response to antileprotic treatment, which was administered according to the WHO multidrug therapy guidelines for multibacillary leprosy,35 combined with low-dose prednisolone, acetylsalicylic acid, and anticoagulant treatment. Thalidomide and high-dose prednisolone (60 mg/d) combined with antileprotic treatment also have been reported to be successful in managing recurrent infarctions in leprosy.36 The Fournier-like gangrenous ulcer of the scrotum was managed by surgical debridement and vacuum therapy.
It is noteworthy that the WHO elimination goal for leprosy was to reduce the prevalence to less than 1 case per 10,000 population. Egypt is among the first countries in North Africa and the Middle East regions to achieve this target supervised by the National Leprosy Control Program as early as 1994; this was further reduced to 0.33 cases per 10,000 population in 2004, and reduced again in 2009; however, certain foci showed a prevalence rate more than the elimination target, particularly in the cities of Qena (1.12) and Sohag (2.47).37 Esna, where our patient is from, is an endemic area in Egypt.38
Conclusion
Case Report
A 70-year-old man living in Esna, Luxor, Egypt presented to the Department of Rheumatology and Rehabilitation with widespread gangrenous skin lesions associated with ulcers of 2 weeks’ duration. One year prior, the patient had an insidious onset of nocturnal fever, bilateral leg edema, and numbness and a tingling sensation in both hands. He presented some laboratory and radiologic investigations that were performed at another hospital prior to the current presentation, which revealed thrombocytopenia, mild splenomegaly, and generalized lymphadenopathy. An excisional left axillary lymph node biopsy was performed at another hospital prior to the current presentation, and the pathology report provided by the patient described a reactive, foamy, histiocyte-rich lesion, suggesting a diagnosis of hemophagocytic lymphohistiocytosis. The patient had no diabetes or hypertension and no history of deep vein thrombosis, stroke, or unintentional weight loss. No medications were taken prior to the onset of the skin lesions, and his family history was irrelevant.
General examination at the current presentation revealed a fever (temperature, 101.3 °F [38.5 °C]), a normal heart rate (90 beats per minute), normal blood pressure (120/80 mmHg), normal respiratory rate (14 breaths per minute), accentuated heart sounds, and normal vesicular breathing without adventitious sounds. He had saddle nose, loss of the outer third of the eyebrows, and marked reduction in the density of the eyelashes (madarosis). Bilateral pitting edema of the legs also was present. Neurologic examination revealed hypoesthesia in a glove-and-stocking pattern, thickened peripheral nerves, and trophic changes over both hands; however, he had normal muscle power and deep reflexes. Joint examination revealed no abnormalities. Skin examination revealed widespread, reticulated, necrotizing, purpuric lesions on the arms, legs, abdomen, and ears, some associated with gangrenous ulcerations and hemorrhagic blisters. Scattered vasculitic ulcers and gangrenous patches were seen on the fingers. A gangrenous ulcer mimicking Fournier gangrene was seen involving the scrotal skin in addition to a gangrenous lesion on the glans penis (Figure 1–3). Unaffected skin appeared smooth, shiny, and edematous and showed no nodular lesions. Peripheral pulsations were intact.
Positive findings from a wide panel of laboratory investigations included an elevated erythrocyte sedimentation rate (103 mm for the first hour [reference range, 0–22 mm]), high C-reactive protein (50.7 mg/L [reference range, up to 6 mg/L]), anemia (hemoglobin count, 7.3 g/dL [reference range, 13.5–17.5 g/dL]), thrombocytopenia (45×103/mm3 [reference range, 150×103/mm3), low serum albumin (2.3 g/dL [reference range, 3.4–5.4 g/dL]), elevated IgG and IgM anticardiolipin antibodies (IgG, 21.4 IgG phospholipid [GPL] units [reference range, <10 IgG phospholipid (GPL) units]; IgM, 59.4 IgM phospholipid (MPL) units [reference range, <7 IgM phospholipid (MPL) units]), positive lupus anticoagulant panel test, elevated anti-β2 glycoprotein antibodies (IgG, 17.5
Nerve conduction velocity showed axonal sensory polyneuropathy. Motor nerve conduction studies for median and ulnar nerves were within normal range. Lower-limb nerves assessment was limited by the ulcerated areas and marked edema. Echocardiography was unremarkable. Arterial Doppler studies were only available for the upper limbs and were unremarkable.
A punch biopsy was taken from one of the necrotizing purpuric lesions on the legs, and histopathologic examination revealed foci of epidermal necrosis and subepidermal separation and superficial and deep perivascular and periadnexal infiltrates extending into the fat lobules. The infiltrates were mainly made up of foamy macrophages, and some contained globi (lepra cells), in addition to lymphocytes and many neutrophils with nuclear dust. Blood vessels in the superficial and deep dermis and in the subcutaneous fat showed fibrinoid necrosis in their walls with neutrophils infiltrating the walls and thrombi in the lumens (Figure 4). Modified Ziehl-Neelsen staining revealed clumps of acid-fast lepra bacilli inside vascular lumina and endothelial cell lining and within the foamy macrophages (Figure 5). Slit-skin smear examination was performed twice and yielded negative results. The slide and paraffin block of the already performed lymph node biopsy were retrieved. Examination revealed aggregates of foamy histiocytes surrounded by lymphocytes and plasma cells replacing normal lymphoid follicles. Modified Ziehl-Neelsen stain was performed, and clusters of acid-fast bacilli were detected within the foamy histiocytic infiltrate (Figure 6).
According to the results of the skin biopsy, the revised result of the lymph node biopsy, and the pattern of neurologic deficit together with clinical and laboratory correlation, the patient was diagnosed with diffuse nonnodular lepromatous leprosy presenting with Lucio phenomenon (Lucio leprosy) and associated with lepromatous lymphadenitis.
The patient received the following treatment: methylprednisolone 500 mg (intravenous pulse therapy) followed by daily oral administration of prednisolone 10 mg, rifampicin 300 mg, dapsone 100 mg, clofazimine 100 mg, acetylsalicylic acid 150 mg, and enoxaparin sodium 80 mg. In addition, the scrotal Fournier gangrene–like lesion was treated by surgical debridement followed by vacuum therapy. By the second week after treatment, the gangrenous lesions of the fingers developed a line of demarcation, and the skin infarctions started to recede.
Comment
Despite a decrease in its prevalence through a World Health Organization (WHO)–empowered eradication program, leprosy still represents a health problem in endemic areas.1,2 It is characterized by a wide range of immune responses to Mycobacterium leprae, displaying a spectrum of clinical and histopathologic manifestations that vary from the tuberculoid or paucibacillary pole with a strong cell-mediated immune response and fewer organisms to the lepromatous or multibacillary pole with weaker cell-mediated immune response and higher loads of organisms.3 In addition to its well-known cutaneous and neurologic manifestations, leprosy can present with a variety of manifestations, including constitutional symptoms, musculoskeletal manifestations, and serologic abnormalities; thus, leprosy can mimic rheumatoid arthritis, spondyloarthritis, and vasculitis—a pitfall that may result in misdiagnosis as a rheumatologic disorder.3-7
The chronic course of leprosy can be disrupted by acute, immunologically mediated reactions known as lepra reactions, of which there are 3 types.8 Type I lepra reactions are cell mediated and occur mainly in patients with borderline disease, often representing an upgrade toward the tuberculoid pole; less often they represent a downgrade reaction. Nerves become painful and swollen with possible loss of function, and skin lesions become edematous and tender; sometimes arthritis develops.9 Type II lepra reactions, also known as erythema nodosum leprosum (ENL), occur in borderline lepromatous and lepromatous patients with a high bacillary load. They are characterized by fever, body aches, tender cutaneous/subcutaneous nodules that may ulcerate, possible bullous lesions, painful nerve swellings, swollen joints, iritis, lymphadenitis, glomerulonephritis, epididymo-orchitis, and hepatic affection. Both immune-complex and delayed hypersensitivity reactions play a role in ENL.8,10 The third reaction is a rare aggressive type known as Lucio phenomenon or Lucio leprosy, which presents with irregular-shaped, angulated, or stellar necrotizing purpuric lesions (hemorrhagic infacrtions) developing mainly on the extremities. The lesions evolve into ulcers that heal with atrophic scarring.2,11 Lucio phenomenon develops as a result of thrombotic vascular occlusion secondary to massive invasion of vascular endothelial cells by lepra bacilli.2,11-14 Involvement of the scrotal skin, such as in our patient, is rare.
Lucio phenomenon mainly is seen in Mexico and Central America, and few cases have been documented in Cuba, South America, the United States, India, Polynesia, South Africa, and Southeast Asia.15-17 It specifically occurs in patients with untreated, diffuse, nonnodular lepromatous leprosy (pure and primitive diffuse lepromatous leprosy (DLL)/diffuse leprosy of Lucio and Latapí). This type of leprosy was first described by Lucio and Alvarado18 in 1852 as a distinct form of lepromatous leprosy characterized by widespread and dense infiltration of the whole skin by lepra bacilli without the typical nodular lesions of leprosy, rendering its diagnosis challenging, especially in sporadic cases. Other manifestations of DLL include complete alopecia of the eyebrows and eyelashes, destructive rhinitis, and areas of anhidrosis and dyesthesia.2
Latapí and Chévez-Zomora19 defined Lucio phenomenon in 1948 as a form of histopathologic vasculitis restricted to patients with DLL. Histopathologically, in addition to the infiltration of the skin with acid-fast bacilli–laden foamy histiocytes, lesions of Lucio phenomenon show features of necrotizing (leukocytoclastic) vasculitis with fibrinoid necrosis20 or vascular thrombi with minimal perivascular lymphocytic infiltrate and no evidence of vasculitis.11 Medium to large vessels in the deep dermis and subcutaneous tissue show infiltration of their walls with a large number of macrophages laden with acid-fast bacilli.11 Cases with histopathologic features mimicking antiphospholipid syndrome with endothelial cell proliferation, thrombosis, and mild mononuclear cell infiltrate also may be seen.20 In all cases, ischemic epidermal necrosis is seen, as well as acid-fast bacilli, both singly and in clusters (globi) within endothelial cells and inside blood vessel lumina.
Although Lucio phenomenon initially was thought to be immune-complex mediated like ENL, it has been suggested that the main trigger is thrombotic vascular occlusion secondary to massive invasion of the vascular endothelial cells by the lepra bacilli, resulting in necrosis.14 Bacterial lipopolysaccharides promote the release of IL-1 and tumor necrosis factor α, which in turn stimulate the production of prostaglandins, IL-6, and coagulation factor III, leading to vascular thrombosis and tissue necrosis.21,22 Moreover, antiphospholipid antibodies, which have been found to be induced in response to certain infectious agents in genetically predisposed individuals,23 have been reported in patients with leprosy, mainly in association with lepromatous leprosy. The reported prevalence of anticardiolipin antibodies ranged from 37% to 98%, whereas anti-β2-glycoprotein I antibodies ranged from 3% to 19%, and antiprothrombin antibodies ranged from 6% to 45%.24,25 Antiphospholipid antibodies have been reported to play a role in the pathogenesis of Lucio phenomenon.11,13,15,26 Our case supports this hypothesis with positive anticardiolipin antibodies, anti-β2 glycoprotein antibodies, and positive lupus anticoagulant.
In accordance with Curi et al,2 who reported 5 cases of DLL with Lucio phenomenon, our patient showed a similar presentation with positive inflammatory markers in association with a negative autoimmune profile (ANA, ANCA-C&P) and negative venereal disease research laboratory test. It is important to mention that a positive autoimmune profile (ANA, ANCA-C&P) can be present in leprotic patients, causing possible diagnostic confusion with collagen diseases.27,28
An interesting finding in our case was the negative slit-skin smear results. Although the specificity of slit-skin smear is 100%, as it directly demonstrates the presence of acid-fast bacilli,29 its sensitivity is low and varies from 10% to 50%.30 The detection of acid-fast bacilli in tissue sections is reported to be a better method for confirming the diagnosis of leprosy.31
The provisional impression of hemophagocytic lymphohistiocytosis in the lymph node biopsy in our patient was excluded upon detection of acid-fast bacilli in the foamy histiocytes infiltrating the lymph node; moreover, the normal serum lipids and serum ferritin argued against this diagnosis.32 Leprosy tends to involve the lymph nodes, particularly in borderline, borderline lepromatous, and lepromatous forms.33 The incidence of lymph node involvement accompanied by skin lesions with the presence of acid-fast bacilli in the lymph nodes is 92.2%.34
Our patient showed an excellent response to antileprotic treatment, which was administered according to the WHO multidrug therapy guidelines for multibacillary leprosy,35 combined with low-dose prednisolone, acetylsalicylic acid, and anticoagulant treatment. Thalidomide and high-dose prednisolone (60 mg/d) combined with antileprotic treatment also have been reported to be successful in managing recurrent infarctions in leprosy.36 The Fournier-like gangrenous ulcer of the scrotum was managed by surgical debridement and vacuum therapy.
It is noteworthy that the WHO elimination goal for leprosy was to reduce the prevalence to less than 1 case per 10,000 population. Egypt is among the first countries in North Africa and the Middle East regions to achieve this target supervised by the National Leprosy Control Program as early as 1994; this was further reduced to 0.33 cases per 10,000 population in 2004, and reduced again in 2009; however, certain foci showed a prevalence rate more than the elimination target, particularly in the cities of Qena (1.12) and Sohag (2.47).37 Esna, where our patient is from, is an endemic area in Egypt.38
Conclusion
1. World Health Organization. World Health Statistics: 2011. World Health Organization; 2011. https://www.who.int/gho/publications/world_health_statistics/EN_WHS2011_Full.pdf
2. Curi PF, Villaroel JS, Migliore N, et al. Lucio’s phenomenon: report of five cases. Clin Rheumatol. 2016;35:1397-1401.
3. Shrestha B, Li YQ, Fu P. Leprosy mimics adult onset Still’s disease in a Chinese patient. Egypt Rheumatol. 2018;40:217-220.
4. Prasad S, Misra R, Aggarwal A, et al. Leprosy revealed in a rheumatology clinic: a case series. Int J Rheum Dis. 2013;16:129-133.
5. Chao G, Fang L, Lu C. Leprosy with ANA positive mistaken for connective tissue disease. Clin Rheumatol. 2013;32:645-648.
6. Chauhan S, Wakhlu A, Agarwal V. Arthritis in leprosy. Rheumatology. 2010;49:2237-2242.
7. Rath D, Bhargava S, Kundu BK. Leprosy mimicking common rheumatologic entities: a trial for the clinician in the era of biologics. Case Rep Rheumatol. 2014;2014:429698.
8. Cuevas J, Rodríguez-Peralto JL, Carrillo R, et al. Erythema nodosum leprosum: reactional leprosy. Semin Cutan Med Surg. 2007;26:126-130.
9. Henriques CC, Lopéz B, Mestre T, et al. Leprosy and rheumatoid arthritis: consequence or association? BMJ Case Rep. 2012;13:1-4.
10. Vázquez-Botet M, Sánchez JL. Erythema nodosum leprosum. Int J Dermatol. 1987;26:436-437.
11. Nunzie E, Ortega Cabrera LV, Macanchi Moncayo FM, et al. Lucio leprosy with Lucio’s phenomenon, digital gangrene and anticardiolipin antibodies. Lepr Rev. 2014;85:194-200.
12. Salvi S, Chopra A. Leprosy in a rheumatology setting: a challenging mimic to expose. Clin Rheumatol. 2013;32:1557-1563.
13. Azulay-Abulafia L, Pereira SL, Hardmann D, et al. Lucio phenomenon. vasculitis or occlusive vasculopathy? Hautarzt. 2006;57:1101-1105.
14. Benard G, Sakai-Valente NY, Bianconcini Trindade MA. Concomittant Lucio phenomenon and erythema nodosum in a leprosy patient: clues for their distinct pathogenesis. Am J Dermatopathol. 2009;31:288-292.
15. Rocha RH, Emerich PS, Diniz LM, et al. Lucio’s phenomenon: exuberant case report and review of Brazilian cases. An Bras Dermatol. 2016;91(suppl 5):S60-S63.
16. Costa IM, Kawano LB, Pereira CP, et al. Lucio’s phenomenon: a case report and review of the literature. Int J Dermatol. 2005;44:566-571.
17. Kumari R, Thappa DM, Basu D. A fatal case of Lucio phenomenon from India. Dermatol Online J. 2008;14:10.
18. Lucio R, Alvarado I. Opúsculo Sobre el Mal de San Lázaro o Elefantiasis de los Griegos. M. Murguía; 1852.
19. Latapí F, Chévez-Zamora A. The “spotted” leprosy of Lucio: an introduction to its clinical and histological study. Int J Lepr. 1948;16:421-437.
20. Vargas OF. Diffuse leprosy of Lucio and Latapí: a histologic study. Lepr Rev. 2007;78:248-260.
21. Latapí FR, Chevez-Zamora A. La lepra manchada de Lucio. Rev Dermatol Mex. 1978;22:102-107.
22. Monteiro R, Abreu MA, Tiezzi MG, et al. Fenômeno de Lúcio: mais um caso relatado no Brasil. An Bras Dermatol. 2012;87:296-300.
23. Gharavi EE, Chaimovich H, Cucucrull E, et al. Induction of antiphospholipid antibodies by immunization with synthetic bacterial & viral peptides. Lupus. 1999;8:449-455.
24. de Larrañaga GF, Forastiero RR, Martinuzzo ME, et al. High prevalence of antiphospholipid antibodies in leprosy: evaluation of antigen reactivity. Lupus. 2000;9:594-600.
25. Loizou S, Singh S, Wypkema E, et al. Anticardiolipin, anti-beta(2)-glycoprotein I and antiprothrombin antibodies in black South African patients with infectious disease. Ann Rheum Dis. 2003;62:1106-1111.
26. Akerkar SM, Bichile LS. Leprosy & gangrene: a rare association; role of antiphospholipid antibodies. BMC Infect Dis. 2005,5:74.
27. Horta-Baas G, Hernández-Cabrera MF, Barile-Fabris LA, et al. Multibacillary leprosy mimicking systemic lupus erythematosus: case report and literature review. Lupus. 2015;24:1095-1102.
28. Pradhan V, Badakere SS, Shankar KU. Increased incidence of cytoplasmic ANCA (cANCA) and other auto antibodies in leprosy patients from western India. Lepr Rev. 2004;75:50-56.
29. Oskam L. Diagnosis and classification of leprosy. Lepr Rev. 2002;73:17-26.
30. Rao PN. Recent advances in the control programs and therapy of leprosy. Indian J Dermatol Venereol Leprol. 2004;70:269-276.
