User login
Raynaud Phenomenon of the Nipple Successfully Treated With Nifedipine and Gabapentin
To the Editor:
Raynaud phenomenon is characterized by vasospasm of arterioles causing intermittent ischemia of the digits. The characteristic triphasic color change presents first as a dramatic change in skin color from normal to white, as the vasoconstriction causes pallor secondary to ischemia. This change is followed by a blue appearance, as cyanosis results from the deoxygenated venous blood. Finally, reflex vasodilation and reperfusion manifest as a red color from erythema. Several cases have been reported describing Raynaud phenomenon affecting the nipples of breastfeeding women.1-5 This vasospasm results in episodic nipple pain manifesting from breastfeeding and exposure to cold. If it is not appropriately treated, the pain’s severity causes affected women to stop breastfeeding. We report a case of vasospasm of the nipple in which the patient experienced nipple pain and a separate lancinating pain that radiated through the breasts.
A 36-year-old woman presented with excruciating nipple and breast pain 3 weeks after delivering her first child. She had no history of smoking or Raynaud phenomenon. The nipple pain was triggered upon breastfeeding and exposure to cold. During these episodes, the nipples would initially blanch white, then turn purple and finally a deep red. The patient also experienced an episodic excruciating lancinating pain of the breast that would randomly and spontaneously radiate through either breast several times per day for 15 to 30 seconds. A workup including an antinuclear antibody test, complete blood cell count with differential, and comprehensive metabolic panel all were within reference range.
The patient was diagnosed with nipple vasospasm. Partial relief of nipple pain occurred after treatment with 30 mg daily of nifedipine; 60 mg daily resulted in complete control, allowing the patient to breastfeed without discomfort, but the lancinating pain continued unabated. The patient could not discontinue breastfeeding because her child was intolerant to formula. She became despondent, as she could find no relief from the pain that she found to be intolerable. Because the patient’s description was reminiscent of the lancinating pain seen in postherpetic neuralgia, a trial of pregabalin was prescribed. A dosage of 75 mg twice daily resulted in near-complete resolution of the pain. After 3 months, the patient successfully weaned her child from breast milk to formula, and the nipple and breast pain promptly resolved. The baby experienced no adverse effects from the patient’s use of pregabalin.
This condition was first described by Gunther1 in 1970 as initial blanching of the nipple followed by a mulberry color. It was termed psychosomatic sore nipples.1 Lawlor-Smith and Lawlor-Smith2 described the condition in 1997 and termed it vasospasm of the nipple. They reported 5 patients who experienced debilitating nipple pain as well as the triphasic color change of Raynaud phenomenon or a biphasic color change (white and blue). Two patients had a history of Raynaud phenomenon affecting the digits before their first pregnancy.2 Anderson et al3 presented 12 breastfeeding women with Raynaud phenomenon of the nipple; only 1 patient had a history of Raynaud phenomenon. In this series, all 6 women who chose to try nifedipine responded well to the drug.3
Raynaud phenomenon of the nipple also has been reported to be associated with the use of labetalol.4 In this case, the patient had a history of Raynaud phenomenon affecting the toes and nipples on cold days. In 2 subsequent pregnancies she was treated with labetalol for pregnancy-induced hypertension, which resulted in severe nipple pain with each pregnancy unrelated to cold weather. Unlike other cases, this patient experienced antenatal symptoms in addition to the typical postnatal symptoms. The nipple pain resolved with discontinuation of the labetalol.4
Barrett et al5 conducted a retrospective review of medical records of 88 breastfeeding mothers who presented with nipple pain and dermatitis. They defined the criteria for Raynaud phenomenon of the nipple as chronic deep breast pain (in general lasting >4 weeks) that responded to therapy for the condition and had at least 2 of the following characteristics: (1) observed or self-reported color changes of the nipple, especially with cold exposure (white, blue, or red); (2) cold sensitivity or color changes of the hands or feet with cold exposure; or (3) failed therapy with oral antifungals. Using these criteria, they diagnosed 22 women (25%) with Raynaud phenomenon of the nipple; 20 (91%) reported a history of cold sensitivity or color change of acral surfaces. Of 12 patients who received and tolerated nifedipine use, 10 (83%) reported decreased pain or complete resolution. This series described breast or nipple pain, whereas other reported cases only described nipple pain. The authors described a sharp, shooting, or stabbing pain—qualifications not previously noted.5 Our patient experienced both nipple pain and a lancinating breast pain consistent with the cases reported by Barrett et al.5
The nipple pain and treatment response in our patient was typical of previously reported cases of vasospasm of the nipple in breastfeeding women; however, Barrett et al5 did not describe individual patients who exhibited the dual nature of the pain described in our patient. The nipple pain experienced during breastfeeding in our patient was successfully treated with nifedipine. We report the successful treatment of the separate lancinating pain with pregabalin.
- Gunther M. Infant Feeding. London, United Kingdom: Methuen; 1970.
- Lawlor-Smith L, Lawlor-Smith C. Vasospasm of the nipple—a manifestation of Raynaud’s phenomenon: case reports. BMJ. 1997;314:644-645.
- Anderson JE, Held N, Wright K. Raynaud phenomenon of the nipple: a treatable cause of painful breastfeeding. Pediatrics. 2004;113:360-364.
- McGuinness N, Cording V. Raynaud’s phenomenon of the nipple associated with labetalol use. J Hum Lact. 2013;29:17-19.
- Barrett ME, Heller MM, Stone HF, et al. Raynaud phenomenon of the nipple in breastfeeding mothers: an underdiagnosed cause of nipple pain. JAMA Dermatol. 2013;149:300-306.
To the Editor:
Raynaud phenomenon is characterized by vasospasm of arterioles causing intermittent ischemia of the digits. The characteristic triphasic color change presents first as a dramatic change in skin color from normal to white, as the vasoconstriction causes pallor secondary to ischemia. This change is followed by a blue appearance, as cyanosis results from the deoxygenated venous blood. Finally, reflex vasodilation and reperfusion manifest as a red color from erythema. Several cases have been reported describing Raynaud phenomenon affecting the nipples of breastfeeding women.1-5 This vasospasm results in episodic nipple pain manifesting from breastfeeding and exposure to cold. If it is not appropriately treated, the pain’s severity causes affected women to stop breastfeeding. We report a case of vasospasm of the nipple in which the patient experienced nipple pain and a separate lancinating pain that radiated through the breasts.
A 36-year-old woman presented with excruciating nipple and breast pain 3 weeks after delivering her first child. She had no history of smoking or Raynaud phenomenon. The nipple pain was triggered upon breastfeeding and exposure to cold. During these episodes, the nipples would initially blanch white, then turn purple and finally a deep red. The patient also experienced an episodic excruciating lancinating pain of the breast that would randomly and spontaneously radiate through either breast several times per day for 15 to 30 seconds. A workup including an antinuclear antibody test, complete blood cell count with differential, and comprehensive metabolic panel all were within reference range.
The patient was diagnosed with nipple vasospasm. Partial relief of nipple pain occurred after treatment with 30 mg daily of nifedipine; 60 mg daily resulted in complete control, allowing the patient to breastfeed without discomfort, but the lancinating pain continued unabated. The patient could not discontinue breastfeeding because her child was intolerant to formula. She became despondent, as she could find no relief from the pain that she found to be intolerable. Because the patient’s description was reminiscent of the lancinating pain seen in postherpetic neuralgia, a trial of pregabalin was prescribed. A dosage of 75 mg twice daily resulted in near-complete resolution of the pain. After 3 months, the patient successfully weaned her child from breast milk to formula, and the nipple and breast pain promptly resolved. The baby experienced no adverse effects from the patient’s use of pregabalin.
This condition was first described by Gunther1 in 1970 as initial blanching of the nipple followed by a mulberry color. It was termed psychosomatic sore nipples.1 Lawlor-Smith and Lawlor-Smith2 described the condition in 1997 and termed it vasospasm of the nipple. They reported 5 patients who experienced debilitating nipple pain as well as the triphasic color change of Raynaud phenomenon or a biphasic color change (white and blue). Two patients had a history of Raynaud phenomenon affecting the digits before their first pregnancy.2 Anderson et al3 presented 12 breastfeeding women with Raynaud phenomenon of the nipple; only 1 patient had a history of Raynaud phenomenon. In this series, all 6 women who chose to try nifedipine responded well to the drug.3
Raynaud phenomenon of the nipple also has been reported to be associated with the use of labetalol.4 In this case, the patient had a history of Raynaud phenomenon affecting the toes and nipples on cold days. In 2 subsequent pregnancies she was treated with labetalol for pregnancy-induced hypertension, which resulted in severe nipple pain with each pregnancy unrelated to cold weather. Unlike other cases, this patient experienced antenatal symptoms in addition to the typical postnatal symptoms. The nipple pain resolved with discontinuation of the labetalol.4
Barrett et al5 conducted a retrospective review of medical records of 88 breastfeeding mothers who presented with nipple pain and dermatitis. They defined the criteria for Raynaud phenomenon of the nipple as chronic deep breast pain (in general lasting >4 weeks) that responded to therapy for the condition and had at least 2 of the following characteristics: (1) observed or self-reported color changes of the nipple, especially with cold exposure (white, blue, or red); (2) cold sensitivity or color changes of the hands or feet with cold exposure; or (3) failed therapy with oral antifungals. Using these criteria, they diagnosed 22 women (25%) with Raynaud phenomenon of the nipple; 20 (91%) reported a history of cold sensitivity or color change of acral surfaces. Of 12 patients who received and tolerated nifedipine use, 10 (83%) reported decreased pain or complete resolution. This series described breast or nipple pain, whereas other reported cases only described nipple pain. The authors described a sharp, shooting, or stabbing pain—qualifications not previously noted.5 Our patient experienced both nipple pain and a lancinating breast pain consistent with the cases reported by Barrett et al.5
The nipple pain and treatment response in our patient was typical of previously reported cases of vasospasm of the nipple in breastfeeding women; however, Barrett et al5 did not describe individual patients who exhibited the dual nature of the pain described in our patient. The nipple pain experienced during breastfeeding in our patient was successfully treated with nifedipine. We report the successful treatment of the separate lancinating pain with pregabalin.
To the Editor:
Raynaud phenomenon is characterized by vasospasm of arterioles causing intermittent ischemia of the digits. The characteristic triphasic color change presents first as a dramatic change in skin color from normal to white, as the vasoconstriction causes pallor secondary to ischemia. This change is followed by a blue appearance, as cyanosis results from the deoxygenated venous blood. Finally, reflex vasodilation and reperfusion manifest as a red color from erythema. Several cases have been reported describing Raynaud phenomenon affecting the nipples of breastfeeding women.1-5 This vasospasm results in episodic nipple pain manifesting from breastfeeding and exposure to cold. If it is not appropriately treated, the pain’s severity causes affected women to stop breastfeeding. We report a case of vasospasm of the nipple in which the patient experienced nipple pain and a separate lancinating pain that radiated through the breasts.
A 36-year-old woman presented with excruciating nipple and breast pain 3 weeks after delivering her first child. She had no history of smoking or Raynaud phenomenon. The nipple pain was triggered upon breastfeeding and exposure to cold. During these episodes, the nipples would initially blanch white, then turn purple and finally a deep red. The patient also experienced an episodic excruciating lancinating pain of the breast that would randomly and spontaneously radiate through either breast several times per day for 15 to 30 seconds. A workup including an antinuclear antibody test, complete blood cell count with differential, and comprehensive metabolic panel all were within reference range.
The patient was diagnosed with nipple vasospasm. Partial relief of nipple pain occurred after treatment with 30 mg daily of nifedipine; 60 mg daily resulted in complete control, allowing the patient to breastfeed without discomfort, but the lancinating pain continued unabated. The patient could not discontinue breastfeeding because her child was intolerant to formula. She became despondent, as she could find no relief from the pain that she found to be intolerable. Because the patient’s description was reminiscent of the lancinating pain seen in postherpetic neuralgia, a trial of pregabalin was prescribed. A dosage of 75 mg twice daily resulted in near-complete resolution of the pain. After 3 months, the patient successfully weaned her child from breast milk to formula, and the nipple and breast pain promptly resolved. The baby experienced no adverse effects from the patient’s use of pregabalin.
This condition was first described by Gunther1 in 1970 as initial blanching of the nipple followed by a mulberry color. It was termed psychosomatic sore nipples.1 Lawlor-Smith and Lawlor-Smith2 described the condition in 1997 and termed it vasospasm of the nipple. They reported 5 patients who experienced debilitating nipple pain as well as the triphasic color change of Raynaud phenomenon or a biphasic color change (white and blue). Two patients had a history of Raynaud phenomenon affecting the digits before their first pregnancy.2 Anderson et al3 presented 12 breastfeeding women with Raynaud phenomenon of the nipple; only 1 patient had a history of Raynaud phenomenon. In this series, all 6 women who chose to try nifedipine responded well to the drug.3
Raynaud phenomenon of the nipple also has been reported to be associated with the use of labetalol.4 In this case, the patient had a history of Raynaud phenomenon affecting the toes and nipples on cold days. In 2 subsequent pregnancies she was treated with labetalol for pregnancy-induced hypertension, which resulted in severe nipple pain with each pregnancy unrelated to cold weather. Unlike other cases, this patient experienced antenatal symptoms in addition to the typical postnatal symptoms. The nipple pain resolved with discontinuation of the labetalol.4
Barrett et al5 conducted a retrospective review of medical records of 88 breastfeeding mothers who presented with nipple pain and dermatitis. They defined the criteria for Raynaud phenomenon of the nipple as chronic deep breast pain (in general lasting >4 weeks) that responded to therapy for the condition and had at least 2 of the following characteristics: (1) observed or self-reported color changes of the nipple, especially with cold exposure (white, blue, or red); (2) cold sensitivity or color changes of the hands or feet with cold exposure; or (3) failed therapy with oral antifungals. Using these criteria, they diagnosed 22 women (25%) with Raynaud phenomenon of the nipple; 20 (91%) reported a history of cold sensitivity or color change of acral surfaces. Of 12 patients who received and tolerated nifedipine use, 10 (83%) reported decreased pain or complete resolution. This series described breast or nipple pain, whereas other reported cases only described nipple pain. The authors described a sharp, shooting, or stabbing pain—qualifications not previously noted.5 Our patient experienced both nipple pain and a lancinating breast pain consistent with the cases reported by Barrett et al.5
The nipple pain and treatment response in our patient was typical of previously reported cases of vasospasm of the nipple in breastfeeding women; however, Barrett et al5 did not describe individual patients who exhibited the dual nature of the pain described in our patient. The nipple pain experienced during breastfeeding in our patient was successfully treated with nifedipine. We report the successful treatment of the separate lancinating pain with pregabalin.
- Gunther M. Infant Feeding. London, United Kingdom: Methuen; 1970.
- Lawlor-Smith L, Lawlor-Smith C. Vasospasm of the nipple—a manifestation of Raynaud’s phenomenon: case reports. BMJ. 1997;314:644-645.
- Anderson JE, Held N, Wright K. Raynaud phenomenon of the nipple: a treatable cause of painful breastfeeding. Pediatrics. 2004;113:360-364.
- McGuinness N, Cording V. Raynaud’s phenomenon of the nipple associated with labetalol use. J Hum Lact. 2013;29:17-19.
- Barrett ME, Heller MM, Stone HF, et al. Raynaud phenomenon of the nipple in breastfeeding mothers: an underdiagnosed cause of nipple pain. JAMA Dermatol. 2013;149:300-306.
- Gunther M. Infant Feeding. London, United Kingdom: Methuen; 1970.
- Lawlor-Smith L, Lawlor-Smith C. Vasospasm of the nipple—a manifestation of Raynaud’s phenomenon: case reports. BMJ. 1997;314:644-645.
- Anderson JE, Held N, Wright K. Raynaud phenomenon of the nipple: a treatable cause of painful breastfeeding. Pediatrics. 2004;113:360-364.
- McGuinness N, Cording V. Raynaud’s phenomenon of the nipple associated with labetalol use. J Hum Lact. 2013;29:17-19.
- Barrett ME, Heller MM, Stone HF, et al. Raynaud phenomenon of the nipple in breastfeeding mothers: an underdiagnosed cause of nipple pain. JAMA Dermatol. 2013;149:300-306.
Practice Points
- Raynaud phenomenon of the nipple may be accompanied by lancinating pain of the breast in addition to nipple pain reminiscent of postherpetic neuralgia.
- Associated breast pain is particularly distressing for breastfeeding women, particularly primiparous mothers with children intolerant to formula.
- In women with Raynaud phenomenon accompanied by lancinating breast pain, consider a trial of pregabalin.
Radiation Recall Dermatitis Triggered by Prednisone
To the Editor:
A 69-year-old woman presented to the allergy clinic for evaluation of a rash on the left breast. The patient had a history of breast cancer that was treated with a lumpectomy followed by external beam radiation therapy (total dose, 6000 cGy) to the lateral aspect of the left breast approximately 4 years prior. She developed acute breast dermatitis from the radiation, which was self-treated with over-the-counter hydrocortisone cream. The patient subsequently developed a blistering skin eruption over the area where she applied the cream. She did not recall the subtype of hydrocortisone she used (butyrate and acetate are available over-the-counter). She discontinued the hydrocortisone and was started on triamcinolone cream 0.1%, which was well tolerated, and the rash resolved.
The patient had a history of a similar reaction to hydrocortisone butyrate after blepharoplasty approximately 10 years prior to the current presentation, characterized by facial erythema, pruritus, and blistering. A patch test confirmed reactivity to hydrocortisone-17-butyrate and tixocortol pivalate. However, a skin-prick test for hydrocortisone acetate cream 1% was negative.
Subsequently, the patient developed acute-onset dyspepsia, gnawing epigastric pain, regurgitation, and bloating. A diagnosis of eosinophilic gastritis was established via biopsy, which found increased eosinophils in the lamina propria (>50 eosinophils per high-power field). Helicobacter pylori was not identified. She was started on the proton-pump inhibitor dexlansoprazole but symptoms did not improve. Her other medications included benazepril, alprazolam as needed, vitamin D, and magnesium. The patient subsequently was started on a trial of oral prednisone 40 mg/d. Three days after initiation, she developed an erythematous macular rash over the left breast.
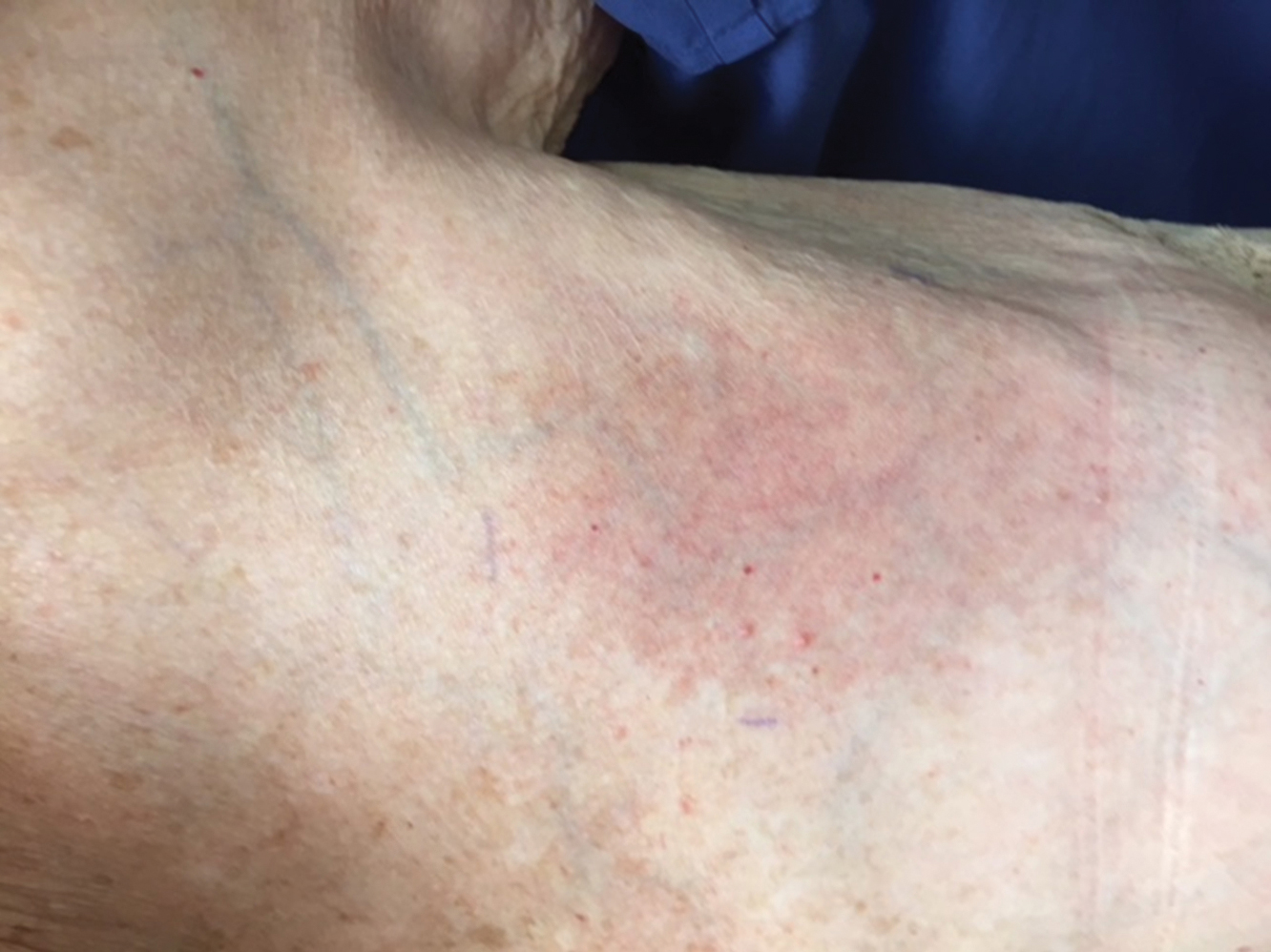
The next day she presented to the allergy clinic. Physical examination of the left breast revealed a 20×10-cm, nipple-sparing patch of well-demarcated erythema without fluctuance or overlying lesions. The area of erythema overlapped with the prior radiation field based on radiation marker tattoos and the lumpectomy scar (Figure). There was no evidence to suggest inflammation of deeper tissue or the pectoral muscles. Vital signs were normal, and the remainder of the examination was unremarkable, including breast, lymph node, and complete skin examinations.
At evaluation, the differential diagnosis included contact dermatitis, fixed drug eruption, infection, tumor recurrence with overlying skin changes, and radiation recall dermatitis. Given that the dermatitis had developed at the site of previously irradiated skin in the absence of fever or an associated mass, the presentation was thought to be most consistent with radiation recall dermatitis.
Oral prednisone was discontinued, and the dermatitis spontaneously improved in a few weeks. Given the patient’s test results and prior tolerance to triamcinolone, eosinophilic gastroenteritis was treated with triamcinolone acetonide 40 mg via intramuscular injection, which was well tolerated.
Radiation recall dermatitis is an acute inflammatory reaction over an area of skin that was previously irradiated. It is most often triggered by chemotherapy agents and occurs in as many as 9% of patients who receive chemotherapy after radiation.1 Commonly implicated chemotherapy agents include anthracyclines, taxanes, antimetabolites, and alkylating agents. Newer targeted cancer treatments also have been reported to trigger radiation recall dermatitis, including epidermal growth factor receptor inhibitors, vascular endothelial growth factor receptor inhibitors, mammalian target of rapamycin inhibitors, and anti–programmed cell death protein 1 monoclonal antibodies.2-5 Radiation recall dermatitis also has been reported to be triggered by intravenous contrast dye.6
The clinical presentation of radiation recall dermatitis ranges from mild rash to skin necrosis and desquamation. Patients often report pruritus or pain in the affected area. The US National Cancer Institute’s Common Terminology Criteria for Adverse Events (CTCAE) includes a 5-point scale for grading the severity of radiation recall dermatitis: grade 1, faint erythema or dry desquamation; grade 2, moderate to brisk erythema or patchy moist desquamation, mostly confined to skin folds and creases; grade 3, moist desquamation in areas other than skin folds and creases, with bleeding induced by minor trauma or abrasion; grade 4, skin necrosis or ulceration of full-thickness dermis, with spontaneous bleeding; grade 5, death.7 Based on these criteria, our patient had grade 2 radiation recall dermatitis.
In addition to cutaneous inflammation, additional sites can be inflamed, including the gastrointestinal tract, lungs, and oral mucosa. Cases of myocarditis, sialadenitis, and cystitis also have been reported.⁷
Radiation recall dermatitis can occur even if dermatitis did not occur upon initial treatment. The inflammatory reaction can occur weeks or years after initial irradiation. A study evaluating targeted chemotherapy agents found the median time from initiation of chemotherapy to radiation recall dermatitis was 16.9 weeks (range, 1–86.9 weeks). Inflammation usually lasts approximately 1 to 2 weeks but has been reported to persist as long as 14 weeks.8 Withdrawal of the offending agent in addition to administration of corticosteroids or nonsteroidal anti-inflammatory agents typically results in clinical improvement. Histology on skin biopsy is nonspecific and can reveal mixed infiltrates.7
The pathophysiology of radiation recall dermatitis remains unknown; the condition might be an idiosyncratic drug reaction. It has been hypothesized that prior radiation lowers the threshold for an inflammatory reaction, an example of Ruocco immunocompromised cutaneous districts, in which a prior injury at a cutaneous site increases the likelihood of opportunistic infection, tumor, and immune reactions.9 Because radiation can induce expression of inflammatory cytokines, such as IL-1, IL-6, platelet-derived growth factor β, and tumor necrosis factor α, cells in irradiated areas can continue to secrete low levels of these cytokines after radiation therapy, thus priming an inflammatory reaction in the future.10 An alternative theory is that radiation induces mutations within surviving stem cells, rendering them unable to tolerate or unusually sensitive to subsequent chemotherapy and cytotoxic drugs. However, this premise would not explain how noncytotoxic drugs also can trigger radiation recall dermatitis, as described in our case.11
Prednisone-triggered radiation recall dermatitis is curious, as corticosteroids are used to treat the condition. Corticosteroids are classified by their chemical structure, and patch testing can be used to distinguish allergies across the various classes. Hydrocortisone acetate,
In contrast, triamcinolone is a class B steroid, which has a C16,17-cis-diol or -ketal. Other than budesonide, which can cross-react with D2 steroids, class B steroids do not cross-react with hydrocortisone or prednisone. Triamcinolone does not usually cross-react with D2 corticosteroids, which likely explains why our patient was later able to tolerate triamcinolone to treat eosinophilic gastrointestinal tract disease.
In summary, we present a case of radiation recall dermatitis triggered by prednisone. Radiation can prime an area for a future inflammatory response by upregulating proinflammatory cytokines or triggering stem cell mutation. In our case, clinical reactivity to hydrocortisone-17-butyrate and sensitization to tixocortol pivalate via patch testing could have increased the likelihood of a reaction with prednisone use due to cross-reactivity. This case instructs dermatologists, allergists, and oncologists to be aware of prednisone as a potential trigger of radiation recall dermatitis.
- Kodym E, Kalinska R, Ehringfeld C, et al. Frequency of radiation recall dermatitis in adult cancer patients. Onkologie. 2005;28:18-21.
- Seidel C, Janssen S, Karstens JH, et al. Recall pneumonitis during systemic treatment with sunitinib. Ann Oncol. 2010;21:2119-2120.
- Togashi Y, Masago K, Mishima M, et al. A case of radiation recall pneumonitis induced by erlotinib, which can be related to high plasma concentration. J Thorac Oncol. 2010;5:924-925.
- Bourgier C, Massard C, Moldovan C, et al. Total recall of radiotherapy with mTOR inhibitors: a novel and potentially frequent side-effect? Ann Oncol. 2011;22:485-486.
- Korman AM, Tyler KH, Kaffenberger BH. Radiation recall dermatitis associated with nivolumab for metastatic malignant melanoma. Int J Dermatol. 2017;56:e75-e77.
- Lau SKM, Rahimi A. Radiation recall precipitated by iodinated nonionic contrast. Pract Radiat Oncol. 2015;5:263-266.
- US Department of Health and Human Services. Common Terminology Criteria for Adverse Events (CTCAE) Version 5.0. https://ctep.cancer.gov/protocoldevelopment/electronic
_applications/docs/CTCAE_v5_Quick_Reference_8.5x11.pdf. Published November 27, 2017. Accessed June 10, 2020.] - Levy A, Hollebecque A, Bourgier C, et al. Targeted therapy-induced radiation recall. Eur J Cancer. 2013;49:1662-1668.
- Piccolo V, Baroni A, Russo T, et al. Ruocco’s immunocompromised cutaneous district. Int J Dermatol. 2016;55:135-141.
- Johnson CJ, Piedboeuf P, Rubin P, et al. Early and persistent alterations in the expression of interleukin-1 alpha, interleukin-1 beta and tumour necrosis factor alpha mRNA levels in fibrosis-resistant and sensitive mice after thoracic irradiation. Radiat Res. 1996;145:762-767.
- Azira D, Magné N, Zouhair A, et al. Radiation recall: a well recognized but neglected phenomenon. Cancer Treat Rev. 2005;31:555-570.
- Jacob SE, Steele T. Corticosteroid classes: a quick reference guide including patch test substances and cross-reactivity. J Am Acad Dermatol. 2006;54:723-727.
To the Editor:
A 69-year-old woman presented to the allergy clinic for evaluation of a rash on the left breast. The patient had a history of breast cancer that was treated with a lumpectomy followed by external beam radiation therapy (total dose, 6000 cGy) to the lateral aspect of the left breast approximately 4 years prior. She developed acute breast dermatitis from the radiation, which was self-treated with over-the-counter hydrocortisone cream. The patient subsequently developed a blistering skin eruption over the area where she applied the cream. She did not recall the subtype of hydrocortisone she used (butyrate and acetate are available over-the-counter). She discontinued the hydrocortisone and was started on triamcinolone cream 0.1%, which was well tolerated, and the rash resolved.
The patient had a history of a similar reaction to hydrocortisone butyrate after blepharoplasty approximately 10 years prior to the current presentation, characterized by facial erythema, pruritus, and blistering. A patch test confirmed reactivity to hydrocortisone-17-butyrate and tixocortol pivalate. However, a skin-prick test for hydrocortisone acetate cream 1% was negative.
Subsequently, the patient developed acute-onset dyspepsia, gnawing epigastric pain, regurgitation, and bloating. A diagnosis of eosinophilic gastritis was established via biopsy, which found increased eosinophils in the lamina propria (>50 eosinophils per high-power field). Helicobacter pylori was not identified. She was started on the proton-pump inhibitor dexlansoprazole but symptoms did not improve. Her other medications included benazepril, alprazolam as needed, vitamin D, and magnesium. The patient subsequently was started on a trial of oral prednisone 40 mg/d. Three days after initiation, she developed an erythematous macular rash over the left breast.
The next day she presented to the allergy clinic. Physical examination of the left breast revealed a 20×10-cm, nipple-sparing patch of well-demarcated erythema without fluctuance or overlying lesions. The area of erythema overlapped with the prior radiation field based on radiation marker tattoos and the lumpectomy scar (Figure). There was no evidence to suggest inflammation of deeper tissue or the pectoral muscles. Vital signs were normal, and the remainder of the examination was unremarkable, including breast, lymph node, and complete skin examinations.
At evaluation, the differential diagnosis included contact dermatitis, fixed drug eruption, infection, tumor recurrence with overlying skin changes, and radiation recall dermatitis. Given that the dermatitis had developed at the site of previously irradiated skin in the absence of fever or an associated mass, the presentation was thought to be most consistent with radiation recall dermatitis.
Oral prednisone was discontinued, and the dermatitis spontaneously improved in a few weeks. Given the patient’s test results and prior tolerance to triamcinolone, eosinophilic gastroenteritis was treated with triamcinolone acetonide 40 mg via intramuscular injection, which was well tolerated.
Radiation recall dermatitis is an acute inflammatory reaction over an area of skin that was previously irradiated. It is most often triggered by chemotherapy agents and occurs in as many as 9% of patients who receive chemotherapy after radiation.1 Commonly implicated chemotherapy agents include anthracyclines, taxanes, antimetabolites, and alkylating agents. Newer targeted cancer treatments also have been reported to trigger radiation recall dermatitis, including epidermal growth factor receptor inhibitors, vascular endothelial growth factor receptor inhibitors, mammalian target of rapamycin inhibitors, and anti–programmed cell death protein 1 monoclonal antibodies.2-5 Radiation recall dermatitis also has been reported to be triggered by intravenous contrast dye.6
The clinical presentation of radiation recall dermatitis ranges from mild rash to skin necrosis and desquamation. Patients often report pruritus or pain in the affected area. The US National Cancer Institute’s Common Terminology Criteria for Adverse Events (CTCAE) includes a 5-point scale for grading the severity of radiation recall dermatitis: grade 1, faint erythema or dry desquamation; grade 2, moderate to brisk erythema or patchy moist desquamation, mostly confined to skin folds and creases; grade 3, moist desquamation in areas other than skin folds and creases, with bleeding induced by minor trauma or abrasion; grade 4, skin necrosis or ulceration of full-thickness dermis, with spontaneous bleeding; grade 5, death.7 Based on these criteria, our patient had grade 2 radiation recall dermatitis.
In addition to cutaneous inflammation, additional sites can be inflamed, including the gastrointestinal tract, lungs, and oral mucosa. Cases of myocarditis, sialadenitis, and cystitis also have been reported.⁷
Radiation recall dermatitis can occur even if dermatitis did not occur upon initial treatment. The inflammatory reaction can occur weeks or years after initial irradiation. A study evaluating targeted chemotherapy agents found the median time from initiation of chemotherapy to radiation recall dermatitis was 16.9 weeks (range, 1–86.9 weeks). Inflammation usually lasts approximately 1 to 2 weeks but has been reported to persist as long as 14 weeks.8 Withdrawal of the offending agent in addition to administration of corticosteroids or nonsteroidal anti-inflammatory agents typically results in clinical improvement. Histology on skin biopsy is nonspecific and can reveal mixed infiltrates.7
The pathophysiology of radiation recall dermatitis remains unknown; the condition might be an idiosyncratic drug reaction. It has been hypothesized that prior radiation lowers the threshold for an inflammatory reaction, an example of Ruocco immunocompromised cutaneous districts, in which a prior injury at a cutaneous site increases the likelihood of opportunistic infection, tumor, and immune reactions.9 Because radiation can induce expression of inflammatory cytokines, such as IL-1, IL-6, platelet-derived growth factor β, and tumor necrosis factor α, cells in irradiated areas can continue to secrete low levels of these cytokines after radiation therapy, thus priming an inflammatory reaction in the future.10 An alternative theory is that radiation induces mutations within surviving stem cells, rendering them unable to tolerate or unusually sensitive to subsequent chemotherapy and cytotoxic drugs. However, this premise would not explain how noncytotoxic drugs also can trigger radiation recall dermatitis, as described in our case.11
Prednisone-triggered radiation recall dermatitis is curious, as corticosteroids are used to treat the condition. Corticosteroids are classified by their chemical structure, and patch testing can be used to distinguish allergies across the various classes. Hydrocortisone acetate,
In contrast, triamcinolone is a class B steroid, which has a C16,17-cis-diol or -ketal. Other than budesonide, which can cross-react with D2 steroids, class B steroids do not cross-react with hydrocortisone or prednisone. Triamcinolone does not usually cross-react with D2 corticosteroids, which likely explains why our patient was later able to tolerate triamcinolone to treat eosinophilic gastrointestinal tract disease.
In summary, we present a case of radiation recall dermatitis triggered by prednisone. Radiation can prime an area for a future inflammatory response by upregulating proinflammatory cytokines or triggering stem cell mutation. In our case, clinical reactivity to hydrocortisone-17-butyrate and sensitization to tixocortol pivalate via patch testing could have increased the likelihood of a reaction with prednisone use due to cross-reactivity. This case instructs dermatologists, allergists, and oncologists to be aware of prednisone as a potential trigger of radiation recall dermatitis.
To the Editor:
A 69-year-old woman presented to the allergy clinic for evaluation of a rash on the left breast. The patient had a history of breast cancer that was treated with a lumpectomy followed by external beam radiation therapy (total dose, 6000 cGy) to the lateral aspect of the left breast approximately 4 years prior. She developed acute breast dermatitis from the radiation, which was self-treated with over-the-counter hydrocortisone cream. The patient subsequently developed a blistering skin eruption over the area where she applied the cream. She did not recall the subtype of hydrocortisone she used (butyrate and acetate are available over-the-counter). She discontinued the hydrocortisone and was started on triamcinolone cream 0.1%, which was well tolerated, and the rash resolved.
The patient had a history of a similar reaction to hydrocortisone butyrate after blepharoplasty approximately 10 years prior to the current presentation, characterized by facial erythema, pruritus, and blistering. A patch test confirmed reactivity to hydrocortisone-17-butyrate and tixocortol pivalate. However, a skin-prick test for hydrocortisone acetate cream 1% was negative.
Subsequently, the patient developed acute-onset dyspepsia, gnawing epigastric pain, regurgitation, and bloating. A diagnosis of eosinophilic gastritis was established via biopsy, which found increased eosinophils in the lamina propria (>50 eosinophils per high-power field). Helicobacter pylori was not identified. She was started on the proton-pump inhibitor dexlansoprazole but symptoms did not improve. Her other medications included benazepril, alprazolam as needed, vitamin D, and magnesium. The patient subsequently was started on a trial of oral prednisone 40 mg/d. Three days after initiation, she developed an erythematous macular rash over the left breast.
The next day she presented to the allergy clinic. Physical examination of the left breast revealed a 20×10-cm, nipple-sparing patch of well-demarcated erythema without fluctuance or overlying lesions. The area of erythema overlapped with the prior radiation field based on radiation marker tattoos and the lumpectomy scar (Figure). There was no evidence to suggest inflammation of deeper tissue or the pectoral muscles. Vital signs were normal, and the remainder of the examination was unremarkable, including breast, lymph node, and complete skin examinations.
At evaluation, the differential diagnosis included contact dermatitis, fixed drug eruption, infection, tumor recurrence with overlying skin changes, and radiation recall dermatitis. Given that the dermatitis had developed at the site of previously irradiated skin in the absence of fever or an associated mass, the presentation was thought to be most consistent with radiation recall dermatitis.
Oral prednisone was discontinued, and the dermatitis spontaneously improved in a few weeks. Given the patient’s test results and prior tolerance to triamcinolone, eosinophilic gastroenteritis was treated with triamcinolone acetonide 40 mg via intramuscular injection, which was well tolerated.
Radiation recall dermatitis is an acute inflammatory reaction over an area of skin that was previously irradiated. It is most often triggered by chemotherapy agents and occurs in as many as 9% of patients who receive chemotherapy after radiation.1 Commonly implicated chemotherapy agents include anthracyclines, taxanes, antimetabolites, and alkylating agents. Newer targeted cancer treatments also have been reported to trigger radiation recall dermatitis, including epidermal growth factor receptor inhibitors, vascular endothelial growth factor receptor inhibitors, mammalian target of rapamycin inhibitors, and anti–programmed cell death protein 1 monoclonal antibodies.2-5 Radiation recall dermatitis also has been reported to be triggered by intravenous contrast dye.6
The clinical presentation of radiation recall dermatitis ranges from mild rash to skin necrosis and desquamation. Patients often report pruritus or pain in the affected area. The US National Cancer Institute’s Common Terminology Criteria for Adverse Events (CTCAE) includes a 5-point scale for grading the severity of radiation recall dermatitis: grade 1, faint erythema or dry desquamation; grade 2, moderate to brisk erythema or patchy moist desquamation, mostly confined to skin folds and creases; grade 3, moist desquamation in areas other than skin folds and creases, with bleeding induced by minor trauma or abrasion; grade 4, skin necrosis or ulceration of full-thickness dermis, with spontaneous bleeding; grade 5, death.7 Based on these criteria, our patient had grade 2 radiation recall dermatitis.
In addition to cutaneous inflammation, additional sites can be inflamed, including the gastrointestinal tract, lungs, and oral mucosa. Cases of myocarditis, sialadenitis, and cystitis also have been reported.⁷
Radiation recall dermatitis can occur even if dermatitis did not occur upon initial treatment. The inflammatory reaction can occur weeks or years after initial irradiation. A study evaluating targeted chemotherapy agents found the median time from initiation of chemotherapy to radiation recall dermatitis was 16.9 weeks (range, 1–86.9 weeks). Inflammation usually lasts approximately 1 to 2 weeks but has been reported to persist as long as 14 weeks.8 Withdrawal of the offending agent in addition to administration of corticosteroids or nonsteroidal anti-inflammatory agents typically results in clinical improvement. Histology on skin biopsy is nonspecific and can reveal mixed infiltrates.7
The pathophysiology of radiation recall dermatitis remains unknown; the condition might be an idiosyncratic drug reaction. It has been hypothesized that prior radiation lowers the threshold for an inflammatory reaction, an example of Ruocco immunocompromised cutaneous districts, in which a prior injury at a cutaneous site increases the likelihood of opportunistic infection, tumor, and immune reactions.9 Because radiation can induce expression of inflammatory cytokines, such as IL-1, IL-6, platelet-derived growth factor β, and tumor necrosis factor α, cells in irradiated areas can continue to secrete low levels of these cytokines after radiation therapy, thus priming an inflammatory reaction in the future.10 An alternative theory is that radiation induces mutations within surviving stem cells, rendering them unable to tolerate or unusually sensitive to subsequent chemotherapy and cytotoxic drugs. However, this premise would not explain how noncytotoxic drugs also can trigger radiation recall dermatitis, as described in our case.11
Prednisone-triggered radiation recall dermatitis is curious, as corticosteroids are used to treat the condition. Corticosteroids are classified by their chemical structure, and patch testing can be used to distinguish allergies across the various classes. Hydrocortisone acetate,
In contrast, triamcinolone is a class B steroid, which has a C16,17-cis-diol or -ketal. Other than budesonide, which can cross-react with D2 steroids, class B steroids do not cross-react with hydrocortisone or prednisone. Triamcinolone does not usually cross-react with D2 corticosteroids, which likely explains why our patient was later able to tolerate triamcinolone to treat eosinophilic gastrointestinal tract disease.
In summary, we present a case of radiation recall dermatitis triggered by prednisone. Radiation can prime an area for a future inflammatory response by upregulating proinflammatory cytokines or triggering stem cell mutation. In our case, clinical reactivity to hydrocortisone-17-butyrate and sensitization to tixocortol pivalate via patch testing could have increased the likelihood of a reaction with prednisone use due to cross-reactivity. This case instructs dermatologists, allergists, and oncologists to be aware of prednisone as a potential trigger of radiation recall dermatitis.
- Kodym E, Kalinska R, Ehringfeld C, et al. Frequency of radiation recall dermatitis in adult cancer patients. Onkologie. 2005;28:18-21.
- Seidel C, Janssen S, Karstens JH, et al. Recall pneumonitis during systemic treatment with sunitinib. Ann Oncol. 2010;21:2119-2120.
- Togashi Y, Masago K, Mishima M, et al. A case of radiation recall pneumonitis induced by erlotinib, which can be related to high plasma concentration. J Thorac Oncol. 2010;5:924-925.
- Bourgier C, Massard C, Moldovan C, et al. Total recall of radiotherapy with mTOR inhibitors: a novel and potentially frequent side-effect? Ann Oncol. 2011;22:485-486.
- Korman AM, Tyler KH, Kaffenberger BH. Radiation recall dermatitis associated with nivolumab for metastatic malignant melanoma. Int J Dermatol. 2017;56:e75-e77.
- Lau SKM, Rahimi A. Radiation recall precipitated by iodinated nonionic contrast. Pract Radiat Oncol. 2015;5:263-266.
- US Department of Health and Human Services. Common Terminology Criteria for Adverse Events (CTCAE) Version 5.0. https://ctep.cancer.gov/protocoldevelopment/electronic
_applications/docs/CTCAE_v5_Quick_Reference_8.5x11.pdf. Published November 27, 2017. Accessed June 10, 2020.] - Levy A, Hollebecque A, Bourgier C, et al. Targeted therapy-induced radiation recall. Eur J Cancer. 2013;49:1662-1668.
- Piccolo V, Baroni A, Russo T, et al. Ruocco’s immunocompromised cutaneous district. Int J Dermatol. 2016;55:135-141.
- Johnson CJ, Piedboeuf P, Rubin P, et al. Early and persistent alterations in the expression of interleukin-1 alpha, interleukin-1 beta and tumour necrosis factor alpha mRNA levels in fibrosis-resistant and sensitive mice after thoracic irradiation. Radiat Res. 1996;145:762-767.
- Azira D, Magné N, Zouhair A, et al. Radiation recall: a well recognized but neglected phenomenon. Cancer Treat Rev. 2005;31:555-570.
- Jacob SE, Steele T. Corticosteroid classes: a quick reference guide including patch test substances and cross-reactivity. J Am Acad Dermatol. 2006;54:723-727.
- Kodym E, Kalinska R, Ehringfeld C, et al. Frequency of radiation recall dermatitis in adult cancer patients. Onkologie. 2005;28:18-21.
- Seidel C, Janssen S, Karstens JH, et al. Recall pneumonitis during systemic treatment with sunitinib. Ann Oncol. 2010;21:2119-2120.
- Togashi Y, Masago K, Mishima M, et al. A case of radiation recall pneumonitis induced by erlotinib, which can be related to high plasma concentration. J Thorac Oncol. 2010;5:924-925.
- Bourgier C, Massard C, Moldovan C, et al. Total recall of radiotherapy with mTOR inhibitors: a novel and potentially frequent side-effect? Ann Oncol. 2011;22:485-486.
- Korman AM, Tyler KH, Kaffenberger BH. Radiation recall dermatitis associated with nivolumab for metastatic malignant melanoma. Int J Dermatol. 2017;56:e75-e77.
- Lau SKM, Rahimi A. Radiation recall precipitated by iodinated nonionic contrast. Pract Radiat Oncol. 2015;5:263-266.
- US Department of Health and Human Services. Common Terminology Criteria for Adverse Events (CTCAE) Version 5.0. https://ctep.cancer.gov/protocoldevelopment/electronic
_applications/docs/CTCAE_v5_Quick_Reference_8.5x11.pdf. Published November 27, 2017. Accessed June 10, 2020.] - Levy A, Hollebecque A, Bourgier C, et al. Targeted therapy-induced radiation recall. Eur J Cancer. 2013;49:1662-1668.
- Piccolo V, Baroni A, Russo T, et al. Ruocco’s immunocompromised cutaneous district. Int J Dermatol. 2016;55:135-141.
- Johnson CJ, Piedboeuf P, Rubin P, et al. Early and persistent alterations in the expression of interleukin-1 alpha, interleukin-1 beta and tumour necrosis factor alpha mRNA levels in fibrosis-resistant and sensitive mice after thoracic irradiation. Radiat Res. 1996;145:762-767.
- Azira D, Magné N, Zouhair A, et al. Radiation recall: a well recognized but neglected phenomenon. Cancer Treat Rev. 2005;31:555-570.
- Jacob SE, Steele T. Corticosteroid classes: a quick reference guide including patch test substances and cross-reactivity. J Am Acad Dermatol. 2006;54:723-727.
Practice Points
- Consider the diagnosis of radiation recall dermatitis for a skin eruption that occurs in the same location as prior radiation exposure.
- Prednisone may be a trigger for radiation recall dermatitis in patients with sensitization to cross-reactive topical steroids such as tixocortol pivalate.
- Radiation therapy may prime the skin for a future inflammatory response by upregulating proinflammatory cytokines that persist after the conclusion of treatment.
Mycosis Fungoides Manifesting as a Morbilliform Eruption Mimicking a Viral Exanthem
To the Editor:
Mycosis fungoides (MF) is the most common type of primary cutaneous lymphoma, occurring in approximately 4 of 1 million individuals per year in the United States.1 It classically occurs in patch, plaque, and tumor stages with lesions preferentially occurring on regions of the body spared from sun exposure2; however, MF is known to have variable presentations and has been reported to imitate at least 25 other dermatoses.3 This case describes MF as a morbilliform eruption mimicking a viral exanthem.
A 30-year-old man with a 12-year history of nodular sclerosing Hodgkin lymphoma (HL) presented with a widespread rash of 2 weeks’ duration. At the time of diagnosis of HL, the patient had several slightly enlarged, hyperdense, bilateral inguinal lymph nodes seen on positron emission tomography–computed tomography. He achieved complete remission 11 years prior after 6 cycles of ABVD (doxorubicin-bleomycin-vinblastine-dacarbazine) chemotherapy. He initially presented to us prior to starting chemotherapy for evaluation of what he described as eczema on the bilateral arms and legs that had been present for 10 years. Findings from a skin biopsy of an erythematous scaling patch on the left lateral thigh were consistent with MF. One year later, new lesions on the left lateral thigh were clinically and histologically consistent with lymphomatoid papulosis (LyP).
At the current presentation, the patient denied any changes in medications, which consisted of topical clobetasol, triamcinolone, and mupirocin; however, he reported that his young child had recently been diagnosed with bronchitis and impetigo. Physical examination revealed pink-orange macules and papules on the anterior and posterior trunk, medial upper arms, and bilateral legs involving 18% of the body surface area. A complete blood cell count showed no leukocytosis or left shift. A respiratory viral panel was positive for human metapneumovirus. Two weeks later, the patient noted improvement of the rash with use of topical triamcinolone.
Four months later, the rash still had not completely resolved and now involved 50% of the body surface area. A punch biopsy of the left lower abdomen demonstrated an atypical lymphoid infiltrate with focal epidermotropism and predominance of CD4 over CD8 cells (approximately 4:1 ratio), and CD30 labeled rare cells. Polymerase chain reaction analysis of the biopsy revealed monoclonal T-cell receptor gamma chain gene rearrangement. Taken together, the findings were consistent with MF. The patient started narrowband UVB phototherapy and completed a total of 25 treatments, reaching a maximum 4-minute dose, with minimal improvement.
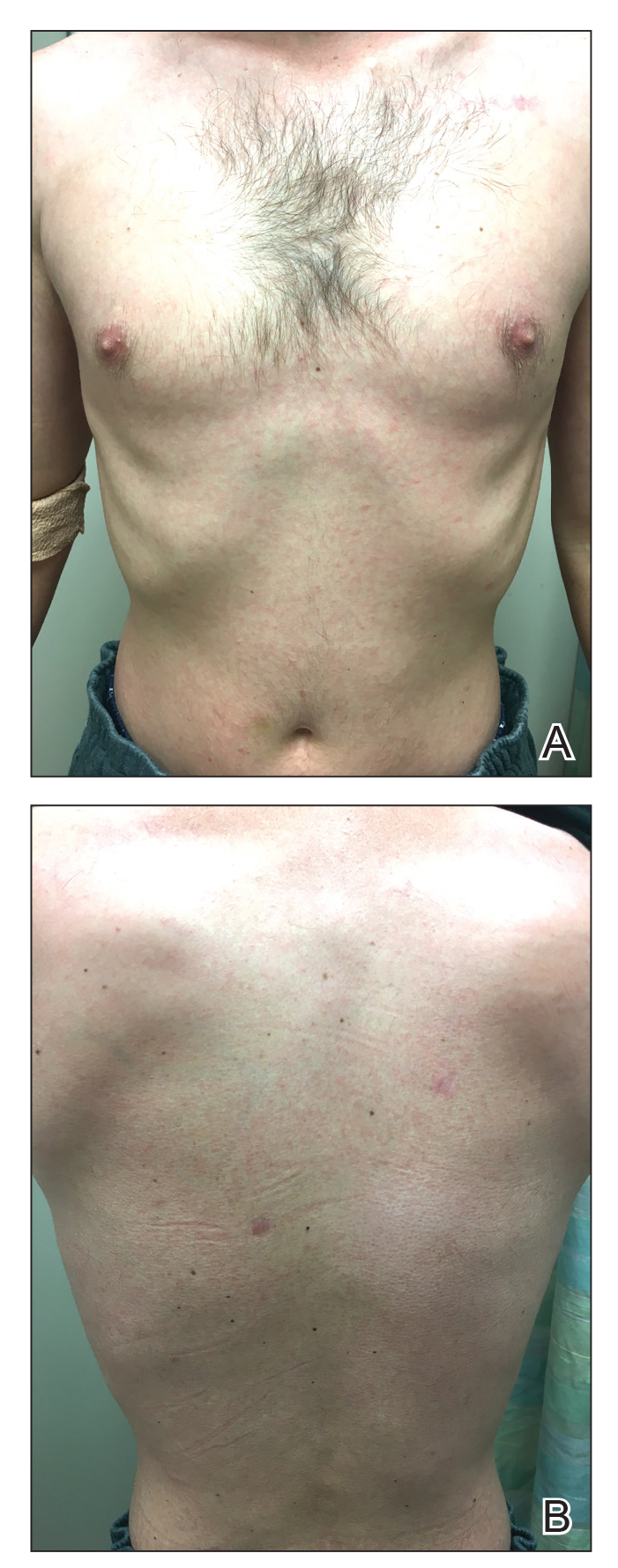
Three months later, the patient had 90% body surface area involvement and started treatment with intramuscular interferon alfa-2b at 1 million units 3 times weekly. He noticed improvement within the first week of treatment and reported that his skin was clear until 5 months later when he woke up one morning with a morbilliform eruption on the anterior trunk, thighs, and upper arms (Figure 1). Biopsy from the right thigh showed an infiltrate of CD3+ lymphocytes with a predominance of CD4 over CD8 cells (approximately 6:1 ratio), both in the dermis and epidermis (Figure 2). CD30 highlighted approximately 10% of cells (Figure 3). Findings again were consistent with MF. Flow cytometry was negative for peripheral blood involvement.
Three months later, the patient reported enlargement of several left inguinal nodes. Fine needle aspiration of 1 node demonstrated an atypical lymphoid proliferation consistent with MF. Positron emission tomography–computed tomography showed several mildly enlarged inguinal lymph nodes, which were unchanged from the initial diagnosis of HL. There were no hypermetabolic lesions. One month later, the patient started extracorporeal electrophoresis in addition to interferon alfa-2b with notable improvement of the rash. The rash later recurred after completion of these treatments and continues to have a waxing and waning course. It is currently managed with triamcinolone cream only.
At the time of the initial diagnosis of MF, the patient’s lesions appeared as eczematous patches on the face, abdomen, buttocks, and legs. Based on the history of a sick child at home, viral panel positive for human metapneumovirus, and clinical appearance, a viral exanthem was considered to be a likely explanation for the patient’s new-onset morbilliform eruption rash occurring 12 years later. A drug reaction also was considered in the differential based on the appearance of the rash; however, it was deemed less likely because the patient reported no changes in his medications at the time of rash onset. Persistence of the eruption for many months was less consistent with a reactive condition. A biopsy demonstrated the rash to be histologically consistent with MF. This patient was a rare case of MF manifesting as a morbilliform eruption mimicking a viral exanthem.
Various inflammatory conditions, including drug eruptions and lichen sclerosus et atrophicus, may mimic MF, not only based on their histophenotypic findings but also occasionally clonal proliferation by molecular study.4,5 In our patient, one consideration was the possibility of a viral infection mimicking MF; however, biopsies showed both definite histophenotypic features of MF and clonality. More importantly, subsequent biopsy also revealed similar findings by morphology, immunohistochemical study, and T-cell gene rearrangement study, confirming the diagnosis of MF.
Another interesting feature of our case was the occurrence of HL, LyP, and MF in the same patient. Lymphomatoid papulosis is a chronic condition characterized by self-healing lesions and histologic features suggestive of malignancy that lies within a spectrum of primary cutaneous CD30+ lymphoproliferative disorders. There is a known association between LyP and an increased incidence of lymphomas, including MF and HL.1 In a 2016 study, lymphomas occurred in 52% of patients with LyP (N=180), with MF being the most frequently associated lymphoma.6 Notably, biopsies consistent with both HL and MF, respectively, in our patient were positive for the CD30 marker. Patients with HL also are at increased risk for developing other malignancies, with the risk of leukemias and non-HLs greater than that of solid tumors.5 There have been multiple reported cases of HL and MF occurring in the same patient and at least one prior reported case of LyP, HL, and MF occurring in the same patient.6,7
This case highlights the myriad presentations of MF and describes an unusual case of MF manifesting as a morbilliform eruption mimicking a viral exanthem.
- de la Garza Bravo MM, Patel KP, Loghavi S, et al. Shared clonality in distinctive lesions of lymphomatoid papulosis and mycosis fungoides occurring in the same patients suggests a common origin [published online December 31, 2014]. Hum Pathol. 2015;46:558-569.
- Howard MS, Smoller BR. Mycosis fungoides: classic disease and variant presentations. Semin Cutan Med Surg. 2000;19:91-99.
- Zackheim HS, Mccalmont TH. Mycosis fungoides: the great imitator. J Am Acad Dermatol. 2002;47:914-918.
- Suchak R, Verdolini R, Robson A, et al. Extragenital lichen sclerosus et atrophicus mimicking cutaneous T-cell lymphoma: report of a case. J Cutan Pathol. 2010;37:982-986.
- Sarantopoulos GP, Palla B, Said J, et al. Mimics of cutaneous lymphoma: report of the 2011 Society for Hematopathology/European Association for Haematopathology workshop. Am J Clin Pathol. 2013;139:536-551.
- Wieser I, Oh CW, Talpur R, et al. Lymphomatoid papulosis: treatment response and associated lymphomas in a study of 180 patients. J Am Acad Dermatol. 2016;74:59-67.
- Sont JK, van Stiphout WA, Noordijk EM, et al. Increased risk of second cancers in managing Hodgkins disease: the 20-year Leiden experience. Ann Hematol. 1992;65:213-218.
To the Editor:
Mycosis fungoides (MF) is the most common type of primary cutaneous lymphoma, occurring in approximately 4 of 1 million individuals per year in the United States.1 It classically occurs in patch, plaque, and tumor stages with lesions preferentially occurring on regions of the body spared from sun exposure2; however, MF is known to have variable presentations and has been reported to imitate at least 25 other dermatoses.3 This case describes MF as a morbilliform eruption mimicking a viral exanthem.
A 30-year-old man with a 12-year history of nodular sclerosing Hodgkin lymphoma (HL) presented with a widespread rash of 2 weeks’ duration. At the time of diagnosis of HL, the patient had several slightly enlarged, hyperdense, bilateral inguinal lymph nodes seen on positron emission tomography–computed tomography. He achieved complete remission 11 years prior after 6 cycles of ABVD (doxorubicin-bleomycin-vinblastine-dacarbazine) chemotherapy. He initially presented to us prior to starting chemotherapy for evaluation of what he described as eczema on the bilateral arms and legs that had been present for 10 years. Findings from a skin biopsy of an erythematous scaling patch on the left lateral thigh were consistent with MF. One year later, new lesions on the left lateral thigh were clinically and histologically consistent with lymphomatoid papulosis (LyP).
At the current presentation, the patient denied any changes in medications, which consisted of topical clobetasol, triamcinolone, and mupirocin; however, he reported that his young child had recently been diagnosed with bronchitis and impetigo. Physical examination revealed pink-orange macules and papules on the anterior and posterior trunk, medial upper arms, and bilateral legs involving 18% of the body surface area. A complete blood cell count showed no leukocytosis or left shift. A respiratory viral panel was positive for human metapneumovirus. Two weeks later, the patient noted improvement of the rash with use of topical triamcinolone.
Four months later, the rash still had not completely resolved and now involved 50% of the body surface area. A punch biopsy of the left lower abdomen demonstrated an atypical lymphoid infiltrate with focal epidermotropism and predominance of CD4 over CD8 cells (approximately 4:1 ratio), and CD30 labeled rare cells. Polymerase chain reaction analysis of the biopsy revealed monoclonal T-cell receptor gamma chain gene rearrangement. Taken together, the findings were consistent with MF. The patient started narrowband UVB phototherapy and completed a total of 25 treatments, reaching a maximum 4-minute dose, with minimal improvement.
Three months later, the patient had 90% body surface area involvement and started treatment with intramuscular interferon alfa-2b at 1 million units 3 times weekly. He noticed improvement within the first week of treatment and reported that his skin was clear until 5 months later when he woke up one morning with a morbilliform eruption on the anterior trunk, thighs, and upper arms (Figure 1). Biopsy from the right thigh showed an infiltrate of CD3+ lymphocytes with a predominance of CD4 over CD8 cells (approximately 6:1 ratio), both in the dermis and epidermis (Figure 2). CD30 highlighted approximately 10% of cells (Figure 3). Findings again were consistent with MF. Flow cytometry was negative for peripheral blood involvement.
Three months later, the patient reported enlargement of several left inguinal nodes. Fine needle aspiration of 1 node demonstrated an atypical lymphoid proliferation consistent with MF. Positron emission tomography–computed tomography showed several mildly enlarged inguinal lymph nodes, which were unchanged from the initial diagnosis of HL. There were no hypermetabolic lesions. One month later, the patient started extracorporeal electrophoresis in addition to interferon alfa-2b with notable improvement of the rash. The rash later recurred after completion of these treatments and continues to have a waxing and waning course. It is currently managed with triamcinolone cream only.
At the time of the initial diagnosis of MF, the patient’s lesions appeared as eczematous patches on the face, abdomen, buttocks, and legs. Based on the history of a sick child at home, viral panel positive for human metapneumovirus, and clinical appearance, a viral exanthem was considered to be a likely explanation for the patient’s new-onset morbilliform eruption rash occurring 12 years later. A drug reaction also was considered in the differential based on the appearance of the rash; however, it was deemed less likely because the patient reported no changes in his medications at the time of rash onset. Persistence of the eruption for many months was less consistent with a reactive condition. A biopsy demonstrated the rash to be histologically consistent with MF. This patient was a rare case of MF manifesting as a morbilliform eruption mimicking a viral exanthem.
Various inflammatory conditions, including drug eruptions and lichen sclerosus et atrophicus, may mimic MF, not only based on their histophenotypic findings but also occasionally clonal proliferation by molecular study.4,5 In our patient, one consideration was the possibility of a viral infection mimicking MF; however, biopsies showed both definite histophenotypic features of MF and clonality. More importantly, subsequent biopsy also revealed similar findings by morphology, immunohistochemical study, and T-cell gene rearrangement study, confirming the diagnosis of MF.
Another interesting feature of our case was the occurrence of HL, LyP, and MF in the same patient. Lymphomatoid papulosis is a chronic condition characterized by self-healing lesions and histologic features suggestive of malignancy that lies within a spectrum of primary cutaneous CD30+ lymphoproliferative disorders. There is a known association between LyP and an increased incidence of lymphomas, including MF and HL.1 In a 2016 study, lymphomas occurred in 52% of patients with LyP (N=180), with MF being the most frequently associated lymphoma.6 Notably, biopsies consistent with both HL and MF, respectively, in our patient were positive for the CD30 marker. Patients with HL also are at increased risk for developing other malignancies, with the risk of leukemias and non-HLs greater than that of solid tumors.5 There have been multiple reported cases of HL and MF occurring in the same patient and at least one prior reported case of LyP, HL, and MF occurring in the same patient.6,7
This case highlights the myriad presentations of MF and describes an unusual case of MF manifesting as a morbilliform eruption mimicking a viral exanthem.
To the Editor:
Mycosis fungoides (MF) is the most common type of primary cutaneous lymphoma, occurring in approximately 4 of 1 million individuals per year in the United States.1 It classically occurs in patch, plaque, and tumor stages with lesions preferentially occurring on regions of the body spared from sun exposure2; however, MF is known to have variable presentations and has been reported to imitate at least 25 other dermatoses.3 This case describes MF as a morbilliform eruption mimicking a viral exanthem.
A 30-year-old man with a 12-year history of nodular sclerosing Hodgkin lymphoma (HL) presented with a widespread rash of 2 weeks’ duration. At the time of diagnosis of HL, the patient had several slightly enlarged, hyperdense, bilateral inguinal lymph nodes seen on positron emission tomography–computed tomography. He achieved complete remission 11 years prior after 6 cycles of ABVD (doxorubicin-bleomycin-vinblastine-dacarbazine) chemotherapy. He initially presented to us prior to starting chemotherapy for evaluation of what he described as eczema on the bilateral arms and legs that had been present for 10 years. Findings from a skin biopsy of an erythematous scaling patch on the left lateral thigh were consistent with MF. One year later, new lesions on the left lateral thigh were clinically and histologically consistent with lymphomatoid papulosis (LyP).
At the current presentation, the patient denied any changes in medications, which consisted of topical clobetasol, triamcinolone, and mupirocin; however, he reported that his young child had recently been diagnosed with bronchitis and impetigo. Physical examination revealed pink-orange macules and papules on the anterior and posterior trunk, medial upper arms, and bilateral legs involving 18% of the body surface area. A complete blood cell count showed no leukocytosis or left shift. A respiratory viral panel was positive for human metapneumovirus. Two weeks later, the patient noted improvement of the rash with use of topical triamcinolone.
Four months later, the rash still had not completely resolved and now involved 50% of the body surface area. A punch biopsy of the left lower abdomen demonstrated an atypical lymphoid infiltrate with focal epidermotropism and predominance of CD4 over CD8 cells (approximately 4:1 ratio), and CD30 labeled rare cells. Polymerase chain reaction analysis of the biopsy revealed monoclonal T-cell receptor gamma chain gene rearrangement. Taken together, the findings were consistent with MF. The patient started narrowband UVB phototherapy and completed a total of 25 treatments, reaching a maximum 4-minute dose, with minimal improvement.
Three months later, the patient had 90% body surface area involvement and started treatment with intramuscular interferon alfa-2b at 1 million units 3 times weekly. He noticed improvement within the first week of treatment and reported that his skin was clear until 5 months later when he woke up one morning with a morbilliform eruption on the anterior trunk, thighs, and upper arms (Figure 1). Biopsy from the right thigh showed an infiltrate of CD3+ lymphocytes with a predominance of CD4 over CD8 cells (approximately 6:1 ratio), both in the dermis and epidermis (Figure 2). CD30 highlighted approximately 10% of cells (Figure 3). Findings again were consistent with MF. Flow cytometry was negative for peripheral blood involvement.
Three months later, the patient reported enlargement of several left inguinal nodes. Fine needle aspiration of 1 node demonstrated an atypical lymphoid proliferation consistent with MF. Positron emission tomography–computed tomography showed several mildly enlarged inguinal lymph nodes, which were unchanged from the initial diagnosis of HL. There were no hypermetabolic lesions. One month later, the patient started extracorporeal electrophoresis in addition to interferon alfa-2b with notable improvement of the rash. The rash later recurred after completion of these treatments and continues to have a waxing and waning course. It is currently managed with triamcinolone cream only.
At the time of the initial diagnosis of MF, the patient’s lesions appeared as eczematous patches on the face, abdomen, buttocks, and legs. Based on the history of a sick child at home, viral panel positive for human metapneumovirus, and clinical appearance, a viral exanthem was considered to be a likely explanation for the patient’s new-onset morbilliform eruption rash occurring 12 years later. A drug reaction also was considered in the differential based on the appearance of the rash; however, it was deemed less likely because the patient reported no changes in his medications at the time of rash onset. Persistence of the eruption for many months was less consistent with a reactive condition. A biopsy demonstrated the rash to be histologically consistent with MF. This patient was a rare case of MF manifesting as a morbilliform eruption mimicking a viral exanthem.
Various inflammatory conditions, including drug eruptions and lichen sclerosus et atrophicus, may mimic MF, not only based on their histophenotypic findings but also occasionally clonal proliferation by molecular study.4,5 In our patient, one consideration was the possibility of a viral infection mimicking MF; however, biopsies showed both definite histophenotypic features of MF and clonality. More importantly, subsequent biopsy also revealed similar findings by morphology, immunohistochemical study, and T-cell gene rearrangement study, confirming the diagnosis of MF.
Another interesting feature of our case was the occurrence of HL, LyP, and MF in the same patient. Lymphomatoid papulosis is a chronic condition characterized by self-healing lesions and histologic features suggestive of malignancy that lies within a spectrum of primary cutaneous CD30+ lymphoproliferative disorders. There is a known association between LyP and an increased incidence of lymphomas, including MF and HL.1 In a 2016 study, lymphomas occurred in 52% of patients with LyP (N=180), with MF being the most frequently associated lymphoma.6 Notably, biopsies consistent with both HL and MF, respectively, in our patient were positive for the CD30 marker. Patients with HL also are at increased risk for developing other malignancies, with the risk of leukemias and non-HLs greater than that of solid tumors.5 There have been multiple reported cases of HL and MF occurring in the same patient and at least one prior reported case of LyP, HL, and MF occurring in the same patient.6,7
This case highlights the myriad presentations of MF and describes an unusual case of MF manifesting as a morbilliform eruption mimicking a viral exanthem.
- de la Garza Bravo MM, Patel KP, Loghavi S, et al. Shared clonality in distinctive lesions of lymphomatoid papulosis and mycosis fungoides occurring in the same patients suggests a common origin [published online December 31, 2014]. Hum Pathol. 2015;46:558-569.
- Howard MS, Smoller BR. Mycosis fungoides: classic disease and variant presentations. Semin Cutan Med Surg. 2000;19:91-99.
- Zackheim HS, Mccalmont TH. Mycosis fungoides: the great imitator. J Am Acad Dermatol. 2002;47:914-918.
- Suchak R, Verdolini R, Robson A, et al. Extragenital lichen sclerosus et atrophicus mimicking cutaneous T-cell lymphoma: report of a case. J Cutan Pathol. 2010;37:982-986.
- Sarantopoulos GP, Palla B, Said J, et al. Mimics of cutaneous lymphoma: report of the 2011 Society for Hematopathology/European Association for Haematopathology workshop. Am J Clin Pathol. 2013;139:536-551.
- Wieser I, Oh CW, Talpur R, et al. Lymphomatoid papulosis: treatment response and associated lymphomas in a study of 180 patients. J Am Acad Dermatol. 2016;74:59-67.
- Sont JK, van Stiphout WA, Noordijk EM, et al. Increased risk of second cancers in managing Hodgkins disease: the 20-year Leiden experience. Ann Hematol. 1992;65:213-218.
- de la Garza Bravo MM, Patel KP, Loghavi S, et al. Shared clonality in distinctive lesions of lymphomatoid papulosis and mycosis fungoides occurring in the same patients suggests a common origin [published online December 31, 2014]. Hum Pathol. 2015;46:558-569.
- Howard MS, Smoller BR. Mycosis fungoides: classic disease and variant presentations. Semin Cutan Med Surg. 2000;19:91-99.
- Zackheim HS, Mccalmont TH. Mycosis fungoides: the great imitator. J Am Acad Dermatol. 2002;47:914-918.
- Suchak R, Verdolini R, Robson A, et al. Extragenital lichen sclerosus et atrophicus mimicking cutaneous T-cell lymphoma: report of a case. J Cutan Pathol. 2010;37:982-986.