31. Rao PN, Pratap D, Ramana Reddy AV, et al. Evaluation of leprosy patients with 1 to 5 skin lesions with relevance to their grouping into paucibacillary or multibacillary disease. Indian J Dermatol Venereol Leprol. 2006;72:207-210.
32. Rosado FGN, Kim AS. Hemophagocytic lymphohistiocytosis. an update on diagnosis and pathogenesis. Am J Clin Pathol. 2013;139:713-727.
33. Kar HK, Mohanty HC, Mohanty GN, et al. Clinicopathological study of lymph node involvement in leprosy. Lepr India. 1983;55:725-738.
34. Gupta JC, Panda PK, Shrivastava KK, et al. A histopathologic study of lymph nodes in 43 cases of leprosy. Lepr India. 1978;50:196-203.
35. WHO Expert Committee on Leprosy. Seventh Report. World Health Organization; 1998. https://apps.who.int/iris/bitstream/handle/10665/42060/WHO_TRS_874.pdf?sequence=1&isAllowed=y
36. Misra DP, Parida JR, Chowdhury AC, et al. Lepra reaction with Lucio phenomenon mimicking cutaneous vasculitis. Case Rep Immunol. 2014;2014:641989.
37. Amer A, Mansour A. Epidemiological study of leprosy in Egypt: 2005-2009. Egypt J Dermatol Venereol. 2014;34:70-73.
38. World Health Organization. Screening campaign aims to eliminate leprosy in Egypt. Published May 9, 2018. Accessed September 8, 2021. http://www.emro.who.int/egy/egypt-events/last-miless-activities-on-eliminating-leprosy-from-egypt.html
1. World Health Organization. World Health Statistics: 2011. World Health Organization; 2011. https://www.who.int/gho/publications/world_health_statistics/EN_WHS2011_Full.pdf
2. Curi PF, Villaroel JS, Migliore N, et al. Lucio’s phenomenon: report of five cases. Clin Rheumatol. 2016;35:1397-1401.
3. Shrestha B, Li YQ, Fu P. Leprosy mimics adult onset Still’s disease in a Chinese patient. Egypt Rheumatol. 2018;40:217-220.
4. Prasad S, Misra R, Aggarwal A, et al. Leprosy revealed in a rheumatology clinic: a case series. Int J Rheum Dis. 2013;16:129-133.
5. Chao G, Fang L, Lu C. Leprosy with ANA positive mistaken for connective tissue disease. Clin Rheumatol. 2013;32:645-648.
6. Chauhan S, Wakhlu A, Agarwal V. Arthritis in leprosy. Rheumatology. 2010;49:2237-2242.
7. Rath D, Bhargava S, Kundu BK. Leprosy mimicking common rheumatologic entities: a trial for the clinician in the era of biologics. Case Rep Rheumatol. 2014;2014:429698.
8. Cuevas J, Rodríguez-Peralto JL, Carrillo R, et al. Erythema nodosum leprosum: reactional leprosy. Semin Cutan Med Surg. 2007;26:126-130.
9. Henriques CC, Lopéz B, Mestre T, et al. Leprosy and rheumatoid arthritis: consequence or association? BMJ Case Rep. 2012;13:1-4.
10. Vázquez-Botet M, Sánchez JL. Erythema nodosum leprosum. Int J Dermatol. 1987;26:436-437.
11. Nunzie E, Ortega Cabrera LV, Macanchi Moncayo FM, et al. Lucio leprosy with Lucio’s phenomenon, digital gangrene and anticardiolipin antibodies. Lepr Rev. 2014;85:194-200.
12. Salvi S, Chopra A. Leprosy in a rheumatology setting: a challenging mimic to expose. Clin Rheumatol. 2013;32:1557-1563.
13. Azulay-Abulafia L, Pereira SL, Hardmann D, et al. Lucio phenomenon. vasculitis or occlusive vasculopathy? Hautarzt. 2006;57:1101-1105.
14. Benard G, Sakai-Valente NY, Bianconcini Trindade MA. Concomittant Lucio phenomenon and erythema nodosum in a leprosy patient: clues for their distinct pathogenesis. Am J Dermatopathol. 2009;31:288-292.
15. Rocha RH, Emerich PS, Diniz LM, et al. Lucio’s phenomenon: exuberant case report and review of Brazilian cases. An Bras Dermatol. 2016;91(suppl 5):S60-S63.
16. Costa IM, Kawano LB, Pereira CP, et al. Lucio’s phenomenon: a case report and review of the literature. Int J Dermatol. 2005;44:566-571.
17. Kumari R, Thappa DM, Basu D. A fatal case of Lucio phenomenon from India. Dermatol Online J. 2008;14:10.
18. Lucio R, Alvarado I. Opúsculo Sobre el Mal de San Lázaro o Elefantiasis de los Griegos. M. Murguía; 1852.
19. Latapí F, Chévez-Zamora A. The “spotted” leprosy of Lucio: an introduction to its clinical and histological study. Int J Lepr. 1948;16:421-437.
20. Vargas OF. Diffuse leprosy of Lucio and Latapí: a histologic study. Lepr Rev. 2007;78:248-260.
21. Latapí FR, Chevez-Zamora A. La lepra manchada de Lucio. Rev Dermatol Mex. 1978;22:102-107.
22. Monteiro R, Abreu MA, Tiezzi MG, et al. Fenômeno de Lúcio: mais um caso relatado no Brasil. An Bras Dermatol. 2012;87:296-300.
23. Gharavi EE, Chaimovich H, Cucucrull E, et al. Induction of antiphospholipid antibodies by immunization with synthetic bacterial & viral peptides. Lupus. 1999;8:449-455.
24. de Larrañaga GF, Forastiero RR, Martinuzzo ME, et al. High prevalence of antiphospholipid antibodies in leprosy: evaluation of antigen reactivity. Lupus. 2000;9:594-600.
25. Loizou S, Singh S, Wypkema E, et al. Anticardiolipin, anti-beta(2)-glycoprotein I and antiprothrombin antibodies in black South African patients with infectious disease. Ann Rheum Dis. 2003;62:1106-1111.
26. Akerkar SM, Bichile LS. Leprosy & gangrene: a rare association; role of antiphospholipid antibodies. BMC Infect Dis. 2005,5:74.
27. Horta-Baas G, Hernández-Cabrera MF, Barile-Fabris LA, et al. Multibacillary leprosy mimicking systemic lupus erythematosus: case report and literature review. Lupus. 2015;24:1095-1102.
28. Pradhan V, Badakere SS, Shankar KU. Increased incidence of cytoplasmic ANCA (cANCA) and other auto antibodies in leprosy patients from western India. Lepr Rev. 2004;75:50-56.
29. Oskam L. Diagnosis and classification of leprosy. Lepr Rev. 2002;73:17-26.
30. Rao PN. Recent advances in the control programs and therapy of leprosy. Indian J Dermatol Venereol Leprol. 2004;70:269-276.
31. Rao PN, Pratap D, Ramana Reddy AV, et al. Evaluation of leprosy patients with 1 to 5 skin lesions with relevance to their grouping into paucibacillary or multibacillary disease. Indian J Dermatol Venereol Leprol. 2006;72:207-210.
32. Rosado FGN, Kim AS. Hemophagocytic lymphohistiocytosis. an update on diagnosis and pathogenesis. Am J Clin Pathol. 2013;139:713-727.
33. Kar HK, Mohanty HC, Mohanty GN, et al. Clinicopathological study of lymph node involvement in leprosy. Lepr India. 1983;55:725-738.
34. Gupta JC, Panda PK, Shrivastava KK, et al. A histopathologic study of lymph nodes in 43 cases of leprosy. Lepr India. 1978;50:196-203.
35. WHO Expert Committee on Leprosy. Seventh Report. World Health Organization; 1998. https://apps.who.int/iris/bitstream/handle/10665/42060/WHO_TRS_874.pdf?sequence=1&isAllowed=y
36. Misra DP, Parida JR, Chowdhury AC, et al. Lepra reaction with Lucio phenomenon mimicking cutaneous vasculitis. Case Rep Immunol. 2014;2014:641989.
37. Amer A, Mansour A. Epidemiological study of leprosy in Egypt: 2005-2009. Egypt J Dermatol Venereol. 2014;34:70-73.
38. World Health Organization. Screening campaign aims to eliminate leprosy in Egypt. Published May 9, 2018. Accessed September 8, 2021. http://www.emro.who.int/egy/egypt-events/last-miless-activities-on-eliminating-leprosy-from-egypt.html
Practice Points
- Leprosy is a great mimicker of many connective tissue diseases, including vasculitis.
- Antiphospholipid antibodies are involved in Lucio phenomenon.
- Prompt treatment is important in Lucio phenomenon to avoid morbidity and mortality.
The Role of Diagnostic Imaging in Macular Telangiectasia Type 2
While uncommon with subtle findings, macular telangiectasia type 2 can be diagnosed with careful retinal examination and selective use of diagnostic imaging.
Macular telangiectasia type 2 (MacTel2) is an uncommon, bilateral, and asymmetric condition that typically presents between the ages of 40 and 60 years without sex predilection.1-9 Its estimated prevalence ranges from 0.02 to 0.10%.2,8 The disease can manifest in either a nonproliferative or proliferative phase; the latter is far less common. The etiology of MacTel2 is poorly understood, but it is believed to have neurodegenerative as well as vascular components.1-6,8-10 We present a case of MacTel2 and highlight the role of diagnostic imaging in early diagnosis prior to development of classic funduscopic features.
Case Presentation
A 66-year-old White male with a 10-year history of type 2 diabetes mellitus (T2DM) presented to the eye clinic for an annual eye examination. The patient was taking metformin, and 6 months prior to presentation, his hemoglobin A1c was 7.4%. He had a history of mild nonproliferative diabetic retinopathy in the left eye without diabetic macular edema. He reported no ocular concerns.
On examination, best-corrected visual acuity (VA) was 20/20 in each eye. Slit-lamp examination was notable only for bilateral mild nuclear sclerosis. Dilated fundus examination showed a blunted foveal reflex consistent with the appearance of a macular pseudo-hole in the right eye and was unremarkable in the left eye (Figure 1).
Macular optical coherence tomography (OCT) revealed an intraretinal cyst without thickening in the temporal fovea of both eyes with mild disruption of the underlying ellipsoid zone in the right eye (Figure 2). A presumptive diagnosis of MacTel2 vs diabetic macular edema was made, and the patient was referred to the retina clinic for further evaluation.
At the 1-month follow-up in the retina clinic, VA, macula OCT, and fundus examination were stable. Fundus autofluorescence (FAF), optical coherence tomography angiography (OCT-A), and fluorescein angiography (FA) were performed. The FAF revealed a hyperreflective crescent in the temporal aspect of the fovea of both eyes, greater in the right eye than the left (Figure 3). The OCT-A showed abnormal dilation of the vessels in the deep capillary plexus of the temporal fovea of both eyes (Figure 4). This area of abnormality correlated to the area of hyperreflectivity seen on FAF. The early- phase FA revealed telangiectatic vessels in the temporal fovea in both eyes; in the late phase, there was leakage of telangiectatic vessels, which remained localized to the temporal perifovea and spared the central fovea of both eyes (Figure 5). The patient was diagnosed with MacTel2.
Discussion
This case highlights several important management considerations in MacTel2. These include symptoms, disease stage, and diagnostic imaging, which can allow more precise staging of the disease.
The etiology of MacTel2 is unknown.6 It is believed to be primarily a neurodegenerative condition that damages Müller cells and photoreceptors, leading to vascular changes.1-6,8-10 Müller cells may play a role in creating and maintaining the integrity of the blood-retinal barrier, particularly in the deep capillary plexus where the vascular abnormalities begin.6,10 These early changes in the deep capillary plexus may evolve to include the superficial capillary plexus in intermediate stages with anastomoses forming between the 2 layers.2,6-10 Late proliferative stages show significant alterations of the juxtafoveal capillary network, subretinal neovascularization and retinochoroidal anastomoses.6,7,9,11 In one cohort study, 81% of patients with MacTel2 were White, and a genetic link is still under investigation.2,4-9
Presentation
The most common symptoms of MacTel2 include blurred vision, microscotoma, metamorphopsia, and difficulty reading, with missing or distorted letters a common concern.1,2,4-8 Best-corrected VA at presentation is usually better than 20/30, and disease progression tends to be slow.2,6 Microscotomata are best mapped with microperimetry.1-3,5-7
There are several classic fundus findings (Table). In the early stages, these findings are subtle or entirely absent funduscopically.1,2,4-10 In intermediate stages, fundus findings become apparent and include a loss of retinal clarity (grayish perifoveal sheen), telangiectatic macular vessels, retinal pigment epithelium hypertrophy, blunted right-angled vessels, and superficial retinal crystalline deposits.2,4-11 Right-angled vessels may have a greater association with choroidal neovascularization, with growth into the outer retina in particular being a marker of disease progression.9 The crystalline deposits have been hypothesized to be the footplates of degenerated Müller cells.6

An important vision-threatening complication of MacTel2 is progression to proliferative disease.1,2,5-10 Choroidal neovascularization is present in a minority of cases and is associated with rapid vision loss.2,6 It is often accompanied by subretinal hemorrhage and lipid exudation.6,7,9 If untreated, the result can be disciform scarring and fibrosis.2,5,6 Additional complications of MacTel2 are foveal atrophy and full thickness macular holes.1,2,4-8, Macular holes secondary to MacTel2 respond poorly to pars plana vitrectomy with inner limiting membrane (ILM) peel.2,6
Diagnostic Testing
Diagnostic retinal imaging is invaluable in the diagnosis of MacTel2. The OCT can detect hyporeflectivity within the ellipsoid zone in early disease corresponding to ellipsoid zone loss, which increases as the disease progresses.1-8,10 This loss most often begins in the temporal parafoveal region and correlates with the progression of both relative and absolute scotomas perceived by affected individuals.2,3,5,8
Intraretinal foveal hyporeflective spaces on the OCT represent cavity formation after Müller cell and photoreceptor loss and do not correlate with increased thickness.1,2,4,6,7 This is important in differentiating from diabetic macular edema, which will often show thickening.6 In most cases of MacTel2, foveal thickness is decreased.4-6 The ILM remains intact overlying this space and is referred to as ILM drape.6,7 This can cause blunting or absence of the foveal light reflex and mimic the appearance of a macular pseudohole.4
The OCT-A allows visualization of capillary changes through every layer of the retina, which could not otherwise be appreciated, allowing early detection as well as precise staging of the disease.2,6-10 Anastomoses present in late-stage disease also can be imaged using OCT-A.7,9 These anastomoses can be seen as hyperreflective vasculature present between the retinal layers where there is little to no vasculature visible in normal eyes.7
A lesser-known occurrence in MacTel2 is the depletion of macular luteal pigment, with many eyes possessing an abnormal distribution.2,4,6-8,10 This depletion and abnormal distribution can be visualized with FAF. In particular, short wavelength fundus autofluorescence (SW-FAF) is the most effective at highlighting these changes.10 The characteristic finding is a hyperreflective halo surrounding the fovea.2,6 Fluorescence life imaging ophthalmoscopy (FLIO) is a recent development in FAF that measures FAF lifetime, which is the duration of time a structure autofluoresces.8 A cross-sectional study published in 2018 showed prolonged FAF lifetime in the temporal fovea of patients with early and moderate stage MacTel2 when compared with normal patients.8 More advanced stages showed a ring encircling the entire fovea.8
Classic FA findings in MacTel2 include early hyperfluorescence of temporal foveal telangiectatic capillaries and late-stage leakage with sparing of the central fovea.1,2,4,6,7,11
Management and Prognosis
Management of MacTel2 depends on the stage of the disease. In the absence of proven treatment, management in nonproliferative stages is conservative.2,6 Intravitreal anti-VEGF does not offer any benefit in nonproliferative disease.2,5,6 Indeed, as VEGF may have a neuroprotective effect on the retina, anti-VEGF may result in more harm than benefit in earlier disease stages.5 In proliferative stages, intravitreal anti-VEGF can help limit scarring and prevent vision loss.2,5
Long-term prognosis of MacTel2 is variable with VA typically better than 20/100.2 Vision loss in MacTel2 most often begins paracentrally; it can then progress centrally, leading to significant reduction in VA.12 The progression of this functional vision loss and corresponding structural damage is typically slow.3 VA worse than 20/100 is usually a result of proliferative disease; in such cases, there is potential for severe central vision loss and legal blindness.1
Conclusions
This case of MacTel2 underscores the subtle retinal findings in the earliest stages of the disease and the importance of a complete retinal examination and diagnostic imaging with macula OCT, OCT-A, and FAF to establish the correct diagnosis.
1. Chew EY, Clemons TE, Jaffe GJ, et al. Effect of ciliary neurotrophic factor on retinal neurodegeneration in patients with macular telangiectasia type 2: a randomized clinical trial. Ophthalmology. 2019;126(4):540-549. doi:10.1016/j.ophtha.2018.09.041
2. Christakis PG, Fine HF, Wiley HE. The diagnosis and management of macular telangiectasia. Ophthalmic Surg Lasers Imaging Retina. 2019;50(3):139-144. doi:10.3928/23258160-20190301-02
3. Heeren TFC, Kitka D, Florea D, et al. Longitudinal correlation of ellipsoid zone loss and functional loss in macular telangiectasia type 2. Retina. 2018;38 Suppl 1(suppl 1):S20-S26. doi:10.1097/IAE.0000000000001715
4. Charbel Issa P, Heeren TF, Kupitz EH, Holz FG, Berendschot TT. Very early disease manifestations of macular telangiectasia type 2. Retina. 2016;36(3):524-534. doi:10.1097/IAE.0000000000000863
5. Khodabande A, Roohipoor R, Zamani J, et al. Management of idiopathic macular telangiectasia type 2. Ophthalmol Ther. 2019;8(2):155-175. doi:10.1007/s40123-019-0170-1
6. Wu L. When is macular edema not macular edema? An update on macular telangiectasia type 2. Taiwan J Ophthalmol. 2015;5(4):149-155. doi:10.1016/j.tjo.2015.09.001
7. Roisman L, Rosenfeld PJ. Optical Coherence Tomography Angiography of Macular Telangiectasia Type 2. Dev Ophthalmol. 2016;56:146-158. doi:10.1159/000442807
8. Sauer L, Gensure RH, Hammer M, Bernstein PS. Fluorescence lifetime imaging ophthalmoscopy: a novel way to assess macular telangiectasia type 2. Ophthalmol Retina. 2018;2(6):587-598. doi:10.1016/j.oret.2017.10.008
9. Tzaridis S, Heeren T, Mai C, et al. Right-angled vessels in macular telangiectasia type 2. Br J Ophthalmol. 2021;105(9):1289-1296. doi:10.1136/bjophthalmol-2018-313364
10. Micevych PS, Lee HE, Fawzi AA. Overlap between telangiectasia and photoreceptor loss increases with progression of macular telangiectasia type 2. PLoS One. 2019;14(10):e0224393. Published 2019 Oct 28. doi:10.1371/journal.pone.0224393
11. Gass JD, Oyakawa RT. Idiopathic juxtafoveolar retinal telangiectasis. Arch Ophthalmol. 1982;100(5):769-780. doi:10.1001/archopht.1982.01030030773010
12. Heeren TF, Clemons T, Scholl HP, Bird AC, Holz FG, Charbel Issa P. Progression of vision loss in macular telangiectasia type 2. Invest Ophthalmol Vis Sci. 2015;56(6):3905-3912. doi:10.1167/iovs.15-16915
While uncommon with subtle findings, macular telangiectasia type 2 can be diagnosed with careful retinal examination and selective use of diagnostic imaging.