- Sarantopoulos GP, Palla B, Said J, et al. Mimics of cutaneous lymphoma: report of the 2011 Society for Hematopathology/European Association for Haematopathology workshop. Am J Clin Pathol. 2013;139:536-551.
- Wieser I, Oh CW, Talpur R, et al. Lymphomatoid papulosis: treatment response and associated lymphomas in a study of 180 patients. J Am Acad Dermatol. 2016;74:59-67.
- Sont JK, van Stiphout WA, Noordijk EM, et al. Increased risk of second cancers in managing Hodgkins disease: the 20-year Leiden experience. Ann Hematol. 1992;65:213-218.
Practice Points
- Mycosis fungoides classically occurs in patch, plaque, and tumor stages, with lesions preferentially occurring on regions of the body spared from sun exposure; however, the condition may present atypically, mimicking a variety of other conditions.
- Lymphomatoid papulosis exists within a spectrum of primary cutaneous CD30+ lymphoproliferative disorders and is associated with increased incidence of lymphomas.
Disseminated Erythema Induratum in a Patient With a History of Tuberculosis
To the Editor:
Erythema induratum, also known as nodular vasculitis, is a panniculitis that usually affects the lower extremities in middle-aged women. Classically, it has been described as a delayed-type hypersensitivity reaction to Mycobacterium tuberculosis, also known as a tuberculid.1,2 Other infections, however, also have been implicated as causes of erythema induratum, including bacillus Calmette-Guérin (BCG), the attenuated form of Mycobacterium bovis, which commonly is used for tuberculosis vaccination. Medications also may cause erythema induratum. The characteristic distribution of the nodules on the posterior calves helps to distinguish erythema induratum from other panniculitides. A PubMed search of articles indexed for MEDLINE using the term disseminated erythema induratum revealed few case reports documenting nodules on the arms, thighs, or chest, and only 1 case report of disseminated erythema induratum.3-8 We describe a rare combination of disseminated erythema induratum in a patient with remote exposure to tuberculosis and recent BCG exposure.
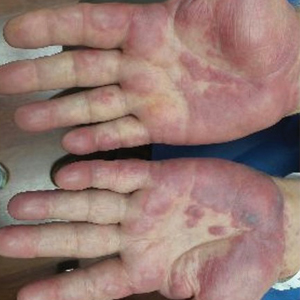
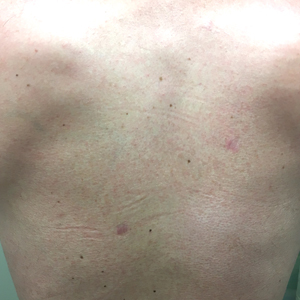
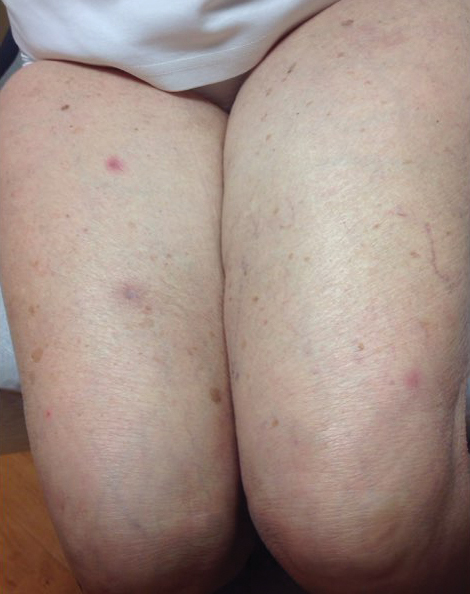
An 88-year-old woman presented for evaluation of violaceous, minimally tender, nonulcerated, subcutaneous nodules on the legs, arms, and trunk of several weeks’ duration (Figure 1). She had a remote history of tuberculosis as a child, prior to the advent of modern antituberculosis regimens. Her medical history also included hypertension, breast cancer treated with lymph node dissection, gastroesophageal reflux disease, and bladder cancer treated with intravesical BCG 10 years prior to the onset of the nodules. She reported minimal coughing and a 25-lb weight loss over the last year, but she denied night sweats, fever, or chills.
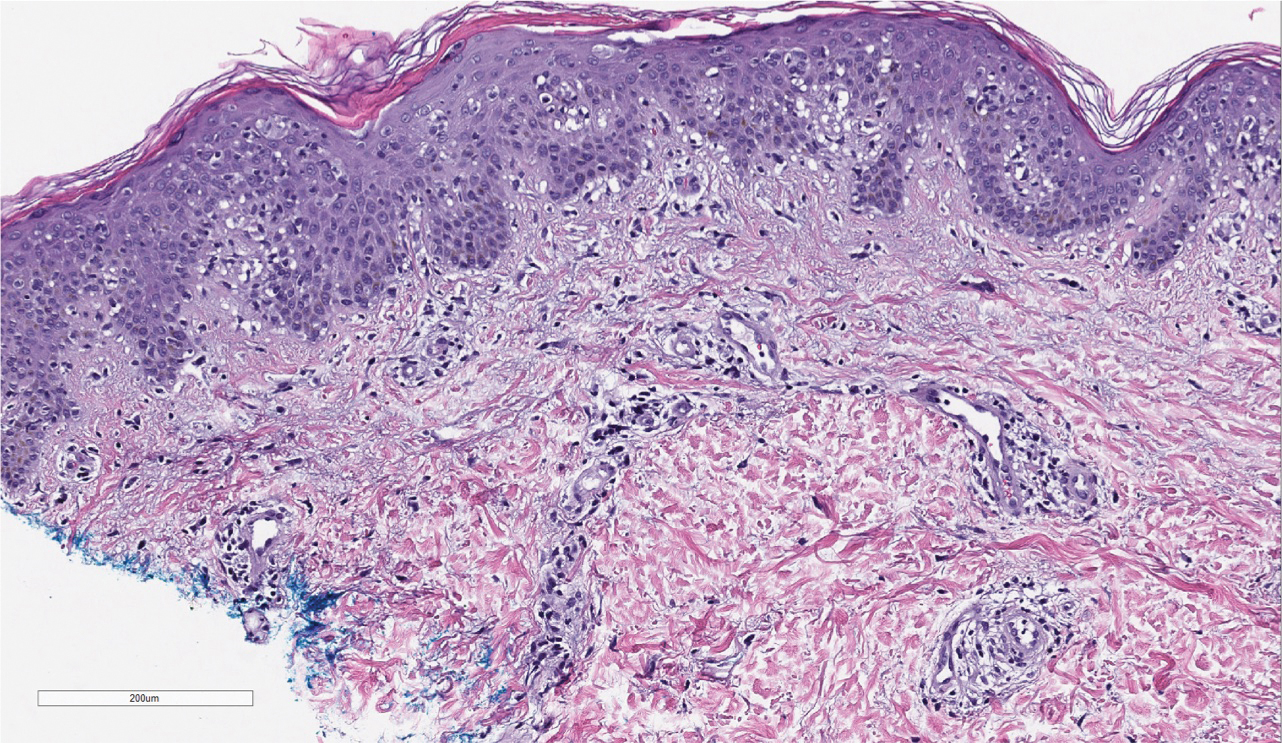
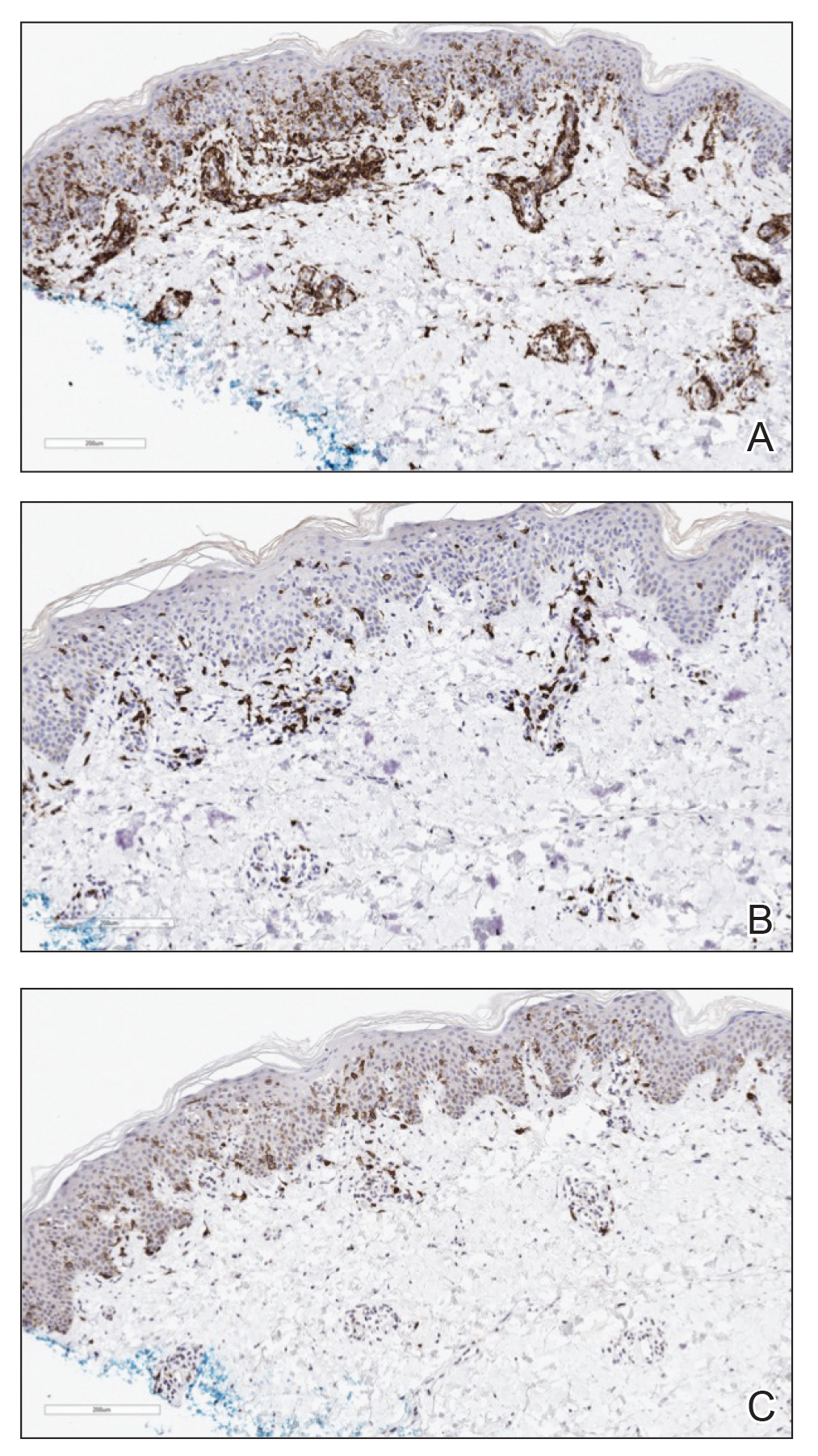
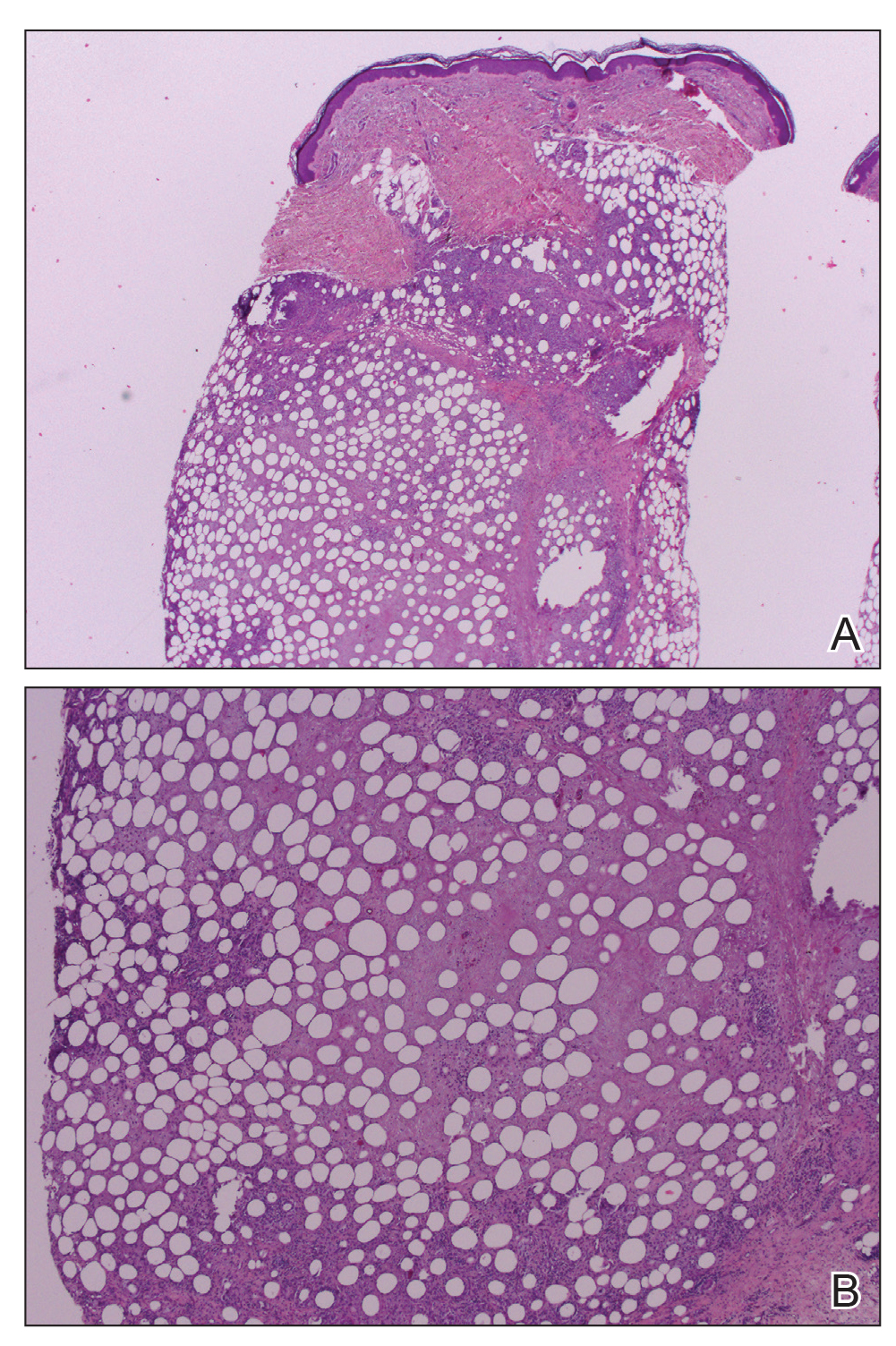
Workup included a biopsy, which showed a dense inflammatory infiltrate within the septae and lobules of the subcutaneous tissue (Figure 2A). Foci of necrosis were seen within the fat lobules (Figure 2B). The histologic diagnosis was erythema induratum. Tissue cultures for bacteria, fungi, and atypical mycobacteria were negative. Mycobacterium tuberculosis polymerase chain reaction (PCR) analysis also was negative. An IFN-γ release assay test was positive for infection with M tuberculosis, suggesting that the erythema induratum was due to tuberculosis rather than BCG exposure. A chest radiograph demonstrated a 22-mm nodule in the left lung (unchanged from a prior film) and a new 10-mm nodule in the left upper lobe.
The patient was referred to an infectious disease specialist who concurred that the erythema induratum and the new lung nodule likely represented a reactivation of tuberculosis. Sputum samples were found to be smear and culture negative for mycobacteria, but due to high clinical suspicion, she was started on a 4-drug tuberculosis regimen of isoniazid, rifampin, pyrazinamide, and ethambutol. Some lesions had started to improve prior to the institution of therapy; after initiation of treatment, all lesions resolved within 4 weeks of starting treatment without recurrence.
Erythema induratum was first described by Bazin9 in 1861. The disorder usually occurs in middle-aged women and is characterized by violaceous ulcerative plaques that classically present on the lower extremities, especially the calves. When the eruption occurs due to a nontuberculous etiology, the term nodular vasculitis is used.1,5 The distinction largely is historical, as most dermatologists today recognize erythema induratum and nodular vasculitis to be the same entity. Examples of nontuberculous causes include infections such as Nocardia, Pseudomonas, Fusarium, or other Mycobacterium species.10 Medications such as propylthiouracil also have been implicated.11 The classification of erythema induratum as a tuberculid suggests that the nodules are a reaction pattern rather than a primary infection, though the term tuberculid may be imprecise. The differential diagnosis of violaceous nodules on the lower extremities and trunk is broad and includes erythema nodosum, cutaneous polyarteritis nodosa, pancreatic panniculitis, subcutaneous T-cell lymphoma, and lupus profundus.1,11,12
Histologically, lesions classically demonstrate a mostly lobular panniculitis with varying degrees of septal fibrosis and focal necrosis. Neutrophils may predominate early, while adipocyte necrosis, epithelioid histiocytes, multinucleated giant cells, and lymphocytes may be found in older lesions. The presence of vasculitis as a requisite diagnostic criterion remains controversial.1,12
The incidence of erythema induratum has decreased since multidrug tuberculosis treatment has become more widespread.3 Our case displayed the disseminated variant of erythema induratum, an even rarer clinical entity.8 Interestingly, our patient had a history of tuberculosis and exposure to BCG prior to the development of lesions. Case reports have documented erythema induratum after BCG exposure but less frequently than in cases associated with tuberculosis.3,13
The use of BCG vaccines has necessitated the need for a more precise method of determining tuberculosis activity. The tuberculin skin test reacts positively with a history of BCG exposure, rendering it an inadequate test in a patient who is suspected of having an active or latent M tuberculosis infection.13,14 IFN-γ release assays are more specific in detecting latent or active tuberculosis than the tuberculin skin test. Such assays use early secretory antigenic target 6 and cultured filtrate protein 10 as antigens to determine sensitization to M tuberculosis.13,15 These antigens are not produced by BCG or Mycobacterium avium; however, other mycobacteria such as Mycobacterium marinum, Mycobacterium kansasii, and some strains of M bovis produce the aforementioned antigens, and exposure to these microbes may be confounding.13 Importantly, positive IFN-γ release assay results also have been documented after BCG exposure but occur at a much lower frequency than for tuberculosis.15 Thus, the combination of the positive IFN-γ release assay and new chest radiograph nodule in our patient provided strong evidence of reactivated tuberculosis as the precipitating cause of her skin disease.
Despite her negative PCR study, our patient’s presentation remains consistent with the diagnosis of disseminated erythema induratum.13,15 The value of PCR studies in establishing the diagnosis remains to be determined. Case reports have described positive PCR results detecting M tuberculosis in panniculitic nodules, suggesting that trace amounts of the organism are present in lesional tissue despite the negative culture result and immunostains.1 Tuberculid reactions, including lichen scrofulosorum, papulonecrotic tuberculid, and erythema induratum, historically are defined by the lack of positive cultures and immunostains, making positive PCR results difficult to reconcile pathophysiologically.1,13 Therefore, use of the term tuberculid altogether as a descriptor for pathogenesis of this disease may need to be avoided.16 Postulated explanations for the relationship of tuberculid diseases and negative cultures and immunostains include the presence of a small number of bacilli that escape routine laboratory detection, early destruction of organisms, or a reaction to circulating M tuberculosis fragments.2 Regardless, until the pathophysiology of erythema induratum has been fully elucidated, the value of PCR remains unclear.
Disseminated erythema induratum, an exceptionally rare variant of panniculitis, may be seen in patients with a remote history of M tuberculosis exposure and/or recent therapeutic BCG exposure. It is imperative to rule out active tuberculosis, especially in elderly patients whose disease predated the advent of modern antituberculosis therapy. Using an IFN-γ release assay in addition to chest radiographs and other clinical stigmata allows differentiation of the etiology of erythema induratum in those patients with tuberculosis who also were treated with BCG.
- Mascaro JM, Basalga E. Erythema induratum of Bazin. Dermatol Clin. 2008;28:439-445.
- Lighter J, Tse DB, Li Y, et al. Erythema induratum of Bazin in a child: evidence for a cell-mediated hyper-response to Mycobacterium tuberculosis. Pediatr Infect Dis J. 2009;28:326-328.
- Inoue T, Fukumoto T, Ansai S, et al. Erythema induratum of Bazin in an infant after bacilli Calmette-Guerin vaccination. J Dermatol. 2006;33:268-272.
- Degonda Halter M, Nebiker P, Hug B, et al. Atypical erythema induratum Bazin with tuberculous osteomyelitis. Internist. 2006;47:853-856.
- Gilchrist H, Patterson JW. Erythema nodosum and erythema induratum (nodular vasculitis): diagnosis and management. Dermatol Ther. 2010;23:320-327.
- Sharma S, Sehgal VN, Bhattacharya SN, et al. Clinicopathologic spectrum of cutaneous tuberculosis: a retrospective analysis of 165 Indians. Am J Dermatopathol. 2015;37:444-450.
- Sethuraman G, Ramesh V. Cutaneous tuberculosis in children. Pediatr Dermatol. 2013;30:7-16.
- Teramura K, Fujimoto N, Nakanishi G, et al. Disseminated erythema induratum of Bazin. Eur J Dermatol. 2014;24:697-698.
- Bazin E. Extrait des Lecons Théoretiques et Cliniques sur le Scrofule. 2nd ed. Paris, France: Delhaye; 1861.
- Campbell SM, Winkelmann RR, Sammons DL. Erythema induratum caused by Mycobacterium chelonei in an immunocompetent patient. J Clin Aesthet Dermatol. 2013;6:38-40.
- Patterson JW. Panniculitis. In: Bolognia JL, Jorizzo J, Rapini RP, et al, eds. Dermatology. Barcelona, Spain: Mosby Elsevier; 2012:1641-1662.
- Segura S, Pujol R, Trinidade F, et al. Vasculitis in erythema induratum of Bazin: a histopathologic study of 101 biopsy specimens from 86 patients. J Am Acad Dermatol. 2008;59:839-851.
- Vera-Kellet C, Peters L, Elwood K, et al. Usefulness of interferon-γ release assays in the diagnosis of erythema induratum. Arch Dermatol. 2011;147:949-952.
- Prajapati V, Steed M, Grewal P, et al. Erythema induratum: case series illustrating the utility of the interferon-γ release assay in determining the association with tuberculosis. J Cutan Med Surg. 2013;17:S6-S11.
- Sim JH, Whang KU. Application of the QuantiFERON-Gold TB test in erythema induratum. J Dermatolog Treat. 2014;25:260-263.
- Wiebels D, Turnbull K, Steinkraus V, et al. Erythema induratum Bazin.”tuberculid” or tuberculosis? [in German]. Hautarzt. 2007;58:237-240.
To the Editor:
Erythema induratum, also known as nodular vasculitis, is a panniculitis that usually affects the lower extremities in middle-aged women. Classically, it has been described as a delayed-type hypersensitivity reaction to Mycobacterium tuberculosis, also known as a tuberculid.1,2 Other infections, however, also have been implicated as causes of erythema induratum, including bacillus Calmette-Guérin (BCG), the attenuated form of Mycobacterium bovis, which commonly is used for tuberculosis vaccination. Medications also may cause erythema induratum. The characteristic distribution of the nodules on the posterior calves helps to distinguish erythema induratum from other panniculitides. A PubMed search of articles indexed for MEDLINE using the term disseminated erythema induratum revealed few case reports documenting nodules on the arms, thighs, or chest, and only 1 case report of disseminated erythema induratum.3-8 We describe a rare combination of disseminated erythema induratum in a patient with remote exposure to tuberculosis and recent BCG exposure.
An 88-year-old woman presented for evaluation of violaceous, minimally tender, nonulcerated, subcutaneous nodules on the legs, arms, and trunk of several weeks’ duration (Figure 1). She had a remote history of tuberculosis as a child, prior to the advent of modern antituberculosis regimens. Her medical history also included hypertension, breast cancer treated with lymph node dissection, gastroesophageal reflux disease, and bladder cancer treated with intravesical BCG 10 years prior to the onset of the nodules. She reported minimal coughing and a 25-lb weight loss over the last year, but she denied night sweats, fever, or chills.
Workup included a biopsy, which showed a dense inflammatory infiltrate within the septae and lobules of the subcutaneous tissue (Figure 2A). Foci of necrosis were seen within the fat lobules (Figure 2B). The histologic diagnosis was erythema induratum. Tissue cultures for bacteria, fungi, and atypical mycobacteria were negative. Mycobacterium tuberculosis polymerase chain reaction (PCR) analysis also was negative. An IFN-γ release assay test was positive for infection with M tuberculosis, suggesting that the erythema induratum was due to tuberculosis rather than BCG exposure. A chest radiograph demonstrated a 22-mm nodule in the left lung (unchanged from a prior film) and a new 10-mm nodule in the left upper lobe.
The patient was referred to an infectious disease specialist who concurred that the erythema induratum and the new lung nodule likely represented a reactivation of tuberculosis. Sputum samples were found to be smear and culture negative for mycobacteria, but due to high clinical suspicion, she was started on a 4-drug tuberculosis regimen of isoniazid, rifampin, pyrazinamide, and ethambutol. Some lesions had started to improve prior to the institution of therapy; after initiation of treatment, all lesions resolved within 4 weeks of starting treatment without recurrence.
Erythema induratum was first described by Bazin9 in 1861. The disorder usually occurs in middle-aged women and is characterized by violaceous ulcerative plaques that classically present on the lower extremities, especially the calves. When the eruption occurs due to a nontuberculous etiology, the term nodular vasculitis is used.1,5 The distinction largely is historical, as most dermatologists today recognize erythema induratum and nodular vasculitis to be the same entity. Examples of nontuberculous causes include infections such as Nocardia, Pseudomonas, Fusarium, or other Mycobacterium species.10 Medications such as propylthiouracil also have been implicated.11 The classification of erythema induratum as a tuberculid suggests that the nodules are a reaction pattern rather than a primary infection, though the term tuberculid may be imprecise. The differential diagnosis of violaceous nodules on the lower extremities and trunk is broad and includes erythema nodosum, cutaneous polyarteritis nodosa, pancreatic panniculitis, subcutaneous T-cell lymphoma, and lupus profundus.1,11,12
Histologically, lesions classically demonstrate a mostly lobular panniculitis with varying degrees of septal fibrosis and focal necrosis. Neutrophils may predominate early, while adipocyte necrosis, epithelioid histiocytes, multinucleated giant cells, and lymphocytes may be found in older lesions. The presence of vasculitis as a requisite diagnostic criterion remains controversial.1,12
The incidence of erythema induratum has decreased since multidrug tuberculosis treatment has become more widespread.3 Our case displayed the disseminated variant of erythema induratum, an even rarer clinical entity.8 Interestingly, our patient had a history of tuberculosis and exposure to BCG prior to the development of lesions. Case reports have documented erythema induratum after BCG exposure but less frequently than in cases associated with tuberculosis.3,13
The use of BCG vaccines has necessitated the need for a more precise method of determining tuberculosis activity. The tuberculin skin test reacts positively with a history of BCG exposure, rendering it an inadequate test in a patient who is suspected of having an active or latent M tuberculosis infection.13,14 IFN-γ release assays are more specific in detecting latent or active tuberculosis than the tuberculin skin test. Such assays use early secretory antigenic target 6 and cultured filtrate protein 10 as antigens to determine sensitization to M tuberculosis.13,15 These antigens are not produced by BCG or Mycobacterium avium; however, other mycobacteria such as Mycobacterium marinum, Mycobacterium kansasii, and some strains of M bovis produce the aforementioned antigens, and exposure to these microbes may be confounding.13 Importantly, positive IFN-γ release assay results also have been documented after BCG exposure but occur at a much lower frequency than for tuberculosis.15 Thus, the combination of the positive IFN-γ release assay and new chest radiograph nodule in our patient provided strong evidence of reactivated tuberculosis as the precipitating cause of her skin disease.
Despite her negative PCR study, our patient’s presentation remains consistent with the diagnosis of disseminated erythema induratum.13,15 The value of PCR studies in establishing the diagnosis remains to be determined. Case reports have described positive PCR results detecting M tuberculosis in panniculitic nodules, suggesting that trace amounts of the organism are present in lesional tissue despite the negative culture result and immunostains.1 Tuberculid reactions, including lichen scrofulosorum, papulonecrotic tuberculid, and erythema induratum, historically are defined by the lack of positive cultures and immunostains, making positive PCR results difficult to reconcile pathophysiologically.1,13 Therefore, use of the term tuberculid altogether as a descriptor for pathogenesis of this disease may need to be avoided.16 Postulated explanations for the relationship of tuberculid diseases and negative cultures and immunostains include the presence of a small number of bacilli that escape routine laboratory detection, early destruction of organisms, or a reaction to circulating M tuberculosis fragments.2 Regardless, until the pathophysiology of erythema induratum has been fully elucidated, the value of PCR remains unclear.
Disseminated erythema induratum, an exceptionally rare variant of panniculitis, may be seen in patients with a remote history of M tuberculosis exposure and/or recent therapeutic BCG exposure. It is imperative to rule out active tuberculosis, especially in elderly patients whose disease predated the advent of modern antituberculosis therapy. Using an IFN-γ release assay in addition to chest radiographs and other clinical stigmata allows differentiation of the etiology of erythema induratum in those patients with tuberculosis who also were treated with BCG.
To the Editor:
Erythema induratum, also known as nodular vasculitis, is a panniculitis that usually affects the lower extremities in middle-aged women. Classically, it has been described as a delayed-type hypersensitivity reaction to Mycobacterium tuberculosis, also known as a tuberculid.1,2 Other infections, however, also have been implicated as causes of erythema induratum, including bacillus Calmette-Guérin (BCG), the attenuated form of Mycobacterium bovis, which commonly is used for tuberculosis vaccination. Medications also may cause erythema induratum. The characteristic distribution of the nodules on the posterior calves helps to distinguish erythema induratum from other panniculitides. A PubMed search of articles indexed for MEDLINE using the term disseminated erythema induratum revealed few case reports documenting nodules on the arms, thighs, or chest, and only 1 case report of disseminated erythema induratum.3-8 We describe a rare combination of disseminated erythema induratum in a patient with remote exposure to tuberculosis and recent BCG exposure.
An 88-year-old woman presented for evaluation of violaceous, minimally tender, nonulcerated, subcutaneous nodules on the legs, arms, and trunk of several weeks’ duration (Figure 1). She had a remote history of tuberculosis as a child, prior to the advent of modern antituberculosis regimens. Her medical history also included hypertension, breast cancer treated with lymph node dissection, gastroesophageal reflux disease, and bladder cancer treated with intravesical BCG 10 years prior to the onset of the nodules. She reported minimal coughing and a 25-lb weight loss over the last year, but she denied night sweats, fever, or chills.
Workup included a biopsy, which showed a dense inflammatory infiltrate within the septae and lobules of the subcutaneous tissue (Figure 2A). Foci of necrosis were seen within the fat lobules (Figure 2B). The histologic diagnosis was erythema induratum. Tissue cultures for bacteria, fungi, and atypical mycobacteria were negative. Mycobacterium tuberculosis polymerase chain reaction (PCR) analysis also was negative. An IFN-γ release assay test was positive for infection with M tuberculosis, suggesting that the erythema induratum was due to tuberculosis rather than BCG exposure. A chest radiograph demonstrated a 22-mm nodule in the left lung (unchanged from a prior film) and a new 10-mm nodule in the left upper lobe.
The patient was referred to an infectious disease specialist who concurred that the erythema induratum and the new lung nodule likely represented a reactivation of tuberculosis. Sputum samples were found to be smear and culture negative for mycobacteria, but due to high clinical suspicion, she was started on a 4-drug tuberculosis regimen of isoniazid, rifampin, pyrazinamide, and ethambutol. Some lesions had started to improve prior to the institution of therapy; after initiation of treatment, all lesions resolved within 4 weeks of starting treatment without recurrence.
Erythema induratum was first described by Bazin9 in 1861. The disorder usually occurs in middle-aged women and is characterized by violaceous ulcerative plaques that classically present on the lower extremities, especially the calves. When the eruption occurs due to a nontuberculous etiology, the term nodular vasculitis is used.1,5 The distinction largely is historical, as most dermatologists today recognize erythema induratum and nodular vasculitis to be the same entity. Examples of nontuberculous causes include infections such as Nocardia, Pseudomonas, Fusarium, or other Mycobacterium species.10 Medications such as propylthiouracil also have been implicated.11 The classification of erythema induratum as a tuberculid suggests that the nodules are a reaction pattern rather than a primary infection, though the term tuberculid may be imprecise. The differential diagnosis of violaceous nodules on the lower extremities and trunk is broad and includes erythema nodosum, cutaneous polyarteritis nodosa, pancreatic panniculitis, subcutaneous T-cell lymphoma, and lupus profundus.1,11,12
Histologically, lesions classically demonstrate a mostly lobular panniculitis with varying degrees of septal fibrosis and focal necrosis. Neutrophils may predominate early, while adipocyte necrosis, epithelioid histiocytes, multinucleated giant cells, and lymphocytes may be found in older lesions. The presence of vasculitis as a requisite diagnostic criterion remains controversial.1,12
The incidence of erythema induratum has decreased since multidrug tuberculosis treatment has become more widespread.3 Our case displayed the disseminated variant of erythema induratum, an even rarer clinical entity.8 Interestingly, our patient had a history of tuberculosis and exposure to BCG prior to the development of lesions. Case reports have documented erythema induratum after BCG exposure but less frequently than in cases associated with tuberculosis.3,13
The use of BCG vaccines has necessitated the need for a more precise method of determining tuberculosis activity. The tuberculin skin test reacts positively with a history of BCG exposure, rendering it an inadequate test in a patient who is suspected of having an active or latent M tuberculosis infection.13,14 IFN-γ release assays are more specific in detecting latent or active tuberculosis than the tuberculin skin test. Such assays use early secretory antigenic target 6 and cultured filtrate protein 10 as antigens to determine sensitization to M tuberculosis.13,15 These antigens are not produced by BCG or Mycobacterium avium; however, other mycobacteria such as Mycobacterium marinum, Mycobacterium kansasii, and some strains of M bovis produce the aforementioned antigens, and exposure to these microbes may be confounding.13 Importantly, positive IFN-γ release assay results also have been documented after BCG exposure but occur at a much lower frequency than for tuberculosis.15 Thus, the combination of the positive IFN-γ release assay and new chest radiograph nodule in our patient provided strong evidence of reactivated tuberculosis as the precipitating cause of her skin disease.
Despite her negative PCR study, our patient’s presentation remains consistent with the diagnosis of disseminated erythema induratum.13,15 The value of PCR studies in establishing the diagnosis remains to be determined. Case reports have described positive PCR results detecting M tuberculosis in panniculitic nodules, suggesting that trace amounts of the organism are present in lesional tissue despite the negative culture result and immunostains.1 Tuberculid reactions, including lichen scrofulosorum, papulonecrotic tuberculid, and erythema induratum, historically are defined by the lack of positive cultures and immunostains, making positive PCR results difficult to reconcile pathophysiologically.1,13 Therefore, use of the term tuberculid altogether as a descriptor for pathogenesis of this disease may need to be avoided.16 Postulated explanations for the relationship of tuberculid diseases and negative cultures and immunostains include the presence of a small number of bacilli that escape routine laboratory detection, early destruction of organisms, or a reaction to circulating M tuberculosis fragments.2 Regardless, until the pathophysiology of erythema induratum has been fully elucidated, the value of PCR remains unclear.
Disseminated erythema induratum, an exceptionally rare variant of panniculitis, may be seen in patients with a remote history of M tuberculosis exposure and/or recent therapeutic BCG exposure. It is imperative to rule out active tuberculosis, especially in elderly patients whose disease predated the advent of modern antituberculosis therapy. Using an IFN-γ release assay in addition to chest radiographs and other clinical stigmata allows differentiation of the etiology of erythema induratum in those patients with tuberculosis who also were treated with BCG.
- Mascaro JM, Basalga E. Erythema induratum of Bazin. Dermatol Clin. 2008;28:439-445.
- Lighter J, Tse DB, Li Y, et al. Erythema induratum of Bazin in a child: evidence for a cell-mediated hyper-response to Mycobacterium tuberculosis. Pediatr Infect Dis J. 2009;28:326-328.
- Inoue T, Fukumoto T, Ansai S, et al. Erythema induratum of Bazin in an infant after bacilli Calmette-Guerin vaccination. J Dermatol. 2006;33:268-272.
- Degonda Halter M, Nebiker P, Hug B, et al. Atypical erythema induratum Bazin with tuberculous osteomyelitis. Internist. 2006;47:853-856.
- Gilchrist H, Patterson JW. Erythema nodosum and erythema induratum (nodular vasculitis): diagnosis and management. Dermatol Ther. 2010;23:320-327.
- Sharma S, Sehgal VN, Bhattacharya SN, et al. Clinicopathologic spectrum of cutaneous tuberculosis: a retrospective analysis of 165 Indians. Am J Dermatopathol. 2015;37:444-450.
- Sethuraman G, Ramesh V. Cutaneous tuberculosis in children. Pediatr Dermatol. 2013;30:7-16.
- Teramura K, Fujimoto N, Nakanishi G, et al. Disseminated erythema induratum of Bazin. Eur J Dermatol. 2014;24:697-698.
- Bazin E. Extrait des Lecons Théoretiques et Cliniques sur le Scrofule. 2nd ed. Paris, France: Delhaye; 1861.
- Campbell SM, Winkelmann RR, Sammons DL. Erythema induratum caused by Mycobacterium chelonei in an immunocompetent patient. J Clin Aesthet Dermatol. 2013;6:38-40.
- Patterson JW. Panniculitis. In: Bolognia JL, Jorizzo J, Rapini RP, et al, eds. Dermatology. Barcelona, Spain: Mosby Elsevier; 2012:1641-1662.
- Segura S, Pujol R, Trinidade F, et al. Vasculitis in erythema induratum of Bazin: a histopathologic study of 101 biopsy specimens from 86 patients. J Am Acad Dermatol. 2008;59:839-851.
- Vera-Kellet C, Peters L, Elwood K, et al. Usefulness of interferon-γ release assays in the diagnosis of erythema induratum. Arch Dermatol. 2011;147:949-952.
- Prajapati V, Steed M, Grewal P, et al. Erythema induratum: case series illustrating the utility of the interferon-γ release assay in determining the association with tuberculosis. J Cutan Med Surg. 2013;17:S6-S11.
- Sim JH, Whang KU. Application of the QuantiFERON-Gold TB test in erythema induratum. J Dermatolog Treat. 2014;25:260-263.
- Wiebels D, Turnbull K, Steinkraus V, et al. Erythema induratum Bazin.”tuberculid” or tuberculosis? [in German]. Hautarzt. 2007;58:237-240.
- Mascaro JM, Basalga E. Erythema induratum of Bazin. Dermatol Clin. 2008;28:439-445.
- Lighter J, Tse DB, Li Y, et al. Erythema induratum of Bazin in a child: evidence for a cell-mediated hyper-response to Mycobacterium tuberculosis. Pediatr Infect Dis J. 2009;28:326-328.
- Inoue T, Fukumoto T, Ansai S, et al. Erythema induratum of Bazin in an infant after bacilli Calmette-Guerin vaccination. J Dermatol. 2006;33:268-272.
- Degonda Halter M, Nebiker P, Hug B, et al. Atypical erythema induratum Bazin with tuberculous osteomyelitis. Internist. 2006;47:853-856.
- Gilchrist H, Patterson JW. Erythema nodosum and erythema induratum (nodular vasculitis): diagnosis and management. Dermatol Ther. 2010;23:320-327.
- Sharma S, Sehgal VN, Bhattacharya SN, et al. Clinicopathologic spectrum of cutaneous tuberculosis: a retrospective analysis of 165 Indians. Am J Dermatopathol. 2015;37:444-450.
- Sethuraman G, Ramesh V. Cutaneous tuberculosis in children. Pediatr Dermatol. 2013;30:7-16.
- Teramura K, Fujimoto N, Nakanishi G, et al. Disseminated erythema induratum of Bazin. Eur J Dermatol. 2014;24:697-698.
- Bazin E. Extrait des Lecons Théoretiques et Cliniques sur le Scrofule. 2nd ed. Paris, France: Delhaye; 1861.
- Campbell SM, Winkelmann RR, Sammons DL. Erythema induratum caused by Mycobacterium chelonei in an immunocompetent patient. J Clin Aesthet Dermatol. 2013;6:38-40.
- Patterson JW. Panniculitis. In: Bolognia JL, Jorizzo J, Rapini RP, et al, eds. Dermatology. Barcelona, Spain: Mosby Elsevier; 2012:1641-1662.
- Segura S, Pujol R, Trinidade F, et al. Vasculitis in erythema induratum of Bazin: a histopathologic study of 101 biopsy specimens from 86 patients. J Am Acad Dermatol. 2008;59:839-851.
- Vera-Kellet C, Peters L, Elwood K, et al. Usefulness of interferon-γ release assays in the diagnosis of erythema induratum. Arch Dermatol. 2011;147:949-952.
- Prajapati V, Steed M, Grewal P, et al. Erythema induratum: case series illustrating the utility of the interferon-γ release assay in determining the association with tuberculosis. J Cutan Med Surg. 2013;17:S6-S11.
- Sim JH, Whang KU. Application of the QuantiFERON-Gold TB test in erythema induratum. J Dermatolog Treat. 2014;25:260-263.
- Wiebels D, Turnbull K, Steinkraus V, et al. Erythema induratum Bazin.”tuberculid” or tuberculosis? [in German]. Hautarzt. 2007;58:237-240.
Practice Points
- Erythema induratum is an uncommon panniculitis attributed to a delayed-type hypersensitivity reaction, classically to Mycobacterium tuberculosis.
- The workup for such patients with exposure to both M tuberculosis and bacillus Calmette-Guérin should include IFN-11γ release assays.
- Clinicians should be aware of the disseminated variant of erythema induratum and the laboratory testing needed to establish a cause and help direct treatment.
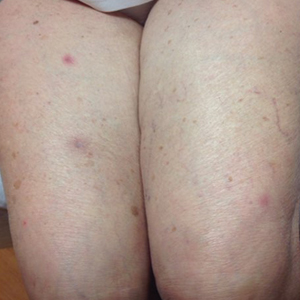
Follicular Traction Urticaria Induced by Electric Epilation
To the Editor:
A 33-year-old woman who was otherwise healthy presented with itchy wheals that developed within 15 to 20 minutes of removing leg hair with an electric epilator. Furthermore, she reported that small hives often developed after waxing the legs with warm wax. All lesions spontaneously disappeared within 3 hours; depilatory creams and shaving did not trigger urticarial lesions. She had no history of atopy or prior episodes of spontaneous urticaria. Symptomatic dermographism also was not reported. Classic physical stimuli that could be associated with the use of an electric epilator, such as heat, vibration, and pressure, did not elicit lesions.
Physical examination showed no active lesions. Dermographism was not inducible by stroking the patient’s skin with a blunt object. She brought personal photographs that showed erythematous follicular hives measuring 1 to 3 mm in diameter located on the distal legs (Figure). In accordance with these findings, she was diagnosed with an unusual form of physical urticaria likely resulting from hair traction and was prescribed oral H1 antihistamines to be taken a few days before and after hair removal.
Physical urticaria are characterized by the presence of reddish, edematous, and pruritic wheals developing in response to a variety of exogenous physical stimuli such as heat, cold, vibration, dermographism, and pressure. These variants are widely described; nonetheless, follicular traction urticaria has been proposed as a new form of physical urticaria elicited by traction of hair, which would cause tension on and around hair follicles on a secondary basis.1 A PubMed search of articles indexed for MEDLINE using the term traction urticaria revealed 6 other cases. In 3 cases, hives were triggered by waxing or using an electric epilator.1-3 In 1 case, urticaria was elicited by shaving with a wet straight razor,whereas the other 2 cases were induced by the removal of patch tests.4-6 Sheraz et al7 investigated the role of dermographism in erythematous reactions during patch testing and concluded that some of these reactions might be caused by traction urticaria instead of being a form of dermographism.
Özkaya and Yazganog˘lu1 proposed that follicular dermographism should be differentiated from physical urticaria. This variant of dermographism is characterized by discrete urticarial papules appearing at the location of hair follicles after having stroked the skin with a blunt object.1,8 These lesions usually disappear within 30 minutes.8 Given that none of the reported cases presented dermographism on examination tests, we agree with Özkaya and Yazganog˘lu1 that this phenomenon of traction urticaria likely is a different condition than follicular dermographism, even though intraindividual variability sometimes can be seen in dermographism skin tests.7
We present a unique form of urticaria that easily can be misdiagnosed as pseudofolliculitis, which tends to be more commonly associated with the use of electric epilators.
- Özkaya E, Yazganog˘lu KD. Follicular traction urticaria. J Am Acad Dermatol. 2012;67:E234-E236.
- Duman H, Topal IO, Kocaturk E. Follicular traction urticaria. An Bras Dermatol. 2016;91:64-65.
- Raison-Peyron N, Reymann V, Bessis D. Follicular traction urticaria: a new form of chronic inducible urticaria? Acta Derm Venereol. 2017;97:522-523.
- Patel SS, Lockey RF. Follicular traction urticaria. J Allergy Clin Immunol Pract. 2018;6:1383.
- Gallo R, Fausti V, Parodi A. Traction urticaria. Contact Dermatitis. 2009;61:301-302.
- Özkaya E. Follicular traction urticaria: an occult case diagnosed by patch testing. Dermatitis. 2019;30:171-173.
- Sheraz A, Simms MJ, White IR, et al. Erythematous reactions on removal of Scanpor® tape in patch testing are not necessarily caused by dermographism. Contact Dermatitis. 2014;71:62-64.
- Bhute D, Doshi B, Pande S, et al. Dermatographism. Indian J Dermatol Venereol Leprol. 2008;74:177-179.
To the Editor:
A 33-year-old woman who was otherwise healthy presented with itchy wheals that developed within 15 to 20 minutes of removing leg hair with an electric epilator. Furthermore, she reported that small hives often developed after waxing the legs with warm wax. All lesions spontaneously disappeared within 3 hours; depilatory creams and shaving did not trigger urticarial lesions. She had no history of atopy or prior episodes of spontaneous urticaria. Symptomatic dermographism also was not reported. Classic physical stimuli that could be associated with the use of an electric epilator, such as heat, vibration, and pressure, did not elicit lesions.
Physical examination showed no active lesions. Dermographism was not inducible by stroking the patient’s skin with a blunt object. She brought personal photographs that showed erythematous follicular hives measuring 1 to 3 mm in diameter located on the distal legs (Figure). In accordance with these findings, she was diagnosed with an unusual form of physical urticaria likely resulting from hair traction and was prescribed oral H1 antihistamines to be taken a few days before and after hair removal.
Physical urticaria are characterized by the presence of reddish, edematous, and pruritic wheals developing in response to a variety of exogenous physical stimuli such as heat, cold, vibration, dermographism, and pressure. These variants are widely described; nonetheless, follicular traction urticaria has been proposed as a new form of physical urticaria elicited by traction of hair, which would cause tension on and around hair follicles on a secondary basis.1 A PubMed search of articles indexed for MEDLINE using the term traction urticaria revealed 6 other cases. In 3 cases, hives were triggered by waxing or using an electric epilator.1-3 In 1 case, urticaria was elicited by shaving with a wet straight razor,whereas the other 2 cases were induced by the removal of patch tests.4-6 Sheraz et al7 investigated the role of dermographism in erythematous reactions during patch testing and concluded that some of these reactions might be caused by traction urticaria instead of being a form of dermographism.
Özkaya and Yazganog˘lu1 proposed that follicular dermographism should be differentiated from physical urticaria. This variant of dermographism is characterized by discrete urticarial papules appearing at the location of hair follicles after having stroked the skin with a blunt object.1,8 These lesions usually disappear within 30 minutes.8 Given that none of the reported cases presented dermographism on examination tests, we agree with Özkaya and Yazganog˘lu1 that this phenomenon of traction urticaria likely is a different condition than follicular dermographism, even though intraindividual variability sometimes can be seen in dermographism skin tests.7
We present a unique form of urticaria that easily can be misdiagnosed as pseudofolliculitis, which tends to be more commonly associated with the use of electric epilators.
To the Editor:
A 33-year-old woman who was otherwise healthy presented with itchy wheals that developed within 15 to 20 minutes of removing leg hair with an electric epilator. Furthermore, she reported that small hives often developed after waxing the legs with warm wax. All lesions spontaneously disappeared within 3 hours; depilatory creams and shaving did not trigger urticarial lesions. She had no history of atopy or prior episodes of spontaneous urticaria. Symptomatic dermographism also was not reported. Classic physical stimuli that could be associated with the use of an electric epilator, such as heat, vibration, and pressure, did not elicit lesions.
Physical examination showed no active lesions. Dermographism was not inducible by stroking the patient’s skin with a blunt object. She brought personal photographs that showed erythematous follicular hives measuring 1 to 3 mm in diameter located on the distal legs (Figure). In accordance with these findings, she was diagnosed with an unusual form of physical urticaria likely resulting from hair traction and was prescribed oral H1 antihistamines to be taken a few days before and after hair removal.
Physical urticaria are characterized by the presence of reddish, edematous, and pruritic wheals developing in response to a variety of exogenous physical stimuli such as heat, cold, vibration, dermographism, and pressure. These variants are widely described; nonetheless, follicular traction urticaria has been proposed as a new form of physical urticaria elicited by traction of hair, which would cause tension on and around hair follicles on a secondary basis.1 A PubMed search of articles indexed for MEDLINE using the term traction urticaria revealed 6 other cases. In 3 cases, hives were triggered by waxing or using an electric epilator.1-3 In 1 case, urticaria was elicited by shaving with a wet straight razor,whereas the other 2 cases were induced by the removal of patch tests.4-6 Sheraz et al7 investigated the role of dermographism in erythematous reactions during patch testing and concluded that some of these reactions might be caused by traction urticaria instead of being a form of dermographism.
Özkaya and Yazganog˘lu1 proposed that follicular dermographism should be differentiated from physical urticaria. This variant of dermographism is characterized by discrete urticarial papules appearing at the location of hair follicles after having stroked the skin with a blunt object.1,8 These lesions usually disappear within 30 minutes.8 Given that none of the reported cases presented dermographism on examination tests, we agree with Özkaya and Yazganog˘lu1 that this phenomenon of traction urticaria likely is a different condition than follicular dermographism, even though intraindividual variability sometimes can be seen in dermographism skin tests.7
We present a unique form of urticaria that easily can be misdiagnosed as pseudofolliculitis, which tends to be more commonly associated with the use of electric epilators.
- Özkaya E, Yazganog˘lu KD. Follicular traction urticaria. J Am Acad Dermatol. 2012;67:E234-E236.
- Duman H, Topal IO, Kocaturk E. Follicular traction urticaria. An Bras Dermatol. 2016;91:64-65.
- Raison-Peyron N, Reymann V, Bessis D. Follicular traction urticaria: a new form of chronic inducible urticaria? Acta Derm Venereol. 2017;97:522-523.
- Patel SS, Lockey RF. Follicular traction urticaria. J Allergy Clin Immunol Pract. 2018;6:1383.
- Gallo R, Fausti V, Parodi A. Traction urticaria. Contact Dermatitis. 2009;61:301-302.
- Özkaya E. Follicular traction urticaria: an occult case diagnosed by patch testing. Dermatitis. 2019;30:171-173.
- Sheraz A, Simms MJ, White IR, et al. Erythematous reactions on removal of Scanpor® tape in patch testing are not necessarily caused by dermographism. Contact Dermatitis. 2014;71:62-64.
- Bhute D, Doshi B, Pande S, et al. Dermatographism. Indian J Dermatol Venereol Leprol. 2008;74:177-179.
- Özkaya E, Yazganog˘lu KD. Follicular traction urticaria. J Am Acad Dermatol. 2012;67:E234-E236.
- Duman H, Topal IO, Kocaturk E. Follicular traction urticaria. An Bras Dermatol. 2016;91:64-65.
- Raison-Peyron N, Reymann V, Bessis D. Follicular traction urticaria: a new form of chronic inducible urticaria? Acta Derm Venereol. 2017;97:522-523.
- Patel SS, Lockey RF. Follicular traction urticaria. J Allergy Clin Immunol Pract. 2018;6:1383.
- Gallo R, Fausti V, Parodi A. Traction urticaria. Contact Dermatitis. 2009;61:301-302.
- Özkaya E. Follicular traction urticaria: an occult case diagnosed by patch testing. Dermatitis. 2019;30:171-173.
- Sheraz A, Simms MJ, White IR, et al. Erythematous reactions on removal of Scanpor® tape in patch testing are not necessarily caused by dermographism. Contact Dermatitis. 2014;71:62-64.
- Bhute D, Doshi B, Pande S, et al. Dermatographism. Indian J Dermatol Venereol Leprol. 2008;74:177-179.
Practice Points
- Follicular traction urticaria is an unusual form of chronic inducible urticaria.
- Follicular traction urticaria consists of follicular hives that develop after being triggered by hair traction.
Cutaneous Metastatic Breast Adenocarcinoma
To the Editor:
Cutaneous metastases occur more often in the setting of breast carcinoma than other malignancies in women.1 Although interventions are aimed at halting disease progression, cutaneous metastases indicate widespread disease and are associated with poor prognosis. We present the case of a patient with metastatic breast adenocarcinoma who developed cutaneous metastasis on the trunk after a double mastectomy. The widespread distribution and wide range of clinical manifestations are unique.
An 81-year-old woman presented to the dermatology office for evaluation of a skin eruption that started along a mastectomy scar on the left breast a few months postoperatively. She had a history of stage IV breast adenocarcinoma metastatic to the chest wall that was treated with a double mastectomy 2 years prior. The patient denied associated pain or pruritus and mainly was concerned with the cosmetic appearance. At the time of the initial diagnosis of breast adenocarcinoma, the patient was offered chemotherapy, which she did not tolerate. The patient opted against radiation therapy, as she preferred a more natural approach, such as anticancer shakes, which she was taking from a homeopathic source. She was unaware of the ingredients used in the shakes.
Physical examination revealed multiple grouped, firm, purpuric papules, nodules, and pseudovesicles on a background of violaceous erythema on the chest, abdomen, and flank (Figure 1). The background erythema had a mosaic pattern that extended toward the central back (Figure 2). A scoop shave biopsy of one of the purpuric nodules revealed highly atypical cells with abundant cytoplasm, large nuclei, and prominent nucleoli (Figure 3). Focally, the cells appeared to form glandular structures. Numerous atypical mitotic figures were present. Lymphatic invasion and microcalcifications were identified (Figure 3 [inset]). Immunohistochemical staining for cytokeratin 7 and gross cystic disease fluid protein 15 were strongly positive (Figure 4). Based on the histopathologic and immunohistochemical findings, a diagnosis of cutaneous metastatic breast adenocarcinoma was made. The patient opted to continue the homeopathic anticancer shakes and was subsequently lost to follow-up.
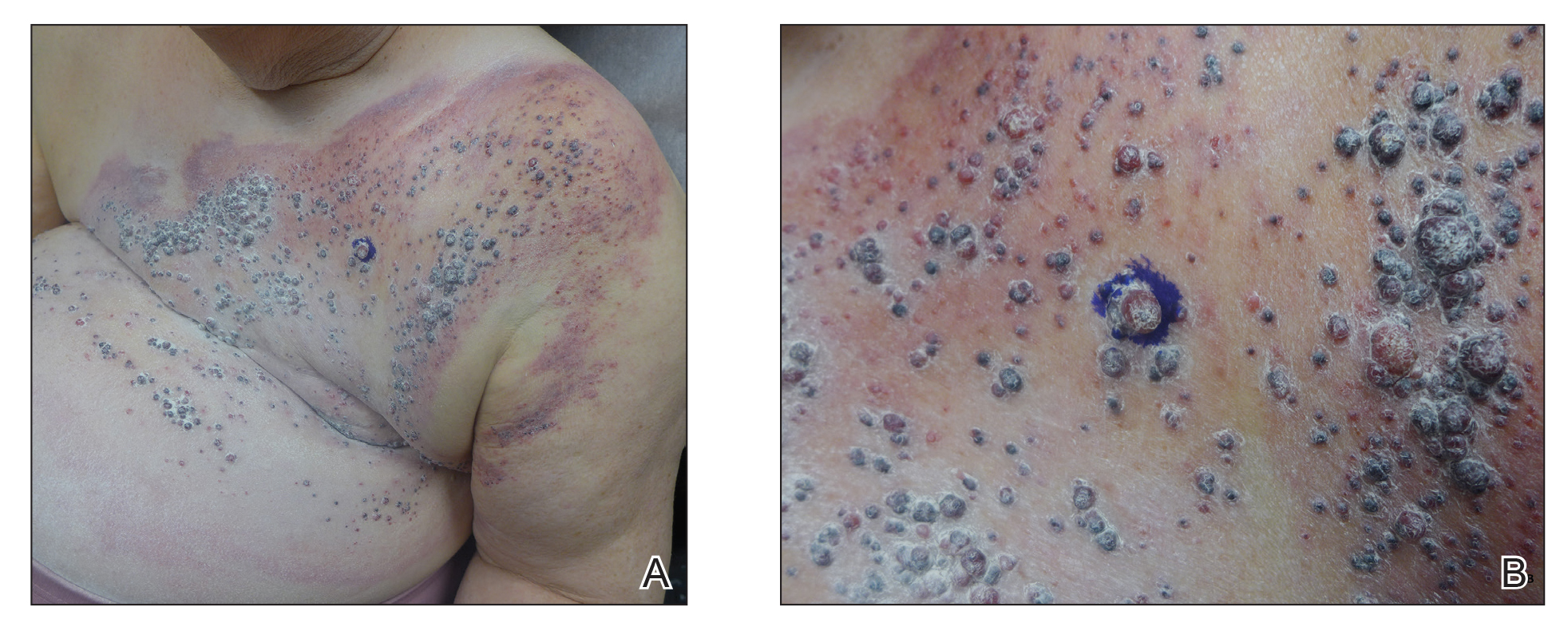

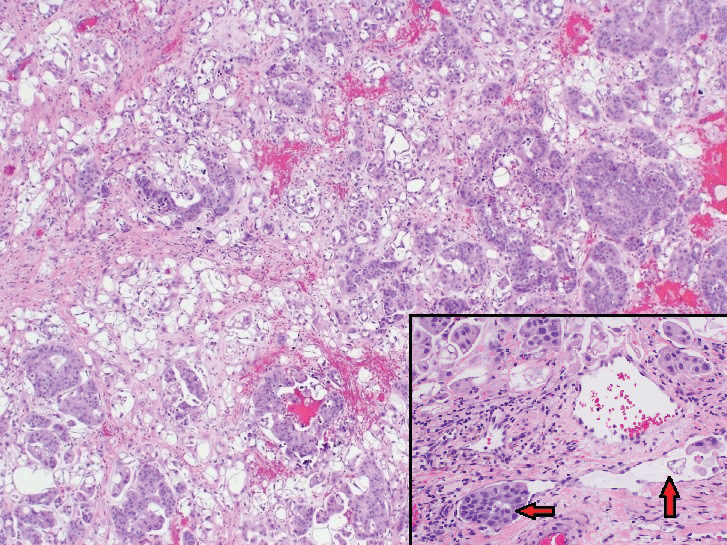
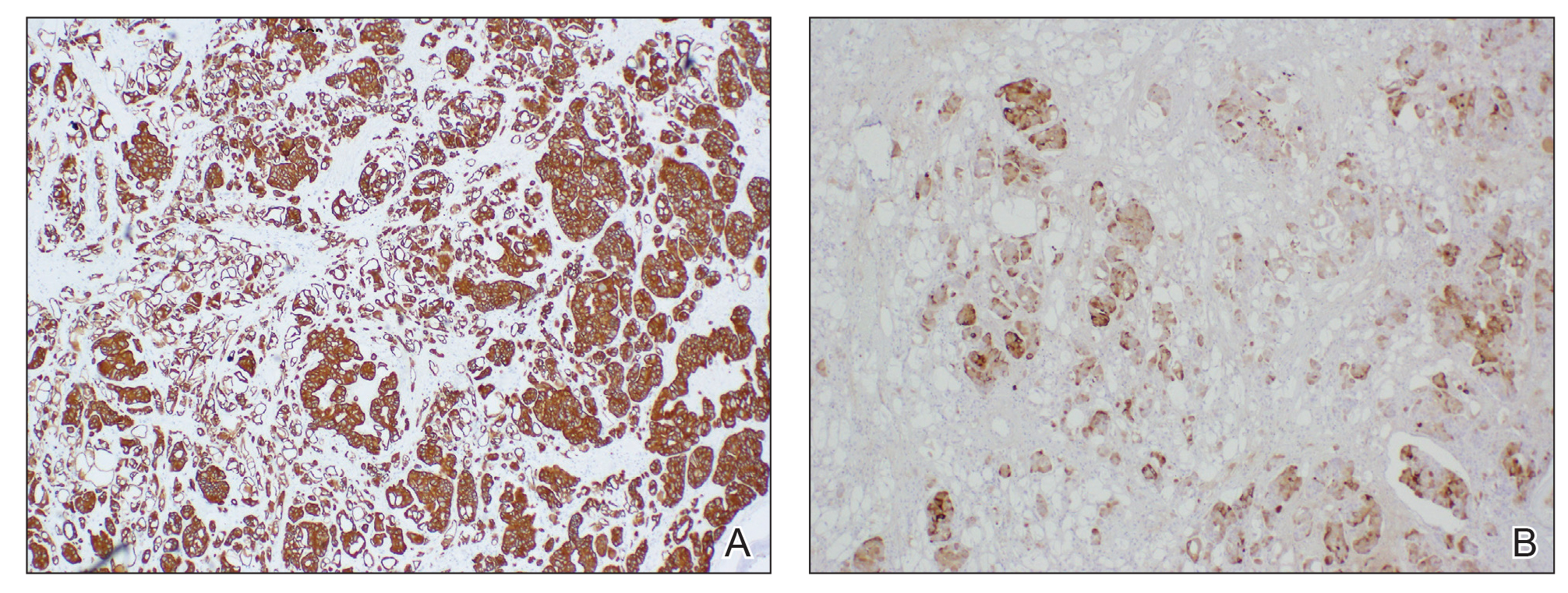
Cutaneous metastases of internal malignancies make up only 2% of all skin tumors,1 making them relatively uncommon in the dermatologic setting. However, cutaneous metastasis occurs in 23.9% of patients with breast carcinoma, making it the most common tumor after malignant melanoma to metastasize to the skin.2 The most common sites for breast carcinoma cutaneous metastasis (BCCM) are the chest wall and abdomen; other sites include the head/neck region and the extremities. The clinical presentation of BCCM varies depending on the mode of dissemination—lymphatic, hematogenous, contiguous growth, or iatrogenic implantation. The most common presentation is nodular carcinoma (47%–80%).2,3 Other presentations include carcinoma telangiectoides (8%–11%),2,3 alopecia neoplastica (2%–12%),2,3 and carcinoma erysipeloides (3%–6%).2,3 Carcinoma en cuirasse is rare.3
Nodular BCCM may present as firm solitary or grouped papules and nodules that are painless and range in color from flesh colored or pink to red-brown. Histologically, they are composed of atypical neoplastic cells arranged in small nests and cords, usually in a single-file line within the collagen bundles of the dermis.4 Carcinoma telangiectoides is characterized by its violaceous hue due to the dilated vascular channels. The lesions are purpuric papules and pseudovesicles appearing on an erythematous base, most commonly contiguous with the surgical scar. Histologically, collections of atypical tumor cells and erythrocytes are present along with dilated blood vessels in the papillary dermis.2 Alopecia neoplastica presents as singular or grouped cicatricial patches of hair loss. Lesions of carcinoma erysipeloides present as warm, erythematous, tender, well-defined patches or plaques. Carcinoma en cuirasse is characterized by an erythematous sclerodermoid plaque on the chest wall.2
Our patient’s presentation was unique due to the widespread distribution, unusual pattern, and variable clinical morphologies of the cutaneous metastases. Our patient had findings of both carcinoma telangiectoides and nodular carcinoma. The mosaic violaceous erythema extending toward the mid-back rarely is reported in the literature and indicates extensive intravascular spread of tumor cells in the dermis.
Metastatic breast cancer is associated with a poor prognosis because it typically occurs in advanced stages and often does not respond to treatment.5 Although chemotherapy, hormonal therapy, and/or radiation therapy may improve survival, the choice in regimen is guided by cancer histology as well as prior treatments. In our case, the patient chose to continue her homeopathic therapy.
- Nashan D, Meiss F, Braun-Falco M, et al. Cutaneous metastasis from internal malignancies. Dermatol Ther. 2010;23:567-580.
- De Giorgi V, Grazzini M, Alfaioli B, et al. Cutaneous manifestations of breast carcinoma. Dermatol Ther. 2010;23:581-589.
- Mordenti C, Peris K, Concetta Fargnoli M, et al. Cutaneous metastatic breast carcinoma. Acta Dermatovenerologica. 2000;9:143-148.
- Nava G, Greer K, Patterson J, et al. Metastatic cutaneous breast carcinoma: a case report and review of the literature. Can J Plast Surg. 2009;17:25-27.
- Kalmykow B, Walker S. Cutaneous metastasis in breast cancer. Clin J Oncol Nurs. 2001;15:99-101.
To the Editor:
Cutaneous metastases occur more often in the setting of breast carcinoma than other malignancies in women.1 Although interventions are aimed at halting disease progression, cutaneous metastases indicate widespread disease and are associated with poor prognosis. We present the case of a patient with metastatic breast adenocarcinoma who developed cutaneous metastasis on the trunk after a double mastectomy. The widespread distribution and wide range of clinical manifestations are unique.
An 81-year-old woman presented to the dermatology office for evaluation of a skin eruption that started along a mastectomy scar on the left breast a few months postoperatively. She had a history of stage IV breast adenocarcinoma metastatic to the chest wall that was treated with a double mastectomy 2 years prior. The patient denied associated pain or pruritus and mainly was concerned with the cosmetic appearance. At the time of the initial diagnosis of breast adenocarcinoma, the patient was offered chemotherapy, which she did not tolerate. The patient opted against radiation therapy, as she preferred a more natural approach, such as anticancer shakes, which she was taking from a homeopathic source. She was unaware of the ingredients used in the shakes.
Physical examination revealed multiple grouped, firm, purpuric papules, nodules, and pseudovesicles on a background of violaceous erythema on the chest, abdomen, and flank (Figure 1). The background erythema had a mosaic pattern that extended toward the central back (Figure 2). A scoop shave biopsy of one of the purpuric nodules revealed highly atypical cells with abundant cytoplasm, large nuclei, and prominent nucleoli (Figure 3). Focally, the cells appeared to form glandular structures. Numerous atypical mitotic figures were present. Lymphatic invasion and microcalcifications were identified (Figure 3 [inset]). Immunohistochemical staining for cytokeratin 7 and gross cystic disease fluid protein 15 were strongly positive (Figure 4). Based on the histopathologic and immunohistochemical findings, a diagnosis of cutaneous metastatic breast adenocarcinoma was made. The patient opted to continue the homeopathic anticancer shakes and was subsequently lost to follow-up.
Cutaneous metastases of internal malignancies make up only 2% of all skin tumors,1 making them relatively uncommon in the dermatologic setting. However, cutaneous metastasis occurs in 23.9% of patients with breast carcinoma, making it the most common tumor after malignant melanoma to metastasize to the skin.2 The most common sites for breast carcinoma cutaneous metastasis (BCCM) are the chest wall and abdomen; other sites include the head/neck region and the extremities. The clinical presentation of BCCM varies depending on the mode of dissemination—lymphatic, hematogenous, contiguous growth, or iatrogenic implantation. The most common presentation is nodular carcinoma (47%–80%).2,3 Other presentations include carcinoma telangiectoides (8%–11%),2,3 alopecia neoplastica (2%–12%),2,3 and carcinoma erysipeloides (3%–6%).2,3 Carcinoma en cuirasse is rare.3
Nodular BCCM may present as firm solitary or grouped papules and nodules that are painless and range in color from flesh colored or pink to red-brown. Histologically, they are composed of atypical neoplastic cells arranged in small nests and cords, usually in a single-file line within the collagen bundles of the dermis.4 Carcinoma telangiectoides is characterized by its violaceous hue due to the dilated vascular channels. The lesions are purpuric papules and pseudovesicles appearing on an erythematous base, most commonly contiguous with the surgical scar. Histologically, collections of atypical tumor cells and erythrocytes are present along with dilated blood vessels in the papillary dermis.2 Alopecia neoplastica presents as singular or grouped cicatricial patches of hair loss. Lesions of carcinoma erysipeloides present as warm, erythematous, tender, well-defined patches or plaques. Carcinoma en cuirasse is characterized by an erythematous sclerodermoid plaque on the chest wall.2
Our patient’s presentation was unique due to the widespread distribution, unusual pattern, and variable clinical morphologies of the cutaneous metastases. Our patient had findings of both carcinoma telangiectoides and nodular carcinoma. The mosaic violaceous erythema extending toward the mid-back rarely is reported in the literature and indicates extensive intravascular spread of tumor cells in the dermis.
Metastatic breast cancer is associated with a poor prognosis because it typically occurs in advanced stages and often does not respond to treatment.5 Although chemotherapy, hormonal therapy, and/or radiation therapy may improve survival, the choice in regimen is guided by cancer histology as well as prior treatments. In our case, the patient chose to continue her homeopathic therapy.
To the Editor:
Cutaneous metastases occur more often in the setting of breast carcinoma than other malignancies in women.1 Although interventions are aimed at halting disease progression, cutaneous metastases indicate widespread disease and are associated with poor prognosis. We present the case of a patient with metastatic breast adenocarcinoma who developed cutaneous metastasis on the trunk after a double mastectomy. The widespread distribution and wide range of clinical manifestations are unique.
An 81-year-old woman presented to the dermatology office for evaluation of a skin eruption that started along a mastectomy scar on the left breast a few months postoperatively. She had a history of stage IV breast adenocarcinoma metastatic to the chest wall that was treated with a double mastectomy 2 years prior. The patient denied associated pain or pruritus and mainly was concerned with the cosmetic appearance. At the time of the initial diagnosis of breast adenocarcinoma, the patient was offered chemotherapy, which she did not tolerate. The patient opted against radiation therapy, as she preferred a more natural approach, such as anticancer shakes, which she was taking from a homeopathic source. She was unaware of the ingredients used in the shakes.