While uncommon with subtle findings, macular telangiectasia type 2 can be diagnosed with careful retinal examination and selective use of diagnostic imaging.
Macular telangiectasia type 2 (MacTel2) is an uncommon, bilateral, and asymmetric condition that typically presents between the ages of 40 and 60 years without sex predilection.1-9 Its estimated prevalence ranges from 0.02 to 0.10%.2,8 The disease can manifest in either a nonproliferative or proliferative phase; the latter is far less common. The etiology of MacTel2 is poorly understood, but it is believed to have neurodegenerative as well as vascular components.1-6,8-10 We present a case of MacTel2 and highlight the role of diagnostic imaging in early diagnosis prior to development of classic funduscopic features.
Case Presentation
A 66-year-old White male with a 10-year history of type 2 diabetes mellitus (T2DM) presented to the eye clinic for an annual eye examination. The patient was taking metformin, and 6 months prior to presentation, his hemoglobin A1c was 7.4%. He had a history of mild nonproliferative diabetic retinopathy in the left eye without diabetic macular edema. He reported no ocular concerns.
On examination, best-corrected visual acuity (VA) was 20/20 in each eye. Slit-lamp examination was notable only for bilateral mild nuclear sclerosis. Dilated fundus examination showed a blunted foveal reflex consistent with the appearance of a macular pseudo-hole in the right eye and was unremarkable in the left eye (Figure 1).
Macular optical coherence tomography (OCT) revealed an intraretinal cyst without thickening in the temporal fovea of both eyes with mild disruption of the underlying ellipsoid zone in the right eye (Figure 2). A presumptive diagnosis of MacTel2 vs diabetic macular edema was made, and the patient was referred to the retina clinic for further evaluation.
At the 1-month follow-up in the retina clinic, VA, macula OCT, and fundus examination were stable. Fundus autofluorescence (FAF), optical coherence tomography angiography (OCT-A), and fluorescein angiography (FA) were performed. The FAF revealed a hyperreflective crescent in the temporal aspect of the fovea of both eyes, greater in the right eye than the left (Figure 3). The OCT-A showed abnormal dilation of the vessels in the deep capillary plexus of the temporal fovea of both eyes (Figure 4). This area of abnormality correlated to the area of hyperreflectivity seen on FAF. The early- phase FA revealed telangiectatic vessels in the temporal fovea in both eyes; in the late phase, there was leakage of telangiectatic vessels, which remained localized to the temporal perifovea and spared the central fovea of both eyes (Figure 5). The patient was diagnosed with MacTel2.
Discussion
This case highlights several important management considerations in MacTel2. These include symptoms, disease stage, and diagnostic imaging, which can allow more precise staging of the disease.
The etiology of MacTel2 is unknown.6 It is believed to be primarily a neurodegenerative condition that damages Müller cells and photoreceptors, leading to vascular changes.1-6,8-10 Müller cells may play a role in creating and maintaining the integrity of the blood-retinal barrier, particularly in the deep capillary plexus where the vascular abnormalities begin.6,10 These early changes in the deep capillary plexus may evolve to include the superficial capillary plexus in intermediate stages with anastomoses forming between the 2 layers.2,6-10 Late proliferative stages show significant alterations of the juxtafoveal capillary network, subretinal neovascularization and retinochoroidal anastomoses.6,7,9,11 In one cohort study, 81% of patients with MacTel2 were White, and a genetic link is still under investigation.2,4-9
Presentation
The most common symptoms of MacTel2 include blurred vision, microscotoma, metamorphopsia, and difficulty reading, with missing or distorted letters a common concern.1,2,4-8 Best-corrected VA at presentation is usually better than 20/30, and disease progression tends to be slow.2,6 Microscotomata are best mapped with microperimetry.1-3,5-7
There are several classic fundus findings (Table). In the early stages, these findings are subtle or entirely absent funduscopically.1,2,4-10 In intermediate stages, fundus findings become apparent and include a loss of retinal clarity (grayish perifoveal sheen), telangiectatic macular vessels, retinal pigment epithelium hypertrophy, blunted right-angled vessels, and superficial retinal crystalline deposits.2,4-11 Right-angled vessels may have a greater association with choroidal neovascularization, with growth into the outer retina in particular being a marker of disease progression.9 The crystalline deposits have been hypothesized to be the footplates of degenerated Müller cells.6
An important vision-threatening complication of MacTel2 is progression to proliferative disease.1,2,5-10 Choroidal neovascularization is present in a minority of cases and is associated with rapid vision loss.2,6 It is often accompanied by subretinal hemorrhage and lipid exudation.6,7,9 If untreated, the result can be disciform scarring and fibrosis.2,5,6 Additional complications of MacTel2 are foveal atrophy and full thickness macular holes.1,2,4-8, Macular holes secondary to MacTel2 respond poorly to pars plana vitrectomy with inner limiting membrane (ILM) peel.2,6
Diagnostic Testing
Diagnostic retinal imaging is invaluable in the diagnosis of MacTel2. The OCT can detect hyporeflectivity within the ellipsoid zone in early disease corresponding to ellipsoid zone loss, which increases as the disease progresses.1-8,10 This loss most often begins in the temporal parafoveal region and correlates with the progression of both relative and absolute scotomas perceived by affected individuals.2,3,5,8
Intraretinal foveal hyporeflective spaces on the OCT represent cavity formation after Müller cell and photoreceptor loss and do not correlate with increased thickness.1,2,4,6,7 This is important in differentiating from diabetic macular edema, which will often show thickening.6 In most cases of MacTel2, foveal thickness is decreased.4-6 The ILM remains intact overlying this space and is referred to as ILM drape.6,7 This can cause blunting or absence of the foveal light reflex and mimic the appearance of a macular pseudohole.4
The OCT-A allows visualization of capillary changes through every layer of the retina, which could not otherwise be appreciated, allowing early detection as well as precise staging of the disease.2,6-10 Anastomoses present in late-stage disease also can be imaged using OCT-A.7,9 These anastomoses can be seen as hyperreflective vasculature present between the retinal layers where there is little to no vasculature visible in normal eyes.7
A lesser-known occurrence in MacTel2 is the depletion of macular luteal pigment, with many eyes possessing an abnormal distribution.2,4,6-8,10 This depletion and abnormal distribution can be visualized with FAF. In particular, short wavelength fundus autofluorescence (SW-FAF) is the most effective at highlighting these changes.10 The characteristic finding is a hyperreflective halo surrounding the fovea.2,6 Fluorescence life imaging ophthalmoscopy (FLIO) is a recent development in FAF that measures FAF lifetime, which is the duration of time a structure autofluoresces.8 A cross-sectional study published in 2018 showed prolonged FAF lifetime in the temporal fovea of patients with early and moderate stage MacTel2 when compared with normal patients.8 More advanced stages showed a ring encircling the entire fovea.8
Classic FA findings in MacTel2 include early hyperfluorescence of temporal foveal telangiectatic capillaries and late-stage leakage with sparing of the central fovea.1,2,4,6,7,11
Management and Prognosis
Management of MacTel2 depends on the stage of the disease. In the absence of proven treatment, management in nonproliferative stages is conservative.2,6 Intravitreal anti-VEGF does not offer any benefit in nonproliferative disease.2,5,6 Indeed, as VEGF may have a neuroprotective effect on the retina, anti-VEGF may result in more harm than benefit in earlier disease stages.5 In proliferative stages, intravitreal anti-VEGF can help limit scarring and prevent vision loss.2,5
Long-term prognosis of MacTel2 is variable with VA typically better than 20/100.2 Vision loss in MacTel2 most often begins paracentrally; it can then progress centrally, leading to significant reduction in VA.12 The progression of this functional vision loss and corresponding structural damage is typically slow.3 VA worse than 20/100 is usually a result of proliferative disease; in such cases, there is potential for severe central vision loss and legal blindness.1
Conclusions
This case of MacTel2 underscores the subtle retinal findings in the earliest stages of the disease and the importance of a complete retinal examination and diagnostic imaging with macula OCT, OCT-A, and FAF to establish the correct diagnosis.
Macular telangiectasia type 2 (MacTel2) is an uncommon, bilateral, and asymmetric condition that typically presents between the ages of 40 and 60 years without sex predilection.1-9 Its estimated prevalence ranges from 0.02 to 0.10%.2,8 The disease can manifest in either a nonproliferative or proliferative phase; the latter is far less common. The etiology of MacTel2 is poorly understood, but it is believed to have neurodegenerative as well as vascular components.1-6,8-10 We present a case of MacTel2 and highlight the role of diagnostic imaging in early diagnosis prior to development of classic funduscopic features.
Case Presentation
A 66-year-old White male with a 10-year history of type 2 diabetes mellitus (T2DM) presented to the eye clinic for an annual eye examination. The patient was taking metformin, and 6 months prior to presentation, his hemoglobin A1c was 7.4%. He had a history of mild nonproliferative diabetic retinopathy in the left eye without diabetic macular edema. He reported no ocular concerns.
On examination, best-corrected visual acuity (VA) was 20/20 in each eye. Slit-lamp examination was notable only for bilateral mild nuclear sclerosis. Dilated fundus examination showed a blunted foveal reflex consistent with the appearance of a macular pseudo-hole in the right eye and was unremarkable in the left eye (Figure 1).
Macular optical coherence tomography (OCT) revealed an intraretinal cyst without thickening in the temporal fovea of both eyes with mild disruption of the underlying ellipsoid zone in the right eye (Figure 2). A presumptive diagnosis of MacTel2 vs diabetic macular edema was made, and the patient was referred to the retina clinic for further evaluation.
At the 1-month follow-up in the retina clinic, VA, macula OCT, and fundus examination were stable. Fundus autofluorescence (FAF), optical coherence tomography angiography (OCT-A), and fluorescein angiography (FA) were performed. The FAF revealed a hyperreflective crescent in the temporal aspect of the fovea of both eyes, greater in the right eye than the left (Figure 3). The OCT-A showed abnormal dilation of the vessels in the deep capillary plexus of the temporal fovea of both eyes (Figure 4). This area of abnormality correlated to the area of hyperreflectivity seen on FAF. The early- phase FA revealed telangiectatic vessels in the temporal fovea in both eyes; in the late phase, there was leakage of telangiectatic vessels, which remained localized to the temporal perifovea and spared the central fovea of both eyes (Figure 5). The patient was diagnosed with MacTel2.
Discussion
This case highlights several important management considerations in MacTel2. These include symptoms, disease stage, and diagnostic imaging, which can allow more precise staging of the disease.
The etiology of MacTel2 is unknown.6 It is believed to be primarily a neurodegenerative condition that damages Müller cells and photoreceptors, leading to vascular changes.1-6,8-10 Müller cells may play a role in creating and maintaining the integrity of the blood-retinal barrier, particularly in the deep capillary plexus where the vascular abnormalities begin.6,10 These early changes in the deep capillary plexus may evolve to include the superficial capillary plexus in intermediate stages with anastomoses forming between the 2 layers.2,6-10 Late proliferative stages show significant alterations of the juxtafoveal capillary network, subretinal neovascularization and retinochoroidal anastomoses.6,7,9,11 In one cohort study, 81% of patients with MacTel2 were White, and a genetic link is still under investigation.2,4-9
Presentation
The most common symptoms of MacTel2 include blurred vision, microscotoma, metamorphopsia, and difficulty reading, with missing or distorted letters a common concern.1,2,4-8 Best-corrected VA at presentation is usually better than 20/30, and disease progression tends to be slow.2,6 Microscotomata are best mapped with microperimetry.1-3,5-7
There are several classic fundus findings (Table). In the early stages, these findings are subtle or entirely absent funduscopically.1,2,4-10 In intermediate stages, fundus findings become apparent and include a loss of retinal clarity (grayish perifoveal sheen), telangiectatic macular vessels, retinal pigment epithelium hypertrophy, blunted right-angled vessels, and superficial retinal crystalline deposits.2,4-11 Right-angled vessels may have a greater association with choroidal neovascularization, with growth into the outer retina in particular being a marker of disease progression.9 The crystalline deposits have been hypothesized to be the footplates of degenerated Müller cells.6
An important vision-threatening complication of MacTel2 is progression to proliferative disease.1,2,5-10 Choroidal neovascularization is present in a minority of cases and is associated with rapid vision loss.2,6 It is often accompanied by subretinal hemorrhage and lipid exudation.6,7,9 If untreated, the result can be disciform scarring and fibrosis.2,5,6 Additional complications of MacTel2 are foveal atrophy and full thickness macular holes.1,2,4-8, Macular holes secondary to MacTel2 respond poorly to pars plana vitrectomy with inner limiting membrane (ILM) peel.2,6
Diagnostic Testing
Diagnostic retinal imaging is invaluable in the diagnosis of MacTel2. The OCT can detect hyporeflectivity within the ellipsoid zone in early disease corresponding to ellipsoid zone loss, which increases as the disease progresses.1-8,10 This loss most often begins in the temporal parafoveal region and correlates with the progression of both relative and absolute scotomas perceived by affected individuals.2,3,5,8
Intraretinal foveal hyporeflective spaces on the OCT represent cavity formation after Müller cell and photoreceptor loss and do not correlate with increased thickness.1,2,4,6,7 This is important in differentiating from diabetic macular edema, which will often show thickening.6 In most cases of MacTel2, foveal thickness is decreased.4-6 The ILM remains intact overlying this space and is referred to as ILM drape.6,7 This can cause blunting or absence of the foveal light reflex and mimic the appearance of a macular pseudohole.4
The OCT-A allows visualization of capillary changes through every layer of the retina, which could not otherwise be appreciated, allowing early detection as well as precise staging of the disease.2,6-10 Anastomoses present in late-stage disease also can be imaged using OCT-A.7,9 These anastomoses can be seen as hyperreflective vasculature present between the retinal layers where there is little to no vasculature visible in normal eyes.7
A lesser-known occurrence in MacTel2 is the depletion of macular luteal pigment, with many eyes possessing an abnormal distribution.2,4,6-8,10 This depletion and abnormal distribution can be visualized with FAF. In particular, short wavelength fundus autofluorescence (SW-FAF) is the most effective at highlighting these changes.10 The characteristic finding is a hyperreflective halo surrounding the fovea.2,6 Fluorescence life imaging ophthalmoscopy (FLIO) is a recent development in FAF that measures FAF lifetime, which is the duration of time a structure autofluoresces.8 A cross-sectional study published in 2018 showed prolonged FAF lifetime in the temporal fovea of patients with early and moderate stage MacTel2 when compared with normal patients.8 More advanced stages showed a ring encircling the entire fovea.8
Classic FA findings in MacTel2 include early hyperfluorescence of temporal foveal telangiectatic capillaries and late-stage leakage with sparing of the central fovea.1,2,4,6,7,11
Management and Prognosis
Management of MacTel2 depends on the stage of the disease. In the absence of proven treatment, management in nonproliferative stages is conservative.2,6 Intravitreal anti-VEGF does not offer any benefit in nonproliferative disease.2,5,6 Indeed, as VEGF may have a neuroprotective effect on the retina, anti-VEGF may result in more harm than benefit in earlier disease stages.5 In proliferative stages, intravitreal anti-VEGF can help limit scarring and prevent vision loss.2,5
Long-term prognosis of MacTel2 is variable with VA typically better than 20/100.2 Vision loss in MacTel2 most often begins paracentrally; it can then progress centrally, leading to significant reduction in VA.12 The progression of this functional vision loss and corresponding structural damage is typically slow.3 VA worse than 20/100 is usually a result of proliferative disease; in such cases, there is potential for severe central vision loss and legal blindness.1
Conclusions
This case of MacTel2 underscores the subtle retinal findings in the earliest stages of the disease and the importance of a complete retinal examination and diagnostic imaging with macula OCT, OCT-A, and FAF to establish the correct diagnosis.