Physical examination revealed multiple grouped, firm, purpuric papules, nodules, and pseudovesicles on a background of violaceous erythema on the chest, abdomen, and flank (Figure 1). The background erythema had a mosaic pattern that extended toward the central back (Figure 2). A scoop shave biopsy of one of the purpuric nodules revealed highly atypical cells with abundant cytoplasm, large nuclei, and prominent nucleoli (Figure 3). Focally, the cells appeared to form glandular structures. Numerous atypical mitotic figures were present. Lymphatic invasion and microcalcifications were identified (Figure 3 [inset]). Immunohistochemical staining for cytokeratin 7 and gross cystic disease fluid protein 15 were strongly positive (Figure 4). Based on the histopathologic and immunohistochemical findings, a diagnosis of cutaneous metastatic breast adenocarcinoma was made. The patient opted to continue the homeopathic anticancer shakes and was subsequently lost to follow-up.
Cutaneous metastases of internal malignancies make up only 2% of all skin tumors,1 making them relatively uncommon in the dermatologic setting. However, cutaneous metastasis occurs in 23.9% of patients with breast carcinoma, making it the most common tumor after malignant melanoma to metastasize to the skin.2 The most common sites for breast carcinoma cutaneous metastasis (BCCM) are the chest wall and abdomen; other sites include the head/neck region and the extremities. The clinical presentation of BCCM varies depending on the mode of dissemination—lymphatic, hematogenous, contiguous growth, or iatrogenic implantation. The most common presentation is nodular carcinoma (47%–80%).2,3 Other presentations include carcinoma telangiectoides (8%–11%),2,3 alopecia neoplastica (2%–12%),2,3 and carcinoma erysipeloides (3%–6%).2,3 Carcinoma en cuirasse is rare.3
Nodular BCCM may present as firm solitary or grouped papules and nodules that are painless and range in color from flesh colored or pink to red-brown. Histologically, they are composed of atypical neoplastic cells arranged in small nests and cords, usually in a single-file line within the collagen bundles of the dermis.4 Carcinoma telangiectoides is characterized by its violaceous hue due to the dilated vascular channels. The lesions are purpuric papules and pseudovesicles appearing on an erythematous base, most commonly contiguous with the surgical scar. Histologically, collections of atypical tumor cells and erythrocytes are present along with dilated blood vessels in the papillary dermis.2 Alopecia neoplastica presents as singular or grouped cicatricial patches of hair loss. Lesions of carcinoma erysipeloides present as warm, erythematous, tender, well-defined patches or plaques. Carcinoma en cuirasse is characterized by an erythematous sclerodermoid plaque on the chest wall.2
Our patient’s presentation was unique due to the widespread distribution, unusual pattern, and variable clinical morphologies of the cutaneous metastases. Our patient had findings of both carcinoma telangiectoides and nodular carcinoma. The mosaic violaceous erythema extending toward the mid-back rarely is reported in the literature and indicates extensive intravascular spread of tumor cells in the dermis.
Metastatic breast cancer is associated with a poor prognosis because it typically occurs in advanced stages and often does not respond to treatment.5 Although chemotherapy, hormonal therapy, and/or radiation therapy may improve survival, the choice in regimen is guided by cancer histology as well as prior treatments. In our case, the patient chose to continue her homeopathic therapy.
- Nashan D, Meiss F, Braun-Falco M, et al. Cutaneous metastasis from internal malignancies. Dermatol Ther. 2010;23:567-580.
- De Giorgi V, Grazzini M, Alfaioli B, et al. Cutaneous manifestations of breast carcinoma. Dermatol Ther. 2010;23:581-589.
- Mordenti C, Peris K, Concetta Fargnoli M, et al. Cutaneous metastatic breast carcinoma. Acta Dermatovenerologica. 2000;9:143-148.
- Nava G, Greer K, Patterson J, et al. Metastatic cutaneous breast carcinoma: a case report and review of the literature. Can J Plast Surg. 2009;17:25-27.
- Kalmykow B, Walker S. Cutaneous metastasis in breast cancer. Clin J Oncol Nurs. 2001;15:99-101.
- Nashan D, Meiss F, Braun-Falco M, et al. Cutaneous metastasis from internal malignancies. Dermatol Ther. 2010;23:567-580.
- De Giorgi V, Grazzini M, Alfaioli B, et al. Cutaneous manifestations of breast carcinoma. Dermatol Ther. 2010;23:581-589.
- Mordenti C, Peris K, Concetta Fargnoli M, et al. Cutaneous metastatic breast carcinoma. Acta Dermatovenerologica. 2000;9:143-148.
- Nava G, Greer K, Patterson J, et al. Metastatic cutaneous breast carcinoma: a case report and review of the literature. Can J Plast Surg. 2009;17:25-27.
- Kalmykow B, Walker S. Cutaneous metastasis in breast cancer. Clin J Oncol Nurs. 2001;15:99-101.
Practice Points
- Breast carcinoma is one of the most common malignancies to metastasize to the skin in women.
- Although interventions are aimed at halting disease progression, cutaneous metastases indicate widespread disease and are associated with a poor prognosis.

Penile Paraffinoma: Dramatic Recurrence After Surgical Resection
To the Editor:
The term paraffinoma refers to a chronic granulomatous response to injection of paraffin, silicone, or other mineral oils into skin and soft tissue. Paraffinomas develop when the material is injected into the skin for cosmetic purposes to augment or enhance one’s appearance. Although they may occur in any location, the most common sites include the breasts and buttocks. The penis is a rare but emerging site for paraffinomas.1-3 We present a rare case of recurrence of a penile paraffinoma following surgical resection.
A 26-year-old uncircumcised Trinidadian man presented with a 5-cm, exquisitely tender tumor involving the penile shaft and median raphe that rapidly evolved over the course of 3 weeks (Figure 1). He presented with inability to urinate, attain an erection, or ambulate without notable tenderness. Additionally, he developed swelling of the penis and surrounding tissue. He had no other medical comorbidities; however, 1 year prior he presented to a urologist with a 1-cm nodule involving the median raphe that was surgically resected and required circumcision. Biopsy at the time of his surgical procedure revealed an exuberant foreign body giant cell reaction with surrounding empty spaces in the dermis resembling Swiss cheese, consistent with a paraffinoma (Figure 2). The recurrent tumor, which was 5 times the size of the initial nodule, was biopsied. Again, histopathologic findings were consistent with a paraffinoma with extensive dermal fibrosis and absence of polarizable material.
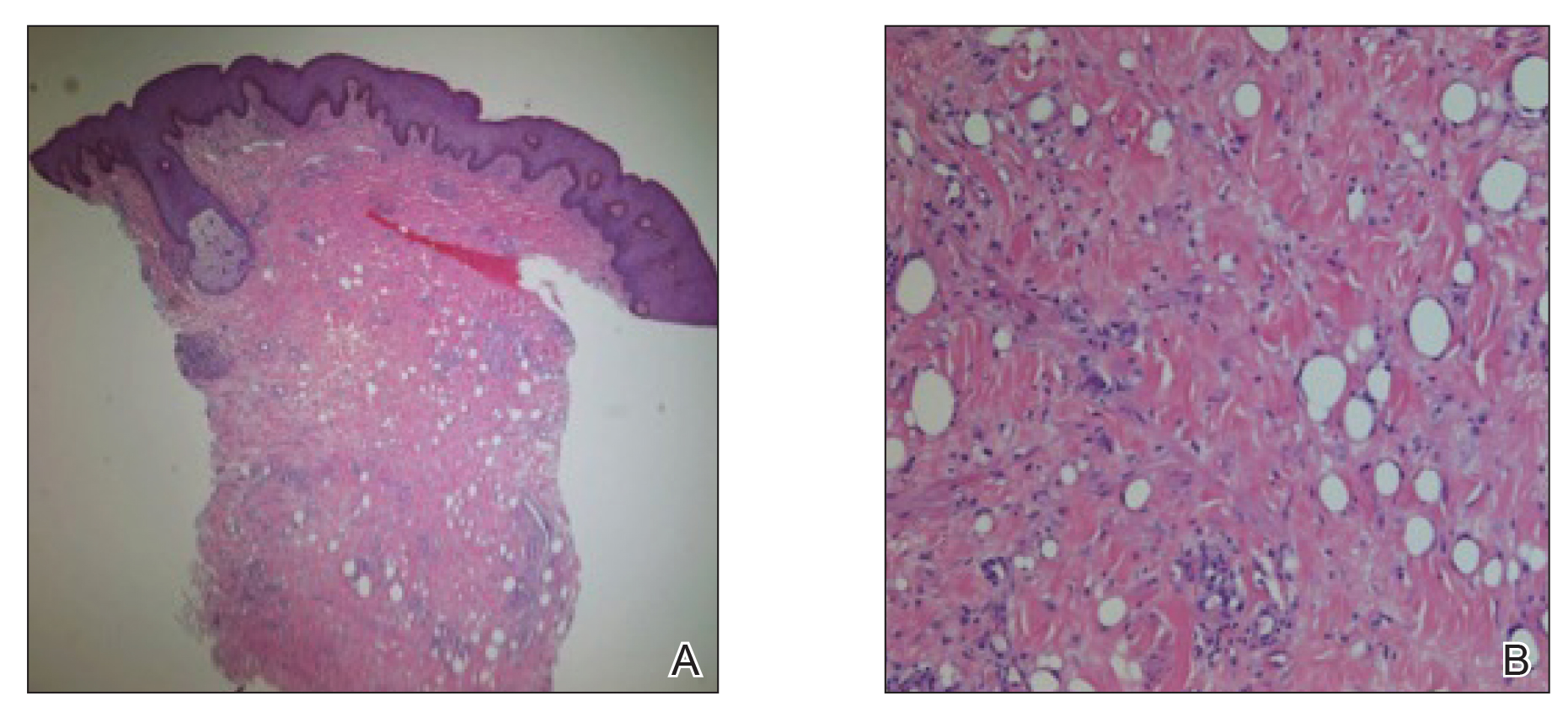
The patient underwent extensive reconstructive surgery requiring skin grafting to the penile shaft. Given the size and location of this recurrent tumor with the ability to destroy vital urologic and reproductive function, consideration for prevention of recurrent episodes included novel therapeutic treatment options to suppress inflammation and fibrosis with doxycycline and nicotinamide.
Paraffin injections are used for cosmetic enhancement and most often occur in a nonclinical setting without medical supervision, as they are not US Food and Drug Administration–approved medical injectable materials. Examples of oils injected include paraffin, camphorated oil, cottonseed or sesame oil, mineral oil, petroleum jelly, and beeswax. These oils are not hydrolyzed by tissue lipases but are instead treated as a foreign body substance with subsequent granuloma formation (also known as sclerosing lipogranuloma), which can occur many years after injection.4 The granulomatous response may be observed months to years after injection. The paraffinoma normally affects the injection site; however, regional lymphadenopathy and systemic disease has been reported.2 Histopathologic findings are characteristic and consist of a foreign body giant cell reaction, variably sized round to oval cavities within the dermis, and varying degrees of dermal fibrosis.5
In 1899, mineral oil was first injected into male genitalia to restore architecture in a patient’s testicles following bilateral orchiectomy. After the success of this endeavor, mineral oil injections were used as filler for other defects.3 However, by 1906 the complications of these injections became public knowledge when 2 patients developed subcutaneous nodules after receiving injections for facial wrinkles.2 Despite public knowledge of these complications, penile paraffin injections continued to occur both in medical and eventually nonmedical settings.
In 1947, Quérnu and Pérol6 described 6 penile paraffinoma cases outside the United States. Patients had petroleum jelly injections that eventuated in penile paraffinomas, and all of them lost the ability to attain an erection.6 Four years later, Bradley and Ehrgott7 described a case of penile paraffinoma likely caused by application of paraffin in association with occupational exposure. In 1956, May and Pickering8 cited a case of penile paraffinoma affecting the entire penile shaft in which the patient had undergone paraffin injection 7 years prior to treat premature ejaculation. Unfortunately, the injection resulted in a painful and unsatisfactory erection without resolution of premature ejaculation.8 Lee et al9 analyzed 26 cases of penile paraffinomas that occurred from 1981 to 1993. They found that all patients underwent injections of paraffin or petroleum jelly performed by nonmedical personnel with the predominant goal of enhancing penis size. Within 18.5 months of injection, 19 patients already experienced tenderness at the injection site. The remaining 7 patients experienced penile skin discoloration and abnormal contouring of the penis. Biopsy specimens revealed hyaline necrosis of subcutaneous adipose septa, cystlike spaces throughout involved tissue, and macrophages engulfing adipose tissue were found near blood vessels.9 In 2007, Eandi et al4 reported a case of penile paraffinoma with a 40-year delay of onset. Four years later, Manny et al10 reported penile paraffinomas in 3 Laotian men who injected a mineral oil.
Currently, paraffin injections are uncommon but still are being performed in some countries in Eastern Europe and the Far East11; they rarely are reported in the United States. Injections can occur in unusual sites such as the knee, and paraffinomas can develop many years after the procedure.12 Additionally, paraffinomas can obscure proper diagnosis of carcinomas, as described by Lee et al13 in a case in which a cervical paraffin injection confounded the diagnosis of a thyroid tumor. Furthermore, these injections usually are performed by nonmedical personnel and typically are repeated multiple times to reach cosmetic goals, rendering the patient vulnerable to early complications including allergic reactions, paraphimosis, infection, and inflammation.3
The clinical presentation of a penile paraffinoma may be a mimicker of several different entities, which are important to consider in the evaluation of a presenting patient. Infectious etiologies must be considered including lymphogranuloma venereum, granuloma inguinale, atypical mycobacteria, lupus vulgaris, and sexually transmitted infections. Importantly, neoplasms must be ruled out including squamous cell carcinoma, soft tissue sarcomas, melanoma, adenocarcinoma, or metastasis. Lymphedema, prior surgical procedures, trauma, and inflammatory etiologies also are in the differential diagnosis.14 Nonetheless, physicians must have a high clinical suspicion in the evaluation of a possible paraffinoma, as patients may not be forthcoming with relevant clinical history regarding a prior injection to the affected site, particularly if the injection occurred many years ago. As such, the patient may not consider this history relevant or may not even remember the event occurred, as was observed in our case. Furthermore, embarrassment, social taboo, and stigma may be associated with the behavior of undergoing injections in nonclinical settings without medical supervision.15
Patients may be motivated to undergo dangerous procedures to potentially alter their appearance due to perceived enhanced sexual ability, influence by loved ones, cultural rituals, and societal pressure.15,16 Furthermore, patients may not be aware of the material being injected or the volume. Given that these injections often are used with the goal of cosmetic enhancement, biopsies in cosmetically sensitive areas must be given careful consideration, and a thorough clinical history must support the decision to pursue a biopsy to obtain a definitive diagnosis.
The definitive diagnosis of a paraffinoma is determined by histopathology. However, the use of imaging modalities such as magnetic resonance imaging and computed tomography have been employed to delineate the extent of involvement. Imaging studies allow for surgical planning and may assist in narrowing a differential diagnosis.17 Currently, wide and complete surgical resection is the only definitive treatment of paraffinomas, including penile paraffinomas, as there is no evidence of spontaneous regression.3 A report of a reconstructive surgery involving penile resurfacing without T-style anastomosis has been found effective at preventing necrosis of the ventral penile skin. Not all paraffinomas behave similarly, and there is no reliable method to determine which paraffinoma may possess a more aggressive clinical course compared to those which have a more indolent course.18 As such, early detection is critical in the management of paraffinomas, especially in anatomic locations where tissue preservation is of utmost importance. In the case of a large penile paraffinoma with the ability to destroy vital urologic and reproductive function, physicians must consider prevention of recurrent episodes through suppression of inflammation and fibrosis with doxycycline and nicotinamide.19 Other medical treatments reported with varying success include corticosteroids, imiquimod, and isotretinoin.19-24 Employing adjunctive medical treatment may decrease the size of the mass, reducing the surgical defect size and preserving tissue vitality. Ultimately, the most crucial aspect in treatment is prevention, as injection of foreign materials elicits a foreign body response and can lead to notable morbidity.
- De Siati M, Selvaggio O, Di Fino G, et al. An unusual delayed complication of paraffin self-injection for penile girth augmentation. BMC Urol. 2013;13:66.
- Sejben I, Rácz A, Svébis M, et al. Petroleum jelly-induced penile paraffinoma with inguinal lymphadenitis mimicking incarcerated inguinal hernia. Can Urol Assoc J. 2012;6:E137-E139.
- Bayraktar N, Basar I. Penile paraffinoma [published online September 17, 2012]. Case Rep Urol. 2012;2012:202840.
- Eandi JA, Yao AP, Javidan J. Penile paraffinoma: the delayed presentation. Int Urol Nephrol. 2007;29:553-555.
- HirshBCJohnsonWC. Pathology of granulomatous diseases. foreign body granulomas. Int J Dermatol. 1984;23:531-538.
- Quérnu J, Pérol E. Paraffinomas of the penis. J Chir Par. 1947;63:345.
- Bradley, RH, Ehrgott WA. Paraffinoma of the penis: case report. J Urol. 1951;65:453.
- May JA, Pickering PP. Paraffinoma of the penis. Calif Med. 1956;85:42-44.
Yonsei Med J. 1994;35:344-348. - Lee T, Choi HR, Lee YT, et al. Paraffinoma of the penis.
- Manny T, Pettus J, Hemal A, et al. Penile sclerosing lipogranulomas and disfigurement from use of “1Super Extenze” among Laotian immigrants. J Sex Med. 2011;8:3505-3510.
- Akkus E Paraffinoma and ulcer of the external genitalia after self-injection of vaseline. J Sex Med. 2006;3:170-172.
- Grassetti L, Lazzeri D, Torresetti M, et al. Paraffinoma of the knee 60 years after primary infection. Arch Plast Surg. 2013;40:789-790.
- Lee YS, Son EJ, Kim BW, et al. Difficult evaluation of thyroid cancer due to cervical paraffin injection. J Korean Surg Soc. 2011;81(suppl 1):S17-S20.
- Gómez-Armayones S, Penín R, Marcoval J. Penile paraffinoma [in Spanish]. Actas Dermosifiliogr. 2014;105:957-959.
- Moon DG, Yoo JW, Bae JH, et al. Sexual function and psychological characteristics of penile paraffinoma. Asian J Androl. 2003;5:191-194.
- Pehlivanov G, Kavaklieva S, Kazandjieva J, et al. Foreign-body granuloma of the penis in sexually active individuals (penile paraffinoma). J Eur Acad Dermatol Venereol. 2008;22:845-851.
- Cormio L, Di Fino G, Scavone C, et al. Magnetic resonance imaging of penile paraffinoma: case report. BMC Med Imaging. 2014;14:39.
- Shin YS, Zhao C, Park JK. New reconstructive surgery for penile paraffinoma to prevent necrosis of ventral penile skin. Urology. 2013;81:437-441.
- Feldmann R, Harms M, Chavaz P, et al. Orbital and palpebral paraffinoma. J Am Acad Dermatol. 1992;26:833-835.
- MastruserioDNPesqueiraMJCobbMW. Severe granulomatous reaction and facial ulceration occurring after subcutaneous silicone injection. J Am Acad Dermatol. 1996;34:849-852.
- HoWS Management of paraffinoma of the breast. Br J Plast Surg. 2001;54:232-234.
- LloretPSuccessful treatment of granulomatous reactions secondary to injection of esthetic implants. Dermatol Surg. 2005;31:486-490.
- RosenbergEThree cases of penile paraffinoma. Urology. 2007;70:372.
- Baumann LS, Halem ML. Lip silicone granulomatous foreign body reaction treated with Aldara (imiquimod 5%). Dermatol Surg. 2003;29:429-432.
To the Editor:
The term paraffinoma refers to a chronic granulomatous response to injection of paraffin, silicone, or other mineral oils into skin and soft tissue. Paraffinomas develop when the material is injected into the skin for cosmetic purposes to augment or enhance one’s appearance. Although they may occur in any location, the most common sites include the breasts and buttocks. The penis is a rare but emerging site for paraffinomas.1-3 We present a rare case of recurrence of a penile paraffinoma following surgical resection.
A 26-year-old uncircumcised Trinidadian man presented with a 5-cm, exquisitely tender tumor involving the penile shaft and median raphe that rapidly evolved over the course of 3 weeks (Figure 1). He presented with inability to urinate, attain an erection, or ambulate without notable tenderness. Additionally, he developed swelling of the penis and surrounding tissue. He had no other medical comorbidities; however, 1 year prior he presented to a urologist with a 1-cm nodule involving the median raphe that was surgically resected and required circumcision. Biopsy at the time of his surgical procedure revealed an exuberant foreign body giant cell reaction with surrounding empty spaces in the dermis resembling Swiss cheese, consistent with a paraffinoma (Figure 2). The recurrent tumor, which was 5 times the size of the initial nodule, was biopsied. Again, histopathologic findings were consistent with a paraffinoma with extensive dermal fibrosis and absence of polarizable material.
The patient underwent extensive reconstructive surgery requiring skin grafting to the penile shaft. Given the size and location of this recurrent tumor with the ability to destroy vital urologic and reproductive function, consideration for prevention of recurrent episodes included novel therapeutic treatment options to suppress inflammation and fibrosis with doxycycline and nicotinamide.
Paraffin injections are used for cosmetic enhancement and most often occur in a nonclinical setting without medical supervision, as they are not US Food and Drug Administration–approved medical injectable materials. Examples of oils injected include paraffin, camphorated oil, cottonseed or sesame oil, mineral oil, petroleum jelly, and beeswax. These oils are not hydrolyzed by tissue lipases but are instead treated as a foreign body substance with subsequent granuloma formation (also known as sclerosing lipogranuloma), which can occur many years after injection.4 The granulomatous response may be observed months to years after injection. The paraffinoma normally affects the injection site; however, regional lymphadenopathy and systemic disease has been reported.2 Histopathologic findings are characteristic and consist of a foreign body giant cell reaction, variably sized round to oval cavities within the dermis, and varying degrees of dermal fibrosis.5
In 1899, mineral oil was first injected into male genitalia to restore architecture in a patient’s testicles following bilateral orchiectomy. After the success of this endeavor, mineral oil injections were used as filler for other defects.3 However, by 1906 the complications of these injections became public knowledge when 2 patients developed subcutaneous nodules after receiving injections for facial wrinkles.2 Despite public knowledge of these complications, penile paraffin injections continued to occur both in medical and eventually nonmedical settings.
In 1947, Quérnu and Pérol6 described 6 penile paraffinoma cases outside the United States. Patients had petroleum jelly injections that eventuated in penile paraffinomas, and all of them lost the ability to attain an erection.6 Four years later, Bradley and Ehrgott7 described a case of penile paraffinoma likely caused by application of paraffin in association with occupational exposure. In 1956, May and Pickering8 cited a case of penile paraffinoma affecting the entire penile shaft in which the patient had undergone paraffin injection 7 years prior to treat premature ejaculation. Unfortunately, the injection resulted in a painful and unsatisfactory erection without resolution of premature ejaculation.8 Lee et al9 analyzed 26 cases of penile paraffinomas that occurred from 1981 to 1993. They found that all patients underwent injections of paraffin or petroleum jelly performed by nonmedical personnel with the predominant goal of enhancing penis size. Within 18.5 months of injection, 19 patients already experienced tenderness at the injection site. The remaining 7 patients experienced penile skin discoloration and abnormal contouring of the penis. Biopsy specimens revealed hyaline necrosis of subcutaneous adipose septa, cystlike spaces throughout involved tissue, and macrophages engulfing adipose tissue were found near blood vessels.9 In 2007, Eandi et al4 reported a case of penile paraffinoma with a 40-year delay of onset. Four years later, Manny et al10 reported penile paraffinomas in 3 Laotian men who injected a mineral oil.
Currently, paraffin injections are uncommon but still are being performed in some countries in Eastern Europe and the Far East11; they rarely are reported in the United States. Injections can occur in unusual sites such as the knee, and paraffinomas can develop many years after the procedure.12 Additionally, paraffinomas can obscure proper diagnosis of carcinomas, as described by Lee et al13 in a case in which a cervical paraffin injection confounded the diagnosis of a thyroid tumor. Furthermore, these injections usually are performed by nonmedical personnel and typically are repeated multiple times to reach cosmetic goals, rendering the patient vulnerable to early complications including allergic reactions, paraphimosis, infection, and inflammation.3
The clinical presentation of a penile paraffinoma may be a mimicker of several different entities, which are important to consider in the evaluation of a presenting patient. Infectious etiologies must be considered including lymphogranuloma venereum, granuloma inguinale, atypical mycobacteria, lupus vulgaris, and sexually transmitted infections. Importantly, neoplasms must be ruled out including squamous cell carcinoma, soft tissue sarcomas, melanoma, adenocarcinoma, or metastasis. Lymphedema, prior surgical procedures, trauma, and inflammatory etiologies also are in the differential diagnosis.14 Nonetheless, physicians must have a high clinical suspicion in the evaluation of a possible paraffinoma, as patients may not be forthcoming with relevant clinical history regarding a prior injection to the affected site, particularly if the injection occurred many years ago. As such, the patient may not consider this history relevant or may not even remember the event occurred, as was observed in our case. Furthermore, embarrassment, social taboo, and stigma may be associated with the behavior of undergoing injections in nonclinical settings without medical supervision.15
Patients may be motivated to undergo dangerous procedures to potentially alter their appearance due to perceived enhanced sexual ability, influence by loved ones, cultural rituals, and societal pressure.15,16 Furthermore, patients may not be aware of the material being injected or the volume. Given that these injections often are used with the goal of cosmetic enhancement, biopsies in cosmetically sensitive areas must be given careful consideration, and a thorough clinical history must support the decision to pursue a biopsy to obtain a definitive diagnosis.
The definitive diagnosis of a paraffinoma is determined by histopathology. However, the use of imaging modalities such as magnetic resonance imaging and computed tomography have been employed to delineate the extent of involvement. Imaging studies allow for surgical planning and may assist in narrowing a differential diagnosis.17 Currently, wide and complete surgical resection is the only definitive treatment of paraffinomas, including penile paraffinomas, as there is no evidence of spontaneous regression.3 A report of a reconstructive surgery involving penile resurfacing without T-style anastomosis has been found effective at preventing necrosis of the ventral penile skin. Not all paraffinomas behave similarly, and there is no reliable method to determine which paraffinoma may possess a more aggressive clinical course compared to those which have a more indolent course.18 As such, early detection is critical in the management of paraffinomas, especially in anatomic locations where tissue preservation is of utmost importance. In the case of a large penile paraffinoma with the ability to destroy vital urologic and reproductive function, physicians must consider prevention of recurrent episodes through suppression of inflammation and fibrosis with doxycycline and nicotinamide.19 Other medical treatments reported with varying success include corticosteroids, imiquimod, and isotretinoin.19-24 Employing adjunctive medical treatment may decrease the size of the mass, reducing the surgical defect size and preserving tissue vitality. Ultimately, the most crucial aspect in treatment is prevention, as injection of foreign materials elicits a foreign body response and can lead to notable morbidity.
To the Editor:
The term paraffinoma refers to a chronic granulomatous response to injection of paraffin, silicone, or other mineral oils into skin and soft tissue. Paraffinomas develop when the material is injected into the skin for cosmetic purposes to augment or enhance one’s appearance. Although they may occur in any location, the most common sites include the breasts and buttocks. The penis is a rare but emerging site for paraffinomas.1-3 We present a rare case of recurrence of a penile paraffinoma following surgical resection.
A 26-year-old uncircumcised Trinidadian man presented with a 5-cm, exquisitely tender tumor involving the penile shaft and median raphe that rapidly evolved over the course of 3 weeks (Figure 1). He presented with inability to urinate, attain an erection, or ambulate without notable tenderness. Additionally, he developed swelling of the penis and surrounding tissue. He had no other medical comorbidities; however, 1 year prior he presented to a urologist with a 1-cm nodule involving the median raphe that was surgically resected and required circumcision. Biopsy at the time of his surgical procedure revealed an exuberant foreign body giant cell reaction with surrounding empty spaces in the dermis resembling Swiss cheese, consistent with a paraffinoma (Figure 2). The recurrent tumor, which was 5 times the size of the initial nodule, was biopsied. Again, histopathologic findings were consistent with a paraffinoma with extensive dermal fibrosis and absence of polarizable material.
The patient underwent extensive reconstructive surgery requiring skin grafting to the penile shaft. Given the size and location of this recurrent tumor with the ability to destroy vital urologic and reproductive function, consideration for prevention of recurrent episodes included novel therapeutic treatment options to suppress inflammation and fibrosis with doxycycline and nicotinamide.
Paraffin injections are used for cosmetic enhancement and most often occur in a nonclinical setting without medical supervision, as they are not US Food and Drug Administration–approved medical injectable materials. Examples of oils injected include paraffin, camphorated oil, cottonseed or sesame oil, mineral oil, petroleum jelly, and beeswax. These oils are not hydrolyzed by tissue lipases but are instead treated as a foreign body substance with subsequent granuloma formation (also known as sclerosing lipogranuloma), which can occur many years after injection.4 The granulomatous response may be observed months to years after injection. The paraffinoma normally affects the injection site; however, regional lymphadenopathy and systemic disease has been reported.2 Histopathologic findings are characteristic and consist of a foreign body giant cell reaction, variably sized round to oval cavities within the dermis, and varying degrees of dermal fibrosis.5
In 1899, mineral oil was first injected into male genitalia to restore architecture in a patient’s testicles following bilateral orchiectomy. After the success of this endeavor, mineral oil injections were used as filler for other defects.3 However, by 1906 the complications of these injections became public knowledge when 2 patients developed subcutaneous nodules after receiving injections for facial wrinkles.2 Despite public knowledge of these complications, penile paraffin injections continued to occur both in medical and eventually nonmedical settings.
In 1947, Quérnu and Pérol6 described 6 penile paraffinoma cases outside the United States. Patients had petroleum jelly injections that eventuated in penile paraffinomas, and all of them lost the ability to attain an erection.6 Four years later, Bradley and Ehrgott7 described a case of penile paraffinoma likely caused by application of paraffin in association with occupational exposure. In 1956, May and Pickering8 cited a case of penile paraffinoma affecting the entire penile shaft in which the patient had undergone paraffin injection 7 years prior to treat premature ejaculation. Unfortunately, the injection resulted in a painful and unsatisfactory erection without resolution of premature ejaculation.8 Lee et al9 analyzed 26 cases of penile paraffinomas that occurred from 1981 to 1993. They found that all patients underwent injections of paraffin or petroleum jelly performed by nonmedical personnel with the predominant goal of enhancing penis size. Within 18.5 months of injection, 19 patients already experienced tenderness at the injection site. The remaining 7 patients experienced penile skin discoloration and abnormal contouring of the penis. Biopsy specimens revealed hyaline necrosis of subcutaneous adipose septa, cystlike spaces throughout involved tissue, and macrophages engulfing adipose tissue were found near blood vessels.9 In 2007, Eandi et al4 reported a case of penile paraffinoma with a 40-year delay of onset. Four years later, Manny et al10 reported penile paraffinomas in 3 Laotian men who injected a mineral oil.
Currently, paraffin injections are uncommon but still are being performed in some countries in Eastern Europe and the Far East11; they rarely are reported in the United States. Injections can occur in unusual sites such as the knee, and paraffinomas can develop many years after the procedure.12 Additionally, paraffinomas can obscure proper diagnosis of carcinomas, as described by Lee et al13 in a case in which a cervical paraffin injection confounded the diagnosis of a thyroid tumor. Furthermore, these injections usually are performed by nonmedical personnel and typically are repeated multiple times to reach cosmetic goals, rendering the patient vulnerable to early complications including allergic reactions, paraphimosis, infection, and inflammation.3
The clinical presentation of a penile paraffinoma may be a mimicker of several different entities, which are important to consider in the evaluation of a presenting patient. Infectious etiologies must be considered including lymphogranuloma venereum, granuloma inguinale, atypical mycobacteria, lupus vulgaris, and sexually transmitted infections. Importantly, neoplasms must be ruled out including squamous cell carcinoma, soft tissue sarcomas, melanoma, adenocarcinoma, or metastasis. Lymphedema, prior surgical procedures, trauma, and inflammatory etiologies also are in the differential diagnosis.14 Nonetheless, physicians must have a high clinical suspicion in the evaluation of a possible paraffinoma, as patients may not be forthcoming with relevant clinical history regarding a prior injection to the affected site, particularly if the injection occurred many years ago. As such, the patient may not consider this history relevant or may not even remember the event occurred, as was observed in our case. Furthermore, embarrassment, social taboo, and stigma may be associated with the behavior of undergoing injections in nonclinical settings without medical supervision.15
Patients may be motivated to undergo dangerous procedures to potentially alter their appearance due to perceived enhanced sexual ability, influence by loved ones, cultural rituals, and societal pressure.15,16 Furthermore, patients may not be aware of the material being injected or the volume. Given that these injections often are used with the goal of cosmetic enhancement, biopsies in cosmetically sensitive areas must be given careful consideration, and a thorough clinical history must support the decision to pursue a biopsy to obtain a definitive diagnosis.
The definitive diagnosis of a paraffinoma is determined by histopathology. However, the use of imaging modalities such as magnetic resonance imaging and computed tomography have been employed to delineate the extent of involvement. Imaging studies allow for surgical planning and may assist in narrowing a differential diagnosis.17 Currently, wide and complete surgical resection is the only definitive treatment of paraffinomas, including penile paraffinomas, as there is no evidence of spontaneous regression.3 A report of a reconstructive surgery involving penile resurfacing without T-style anastomosis has been found effective at preventing necrosis of the ventral penile skin. Not all paraffinomas behave similarly, and there is no reliable method to determine which paraffinoma may possess a more aggressive clinical course compared to those which have a more indolent course.18 As such, early detection is critical in the management of paraffinomas, especially in anatomic locations where tissue preservation is of utmost importance. In the case of a large penile paraffinoma with the ability to destroy vital urologic and reproductive function, physicians must consider prevention of recurrent episodes through suppression of inflammation and fibrosis with doxycycline and nicotinamide.19 Other medical treatments reported with varying success include corticosteroids, imiquimod, and isotretinoin.19-24 Employing adjunctive medical treatment may decrease the size of the mass, reducing the surgical defect size and preserving tissue vitality. Ultimately, the most crucial aspect in treatment is prevention, as injection of foreign materials elicits a foreign body response and can lead to notable morbidity.
- De Siati M, Selvaggio O, Di Fino G, et al. An unusual delayed complication of paraffin self-injection for penile girth augmentation. BMC Urol. 2013;13:66.
- Sejben I, Rácz A, Svébis M, et al. Petroleum jelly-induced penile paraffinoma with inguinal lymphadenitis mimicking incarcerated inguinal hernia. Can Urol Assoc J. 2012;6:E137-E139.
- Bayraktar N, Basar I. Penile paraffinoma [published online September 17, 2012]. Case Rep Urol. 2012;2012:202840.
- Eandi JA, Yao AP, Javidan J. Penile paraffinoma: the delayed presentation. Int Urol Nephrol. 2007;29:553-555.
- HirshBCJohnsonWC. Pathology of granulomatous diseases. foreign body granulomas. Int J Dermatol. 1984;23:531-538.
- Quérnu J, Pérol E. Paraffinomas of the penis. J Chir Par. 1947;63:345.
- Bradley, RH, Ehrgott WA. Paraffinoma of the penis: case report. J Urol. 1951;65:453.
- May JA, Pickering PP. Paraffinoma of the penis. Calif Med. 1956;85:42-44.
Yonsei Med J. 1994;35:344-348. - Lee T, Choi HR, Lee YT, et al. Paraffinoma of the penis.
- Manny T, Pettus J, Hemal A, et al. Penile sclerosing lipogranulomas and disfigurement from use of “1Super Extenze” among Laotian immigrants. J Sex Med. 2011;8:3505-3510.
- Akkus E Paraffinoma and ulcer of the external genitalia after self-injection of vaseline. J Sex Med. 2006;3:170-172.