1. Chew EY, Clemons TE, Jaffe GJ, et al. Effect of ciliary neurotrophic factor on retinal neurodegeneration in patients with macular telangiectasia type 2: a randomized clinical trial. Ophthalmology. 2019;126(4):540-549. doi:10.1016/j.ophtha.2018.09.041
2. Christakis PG, Fine HF, Wiley HE. The diagnosis and management of macular telangiectasia. Ophthalmic Surg Lasers Imaging Retina. 2019;50(3):139-144. doi:10.3928/23258160-20190301-02
3. Heeren TFC, Kitka D, Florea D, et al. Longitudinal correlation of ellipsoid zone loss and functional loss in macular telangiectasia type 2. Retina. 2018;38 Suppl 1(suppl 1):S20-S26. doi:10.1097/IAE.0000000000001715
4. Charbel Issa P, Heeren TF, Kupitz EH, Holz FG, Berendschot TT. Very early disease manifestations of macular telangiectasia type 2. Retina. 2016;36(3):524-534. doi:10.1097/IAE.0000000000000863
5. Khodabande A, Roohipoor R, Zamani J, et al. Management of idiopathic macular telangiectasia type 2. Ophthalmol Ther. 2019;8(2):155-175. doi:10.1007/s40123-019-0170-1
6. Wu L. When is macular edema not macular edema? An update on macular telangiectasia type 2. Taiwan J Ophthalmol. 2015;5(4):149-155. doi:10.1016/j.tjo.2015.09.001
7. Roisman L, Rosenfeld PJ. Optical Coherence Tomography Angiography of Macular Telangiectasia Type 2. Dev Ophthalmol. 2016;56:146-158. doi:10.1159/000442807
8. Sauer L, Gensure RH, Hammer M, Bernstein PS. Fluorescence lifetime imaging ophthalmoscopy: a novel way to assess macular telangiectasia type 2. Ophthalmol Retina. 2018;2(6):587-598. doi:10.1016/j.oret.2017.10.008
9. Tzaridis S, Heeren T, Mai C, et al. Right-angled vessels in macular telangiectasia type 2. Br J Ophthalmol. 2021;105(9):1289-1296. doi:10.1136/bjophthalmol-2018-313364
10. Micevych PS, Lee HE, Fawzi AA. Overlap between telangiectasia and photoreceptor loss increases with progression of macular telangiectasia type 2. PLoS One. 2019;14(10):e0224393. Published 2019 Oct 28. doi:10.1371/journal.pone.0224393
11. Gass JD, Oyakawa RT. Idiopathic juxtafoveolar retinal telangiectasis. Arch Ophthalmol. 1982;100(5):769-780. doi:10.1001/archopht.1982.01030030773010
12. Heeren TF, Clemons T, Scholl HP, Bird AC, Holz FG, Charbel Issa P. Progression of vision loss in macular telangiectasia type 2. Invest Ophthalmol Vis Sci. 2015;56(6):3905-3912. doi:10.1167/iovs.15-16915
1. Chew EY, Clemons TE, Jaffe GJ, et al. Effect of ciliary neurotrophic factor on retinal neurodegeneration in patients with macular telangiectasia type 2: a randomized clinical trial. Ophthalmology. 2019;126(4):540-549. doi:10.1016/j.ophtha.2018.09.041
2. Christakis PG, Fine HF, Wiley HE. The diagnosis and management of macular telangiectasia. Ophthalmic Surg Lasers Imaging Retina. 2019;50(3):139-144. doi:10.3928/23258160-20190301-02
3. Heeren TFC, Kitka D, Florea D, et al. Longitudinal correlation of ellipsoid zone loss and functional loss in macular telangiectasia type 2. Retina. 2018;38 Suppl 1(suppl 1):S20-S26. doi:10.1097/IAE.0000000000001715
4. Charbel Issa P, Heeren TF, Kupitz EH, Holz FG, Berendschot TT. Very early disease manifestations of macular telangiectasia type 2. Retina. 2016;36(3):524-534. doi:10.1097/IAE.0000000000000863
5. Khodabande A, Roohipoor R, Zamani J, et al. Management of idiopathic macular telangiectasia type 2. Ophthalmol Ther. 2019;8(2):155-175. doi:10.1007/s40123-019-0170-1
6. Wu L. When is macular edema not macular edema? An update on macular telangiectasia type 2. Taiwan J Ophthalmol. 2015;5(4):149-155. doi:10.1016/j.tjo.2015.09.001
7. Roisman L, Rosenfeld PJ. Optical Coherence Tomography Angiography of Macular Telangiectasia Type 2. Dev Ophthalmol. 2016;56:146-158. doi:10.1159/000442807
8. Sauer L, Gensure RH, Hammer M, Bernstein PS. Fluorescence lifetime imaging ophthalmoscopy: a novel way to assess macular telangiectasia type 2. Ophthalmol Retina. 2018;2(6):587-598. doi:10.1016/j.oret.2017.10.008
9. Tzaridis S, Heeren T, Mai C, et al. Right-angled vessels in macular telangiectasia type 2. Br J Ophthalmol. 2021;105(9):1289-1296. doi:10.1136/bjophthalmol-2018-313364
10. Micevych PS, Lee HE, Fawzi AA. Overlap between telangiectasia and photoreceptor loss increases with progression of macular telangiectasia type 2. PLoS One. 2019;14(10):e0224393. Published 2019 Oct 28. doi:10.1371/journal.pone.0224393
11. Gass JD, Oyakawa RT. Idiopathic juxtafoveolar retinal telangiectasis. Arch Ophthalmol. 1982;100(5):769-780. doi:10.1001/archopht.1982.01030030773010
12. Heeren TF, Clemons T, Scholl HP, Bird AC, Holz FG, Charbel Issa P. Progression of vision loss in macular telangiectasia type 2. Invest Ophthalmol Vis Sci. 2015;56(6):3905-3912. doi:10.1167/iovs.15-16915
Cisplatin-Induced Acute Kidney Injury and Renal Salt Wasting Syndrome
A treatment strategy that incorporates both water restrictions and sodium supplementation may be appropriate when differentiating between diagnoses of renal salt wasting syndrome and syndrome of inappropriate antidiuretic hormone secretion.
Cisplatin is a potent antineoplastic agent derived from platinum and commonly used in the treatment of head and neck, bladder, ovarian, and testicular malignancies.1,2 Approximately 20% of all cancer patients are prescribed platinum-based chemotherapeutics.3 Although considered highly effective, cisplatin is also a dose-dependent nephrotoxin, inducing apoptosis in the proximal tubules of the nephron and reducing glomerular filtration rate. This nephron injury leads to inflammation and reduced medullary blood flow, causing further ischemic damage to the tubular cells.4 Given that the proximal tubule reabsorbs 67% of all sodium, cisplatin-induced nephron injuries can also lead to hyponatremia.5
The primary mechanisms of hyponatremia following cisplatin chemotherapy are syndrome of inappropriate antidiuretic hormone secretion (SIADH) and renal salt wasting syndrome (RSWS). Though these diagnoses have similar presentations, the treatment recommendations are different due to pathophysiologic differences. Fluid restriction is the hallmark of SIADH treatment, while increased sodium intake remains the hallmark of RSWS treatment.6 This patient presented with a combination of cisplatin-induced acute kidney injury (AKI) and hyponatremia secondary to RSWS. While RSWS and AKI are known complications of cisplatin chemotherapy, the combination is underreported in the literature. Therefore, this case report highlights the combination of these cisplatin-induced complications, emphasizes the clinical challenges in differentiating SIADH from RSWS, especially in the presence of a concomitant AKI, and suggests a treatment approach during diagnostic uncertainty.
Case Presentation
A 71-year-old man with a medical history of squamous cell carcinoma (SCC) of the left neck on cycle 1, day 8 of cisplatin-based chemotherapy and ongoing radiation therapy (720 cGy of 6300 cGy), lung adenocarcinoma status postresection, and hyperlipidemia presented to the emergency department (ED) at the request of his oncologist for abnormal laboratory values. In the ED, his metabolic panel showed a 131-mmol/L serum sodium, 3.3 mmol/L potassium, 83 mmol/L chloride, 29 mmol/L bicarbonate, 61 mg/dL blood urea nitrogen (BUN), and 8.8 mg/dL creatinine (baseline, 0.9 mg/dL). The patient reported throbbing headaches, persistent nausea, and multiple episodes of nonbloody emesis for several days that he attributed to his chemotherapy. He noted decreased urination without discomfort or changes in color or odor and no fatigue, fevers, chills, hematuria, flank, abdominal pain, thirst, or polydipsia. He reported no toxic ingestions or IV drug use. The patient had no relevant family history or additional social history. His outpatient medications included 10 mg cetirizine, 8 mg ondansetron, and 81 mg aspirin. On initial examination, his 137/66 mm Hg blood pressure was mildly elevated. The physical examination findings were notable for a 5-cm mass in the left neck that was firm and irregularly-shaped. His physical examination was otherwise unremarkable. He was admitted to the inpatient medicine service for an AKI complicated by symptomatic hyponatremia.
Investigations
We evaluated the patient’s AKI based on treatment responsiveness, imaging, and laboratory testing. Renal and bladder ultrasound showed no evidence of hydronephrosis or obstruction. He had a benign urinalysis with microscopy absent for protein, blood, ketones, leukocyte esterase, nitrites, and cellular casts. His urine pH was 5.5 (reference range, 5.0-9.0) and specific gravity was 1.011 (reference range, 1.005-1.030). His urine electrolytes revealed 45-mmol/L urine sodium (reference range, 40-220), 33-mmol/L urine chloride (reference range, 110-250), 10-mmol/L urine potassium (reference range, 25-120), 106.7-mg/dL urine creatinine (reference range, 10-400) and a calculated 2.7% fractional excretion of sodium (FENa) and 22.0-mEq/L elevated urine anion gap. As a fluid challenge, he was treated with IV 0.9% sodium chloride at 100-125 mL/h, receiving 3 liters over the first 48 hours of his hospitalization. His creatinine peaked at 9.2 mg/dL and stabilized before improving later in his hospitalization (Figure 1). The patient initially had oliguria (< 0.5 mL/kg/h), which slowly improved over his hospital course. Unfortunately, due to multiple system and clinical factors, accurate inputs and outputs were not adequately maintained during his hospitalization.
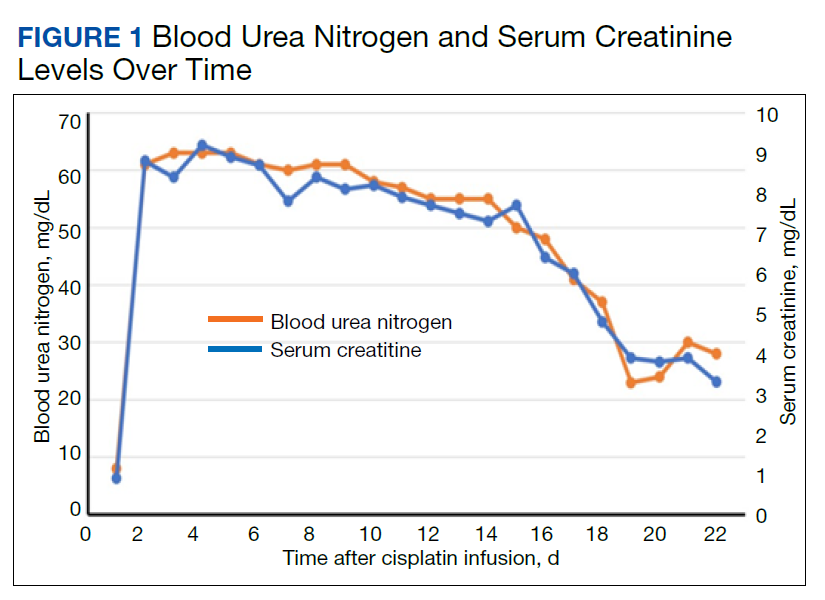
We evaluated hyponatremia with a combination of serum and urine laboratory tests. In addition to urine electrolytes, the initial evaluation focused on trending his clinical trajectory. We repeated a basic metabolic panel every 4 to 6 hours. He had 278-mOsm/kg serum osmolality (reference range, 285-295) with an effective 217-mOsm/kg serum tonicity. His urine osmolality was 270.5 mOsm/kg.
Despite administering 462 mEq sodium via crystalloid, his sodium worsened over the first 48 hours, reaching a nadir at 125 mmol/L on hospital day 3 (Figure 2). While he continued to appear euvolemic on physical examination, his blood pressure became difficult to control with 160- to 180-mm Hg systolic blood pressure readings. His thyroid stimulating hormone (TSH) was normal and aldosterone was low (4 ng/dL). Additional urine studies, including a 24-hour urine sample, were collected for further evaluation. His urine uric acid was 140 mg/d (reference range, 120-820); his serum uric acid level was 8.2 mg/dL (reference range, 3.0-9.0). His 24-hour urine creatinine was 0.57 g/d (reference range, 0.50-2.15) and uric acid to creatinine ratio was 246 mg/g (reference range, 60-580). His serum creatinine collected from the same day as his 24-hour urine sample was 7.3 mg/dL. His fractional excretion of uric acid (FEurate) was 21.9%.
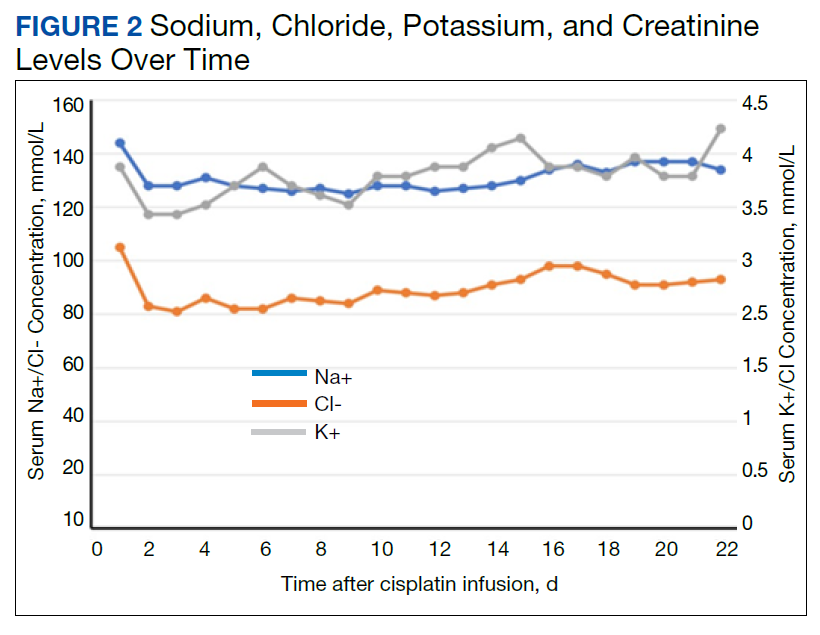
Differential Diagnosis
The patient’s recent administration of cisplatin raised clinical suspicion of cisplatin-induced AKI. To avoid premature diagnostic closure, we used a systematic approach for thinking about our patient’s AKI, considering prerenal, intrarenal, and postrenal etiologies. The unremarkable renal and bladder ultrasound made a postrenal etiology unlikely. The patient’s 2.7% FENa in the absence of a diuretic, limited responsiveness to crystalloid fluid resuscitation, 7.5 serum BUN/creatinine ratio, and 270.5 mOsm/kg urine osmolality suggested an intrarenal etiology, which can be further divided into problems with glomeruli, tubules, small vessels, or interstitial space. The patient’s normal urinary microscopy with no evidence of protein, blood, ketones, leukocyte esterase, nitrites, or cellular casts made a glomerular etiology less likely. The acute onset and lack of additional systemic features, other than hypertension, made a vascular etiology less likely. A tubular etiology, such as acute tubular necrosis (ATN), was highest on the differential and was followed by an interstitial etiology, such as acute interstitial nephritis (AIN).
Patients with drug-induced AIN commonly present with signs and symptoms of an allergic-type reaction, including fever, rash, hematuria, pyuria, and costovertebral angle tenderness. The patient lacked these symptoms. However, cisplatin is known to cause ATN in up to 20-30% of patients.7 Therefore, despite the lack of the classic muddy-brown, granular casts on urine microscopy, cisplatin-induced ATN remained the most likely etiology of his AKI. Moreover, ATN can cause hyponatremia. ATN is characterized by 3 phases: initiation, maintenance, and recovery phases.8 Hyponatremia occurs during the recovery phase, typically starting weeks after renal insult and associated with high urine output and diuresis. This patient presented 1 week after injury and had persistent oliguria, making ATN an unlikely culprit of his hyponatremia.
Our patient presented with hypotonic hyponatremia with a 131 mmol/L initial sodium level and an < 280 mOsm/kg effective serum osmolality, or serum tonicity. The serum tonicity is equivalent to the difference between the measured serum osmolality and the BUN. In the setting of profound AKI, this adjustment is essential for correctly categorizing a patient’s hyponatremia as hyper-, iso-, or hypotonic. The differential diagnosis for this patient’s hypotonic hyponatremia included dilutional effects of hypervolemia, SIADH, hyperthyroidism, adrenal insufficiency, and RSWS. The patient’s volume examination, lack of predisposing comorbidities or suggestive biomarkers, and > 20 mmol/L urinary sodium made hypervolemia unlikely. His urinary osmolality and specific gravity made primary polydipsia unlikely. We worked up his hyponatremia according to a diagnostic algorithm (eAppendix available at doi:10.12788/fp.0198).
The patient had a 217 mOsm/kg serum tonicity and a 270.5 mOsm/kg urine osmolality, consistent with impaired water excretion. His presentation, TSH, and concordant decrease in sodium and potassium made an endocrine etiology of his hyponatremia less likely. In hindsight, a serum cortisol would have been beneficial to more completely exclude adrenal insufficiency. His urine sodium was elevated at 45 mmol/L, raising concern for RSWS or SIADH. The FEurate helped to distinguish between SIADH and RSWS. While FEurate is often elevated in both SIADH and RSWS initially, the FEurate normalizes in SIADH with normalization of the serum sodium. The ideal cutoff for posthyponatremia correction FEurate is debated; however, a FEurate value after sodium correction < 11% suggests SIADH while a value > 11% suggests RSWS.9 Our patient’s FEurate following the sodium correction (serum sodium 134 mmol/L) was 21.9%, most suggestive of RSWS.
Treatment
Upon admission, initial treatment focused on resolving the patient’s AKI. The oncology team discontinued the cisplatin-based chemotherapy. His medication dosages were adjusted for his renal function and additional nephrotoxins avoided. In consultation, the nephrology service recommended 100 mL/h fluid resuscitation. After the patient received 3 L of 0.9% sodium chloride, his creatinine showed limited improvement and his sodium worsened, trending from 131 mmol/L to a nadir of 125 mmol/L. We initiated oral free-water restriction while continuing IV infusion of 0.9% sodium chloride at 125 mL/h.
We further augmented his sodium intake with 1-g sodium chloride tablets with each meal. By hospital day 6, the patient’s serum sodium, BUN, and creatinine improved to 130 mEq/L, 50 mg/dL, and 7.7 mg/dL, respectively. We then discontinued the oral sodium chloride tablets, fluid restriction, and IV fluids in a stepwise fashion prior to discharge. At discharge, the patient’s serum sodium was 136 mEq/L and creatinine, 4.8 mg/dL. The patient’s clinical course was complicated by symptomatic hypertension with systolic blood pressures about 180 mm Hg, requiring intermittent IV hydralazine, which was transitioned to daily nifedipine. Concerned that fluid resuscitation contributed to his hypertension, the patient also received several doses of furosemide. At time of discharge, the patient remained hypertensive and was discharged with nifedipine 90 mg daily.
Outcome and Follow-up
The patient has remained stable clinically since discharge. One week after discharge, his serum sodium and creatinine were 138 mmol/L and 3.8 mg/dL, respectively. More than 1 month after discharge, his sodium remains in the reference range and his creatinine was stable at about 3.5 mg/dL. He continues to follow-up with nephrology, oncology, and radiation oncology. He has restarted chemotherapy with a carboplatin-based regimen without recurrence of hyponatremia or AKI. His blood pressure has gradually improved to the point where he no longer requires nifedipine.