- Grassetti L, Lazzeri D, Torresetti M, et al. Paraffinoma of the knee 60 years after primary infection. Arch Plast Surg. 2013;40:789-790.
- Lee YS, Son EJ, Kim BW, et al. Difficult evaluation of thyroid cancer due to cervical paraffin injection. J Korean Surg Soc. 2011;81(suppl 1):S17-S20.
- Gómez-Armayones S, Penín R, Marcoval J. Penile paraffinoma [in Spanish]. Actas Dermosifiliogr. 2014;105:957-959.
- Moon DG, Yoo JW, Bae JH, et al. Sexual function and psychological characteristics of penile paraffinoma. Asian J Androl. 2003;5:191-194.
- Pehlivanov G, Kavaklieva S, Kazandjieva J, et al. Foreign-body granuloma of the penis in sexually active individuals (penile paraffinoma). J Eur Acad Dermatol Venereol. 2008;22:845-851.
- Cormio L, Di Fino G, Scavone C, et al. Magnetic resonance imaging of penile paraffinoma: case report. BMC Med Imaging. 2014;14:39.
- Shin YS, Zhao C, Park JK. New reconstructive surgery for penile paraffinoma to prevent necrosis of ventral penile skin. Urology. 2013;81:437-441.
- Feldmann R, Harms M, Chavaz P, et al. Orbital and palpebral paraffinoma. J Am Acad Dermatol. 1992;26:833-835.
- MastruserioDNPesqueiraMJCobbMW. Severe granulomatous reaction and facial ulceration occurring after subcutaneous silicone injection. J Am Acad Dermatol. 1996;34:849-852.
- HoWS Management of paraffinoma of the breast. Br J Plast Surg. 2001;54:232-234.
- LloretPSuccessful treatment of granulomatous reactions secondary to injection of esthetic implants. Dermatol Surg. 2005;31:486-490.
- RosenbergEThree cases of penile paraffinoma. Urology. 2007;70:372.
- Baumann LS, Halem ML. Lip silicone granulomatous foreign body reaction treated with Aldara (imiquimod 5%). Dermatol Surg. 2003;29:429-432.
- De Siati M, Selvaggio O, Di Fino G, et al. An unusual delayed complication of paraffin self-injection for penile girth augmentation. BMC Urol. 2013;13:66.
- Sejben I, Rácz A, Svébis M, et al. Petroleum jelly-induced penile paraffinoma with inguinal lymphadenitis mimicking incarcerated inguinal hernia. Can Urol Assoc J. 2012;6:E137-E139.
- Bayraktar N, Basar I. Penile paraffinoma [published online September 17, 2012]. Case Rep Urol. 2012;2012:202840.
- Eandi JA, Yao AP, Javidan J. Penile paraffinoma: the delayed presentation. Int Urol Nephrol. 2007;29:553-555.
- HirshBCJohnsonWC. Pathology of granulomatous diseases. foreign body granulomas. Int J Dermatol. 1984;23:531-538.
- Quérnu J, Pérol E. Paraffinomas of the penis. J Chir Par. 1947;63:345.
- Bradley, RH, Ehrgott WA. Paraffinoma of the penis: case report. J Urol. 1951;65:453.
- May JA, Pickering PP. Paraffinoma of the penis. Calif Med. 1956;85:42-44.
Yonsei Med J. 1994;35:344-348. - Lee T, Choi HR, Lee YT, et al. Paraffinoma of the penis.
- Manny T, Pettus J, Hemal A, et al. Penile sclerosing lipogranulomas and disfigurement from use of “1Super Extenze” among Laotian immigrants. J Sex Med. 2011;8:3505-3510.
- Akkus E Paraffinoma and ulcer of the external genitalia after self-injection of vaseline. J Sex Med. 2006;3:170-172.
- Grassetti L, Lazzeri D, Torresetti M, et al. Paraffinoma of the knee 60 years after primary infection. Arch Plast Surg. 2013;40:789-790.
- Lee YS, Son EJ, Kim BW, et al. Difficult evaluation of thyroid cancer due to cervical paraffin injection. J Korean Surg Soc. 2011;81(suppl 1):S17-S20.
- Gómez-Armayones S, Penín R, Marcoval J. Penile paraffinoma [in Spanish]. Actas Dermosifiliogr. 2014;105:957-959.
- Moon DG, Yoo JW, Bae JH, et al. Sexual function and psychological characteristics of penile paraffinoma. Asian J Androl. 2003;5:191-194.
- Pehlivanov G, Kavaklieva S, Kazandjieva J, et al. Foreign-body granuloma of the penis in sexually active individuals (penile paraffinoma). J Eur Acad Dermatol Venereol. 2008;22:845-851.
- Cormio L, Di Fino G, Scavone C, et al. Magnetic resonance imaging of penile paraffinoma: case report. BMC Med Imaging. 2014;14:39.
- Shin YS, Zhao C, Park JK. New reconstructive surgery for penile paraffinoma to prevent necrosis of ventral penile skin. Urology. 2013;81:437-441.
- Feldmann R, Harms M, Chavaz P, et al. Orbital and palpebral paraffinoma. J Am Acad Dermatol. 1992;26:833-835.
- MastruserioDNPesqueiraMJCobbMW. Severe granulomatous reaction and facial ulceration occurring after subcutaneous silicone injection. J Am Acad Dermatol. 1996;34:849-852.
- HoWS Management of paraffinoma of the breast. Br J Plast Surg. 2001;54:232-234.
- LloretPSuccessful treatment of granulomatous reactions secondary to injection of esthetic implants. Dermatol Surg. 2005;31:486-490.
- RosenbergEThree cases of penile paraffinoma. Urology. 2007;70:372.
- Baumann LS, Halem ML. Lip silicone granulomatous foreign body reaction treated with Aldara (imiquimod 5%). Dermatol Surg. 2003;29:429-432.
Practice Points
- Taking a thorough history in patients with possible paraffinomas is vital, including a history of injectables even in the genital region.
- Biopsies in cosmetically sensitive areas must be given careful consideration. Clinical history must support the decision to pursue a definitive diagnosis.
- Early detection is critical in the management of paraffinomas, especially in anatomic locations where tissue preservation is of utmost importance.
Complex Regional Pain Syndrome Type II After a Brachial Plexus and C6 Nerve Root Injury
To the Editor:
A 62-year-old man presented with an atrophied painful left arm of 17 years’ duration that began when he was hit by a car as a pedestrian. He sustained severe multisystem injuries from the accident, including left brachial plexus and C6 nerve root avulsion injury. When he regained consciousness after 6 weeks in the intensive care unit, he immediately noted diffuse pain throughout the body, especially in the left arm. Since the accident, the patient continued to have diminished sensation to touch and temperature in the left arm. He also had burning, throbbing, and electrical pain in the left arm with light touch as well as spontaneously. He was thoroughly evaluated by a neurologist and was diagnosed with complex regional pain syndrome (CRPS) type II. For the treatment of pain, dorsal column stimulation and hemilaminectomy with exploration of the avulsed nerve root were attempted, both of which had minimal effect. He was maintained on hydromorphone, methadone, and oxazepam. He reported that for many years he was unable move out of bed due to the unbearable pain. With pain medications, he was able to regain most of his independence in his daily life, though the pain and other clinical aspects of CRPS still completely limited his use of the left arm.
Physical examination revealed glossy, cold, hairless skin with hypohidrosis of the left arm, forearm, and hand (Figures 1 and 2A). The left arm was conspicuously atrophied, with the forearm and hand erythematous. The fingers were taut, contracted, and edematous (Figure 2B), and the skin was unable to be pinched. The fingernails on the left hand had dystrophic changes including yellow color and brittleness with longitudinal ridges (Figure 3). The patient could activate the left bicep and tricep muscles against gravity but had minimal function of the deltoid muscle. He also had minimal movement of the left index finger and was unable to move any other digits of the left hand. The patient was continued on pain management treatments and physical therapy for his condition.

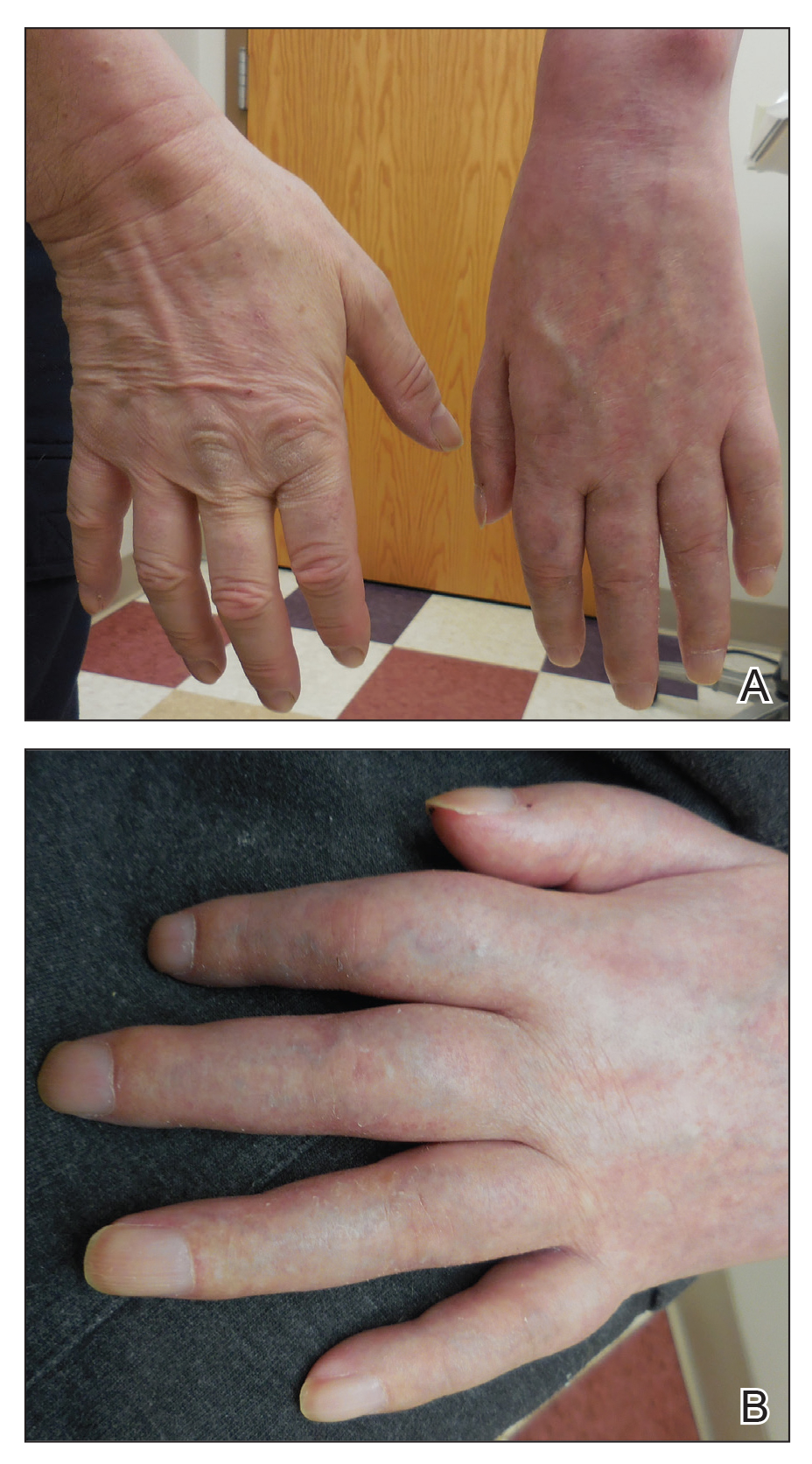
Complex regional pain syndrome is a neuropathic disorder of the extremities characterized by pain and a variety of autonomic and motor disturbances such as local edema, limited active range of motion, and vasomotor and trophic skin changes. There are 2 types of CRPS: type II is marked by explicit nerve injury and type I is not. The pathophysiology of CRPS is unknown.1-3
There is no definite set of diagnostic criteria for CRPS. The lack of any gold-standard diagnostic test for CRPS has made arriving at one valid, widely accepted set of diagnostic criteria impossible.1 There are 4 widely used sets of diagnostic criteria. One is the International Association for the Study of Pain diagnostic criteria defined in 1994.4 However, the criteria rely entirely on subjective symptoms and have been under great scrutiny due to their questionable validity.2 Veldman et al5 presented other widely used CRPS diagnostic criteria in their prospective study of 829 reflex sympathetic dystrophy patients, which paid particular attention to the early clinical manifestations of CRPS. In 1999, Bruehl et al2 proposed their own modified diagnostic criteria, which required physician-assessed signs in 2 of 4 categories to avoid the practice of exclusively relying on subjective symptoms. In addition, during a consensus meeting in Budapest, Hungary, a modified version of the Bruehl criteria was proposed.6 All 4 criteria rely solely on detailed history and physical examination, and the choice of diagnostic criteria remains subjective.
The pathophysiology of CRPS also remains unclear. There are several proposed mechanisms such as sympathetic nervous system dysfunction, abnormal inflammatory response, and central nervous system involvement.1 Psychologic factors, sequelae of nerve injury, and genetic predisposition also have been implicated in the pathophysiology of CRPS.1 It is likely that several mechanisms variably contribute to each presentation of CRPS.
Many dermatologic findings, in addition to neuromuscular symptoms, accompany CRPS and serve as important clues to making the clinical diagnosis. Complex regional pain syndrome has been thought to have 3 distinct sequential stages of CRPS.1,3,7 Stage 1—the acute stage—is marked by hyperalgesia, allodynia, sudomotor disturbances, and prominent edema. Stage 2—the dystrophic stage—is characterized by more marked pain and sensory dysfunction, vasomotor dysfunction, development of motor dysfunction, soft tissue edema, skin and articular soft tissue thickening, and development of dystrophic nail changes. Stage 3—the atrophic stage—is marked by decreased pain and sensory disturbances, markedly increased motor dysfunction, waxy atrophic skin changes, progression of dystrophic nail changes, and skeletal cystic and subchondral erosions with diffuse osteoporosis.1,3,7
The staging model, however, has been called into question.3 In a cluster analysis, Bruehl et al3 arrived at 3 relatively consistent CRPS patient subgroups that did not have notably different pain duration, suggesting the existence of 3 CRPS subtypes, not stages. Their study found that one of the subgroups best represented the clinical presentation of CRPS type II. This subgroup had the greatest pain and sensory abnormalities and the least vasomotor dysfunction of all 3 subgroups. Nonetheless, this study has not settled the discussion, as it only included 113 patients.3 Thus, with future studies, our understanding of CRPS in stages may change, which likely will impact how the clinical diagnosis is made.
There is a lack of high-quality evidence for most treatment interventions for CRPS8; however, the current practice is to use an interdisciplinary approach.1,9,10 The main therapeutic arm of this approach is rehabilitation; physical and occupational therapy can help improve range of motion, contracture, and atrophy. The other 2 arms of the approach are psychologic therapy to improve quality of life and pain management with pharmacologic therapy and/or invasive interventions. The choice of therapy remains empirical; trial and error should be expected in developing an adequate treatment plan for each individual patient.
Many aspects of CRPS remain unclear, and even our current understanding of the disease will inevitably change over time. The syndrome can cause life-changing morbidities in patients, and late diagnosis and treatment are associated with poor prognosis. Because there are many dermatologic findings associated with the disorder, it is crucial for dermatologists to clinically recognize the disorder and to refer patients to appropriate channels so that treatment can be started as soon as possible.
- Borchers A, Gershwin M. Complex regional pain syndrome: a comprehensive and critical review. Autoimmun Rev. 2014;13:242-265.
- Bruehl S, Harden RN, Galer BS, et al. External validation of IASP diagnostic criteria for complex regional pain syndrome and proposed research diagnostic criteria. International Association for the Study of Pain. Pain. 1999;81:147-154.
- Bruehl S, Harden RN, Gaker BS, et al. Complex regional pain syndrome: are there distinct subtypes and sequential stages of the syndrome? Pain. 2002;95:119-124.
- Merskey H, Bogduk N. Classification of Chronic Pain: Descriptions of Chronic Pain Syndromes and Definitions of Pain Terms. 2nd ed. Seattle, WA: IASP Press; 1994.
- Veldman PH, Reynen HM, Arntz IE, et al. Signs and symptoms of reflex sympathetic dystrophy: prospective study of 829 patients. Lancet. 1993;342:1012-1016.
- Harden RN, Bruehl S, Perez RS, et al. Validation of proposed diagnostic criteria (the “Budapest Criteria”) for complex regional pain syndrome. Pain. 2010;150:268-274.
- Sebastin SJ. Complex regional pain syndrome. Indian J Plast Surg. 2011;44:298-307.
- O’Connell NE, Wand BM, McAuley J, et al. Interventions for treating pain and disability in adults with complex regional pain syndrome. Cochrane Database Syst Rev. 2013;4:CD009416.
- Hsu ES. Practical management of complex regional pain syndrome. Am J Ther. 2009;16:147-154.
- Stanton-Hicks MD, Burton AW, Bruehl SP, et al. An updated interdisciplinary clinical pathway for CRPS: report of an expert panel. Pain Pract. 2002;2:1-16.
To the Editor:
A 62-year-old man presented with an atrophied painful left arm of 17 years’ duration that began when he was hit by a car as a pedestrian. He sustained severe multisystem injuries from the accident, including left brachial plexus and C6 nerve root avulsion injury. When he regained consciousness after 6 weeks in the intensive care unit, he immediately noted diffuse pain throughout the body, especially in the left arm. Since the accident, the patient continued to have diminished sensation to touch and temperature in the left arm. He also had burning, throbbing, and electrical pain in the left arm with light touch as well as spontaneously. He was thoroughly evaluated by a neurologist and was diagnosed with complex regional pain syndrome (CRPS) type II. For the treatment of pain, dorsal column stimulation and hemilaminectomy with exploration of the avulsed nerve root were attempted, both of which had minimal effect. He was maintained on hydromorphone, methadone, and oxazepam. He reported that for many years he was unable move out of bed due to the unbearable pain. With pain medications, he was able to regain most of his independence in his daily life, though the pain and other clinical aspects of CRPS still completely limited his use of the left arm.
Physical examination revealed glossy, cold, hairless skin with hypohidrosis of the left arm, forearm, and hand (Figures 1 and 2A). The left arm was conspicuously atrophied, with the forearm and hand erythematous. The fingers were taut, contracted, and edematous (Figure 2B), and the skin was unable to be pinched. The fingernails on the left hand had dystrophic changes including yellow color and brittleness with longitudinal ridges (Figure 3). The patient could activate the left bicep and tricep muscles against gravity but had minimal function of the deltoid muscle. He also had minimal movement of the left index finger and was unable to move any other digits of the left hand. The patient was continued on pain management treatments and physical therapy for his condition.
Complex regional pain syndrome is a neuropathic disorder of the extremities characterized by pain and a variety of autonomic and motor disturbances such as local edema, limited active range of motion, and vasomotor and trophic skin changes. There are 2 types of CRPS: type II is marked by explicit nerve injury and type I is not. The pathophysiology of CRPS is unknown.1-3
There is no definite set of diagnostic criteria for CRPS. The lack of any gold-standard diagnostic test for CRPS has made arriving at one valid, widely accepted set of diagnostic criteria impossible.1 There are 4 widely used sets of diagnostic criteria. One is the International Association for the Study of Pain diagnostic criteria defined in 1994.4 However, the criteria rely entirely on subjective symptoms and have been under great scrutiny due to their questionable validity.2 Veldman et al5 presented other widely used CRPS diagnostic criteria in their prospective study of 829 reflex sympathetic dystrophy patients, which paid particular attention to the early clinical manifestations of CRPS. In 1999, Bruehl et al2 proposed their own modified diagnostic criteria, which required physician-assessed signs in 2 of 4 categories to avoid the practice of exclusively relying on subjective symptoms. In addition, during a consensus meeting in Budapest, Hungary, a modified version of the Bruehl criteria was proposed.6 All 4 criteria rely solely on detailed history and physical examination, and the choice of diagnostic criteria remains subjective.
The pathophysiology of CRPS also remains unclear. There are several proposed mechanisms such as sympathetic nervous system dysfunction, abnormal inflammatory response, and central nervous system involvement.1 Psychologic factors, sequelae of nerve injury, and genetic predisposition also have been implicated in the pathophysiology of CRPS.1 It is likely that several mechanisms variably contribute to each presentation of CRPS.
Many dermatologic findings, in addition to neuromuscular symptoms, accompany CRPS and serve as important clues to making the clinical diagnosis. Complex regional pain syndrome has been thought to have 3 distinct sequential stages of CRPS.1,3,7 Stage 1—the acute stage—is marked by hyperalgesia, allodynia, sudomotor disturbances, and prominent edema. Stage 2—the dystrophic stage—is characterized by more marked pain and sensory dysfunction, vasomotor dysfunction, development of motor dysfunction, soft tissue edema, skin and articular soft tissue thickening, and development of dystrophic nail changes. Stage 3—the atrophic stage—is marked by decreased pain and sensory disturbances, markedly increased motor dysfunction, waxy atrophic skin changes, progression of dystrophic nail changes, and skeletal cystic and subchondral erosions with diffuse osteoporosis.1,3,7
The staging model, however, has been called into question.3 In a cluster analysis, Bruehl et al3 arrived at 3 relatively consistent CRPS patient subgroups that did not have notably different pain duration, suggesting the existence of 3 CRPS subtypes, not stages. Their study found that one of the subgroups best represented the clinical presentation of CRPS type II. This subgroup had the greatest pain and sensory abnormalities and the least vasomotor dysfunction of all 3 subgroups. Nonetheless, this study has not settled the discussion, as it only included 113 patients.3 Thus, with future studies, our understanding of CRPS in stages may change, which likely will impact how the clinical diagnosis is made.
There is a lack of high-quality evidence for most treatment interventions for CRPS8; however, the current practice is to use an interdisciplinary approach.1,9,10 The main therapeutic arm of this approach is rehabilitation; physical and occupational therapy can help improve range of motion, contracture, and atrophy. The other 2 arms of the approach are psychologic therapy to improve quality of life and pain management with pharmacologic therapy and/or invasive interventions. The choice of therapy remains empirical; trial and error should be expected in developing an adequate treatment plan for each individual patient.
Many aspects of CRPS remain unclear, and even our current understanding of the disease will inevitably change over time. The syndrome can cause life-changing morbidities in patients, and late diagnosis and treatment are associated with poor prognosis. Because there are many dermatologic findings associated with the disorder, it is crucial for dermatologists to clinically recognize the disorder and to refer patients to appropriate channels so that treatment can be started as soon as possible.
To the Editor:
A 62-year-old man presented with an atrophied painful left arm of 17 years’ duration that began when he was hit by a car as a pedestrian. He sustained severe multisystem injuries from the accident, including left brachial plexus and C6 nerve root avulsion injury. When he regained consciousness after 6 weeks in the intensive care unit, he immediately noted diffuse pain throughout the body, especially in the left arm. Since the accident, the patient continued to have diminished sensation to touch and temperature in the left arm. He also had burning, throbbing, and electrical pain in the left arm with light touch as well as spontaneously. He was thoroughly evaluated by a neurologist and was diagnosed with complex regional pain syndrome (CRPS) type II. For the treatment of pain, dorsal column stimulation and hemilaminectomy with exploration of the avulsed nerve root were attempted, both of which had minimal effect. He was maintained on hydromorphone, methadone, and oxazepam. He reported that for many years he was unable move out of bed due to the unbearable pain. With pain medications, he was able to regain most of his independence in his daily life, though the pain and other clinical aspects of CRPS still completely limited his use of the left arm.
Physical examination revealed glossy, cold, hairless skin with hypohidrosis of the left arm, forearm, and hand (Figures 1 and 2A). The left arm was conspicuously atrophied, with the forearm and hand erythematous. The fingers were taut, contracted, and edematous (Figure 2B), and the skin was unable to be pinched. The fingernails on the left hand had dystrophic changes including yellow color and brittleness with longitudinal ridges (Figure 3). The patient could activate the left bicep and tricep muscles against gravity but had minimal function of the deltoid muscle. He also had minimal movement of the left index finger and was unable to move any other digits of the left hand. The patient was continued on pain management treatments and physical therapy for his condition.
Complex regional pain syndrome is a neuropathic disorder of the extremities characterized by pain and a variety of autonomic and motor disturbances such as local edema, limited active range of motion, and vasomotor and trophic skin changes. There are 2 types of CRPS: type II is marked by explicit nerve injury and type I is not. The pathophysiology of CRPS is unknown.1-3
There is no definite set of diagnostic criteria for CRPS. The lack of any gold-standard diagnostic test for CRPS has made arriving at one valid, widely accepted set of diagnostic criteria impossible.1 There are 4 widely used sets of diagnostic criteria. One is the International Association for the Study of Pain diagnostic criteria defined in 1994.4 However, the criteria rely entirely on subjective symptoms and have been under great scrutiny due to their questionable validity.2 Veldman et al5 presented other widely used CRPS diagnostic criteria in their prospective study of 829 reflex sympathetic dystrophy patients, which paid particular attention to the early clinical manifestations of CRPS. In 1999, Bruehl et al2 proposed their own modified diagnostic criteria, which required physician-assessed signs in 2 of 4 categories to avoid the practice of exclusively relying on subjective symptoms. In addition, during a consensus meeting in Budapest, Hungary, a modified version of the Bruehl criteria was proposed.6 All 4 criteria rely solely on detailed history and physical examination, and the choice of diagnostic criteria remains subjective.
The pathophysiology of CRPS also remains unclear. There are several proposed mechanisms such as sympathetic nervous system dysfunction, abnormal inflammatory response, and central nervous system involvement.1 Psychologic factors, sequelae of nerve injury, and genetic predisposition also have been implicated in the pathophysiology of CRPS.1 It is likely that several mechanisms variably contribute to each presentation of CRPS.
Many dermatologic findings, in addition to neuromuscular symptoms, accompany CRPS and serve as important clues to making the clinical diagnosis. Complex regional pain syndrome has been thought to have 3 distinct sequential stages of CRPS.1,3,7 Stage 1—the acute stage—is marked by hyperalgesia, allodynia, sudomotor disturbances, and prominent edema. Stage 2—the dystrophic stage—is characterized by more marked pain and sensory dysfunction, vasomotor dysfunction, development of motor dysfunction, soft tissue edema, skin and articular soft tissue thickening, and development of dystrophic nail changes. Stage 3—the atrophic stage—is marked by decreased pain and sensory disturbances, markedly increased motor dysfunction, waxy atrophic skin changes, progression of dystrophic nail changes, and skeletal cystic and subchondral erosions with diffuse osteoporosis.1,3,7
The staging model, however, has been called into question.3 In a cluster analysis, Bruehl et al3 arrived at 3 relatively consistent CRPS patient subgroups that did not have notably different pain duration, suggesting the existence of 3 CRPS subtypes, not stages. Their study found that one of the subgroups best represented the clinical presentation of CRPS type II. This subgroup had the greatest pain and sensory abnormalities and the least vasomotor dysfunction of all 3 subgroups. Nonetheless, this study has not settled the discussion, as it only included 113 patients.3 Thus, with future studies, our understanding of CRPS in stages may change, which likely will impact how the clinical diagnosis is made.
There is a lack of high-quality evidence for most treatment interventions for CRPS8; however, the current practice is to use an interdisciplinary approach.1,9,10 The main therapeutic arm of this approach is rehabilitation; physical and occupational therapy can help improve range of motion, contracture, and atrophy. The other 2 arms of the approach are psychologic therapy to improve quality of life and pain management with pharmacologic therapy and/or invasive interventions. The choice of therapy remains empirical; trial and error should be expected in developing an adequate treatment plan for each individual patient.
Many aspects of CRPS remain unclear, and even our current understanding of the disease will inevitably change over time. The syndrome can cause life-changing morbidities in patients, and late diagnosis and treatment are associated with poor prognosis. Because there are many dermatologic findings associated with the disorder, it is crucial for dermatologists to clinically recognize the disorder and to refer patients to appropriate channels so that treatment can be started as soon as possible.
- Borchers A, Gershwin M. Complex regional pain syndrome: a comprehensive and critical review. Autoimmun Rev. 2014;13:242-265.
- Bruehl S, Harden RN, Galer BS, et al. External validation of IASP diagnostic criteria for complex regional pain syndrome and proposed research diagnostic criteria. International Association for the Study of Pain. Pain. 1999;81:147-154.
- Bruehl S, Harden RN, Gaker BS, et al. Complex regional pain syndrome: are there distinct subtypes and sequential stages of the syndrome? Pain. 2002;95:119-124.
- Merskey H, Bogduk N. Classification of Chronic Pain: Descriptions of Chronic Pain Syndromes and Definitions of Pain Terms. 2nd ed. Seattle, WA: IASP Press; 1994.
- Veldman PH, Reynen HM, Arntz IE, et al. Signs and symptoms of reflex sympathetic dystrophy: prospective study of 829 patients. Lancet. 1993;342:1012-1016.
- Harden RN, Bruehl S, Perez RS, et al. Validation of proposed diagnostic criteria (the “Budapest Criteria”) for complex regional pain syndrome. Pain. 2010;150:268-274.
- Sebastin SJ. Complex regional pain syndrome. Indian J Plast Surg. 2011;44:298-307.
- O’Connell NE, Wand BM, McAuley J, et al. Interventions for treating pain and disability in adults with complex regional pain syndrome. Cochrane Database Syst Rev. 2013;4:CD009416.
- Hsu ES. Practical management of complex regional pain syndrome. Am J Ther. 2009;16:147-154.
- Stanton-Hicks MD, Burton AW, Bruehl SP, et al. An updated interdisciplinary clinical pathway for CRPS: report of an expert panel. Pain Pract. 2002;2:1-16.
- Borchers A, Gershwin M. Complex regional pain syndrome: a comprehensive and critical review. Autoimmun Rev. 2014;13:242-265.
- Bruehl S, Harden RN, Galer BS, et al. External validation of IASP diagnostic criteria for complex regional pain syndrome and proposed research diagnostic criteria. International Association for the Study of Pain. Pain. 1999;81:147-154.
- Bruehl S, Harden RN, Gaker BS, et al. Complex regional pain syndrome: are there distinct subtypes and sequential stages of the syndrome? Pain. 2002;95:119-124.
- Merskey H, Bogduk N. Classification of Chronic Pain: Descriptions of Chronic Pain Syndromes and Definitions of Pain Terms. 2nd ed. Seattle, WA: IASP Press; 1994.
- Veldman PH, Reynen HM, Arntz IE, et al. Signs and symptoms of reflex sympathetic dystrophy: prospective study of 829 patients. Lancet. 1993;342:1012-1016.
- Harden RN, Bruehl S, Perez RS, et al. Validation of proposed diagnostic criteria (the “Budapest Criteria”) for complex regional pain syndrome. Pain. 2010;150:268-274.
- Sebastin SJ. Complex regional pain syndrome. Indian J Plast Surg. 2011;44:298-307.
- O’Connell NE, Wand BM, McAuley J, et al. Interventions for treating pain and disability in adults with complex regional pain syndrome. Cochrane Database Syst Rev. 2013;4:CD009416.
- Hsu ES. Practical management of complex regional pain syndrome. Am J Ther. 2009;16:147-154.
- Stanton-Hicks MD, Burton AW, Bruehl SP, et al. An updated interdisciplinary clinical pathway for CRPS: report of an expert panel. Pain Pract. 2002;2:1-16.
Practice Points
- Complex regional pain syndrome (CRPS) is a neuropathic disorder of the extremities characterized by pain, a variety of autonomic and motor disturbances, and dermatologic findings.
- Early recognition of CRPS is critical, as it presents life-changing morbidities to patients.
- A multidisciplinary treatment approach with physical therapy, occupational therapy, psychological support, and pain control is needed for the management of CRPS.

Vulvar Syringoma
To the Editor:
Syringomas are common benign tumors of the eccrine sweat glands that usually manifest clinically as multiple flesh-colored papules. They are most commonly seen on the face, neck, and chest of adolescent girls. Syringomas may appear at any site of the body but are rare in the vulva. We present a case of a 51-year-old woman who was referred to the Division of Gynecologic Oncology at the University of Alabama at Birmingham for further management of a tumor carrying a differential diagnosis of vulvar syringoma vs microcystic adnexal carcinoma (MAC).
A 51-year-old woman presented to dermatology (G.G.) and was referred to the Division of Gynecologic Oncology at the University of Alabama at Birmingham for further management of possible vulvar syringoma vs MAC. The patient previously had been evaluated at an outside community practice due to dyspareunia, vulvar discomfort, and vulvar irregularities of 1 month’s duration. At that time, a small biopsy was performed, and the histologic differential diagnosis included syringoma vs an adnexal carcinoma. Consequently, she was referred to gynecologic oncology for further management.
Pelvic examination revealed multilobular nodular areas overlying the clitoral hood that extended down to the labia majora. The nodular processes did not involve the clitoris, labia minora, or perineum. A mobile isolated lymph node measuring 2.0×1.0 cm in the right inguinal area also was noted. The patient’s clinical history was notable for right breast carcinoma treated with a right mastectomy with axillary lymph node dissection that showed metastatic disease. She also underwent adjuvant chemotherapy with paclitaxel and doxorubicin for breast carcinoma.
After discussing the diagnostic differential and treatment options, the patient elected to undergo a bilateral partial radical vulvectomy with reconstruction and resection of the right inguinal lymph node. Gross examination of the vulvectomy specimen showed multiple flesh-colored papules (Figure 1). Histologic examination revealed a neoplasm with sweat gland differentiation that was broad and poorly circumscribed but confined to the dermis (Figures 2A and 2B). The neoplasm was composed of epithelial cells that formed ductlike structures, lined by 2 layers of cuboidal epithelium within a fibrous stroma (Figure 2C). A toluidine blue special stain was performed and demonstrated an increased amount of mast cells in the tissue (Figure 3). Immunohistochemical stains for gross cystic disease fluid protein, estrogen receptor (ER), and progesterone receptor (PR) were negative in the tumor cells. The lack of cytologic atypia, perineural invasion, and deep infiltration into the subcutis favored a syringoma. One month later, the case was presented at the Tumor Board Conference at the University of Alabama at Birmingham where a final diagnosis of vulvar syringoma was agreed upon and discussed with the patient. At that time, no recurrence was evident and follow-up was recommended.