Discussion
The US Food and Drug Administration first approved the use of cisplatin, an alkylating agent that inhibits DNA replication, in 1978 for the treatment of testicular cancer.10 Since its approval, cisplatin has increased in popularity and is now considered one of the most effective antineoplastic agents for the treatment of solid tumors.1 Unfortunately, cisplatin has a well-documented adverse effect profile that includes neurotoxicity, gastrointestinal toxicity, nephrotoxicity, and ototoxicity.4 Despite frequent nephrotoxicity, cisplatin only occasionally causes hyponatremia and rarely causes RSWS, a known but potentially fatal complication. Moreover, the combination of AKI and RSWS is unique. Our patient presented with the unique combination of AKI and hyponatremia, most consistent with RSWS, likely precipitated from cisplatin chemotherapy. Through this case, we review cisplatin-associated electrolyte abnormalities, highlight the challenge of differentiating SIADH and RSWS, and suggest a treatment approach for hyponatremia during the period of diagnostic uncertainty.
Blachley and colleagues first discussed renal and electrolyte disturbances, specifically magnesium wasting, secondary to cisplatin use in 1981. In 1984, Kurtzberg and colleagues noted salt wasting in 2 patients receiving cisplatin therapy. The authors suggested that cisplatin inhibits solute transport in the thick ascending limb, causing clinically significant electrolyte abnormalities, coining the term cisplatin-induced salt wasting.11
The prevalence of cisplatin-induced salt wasting is unclear and likely underreported. In 1988, Hutchinson and colleagues conducted a prospective cohort study and noted 10% of patients (n = 70) developed RSWS at some point over 18 months of cisplatin therapy—a higher rate than previously estimated.12 In 1992, another prospective cohort study evaluated the adverse effects of 47 patients with non-small cell lung cancer treated with cisplatin and reported hyponatremia in 43% of its 93 courses of chemotherapy. The authors did not report the etiology of these hyponatremia cases.13 Given the diagnostic challenge, RSWS may be underrepresented as a confirmed etiology of hyponatremia in cisplatin treatment.
Hyponatremia from cisplatin may present as either SIADH or RSWS, complicating treatment decisions. Both conditions lead to hypotonic hyponatremia with urine osmolality > 100 mOSm/kg and urine sodium levels > 40 mmol/L. However, pathophysiology behind SIADH and RSWS is different. In RSWS, proximal tubule damage causes hyponatremia, decreasing sodium reabsorption, and leading to impaired concentration gradient in every segment of the nephron. As a result, RSWS can lead to profound hyponatremia. Treatment typically consists of increasing sodium intake to correct serum sodium with salt tablets and hypertonic sodium chloride while treating the underlying etiology, in our case removing the offending agent, and waiting for proximal tubule function to recover.6 On the other hand, in SIADH, elevated antidiuretic hormone (ADH) increases water reabsorption in the collecting duct, which has no impact on concentration gradients of the other nephron segments.14 Free-water restriction is the hallmark of SIADH treatment. Severe SIADH may require sodium repletion and/or the initiation of vaptans, ADH antagonists that competitively inhibit V2 receptors in the collecting duct to prevent water reabsorption.15
Our patient had an uncertain etiology of his hyponatremia throughout most of his treatment course, complicating our treatment decision-making. Initially, his measured serum osmolality was 278 mOsm/kg; however, his effective tonicity was lower. His AKI elevated his BUN, which in turnrequired us to calculate his serum tonicity (217 mOsm/kg) that was consistent with hypotonic hyponatremia. His elevated urine osmolality and urine sodium levels made SIADH and RSWS the most likely etiologies of his hyponatremia. To confirm the etiology, we waited for correction of his serum sodium. Therefore, we treated him with a combination of sodium repletion with 0.9% sodium chloride (154 mEq/L), hypertonic relative to his serum sodium, sodium chloride tablets, and free-water restriction. In this approach, we attempted to harmonize the treatment strategies for both SIADH and RSWS and effectively corrected his serum sodium. We evaluated his response to our treatment with a basic metabolic panel every 6 to 8 hours. Had his serum sodium decreased < 120 mmol/L, we planned to transfer the patient to the intensive care unit for 3% sodium chloride and/or intensification of his fluid restriction. A significant worsening of his hyponatremia would have strongly suggested hyponatremia secondary to SIADH since isotonic saline can worsen hyponatremia due to increased free-water reabsorption in the collecting duct.16
To differentiate between SIADH and RSWS, we relied on the FEurate after sodium correction. Multiple case reports from Japan have characterized the distinction between the processes through FEurate and serum uric acid. While the optimal cut-off values for FEurate require additional investigation, values < 11% after serum sodium correction suggests SIADH, while a value > 11% suggests RSWS.17 Prior cases have also emphasized serum hypouricemia as a distinguishing characteristic in RSWS. However, our case illustrates that serum hypouricemia is less reliable in the setting of AKI. Due to his severe AKI, our patient could not efficiently clear uric acid, likely contributing to his hyperuricemia.
Ultimately, our patient had an FEurate > 20%, which was suggestive of RSWS. Nevertheless, we recognize limitations and confounders in our diagnosis and have reflected on our diagnostic and management choices. First, the sensitivity and specificity of postsodium correction FEurate is unknown. Tracking the change in FEurate with our interventions would have increased our diagnostic utility, as suggested by Maesaka and colleagues.14 Second, our patient’s serum sodium was still at the lower end of the reference range after treatment, which may decrease the specificity of FEurate. Third, a plasma ADH collected during the initial phase of symptomatic hyponatremia would have helped differentiate between SIADH and RSWS.
Other diagnostic tests that could have excluded alternative diagnoses with even greater certainty include plasma adrenocorticotropic hormone, B-type natriuretic peptide, renin, cortisol, and thyroid function tests. From a practical standpoint, these laboratory results (excluding thyroid function test and brain natriuretic peptide) would have taken several weeks to result at our institution, limiting their clinical utility. Similarly, FEurate also has limited clinical utility, requiring effective treatment as part of the diagnostic test. Therefore, we recommend focusing on optimal treatment for hyponatremia of uncertain etiology, especially where SIADH and RSWS are the leading diagnoses.
Conclusions
We described a rare case of concomitant cisplatin-induced severe AKI and RSWS. We have emphasized the diagnostic challenge of distinguishing between SIADH and RSWS, especially with concomitant AKI, and have acknowledged that optimal treatment relies on accurate differentiation. However, differentiation may not be clinically feasible. Therefore, we suggest a treatment strategy that incorporates both free-water restriction and sodium supplementation via IV and/or oral administration.
1. Dasari S, Tchounwou PB. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol. 2014;740:364-378. doi:10.1016/j.ejphar.2014.07.025
2. Holditch SJ, Brown CN, Lombardi AM, Nguyen KN, Edelstein CL. Recent advances in models, mechanisms, biomarkers, and interventions in cisplatin-induced acute kidney injury. Int J Mol Sci. 2019;20(12):3011. Published 2019 Jun 20. doi:10.3390/ijms20123011
3. National Institutes of Health, National Cancer Institute. The “accidental” cure—platinum-based treatment for cancer: the discovery of cisplatin. Published May 30, 2014. Accessed November 10, 2021. https://www.cancer.gov/research/progress/discovery/cisplatin
4. Ozkok A, Edelstein CL. Pathophysiology of cisplatin-induced acute kidney injury. Biomed Res Int. 2014;2014:967826. doi:10.1155/2014/967826
5. Palmer LG, Schnermann J. Integrated control of Na transport along the nephron. Clin J Am Soc Nephrol. 2015;10(4):676-687. doi:10.2215/CJN.12391213
6. Bitew S, Imbriano L, Miyawaki N, Fishbane S, Maesaka JK. More on renal salt wasting without cerebral disease: response to saline infusion. Clin J Am Soc Nephrol. 2009;4(2):309-315. doi:10.2215/CJN.02740608
7. Shirali AC, Perazella MA. Tubulointerstitial injury associated with chemotherapeutic agents. Adv Chronic Kidney Dis. 2014;21(1):56-63. doi:10.1053/j.ackd.2013.06.010
8. Agrawal M, Swartz R. Acute renal failure [published correction appears in Am Fam Physician 2001 Feb 1;63(3):445]. Am Fam Physician. 2000;61(7):2077-2088.
9. Milionis HJ, Liamis GL, Elisaf MS. The hyponatremic patient: a systematic approach to laboratory diagnosis. CMAJ. 2002;166(8):1056-1062.
10. Monneret C. Platinum anticancer drugs. From serendipity to rational design. Ann Pharm Fr. 2011;69(6):286-295. doi:10.1016/j.pharma.2011.10.001
11. Kurtzberg J, Dennis VW, Kinney TR. Cisplatinum-induced renal salt wasting. Med Pediatr Oncol. 1984;12(2):150-154. doi:10.1002/mpo.2950120219
12. Hutchison FN, Perez EA, Gandara DR, Lawrence HJ, Kaysen GA. Renal salt wasting in patients treated with cisplatin. Ann Intern Med. 1988;108(1):21-25. doi:10.7326/0003-4819-108-1-21
13. Lee YK, Shin DM. Renal salt wasting in patients treated with high-dose cisplatin, etoposide, and mitomycin in patients with advanced non-small cell lung cancer. Korean J Intern Med. 1992;7(2):118-121. doi:10.3904/kjim.1992.7.2.118
14. Maesaka JK, Imbriano L, Mattana J, Gallagher D, Bade N, Sharif S. Differentiating SIADH from cerebral/renal salt wasting: failure of the volume approach and need for a new approach to hyponatremia. J Clin Med. 2014;3(4):1373-1385. Published 2014 Dec 8. doi:10.3390/jcm3041373
15. Palmer BF. The role of v2 receptor antagonists in the treatment of hyponatremia. Electrolyte Blood Press. 2013;11(1):1-8. doi:10.5049/EBP.2013.11.1.1
16. Verbalis JG, Goldsmith SR, Greenberg A, Schrier RW, Sterns RH. Hyponatremia treatment guidelines 2007: expert panel recommendations. Am J Med. 2007;120(11 Suppl 1):S1-S21. doi:10.1016/j.amjmed.2007.09.001
17. Maesaka JK, Imbriano LJ, Miyawaki N. High prevalence of renal salt wasting without cerebral disease as cause of hyponatremia in general medical wards. Am J Med Sci. 2018;356(1):15-22. doi:10.1016/j.amjms.2018.03.02
A treatment strategy that incorporates both water restrictions and sodium supplementation may be appropriate when differentiating between diagnoses of renal salt wasting syndrome and syndrome of inappropriate antidiuretic hormone secretion.
A treatment strategy that incorporates both water restrictions and sodium supplementation may be appropriate when differentiating between diagnoses of renal salt wasting syndrome and syndrome of inappropriate antidiuretic hormone secretion.
Cisplatin is a potent antineoplastic agent derived from platinum and commonly used in the treatment of head and neck, bladder, ovarian, and testicular malignancies.1,2 Approximately 20% of all cancer patients are prescribed platinum-based chemotherapeutics.3 Although considered highly effective, cisplatin is also a dose-dependent nephrotoxin, inducing apoptosis in the proximal tubules of the nephron and reducing glomerular filtration rate. This nephron injury leads to inflammation and reduced medullary blood flow, causing further ischemic damage to the tubular cells.4 Given that the proximal tubule reabsorbs 67% of all sodium, cisplatin-induced nephron injuries can also lead to hyponatremia.5
The primary mechanisms of hyponatremia following cisplatin chemotherapy are syndrome of inappropriate antidiuretic hormone secretion (SIADH) and renal salt wasting syndrome (RSWS). Though these diagnoses have similar presentations, the treatment recommendations are different due to pathophysiologic differences. Fluid restriction is the hallmark of SIADH treatment, while increased sodium intake remains the hallmark of RSWS treatment.6 This patient presented with a combination of cisplatin-induced acute kidney injury (AKI) and hyponatremia secondary to RSWS. While RSWS and AKI are known complications of cisplatin chemotherapy, the combination is underreported in the literature. Therefore, this case report highlights the combination of these cisplatin-induced complications, emphasizes the clinical challenges in differentiating SIADH from RSWS, especially in the presence of a concomitant AKI, and suggests a treatment approach during diagnostic uncertainty.
Case Presentation
A 71-year-old man with a medical history of squamous cell carcinoma (SCC) of the left neck on cycle 1, day 8 of cisplatin-based chemotherapy and ongoing radiation therapy (720 cGy of 6300 cGy), lung adenocarcinoma status postresection, and hyperlipidemia presented to the emergency department (ED) at the request of his oncologist for abnormal laboratory values. In the ED, his metabolic panel showed a 131-mmol/L serum sodium, 3.3 mmol/L potassium, 83 mmol/L chloride, 29 mmol/L bicarbonate, 61 mg/dL blood urea nitrogen (BUN), and 8.8 mg/dL creatinine (baseline, 0.9 mg/dL). The patient reported throbbing headaches, persistent nausea, and multiple episodes of nonbloody emesis for several days that he attributed to his chemotherapy. He noted decreased urination without discomfort or changes in color or odor and no fatigue, fevers, chills, hematuria, flank, abdominal pain, thirst, or polydipsia. He reported no toxic ingestions or IV drug use. The patient had no relevant family history or additional social history. His outpatient medications included 10 mg cetirizine, 8 mg ondansetron, and 81 mg aspirin. On initial examination, his 137/66 mm Hg blood pressure was mildly elevated. The physical examination findings were notable for a 5-cm mass in the left neck that was firm and irregularly-shaped. His physical examination was otherwise unremarkable. He was admitted to the inpatient medicine service for an AKI complicated by symptomatic hyponatremia.
Investigations
We evaluated the patient’s AKI based on treatment responsiveness, imaging, and laboratory testing. Renal and bladder ultrasound showed no evidence of hydronephrosis or obstruction. He had a benign urinalysis with microscopy absent for protein, blood, ketones, leukocyte esterase, nitrites, and cellular casts. His urine pH was 5.5 (reference range, 5.0-9.0) and specific gravity was 1.011 (reference range, 1.005-1.030). His urine electrolytes revealed 45-mmol/L urine sodium (reference range, 40-220), 33-mmol/L urine chloride (reference range, 110-250), 10-mmol/L urine potassium (reference range, 25-120), 106.7-mg/dL urine creatinine (reference range, 10-400) and a calculated 2.7% fractional excretion of sodium (FENa) and 22.0-mEq/L elevated urine anion gap. As a fluid challenge, he was treated with IV 0.9% sodium chloride at 100-125 mL/h, receiving 3 liters over the first 48 hours of his hospitalization. His creatinine peaked at 9.2 mg/dL and stabilized before improving later in his hospitalization (Figure 1). The patient initially had oliguria (< 0.5 mL/kg/h), which slowly improved over his hospital course. Unfortunately, due to multiple system and clinical factors, accurate inputs and outputs were not adequately maintained during his hospitalization.
We evaluated hyponatremia with a combination of serum and urine laboratory tests. In addition to urine electrolytes, the initial evaluation focused on trending his clinical trajectory. We repeated a basic metabolic panel every 4 to 6 hours. He had 278-mOsm/kg serum osmolality (reference range, 285-295) with an effective 217-mOsm/kg serum tonicity. His urine osmolality was 270.5 mOsm/kg.
Despite administering 462 mEq sodium via crystalloid, his sodium worsened over the first 48 hours, reaching a nadir at 125 mmol/L on hospital day 3 (Figure 2). While he continued to appear euvolemic on physical examination, his blood pressure became difficult to control with 160- to 180-mm Hg systolic blood pressure readings. His thyroid stimulating hormone (TSH) was normal and aldosterone was low (4 ng/dL). Additional urine studies, including a 24-hour urine sample, were collected for further evaluation. His urine uric acid was 140 mg/d (reference range, 120-820); his serum uric acid level was 8.2 mg/dL (reference range, 3.0-9.0). His 24-hour urine creatinine was 0.57 g/d (reference range, 0.50-2.15) and uric acid to creatinine ratio was 246 mg/g (reference range, 60-580). His serum creatinine collected from the same day as his 24-hour urine sample was 7.3 mg/dL. His fractional excretion of uric acid (FEurate) was 21.9%.
Differential Diagnosis
The patient’s recent administration of cisplatin raised clinical suspicion of cisplatin-induced AKI. To avoid premature diagnostic closure, we used a systematic approach for thinking about our patient’s AKI, considering prerenal, intrarenal, and postrenal etiologies. The unremarkable renal and bladder ultrasound made a postrenal etiology unlikely. The patient’s 2.7% FENa in the absence of a diuretic, limited responsiveness to crystalloid fluid resuscitation, 7.5 serum BUN/creatinine ratio, and 270.5 mOsm/kg urine osmolality suggested an intrarenal etiology, which can be further divided into problems with glomeruli, tubules, small vessels, or interstitial space. The patient’s normal urinary microscopy with no evidence of protein, blood, ketones, leukocyte esterase, nitrites, or cellular casts made a glomerular etiology less likely. The acute onset and lack of additional systemic features, other than hypertension, made a vascular etiology less likely. A tubular etiology, such as acute tubular necrosis (ATN), was highest on the differential and was followed by an interstitial etiology, such as acute interstitial nephritis (AIN).
Patients with drug-induced AIN commonly present with signs and symptoms of an allergic-type reaction, including fever, rash, hematuria, pyuria, and costovertebral angle tenderness. The patient lacked these symptoms. However, cisplatin is known to cause ATN in up to 20-30% of patients.7 Therefore, despite the lack of the classic muddy-brown, granular casts on urine microscopy, cisplatin-induced ATN remained the most likely etiology of his AKI. Moreover, ATN can cause hyponatremia. ATN is characterized by 3 phases: initiation, maintenance, and recovery phases.8 Hyponatremia occurs during the recovery phase, typically starting weeks after renal insult and associated with high urine output and diuresis. This patient presented 1 week after injury and had persistent oliguria, making ATN an unlikely culprit of his hyponatremia.
Our patient presented with hypotonic hyponatremia with a 131 mmol/L initial sodium level and an < 280 mOsm/kg effective serum osmolality, or serum tonicity. The serum tonicity is equivalent to the difference between the measured serum osmolality and the BUN. In the setting of profound AKI, this adjustment is essential for correctly categorizing a patient’s hyponatremia as hyper-, iso-, or hypotonic. The differential diagnosis for this patient’s hypotonic hyponatremia included dilutional effects of hypervolemia, SIADH, hyperthyroidism, adrenal insufficiency, and RSWS. The patient’s volume examination, lack of predisposing comorbidities or suggestive biomarkers, and > 20 mmol/L urinary sodium made hypervolemia unlikely. His urinary osmolality and specific gravity made primary polydipsia unlikely. We worked up his hyponatremia according to a diagnostic algorithm (eAppendix available at doi:10.12788/fp.0198).
The patient had a 217 mOsm/kg serum tonicity and a 270.5 mOsm/kg urine osmolality, consistent with impaired water excretion. His presentation, TSH, and concordant decrease in sodium and potassium made an endocrine etiology of his hyponatremia less likely. In hindsight, a serum cortisol would have been beneficial to more completely exclude adrenal insufficiency. His urine sodium was elevated at 45 mmol/L, raising concern for RSWS or SIADH. The FEurate helped to distinguish between SIADH and RSWS. While FEurate is often elevated in both SIADH and RSWS initially, the FEurate normalizes in SIADH with normalization of the serum sodium. The ideal cutoff for posthyponatremia correction FEurate is debated; however, a FEurate value after sodium correction < 11% suggests SIADH while a value > 11% suggests RSWS.9 Our patient’s FEurate following the sodium correction (serum sodium 134 mmol/L) was 21.9%, most suggestive of RSWS.
Treatment
Upon admission, initial treatment focused on resolving the patient’s AKI. The oncology team discontinued the cisplatin-based chemotherapy. His medication dosages were adjusted for his renal function and additional nephrotoxins avoided. In consultation, the nephrology service recommended 100 mL/h fluid resuscitation. After the patient received 3 L of 0.9% sodium chloride, his creatinine showed limited improvement and his sodium worsened, trending from 131 mmol/L to a nadir of 125 mmol/L. We initiated oral free-water restriction while continuing IV infusion of 0.9% sodium chloride at 125 mL/h.
We further augmented his sodium intake with 1-g sodium chloride tablets with each meal. By hospital day 6, the patient’s serum sodium, BUN, and creatinine improved to 130 mEq/L, 50 mg/dL, and 7.7 mg/dL, respectively. We then discontinued the oral sodium chloride tablets, fluid restriction, and IV fluids in a stepwise fashion prior to discharge. At discharge, the patient’s serum sodium was 136 mEq/L and creatinine, 4.8 mg/dL. The patient’s clinical course was complicated by symptomatic hypertension with systolic blood pressures about 180 mm Hg, requiring intermittent IV hydralazine, which was transitioned to daily nifedipine. Concerned that fluid resuscitation contributed to his hypertension, the patient also received several doses of furosemide. At time of discharge, the patient remained hypertensive and was discharged with nifedipine 90 mg daily.
Outcome and Follow-up
The patient has remained stable clinically since discharge. One week after discharge, his serum sodium and creatinine were 138 mmol/L and 3.8 mg/dL, respectively. More than 1 month after discharge, his sodium remains in the reference range and his creatinine was stable at about 3.5 mg/dL. He continues to follow-up with nephrology, oncology, and radiation oncology. He has restarted chemotherapy with a carboplatin-based regimen without recurrence of hyponatremia or AKI. His blood pressure has gradually improved to the point where he no longer requires nifedipine.
Discussion
The US Food and Drug Administration first approved the use of cisplatin, an alkylating agent that inhibits DNA replication, in 1978 for the treatment of testicular cancer.10 Since its approval, cisplatin has increased in popularity and is now considered one of the most effective antineoplastic agents for the treatment of solid tumors.1 Unfortunately, cisplatin has a well-documented adverse effect profile that includes neurotoxicity, gastrointestinal toxicity, nephrotoxicity, and ototoxicity.4 Despite frequent nephrotoxicity, cisplatin only occasionally causes hyponatremia and rarely causes RSWS, a known but potentially fatal complication. Moreover, the combination of AKI and RSWS is unique. Our patient presented with the unique combination of AKI and hyponatremia, most consistent with RSWS, likely precipitated from cisplatin chemotherapy. Through this case, we review cisplatin-associated electrolyte abnormalities, highlight the challenge of differentiating SIADH and RSWS, and suggest a treatment approach for hyponatremia during the period of diagnostic uncertainty.
Blachley and colleagues first discussed renal and electrolyte disturbances, specifically magnesium wasting, secondary to cisplatin use in 1981. In 1984, Kurtzberg and colleagues noted salt wasting in 2 patients receiving cisplatin therapy. The authors suggested that cisplatin inhibits solute transport in the thick ascending limb, causing clinically significant electrolyte abnormalities, coining the term cisplatin-induced salt wasting.11
The prevalence of cisplatin-induced salt wasting is unclear and likely underreported. In 1988, Hutchinson and colleagues conducted a prospective cohort study and noted 10% of patients (n = 70) developed RSWS at some point over 18 months of cisplatin therapy—a higher rate than previously estimated.12 In 1992, another prospective cohort study evaluated the adverse effects of 47 patients with non-small cell lung cancer treated with cisplatin and reported hyponatremia in 43% of its 93 courses of chemotherapy. The authors did not report the etiology of these hyponatremia cases.13 Given the diagnostic challenge, RSWS may be underrepresented as a confirmed etiology of hyponatremia in cisplatin treatment.
Hyponatremia from cisplatin may present as either SIADH or RSWS, complicating treatment decisions. Both conditions lead to hypotonic hyponatremia with urine osmolality > 100 mOSm/kg and urine sodium levels > 40 mmol/L. However, pathophysiology behind SIADH and RSWS is different. In RSWS, proximal tubule damage causes hyponatremia, decreasing sodium reabsorption, and leading to impaired concentration gradient in every segment of the nephron. As a result, RSWS can lead to profound hyponatremia. Treatment typically consists of increasing sodium intake to correct serum sodium with salt tablets and hypertonic sodium chloride while treating the underlying etiology, in our case removing the offending agent, and waiting for proximal tubule function to recover.6 On the other hand, in SIADH, elevated antidiuretic hormone (ADH) increases water reabsorption in the collecting duct, which has no impact on concentration gradients of the other nephron segments.14 Free-water restriction is the hallmark of SIADH treatment. Severe SIADH may require sodium repletion and/or the initiation of vaptans, ADH antagonists that competitively inhibit V2 receptors in the collecting duct to prevent water reabsorption.15
Our patient had an uncertain etiology of his hyponatremia throughout most of his treatment course, complicating our treatment decision-making. Initially, his measured serum osmolality was 278 mOsm/kg; however, his effective tonicity was lower. His AKI elevated his BUN, which in turnrequired us to calculate his serum tonicity (217 mOsm/kg) that was consistent with hypotonic hyponatremia. His elevated urine osmolality and urine sodium levels made SIADH and RSWS the most likely etiologies of his hyponatremia. To confirm the etiology, we waited for correction of his serum sodium. Therefore, we treated him with a combination of sodium repletion with 0.9% sodium chloride (154 mEq/L), hypertonic relative to his serum sodium, sodium chloride tablets, and free-water restriction. In this approach, we attempted to harmonize the treatment strategies for both SIADH and RSWS and effectively corrected his serum sodium. We evaluated his response to our treatment with a basic metabolic panel every 6 to 8 hours. Had his serum sodium decreased < 120 mmol/L, we planned to transfer the patient to the intensive care unit for 3% sodium chloride and/or intensification of his fluid restriction. A significant worsening of his hyponatremia would have strongly suggested hyponatremia secondary to SIADH since isotonic saline can worsen hyponatremia due to increased free-water reabsorption in the collecting duct.16
To differentiate between SIADH and RSWS, we relied on the FEurate after sodium correction. Multiple case reports from Japan have characterized the distinction between the processes through FEurate and serum uric acid. While the optimal cut-off values for FEurate require additional investigation, values < 11% after serum sodium correction suggests SIADH, while a value > 11% suggests RSWS.17 Prior cases have also emphasized serum hypouricemia as a distinguishing characteristic in RSWS. However, our case illustrates that serum hypouricemia is less reliable in the setting of AKI. Due to his severe AKI, our patient could not efficiently clear uric acid, likely contributing to his hyperuricemia.
Ultimately, our patient had an FEurate > 20%, which was suggestive of RSWS. Nevertheless, we recognize limitations and confounders in our diagnosis and have reflected on our diagnostic and management choices. First, the sensitivity and specificity of postsodium correction FEurate is unknown. Tracking the change in FEurate with our interventions would have increased our diagnostic utility, as suggested by Maesaka and colleagues.14 Second, our patient’s serum sodium was still at the lower end of the reference range after treatment, which may decrease the specificity of FEurate. Third, a plasma ADH collected during the initial phase of symptomatic hyponatremia would have helped differentiate between SIADH and RSWS.
Other diagnostic tests that could have excluded alternative diagnoses with even greater certainty include plasma adrenocorticotropic hormone, B-type natriuretic peptide, renin, cortisol, and thyroid function tests. From a practical standpoint, these laboratory results (excluding thyroid function test and brain natriuretic peptide) would have taken several weeks to result at our institution, limiting their clinical utility. Similarly, FEurate also has limited clinical utility, requiring effective treatment as part of the diagnostic test. Therefore, we recommend focusing on optimal treatment for hyponatremia of uncertain etiology, especially where SIADH and RSWS are the leading diagnoses.
Conclusions
We described a rare case of concomitant cisplatin-induced severe AKI and RSWS. We have emphasized the diagnostic challenge of distinguishing between SIADH and RSWS, especially with concomitant AKI, and have acknowledged that optimal treatment relies on accurate differentiation. However, differentiation may not be clinically feasible. Therefore, we suggest a treatment strategy that incorporates both free-water restriction and sodium supplementation via IV and/or oral administration.
Cisplatin is a potent antineoplastic agent derived from platinum and commonly used in the treatment of head and neck, bladder, ovarian, and testicular malignancies.1,2 Approximately 20% of all cancer patients are prescribed platinum-based chemotherapeutics.3 Although considered highly effective, cisplatin is also a dose-dependent nephrotoxin, inducing apoptosis in the proximal tubules of the nephron and reducing glomerular filtration rate. This nephron injury leads to inflammation and reduced medullary blood flow, causing further ischemic damage to the tubular cells.4 Given that the proximal tubule reabsorbs 67% of all sodium, cisplatin-induced nephron injuries can also lead to hyponatremia.5
The primary mechanisms of hyponatremia following cisplatin chemotherapy are syndrome of inappropriate antidiuretic hormone secretion (SIADH) and renal salt wasting syndrome (RSWS). Though these diagnoses have similar presentations, the treatment recommendations are different due to pathophysiologic differences. Fluid restriction is the hallmark of SIADH treatment, while increased sodium intake remains the hallmark of RSWS treatment.6 This patient presented with a combination of cisplatin-induced acute kidney injury (AKI) and hyponatremia secondary to RSWS. While RSWS and AKI are known complications of cisplatin chemotherapy, the combination is underreported in the literature. Therefore, this case report highlights the combination of these cisplatin-induced complications, emphasizes the clinical challenges in differentiating SIADH from RSWS, especially in the presence of a concomitant AKI, and suggests a treatment approach during diagnostic uncertainty.
Case Presentation
A 71-year-old man with a medical history of squamous cell carcinoma (SCC) of the left neck on cycle 1, day 8 of cisplatin-based chemotherapy and ongoing radiation therapy (720 cGy of 6300 cGy), lung adenocarcinoma status postresection, and hyperlipidemia presented to the emergency department (ED) at the request of his oncologist for abnormal laboratory values. In the ED, his metabolic panel showed a 131-mmol/L serum sodium, 3.3 mmol/L potassium, 83 mmol/L chloride, 29 mmol/L bicarbonate, 61 mg/dL blood urea nitrogen (BUN), and 8.8 mg/dL creatinine (baseline, 0.9 mg/dL). The patient reported throbbing headaches, persistent nausea, and multiple episodes of nonbloody emesis for several days that he attributed to his chemotherapy. He noted decreased urination without discomfort or changes in color or odor and no fatigue, fevers, chills, hematuria, flank, abdominal pain, thirst, or polydipsia. He reported no toxic ingestions or IV drug use. The patient had no relevant family history or additional social history. His outpatient medications included 10 mg cetirizine, 8 mg ondansetron, and 81 mg aspirin. On initial examination, his 137/66 mm Hg blood pressure was mildly elevated. The physical examination findings were notable for a 5-cm mass in the left neck that was firm and irregularly-shaped. His physical examination was otherwise unremarkable. He was admitted to the inpatient medicine service for an AKI complicated by symptomatic hyponatremia.
Investigations
We evaluated the patient’s AKI based on treatment responsiveness, imaging, and laboratory testing. Renal and bladder ultrasound showed no evidence of hydronephrosis or obstruction. He had a benign urinalysis with microscopy absent for protein, blood, ketones, leukocyte esterase, nitrites, and cellular casts. His urine pH was 5.5 (reference range, 5.0-9.0) and specific gravity was 1.011 (reference range, 1.005-1.030). His urine electrolytes revealed 45-mmol/L urine sodium (reference range, 40-220), 33-mmol/L urine chloride (reference range, 110-250), 10-mmol/L urine potassium (reference range, 25-120), 106.7-mg/dL urine creatinine (reference range, 10-400) and a calculated 2.7% fractional excretion of sodium (FENa) and 22.0-mEq/L elevated urine anion gap. As a fluid challenge, he was treated with IV 0.9% sodium chloride at 100-125 mL/h, receiving 3 liters over the first 48 hours of his hospitalization. His creatinine peaked at 9.2 mg/dL and stabilized before improving later in his hospitalization (Figure 1). The patient initially had oliguria (< 0.5 mL/kg/h), which slowly improved over his hospital course. Unfortunately, due to multiple system and clinical factors, accurate inputs and outputs were not adequately maintained during his hospitalization.
We evaluated hyponatremia with a combination of serum and urine laboratory tests. In addition to urine electrolytes, the initial evaluation focused on trending his clinical trajectory. We repeated a basic metabolic panel every 4 to 6 hours. He had 278-mOsm/kg serum osmolality (reference range, 285-295) with an effective 217-mOsm/kg serum tonicity. His urine osmolality was 270.5 mOsm/kg.
Despite administering 462 mEq sodium via crystalloid, his sodium worsened over the first 48 hours, reaching a nadir at 125 mmol/L on hospital day 3 (Figure 2). While he continued to appear euvolemic on physical examination, his blood pressure became difficult to control with 160- to 180-mm Hg systolic blood pressure readings. His thyroid stimulating hormone (TSH) was normal and aldosterone was low (4 ng/dL). Additional urine studies, including a 24-hour urine sample, were collected for further evaluation. His urine uric acid was 140 mg/d (reference range, 120-820); his serum uric acid level was 8.2 mg/dL (reference range, 3.0-9.0). His 24-hour urine creatinine was 0.57 g/d (reference range, 0.50-2.15) and uric acid to creatinine ratio was 246 mg/g (reference range, 60-580). His serum creatinine collected from the same day as his 24-hour urine sample was 7.3 mg/dL. His fractional excretion of uric acid (FEurate) was 21.9%.
Differential Diagnosis
The patient’s recent administration of cisplatin raised clinical suspicion of cisplatin-induced AKI. To avoid premature diagnostic closure, we used a systematic approach for thinking about our patient’s AKI, considering prerenal, intrarenal, and postrenal etiologies. The unremarkable renal and bladder ultrasound made a postrenal etiology unlikely. The patient’s 2.7% FENa in the absence of a diuretic, limited responsiveness to crystalloid fluid resuscitation, 7.5 serum BUN/creatinine ratio, and 270.5 mOsm/kg urine osmolality suggested an intrarenal etiology, which can be further divided into problems with glomeruli, tubules, small vessels, or interstitial space. The patient’s normal urinary microscopy with no evidence of protein, blood, ketones, leukocyte esterase, nitrites, or cellular casts made a glomerular etiology less likely. The acute onset and lack of additional systemic features, other than hypertension, made a vascular etiology less likely. A tubular etiology, such as acute tubular necrosis (ATN), was highest on the differential and was followed by an interstitial etiology, such as acute interstitial nephritis (AIN).
Patients with drug-induced AIN commonly present with signs and symptoms of an allergic-type reaction, including fever, rash, hematuria, pyuria, and costovertebral angle tenderness. The patient lacked these symptoms. However, cisplatin is known to cause ATN in up to 20-30% of patients.7 Therefore, despite the lack of the classic muddy-brown, granular casts on urine microscopy, cisplatin-induced ATN remained the most likely etiology of his AKI. Moreover, ATN can cause hyponatremia. ATN is characterized by 3 phases: initiation, maintenance, and recovery phases.8 Hyponatremia occurs during the recovery phase, typically starting weeks after renal insult and associated with high urine output and diuresis. This patient presented 1 week after injury and had persistent oliguria, making ATN an unlikely culprit of his hyponatremia.
Our patient presented with hypotonic hyponatremia with a 131 mmol/L initial sodium level and an < 280 mOsm/kg effective serum osmolality, or serum tonicity. The serum tonicity is equivalent to the difference between the measured serum osmolality and the BUN. In the setting of profound AKI, this adjustment is essential for correctly categorizing a patient’s hyponatremia as hyper-, iso-, or hypotonic. The differential diagnosis for this patient’s hypotonic hyponatremia included dilutional effects of hypervolemia, SIADH, hyperthyroidism, adrenal insufficiency, and RSWS. The patient’s volume examination, lack of predisposing comorbidities or suggestive biomarkers, and > 20 mmol/L urinary sodium made hypervolemia unlikely. His urinary osmolality and specific gravity made primary polydipsia unlikely. We worked up his hyponatremia according to a diagnostic algorithm (eAppendix available at doi:10.12788/fp.0198).
The patient had a 217 mOsm/kg serum tonicity and a 270.5 mOsm/kg urine osmolality, consistent with impaired water excretion. His presentation, TSH, and concordant decrease in sodium and potassium made an endocrine etiology of his hyponatremia less likely. In hindsight, a serum cortisol would have been beneficial to more completely exclude adrenal insufficiency. His urine sodium was elevated at 45 mmol/L, raising concern for RSWS or SIADH. The FEurate helped to distinguish between SIADH and RSWS. While FEurate is often elevated in both SIADH and RSWS initially, the FEurate normalizes in SIADH with normalization of the serum sodium. The ideal cutoff for posthyponatremia correction FEurate is debated; however, a FEurate value after sodium correction < 11% suggests SIADH while a value > 11% suggests RSWS.9 Our patient’s FEurate following the sodium correction (serum sodium 134 mmol/L) was 21.9%, most suggestive of RSWS.
Treatment
Upon admission, initial treatment focused on resolving the patient’s AKI. The oncology team discontinued the cisplatin-based chemotherapy. His medication dosages were adjusted for his renal function and additional nephrotoxins avoided. In consultation, the nephrology service recommended 100 mL/h fluid resuscitation. After the patient received 3 L of 0.9% sodium chloride, his creatinine showed limited improvement and his sodium worsened, trending from 131 mmol/L to a nadir of 125 mmol/L. We initiated oral free-water restriction while continuing IV infusion of 0.9% sodium chloride at 125 mL/h.