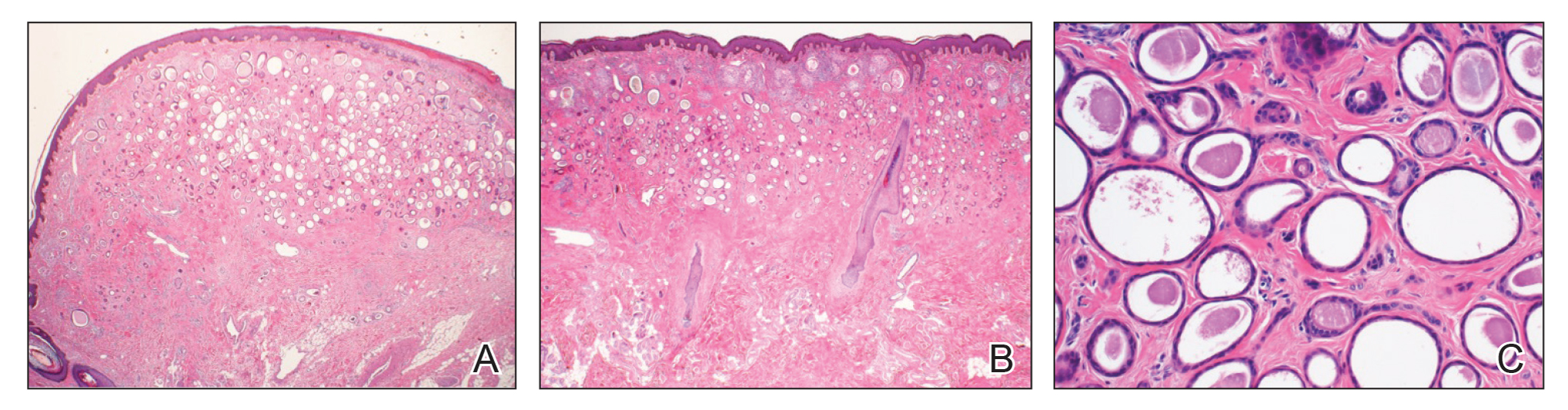
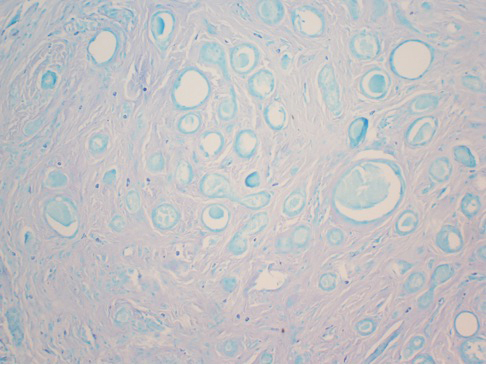
Syringomas are benign tumors of the sweat glands that are fairly common and appear to have a predilection for women. Although most of the literature classifies them as eccrine neoplasms, the term syringoma can be used to describe neoplasms of either apocrine or eccrine lineage.1 To rule out an apocrine lineage of the tumor in our patient, we performed immunohistochemistry for gross cystic disease fluid protein, a marker of apocrine differentiation. This stain highlighted normal apocrine glands that were not involved in the tumor proliferation.
Syringomas may occur at any site on the body but are prone to occur on the periorbital area, especially the eyelids.1 Some of the atypical locations for a syringoma include the anterior neck, chest, abdomen, genitals, axillae, groin, and buttocks.2 Vulvar syringomas were first reported by Carneiro3 in 1971 as usually affecting adolescent girls and middle-aged women. There have been approximately 40 reported cases affecting women aged 8 to 78 years.4,5 Vulvar syringomas classically appear as firm or soft, flesh-colored to transparent, papular lesions. The 2 other clinical variants are miliumlike, whitish, cystic papules as well as lichenoid papules.6 Pérez-Bustillo et al5 reported a case of the lichenoid papule variant on the labia majora of a 78-year-old woman who presented with intermittent vulvar pruritus of 4 years’ duration. Due to this patient’s 9-year history of urinary incontinence, the lesions had been misdiagnosed as irritant dermatitis and associated lichen simplex chronicus (LSC). This case is a reminder to consider vulvar syringoma in patients with LSC who respond poorly to oral antihistamines and topical steroids.5 Rarely, multiple clinical variants may coexist. In a case reported by Dereli et al,7 a 19-year-old woman presented with concurrent classical and miliumlike forms of vulvar syringoma.
Vulvar syringomas usually present as multiple lesions involving both sides of the labia majora; however, Blasdale and McLelland8 reported a single isolated syringoma of the vulva on the anterior right labia minora that measured 1.0×0.5 cm, leading the lesion to be described as a giant syringoma.
Vulvar syringomas usually are asymptomatic and noticed during routine gynecologic examination. Therefore, it is believed that they likely are underdiagnosed.5 When symptomatic, they commonly present with constant9 or intermittent5 pruritus, which may intensify during menstruation, pregnancy, and summertime.6,10-12 Gerdsen et al10 documented a 27-year-old woman who presented with a 2-year history of pruritic vulvar skin lesions that became exacerbated during menstruation, which raised the possibility of cyclical hormonal changes being responsible for periodic exacerbation of vulvar pruritus during menstruation. In addition, patients may experience an increase in size and number of the lesions during pregnancy. Bal et al11 reported a 24-year-old primigravida with vulvar papular lesions that intensified during pregnancy. She had experienced intermittent vulvar pruritus for 12 years but had no change in symptoms during menstruation.11 Few studies have attempted to evaluate the presence of ER and PR in the syringomas. A study of 9 nonvulvar syringomas by Wallace and Smoller13 showed ER positivity in 1 case and PR positivity in 8 cases, lending support to the hormonal theory; however, in another case series of 15 vulvar syringomas, Huang et al6 failed to show ER and PR expression by immunohistochemical staining. A case report published 3 years earlier documented the first case of PR positivity on a vulvar syringoma.14 Our patient also was negative for ER and PR, which suggested that hormonal status is important in some but not all syringomas.
Patients with vulgar syringomas also might have coexisting extragenital syringomas in the neck,4 eyelids,6,7,10 and periorbital area,6 and thorough examination of the body is essential. If an extragenital syringoma is diagnosed, a vulvar syringoma should be considered, especially when the patient presents with unexplained genital symptoms. Although no proven hereditary transmission pattern has been established, family history of syringomas has been established in several cases.15 In a case series reported by Huang et al,6 4 of 18 patients reported a family history of periorbital syringomas. In our case, the patient did not report a family history of syringomas.
The differential diagnosis of vulvar lesions with pruritus is broad and includes Fox-Fordyce disease, lichen planus, LSC, epidermal cysts, senile angiomas, dystrophic calcinosis, xanthomas, steatocytomas, soft fibromas, condyloma acuminatum, and candidiasis. Vulvar syringomas might have a nonspecific appearance, and histologic examination is essential to confirm the diagnosis and rule out any malignant process such as MAC, vulvar intraepithelial neoplasia, extramammary Paget disease, or other glandular neoplasms of the vulva.
Microcystic adnexal carcinoma was first reported in 1982 by Goldstein et al16 as a locally aggressive neoplasm that can be confused with benign adnexal neoplasms, particularly desmoplastic trichoepithelioma, trichoadenoma, and syringoma. Microcystic adnexal carcinomas present as slow-growing, flesh-colored papules that may resemble syringomas and appear in similar body sites. Histologic examination is essential to differentiate between these two entities. Syringomas are tumors confined to the dermis and are composed of multiple small ducts lined by 2 layers of cuboidal epithelium within a dense fibrous stroma. Unlike syringomas, MACs usually infiltrate diffusely into the dermis and subcutis and may extend into the underlying muscle. Although bland cytologic features predominate, perineural invasion frequently is present in MACs. A potential pitfall of misdiagnosis can be caused by a superficial biopsy that may reveal benign histologic appearance, particularly in the upper level of the tumor where it may be confused with a syringoma or a benign follicular neoplasm.17
The initial biopsy performed on our patient was possibly not deep enough to render an unequivocal diagnosis and therefore bilateral partial radical vulvectomy was considered. After surgery, histologic examination of the resection specimen revealed a poorly circumscribed tumor confined to the dermis. The tumor was broad and the lack of deep infiltration into the subcutis and perineural invasion favored a syringoma (Figures 2A and 2B). These findings were consistent with case reports that documented syringomas as being more wide than deep on microscopic examination, whereas the opposite pertained to MAC.18 Cases of plaque-type syringomas that initially were misdiagnosed as MACs also have been reported.19 Because misdiagnosis may affect the treatment plan and potentially result in unnecessary surgery, caution should be taken when differentiating between these two entities. When a definitive diagnosis cannot be rendered on a superficial biopsy, a recommendation should be made for a deeper biopsy sampling the subcutis.
For the majority of the patients with vulvar syringomas, treatment is seldom required due to their asymptomatic nature; however, patients who present with symptoms usually report pruritus of variable intensities and patterns. A standardized treatment does not exist for vulvar syringomas, and oral or topical treatment might be used as an initial approach. Commonly prescribed medications with variable results include topical corticosteroids, oral antihistamines, and topical retinoids. In a case reported by Iwao et al,20 vulvar syringomas were successfully treated with tranilast, which has anti-inflammatory and immunomodulatory effects. This medication could have a possible dual action—inhibiting the release of chemical mediators from the mast cells and inhibiting the release of IL-1β from the eccrine duct, which could suppress the proliferation of stromal connective tissue. Our case was stained with toluidine blue and showed an increased number of mast cells in the tissue (Figure 3). Patients who are unresponsive to tranilast or have extensive disease resulting in cosmetic disfigurement might benefit from more invasive treatment methods including a variety of lasers, cryotherapy, electrosurgery, and excision. Excisions should include the entire tumor to avoid recurrence. In a case reported by Garman and Metry,21 the lesions were surgically excised using small 2- to 3-mm punches; however, several weeks later the lesions recurred. Our patient presented with a 1-month evolution of dyspareunia, vulvar discomfort, and vulvar irregularities that were probably not treated with oral or topical medications before being referred for surgery.
We report a case of a vulvar syringoma that presented diagnostic challenges in the initial biopsy, which prevented the exclusion of an MAC. After partial radical vulvectomy, histologic examination was more definitive, showing lack of deep infiltration into the subcutis or perineural invasion that are commonly seen in MAC. This case is an example of a notable pitfall in the diagnosis of vulvar syringoma on a limited biopsy leading to overtreatment. Raising awareness of this entity is the only modality to prevent misdiagnosis. We encourage reporting of further cases of syringomas, particularly those with atypical locations or patterns that may cause diagnostic problems.
- Bolognia JL, Jorizzo JL, Rapini RP. Dermatology. 2nd ed. Spain: Mosby Elsevier; 2008.
- Weedon D. Skin Pathology. 3rd ed. China: Churchill Livingstone Elsevier; 2010.
- Carneiro SJ, Gardner HL, Knox JM. Syringoma of the vulva. Arch Dermatol. 1971;103:494-496.
- Trager JD, Silvers J, Reed JA, et al. Neck and vulvar papules in an 8-year-old girl. Arch Dermatol. 1999;135:203, 206.
- Pérez-Bustillo A, Ruiz-González I, Delgado S, et al. Vulvar syringoma: a rare cause of vulvar pruritus. Actas Dermo-Sifiliográficas. 2008;99:580-581.
- Huang YH, Chuang YH, Kuo TT, et al. Vulvar syringoma: a clinicopathologic and immunohistologic study of 18 patients and results of treatment. J Am Acad Dermatol. 2003;48:735-739.
- Dereli T, Turk BG, Kazandi AC. Syringomas of the vulva. Int J Gynaecol Obstet. 2007;99:65-66.
- Blasdale C, McLelland J. Solitary giant vulval syringoma. Br J Dermatol. 1999;141:374-375.
- Kavala M, Can B, Zindanci I, et al. Vulvar pruritus caused by syringoma of the vulva. Int J Dermatol. 2008;47:831-832.
- Gerdsen R, Wenzel J, Uerlich M, et al. Periodic genital pruritus caused by syringoma of the vulva. Acta Obstet Gynecol Scand. 2002;81:369-370.
- Bal N, Aslan E, Kayaselcuk F, et al. Vulvar syringoma aggravated by pregnancy. Pathol Oncol Res. 2003;9:196-197.
- Turan C, Ugur M, Kutluay L, et al. Vulvar syringoma exacerbated during pregnancy. Eur J Obstet Gynecol Reprod Biol. 1996;64:141-142.
- Wallace ML, Smoller BR. Progesterone receptor positivity supports hormonal control of syringomas. J Cutan Pathol. 1995;22:442-445.
- Yorganci A, Kale A, Dunder I, et al. Vulvar syringoma showing progesterone receptor positivity. BJOG. 2000;107:292-294.
- Draznin M. Hereditary syringomas: a case report. Dermatol Online J. 2004;10:19.
- Goldstein DJ, Barr RJ, Santa Cruz DJ. Microcystic adnexal carcinoma: a distinct clinicopathologic entity. Cancer. 1982;50:566-572.
- Hamsch C, Hartschuh W. Microcystic adnexal carcinoma - aggressive infiltrative tumor often with innocent clinical appearance. J Dtsch Dermatol Ges. 2010;8:275-278.
- Henner MS, Shapiro PE, Ritter JH, et al. Solitary syringoma. report of five cases and clinicopathologic comparison with microcystic adnexal carcinoma of the skin. Am J Dermatopathol. 1995;17:465-470.
- Suwattee P, McClelland MC, Huiras EE, et al. Plaque-type syringoma: two cases misdiagnosed as microcystic adnexal carcinoma. J Cutan Pathol. 2008;35:570-574.
- Iwao F, Onozuka T, Kawashima T. Vulval syringoma successfully treated with tranilast. Br J Dermatol. 2005;153:1228-1230.
- Garman M, Metry D. Vulvar syringomas in a 9-year-old child with review of the literature. Pediatr Dermatol. 2006;23:369-372.
To the Editor:
Syringomas are common benign tumors of the eccrine sweat glands that usually manifest clinically as multiple flesh-colored papules. They are most commonly seen on the face, neck, and chest of adolescent girls. Syringomas may appear at any site of the body but are rare in the vulva. We present a case of a 51-year-old woman who was referred to the Division of Gynecologic Oncology at the University of Alabama at Birmingham for further management of a tumor carrying a differential diagnosis of vulvar syringoma vs microcystic adnexal carcinoma (MAC).
A 51-year-old woman presented to dermatology (G.G.) and was referred to the Division of Gynecologic Oncology at the University of Alabama at Birmingham for further management of possible vulvar syringoma vs MAC. The patient previously had been evaluated at an outside community practice due to dyspareunia, vulvar discomfort, and vulvar irregularities of 1 month’s duration. At that time, a small biopsy was performed, and the histologic differential diagnosis included syringoma vs an adnexal carcinoma. Consequently, she was referred to gynecologic oncology for further management.
Pelvic examination revealed multilobular nodular areas overlying the clitoral hood that extended down to the labia majora. The nodular processes did not involve the clitoris, labia minora, or perineum. A mobile isolated lymph node measuring 2.0×1.0 cm in the right inguinal area also was noted. The patient’s clinical history was notable for right breast carcinoma treated with a right mastectomy with axillary lymph node dissection that showed metastatic disease. She also underwent adjuvant chemotherapy with paclitaxel and doxorubicin for breast carcinoma.
After discussing the diagnostic differential and treatment options, the patient elected to undergo a bilateral partial radical vulvectomy with reconstruction and resection of the right inguinal lymph node. Gross examination of the vulvectomy specimen showed multiple flesh-colored papules (Figure 1). Histologic examination revealed a neoplasm with sweat gland differentiation that was broad and poorly circumscribed but confined to the dermis (Figures 2A and 2B). The neoplasm was composed of epithelial cells that formed ductlike structures, lined by 2 layers of cuboidal epithelium within a fibrous stroma (Figure 2C). A toluidine blue special stain was performed and demonstrated an increased amount of mast cells in the tissue (Figure 3). Immunohistochemical stains for gross cystic disease fluid protein, estrogen receptor (ER), and progesterone receptor (PR) were negative in the tumor cells. The lack of cytologic atypia, perineural invasion, and deep infiltration into the subcutis favored a syringoma. One month later, the case was presented at the Tumor Board Conference at the University of Alabama at Birmingham where a final diagnosis of vulvar syringoma was agreed upon and discussed with the patient. At that time, no recurrence was evident and follow-up was recommended.
Syringomas are benign tumors of the sweat glands that are fairly common and appear to have a predilection for women. Although most of the literature classifies them as eccrine neoplasms, the term syringoma can be used to describe neoplasms of either apocrine or eccrine lineage.1 To rule out an apocrine lineage of the tumor in our patient, we performed immunohistochemistry for gross cystic disease fluid protein, a marker of apocrine differentiation. This stain highlighted normal apocrine glands that were not involved in the tumor proliferation.
Syringomas may occur at any site on the body but are prone to occur on the periorbital area, especially the eyelids.1 Some of the atypical locations for a syringoma include the anterior neck, chest, abdomen, genitals, axillae, groin, and buttocks.2 Vulvar syringomas were first reported by Carneiro3 in 1971 as usually affecting adolescent girls and middle-aged women. There have been approximately 40 reported cases affecting women aged 8 to 78 years.4,5 Vulvar syringomas classically appear as firm or soft, flesh-colored to transparent, papular lesions. The 2 other clinical variants are miliumlike, whitish, cystic papules as well as lichenoid papules.6 Pérez-Bustillo et al5 reported a case of the lichenoid papule variant on the labia majora of a 78-year-old woman who presented with intermittent vulvar pruritus of 4 years’ duration. Due to this patient’s 9-year history of urinary incontinence, the lesions had been misdiagnosed as irritant dermatitis and associated lichen simplex chronicus (LSC). This case is a reminder to consider vulvar syringoma in patients with LSC who respond poorly to oral antihistamines and topical steroids.5 Rarely, multiple clinical variants may coexist. In a case reported by Dereli et al,7 a 19-year-old woman presented with concurrent classical and miliumlike forms of vulvar syringoma.
Vulvar syringomas usually present as multiple lesions involving both sides of the labia majora; however, Blasdale and McLelland8 reported a single isolated syringoma of the vulva on the anterior right labia minora that measured 1.0×0.5 cm, leading the lesion to be described as a giant syringoma.
Vulvar syringomas usually are asymptomatic and noticed during routine gynecologic examination. Therefore, it is believed that they likely are underdiagnosed.5 When symptomatic, they commonly present with constant9 or intermittent5 pruritus, which may intensify during menstruation, pregnancy, and summertime.6,10-12 Gerdsen et al10 documented a 27-year-old woman who presented with a 2-year history of pruritic vulvar skin lesions that became exacerbated during menstruation, which raised the possibility of cyclical hormonal changes being responsible for periodic exacerbation of vulvar pruritus during menstruation. In addition, patients may experience an increase in size and number of the lesions during pregnancy. Bal et al11 reported a 24-year-old primigravida with vulvar papular lesions that intensified during pregnancy. She had experienced intermittent vulvar pruritus for 12 years but had no change in symptoms during menstruation.11 Few studies have attempted to evaluate the presence of ER and PR in the syringomas. A study of 9 nonvulvar syringomas by Wallace and Smoller13 showed ER positivity in 1 case and PR positivity in 8 cases, lending support to the hormonal theory; however, in another case series of 15 vulvar syringomas, Huang et al6 failed to show ER and PR expression by immunohistochemical staining. A case report published 3 years earlier documented the first case of PR positivity on a vulvar syringoma.14 Our patient also was negative for ER and PR, which suggested that hormonal status is important in some but not all syringomas.
Patients with vulgar syringomas also might have coexisting extragenital syringomas in the neck,4 eyelids,6,7,10 and periorbital area,6 and thorough examination of the body is essential. If an extragenital syringoma is diagnosed, a vulvar syringoma should be considered, especially when the patient presents with unexplained genital symptoms. Although no proven hereditary transmission pattern has been established, family history of syringomas has been established in several cases.15 In a case series reported by Huang et al,6 4 of 18 patients reported a family history of periorbital syringomas. In our case, the patient did not report a family history of syringomas.
The differential diagnosis of vulvar lesions with pruritus is broad and includes Fox-Fordyce disease, lichen planus, LSC, epidermal cysts, senile angiomas, dystrophic calcinosis, xanthomas, steatocytomas, soft fibromas, condyloma acuminatum, and candidiasis. Vulvar syringomas might have a nonspecific appearance, and histologic examination is essential to confirm the diagnosis and rule out any malignant process such as MAC, vulvar intraepithelial neoplasia, extramammary Paget disease, or other glandular neoplasms of the vulva.
Microcystic adnexal carcinoma was first reported in 1982 by Goldstein et al16 as a locally aggressive neoplasm that can be confused with benign adnexal neoplasms, particularly desmoplastic trichoepithelioma, trichoadenoma, and syringoma. Microcystic adnexal carcinomas present as slow-growing, flesh-colored papules that may resemble syringomas and appear in similar body sites. Histologic examination is essential to differentiate between these two entities. Syringomas are tumors confined to the dermis and are composed of multiple small ducts lined by 2 layers of cuboidal epithelium within a dense fibrous stroma. Unlike syringomas, MACs usually infiltrate diffusely into the dermis and subcutis and may extend into the underlying muscle. Although bland cytologic features predominate, perineural invasion frequently is present in MACs. A potential pitfall of misdiagnosis can be caused by a superficial biopsy that may reveal benign histologic appearance, particularly in the upper level of the tumor where it may be confused with a syringoma or a benign follicular neoplasm.17
The initial biopsy performed on our patient was possibly not deep enough to render an unequivocal diagnosis and therefore bilateral partial radical vulvectomy was considered. After surgery, histologic examination of the resection specimen revealed a poorly circumscribed tumor confined to the dermis. The tumor was broad and the lack of deep infiltration into the subcutis and perineural invasion favored a syringoma (Figures 2A and 2B). These findings were consistent with case reports that documented syringomas as being more wide than deep on microscopic examination, whereas the opposite pertained to MAC.18 Cases of plaque-type syringomas that initially were misdiagnosed as MACs also have been reported.19 Because misdiagnosis may affect the treatment plan and potentially result in unnecessary surgery, caution should be taken when differentiating between these two entities. When a definitive diagnosis cannot be rendered on a superficial biopsy, a recommendation should be made for a deeper biopsy sampling the subcutis.
For the majority of the patients with vulvar syringomas, treatment is seldom required due to their asymptomatic nature; however, patients who present with symptoms usually report pruritus of variable intensities and patterns. A standardized treatment does not exist for vulvar syringomas, and oral or topical treatment might be used as an initial approach. Commonly prescribed medications with variable results include topical corticosteroids, oral antihistamines, and topical retinoids. In a case reported by Iwao et al,20 vulvar syringomas were successfully treated with tranilast, which has anti-inflammatory and immunomodulatory effects. This medication could have a possible dual action—inhibiting the release of chemical mediators from the mast cells and inhibiting the release of IL-1β from the eccrine duct, which could suppress the proliferation of stromal connective tissue. Our case was stained with toluidine blue and showed an increased number of mast cells in the tissue (Figure 3). Patients who are unresponsive to tranilast or have extensive disease resulting in cosmetic disfigurement might benefit from more invasive treatment methods including a variety of lasers, cryotherapy, electrosurgery, and excision. Excisions should include the entire tumor to avoid recurrence. In a case reported by Garman and Metry,21 the lesions were surgically excised using small 2- to 3-mm punches; however, several weeks later the lesions recurred. Our patient presented with a 1-month evolution of dyspareunia, vulvar discomfort, and vulvar irregularities that were probably not treated with oral or topical medications before being referred for surgery.
We report a case of a vulvar syringoma that presented diagnostic challenges in the initial biopsy, which prevented the exclusion of an MAC. After partial radical vulvectomy, histologic examination was more definitive, showing lack of deep infiltration into the subcutis or perineural invasion that are commonly seen in MAC. This case is an example of a notable pitfall in the diagnosis of vulvar syringoma on a limited biopsy leading to overtreatment. Raising awareness of this entity is the only modality to prevent misdiagnosis. We encourage reporting of further cases of syringomas, particularly those with atypical locations or patterns that may cause diagnostic problems.
To the Editor:
Syringomas are common benign tumors of the eccrine sweat glands that usually manifest clinically as multiple flesh-colored papules. They are most commonly seen on the face, neck, and chest of adolescent girls. Syringomas may appear at any site of the body but are rare in the vulva. We present a case of a 51-year-old woman who was referred to the Division of Gynecologic Oncology at the University of Alabama at Birmingham for further management of a tumor carrying a differential diagnosis of vulvar syringoma vs microcystic adnexal carcinoma (MAC).
A 51-year-old woman presented to dermatology (G.G.) and was referred to the Division of Gynecologic Oncology at the University of Alabama at Birmingham for further management of possible vulvar syringoma vs MAC. The patient previously had been evaluated at an outside community practice due to dyspareunia, vulvar discomfort, and vulvar irregularities of 1 month’s duration. At that time, a small biopsy was performed, and the histologic differential diagnosis included syringoma vs an adnexal carcinoma. Consequently, she was referred to gynecologic oncology for further management.
Pelvic examination revealed multilobular nodular areas overlying the clitoral hood that extended down to the labia majora. The nodular processes did not involve the clitoris, labia minora, or perineum. A mobile isolated lymph node measuring 2.0×1.0 cm in the right inguinal area also was noted. The patient’s clinical history was notable for right breast carcinoma treated with a right mastectomy with axillary lymph node dissection that showed metastatic disease. She also underwent adjuvant chemotherapy with paclitaxel and doxorubicin for breast carcinoma.
After discussing the diagnostic differential and treatment options, the patient elected to undergo a bilateral partial radical vulvectomy with reconstruction and resection of the right inguinal lymph node. Gross examination of the vulvectomy specimen showed multiple flesh-colored papules (Figure 1). Histologic examination revealed a neoplasm with sweat gland differentiation that was broad and poorly circumscribed but confined to the dermis (Figures 2A and 2B). The neoplasm was composed of epithelial cells that formed ductlike structures, lined by 2 layers of cuboidal epithelium within a fibrous stroma (Figure 2C). A toluidine blue special stain was performed and demonstrated an increased amount of mast cells in the tissue (Figure 3). Immunohistochemical stains for gross cystic disease fluid protein, estrogen receptor (ER), and progesterone receptor (PR) were negative in the tumor cells. The lack of cytologic atypia, perineural invasion, and deep infiltration into the subcutis favored a syringoma. One month later, the case was presented at the Tumor Board Conference at the University of Alabama at Birmingham where a final diagnosis of vulvar syringoma was agreed upon and discussed with the patient. At that time, no recurrence was evident and follow-up was recommended.
Syringomas are benign tumors of the sweat glands that are fairly common and appear to have a predilection for women. Although most of the literature classifies them as eccrine neoplasms, the term syringoma can be used to describe neoplasms of either apocrine or eccrine lineage.1 To rule out an apocrine lineage of the tumor in our patient, we performed immunohistochemistry for gross cystic disease fluid protein, a marker of apocrine differentiation. This stain highlighted normal apocrine glands that were not involved in the tumor proliferation.
Syringomas may occur at any site on the body but are prone to occur on the periorbital area, especially the eyelids.1 Some of the atypical locations for a syringoma include the anterior neck, chest, abdomen, genitals, axillae, groin, and buttocks.2 Vulvar syringomas were first reported by Carneiro3 in 1971 as usually affecting adolescent girls and middle-aged women. There have been approximately 40 reported cases affecting women aged 8 to 78 years.4,5 Vulvar syringomas classically appear as firm or soft, flesh-colored to transparent, papular lesions. The 2 other clinical variants are miliumlike, whitish, cystic papules as well as lichenoid papules.6 Pérez-Bustillo et al5 reported a case of the lichenoid papule variant on the labia majora of a 78-year-old woman who presented with intermittent vulvar pruritus of 4 years’ duration. Due to this patient’s 9-year history of urinary incontinence, the lesions had been misdiagnosed as irritant dermatitis and associated lichen simplex chronicus (LSC). This case is a reminder to consider vulvar syringoma in patients with LSC who respond poorly to oral antihistamines and topical steroids.5 Rarely, multiple clinical variants may coexist. In a case reported by Dereli et al,7 a 19-year-old woman presented with concurrent classical and miliumlike forms of vulvar syringoma.
Vulvar syringomas usually present as multiple lesions involving both sides of the labia majora; however, Blasdale and McLelland8 reported a single isolated syringoma of the vulva on the anterior right labia minora that measured 1.0×0.5 cm, leading the lesion to be described as a giant syringoma.
Vulvar syringomas usually are asymptomatic and noticed during routine gynecologic examination. Therefore, it is believed that they likely are underdiagnosed.5 When symptomatic, they commonly present with constant9 or intermittent5 pruritus, which may intensify during menstruation, pregnancy, and summertime.6,10-12 Gerdsen et al10 documented a 27-year-old woman who presented with a 2-year history of pruritic vulvar skin lesions that became exacerbated during menstruation, which raised the possibility of cyclical hormonal changes being responsible for periodic exacerbation of vulvar pruritus during menstruation. In addition, patients may experience an increase in size and number of the lesions during pregnancy. Bal et al11 reported a 24-year-old primigravida with vulvar papular lesions that intensified during pregnancy. She had experienced intermittent vulvar pruritus for 12 years but had no change in symptoms during menstruation.11 Few studies have attempted to evaluate the presence of ER and PR in the syringomas. A study of 9 nonvulvar syringomas by Wallace and Smoller13 showed ER positivity in 1 case and PR positivity in 8 cases, lending support to the hormonal theory; however, in another case series of 15 vulvar syringomas, Huang et al6 failed to show ER and PR expression by immunohistochemical staining. A case report published 3 years earlier documented the first case of PR positivity on a vulvar syringoma.14 Our patient also was negative for ER and PR, which suggested that hormonal status is important in some but not all syringomas.
Patients with vulgar syringomas also might have coexisting extragenital syringomas in the neck,4 eyelids,6,7,10 and periorbital area,6 and thorough examination of the body is essential. If an extragenital syringoma is diagnosed, a vulvar syringoma should be considered, especially when the patient presents with unexplained genital symptoms. Although no proven hereditary transmission pattern has been established, family history of syringomas has been established in several cases.15 In a case series reported by Huang et al,6 4 of 18 patients reported a family history of periorbital syringomas. In our case, the patient did not report a family history of syringomas.
The differential diagnosis of vulvar lesions with pruritus is broad and includes Fox-Fordyce disease, lichen planus, LSC, epidermal cysts, senile angiomas, dystrophic calcinosis, xanthomas, steatocytomas, soft fibromas, condyloma acuminatum, and candidiasis. Vulvar syringomas might have a nonspecific appearance, and histologic examination is essential to confirm the diagnosis and rule out any malignant process such as MAC, vulvar intraepithelial neoplasia, extramammary Paget disease, or other glandular neoplasms of the vulva.
Microcystic adnexal carcinoma was first reported in 1982 by Goldstein et al16 as a locally aggressive neoplasm that can be confused with benign adnexal neoplasms, particularly desmoplastic trichoepithelioma, trichoadenoma, and syringoma. Microcystic adnexal carcinomas present as slow-growing, flesh-colored papules that may resemble syringomas and appear in similar body sites. Histologic examination is essential to differentiate between these two entities. Syringomas are tumors confined to the dermis and are composed of multiple small ducts lined by 2 layers of cuboidal epithelium within a dense fibrous stroma. Unlike syringomas, MACs usually infiltrate diffusely into the dermis and subcutis and may extend into the underlying muscle. Although bland cytologic features predominate, perineural invasion frequently is present in MACs. A potential pitfall of misdiagnosis can be caused by a superficial biopsy that may reveal benign histologic appearance, particularly in the upper level of the tumor where it may be confused with a syringoma or a benign follicular neoplasm.17
The initial biopsy performed on our patient was possibly not deep enough to render an unequivocal diagnosis and therefore bilateral partial radical vulvectomy was considered. After surgery, histologic examination of the resection specimen revealed a poorly circumscribed tumor confined to the dermis. The tumor was broad and the lack of deep infiltration into the subcutis and perineural invasion favored a syringoma (Figures 2A and 2B). These findings were consistent with case reports that documented syringomas as being more wide than deep on microscopic examination, whereas the opposite pertained to MAC.18 Cases of plaque-type syringomas that initially were misdiagnosed as MACs also have been reported.19 Because misdiagnosis may affect the treatment plan and potentially result in unnecessary surgery, caution should be taken when differentiating between these two entities. When a definitive diagnosis cannot be rendered on a superficial biopsy, a recommendation should be made for a deeper biopsy sampling the subcutis.
For the majority of the patients with vulvar syringomas, treatment is seldom required due to their asymptomatic nature; however, patients who present with symptoms usually report pruritus of variable intensities and patterns. A standardized treatment does not exist for vulvar syringomas, and oral or topical treatment might be used as an initial approach. Commonly prescribed medications with variable results include topical corticosteroids, oral antihistamines, and topical retinoids. In a case reported by Iwao et al,20 vulvar syringomas were successfully treated with tranilast, which has anti-inflammatory and immunomodulatory effects. This medication could have a possible dual action—inhibiting the release of chemical mediators from the mast cells and inhibiting the release of IL-1β from the eccrine duct, which could suppress the proliferation of stromal connective tissue. Our case was stained with toluidine blue and showed an increased number of mast cells in the tissue (Figure 3). Patients who are unresponsive to tranilast or have extensive disease resulting in cosmetic disfigurement might benefit from more invasive treatment methods including a variety of lasers, cryotherapy, electrosurgery, and excision. Excisions should include the entire tumor to avoid recurrence. In a case reported by Garman and Metry,21 the lesions were surgically excised using small 2- to 3-mm punches; however, several weeks later the lesions recurred. Our patient presented with a 1-month evolution of dyspareunia, vulvar discomfort, and vulvar irregularities that were probably not treated with oral or topical medications before being referred for surgery.
We report a case of a vulvar syringoma that presented diagnostic challenges in the initial biopsy, which prevented the exclusion of an MAC. After partial radical vulvectomy, histologic examination was more definitive, showing lack of deep infiltration into the subcutis or perineural invasion that are commonly seen in MAC. This case is an example of a notable pitfall in the diagnosis of vulvar syringoma on a limited biopsy leading to overtreatment. Raising awareness of this entity is the only modality to prevent misdiagnosis. We encourage reporting of further cases of syringomas, particularly those with atypical locations or patterns that may cause diagnostic problems.
- Bolognia JL, Jorizzo JL, Rapini RP. Dermatology. 2nd ed. Spain: Mosby Elsevier; 2008.
- Weedon D. Skin Pathology. 3rd ed. China: Churchill Livingstone Elsevier; 2010.
- Carneiro SJ, Gardner HL, Knox JM. Syringoma of the vulva. Arch Dermatol. 1971;103:494-496.
- Trager JD, Silvers J, Reed JA, et al. Neck and vulvar papules in an 8-year-old girl. Arch Dermatol. 1999;135:203, 206.
- Pérez-Bustillo A, Ruiz-González I, Delgado S, et al. Vulvar syringoma: a rare cause of vulvar pruritus. Actas Dermo-Sifiliográficas. 2008;99:580-581.
- Huang YH, Chuang YH, Kuo TT, et al. Vulvar syringoma: a clinicopathologic and immunohistologic study of 18 patients and results of treatment. J Am Acad Dermatol. 2003;48:735-739.
- Dereli T, Turk BG, Kazandi AC. Syringomas of the vulva. Int J Gynaecol Obstet. 2007;99:65-66.
- Blasdale C, McLelland J. Solitary giant vulval syringoma. Br J Dermatol. 1999;141:374-375.
- Kavala M, Can B, Zindanci I, et al. Vulvar pruritus caused by syringoma of the vulva. Int J Dermatol. 2008;47:831-832.
- Gerdsen R, Wenzel J, Uerlich M, et al. Periodic genital pruritus caused by syringoma of the vulva. Acta Obstet Gynecol Scand. 2002;81:369-370.
- Bal N, Aslan E, Kayaselcuk F, et al. Vulvar syringoma aggravated by pregnancy. Pathol Oncol Res. 2003;9:196-197.
- Turan C, Ugur M, Kutluay L, et al. Vulvar syringoma exacerbated during pregnancy. Eur J Obstet Gynecol Reprod Biol. 1996;64:141-142.
- Wallace ML, Smoller BR. Progesterone receptor positivity supports hormonal control of syringomas. J Cutan Pathol. 1995;22:442-445.
- Yorganci A, Kale A, Dunder I, et al. Vulvar syringoma showing progesterone receptor positivity. BJOG. 2000;107:292-294.
- Draznin M. Hereditary syringomas: a case report. Dermatol Online J. 2004;10:19.
- Goldstein DJ, Barr RJ, Santa Cruz DJ. Microcystic adnexal carcinoma: a distinct clinicopathologic entity. Cancer. 1982;50:566-572.