We further augmented his sodium intake with 1-g sodium chloride tablets with each meal. By hospital day 6, the patient’s serum sodium, BUN, and creatinine improved to 130 mEq/L, 50 mg/dL, and 7.7 mg/dL, respectively. We then discontinued the oral sodium chloride tablets, fluid restriction, and IV fluids in a stepwise fashion prior to discharge. At discharge, the patient’s serum sodium was 136 mEq/L and creatinine, 4.8 mg/dL. The patient’s clinical course was complicated by symptomatic hypertension with systolic blood pressures about 180 mm Hg, requiring intermittent IV hydralazine, which was transitioned to daily nifedipine. Concerned that fluid resuscitation contributed to his hypertension, the patient also received several doses of furosemide. At time of discharge, the patient remained hypertensive and was discharged with nifedipine 90 mg daily.
Outcome and Follow-up
The patient has remained stable clinically since discharge. One week after discharge, his serum sodium and creatinine were 138 mmol/L and 3.8 mg/dL, respectively. More than 1 month after discharge, his sodium remains in the reference range and his creatinine was stable at about 3.5 mg/dL. He continues to follow-up with nephrology, oncology, and radiation oncology. He has restarted chemotherapy with a carboplatin-based regimen without recurrence of hyponatremia or AKI. His blood pressure has gradually improved to the point where he no longer requires nifedipine.
Discussion
The US Food and Drug Administration first approved the use of cisplatin, an alkylating agent that inhibits DNA replication, in 1978 for the treatment of testicular cancer.10 Since its approval, cisplatin has increased in popularity and is now considered one of the most effective antineoplastic agents for the treatment of solid tumors.1 Unfortunately, cisplatin has a well-documented adverse effect profile that includes neurotoxicity, gastrointestinal toxicity, nephrotoxicity, and ototoxicity.4 Despite frequent nephrotoxicity, cisplatin only occasionally causes hyponatremia and rarely causes RSWS, a known but potentially fatal complication. Moreover, the combination of AKI and RSWS is unique. Our patient presented with the unique combination of AKI and hyponatremia, most consistent with RSWS, likely precipitated from cisplatin chemotherapy. Through this case, we review cisplatin-associated electrolyte abnormalities, highlight the challenge of differentiating SIADH and RSWS, and suggest a treatment approach for hyponatremia during the period of diagnostic uncertainty.
Blachley and colleagues first discussed renal and electrolyte disturbances, specifically magnesium wasting, secondary to cisplatin use in 1981. In 1984, Kurtzberg and colleagues noted salt wasting in 2 patients receiving cisplatin therapy. The authors suggested that cisplatin inhibits solute transport in the thick ascending limb, causing clinically significant electrolyte abnormalities, coining the term cisplatin-induced salt wasting.11
The prevalence of cisplatin-induced salt wasting is unclear and likely underreported. In 1988, Hutchinson and colleagues conducted a prospective cohort study and noted 10% of patients (n = 70) developed RSWS at some point over 18 months of cisplatin therapy—a higher rate than previously estimated.12 In 1992, another prospective cohort study evaluated the adverse effects of 47 patients with non-small cell lung cancer treated with cisplatin and reported hyponatremia in 43% of its 93 courses of chemotherapy. The authors did not report the etiology of these hyponatremia cases.13 Given the diagnostic challenge, RSWS may be underrepresented as a confirmed etiology of hyponatremia in cisplatin treatment.
Hyponatremia from cisplatin may present as either SIADH or RSWS, complicating treatment decisions. Both conditions lead to hypotonic hyponatremia with urine osmolality > 100 mOSm/kg and urine sodium levels > 40 mmol/L. However, pathophysiology behind SIADH and RSWS is different. In RSWS, proximal tubule damage causes hyponatremia, decreasing sodium reabsorption, and leading to impaired concentration gradient in every segment of the nephron. As a result, RSWS can lead to profound hyponatremia. Treatment typically consists of increasing sodium intake to correct serum sodium with salt tablets and hypertonic sodium chloride while treating the underlying etiology, in our case removing the offending agent, and waiting for proximal tubule function to recover.6 On the other hand, in SIADH, elevated antidiuretic hormone (ADH) increases water reabsorption in the collecting duct, which has no impact on concentration gradients of the other nephron segments.14 Free-water restriction is the hallmark of SIADH treatment. Severe SIADH may require sodium repletion and/or the initiation of vaptans, ADH antagonists that competitively inhibit V2 receptors in the collecting duct to prevent water reabsorption.15
Our patient had an uncertain etiology of his hyponatremia throughout most of his treatment course, complicating our treatment decision-making. Initially, his measured serum osmolality was 278 mOsm/kg; however, his effective tonicity was lower. His AKI elevated his BUN, which in turnrequired us to calculate his serum tonicity (217 mOsm/kg) that was consistent with hypotonic hyponatremia. His elevated urine osmolality and urine sodium levels made SIADH and RSWS the most likely etiologies of his hyponatremia. To confirm the etiology, we waited for correction of his serum sodium. Therefore, we treated him with a combination of sodium repletion with 0.9% sodium chloride (154 mEq/L), hypertonic relative to his serum sodium, sodium chloride tablets, and free-water restriction. In this approach, we attempted to harmonize the treatment strategies for both SIADH and RSWS and effectively corrected his serum sodium. We evaluated his response to our treatment with a basic metabolic panel every 6 to 8 hours. Had his serum sodium decreased < 120 mmol/L, we planned to transfer the patient to the intensive care unit for 3% sodium chloride and/or intensification of his fluid restriction. A significant worsening of his hyponatremia would have strongly suggested hyponatremia secondary to SIADH since isotonic saline can worsen hyponatremia due to increased free-water reabsorption in the collecting duct.16
To differentiate between SIADH and RSWS, we relied on the FEurate after sodium correction. Multiple case reports from Japan have characterized the distinction between the processes through FEurate and serum uric acid. While the optimal cut-off values for FEurate require additional investigation, values < 11% after serum sodium correction suggests SIADH, while a value > 11% suggests RSWS.17 Prior cases have also emphasized serum hypouricemia as a distinguishing characteristic in RSWS. However, our case illustrates that serum hypouricemia is less reliable in the setting of AKI. Due to his severe AKI, our patient could not efficiently clear uric acid, likely contributing to his hyperuricemia.
Ultimately, our patient had an FEurate > 20%, which was suggestive of RSWS. Nevertheless, we recognize limitations and confounders in our diagnosis and have reflected on our diagnostic and management choices. First, the sensitivity and specificity of postsodium correction FEurate is unknown. Tracking the change in FEurate with our interventions would have increased our diagnostic utility, as suggested by Maesaka and colleagues.14 Second, our patient’s serum sodium was still at the lower end of the reference range after treatment, which may decrease the specificity of FEurate. Third, a plasma ADH collected during the initial phase of symptomatic hyponatremia would have helped differentiate between SIADH and RSWS.
Other diagnostic tests that could have excluded alternative diagnoses with even greater certainty include plasma adrenocorticotropic hormone, B-type natriuretic peptide, renin, cortisol, and thyroid function tests. From a practical standpoint, these laboratory results (excluding thyroid function test and brain natriuretic peptide) would have taken several weeks to result at our institution, limiting their clinical utility. Similarly, FEurate also has limited clinical utility, requiring effective treatment as part of the diagnostic test. Therefore, we recommend focusing on optimal treatment for hyponatremia of uncertain etiology, especially where SIADH and RSWS are the leading diagnoses.
Conclusions
We described a rare case of concomitant cisplatin-induced severe AKI and RSWS. We have emphasized the diagnostic challenge of distinguishing between SIADH and RSWS, especially with concomitant AKI, and have acknowledged that optimal treatment relies on accurate differentiation. However, differentiation may not be clinically feasible. Therefore, we suggest a treatment strategy that incorporates both free-water restriction and sodium supplementation via IV and/or oral administration.
1. Dasari S, Tchounwou PB. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol. 2014;740:364-378. doi:10.1016/j.ejphar.2014.07.025
2. Holditch SJ, Brown CN, Lombardi AM, Nguyen KN, Edelstein CL. Recent advances in models, mechanisms, biomarkers, and interventions in cisplatin-induced acute kidney injury. Int J Mol Sci. 2019;20(12):3011. Published 2019 Jun 20. doi:10.3390/ijms20123011
3. National Institutes of Health, National Cancer Institute. The “accidental” cure—platinum-based treatment for cancer: the discovery of cisplatin. Published May 30, 2014. Accessed November 10, 2021. https://www.cancer.gov/research/progress/discovery/cisplatin
4. Ozkok A, Edelstein CL. Pathophysiology of cisplatin-induced acute kidney injury. Biomed Res Int. 2014;2014:967826. doi:10.1155/2014/967826
5. Palmer LG, Schnermann J. Integrated control of Na transport along the nephron. Clin J Am Soc Nephrol. 2015;10(4):676-687. doi:10.2215/CJN.12391213
6. Bitew S, Imbriano L, Miyawaki N, Fishbane S, Maesaka JK. More on renal salt wasting without cerebral disease: response to saline infusion. Clin J Am Soc Nephrol. 2009;4(2):309-315. doi:10.2215/CJN.02740608
7. Shirali AC, Perazella MA. Tubulointerstitial injury associated with chemotherapeutic agents. Adv Chronic Kidney Dis. 2014;21(1):56-63. doi:10.1053/j.ackd.2013.06.010
8. Agrawal M, Swartz R. Acute renal failure [published correction appears in Am Fam Physician 2001 Feb 1;63(3):445]. Am Fam Physician. 2000;61(7):2077-2088.
9. Milionis HJ, Liamis GL, Elisaf MS. The hyponatremic patient: a systematic approach to laboratory diagnosis. CMAJ. 2002;166(8):1056-1062.
10. Monneret C. Platinum anticancer drugs. From serendipity to rational design. Ann Pharm Fr. 2011;69(6):286-295. doi:10.1016/j.pharma.2011.10.001
11. Kurtzberg J, Dennis VW, Kinney TR. Cisplatinum-induced renal salt wasting. Med Pediatr Oncol. 1984;12(2):150-154. doi:10.1002/mpo.2950120219
12. Hutchison FN, Perez EA, Gandara DR, Lawrence HJ, Kaysen GA. Renal salt wasting in patients treated with cisplatin. Ann Intern Med. 1988;108(1):21-25. doi:10.7326/0003-4819-108-1-21
13. Lee YK, Shin DM. Renal salt wasting in patients treated with high-dose cisplatin, etoposide, and mitomycin in patients with advanced non-small cell lung cancer. Korean J Intern Med. 1992;7(2):118-121. doi:10.3904/kjim.1992.7.2.118
14. Maesaka JK, Imbriano L, Mattana J, Gallagher D, Bade N, Sharif S. Differentiating SIADH from cerebral/renal salt wasting: failure of the volume approach and need for a new approach to hyponatremia. J Clin Med. 2014;3(4):1373-1385. Published 2014 Dec 8. doi:10.3390/jcm3041373
15. Palmer BF. The role of v2 receptor antagonists in the treatment of hyponatremia. Electrolyte Blood Press. 2013;11(1):1-8. doi:10.5049/EBP.2013.11.1.1
16. Verbalis JG, Goldsmith SR, Greenberg A, Schrier RW, Sterns RH. Hyponatremia treatment guidelines 2007: expert panel recommendations. Am J Med. 2007;120(11 Suppl 1):S1-S21. doi:10.1016/j.amjmed.2007.09.001
17. Maesaka JK, Imbriano LJ, Miyawaki N. High prevalence of renal salt wasting without cerebral disease as cause of hyponatremia in general medical wards. Am J Med Sci. 2018;356(1):15-22. doi:10.1016/j.amjms.2018.03.02
1. Dasari S, Tchounwou PB. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol. 2014;740:364-378. doi:10.1016/j.ejphar.2014.07.025
2. Holditch SJ, Brown CN, Lombardi AM, Nguyen KN, Edelstein CL. Recent advances in models, mechanisms, biomarkers, and interventions in cisplatin-induced acute kidney injury. Int J Mol Sci. 2019;20(12):3011. Published 2019 Jun 20. doi:10.3390/ijms20123011
3. National Institutes of Health, National Cancer Institute. The “accidental” cure—platinum-based treatment for cancer: the discovery of cisplatin. Published May 30, 2014. Accessed November 10, 2021. https://www.cancer.gov/research/progress/discovery/cisplatin
4. Ozkok A, Edelstein CL. Pathophysiology of cisplatin-induced acute kidney injury. Biomed Res Int. 2014;2014:967826. doi:10.1155/2014/967826
5. Palmer LG, Schnermann J. Integrated control of Na transport along the nephron. Clin J Am Soc Nephrol. 2015;10(4):676-687. doi:10.2215/CJN.12391213
6. Bitew S, Imbriano L, Miyawaki N, Fishbane S, Maesaka JK. More on renal salt wasting without cerebral disease: response to saline infusion. Clin J Am Soc Nephrol. 2009;4(2):309-315. doi:10.2215/CJN.02740608
7. Shirali AC, Perazella MA. Tubulointerstitial injury associated with chemotherapeutic agents. Adv Chronic Kidney Dis. 2014;21(1):56-63. doi:10.1053/j.ackd.2013.06.010
8. Agrawal M, Swartz R. Acute renal failure [published correction appears in Am Fam Physician 2001 Feb 1;63(3):445]. Am Fam Physician. 2000;61(7):2077-2088.
9. Milionis HJ, Liamis GL, Elisaf MS. The hyponatremic patient: a systematic approach to laboratory diagnosis. CMAJ. 2002;166(8):1056-1062.
10. Monneret C. Platinum anticancer drugs. From serendipity to rational design. Ann Pharm Fr. 2011;69(6):286-295. doi:10.1016/j.pharma.2011.10.001
11. Kurtzberg J, Dennis VW, Kinney TR. Cisplatinum-induced renal salt wasting. Med Pediatr Oncol. 1984;12(2):150-154. doi:10.1002/mpo.2950120219
12. Hutchison FN, Perez EA, Gandara DR, Lawrence HJ, Kaysen GA. Renal salt wasting in patients treated with cisplatin. Ann Intern Med. 1988;108(1):21-25. doi:10.7326/0003-4819-108-1-21
13. Lee YK, Shin DM. Renal salt wasting in patients treated with high-dose cisplatin, etoposide, and mitomycin in patients with advanced non-small cell lung cancer. Korean J Intern Med. 1992;7(2):118-121. doi:10.3904/kjim.1992.7.2.118
14. Maesaka JK, Imbriano L, Mattana J, Gallagher D, Bade N, Sharif S. Differentiating SIADH from cerebral/renal salt wasting: failure of the volume approach and need for a new approach to hyponatremia. J Clin Med. 2014;3(4):1373-1385. Published 2014 Dec 8. doi:10.3390/jcm3041373
15. Palmer BF. The role of v2 receptor antagonists in the treatment of hyponatremia. Electrolyte Blood Press. 2013;11(1):1-8. doi:10.5049/EBP.2013.11.1.1
16. Verbalis JG, Goldsmith SR, Greenberg A, Schrier RW, Sterns RH. Hyponatremia treatment guidelines 2007: expert panel recommendations. Am J Med. 2007;120(11 Suppl 1):S1-S21. doi:10.1016/j.amjmed.2007.09.001
17. Maesaka JK, Imbriano LJ, Miyawaki N. High prevalence of renal salt wasting without cerebral disease as cause of hyponatremia in general medical wards. Am J Med Sci. 2018;356(1):15-22. doi:10.1016/j.amjms.2018.03.02
25-year-old woman • abdominal pain • urticarial rash • recent influenza immunization • Dx?
THE CASE
A 25-year-old woman presented to an infectious diseases (ID) physician with a 4-day history of symptoms following receipt of a quadrivalent influenza vaccine. Two hours after receiving the vaccine, the patient experienced abdominal pain. One hour later, she felt warm and developed diffuse urticaria and rigors. Because of her worsening condition, she presented to the emergency department, where she was given intravenous methylprednisolone 40 mg, ondansetron 8 mg, diphenhydramine 25 mg, and normal saline. Her urticarial rash resolved within 45 minutes, and she was discharged home.
Three days later, she sought additional medical care because of persistent chest tightness, new-onset bronchospasm, pleuritic chest pain, nausea, diarrhea, facial swelling, urticaria, and anorexia. The patient’s vital signs were within normal limits. The oropharynx lacked erythema or obstruction. The lungs were clear to auscultation bilaterally, and heart sounds were regular, with no ectopy or murmurs. Her abdomen was soft, nontender, and nondistended. The patient demonstrated dermatographism on her back.
Historically, the patient had received the influenza vaccine without difficulty. She tolerated latex but had concerns about egg allergy due to vomiting with egg-yolk exposure.
THE DIAGNOSIS
The ID physician, suspecting anaphylaxis and sustained allergic response to the influenza vaccine, arranged for immediate follow-up with an allergist. Multiple tests were done. A negative result on epicutaneous testing to egg was inconsistent with an immunoglobulin (Ig) E-mediated food allergy.
Intradermal testing with the flu vaccine (diluted 1:100) was subsequently performed with appropriate controls. A positive intradermal result is typically a wheal ≥ 5 mm larger than the control. The patient had a 5-mm/15-mm wheal-and-flare response to the flu vaccine, compared to a negative response to saline (FIGURE). (Since the vaccine did not contain gelatin, this was not tested.)