- Hamsch C, Hartschuh W. Microcystic adnexal carcinoma - aggressive infiltrative tumor often with innocent clinical appearance. J Dtsch Dermatol Ges. 2010;8:275-278.
- Henner MS, Shapiro PE, Ritter JH, et al. Solitary syringoma. report of five cases and clinicopathologic comparison with microcystic adnexal carcinoma of the skin. Am J Dermatopathol. 1995;17:465-470.
- Suwattee P, McClelland MC, Huiras EE, et al. Plaque-type syringoma: two cases misdiagnosed as microcystic adnexal carcinoma. J Cutan Pathol. 2008;35:570-574.
- Iwao F, Onozuka T, Kawashima T. Vulval syringoma successfully treated with tranilast. Br J Dermatol. 2005;153:1228-1230.
- Garman M, Metry D. Vulvar syringomas in a 9-year-old child with review of the literature. Pediatr Dermatol. 2006;23:369-372.
- Bolognia JL, Jorizzo JL, Rapini RP. Dermatology. 2nd ed. Spain: Mosby Elsevier; 2008.
- Weedon D. Skin Pathology. 3rd ed. China: Churchill Livingstone Elsevier; 2010.
- Carneiro SJ, Gardner HL, Knox JM. Syringoma of the vulva. Arch Dermatol. 1971;103:494-496.
- Trager JD, Silvers J, Reed JA, et al. Neck and vulvar papules in an 8-year-old girl. Arch Dermatol. 1999;135:203, 206.
- Pérez-Bustillo A, Ruiz-González I, Delgado S, et al. Vulvar syringoma: a rare cause of vulvar pruritus. Actas Dermo-Sifiliográficas. 2008;99:580-581.
- Huang YH, Chuang YH, Kuo TT, et al. Vulvar syringoma: a clinicopathologic and immunohistologic study of 18 patients and results of treatment. J Am Acad Dermatol. 2003;48:735-739.
- Dereli T, Turk BG, Kazandi AC. Syringomas of the vulva. Int J Gynaecol Obstet. 2007;99:65-66.
- Blasdale C, McLelland J. Solitary giant vulval syringoma. Br J Dermatol. 1999;141:374-375.
- Kavala M, Can B, Zindanci I, et al. Vulvar pruritus caused by syringoma of the vulva. Int J Dermatol. 2008;47:831-832.
- Gerdsen R, Wenzel J, Uerlich M, et al. Periodic genital pruritus caused by syringoma of the vulva. Acta Obstet Gynecol Scand. 2002;81:369-370.
- Bal N, Aslan E, Kayaselcuk F, et al. Vulvar syringoma aggravated by pregnancy. Pathol Oncol Res. 2003;9:196-197.
- Turan C, Ugur M, Kutluay L, et al. Vulvar syringoma exacerbated during pregnancy. Eur J Obstet Gynecol Reprod Biol. 1996;64:141-142.
- Wallace ML, Smoller BR. Progesterone receptor positivity supports hormonal control of syringomas. J Cutan Pathol. 1995;22:442-445.
- Yorganci A, Kale A, Dunder I, et al. Vulvar syringoma showing progesterone receptor positivity. BJOG. 2000;107:292-294.
- Draznin M. Hereditary syringomas: a case report. Dermatol Online J. 2004;10:19.
- Goldstein DJ, Barr RJ, Santa Cruz DJ. Microcystic adnexal carcinoma: a distinct clinicopathologic entity. Cancer. 1982;50:566-572.
- Hamsch C, Hartschuh W. Microcystic adnexal carcinoma - aggressive infiltrative tumor often with innocent clinical appearance. J Dtsch Dermatol Ges. 2010;8:275-278.
- Henner MS, Shapiro PE, Ritter JH, et al. Solitary syringoma. report of five cases and clinicopathologic comparison with microcystic adnexal carcinoma of the skin. Am J Dermatopathol. 1995;17:465-470.
- Suwattee P, McClelland MC, Huiras EE, et al. Plaque-type syringoma: two cases misdiagnosed as microcystic adnexal carcinoma. J Cutan Pathol. 2008;35:570-574.
- Iwao F, Onozuka T, Kawashima T. Vulval syringoma successfully treated with tranilast. Br J Dermatol. 2005;153:1228-1230.
- Garman M, Metry D. Vulvar syringomas in a 9-year-old child with review of the literature. Pediatr Dermatol. 2006;23:369-372.
Practice Points
- Ensure adequate depth of biopsy to assist in the histologic diagnosis of syringoma vs microcystic adnexal carcinoma.
- Vulvar syringomas also may contribute to notable pruritus and ultimately be the underlying etiology for secondary skin changes leading to a lichen simplex chronicus–like phenotype.

Sweet Syndrome With Marked Eosinophilic Infiltrate
To the Editor:
Sweet syndrome (SS), also known as acute febrile neutrophilic dermatosis, is an uncommon inflammatory skin disorder characterized by sudden onset of fever, leukocytosis, neutrophilia, and tender erythematous papules or plaques or both. Skin biopsy usually reveals extensive infiltration of neutrophils into the epidermis and dermis.1-3 Although rare, cases of eosinophil-rich SS have been reported in patients with drug-induced and malignancy-associated SS.4,5 We report a case of a patient with classical SS with dermal eosinophilic infiltration.
An 80-year-old Hispanic man presented with abrupt onset of a rash on the posterior scalp, left ear, back, and hands of 5 days’ duration. The lesions were painful and had progressed to the point of impairing hand grip. The patient’s medical history included a reported common cold the week prior, hyperlipidemia, and hypertension, for which he took metoprolol, simvastatin, aspirin, and clopidogrel. He denied oral lesions and medication changes. He was afebrile and did not experience dietary changes, weight loss, or fatigue. He recently returned from travel to the Dominican Republic.
Physical examination revealed tender, well demarcated, pink to violaceous, pseudovesicular papules and plaques on the palms and dorsal hands (Figure 1), the posterior scalp, left ear, proximal left arm, and back. Pink, juicy, targetoid papules were also found on the scalp, back, and left arm. There was no evidence of lymphadenopathy. Laboratory test results revealed an elevated white blood cell count (11,500/µL [reference range, 3800-10,800/µL]), absolute neutrophil count (8073/µL [reference range, 1500–7800/µL]), and eosinophil count (610/µL [reference range, 15–500/µL]). These results indicated leukocytosis with neutrophilia and mild eosinophilia. The patient also was anemic (hemoglobin, 11.5 g/dL [reference range, 13.2–17.1 g/dL]; hematocrit, 35.1% [reference range, 38.5%–50%]). Urine testing revealed altered renal function (serum creatinine, 2.42 mg/dL [reference range, 0.7–1.1 mg/dL]; blood urea nitrogen, 34 mg/dL [reference range, 7–25 mg/dL]; glomerular filtration rate, 4 mL/min/1.73 m2 (reference range, ≥60 mL/min/1.73 m2]), suggesting stage 4 chronic kidney disease. Urinalysis showed mild hematuria and proteinuria.
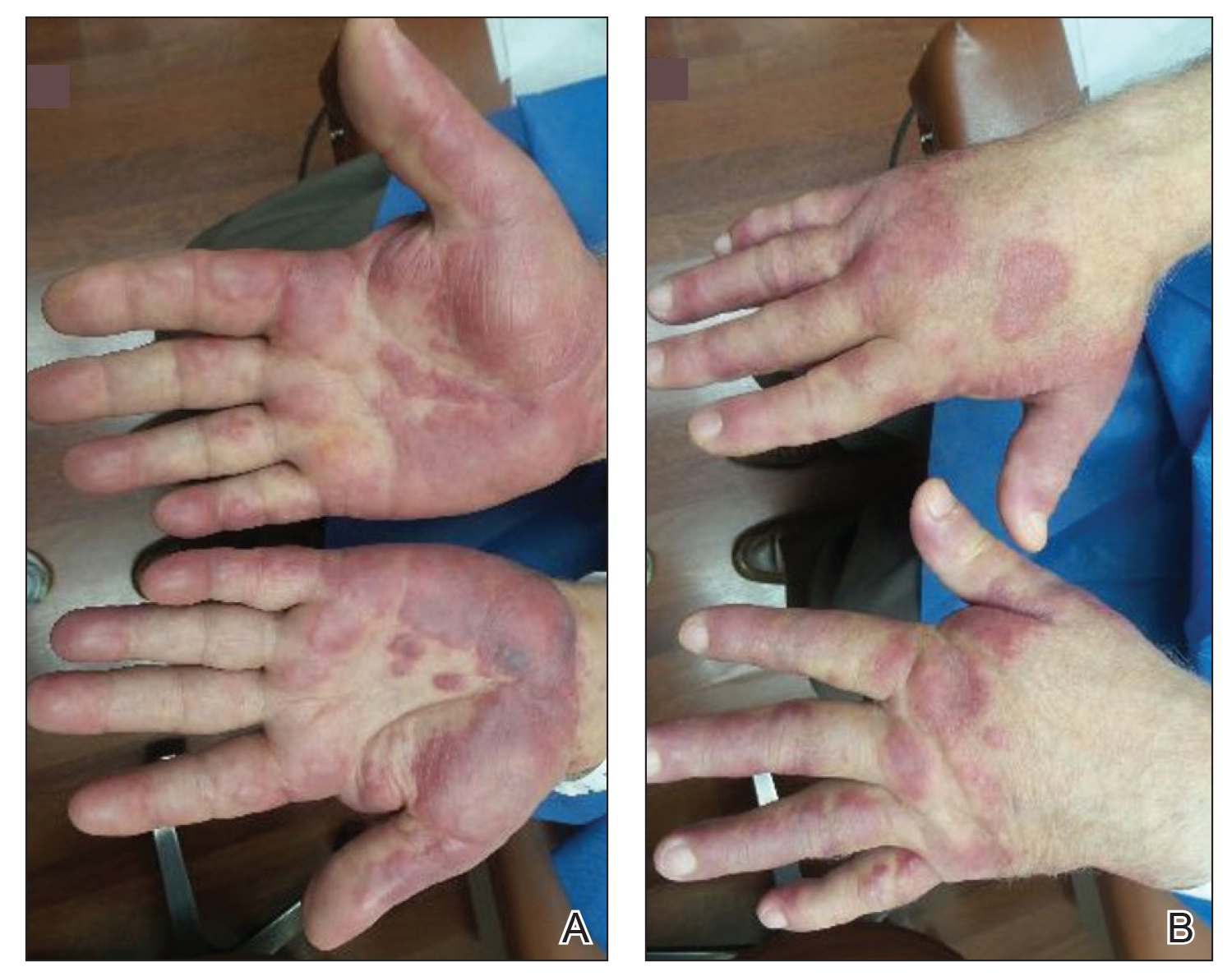
Histopathology of biopsies taken from plaques on the left arm and lower back revealed a dense neutrophilic infiltrate with numerous scattered eosinophils in the dermis. Some neutrophils were intact; others were fragmented without evidence of vasculitis. A subtle subepidermal edema also was noted (Figure 2). A diagnosis of SS was made.
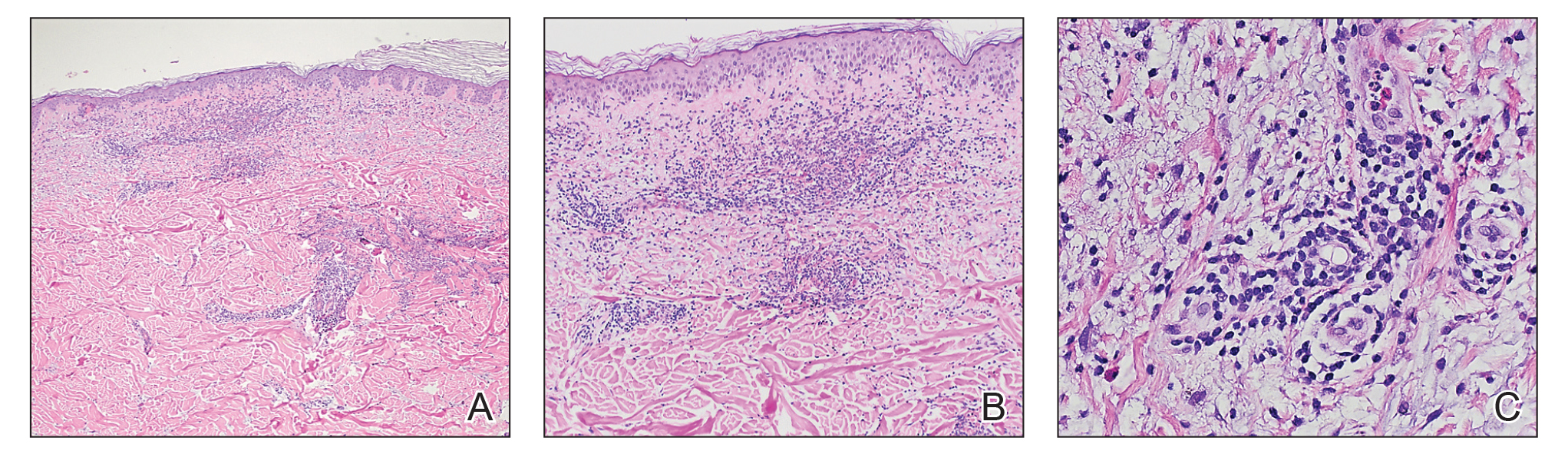
Initial treatment included prednisone (40 mg daily, tapered by 5 mg every 3 days) and erythromycin (500 mg 4 times daily) for 7 days because of suspected Mycoplasma infection. The rash resolved in 1 week. No recurrence was noted during 4 months of follow-up. The white blood cell count returned to within reference range (8400/µL), ruling out the possibility of a smoldering myeloid process.
Acute febrile neutrophilic dermatosis was first described in a case series of 8 women by Sweet6 in 1964. Patients typically present first with fever, which can precede cutaneous symptoms for days or weeks. Skin lesions generally are asymmetric and located on the face, neck, and upper extremities. Lesions can be described as painful, purple to red papules, plaques, or nodules. Sweet syndrome can present as 3 subtypes based on cause7: (1) classical SS, also known as idiopathic SS, can be preceded by an upper respiratory tract or gastrointestinal tract infection or vaccination, or can be pregnancy associated2; (2) drug-induced SS usually follows use of granulocyte colony-stimulating factor, or other causative drugs including trimethoprim-sulfamethoxazole, nitrofurantoin, quinolones, oral contraceptives, furosemide, hydralazine, diazepam, clozapine, abacavir, imatinib, bortezomib, azathioprine, and celecoxib2,3,8; and (3) malignancy-associated SS can occur as a paraneoplastic syndrome and generally is associated with hematologic malignancy or a solid tumor.1,9
In our patient, the observed clinical and histological findings were consistent with a diagnosis of SS,2,10 specifically tender erythematous plaques of sudden onset, fast response to systemic corticosteroid therapy, a dermal neutrophilic infiltrate without evidence of leukocytoclastic vasculitis, and leukocytosis greater than 8000/µL with more than 70% neutrophils. He also exhibited targetoid lesions, which have been reported in 7% to 12% of SS patients.10,11
The predominant cells involved in the dermis of SS lesions are mature neutrophils; however, eosinophils have been observed in small numbers within dermal infiltrates in skin lesions of patients with either classical SS or drug-induced dermatosis.2 In 2 studies of cases of SS (N=73 and N=31), eosinophils were reported in 35% and 41% of skin biopsies, respectively.4,5 Nevertheless, cases with dense eosinophilic infiltrates are rare. Furthermore, Masuda et al12 reported a case of eosinophil-rich SS in a 29-year-old woman after treatment of an upper respiratory tract infection with an antibiotic, and Soon et al13 described an eosinophil-rich case of SS in the setting of new-onset enteropathy-associated T-cell lymphoma.
Our patient was considered to have classical SS because he had an episode of an upper respiratory tract infection 1 week prior to onset of clinical manifestations. The histologic finding of numerous eosinophils in our case was unusual for idiopathic SS. This finding might suggest a drug hypersensitivity reaction, but the lack of any change in the patient’s long-term medication list and the lack of any other episodes made a diagnosis of drug-induced SS less likely in our patient.
Eosinophilic dermatosis of hematologic malignancy is a rare cutaneous condition in which nodules, pruritic papules, and vesicles arise in patients with a hematologic malignancy, such as chronic lymphocytic leukemia and mantle cell lymphoma,13 in which a deep perivascular lymphocytic infiltrate and numerous eosinophils are observed. Malignancy was ruled out in our patient because of the lack of characteristic abnormalities in blood testing, the fast response to corticosteroid therapy, and the lack of recurrence posttreatment or additional systemic concerns.
The typical pathology findings of SS consist of mature neutrophils found in the dermis without evidence of leukocytoclastic vasculitis. Eosinophil-rich infiltration, however rare, has been reported in SS. This report highlights a case of classical SS with a particularly dense eosinophilic infiltrate, which could be mistaken for other eosinophilic dermatoses. Dermatologists should be aware of the possibility of marked eosinophilic infiltration in all subtypes of this disorder.
- Herbert-Cohen D, Jour G, Saul T. Sweet’s syndrome. J Emerg Med. 2015;49:e95-e97.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Villarreal-Villarreal CD, Ocampo-Candiani J, Villarreal-Martínez A. Sweet syndrome: a review and update. Actas Dermosifiliogr. 2016;107:369-378.
- Rochael MC, Pantaleão L, Vilar EA, et al. Sweet’s syndrome: study of 73 cases, emphasizing histopathological findings. An Bras Dermatol. 2011;86:702-707.
- Ratzinger G, Burgdorf W, Zelger BG, et al. Acute febrile neutrophilic dermatosis: a histopathologic study of 31 cases with review of literature. Am J Dermatopathol. 2007;29:125-133.
- Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349-356.
- Cohen PR, Kurzrock R. Sweet’s syndrome revisited: a review of disease concepts. Int J Dermatol. 2003;42:761-778.
- Polimeni G, Cardillo R, Garaffo E, et al. Allopurinol-induced Sweet’s syndrome. Int J Immunopathol Pharmacol. 2016;29:329-332.
- Paydas S. Sweet’s syndrome: a revisit for hematologists and oncologists. Crit Rev Oncol Hematol. 2013;86:85-95.
- Amouri M, Masmoudi A, Ammar M, et al. Sweet’s syndrome: a retrospective study of 90 cases from a tertiary care center. Int J Dermatol. 2016;55:1033-1039.
- Marcoval J, Martín-Callizo C, Valentí-Medina F, et al. Sweet syndrome: long-term follow-up of 138 patients. Clin Exp Dermatol. 2016;41:741-746.
- Masuda T, Abe Y, Arata J, et al. Acute febrile neutrophilic dermatosis (Sweet’s syndrome) associated with extreme infiltration of eosinophils. J Dermatol. 1994;21:341-346.
- Soon CW, Kirsch IR, Connolly AJ, et al. Eosinophil-rich acute febrile neutrophilic dermatosis in a patient with enteropathy-associated T-cell lymphoma, type 1. Am J Dermatopathol. 2016;38:704-708.
To the Editor:
Sweet syndrome (SS), also known as acute febrile neutrophilic dermatosis, is an uncommon inflammatory skin disorder characterized by sudden onset of fever, leukocytosis, neutrophilia, and tender erythematous papules or plaques or both. Skin biopsy usually reveals extensive infiltration of neutrophils into the epidermis and dermis.1-3 Although rare, cases of eosinophil-rich SS have been reported in patients with drug-induced and malignancy-associated SS.4,5 We report a case of a patient with classical SS with dermal eosinophilic infiltration.
An 80-year-old Hispanic man presented with abrupt onset of a rash on the posterior scalp, left ear, back, and hands of 5 days’ duration. The lesions were painful and had progressed to the point of impairing hand grip. The patient’s medical history included a reported common cold the week prior, hyperlipidemia, and hypertension, for which he took metoprolol, simvastatin, aspirin, and clopidogrel. He denied oral lesions and medication changes. He was afebrile and did not experience dietary changes, weight loss, or fatigue. He recently returned from travel to the Dominican Republic.
Physical examination revealed tender, well demarcated, pink to violaceous, pseudovesicular papules and plaques on the palms and dorsal hands (Figure 1), the posterior scalp, left ear, proximal left arm, and back. Pink, juicy, targetoid papules were also found on the scalp, back, and left arm. There was no evidence of lymphadenopathy. Laboratory test results revealed an elevated white blood cell count (11,500/µL [reference range, 3800-10,800/µL]), absolute neutrophil count (8073/µL [reference range, 1500–7800/µL]), and eosinophil count (610/µL [reference range, 15–500/µL]). These results indicated leukocytosis with neutrophilia and mild eosinophilia. The patient also was anemic (hemoglobin, 11.5 g/dL [reference range, 13.2–17.1 g/dL]; hematocrit, 35.1% [reference range, 38.5%–50%]). Urine testing revealed altered renal function (serum creatinine, 2.42 mg/dL [reference range, 0.7–1.1 mg/dL]; blood urea nitrogen, 34 mg/dL [reference range, 7–25 mg/dL]; glomerular filtration rate, 4 mL/min/1.73 m2 (reference range, ≥60 mL/min/1.73 m2]), suggesting stage 4 chronic kidney disease. Urinalysis showed mild hematuria and proteinuria.
Histopathology of biopsies taken from plaques on the left arm and lower back revealed a dense neutrophilic infiltrate with numerous scattered eosinophils in the dermis. Some neutrophils were intact; others were fragmented without evidence of vasculitis. A subtle subepidermal edema also was noted (Figure 2). A diagnosis of SS was made.
Initial treatment included prednisone (40 mg daily, tapered by 5 mg every 3 days) and erythromycin (500 mg 4 times daily) for 7 days because of suspected Mycoplasma infection. The rash resolved in 1 week. No recurrence was noted during 4 months of follow-up. The white blood cell count returned to within reference range (8400/µL), ruling out the possibility of a smoldering myeloid process.
Acute febrile neutrophilic dermatosis was first described in a case series of 8 women by Sweet6 in 1964. Patients typically present first with fever, which can precede cutaneous symptoms for days or weeks. Skin lesions generally are asymmetric and located on the face, neck, and upper extremities. Lesions can be described as painful, purple to red papules, plaques, or nodules. Sweet syndrome can present as 3 subtypes based on cause7: (1) classical SS, also known as idiopathic SS, can be preceded by an upper respiratory tract or gastrointestinal tract infection or vaccination, or can be pregnancy associated2; (2) drug-induced SS usually follows use of granulocyte colony-stimulating factor, or other causative drugs including trimethoprim-sulfamethoxazole, nitrofurantoin, quinolones, oral contraceptives, furosemide, hydralazine, diazepam, clozapine, abacavir, imatinib, bortezomib, azathioprine, and celecoxib2,3,8; and (3) malignancy-associated SS can occur as a paraneoplastic syndrome and generally is associated with hematologic malignancy or a solid tumor.1,9
In our patient, the observed clinical and histological findings were consistent with a diagnosis of SS,2,10 specifically tender erythematous plaques of sudden onset, fast response to systemic corticosteroid therapy, a dermal neutrophilic infiltrate without evidence of leukocytoclastic vasculitis, and leukocytosis greater than 8000/µL with more than 70% neutrophils. He also exhibited targetoid lesions, which have been reported in 7% to 12% of SS patients.10,11
The predominant cells involved in the dermis of SS lesions are mature neutrophils; however, eosinophils have been observed in small numbers within dermal infiltrates in skin lesions of patients with either classical SS or drug-induced dermatosis.2 In 2 studies of cases of SS (N=73 and N=31), eosinophils were reported in 35% and 41% of skin biopsies, respectively.4,5 Nevertheless, cases with dense eosinophilic infiltrates are rare. Furthermore, Masuda et al12 reported a case of eosinophil-rich SS in a 29-year-old woman after treatment of an upper respiratory tract infection with an antibiotic, and Soon et al13 described an eosinophil-rich case of SS in the setting of new-onset enteropathy-associated T-cell lymphoma.
Our patient was considered to have classical SS because he had an episode of an upper respiratory tract infection 1 week prior to onset of clinical manifestations. The histologic finding of numerous eosinophils in our case was unusual for idiopathic SS. This finding might suggest a drug hypersensitivity reaction, but the lack of any change in the patient’s long-term medication list and the lack of any other episodes made a diagnosis of drug-induced SS less likely in our patient.
Eosinophilic dermatosis of hematologic malignancy is a rare cutaneous condition in which nodules, pruritic papules, and vesicles arise in patients with a hematologic malignancy, such as chronic lymphocytic leukemia and mantle cell lymphoma,13 in which a deep perivascular lymphocytic infiltrate and numerous eosinophils are observed. Malignancy was ruled out in our patient because of the lack of characteristic abnormalities in blood testing, the fast response to corticosteroid therapy, and the lack of recurrence posttreatment or additional systemic concerns.
The typical pathology findings of SS consist of mature neutrophils found in the dermis without evidence of leukocytoclastic vasculitis. Eosinophil-rich infiltration, however rare, has been reported in SS. This report highlights a case of classical SS with a particularly dense eosinophilic infiltrate, which could be mistaken for other eosinophilic dermatoses. Dermatologists should be aware of the possibility of marked eosinophilic infiltration in all subtypes of this disorder.
To the Editor:
Sweet syndrome (SS), also known as acute febrile neutrophilic dermatosis, is an uncommon inflammatory skin disorder characterized by sudden onset of fever, leukocytosis, neutrophilia, and tender erythematous papules or plaques or both. Skin biopsy usually reveals extensive infiltration of neutrophils into the epidermis and dermis.1-3 Although rare, cases of eosinophil-rich SS have been reported in patients with drug-induced and malignancy-associated SS.4,5 We report a case of a patient with classical SS with dermal eosinophilic infiltration.
An 80-year-old Hispanic man presented with abrupt onset of a rash on the posterior scalp, left ear, back, and hands of 5 days’ duration. The lesions were painful and had progressed to the point of impairing hand grip. The patient’s medical history included a reported common cold the week prior, hyperlipidemia, and hypertension, for which he took metoprolol, simvastatin, aspirin, and clopidogrel. He denied oral lesions and medication changes. He was afebrile and did not experience dietary changes, weight loss, or fatigue. He recently returned from travel to the Dominican Republic.
Physical examination revealed tender, well demarcated, pink to violaceous, pseudovesicular papules and plaques on the palms and dorsal hands (Figure 1), the posterior scalp, left ear, proximal left arm, and back. Pink, juicy, targetoid papules were also found on the scalp, back, and left arm. There was no evidence of lymphadenopathy. Laboratory test results revealed an elevated white blood cell count (11,500/µL [reference range, 3800-10,800/µL]), absolute neutrophil count (8073/µL [reference range, 1500–7800/µL]), and eosinophil count (610/µL [reference range, 15–500/µL]). These results indicated leukocytosis with neutrophilia and mild eosinophilia. The patient also was anemic (hemoglobin, 11.5 g/dL [reference range, 13.2–17.1 g/dL]; hematocrit, 35.1% [reference range, 38.5%–50%]). Urine testing revealed altered renal function (serum creatinine, 2.42 mg/dL [reference range, 0.7–1.1 mg/dL]; blood urea nitrogen, 34 mg/dL [reference range, 7–25 mg/dL]; glomerular filtration rate, 4 mL/min/1.73 m2 (reference range, ≥60 mL/min/1.73 m2]), suggesting stage 4 chronic kidney disease. Urinalysis showed mild hematuria and proteinuria.
Histopathology of biopsies taken from plaques on the left arm and lower back revealed a dense neutrophilic infiltrate with numerous scattered eosinophils in the dermis. Some neutrophils were intact; others were fragmented without evidence of vasculitis. A subtle subepidermal edema also was noted (Figure 2). A diagnosis of SS was made.
Initial treatment included prednisone (40 mg daily, tapered by 5 mg every 3 days) and erythromycin (500 mg 4 times daily) for 7 days because of suspected Mycoplasma infection. The rash resolved in 1 week. No recurrence was noted during 4 months of follow-up. The white blood cell count returned to within reference range (8400/µL), ruling out the possibility of a smoldering myeloid process.
Acute febrile neutrophilic dermatosis was first described in a case series of 8 women by Sweet6 in 1964. Patients typically present first with fever, which can precede cutaneous symptoms for days or weeks. Skin lesions generally are asymmetric and located on the face, neck, and upper extremities. Lesions can be described as painful, purple to red papules, plaques, or nodules. Sweet syndrome can present as 3 subtypes based on cause7: (1) classical SS, also known as idiopathic SS, can be preceded by an upper respiratory tract or gastrointestinal tract infection or vaccination, or can be pregnancy associated2; (2) drug-induced SS usually follows use of granulocyte colony-stimulating factor, or other causative drugs including trimethoprim-sulfamethoxazole, nitrofurantoin, quinolones, oral contraceptives, furosemide, hydralazine, diazepam, clozapine, abacavir, imatinib, bortezomib, azathioprine, and celecoxib2,3,8; and (3) malignancy-associated SS can occur as a paraneoplastic syndrome and generally is associated with hematologic malignancy or a solid tumor.1,9
In our patient, the observed clinical and histological findings were consistent with a diagnosis of SS,2,10 specifically tender erythematous plaques of sudden onset, fast response to systemic corticosteroid therapy, a dermal neutrophilic infiltrate without evidence of leukocytoclastic vasculitis, and leukocytosis greater than 8000/µL with more than 70% neutrophils. He also exhibited targetoid lesions, which have been reported in 7% to 12% of SS patients.10,11
The predominant cells involved in the dermis of SS lesions are mature neutrophils; however, eosinophils have been observed in small numbers within dermal infiltrates in skin lesions of patients with either classical SS or drug-induced dermatosis.2 In 2 studies of cases of SS (N=73 and N=31), eosinophils were reported in 35% and 41% of skin biopsies, respectively.4,5 Nevertheless, cases with dense eosinophilic infiltrates are rare. Furthermore, Masuda et al12 reported a case of eosinophil-rich SS in a 29-year-old woman after treatment of an upper respiratory tract infection with an antibiotic, and Soon et al13 described an eosinophil-rich case of SS in the setting of new-onset enteropathy-associated T-cell lymphoma.
Our patient was considered to have classical SS because he had an episode of an upper respiratory tract infection 1 week prior to onset of clinical manifestations. The histologic finding of numerous eosinophils in our case was unusual for idiopathic SS. This finding might suggest a drug hypersensitivity reaction, but the lack of any change in the patient’s long-term medication list and the lack of any other episodes made a diagnosis of drug-induced SS less likely in our patient.
Eosinophilic dermatosis of hematologic malignancy is a rare cutaneous condition in which nodules, pruritic papules, and vesicles arise in patients with a hematologic malignancy, such as chronic lymphocytic leukemia and mantle cell lymphoma,13 in which a deep perivascular lymphocytic infiltrate and numerous eosinophils are observed. Malignancy was ruled out in our patient because of the lack of characteristic abnormalities in blood testing, the fast response to corticosteroid therapy, and the lack of recurrence posttreatment or additional systemic concerns.
The typical pathology findings of SS consist of mature neutrophils found in the dermis without evidence of leukocytoclastic vasculitis. Eosinophil-rich infiltration, however rare, has been reported in SS. This report highlights a case of classical SS with a particularly dense eosinophilic infiltrate, which could be mistaken for other eosinophilic dermatoses. Dermatologists should be aware of the possibility of marked eosinophilic infiltration in all subtypes of this disorder.
- Herbert-Cohen D, Jour G, Saul T. Sweet’s syndrome. J Emerg Med. 2015;49:e95-e97.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Villarreal-Villarreal CD, Ocampo-Candiani J, Villarreal-Martínez A. Sweet syndrome: a review and update. Actas Dermosifiliogr. 2016;107:369-378.
- Rochael MC, Pantaleão L, Vilar EA, et al. Sweet’s syndrome: study of 73 cases, emphasizing histopathological findings. An Bras Dermatol. 2011;86:702-707.
- Ratzinger G, Burgdorf W, Zelger BG, et al. Acute febrile neutrophilic dermatosis: a histopathologic study of 31 cases with review of literature. Am J Dermatopathol. 2007;29:125-133.
- Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349-356.
- Cohen PR, Kurzrock R. Sweet’s syndrome revisited: a review of disease concepts. Int J Dermatol. 2003;42:761-778.
- Polimeni G, Cardillo R, Garaffo E, et al. Allopurinol-induced Sweet’s syndrome. Int J Immunopathol Pharmacol. 2016;29:329-332.
- Paydas S. Sweet’s syndrome: a revisit for hematologists and oncologists. Crit Rev Oncol Hematol. 2013;86:85-95.
- Amouri M, Masmoudi A, Ammar M, et al. Sweet’s syndrome: a retrospective study of 90 cases from a tertiary care center. Int J Dermatol. 2016;55:1033-1039.
- Marcoval J, Martín-Callizo C, Valentí-Medina F, et al. Sweet syndrome: long-term follow-up of 138 patients. Clin Exp Dermatol. 2016;41:741-746.
- Masuda T, Abe Y, Arata J, et al. Acute febrile neutrophilic dermatosis (Sweet’s syndrome) associated with extreme infiltration of eosinophils. J Dermatol. 1994;21:341-346.
- Soon CW, Kirsch IR, Connolly AJ, et al. Eosinophil-rich acute febrile neutrophilic dermatosis in a patient with enteropathy-associated T-cell lymphoma, type 1. Am J Dermatopathol. 2016;38:704-708.
- Herbert-Cohen D, Jour G, Saul T. Sweet’s syndrome. J Emerg Med. 2015;49:e95-e97.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Villarreal-Villarreal CD, Ocampo-Candiani J, Villarreal-Martínez A. Sweet syndrome: a review and update. Actas Dermosifiliogr. 2016;107:369-378.
- Rochael MC, Pantaleão L, Vilar EA, et al. Sweet’s syndrome: study of 73 cases, emphasizing histopathological findings. An Bras Dermatol. 2011;86:702-707.
- Ratzinger G, Burgdorf W, Zelger BG, et al. Acute febrile neutrophilic dermatosis: a histopathologic study of 31 cases with review of literature. Am J Dermatopathol. 2007;29:125-133.
- Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349-356.
- Cohen PR, Kurzrock R. Sweet’s syndrome revisited: a review of disease concepts. Int J Dermatol. 2003;42:761-778.
- Polimeni G, Cardillo R, Garaffo E, et al. Allopurinol-induced Sweet’s syndrome. Int J Immunopathol Pharmacol. 2016;29:329-332.
- Paydas S. Sweet’s syndrome: a revisit for hematologists and oncologists. Crit Rev Oncol Hematol. 2013;86:85-95.
- Amouri M, Masmoudi A, Ammar M, et al. Sweet’s syndrome: a retrospective study of 90 cases from a tertiary care center. Int J Dermatol. 2016;55:1033-1039.
- Marcoval J, Martín-Callizo C, Valentí-Medina F, et al. Sweet syndrome: long-term follow-up of 138 patients. Clin Exp Dermatol. 2016;41:741-746.
- Masuda T, Abe Y, Arata J, et al. Acute febrile neutrophilic dermatosis (Sweet’s syndrome) associated with extreme infiltration of eosinophils. J Dermatol. 1994;21:341-346.
- Soon CW, Kirsch IR, Connolly AJ, et al. Eosinophil-rich acute febrile neutrophilic dermatosis in a patient with enteropathy-associated T-cell lymphoma, type 1. Am J Dermatopathol. 2016;38:704-708.
Practice Points
- This report highlights a case of classical Sweet syndrome (SS) with a particularly dense eosinophilic infiltrate, which could be mistaken for other eosinophilic dermatoses.
- Dermatologists should be aware of the possibility of marked eosinophilic infiltration in all subtypes of SS.