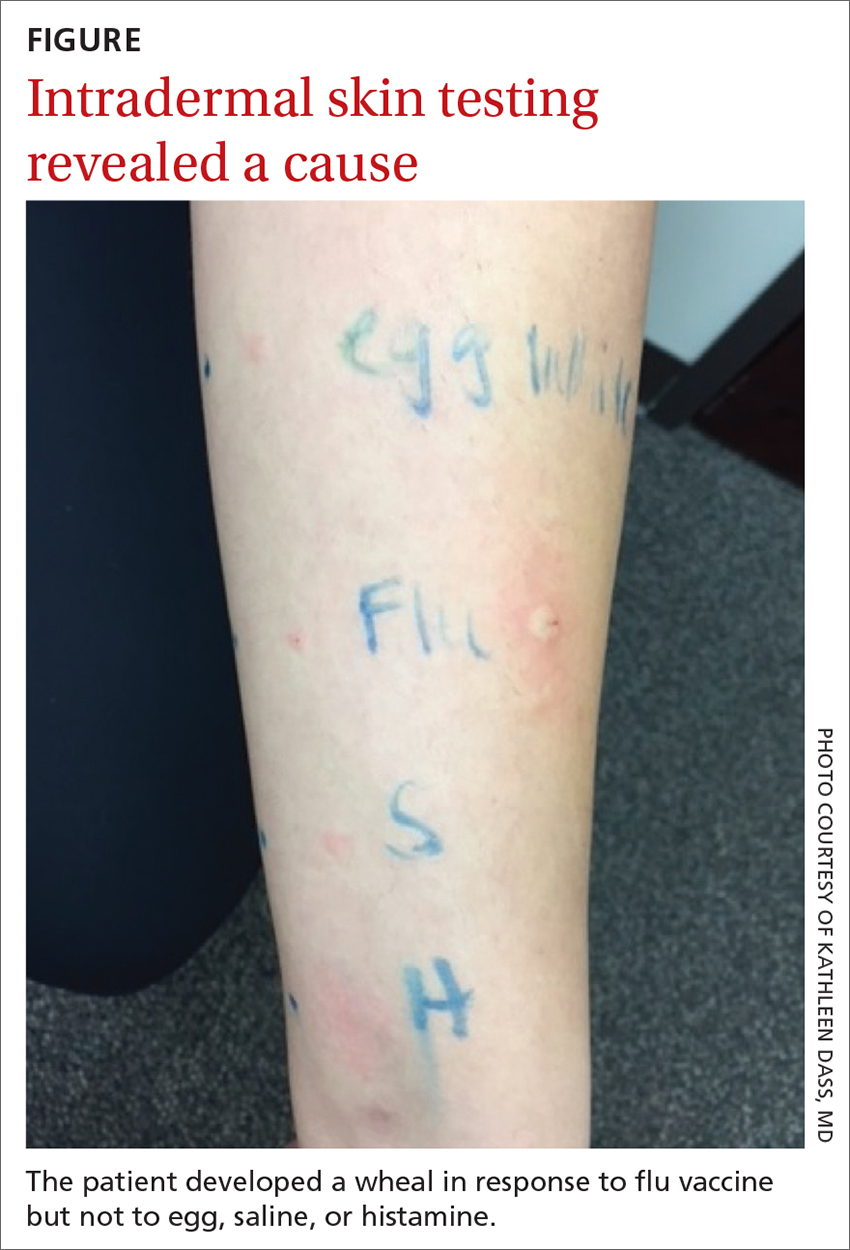
Based on the positive response to flu vaccine and negative response to egg, it was determined that the patient had experienced an anaphylactic reaction to the vaccine itself.
DISCUSSION
In adults, the most common adverse reactions to quadrivalent flu vaccine include pain, headache, and fatigue. IgE-mediated reactions to the influenza vaccine, especially anaphylactic reactions, are rare. A Vaccine Safety Datalink study found 10 cases of anaphylaxis after more than 7.4 million doses of inactivated flu vaccine were given, for a rate of 1.35 per 1 million doses.1
Continue to: Don't blame eggs
Don’t blame eggs. It was previously believed that reactions to the flu vaccine were due to egg allergies, because the vaccine may contain a tiny amount of ovalbumin, a protein found in egg. However, multiple studies have supported the safety of injectable influenza vaccine in patients with an egg allergy because the amount of ovalbumin contained in each dose is very low and thus not likely to evoke an allergic response.2,3
How and when to test for allergy. For patients who have a severe allergic reaction or anaphylaxis after immunization, immediate-type allergy skin testing should be performed by an allergist to establish whether the reaction was IgE mediated and to determine the causative agent.
It’s best to wait 4 to 6 weeks after an anaphylactic reaction before doing skin testing, as earlier testing can lead to false-negative results.4 The vaccine should first be tested by using the prick method. If this test is negative, an intradermal test with the vaccine diluted 1:100 should be performed with appropriate controls.5
Should the patient receive future vaccinations?
If skin testing is positive, there are several ways to proceed. A vaccine to which the patient has previously had an allergic reaction and positive skin test can still be administered, with caution.5 With emergency supplies, medication, and equipment immediately available, medical personnel can administer the influenza vaccine in titrated doses. If the full vaccine dose is normally a volume of 0.5 mL, the patient is first given 0.05 mL of a 1:10 dilution and then, at 15-minute intervals, given full-strength vaccine at doses of 0.05, 0.1, 0.15, and finally 0.2 mL, for a cumulative dose of 0.5 mL.5
Alternatively, the patient can forego the vaccination, although this decision has its own risks. In a patient who has previously had an anaphylactic reaction but has negative skin tests—meaning it is unlikely that the patient has IgE antibody to the vaccine—the vaccine can be administered and followed with an observation period of at least 30 minutes.5z Our patient was counseled on both options and decided to forego the vaccine.
THE TAKEAWAY
Anaphylaxis is a life-threatening allergic reaction requiring immediate treatment. Anaphylaxis after vaccine receipt is exceedingly rare.6 Most IgE-mediated allergic reactions post vaccination are attributed to added or residual substances in the vaccine, rather than the immunizing agent itself.6 While common local reactions and fever post vaccination do not contraindicate future vaccination, rare anaphylactic reactions need to be further evaluated, with a referral to an allergist to determine if the patient is, in fact, allergic to additive ingredients within the vaccine vs allergic to the vaccine itself.
CORRESPONDENCE
Kathleen Dass, MD, 24601 Coolidge Highway, Oak Park, MI 48237; [email protected]
1. Fluarix [package insert]. GlaxoSmithKline Biologicals. Dresden, Germany. 2016. Accessed November 9, 2021. www.fda.gov/downloads/BiologicsBloodVaccines/Vaccines/ApprovedProducts/UCM220624.pdf
2. Webb L, Petersen M, Boden S, et al. Single-dose influenza vaccination of patients with egg allergy in a multicenter study. J Allergy Clin Immunol. 2011;128:218-219. doi: 10.1016/j.jaci.2011.02.013
3. Howe LE, Conlon ASC, Greenhawt MJ, et al. Safe administration of seasonal influenza vaccine to children with egg allergy of all severities. Ann Allergy Asthma Immunol. 2011;106:446-447. doi: 10.1016/j.anai.2011.01.024
4. Soetens F, Rose M, Fisher M. Timing of skin testing after a suspected anaphylactic reaction during anaesthesia. Acta Anaesthesiol Scand. 2012;56:1042-1046. doi: 10.1111/j.1399-6576.2011.02643.x
5. Kelso JM, Greenhawt MJ, Li JT, et al. Adverse reactions to vaccines practice parameter 2012 update. J Allergy Clin Immunol. 2012;130:25-43. doi: 10.1016/j.jaci.2012.04.003
6. McNeil MM, Weintraub ES, Duffy J, et al. Risk of anaphylaxis after vaccination in children and adults. J Allergy Clin Immunol. 2016;137:868-878. doi: 10.1016/j.jaci.2015.07.048
THE CASE
A 25-year-old woman presented to an infectious diseases (ID) physician with a 4-day history of symptoms following receipt of a quadrivalent influenza vaccine. Two hours after receiving the vaccine, the patient experienced abdominal pain. One hour later, she felt warm and developed diffuse urticaria and rigors. Because of her worsening condition, she presented to the emergency department, where she was given intravenous methylprednisolone 40 mg, ondansetron 8 mg, diphenhydramine 25 mg, and normal saline. Her urticarial rash resolved within 45 minutes, and she was discharged home.
Three days later, she sought additional medical care because of persistent chest tightness, new-onset bronchospasm, pleuritic chest pain, nausea, diarrhea, facial swelling, urticaria, and anorexia. The patient’s vital signs were within normal limits. The oropharynx lacked erythema or obstruction. The lungs were clear to auscultation bilaterally, and heart sounds were regular, with no ectopy or murmurs. Her abdomen was soft, nontender, and nondistended. The patient demonstrated dermatographism on her back.
Historically, the patient had received the influenza vaccine without difficulty. She tolerated latex but had concerns about egg allergy due to vomiting with egg-yolk exposure.
THE DIAGNOSIS
The ID physician, suspecting anaphylaxis and sustained allergic response to the influenza vaccine, arranged for immediate follow-up with an allergist. Multiple tests were done. A negative result on epicutaneous testing to egg was inconsistent with an immunoglobulin (Ig) E-mediated food allergy.
Intradermal testing with the flu vaccine (diluted 1:100) was subsequently performed with appropriate controls. A positive intradermal result is typically a wheal ≥ 5 mm larger than the control. The patient had a 5-mm/15-mm wheal-and-flare response to the flu vaccine, compared to a negative response to saline (FIGURE). (Since the vaccine did not contain gelatin, this was not tested.)
Based on the positive response to flu vaccine and negative response to egg, it was determined that the patient had experienced an anaphylactic reaction to the vaccine itself.
DISCUSSION
In adults, the most common adverse reactions to quadrivalent flu vaccine include pain, headache, and fatigue. IgE-mediated reactions to the influenza vaccine, especially anaphylactic reactions, are rare. A Vaccine Safety Datalink study found 10 cases of anaphylaxis after more than 7.4 million doses of inactivated flu vaccine were given, for a rate of 1.35 per 1 million doses.1
Continue to: Don't blame eggs
Don’t blame eggs. It was previously believed that reactions to the flu vaccine were due to egg allergies, because the vaccine may contain a tiny amount of ovalbumin, a protein found in egg. However, multiple studies have supported the safety of injectable influenza vaccine in patients with an egg allergy because the amount of ovalbumin contained in each dose is very low and thus not likely to evoke an allergic response.2,3
How and when to test for allergy. For patients who have a severe allergic reaction or anaphylaxis after immunization, immediate-type allergy skin testing should be performed by an allergist to establish whether the reaction was IgE mediated and to determine the causative agent.
It’s best to wait 4 to 6 weeks after an anaphylactic reaction before doing skin testing, as earlier testing can lead to false-negative results.4 The vaccine should first be tested by using the prick method. If this test is negative, an intradermal test with the vaccine diluted 1:100 should be performed with appropriate controls.5
Should the patient receive future vaccinations?
If skin testing is positive, there are several ways to proceed. A vaccine to which the patient has previously had an allergic reaction and positive skin test can still be administered, with caution.5 With emergency supplies, medication, and equipment immediately available, medical personnel can administer the influenza vaccine in titrated doses. If the full vaccine dose is normally a volume of 0.5 mL, the patient is first given 0.05 mL of a 1:10 dilution and then, at 15-minute intervals, given full-strength vaccine at doses of 0.05, 0.1, 0.15, and finally 0.2 mL, for a cumulative dose of 0.5 mL.5
Alternatively, the patient can forego the vaccination, although this decision has its own risks. In a patient who has previously had an anaphylactic reaction but has negative skin tests—meaning it is unlikely that the patient has IgE antibody to the vaccine—the vaccine can be administered and followed with an observation period of at least 30 minutes.5z Our patient was counseled on both options and decided to forego the vaccine.
THE TAKEAWAY
Anaphylaxis is a life-threatening allergic reaction requiring immediate treatment. Anaphylaxis after vaccine receipt is exceedingly rare.6 Most IgE-mediated allergic reactions post vaccination are attributed to added or residual substances in the vaccine, rather than the immunizing agent itself.6 While common local reactions and fever post vaccination do not contraindicate future vaccination, rare anaphylactic reactions need to be further evaluated, with a referral to an allergist to determine if the patient is, in fact, allergic to additive ingredients within the vaccine vs allergic to the vaccine itself.
CORRESPONDENCE
Kathleen Dass, MD, 24601 Coolidge Highway, Oak Park, MI 48237; [email protected]
THE CASE
A 25-year-old woman presented to an infectious diseases (ID) physician with a 4-day history of symptoms following receipt of a quadrivalent influenza vaccine. Two hours after receiving the vaccine, the patient experienced abdominal pain. One hour later, she felt warm and developed diffuse urticaria and rigors. Because of her worsening condition, she presented to the emergency department, where she was given intravenous methylprednisolone 40 mg, ondansetron 8 mg, diphenhydramine 25 mg, and normal saline. Her urticarial rash resolved within 45 minutes, and she was discharged home.
Three days later, she sought additional medical care because of persistent chest tightness, new-onset bronchospasm, pleuritic chest pain, nausea, diarrhea, facial swelling, urticaria, and anorexia. The patient’s vital signs were within normal limits. The oropharynx lacked erythema or obstruction. The lungs were clear to auscultation bilaterally, and heart sounds were regular, with no ectopy or murmurs. Her abdomen was soft, nontender, and nondistended. The patient demonstrated dermatographism on her back.
Historically, the patient had received the influenza vaccine without difficulty. She tolerated latex but had concerns about egg allergy due to vomiting with egg-yolk exposure.
THE DIAGNOSIS
The ID physician, suspecting anaphylaxis and sustained allergic response to the influenza vaccine, arranged for immediate follow-up with an allergist. Multiple tests were done. A negative result on epicutaneous testing to egg was inconsistent with an immunoglobulin (Ig) E-mediated food allergy.
Intradermal testing with the flu vaccine (diluted 1:100) was subsequently performed with appropriate controls. A positive intradermal result is typically a wheal ≥ 5 mm larger than the control. The patient had a 5-mm/15-mm wheal-and-flare response to the flu vaccine, compared to a negative response to saline (FIGURE). (Since the vaccine did not contain gelatin, this was not tested.)
Based on the positive response to flu vaccine and negative response to egg, it was determined that the patient had experienced an anaphylactic reaction to the vaccine itself.
DISCUSSION
In adults, the most common adverse reactions to quadrivalent flu vaccine include pain, headache, and fatigue. IgE-mediated reactions to the influenza vaccine, especially anaphylactic reactions, are rare. A Vaccine Safety Datalink study found 10 cases of anaphylaxis after more than 7.4 million doses of inactivated flu vaccine were given, for a rate of 1.35 per 1 million doses.1
Continue to: Don't blame eggs
Don’t blame eggs. It was previously believed that reactions to the flu vaccine were due to egg allergies, because the vaccine may contain a tiny amount of ovalbumin, a protein found in egg. However, multiple studies have supported the safety of injectable influenza vaccine in patients with an egg allergy because the amount of ovalbumin contained in each dose is very low and thus not likely to evoke an allergic response.2,3
How and when to test for allergy. For patients who have a severe allergic reaction or anaphylaxis after immunization, immediate-type allergy skin testing should be performed by an allergist to establish whether the reaction was IgE mediated and to determine the causative agent.
It’s best to wait 4 to 6 weeks after an anaphylactic reaction before doing skin testing, as earlier testing can lead to false-negative results.4 The vaccine should first be tested by using the prick method. If this test is negative, an intradermal test with the vaccine diluted 1:100 should be performed with appropriate controls.5
Should the patient receive future vaccinations?
If skin testing is positive, there are several ways to proceed. A vaccine to which the patient has previously had an allergic reaction and positive skin test can still be administered, with caution.5 With emergency supplies, medication, and equipment immediately available, medical personnel can administer the influenza vaccine in titrated doses. If the full vaccine dose is normally a volume of 0.5 mL, the patient is first given 0.05 mL of a 1:10 dilution and then, at 15-minute intervals, given full-strength vaccine at doses of 0.05, 0.1, 0.15, and finally 0.2 mL, for a cumulative dose of 0.5 mL.5
Alternatively, the patient can forego the vaccination, although this decision has its own risks. In a patient who has previously had an anaphylactic reaction but has negative skin tests—meaning it is unlikely that the patient has IgE antibody to the vaccine—the vaccine can be administered and followed with an observation period of at least 30 minutes.5z Our patient was counseled on both options and decided to forego the vaccine.
THE TAKEAWAY
Anaphylaxis is a life-threatening allergic reaction requiring immediate treatment. Anaphylaxis after vaccine receipt is exceedingly rare.6 Most IgE-mediated allergic reactions post vaccination are attributed to added or residual substances in the vaccine, rather than the immunizing agent itself.6 While common local reactions and fever post vaccination do not contraindicate future vaccination, rare anaphylactic reactions need to be further evaluated, with a referral to an allergist to determine if the patient is, in fact, allergic to additive ingredients within the vaccine vs allergic to the vaccine itself.
CORRESPONDENCE
Kathleen Dass, MD, 24601 Coolidge Highway, Oak Park, MI 48237; [email protected]
1. Fluarix [package insert]. GlaxoSmithKline Biologicals. Dresden, Germany. 2016. Accessed November 9, 2021. www.fda.gov/downloads/BiologicsBloodVaccines/Vaccines/ApprovedProducts/UCM220624.pdf
2. Webb L, Petersen M, Boden S, et al. Single-dose influenza vaccination of patients with egg allergy in a multicenter study. J Allergy Clin Immunol. 2011;128:218-219. doi: 10.1016/j.jaci.2011.02.013
3. Howe LE, Conlon ASC, Greenhawt MJ, et al. Safe administration of seasonal influenza vaccine to children with egg allergy of all severities. Ann Allergy Asthma Immunol. 2011;106:446-447. doi: 10.1016/j.anai.2011.01.024
4. Soetens F, Rose M, Fisher M. Timing of skin testing after a suspected anaphylactic reaction during anaesthesia. Acta Anaesthesiol Scand. 2012;56:1042-1046. doi: 10.1111/j.1399-6576.2011.02643.x
5. Kelso JM, Greenhawt MJ, Li JT, et al. Adverse reactions to vaccines practice parameter 2012 update. J Allergy Clin Immunol. 2012;130:25-43. doi: 10.1016/j.jaci.2012.04.003
6. McNeil MM, Weintraub ES, Duffy J, et al. Risk of anaphylaxis after vaccination in children and adults. J Allergy Clin Immunol. 2016;137:868-878. doi: 10.1016/j.jaci.2015.07.048
1. Fluarix [package insert]. GlaxoSmithKline Biologicals. Dresden, Germany. 2016. Accessed November 9, 2021. www.fda.gov/downloads/BiologicsBloodVaccines/Vaccines/ApprovedProducts/UCM220624.pdf
2. Webb L, Petersen M, Boden S, et al. Single-dose influenza vaccination of patients with egg allergy in a multicenter study. J Allergy Clin Immunol. 2011;128:218-219. doi: 10.1016/j.jaci.2011.02.013
3. Howe LE, Conlon ASC, Greenhawt MJ, et al. Safe administration of seasonal influenza vaccine to children with egg allergy of all severities. Ann Allergy Asthma Immunol. 2011;106:446-447. doi: 10.1016/j.anai.2011.01.024
4. Soetens F, Rose M, Fisher M. Timing of skin testing after a suspected anaphylactic reaction during anaesthesia. Acta Anaesthesiol Scand. 2012;56:1042-1046. doi: 10.1111/j.1399-6576.2011.02643.x
5. Kelso JM, Greenhawt MJ, Li JT, et al. Adverse reactions to vaccines practice parameter 2012 update. J Allergy Clin Immunol. 2012;130:25-43. doi: 10.1016/j.jaci.2012.04.003
6. McNeil MM, Weintraub ES, Duffy J, et al. Risk of anaphylaxis after vaccination in children and adults. J Allergy Clin Immunol. 2016;137:868-878. doi: 10.1016/j.jaci.2015.07.048